
Home » Posts tagged 'FDA 2022'
Tag Archives: FDA 2022
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Adagrasib
Adagrasib
| Formula | C32H35ClFN7O2 |
|---|---|
| cas | 2326521-71-3 |
| Mol weight | 604.1174 |
| Antineoplastic | |
| Disease | Non-small cell lung cancer |
|---|
| 2022/12/12 |
FDA APPROVED, KRAZATI (Mirati Therapeutics)
- MRTX-849
- MRTX849
- KRAS G12C inhibitor MRTX849
Adagrasib, sold under the brand name Krazati, is an anticancer medication used to treat non-small cell lung cancer.[1][2] Adagrasib is an inhibitor of the RAS GTPase family.[1] It is taken by mouth.[1] It is being developed by Mirati Therapeutics.[1][3]
The most common adverse reactions include diarrhea, nausea, fatigue, vomiting, musculoskeletal pain, hepatotoxicity, renal impairment, dyspnea, edema, decreased appetite, cough, pneumonia, dizziness, constipation, abdominal pain, and QTc interval prolongation.[2] The most common laboratory abnormalities include decreased lymphocytes, increased aspartate aminotransferase, decreased sodium, decreased hemoglobin, increased creatinine, decreased albumin, increased alanine aminotransferase, increased lipase, decreased platelets, decreased magnesium, and decreased potassium.[2]
It was approved for medical use in the United States in December 2022.[1][3]
Synthesis Reference
Fell, Jay B et al. “Identification of the Clinical Development Candidate MRTX849, a Covalent KRASG12C Inhibitor for the Treatment of Cancer.” Journal of medicinal chemistry vol. 63,13 (2020): 6679-6693. doi:10.1021/acs.jmedchem.9b02052
Journal of Medicinal Chemistry (2020), 63(13), 6679-6693
PATENT
WO2020101736 https://patents.google.com/patent/WO2020101736A1/en
EXAMPLE 7

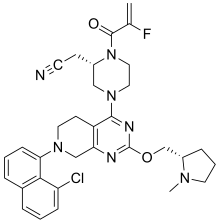
2-[(2S)-4-[7-(8-chloro-1-naphthyl)-2-[[(2S)-1-methylpyrrolidin-2-yl]methoxy]-6,8-dihydro-5H- pyrido[3,4-d]pyrimidin-4-yl]-1-(2-fluoroprop-2-enoyl)piperazin-2-yl]acetonitrile
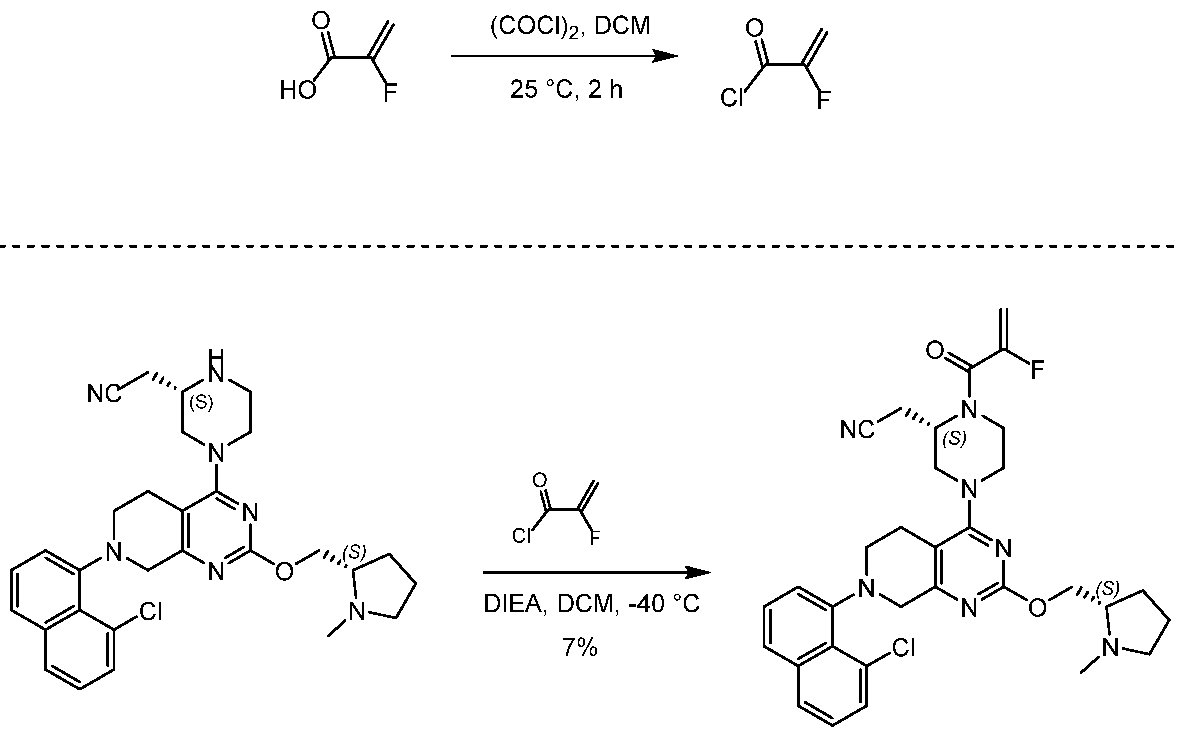
[0432] 2-fluoroprop-2-enoyl chloride. To a solution of 2-fluoroprop-2-enoic acid (400 mg, 4.44 mmol, 1 eq) in DCM (4 mL) was added (COCl)2 (846 mg, 6.66 mmol, 583 µL, 1.5 eq) and DMF (32.5 mg, 444 umol, 34.2 µL, 0.1 eq). The mixture was stirred at 25 °C for 2 hrs. The reaction mixture was concentrated under reduced pressure to remove a part of solvent and give a residue in DCM. Compound 2-fluoroprop-2-enoyl chloride (400 mg, crude) was obtained as a yellow liquid and used into the next step without further purification. [0433] Step A: 2-[(2S)-4-[7-(8-chloro-1-naphthyl)-2-[[(2S)-1- methylpyrrolidin-2-yl]methoxy]- 6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-4-yl]-1-(2-fluoroprop-2-enoyl)piperazin-2- yl]acetonitrile. To a solution of 2-[(2S)-4-[7-(8-chloro-1-naphthyl)-2-[[(2S)- 1-methylpyrrolidin- 2-yl]methoxy]-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-4-yl]piperazin-2-yl]acetonitrile (300 mg, 528 umol, 1 eq, HCl) in DCM (5 mL) was added DIEA (1.73 g, 13.4 mmol, 2.33 mL, 25.4 eq) and 2-fluoroprop-2-enoyl chloride (286 mg, 2.64 mmol, 5 eq) in DCM (5 mL). The mixture was stirred at 0 °C for 1 hour. The reaction mixture was concentrated under reduced pressure to give a residue. The residue was purified by column chromatography (Al2O3, Dichloromethane/Methanol = 10/1 to 10/1). The residue was purified by prep-HPLC (column: Gemini 150 * 25 5u; mobile phase: [water (0.05% ammonia hydroxide v / v) – ACN]; B%: 55% – 85%, 12min). The residue was purified by prep-HPLC (column: Phenomenex Synergi C18 150 * 30mm * 4um; mobile phase: [water (0.225% FA) – ACN]; B%: 20% – 50%, 10.5min). The residue was concentrated under reduced pressure to remove ACN, and then lyophlization. Title compound 2-[(2S)-4-[7-(8-chloro- 1-naphthyl)-2-[[(2S)-1- methylpyrrolidin-2-yl]methoxy]-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin- 4-yl]-1-(2-fluoroprop-2-enoyl)piperazin-2-yl]acetonitrile (EXAMPLE 7, 24.1 mg, 36.7 umol, 7% yield, 99.1% purity, FA) was obtained as a brown solid. [0434] SFC condition: “AD – 3S_3_5_40_3ML Column: Chiralpak AD – 3 100 × 4.6mm I.D., 3um Mobile phase: methanol (0.05% DEA) in CO2 from 5% to 40% Flow rate: 3mL/min Wavelength: 220nm”. [0435] 1H NMR (400 MHz, Acetic) d = 7.82 (d, J = 8.0 Hz, 1H), 7.69 (d, J = 8.0 Hz, 1H), 7.56 (d, J = 7.6 Hz, 1H), 7.49 (t, J = 7.6 Hz, 1H), 7.41 – 7.30 (m, 2H), 5.58 – 5.25 (m, 2H), 5.17 – 4.59 (m, 4H), 4.57 – 4.28 (m, 3H), 4.24 – 3.78 (m, 4H), 3.67 – 3.13 (m, 7H), 3.08 (br d, J = 2.4 Hz, 3H), 2.98 (br d, J = 6.4 Hz, 1H), 2.83 – 2.61 (m, 1H), 2.45 – 2.29 (m, 1H), 2.24 – 2.08 (m, 3H).
PATENT
US20190144444 https://patents.google.com/patent/US20190144444A1/en
////////

AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
Adagrasib (MRTX849) is an oral, small-molecule KRAS inhibitor developed by Mirati Therapeutics. KRAS mutations are highly common in cancer and account for approximately 85% of all RAS family mutations.5 However, the development of KRAS inhibitors has been challenging due to their high affinity for guanosine triphosphate (GTP) and guanosine diphosphate (GDP), as well as the lack of a clear binding pocket.1 Adagrasib targets KRASG12C, one of the most common KRAS mutations, at the cysteine 12 residue and inhibits KRAS-dependent signalling.2 In a phase I/IB clinical study that included patients with KRASG12C-mutated advanced solid tumors (NCT03785249), adagrasib exhibited anti-tumor activity. The phase II of the same study showed that in patients with KRASG12C-mutated non-small-cell lung cancer (NSCLC), adagrasib was efficient without new safety signals.2,3,6
In February 2022, the FDA accepted a new drug application (NDA) for adagrasib for the treatment of patients with previously treated KRASG12C–positive NSCLC.7 In December 2022, the FDA granted accelerated approval to adagrasib for the treatment of KRASG12C-mutated locally advanced or metastatic NSCLC who have received at least one prior systemic therapy.8,9 Adagrasib joins sotorasib as another KRASG12C inhibitor approved by the FDA.4
Medical uses
Adagrasib is indicated for the treatment of adults with KRAS G12C-mutated locally advanced or metastatic non-small cell lung cancer (NSCLC), as determined by an FDA approved test, who have received at least one prior systemic therapy.[1][2][4]
History
Approval by the US Food and Drug Administration (FDA) was based on KRYSTAL-1, a multicenter, single-arm, open-label clinical trial (NCT03785249) which included participants with locally advanced or metastatic non-small cell lung cancer with KRAS G12C mutations.[2] Efficacy was evaluated in 112 participants whose disease has progressed on or after platinum-based chemotherapy and an immune checkpoint inhibitor, given either concurrently or sequentially.[2]
The FDA granted the application for adagrasib fast-track, breakthrough therapy, and orphan drug designations.[2]
Research
It is undergoing clinical trials.[5][6][7][8][9][10]
References
- ^ Jump up to:a b c d e f g https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/216340s000lbl.pdf
- ^ Jump up to:a b c d e f g h “FDA grants accelerated approval to adagrasib for KRAS G12C-mutated NSC”. U.S. Food and Drug Administration (FDA). 12 December 2022. Retrieved 14 December 2022.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ Jump up to:a b “Mirati Therapeutics Announces U.S. FDA Accelerated Approval of Krazati (adagrasib) as a Targeted Treatment Option for Patients with Locally Advanced or Metastatic Non-Small Cell Lung Cancer (NSCLC) with a KRASG12C Mutation” (Press release). Mirati Therapeutics Inc. 12 December 2022. Retrieved 13 December 2022 – via MultiVu.
- ^ https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2022/216340Orig1s000ltr.pdf This article incorporates text from this source, which is in the public domain.
- ^ Hallin J, Engstrom LD, Hargis L, Calinisan A, Aranda R, Briere DM, et al. (January 2020). “The KRASG12C Inhibitor MRTX849 Provides Insight toward Therapeutic Susceptibility of KRAS-Mutant Cancers in Mouse Models and Patients”. Cancer Discovery. 10 (1): 54–71. doi:10.1158/2159-8290.CD-19-1167. PMC 6954325. PMID 31658955.
- ^ Fell JB, Fischer JP, Baer BR, Blake JF, Bouhana K, Briere DM, et al. (July 2020). “Identification of the Clinical Development Candidate MRTX849, a Covalent KRASG12C Inhibitor for the Treatment of Cancer”. Journal of Medicinal Chemistry. 63 (13): 6679–6693. doi:10.1021/acs.jmedchem.9b02052. PMID 32250617.
- ^ Thein KZ, Biter AB, Hong DS (January 2021). “Therapeutics Targeting Mutant KRAS”. Annual Review of Medicine. 72: 349–364. doi:10.1146/annurev-med-080819-033145. PMID 33138715. S2CID 226242453.
- ^ Christensen JG, Olson P, Briere T, Wiel C, Bergo MO (August 2020). “Targeting Krasg12c -mutant cancer with a mutation-specific inhibitor”. Journal of Internal Medicine. 288 (2): 183–191. doi:10.1111/joim.13057. PMID 32176377.
- ^ Dunnett-Kane V, Nicola P, Blackhall F, Lindsay C (January 2021). “Mechanisms of Resistance to KRASG12C Inhibitors”. Cancers. 13 (1): 151. doi:10.3390/cancers13010151. PMC 7795113. PMID 33466360.
- ^ Jänne PA, Riely GJ, Gadgeel SM, Heist RS, Ou SI, Pacheco JM, et al. (July 2022). “Adagrasib in Non–Small-Cell Lung Cancer Harboring a KRASG12C Mutation”. New England Journal of Medicine. 387 (2): 120–131. doi:10.1056/NEJMoa2204619. PMID 35658005. S2CID 249352736.
External links
- “Adagrasib”. Drug Information Portal. U.S. National Library of Medicine.
- Clinical trial number NCT03785249 for “Phase 1/2 Study of MRTX849 in Patients With Cancer Having a KRAS G12C Mutation KRYSTAL-1” at ClinicalTrials.gov
///////Adagrasib, KRAZATI, FDA 2022, APPROVALS 2022, MRTX-849, MRTX849, Mirati Therapeutics
[H][C@@]1(COC2=NC3=C(CCN(C3)C3=CC=CC4=C3C(Cl)=CC=C4)C(=N2)N2CCN(C(=O)C(F)=C)[C@@]([H])(CC#N)C2)CCCN1C

NEW DRUG APPROVALS
ONE TIME
$10.00
Olutasidenib

Olutasidenib
- FT-2102
- FT2102
C18H15ClN4O2
354.79
CAS1887014-12-1
Rezlidhia (Forma Therapeutics)
SYN Caravella JA, et al. Structure-Based Design and Identification of FT-2102 (Olutasidenib), a Potent Mutant-Selective IDH1 Inhibitor. J Med Chem. 2020 Feb 27;63(4):1612-1623. doi: 10.1021/acs.jmedchem.9b01423. Epub 2020 Feb 12.
FDA 12/1/2022, To treat adults with relapsed or refractory acute myeloid leukemia with a susceptible isocitrate dehydrogenase-1 (IDH1) mutation, Rezlidhia
Olutasidenib, sold under the brand name Rezlidhia, is an anticancer medication used to treat relapsed or refractory acute myeloid leukemia.[1][2] Olutasidenib is an isocitrate dehydrogenase-1 (IDH1) inhibitor.[1] It is taken by mouth.[1]
Olutasidenib was approved for medical use in the United States in December 2022.[1][2][3][4]
Medical uses
Olutasidenib is indicated for the treatment of adults with relapsed or refractory acute myeloid leukemia with a susceptible isocitrate dehydrogenase-1 (IDH1) mutation as detected by an FDA-approved test.[1][2]
Society and culture
Names
Olutasidenib is the international nonproprietary name.[5]
Olutasidenib is an isocitrate dehydrogenase-1 (IDH1) inhibitor indicated for the treatment of patients with relapsed or refractory acute myeloid leukemia with a susceptible IDH1 mutation as detected by an FDA-approved test.
Olutasidenib (FT-2102) is a selective and potent isocitrate dehydrogenase-1 (IDH1) inhibitor approved by the FDA in December 2022.5,6 It is indicated for the treatment of relapsed or refractory acute myeloid leukemia (AML) in patients with a susceptible IDH1 mutation as determined by an FDA-approved test.5 IDH1 mutations are common in different types of cancer, such as gliomas, AML, intrahepatic cholangiocarcinoma, chondrosarcoma, and myelodysplastic syndromes (MDS), and they lead to an increase in 2-hydroxyglutarate (2-HG), a metabolite that participates in tumerogenesis.1,2 Olutasidenib inhibits the mutated IDH1 specifically, and provides a therapeutic benefit in IDH1-mutated cancers.1,5
Other IDH1 inhibitors, such as ivosidenib, have also been approved for the treatment of relapsed or refractory AML.3,4 Olutasidenib is orally bioavailable and capable of penetrating the blood-brain barrier, and is also being evaluated for the treatment of myelodysplastic syndrome (MDS), as well as solid tumors and gliomas (NCT03684811).4
SYN
https://pubs.acs.org/doi/10.1021/acs.jmedchem.9b01423

