
Home » 2017 (Page 11)
Yearly Archives: 2017
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
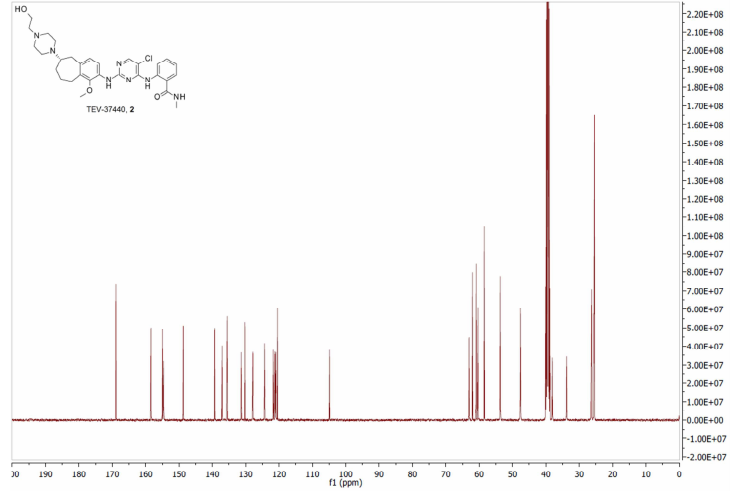
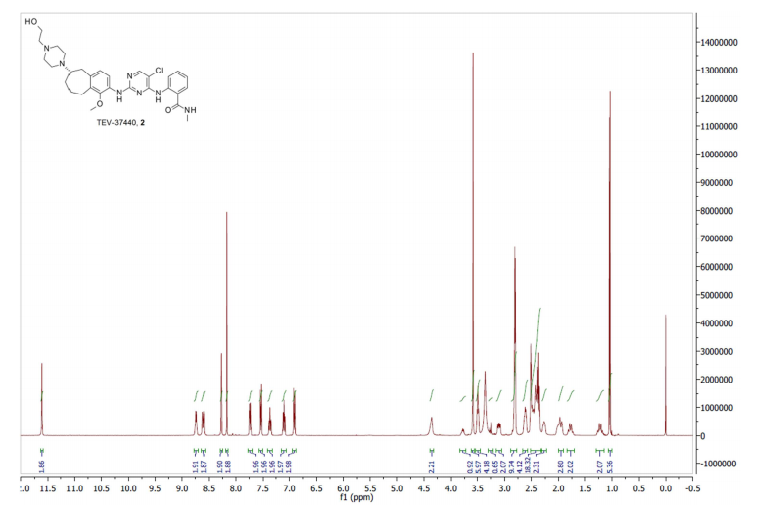
TEV-37440, CEP-37440
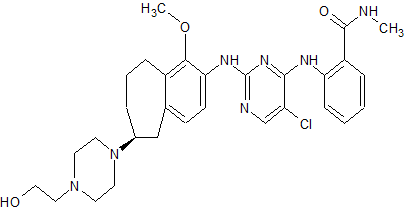
- Molecular Weight, 580.12
2-[[5-chloro-2-[[(6S)-6-[4-(2-hydroxyethyl)piperazin- 1 -yl]- 1 -methoxy-6,7,8,9-tetrahydro-5H-benzo[7]annulen-2-yl]amino]pyrimidin-4-yl]amino]-N-methyl-benzamide, and is also known as CEP-37440.
![]()
| Applicants: | CEPHALON, INC. [US/US]; 41 Moores Road P.O. Box 4011 Frazer, Pennsylvania 19355 (US) |
| Inventors: | COURVOISIER, Laurent; (US). JACOBS, Martin J.; (US). OTT, Gregory R.; (US). ALLWEIN, Shawn P.; (US) |
Anaplastic Lymphoma Kinase (ALK) is a cell membrane-spanning receptor tyrosine kinase, which belongs to the insulin receptor subfamily. The most abundant expression of ALK occurs in the neonatal brain, suggesting a possible role for ALK in brain development (Duyster, J. et al, Oncogene, 2001, 20, 5623-5637).
ALK is also implicated in the progression of certain tumors. For example, approximately sixty percent of anaplastic large cell lymphomas (ALCL) are associated with a chromosome mutation that generates a fusion protein consisting of nucleophosmin (NPM) and the intracellular domain of ALK. (Armitage, J.O. et al., Cancer: Principle and Practice of Oncology, 6th edition, 2001, 2256-2316; Kutok J.L. & Aster J.C., J. Clin. Oncol, 2002, 20, 3691-3702). This mutant protein, NPM- ALK, possesses a constitutively active tyrosine kinase domain that is responsible for its oncogenic property through activation of downstream effectors. (Falini, B. et al, Blood, 1999, 94, 3509-3515; Morris, S.W. et al, Brit. J. Haematol, 2001, 113, 275-295; Duyster et al; Kutok & Aster). In addition, the transforming EML4-ALK fusion gene has been identified in non-small-cell lung cancer (NSCLC) patients (Soda, M., et al, Nature, 2007, 448, 561 – 566) and represents another in a list of ALK fusion proteins that are promising targets for ALK inhibitor therapy. Experimental data have demonstrated that the aberrant expression of constitutively active ALK is directly implicated in the pathogenesis of ALCL and that inhibition of ALK can markedly impair the growth of ALK+ lymphoma cells (Kuefer, Mu et al. Blood, 1997, 90, 2901-2910; Bai, R.Y. et al, Mol. Cell Biol, 1998, 18, 6951-6961; Bai, R.Y. et al, Blood, 2000, 96, 4319-4327; Ergin, M. et al, Exp. Hematol, 2001, 29, 1082-1090; Slupianek, A. et al, Cancer Res., 2001, 61, 2194-2199; Turturro, F. et al, Clin. Cancer Res., 2002, 8, 240-245). The constitutively activated chimeric ALK has also been demonstrated in about 60% of inflammatory myofibroblastic tumors (IMTs), a slow-growing sarcoma that mainly affects children and young adults. (Lawrence, B. et al., Am. J. Pathol, 2000, 157, 377-384; Duyster et al).
In addition, ALK and its putative ligand, pleiotrophin, are overexpressed in human glioblastomas (Stoica, G. et al, J. Biol. Chem., 2001, 276, 16772-16779). In mouse studies, depletion of ALK reduced glioblastoma tumor growth and prolonged animal
survival (Powers, C. et al, J. Biol. Chem., 2002, 277, 14153-14158; Mentlein, R. et al, J. Neurochem., 2002, 83, 747-753).
An ALK inhibitor would be expected to either permit durable cures when combined with current chemotherapy for ALCL, IMT, proliferative disorders,
glioblastoma and possible other solid tumors, or, as a single therapeutic agent, could be used in a maintenance role to prevent cancer recurrence in those patients. Various ALK inhibitors have been reported, such as indazoloisoquinolines (WO 2005/009389), thiazole amides and oxazole amides (WO 2005/097765), pyrrolopyrimidines (WO 2005080393), and pyrimidinediamines (WO 2005/016894).
WO 2008/051547 discloses fused bicyclic derivatives of 2,4-diaminopyrimidine as ALK and c-Met inhibitors. The lead drug candidate disclosed in the ‘547 application is CEP-28122, a potent ALK inhibitor with oral efficacy against SUP-M2 and Karpas-299 ALK-dependent tumors in mouse xenograft models. CEP-28122 progressed to IND-enabling studies until its development was terminated due to the unexpected occurrence of severe lung toxicity in CEP-28122-treated monke s.

CEP-28122
Focal adhesion kinase (FAK) is an evolutionarily conserved non-receptor tyrosine kinase localized at focal adhesions, sites of cellular contact with the ECM (extra-cellular matrix) that functions as a critical transducer of signaling from integrin receptors and multiple receptor tyrosine kinases, including EGF-R, HER2, IGF-R1, PDGF-R and VEGF-R2 and TIE-2 (Parsons, JT; Slack-Davis, J; Tilghman, R; Roberts, WG. Focal adhesion kinase: targeting adhesion signaling pathways for therapeutic intervention. Clin. Cancer Res., 2008, 14, 627-632; Kyu-Ho Han, E; McGonigal, T. Role of focal adhesion kinase in human cancer – a potential target for drug discovery. Anti-cancer Agents Med. Chem., 2007, 7, 681-684). The integrin-activated FAK forms a binary complex with Src which can phosphorylate other substrates and trigger multiple signaling pathways. Given the central role of FAK binding and phosphorylation in mediating signal transduction with multiple SH2- and SH3- domain effector proteins (Mitra, SK; Hanson, DA; Schlaeper, DD. Focal adhesion kinase: in command and control of cell motility. Nature Rev. Mol.
Cell Biol, 2005, 6, 56-68), activated FAK plays a central role in mediating cell adhesion, migration, morphogenesis, proliferation and survival in normal and malignant cells (Mitra et al. 2005; McLean, GW; Carragher, NO; Avizzienyte, E; et al. The role of focal adhesion kinase in cancer – a new therapeutic opportunity. Nature Reviews Cancer, 2005, 5, 505-515; and Kyu-Ho Han and McGonigal, 2007). In tumors, FAK activation mediates anchorage-independent cell survival, one of the hallmarks of cancer cells. Moreover, FAK over expression and activation appear to be associated with an enhanced invasive and metastatic phenotype and tumor angiogenesis in these malignancies (Owens, LV; Xu, L; Craven, RJ; et al. Over expression of the focal adhesion kinase (pi 25 FAK) in invasive human tumors. Cancer Res., 1995, 55, 2752-2755; Tremblay, L; Hauck, W. Focal adhesion kinase (ppl25FAK) expression, activation and association with paxillin and p50CSK in human metastatic prostate carcinoma. Int. J. Cancer, 1996, 68, 164-171; Kornberg, IJ. Focal adhesion kinase in oral cancers. Head and Neck, 1998, 20: 634-639; Mc Clean et al 2005; Kyu-Ho Han and McGonigal, 2007) and correlated with poor prognosis and shorter metastasis-free survival.
Multiple proof-of-concept studies conducted in various solid tumors using siRNA
(Haider, J; Kamat ,AA; Landen, CN; et al. Focal adhesion kinase targeting using in vivo short interfering RNA delivery in neutral liposomes for ovarian carcinoma therapy. Clin. Cancer Res., 2006, 12, 4916-4924), dominant-negative FAK, and small molecule FAK inhibitors (Haider, J; Lin, YG; Merritt, WM; et al. Therapeutic efficacy of a novel focal adhesion kinase inhibitor, TAE226 in ovarian carcinoma. Cancer Res., 2007, 67, 10976-10983; Roberts, WG; Ung, E; Whalen, P; et al. Anti-tumor activity and pharmacology of a selective focal adhesion kinase inhibitor, PF-562,271. Cancer Res., 2008, 68, 1935-1944; Bagi CM; Roberts GW; and Andersen CJ. Dual focal adhesion kinse/Pyk2 inhibitor has positive effects on bone tumors – implications for bone metastases. Cancer, 2008, 112, 2313-2321) have provided pre-clinical support for the therapeutic utility of FAK inhibition as an anti-tumor/anti-angiogenic strategy, particularly for androgen-independent prostate cancers, breast cancers, and HNSCCs. In preclinical models of human breast cancer (MDA-MB-231) in nude rats, administration of a small molecule FAK inhibitor (PF-562,271) inhibited primary tumor growth and intra-tibial tumor spread, and restored tumor-induced bone loss (Bagi et al, 2008). Roberts et al, (2008) showed that PF-562,271 inhibited bone metastases, prevented bone resorption, and increased osteogenesis in breast and androgen-independent prostate cancer patients with and without bone metastases, supporting an additional benefit of FAK inhibition in these specific malignancies.
In summary, there is clear genetic and biological evidence that links aberrant ALK activation and constitutive activation of FAK with the onset and progression of certain types of cancer in humans. Considerable evidence indicates that ALK- and FAK-positive tumor cells require these oncogenes to proliferate and survive, and in the case of FAK, to invade and metastasize to distant sites, while inhibition of both ALK and FAK signaling leads to tumor cell growth arrest or apoptosis, resulting in objective cytoreductive effects. Inhibition of FAK also results in attenuation of tumor motility, invasiveness, and metastatic spread, particularly in specific cancers characterized by bone metastatic dissemination and osteolytic disease. FAK activation protects tumor cells from
chemotherapy-induced apoptosis, contributing to tumor resistance; modulation of FAK activity (by siRNA or pharmacologically) potentiates efficacy of chemotherapeutic agents in vivo (e.g., doxorubicin, docetaxel and gemcitabine), suggesting the utility for rational combination therapies in specific cancers. ALK and FAK are minimally expressed in most normal tissues in the healthy adult and are activated and/or dysregulated in specific cancers during oncogenesis and/or during early stages of malignant progression.
Consequently, the on-target effects of treatment with a dual ALK and FAK inhibitor against normal cells should be minimal, creating a favorable therapeutic index.
A need exists for additional safe and effective ALK and/or FAK inhibitors for the treatment of cancer.




The present invention rovides a compound of formula (I)

or a salt form thereof.
The compound of formula (I) has ALK and FAK inhibitory activity, and may be used to treat ALK- or FAK-mediated disorders or conditions.
The present invention further provides a pharmaceutical composition comprising at least one compound of the present invention together with at least one pharmaceutically acceptable excipient.
CEP-37440
The synthesis of 2-(5-chloro-2-{(S)-6-[4-(2-hydroxy-ethyl)-piperazin-l-yl]-l-methoxy-6,7,8,9-tetrahydro-5H-benzocyclohepten-2-ylamino}-pyrimidin-4-ylamino)-N-methyl-benzamide can be carried out according Fig. 1, following the procedures outlined in Steps 1-8.
Stepl : 5-Methoxy-l-methylene-l,2,3,4-tetrahydro-naphthalene: To a slurry of 5- Methoxy-3,4-dihydro-2H-naphthalen-l-one (25 g, 0.14 mol) and methyltriphenylphos-phonium iodide (1.13 eq) in THF (250 mL) at RT was added potassium t-butoxide (1.6 eq) at such a rate as to maintain a temperature no higher than warm to the touch. The reaction was stirred for one hour and concentrated. The reaction was then azeotroped with three volumes of hexane to remove excess t-butanol. Fresh hexane was added the solution was let to stand overnight to effect trituration. The red-brown solid was removed by filtration and the filtratewas washed twice with water and was concentrated. Purification by
chromatography on ISCO (330g Si02 cartridge: stepwise hexane and then DCM) affords the title compound as a pale yellow oil (24 g, 99%). 1H-NMR (400 MHz, CDC13) 7.29 (d, J = 8.0 Hz, 1H), 7.15 (t, J = 8.0 Hz, 1H), 6.76 (d, J = 8.0 Hz, 1H), 5.49 (s, 1H), 4.98 (s, 1H), 3.85 (s, 3H), 2.77 (t, J = 6.4 Hz, 2H), 2.53-2.50 (m, 2H), 1.93-1.87 (m, 2H).
Step 2: l-Methoxy-5,7,8,9-tetrahydro-benzocyclohepten-6-one: 5-Methoxy-l-methylene-l,2,3,4-tetrahydro-naphthalene (23.8 g, 0.137 mol) in 150 mL MeOH added in one portion to freshly prepared solution of thallium(III)nitrate trihydrate (1.0 eq) in 300 mL MeOH. Stirred one minute and 400 mL chloroform added. The solution was filtered and the organics partitioned between dichloromethane and water. The organics were dried (MgS04) and concentrated. Purification by chromatography (ISCO, 330g silica cartridge; stepwise elution hexane (5 min) then 7 minute gradient to 100% dicloromethane (20 min) affords the title compound as the most polar of the products as a pale yellow oil (26g, 97%). 1H-NMR (400 MHz, CDC13) 7.16 (t, J = 7.9 Hz, 1H), 7.84 (d, J = 8.3 Hz, 1H), 6.79 (d, J = 7.5 Hz, 1H), 3.84 (s, 3H), 3.73 (s, 2H), 3.05-3.01 (m, 2H), 2.55 (t, J = 7.0 Hz, 2H), 2.01-1.96 (m, 2H). LC/MS (ESI+) m/z = 191 (M+H)+
Step 3: l-Methoxy-2-nitro-5,7,8,9-tetrahydro-benzocyclohepten-6-one: To potassium nitrate in acetonitrile (50 mL) and trifluoroacetic anhydride (100 mL) at 0°C was added dropwise l-methoxy-5,7,8,9-tetrahydro-benzocyclohepten-6-one (25 g, 0.131 mol) in 50 mL acetonitrile. The reaction was stirred for 2.5 hours while warming to RT. The reaction was concentrated without heat on a rotary evaporator. MeOHwas added and stirred briefly. Reconcentrated and worked-up by partitioning between dichloromethane and sat. sq. sodium bicarbonate solution. The organic layer was separated and dried (Mg2S04), concentrated and purified by chromatography ISCO (330g silica cartridge: gradient elution – 10 to 50% EA:HEX over 60 minutes) affording two isomers. The title compound was the later eluting (10.7 grams, 34.6% yield). 1H-NMR (400 MHz, CDC13) 7.70 (d, J = 8.3 Hz, 1H), 7.06 (d, J = 8.3 Hz, 1H), 3.92 (s, 3H), 3.80 (s, 2H), 3.13-3.09 (m, 2H), 2.60 (t, J = 7.0 Hz, 2H), 2.10-2.03 (m, 2H).
Step 4: 2-[4-(l-Methoxy-2-nitro-6,7,8,9-tetrahydro-5H-benzocyclohepten-6-yl)-piperazin-l-yl]-ethanol: l-Methoxy-2-nitro-5,7,8,9-tetrahydro-benzocyclohepten-6-one (15.09 g, 64.15 mmol) in methylene chloride (870 ml)treated with 2-Piperazin-l-yl-ethanol (3 eq) followed by acetic acid (10 eq). The mixture was stirred at 50°C for 2 hrs and cooled to 0°C and sodium triacetoxyborohydride (4 eq) was added, then warmed to RT and stirred. After a few hours starting material was still present. Added
0.4 eq further of sodium triacetoxyborohydride, then again after 6 hours. Stirred overnight. Poured into a solution of sat. aq. Sodium bicarbonate and ice and made basic to pH 10 with IN sodium hydroxide, extracted 2X dichloromethane, dried MgS04, filtered and concentrated. This material was taken up into ethanol and HC1/
ethanol was added. The resulting precipitate was triturated for 2 hours then filtered. The solid was free-based using NaOH followed by sodium bicarbonate and extracted into dichloromethane to give the title compound (19g, 85% yield). 1H-NMR (400 MHz, CDCI3) 7.56 (d, J = 8.2 Hz, 1H), 7.00 (d, J = 8.2 Hz, 1H), 3.82 (s, 3H), 3.63-3.06 (m, 2H), 3.29-3.24 (m, 1H), 3.00-2.86 (m, 3H), 2.72-2.67 (m, 2H), 2.60-2.51 (m, 8H), 2.46-2.37 (m, 2H), 2.12-2.07 (m, 2H), 1.87-1.78 (m,lH), 1.37-1.29 (m, 1H). LC/MS (ESI+) m/z = 350 (M+H)+
Step 5 : 2-[4-(2-Amino- 1 -methoxy-6,7,8,9-tetrahydro-5H-benzocyclohepten-6-yl)-piperazin- 1 -yl]-ethanol. 2-[4-(l -Methoxy-2-nitro-6,7,8,9-tetrahydro-5H-benzocyclohepten-6-yl)-piperazin-l-yl]-ethanol (19.0 g, 54.4 mmol) was
split into two batches and dissolved in a total of Ethanol (232 mL). 10 % Pd/C (
1.74 g, 1.64 mmol) was divided in half and the reaction was hydrogenated for 3-4 hours at 50 psi. Each reaction mixture was filtered through celite to remove Pd. The filtrates were combined and then concentrated and the title compound isolated as a foamy solid (17.25g, 99% yield). 1H-NMR (400 MHz, CDC13) 6.76 (d, J = 7.9 Hz, 1H), 6.53 (d, J = 7.9 Hz, 1H), 3.72 (broad s, 3H), 3.71 (s, 3H),3.64 (t, J = 5.4 Hz, 2H), 3.26-3.20 (m, 1H), 2.84- 2.72 (m, 5H), 2.62-2.56 (m, 8H), 2.42-2.35 (m, 2H), 2.40-2.37 (m, 1H), 1.81-1.74 (m, 1H), 1.70 (broad s,lH), 1.41-1.33 (m, 1H). LC/MS (ESI+) m/z = 320 (M+H)+
Step 6 : 2-[4-((S)-2-Amino- 1 -methoxy-6,7,8,9-tetrahydro-5H-benzocyclohepten-6-yl)-piperazin-l-yl] -ethanol: 34 grams of racemic 2-[4-(2 -Amino- l-methoxy-6, 7,8,9-tetrahydro-5H-benzocyclohepten-6-yl)-piperazin-l-yl]-ethanol were separated using SFC (supercritical fluid C02) chromatography using a Chiralcel OJ-H (3 x 15 cm) 808041 column with 15% methanol(0.2% DEA)/C02, 100 bar eluent at 80 mL/min flow rate monitoring the wavelength of 220 nm with an injection volume: 0.5 mL, 20 mg/mL ethanol. 16.9 grams of the (R)-enantiomer and 17 grams of the titled compound were isolated with a chemical purity >99% and an enantiomeric excess (ee) >99% (measured using a Chiralcel OJ-H analytical column). NMR and mass were equivalent to the racemic material. The absolute configurationof the first eluting isomer was unambiguously assigned as the (R)-configuration via small-molecule X-ray using anomalous dispersion of the bis-p-bromobenzyl derivative: 4-bromo-benzoic acid 2-{4-[(R)-2-(4-bromo-
benzoylamino)- 1 -methoxy-6,7,8,9-tetrahydro-5H-benzocyclohepten-6-yl]-piperazin-l -yl} -ethyl ester. Thus, the second eluting enantiomer was determined to be (S)-configuration.
Step 7: 2-(2,5-Dichloro-pyrimidin-4-ylamino)-N-methyl-benzamide: 2-Amino-N-methyl-benzamide (24.4 g, 0.16 mol) in DMF (0.5 L) was added 2,4,5-Trichloro-pyrimidine (39 g, 1.3 eq) and Potassium carbonate (1.3 eq). Stired under argon at 75 °C for 5 hrs and then at RT overnight. Poured into 1 L water and precipitate isolated by filtration and washed 1 : 1 acetonitrile: water followed by drying in air stream and under vacuum to afford the title compound as a yellow solid (38 g, 78% yield). 11.70 (s, 1H), 8.74 (d, J = 8.2 Hz, 1H), 8.24 (s, 1H), 7.59 (d, J = 8.4 Hz, 1H), 7.53 (d, J = 8.8 Hz, 1H), 7.16 (t, J = 8.4 Hz, 1H), 6.28 (s, 1H), 3.06 (d, J = 4.7 Hz, 3H).
Step 8 : 2-(5-Chloro-2- {(S)-6-[4-(2-hydroxy-ethyl)-piperazin-l -yl]- 1 -methoxy-6,7,8,9-tetrahydro-5H-benzocyclohepten-2-ylamino}-pyrimidin-4-ylamino)-N-methyl-benzamide: To a sealed vessel 2-[4-((S)-2-Amino-l-methoxy-6,7,8,9-tetrahydro-5H-benzocyclohepten-6-yl)-piperazin-l-yl]-ethano (2.69 g, 8.41 mmol) and 2-(2,5-Dichloro-pyrimidin-4-ylamino)-N-methyl-benzamide (2.00 g, 6.73 mmol) were combined in 1-methoxy-2-propanol (120 mL, 1200 mmol) followed by the addition of Methanesulfonic acid (2.44 mL, 37.7 mmol). The reaction was then heated at 90°C for 18 hours.
The reaction mixture was added to a separatory funnel and diluted with sat. bicarb until a cloudy ppt formed. This was extracted with dichloromethane 3x. The organic
layer was then washed with brine, dried over MgS04, filtered and concentrated. The residue was pumped dry then chromatographed on ISCO flash column. It was injected in dichloromethane onto a normal phase column and eluted on a gradient of 0-10%
(dichloromethane: 10%NH4OH in MeOH). The desired product eluted around 9-10% and the 10% gradient was held until product eluted completely. Mixed fractions concentrated and were chromatographed on the Gilson reverse-phase HPLC gradient elution 0-40%
CH3CN. Chromatogrpahy was repeated using normal phase silica and reverse phase HPLC to effect further purification as desired. Following neutralization and concentration of all the material, the resulting solid was obtained by taking the foam up into EtOAc and concentrating to dryness several times to give the title compound (1.1 g, 28%>). 11.02 (s, 1H), 8.69 (d, J = 8.9 Hz, 1H), 8.13 (s, 1H), 8.08 (d, J = 8.4 Hz, 1Η),7.59-7.50 (m, 2H),
7.41 (s, 1H), 7.13 (t, J = 7.4 Hz, 1H), 6.91 (d, J = 8.1 Hz, 1H), 6.21 (s, 1H), 3.74 (m, 3H), 3.66-3.63 (m, 2H), 3.29-3.23 (m, 1H), 3.06 (d, J = 4.3 Hz, 3H), 2.92-2.72 (m, 5H), 2.66-2.55 (m, 8H), 2.48-2.39 (m, 2H), 2.16-2.10 (m, 2H), 1.87-1.77 (m, 1H), 1.42-1.32 (m, 1H).LC/MS (ESI+) m/z = 580 (M+H)+
CEP-37440 amorphous HC1 salt
2-(5-Chloro-2- {6-[4-(2-hydroxy-ethyl)-piperazin- 1 -yl]- 1 -methoxy-6,7,8,9-tetrahydro-5H-benzocyclohepten-2-ylamino}-pyrimidin-4-ylamino)-N-methyl-benzamide hydrochloride: 2-(5-Chloro-2-{6-[4-(2-hydroxy-ethyl)-piperazin-l-yl]-l-methoxy-6, 7,8,9-tetrahydro-5H-benzocyclohepten-2-ylamino}-pyrimidin-4-ylamino)-N-methyl-benzamide (4.90 g, 8.45 mmol) and 2.5 M of HC1 in ethanol (13.5 niL, 33.8 mmol) were heated until they dissolved in ethanol (164 mL). The reaction was concentrated two times from ethanol, then warmed in a small amount of ethanol until completely dissolved. This solution was allowed to cool slowly with a stirring (<100 rpm). A solid preciptate formed quickly before the solution had cooled. This mixture was allowed to stir until ambient temperature was achieved and then filtered. The solid was washed with ethanol followed by ether then directly pumped dry under high vac to give 2-(5-chloro-2-{6-[4-(2-hydroxy-ethyl)-piperazin-l -yl]- 1 -methoxy-6,7,8,9-tetrahydro-5H-benzocyclohepten-2-ylamino} -pyrimidin-4-ylamino)-N-methyl-benzamide hydrochloride (5.3 grams, quantitative yield). 1H-NMR (MeOD, 400 MHz) δ 8.55 (s, 1H), 8.17 (s, 1H), 7.80 (d, J = 7.7 Hz, 1H), 7.55 (t, J = 6.8 Hz, 1H), 7.46 (broad s, 1H), 7.36 (t, J = 7.2 Hz, 1H), 7.23 (d, J = 8.5 Hz, 1H), 4.00-3.95 (m, 4H), 3.83-3.72 (m, 5H), 3.73 (s, 3H), 3.65-3.59 (m, 2H), 3.47-3.38 (m, 5H), 2.95 (s, 3H), 2.72-2.65 (m, 1H), 2.44-2.38 (m, 1H), 2.29-2.28 (m, 1H), 2.19-2.12 (m, 1H), 1.59-1.49 (m, 1H). LC/MS (ESI+) m/z = 580 (M+H)+.
2-[[5-chloro-2-[[(6S)-6-[4-(2-hydroxyethyl)piperazin-l-yl]-l-methoxy-6,7,8,9-tetrahydro-5Hbenzo[7]annulen-2-yl]amino]pyrimidin-4-yl]amino]-N-methyl-benzamide (CEP-37440) is an orally available dual kinase inhibitor of the receptor tyrosine kinase anaplastic lymphoma kinase (ALK) and focal adhesion kinase (FAK) with antineoplastic activity. See, e.g., WO 2013/134353.
H

