
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....

Droxidropa
FDA Deems Resubmission a Complete Response; PDUFA Date Set as
February 14, 2014
CHARLOTTE, N.C., Sept. 4, 2013 (GLOBE NEWSWIRE) — Chelsea Therapeutics International, Ltd. (Nasdaq:CHTP) today announced that the U.S. Food and Drug Administration (FDA) has acknowledged receipt of the New Drug Application (NDA) resubmission seeking approval to market NORTHERA(TM) (droxidopa), an orally active synthetic precursor of norepinephrine
read all at
http://www.pharmalive.com/chelsea-therapeutics-announces-fda-acceptance-of-northera-nda-resubmission
UPDATE………………….

DROXIDOPA
ORPHAN DRUG,
CAS 23651-95-8, 3916-18-5
ROTATION –FORM
|
(2S,3R)-2-amino-3-(3,4-dihydroxyphenyl)-3-hydroxypropanoic acid
|
Proprietary Name: NORTHERA
Dosage Form; Route of Administration: CAPSULE; ORAL
Strength: 100MG
Reference Listed Drug: No
TE Code:
Application Number: N203202
Product Number: 001
Approval Date: Feb 18, 2014
Applicant Holder Full Name: LUNDBECK NA LTD
Marketing Status: Prescription


Droxidopa (INN; trade name Northera; also known as L-DOPS, L–threo-dihydroxyphenylserine, L-threo-DOPS and SM-5688) is a synthetic amino acid precursor which acts as a prodrug to the neurotransmitter norepinephrine (noradrenaline).[1] Unlike norepinephrine, droxidopa is capable of crossing the protective blood–brain barrier (BBB).[1]
CLIP