a Reagents and conditions: (a) DIEA, DMSO, 80−110 °C, 16 h, 67%; (b) (R)-2-methylpropane-2-sulfinamide, CuSO4, 55 °C, DCE, 16 h, 81%; (c) MeMgBr, DCM, −50 to −60 °C, 3 h, 63%; (d) 1 N HCl, dioxane, reflux, 16 h, >98%, 98.4% ee; (e) m-CPBA, CHCl3, reflux, 4 days, 52%; (f) Ac2O, reflux, 3 days, 60%; (g) K2CO3, MeOH, 4 h, 92%; (h) MeI, K2CO3, DMF, 45 min, 67%.
1H NMR (300 MHz,
DMSO-d6) δ 12.07 (s, 1 H), 7.71−7.76 (m, 2 H), 7.51 (dd, J = 8.79,
2.35 Hz, 1 H), 7.31 (d, J = 8.79 Hz, 1 H), 6.97 (d, J = 7.92 Hz, 1 H),
6.93 (d, J = 7.92 Hz, 1 H), 5.95 (d, J = 7.92 Hz, 1 H), 4.62−4.75 (m,
1 H), 3.58 (s, 3 H), 1.50 (d, J = 6.74 Hz, 3 H); 13C NMR (75 MHz,
DMSO-d6) δ 161.0, 155.9, 141.4, 136.6, 135.0, 133.4, 129.8, 126.7,
125.8, 120.1, 119.4, 116.7, 115.1, 104.5, 103.7, 47.4, 34.0, 20.3; LCMS
(method 2) >95% purity; tR 10.18 min; m/z 355, 357 [M + H]+
;
HRMS (ESI) calcd for C18H16ClN4O2 [M + H]+ 355.0962 found
356.0956.
////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
| Clinical data | |
|---|---|
| Trade names | Rezlidhia |
| Other names | FT-2102 |
| License data | US DailyMed: Olutasidenib |
| Routes of administration | By mouth |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only [1][2] |
| Identifiers | |
| CAS Number | 1887014-12-1 |
| PubChem CID | 118955396 |
| IUPHAR/BPS | 10319 |
| DrugBank | DB16267 |
| ChemSpider | 72380144 |
| UNII | 0T4IMT8S5Z |
| KEGG | D12483 |
| ChEMBL | ChEMBL4297610 |
| PDB ligand | PWV (PDBe, RCSB PDB) |
| Chemical and physical data | |
| Formula | C18H15ClN4O2 |
| Molar mass | 354.79 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
References
- ^ Jump up to:a b c d e f https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/215814s000lbl.pdf
- ^ Jump up to:a b c d https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2022/215814Orig1s000ltr.pdf This article incorporates text from this source, which is in the public domain.
- ^ “Rigel Announces U.S. FDA Approval of Rezlidhia (olutasidenib) for the Treatment of Adult Patients with Relapsed or Refractory Acute Myeloid Leukemia with a Susceptible IDH1 Mutation”. Rigel Pharmaceuticals, Inc. (Press release). 1 December 2022. Retrieved 2 December 2022.
- ^ “Rigel Announces U.S. FDA Approval of Rezlidhia (olutasidenib) for the Treatment of Adult Patients with Relapsed or Refractory Acute Myeloid Leukemia with a Susceptible IDH1 Mutation” (Press release). Rigel Pharmaceuticals. 1 December 2022. Retrieved 2 December 2022 – via PR Newswire.
- ^ World Health Organization (2019). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 82”. WHO Drug Information. 33 (3). hdl:10665/330879.
Further reading
- Liu X, Gong Y (2019). “Isocitrate dehydrogenase inhibitors in acute myeloid leukemia”. Biomarker Research. 7: 22. doi:10.1186/s40364-019-0173-z. PMC 6806510. PMID 31660152.
- Watts JM, Baer MR, Yang J, Prebet T, Lee S, Schiller GJ, et al. (November 2022). “Olutasidenib alone or with azacitidine in IDH1-mutated acute myeloid leukaemia and myelodysplastic syndrome: phase 1 results of a phase 1/2 trial”. The Lancet Haematology. doi:10.1016/S2352-3026(22)00292-7. PMID 36370742. S2CID 253471380.
External links
- “Olutasidenib”. Drug Information Portal. U.S. National Library of Medicine.
- Clinical trial number NCT02719574 for “Open-label Study of FT-2102 With or Without Azacitidine or Cytarabine in Patients With AML or MDS With an IDH1 Mutation” at ClinicalTrials.gov
/////////////Olutasidenib, FDA 2022, APPROVALS 2022, Rezlidhia, FT-2102, FT 2102

NEW DRUG APPROVALS
ONE TIME
$10.00
Mirvetuximab soravtansine-gynx
Mirvetuximab soravtansine-gynx
FDA 11/14/2022,To treat patients with recurrent ovarian cancer that is resistant to platinum therapy
| Elahere |
FDA Approves Mirvetuximab Soravtansine-gynx for FRα+ Platinum-resistant Ovarian Cancer
https://www.biochempeg.com/article/315.html
4846-85a8-48171ab38275
FDA Approves Mirvetuximab Soravtansine-gynx for FRα+ Platinum-resistant Ovarian Cancer
November 15, 2022
The FDA has granted accelerated approval to mirvetuximab soravtansine-gynx (Elahere) for the treatment of select patients with folate receptor α–positive, platinum-resistant epithelial ovarian, fallopian tube, or primary peritoneal cancer.
The FDA has granted accelerated approval to mirvetuximab soravtansine-gynx (Elahere) for the treatment of adult patients with folate receptor α (Frα)–positive, platinum-resistant epithelial ovarian, fallopian tube, or primary peritoneal cancer, who have received 1 to 3 prior systemic treatment regimens.1-3
The regulatory agency also gave the green light to the VENTANA FOLR1 (FOLR-2.1) RxDx Assay for use as a companion diagnostic device to identify patients who are eligible to receive the agent. Testing can be done on fresh or archived tissue. Newly diagnosed patients can be tested at diagnosis to determine whether this agent will be an option for them at the time of progression to platinum resistance.
The decision was supported by findings from the phase 3 SORAYA trial (NCT04296890), in which mirvetuximab soravtansine elicited a confirmed investigator-assessed objective response rate (ORR) of 31.7% (95% CI, 22.9%-41.6%); this included a complete response rate of 4.8% and a partial response rate of 26.9%. Moreover, the median duration of response (DOR) was 6.9 months (95% CI, 5.6-9.7) per investigator assessment.
“The approval of Elahere is significant for patients with FRα-positive platinum-resistant ovarian cancer, which is characterized by limited treatment options and poor outcomes,” Ursula Matulonis, MD, chief of the Division of Gynecologic Oncology at the Dana-Farber Cancer Institute, professor of medicine at the Harvard Medical School, and SORAYA co-principal investigator, stated in a press release. “Elahere impressive anti-tumor activity, durability of response, and overall tolerability observed in SORAYA demonstrate the benefit of this new therapeutic option, and I look forward to treating patients with Elahere.”
The global, single-arm SORAYA trial enrolled a total of 106 patients with platinum-resistant ovarian cancer whose tumors expressed high levels of FRα. Patients were allowed to have received up to 3 prior lines of systemic treatment, and all were required to have received bevacizumab (Avastin).
If patients had corneal disorders, ocular conditions in need of ongoing treatment, peripheral neuropathy that was greater than grade 1 in severity, or noninfectious interstitial lung disease, they were excluded.
Study participants received intravenous mirvetuximab soravtansine at 6 mg/kg once every 3 weeks until progressive disease or unacceptable toxicity. Investigators conducted tumor response assessments every 6 weeks for the first 36 weeks, and every 12 weeks thereafter.
Confirmed investigator-assessed ORR served as the primary end point for the research, and the key secondary end point was DOR by RECIST v1.1 criteria.
In the efficacy-evaluable population (n = 104), the median age was 62 years (range, 35-85). Ninety-six percent of patients were White, 2% were Asian, and 2% did not have their race information reported; 2% of patients were Hispanic or Latino. Regarding ECOG performance status, 57% of patients had a status of 0 and the remaining 43% had a status of 1.
Ten percent of patients received 1 prior line of systemic treatment, 39% received 2 prior lines, and 50% received 3 or more prior lines. All patients previously received bevacizumab, as required, and 47% previously received a PARP inhibitor.
The safety of mirvetuximab soravtansine was evaluated in all 106 patients. The median duration of treatment with the agent was 4.2 months (range, 0.7-13.3).
The all-grade toxicities most commonly experienced with mirvetuximab soravtansine included vision impairment (50%), fatigue (49%), increased aspartate aminotransferase (50%), nausea (40%), increased alanine aminotransferase (39%), keratopathy (37%), abdominal pain (36%), decreased lymphocytes (35%), peripheral neuropathy (33%), diarrhea (31%), decreased albumin (31%), constipation (30%), increased alkaline phosphatase (30%), dry eye (27%), decreased magnesium (27%), decreased leukocytes (26%), decreased neutrophils (26%), and decreased hemoglobin (25%).
Thirty-one percent of patients experienced serious adverse reactions with the agent, which included intestinal obstruction (8%), ascites (4%), infection (3%), and pleural effusion (3%). Toxicities proved to be fatalfor 2% of patients, and these included small intestinal obstruction (1%) and pneumonitis (1%).
Twenty percent of patients required dose reductions due to toxicities. Eleven percent of patients discontinued treatment with mirvetuximab soravtansine because of adverse reactions. Toxicities that resulted in more than 2% of patients discontinuing treatment included intestinal obstruction (2%) and thrombocytopenia (2%). One patient discontinued because of visual impairment.
References
- ImmunoGen announces FDA accelered approval of Elahere (mirvetuximab soravtansine-gynx) for the treatment of platinum-resistant ovarian cancer. News release. ImmunoGen Inc. November 14, 2022. Accessed November 14, 2022. http://bit.ly/3GgrCwL
- FDA grants accelerated approval to mirvetuximab soravtansine-gynx for FRα positive, platinum-resistant epithelial ovarian, fallopian tube, or peritoneal cancer. News release. FDA. November 14, 2022. Accessed November 14, 2022. http://bit.ly/3UP742w
- Elahere (mirvetuximab soravtansine-gynx). Prescribing information; ImmunoGen Inc; 2022. Accessed November 14, 2022. https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/761310s000lbl.pdf

NEW DRUG APPROVALS
ONE TIME
$10.00
////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
//////////Mirvetuximab soravtansine-gynx, FDA 2022, APPROVALS 2022, recurrent ovarian cancer,
| Elahere |
Tremelimumab
(Light chain)
DIQMTQSPSS LSASVGDRVT ITCRASQSIN SYLDWYQQKP GKAPKLLIYA ASSLQSGVPS
RFSGSGSGTD FTLTISSLQP EDFATYYCQQ YYSTPFTFGP GTKVEIKRTV AAPSVFIFPP
SDEQLKSGTA SVVCLLNNFY PREAKVQWKV DNALQSGNSQ ESVTEQDSKD STYSLSSTLT
LSKADYEKHK VYACEVTHQG LSSPVTKSFN RGEC
(Heavy chain)
QVQLVESGGG VVQPGRSLRL SCAASGFTFS SYGMHWVRQA PGKGLEWVAV IWYDGSNKYY
ADSVKGRFTI SRDNSKNTLY LQMNSLRAED TAVYYCARDP RGATLYYYYY GMDVWGQGTT
VTVSSASTKG PSVFPLAPCS RSTSESTAAL GCLVKDYFPE PVTVSWNSGA LTSGVHTFPA
VLQSSGLYSL SSVVTVPSSN FGTQTYTCNV DHKPSNTKVD KTVERKCCVE CPPCPAPPVA
GPSVFLFPPK PKDTLMISRT PEVTCVVVDV SHEDPEVQFN WYVDGVEVHN AKTKPREEQF
NSTFRVVSVL TVVHQDWLNG KEYKCKVSNK GLPAPIEKTI SKTKGQPREP QVYTLPPSRE
EMTKNQVSLT CLVKGFYPSD IAVEWESNGQ PENNYKTTPP MLDSDGSFFL YSKLTVDKSR
WQQGNVFSCS VMHEALHNHY TQKSLSLSPG K
(Disulfide bridge: L23-L88, L134-L194, L214-H139, H22-H96, H152-H208, H265-H325, H371-H429, H227-H’227, H228-H’228, H231-H’231, H234-H’234)

