
Home » SPOTLIGHT (Page 3)
Category Archives: SPOTLIGHT
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Welcome Scientific update to Pune, India 2-3 and 4-5 Dec 2014 for celebrating Process chemistry
WEBSITE http://www.scientificupdate.co.uk/
SCIENTIFIC UPDATE HAS A REPUTATION FOR ITS HIGH QUALITY EVENTS, BOTH FOR THE SCIENTIFIC CONTENT AND ALSO FOR THE EFFICIENCY OF ITS ORGANISATION. KEEP YOUR SKILLS UP TO DATE AND INVEST IN YOUR CONTINUING PERSONAL PROFESSIONAL DEVELOPMENT.



TRAINING COURSE 2-3 DEC 2014
Process Development for Low Cost Manufacturing
When:02.12.2014 – 03.12.2014
Tutors:
Where: National Chemical Laboratory – Pune, India
Brochure:View Brochure
Register http://scientificupdate.co.uk/training/scheduled-training-courses.html
DESCRIPTION
Chemical process research and development is recognised as a key function during the commercialisation of a new product particularly in the generic and contract manufacturing arms of the chemical, agrochemical and pharmaceutical industries.
The synthesis and individual processes must be economic, safe and must generate product that meets the necessary quality requirements.
This 2-day course presented by highly experienced process chemists will concentrate on the development and optimisation of efficient processes to target molecules with an emphasis on raw material cost, solvent choice, yield improvement, process efficiency and work up, and waste minimisation.
Process robustness testing and reaction optimisation via stastical methods will also be covered.
A discussion of patent issues and areas where engineering and technology can help reduce operating costs.
The use of engineering and technology solutions to reduce costs will be discussed and throughout the course the emphasis will be on minimising costs and maximising returns.


Conference 4-5 DEC 2014
TITLE . Organic Process Research & Development – India
Subtitle:The 32nd International Conference and Exhibition
When:04.12.2014 – 05.12.2014
Where:National Chemical Laboratory – Pune, India
Brochure:View Brochure
Register..http://scientificupdate.co.uk/conferences/conferences-and-workshops.html

for
- Process Research & Development Chemists
- Chemical Engineers in Industry
- Heads of Departments & Team Leaders
Benefits
- Invest in yourself: keeping up to date on current developments and future trends could mean greater job security.
- Learn from a wide range of industrial case studies given by hand-picked industrial speakers.
- Take home relevant ideas and information that are directly applicable to your own work with the full proceedings and a CD of the talks.
- Save time. Our intensive, commercial-free programme means less time away from work.
- Meet and network with the key people in the industry in a relaxed and informal atmosphere.

Do you want to improve efficiency and innovation in your synthetic route design, development and optimisation?
The efficient conversion of a chemical process into a process for manufacture on tonnage scale has always been of importance in the chemical and pharmaceutical industries. However, in the current economic and regulatory climate, it has become increasingly vital and challenging to do so efficiently. Indeed, it has never been so important to keep up to date with the latest developments in this dynamic field.
At this Organic Process Research & Development Conference, you will hear detailed presentations and case studies from top international chemists. The hand-picked programme of speakers has been put together specifically for an industrial audience. They will discuss the latest issues relating to synthetic route design, development and optimisation in the pharmaceutical, fine chemical and allied fields. Unlike other conferences, practically all our speakers are experts from industry, which means the ideas and information you take home will be directly applicable to your own work.
The smaller numbers at our conferences create a more intimate atmosphere. You will enjoy plenty of opportunities to meet and network with speakers and fellow attendees during the reception, sit-down lunches and extended coffee breaks in a relaxed and informal environment. Together, you can explore the different strategies and tactics evolving to meet today’s challenges.
This is held in Pune, close proximity to Mumbai city, very convenient to stay and travel to either in Pune or Mumbai. I feel this should be an opportunity to be grabbed before the conference is full and having no room
Hurry up rush


References
1 https://newdrugapprovals.org/scientificupdate-uk-on-a-roll/
2 http://scientificupdate.co.uk/conferences/conferences-and-workshops.html
3 http://en.wikipedia.org/wiki/Pune
PROFILES
Will Watson

Dr Will Watson gained his PhD in Organic Chemistry from the University of Leeds in 1980. He joined the BP Research Centre at Sunbury-on-Thames and spent five and a half years working as a research chemist on a variety of topics including catalytic dewaxing, residue upgrading, synthesis of novel oxygenates for use as gasoline supplements, surfactants for use as gasoline detergent additives and non-linear optical compounds.
In 1986 he joined Lancaster Synthesis and during the next 7 years he was responsible for laboratory scale production and process research and development to support Lancaster’s catalogue, semi-bulk and custom synthesis businesses.
In 1993 he was appointed to the position of Technical Director, responsible for all Production (Laboratory and Pilot Plant scale), Process Research and Development, Engineering and Quality Control. He helped set up and run the Lancaster Laboratories near Chennai, India and had technical responsibility for the former PCR laboratories at Gainesville, Florida.
He joined Scientific Update as Technical Director in May 2000. He has revised and rewritten the ‘Chemical Development and Scale Up in the Fine Chemical & Pharmaceutical Industries’ course and gives this course regularly around the world. He has been instrumental in setting up and developing new courses such as ‘Interfacing Chemistry with Patents’ and ‘Making and Using Fluoroorganic Molecules’.
He is also involved in an advisory capacity in setting up conferences and in the running of the events. He is active in the consultancy side of the business and sits on the Scientific Advisory Boards of various companies.
………………………………………………………………………………………………….
John Knight

Dr John Knight gained a first class honours degree in chemistry at the University of Southampton, UK. John remained at Southampton to study for his PhD in synthetic methodology utilizing radical cyclisation and dipolar cyloaddition chemistry.
After gaining his PhD, John moved to Columbia University, New York, USA where he worked as a NATO Postdoctoral Fellow with Professor Gilbert Stork. John returned to the UK in 1987 joining Glaxo Group Research (now GSK) as a medicinal chemist, where he remained for 4 years before moving to the process research and development department at Glaxo, where he remained for a further 3½ years.
During his time at Glaxo, John worked on a number of projects and gained considerable plant experience (pilot and manufacturing). In 1994 John moved to Oxford Asymmetry (later changing its name to Evotec and most recently to Aptuit) when it had just 25 staff. John’s major role when first at Oxford Asymmetry was to work with a consultant project manager to design, build and commission a small pilot plant, whilst in parallel developing the chemistry PRD effort at Oxford Asymmetry.
The plant was fully operational within 18 months, operating to a 24h/7d shift pattern. John continued to run the pilot plant for a further 3 years, during which time he had considerable input into the design of a second plant, which was completed and commissioned in 2000. After an 18-month period at a small pharmaceutical company, John returned to Oxford in 2000 (by now called Evotec) to head the PRD department. John remained in this position for 6.5 years, during which time he assisted in its expansion, established a team to perform polymorph and salt screening studies and established and maintained high standards of development expertise across the department.
John has managed the chemical development and transfer of numerous NCE’s into the plant for clients and been involved in process validations. He joined Scientific Update in January 2008 as Scientific Director.
Pune images

From top:1 Fergusson College, 2 Mahatma Gandhi Road, Shaniwarwada 3 the HSBC Global Technology India Headquarters, and the 4National War Memorial Southern Command



NCL PUNE
 The National Chemical Laboratory is located in the state of Maharashtra in India. Maharashtra state is the largest contributor to India’s GDP. The National Chemical Laboratory is located in Pune city, and is the cultural capital of Maharashtra. Pune city is second only to Mumbai (the business capital of India) in size and industrial strength. Pune points of interest include: The tourist places in Pune include: Lal Deval Synagogue, Bund Garden, Osho Ashram, Shindyanchi Chhatri and Pataleshwar Cave Temple.
The National Chemical Laboratory is located in the state of Maharashtra in India. Maharashtra state is the largest contributor to India’s GDP. The National Chemical Laboratory is located in Pune city, and is the cultural capital of Maharashtra. Pune city is second only to Mumbai (the business capital of India) in size and industrial strength. Pune points of interest include: The tourist places in Pune include: Lal Deval Synagogue, Bund Garden, Osho Ashram, Shindyanchi Chhatri and Pataleshwar Cave Temple.

MAKE IN INDIA
http://makeinindia.com/sector/pharmaceuticals/
Read all about Organic Spectroscopy on ORGANIC SPECTROSCOPY INTERNATIONAL 


KEYWORDS
JOHN KNIGHT, WILL WATSON, SCIENTIFIC UPDATE, PROCESS, COURSE, CONFERENCE, INDIA, PUNE, PROCESS DEVELOPMENT, LOW COST, MANUFACTURING, SCALEUP
Selective inhibitors of the Janus kinase Jak3—Are they effective?

Selective inhibitors of the Janus kinase Jak3—Are they effective?
Volume 24, Issue 19, 1 October 2014, Pages 4617–4621
- DOI: 10.1016/j.bmcl.2014.08.046
Abstract
Jak3, together with Jak1, is involved in signal transduction initiated by cytokines signaling through the common gamma chain which are important in immune homeostasis and immune pathologies. Based on genetic evidence Jak3 has been considered to be an attractive target for immunosuppression. The Jak inhibitor tofacitinib (CP-690,550) which is an approved drug for rheumatoid arthritis was originally introduced as a selective Jak3 inhibitor, however, it also inhibits Jak1 and Jak2. The search for new selective Jak3 inhibitors has yielded several compounds whose profiles will be reviewed here. Implications on Jak3 as a therapeutic target are also discussed.
notes
JAnus Kinase 3 (JAK3) is a member of the JAK family of non-receptor protein tyrosine kinases (PTKs) that include the closely related isoforms—namely, JAK1, JAK2 and tyrosine kinase 2 (TYK2). The realization that human defects in JAK3 signaling result in the clinical manifestation of a severe combined immunodeficiency (SCID) phenotype has suggested that selective JAK3 inhibitors may be useful as therapeutic agents in the areas of organ transplantation and autoimmune diseases. In addition, the promising clinical efficacy reported for the JAK3 inhibitor CP-690,550 in rheumatoid arthritis patients is noteworthy and suggests that obtaining efficacy comparable to, or perhaps better than, the current marketed biologic therapies in this disease may be possible with a small molecule. Although the highly selective inhibition of JAK3 for immunosuppression is particularly attractive from a safety perspective, it remains to be convincingly demonstrated in the clinic. While CP-690,550 does potently inhibit JAK3, it has been shown to inhibit to some extent other JAK family members—namely, JAK1 and JAK2, which may contribute to enhance efficacy in the clinic relative to purely selective JAK3 inhibition.
Anthony crasto’s blog New drug approvals touches 3 lakh views…….Helping millions

link is https://newdrugapprovals.org/
All about Drugs, live, by DR ANTHONY MELVIN CRASTO, Worlddrugtracker, Helping millions, 7 million hits on google, pushing boundaries, one lakh plus connections worldwide, 3 lakh plus VIEWS on this blog in 193 countries

THANKS AND REGARD’S
DR ANTHONY MELVIN CRASTO Ph.D
web link

New Drug Approvals, ALL ABOUT DRUGS, WORLD DRUG TRACKER
MEDICINAL CHEM INTERNATIONAL, DRUG SYN INTERNATIONAL
SCALEUP OF DRUGS, ALL FOR DRUGS ON WEB,
MY CHINA, VIETNAM AND JAPAN BLOGS
ICELAND, RUSSIA, ARAB
BOBRDOBR, BLAND ICELAND, 100zakladok, adfty
GROUPS
you can post articles and will be administered by me on the google group which is very popular across the world
OPD GROUPSPACES, SCOOP OCI, organic-process-development GOOGLE, TVINX, MENDELEY WDT, SCIPEOPLE OPD, EPERNICUS OPD, SYNTHETIC ORGANIC CHEMISTRYLinkedIn group, DIIGO OPD, LINKEDIN OPD, WDT LINKEDIN, WDTI ZING


Ann-Teresa Cusenza…..Managing Editor, Orphan Druganaut Blog

Ann-Teresa Cusenza
It is a great pleasure to write about ANN…. I read her blog everyday……………………….
Medical Information Specialist | Medical Librarian | Managing Editor, Orphan Druganaut Blog
| Current | |
|---|---|
| Previous | |
| Education |
- Ann-Teresa Cusenza, MLS, MBA
She is Managing Editor, Orphan Druganaut Blog
- read at
http://orphandruganaut.wordpress.com/this is all about orphan drugs, great work ANN
![]()

-
ABOUT | Orphan Druganaut Blog
orphandruganaut.wordpress.com/about/Ann-Teresa Cusenza, MLS, MBA. Managing Editor, Orphan Druganaut Blog. Medical Information Specialist/Pharmaceutical Competitive Intelligence Consultant.
- SPECIALITIES :
- • Providing medical library information services :
- 1. Creation of Scientific Publication Plans across therapy areas
- 2. Performed searching of medical/pharmaceutical & business databases
- 3. Performed document delivery services
- 4. Scientific literature searching and analysis
- 5. Medical fact checking
- 6. Responsible for completing research requests, adhoc requests, and large projects via phone inquiries, E-Mail, and face-to-face meetings
- • Consulting services through full Information Life Cycle :
- 1. Client consultation
- 2. Search strategy
- 3. Research
4. Information analysis and organization5. Presentation to clients
- • Literature searches and analysis using pharmaceutical/medical/healthcare and business databases, search engines, and other electronic and print resources
- • Monitoring on a daily basis, competitor products in the Drug Development Pipeline
- • Providing competitive intelligence, case scenarios, and strategic recommendations on Product Lifecycle Management in the pharmaceutical industry
- • Creating Daily Newsletters with timely information, analytic overview of pharmaceutical marketplace, analysis of medical meeting abstracts and presentations across therapy areas
- • Providing research, analysis, and identification of Domestic and International Key Opinion Leaders (KOLs) across therapy areas
- • Creation, research, writing and editing pharmaceutical/medical/healthcare Blogs using WordPress.
- COMMITMENT TO LIFELONG CONTINUING EDUCATION :
- • Emerging Web Technologies & Social Media
- • Blogging Using WordPress.
- FELLOWSHIPS :
- • National Library of Medicine (NLM) Fellowship for BioMedical Informatics at the Marine Biological Laboratory (MBL), Woods Hole, MA.
- YOU CAN CONNECT WITH HER ON TWITTER AND LINKEDIN
| Websites |
|---|
-

- Medical Librarian at HackensackUMC … ·
- Pharmaceuticals
View Ann-Teresa Cusenza’s professional profile on LinkedIn. LinkedIn is the world’s largest business network, helping professionals like Ann-Teresa Cusenza …
Check out her linkedin group

http://www.linkedin.com/groups/AnnTeresa-Cusenza-2013-Orphan-Drug-2179312.S.223691622
Thankyou Ann-Teresa Cusenza

Flow chemistry approaches directed at improving chemical synthesis

The true potential of flow chemistry as an enabling technology can really only be fully appreciated when seen in the context of a target driven multi-step synthesis, aimed at the delivery of advanced chemical structures such as active pharmaceutical ingredients (APIs) .
As most pharmaceutical syntheses typically require between 8 and 10 chemical transformations (this is often somewhat reduced to 5/6 steps when analogue/library syntheses are being conducted), excluding protecting group manipulations, to realize the target molecule, this is a good foundation from which to explore the advantages of flow chemistry. We have generated a flow protocol for the synthesis of imatinib, the API of the Novartis block buster anticancer therapeutic Gleevec (imatinib mesylate), including a series of analogues (Scheme 11)
Furthermore, we aimed to create a route which would allow each of the three main fragments to be exchanged to address maximum variation in subsequent analogue synthesis. This requires additional planning to build flexibility into the sequence where this desired diversity can be easily introduced. Again, prior consideration of the generated intermediates, and any potential by-products that may arise, is critical and should be addressed prior to embarking on the synthesis.
Consequently, the extensive profiling of the reaction in terms of its purity profile is more closely analogous to process chemistry than traditional Medicinal Chemistry, even at the development stage. So, although more time consuming in the planning stage, having a greater understanding of the chemistry, does then enable a smoother up scaling and more rapid optimization of the route.

read all this at
http://www.degruyter.com/view/j/gps.2013.2.issue-3/gps-2013-0029/gps-2013-0029.xml
Flow chemistry approaches directed at improving chemical synthesis
1Department of Chemistry, Durham University, South Road, Durham, DH1 3LE, UK
Corresponding author: Ian R. Baxendale, Department of Chemistry, Durham University, South Road, Durham, DH1 3LE, UK
Citation Information: Green Processing and Synthesis. Volume 2, Issue 3, Pages 211–230, ISSN (Online) 2191-9550, ISSN (Print) 2191-9542, DOI: 10.1515/gps-2013-0029, May 2013
IRBESARTAN

IRBESARTAN, SR 47436, BMS-186295
Avapro® (Bristol-Myers Squibb) and Karvea®
(Sanofi-Winthrop)
2-butyl-3-({4-[2-(2H-1,2,3,4-tetrazol-5-yl)phenyl]phenyl}methyl)-1,3-diazaspiro[4.4]non-1-en-4-one
138402-11-6 CAS NO
U.S. Patents 5,270,317 and 5,352,788, 6,162,922
The compound prepared according to US 5270317 is polymorph A
-
Irbesartan is known by following chemical names:
- (a) 2-Butyl-3-[[2′-(1H-tetrazol-5-yl)[1,1′-biphenyl]-4-yl]methyl]-1,3-diazaspiro[4,4]non-1-en-4-one
- (b) 2-Butyl-3-[p-(o-1H-tetrazol-5-ylphenyl)benzyl]-1,3-diazaspiro[4,4]non-1-en-4-one
- (c) 2-n-butyl-4-spirocyclopentane-1-[(2′-(tetrazol-5-yl)biphenyl-4-yl) methyl]-2-imidazolin-5-one.
-
The structural formula of Irbesartan is represented below.

