
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
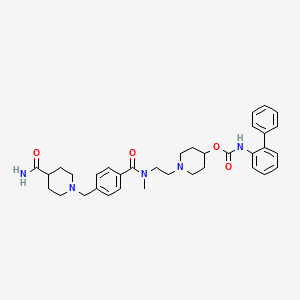
Revefenacin; 864750-70-9; TD-4208; UNII-G2AE2VE07O; G2AE2VE07O; TD-4208; GSK-1160724;
160724; GSK 1160724; TD-4028; YUPELRI
| Molecular Formula: | C35H43N5O4 |
|---|---|
| Molecular Weight: | 597.76 g/mol |
[1-[2-[[4-[(4-carbamoylpiperidin-1-yl)methyl]benzoyl]-methylamino]ethyl]piperidin-4-yl] N-(2-phenylphenyl)carbamate
Revefenacin is under investigation for the treatment of Chronic Obstructive Pulmonary Disease (COPD).
- Originator Theravance
- Developer Theravance Biopharma
- Class Antiasthmatics; Biphenyl compounds; Carbamates; Piperidines
- Mechanism of Action Muscarinic receptor antagonists
- Preregistration Chronic obstructive pulmonary disease
- 17 Sep 2018 Efficacy data from two replicate 12-week phase III trials and a 12-month safety trial in Chronic obstructive pulmonary disease (COPD) presented at the European Respiratory Society International Congress (ERS-2018)
- 31 May 2018 Theravance Biopharma in collaboration with Theravance Biopharma initiates enrolment in a phase III trial for Chronic obstructive pulmonary disease in USA (NCT03573817)
- 18 May 2018Efficacy and adverse events data from a phase I trial in Chronic obstructive pulmonary disease presented at the 114th International Conference of the American Thoracic Society
The compound was licensed to GlaxoSmithKline by Theravance for the inhalation treatment of chronic obstructive pulmonary disease in 2004. The rights were returned in 2009. In 2014, Theravance Biopharma spun-off from Theravance. In 2015, Theravance Biopharma and Mylan enter in a co development agreement for the global development and commercialization of the once-daily nebulizer for the treatment of chronic obstructive pulmonary disease and other respiratory diseases.
SYN
WO 2012009166
SYN OF INT
FINAL
PAPER
Discovery of (R)-1-(3-((2-Chloro-4-(((2-hydroxy-2-(8-hydroxy-2-oxo-1,2-dihydroquinolin-5-yl)ethyl)amino)methyl)-5-methoxyphenyl)amino)-3-oxopropyl)piperidin-4-yl (1,1′-biphenyl)-2-ylcarbamate (TD-5959, GSK961081, batefenterol): First-in-class dual pharmacology multivalent muscarinic antagonist and 2 agonist (MABA) for the treatment of chronic obstructive pulmonary disease (COPD)
J Med Chem 2015, 58(6): 2609
Discovery of (R)-1-(3-((2-Chloro-4-(((2-hydroxy-2-(8-hydroxy-2-oxo-1,2-dihydroquinolin-5-yl)ethyl)amino)methyl)-5-methoxyphenyl)amino)-3-oxopropyl)piperidin-4-yl [1,1′-Biphenyl]-2-ylcarbamate (TD-5959, GSK961081, Batefenterol): First-in-Class Dual Pharmacology Multivalent Muscarinic Antagonist and β2 Agonist (MABA) for the Treatment of Chronic Obstructive Pulmonary Disease (COPD)

Through application of our multivalent approach to drug discovery we previously reported the first discovery of dual pharmacology MABA bronchodilators, exemplified by 1. Herein we describe the subsequent lead optimization of both muscarinic antagonist and β2 agonist activities, through modification of the linker motif, to achieve 24 h duration of action in a guinea pig bronchoprotection model. Concomitantly we targeted high lung selectivities, low systemic exposures and identified crystalline forms suitable for inhalation devices. This article culminates with the discovery of our first clinical candidate 12f (TD-5959, GSK961081, batefenterol). In a phase 2b trial, batefenterol produced statistical and clinically significant differences compared to placebo and numerically greater improvements in the primary end point of trough FEV1 compared to salmeterol after 4 weeks of dosing in patients with moderate to severe chronic obstructive pulmonary disease (COPD).
PATENT
WO 2006099165
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2006099165
FIG. 18 shows a PXRD pattern of Form I of the crystalline freebase of the compound of formula I. This crystalline freebase is further characterized by the DSC trace in FIG. 19, the TGA trace in FIG. 20, the DMS trace in FIG. 21, and the micrographic image in FIG. 22.
FIG. 23 shows a PXRD pattern of Form II of the crystalline freebase of the compound of formula I. This crystalline freebase is further characterized by the DSC trace in FIG. 24, the TGA trace in FIG. 25, and the DMS trace in FIG. 26.
PREPARATION 1
Biphenyl-2-ylcarbamic Acid Piperidin-4-yl Ester
Biphenyl-2-isocyanate (97.5 g, 521 mmol) and 4-hydroxy-N-benzylpiperidine (105 g, 549 mmol) were heated together at 70 0C for 12 hours. The reaction mixture was then cooled to 50 0C and ethanol (1 L) was added and then 6M HCl (191 mL) was added slowly. The resulting mixture was then cooled to ambient temperature and ammonium formate (98.5 g, 1.56 mol) was added and then nitrogen gas was bubbled through the solution vigorously for 20 minutes. Palladium on activated carbon (20 g, 10 wt% dry basis) was then added and the reaction mixture was heated at 40 0C for 12 hours, and then filtered through a pad of Celite. The solvent was then removed under reduced pressure and IM HCl (40 mL) was added to the crude residue. The pH of the mixture was then adjusted with IO N NaOH to pH 12. The aqueous layer was extracted with ethyl acetate (2 x 150 mL) and the organic layer was dried (magnesium sulfate), filtered and the solvent removed under reduced pressure to give 155 g of the title intermediate (100% yield). HPLC (10-70) Rt = 2.52; m/z: [M + H+] calc’d for C18H20N2O2 297.15; found 297.31
PREPARATION 2
iV-Benzyl-iV-methylaminoacetaldehvde
To a 3-necked 2-L flask was added N-benzyl-N-methylethanolamine (30.5 g, 0.182 mol), DCM (0.5 L), DIPEA (95 mL, 0.546 mol) and DMSO (41 mL, 0.728 mol).
Using an ice bath, the mixture was cooled to about -10 °C and sulfur trioxide pyridine-complex (87 g, 0.546 mol) was added in 4 portions over 5 minute intervals. The reaction was stirred at -10 0C for 2 hours. Before removing the ice-bath, the reaction was quenched by adding water (0.5 L). The aqueous layer was separated and the organic layer was washed with water (0.5 L) and brine (0.5 L) and then dried over magnesium sulfate and filtered to provide the title compound which was used without further purification.
PREPARATION 3
Biphenyl-2-ylcarbamic Acid l-[2-(Εenzylmethylammo)ethyllpiperidin-4-yl Ester
To a 2-L flask, containing the product of Preparation 2 in DCM (0.5 L) was added the product of Preparation 1 (30 g, 0.101 mol) followed by sodium triacetoxyborohydride (45 g, 0.202 mol). The reaction mixture was stirred overnight and then quenched by the addition of 1 N hydrochloric acid (0.5 L) with vigorous stirring. Three layers were observed and the aqueous layer was removed. After washing with IN NaOH (0.5 L)3 a homogenous organic layer was obtained which was then washed with a saturated solution of aqueous NaCl (0.5 L), dried over magnesium sulfate, filtered and the solvent removed under reduced pressure. The residue was purified by dissolving it in a minimal amount of isopropanol and cooling this solution to 0 °C to form a solid which was collected and washed with cool isopropanol to provide 42.6 g of the title compound (95% yield). MS m/z: [M + H+] calc’d f for C28H33N3O2444.3; found 444.6. Rf=3.5l min (10-70 ACN:H2O, reverse phase HPLC).
PREPARATION 3 A
Biphenyl-2-ylcarbamic Acid l-f2-(Benzylmethylammo)ethyllpiperidin-4-yl Ester
The title compound was prepared by mesylation of iV-benzyl-N-methyl
ethanolamine, which was then reacted with biphenyl-2-ylcarbamic acid piperidin-4-yl ester in an alkylation reaction.
A 500 mL flask (reactor flask) was charged with N-benzyl-iV-methylethanolamine (24.5 mL), DCM (120 mL), NaOH (80 mL; 30wt%) and tetrabutylammonium chloride. Mixing at low speed throughout the reaction, the mixture was cooled to -10 °C (cooling bath), and the addition funnel charged with DCM (30 mL) and mesyl chloride (15.85 mL), which was added drop wise at a constant rate over 30 minutes. The addition was exothermic, and stirring was continued for 15 minutes while the temperature equilibrated back to -10 0C. The reaction was held for at least 10 minutes to ensure full hydrolysis of the excess mesyl chloride.
