
Home » Posts tagged 'FDA 2022' (Page 2)
Tag Archives: FDA 2022
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Vutrisiran sodium, ALN 65492, Votrisiran
RNA, (Um-sp-(2′-deoxy-2′-fluoro)C-sp-Um-Um-Gm-(2′-deoxy-2′-fluoro)G-Um-Um-(2′-deoxy-2′-fluoro)A-Cm-Am-Um-Gm-(2′-deoxy-2′-fluoro)A-Am-(2′-deoxy-2′-fluoro)A-Um-Cm-Cm-Cm-Am-sp-Um-sp-Cm), complex with RNA (Um-sp-Gm-sp-Gm-Gm-Am-Um-(2′-deoxy-2′-fluoro)U-Um-(2′-deoxy-2′-fluoro)C-(2′-deoxy-2′-fluoro)A-(2′-deoxy-2′-fluoro)U-Gm-Um-Am-Am-Cm-Cm-Am-Am-Gm-Am) 3′-[[(2S,4R)-1-[29-[[2-(acetylamino)-2-deoxy-β-D-galactopyranosyl]oxy]-14,14-bis[[3-[[3-[[5-[[2-(acetylamino)-2-deoxy-β-D-galactopyranosyl]oxy]-1-oxopentyl]amino]propyl]amino]-3-oxopropoxy]methyl]-1,12,19,25-tetraoxo-16-oxa-13,20,24-triazanonacos-1-yl]-4-hydroxy-2-pyrrolidinyl]methyl hydrogen phosphate] (1:1)
Vutrisiran Sodium
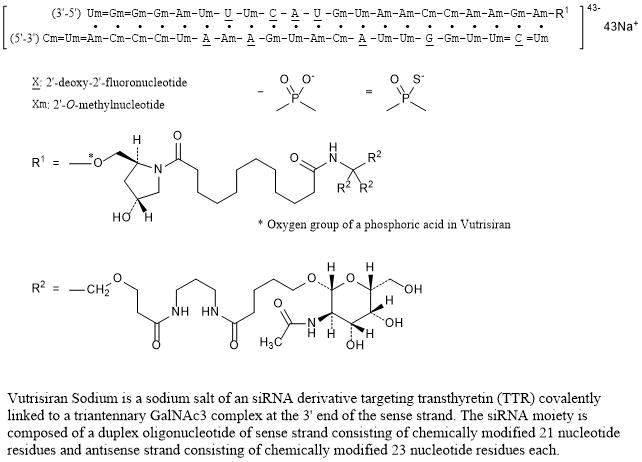
Nucleic Acid Sequence
Sequence Length: 44, 23, 2113 a 9 c 8 g 14 umultistranded (2); modified
Vutrisiran sodium
- ALN 65492
- Votrisiran
C530H672F9N171Na43O323P43S6 : 17289.77
[1867157-35-4 , Vutrisiran]
| Formula | C530H672F9N171O323P43S6.43Na ORC530H672F9N171Na43O323P43S6 |
|---|---|
| CAS | 1867157-35-4 , VURISIRAN |
| Mol weight | 17289.7661 |
FDA APPROVED, AMVUTTRA, 2022/6/13
| ブトリシランナトリウム |
| Efficacy | Gene expression regulator |
|---|---|
| Disease | Polyneuropathy of hereditary transthyretin-mediated amyloidosis [D |
| Comment | RNA interference (RNAi) drug Treatment of transthyretin (TTR)-mediated amyloidosis (ATTR amyloidosis) |
UNII28O0WP6Z1P UNII
Vutrisiran
Vutrisiran Sodium is a sodium salt of an siRNA derivative targeting transthyretin (TTR) covalently linked to a triantennary GalNAc3 complex at the 3’ end of the sense strand. The siRNA moiety is composed of a duplex oligonucleotide of sense strand consisting of chemically modified 21 nucleotide residues and antisense strand consisting of chemically modified 23 nucleotide residues each.
Vutrisiran is a double-stranded small interfering ribonucleic acid (siRNA) that targets wild-type and mutant transthyretin (TTR) messenger RNA (mRNA).7 This siRNA therapeutic is indicated for the treatment of neuropathies associated with hereditary transthyretin-mediated amyloidosis (ATTR), a condition caused by mutations in the TTR gene.2 More than 130 TTR mutations have been identified so far,3 but the most common one is the replacement of valine with methionine at position 30 (Val30Met).2 The Val30Met variant is the most prevalent among hereditary ATTR patients with polyneuropathy, especially in Portugal, France, Sweden, and Japan.2
TTR mutations lead to the formation of misfolded TTR proteins, which form amyloid fibrils that deposit in different types of tissues. By targeting TTR mRNA, vutrisiran reduces the serum levels of TTR.6,7 Vutrisiran is commercially available as a conjugate of N-acetylgalactosamine (GalNAc), a residue that enables the delivery of siRNA to hepatocytes.5,7 This delivery platform gives vutrisiran high potency and metabolic stability, and allows for subcutaneous injections to take place once every three months.8 Another siRNA indicated for the treatment of polyneuropathy associated with hereditary ATTR is patisiran.2 Vutrisiran was approved by the FDA in June 2022.
CLIP
https://www.nature.com/articles/s41392-020-0207-x
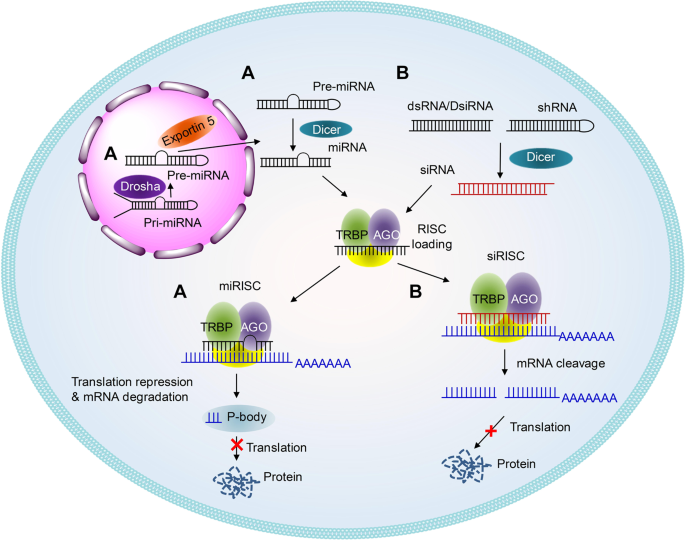
Schematic illustrations of the working mechanisms of miRNA (a) and siRNA (b)
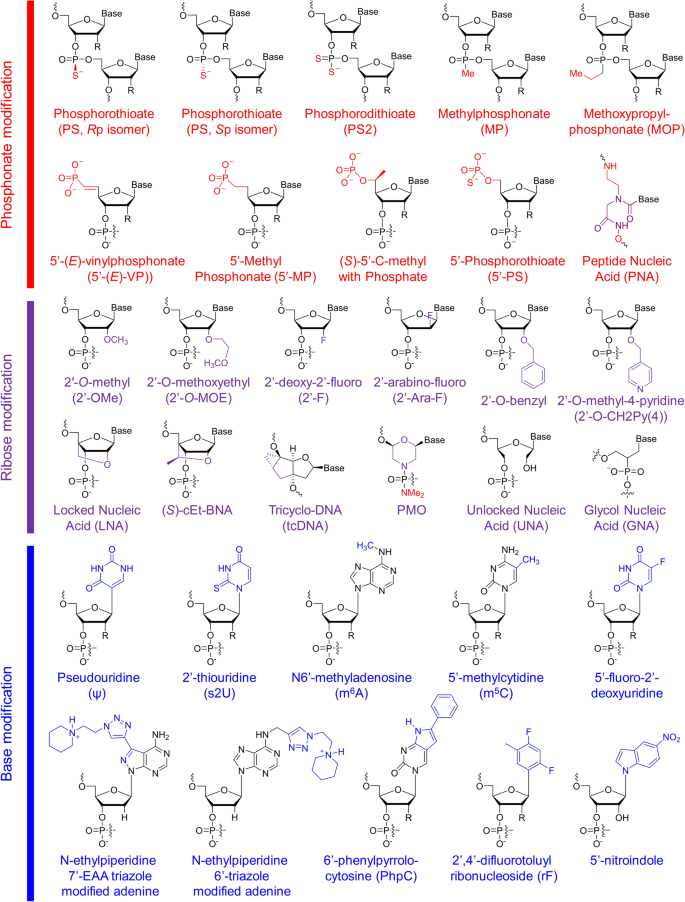
Structures of chemical modifications and analogs used for siRNA and ASO decoration. According to the modification site in the nucleotide acid, these structures can be divided into three classes: phosphonate modification, ribose modification and base modification, which are marked in red, purple and blue, respectively. R = H or OH, for RNA or DNA, respectively. (S)-cEt-BNA (S)-constrained ethyl bicyclic nucleic acid, PMO phosphorodiamidate morpholino oligomer
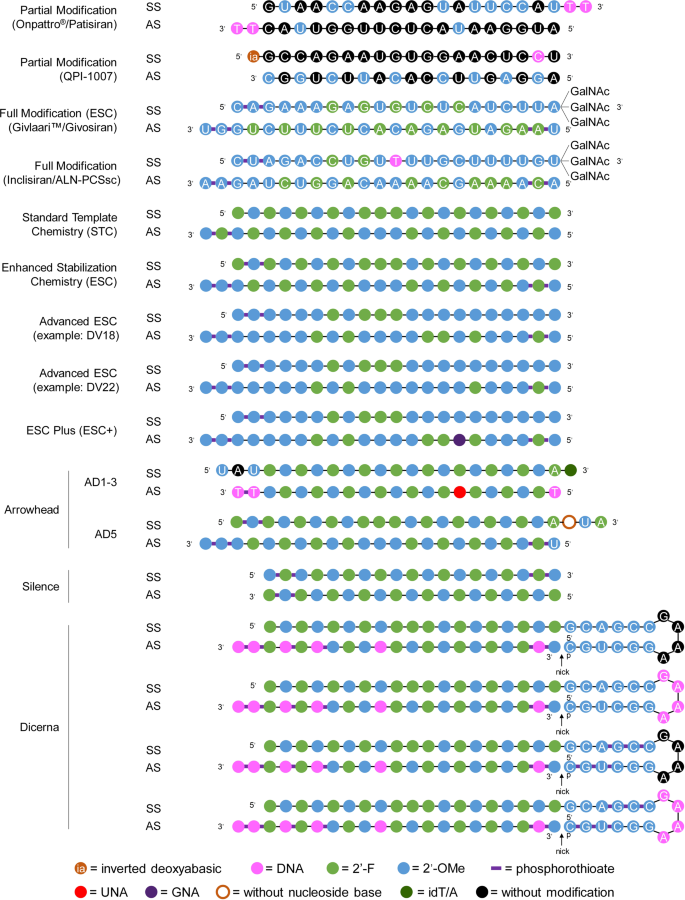
Representative designs for the chemical modification of siRNA. The sequences and modification details for ONPATTRO®, QPI-1007, GIVLAARI™ and inclisiran are included. The representative siRNA modification patterns developed by Alnylam (STC, ESC, advanced ESC and ESC+) and arrowhead (AD1-3 and AD5) are shown. Dicerna developed four GalNAc moieties that can be positioned at the unpaired G–A–A–A nucleotides of the DsiRNA structure. 2′-OMe 2′-methoxy, 2′-F 2′-fluoro, GNA glycol nucleic acid, UNA unlocked nucleic acid, SS sense strand, AS antisense strand
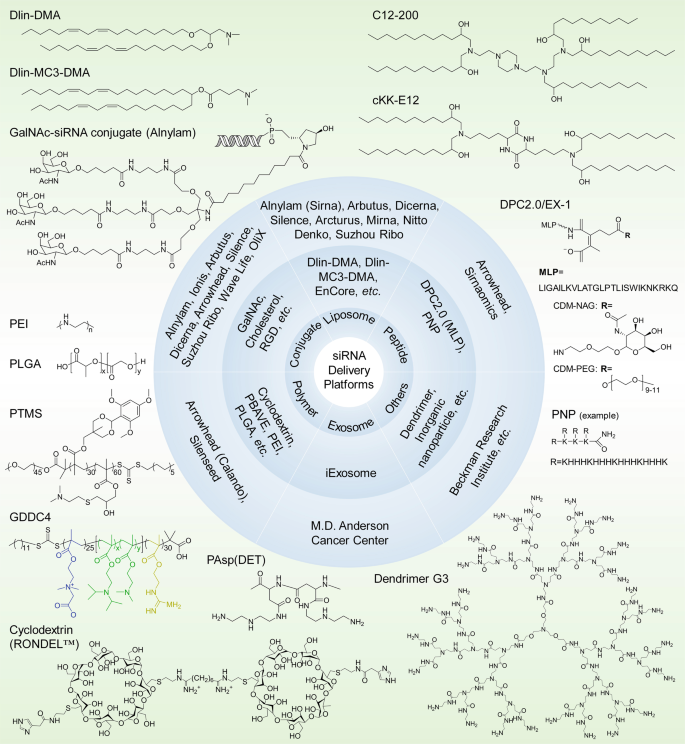
siRNA delivery platforms that have been evaluated preclinically and clinically. Varieties of lipids or lipidoids, siRNA conjugates, peptides, polymers, exosomes, dendrimers, etc. have been explored and employed for siRNA therapeutic development by biotech companies or institutes. The chemical structures of the key component(s) of the discussed delivery platforms, including Dlin-DMA, Dlin-MC3-DMA, C12-200, cKK-E12, GalNAc–siRNA conjugates, MLP-based DPC2.0 (EX-1), PNP, PEI, PLGA-based LODER, PTMS, GDDC4, PAsp(DET), cyclodextrin-based RONDEL™ and dendrimer generation 3 are shown. DLin-DMA (1,2-dilinoleyloxy-3-dimethylaminopropane), DLin-MC3-DMA (6Z,9Z,28Z,31Z)-heptatriaconta-6,9,28,31-tetraen-19-yl-4-(dimethylamino) butanoate, DPC Dynamic PolyConjugates, MLP membrane-lytic peptide, CDM carboxylated dimethyl maleic acid, PEG polyethylene glycol, NAG N-acetylgalactosamine, PNP polypeptide nanoparticle, PEI poly(ethyleneimine), LODER LOcal Drug EluteR, PLGA poly(lactic-co-glycolic) acid, PTMS PEG-PTTMA-P(GMA-S-DMA) poly(ethylene glycol)-co-poly[(2,4,6-trimethoxybenzylidene-1,1,1-tris(hydroxymethyl))] ethane methacrylate-co-poly(dimethylamino glycidyl methacrylate), GDDC4 PG-P(DPAx-co-DMAEMAy)-PCB, where PG is guanidinated poly(aminoethyl methacrylate) PCB is poly(carboxybetaine) and P(DPAx-co-DMAEMAy) is poly(dimethylaminoethyl methacrylate-co-diisopropylethyl methacrylate), PEG-PAsp(DET) polyethylene glycol-b-poly(N′-(N-(2-aminoethyl)-2-aminoethyl) aspartamide), PBAVE polymer composed of butyl and amino vinyl ether, RONDEL™ RNAi/oligonucleotide nanoparticle delivery
REF
Nucleic Acids Research (2019), 47(7), 3306-3320.
Drug Metabolism & Disposition (2019), 47(10), 1183-1201.
PATENT
WO 2020128816
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2020128816
The present invention relates to pharmaceutical compositions and methods of treatment comprising administering to a patient in need thereof a combination of a benzoxazole derivative transthyretin stabilizer or a pharmaceutically acceptable salt or prodrug thereof and an additional therapeutic agent for the treatment of transthyretin amyloidosis. Particularly, the present invention relates to pharmaceutical compositions and methods of treatment comprising administering to a patient in need thereof 2-(3,5-dichlorophenyl)-1,3-benzoxazole-6-carboxylic acid or a pharmaceutically acceptable salt or prodrug thereof and one or more additional therapeutic agent for the treatment of transthyretin amyloidosis.
The present invention relates to pharmaceutical compositions and methods of treatment comprising administering to a patient in need thereof a combination of a benzoxazole derivative transthyretin stabilizer or a pharmaceutically acceptable salt or prodrug thereof and one or more additional therapeutic agent. Particularly, the present invention relates to pharmaceutical compositions and methods of treatment comprising administering to a patient in need thereof 2-(3,5-dichlorophenyl)-1,3-benzoxazole-6-carboxylic acid or a pharmaceutically acceptable salt or prodrug thereof and one or more additional therapeutic agent. The compositions and methods of the invention are useful in stabilizing transthyretin, inhibiting transthyretin misfolding, proteolysis, and treating amyloid diseases associated thereto.
Transthyretin (TTR) is a 55 kDa homotetrameric protein present in serum and cerebral spinal fluid and which functions as a transporter of L-thyroxine (T4) and holo-retinol binding protein (RBP). TTR has been found to be an amyloidogenic protein that, under certain conditions, can be transformed into fibrils and other aggregates which can lead to disease pathology such as polyneuropathy or cardiomyopathy in humans.
US Patent Nos. 7,214,695; 7,214,696; 7,560,488; 8, 168.683; and 8,653,119 each of which is incorporated herein by reference, discloses benzoxazole derivatives which act as transthyretin stabilizers and are of the formula
or a pharmaceutically acceptable salt thereof; wherein Ar is 3,5-difluorophenyl, 2,6-difluorophenyl, 3,5-dichlorophenyl, 2,6-dichlorophenyl, 2-(trifluoromethyl)phenyl or 3-(trifluoromethyl)phenyl. Particularly, 2-(3,5-dichlorophenyl)-1,3-benzoxazole-6-carboxylic acid (tafamidis) of the formula
is disclosed therein. Tafamidis is an orally active transthyretin stabilizer that inhibits tetramer dissociation and proteolysis that has been approved in certain jurisdictions for the treatment of transthyretin polyneuropathy (TTR-PN) and is currently in development for the treatment of transthyretin cardiomyopathy (TTR-CM). US Patent No. 9,249, 112, also incorporated herein by reference, discloses polymorphic forms of the meglumine salt of 2-(3,5-dichlorophenyl)-1,3-benzoxazole-6-carboxylic acid (tafamidis meglumine). US Patent No. 9,770,441 discloses polymorphic forms of the free acid of 2-(3,5-dichlorophenyl)-1,3-benzoxazole-6-carboxylic acid (tafamidis), and is also incorporated by reference herein.
Summary of the Invention
The present invention provides pharmaceutical compositions and methods comprising the compound 2-(3,5-dichlorophenyl)-1,3-benzoxazole-6-carboxylic acid or a pharmaceutically acceptable salt or prodrug thereof, and one or more additional therapeutic agent. Particular embodiments of this invention are pharmaceutical compositions and methods comprising 2-(3,5-dichlorophenyl)-1,3-benzoxazole-6-carboxylic acid or a pharmaceutically acceptable salt or prodrug thereof, and one or more additional therapeutic agents selected from the group consisting of agents that lower plasma levels of TTR such as an antisense therapy, TTR gene editing therapy, transcriptional modulators, translational modulators, TTR protein degraders and antibodies that bind and reduce TTR levels; amyloid reduction therapies such as anti amyloid antibodies (either TTR selective or general), stimulators of amyloid clearance, fibril disruptors and therapies that inhibit amyloid nucleation; other TTR stabilizers; and TTR modulators such as therapeutics which inhibit TTR cleavage. Particularly, the present invention provides pharmaceutical compositions and methods comprising tafamidis or tafamidis meglumine salt with one or more additional therapeutic agents. More particularly, the present invention provides pharmaceutical compositions and the present invention provides pharmaceutical compositions and methods comprising tafamidis or tafamidis meglumine salt with one or more additional therapeutic agents. More particularly, the present invention provides pharmaceutical compositions and the present invention provides pharmaceutical compositions and methods comprising tafamidis or tafamidis meglumine salt with one or more additional therapeutic agents. More particularly, the present invention provides pharmaceutical compositions and
methods comprising a polymorphic form of tafamidis free acid or a polymorphic form of tafamidis meglumine salt with one or more additional therapeutic agents.
The present invention also provides a method of treating or preventing transthyretin amyloidosis in a patient, the method comprising administering to a patient in need thereof a therapeutically or prophylactically effective amount of 2-(3,5-dichlorophenyl)-1,3-benzoxazole- 6-carboxylic acid or a pharmaceutically acceptable salt or prodrug thereof, and one or more additional therapeutic agents.
A particular embodiment of the present method of treatment is the method comprising a pharmaceutical composition comprising 2-(3,5-dichlorophenyl)-1,3-benzoxazole-6-carboxylic acid or a pharmaceutically acceptable salt or prodrug thereof, and one or more additional therapeutic agent are administered orally. Additional embodiments of this invention are methods of treatment as described above wherein the 2-(3,5-dichlorophenyl)-1,3-benzoxazole-6-carboxylic acid or a pharmaceutically acceptable salt or prodrug thereof, and one or more additional therapeutic agent are administered parenterally (intravenously or subcutaneously). Further embodiments of this invention are methods of treatment wherein the 2-(3,5-dichlorophenyl)-1, 3-benzoxazole-6-carboxylic acid or a pharmaceutically acceptable salt or prodrug thereof is administered orally and the one or more additional therapeutic agent is administered either orally or parenterally. Another embodiment of the present invention is wherein a pharmaceutical composition comprising 2-(3,5-dichlorophenyl)-1,3-benzoxazole-6-carboxylic acid or a pharmaceutically acceptable salt or prodrug thereof in combination with one or more additional therapeutic agent is administered parenterally and then 2-(3,5-dichlorophenyl)-1,3-benzoxazole-6-carboxylic acid or a pharmaceutically acceptable salt or prodrug thereof is administered orally. A particular method of treatment is a method of treating TTR amyloidosis such as TTR polyneuropathy or TTR Another embodiment of the present invention is wherein a pharmaceutical composition comprising 2-(3,5-dichlorophenyl)-1,3-benzoxazole-6-carboxylic acid or a pharmaceutically acceptable salt or prodrug thereof in combination with one or more additional therapeutic agent is administered parenterally and then 2-(3,5-dichlorophenyl)-1,3-benzoxazole-6-carboxylic acid or a pharmaceutically acceptable salt or prodrug thereof is administered orally. A particular method of treatment is a method of treating TTR amyloidosis such as TTR polyneuropathy or TTR Another embodiment of the present invention is wherein a pharmaceutical composition comprising 2-(3,5-dichlorophenyl)-1,3-benzoxazole-6-carboxylic acid or a pharmaceutically acceptable salt or prodrug thereof in combination with one or more additional therapeutic agent is administered parenterally and then 2-(3,5-dichlorophenyl)-1,3-benzoxazole-6-carboxylic acid or a pharmaceutically acceptable salt or prodrug thereof is administered orally. A particular method of treatment is a method of treating TTR amyloidosis such as TTR polyneuropathy or TTR 5-dichlorophenyl)-1,3-benzoxazole-6-carboxylic acid or a pharmaceutically acceptable salt or prodrug thereof is administered orally. A particular method of treatment is a method of treating TTR amyloidosis such as TTR polyneuropathy or TTR 5-dichlorophenyl)-1,3-benzoxazole-6-carboxylic acid or a pharmaceutically acceptable salt or prodrug thereof is administered orally. A particular method of treatment is a method of treating TTR amyloidosis such as TTR polyneuropathy or TTR
cardiomyopathy, the method comprising administering to a patient in need thereof a therapeutically effective amount of 2-(3,5-dichlorophenyl)-1,3-benzoxazole-6-carboxylic acid or a pharmaceutically acceptable salt or prodrug thereof in combination with one or more additional therapeutic agents.
Brief Description of the Drawings
REF
Biochemical Pharmacology (Amsterdam, Netherlands) (2021), 189, 114432.
PATENT
WO 2021041884
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2021041884
Exemplary RNAi agents that reduce the expression of TTR include patisiran and vutrisiran.
The ter s “antisense polynucleotide agent”, “antisense oligonucleotide”, “antisense compound”, and “antisense agent” as used interchangeably herein, refer to an agent comprising a single-stranded oligonucleotide that specifically binds to the target nucleic acid molecules via hydrogen bonding (e.g., Watson-Crick, Hoogsteen, or reversed Hoogsteen hydrogen bonding) and inhibits the expression of the targeted nucleic acid by an antisense mechanism of action, e.g., by RNase H. In some embodiments, an antisense agent is a nucleic acid therapeutic that acts by reducing the expression of a target gene, thereby reducing the expression of the polypeptide encoded by the target gene. Exemplary antisense agents that reduce the expression of TTR include inotersen and Ionis 682884/ ION-TTR-LRx (see, e.g., WO2014179627 which is incorporated by reference in its entirety). Further antisense agents that reduce the expression of TTR are provided, for example in WO2011139917 and WO2014179627, each of which is incorporated by reference in its entirety.
REF
Clinical Pharmacology & Therapeutics (Hoboken, NJ, United States) (2021), 109(2), 372-382
Annals of Plastic Surgery (2021), 86(2S_Suppl_1), S23-S29.
Journal of Cardiovascular Pharmacology (2021), 77(5), 544-548.
Annals of Pharmacotherapy (2021), 55(12), 1502-1514.
Kidney International (2022), 101(2), 208-211
//////////

AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
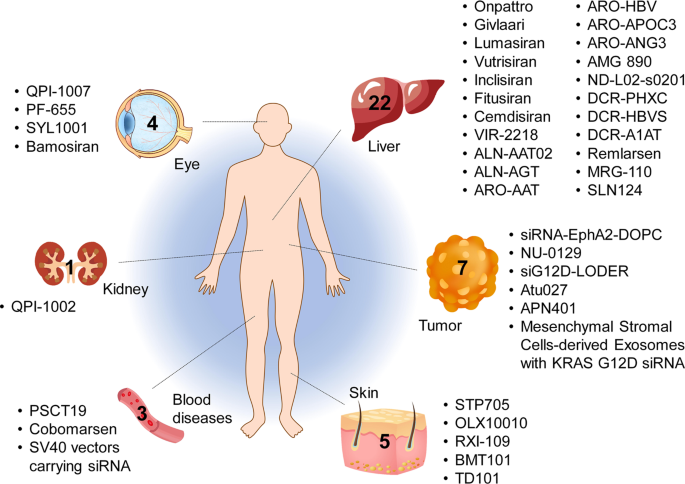
Tissues targeted by siRNA and miRNA therapeutics currently being investigated at the clinical stage. The corresponding therapeutic names are shown beside the tissues
CLIP
Vutrisiran An Investigational RNAi Therapeutic for ATTR Amyloidosis Vutrisiran has not been approved by the U.S. Food and Drug Administration, European Medicines Agency, or any other regulatory authority and no conclusions can or should be drawn regarding the safety or effectiveness of this investigational therapeutic. Overview • Vutrisiran is an investigational RNAi therapeutic in development for the treatment of transthyretin-mediated (ATTR) amyloidosis, which encompasses both hereditary ATTR (hATTR) amyloidosis and wild-type ATTR (wtATTR) amyloidosis.1, 2 • Vutrisiran inhibits the production of disease-causing transthyretin (TTR) protein by the liver, leading to a reduction in the level of TTR in the blood.1, 2 • Vutrisiran is administered subcutaneously (under the skin) and utilizes one of Alnylam’s delivery platforms known as the Enhanced Stabilization Chemistry (ESC)-GalNAc-conjugate delivery platform.1, 2 • Vutrisiran is administered every three months.2 • Vutrisiran is under review by the U.S. Food and Drug Administration (FDA), the European Medicines Agency (EMA), and the Brazilian Health Regulatory Agency (ANVISA). Vutrisiran has been granted Orphan Drug Designation in the U.S. and the European Union (EU) for the treatment of ATTR amyloidosis. Vutrisiran has also been granted a Fast Track designation in the U.S. for the treatment of the polyneuropathy of hATTR amyloidosis in adults. In the U.S. vutrisiran has received an action date under the Prescription Drug User Fee Act (PDUFA) of April 14, 2022. The Company received orphan drug designation in Japan. Alnylam has global commercial rights to vutrisiran, assuming regulatory approvals. Clinical Development • A Phase 1 clinical study of vutrisiran was conducted in 80 healthy volunteers (60 received vutrisiran and 20 received placebo). Vutrisiran demonstrated an acceptable safety profile and a single dose reduced serum TTR for a period of at least 90 days.2 • The safety and efficacy of vutrisiran are being evaluated in the HELIOS Phase 3 clinical program, currently consisting of two clinical trials: HELIOS-A and HELIOS-B. • HELIOS-A is a randomized, open-label, global multi-center Phase 3 study of 164 adult patients with hATTR amyloidosis with polyneuropathy.1 • The primary endpoint of HELIOS-A is change from baseline in the modified Neuropathy Impairment Score +7 (mNIS+7) at 9 months. • Secondary endpoints at 9 months include the Norfolk Quality of Life-Diabetic Neuropathy (Norfolk QoL-DN) Total Score and the 10-Meter Walk Test (10-MWT). • The 9-month endpoints will be analyzed at 18 months with the addition of other secondary endpoints. • HELIOS-B is a randomized, double-blind, placebo-controlled Phase 3 study of 655 adult patients with ATTR amyloidosis with cardiomyopathy (including both hATTR and wtATTR amyloidosis).3 • The primary endpoint will evaluate the efficacy of vutrisiran versus placebo for the composite outcome of all-cause mortality and recurrent cardiovascular (CV) events (CV hospitalizations and urgent heart failure (HF) visits) at 30-36 months. • Secondary endpoints include the change from baseline in the 6-minute walk test (6-MWT), health status measured using the Kansas City Cardiomyopathy Questionnaire Overall Summary (KCCQ-OS), echocardiographic assessments of mean left ventricular wall thickness and global longitudinal strain, the N-terminal prohormone B-type natriuretic peptide (NT-proBNP) as a cardiac biomarker, and all-cause mortality, rate of recurrent CV events, and composite of all-cause mortality and recurrent all-cause hospitalizations and urgent HF visits at month 30 or 30-36 months. Page 2 © 2021 Alnylam Pharmaceuticals, Inc. All rights reserved. TTRsc02-USA-00012 v4 About ATTR Amyloidosis • ATTR amyloidosis is a rare, underdiagnosed, rapidly progressive, debilitating, and fatal disease caused by misfolded TTR that accumulates as amyloid fibrils in multiple tissues including the nerves, heart, and GI tract. There are two types of ATTR amyloidosis: hATTR amyloidosis and wtATTR amyloidosis.4,5,6 • hATTR amyloidosis is an inherited condition that is caused by variants (i.e., mutations) in the transthyretin (TTR) gene.5,7,8 TTR protein is produced primarily in the liver and is normally a carrier of vitamin A.9 The variant results in misfolded TTR proteins that accumulate as amyloid deposits in multiple tissues, including the nerves, heart and gastrointestinal (GI) tract.5, 6, 7 It is a multisystem disease that can include sensory and motor, autonomic, and cardiac symptoms. The condition can have a debilitating impact on a patient’s life and may lead to premature death with a median survival of 4.7 years following diagnosis.8,10 It is estimated that there are approximately 50,000 patients with hATTR amyloidosis worldwide.11 • wtATTR amyloidosis is a non-hereditary condition that occurs when misfolded wild-type TTR accumulates as amyloid deposits in multiple organs. It predominantly manifests as cardiac symptoms, but other systems are also involved, and commonly leads to heart failure and mortality within 2.5 to 5.5 years.12,13,14,15,16,17,18,19 wtATTR amyloidosis affects an estimated 200,000-300,000 people worldwide.20 • Alnylam is committed to developing multiple treatment options for people who are living with ATTR amyloidosis to help manage the debilitating and progressive nature of the disease. For more information about vutrisiran, please contact media@alnylam.com. For more information on HELIOS-A (NCT03759379) and HELIOS-B (NCT04153149) please visit http://www.clinicaltrials.gov or contact media@alnylam.com. Current information as of November 2021
CLIP
Alnylam announces extension of review period for new drug vutrisiran to treat ATTR amyloidosis
Alnylam announces 3-month extension of review period for new drug application for vutrisiran to treat ATTR amyloidosis.
Alnylam Pharmaceuticals, Inc., a RNAi therapeutics company, announced that the FDA has extended the review timeline of the New Drug Application (NDA) for vutrisiran, an investigational RNAi therapeutic in development for the treatment of transthyretin-mediated (ATTR) amyloidosis, to allow for the review of newly added information related to the new secondary packaging and labelling facility.
Alnylam recently learned that the original third-party secondary packaging and labelling facility the Company planned to use for the vutrisiran launch was recently inspected and the inspection requires classification for the FDA to take action on the vutrisiran NDA. The inspection observations were not directly related to vutrisiran. In order to minimize delays to approval, Alnylam has identified a new facility to pack and label vutrisiran and submitted an amendment to the NDA for review by the FDA. The updated Prescription Drug User Fee Act (PDUFA) goal date to allow for this review is July 14, 2022. No additional clinical data have been requested by the FDA.
////////////Vutrisiran sodium, APPROVALS 2022, FDA 2022, FDA APPROVED, AMVUTTRA, 2022/6/13, ブトリシランナトリウム , ALN 65492, Votrisiran, siRNA

NEW DRUG APPROVALS
ONE TIME TO SUSTAIN AND MAINTAIN THIS BLOG
$10.00
Tirzepatide
YXEGTFTSDY SIXLDKIAQK AFVQWLIAGG PSSGAPPPS



Tirzepatide
チルゼパチド
LY3298176,
| Formula | C225H348N48O68 |
|---|---|
| CAS | 2023788-19-2 |
| Mol weight | 4813.4514 |
FDA APPROVED 2022/5/13, Mounjaro
| Class | Antidiabetic agent GLP-1 receptor agonist |
|---|---|
| Efficacy | Antidiabetic, Gastric inhibitory polypeptide receptor agonist, Glucagon-like peptide 1 (GLP-1) receptor agonist |
| Disease | Type 2 diabetes mellitus |

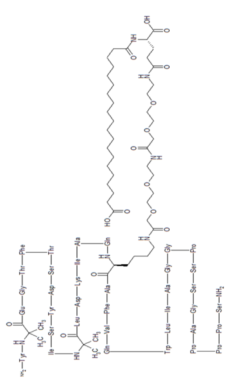
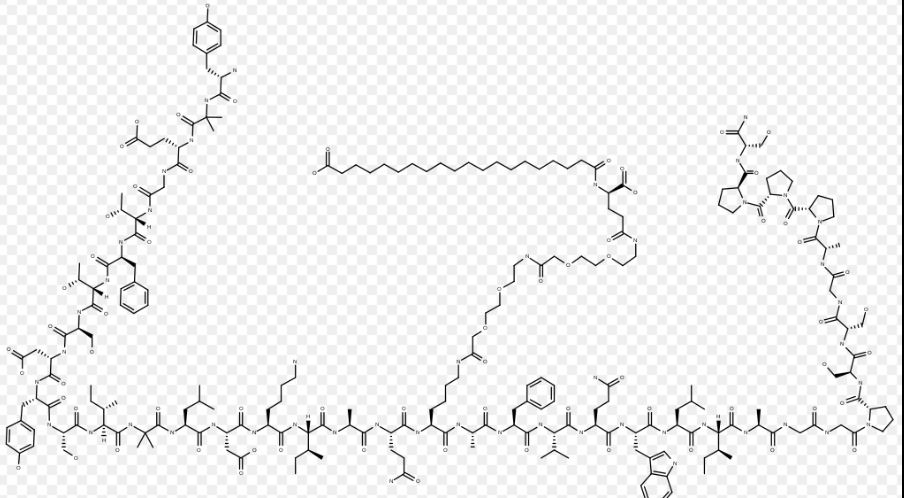
Tirzepatide is an agonist of human glucose-dependent insulinotropic polypeptide (GIP) and human glucagon-like peptide-1 (GLP-1) receptors, whose amino acid residues at positions 2 and 13 are 2-methylAla, and the C-terminus is amidated Ser. A 1,20-icosanedioic acid is attached to Lys at position 20 via a linker which consists of a Glu and two 8-amino-3,6-dioxaoctanoic acids. Tirzepatide is a synthetic peptide consisting of 39 amino acid residues.
C225H348N48O68 : 4813.45
[2023788-19-2]
L-Serinamide, L-tyrosyl-2-methylalanyl-L-α-glutamylglycyl-L-threonyl-L-phenylalanyl-L-threonyl-L-seryl-L-α-aspartyl-L-tyrosyl-L-seryl-L-isoleucyl-2-methylalanyl-L-leucyl-L-α-aspartyl-L-lysyl-L-isoleucyl-L-alanyl-L-glutaminyl-N6-[(22S)-22,42-dicarboxy-1,10,19,24-tetraoxo-3,6,12,15-tetraoxa-9,18,23-triazadotetracont-1-yl]-L-lysyl-L-alanyl-L-phenylalanyl-L-valyl-L-glutaminyl-L-tryptophyl-L-leucyl-L-isoleucyl-L-alanylglycylglycyl-L-prolyl-L-seryl-L-serylglycyl-L-alanyl-L-prolyl-L-prolyl-L-prolyl-
Other Names
- L-Tyrosyl-2-methylalanyl-L-α-glutamylglycyl-L-threonyl-L-phenylalanyl-L-threonyl-L-seryl-L-α-aspartyl-L-tyrosyl-L-seryl-L-isoleucyl-2-methylalanyl-L-leucyl-L-α-aspartyl-L-lysyl-L-isoleucyl-L-alanyl-L-glutaminyl-N6-[(22S)-22,42-dicarboxy-1,10,19,24-tetraoxo-3,6,12,15-tetraoxa-9,18,23-triazadotetracont-1-yl]-L-lysyl-L-alanyl-L-phenylalanyl-L-valyl-L-glutaminyl-L-tryptophyl-L-leucyl-L-isoleucyl-L-alanylglycylglycyl-L-prolyl-L-seryl-L-serylglycyl-L-alanyl-L-prolyl-L-prolyl-L-prolyl-L-serinamide
Tirzepatide, sold under the brand name Mounjaro,[1] is a medication used for the treatment type 2 diabetes.[2][3][4] Tirzepatide is given by injection under the skin.[2] Common side effects may include nausea, vomiting, diarrhea, decreased appetite, constipation, upper abdominal discomfort and abdominal pain.[2]
Glucagon-like peptide-1 (GLP-1) and glucose-dependent insulinotropic polypeptide (GIP) are hormones involved in blood sugar control.[2] Tirzepatide is a first-in-class medication that activates both the GLP-1 and GIP receptors, which leads to improved blood sugar control.[2] Tirzepatide was approved for medical use in the United States in May 2022.[2]
SYN
https://pubs.acs.org/doi/10.1021/acs.oprd.1c00108