REF http://www.accessdata.fda.gov/drugsatfda_docs/nda/2014/203202Orig1s000ChemR.pdf
Distribution
Droxidopa exhibits plasma protein binding of 75% at 100 ng/mL and 26% at 10,000 ng/mL with an apparent volume of distribution of about 200 L.
Droxidopa starting dose is 100mg three times daily (which can be titrated to a maximum of 600 mg three times daily). One dose should be taken in late afternoon at least 3 hours prior to bedtime to reduce the potential for supine hypertension during sleep.
Indications
- Neurogenic orthostatic hypotension (NOH) dopamine beta hydrolase deficiency,[2] as well as NOH associated with multiple system atrophy (MSA), familial amyloid polyneuropathy (FAP), pure autonomic failure (PAF).
- Intradialytic hypotension (IDH) or hemodialysis-induced hypotension.
- Freezing of gait in Parkinson’s disease (off-label)
History
Droxidopa was developed by Sumitomo Pharmaceuticals for the treatment of hypotension, including NOH,[2] and NOH associated with various disorders such as MSA, FAP, and PD, as well as IDH. The drug has been used in Japan and some surrounding Asianareas for these indications since 1989. Following a merge with Dainippon Pharmaceuticals in 2006, Dainippon Sumitomo Pharmalicensed droxidopa to Chelsea Therapeutics to develop and market it worldwide except in Japan, Korea, China, and Taiwan. In February 2014, the Food and Drug Administration approved droxidopa for the treatment of symptomatic neurogenic orthostatic hypotension.[3]
Clinical trials
Chelsea Therapeutics obtained orphan drug status (ODS) for droxidopa in the U.S. for NOH, and that of which associated with PD, PAF, and MSA. In 2014, Chelsea Therapeutics was acquired by Lundbeck along with the rights to droxidopa which was launched in the US in Sept 2014.[4]
REGULATORY
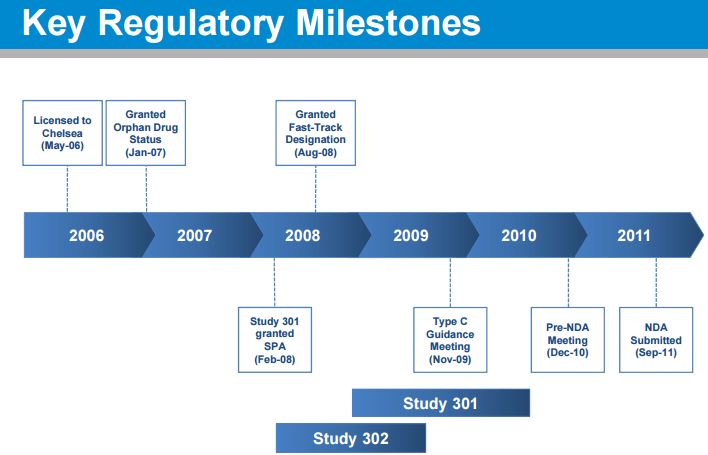
CLICK ON IMAGE TO VIEW
FDA agreement on overall development program (Sep 2007)
• FDA agreement on Study 301 design under a Special Protocol Assessment (Feb 2008) – Included agreement: length of patient exposure was adequate for the safety evaluation
• FDA agreement on changing primary endpoint of Study 301 while it was ongoing and prior to any unblinding (Nov 2009) – From dizziness to the OHQ – SPA remained intact
• FDA agrees to NDA package (Dec 2010) – Studies 301, 302, 303, 304 and 305 – Renal safety study conducted post-marketing
• FDA accepts droxidopa NDA and grants priority review (Sep 2011)
Pharmacology
Droxidopa is a prodrug of norepinephrine used to increase the concentrations of these neurotransmitters in the body and brain.[1][What, if any, are the other neurotransmitters droxidopa increases concentrations of? “These neurotransmitters” implies multiple(see above)] It ismetabolized by aromatic L-amino acid decarboxylase (AAAD), also known as DOPA decarboxylase (DDC). Patients with NOH have depleted levels of norepinephrine which leads to decreased blood pressure or hypotension upon orthostatic challenge.[5] Droxidopa works by increasing the levels of norepinephrine in the peripheral nervous system (PNS), thus enabling the body to maintain blood flow upon and while standing.
Droxidopa can also cross the blood–brain barrier (BBB) where it is converted to norepinephrine from within the brain.[1] Increased levels of norepinephrine in the central nervous system (CNS) may be beneficial to patients in a wide range of indications. Droxidopa can be coupled with a peripheral aromatic L-amino acid decarboxylase inhibitor (AAADI) orDOPA decarboxylase inhibitor (DDC) such as carbidopa (Lodosyn) to increase central norepinephrine concentrations while minimizing increases of peripheral levels.
Side effects
With over 20 years on the market, droxidopa has proven to have few side effects of which most are mild. The most common side effects reported in clinical trials include headache, dizziness nausea, hypertension and fatigue.[6][7][8][8]
L-threo-dihydroxyphenylserine, also known as droxidopa, L-threo-DOPS, or L-DOPS, is an orally active synthetic precursor of norepinephrine. Droxidopa thus replenishes depleted norepinephrine, allowing for re-uptake of norepinephrine into peripheral nervous system neurons. This reuptake, in turn, stimulates receptors for vasoconstriction, providing physiological improvement in symptomatic neurogenic orthostatic hypotension patients. It has also shown efficacy in other diseases, such as Parkinson’s disease and depression.
Droxidopa has been used in Japan for many years for the treatment of orthostatic hypotension. It was originally approved in 1989 for the treatment of frozen gait or dizziness associated with Parkinson’s disease and for the treatment of orthostatic hypotension, syncope or dizziness associated with Shy-Drager syndrome and Familial Amyloidotic Polyneuropathy.
Marketing approval was later expanded to include treatment of vertigo, dizziness and weakness associated with orthostatic hypotension in hemodialysis patients.
The preparation of droxidopa generally involves a multi-step synthesis. Typically, one or more of the necessary steps in the synthesis requires that reactive sites other than that site targeted for reaction are temporarily protected. Thus, the synthesis of droxidopa typically comprises at least one protecting and associated deprotecting step. For example, the catechol moiety, the amine moiety, and/or the carboyxyl moiety may require protection and subsequent deprotection, depending upon the synthetic route and the reagents used in the preparation of droxidopa.
U.S. Patent Nos. 4,319,040 and 4,480,109 to Ohashi et al. describe processes for the preparation of optically active D- and L- threo-DOPS by optically resolving a racemic mixture of threo-2-(3,4-methylenedioxyphenyl)-N-carbobenzyloxyserine or threo-2-(3,4-dibenzyloxy-phenyl)- N-carbobenzyloxyserine, respectively. Following optical resolution of these racemic mixtures to give the desired L-enantiomer, the methylene or benzyl groups must be removed from the catechol moiety and the carbobenzyloxy (CBZ) group must be removed from the amine group to give droxidopa. The methylene group can be readily removed by reaction with a Lewis acid {e.g., aluminum chloride). The CBZ group (and the benzyl catechol protecting groups, where applicable) is removed from the amine by hydrogenolysis to give the desired compound. The hydrogenolysis step is noted to be carried out by treating the optically resolved salt with hydrogen in the presence of a catalyst, e.g., palladium, platinum, nickel, or the like.
However, for large-scale production of pharmaceutical compounds, hydrogenolysis may not be desirable. For example, hydrogenolysis requires expensive, specialized equipment, which represents a large capital investment. Labor costs are also high, as the process requires careful handling and disposal of certain compounds (e.g., the pyrophoric catalyst). Further, due to the hazards associated with both the reagents and the high pressure system required for hydrogenolysis, it is desirable to avoid synthetic methods that require hydrogenolysis.
In an alternative method for the production of droxidopa, taught by U.S. Patent No.
4,562,263 to Ohashi et al, hydrogenation is not required. In this process, the amine group is protected via a phthaloyl group. Following optical resolution, the phthaloyl group is removed from the droxidopa precursor by hydrazine.
However, hydrazine is known to be genotoxic and has been classified by the EPA as a
Group B2 probable human carcinogen. Thus, it is desirable to remove even trace amounts of hydrazine from pharmaceutical compounds. In practice, the method described in the ‘263 patent suffers from the inability to remove 100% of the hydrazine from the final product. Thus, there is some level of contamination by hydrazine using this method. The Food and Drug Administration has established a maximum genotoxic impurity level of 1.5 micrograms per day. Therefore, based on the maximum daily dose of droxidopa (1.8 g), the maximum allowable hydrazine level therein is 0.8 ppm. Accordingly, it would be advantageous to find a new synthetic route for the preparation of droxidopa that avoids the use of hydrogenolysis and also avoids the use of hydrazine
PATENT
https://www.google.ch/patents/WO2013142093A1?cl=en
The synthetic route for the preparation of droxidopa comprises the following steps: a) converting piperonal to 2-amino-3-(benzo-l,3-diox-5-yl)-3-hydroxypropanoic acid

c) optical resolution and separation of the desired isomer



Experimental Section
Example 1 : Screening of Deprotection Strategies for Phthaloyl Group

Example 2: Exemplary Synthesis of Droxidopa
The synthesis of droxidopa according to the methods provided herein can be conducted as a continuous process or can be conducted in a series of individual steps. Both processes are intended to be encompassed by the present disclosure.
Synthesis of N-carbomethoxy phthalimide

3-Methoxy phthalimide 1 (120 kg) is added to a vessel containing dimethylformamide (420 L) and stirred (95 ± 10 RPM) at 25 – 30 °C for 30 min. The contents are cooled to 18 – 20 °C and triethylamine (124 L) is added. The contents are further cooled to -10 °C to -5 °C and
methylchloroformate 2 (85 kg) is added. The reaction temperature is maintained in the range of -10 °C to 0 °C to control the exothermicity during the addition of methylchloroformate. The temperature of the mixture is maintained at 0 – 5 °C for 1 h after the addition of
methylchloroformate .
The reaction mixture is then heated to 25 – 30 °C for 1 h. An in-process sample is taken to confirm a phthalimide content limit <2.5%. The mixture is sampled again to confirm a phthalimide content <0.5%. The mixture is transferred to another reactor, cooled to 0 – 5 °C, and the reaction is quenched with the addition of demineralized water (1260 ± 10 L) at a temperature of 10 ± 5 °C. The mixture is then heated at 25 – 30 °C for 1 h.
The material is centrifuged for 2 h and the wet cake is washed three times with
demineralized water (360 L). The wet cake is dried at a temperature of 55 – 60 °C and a sample is taken after 12 h of drying to confirm water content <1.0% w/w.
Expected yield of N-carbomethoxy phthalimide (3): 144-158 kg. This material is not isolated and is used directly in the next step.
Synthesis of 2-amino-3-(benzo-l ,3-dioxol-5-yl)-3-hydroxypropanoic acid