Fab fragment of tremelimumab (blue) binding CTLA-4 (green). From PDB entry 5GGV.
Tremelimumab
| Formula | C6500H9974N1726O2026S52 |
|---|---|
| CAS | 745013-59-6 |
| Mol weight | 146380.4722 |
FDA APPROVED2022/10/21, Imjudo
PEPTIDE, CP 675206
| Antineoplastic, Immune checkpoint inhibitor, Anti-CTLA4 antibody | |
| Disease | Hepatocellular carcinoma |
|---|
Tremelimumab (formerly ticilimumab, CP-675,206) is a fully human monoclonal antibody against CTLA-4. It is an immune checkpoint blocker. Previously in development by Pfizer,[1] it is now in investigation by MedImmune, a wholly owned subsidiary of AstraZeneca.[2] It has been undergoing human trials for the treatment of various cancers but has not attained approval for any.
Imjudo (tremelimumab) in combination with Imfinzi approved in the US for patients with unresectable liver cancer
PUBLISHED24 October 2022
24 October 2022 07:00 BST
Approval based on HIMALAYA Phase III trial results which showed single priming dose of Imjudo added to Imfinzi reduced risk of death by 22% vs. sorafenib
AstraZeneca’s Imjudo (tremelimumab) in combination with Imfinzi (durvalumab) has been approved in the US for the treatment of adult patients with unresectable hepatocellular carcinoma (HCC), the most common type of liver cancer. The novel dose and schedule of the combination, which includes a single dose of the anti-CTLA-4 antibody Imjudo 300mg added to the anti-PD-L1 antibody Imfinzi 1500mg followed by Imfinzi every four weeks, is called the STRIDE regimen (Single Tremelimumab Regular Interval Durvalumab).
The approval by the US Food and Drug Administration (FDA) was based on positive results from the HIMALAYA Phase III trial. In this trial, patients treated with the combination of Imjudo and Imfinzi experienced a 22% reduction in the risk of death versus sorafenib (based on a hazard ratio [HR] of 0.78, 95% confidence interval [CI] 0.66-0.92 p=0.0035).1 Results were also published in the New England Journal of Medicine Evidence showing that an estimated 31% of patients treated with the combination were still alive after three years, with 20% of patients treated with sorafenib still alive at the same duration of follow-up.2
Liver cancer is the third-leading cause of cancer death and the sixth most commonly diagnosed cancer worldwide.3,4 It is the fastest rising cause of cancer-related deaths in the US, with approximately 36,000 new diagnoses each year.5,6
Ghassan Abou-Alfa, MD, MBA, Attending Physician at Memorial Sloan Kettering Cancer Center (MSK), and principal investigator in the HIMALAYA Phase III trial, said: “Patients with unresectable liver cancer are in need of well-tolerated treatments that can meaningfully extend overall survival. In addition to this regimen demonstrating a favourable three-year survival rate in the HIMALAYA trial, safety data showed no increase in severe liver toxicity or bleeding risk for the combination, important factors for patients with liver cancer who also have advanced liver disease.”
Dave Fredrickson, Executive Vice President, Oncology Business Unit, AstraZeneca, said: “With this first regulatory approval for Imjudo, patients with unresectable liver cancer in the US now have an approved dual immunotherapy treatment regimen that harnesses the potential of CTLA-4 inhibition in a unique combination with a PD-L1 inhibitor to enhance the immune response against their cancer.”
Andrea Wilson Woods, President & Founder, Blue Faery: The Adrienne Wilson Liver Cancer Foundation, said: “In the past, patients living with liver cancer had few treatment options and faced poor prognoses. With today’s approval, we are grateful and optimistic for new, innovative, therapeutic options. These new treatments can improve long-term survival for those living with unresectable hepatocellular carcinoma, the most common form of liver cancer. We appreciate the patients, their families, and the broader liver cancer community who continue to fight for new treatments and advocate for others.”
The safety profiles of the combination of Imjudo added to Imfinzi and for Imfinzi alone were consistent with the known profiles of each medicine, and no new safety signals were identified.
Regulatory applications for Imjudo in combination with Imfinzi are currently under review in Europe, Japan and several other countries for the treatment of patients with advanced liver cancer based on the HIMALAYA results.
Notes
Liver cancer
About 75% of all primary liver cancers in adults are HCC.3 Between 80-90% of all patients with HCC also have cirrhosis.7 Chronic liver diseases are associated with inflammation that over time can lead to the development of HCC.7
More than half of patients are diagnosed at advanced stages of the disease, often when symptoms first appear.8 A critical unmet need exists for patients with HCC who face limited treatment options.8 The unique immune environment of liver cancer provides clear rationale for investigating medications that harness the power of the immune system to treat HCC.8
HIMALAYA
HIMALAYA was a randomised, open-label, multicentre, global Phase III trial of Imfinzi monotherapy and a regimen comprising a single priming dose of Imjudo 300mg added to Imfinzi 1500mg followed by Imfinzi every four weeks versus sorafenib, a standard-of-care multi-kinase inhibitor.
The trial included a total of 1,324 patients with unresectable, advanced HCC who had not been treated with prior systemic therapy and were not eligible for locoregional therapy (treatment localised to the liver and surrounding tissue).
The trial was conducted in 181 centres across 16 countries, including in the US, Canada, Europe, South America and Asia. The primary endpoint was overall survival (OS) for the combination versus sorafenib and key secondary endpoints included OS for Imfinzi versus sorafenib, objective response rate and progression-free survival (PFS) for the combination and for Imfinzi alone.
Imfinzi
Imfinzi (durvalumab) is a human monoclonal antibody that binds to the PD-L1 protein and blocks the interaction of PD-L1 with the PD-1 and CD80 proteins, countering the tumour’s immune-evading tactics and releasing the inhibition of immune responses.
Imfinzi was recently approved to treat patients with advanced biliary tract cancer in the US based on results from the TOPAZ-1 Phase III trial. It is the only approved immunotherapy in the curative-intent setting of unresectable, Stage III non-small cell lung cancer (NSCLC) in patients whose disease has not progressed after chemoradiotherapy and is the global standard of care in this setting based on the PACIFIC Phase III trial.
Imfinzi is also approved in the US, EU, Japan, China and many other countries around the world for the treatment of extensive-stage small cell lung cancer (ES-SCLC) based on the CASPIAN Phase III trial. In 2021, updated results from the CASPIAN trial showed Imfinzi plus chemotherapy tripled patient survival at three years versus chemotherapy alone.
Imfinzi is also approved for previously treated patients with advanced bladder cancer in several countries.
Since the first approval in May 2017, more than 100,000 patients have been treated with Imfinzi.
As part of a broad development programme, Imfinzi is being tested as a single treatment and in combinations with other anti-cancer treatments for patients with SCLC, NSCLC, bladder cancer, several gastrointestinal (GI) cancers, ovarian cancer, endometrial cancer, and other solid tumours.
Imfinzi combinations have also demonstrated clinical benefit in metastatic NSCLC in the POSEIDON Phase III trial.
Imjudo
Imjudo (tremelimumab) is a human monoclonal antibody that targets the activity of cytotoxic T-lymphocyte-associated protein 4 (CTLA-4). Imjudo blocks the activity of CTLA-4, contributing to T-cell activation, priming the immune response to cancer and fostering cancer cell death.
Beyond HIMALAYA, Imjudo is being tested in combination with Imfinzi across multiple tumour types including locoregional HCC (EMERALD-3), SCLC (ADRIATIC) and bladder cancer (VOLGA and NILE).
Imjudo is also under review by global regulatory authorities in combination with Imfinzi and chemotherapy in 1st-line metastatic NSCLC based on the results of the POSEIDON Phase III trial, which showed the addition of a short course of Imjudo to Imfinzi plus chemotherapy improved both overall and progression-free survival compared to chemotherapy alone.
AstraZeneca in GI cancers
AstraZeneca has a broad development programme for the treatment of GI cancers across several medicines spanning a variety of tumour types and stages of disease. In 2020, GI cancers collectively represented approximately 5.1 million new diagnoses leading to approximately 3.6 million deaths.9
Within this programme, the Company is committed to improving outcomes in gastric, liver, biliary tract, oesophageal, pancreatic, and colorectal cancers.
Imfinzi (durvalumab) is being assessed in combinations in oesophageal and gastric cancers in an extensive development programme spanning early to late-stage disease across settings.
The Company aims to understand the potential of Enhertu (trastuzumab deruxtecan), a HER2-directed antibody drug conjugate, in the two most common GI cancers, colorectal and gastric cancers. Enhertu is jointly developed and commercialised by AstraZeneca and Daiichi Sankyo.
Lynparza (olaparib) is a first-in-class PARP inhibitor with a broad and advanced clinical trial programme across multiple GI tumour types including pancreatic and colorectal cancers. Lynparza is developed and commercialised in collaboration with MSD (Merck & Co., Inc. inside the US and Canada).
AstraZeneca in immuno-oncology (IO)
Immunotherapy is a therapeutic approach designed to stimulate the body’s immune system to attack tumours. The Company’s immuno-oncology (IO) portfolio is anchored in immunotherapies that have been designed to overcome evasion of the anti-tumour immune response. AstraZeneca is invested in using IO approaches that deliver long-term survival for new groups of patients across tumour types.
The Company is pursuing a comprehensive clinical trial programme that includes Imfinzi as a single treatment and in combination with Imjudo (tremelimumab) and other novel antibodies in multiple tumour types, stages of disease, and lines of treatment, and where relevant using the PD-L1 biomarker as a decision-making tool to define the best potential treatment path for a patient.
In addition, the ability to combine the IO portfolio with radiation, chemotherapy, and targeted small molecules from across AstraZeneca’s oncology pipeline, and from research partners, may provide new treatment options across a broad range of tumours.
AstraZeneca in oncology
AstraZeneca is leading a revolution in oncology with the ambition to provide cures for cancer in every form, following the science to understand cancer and all its complexities to discover, develop and deliver life-changing medicines to patients.
The Company’s focus is on some of the most challenging cancers. It is through persistent innovation that AstraZeneca has built one of the most diverse portfolios and pipelines in the industry, with the potential to catalyse changes in the practice of medicine and transform the patient experience.
AstraZeneca has the vision to redefine cancer care and, one day, eliminate cancer as a cause of death.
////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
Mechanism of action
Tremelimumab aims to stimulate an immune system attack on tumors. Cytotoxic T lymphocytes (CTLs) can recognize and destroy cancer cells. However, there is also an inhibitory mechanism (immune checkpoint) that interrupts this destruction. Tremelimumab turns off this inhibitory mechanism and allows CTLs to continue to destroy the cancer cells.[3] This is immune checkpoint blockade.
Tremelimumab binds to the protein CTLA-4, which is expressed on the surface of activated T lymphocytes and inhibits the killing of cancer cells. Tremelimumab blocks the binding of the antigen-presenting cell ligands B7.1 and B7.2 to CTLA-4, resulting in inhibition of B7-CTLA-4-mediated downregulation of T-cell activation; subsequently, B7.1 or B7.2 may interact with another T-cell surface receptor protein, CD28, resulting in a B7-CD28-mediated T-cell activation unopposed by B7-CTLA-4-mediated inhibition.
Unlike Ipilimumab (another fully human anti-CTLA-4 monoclonal antibody), which is an IgG1 isotype, tremelimumab is an IgG2 isotype.[4][5]
Clinical trials
Melanoma
Phase 1 and 2 clinical studies in metastatic melanoma showed some responses.[6] However, based on early interim analysis of phase III data, Pfizer designated tremelimumab as a failure and terminated the trial in April 2008.[1][7]
However, within a year, the survival curves showed separation of the treatment and control groups.[8] The conventional Response Evaluation Criteria in Solid Tumors (RECIST) may underrepresent the merits of immunotherapies. Subsequent immunotherapy trials (e.g. ipilimumab) have used the Immune-Related Response Criteria (irRC) instead.
Mesothelioma
Although it was designated in April 2015 as orphan drug status in mesothelioma,[9] tremelimumab failed to improve lifespan in the phase IIb DETERMINE trial, which assessed the drug as a second or third-line treatment for unresectable malignant mesothelioma.[10][11]
Non-small cell lung cancer
In a phase III trial, AstraZeneca paired tremelimumab with a PD-L1 inhibitor, durvalumab, for the first-line treatment of non-small cell lung cancer.[12] The trial was conducted across 17 countries, and in July 2017, AstraZeneca announced that it had failed to meet its primary endpoint of progression-free survival.[13]
References
- ^ Jump up to:a b “Pfizer Announces Discontinuation of Phase III Clinical Trial for Patients with Advanced Melanoma”. Pfizer.com. 1 April 2008. Retrieved 5 December 2015.
- ^ Mechanism of Pathway: CTLA-4 Inhibition[permanent dead link]
- ^ Antoni Ribas (28 June 2012). “Tumor immunotherapy directed at PD-1”. New England Journal of Medicine. 366 (26): 2517–9. doi:10.1056/nejme1205943. PMID 22658126.
- ^ Tomillero A, Moral MA (October 2008). “Gateways to clinical trials”. Methods Find Exp Clin Pharmacol. 30 (8): 643–72. doi:10.1358/mf.2008.30.5.1236622. PMID 19088949.
- ^ Poust J (December 2008). “Targeting metastatic melanoma”. Am J Health Syst Pharm. 65 (24 Suppl 9): S9–S15. doi:10.2146/ajhp080461. PMID 19052265.
- ^ Reuben, JM; et al. (1 Jun 2006). “Biologic and immunomodulatory events after CTLA-4 blockade with tremelimumab in patients with advanced malignant melanoma”. Cancer. 106 (11): 2437–44. doi:10.1002/cncr.21854. PMID 16615096. S2CID 751366.
- ^ A. Ribas, A. Hauschild, R. Kefford, C. J. Punt, J. B. Haanen, M. Marmol, C. Garbe, J. Gomez-Navarro, D. Pavlov and M. Marsha (May 20, 2008). “Phase III, open-label, randomized, comparative study of tremelimumab (CP-675,206) and chemotherapy (temozolomide [TMZ] or dacarbazine [DTIC]) in patients with advanced melanoma”. Journal of Clinical Oncology. 26 (15S): LBA9011. doi:10.1200/jco.2008.26.15_suppl.lba9011.[permanent dead link]
- ^ M.A. Marshall, A. Ribas, B. Huang (May 2010). “Evaluation of baseline serum C-reactive protein (CRP) and benefit from tremelimumab compared to chemotherapy in first-line melanoma”. Journal of Clinical Oncology. 28 (15S): 2609. doi:10.1200/jco.2010.28.15_suppl.2609.[permanent dead link]
- ^ FDA Grants AstraZeneca’s Tremelimumab Orphan Drug Status for Mesothelioma [1]
- ^ “Tremelimumab Fails Mesothelioma Drug Trial”. Archived from the original on 2016-03-06. Retrieved 2016-03-06.
- ^ AZ’ tremelimumab fails in mesothelioma trial
- ^ “AstraZeneca’s immuno-oncology combo fails crucial Mystic trial in lung cancer | FierceBiotech”.
- ^ “AstraZeneca reports initial results from the ongoing MYSTIC trial in Stage IV lung cancer”.
///////////Tremelimumab, Imjudo, APPROVALS 2022, FDA 2022, PEPTIDE, CP 675206, Antineoplastic, Immune checkpoint inhibitor, Anti-CTLA4 antibody

NEW DRUG APPROVALS
ONE TIME
$10.00
Futibatinib
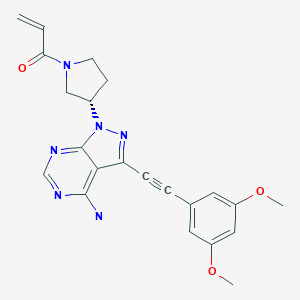

Futibatinib
フチバチニブ
| Formula | C22H22N6O3 |
|---|---|
| CAS | 1448169-71-8 |
| Mol weight | 418.4485 |
2022/9/30 FDA APPROVED, Lytgobi
| Antineoplastic, Receptor tyrosine kinase inhibitor | |
| Disease | Cholangiocarcinoma (FGFR2 gene fusion) |
|---|
1-[(3S)-3-[4-amino-3-[2-(3,5-dimethoxyphenyl)ethynyl]-1H-pyrazolo[3,4-d]pyrimidin-1-yl]-1-pyrrolidinyl]-2-propen-1-one
TAS-120, TAS 120, TAS120; Futibatinib
Futibatinib, also known as TAS-120 is an orally bioavailable inhibitor of the fibroblast growth factor receptor (FGFR) with potential antineoplastic activity. FGFR inhibitor TAS-120 selectively and irreversibly binds to and inhibits FGFR, which may result in the inhibition of both the FGFR-mediated signal transduction pathway and tumor cell proliferation, and increased cell death in FGFR-overexpressing tumor cells. FGFR is a receptor tyrosine kinase essential to tumor cell proliferation, differentiation and survival and its expression is upregulated in many tumor cell types.

SYN
Patent Document 1: International Publication WO 2007/087395 pamphlet
Patent Document 2: International Publication WO 2008/121742 pamphlet
Patent Document 3: International Publication WO 2010/043865 pamphlet
Patent Document 4: International Publication WO 2011/115937 pamphlet
Unlicensed Document 1 : J. Clin. Oncol. 24, 3664-3671 (2006)
Non-licensed Document 2: Mol. Cancer Res. 3, 655-667 (2005)
Non-licensed Document 3: Cancer Res. 70, 2085-2094 (2010)
Non-licensed Document 4: Clin. Cancer Res. 17, 6130-6139 (2011)
Non-licensed Document 5: Nat. Med. 1, 27-31 (1995)
WO2020095452
WO2020096042
WO2020096050
WO2019034075
WO2015008844
WO2015008839
WO2013108809
SYN
US9108973
SYN
Reference Example 1: WXR1
Compound WXR1 was synthesized according to the route reported in patent WO2015008844. 1 H NMR(400MHz, DMSO-d 6 )δ8.40(d,J=3.0Hz,1H),6.93(d,J=2.5Hz,2H),6.74-6.52(m,2H),6.20-6.16( m,1H), 5.74-5.69(m,1H), 5.45-5.61(m,1H), 4.12-3.90(m,2H), 3.90-3.79(m,8H), 2.47-2.30(m,2H). MS m/z: 419.1[M+H] +
PAPER
Bioorg Med Chem, March 2013, Vol.21, No.5, pp.1180-1189
SYN
WO2015008844
PATENT
////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
| Clinical data | |
|---|---|
| Trade names | Lytgobi |
| Other names | TAS-120 |
| License data | US DailyMed: Futibatinib |
| Routes of administration | By mouth |
| Drug class | Antineoplastic |
| ATC code | L01EN04 (WHO) |
| Legal status | |
| Legal status | US: ℞-only [1] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 1448169-71-8 |
| PubChem CID | 71621331 |
| IUPHAR/BPS | 9786 |
| DrugBank | DB15149 |
| ChemSpider | 58877816 |
| UNII | 4B93MGE4AL |
| KEGG | D11725 |
| ChEMBL | ChEMBL3701238 |
| PDB ligand | TZ0 (PDBe, RCSB PDB) |
| Chemical and physical data | |
| Formula | C22H22N6O3 |
| Molar mass | 418.457 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI |
Futibatinib, sold under the brand name Lytgobi, is a medication used for the treatment of cholangiocarcinoma (bile duct cancer).[1][2] It is a kinase inhibitor.[1][3] It is taken by mouth.[1]
Futibatinib was approved for medical use in the United States in September 2022.[1][2][4]
Medical uses
Futibatinib is indicated for the treatment of adults with previously treated, unresectable, locally advanced or metastatic intrahepatic cholangiocarcinoma harboring fibroblast growth factor receptor 2 (FGFR2) gene fusions or other rearrangements.[1][2]
Names
Futibatinib is the international nonproprietary name (INN).[5]
References
- ^ Jump up to:a b c d e f “Lytgobi (futibatinib) tablets, for oral use” (PDF). Archived (PDF) from the original on 4 October 2022. Retrieved 4 October 2022.
- ^ Jump up to:a b c https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2022/214801Orig1s000ltr.pdf Archived 4 October 2022 at the Wayback Machine This article incorporates text from this source, which is in the public domain.
- ^ “Lytgobi (Futibatinib) FDA Approval History”. Archived from the original on 4 October 2022. Retrieved 4 October 2022.
- ^ “FDA Approves Taiho’s Lytgobi (futibatinib) Tablets for Previously Treated, Unresectable, Locally Advanced or Metastatic Intrahepatic Cholangiocarcinoma” (Press release). Taiho Oncology. 30 September 2022. Archived from the original on 4 October 2022. Retrieved 4 October 2022 – via PR Newswire.
- ^ World Health Organization (2019). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 81”. WHO Drug Information. 33 (1). hdl:10665/330896.
External links
- “Futibatinib”. Drug Information Portal. U.S. National Library of Medicine.
//////////Futibatinib, Lytgobi, FDA 2022, APPROVALS 2022, フチバチニブ , ANTINEOPLASTIC, TAS 120
C=CC(N1C[C@@H](N2N=C(C#CC3=CC(OC)=CC(OC)=C3)C4=C(N)N=CN=C42)CC1)=O