The large-scale manufacture of complex synthetic peptides is challenging due to many factors such as manufacturing risk (including failed product specifications) as well as processes that are often low in both yield and overall purity. To overcome these liabilities, a hybrid solid-phase peptide synthesis/liquid-phase peptide synthesis (SPPS/LPPS) approach was developed for the synthesis of tirzepatide. Continuous manufacturing and real-time analytical monitoring ensured the production of high-quality material, while nanofiltration provided intermediate purification without difficult precipitations. Implementation of the strategy worked very well, resulting in a robust process with high yields and purity.
PATENT
- WO2016111971
- US2020023040
- WO2019245893
- US2020155487
- US2020155650
- WO2020159949CN112592387
- WO2021066600CN112661815
- WO2021154593
- US2021338769

NEW DRUG APPROVALS
ONE TIME TO MAINTAIN THIS BLOG SUBSCRIPTION
$10.00
//////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
Medical uses
Tirzepatide in indicated to improve blood sugar control in adults with type 2 diabetes, as an addition to diet and exercise.[2]
Contraindications
Tirzepatide should not be used in people with a personal or family history of medullary thyroid cancer or in people with multiple endocrine neoplasia syndrome type 2.[2]
Adverse effects
Preclinical, phase I, and phase II trials have indicated that tirzepatide exhibits similar adverse effects to other established GLP-1 receptor agonists, such as GLP-1 receptor agonist dulaglutide. These effects occur largely within the gastrointestinal tract.[5] The most frequently observed adverse effects are nausea, diarrhoea and vomiting, which increased in incidence with the dosage amount (i.e. higher likelihood the higher the dose). The number of patients who discontinued taking tirzepatide also increased as dosage increased, with patients taking 15 mg having a 25% discontinuation rate vs 5.1% for 5 mg patients and 11.1% for dulaglutide.[6] To a slightly lesser extent, patients also reported reduced appetite.[5] Other side effects reported were dyspepsia, constipation, abdominal pain, dizziness and hypoglycaemia.[7][8]
Pharmacology
Tirzepatide is an analogue of gastric inhibitory polypeptide (GIP), a human hormone which stimulates the release of insulin from the pancreas. Tirzepatide is a linear polypeptide of 39 amino acids which has been chemically modified by lipidation to improve its uptake into cells and its stability to metabolism.[9] The compound is administered as a weekly subcutaneous injection.[10] It completed phase III trials globally in 2021.[11][12]
Mechanism of action
Tirzepatide has a greater affinity to GIP receptors than to GLP-1 receptors, and this dual agonist behaviour has been shown to produce greater reductions of hyperglycemia compared to a selective GLP-1 receptor agonist.[3] Signaling studies have shown that this is due to tirzepatide mimicking the actions of natural GIP at the GIP receptor.[13] However, at the GLP-1 receptor, tirzepatide shows bias towards cAMP (a messenger associated with regulation of glycogen, sugar and lipid metabolism) generation, rather than β-arrestin recruitment. This combination of preference towards GIP receptor and distinct signaling properties at GLP-1 suggest this biased agonism increases insulin secretion.[13] Tirzepatide has also been shown to increase levels of adiponectin, an adipokine involved in the regulation of both glucose and lipid metabolism, with a maximum increase of 26% from baseline after 26 weeks, at the 10 mg dosage.[3]
Chemistry
Structure
Tirzepatide is an analog of the human GIP hormone with a C20 fatty-diacid portion attached, used to optimise the uptake and metabolism of the compound.[9] The fatty-diacid section (eicosanedioic acid) is linked via a glutamic acid and two (2-(2-aminoethoxy)ethoxy)acetic acid units to the side chain of the lysine residue. This arrangement allows for a much longer half life, extending the time between doses, because of its high affinity to albumin.[14]
Synthesis
The synthesis of tirzepatide was first disclosed in patents filed by Eli Lilly and Company.[15] This uses standard solid phase peptide synthesis, with an allyloxycarbonyl protecting group on the lysine at position 20 of the linear chain of amino acids, allowing a final set of chemical transformations in which the sidechain amine of that lysine is derivatized with the lipid-containing fragment.
Large-scale manufacturing processes have been reported for this compound.[16]
History
Indiana-based pharmaceutical company Eli Lilly and Company first applied for a patent for a method of glycemic control using tirzepatide in early 2016.[15] The patent was published late that year. After passing phase 3 clinical trials, Lilly applied for FDA approval in October 2021 with a priority review voucher.[17]
Following the completion of the pivotal SURPASS-2 trial no. NCT03987919, the company announced on 28 April that tirzepatide had successfully met their endpoints in obese and overweight patients without diabetes.[18] Alongside results from the SURMOUNT-1 trial no. NCT04184622, they suggest that tirzepatide may potentially be a competitor for existing diabetic medication semaglutide, manufactured by Novo Nordisk.[19][20]
In industry-funded preliminary trials comparing tirzepatide to the existing diabetes medication semaglutide (an injected analogue of the hormone GLP-1), tirzepatide showed minor improvement of reductions (2.01%–2.30% depending on dosage) in glycated hemoglobin tests relative to semaglutide (1.86%).[21] A 10 mg dose has also been shown to be effective in reducing insulin resistance, with a reduction of around 8% from baseline, measured using HOMA2-IR (computed with fasting insulin).[3] Fasting levels of IGF binding proteins like IGFBP1 and IGFBP2 increased following tirzepatide treatment, increasing insulin sensitivity.[3] A meta-analysis published by Dutta et al. showed that over 1-year clinical use, tirzepatide was observed to be superior to dulaglutide, semaglutide, degludec, and insulin glargine with regards to glycemic efficacy and obesity reduction. Tirzepatide is perhaps the most potent agent developed to date to tackle the global problem of “diabesity“.[22]
Society and culture
Names
Tirzepatide is the international nonproprietary name (INN).[23]
References
- ^ Jump up to:a b “Highlights of prescribing information” (PDF). accessdata.fda.gov. FDA. May 2022. Retrieved 14 May 2022.
- ^ Jump up to:a b c d e f g h i “FDA Approves Novel, Dual-Targeted Treatment for Type 2 Diabetes”. U.S. Food and Drug Administration (FDA) (Press release). 13 May 2022. Retrieved 13 May 2022.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ Jump up to:a b c d e Thomas MK, Nikooienejad A, Bray R, Cui X, Wilson J, Duffin K, et al. (January 2021). “Dual GIP and GLP-1 Receptor Agonist Tirzepatide Improves Beta-cell Function and Insulin Sensitivity in Type 2 Diabetes”. The Journal of Clinical Endocrinology and Metabolism. 106 (2): 388–396. doi:10.1210/clinem/dgaa863. PMC 7823251. PMID 33236115.
- ^ Coskun T, Sloop KW, Loghin C, Alsina-Fernandez J, Urva S, Bokvist KB, et al. (December 2018). “LY3298176, a novel dual GIP and GLP-1 receptor agonist for the treatment of type 2 diabetes mellitus: From discovery to clinical proof of concept”. Molecular Metabolism. 18: 3–14. doi:10.1016/j.molmet.2018.09.009. PMC 6308032. PMID 30473097.
- ^ Jump up to:a b Min T, Bain SC (January 2021). “The Role of Tirzepatide, Dual GIP and GLP-1 Receptor Agonist, in the Management of Type 2 Diabetes: The SURPASS Clinical Trials”. Diabetes Therapy. 12 (1): 143–157. doi:10.1007/s13300-020-00981-0. PMC 7843845. PMID 33325008.
- ^ Frias JP, Nauck MA, Van J, Kutner ME, Cui X, Benson C, et al. (November 2018). “Efficacy and safety of LY3298176, a novel dual GIP and GLP-1 receptor agonist, in patients with type 2 diabetes: a randomised, placebo-controlled and active comparator-controlled phase 2 trial”. The Lancet. 392 (10160): 2180–2193. doi:10.1016/S0140-6736(18)32260-8. PMID 30293770.
- ^ Frias JP, Nauck MA, Van J, Benson C, Bray R, Cui X, et al. (June 2020). “Efficacy and tolerability of tirzepatide, a dual glucose-dependent insulinotropic peptide and glucagon-like peptide-1 receptor agonist in patients with type 2 diabetes: A 12-week, randomized, double-blind, placebo-controlled study to evaluate different dose-escalation regimens”. Diabetes, Obesity & Metabolism. 22 (6): 938–946. doi:10.1111/dom.13979. PMC 7318331. PMID 31984598.
- ^ Dahl D, Onishi Y, Norwood P, Huh R, Bray R, Patel H, Rodríguez Á (February 2022). “Effect of Subcutaneous Tirzepatide vs Placebo Added to Titrated Insulin Glargine on Glycemic Control in Patients With Type 2 Diabetes: The SURPASS-5 Randomized Clinical Trial”. JAMA. 327 (6): 534–545. doi:10.1001/jama.2022.0078. PMID 35133415.
- ^ Jump up to:a b Ahangarpour M, Kavianinia I, Harris PW, Brimble MA (January 2021). “Photo-induced radical thiol-ene chemistry: a versatile toolbox for peptide-based drug design”. Chemical Society Reviews. Royal Society of Chemistry. 50 (2): 898–944. doi:10.1039/d0cs00354a. PMID 33404559. S2CID 230783854.
- ^ Bastin M, Andreelli F (2019). “Dual GIP-GLP1-Receptor Agonists In The Treatment Of Type 2 Diabetes: A Short Review On Emerging Data And Therapeutic Potential”. Diabetes, Metabolic Syndrome and Obesity: Targets and Therapy. 12: 1973–1985. doi:10.2147/DMSO.S191438. PMC 6777434. PMID 31686879.
- ^ “Tirzepatide significantly reduced A1C and body weight in people with type 2 diabetes in two phase 3 trials from Lilly’s SURPASS program” (Press release). Eli Lilly and Company. 17 February 2021. Retrieved 28 October 2021 – via PR Newswire.
- ^ “Lilly : Phase 3 Tirzepatide Results Show Superior A1C And Body Weight Reductions In Type 2 Diabetes”. Business Insider. RTTNews. 19 October 2021. Retrieved 28 October 2021.
- ^ Jump up to:a b Willard FS, Douros JD, Gabe MB, Showalter AD, Wainscott DB, Suter TM, et al. (September 2020). “Tirzepatide is an imbalanced and biased dual GIP and GLP-1 receptor agonist”. JCI Insight. 5 (17). doi:10.1172/jci.insight.140532. PMC 7526454. PMID 32730231.
- ^ Østergaard S, Paulsson JF, Kofoed J, Zosel F, Olsen J, Jeppesen CB, et al. (October 2021). “The effect of fatty diacid acylation of human PYY3-36 on Y2 receptor potency and half-life in minipigs”. Scientific Reports. 11 (1): 21179. Bibcode:2021NatSR..1121179O. doi:10.1038/s41598-021-00654-3. PMC 8551270. PMID 34707178.
- ^ Jump up to:a b US patent 9474780, Bokvist BK, Coskun T, Cummins RC, Alsina-Fernandez J, “GIP and GLP-1 co-agonist compounds”, issued 2016-10-25, assigned to Eli Lilly and Co
- ^ Frederick MO, Boyse RA, Braden TM, Calvin JR, Campbell BM, Changi SM, et al. (2021). “Kilogram-Scale GMP Manufacture of Tirzepatide Using a Hybrid SPPS/LPPS Approach with Continuous Manufacturing”. Organic Process Research & Development. 25 (7): 1628–1636. doi:10.1021/acs.oprd.1c00108. S2CID 237690232.
- ^ Sagonowsky, Eric (26 October 2021). “As Lilly gears up for key 2022 launches, Trulicity, Taltz and more drive solid growth”. Fierce Pharma. Retrieved 9 April 2022.
- ^ Kellaher, Colin (28 April 2022). “Eli Lilly’s Tirzepatide Meets Main Endpoints in Phase 3 Obesity Study >LLY”. Dow Jones Newswires. Retrieved 29 April 2022 – via MarketWatch.
- ^ Kahan, Scott; Garvey, W. Timothy (28 April 2022). “SURMOUNT-1: Adults achieve weight loss of 16% or more at 72 weeks with tirzepatide”. healio.com. Retrieved 29 April 2022.
- ^ Taylor, Nick Paul (28 April 2022). “SURMOUNT-able: Lilly’s tirzepatide clears high bar set by Novo’s Wegovy in obesity”. FierceBiotech. Retrieved 29 April 2022.
- ^ Frías JP, Davies MJ, Rosenstock J, Pérez Manghi FC, Fernández Landó L, Bergman BK, et al. (August 2021). “Tirzepatide versus Semaglutide Once Weekly in Patients with Type 2 Diabetes”. The New England Journal of Medicine. 385 (6): 503–515. doi:10.1056/NEJMoa2107519. PMID 34170647. S2CID 235635529.
- ^ Dutta D, Surana V, Singla R, Aggarwal S, Sharma M (November–December 2021). “Efficacy and safety of novel twincretin tirzepatide a dual GIP and GLP-1 receptor agonist in the management of type-2 diabetes: A Cochrane meta-analysis”. Indian Journal of Endocrinology and Metabolism. 25 (6): 475–489. doi:10.4103/ijem.ijem_423_21.
- ^ World Health Organization (2019). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 81”. WHO Drug Information. 33 (1). hdl:10665/330896.
Further reading
- Bhagavathula AS, Vidyasagar K, Tesfaye W (September 2021). “Efficacy and Safety of Tirzepatide in Patients with Type 2 Diabetes Mellitus: A Systematic Review and Meta-Analysis of Randomized Phase II/III Trials”. Pharmaceuticals (Basel). 14 (10). doi:10.3390/ph14100991. PMC 8537322. PMID 34681215.
- Frías JP (November 2020). “Tirzepatide: a glucose-dependent insulinotropic polypeptide (GIP) and glucagon-like peptide-1 (GLP-1) dual agonist in development for the treatment of type 2 diabetes”. Expert Rev Endocrinol Metab. 15 (6): 379–394. doi:10.1080/17446651.2020.1830759. PMID 33030356.
- Ryan DH (September 2021). “Next Generation Antiobesity Medications: Setmelanotide, Semaglutide, Tirzepatide and Bimagrumab: What do They Mean for Clinical Practice?”. J Obes Metab Syndr. 30 (3): 196–208. doi:10.7570/jomes21033. PMC 8526285. PMID 34518444.
External links
- “Tirzepatide”. Drug Information Portal. U.S. National Library of Medicine.
- Clinical trial number NCT03954834 for “A Study of Tirzepatide (LY3298176) in Participants With Type 2 Diabetes Not Controlled With Diet and Exercise Alone (SURPASS-1)” at ClinicalTrials.gov
- Clinical trial number NCT03987919 for “A Study of Tirzepatide (LY3298176) Versus Semaglutide Once Weekly as Add-on Therapy to Metformin in Participants With Type 2 Diabetes (SURPASS-2)” at ClinicalTrials.gov
- Clinical trial number NCT03882970 for “A Study of Tirzepatide (LY3298176) Versus Insulin Degludec in Participants With Type 2 Diabetes (SURPASS-3)” at ClinicalTrials.gov
- Clinical trial number NCT03730662 for “A Study of Tirzepatide (LY3298176) Once a Week Versus Insulin Glargine Once a Day in Participants With Type 2 Diabetes and Increased Cardiovascular Risk (SURPASS-4)” at ClinicalTrials.gov
- Clinical trial number NCT04039503 for “A Study of Tirzepatide (LY3298176) Versus Placebo in Participants With Type 2 Diabetes Inadequately Controlled on Insulin Glargine With or Without Metformin (SURPASS-5)” at ClinicalTrials.gov
CLIP
FDA approves Lilly’s Mounjaro™ (tirzepatide) injection, the first and only GIP and GLP-1 receptor agonist for the treatment of adults with type 2 diabetes
May 13, 2022
Mounjaro delivered superior A1C reductions versus all comparators in phase 3 SURPASS clinical trials
While not indicated for weight loss, Mounjaro led to significantly greater weight reductions versus comparators in a key secondary endpoint
Mounjaro represents the first new class of diabetes medicines introduced in nearly a decade and is expected to be available in the U.S. in the coming weeks
INDIANAPOLIS, May 13, 2022 /PRNewswire/ — The U.S. Food and Drug Administration (FDA) approved Mounjaro™ (tirzepatide) injection, Eli Lilly and Company’s (NYSE: LLY) new once-weekly GIP (glucose-dependent insulinotropic polypeptide) and GLP-1 (glucagon-like peptide-1) receptor agonist indicated as an adjunct to diet and exercise to improve glycemic control in adults with type 2 diabetes. Mounjaro has not been studied in patients with a history of pancreatitis and is not indicated for use in patients with type 1 diabetes mellitus.
As the first and only FDA-approved GIP and GLP-1 receptor agonist, Mounjaro is a single molecule that activates the body’s receptors for GIP and GLP-1, which are natural incretin hormones.1
“Mounjaro delivered superior and consistent A1C reductions against all of the comparators throughout the SURPASS program, which was designed to assess Mounjaro’s efficacy and safety in a broad range of adults with type 2 diabetes who could be treated in clinical practice. The approval of Mounjaro is an exciting step forward for people living with type 2 diabetes given the results seen in these clinical trials,” said Juan Pablo Frías, M.D., Medical Director, National Research Institute and Investigator in the SURPASS program.
Mounjaro will be available in six doses (2.5 mg, 5 mg, 7.5 mg, 10 mg, 12.5 mg, 15 mg) and will come in Lilly’s well-established auto-injector pen with a pre-attached, hidden needle that patients do not need to handle or see.
The approval was based on results from the phase 3 SURPASS program, which included active comparators of injectable semaglutide 1 mg, insulin glargine and insulin degludec. Efficacy was evaluated for Mounjaro 5 mg, 10 mg and 15 mg used alone or in combination with commonly prescribed diabetes medications, including metformin, SGLT2 inhibitors, sulfonylureas and insulin glargine. Participants in the SURPASS program achieved average A1C reductions between 1.8% and 2.1% for Mounjaro 5 mg and between 1.7% and 2.4% for both Mounjaro 10 mg and Mounjaro 15 mg. While not indicated for weight loss, mean change in body weight was a key secondary endpoint in all SURPASS studies. Participants treated with Mounjaro lost between 12 lb. (5 mg) and 25 lb. (15 mg) on average.1
Side effects reported in at least 5% of patients treated with Mounjaro include nausea, diarrhea, decreased appetite, vomiting, constipation, indigestion (dyspepsia), and stomach (abdominal) pain. The labeling for Mounjaro contains a Boxed Warning regarding thyroid C-cell tumors. Mounjaro is contraindicated in patients with a personal or family history of medullary thyroid carcinoma or in patients with Multiple Endocrine Neoplasia syndrome type 2.1
“Lilly has a nearly 100-year heritage of advancing care for people living with diabetes – never settling for current outcomes. We’re not satisfied knowing that half of the more than 30 million Americans living with type 2 diabetes are not reaching their target blood glucose levels,” said Mike Mason, president, Lilly Diabetes. “We are thrilled to introduce Mounjaro, which represents the first new class of type 2 diabetes medication introduced in almost a decade and embodies our mission to bring innovative new therapies to the diabetes community.”
Mounjaro is expected to be available in the United States in the coming weeks. Lilly is committed to helping people access the medicines they are prescribed and will work with insurers, health systems and providers to help enable patient access to Mounjaro. Lilly plans to offer a Mounjaro savings card for people who qualify. Patients or healthcare professionals with questions about Mounjaro can visit www.Mounjaro.com or call The Lilly Answers Center at 1-800-LillyRx (1-800-545-5979).
Tirzepatide is also under regulatory review for the treatment of type 2 diabetes in Europe, Japan and several additional markets. A multimedia gallery is available on Lilly.com.
About the SURPASS clinical trial program
The SURPASS phase 3 global clinical development program for tirzepatide began in late 2018 and included five global registration trials and two regional trials in Japan. These studies ranged from 40 to 52 weeks and evaluated the efficacy and safety of Mounjaro 5 mg, 10 mg and 15 mg as a monotherapy and as an add-on to various standard-of-care medications for type 2 diabetes. The active comparators in the studies were injectable semaglutide 1 mg, insulin glargine and insulin degludec. Collectively, the five global registration trials consistently demonstrated A1C reductions for participants taking Mounjaro across multiple stages of their type 2 diabetes journeys, from an average around five to 13 years of having diabetes.2-8
- SURPASS-1 (NCT03954834) was a 40-week study comparing the efficacy and safety of Mounjaro 5 mg (N=121), 10 mg (N=121) and 15 mg (N=120) as monotherapy to placebo (N=113) in adults with type 2 diabetes inadequately controlled with diet and exercise alone. From a baseline A1C of 7.9%, Mounjaro reduced participants’ A1C by a mean of 1.8%* (5 mg) and 1.7%* (10 mg and 15 mg) compared to 0.1% for placebo. In a key secondary endpoint, from a baseline weight of 189 lb., Mounjaro reduced participants’ weight by a mean of 14 lb.* (5 mg), 15 lb.* (10 mg) and 17 lb.* (15 mg) compared to 2 lb. for placebo.2,3
- SURPASS-2 (NCT03987919) was a 40-week study comparing the efficacy and safety of Mounjaro 5 mg (N=470), 10 mg (N=469) and 15 mg (N=469) to injectable semaglutide 1 mg (N=468) in adults with type 2 diabetes inadequately controlled with ≥1500 mg/day metformin alone. From a baseline A1C of 8.3%, Mounjaro reduced participants’ A1C by a mean of 2.0%ꝉ (5 mg), 2.2%* (10 mg) and 2.3%* (15 mg) compared to 1.9% for semaglutide. In a key secondary endpoint, from a baseline weight of 207 lb., Mounjaro reduced participants’ weight by a mean of 17 lb.ꝉ (5 mg), 21 lb.* (10 mg) and 25 lb.* (15 mg) compared to 13 lb. for semaglutide.4,5
- SURPASS-3 (NCT03882970) was a 52-week study comparing the efficacy of Mounjaro 5 mg (N=358), 10 mg (N=360) and 15 mg (N=358) to titrated insulin degludec (N=359) in adults with type 2 diabetes treated with metformin with or without an SGLT-2 inhibitor. From a baseline A1C of 8.2%, Mounjaro reduced participants’ A1C by a mean of 1.9%* (5 mg), 2.0%* (10 mg) and 2.1%* (15 mg) compared to 1.3% for insulin degludec. From a baseline weight of 208 lb., Mounjaro reduced participants’ weight by a mean of 15 lb.* (5 mg), 21 lb.* (10 mg) and 25 lb.* (15 mg) compared to an increase of 4 lb. for insulin degludec.6
- SURPASS-4 (NCT03730662) was a 104-week study comparing the efficacy and safety of Mounjaro 5 mg (N=328), 10 mg (N=326) and 15 mg (N=337) to insulin glargine (N=998) in adults with type 2 diabetes inadequately controlled with at least one and up to three oral antihyperglycemic medications (metformin, sulfonylureas or SGLT-2 inhibitors), who have increased cardiovascular (CV) risk. The primary endpoint was measured at 52 weeks. From a baseline A1C of 8.5%, Mounjaro reduced participants’ A1C by a mean of 2.1%* (5 mg), 2.3%* (10 mg) and 2.4%* (15 mg) compared to 1.4% for insulin glargine. From a baseline weight of 199 lb., Mounjaro reduced weight by a mean of 14 lb.* (5 mg), 20 lb.* (10 mg) and 23 lb.* (15 mg) compared to an increase of 4 lb. for insulin glargine.7
- SURPASS-5 (NCT04039503) was a 40-week study comparing the efficacy and safety of Mounjaro 5 mg (N=116), 10 mg (N=118) and 15 mg (N=118) to placebo (N=119) in adults with inadequately controlled type 2 diabetes already being treated with insulin glargine, with or without metformin. From a baseline A1C of 8.3%, Mounjaro reduced A1C by a mean of 2.1%* (5 mg), 2.4%* (10 mg) and 2.3%* (15 mg) compared to 0.9% for placebo. From a baseline weight of 210 lb., Mounjaro reduced participants’ weight by a mean of 12 lb.* (5 mg), 17 lb.* (10 mg) and 19 lb.* (15 mg) compared to an increase of 4 lb. for placebo.8
*p<0.001 for superiority vs. placebo or active comparator, adjusted for multiplicity
ꝉp<0.05 for superiority vs. semaglutide 1 mg, adjusted for multiplicity
About Mounjaro™ (tirzepatide) injection1
Mounjaro™ (tirzepatide) injection is FDA-approved as an adjunct to diet and exercise to improve glycemic control in adults with type 2 diabetes mellitus. As the first and only FDA-approved GIP and GLP-1 receptor agonist, Mounjaro is a single molecule that activates the body’s receptors for GIP (glucose-dependent insulinotropic polypeptide) and GLP-1 (glucagon-like peptide-1). Mounjaro will be available in six doses (2.5 mg, 5 mg, 7.5 mg, 10 mg, 12.5 mg, 15 mg) and will come in Lilly’s well-established auto-injector pen with a pre-attached, hidden needle that patients do not need to handle or see.
PURPOSE AND SAFETY SUMMARY WITH WARNINGS
Important Facts About MounjaroTM (mown-JAHR-OH). It is also known as tirzepatide.
- Mounjaro is an injectable prescription medicine for adults with type 2 diabetes used along with diet and exercise to improve blood sugar (glucose).
- It is not known if Mounjaro can be used in people who have had inflammation of the pancreas (pancreatitis). Mounjaro is not for use in people with type 1 diabetes. It is not known if Mounjaro is safe and effective for use in children under 18 years of age.
Warnings
Mounjaro may cause tumors in the thyroid, including thyroid cancer. Watch for possible symptoms, such as a lump or swelling in the neck, hoarseness, trouble swallowing, or shortness of breath. If you have a symptom, tell your healthcare provider.
- Do not use Mounjaro if you or any of your family have ever had a type of thyroid cancer called medullary thyroid carcinoma (MTC).
- Do not use Mounjaro if you have Multiple Endocrine Neoplasia syndrome type 2 (MEN 2).
- Do not use Mounjaro if you are allergic to tirzepatide or any of the ingredients in Mounjaro.
Mounjaro may cause serious side effects, including:
Inflammation of the pancreas (pancreatitis). Stop using Mounjaro and call your healthcare provider right away if you have severe pain in your stomach area (abdomen) that will not go away, with or without vomiting. You may feel the pain from your abdomen to your back.
Low blood sugar (hypoglycemia). Your risk for getting low blood sugar may be higher if you use Mounjaro with another medicine that can cause low blood sugar, such as a sulfonylurea or insulin. Signs and symptoms of low blood sugar may include dizziness or light-headedness, sweating, confusion or drowsiness, headache, blurred vision, slurred speech, shakiness, fast heartbeat, anxiety, irritability, or mood changes, hunger, weakness and feeling jittery.
Serious allergic reactions. Stop using Mounjaro and get medical help right away if you have any symptoms of a serious allergic reaction, including swelling of your face, lips, tongue or throat, problems breathing or swallowing, severe rash or itching, fainting or feeling dizzy, and very rapid heartbeat.
Kidney problems (kidney failure). In people who have kidney problems, diarrhea, nausea, and vomiting may cause a loss of fluids (dehydration), which may cause kidney problems to get worse. It is important for you to drink fluids to help reduce your chance of dehydration.
Severe stomach problems. Stomach problems, sometimes severe, have been reported in people who use Mounjaro. Tell your healthcare provider if you have stomach problems that are severe or will not go away.
Changes in vision. Tell your healthcare provider if you have changes in vision during treatment with Mounjaro.
Gallbladder problems. Gallbladder problems have happened in some people who use Mounjaro. Tell your healthcare provider right away if you get symptoms of gallbladder problems, which may include pain in your upper stomach (abdomen), fever, yellowing of skin or eyes (jaundice), and clay-colored stools.
Common side effects
The most common side effects of Mounjaro include nausea, diarrhea, decreased appetite, vomiting, constipation, indigestion, and stomach (abdominal) pain. These are not all the possible side effects of Mounjaro. Talk to your healthcare provider about any side effect that bothers you or doesn’t go away.
Tell your healthcare provider if you have any side effects. You can report side effects at 1-800-FDA-1088 or www.fda.gov/medwatch.
Before using
- Your healthcare provider should show you how to use Mounjaro before you use it for the first time.
- Before you use Mounjaro, talk to your healthcare provider about low blood sugar and how to manage it.
Review these questions with your healthcare provider:
- Do you have other medical conditions, including problems with your pancreas or kidneys, or severe problems with your stomach, such as slowed emptying of your stomach (gastroparesis) or problems digesting food?
- Do you take other diabetes medicines, such as insulin or sulfonylureas?
- Do you have a history of diabetic retinopathy?
- Are you pregnant or plan to become pregnant or breastfeeding or plan to breastfeed? It is not known if Mounjaro will harm your unborn baby.
- Do you take birth control pills by mouth? These may not work as well while using Mounjaro. Your healthcare provider may recommend another type of birth control when you start Mounjaro or when you increase your dose.
- Do you take any other prescription medicines or over-the-counter drugs, vitamins, or herbal supplements?
How to take
- Read the Instructions for Use that come with Mounjaro.
- Use Mounjaro exactly as your healthcare provider says.
- Mounjaro is injected under the skin (subcutaneously) of your stomach (abdomen), thigh, or upper arm.
- Use Mounjaro 1 time each week, at any time of the day.
- Do not mix insulin and Mounjaro together in the same injection.
- If you take too much Mounjaro, call your healthcare provider or seek medical advice promptly.
Learn more
For more information, call 1-800-LillyRx (1-800-545-5979) or go to www.mounjaro.com.
This information does not take the place of talking with your healthcare provider. Be sure to talk to your healthcare provider about Mounjaro and how to take it. Your healthcare provider is the best person to help you decide if Mounjaro is right for you.
MounjaroTM and its delivery device base are trademarks owned or licensed by Eli Lilly and Company, its subsidiaries, or affiliates.
Please click to access full Prescribing Information and Medication Guide.
TR CON CBS MAY2022
About Lilly
Lilly unites caring with discovery to create medicines that make life better for people around the world. We’ve been pioneering life-changing discoveries for nearly 150 years, and today our medicines help more than 47 million people across the globe. Harnessing the power of biotechnology, chemistry and genetic medicine, our scientists are urgently advancing new discoveries to solve some of the world’s most significant health challenges, redefining diabetes care, treating obesity and curtailing its most devastating long-term effects, advancing the fight against Alzheimer’s disease, providing solutions to some of the most debilitating immune system disorders, and transforming the most difficult-to-treat cancers into manageable diseases. With each step toward a healthier world, we’re motivated by one thing: making life better for millions more people. That includes delivering innovative clinical trials that reflect the diversity of our world and working to ensure our medicines are accessible and affordable. To learn more, visit Lilly.com and Lilly.com/newsroom or follow us on Facebook, Instagram, Twitter and LinkedIn. P-LLY
Lilly Cautionary Statement Regarding Forward-Looking Statements
This press release contains forward-looking statements (as that term is defined in the Private Securities Litigation Reform Act of 1995) about Mounjaro™ (tirzepatide 2.5 mg, 5 mg, 7.5 mg, 10 mg, 12.5 mg and 15 mg) injection as a treatment to improve glycemic control in adults with type 2 diabetes, the timeline for supply of Mounjaro to become available, and certain other milestones and ongoing clinical trials of Mounjaro and reflects Lilly’s current beliefs and expectations. However, as with any pharmaceutical product or medical device, there are substantial risks and uncertainties in the process of research, development and commercialization. Among other things, there can be no guarantee that Mounjaro will be commercially successful, that future study results will be consistent with results to date, or that we will meet our anticipated timelines for the commercialization of Mounjaro. For further discussion of these and other risks and uncertainties, see Lilly’s most recent Form 10-K and Form 10-Q filings with the United States Securities and Exchange Commission. Except as required by law, Lilly undertakes no duty to update forward-looking statements to reflect events after the date of this release.
References
- Mounjaro. Prescribing Information. Lilly USA, LLC.
- Rosenstock, J, et. al. Efficacy and Safety of Once Weekly Tirzepatide, a Dual GIP/GLP-1 Receptor Agonist Versus Placebo as Monotherapy in People with Type 2 Diabetes (SURPASS-1). Abstract 100-OR. Presented virtually at the American Diabetes Association’s 81st Scientific Sessions; June 25-29.
- Rosenstock, J, et. al. (2021). Efficacy and safety of a novel dual GIP and GLP-1 receptor agonist tirzepatide in patients with type 2 diabetes (SURPASS-1): a double-blind, randomised, phase 3 trial. Lancet. 2021;398(10295):143-155. doi: 10.1016/S0140-6736(21)01324-6.
- Frías JP, Davies MJ, Rosenstock J, et al; for the SURPASS-2 Investigators. Tirzepatide versus semaglutide once weekly in patients with type 2 diabetes. N Engl J Med. 2021;385(6)(suppl):503-515. doi: 10.1056/NEJMoa2107519
- Frias, J.P. Efficacy and Safety of Tirzepatide vs. Semaglutide Once Weekly as Add-On Therapy to Metformin in Patients with Type 2 Diabetes. Abstract 84-LB. Presented virtually at the American Diabetes Association’s 81st Scientific Sessions; June 25-29.
- Ludvik B, Giorgino F, Jódar E, et al. Once-weekly tirzepatide versus once-daily insulin degludec as add-on to metformin with or without SGLT2 inhibitors in patients with type 2 diabetes (SURPASS-3): a randomised, open-label, parallel-group, phase 3 trial. Lancet. 2021;398(10300):583-598. doi: 10.1016/S0140-6736(21)01443-4
- Del Prato S, Kahn SE, Pavo I, et al; for the SURPASS-4 Investigators. Tirzepatide versus insulin glargine in type 2 diabetes and increased cardiovascular risk (SURPASS-4): a randomised, open-label, parallel-group, multicentre, phase 3 trial. Lancet. 2021;398(10313):1811-1824. doi: 10.1016/S0140-6736(21)02188-7
- Dahl D, Onishi Y, Norwood P, et al. Effect of subcutaneous tirzepatide vs placebo added to titrated insulin glargine on glycemic control in patients with type 2 diabetes: the SURPASS-5 randomized clinical trial. JAMA. 2022;327(6):534-545. doi:10.1001/jama.2022.0078
CLIP
Lilly’s tirzepatide delivered up to 22.5% weight loss in adults with obesity or overweight in SURMOUNT-1
April 28, 2022
Participants taking tirzepatide lost up to 52 lb. (24 kg) in this 72-week phase 3 study
63% of participants taking tirzepatide 15 mg achieved at least 20% body weight reductions as a key secondary endpoint
INDIANAPOLIS, April 28, 2022 /PRNewswire/ — Tirzepatide (5 mg, 10 mg, 15 mg) achieved superior weight loss compared to placebo at 72 weeks of treatment in topline results from Eli Lilly and Company’s (NYSE: LLY) SURMOUNT-1 clinical trial, with participants losing up to 22.5% (52 lb. or 24 kg) of their body weight for the efficacy estimandi. This study enrolled 2,539 participants and was the first phase 3 global registration trial evaluating the efficacy and safety of tirzepatide in adults with obesity, or overweight with at least one comorbidity, who do not have diabetes. Tirzepatide met both co-primary endpoints of superior mean percent change in body weight from baseline and greater percentage of participants achieving body weight reductions of at least 5% compared to placebo for both estimandsii. The study also achieved all key secondary endpoints at 72 weeks.
For the efficacy estimand, participants taking tirzepatide achieved average weight reductions of 16.0% (35 lb. or 16 kg on 5 mg), 21.4% (49 lb. or 22 kg on 10 mg) and 22.5% (52 lb. or 24 kg on 15 mg), compared to placebo (2.4%, 5 lb. or 2 kg). Additionally, 89% (5 mg) and 96% (10 mg and 15 mg) of people taking tirzepatide achieved at least 5% body weight reductions compared to 28% of those taking placebo.
In a key secondary endpoint, 55% (10 mg) and 63% (15 mg) of people taking tirzepatide achieved at least 20% body weight reductions compared to 1.3% of those taking placebo. In an additional secondary endpoint not controlled for type 1 error, 32% of participants taking tirzepatide 5 mg achieved at least 20% body weight reductions. The mean baseline body weight of participants was 231 lb. (105 kg).
“Obesity is a chronic disease that often does not receive the same standard of care as other conditions, despite its impact on physical, psychological and metabolic health, which can include increased risk of hypertension, heart disease, cancer and decreased survival,” said Louis J. Aronne, MD, FACP, DABOM, director of the Comprehensive Weight Control Center and the Sanford I. Weill Professor of Metabolic Research at Weill Cornell Medicine, obesity expert at NewYork-Presbyterian/Weill Cornell Medical Center and Investigator of SURMOUNT-1. “Tirzepatide delivered impressive body weight reductions in SURMOUNT-1, which could represent an important step forward for helping the patient and physician partnership treat this complex disease.”
For the treatment-regimen estimandiii, results showed:
- Average body weight reductions: 15.0% (5 mg), 19.5% (10 mg), 20.9% (15 mg), 3.1% (placebo)
- Percentage of participants achieving body weight reductions of ≥5%: 85% (5 mg), 89% (10 mg), 91% (15 mg), 35% (placebo)
- Percentage of participants achieving body weight reductions of ≥20%: 30% (5 mg, not controlled for type 1 error), 50% (10 mg), 57% (15 mg), 3.1% (placebo)
The overall safety and tolerability profile of tirzepatide was similar to other incretin-based therapies approved for the treatment of obesity. The most commonly reported adverse events were gastrointestinal-related and generally mild to moderate in severity, usually occurring during the dose escalation period. For those treated with tirzepatide (5 mg, 10 mg and 15 mg, respectively), nausea (24.6%, 33.3%, 31.0%), diarrhea (18.7%, 21.2%, 23.0%), vomiting (8.3%, 10.7%, 12.2%) and constipation (16.8%, 17.1%, 11.7%) were more frequently experienced compared to placebo (9.5% [nausea], 7.3% [diarrhea], 1.7% [vomiting], 5.8% [constipation]).
Treatment discontinuation rates due to adverse events were 4.3% (5 mg), 7.1% (10 mg), 6.2% (15 mg) and 2.6% (placebo). The overall treatment discontinuation rates were 14.3% (5 mg), 16.4% (10 mg), 15.1% (15 mg) and 26.4% (placebo).
Participants who had pre-diabetes at study commencement will remain enrolled in SURMOUNT-1 for an additional 104 weeks of treatment following the initial 72-week completion date to evaluate the impact on body weight and the potential differences in progression to type 2 diabetes at three years of treatment with tirzepatide compared to placebo.
“Tirzepatide is the first investigational medicine to deliver more than 20 percent weight loss on average in a phase 3 study, reinforcing our confidence in its potential to help people living with obesity,” said Jeff Emmick, MD, Ph.D., vice president, product development, Lilly. “Obesity is a chronic disease that requires effective treatment options, and Lilly is working relentlessly to support people with obesity and modernize how this disease is approached. We’re proud to research and develop potentially innovative treatments like tirzepatide, which helped nearly two thirds of participants on the highest dose reduce their body weight by at least 20 percent in SURMOUNT-1.”
Tirzepatide is a novel investigational once-weekly GIP (glucose-dependent insulinotropic polypeptide) receptor and GLP-1 (glucagon-like peptide-1) receptor agonist, representing a new class of medicines being studied for the treatment of obesity. Tirzepatide is a single peptide that activates the body’s receptors for GIP and GLP-1, two natural incretin hormones. Obesity is a chronic, progressive disease caused by disruptions in the mechanisms that control body weight, often leading to an increase in food intake and/or a decrease in energy expenditure. These disruptions are multifactorial and can be related to genetic, developmental, behavioral, environmental and social factors. To learn more, visit Lilly.com/obesity.
Lilly will continue to evaluate the SURMOUNT-1 results, which will be presented at an upcoming medical meeting and submitted to a peer-reviewed journal. Additional studies are ongoing for tirzepatide as a potential treatment for obesity or overweight.
About tirzepatide
Tirzepatide is a once-weekly GIP (glucose-dependent insulinotropic polypeptide) receptor and GLP-1 (glucagon-like peptide-1) receptor agonist that integrates the actions of both incretins into a single novel molecule. GIP is a hormone that may complement the effects of GLP-1 receptor agonists. In preclinical models, GIP has been shown to decrease food intake and increase energy expenditure therefore resulting in weight reductions, and when combined with GLP-1 receptor agonism, may result in greater effects on markers of metabolic dysregulation such as body weight, glucose and lipids. Tirzepatide is in phase 3 development for adults with obesity or overweight with weight-related comorbidity and is currently under regulatory review as a treatment for adults with type 2 diabetes. It is also being studied as a potential treatment for non-alcoholic steatohepatitis (NASH) and heart failure with preserved ejection fraction (HFpEF). Studies of tirzepatide in obstructive sleep apnea (OSA) and in morbidity/mortality in obesity are planned as well.
About SURMOUNT-1 and the SURMOUNT clinical trial program
SURMOUNT-1 (NCT04184622) is a multi-center, randomized, double-blind, parallel, placebo-controlled trial comparing the efficacy and safety of tirzepatide 5 mg, 10 mg and 15 mg to placebo as an adjunct to a reduced-calorie diet and increased physical activity in adults without type 2 diabetes who have obesity, or overweight with at least one of the following comorbidities: hypertension, dyslipidemia, obstructive sleep apnea or cardiovascular disease. The trial randomized 2,539 participants across the U.S., Argentina, Brazil, China, India, Japan, Mexico, Russia and Taiwan in a 1:1:1:1 ratio to receive either tirzepatide 5 mg, 10 mg or 15 mg or placebo. The co-primary objectives of the study were to demonstrate that tirzepatide 10 mg and/or 15 mg is superior in percentage of body weight reductions from baseline and percentage of participants achieving ≥5% body weight reduction at 72 weeks compared to placebo. Participants who had pre-diabetes at study commencement will remain enrolled in SURMOUNT-1 for an additional 104 weeks of treatment following the initial 72-week completion date to evaluate the impact on body weight and potential differences in progression to type 2 diabetes at three years of treatment with tirzepatide compared to placebo.
All participants in the tirzepatide treatment arms started the study at a dose of tirzepatide 2.5 mg once-weekly and then increased the dose in a step-wise approach at four-week intervals to their final randomized maintenance dose of 5 mg (via a 2.5 mg step), 10 mg (via steps at 2.5 mg, 5 mg and 7.5 mg) or 15 mg (via steps at 2.5 mg, 5 mg, 7.5 mg, 10 mg and 12.5 mg).
The SURMOUNT phase 3 global clinical development program for tirzepatide began in late 2019 and has enrolled more than 5,000 people with obesity or overweight across six clinical trials, four of which are global studies. Results from SURMOUNT-2, -3, and -4 are anticipated in 2023.
About Lilly
Lilly unites caring with discovery to create medicines that make life better for people around the world. We’ve been pioneering life-changing discoveries for nearly 150 years, and today our medicines help more than 47 million people across the globe. Harnessing the power of biotechnology, chemistry and genetic medicine, our scientists are urgently advancing new discoveries to solve some of the world’s most significant health challenges, redefining diabetes care, treating obesity and curtailing its most devastating long-term effects, advancing the fight against Alzheimer’s disease, providing solutions to some of the most debilitating immune system disorders, and transforming the most difficult-to-treat cancers into manageable diseases. With each step toward a healthier world, we’re motivated by one thing: making life better for millions more people. That includes delivering innovative clinical trials that reflect the diversity of our world and working to ensure our medicines are accessible and affordable. To learn more, visit Lilly.com and Lilly.com/newsroom or follow us on Facebook, Instagram, Twitter and LinkedIn. P-LLY
CLIP
Tirzepatide results superior A1C and body weight reductions compared to insulin glargine in adults with type 2 diabetes
Newly published data show that participants maintained A1C and weight control up to two years in SURPASS-4, the largest and longest SURPASS trial completed to dateNo increased cardiovascular risk identified with tirzepatide; hazard ratio of 0.74 observed for MACE-4 events
SURPASS-4 is the largest and longest clinical trial completed to date of the phase 3 program studying tirzepatide as a potential treatment for type 2 diabetes. The primary endpoint was measured at 52 weeks, with participants continuing treatment up to 104 weeks or until study completion. The completion of the study was triggered by the accrual of major adverse cardiovascular events (MACE) to assess CV risk. In newly published data from the treatment period after 52 weeks, participants taking tirzepatide maintained A1C and weight control for up to two years.
The overall safety profile of tirzepatide, assessed over the full study period, was consistent with the safety results measured at 52 weeks, with no new findings up to 104 weeks. Gastrointestinal side effects were the most commonly reported adverse events, usually occurring during the escalation period and then decreasing over time.
“We are encouraged by the continued A1C and weight control that participants experienced past the initial 52 week treatment period and up to two years as we continue to explore the potential impact of tirzepatide for the treatment of type 2 diabetes,” said John Doupis, M.D., Ph.D., Director, Diabetes Division and Clinical Research Center, Iatriko Paleou Falirou Medical Center, Athens, Greece and Senior Investigator for SURPASS-4.
Tirzepatide is a novel investigational once-weekly dual glucose-dependent insulinotropic polypeptide (GIP) and glucagon-like peptide-1 (GLP-1) receptor agonist that integrates the actions of both incretins into a single molecule, representing a new class of medicines being studied for the treatment of type 2 diabetes.
SURPASS-4 was an open-label global trial comparing the safety and efficacy of three tirzepatide doses (5 mg, 10 mg and 15 mg) to titrated insulin glargine in 2,002 adults with type 2 diabetes with increased CV risk who were treated with between one and three oral antihyperglycemic medicines (metformin, a sulfonylurea or an SGLT-2 inhibitor). Of the total participants randomized, 1,819 (91%) completed the primary 52-week visit and 1,706 (85%) completed the study on treatment. The median study duration was 85 weeks and 202 participants (10%) completed two years.
Study participants had a mean duration of diabetes of 11.8 years, a baseline A1C of 8.52 percent and a baseline weight of 90.3 kg. More than 85 percent of participants had a history of cardiovascular events. In the insulin glargine arm, the insulin dose was titrated following a treat-to-target algorithm with the goal of fasting blood glucose below 100 mg/dL. The starting dose of insulin glargine was 10 units per day, and the mean dose of insulin glargine at 52 weeks was 43.5 units per day.
About tirzepatide
Tirzepatide is a once-weekly dual glucose-dependent insulinotropic polypeptide (GIP) and glucagon-like peptide-1 (GLP-1) receptor agonist that integrates the actions of both incretins into a single novel molecule. GIP is a hormone that may complement the effects of GLP-1. In preclinical models, GIP has been shown to decrease food intake and increase energy expenditure therefore resulting in weight reductions, and when combined with a GLP-1 receptor agonist, may result in greater effects on glucose and body weight. Tirzepatide is in phase 3 development for blood glucose management in adults with type 2 diabetes, for chronic weight management and heart failure with preserved ejection fraction (HFpEF). It is also being studied as a potential treatment for non-alcoholic steatohepatitis (NASH).
About SURPASS-4 and the SURPASS clinical trial program
SURPASS-4 (NCT03730662) is a randomized, parallel, open-label trial comparing the efficacy and safety of tirzepatide 5 mg, 10 mg and 15 mg to insulin glargine in adults with type 2 diabetes inadequately controlled with at least one and up to three oral antihyperglycemic medications (metformin, sulfonylureas or SGLT-2 inhibitors), who have increased cardiovascular (CV) risk. The trial randomized 2,002 study participants in a 1:1:1:3 ratio to receive either tirzepatide 5 mg, 10 mg or 15 mg or insulin glargine. Participants were located in the European Union, North America (Canada and the United States), Australia, Israel, Taiwan and Latin America (Brazil, Argentina and Mexico). The primary objective of the study was to demonstrate that tirzepatide (10 mg and/or 15 mg) is non-inferior to insulin glargine for change from baseline A1C at 52 weeks in people with type 2 diabetes and increased CV risk. The primary and key secondary endpoints were measured at 52 weeks, with participants continuing treatment up to 104 weeks or until study completion. The completion of the study was triggered by the accrual of major adverse cardiovascular events (MACE). Study participants enrolled had to have a mean baseline A1C between 7.5 percent and 10.5 percent and a BMI greater than or equal to 25 kg/m2 at baseline. All participants in the tirzepatide treatment arms started the study at a dose of tirzepatide 2.5 mg once-weekly and then increased the dose in a step-wise approach at four-week intervals to their final randomized maintenance dose of 5 mg (via a 2.5 mg step), 10 mg (via steps at 2.5 mg, 5 mg and 7.5 mg) or 15 mg (via steps at 2.5 mg, 5 mg, 7.5 mg, 10 mg and 12.5 mg). All participants in the titrated insulin glargine treatment arm started with a baseline dose of 10 units per day and titrated following a treat-to-target algorithm to reach a fasting blood glucose below 100 mg/dL.
The SURPASS phase 3 global clinical development program for tirzepatide has enrolled more than 20,000 people with type 2 diabetes across 10 clinical trials, five of which are global registration studies. The program began in late 2018, and all five global registration trials have been completed.
About Diabetes
Approximately 34 million Americans2 (just over 1 in 10) and an estimated 463 million adults worldwide3 have diabetes. Type 2 diabetes is the most common type internationally, accounting for an estimated 90 to 95 percent of all diabetes cases in the United States alone2. Diabetes is a chronic disease that occurs when the body does not properly produce or use the hormone insulin.
| Clinical data | |
|---|---|
| Trade names | Mounjaro |
| Other names | LY3298176, GIP/GLP-1 RA |
| License data | US DailyMed: Tirzepatide |
| Routes of administration | subcutaneous |
| Drug class | Antidiabetic, GLP-1 receptor agonist |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only [1][2] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 2023788-19-2 |
| PubChem CID | 156588324 |
| IUPHAR/BPS | 11429 |
| DrugBank | DB15171 |
| ChemSpider | 76714503 |
| UNII | OYN3CCI6QE |
| KEGG | D11360 |
| ChEMBL | ChEMBL4297839 |
| Chemical and physical data | |
| Formula | C225H348N48O68 |
| Molar mass | 4813.527 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI |
////////////Tirzepatide, FDA 2022, APPROVALS 2022, Mounjaro, PEPTIDE, チルゼパチド , LY3298176,
UNIIOYN3CCI6QE
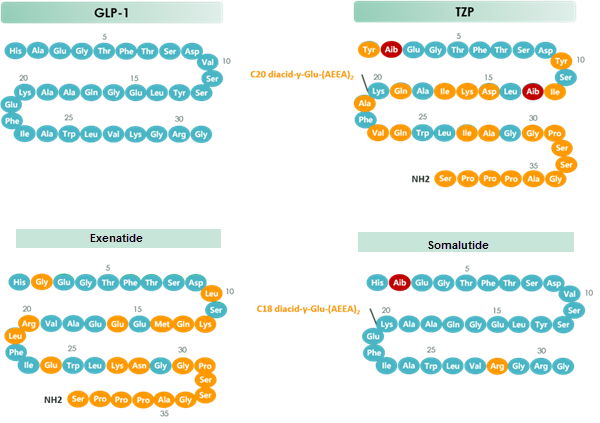
chart 1 Structure of GLP-1 & TZP & Exenatide & Somalutide
Lutetium Lu 177 vipivotide tetraxetan


