
Home » FDA 2014 (Page 6)
Category Archives: FDA 2014
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
TAVABOROLE, AN 2690, 他伐硼罗 Таваборол تافابورول
TAVABOROLE
- AN 2690
- AN-2690
- AN2690
- UNII-K124A4EUQ3
5-Fluoro-1,3-dihydro-1-hydroxy-2,1-benzoxaborole
5-Fluoro-2,1-benzoxaborol-1(3H)-ol;
1,3-Dihydro-5-fluoro-1-hydroxy-2,1-benzoxaborole
MOLECULAR FORMULA C7H6BFO2
MOLECULAR WEIGHT 151.9
SPONSOR Anacor Pharmaceuticals, Inc.
CAS REGISTRY NUMBER 174671-46-6
Mp 118-120° C…..US20070265226
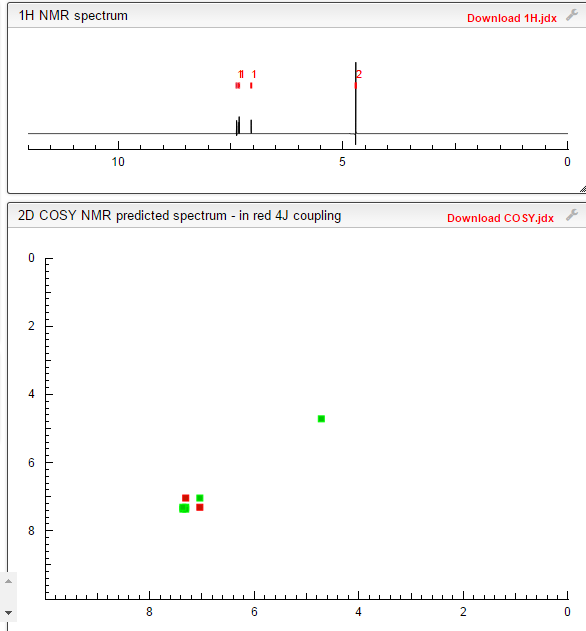
1H NMR (300 MHz, DMSO-d6) δ (ppm) 4.95 (s, 2H), 7.15 (m, 1H), 7.24 (dd, J=9.7, 1.8 Hz, 1H), 7.74 (dd, J=8.2, 6.2 Hz, 1H), 9.22 (s, 1H)
FDA APPROVED JULY 2 2014………..“FDA Approves Anacor Pharmaceuticals’ KERYDIN™ (Tavaborole) Topical Solution, 5% for the Treatment of Onychomycosis of the Toenails”. Market Watch. July 8, 2014.
Has antifungal activity.
The US Food and Drug Administration (FDA) 2014 JULY 8 ratified the Anacor’s Kerydin (5% Tavaborole solution) for the topical treatment of nail fungal infections. Tavaboroleindications of toenail fungus Trichophyton rubrum or Trichophyton rubrum infections.Instructions recommended once a day for toenail infections, treatment for 48 weeks, on the recommendation of Anacor, and do not need to nail debridement.
I tis an oxaborole antifungal used topically, as a 5% w/w solution, for the treatment of onychomycosis of the toenails due to Trichophyton rubrumor T. mentagrophytes. It is applied to the affected toenail once daily for 48 weeks.
Ingrowing toenails and application site reactions including exfoliation, erythema, and dermatitis have been reported during use.
1H NMR FROM NET

CLICK ON IMAGE FOR CLEAR VIEW
COSY NMR PREDICT
Tavaborole (AN2690, trade name Kerydin) is a topical antifungal medication for the treatment of onychomycosis, a fungal infectionof the nail and nail bed. Tavaborole began its Phase 3 trials in December 2010[1] and was approved in July 2014.[2] Tavaborole inhibits an essential fungal enzyme, Leucyl-tRNA synthetase, or LeuRS, required for protein synthesis. The inhibition of protein synthesis leads to termination of cell growth and cell death, eliminating the fungal infection. No treatment-related systemic side effects were observed in any of its clinical trials.
Tavaborole is the first oxygen boron used to treat toenail infections dioxolane (oxaborole) antifungal agents, located in Palo Alto, Anacor focuses on boron-based drug development and production, according to the latest news, Tavaborole future also be used to infect fingernails. Wedbush Securities analyst predicts that next year the drug sales in the United States for $ 16 million, by 2021 will reach peak sales of $ 347 million.
Gram-negative bacteria cause approximately 70% of the infections in intensive care units. A growing number of bacterial isolates responsible for these infections are resistant to currently available antibiotics and to many in development. Most agents under development are modifications of existing drug classes, which only partially overcome existing resistance mechanisms. Therefore, new classes of Gram-negative antibacterials with truly novel modes of action are needed to circumvent these existing resistance mechanisms. We have previously identified a new a way to inhibit an aminoacyl-tRNA synthetase, leucyl-tRNA synthetase (LeuRS), in fungi via the oxaborole tRNA trapping (OBORT) mechanism.
Herein, we show how we have modified the OBORT mechanism using a structure-guided approach to develop a new boron-based antibiotic class, the benzoxaboroles, which inhibit bacterial leucyl-tRNA synthetase and have activity against Gram-negative bacteria by largely evading the main efflux mechanisms in Escherichia coli and Pseudomonas aeruginosa. The lead analogue, is active against Gram-negative bacteria, including Enterobacteriaceaebearing NDM-1 and KPC carbapenemases, as well as P. aeruginosa. This novel boron-based antibacterial, has good mouse pharmacokinetics and was efficacious against E. coli and P. aeruginosa in murine thigh infection models, which suggest that this novel class of antibacterials has the potential to address this unmet medical need.
Anacor continued development on that drug, tavaborole, and filed for FDA approval in July. The FDA will review the phase 3 trial data and issue a decision on July 29, 2014.
If approved, Anacor hopes tavaborole’s ability to clear onychomycosis in 10% of treated patients will be enough to win market share away from generic Lamisil and generic topical Pentac. While Lamisil cleared the fungus in 38% of patients, it’s been associated with rare cases of liver failure. And Pentac requires frequent debridement of the nail and only clears the fungus in 5.5% to 8.5% of patients.
Tavaborole is a novel, topical antifungal medication being developed for the topical treatment of onychomycosis, a nail fungus infection, which affects seven to ten percent of the U.S. population. Early studies show AN-2690 penetrates the nail effectively and has robust activity against dermatophytes, which cause onychomycosis.

1H NMR PREDICT

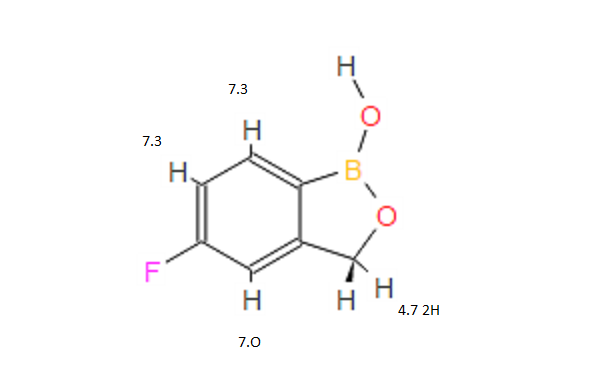
……………………………………………………………………………
13 C NMR PREDICT

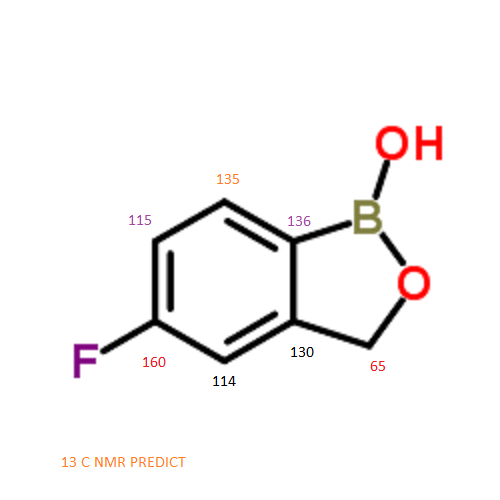
ARTICLE
Anacor Pharmaceuticals to Present Pivotal Phase 3 Data of Tavaborole for the Topical Treatment of Toenail Onychomycosis
Abstract Accepted for Oral Presentation at the 2013 American Podiatric Medical Association Annual Scientific Meeting
PALO ALTO, Calif.–(BUSINESS WIRE)– Anacor Pharmaceuticals (NASDAQ:ANAC) announced today that its abstract “Pivotal Phase 3 Safety and Efficacy Results of Tavaborole (Formerly AN2690), a Novel Boron-Based Molecule for the Topical Treatment of Toenail Onychomycosis” was accepted for oral presentation at the 2013 APMA Annual Scientific Meeting (The National) to be held in Las Vegas, Nevada. Max Weisfeld, DPM, will present the data from tavaborole’s Phase 3 studies on Monday, July 22, 2013 during the Evidence-Based Medicine and Oral Abstracts session.
As announced earlier this year, tavaborole achieved statistically significant and clinically meaningful results on all primary and secondary endpoints in two Phase 3 pivotal studies without concomitant debridement. Anacor is seeking approval for tavaborole from the Food and Drug Administration (FDA) and will file a New Drug Application imminently. Currently, there is only one FDA-approved topical treatment for onychomycosis, a fungal infection of the nail and nail bed, which affects approximately 35 million people in the United States.
“I’m impressed with tavaborole’s safety and efficacy data. There is no FDA-approved topical treatment for onychomycosis with tavaborole’s range of efficacy and ability to penetrate the nail to reach the site of the infection,” said Dr. Weisfeld. “Tavaborole’s Phase 3 results demonstrate its ability to clear the nail and eliminate the infection which is important to both patients and the physicians who treat them. In addition, tavaborole is easy to apply and dries quickly which makes it convenient for patients to use.”
“We are pleased to present these positive data at the APMA’s Annual Scientific Meeting, the leading annual meeting of podiatrists. As we seek FDAapproval for tavaborole, we look forward to developing relationships with podiatrists to potentially offer them a new treatment option for the large number of patients who seek treatment for onychomycosis,” said David Perry, Chief Executive Officer of Anacor Pharmaceuticals.
About the Studies
Anacor conducted two separate Phase 3 studies of tavaborole on patients with distal subungual onychomycosis affecting 20 to 60 percent of the target great toenail. Approximately 600 patients aged 18 years and older with no upper age limit (the oldest subject was 88 years old) were enrolled in each study and randomized two-to-one to receive either tavaborole or the vehicle control. Patients were instructed to apply tavaborole solution or the vehicle to the toenail once daily for 48 weeks.
A copy of the presentation will be available on Anacor’s website following the oral session.
About Anacor Pharmaceuticals
Anacor is a biopharmaceutical company focused on discovering, developing and commercializing novel small-molecule therapeutics derived from its boron chemistry platform. Anacor has discovered eight compounds that are currently in development. Its two lead product candidates are topically administered dermatologic compounds — tavaborole, a topical antifungal for the treatment of onychomycosis, and AN2728, a topical anti-inflammatory PDE-4 inhibitor for the treatment of atopic dermatitis and psoriasis. In addition to its two lead programs, Anacor has discovered three other wholly-owned clinical product candidates — AN2718 and AN2898, which are backup compounds to tavaborole and AN2728, respectively, and AN3365 an antibiotic for the treatment of infections caused by Gram-negative bacteria. We have discovered three other compounds that we have out-licensed for further development — two compounds for the treatment of animal health indications that are licensed to Eli Lilly and Company and AN5568, also referred to as SCYX-7158, for human African trypanosomiasis (HAT, or sleeping sickness), which is licensed to Drugs for Neglected Diseases initiative, or DNDi. We also have a pipeline of other internally discovered topical and systemic boron-based compounds in development. For more information, visit http://www.anacor.com.
Patents
WO 1995033754
WO 2004009578….
WO 2006089067
WO 2008025543
…………………………..
SYNTHESIS

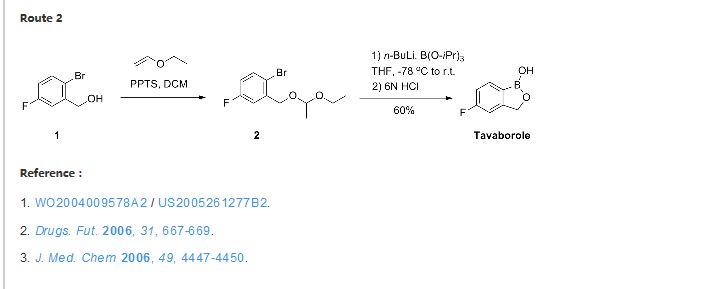
Reference:
ELI LILLY AND COMPANY Patent: WO2004/9578 A2, 2004 ; Location in patent: Page 36-37 ; WO 2004/009578 A2
PATENT
Anacor Pharmaceuticals Patent: US2007/265226 A1, 2007 ; Location in patent: Page/Page column 59 ;
http://www.google.com/patents/US20070265226

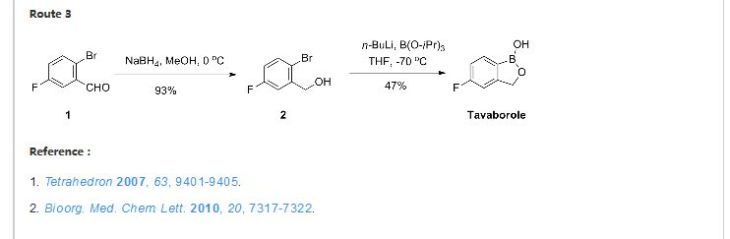
1,3-Dihydro-5-fluoro-1-hydroxy-2,1-benzoxaborole (19b)
To a solution of 5b (73.2 g, 293 mmol) in dry THF (400 mL) was added n-butyllithium (1.6 M in hexanes; 200 mL) over 45 min at −78° C. under nitrogen atmosphere. Anion precipitated. After 5 min, (i-PrO)3B (76.0 mL, 330 mmol) was added over 10 min, and the mixture was allowed to warm to room temperature over 1.5 h. Water and 6 N HCl (55 mL) were added, and the solvent was removed under reduced pressure to about a half volume. The mixture was poured into ethyl acetate and water. The organic layer was washed with brine and dried over anhydrous Na2SO4. The solvent was removed under reduced pressure. To a solution of the residue in tetrahydrofuran (360 mL) was added 6 N HCl (90 mL), and the mixture was stirred at 30° C. overnight. The solvent was removed under reduced pressure to about a half volume. The mixture was poured into ethyl acetate and water. The organic layer was washed with brine and dried over anhydrous Na2SO4. The solvent was removed under reduced pressure, and the residue was treated with i-Pr2O/hexane to give 19b (26.9 g, 60%) as a white powder:
mp 118-120° C.;
1H NMR (300 MHz, DMSO-d6) δ (ppm) 4.95 (s, 2H), 7.15 (m, 1H), 7.24 (dd, J=9.7, 1.8 Hz, 1H), 7.74 (dd, J=8.2, 6.2 Hz, 1H), 9.22 (s, 1H);
ESI-MS m/z 151 (M−H)−;
HPLC purity 97.8%; Anal (C7H6BFO2) C, H.
…………………
Gunasekera, Dinara S.; Gerold, Dennis J.; Aalderks, Nathan S.; Chandra, J. Subash; Maanu, Christiana A.; Kiprof, Paul; Zhdankin, Viktor V.; Reddy, M. Venkat Ram Tetrahedron, 2007 , vol. 63, # 38 p. 9401 – 9405
…………………….
Baker, Stephen J.; Zhang, Yong-Kang; Akama, Tsutomu; Lau, Agnes; Zhou, Huchen; Hernandez, Vincent; Mao, Weimin; Alley; Sanders, Virginia; Plattner, Jacob J. Journal of Medicinal Chemistry, 2006 , vol. 49, # 15 p. 4447 – 4450
………………..
Ding, Charles Z.; Zhang, Yong-Kang; Li, Xianfeng; Liu, Yang; Zhang, Suoming; Zhou, Yasheen; Plattner, Jacob J.; Baker, Stephen J.; Liu, Liang; Duan, Maosheng; Jarvest, Richard L.; Ji, Jingjing; Kazmierski, Wieslaw M.; Tallant, Matthew D.; Wright, Lois L.; Smith, Gary K.; Crosby, Renae M.; Wang, Amy A.; Ni, Zhi-Jie; Zou, Wuxin; Wright, Jon Bioorganic and Medicinal Chemistry Letters, 2010 , vol. 20, # 24 p. 7317 – 7322
…………..
PATENT
PREPARATION 13 5-Fluoro-3H-benzo[c][1,2)oxaborol-1-ol