NEW DRUG APPROVALS
ONE TIME
$10.00
Gadopiclenol

![Chemical structure of gadopiclenol [gadolinium chelate of 2,2′,2″-(3,6,9-triaza-1(2,6)-pyridinacyclodecaphane-3,6,9-triyl)tris(5-((2,3-dihydroxypropyl)amino)-5-oxopentanoic acid)]. The PCTA parent structure is shown in red. Two water molecules are included to show the coordination in solution.](https://www.researchgate.net/profile/Jean-Marc-Idee/publication/334838366/figure/fig1/AS:797490152476678@1567147877999/Chemical-structure-of-gadopiclenol-gadolinium-chelate-of.jpg)
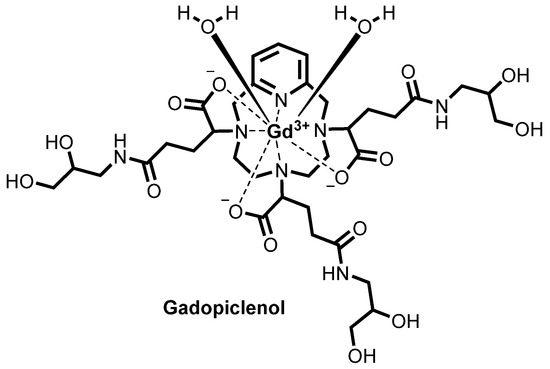
Gadopiclenol
ガドピクレノール;
| Formula | C35H54N7O15. Gd |
|---|---|
| CAS | 933983-75-6 |
| Mol weight | 970.0912 |
FDA APPROVED 2022/9/21, Elucirem
Diagnostic agent (MR imaging), WHO 10744, P 03277, UNII: S276568KOY
EluciremTM; G03277; P03277; VUEWAY
(alpha3,alpha6,alpha9-Tris(3-((2,3-dihydroxypropyl)amino)-3-oxopropyl)-3,6,9,15-tetraazabicyclo(9.3.1)pentadeca-1(15),11,13-triene-3,6,9-triacetato(3-)-kappaN3,kappaN6,kappaN9,kappaN15,kappaO3,kappaO6,kappaO9)gadolinium
- OriginatorGuerbet
- ClassDiagnostic agents; Gadolinium-containing contrast agents; Macrocyclic compounds; Propylamines; Pyridines
- Mechanism of ActionMagnetic resonance imaging enhancers
- RegisteredCNS disorders
- Phase IIIUnspecified
- Phase IILiver cancer
- 21 Sep 2022Registered for CNS disorders (Diagnosis) in USA (IV)
- 13 Jun 2022Guerbet plans to launch Gadopiclenol in Europe
- 13 Jun 2022The European Medicines Agency (EMA) accepts brand name EluciremTM for Gadopiclenol
PATENT
https://patents.google.com/patent/WO2020030618A1/en
MRI contrast agents used in daily diagnostic practice typically include gadolinium complex compounds characterized by high stability constants that guarantee against the in vivo release of the free metal ion (that is known to be extremely toxic for living organisms).
Another key parameter in the definition of the tolerability of a gadolinium-based contrast agent is the kinetic inertness (or kinetic stability) of Gd(III)-complex, that is estimated through the half-life (ti/2) of the dissociation (i.e. decomplexation) of the complex.
A high inertness becomes crucial in particular for those complex compounds having lower thermodynamic stability and/or longer retention time before excretion, in order to avoid or minimize possible decomplexation or transmetallation reactions.
EP1931673 (Guerbet) discloses PCTA derivatives of formula

and a synthetic route for their preparation.
EP 2988756 (same Applicant) discloses a pharmaceutical composition comprising the above derivatives together with a calcium complex of 1,4,7, 10-tetraazacyclododecane- 1,4,7, 10-tetraacetic acid. According to the EP 2988756, the calcium complex compensates the weak thermodynamic stability observed for PCTA-based gadolinium complexes, by forming, through transmetallation, a strong complex with free lanthanide ion, thereby increasing the tolerability of the contrast agent.
Both EP1931673 and EP 2988756 further refer to enantiomers or diastereoisomers of the claimed compounds, or mixture thereof, preferentially chosen from the RRS, RSR, and RSS diastereoisomers. Both the above patents disclose, among the specific derivatives, (a3, a6, a9)-tris(3- ((2,3-dihydroxypropyl)amino)-3-oxopropyl)-3,6,9,15-tetraazabicyclo(9.3.1)pentadeca- l(15),l l,13-triene-3,6,9-triacetato(3-)-(KN3,KN6,KN9,KN15,K03,K06,K09)gadolinium, more recently identified as gadolinium chelate of 2,2′,2″-(3,6,9-triaza-l(2,6)- pyridinacyclodecaphane-3,6,9-triyl)tris(5-((2,3-dihydroxypropyl)amino)-5-oxopentanoic acid), (CAS registry number: 933983-75-6), having the following formula

otherwise identified as P03277 or Gadopiclenol.
For Gadopiclenol, EP1931673 reports a relaxivity of 11 mM _1s _1Gd 1 (in water, at 0.5 T, 37°C) while EP 2988756 reports a thermodynamic equilibrium constant of 10 14 9 (log Kterm
= 14.9).
Furthermore, for this same compound a relaxivity value of 12.8 mM _1s 1 in human serum (37°C, 1.41 T), stability (log Kterm) of 18.7, and dissociation half-life of about 20 days (at pH 1.2; 37°C) have been reported by the proprietor (Investigative Radiology 2019, Vol 54, (8), 475-484).
The precursor for the preparation of the PCTA derivatives disclosed by EP1931673 (including Gadopiclenol) is the Gd complex of the 3,6,9,15-tetraazabicyclo- [9.3.1]pentadeca-l(15),l l,13-triene-tri(a-glutaric acid) having the following formula

Gd(PCTA-tris-glutaric acid)
herein identified as “Gd(PCTA-tris-glutaric acid)”. In particular, Gadopiclenol is obtained by amidation of the above compound with isoserinol.
As observed by the Applicant, Gd(PCTA-tris-qlutaric acid) has three stereocenters on the glutaric moieties (identified with an asterisk (*) in the above structure) that lead to a 23 = 8 possible stereoisomers. More particularly, the above structure can generate four pairs of enantiomers, schematized in the following Table 1
Table 1

Isomer RRR is the mirror image of isomer SSS and that is the reason why they are called enantiomers (or enantiomer pairs). As known, enantiomers display the same physicochemical properties and are distinguishable only using chiral methodologies, such as chiral chromatography or polarized light.
On the other hand, isomer RRR is neither equal to nor is it the mirror image of any of the other above six isomers; these other isomers are thus identified as diastereoisomers of the RRR (or SSS) isomer. Diastereoisomers may display different physicochemical properties, (e.g., melting point, water solubility, relaxivity, etc.).
Concerning Gadopiclenol, its chemical structure contains a total of six stereocenters, three on the glutaric moieties of the precursor as above discussed and one in each of the three isoserinol moieties attached thereto, identified in the following structure with an asterisk (*) and with an empty circle (°), respectively:

This leads to a total theoretical number of 26 = 64 stereoisomers for this compound. However, neither EP1931673 nor EP 2988756 describe the exact composition of the isomeric mixture obtained by following the reported synthetic route, nor does any of them provide any teaching for the separation and characterization of any of these isomers, or disclose any stereospecific synthesis of Gadopiclenol. Summary of the invention
The applicant has now found that specific isomers of the above precursor Gd(PCTA- tris-glutaric acid) and of its derivatives (in particular Gadopiclenol) possess improved physico-chemical properties, among other in terms of relaxivity and kinetic inertness.
An embodiment of the invention relates to a compound selected from the group consisting of:
the enantiomer [(aR,a’R,a”R)-a,a’,a”-tris(2-carboxyethyl)-3,6,9,15- tetraazabicyclo[9.3.1]pentadeca-l(15),l l,13-triene-3,6,9-triacetato(3-)- Kl\l3,Kl\l6,Kl\l9,Kl\ll5,K03,K06,K09]-gadolinium (RRR enantiomer) having the formula (la):

the enantiomer [(aS,a’S,a”S)-a,a’,a”-tris(2-carboxyethyl)-3,6,9,15-tetraazabicyclo- [9.3.1]pentadeca-l(15),ll,13-triene-3,6,9-triacetato(3-)KN3,KN6,KN9,KN15,K03,K06,K09]- gadolinium (SSS enantiomer) having the formula (lb):

the mixtures of such RRR and SSS enantiomers, and a pharmaceutically acceptable salt thereof.
Another embodiment of the invention relates to an isomeric mixture of Gd(PCTA-tris- glutaric acid) comprising at least 50% of the RRR isomer [(aR,a’R,a”R)-a,a’,a”-tris(2- carboxyethyl)-3,6,9,15-tetraazabicyclo[9.3.1]pentadeca-l(15),l l,13-triene-3,6,9- triacetato(3-)-KN3,KN6,KN9,KN15,K03,K06,K09]-gadolinium, of formula (la), or of the SSS isomer [(aS,a’S,a”S)-a,a’,a”-tris(2-carboxyethyl)-3,6,9,15- tetraazabicyclo[9.3.1]pentadeca-l(15),l l,13-triene-3,6,9-triacetato(3-)- Kl\l3,Kl\l6,Kl\l9,Kl\ll5,K03,K06,K09]-gadolinium of formula (lb), or of a mixture thereof, or a pharmaceutically acceptable salt thereof. Another aspect of the invention relates to the amides obtained by conjugation of one of the above compounds or isomeric mixture with an amino group, e.g. preferably, serinol or isoserinol.
An embodiment of the invention relates to an amide derivative of formula (II A)
F( N RI R2)3 (II A)
in which :
F is:
a RRR enantiomer residue of formula Ilia

a SSS enantiomer residue of formula Illb

or a mixture of such RRR and SSS enantiomer residues;
and each of the three -NRIR2 group is bound to an open bond of a respective carboxyl moiety of F, identified with a full circle (·) in the above structures;
Ri is H or a Ci-Ce alkyl, optionally substituted by 1-4 hydroxyl groups;
R2 is a Ci-Ce alkyl optionally substituted by 1-4 hydroxyl groups, and preferably a C1-C3 alkyl substituted by one or two hydroxyl groups.
Another embodiment of the invention relates to an isomeric mixture of an amide derivative of Gd(PCTA-tris-glutaric acid) having the formula (II B)
F'( N RI R2)3 (II B)
in which :
F’ is an isomeric mixture of Gd(PCTA-tris-glutaric acid) residue of formula (III)

said isomeric mixture of the Gd(PCTA-tris-glutaric acid) residue comprising at least 50 % of an enantiomer residue of the above formula (Ilia), of the enantiomer residue of the above formula (Illb), or of a mixture thereof; and each of the -NR1R2 groups is bound to an open bond of a respective carboxyl moiety of F’, identified with a full circle (·) in the above structure, and is as above defined for the compounds of formula (II A).
EXPERIMENTAL PART
HPLC characterization of the obtained compounds.
General procedures
Procedure 1: HPLC Characterization of Gd(PCTA-tris-glutaric acid) (isomeric mixture and individual/enriched isomers).
The HPLC characterization of the Gd(PCTA-tris-glutaric acid) obtained as isomeric mixture from Example 1 was performed with Agilent 1260 Infinity II system. The experimental setup of the HPLC measurements are summarized below.
Analytical conditions
HPLC system HPLC equipped with quaternary pump, degasser, autosampler,
PDA detector ( Agilent 1260 Infinity II system)
Stationary phase: Phenomenex Gemini® 5pm C18 lloA
Mobile phase: H2O/HCOOH 0.1% : Methanol
Elution : Gradient Time (min) H2O/HCOOH 0.1% Methanol
0 95 5
5 95 5
30 50 50
35 50 50
40 95 5
Flow 0.6 mL/min
Temperature 25 °C
Detection PDA scan wavelenght 190-800nm
Injection volume 50 pL
Sample Cone. 0.2 mM Gd(PCTA-tris-glutaric acid) complex
Stop time 40 min
Retention time GdL = 18-21 min.
Obtained HPLC chromatogram is shown in Figure 1
The HPLC chromatogram of the enriched enantiomers pair C is shown in Figure 2.
Procedure 2: HPLC Characterization of Gadopiclenol (isomeric mixture) and compounds obtained by coupling of enantiomers pair C with R, S, or racemic isoserinol.
The HPLC characterization of Gadopiclenol either as isomeric mixture obtained from Example 2, or as the compound obtained by conjugation of enantiomers pair C of the Gd(PCTA-tris-glutaric acid) with R, S, or racemic isoserinol was performed with Thermo Finnigan LCQ DECA XPPIus system. The experimental setup of the HPLC measurements are summarized below.
Analytical conditions
HPLC system HPLC equipped with quaternary pump, degasser, autosampler,
PDA and MS detector (LCQ Deca XP-Plus – Thermo Finnigan )
Stationary phase: Phenomenex Gemini 5u C18 110A
Mobile phase: H2O/TFA 0.1% : Acetonitrile/0.1%TFA
Elution : Gradient Time (min) H2O/TFA 0.1% Acetonitrile/0.1%TFA
0 100 0
5 100 0
22 90 10
26 90 10
Flow 0.5 mL/min
Temperature 25 °C
Detection PDA scan wavelenght 190-800nm
MS positive mode – Mass range 100-2000
Injection volume 50 pL
Sample cone. 0.2 mM Gd complex
Stop time 26 min
Retention time GdL = 20-22min.
Obtained HPLC chromatograms are shown in Figure 6.
Procedure 3: Chiral HPLC method for the separation of enantiomers of the compound C
A specific chiral HPLC method was set up in order to separate the RRR and SSS enantiomers of the enantiomers pair C (compound VI), prepared as described in Example 3. The separation and characterization of the enantiomers were performed with Agilent 1200 system or Waters Alliance 2695 system. The experimental setup of the HPLC measurements are summarized below.
Analytical conditions
HPLC System HPLC equipped with quaternary pump, degasser, autosampler,
PDA detector
Stationary phase SUPELCO Astec CHIROBIOTIC 5 pm 4.6x250mm
Mobile phase H2O/HCOOH 0.025% : Acetonitrile
Elution : isocratic 2% Acetonitrile for 30 minutes
Flow 1 mL/min
Column Temperature 40°C
Detection 210-270 nm. Obtained HPLC chromatogram is shown in Figure 5a) compared to the chromatograms of the pure RRR enantiomer (compound XII of Example 5, Tr. 7.5 min.) and the pure SSS enantiomer (Compound XVII of Example 6, Tr. 8.0 min), shown in figure 5b) and 5c), respectively.
Example 1: Synthesis of Gd(PCTA-tris-glutaric acid) (isomeric mixture)
Gd(PCTA-tris-glutaric acid) as an indiscriminate mixture of stereoisomers has been prepared by using the procedure reported in above mentioned prior-art, according to the following synthetic Scheme 1 :
Scheme 1

a) Preparation of Compound II
Racemic glutamic acid (33.0 g, 0.224 mol) and sodium bromide (79.7 g, 0.782 mol) were suspended in 2M HBr (225 ml_). The suspension was cooled to -5°C and NaN02 (28.0 g, 0.403 mol) was slowly added in small portions over 2.5 hours, maintaining the inner temperature lower than 0 °C. The yellow mixture was stirred for additional 20 minutes at a temperature of -5°C; then concentrated sulfuric acid (29 ml.) was dropped in the mixture. The obtained dark brown mixture was warmed to RT and then extracted with diethyl ether (4×150 ml_). The combined organic phases were washed with brine, dried over Na2S04 and concentrated to a brown oil (21.2 g), used in the following step without further purification. The oil was dissolved in ethanol (240 ml_), the resulting solution was cooled in ice and thionyl chloride (14.5 ml_, 0.199 mol) was slowly added. The slightly yellow solution was stirred at RT for 2 days. Then the solvent was removed in vacuum and the crude oil was dissolved in dichloromethane (200 ml.) and washed with 5% aq. NaHCC>3 (4×50 ml_), water (1×50 ml.) and brine (1×50 ml_). The organic phase was concentrated and purified on silica eluting with petroleum ether-ethyl acetate 3: 1, obtaining 19.5 g of pure product. (Yield 33%).
b) Preparation of Compound IV
A solution of Compound II (17.2 g, 0.0645 mol) in acetonitrile (40 ml.) was added to a suspension of 3,6,9,15-tetraazabicyclo[9.3.1]pentadeca-l(15),l l,13-triene (pyclen) Compound (III) (3.80 g, 0.018 mol) and K2CO3 (11.2 g, 0.0808 mol) in acetonitrile (150 ml_). The yellow suspension was heated at 65 °C for 24 h, then the salts were filtered out and the organic solution was concentrated. The orange oil was dissolved in dichloromethane and the product was extracted with 1M HCI (4 x 50 ml_). The aqueous phases were combined, cooled in ice and brought to pH 7-8 with 30% aq. NaOH. The product was then extracted with dichloromethane (4 x 50 ml.) and concentrated to give a brown oil (10.1 g, yield 73%). The compound was used in the following step without further purification.
c) Preparation of compound V
Compound IV (9.99 g, 0.013 mol) was dissolved in Ethanol (40 ml.) and 5M NaOH (40 ml_). The brown solution was heated at 80 °C for 23 h. Ethanol was concentrated; the solution was cooled in ice and brought to pH 2 with cone HCI. The ligand was purified on resin Amberlite XAD 1600, eluting with water-acetonitrile mixture, obtaining after freeze- drying 5.7 g as white solid (yield 73%). The product was characterized in HPLC by several peaks.
d) Preparation of compound VI
Compound V (5.25 g, 0.0088 mol) was dissolved in deionized water (100 ml.) and the solution was brought to pH 7 with 2M NaOH (20 ml_). A GdCh solution (0.0087 mol) was slowly added at RT, adjusting the pH at 7 with 2M NaOH and checking the complexation with xylenol orange. Once the complexation was completed, the solution was concentrated and purified on resin Amberlite XAD 1600 eluting with water-acetonitrile gradient, in order to remove salts and impurities. After freeze-drying the pure compound was obtained as white solid (6.79 g, yield 94%). The product was characterized in HPLC; the obtained HPLC chromatogram, characterized by several peaks, is shown in Figure 1 A compound totally equivalent to compound VI, consisting of an isomeric mixture with a HPLC chromatogram substantially superimposable to that of Figure 1 is obtained even by using (S)-methyl a-bromoglutarate obtained starting from L-glutamic acid.
Example 2: Synthesis of Gadopiclenol (isomeric mixture)
Gadopiclenol as an indiscriminate mixture of stereoisomers has been prepared as disclosed in EP11931673 B1 by coupling the isomeric mixture of Gd(PCTA-tris-glutaric acid) obtained from Example 1 with racemic isoserinol according to the following synthetic Scheme 2:
Scheme 2