Lutetium Lu 177 vipivotide tetraxetan
FDA APPROVED 2022/3/23, Pluvicto
To treat prostate-specific membrane antigen-positive metastatic castration-resistant prostate cancer following other therapies
| Formula | C49H65N9O16. Lu. 3H |
|---|---|
| CAS | 1703749-62-5 |
| Mol weight | 1214.0819 |
| Antineoplastic, Radioactive agent | |
| Disease | Prostate cancer (PSMA positive) |
|---|
ルテチウム(177Lu)ビピボチドテトラキセタン;
UNII-G6UF363ECX, WHO 11429
G6UF363ECX
177Lu-Psma-617
Vipivotide tetraxetan Lu-177
177Lu-Labeled PSMA-617
2-[4-[2-[[4-[[(2S)-1-[[(5S)-5-carboxy-5-[[(1S)-1,3-dicarboxypropyl]carbamoylamino]pentyl]amino]-3-naphthalen-2-yl-1-oxopropan-2-yl]carbamoyl]cyclohexyl]methylamino]-2-oxoethyl]-7,10-bis(carboxylatomethyl)-1,4,7,10-tetrazacyclododec-1-yl]acetate;lutetium-177(3+)
(177Lu)Lutetium 2,2′,2”-[10-(2-{[(trans-4-{[(2S)-1-{[(5S)-5-carboxy-5-({[(1S)-1,3-dicarboxypropyl]carbamoyl}amino)pentyl]amino}-3-(2-naphthyl)-1-oxo-2-propanyl]carbamoyl}cyclohexyl)methyl]amino}-2- oxoethyl)-1,4,7,10-tetraazacyclododecane-1,4,7-triyl]triacetate (non-preferred name)
1983157-55-6[RN]
PSMA-617 LU-177
Lutetium Lu 177 Vipivotide Tetraxetan is a radioconjugate composed of PSMA-617, a human prostate-specific membrane antigen (PSMA)-targeting ligand, conjugated to the beta-emitting radioisotope lutetium Lu 177 (177Lu), with potential antineoplastic activity against PSMA-expressing tumor cells. Upon intravenous administration of lutetium Lu 177 vipivotide tetraxetan, vipivotide tetraxetan targets and binds to PSMA-expressing tumor cells. Upon binding, PSMA-expressing tumor cells are destroyed by 177Lu through the specific delivery of beta particle radiation. PSMA, a tumor-associated antigen and type II transmembrane protein, is expressed on the membrane of prostatic epithelial cells and overexpressed on prostate tumor cells.
Lutetium (177Lu) vipivotide tetraxetan, sold under the brand name Pluvicto, is a radiopharmaceutical medication used for the treatment of prostate-specific membrane antigen (PSMA)-positive metastatic castration-resistant prostate cancer (mCRPC).[2] Lutetium (177Lu) vipivotide tetraxetan is a targeted radioligand therapy.[2][3]
The most common adverse reactions include fatigue, dry mouth, nausea, anemia, decreased appetite, and constipation.[2]
Lutetium (177Lu) vipivotide tetraxetan is a radioconjugate composed of PSMA-617, a human prostate-specific membrane antigen (PSMA)-targeting ligand, conjugated to the beta-emitting radioisotope lutetium Lu 177 (177Lu), with potential antineoplastic activity against PSMA-expressing tumor cells.[4] Upon intravenous administration of lutetium Lu 177 vipivotide tetraxetan, vipivotide tetraxetan targets and binds to PSMA-expressing tumor cells.[4] Upon binding, PSMA-expressing tumor cells are destroyed by 177Lu through the specific delivery of beta particle radiation.[4] PSMA, a tumor-associated antigen and type II transmembrane protein, is expressed on the membrane of prostatic epithelial cells and overexpressed on prostate tumor cells.[4]
Lutetium (177Lu) vipivotide tetraxetan was approved for medical use in the United States in March 2022.[2][5]
///////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
History[edit]
Efficacy was evaluated in VISION (NCT03511664), a randomized (2:1), multicenter, open-label trial that evaluated lutetium (177Lu) vipivotide tetraxetan plus best standard of care (BSoC) (n=551) or BSoC alone (n=280) in men with progressive, prostate-specific membrane antigen (PSMA)-positive metastatic castration-resistant prostate cancer (mCRPC).[2] All participants received a GnRH analog or had prior bilateral orchiectomy.[2] Participants were required to have received at least one androgen receptor pathway inhibitor, and 1 or 2 prior taxane-based chemotherapy regimens.[2] Participants received lutetium (177Lu) vipivotide tetraxetan 7.4 GBq (200 mCi) every 6 weeks for up to a total of 6 doses plus BSoC or BSoC alone.[2]
The U.S. Food and Drug Administration granted the application for lutetium (177lu) vipivotide tetraxetan priority review and breakthrough therapy designations.[2]
References
- ^ “Highlights of prescribing information: PLUVICTOTM (lutetium Lu 177 vipivotide tetraxetan) injection, for intravenous use” (PDF). Advanced Accelerator Applications USA, Inc. Novartis. March 2022.
- ^ Jump up to:a b c d e f g h i j “FDA approves Pluvicto for metastatic castration-resistant prostate can”. U.S. Food and Drug Administration. 23 March 2022. Retrieved 23 March 2022. This article incorporates text from this source, which is in the public domain.
- ^ Neels OC, Kopka K, Liolios C, Afshar-Oromieh A (December 2021). “Radiolabeled PSMA Inhibitors”. Cancers. 13 (24): 6255. doi:10.3390/cancers13246255. PMC 8699044. PMID 34944875.
- ^ Jump up to:a b c d “Lutetium Lu 177 Vipivotide Tetraxetan (Code C148145)”. NCI Thesaurus. 28 February 2022. Retrieved 23 March 2022. This article incorporates text from this source, which is in the public domain.
- ^ “Novartis Pluvicto approved by FDA as first targeted radioligand therapy for treatment of progressive, PSMA positive metastatic castration-resistant prostate cancer” (Press release). Novartis. 23 March 2022. Retrieved 23 March 2022.
External links
- “Lutetium lu 177 vipivotide tetraxetan”. Drug Information Portal. U.S. National Library of Medicine.
| Clinical data | |
|---|---|
| Trade names | Pluvicto |
| Other names | 177Lu-PSMA-617, Lutetium Lu 177 vipivotide tetraxetan (USAN US) |
| License data | US DailyMed: Pluvicto |
| Routes of administration | Intravenous |
| Drug class | Radiopharmaceutical |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only [1][2] |
| Identifiers | |
| CAS Number | 1703749-62-5 |
| PubChem CID | 122706785 |
| ChemSpider | 58828499 |
| UNII | G6UF363ECX |
| KEGG | D12335 |
| Chemical and physical data | |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| show |
////////////Lutetium Lu 177 vipivotide tetraxetan, ルテチウム(177Lu)ビピボチドテトラキセタン, FDA 2022, APPROVALS 2022, PROSTRATE CANCER, WHO 11429
C1CC(CCC1CNC(=O)CN2CCN(CCN(CCN(CC2)CC(=O)[O-])CC(=O)[O-])CC(=O)[O-])C(=O)NC(CC3=CC4=CC=CC=C4C=C3)C(=O)NCCCCC(C(=O)O)NC(=O)NC(CCC(=O)O)C(=O)O.[Lu+3]
Vipivotide tetraxetan (Synonyms: PSMA-617)
CAS No. : 1702967-37-0
Vipivotide tetraxetan (PSMA-617) is a high potent prostate-specific membrane antigen (PSMA) inhibitor, with a Ki of 0.37 nM.

NEW DRUG APPROVALS
one time
$10.00
Sutimlimab-jome
(Heavy chain)
EVQLVESGGG LVKPGGSLRL SCAASGFTFS NYAMSWVRQA PGKGLEWVAT ISSGGSHTYY
LDSVKGRFTI SRDNSKNTLY LQMNSLRAED TALYYCARLF TGYAMDYWGQ GTLVTVSSAS
TKGPSVFPLA PCSRSTSEST AALGCLVKDY FPEPVTVSWN SGALTSGVHT FPAVLQSSGL
YSLSSVVTVP SSSLGTKTYT CNVDHKPSNT KVDKRVESKY GPPCPPCPAP EFEGGPSVFL
FPPKPKDTLM ISRTPEVTCV VVDVSQEDPE VQFNWYVDGV EVHNAKTKPR EEQFNSTYRV
VSVLTVLHQD WLNGKEYKCK VSNKGLPSSI EKTISKAKGQ PREPQVYTLP PSQEEMTKNQ
VSLTCLVKGF YPSDIAVEWE SNGQPENNYK TTPPVLDSDG SFFLYSRLTV DKSRWQEGNV
FSCSVMHEAL HNHYTQKSLS LSLGK
(Light chain)
QIVLTQSPAT LSLSPGERAT MSCTASSSVS SSYLHWYQQK PGKAPKLWIY STSNLASGVP
SRFSGSGSGT DYTLTISSLQ PEDFATYYCH QYYRLPPITF GQGTKLEIKR TVAAPSVFIF
PPSDEQLKSG TASVVCLLNN FYPREAKVQW KVDNALQSGN SQESVTEQDS KDSTYSLSST
LTLSKADYEK HKVYACEVTH QGLSSPVTKS FNRGEC
(Disulfide bridge: H22-H96, H132-L216, H145-H201, H224-H’224, H227-H’227, H259-H319, H365-H423, H’22-H’96, H’132-L’216, H’145-H’201, H’259-H’319, H’365-H’423, L23-L89, L136-L196, L’23-L’89, L’136-L’196)
Sutimlimab-jome
スチムリマブ (遺伝子組換え)
| Formula | C6436H9912N1700O2016S46 |
|---|---|
| CAS | 2049079-64-1 |
| Mol weight | 144832.7369 |
- BIVV009
- Sutimlimab
- Sutimlimab [INN]
- Sutimlimab [WHO-DD]
- TNT009
- UNII-GNWE7KJ995
- WHO 10757
| Efficacy | Anti-anemic, Anti-complement C1s antibody |
|---|---|
| Comment | Monoclonal antibody |
FDA APPROVED 2/4/2022, To decrease the need for red blood cell transfusion due to hemolysis in cold agglutinin disease, Enjaymo
A Humanized Antibody for the Specific Inhibition of the Classical Complement Pathway.

Sutimlimab, sold under the brand name Enjaymo, is a monoclonal antibody that is used to treat adults with cold agglutinin disease (CAD).[1][2][3] It is given by intravenous infusion.[1]
The most common side effects include respiratory tract infection, viral infection, diarrhea, dyspepsia (indigestion), cough, arthralgia (joint stiffness), arthritis, and swelling in the lower legs and hands.[2]
Sutimlimab prevents complement-enhanced activation of autoimmune human B cells in vitro.[4]
This drug is being developed by Bioverativ, a Sanofi company.[5] Sutimlimab was approved for medical use in the United States in February 2022.[2][6]
Sutimlimab-jome, a classical complement inhibitor, is a humanized monoclonal antibody expressed by recombinant in Chinese hamster ovary (CHO) cells and produced in vitro using standard mammalian cell culture methods. Sutimlimab-jome is composed of two heterodimers. Each heterodimer is composed of a heavy and a light polypeptide chain. Each heavy chain (H-chain) is composed of 445 amino acids and each light chain (L-chain) contains 216 amino acids. Sutimlimab-jome has a molecular weight of approximately 147 kDa.
ENJAYMO (sutimlimab-jome) injection is a sterile, clear to slightly opalescent, colorless to slightly yellow, preservative-free solution for intravenous use. Each single-dose vial contains 1,100 mg sutimlimab-jome at a concentration of 50 mg/mL with a pH of 6.1. Each mL contains 50 mg of sutimlimab-jome and also contains polysorbate 80 (0.2 mg), sodium chloride (8.18 mg), sodium phosphate dibasic heptahydrate (0.48 mg), sodium phosphate monobasic monohydrate (1.13 mg), and Water for Injection, USP. https://www.rxlist.com/enjaymo-drug.htm#clinpharm
Medical uses
Sutimlimab is indicated to decrease the need for red blood cell transfusion due to hemolysis (red blood cell destruction) in adults with cold agglutinin disease (CAD).[1][2]
History
The effectiveness of sutimlimab was assessed in a study of 24 adults with cold agglutinin disease who had a blood transfusion within the past six months.[2] All participants received sutimlimab for up to six months and could choose to continue therapy in a second part of the trial.[2] Based on body weight, participants received either a 6.5g or 7.5g infusion of sutimlimab into their vein on day 0, day 7, and every 14 days through week 25.[2]
In total, 54% of participants responded to sutimlimab.[2] The response was defined in the study as an increase in hemoglobin (an indirect measurement of the amount of red blood cells that are not destroyed) of 2 g/dL or greater (or to 12 g/dL or greater), and no red blood cell transfusions after the first five weeks of treatment; and no other therapies for cold agglutinin disease as defined in the study.[2]
The application for sutimlimab received orphan drug,[2][7] breakthrough therapy,[2] and priority review designations.[2]
Society and culture
Names
Sutimlimab is the International nonproprietary name (INN).[8]
//////////////////////////////////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
CLIP
https://www.sanofi.com/en/media-room/press-releases/2022/2022-02-04-23-00-00-2379517
FDA approves Enjaymo™ (sutimlimab-jome), first treatment for use in patients with cold agglutinin disease
- Enjaymo is the only approved treatment to decrease the need for red blood cell transfusion due to hemolysis, the destruction of red blood cells, in adults with cold agglutinin disease (CAD)
- Enjaymo addresses a serious and chronic unmet medical need for adults living with CAD, a rare blood disorder
Paris, February 4, 2022. The U.S. Food and Drug Administration (FDA) has approved Enjaymo™ (sutimlimab-jome) to decrease the need for red blood cell transfusion due to hemolysis in adults with cold agglutinin disease (CAD). Enjaymo is the first and only approved treatment for people with CAD and works by inhibiting the destruction of red blood cells (hemolysis).
Bill Sibold
Executive Vice President, Head of Specialty Care
“Until now, people living with cold agglutinin disease haven’t had an approved treatment option to manage the constant destruction of red blood cells. Without healthy, viable red blood cells, a chain reaction of debilitating signs and symptoms can be triggered, starting with severe anemia. Enjaymo is the only approved treatment to inhibit red blood cell destruction in CAD and help stop the chain reaction from the start.”
CAD, a rare autoimmune hemolytic anemia, is caused by antibodies called cold agglutinins binding to the surface of red blood cells, which starts a process that causes the body’s immune system to mistakenly attack healthy red blood cells and cause their rupture (hemolysis). As red blood cells have the vital job of carrying oxygen throughout the body, patients with CAD may experience severe anemia, which can result in fatigue, weakness, shortness of breath, light-headedness, chest pain, irregular heartbeat, and other potential complications. CAD is a chronic and rare blood disorder that impacts the lives of an estimated 5,000 people in the U.S.
Enjaymo, targeting C1s in the classical complement pathway
Enjaymo is a humanized monoclonal antibody that is designed to selectively target and inhibit C1s in the classical complement pathway, which is part of the innate immune system. By blocking C1s, Enjaymo inhibits the activation of the complement cascade in the immune system and inhibits C1-activated hemolysis in CAD to prevent the abnormal destruction of healthy red blood cells. Enjaymo does not inhibit the lectin and alternative pathways.
Enjaymo Phase 3 pivotal CARDINAL study results supporting approval
The approval of Enjaymo in the U.S. is based on positive results from the 26-week open label, single arm pivotal Phase 3 study in patients with CAD (n=24) who have a recent history of blood transfusion, also known as the CARDINAL study.
Catherine Broome, MD
Associate professor of medicine at Georgetown University Lombardi Comprehensive Cancer Center, and a principal investigator in the CARDINAL study
“For people living with cold agglutinin disease, it is as if their body’s immune system is waging a war on itself. The relentless destruction of healthy red blood cells is a daily, silent reality for people with CAD. For the first time, we have a treatment that targets complement-mediated hemolysis, which is the underlying cause of the red blood cell destruction in many CAD patients. In the pivotal study, patients treated with sutimlimab had an improvement in anemia as measured by hemoglobin and bilirubin levels during the 26-week study.”
In the study, Enjaymo met its primary efficacy endpoint, which was a composite endpoint defined as the proportion of patients who achieved normalization of hemoglobin (Hgb) level ≥12 g/dL or demonstrated an increase from baseline in Hgb level ≥2 g/dL at the treatment assessment time point (mean value from weeks 23, 25, and 26) and no blood transfusion from weeks 5 through 26 or medications prohibited per the protocol from weeks 5 through 26. Secondary endpoints were also met, including improvements in hemoglobin and normalization of bilirubin.
- The majority of patients (54%; n=13) met the composite primary endpoint criteria with 63% (n=15) of patients achieving a hemoglobin ≥ 12 g/dL or an increase of at least 2 g/dL; 71% (n=17) of patients remaining transfusion-free after week five; and 92% (n=22) of patients did not use other CAD-related treatments.
- For the secondary measures on disease process, patients enrolled experienced a mean increase in hemoglobin level of 2.29 g/dL (SE: 0.308) at week 3 and 3.18 g/dL (SE: 0.476) at the 26-week treatment assessment timepoint from the mean baseline level of 8.6 g/dL. The mean reduction in bilirubin levels (n=14) was by -2.23 mg/dL (95% CI: -2.49 to -1.98) from a mean baseline level of 3.23 mg/dL (2.7-fold ULN).
In the CARDINAL study, the most common adverse reactions occurring in 10 percent or more of patients were respiratory tract infection, viral infection, diarrhea, dyspepsia, cough, arthralgia, arthritis, and peripheral edema. Serious adverse reactions were reported in 13 percent (3/24) of patients who received Enjaymo. These serious adverse reactions were streptococcal sepsis and staphylococcal wound infection (n=1), arthralgia (n=1), and respiratory tract infection (n=1). None of the adverse reactions led to discontinuation of Enjaymo in the study. Dosage interruptions due to an adverse reaction occurred in 17 percent (4/24) of patients who received Enjaymo.
Following the completion of the 26-week treatment period of CARDINAL (Part A), eligible patients continued to receive Enjaymo in an extension study.
The recommended dose of Enjaymo is based on body weight (6,500 mg for people 39-75 kg and 7,500 mg for people >75 kg). Enjaymo is administered intravenously weekly for the first two weeks with administration every two weeks thereafter.
Enjaymo is expected to be available in the U.S. in the coming weeks. The U.S. list price, or wholesale acquisition cost, of Enjaymo is $1,800 per vial. Actual costs to patients are generally anticipated to be lower as the list price does not reflect insurance coverage, co-pay support, or financial assistance from patient support programs. As part of our commitment to ensure treatment access and affordability for innovative therapies, Enjaymo Patient Solutions provides disease education, financial and co-pay assistance programs and other support services to eligible patients. For more information, please call 1-833-223-2428.
Enjaymo received FDA Breakthrough Therapy and Orphan Drug designation, and priority review, which is reserved for medicines that, if approved, would represent significant improvements in safety or efficacy in treating serious conditions. Outside of the U.S., sutimlimab has been submitted to regulatory authorities in Europe and Japan and reviews are ongoing.
About Sanofi
We are an innovative global healthcare company, driven by one purpose: we chase the miracles of science to improve people’s lives. Our team, across some 100 countries, is dedicated to transforming the practice of medicine by working to turn the impossible into the possible. We provide potentially life-changing treatment options and life-saving vaccine protection to millions of people globally, while putting sustainability and social responsibility at the center of our ambitions.
Sanofi is listed on EURONEXT: SAN and NASDAQ: SNY
References
- ^ Jump up to:a b c d https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/761164s000lbl.pdf
- ^ Jump up to:a b c d e f g h i j k l “FDA approves treatment for adults with rare type of anemia”. U.S. Food and Drug Administration. 4 February 2022. Retrieved 6 February 2022. This article incorporates text from this source, which is in the public domain.
- ^ Tvedt TH, Steien E, Øvrebø B, Haaverstad R, Hobbs W, Wardęcki M, et al. (February 2022). “Sutimlimab, an investigational C1s inhibitor, effectively prevents exacerbation of hemolytic anemia in a patient with cold agglutinin disease undergoing major surgery”. American Journal of Hematology. 97 (2): E51–E54. doi:10.1002/ajh.26409. PMID 34778998. S2CID 244116614.
- ^ Nikitin PA, Rose EL, Byun TS, Parry GC, Panicker S (February 2019). “C1s Inhibition by BIVV009 (Sutimlimab) Prevents Complement-Enhanced Activation of Autoimmune Human B Cells In Vitro”. Journal of Immunology. 202 (4): 1200–1209. doi:10.4049/jimmunol.1800998. PMC 6360260. PMID 30635392.
- ^ “Sutimlimab FDA Approval Status”. FDA. 19 May 2020.
- ^ “FDA approves Enjaymo (sutimlimab-jome), first treatment for use in patients with cold agglutinin disease”. Sanofi (Press release). 4 February 2022. Retrieved 6 February 2022.
- ^ “Sutimlimab Orphan Drug Designations and Approvals”. U.S. Food and Drug Administration (FDA). 27 July 2016. Retrieved 6 February 2022.
- ^ World Health Organization (2018). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 80”. WHO Drug Information. 32 (3). hdl:10665/330907.
External links
- “Sutimlimab”. Drug Information Portal. U.S. National Library of Medicine.
- Clinical trial number NCT03347396 for “A Study to Assess the Efficacy and Safety of BIVV009 (Sutimlimab) in Participants With Primary Cold Agglutinin Disease Who Have a Recent History of Blood Transfusion (Cardinal Study)” at ClinicalTrials.gov
//////////////Sutimlimab-jome, Enjaymo, FDA 2022, APPROVALS 2022, agglutinin disease, BIVV009, TNT009, UNII-GNWE7KJ995, WHO 10757, PEPTIDE, MONOCLONAL ANTIBODY, スチムリマブ (遺伝子組換え),