A 250 mL flask was charged with biphenyl-2-ylcarbamic acid piperidin-4-yl ester (26 g; prepared as described in Preparation 1) and DCM (125 mL), stirred for 15 minutes at room temperature, and the mixture chilled briefly to 10 0C to form a slurry. The slurry was then charged into the reactor flask via the addition funnel. The cooling bath was removed and the reaction mixture was warmed to 5 °C. The mixture was transferred to a separatory funnel, the layers allowed to settle, and the aqueous layer removed. The organic layer was transferred back to the reactor flask, stirring resumed, the mixture held to room
temperature, and the reaction monitored by HPLC for a total of 3.5 hours.
The reactor flask was charged with NaOH (IM solution; 100 mL), stirred, and the layers allowed to settle. The organic layer was separated, washed (NaCl satd. solution), its volume partially reduced under vacuum, and subjected to repeated IPA washings. The solids were collected and allowed to air-dry (25.85 g, 98% purity). Additional solids were obtained from further processing of the mother liquor (volume reduction, EPA, cooling).
PREPARATION 4
Biphenyl-2-ylcarbamic Acid l-(2-Methylaminoethyl)piperidin-4-yl Ester
To a Parr hydrogenation flask was added the product of Preparation 3 (40 g, 0.09 mol) and ethanol (0.5 L). The flask was flushed with nitrogen gas and palladium on activated carbon (15g, 10 wt% (dry basis), 37% wt/wt) was added along with acetic acid (20 mL). The mixture was kept on the Parr hydrogenator under a hydrogen atmosphere (-50 psi) for 3 hours. The mixture was then filtered and washed with ethanol. The filtrate was condensed and the residue was dissolved in a minimal amount of DCM. Isopropyl acetate (10 volumes) was added slowly to form a solid which was collected to provide 22.0 g of the title compound (70% yield). MS m/z: [M + H+] calc’d for C21H27N3O2 354.2; found 354.3. R/=2.96 min (10-70 ACNrH2O, reverse phase HPLC).
PREPARATION 5
Biphenyl-2-ylcarbamic Acid l-{2-[(4-Formylbenzoyr)
methylaminol ethyll piperidin-4- yl Ester
To a three-necked 1-L flask was added 4-carboxybenzaldehyde (4.77 g,
31.8 mmol), EDC (6.64 g, 34.7 mmol), HOBT (1.91 g, 31.8 mmol), and DCM (200 mL). When the mixture was homogenous, a solution of the product of Preparation 4 (10 g, 31.8 mmol) in DCM (100 mL) was added slowly. The reaction mixture was stirred at room temperature for approximately 16 hours and then washed with water (1 x 100 mL), IN HCl (5 x 60 mL), IN NaOH (1 x 100 mL) brine (1 x 5OmL)3 dried over sodium sulfate, filtered and concentrated to afford 12.6 g of the title compound (92% yield; 85% purity based on HPLC). MS m/z: [M + H+] calc’d for C29H31N3O4 486.2; found 486.4. i?y=3.12 min (10-70 ACNiH2O, reverse phase HPLC).
EXAMPLE 1
Biphenyl-2-ylcarbamic Acid 1 -(2- { |“4-(4-Carbamoylpiperidin- 1 -ylmethvD
benzoylimethylamino) ethyl’)piperidin-4-vl Ester
To a three-necked 2-L flask was added isonipecotamide (5.99 g, 40.0 mmol), acetic acid (2.57 mL), sodium sulfate (6.44 g) and isopropanol (400 mL). The reaction mixture was cooled to 0-10 0C with an ice bath and a solution of biphenyl-2-ylcarbamic acid l-{2-[(4-formylbenzoyl)methylamino]ethyl}piperidin-4-yl ester (11 g, 22.7 mmol; prepared as described in Preparation 5) in isopropanol (300 mL) was slowly added. The reaction mixture was stirred at room temperature for 2 hours and then cooled to 0-10 0C. Sodium triacetoxyborohydride (15.16 g, 68.5 mmol) was added portion wise and this mixture was stirred at room temperature for 16 hours. The reaction mixture was then concentrated under reduced pressure to a volume of about 50 mL and this mixture was acidified with IN HCl (200 mL) to pH 3. The resulting mixture was stirred at room temperature for 1 hour and then extracted with DCM (3 x 250 mL). The aqueous phase was then cooled to 0-5 °C with an ice bath and 50% aqueous NaOH solution was added to adjust the pH of the mixture to 10. This mixture was then extracted with isopropyl acetate (3 x 300 mL) and the combined organic layers were washed with water (100 mL), brine (2 x 50 mL), dried over sodium sulfate, filtered and concentrated to afford 10.8 g of the title compound (80% yield. MS m/z: [M + H+] calc’d for C35H43N5O4 598.3; found 598.6. Rj=232 min (10-70 ACNiH2O, reverse phase HPLC).
EXAMPLE 2
Crystalline Diphosphate Salt of Biphenyl-2-ylcarbamic Acid l-(2-{[4-(4- Carbamoylpiperidin-l-ylmethyl)benzoyl1methylamino>ethyDpiperidin-4-yl Ester
500 mg of biphenyl-2-ylcarbamic acid l-(2-{[4-(4-carbamoylpiρeridin-l-ylmethyl) benzoyl]methylamino}ethyl)piperidin-4-yl ester (0.826 mmol of 96% pure material;
prepared as described in Example 1) was taken up in 5 ml of water and 1.5 ml of IM phosphoric acid. The pH was adjusted to approximately pH 5.3 with an additional 0.25ml of IM phosphoric acid (equaling 2.1 molar equivalents). The clear solution was filtered through a 0.2 micron filter, frozen and lyophilized to dryness to yield an amorphous diphosphate salt.
20 mg of the amorphous diphosphate salt was dissolved in 2 ml of IPA: ACN (1:1). 0.1 ml of water was added and the mixture heated to 60 °C under stirring. Almost all of the solids dissolved. The suspension was allowed to cool to ambient temperature, under stirring, overnight. The resulting crystals were collected by filtration and air-dried for 20 minutes to give the title compound (18.5 mg, 93% yield) as a white crystalline solid.
When examined under a microscope using polarized light, the crystals exhibited some birefringence.
EXAMPLE 3
Crystalline Diphosphate Salt of Biphenyl-2-ylcarbamic Acid l-(2-{|“4-(4- Carbamoylpiperidin-l-vhτiethyl‘)benzoyl]methylamino}ethyl)piperidin-4-yl Ester
5.0 g of biphenyl-2-ylcarbamic acid l-(2-{[4-(4-carbamoylpiperidin-l-ylmethyl)benzoyl]methylamino}ethyl)piperidin-4-yl ester (freebase; prepared as described in Example 1) was combined with 80 ml of IPA:ACN (1:1). 4.0 ml of water was added and the mixture heated to 50 °C under stirring, forming a clear solution. To this was added dropwise at 50 °C, 16 ml IM phosphoric acid. The resulting cloudy solution was stirred at 50 °C for 5 hours, then allowed to cool to ambient temperature, under slow stirring, overnight. The resulting crystals were collected by filtration and air-dried for 1 hour, then under vacuum for 18 hours, to give the title compound (5.8 g, 75% yield) as a white crystalline solid (98.3% purity by HPLC).
EXAMPLE 4
Crystalline Monosulfate Salt of Biphenyl-2-ylcarbamic Acid l-(2-{[4-(4- Carbamoylpiperidm-l-ylmethvπbenzoyllmethylamino>ethyl)piperidm-4-yl Ester
442 mg of biphenyl-2-ylcarbamic acid l-(2-{[4-(4-Carbamoylpiperidin-l-ylmethyl) benzoyl]methylamino} ethyl)piperidin-4-yl ester (0.739 mmol of 96% pure material;
prepared as described in Example 1) was taken up in 5 ml of H2OrACN (1 : 1) and 1.45 ml of IN sulfuric acid was added slowly, while monitoring the pH. The pH was adjusted to approx. pH 3.3. The clear solution was filtered through a 0.2 micron filter, frozen and lyophilized to dryness to yield a monosulfate salt.
30.3 mg of the monosulfate salt was dissolved in 1.65 ml of IPA:ACN (10:1). The suspension was heated by placing the vial in a pre-heated 60 °C water bath for 30 minutes. A viscous material was formed and the heat increased to 70 °C for 30 minutes. Since the material remained viscous, the heat was lowered to 60 0C and the mixture heated for an additional hour. The heat was turned off and the mixture was allowed to cool to room temperature. After 4 days, the material appeared to be solid, and the sample was allowed to sit for an additional nine days. The solid was then filtered and dried using a vacuum pump for 1 hour to give the title compound (23 mg, 76% yield).
EXAMPLE 5
Crystalline Monosulfate Salt of Biphenyl-2-ylcarbamic Acid l-(2-{[~4-(4- Carbamoylpiperidin-l-ylmethyl)benzoyl]methylamino>ethyl)piperidin-4-yl Ester
161 g of the monosulfate salt (prepared as described in Example 4) was dissolved in 8.77 ml of IPA:ACN (10:1). The suspension was heated by placing the vial in a pre-heated 70 °C water bath for 1.5 hours. Oil droplets formed within 5 minutes. The heat was lowered to 60 °C and the mixture heated for an additional 1.5 hours, followed by heating at 50 °C for 40 minutes, at 40 °C for 40 minutes, then at 30 0C for 45 minutes. The heat was turned off and the mixture was allowed to slowly cool to room temperature. The next day, the material was viewed under a microscope and indicated needles and plates. The material was then heated at 40 °C for 2 hours, at 35 0C for 30 minutes, and then at 30 °C for 30 minutes. The heat was turned off and the mixture was allowed to slowly cool to room temperature. The solid was then filtered and dried using a vacuum pump for 1 hour to give the title compound (117 mg, 73% yield).
