
Home » Posts tagged 'manufacturing' (Page 2)
Tag Archives: manufacturing
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
ALIROCUMAB
ALIROCUMAB
http://www.ama-assn.org/resources/doc/usan/alirocumab.pdf
Immunoglobulin G1, anti-(human neural apoptosis-regulated proteinase 1) (human REGN727 heavy chain), disulfide with human REGN727 κ-chain, dimer
Immunoglobulin G1, anti-(human proprotein convertase subtilisin/kexin type 9
(EC=3.4.21.-, neural apoptosis-regulated convertase 1, proprotein convertase 9,
subtilisin/kexin-like protease PC9)); human monoclonal REGN727 des-448-
lysine(CH3-K107)-1 heavy chain (221-220′)-disulfide with human monoclonal
REGN727 light chain dimer (227-227”:230-230”)-bisdisulfide
Clinical Trials for Compound
| Number of clinical trials registered at clinicaltrials.gov | 30 |
Biological Sequence
| Description | Sequence |
| Alirocumab heavy chain | EVQLVESGGGLVQPGGSLRLSCAASGFTFNNYAMNWVRQAPGKGLDWVSTISGSGGTTNY ADSVKGRFIISRDSSKHTLYLQMNSLRAEDTAVYYCAKDSNWGNFDLWGRGTLVTVSSAS TKGPSVFPLAPSSKSTSGGTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGL YSLSSVVTVPSSSLGTQTYICNVNHKPSNTKVDKKVEPKSCDKTHTCPPCPAPELLGGPS VFLFPPKPKDTLMISRTPEVTCVVVDVSHEDPEVKFNWYVDGVEVHNAKTKPREEQYNST YRVVSVLTVLHQDWLNGKEYKCKVSNKALPAPIEKTISKAKGQPREPQVYTLPPSRDELT KNQVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTPPVLDSDGSFFLYSKLTVDKSRWQQ GNVFSCSVMHEALHNHYTQKSLSLSPG |
| Alirocumab light chain | DIVMTQSPDSLAVSLGERATINCKSSQSVLYRSNNRNFLGWYQQKPGQPPNLLIYWASTR ESGVPDRFSGSGSGTDFTLTISSLQAEDVAVYYCQQYYTTPYTFGQGTKLEIKRTVAAPS VFIFPPSDEQLKSGTASVVCLLNNFYPREAKVQWKVDNALQSGNSQESVTEQDSKDSTYS LSSTLTLSKADYEKHKVYACEVTHQGLSSPVTKSFNRGEC |
1245916-14-6 CAS
C6472H9996N1736O2032S42
Alirocumab is a human monoclonal antibody designed for the treatment of hypercholesterolemia.[1]
This drug was discovered by Regeneron Pharmaceuticals and is being co-developed by Regeron and Sanofi.
When the results from Phase II trials of Sanofi and Regeneron’s proprotein convertase subtilisin kexin 9 (PCSK9) inhibitor alirocumab were presented in March, they stunned even the company representatives working on the trials. “I’m still amazed by the reduction in low-density lipoprotein cholesterol (LDL-C) that we saw with our drug,” says Bill Sasiela, vice president of cardiovascular and metabolic research at Regeneron. The monoclonal antibody (mAb) reduced LDL-C levels by up to 73% in three mid-stage trials, irrespective of baseline LDL-C levels or background treatment, offering hope for millions of patients who can’t hit the recommended cholesterol targets with statins — the standard therapies for lowering LDL-C levels in patients with cardiovascular disease. Spurred on by these results, Sanofi and Regeneron geared up into Phase III trials of the first-in-class alirocumab (also known as REGN727 and SAR236553) over the summer, and initiated the latest and largest trial — an 18,000-patient outcomes study
It is a Proprotein convertase subtilisin/kexin type 9, (also known as PCSK9) inhibitor . Phase III trials showed a 47% reduction in LDL-C. There was a high rate of adverse events with 69% experiencing side effects (most common problem was infection).
About PCSK9 PCSK9 is known to be a determinant of circulating LDL levels, as it binds to LDL receptors resulting in their degradation so that fewer are available on liver cells to remove excess LDL-cholesterol from the blood. Moreover, traditional LDL-lowering therapies such as statins actually stimulate the production of PCSK9, which limits their own ability to lower LDL-cholesterol. Blocking the PCSK9 pathway is therefore a potentially novel mechanism for lowering LDL-cholesterol.
Alirocumab is an investigational, fully-human monoclonal antibody that targets and blocks PCSK9. It is administered via subcutaneous injection. By inhibiting PCSK9, a determinant of circulating LDL-C levels in the blood, alirocumab has been shown in pre-clinical studies to increase the number of LDL receptors on hepatocytes, thereby lowering LDL-C.
The investigational agent described above is currently under clinical development and its safety and efficacy have not been fully evaluated by any regulatory authority
References
- Statement On A Nonproprietary Name Adopted By The USAN Council – Alirocumab, American Medical Association.

PARIS and TARRYTOWN, N.Y., Oct. 16, 2013 /PRNewswire via COMTEX/ — Sanofi and Regeneron Pharmaceuticals, Inc. REGN -1.73% today announced that the Phase 3 ODYSSEY MONO trial with alirocumab, an investigational monoclonal antibody targeting PCSK9 (proprotein convertase subtilisin/kexin type 9), met its primary efficacy endpoint. The mean low-density lipoprotein-cholesterol (LDL-C, or “bad” cholesterol) reduction from baseline to week 24, the primary efficacy endpoint of the study, was significantly greater in patients randomized to alirocumab, as compared to patients randomized to ezetimibe (47.2% vs. 15.6%, p<0.0001). In the trial, which employed a dose increase (up-titration) for patients who did not achieve an LDL-C level of 70 milligrams/deciliter (mg/dL), the majority of patients remained on the initial low dose of alirocumab of 75 milligrams (mg). read at
Pipeline of selected PCSK9 inhibitors
| Drug name | Companies | Modality | Clinical phase |
|---|---|---|---|
| Alirocumab (also known as REGN727 and SAR236553) | Regeneron/Sanofi | Monoclonal antibody | III |
| AMG145 | Amgen | Monoclonal antibody | II |
| LGT209 | Novartis | Monoclonal antibody | II |
| RG7652 | Roche/Genentech | Monoclonal antibody | II |
| RN316 | Pfizer | Monoclonal antibody | II |
| BMS-962476 | Bristol-Myers Squibb | Adnectin | I |
| ALN-PCS | Alnylam | RNA interference | I |
| ISIS-405879/BMS-844421 | Isis/Bristol-Myers Squibb | Antisense | Discontinued |
| PCSK9, proprotein convertase subtilisin kexin 9. | |||

Finerenone (BAY 94-8862), BAYER’S next generation oral, non-steroidal Mineralocorticoid Receptor antagonist which blocks the deleterious effects of aldosterone
CAS Number: 1050477-31-0, UNII-DE2O63YV8R
MW: 378.4298, C21-H22-N4-O3
Finerenone (BAY 94-8862) is a next generation oral, non-steroidal Mineralocorticoid Receptor antagonist which blocks the deleterious effects of aldosterone.
Currently available steroidal MR antagonists have proven to be effective in reducing cardiovascular mortality in patients with heart failure but have significant side effects that limit their utilization.
Finerenone is currently in clinical Phase IIb development for the treatment of worsening chronic heart failure, as well as diabetic nephropathy.
The U.S. Food and Drug Administration approved Adempas (riociguat) to treat adults with two forms of pulmonary hypertension.

October 8, 2013 — The U.S. Food and Drug Administration today approved Adempas (riociguat) to treat adults with two forms of pulmonary hypertension.
Pulmonary hypertension is caused by abnormally high blood pressure in the arteries of the lungs. It makes the right side of the heart work harder than normal. In its various forms, pulmonary hypertension is a chronic, progressive, debilitating disease, often leading to death or need for lung transplantation
read all at
http://www.drugs.com/newdrugs/fda-approves-adempas-pulmonary-hypertension-3927.html
In the area of pulmonary hypertension Adempas (Riociguat) is the first member of a novel class of compounds – so-called ‘soluble guanylate cyclase (sGC) stimulators’ – being investigated as a new and specific approach to treating different types of pulmonary hypertension (PH). Adempas has the potential to overcome a number of limitations of currently approved treatments for pulmonary arterial hypertension (PAH) and addresses the unmet medical need in patients with chronic thromboembolic pulmonary hypertension (CTEPH). It was approved for the treatment of CTEPH in Canada in September 2013, making it the world’s first drug approved in this deadly disease.
Riociguat has already shown promise as a potential treatment option beyond these two PH indications. An early clinical study was conducted in PH-ILD (interstitial lung disease), a disease characterized by lung tissue scarring (fibrosis) or lung inflammation which can lead to pulmonary hypertension, and, based on positive data, the decision was taken to initiate Phase IIb studies in PH-IIP (idiopathic pulmonary fibrosis), a subgroup of PH-ILD. Moreover, scientific evidence was demonstrated in preclinical models that the activity may even go beyond vascular relaxation. To prove the hypothesis Bayer is initiating clinical studies in the indication of systemic sclerosis (SSc), an orphan chronic autoimmune disease of the connective tissue affecting several organs and associated with high morbidity and mortality. If successful, Riociguat has the potential to become the first approved treatment for this devastating disease.
synthesis
Generic Name: Riociguat
Trade Name: Adempas
Synonym: BAY 63-2521
CAS number: 625115-55-1
Chemical Name: Methyl N-[4,6-Diamino-2-[1-[(2-fluorophenyl)methyl]-1H-pyrazolo[3,4-b]pyridin-3-yl]-5-pyrimidinyl]-N-methyl-carbaminate
Mechanism of Action: soluble guanylyl cyclase (sGC) stimulator
Date of Approval: October 8, 2013(US)
Indication: Pulmonary Hypertension
Company: Bayer AG