Preparation of compound VII
Compound VI (0.90 g, 0.0011 mol) obtained from Example 1 was added to a solution of racemic isoserinol (0.40 g, 0.0044 mol) in water adjusted to pH 6 with cone. HCI. Then N- ethyl-N’-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDCI-HCI) (1.0 g, 0.0055 mol) and hydroxybenzotriazole (HOBT) (0.12 g, 0.00088 mol) were added and the resulting solution was stirred at pH 6 and RT for 24 h. The product was then purified on preparative HPLC on silica C18, eluting with water/acetonitrile gradient. Fractions containing the pure compound were concentrated and freeze-dried, obtaining a white solid (0.83 g, yield 78%). The product was characterized in HPLC; the obtained HPLC chromatogram is shown in Figure 4a.
Example 3: Isolation of the enantiomers pair related to the peak C.
Compound VI obtained as described in Example 1 (step d) (1.0 g, 0.0013 mol) was dissolved in water (4 ml.) and the solution was acidified to pH 2-3 with cone. HCI. The obtained solution was loaded into a pre-packed column of silica C18 (Biotage® SNAP ULTRA C18 120 g, HP-sphere C18 25 pm) and purified with an automated flash chromatography system eluting with deionized water (4 CV) and then a very slow gradient of acetonitrile. Fractions enriched of the enantiomers pair related to the peak C were combined, concentrated and freeze-dried obtaining a white solid (200 mg).
The HPLC chromatogram of the obtained enriched enantiomers pair C is shown in Figure 2.
Corresponding MS spectrum (Gd(H4L)+:752.14 m/z) is provided in Figure 3
Example 4: Coupling of the enantiomers pair C with isoserinol.
a) Coupling of the enantiomers pair C with R-isoserinol.
Enriched enantiomers pair C collected e.g. as in Example 3 (34 mg, titer 90%, 0.040 mmol) was dissolved in deionized water (5 ml_), and R-isoserinol (16 mg, 0.17 mmol) was added adjusting the pH at 6 with HCI 1M. Then, EDCI-HCI (39 mg, 0.20 mmol) and HOBT (3 mg, 0.02 mmol) were added and the solution was stirred at RT at pH 6 for 48 h. The solution was concentrated and loaded to pre-packed silica C18 column (Biotage® SNAP ULTRA C18 12 g, HP-sphere C18 25 pm), eluting with water/acetonitrile gradient using an automated flash chromatography system. Fractions containing the pure product, or showing a major peak at the HPLC with area greater than 90%, were combined, concentrated and freeze-dried giving a white solid (21 mg, yield 54%).
The HPLC chromatogram of the obtained product is shown in Figure 6b.
b) Coupling of the enantiomers pair C with S-isoserinol
Enriched enantiomers pair C collected e.g. as in Example 3 (55 mg, titer 90%, 0.066 mmol) was dissolved in deionized water (5 mL), and S-isoserinol (34 mg, 0.29 mmol) was added adjusting the pH at 6 with 1M HCI. Then, EDCI-HCI (64 mg, 0.33 mmol) and HOBT (4.5 mg, 0.033 mmol) were added and the solution was stirred at RT at pH 6 for 48 h. The solution was concentrated and loaded to pre-packed silica C18 column (Biotage® SNAP ULTRA C18 12 g, HP-sphere C18 25 pm), eluting with water/acetonitrile gradient using an automated flash chromatography system. Fractions containing the pure product, or showing a major peak at the HPLC with area greater than 90%, were combined, concentrated and freeze-dried giving a white solid (52 mg, yield 81%).
HPLC chromatogram of the obtained product is shown in Figure 6c.
c) Coupling of the enantiomers pair C with racemic isoserinol.
The enriched enantiomers pair C collected e.g. as in Example 3 (54 mg, titer 90%, 0.065 mmol) was dissolved in deionized water (5 mL), and racemic isoserinol (27 mg, 0.29 mmol) was added adjusting the pH at 6 with 1M HCI. Then, EDCI-HCI (62 mg, 0.32 mmol) and HOBT (4.3 mg, 0.032 mmol) were added and the solution was stirred at RT at pH 6 for 24 h. The solution was concentrated and loaded to pre-packed silica C18 column (Biotage® SNAP ULTRA C18 12 g, HP-sphere C18 25 pm), eluting with water/acetonitrile gradient using an automated flash chromatography system. Fractions containing the pure product, or showing a major peak at the HPLC with area greater than 90%, were combined, concentrated and freeze-dried giving a white solid (60 mg, yield 95%).
HPLC chromatogram of the obtained product is shown in Figure 6d. Example 5: Stereoselective synthesis of the RRR Gd(PCTA-tris-glutaric acid) (compound XII).
RRR enriched Gd(PCTA-tris-glutaric acid) acid has been prepared by following the synthetic Scheme 3 below
Scheme 3

comprising :
a) Preparation of Compound VIII
The preparation was carried out as reported in Tetrahedron 2009, 65, 4671-4680.
In particular: 37% aq. HCI (50 pL) was added to a solution of (S)-(+)-5- oxotetrahydrofuran-2-carboxylic acid (2.48 g, 0.019 mol) (commercially available) in anhydrous methanol (20 ml_). The solution was refluxed under N2 atmosphere for 24 h. After cooling in ice, NaHCC>3 was added, the suspension was filtered, concentrated and purified on silica gel with hexanes/ethyl acetate 1 : 1. Fractions containing the pure product were combined and concentrated, giving a colorless oil (2.97 g, yield 89%).
b) Preparation of Compounds IX and X
Compound VIII (445 mg, 2.52 mmol) obtained at step a) was dissolved in anhydrous dichloromethane (6 ml.) and triethylamine (0.87 ml_, 6.31 mmol) was added. The solution was cooled at -40°C and then (triflic) trifluoromethansulfonic anhydride (0.49 ml_,2.91 mmol) was slowly added. The dark solution was stirred at -40°C for 1 h, then a solution of Compound III (104 mg, 0.506 mmol) in anhydrous dichloromethane (3 ml.) and triethylamine (1 ml_, 7.56 mmol) were added and the solution was slowly brought to RT and stirred at RT overnight. The organic solution was then washed with 2M HCI (4x 10 ml_), the aqueous phase was extracted again with dichloromethane (3 x 10 ml_). The organic phases were combined and concentrated in vacuum, obtaining 400 mg of a brown oil that was used in the following step with no further purification.
c) Preparation of Compound XI
Compound X (400 mg, 0.59 mmol) was dissolved in methanol (2.5 ml.) and 5M NaOH (2.5 ml_). The brown solution was heated at 80°C for 22 h to ensure complete hydrolysis. Methanol was concentrated, the solution was brought to pH 1 with concentrated HCI and purified through an automated flash chromatography system with a silica C18 pre-packed column (Biotage® SNAP ULTRA C18 12 g, HP-sphere C18 25 pm), eluting with deionized water/acetonitrile gradient. Fractions containing the pure product were combined, concentrated and freeze-dried (64 mg, yield 18 %). The HPLC showed a major peak.
d) Compound XII
Compound XI (32 mg, 0.054 mmol) was dissolved in deionized water (4 mL) and the pH was adjusted to 7 with 1M NaOH. GdCl3-6H20 (20 mg, 0.054 mmol) was added and the pH was adjusted to 7 with 0.1 M NaOH. The clear solution was stirred at RT overnight and the end of the complexation was checked by xylenol orange and HPLC. The HPLC of the crude showed the desired RRR isomer as major peak: about 80% in area %. The mixture was brought to pH 2 with concentrated HCI and purified through an automated flash chromatography system with a silica C18 pre-packed column (Biotage® SNAP ULTRA C18 12 g, HP-sphere C18 25 pm), eluting with deionized water/acetonitrile gradient. Fractions containing the pure product were combined, concentrated and freeze-dried (36 mg, yield 90%).
By reaction of the collected compound with isoserinol e.g. by using the procedure of the Example 2, the corresponding RRR amide derivative can then be obtained.
Example 6: stereoselective synthesis of the SSS Gd(PCTA-tris-glutaric acid) (compound XVII).
SSS enriched Gd(PCTA-tris-glutaric acid) acid has been similarly prepared by following the synthetic Scheme 4 below Scheme 4

comprising :
a) Preparation of Compound XIII
37% aq. HCI (100 pl_) was added to a solution of (R)-(-)-5-oxotetrahydrofuran-2- carboxylic acid (5.0 g, 0.038 mol) (commercially available) in anhydrous methanol (45 ml_). The solution was refluxed under N2 atmosphere for 24 h. After cooling in ice, NaHC03 was added, the suspension was filtered, concentrated and purified on silica gel with hexanes/ethyl acetate 1 : 1. Fractions containing the pure product were combined and concentrated, giving a colorless oil (6.7 g, yield 99%).
b) Preparation of Compounds XIV and XV
Compound XIII (470 mg, 2.67 mmol) was dissolved in anhydrous dichloromethane (6 ml.) and trimethylamine (0.93 ml_, 6.67 mmol) was added. The solution was cooled down at -40°C and then trifluoromethanesulfonic anhydride (0.50 ml_, 3.07 mmol) was slowly dropped. The dark solution was stirred at -40°C for 1 h, then Compound III (140 mg, 0.679 mmol) and trimethylamine (0.93 ml_, 6.67 mmol) were added and the solution was slowly brought to RT overnight. The organic solution was then washed with water (3 x 5 ml.) and 2M HCI (4 x 5 ml_). The aqueous phase was extracted again with dichloromethane (3 x 10 ml_). the organic phases were combined and concentrated in vacuum, obtaining 350 mg of a brown oil that was used in the following step with no further purification. c) Preparation of Compound XVI
Compound XV (350 mg, 0.514 mmol) was dissolved in methanol (4.5 ml.) and 5M NaOH (4.5 ml_). The obtained brown solution was heated at 80°C for 16 h to ensure complete hydrolysis. Methanol was concentrated, the solution was brought to pH 2 with concentrated HCI and purified through an automated flash chromatography system with a silica C18 pre-packed column (Biotage® SNAP ULTRA C18 12 g, HP-SPHERE C18 25 pm), eluting with a water/acetonitrile gradient. Fractions containing the pure product were combined, concentrated and freeze-dried (52 mg, yield 17%). The HPLC showed a major peak.
d) Preparation of Compound XVII
Compound XVI (34 mg, 0.057 mmol) was dissolved in deionized water (5 mL) and the pH was adjusted to 7 with 1 M HCI. GdCl3-6H20 (20 mg, 0.0538 mmol) was added and the pH was adjusted to 7 with 0.1 M NaOH. The solution was stirred at RT overnight and the end of complexation was checked by xylenol orange and HPLC. The HPLC of the crude showed the desired SSS isomer as major peak: about 85% in area %. The solution was brought to pH 2.5 with concentrated HCI and purified through an automated flash chromatography system with a silica C18 pre-packed column (Biotage® SNAP ULTRA C18 12 g, HP-SPHERE C18 25 pm), eluting with a water/acetonitrile gradient. Fractions containing the pure product SSS were combined, concentrated and freeze-dried (39 mg, yield 87%).
Example 7: Kinetic studies of the dissociation reactions of Gd(PCTA-tris- glutaric acid) (isomeric mixture) in 1.0 M HCI solution (25°C)
The kinetic inertness of a Gd(III)-complex is characterized either by the rate of dissociation measured in 0.1-1.0 M HCI or by the rate of the transmetallation reaction, occurring in solutions with Zn(II) and Cu(II) or Eu(III) ions. However, the dissociation of lanthanide(III)-complexes formed with macrocyclic ligands is very slow and generally proceeds through a proton-assisted pathway without the involvement of endogenous metal ions like Zn2+ and Cu2+.
We characterized the kinetic inertness of the complex Gd(PCTA-tris-glutaric acid) by the rates of the dissociation reactions taking place in 1.0 M HCI solution. The complex (isomeric mixture from Example 1) (0.3 mg) was dissolved in 2.0 mL of 1.0 M HCI solution and the evolution of the solution kept at 25 °C was followed over time by HPLC. The HPLC measurements were performed with an Agilent 1260 Infinity II system by use of the analytical Procedure 1.
The presence of a large excess of H+ ([HCI] = 1.0 M), guarantees the pseudo-first order kinetic conditions.
GdL + yH÷ ^ Gd3+ + HyL y=7 and 8 (Eg. 1) where L is the protonated PCTA-tri-glutaric acid, free ligand, and y is the number of protons attached to the ligand.
The HPLC chromatogram of Gd(PCTA-tris-glutaric acid) is characterized by the presence of four signals (A, B, C and D) having the same m/z ratio (Gd(H4L)+ :752.14 m/z) in the MS spectrum. Each of these peaks is reasonably ascribable to one of the 4 pairs of enantiomers generated by the three stereocenters on the three glutaric arms of the molecule, formerly identified in Table 1. The HPLC chromatogram of this complex in the presence of 1.0 M HCI changes over time: in particular, the areas of peaks A, B, C, and D decrease, although not in the same way for the different peaks, while new signals corresponding to non-complexed diastereoisomers are formed and grow over time. Differences in the decrease of the integral areas of the peaks can be interpreted by a different dissociation rate of the enantiomer pairs associated to the different peaks.
In the presence of [H + ] excess the dissociation reaction of enantiomer pairs of Gd(PCTA-tris-glutaric acid) can be treated as a pseudo-first-order process, and the rate of the reactions can be expressed with the following Eq. 2, where kA, kB, kc and kD are the pseudo-first-order rate constants that are calculated by fitting the area-time data pair, and [A]t, [B]t, [C]t and [D]t are the total concentration of A, B, C and D compounds at time t.

The decrease of the area values of signals of A, B, C, and D has been assessed and plotted over time. Area values of A, B, C and D signals as a function of time are shown in Figure 7.
Area value at time t can be expressed by the following equation:
A. = A + (A0 – A )e kxt
(Eg. 3)
where At, A0 and Ae are the area values at time t, at the beginning and at the end of the reactions, respectively, kx pseudo-first-order rate constants (/fX=/fA, kB, kc and kD) characterizing the dissociation rate of the different enantiomer pairs of Gd(PCTA-tris-glutaric acid) complex were calculated by fitting the area – time data pairs of Figure 7 to the above equation 3. kx rate constants and half-lives (ti/2= In2/ x) are thus obtained, as well as the average the half-life value for the isomeric mixture of Gd(PCTA-tris-glutaric acid), calculated by considering the percentage composition of the mixture. Obtained values are summarized in the following Table 2, and compared with corresponding values referred in the literature for some reference contrast agents. (Gd-DOTA or DOTAREM™). Table 2. Rate constants ( kx ) and half-lives (ti/2= In2/ x) characterizing the acid catalyzed dissociation of the different stereoisomers of Gd(PCTA-tris-glutaric acid), Dotarem® and Eu(PCTA) in 1.0 M HCI (pH 0) ( 25°C)
A B C D
Ms 1) (4.5±0.1) x105 (1.1±0.1)x104 (1.6±0.1)x10-6 (1.2±0.1)x10-5 fi/2 (hour) 4.28 ± 0.03 1.76 ± 0.02 120 ± 3 15.8 ± 0.5
fi/2 (hour)

average
Dotarem a
k, (S‘1) 8.0×10-6
fi/2 (hour) 23 hour
Eu(PCTA) b
*1 (s·1) 5.08X10·4
fi/2 (hour) 0.38 hour
a) Inorg. Chem. 1992, 31 ,1095-1099.
b) Tircso, G. et al. Inorg Chem 2006, 45 (23), 9269-80.
/////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
A gadolinium-based paramagnetic contrast agent, with potential imaging enhancing activity upon magnetic resonance imaging (MRI). Upon administration of gadopiclenol and placement in a magnetic field, this agent produces a large magnetic moment and creates a large local magnetic field, which can enhance the relaxation rate of nearby protons. This change in proton relaxation dynamics, increases the MRI signal intensity of tissues in which this agent has accumulated; therefore, contrast and visualization of those tissues is enhanced compared to unenhanced MRI.
FDA Approves New MRI Contrast Agent Gadopiclenol
September 22, 2022
https://www.diagnosticimaging.com/view/fda-approves-new-mri-contrast-agent-gadopiclenol
Requiring only half of the gadolinium dose of current non-specific gadolinium-based contrast agents (GBCAs), gadopiclenol can be utilized with magnetic resonance imaging (MRI) to help detect lesions with abnormal vascularity in the central nervous system and other areas of the body.
Gadopiclenol, a new magnetic resonance imaging (MRI) contrast agent that offers high relaxivity and reduced dosing of gadolinium, has been approved by the Food and Drug Administration (FDA).1
Approved for use with MRI in adults and pediatric patients two years of age or older, gadopiclenol is a macrocyclic gadolinium-based contrast agent that aids in the diagnosis of lesions with abnormal vascularity in the brain, spine, abdomen, and other areas of the body.
Recently published research demonstrated that gadopiclenol provides contrast enhancement and diagnostic efficacy at half of the gadolinium dosing of other gadolinium-based contrast agents (GBCAs) such as gadobutrol and gadobenate dimeglumine.2
Co-developed by Bracco Diagnostics and Guerbet, gadopiclenol will be manufactured and marketed as Vueway™ (Bracco Diagnostics) and Elucirem™ (Guerbet).1,3
Alberto Spinazzi, M.D., the chief medical and regulatory officer at Bracco Diagnostics, said gadopiclenol is “a first of its kind MRI agent that delivers the highest relaxivity and highest kinetic stability of all GBCAs on the market today.”
Reference
1. Bracco Diagnostics. Bracco announces FDA approval of gadopiclenol injection, a new macrocyclic high-relaxivity gadolinium-based contrast agent which will be commercialized as VUEWAY™ (gadopiclenol) injection and VUEWAY™ (gadopiclenol) phamarcy bulk package by Bracco. Cision PR Newswire. Available at: https://www.prnewswire.com/news-releases/bracco-announces-fda-approval-of-gadopiclenol-injection-a-new-macrocyclic-high-relaxivity-gadolinium-based-contrast-agent-which-will-be-commercialized-as-vueway-gadopiclenol-injection-and-vueway-gadopiclenol-pharmacy-bulk-p-301630124.html . Published September 21, 2022. Accessed September 21, 2022.
2. Bendszus M, Roberts D, Kolumban B, et al. Dose finding study of gadopiclenol, a new macrocyclic contrast agent, in MRI of central nervous system. Invest Radiol. 2020;55(3):129-137.
3. Guerbet. Guerbet announces U.S. Food and Drug Administration (FDA) approval of Elucirem™ (gadopiclenol) injection for use in contrast-enhanced MRI. Cision PR Newswire. Available at: https://www.prnewswire.com/news-releases/guerbet-announces-us-food-and-drug-administration-fda-approval-of-elucirem-gadopiclenol-injection-for-use-in-contrast-enhanced-mri-301630085.html . Published September 21, 2022. Accessed September 21, 2022.
////Gadopiclenol, FDA 2022, APPROVALS 2022, ガドピクレノール, WHO 10744, P 03277, EluciremTM, G03277; P03277, VUEWAY, Guerbet