CEP-37440
[0003] In view of the surprising and unexpected properties observed with CEP-37440, improved methods for its preparation in high enantiomeric purity are needed.

CEP-37825 CEP-38063-tartrate salt
Scheme 2

CEP-37440
[0059] Preferred methods for asymmetrically producing an intermediate for the formation of CEP-37440 according to the disclosure are depicted in Scheme 3.
Scheme 3

4
[0060] Preferred methods for asymmetrically producing an intermediate for the formation of CEP-37440 according to the disclosure are depicted in Scheme 4.

[0061] The following examples are provided to illustrate the compositions, processes, and properties of the present disclosure. The examples are merely illustrative and are not intended to limit the disclosure to the materials, conditions, or process parameters set forth therein.
EXAMPLES
Chiral HPLC method:
CHIRALCEL AD column
eluent: heptane/isopropanol (90:10)
flow: 1.2 mL/min
detection: 220 nm and 254 nm.
Example 1

5-methoxytetralone CEP-41158
[0063] To a 12L 4-neck round bottom flask was added methoxytetralone (500 g, 2830 mmol, 1 eq) and Pti3PCH3I (1320 g, 3264 mmol, 1.15 eq) as solids via a powder funnel. THF (5 L) was added and the mixture was stirred with overhead stirring at 19 °C. tBuO (525 g, 4683, 1.65 eq) was added portion-wise over 2 hours to maintain a maximum reaction temperature of 47 °C. A THF solution of tBuOK can also be used with similar results. The funnel was washed with additional THF as needed. The resulting slurry was stirred for an additional hour at 30 °C. HPLC analysis showed < 0.5% starting material.
[0064] The reaction was transferred to a single neck flask and concentrated by roto-evaporator and solvent switched to heptanes (approximately 2.5L, removing as much THF as possible). The slurry was filtered, washing with an additional 0.5 L heptanes. The combined filtrant and wash (approximately 3 L) was washed 2 times with water (2 x 250 mL). The water layers were back extracted 1 time with 0.5 L heptanes which was combined with the other organic layers (total volume was approximately 3.5L).
[0065] To remove the residual amounts of Pl^PO, the solution can be dried and cooled (overnight in the cold room) to precipitate out the triphenylphosphine oxide. Filtration results in a clean product stream that can be concentrated to an oil. Alternatively, the initial heptane product stream may be passed through a plug of S1O2 followed by concentration to an oil.
Example 2
![]()
[0066] CEP-41 158 (Limiting Reagent, see Example 1 ) was charged to a reaction vessel followed by methyl tertiary butyl ether (MTBE) (6.3 volumes), isopropanol (3.3 volumes),
Triton X (0.1 volumes), water (6.3 volumes), and 2-iodo-5-methylbenzenesulfonic acid (0.125 eq). While holding the batch at 20-25 °C, oxone (0.65 eq) was added portion-wise over
approximately 1.5 h and then continued to stir at room temperature. HPLC was used to monitor reaction conversion. A typical reaction time was 4 h but the reaction can be stirred overnight, as well.
[0067] The reaction was quenched by the slow addition of sodium sulfite (0.5 eq) over 10 minutes. Peroxide test strips were used to confirm no peroxides remained. NaOH (5N) was then added at <30 °C to obtain a pH of 8 (typically this was approximately 2 volumes of 5N NaOH). Celite was added and the solution was filtered. The aqueous layer was removed and the organics were washed with brine before being concentrated to an oil.
[0068] Bisulfite adduct formation: The above oil was taken up in isopropanol (7 volumes) before adding water (4 volumes). To this solution was slowly added a freshly prepared aqueous sodium bisulfite solution (6.4 M, 2 eq) at ambient temperature. The resulting slurry was stirred overnight then filtered. The solids were washed with isopropanol and then dried at 40 °C in a vacuum oven with a nitrogen bleed.
Example 3

CEP -41609 CEP-41159
[0069] Unless otherwise noted all volumes and equivalents are based on the wt% corrected charge of CEP-41609. To a nitrogen purged 2.0 L jacketed reactor equipped with an anchor overhead stirrer, and a thermocouple probe was charged 1 17.8 g of CEP-41609 (84.9 wt%, 100.0 g, 0.3398 mol). 500 mL of acetonitrile (5 volumes) was then introduced and the jacket was set to 15 °C. 300 mL of deionized H2O (3 volumes) was then charged and the slurry cooled from -17 °C to 10 °C with stirring at 130 RPM. HC1 (86.4 mL, 1 1.8M, 1.019 mol, 3 equiv.) was added in one portion with the slurry at 10 °C. The mixture exothermed to 17.3 °C and the jacket was adjusted to 22 °C. The slurry cooled to 13.8 °C before being warmed back up to 20 °C in 13 minutes. The stirring was turned up to 175 RPM to improve mixing while completing a 30 minute age. The solids had dissolved after 30 minutes. At 57 minutes the stirring was stopped and the biphasic solution was allowed to settle and sit overnight (15 h).
[0070] After sitting overnight, stirring (175 RPM) was reinitiated and the jacket was set to -40 °C. It took 28 minutes for the mixture to reach -15 °C and in that time the jacket was adjusted to -20 °C. With the jacket at -20 and the reaction at -15 °C the first charge of N-chlorosuccinimide (NCS) was done (28.4 g, 0.2127 mol, 0.626 equiv.). The reaction exothermed to -1.2 °C in 1.5 minutes. 4 minutes later with the reaction cooled to -6.9 °C and the jacket still at -20 °C the second charge of NCS was done (85.0 g, 0.6367 mol, 1.874 equiv.). The reaction exothermed to 2.2 °C in 1.5 minutes. The reaction was then cooled to 0 °C in 1 minute and held at 0 ± 2 °C. Acetonitrile (ACN) (25 mL, 0.25 volumes vs wt% corrected CEP-41609) was used to rinse residual NCS off the wall of the reactor and into the reaction just after the second charge. After 2 hours at 0 ± 2 °C an IPC was taken from the organic layer and the HPLC showed undetectable levels of CEP-41608.
[0071] At 2 hours 22 minutes the work up began with the addition of MTBE (650 mL, 6.5 volumes) over 6 minutes at 0 to 1.7 °C. The mixture was stirred at 175 RPM (complete mixing achieved) for 2.5 minutes then stirring was stopped and the layers settled in 2.5 minutes. 280 mL of an aqueous layer was then cut at -1.6 °C. To the remaining 1500 mL of organic solution was charged 650 mL NaCl solution (6.5 volumes, 24 wt% NaCl; 189.2 g NaCl mixed with 599.25 g D.I. H20) over 8 minutes at -1.3 to 1.7 °C. Stirring was increased to 325 RPM to achieve complete mixing. The solution was allowed to stir for 1.5 minutes before stopping the stirring and allowing the layers settle in 3 minutes. 1000 mL of aqueous layer was cut at -0.4 °C. To the remaining 1 100 mL of organic layer was charged 650 mL NaHCC solution (6.5 volumes, 7.5 wt% NaHC03; 53 g NaHC03 mixed with 655.5 g D.I. H20) over 9 minutes at -0.3 to 2.6 °C with stirring at 325 RPM to achieve complete mixing. The solution was allowed to stir for 5 minutes and then the stirring was stopped and the layers settled in 4 minutes. 800 mL of an aqueous layer (pH = 8) was cut at 0.2 °C.
[0072] The remaining organic layer (1000 mL) was checked by HPLC (70.2 A% CEP-41 159, 17.5 A% impurity 1, 5.9 A% impurity 2, 2.9 A% impurity 3). Thejacket was set to 10 °C while a solution of Na2S204 (33.4 g @ 85 wt%, 0.1631 mol, in 326 mL DI H20) was prepared in a capped imax jar. 50 minutes after cutting the NaHCC layer the organic layer was at 8.8 °C. With thejacket at 10 °C the Na2S204 solution was added in one portion with the stirring at 250 RPM. The mixture warms to 15.4 °C and the jacket was adjusted to maintain the reaction at 15 ± 1 °C for 15 minutes. Stirring was then stopped and an HPLC was taken from the organic layer while holding the biphasic solution at 15 °C. The HPLC showed no detectable impurity 1 (76.1 A% CEP-41 159, 6.4 A% impurity 2, 3.1 A% impurity 3, 0.54 A% CEP-41608). After a total of 39 minutes contact time with Na2S204 solution the aqueous layer was cut leaving 925 mL of organic layer. This solution was held overnight with the jacket at 20 °C.
[0073] After 16 hours 1 1 minutes the organic solution was drained into a round bottom flask, rinsing with 100 mL MTBE (1 volume). The solution was then concentrated at 120 mbar
with the bath temperature at 35 °C. Once 590 mL (5.9 volumes vs. wt% corrected CEP-41609) was removed the remaining solution was diluted with 550 mL AcOH (5.5 volumes vs. wt% corrected CEP-41609). This solution was then concentrated again at 65 mbar with the bath temperature at 45 °C. Once 320 mL (3.2 volumes vs. wt% corrected CEP-41609) was removed the distillation was stopped and the remaining solution was weighted (523.39 g) and checked by HPLC and Ή NMR. HPLC showed 66.31 g (87.0% yield) of CEP-41 159 in solution, and the NMR showed 96.0 wt% AcOH (3.1 wt% ACN, 0.9 wt% MTBE; 93 wt% AcOH desired). The desired AcOH solution mass was calculated from the HPLC assay of CEP-41 159 (9 x 66.3 lg = 596.79 g) and the amount of AcOH needed to dilute to this mass was also calculated (596.79-523.39 = 73.4 g x (lmL/1.049g) = 70 mL AcOH). The AcOH solution was then transferred back to the 2 L JLR (which had been rinsed with H2O and dried with an N2 sweep and 40 °C jacket) and 70 mL of AcOH was used to rinse out the round bottom flask and dilute the solution.
[0074] After sitting at 20 °C for 2.5 hours this solution was diluted with 199 mL DI H20 (3 volumes vs. CEP-41 159) while setting the stirring at 225 RPM and the jacket at -30 °C. The resulting homogeneous solution was cooled to -10 °C in 21 minutes. The stirring was set to 324 RPM and after 1 minute at -10 °C seed (249.4 mg, Lot # 3292-1 1 1-Pl ) was added to the solution. A thick slurry formed in <2 minutes (exotherms to -8.6 °C) but the stir speed allowed for good mixing. After 29 minutes added 332 mL DI H2O (5 volumes vs. CEP-41 159) over 12 minutes at -10 to -8 °C. Held the slurry for 28 minutes at -10 °C and then filtered a sample to check the mother liquor losses. Losses looked good at 7.7 mg/g (desired <9 mg g). After 50 minutes at -10 °C with all water added the slurry was filtered. Filtration was quick, taking <1 minute. The flask and cake were washed with 265.3 mL of ambient temperature 25 vol% ACOH H2O (4 volumes vs. CEP-41 159). The resulting mother liquors and wash were assayed and found to contain 6.7% losses (4.0 mg/g CEP-41 159). The solids were held on the filter with the vacuum pulling air across them and with aluminum foil keeping light from them.
[0075] After 67 hours and the solids were then left on the filter for another 48 hours. A total of 58.08 g of white cottony solids were recovered and were checked by HPLC and Ή NMR. HPLC indicates the solids were 100 A% and 102.7 wt% while the NMR showed only trace levels of AcOH. Based on this data the solids were deemed 100% pure and the yield was 76.2%.
Example 4

CEP -41159 CEP-41160
All volumes and equivalents are based on the wt% corrected charge of CEP-41 159 unless other wise stated. Potassium nitrate (22.99 g, 0.2274 mol, 1.02 equivalents) was dissolved in trifluoroacetic acid (TFA) (125 mL, 2.5 volumes). This dissolution took ~5 minutes with vigorous stirring. CEP-41 159 (50.0 g, 0.2229 mol, 1.00 equivalents) was taken up in TFA (125 mL, 2.5 volumes) at 22 °C. This solution was cooled to -13 °C over 21 minutes and then the addition of the potassium nitrate/TFA solution was started. This solution was added in four equal portions. After each portion was added an HPLC was run on a sample of the reaction to evaluate if the reaction had progressed. The sample was diluted in ACN before it could warm up as a temperature rise would likely cause further reaction to occur. The HPLC after the first addition was completed (addition took 13 min at -13 to -6.2 °C) showed 20.9 % conversion. The batch was cooled to -13 °C and the second addition was done over 5 minutes at -13 to -1.2 °C. HPLC after the second addition showed 43.6% conversion. After the reaction was cooled back to -13 °C the third addition was started. This third addition was done over 5 minutes at -13 to -2.7 °C, and the HPLC showed 70.1% conversion. After the reaction was cooled back to -13 °C the final addition was started. This final addition was done over 3 minutes at -13 to -7.5 °C, and the HPLC showed 98.9% conversion. The reaction was held at -13 °C while sodium acetate (22.85 g, 0.2787 mol, 1.25 equivalents) was taken up in DI water (600 mL, 12.0 volumes). After 19 minutes at -14 to -13 °C the reaction was diluted with half of the sodium acetate solution over 3 minutes while allowing the reaction to warm from -14 °C to 14.5 °C. The resulting solution was warmed to 22 °C over 14 minutes and seeded with CEP-41 160 (50 mg, 0.001 x CEP-41 159). The resulting slurry was allowed to stir overnight. After 15 hours 51 minutes the second half of the sodium acetate solution was added over 12 minutes at 21.6-22.6 °C. The resulting slurry was stirred for 34 minutes and then assayed for losses. The losses were at 2.88mg/g. The slurry was then filtered 1 hour 07 minutes after final sodium acetate solution addition was done. The reaction vessel and cake were washed with Dl water (200 mL, 4.0 volumes). The mother liquors and wash were combined and assayed by HPLC to determine they contain 4.3% of the product. The solids were dried in the filter open to air with the vacuum pulling on the bottom of the filter. After 4 hours 52 minutes of drying 52.985g of CEP-41 160 was recovered. These were bright
yellow sandy solids which were 100 A% and 100.8 wt% (@ 238 nm). This is 88.3% yield of CEP-41 160.
Example 5

CEP-37825 CEP-3B063-Tartrate Salt
(A-Ring)
[0076] A glass jar was charged with CEP-37825 (17.0 g physical, 16.7 g corrected, 52.3m mol, 1.00 equiv, Johnson Matthey 4239.A.13.2), MeOH (167 mL) and H20 (40.5 g). The mixture was stirred at ambient temperature until complete dissolution occurred. The resulting solution was filtered over a sintered glass funnel into a 500 mL jacketed reactor, rinsing with MeOH (85 mL). The reactor was evacuated and filled with N2. The solution was heated to 35 °C (internal temperature). A solution of L-tartaric acid (7.85 g, 52.3 mmol, 1.00 equiv) in MeOH (84 mL) was added via addition funnel over 1 1 minutes. Additional MeOH (30 mL) was used as a rinse. The solution was seeded with a slurry of CEP-38063 L-tartrate (50.0 mg) in MeOH (2.5 mL). The vial containing the slurry was rinsed with additional MeOH (1.2 mL). A slurry gradually formed, and the mixture was aged at 33-34 °C for 90 min. The internal temperature was decreased to 29 °C and agitated for 63 min after which the mixture was cooled to 20-25 °C and stirred overnight.
[0077] After stirring overnight at ambient temperature, the mixture was filtered, rinsing with a solution of H20 (2.6 mL) in MeOH (49 mL). The mixture was dried at room temperature overnight under vacuum. Crude CEP-38063 L-tartrate (10.07 g) was obtained in 87.6% de (93.8% dr). The CEP-38063 free base content was 64.2%. The yield was 36% from CEP-37825 racemate.
[0078] Recrystallization: A 1 L OptiMax vessel was charged with two lots of crude CEP-38063 L-tartrate (18.9 g, 89.3% dr, 9.47 g, 88.6% de, 57.1 mmol total). Methanol (346 mL) and H20 (38.8 g) were added and the reactor was placed under nitrogen. The slurry was heated to 66.5 °C during which time the solids dissolved. The solution was then cooled to 55 °C and seeded with a slurry of CEP-38063 L-tartrate (106.9 mg) in MeOH (5.4 mL). The vial containing the slurry was rinsed with additional MeOH (2.6 mL). The slurry was agitated at 53- 54 °C for 82 min. It was then cooled to 48.5 °C over 60 minutes. It was agitated for 1 h, then cooled to 20 °C over 60 minutes.
[0079] After stirring overnight at ambient temperature, the mixture was filtered, rinsing with a solution of H2O (5.7 mL) in MeOH (107 mL). The mixture was dried under vacuum at room temperature overnight. CEP-38063 L-tartrate (21.4 g) was obtained in >99 A%, 99.4% de (99.7% dr) . The CEP-38063 free base content was 65.3%.
Example 6: Procedure for CEP-19036 (amidation and coupling step)
[0084] Into a 20-L jacketed glass reactor were charged isatoic anhydride (500 g, 3.06 mol,) and ethanol (2500 mL). This was followed by the controlled addition of 30 wt% methylamine in ethanol (378.5 g, 3.98 mol) further diluted with ethanol (330 mL) over 70 minutes from an addition funnel at 20±5 °C. The resulting mixture was stirred at 20±5 °C for 60 min and checked by HPLC for reaction completion (<1 A% S ). Upon completion, 13% NaCl
(prepared by dissolving 390 g of NaCl in 2610 mL of DI water) was added over 20.0 min. The resulting mixture was agitated at 20±5 °C for 10 min then allowed to settle for 10 min. The bottom aqueous layer was removed. The organic layer was washed with 26% NaCl (prepared by dissolving 792 g of NaCl in 2210 mL of DI water). The combined aqueous washes were extracted with ethyl acetate (5100 mL). The ethyl acetate extract was combined with the batch and concentrated under reduced pressure (50-80 mmHg) at 30-40 °C in a 12-L round-bottomed flask until the batch volume was approximately 1.5 L (3x the weight of isatoic anhydride). To the residue was added ethyl acetate (5100 mL), which was then concentrated under reduced pressure (50-80 mmHg), a second time, to approximately 1.5L (3 x the weight of isatoic anhydride). To the concentrated batch was added 5.0 L of acetonitrile. The resulting mixture was stirred at room temperature for 60 min, and filtered through a pad of Celite in a sintered glass funnel with fine porosity. The pad was rinsed with acetonitrile (500 mL) and the rinse was combined with the batch. The clear filtrate was transferred to a 20-L jacketed glass reactor. Hunig’s base (712.6 g) and 2,4,5-trichloropyrimidine (657.3 g) were added. The resulting solution was heated to 73±3 °C and agitated at 73±3 °C until the reaction was complete as indicated by an in-process test. The reaction mixture was then cooled to 0±5 °C over 30 min and stirred at this temperature for 1 -2 hrs. The product was collected by vacuum filtration on a sintered glass funnel. The cake was rinsed with acetonitrile (1240 mL) and pulled dry under vacuum with a nitrogen bleed until the residual water and acetonitrile content were less than 1 wt% by NMR analysis and KF titration. A total of 746.3 g (81.9% overall yield) of CEP- 19036 was obtained as a light tan solid with the following quality attributes: 99.8 A%, 98.99 wt%, 0.1 wt% NaCl, 0.1 wt% H20, 0.1 wt% acetonitrile.
Example 7