Irbesartan
-
The synthesis of irbesartan is first disclosed in US5270317 (equivalentEP0454511 ) and subsequently, several other patents disclose the synthesis of irbesartan by different methods. Basically the synthesis of this molecule involves two common intermediates namely spiroimidazole and substituted 4′-bromomethylbiphenyl.
-
US 5270317 describes preparation of irbesartan wherein 1-[(2′-cyanobiphenyl-4-yl)methyl]-2-n-butyl-4-spirocyclopentane-2-imidazolin -5-one which is reacted with tributyltin azide in xylene at reflux temperature for 66 hours to give a product which is isolated from the reaction mass as trityl irbesartan and then deprotected in methanol/THF mixture using 4N hydrochloric acid to get irbesartan.
-
US5629331 describes a process for the preparation of irbesartan from 1-[(2′-cyanobiphenyl)4-yl)methyl]-2-n-butyl-4-spirocyclopentane-2-imidazolin-5-one using sodium azide, TEA.HCl in N-methylpyrrolidone. The product is isolated from the alkaline reaction mass after acidification to pH 4.7 to 5.8 and the crude product is recrystallised from IPA/water to get Form A and ethanol/water to get Form B.
Irbesartan (INN) /ɜrbəˈsɑrtən/ is an angiotensin II receptor antagonist used mainly for the treatment of hypertension. Irbesartan was developed by Sanofi Research (now part ofsanofi-aventis). It is jointly marketed by sanofi-aventis and Bristol-Myers Squibb under thetrade names Aprovel, Karvea, and Avapro.
It is marketed in Brazil by Sanofi-Aventis under the trade name Aprovel .

As with all angiotensin II receptor antagonists, irbesartan is indicated for the treatment ofhypertension. Irbesartan may also delay progression of diabetic nephropathy and is also indicated for the reduction of renal disease progression in patients with type 2 diabetes,[1]hypertension and microalbuminuria (>30 mg/24 hours) or proteinuria (>900 mg/24 hours).[2]
Irbesartan is also available in a combination formulation with a low dose thiazide diuretic, invariably hydrochlorothiazide, to achieve an additive antihypertensive effect. Irbesartan/hydrochlorothiazide combination preparations are marketed under similar trade names to irbesartan preparations, including Irda, CoIrda, CoAprovel, Karvezide,Avalide and Avapro HCT.
A large randomized trial following 4100+ men and women with heart failure and normal ejection fraction (>=45%) over 4+ years found no improvement in study outcomes or survival with irbesartan as compared to placebo.[3]

BMS annual sales approx $1.3bn. Sanofi-aventis annual sales approx $2.1bn. In the United States, a generic version is available. Patent expired March 2012.
- Lewis EJ, Hunsicker LG, Clarke WR, Berl T, Pohl MA, Lewis JB, Ritz E, Atkins RC, Rohde R, Raz I; Collaborative Study Group. (2001). “Renoprotective effect of the angiotensin-receptor antagonist irbesartan in patients with nephropathy due to type 2 diabetes”. N Engl J Med 345 (12): 851–60. doi:10.1056/NEJMoa011303.PMID 11565517.
- Rossi S, editor. Australian Medicines Handbook 2006. Adelaide: Australian Medicines Handbook; 2006. ISBN 0-9757919-2-3
- Massie BM, Carson PE, McMurray JJ, Komajda M, McKelvie R, Zile MR, Anderson S, Donovan M, Iverson E, Staiger C, Ptaszynska A (December 2008). “Irbesartan in patients with heart failure and preserved ejection fraction”. N. Engl. J. Med. 359 (23): 2456–67.doi:10.1056/NEJMoa0805450. PMID 19001508.
4……….C. A. Bernhart, P. M. Perreaut, B. P. Ferrari, Y. A. Muneaux,
J.-L. A. Assens, J. Clement, F. Haudricourt, C. F. Muneaux,
J. E. Taillades, M.-A. Vignal, J. Gougat, P. R. Guiraudou, C.
A. Lacour, A. Roccon, C. F. Cazaubon, J.-C. Brelihre, G. Le
Fur, D. Nisato, J. Med. Chem. 1993, 36, 3371–3380.
5…. K. F. Croom, M. P. Curran, K. L. Goa, Drugs 2004 64,
999–1028.
6… C. Bernhard, J.-C. Breliere, J. Clement, D. Nisato, P. M. Perreaut, C. F. Muneaux, (Elf Sanofi) US 5 270 317; Chem. Abstr. 1993, 119, 95560.
7. S. Chava, M. Bandari, K. S. Mathuresh, (Matrix Laboratories) WO 2005/122699; Chem. Abstr. 2005, 144, 88292.
5. S. Zupan~i~, A. Pe~avar, R. Zupet, (Krka) WO 2006/073376;
Chem. Abstr. 2006, 145, 124576.
8. C. V. Kavitha, S. L. Gaonkar, J. N. Chandra, S. Narendra, C.
T. Sadashiva, K. S. Rangappa, Bioorg. Med. Chem. 2007, 15,
7391–7398.
9. S. Rádl, J. Stach, O. Klecán, (Zentiva) WO 2005/021535;
Chem. Abstr. 2005, 142, 298118.
10. B. Satyanarayana, Y. Anjaneyulu, P. Veerasomaiah, P. P.
Reddy, Heterocycl. Commun. 2007, 13, 223–228.
11. V. V. Korrapati, P. Rao, R. Dandala, V. K. Handa, I. V. S. Rao,
A. Rani, A. Naidu, Synth. Commun. 2007, 37, 2897–2905.
12. J. Havlí~ek, Z. Mandelová, R. Weisemann, I. Strˇelec, S.
Rádl, Collect. Czech. Chem. Commun. 2009, 77, 347.
Irbesartan of formula (I).

The chemical name of Irbesartan is 2-Butyl-3-[[2′-(lH-tetrazol-5-yl)[l,l’-biphenyl]-4- yl]methyl]-l,3-diazaspiro[4,4]non-l-en-4-one and formula is C2SH2SN6O and molecular weight is 428.53. The current pharmaceutical product containing this drug is being sold by Sanofi Synthelabo using the tradename AVAPRO, in the form of tablets. Irbesartan is useful in the treatment of diabetic neuropathy, heart failure therapy and hypertension. Irbesartan is angiotension II type I (AΙIi)-receptor antagonist. Angiotension II is the principal pressor agent of the rennin-angiotension system and also stimulates aldosterone synthesis and secretion by adrenal cortex, cardiac contraction, renal resorption of sodium, activity of the sympathetic nervous system and smooth muscle cell growth. Irbesartan blocks the vasoconstrictor and aldosterone- secreting effects of angiotension II by selectively binding to the ATi angiotension II receptor. U.S. Pat. Nos. 5,270,317 and 5,559,233 describes a process for the preparation of N- substituted heterocyclic derivatives which involves reacting a heterocyclic compound of the formula

with a (biphenyl-4-yl)methyl derivative of the formula

wherein R1, R2, R3, R4, R5, and t, z and Hal have the meanings given in said U.S. Pat. No.
5,270,317, in the presence of an inert solvent such as DMF, DMSO or THF, with a basic reagent, for example KOH, a metal alcoholate, a metal hydride, calcium carbonate or triethylamine. The products of the reaction were purified by chromatography.
U.S. Pat. Nos. 5,352,788, and 5,559,233, and WO 91/14679 also describe identical alkylation of the nitrogen atom of the heterocyclic compound with the halo-biphenyl compound using the same inert solvent and the same basic reagents.
-
US5629331 describes a process for the preparation of irbesartan from 1-[(2′-cyanobiphenyl)4-yl)methyl]-2-n-butyl-4-spirocyclopentane-2-imidazolin-5-one using sodium azide, TEA.HCl in N-methylpyrrolidone. The product is isolated from the alkaline reaction mass after acidification to pH 4.7 to 5.8 and the crude product is recrystallised from IPA/water to get Form A and ethanol/water to get Form B.
-
WO 2005/051943 A1 describes a process for the preparing irbesartan wherein 1-[(2′-cyanobiphenyl-4-yl)methyl]-2-n-butyl-4-spirocyclopentane-2-imidazolin-5-one is reacted with tributyltin chloride, sodium azide and TBAB in toluene at reflux temperature for 20 hours. Product is isolated from the reaction mass as trityl irbesartan and then deprotected in methanol and formic acid to get irbesartan.
-
WO 2006/023889 describes a method for preparing irbesartan, wherein 1-(2′-cyanobiphenyl-4-yl)methyl)-2-n-butyl-4-spirocyclopentane-2-imidazolin-5-one is reacted with sodium azide and triethylamine hydrochloride in N-methyl-2-pyrrolidone to give irbesartan.
-
WO 2005/113518 describes a process for preparing irbesartan wherein cyano irbesartan in xylene, is reacted with tributyltin chloride and sodium azide at reflux temperature till reaction is completed followed by aqueous work-up and recrystallization to give irbesartaN
-
The process involving use of zinc salt for the transformation of nitrile to tetrazole is a safe and efficient process as reported in JOC (2001) 66, 7945-50. The use of zinc salt for transforming nitrile to tetrazole has also been published in WO9637481 and US5502191
Also Canadian Patent No. 2050769 describes the alkylation of the nitrogen atom of the heterocycle of the formula
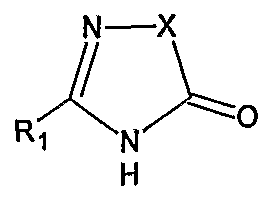
with a compound of the formula

wherein X, R1, Z1 and Z6 have the meanings given therein, in the presence of N,N- dimethylformamide and a basic reagent, such as alkali metal hydrides for example sodium or potassium hydride.
All of the above identified patents describe alkylation in solvents, such as N5N- dimethylformamide or DMSO, etc. in the presence of a basic reagent, for example, a metal hydride or a metal alcoholate etc. The strong bases, such as metal hydride or a metal alcoholate require anhydrous reaction conditions. Since N,N-dimethylformamide is used as a solvent, its removal requires high temperature concentration by distillation, which can result in degradation of the final product. The product intermediate is also purified by chromatography which is commercially not feasible and cumbersome on large scale. Another process given in Canadian Patent No. 2050769 provides synthetic scheme as herein given below.

This process comprises the steps of protecting carboxylic group present on cyclopentane ring which is deprotected in consecutive step by vigourous hydrogenation condition in autoclave which is operationally difficult at a large scale.
US Patent No. 2004242894 also discloses the process of preparation of lrbesartan from 4- bromomethyl biphenyl 2′-(lH-tetrazol (2-triphenylmethyl) 5-yl) and Ethyl ester of 1- Valeramido cyclopentanecarboxylic acid in toluene in presence of base and PTC, and then hydrolyzing the protecting group. However this requires chromatographic purification.
This patent also discloses the process of preparation of tetrazolyl protected lrbesartan using 2,6 lutidine and oxalylchloride in toluene. However in this process the yield is as low as 30%.
US Patent No. 2004192713 discloses the process of preparation of lrbesartan by condensing the two intermediates via Suzuki coupling reaction. The reaction scheme is as given herein below.

However, this process has several disadvantages such as use of the reagents like butyl lithium and triisobutyl borate at low temp such as -20 to -30°C under Argon atmosphere condition which is difficult to maintain at commercial scale.
WO2005113518 discloses the process of preparation of Irbesartan by condensing n- pentanoyl cycloleucine (V) with 2-(4-aminomethyl phenyl) benzonitrile (VI) using dicyclocarbodiimide (DCC) and 1 -hydroxy benzotriazole as catalyst to give an open chain intermediate of formula (VIII) which is then cyclized in the presence of an acid, preferably trifluoro acetic acid to give cyano derivative of formula (VII) and which in turn is converted to Irbesartan by treating it with tributyl tin chloride and sodium azide.

In this application further describes another process comprising the steps of reacting 2- butyl-l,3-diazasρiro[4,4]non-l-en-4-one monohydrochloride (A) with 4-bromobenzyl bromide (B) in presence of base and solvent to give 3-[4-bromobenzyl]-2-butyl-l,3- diazaspiro[4,4]non-l-en-4-one (C) which is condensed with 2-[2′-(triphenylmethyl-2’H- tetrazol-5′-yl)phenyl boronic acid in the presence of tetrakis triphenyl phosphine palladium and base to give lrbesartan (I). However these processes suffer with several disadvantages such as it uses trifluoroacetic acid for the cyclization step which is highly corrosive material. The process requires an additional step of activation by DCC. This step not only increases number of steps but also create problem in handling DCC at an industrial scale as it is highly prone to hazard which makes the process least preferred on a large scale production of lrbesartan. Further it uses phenyl boronic acid derivative and triphenyl phosphine complex which are harmful for the skin and eye tissue and also harmful for respiratory system. Tetrakis triphenyl phosphine palladium is also a costly material which increases overall cost for the production of lrbesartan. Moreover the yield is as low as 22%. All the above patents/applications are incorporated herein as reference. In summary, prior art relating to the process for the preparation of lrbesartan suffers with several drawbacks such as i) It requires chromatographic purification of intermediates at various stages. ii) It requires specific autoclave conditions for a deprotection of protecting group. iii) It requires maintaining low temperature conditions such as -300C and requires special handling care and air and moisture tight condition with the reagents such as butyl lithium and triisobutyl borate. iv) It uses hazardous and highly corrosive reagents, v) It suffers low yield problem. vi) All the process is having more number of reaction steps.
- Irbesartan is described in Bernhart et al., U.S. Patent No. 5,270,317
-
Irbesartan, is a potent, long-acting angiotensin II receptor antagonist which is particularly useful in the treatment of cardiovascular ailments such as hypertension and heart failure. Its chemical name is2-n-butyl-4-spirocyclopentane-1-[(2′-(tetrazol-5-yl)biphenyl-4-yl)methyl]-2-imidazolin-5-one.
Irbesartan is an antihypertensive agent known from EP 454511. From EP 708103, which discloses their X-ray spectra, two polymorphs are known where form A can be produced form a solvent system containing less than 10% of water, while Form B from a system with more than 10% of water. The specific morphological variant of form A can be prepared having properties as disclosed in EP 1089994. Additional form has been disclosed in WO 04089938. Amorphous irbesartan is known from WO 03050110. It is said that Irbesartan produced as taught in EP 454511 is a fluffy material with relatively low bulk and tap densities and undesirable flow characteristics, which consequently has unadvantageous electrostatic properties, among them a high chargeability as measured by tribugeneration between -30 and -40 nanocoulomb/g (10‘9As/g). Alternativelyirbesartan could be prepared by complex process using sonifications and/or temperature oscillations according to EP 1089994 to exhibit a chargeability as measured by tribugeneration between -0 and -10 nanocoulomb/g.
According to EP 454511 a solid composition in form of tablets is prepared by mixing the active ingredient with a vehicle such as gelatine, starch, lactose, magnesium stearate, talc, gum Arabic or the like and can be optionally coated. The compositions containing from 20% to 70% by weight of irbesartan are known from EP 747050.
WO 04/007482 teaches the acidification to pH 2 – 3,5 of trityl irbesartan, which is sufficient to remove the protecting group, but not to convert into an acid addition salt; WO 04/065383 is likewise silent on hydrohalide acid addition salts. WO
06/011859 relates to the preparation of a hydrochloride salt of irbesartan in order to incorporate it into a pharmaceutical formulation. W099/38847 mentions optional conversion of irbesartan into hydrochloride, hydrobromide or hydrogen sulfate salts
……………………………………………
…………………