Dissolve 1-bromo-2-(1-ethoxy-ethoxymethyl)-4-fluoro-benzene(5.4 g, 19.5 mmol) in dry THF (100 mL) and cool to −78° C. under nitrogen. Add butyl lithium (2.5M in Hexanes, 10.2 mL, 25.4 mmol) dropwise at −78° C. Upon complete addition, stir the reaction at −78° C. for 10 minutes and then add trimethyl borate (4.4 mL, 39 mmol) and warm the reaction to room temperature. Pour the reaction into 1N HCl (100 mL) and stir for 1 hour. Extract the biphasic mixture with ether three times. Dry the combined organic layers with sodium sulfate, filter and concentrate in vacuo. Triturate the oily residue with cold hexanes to yield 2.1 g (70%) of the title compoud as a white solid.
1H NMR (d6-DMSO)
9.18 (s, 1H),
7.70 (dd, J=8.2, 5.8 Hz, 1H),
7.20 (dd, J=9.5, 2.7 Hz, 1H),
7.11 (m, 1H), 4.92 (s, 1H).
…………………
SEE
http://jpet.aspetjournals.org/content/early/2012/11/28/jpet.112.200030.full.pdf
………………………………..
SEE
Baker SJ, Zhang YK, Akama T, Lau A, Zhou H, Hernandez V, Mao W, Alley MR, Sanders V, Plattner JJ.
J Med Chem. 2006 Jul 27;49(15):4447-50.
Boron-containing inhibitors of synthetases.
Baker SJ, Tomsho JW, Benkovic SJ.
Chem Soc Rev. 2011 Aug;40(8):4279-85. doi: 10.1039/c0cs00131g. Epub 2011 Feb 7. Review.
- Benzoxaborole antimalarial agents. Part 2: Discovery of fluoro-substituted 7-(2-carboxyethyl)-1,3-dihydro-1-hydroxy-2,1-benzoxaboroles.
Zhang YK, Plattner JJ, Freund YR, Easom EE, Zhou Y, Ye L, Zhou H, Waterson D, Gamo FJ, Sanz LM, Ge M, Li Z, Li L, Wang H, Cui H.
Bioorg Med Chem Lett. 2012 Feb 1;22(3):1299-307. doi: 10.1016/j.bmcl.2011.12.096. Epub 2011 Dec 28.
Tavaborole Market Opportunity
Anacor is developing tavaborole specifically to address the current limitations of existing treatment options for onychomycosis. This includes designed leaps forward in both the potential safety and efficacy profile aimed to make the drug a best-in-class therapy. Additionally, management has used the company’s expertise in medicinal chemistry to improve delivery of the compound through the nail plate to the nail bed, the site of onychomycosis infection. For example, preclinical studies indicate that tavaborole is able to penetrate the nail plate 250 times more effectively than ciclopirox.
Tavaborole novel mechanism of action inhibits an essential fungal enzyme, leucyl transfer RNA synthetase, or LeuRS required for protein synthesis. The inhibition of protein synthesis leads to termination of cell growth and cell death, eliminating the fungal infection.
Likewise, the topical dosing was designed to eliminate systemic absorption. Previous preclinical and clinical data shows topical treatment with tavaborole resulted in little or no detectable levels of drug in the blood or urine. No treatment related systemic side effects have been observed in any clinical trials to date. Safety data from the company’s studies to date was recently presented at the 100th National APMA meeting in Washington, DC.
Anacor’s topical solution currently in two phase III trials for onychomycosis. Phase II data with tavaborole suggests efficacy superior to ciclopirox with little to no systemic exposure.
Data from an open-label phase 2 program with tavaborole showed 50% patients using a 7.5% solution saw 2 mm clear nail growth and negative fungal cultures after six months. Roughly 25% of the patients saw 5 mm clear nail growth and negative fungal cultures after six months.
Anacor and partner Merck (NYSE:MRK) met with the U.S. FDA in 2009 to discuss the phase II data. Merck has since returned the rights to tavaborole to Anacor. The original deal was with Schering-Plough in 2007. Merck most likely felt as though tavaborole clashed with existing products or did not have peak sales potential large enough to continue the partnership with Anacor. We see tavaborole as a specialty promoted product, into podiatrists and dermatologists. For a company like Anacor, it’s an attractive first product.
Anacor’s first phase III trial completed enrollment in November 2011. The second phase III trial completed enrollment in December 2011. Data from these trials are expected around the middle of January 2013. Data from the second study is expected six weeks later. Given the positive phase II data noted above, we think odds favor a positive outcome. A benchmark for the trial is the efficacy of Lamisil, which is a complete cure rate of around 35% to 40%, and a mycological cure of around 70% after a typical course of treatment.
I note that on Anacor’s third quarter conference call management noted that they are pleased with the conduct of the trial to date. Specifically, the compliance rate appears to better than management had expected. The trial was designed with a 20% drop-out rate. It looks as though the drop-out rate is only around 13%, at a minimum suggestive of good safety and tolerability, but potentially also a sign that the drug is working.
I see onychomycosis as a significant market opportunity for Anacor. An estimated 35 million Americans have nail fungus, with about 95% of the infections in the toenail. With efficacy similar to Lamisil, we think Anacor can capture 20% of the market. With a price per course of treatment at around $1,200, I think peak sales of tavaborole are $500 million.
Conclusion
I’ll note two more important pieces of information for investors. Firstly, besides optimism for tavaborole, Anacor has apipeline of anti-infectant drugs. For this article I discussed only tavaborole. A second article can be dedicated entirely to AN2728 for the treatment of psoriasis and atopic dermatitis. Anacor also has an animal health collaboration with Eli Lilly (NYSE:LLY).
The second important thing to note is Anacor’s cash position. The company reported financial results on November 7, 2012. The company held $36.6 million in cash on the balance sheet as of September 30, 2012. However, in October 2012, the company completed an underwritten public offering of 4.0 million shares of common stock at $6.00 per share to raise net proceeds of $22.7 million. I view the current cash position as sufficient to report data from both phase 3 trials and, if positive, file the new drug application (NDA) around the middle of 2013.
With phase 3 data expected in less than two months, good prior evidence of both safety and efficacy, and a solid cash position, I think Anacor could be an attractive investment at today’s price. The stock is down meaningfully over the past month and investors can buy sizably below the October offering.


SYNTHESIS

References
- Clinical trial number NCT01270971 at ClinicalTrials.gov
- “FDA Approves Anacor Pharmaceuticals’ KERYDIN™ (Tavaborole) Topical Solution, 5% for the Treatment of Onychomycosis of the Toenails”. Market Watch. July 8, 2014.
- http://www.accessdata.fda.gov/drugsatfda_docs/label/2014/204427s000lbl.pdf
- http://www.molbase.com/en/hnmr_174671-46-6-moldata-1568017.html#tabs
 |
|
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
5-Fluoro-2,1-benzoxaborol-1(3H)-ol
|
|
| Clinical data | |
| Trade names | Kerydin |
| Legal status |
|
| Routes of administration |
Topical use only |
| Identifiers | |
| CAS Registry Number | 174671-46-6 |
| ATC code | None |
| PubChem | CID: 11499245 |
| ChemSpider | 9674047 |
| Synonyms | AN2690 |
| Chemical data | |
| Formula | C7H6BFO2 |
| Molecular mass | 151.93 g/mol |
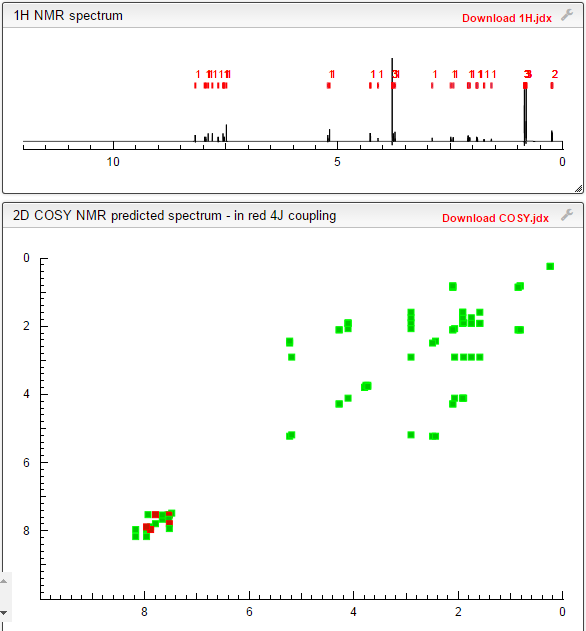
NMR PREDICT
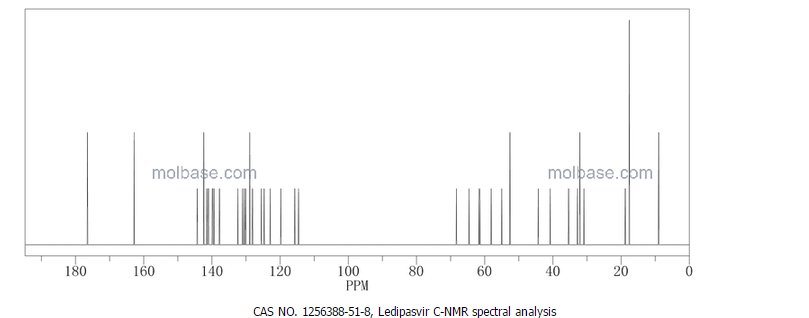
 CAS NO. 174671-46-6, 5-fluoro-1-hydroxy-3H-2,1-benzoxaborole H-NMR spectral analysis |

 CAS NO. 174671-46-6, 5-fluoro-1-hydroxy-3H-2,1-benzoxaborole C-NMR spectral analysis |

more

 Anacor
Anacor
Anacor Pharmaceuticals is out to change that. The Palo Alto, Calif.-based biotechnology company is developing a family of boron-containing small-molecule drugs. And with the assistance of Naeja Pharmaceutical, a Canadian contract research organization, Anacor has licensed one of those molecules to GlaxoSmithKline and taken another one into Phase III clinical trials.
Anacor was founded in 2002 to develop technology created by Lucy Shapiro, a Stanford University bacterial geneticist, and Stephen J. Benkovic, a Pennsylvania State University organic chemist. Through a long-standing scientific collaboration, the two researchers had discovered boron-containing compounds that inhibited specific bacterial targets………..https://pubs.acs.org/cen/coverstory/89/8912cover3.html
mp 118-120….http://www.syninnova.com/catalog/product/SL-264
antifugal AN2690 by Anacor
Tavaborole inhibits an essential fungal enzyme, Leucyl-tRNA synthetase, or LeuRS, required for protein synthesis.
Minimum Inhibitory Concentration: 1, 1, 0.5, 0.25, and 0.25 μg/mL for T.rubrum, T.mentagrophytes, C.albicans, C.neoformans, A.fumigatus, respectivley.
AN2690 is a new boron-containing antifungal agent for the potential treatment of onychomycosis. Onychomycosis is caused mainly by dermatophytes, a class of fungus that dwells on skin, hair, and nails and is the cause of other cutaneous fungal infections such as athlete’s foot.
In vitro: AN2690 showed the most active against fungi and especially against the dermatophytes T. rubrum and T. mentagrophytes, the primary fungal pathogens causing onychomycosis. In addition, AN2690 was identified as having a unique profile of in vitro antidermatophyte activity, maintenance of this activity in the presence of keratin, and exceedingly good penetration of human nails [1].
Ex vivo: AN2690 was found to have superior penetration compared to ciclopirox, and achieves levels within and under the nail plate that suggest it has the potential to be an effective topical treatment for onychomycosis [2].
Clinical trial: The efficacy of tavaborole as a topical treatment for onychomycosis has been evaluated in two identical randomised, double-blind phase III studies, NCT01270971 (301) and NCT01302119 (302), enrolling 593 and 601 patients, respectively. Completely or almost clear nail and negative mycology was achieved in 15.3 and 17.9 % of tavaborole recipients compared with 1.5 and 3.9 % of vehicle recipients [3]
References:
[1] Baker SJ, Zhang YK, Akama T, Lau A, Zhou H, Hernandez V, Mao W, Alley MR, Sanders V, Plattner JJ. Discovery of a new boron-containing antifungal agent, 5-fluoro-1,3-dihydro-1-hydroxy-2,1- benzoxaborole (AN2690), for the potential treatment of onychomycosis. J Med Chem. 2006;49(15):4447-50.
[2] Hui X, Baker SJ, Wester RC, Barbadillo S, Cashmore AK, Sanders V, Hold KM, Akama T, Zhang YK, Plattner JJ, Maibach HI. In Vitro penetration of a novel oxaborole antifungal (AN2690) into the human nail plate. J Pharm Sci. 2007;96(10):2622-31.
[3] Markham A. Tavaborole: first global approval. Drugs. 2014;74(13):1555-8.
UPDATE
http://www.google.im/patents/EP1976536A2?cl=en
EXAMPLE 23
Alternative Preparation of 4 from 3
A 22.0 L 3-neck flask was equipped with a stir motor, N2 inlet, addition funnel, heating mantle, and condenser. The flask was charged with 3500 g (17.1 moi) of 2-bromo-5-fluorobenzyl alcohol followed by the addition of 3556 g of tetrahydrofuran and 16.4 g (0.17 mol) of methanesulfonic acid. Next, 400 g (4.7 mol) of 3,4-dihydro-2H-pyran was added at 100C. This step is exothermic so no additional charges should be made until exotherm subsides. The temperature was increased to 27°C, stirred for 15 min and then charged with 400 g (4.7 mol) of 3,4-dihydro-2H- pyran at 240C. Again the temperature increased (24°C to 380C). The mixture was stirred for 15 min. Once the exotherm subsided, the flask was again charged with 40Og (4.7 mol) of 3,4-dihydro-2H-pyran at 350C. The temperature again increased to 470C over a 20 min period. Once the exotherm subsided, the mixture was stirred for 15 min. Finally the remaining 400 g (4.7 mol) of 3,4-dihydro-2H-pyran was added at 440C. The temperature increased to 510C. After stirring for one hour, a sample was removed to check for removal of starting material. Upon reaction completion, contents were cooled to 20 ± 5 0C.
EXAMPLE 24

Alternative Preparation of 5 from 4
To a 22.0 L 3-neck flask equipped with a stir motor, N2 inlet, addition funnel, cooling bath, and condenser was charged 436 g (17.96 mol) of magnesium turnings. 5334 g of tetrahydrofuran was then added followed by 291 g (0.51 mol) of diisobutylaluminum hydride (DIBAL) (25%wt) in toluene. The mixture was stirred for 60 min at 20 ± 5 0C. Some gas evolution was seen. Next, 260-430 g -3-5% (by weight if solution of 4 was dropped to drums) of 4 in THF was added. The mixture was stirred for 15-30 min at which time a slight exotherm should be seen (ΔT = 10- 150C). Once the exotherm was observed, the reaction mixture was cooled to 5 ± 5 0C. To this mixture, the remaining 8.22-8.39 kg of 4 in THF was added at a rate such that the temperature was kept below 300C (t = 3h). The reaction was stirred at 20-25 0C for 30 min, at which time an aliquot was removed, quench with 3 N HCl (10 mL), and analyzed.
Upon completion, the contents were cooled to -25 ± 5°C. A solution of trimethylborate in THF was prepared by mixing 2665 g (25.7 mol) of trimethyl borate and 6666 g of tetrahydrofuran. This solution can be prepared in a drum with stirring. [0618] Next, the 9331 g of trimethyl borate in THF was added at a rate such that the temperature was kept between -35 and -20 °C (t = 2.5h). The mixture became very thick so THF was added. After stirring at -25 ± 5°C for 10 min, 50 mL aliquot was removed, quenched with 25 mL of 3N HCl, and submitted for CoR. Stirring continued at -25 ± 50C for Ih, and then the mixture was allowed to warm to ambient temperature, where it was stirred for at least 12h. Pull two samples (one at 6h and the other at 12h).
Results:
1H-NMR (300 MHz, DMSO-d6) δ (ppm) 1.45-1.75 (m, 6H), 3.53 (s, 6H), 3.45 (m, IH), 3.75 (m, IH), 4.69 (t, J=3 Hz, IH), 4.97 (d, J=14.1 Hz, IH), 5.14 (d, J=14.1 Hz, IH), 7.03 ((td, J=8.4, 2.7 Hz, IH), 7.24 (dd, J=10.8, 2.1 Hz, IH), 7.89 (t, J=7.8 Hz, IH), 8.76 (s, IH).
EXAMPLE 25
Alternative Preparation of I from 5