1)J. Mittendorf.; S. Weigand.; C. Alonso-Alija.; E. Bischoff.; A. Feurer.; M. Gerisch.; A. Kern.; A. Knorr.; D. Lang.; K. Muenter.; M. Radtke.; H. Schirok.; K.-H. Schlemmer.; E. Stahl.; A. Straub.; F. Wunder.; J.-P. Stasch. Discovery of Riociguat (BAY 63-2521): A Potent, Oral Stimulator of Soluble Guanylate Cyclase for the Treatment of Pulmonary Hypertension, ChemMedChem. 2009, 4, 853-865.
2)Cristina Alonso-Alija, Bayer Ag, Erwin Bischoff, Achim Feurer, Klaus Muenter, Elke Stahl, Johannes-Peter Stasch, Stefan Weigand, Carbamate-substituted pyrazolopyridines, WO2003095451 A1
3)Franz-Josef Mais, Joachim Rehse, Winfried Joentgen, Konrad SIEGEL, Process for preparing methyl methylcarbamate and its purification for use as pharmaceutically active compound,US20110130410
4)Claudia Hirth-Dietrich, Peter Sandner, Johannes-Peter Stasch, Andreas Knorr, Degenfeld Georges Von, Michael Hahn, Markus Follmann, The use of sGC stimulators, sGC activators, alone and combinations with PDE5 inhibitors for the treatment of systemic sclerosis (SSc), WO 2011147810A1
5)Li Liang, Li Xing-zhou, Liu Ya-dan, Zheng Zhi-bing, Li Song, Synthesis of riociguat in treatment of pulmonary hypertension, Chinese Journal of Medicinal Chemistry(Zhongguo Yaowu Huaxue Zazhi), 21(2),120-125; 2011

Jens Ackerstaff, Lars BÄRFACKER, Markus Follmann, Nils Griebenow, Andreas Knorr, Volkhart Min-Jian Li, Gorden Redlich, Johannes-Peter Stasch, Stefan Weigand, Frank Wunder, Bicyclic aza heterocycles, and use thereof, WO2012028647 A1
2)Claudia Hirth-Dietrich, Peter Sandner, Johannes-Peter Stasch, Andreas Knorr, Degenfeld Georges Von, Michael Hahn, Markus Follmann, The use of sGC stimulators, sGC activators, alone and combinations with PDE5 inhibitors for the treatment of systemic sclerosis (SSc), WO 2011147810A1

Jin Li, Xiaoyu Yang, Jingwei ZHU, Minmin Yang, Xihan Wu, Method for synthesizing 1-(2-fluorobenzyl)-1H -pyrazolo[3,4-b]pyridin -3-formamidine hydrochloride, WO2013086935 A1

veerareddy Arava, Surendrareddy Gogireddy, An expeditious synthesis of riociguat, A pulmonary hypertension drug, Der Pharma Chemica, 2013, 5(4):232-239
cut paste from my earlier post

RIOCIQUAT
CAS NO 625115-55-1
Methyl N-[4,6-Diamino-2-[1-[(2-fluorophenyl)methyl]-1H-pyrazolo[3,4-b]pyridin-3-yl]-5-pyrimidinyl]-N-methyl-carbaminate
9 APRIL2013
Bayer has been boosted by the news that regulators in the USA are fast-tracking the German group’s investigational pulmonary arterial hypertension riociguat.
The US Food and Drug Administration has granted priority review to the New Drug Application for riociguat, which Bayer filed in February on both sides of the Atlantic for PAH and a related condition, inoperable chronic thromboembolic pulmonary hypertension (CTEPH). The FDA bestows a priority review on medicines that offer major advances in care or that provide a treatment where no adequate therapy exists. The agency aims to complete its assessment within eight months from the submission of the NDA, rather than the standard 12 months.
Riociguat (BAY 63-2521) is a novel drug that is currently in clinical development by Bayer. It is a stimulator of soluble guanylate cyclase (sGC). At the moment Phase III clinical trialsinvestigate the use of riociguat as a new approach to treat two forms of pulmonary hypertension (PH): chronic thromboembolic pulmonary hypertension (CTEPH) andpulmonary arterial hypertension (PAH). Riociguat constitutes the first drug of a novel class of sGC stimulators
The submissions are based on two Phase III studies and riociguat, the first member of a novel class of compounds called stimulators of soluble guanylate cyclase (sGC), met its primary endpoint in both trials, a change in exercise capacity after 12- or 16 weeks respectively. The drug was generally well tolerated, with a good safety profile.

If approved, riociguat would be going up against Actelion’s Tracleer (bosentan) and Gilead Sciences/GlaxoSmithKline’s Letairis/Volibris (ambrisentan). Actelion, which has dominated the PAH market, has already filed its follow-up to Tracleer, Opsumit (macitentan).

Epratuzumab
Epratuzumab
Epratuzumab is a humanised anti-CD22 monoclonal antibody under investigation (clinical development phase III) for its efficacy in SLE. CD22 is a B cell specific surface protein that is considered to be involved in B cell function.
| Expected indication | Systemic lupus erythematosus |
| R&D stage | Phase 3 ongoing (started in December 2010) |
| Next milestone | Phase 3 results (H1 2014) |
| Quick facts |
|
Epratuzumab is a humanized monoclonal antibody. Potential uses may be found inoncology and in treatment of inflammatory autoimmune disorders, such as lupus (SLE).[1][2] The manufacturers in August 2009 announced success in early trials against SLE.[3]
Epratuzumab binds to the glycoprotein CD22 of mature and malignant B-cells.
- Epratuzumab, a humanized monoclonal antibody targeting CD22: characterization of in vitro properties Clinical Cancer Research Vol. 9, September 1, 2003 free full text
- Dose-Fractionated Radioimmunotherapy in Non-Hodgkin’s Lymphoma Using DOTA-Conjugated, 90Y-Radiolabeled, Humanized Anti-CD22 Monoclonal Antibody, Epratuzumab Clinical Cancer Research Vol. 11, July 15, 2005 free full text
- Reuters: UCB and Immunomedics Announce Positive Results for Epratuzumab Phase IIb Study in Systemic Lupus Erythematosus (SLE)
Epratuzumab is a humanized IgG1 antibody that acts as an antagonist of the CD22 receptor present on B cells. UCB is currently enrolling patients for the 2 Phase III trials, EMBODY-1 and EMBODY-2. The primary objective of both studies is to measure the percent of subjects meeting treatment response criteria at week 48 among those patients with moderate to severe SLE. Epratuzumab is dosed at either 600 mg per week or 1200 mg every other week administered over four 12-week treatment cycles.
The cumulative dose for both treatment arms is 2400 mg for each of the 4-week dosing periods. The estimated primary completion date is January 2014 for both EMBODY-1 and EMBODY-2. –
UCB pipeline. UCB Web site. www.ucb.com/rd/pipeline/new-development/epratuzumab. Published July 10, 2010. Accessed June 18, 2011
Brussels (Belgium), June 13th 2013, 0700 CEST – UCB today announced new data from an open-label extension (SL0008) of the EMBLEM™ phase 2b study evaluating the long-term effects of epratuzumab treatment in adult patients with moderate-to-severe systemic lupus erythematosus (SLE). The primary outcome of the open-label extension was to assess the safety of epratuzumab in patients with SLE.4
Relative to the 12 week, double-blind, placebo-controlled EMBLEM™ study, data from the open-label, long-term extension identified no new safety or tolerability signals.1 In addition, relative to EMBLEM™ baseline values, secondary outcome data indicated that the efficacy of epratuzumab as measured by reduction in disease activity was maintained over two years.2 Secondary outcome data also indicated that relative to EMBLEM™ baseline values, treatment over two years with epratuzumab was associated with decreases in corticosteroid use in patients receiving >7.5 mg/day.1 These data were presented this week at the European League Against Rheumatism 2013 Congress in Madrid, Spain.
Epratuzumab, licensed from Immunomedics Inc. (NASDAQ: IMMU), is an investigational medicine and the first CD-22/B-Cell receptor (BCR) targeted monoclonal antibody to be evaluated in clinical studies for the treatment of SLE. Also known as lupus, SLE is a complex, systemic autoimmune disease that affects many different organ systems, including the skin, joints, lungs, kidneys and blood.3,5
“In EMBLEM™, a dose-ranging, phase 2b study, reduction in disease activity was observed in patients treated with epratuzumab,” said Professor Daniel J Wallace MD, Clinical Professor of Medicine, Cedars-Sinai Medical Center, California, US. “This double-blind study had a relatively short 12-week, placebo-controlled, treatment period and it was important to accumulate long-term data on epratuzumab in the treatment of SLE. The phase 2b extension study adds new two year open-label data on epratuzumab to that already available from the 12-week, randomized, controlled study.”
EMBLEM™ was designed to identify a suitable dosing regimen for epratuzumab.6 A total of 227 patients with moderate-to-severe SLE received either: placebo, epratuzumab cumulative dose of 200 mg (100 mg every other week), 800 mg (400 mg every other week), 2400 mg (600 mg weekly), 2400 mg (1200 mg every other week) or 3600 mg (1800 mg every other week).3,6 In the open-label extension 203 patients from any arm of the EMBELM™ study received 1200 mg epratuzumab at weeks 0 and 2 of 12-week cycles.1,2,7
Data on epratuzumab presented at EULAR 2013
Evaluation of the safety profile of long-term epratuzumab treatment in patients with moderate-to-severe SLE1
Safety variables were primary outcome measures in SL0008 and included duration of exposure, adverse events, infusion reactions and infections.
Exposure to epratuzumab was a median 845 days over a median 10 treatment cycles. Adverse events (AEs) caused discontinuation in 29 (14.3%) patients. The most common serious AEs were SLE flare (3.4%), lupus nephritis (2%) and symptomatic cholelithiasis (1.5%). The most common infections/infestations were urinary tract infection (24.6%) and upper respiratory tract infection (23.2%). There were no opportunistic infections and no patterns of specific serious or severe infections.
Evaluation of long-term efficacy of epratuzumab as measured by reduction in disease activity in patients with moderate-to-severe SLE2
Secondary outcome measures in SL0008 included efficacy as measured by reduction in disease activity, and assessed by: British Isles Lupus Assessment Group (BILAG) improvement, SLE disease activity index (SLEDAI) score, Physician Global Assessment (PGA) score and combined treatment response defined as BILAG improvement without worsening, no SLEDAI worsening and no PGA worsening, relative to EMBLEM™ baseline.
The median BILAG total score was 25.0 at EMBLEM™ baseline and 9.0 at week 108. The score was 14.0 at SL0008 screening. Median SLEDAI score was 12.0 at EMBLEM™ baseline and 4.0 at week 108. The score was 10.0 at SL0008 screening. The median PGA score was 50.0 at EMBLEM™ baseline and 17.5 at week 108 with a score of 31.0 at SL0008 screening.
The proportion of patients achieving the combined treatment response was 32.5% at SL0008 screening (n=203) and 60.3% at week 108 (n=116).
Effect of corticosteroid use of long-term epratuzumab treatment in patients with moderate-to-severe SLE1
Corticosteroid doses were monitored throughout SL0008 and was a secondary outcome measure.
Median corticosteroid dose at EMBLEM™ baseline and SL0008 screening was 10.0 mg/day. At week 116, this was 5 mg/day (n=112). Data indicated that treatment over two years with epratuzumab was associated with decreases in corticosteroid use in patients receiving >7.5 mg/day with a corresponding increase in the proportion of patients receiving lower doses or no longer receiving corticosteroids.
The proportion of patients requiring 7.5-20 mg/day and >20 mg/day decreased (49.8% and 10.8% at baseline and 33.9% and 8.0% respectively, at week 116) and the proportion of patients receiving >0–7.5mg/day or no longer receiving corticosteroids increased (33.5% and 5.9% at baseline and 45.5% and 12.5% respectively, at week 116).
Aerial Biopharma announce positive Phase II results for narcolepsy drug
ADX-N05, ARL-N05, SKL-N05
Aerial Biopharma announce positive Phase II results for narcolepsy drug
A new drug to treat excessive daytime sleepiness associated with narcolepsy has shown positive results from a phase 2b clinical trial, US-based Aerial Biopharma has announced this week.
read all at
Secukinumab
Secukinumab is an anti-IL17A drug being investigated for a number of inflammatory conditions. For plaque psoriasis, Novartis is planning to evaluate a dose of 150 mg subcutaneously compared with placebo.
The primary outcome measure of the planned Phase III trial named ERASURE is to evaluate the efficacy in patients with moderate to severe chronic plaque-type psoriasis. Novartis is also planning to evaluate secukinumab dosed at either 150 or 300 mg versus Enbrel (enterecept) 50 mg in a Phase III trial entitled FIXTURE.
Final data collection for the primary outcome measures in both ERASURE and FIXTURE are anticipated in March 2013.
Secukinumab is a human monoclonal antibody designed for the treatments of uveitis,rheumatoid arthritis, and psoriasis. It targets member A from the cytokine family ofinterleukin 17.[1][2]
Secukinumab was developed by Novartis Pharma AG and has completed Phase II clinical trials for plaque psoriasis in 2011.[3]
CAS registry numbers
- 875356-43-7 (heavy chain)
- 875356-44-8 (light chain)
- ^ “Statement On A Nonproprietary Name Adopted By The USAN Council: Secukinumab”. American Medical Association.
- ^ Hueber, W.; Patel, D. D.; Dryja, T.; Wright, A. M.; Koroleva, I.; Bruin, G.; Antoni, C.; Draelos, Z.; Gold, M. H.; Psoriasis Study, P.; Durez, P. P.; Tak, J. J.; Gomez-Reino, C. S.; Rheumatoid Arthritis Study, R. Y.; Foster, C. M.; Kim, N. S.; Samson, D. S.; Falk, D.; Chu, Q. D.; Callanan, K.; Nguyen, A.; Uveitis Study, F.; Rose, K.; Haider, A.; Di Padova, F. (2010). “Effects of AIN457, a Fully Human Antibody to Interleukin-17A, on Psoriasis, Rheumatoid Arthritis, and Uveitis”. Science Translational Medicine 2 (52): 52ra72.doi:10.1126/scitranslmed.3001107. PMID 20926833. edit
- ^ Papp K.A. et al. ‘Secukinumab efficacy and safety preliminary results from a phase II subcutaneous dose-ranging study in the treatment of moderate-to-severe plaque psoriasis.’ Presented at: 20th Congress of the European Academy of Dermatology and Venereology; 20-24 October, 2011; Lisbon, Portugal.