NEW DRUG APPROVALS
ONE TIME TO MAINTAIN THIS BLOG SUBSCRIPTIONS
$10.00
Faricimab-svoa

(A chain)
QVQLVQSGAE VKKPGASVKV SCKASGYTFT GYYMHWVRQA PGQGLEWMGW INPNSGGTNY
AQKFQGRVTM TRDTSISTAY MELSRLRSDD TAVYYCARSP NPYYYDSSGY YYPGAFDIWG
QGTMVTVSSA SVAAPSVFIF PPSDEQLKSG TASVVCLLNN FYPREAKVQW KVDNALQSGN
SQESVTEQDS KDSTYSLSST LTLSKADYEK HKVYACEVTH QGLSSPVTKS FNRGECDKTH
TCPPCPAPEA AGGPSVFLFP PKPKDTLMAS RTPEVTCVVV DVSHEDPEVK FNWYVDGVEV
HNAKTKPREE QYNSTYRVVS VLTVLAQDWL NGKEYKCKVS NKALGAPIEK TISKAKGQPR
EPQVCTLPPS RDELTKNQVS LSCAVKGFYP SDIAVEWESN GQPENNYKTT PPVLDSDGSF
FLVSKLTVDK SRWQQGNVFS CSVMHEALHN AYTQKSLSLS PGK
(B chain)
EVQLVESGGG LVQPGGSLRL SCAASGYDFT HYGMNWVRQA PGKGLEWVGW INTYTGEPTY
AADFKRRFTF SLDTSKSTAY LQMNSLRAED TAVYYCAKYP YYYGTSHWYF DVWGQGTLVT
VSSASTKGPS VFPLAPSSKS TSGGTAALGC LVKDYFPEPV TVSWNSGALT SGVHTFPAVL
QSSGLYSLSS VVTVPSSSLG TQTYICNVNH KPSNTKVDKK VEPKSCDKTH TCPPCPAPEA
AGGPSVFLFP PKPKDTLMAS RTPEVTCVVV DVSHEDPEVK FNWYVDGVEV HNAKTKPREE
QYNSTYRVVS VLTVLAQDWL NGKEYKCKVS NKALGAPIEK TISKAKGQPR EPQVYTLPPC
RDELTKNQVS LWCLVKGFYP SDIAVEWESN GQPENNYKTT PPVLDSDGSF FLYSKLTVDK
SRWQQGNVFS CSVMHEALHN AYTQKSLSLS PGK
(C chain)
DIQLTQSPSS LSASVGDRVT ITCSASQDIS NYLNWYQQKP GKAPKVLIYF TSSLHSGVPS
RFSGSGSGTD FTLTISSLQP EDFATYYCQQ YSTVPWTFGQ GTKVEIKRTV AAPSVFIFPP
SDEQLKSGTA SVVCLLNNFY PREAKVQWKV DNALQSGNSQ ESVTEQDSKD STYSLSSTLT
LSKADYEKHK VYACEVTHQG LSSPVTKSFN RGEC
(D chain)
SYVLTQPPSV SVAPGQTARI TCGGNNIGSK SVHWYQQKPG QAPVLVVYDD SDRPSGIPER
FSGSNSGNTA TLTISRVEAG DEADYYCQVW DSSSDHWVFG GGTKLTVLSS ASTKGPSVFP
LAPSSKSTSG GTAALGCLVK DYFPEPVTVS WNSGALTSGV HTFPAVLQSS GLYSLSSVVT
VPSSSLGTQT YICNVNHKPS NTKVDKKVEP KSC
(Disulfide bridge: A22-A96, A156-A216, A236-D213, A242-B232, A245-B235, A277-A337, A365-A441, B22-B96, B150-B206, B226-C214, B267-B327, B360-B431, B23-B88, B134-B194, D22-D87, D137-D193)
Faricimab
| Formula | C6506H9968N1724O1026S45 |
|---|---|
| CAS | 1607793-29-2 |
| Mol weight | 130194.6203 |
Faricimab-svoa
FDA APPROVED 1/28/2022, Vabysmo
To treat neovascular (wet) aged-related macular degeneration and diabetic macular edema
RO6867461
- Faricimab
- Faricimab [INN]
- RG-7716
- RG7716
- RO-6867461
- RO6867461
- UNII-QC4F7FKK7I
- WHO 10563

| Efficacy | Angiogenesis inhibitor, Anti-angiopoietin 2 antibody, Anti-VEGF antibody |
|---|---|
| Comment | Antibody Opthamology indications in patients susceptible to blocking of vascular endothelial growth factor A (VEGF-A) and angiopoietin-2 (Ang-2) |
Faricimab, sold under the brand name Vabysmo, is a monoclonal antibody used for the treatment of neovascular age-related macular degeneration (nAMD) and diabetic macular edema (DME).[1] Faricimab is a bispecific monoclonal antibody.[2]
Faricimab was developed by Roche. Faricimab completed Phase III trials[3] and was approved for use in the United States by the Food and Drug Administration in January 2022.[1][4]
FDA Approves Faricimab to Treat Wet AMD and DME\
FDA Approves Faricimab to Treat Wet AMD and DMEFebruary 1, 2022
This represents the approval of the first bispecific antibody to treat wet age-related macular degeneration (AMD) and diabetic macular edema (DME).
https://www.ajmc.com/view/fda-approves-fariximab-to-treat-wet-amd-and-dme
The FDA has approved faricimab-svoa (Vabysmo; Genentech) to treat 2 leading causes of vision loss: wet, or neovascular, age-related macular degeneration (AMD) and diabetic macular edema (DME).
After 4 initial monthly doses, faricimab is delivered as injections from 1 to 4 months apart in the first year while the current standard of care for wet AMD and DME requires injections every 1 to 2 months. In wet AMD, patients receive the 4 monthly injections first and then based on outcomes may receive their subsequent treatments every 2, 3, or 4 months. For DME, after the 4 initial monthly injections, treatment is extended or reduced based on outcomes, with a range of 1 to 4 months between doses.
The treatment targets and inhibits pathways involving angiopoietin-2 and vascular endothelial growth factor-A (VEGF-A), which are thought to contribute to vision loss by destabilizing blood vessels.
“Vabysmo represents an important step forward for ophthalmology. It is the first bispecific antibody approved for the eye and a major advance in treating retinal conditions such as wet AMD and diabetic macular edema,” Charles Wykoff, MD, PhD, director of research at Retina Consultants of Texas in Houston and a Vabysmo phase 3 investigator, said in a statement. “With Vabysmo, we now have the opportunity to offer patients a medicine that could improve their vision, potentially lowering treatment burden with fewer injections over time.”
The FDA approved faricimab on the results from 4 phase 3 studies: TENAYA and LUCERNE for wet AMD and YOSEMITE and RHINE for DME. All 4 studies were randomized, multicenter, double-masked, global trials.
TENAYA and LUCERNE were identical: 1329 treatment-naive patients with wet AMD, aged 50 and older, were assigned 1:1 to faricimab up to every 16 weeks or aflibercept every 8 weeks. YOSEMITE and RHINE were also identical: 1891 patients with vision loss due to DME were randomly assigned 1:1:1 to faricimab every 8 weeks, faricimab per personalized treatment interval, or aflibercept every 8 weeks.
For all trials, faricimab was noninferior to aflibercept and the incidence of ocular adverse events was comparable. The researchers determined that the longer time between dosing intervals combined with the visual benefits of faricimab reduced the burden in patients.
The 1-year results from these studies were published January 24 in The Lancet.1,2
“These data published in The Lancet reinforce the potential of faricimab as an important treatment option that may help improve and maintain vision while extending the time between treatments up to 4 months,” Levi Garraway, MD, PhD, chief medical officer and head of Global Product Development, said in a statement. “We remain deeply committed to developing new medicines such as faricimab that may help preserve sight in many people living with serious retinal conditions.”
Now that faricimab is approved, Genentech expects it to become available in the United States within weeks. Meanwhile, the European Medicines Agency is currently evaluating a Marketing Authorization Application for faricimab to treat wet AMD and DME.
There are additional trials—COMINO and BALATON—underway to evaluate the efficacy and safety of faricimab in people with macular edema following retinal vein occlusion. In addition, 2-year results for faricimab in DME will be presented at the Angiogeneisis, Exudation, and Degeneration 2022 meeting in February.
References
1. Heier JS, Khanani AM, Quezada Ruiz C, et al; TENAYA and LUCERNE Investigators. Efficacy, durability, and safety of intravitreal faricimab up to every 16 weeks for neovascular age-related macular degeneration (TENAYA and LUCERNE): two randomised, double-masked, phase 3, non-inferiority trials. Lancet. Published January 24, 2022. doi:10.1016/S0140-6736(22)00010-1
2. Wykoff CC, Abreu F, Adamis AP, et al. Efficacy, durability, and safety of intravitreal faricimab with extended dosing up to every 16 weeks in patients with diabetic macular oedema (YOSEMITE and RHINE): two randomised, double-masked, phase 3 trials. Lancet. Published online January 24, 2022. doi:10.1016/S0140-6736(22)00018-6
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
| Monoclonal antibody | |
|---|---|
| Type | Whole antibody |
| Source | Humanized |
| Target | VEGF-A, angiopoietin 2 |
| Clinical data | |
| Trade names | Vabysmo |
| Other names | RO6867461; faricimab-svoa |
| License data | US DailyMed: Faricimab |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only |
| Identifiers | |
| CAS Number | 1607793-29-2 |
| UNII | QC4F7FKK7I |
| KEGG | D11516 |
| Chemical and physical data | |
| Formula | C6506H9968N1724O1026S45 |
| Molar mass | 130197.05 g·mol−1 |
Society and culture
Names
Faricimab is the International Nonproprietary Name (INN).[5]
References
- ^ Jump up to:a b “FDA approves Roche’s Vabysmo, the first bispecific antibody for the eye, to treat two leading causes of vision loss”. Roche (Press release). 31 January 2022. Retrieved 31 January 2022.
- ^ Nicolò M, Ferro Desideri L, Vagge A, Traverso CE (March 2021). “Faricimab: an investigational agent targeting the Tie-2/angiopoietin pathway and VEGF-A for the treatment of retinal diseases”. Expert Opinion on Investigational Drugs. 30 (3): 193–200. doi:10.1080/13543784.2021.1879791. PMID 33471572. S2CID 231665201.
- ^ Khan M, Aziz AA, Shafi NA, Abbas T, Khanani AM (August 2020). “Targeting Angiopoietin in Retinal Vascular Diseases: A Literature Review and Summary of Clinical Trials Involving Faricimab”. Cells. 9 (8): 1869. doi:10.3390/cells9081869. PMC 7464130. PMID 32785136.
- ^ “FDA approves faricimab for treatment of wet AMD, DME”. Ophthalmology Times. 28 January 2022.
- ^ World Health Organization (2018). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 80”. WHO Drug Information. 32 (3). hdl:10665/330907.
External links
- “Faricimab”. Drug Information Portal. U.S. National Library of Medicine.
////////////Faricimab-svoa, APPROVALS 2022, FDA 2022, RO6867461, RO 6867461, PEPTIDE, MONOCLONAL ANTIBODY, RG 7716, WHO 10563, peptide

NEW DRUG APPROVALS
one time
$10.00
Tebentafusp-tebn
Tebentafusp-tebn
- IMCGP100
UNIIN658GY6L3E
CAS number1874157-95-5
FDA APPROVED 1/25/2022, Kimmtrak, To treat unresectable or metastatic uveal melanoma
Immunocore Limited
- T cell receptor α chain (synthetic human) fusion protein with T cell receptor β chain (synthetic human) fusion protein with immunoglobulin, anti-(human CD3 antigen) (synthetic scFv fragment)
- Protein Sequence
- Sequence Length: 695, 500, 195
Sequence:
1AIQMTQSPSS LSASVGDRVT ITCRASQDIR NYLNWYQQKP GKAPKLLIYY51TSRLESGVPS RFSGSGSGTD YTLTISSLQP EDFATYYCQQ GNTLPWTFGQ101GTKVEIKGGG GSGGGGSGGG GSGGGGSGGG SEVQLVESGG GLVQPGGSLR151LSCAASGYSF TGYTMNWVRQ APGKGLEWVA LINPYKGVST YNQKFKDRFT201ISVDKSKNTA YLQMNSLRAE DTAVYYCARS GYYGDSDWYF DVWGQGTLVT251VSSGGGGSDG GITQSPKYLF RKEGQNVTLS CEQNLNHDAM YWYRQDPGQG301LRLIYYSWAQ GDFQKGDIAE GYSVSREKKE SFPLTVTSAQ KNPTAFYLCA351SSWGAPYEQY FGPGTRLTVT EDLKNVFPPE VAVFEPSEAE ISHTQKATLV401CLATGFYPDH VELSWWVNGK EVHSGVCTDP QPLKEQPALN DSRYALSSRL451RVSATFWQDP RNHFRCQVQF YGLSENDEWT QDRAKPVTQI VSAEAWGRAD
Sequence:
1AQQGEEDPQA LSIQEGENAT MNCSYKTSIN NLQWYRQNSG RGLVHLILIR51SNEREKHSGR LRVTLDTSKK SSSLLITASR AADTASYFCA TDGSTPMQFG101KGTRLSVIAN IQKPDPAVYQ LRDSKSSDKS VCLFTDFDSQ TNVSQSKDSD151VYITDKCVLD MRSMDFKSNS AVAWSNKSDF ACANAFNNSI IPEDT
Sequence Modifications
| Type | Location | Description |
|---|---|---|
| bridge | Cys-23 – Cys-88 | disulfide bridge |
| bridge | Cys-153 – Cys-227 | disulfide bridge |
| bridge | Cys-281 – Cys-349 | disulfide bridge |
| bridge | Cys-401 – Cys-466 | disulfide bridge |
| bridge | Cys-427 – Cys-157′ | disulfide bridge |
| bridge | Cys-23′ – Cys-89′ | disulfide bridge |
| bridge | Cys-132′ – Cys-182′ | disulfide bridge |
Tebentafusp, sold under the brand name Kimmtrak, is an anti-cancer medication used to treat uveal melanoma (eye cancer).[1][2]
The most common side effects include cytokine release syndrome, rash, pyrexia (fever), pruritus (itching), fatigue, nausea, chills, abdominal pain, edema, hypotension, dry skin, headache, and vomiting.[1][2]
Tebentafusp is a bispecific gp100 peptide-HLA-directed CD3 T cell engager.[1][2] It was approved for medical use in the United States in January 2022.[1][2]
Tebentafusp is a bispecific gp100 peptide-HLA-directed CD3 T cell engager used to treat unresectable or metastatic uveal melanoma.
Tebentafusp is a gp100 peptide-HLA-directed CD3 T cell engager.5 It is a bispecific, fusion protein and first-in-class drug of immune-mobilizing monoclonal T cell receptors against cancer (ImmTACs), a recently developed cancer immunotherapy with a novel mechanism of action. ImmTACs bind to target cancer cells that express a specific antigen of interest and recruit cytotoxic T cells to lyse the cells, such as melanocytes.1,2
Uveal melanoma is a rare ocular tumour with often poor prognosis and limited treatment options. Even after surgical ablation or removal of the ocular tumour, almost 50% of patients with uveal melanoma develop metastatic disease.1 On January 26, 2022, tebentafusp was first approved by the FDA for the treatment of HLA-A*02:01-positive adults with unresectable or metastatic uveal melanoma. This approval marks the first bispecific T cell engager to be approved by the FDA to treat a solid tumour and being the first and only therapy for the treatment of unresectable or metastatic uveal melanoma to be approved by the FDA.5
FDA approves tebentafusp-tebn for unresectable or metastatic uveal melanoma
On January 25, 2022, the Food and Drug Administration approved tebentafusp-tebn (Kimmtrak, Immunocore Limited), a bispecific gp100 peptide-HLA-directed CD3 T cell engager, for HLA-A*02:01-positive adult patients with unresectable or metastatic uveal melanoma.
Efficacy was evaluated in IMCgp100-202 (NCT03070392), a randomized, open-label, multicenter trial of 378 patients with metastatic uveal melanoma. Patients were required to be HLA-A*02:01 genotype positive identified by a central assay. Patients were excluded if prior systemic therapy or localized liver-directed therapy were administered. Prior surgical resection of oligometastatic disease was permitted. Patients with clinically significant cardiac disease or symptomatic, untreated brain metastases were excluded.
Patients were randomized (2:1) to receive tebentafusp-tebn (N=252) or investigator’s choice (N=126) of either pembrolizumab, ipilimumab, or dacarbazine. Tebentafusp-tebn was administered weekly by intravenous infusion at 20 mcg on day 1, 30 mcg on day 8, 68 mcg on day 15 and every subsequent week until disease progression or unacceptable toxicity. The main efficacy outcome measure was overall survival (OS). An additional efficacy outcome was investigator-assessed progression-free survival (PFS) per RECIST 1.1. Median OS was 21.7 months (95% CI: 18.6, 28.6) for patients treated with tebentafusp-tebn and 16 months (95% CI: 9.7, 18.4) in the investigator’s choice arm (HR=0.51, 95% CI: 0.37, 0.71, p<0.0001) PFS was 3.3 months (95% CI: 3, 5) for those receiving tebentafusp-tebn and 2.9 months (95% CI: 2.8, 3) in the investigator’s choice arm (HR=0.73, 95% CI: 0.58, 0.94, p=0.0139).
The most common adverse reactions (≥30%) were cytokine release syndrome, rash, pyrexia, pruritus, fatigue, nausea, chills, abdominal pain, edema, hypotension, dry skin, headache, and vomiting. The most common laboratory abnormalities (≥50%) were decreased lymphocyte count, increased creatinine, increased glucose, increased aspartate aminotransferase, increased alanine aminotransferase, decreased hemoglobin, and decreased phosphate.
The recommended tebentafusp-tebn dose administered intravenously is:
- 20 mcg on day 1,
- 30 mcg on day 8,
- 68 mcg on day 15, and
- 68 mcg once weekly thereafter.
View full prescribing information for Kimmtrak.
This review was conducted under Project Orbis, an initiative of the FDA Oncology Center of Excellence. Project Orbis provides a framework for concurrent submission and review of oncology drugs among international partners. For this review, FDA collaborated with the Australian Therapeutic Goods Administration (TGA), Health Canada, and the United Kingdom’s Medicines and Healthcare product Regulatory Agency (MHRA). The application reviews may be ongoing at the other regulatory agencies.
This review used the Real-Time Oncology Review (RTOR) pilot program, which streamlined data submission prior to the filing of the entire clinical application, and the Assessment Aid, a voluntary submission from the applicant to facilitate the FDA’s assessment.
This application was granted priority review, breakthrough designation and orphan drug designation. A description of FDA expedited programs is in the Guidance for Industry: Expedited Programs for Serious Conditions-Drugs and Biologics.
//////////////////////////////////////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
| Clinical data | |
|---|---|
| Trade names | Kimmtrak |
| Other names | IMCgp100, tebentafusp-tebn |
| License data | US DailyMed: Tebentafusp |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only [1][2] |
| Identifiers | |
| CAS Number | 1874157-95-5 |
| DrugBank | DB15283 |
| UNII | N658GY6L3E |
Medical uses
Tebentafusp is indicated for HLA-A*02:01-positive adults with unresectable or metastatic uveal melanoma.[1][2]
History
Efficacy was evaluated in IMCgp100-202 (NCT03070392), a randomized, open-label, multicenter trial of 378 participants with metastatic uveal melanoma.[2] Participants were required to be HLA-A*02:01 genotype positive identified by a central assay.[2] Participants were excluded if prior systemic therapy or localized liver-directed therapy were administered.[2] Prior surgical resection of oligometastatic disease was permitted.[2] Participants with clinically significant cardiac disease or symptomatic, untreated brain metastases were excluded.[2]
The U.S. Food and Drug Administration (FDA) granted Immunocore‘s application for tebentafusp priority review, breakthrough therapy, and orphan drug designations.[2]
References
- ^ Jump up to:a b c d e f https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/761228s000lbl.pdf
- ^ Jump up to:a b c d e f g h i j k l “FDA approves tebentafusp-tebn for unresectable”. U.S. Food and Drug Administration (FDA). 25 January 2022. Retrieved 28 January 2022. This article incorporates text from this source, which is in the public domain.
External links
- “Tebentafusp”. Drug Information Portal. U.S. National Library of Medicine.
- Clinical trial number NCT03070392 for “Safety and Efficacy of IMCgp100 Versus Investigator Choice in Advanced Uveal Melanoma” at ClinicalTrials.gov
/////////////////Tebentafusp-tebn, Kimmtrak, priority review, breakthrough designation, orphan drug designation, Immunocore Limited, IMCGP100, APPROVALS 2022, FDA 2022

NEW DRUG APPROVALS
ONE TIME
$10.00
Daridorexant

Daridorexant
- Molecular FormulaC23H23ClN6O2
- Average mass450.921 Da
[(2S)-2-(5-Chloro-4-methyl-1H-benzimidazol-2-yl)-2-methyl-1-pyrrolidinyl][5-methoxy-2-(2H-1,2,3-triazol-2-yl)phenyl]methanone
1505484-82-1[RN]
Methanone, [(2S)-2-(5-chloro-4-methyl-1H-benzimidazol-2-yl)-2-methyl-1-pyrrolidinyl][5-methoxy-2-(2H-1,2,3-triazol-2-yl)phenyl]-
ACT-541468, , Nemorexant
FDA APPROVED 2022, 1/7/2022, To treat insomnia,

Daridorexant HCl
CAS#: 1792993-84-0 (HCl)
Chemical Formula: C23H24Cl2N6O2
Molecular Weight: 487.39
Elemental Analysis: C, 56.68; H, 4.96; Cl, 14.55; N, 17.24; O, 6.57
Methanone, ((2S)-2-(6-chloro-7-methyl-1H-benzimidazol-2-yl)-2-methyl-1-pyrrolidinyl)(5-methoxy-2-(2H-1,2,3-triazol-2-yl)phenyl)-, hydrochloride (1:1)
Daridorexant HCl; Daridorexant hydrochloride; ACT541468A; ACT 541468A; ACT-541468A; ACT541468 hydrochloride; ACT 541468 hydrochloride; ACT-541468 hydrochloride
Daridorexant HCl is used in the treat of Insomnia Disorder in Adult Patients
Daridorexant, sold under the brand name Quviviq, is a medication used for the treatment of insomnia.[1] Daridorexant is a dual orexin receptor antagonist (DORA) which was originated by Actelion Pharmaceuticals and is under development by Idorsia Pharmaceuticals.[3][4] It acts as a selective dual antagonist of the orexin receptors OX1 and OX2.[3][4] The medication has a relatively short elimination half-life of 6 to 10 hours.[2] As of April 2020, daridorexant has passed its first phase III clinical trial for the treatment of insomnia.[3]Daridorexant was approved for medical use in the United States in January 2022.[1][5][6]
Daridorexant, formerly known as nemorexant, is a selective dual orexin receptor antagonist used to treat insomnia. Insomnia is characterized by difficulties with sleep onset and/or sleep maintenance and impairment of daytime functioning. It chronically affects the person’s daily functioning and long-term health effects, as insomnia is often associated with comorbidities such as hypertension, diabetes, and depression. Conventional treatments for insomnia include drugs targeting gamma-aminobutyric acid type-A (GABA-A), serotonin, histamine, or melatonin receptors; however, undesirable side effects are frequently reported, such as next-morning residual sleepiness, motor incoordination, falls, memory and cognitive impairment. Novel drugs that target orexin receptors gained increasing attention after discovering the role of orexin signalling pathway in wakefulness and almorexant, an orexin receptor antagonist that improved sleep. Daridorexant was designed via an intensive drug discovery program to improve the potency and maximize the duration of action while minimizing next-morning residual activity.1
Daridorexant works on orexin receptors OX1R and OX2R to block the binding of orexins, which are wake-promoting neuropeptides and endogenous ligands to these receptors. Daridorexant reduces overactive wakefulness: in the investigational trials, daridorexant reportedly improved sleep and daytime functioning in patients with insomnia.1 It was approved by the FDA on January 10, 2022, under the name QUVIVIQ.6 as the second orexin receptor antagonist approved to treat insomnia following suvorexant.2
QUVIVIQ
- Generic Name: daridorexant tablets
- Brand Name: Quviviq
QUVIVIQ contains daridorexant, an orexin receptor antagonist. The chemical name of daridorexant hydrochloride is (S)-(2-(5-chloro-4-methyl-1H-benzo[d]imidazol-2-yl)-2-methylpyrrolidin-1-yl)(5- methoxy-2-(2H-1,2,3-triazol-2-yl)phenyl)methanone hydrochloride. The molecular formula is C23H23N6O2Cl * HCl. The molecular weight is 487.38 g/mol.
The structural formula is:
 |
Daridorexant hydrochloride is a white to light yellowish powder that is very slightly soluble in water.
QUVIVIQ tablets are intended for oral administration. Each film-coated tablet contains 27 mg or 54 mg of daridorexant hydrochloride equivalent to 25 mg or 50 mg of daridorexant, respectively. The inactive ingredients are croscarmellose sodium, magnesium stearate, mannitol, microcrystalline cellulose, povidone, and silicon dioxide.
In addition, the film coating contains the following inactive ingredients: glycerin, hypromellose, iron oxide black, iron oxide red, microcrystalline cellulose, talc, titanium dioxide, and, in the 50 mg tablet only, iron oxide yellow.
Dosage Forms And Strengths
QUVIVIQ (daridorexant) tablets are available as:
25 mg: light purple, arc-triangle shaped, film-coated tablet debossed with “25” on one side and “i” (Idorsia logo) on the other side, containing 25 mg daridorexant.
50 mg: light orange, arc-triangle shaped, film-coated tablet debossed with “50” on one side and “i” (Idorsia logo) on the other side, containing 50 mg daridorexant.
QUVIVIQ tablets are available as:
25 mg, light purple, arc-triangle shaped film-coated tablets debossed with “25” on one side, and “i” on the other side. NDC 80491-7825-3, bottle of 30 with child-resistant closure
50 mg: light orange, arc-triangle shaped film-coated tablets debossed with “50” on one side, and “i” on the other side. NDC 80491-7850-3, bottle of 30 with child-resistant closure
SYN
https://chemistry-europe.onlinelibrary.wiley.com/doi/10.1002/cmdc.202000453
Since its discovery in 1998, the orexin system has been of interest to the research community as a potential therapeutic target for the treatment of sleep/wake disorders. Herein we describe our efforts leading to the identification of daridorexant, which successfully finished two pivotal phase 3 clinical trials for the treatment of insomnia disorders.

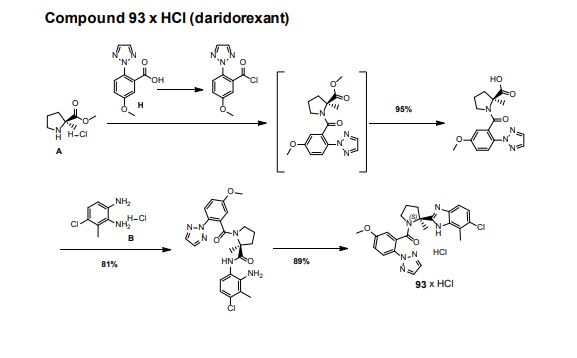
Step 3. Amide (S7) (1000 g, 2.13 mmol) was dissolved in EtOH (5 L) and 32% aqueous HCl (500 mL) was added at 23 °C. The solution was filtered through a Whatman filter (5 µm). The filtrate was heated to 75 °C for 4h. The resulting suspension was cooled to 0 °C and filtered. The product was dried under reduced pressure to yield 93 x HCl (922 g, 89%) as a white solid.
LC-MS B: tR = 0.78 min; [M+H]+ = 451.19, mp 280 °C.
1H NMR (500 MHz, D6-DMSO) δ: 15.05- 15.65 (m, 1 H), 8.06 (s, 2 H), 7.79 (s, 1 H), 7.75 (d, J = 8.9 Hz, 2 H), 7.66 (m, 1 H), 7.57 (d, J = 8.7 Hz, 1 H), 7.15 (dd, J1 = 2.9 Hz, J2 = 8.9 Hz, 1 H), 4.06-4.10 (m, 1 H), 3.92 (s, 3 H), 3.35 (s, 1 H), 2.78 (s, 3 H), 2.54-2.67 (m, 1 H), 2.23-2.31 (m, 1 H), 2.06-2.20 (m, 2 H), 1.97 (s, 3 H),
13C NMR (125 MHz, D6-DMSO) δ: 166.2, 159.3, 158.6, 136.5, 132.7, 131.9, 130.4, 130.3, 129.4, 126.8, 124.5, 123.4, 116.4, 113.7, 113.0, 61.6, 56.8, 49.7, 41.1, 23.9, 20.2, 15.7.

SYN
https://chemistry-europe.onlinelibrary.wiley.com/doi/10.1002/cmdc.201900618
Abstract
DORA explorers: The orexin system plays an important role in regulating the sleep-wake cycle. Herein we report our optimization efforts toward a novel dual orexin receptor antagonist (DORA) with improved properties over compound 6. Replacing the oxadiazole by a triazole resulted in compounds (e. g. compound 33) with improved properties, such as higher intrinsic metabolic stability, lower plasma protein binding, higher brain free fraction, and increased solubility. Further optimization was needed to decrease the compounds P-glycoprotein susceptibility. Our work led to the identification of compound 42, a potent, brain-penetrating DORA with improved in vivo efficacy in dogs compared with compound 6.