To the reaction mixture above was added 5.3 kg of USP water. After stirring for 30 min, the mixture was charged 5.3 kg of acetic acid. Gas evolution was seen. After stirring for 30 min, an aliquot was removed for analysis. Mixture was then heated to reflux for 36-48 hours. During the reflux period, 12-13 L of THF were removed.
When the reaction was complete, the contents were cooled by the reactor to <40°C by setting jacket and by charging 10.5 kg of USP water. THF was removed until distillate did not remain. Contents of the reactor were transferred to Rosenmund filter dryer and allowed to cool to 20 ± 5°C. Reactor was rinsed with water, filtered, and then washed again with 10.5 kg of USP water. The flask was charged with 10.5 kg of 10% ACN in water (v/v) and agitated for Ih. After filtering, the cake was washed with 10.5 kg of 10% ACN in water (v/v), and then charged with 10.5 kg 10% ACN in water (v/v). The contents were agitated for Ih. The contents were subsequently washed with 10.5 kg of USP water, charged with 7.0 L of 5% Methyl t- Butyl Ether (MTBE)/Heptane (v/v), agitated for Ih, filtered, charged with 7.0 L of 5% MTBE/Heptanes (v/v) and again agitated for Ih. After filtering, the contents were charged again with 7.0 L of heptane and filtered. Solids were dried at <45°C to constant weight. Solids were recrystallized from toluene :heptane 75:25.
see full series on boroles
http://apisynthesisint.blogspot.in/p/borole-compds.html
http://apisynthesisint.blogspot.in/p/borole-compds.html
http://apisynthesisint.blogspot.in/p/borole-compds.html
do not miss out
see full series on boroles
http://apisynthesisint.blogspot.in/p/borole-compds.html
http://apisynthesisint.blogspot.in/p/borole-compds.html
http://apisynthesisint.blogspot.in/p/borole-compds.html
do not miss out
///////
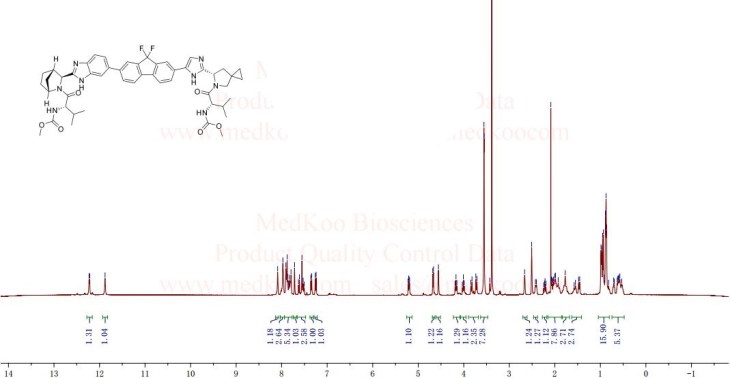
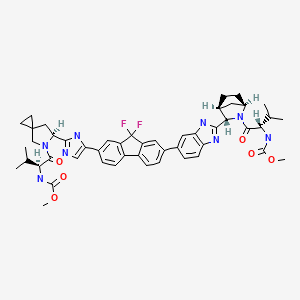
LEDIPASVIR , 来迪派韦 , Ледипасвир , ليديباسفير


Carbamic acid, N-((1S)-1-(((6S)-6-(5-(9,9-difluoro-7-(2-((1R,3S,4S)-2-((2S)-2-((methoxycarbonyl)amino)-3-methyl-1-oxobutyl)-2-azabicyclo(2.2.1)hept-3-yl)-1H-benzimidazol-6-yl)-9H-fluoren-2-yl)-1H-imidazol-2-yl)-5-azaspiro(2.4)hept-5-yl)carbonyl)-2-me
Chemical Formula:C52H60F2N8O7
Molecular Weight:947.08


The structure of ledipasvir was unambiguously confirmed by 1 H, 13C and 19F NMR spectroscopy, UV spectroscopy, IR spectroscopy, high resolution mass spectrometry, elemental analysis and X-ray crystallography. LDV-AS is a white to tinted (off-white, tan, yellow, orange, or pink), slightly hygroscopic crystalline solid. It shows pH dependent solubility in aqueous media: it is slightly soluble in pH 2.3 buffer but practically insoluble in pH 4-7.5 buffers. It is freely soluble in ethanol and DMSO and slightly soluble in acetone. Ledipasvir is chiral and possesses 6 stereogenic centres and enantiomeric purity is controlled in starting material specifications. Three crystalline forms are known and ledipasvir acetone solvate is the designated commercial form. The first step for finished product manufacture involves the dissolution of ledipasvir in ethanol followed by spray-drying and thus precise control of morphology and particle size is not considered important. Ledipasvir is a chemical substance not previously authorised as a medicinal product in the European Union. Furthermore, it is not a salt, complex, derivative or isomer, (nor mixture of isomers), of a previously authorised substance. Whilst it contains some structural features in common with daclastavir, it is metabolically stable and the applicant presented data indicating that there are no common active metabolites. Therefore, the therapeutic moieties are not the same. Ledipasvir thus meets the definition of a New Active Substance according to the Notice to Applicants (NtA), Vol 2A, Chapter 1, Annex 3.
The mode of action of ledipasvir has not been directly established but indirect evidence is consistent with the compound targeting the NS5A molecule. In vitro resistance selection and cross-resistance studies, and the lack of HCV enzyme or kinase inhibition was taken to support the conclusion that ledipasvir targets NS5A as its mode of action. Ledipasvir has shown antiviral activity against HCV genotypes 1a and 1b with mean EC50 values of 0.031 and 0.004 nM, respectively. Antiviral activity determined as EC50 against genotypes 2 to 6 ranged from 0.15 to 530 nM. Ledipasvir showed no relevant antiviral activity at the highest concentration tested, or the highest concentration without cytotoxicity, against other virus such as bovine viral diarrhea virus (BVDV), RSV, HBV, HIV-1, HRV, influenza A and B, and a panel of flaviviruses (including West Nile virus, yellow fever virus, dengue virus, and banzai virus). Cytotoxicity of ledipasvir was characterised by CC50 of 4029 to >50000 nM using different cell lines (1b-Rluc-2, Huh-luc, 1a-HRlucp, Hep G2, SL3, Huh7, Hep-2, AD-38 and MT4 cells). Ledipasvir at 10 µM showed significant binding to 3 ion channels and 1 receptor in a radioligand binding assay screen against a panel of 68 mammalian ion channels and receptors. The IC50s of ledipasvir were 0.210 and 3.47 μM against sodium channel site 2 and calcium channel L-type (dihydropyridine), respectively. A 50% inhibition of androgen receptor was noted at 10 μM. Ledipasvir activity against 442 kinases was assessed using a quantitative polymerase chain reaction (qPCR)-based competition assay. Results showed weak competition for binding of 2 kinases, Bruton’s tyrosine kinase (BTK) and homeodomain-interacting protein kinase 1 (HIPK1) at 0.1 and 1 μM, respectively. Taking into account the high protein binding, >99.5%, of ledipasvir the large margin between unbound maximum clinical plasma levels (0.8 nM) and potential ion channel/receptor inhibition indicates limited clinical relevance.
Ledipasvir (formerly GS-5885) is a drug for the treatment of hepatitis C that was developed by Gilead Sciences.[1] After completingPhase III clinical trials, on February 10, 2014 Gilead filed for U.S. approval of a ledipasvir/sofosbuvirfixed-dose combination tablet for genotype 1 hepatitis C.[2][3] The ledipasvir/sofosbuvir combination is a direct-acting antiviral agent that interferes with HCV replication and can be used to treat patients with genotypes 1a or 1b without PEG-interferon or ribavirin.
Ledipasvir is an inhibitor of the hepatitis C virusNS5A protein.
Data presented at the 20th Conference on Retroviruses and Opportunistic Infections in March 2013 showed that a triple regimen of the nucleotide analog inhibitor sofosbuvir, ledipasvir, and ribavirin produced a 12-week post-treatment sustained virological response (SVR12) rate of 100% for both treatment-naive patients and prior non-responders with HCV genotype 1.[4][5] The sofosbuvir/ledipasvir coformulation is being tested with and without ribavirin. In February 2014 Gilead has filed for United StatesFood and Drug Administration (FDA) approval of ledipasvir/sofosbuvir oral treatment, without interferon and ribavirin.[6]
On October 10, 2014 the FDA approved the combination product ledipasvir 90 mg/sofosbuvir 400 mg called Harvoni.[7]


https://www.google.co.in/patents/WO2013184698A1
CLIP
SYN


https://www.google.co.in/patents/WO2013184702A1









PATENT
https://www.google.co.in/patents/US8088368
Example ED Preparation of Intermediate 5-Aza-spiro[2.4]heptane-5,6-dicarboxylic acid 5-benzyl ester 6-methyl ester

4-Methylene-pyrrolidine-1,2-dicarboxylic acid 1-benzyl ester 2-methyl ester
5-Aza-spiro[2.4]heptane-5,6-dicarboxylic acid 5-benzyl ester
Example ED′


2,7-Dibromo-9,9-difluoro-9H-fluorene
5-Aza-spiro[2.4]heptane-5,6-dicarboxylic acid 5-benzyl ester 6-[2-(7-bromo-9,9-difluoro-9H-fluoren-2-yl)-2-oxo-ethyl]ester
6-[5-(7-Bromo-9,9-difluoro-9H-fluoren-2-yl)-1H-imidazol-2-yl]-5-aza-spiro[2.4]heptane-5-carboxylic acid benzyl ester
(1-{6-[5-(7-Bromo-9,9-difluoro-9H-fluoren-2-yl)-1H-imidazol-2-yl]-5-aza-spiro[2.4]heptane-5-carbonyl}-2-methyl-propyl)-carbamic acid methyl ester
3-[6-(9,9-Difluoro-7-{2-[5-(2-methoxycarbonylamino-3-methyl-butyryl)-5-aza-spiro[2.4]hept-6-yl]-3H-imidazol-4-yl}-9H-fluoren-2-yl)-1H-benzoimidazol-2-yl]-2-aza-bicyclo[2.2.1]heptane-2-carboxylic acid tert-butyl ester
(1-{3-[6-(9,9-Difluoro-7-{2-[5-(2-methoxycarbonylamino-3-methyl-butyryl)-5-aza-spiro[2.4]hept-6-yl]-3H-imidazol-4-yl}-9H-fluoren-2-yl)-1H-benzoimidazol-2-yl]-2-aza-bicyclo[2.2.1]heptane-2-carbonyl}-2-methyl-propyl)-carbamic acid methyl ester
https://www.google.co.in/patents/US8088368

2-(5-{9,9-Difluoro-7-[2-(2-Boc-2-aza-bicyclo[2.2.1]hept-3-yl)-3H-benzoimidazol-5-yl]-9H-fluoren-2-yl}-1H-imidazol-2-yl)-pyrrolidine-1-carboxylic acid tert-butyl ester: A mixture of 2-[5-(7-Bromo-9,9-difluoro-9H-fluoren-2-yl)-1H-imidazol-2-yl]-pyrrolidine-1-carboxylic acid tert-butyl ester (324 mg, 0.627 mmol), 3-[6-(4,4,5,5-Tetramethyl-[1,3,2]dioxaborolan-2-yl)-1H-benzoimidazol-2-yl]-2-aza-bicyclo[2.2.1]heptane-2-carboxylic acid tert-butyl ester (1.1 eq., 304 mg), [1,1′ bis(diphenylphosphino)ferrocene]dichloropalladium(II)(3%, 15 mg), tetrakis(triphenylphosphine)palladium (3%, 22 mg) and potassium carbonate (3.3 eq., 285 mg) in 10 mL DME and 3 mL water was heated to 90° C. under Argon for 3 hours. The reaction mixture was cooled and diluted with ethyl acetate and washed with saturated sodium bicarbonate solution. The organic layer was dried (MgSO4), concentrated and purified by flash column chromatography (silica gel, 20 to 100% ethyl acetate/hexane) to give 2-(5-{9,9-Difluoro-7-[2-(2-Boc-2-aza-bicyclo[2.2.1]hept-3-yl)-3H-benzoimidazol-5-yl]-9H-fluoren-2-yl}-1H-imidazol-2-yl)-pyrrolidine-1-carboxylic acid tert-butyl ester (361 mg, yield 77%). LCMS-ESI−: calc’d for C43H46F2N6O4: 748.86. Found: 749.2 (M+H+).
PATENTS
SEE
WO 2010132601
WO 2013040492
WO 2013059630
WO 2013059638
CLIP
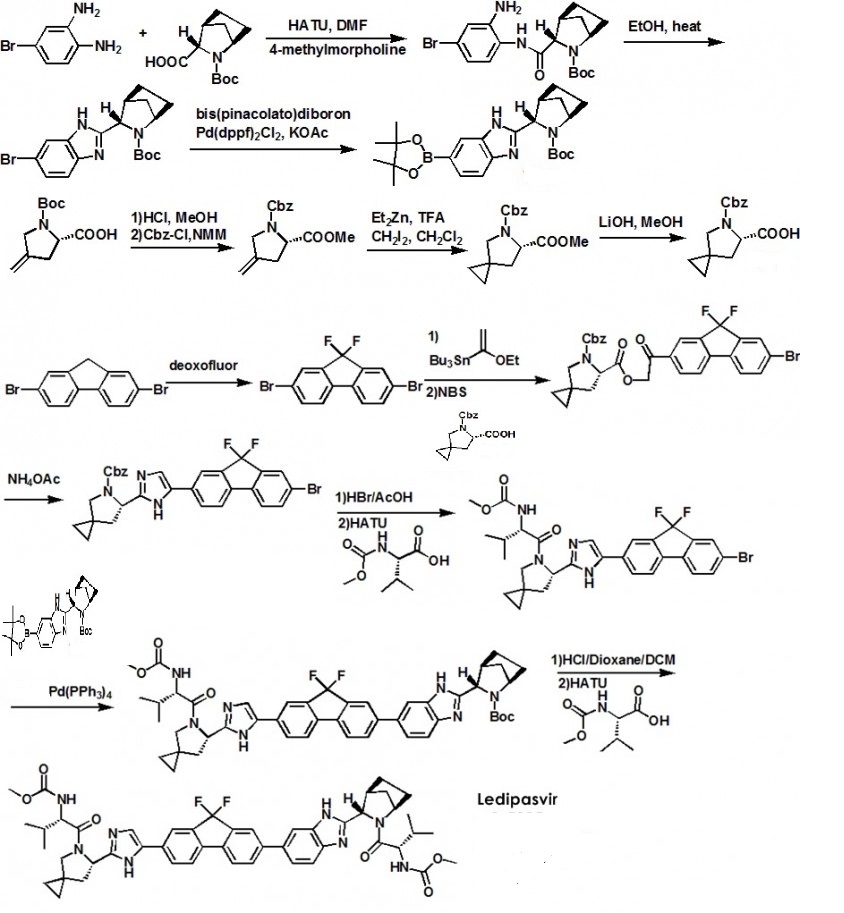
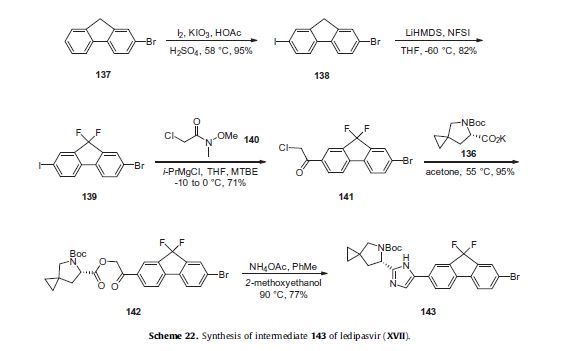
Ledipasvir (Harvoni) Ledipasvir is a potent NS5A inhibitor that is approved for use in combination with sofosbuvir, a nucleotide inhibitor of viral polymerase, for the treatment of chronic hepatitis C virus genotype 1 infection.14,130,131 This combination was discovered and developed at Gilead Sciences and is marketed as the fixed combination with brand name of Harvoni. The synthesis of ledipasvir has been reported in the literature132 and the routes shown in Schemes 22–24 below represent the most efficient and largest scale sequence reported in the patent literature.133,134 The synthesis of the spirocyclopropane proline intermediate 136 is described in Scheme 21. Bis-iodination of cyclopropane-1,1-diyldimethanol (131) in the presence of triphenylphosphine gave diiodide 132 in 70% yield. N-Boc-glycine ethyl ester (133) was then treated with sodium hydride followed by diiodide 132 to give the protected proline analog 134 in 61% yield. Saponification of the ester followed by a classical resolution with (1S,2R)-amino-indanol gave enantomerically pure salt 135. Liberation of the free acid with 1 M HCl followed by treatment with potassium tert-butoxide provided enantiopure potassium salt 136 in high yield. The synthesis of the difluoro-fluorene Suzuki coupling intermediate 143 is described in Scheme 22. Iodination of 2-bromofluorene (137) produced aryl iodide 138 in 95% yield, which was then treated with lithium hexamethyldisilazide and N-fluorobenzenesulfonimide (NFSI) to give the difluoro intermediate 139 in 82% yield. Formation of the Grignard reagent of 139 through reaction with isopropylmagnesium chloride followed by condensation with Weinreb amide 140 gave chloroketone 141 in 71% yield. The potassium salt of the cyclopropyl proline intermediate 136 (described in Scheme 21) was coupled with 141 to give keto ester 142 in high yield. Heating 142 with ammonium acetate resulted in formation of the imidazole ring in intermediate 143 in 77% yield. The completion of the synthesis of ledipasvir is described in Scheme 23. Commercially available (1R,3S,4S)-N-Boc-2-azabicyclo [2.2.1]heptane-3-carboxylic acid (144) was coupled to 4-bromo- 1,2-benzenediamine (145) using EDC/HOBt to give a mixture ofamides 146a/146b in 72% yield. Heating mixture 146a/146b with acetic acid affected cyclization to benzimidazole 147 in 94% yield. Palladium mediated coupling of bromide 147 to bis(pinacolato)diboron gave intermediate148 which was then coupled in the same reaction vessel to bromide 143 generated in Scheme 22. This was followed by formation of the oxalate salt to give the protected central core of ledipasvir (149) in good overall yield. Removal of the amine protecting groups gave diamine 150 which was coupled to two equivalents of Moc-valine (151) via EDC/HOBt to give ledipasvir XVII in 73% yield. 19. Lobeglitazone sulfate