EXAMPLE 6
Crystalline Dioxalate Salt of Biphenyl-2-ylcarbamic Acid l-(“2-{|“4-(4-Carbamoylpiperidin- 1 -ylmethyl)benzoyl]methylamino> ethyl)piperidin-4-yl Ester
510 mg of biphenyl-2-ylcarbamic acid l-(2-{[4-(4-carbamoylpiperidin-l-ylmethyl)benzoyl]methylamino} ethyl)piperidin-4-yl ester (0.853 mmol of 96% pure material; prepared as described in Example 1) was taken up in 5 ml of H2O:ACN (1:1) and 1.7 ml of IM aqueous oxalic acid was added slowly, while monitoring the pH. The pH was adjusted to approx. pH 3.0. The clear solution was filtered through a 0.2 micron filter, frozen and lyophilized to dryness to yield a dioxalate salt.
31.5 mg of the dioxalate salt was dissolved in 2.76 ml of 94%IPA/6%H20. The mixture was stirred in a pre-heated 60 °C water bath for 2.5 hours. After 25 minutes, all of the sample was in solution. The heat was turned off and the mixture was allowed to cool to room temperature. The next day, a small amount of viscous material was present. The vial was refrigerated at 4 °C. After 4 days, the viscous material was still present. The vial was then placed at room temperature and observed one month later. The material appeared to be solid, and was observed to be crystalline under a microscope. The solid was then filtered and dried using a vacuum pump for 1 hour to give the title compound (20 mg, 63.5% yield).
EXAMPLE 7
Crystalline Dioxalate Salt of Biphenyl-2-ylcarbamic Acid l-(2-{T4-(4-Carbamoylpiperidin- 1 -ylmethyl)benzoyl]methylammo) ethvDpiperidin-4-yl Ester
150 mg of the dioxalate salt (prepared as described in Example 6) was dissolved in 13.1 ml of 94%IPA/6%H20. The mixture was stirred in a pre-heated 60 °C water bath for 2.5 hours. The heat was turned off and the mixture was allowed to cool to room
temperature. The vial was refrigerated at 4 °C. After 6 days, an oily material was observed with what appeared to be a crystal on the side of the vial. The vial was then allowed to reach room temperature, at which point seeds (crystalline material from Example 6) were added and allowed to sit for 16 days. During this time, more crystals were observed to come out of solution. The solid was then filtered and dried using a vacuum pump for 14 hours to give the title compound (105 mg, 70% yield).
EXAMPLE 8
Crystalline Freebase Biphenyl-2-ylcarbamic Acid l-(2-(f4-(‘4-Carbamoylpiperidin-l- ylmethvDbenzoyl]methylaniino}ethyl)piperidin-4-yl Ester (Form T)
109 mg of biphenyl-2-ylcarbamic acid l-(2-{[4-(4-carbamoylpiperidin-l-ylmethyl)benzoyl]methylamino}ethyl)piperidin-4-yl ester (prepared as described in
Example 1) was dissolved in 0.56 ml of H2O: ACN (1:1). The suspension was left in a vial (cap loosely placed on top) to allow for a slower evaporation time. The vial was placed under a nitrogen flow environment, although the nitrogen was not used for evaporation, only for the environment. A precipitate was visible within 1 day, which was observed to be crystalline under a microscope. The solid was then placed on a high vacuum line to remove all solvent to give the title compound. Quantitative recovery, 97.8% pure by HPLC.
In an alternate procedure, after dissolving in H2O: ACN (1:1) (approximately 350 mg/mL), the vial was stored at 5 0C, and the precipitate was visible at day 2. The solid was filtered, rinsed with water, and dried on high vacuum overnight. Recovery was 55%, with the solid having 98.2% purity and the liquid having 92.8% purity.
EXAMPLE 9
Crystalline Freebase Biphenyl-2-ylcarbamic Acidl-(“2-{J4-(4-Carbamoylpiperidin- l-yhiaethyl)benzoyllmethylammo|ethvDpiperidin-4-yl Ester (Form T)
50.4 mg of biphenyl-2-ylcarbamic acid l-(2-{[4-(4-carbamoylpiperidin-l-ylmethyl)benzoyl]methylamino}ethyl)piperidin-4-yl ester (prepared as described in
Example 1) was dissolved in 0.144 ml of H2O:ACN (1:1). The suspension was left in vial (cap loosely placed on top) to allow for a slower evaporation time. The vial was refrigerated at 4 0C for 6 days. A precipitate was visible after 2 days. The solid was filtered and placed on a high vacuum line to remove all solvent and give the title compound as a white solid (27.8 mg, 55.2 % yield).
EXAMPLE 10
Crystalline Freebase Biphenyl-2-ylcarbamic Acid l-(2-{[4-(4-Carbamoylpiperidin- l-vhnethvDbenzoyl]methylamino>ethvDpiperidin-4-yl Ester (Form T)
230 mg of biphenyl-2-ylcarbamic acid l-(2-{[4-(4-carbamoylpiperidin-l-yhnethyl)benzoyl]methylamino}ethyl)piρeridin-4-yl ester (prepared as described in
Example 1) was dissolved in 0.2 ml of H2O:ACN (1:1), using slight heat. The mixture was then heated in a 70 °C water bath for 2 hours. The heat was turned off and the mixture was allowed to cool to room temperature, then refrigerated at 4 °C for 1 hour. 50 μl of water was then added (oiled out), followed by the addition of 40 μl of ACN to get the sample back into solution. Seeds (crystalline material from Example 8) were added under slow stirring at room temperature. Crystals started to form ,and the mixture was allowed to sit overnight, with slow stirring. The next day, a heat cool cycle was applied (30 °C for 10 minutes, 40 0C for 10 minutes, then 50 °C for 20 minutes). The heat was turned off and the mixture allowed to cool overnight, with slow stirring. The next day, a second heat/cool cycle was applied (60 0C for 1 hour, with dissolving observed at 70 °C). The heat was turned off and the mixture allowed to cool overnight, with slow stirring. The next day, crystals were present and a third heat cool cycle was applied (60 0C for 3 hours). The heat was turned off and the mixture allowed to cool overnight, with slow stirring. The next day, a heat cool cycle was applied (60 °C for 3 hours, slow cool, then 60 °C for 3 hours). The heat was turned off and the mixture allowed to cool overnight, with slow stirring. After 3 days, the solid was filtered and placed on a high vacuum line to remove all solvent and give the title compound.
EXAMPLE 11
Crystalline Freebase Biphenyl-2-ylcarbamic Acid l-(2-{[4-(4-Carbamoylpiperidin- l-ylmethyl)benzoyl]methylamino|ethyl‘)piperidin-4-yl Ester (Form JD
70 mg of biphenyl-2-ylcarbamic acid l-(2-{[4-(4-carbamoylpiperidin-l-yhnethyl)benzoyl]methylamino}ethyl)piperidin-4-yl ester (prepared as described in
Example 1) was dissolved in 0.1 mL ACN. After addition of 0.3 ml MTBE, the solution appeared cloudy. An additional 50 μl of ACN was added to clarify the solution (155 mg/ml ACN:MTBE = 1 :2). The mixture was left in the vial and capped. Crystals appeared by the next day. The solid was then filtered and placed on a high vacuum line to remove all solvent and give the title compound.
PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2011008809
U.S. Patent Publication No. 2005/0203133 to Mammen et al. discloses novel biphenyl compounds that are expected to be useful for treating pulmonary disorders such as chronic obstructive pulmonary disease (COPD) and asthma. In particular, the compound biphenyl-2-ylcarbamic acid l-(2-{[4-(4-carbamoylpiperidin-l-ylmethyl) benzoyl]methylamino}ethyl)piperidin-4-yl ester is specifically described in this application as possessing muscarinic receptor antagonist or anticholinergic activity.
The chemical structure of biphenyl-2-ylcarbamic acid l-(2-{[4-(4-carbamoyl piperidin- 1 -ylmethyl)benzoyl]methylamino } ethyl)piperidin-4-yl ester is represented by formula I:
I
The compound of formula I has been named using the commercially-available AutoNom software (MDL, San Leandro, California).
Therapeutic agents useful for treating pulmonary or respiratory disorders are advantageously administered directly into the respiratory tract by inhalation. In this regard, several types of pharmaceutical inhalation devices have been developed for administering therapeutic agents by inhalation including dry powder inhalers (DPI),
metered-dose inhalers (MDI) and nebulizer inhalers. When preparing pharmaceutical compositions and formulations for use in such devices, it is highly desirable to have a crystalline form of the therapeutic agent that is neither hygroscopic nor deliquescent and which has a relatively high melting point thereby allowing the material to be micronized without significant decomposition. Although crystalline freebase forms of the compound of formula I have been reported in U.S. Patent Publication No. 2007/0112027 to Axt et al. as Form I and Form II, the crystalline freebase forms of the present invention have different and particularly useful properties, including higher melting points
One aspect of the invention relates to crystalline freebase forms of biphenyl-2-ylcarbamic acid 1 -(2- { [4-(4-carbamoylpiperidin- 1 -ylmethyl)benzoyl]methy lamino } ethyl) piperidin-4-yl ester characterized by a powder x-ray diffraction pattern comprising diffraction peaks at 2Θ values of 6.6±0.1, 13.1±0.1, 18.6±0.1, 19.7±0.1, and 20.2±0.1.