Piperonal 4 (229 ± 1 kg) is added to toluene (310 ± 5 L) in a reactor and the mixture is stirred (85 – 95 RPM) until a clear solution is obtained (approximately 30 min). The piperonal solution is transferred to a vessel for later use. Methanol (415 ± 5 L) is added to the reactor followed by the addition of potassium hydroxide (85 kg). The mixture is stirred for approximately 30 min at 25 – 30 °C to provide a clear solution. The potassium hydroxide solution is cooled to 20 – 25 °C, and then glycine 5 (52 ± 1 kg) and toluene (310 ± 5 L) are added while stirring at 20 – 25 °C. The contents of the reactor are cooled to 15 – 20 °C. The solution of piperonal in toluene is slowly added to the reactor while maintaining the temperature at 15 – 20 °C. The reactor temperature is increased to 20 – 25 °C and maintained for 18 h. An in-process sample is taken to determine glycine content by TLC (limit <5.0%).
The reaction mass is transferred to another reactor, the temperature is increased to 40 °C, and the solvents (toluene and methanol) are distilled off under vacuum until the mixture becomes thick. Additional toluene (210 ± 5L) is added to the reaction mass three times and distilled out for complete removal of methanol and toluene. The reaction mixture is kept under vacuum at 40 °C. After 3 h, the reaction mixture is cooled to 18 – 22 °C and a dilute hydrochloric acid solution (230 ± 5L hydrochloric acid and 1145 ± 10 L demineralized water) is added and mixed for 30 min.
The mixture is allowed to settle for 30 min to separate into organic and aqueous layers. The aqueous layer is washed with toluene (310 ± 5 L) and separated. Glacial acetic acid (218 ± 2 kg) is added to the washed aqueous layer at 20-25 °C. Caustic solution (580 ± 5 L DM Water and 200 ± 1 kg caustic flakes) is slowly added into the reaction mass to bring the pH 5.0 to 5.1 while maintaining the temperature at 25 – 30 °C. The pH of the mixture is brought to 5.45 – 5.50 at 25 – 30 °C, while stirring for 30 min. The mixture is centrifuged for 8 h 30 min to 9 h and the resulting wet cake is washed with demineralized water (50 ± 5 L). The cake is dried at 50 – 55 °C under vacuum, and a sample is taken after 12 h to confirm that water content is <10% w/w. The purity is analyzed by HPLC (limit < 10%).
Expected yield of 2-amino-3-(benzo-l ,3-dioxol-5-yl)-3-hydroxypropanoic acid (6):
135 – 145 kg.
Synthesis of 2-phthalimido-3-hydroxy-3-(3,4-methylenedioxyphenvnpropionic acid

Intermediate 6 (140 kg) is added to a reactor containing demineralized water (1 120 L) and stirred (85-95 RPM) for 10 min at 20-25 °C. The contents are cooled to 15-20 °C and compound 3 (140 kg) is added followed by a sodium carbonate solution (63.5-68.3 kg sodium carbonate in 189-203 L demineralized water) within 45-60 min. The mixture is heated to 30-35 °C and held for 90 min. An in-process sample is taken to measure for Stage II (<2.5%) and Stage-I intermediate (<2.5 %). After acceptance criteria are met, the mixture is cooled to 15-20 °C. A dilute sulfuric acid solution (134 kg sulfuric acid in 1120 L demineralized water) at 15-20 °C is added to the mixture to bring the pH to 1.0-2.0. The mixture is maintained at this temperature and pH for 30 min, and then the mixture is heated to 20-25 °C for 2 h.
The mixture is centrifuged for 9 h and the resulting wet cake is washed twice with 518 L of demineralized water. The wet cake is removed from the centrifuge, washed in a reactor containing demineralized water (2590 L), and stirred for 1 h at 25-30 °C. The material is centrifuged for 9 h and the wet cake is washed twice with demineralized water (518 L). The final wet cake is dried at 45-50 °C under vacuum until water content is <1.0% w/w. Intermediate (6) output is considered as standard input and a mean of 140 kg is taken for all inputs.
Expected yield of 2-Phthalimido-3-hydroxy-3-(3,4-methylenedioxyphenyl)propionic acid (7): 187 – 208 kg.
Synthesis of L-threo (N-phthaloyl-3-(3,4-methylenedioxyphenyl)serine) norephedrine salt

L-Norephedrine 8 (89 kg) is added to a reactor containing methanol (296 L) and stirring (45-50 RPM) is started. The mixture is maintained at 25 – 30 °C for 15 – 20 min, and then transferred into a vessel for later use.
2-Phthalimido-3-hydroxy-3-(3,4-methylenedioxyphenyl)propionic acid 7 (197.5 kg) is added to a reactor containing methanol (395 L). The material is stirred for 15 – 20 min at 25 – 30 °C. The L-norephedrine solution is added and mixed for 3 h. If precipitation is not observed within 30 min of adding the L-norephedrine solution, it is seeded with L-threo(N-phthaloyl-3-(3,4- methylenedioxyphenyl) serine (approximately 50 g). After 3 h of mixing, the mixture is cooled to 10 – 15 °C and maintained for 1 h. An in-process sample is taken to check for purity by HPLC (>99.0% a/a). The mixture is centrifuged for 1 h to 1 h 30 min and the wet cake is washed with methanol (49 L) followed by isopropyl alcohol (197.5 L). The wet cake is checked for purity. If purity is <99% a/a, the wet cake is washed with a prechilled solution of methanol (197.5 L) followed by isopropyl alcohol (99 L). After achieving the required purity level, as measured by HPLC, the wet cake is removed from the centrifuge. The cake is dried at 45 – 50 °C until loss on drying <1.0% w/w.
Expected yield of L-threo (N-phthalpyl-3-(3,4-methylenedioxyphenyl)serine) norephedrine (9) salt: 85-99 kg.
Synthesis of L-threo flSi-phthaloyl-S-CS^-methylenedioxyphenvDserine)