130. Gentile, I.; Buonomo, A. R.; Borgia, F.; Castaldo, G.; Borgia, G. Expert Opin.Invest. Drugs 2014, 23, 561.
131. Smith, M. A.; Chan, J.; Mohammad, R. A. Ann. Pharmacother. 2015, 49, 343.132. Link, J. O.; Taylor, J. G.; Xu, L.; Mitchell, M.; Guo, H.; Liu, H.; Kato, D.;Kirschberg, T.; Sun, J.; Squires, N.; Parrish, J.; Keller, T.; Yang, Z. Y.; Yang, C.;Matles, M.; Wang, Y.; Wang, K.; Cheng, G.; Tian, Y.; Mogalian, E.; Mondou, E.;Cornpropst, M.; Perry, J.; Desai, M. C. J. Med. Chem. 2014, 57, 2033.
133. Guo, H.; Kato, D.; Kirschberg, T. A.; Liu, H.; Link, J. O.; Mitchell, M. L.; Parrish, J.P.; Squires, N.; Sun, J.; Taylor, J.; Bacon, E. M.; Canales, E.; Cho, A.; Cottell, J. J.;Desai, M. C.; Halcomb, R. L.; Krygowski, E. S.; Lazerwith, S. E.; Liu, Q.;Mackman, R.; Pyun, H. J.; Saugier, J. H.; Trenkle, J. D.; Tse, W. C.; Vivian, R. W.;Schroeder, S. D.; Watkins, W. J.; Xu, L.; Yang, Z. Y.; Kellar, T.; Sheng, X.; Clarke,M. O. N. H.; Chou, C. H.; Graupe, M.; Jin, H.; McFadden, R.; Mish, M. R.;Metobo, S. E.; Phillips, B. W.; Venkataramani, C. WO Patent 2010132601A1,2010.
134. Scott, R. W.; Vitale, J. P.; Matthews, K. S.; Teresk, M. G.; Formella, A.; Evans, J.W. US Patent 2013324740A1, 2013.
135. Jin, S. M.; Park, C. Y.; Cho, Y. M.; Ku, B. J.; Ahn, C. W.; Cha, B.-S.; Min, K. W.;Sung, Y. A.; Baik, S. H.; Lee, K. W.; Yoon, K.-H.; Lee, M.-K.; Park, S. W. Diab.Obes. Metab. 2015, 17, 599.
136. Lee, H. W.; Ahn, J. B.; Kang, S. K.; Ahn, S. K.; Ha, D.-C. Org. Process Res. Dev.2007, 11, 190.
137. Lee, H. W.; Kim, B. Y.; Ahn, J. B.; Kang, S. K.; Lee, J. H.; Shin, J. S.; Ahn, S. K.; Lee,S. J.; Yoon, S. S. Eur. J. Med. Chem. 2005,
PAPER
The Discovery of Ledipasvir (GS-5885), a Potent Once-Daily Oral NS5A Inhibitor for the Treatment of Hepatitis C Virus Infection
http://pubs.acs.org/doi/abs/10.1021/jm401499g?prevSearch=LEDIPASVIR&searchHistoryKey=
http://pubs.acs.org/doi/pdf/10.1021/jm401499g
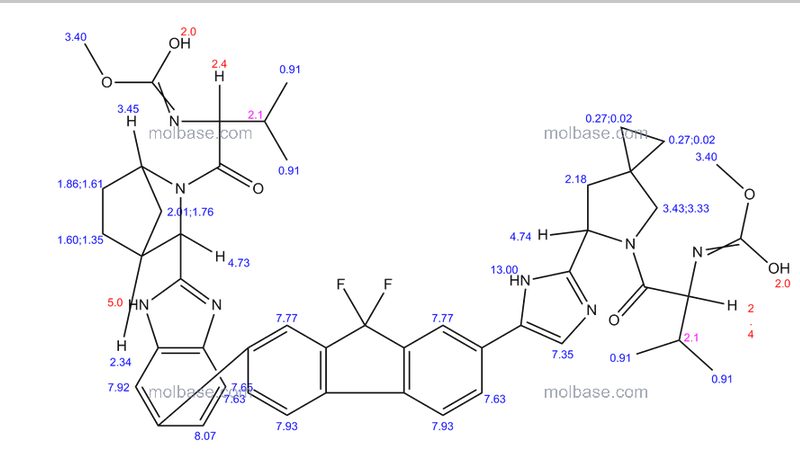
1H-NMR: 300 MHz, (dmso-d6) δ: 8.20-7.99 (m, 8H), 7.73 (s, 2H), 7.37 – 7.27
(m, 2H), 5.25 (dd, J = 7.2 Hz, 1H), 4.78 (s, 1H) 4.54 (s, 1H), 4.16 (m, 1H), 4.02 (m,
1H), 3.87 (m,1H), 3.74 (m, 1H), 3.55 (s, 3H), 3.53 (s, 3H), 2.75 (m, 1H), 2.25 (m,
2H), 2.09 – 2.04 (m, 2H), 1.88 – 1.79 (m, 2H), 1.54 (m, 1H), 0.94 – 0.77 (m, 15H)
0.63 (m, 4H) ppm.
19F-NMR: 282 MHz, (dmso-d6) δ: -109.1 ppm [-74.8 ppm TFA].
HRMS (ESI-TOF) m/z: [M + H]+
calc’d for C49H55F2N8O6: 889.4207; Found: 889.4214.
methyl [(2S)-1-{(6S)-6-[5-(9,9-difluoro-7-{2-[(1R,3S,4S)-2-{(2S)-2-[(methoxycarbonyl)amino]-3-methylbutanoyl}-2-azabicyclo[2.2.1]hept-3-yl]-1H-benzimidazol-6-yl}-9H-fluoren-2-yl)-1H-imidazol-2-
yl]-5-azaspiro[2.4]hept-5-yl}-3-methyl-1-oxobutan-2-yl]carbamate (39 NOS IS LEDISPAVIR
PATENT
Synthesis of 25
25
B. Synthesis of 26 and 27
25 26 27
[0186] To a flask was charged 25 (20.00 g, 0.083 mol), 4-bromo-l,2-benzenediamine (16.74 g, 0.089 mol, 1.08 equiv.), hydroxybenzotriazole (HOBt) (13.96 g, 0.091 mol, 1.1 equiv.), and l-ethyl-3-(3-dimethylaminopropyl) carbodiimide HC1 (EDC.HC1) (17.48 g, 0.091 mol, 1.1 equiv.). The flask was cooled in an ice bath, and was charged with N,N- dimethylacetamide (DMAc, 80 mL). The reaction was allowed to cool to ca. 10 °C with stirring. N-methylmorpholine (NMM) (27.34 mL, 0.249 mol, 3 equiv.) was added over 5 minutes keeping the internal temperature below 20 °C. The reaction was stirred at rt for 20 h. Upon reaction completion, the reaction mixture was added to MTBE (200 mL) and water (600 mL) in a separatory funnel and was gently shaken. The layers were allowed to separate, and the aqueous layer was removed. The aqueous layer was extracted twice with MTBE (50 mL), and the organic extracts were combined. The combined organic extracts were then extracted with water (500 mL), forming a mixture that did not separate well. The mixture was filtered over an appropriate solid support and the layers were separated. The organic phase was concentrated under vacuum, and the resulting residue was dissolved in diisopropyl ether (100 mL). The solution was cooled to ca. 5 °C with stirring. Acetic acid (5.22 mL, 0.091 mol, 1.1 equiv.) was added slowly keeping the internal temperature below 10 °C, and the resulting suspension was stirred 2 h at 5 °C. The thick suspension was then filtered, and the solid was rinsed with diisopropyl ether (100 mL), followed by heptane (100 mL). The cake was dried under vacuum to give the product as a light-beige solid as a mixture of regioisomers 26 and 27 (28.19 g, 72%, >99% AN). 1H NMR (400 MHz, DMSO) mixture of 26 & 27 (data is for the two rotamers of the major regioisomer): δ 9.25 (s, 0.5H), 9.13 (s, 0.5H), 7.08 (d, J= 8.3 Hz, 0.5H); 7.06 (d, J= 8.2 Hz, 0.5H), 6.92 (d, J= 2.2 Hz, 0.5H), 6.89 (d, J= 2.1 Hz, 0.5H), 6.71 (dd, J= 8.4, 2.2, 0.5H), 6.66 (dd, J= 8.4, 2.2, 0.5H), 5.10 (br s, 1H), 5.05 (br s, 1H), 4.15 (br s, 0.5H), 4.10 (br s, 0.5H), 3.76 (s, 1H), 2.64 (br s, 1H), 1.96- 1.88 (m, 1H), 1.77-1.67 (m, 1H), 1.67-1.19 (m, 4H), 1.41 (s, 4.5H), 1.33 (s, 4.5H). MS-ESI+: [M + H]+ calcd for Ci8H25Br03N3, 410.1, 412.1; found, 410.0, 412.0
[0187] The disclosure provides in some embodiments the use of other coupling reagents. These include but are not limited to N,N”-dicyclohexylcarbodiimide (DCC), NJV- diisopropylcarbodiimide (DIC), 6-chloro-2,4-dimethoxy-s-triazine (CDMT), O- benzotriazole-N^N^A^-tetramethyl-uronium-hexafluoro-phosphate (HBTU), and 2-(7-Aza- 1H- benzotriazole-l-yl)-l,l,3,3-tetramethyluronium hexafluorophosphate (HATU).
[0188] The amine base also can be varied or omitted completely. For instance the amine is selected from tertiary amines (R3N), 2,6-lutidine, pyridine, dicyclohexylmethylamme, and N- methylmorpholine (NMM).
[0189] Suitable solvent alternatives are selected from DMF, NMP, dialkyl and cyclic ethers R20, THF, 2-MeTHF, DCM, DCE, toluene, EtOAc, IP Ac, acetone, MIBK, and MEK.
[0190] Suitable temperatures for the reaction range from about -20 °C to 80 °C.
NMR PREDICT

1H/13C NMR PREDICT


COSY
Links
1)Link, John O.et al; The Discovery of Ledipasvir (GS-5885), a Potent Once-Daily Oral NS5A Inhibitor for the Treatment of Hepatitis C Virus Infection; Journal of Medicinal Chemistry (2013), Ahead of Print.DOI:10.1021/jm401499g
2)Ray, Adrian S. et al; Preparation of pyridazinylmethylimidazopyridine derivatives and analogs for use in the treatment of hepatitis C virus using combination chemotherapy, PCT Int. Appl., WO2013040492
3) Delaney, William E. et al ; Preparation of pyridazinylmethylimidazopyridine derivatives and analogs for use in the treatment of hepatitis C virus using combination chemotherapy, PCT Int. Appl., wo2012087596
4) Delaney, William E., IV et al; Preparation of quinoline derivatives and analogs for use in the treatment of hepatitis C virus infection in combination with ribavirin; PCT Int. Appl., wo2011156757
5) Guo, Hongyan et al; Preparation of biaryls, arylheteroaryls, heteroaryls, biarylacetylenes and related compounds end-capped with amino acid or peptide derivatives as antiviral agents; PCT Int. Appl., WO2010132601
6)Phase III (Sofosbuvir + Ledipasvir) ION-1 study: (Clinical Trial number: NCT01701401):
Title:A Phase 3, Multicenter, Randomized, Open-Label Study to Investigate the Efficacy and Safety of Sofosbuvir/Ledipasvir Fixed-Dose Combination (FDC) +/- Ribavirin for 8 Weeks and Sofosbuvir/Ledipasvir Fixed-Dose Combination (FDC) for 12 Weeks in Treatment-Naive Subjects With Chronic Genotype 1 HCV Infection
7) Phase III (Sofosbuvir + Ledipasvir) ION-2 study: (Clinical Trial number: NCT01768286)
Title:A Phase 3, Multicenter, Randomized, Open-Label Study to Investigate the Efficacy and Safety of Sofosbuvir/GS-5885 Fixed-Dose Combination ± Ribavirin for 12 and 24 Weeks in Treatment-Experienced Subjects With Chronic Genotype 1 HCV Infection
8) Phase III (Sofosbuvir + Ledipasvir) ION-3 study: (Clinical trial number: NCT01851330)
Title:A Phase 3, Multicenter, Randomized, Open-Label Study to Investigate the Efficacy and Safety of Sofosbuvir/Ledipasvir Fixed-Dose Combination (FDC) +/- Ribavirin for 8 Weeks and Sofosbuvir/Ledipasvir Fixed-Dose Combination (FDC) for 12 Weeks in Treatment-Naive Subjects With Chronic Genotype 1 HCV Infection