Dimeric Thymosin beta-4……..accelerates the rate of wound healing

Structure of a Longitudinal Actin Dimer Assembled by Tandem W Domains
Thymosin beta 4 (Tβ4) is a peptide with 43 amino acids that is critical for repair and remodeling tissues on the skin, eye, heart, and neural system following injury
Thymosin beta-4 is a protein that in humans is encoded by the TMSB4X gene.
The protein consists (in humans) of 43 amino acids (msdkpdmaei ekfdksklkk tetqeknplp sketieqekq ages) molWt 4921

NMR structure of a β-thymosin. Both thymosin α1 and β-thymosins areintrinsically unstructured proteins, i.e. they lack a stable fold when free in aqueous solution. This structure, mostly alpha helix, was artificially stabilised by an organic solvent. The thymosin illustrated, originally named β9 is the cow orthologue of human β10
It has been studied in a number of clinical trials.
The thymosin beta-4 peptide, if used after a heart attack, might reactivate cardiacprogenitor cells to repair damaged heart tissue.
Doping in Sports
Thymosin beta-4 was allegedly used by some players in various Australian football codes and is under investigation by the Australian Sports Anti-Doping Authority for anti-doping violations (Feb/Mar 2013):
https://theconversation.edu.au/cronulla-sharks-and-thymosin-beta-4-is-it-doping-12694
FDA, EMA Accept Omeros Ophthalmology Product NDA
OMS302
US and European Regulators Accept for Review OMS302 Marketing Applications
— OMS302 Remains on Track for Planned 2014 Commercial Launch —
SEATTLE, Oct. 2, 2013 /PRNewswire/ — Omeros Corporation (NASDAQ: OMER) announced today that the New Drug Application (NDA) for its ophthalmology product, OMS302, has been confirmed for filing by the U.S. Food and Drug Administration (FDA), which means that the application, submitted in July of this year, is sufficiently complete to permit a substantive review. The company also announced that its Marketing Authorization Application (MAA) for OMS302, submitted last month, has been validated by the European Medicines Agency (EMA). Validation of the MAA confirms that the submission package is administratively complete and is ready for formal review by Europe’s Committee for Medicinal Products for Human Use (CHMP).
read all at
http://www.pharmalive.com/fda-ema-accept-omeros-opthamology-product-nda
Isavuconazole – Basilea reports positive results from study

This post is updated in sept 2015……..
Isavuconazole (BAL4815; trade name Cresemba) is a triazole antifungal drug. Its prodrug, isavuconazonium sulfate (BAL8557), was granted approval by the U.S. Food and Drug Administration (FDA) on March 6, 2015[1]
During its Phase III drug trials, Astellas partnered with Basilea Pharmaceutica, the developer of the drug, for rights to co-development and marketing of isavuconazole. [2]
On May 28, 2013, Basilea Pharmaceutica announced it had been granted orphan drug status by the FDA for treatment of aspergillosis.[3] Since then, it has also been granted orphan drug status for the treatment of invasive candidiasis.[4]

Isavuconazonium sulfate (BAL8557)—a prodrug of isavuconazole.
CLINICAL TRIALS…LINK
PATENTS
|
6-27-2012
|
Process for the manufacture of enantiomerically pure antifungal azoles as ravuconazole and isavuconazole
|
|
|
11-18-2011
|
Antifungal Composition
|
|
|
9-29-2010
|
PROCESS FOR PREPARATION OF WATER-SOLUBLE AZOLE PRODRUGS
|
|
|
12-3-2008
|
N-substituted carbamoyloxyalkyl-azolium derivatives
|
|
|
3-14-2007
|
N-phenyl substituted carbamoyloxyalkyl-azolium derivatives
|
|
|
11-3-2004
|
N-substituted carbamoyloxyalkyl-azolium derivatives
|
|
|
10-10-2001
|
Azoles for treatment of fungal infections
|
Several azoles are currently used for systemic mycoses. However, none of them fulfills the needs of clinical requirement in full extent, particularly with regard 0 to broad antifungal spectrum including aspergillus fumigatus, less drug-drug interaction, and appropriate plasma half-life for once a day treatment. Other clinical requirements which are not fulfilled by the azoles currently used, are efficacy against major systemic mycoses including disseminated aspergillosis, safety, and oral or parenteral formulations. Particularly, demand of a 5 parenteral administration of the azoles is increasing for the treatment of serious systemic mycoses. Most of the azoles on the market as well as under development are highly lipophilic molecules that make the parenteral formulation difficult.

Isavuconazole [(2R,3R)-3-[4-(4-cyanophenyl)thiazol-2-yl)]-1-(1H-1,2,4-triazol-1-yl)-2-(2,5-difluorophenyl)-butan-2-ol; formula I, R1 and R3 represent fluorine and R2 represents hydrogen] as well as Ravuconazole [(2R,3R)-3-[4-(4-cyanophenyl)thiazol-2-yl)]-1-(1H-1,2,4-triazol-1-yl)-2-(2,4-difluorophenyl)-butan-2-ol; formula I, R1 and R2 represent fluorine and R3 represents hydrogen] are useful antifungal drugs as reported in U.S. Pat. No. 5,648,372 from Feb. 1, 1995 or in U.S. Pat. No. 5,792,781 from Sep. 18, 1996 or in U.S. Pat. No. 6,300,353 from Oct. 9, 2001 (WO99/45008).
Since compounds of general formula I contain two adjacent chiral centers, synthesis of enantiomerically pure compound is complex and until now, all patented syntheses are not efficient enough and do not allow cost effective manufacturing on a technical scale:
Thus, U.S. Pat. Nos. 5,648,372 or 5,792,781 describe enantioselective synthesis of compounds of formula I (specifically Ravuconazole) from chiral 3-hydroxy-2-methyl propionic acid in 12 steps with overall yield lower than 5%. In another approach including 13 steps and low overall yield, (R)-lactic acid was used as the starting material (Chem. Pharm. Bull. 46(4), 623 (1998) and ibid. 46(7), 1125 (1998)).
Because both starting materials contain only one chiral center, in a number of inefficient steps, the second, adjacent chiral center has to be created by a diastereoselective reaction (using either Corey or Sharpless epoxidation method) which is not sufficiently selective leading mostly to a mixture of two diastereomers which have to be separated.
The second approach, based on (R)-methyl lactate, was recently very thoroughly optimized by BMS on a multi kilogram scale but it still does not fulfill requirements for cost effective manufacturing process (Organic Process Research & Development 13, 716 (2009)). The overall yield of this optimized 11 steps process is still only 16% (Scheme 1).