Abstract
The orexin system is responsible for regulating the sleep-wake cycle. Suvorexant, a dual orexin receptor antagonist (DORA) is approved by the FDA for the treatment of insomnia disorders. Herein, we report the optimization efforts toward a DORA, where our starting point was (5-methoxy-4-methyl-2-[1,2,3]triazol-2-yl-phenyl)-{(S)-2-[5-(2-trifluoromethoxy-phenyl)-[1,2,4]oxadiazol-3-yl]-pyrrolidin-1-yl}methanone (6), a compound which emerged from our in-house research program. Compound 6 was shown to be a potent, brain-penetrating DORA with in vivo efficacy similar to suvorexant in rats. However, shortcomings from low metabolic stability, high plasma protein binding (PPB), low brain free fraction (fu brain), and low aqueous solubility, were identified and hence, compound 6 was not an ideal candidate for further development. Our optimization efforts addressing the above-mentioned shortcomings resulted in the identification of (4-chloro-2-[1,2,3]triazol-2-yl-phenyl)-{(S)-2-methyl-2-[5-(2-trifluoromethoxy-phenyl)-4H-[1,2,4]triazol-3-yl]-pyrrolidin-1-yl}l-methanone (42), a DORA with improved in vivo efficacy compared to 6.
PAT
WO 2015083071
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015083071
Reference Example 1
1) Synthesis of 5-methoxy-2-(2H-1 ,2,3-triazol-2-yl)benzoic acid
2-lodo-5-methoxy benzoic acid (15.0 g; 53.9 mmol) is dissolved in anhydrous DMF (45 ml) followed by the addition of 1 H-1 ,2,3-triazole (7.452 g; 108 mmol) and cesium carbonate (35.155 g; 108 mmol). By the addition of cesium carbonate the temperature of the reaction mixture increases to 40°C and gas evolved from the reaction mixture. Copper(l)iodide (514 mg; 2.7 mmol) is added. This triggers a strongly exothermic reaction and the temperature of the reaction mixture reaches 70°C within a few seconds. Stirring is continued for 30 minutes. Then the DMF is evaporated under reduced pressure followed by the addition of water (170 ml) and EtOAc (90 ml). The mixture is vigorously stirred and by the addition of citric acid monohydrate the pH is adjusted to 3-4. The precipitate is filtered off and washed with water and EtOAc and discarded. The filtrate is poured into a separation funnel and the phases are separated. The water phase is extracted again with EtOAc. The combined organic layers are dried over MgS04, filtered and the solvent is evaporated to give 7.1 g of 5-methoxy-2-(2H-1 ,2,3-triazol-2-yl)benzoic acid as a white powder of 94% purity (6 % impurity is the regioisomerically N1-linked triazolo-derivative); tR [min] = 0.60; [M+H]+ = 220.21
2) Synthesis of (S)-1 -(tert-butoxycarbonyl)-2-methylpyrrolidine-2-carboxylic acid
2-Methyl-L-proline hydrochloride (99.7 g; 602 mmol) is dissolved in a 1/1-mixture of MeCN and water (800 ml) and triethylamine (254 ml; 1810 mmol) is added. The temperature of the reaction mixture slightly rises. The reaction mixture is cooled to 10°C to 15°C followed by careful addition of a solution of Boc20 (145 g; 662 mmol) in MeCN (200 ml) over 10 minutes.
Stirring at RT is continued for 2 hours. The MeCN is evaporated under reduced pressure and aq. NaOH solution (2M; 250 ml) is added to the residual aq. part of the reaction mixture. The water layer is washed with Et20 (2x 300 ml) then cooled to 0°C followed by slow and careful addition of aq. HCI (25%) to adjust the pH to 2. During this procedure a suspension forms.
The precipitate is filtered off and dried at HV to give 1 10.9 g of the title compound as a beige powder; tR [min] = 0.68; [M+H]+ = 230.14
3) Synthesis of (S)-tert-butyl 2-((2-amino-4-chloro-3-methylphenyl)carbamoyl)-2-
(S)-1-(tert-butoxycarbonyl)-2-methylpyrrolidine-2-carboxylic acid (60 g; 262 mmol) and HATU (100 g; 264 mmol) is suspended in DCM (600 ml) followed by the addition of DIPEA (84.6 g; 654 mmol) and 6-chloro-2,3-diaminotoluene (41 g; 262 mmol). The reaction mixture is stirred at rt for 14 hours then concentrated under reduced pressure and to the residue is added water followed by the extraction of the product with EtOAc (3x). The combined organic layers are washed with brine, dried over MgS04, filtered and the solvent is evaporated under
reduced pressure to give 185 g of the title compound as a dark brownish oil, which is used in the next step without further purification; tR [min] = 0.89; [M+H]+ = 368.01
4) Synthesis of (S)-tert-butyl 2-(5-chloro-4-methyl-1 H-benzo[d]imidazol-2-yl)-2-methylpyrrolidine-1 -carboxylate
(S)-tert-butyl 2-((2-amino-4-chloro-3-methylphenyl)carbamoyl)-2-methylpyrrolidine-1-carboxylate (185 g; 427 mmol) are dissolved in AcOH (100%; 611 ml), heated to 100°C and stirring continued for 90 minutes. The AcOH is evaporated under reduced pressure and the residue is dissolved in DCM followed by careful addition of saturated sodium bicarbonate solution. The phases are separated, the aq. phase is extracted once more with DCM, the combined aq. phases are dried over MgS04, filtered and the solvent is evaporated under reduced pressure to give 142.92 g of the title compound as a dark brown oil which is used in the next step without further purification; tR [min] = 0.69; [M+H]+ = 350.04
5) Synthesis of (S)-5-chloro-4-methyl-2-(2-methylpyrrolidin-2-yl)-1 H-benzo[d]imidazole hydrochloride
(S)-tert-butyl 2-(5-chloro-4-methyl-1 H-benzo[d]imidazol-2-yl)-2-methylpyrrolidine-1-carboxylate (355.53 g; 1.02 mol) are dissolved in dioxane (750 ml) followed by careful addition of HCI solution in dioxane (4M; 750 ml; 3.05 mol). The reaction mixture is stirred for 3 hours followed by the addition of Et20 (800 ml) which triggered precipitation of the product. The solid is filtered off and dried at high vacuum to give 298.84 g of the title compound as a redish powder; tR [min] = 0.59; [M+H]+ = 250.23
6) Synthesis of [(S)-2-(5-chloro-4-methyl-1 H-benzoimidazol-2-yl)-2-methyl-pyrrolidin-1- -(5-methoxy-2-[1,2,3]triazol-2-yl-phenyl)-methanone
(S)-5-chloro-4-methyl-2-(2-methylpyrrolidin-2-yl)-1 H-benzo[d]imidazole hydrochloride (62.8 g; 121 mmol) is dissolved in DCM (750 ml) followed by the addition of 5-methoxy-2-(2H-1 ,2,3-triazol-2-yl)benzoic acid (62.8 g; 121 mmol) and DIPEA (103 ml; 603 mmol). Stirring is continued for 10 minutes followed by the addition of HATU (47 g; 124 mmol). The reaction mixture is stirred for 16 hours at RT. The solvents are evaporated under reduced pressure and the residue is dissolved in EtOAc (1000 ml) and washed with water (3x 750 ml). The organic phase is dried over MgS04, filtered and the solvent is evaporated under reduced pressure. The residue is purified by CC with EtOAc / hexane = 2 / 1to give 36.68 g of the title compound as an amorphous white powder. tR [min] = 0.73; [M+H]+ = 450.96
Table 1 : Characterisation data for COMPOUND as free base in amorphous form
II. Preparation of crystalline forms of COMPOUND
Example 1 :
Preparation of seeding material of COMPOUND hydrochloride in crystalline Form 1
10 mg COMPOUND is mixed with 0.2 mL 0.1 M aq. HCI and 0.8 mL EtOH. The solvent is fully evaporated and 0.05 mL isopropanol is added. Alternatively 0.05 mL methyl-isobutylketone can be added. The sample is stored closed at room temperature for 4 days and crystalline material of COMPOUND hydrochloride in crystalline Form 1 is obtained. This material can be used as seeding material for further crystallization of COMPOUND hydrochloride in crystalline Form 1.
Example 2: Preparation and characterization of COMPOUND hydrochloride in crystalline form 1
5g COMPOUND is mixed with 0.9 mL 1 M aq. HCI and 20 mL EtOH. The solvent is evaporated and 25 mL isopropanol is added. Seeds of COMPOUND hydrochloride are added and the sample is allowed to stand at room temperature. After about 2 days the suspension is filtered and the solid residue is dried at reduced pressure (2 mbar for 1 hour) and allowed to equilibrate open for 2 hours at 24°C/46% relative humidity. The obtained solid is COMPOUND hydrochloride in crystalline Form 1
Table 2: Characterisation data for COMPOUND hydrochloride in crystalline form 1
PAT
WO 2018202689
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2018202689
Examples
Reference Example 1
Synthesis of 5-methoxy-2-(2H-1,2,3-triazol-2-yl)benzoic acid
4,5-dibromo-2-(4-methoxy-2-nitrophenyl)-2H-1,2,3-triazole
4- Fluoro-3-nitroanisole (3.44 g, 1 eq.), 4,5-dibromo-2/-/-1 ,2,3-triazole (4.56 g, 1 eq.)1, K2C03 (2.78 g, 1 eq.) and DMF (30 mL) are heated to 1 10 °C for 32 h. The reaction mixture is cooled to 22 °C and treated with water (70 mL). The resulting suspension is filtered, washed with water (15 mL). The product is slurried in isopropanol (40 mL), filtered and dried under reduced pressure to yield a white solid. Yield: 6.42 g, 84%. Purity: 100% a/a (LC-MS method 2). 1H NMR (400 MHz, CDCI3) δ: 7.71 (d, J = 8.9 Hz, 1 H), 7.47 (d, J = 2.8 Hz, 1 H), 7.25 (dd, Ji = 2.8 Hz, J2 = 8.9 Hz, 1 H), 3.97 (s, 3 H).
1 X. Wang, L. Zhang, D. Krishnamurthy, C. H. Senanayake, P. Wipf Organic Letters 2010 12 (20), 4632-4635.
5- methoxy-2-(2H-1 ,2,3-triazol-2-yl)aniline
4, 5-Dibromo-2-(4-methoxy-2-nitrophenyl)-2/-/-1 ,2,3-triazole (2 g, 1 eq.), sodium acetate (1.3 g, 3 eq.), and 10% Pd/C 50% water wet (0.3 g) is suspended in EtOAc (10 mL). The mixture is heated to 50 °C and set under hydrogen until conversion is complete. The reaction mixture is filtered over Celite. The filtrate is washed with 1 N NaOH (10 mL) and water (15 mL). The organic layer is concentrated under reduced pressure to yield an oil. Yield: 0.95 g, 94%. Purity: 96% a/a (LC-MS method 2). 1H NMR (400 MHz, DMSO) <5: 8.05 (s, 2 H), 7.53 (d, J = 8.9 Hz, 1 H), 6.49 (d, J = 2.7 Hz, 1 H), 6.30 (dd, Ji = 2.7 Hz, J2 = 8.9 Hz, 1 H), 5.94 (s, 2 H), 3.74 (s, 3 H).
5-methoxy-2-(2H-1,2,3-triazol-2-yl)aniline monosulfate
5-Methoxy-2-(2/-/-1 ,2,3-triazol-2-yl)aniline (455 g, 1 eq ) is dissolved in isopropanol (3 L). To the solution is added cone. H2SO4 (235 g, 1 eq.) below 40 °C. The suspension is cooled to
20 °C and filtered. The cake is washed with isopropanol (700 mL) and TBME (1.5 L). The product is dried to obtain a white solid. Yield: 627 g, 91 %. Purity: 100% a/a (LC-MS method 2).
2-(2-iodo-4-methoxyphenyl)-2H-1,2,3-triazole
5-Methoxy-2-(2/-/-1 ,2,3-triazol-2-yl)aniline monosulfate (200 g, 1 eq.) is dissolved in 2 M aq. H2SO4 soln. (1.4 L) and cooled to -5 °C. To the solution is added a solution of sodium nitrite (62 g, 1.3 eq.) in water (600 mL) at -5 to 0 °C. The mixture is stirred at 0 °C for 30 min and then added to a preheated mixture of Kl (161 g, 1.4 eq.) in water (700 mL) at 65 °C. The resulting solution is stirred at 60 °C for 20 min, cooled to 20 °C and treated with a soln. of sulfamic acid (27 g, 0.4 eq.) in water (120 mL). The mixture is extracted with isopropyl acetate (2 L). The organic layer is washed with a mixture of 2 N NaOH (500 mL) and 40% NaHS03 soln. (100 mL), and a mixture of 1 N HCI (50 mL) and water (500 mL). The organic layer is concentrated to dryness. The residue is dissolved in isopropanol (700 mL) and cooled to 0 °C. The resulting suspension is filtered. The solid is dried under reduced pressure. Yield: 164 g, 79%. Purity: 100% a/a (LC-MS method 2). 1H NMR (400 MHz, DMSO) <5: 8.08 (s, 2 H), 7.57 (d, J = 2.8 Hz, 1 H), 7.43 (d, J = 8.8 Hz, 1 H), 7.13 (dd, Ji = 2.8 Hz, J2 = 8.8 Hz, 1 H), 3.85 (s, 3 H).
5-methoxy-2-(2H-1 ,2,3-triazol-2-yl)benzoic acid
2-(2-lodo-4-methoxyphenyl)-2/-/-1 ,2,3-triazole (200 g, 1 eq.) is dissolved in THF (2 L) and cooled to 0 °C. 2 M iPrMgCI soln. in THF (350 mL, 1.05 eq.) is added at 0 °C. The mixture is cooled to -20 °C and C02 (gas) is bubbled into the solution over 30 min until the exothermicity is ceased. To the mixture is added 2 N HCI (600 mL) at 8 °C and concentrated under reduced pressure to remove 2.4 L solvent. The residue is extracted with TBME (1.6 L). The organic layer is washed with 1 N HCI (200 mL) and extracted with 1 N NaOH (600 mL and 200 mL). The aq. layer is filtered over charcoal (15 g), diluted with water (200 mL) and treated with 32% HCI (160 mL). The resulting suspension is filtered and washed with water (200 mL). Yield: 127 g, 87%. Purity: 100% a/a (LC-MS method 2); MP: 130 °C (DSC goldpan). The obtained product may be re-crystallized from toluene (MP: 130.9 °C) or water (MP: 130 °C).
Table Ref 1 : Characterisation data for 5-methoxy-2-(2H-1,2,3-triazol-2-yl)benzoic acid in crystalline form 2 (recrystallization from toluene)
Technique Data Summary Remarks
XRPD Crystalline see Fig. 8
Reference Example 2
Synthesis of 4-methyl-2-(2H-1 ,2,3-triazol-2-yl)benzoic acid
4,5-Dibromo-2-(5-methyl-2-nitrophenyl)-2H-1 ,2,3-triazole
3- Fluoro-4-nitrotoluene (1367 g, 1 eq.), 4,5-dibromo-2/-/-1 ,2,3-triazole (1999 g, 1 eq.), K2C03 (1340 g, 1.1 eq.) and DMF (1 1 L) is heated to 75 °C for 15 h. The reaction mixture is cooled to 22 °C and treated with water (18 L). The resulting suspension is filtered, washed with water (4 L). The product is washed with isopropanol (5 L), and dried under reduced pressure to yield a white solid. Yield: 281 1 g, 88%. Purity: 100% a/a (LC-MS method 2). 1H NMR (400 MHz, DMSO) <5: 8.10 (d, J = 8.3 Hz, 1 H), 7.86 (d, J = 1.0 Hz, 1 H), 7.66 (dd, J1 = 0.9 Hz, J2 = 8.3 Hz, 1 H), 2.51 (s, 3 H).
4- Methyl-2-(2H-1 ,2,3-triazol-2-yl)aniline
4, 5-Dibromo-2-(5-methyl-2-nitrophenyl)-2/-/-1 ,2,3-triazole (205 g, 1 eq.), sodium acetate (149 g, 3.2 eq.), and 5% Pd/C 50% water wet (37.8 g) is suspended in EtOAc (0.8 L). The mixture is heated to 40-50 °C and set under hydrogen (2 bar) until conversion is complete. The reaction mixture is filtered over Celite. The filtrate is washed with water (300 mL), 2N NaOH (300 ml_+250 mL) and water (300 mL). The organic layer is concentrated under reduced pressure to yield a yellow oil. Yield: 132 g, 90%. Purity: 100% a/a (LC-MS method 2). 1H NMR (400 MHz, DMSO) <5: 8.09 (s, 2 H), 7.48 (d, J = 1.3 Hz, 1 H), 6.98 (dd, J1 = 1.8 Hz, J2 = 8.3 Hz, 1 H), 6.85 (d, J = 8.2 Hz, 1 H), 5.79 (s, 2 H), 2.23 (s, 3 H).
4-Methyl-2-(2H-1,2,3-triazol-2-yl)aniline monosulfate
4-Methyl-2-(2/-/-1 ,2,3-triazol-2-yl) aniline (199 g, 1 eq ) is dissolved in isopropanol (1.7 L). To the solution is added cone. H2SO4 (118 g, 1.05 eq.) below 40 °C. The suspension is cooled to 20 °C and filtered. The cake is washed with isopropanol (500 mL). The product is dried to obtain a white solid. Yield: 278 g, 89%. Purity: 100% a/a (LC-MS method 2). 1H NMR (400 MHz, DMSO) <5: 8.21 (s, 2 H), 7.70 (s, 1 H), 7.23 (s, 2 H), 2.35 (s, 3 H).
2-(2-iodo-5-methylphenyl)-2H-1 ,2,3-triazole
4-Methyl-2-(2/-/-1 ,2,3-triazol-2-yl)aniline monosulfate (1553 g, 1 eq.) is dissolved in 1 M aq. H2S04 Soln. (1 1 L) and cooled to -5 °C. To the solution is added a solution of sodium nitrite (433 g, 1.1 eq.) in water (4 L) at -5 to 0 °C. The mixture is stirred at 0 °C for 30 min and then added to a preheated mixture of potassium iodide (1325 g, 1.4 eq.) in water (4 L) at 55-70 °C. The resulting solution is stirred at 60 °C for 20 min, cooled to 20 °C and treated with a soln. of sulfamic acid (220 g, 0.4 eq.) in water (900 mL). The mixture is extracted with isopropyl acetate (13 L). The organic layer is washed with a mixture of 2 N NaOH (3.5 L) and 40% NaHSOs soln. (330 g), and a mixture of 1 N HCI (280 mL) and water (3.5 L). The
organic layer is concentrated to dryness. Yield: 1580 g, 97%. Purity: 91 % a/a (LC-MS method 2). 1 H NMR (400 MHz, CDCI3) <5: 7.90 (s, 2 H), 7.87 (d, J = 8.1 Hz, 1 H), 7.34 (d, J = 1 .6 Hz, 1 H), 7.03-7.06 (m, 1 H), 2.40 (s, 3 H).
The crude product, together with a second batch (141 1 g) is purified by distillation on a short path distillation equipment at 120 °C jacket temperature, feeding tank (70 °C), cooling finger (20 °C) and at a pressure of 0.004 mbar. Yield: 2544 g (78%), Purity: 100 % a/a ()LC-MS method 2).
4-Methyl-2-(2H-1 ,2,3-triazol-2-yl)benzoic acid
2-(2-lodo-5-methylphenyl)-2/-/-1 ,2,3-triazole (1250 g, 1 eq.) is dissolved in THF (13 L) and cooled to 0 °C. 2 M iPrMgCI soln. in THF (2.2 L, 1 eq.) is added at 0 °C. The mixture is cooled to -25 °C and CO2 (gas) is bubbled into the solution over 60 min until the exothermicity is ceased. To the mixture is added 2 N HCI (5 L) at 4 °C and concentrated under reduced pressure to remove 14.5 L solvent. The residue is extracted with TBME (10 L). The organic layer is extracted with 1 N NaOH (6 L and 3 L). The aq. layer is filtered over charcoal (15 g), diluted with water (200 mL) and treated with 32% HCI (1 .23 L). The resulting suspension is filtered and washed with water (5 L). Yield: 796 g, 89%. Purity: 100% a/a (LC-MS method 2); MP: 125 °C (DSC goldpan).
The following examples illustrate the invention.
Example 1 :
Example 1.1: Crystalline 5-methoxy-2-(2H-1,2,3-triazol-2-yl)benzoic acid potassium salt (potassium 5-methoxy-2-(2H-1,2,3-triazol-2-yl)benzoate)
2-Bromo-5-methoxybenzoic acid (21 .5 g, 0.093 mol, 1 eq.) copper (I) iodide (0.886 g, 0.05 eq.), and K2CO3 powder (32.2 g, 2.5 eq.) were suspended in dioxane (600 mL) and water (8.4 mL). To the mixture were added 1 H-1 ,2,3-triazole (10.8 mL, 2 eq.) and trans-/V,/V-dimethylcyclohexane-1 ,2-diamine (1 .32 g, 0.1 eq.). The mixture was heated at reflux for 3.5 h. IPC showed full conversion. The ratio of the desired N(2) to the regioisomeric Λ/(1 ) isomer was 84: 16. The mixture was cooled to 40 °C and filtered. The cake was washed with dioxane (100 mL). The solid was dried to obtain 50.6 g of a blue solid. The ratio of N{2) to Λ/(1 ) isomer of was 98.6: 1 .4.
Table 1 : Characterisation data for 5-methoxy-2-(2H-1,2,3-triazol-2-yl)benzoic acid potassium salt in crystalline form 1
Example 1.2: Crystalline 5-methoxy-2-(2H-1,2,3-triazol-2-yl)benzoic acid
The solid of Example 1.1 was dissolved in water (300 mL). TBME (200 mL) and 32% aq. HCI (35 mL) was added. The aq. layer was separated and discarded. The organic layer was washed with a mixture of 2N aq. HCI (100 mL) and 32% aq. HCI (20 mL). The organic layer was washed with 1 N aq. HCI (50 mL). The organic layer was extracted with 1 N aq. NaOH (200 mL). The aq. layer was heated to 45 °C and traces of TBME were removed under reduced pressure. To the aq. layer was added at 45 °C 32% aq. HCI (20 mL). At a pH of 6 optionally seed crystals were added. The resulting suspension was filtered at 40 °C. The cake was washed with water (30 mL). The product was dried at 60 °C and 5 mbar. Yield: 12.4 g, 61 %. Purity: 100% a/a, tR 0.63 min. Seed crystals may be obtained by careful crystallization according to the above procedure.
MP: 80 °C (DSC).
1H NMR (400 MHz, DMSO) & 3.87 (s, 3 H), 7.26 (m, 2 H), 7.64 (d, J = 8.7 Hz, 1 H), 8.02 (s, 2 H), 13.01-13.22 (br, 1 H).
Table 2: Characterisation data for 5-methoxy-2-(2H-1,2,3-triazol-2-yl)benzoic acid in crystalline form 1
Example 1.3: Crystalline 5-methoxy-2-(2H-1,2,3-triazol-2-yl)benzoic acid potassium salt
5-Methoxy-2-(2/-/-1 ,2,3-triazol-2-yl)benzoic acid, e.g. obtained according to the procedure of Reference Example 1 (5 g, 0.0228 mol) and KHCO3 (1.61 g, 0.7 eq) were suspended in dioxane (100 mL) and water (1 mL). The mixture was heated at reflux for 40 min. The mixture was cooled to 20 °C and filtered. Yield: 2.56 g, 44%. 1H NMR (400 MHz, D20) & 3.80 (s, 3 H), 7.04 (m, 2 H), 7.46 (d, J = 8.7 Hz, 1 H), 7.82 (s, 2 H). MP: 279.5°C (DSC shows additionally a broad endothermic event at about 153 °C to 203 °C which may be attributed to endothermic desolvations; melting is immediately followed by exothermic degradation).
Table 3: Characterisation data for 5-methoxy-2-(2H-1,2,3-triazol-2-yl)benzoic acid potassium salt in crystalline form 2
Example 1.4: Crystalline 5-methoxy-2-(2H-1,2,3-triazol-2-yl)benzoic acid potassium salt
In an alternative procedure, 2-Bromo-5-methoxybenzoic acid (20 g, 0.086 mol, 1 eq.) copper (I) iodide (0.824 g, 0.05 eq.), and K2C03 powder (26.9 g, 2.25 eq.) were suspended in dioxane (494 mL). To the mixture was added 1 H-1 ,2,3-triazole (12 g, 2 eq.). The mixture was heated at reflux for 1 h. To the mixture was added water (12.5 g, 8 eq.). The mixture was heated at reflux for 2 h. Solvent (100 mL) was removed by distillation. The residue was cooled to 45 °C in 8 min, filtered and washed with dioxane (50 mL).
XRPD corresponds to crystalline form 1 (see Fig. 1 , Example 1.1 ).
Example 1.5: Crystalline 5-methoxy-2-(2H-1,2,3-triazol-2-yl)benzoic acid
The solid of Example 1.4 was dissolved in water (200 mL). The mixture was heated to 50 °C and 20% aq. H2SO4 (40 mL) was added to adjust the pH to 5. The mixture was filtered over Celite. The filtrate was treated at 45 °C with 20% aq. H2S04 (40 mL). At pH 3 seeds (obtained for example using the procedure of reference example 1 ) were added. The suspension was stirred at 45 °C and filtered. The product was washed with water (20 mL) and dried at 60 °C and 10 mbar to yield a white solid. Yield: 10.8 g, 57%. Purity: 100% a/a, tR 0.63 min.
Characterisation of 5-methoxy-2-(2/-/-1 ,2,3-triazol-2-yl)benzoic acid obtained according to Example 1.5:
XRPD corresponds to crystalline form 1 (see Fig. 2, Example 1.2).
Example 2:
Example 2.1: Crystalline 4-methyl-2-(2H-1,2,3-triazol-2-yl)benzoic acid potassium salt (potassium 4-methyl-2-(2H-1,2,3-triazol-2-yl)benzoate)
2-Bromo-4-methylbenzoic acid (20 g, 0.093 mol, 1 eq.) copper (I) iodide (0.886 g, 0.05 eq.), and K2CO3 powder (32.2 g, 2.5 eq.) were suspended in dioxane (300 mL) and water (10.1 mL). To the mixture was added 1 A7-1 ,2,3-triazole (10.8 mL, 2 eq.) and trans-Λ/,ΛΑ-
dimethylcyclohexane-1 ,2-diamine (1 .32 g, 0.1 eq.). The mixture was heated at reflux for 4 h. IPC showed a conversion of 98.5%. The ratio of the desired N(2) to the regioisomeric Λ/(1 ) isomer was 75:25. The mixture was concentrated at normal pressure and external temperature of 130 °C. Solvent (100 mL) was removed. To the residue was added dioxane (100 mL) and the mixture was cooled to 45 °C and filtered. The cake was washed with dioxane (80 mL). The solid was dried to obtain 48.8 g of a blue solid. The ratio of N(2) to Λ/(1 ) isomer was 98.7: 1 .3.
Table 4: Characterisation data for 4-methyl-2-(2H-1 ,2,3-triazol-2-yl)benzoic acid potassium salt in crystalline form 1
Example 2.2: Crystalline 4-methyl-2-(2H-1,2,3-triazol-2-yl) benzoic acid
The solid of Example 2.1 was dissolved in water (300 mL) and filtered. To the filtrate were added TBME (200 mL) and 32% aq. HCI (30 mL). The aq. layer was separated and discarded. The organic layer was washed with a mixture of 2N aq. HCI (100 mL) and 32% aq. HCI (10 mL). The organic layer was washed with 1 N aq. HCI (50 mL). The organic layer was extracted with 1 N aq. NaOH (200 mL). The aq. layer was heated to 45 °C and traces of TBME were removed under reduced pressure. To the aq. layer was added at 45 °C 32% aq. HCI (20 mL). At a pH of 6 seed crystals (obtained for example using the procedure of reference example 2) were added. The resulting suspension was filtered at 40 °C. The cake was washed with water (30 mL). The product was dried at 60 °C and 5 mbar. Yield: 1 1 .7 g, 62%. Purity: 100% a/a. tR 0.66 min.
MP: 125 °C (DSC).
1H NMR (400 MHz, DMSO) & 2.44 (s, 3 H), 7.41 (d, J = 7.9 Hz, 1 H), 7.56 (s, 1 H), 7.68 (d, J = 7.9 Hz, 1 H), 8.06 (s, 2 H), 12.53-13.26 (br, 1 H)
Table 5: Characterisation data for 4-methyl-2-(2H-1 ,2,3-triazol-2-yl)benzoic acid in crystalline form 1
Technique Data Summary Remarks
XRPD Crystalline see Fig. 5
Example 2.3: Crystalline 4-methyl-2-(2H-1,2,3-triazol-2-yl)benzoic acid potassium salt
4-Methyl-2-(2H-1 ,2,3-triazol-2-yl)benzoic acid (5 g, 0.0246 mol) and KHC03 (1 .74 g , 0.7 eq) were suspended in dioxane ( 100 mL) and water (1 mL). The mixture was heated at reflux for 40 min. The mixture was cooled to 20 °C and filtered. Yield: 2.47 g, 42% . MP: 277 °C (DSC Alupan) 1 H NMR (400 MHz, D20) & 2.32 (s, 3 H), 7.28 (d, J = 7.9 Hz, 1 H), 7.39 (m, 2 H), 7.84 (s, 2 H).
MP: 276.8 °C (DSC shows additionally a broad endothermic event at about 140 °C to 208 °C which may be attributed to endothermic desolvations; melting is immediately followed by exothermic degradation).
XRPD corresponds to crystalline form 1 (see Fig. 4, Example 2.1 ).
Reference Example 3:
Reference Example 3.1: Crystalline 5-methyl-2-(2H-1,2,3-triazol-2-yl)benzoic acid sodium salt (sodium 5-methyl-2-(2H-1,2,3-triazol-2-yl)benzoate)
2-Bromo-5-methylbenzoic acid (20 g, 0.093 mol, 1 eq. ) copper (I) iodide (0.886 g, 0.05 eq.), Na2CC>3 powder (24.6 g, 2.5 eq.) were suspended in dioxane (300 mL) and water (10.1 mL). To the mixture was added 1 /-/-1 ,2,3-triazole ( 10.8 mL, 2 eq.) and 8-hydroxy quinoline ( 1 .35 g, 0.1 eq.). The mixture was heated at reflux for 5 h. IPC showed a conversion of >99%. The ratio of the desired N(2) to the regioisomeric Λ/(1 ) isomer was 78:22. The mixture was concentrated at normal pressure and external temperature of 135 °C. Solvent (100 mL) was removed. To the residue was added dioxane (100 mL) and the mixture was cooled to 45 °C and filtered. The cake was washed with dioxane (80 mL). The solid was dried to obtain 36.2 g of a yellow solid. The ratio of N(2) to Λ/( 1 ) isomer of was 99: 1 .
Table 6: Characterisation data for 5-methyl-2-(2H-1 ,2,3-triazol-2-yl)benzoic acid sodium salt in crystalline form 1
Reference Example 3.2: Crystalline 5-methyl-2-(2H-1,2,3-triazol-2-yl)benzoic acid
The solid obtaind in Reference Example 3.1 was dissolved in water (300 mL) and filtered. To the filtrate was added TBME (200 mL) and 32% aq. HCI (30 mL) was added. The aq. layer was separated and discarded. The organic layer was washed with 1 N aq. HCI ( 100 mL). The organic layer was washed with 1 N aq. HCI (50 mL). The organic layer was extracted with 1 N aq. NaOH (200 mL). The aq. layer was heated to 45 °C and traces of TBME were removed
under reduced pressure. To the aq. layer was added at 45 °C 32% aq. HCI (20 mL). At a pH of 6 seed crystals (obtained for example using the procedure of Reference example 2) were added. The resulting suspension was filtered at 40 °C. The cake was washed with water (30 mL). The product was dried at 60 °C and 5 mbar. Yield: 12.1 g, 64%. Purity: 100% a/a. tR 0.67 min.
MP: 173 °C (DSC)
1 H NMR (400 MHz, DMSO) & 2.42 (s, 3 H), 7.50-7.52 (m, 1 H), 7.58 (s, 1 H), 7.63 (m, 1 H), 8.05 (s, 2 H), 13.01 (s, 1 H).
Table 7: Characterisation data for 5-methyl-2-(2H-1 ,2,3-triazol-2-yl)benzoic acid in crystalline form 1
Reference Example 3.3: Crystalline 5-methyl-2-(2H-1,2,3-triazol-2-yl) benzoic acid sodium salt
5-Methyl-2-(2/-/-1 ,2,3-triazol-2-yl)benzoic acid (5 g, 0.0246 mol) and Na2C03 (1 .05 g, 0.4 eq) were suspended in dioxane ( 100 mL) and water (1 mL). The mixture was heated at reflux for 40 min. The mixture was cooled to 20 °C and filtered. Yield: 2.79 g, 50%. MP: 341 °C (DSC Alupan) 1 H NMR (400 MHz, D20) & 2.32 (s, 3 H), 7.30 (m, 2 H), 7.43 (m, 1 H), 7.83 (s, 2 H).
XRPD corresponds to crystalline form 1 (see Fig. 6, Reference Example 3.1 ).
Reference Example 3.4: 5-methyl-2-(2H-1,2,3-triazol-2-yl)benzoic acid potassium salt
2-Bromo-5-methylbenzoic acid (20 g, 0.093 mol, 1 eq. ) copper (I) iodide (0.886 g, 0.05 eq.), and K2CO3 powder (32.1 g, 2.5 eq.) were suspended in dioxane (600 mL). To the mixture was added 1 /-/-1 ,2,3-triazole ( 10.8 mL, 2 eq.) and 8-hydroxy quinoline ( 1 .35 g, 0.1 eq.). The mixture was heated at reflux for 4 h. IPC showed a conversion of >94%. The ratio of the desired N(2) to the regioisomeric Λ/( 1 ) isomer was 78:22. The mixture was cooled to 35 °C and filtered. The cake was washed with dioxane (100 mL). The products were dissolved in water and a LC-MS was recorded. The ratio of N(2) to Λ/(1 ) isomer of was 83: 17.
Reference Example 4.1: Methyl (S)-1-(5-methoxy-2-(2H-1,2,3-triazol-2-yl) benzoyl)-2-methylpyrrolidine-2-carboxylate
5-Methoxy-2-(2/-/-1 ,2,3-triazol-2-yl) benzoic acid (100 g, 0.46 mol) was suspended in DCM (650 mL) and DMF (10 mL) at 20 °C. To this suspension was added oxalyl chloride (51 mL, 0.59 mol) over a period of 30 min. LC-MS showed 60% conversion to acid chloride intermediate. Oxalyl chloride (17.6 mL, 0.45 eq.) was added dropwise. LC-MS showed full conversion to acid chloride intermediate.
Methyl (S)-2-methylpyrrolidine-2-carboxylate hydrochloride (84 g, 0.47 mol) was suspended in DCM (800 mL) in a second flask. The suspension was cooled to 10 °C. Triethylamine (200 mL, 1.41 mol) was added over 15 min. The acid chloride solution was added to the reaction mixture at 10-20 °C over at least 15 min. The reaction mixture was washed with 1 M HCI (500 mL), 1 N NaOH (500 mL) and water (500 mL). The organic layer was concentrated to dryness to give a light-yellow solid as product. Yield: 157 g, 100%, 99% a/a (LC-MS), M+1 =345. 1H NMR (400 MHz, DMSO) δ: 8.06 (s, 2 H), 7.79 (d, J = 8.9 Hz, 1 H), 7.21 (dd, J1 = 2.9 Hz, J2 = 8.9 Hz, 1 H), 6.85 (d, J = 1.9 Hz, 1 H), 3.89 (s, 3 H), 3.66 (s, 3 H), 3.29 (m, 1 H), 3.03 (m, 1 H), 2.08 (m, 1 H), 1.82 (m, 3 H), 1.50 (s, 3 H).
Reference Example 4.2: (S)-1-(5-methoxy-2-(2H-1,2,3-triazol-2-yl) benzoyl)-2-methylpyrrolidine-2-carboxylic acid
Methyl (S)-1-(5-methoxy-2-(2/-/-1 ,2,3-triazol-2-yl) benzoyl)-2-methylpyrrolidine-2-carboxylate (157 g, 0.46 mol) was dissolved in MeOH (750 mL) at 20 °C. To this solution was added 16% NaOH (300 mL). The resulting solution was heated up to 80 °C and stirred for 60 min. Solvent was distilled off under reduced pressure (850 mL). The residue was taken up in DCM (1500 mL) and water (450 ml) at 20 °C. 32% HCI (200 mL) was added. Layers were separated and the organic layer was washed with water (450 mL). The organic layer was concentrated to the minimum stirring volume under reduced pressure. Toluene (750 mL) was added and solvent was further distilled under vacuum (150 mL distilled). The mixture was cooled to 20 °C and stirred for 15 min. The suspension was filtered at 20 °C. The cake was rinsed with toluene (150 mL) and then dried under reduced pressure at 50 °C to give a white solid as product. Yield: 128 g, 85%, 94% a/a (LC-MS), M+1 =331. Melting point: 178 °C (DSC). 1H NMR (400 MHz, DMSO) δ: 12.3 (s, 1 H), 8.04 (s, 2 H), 7.79 (d, 1 H), 7.20 (dd, J1 = 2.8 Hz, J2 = 8.9 Hz, 1 H), 6.84 (m, 1 H), 3.88 (s, 3 H), 3.29 (m, 1 H), 2.99 (m, 1 H), 2.1 1 (m, 1 H), 1.81 (m, 3 H), 1.47 (s, 3 H).
Reference Example 4.3: (S)-N-(2-amino-4-chloro-3-methylphenyl)-1-(5-methoxy-2-(2H-1,2,3-triazol-2-yl) benzoyl)-2 methylpyrrolidine-2-carboxamide
(S)-1-(5-Methoxy-2-(2/-/-1 ,2,3-triazol-2-yl) benzoyl)-2-methylpyrrolidine-2-carboxylic acid (128 g, 0.39 mol) was suspended in DCM (850 mL) and DMF (6 mL) at 20 °C. To this suspension was added oxalyl chloride (39 mL, 0.45 mol) over a period of 30 min. 4-Chloro-3-methylbenzene-1 ,2-diamine hydrochloride (75 g, 0.39 mol) was suspended in DCM (1300 mL) in a second flask. The suspension was cooled down to 10 °C. Triethylamine (180 mL, 1.27 mol) was added. The acid chloride solution was added to the reaction mixture at 10-20 °C over at least 15 min. Water (650 mL) was added to the reaction mixture. Layers were separated and the organic phase was concentrated under reduced pressure (1900 mL distilled out). TBME (1000 mL) was added and solvent was further distilled under vacuum (400 mL distilled). The mixture was finally cooled down to 20 °C and stirred for 15 min. The resulting suspension was filtered off at 20 °C. The cake was rinsed with TBME (250 mL) and then dried under reduced pressure at 50 °C to give a white solid as product. Yield: 145 g, 80%, 97% a/a (LC-MS), M+1=469. Melting point: 185 °C (DSC). 1H NMR (400 MHz, DMSO) δ: 9.10-9.14 (m, 1 H), 7.88-8.12 (m, 2 H), 7.81-7.82 (m, 1 H), 7.38-7.44 (m, 1 H), 7.21 (dd, J1 = 2.7 Hz, J2 = 8.9 Hz, 1 H), 6.84 (d, J = 7.8 Hz, 1 H), 6.64 (d, J = 8.3 Hz, 1 H), 5.01 (brs, 2 H), 3.88 (s, 3 H), 3.61-3.73 (m, 1 H), 3.14-3.26 (m, 1 H), 2.25-2.30 (m, 1 H), 2.13 (s, 3 H), 1.97 (m, 3 H), 1.47-1.61 (m, 3 H).
Reference Example 4.4: (S)-(2-(5-chloro-4-methyl-1H-benzo[d]imidazol-2-yl)-2-methylpyrrolidin-1-yl) (5-methoxy-2-(2H-1,2,3-triazol-2-yl)phenyl)methanone hydrochloride
(S)-/V-(2-amino-4-chloro-3-methylphenyl)-1-(5-methoxy-2-(2H-1 ,2,3-triazol-2-yl) benzoyl)-2 methylpyrrolidine-2-carboxamide (145 g, 0.31 mol) was dissolved in isopropanol (870 mL) at 20 °C. To this solution was added carefully 5-6 N HCI in isopropanol (260 mL) over 10 min. the reaction mixture was then heated up to 90 °C and stirred for 4 hours. Water (28 mL) was added and the reaction mixture was stirred for an additional one hour. The reaction mixture was cooled to 20 °C. A light brown suspension was obtained which was filtered. The cake was rinsed with isopropanol (220 mL). The solid was finally dried under reduced pressure at 60 °C to give a beige solid. Yield: 133 g, 88%, 100% a/a (LC-MS), M+1 =451. Melting point: 277 °C (DSC). Ή NMR (400 MHz, DMSO) δ: 8.06 (s, 2 H), 7.76 (d, J = 8.9 Hz, 1 H), 7.63 (d, J = 8.8 Hz, 2 H), 7.55 (m, 1 H), 7.16 (dd, J1 = 2.7 Hz, J2 = 8.9 Hz, 1 H), 3.98 (m, 1 H), 3.90 (s, 3 H), 3.33 (m, 2H), 3.32 (m, 1 H), 2.74 (s, 3 H), 2.55 (m, 1 H), 2.23 (m, 1 H), 2.10 (m, 2 H), 1.95 (s, 3 H).
/////////////////////////////////////////////////////////////////////

AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
| Clinical data | |
|---|---|
| Trade names | Quviviq |
| Other names | Nemorexant; ACT-541468 |
| License data | US DailyMed: Daridorexant |
| Routes of administration | By mouth |
| Drug class | Orexin antagonist |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only [1] |
| Pharmacokinetic data | |
| Elimination half-life | 6–10 hours[2] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 1505484-82-1 |
| PubChem CID | 91801202 |
| DrugBank | DB15031 |
| ChemSpider | 64854514 |
| UNII | LMQ24G57E9 |
| KEGG | D11886 |
| PDB ligand | NS2 (PDBe, RCSB PDB) |
| Chemical and physical data | |
| Formula | C23H23ClN6O2 |
| Molar mass | 450.93 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI |
REF
References
- ^ Jump up to:a b c https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/214985s000lbl.pdf
- ^ Jump up to:a b Muehlan C, Vaillant C, Zenklusen I, Kraehenbuehl S, Dingemanse J (November 2020). “Clinical pharmacology, efficacy, and safety of orexin receptor antagonists for the treatment of insomnia disorders”. Expert Opin Drug Metab Toxicol. 16 (11): 1063–1078. doi:10.1080/17425255.2020.1817380. PMID 32901578.
- ^ Jump up to:a b c “Daridorexant – Idorsia Pharmaceuticals – AdisInsight”.
- ^ Jump up to:a b Equihua-Benítez AC, Guzmán-Vásquez K, Drucker-Colín R (July 2017). “Understanding sleep-wake mechanisms and drug discovery”. Expert Opin Drug Discov. 12 (7): 643–657. doi:10.1080/17460441.2017.1329818. PMID 28511597.
- ^ “Daridorexant: FDA-Approved Drugs”. U.S. Food and Drug Administration (FDA). Retrieved 11 January 2022.
- ^ “Idorsia receives US FDA approval of Quviviq (daridorexant)” (Press release). Idorsia Pharmaceuticals. 10 January 2022. Retrieved 11 January 2022 – via GlobeNewswire.
External links
- “Daridorexant”. Drug Information Portal. U.S. National Library of Medicine.
- Clinical trial number NCT03545191 for “Study to Assess the Efficacy and Safety of ACT-541468 in Adult and Elderly Subjects With Insomnia Disorder” at ClinicalTrials.gov
- Clinical trial number NCT03575104 for “Study to Assess the Efficacy and Safety of ACT-541468 in Adult and Elderly Subjects Suffering From Difficulties to Sleep” at ClinicalTrials.gov
- Clinical trial number NCT03679884 for “Study to Assess the Long Term Safety and Tolerability of ACT-541468 in Adult and Elderly Subjects Suffering From Difficulties to Sleep” at ClinicalTrials.gov
///////////////Daridorexant, Quviviq, FDA 2022, APPROVALS 2022, INSOMNIA, ACT541468A, ACT 541468A. ACT-541468A, ACT541468, FDA 2022, APPROVALS 2022
O=C(N1[C@](C)(C2=NC3=CC=C(Cl)C(C)=C3N2)CCC1)C4=CC(OC)=CC=C4N5N=CC=N5.[H]Cl

NEW DRUG APPROVALS
ONE TIME
$10.00
OTESECONAZOLE

OTESECONAZOLE
VT 1161
オテセコナゾール;
(2R)-2-(2,4-difluorophenyl)-1,1-difluoro-3-(tetrazol-1-yl)-1-[5-[4-(2,2,2-trifluoroethoxy)phenyl]pyridin-2-yl]propan-2-ol
| C23H16F7N5O2 527.4 | |
| Synonyms | VT 1161 Oteseconazole CAS1340593-59-0 |
|---|
Other Names
- (αR)-α-(2,4-Difluorophenyl)-β,β-difluoro-α-(1H-tetrazol-1-ylmethyl)-5-[4-(2,2,2-trifluoroethoxy)phenyl]-2-pyridineethanol
- (2R)-2-(2,4-difluorophenyl)-1,1-difluoro-3-(1H-1,2,3,4-tetrazol-1-yl)- 1-{5-[4-(2,2,2-trifluoroethoxy)phenyl]pyridin-2-yl}propan-2-ol
UPDATE MAY 2022… FDA APPROVED 2022/4/26, Vivjoa
Oteseconazole, sold under the brand name Vivjoa, is a medication used for the treatment of vaginal yeast infections.[1]
It was approved for medical use in the United States in April 2022.[2][3] It was developed by Mycovia Pharmaceuticals.[3]
Names
Oteseconazole is the international nonproprietary name (INN).[4]
Oteseconazole is an azole antifungal used to prevent recurrent vulvovaginal candidiasis in females who are not of reproductive potential.
Oteseconazole, also known as VT-1161, is a tetrazole antifungal agent potentially for the treatment of candidal vaginal infection. VT-1161 Protects Immunosuppressed Mice from Rhizopus arrhizus var. arrhizus Infection. VT-1161 dosed once daily or once weekly exhibits potent efficacy in treatment of dermatophytosis in a guinea pig model.
Oteseconazole has been used in trials studying the treatment of Tinea Pedis, Onychomycosis, Candidiasis, Vulvovaginal, and Recurrent Vulvovaginal Candidiasis.
Mycovia Pharmaceuticals is developing oteseconazole, the lead from a program of metalloenzyme Cyp51 (lanosterol demethylase) inhibitors, developed using the company’s Metallophile technology, for treating fungal infections including onychomycosis and recurrent vulvovaginal candidiasis (RVVC). In July 2021, oteseconazole was reported to be in phase 3 clinical development. Licensee Jiangsu Hengrui Medicine is developing otesaconazole, as an oral capsule formulation, for treating fungal conditions, including RVVC, onychomycosis and invasive fungal infections, in Greater China and planned for a phase 3 trial in April 2021 for treating VVC.
- OriginatorViamet Pharmaceuticals
- DeveloperMycovia Pharmaceuticals; Viamet Pharmaceuticals
- ClassAntifungals; Foot disorder therapies; Pyridines; Small molecules; Tetrazoles
- Mechanism of Action14-alpha demethylase inhibitors
- PreregistrationVulvovaginal candidiasis
- Phase IIOnychomycosis
- No development reportedTinea pedis
- 01 Jun 2021Preregistration for Vulvovaginal candidiasis (In adolescents, In adults, In children, Recurrent) in USA (PO)
- 01 Jun 2021Mycovia intends to launch otesaconazole (Recurrent) for Vulvovaginal candidiasis in the US in early 2022
- 06 Jan 2021Interim efficacy and adverse events data from a phase III ultraVIOLET trial in Vulvovaginal candidiasis released by Mycovia Pharmaceuticals