Demineralized water (552 L) is added to a reactor and cooled to 10 – 15 °C. Sulfuric acid (20 kg) is added while maintaining the temperature below 30 °C and stirring for 15 – 20 min. The solution is cooled to 15 – 20 °C and 9 (92 kg) is slowly added while stirring and maintaining temperature. The solution is heated to 45 – 50 °C for 6 h, cooled to 25 – 30 °C, and held for 1 h. The pH is checked to confirm the solution is <2.0.
The mixture is centrifuged for 1 h and the wet cake is washed two times with demineralized water (138 L). The wet cake is removed and added to a reactor containing demineralized water (460 L). The temperature is maintained at 25 – 30 °C and stirred for 1 h. The material is centrifuged for 30 min and the wet cake is washed two times with demineralized water (138 L). The material is collected and placed into preweighed containers.
Expected yield of L-threo(N-phthaloyl-3-(3,4-methylenedioxyphenyl)serine) (10): 60-64 kg.
Synthesis of L-threo(N-phthaloyl-3-(3 ,4-dihydroxyphenyl serine)

Compound 10 (62 kg) wet cake is added to a reactor containing methylene chloride (1240 L) and stirred for 10 min. The mixture is heated to remove methylene chloride and water under azeotropic reflux. After methylene chloride (1550 L) is removed and no water remains in the distillate, the mixture is cooled to 25 – 30 °C. An in-process sample is taken to determine water content (limit <0.1 %).
Methylene chloride (186 L) is added to another reactor at 25-30 °C. An in-process sample is taken to check for water content (limit <0.2% w/w). Aluminum chloride (81 kg) is added and the contents are stirred at 25 – 30 °C for 10 – 15 min. The mixture is cooled to 10 – 15 °C and octanethiol (78 kg) is added. The mixture is cooled to -20 to -10 °C.
The slurry of 10 in methylene chloride controlled at -20 to -7 °C is added to the stirred mixture that is temperature controlled at -15 to -10 °C for 20 – 30 min. The mixture is heated to 10-15 °C for 1.5-2.5 h. An in-process sample is taken to determine 10 content (limit <3.5%). The mixture is further cooled to -20 to -10 °C and then transferred to another reactor containing oxalic acid (62 kg), methylene chloride (186 L), and demineralized water (744 L) while maintaining the temperature below -3 °C to quench the reaction. The quenched material is slowly heated to 25 – 30 °C and maintained at this temperature for 12 h. Methylene chloride is distilled out at 25 – 30 °C under vacuum until the mixture volume is reduced to 1364 L.
The mixture is centrifuged for 3 h and the wet cake is washed with demineralized water (62 L). The wet cake is added to a reactor containing oxalic acid (2.5 kg) and demineralized water (248 L) and the contents are stirred at 25-30 °C for 2 h to obtain a clear solution. The material is centrifuged for 1 h 15 min to 1 h 30 min and the wet cake is washed twice with demineralized water (186 L). The wet cake is added to a reactor containing demineralized water (248 L) at 25 – 30 °C and the contents are stirred for 2 h. The material is centrifuged for 1 h 30 min to 2 h and the wet cake is washed twice with demineralized water (186 L). The material is collected and placed into preweighed containers.
Expected yield of L-threo(N-phthaloyl-3-(3,4-dihydroxyphenyl)serine (11): 40-50 kg. Synthesis of L-threo (3,4-dihydroxyphenvP)serine

Methanol (360 L) is added to a reactor and cooled to 20 – 25 °C. Compound 11 (45 kg) is added to the reactor while stirring at 25 – 30 °C for 15 – 20 min. Demineralized water (225 L) and sodium bicarbonate (17 kg) are added to another reactor and cooled to 20 – 25 °C. Hydroxylamine hydrochloride (14 kg) is added and mixed for 15 – 20 min at 20 – 25 °C to obtain a clear solution. The solution of 11 in methanol is transferred through a sparkler filter into a reactor. The hydroxylamine and sodium bicarbonate solution is added to the reactor while maintaining the temperature at 25 – 30 °C. The reaction mixture is heated to 65 – 70 °C and refluxed for 16 h. An in-process sample is taken to determine 11 content (limit <3%). The material is cooled to 25-30 °C with mixing for 2 h.
The material is centrifuged for 1 h and the wet cake is washed three times with methanol (23 L). The wet cake is dried at 40 – 45 °C until water content is <1.0% w/w.
Expected yield of L-threo(3,4-dihydroxyphenyl)serine (12): 20-24 kg. Synthesis of L-threo(3,4-dihvdroxyphenyl)serine hydrochloride

L-threo (3,4-dihydroxyphenyl)serine 12 (22 kg) material is added to a reactor containing demineralized water (55 L) and stirred for 15-30 min. The material is cooled to 20-25 °C and concentrated hydrochloric acid (13 L) is added to form L-threo(3,4-dihydroxyphenyl)serine hydrochloride) (13). The mixture is stirred for 30^15 min until a white thick suspension is observed. The mixture is stirred for an additional 2.0 h ± 15 min. Isopropyl alcohol (132 L) is slowly added and the mixture is stirred for 5 hr ± 15 min. The mixture is cooled to 15-20 °C and stirred for 30^5 min. The mixture is centrifuged for 30 min and the wet cake is washed twice with chilled isopropyl alcohol (22 L) at 15-20 °C. The material is unloaded from the centrifuge and a sample is taken to check the individual impurity by HPLC (limit <0.05%) and purity by HPLC (limit >99.0%).
Reprocessing: If the individual impurity by HPLC does not meet the limit <0.05%, compound 13 is reprocessed by adding the material to a reactor containing demineralized water (28 L) and stirring for 15 – 30 min. Concentrated hydrochloric acid (3 L) is added at 20-25 °C and mixing is continued for 15 – 30 min. Continue mixing for 2 h ± 15 min at the same temperature. Isopropyl alcohol (74 L) is added over a period of 2 – 3 h at 25 – 30 °C. Mixing is continued at 25 – 30 °C for 5 h ± 15 min followed by cooling to 15 – 20 °C and mixing for 30 – 45 min. The mixture is centrifuged for 30 min and washed twice with chilled isopropyl alcohol (22 L) and checked for the individual impurity by HPLC (limit <0.05%).
Expected yield of L-threo(3,4-dihydroxyphenyl)serine hydrochloride) (13): 19-20 kg. Synthesis of L-threo(3,4-dihvdroxyphenyl)serine