[0085] CEP-38063-tartrate salt (30.1 g of salt, Limiting Reagent) was charged to a vessel along with 10 volumes of water and 10 volumes of dichloromethane. At room temperature NaOH (10 N aqueous solution, 2 equiv) was added and stirred for 15 minutes. The bottom organic layer was removed and the aqueous layer was washed a second time with 10 volumes of dichloromethane. The combined organics were washed with brine (5 volumes) then concentrated under vacuum by distillation. To the concentrate, l-methoxy-2-propanol (12.7 volumes) was added along with CEP- 19036 (1.25 equiv) and methanesulfonic acid (2.75 eq). The resulting mixture was heated to 70 °C for approximately 48 hours then cooled to room temperature. Water (14 volumes) and dichloromethane (14 volumes) was added and stirred for 15 minutes. The bottom organic layer was cut to waste prior to adding an additional 14 volumes of dichloromethane and NaOH (ION, 3.6 equiv). The bottom organic layer was removed and the aqueous was washed with an additional 14 volumes of dichloromethane. The organic layers were combined, washed with brine and concentrated under vacuum by distillation. Isopropanol (30 volumes) and water (0.6 volumes) was added and heated to 70 °C. The resulting solution was cooled to 50 °C before adding additional water (5.8 volumes) which results in
crystallization. The slurry was then cooled to room temperature, filtered and dried at 60 °C to afford a 74% yield of product with >99% purity.
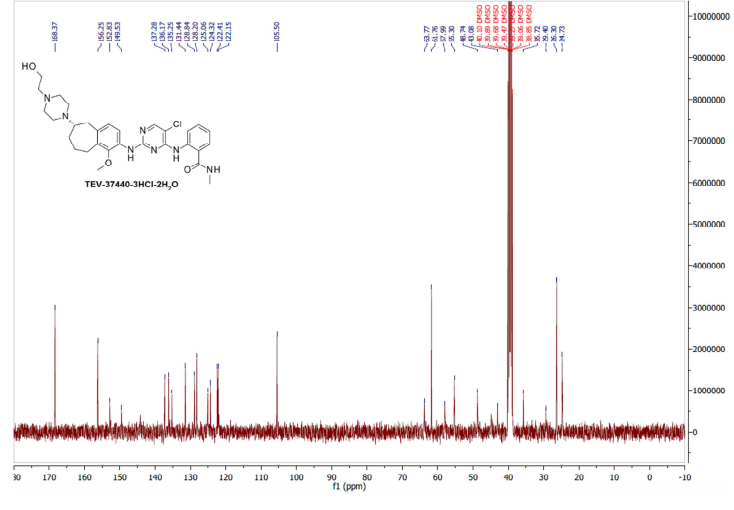
Example 8: Procedure for CEP-37440-3HCl-2H2O (salt formation)
[0086] Combined free base (145.83 g, 251.4 mmol) into a 5 L 3-neck round bottom flask equipped with overhead stirring and an addition funnel. To these solids was added nBuOH which resulted in a yellow cloudy solution (free of large solids) after stirring for 40 minutes. Precipitation occurred immediately and a slight exotherm to 23 °C was observed. The resulting slurry was heated to 85 °C over 45 minutes. Near 60 °C, the solids went into solution. Once the heating reached 80 °C, HC1 (2.5 M in 1 : 1 : 1 MeOH/EtOH/water, 307 mL, 767 mmol) was added via addition over 4 minutes and seed crystals (CEP-37440 H2A3, 1.7 g) were added. The seed held and more solids formed during a 1 h age at 85 °C. The solution was then cooled to room temperature over 1 h then further cooled to 2 °C over an additional 30 minutes. The slurry was stirred at 0-5 °C for lh then filtered, washing with 0 °C nBuOH (400 mL). Initial cake dimensions (cylinder) were 6.0 cm high with a diameter of 13.5 cm. After wash, compressed cake was 4.3 cm high. Losses to the mother liquor were approximately 0.5%. The resulting solids were dried in a vacuum oven at 60 °C for 24 h (with N2 purging after the first 16 h) to give 162 g of the desired product (98.8 HPLC purity, 88% isolated yield assuming 100 wt% starting material, 0.5% residual nBuOH by NMR, XRPD and m.p. confirmed H2A3 salt form).
Example 9: Enamine formation and hydrogenatlon

[0087] To CEP-41 160 (10 g) was added MgS04 (2.5 g, 25 wt%) and dichloromethane (8 volumes wrt CEP-41 160) at room temperature and stirred for 4 days. The resulting slurry was filtered and the filtrant was concentrated to an oil. The oil was then taken up into MTBE (70 mL, 7 volumes) which resulted in crystallization. The slurry was cooled to 0 °C and the product was filtered washing with cold MTBE. The product was dried with nitrogen under vacuum to afford 10.5 g of the product (68%). Additional MTBE crystallizations could be applied to increase purity as desired.
[0088] Prior to the reaction, the NaBArF was dried by co-evaporation with dry toluene (3 times) to remove traces of water. Trifluoroethanol was dried over molecular sieves (Union Carbide, Type 13X). In a pre-dried Schlenk, the appropriate amount of [Ir(COE)2Cl]2 precursor and RD81 was dissolved in dry DCM. After stirring the solution for 30 minutes, the NaBArF was added and stirred for an additional 30 minutes. In a separate Schlenk, the appropriate amount of substrate was dissolved in dry trifluoroethanol and stirred for 30 minutes. To a pre-dried high-pressure autoclave both the catalyst solution and substrate solution were transferred under a gentle stream of dry nitrogen. The autoclave was closed and pressurized to 50 bars of hydrogen and stirred for the desired reaction time. Then, the autoclave was vented and the reaction mixture collected. Work-up of the samples: all volatiles were removed in vacuo.
[
Example 10

I II
[0090] Preparation of hydrogenation substrate was accomplished by condensation of the ketone (1 eq.) and amide (1.1 eq. – 1.3 eq) catalyzed by TsOH (0.05 eq. to 0.1 eq.) in toluene.
[0091] To a solution of I (4.7 g, 20 mmol) in toluene (30 mL, 7 vol) was added acetamide (1.54 g, 26 mmol, 1.3 equiv) and TsOH.H20 (0.02 g, 1 mmol, 0.05 equiv.). The mixture was refluxed under N2 equipped with a Dean-Stark apparatus to remove H20. The progress of the reaction was monitored by TLC. When the reaction was completed, the mixture was cooled down. The solution was washed with H20, dried over Na2S04 and concentrated. The residue was purified by silica gel column chromatography with hexane/ethyl acetate (5/l→ 1/1, v/v) as eluent to give the desired product as a yellow solid (5.0 g, 90% yield).
Example 11

Π m
[0092] Asymetric hydrogenations were conducted using catalytic Ru(R-C3-TunePhos)(acac)2 in methanol with 0.25 eq. of H3P04 (20 mg/mL), at about 30, 50, and 70 °C using 10, 20, 35, 50, and 70 atm of H2. Reaction times were about 15 – 18 h.Conversions of >99% were achieved. Enantiomeric excesses (%) of 67% – 82% were observed.
Example 12

Π m
[0093] Asymetric hydrogenations were conducted using catalytic Ru(S-Cs-TunePhos)(acac)2 in methanol or ethanol with 0.063 eq., 0.125 eq. 0.25 eq., and 0.5 eq. of H3PO4 (20 mg/mL), at about 50, 70, and 90 °C using 20, 50, 70,and 75 atm of H2. Reaction times were about 15 – 18 h.Conversions of >99% were achieved. Enantiomeric excesses (%) of 79% – 86% were observed.
Example 13

π m
[0094] In a glove box, to a hydrogenator (with glass liner) were added the substrate (4.14 g, 15 mmol), Ru(S-C5-TunePhos)(acac)2 (13.8 mg, 0.015 mmol, TONI OOO), Η3ΡΟ» (9 mL, 20 mg/mL in CH3OH, 0.125 equiv), CH3OH (19 mL, 7 vol) and a magnetic stirring bar. The hydrogenator was sealed and taken out of the glove box. The hydrogenator was charged with hydrogen to 50 atm. and put in an oil bath. The reaction mixture was stirred under 50 atm. at 50 °C for 48 h. After hydrogen was released carefully, the reaction mixture was concentrated.
Recrystallization from CH3OH (55 mL) gave the desired product as an off-white solid (2.29 g, 55% yield).
References:
Allwein, S.P et al., Org. Process Res. Dev. 2012, 16, 148-155.
Purohit, V.C. Org. Lett. 2013, 15, 1650-1653.
WO2008/051547
Discovery of Clinical Candidate CEP-37440, a Selective Inhibitor of Focal Adhesion Kinase (FAK) and Anaplastic Lymphoma Kinase (ALK)

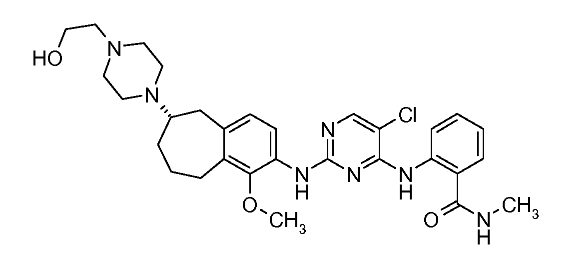
(27b) 2-[[5-Chloro-2-[[(6S)-6-[4-(2-hydroxyethyl)piperazin-1-yl]-1-methoxy-6,7,8,9-tetrahydro-5H-benzo[7]annulen-2-yl]amino]pyrimidin-4-yl]amino]-N-methylbenzamide
Development of a Process Route to the FAK/ALK Dual Inhibitor TEV-37440
 , Dale R. Mowrey†, Daniel E. Petrillo†, James J. Reif†, Vikram C. Purohit†, Karen L. Milkiewicz†, Roger P. Bakale†, Michael A. Christie†, Mark A. Olsen‡, Christopher J. Neville‡, and Gregory J. Gilmartin‡
, Dale R. Mowrey†, Daniel E. Petrillo†, James J. Reif†, Vikram C. Purohit†, Karen L. Milkiewicz†, Roger P. Bakale†, Michael A. Christie†, Mark A. Olsen‡, Christopher J. Neville‡, and Gregory J. Gilmartin‡
The development of a scalable route to TEV-37440, a dual inhibitor of focal adhesion kinase (FAK) and anaplastic lymphoma kinase (ALK), is presented. The medicinal chemistry route used to support this target through nomination is reviewed, along with the early process chemistry route to support IND (inversigational new drug) enabling activities within CMC (Chemistry, Manufacturing, and Controls). The identification and development of an improved route that was performed in the pilot plant to supply early phase clinical supplies are discussed. Details surrounding the use of a novel ring expansion, a selective nitration through a para-blocking group strategy, a single-pot amination–hydrogenation, a diastereomeric salt resolution, a through-process step to avoid a hazardous intermediate, and a practical formation of a trihydrochloride dihydrate salt are disclosed.
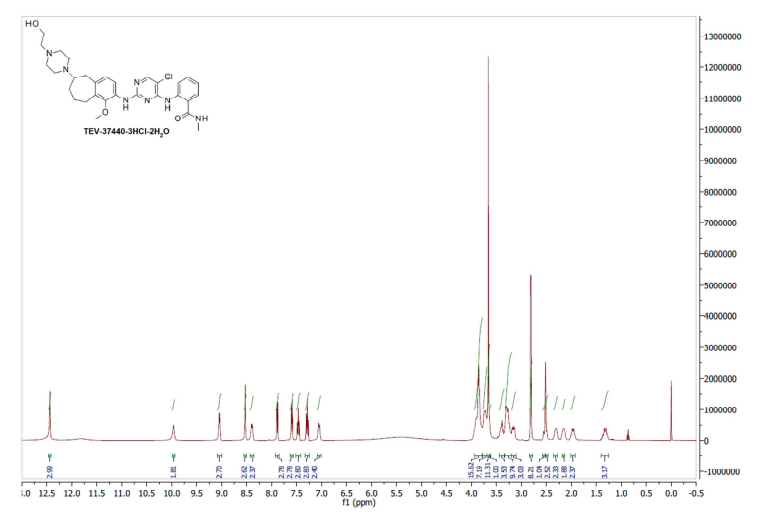
2-[(5-Chloropyrimidin-4-yl)amino]-N-methyl-benzamide; 2-[4-[(6S)-1-Methoxy-2-(methylamino)-6,7,8,9-tetrahydro-5H-benzo[7]annulen-6-yl]piperazin-1-yl]ethanol (2, Free Base)
2-[(5-Chloropyrimidin-4-yl)amino]-N-methyl-benzamide; 2-[4-[(6S)-1-Methoxy-2-(methylamino)-6,7,8,9-tetrahydro-5H-benzo[7]annulen-6-yl]piperazin-1-yl]ethanol Trihydrochloride Dihydrate Salt (2, Salt)
-
Allwein, S. P.; Roemmele, R. C.; Haley, J. J.; Mowrey, D. R.; Petrillo, D. E.; Reif, J. J.; Gingrich, D. E.;Bakale, R. P. Org. Process Res. Dev. 2012, 16, 148– 155, DOI: 10.1021/op200313v
-
2.Ott, G. R.; Cheng, M.; Learn, K. S.; Wagner, J.; Gingrich, D. E.; Lisko, J. G.; Curry, M.; Mesaros, E. F.;Ghose, A. K.; Quail, M. R.; Wan, W.; Lu, L.; Dobrzanski, P.; Albom, M. S.; Angeles, T. S.; Wells-Knecht, K.;Huang, Z.; Aimone, L. D.; Bruckheimer, E.; Anderson, N.; Friedman, J.; Fernandez, S. V.; Ator, M. A.;Ruggeri, B. A.; Dorsey, B. D. J. Med. Chem. 2016, 59, 7478– 7496, DOI: 10.1021/acs.jmedchem.6b00487
///////////////////TEV-37440, TEV 37440, CEP-37440, CEP 37440
CNC(=O)c5ccccc5Nc1nc(ncc1Cl)Nc3ccc2C[C@H](CCCc2c3OC)N4CCN(CC4)CCO
NVP-LXS196

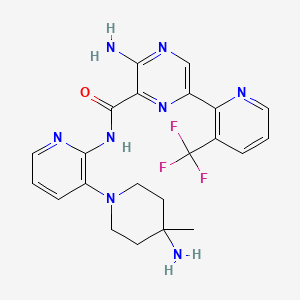
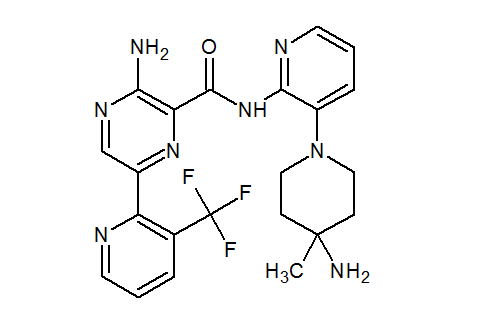
NVP-LXS196
CAS 1874276-76-2
3-amino-N-[3-(4-amino-4-methylpiperidin-1-yl)pyridin-2-yl]-6-[3-(trifluoromethyl)pyridin-2-yl]pyrazine-2-carboxamide
- 3-Amino-N-[3-(4-amino-4-methylpiperidin-1-yl)pyridin-2-yl]-6-[3-(trifluoromethyl)pyridin-2-yl]pyrazine-2-carboxamide
| Molecular Formula: | C22H23F3N8O |
|---|---|
| Molecular Weight: | 472.476 g/mol |
| Inventors | Michael Joseph Luzzio, Julien Papillon,Michael Scott Visser |
| Applicant | Novartis Ag |



SYNTHESIS

Uveal melanoma (UM) is the most common cancer of the eye in adults (Singh AD. et al., Ophthalmology. 201 1 ; 1 18: 1881-5). Most UM patients develop metastases for which no curative treatment has been identified so far. The majority of UM tumors have mutations in the genes GNAQ (guanine nucleotide-binding protein G(q) subunit alpha) and GNA11 (guanine nucleotide-binding protein G(q) subunit 1 1 ), which encode for small GTPases (Harbour JW. Pigment Cell Melanoma Res. 2012;25: 171-81). Both of these mutations lead to activation of the protein kinase C (PKC) pathway. The up-regulation of PKC pathway has downstream effects which leads to constitutive activation of the mitogen-activated protein kinase (MAPK) signaling pathway that has been implicated in causing uncontrolled cell growth in a number of proliferative diseases.
Whilst anti-proliferative effects have been observed with certain PKC pathway inhibitors, no sustained MAPK pathway inhibition has been observed. Thus far, PKC inhibitors (PKCi) have had limited efficacy as single agents in patients (Mochly-Rosen D et al., Nat Rev Drug Discov. 2012 Dec;1 1 (12):937-57). Moreover, inhibition of PKC alone was unable to trigger cell death in vitro and/or tumor regression in vivo (Chen X, et al., Oncogene. 2014;33:4724-34).
The protein p53 is a transcription factor that controls the expression of a multitude of target genes involved in DNA damage repair, apoptosis and cell cycle arrest, which are all important phenomena counteracting the malignant growth of tumors. The TP53 gene is one of the most frequently mutated genes in human cancers, with approximately half of all cancers having inactivated p53. Furthermore, in cancers with a non-mutated TP53 gene, typically the p53 is functionally inactivated at the protein level. One of the mechanisms of p53 inactivation is through its interaction with human homolog of MDM2 (Mouse double minute 2) protein. MDM2 protein functions both as an E3 ubiquitin ligase, that leads to proteasomal degradation of p53, and an inhibitor of p53 transcriptional activation. Therefore, MDM2 is an important negative regulator of the p53 tumor suppressor. MDM2 inhibitors can prevent interaction between MDM2 and p53 and thus allow the p53 protein to exert its effector functions. Whilst TP53 mutations are not common in UM, there are reports suggesting the p53 pathway is inactivated by either high expression of MDM2 protein or downregulation of the PERP protein in UM patients.
A combination of an MDM2 inhibitor (Nutlin-3) has been shown to act synergistically with reactivation of p53 and induction of tumor cell apoptosis (RITA) and Topotecan to cause growth inhibition in UM cell lines (De Lange J. et al., Oncogene. 2012;31 :1 105-16). However, Nutlin-3 and Topotecan delayed in vivo tumor growth only in a limited manner.
PATENT
PATENT
Example 9: 3-amino-N-(3-(4-amino-4-methylpiperidin-l-yl)pyridin-2-yl)- 6-(3-(trifluoromethyl)pyridin-2-yl)pyrazine-2-carboxamide

Synthesis of tert-butyl (4-meth l-l-(2-nitropyridin-3-yl)piperidin-4-yl)carbamate

To a solution of 3-fluoro-2-nitropyridine (11.2 g, 81 mmol) in dioxane (200 mL) was added tert-butyl (4-methylpiperidin-4-yl)carbamate (26 g, 121 mmol). Huenig’s Base (28.3 mL,
162 mmol) was added and the mixture was heated to 85 °C for 18 hrs. The reaction was cooled to RT and concentrated to give a brown solid. The solids were washed with 200 mL of 4: 1 heptane:EtOAc. Slurry was concentrated to half volume and filtered to collect (26.2 g, 78 mmol, 96%) brown solid. LC-MS (Acidic Method): ret.time= 1.46 min, M+H = 337.4
Step 2: Synthesis of tert-butyl (4-meth l-l-(2-nitropyridin-3-yl)piperidin-4-yl)carbamate

To a solution of tert-butyl (4-methyl-l-(2-nitropyridin-3-yl)piperidin-4-yl)carbamate (11.6 g, 37.2 mmol) in ethyl acetate (200 mL). 10% Pd-C (3.48 g) was added and stirred under H2 balloon pressure at RT for 4h. A small amount of MgS04 was added to the reaction and then the reaction mixture was filtered through a pad of cellite, then washed with ethyl acetate (100 mL) and the filtrate was concentrated to afford a brown solid (8.54 g, 27.9 mmol, 85%). LC-MS (Acidic Method): ret.time= 0.91 min, M+H = 307.4.
Step 3: Synthesis of tert-butyl (l-(2-(3-amino-6-(3-(trifluoromethyl)pyridin-2-yl)pyrazine-2-carboxamido)pyridin-3-yl)-4-meth lpiperidin-4-yl)carbamate