References
- “Ledipasvir” (PDF). United States Adopted Name.
- “Ledipasvir-submitted-to-FDA”.
- “GS-5885”. Gilead Sciences.
- ELECTRON: 100% Suppression of Viral Load through 4 Weeks’ Post-treatment for Sofosbuvir + Ledipasvir (GS-5885) + Ribavirin for 12 Weeks in Treatment-naïve and -experienced Hepatitis C Virus GT 1 Patients. Gane, Edward et al. 20th Conference on Retroviruses and Opportunistic Infections. March 3–6, 2013. Abstract 41LB.
- CROI 2013: Sofosbuvir + Ledipasvir + Ribavirin Combo for HCV Produces 100% Sustained Response. Highleyman, Liz. HIVandHepatitis.com. 4 March 2013.
- “Gilead Files for U.S. Approval of Ledipasvir/Sofosbuvir Fixed-Dose Combination Tablet for Genotype 1 Hepatitis C”. Gilead Sciences. 10 February 2014.
- “U.S. Food and Drug Administration Approves Gilead’s Harvoni® (Ledipasvir/Sofosbuvir), the First Once-Daily Single Tablet Regimen for the Treatment of Genotype 1 Chronic Hepatitis C”. 10 October 2014. Retrieved 10 October 2014.
- Afdhal, N; Zeuzem, S; Kwo, P; Chojkier, M; Gitlin, N; Puoti, M; Romero-Gomez, M; Zarski, J. P.; Agarwal, K; Buggisch, P; Foster, G. R.; Bräu, N; Buti, M; Jacobson, I. M.; Subramanian, G. M.; Ding, X; Mo, H; Yang, J. C.; Pang, P. S.; Symonds, W. T.; McHutchison, J. G.; Muir, A. J.; Mangia, A; Marcellin, P; Ion-1, Investigators (2014). “Ledipasvir and sofosbuvir for untreated HCV genotype 1 infection”. New England Journal of Medicine 370 (20): 1889–98. doi:10.1056/NEJMoa1402454. PMID 24725239.
- http://www.gilead.com/~/media/Files/pdfs/medicines/liver-disease/harvoni/harvoni_pi.pdf
- http://www.hepatitisc.uw.edu/page/treatment/drugs/ledipasvir-sofosbuvir
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
Methyl N-[(2S)-1-[(6S)-6-[5-[9,9-Difluoro-7-[2-[(1S,2S,4R)-3-[(2S)-2-(methoxycarbonylamino)-3-methylbutanoyl]-3-azabicyclo[2.2.1]heptan-2-yl]-3H-benzimidazol-5-yl]fluoren-2-yl]-1H-imidazol-2-yl]-5-azaspiro[2.4]heptan-5-yl]-3-methyl-1-oxobutan-2-yl]carbamate
|
|
| Clinical data | |
| Legal status |
|
| Routes of administration |
Oral |
| Pharmacokinetic data | |
| Bioavailability | 76% |
| Protein binding | >99% |
| Metabolism | No cytochromemetabolism |
| Biological half-life | 47 hrs |
| Identifiers | |
| CAS Registry Number | 1256388-51-8 |
| ATC code | None |
| ChemSpider | 29271894 |
| ChEBI | CHEBI:85089 |
| Chemical data | |
| Formula | C49H54F2N8O6 |
| Molecular mass | 889.00 g/mol |
click
http://drugsynthesisint.blogspot.in/p/vir-series-hep-c-virus-22.html
AND
http://medcheminternational.blogspot.in/p/vir-series-hep-c-virus.html
THE VIEWS EXPRESSED ARE MY PERSONAL AND IN NO-WAY SUGGEST THE VIEWS OF THE PROFESSIONAL BODY OR THE COMPANY THAT I REPRESENT
////////////////GS-5885, LEDIPASVIR
PATENT 1
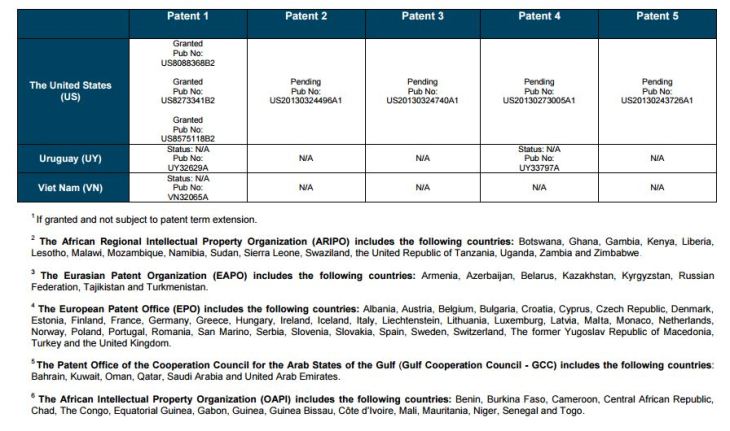
Patent application WO2010132601A1 (primary patent) discloses the base compound of ledipasvir. The application claims a general structural formula (Markush) of new amide compounds useful for treating disorders associated with HCV. This patent, if granted, serves as a blocking patent preventing competitors from making the product. The claims are very broad, using a Markush structure of antiviral agents. As per the WIPO ISR, claims 1-19 are novel and inventive. However, according to the ISR, all remaining claims (claims 20 to 173), covering a large number of compounds, lack both novelty and inventive step, due to lack of support from the patent specification and in the light of prior art. Prosecution at the USPTO Three patents have been granted in the United States: US8088368B2, claiming the base compound by general structural formula; US8273341B2 (a division of US8088368B2), claiming a method of inhibiting HCV; and US8575118B2 (a continuation of US8273341B2 and a division of US8088368B2), claiming specific amide compounds not covered in the other two related patents. The examination report of US8088368B2 reveals that the application was allowed after the applicant cancelled and amended claims on Markush substuents. The examination report of US8273341B2 reveals that the application was allowed after the applicant amended a claim ‘A method of treating HCV’ to ‘A method of inhibiting HCV´. The examination report of US8575118B2 reveals that the application was allowed after the applicant cancelled claims already covered by the related patents, and limited claims to four specific compounds. Patent 1 has been filed in various jurisdictions: The patent has been granted by the ARIPO, in South Africa, and the United States. The patent (or a related patent) is pending in Argentina, Australia, Canada, China, as well as China, Hong Kong SAR, the EAPO, the EPO, Israel, India, Japan, New Zealand, Singapore, and Ukraine. Legal status is not available for Colombia, Ecuador, Mexico, Peru, Uruguay, and Viet Nam. 13 Litigation / Opposition on Patent 1 In December 2013, Gilead Sciences filed apatent infringement lawsuit against Abbott Laboratories and AbbVie Inc., in the United States District Court for the District of Delaware (case Number: 1:13cv02034). The case involves Gilead Sciences patents US8088368B2, US8273341B2, and US8575118B2.
PATENT 2 Patent application WO2013184698A1 is a product and process patent, claiming new crystalline solvate forms of ledipasvir useful for treating a subject suffering from HCV infection. The application also claims processes of manufacture of such amorphous and crystalline forms with specific X-ray diffraction peaks, and compositions and combinations comprising them. The application has just recently been published and no written opinion on patentability is available at this stage. As per the available information (details available in the Annex): The patent is pending at the EPO and the United States. There are no litigation or opposition procedures reported.
PATENT 3 Patent application WO2013184702A1 is a process patent, claiming processes for the preparation of ledipasvir. The disclosure also provides compounds that are synthetic intermediates to compounds of ledipasvir. The claims are moderately narrow covering crystalline and amorphous forms of ledipasvir with specific X-ray diffraction peaks. The application has just recently been published and no written opinion on patentability is available at this stage. As per the available information (details available in the Annex): The patent is pending at the EPO and the United States. There are no litigation or opposition procedures reported.
PATENT 4 Patent application WO2012087596A1 is a formulation patent, claiming various formulations comprising a combination of ledipasvir with GS-9256, or tegobuvir or with other compounds. The application also claims methods of treatment with the said combinations for reducing viral load in a person infected with HCV. 14 As per the WIPO ISR, the application is novel but not inventive in comparison to the closest prior art retrieved during the search. The combinations claimed in the instant application are not disclosed in the prior art, thus the combinations are novel. However, the prior art discloses various combinations, therefore, the problem to be solved through the invention should be new combinations with fewer side effects. Further, no experimental data of synergism has been provided to support double, triple, or quadruple combinations. Thus, according to the ISR, the instant invention cannot be regarded as inventive. As per the available information (details available in the Annex): The patent has been granted in Argentina. The patent is pending in Australia, Canada, the EPO, and the United States. Legal status is not available for Japan and Uruguay. There are no litigation or opposition procedures reported.
PATENT 5 Patent application WO2013040492A2 is a formulation and method of use patent, claiming compositions and a method of using the combination for the treatment of HCV. Drug combinations are used, and the compositions include sofosbuvir, PSI-7851 and ledipasvir. Since the application claims a group of compounds of Markush structure, it gives the claims a broad scope. As per the WIPO ISR the application is novel but lacks the inventive step in light of prior art. The invention lacks an inventive step as it would be obvious to a person skilled in the art to combine the diastereoisomer of the present invention, disclosed in the prior art, with other antiviral agents to provide an alternative HCV therapy. As per the available information (details available in the Annex): The patent is pending in Australia, Canada, the EPO, and the United States. There are no litigation or opposition procedures reported. This patent is listed in the sofosbuvir report as Patent No. 7
http://www.who.int/phi/implementation/ip_trade/ledipasvir_report_2014-09-02.pdf
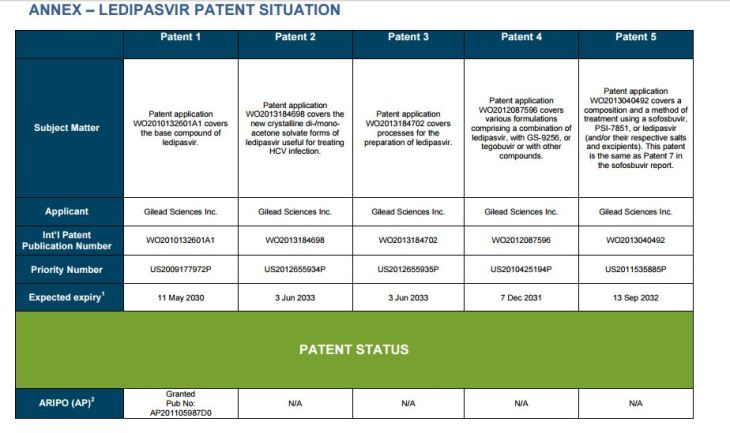
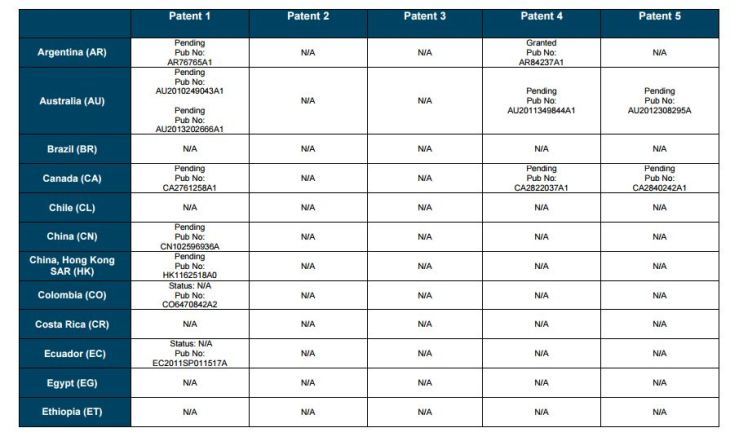


SUMMARY The search revealed patents filed with respect to ledipasvir by the Sponsor as well as a nonSponsor. The ledipasvir Sponsor patent collection comprises 5 different patents (patent families) with 47 family members published in 23 jurisdictions. The majority of these patent applications are still pending in the respective patent offices (see Patents 1 to 5 in the Annex). Patent 1 is the primary patent, claiming the base compound through a Markush claim, along with various substituents. Where granted, this patent can prevent competitors from making ledipasvir. Patents 2 and 3 claim processes to make ledipasvir and thus if granted will require competitors to design around these patents and use other production processes. The chemical product itself is not protected. Patents 4 and 5 claim combinations of different HCV drugs with ledipasvir, and their formulations. There is competition in the field by AbbVie, Inc., which filed formulation patents. Note: The search also revealed two patents that are relevant for all seven reports. Patent applications WO2013059630A1 and WO2013059638A1 inter alia claim the use of combinations of unnamed direct-acting antiviral agents for treating HCV, where the treatment does not include administration of interferon or ribavirin, and the treatment lasts between 8-12 weeks. The description and the dataset for these two patents can be found in the Working Paper on ombitasvir (Patents No 3 and 4). These patents are in litigation. Detailed information can be found in the Working Paper on sofosbuvir under Patent No 2.
World Drug Tracker: LEDIPASVIR
click
http://drugsynthesisint.blogspot.in/p/vir-series-hep-c-virus-22.html
AND
http://medcheminternational.blogspot.in/p/vir-series-hep-c-virus.html
LEDIPASVIR
Biological Activity of Ledipasvir
Ledipasvir(GS5885) is an inhibitor of the hepatitis C virus NS5A protein. Ledipasvir is an experimental drug for the treatment of hepatitis C.
IC50 Value: 141 nM (EC50, JFH1/3a-NS5A hybrid replicon) [1]
Target: HCV NS5A
in vitro: Against JFH1/3a-NS5A, DCV was more potent (EC(50) = 0.52 nM) than GS-5885 (EC(50) = 141 nM). DCV sensitivity was increased against JFH1/3a-NS5A-M28V (EC50 = 0.006 nM), A30V (EC(50) = 0.012 nM), and E92A (EC(50) = 0.004 nM) while the NS5A-A30K and -Y93H variants exhibited reduced sensitivity to DCV (EC50 values of 23 nM and 1120 nM, respectively) and to GS-5885 (EC50 values of 1770 nM and 4300 nM, respectively) [1].
in vivo: GS-5885 was well tolerated and resulted in median maximal reductions in HCV RNA ranging from 2.3 log(10) IU/ml (1 mg QD) to 3.3 log(10) IU/ml (10 mg QD in genotype 1b and 30 mg QD). E(max) modeling indicated GS-5885 30 mg was associated with>95% of maximal antiviral response to HCV genotype 1a. HCV RNA reductions were generally more sustained among patients with genotype 1b vs. 1a. Three of 60 patients had a reduced response and harbored NS5A-resistant virus at baseline. NS5A sequencing identified residues 30 and 31 in genotype 1a, and 93 in genotype 1b as the predominant sites of mutation following GS-5885 dosing. Plasma pharmacokinetics was consistent with QD dosing [2].
Toxicity:
Clinical trial: Combination Therapy for Chronic Hepatitis C Infection. Phase 2
Clinical Information of Ledipasvir
| Product Name | Sponsor Only | Condition | Start Date | End Date | Phase | Last Change Date |
|---|---|---|---|---|---|---|
| Ledipasvir | Gilead Sciences Inc | Hepatitis C virus infection | 31-OCT-12 | 31-DEC-14 | Phase 3 | 12-SEP-13 |
| Gilead Sciences Inc | Hepatitis C virus infection | 31-OCT-13 | 31-JAN-15 | Phase 3b | 11-NOV-13 | |
| Gilead Sciences Inc | Hepatitis C virus infection | 31-MAY-13 | 31-DEC-14 | Phase 3 | 12-SEP-13 | |
| Gilead Sciences Inc | Hepatitis C virus infection | 31-DEC-10 | 30-APR-14 | Phase 2b | 28-AUG-13 | |
| Gilead Sciences Inc | Hepatitis C virus infection | 31-JUL-11 | 30-JUN-13 | Phase 2 | 22-AUG-13 | |
| Gilead Sciences Inc | Hepatitis C virus infection | 31-JUL-11 | 30-APR-13 | Phase 2b | 03-OCT-12 | |
| Gilead Sciences Inc | Hepatitis C virus infection | 31-OCT-13 | 31-JAN-15 | Phase 3 | 11-NOV-13 | |
| Gilead Sciences Inc | Hepatitis C virus infection | 31-MAY-13 | 31-DEC-14 | Phase 3 | 12-SEP-13 | |
| Gilead Sciences Inc | Hepatitis C virus infection | 31-OCT-12 | 31-DEC-14 | Phase 3 | 12-SEP-13 | |
| Gilead Sciences Inc | Hepatitis C virus infection | 31-JUL-11 | 30-APR-13 | Phase 2 | 03-OCT-12 | |
| Gilead Sciences Inc | Hepatitis C virus infection | 31-JUL-11 | 30-JUN-13 | Phase 2b | 22-AUG-13 |
update………..
![]()