Example 1Preparation of Compounds of formula IVa and IVb:
-

-
A jacketed 1,000 mL 3-neck flask was charged with 4′-methylbiphenyl-2-carbonitrile (Compound 1, 100.0 g) and CH2CI2 (500 mL) under nitrogen. To a 500 mL Erlenmeyer flask with magnetic stirrer, sodium bromate (NaBrO3; 31.2 g) was dissolved in water (170 mL). The NaBrO3 solution was transferred to the 1,000 mL flask and the reaction mixture was cooled to about 5 °C or less. Aqueous HBr solution (48 %, 105.0 g) was added to the 1,000 mL flask and the resulting reaction mixture was recycled though a UV lamp reactor. The reaction mixture was kept at 0-20 °C and the recycling was continued until the reaction was deemed complete by HPLC. Optionally, additional sodium bromate and hydrogen bromide may be added. The relative amounts of Compound 2 and Compound 3 were about 80-90% and about 10-20% respectively. Aqueous sodium metabisulfite solution (2.0 g of in 10 mL water) was added to the reaction mixture. Allow the phases to settle and the methylene chloride phase was washed with water and used in the next step without further purification.
Example 2Preparation of Compound II:
-

-
A 1L 3-neck flask was charged with Compound V (134.0 g), MTBAC (5.0 g) and CH2Cl2 (170 mL) and cool to -5 to 5 °C. An aqueous solution of KOH (182.6 g in 212 mL water) was added slowly to the 1L flask and the reaction temperature was kept at ≤ 5 °C. The methylene chloride solution of Compound IVa and Compound IVb from Example 1 was added to the reaction mixture slowly, while maintaining the temperature at 0-10 °C. Diethyl phosphite (39.66g) was added drop wise at 0-10 °C. Check the reaction mixture for completion of the reduction reaction, and additional diethyl phosphite may be added.
-
The reaction mixture was allowed to warm to ambient (20-30 °C) and agitated until the reaction was deemed complete by HPLC. Water (150 mL) was added and the phases were separated. The organic layer was extracted with water (230 mL) and polish filtered.
-
The methylene chloride (which contained the crude Compound II) was distilled off and exchanged with about 400 mL of methyl tert-butyl ether (MTBE) (optionally, the MTBE recycled from washing below can be used here). Upon cooling, crystallization occurred (optionally seeds were added) and after further cooling to below 25°C, crystals of Compound II were isolated, washed with MTBE and dried in vacuum at a temperature of less than 60°C. HPLC retention time: 18.126 min. Typically, the yield was about 85 to about 88%. Alternatively, IPA could be used as the crystallization and washing solvent
-
Optionally, the solvent (i.e., MTBE or IPA) used to wash the crystals of Compound II above can be recycled and used to crystallize the crude Compound II in the next batch. Since the washed solvent contains Compound II as well as impurities, it was surprisingly found that the washed solvent can be recovered and used again in crystallizing the crude compound of formula II in the next batch without sacrificing its purity while increasing its yield.
Example 3Preparation of Compound I:
-

-
A reactor was charged with Compound II (1 kg), triethylamine chlorhydrate (0.713 kg), sodium azide (0.337 kg) and N-methyl pyrrolidinone (2.07 kg), and the reaction mixture was heated to about 122°C under stirring. After completion of the reaction as determined by HPLC, the reaction mixture was cooled to about 45°C, and an aqueous solution of sodium hydroxide (35%, 5.99 kg) and water (3.0 kg) were added, the resulting mixture was stirred at a temperature between about 20 and about 40°C for about 0.5 hours. The aqueous phase was discarded and the organic phase was treated with toluene (1.73 kg) and water (5.0 kg), and stirred for about 0.5 hours at about 20 – about 30°C. The toluene phase was discarded and the aqueous phase was washed with ethyl acetate (1.8 kg) and treated with aqueous HCl until pH was adjusted to about 4.8 – about 5.2. Precipitation occurred and the resulting suspension was stirred for about 1 hour at about 20 – about 25°C. The precipitation was collected and washed with water three times (1.0 kg x 3). The crude wet product was recrystallized using a mixture of iso-propanol (0.393 kg) and water (4.5 kg). HPLC retention time: 11.725 min. The yield for Compound I was about 87%.
…………………………………………….
SPECTRAL DATA
The ESI mass spectrum of irbesartan showed a protonated molecular ion peak at m/z 429.3 confirming the molecular weight 428. The fragmentation pattern of parent ion 429.3 showed the fragment ions at m/z 385.9, 235.1, 207, 195.4, 192.1, 180.2 and 84
The FT-IR spectrum exhibited a characteristic stretching absorption band at 1732 cm-1 for the carbonyl group of amide functionality. The presence of this band at higher frequency was due to the ring stretching due to five member ring system. Another band at 1614cm-1 was due to C=N stretching vibrations
1H and 13C- NMR were recorded using DMSO-d6 as a solvent. In 1H-NMR the signal due to tetrazole NH proton was not detected may probably due to the tautomerism.
SEE
http://orgspectroscopyint.blogspot.in/2013/12/irbesartan-spectral-data.html
DP 1 IS IMPURITY
………………………………………….
NMR
1H-NMR (DMSO d6): δppm 0.78 (t, 3H); 1.17-1.30 (sex, 2H); 1.40-1.50 (quent, 2H); 1.64-1.66 (m, 2H); 1.80-1.82 (m, 6H); 2.22-2.29 (t, 2H); 4.67 (s, 2H); 7.07 (s, 4H); 7.50- 7.68 (m, 4H) M+: 429.6
,…………………..
m.p:181-182oC,
IR (KBr, cm-1) 1732 (C=O), 1616 (C=N); 1H NMR (DMSO-d6): δ 7.95–7.32 (m, 8 H), 4.80 –4.60 (s, 2 H), 3.60– 3.00 (br s, 1 H), 2.40– 2.20 (t, 2 H , J = 6.04 Hz), 2.00– 1.60 (m, 8 H),1.60–1.45 (quint, 2 H), 1.40– 1.20 (sext, 2 H), 0.91–0.70 (t, 3H, J = 7.41 Hz);
13C-NMR (DMSOd6): δ 186.5, 162.0,155.9, 141.9, 139.2, 137.2. 131.9, 131.4, 130.1, 128.7, 127.1, 124.3, 76.7, 43.1,
37.7, 28.3, 27.4, 26.3, 22.4, 14.5;
MS: m/z= 429 [M+1];
Anal. Calcd for C25H28N6O : C, 70.07; H,
6.59; N, 19.61. Found: C, 70.04; H, 6.57; N, 19.58.
http://www.acgpubs.org/OC/2011/Volume%204/Issue%201/13-OC-1106-199.pdf
………………………………………………..
1H NMR in DMSO-D6 : 7.68 (d. 2H, Ar-H), 7.52 (d, 2 H, Ar-H), 7.08 (s, 4 H, Ar-H), 4.68(s, 2H, -CH2), 2.69(t,2H,-CH2),2.18(m,2H,-CH2),1.83(m,2H,-CH2),1.81 (t, 2H, -CH2), 1.65 (t, 2H, -CH2), 1.45 (m, 2 H, -CH2), 1.24(m , 2H, -CH2), 0.77 (t, 3H, -CH3),
IR (KBR): 3061 (Aromatic C-H stretching), 2960 (Aliphatic C-H stretching), 3443 (N-H stretching), 1733 (C=0 stretching), 1617(CN stretching), 1337.99(CN stretching), 1407(N=N stretching) cm“1.
……………………….
HPLC condition:
Column: Alltima C18 (Alltech 88050) 15.0cm in length x 4.6mm in internal diameter and 5 micron particle size;
Column temperature: 40 C;
Solvent A: Buffer solution A 1.1 g of heptanesulfonic acid in 1 liter of water and adjust the pH to 2.5;
Solvent B: Methanol Flow rate: 1.2mL/min;
Gradient Elution Condition:
Time% A % %B
0 min 50 50
35 min 15 85
Detector: 240 nm;
Injection volume: 10 uL.
The chromatographic purity of
the compounds was analyzed using Agilent 1200 series HPLC instrument under the following conditions:
Column : Symmetry C18, 4.6 × 75 mm, 3.5 µm
Mobile phase : Eluent A: Deionized water, Eluent B: HPLC grade Methanol
Chromatographic Conditions
a. Column temperature : Ambient
b. Sample compartment : Ambient
c. Detector : 225 nm
d. Injection volume : 10 µL
e. Run time : 45 minutes
f. Flow rate :1.0 mL/min
g. Injector :Auto sampler with variable volume injector
h. Diluent : HPLC grade Acetonitrile
DRUG SPOTLIGHT …… DOXOFYLLINE

DOXOFYLLINE
LAUNCHED 1987, Istituto Biologico Chemioterapico ABC
69975-86-6 CAS NO
7-(1,3-dioxolan-2-ylmethyl)-1,3-dimethylpurine-2,6-dione
1H-Purine-2,6-dione, 3,7-dihydro-7-(1,3-dioxolan-2-ylmethyl)-1,3-dimethyl- (9CI)
7-(1,3-Dioxolan-2-ylmethyl)-3,7-dihydro-1,3-dimethyl-1H-purine-2,6-dione; 7-[1,3-(Dioxolan-d4)-2-ylmethyl)]theophylline; 2-(7�-Theophyllinemethyl)-1,3- dioxolane; ABC 12/3; ABC 1213; Ansimar; Dioxyfilline; Doxophylline; Maxivent; Ventax;
Synonyms
-
2-(7′-Teofillinmetil)-1,3-diossolano
-
2-(7′-Teofillinmetil)-1,3-diossolano [Italian]
-
2-(7′-Theophyllinemethyl)-1,3-dioxolane
-
5-26-14-00120 (Beilstein Handbook Reference)
-
7-(1,3-Dioxolan-2-ylmethyl)theophylline
| Formula | C11H14N4O4 |
|---|---|
| Mol. mass | 266.25 g/mol |
- ABC 12/3
- Ansimar
- BRN 0561195
- Dioxyfilline
- Doxofilina
- Doxofilina [INN-Spanish]
- Doxofylline
- Doxofyllinum
- Doxofyllinum [INN-Latin]
- Doxophylline
- EINECS 274-239-6
- Maxivent
- UNII-MPM23GMO7Z
- Ventax
Doxofylline (INN), (also known as doxophylline) is a xanthine derivative drug used in the treatment of asthma.[1]
Doxofylline is a xanthine molecule that appears to be both bronchodilator and anti-inflammatory with an improved therapeutic window over conventional xanthines such as Theophylline and the evidence supporting the effects of Doxofylline in the treatment of lung diseases
It has antitussive and bronchodilator[2] effects, and acts as aphosphodiesterase inhibitor.[3]
In animal and human studies, it has shown similar efficacy to theophylline but with significantly fewer side effects.[4]
Unlike other xanthines, doxofylline lacks any significant affinity for adenosine receptorsand does not produce stimulant effects. This suggests that its antiasthmatic effects are mediated by another mechanism, perhaps its actions on phosphodiesterase.[1]
Doxofylline, [7-(1, 3-dioxolan-2-ylmethyl)-3, 7-dihydro-1, 3-dimethyl-1H-purine-2, 6-dione] is a new bronchodilator xanthine based drug which differs from theophylline by the presence of dioxalane group at position 7. It is used in the treatment of bronchial asthma, chronic obstructive pulmonary disease (COPD), and chronic bronchitis . The mechanism of action is similar to that of theophylline in that it inhibits phosphodiesterase (PDE-IV), thereby preventing breakdown of cyclic adenosine monophosphate (cAMP). Increase in cAMP inhibits activation of inflammatory cells resulting in bronchodilating effect [52]. In contrast to theophylline, doxofylline has very low affinity towards adenosine A1 and A2 receptors which explain its better safety profile
Doxofylline (7-(l,3-dioxalan-2-ylmethyl)-theophylline) is a drug derived from theophylline which is used in therapy as a bronchodilator, with anti-inflammatory action, in reversible airway obstruction. It is commonly administered in doses ranging from 800 to 1200 mg per day, orally, according to a dosage which provides for the intake of two to three dosage units per day in order to maintain therapeutically effective haematic levels. The doxofylline tablets commercially available generally contain 400 mg of active ingredient and release almost all the drug within one hour from intake. The half- life of the drug is around 6-7 hours and for this reason several administrations are required during the 24-hour period.
Obviously a drop in haematic concentration of the drug in an asthmatic patient or patient suffering from COPD (chronic obstructive pulmonary disease) can result in serious consequences, in which case the patient must have recourse to rescue medication, such as salbutamol inhalers.
Pharmaceutical techniques for obtaining the modified release of drugs have been known for some time, but no modified release formulation of doxofylline is known. In fact the present inventors have observed that there are significant difficulties in the production of a doxofylline formula that can be administered only once a day and in particular have encountered problems correlated with bioequivalence.
Various attempts to formulate doxofylline in modified release systems, with different known polymers, have not provided the desired results, i.e. a composition that can be administered once a day, bio equivalent to the plasmatic concentration obtained with the traditional compositions currently on sale. In fact currently, dosage units containing 400 mg of active ingredient are currently administered two/three times a day for a daily average of approximately 1000 mg of active ingredient, a dosage considered necessary to maintain the therapeutic haematic levels of doxofylline.
Such a dosage unit is currently marketed by Dr. Reddy’s Laboratories Ltd as DOXOBID and has the following quali-quantitative composition: doxofylline (400 mg), colloidal silicon dioxide (13 mg), corn starch (63 mg), mannitol (40 mg), povidone (7 mg), microcrystalline cellulose (64 mg), talc (30 mg), magnesium stearate (3 mg) and water (0.08 ml).
Xanthine is a dioxypurine that is structurally related to uric acid. Xanthine can be represented by the following structure:

Caffeine, theophylline and theobromine are methylated xanthines. Methylated xanthines such as caffeine and theophylline are typically used for their bronchodilating action in the management of obstructive airways diseases such as asthma. The bronchodilator effects of methylxanthines are thought to be mediated by relaxation of airway smooth muscle. Generally, methylxanthines function by inhibiting cyclic nucleotide phosphodiesterases and antagonizing receptor-mediated actions of adenosine.
Theophylline can be represented by the following structure:

However, when administered intravenously or orally, theophylline has numerous undesired or adverse effects that are generally systemic in nature. It has a number of adverse side effects, particularly gastrointestinal disturbances and CNS stimulation. Nausea and vomiting are the most common symptoms of theophylline toxicity. Moderate toxicity is due to relative epinephrine excess, and includes tachycardia, arrhythmias, tremors, and agitation. Severe toxicity results in hallucinations, seizures, dysrhythmias and hypotension. The spectrum of theophylline toxicity can also include death.
Furthermore, theophylline has a narrow therapeutic range of serum concentrations above which serious side effects can occur. The pharmacokinetic profile of theophylline is dependent on liver metabolism, which can be affected by various factors including smoking, age, disease, diet, and drug interactions.
Generally, the solubility of methylxanthines is low and is enhanced by the formation of complexes, such as that between theophylline and ethylenediamine (to form aminophylline). The formation of complex double salts (such as caffeine and sodium benzoate) or true salts (such as choline theophyllinate) also enhances aqueous solubility. These salts or complexes dissociate to yield the parent methylxanthine when dissolved in aqueous solution. Although salts such as aminophylline have improved solubility over theophylline, they dissociate in solution to form theophylline and hence have similar toxicities.
Dyphylline is a covalently modified derivative of xanthine (1,3, -dimethyl-7-(2,3-dihydroxypropl)xanthine. Because it is covalently modified, dyphylline is not converted to free theophylline in vivo. Instead, it is absorbed rapidly in therapeutically active form. Dyphylline has a lower toxicity than theophylline. Dyphylline can be represented by the following structure:

Dyphylline is an effective bronchodilator that is available in oral and intramuscular preparations. Generally, dyphylline possesses less of the toxic side effects associated with theophylline.
U.S. Pat. No. 4,031,218 (E1-Antably) discloses the use of 7-(2,3-dihydroxypropyl)-1,3-di-n-propylxanthine, a derivative of theophylline, as a bronchodilator. U.S. Pat. No. 4,341,783 (Scheindlin) discloses the use of dyphylline in the treatment of psoriasis and other diseases of the skin by topical administration of dyphylline. U.S. Pat. No. 4,581,359 (Ayres) discloses methods for the management of bronchopulmonary insufficiency by administering an N-7-substituted derivative of theophylline, including dyphylline, etophylline, and proxyphylline.
At present, domestic synthetic Doxofylline composed of two main methods: one is by the condensation of theophylline prepared from acetaldehyde and ethylene glycol, but this method is more complex synthesis of acetaldehyde theophylline, require high periodate oxidation operation. Another is a halogenated acetaldehyde theophylline and ethylene glycol is prepared by reaction of an organic solvent, the method were carried out in an organic solvent, whereby the product Theophylline caused some pollution, conducive to patients taking.
current domestic Doxofylline synthetic methods reported in the literature are: 1, CN Application No. 94113971.9, the name “synthetic drugs Doxofyllinemethod” patents, the patent is determined by theophylline with a 2 – (halomethyl) -1,3 – dimethoxy-dioxolane in a polar solvent, with a base made acid absorbent,Doxofylline reaction step. 2, CN Application No. 97100911.2, entitled “Synthesis of Theophylline,” the patent, the patent is obtained from 7 – (2,2 – dialkoxy-ethyl) theophylline with ethylene glycol in N, N-dimethylformamide solvent with an alkali metal carbonate to make the condensing agent, p-toluenesulfonic acid catalyst in the condensation Doxofylline.
Doxofylline of xanthine asthma drugs, and its scientific name is 7 – (1,3 – dioxolan – ethyl methyl) -3,7 – dihydro-1,3 – dimethyl-1H – purine-2 ,6 – dione. The drug developed by the Italian Roberts & Co. in 1988, listed its tablet tradename Ansimar. This product is compared with similar asthma drugs, high efficacy, low toxicity, oral LD50 in mice is 1.5 times aminophylline, non-addictive. Adenosine and its non-blocking agents, it does not produce bronchial pulmonary side effects, no aminophylline like central and cardiovascular system. U.S. patent (US4187308) reported the synthesis of doxofylline, theophylline and acetaldehyde from ethylene glycol p-toluenesulfonic acid catalyst in the reaction of benzene as a solvent Doxofylline. Theophylline acetaldehyde by the method dyphylline derived reaction with a peroxy periodate or 7 – (2,2 – dialkoxy-ethyl) ammonium chloride aqueous solution in the decomposition of theophylline converted to acetaldehyde theophylline . Former method is relatively complex, and the high cost of using periodic acid peroxide, low yield after France. And theophylline acetaldehyde and ethylene glycol solvent used in the reaction of benzene toxicity, harm to health, and the yield is low, with an average around 70%, not suitable for industrial production.
SYN 1

Theophylline-7-acetaldehyde (I) could react with ethylene glycol (II) in the presence of p-toluenesulfonic acid in refluxing benzene to produce Doxofylline.
SYN 2

Doxofylline can be prepared by N-alkylation of theophylline (I) with bromoacetaldehyde ethylene glycol acetal (II) using Na2CO3 in refluxing H2O (1).
.…………………………………….
Synthesis
EXAMPLE
A mixture of 15 g of theophyllineacetic aldehyde, 30 ml of ethylene glycol and 1.5 g of p-toluenesulphonic acid in 600 ml of benzene is heated under reflux in a flask provided with a Marcusson apparatus.
After two hours the separation of the water is complete.
The reaction mixture is washed with 200 ml of a 3.5% aqueous solution of sodium bicarbonate.
The organic phase is dried and concentrated to dryness under reduced pressure, to leave a product residue which is taken up in ethyl ether, separated by filtration and purified by ethanol.
2-(7′-theophyllinemethyl)-1,3-dioxolane is obtained.
M.P. 144
Average yield 70%
Analysis: C.sub.11 H.sub.14 N.sub.4 O.sub.4 : M.W. 266.26: Calculated: C%, 49.62; H%, 5.30; N%, 21.04. Found: C%, 49.68; H%, 5.29; N%, 21.16.
………………………………..
the reaction is:
a, anhydrous theophylline and bromoacetaldehyde ethylene glycol as the basic raw material, purified water as a solvent with anhydrous sodium carbonate as acid-binding agent;
Doxofylline
UV (95% C2H5OH, nm) λmax273 (ε9230); λmin244 (ε2190)
IR (KBr, cm-1) 1134 (CO); 1233 (CN) ; 1547 (C = N); 1656 (C = C); 1700 (C = O); 2993 (CH)
1H-NMR [CDCl3, δ (ppm)] 3.399 (s, 3H, N-CH3); 3.586 (S, 3H, N-CH3); 3.815-3.885 (m, 4H, OCH2 × 2); 4.581 (d, 2H, CH2); 5.211 (t, 1H, CH ); 7.652 (S, 1H, CH = N)
13C-NMR [CDCL3, δ (ppm)] 27.88 (CH3); 29.69 (CH3); 47.87 (CH2); 65.37 ( OCH2); 100.76 (CH); 107.26 (C = C); 142.16 (CH = N); 148.22 (C = C); 151.59 (C = O); 155.25 ( C
……………………………
HPLC
http://www.scipharm.at/download.asp?id=1401
…………………..
- Cirillo R, Barone D, Franzone JS (1988). “Doxofylline, an antiasthmatic drug lacking affinity for adenosine receptors”. Arch Int Pharmacodyn Ther 295: 221–37.PMID 3245738.
- Poggi R, Brandolese R, Bernasconi M, Manzin E, Rossi A (October 1989). “Doxofylline and respiratory mechanics. Short-term effects in mechanically ventilated patients with airflow obstruction and respiratory failure”. Chest 96 (4): 772–8.doi:10.1378/chest.96.4.772. PMID 2791671.
- Dini FL, Cogo R (2001). “Doxofylline: a new generation xanthine bronchodilator devoid of major cardiovascular adverse effects”. Curr Med Res Opin 16 (4): 258–68.doi:10.1185/030079901750120196. PMID 11268710.
- Sankar J, Lodha R, Kabra SK (March 2008). “Doxofylline: The next generation methylxanthine”. Indian J Pediatr 75 (3): 251–4. doi:10.1007/s12098-008-0054-1.PMID 18376093.
- Dali Shukla, Subhashis Chakraborty, Sanjay Singh & Brahmeshwar Mishra. Doxofylline: a promising methylxanthine derivative for the treatment of asthma and chronic obstructive pulmonary disease. Expert Opinion on Pharmacotherapy. 2009; 10(14): 2343-2356, DOI 10.1517/14656560903200667, PMID 19678793
- Farmaco, Edizione Scientifica, 1981 , vol. 36, 3 pg. 201 – 219, mp 144 – 144.5 °C
- Drugs Fut 1982, 7(5): 301
| US6313131 | 16 feb 2000 | 6 nov 2001 | Upsher-Smith Laboratories, Inc. | Method of kidney treatment |
| US6348470 * | 20 maart 1998 | 19 feb 2002 | Korbonits Dezsoe | Antitussive compositions |
| US6423719 | 16 feb 2000 | 23 juli 2002 | Upsher-Smith Laboratories, Inc. | Method for treating benign prostate hyperplasia |
| CN101647776B | 2 sept 2009 | 20 april 2011 | 吴光彦 | Doxofylline venous injection with small volume as well as preparation method and quality control method thereof |
| DE3114130A1 * | 8 april 1981 | 28 jan 1982 | Abc Ist Biolog Chem Spa | Neue theophyllinylmethyldioxolan-derivate, verfahren zu ihrer herstellung und sie enthaltende pharmazeutische ansaetze |
| EP0272596A2 * | 16 dec 1987 | 29 juni 1988 | ISTITUTO BIOLOGICO CHEMIOTERAPICO “ABC” S.p.A. | Theophyllinemethyldithiolan and theophyllinemethyldithianyl derivates, a method for their preparation and pharmaceutical compositions in which they are included |
| WO2011146031A1 | 16 mei 2011 | 24 nov 2011 | Bilgic Mahmut | Pharmaceutical composition comprising n- acetylcysteine and a xanthine |
| WO2013055302A1 | 14 mei 2012 | 18 april 2013 | Mahmut Bilgic | Effervescent composition comprising n- acetylcysteine and doxophylline or theophylline |
………………………………………………………………………………………..
I n case Images are blocked on your computer, VIEW AT
14-chapter 4.pdf – Shodhganga
Although various bioanalytical methods for estimation of doxofylline in …. 1H and 13C-NMR spectra of doxofylline and its degradation products were recorded by….. CLICK ABOVE
SPECTRAL DATA

The ESI mass spectrum exhibited a protonated molecular ion peak at m/z 267 in positive ion mode indicating the molecular weight of 266. The tandem mass spectrum showed the fragment ions m/z 223, 181.2, 166.2, 138.1, 124.1 and 87.1.
The FT-IR spectrum, two strong peaks at 1697cm-1 and 1658cm-1 indicated presence of two carbonyl groups. A strong peak at frequency 1546cm-1 indicated presence of C=N stretch. A medium peak at 1232cm-1 was due to C-O stretch
FT IR
1H and 13C-NMR spectra of doxofylline and its degradation products were recorded by using Bruker NMR 300MHz instrument with a dual broad band probe and z-axis gradients. Spectra were recorded using DMSO-d6 as a solvent and tetramethylsilane as an internal standard.
4.2.6 Validation
1H NMR
13 C NMR
COMPARISONS
Drug spotlight- Zafirlukast

ZAFIRLIKAST 
cyclopentyl 3-{2-methoxy-4-[(o-tolylsulfonyl)carbamoyl]benzyl}-1-methyl-1H-indol-5-ylcarbamate 107753-78-6
Matassa, V.G. et al, J. Med. Chem., v. 33, 1781 (1990);
U. S. Patent No. 4,859,692;
U. S. Patent No. 5,993,859;
http://www.accessdata.fda.gov/drugsatfda_docs/label/2011/020547s031lbl.pdf
Zafirlukast is an oral leukotriene receptor antagonist (LTRA) for the maintenance treatment of asthma, often used in conjunction with an inhaled steroid and/or long-acting bronchodilator. It is available as a tablet and is usually dosed twice daily. Another leukotriene receptor antagonist is montelukast (Singulair), taken once daily. Zileuton (Zyflo), also used in the treatment of asthma via its inhibition of 5-lipoxygenase, is taken four times per day.
Zafirlukast blocks the action of the cysteinyl leukotrienes on the CysLT1 receptors, thus reducing constriction of the airways, build-up of mucus in the lungs andinflammation of the breathing passages.
Zafirlukast is marketed by Astra Zeneca with the brand names Accolate, Accoleit, and Vanticon. It was the first LTRA to be marketed in the USA and is now approved in over 60 countries, including the UK, Japan, Taiwan, Italy, Spain, Canada, Brazil, China and Turkey
Healthy young men who received a single oral 40 mg dose attained peak plasma zafirlukast concentrations that averaged 607 μg/L at 3.4 hours. The elimination half-life ranged from 12 to 20 hours. In another study involving a 20 mg single oral dose in healthy men, the elimination half-life averaged 5.6 hours.[1][2]
A letter was submitted to the FDA by Zeneca Pharmaceuticals on July 22, 1997, notifying them of a change in product labeling that includes the following potential reaction in patients undergoing a dosage reduction of oral steroids who are currently taking zafirlukast:
PRECAUTIONS-Eosinophilic Conditions: The reduction of the oral steroid dose, in some patients on ACCOLATE therapy, has been followed in rare cases by the occurrence of eosinophilia, vasculitic rash, worsening pulmonary symptoms, cardiac complications, and/or neuropathy sometimes presenting as Churg–Strauss syndrome, a systemic eosinophilic vasculitis. Although a causal relationship with ACCOLATE has not been established, caution is required when oral steroid reduction is being considered.1
NDA..020547 26/09/1996, ACCOLATE, ASTRAZENECA, 20MG TABLET
| US Patent No | Expirey Date | patent use code |
|---|---|---|
| 5482963 | Jan 9, 2013 | |
| 5612367 | Mar 18, 2014 | U-189 |
Brief background information
| Salt | ATC | Formula | MM | CAS |
|---|---|---|---|---|
| – | R03DC01 | C 31 H 33 N 3 O 6 S | 575.69 g / mol | 107753-78-6 |
| monohydrate | R03DC01 | C 31 H 33 N 3 O 6 S · H 2 O | 593.70 g / mol | 143052-93-1 |
| calcium (2: 1) | R03DC01 | C 62 H 64 CaN 6 O 12 S 2 | 1189.43 g / mol | 107753-86-6 |
Application
-
antihistamine effect
-
LTD4-antagonist
Classes of substances
-
Benzenesulfonamide (s -imidy), as well as their derivatives
-
Esters of carbamic acid
-
Cyclopentanes
-
Hydroxybenzoic acid amides, and hydroxy acids alkoksibenzoynyh
-
Indoles
-
-
-
-
Zafirlukast is a synthetic, selective peptide leukotriene receptor antagonist (LTRA), with the chemical name 4(5-cyclopentyloxy-carbonylamino-1-methyl-indol-3ylmethyl)-3-methoxy-N-o-tolylsulfonylbenzamide. The molecular weight of zafirlukast is 575.7 and the structural formula is:

Zafirlukast, a fine white to pale yellow amorphous powder, is practically insoluble in water. It is slightly soluble in methanol and freely soluble in tetrahydrofuran, dimethylsulfoxide, and acetone.The empirical formula is: C31H33N3O6S
- Fischer JD, Song MH, Suttle AB, Heizer WD, Burns CB, Vargo DL, Brouwer KL. Comparison of zafirlukast (Accolate) absorption after oral and colonic administration in humans. Pharmaceut. Res. 17: 154-159, 2000.
- Bharathi DV, Naidu A, Jagadeesh B, Laxmi KN, Laxmi PR, Reddy PR, Mullangi R. Development and validation of a sensitive LC-MS/MS method with electrospray ionization for quantitation of zafirlukast, a selective leukotriene antagonist in human plasma: application to a clinical pharmacokinetic study. Biomed. Chromatogr. 22: 645-653, 2008.
- Zafirlukast (U.S. National Library of Medicine)
- Zafirlukast (patient information)

 |
|
 |
|
| Systematic (IUPAC) name | |
|---|---|
| cyclopentyl 3-{2-methoxy-4-[(o-tolylsulfonyl)carbamoyl]benzyl}-1-methyl-1H-indol-5-ylcarbamate | |
| Clinical data | |
| Trade names | Accolate |
| AHFS/Drugs.com | monograph |
| MedlinePlus | a697007 |
| Pregnancy cat. | B1 (Australia), B (United States) |
| Legal status | POM (UK) |
| Routes | Oral |
| Pharmacokinetic data | |
| Bioavailability | Unknown |
| Protein binding | 99% |
| Metabolism | Hepatic (CYP2C9-mediated) |
| Half-life | 10 hours |
| Excretion | Biliary |
| Identifiers | |
| CAS number | 107753-78-6 |
| ATC code | R03DC01 |
| PubChem | CID 5717 |
| IUPHAR ligand | 3322 |
| DrugBank | DB00549 |
| ChemSpider | 5515 |
| UNII | XZ629S5L50 |
| KEGG | D00411 |
| ChEBI | CHEBI:10100 |
| ChEMBL | CHEMBL603 |
| Chemical data | |
| Formula | C31H33N3O6S |
| Mol. mass | 575.676 g/mol |
Trade Names
| Country | Trade name | Manufacturer |
|---|---|---|
| United Kingdom | Akkolat | AstraZeneca |
| Italy | Akkoleit | – “- |
| Zafirst | Chiesi | |
| Japan | Akkolat | AstraZeneca |
| USA | – “- | Zeneca |
| Ukraine | No | No |
Formulations
-
Tablets of 20 mg, 40 mg

is a first anti-asthmatic leukotriene antagonist (Matassa, V.G. et al, J. Med. Chem., v. 33, 1781 ‘(1990); U. S. Patent No. 4,859,692 and The Merck Index, 12th Edition, 10241). Methods for the preparation of Zafirlukast are described in J. Med. Chem., v. 33, 1781 (1990), U. S. Patent 4,859,692 and U.S. Patent 5,993,859 starting from methyl 3-methoxy-4-(l-methyl-5-nitroindol-3-ylmethyl)benzoate [la]



in the presence of an equivalent quantity of silver(I) oxide,

The above process has serious disadvantages in the isolation of the product [4] in step (b) which is due to the fact that alkylation of indole, that is unsubstituted at positions 1-, 2- and 3-, at the 3-position, is accompanied by the undesired process of poly alkylation, to form polysubstituted indoles of formula [7] and/or formula [8] :

while at the same time some quantity of the starting unreacted indole remains in the reaction mixture. Most common methods for the separation of alkyl (indol-3-ylmethyl)benzoate of formula [4] from by-products of polyalkylation and starting unreacted indole, which are all covalent compounds with similar physical properties, include column chromatography that is an unpractical method for industrial scale applications.
Formula (I) compound for the synthesis of an important intermediate of zafirlukast.Reported in the patent EP199543 synthesized compound (I) of the conventional method, the following formula:

(A) (I)
In this method, Intermediate A and 5 – nitro-indole silver oxide in the presence of a catalyst, for docking composite formula (I) compound. Reported only 45% of the reaction yield, the reaction is difficult to complete the reaction and post-treatment using chromatographic methods, resulting in product purification more difficult. And the use of more expensive silver oxide catalysts, high cost.
W00246153 reported a catalyst for the above reaction to zinc bromide, Compound (I), after treatment of the compound (I) with sodium hydroxide hydrolysis of the intermediate (B), separating the product and raw materials purification products.

The method reported in the literature a yield of 60%, but the actual operation is repeated only about 30% yield, and the operation is complicated, cumbersome and costly.
zaafirlukast is a selective and competitive receptor antagonist of leukotriene D4 and E4 (LTD4 and LTE4), components of slow-reacting substance of anaphylaxis (SRSA). Cysteinyl leukotriene production and receptor occupation have been correlated with the pathophysiology of asthma, including airway edema, smooth muscle constriction, and altered cellular activity associated with the inflammatory process, which contribute to the signs and symptoms of asthma.
The cysteinyl leukotrienes (LTC4 LTD4, LTE4) are the products of arachidonic acid metabolism and are various cells, including mast cells and eosinophills, these eicosinoids bind to cysteinyl leukotriene (CysLT) receptors. The CysLT type-1 (CysLT1) receptor is found in human airway and other pro-inflammatory cells. CysLTs have been correlated with the pathophysiology of asthma.
Zafirlukast is a synthetic, selective peptide leukotriene receptor antagonist (LTRA), useful for the treatment of asthma and is commercially available in products sold under the brand name ACCOLATE™ as 10 and 20 mg tablets for oral administration. ACCOLATE™ is indicated for the prophylaxis and treatment of asthma in adults and children 5 years of age and older.
ACCOLATE™ film coated tablets contain amorphous zafirlukast as the active ingredient and the excipients croscarmellose sodium, lactose, magnesium stearate, microcrystalline cellulose, povidone, hypromellose, and titanium dioxide.
The greatest prevalence of asthma is in preschool children; however, the clinical utility of asthma therapy for this age group is limited by a narrow therapeutic index, long-term tolerability, and frequency and/or difficulty of administration. Asthma treatment requires an immediate perceivable effect. Inhalation therapy is a very common therapy prescribed for young children; inhalation therapy has the disadvantage of high dose variability.
 Process for the preparation of zafirlukast
Process for the preparation of zafirlukastUS 20040186300 A1



An Improved and Scalable Process for Zafirlukast: An Asthma Drug
Melting range: 142−145 °C; MS (m/z): 576 (M+ + H); IR (KBr, cm−1): 3326 (NH), 1679 (−C═O), 1H NMR (CDCl3) δ 7.0−8.0 (m, 11H), 3.7 (s, 3H), 4.0 (s, 2H), 3.9 (s, 3H), 2.6 (s, 3H), 1.45−1.8 (s, 9H). ……………………………………………………………….. US 20040186300 A1 http://www.google.com/patents/US20040186300 zafirlukast ethanolate as white powder with mp 132-133° C. (dec.) and 99.8% purity by HPLC. 1H NMR (CDCl3, δ, ppm): 1.22 (t, J 7.05 Hz, 3H), 1.45-1.87 (m, 8H), 2.66 (s, 3H), 3.67 (s, 3H), 3.73 (q, J 7.05 Hz, 4H), 3.79 (s, 3H), 3.98 (s, 2H), 5.08-5.23 (m, 1H), 6.58 (s, 1H), 6.73 (s, 1H), 7.01-7.51 (m, 9H), 8.23 (d, J 7.52 Hz, 1H), 9.67 (s, 1H).
Synthesis pathway
| Synthesis a) |
|---|
      |
| Synthesis of b) |
   |
-
Synthesis a)
-
US 4,859,692 (ICI; 08/22/1989; GB -prior. 4/17/1985; 17.10.1985).
-
EP 199 543 (ICI, Zeneca; appl. 16.4.1986; GB -prior. 4/17/1985).
-
-
Synthesis of b)
-
EP 490 649 (ICI, Zeneca; 11.12.1991; GB -prior. 12.12.1990).
-
Matassa, G. et al .: J. Med. Chem. (JMCMAR) 33, 1781 (1990).
-
Srinivas, K. et al .: Org. Process Res. Dev. (OPRDFK) 8 (6), 952 (2004).
-
added info Asthma is a disease that causes swelling and narrowing the airways of the lungs. Airways are air carriers to and from lungs. Swollen and narrower airways affect the air flow to and from the lungs and this lead to tightness of chest, wheezing, shortness of breath and cough. These symptoms are often occurs in early morning and in night. Asthma is caused by genetic and environmental factors, it was not curable completely but this can be controlled with good medical care. Leukotriene antagonists also known as leukast are the medicaments that are used to reduce leukotrienes, which are produced by several types of cells and causes inflammation in asthma and bronchitis. Leukotriene antagonists that are available in market are Montelukast, Zafirlukast and Pranlukast. Zafirlukast is the first leukast compound approved for management of Asthma. US FDA approved zafirlukast in the form of 10 mg and 20 mg tablet with the brand name of Accolate®.1 Subsequently this was approved and launched by innovator in few other countries. There are many synthetic routes for the preparation of Zafirlukast 4 is well documented in literature. Some of the key approaches are discussed here under. Scientists from ICI Americas Inc2 have reported process for the synthesis of 4, which starts with esterification of 3-methoxy-4-methyl benzoic acid 53 using methanol in presence of acetyl chloride PRODUCT PATENT ROUTE Allylic bromination of methyl ester 54 using bromine in presence of CCl4 resulted bromo compound 55, which was reacted with 5-nitro indole 124 using silver oxide as catalyst to obtain condensed compound 125. N-methylation of 125 utilizing methyl iodide in presence of NaH afforded N-methyl indole derivative 57. Thus obtained 57 was subjected to reduction using palladium carbon (Pd/C) in methanol followed by reacted with cyclopentyl chloroformate to obtain compound 59. Hydrolysis of 59 using LiOH.H2O subsequently reaction with o-toluene sulfonamide (OTSA) in presence of 1-[3-(dimethylamino)propyl]-3-ethyl carbodiimide hydrochloride (DMAPEC) and DMAP furnished zafirlukast 4. Matassa et al3 also reported similar procedure for the synthesis of Zafirlukast 4. 


DRUG SPOTLIGHT…LEVETIRACETAM

LEVETIRACETAM, etiracetam
(-)-(S)-α-ethyl-2-oxo-1-pyrrolidine acetamide
(−)-(S)-alpha-ethyl-2-oxo-1-pyrrolidineacetamide
CAS…102767-28-2
Crystals from ethyl acetate, mp 117°. [a]25D -90.0° (c = 1 in acetone). Soly (g/100 ml): water 104.0; chloroform 65.3; methanol 53.6; ethanol 16.5; acetonitrile 5.7. Practically insol in n-hexane. LD50 in male mice, male rats (mg/kg): 1081, 1038 i.v. (Gobert, 1990).
Mp: mp 117°C
| Active Ingredient: | LEVETIRACETAM |
| Dosage Form;Route: | INJECTABLE;IV (INFUSION) |
| Proprietary Name: | KEPPRA |
| Applicant: | UCB INC |
| Strength: | 500MG/5ML (100MG/ML) |
| Application Number: | N021872 |
| Product Number: | 001 |
| Approval Date: | Jul 31, 2006 |
| Reference Listed Drug | Yes |
| RX/OTC/DISCN: | RX |
Levetiracetam is an anticonvulsant medication used to treat epilepsy. Levetiracetam may selectively prevent hypersynchronization of epileptiform burst firing and propagation of seizure activity. Levetiracetam binds to the synaptic vesicle protein SV2A, which is thought to be involved in the regulation of vesicle exocytosis. Although the molecular significance of levetiracetam binding to synaptic vesicle protein SV2A is not understood, levetiracetam and related analogs showed a rank order of affinity for SV2A which correlated with the potency of their antiseizure activity in audiogenic seizure-prone mice.

Epilepsy is a chronic neurological disorder that consists of repeated occurrences of spontaneous seizures. Levetiracetam, [(S)-a-ethyl-2-oxopyrrolidine acetamide], has recently been approved as an add-on therapy for the treatment of refractory epilepsy . The (S)-enantiomer of etiracetam (levetiracetam) has shown remarkable pharmacokinetic and pharmacological activity which has led to the quick approval of this antiepileptic drug by the FDA.
Levetiracetam offers several advantages over traditional therapy, including twice-daily dosing, a wide margin of safety with no requirements for serum drug concentration monitoring and no interactions with other anticonvulsants, and less adverse effects than traditional treatments
Levetiracetam (INN) /lɛvɨtɪˈræsɨtæm/ is an anticonvulsant medication used to treatepilepsy. It is the S-enantiomer of etiracetam, structurally similar to the prototypicalnootropic drug piracetam.
Levetiracetam is marketed under the trade name Keppra. Keppra is manufactured by UCB Pharmaceuticals Inc. Since November 2008 the drug has been available as a genericbrand in the United States.
Levetiracetam has been approved in the European Union as a monotherapy treatment for epilepsy in the case of partial seizures, or as an adjunctive therapy for partial, myoclonicand tonic-clonic seizures. It is also used in veterinary medicine for similar purposes.
Levetiracetam has potential benefits for other psychiatric and neurologic conditions such as Tourette syndrome, autism, bipolar disorder and anxiety disorder, as well asAlzheimer’s disease. However, its most serious adverse effects are behavioral, and its benefit-risk ratio in these conditions is not well understood.
Along with other anticonvulsants like gabapentin, it is also sometimes used to treatneuropathic pain. It has not been found to be useful for essential tremors.
Levetiracetam (LEV) is a novel antiepileptic drug (AED) which was discovered in early 1980s and soon, in 1999 FDA approved LEV for the management of partial onset seizure. In India, LEV tablet was approved in April 2005. It acts by binding to the synaptic vesicle protein SV2A, which is present on synaptic vesicles and some neuroendocrine cells. Pharmacokinetics of LEV such as, less protein binding and lack of hepatic metabolism makes LEV less susceptible to drug interactions with other anticonvulsants. Evidence also suggests that LEV is much better than other AEDs in the way of broad therapeutic window, convenient dosing and less adverse effect. Besides the pharmacological effects, pharmacoeconomically also, LEV is a beneficial drug. All these valuable pharmacological and pharmacoeconomic aspect makes LEV an important option in management of various types of epilepsy.

- PubMed Health A division of the National Library of Medicine at the National Institutes of Health.
- Keppra (levetiracetam) Final Printed Label April 2009. Center for Drug Evaluation and Research, U.S. Food and Drug Administration. Accessed 29 July 2011.
- Keppra UCB (manufacturer’s website)
- NIH MedLine drug information
KEPPRA injection is an antiepileptic drug available as a clear, colorless, sterile solution (100 mg/mL) for intravenous administration.
The chemical name of levetiracetam, a single enantiomer, is (-)-(S)-α-ethyl-2-oxo-1-pyrrolidine acetamide, its molecular formula is C8H14N2O2 and its molecular weight is 170.21. Levetiracetam is chemically unrelated to existing antiepileptic drugs (AEDs). It has the following structural formula:
 |
Levetiracetam is a white to off-white crystalline powder with a faint odor and a bitter taste. It is very soluble in water (104.0 g/100 mL). It is freely soluble inchloroform (65.3g/100 mL) and in methanol (53.6 g/100 mL), soluble in ethanol (16.5 g/100 mL), sparingly soluble in acetonitrile (5.7 g/100 mL) and practically insoluble in n-hexane. (Solubility limits are expressed as g/100 mL solvent.)
KEPPRA injection contains 100 mg of levetiracetam per mL. It is supplied in single-use 5 mL vials containing 500 mg levetiracetam, water for injection, 45 mg sodium chloride, and buffered at approximately pH 5.5 with glacial acetic acid and 8.2 mg sodium acetate trihydrate. KEPPRA injection must be diluted prior to intravenous infusion

(S)-(−)-α-ethyl-2-oxo-1-pyrrolidine acetamide, which is referred under the International Nonproprietary Name of Levetiracetam, its dextrorotatory enantiomer and related compounds. Levetiracetam is shown as having the following structure:

Levetiracetam, a laevorotary compound is disclosed as a protective agent for the treatment and the prevention of hypoxic and ischemic type aggressions of the central nervous system in the European patent No. 162036. This compound is also effective in the treatment of epilepsy, a therapeutic indication for which it has been demonstrated that its dextrorotatory enantiomer (R)-(+)-α-ethyl-2-oxo-1-pyrrolidine acetamide completely lacks activity (A. J. GOWER et al., Eur. J. Pharmacol., 222, (1992), 193-203). Finally, in the European patent application No. 0 645 139 this compound has been disclosed for its anxiolytic activity.
The asymmetric carbon atom carries a hydrogen atom (not shown) positioned above the plane of the paper. The preparation of Levetiracetam has been described in the European patent No. 0162 036 and in the British patent No. 2 225 322, both of which are assigned to the assignee of the present invention. The preparation of the dextrorotatory enantiomer (R)-(+)-α-ethyl-2-oxo-1-pyrrolidine acetamide has been described in the European patent No. 0165 919.
-
Levetiracetam was first disclosed in EP 162036 indicating particular therapeutic properties distinguishing it from the racemic form.

-
Several processes for obtaining levetiracetam have been disclosed. One promising approach is the reaction of (S)-2-aminobutyramide (5) with an alkyl 4-halobutyrate or with a 4-halobutyryl halide followed by cyclization as outlined in EP 162036 . Clearly, said (S)-2-aminobutyramide (5) is a key intermediate in the preparation of levetiracetam and given the importance of the correct stereochemistry of levetiracetam also the correct stereochemistry in the key intermediates is of importance.
-
The separation of stereoisomers is considered to be one of the difficult tasks in chemistry since chiral compounds exhibit identical physical properties in non-chiral environments. Although several approaches for the preparation of optically pure (S)-2-aminobutyramide (5) have been reported, many of these are related to resolution of racemic (R,S)-2-aminobutyramide (e.g. WO 2006/103696 ), optionally using catalytic amounts of an aldehyde such as described in JP 2007/191470 . However, an approach directly starting from the Schiff base of racemic (R,S)-2-aminobutyramide (i.e. compound (1)) is unavailable whereas there is a need for this as said Schiff bases are highly suitable from a preparative point of view as these compounds may be conveniently isolated from the aqueous media that they are usually prepared in. This is in contrast with the parent 2-aminobutyramide which is highly soluble in water and consequently difficult to obtain in sufficient purity.