The manufacturing process for Isavuconazole is similar: Since Isavuconazole differentiates from Ravuconazole by only another fluorine substitution on the aromatic ring (2,5- instead of 2,4-difluorophenyl), the identical synthesis has been used (U.S. Pat. No. 6,300,353 from Oct. 9, 2001 and Bioorg. & Med. Chem. Lett. 13, 191 (2003)). Consequently, also this manufacturing process, based on (R)-lactic acid, faces the same problems: to many steps, extremely low overall yield and in addition to U.S. Pat. No. 6,300,353 claims even already known step as novel (claim 36).
Recent attempts to improve this concept as reported in WO 2007/062542 (Dec. 1, 2005), using less expensive, natural configured (S)-lactic acid, also failed: As already reported in U.S. Pat. No. 6,133,485 and in US 2003/0236419, the second chiral center was formed from an optically active allyl alcohol prepared in a few steps from (S)-lactic acid.
This allyl alcohol was subjected to Sharpless diastereoselective epoxidation providing first an opposite configured, epimeric epoxy alcohol which had to be then epimerized in an additional inversion step yielding finally the desired epoxy alcohol as the known precursor for Isavuconazole (U.S. Pat. No. 6,300,353). It is obvious that this process using less expensive (S)-lactic acid makes the entire process with an inversion step even more complex than the original approach.
Elegant and more efficient process has been claimed in US 2004/0176432 from Jun. 26, 2001) in which both chiral centers have been formed simultaneously, diastereo- and enantio-selectively pure in one single reaction step using chiral (R)-2-butynol as a chiral precursor in the presence of Pd(II)-catalyst and diethyl zinc (Scheme 2).

Since water soluble, (R)-2-butynol is expensive, recently identical process has been published, in which instead of (R)-2-butynol less water soluble and therefore, less expensive (R)-4-phenyl-3-butyn-2-ol was used (Synthetic Commun. 39, 1611 (2009)). Nevertheless, as incorrectly stated there, this process does not provide better diastereoselectivity than the original process using (R)-2-butynol: On the contrary disadvantage of this process is a very bad atom economy because huge phenyl group of (R)-4-phenyl-3-butyn-2-ol has to be “disposed” in oxidation step by the conversion of triple bond into carboxylic acid function.
All known processes for enantiomerically pure compounds of formula I have definitely too many operation steps and specifically very low overall yield. The chiral starting materials used, either 3-hydroxy-2-methyl propionic acid or (S)- or (R)-methyl lactate, contain only one chiral center and consequently, in number of steps, the second adjacent chiral center has to be ineffectively generated which makes the entire process long and expensive. The only known process, which generates both chiral centers simultaneously, requires again expensive chiral starting material (R)-2-butynol.
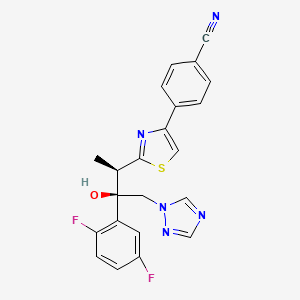
ISAVUCONAZOLE
…………………………………………….
synthetic scheme A, starting from 4-[(2R)-2-(3,4,5,6-tetrahydro-2H-pyran-2-yloxy)-propionyl]morpholine [which can be prepared by a same procedure as described in Chem. Pharm. Bull. 41, 1035, 1993.]. This synthesis route has been described for example in European Patent Application No. 99101360.8.


(a)
………………………………………………………………………
Example 1 (2R,3R)-3-[4-(4-cyanophenyl)thiazol-2-yl]-1-(1H-1,2,4-triazol-1-yl)-2-(2,4-difluorophenyl)-butan-2-ol
To a solution of racemic 3-[4-(4-cyanophenyl)thiazol-2-yl]-1-(1H-1,2,4-triazol-1-yl)-2-(2,4-difluorophenyl)-butan-2-ol (43.7 g) in acetone (800 ml) a solution of (1R)-10-camphorsulfonic acid (23 g) in methanol (300 ml) was added and the mixture was heated under reflux until a clear solution was obtained. The solution was slowly cooled to rt, seeded with crystals of the title enantiomeric salt and let overnight. The solid was collected by filtration, washed with acetone and dried to provide (2R,3R)-3-[4-(4-cyanophenyl)thiazol-2-yl]-1-(1H-1,2,4-triazol-1-yl)-2-(2,4-difluorophenyl)-butan-2-ol (1R)-10-camphorsulfonate as white solid. This crude salt was then taken up in methylenechloride (100 ml) and water (ca. 100 ml) and the mixture was basified with aqueous sodium hydroxide solution. The organic layer was separated and the aqueous phase washed twice with methylenechloride (50 ml) and combined. The organic phases were then washed twice with water (2×50 ml), dried with sodium sulfate, filtrated and the solvent removed under reduced pressure. The crude product was then mixed with isopropanol (ca. 150 ml), heated for 10 min, cooled to 0° C. and stirred for ca. 2 hrs. The product was collected, washed with isopropanol and dried under reduced pressure to provide the enantiomerically pure title compound (17.5 g, 41% yield, 99.1% ee);
m.p. 164-166° C.; [α]=−30° (c=1, methanol, 25° C.);
NMR (CDCl3): 1.23 (3H, d, J=8 Hz), 4.09 (1H, q, J=8 Hz), 4.26 (1H, d, J=14 Hz), 4.92 (1H, d, J=14 Hz), 5.75 (1H, s), 6.75-6.85 (2H, m), 7.45-7.54 (2H, m), 7.62 (1H, s), 7.69 (1H, s), 7.75 (1H, d, J=8 Hz), 7.86 (1H, s), 8.03 (1H, d, J=8 Hz).
The analytical data were identical with published (U.S. Pat. No. 5,648,372 and Chem. Pharm. Bull. 1998, 46, 623-630).
Example 2 (2R,3R)-3-[4-(4-cyanophenyl)thiazol-2-yl]-1-(1H-1,2,4-triazol-1-yl)-2-(2,4-difluorophenyl)-butan-2-ol
Racemic 3-[4-(4-cyanophenyl)thiazol-2-yl]-1-(1H-1,2,4-triazol-1-yl)-2-(2,4-difluorophenyl)-butan-2-ol (44 g) and (1R)-10-camphorsulfonic acid (20 g) were suspended in methanol (ca. 300 ml), the slurry was stirred intensively, warmed up to ca. 70° C. and a small addition of acetic acid was added to obtain a clear solution. After cooling of the solution to rt and then to 0° C., the mixture was seeded with enantiomerically pure salt and stirred for another 2 hrs. The crystalline solid was collected by filtration, washed with cooled methanol and dried under reduced pressure. The crystals were partitioned between methylenechloride (300 ml) and saturated aqueous sodium bicarbonate solution (200 ml). The organic layer was washed twice with water (50 ml), dried with magnesium sulphate, filtrated and evaporated under reduced pressure to give the title compound (16.9 g, 38% yield, 95% ee). The analytical data were identical with published (U.S. Pat. No. 5,648,372 or Chem. Pharm. Bull. 1998, 46, 623).
Example 3 (2R,3R)-3-[4-(4-cyanophenyl)thiazol-2-yl]-1-(1H-1,2,4-triazol-1-yl)-2-(2,5-difluorophenyl)-butan-2-ol
To a solution of racemic 3-[4-(4-cyanophenyl)thiazol-2-yl]-1-(1H-1,2,4-triazol-1-yl)-2-(2,5-difluorophenyl)-butan-2-ol (10 g) in acetone (ca. 200 ml) a solution of (1R)-10-camphorsulfonic acid (3.9 g) in methanol (50 ml) was added and the mixture was heated shortly under reflux until a clear solution was obtained. The solution was then slowly cooled to rt, seeded with crystals of the desired enantiomeric salt and let overnight. The solid precipitate was collected by filtration, washed with acetone and dried to provide (2R,3R)-3-[4-(4-cyanophenyl)thiazol-2-yl]-1-(1H-1,2,4-triazol-1-yl)-2-(2,5-difluorophenyl)-butan-2-ol (1R)-10-camphorsulfonate as white solid. This salt was then taken up in methylenechloride and water and basified with aqueous sodium bicarbonate solution. The organic layer was separated and the aqueous phase washed twice with methylenechloride. The organic phases were combined, dried with sodium sulphate, filtrated and the solvent removed under reduced pressure. The crude product was then dissolved in ethanol, the slurry heated for 20 min, small amount of water was added, the solution slowly cooled to 0° C. and stirred for ca. 2 hrs. The product was collected, washed with cold ethanol and dried under reduced pressure to provide the title enantiomerically pure compound (3.9 g, 39% yield, 96% ee). The analytical date were identical with published in U.S. Pat. No. 6,300,353 B1 and WO 99/45008.
Example 4 (2R,3R)-3-[4-(4-cyanophenyl)thiazol-2-yl]-1-(1H-1,2,4-triazol-1-yl)-2-(2,5-difluorophenyl)-butan-2-ol
To a solution of racemic 3-[4-(4-cyanophenyl)thiazol-2-yl]-1-(1H-1,2,4-triazol-1-yl)-2-(2,5-difluorophenyl)-butan-2-ol (100 g) in acetone (1000 ml) a solution of (1R)-10-camphorsulfonic acid (47 g) in methanol (500 ml) was added at rt, then slurry was heated under stirring to almost reflux for ca. 30 min, then cooled slowly to rt, seeded with the pure enantiomeric salt and stirred over night. The solid was collected by filtration, washed with methanol/acetone mixture, dried under reduced pressure. The residue was taken up with a solvent mixture of methylenechloride/water and after addition of saturated aqueous sodium bicarbonate solution the organic phase was separated and aqueous phase washed twice with methylenechloride. The combined organic phases were filtrated, the solvent removed under reduced pressure. Recrystallization of the crude product from aqueous ethanol provided enantiomerically pure title compound: 39 g (39% yield, 92% ee). The analytical data were identical with published: U.S. Pat. No. 6,300,353 and WO 99/45008.
Example 5 (2R,3R)-3-[4-(4-cyanophenyl)thiazol-2-yl]-1-(1H-1,2,4-triazol-1-yl)-2-(2,5-difluorophenyl)-butan-2-ol
A solution of the racemic 3-[4-(4-cyanophenyl)thiazol-2-yl]-1-(1H-1,2,4-triazol-1-yl)-2-(2,5-difluorophenyl)-butan-2-ol (4.4 g) and (1R)-10-camphorsulfonic acid (2 g) in toluene (40 ml) containing glacial acetic acid (0.6 ml) was warmed up to approximately 70° C., then allowed to cool slowly to 20° C., seeded with the pure enantiomeric salt whereupon the pure enantiomeric salt start to crystallize out. After ca. 2 hrs at this temperature the solid was collected, washed with cold toluene and dried. The crystals were taken with a solvent mixture of methylenechloride/water and after addition of aqueous saturated sodium bicarbonate solution the organic phase was separated and aqueous phase washed twice with methylenechloride. The combined organic phases were filtrated and the solvent removed under reduced pressure. Recrystallization of the crude product from aqueous ethanol provided enantiomerically pure title compound: 2 g (45% yield, 99% ee). The analytical data were identical with published: U.S. Pat. No. 6,300,353 and WO 99/45008.
…………………………………..
WO 1999045008
The following synthetic scheme 1 illustrates the manufacture of one of the compounds of formula I′:



……………………………….
Bioorganic and medicinal chemistry letters, 2003 , vol. 13, 2 p. 191 – 196
http://www.sciencedirect.com/science/article/pii/S0960894X02008922
A highly potent water soluble triazole antifungal prodrug, RO0098557 (1), has been identified from its parent, the novel antifungal agent RO0094815 (2). The prodrug includes a triazolium salt linked to an aminocarboxyl moiety, which undergoes enzymatic activation followed by spontaneous chemical degradation to release 2. Prodrug 1 showed high chemical stability and water solubility and exhibited strong antifungal activity against systemic candidiasis and aspergillosis as well as pulmonary aspergillosis in rats.
A highly potent water soluble triazole antifungal prodrug, RO0098557 (1), has been identified from its parent, the novel antifungal agent RO0094815 (2). The prodrug includes a triazolium salt linked to an aminocarboxyl moiety, which undergoes enzymatic activation followed by spontaneous chemical degradation to release 2. Prodrug 1 showed high chemical stability and water solubility and exhibited strong antifungal activity against systemic candidiasis and aspergillosis as well as pulmonary aspergillosis in rats.

-

-
Scheme 1.
Chemistry
Scheme 1.We synthesized a series of new triazolium derivatives of Figure 1, Figure 3 and Scheme 1. CompoundsScheme 1 and Scheme 2, 6, 9, 10 and 11 were first prepared as outlined in Scheme 2 in order to analyze their stability and ability to release Figure 1, Figure 3 and Scheme 1. Next, aromatic analogues 18, 19, 20,21 and Figure 1, Figure 3 and Scheme 3 were synthesized for optimization of 11 to increase its water solubility and conversion rate. Compounds in the second series had sarcosine esters6 to make them water soluble, and they were also designed to generate acetaldehyde7 instead of formaldehyde for a better safety profile. The synthetic procedures for the second series of the derivatives are outlined in Scheme 3.

-
Scheme 2.
(a) ClCOOCH2Cl, diisopropylethylamine, CH2Cl2, rt (quant); (b) Figure 1, Figure 3 and Scheme 1, CH3CN, 80 °C (60%); (c) (1) ClCOOCH2Cl, Et3N, CH2Cl2, rt; (2) Ac2O, pyridine, rt (30%, two steps); (d) (1) NaI, CH3CN, 50 °C ; (2) Figure 1, Figure 3 and Scheme 1, CH3CN, 50 °C (88%, two steps); Synthesis of Scheme 1 and Scheme 2: (1) N-3-hydroxypropyl-N-methylamine, ClCOOCH2Cl, Et3N, CH2Cl2, rt; (2) AcCl, Et3N, CH2Cl2, rt (20%, two steps); (3) Figure 1, Figure 3 and Scheme 1, NaI, CH3CN, 50 °C (82%); Synthesis of 10: (1) l-prolinol, ClCOOCH2Cl, Et3N, CH2Cl2, rt; (2) Ac2O, pyridine, rt (<10%, 2 steps); (3) Figure 1, Figure 3 and Scheme 1, NaI, CH3CN, 50 °C (92%); Synthesis of 11: (1) 2-hydroxymethyl-N-methylaniline, ClCOOCH2Cl, diisopropylethylamine, CH2Cl2, rt; (2) Ac2O, diisopropylethylamine, rt (20%, two steps); (3)Figure 1, Figure 3 and Scheme 1, cat. NaI, CH3CN, reflux (63%).
-
Figure options

-
Scheme 3.
(a) (1) oxalyl chloride, DMF, 0 °C; (2) KOtBu, THF, −5 °C (97%, two steps); (b) CH3NH2, MeOH, rt (90%); (c) LiAlH4, THF, 0 °C (80%); (d) (1) ClCOOCH(CH3)Cl, diisopropylethylamine, CH2Cl2, 0 °C; (2) Boc-Sarcosine, WSCI, DMAP, CH2Cl2, 0 °C (84%, two steps); (e) (1) Figure 1, Figure 3 and Scheme 1, NaI, CH3CN, 50 °C; (2) DOWEX-1 Cl− form, aqueous MeOH, rt (65%, two steps); (f) (1) HCl, EtOAc, rt; (2) lyophilization (69%, two steps); Synthesis of 18: (1) (i) (4,5-difluoro-2-methylaminophenyl)methanol, ClCOOCH(CH3)Cl, diisopropylethylamine, CH2Cl2, 0 °C; (ii) Boc-Sarcosine, WSCI, DMAP, CH2Cl2, 0 °C (quant, two steps); (2) Figure 1, Figure 3 and Scheme 1, cat. NaI, CH3CN, 80 °C; (50%,); (3) HCl, EtOAc, rt (90%); Synthesis of 19: (1) (i) 2-fluoro-6-methylaminophenyl)methanol, ClCOOCH(CH3)Cl, diisopropylethylamine, CH2Cl2, 0 °C; (ii) Boc-Sarcosine, WSCI, DMAP, CH2Cl2, 0 °C (74%, two steps); (2) Figure 1, Figure 3 and Scheme 1, cat. NaI, CH3CN, reflux; (3) HCl, EtOAc, rt (29%, two steps); Synthesis of 20: (1) (i) (5-fluoro-2-methylaminophenyl)methanol, ClCOOCH(CH3)Cl, diisopropylethylamine, CH2Cl2, 0 °C; (ii) Boc-Sarcosine, WSCI, DMAP, CH2Cl2, 0 °C (91%, two steps); (2) Figure 1, Figure 3 and Scheme 1, cat. NaI, CH3CN, 70 °C (72%); (3) HCl, EtOAc, rt (88%); Synthesis of 21: (1) (i) (4-chloro-2-methylaminophenyl)methanol, ClCOOCH(CH3)Cl, diisopropylethylamine, CH2Cl2, 0 °C; (ii) Boc-Sarcosine, WSCI, DMAP, CH2Cl2, 0 °C (71%, two steps); (2) Figure 1, Figure 3 and Scheme 1, CH3CN, 65 °C; (3) HCl, EtOAc, rt (65%, two steps).