Compound 13 (19.5 kg) is added to a reactor containing demineralized water (195 L) while stirring at 25 – 30 °C. Concentrated hydrochloric acid (6 L) is added and mixed for 25 – 30 min. For complete dissolution, the contents can be mixed for another 15 – 20 min. Activated carbon (1 kg) and celite (0.2 kg) are added and mixed for 30 – 40 min. The mixture is filtered through a sparkler filter and the filter is washed with demineralized water (1 X L). The filtrate is transferred to another reactor. A solution containing triethylamine (14 kg) and methanol (41 L) is slowly added to the reaction mass (reactor) while mixing. The pH of the filtrate is adjusted to 7.0 – 7.25 over a period of 3 h at 25 – 30 °C. The contents are stirred for 20-30 min. An in-process sample is taken to confirm the pH is 7.0 – 7.25. The mixture is stirred for 3 h. The mixture is centrifuged for 1 h and the wet cake is washed twice with demineralized water (19.5 L). The wet cake is removed from the centrifuge and kept for a slurry wash. The wet cake is added to a reactor containing methanol (58.5 L) while stirring at 25 – 30 °C for 30 – 40 min. The material is centrifuged for 1 h and the wet cake is washed with methanol (19.5 L). The wet cake is unloaded from the centrifuge and retained for water washing.
The wet cake is added to a reactor containing demineralized water (39 L) while stirring for 30 – 40 min. The material is centrifuged for 10 min and the wet cake is washed twice with methanol (19.5 L). The wet cake is unloaded and a sample is taken to check the chloride content (<200 ppm). The wet cake is dried at 40 – 45 °C until the water content is <0.1 % w/w. A sample is taken after 16 h of drying to confirm loss on drying is <0.1% w/w. The dry material is sieved through a sifter (400 micron) and packed. A sample is taken for quality control testing.
Expected yield of L-threo(3,4-dihydroxyphenyl)serine: 14-15 kg.
Scheme 1: Overview of Droxidopa Synthesis
a) converting piperonal to 2-amino-3-(benzo-l,3-diox-5-yl)-3-hydroxypropanoic acid

d) removal of the catechol protecting group


PATENT
https://www.google.com/patents/CN103086906A?cl=en
droxidopa, the English name Droxidopa, chemical name (_) _ (2S, 3R) _2_ amino _3_ hydroxy-3- (3,4-hydroxyphenyl) propionic acid, the formula is as follows:

· It is a synthetic (-) _ norepinephrine precursor amino acids by intestinal absorption and metabolism of norepinephrine, in patients with Parkinson’s disease is mainly used to improve gait stiffness and postural dizziness, and for Treatment of orthostatic hypotension drugs.
JP 09-031038 discloses droxidopa preparation, preparation process is as follows:

That structural formula I obtained in the presence of a copper catalyst structure formulas II, and then the open-loop, elimination of R and X groups of structure III of droxidopa.
JP05 – 239025 also provides a droxidopa preparation, the preparation process is as follows.

JP 59-055861 discloses a method of droxidopa preparation of optically active substances: Xi Qu racemic 多巴加 heat in water to form a saturated solution, the first addition to control the amount of optical after cooling Activity droxidopa as a seed, heat after the second cooling crystallize. JP 64-022849 discloses a method for purifying droxidopa: A mixture of alcohol solvents crude droxidopa was dissolved in water and inorganic acid, and then with an organic or inorganic base to neutralize, to precipitate crystals obtained purification. JP 59-055861 claims to obtain optically active by the method droxidopa, and JP64-022849 purification process is not considered to be heated, to avoid caused by heating droxidopa degradation.
Example 1
Preparation L- threo-3- (3,4-dihydroxyphenyl) serine 1: 300kg methanol successively and LN- carbonyl benzyloxy-3- (3,4-benzyloxyphenyl) serine Add 30kg 500L hydrogenation reactor, added dropwise 3mol / L hydrochloric acid to dissolve the solid, was added 5% palladium carbon 8kg, introducing hydrogen pressure was maintained at 0.02Mpa, the reaction temperature is controlled at 40 ± 5 ° C, 6 hours After discharge, the addition of concentrated hydrochloric acid 7kg and 0.3kg activated carbon and stirred for 20 minutes, filtration, the filtrate with 30% NaOH aqueous solution adjusted to pH 6-7, filtered crystallization two hours, that was (reaction formula below).

L- Su _3_ (3,4-light-phenyl) Preparation of silk atmosphere acid 2:
Dad was added to the reaction in ethanol 100kg, was added under stirring L- threo-3- (3,4-light-phenyl) -3-light-2 phthalamide imino acid, stirred at room temperature until The solid was dissolved clear, liquid solution of hydrazine hydrate at 40-45 ° C, after completion of the addition of hydrazine hydrate, the reaction was refluxed for 16 hours, cooled to below 30 ° C, concentrated hydrochloric acid was added dropwise 30kg, maintaining 30 ° C under stirring for I hour, Rejection filter cake washed once with aqueous hydrochloric acid, and the filtrate with 30% NaOH solution pH was adjusted to 6_7, filtered crystallization two hours, that was (reaction formula below).