British Pat. No. 1,309,692 describes the compound α-ethyl-2-oxo-l- pyrrolidineactamide (melting point 122 degrees C.) and states that compounds of this type can be used for therapeutic purposes, for example for the treatment of motion sickness, hyperkinesia, hypertonia and epilepsy.
-
Several processes for obtaining levetiracetam have been disclosed in the art. Patent application EP 162,036-A1 discloses obtaining levetiracetam by reacting (S)-α-ethyl-2-oxo-1-pyrrolidineacetic acid with an alkyl haloformate and subsequently with ammonia, as summarized in the following scheme:

-
The same document discloses obtaining levetiracetam by reacting (S)-2-aminobutanamide with an alkyl 4-halobutyrate or with a 4-halobutyryl halide, and subsequent cyclization of alkyl (S)-4-[[1-(aminocarbonyl)propyl]amino]butyrate or of (S)-N-[1-(aminocarbonyl)propyl]-4-halobutanamide thus obtained, as summarized in the attached scheme:

-
The two previous processes have the drawback of operating at temperatures between -10°C and -60°C and the drawback of using intermediates for cyclization that are not readily obtained.
-
Patent application GB 2,225,322-A1 discloses a process for obtaining levetiracetam by hydrogenolysis of (S)-α-[2-(methylthio)ethyl]-(2-oxo-1-pyrrolidine)acetamide by means of a desulfurizing reagent such as Raney nickel or NaBH4.NiCl2.6H2O, according to the following scheme:

-
A drawback of this industrial-scale process is that it requires special equipment and special precautions for handling the products.
-
Other processes are known (for example US patents No 6,107,492 and6,124,473 ) in which levetiracetam is obtained by means of optical resolution of racemic etiracetam of formula (I). InUS patent No 6,107,492 resolution is performed by means of preparative high performance liquid chromatography or by means of a continuous simulated fluid bed chromatographic system with a chiral stationary phase. US patent No 6,124,473 discloses a continuous simulated fluid bed chromatographic system consisting of at least three chiral stationary phase columns. These industrial-scale resolution processes are affected by drawbacks related to using chromatography.
-
Patent applications WO 01/64,637-A1 and WO 02/26,705-A2 disclose processes for preparing levetiracetam by asymmetric hydrogenation of intermediates with a double bond, the hydrogenation of which gives the levetiracetam ethyl group, according to the following scheme:

-
The industrial-scale difficulties and hazard of hydrogenation can be mentioned in relation to these processes.
-
Finally, patent application ES 447,346 describes a process for the preparation of a pyrrolidone derivative, in particular the 2-oxo-1-pyrrolidinylacetamide, which comprises first reacting pyrrolidone with formaldehyde and a secondary amine, then reacting the compound obtained with an alkylating agent such as dimethyl sulfate, followed by treating the compound obtained with sodium or potassium cyanide, and finally reacting the compound obtained with hydrogen peroxide in basic medium.




Moreover, it also mentions that these compounds can be applied in the field of memory disorders in normal or pathological conditions.
It is also known that α-ethyl-2-oxo-l-pyrrolidineacetamide possesses a protective activity against aggressions of the central nervous system caused by hypoxias, cerebral ischemia, etc. (Pharmazie, 37/11, (1982), 753-765).
U.S. patent 4,969,943 discloses the levorotatory isomer of α-ethyl-2-oxo-l- pyrrolidineacetamide, which has the absolute S configuration, a method for making the isomer and pharmaceutical compositions containing the same. U.S. patent 4,696,943 discloses that the levorotatory isomer has a 10 times higher protective activity against hypoxia and a 4 times higher protective activity against ischemia compared to the known racemic form.

 http://oasys2.confex.com/acs/229nm/techprogram/P831117.HTM
http://oasys2.confex.com/acs/229nm/techprogram/P831117.HTM
Parallel synthesis of Levetiracetam (Keppra®) and its analogs via an Ugi-RCM strategy
ORGN 61 |
As part of our continued efforts to develop protocols for parallel synthesis of bioactive small molecules for drug discovery, we have recently developed a synthetic strategy using the tandem Ugi/Ring closing metathesis (RCM) sequence to construct the pyrrolidone scaffold, which has been known as an attractive pharmacophoric motif in medicinal chemistry. This method allows the use of readily available acrylic acid, and diversity elements of ketones, aldehydes and isocynides. To demonstrate its utility, parallel synthesis of Levetiractam (Keppra®, alpha-ethyl-2-oxopyrrolidine acetamide, 1), a drug on the market developed by UCB for add-on treatment of refractory partial seizures, and its diverse analogs, have been achieved. |
…………………………
http://www.google.com/patents/US7939676
Levetiracetam, (−)-(S)-alpha-ethyl-2-oxo-1-pyrrolidineacetamide, is a drug useful as a protective agent for treating and preventing hypoxic and ischemic type aggressions of the central nervous system. It is the active ingredient of KEPPRA®, tablets and flavored liquid, indicated as adjunctive therapy in the treatment of partial onset seizures in adults and children four years of age and older with epilepsy.
Levetiracetam was first described in U.S. Pat. No. 4,837,223 (UCB Societe Anonyme) where it is stated that it has particular therapeutic properties compared to the known racemic form (non proprietary name etiracetam). The S-enantiomer, for example, has a ten times higher protective activity against hypoxia and a four times higher protective activity against cerebral ischemia than the racemic mixture.
The U.S. Pat. No. 4,837,223 describes a method for the preparation of levetiracetam which comprises reacting (−)-(S)-alpha-ethyl-2-oxo-1-pyrrolidineacetic acid successively with alkylhaloformate and with ammonia. Said acid intermediate is, in turn, obtained from racemic (±)-alpha-ethyl-2-oxo-1-pyrrolidine acetic acid by a classic optical resolution according to known methods. In example 1, ethyl (±)-alpha-ethyl-2-oxo-1-pyrrolidine acetate is hydrolyzed to give the corresponding racemic acid in the presence of sodium hydroxide; said acid is subjected to chemical resolution by reaction with an optically active base, (+)-(R)-(1-phenyl ethyl)-amine, selective crystallization of diastereoisomeric salts thereof and isolation of the desired enantiomeric form; finally, the resultant (−)-(S)-alpha-ethyl-2-oxo-1-pyrrolidineacetic acid is converted into the corresponding amide via activation of the carboxyl residue with ethyl chloroformate.
Several alternative processes for the preparation of levetiracetam have been disclosed in the art.
GB 1,309,692 (UCB S.A.) describes the preparation of several N-substituted lactams, including, inter alia, 2-(2-oxo-pyrrolidino)-butyramide, i.e. the racemic form of levetiracetam, by converting the corresponding ester, obtained by reacting the appropriate pyrrolidin-2-one with an appropriate alkyl haloalkylcarboxylate, with gaseous ammonia in methanol (example 2) or by converting the corresponding acid chloride, obtained by reacting the corresponding acid with thionyl chloride, with gaseous ammonia (example 3).
WO 01/64637 (UCB Farchim) describes the preparation of levetiracetam by asymmetric hydrogenation in the presence of a chiral catalyst of (Z) or (E)-2-(2-oxotetrahydro-1H-1-pyrrolyl)-2-butenamide, which in turn is obtained by reacting the corresponding acid with PCl5 to give the corresponding acid chloride, and then with gaseous ammonia.
WO 03/014080 (UCB S.A.) describes a process for the preparation of levetiracetam and analogues thereof comprising the synthesis of the corresponding ester derivative, methyl-(S)-alpha-ethyl-2-oxo-1-pyrrolidine-acetate, and the subsequent ammonolysis reaction in the presence of water.
EP 1,566,376 (REDDYS LAB LTD DR) discloses a process for the preparation of levetiracetam by reacting 4-chlorobutyl chloride with (S)-2-Aminobutyramide hydrochloride, this latter being obtained by first reacting (5)-2-aminobutyric acid hydrochloride with thionyl chloride in methanol to give the corresponding ester hydrochloride, and then reacting the corresponding ester with ammonia in isopropanol.
Several other patents and patent applications describe other approaches to the synthesis of levetiracetam, such as, for example, U.S. Pat. Nos. 6,107,492 and 6,124,473 which describe the preparation of levetiracetam by optical resolution of etiracetam by means of preparative high performance liquid chromatography or continuous simulated moving bed chromatographic system, GB 2,225,322, which describes a process for the preparation of levetiracetam by hydrogenolysis of (S)-alpha-[2-(methylthio)-ethyl]-(2-oxo-1-pyrrolidine)-acetamide in the presence of a desulfurizing agent, and WO 2004/069796, which describes a process for preparing levetiracetam which comprises reacting (S)-2-aminobutyrramide hydrochloride and 4-chlorobutyl chloride in a solvent selected from acetonitrile and methyl tertbutyl ether in the presence of a strong base and recovering the crude product.
EXAMPLE 1Invention
Step 1
(−)-(S)-alpha-ethyl-2-oxo-1-pyrrolidine acetic acid (150 g, 0.87 mol) was dissolved in methanol (235 g, 300 ml) at 45° C. and thionyl chloride (56 g, 0.47 mol) was added dropwise over 30 min.
The reaction mixture was stirred at 45° C. for additional 15-30 min until complete conversion of (−)-(S)-alpha-ethyl-2-oxo-1-pyrrolidine acetic acid was observed via HPLC (unreacted (−)-(S)-alpha-ethyl-2-oxo-1-pyrrolidine acetic acid ≦2%, by HPLC % area).
At reaction completed, the volatiles were distilled off at moderate temperature and reduced pressure (35°-40° C., 150-200 mbar) until 10% of the whole volume was eliminated, then the mixture was reintegrated with fresh methanol up to initial volume.
After that, the reaction mixture was neutralized by bubbling ammonia gas at 20° C. up to a pH value equal to about 5, and stirred at 20° C. for 1 h. A limited amount of salts (about 44 g) precipitated and was filtered off. The resulting methanol solution was directly transferred to the autoclave.
Step 2
The reaction mixture was pressurized up to about 3 bar with ammonia gas at 20° C., and stirred until complete conversion to (−)-(S)-alpha-ethyl-2-oxo-1-pyrrolidineacetamide was observed via HPLC.
Then, once the reaction mixture was taken out of the autoclave, the residual salts formed (about 20 grams) were filtered off and the methanol solution was distilled up to a minimum volume at moderate temperature and reduced pressure (35°-40° C., 150-200 mbar).
Acetone (115 ml) was added and the mixture was distilled again at moderate temperature and reduced pressure (35°-40° C., 150-200 mbar) to minimum volume. After that acetone (300 ml) was charged over the residue and the mixture was heated and refluxed for 30 minutes. Finally, the solution was cooled down slowly to 0° C. and crude levetiracetam was isolated by filtration.
Crude levetiracetam (molar yield 73.1%, (R)-enantiomer: 1.171%) was then submitted to a final purification process in one step to give pure levetiracetam.
Acetone (750 ml) was charged over crude levetiracetam and the mixture was again stirred and heated to reflux. Once refluxed for about 30 minutes the hot mixture was filtered to remove residual salts and cooled slowly to 0° C.
Pure levetiracetam ((R)-enantiomer: 0.01%) was obtained by filtration and drying under vacuum at 40° C. Overall molar yield was 60.0% by mole of the starting amount of (−)-(S)-alpha-ethyl-2-oxo-1-pyrrolidine acetic acid (ponderal yield 78.4% by weight).
EXAMPLE 2
Example 1 was repeated, but the neutralization step with ammonia at the end of step 1 was omitted. At the end of step 2, crude levetiracetam was isolated (molar yield 73.1%, (R)-enantiomer: 2.21%). After purification step, pure levetiracetam (molar yield 64.4%, (R)-enantiomer: 0.58%) was obtained.
EXAMPLE 3Comparison
Step 1 of Example 1 was repeated using an excess of thionyl chloride (114 g, 0.96 mol) with respect to (−)-(S)-alpha-ethyl-2-oxo-1-pyrrolidine acetic acid. Further, when the complete conversion of (−)-(S)-alpha-ethyl-2-oxo-1-pyrrolidine acetic acid was observed, the reaction mixture was distilled off at moderate temperature and reduced pressure (35°-40° C., 150-200 mbar) until dryness. Decomposition of about 13% by weight of the intermediate product to starting product (−)-(S)-alpha-ethyl-2-oxo-1-pyrrolidine acetic acid was observed.
EXAMPLE 4Effect of Activating Agent Amount
Example 1 was repeated using different amount of thionyl chloride as reported in the following Table 1. The amount of thionyl chloride is expressed in terms of equivalent with respect to (−)-(S)-alpha-ethyl-2-oxo-1-pyrrolidine acetic acid.
| TABLE 1 | ||||
| Conversion | Unreacted (% | Converted (% | ||
| Sample | SOCl2 | Time (hours) | w/w) | w/w) |
| 1 | 1.10 | 1 | 0.1 | 99.9 |
| 2 | 0.78 | 0.5 | 0.4 | 99.7 |
| 3 | 0.60 | 1 | 1.1 | 99.3 |
| 4 | 0.54 | 0.5 | 1.1 | 99.3 |
| 5 | 0.29 | 2 | 2.2 | 98.4 |
| 6 | 0.09 | 5 | 4.9 | 96.7 |
| 7 | 0.05 | 24 | 1.7 | 99.0 |
The data of Table 1 clearly show that the use of a substoichiometric amount of thionyl chloride (samples 2 to 5) does not substantially affect the conversion time and conversion yield of (−)-(S)-alpha-ethyl-2-oxo-1-pyrrolidine acetic acid. On the contrary, the use of catalytic amount of thionyl chloride (samples 6 and 7) substantially increases the conversion time and/or the conversion yield.
………………………..

Compound (I) can also be condensed with 4-chlorobutyryl chloride (IV) either directly in the presence of tetrabutylammonium bromide (TBAB) in dichloromethane, followed by in situ treatment with potassium hydroxide, or via the isolation of intermediate (S)-N-[1-(carbamol)propyl]-4-chlorobutyramide (V).
……………..

An alternative procedure involves hydrolysis of racemic ethyl 2-(2-oxopyrrolidin-1-yl)burytate (VI) with sodium hydroxide to produce racemic 2-(2-oxopyrrolidin-1-yl)butyric acid (VII), which is resolved by fractional crystallization with (R)-(+)-alpha-methylbenzylamine in benzene, followed by acid-base treatment to give (S)-2-(2-oxopyrrolidin-1-yl)butyric acid (VIII). Compound (VIII) is finally treated with ethyl chloroformiate and ammonia in dichloromethane
………………………….
US Patent 8,338,621
J. Surtees and co-inventors disclose alternative processes for making active pharmaceutical ingredients (APIs) that are used to treat epilepsy and seizures. One compound that can be prepared by their processes is the established drug levetiracetam (1, Figure 1), marketed under the trade name Keppra. Because 1 is now off-patent, there is obvious interest in new drugs.

The inventors also claim that seletracetam (2) and brivaracetam (3) (Figure 2) can be prepared by their processes. These drugs are apparently much more active than 1.
All of the drugs are used as single isomers, so a stereoselective synthesis is desirable. The inventors describe two routes for preparing the molecules; the first, shown in Figure 1, is the synthesis of 1 by the reaction between pyrrolidone (4) and chiral bromo amide 5 in the presence of a base. GC analysis showed that the conversion is 40.3% and that the product contains 51% of the (S)-enantiomer and 49% of the (R)-isomer. No details of their separation are given, although the use of chiral HPLC is discussed.
The same reaction is used to prepare derivative 6 of 1. Compound 7 is prepared from the corresponding hydroxy ester and then condensed with 4 to give 6. Chiral HPLC showed that the product is a mixture of 89.3% (S)-enantiomer 6and 10.7% of its (R)-isomer.
The inventors do not describe the detailed preparation of 2, but they report that acid 8 is prepared in 41% yield from pyrrolidone 9 and acid 10 in the presence of NaH (Figure 2). Ammonolysis of 8 produces 2; no reaction details are provided.