To a solution of 3-amino-6-(3-(trifluoromethyl)pyridin-2-yl)pyrazine-2-carboxylic acid in dimethyl formamide (125 mL) was added ((lH-benzo[d][l,2,3]triazol-l- yl) oxy)
tris(dimethylamino) phosphonium hexafluorophosphate(V) (1.8g, 4.24 mmol) and 4-methylmorpholine (1 mL, 9.79 mmol). Reaction stirred at RT for 40 minutes. Tert-butyl (l-(2-aminopyridin-3-yl)-4-methylpiperidin-4-yl) carbamate in dimethylformamide (25 mL) was added and reaction stirred for 16 hrs at RT. The reaction mixture was diluted with EtOAc and was washed with NaHC03(aq) (3 x 200mL) and brine (lx 200mL). The organic phase was dried with Na2S04, filtered and concentrated. The crude product was taken up in acetonitrile (30 mL) and mixture was allowed to stand at RT for a period of time. Yellow solid collected by filtration (1.39g, 74%). LC-MS (Acidic Method): ret.time= 1.13 mm, M+H = 573.3.
Step 4: Synthesis of 3-amino-N-(3-(4-amino-4-methylpiperidin-l-yl)pyridin-2-yl)-6-(3-(trifluoromethyl)pyridin-2-yl)pyrazine-2-carboxamide
A solution of tert-butyl (l-(2-(3-amino-6-(3-(trifluoromethyl)pyridin-2-yl)pyrazine-2-carboxamido)pyridin-3-yl)-4-methylpiperidin-4-yl)carbamate (l -39g, 2.06 mmol) in dichloromethane (10 mL) was cooled to 0 °C. 2,2,2-trifluoroacetic acid (2.4 ml, 31 mmol) was added dropwise to the solution. The mixture was allowed to warm to 22 °C and stirred for 4 hrs. Reaction mixture was concentrated to remove DCM and excess TFA. A red oil was produced, which was taken up in 100 mL CHCI3/IPA 3: 1 and saturated aq. NaHCC was added to neutralize the solution. The mixture was then stirred at 22°C for 16 hrs. The mixture transfered to separatory funnel and aqueous layers were washed with CHCI3/IPA 3: 1 (3X 100 mL). Combined organic phases were dried with Na2S04, filtered and concentrated to afford a yellow solid. The crude product was recrystallized from acetonitrile. A yellow solid was collected by filtration (0.82g, 83%). LC-MS (Acidic Method ): ret.time= 0.75 mm, M+H = 473.2. 1H NMR (400 MHz, Methanol-^) δ 8.92 (dd, J = 5.1, 1.4 Hz, 1H), 8.68 (s, 1H), 8.47 – 8.27 (m, 1H), 8.12 (dd, J = 4.9, 1.6 Hz, 1H), 7.83 – 7.50 (m, 2H), 7.18 (dd, J = 7.9, 4.9 Hz, 1H), 3.02 – 2.65 (m, 4H), 1.54 – 1.24 (m, 4H), 0.74 (s, 3H).
REFERENCES
Visser, M.; Papillon, J.; Fan, J.; et al.
NVP-LXS196, a novel PKC inhibitor for the treatment of uveal melanoma
253rd Am Chem Soc (ACS) Natl Meet (April 2-6, San Francisco) 2017, Abst MEDI 366
| Patent ID | Patent Title | Submitted Date | Granted Date |
|---|---|---|---|
| US2016046605 | PROTEIN KINASE C INHIBITORS AND METHODS OF THEIR USE | 2015-08-05 | 2016-02-18 |
//////////////NVP-LXS196, NVP-LXS 196, 1874276-76-2, Michael Joseph Luzzio, Julien Papillon,Michael Scott Visser, NOVARTIS, PKC inhibitor, uveal melanoma
FC(F)(F)c1cccnc1c2cnc(N)c(n2)C(=O)Nc3ncccc3N4CCC(C)(N)CC4
http://sanfrancisco2017.acs.org/i/803418-253rd-american-chemical-society-national-meeting-expo/289
Michael Visser of @Novartis talking now in 1st time disclosures about a PKC inhibitor to treat uveal melanoma #ACSSanFran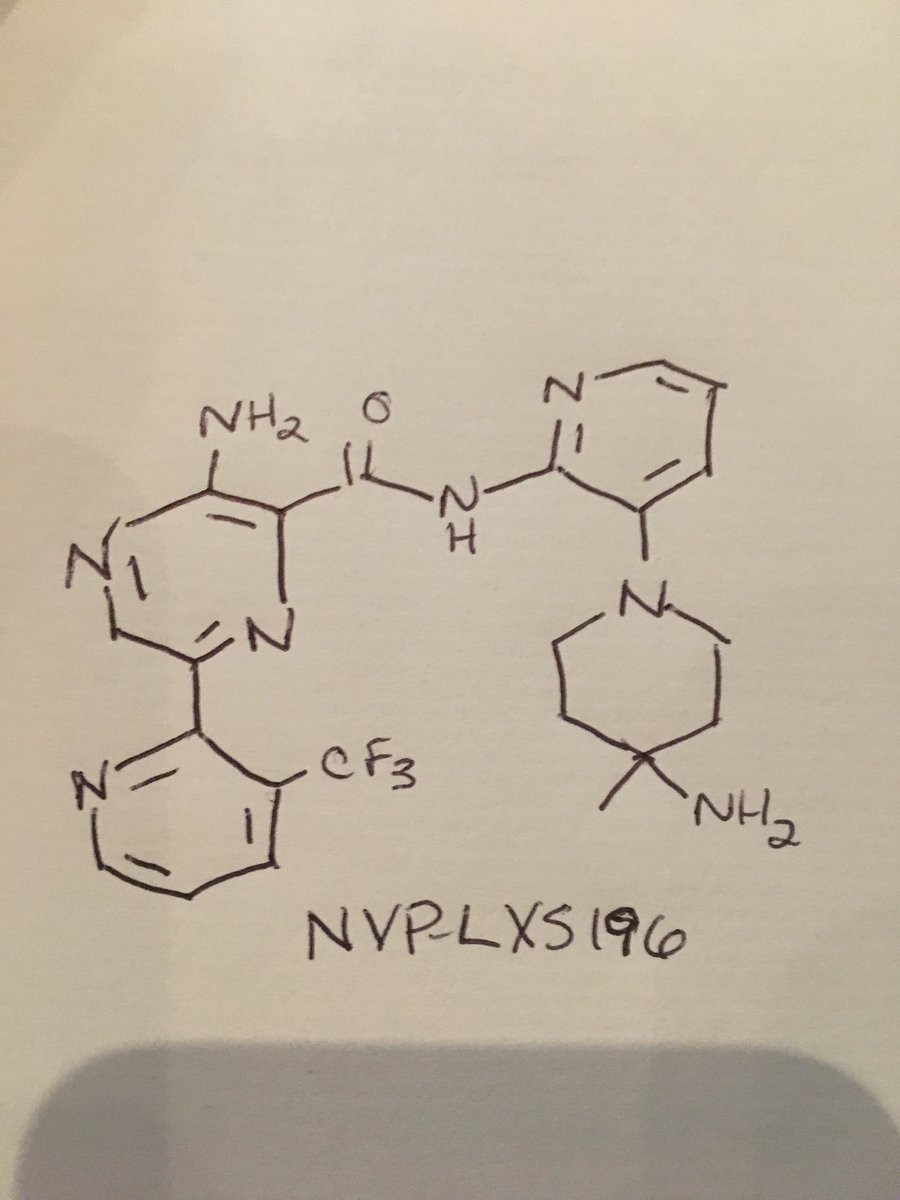
FDA approves new combination treatment for acute myeloid leukemia, Rydapt (midostaurin)
MIDOSTAURIN
April 28, 2017
Release
The U.S. Food and Drug Administration today approved Rydapt (midostaurin) for the treatment of adult patients with newly diagnosed acute myeloid leukemia (AML) who have a specific genetic mutation called FLT3, in combination with chemotherapy. The drug is approved for use with a companion diagnostic, the LeukoStrat CDx FLT3 Mutation Assay, which is used to detect the FLT3 mutation in patients with AML.
AML is a rapidly progressing cancer that forms in the bone marrow and results in an increased number of white blood cells in the bloodstream. The National Cancer Institute estimated that approximately 19,930 people would be diagnosed with AML in 2016 and 10,430 were projected to die of the disease.
“Rydapt is the first targeted therapy to treat patients with AML, in combination with chemotherapy,” said Richard Pazdur, M.D., acting director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research and director of the FDA’s Oncology Center of Excellence. “The ability to detect the gene mutation with a diagnostic test means doctors can identify specific patients who may benefit from this treatment.”
Rydapt is a kinase inhibitor that works by blocking several enzymes that promote cell growth. If the FLT3 mutation is detected in blood or bone marrow samples using the LeukoStrat CDx FLT3 Mutation Assay, the patient may be eligible for treatment with Rydapt in combination with chemotherapy.
The safety and efficacy of Rydapt for patients with AML were studied in a randomized trial of 717 patients who had not been treated previously for AML. In the trial, patients who received Rydapt in combination with chemotherapy lived longer than patients who received chemotherapy alone, although a specific median survival rate could not be reliably estimated. In addition, patients who received Rydapt in combination with chemotherapy in the trial went longer (median 8.2 months) without certain complications (failure to achieve complete remission within 60 days of starting treatment, progression of leukemia or death) than patients who received chemotherapy alone (median three months).
Common side effects of Rydapt in patients with AML include low levels of white blood cells with fever (febrile neutropenia), nausea, inflammation of the mucous membranes (mucositis), vomiting, headache, spots on the skin due to bleeding (petechiae), musculoskeletal pain, nosebleeds (epistaxis), device-related infection, high blood sugar (hyperglycemia) and upper respiratory tract infection. Rydapt should not be used in patients with hypersensitivity to midostaurin or other ingredients in Rydapt. Women who are pregnant or breastfeeding should not take Rydapt because it may cause harm to a developing fetus or a newborn baby. Patients who experience signs or symptoms of lung damage (pulmonary toxicity) should stop using Rydapt.
Rydapt was also approved today for adults with certain types of rare blood disorders (aggressive systemic mastocytosis, systemic mastocytosis with associated hematological neoplasm or mast cell leukemia). Common side effects of Rydapt in these patients include nausea, vomiting, diarrhea, swelling (edema), musculoskeletal pain, abdominal pain, fatigue, upper respiratory tract infection, constipation, fever, headache and shortness of breath.
The FDA granted this application Priority Review, Fast Track (for the mastocytosis indication) and Breakthrough Therapy (for the AML indication) designations.
The FDA granted the approval of Rydapt to Novartis Pharmaceuticals Corporation. The FDA granted the approval of the LeukoStrat CDx FLT3 Mutation Assay to Invivoscribe Technologies Inc.
MIDOSTAURIN
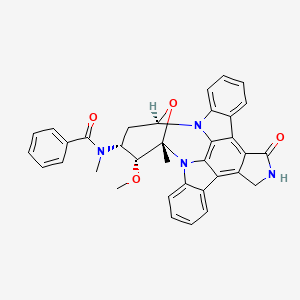
(9S,10R,11R,13R)-2,3,10,11,12,13-Hexahydro-10-methoxy-9-methyl-11-(methylamino)-9,13-epoxy-1H,9H-diindolo[1,2,3-gh:3′,2′,1′-lm]pyrrolo[3,4-j][1,7]benzodiamzonine-1-one
N-[(9S,10R,11R,13R)-2,3,10,11,12,13-Hexahydro-10-methoxy-9-methyl-1-oxo-9,13-epoxy-1H,9H-diindolo[1,2,3-gh:3′,2′,1′-lm]pyrrolo[3,4-j][1,7]benzodiazonin-11-yl]-N-methylbenzamide
N-((9S,10R,11R,13R)-2,3,9,10,11,12-hexahydro-10-methoxy-9-methyl-1-oxo-9,13-epoxy-1H,9H-diindolo(1,2,3-gh:3′,2′,1′-lm)pyrrolo(3,4-j)(1,7)benzodiazonin-11-yl)-N-methyl-,
N-[(2R,4R,5R,6S)-5-methoxy-6-methyl-18-oxo-29-oxa-1,7,17-triazaoctacyclo[12.12.2.12,6.07,28.08,13.015,19.020,27.021,26]nonacosa-8,10,12,14(28),15(19),20(27),21(26),22,24-nonaen-4-yl]-N-methylbenzamide hydrate
N-benzoyl staurosporine
NOVARTIS ONCOLOGY ORIGINATOR
Chemical Formula: C35H30N4O4
Exact Mass: 570.22671
Molecular Weight: 570.63710
Elemental Analysis: C, 73.67; H, 5.30; N, 9.82; O, 11.22
Tyrosine kinase inhibitors
PKC 412。PKC412A。CGP 41251。Benzoylstaurosporine;4′-N-Benzoylstaurosporine;Cgp 41251;Cgp 41 251.
120685-11-2 CAS
PHASE 3
- 4′-N-Benzoylstaurosporine
- Benzoylstaurosporine
- Cgp 41 251
- CGP 41251
- CGP-41251
- Midostaurin
- PKC 412
- PKC412
- UNII-ID912S5VON
Midostaurin is an inhibitor of tyrosine kinase, protein kinase C, and VEGF. Midostaurin inhibits cell growth and phosphorylation of FLT3, STAT5, and ERK. It is a potent inhibitor of a spectrum of FLT3 activation loop mutations.

it is prepared by acylation of the alkaloid staurosporine (I) with benzoyl chloride (II) in the presence of diisopropylethylamine in chloroform.
Midostaurin is a synthetic indolocarbazole multikinase inhibitor with potential antiangiogenic and antineoplastic activities. Midostaurin inhibits protein kinase C alpha (PKCalpha), vascular endothelial growth factor receptor 2 (VEGFR2), c-kit, platelet-derived growth factor receptor (PDGFR) and FMS-like tyrosine kinase 3 (FLT3) tyrosine kinases, which may result in disruption of the cell cycle, inhibition of proliferation, apoptosis, and inhibition of angiogenesis in susceptible tumors.
MIDOSTAURIN
Derivative of staurosporin, orally active, potent inhibitor of FLT3 tyrosine kinase (fetal liver tyrosine kinase 3). In addition Midostaurin inhibits further molecular targets such as VEGFR-1 (Vascular Endothelial Growth Factor Receptor 1), c-kit (stem cell factor receptor), H-and K-RAS (Rat Sarcoma Viral homologue) and MDR (multidrug resistance protein).
Midostaurin inhibits both wild-type FLT3 and FLT3 mutant, wherein the internal tandem duplication mutations (FLT3-ITD), and the point mutation to be inhibited in the tyrosine kinase domain of the molecule at positions 835 and 836.Midostaurin is tested in patients with AML.
Midostaurin, a protein kinase C (PKC) and Flt3 (FLK2/STK1) inhibitor, is in phase III clinical development at originator Novartis for the oral treatment of acute myeloid leukemia (AML).
Novartis is conducting phase III clinical trials for the treatment of aggressive systemic mastocytosis or mast cell leukemia. The National Cancer Institute (NCI) is conducting phase I/II trials with the drug for the treatment of chronic myelomonocytic leukemia (CMML) and myelodysplastic syndrome (MDS).
Massachusetts General Hospital is conducting phase I clinical trials for the treatment of adenocarcinoma of the rectum in combination with radiation and standard chemotherapy.
 MIDOSTAURIN
MIDOSTAURIN
Midostaurin (PKC412) is a multi-target protein kinase inhibitor being investigated for the treatment of acute myeloid leukemia (AML) and myelodysplastic syndrome (MDS). It is a semi-synthetic derivative of staurosporine, an alkaloid from the bacterium Streptomyces staurosporeus, and is active in patients with mutations of CD135 (FMS-like tyrosine kinase 3 receptor).[1]
After successful Phase II clinical trials, a Phase III trial for AML has started in 2008. It is testing midostaurin in combination with daunorubicin and cytarabine.[2] In another trial, the substance has proven ineffective in metastatic melanoma.[3]
Midostaurin has also been studied at Johns Hopkins University for the treatment of age-related macular degeneration (AMD), but no recent progress reports for this indication have been made available. Trials in macular edema of diabetic origin were discontinued at Novartis.
In 2004, orphan drug designation was received in the E.U. for the treatment of AML. In 2009 and 2010, orphan drug designation was assigned for the treatment of acute myeloid leukemia and for the treatment of mastocytosis, respectively, in the U.S. In 2010, orphan drug designation was assigned in the E.U. for the latter indication.
References
- Fischer, T.; Stone, R. M.; Deangelo, D. J.; Galinsky, I.; Estey, E.; Lanza, C.; Fox, E.; Ehninger, G.; Feldman, E. J.; Schiller, G. J.; Klimek, V. M.; Nimer, S. D.; Gilliland, D. G.; Dutreix, C.; Huntsman-Labed, A.; Virkus, J.; Giles, F. J. (2010). “Phase IIB Trial of Oral Midostaurin (PKC412), the FMS-Like Tyrosine Kinase 3 Receptor (FLT3) and Multi-Targeted Kinase Inhibitor, in Patients with Acute Myeloid Leukemia and High-Risk Myelodysplastic Syndrome with Either Wild-Type or Mutated FLT3”. Journal of Clinical Oncology 28 (28): 4339–4345. doi:10.1200/JCO.2010.28.9678. PMID 20733134. edit
- ClinicalTrials.gov NCT00651261 Daunorubicin, Cytarabine, and Midostaurin in Treating Patients With Newly Diagnosed Acute Myeloid Leukemia
- Millward, M. J.; House, C.; Bowtell, D.; Webster, L.; Olver, I. N.; Gore, M.; Copeman, M.; Lynch, K.; Yap, A.; Wang, Y.; Cohen, P. S.; Zalcberg, J. (2006). “The multikinase inhibitor midostaurin (PKC412A) lacks activity in metastatic melanoma: a phase IIA clinical and biologic study”. British Journal of Cancer 95 (7): 829–834. doi:10.1038/sj.bjc.6603331. PMC 2360547. PMID 16969355.
-
- Midostaurin product page, Fermentek
- Wang, Y; Yin, OQ; Graf, P; Kisicki, JC; Schran, H (2008). “Dose- and Time-Dependent Pharmacokinetics of Midostaurin in Patients With Diabetes Mellitus”. J Clin Pharmacol 48 (6): 763–775. doi:10.1177/0091270008318006. PMID 18508951.
- Ryan KS (2008). “Structural studies of rebeccamycin, staurosporine, and violacein biosynthetic enzymes”. Ph.D. Thesis. Massachusetts Institute of Technology.
Bioorg Med Chem Lett 1994, 4(3): 399
US 5093330
EP 0657164
EP 0711556
EP 0733358
WO 1998007415
WO 2002076432
WO 2003024420
WO 2003037347
WO 2004112794
WO 2005027910
WO 2005040415
WO 2006024494
WO 2006048296
WO 2006061199
WO 2007017497
WO 2013086133
WO 2012016050
WO 2011000811
|
3-1-2010
|
Colony stimulating factor-1 receptor as a target for small molecule inhibitors.
|
Bioorganic & medicinal chemistry
|
|
7-18-2012
|
Staurosporine Derivatives as Inhibitors of FLT3 Receptor Tyrosine Kinase Activity
|
|
|
6-13-2012
|
Crystal form of N-benzoyl-staurosporine
|
|
|
12-14-2011
|
COMPOSITIONS FOR TREATMENT OF SYSTEMIC MASTOCYTOSIS
|
|
|
7-6-2011
|
Staurosporine derivatives as inhibitors of flt3 receptor tyrosine kinase activity
|
|
|
7-6-2011
|
Staurosporine Derivatives for Use in Alveolar Rhabdomyosarcoma
|
|
|
12-10-2010
|
Pharmaceutical Compositions for treating wouds and related methods
|
|
|
11-5-2010
|
COMBINATIONS OF JAK INHIBITORS
|
|
|
7-23-2010
|
COMBINATIONS COMPRISING STAUROSPORINES
|
|
|
3-5-2010
|
COMBINATION OF IAP INHIBITORS AND FLT3 INHIBITORS
|
|
|
1-29-2010
|
ANTI-CANCER PHOSPHONATE ANALOGS
|
|
1-13-2010
|
Therapeutic phosphonate compounds
|
|
|
11-20-2009
|
Use of Staurosporine Derivatives for the Treatment of Multiple Myeloma
|
|
|
7-17-2009
|
KINASE INHIBITORY PHOSPHONATE ANALOGS
|
|
|
6-19-2009
|
Organic Compounds
|
|
|
3-20-2009
|
Use of Midostaurin for Treating Gastrointestinal Stromal Tumors
|
|
|
11-21-2008
|
PHARMACEUTICAL COMPOSITIONS COMPRISING A POORLY WATER-SOLUBLE ACTIVE INGREDIENT, A SURFACTANT AND A WATER-SOLUBLE POLYMER
|
|
|
11-19-2008
|
Anti-cancer phosphonate analogs
|
|
|
9-12-2008
|
Multi-Functional Small Molecules as Anti-Proliferative Agents
|
|
|
9-5-2008
|
Sensitization of Drug-Resistant Lung Caners to Protein Kinase Inhibitors
|
|
|
8-29-2008
|
Organic Compounds
|
|
8-27-2008
|
Kinase inhibitory phosphonate analogs
|
|
|
4-25-2008
|
Treatment Of Gastrointestinal Stromal Tumors With Imatinib And Midostaurin
|
|
|
12-28-2007
|
Pharmaceutical Uses of Staurosporine Derivatives
|
|
|
12-7-2007
|
Kinase Inhibitor Phosphonate Conjugates
|
|
|
8-17-2007
|
Combinations comprising staurosporines
|
|
|
10-13-2006
|
Staurosporine derivatives for hypereosinophilic syndrome
|
|
|
7-15-2005
|
Phosphonate substituted kinase inhibitors
|
|
|
10-20-2004
|
Staurosporin derivatives
|

MIDOSTAURIN HYDRATE
Midostaurin according to the invention is N-[(9S,10R,11R,13R)-2,3,10,11,12,13-hexahydro-10-methoxy-9-methyl-1-oxo-9,13-epoxy-1H,9H-diindolo[1,2,3-gh:3′,2′,1′-lm]pyrrolo[3,4-j][1,7]benzodiazonin-11-yl]-N-methylbenzamide of the formula (II):

or a salt thereof, hereinafter: “Compound of formula II or midostaurin”.
Compound of formula II or midostaurin [International Nonproprietary Name] is also known as PKC412.
Midostaurin is a derivative of the naturally occurring alkaloid staurosporine, and has been specifically described in the European patent No. 0 296 110 published on Dec. 21, 1988, as well as in U.S. Pat. No. 5093330 published on Mar. 3, 1992, and Japanese Patent No. 2 708 047.
………………….
https://www.google.co.in/patents/EP0296110B1
The nomenclature of the products is, on the complete structure of staurosporine ([storage]-NH-CH ₃derived, and which is designated by N-substituent on the nitrogen of the methylamino group

Example 18:
- N-Benzoyl-staurospor
-
A solution of 116.5 mg (0.25 mmol) of staurosporine and 0.065 ml (0.38 mmol) of N, N-diisopropylethylamine in 2 ml of chloroform is added at room temperature with 0.035 ml (0.3 mmol) of benzoyl chloride and 10 stirred minutes.The reaction mixture is diluted with chloroform, washed with sodium bicarbonate, dried over magnesium sulfate and evaporated. The crude product is chromatographed on silica gel (eluent methylene chloride / ethanol 30:1), mp 235-247 ° with brown coloration.
- cut paste may not be ok below
Staurosporine the formula [storage]-NH-CH ₃ (II) (for the meaning of the rest of [storage] see above) as the basic material of the novel compounds was already in 1977, from the cultures of Streptomyces staurosporeus AWAYA, and TAKAHASHI
O ¯