Synthesis Reference
Hoekstra, WJ., et al. (2020). Antifungal compound process (U.S. Patent No. US 10,745,378 B2). U.S. Patent and Trademark Office. https://patentimages.storage.googleapis.com/f4/62/19/5ba525b1caad0e/US10745378.pdf
PATENT
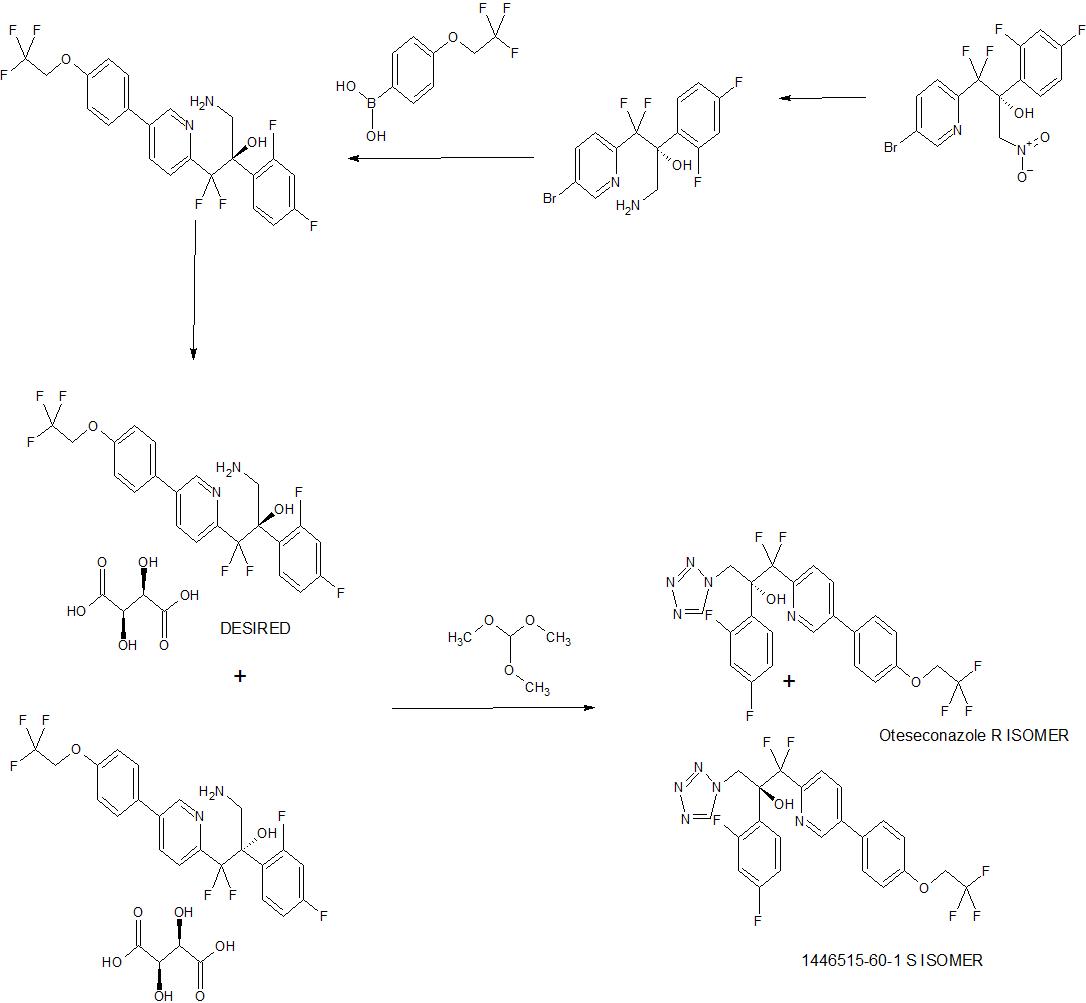
WO 2017049080
WO 2016149486
US 20150024938
WO 2015143172
WO 2015143184
WO 2015143180
WO 2015143142
WO 2013110002
WO 2013109998
WO 2011133875
PATENT
WO 2017049080,
Syn
J. Med. Chem. 2024, 67, 4376−4418
Oteseconazole was approved by the USFDA in April 2022 for the treatment of recurrent vulvovaginal candidiasis in women with a history of vulvovaginal candidiasis and who are not of reproductive
potential. Additional studies for other invasive and opportunistic infections and for onychomycosis are underway.40, The design and discovery of oteseconazole is published by a group from Viamet Pharmaceuticals, now part of Mycovia Pharmaceuticals. It details the racemic synthesis of the drug on
<1 g scale in which the metal-binding tetrazole is installed by treatment of ester 5.2 (Scheme 10) with diazomethane and tetrazole.42
A more scale-friendly asymmetric route that avoided the use of diazomethane was subsequently disclosed in patents and is detailed in Scheme 10 and Scheme 11.43
First, a mixture of ethyl bromodifluoroacetate, stoichiometric copper
powder, and 2,5-dibromopyridine (5.1) in DMSO provided ester 5.2 as an oil that was purified via distillation (Scheme10). Conversion to the aryl ketone 5.5 was achieved via direct addition of lithiated 5.3 or via a two-step process by first conversion to morpholine amide 5.4 followed by addition of
the Grignard generated from aryl bromide 5.3. The resulting ketone 5.5 was a liquid that was carried into the next step without purification.
The key step in the synthesis of 5 is an asymmetric Henry reaction using cinchona alkaloid catalyst 5.6. Addition of nitromethane to ketone 5.5 furnished alcohol 5.7 in 75% yield and ∼90:10 ratio of enantiomers. Next, reduction of the nitro group to the primary amine was accomplished using Pt
catalyzed hydrogenation. The chiral purity of the resulting amine was upgraded by classical resolution using di-p-toluoyl L-tartaric acid to provide 5.8·L-DTTA in 33% yield and >99% chiral purity.Conversion of amino alcohol 5.8 to oteseconazole (5) required two steps: cross coupling to introduce the aryltrifluoroethyl ether fragment and tetrazole formation. These steps were performed in either sequence in the patent. The route shown in Scheme 11 represents the largest scale demonstrated (>100 g input of 5.8). While the use of azide containing reagents presents significant safety risks, no information was provided on safe operation of the tetrazole forming step in the laboratory or on plant scale. Some of the
procedures for tetrazole formation described in the patent would likely require modification for safe scale-up.
To complete the synthesis of oteseconazole, resolved amino alcohol 5.8 first underwent a salt break followed by Suzuki coupling using boronic acid 5.9 to provide biaryl product 5.10 as the L-tartrate salt (Scheme 11). Conversion of 5.10 to 5 was accomplished using TMSN3 in acetic acid with sodium acetate and trimethoxy orthoformate. Treatment of the resulting solution with a Pd scavenger preceded crystallization of the product from EtOH and water after pH adjustment with potassium carbonate. The product was isolated in 85% yield as a hydrated form. Another patent described conversion of the oteseconazolehydrate totheanhydrous form byrecrystallizationfrom EtOHandn-heptanetofurnish5 in90%yield.45
(40) Hoy, S. M. Oteseconazole: First approval. Drugs 2022, 82,1017−1023.
(41) Sobel, J. D.; Nyirjesy, P. Oteseconazole: an advance in
treatment of recurrent vulvovaginal candidiasis. Future Microbiol 2021,
16, 1453−1461.
(42) Hoekstra, W. J.; Garvey, E. P.; Moore, W. R.; Rafferty, S. W.;
Yates, C. M.; Schotzinger, R. J. Design and optimization of highly
selective fungal CYP51 inhibitors. Bioorg. Med. Chem. Lett. 2014, 24,
3455−3458.
(43) Wirth, D. D.; Yates, C. M.; Hoekstra, W. J.; Bindl, M. F.;
Hartmann, E. Process for enantioselective preparation of tetrazolyl
pyridinyl diaryl propanols as antifungal drugs and their precursors.
WO 2017049080, 2017.
(44) González-Bobes, F.; Kopp, N.; Li, L.; Deerberg, J.; Sharma, P.;
Leung, S.; Davies, M.; Bush, J.; Hamm, J.; Hrytsak, M. Scale-up of
Azide Chemistry: A Case Study. Org. Process Res. Dev. 2012, 16,
2051−2057.
(45) Hoekstra, W. J.; Wirth, D. D.; Ehiwe, T.; Bonnaud, T.
Antifungal compounds and processes for making. WO 2016149486,
2016.


.
PATENT
WO-2021143811
Novel crystalline polymorphic form of VT-1161 (also known as oteseconazole) phosphate disodium salt, useful as a prodrug of oteseconazole, for treating systemic fungal infection (eg Candida albicans infection) or onychomycosis.The function of metalloenzymes is highly dependent on the presence of metal ions in the active site of the enzyme. It is recognized that reagents that bind to and inactivate metal ions at the active site greatly reduce the activity of the enzyme. Nature uses this same strategy to reduce the activity of certain metalloenzymes during periods when enzyme activity is not needed. For example, the protein TIMP (tissue inhibitor of metalloproteinases) binds to zinc ions in the active sites of various matrix metalloproteinases, thereby inhibiting enzyme activity. The pharmaceutical industry has used the same strategy in the design of therapeutic agents. For example, the azole antifungal agents fluconazole and voriconazole contain 1-(1,2,4-triazole) group, which exists in the active site of the target enzyme lanosterol demethylase The heme iron binds, thereby inactivating the enzyme. Another example includes zinc-bound hydroxamic acid groups, which have been introduced into most of the published inhibitors of matrix metalloproteinases and histone deacetylases. Another example is the zinc-binding carboxylic acid group, which has been introduced into most of the published angiotensin converting enzyme inhibitors.
VT-1161, the compound 2-(2,4-difluorophenyl)-1,1-difluoro-3-(1H-tetrazol-1-yl)-1-(5-(4-(2, 2,2-Trifluoroethoxy)phenyl)pyridin-2-yl)propan-2-ol, is an antifungal drug developed by VIAMET, currently in the clinical research stage, its structure is as follows Shown:
This compound mainly acts on the CYP51 target of fungal cells. Compared with the previous triazole antifungal drugs, it has the advantages of wider antibacterial spectrum, low toxicity, high safety and good selectivity. However, this compound is not suitable for Liquid preparations (including or excluding the parenteral delivery carrier) are used to treat patients in need thereof.
2-(2,4-Difluorophenyl)-1,1-difluoro-3-(1H-tetrazol-1-yl)-1-(5-(4-(2,2,2-trifluoro Ethoxy)phenyl)pyridin-2-yl)propan-2-yl dihydrogen phosphate is a prodrug of VT-1161.
On the other hand, nearly half of the drug molecules are in the form of salts, and salt formation can improve certain undesirable physicochemical or biological properties of the drug. Relative to 2-(2,4-difluorophenyl)-1,1-difluoro-3-(1H-tetrazol-1-yl)-1-(5-(4-(2,2,2- Trifluoroethoxy)phenyl)pyridin-2-yl)propan-2-yl dihydrogen phosphate, it is of great significance to develop salts with more excellent properties in terms of physical and chemical properties or pharmaceutical properties.To this end, the present disclosure provides a new pharmaceutically acceptable salt form of a metalloenzyme inhibitor.Example 1:[0161](R)-2-(2,4-Difluorophenyl)-1,1-difluoro-3-(1H-tetrazol-1-yl)-1-(5-(4-(2,2, 2-Trifluoroethoxy)phenyl)pyridin-2-yl)propan-2-yl phosphate disodium salt (Compound 1)[0162]
[0163](R)-2-(2,4-Difluorophenyl)-1,1-difluoro-3-(1H-tetrazol-1-yl)-1-(5-(4-(2,2 ,2-Trifluoroethoxy)phenyl)pyridin-2-yl)propan-2-yl phosphate (compound 1a, prepared according to the method of patent WO2013110002, 0.28g, 0.46mmol, 1.0eq) and ethanol (5mL ) Add to the reaction flask and stir evenly. A solution of NaOH (36.90 mg, 2.0 eq) dissolved in water (1 mL) was added dropwise into the above reaction flask, stirring was continued for 2 h, and concentrated to obtain compound 1, 300 mg of white solid.[0164]After X-ray powder diffraction detection, the XRPD spectrum has no sharp diffraction peaks, as shown in FIG. 10.[0165]Ms:608.10[M-2Na+3H] + .[0166]Ion chromatography detected that the sodium ion content was 6.23%.[0167]Example 2: (R)-((2-(2,4-Difluorophenyl)-1,1-difluoro-3-(1H-tetrazol-1-yl)-1-(5-(4 -(2,2,2-Trifluoroethoxy)phenyl)pyridin-2-yl)prop-2-yl)oxy)methyl phosphate disodium salt (compound 2)
[0169]Under ice-cooling, NaH (58mg, 0.87mmol) was added to the reaction flask, 1.5mL of N,N-dimethylformamide and 0.6mL of tetrahydrofuran were added, followed by iodine (38mg, 0.15mmol), and then Compound 2-(2,4-difluorophenyl)-1,1-difluoro-3-(1H-tetrazol-1-yl)-1-(5-(4-(2,2,2-tri Fluoroethoxy)phenyl)pyridin-2-yl)propan-2-ol (2b, prepared according to the method of patent WO2013110002, 158mg, 0.3mmol) tetrahydrofuran (1ml) solution was added to the reaction solution, stirred and reacted for 1-4h , And then add compound 2a (519mg, 2.01mmol) in tetrahydrofuran (1ml) solvent to the reaction, stir until the reaction is complete, 10% aqueous ammonium chloride solution to quench the reaction, extract, concentrate and drain, the crude product 2c is directly used for the next One-step reaction, Ms: 750.0[M+H] + .[0170]
[0171]Under ice-bath cooling, add trifluoroacetic acid (0.5mL) to the crude product 2c (300mg) in dichloromethane (2mL) solution, stir until the reaction is complete, and after concentration, the target compound 2d, 82mg, Ms was separated by high performance liquid phase separation. :638.0[M+H] + .[0172]
Add compound 2d (0.29g, 0.46mmol, 1.0eq) and ethanol (5mL) obtained in the previous step into the reaction flask, stir, and add NaOH (36.90mg, 2.0eq) water (1ml) solution dropwise to the aforementioned reaction solution , Stirred for 2-5 h, and concentrated to obtain 2,313 mg of the target compound.
Ms:638.10[M-2Na+3H] + .
PATENT
WO2011133875
https://patents.google.com/patent/WO2011133875A2/en
Product pat, WO2011133875 , protection in the EU states and the US April 2031.
PATENT
WO2015143184 ,
https://patents.google.com/patent/WO2015143184A1/en
Mycovia, claiming a process for preparing antifungal compounds, particularly oteseconazole.EXAMPLE 11

2-(2,4-Difluorophenyl)-l,l-difluoro-3-(lH-tetrazol-l-yl)-l-(5-(4-(2,2,2- trifluoroethoxy)phenyl)pyridin-2-yl)propan-2-ol (11)Compound 11 was prepared using the conditions employed for 1: 0.33 g as a solid. The precursor l-bromo-4-(2,2,2-trifluoroethoxy)benzene was prepared as described below in one step.1H NMR (500 MHz, CDC13): δ 8.76 (s, 1 H), 8.70 (s, 1 H), 7.95 (d, / = 8.0 Hz, 1 H), 7.70 (s, 1 H), 7.64 (d, / = 8.5 Hz, 1 H), 7.54 (d, / = 8.5 Hz, 2 H), 7.42- 7.37 (m, 1 H), 7.08 (d, / = 8.5 Hz, 2 H), 6.79- 6.75 (m, 1 H), 6.69- 6.66 (m, 1 H), 5.58 (d, / = 14.0 Hz, 1 H), 5.14 (d, / = 14.0 Hz, 1 H), 4.44 – 4.39 (m, 2 H). HPLC: 99.1%. MS (ESI): m/z 528 [M++l].Chiral preparative HPLC Specifications for (+)-ll:Column: Chiralpak IA, 250 x 4.6mm, 5uMobile Phase: A) w-Hexane, B) IPAIsocratic: A: B (65:35)Flow Rte: l.OO mL/minOptical rotation [a]D: + 24° (C = 0.1 % in MeOH). 1 -Bromo-4-( 2,2,2-trifluoroethoxy )benzeneTo a stirred solution of trifluoroethyl tosylate (1.5 g, 5.8 mmol) in DMF (20 mL) was added K2CO3 (4 g, 29.4 mmol) followed by addition of p-bromo phenol (1.1 g, 6.46 mmol) at RT under inert atmosphere. The reaction mixture was stirred at 120 °C for 6 h. The volatiles were evaporated under reduced pressure; the residue was diluted with water (5 mL) and extracted with ethyl acetate (3 x 30 mL). The organic layer was washed with water, brine and dried over anhydrous Na2S04, filtered and concentrated in vacuo. The crude compound was purified by silica gel column chromatography eluting with 5% EtOAc/hexane to afford the desired product (0.8 g, 3.13 mmol, 53.3%) as semi solid. 1H NMR (200 MHz, CDC13): δ 7.44 – 7.38 (m, 2 H), 6.86-6.80 (m, 2 H), 4.38- 4.25 (m, 2 H).ExamplesThe present invention will now be demonstrated using specific examples that are not to be construed as limiting.General Experimental ProceduresDefinitions of variables in the structures in schemes herein are commensurate with those of corresponding positions in the formulae delineated herein.Synthesis of 1 or la

A process to prepare enantiopure compound 1 or la is disclosed. Syntheses of lor la may be accomplished using the example syntheses that are shown below (Schemes 1-4). The preparation of precursor ketone 3-Br is performed starting with reaction of 2,5-dibromo- pyridine with ethyl 2-bromo-difluoroacetate to produce ester 2-Br. This ester can be reacted with morpholine to furnish morpholine amide 2b-Br, followed by arylation to provide ketone 3-Br. Alternatively, ketone 3-Br can be afforded directly from ester 2-Br as shown in Scheme 1. Scheme 1. Synthesis of ketone 3-Br r

Ketone 3 may be prepared in an analogous fashion as described in Scheme 1 starting from corresponding substituted 2-bromo-pyridines, which can be prepared according to synthetic transformations known in the art and contained in the references cited herein (Scheme 2).Scheme 2. Synthesis of ketone 3

R-i = halo, -0(C=0)-alkyl, -0(C=0)-substituted alkyl, -0(C=0)-aryl, -0(C=0)-substituted aryl, -0(C=0)-0-alkyl, – 0(C=0)-0-substituted alkyl, -0(C=0)-0-aryl, -0(C=0)-0-substituted aryl, -0(S02)-alkyl, -0(S02)-substituted alkyl, – 0(S02)-aryl, or -0(S02)-substituted aryl.Alternatively, compound 1 can be prepared according to Scheme 3 utilizing diols 2-6b (or 2- 6d, the enantiomer of 2-6b, or mixtures thereof) or 2-6a (or 2-6c, the enantiomer of 2-6a, or mixtures thereof). Olefins 2-5a and 2-5 can be prepared by reacting ketones 3 and 1-4 under Wittig olefination conditions (e.g., Ph3PCH3Br and BuLi). Also, as indicated in Scheme 5, any of pyridine compounds, 3, 2-5a, 2-6b, 2-7b, 4*, 4b, or 6 can be converted to the corresponding 4-CF3CH2O-PI1 analogs (e.g., 1-4, 2-5, 2-6a, 2-7a, 5*, 1-6*, or 1 or the corresponding enantiomers, or mixtures thereof) by cross-coupling with 4,4,5, 5-tetramethyl-2- (4-(2,2,2-trifluoroethoxy)phenyl)-l,3,2-dioxaborolane (or the corresponding alkyl boronates or boronic acid or the like), in a suitable solvent system (e.g., an organic-aqueous solvent mixture), in the presence of a transition metal catalyst (e.g., (dppf)PdCl2), and in the presence of a base (e.g., KHCO3, K2C03, Cs2C03, or Na2C03, or the like). Olefins 2-5a and 2-5 can be transformed to the corresponding chiral diols, 2-6b (or 2-6d, the enantiomer of 2-6b, or mixtures thereof) or 2-6a (or 2-6c, the enantiomer of 2-6a, or mixtures thereof), through exposure to Sharpless asymmetric dihydroxylation conditions: 1) commercially available AD- mix alpha or AD-mix beta with or without additional osmium oxidant and methanesulfonamide, 2) combination of a catalytic osmium oxidant (e.g., Os04 or K20sC>2(OH)4), a stoichiometric iron oxidant (e.g., K3Fe(CN)6), a base (e.g., KHCO3, K2CO3, Cs2C03, or Na2C03, or the like), and a chiral ligand (e.g., (DHQ)2PHAL, (DHQD)2PHAL, (DHQD)2AQN, (DHQ)2AQN, (DHQD)2PYR, or (DHQ)2PYR; preferably (DHQ)2PHAL, (DHQD)2PHAL, (DHQD)2AQN, and (DHQD)2PYR), or 3) option 2) with methanesulfonamide. The primary alcohol of the resultant chiral diols, 2-6b (or 2-6d, the enantiomer of 2-6b, or mixtures thereof) or 2-6a (or 2-6c, the enantiomer of 2-6a, or mixtures thereof), can then be activated to afford compounds 2-7b (or 2-7d, the enantiomer of 2-7b, or mixtures thereof) or 2-7a (or 2-7c, the enantiomer of 2-7a, or mixtures thereof). For example, the mesylates can be prepared by exposing chiral diols, 2-6b (or 2-6d, the enantiomer of 2-6b, or mixtures thereof) or 2-6a (or 2-6c, the enantiomer of 2-6a, or mixtures thereof), to methanesulfonyl chloride and a base. Epoxide formation can be affected by the base-mediated (e.g., KHCO3, K2CO3, CS2CO3, or Na2CC>3, or the like) ring closure of compounds 2-7b (or 2- 7d, the enantiomer of 2-7b, or mixtures thereof) or 2-7a (or 2-7c, the enantiomer of 2-7a, or mixtures thereof) to provide epoxides 4* (or 4c*, the enantiomer of 4*, or mixtures thereof) and 5* (or 5-b*, the enantiomer of 5*, or mixtures thereof). The epoxides can then be converted into amino-alcohols 4b (or 4c, the enantiomer of 4b, or mixtures thereof) and 1-6* (or 1-7*, the enantiomer of 1-6*, or mixtures thereof) through ammonia-mediated epoxide opening using ammonia in a suitable solvent (e.g., MeOH, EtOH, or water). Subsequent treatment with TMS-azide in the presence of trimethylorthoformate and sodium acetate in acetic acid would yield compounds 6 (or 6a, the enantiomer of 6, or mixtures thereof) or 1 (or la, the enantiomer of 1, or mixtures thereof) (US 4,426,531).Scheme 3. Synthesis of 1 via Asymmetric Dihydroxylation Method


Y is -OS02-alkyl, -OS02-substituted alkyl, -OS02-aryl, -OS02- substituted aryl, -0(C=0)-alkyl, -0(C=0)-substituted alkyl, – 0(C=0)-aryl, -0(C=0)-substituted aryl, or halogen

R-i = halo, -0(C=0)-alkyl, -0(C=0)-substituted alkyl, -0(C=0)-aryl, -0(C=0)-substituted aryl, -0(C=0)-0-alkyl, -0(C=0)-0-substituted alkyl, -0(C=0)-0-aryl, -0(C=0)-0-substituted aryl, -0(S02)-alkyl, -0(S02)-substituted alkyl, -0(S02)-aryl, or -0(S02)-substituted aryl.Compound 1 (or la, the enantiomer of 1, or mixtures thereof) prepared by any of the methods presented herein can be converted to a sulfonic salt of formula IX (or IXa, the enantiomer of IX, or mixtures thereof), as shown in Scheme 4. This can be accomplished by a) combining compound 1 (or la, the enantiomer of 1, or mixtures thereof), a crystallization solvent or crystallization solvent mixture (e.g., EtOAc, i‘PrOAc, EtOH, MeOH, or acetonitrile, or oZ-S-OHcombinations thereof), and a sulfonic acid o (e.g., Z = Ph, p-tolyl, Me, or Et), b) diluting the mixture with an appropriate crystallization co-solvent or crystallization co-solvent mixture (e.g., pentane, methyl i-butylether, hexane, heptane, or toluene, or combinations thereof), and c) filtering the mixture to obtain a sulfonic acid salt of formula IX (or IXa, the enantiomer of IX, or mixtures thereof). cheme 4. Synthesis of a Sulfonic Acid Salt of Compound 1 or la

The following describes the HPLC method used in assessing HPLC purity of the examples and intermediates presented below:Column: Waters XBridge Shield RP18, 4.6 x 150 mm, 3.5 μιηMobile Phase: A = 0.05% TFA/H20, B = 0.05% TFA/ACNAutosampler flush: 1 : 1 ACN/H20Diluent: 1:1 ACN/H20Flow Rate: 1.0 ml/minTemperature: 45 °CDetector: UV 275 nmPump Parameters:

EXAMPLE 1Preparation of ethyl 2-(5-bromopyridin-2-yl)-2,2-difluoroacetate (2-Br)

2-Br Dialkylated impurity In a clean multi-neck round bottom flask, copper powder (274.7 g, 2.05 eq) was suspended in dimethyl sulfoxide (3.5 L, 7 vol) at 20 – 35 °C. Ethyl bromodifluoroacetate (449 g, 1.05 eq) was slowly added to the reaction mixture at 20 – 25 °C and stirred for 1 – 2 h. 2, 5- dibromopyridine (500 g, 1 eq) was added to the reaction mixture and the temperature was increased to 35 – 40 °C. The reaction mixture was maintained at this temperature for 18 – 24 h and the reaction progress was monitored by GC.After the completion of the reaction, ethyl acetate (7 L, 14 vol) was added to the reaction mixture and stirring was continued for 60 – 90 min at 20 – 35 °C. The reaction mixture was filtered through a Celite bed (100 g; 0.2 times w/w Celite and 1L; 2 vol ethyl acetate). The reactor was washed with ethyl acetate (6 L, 12 vol) and the washings were filtered through a Celite bed. The Celite bed was finally washed with ethyl acetate (1 L, 2 vol) and all the filtered mother liquors were combined. The pooled ethyl acetate solution was cooled to 8 – 10 °C, washed with the buffer solution (5 L, 10 vol) below 15 °C (Note: The addition of buffer solution was exothermic in nature. Controlled addition of buffer was required to maintain the reaction mixture temperature below 15 °C). The ethyl acetate layer was washed again with the buffer solution until (7.5 L; 3 x 5 vol) the aqueous layer remained colorless. The organic layer was washed with a 1: 1 solution of 10 % w/w aqueous sodium chloride and the buffer solution (2.5 L; 5 vol). The organic layer was then transferred into a dry reactor and the ethyl acetate was distilled under reduced pressure to get crude 2-Br.The crude 2-Br was purified by high vacuum fractional distillation and the distilled fractions having 2-Br purity greater than 93 % (with the dialkylated not more than 2 % and starting material less than 0.5 %) were pooled together to afford 2-Br.Yield after distillation: 47.7 % with > 93 % purity by GC (pale yellow liquid). Another 10 % yield was obtained by re-distillation of impure fractions resulting in overall yield of ~ 55 – 60 %.*H NMR: δ values with respect to TMS (DMSO-d6; 400 MHz): 8.85 (1H, d, 1.6 Hz), 8.34 (1H, dd, J = 2.0 Hz, 6.8 Hz), 7.83 (1H, d, J = 6.8 Hz), 4.33 (2H, q, J = 6.0 Hz), 1.22 (3H, t, J = 6.0 Hz). 13C NMR: 162.22 (i, -C=0), 150.40 (Ar-C-), 149.35 (t, Ar-C), 140.52 (Ar-C), 123.01 (Ar-C), 122.07 (Ar-C), 111.80 (t, -CF2), 63.23 (-OCH2-), 13.45 (-CH2CH3).EXAMPLE 2
Preparation of2-( 5-bromopyridin-2-yl )-l -(2,4-difluorophenyl )-2, 2-difluoroethanone ( 3-Br ) A. One-step Method

l-Bromo-2,4-difluorobenzene (268.7 g; 1.3 eq) was dissolved in methyl tert butyl ether (MTBE, 3.78 L, 12.6 vol) at 20 – 35 °C and the reaction mixture was cooled to -70 to -65 °C using acetone/dry ice bath. n-Butyl lithium (689 rriL, 1.3 eq; 2.5 M) was then added to the reaction mixture maintaining the reaction temperature below -65 °C (Note: Controlled addition of the n-Butyl Lithium to the reaction mixture was needed to maintain the reaction mixture temperature below – 65 °C). After maintaining the reaction mixture at this temperature for 30 – 45 min, 2-Br (300 g, 1 eq) dissolved in MTBE (900 rriL, 3 vol) was added to the reaction mixture below – 65 °C. The reaction mixture was continued to stir at this temperature for 60 – 90 min and the reaction progress was monitored by GC.The reaction was quenched by slow addition of 20 % w/w ammonium chloride solution (750 mL, 2.5 vol) below -65 °C. The reaction mixture was gradually warmed to 20 – 35 °C and an additional amount of 20 % w/w ammonium chloride solution (750 mL, 2.5 vol) was added. The aqueous layer was separated, the organic layer was washed with a 10 % w/w sodium bicarbonate solution (600 mL, 2 vol) followed by a 5 % sodium chloride wash (600 mL, 2 vol). The organic layer was dried over sodium sulfate (60 g; 0.2 times w/w), filtered and the sodium sulfate was washed with MTBE (300 mL, 1 vol). The organic layer along with washings was distilled below 45 °C under reduced pressure until no more solvent was collected in the receiver. The distillation temperature was increased to 55 – 60 °C, maintained under vacuum for 3 – 4 h and cooled to 20 – 35 °C to afford 275 g (73.6 % yield, 72.71 % purity by HPLC) of 3-Br as a pale yellow liquid.*H NMR: δ values with respect to TMS (DMSO-d6; 400 MHz):8.63 (1H, d, 1.6 Hz, Ar-H), 8.07 – 8.01 (2H, m, 2 x Ar-H), 7.72 (1H, d, J = 6.8 Hz, Ar-H), 7.07 – 6.82 (1H, m, Ar-H), 6.81 – 6.80 (1H, m, Ar-H). 13C NMR: 185.60 (t, -C=0), 166.42 (dd, Ar-C-), 162.24 (dd, Ar-C),150.80 (Ar-C), 150.35 (Ar-C), 140.02 (Ar-C), 133.82 (Ar-C), 123.06 (Ar-C), 1122.33 (Ar-C), 118.44 (Ar-C), 114.07 (-CF2-), 122.07 (Ar-C), 105.09 (Ar-C).
B. Two-step Method via 2b-Br

2-Br (147.0 g) was dissolved in n-heptane (1.21 L) and transferred to a 5-L reactor equipped with overhead stirrer, thermocouple, condenser and addition funnel. Morpholine (202 ml) was added. The solution was heated to 60 °C and stirred overnight. The reaction was complete by HPLC analysis (0.2% 2-Br; 94.7% 2b-Br). The reaction was cooled to room temperature and 1.21 L of MTBE was added. The solution was cooled to ~4 °C and quenched by slow addition of 30% citric acid (563 ml) to maintain the internal temperature <15 °C. After stirring for one hour the layers were allowed to settle and were separated (Aq. pH=5). The organic layer was washed with 30% citric acid (322 ml) and 9% NaHC03 (322 ml, aq. pH 7+ after separation). The organic layer was concentrated on the rotary evaporator (Note 1) to 454 g (some precipitation started immediately and increased during concentration). After stirring at room temperature the suspension was filtered and the product cake was washed with n-heptane (200 ml). The solid was dried in a vacuum oven at room temperature to provide 129.2 g (77%) dense powder. The purity was 96.5% by HPLC analysis.To a 1-L flask equipped with overhead stirring, thermocouple, condenser and addition funnel was added magnesium turnings (14.65 g), THF (580 ml) and l-bromo-2,4-difluorobenzene (30.2 g, 0.39 equiv). The mixture was stirred until the reaction initiated and self-heating brought the reaction temperature to 44 °C. The temperature was controlled with a cooling bath as the remaining l-bromo-2,4-difluorobenzene (86.1 g, 1.11 equiv) was added over about 30 min. at an internal temperature of 35-40 °C. The reaction was stirred for 2 hours while gradually cooling to room temperature. The dark yellow solution was further cooled to 12 °C.During the Grignard formation, a jacketed 2-L flask equipped with overhead stirring, thermocouple, and addition funnel was charged with morpholine amide 2b-Br (129.0 g) and THF (645 ml). The mixture was stirred at room temperature until the solid dissolved, and then the solution was cooled to -8.7 °C. The Grignard solution was added via addition funnel over about 30 min. at a temperature of -5 to 0 °C. The reaction was stirred at 0 °C for 1 hour and endpointed by HPLC analysis. The reaction mixture was cooled to -5 °C and quenched by slow addition of 2N HC1 over 1 hour at <10 °C. The mixture was stirred for 0.5 h then the layers were allowed to settle and were separated. The aqueous layer was extracted with MTBE (280 ml). The combined organic layers were washed with 9% NaHCC>3 (263 g) and 20% NaCl (258 ml). The organic layer was concentrated on the rotary evaporator with THF rinses to transfer all the solution to the distillation flask. Additional THF (100 ml) and toluene (3 x 100 ml) were added and distilled to remove residual water from the product. After drying under vacuum, the residue was 159.8 g of a dark brown waxy solid (>theory). The purity was approximately 93% by HPLC analysis.EXAMPLE 3Preparation of 3-amino-l-(5-bromopyridin-2-yl)-2-(2,4-difluorophenyl)-l,l-difluoropropan- -ol (±ib-Br)

4-Br (200g, 1 eq) was added into methanolic ammonia (8.0 L; 40 vol; ammonia content: 15 – 20 % w/v) in an autoclave at 10 – 20 °C. The reaction mixture was gradually heated to 60 – 65 °C and at 3 – 4 kg/cm2 under sealed conditions for 10 – 12 h. The reaction progress was monitored by GC. After completion of the reaction, the reaction mixture was cooled to 20 – 30 °C and released the pressure gradually. The solvent was distilled under reduced pressure below 50 °C and the crude obtained was azeotroped with methanol (2 x 600 mL, 6 vol) followed by with isopropanol (600 mL, 2 vol) to afford 203 g (96.98 % yield, purity by HPLC: 94.04 %) of +4b-Br. EXAMPLE 4Preparation of3-amino-l-(5-bromopyridin-2-yl)-2-(2,4-difluorophenyl)-l,l-difluoropropan- -ol (4b-Br or 2c-Br)

Amino alcohol ±4b-Br (150 g, 1 eq) was dissolved in an isopropanol /acetonitrile mixture (1.5L, 8:2 ratio, 10 vol) and Di-p-toluoyl-L-tartaric acid (L-DPTTA) (84.05 g, 0.55 eq) was added into the reactor at 20 – 30 °C. The reaction mixture was heated to 45 – 50 °C for 1 – 1.5 h (Note: The reaction mixture becomes clear and then became heterogeneous). The reaction mixture was gradually cooled to 20 – 30 °C and stirred for 16 – 18 h. The progress of the resolution was monitored by chiral HPLC analysis.After the completion of the resolution, the reaction mixture was gradually cooled to 20 – 35 °C. The reaction mixture was filtered and the filtered solid was washed with a mixture of acetonitrile and isopropanol (8:2 mixture, 300 mL, 2 vol) and dried to afford 75 g of the L- DPTTA salt (95.37 % ee). The L-DPTTA salt obtained was chirally enriched by suspending the salt in isopropanol /acetonitrile (8:2 mixture; 750 mL, 5 vol) at 45 – 50 °C for 24 – 48 h. The chiral enhancement was monitored by chiral HPLC; the solution was gradually cooled to 20 – 25 °C, filtered and washed with an isoporpanol /acetonitrile mixture (8:2 mixture; 1 vol). The purification process was repeated and after filtration, the salt resulted in chiral purity greater than 96 % ee. The filtered compound was dried under reduced pressure at 35 – 40 °C to afford 62 g of the enantio-enriched L-DPPTA salt with 97.12% ee as an off-white solid. The enantio-enriched L-DPTTA salt (50 g, 1 eq) was dissolved in methanol (150 mL, 3 vol) at 20 – 30 °C and a potassium carbonate solution (18.05 g K2CO3 in 150 mL water) was slowly added at 20 – 30 °C under stirring. The reaction mixture was maintained at this temperature for 2 – 3 h (pH of the solution at was maintained at 9). Water (600 mL, 12 vol) was added into the reaction mixture through an additional funnel and the reaction mixture was stirred for 2 – 3 h at 20 – 30 °C. The solids were filtered; washed with water (150 mL, 3 vol) and dried under vacuum at 40 – 45 °C to afford 26.5 g of amino alcohol 4b-Br or 4c-Br with 99.54 % chemical purity, 99.28 % ee as an off-white solid. (Water content of the chiral amino alcohol is below 0.10 % w/w).1H NMR: δ values with respect to TMS (DMSO-d6; 400 MHz):8.68 (1H, d, J = 2.0 Hz, Ar- H), 8.16 (1H, dd, J = 8.0 Hz, 2.0 Hz, Ar-H), 7.49 – 7.43 (1H, m, Ar-H), 7.40 (1H, d, J = 8 Hz, Ar-H), 7.16 – 7.11 (1H, m, Ar-H), 7.11 – 6.99 (1H, m, Ar-H), 3.39 – 3.36 (1H, m, -OCHAHB– ), 3.25 – 3.22 (1H, m, -OCHAHB-).13C NMR: 163.87 -158.52 (dd, 2 x Ar-C-), 150.88 (Ar-C), 149.16 (Ar-C), 139.21 (Ar-C), 132.39 (Ar-C), 124.49 (Ar-C), 122.17 (Ar-C), 121.87 (d, Ar- C), 119.91 (t, -CF2-), 110.68 (Ar-C), 103.97 (i, Ar-C), 77.41 (i,-C-OH), 44.17 (-CH2-NH2).EXAMPLE 5
Preparation of l-(5-bromopyridin-2-yl)-2-(2,4-difluorophenyl)-l,l-difluoro-3-(lH-tetrazol-l- yl)propan-2-ol (l-6*-Br or l-7*-Br)