WO 2016145990, Ledipasvir, New patent, SHANGHAI FOREFRONT PHARMACEUTICAL CO., LTD
(WO2016145990) METHOD OF PREPARATION FOR LEDIPASVIR AND DERIVATIVE THEREOF, AND INTERMEDIATE COMPOUND FOR PREPARATION OF LEDIPASVIR
SHANGHAI FOREFRONT PHARMCEUTICAL CO., LTD [CN/CN]; Room 1306, No.781 Cailun Road China (Shanghai) Pilot Free Trade Zone, Pudong New Area Shanghai 201203 (CN)
HUANG, Chengjun; (CN).
FU, Gang; (CN).
FU, Shaojun; (CN).
WEI, Zhewen; (CN).
LI, Wei; (CN).
ZHANG, Xixuan; (CN)
chinese machine translation please bear………..
SMILES COC(=O)N[C@@H](C(C)C)C(=O)N1CC2(CC2)C[C@H]1c3ncc([nH]3)c4ccc5c6ccc(cc6C(F)(F)c5c4)c7ccc8nc([nH]c8c7)[C@@H]9[C@H]%10CC[C@H](C%10)N9C(=O)[C@@H](NC(=O)OC)C(C)C
Chelsea Therapeutics Announces FDA Acceptance of NORTHERA(TM) (droxidopa) NDA Resubmission

Droxidropa
FDA Deems Resubmission a Complete Response; PDUFA Date Set as
February 14, 2014
CHARLOTTE, N.C., Sept. 4, 2013 (GLOBE NEWSWIRE) — Chelsea Therapeutics International, Ltd. (Nasdaq:CHTP) today announced that the U.S. Food and Drug Administration (FDA) has acknowledged receipt of the New Drug Application (NDA) resubmission seeking approval to market NORTHERA(TM) (droxidopa), an orally active synthetic precursor of norepinephrine
read all at
http://www.pharmalive.com/chelsea-therapeutics-announces-fda-acceptance-of-northera-nda-resubmission
UPDATE………………….

DROXIDOPA
ORPHAN DRUG,
CAS 23651-95-8, 3916-18-5
ROTATION –FORM
|
(2S,3R)-2-amino-3-(3,4-dihydroxyphenyl)-3-hydroxypropanoic acid
|
Proprietary Name: NORTHERA
Dosage Form; Route of Administration: CAPSULE; ORAL
Strength: 100MG
Reference Listed Drug: No
TE Code:
Application Number: N203202
Product Number: 001
Approval Date: Feb 18, 2014
Applicant Holder Full Name: LUNDBECK NA LTD
Marketing Status: Prescription


Droxidopa (INN; trade name Northera; also known as L-DOPS, L–threo-dihydroxyphenylserine, L-threo-DOPS and SM-5688) is a synthetic amino acid precursor which acts as a prodrug to the neurotransmitter norepinephrine (noradrenaline).[1] Unlike norepinephrine, droxidopa is capable of crossing the protective blood–brain barrier (BBB).[1]
CLIP




REF http://www.accessdata.fda.gov/drugsatfda_docs/nda/2014/203202Orig1s000ChemR.pdf
Distribution
Droxidopa exhibits plasma protein binding of 75% at 100 ng/mL and 26% at 10,000 ng/mL with an apparent volume of distribution of about 200 L.
Droxidopa starting dose is 100mg three times daily (which can be titrated to a maximum of 600 mg three times daily). One dose should be taken in late afternoon at least 3 hours prior to bedtime to reduce the potential for supine hypertension during sleep.
Indications
- Neurogenic orthostatic hypotension (NOH) dopamine beta hydrolase deficiency,[2] as well as NOH associated with multiple system atrophy (MSA), familial amyloid polyneuropathy (FAP), pure autonomic failure (PAF).
- Intradialytic hypotension (IDH) or hemodialysis-induced hypotension.
- Freezing of gait in Parkinson’s disease (off-label)
History
Droxidopa was developed by Sumitomo Pharmaceuticals for the treatment of hypotension, including NOH,[2] and NOH associated with various disorders such as MSA, FAP, and PD, as well as IDH. The drug has been used in Japan and some surrounding Asianareas for these indications since 1989. Following a merge with Dainippon Pharmaceuticals in 2006, Dainippon Sumitomo Pharmalicensed droxidopa to Chelsea Therapeutics to develop and market it worldwide except in Japan, Korea, China, and Taiwan. In February 2014, the Food and Drug Administration approved droxidopa for the treatment of symptomatic neurogenic orthostatic hypotension.[3]
Clinical trials
Chelsea Therapeutics obtained orphan drug status (ODS) for droxidopa in the U.S. for NOH, and that of which associated with PD, PAF, and MSA. In 2014, Chelsea Therapeutics was acquired by Lundbeck along with the rights to droxidopa which was launched in the US in Sept 2014.[4]
REGULATORY
CLICK ON IMAGE TO VIEW
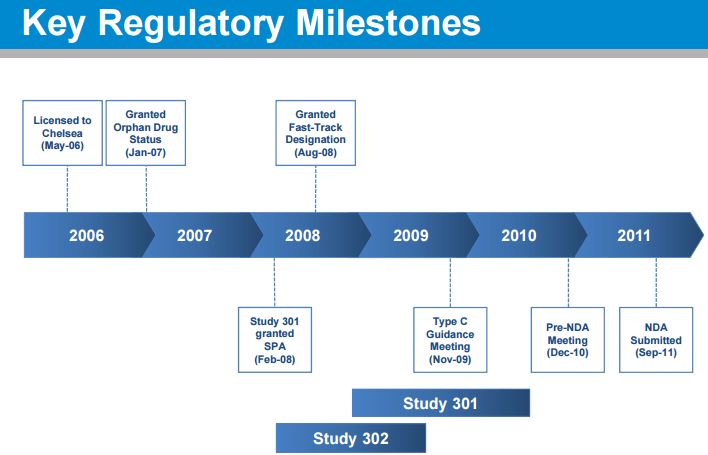
FDA agreement on overall development program (Sep 2007)
• FDA agreement on Study 301 design under a Special Protocol Assessment (Feb 2008) – Included agreement: length of patient exposure was adequate for the safety evaluation
• FDA agreement on changing primary endpoint of Study 301 while it was ongoing and prior to any unblinding (Nov 2009) – From dizziness to the OHQ – SPA remained intact
• FDA agrees to NDA package (Dec 2010) – Studies 301, 302, 303, 304 and 305 – Renal safety study conducted post-marketing
• FDA accepts droxidopa NDA and grants priority review (Sep 2011)
Pharmacology
Droxidopa is a prodrug of norepinephrine used to increase the concentrations of these neurotransmitters in the body and brain.[1][What, if any, are the other neurotransmitters droxidopa increases concentrations of? “These neurotransmitters” implies multiple(see above)] It ismetabolized by aromatic L-amino acid decarboxylase (AAAD), also known as DOPA decarboxylase (DDC). Patients with NOH have depleted levels of norepinephrine which leads to decreased blood pressure or hypotension upon orthostatic challenge.[5] Droxidopa works by increasing the levels of norepinephrine in the peripheral nervous system (PNS), thus enabling the body to maintain blood flow upon and while standing.
Droxidopa can also cross the blood–brain barrier (BBB) where it is converted to norepinephrine from within the brain.[1] Increased levels of norepinephrine in the central nervous system (CNS) may be beneficial to patients in a wide range of indications. Droxidopa can be coupled with a peripheral aromatic L-amino acid decarboxylase inhibitor (AAADI) orDOPA decarboxylase inhibitor (DDC) such as carbidopa (Lodosyn) to increase central norepinephrine concentrations while minimizing increases of peripheral levels.
Side effects
With over 20 years on the market, droxidopa has proven to have few side effects of which most are mild. The most common side effects reported in clinical trials include headache, dizziness nausea, hypertension and fatigue.[6][7][8][8]
L-threo-dihydroxyphenylserine, also known as droxidopa, L-threo-DOPS, or L-DOPS, is an orally active synthetic precursor of norepinephrine. Droxidopa thus replenishes depleted norepinephrine, allowing for re-uptake of norepinephrine into peripheral nervous system neurons. This reuptake, in turn, stimulates receptors for vasoconstriction, providing physiological improvement in symptomatic neurogenic orthostatic hypotension patients. It has also shown efficacy in other diseases, such as Parkinson’s disease and depression.
Droxidopa has been used in Japan for many years for the treatment of orthostatic hypotension. It was originally approved in 1989 for the treatment of frozen gait or dizziness associated with Parkinson’s disease and for the treatment of orthostatic hypotension, syncope or dizziness associated with Shy-Drager syndrome and Familial Amyloidotic Polyneuropathy.
Marketing approval was later expanded to include treatment of vertigo, dizziness and weakness associated with orthostatic hypotension in hemodialysis patients.
The preparation of droxidopa generally involves a multi-step synthesis. Typically, one or more of the necessary steps in the synthesis requires that reactive sites other than that site targeted for reaction are temporarily protected. Thus, the synthesis of droxidopa typically comprises at least one protecting and associated deprotecting step. For example, the catechol moiety, the amine moiety, and/or the carboyxyl moiety may require protection and subsequent deprotection, depending upon the synthetic route and the reagents used in the preparation of droxidopa.
U.S. Patent Nos. 4,319,040 and 4,480,109 to Ohashi et al. describe processes for the preparation of optically active D- and L- threo-DOPS by optically resolving a racemic mixture of threo-2-(3,4-methylenedioxyphenyl)-N-carbobenzyloxyserine or threo-2-(3,4-dibenzyloxy-phenyl)- N-carbobenzyloxyserine, respectively. Following optical resolution of these racemic mixtures to give the desired L-enantiomer, the methylene or benzyl groups must be removed from the catechol moiety and the carbobenzyloxy (CBZ) group must be removed from the amine group to give droxidopa. The methylene group can be readily removed by reaction with a Lewis acid {e.g., aluminum chloride). The CBZ group (and the benzyl catechol protecting groups, where applicable) is removed from the amine by hydrogenolysis to give the desired compound. The hydrogenolysis step is noted to be carried out by treating the optically resolved salt with hydrogen in the presence of a catalyst, e.g., palladium, platinum, nickel, or the like.
However, for large-scale production of pharmaceutical compounds, hydrogenolysis may not be desirable. For example, hydrogenolysis requires expensive, specialized equipment, which represents a large capital investment. Labor costs are also high, as the process requires careful handling and disposal of certain compounds (e.g., the pyrophoric catalyst). Further, due to the hazards associated with both the reagents and the high pressure system required for hydrogenolysis, it is desirable to avoid synthetic methods that require hydrogenolysis.
In an alternative method for the production of droxidopa, taught by U.S. Patent No.
4,562,263 to Ohashi et al, hydrogenation is not required. In this process, the amine group is protected via a phthaloyl group. Following optical resolution, the phthaloyl group is removed from the droxidopa precursor by hydrazine.
However, hydrazine is known to be genotoxic and has been classified by the EPA as a
Group B2 probable human carcinogen. Thus, it is desirable to remove even trace amounts of hydrazine from pharmaceutical compounds. In practice, the method described in the ‘263 patent suffers from the inability to remove 100% of the hydrazine from the final product. Thus, there is some level of contamination by hydrazine using this method. The Food and Drug Administration has established a maximum genotoxic impurity level of 1.5 micrograms per day. Therefore, based on the maximum daily dose of droxidopa (1.8 g), the maximum allowable hydrazine level therein is 0.8 ppm. Accordingly, it would be advantageous to find a new synthetic route for the preparation of droxidopa that avoids the use of hydrogenolysis and also avoids the use of hydrazine
PATENT
https://www.google.ch/patents/WO2013142093A1?cl=en
The synthetic route for the preparation of droxidopa comprises the following steps: a) converting piperonal to 2-amino-3-(benzo-l,3-diox-5-yl)-3-hydroxypropanoic acid

c) optical resolution and separation of the desired isomer



Experimental Section
Example 1 : Screening of Deprotection Strategies for Phthaloyl Group

Example 2: Exemplary Synthesis of Droxidopa
The synthesis of droxidopa according to the methods provided herein can be conducted as a continuous process or can be conducted in a series of individual steps. Both processes are intended to be encompassed by the present disclosure.
Synthesis of N-carbomethoxy phthalimide

3-Methoxy phthalimide 1 (120 kg) is added to a vessel containing dimethylformamide (420 L) and stirred (95 ± 10 RPM) at 25 – 30 °C for 30 min. The contents are cooled to 18 – 20 °C and triethylamine (124 L) is added. The contents are further cooled to -10 °C to -5 °C and
methylchloroformate 2 (85 kg) is added. The reaction temperature is maintained in the range of -10 °C to 0 °C to control the exothermicity during the addition of methylchloroformate. The temperature of the mixture is maintained at 0 – 5 °C for 1 h after the addition of
methylchloroformate .
The reaction mixture is then heated to 25 – 30 °C for 1 h. An in-process sample is taken to confirm a phthalimide content limit <2.5%. The mixture is sampled again to confirm a phthalimide content <0.5%. The mixture is transferred to another reactor, cooled to 0 – 5 °C, and the reaction is quenched with the addition of demineralized water (1260 ± 10 L) at a temperature of 10 ± 5 °C. The mixture is then heated at 25 – 30 °C for 1 h.
The material is centrifuged for 2 h and the wet cake is washed three times with
demineralized water (360 L). The wet cake is dried at a temperature of 55 – 60 °C and a sample is taken after 12 h of drying to confirm water content <1.0% w/w.
Expected yield of N-carbomethoxy phthalimide (3): 144-158 kg. This material is not isolated and is used directly in the next step.
Synthesis of 2-amino-3-(benzo-l ,3-dioxol-5-yl)-3-hydroxypropanoic acid