Another aspect of the invention relates to a crystalline freebase of biphenyl-2-ylcarbamic acid 1 -(2- { [4-(4-carbamoylpiperidin- 1 -ylmethyl)benzoyl]methy lamino } ethyl) piperidin-4-yl ester, designated as form III, which is characterized by a powder x-ray diffraction pattern comprising diffraction peaks at 2Θ values of 6.6±0.1, 13. l±O.l,
18.6±0.1, 19.7±0.1, and 20.2±0.1; and further characterized by having five or more additional diffraction peaks at 2Θ values selected from 8.8=1=0.1, 10. l±O.l, 11.4±0.1, l l.β±O.l, 14.8±0.1, 15.2±0.1, lβ.l±O.l, 16.4±0.1, 16.9±0.1, 17.5±0.1, 18.2±0.1, 19.3±0.1, 19.9±0.1, 20.8±0.1, 21. l±O.l, 21.7±0.1, and 22.3±0.1.
Still another aspect of the invention relates to a crystalline freebase of biphenyl-2-ylcarbamic acid 1 -(2- { [4-(4-carbamoylpiperidin- 1 -ylmethyl)benzoyl]methy lamino } ethyl) piperidin-4-yl ester, designated as form IV, which is characterized by a powder x-ray diffraction pattern comprising diffraction peaks at 2Θ values of 6.6±0.1 , 13. l±O.1 ,
18.6=1=0.1, 19.7=1=0.1, and 20.2±0.1; and further characterized by having five or more additional diffraction peaks at 2Θ values selected from 10.6±0.1, 15.0=1=0.1, lβ.O±O.l, 17.3±0.1, 17.7±0.1, 20.9±0.1, 21.4±0.1, 22.6±0.1, 24.6±0.1, and 27.8±0.1.
Preparation 1
Biphenyl-2-ylcarbamic acid l-(2-{[4-(4-carbamoylpiperidin-l- ylmethvDbenzovHmethylaminol ethyDpiperidin-4-yl Ester The diphosphate salt of biphenyl-2-ylcarbamic acid l-(2-{[4-(4-carbamoylpiperidin-l-ylmethyl)benzoyl]methylamino}ethyl)piperidin-4-yl ester (16 g) was dissolved in a biphasic mixture of water (100 mL) and EtOAc (200 mL). NaOH (2 N, 75 mL) was added over a period of 5 minutes. The mixture was then stirred for 30 minutes. The phases were separated and the aqueous phase was extracted with EtOAc (200 mL). The combined organic phases were concentrated. DCM (100 mL) was added, and the mixture evaporated to dryness. The solids were dried in an oven for about 48 hours to yield the title compound (9.6 g).
EXAMPLE 1
Crystalline Freebase of Biphenyl-2-ylcarbamic Acid l-(2-{r4-(4-Carbamoylpiperidin-l- ylmethyl)benzoyllmethylamino|ethyl)piperidin-4-yl Ester (Form III) Biphenyl-2-ylcarbamic acid l-(2-{[4-(4-carbamoylpiperidin-l-ylmethyl)benzoyl]methylamino}ethyl)piperidin-4-yl ester (102.4 mg) was dissolved in MeCN (500 μL). The solution was stirred at room temperature for 80 minutes and a white solid precipitate formed. The mixture was placed in the shaker block to thermocycle (0-40 0C in one hour blocks) for 48 hours. A white, dense, immobile solid was observed. MeCN (500 μL) was added to mobilize the slurry. The mixture was then placed back in the shaker block for 2 hours. The solids were isolated by vacuum filtration using a sinter funnel, then placed in the piston dryer at 40 0C under full vacuum for 15.5 hours, to yield 76.85 mg of the title crystalline compound.
EXAMPLE 2
Crystalline Freebase of Biphenyl-2-ylcarbamic Acid l-(2-{r4-(4-Carbamoylpiperidin-l- ylmethyl)benzoyllmethylamino|ethyl)piperidin-4-yl Ester (Form III) Diphosphate salt of biphenyl-2-ylcarbamic acid l-(2-{[4-(4-carbamoyl-piperidin-l-ylmethyl)benzoyl]methylamino}ethyl)piperidin-4-yl ester (C3sH43NsO4»2H3PO4; MW 793.75; 632.9 g) was slurried in isopropyl acetate (11.08 L) and water (6.33 L) at room temperature under nitrogen. The suspension was warmed to 53±3 0C and 1OM NaOH (317 mL) was added to the stirred mixture, while maintaining the temperature of the mixture above 50 0C. The mixture was stirred for approximately 5 minutes at 53±3 0C before allowing the layers to settle. The layers were then separated and the aqueous layer was removed. Water (3.16 L) was added to the organic layer while maintaining the temperature of the mixture above 50 0C. The mixture was stirred for 5 minutes at 53±3 0C before allowing the layers to settle. The layers were separated and the water layer was removed. Isopropyl acetate (6.33 L) was added and then about 10 volumes of distillate were collected by atmospheric distillation. This step was repeated with additional isopropyl acetate (3.2 L). After the second distillation, the temperature of the clear solution was reduced to 53±3 0C, then seeded with a suspension of the biphenyl-2-ylcarbamic acid l-(2-{[4-(4-carbamoylpiperidin-l-ylmethyl)benzoyl]methylamino}ethyl)piperidin-4-yl ester crystalline freebase (Form III; 3.2 g) in isopropyl acetate (51 mL). The resulting suspension was stirred at 53±3 0C for 2 hours, then cooled to 10±3 0C over 4 hours. The suspension was stirred at 10±3 0C for at least 2 hours and then the solids were collected by filtration. The resulting filter cake was washed with isopropyl acetate (2 x 1.9 L) and the product was dried in vacuo at 50 0C to yield the title crystalline compound (C3SH43NsO4; MW 597.76; 382.5 g, 80.3% yield).
EXAMPLE 3
Recrystallization of Crystalline Freebase of Biphenyl-2-ylcarbamic Acid l-(2-{[4-(4- Carbamoylpiperidin- 1 -ylmethyDbenzoyllmethylaminol ethyl)piperidin-4-yl Ester (Form
III)
Crystalline freebase of biphenyl-2-ylcarbamic acid l-(2-{[4-(4-carbamoylpiperidin-l-ylmethyl)benzoyl]methylamino}ethyl)piperidin-4-yl ester (Form III; C35H43N5O4; MW 597.76; 372.5 g) was slurried in toluene (5.6 L) at 20±3 0C under nitrogen. The suspension was warmed to 82±3 0C, and held at this temperature until complete dissolution was observed. The solution was then clarified into the crystallizer vessel, followed by rinsing with toluene (373 μL). Solids were observed in the crystallizer vessel, and the vessel was re-heated to 82±3 0C to effect dissolution, then cooled to 58±3 0C and seeded with a pre-sonicated (approximately 1 minute) of crystalline freebase (Form III; 1.9 g) in toluene (8 μL). The resulting suspension was allowed to stand at 58±3 0C for at least 4 hours, then cooled to 20±3 0C over 2 hours (approximate cooling rate of 0.33 °C/min). The suspension was stirred at 20±3 0C for at least 1 hour, then the solids were collected by filtration. The resulting filter cake was washed with toluene (2 x 1.2 L) and the product was dried in vacuo at 52±3 0C to yield the title crystalline compound (345.3 g, 92.7% yield).
EXAMPLE 4
Crystalline Freebase of Biphenyl-2-ylcarbamic Acid l-(2-{r4-(4-Carbamoylpiperidin-l- ylmethyl)benzoyllmethylamino|ethyl)piperidin-4-yl Ester (Form IV) Biphenyl-2-ylcarbamic acid l-(2-{[4-(4-carbamoylpiperidin-l-ylmethyl)benzoyl]methylamino}ethyl)piperidin-4-yl ester (prepared as described in Preparation 1; 2.5 g) was dissolved in MeCN (10 mL) to yield a viscous oily pale yellow material. Additional MeCN (5 mL) was added to dilute the material. The solution was seeded with crystalline freebase (20 mg; Form III prepared as described in Example 1) and stirred at room temperature for 90 minutes. A large amount of white precipitate (small crystals) was observed. The slurry was analyzed under a polarized light microscope and found to be birefringent.
Additional MeCN (3 mL) was added and the slurry was placed in a Metz SynlO block to thermocycle (0-40 0C in one hour blocks) at 800 rpm overnight. The Metz SynlO is a 10 position parallel reaction station that is static. Agitation of the solution/slurry was by a cross magnetic stirrer bar. The shaker block was a separate piece of equipment that was heated and cooled by an external Julabo bath. The material was removed at 0 0C. It was observed that the slurry had settled out, leaving a pale yellow solution above the white precipitate. The slurry was stirred and placed back in the shaker block to thermocycle.