read more at
Boyd, B.; Castaner, J. BAL-4815/BAL-8557
Drugs Fut 2006, 31(3): 187
Antimicrobial Agents and Chemotherapy, 2008 , vol. 52, 4 p. 1396 – 1400
Ohwada, J.; Tsukazaki, M.; Hayase, T.; Oikawa, N.; Isshiki, Y.; Umeda, I.; Yamazaki, T.; Ichihara, S.; Shimma, N.Development of novel water antifungal, RO0098557
21st Med Chem Symp (November 28-30, Kyoto) 2001, Abst 1P-06
Ohwada, J.; Tsukazaki, M.; Hayase, T.; et al.
RO0098557, a novel water soluble azole prodrug for parenteral and oral administration (I). Design, synthesis, physicochemical properties and bioconversion42nd Intersci Conf Antimicrob Agents Chemother (ICAAC) (September 27-30, San Diego) 2002, Abst F-820
Tasaka et al., Chem. Pharm. Bull. 41(6) pp. 1035-1042 (1993).
Clinical trials
There have been three phase III clinical trials of isavuconazole, ACTIVE, VITAL and SECURE. As of June 2015, SECURE and VITAL have been presented in abstract form and results from ACTIVE have not been released.[9]
The SECURE trial compared voriconazole and isavuconazole in invasive fungal infections due to aspergillus. Isuvaconazole was found to be non-inferior to voriconazole, anothertriazole antifungal, with all cause mortality at 18.6%, compared to 20.2% in the voriconazole group. It additionally demonstrated a similar side effect profile.[10]
Data from the VITAL study showed that isavuconazole could be used in treatment of invasive mucormycosis, but did not evaluate its clinical efficacy for this indication.[11]
The ACTIVE trial is a comparison of isuvaconazole and caspofungin for invasive candida infections and results are anticipated in the second half of 2015.[12][13]
References
- [1]
- Saboo, Alok. “Basilea Announces Global Partnership With Astellas for Its Antifungal Isavuconazole.” FierceBiotech. N.p., 24 Feb. 2010. Web.
- “Basilea reports isavuconazole orphan drug designation by U.S. FDA.” Market Wired. 28 May 2013.
- “FDA Grants Orphan Drug Designation to Astellas for Isavuconazole for the Treatment of Invasive Candidiasis.” News Releases. Astellas. 3 Nov 2014.
- Cresemba (isovuconazole sulfate) [prescribing information]. Astella Pharma US, Inc. Revised March 2015.
- Jump up^ “Aspergillosis.” Centers for Disease Control and Prevention. Centers for Disease Control and Prevention, 08 Sept. 2014.
- Jump up^ “Astellas Receives FDA Approval for CRESEMBA® (isavuconazonium Sulfate) for the Treatment of Invasive Aspergillosis and Invasive Mucormycosis.” PR Newswire. N.p., 6 Mar. 2015.
- Jump up^ “Isavuconazonium.” Micromedex Solutions. Truven Health Analytics, n.d. Web. <www.micromedexsolutions.com>.
- Jump up^ Pettit, Natasha N.; Carver, Peggy L. (2015-07-01). “Isavuconazole A New Option for the Management of Invasive Fungal Infections”. Annals of Pharmacotherapy 49 (7): 825–842.doi:10.1177/1060028015581679. ISSN 1060-0280. PMID 25940222.
- Mujais, A. “2014: M-1756. A Phase 3 Randomized, Double-Blind, Non-Inferiority Trial Evaluating Isavuconazole (ISA) vs. Voriconazole (VRC) for the Primary Treatment of Invasive Fungal Disease (IFD) Caused by Aspergillus spp. or other Filamentous Fungi (SECURE): Outcomes by Malignancy Status”. http://www.icaaconline.com. Retrieved 2015-06-19.
- “Abstract: An Open-Label Phase 3 Study of Isavuconazole (VITAL): Focus on Mucormycosis (IDWeek 2014)”. idsa.confex.com. Retrieved 2015-06-19.
- Ltd., Basilea. “Basilea Pharmaceutica – Portfolio – Isavuconazole”. http://www.basilea.com. Retrieved 2015-06-19.
- “Isavuconazole (BAL8557) in the Treatment of Candidemia and Other Invasive Candida Infections – Full Text View – ClinicalTrials.gov”. clinicaltrials.gov. Retrieved 2015-06-19.
| US4861879 | Feb 9, 1988 | Aug 29, 1989 | Janssen Pharmaceutica N.V. | [[4-[4-Phenyl-1-piperazinyl)phenoxymethyl]-1-3-dioxolan-2-yl]-methyl]-1H-imidazoles and 1H-1,2,4-triazoles |
| US5900486 | Sep 9, 1997 | May 4, 1999 | Hoffmann-La Roche Inc. | N-benzylazolium derivatives |
| AU4536497A | Title not available | |||
| EP0667346A2 | Feb 3, 1995 | Aug 16, 1995 | Eisai Co., Ltd. | Azole antifungal agents, process for the preparation there of and intermediates |
| WO1992017474A1 | Mar 26, 1992 | Oct 15, 1992 | Pfizer | Triazole antifungal agents |
| US5648372 | Feb 1, 1995 | Jul 15, 1997 | Eisai Co., Ltd. | Antifungal agents, and compositions |
| US5686646 * | May 23, 1995 | Nov 11, 1997 | Schering-Plough Corporation | Chiral hydrazine derivatives |
| US5746840 * | Mar 28, 1997 | May 5, 1998 | Janssen Pharmaceutica, N.V. | Process for preparing enantiomerically pure 6-{4-chlorophenyl) (1 H-1,2,4-triazol-1-YL) methyl}-1-methyl-1 H-benzotriazole |
| US5792781 | Sep 18, 1996 | Aug 11, 1998 | Eisai Co., Ltd. | Antifungal agents, processes for the preparation thereof, and intermediates |
| US6020497 | Oct 9, 1998 | Feb 1, 2000 | Merck & Co., Inc. | 3-substitutes isoxazolidines as chiral auxiliary agents |
| US6133485 | Apr 15, 1998 | Oct 17, 2000 | Synphar Laboratories, Inc. | Asymmetric synthesis of 2-(2,4-difluorophenyl)-1-heterocycl-1-yl butan-2,3-diols |
| US6300353 | Mar 5, 1999 | Oct 9, 2001 | Basilea Pharmaceutica Ag, A Swiss Company | Azoles for treatment of fungal infections |
| US6383233 | Mar 7, 1997 | May 7, 2002 | Reuter Chemicscher Apparatebau Kg | Separation process |
| US6812238 * | Oct 31, 2000 | Nov 2, 2004 | Basilea Pharmaceutica Ag | N-substituted carbamoyloxyalkyl-azolium derivatives |
| US7151182 * | Sep 3, 2004 | Dec 19, 2006 | Basilea Pharmaceutica Ag | Intermediates for N-substituted carbamoyloxyalkyl-azolium derivatives |
| US7803949 * | Dec 20, 2006 | Sep 28, 2010 | Eisai R&D Management Co., Ltd. | Process for preparation of water-soluble azole prodrugs |
| US20030236419 | Dec 31, 2002 | Dec 25, 2003 | Sumika Fine Chemicals Co., Ltd. | Production methods of epoxytriazole derivative and intermediate therefor |
| US20040176432 | Jun 17, 2002 | Sep 9, 2004 | Milan Soukup | Intermediate halophenyl derivatives and their use in a process for preparing azole derivatives |
| WO2003002498A1 * | Jun 17, 2002 | Jan 9, 2003 | Basilea Pharmaceutica Ag | Intermediate halophenyl derivatives and their use in a process for preparing azole derivatives |
 |
|
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
4-{2-[(1R,2R)-(2,5-Difluorophenyl)-2-hydroxy-1-methyl-3-(1H-1,2,4-triazol-1-yl)propyl]-1,3-thiazol-4-yl}benzonitrile
|
|
| Clinical data | |
| Trade names | Cresemba (prodrug form) |
| AHFS/Drugs.com | entry |
| Pregnancy category |
|
| Legal status |
|
| Routes of administration |
Oral, intravenous |
| Identifiers | |
| ATC code | None |
| PubChem | CID: 6918485 |
| ChemSpider | 5293682 |
| UNII | 60UTO373KE |
| ChEBI | CHEBI:85979 |
| ChEMBL | CHEMBL409153 |
| NIAID ChemDB | 416566 |
| Chemical data | |
| Formula | C22H17F2N5OS |
| Molecular mass | 437.47 g/mol |


/////
VINCRISTINE……..Chemistry, Isolation

VINCRISTINE
(3aR,3a1R,4R,5S,5aR,10bR)-methyl 4-acetoxy-3a-ethyl-9-((5S,7S,9S)-5-ethyl-5-hydroxy-9-(methoxycarbonyl)-2,4,5,6,7,8,9,10-octahydro-1H-3,7-methano[1]azacycloundecino[5,4-b]indol-9-yl)-6-formyl-5-hydroxy-8-methoxy-3a,3a1,4,5,5a,6,11,12-octahydro-1H-indolizino[8,1-cd]carbazole-5-carboxylate
…………………………………………………………..
………………………………………………………………….


Vincristine (brand name, Oncovin), formally known as leurocristine, sometimes abbreviated “VCR”, is a vinca alkaloid from the Catharanthus roseus (Madagascar periwinkle), formerly Vinca rosea and hence its name. It is amitotic inhibitor, and is used in cancer chemotherapy. Vincristine is created by the coupling of indole alkaloids vindoline and catharanthine in the vinca plant.[1]
Mechanism
Tubulin is a structural protein that polymerizes to microtubules. The cell cytoskeleton and mitotic spindle, among other things, are made of microtubules. Vincristine binds to tubulin dimers, inhibiting assembly of microtubule structures. Disruption of the microtubules arrests mitosis in metaphase. Therefore, the vinca alkaloids affect all rapidly dividing cell types including cancer cells, but also those of intestinal epithelium and bone marrow.

Uses
Vincristine is delivered via intravenous infusion for use in various types of chemotherapy regimens. Its main uses are in non-Hodgkin’s lymphoma as part of the chemotherapy regimen CHOP, Hodgkin’s lymphoma as part of MOPP, COPP, BEACOPP, or the less popular Stanford V chemotherapy regimen, in acute lymphoblastic leukemia, and in treatment for nephroblastoma (Wilms tumor, a kidney tumor most common in young children). It is also used to induce remission in ALL with Dexamethasone and L-Asparaginase. Vincristine is occasionally used as an immunosuppressant, for example, in treating thrombotic thrombocytopenic purpura (TTP) or chronic idiopathic thrombocytopenic purpura (ITP). It is used in combination with prednisone to treat childhood leukemia.
The main side-effects of vincristine are peripheral neuropathy, hyponatremia, constipation, and hair loss.
Peripheral neuropathy can be severe, and hence a reason to avoid, reduce, or stop the use of vincristine. One of the first symptoms of peripheral neuropathy is foot drop: A person with a family history of foot drop and/or Charcot-Marie-Tooth disease (CMT) should avoid the taking of vincristine.[2]
Accidental injection of vinca alkaloids into the spinal canal (intrathecal administration) is highly dangerous, with a mortality rate approaching 100 percent. The medical literature documents cases of ascending paralysis due to massive encephalopathy and spinal nerve demyelination, accompanied by intractable pain, almost uniformly leading to death; a handful of survivors were left with devastating neurological damage with no hope of recovery. Rescue treatments consist of washout of the cerebrospinal fluid and administration of protective medications.[3] A significant series of inadvertent intrathecal vincristine administration occurred in China in 2007 when batches of cytarabine andmethotrexate (both often used intrathecally) manufactured by the company Shanghai Hualian were found to be contaminated with vincristine.[4]