Piperonal 4 (229 ± 1 kg) is added to toluene (310 ± 5 L) in a reactor and the mixture is stirred (85 – 95 RPM) until a clear solution is obtained (approximately 30 min). The piperonal solution is transferred to a vessel for later use. Methanol (415 ± 5 L) is added to the reactor followed by the addition of potassium hydroxide (85 kg). The mixture is stirred for approximately 30 min at 25 – 30 °C to provide a clear solution. The potassium hydroxide solution is cooled to 20 – 25 °C, and then glycine 5 (52 ± 1 kg) and toluene (310 ± 5 L) are added while stirring at 20 – 25 °C. The contents of the reactor are cooled to 15 – 20 °C. The solution of piperonal in toluene is slowly added to the reactor while maintaining the temperature at 15 – 20 °C. The reactor temperature is increased to 20 – 25 °C and maintained for 18 h. An in-process sample is taken to determine glycine content by TLC (limit <5.0%).
The reaction mass is transferred to another reactor, the temperature is increased to 40 °C, and the solvents (toluene and methanol) are distilled off under vacuum until the mixture becomes thick. Additional toluene (210 ± 5L) is added to the reaction mass three times and distilled out for complete removal of methanol and toluene. The reaction mixture is kept under vacuum at 40 °C. After 3 h, the reaction mixture is cooled to 18 – 22 °C and a dilute hydrochloric acid solution (230 ± 5L hydrochloric acid and 1145 ± 10 L demineralized water) is added and mixed for 30 min.
The mixture is allowed to settle for 30 min to separate into organic and aqueous layers. The aqueous layer is washed with toluene (310 ± 5 L) and separated. Glacial acetic acid (218 ± 2 kg) is added to the washed aqueous layer at 20-25 °C. Caustic solution (580 ± 5 L DM Water and 200 ± 1 kg caustic flakes) is slowly added into the reaction mass to bring the pH 5.0 to 5.1 while maintaining the temperature at 25 – 30 °C. The pH of the mixture is brought to 5.45 – 5.50 at 25 – 30 °C, while stirring for 30 min. The mixture is centrifuged for 8 h 30 min to 9 h and the resulting wet cake is washed with demineralized water (50 ± 5 L). The cake is dried at 50 – 55 °C under vacuum, and a sample is taken after 12 h to confirm that water content is <10% w/w. The purity is analyzed by HPLC (limit < 10%).
Expected yield of 2-amino-3-(benzo-l ,3-dioxol-5-yl)-3-hydroxypropanoic acid (6):
135 – 145 kg.
Synthesis of 2-phthalimido-3-hydroxy-3-(3,4-methylenedioxyphenvnpropionic acid

Intermediate 6 (140 kg) is added to a reactor containing demineralized water (1 120 L) and stirred (85-95 RPM) for 10 min at 20-25 °C. The contents are cooled to 15-20 °C and compound 3 (140 kg) is added followed by a sodium carbonate solution (63.5-68.3 kg sodium carbonate in 189-203 L demineralized water) within 45-60 min. The mixture is heated to 30-35 °C and held for 90 min. An in-process sample is taken to measure for Stage II (<2.5%) and Stage-I intermediate (<2.5 %). After acceptance criteria are met, the mixture is cooled to 15-20 °C. A dilute sulfuric acid solution (134 kg sulfuric acid in 1120 L demineralized water) at 15-20 °C is added to the mixture to bring the pH to 1.0-2.0. The mixture is maintained at this temperature and pH for 30 min, and then the mixture is heated to 20-25 °C for 2 h.
The mixture is centrifuged for 9 h and the resulting wet cake is washed twice with 518 L of demineralized water. The wet cake is removed from the centrifuge, washed in a reactor containing demineralized water (2590 L), and stirred for 1 h at 25-30 °C. The material is centrifuged for 9 h and the wet cake is washed twice with demineralized water (518 L). The final wet cake is dried at 45-50 °C under vacuum until water content is <1.0% w/w. Intermediate (6) output is considered as standard input and a mean of 140 kg is taken for all inputs.
Expected yield of 2-Phthalimido-3-hydroxy-3-(3,4-methylenedioxyphenyl)propionic acid (7): 187 – 208 kg.
Synthesis of L-threo (N-phthaloyl-3-(3,4-methylenedioxyphenyl)serine) norephedrine salt

L-Norephedrine 8 (89 kg) is added to a reactor containing methanol (296 L) and stirring (45-50 RPM) is started. The mixture is maintained at 25 – 30 °C for 15 – 20 min, and then transferred into a vessel for later use.
2-Phthalimido-3-hydroxy-3-(3,4-methylenedioxyphenyl)propionic acid 7 (197.5 kg) is added to a reactor containing methanol (395 L). The material is stirred for 15 – 20 min at 25 – 30 °C. The L-norephedrine solution is added and mixed for 3 h. If precipitation is not observed within 30 min of adding the L-norephedrine solution, it is seeded with L-threo(N-phthaloyl-3-(3,4- methylenedioxyphenyl) serine (approximately 50 g). After 3 h of mixing, the mixture is cooled to 10 – 15 °C and maintained for 1 h. An in-process sample is taken to check for purity by HPLC (>99.0% a/a). The mixture is centrifuged for 1 h to 1 h 30 min and the wet cake is washed with methanol (49 L) followed by isopropyl alcohol (197.5 L). The wet cake is checked for purity. If purity is <99% a/a, the wet cake is washed with a prechilled solution of methanol (197.5 L) followed by isopropyl alcohol (99 L). After achieving the required purity level, as measured by HPLC, the wet cake is removed from the centrifuge. The cake is dried at 45 – 50 °C until loss on drying <1.0% w/w.
Expected yield of L-threo (N-phthalpyl-3-(3,4-methylenedioxyphenyl)serine) norephedrine (9) salt: 85-99 kg.
Synthesis of L-threo flSi-phthaloyl-S-CS^-methylenedioxyphenvDserine)

Demineralized water (552 L) is added to a reactor and cooled to 10 – 15 °C. Sulfuric acid (20 kg) is added while maintaining the temperature below 30 °C and stirring for 15 – 20 min. The solution is cooled to 15 – 20 °C and 9 (92 kg) is slowly added while stirring and maintaining temperature. The solution is heated to 45 – 50 °C for 6 h, cooled to 25 – 30 °C, and held for 1 h. The pH is checked to confirm the solution is <2.0.
The mixture is centrifuged for 1 h and the wet cake is washed two times with demineralized water (138 L). The wet cake is removed and added to a reactor containing demineralized water (460 L). The temperature is maintained at 25 – 30 °C and stirred for 1 h. The material is centrifuged for 30 min and the wet cake is washed two times with demineralized water (138 L). The material is collected and placed into preweighed containers.
Expected yield of L-threo(N-phthaloyl-3-(3,4-methylenedioxyphenyl)serine) (10): 60-64 kg.
Synthesis of L-threo(N-phthaloyl-3-(3 ,4-dihydroxyphenyl serine)

Compound 10 (62 kg) wet cake is added to a reactor containing methylene chloride (1240 L) and stirred for 10 min. The mixture is heated to remove methylene chloride and water under azeotropic reflux. After methylene chloride (1550 L) is removed and no water remains in the distillate, the mixture is cooled to 25 – 30 °C. An in-process sample is taken to determine water content (limit <0.1 %).
Methylene chloride (186 L) is added to another reactor at 25-30 °C. An in-process sample is taken to check for water content (limit <0.2% w/w). Aluminum chloride (81 kg) is added and the contents are stirred at 25 – 30 °C for 10 – 15 min. The mixture is cooled to 10 – 15 °C and octanethiol (78 kg) is added. The mixture is cooled to -20 to -10 °C.
The slurry of 10 in methylene chloride controlled at -20 to -7 °C is added to the stirred mixture that is temperature controlled at -15 to -10 °C for 20 – 30 min. The mixture is heated to 10-15 °C for 1.5-2.5 h. An in-process sample is taken to determine 10 content (limit <3.5%). The mixture is further cooled to -20 to -10 °C and then transferred to another reactor containing oxalic acid (62 kg), methylene chloride (186 L), and demineralized water (744 L) while maintaining the temperature below -3 °C to quench the reaction. The quenched material is slowly heated to 25 – 30 °C and maintained at this temperature for 12 h. Methylene chloride is distilled out at 25 – 30 °C under vacuum until the mixture volume is reduced to 1364 L.
The mixture is centrifuged for 3 h and the wet cake is washed with demineralized water (62 L). The wet cake is added to a reactor containing oxalic acid (2.5 kg) and demineralized water (248 L) and the contents are stirred at 25-30 °C for 2 h to obtain a clear solution. The material is centrifuged for 1 h 15 min to 1 h 30 min and the wet cake is washed twice with demineralized water (186 L). The wet cake is added to a reactor containing demineralized water (248 L) at 25 – 30 °C and the contents are stirred for 2 h. The material is centrifuged for 1 h 30 min to 2 h and the wet cake is washed twice with demineralized water (186 L). The material is collected and placed into preweighed containers.
Expected yield of L-threo(N-phthaloyl-3-(3,4-dihydroxyphenyl)serine (11): 40-50 kg. Synthesis of L-threo (3,4-dihydroxyphenvP)serine

Methanol (360 L) is added to a reactor and cooled to 20 – 25 °C. Compound 11 (45 kg) is added to the reactor while stirring at 25 – 30 °C for 15 – 20 min. Demineralized water (225 L) and sodium bicarbonate (17 kg) are added to another reactor and cooled to 20 – 25 °C. Hydroxylamine hydrochloride (14 kg) is added and mixed for 15 – 20 min at 20 – 25 °C to obtain a clear solution. The solution of 11 in methanol is transferred through a sparkler filter into a reactor. The hydroxylamine and sodium bicarbonate solution is added to the reactor while maintaining the temperature at 25 – 30 °C. The reaction mixture is heated to 65 – 70 °C and refluxed for 16 h. An in-process sample is taken to determine 11 content (limit <3%). The material is cooled to 25-30 °C with mixing for 2 h.
The material is centrifuged for 1 h and the wet cake is washed three times with methanol (23 L). The wet cake is dried at 40 – 45 °C until water content is <1.0% w/w.
Expected yield of L-threo(3,4-dihydroxyphenyl)serine (12): 20-24 kg. Synthesis of L-threo(3,4-dihvdroxyphenyl)serine hydrochloride

L-threo (3,4-dihydroxyphenyl)serine 12 (22 kg) material is added to a reactor containing demineralized water (55 L) and stirred for 15-30 min. The material is cooled to 20-25 °C and concentrated hydrochloric acid (13 L) is added to form L-threo(3,4-dihydroxyphenyl)serine hydrochloride) (13). The mixture is stirred for 30^15 min until a white thick suspension is observed. The mixture is stirred for an additional 2.0 h ± 15 min. Isopropyl alcohol (132 L) is slowly added and the mixture is stirred for 5 hr ± 15 min. The mixture is cooled to 15-20 °C and stirred for 30^5 min. The mixture is centrifuged for 30 min and the wet cake is washed twice with chilled isopropyl alcohol (22 L) at 15-20 °C. The material is unloaded from the centrifuge and a sample is taken to check the individual impurity by HPLC (limit <0.05%) and purity by HPLC (limit >99.0%).
Reprocessing: If the individual impurity by HPLC does not meet the limit <0.05%, compound 13 is reprocessed by adding the material to a reactor containing demineralized water (28 L) and stirring for 15 – 30 min. Concentrated hydrochloric acid (3 L) is added at 20-25 °C and mixing is continued for 15 – 30 min. Continue mixing for 2 h ± 15 min at the same temperature. Isopropyl alcohol (74 L) is added over a period of 2 – 3 h at 25 – 30 °C. Mixing is continued at 25 – 30 °C for 5 h ± 15 min followed by cooling to 15 – 20 °C and mixing for 30 – 45 min. The mixture is centrifuged for 30 min and washed twice with chilled isopropyl alcohol (22 L) and checked for the individual impurity by HPLC (limit <0.05%).
Expected yield of L-threo(3,4-dihydroxyphenyl)serine hydrochloride) (13): 19-20 kg. Synthesis of L-threo(3,4-dihvdroxyphenyl)serine

Compound 13 (19.5 kg) is added to a reactor containing demineralized water (195 L) while stirring at 25 – 30 °C. Concentrated hydrochloric acid (6 L) is added and mixed for 25 – 30 min. For complete dissolution, the contents can be mixed for another 15 – 20 min. Activated carbon (1 kg) and celite (0.2 kg) are added and mixed for 30 – 40 min. The mixture is filtered through a sparkler filter and the filter is washed with demineralized water (1 X L). The filtrate is transferred to another reactor. A solution containing triethylamine (14 kg) and methanol (41 L) is slowly added to the reaction mass (reactor) while mixing. The pH of the filtrate is adjusted to 7.0 – 7.25 over a period of 3 h at 25 – 30 °C. The contents are stirred for 20-30 min. An in-process sample is taken to confirm the pH is 7.0 – 7.25. The mixture is stirred for 3 h. The mixture is centrifuged for 1 h and the wet cake is washed twice with demineralized water (19.5 L). The wet cake is removed from the centrifuge and kept for a slurry wash. The wet cake is added to a reactor containing methanol (58.5 L) while stirring at 25 – 30 °C for 30 – 40 min. The material is centrifuged for 1 h and the wet cake is washed with methanol (19.5 L). The wet cake is unloaded from the centrifuge and retained for water washing.
The wet cake is added to a reactor containing demineralized water (39 L) while stirring for 30 – 40 min. The material is centrifuged for 10 min and the wet cake is washed twice with methanol (19.5 L). The wet cake is unloaded and a sample is taken to check the chloride content (<200 ppm). The wet cake is dried at 40 – 45 °C until the water content is <0.1 % w/w. A sample is taken after 16 h of drying to confirm loss on drying is <0.1% w/w. The dry material is sieved through a sifter (400 micron) and packed. A sample is taken for quality control testing.
Expected yield of L-threo(3,4-dihydroxyphenyl)serine: 14-15 kg.
Scheme 1: Overview of Droxidopa Synthesis
a) converting piperonal to 2-amino-3-(benzo-l,3-diox-5-yl)-3-hydroxypropanoic acid

d) removal of the catechol protecting group


PATENT
https://www.google.com/patents/CN103086906A?cl=en
droxidopa, the English name Droxidopa, chemical name (_) _ (2S, 3R) _2_ amino _3_ hydroxy-3- (3,4-hydroxyphenyl) propionic acid, the formula is as follows:

· It is a synthetic (-) _ norepinephrine precursor amino acids by intestinal absorption and metabolism of norepinephrine, in patients with Parkinson’s disease is mainly used to improve gait stiffness and postural dizziness, and for Treatment of orthostatic hypotension drugs.
JP 09-031038 discloses droxidopa preparation, preparation process is as follows:

That structural formula I obtained in the presence of a copper catalyst structure formulas II, and then the open-loop, elimination of R and X groups of structure III of droxidopa.
JP05 – 239025 also provides a droxidopa preparation, the preparation process is as follows.

JP 59-055861 discloses a method of droxidopa preparation of optically active substances: Xi Qu racemic 多巴加 heat in water to form a saturated solution, the first addition to control the amount of optical after cooling Activity droxidopa as a seed, heat after the second cooling crystallize. JP 64-022849 discloses a method for purifying droxidopa: A mixture of alcohol solvents crude droxidopa was dissolved in water and inorganic acid, and then with an organic or inorganic base to neutralize, to precipitate crystals obtained purification. JP 59-055861 claims to obtain optically active by the method droxidopa, and JP64-022849 purification process is not considered to be heated, to avoid caused by heating droxidopa degradation.
Example 1
Preparation L- threo-3- (3,4-dihydroxyphenyl) serine 1: 300kg methanol successively and LN- carbonyl benzyloxy-3- (3,4-benzyloxyphenyl) serine Add 30kg 500L hydrogenation reactor, added dropwise 3mol / L hydrochloric acid to dissolve the solid, was added 5% palladium carbon 8kg, introducing hydrogen pressure was maintained at 0.02Mpa, the reaction temperature is controlled at 40 ± 5 ° C, 6 hours After discharge, the addition of concentrated hydrochloric acid 7kg and 0.3kg activated carbon and stirred for 20 minutes, filtration, the filtrate with 30% NaOH aqueous solution adjusted to pH 6-7, filtered crystallization two hours, that was (reaction formula below).

L- Su _3_ (3,4-light-phenyl) Preparation of silk atmosphere acid 2:
Dad was added to the reaction in ethanol 100kg, was added under stirring L- threo-3- (3,4-light-phenyl) -3-light-2 phthalamide imino acid, stirred at room temperature until The solid was dissolved clear, liquid solution of hydrazine hydrate at 40-45 ° C, after completion of the addition of hydrazine hydrate, the reaction was refluxed for 16 hours, cooled to below 30 ° C, concentrated hydrochloric acid was added dropwise 30kg, maintaining 30 ° C under stirring for I hour, Rejection filter cake washed once with aqueous hydrochloric acid, and the filtrate with 30% NaOH solution pH was adjusted to 6_7, filtered crystallization two hours, that was (reaction formula below).