CLIP

The condensation of 3,4-dimethoxybenzaldehyde (I ) with glycine (II) by means of KOH in hot methanol gives racemic threo-3- (3,4-dimethoxyphenyl) serine (III), which is acylated with N – (ethoxycarbonyl) phthalimide (IV) by means of Na2CO3 in water yielding the corresponding N-phthaloyl derivative (V) The reaction of (V) with AlCl3 and ethyl mercaptan in dichloromethane affords N-phthaloyl-3- (3,4-. dihydroxyphenyl) serine (VI), which is deprotected with hydrazine in refluxing ethanol to racemic threo-3- (3,4-dihydroxyphenyl) serine (VII). The resolution of the racemic form (VII) is performed through its benzyloxy derivative.
References
- Goldstein, DS (2006). “L-Dihydroxyphenylserine (L-DOPS): a norepinephrine prodrug”. Cardiovasc Drug Rev. 24 (3-4): 189–203. doi:10.1111/j.1527-3466.2006.00189.x. PMID 17214596.
- Mathias, Christopher J (2008). “L-dihydroxyphenylserine (Droxidopa) in the treatment of orthostatic hypotension”. Clin Auton Res. 18 (Supplement 1): 25–29.doi:10.1007/s10286-007-1005-z.
- “FDA grants accelerated approval to NORTHERA (droxidopa) for patients with symptomatic NOH”. news-medical.net. February 18, 2014.
- http://investor.lundbeck.com/releasedetail.cfm?ReleaseID=846443http://lundbeck.com/upload/us/files/pdf/2014_Releases/NORTHERA%20Availability%20press%20release%209.2.14.pdf
- Robertson, David (2008). “The pathophysiology and diagnosis of orthostatic hypotension”. Clin Auton Res. 18 (Supplement 1): 2–7. doi:10.1007/s10286-007-1004-0.
- Kaufmann, Horacio; Freeman, Roy; Biaggioni, Italo; Low, Phillip; Pedder, Simon; Hewitt, L. Arthur; Mauney, Joe; Feirtag, Michael; Mathias, Christopher J. (2014). “Droxidopa for neurogenic orthostatic hypotension: a randomized placebo-controlled Phase 3 trial”.Neurology. 83 (4): 328–335. doi:10.1212/WNL.0000000000000615. PMC 4115605
 .PMID 24944260.
.PMID 24944260. - Hauser, Robert A.; Isaacson, Stuart; Lisk, Jerome P.; Hewitt, L. Arthur; Rowse, Gerry (2015). “Droxidopa for the Short-Term Treatment of Symptomatic Neurogenic Orthostatic Hypotension in Parkinson’s Disease (nOH306B)”. Movement Disorders. 30 (5): 646–654.doi:10.1002/mds.26086.
- http://www.accessdata.fda.gov/drugsatfda_docs/label/2014/203202lbl.pdf
- http://www.accessdata.fda.gov/drugsatfda_docs/nda/2014/203202Orig1s000ChemR.pdf
| CN101657193A * | Mar 7, 2008 | Feb 24, 2010 | 切尔西治疗公司 | Droxidopa and pharmaceutical composition thereof for the treatment of fibromyalgia |
| EP0141613A2 * | Oct 24, 1984 | May 15, 1985 | Zaidan Hojin Biseibutsu Kagaku Kenkyu Kai | A process for producing an optically active 3-(3,4-dihydroxphenyl) serine and a proteced derivative thereof |
| JPS6422849A * | Title not available | |||
| JPS60160895A * | Title not available | |||
| WO2005085178A1 * | Mar 4, 2005 | Sep 15, 2005 | Estechpharma Co., Ltd. | Method of preparing optically active serine derivative |
| WO2006123678A1 * | May 17, 2006 | Nov 23, 2006 | Dainippon Sumitomo Pharma Co., Ltd. | Stable tablet containing droxidopa |
| WO2011001976A1 * | 29. Juni 2010 | 6. Jan. 2011 | Dainippon Sumitomo Pharma Co., Ltd. | Method for producing threo-3-(3,4-dihydroxyphenyl)-l-serine |
| US4319040 | 18. Juli 1980 | 9. März 1982 | Sumitomo Chemical Company, Limited | Process for the production of optically active threo-3-(3,4-dihydroxyphenyl)serine |
| US4480109 | 3. Jan. 1983 | 30. Okt. 1984 | Sumitomo Chemical Company, Limited | Process for producing threo-3-(3,4-dihydroxyphenyl)serine |
| US4562263 | 25. Mai 1984 | 31. Dez. 1985 | Sumitomo Chemical Company, Limited | Process for producing 3-(3,4-dihydroxyphenyl) serine |
| US20080015181 | 28. Juni 2007 | 17. Jan. 2008 | Chelsea Therapeutics, Inc. | Pharmaceutical Compositions Comprising Droxidopa |
| US20080221170 | 7. März 2008 | 11. Sept. 2008 | Chelsea Therapeutics, Inc. | Droxidopa and pharmaceutical composition thereof for the treatment of fibromyalgia |
| US20080227830 | 12. März 2008 | 18. Sept. 2008 | Chelsea Therapeutics, Inc. | Droxidopa and pharmaceutical composition thereof for the treatment of neurally mediated hypotension |
| US20090023705 | 7. Mai 2008 | 22. Jan. 2009 | Chelsea Therapeutics, Inc. | Droxidopa and pharmaceutical composition thereof for the treatment of mood disorders, sleep disorders or attention deficit disorders |
| Referenz | ||
|---|---|---|
| 1 | * | “Protection for the amino group” In: PETER G M WUTS; THEODORA W GREENE: “GREENE’S PROTECTIVE GROUPS IN ORGANIC SYNTHESIS,“, 2007, WILEY-Interscience,, HOBOKEN, NJ, USA, XP002685963, ISBN: 978-0-471-69754-1 page 696-700, 790-793, 799-802, the whole document |
| 2 | * | A. ARIFFIN ET AL.: “Suggested Improved Method for the Ing-Manske and Related Reactions for the Second Step of Gabriel Synthesis of Primary Amines“, SYNTHETIC COMMUNICATIONS: AN INTERNATIONAL JOURNAL FOR RAPID COMMUNICATION OF SYNTHETIC ORGANIC CHEMISTRY, vol. 34, no. 24, 2004, pages 4439-4445, XP002695538, Taylor & Francis Inc. ISSN: 0039-7911 |
| 3 | T. W. GREEN; P. G. M. WUTS: ‘Protective Groups in Organic Synthesis‘, vol. 583-584, 1999, WILEY-INTERSCIENCE pages 744 – 747 | |
FDA NEWS RELEASE
For Immediate Release: Feb. 18, 2014
FDA approves Northera to treat neurogenic orthostatic hypotension
The U.S. Food and Drug Administration today approved Northera capsules (droxidopa) for the treatment of neurogenic orthostatic hypotension (NOH). NOH is a rare, chronic and often debilitating drop in blood pressure upon standing that is associated with Parkinson’s disease, multiple-system atrophy, and pure autonomic failure.
Symptoms of NOH include dizziness, lightheadedness, blurred vision, fatigue and fainting when a person stands.