NEW DRUG APPROVALS
ONE TIME
$10.00
Eflapegrastim
Eflapegrastim
エフラペグラスチム;
Molecular Formula
- C15-H28-N2-O6(C2-H4-O)n
Molecular Weight
- 376.4468
| Formula | C3070H4764N806O927S23.(C2H4O)n |
|---|
UNII: UT99UG9QJX
HM10460A
SPI-2012
- HNK460
Reducing neutropenia and the incidence of infecton in patients with cancer
(2S)-1-{3-[2-(3-{[(1S,2R)-1-carboxy-2-hydroxypropyl]amino}propoxy)ethoxy]propyl}pyrrolidine-2-carboxylic acid
APPROVED FDA 2022/9/9, Rolvedon
CAS: 1384099-30-2
LAPS-GCSF, ROLONTIS
Antineutropenic, Leukocyte growth factor
Poly(oxy-1,2-ethanediyl), α-hydro-ω-hydroxy-, 1-ether with immunoglobulin G4 [1-[1-(3-hydroxypropyl)proline]] (human Fc fragment), (3→3′)-disulfide with immunoglobulin G4 (human Fc fragment), 1′′-ether with granulocyte colony-stimulating factor [N-(3-hydroxypropyl),17-serine,65-serine] (human) (ACI)
A long-acting, recombinant analog of the endogenous human granulocyte colony-stimulating factor (G-CSF) with hematopoietic activity. Similar to G-CSF, eflapegrastim binds to and activates specific cell surface receptors and stimulates neutrophil progenitor proliferation and differentiation, as well as selected neutrophil functions. Therefore, this agent may decrease the duration and incidence of chemotherapy-induced neutropenia. Eflapegrastim extends the half-life of G-CSF, allowing for administration once every 3 weeks.
- A long-acting GCSF that consists of 17th serine-G-CSF conjugated to the G4 fragment HMC001 via a PEG linker.
PATENT
WO2021113597
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2021113597
Neutropenia is a relatively common disorder most often due to chemotherapy treatments, adverse drug reactions, or autoimmune disorders. Chemotherapy-induced neutropenia is a common toxicity caused by the administration of anticancer drugs. It is associated with life-threatening infections and may alter the chemotherapy schedule, thus impacting on early and long term outcome. Febrile Neutropenia (FN) is a major dose-limiting toxicity of myelosuppressive chemotherapy regimens such as docetaxel, doxorubicin, cyclophosphamide (TAC); dose-dense doxorubicin plus cyclophosphamide (AC), with or without subsequent weekly or semiweekly paclitaxel; and docetaxel plus cyclophosphamide (TC). It usually leads to prolonged hospitalization, intravenous administration of broad-spectrum antibiotics, and is often associated with significant morbidity and mortality.
Current therapeutic modalities employ granulocyte colony-stimulating factor (G-CSF) and/or antibiotic agents to combat this condition. G-CSF or its other polypeptide derivatives are easy to denature or easily de-composed by proteolytic enzymes in blood to be readily removed through the kidney or liver. Therefore, to maintain the blood concentration and titer of the G-CSF containing drugs, it is necessary to frequently administer the protein drug to patients, which causes excessive suffering in patients. To solve such problems, G-CSF was chemically attached to polymers having a high solubility such as polyethylene glycol (“PEG”), thereby increasing its blood stability and maintaining suitable blood concentration for a longer time.
Filgrastim, tbo-filgrastim, and pegfilgrastim are G-CSFs currently approved by the US Food and Drug Administration (FDA) for the prevention of chemotherapy-induced neutropenia, While the European guidelines also include lenograstim as a recommended G-CSF in solid tumors and non-myeloid malignancies, it is not approved for use in the US. Binding of PEG to G-CSF, even though may increase blood stability, does dramatically reduce the titer needed for optimal physiologic effect. Thus there is a need to address this shortcoming in the art.
PATENT
WO2021112654
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2021112654
Eflapegrastim
[54]
Eflapegrastim, as known as Rolontis ®, SPI-2012, HM10460A, and 17,65S-G-CSF, is a long-acting granulocyte-colony stimulating factor (G-CSF) that has been developed to reduce the severity and duration of severe neutropenia, as well as complications of neutropenia, associated with the use of myelosuppressive anti-cancer drugs or radiotherapy. Eflapegrastim consists of a recombinant human G-CSF analog (ef-G-CSF) and a recombinant fragment of the Fc region of human immunoglobulin G4 (IgG4), linked by a Bifunctional polyethylene glycol linker. In certain embodiments, the recombinant human G-CSF analog (ef-G-CSF) varies from human G-CSF (SED ID NO: 1) at positions 17 and 65 which are substituted with serine (SED ID NO: 2). Without wishing to be bound by theory, it is believed that the Fc region of human IgG4 increases the serum half-life of ef-G-CSF.
[55]
ef-G-CSF is produced by transformed E. coli in soluble form in the periplasmic space. Separately, the Fc fragment is produced in transformed E. coli as an inclusion body. The ef-G-CSF and the Fc fragment are independently isolated and purified through successive purification steps. The purified ef-G-CSF (SEQ ID NO: 2) and Fc fragment (SEQ ID NOs: 3 and 4) are then linked via a 3.4 kDa PEG molecule that was designed with reactive groups at both ends. Eflapegrastim itself is the molecule resulting from the PEG linker binding at each of the N-termini of ef-G-CSF and the Fc fragment. The G-CSF analog is conjugated to the 3.4 kDa polyethylene glycol analogue with propyl aldehyde end groups at both ends, (OHCCH 2CH 2(OCH 2CH 2) nOCH 2CH 2CHO) at the nitrogen atom of its N-terminal Thr residue via reductive amination to form a covalent bond. The resulting G-CSF-PEG complex is then linked to the N-terminal Pro at the nitrogen of the recombinant Fc fragment variant produced in E. coli via reductive amination to yield the final conjugate of Eflapegrastim.
[56]
Example 1: Preparation of Eflapegrastim ( 17,65S-G-CSF-PEG-Fc)
[120]
Step 1: Preparation of Immunoglobulin Fc Fragment Using Immunoglobulin
[121]
Preparation of an immunoglobulin Fc fragment was prepared as follows.
[122]
200 mg of 150-kDa immunoglobulin G (IgG) (Green Cross, Korea) dissolved in 10 mM phosphate buffer was treated with 2 mg of a proteolytic enzyme, papain (Sigma) at 37℃ for 2 hrs with gentle agitation.
[123]
After the enzyme reaction, the immunoglobulin Fc fragment regenerated thus was subjected to chromatography for purification using sequentially a Superdex column, a protein A column and a cation exchange column. In detail, the reaction solution was loaded onto a Superdex 200 column (Pharmacia) equilibrated with 10 mM sodium phosphate buffer (PBS, pH 7.3), and the column was eluted with the same buffer at a flow rate of 1 ml/min. Unreacted immunoglobulin molecules (IgG) and F(ab’)2, which had a relatively high molecular weight compared to the immunoglobulin Fc fragment, were removed using their property of being eluted earlier than the Ig Fc fragment. Fab fragments having a molecular weight similar to the Ig Fc fragment were eliminated by protein A column chromatography (FIGURE 1). The resulting fractions containing the Ig Fc fragment eluted from the Superdex 200 column were loaded at a flow rate of 5 ml/min onto a protein A column (Pharmacia) equilibrated with 20 mM phosphate buffer (pH 7.0), and the column was washed with the same buffer to remove proteins unbound to the column. Then, the protein A column was eluted with 100 mM sodium citrate buffer (pH 3.0) to obtain highly pure immunoglobulin Fc fragment. The Fc fractions collected from the protein A column were finally purified using a cation exchange column (polyCAT, PolyLC Company), wherein this column loaded with the Fc fractions was eluted with a linear gradient of 0.15-0.4 M NaCl in 10 mM acetate buffer (pH 4.5), thus providing highly pure Fc fractions. The highly pure Fc fractions were analyzed by 12% SDS-PAGE (lane 2 in FIGURE 2).
[124]
Step 2: Preparation of 17,65S-G-CSF-PEG Complex
[125]
3.4-kDa polyethylene glycol having an aldehyde reactive group at both ends, ALD-PEG-ALD (Shearwater), was mixed with human granulocyte colony stimulating factor ( 17,65S-G-CSF, MW: 18.6 kDa) dissolved in 100 mM phosphate buffer in an amount of 5 mg/ml at a 17,65S-G-CSF: PEG molar ratio of 1:5. To this mixture, a reducing agent, sodium cyanoborohydride (NaCNBH 3, Sigma), was added at a final concentration of 20 mM and was allowed to react at 4℃ for 3 hrs with gentle agitation to allow PEG to link to the amino terminal end of 17,65S-G-CSF. To obtain a 1:1 complex of PEG and 17,65S-G-CSF, the reaction mixture was subjected to size exclusion chromatography using a Superdex R column (Pharmacia). The 17,65S-G-CSF-PEG complex was eluted from the column using 10 mM potassium phosphate buffer (pH 6.0) as an elution buffer, and 17,65S-G-CSF not linked to PEG, unreacted PEG and dimer byproducts where PEG was linked to 17,65S-G-CSF molecules were removed. The purified 17,65S-G-CSF-PEG complex was concentrated to 5 mg/ml. Through this experiment, the optimal reaction molar ratio for 17,65S-G-CSF to PEG, providing the highest reactivity and generating the smallest amount of byproducts such as dimers, was found to be 1:5.
[126]
Step 3: Preparation of the 17,65S-G-CSF-PEG-Fc Conjugate
[127]
To link the 17,65S-G-CSF-PEG complex purified in the above step 2 to the N-terminus of an immunoglobulin Fc fragment, the immunoglobulin Fc fragment (about 53 kDa) prepared in Step 1 was dissolved in 10 mM phosphate buffer and mixed with the 17,65S-G-CSF-PEG complex at an 17,65S-G-CSF-PEG complex:Fc molar ratio of 1:1, 1:2, 1:4 and 1:8. After the phosphate buffer concentration of the reaction solution was adjusted to 100 mM, a reducing agent, NaCNBH 3, was added to the reaction solution at a final concentration of 20 mM and was allowed to react at 4℃ for 20 hrs with gentle agitation. Through this experiment, the optimal reaction molar ratio for 17,65S-G-CSF-PEG complex to Fc, providing the highest reactivity and generating the fewest byproducts such as dimers, was found to be 1:2.
[128]
Step 4: Isolation and Purification of the G-CSF-PEG-Fc Conjugate
[129]
After the reaction of the above step 3, the reaction mixture was subjected to Superdex size exclusion chromatography so as to eliminate unreacted substances and byproducts and purify the 17,65S-G-CSF-PEG-Fc protein conjugate produced. After the reaction mixture was concentrated and loaded onto a Superdex column, 10 mM phosphate buffer (pH 7.3) was passed through the column at a flow rate of 2.5 ml/min to remove unbound Fc and unreacted substances, followed by column elution to collect 17,65S-G-CSF-PEG-Fc protein conjugate fractions. Since the collected 17,65S-G-CSF-PEG-Fc protein conjugate fractions contained a small amount of impurities, unreacted Fc and interferon alpha dimers, cation exchange chromatography was carried out to remove the impurities. The 17,65S-G-CSF-PEG-Fc protein conjugate fractions were loaded onto a PolyCAT LP column (PolyLC) equilibrated with 10 mM sodium acetate (pH 4.5), and the column was eluted with a linear gradient of 0-0.5 M NaCl in 10 mM sodium acetate buffer (pH 4.5) using 1 M NaCl. Finally, the 17,65S-G-CSF-PEG-Fc protein conjugate was purified using an anion exchange column. The 17,65S-G-CSF-PEG-Fc protein conjugate fractions were loaded onto a PolyWAX LP column (PolyLC) equilibrated with 10 mM Tris-HCl (pH 7.5), and the column was then eluted with a linear gradient of 0-0.3 M NaCl in 10 mM Tris-HCl (pH 7.5) using 1 M NaCl, thus isolating the 17,65S-G-CSF-PEG-Fc protein conjugate in a highly pure form.
[130]
[131]
Example 2: Efficacy Study of Eflapegrastim by Different Dosing Regimens in Rats with Docetaxel/Cyclophosphamide induced Neutropenia
[132]
The efficacy of Eflapegrastim (HM10460A), a long acting G-CSF analogue, was compared with Pegfilgrastim by different dosing regimens in a chemotherapy-induced neutropenic rat model.
[133]
In the following study, the Eflapegrastim was created essentially as described in Example 1.
[134]
(i) Materials for Study
[135]
[Table 1] Test Articles
| Name | Batch/Lot No. | Storage Condition | Purity (%) | Expiration Date | Supplier |
| HM10460A | 906617001 | 2~8 ℃ | RP-HPLC: 98.6% IE-HPLC: 97.4% SE-HPLC: 98.6% | 01/31/2019 | – |
| Pegfilgrastim | 1070334 | 2~8 ℃ | – | – | Amgen |
[136]
[Table 2] Vehicles
| Name | Composition | Storage Condition | Supplier |
| Dulbecco’s phosphate buffered saline (DPBS) | – | 2~8 ℃ | Sigma-Aldrich |
[137]
[Table 3] Neutropenia-Inducing Agents
| Name | Batch/Lot No. | Storage Condition | Purity (%) | Expiration Date | Supplier |
| Cyclo-phosphamide | C3250000 | 2~8 ℃ | – | – | Sigma-Aldrich |
| Docetaxel | 17006 | RT (20 – 25 ℃) | – | 10/31/2020 | Hanmi Pharmaceutical Co. |
[138]
Preparing HM10460A Solutions for Subcutaneous Administration
[139]
Preparation of a 61.8 ㎍/kg HM10460A solution for subcutaneous administration: a stock solution of HM10460A (6.0 mg/mL) 92.7 μL was diluted with DPBS 17907.3 μL.
[140]
Preparation of a 372.0 ㎍/kg HM10460A solution for subcutaneous administration: a stock solution of HM10460A (6.0 mg/mL) 558.0 μL was diluted with DPBS 17442.0 μL.
[141]
Preparation of a 496.0 ㎍/kg HM10460A solution for subcutaneous administration: a stock solution of HM10460A (6.0 mg/mL) 744.0μL was diluted with DPBS 17256.0 μL.
[142]
The test article was prepared based on G-CSF protein dosage on drug label(HM10460A.)
[143]
The HM10460A solution for subcutaneous administration was then diluted with DPBS to a final dose concentration of 2 mL/kg.
[144]
Preparing Pegfilgrastim Solutions for Subcutaneous Administration
[145]
Preparation of a 103.3 ㎍/kg Pegfilgrastim solution for subcutaneous administration: a stock solution of Pegfilgrastim (10 mg/mL) 93.0 μL was diluted with DPBS 17907.0 μL.
[146]
Preparation of a 620.0 ㎍/k Pegfilgrastim solution for subcutaneous administration: a stock solution of Pegfilgrastim (10 mg/mL) 558.0 μL was diluted with DPBS 17442.0 μL.
[147]
The Pegfilgrastim solution for subcutaneous administration was then diluted with DPBS to a final dose concentration of 2 mL/kg.
[148]
Preparing Solutions of Neutropenia-Inducing Agents
[149]
To induce neutropenia in rats, Docetaxel/cyclophosphamide was administered using a 1/3 human equivalent dose (Docetaxel 4 mg/kg and CPA 32 mg/kg) (“TC”).
[150]
Preparation of a 32 mg/kg cyclophosphamide solution for subcutaneous administration: cyclophosphamide powder (CPA, Sigma, USA) 2560.0 g was diluted with distilled water (DW, Daihan, Korea) 80000.0 μL.
[151]
Preparation of a 4 mg/kg docetaxel solution for subcutaneous administration: Docel inj. (Hanmi Pharmaceutical, Korea) (42.68 mg/mL) 29070.0 μL was diluted with a commercial formulation buffer (FB, Etahnol 127.4mg/mL in DW) 30930.0 μL.
[152]
The docetaxel and cyclophosphamide solutions for subcutaneous administration were then diluted with FB to a final dose concentration of 1 mL/kg. HM10460A and Pegfilgrastim were diluted with DPBS to a final dose concentration of 2 mL/kg.
[153]
(ii) Methods
[154]
Test System
[155]
[Table 4]
| Species and Strain | Rats Crl: CD Sprague Dawley (SD) |
| Justification for Species | SD rats were chosen due to their extensive characterization collected from various preclinical studies, especially with the study done to test G-CSF analogue1), 2). |
| Supplier | Orient Bio corp. Korea 143-1, Sangdaewondong, Jungwon-gu, Seongnam-si, Gyeonggi-do, Korea |
| Number of animals | Male 125 (at group allocation) |
| Age | 8 weeks (at group allocation) |
| Body weight range | 239.54 ~ 316.46 g (at start of dosing) |
| Neutropenia induction with chemotherapy | Normal SD rats were administered with Docetaxel 4 mg/kg and CPA 32 mg/kg once intraperitoneally to induce neutropenia. Docetaxel and CPA were injected to induce neutropenia in a rat model according to 4 different regimens: Concomitant (G2-G7), 2 hour (G8-G13), 5 hour (G14-G19), and 24 hour (G20-G25) prior to test article administration. |
[156]
Animal Care and Identification
/////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
Eflapegrastim
25/10/2019by Christian Hilscher
Neutropenia in Breast Cancer: Spectrum Pharmaceuticals has submitted an updated regulatory submission to the US FDA for its biologic Rolontis
10/25/2019 Spectrum Pharmaceutical announced that it has filed an updated Biologics License Application (BLA) with the US Food and Drug Administration (FDA) for Rolontis (eflapegrastim).
The BLA for Rolontis is supported by data from two identically designed Phase 3 clinical trials – ADVANCE and RECOVER – that evaluated the safety and efficacy of eflapegrastim in 643 patients with early breast cancer for the treatment of neutropenia with myelosuppressive chemotherapy.
In both studies, eflapegrastim demonstrated the pre-specified hypothesis of non-inferiority (NI) in Duration of Severe Neutropenia (DSN) and a similar safety profile to pegfilgrastim .
Eflapegrastim also demonstrated non-inferiority to pegfilgrastim in DSN across all 4 cycles in both studies (all NI p<0.0001), the company writes.
© arznei-news.de – Source: Spectrum Pharmaceuticals
Eflapegrastim, sold under the brand names Rolvedon among others, is a long-acting G-CSF analog developed by Hanmi Pharmaceutical and licensed to Spectrum Pharmaceuticals.[2] Eflapegrastim is a leukocyte growth factor.[1] It is used to reduce the risk of febrile neutropenia in people with non-myeloid malignancies receiving myelosuppressive anti-cancer agents.[1]
Eflapegrastim was approved for medical use in the United States in September 2022.[1][3][4]
Medical uses
Eflapegrastim is indicated to decrease the incidence of infection, as manifested by febrile neutropenia, in adults with non-myeloid malignancies receiving myelosuppressive anti-cancer drugs associated with clinically significant incidence of febrile neutropenia.[1]
Its efficacy has been shown to be non-inferior to pegfilgrastim.[1]
References
- ^ Jump up to:a b c d e f “Archived copy” (PDF). Archived (PDF) from the original on 19 September 2022. Retrieved 19 September 2022.
- ^ pharmaceutical, hanmi. “Pipeline – R&D”. Hanmi Pharmaceutical. Archived from the original on 2 February 2017. Retrieved 23 January 2017.
- ^ “Rolvedon: FDA-Approved Drugs”. U.S. Food and Drug Administration (FDA). Archived from the original on 19 September 2022. Retrieved 18 September 2022.
- ^ “Spectrum Pharmaceuticals Receives FDA Approval for Rolvedon (eflapegrastim-xnst) Injection”. Business Wire (Press release). 9 September 2022. Archived from the original on 9 September 2022. Retrieved 18 September 2022.
External links
- “Eflapegrastim”. Drug Information Portal. U.S. National Library of Medicine.
- Clinical trial number NCT02643420 for “SPI-2012 vs Pegfilgrastim in the Management of Neutropenia in Participants With Breast Cancer With Docetaxel and Cyclophosphamide (ADVANCE) (ADVANCE)” at ClinicalTrials.gov
- Clinical trial number NCT02953340 for “SPI-2012 vs Pegfilgrastim in Management of Neutropenia in Breast Cancer Participants With Docetaxel and Cyclophosphamide” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | Rolvedon |
| Other names | Eflapegrastim-xnst, HM-10460A, SPI-2012 |
| Routes of administration | Subcutaneous |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only [1] |
| Identifiers | |
| CAS Number | 1384099-30-2 |
| ChemSpider | None |
| UNII | UT99UG9QJX |
| KEGG | D11188 |
////////////Eflapegrastim, Rolvedon, APPROVALS 2022, FDA 2022, エフラペグラスチム , HM10460A, SPI-2012, HNK460, ROLONTIS