In a reaction similar to the preparation of 8, acid 11 is prepared from 10 and pyrrolidone 12. The product is isolated in 77% yield and can be converted to 3by ammonolysis. Again, no details are provided for this reaction.
The second route for preparing the substituted pyrrolidones does not start with simple pyrrolidones and is the subject of additional claims. The route involves a cyclization reaction, shown in Figure 3. The preparation of enantiomer 13 begins with the reaction of racemic salt 14 and optically pure bromo ester 15. This step produces intermediate 16, isolated as a yellow oil. The crude material is treated with 2-hydroxypyridine (2-HP) to cyclize it to 17. This ester is hydrolyzed to give acid 18. Conversion to 13 is carried out by adding ClCO2Et, followed by reaction with liquid NH3 in the presence of K2CO3. The overall yield of 13 is 32%.

This route is also used to prepare levetiracetam (1) by treating 5 with the HCl salt of amino ester 19 to give 20, recovered as its HCl salt in 49% yield. The salt is basified with Et3N and treated with 2-HP to cyclize it to 1, initially isolated as an oil. GC analysis showed 100% conversion, and chiral HPLC showed that the product contains 98.6% (S)-isomer and 1.4% (R)-isomer.
The inventors also prepared 1 and its (R)-enantiomer 21 by using a similar reaction scheme with alternative substrates to 5. Figure 4 outlines the route, which starts from protected hydroxy amide 22 and amino ester 23. When the reaction is carried out in the presence of Cs2CO3, the product is (R)-enantiomer24, which is used without purification to prepare 21 by treating it with 2-HP. Chiral HPLC showed that the product is 94% (R) and 6% (S).

When the reaction between 22 and 23 is run with K2CO3, the product is (S)-enantiomer 25. This is used to prepare 1, but the product contains only 79% (S)-isomer.
The inventors do not comment on the apparent stereoselectivity of the carbonate salts in the reaction of 22 with 23. This is an intriguing finding and worthy of investigation. (UCB S.A. [Brussels]. US Patent 8,338,621,
…………………
Production of Levetiracetam
(1)H-MET-NH2 can be used to manufacture Levetiracetam. The detail is as follows:

(2)A reaction flask was added 500ml of methanol and deionized water 33ml, cooled to 0 ° C. Then add with stirring 50.0g (0.27mol), pass ammonia and dissolve to saturation, and seale reaction flask 0 to 5 º C reaction was stirred 96h TLC tracking,eluent, ethyl acetate / acetone (3:1) product Rf = 0.28, raw material Rf = 0.6]. feedstock point disappears, and the end of the reaction. Finanly, it was distilled under reduced pressure to obtain a yellow solid levetiracetam crude product 41.5g and the yield is 90.2%.

SYNTHESIS
SYN 1
UCB PHARMA, S.A. Patent: WO2007/65634 A1, 2007 ; Location in patent: Page/Page column 16-17 ;
941289-97-0![]()
LEVETIRACETAM
SYN 2
TEVA PHARMACEUTICAL INDUSTRIES LTD.; TEVA PHARMACEUTICALS USA, INC. Patent: WO2004/69796 A2, 2004 ; Location in patent: Page 9 ;
 AND
AND  GIVES PDT
GIVES PDT
SYN 3
 GIVES PDT
GIVES PDT
ZACH SYSTEM S.P.A. Patent: US2011/65932 A1, 2011 ; Location in patent: Page/Page column 3 ;
SYN 4

ZaCh System S.p.A. Patent: EP2147911 A1, 2010 ; Location in patent: Page/Page column 5 ;
SYN 5
 AND GIVES PDT
AND GIVES PDT
U C B Societe Anonyme Patent: US4696943 A1, 1987 ;
SYN 6
 AND
AND
UCB, S.A. Patent: WO2005/28435 A1, 2005 ; Location in patent: Page/Page column 10 ;
SYN 7

Tetrahedron Letters, , vol. 47, # 38 p. 6813 – 6815
SYN 8

WO2004/69796 A2, ; Page 11 ;
SEE ALSO
Tetrahedron Asymmetry, , vol. 16, # 22 p. 3739 – 3745
WO2008/77035 A2, ; Page/Page column 15 ;
EP1806339 A1, ; Page/Page column 22 ;
……………
Green Chemistry Letters and Reviews Vol. 3, No. 3, September 2010, 225230
http://www.tandfonline.com/doi/pdf/10.1080/17518251003716568
The desired compound 1 was re-crystallized in hot ethyl acetate (2106 ml) at 60degC, subsequently cooled to 25-30degC, filtered and dried at 35-40degC to obtain product in 65% yield (13 g) and 99.9% purity (by chiral HPLC) as a white solid: mp 116degC (lit3c 117degC); Rf: 0.34 [3:7 (EtOAc: Hexane)]; IR (KBr) nmax 3362, 3200, 2991, 2911, 1676, 1491, 1457, and 1383 cm1 ; 1 H NMR (400 MHz, CDCl3) d 0.91 (t, 3H, J7.5 Hz), 1.601.75 (m, 1H), 1.902.09 (m, 3H), 2.382.47 (m, 2H), 3.343.55 (m, 2H), 4.44 (dd, 1H, J6.7, 8.6 Hz) 5.74 (br, 1H s), 6.45 (br, 1H, s).
3 ) Kotkar, S.P.; Sudalai, A. Tetrahed. Lett. 2006, 47, 68136815.
……..
http://www.google.com/patents/WO2008012268A1?cl=en
Levetiracetam, (-)-(S)-alpha-ethyl-2-oxo- 1 -pyrrolidineacetamide, is a drug useful as a protective agent for treating and preventing hypoxic and ischemic type aggressions of the central nervous system. It is the active ingredient of KEPPRA®, tablets and flavored liquid, indicated as adjunctive therapy in the treatment of partial onset seizures in adults and children four years of age and older with epilepsy. Levetiracetam was first described in US 4,837,223 (UCB Societe Anonyme) where it is stated that it has particular therapeutic properties compared to the known racemic form (non proprietary name etiracetam). The S-enantiomer, for example, has a ten times higher protective activity against hypoxia and a four times higher protective activity against cerebral ischemia than the racemic mixture US ‘223 describes a method for the preparation of levetiracetam which comprises reacting (-)-(S)-alpha-ethyl-2-oxo-l-pyrrolidineacetic acid successively with alkylhaloformate and with ammonia. Said acid intermediate is, in turn, obtained from racemic (±)-alpha-ethyl-2-oxo-l -pyrrolidine acetic acid by a classic optical resolution according to known methods. In example 1 of the above US patent, ethyl (±)-alpha-ethyl-2-oxo-l -pyrrolidine acetate is hydrolyzed to give the corresponding racemic acid in the presence of sodium hydroxide; said acid is subjected to chemical resolution by reaction with an optically active base, (+)-(R)-(l -phenyl ethyl)-amine, selective crystallization of diastereoisomeric salts thereof and isolation of the desired enantiomeric form; finally, the resultant (-)-(S)-alpha-ethyl-2-oxo-l-pyrrolidineacetic acid is converted into the corresponding amide via activation of the carboxyl residue with ethyl chloroformate, in accordance with the following reaction scheme:

Several alternative processes for the preparation of levetiracetam have been disclosed in the art. WO 03/014080 (UCB S.A.) describes an improved process for the preparation of levetiracetam and analogues thereof comprising the ammonolysis reaction of the corresponding ester derivatives in the presence of water.
US 6,107,492 (Daicel Chem; UCB) and US 6,124,473 (UCB) describe the preparation of levetiracetam by optical resolution of etiracetam by means of preparative high performance liquid chromatography or continuous simulated moving bed chromatographic system.
GB 2,225,322 (UCB) describes a process for the preparation of levetiracetam by hydrogenolysis of (S)-alpha-[2-(methylthio)-ethyl]-(2-oxo-l-pyrrolidine)-acetamide in the presence of a desulfurizing agent such as NaBH4/NiC12 6 H2O, nickel Raney W-2 or nickel Raney T- 1.
WO 01/64637 (UCB Farchim) describes the preparation of levetiracetam by asymmetric hydrogenation of (Z) or (E)-2-(2-oxotetrahydro-lH-l-pyrrolyl)-2- butenamide by using a chiral catalyst. EP 162,036 (UCB) describes the preparation of levetiracetam by reacting (S)-2- aminobutanamide with an alkyl 4-halobutyrate or with a 4-halobutyryl halide, and subsequent cyclization of alkyl (S)-4-[[l-(aminocarbonyl)-propyl]-amino-butyrate or of (S)-N-[l-(aminocarbonyl)-propyl]-4-halobutanamide thus obtained. WO 2004/069796 (Teva Pharmaceutical Industries) describes a process for preparing levetiracetam which comprises reacting (S)-2-aminobutyrramide hydrochloride and 4-chlorobutyl chloride in a solvent selected from acetonitrile and methyl tertbutyl ether in the presence of a strong base and recovering the crude product. US 2005/0182262 (Dr. Reddy’s Laboratories) describes the preparation of (S)-2- aminobutyrramide hydrochloride, intermediate useful for the manufacture of levetiracetam via reaction with 4-chlorobutyl chloride.
WO 2004/076416 (Farma Lepori S.A.) describes a process to levetiracetam by means of deaminomethylation of a sufficiently pure enantiomer S-intermediate of formula

or a salt thereof.
In accordance with US ‘223, (-)-(S)-alpha-ethyl-2-oxo-l-pyrrolidineacetamide can not be obtained directly from the racemic mixture by separating the desired enantiomer.
Thus, as underlined above, in US ‘223 the resolution step is carried out on the intermediate (±)-alpha-ethyl-2-oxo-l-pyrrolidineacetic acid.
Said procedure has an intrinsic drawback due to separation of the S-enantiomer from the corresponding racemic mixture by classic optical resolution which, necessarily, leads to a loss of 50% of the acid substrate used.
Example 6 (-)-(S)-alpha-ethyl-2-oxo-l-pyrrolidineacetamide (levetiracetam).
In a 25 ml flask equipped with thermometer, mechanical stirring and bubble condenser, 3.344 g of (-)-(S)-alpha-ethyl-2-oxo-l-pyrrolidineacetic acid (19.58 mmol, e.e.= 95.0%), 0.11 ml of concentrated sulfuric acid (95.6% m/m, 1.97 mmol) and 17 ml of methanol were charged under nitrogen atmosphere at room temperature. Reaction mixture was heated up to 65°C temperature by oil bath and maintained at reflux temperature up to complete disappearing of starting material (about 2.5 h; checked by TLC, Rf = 0.58 CH2Cl2:Me0H:Ac0H 80:20: 1/silica gel). Reaction mixture was concentrated under vacuum up to a residue was formed then water (2.0 ml) was added. In a 25 ml flask equipped with magnetic stirring and condenser, 7.5 ml of 30% aqueous ammonia solution was charged and cooled to 00C temperature and, keeping under stirring, the aqueous solution of crude (-)-(S)-alpha-ethyl-2-oxo- 1-pyrrolidineacetic acid methyl ester was charged dropwise. When addition was completed, reaction mixture was thermostabilized at 200C and said conditions were maintained overnight.
At complete conversion (about 10 h) excess of ammonia was eliminated by vacuum evaporator. Reaction mixture was extracted with dichloromethane (2 x 3.5 ml), transferred into a continuous liquid-liquid extractor and then refluxed with 7 ml of dichloromethane for 6 hours. Collected organic phases were concentrated under vacuum up to a residue was formed. 2.666 g of a yellow solid was obtained which was suspended in 15.0 ml of acetone. Reaction mixture was heated up to 600C temperature so that complete dissolution of the solid was reached. Then, mixture was slowly cooled. White solid was isolated by filtration, washed with mother liquors and then with 3 ml of cold acetone and, finally, dried in oven under vacuum at 400C temperature for 4 hours to give 2.259 g of levetiracetam (13.274 mmol, 67.8% yield, e.e. 99.9%).
Example 7
(-)-(S)-alpha-ethyl-2-oxo-l-pyrrolidineacetic acid methyl ester. In a 250 ml reactor equipped with mechanical stirring, thermometer and condenser, 2.5 g of (-)-(S)-alpha-ethyl-2-oxo-l-pyrrolidineacet-N-(+)-(R)-(l-phenylethyl)-amide (9.112 mmol, d.e.= 99.3%), 24.85 g (6 eq.) of p-toluensulfonic acid supported by polymeric matrix (30.00-60.00 mesh, 2.2 mmol/g) and 75 ml of toluene were charged. To the reaction mixture was added 0.660 ml (36.64 mmol) of water under stirring and mixture was heated up to reflux temperature. Reaction was monitored by HPLC and at complete conversion of starting material (about 6 h), mixture was cooled to 600C temperature and 75 ml of methanol added. Reaction mixture was maintained at that temperature for 3 h up to complete formation of (-)-(S)-alpha- ethyl-2-oxo-l-pyrrolidineacetic acid methyl ester. Reaction mixture was permitted to cool and then it was filtered on gootch in order to separate the product from the resin. Resin was washed with methanol (2 x 75 ml) and organic phases were collected to give 365.1 g of a 0.462% organic solution of (-)-(S)-alpha-ethyl-2-oxo-l- pyrrolidineacetic acid methyl ester (1.69 g, 9.110 mmol, 100.0% yield) which was used in the following synthetic step. In order to recover (+)-(R)-(l-phenylethyl)-amine, resin was treated with 100 ml of 30% aqueous ammonia solution, 100 ml of methanol, 100 ml of 30% aqueous soda and again with 100 ml of methanol. Resin was then regenerated by washing with HCl 6 M (100 ml) and water up to neuter pH of the eluted phase. Finally, resin was washed with 100 ml of methanol and dried in oven at 500C temperature under vacuum overnight.
Example 8
(-)-(S)-alpha-ethyl-2-oxo-l-pyrrolidineacetamide (levetiracetam) (alternative 1). 365.1 g of the solution of (-)-(S)-alpha-ethyl-2-oxo-l-pyrrolidineacetic acid methyl ester (0.462%, 1.69 g, 9.110 mmol) obtained in Example 7 was charged in a flask and concentrated up to a residue was formed. 2.482 g of a brown oil was obtained. Residue was charged in a 10 ml flask equipped with magnetic stirring and condenser. Reaction mixture was cooled to 00C temperature and, keeping under stirring, 0.8 ml of water and 3.2 ml of 30% aqueous ammonia solution were charged dropwise in about 10 minutes. When addition was completed, reaction mixture was thermostabilized at 200C and said conditions were maintained overnight.
At complete conversion (about 14 h) excess of ammonia was eliminated by vacuum evaporator. Reaction mixture was then extracted with dichloromethane (10 x 5 ml). Collected organic phases were dried on Na2SO4, and concentrated under vacuum up to a residue was formed. 1.999 g of a yellow solid was obtained which was suspended in 5 ml of acetone. Reaction mixture was heated up to 600C temperature so that complete dissolution of the solid was reached. Then, mixture was slowly cooled. White solid was isolated by filtration, washed with mother liquors and then with 1 ml of cold acetone and, finally, dried in oven under vacuum at 25°C temperature for 1 night to give 0.965 g of levetiracetam (5.669 mmol, 62.2% yield, e.e. 94.2%). Example 9
(-)-(S)-alpha-ethyl-2-oxo-l-pyrrolidineacetamide (levetiracetam) (alternative 2). In a 50 ml reactor equipped with mechanical stirring, thermometer and condenser, 0.275 g of (-)-(S)-alpha-ethyl-2-oxo-l-pyrrolidineacet-N-(+)-(R)-(l-phenylethyl)- amide (1.0 mmol, d.e.= 99.3%), 10.0 g of ethyl-thiophenyl-sulfonic acid supported on silica (0.6 mmol/g, supplied by Phosphonics ®) and 15 ml of toluene were charged. To the reaction mixture was added 0.075 ml (4.0 mmol) of water under stirring and mixture was heated up to reflux temperature. Reaction is monitored by HPLC and at complete conversion of starting material (about 5 h), reaction mixture was cooled to 600C temperature and 10 ml of methanol added. Reaction mixture was maintained at that temperature for 3 h up to complete formation of (-)-(S)-alpha-ethyl-2-oxo-l- pyrrolidineacetic acid methyl ester. Reaction mixture was permitted to cool and then worked up according to the procedure described in example 7. 57.9 g of a 0.280% organic solution of (-)-(S)-alpha-ethyl-2-oxo-l-pyrrolidineacetic acid methyl ester (0.162 g, 0.875 mmol, 87.5% yield) was thus obtained. Such solution was charged in a flask and concentrated up to a residue was formed. 0.486 g of a brown oil was obtained. Residue was charged in a 5 ml flask equipped with magnetic stirring and condenser. Reaction mixture was cooled to 00C temperature and, keeping under stirring, 1.5 ml of 30% aqueous ammonia solution were charged dropwise. When addition was completed, reaction mixture was thermostabilized at 200C and said conditions were maintained overnight.
At complete conversion (about 15 h) excess of ammonia was eliminated by vacuum evaporator. Reaction mixture was then extracted with dichloromethane as described in example 8. Recrystallization of the crude product from refluxing acetone afforded 0.076 g of levetiracetam (0.447 mmol, 44.6% yield compared to the starting amide, e.e. 99.9%).
……………