MURA, sp. nov. AM 2282, see Omura, S., Iwai, Y., Hirano, A., Nakagawa, A.; awayâ, J., Tsuchiya, H., Takahashi, Y., and Masuma, R. J. Antibiot. 30, 275-281 (1977) isolated and tested for antimicrobial activity. It was also found here that the compound against yeast-like fungi and microorganisms is effective (MIC of about 3-25 mcg / ml), taking as the hydrochloride = having a LD ₅ ₀ 6.6 mg / kg (mouse, intraperitoneal). Stagnated recently it has been shown in extensive screening, see Tamaoki, T., Nomoto, H., Takahashi, I., Kato, Y, Morimoto, M. and Tomita, F.: Biochem. and Biophys. Research Commun. 135 (No. 2), 397-402 (1986) that the compound exerts a potent inhibitory effect on protein kinase C (rat brain)
…………………
https://www.google.co.in/patents/US5093330
EXAMPLE 18 N-benzoyl-staurosporine
0.035 ml (0.3 mmol) of benzoyl chloride is added at room temperature to a solution of 116.5 mg (0.25 mmol) of staurosporine and 0.065 ml (0.38 mmol) of N,N-diisopropylethylamine in 2 ml of chloroform and the whole is stirred for 10 minutes. The reaction mixture is diluted with chloroform, washed with sodium bicarbonate solution, dried over magnesium sulphate and concentrated by evaporation. The crude product is chromatographed on silica gel (eluant:methylene chloride/ethanol 30:1); m.p. 235
…………………….
Bioorg Med Chem Lett 1994, 4(3): 399
http://www.sciencedirect.com/science/article/pii/0960894X94800049

……………………
http://www.google.com/patents/WO1998007415A2
A variety of PKC inhibitors are available in the art for use in the invention. These include bryostatin (U.S. Patent 4,560,774), safinogel (WO 9617603), fasudil (EP 187371), 7- hydoxystaurosporin (EP 137632B), various diones described in EP 657458, EP 657411 and WO9535294, phenylmethyl hexanamides as described in WO9517888, various indane containing benzamides as described in WO9530640, various pyrrolo [3,4-c]carbazoles as described in EP 695755, LY 333531 (IMSworld R & D Focus 960722, July 22, 1996 and Pharmaprojects Accession No. 24174), SPC-104065 (Pharmaprojects Accession No. 22568), P-10050 (Pharmaprojects Accession No. 22643), No. 4432 (Pharmaprojects Accession No. 23031), No. 4503 (Pharmaprojects Accession No. 23252), No. 4721 (Pharmaprojects Accession No. 23890), No. 4755 (Pharmaprojects Accession No. 24035), balanol (Pharmaprojects Accession No. 20376), K-7259 (Pharmaprojects Accession No. 16649), Protein kinase C inhib, Lilly (Pharmaprojects Accession No. 18006), and UCN-01 (Pharmaprojects Accession No. 11915). Also see, for example, Tamaoki and Nakano (1990) Biotechnology 8:732-735; Posada et al. (1989) Cancer Commun. 1:285-292; Sato et al. (1990) Biochem Biophys. Res. Commun. 173:1252-1257; Utz et al. (1994) Int. J. Cancer 57:104-110; Schwartz et al. (1993) J. Na . Cancer lnst. 85:402-407; Meyer et al. (1989) Int. J. Cancer 43:851-856; Akinaga et al. (1991) Cancer Res. 51:4888-4892, which disclosures are herein incorporated by reference. Additionally, antisense molecules can be used as PKC inhibitors. Although such antisense molecules inhibit mRNA translation into the PKC protein, such antisense molecules are considered PKC inhibitors for purposes of this invention. Such antisense molecules against PKC inhibitors include those described in published PCT patent applications WO 93/19203, WO 95/03833 and WO 95/02069, herein incorporated by reference. Such inhibitors can be used in formulations for local delivery to prevent cellular proliferation. Such inhibitors find particular use in local delivery for preventing rumor growth and restenosis.
N-benzoyl staurosporine is a benzoyl derivative of the naturally occurring alkaloid staurosporine. It is chiral compound ([a]D=+148.0+-2.0°) with the formula C35H30R1O4 (molecular weight 570.65). It is a pale yellow amorphous powder which remains unchanged up to 220°C. The compound is very lipophilic (log P>5.48) and almost insoluble in water (0.068 mg/1) but dissolves readily in DMSO.
……………………….
staurosporine
Staurosporine (antibiotic AM-2282 or STS) is a natural product originally isolated in 1977 from the bacterium Streptomyces staurosporeus. It was the first of over 50 alkaloids to be isolated with this type of bis-indole chemical structure. The chemical structure of staurosporine was elucidated by X-ray analysis of a single crystal and the absolute stereochemical configuration by the same method in 1994.
Staurosporine was discovered to have biological activities ranging from anti-fungal to anti-hypertensive. The interest in these activities resulted in a large investigative effort in chemistry and biology and the discovery of the potential for anti-cancer treatment

Staurosporine is the precursor of the novel protein kinase inhibitor midostaurin(PKC412). Besides midostaurin, staurosporine is also used as a starting material in the commercial synthesis of K252c (also called staurosporine aglycone). In the natural biosynthetic pathway, K252c is a precursor of staurosporine.
Indolocarbazoles belong to the alkaloid sub-class of bisindoles. Of these carbazoles the Indolo(2,3-a)carbazoles are the most frequently isolated; the most common subgroup of the Indolo(2,3-a)carbazoles are the Indolo(2,3-a)pyrrole(3,4-c)carbazoles which can be divided into two major classes – halogenated (chlorinated) with a fully oxidized C-7 carbon with only one indole nitrogen containing a β-glycosidic bond and the second class consists of both indole nitrogen glycosilated, non-halogenated, and a fully reduced C-7 carbon. Staurosporine is part of the second non-halogenated class.
The biosynthesis of staurosporine starts with the amino acid L-tryptophan in its zwitterionic form. Tryptophan is converted to an imineby enzyme StaO which is an L-amino acid oxidase (that may be FAD dependent). The imine is acted upon by StaD to form an uncharacterized intermediate proposed to be the dimerization product between 2 imine molecules. Chromopyrrolic acid is the molecule formed from this intermediate after the loss of VioE (used in the biosynthesis of violacein – a natural product formed from a branch point in this pathway that also diverges to form rebeccamycin. An aryl aryl coupling thought to be catalyzed by a cytochrome P450enzyme to form an aromatic ring system occurs

This is followed by a nucleophilic attack between the indole nitrogens resulting in cyclization and then decarboxylation assisted by StaC exclusively forming staurosporine aglycone or K252c. Glucose is transformed to NTP-L-ristoamine by StaA/B/E/J/I/K which is then added on to the staurosporine aglycone at 1 indole N by StaG. The StaN enzyme reorients the sugar by attaching it to the 2nd indole nitrogen into an unfavored conformation to form intermediated O-demethyl-N-demethyl-staurosporine. Lastly, O-methylation of the 4’amine by StaMA and N-methylation of the 3′-hydroxy by StaMB leads to the formation of staurosporine
| US4107297 * | 28 Nov 1977 | 15 Aug 1978 | The Kitasato Institute | Antibiotic compound |
| US4735939 * | 27 Feb 1987 | 5 Apr 1988 | The Dow Chemical Company | Insecticidal activity of staurosporine |
| ZA884238A * | Title not available |
MIDOSTAURIN
NOVARTIS
- Chemistry Review(s) (PDF)

| Rydapt | FDA
4/28/2017 |
To treat acute myeloid leukemia Press Release Drug Trials Snapshot |
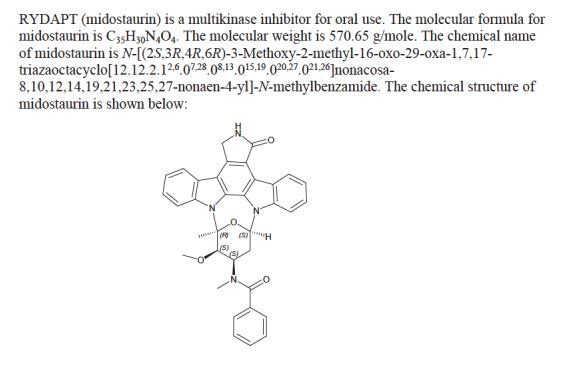

An insight into the therapeutic potential of quinazoline derivatives as anticancer agents


The Food and Drug Administration (FDA) has approved several quinazoline derivatives for clinical use as anticancer drugs. These include gefitinib, erlotinib, lapatinib, afatinib, and vandetanib (Fig.1) [43]. Gefitinib (Iressa®) was approved by the FDA in 2003 for the treatment of locally advanced or metastatic non-small-cell lung cancer (NSCLC) in patients after failure of both platinum-based and/or docetaxel chemotherapies. In 2004, erlotinib (Tarceva®) was approved by the FDA for treating NSCLC. Furthermore, in 2005, the FDA approved erlotinib in combination with gemcitabine for treatment of locally advanced, unrespectable, or metastatic pancreatic cancer. Erlotinib acts as a reversible tyrosine kinase inhibitor. Lapatinib (Tykreb®) was approved by the FDA in 2012 for breast cancer treatment. It inhibits the activity of both human epidermal growth factor receptor-2 (HER2/neu) and epidermal growth factor receptor (EGFR) pathways. Vandetanib (Caprelsa®) was approved by the FDA in 2011 for the treatment of metastatic medullary thyroid cancer. It acts as a kinase inhibitor of a number of cell receptors, mainly the vascular endothelial growth factor receptor (VEGFR), EGFR, and rearranged during transfection (RET)-tyrosine kinase (TK). Afatinib (Gilotrif®) was approved by the FDA in 2013 for NSCLC treatment. It acts as an irreversible covalent inhibitor of the receptor tyrosine kinases (RTK) for EGFR and erbB-2 (HER2).
An insight into the therapeutic potential of quinazoline derivatives as anticancer agents
*Corresponding authors
Abstract
Cancer is one of the major causes of worldwide human mortality. A wide range of cytotoxic drugs are available on the market, and several compounds are in different phases of clinical trials. Many studies suggest that these cytotoxic molecules are also associated with different types of adverse side effects; therefore researchers around the globe are involved in the development of more efficient and safer anticancer drugs. In recent years, quinazoline and its derivatives have been considered as a novel class of cancer chemotherapeutic agents that show promising activity against different tumors. The aim of this article is to comprehensively review and highlight the recent developments concerning the anticancer activity of quinazoline derivatives as well as offer perspectives on the development of novel quinazoline derivatives as anticancer agents in the near future.
An insight into the therapeutic potential of quinazoline derivatives as anticancer agents
DOI: 10.1039/C7MD00097A, Review Article
This article reviews the recent advances in the development of quinazoline derivatives as anticancer agents.


Dr. Shagufta Waseem
ASSISTANT PROFESSOR – CHEMISTRY

Biography
Dr. Shagufta joined the American University of Ras Al Khaimah as an Assistant Professor of Chemistry in the School of Arts and Sciences in August 2014. Prior to joining AURAK, Dr. Shagufta worked as an Adjunct Assistant Professor of Chemistry at the University of Modern Sciences, Dubai and American University of Ras Al Khaimah, UAE.
Dr. Shagufta also worked as a Postdoctoral Researcher Associate at the Department of Chemistry and Biochemistry, Oklahoma University, USA. She developed the noble drug delivery system for breast cancer drugs using carbon nanotubes and acquired the significant experience in nanotechnology and synthetic organic chemistry. She was appointed as a Postdoctoral Research Fellow and Visiting Scientist at Leiden/Amsterdam Centre for Drug Research (LACDR), Leiden, The Netherlands. Her research interest was In silico prediction and clinical evaluation of the cardiotoxicity of drug candidates. She was focused to identify chemical substructures as ‘chemical alerts’ that interact with this hERG channel. Dr. Shagufta received a Ph.D. under the prestigious CSIR-JRF and SRF research fellowship in Chemistry from Central Drug Research Institute (CDRI)/Lucknow University, India in 2008, her PhD research work was in the field of estrogens and antiestrogens, design and synthesis of steroidal and non-steroidal tissue selective estrogen receptor modulators (SERMs) for breast cancer, 3D-QSAR CoMFA and CoMSIA studies and analysis of pharmaceutical important molecules.
Dr. Shagufta has published 20 articles in peer-reviewed International journals of Royal Society of Chemistry, Elsevier, Wiley and Springer. Dr. Shagufta teaches courses such as General chemistry, Organic Chemistry, Chemistry in Everyday Life, and Spectroscopy along with laboratory courses.
Research and Publication
Research Interest-Dr. Shagufta
Organic Chemistry, Medicinal Chemistry focused on Breast Cancer and Osteoporosis, Heterogeneous catalysis and Nanotechnology.
Publications- Dr. Shagufta
- Irshad Ahmad and Shagufta. 2015. Recent developments in steroidal and nonsteroidal aromatase inhibitors for the chemoprevention of estrogen-dependent breast cancer. European Journal of Medicinal Chemistry, 102, 375-386.
- Irshad Ahmad and Shagufta. 2015. Sulfones: An important class of organic compounds with diverse biological activities. International Journal of Pharmacy and Pharmaceutical Sciences, 7 (3), 19-27.
- Priyanka Singh, Subal Kumar Dinda, Shagufta, Gautam Panda. 2013. Synthetic approach towards trisubstituted methanes and a chiral tertiary α-hydroxyaldehyde, possible intermediate for tetrasubstituted methanes. RSC Adv.(Royal Society of Chemistry) 3, 12100-12103. [ISSN: 2046-2069]
- Donna J. Nelson, Shagufta, Ravi Kumar. 2012. Characterization of a tamoxifen-tethered single-walled carbon nanotube conjugate by using NMR spectroscopy. Anal. Bioanal. Chem.[Springer] 404:771–776. [ISSN: 1618-2642]
- Donna J. Nelson, Ravi Kumar, Shagufta. 2012. Regiochemical reversals in nitrosobenzene reactions with carbonyl compounds – α-aminooxy ketone versus α-hydroxyamino ketone products. Eur. J. Org. Chem.(Wiley-VCH) 6013-6020. [ISSN: 1099-0690]
- Munikumar R. Reddy, Elisabeth Klaasse, Shagufta, Adriaan P. IJzerman, Andreas Bender. 2010. Validation of an in silico hERG model and its applications to the virtual screening of commercial compound databases. Chem. Med. Chem. (Wiley-VCH)5: 716-729. [ISSN: 1860-7187]
- Shagufta, Dong Guo, Elisabeth Klaasse, Henk de Vries, Johannes Brussee, Lukas Nalos, Martin B Rook, Marc A Vos, Marcel AG van der Heyden and Adriaan P. IJzerman. 2009. Exploring the chemical substructures essential for hERG K+ channel blockade by synthesis and biological evaluation of dofetilide analogues. Chem. Med. Chem.(Wiley-VCH) 4:1722-1732. [ISSN: 1860-7187]
- Shagufta, Ritesh Singh and Gautam Panda. 2009, Synthetic studies towards steroid-amino acid hybrids. Indian Journal of Chemistry.(Indian Science) 48B: 989-995. [ISSN: 0975-0983]
- Maloy K. Parai, Shagufta, Ajay K. Srivastava, Matthias Kassack, Gautam Panda. 2008. An unexpected reaction of phosphorous tribromide on chromanone, thiochromanone, 3,4-dihydro-2H-benzo[b]thiepin-5-one, 3,4-dihydro-2H-benzo[b]oxepin-5-one and tetralone derived allylic alcohols: a case study. Tetrahedron (Elsevier)64: 9962-9976. [ISSN: 0040-4020]
- Gautam Panda, Maloy Kumar Parai, Sajal Kumar Das, Shagufta, Manish Sinha, Vinita Chaturvedi, Anil K. Srivastava, Anil N. Gaikwad, Sudhir Sinha. 2007. Effect of substituents on diarymethanes for antitubercular activity. European Journal of Medicinal Chemistry (Elsevier) 42: 410-419. [ISSN: 0223-5234]
- Shagufta and Gautam Panda. 2007. A new example of a steroid-amino acid hybrid: Construction of constrained nine membered D-ring steroids. Organic and Biomolecular Chemistry (Royal Society of Chemistry) 5 : 360- 366. [ISSN 1477-0539]
- Shagufta, Ashutosh Kumar, Gautam Panda and Mohammad Imran Siddiqi. 2007. CoMFA and CoMSIA 3D-QSAR analysis of diaryloxy methano phenanthrene derivatives as anti- tubercular agents. Journal of Molecular Modeling (Springer) 13: 99-107. [ISSN:0948-5023]
- Shagufta, Ajay Kumar Srivastava, Ramesh Sharma, Rajeev Mishra, Anil K. Balapure, Puvvada S. R. Murthy and Gautam Panda. 2006. Substituted phenanthrenes with basic amino side chains: A new series of anti-breast cancer agents. Bioorganic and Medicinal Chemistry (Elsevier) 14: 1497-1505. [ISSN: 0968-0896]
- Shagufta, Ajay Kumar Srivastava and Gautam Panda. 2006. Isomerization of allylic alcohols into saturated carbonyls using phosphorus tribromide. Tetrahedron Letters (Elsevier) 47: 1065-1070. [ISSN: 0040-4039]
- Gautam Panda, Jitendra K. Mishra, Shagufta, T. C. Dinadayalane and G. Narahari Sastry & Devendra S Negi. 2006. Hard-soft acid-base (HSAB) principle and difference in d-orbital configurations of metals explain the regioselectivity of nucleophilic attack to a carbinol in Friedel-Crafts reaction catalyzed by Lewis and protonic acids. Indian Journal of Chemistry (Indian Science)45B: 276-287. [ISSN: 0975-0983]
- Shagufta, Maloy Kumar Parai and Gautam Panda. 2005. A new strategy for the synthesis of aryl- and heteroaryl-substituted exocyclic olefins from allyl alcohols using PBr3. Terahedron Letters (Elsevier) 46: 8849-8852. [ISSN: 0040-4039]
- Shagufta, Resmi Raghunandan, Prakash R. Maulik and Gautam Panda. 2005. Convenient phosphorus tribromide induced syntheses of substituted 1-arylmethylnaphthalenes from 1-tetralone derivatives. Tetrahedron Letters (Elsevier) 46: 5337-5341. [ISSN: 0040-4039]
- Gautam Panda, Shagufta, Anil K. Srivastava and Sudhir Sinha. 2005. Synthesis and antitubercular activity of 2-hydroxy-aminoalkyl derivatives of diaryloxy methano phenanthrenes. Bioorganic and Medicinal Chemistry Letters (Elsevier) 15: 5222-5225. [ISSN: 0960-894X]
- Sajal Kumar Das, Shagufta, and Gautam Panda. 2005. An easy access to unsymmetric trisubstituted methane derivatives (TRSMs). Tetrahedron Letters (Elsevier) 46: 3097-3102. [ISSN: 0040-4039]
- Shagufta, Jitendra Kumar Mishra, Vinita Chaturvedi, Anil K. Srivastava, Ranjana Srivastava and Brahm S. Srivastava. 2004. Diaryloxy methano phenanthrenes: a new class of antituberculosis agents. Bioorganic and Medicinal Chemistry (Elsevier) 12: 5269-5276. [ISSN: 0968-0896]