NEW DRUG APPROVALS
ONE TIME
$10.00
Terlipressin acetate

Terlipressin acetate
テルリプレシン酢酸塩
C52H74N16O15S2. (C2H4O2)x
| CAS: 914453-96-6 ACETATEFREE FORM 14636-12-5 |
Terlipressin acetate (JAN);
Heamopressin (TN);
Terlivaz (TN)
Cardiovascular agent
Antidiuretic, Vasoconstrictor, Arginine vasopressin receptor agonist
USFDA APPROVED 2022/9/14
An inactive peptide prodrug that is slowly converted in the body to lypressin. It is used to control bleeding of ESOPHAGEAL VARICES and for the treatment of HEPATORENAL SYNDROME.
- EINECS 238-680-8
- Terlipressin
- Terlipressina
- Terlipressina [INN-Spanish]
- Terlipressine
- Terlipressine [INN-French]
- Terlipressinum
- Terlipressinum [INN-Latin]
- UNII-7Z5X49W53P
acetic acid;(2S)-1-[(4R,7S,10S,13S,16S,19R)-19-[[2-[[2-[(2-aminoacetyl)amino]acetyl]amino]acetyl]amino]-7-(2-amino-2-oxoethyl)-10-(3-amino-3-oxopropyl)-13-benzyl-16-[(4-hydroxyphenyl)methyl]-6,9,12,15,18-pentaoxo-1,2-dithia-5,8,11,14,17-pentazacycloicosane-4-carbonyl]-N-[(2S)-6-amino-1-[(2-amino-2-oxoethyl)amino]-1-oxohexan-2-yl]pyrrolidine-2-carboxamide
FREE FORM
| Formula: | C52H74N16O15S2 |
|---|---|
| Molecular Weight: | 1227.39 |
(2S)-1-[(4R,7S,10S,13S,16S,19R)-19-[[2-[[2-[(2-aminoacetyl)amino]acetyl]amino]acetyl]amino]-13-benzyl-10-(2-carbamoylethyl)-7-(carbamoylmethyl)-16-[(4-hydroxyphenyl)methyl]-6,9,12,15,18-pentaoxo-1,2-dithia-5,8,11,14,17-pentazacycloicosane-4-carbonyl]-N-[(1S)-5-amino-1-(carbamoylmethylcarbamoyl)pentyl]pyrrolidine-2-carboxamide;N-(N-(N-Glycylglycyl)glycyl)-8-L-lysinevasopressin;Glypressin;Terlipressin Acetate;Remestyp;Thymosin α1 Acetate;Gly-Gly-Gly-Cys-Tyr-Phe-Gln-Asn-Cys-Pro-Lys-Gly-NH2 (disulfide bridge 4:9);Glycylpressin;
/////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
Terlipressin, sold under the brand name Terlivaz among others, is an analogue of vasopressin used as a vasoactive drug in the management of low blood pressure. It has been found to be effective when norepinephrine does not help. Terlipressin is a vasopressin receptor agonist.[1]
Medical uses
Terlipressin is indicated to improve kidney function in adults with hepatorenal syndrome with rapid reduction in kidney function.[1]
Indications for use include norepinephrine-resistant septic shock[2] and hepatorenal syndrome.[3] In addition, it is used to treat bleeding esophageal varices.[4]
Contraindications
Terlipressin is contraindicated in people experiencing hypoxia or worsening respiratory symptoms and in people with ongoing coronary, peripheral or mesenteric ischemia.[1] Terlipressin may cause fetal harm when used during pregnancy.[1]
Society and culture
Terlipressin is available in New Zealand,[5] Australia, the European Union,[6] India, Pakistan & UAE. It is sold under various brand names including Glypressin.
| Clinical data | |
|---|---|
| Trade names | Terlivaz |
| AHFS/Drugs.com | International Drug Names |
| Routes of administration | Intravenous |
| ATC code | H01BA04 (WHO) |
| Legal status | |
| Legal status | US: ℞-only [1] |
| Pharmacokinetic data | |
| Protein binding | ~30% |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 14636-12-5 |
| PubChem CID | 72081 |
| DrugBank | DB02638 |
| ChemSpider | 65067 |
| UNII | 7Z5X49W53P |
| KEGG | D06672 |
| CompTox Dashboard (EPA) | DTXSID7048952 |
| ECHA InfoCard | 100.035.149 |
| Chemical and physical data | |
| Formula | C52H74N16O15S2 |
| Molar mass | 1227.38 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
| (verify) |
References
- ^ Jump up to:a b c d e “Archived copy” (PDF). Archived (PDF) from the original on 2022-09-19. Retrieved 2022-09-19.
- ^ O’Brien A, Clapp L, Singer M (2002). “Terlipressin for norepinephrine-resistant septic shock”. Lancet. 359 (9313): 1209–10. doi:10.1016/S0140-6736(02)08225-9. PMID 11955542. S2CID 38463837.
- ^ Uriz J, Ginès P, Cárdenas A, Sort P, Jiménez W, Salmerón J, Bataller R, Mas A, Navasa M, Arroyo V, Rodés J (2000). “Terlipressin plus albumin infusion: an effective and safe therapy of hepatorenal syndrome”. J Hepatol. 33 (1): 43–8. doi:10.1016/S0168-8278(00)80158-0. PMID 10905585.
- ^ Ioannou G, Doust J, Rockey D (2003). Ioannou GN (ed.). “Terlipressin for acute esophageal variceal hemorrhage”. Cochrane Database Syst Rev (1): CD002147. doi:10.1002/14651858.CD002147. PMC 7017851. PMID 12535432.
- ^ http://www.medsafe.govt.nz/profs/datasheet/g/Glypressin01mgmlFerringinj.pdf Archived 2021-12-20 at the Wayback Machine[bare URL PDF]
- ^ “Terlipressin”. Archived from the original on 2019-06-26. Retrieved 2018-01-23.
External links
- “Terlipressin”. Drug Information Portal. U.S. National Library of Medicine.
////Terlipressin acetate, テルリプレシン酢酸塩 , FDA 2022, APPROVALS
2022, CC(=O)O.C1CC(N(C1)C(=O)C2CSSCC(C(=O)NC(C(=O)NC(C(=O)NC(C(=O)NC(C(=O)N2)CC(=O)N)CCC(=O)N)CC3=CC=CC=C3)CC4=CC=C(C=C4)O)NC(=O)CNC(=O)CNC(=O)CN)C(=O)NC(CCCCN)C(=O)NCC(=O)N
NEW DRUG APPROVALS
ONE TIME
$10.00
Betibeglogene autotemcel

Betibeglogene autotemcel
ベチベグロゲンアウトテムセル
2022/8/17, FDA APPROVED Zynteglo
Cellular therapy product
Treatment of betathalassemia
BB305 LVV
bb 1111
BB305 transduced SCD CD34+ HSCs bb1111
LentiGlobin BB305 LVV-transduced autologous SCD CD34+ HSCs bb1111
LentiGlobin drug product for SCD
LentiGlobin drug product for sickle cell disease
LentiGlobin for SCD bb1111
Betibeglogene autotemcel, sold under the brand name Zynteglo, is a medication for the treatment for beta thalassemia.[1][5][2] It was developed by Bluebird Bio and was given breakthrough therapy designation by the U.S. Food and Drug Administration in February 2015.[6][7]
The most common adverse reactions include reduced platelet and other blood cell levels, as well as mucositis, febrile neutropenia, vomiting, pyrexia (fever), alopecia (hair loss), epistaxis (nosebleed), abdominal pain, musculoskeletal pain, cough, headache, diarrhea, rash, constipation, nausea, decreased appetite, pigmentation disorder and pruritus (itch).[5]
It was approved for medical use in the European Union in May 2019,[2] and in the United States in August 2022.[5]
FDA Approves First Cell-Based Gene Therapy to Treat Adult and Pediatric Patients with Beta-thalassemia Who Require Regular Blood Transfusions
https://www.fda.gov/news-events/press-announcements/fda-approves-first-cell-based-gene-therapy-treat-adult-and-pediatric-patients-beta-thalassemia-whoFor Immediate Release:August 17, 2022
Today, the U.S. Food and Drug Administration approved Zynteglo (betibeglogene autotemcel), the first cell-based gene therapy for the treatment of adult and pediatric patients with beta-thalassemia who require regular red blood cell transfusions.
“Today’s approval is an important advance in the treatment of beta-thalassemia, particularly in individuals who require ongoing red blood cell transfusions,” said Peter Marks, M.D., Ph.D., director of the FDA’s Center for Biologics Evaluation and Research. “Given the potential health complications associated with this serious disease, this action highlights the FDA’s continued commitment to supporting development of innovative therapies for patients who have limited treatment options.”
Beta-thalassemia is a type of inherited blood disorder that causes a reduction of normal hemoglobin and red blood cells in the blood, through mutations in the beta-globin subunit, leading to insufficient delivery of oxygen in the body. The reduced levels of red blood cells can lead to a number of health issues including dizziness, weakness, fatigue, bone abnormalities and more serious complications. Transfusion-dependent beta-thalassemia, the most severe form of the condition, generally requires life-long red blood cell transfusions as the standard course of treatment. These regular transfusions can be associated with multiple health complications of their own, including problems in the heart, liver and other organs due to an excessive build-up of iron in the body.
Zynteglo is a one-time gene therapy product administered as a single dose. Each dose of Zynteglo is a customized treatment created using the patient’s own cells (bone marrow stem cells) that are genetically modified to produce functional beta-globin (a hemoglobin component).
The safety and effectiveness of Zynteglo were established in two multicenter clinical studies that included adult and pediatric patients with beta-thalassemia requiring regular transfusions. Effectiveness was established based on achievement of transfusion independence, which is attained when the patient maintains a pre-determined level of hemoglobin without needing any red blood cell transfusions for at least 12 months. Of 41 patients receiving Zynteglo, 89% achieved transfusion independence.
The most common adverse reactions associated with Zynteglo included reduced platelet and other blood cell levels, as well as mucositis, febrile neutropenia, vomiting, pyrexia (fever), alopecia (hair loss), epistaxis (nosebleed), abdominal pain, musculoskeletal pain, cough, headache, diarrhea, rash, constipation, nausea, decreased appetite, pigmentation disorder and pruritus (itch).
There is a potential risk of blood cancer associated with this treatment; however, no cases have been seen in studies of Zynteglo. Patients who receive Zynteglo should have their blood monitored for at least 15 years for any evidence of cancer. Patients should also be monitored for hypersensitivity reactions during Zynteglo administration and should be monitored for thrombocytopenia and bleeding.
This application was granted a rare pediatric disease voucher, in addition to receiving Priority Review, Fast Track, Breakthrough Therapy, and Orphan designations.
The FDA granted approval of Zynteglo to bluebird bio, Inc.
/////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
| Clinical data | |
|---|---|
| Trade names | Zynteglo |
| Other names | LentiGlobin BB305, autologous CD34+ cells encoding βA-T87Q-globin gene |
| License data | EU EMA: by INNUS DailyMed: Betibeglogene autotemcel |
| Pregnancy category | Contraindicated[1][2] |
| Routes of administration | Intravenous[3] |
| ATC code | B06AX02 (WHO) |
| Legal status | |
| Legal status | UK: POM (Prescription only) [1]US: ℞-only [3][4][5]EU: Rx-only [2]In general: ℞ (Prescription only) |
| Identifiers | |
| UNII | MEE8487RTP |
| KEGG | D11930 |
Medical uses
Betibeglogene autotemcel is indicated for the treatment of people twelve years and older with transfusion-dependent beta thalassemia (TDT) who do not have a β0/β0 genotype, for whom hematopoietic stem cell (HSC) transplantation is appropriate but a human leukocyte antigen (HLA)-matched related HSC donor is not available.[2]
Betibeglogene autotemcel is made individually for each recipient out of stem cells collected from their blood, and must only be given to the recipient for whom it is made.[2] It is given as an autologous intravenous infusion and the dose depends on the recipient’s body weight.[3][2]
Before betibeglogene autotemcel is given, the recipient receives conditioning chemotherapy to clear their bone marrow of cells (myeloablation).[2]
To make betibeglogene autotemcel, the stem cells taken from the recipient’s blood are modified by a virus that carries working copies of the beta globin gene into the cells.[2] When these modified cells are given back to the recipient, they are transported in the bloodstream to the bone marrow where they start to make healthy red blood cells that produce beta globin.[2] The effects of betibeglogene autotemcel are expected to last for the recipient’s lifetime.[2]
Mechanism of action
Beta thalassemia is caused by mutations to or deletions of the HBB gene leading to reduced or absent synthesis of the beta chains of hemoglobin that result in variable outcomes ranging from severe anemia to clinically asymptomatic individuals.[8] LentiGlobin BB305 is a lentiviral vector which inserts a functioning version of the HBB gene into a recipient’s blood-producing hematopoietic stem cells (HSC) ex vivo. The resulting engineered HSCs are then reintroduced to the recipient.[9][10]
History
In early clinical trials several participants with beta thalassemia, who usually require frequent blood transfusions to treat their disease, were able to forgo blood transfusions for extended periods of time.[11][12][13] In 2018, results from phase 1-2 trials suggested that of 22 participants receiving Lentiglobin gene therapy, 15 were able to stop or reduce regular blood transfusions.[14][15]
In February 2021, a clinical trial[16] of betibeglogene autotemcel in sickle cell anemia was suspended following an unexpected instance of acute myeloid leukemia.[17] The HGB-206 Phase 1/2 study is expected to conclude in March 2023.[16]
It was designated an orphan drug by the European Medicines Agency (EMA) and by the U.S. Food and Drug Administration (FDA) in 2013.[2][18] The Food and Drug Administration has also declared betibeglogene autotemcel a Regenerative Medicine Advanced Therapy.[19]
The safety and effectiveness of betibeglogene autotemcel were established in two multicenter clinical studies that included adult and pediatric particpiants with beta-thalassemia requiring regular transfusions.[5] Effectiveness was established based on achievement of transfusion independence, which is attained when the particpiant maintains a pre-determined level of hemoglobin without needing any red blood cell transfusions for at least 12 months. Of 41 particpiants receiving betibeglogene autotemcel, 89% achieved transfusion independence.[5]
Society and culture
Legal status
It was approved for medical use in the European Union in May 2019,[2] and in the United States in August 2022.[5]
Names
The international nonproprietary name (INN) is betibeglogene autotemcel.[20]
References
- ^ Jump up to:a b c “Zynteglo dispersion for infusion – Summary of Product Characteristics (SmPC)”. (emc). 12 May 2020. Retrieved 3 January 2021.[permanent dead link]
- ^ Jump up to:a b c d e f g h i j k l m “Zynteglo EPAR”. European Medicines Agency (EMA). 25 March 2019. Archived from the original on 16 August 2019. Retrieved 16 August 2019. Text was copied from this source which is © European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ Jump up to:a b c “Archived copy”. Archived from the original on 26 August 2022. Retrieved 26 August 2022.
- ^ “Zynteglo”. U.S. Food and Drug Administration. 17 August 2022. Archived from the original on 26 August 2022. Retrieved 26 August 2022.
- ^ Jump up to:a b c d e f g “FDA Approves First Cell-Based Gene Therapy to Treat Adult and Pediatric Patients with Beta-thalassemia Who Require Regular Blood Transfusions”. U.S. Food and Drug Administration (FDA) (Press release). 17 August 2022. Archived from the original on 21 August 2022. Retrieved 20 August 2022. This article incorporates text from this source, which is in the public domain.
- ^ “Ten things you might have missed Monday from the world of business”. The Boston Globe. 3 February 2015. Archived from the original on 1 August 2020. Retrieved 13 February 2015.
- ^ “Lentiviral vectors”. 27 June 2019. Archived from the original on 21 August 2022. Retrieved 8 July 2019.
- ^ Cao A, Galanello R (February 2010). “Beta-thalassemia”. Genetics in Medicine. 12 (2): 61–76. doi:10.1097/GIM.0b013e3181cd68ed. PMID 20098328.
- ^ Negre O, Bartholomae C, Beuzard Y, Cavazzana M, Christiansen L, Courne C, et al. (2015). “Preclinical evaluation of efficacy and safety of an improved lentiviral vector for the treatment of β-thalassemia and sickle cell disease” (PDF). Current Gene Therapy. 15 (1): 64–81. doi:10.2174/1566523214666141127095336. PMC 4440358. PMID 25429463. Archived (PDF) from the original on 19 July 2018. Retrieved 19 June 2018.
- ^ Thompson AA, Rasko JE, Hongeng S, Kwiatkowski JL, Schiller G, von Kalle C, et al. (2014). “Initial Results from the Northstar Study (HGB-204): A Phase 1/2 Study of Gene Therapy for β-Thalassemia Major Via Transplantation of Autologous Hematopoietic Stem Cells Transduced Ex Vivo with a Lentiviral βΑ-T87Q -Globin Vector (LentiGlobin BB305 Drug Product)”. Blood. 124 (21): 549. doi:10.1182/blood.V124.21.549.549. Archived from the original on 18 October 2019. Retrieved 13 February 2015.
- ^ Cavazzana-Calvo M, Payen E, Negre O, Wang G, Hehir K, Fusil F, et al. (September 2010). “Transfusion independence and HMGA2 activation after gene therapy of human β-thalassaemia”. Nature. 467 (7313): 318–322. Bibcode:2010Natur.467..318C. doi:10.1038/nature09328. PMC 3355472. PMID 20844535.
- ^ Winslow R (8 December 2015). “New Gene Therapy Shows Promise for Lethal Blood Disease”. The Wall Street Journal. Archived from the original on 2 March 2020. Retrieved 13 February 2015.
- ^ (8 December 2014) bluebird bio Announces Data Demonstrating First Four Patients with β-Thalassemia Major Treated with LentiGlobin are Transfusion-Free Archived 26 September 2015 at the Wayback Machine Yahoo News, Retrieved 17 May 2015
- ^ Thompson AA, Walters MC, Kwiatkowski J, Rasko JE, Ribeil JA, Hongeng S, et al. (April 2018). “Gene Therapy in Patients with Transfusion-Dependent β-Thalassemia”. The New England Journal of Medicine. 378 (16): 1479–1493. doi:10.1056/NEJMoa1705342. PMID 29669226.
- ^ Stein R (18 April 2018). “Gene Therapy For Inherited Blood Disorder Reduced Transfusions”. NPR. Archived from the original on 21 August 2022. Retrieved 4 March 2019.
- ^ Jump up to:a b Clinical trial number NCT02140554 for “A Phase 1/2 Study Evaluating Gene Therapy by Transplantation of Autologous CD34+ Stem Cells Transduced Ex Vivo With the LentiGlobin BB305 Lentiviral Vector in Subjects With Severe Sickle Cell Disease” at ClinicalTrials.gov
- ^ “Bluebird bio Halts Sickle Cell Trials After Leukemia Diagnosis”. BioSpace. Archived from the original on 27 June 2021. Retrieved 27 June 2021.
- ^ “Autologous CD34+ hematopoietic stem cells transduced with LentiGlobin BB305 lentiviral vector encoding the human BA-T87Q-globin gene Orphan Drug Designations and Approvals”. U.S. Food and Drug Administration (FDA). 18 March 2013. Archived from the original on 9 June 2020. Retrieved 8 June 2020.
- ^ “bluebird bio Announces Temporary Suspension on Phase 1/2 and Phase 3 Studies of LentiGlobin Gene Therapy for Sickle Cell Disease (bb1111)”. Bluebird Bio (Press release). 16 February 2021. Archived from the original on 27 June 2021. Retrieved 27 June 2021.
- ^ World Health Organization (2020). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 83”. WHO Drug Information. 34 (1): 34. Archived from the original on 15 July 2020.
////////////Betibeglogene autotemcel, FDA 2022, APPROVALS 2022, ベチベグロゲンアウトテムセル , Zynteglo, bluebird bio, bb 1111
BB305 transduced SCD CD34+ HSCs bb1111
LentiGlobin BB305 LVV-transduced autologous SCD CD34+ HSCs bb1111
LentiGlobin drug product for SCD
LentiGlobin drug product for sickle cell disease
LentiGlobin for SCD bb1111