The material was removed at 40 0C, and stirred at a high agitation rate at room temperature for 80 minutes. The slurry was again analyzed and found to be birefringent. The filter cake was isolated by vacuum filtration using a sinter funnel. MeCN (3 mL) was used to wet the filter paper and the filter cake was washed with MeCN prior to filtration. The cake was deliquored under vacuum for 40 minutes to yield 2.3 g of a flowing white powder. The material was placed in a piston dryer at 400C for 65 hours, to yield 2.2 g of the title crystalline compound as a white powder (99.6% purity).
PATENT
Example 1
Biphenyl-2-ylcarbamic Acid l-(2-{[4-(4-Carbamoylpiperidin-l- ylmethyl)benzoyl]methylamino}ethyl)piperidin-4-yl Ester
To a three-necked 2-L flask was added isonipecotamide (5.99 g, 40.0 mmol), acetic acid (2.57 mL), sodium sulfate (6.44 g) and LPA (400 mL). The reaction mixture was cooled to 0-10°C with an ice bath and a solution ofthe product of Preparation 5 (11 g, 22.7 mmol) in LPA (300 mL) was slowly added. The reaction mixture was stined at room temperature for 2 hours and then cooled to 0-10°C. Sodium triacetoxyborohydride (15.16 g, 68.5 mmol) was added portion wise and this mixture was stined at room temperature for 16 h. The reaction mixture was then concentrated under reduced pressure to a volume of about 50 mL and this mixture was acidified with IN HCl (200 mL) to pH 3. The resulting mixture was stined at room temperature for 1 hour and then extracted with DCM (3 x 250 mL). The aqueous phase was then cooled to 0-5°C with an ice bath and 50% aqueous NaOH solution was added to adjust the pH ofthe mixture to 10. This mixture was then extracted with isopropyl acetate (3 x 300 mL) and the combined organic layers were washed with water (100 mL), brine (2 x 50 mL), dried over sodium sulfate, filtered and concentrated to afford 10.8 g ofthe title compound (80% yield. MS m/z: [M + H“1”] calcd for C35H43N5O4, 598.3; found, 598.6. Rf = 2.32 min (10-70 ACN: H2O, reverse phase HPLC).
Example 1A
Biphenyl-2-ylcarbamic acid l-(2- {[4-(4-carbamoylpiperidin-l-ylmethyl)benzoyl] methylamino} ethyl)piperidin-4-yl ester was also prepared as a diphosphate salt using the following procedure :
5.0 g ofthe product of Example 1 was combined with 80 ml of IPA:ACN (1:1). 4.0 ml of water was added and the mixture heated to 50°C under stining, forming a clear solution. To this was added dropwise at 50°C, 16 ml 1M phosphoric acid. The resulting cloudy solution was stined at 50°C for 5 hours, then allowed to cool to ambient temperature, under slow stirring, overnight. The resulting crystals were collected by filtration and air-dried for 1 hour, then under vacuum for 18 hours, to give the diphosphate salt ofthe title compound (5.8 g, 75% yield) as a white crystalline solid (98.3% purity by HPLC).
Example IB
Biphenyl-2-ylcarbamic acid 1 -(2- { [4-(4-carbamoylpiperidin- 1 -ylmethyl)benzoyl] methylamino }ethyl)piperidin-4-yl ester was also prepared as a monosulfate salt using the following procedure.
442 mg ofthe product of Example 1 (0.739 mmol of 96% pure material) was taken up in 5 ml of H2O:ACN (1:1) and 1.45 ml of IN sulfuric acid was added slowly, while monitoring the pH. The pH was adjusted to approx. pH 3.3. The clear solution was filtered through a 0.2 micron filter, frozen and lyophilized to dryness. 161 g of the lyophilized material was dissolved in 8.77 ml of IPA:ACN (10:1). The suspension was heated by placing the vial in a pre-heated 70°C water bath for 1.5 hours. Oil droplets formed within 5 minutes. The heat was lowered to 60°C and the mixture heated for an additional 1.5 hours, followed by heating at 50°C for 40 minutes, at 40°C for 40 minutes, then at 30°C for 45 minutes. The heat was turned off and the mixture was allowed to slowly cool to room temperature. The next day, the material was viewed under a microscope and indicated needles and plates. The material was then heated at 40°C for 2 hours, at 35°C for 30 minutes, and then at 30°C for 30 minutes. The heat was turned off and the mixture was allowed to slowly cool to room temperature. The solid was then filtered and dried using a vacuum pump for 1 hour to give the monosulfate salt ofthe title compound (117 mg, 73% yield).
Example IC
Biphenyl-2-ylcarbamic acid l-(2- {[4-(4-carbamoylpiperidin-l-ylmethyl)benzoyl] methylamino} ethyl)piperidin-4-yl ester was also prepared as a dioxalate salt using the following procedure.
510 mg ofthe product of Example 1 (0.853 mmol of 96% pure material) was taken up in 5 ml of H2O:ACN (1:1) and 1.7 ml of 1M aqueous oxalic acid was added slowly, while monitoring the pH. The pH was adjusted to approx. pH 3.0. The clear solution was filtered through a 0.2 micron filter, frozen and lyophilized to dryness. 150 mg ofthe lyophilized material was dissolved in 13.1 ml of 94%IPA/6%H20. The mixture was stined in a pre-heated 60°C water bath for 2.5 hours. The heat was turned off and the mixture was allowed to cool to room temperature. The vial was refrigerated at 4°C. After 6 days, an oily material was observed with what appeared to be a crystal on the side ofthe vial. The vial was then allowed to reach room temperature, at which point seeds (synthesis described below) were added and allowed to sit for 16 days. During this time, more crystals were observed to come out of solution. The solid was then filtered and dried using a vacuum pump for 14 hours to give the dioxalate salt ofthe title compound (105 mg, 70% yield).
Seed Synthesis
510 mg ofthe product of Example 1 (0.853 mmol of 96% pure material) was taken up in 5 ml of H2O:ACN (1:1) and 1.7 ml of 1M aqueous oxalic acid was added slowly, while monitoring the pH. The pH was adjusted to approx. pH 3.0. The clear solution was filtered through a 0.2 micron filter, frozen and lyophilized to dryness to yield a dioxalate salt. 31.5 mg of this dioxalate salt was dissolved in 2.76 ml of 94%IPA/6%H20. The mixture was stined in a pre-heated 60°C water bath for 2.5 hours. After 25 minutes, all of the sample was in solution. The heat was turned off and the mixture was allowed to cool to room temperature. The next day, a small amount of viscous material was present. The vial was refrigerated at 4°C. After 4 days, the viscous material was still present. The vial was then placed at room temperature and observed one month later. The material appeared to be solid, and was observed to be crystalline under a microscope. The solid was then » filtered and dried using a vacuum pump for 1 hour to give the dioxalate salt (20 mg, 63.5% yield).
Example ID
Biphenyl-2-ylcarbamic acid 1 -(2- { [4-(4-carbamoylpiperidin- 1 -ylmethyl)benzoyl] methylamino} ethyl)piperidin-4-yl ester was also prepared as a freebase crystal using the following procedure.
230 mg ofthe product of Example 1 was dissolved in 0.2 ml of H O:ACN (1:1), using slight heat. The mixture was then heated in a 70°C water bath for 2 hours. The heat was turned off and the mixture was allowed to cool to room temperature, then refrigerated at 4°C for 1 hour. 50 μl of water was then added (oiled out), followed by the addition of 40 μl of ACN to get the sample back into solution. Seeds (synthesis described below) were added under slow stirring at room temperature. Crystals started to form ,and the mixture was allowed to sit overnight, with slow stirring. The next day, a heat cool cycle was applied (30°C for 10 minutes, 40°C for 10 minutes, then 50°C for 20 minutes). The heat was turned off and the mixture allowed to cool overnight, with slow stirring. The next day, a second heat/cool cycle was applied (60°C for 1 hour, with dissolving observed at 70°C). The heat was turned off and the mixture allowed to cool overnight, with slow stirring. The next day, crystals were present and a third heat cool cycle was applied (60°C for 3 hours). The heat was turned off and the mixture allowed to cool overnight, with slow stirring. The next day, a heat cool cycle was applied (60°C for 3 hours, slow cool, then 60°C for 3 hours). The heat was turned off and the mixture allowed to cool overnight, with slow stirring. After 3 days, the solid was filtered and placed on a high vacuum line to remove all solvent and give a freebase crystal ofthe title compound.
Seed Synthesis
109 mg ofthe product of Example 1 was dissolved in 0.56 ml of H2O:ACN (1:1). The suspension was left in a vial (cap loosely placed on top) to allow for a slower evaporation time. The vial was placed under a nitrogen flow environment, although the nitrogen was not used for evaporation, only for the environment. A precipitate was visible within 1 day, which was observed to be crystalline under a microscope. The solid was then placed on a high vacuum line to remove all solvent to give the freebase crystal.
Quantitative recovery, 97.8% pure by HPLC.
Example IE
Biphenyl-2-ylcarbamic acid 1 -(2- { [4-(4-carbamoylpiperidin- 1 -ylmethyl)benzoyl] methylamino} ethyl)piperidin-4-yl ester was also prepared as a freebase crystal using the following alternate procedure.