Having been used as a folk remedy for centuries, studies in the 1950s revealed that C. roseus contained 70 alkaloids, many of which are biologically active. While initial studies for its use in diabetes mellitus were disappointing, the discovery that it caused myelosuppression (decreased activity of the bone marrow) led to its study in mice withleukemia, whose lifespan was prolonged by the use of a vinca preparation. Treatment of the ground plant with Skelly-B defatting agent and an acid benzene extract led to a fraction termed “fraction A”. This fraction was further treated withaluminium oxide, chromatography, trichloromethane, benz-dichloromethane, and separation by pH to yield vincristine.[5]
Vincristine was approved by the United States Food and Drug Administration (FDA) in July 1963 as Oncovin. The drug was initially discovered by a team led by Dr. J.G. Armstrong, then marketed by Eli Lilly and Company.
Like LSD, the microtubule toxin vincristine allegedly causes not-unpleasant visual hallucinations in humans. Other side-effects of vincristine include depression, agitation, and insomnia. Very small doses are needed for the effects of LSD or vincristine, for example, these drugs are active at concentrations of 4.3E-7 M-1 vincristine and 1.0E-8 M-1 LSD.
Many researchers have favored the drug-receptor theory to explain drug-induced hallucinations, usually at the 5-HT2A receptor. In the drug-receptor theory, signal amplification takes place when one molecule of drug binds to a receptor, which activates G-proteins, which affects more proteins, thus signaling cascades explain how a small amount of LSD can lead to widespread changes in the cell.
Van Woerkom suggests instead that LSD binds an element of the cytoskeleton, in a fashion similar to colchicine or vinblastine, which directly bind tubulin. The amount of LSD needed to produce hallucinations is so vanishly small, that it seems hard to believe that a submicromolar dosage of LSD could act on a substrate as vast as the cytoskeleton. However, some microtubule inhibitors such as vincristine are effective at very low dosages. The potency of vincristine may partly explain the success of this drug as a chemotherapeutic drug.
Three generic drug makers supply vincristine in the United States – APP, Mayne, and Sicor (Teva).
- ^ “Pharmacognosy of Vinca Alkaloids”.
- Graf, W. D.; Chance, P. F.; Lensch, M. W.; Eng, L. J.; Lipe, H. P.; Bird, T. D. (1996). “Severe Vincristine Neuropathy in Charcot-Marie-Tooth Disease Type 1A”. Cancer 77 (7): 1356–1362. doi:10.1002/(SICI)1097-0142(19960401)77:7<1356::AID-CNCR20>3.0.CO;2-#. PMID 8608515.
- Qweider, M.; Gilsbach, J. M.; Rohde, V. (2007). “Inadvertent Intrathecal Vincristine Administration: A Neurosurgical Emergency. Case Report”. Journal of Neurosurgery: Spine 6 (3): 280–283. doi:10.3171/spi.2007.6.3.280. PMID 17355029.
- Jake Hooker and Walt Bogdanich (January 31, 2008). “Tainted Drugs Tied to Maker of Abortion Pill”. New York Times.
- Johnson, I. S.; Armstrong, J. G.; Gorman, M.; Burnett, J. P. (1963). “The Vinca Alkaloids: A New Class of Oncolytic Agents” (pdf). Cancer Research 23 (8 Part 1): 1390–1427.PMID 14070392.
External links
- Vincristine chemotherapy
- Vincristine and vinblastine
- Description and Natural History of the Periwinkle
- The Boger Route to (-)-Vindoline
- U.S. National Library of Medicine: Drug Information Portal – Vincristine
-
Cytostatic Vinca alkaloids rosea L. Catharanthus roseus G.Don) are now well known anticancer and particularly useful. Given the small amount of vincristine in Catharanthus present, quite a number of ways of preparation have been proposed by chemists. Thus FR-A-2296418 describes the synthesis of vincristine by coupling Catha-ranthine and vindoline. Other laboratories have achieved the transformation of vinblastine vincristine oxidation under controlled conditions, very strict.
-
FR-A-2210393 and US-A-3899493 perform the oxidation by chromic acid at -30, -90 ° C in a mixture of acetic acid-acetone or chloroform-acetic acid at -55 ° C.
-
In U.S. 4,375,432, chromic compound is also used in acid medium at -65 ° C, -50 ° C in a medium based solvent THF. In addition, EP-A-37289 boasts an oxidation mixture ferrous salt, hydrogen peroxide, perchlorate in acetonitrile. ZA-A-82 08939 discloses a method with chromic acid and an ether-chloroform.
-
HU-A-23638 offers diterbutylchromate in pelargonic acid, and finally EP-A-117861 gets vinblastinel transformation vincristine oxidant potassium permanganate in acetic acid medium. It is clear that these dimeric alkaloids are a valuable material because of their low levels in vegetable raw materials, and therefore the processes of synthesis or semi-synthesis performance are of extreme interest.
-
Vincristine is used in cancer chemotherapy, particularly for the treatment of certain acute leukemias.
-
This alkaloid is obtained mainly by extraction from leaves of Catharanthus Ro-seus (U.S. Patent No. 3,205,220) where it is accompanied by other alkaloids bis-Indo-holic, especially vinblastine.Vinblastine (I, R = CH 3), however, is present at a concentration much higher than that of vincristine and is therefore a precursor of choice for the semisynthesis of the latter.
-
Several processes of vincristine from vinblastine were disclosed. We note in particular patents or patent applications include:
- a) Belgian Patent 739,337 (Gedeon Richter) which describes a method for the oxidation of vinblastine vincristine in a mixture chromic acid, acetic acid and acetone.
- b) Belgian Patent 823560 (Gedeon Richter) the oxidation is performed with oxygen in the presence of formic acid and of a catalyst based on platinum at room temperature.
- c) European Patent Application 18231 (Gedeon Richter): is carried out by oxidation with chromic acid or an alkali metal dichromate in the presence of acetic anhydride and, optionally, of ethanol and an organic solvent immis target with water.
- d) European Patent Application 37289 (Eli Lil-ly): the oxidation is effected by the perchlorate of iron (II) in the presence of hydrogen peroxide and acetonitrile.
-
In addition, the European patent application 37. 290 discloses a process for the oxidation of vinblastine base with Na 2 Cr 2 O 7 in the presence of sulfuric acid in tetrahydrofuran. This reaction led to -50 ° C, is achieved with a yield of 80-92% calculated for each estimation.
-
Observed yields or purity of the products obtained characterizing the processes described above are, however, significant disadvantages.
-
Frequently a secondary product formed is N-demethyl vinblastine need then reformulate for vincristine.
Thus Potier and Kutney obtained products with the C18’S-C2’R absolute configuration, which is critical for anti-tumor activity, by a coupling reaction of the N.sup.b -oxide of catharanthine, or its derivatives, with vindoline, in the presence of trifluoroacetic anhydride, followed by a reduction reaction. [See Potier et. al. J. Am. Chem. Soc. 98. 7017 (1976) and Kutney et. al. Helv. Chim. Acta, 59, 2858 (1976)].
The Potier and Kutney coupling process has disadvantages. The yields are not satisfactory except for the coupling of catharanthine N-oxide with vindoline and even there the preparative yield is low. While vindoline is the most abundant alkaloid of Vinca rosea and is thus readily available, the other possible components of the Potier-Kutney coupling process (catharanthine, allocatharanthine, voacangine,) are relatively inaccessible, costly, and they do not allow a wide range of structural variation of that component of the coupling process.
- …………………………………………………………………………………………………………………………………………………………………………………………………………..
-
EP 0117861 B1
-
clips
-
The process of the present invention produces a simple vincristine, in quantity and purity requiring little or no additional purification by recrystallization or chromatography.
-
[0009]The reagent used is oxidation permanganate ion dissolved in toluene or dichloromethane as solvent. An alternative consists in immobilizing the resin on a permanganate anion, for example a polymer such as polystyrene comprising ammonium groups. Solubilization can be achieved by the action of a complexing agent crown ether (“crown-ether”) of potassium permanganate.
-
[0010]The permanganate anion can also be solubilized by preparing an ammonium salt or quaternary phosphonium corresponding which is soluble in methylene chloride or toluene. For this purpose, it is preferable to use potassium permanganate benzyltriethylammonium.
-
[0011]Obtaining from vincristine vinblastine using a permanganate salt is unexpected since the potassium permanganate used in some acetone oxide derivatives of vinblastine at the portion of the molecule velbanamine (Kutney, Balsevich and Worth, Heterocycles, 11, 69, 1978). The N-methyl group of the vindoline part intact.
-
[0012]The formation of N-CHO indoline skeleton on a bis-indole group vinblastine using a permanganate salt has never been reported.
-
[0013]According to one embodiment of the method of the present invention, vinblastine, preferably in the form of sulphate, is treated in the presence of an organic acid such as acetic acid, with an excess of potassium permanganate dissolved in dichloromethane or toluene in the presence of “18-crown-6” or ether derivatives dibenzo-or di-cyclohexylcorrespondants. The reaction is conducted at a temperature between -40 ° C and -75 ° C and is preferably followed by thin layer chromatography. The reaction time generally ranges from 5 minutes to 3 hours.
-
[0014]Potassium permanganate is preferably dissolved in dichloromethane and the oxidation reaction is then carried out at -70 ° C.
-
[0015]The solubility of potassium permanganate is indeed substantially increased in the presence of a macrocyclic polyether as the “18-crown-6” ether (1, 4, 7, 10, 13, 16-hexaoxacy-clooctadécane) or derivative dibenzo – or corresponding dicyclohexyl-hexyl.
-
[0016]The reaction mixture is then treated simultaneously by a mild reducing and alkaline. For this purpose, use is preferably an aqueous solution of bisulfite, disulfite or sodium metabisulfite and ammonia.
-
[0017]The organic phase was separated and the aqueous phase is extracted several times with methylene chloride. The combined organic phases were concentrated in vacuo to give a residue containing 80-85% of base vincristine, a 90-95% yield.
-
[0018]Alternatively, you can proceed with the extraction of the reaction mixture after reduction without conducting a simultaneous alkalinization. The acidic aqueous solution was then extracted with dichloromethane. This route is a novel process for purification of vincristine formed in the reaction medium.
-
[0019]According to another embodiment of the present invention, vincristine is obtained by oxidation of vinblastine by reacting a quaternary ammonium permanganate. The ammonium cation is preferably benzyltriethylammonium group or benzyl trimethyl ammonium (see eg Angew. Chem., Intern. Ed. 13, 170, 1974). The reaction is carried out in 2 to 6 hours at -60 ° C in an inert solvent wherein the ammonium salt is soluble, and an acid, preferably an organic acid of low molecular weight. A mixture of dichloromethane and glacial acetic acid can be used. After treatment with a mild reducing agent in aqueous medium, the resulting acidic solution is extracted with dichloromethane, and the organic phase is made alkaline by washing with a basic aqueous solution and concentrated. Vincristine solvate is isolated with a yield higher than 90%.
-
[0020]The latest variant of the method of the invention is particularly advantageous in terms of economic and technical.
-
[0021]Purification or separation may be effected by crystallization and chromatography using techniques well known this from the crude product of the reaction. The product can also be lyophilized.
-
[0022]In most cases, vincristine thus obtained can be converted directly into an addition salt with an organic or inorganic acid, preferably pharmaceutically acceptable. This salt is preferably a sulfate that may arise in a more or less solvated or hydrated.
-
[0023]We can also prepare vincristine dissolved in a physiologically acceptable solvent and ready to be injected.
-
[0024]In particular, vincristine sulfate is obtained by addition of H 2 S0 4 to a solution of vincristine gross or recrystallized from ethanol, dissolved in a mixture of methylene chloride and anhydrous ethanol, partial removal in vacuo chloride methylene and crystallization.
-
[0025]Vincristine sulfate thus obtained has a purity sufficient for use as a medicament, particularly in the form of injectable solutions.