Dr. Irshad Ahmad
ASSOCIATE PROFESSOR – CHEMISTRY
Biography
Dr. Irshad Ahmad joined the American University of Ras Al Khaimah in spring 2011 as an Assistant Professor of Chemistry. He received the master’s degree in chemistry from Jiwaji University in 1999. Subsequently acquired significant pharmaceutical industrial experience and developed cardio-selective beta-blocker drug molecule. He joined Central Salt and Marine Chemical Research Institute and Bhavnagar University under the sponsored project of DST and CSIR as a senior research fellow and received his PhD degree in chemistry in 2006. Subsequently, he accepted an invited scientist position in Korea Research Institute of Chemical Technology, South Korea and contributed his expertise in the field of Nanotechnology. Dr. Irshad is a recipient of prestigious European fellowships (NWO-Rubicon & FCT) and he joined Van’t Hoff Institute for Molecular Sciences, University of Amsterdam, The Netherlands as a NWO Rubicon fellow (Netherlands Organization for Scientific Research, the Dutch Science Foundation), he acquired expertise in the field of supramolecular chemistry.
Afterward, he moved to the Leibniz Institute for Surface Modification, Leipzig, Germany under the Deutsche Forschungsgemeinschaft Grant. Dr. Irshad developed “Novel ultra-fast metathesis catalyst” for the production of high quality alternating copolymers. Subsequently Dr. Irshad, joined Department of Chemistry and Biochemistry, Stephenson Life Science Research Center, University of Oklahoma, USA as a postdoctoral research associate. He developed strategies for the novel environmentally friendly reactions for the production of value added chemicals from biomass.
Dr. Irshad specialized in the area of chemistry, bridging the traditional disciplines of inorganic, organic and bio-organic chemistry. He contributed US and European patent for green and clean technology development. He has published peer-reviewed international research articles in the American Chemical Society (ACS), Royal Society of Chemistry (RSC) Cambridge, Elsevier Science, Wiley, and Springer journals. He has presented his research at several scientific conferences worldwide and received awards.
Research and Publication
Research Interest:
Asymmetric catalysis, Biotechnology, Metathesis, Material science, Nanotechnology, Pharmaceutical, Renewable energy and Supramolecular chemistry
Book:
Asymmetric Homogeneous and Heterogeneous Catalysts: An Approach to the Synthesis of Chiral Drug Intermediates by Scholars Press, Germany. 2013, ISBN: 978-3-639-51138-3
Membership:
- American Chemical Society (ACS), USA
- The Royal Society of Chemistry, Cambridge, UK
Patents:
- United States Patent 7,235,676, H. Khan, S. H. R. Abdi, R. I. Kureshy, S. Singh, I. Ahmad, R. V. Jasra, P. K. Ghosh, ‘Catalytic process for the preparation of epoxides from alkenes.
- Patent Cooperation Treaty (PCT) WO/2005/095370, N. H. Khan, S. H. R. Abdi, R. I. Kureshy, S. Singh, I. Ahmad, R. V. Jasra, P. K. Ghosh. An improved catalytic process for the preparation of epoxides from alkenes.
- European Patent EP 1732910 A1, N. H. Khan, S. H. R. Abdi, R. I. Kureshy, S. SinghA, I. Ahmad, R. V. Jasra, P. K. Ghosh, An improved catalytic process for the preparation of epoxides from alkenes.
Publications:
- Pramoda, U. Gupta, I. Ahmad, R. Kumar, C.N.R. Rao, Assemblies of Covalently Cross-linked Nanosheets of MoS2 and of MoS2-RGO: Synthesis and Novel Properties, Journal of Materials Chemistry A, 4, 2016, 8989.
- Shagufta, I. Ahmad, Recent insight into the biological activities of synthetic xanthone derivatives, European Journal of Medicinal Chemistry, 116, 2016, 267.
- Ahmad, Shagufta, Recent Development in Steroidal and Non-steroidal Aromatase Inhibitors for the Chemoprevention of Estrogen dependent Breast Cancer, European Journal of Medicinal Chemistry, 102, 2015, 375.
- Ahmad, Shagufta, Sulfones: An important class of organic compounds with diverse biological activities, International Journal of Pharmacy and Pharmaceutical Sciences, 7, 3, 2015, 19.
- Kumar, K. Gopalakrishnan, I. Ahmad, and C. N. R. Rao, BN-Graphene Composites Generated by Covalent Cross-Linking with Organic Linkers, Advanced Functional Materials, 25, 37, 2015, 5910.
- Kumar, D. Raut, I. Ahmad, U. Ramamurty, T. K. Maji and C. N. R. Rao. Functionality preservation with enhanced mechanical integrity in the nanocomposites of the metal–organic framework, ZIF-8, with BN nanosheets, Materials Horizons, 1, 2014, 513.
- R. Buchmeiser, I. Ahmad, V. Gurram and P. S. Kumar, Pseudo-Halide and Nitrate Derivatives of Grubbs and Grubbs_Hoveyda Initiators: Some Structural Features Related to the Alternating Ring-Opening Metathesis Copolymerization of Norborn-2-ene with Cyclic Olefins, Macromolecule, 44 (11), 2011, 4098.
- Ahmad, G. Chapman and K. M. Nicholas, Sulfite-Driven, Oxorhenium-Catalyzed Deoxydehydration of Glycols, Organometallics, 30 (10), 2011, 2810.
- Vkuturi, G. Chapman, I. Ahmad, K. M. Nicholas, Rhenium-Catalyzed Deoxydehydration of Glycols by Sulfite, Inorganic Chemistry, 49, 2010, 4744.
- I. Kureshy, I. Ahmad, K. Pathak, N. H. Khan, S. H. R. Abdi, H. C. Bajaj, Solvent- free microwave synthesis of aryloxypropanolamines by ring opening of aryloxy epoxides, Research Letters in Organic Chemistry, 2009, Article ID 109717, doi:10.1155/2009/109717.
- I. Kureshy, I. Ahmad, K. Pathak, N. H. Khan, S. H. R. Abdi, R. V. Jasra, Sulfonic acid functionalized mesoporous SBA-15 as an efficient and recyclable catalyst for the synthesis of chromenes from chromanols, Catalysis Communications 10, 2009, 572.
- Pathak, I. Ahmad, S. H. R. Abdi, R. I. Kureshy, N. H. Khan, R. V. Jasra, The synthesis of silica-supported chiral BINOL: Application in Ti-catalyzed asymmetric addition of diethylzinc to aldehydes, Journal of Molecular Catalysis A-Chemical 280, 2008, 106.
- Kluwer, I. Ahmad, J. N. H. Reek, Improved synthesis of monodentate and bidentate 2- and 3-pyridylphosphines, Tetrahedron Letter 48, 2007, 2999.
- Pathak, I. Ahmad, S. H. R. Abdi, R. I. Kureshy, N. H. Khan, R. V. Jasra, Oxidative Kinetic Resolution of racemic Secondary Alcohols catalyzed by recyclable Dimeric Mn(III) salen catalysts, Journal of Molecular Catalysis A-Chemical 274, 2007, 120.
- I. Kureshy, I. Ahmad, N. H. Khan, S. H. R. Abdi, K. Pathak, R. V. Jasra, Easily Recyclable Chiral Polymeric Mn (salen) Complex for Oxidative Kinetic resolution of Racemic Secondary Alcohols, Chirality, 19, 2007, 352.
- Pathak, A. P. Bhatt, S. H. R. Abdi, R. I. Kureshy, N. H. Khan, I. Ahmad, R. V. Jasra, Enantioselective phenylacetylene addition to aromatic aldehydes and ketones catalyzed by recyclable polymeric Zn(II) salen complex, Chirality, 19, 2007, 1.
- I. Kureshy, I. Ahmad, N. H. Khan, S. H. R. Abdi, K. Pathak, R. V. Jasra, Chiral Mn (III) salen complexes covalently bonded on modified MCM-41 and SBA-15 as efficient catalysts for enantioselective epoxidation of non- functionalized alkenes, Journal of Catalysis A-Chemical, 238, 2006, 134.
- Pathak, A. P. Bhatt, S. H. R. Abdi, R. I. Kureshy, N. H. Khan, I. Ahmad, R. V. Jasra Enantioselective addition of diethylzinc to aldehydes using immobilized chiral BINOL-Ti complex on ordered mesoporous silicas, Tetrahedron: Asymmetry,17, 2006, 1506.
- I. Kureshy, I. Ahmad, N. H. Khan, S. H. R. Abdi, K. Pathak, R. V. Jasra, Encapsulation of chiral MnIII (salen) complex in ordered mesoporous silicas: An approach Towards heterogenizing asymmetric Epoxidation catalysts for non-Functionalized alkenes, Tetrahedron: Asymmetry 16, 2005, 3562.
- I. Kureshy, I. Ahmad, N. H. Khan, S. H. R. Abdi, S. Singh, P. H. Pandia, R. V. Jasra, New immobilized chiral Mn(III) salen complexes on pyridine N-Oxide Modified MCM-41as effective catalysts for epoxidation of nonfunctionalized Alkenes, Journal of Catalysis A- Chemical 235 , 2005, 28.
- Pathak, A. P. Bhatt, S. H. R. Abdi, R. I. Kureshy, N. H. Khan, I. Ahmad, R. V. Jasra Enantioselective addition of diethylzinc to aldehydes using immobilized chiral BINOL-Ti complex on ordered mesoporous silicas, Tetrahedron: Asymmetry,17, 2006, 1506.
- I. Kureshy, S. Singh, N. H. Khan, S. H. R. Abdi, I. Ahmad, A. Bhatt, R. V. Jasra, Improved catalytic activity of homochiral dimeric cobalt salen hydrolytic kinetic resolution of terminal racemic epoxides, Chirality, 17, 2005, 1.
- I. Kureshy, S. Singh, N. H. Khan, S. H. R. Abdi , I. Ahmad, .Bhatt, R. V. Jasra, Environment friendly protocol for enantioselective epoxidation of non-functionalized Alkenes catalyzed by recyclable homochiral dimeric Mn(III)salen complexes with hydrogen peroxide and UHP adduct as Oxidants, Catalysis Letters, 107, 2005, 127.
- I. Kureshy, N. H. Khan, S. H. R. Abdi, I. Ahmad, S. Singh, and R. V. Jasra, Dicationic chiral Mn (III) Salen complex exchange in the interlayers of Montmorillonite clay: a heterogeneous enantioselective catalyst for epoxidation of non-functionalised alkenes, Journal of Catalysis, 221, 2004, 234.
- I. Kureshy, N. H. Khan, S. H. R. Abdi, S. Singh, I. Ahmad, R. V. Jasra, Catalytic asymmetric epoxidation of non-functionalised alkenes using polymeric Mn(III)Salen as catalysts and NaOCl as oxidant, Journal of Molecular Catalysis A-Chemical, 218, 2004, 141.
- I. Kureshy, N.H. Khan, S.H. R. Abdi, A. P. Vyas, I. Ahmad, S. Singh, R. V. Jasra, Enantioselective Epoxidation of Non-Functionalised Alkenes catalysed by recyclable new Homo Chiral Dimeric Mn(III) Salen complexes, Journal of Catalysis, 224, 2004, 229.
- I. Kureshy, N. H. Khan, S. H. R. Abdi, I. Ahmad, S. Singh, and R. V. Jasra, Immobilization of dicationic Mn(III) salen in the interlayers of montmorrillonite Clay for enantioselective epoxidation of non-functionalised alkenes, Catalysis Letters, 91, 2003, 207.
Selected International Events:
- Applied Nanotechnology and Nanoscience International Conference (ANNIC), November 9-11, 2016, Barcelona, SPAIN.
- 2nd International Conference on Smart Material Research (ICSMR), September 22-24, 2016, Istanbul, TURKEY.
- Emirates Foundation’s Think Science Competition, April 17-19, 2016, World Trade Center, Dubai, UAE.
- SSL Visiting Fellow 2013-15 at the International Centre for Materials Science, JNCASR, SSL, Bangalore, INDIA.
- Global Conference on Materials Sciences (GC-MAS-2014), November 13-15, 2014, Antalya, TURKEY.
- 5th Annual International Workshop on Advanced Material (IWAM 2013), organized by Ras Al Khaimah Center for Advance Materials (RAK CAM), Feb. 24-26, 2013 at Al Hamra Fort Hotel, Ras Al Khaimah, UAE.
- Internal Quality Assurance in Higher Education Institutions workshop organized by the Commission for Academic Accreditation (CAA)- 2nd May 2011, Alghurair University campus, Dubai, UAE.
- 45th American Chemical Society (ACS) Midwest Regional meeting, Oct. 27-30, 2010, Wichita, Kansas, USA.
- 55th Annual American Chemical Society (ACS) PentaSectional Meeting- Biofuel, April 10, 2010, organized by American Chemical Society (ACS), Norman, Oklahoma, USA.
- 18th International Symposium on Olefin Metathesis and Related Chemistry (ISOM XVIII), Organized by the Leibniz-Institute for Surface modification (IOM), August 2-7, 2009, Leipzig, GERMANY.
- 16th International Symposium on Homogeneous Catalysis (ISHC-XVI), July 6-11, 2008, Organized by the Institute of Chemistry of Organometallic Compounds (ICCOM) of the Italian Research Council (CNR) held in Florence, ITALY.
- European IDECAT Summer School on Computational Methods for Catalysis and Materials Science, 15-22 September 2007, Porquerolles, FRANCE.
- 8th Netherland’s Catalysis and Chemistry Conference (NCCC), March 5-7, 2007, Noordwijkerhout, The NETHERLANDS.
- 7th International Symposium on Catalysis Applied to Fine Chemicals organized by German Catalysis Society and Dechema. Oct 23-27, 2005, Bingen -Mainz, GERMANY.
- 1st Indo- German Conference on Catalysis-A Cross Disciplinary Vision, February 6-8, 2003, Indian Institute of Chemical Technology (IICT), Hyderabad, INDIA.
FDA approves first treatment for a form of Batten disease, Brineura (cerliponase alfa)
The U.S. Food and Drug Administration today approved Brineura (cerliponase alfa) as a treatment for a specific form of Batten disease. Brineura is the first FDA-approved treatment to slow loss of walking ability (ambulation) in symptomatic pediatric patients 3 years of age and older with late infantile neuronal ceroid lipofuscinosis type 2 (CLN2), also known as tripeptidyl peptidase-1 (TPP1) deficiency.
“The FDA is committed to approving new and innovative therapies for patients with rare diseases, particularly where there are no approved treatment options,” said Julie Beitz, M.D., director of the Office of Drug Evaluation III in the FDA’s Center for Drug Evaluation and Research. “Approving the first drug for the treatment of this form of Batten disease is an important advance for patients suffering with this condition.”
CLN2 disease is one of a group of disorders known as neuronal ceroid lipofuscinoses (NCLs), collectively referred to as Batten disease. CLN2 disease is a rare inherited disorder that primarily affects the nervous system. In the late infantile form of the disease, signs and symptoms typically begin between ages 2 and 4. The initial symptoms usually include language delay, recurrent seizures (epilepsy) and difficulty coordinating movements (ataxia). Affected children also develop muscle twitches (myoclonus) and vision loss. CLN2 disease affects essential motor skills, such as sitting and walking. Individuals with this condition often require the use of a wheelchair by late childhood and typically do not survive past their teens. Batten disease is relatively rare, occurring in an estimated two to four of every 100,000 live births in the United States.
Brineura is an enzyme replacement therapy. Its active ingredient (cerliponase alfa) is a recombinant form of human TPP1, the enzyme deficient in patients with CLN2 disease. Brineura is administered into the cerebrospinal fluid (CSF) by infusion via a specific surgically implanted reservoir and catheter in the head (intraventricular access device). Brineura must be administered under sterile conditions to reduce the risk of infections, and treatment should be managed by a health care professional knowledgeable in intraventricular administration. The recommended dose of Brineura in pediatric patients 3 years of age and older is 300 mg administered once every other week by intraventricular infusion, followed by an infusion of electrolytes. The complete Brineura infusion, including the required infusion of intraventricular electrolytes, lasts approximately 4.5 hours. Pre-treatment of patients with antihistamines with or without antipyretics (drugs for prevention or treatment of fever) or corticosteroids is recommended 30 to 60 minutes prior to the start of the infusion.
The efficacy of Brineura was established in a non-randomized, single-arm dose escalation clinical study in 22 symptomatic pediatric patients with CLN2 disease and compared to 42 untreated patients with CLN2 disease from a natural history cohort (an independent historical control group) who were at least 3 years old and had motor or language symptoms. Taking into account age, baseline walking ability and genotype, Brineura-treated patients demonstrated fewer declines in walking ability compared to untreated patients in the natural history cohort.
The safety of Brineura was evaluated in 24 patients with CLN2 disease aged 3 to 8 years who received at least one dose of Brineura in clinical studies. The safety and effectiveness of Brineura have not been established in patients less than 3 years of age.
The most common adverse reactions in patients treated with Brineura include fever, ECG abnormalities including slow heart rate (bradycardia), hypersensitivity, decrease or increase in CSF protein, vomiting, seizures, hematoma (abnormal collection of blood outside of a blood vessel), headache, irritability, increased CSF white blood cell count (pleocytosis), device-related infection, feeling jittery and low blood pressure.
Brineura should not be administered to patients if there are signs of acute intraventricular access device-related complications (e.g., leakage, device failure or signs of device-related infection such as swelling, erythema of the scalp, extravasation of fluid, or bulging of the scalp around or above the intraventricular access device). In case of intraventricular access device complications, health care providers should discontinue infusion of Brineura and refer to the device manufacturer’s labeling for further instructions. Additionally, health care providers should routinely test patient CSF samples to detect device infections. Brineura should also not be used in patients with ventriculoperitoneal shunts (medical devices that relieve pressure on the brain caused by fluid accumulation).
Health care providers should also monitor vital signs (blood pressure, heart rate, etc.) before the infusion starts, periodically during infusion and post-infusion in a health care setting. Health care providers should perform electrocardiogram (ECG) monitoring during infusion in patients with a history of slow heart rate (bradycardia), conduction disorder (impaired progression of electrical impulses through the heart) or structural heart disease (defect or abnormality of the heart), as some patients with CLN2 disease can develop conduction disorders or heart disease. Hypersensitivity reactions have also been reported in Brineura-treated patients. Due to the potential for anaphylaxis, appropriate medical support should be readily available when Brineura is administered. If anaphylaxis occurs, infusion should be immediately discontinued and appropriate treatment should be initiated.
The FDA will require the Brineura manufacturer to further evaluate the safety of Brineura in CLN2 patients below the age of 2 years, including device related adverse events and complications with routine use. In addition, a long-term safety study will assess Brineura treated CLN2 patients for a minimum of 10 years.
The FDA granted this application Priority Review and Breakthrough Therapydesignations. Brineura also received Orphan Drug designation, which provides incentives to assist and encourage the development of drugs for rare diseases.
The sponsor is also receiving a Rare Pediatric Disease Priority Review Voucherunder a program intended to encourage development of new drugs and biologics for the prevention and treatment of rare pediatric diseases. A voucher can be redeemed by a sponsor at a later date to receive Priority Review of a subsequent marketing application for a different product. This is the tenth rare pediatric disease priority review voucher issued by the FDA since the program began.
The FDA granted approval of Brineura to BioMarin Pharmaceutical Inc.
BLU 285


BLU-285
CAS 1703793-34-3
- Molecular FormulaC26H27FN10
- Average mass498.558 Da
- 5-Pyrimidinemethanamine, α-(4-fluorophenyl)-α-methyl-2-[4-[6-(1-methyl-1H-pyrazol-4-yl)pyrrolo[2,1-f][1,2,4]triazin-4-yl]-1-piperazinyl]-, (αS)-
- Originator Blueprint Medicines
- Class Antineoplastics; Skin disorder therapies; Small molecules
- Mechanism of Action Platelet-derived growth factor alpha receptor modulators; Proto oncogene protein c-kit inhibitors
- Orphan Drug Status Yes – Systemic mastocytosis; Gastrointestinal stromal tumours
- Phase I Gastrointestinal stromal tumours; Solid tumours; Systemic mastocytosis
- 04 Dec 2016 Proof-of-concept data from phase I trial in Systemic mastocytosis presented at the 58thAnnual Meeting and Exposition of the American Society of Hematology (ASH Hem-2016)
- 03 Dec 2016 Pharmacodynamics data from preclinical studies in Systemic mastocytosis presented at the 58th Annual Meeting and Exposition of the American Society of Hematology (ASH-Hem-2016)
- 03 Dec 2016 Preliminary pharmacokinetic data from a phase I trial in Systemic mastocytosis presented at the 58th Annual Meeting and Exposition of the American Society of Hematology (ASH Hem-2016)
BLU 285
(S)- 1 – (4- fluorophenyl)- l-(2-(4-(6-(l-methyl-lH-pyrazol-4-yl)pyrrolo[2, l-/] [l,2,4]triazin-4-yl)piperazin-l-yl)pyrimidin-5-yl)ethanamine (Compounds 44) WO2015057873
| Inventors | Yulian Zhang, Brian L. Hodous, Joseph L. Kim, Kevin J. Wilson, Douglas Wilson |
| Applicant | Blueprint Medicines Corporation |


Yulian Zhang,
Blueprint Medicines Corporation
ΚΓΓ and PDGFR.
The enzyme KIT (also called CD117) is a receptor tyrosine kinase expressed on a wide variety of cell types. The KIT molecule contains a long extracellular domain, a transmembrane segment, and an intracellular portion. The ligand for KIT is stem cell factor (SCF), whose binding to the extracellular domain of KIT induces receptor dimerization and activation of downstream signaling pathways. KIT mutations generally occur in the DNA encoding the juxtumembrane domain (exon 11). They also occur, with less frequency, in exons 7, 8, 9, 13, 14, 17, and 18. Mutations make KIT function independent of activation by SCF, leading to a high cell division rate and possibly genomic instability. Mutant KIT has been implicated in the pathogenesis of several disorders and conditions including systemic mastocytosis, GIST (gastrointestinal stromal tumors), AML (acute myeloid leukemia), melanoma, and seminoma. As such, there is a need for therapeutic agents that inhibit ΚΓΓ, and especially agents that inhibit mutant ΚΓΓ.Platelet-derived growth factor receptors (PDGF-R) are cell surface tyrosine kinase receptors for members of the platelet-derived growth factor (PDGF) family. PDGF subunits -A and -B are important factors regulating cell proliferation, cellular differentiation, cell growth, development and many diseases including cancer. A PDGFRA D842V mutation has been found in a distinct subset of GIST, typically from the stomach. The D842V mutation is known to be associated with tyrosine kinase inhibitor resistance. As such, there is a need for agents that target this mutation.

CONTD………..

PATENT
Example 7: Synthesis of (R)-l-(4-fluorophenyl)- l-(2-(4-(6-(l-methyl-lH-pyrazol-4-yl)pyrrolo[2, 1 -f\ [ 1 ,2,4] triazin-4-yl)piperazin- 1 -yl)pyrimidin-5-yl)ethanamine and (S)- 1 – (4- fluorophenyl)- l-(2-(4-(6-(l-methyl-lH-pyrazol-4-yl)pyrrolo[2, l-/] [l,2,4]triazin-4-yl)piperazin-l-yl)pyrimidin-5-yl)ethanamine (Compounds 43 and 44)


Step 1 : Synthesis of (4-fluorophenyl)(2-(4-(6-(l-methyl- lH-pyrazol-4-yl)pyrrolo[2,l-f] [ 1 ,2,4] triazin-4-yl)piperazin- 1 -yl)pyrimidin-5-yl)methanone:

4-Chloro-6-(l-methyl- lH-pyrazol-4-yl)pyrrolo[2,l-/] [l,2,4]triazine (180 mg, 0.770 mmol), (4-fluorophenyl)(2-(piperazin-l-yl)pyrimidin-5-yl)methanone, HC1 (265 mg, 0.821 mmol) and DIPEA (0.40 mL, 2.290 mmol) were stirred in 1,4-dioxane (4 mL) at room temperature for 18 hours. Saturated ammonium chloride was added and the products extracted into DCM (x2). The combined organic extracts were dried over Na2S04, filtered through Celite eluting with DCM, and the filtrate concentrated in vacuo. Purification of the residue by MPLC (25- 100% EtOAc-DCM) gave (4-fluorophenyl)(2-(4-(6-(l-methyl-lH-pyrazol-4-yl)pyrrolo[2,l- ] [l,2,4]triazin-4-yl)piperazin- l-yl)pyrimidin-5-yl)methanone (160 mg, 0.331 mmol, 43 % yield) as an off-white solid. MS (ES+) C25H22FN90 requires: 483, found: 484 [M + H]+.
Step 2: Synthesis of (5,Z)-N-((4-fluorophenyl)(2-(4-(6-(l-methyl- lH-p razol-4-yl)p rrolo[2, l- ] [l,2,4]triazin-4- l)piperazin- l-yl)pyrimidin-5-yl)methylene)-2-methylpropane-2-sulfinamide:

(S)-2-Methylpropane-2-sulfinamide (110 mg, 0.908 mmol), (4-fluorophenyl)(2-(4-(6-(l-methyl- lH-pyrazol-4-yl)pyrrolo[2,l-/][l,2,4]triazin-4-yl)piperazin- l-yl)pyrimidin-5-yl)methanone (158 mg, 0.327 mmol) and ethyl orthotitanate (0.15 mL, 0.715 mmol) were stirred in THF (3.2 mL) at 70 °C for 18 hours. Room temperature was attained, water was added, and the products extracted into EtOAc (x2). The combined organic extracts were washed with brine, dried over Na2S04, filtered, and concentrated in vacuo while loading onto Celite. Purification of the residue by MPLC (0- 10% MeOH-EtOAc) gave (5,Z)-N-((4-fluorophenyl)(2-(4-(6-(l-methyl- lH-pyrazol-4-yl)pyrrolo[2, l-/] [l,2,4]triazin-4-yl)piperazin-l-yl)pyrimidin-5-yl)methylene)-2- methylpropane-2-sulfinamide (192 mg, 0.327 mmol, 100 % yield) as an orange solid. MS (ES+) C29H3iFN10OS requires: 586, found: 587 [M + H]+.
Step 3: Synthesis of (lS’)-N-(l-(4-fluorophenyl)- l-(2-(4-(6-(l-methyl- lH-pyrazol-4- l)pyrrolo[2, l-/] [l,2,4]triazin-4-yl)piperazin-l-yl)pyrimidin-5-yl)ethyl)-2-methylpropane-2-

(lS’,Z)-N-((4-Fluorophenyl)(2-(4-(6-(l-methyl-lH-pyrazol-4-yl)pyrrolo[2,l- ] [l,2,4]triazin-4-yl)piperazin- l-yl)pyrimidin-5-yl)methylene)-2-methylpropane-2-sulfinamide (190 mg, 0.324 mmol) was taken up in THF (3 mL) and cooled to 0 °C. Methylmagnesium bromide (3 M solution in diethyl ether, 0.50 mL, 1.500 mmol) was added and the resulting mixture stirred at 0 °C for 45 minutes. Additional methylmagnesium bromide (3 M solution in diethyl ether, 0.10 mL, 0.300 mmol) was added and stirring at 0 °C continued for 20 minutes. Saturated ammonium chloride was added and the products extracted into EtOAc (x2). The combined organic extracts were washed with brine, dried over Na2S04, filtered, and concentrated in vacuo while loading onto Celite. Purification of the residue by MPLC (0-10% MeOH-EtOAc) gave (lS’)-N-(l-(4-fluorophenyl)-l-(2-(4-(6-(l-methyl- lH-pyrazol-4-yl)pyrrolo[2, l- ] [l,2,4]triazin-4-yl)piperazin- l-yl)pyrimidin-5-yl)ethyl)-2-methylpropane-2-sulfinamide (120 mg, 0.199 mmol, 61.5 % yield) as a yellow solid (mixture of diastereoisomers). MS (ES+) C3oH35FN10OS requires: 602, found: 603 [M + H]+.
Step 4: Synthesis of l-(4-fluorophenyl)- l-(2-(4-(6-(l-methyl- lH-pyrazol-4-yl)pyrrolo[2,l-f\ [ 1 ,2,4] triazin-4- l)piperazin- 1 -yl)pyrimidin-5-yl)ethanamine:

(S)-N- ( 1 – (4-Fluorophenyl)- 1 -(2- (4- (6-( 1 -methyl- 1 H-pyrazol-4-yl)pyrrolo [2,1-/] [l,2,4]triazin-4-yl)piperazin- l-yl)pyrimidin-5-yl)ethyl)-2-methylpropane-2-sulfinamide (120 mg, 0.199 mmol) was stirred in 4 M HCl in 1,4-dioxane (1.5 mL)/MeOH (1.5 mL) at room temperature for 1 hour. The solvent was removed in vacuo and the residue triturated in EtOAc to give l-(4-fluorophenyl)- l-(2-(4-(6-(l -methyl- lH-pyrazol-4-yl)pyrrolo[2, l-/][l,2,4]triazin-4-yl)piperazin- l-yl)pyrimidin-5-yl)ethanamine, HCl (110 mg, 0.206 mmol, 103 % yield) as a pale yellow solid. MS (ES+) C26H27FN10 requires: 498, found: 482 [M- 17 + H]+, 499 [M + H]+.
Step 5: Chiral separation of (R)-l-(4-fluorophenyl)- l-(2-(4-(6-(l-methyl- lH-pyrazol-4-yl)pyrrolo[2, l-/] [l,2,4]triazin-4-yl)piperazin-l-yl)pyrimidin-5-yl)ethanamine and (5)-1-(4-fluorophenyl)- l-(2-(4-(6-(l-methyl-lH-pyrazol-4-yl)pyrrolo[2, l-/] [l,2,4]triazin-4-yl)piperazin-1 -yl)pyrimidin- -yl)ethanamine:

The enantiomers of racemic l-(4-fluorophenyl)- l-(2-(4-(6-(l-methyl- lH-pyrazol-4-yl)pyrrolo[2, l-/] [l,2,4]triazin-4-yl)piperazin-l-yl)pyrimidin-5-yl)ethanamine (94 mg, 0.189 mmol) were separated by chiral SFC to give (R)-l-(4-fluorophenyl)- l-(2-(4-(6-(l-methyl-lH-
pyrazol-4-yl)pyrrolo[2, l-/][l,2,4]triazin-4-yl)piperazin- l-yl)pyrimidin-5-yl)ethanamine (34.4 mg, 0.069 mmol, 73.2 % yield) and (lS,)-l-(4-fluorophenyl)- l-(2-(4-(6-(l-methyl-lH-pyrazol-4-yl)pyrrolo[2, l-/] [l,2,4]triazin-4-yl)piperazin-l-yl)pyrimidin-5-yl)ethanamine (32.1 mg, 0.064 mmol, 68.3 % yield). The absolute stereochemistry was assigned randomly. MS (ES+)
C26H27FN10 requires: 498, found: 499 [M + H]+.

/////////BLU-285, 1703793-34-3, PHASE 1, Brian Hodous, BlueprintMeds, KIT & PDGFRalpha inhibitors, Orphan Drug Status
Fc1ccc(cc1)[C@](C)(N)c2cnc(nc2)N3CCN(CC3)c4ncnn5cc(cc45)c6cn(C)nc6
Next in 1st time disclosures Brian Hodous of @BlueprintMeds will talk about KIT & PDGFRalpha inhibitors #ACSSanFran
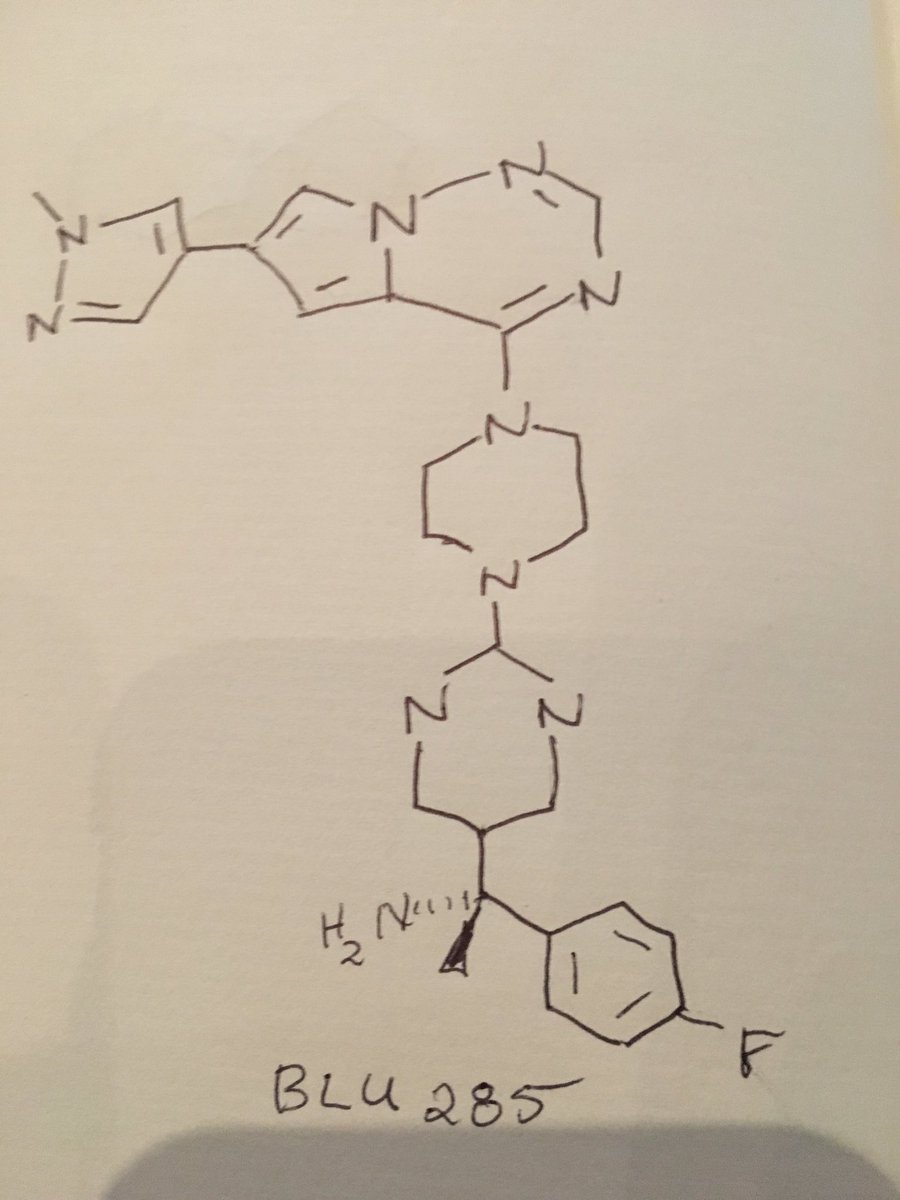
Tamibarotene, тамибаротен , تاميباروتان , 他米巴罗汀 ,

Tamibarotene
тамибаротен , تاميباروتان , 他米巴罗汀 ,
94497-51-5 CAS
- Molecular FormulaC22H25NO3
- Average mass351.439 Da
4-(((5,6,7,8-Tetrahydro-5,5,8,8-tetramethyl-2-naphthalenyl)amino)carbonyl)benzoic Acid, Tamibarotene
- Am 80
- Am 80 (pharmaceutical)
- Amnolake
- NSC 608000
- RR 110
Hisao Ekimoto, “TAMIBAROTENE CAPSULE PREPARATION.” U.S. Patent US20100048708, issued February 25, 2010.

Tamibarotene , a small molecule retinoic acid receptor alpha (RARα) agonists developed by Nippon Shinyaku, was approved in Japan for the treatment of acute promyelocytic leukemia (APL) in 2005. Recently, the drug was in clinical development for the treatment of acute myeloid leukemia (AML), myelodysplasia, pediatric solid tumor, and steroid-refractory
Tamibarotene (brand name: Amnolake), also called retinobenzoic acid, is orally active, synthetic retinoid, developed to overcome all-trans retinoic acid (ATRA) resistance, with potential antineoplastic activity against acute promyelocytic leukaemia (APL) .[1] It is currently marketed only in Japan and early trials have demonstrated that it tends to be better tolerated than ATRA.[2] It has also been investigated as a possible treatment for Alzheimer’s disease, multiple myeloma and Crohn’s disease.[2][3]
Synthesis
The tetralin-based compound tamibarotene (7) has been tested as an agent for treating leukaemias.

Reaction of the diol (1) with hydrogen chloride affords the corresponding dichloro derivative (2). Aluminum chloride mediated Friedel–Crafts alkylation of acetanilide with the dichloride affords the tetralin (3). Basic hydrolysis leads to the primary amine (4). Acylation of the primary amino group with the half acid chloride half ester from terephthalic acid (5) leads to the amide (6). Basic hydrolysis of the ester grouping then affords (7).[4]
PAPER
Synthesis of Tamibarotene via Ullmann-Type Coupling

An effective process was developed for the preparation of tamibarotene via an Ullmann-type coupling in a nonpressurized l-proline/DMSO system. Notable features were the telescoping of reactions, avoiding environmentally hazardous materials, and an acceptable overall yield. The safe scalable process was validated on a 1 kg scale.
Mp 223–225 °C (lit. SEE BELOW mp 231–232 °C); 1H NMR (400 MHz, chloroform-d) δ 8.22 (d, J = 7.7 Hz, 2H), 7.97 (d, J = 7.7 Hz, 2H), 7.80 (s, 1H), 7.54 (s, 1H), 7.46 (d, J = 8.4 Hz, 1H), 7.33 (d, J = 8.3 Hz, 1H), 1.70 (s, 4H), 1.31 (s, 6H), 1.29 (s, 6H); MS (ESI) m/z: 350.0 [M – H]−
…Journal of Medicinal Chemistry (1988), 31 (11), 2182-92

PAPER
Journal of Medicinal Chemistry (1988), 31 (11), 2182-92
PAPER
Chem. Pharm. Bull. 61(8) 846–852 (2013)
https://www.jstage.jst.go.jp/article/cpb/61/8/61_c13-00356/_pdf
References
- Jump up^ “Tamibarotene: AM 80, retinobenzoic acid, Tamibaro”. Drugs in R&D. 5 (6): 359–62. 2004. doi:10.2165/00126839-200405060-00010. PMID 15563242.
- ^ Jump up to:a b Miwako, I; Kagechika, H (August 2007). “Tamibarotene”. Drugs Today (Barc). 43 (8): 563–568. doi:10.1358/dot.2007.43.8.1072615. PMID 17925887.
- Jump up^ Fukasawa, H; Nakagomi, M; Yamagata, N; Katsuki, H; Kawahara, K; Kitaoka, K; Miki, T; Shudo, K (2012). “Tamibarotene: a candidate retinoid drug for Alzheimer’s disease” (PDF). Biological & Pharmaceutical Bulletin. 35 (8): 1206–1212. doi:10.1248/bpb.b12-00314. PMID 22863914.
- Jump up^ Y. Hamada, I. Yamada, M. Uenaka, T. Sakata, U.S. Patent 5,214,202 (1993).
 |
|
| Names | |
|---|---|
| IUPAC name
4-[(1,1,4,4-tetramethyltetralin-6-yl)carbamoyl]benzoic acid
|
|
| Identifiers | |
|
3D model (Jmol)
|
|
| 3564473 | |
| ChEBI | |
| ChemSpider | |
| DrugBank | |
|
PubChem CID
|
|
| UNII | |
| Properties | |
| C22H25NO3 | |
| Molar mass | 351.45 g·mol−1 |
|
Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa).
|
|
////////////Tamibarotene, тамибаротен , تاميباروتان , 他米巴罗汀 ,
CC1(C)CCC(C)(C)C2=C1C=CC(NC(=O)C1=CC=C(C=C1)C(O)=O)=C2
PF 2562
PF 2562
CAS 1609258-91-4
MF C19 H17 N5 O


Jennifer Elizabeth Davoren
Principal Scientist at Pfizer
SYNTHESIS


-
Dopamine acts upon neurons through two families of dopamine receptors, D1-like receptors (D1Rs) and D2-like receptors (D2Rs). The D1-like receptor family consists of D1 and D5 receptors which are expressed in many regions of the brain. D1 mRNA has been found, for example, in the striatum and nucleus accumbens. See e.g., Missale C, Nash S R, Robinson S W, Jaber M, Caron M G “Dopamine receptors: from structure to function”, Physiological Reviews 78:189-225 (1998). Pharmacological studies have reported that D1 and D5 receptors (D1/D5), namely D1-like receptors, are linked to stimulation of adenylyl cyclase, whereas D2, D3, and D4 receptors, namely D2-like receptors, are linked to inhibition of cAMP production.
-
Dopamine D1 receptors are implicated in numerous neuropharmacological and neurobiological functions. For example, D1 receptors are involved in different types of memory function and synaptic plasticity. See e.g., Goldman-Rakic P S et al., “Targeting the dopamine D1 receptor in schizophrenia: insights for cognitive dysfunction”, Psychopharmacology 174(1):3-16 (2004). Moreover, D1 receptors have been implicated in a variety of psychiatric, neurological, neurodevelopmental, neurodegenerative, mood, motivational, metabolic, cardiovascular, renal, ophthalmic, endocrine, and/or other disorders described herein including schizophrenia (e.g., cognitive and negative symptoms in schizophrenia), cognitive impairment associated with D2 antagonist therapy, ADHD, impulsivity, autism spectrum disorder, mild cognitive impairment (MCI), age-related cognitive decline, Alzheimer’s dementia, Parkinson’s disease (PD), Huntington’s chorea, depression, anxiety, treatment-resistant depression (TRD), bipolar disorder, chronic apathy, anhedonia, chronic fatigue, post-traumatic stress disorder, seasonal affective disorder, social anxiety disorder, post-partum depression, serotonin syndrome, substance abuse and drug dependence, Tourette’s syndrome, tardive dyskinesia, drowsiness, sexual dysfunction, migraine, systemic lupus erythematosus (SLE), hyperglycemia, dislipidemia, obesity, diabetes, sepsis, post-ischemic tubular necrosis, renal failure, resistant edema, narcolepsy, hypertension, congestive heart failure, postoperative ocular hypotonia, sleep disorders, pain, and other disorders in a mammal. See e.g., Goulet M, Madras B K “D(1) dopamine receptor agonists are more effective in alleviating advanced than mild parkinsonism in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-treated monkeys”, Journal of Pharmacology and Experimental Therapy 292(2):714-24 (2000); Surmeier D J et al., “The role of dopamine in modulating the structure and function of striatal circuits”, Prog. Brain Res. 183:149-67 (2010).New or improved agents that modulate (such as agonize or partially agonize) D1 are needed for developing new and more effective pharmaceuticals to treat diseases or conditions associated with dysregulated activation of D1, such as those described herein.
PATENT
Example 6
4-[4-(4,6-Dimethylpyrimidin-5-yl)-3-methylphenoxy]-1H-pyrazolo[4,3-c]pyridine (6)

Step 1. Synthesis of 4-[4-(4,6-dimethylpyrimidin-5-yl)-3-methylphenoxy]-1-(tetrahydro-2H-pyran-2-yl)-1H-pyrazolo[4,3-c]pyridine (C31)
Cesium carbonate (1.03 g, 3.16 mmol) and palladium(II) acetate (24 mg, 0.11 mmol) were added to a solution of C28 (225 mg, 1.05 mmol) and P3 (250 mg, 1.05 mmol) in 1,4-dioxane (10 mL) in a sealable reaction vessel, and the solution was purged with nitrogen for 10 minutes. Di-tert-butyl[3,4,5,6-tetramethyl-2′,4′,6-tri(propan-2-yl)biphenyl-2-yl]phosphane (97%, 104 mg, 0.210 mmol) was added, and the reaction mixture was briefly purged with nitrogen. The vessel was sealed and the reaction mixture was stirred at 100° C. for 3 hours. After cooling to room temperature, the mixture was filtered through Celite and the filter pad was washed with ethyl acetate; the combined filtrates were concentrated in vacuo and purified via silica gel chromatography (Eluents: 20%, then 50%, then 100% ethyl acetate in heptane). The product was obtained as an off-white solid. Yield: 272 mg, 0.655 mmol, 62%. LCMS m/z 416.5 [M+H+]. 1H NMR (400 MHz, CDCl3) δ 8.99 (s, 1H), 8.11 (d, J=0.6 Hz, 1H), 7.99 (d, J=6.0 Hz, 1H), 7.25-7.27 (m, 2H, assumed; partially obscured by solvent peak), 7.20-7.24 (m, 1H), 7.10 (d, J=8.4 Hz, 1H), 5.73 (dd, J=9.4, 2.5 Hz, 1H), 4.04-4.10 (m, 1H), 3.74-3.82 (m, 1H), 2.49-2.59 (m, 1H), 2.28 (s, 6H), 2.08-2.21 (m, 2H), 2.04 (s, 3H), 1.66-1.84 (s, 3H).
Step 2. Synthesis of 4-[4-(4,6-dimethylpyrimidin-5-yl)-3-methylphenoxy]-1H-pyrazolo[4,3-c]pyridine (6)
C31 (172 mg, 0.414 mmol) was dissolved in 1,4-dioxane (5 mL) and dichloromethane (5 mL), and cooled to 0° C. A solution of hydrogen chloride in 1,4-dioxane (4 M, 1.04 mL, 4.16 mmol) was added, and the reaction mixture was allowed to stir at room temperature for 45 hours. After removal of solvent in vacuo, the residue was partitioned between saturated aqueous sodium bicarbonate solution and dichloromethane. The aqueous layer was extracted twice with dichloromethane, and the combined organic layers were dried over sodium sulfate, filtered, and concentrated under reduced pressure, affording the product as an off-white solid. Yield: 130 mg, 0.392 mmol, 95%. LCMS m/z 332.3 [M+H+]. 1H NMR (400 MHz, CDCl3) δ 9.00 (s, 1H), 8.20 (br s, 1H), 7.99 (d, J=6.0 Hz, 1H), 7.28-7.30 (m, 1H), 7.23-7.27 (m, 1H), 7.16 (dd, J=6.0, 1.0 Hz, 1H), 7.11 (d, J=8.2 Hz, 1H), 2.28 (s, 6H), 2.05 (s, 3H).
Preparation P8
6-(4-Hydroxy-2-methylphenyl)-1,5-dimethylpyrazin-2(1H)-one (P8)