CLIP

The condensation of 3,4-dimethoxybenzaldehyde (I ) with glycine (II) by means of KOH in hot methanol gives racemic threo-3- (3,4-dimethoxyphenyl) serine (III), which is acylated with N – (ethoxycarbonyl) phthalimide (IV) by means of Na2CO3 in water yielding the corresponding N-phthaloyl derivative (V) The reaction of (V) with AlCl3 and ethyl mercaptan in dichloromethane affords N-phthaloyl-3- (3,4-. dihydroxyphenyl) serine (VI), which is deprotected with hydrazine in refluxing ethanol to racemic threo-3- (3,4-dihydroxyphenyl) serine (VII). The resolution of the racemic form (VII) is performed through its benzyloxy derivative.
References
- Goldstein, DS (2006). “L-Dihydroxyphenylserine (L-DOPS): a norepinephrine prodrug”. Cardiovasc Drug Rev. 24 (3-4): 189–203. doi:10.1111/j.1527-3466.2006.00189.x. PMID 17214596.
- Mathias, Christopher J (2008). “L-dihydroxyphenylserine (Droxidopa) in the treatment of orthostatic hypotension”. Clin Auton Res. 18 (Supplement 1): 25–29.doi:10.1007/s10286-007-1005-z.
- “FDA grants accelerated approval to NORTHERA (droxidopa) for patients with symptomatic NOH”. news-medical.net. February 18, 2014.
- http://investor.lundbeck.com/releasedetail.cfm?ReleaseID=846443http://lundbeck.com/upload/us/files/pdf/2014_Releases/NORTHERA%20Availability%20press%20release%209.2.14.pdf
- Robertson, David (2008). “The pathophysiology and diagnosis of orthostatic hypotension”. Clin Auton Res. 18 (Supplement 1): 2–7. doi:10.1007/s10286-007-1004-0.
- Kaufmann, Horacio; Freeman, Roy; Biaggioni, Italo; Low, Phillip; Pedder, Simon; Hewitt, L. Arthur; Mauney, Joe; Feirtag, Michael; Mathias, Christopher J. (2014). “Droxidopa for neurogenic orthostatic hypotension: a randomized placebo-controlled Phase 3 trial”.Neurology. 83 (4): 328–335. doi:10.1212/WNL.0000000000000615. PMC 4115605
 .PMID 24944260.
.PMID 24944260. - Hauser, Robert A.; Isaacson, Stuart; Lisk, Jerome P.; Hewitt, L. Arthur; Rowse, Gerry (2015). “Droxidopa for the Short-Term Treatment of Symptomatic Neurogenic Orthostatic Hypotension in Parkinson’s Disease (nOH306B)”. Movement Disorders. 30 (5): 646–654.doi:10.1002/mds.26086.
- http://www.accessdata.fda.gov/drugsatfda_docs/label/2014/203202lbl.pdf
- http://www.accessdata.fda.gov/drugsatfda_docs/nda/2014/203202Orig1s000ChemR.pdf
| CN101657193A * | Mar 7, 2008 | Feb 24, 2010 | 切尔西治疗公司 | Droxidopa and pharmaceutical composition thereof for the treatment of fibromyalgia |
| EP0141613A2 * | Oct 24, 1984 | May 15, 1985 | Zaidan Hojin Biseibutsu Kagaku Kenkyu Kai | A process for producing an optically active 3-(3,4-dihydroxphenyl) serine and a proteced derivative thereof |
| JPS6422849A * | Title not available | |||
| JPS60160895A * | Title not available | |||
| WO2005085178A1 * | Mar 4, 2005 | Sep 15, 2005 | Estechpharma Co., Ltd. | Method of preparing optically active serine derivative |
| WO2006123678A1 * | May 17, 2006 | Nov 23, 2006 | Dainippon Sumitomo Pharma Co., Ltd. | Stable tablet containing droxidopa |
| WO2011001976A1 * | 29. Juni 2010 | 6. Jan. 2011 | Dainippon Sumitomo Pharma Co., Ltd. | Method for producing threo-3-(3,4-dihydroxyphenyl)-l-serine |
| US4319040 | 18. Juli 1980 | 9. März 1982 | Sumitomo Chemical Company, Limited | Process for the production of optically active threo-3-(3,4-dihydroxyphenyl)serine |
| US4480109 | 3. Jan. 1983 | 30. Okt. 1984 | Sumitomo Chemical Company, Limited | Process for producing threo-3-(3,4-dihydroxyphenyl)serine |
| US4562263 | 25. Mai 1984 | 31. Dez. 1985 | Sumitomo Chemical Company, Limited | Process for producing 3-(3,4-dihydroxyphenyl) serine |
| US20080015181 | 28. Juni 2007 | 17. Jan. 2008 | Chelsea Therapeutics, Inc. | Pharmaceutical Compositions Comprising Droxidopa |
| US20080221170 | 7. März 2008 | 11. Sept. 2008 | Chelsea Therapeutics, Inc. | Droxidopa and pharmaceutical composition thereof for the treatment of fibromyalgia |
| US20080227830 | 12. März 2008 | 18. Sept. 2008 | Chelsea Therapeutics, Inc. | Droxidopa and pharmaceutical composition thereof for the treatment of neurally mediated hypotension |
| US20090023705 | 7. Mai 2008 | 22. Jan. 2009 | Chelsea Therapeutics, Inc. | Droxidopa and pharmaceutical composition thereof for the treatment of mood disorders, sleep disorders or attention deficit disorders |
| Referenz | ||
|---|---|---|
| 1 | * | “Protection for the amino group” In: PETER G M WUTS; THEODORA W GREENE: “GREENE’S PROTECTIVE GROUPS IN ORGANIC SYNTHESIS,“, 2007, WILEY-Interscience,, HOBOKEN, NJ, USA, XP002685963, ISBN: 978-0-471-69754-1 page 696-700, 790-793, 799-802, the whole document |
| 2 | * | A. ARIFFIN ET AL.: “Suggested Improved Method for the Ing-Manske and Related Reactions for the Second Step of Gabriel Synthesis of Primary Amines“, SYNTHETIC COMMUNICATIONS: AN INTERNATIONAL JOURNAL FOR RAPID COMMUNICATION OF SYNTHETIC ORGANIC CHEMISTRY, vol. 34, no. 24, 2004, pages 4439-4445, XP002695538, Taylor & Francis Inc. ISSN: 0039-7911 |
| 3 | T. W. GREEN; P. G. M. WUTS: ‘Protective Groups in Organic Synthesis‘, vol. 583-584, 1999, WILEY-INTERSCIENCE pages 744 – 747 | |
FDA NEWS RELEASE
For Immediate Release: Feb. 18, 2014
FDA approves Northera to treat neurogenic orthostatic hypotension
The U.S. Food and Drug Administration today approved Northera capsules (droxidopa) for the treatment of neurogenic orthostatic hypotension (NOH). NOH is a rare, chronic and often debilitating drop in blood pressure upon standing that is associated with Parkinson’s disease, multiple-system atrophy, and pure autonomic failure.
Symptoms of NOH include dizziness, lightheadedness, blurred vision, fatigue and fainting when a person stands.
“People with neurogenic orthostatic hypotension are often severely limited in their ability to perform routine daily activities that require walking or standing,” said Norman Stockbridge, M.D., Ph.D, director of the Division of Cardiovascular and Renal Drugs in the FDA’s Center for Drug Evaluation and Research. “There are limited treatment options for people with NOH and we are committed to helping make safe and effective treatments available.”
The FDA is approving Northera under the accelerated approval program, which allows for approval of a drug to treat a serious disease based on clinical data showing that the drug has an effect on an intermediate clinical measure (in this case, short-term relief of dizziness) that is reasonably likely to predict the outcome of ultimate interest (relief of dizziness during chronic treatment). This program provides patient access to promising drugs while the company conducts post-approval clinical trials to verify the drug’s clinical benefit, which for this approval is a long-term effect on patient symptoms in NOH, a chronic disease.
Northera has a boxed warning to alert health care professionals and patients about the risk of increased blood pressure while lying down (supine hypertension), a common problem that affects people with primary autonomic failure and can cause stroke. It is essential that patients be reminded that they must sleep with their head and upper body elevated. Supine blood pressure should be monitored prior to and during treatment and more frequently when increasing doses.
The most common adverse events reported by clinical trial participants taking Northera were headache, dizziness, nausea, high blood pressure (hypertension) and fatigue.
The effectiveness of Northera was shown through two-weeks in two clinical trials in people with NOH. People taking Northera reported a decrease in dizziness, lightheadedness, feeling faint, or feeling as if they might black out compared to those taking an inactive pill (placebo). Durability of the improvement in patient symptoms beyond two weeks has not been demonstrated.
Northera received orphan-product designation from the FDA because it is intended to treat a rare disease or condition.
Northera is made by Charlotte-based Chelsea Therapeutics Inc.
For more information:
FDA: Approved Drugs
FDA: Drug Innovation
National Institute of Neurological Disorders and Stroke: Orthostatic Hypotension
|
|
| Systematic (IUPAC) name | |
|---|---|
|
(2S,3R)-2-Amino-3-(3,4-dihydroxyphenyl)-3-hydroxypropanoic acid
|
|
| Clinical data | |
| Trade names | Northera |
| Routes of administration |
Oral |
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Bioavailability | 90% |
| Metabolism | Hepatic |
| Biological half-life | 1.5 hours |
| Excretion | Renal |
| Identifiers | |
| CAS Number | 23651-95-8 |
| ATC code | C01CA27 (WHO) |
| PubChem | CID 6989215 |
| ChemSpider | 83927 |
| UNII | J7A92W69L7 |
| ChEBI | CHEBI:31524 |
| ChEMBL | CHEMBL2103827 |
| Synonyms | β,3-Dihydroxytyrosine |
| Chemical data | |
| Formula | C9H11NO5 |
| Molar mass | 213.18734 g/mol |
NORTHERA capsules contain droxidopa, which is a synthetic amino acidprecursor of norepinephrine, for oral administration. Chemically, droxidopa is (–)-threo-3-(3,4- Dihydroxyphenyl)-L-serine. It has the following structural formula:
 |
Droxidopa is an odorless, tasteless, white to off-white crystals or crystalline powder. It is slightly soluble in water, and practically insoluble in methanol, glacial acetic acid, ethanol, acetone, ether, and chloroform. It is soluble in dilute hydrochloric acid. It has a molecular weight of 213.19 and a molecular formula of C9H11NO5.
NORTHERA capsules also contain the following inactive ingredients: mannitol, corn starch, and magnesium stearate. The capsule shell is printed with black ink. The black inks contain shellac glaze, ethanol, iron oxide black, isopropyl alcohol, n-butyl alcohol, propylene glycol, and ammonium hydroxide. The capsule shell contains the following inactive ingredients: 100 mg – gelatin, titanium dioxide, FD&C Blue No. 2, black and red iron oxide; 200 mg – gelatin, titanium dioxide, FD&C Blue No. 2, black and yellow iron oxide; 300 mg – gelatin, titanium dioxide, FD&C Blue No. 1, FD&C Yellow No. 5 (tartrazine), and FD&C Red No. 40. NORTHERA capsules differ in size and color by strength
CLIP
//////////DROXIDOPA, Chelsea Therapeutics, orphan drug status, FDA 2015, 3916-18-5, NORTHERA, SUMITOMO, Antiparkinsonian
THESIS
http://ncl.csircentral.net/668/1/th1739.pdf
Review of Literature Literature search showed that there are only few reports available,42-44 which describe the synthesis of L-threo-DOPS (43) as detailed below. Kirk’s approach (2001)43 Kirk et. al have reported the synthesis of fluorinated analogue of L-threo-DOPS (49) starting from reaction of aldehyde 44 with isothiocyanate 45 in presence of LHMDS and Sn(OTf)2 to give ester 46 with d.r = 8.5:1. Subsequently, removal of chiral auxiliary with methoxymagnesium bromide followed by Boc protection of the thiocarbamate nitrogen was carried out. Thiocarbamate 46 was then converted to oxygen analogue 47 in 96% yield by treating it with Hg(OAc)2. This was followed by subsequent cleavage of 47 using Cs2CO3 and its Boc depotection gave the ester 48. Saponification of ester 48 followed by hydrogenation afforded fluoro analogue of L-threo-DOPS (49) (Scheme 13).
Sang-Ho’s approach (2007)44 Sang-Ho et. al have achieved an enzyme-catalyzed synthesis of L-threo-DOPS (43) by reacting in one-pot glycine, 3,4-dihydrobenzaldehyde, 2-mercaptoethanol, pyridoxal-5- phosphate solution, sodium sulfite and Triton X-100 in presence of E.coli at 15 °C

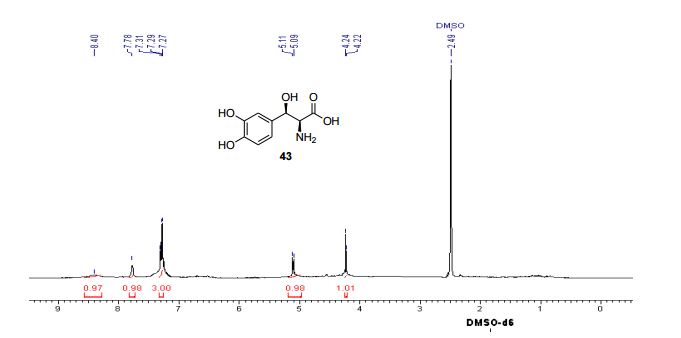
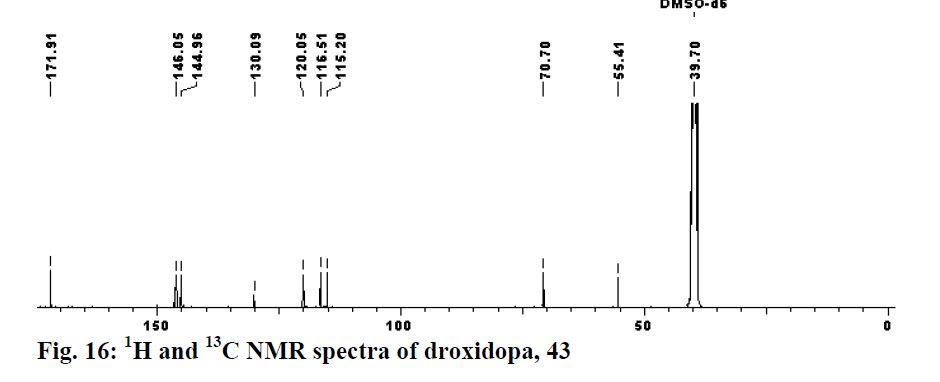
Yield: 94%; mp: 230-233 ºC; (lit.42 mp: 232-235 ºC); [α] 25 D: -38.7 (c 0.4, 1N aq. HCl); {lit.42 [α] 25 D: -39 (c 1, 1N HCl)}; IR (CHCl3, cm-1 ): 3018, 2399, 2366, 2345, 1652, 1519, 1215, 1018, 929, 756, 669; 1H NMR (200 MHz, DMSO-d6): δ 4.23 (d, J = 4.3 Hz, 1H), Droxidopa Chapter IV 226 5.10 (d, J = 3.8 Hz, 1H), 7.27-7.31 (m, 3H), 7.78 (br s, 1H), 8.40 (br s, 1H); 13C NMR (100 MHz, DMSO-d6): δ 55.41, 70.70, 115.21, 116.51, 120.06, 130.09, 144.96, 146.06, 171.91; Analysis: C9H11NO5 requires C, 50.70; H, 5.20; N, 6.57%; found: C, 50.99; H, 5.01; N, 6.33%
42 Hegedus, V. B.; Krasso, A. F.; Noack, K.; Zeller, P. Helv. Chim. Acta 1975, 58, 147.
[PDF]Download (3818Kb) – IR@NCL
ncl.csircentral.net/668/1/th1739.pdf
I here by declare that the thesis entitled “Enantioselective synthesis of bioactive molecules …. Section III Enantioselective synthesis of L-threo-DOPS (droxidopa).