“People with neurogenic orthostatic hypotension are often severely limited in their ability to perform routine daily activities that require walking or standing,” said Norman Stockbridge, M.D., Ph.D, director of the Division of Cardiovascular and Renal Drugs in the FDA’s Center for Drug Evaluation and Research. “There are limited treatment options for people with NOH and we are committed to helping make safe and effective treatments available.”
The FDA is approving Northera under the accelerated approval program, which allows for approval of a drug to treat a serious disease based on clinical data showing that the drug has an effect on an intermediate clinical measure (in this case, short-term relief of dizziness) that is reasonably likely to predict the outcome of ultimate interest (relief of dizziness during chronic treatment). This program provides patient access to promising drugs while the company conducts post-approval clinical trials to verify the drug’s clinical benefit, which for this approval is a long-term effect on patient symptoms in NOH, a chronic disease.
Northera has a boxed warning to alert health care professionals and patients about the risk of increased blood pressure while lying down (supine hypertension), a common problem that affects people with primary autonomic failure and can cause stroke. It is essential that patients be reminded that they must sleep with their head and upper body elevated. Supine blood pressure should be monitored prior to and during treatment and more frequently when increasing doses.
The most common adverse events reported by clinical trial participants taking Northera were headache, dizziness, nausea, high blood pressure (hypertension) and fatigue.
The effectiveness of Northera was shown through two-weeks in two clinical trials in people with NOH. People taking Northera reported a decrease in dizziness, lightheadedness, feeling faint, or feeling as if they might black out compared to those taking an inactive pill (placebo). Durability of the improvement in patient symptoms beyond two weeks has not been demonstrated.
Northera received orphan-product designation from the FDA because it is intended to treat a rare disease or condition.
Northera is made by Charlotte-based Chelsea Therapeutics Inc.
For more information:
FDA: Approved Drugs
FDA: Drug Innovation
National Institute of Neurological Disorders and Stroke: Orthostatic Hypotension
|
|
| Systematic (IUPAC) name | |
|---|---|
|
(2S,3R)-2-Amino-3-(3,4-dihydroxyphenyl)-3-hydroxypropanoic acid
|
|
| Clinical data | |
| Trade names | Northera |
| Routes of administration |
Oral |
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Bioavailability | 90% |
| Metabolism | Hepatic |
| Biological half-life | 1.5 hours |
| Excretion | Renal |
| Identifiers | |
| CAS Number | 23651-95-8 |
| ATC code | C01CA27 (WHO) |
| PubChem | CID 6989215 |
| ChemSpider | 83927 |
| UNII | J7A92W69L7 |
| ChEBI | CHEBI:31524 |
| ChEMBL | CHEMBL2103827 |
| Synonyms | β,3-Dihydroxytyrosine |
| Chemical data | |
| Formula | C9H11NO5 |
| Molar mass | 213.18734 g/mol |
NORTHERA capsules contain droxidopa, which is a synthetic amino acidprecursor of norepinephrine, for oral administration. Chemically, droxidopa is (–)-threo-3-(3,4- Dihydroxyphenyl)-L-serine. It has the following structural formula:
 |
Droxidopa is an odorless, tasteless, white to off-white crystals or crystalline powder. It is slightly soluble in water, and practically insoluble in methanol, glacial acetic acid, ethanol, acetone, ether, and chloroform. It is soluble in dilute hydrochloric acid. It has a molecular weight of 213.19 and a molecular formula of C9H11NO5.
NORTHERA capsules also contain the following inactive ingredients: mannitol, corn starch, and magnesium stearate. The capsule shell is printed with black ink. The black inks contain shellac glaze, ethanol, iron oxide black, isopropyl alcohol, n-butyl alcohol, propylene glycol, and ammonium hydroxide. The capsule shell contains the following inactive ingredients: 100 mg – gelatin, titanium dioxide, FD&C Blue No. 2, black and red iron oxide; 200 mg – gelatin, titanium dioxide, FD&C Blue No. 2, black and yellow iron oxide; 300 mg – gelatin, titanium dioxide, FD&C Blue No. 1, FD&C Yellow No. 5 (tartrazine), and FD&C Red No. 40. NORTHERA capsules differ in size and color by strength
CLIP
//////////DROXIDOPA, Chelsea Therapeutics, orphan drug status, FDA 2015, 3916-18-5, NORTHERA, SUMITOMO, Antiparkinsonian
THESIS
http://ncl.csircentral.net/668/1/th1739.pdf
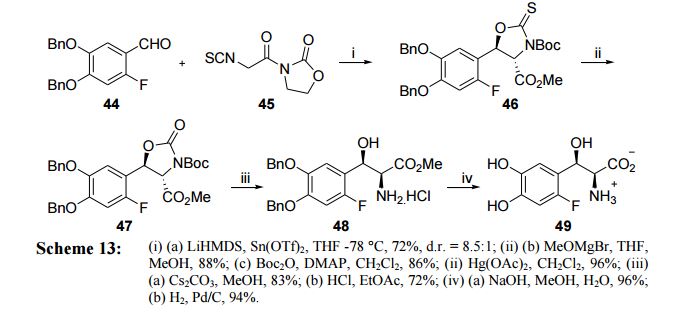
Review of Literature Literature search showed that there are only few reports available,42-44 which describe the synthesis of L-threo-DOPS (43) as detailed below. Kirk’s approach (2001)43 Kirk et. al have reported the synthesis of fluorinated analogue of L-threo-DOPS (49) starting from reaction of aldehyde 44 with isothiocyanate 45 in presence of LHMDS and Sn(OTf)2 to give ester 46 with d.r = 8.5:1. Subsequently, removal of chiral auxiliary with methoxymagnesium bromide followed by Boc protection of the thiocarbamate nitrogen was carried out. Thiocarbamate 46 was then converted to oxygen analogue 47 in 96% yield by treating it with Hg(OAc)2. This was followed by subsequent cleavage of 47 using Cs2CO3 and its Boc depotection gave the ester 48. Saponification of ester 48 followed by hydrogenation afforded fluoro analogue of L-threo-DOPS (49) (Scheme 13).
Sang-Ho’s approach (2007)44 Sang-Ho et. al have achieved an enzyme-catalyzed synthesis of L-threo-DOPS (43) by reacting in one-pot glycine, 3,4-dihydrobenzaldehyde, 2-mercaptoethanol, pyridoxal-5- phosphate solution, sodium sulfite and Triton X-100 in presence of E.coli at 15 °C