4b-Br or 4c-Br (20.0 g, 1 eq.) was added to acetic acid (50 mL, 2.5 vol) at 25 – 35 °C followed by the addition of anhydrous sodium acetate (4.32 g, 1 eq), trimethyl orthoformate (15.08 g, 2.7 eq). The reaction mixture was stirred for 15 – 20 min at this temperature and trimethylsilyl azide (12.74 g, 2.1 eq) was added to the reaction mixture (Chilled water was circulated through the condenser to minimize the loss of trimethylsilyl azide from the reaction mixture by evaporation). The reaction mixture was then heated to 70 – 75 °C and maintained at this temperature for 2 -3 h. The reaction progress was monitored by HPLC. Once the reaction was complete, the reaction mixture was cooled to 25 – 35 °C and water (200 mL, 10 vol) was added. The reaction mixture was extracted with ethyl acetate (400 mL, 20 vol) and the aqueous layer was back extracted with ethyl acetate (100 mL, 5 vol). The combined organic layers were washed with 10 % potassium carbonate solution (3 x 200 mL; 3 x 10 vol) followed by a 10 % NaCl wash (1 x 200 mL, 10 vol). The organic layer was distilled under reduced pressure below 45 °C. The crude obtained was azeotroped with heptanes (3 x 200 mL) to get 21.5g (94 % yield, 99.26 5 purity) of tetrazole 1-6* or 1-7* compound as pale brown solid (low melting solid).1H NMR: δ values with respect to TMS (DMSO-d6; 400 MHz NMR instrument): 9.13 (1H, Ar-H), 8.74 (1H, Ar-H), 8.22 – 8.20 (1H, m, Ar-H), 7.44 (1H, d, J = 7.2 Hz, Ar-H), 7.29 (1H„Ar-H), 7.23 – 7.17 (1H, m, Ar-H), 6.92 – 6.88 (1H, Ar-H), 5.61 (1H, d, J = 1 1.2 Hz, – OCHAHB-), 5.08 (1H, d, J = 5.6 Hz, -OCHAHB-).13C NMR: 163.67 -161.59 (dd, Ar-C-), 160.60 – 158.50 (dd, Ar-C-), 149.65 (Ar-C), 144.99 (Ar-C), 139.75 (Ar-C), 131.65 (Ar-C), 124.26 (Ar-C), 122.32 (d, Ar-C), 119.16 (t, -CF2-), 118.70 (d, Ar-C), 1 11.05 (d, Ar-C) 104.29 (t, Ar-C), 76.79 (i,-C-OH), 59.72 (Ar-C), 50.23 (-OCH2N-). EXAMPLE 6Preparation of 2-(2,4-difluorophenyl)-l , 1 -difluoro-3-( 1 H-tetrazol-1 -yl)-l -(5-(4-(2,2,2- trifluoroethoxy)phenyl)pyridin-2-yl)propan-2-ol (1 or la)A. Preparation of 1 or la via l-6*-Br or l-7*-Br

Synthesis of 4,4,5, 5-tetramethyl-2-(4-(2,2,2-trifluoroethoxy)phenyl)-l,3,2-dioxaborolane Potassium carbonate (59.7 g, 2.2 eq.) was added to a slurry of DMF (190 mL, 3.8 Vol.), 4- Bromo phenol (37.4g, 1.1 eq.) and 2,2,2-trifluroethyl tosylate (50.0 g, 1.0 eq.) at 20 – 35 °C under an inert atmosphere. The reaction mixture was heated to 115 – 120 °C and maintained at this temperature for 15 – 18 h. The reaction progress was monitored by GC. The reaction mixture was then cooled to 20 – 35 °C, toluene (200 mL, 4.0 vol.) and water (365 mL, 7. 3 vol.) were added at the same temperature, stirred for 10 – 15 minutes and separated the layers. The aqueous layer was extracted with toluene (200 mL, 4.0 vol.). The organic layers were combined and washed with a 2M sodium hydroxide solution (175 mL, 3.5 vol.) followed by a 20 % sodium chloride solution (175 mL, 3.5 vol.). The organic layer was then dried over anhydrous sodium sulfate and filtered. The toluene layer was transferred into clean reactor, spurged with argon gas for not less than 1 h. Bis(Pinacolato) diborane (47 g, 1.1 eq.), potassium acetate (49.6 g, 3.0 eq.) and 1,4-dioxane (430 mL, 10 vol.) were added at 20 -35 °C, and spurged the reaction mixture with argon gas for at least 1 h. Pd(dppf)Cl2 (6.88 g, 0.05eq) was added to the reaction mixture and continued the argon spurging for 10 – 15 minutes. The reaction mixture temperature was increased to 70 – 75 °C, maintained the temperature under argon atmosphere for 15 – 35 h and monitored the reaction progress by GC. The reaction mixture was cooled to 20 – 35 °C, filtered the reaction mixture through a Celite pad, and washed with ethyl acetate (86 mL, 2 vol.). The filtrate was washed with water (430 mL, 10 vol.). The aqueous layer was extracted with ethyl acetate (258 mL, 6 vol.) and washed the combined organic layers with a 10 % sodium chloride solution (215 mL, 5 vol.). The organic layer was dried over anhydrous sodium sulfate (43g, 1 time w/w), filtered and concentrated under reduced pressure below 45 °C to afford crude 4,4,5, 5-tetramethyl-2-(4-(2,2,2- trifluoroethoxy)phenyl)-l,3,2-dioxaborolane (65 g; 71 % yield with the purity of 85.18 % by GC). The crude 4,4,5,5-tetramethyl-2-(4-(2,2,2-trifluoroethoxy)phenyl)-l,3,2-dioxaborolane (65 g, 1 eq.) was dissolved in 10 % ethyl acetate – n-Heptane (455 mL, 7 vol.) and stirred for 30 – 50 minutes at 20 – 35 °C. The solution was filtered through a Celite bed and washed with 10 % ethyl acetate in n-Heptane (195 mL, 3 vol.). The filtrate and washings were pooled together, concentrated under vacuum below 45 °C to afford 4,4,5, 5-tetramethyl-2-(4-(2,2,2- trifluoroethoxy)phenyl)-l,3,2-dioxaborolane as a thick syrup (45.5 g; 70 % recovery). This was then dissolved in 3 % ethyl acetate-n-heptane (4 vol.) and adsorbed on 100 – 200 M silica gel (2 times), eluted through silica (4 times) using 3 % ethyl acetate – n- heptane. The product rich fractions were pooled together and concentrated under vacuum. The column purified fractions (> 85 % pure) were transferred into a round bottom flask equipped with a distillation set-up. The compound was distilled under high vacuum below 180 °C and collected into multiple fractions. The purity of fractions was analyzed by GC (should be > 98 % with single max impurity < 1.0 %). The less pure fractions (> 85 % and < 98 % pure fraction) were pooled together and the distillation was repeated to get 19g (32% yield) of 4,4,5, 5-tetramethyl-2-(4- (2,2,2-trifluoroethoxy)phenyl)-l,3,2-dioxaborolane as a pale yellow liquid.*H NMR: δ values with respect to TMS (DMSO-d6; 400 MHz):7.64 (2H, d, 6.8 Hz), 7.06 (2H, d, J = 6.4 Hz), 4.79 (2H, q, J = 6.8 Hz), 1.28 (12H, s).13C NMR: 159.46 (Ar-C-O-), 136.24 (2 x Ar-C-), 127.77 – 120.9 (q, -CF3), 122.0 (Ar-C-B), 114.22 (2 x Ar-C-), 64.75 (q, J = 27.5 Hz).Synthesis of 2-(2.4-difluorophenyl)-l.l-difluoro-3-(lH-tetrazol-l-yl)-l-(5-(4-(2.2.2- trifluoroethoxy)phenyl)pyridin-2-yl)propan-2-ol (1 or la)l-6*-Br or l-7*-Br (14 g, 0.03 mol, 1 eq) was added to tetrahydrofuran (168 mL, 12 vol) at 25 – 35 °C and the resulting solution was heated to 40 – 45 °C. The reaction mixture was maintained at this temperature for 20 – 30 min under argon bubbling. Sodium carbonate (8.59 g, 0.08 mol, 2.5 eq) and water (21 mL, 1.5 vol) were added into the reaction mixture and the bubbling of argon was continued for another 20 – 30 min. 4,4,5, 5-tetramethyl-2-(4-(2,2,2- trifluoroethoxy)phenyl)-l,3,2-dioxaborolane (10.76 g, 1.1 eq) dissolved in tetrahydrofuran (42 mL, 3 vol) was added into the reaction mixture and argon bubbling was continued for 20 – 30 min. Pd(dppf)Cl2 (2.65 g, 0.1 eq) was added to the reaction mixture under argon bubbling and stirred for 20 – 30 min (Reaction mixture turned into dark red color). The reaction mixture was heated to 65 – 70 °C and maintained at this temperature for 3 – 4 h. The reaction progress was monitored by HPLC. The reaction mixture was cooled to 40 – 45 °C and the solvent was distilled under reduced pressure. Toluene (350 mL, 25 vol.) was added to the reaction mixture and stirred for 10 – 15 min followed by the addition of water (140 mL, 10 vol). The reaction mixture was filtered through Hyflo (42 g, 3 times), the layers were separated and the organic layer was washed with water (70 mL, 5 vol) and a 20 % w/w sodium chloride solution (140 mL, 10 vol). The organic layer was treated with charcoal (5.6 g, 0.4 times, neutral chalrcoal), filtered through Hyflo. (lS)-lO-Camphor sulfonic acid (7.2 g, 1 eq.) was added to the toluene layer and the resulting mixture was heated to 70 – 75 °C for 2 – 3 h. The reaction mixture was gradually cooled to 25 – 35 °C and stirred for 1 – 2 h. The solids were filtered, washed with toluene (2 x 5 vol.) and then dried under vacuum below 45 °C to afford 18.0 g of an off white solid. The solids (13.5 g, 1 eq.) were suspended in toluene (135 mL, 10 vol) and neutralized by adding 1M NaOH solution (1.48 vol, 1.1 eq) at 25 – 35 °C and stirred for 20 – 30 min. Water (67.5 mL, 5 vol) was added to the reaction mixture and stirred for 10 – 15 min, and then the layers were separated. The organic layer was washed with water (67.5 mL, 5 vol) to remove the traces of CSA. The toluene was removed under reduced pressure below 45 °C to afford crude 1 or la. Traces of toluene were removed by azeotroping with ethanol (3 x 10 vol), after which light brown solid of crude 1 or la (7.5 g, 80% yield) was obtained.The crude 1 or la (5 g) was dissolved in ethanol (90 mL, 18 vol.) at 20 – 35 °C, and heated to 40 – 45 °C. Water (14 vol) was added to the solution at 40 – 45 °C, the solution was maintained at this temperature for 30 – 45 min and then gradually cooled to 20 – 35 °C. The resulting suspension was continued to stir for 16 – 18 h at 20 – 35 °C, an additional amount of water (4 vol.) was added and the stirring continued for 3 – 4 h. The solids were filtered to afford 4.0 g (80% recovery) of 1 or la (HPLC purity >98%) as an off-white solid.1H NMR: δ values with respect to TMS (DMSO-d6; 400 MHz):9.15 (1H, s, Ar-H), 8.93 (1H, d, J = 0.8 Hz, Ar-H), .8.22 – 8.20 (1H, m, Ar-H), 7.80 (2H, d, J = 6.8 Hz, Ar-H), 7.52 (1H, d, J = 6.8 Hz, Ar-H), 7.29 (1H, d,J = 3.2Hz, Ar-H), 7.27 – 7.21 (1H, m, Ar-H), 7.23 – 7.21 (2H, d, J = 6.8 Hz, Ar-H), 7.19 (1H, d, J = 6.8 Hz, Ar-H), 6.93 – 6.89 (1H, m, Ar-H), 5.68 (1H, / = 12 Hz, -CHAHB), 5.12 (2H, d, J = 11.6 Hz, -CHAHB), 4.85 (2H, q, J = 1.6 Hz).13C NMR: 163.93 – 158.33 (m, 2 x Ar-C), 157.56 (Ar-C), 149.32 (i, Ar-C), 146.40 (Ar-C), 145.02 (Ar-C), 136.20 (Ar-C), 134.26 (2 x Ar-C), 131.88 – 131.74 (m, AR-C), 129.72 (Ar-C), 128.47 (2 x Ar-C), 123.97 (q, -CF2-), 122.41 (Ar-C), 119.30 (-CF3), 118.99 (Ar-C), 115.65 (2 x Ar-C), 110.99 (d, Ar-C), 104.22 (i, Ar-C), 77.41 – 76.80 (m, Ar-C), 64.72 (q, -OCH2-CF3), 50.54 (-CH2-N-).B. Preparation of 1 or la via 4b-Br or 4c-Br


Synthesis of 3-amino-2-(2.4-difluorophenyl)-l.l-difluoro-l-(5-(4-(2.2.2- trifluoroethoxy)phenyl)pyridin-2-yl)propan-2-ol (8a or 8b)Potassium carbonate (30.4 g) and water (53.3 g) were charged to a 1-L flask equipped with overhead stirring, thermocouple, and nitrogen/vacuum inlet valve, and stirred until dissolved. The boronic acid (19.37 g), a solution of 4b-Br or 4c-Br in 2-butanol (103.5 g, 27.8 g theoretical 4b-Br or 4c-Br)) and 2-BuOH (147.1 g) were added and stirred to form a clear mixture. The flask was evacuated and refilled with nitrogen 3 times. Pd(d f)2Cl2 (0.30 g) was added and stirred to form a light orange solution. The flask was evacuated and refilled with nitrogen 4 times. The mixture was heated to 85 °C and stirred overnight and endpointed by HPLC analysis. The reaction mixture was cooled to 60 °C and the layers were allowed to settle. The aqueous layer was separated. The organic layer was washed with 5% NaCl solution (5 x 100 ml) at 30-40 °C. The organic layer was filtered and transferred to a clean flask with rinses of 2-BuOH. The combined solution was 309.7 g, water content 13.6 wt% by KF analysis. The solution was diluted with 2-BuOH (189 g) and water (10 g). Theoretically the solution contained 34.8 g product, 522 ml (15 volumes) of 2-BuOH, and 52.2 ml (1.5 volumes) of water. L-Tartaric acid (13.25 g) was added and the mixture was heated to a target temperature of 70-75 °C. During the heat-up, a thick suspension formed. After about 15 minutes at 70-72 °C the suspension became fluid and easily stirred. The suspension was cooled at a rate of 10 °C/hour to 25 °C then stirred at 25 °C for about 10 hours. The product was collected on a vacuum filter and washed with 10:1 (v/v) 2-BuOH/water (50 ml) and 2- butanol (40 ml). The salt was dried in a vacuum oven at 60 °C with a nitrogen purge for 2 days. The yield was 40.08 g of 8a or 8b as a fluffy, grayish-white solid. The water content was 0.13 wt% by KF analysis. The yield was 87.3% with an HPLC purity of 99.48%. Synthesis of 2-(2,4-difluorophenyl)-l,l-difluoro-3-(lH-tetrazol-l-yl)-l-(5-(4-(2,2,2- trifluoroethoxy)phenyl)pyridin-2-yl)propan-2-ol (1 or la)To a 350 ml pressure bottle were charged acetic acid (73 ml), 8a or 8b (34.8 g), sodium acetate (4.58 g) and trimethylorthoformate (16.0 g). The mixture was stirred for 18 min. at room temperature until a uniform suspension was obtained. Azidotrimethylsilane (8.88 g) was added and the bottle was sealed. The bottle was immersed in an oil bath and magnetically stirred. The oil bath was at 52 °C initially, and was warmed to 62-64 °C over about ½ hour. The suspension was stirred at 62-64 °C overnight. After 20.5 hours the suspension was cooled to room temperature and sampled. The reaction was complete by HPLC analysis. The reaction was combined with three other reactions that used the same raw material lots and general procedure (total of 3.0 g additional starting material). The combined reactions were diluted with ethyl acetate (370 ml) and water (368 ml) and stirred for about ½ hour at room temperature. The layers were settled and separated. The organic layer was washed with 10% K2C03 solution (370 ml/ 397 g) and 20% NaCl solution (370 ml/ 424 g). The organic layer (319 g) was concentrated, diluted with ethanol (202 g) and filtered, rinsed with ethanol (83 g). The combined filtrate was concentrated to 74 g of amber solution.The crude 1 or la solution in ethanol (74 g solution, containing theoretically 31.9 g 1 or la) was transferred to a 2-L flask equipped with overhead stirring, thermocouple, and addition funnel. Ethanol (335 g) was added including that used to complete the transfer of the 1 or la solution. The solution was heated to nominally 50 °C and water (392 g) was added over 12 minutes. The resulting hazy solution was seeded with 1 or la crystals and stirred at 50 °C. After about ½ hour the mixture was allowed to cool to 40 °C over about ½ hour during which time crystallization started. Some darker colored chunky solid separated out from the main suspension. The pH of the crystallizing mixture was adjusted from 4.5 to 6 using 41% KOH (1.7 g). After about 1 hour a good suspension had formed. Additional water (191 g) was added slowly over ½ hour. The suspension was heated to 50 °C and cooled at 5 °C/min to room temperature. After stirring overnight the suspension was cooled in a water bath to 16 °C and filtered after 1 hour. The wet cake was washed with 55:45 (v/v) water/ethanol (2 x 50 ml) and air-dried on the vacuum filter funnel overnight. Further drying at 40 °C in a vacuum oven with a nitrogen bleed resulted in no additional weight loss. The yield was 30.2 g of off-white fine powder plus some darker granular material. By in-process HPLC analysis there was no difference in the chemical purity of the darker and lighter materials. The purity was 99.4%. The water content was 2.16 wt% by KF analysis. The residual ethanol was 1.7 wt% estimated by ‘Ft NMR analysis. The corrected yield was 29.0 g, 91.0% overall yield for tetrazole formation and crystallization. The melting point was 65 °C by DSC analysis.
FDA Approves Mycovia Pharmaceuticals’ VIVJOA™ (oteseconazole), the First and Only FDA-Approved Medication for Recurrent Vulvovaginal Candidiasis (Chronic Yeast Infection)
– Approval of VIVJOA™ marks a significant therapeutic advancement for reducing the incidence of RVVC, a condition with substantial unmet need, in permanently infertile and postmenopausal women
– VIVJOA™ is the first FDA approval in Mycovia’s pipeline of novel treatments for fungal infections
– U.S. commercial launch of VIVJOA™ expected in Q2
April 28, 2022 07:55 AM Eastern Daylight Time
DURHAM, N.C.–(BUSINESS WIRE)–The U.S. Food and Drug Administration (FDA) approved VIVJOA™ (oteseconazole capsules), an azole antifungal indicated to reduce the incidence of recurrent vulvovaginal candidiasis (RVVC) in females with a history of RVVC who are NOT of reproductive potential. VIVJOA is the first and only FDA-approved medication for this condition and provides sustained efficacy demonstrated by significant long-term reduction of RVVC recurrence through 50 weeks versus comparators. VIVJOA is the first FDA-approved product for Mycovia Pharmaceuticals, Inc. (Mycovia), an emerging biopharmaceutical company dedicated to recognizing and empowering those living with unmet medical needs by developing novel therapies.
“We believe the market need for VIVJOA is strong, and we are eager to execute our commercial plans”Tweet this
RVVC, also known as chronic yeast infection, is defined by the Centers for Disease Control and Prevention (CDC) as three or more symptomatic acute episodes of yeast infection in 12 months. RVVC is a distinct condition from vulvovaginal candidiasis (VVC), and until now, there have been no FDA-approved medications specifically indicated for it. Nearly 75% of all adult women will have at least one yeast infection in their lifetime, with approximately half experiencing a recurrence. Of those women, up to 9% develop RVVC.
“After nearly two decades of living with chronic yeast infection and feeling like there was no hope from the itchiness, irritation and constant dread of when the next yeast infection would return, I was overjoyed to even be a part of this clinical trial,” said Leslie Ivey, RVVC patient and clinical trial participant. “It is gratifying to see RVVC finally get the attention it deserves.”
Symptoms of RVVC include vaginal itching, burning, irritation and inflammation. Some women may experience abnormal vaginal discharge and painful sexual intercourse or urination, causing variable but often severe discomfort and pain.
VIVJOA’s FDA approval is based upon the positive results from three Phase 3 trials of oteseconazole – two global, pivotal VIOLET studies and one U.S.-focused ultraVIOLET study, including 875 patients at 232 sites across 11 countries. In the two global VIOLET studies, 93.3% and 96.1% of women with RVVC who received VIVJOA did not have a recurrence for the 48-week maintenance period compared to 57.2% and 60.6% of patients who received placebo (p <0.001). In the ultraVIOLET study, 89.7% of women with RVVC who received VIVJOA cleared their initial yeast infection and did not have a recurrence for the 50-week maintenance period compared to 57.1% of those who received fluconazole followed by placebo (p <0.001). The most common side effects reported in Phase 3 clinical studies were headache (7.4%) and nausea (3.6%). VIVJOA is contraindicated in those with a hypersensitivity to oteseconazole, and based on data from rat studies, also in females who are of reproductive potential, pregnant, or lactating. Please see additional Important Safety Information below.
Patrick Jordan, CEO of Mycovia Pharmaceuticals and Partner at NovaQuest Capital Management, stated, “We celebrate this important milestone for Mycovia, as VIVJOA is the first antifungal in our pipeline to obtain FDA approval and achieves our goal to fulfill a previously unmet medical need among women suffering from RVVC. We are honored to lead this advancement in women’s health.”
“We believe the market need for VIVJOA is strong, and we are eager to execute our commercial plans,” Jordan continued. “As we enter a new chapter of our history as a commercial biopharmaceutical company, we will continue driving our mission forward to develop novel therapies for overlooked conditions.”
Oteseconazole is designed to inhibit fungal CYP51, which is required for fungal cell wall integrity, and this selective interaction is also toxic to fungi, resulting in the inhibition of fungal growth. Due to its chemical structure, oteseconazole has a lower affinity for human CYP enzymes as compared to fungal CYP enzymes. The FDA granted oteseconazole Qualified Infectious Disease Product and Fast Track designations.
“A medicine with VIVJOA’s sustained efficacy combined with the clinical safety profile has been long needed, as until now, physicians and their patients have had no FDA-approved medications for RVVC,” stated Stephen Brand, Ph.D., Chief Development Officer of Mycovia. “We are excited to be the first to offer a medication designed specifically for RVVC, a challenging and chronic condition that is expected to increase in prevalence over the next decade.”
Mycovia is planning its commercial launch of VIVJOA™ in the second quarter of 2022.
About Recurrent Vulvovaginal Candidiasis
RVVC is a debilitating, chronic infectious condition that affects 138 million women worldwide each year. RVVC, also known as chronic yeast infection, is a distinct condition from vulvovaginal candidiasis (VVC) and defined as three or more symptomatic acute episodes of yeast infection in 12 months. Primary symptoms include vaginal itching, burning, irritation and inflammation. Some women may experience abnormal vaginal discharge and painful sexual intercourse or urination, causing variable but often severe discomfort and pain.
About VIVJOA™
VIVJOA™ (oteseconazole) is an azole antifungal indicated to reduce the incidence of recurrent vulvovaginal candidiasis (RVVC) in females with a history of RVVC who are NOT of reproductive potential. VIVJOA is the first and only FDA-approved medication that provides sustained efficacy demonstrated by significant long-term reduction of RVVC recurrence through 50 weeks versus comparators. Oteseconazole is designed to inhibit fungal CYP51, which is required for fungal cell wall integrity, and this selective interaction is also toxic to fungi, resulting in the inhibition of fungal growth. Due to its chemical structure, oteseconazole has a lower affinity for human CYP enzymes as compared to fungal CYP enzymes. The FDA approved VIVJOA based upon the positive results from three Phase 3 clinical trials of oteseconazole – two global, pivotal VIOLET studies and one U.S.-focused ultraVIOLET study, including 875 patients at 232 sites across 11 countries.
References
- ^ Jump up to:a b https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/215888s000lbl.pdf
- ^ “Vivjoa: FDA-Approved Drugs”. U.S. Food and Drug Administration (FDA). Retrieved 27 April 2022.
- ^ Jump up to:a b “FDA Approves Mycovia Pharmaceuticals’ VIVJOA (oteseconazole), the First and Only FDA-Approved Medication for Recurrent Vulvovaginal Candidiasis (Chronic Yeast Infection)” (Press release). Mycovia Pharmaceuticals. 28 April 2022. Retrieved 28 April 2022 – via Business Wire.
- ^ World Health Organization (2016). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 76”. WHO Drug Information. 30 (3). hdl:10665/331020.
Further reading
- Sobel JD, Nyirjesy P (December 2021). “Oteseconazole: an advance in treatment of recurrent vulvovaginal candidiasis”. Future Microbiology. 16: 1453–1461. doi:10.2217/fmb-2021-0173. PMID 34783586.
External links
- “Oteseconazole”. Drug Information Portal. U.S. National Library of Medicine.
- Clinical trial number NCT03562156 for “A Study of Oral Oteseconazole for the Treatment of Patients With Recurrent Vaginal Candidiasis (Yeast Infection) (VIOLET)” at ClinicalTrials.gov
- Clinical trial number NCT03561701 for “A Study of Oral Oteseconazole (VT-1161) for the Treatment of Patients With Recurrent Vaginal Candidiasis (Yeast Infection) (VIOLET)” at ClinicalTrials.gov
- Clinical trial number NCT03840616 for “Study of Oral Oteseconazole (VT-1161) for Acute Yeast Infections in Patients With Recurrent Yeast Infections (ultraVIOLET)” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | Vivjoa |
| Other names | VT-1161 |
| License data | US DailyMed: Oteseconazole |
| Routes of administration | By mouth |
| Drug class | Antifungal |
| ATC code | J02AC06 (WHO) |
| Legal status | |
| Legal status | US: ℞-only [1] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 1340593-59-0 |
| PubChem CID | 77050711 |
| DrugBank | DB13055 |
| ChemSpider | 52083215 |
| UNII | VHH774W97N |
| KEGG | D11785 |
| ChEBI | CHEBI:188153 |
| ChEMBL | ChEMBL3311228 |
| ECHA InfoCard | 100.277.989 |
| Chemical and physical data | |
| Formula | C23H16F7N5O2 |
| Molar mass | 527.403 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI |
/////////OTESECONAZOLE, vt 1161, fungal infection, Candida albicans infection, onychomycosis, PHASE 3,


AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

……
C1=CC(=CC=C1C2=CN=C(C=C2)C(C(CN3C=NN=N3)(C4=C(C=C(C=C4)F)F)O)(F)F)OCC(F)(F)F
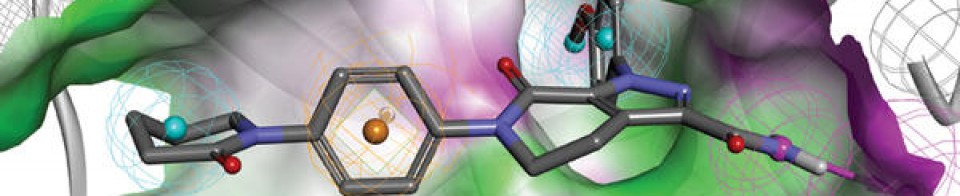
NEW DRUG APPROVALS
ONE TIME
$10.00
DEUCRAVACITINIB
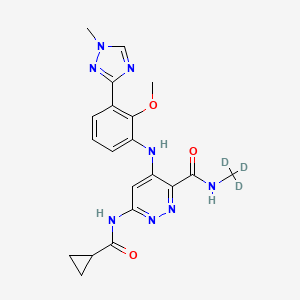
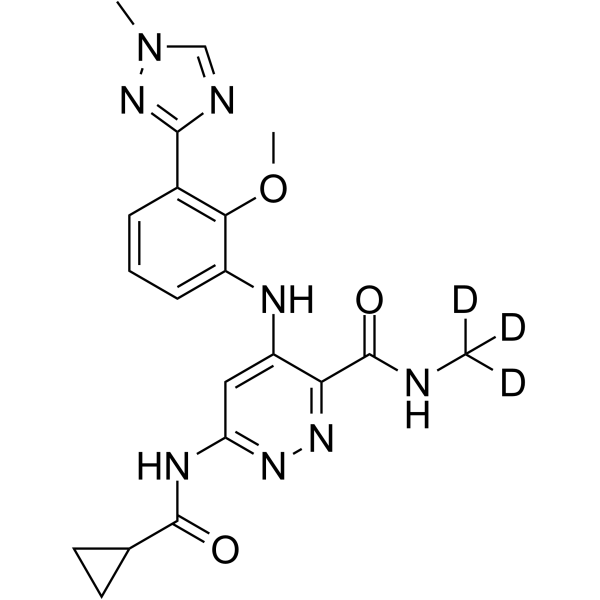
DEUCRAVACITINIB
BMS-986165
CAS 1609392-27-9, C20H22N8O3, 425.46
6-(cyclopropanecarbonylamino)-4-[2-methoxy-3-(1-methyl-1,2,4-triazol-3-yl)anilino]-N-(trideuteriomethyl)pyridazine-3-carboxamide
- OriginatorBristol-Myers Squibb
- ClassAmides; Aniline compounds; Anti-inflammatories; Antipsoriatics; Antirheumatics; Cyclopropanes; Ethers; Hepatoprotectants; Organic deuterium compounds; Pyridazines; Skin disorder therapies; Small molecules; Triazoles
- Mechanism of ActionTYK2 kinase inhibitors
- Phase IIIPlaque psoriasis
- Phase IICrohn’s disease; Lupus nephritis; Psoriatic arthritis; Systemic lupus erythematosus; Ulcerative colitis
- Phase IAutoimmune disorders
- No development reportedInflammatory bowel diseases; Psoriasis
- 02 Jul 2021Bristol-Myers Squibb plans a phase I pharmacokinetics trial (In volunteers) in USA (PO, Tablet) in July 2021 (NCT04949269)
- 14 Jun 2021Bristol-Myers Squibb plans a phase III trial for Psoriatic arthritis (Treatment-naïve) in USA, Brazil, Colombia, Czech republic, Hungary, Italy, Mexico, Romania, Spain and Taiwan in July 2021 (NCT04908202) (EudraCT2020-005097-10)
- 02 Jun 2021Interim efficacy and adverse events data from the phase III POETYK-PSO-1 trial in Psoriatic psoriasis presented at the 22nd Annual Congress of the European League Against Rheumatism (EULAR-2021)
BMS , presumed to be in collaboration with Jinan University and Chinese Academy of Sciences , is developing deucravacitinib, a TYK2 inhibitor, for treating autoimmune diseases, primarily psoriasis. In July 2021, deucravacitinib was reported to be in phase 3 clinical development.
Deucravacitinib (BMS-986165) is a highly selective, orally bioavailable allosteric TYK2 inhibitor for the treatment of autoimmune diseases, which selectively binds to TYK2 pseudokinase (JH2) domain (IC50=1.0 nM) and blocks receptor-mediated Tyk2 activation by stabilizing the regulatory JH2 domain. Deucravacitinib inhibits IL-12/23 and type I IFN pathways.
PAPER
https://pubs.acs.org/doi/abs/10.1021/acs.jmedchem.9b00444