NEW DRUG APPROVALS
one time
$10.00
Spesolimab
(Heavy chain)
QVQLVQSGAE VKKPGASVKV SCKASGYSFT SSWIHWVKQA PGQGLEWMGE INPGNVRTNY
NENFRNKVTM TVDTSISTAY MELSRLRSDD TAVYYCTVVF YGEPYFPYWG QGTLVTVSSA
STKGPSVFPL APSSKSTSGG TAALGCLVKD YFPEPVTVSW NSGALTSGVH TFPAVLQSSG
LYSLSSVVTV PSSSLGTQTY ICNVNHKPSN TKVDKRVEPK SCDKTHTCPP CPAPEAAGGP
SVFLFPPKPK DTLMISRTPE VTCVVVDVSH EDPEVKFNWY VDGVEVHNAK TKPREEQYNS
TYRVVSVLTV LHQDWLNGKE YKCKVSNKAL PAPIEKTISK AKGQPREPQV YTLPPSREEM
TKNQVSLTCL VKGFYPSDIA VEWESNGQPE NNYKTTPPVL DSDGSFFLYS KLTVDKSRWQ
QGNVFSCSVM HEALHNHYTQ KSLSLSPGK
(Light chain)
QIVLTQSPGT LSLSPGERAT MTCTASSSVS SSYFHWYQQK PGQAPRLWIY RTSRLASGVP
DRFSGSGSGT DFTLTISRLE PEDAATYYCH QFHRSPLTFG AGTKLEIKRT VAAPSVFIFP
PSDEQLKSGT ASVVCLLNNF YPREAKVQWK VDNALQSGNS QESVTEQDSK DSTYSLSSTL
TLSKADYEKH KVYACEVTHQ GLSSPVTKSF NRGEC
(Disulfide bridge: H22-H96, H146-H202, H222-L215, H228-H’228, H231-H’231, H263-H323, H369-H427, H’22-H’96, H’146-H’202, H’222-L’215, H’263-H’323, H’369-H’427, L23-L89, L135-L195, L’23-L’89, L’135-L’195)
Spesolimab
スペソリマブ (遺伝子組換え)
| Formula | C6480H9988N1736O2012S46 |
|---|---|
| cas | 2097104-58-8 |
| Mol weight | 145878.0547 |
| Antipsoriatic, Anti-IL-36 receptor antagonist |
fda approved 2022/9/1, spevigo
BI 655130; Spesolimab-sbzo
- OriginatorBoehringer Ingelheim
- ClassAnti-inflammatories; Antipsoriatics; Monoclonal antibodies; Skin disorder therapies
- Mechanism of ActionInterleukin 36 receptor antagonists
- Orphan Drug StatusYes – Generalised pustular psoriasis
- RegisteredGeneralised pustular psoriasis
- Phase II/IIIUlcerative colitis
- Phase IICrohn’s disease; Hidradenitis suppurativa; Palmoplantar pustulosis
- DiscontinuedAtopic dermatitis
- 01 Sep 2022First global approval – Registered for Generalised pustular psoriasis in USA (IV)
- 01 Sep 2022Adverse events data from the Effisayil 1 phase II trial in Generalised pustular psoriasis released by Boehringer Ingelheim
- 03 Aug 2022Boehringer Ingelheim anticipates regulatory approval in Generalised pustular psoriasis by 2022
Spesolimab (BI 655130) is a humanised monoclonal antibody, being developed by Boehringer Ingelheim, for the treatment of generalised pustular psoriasis, Crohn’s disease, palmoplantar pustulosis, ulcerative colitis and hidradenitis suppurativa.
What causes Palmoplantar Pustulosis?
Researchers have found some possible causes including smoking, infections, certain medications and genetics. Smoking: Many patients who have PPP are smokers or have smoked in the past. Smoking may cause sweat glands to become inflamed, especially on the hands and feet, which causes pustules to form.
FDA approves the first treatment option for generalized pustular psoriasis flares in adults
- More than half of patients treated with SPEVIGO® (spesolimab-sbzo) injection, for intravenous use showed no visible pustules one week after receiving treatment
- Spesolimab is a monoclonal antibody that inhibits interleukin-36 (IL-36) signaling
Ridgefield, Conn., September 1, 2022 – Boehringer Ingelheim announced today the U.S. Food and Drug Administration has approved SPEVIGO, the first approved treatment option for generalized pustular psoriasis (GPP) flares in adults. SPEVIGO is a novel, selective antibody that blocks the activation of the interleukin-36 receptor (IL-36R), a key part of a signaling pathway within the immune system shown to be involved in the cause of GPP.
“GPP flares can greatly impact a patient’s life and lead to serious, life-threatening complications,” said Mark Lebwohl, M.D., lead investigator and publication author, and Dean for Clinical Therapeutics, Icahn School of Medicine at Mount Sinai, Kimberly and Eric J. Waldman Department of Dermatology, New York. “The approval of SPEVIGO is a turning point for dermatologists and clinicians. We now have an FDA-approved treatment that may help make a difference for our patients who, until now, have not had any approved options to help manage GPP flares.”
Distinct from plaque psoriasis, GPP is a rare and potentially life-threatening neutrophilic skin disease, which is characterized by flares (episodes of widespread eruptions of painful, sterile pustules). In the United States, it is estimated that 1 out of every 10,000 people has GPP. Given that it is so rare, recognizing the signs and symptoms can be challenging and consequently lead to delays in diagnosis.
“This important approval reflects our successful efforts to accelerate our research with the aim to bring innovative treatments faster to the people most in need,” said Carinne Brouillon, Member of the Board of Managing Directors, responsible for Human Pharma, Boehringer Ingelheim. “We recognize how devastating this rare skin disease can be for patients, their families and caregivers. GPP can be life-threatening and until today there have been no specific approved therapies for treating the devastating GPP flares. It makes me proud that with the approval of SPEVIGO we can now offer the first U.S. approved treatment option for those in need.”
In the 12-week pivotal Effisayil™ 1 clinical trial, patients experiencing a GPP flare (N=53) were treated with SPEVIGO or placebo. After one week, patients treated with SPEVIGO showed no visible pustules (54%) compared to placebo (6%).
In Effisayil™ 1, the most common adverse reactions (≥5%) in patients that received SPEVIGO were asthenia and fatigue, nausea and vomiting, headache, pruritus and prurigo, infusion site hematoma and bruising, and urinary tract infection.
“GPP can have an enormous impact on patients’ physical and emotional wellbeing. With the FDA approval of this new treatment, people living with GPP now have hope in knowing that there is an option to help treat their flares,” said Thomas Seck, M.D., Senior Vice President, Medicine and Regulatory Affairs, Boehringer Ingelheim. “SPEVIGO represents Boehringer Ingelheim’s commitment to delivering meaningful change for patients living with serious diseases with limited treatment options.”
About SPEVIGO
SPEVIGO is indicated for the treatment of GPP flares in adults. SPEVIGO is contraindicated in patients with severe or life-threatening hypersensitivity to spesolimab-sbzo or to any of the excipients in SPEVIGO. Reactions have included drug reaction with eosinophilia and systemic symptoms (DRESS).
What is SPEVIGO?
SPEVIGO is a prescription medicine used to treat generalized pustular psoriasis (GPP) flares in adults. It is not known if SPEVIGO is safe and effective in children.
U.S. FDA grants Priority Review for spesolimab for the treatment of flares in patients with generalized pustular psoriasis (GPP), a rare, life-threatening skin disease
December 15, 2021 – Boehringer Ingelheim today announced that the U.S. Food and Drug Administration (FDA) has accepted a Biologics License Application (BLA) and granted Priority Review for spesolimab for the treatment of generalized pustular psoriasis (GPP) flares.
FDA grants Priority Review to applications for medicines that, if approved, would offer significant improvement over available options in the safety or effectiveness of the treatment, diagnosis, or prevention of serious conditions. The FDA has granted spesolimab Orphan Drug Designation for the treatment of GPP, and Breakthrough Therapy Designation for spesolimab for the treatment of GPP flares in adults.
“The FDA acceptance of our filing for spesolimab is a critical step in our efforts to bring this first-in-class treatment to people living with GPP,” said Matt Frankel, M.D., Vice President, Clinical Development and Medical Affairs, Specialty Care, Boehringer Ingelheim. “There is an urgent unmet need for an approved treatment option that can rapidly clear painful GPP flares.”
GPP is a rare, life-threatening neutrophilic skin disease, which is distinct from plaque psoriasis. It is characterized by episodes of widespread eruptions of painful, sterile pustules (blisters of non-infectious pus). There is a high unmet need for treatments that can rapidly and completely resolve the signs and symptoms of GPP flares. Flares greatly affect a person’s quality of life and can lead to hospitalization with serious complications, including heart failure, renal failure, sepsis, and death.
About spesolimab
Spesolimab is a novel, humanized, selective antibody that blocks the activation of the interleukin-36 receptor (IL-36R), a signaling pathway within the immune system shown to be involved in the pathogeneses of several autoimmune diseases, including GPP. Spesolimab is also under investigation for the prevention of GPP flares and for the treatment of other neutrophilic skin diseases, such as palmoplantar pustulosis (PPP) and hidradenitis suppurativa (HS).
About generalized pustular psoriasis (GPP)
GPP is a rare, heterogenous and potentially life-threatening neutrophilic skin disease, which is clinically distinct from plaque psoriasis. GPP is caused by neutrophils (a type of white blood cell) accumulating in the skin, resulting in painful, sterile pustules all over the body. The clinical course varies, with some patients having a relapsing disease with recurrent flares, and others having a persistent disease with intermittent flares. While the severity of GPP flares can vary, if left untreated they can be life-threatening due to complications such as sepsis and multisystem organ failure. This chronic, systemic disease has a substantial quality of life impact for patients and healthcare burden. GPP has a varied prevalence across different geographical regions and more women are affected than men.
Boehringer Ingelheim Immunology: Pioneering Science, Inspired By Patients
Living with fibrotic and inflammatory diseases greatly impacts patients’ lives emotionally and physically. These patients are our guides, partners and inspiration as we redefine treatment paradigms. As a family-owned company, we can plan long-term. Our goal is to discover and develop first-of-their-kind therapies. With a deep understanding of molecular pathways, we are pioneering scientific breakthroughs that target, repair and prevent many fibrotic and inflammatory diseases. By building on long-term external collaborations, we strive to bring treatment breakthroughs to patients in the shortest time. We won’t rest until we can give people the chance to live the lives they want.
Boehringer Ingelheim
Boehringer Ingelheim is working on breakthrough therapies that improve the lives of humans and animals. As a leading research-driven biopharmaceutical company, the company creates value through innovation in areas of high unmet medical need. Founded in 1885 and family-owned ever since, Boehringer Ingelheim takes a long-term perspective. Around 52,000 employees serve more than 130 markets in the three business areas, Human Pharma, Animal Health, and Biopharmaceutical Contract Manufacturing. Learn more at www.boehringer-ingelheim.com.
MPR-US-101971
////////Spesolimab, monoclonal antibody, fda 2022, approvals 2022, Orphan Drug Status, Generalised pustular psoriasis, BI 655130, Spesolimab-sbzo, peptide, monoclonal antibody

NEW DRUG APPROVALS
ONE TIME
$10.00

{kind=link}
{kind=link}
{kind=link}