PAPER FROM HINDAWI
Journal of Chemistry Volume 2013 (2013), Article ID 176512, 5 pages http://dx.doi.org/10.1155/2013/176512
Enantioselective Synthesis of Antiepileptic Agent, (−)-Levetiracetam, through Evans Asymmetric Strategy
1Department of Research and Development, Inogent Laboratories Private Limited, 28A, IDA Nacharam, Hyderabad 500 076, India
2Centre for Pharmaceutical Sciences, Institute of Science and Technology, Jawaharlal Nehru Technological University, Kukatpally, Hyderabad 500 072, India
3R&D Centre, Orchid Chemicals and Pharmaceuticals Ltd., 476/14, Sholinganallur, Chennai 600 119,
http://www.hindawi.com/journals/jchem/2013/176512/
A practical and efficient enantioselective synthesis of antiepileptic drug, (−)-Levetiracetam, has been described in five steps (33.0% overall yield) and high optical purity (99.0% ee), using Evans asymmetric strategy for -alkylation of carbonyl functionality as the key step. The simplicity of the experimental procedures and high stereochemical outcome make this method synthetically attractive for preparing the target compound on multigram scales.

white solid. Mp: 113–114°C.
S ROT= −95.0 [c = l.0, acetone].
1H NMR , 400 MHz):
δ 6.50 (br s, 1H),
5.70 (br s, 1H),
4.50 (t, J = 8.7, 6.8 Hz, 1H),
3.48 (m, 2H), 2.50 (m, 2H),
1.98–2.20 (m, 3H),
1.70 (m, 1H),
0.98 (t, J = 7.7 Hz, 3H) ppm; CH2–CH3
13C NMR , 75 MHz): δ175.9, 172.7, 55.9, 43.7, 31.0, 21.2, 18.0, 10.4 ppm;
IR : 3200, 1731, 1620 cm−1;
ESI-MS: m/z 171.0 [M++1].
Anal. calcd. for C8H14N2O2: C, 56.45; H, 8.29; N, 16.46; O, 18.80. Found: C, 56.76; H, 8.52; N, 16.87; O, 19.26.
Chiral HPLC purity 99% ee. The enantiomeric excess was determined by HPLC analysis in comparison with authentic racemic material and HPLC conditions: Chiral OD-H column; hexane: i-PrOH (90 : 10 v/v); flow rate 1.0 mL/min; UV −210 nm; column temperature 25°C; CHIRAL HPLC purity: = 14.4 min (S)-isomer (major enantiomer) and 9.3 min (R)-isomer (minor enantiomer).
…………………
Indian Journal of Chemistry -Section B (IJC-B) >
IJC-B Vol.53B [2014] >
IJC-B Vol.53B(09) [September 2014] >
http://nopr.niscair.res.in/handle/123456789/29370

1H nmr predict

13 C NMR PREDICT
…
| US4837223 | Mar 12, 1987 | Jun 6, 1989 | Ucb Societe Anonyme | (S)-alpha-ethyl-2-oxo-1-pyrrolidineacetamide compositions |
| US6107492 | May 7, 1999 | Aug 22, 2000 | Ucb, S.A. | By optical resolution of a racemic mixture of alpha-ethyl-2-oxo-1-pyrrolidine acetamide by chromatography using silica gel supporting amylose tris(3,5-dimethylphenylcarbamate) as a packing material |
| US6124473 | May 7, 1999 | Sep 26, 2000 | Ucb, S.A. | Process for preparing (s)- and (R)-α-ethyl-2-oxo-1-pyrrolidineacetamide |
| US7531673 * | Feb 16, 2005 | May 12, 2009 | Dr. Reddy’s Laboratories Limited | Preparation of amino acid amides |
| US20050182262 * | Feb 16, 2005 | Aug 18, 2005 | Acharyulu Palle V.R. | Reacting an amino acid or acid salt with a halogenating agent (thionyl chloride, phosphorous pentachloride or oxalyl chloride) , to form an intermediate, reacting the intermediate with ammonia; amidation; chemical intermediate to form Levetiracetam |
| EP1566376A1 | Feb 17, 2005 | Aug 24, 2005 | Dr. Reddy’s Laboratories Limited | Preparation of amino acid amides |
| GB1309692A | Title not available | |||
| GB2225322A | Title not available | |||
| WO2001064637A1 | Feb 21, 2001 | Sep 7, 2001 | Edmond Differding | 2-oxo-1-pyrrolidine derivatives, process for preparing them and their uses |
| WO2003014080A2 | Aug 5, 2002 | Feb 20, 2003 | Celal Ates | Oxopyrrolidine compounds, preparation of said compounds and their use in the manufacturing of levetiracetam and analogues |
| WO2004069796A2 | Feb 3, 2004 | Aug 19, 2004 | Ben-Zion Dolitzky | Process for producing levetiracetam |
| WO2006095362A1 * | Jan 20, 2006 | Sep 14, 2006 | Rubamin Ltd | Process for preparing levetiracetam |
| WO2008012268A1 | Jul 20, 2007 | Jan 31, 2008 | Zach System Spa | Process for the preparation of levetiracetam |
……
http://orgspectroscopyint.blogspot.in/2015/03/rs-alpha-ethyl-2-oxo-l-pyrrolidineacet.html
PREPARATION OF KEY INETERMEDIATE

(±)-(R,S)-alpha-ethyl-2- oxo-l-pyrrolidineacet-N-(+)-(R)-(l-phenylethyl)-amide
methyl (±)-(R,S)-alpha-ethyl-2-oxo-l -pyrrolidine acetate with (+)-(R)-(l-phenylethyl)- amine in toluene in the presence of a base such as sodium hydride or methoxide; crystallization- induced dynamic resolution of the resultant (±)-(R,S)-alpha-ethyl-2- oxo-l-pyrrolidineacet-N-(+)-(R)-(l-phenylethyl)-amide

(R)-(+)-1-Phenylethylamine
33978-83-5
1-Pyrrolidineacetic acid, α-ethyl-2-oxo-, methyl ester

1004767-60-5
1-Pyrrolidineacetamide, α-ethyl-2-oxo-N-[(1R)-1-phenylethyl]-
(±)-(R.S)-alpha-ethyl-2-oxo-l-pyrrolidineacet-N-(+)-(R)-(l-phenylethyl)-amide
Example 1
(±)-(R,S)-alpha-ethyl-2-oxo-l-pyrrolidineacet-N-(+)-(R)-(l-phenylethyl)-amide.
In a 100 ml reactor equipped with mechanical stirring, thermometer and bubble condenser, 13.4 g of (±)-(R,S)-alpha-ethyl-2-oxo-l-pyrrolidineacetic acid methyl ester (71.6 mmol), 8.8 g of (+)-(R)-(l-phenylethyl)-amine (72.5 mmol) and 45 ml of tetrahydrofuran were charged. 3.4 g of NaH (60% dispersion in mineral oil, 85.6 mmol) was added in small portions under nitrogen atmosphere. Reaction mixture was maintained at room temperature for about 2 h. Then, it was heated up to 350C and kept under stirring overnight. Reaction was controlled by TLC (Rf = 0.5, AcOEt/silica gel).
At reaction completed, one night at 35°C temperature, reaction mixture was cooled to room temperature and 30 ml of water was slowly charged. It was transferred into a separatory funnel and was diluted with 30 ml of water and 80 ml of dichloromethane. Phases were separated and the aqueous one was washed with 50 ml of dichloromethane. Collected organic phases were washed with an aqueous acid solution, dried on Na2SO4, filtered and concentrated under vacuum. 19.5 g of an oil residue was obtained which slowly solidified. Solid was suspended in 20 ml of a hexane/dichloromethane 9/1 v/v mixture. It was then filtered, washed with 10 ml of the same solvent mixture and dried at 400C to give 12.1 g of the title compound (44.1 mmol, 61.6% yield) as dry solid.
1H NMR (400.13 MHz, CDCl3, 25 0C): δ (ppm, TMS)
7.35-7.19 (1OH, m),
6.49 (2H, br s),
5.09-5.00 (2H, m),
4.41 (IH, dd, J = 8.3, 7.4 Hz),
4.36 (IH, dd, J = 8.6, 7.1 Hz),
3.49 (IH, ddd, J = 9.8, 7.7, 6.6 Hz),
3.41 (IH, ddd, J = 9.8, 7.7, 6.2 Hz),
3.30 (IH, ddd, J = 9.6, 8.3, 5.5 Hz),
3.13 (IH, ddd, 9.7, 8.5, 6.1 Hz), 2.47-2.38 (2H, m), 2.41 (IH, ddd, J = 17.0, 9.6, 6.3 Hz), 2.26 (IH, ddd, 17.0, 9.5, 6.6 Hz), 2.10-1.98 (2H, m), 2.01-1.89 (IH, m), 1.99-1.88 (IH, m), 1.98-1.85 (IH, m), 1.88-1.78 (IH, m), 1.75- 1.62 (IH, m), 1.72-1.59 (IH, m), 1.45 (3H, d, J = 7.1 Hz), 1.44 (3H, d, J = 7.1 Hz), 0.90 (3H, t, J = 7.4 Hz), 0.86 (3H, t, J = 7.4 Hz).


13C NMR (100.62 MHz, CDCl3, 25 0C): δ (ppm, TMS)
176.05 (CO), 176.00 (CO), 169.08 (CO),
168.81 (CO), 143.59 (Cquat),
143.02 (Cquat), 128.66 (2 x CH), 128.55 (2 x CH),
127.33 (CH), 127.19 (CH), 126.05 (2 x CH),
125.80 (2 x CH), 56.98 (CH), 56.61 (CH),
48.90 (CH), 48.84 (CH), 44.08 (CH2),
43.71 (CH2), 31.19 (CH2), 31.07 (CH2), 22.08 (CH3),
22.04 (CH3), 21.21 (CH2), 20.68 (CH2),
18.28 (CH2), 18.08 (CH2), 10.50 (CH3), 10.45 (CH3).


Example 2 (±)-(R.S)-alpha-ethyl-2-oxo-l-pyrrolidineacet-N-(+)-(R)-(l-phenylethyl)-amide (alternative 1).
In a 500 ml reactor equipped with mechanical stirring, thermometer and condenser, 24.2 g of (+)-(R)-(l-phenylethyl)-amine (199.51 mmol) and 40 ml of toluene were charged. By keeping the reaction mixture at 00C temperature under nitrogen atmosphere, 9.5 g of NaH (60% mineral oil suspension, 237.50 mmol) was added in small portions. At the same temperature, 190.0 g of a toluene solution of (±)-(R,S)- alpha-ethyl-2-oxo-l-pyrrolidineacetic acid methyl ester (19.28% equal to 36.63 g, 197.77 mmol) was charged. Reaction mixture was then heated up to 35°C and maintained in that condition till complete disappearing of methyl ester reagent (about 14 h; checked by HPLC).
At reaction completed, reaction mixture was cooled and when room temperature was reached, 100 ml of water was slowly charged. Aqueous phases were separated and extracted with toluene (2 x 75 ml). Collected organic phases were treated with acid water till neuter pH. Solvent was evaporated and residue was suspended in about 100 ml of heptane for about 30 minutes. Product was isolated by filtration and dried in oven at 400C temperature under vacuum overnight to give 45.2 g of the title compound (164.54 mmol, 83.2% yield, d.e. 0.0%) as white dusty solid.
Example 3
(±)-(R,S)-alpha-ethyl-2-oxo-l-pyrrolidineacet-N-(+)-(R)-(l-phenylethyl)-amide (alternative 2).
In a 500 ml reactor equipped with mechanical stirring, thermometer and Dean-Stark distiller, 24.2 g of (+)-(R)-(l-phenylethyl)-amine (199.51 mmol) and 40 ml of toluene were charged. By keeping the reaction mixture at 00C temperature, 42.7 g of sodium methoxide (30% solution in methanol, 237.14 mmol) was added under nitrogen atmosphere. At the same temperature, 190.0 g of a toluene solution of (±)- (R,S)-alpha-ethyl-2-oxo-l-pyrrolidineacetic acid methyl ester (19.28% equal to 36.63 g, 197.77 mmol) was charged. Reaction mixture was then heated up to 65- 700C and maintained in that condition till complete disappearing of methyl ester reagent (about 4 h; checked by HPLC). After a work-up carried out according to the procedure described in example 2, 40.2 g of the title compound (146.53 mmol, 74.1% yield, d.e. 0.0%) as white dusty solid was obtained.
…….
SCRIP Awards 2013 -Best Company in an Emerging Market – Dr Reddy’s Laboratories – India, Novartis’s Bexsero, Best New Drug

The SCRIP Awards 2013 celebrated achievements in the global biopharma industry last night at the Lancaster, London.
Hosted by Justin Webb, the evening was a fantastic mix of dining, entertainment and awards.
Among the winners were:
- Novartis’s Bexsero, Best New Drug
- Genmab, Biotech Company of the Year
- Regeneron Pharmaceuticals and Sanofi’s Phase IIa study dupilumab in asthma, Clinical Advance of the Year
You can view the full roll of honour by clicking on the button below.
It was a great night and we would like to thank all those who entered and attended this year’s awards.
Finally congratulations to our winners and a huge thanks to our sponsors for helping us make it such a fantastic success.
Don’t forget to check our website in the next couple of days for all the pictures from the night.
2013 Winners
Best Company in an Emerging Market – Sponsored by Clinigen Group
- Dr Reddy’s Laboratories – India
Best Technological Development in Clinical Trials
- Quintiles’s Infosario Safety
Best Partnership Alliance
- AstraZeneca with Bristol-Myers Squibb and Amylin in diabetes
Financing Deal of the Year
- Mesoblast’s equity financing of Aus$170m
Best Advance in an Emerging Market
- Novartis’s Jian Kang Kuai Che Healthcare Project in China
Clinical Advance of the Year – Sponsored by Quintiles
- Regeneron Pharmaceuticals and Sanofi’s Phase IIa study dupilumab in in asthma
Licensing Deal of the Year – Sponsored by Hume Brophy
- AstraZeneca and Horizon Discovery for the development and commercialization of the HD-001 kinase target program for multiple cancer types
Executive of the Year
- Roch Doliveux, chairman and chief executive officer of UCB
Biotech Company of the Year
- Genmab
Best Contract Research Organization
- Quintiles
Management Team of the Year
- Regeneron Pharmaceuticals’ CEO Leonard S Schleifer and CSO George D Yancopoulos
Best New Drug – Sponsored by INC Research
- Novartis’ Bexsero (meningococcal group B vaccine)
Pharma Company of the Year – Sponsored by ICON
- Astellas
Lifetime Achievement Award
- Prof Dr Désiré Collen
…….read about bexero at

DR ANTHONY MELVIN CRASTO Ph.D