Madagascar Periwinkle: Public Domain Illustration by Sydenham Edwards
The Madagascar periwinkle, an attractive flowering plant, contains the powerful anti-cancer chemicals vinblastine and vincristine. Velvet beans, which are named from the covering of soft hairs on the young plant, contain L-dopa, a very helpful chemical in the treatment of Parkinson’s disease. The Madagascar periwinkle and the velvet bean are just two of the large number of plants that have been found to contain medicinal chemicals. There are almost certainly many more plants that have undiscovered health benefits.
The Madagascar Periwinkle
The Madagascar periwinkle is native to Madagascar and India, but is now grown in many countries as a garden plant. It has also escaped from gardens and grows as a weed. The red, purple, pink or white flowers often have a center which is a different color from the rest of the flower. Madagascar periwinkles may grow up to one meter tall and have glossy green leaves.
The sap of the Madagascar periwinkle, which has a milky appearance and is poisonous, contains vinblastine, vincristine and many other alkaloids. Researchers are discovering that many of these alkaloids are biologically active inside the human body.
Vinblastine and Vincristine
Vinblastine and vincristine have very similar chemical structures, but their effects on the body are not the same. Vinblastine is used to treat specific types of cancer, such as Hodgkin’s disease, breast cancer, testicular cancer and non-small cell lung cancer. Vincristine is used in the treatment of acute lymphoblastic leukemia (ALL) and has provided a great breakthrough in successful treatment of this disease in children. When vincristine is added to the treatment regimen for children suffering from ALL, the survival rate reaches eighty percent. Vincristine is not so impressive in the treatment of ALL in adults.
Cells contain a supporting network of protein tubules, which are known as microtubules. Microtubules also play a vital role in the process of cell division. Before a cell divides, each chromosome in the cell is replicated. The replicated chromosomes are separated from their partners and pulled to opposite ends of the cell by microtubules during a process called mitosis. The cell then divides down the middle.
Vinblastine and vincristine stop microtubule formation during mitosis and therefore prevent cells from reproducing. This effect is strongest in cells that have a high rate of division, such as cancer cells. However, vinblastine and vincristine also affect cells lining the intestine, the cells in the bone marrow that produce blood cells, and the cells in the hair follicles, since these too have a high rate of cell division.
Possible vinblastine or vincristine side effects include constipation, hair loss, a low platelet count, which can cause increased bleeding, a low white blood cell count, which can lead to increased infections, or a low red blood cell count, resulting in anemia. There may occasionally be nerve damage, possibly due to the effect of the medicines on the microctubules in the nerve cells. Vincristine is more likely to cause nerve damage than vinblastine.

,,,,,,,,,,,,,


Total synthesis of (+)-vincristine (2). TFA, trifluoroacetic acid or trifluoroacetyl; DBU, 1,8-diazabicyclo[5.4.0]undec-7-ene.
Stereocontrolled total synthesis of (+)-vincristine

………………….
see docstoc presentation
click below
Vincristine
var docstoc_docid=”51697405″;var docstoc_title=”Vincristine”;var docstoc_urltitle=”Vincristine”;
…………………….
isolation
Kumar A, Patil D, Rajamohanan PR, Ahmad A (2013)
Isolation, Purification and Characterization of Vinblastine and Vincristine from Endophytic Fungus Fusarium oxysporumIsolated from Catharanthus roseus. PLoS ONE 8(9): e71805. doi:10.1371/journal.pone.0071805
http://www.plosone.org/article/info%3Adoi%2F10.1371%2Fjournal.pone.0071805
Isolation, purification and characterization of vinblastine and vincristine from the endophytic fungus Fusarium oxysporum
A two stage fermentation procedure was employed for the isolation of vinblastine and vincristine by Fusarium oxysporum. In the first stage, 500 ml Erlenmeyer flasks containing 100 ml medium (MGYP, (0.3%) malt extract, (1.0%) glucose, (0.3%) yeast extract and (0.5%) peptone) were inoculated with 7 days old culture and incubated at 28°C on a rotary shaker (240 rpm) for 4–5 days, which was used as seed culture (I stage). Later, 10 ml seed culture was transferred to 500 ml Erlenmeyer flask containing 100 ml production medium called as vinca medium-1 (Glucose: 3%, Succinic acid: 1%, Sodium benzoate: 100 mg, Peptone: 1%, Magnesium sulphate: 3.6 mg, Biotin: 1 mg, Thiamine: 1 mg, Pyridoxal: 1 mg, Calcium pentothenate: 1 mg, Phosphate buffer: 1 ml (pH 6.8), L-Tryptophan: 0.1%, Geranium oil: 0.05%.) which were incubated at 28°C for 20 days as shake culture (II stage), after which it was harvested and used for further study. Culture filtrates and mycelia were separated with the help of muslin cloth and then lyophilized. Lyophilized culture filtrate was extracted using ethyl acetate as a solvent system. The organic layer was separated from the aqueous layer using separating funnel. The extraction was repeated thrice and the solvent was dried using anhydrous sodium sulphate and concentrated under vacuum using rotavapour at 40°C in order to get crude extract. A small amount of crude extract was dissolved in ethyl acetate and subjected to thin layer chromatography (TLC) on silica gel-G (0.5 mm thickness) using chloroform:methanol (8:2) as a solvent system. The TLC plates were sprayed with ceric ammonium sulphate reagent. Vinca alkaloids spots produced brilliant violet color as well as purple color with above spraying reagent. Purification of fungal vinblastine and vincristine were done by silica gel column chromatography. The crude extract was loaded on silica gel column (60–120 mesh size, 40 cm×2 cm length width) pre-equilibrated with chloroform and eluted with a gradient of chloroform:methanol (100% chloroform, 9:1, 8:2, 7:3, 1:1 and 3:7 and 100% methanol). Fractions containing compounds with Rf values similar to that of the standard vinblastine and vincristine were pooled and subjected to preparative TLC on a 0.5 mm thick (20 cm×20 cm) silica plate and developed in chloroform:methanol (8:2) solvent system. The putative bands of fungal vinblastine and vincristine were scraped and eluted out with methanol. Purity of the isolated compounds was checked on TLC in the solvent systems such as (a) chloroform:methanol (8:2) (b) chloroform:methanol (9:1) and (c) ethyl acetate: acetonitrile (8:2).
http://www.plosone.org/article/info%3Adoi%2F10.1371%2Fjournal.pone.0071805
see also
http://www.ncbi.nlm.nih.gov/pubmed/20209002
ALSO
large-scale isolation of native catharantine, vindoline and 3′,4′-anhydrovinblastine whereby the isolation of vincristine, vinblastine, leurosine and the corresponding desacetoxy, desacetyl and N-desmethyl derivatives in a manner known per se can also be accomplished.
For the isolation of the two monoindole alkaloids: vindoline and catharantine from the dried plant Vinca rosea L. Svoboda [J. Am. Pharm. Assoc. 48, (11), 659 (1959)] described a method, which can be accomplished only with a very modest yield. From 1 kg. of the dried plant–subjecting the whole plant to a suitable treatment–approximately 0.6 g. of vindoline and 0.05 g. of catharantine were obtained.
3′,4′-ANHYDROVINBLASTINE UNTIL NOW HAS NEITHER BEEN ISOLATED FROM THE PLANT Vinca rosea L. nor identified in it.
For the preparation of the diindole alkaloid components starting from the leaves of Vinca rosea L. there are more methods known in the art (U.S. Pat. nos. 3,097,137; 3,205,220; 3,225,030 and Hungarian Pat. Nos. 153,200; 154,715; 160,967 and 164,958 as well as Austrian Pat. Nos. 313,435, 313,485, Australian pat. No. 458,629 and Swiss Pat. No. 572,488 and British Pat Nos. 1,412,932, 1,382,460 corresponding to the preceding two patents). According to these known processes from 1 kg. of the dried leaves of Vinca rosea L. about 0.1 to 0.2 g. of leurosine can be obtained and vinblastine, vincristine and optionally the corresponding N-desmethyl, desacetyl and desacetoxy derivatives are also simultaneously isolated.
Further on it is well known that the synthetic catharantine and vindoline may be coupled by the Polonovszky reaction to give 3′,4′-anhydrovinblastine which can thereafter be epoxidized to leurosine [Potier et al. Tetrahedron Letters 3945 (1976); DT-OS 25 58,124; Helv. Chim. Acta 59, 2858 (1976); Heterocycles 4, 997 (1976), Belgian patent specification No. 842,200 equivalent to U.S. patent application Ser. No. 582,372]. Leurosine itself has a valuable tumour growth inhibiting activity and the N-desmethyl-N-formyl derivative thereof is the most promising substance against leukemia (Hungarian Pat. No. 165,986 equivalent to U.S. patent application Ser. No. 422,100, and Austrian Pat. No. 332,566 which has issued as British Pat. No. 1,412,932).