70 mg ofthe product of Example 1 was dissolved in 0.1 mL ACN. After addition of 0.3 ml MTBE, the solution appeared cloudy. An additional 50 μl of ACN was added to clarify the solution (155 mg/ml ACNMTBE = 1 :2). The mixture was left in the vial and capped. A solid appeared by the next day. The solid was then filtered and placed on a high vacuum line to remove all solvent and give a freebase crystal ofthe title compound.
PATENT
https://patents.google.com/patent/WO2012009166A1/en
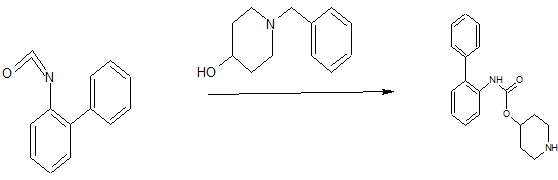
U.S. Patent No. 7,228,657 to Mammen et al. discloses novel biphenyl compounds that are expected to be useful for treating pulmonary disorders such as chronic obstructive pulmonary disease and asthma. In particular, the compound biphenyl-2-ylcarbamic acid 1- (2- {[4-(4-carbamoylpiperidin-l-ylmethyl)benzoyl]methylamino}-ethyl)piperidin-4-yl ester is specifically described in this application as possessing muscarinic receptor antagonist or anticholiner ic activity, and is represented by formula I:

The compound of formula I is synthesized from the compound 8, which is described as being prepared from the oxidation of 2-(benzylmethylamino)ethanol to the aldehyde intermediate followed by reductive amination with biphenyl-2-yl-carbamic acid piperidin- 4-yl ester and debenzylation:


However, while this procedure performs well on small scale, the aldehyde intermediate is difficult to scale up due to its instability, and low yields were typically observed.
Thus, a need exists for an efficient process of preparing compound 8 as a pure material with high chemical purity and good overall yield, without having to isolate intermediates. This invention addresses those needs.
Therapeutic agents useful for treating pulmonary or respiratory disorders are advantageously administered directly into the respiratory tract by inhalation. In this regard, several types of pharmaceutical inhalation devices have been developed for administering therapeutic agents by inhalation including dry powder inhalers, metered- dose inhalers, and nebulizer inhalers. When preparing pharmaceutical compositions and formulations for use in such devices, it is highly desirable to have a crystalline form of the therapeutic agent that is neither hygroscopic nor deliquescent and which has a relatively high melting point thereby allowing the material to be micronized without significant decomposition.
A crystalline diphosphate of the compound of formula I has been reported in U.S. Patent No. 7,700,777 to Axt et al, and a crystalline freebase (identified as Form III) is described in U.S. Patent Application Publication No. 201 1/0015163 to Woollham. All of the aforementioned disclosures are incorporated herein by reference.
The compound of formula I is described as being prepared by reacting compound 8 with 4-carboxybenzaldehyde to form the aldehyde core 10:

which is then isolated prior to being combined with isonipicotamide in the presence of a reducing agent to form the compound of formula I. The crystalline diphosphate is prepared by contacting the separated and purified compound of formula I with phosphoric acid. The crystalline freebase (Form III) can then be prepared from the crystalline diphosphate.
A need also exists for an efficient process of preparing the crystalline freebase (Form III). It is desirable to develop a process that does not first require preparation of the crystalline diphosphate. This invention addresses those needs.




Preparation 1
Biphenyl-2-yl-carbamic acid piperidin-4-yl Ester

Biphenyl-2-isocyanate (97.5 g, 521 mmol) and 1 -benzylpiperidin-4-ol (105 g, 549 mmol) were heated together at 70°C for 12 hours. The mixture was then cooled to 50°C and EtOH (1 L) was added, followed by the slow addition of 6M HC1 (191 mL). The resulting mixture was then cooled to ambient temperature. Ammonium formate (98.5 g, 1.6 mol) was added and then nitrogen gas was bubbled through the solution vigorously for 20 minutes. Palladium on activated carbon (20 g, 10 wt% dry basis) was added and the mixture was heated at 40°C for 12 hours, and then filtered. The solvent was removed under reduced pressure and 1M HC1 (40 mL) was added to the crude residue. The pH of the mixture was adjusted with 10 N NaOH to pH 12. The aqueous layer was extracted with EtOAc (2×150 mL), and the organic layer was dried over MgS04, filtered and the solvent removed under reduced pressure to yield the title compound (155 g). HPLC (10-70) ¾ = 2.52; m/z: [M + H+] calcd for Ci8H2o 202 297.15; found 297.3.
EXAMPLE 1
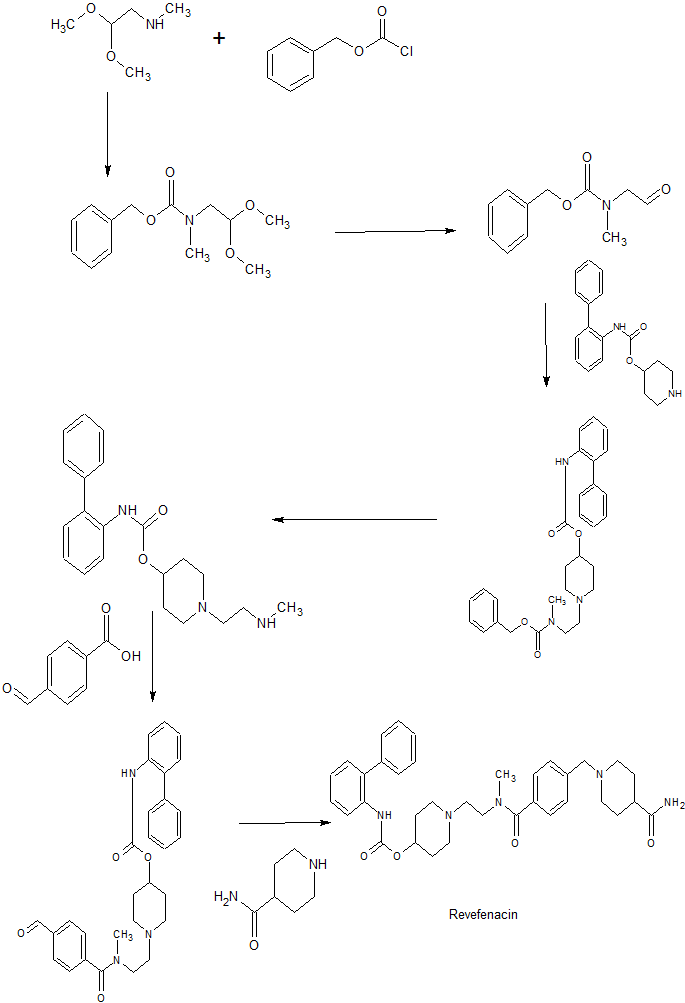
Step A: (2,2-Dimethoxyethyl)methylcarbamic Acid Benzyl Ester

K2CO3 (13.8 g, 100 mmol, 1.76 eq.) and H20 (46 mL) were mixed to form a homogeneous solution. The solution was cooled to 20°C. N-methylaminoacetaldehyde dimethylacetal (12.8 mL, 100 mmol, 1.8 eq) and MeTHF (50 mL) were added. The resulting mixture was cooled to 2°C. Benzyl chloroformate (8.1 mL, 56.7 mmol, 1.0 eq.) was added by syringe over 10 minutes (addition was exothermic). The mixture was maintained at room temperature until completion of the reaction. The layers were separated and the organic layer was washed with IN HC1 (50 mL) and used directly in the next step.
Step B: Methyl-(2-oxoethyl)carbamic Acid Benzyl Ester

The mixture from the previous step was combined with a 3N HC1 solution (70 mL), and the resulting mixture was stirred for 18 hours at 22°C to yield a clear homogeneous pale yellow solution. Solid aHC03 was added to the solution to bring the pH to neutral. The layers were separated and the aqueous layer was back-extracted with MeTHF (20 mL). The organic layers were combined and washed with a saturated aHC03 solution (50 mL). The layers were separated and the organic layer was dried over Na2S04, filtered and concentrated to dryness to afford the title compound (1 1.9 g) as a pale yellow oil.
Step C: Biphenyl-2-yl-carbamic acid l-[2-(benzyloxycarbonyl
methylamino)ethyl]piperidin-4-yl Ester

Biphenyl-2-yl-carbamic acid piperidin-4-yl ester (31.1 g, 105 mmol, 1.0 eq.) and MeTHF (150 mL) were mixed. A solution of methyl-(2-oxoethyl)carbamic acid benzyl ester (23 g, 113.4 mmol, 1.05 eq.) in MeTHF (150 mL) was prepared and added to the ester mixture. The resulting mixture was heated to 30°C for a few minutes, then cooled to room temperature over 1 hour. The mixture was then cooled to 3°C and the temperature maintained for 1 hour. NaHB(OAc)3 (35.1 g, 170 mmol, 2.0 eq.) was added portion-wise while maintaining the internal temperature at 7±1°C. After addition, the mixture was allowed to warm to room temperature until the reaction was complete. A saturated solution of aHC03 (3000 mL) was added, stirred for 20 minutes, and the layers separated. This was repeated, after which the organic layer was dried over a2S04. The material was filtered, concentrated and dried under high vacuum to afford the title compound (43 g) as a thick colorless to pale yellow oil, which was used directly in the next step without purification.