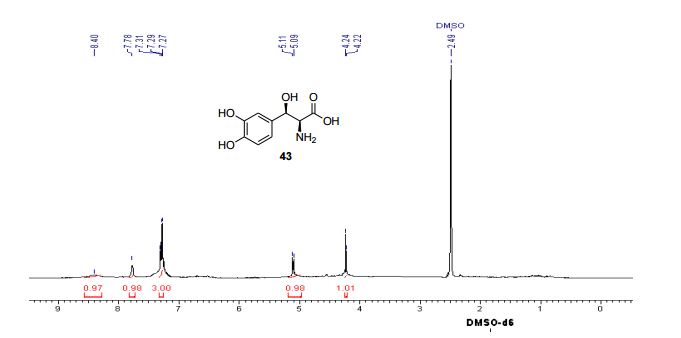
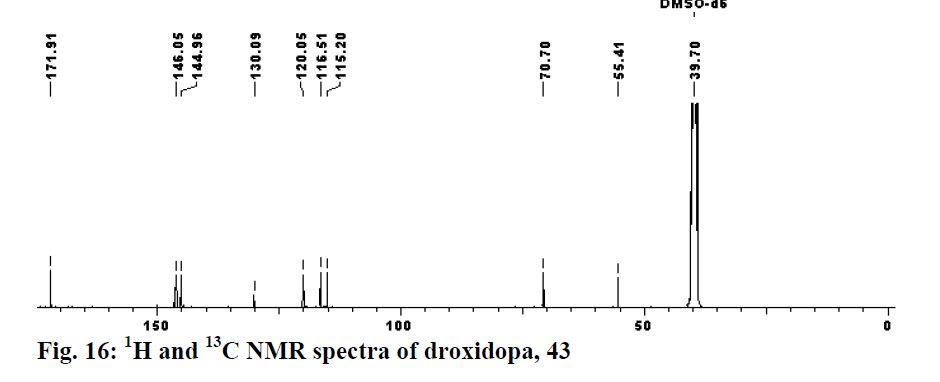
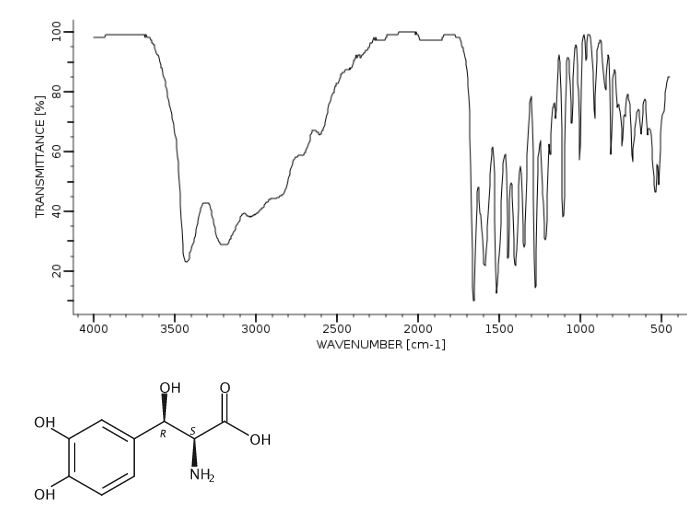
Yield: 94%; mp: 230-233 ºC; (lit.42 mp: 232-235 ºC); [α] 25 D: -38.7 (c 0.4, 1N aq. HCl); {lit.42 [α] 25 D: -39 (c 1, 1N HCl)}; IR (CHCl3, cm-1 ): 3018, 2399, 2366, 2345, 1652, 1519, 1215, 1018, 929, 756, 669; 1H NMR (200 MHz, DMSO-d6): δ 4.23 (d, J = 4.3 Hz, 1H), Droxidopa Chapter IV 226 5.10 (d, J = 3.8 Hz, 1H), 7.27-7.31 (m, 3H), 7.78 (br s, 1H), 8.40 (br s, 1H); 13C NMR (100 MHz, DMSO-d6): δ 55.41, 70.70, 115.21, 116.51, 120.06, 130.09, 144.96, 146.06, 171.91; Analysis: C9H11NO5 requires C, 50.70; H, 5.20; N, 6.57%; found: C, 50.99; H, 5.01; N, 6.33%
42 Hegedus, V. B.; Krasso, A. F.; Noack, K.; Zeller, P. Helv. Chim. Acta 1975, 58, 147.
[PDF]Download (3818Kb) – IR@NCL
ncl.csircentral.net/668/1/th1739.pdf
I here by declare that the thesis entitled “Enantioselective synthesis of bioactive molecules …. Section III Enantioselective synthesis of L-threo-DOPS (droxidopa).


Reblogged this on Med.Chem.in.Iceland.
LikeLike
Reblogged this on Srilanka-Chem.
LikeLike
Reblogged this on Med.Chem in Nepal.
LikeLike