Small molecule JAK inhibitors have emerged as a major therapeutic advancement in treating autoimmune diseases. The discovery of isoform selective JAK inhibitors that traditionally target the catalytically active site of this kinase family has been a formidable challenge. Our strategy to achieve high selectivity for TYK2 relies on targeting the TYK2 pseudokinase (JH2) domain. Herein we report the late stage optimization efforts including a structure-guided design and water displacement strategy that led to the discovery of BMS-986165 (11) as a high affinity JH2 ligand and potent allosteric inhibitor of TYK2. In addition to unprecedented JAK isoform and kinome selectivity, 11 shows excellent pharmacokinetic properties with minimal profiling liabilities and is efficacious in several murine models of autoimmune disease. On the basis of these findings, 11 appears differentiated from all other reported JAK inhibitors and has been advanced as the first pseudokinase-directed therapeutic in clinical development as an oral treatment for autoimmune diseases.
Bristol Myers Squibb Presents Positive Data from Two Pivotal Phase 3 Psoriasis Studies Demonstrating Superiority of Deucravacitinib Compared to Placebo and Otezla® (apremilast)
Significantly more patients treated with deucravacitinib achieved PASI 75 and sPGA 0/1 compared to patients treated with placebo and Otezla at Week 16, with an increased benefit versus Otezla at Week 24 and maintained through Week 52
Deucravacitinib was well tolerated with a low rate of discontinuation due to adverse events
Deucravacitinib is a first-in-class, oral, selective tyrosine kinase 2 (TYK2) inhibitor with a unique mechanism of action
Results presented as late-breaking research at the 2021 American Academy of Dermatology Virtual Meeting Experience
PRINCETON, N.J.–(BUSINESS WIRE)– Bristol Myers Squibb (NYSE:BMY) today announced positive results from two pivotal Phase 3 trials evaluating deucravacitinib, an oral, selective tyrosine kinase 2 (TYK2) inhibitor, for the treatment of patients with moderate to severe plaque psoriasis. The POETYK PSO-1 and POETYK PSO-2 trials, which evaluated deucravacitinib 6 mg once daily, met both co-primary endpoints versus placebo, with significantly more patients achieving Psoriasis Area and Severity Index (PASI) 75 response and a static Physician’s Global Assessment score of clear or almost clear (sPGA 0/1) after 16 weeks of treatment with deucravacitinib. Deucravacitinib was well tolerated with a low rate of discontinuation due to adverse events (AEs).
This press release features multimedia. View the full release here: https://www.businesswire.com/news/home/20210423005134/en(Graphic: Business Wire)
Deucravacitinib demonstrated superior skin clearance compared with Otezla® (apremilast) for key secondary endpoints in both studies, as measured by PASI 75 and sPGA 0/1 responses at Week 16 and Week 24. Findings include:
PASI 75 Response in POETYK PSO-1 and POETYK PSO-2:
- At Week 16, 58.7% and 53.6% of patients receiving deucravacitinib achieved PASI 75 response, respectively, versus 12.7% and 9.4% receiving placebo and 35.1% and 40.2% receiving Otezla.
- At Week 24, 69.0% and 59.3% of patients receiving deucravacitinib achieved PASI 75 response, respectively, versus 38.1% and 37.8% receiving Otezla.
- Among patients who achieved PASI 75 response at Week 24 with deucravacitinib and continued treatment with deucravacitinib, 82.5% and 81.4%, respectively, maintained PASI 75 response at Week 52.
sPGA 0/1 Response in POETYK PSO-1 and POETYK PSO-2:
- At Week 16, 53.6% and 50.3% of patients receiving deucravacitinib achieved sPGA 0/1 response, respectively, versus 7.2% and 8.6% receiving placebo and 32.1% and 34.3% receiving Otezla.
- At Week 24, 58.4% and 50.4% of patients receiving deucravacitinib achieved sPGA 0/1 response, respectively, versus 31.0% and 29.5% receiving Otezla.
“In both pivotal studies, deucravacitinib was superior to Otezla across multiple endpoints, including measures of durability and maintenance of response, suggesting that deucravacitinib has the potential to become a new oral standard of care for patients who require systemic therapy and need a better oral option for their moderate to severe plaque psoriasis,” said April Armstrong, M.D., M.P.H., Associate Dean and Professor of Dermatology at the University of Southern California. “As many patients with moderate to severe plaque psoriasis remain undertreated or even untreated, it is also highly encouraging to see that deucravacitinib improved patient symptoms and outcomes to a greater extent than Otezla.”
Superiority of Deucravacitinib Versus Placebo and Otezla
Deucravacitinib demonstrated a robust efficacy profile, including superiority to placebo for the co-primary endpoints and to Otezla for key secondary endpoints. In addition to PASI 75 and sPGA 0/1 measures, deucravacitinib was superior to Otezla across both studies in multiple other secondary endpoints, demonstrating significant and clinically meaningful efficacy improvements in symptom burden and quality of life measures.
| POETYK PSO-1 and POETYK PSO-2 Results at Week 16 and Week 24 | ||||||
| Endpoint | POETYK PSO-1 (n=666) | POETYK PSO-2 (n=1,020) | ||||
| Deucravacitinib6 mg(n=332) | Otezla30 mg(n=168) | Placebo(n=166) | Deucravacitinib6 mg(n=511) | Otezla30 mg(n=254) | Placebo(n=255) | |
| PASI 75*a | ||||||
| Week 16 | 58.7%*† | 35.1% | 12.7% | 53.6%*‡ | 40.2% | 9.4% |
| Week 24 | 69.0%† | 38.1% | – | 59.3%† | 37.8% | – |
| sPGA 0/1*b | ||||||
| Week 16 | 53.6%*† | 32.1% | 7.2% | 50.3%*† | 34.3% | 8.6% |
| Week 24 | 58.4%† | 31.0% | – | 50.4%† | 29.5% | – |
| (Scalp) ss-PGA 0/1c | ||||||
| Week 16 | 70.8%*† | 39.1% | 17.4% | 60.3%*† | 37.3% | 17.3% |
| Week 24 | 71.8%† | 42.7% | – | 59.7%‡ | 41.6% | – |
| PSSD-Symptoms CFBd | ||||||
| Week 16 | -26.7*† | -17.8 | -3.6 | -28.3*† | -21.1 | -4.7 |
| Week 24 | -31.9† | -20.7 | – | -29.1† | -21.4 | – |
| DLQI 0/1e | ||||||
| Week 16 | 40.7%*† | 28.6% | 10.6% | 38.0%*† | 23.1% | 9.8% |
| Week 24 | 47.8%‡ | 24.2% | – | 41.8%† | 21.5% | – |
| *Co-primary endpoints for POETYK PSO-1 and POETYK PSO-2 were PASI 75 and sPGA 0/1 for deucravacitinib vs placebo at Week 16. |
| a. PASI 75 is defined as at least a 75% improvement from baseline in Psoriasis Area and Severity Index (PASI) scores. *p<0.0001 vs placebo. †p<0.0001 vs Otezla. ‡p=0.0003 vs Otezla. |
| b. sPGA 0/1 is defined as a static Physician’s Global Assessment (sPGA) score of clear or almost clear. *p<0.0001 vs placebo. †p<0.0001 vs Otezla. |
| c. ss-PGA 0/1 is defined as a scalp-specific Physician’s Global Assessment (ss-PGA) score of clear or almost clear in those with ss-PGA of at least 3 (moderate) at baseline. POETYK PSO-1: *p<0.0001 vs placebo. †p<0.0001 vs Otezla. POETYK PSO-2: *p<0.0001 vs placebo. †p<0.0001 vs Otezla. ‡p=0.0002 vs Otezla. |
| d. Change from baseline (CFB) in Psoriasis Symptoms and Signs Diary (PSSD) captures improvement in symptoms of itch, pain, stinging, burning and skin tightness in patient eDiaries. *p<0.0001 vs placebo. †p<0.0001 vs Otezla. |
| e. Dermatology Life Quality Index (DLQI) 0/1 scores reflect no effect at all on patient’s life in patients with a baseline DLQI score of ≥2. POETYK PSO-1: *p<0.0001 vs placebo. †p=0.0106 vs Otezla. ‡p<0.0001 vs Otezla. POETYK PSO-2: *p<0.0001 vs placebo. †p<0.0001 vs Otezla. |
Safety and Tolerability
Deucravacitinib was well-tolerated and had a similar safety profile in both trials. At Week 16, 2.9% of 419 patients on placebo, 1.8% of 842 patients on deucravacitinib and 1.2% of 422 patients on Otezla experienced serious adverse events (SAEs) across both studies. The most common AEs (≥5%) with deucravacitinib treatment at Week 16 were nasopharyngitis and upper respiratory tract infection with low rates of headache, diarrhea and nausea. At Week 16, 3.8% of patients on placebo, 2.4% of patients on deucravacitinib and 5.2% of patients on Otezla experienced AEs leading to discontinuation. Across POETYK PSO-1 and POETYK PSO-2 over 52 weeks, SAEs when adjusted for exposure (exposure adjusted incidence per 100 patient-years [EAIR]) were 5.7 with placebo, 5.7 with deucravacitinib and 4.0 with Otezla. In the same timeframe across both studies, EAIRs for AEs leading to discontinuation were 9.4 with placebo, 4.4 with deucravacitinib and 11.6 with Otezla. No new safety signals were observed during Weeks 16‒52.
Across both Phase 3 trials, rates of malignancy, major adverse cardiovascular events (MACE), venous thromboembolism (VTE) and serious infections were low and generally consistent across active treatment groups. No clinically meaningful changes were observed in multiple laboratory parameters (including anemia, blood cells, lipids and liver enzymes) over 52 weeks.
“The findings from both studies affirm that deucravacitinib – a first-in-class, oral, selective TYK2 inhibitor with a unique mechanism of action that inhibits the IL-12, IL-23 and Type 1 IFN pathways –may become an oral treatment of choice for people living with psoriasis. We believe deucravacitinib has significant potential across a broad range of immune-mediated diseases, and we are committed to further advancing our expansive clinical program with this agent,” said Mary Beth Harler, M.D., head of Immunology and Fibrosis Development, Bristol Myers Squibb. “We are in discussions with health authorities with the goal of bringing this new therapy to appropriate patients as soon as possible. At Bristol Myers Squibb, we are committed to building an immunology portfolio that addresses pressing unmet needs that exist for those impacted by serious dermatologic conditions and other immune-mediated diseases, to ultimately deliver the promise of living a better life.”
These results are available as a late-breaking research presentation (Session S033 – Late-Breaking Research Abstracts) as part of the 2021 American Academy of Dermatology (AAD) Virtual Meeting Experience (VMX). Full results of both studies will be submitted to a medical journal for peer review. In November 2020 and February 2021, respectively, Bristol Myers Squibb announced positive topline results from POETYK PSO-1 and POETYK PSO-2.
Visit www.bms.com/media/medical-meetings/bms-at-aad-vmx.html for more information on Bristol Myers Squibb’s scientific approach and resources on psoriasis and immune-mediated diseases.
About Deucravacitinib
Deucravacitinib (pronounced doo-krav-a-sih-ti-nib) is a first-in-class, oral, selective tyrosine kinase 2 (TYK2) inhibitor with a unique mechanism of action. Deucravacitinib is the first and only TYK2 inhibitor in clinical studies across multiple immune-mediated diseases. Bristol Myers Squibb scientists designed deucravacitinib to selectively target TYK2, thereby inhibiting signaling of interleukin (IL)-12, IL-23 and Type 1 interferon (IFN), key cytokines involved in psoriasis pathogenesis. Deucravacitinib achieves a high degree of selectivity by uniquely binding to the regulatory, rather than the active, domain of TYK2, which is structurally distinct from the regulatory domains of Janus kinase (JAK) 1, 2 and 3. At therapeutic doses, deucravacitinib does not inhibit JAK1, JAK2 or JAK3. Due to the innovative design of deucravacitinib, Bristol Myers Squibb earned recognition with the 2019 Thomas Alva Edison Patent Award for the science underpinning the clinical development of deucravacitinib.
Deucravacitinib is being studied in multiple immune-mediated diseases, including psoriasis, psoriatic arthritis, lupus and inflammatory bowel disease. In addition to POETYK PSO-1 and POETYK PSO-2, Bristol Myers Squibb is evaluating deucravacitinib in three other Phase 3 studies in psoriasis: POETYK PSO-3 (NCT04167462); POETYK PSO-4 (NCT03924427); POETYK PSO-LTE (NCT04036435). Deucravacitinib is not approved for any use in any country.
About the Phase 3 POETYK PSO-1 and POETYK PSO-2 Studies
PrOgram to Evaluate the efficacy and safety of deucravacitinib, a selective TYK2 inhibitor (POETYK) PSO-1 (NCT03624127) and POETYK PSO-2 (NCT03611751) are global Phase 3 studies designed to evaluate the safety and efficacy of deucravacitinib compared to placebo and Otezla® (apremilast) in patients with moderate to severe plaque psoriasis. Both POETYK PSO-1, which enrolled 666 patients, and POETYK PSO-2, which enrolled 1,020 patients, were multi-center, randomized, double-blind trials that evaluated deucravacitinib (6 mg once daily) compared with placebo and Otezla (30 mg twice daily). POETYK PSO-2 included a randomized withdrawal and retreatment period after Week 24.
The co-primary endpoints of both POETYK PSO-1 and POETYK PSO-2 were the percentage of patients who achieved Psoriasis Area and Severity Index (PASI) 75 response and those who achieved static Physician’s Global Assessment (sPGA) score of 0 or 1 at Week 16 versus placebo. Key secondary endpoints of the trials included the percentage of patients who achieved PASI 75 and sPGA 0/1 compared to Otezla at Week 16 and other measures.
About Psoriasis
Psoriasis is a widely prevalent, chronic, systemic immune-mediated disease that substantially impairs patients’ physical health, quality of life and work productivity. Psoriasis is a serious global problem, with at least 100 million people worldwide impacted by some form of the disease, including around 14 million people in Europe and approximately 7.5 million people in the United States. Up to 90 percent of patients with psoriasis have psoriasis vulgaris, or plaque psoriasis, which is characterized by distinct round or oval plaques typically covered by silvery-white scales. Despite the availability of effective systemic therapy, many patients with moderate to severe psoriasis remain undertreated or even untreated and are dissatisfied with current treatments. People with psoriasis report an impact on their emotional well-being, straining both personal and professional relationships and causing a reduced quality of life. Psoriasis is associated with multiple comorbidities that may impact patients’ well-being, including psoriatic arthritis, cardiovascular disease, metabolic syndrome, obesity, diabetes, inflammatory bowel disease and depression.
About Bristol Myers Squibb
Bristol Myers Squibb is a global biopharmaceutical company whose mission is to discover, develop and deliver innovative medicines that help patients prevail over serious diseases. For more information about Bristol Myers Squibb, visit us at BMS.com or follow us on LinkedIn, Twitter, YouTube, Facebook and Instagram.
Celgene and Juno Therapeutics are wholly owned subsidiaries of Bristol-Myers Squibb Company. In certain countries outside the U.S., due to local laws, Celgene and Juno Therapeutics are referred to as, Celgene, a Bristol Myers Squibb company and Juno Therapeutics, a Bristol Myers Squibb company.
Otezla® (apremilast) is a registered trademark of Amgen Inc.
PATENT
WO-2021129467
Novel crystalline polymorphic forms (CSI and CSII) of deucravacitinib (also known as BMS-986165), useful a tyrosine kinase 2 pseudokinase domain (TYK2) inhibitor for treating psoriasis, systemic lupus erythematosus, and Crohn’s disease.Tyrosine kinase 2 (TYK2) is an intracellular signal transduction kinase that can mediate interleukin-23 (IL-23), interleukin-12 (IL-12) and type I interferon (IFN) These cytokines are involved in inflammation and immune response.
BMS-986165 is the first and only new oral selective TYK2 inhibitor, clinically used to treat autoimmune and autoinflammatory diseases (such as psoriasis, psoriatic arthritis, lupus and inflammatory bowel disease, Crowe Graciousness, etc.). The results of a phase III clinical study of the drug announced in November 2020 showed that BMS-986165 has shown positive clinical effects in the treatment of moderate to severe plaque psoriasis. In addition, BMS-986165 also shows good therapeutic effects in the treatment of systemic lupus erythematosus and Crohn’s disease.
The chemical name of BMS-986165 is 6-(cyclopropaneamido)-4-((2-methoxy-3-(1-methyl-1H-1,2,4-triazol-3-yl)benzene (Yl)amino)-N-(methyl-D3)pyridazine-3-carboxamide, the structural formula is shown below, and is hereinafter referred to as “compound I”:
The crystal form is a solid in which the compound molecules are arranged in a three-dimensional order in the microstructure to form a crystal lattice. The phenomenon of drug polymorphism refers to the existence of two or more different crystal forms of the drug. Because of different physical and chemical properties, different crystal forms of the drug may have different dissolution and absorption in the body, which in turn affects the clinical efficacy and safety of the drug to a certain extent. Especially for poorly soluble solid drugs, the crystal form will have a greater impact. Therefore, drug crystal form must be an important content of drug research and also an important content of drug quality control.
WO2018183656A1 discloses compound I crystal form A (hereinafter referred to as “crystal form A”) and a preparation method thereof. The crystalline form A disclosed in WO2018183656A1 is the only known free crystalline form of Compound I. The inventor of the present application repeated the preparation method disclosed in WO2018183656A1 to obtain and characterize the crystal form A. The results show that the crystal form A has poor compressibility and high adhesion. Therefore, there is still a need in the art to develop a compound I crystalline form with good stability, good compressibility, and low adhesion for the development of drugs containing compound I.
The inventor of the present application has paid a lot of creative work and unexpectedly discovered the crystalline form CSI of compound I and the crystalline form CSII of compound I provided by the present invention, which have advantages in physical and chemical properties, preparation processing performance and bioavailability, for example, There are advantages in at least one aspect of melting point, solubility, hygroscopicity, purification, stability, adhesion, compressibility, fluidity, dissolution in vivo and in vitro, and bioavailability, especially good physical and chemical stability and mechanical stability It has good performance, good compressibility, and low adhesion, which solves the problems existing in the prior art, and is of great significance to the development of drugs containing compound I.
PATENT
US9505748 , a family member of WO2014074661 .
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2014074661
Preparation 1
Step l Int1
Step 2 Int2 Step 3 Int3 Step 4 Int4
Example 52
Step 1
[00219] To a solution of 2-methoxy-3-(l-methyl-lH-l ,2,4-triazol-3-yl)aniline (10.26 g, 50.2 mmol) and Int8 (10.5 g, 50.2 mmol) in THF (120 mL) was added lithium bis(trimethylsilyl)amide (LiHMDS, 1M in THF, 151 mL, 151 mmol) in a dropwise manner using a pressure equalized addition funnel. The reaction was run for 10 minutes after the completion of the addition and then quenched with HCl (1M aq., 126 mL, 126 mmol). The reaction was concentrated on a rotary evaporator until the majority of the THF was removed and a precipitate prevailed throughout the vessel. Water (-500 mL) was then added and the slurry sonicated for 5 minutes and stirred for 15 min. The solid was filtered off, rinsing with water and then air dried for 30 minutes. The powder was collected and dissolved in dichloromethane. The organic layer was washed with water and brine and then dried over sodium sulfate, filtered and concentrated to provide the product (12.5 g, 66% yield) (carried on as is). 1H NMR (400MHz, DMSO-d6) δ 11.11 (s, 1H), 9.36 (s, 1H), 8.56 (s, 1H), 7.72 (dd, J=7.8, 1.6 Hz, 1H), 7.60 (dd, J=7.9, 1.5 Hz, 1H), 7.29 (t, J=7.9 Hz, 1H), 7.19 (s, 1H), 3.95 (s, 3H), 3.72 (s, 3H). LC retention time 1.18 [E]. MS(E+) m/z: 377 (MH+).
Step 2
[00220] Intl3 (2.32 g, 6.16 mmol) and cyclopropanecarboxamide (1.048 g, 12.31 mmol) were dissolved in dioxane (62 mL) and Pd2(dba)3 (564 mg, 0.616 mmol), Xantphos (534 mg, 0.924 mmol) and cesium carbonate (4.01 g, 12.3 mmol) were added. The vessel was evacuated three times (backfilling with nitrogen) and then sealed and heated to 130 °C for 140 minutes. The reaction was filtered through CELITE® (eluting with ethyl acetate) and concentrated (on smaller scale this material could then be purified using preparative HPLC). The crude product was adsorbed onto CELITE® using dichloromethane, dried and purified using automated chromatography (100% EtOAc) to provide example 52 (1.22 g, 46% yield). 1H NMR (500MHz, chloroform-d) δ 10.99 (s, 1H), 8.63 (s, 1H), 8.18 (s, 1H), 8.10 (d, J=0.5 Hz, 2H), 7.81 (dd, J=7.9, 1.7 Hz, 1H), 7.51 (dd, J=7.9, 1.4 Hz, 1H), 7.33 – 7.20 (m, 7H), 4.01 (d, J=0.3 Hz, 3H), 3.82 (s, 3H), 1.73 -1.60 (m, 1H), 1.16 – 1.06 (m, 2H), 0.97 – 0.84 (m, 2H). LC retention time 6.84 [N]. MS(E+) m/z: 426 (MH+).
Example 53
[00221] To a homogeneous solution of Example 52 (50 mg, 0.12 mmol) in dichloromethane (3 mL) was added HCI (1M aq., 0.13 mL, 0.13 mmol) resulting in the solution turning yellow. The homogenous solution was concentrated down and then re-concentrated from dichloromethane twice to remove residual water, resulting in a white powder. The powder was suspended in dichloromethane and sonicated for 15 minutes, the powder was then collected via filtration, rinsing with dichloromethane to provide the corresponding HCI salt (38 mg, 70% yield). 1H NMR (500MHz, chloroform-d) δ 12.02 (s, 1H), 8.35 (s, 1H), 8.16 (s, 1H), 8.01 (dd, J=7.9, 1.5 Hz, 1H), 7.57 (br. s., 1H), 7.52 -7.46 (m, 1H), 7.36 (t, J=7.9 Hz, 1H), 4.03 (s, 3H), 3.83 (s, 3H), 2.05 – 1.95 (m, 1H), 1.16 – 1.09 (m, 2H), 1.03 (dd, J=7.4, 3.6 Hz, 2H). LC retention time 0.62 [j]. MS(E+) m/z: 426 (MH+).
[00222] Compare to NMR of parent free base: 1H NMR (500MHz, chloroform-d) δ 10.99 (s, 1H), 8.63 (s, 1H), 8.18 (s, 1H), 8.10 (d, J=0.5 Hz, 2H), 7.81 (dd, J=7.9, 1.7 Hz, 1H), 7.51 (dd, J=7.9, 1.4 Hz, 1H), 7.33 – 7.20 (m, 7H), 4.01 (d, J=0.3 Hz, 3H), 3.82 (s, 3H), 1.73 – 1.60 (m, 1H), 1.16 – 1.06 (m, 2H), 0.97 – 0.84 (m, 2H).
////////////DEUCRAVACITINIB, phase 3, BMS-986165, BMS 986165, psoriasis, systemic lupus erythematosus, Crohn’s disease,
CNC(=O)C1=NN=C(C=C1NC2=CC=CC(=C2OC)C3=NN(C=N3)C)NC(=O)C4CC4

NEW DRUG APPROVALS
one time
$10.00
Benvitimod, Tapinarof, тапинароф , تابيناروف , 他匹那罗 ,
Benvitimod, Tapinarof
- Molecular FormulaC17H18O2
- Average mass254.324 Da
3,5-dihydroxy-4-isopropyl-trans-stilbene
Launched – 2019 CHINA, Psoriasis, Tianji Pharma
тапинароф [Russian] [INN]WBI-1001
DMVT-505
GSK-2894512
RVT-505
WB-1001
WBI-1001
84HW7D0V04 (UNII code)
Benvitimod is in phase III clinical trials, Dermavant Sciences for the treatment of atopic dermatitis and psoriasis.
The compound was co-developed by Welichem Biotech and Stiefel Laboratories (subsidiary of GSK). However, Shenzhen Celestial Pharmaceuticals acquired the developement rights in China, Taiwan, Macao and Hong Kong.
Benvitimod (also known as Tapinarof or 3,5-dihydroxy-4-isopropyl-trans-stilbene) is a bacterial stilbenoid produced in Photorhabdus bacterial symbionts of Heterorhabditis nematodes.It is a product of an alternative ketosynthase-directed stilbenoids biosynthesis pathway. It is derived from the condensation of two β-ketoacyl thioesters. It is produced by the Photorhabdus luminescens bacterial symbiont species of the entomopathogenic nematode, Heterorhabditis megidis.
Benvitimod (also known as tapinarof or 3,5-dihydroxy-4-isopropyl-trans-stilbene) is a bacterial stilbenoid produced in Photorhabdus bacterial symbionts of Heterorhabditis nematodes. It is a product of an alternative ketosynthase-directed stilbenoids biosynthesis pathway. It is derived from the condensation of two β-ketoacyl thioesters .[1] It is produced by the Photorhabdus luminescens bacterial symbiont species of the entomopathogenic nematode, Heterorhabditis megidis. Experiments with infected larvae of Galleria mellonella, the wax moth, support the hypothesis that the compound has antibiotic properties that help minimize competition from other microorganisms and prevents the putrefaction of the nematode-infected insect cadaver.[2]
Tapinarof is a non-steroidal anti-inflammatory drug originated by Welichem Biotech. Dermavant Sciences is developing the product outside China in phase III clinical trials for the treatment of plaque psoriasis. The company is also conducting phase II clinical trials for the treatment of atopic dermatitis. Phase II studies had also been conducted by Welichem Biotech and Stiefel (subsidiary of GlaxoSmithKline) for these indications.
Tapinarof was originated at Welichem Biotech, from which Tianji Pharma and Shenzen Celestial Pharmaceuticals obtained rights to the product in the Greater China region in 2005. In 2012, Welichem licensed development and commercialization rights in all other regions to Stiefel. In 2013, Welichem entered into an asset purchase agreement to regain Greater China rights to the product from Tianji Pharma and Celestial; however, this agreement was terminated in 2014. In 2018, Stiefel transferred its product license to Dermavant Sciences.
_Poinar,_1975.jpg)
Entomopathogenic nematodesemerging from a wax moth cadaver
//////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
Medical research
Benvitimod is being studied in clinical trials for the treatment of plaque psoriasis.[3]
PATENTS

1. US2003171429A1.
2. US2005059733A1.

Reference:1. CN103265412A.


Patent
https://patents.google.com/patent/CN103992212A/en
phenalkenyl Maude (Benvitimod) is a new generation of anti-inflammatory drugs, are useful for treating a variety of major autoimmune diseases, such as psoriasis, eczema, hair and more concentrated colitis allergic diseases.Phenalkenyl Maud stilbene compound, comprising cis and trans isomers, the trans alkenyl benzene Maude has a strong physiological activity, stability and physical and chemical properties, and cis alkenyl benzene Modesto predominantly trans phenalkenyl Maud byproducts during synthesis, conventional methods such as benzene alkenyl Maude Wittig reaction of cis-isomer impurity is inevitable.

[0004] benzyl trans-alkenyl Maude as main impurities in the synthesis, whether a drug is detected, or monitored during the reaction, the synthesis and analysis methods established cis alkenyl benzene Maude has very important significance.Phenalkenyl Maud conventional synthetic methods the impurity content is very low, and the properties of the cis compound is extremely unstable, easily converted to trans-structure, the synthetic method according to the preceding, the cis compound difficult to separate. The synthesis method has not been reported before in the literature. Thus, to find a synthesis route of cis-alkenyl benzene Maude critical.
[0005] The synthesis of compounds of cis-stilbene, in the prior art, there have been many reports, however, the prior art method of synthesizing a reaction product of the cis starting materials and reagents difficult source, the catalyst used is expensive higher costs, operational difficulties, is not conducive to large-scale production, such as:
① Gaukroger K, John A.Hadfield.Novel syntheses of cis and trans isomers ofcombretastatin A-4 [J] .J.0rg.Chemj 2001, (66): 8135-8138, instead of styrene and substituted phenyl bromide boric acid as the raw material, the Suzuki coupling reaction is a palladium catalyst, to give the cis compound, the reaction follows the formula:

Yield and selectivity of the process the structure is good, but the reaction is difficult source of raw materials, catalyst more expensive, limiting the use of this method.
[0006] ② Felix N, Ngassaj Erick A, Lindsey, Brandon Ej Haines.The first Cu- and
amine-free Sonogashira-type cross-coupling in the C_6 -alkynylation of protected
2, -deoxyadenosine [J] .Tetrahedron Letters, 2009, (65): 4085-4091, with a substituted phenethyl m
Alkynyl easily catalyst Pd / CaC03, Fe2 (CO) 9, Pd (OAc) 2 and the like produce cis compound to catalytic reduction. The reaction follows the formula:

Advantage of this method is stereospecific reduction of alkynes in the catalyst, to overcome the phenomenon of cis-trans isomerization of the Wittig reaction, but the reaction requires at _78 ° C, is not conducive to the operation, and the reagent sources difficult, expensive than high cost increase is not conducive to mass production.
[0007] ③ Belluci G, Chiappe C, Moro G L0.Crown ether catalyzed stereospecificsynthesis of Z_and E-stilbenes by Wittig reaction in a solid-liquid two-phasessystem [J] .Tetrahedron Letters, 1996, (37): 4225-4228 using Pd (PPh3) 4 as catalyst, an organic zinc reagent with a halide compound of cis-coupling reaction formula as follows:

The advantage of this method is that selective, high yield to give cis; deficiency is difficult to handle, the catalyst is expensive.
[0008] ④ new Wang, Zhangxue Jing, Zhou Yue, Zouyong Shun, trans-3,4 ‘, 5-trihydroxy-stilbene China Pharmaceutical Synthesis, 2005, 14 (4);. 204-208, reported that the trans compound of formula was dissolved in DMSO solution at a concentration dubbed, ultraviolet irradiation was reacted at 365nm, converted into cis compounds, see the following reaction formula:

However, the concentration of the solution preparation method, the reaction time is more stringent requirements.

The synthesis of cis-alkenyl benzene Maude application embodiments Example 1 A synthesis of cis-alkenyl Maude benzene and benzene-cis-ene prepared Maude, the reaction was carried out according to the following scheme:

Specific preparation process steps performed in the following order:
(O methylation reaction
The 195.12g (Imol) of 3, 5-hydroxy-4-isopropyl benzoic acid, 414.57g (3mol) in DMF was added 5000ml anhydrous potassium carbonate, mixing, stirred at room temperature, then cooled in an ice-salt bath next, slowly added dropwise 425.85g (3mol) of iodomethane, warmed to room temperature after the addition was complete, the reaction 2h, after completion of the reaction was stirred with water, extracted with ethyl acetate, and concentrated to give 3,5-dimethoxy-4- isopropyl benzoate; yield 93%, purity of 99%.
[0033] (2) a reduction reaction
3000ml tetrahydrofuran and 240g (Imol) 3,5-dimethoxy-4-isopropyl benzoate, 151.40g (4mol) mixing at room temperature sodium borohydride was stirred and heated to reflux was slowly added dropwise 400ml methanol, reaction 4h, was added 3L of water was stirred, extracted with ethyl acetate, washed with water, the solvent was removed by rotary evaporation to give a white solid, to give 3,5-dimethoxy-4-isopropylbenzene methanol; 96% yield purity was 99%.
[0034] (3) the oxidation reaction
The 212g (ImoI) of 3,5-dimethoxy-4-isopropylbenzene methanol, DMSO 800ml and 500ml of acetic anhydride were mixed and stirred at rt After 2h, stirred with water, extracted with ethyl acetate, washed with water, dried , and concentrated to give 3,5-dimethoxy-4-isopropyl-benzaldehyde; 94% yield, 99% purity.
[0035] (4) a condensation reaction
The mixture was 209.18g (lmol) of 3,5-dimethoxy-4-isopropyl-benzoic awake and 136.15g (Imol) phenylacetic acid was added 5000ml of acetic anhydride, stirred to dissolve, sodium acetate was added 246.09g , heating to 135 ° C, the reaction after 6h, cooled to room temperature after adjusting the dilute acid 2 was added, extracted with ethyl acetate, the pH was concentrated, added saturated sodium bicarbonate solution adjusted to pH 7, stirred 2h, and extracted with dichloromethane , adding dilute aqueous hydrochloric acid pH 2, the yellow solid was filtered, to obtain 3,5-dimethoxy-4-isopropyl-stilbene acid; 96% yield, 80% purity.
[0036] (5) decarboxylation reaction
The 327g (Imol) of 3,5-dimethoxy-4-isopropyl-stilbene acid and 384g (6mol) of copper powder were added to 5000ml of quinoline, 180 ° C reaction 3h, cooled to room temperature ethyl acetate was added with stirring, filtered, and the filtrate was washed with dilute hydrochloric acid to the aqueous layer was colorless and the aqueous phase was extracted with ethyl acetate inverted, the organic layers were combined, washed with water and saturated brine until neutral, i.e., spin-dried to give 3,5 – dimethoxy-4-isopropyl-stilbene; 92% yield, 77% purity.
[0037] (6) Demethylation
The 282.32g (Imol) of 3,5-dimethoxy-4-isopropyl-stilbene 4000ml toluene was placed in an ice bath and stirring, was cooled to 0 ° C, and dissolved slowly added 605.9g (5mol after) in N, N- dimethylaniline, was added 666.7g (5mol) of anhydrous aluminum chloride. after stirring for 0.5h, warmed to room temperature, the reaction was heated to 100 ° C 2h, cooled to 60 ° C , hot toluene layer was separated, diluted hydrochloric acid was added to the aqueous phase with stirring to adjust the PH value of 2, extracted with ethyl acetate, washed with water, and concentrated to give the cis-alkenyl benzene Modesto; crude yield 95%, purity 74 %.After separation by column chromatography using 300-400 mesh silica gel, benzene-cis-ene was isolated Maude pure, 68% yield, 98.5% purity. The resulting cis-alkenyl benzene Maud NMR shown in Figure 1, NMR data are as follows:
1HNMR (CDCl3, 500 Hz, δ: ppm), 7.255 (m, 5H), 6.558 (d, 1H), 6.402 (d, 1H), 6.218 (s, 2H), 4.872 (s, 2H), 3.423 (m , 1H), 1.359 (q, 6H). Coupling constants / = 12.
[0038] trans-alkenyl benzene Maud NMR shown in Figure 2, the following NMR data:
1HNMR (CDCl3, 500 Hz, δ: ppm), 7.477 (d, 2H), 7.360 (t, 2H), 6.969 (q, 2H), 6.501 (s, 1H), 4.722 (s, 2H), 3.486 (m , 1H), 1.380 (t, 6H). Coupling constants / = 16.
[0039] HPLC conditions a cis alkenyl benzene Maude pure product: column was Nucleosil 5 C18; column temperature was 20 ° C; detection wavelength 318nm; mobile phase consisting of 50:50 by volume of acetonitrile and water; flow rate It was 0.6mL / min, injection volume of 5 μ L; cis phenalkenyl Maude 18.423min retention time of a peak in an amount of 96.39%, see Figure 3. Trans phenalkenyl Maude 17.630min retention time of a peak, the content was 99.8%, see Figure 4.After mixing the two, trans-alkenyl benzene Maude 17.664min retention time of the peak, cis-alkenyl benzene Maude 18.458min retention time of the peak, see Figure 5.
PATENT
https://patents.google.com/patent/CN103172497A/en

phenalkenyl Maude is a natural product, a metabolite as to be symbionts.Phenalkenyl Maud Escherichia coli, Staphylococcus aureus has a very significant inhibitory effect, in addition, there is a styrenic Maude suppression of inflammation and its reactive derivative with immunomodulating activity. Alkenyl benzene Modesto topical ointment as an active ingredient, as a class of drugs has been completed two clinical treatment of psoriasis and eczema, the results of ongoing clinical phase III clinical studies, it has been shown to be completed in both psoriasis and eczema clearly effect, together with a styrenic Maude is a non-hormonal natural small molecule compounds, can be prepared synthetically prepared, therefore, it exhibits good market prospect.
[0004] a styrenic Maude initial synthesis route is as follows:
[0005]

[0006] The reaction conditions for each step: 1) isopropanol, 80% sulfuric acid, 60 ° C, 65% .2) sodium borohydride, boron trifluoride, tetrahydrofuran, 0 ° C, 90% .3). of thionyl chloride, heated under reflux, 85% .4). triethyl phosphate, 120 ° C, 80% .5). benzaldehyde, sodium hydride, 85% .6) pyridine hydrochloride, 190 ° C, 60 %.
[0007] The chemical synthesis route, although ultimately obtained a styrenic Maude, but the overall yield is low, part of the reaction step is not suitable for industrial production, due to process conditions result in the synthesis of certain byproducts produced is difficult to remove impurities, difficult to achieve the quality standard APIs.
Preparation of 4-isopropyl-dimethoxy-benzoic acid [0011] 1,3,5_
[0012] 1000 l reactor 200 liters of 80% sulfuric acid formulation (V / V), the temperature was lowered to room temperature, put 80 kg 3,5_-dimethoxybenzoate ,, stirring gradually warmed to 60 ° C, in was added dropwise within 25 kg of isopropanol I hour, the reaction was complete after 5 hours, 500 liters of hot water, filtered, the filter cake was washed with a small amount of hot water I th, crushed cake was removed and dried. The dried powder was recrystallized from toluene, the product was filtered to give 78 kg `, yield 86%. Preparation 2,3,5_ dimethoxy-4-isopropylbenzene methanol
[0013] 1000 l reactor was added 50 kg 3,5_ _4_ isopropyl dimethoxy benzoic acid, 24 kg of potassium borohydride, 400 l of THF, at room temperature was slowly added dropwise 65 kg BF3.Et2O was stirred 12 hours, the reaction was complete, pure water was added dropwise to destroy excess BF3, filtered, concentrated to dryness, methanol – water to give an off-white recrystallized 40.3 kg, yield 90.1%.
[0014] Preparation of 3,3,5-_ ■ methoxy _4- isopropyl group gas section
[0015] 1000 l autoclave, 100 kg of 3,5-dimethoxy-4-isopropylbenzene methanol, 220 l of DMF, 0 ° C and added dropwise with stirring and 50 l of thionyl chloride, 24 hours after the reaction was complete, 300 liters of water and 300 liters of ethyl acetate, the aqueous phase was stirred layered discharged, and then washed with 200 liters of water was added 3 times, until complete removal of DMF, was added concentrated crystallized from petroleum ether to give 98 kg of white solid was filtered and dried a yield of 91%.
Preparation of methyl-dimethoxy-4-isopropylbenzene of diethyl [0016] 4,3,5_
[0017] 500 l autoclave, 98 kg 3,5_ _4_ isopropyl dimethoxy benzyl chloride and 120 l of triethyl phosphite, the reaction at 120 ° C 5h, fear distilled off under reduced pressure, the collection 145-155 ° C / 4mmHg fear minutes, cured at room temperature to give a colorless light solid was 118 kg, yield 81.6%.
, 3- [0018] 5, E-1 _ ■ methoxy-2-isopropyl-5- (2-phenylethyl lean-yl) – benzene
[0019] 500 l autoclave, 33 kg 3,5_-dimethoxy-4-isopropylbenzene acid diethyl ester, 10.8 kg of benzaldehyde, and 120 l of tetrahydrofuran, at 40 ° C, and nitrogen with stirring, was added dropwise a solution of 11.8 kg potassium tert-butoxide in 50 liters of tetrahydrofuran, the temperature dropping control not to exceed 50 ° C. after the dropwise addition stirring was continued for I h, the reaction was complete, 150 liters of ethyl acetate and extracted , washed twice with 150 liters of water, 100 l I washed with brine, and the organic phase was dried and concentrated, methanol – water (I: D as a white crystalline solid 25.3 kg, yield 91%.
[0020] 6> 1, 3 ~ _ ■ Light-2-isopropyl-5- (2-phenylethyl lean-yl) – benzene (I), (De Dae dilute benzene)
[0021] 100 l autoclave, 10 kg 1,3_-dimethoxy-2-isopropyl-5- (2-styryl) benzene _ pyridine hydrochloride and 25 kg nitrogen atmosphere was heated to 180 -190 ° C, stirred for 3 hours after the reaction was completed, 20 l HCl (2N) cooling to 100 ° C, and 20 liters of ethyl acetate the product was extracted, dried and concentrated to give the product 7.3 kg, 83% yield.
[0022] The method for purifying:
[0023] 100 l added to the reaction vessel 15.5 kg of crude product and 39 liters of toluene, heated to the solid all dissolved completely, filtered hot and left to crystallize, after crystallization, filtration, the crystals with cold toluene 10 washed liter at 60 ° C, protected from light vacuo dried for 24 hours, to obtain 14 kg of white needle crystals, yield 90%.
CLIP
https://www.eosmedchem.com/article/237.html
Design new synthesis of Route of Benvitimod
Benvitimod 79338-84-4 intermediate: 1999-10-5
Benvitimod 79338-84-4 intermediate: 2150-37-0
Benvitimod 79338-84-4 intermediate: 344396-17-4
Benvitimod 79338-84-4 intermediate: 344396-18-5
Benvitimod 79338-84-4 intermediate: 344396-19-6
Benvitimod 79338-84-4 intermediate: 1080-32-6
Benvitimod 79338-84-4 intermediate: 678986-73-7
Benvitimod 79338-84-4 intermediate: 55703-81-6
Benvitimod 79338-84-4 intermediate: 1190122-19-0
Benvitimod 79338-84-4 intermediate: 443982-76-1
Benvitimod 79338-84-4 intermediate: 100-52-72.ROS-Benvitimod
(1)
(2)
Name: Benvitimod
CAS#: 79338-84-4
Chemical Formula: C17H18O2
Exact Mass: 254.1307
Molecular Weight: 254.329
Elemental Analysis: C, 80.28; H, 7.13; O, 12.58
References
- ^ Joyce SA; Brachmann AO; Glazer I; Lango L; Schwär G; Clarke DJ; Bode HB (2008). “Bacterial biosynthesis of a multipotent stilbene”. Angew Chem Int Ed Engl. 47 (10): 1942–5. doi:10.1002/anie.200705148. PMID 18236486.
- ^ Hu, K; Webster, JM (2000). “Antibiotic production in relation to bacterial growth and nematode development in Photorhabdus–Heterorhabditis infected Galleria mellonella larvae”. FEMS Microbiology Letters. 189 (2): 219–23. doi:10.1111/j.1574-6968.2000.tb09234.x. PMID 10930742.
- ^ “New Topical for Mild to Moderate Psoriasis in the Works”. Medscape. March 5, 2017.
- https://onlinelibrary.wiley.com/action/downloadSupplement?doi=10.1002%2Fanie.201814016&file=anie201814016-sup-0001-misc_information.pdf
///Benvitimod, Tapinarof, WBI-1001, тапинароф , تابيناروف , 他匹那罗 , Welichem Biotech, Stiefel Laboratories, Shenzhen Celestial Pharmaceuticals,CHINA 2019 , Psoriasis, Tianji Pharma, Dermavant Sciences, PHASE 3, fda 2022, approvals 2022, vtama, tapinarof
update….
5/23/2022 fda approved, To treat plaque psoriasis, vtama, tapinarof
 |
|
| Names | |
|---|---|
| Preferred IUPAC name
5-[(E)-2-Phenylethen-1-yl]-2-(propan-2-yl)benzene-1,3-diol
|
|
| Other names
|
|
| Identifiers | |
|
3D model (JSmol)
|
|
| ChemSpider | |
|
PubChem CID
|
|
| UNII | |
| Properties | |
| C17H18O2 | |
| Molar mass | 254.329 g·mol−1 |
|
Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa).
|
|
Benvitimod (also known as tapinarof or 3,5-dihydroxy-4-isopropyl-trans-stilbene) is a bacterial stilbenoid produced in Photorhabdus bacterial symbionts of Heterorhabditis nematodes. It is a product of an alternative ketosynthase-directed stilbenoid biosynthesis pathway. It is derived from the condensation of two β-ketoacyl thioesters.[1] It is produced by the Photorhabdus luminescens bacterial symbiont species of the entomopathogenic nematode, Heterorhabditis megidis. Experiments with infected larvae of Galleria mellonella, the wax moth, support the hypothesis that the compound has antibiotic properties that help minimize competition from other microorganisms and prevents the putrefaction of the nematode-infected insect cadaver.[2]
Medical research
Benvitimod is being studied in clinical trials for the treatment of plaque psoriasis.[3]
See also
- Pinosylvin, a molecule produced in pines that does not bear the isopropyl alkylation.
References
- ^ Joyce SA; Brachmann AO; Glazer I; Lango L; Schwär G; Clarke DJ; Bode HB (2008). “Bacterial biosynthesis of a multipotent stilbene”. Angew Chem Int Ed Engl. 47 (10): 1942–5. CiteSeerX 10.1.1.603.247. doi:10.1002/anie.200705148. PMID 18236486.
- ^ Hu, K; Webster, JM (2000). “Antibiotic production in relation to bacterial growth and nematode development in Photorhabdus–Heterorhabditis infected Galleria mellonella larvae”. FEMS Microbiology Letters. 189 (2): 219–23. doi:10.1111/j.1574-6968.2000.tb09234.x. PMID 10930742.
- ^ “New Topical for Mild to Moderate Psoriasis in the Works”. Medscape. March 5, 2017.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}