Step D: Biphenyl-2-yl-carbamic acid l-(2-methylaminoethyl)piperidin-4-yl Ester

Biphenyl-2-yl-carbamic acid l-[2-(benzyloxycarbonyl methylamino)ethyl] piperidin-4-yl ester (53 g, 105 mmol, 1 eq.), MeOH (250 mL), and MeTHF (50 mL) were combined under nitrogen. 10% palladium on carbon (0.8 g) was added and hydrogen was bubbled into the mixture for 1 minute. The reaction vessel was sealed and stirred under hydrogen at atmospheric pressure for three hours. The mixture was then filtered, and the solids were washed MeTHF (10 mL).
The filtrate and washes were combined and concentrated under reduced pressure (250 mL removed). MTBE (100 mL) was added, and the solution again concentrated under reduced pressure (100 mL removed). MTBE (200 mL) was added and the solution was seeded with a few milligrams of biphenyl-2-yl-carbamic acid l-(2-methylaminoethyl) piperidin-4-yl ester, and the mixture was maintained for 3 hours. The solids were collected and the vessel and filter cake were washed with MTBE (2×15 mL). The material was dried to yield 13.2 g of the title compound (99.5% pure). This process was repeated to yield the title compound (12.5 g, 98.6% pure). The filtrate and washes were combined and concentrated under reduced pressure. MTBE (150 mL) was added and the solution was seeded with a few milligrams of biphenyl-2-yl-carbamic acid l-(2-methylaminoethyl) piperidin-4-yl ester, and the mixture was maintained for 20 hours. The solids were collected and the vessel and filter cake were washed with MTBE (2×15 mL). The material was dried to yield the title compound (5 g, 90% pure).
A portion of the three crops (13 g , 12 g, 4.5 g, respectively) were combined taken up in IPA (90 mL). The resulting slurry was heated to 45°C, then cooled to room temperature over 1 hour. The slurry was stirred for 5 hours at 25°C. The solids were collected and washed with IPA (2×15 mL). The solids were then dried for 1 hour to yield the title compound (25 g, >99% pure).
EXAMPLE 2
All volumes and molar equivalents are given relative to biphenyl-2-yl-carbamic acid piperidin-4-yl ester.
Step A: (2,2-Dimethoxyethyl)methylcarbamic Acid Benzyl Ester K2C03 (8.4 kg, 60 mol, 1.8 eq.) and H20 (49.3 kg, 2.6 volumes) were placed in the reaction vessel and stirred. N-methylaminoacetaldehyde dimethylacetal (6.5 kg, 54 mol, 1.6 eq) and MeTHF (20.2 kg, 2.9 volumes) were added. The resulting mixture was cooled to 5°C. Benzyl chloroformate (6.8 kg, 37.6 mol, 1.1 eq.) was added over a period of about 30 minutes, while maintaining the temperature below 10°C. The feed line was rinsed with MeTHF (4.3 kg). The mixture was then maintained at 5°C and stirred for 1 hour. The layers were separated and the organic layer was washed with IN HC1 (14.3 kg, 1 1.7 mol, 1.4 volumes) and used directly in the next step.
Step B: Methyl-(2-oxoethyl)carbamic Acid Benzyl Ester
The mixture from the previous step was combined with water (23.4 kg,
2.9 volumes) and 30% hydrochloric acid (13.1 kg, 107.7 mol, 1.1 volumes). Water (5.1 kg) was used to rinse the feed line. The temperature was adjusted to 25-30°C, and the reaction was run for 16-24 hours. A 25% NaOH solution (1 1.8 kg, 71.1 mol, 2.2 eq.) was added to the solution to adjust the pH and obtain phase separation.
The layers were separated and the aqueous layer was back-extracted with MeTHF
(10.0 kg, 1.1 volumes). The aqueous layer was discarded and the organic layers were combined. MeTHF (4.4 kg) was used to rinse the feed line. The organics were washed with a saturated aHC03 solution (14.6 kg, 15.6 mol, 1.1 volumes). The layers were separated and the organic layer was dried over a2S04 (2.5 kg, 17.6 mol) for 60-90 minutes. The drying agent was filtered off and the remaining solids were washed with
MeTHF (8.8 kg, 1 volume). The reaction vessel was washed with water and MeOH before continuing with the next step.
Step C: Biphenyl-2-yl-carbamic acid l-[2-(benzyloxycarbonyl
methylamino) ethyl Jpiperidin-4-yl Ester
The product from the previous step (in MeTHF) and biphenyl-2-yl-carbamic acid piperidin-4-yl ester (10.0 kg, 32.6 mol, 1.0 eq.) in MeTHF (28.5 kg) were placed in the reaction vessel and heated to 30°C for one hour. The mixture was then cooled to 5°C. NaHB(OAc)3 (10.0 kg, 45.8 mol, 1.4 eq.) was added portion wise over a period of 40 minutes while maintaining the temperature below 20°C. The mixture was then stirred for 30 minutes. Additional NaHB(OAc)3 (0.5 kg) was added the reaction allowed to progress to completion. A saturated solution of NaHCC^ (14.3 kg, 15.3 mol, 1.1 volumes) was added and stirred for 10 minutes. The aqueous phase was separated and discarded. A 33% NaOH solution (15.8 kg, 129.9 mol, 4.0 eq.) was added to the reaction mixture to adjust the H to be in the range of 8-12. Water (40 kg) was added in two portions, after which phase separation occurred. A saturated NaHCC (7.1 kg, 7.6 mol, 0.7 volumes) was added to the reaction mixture and stirred for 10 minutes. The aqueous phase was separated and discarded. Additional water (4.9 kg) was added to dissolve any remaining salts and a vacuum distillation was conducted at a maximum temperature of 45°C to remove part of the solvent (7.2 volumes). MeOH (56.1 kg, 7.2 volumes) was added to the reaction mixture before continuing with the next step.
Step D: Biphenyl-2-yl-carbamic acid l-(2-methylaminoethyl)piperidin-4-yl Ester
10% palladium on carbon (0.4 kg, 0.03 wt%, Degussa type 101 NE/W) was added to the reaction mixture. A hydrogenation reaction was performed to remove the benzyloxycarbonyl protective group, with reaction conditions at 30±5°C and 4 bar pressure. The reaction was run until completion. The mixture was then filtered and the filter cake was washed with MeOH (8.0 kg, 1.0 volume). The reaction was continued in a clean vessel, which was charged with the product solution (in MeTHF/MeOH) from the hydrogenation reaction. 3-Mercaptopropyl silica (0.6 kg, 0.07 wt%, Silicycle) was added. MeOH (4.8 kg) was used to rinse the feed line. The reaction mixture was stirred for 14-72 hours at 25±5°C. Activated carbon (0.7 kg, 0.07 wt%) was added and the mixture stirred for 30 minutes. The mixture was filtered and the filter cake was washed with MeOH (1.0 volume). The reaction was continued in a clean vessel, which was charged with the product solution (in MeTHF/MeOH), and MeOH (4.2 kg) was used to rinse the feed line. The mixture was heated to 40-45°C and a vacuum distillation was performed to bring the final volume to 5.6 volumes (removal of methanol).
2-propanol (40.2 kg, 5.0 volumes) was added and distillation continued until the volume was reduced to 2.5 volumes. The solids were then isolated by filtration and washed with MTBE (1.5 volumes) to yield the product as a wet cake (8.6 kg, 96.8% purity). The cake was charged to the reaction vessel and additional 2-propanol
(1.9 volumes) was added. The mixture was warmed to 40±5°C, and maintained at that temperature for 2 hours. The mixture was then slowly cooled over a minimum of 4 hours to 20°C, then actively cooled to 5-10°C, followed by stirring for 2 hours. The product was filtered and the resulting cake washed with MTBE (1.0 volume). The solids were then dried under atmospheric conditions to yield the title compound (6.6 kg, 98.5% purity).
EXAMPLE 3
Crystalline Freebase of Biphenyl-2-yl-carbamic Acid l- {2-r(4-carbamoylbenzoyl) methylaminolethyllpiperidin-4-yl Ester (Form III)
Biphenyl-2-yl-carbamic acid l-{2-[(4-formylbenzoyl)
methylamino ] ethyl }piperidin-4-yl Ester

4-Carboxybenzaldehyde (9 g, 60 mmol, 1.0 eq.) and biphenyl-2-yl-carbamic acid 1-
(2-methylaminoethyl)piperidin-4-yl ester (21.2 g, 60 mmol, 1.0 eq.) were combined in MeTHF (115 mL). The mixture was stirred for 0.5 hours, forming a thick slurry.
Additional MeTHF (50 mL) was added to form a free-flowing slurry. 4-(4,6-dimethoxy- l,3,5-triazin-2-yl)-4-methylmorpholinium chloride (18 g, 63 mmol, 1.1 eq., 97% pure) was added in two portions and the funnel rinsed with additional MeTHF (50 mL). The mixture was stirred at room temperature overnight. MeCN (50 mL) was added and the mixture was filtered. The solids were washed with MeTHF (30 mL). The filtrate and washes were combined and a saturated aHC03 solution (100 mL) was added and stirred for 10 minutes. The layers were separated and a saturated NaCl solution (100 mL) was added and stirred for 10 minutes. The layers were separated and the aqueous layer discarded. The resulting solution was concentrated under reduced pressure and held at room temperature for three days, then used directly in the next step.
Step B: Biphenyl-2-yl-carbamic acid l-{2-[(4-carbamoylbenzoyl)
meth lamino] ethyl}piperidin-4-yl ester (non-isolated form)

Isonipecotamide (15.4, 120 mmol, 2.0 eq.) and IPA (200 mL) were added to the solution of biphenyl-2-yl-carbamic acid l-{2-[(4-formylbenzoyl)methylamino]ethyl} piperidin-4-yl ester from the previous step. Liquid (200 mL) was distilled off and additional IPA (400 mL) was added under reduced pressure at 60°C. Liquid (400 mL) was distilled off over a period of 1.5 hours and additional IPA (600 mL) was added. Liquid (100 mL) was distilled off and the remaining solution was cooled to 30°C to yield a hazy white mixture, which was then added to Na2S04 (18 g). The flask was rinsed with IPA (100 mL) and added to the solution. The resulting mixture was cooled to room
temperature and AcOH (20 mL, 360 mmol, 6.0 eq.) was added. The mixture was cooled to 18°C with an ice bath and NaHB(OAc)3 (38.2 g, 180 mmol, 3.0 eq.) was added over 5 minutes. The mixture was allowed to warm up to 25°C and was maintained at that temperature for 2 hours. Solvent was removed under reduced pressure, and the remaining material was used directly in the next step.
Step C: Biphenyl-2-yl-carbamic acid l-{2-[(4-carbamoylbenzoyl)
methylamino]ethyl}piperidin-4-yl ester (isolated solid)
iPrOAc (300 mL) was added to the material, followed by the addition of water (200 mL). The pH of the solution was adjusted to pH 1 with 3N HC1 (-150 mL). The layers were separated and the organic layer was discarded. The aqueous layer was collected, and iPrOAc (300 mL) was added. The pH of the solution was adjusted to basic pH with 50 wt% NaOH (-100 mL). The resulting mixture was stirred for 15 minutes and the layers were separated. The organic layer was filtered and seeded with micronized crystalline freebase of biphenyl-2-yl-carbamic acid l- {2-[(4-carbamoylbenzoyl) methylamino]ethyl}piperidin-4-yl ester (Form III; prepared as described in U.S. Patent Application Publication No. 201 1/0015163 to Woollham) and stirred overnight at room temperature to yield a white slurry. Stirring was continued for 8 hours at room temperature and for 16 hours at 5°C (cold room). The mixture was slowly filtered under pressure. The cake was washed with cold iPrOAc (2×20 mL) and dried under nitrogen to yield a white solid (27.5 g). The material was further dried in a vacuum oven at 30°C for 24 hours to yield 25.9 g.
Step D: Crystalline Freebase of Biphenyl-2-yl-carbamic Acid l-{2-[ ( 4- carbamoylbenzoyl)methylamino]ethyl}piperidin-4-yl Ester (Form III) The white solid (5 g, 60 mmol, 1.0 eq.) was dissolved in toluene (75 mL) and the resulting mixture was heated to 82°C to yield a clear solution. The solution was filtered. The solids were washed with toluene (2 x 5 mL), and the filtrate and washes were combined. The mixture was cooled to 60°C and seeded with micronized crystalline freebase of biphenyl-2-yl-carbamic acid l-{2-[(4-carbamoylbenzoyl)methylamino]ethyl} piperidin-4-yl ester (Form III; prepared as described in Example 3 in U.S. Patent
Application Publication No. 201 1/0015163 to Woollham). The mixture was maintained at 55°C for 2 hours, then cooled to room temperature on an oil bath overnight (~16 hours). The resulting slurry was then filtered and the cake was dried for 3 hours to yield a solid while material (4.6 g). The material was further dried in a vacuum oven at 30°C for 24 hours (exhibited no further weight loss) to yield the title compound (4.6 g).
The product was analyzed by powder x-ray diffraction, differential scanning calorimetry and thermal gravimetric analysis, and was determined to be the crystalline freebase (Form III) of biphenyl-2-ylcarbamic acid l-(2-{[4-(4-carbamoylpiperidin-l- ylmethyl)benzoyl]methylamino}ethyl)piperidin-4-yl ester described in U.S. Patent Application Publication No. 201 1/0015163 to Woollham.
| Patent ID | Title | Submitted Date | Granted Date |
|---|---|---|---|
| US9415041 | Crystalline freebase forms of a biphenyl compound |
2015-12-01
|
2016-08-16
|
| US9249099 | CRYSTALLINE FORMS OF A BIPHENYL COMPOUND |
2014-11-25
|
2015-06-04
|
| US8921396 | Crystalline freebase forms of a biphenyl compound |
2013-08-22
|
2014-12-30
|
| US7521041 | Biphenyl compounds useful as muscarinic receptor antagonists |
2008-04-24
|
2009-04-21
|
| US2007112027 | Crystalline forms of a biphenyl compound |
2007-05-17
|
| Patent ID | Title | Submitted Date | Granted Date |
|---|---|---|---|
| US8017783 | Biphenyl compounds useful as muscarinic receptor antagonists |
2008-03-20
|
2011-09-13
|
| US7550595 | Biphenyl compounds useful as muscarinic receptor antagonists |
2007-12-20
|
2009-06-23
|
| US9283183 | BIPHENYL COMPOUNDS USEFUL AS MUSCARINIC RECEPTOR ANTAGONISTS |
2014-11-12
|
2015-06-18
|
| US2010048622 | CRYSTALLINE FORMS OF A BIPHENYL COMPOUND |
2010-02-25
|
|
| US9452161 | Biphenyl compounds useful as muscarinic receptor antagonists |
2016-02-05
|
2016-09-27
|
| Patent ID | Title | Submitted Date | Granted Date |
|---|---|---|---|
| US8754225 | PROCESS FOR PREPARING A BIPHENYL-2-YLCARBAMIC ACID |
2012-01-19
|
|
| US8921395 | Crystalline forms of a biphenyl compound |
2014-03-19
|
2014-12-30
|
| US8716313 | Crystalline forms of a biphenyl compound |
2013-01-14
|
2014-05-06
|
| US8557997 | Biphenyl compounds useful as muscarinic receptor antagonists |
2012-08-23
|
2013-10-15
|
| US8541451 | CRYSTALLINE FREEBASE FORMS OF A BIPHENYL COMPOUND |
2011-01-20
|
| Patent ID | Title | Submitted Date | Granted Date |
|---|---|---|---|
| US8377965 | CRYSTALLINE FORMS OF A BIPHENYL COMPOUND |
2010-10-07
|
|
| US8242137 | CRYSTALLINE FORMS OF A BIPHENYL COMPOUND |
2010-01-28
|
2012-08-14
|
| US2017204061 | BIPHENYL COMPOUNDS USEFUL AS MUSCARINIC RECEPTOR ANTAGONISTS |
2016-08-30
|
|
| US9765028 | CRYSTALLINE FREEBASE FORMS OF A BIPHENYL COMPOUND |
2016-07-11
|
|
| US9035061 | PROCESS FOR PREPARING A BIPHENYL-2-YLCARBAMIC ACID |
2013-11-26
|
2014-05-01
|
| Patent ID | Title | Submitted Date | Granted Date |
|---|---|---|---|
| US7803812 | BIPHENYL COMPOUNDS USEFUL AS MUSCARINIC RECEPTOR ANTAGONISTS |
2009-09-10
|
2010-09-28
|
| US7910608 | Biphenyl compounds useful as muscarinic receptor antagonists |
2009-01-15
|
2011-03-22
|
| US7491736 | Biphenyl compounds useful as muscarinic receptor antagonists |
2007-12-20
|
2009-02-17
|
| US7585879 | Biphenyl compounds useful as muscarinic receptor antagonists |
2007-11-15
|
2009-09-08
|
| US7288657 | Biphenyl compounds useful as muscarinic receptor antagonists |
2005-09-15
|
2007-10-30
|
| Patent ID | Title | Submitted Date | Granted Date |
|---|---|---|---|
| US8912334 | Biphenyl compounds useful as muscarinic receptor antagonists |
2013-09-11
|
2014-12-16
|
| US8273894 | Biphenyl compounds useful as muscarinic receptor antagonists |
2012-04-03
|
2012-09-25
|
| US8173815 | BIPHENYL COMPOUNDS USEFUL AS MUSCARINIC RECEPTOR ANTAGONISTS |
2011-12-29
|
2012-05-08
|
| US8053448 | BIPHENYL COMPOUNDS USEFUL AS MUSCARINIC RECEPTOR ANTAGONISTS |
2011-06-02
|
2011-11-08
|
| US8034946 | BIPHENYL COMPOUNDS USEFUL AS MUSCARINIC RECEPTOR ANTAGONISTS |
2010-09-30
|
2011-10-11
|
/////////TD-4208, UNII:G2AE2VE07O, ревефенацин , ريفيفيناسين , 瑞维那新 , GSK 1160724, revefenacin, PHASE 3
CN(CCN1CCC(CC1)OC(=O)NC2=CC=CC=C2C3=CC=CC=C3)C(=O)C4=CC=C(C=C4)CN5CCC(CC5)C(=O)N