Step 1. Synthesis of 1-(4-methoxy-2-methylphenyl)propan-2-one (C8)
Four batches of this experiment were carried out (4×250 g substrate). Tributyl(methoxy)stannane (400 g, 1.24 mol), 1-bromo-4-methoxy-2-methylbenzene (250 g, 1.24 mol), prop-1-en-2-yl acetate (187 g, 1.87 mol), palladium(II) acetate (7.5 g, 33 mmol) and tris(2-methylphenyl)phosphane (10 g, 33 mmol) were stirred together in toluene (2 L) at 100° C. for 18 hours. After cooling to room temperature, the reaction mixture was treated with aqueous potassium fluoride solution (4 M, 400 mL) and stirred for 2 hours at 40° C. The resulting mixture was diluted with toluene (500 mL) and filtered through Celite; the filter pad was thoroughly washed with ethyl acetate (2×1.5 L). The organic phase from the combined filtrates was dried over sodium sulfate, filtered, and concentrated in vacuo. Purification via silica gel chromatography (Gradient: 0% to 5% ethyl acetate in petroleum ether) provided the product as a yellow oil. Combined yield: 602 g, 3.38 mol, 68%. LCMS m/z 179.0 [M+H+]. 1H NMR (400 MHz, CDCl3) δ 7.05 (d, J=8.3 Hz, 1H), 6.70-6.77 (m, 2H), 3.79 (s, 3H), 3.65 (s, 2H), 2.22 (s, 3H), 2.14 (s, 3H).
Step 2. Synthesis of 1-(4-methoxy-2-methylphenyl)propane-1,2-dione (C9)
C8 (6.00 g, 33.7 mmol) and selenium dioxide (7.47 g, 67.3 mmol) were suspended in 1,4-dioxane (50 mL) and heated at 100° C. for 18 hours. The reaction mixture was cooled to room temperature and filtered through Celite; the filtrate was concentrated in vacuo. Silica gel chromatography (Eluent: 10% ethyl acetate in heptane) afforded the product as a bright yellow oil. Yield: 2.55 g, 13.3 mmol, 39%. LCMS m/z 193.1 [M+H+]. 1H NMR (400 MHz, CDCl3) δ 7.66 (d, J=8.6 Hz, 1H), 6.81 (br d, half of AB quartet, J=2.5 Hz, 1H), 6.78 (br dd, half of ABX pattern, J=8.7, 2.6 Hz, 1H), 3.87 (s, 3H), 2.60 (br s, 3H), 2.51 (s, 3H).
Step 3. Synthesis of 6-(4-methoxy-2-methylphenyl)-5-methylpyrazin-2(1H)-one (C10)
C9 (4.0 g, 21 mmol) and glycinamide acetate (2.79 g, 20.8 mmol) were dissolved in methanol (40 mL) and cooled to −10° C. Aqueous sodium hydroxide solution (12 N, 3.5 mL, 42 mmol) was added, and the resulting mixture was slowly warmed to room temperature. After stirring for 3 days, the reaction mixture was concentrated in vacuo. The residue was diluted with water, and 1 N aqueous hydrochloric acid was added until the pH was approximately 7. The aqueous phase was extracted with ethyl acetate, and the combined organic extracts were washed with saturated aqueous sodium chloride solution, dried over magnesium sulfate, filtered, and concentrated under reduced pressure. The resulting residue was slurried with 3:1 ethyl acetate/heptane, stirred for 5 minutes, filtered, and concentrated in vacuo. Silica gel chromatography (Eluent: ethyl acetate) provided the product as a tan solid that contained 15% of an undesired regioisomer; this material was used without further purification. Yield: 2.0 g. LCMS m/z 231.1 [M+H+]. 1H NMR (400 MHz, CDCl3) δ 8.09 (s, 1H), 7.14 (d, J=8.2 Hz, 1H), 6.82-6.87 (m, 2H), 3.86 (s, 3H), 2.20 (s, 3H), 2.11 (s, 3H).
Step 4. Synthesis of 6-(4-methoxy-2-methylphenyl)-1,5-dimethylpyrazin-2(1H)-one (C11)
C10 (from the previous step, 1.9 g) was dissolved in N,N-dimethylformamide (40 mL). Lithium bromide (0.86 g, 9.9 mmol) and sodium bis(trimethylsilyl)amide (95%, 1.91 g, 9.89 mmol) were added, and the resulting solution was stirred for 30 minutes. Methyl iodide (0.635 mL, 10.2 mmol) was added and stirring was continued at room temperature for 18 hours. The reaction mixture was then diluted with water and brought to a pH of approximately 7 by slow portion-wise addition of 1 N aqueous hydrochloric acid. The aqueous layer was extracted with ethyl acetate and the combined organic layers were washed several times with water, dried over magnesium sulfate, filtered, and concentrated. Silica gel chromatography (Gradient: 75% to 100% ethyl acetate in heptane) afforded the product as a viscous orange oil. Yield: 1.67 g, 6.84 mmol, 33% over two steps. LCMS m/z 245.1 [M+H+]. 1H NMR (400 MHz, CDCl3) δ 8.17 (s, 1H), 7.03 (br d, J=8 Hz, 1H), 6.85-6.90 (m, 2H), 3.86 (s, 3H), 3.18 (s, 3H), 2.08 (br s, 3H), 2.00 (s, 3H).
Step 5. Synthesis of P8
To a −78° C. solution of C11 (1.8 g, 7.37 mmol) in dichloromethane (40 mL) was added a solution of boron tribromide in dichloromethane (1 M, 22 mL, 22 mmol). The cooling bath was removed after 30 minutes, and the reaction mixture was allowed to warm to room temperature and stir for 18 hours. The reaction was cooled to −78° C., and methanol (10 mL) was slowly added; the resulting mixture was slowly warmed to room temperature. The reaction mixture was concentrated in vacuo, methanol (20 mL) was added, and the mixture was again concentrated under reduced pressure. The residue was diluted with ethyl acetate (300 mL) and water (200 mL) and the aqueous layer was brought to pH 7 via portion-wise addition of saturated aqueous sodium carbonate solution. The mixture was extracted with ethyl acetate (3×200 mL). The combined organic extracts were washed with water and with saturated aqueous sodium chloride solution, dried over magnesium sulfate, filtered, and concentrated in vacuo to afford the product as a light tan solid. Yield: 1.4 g, 6.0 mmol, 81%. LCMS m/z 231.1 [M+H+]. 1H NMR (400 MHz, CDCl3) δ 8.21 (s, 1H), 6.98 (d, J=8.2 Hz, 1H), 6.87-6.89 (m, 1H), 6.85 (br dd, J=8.2, 2.5 Hz, 1H), 3.22 (s, 3H), 2.06 (br s, 3H), 2.03 (s, 3H).
Step 1. Synthesis of 5-(4-methoxy-2-methylphenyl)-4,6-dimethylpyrimidine (C27)
1,1′-Bis(diphenylphosphino)ferrocene]dichloropalladium(II)-dichloromethane complex (5 g, 6 mmol) was added to a degassed mixture of 2-(4-methoxy-2-methylphenyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (30 g, 120 mmol), 5-bromo-4,6-dimethylpyrimidine (22.5 g, 120 mmol), and potassium phosphate (76.3 g, 359 mmol) in 1,4-dioxane (300 mL) and water (150 mL). The reaction mixture was heated at reflux for 4 hours, whereupon it was filtered and concentrated in vacuo. Purification via silica gel chromatography (Gradient: ethyl acetate in petroleum ether) provided the product as a brown solid. Yield: 25 g, 110 mmol, 92%. LCMS m/z 229.3 [M+H+]. 1H NMR (300 MHz, CDCl3) δ 8.95 (s, 1H), 6.94 (d, J=8.2 Hz, 1H), 6.87-6.89 (m, 1H), 6.84 (dd, J=8.3, 2.5 Hz, 1H), 3.86 (s, 3H), 2.21 (s, 6H), 1.99 (s, 3H).
Step 2. Synthesis of 4-(4,6-dimethylpyrimidin-5-yl)-3-methylphenol (C28)
Boron tribromide (3.8 mL, 40 mmol) was added drop-wise to a solution of C27 (3.0 g, 13 mmol) in dichloromethane (150 mL) at −70° C. The reaction mixture was stirred at room temperature for 16 hours, then adjusted to pH 8 with saturated aqueous sodium bicarbonate solution. The aqueous layer was extracted with dichloromethane (3×200 mL), and the combined organic layers were dried over sodium sulfate, filtered, and concentrated in vacuo. Silica gel chromatography (Gradient: 60% to 90% ethyl acetate in petroleum ether) afforded the product as a yellow solid. Yield: 1.2 g, 5.6 mmol, 43%. LCMS m/z 215.0 [M+H+]. 1H NMR (400 MHz, CDCl3) δ 8.98 (s, 1H), 6.89 (d, J=8.0 Hz, 1H), 6.86 (d, J=2.3 Hz, 1H), 6.80 (dd, J=8.3, 2.5 Hz, 1H), 2.24 (s, 6H), 1.96 (s, 3H).
//////////////PF 2562, non-catechol dopamine 1 receptor agonist, PFIZER, Jennifer Elizabeth Davoren, Amy Beth Dounay, Ivan Viktorovich Efremov, David Lawrence Firman Gray, Scot Richard Mente, Steven Victor O’Neil, Bruce Nelsen Rogers, Chakrapani Subramanyam, Lei Zhang, 1609258-91-4
Now at #MEDI 1st time disclosures David Gray of @pfizer on a non-catechol dopamine 1 receptor agonist #ACSSanFran
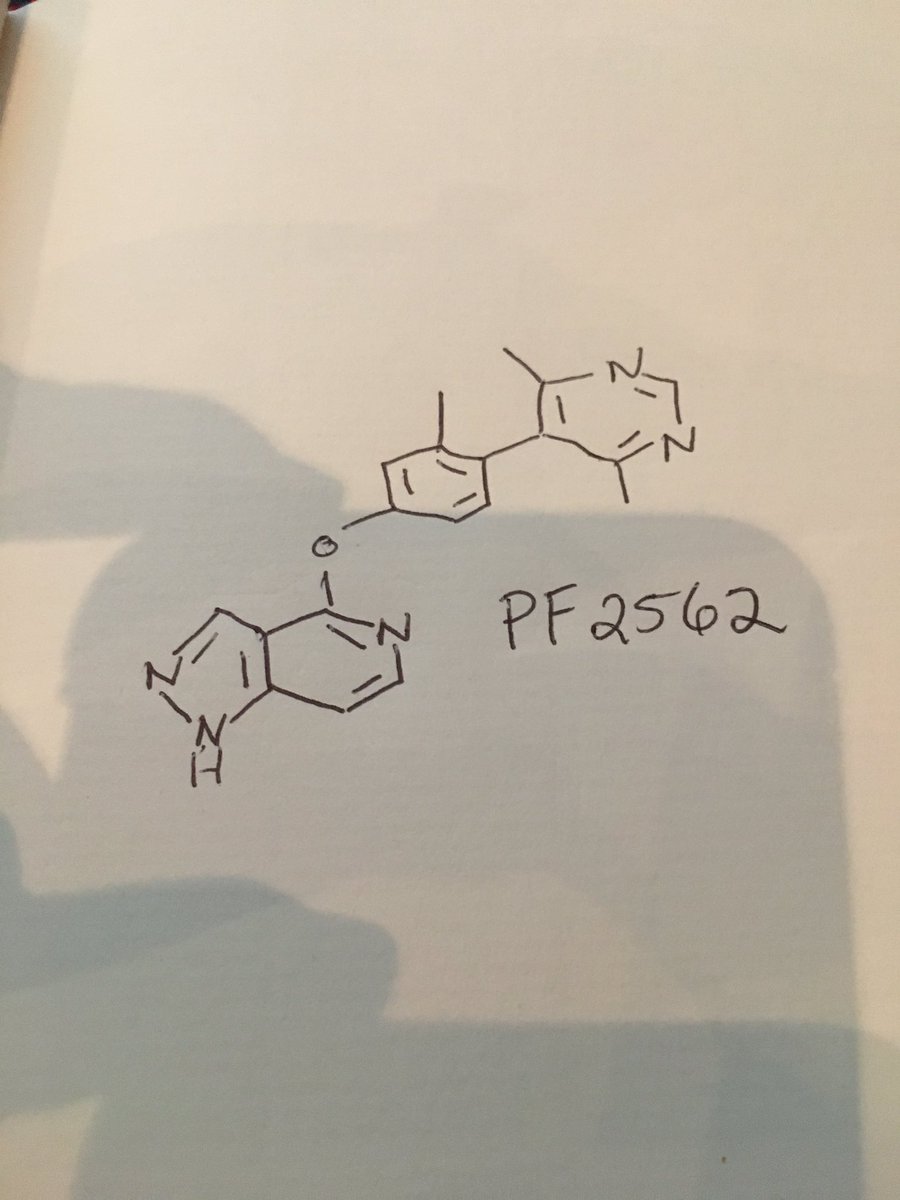
Cc1ncnc(C)c1c2ccc(cc2C)Oc4nccc3nncc34
GSK 3008348
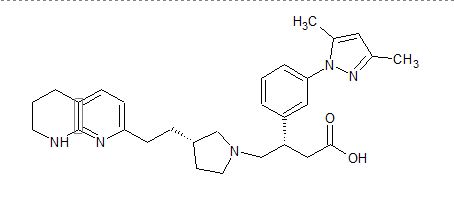
GSK 3008348
(3S)-3-[3-(3,5-Dimethyl-1H-pyrazol-1-yl)phenyl]-4-{(3S)-3-[2-(5,6,7,8-tetrahydro-1,8-naphthyridin-2-yl)ethyl]-1-pyrrolidinyl}butanoic acid
cas 1629249-33-7
1-Pyrrolidinebutanoic acid, β-[3-(3,5-dimethyl-1H-pyrazol-1-yl)phenyl]-3-[2-(5,6,7,8-tetrahydro-1,8-naphthyridin-2-yl)ethyl]-, (βS,3R)-
(S)-3-(3-(3,5-Dimethyl-1H-pyrazol-1-yl)phenyl)-4-((R)-3-(2-(5,6,7,8-tetrahydro-1,8- naphthyridin-2-yl)ethyl)pyrrolidin-1-yl)butanoic acid
- (βS,3R)-β-[3-(3,5-Dimethyl-1H-pyrazol-1-yl)phenyl]-3-[2-(5,6,7,8-tetrahydro-1,8-naphthyridin-2-yl)ethyl]-1-pyrrolidinebutanoic acid
- Molecular Formula C29H37N5O2
- Average mass 487.636 Da
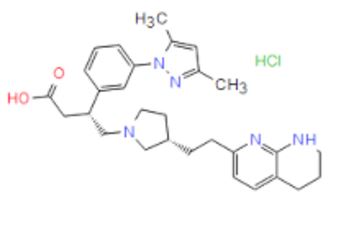
| CAS Number: | 1629249-40-6 |
| Molecular Weight: | 524.1 |
| Molecular Formula: | C29H38ClN5O2 |
- Originator GlaxoSmithKline
- Mechanism of Action Integrin alphaV antagonists
- Phase I Idiopathic pulmonary fibrosis
- 06 Mar 2017 GlaxoSmithKline plans a phase I trial for Idiopathic pulmonary fibrosis (NCT03069989)
- 01 Jun 2016 GlaxoSmithKline completes a first-in-human phase I trial for Idiopathic pulmonary fibrosis in United Kingdom (Inhalation) (NCT02612051)
- 01 Dec 2015 Phase-I clinical trials in Idiopathic pulmonary fibrosis in United Kingdom (Inhalation) (NCT02612051)
Inventors Niall Andrew ANDERSON, Brendan John FALLON, John Martin Pritchard
Applicant Glaxosmithkline Intellectual Property Development Limited

Niall Anderson
![]()
GSK-3008348, an integrin alpha(v)beta6 antagonist, is being developed at GlaxoSmithKline in early clinical studies for the treatment of idiopathic pulmonary fibrosis (IPF).
Idiopathic pulmonary fibrosis (IPF) is a chronic disease characterised by a progressive decline in lung function, due to excessive deposition of extracellular matrix (collagen) within the pulmonary interstitium. It affects approximately 500,000 people in the USA and Europe and is poorly treated. IPF inexorably leads to respiratory failure due to obliteration of functional alveolar units. Patients’ mean life-expectancy is less than 3 years following diagnosis.
IPF therefore represents a major unmet medical need for which novel therapeutic approaches are urgently required.1 Pirfenidone (EsbrietTM from Roche), a non-selective kinase inhibitor, is approved for mild and moderate IPF patients in Japan, Europe, Canada and China and for all IPF patients in USA . Furthermore, nintedanib (OfevTM formerly BIBF-1120 from Boehringer-Ingelheim), a multiple tyrosine-kinase inhibitor targeting vascular endothelial factor receptor, fibroblast growth factor and platelet derived growth factor receptor is approved for all patients with IPF in USA and Europe. Both compounds are administered orally twice or three times per day at high total doses (pirfenidone at 2.4 g/day and nintedanib at 300 mg/day).

Patient compliance is limited by tolerability due to gastro-intenstinal and phototoxicity issues, which require dose titration. (S)-3-(3-(3,5-Dimethyl-1H-pyrazol-1-yl)phenyl)-4-((R)-3-(2-(5,6,7,8-tetrahydro-1,8- naphthyridin-2-yl)ethyl)pyrrolidin-1-yl)butanoic acid hydrochloride is a first in class compound (descovered by GlaxoSmithKline) undergoing currently Phase I clinical trials for the treatment of IPF. It is a non-peptidic αvβ6 integrin inhibitor and in cell adhesion assays has high affinity for the human receptor with a pIC50 of 8.4, and lower affinity for other integrins, such as αvβ3 6.0, αvβ5 5.9 and αvβ8 7.7. Inhibition of integrin αvβ6 is thought to prevent pulmonary fibrosis without exacerbating inflammation.
Integrin superfamily proteins are heterodimeric cell surface receptors, composed of an alpha and beta subunit. 18 alpha and 8 beta subunits have been reported, which have been demonstrated to form 24 distinct alpha/beta heterodimers. Each chain comprises a large extracellular domain (>640 amino acids for the beta subunit, >940 amino acids for the alpha subunit), with a transmembrane spanning region of around 20 amino acids per chain, and generally a short cytoplasmic tail of 30-50 amino acids per chain. Different integrins have been shown to participate in a plethora of cellular biologies, including cell adhesion to the extracellular matrix, cell-cell interactions, and effects on cell migration, proliferation, differentiation and survival (Barczyk et al, Cell and Tissue Research, 2010, 339, 269).
Integrin receptors interact with binding proteins via short protein-protein binding interfaces with ligands and the integrin family can be grouped into sub-families that share similar binding recognition motifs in such ligands. A major subfamily is the RGD-integrins, which recognise ligands that contain an RGD (Arginine-glycine-aspartic acid) motif within their protein sequence. There are 8 integrins in this sub-family, namely ανβι, ανβ3, νβ5ι νβ6ι ανβδ, αι¾β3, α5βι, α8βι, where nomenclature demonstrates that ανβι, ανβ3, νβ5ι νβ6ι & ανβδ share a common V subunit with a divergent β subunit, and ανβι, α5βι & α8βι share a common β!subunit with a divergent a subunit. The βι subunit has been shown to pair with 11 different a subunits, of which only the 3 listed above commonly recognise the RGD peptide motif. (Humphries et al, Journal of Cell Science, 2006, 119, 3901).
Within the 8 RGD-binding integrins are different binding affinities and specificities for different RGD-containing ligands. Ligands include proteins such as fibronectin, vitronectin, osteopontin, and the latency associated peptides (LAPs) of Transforming growth factor βι and β3 (ΤΰΡβι and ΤΰΡβ3). The binding to the LAPs of ΤΰΡβι and ΤΰΡβ3 has been shown in several systems to enable activation of the ΤΰΡβι and ΤΰΡβ3 biological activities, and subsequent ΤΰΡβ- driven biologies (Worthington et al, Trends in Biochemical Sciences, 2011, 36, 47). The specific binding of RGD integrins to such ligands depends on a number of factors, depending on the cell phenotype. The diversity of such ligands, coupled with expression patterns of RGD-binding integrins, generates multiple opportunities for disease intervention. Such diseases include fibrotic diseases (Margadant et al, EMBO reports, 2010, 11, 97), inflammatory disorders, cancer (Desgrosellier et al, Nature Reviews Cancer, 2010, 10, 9), restenosis, and other diseases with an angiogenic component (Weis et al, Cold Spring. Harb. Perspect Med.2011, 1, a006478).
A significant number of av integrin antagonists (Goodman et al, Trends in Pharmacological Sciences, 2012, 33, 405) have been disclosed in the literature including antagonist antibodies, small peptides and compounds. For antibodies these include the pan-av antagonist Intetumumab, the selective ανβ3 antagonist Etaracizumab, and the selective a 6 antagonist STX-100. Cilengitide is a cyclic peptide antagonist that inhibits both ανβ3 and ανβ5, and SB-267268 is an example of a compound (Wilkinson-Berka et al, Invest. Ophthalmol. Vis. Sci, 2006, 47, 1600), which inhibits both ανβ3 and ανβ5. Invention of compounds to act as antagonists of differing combinations of av integrins enables novel agents to be generated and tailored for specific disease indications.
Pulmonary fibrosis represents the end stage of several interstitial lung diseases, including the idiopathic interstitial pneumonias, and is characterised by the excessive deposition of extracellular matrix within the pulmonary interstitium. Among the idiopathic interstitial pneumonias, idiopathic pulmonary fibrosis (IPF) represents the commonest and most fatal condition with a median survival of 3 to 5 years following diagnosis. Fibrosis in IPF is generally progressive, refractory to current pharmacological intervention and inexorably leads to respiratory failure due to obliteration of functional alveolar units. IPF affects approximately 500,000 people in the USA and Europe. This condition therefore represents a major unmet medical need for which novel therapeutic approaches are urgently required (Datta A et al, Novel therapeutic approaches for pulmonary fibrosis, British Journal of ‘Pharmacology’2011163: 141-172).
There are strong in vitro, experimental animal and IPF patient immunohistochemistry data to support a key role for the epithelial-restricted integrin, α 6ι in the activation of TGF-βΙ. Expression of this integrin is low in normal epithelial tissues and is significantly up-regulated in injured and inflamed epithelia including the activated epithelium in IPF. Targeting this integrin therefore reduces the theoretical possibility of interfering with wider TGF-β homeostatic roles. Partial inhibition of the a 6 integrin by antibody blockade has been shown to prevent pulmonary fibrosis without exacerbating inflammation (Horan GS etal Partial inhibition of integrin a 6 prevents pulmonary fibrosis without exacerbating inflammation. Am J Respir Crit Care Med2008177: 56-65)
The ανβ3 integrin is expressed on a number of cell types including vascular endothelium where it has been characterised as a regulator of barrier resistance. Data in animal models of acute lung injury and sepsis have demonstrated a significant role for this integrin in vascular leak since knockout mice show markedly enhanced vessel leak leading to pulmonary oedema or death. Furthermore antibodies capable of inhibiting ανβ3 function caused dramatic increases in monolayer permeability in human pulmonary artery and umbilical vein endothelial cells in response to multiple growth factors. These data suggest a protective role for ανβ3 in the maintenance of vascular endothelial integrity following vessel stimulation and that inhibition of this function could drive pathogenic responses in a chronic disease setting (Su et al Absence of integrin ανβ3 enhances vascular leak in mice by inhibiting endothelial cortical actin formation Am J Respir Crit Care Med 2012 185: 58-66). Thus, selectivity for cl over α 3 may provide a safety advantage.
It is an object of the invention to provide ανβ6 antagonists.
PATENT
| Inventors | Niall Andrew ANDERSON, Brendan John FALLON, John Martin Pritchard |
| Applicant | Glaxosmithkline Intellectual Property Development Limited |
Scheme 1

Reagents and conditions: (a) iodine, imidazole, triphenylphosphine, DCM, 0°C; (b) 2- methyl-[l,8]-naphthyridine, LiN(TMS)2, THF, 0°C; (c) 4M HQ in dioxane.
Scheme 2

Reagents and conditions: (a) isobutylene, cone. H2S04, diethyl ether, 24 h; (b) potassium acetate, acetonitrile, 60 °C, 4 h.


Scheme 3. Reagents and Conditions: (a) LiAIH4, THF; (b) H2, 5% Rh/C, EtOH


Intermediate 42
iate 39


Scheme 6. Reagents and Conditions: (a) EDC, HOBT, NMM, DCM; (b) H2, 5% Rh/C, EtOH; (c) TFA, DCM; (d) BH3.THF; (e) UAIH4, THF, 60°C
Example 1: 3-f3-f3,5-Dimethyl-l pyrazol-l-vnphenvn-4-ff/?)-3-f2-f5,6,7,8- tetrahvdro-l,8-naphthyridin- -vnethvnpyrrolidin-l-vnbutanoic acid

A solution of te/f-butyl 3-(3-(3,5-dimethyl-l pyrazol-l-yl)phenyl)-4-((>?)-3-(2-(5,6,7,8- tetrahydro-l,8-naphthyridin-2-yl)ethyl)pyrrolidin-l-yl)butanoate (Intermediate 14) (100 mg, 0.184 mmol) in 2-methylTHF (0.5 mL) was treated with cone. HCI (12M, 0.077 mL, 0.92 mmol) and stirred at 40 °C for 2 h. The solvent was evaporated in vacuo and the residual oil was dissolved in ethanol (2 mL) and applied to a SCX-2 ion-exchange cartridge (5 g), eluting with ethanol (2 CV) and then 2M ammonia in MeOH (2 CV). The ammoniacal fractions were combined and evaporated in vacuo to give the title compound (79 mg, 88%) as an off-white solid: LCMS (System A) RT= 0.86 min, 100%, ES+ve /77/Z488 (M+H)+; H NMR δ (CDCI3; 600 MHz): 7.42 – 7.37 (m, 1H), 7.31 (d, 7=1.5 Hz, 1H), 7.29 (d, 7=0.9 Hz, 1H), 7.23 (d, 7=7.7 Hz, 1H), 7.21 (d, 7=7.3 Hz, 1H), 6.31 (d, 7=7.3 Hz, 1H), 5.99 (s, 1H), 3.55 (br. s., 1H), 3.60 – 3.52 (m, 1H), 3.45 (t, 7=5.4 Hz, 2H), 3.27 (t, 7=10.6 Hz, 1H), 3.09 (br. S.,1H), 2.93 – 2.86 (m, 1H), 2.82 (d, 7=10.1 Hz, 1H), 2.86 – 2.75 (m, 2H), 2.72 (t, 7=6.2 Hz, 1H), 2.74 – 2.67 (m, 2H), 2.75 (d, 7=9.0 Hz, 1H), 2.61 – 2.50 (m, 1H), 2.31 (s, 3H), 2.29 (s, 3H), 2.33 – 2.26 (m, 1H), 2.24 – 2.11 (m, 1H), 1.94 – 1.86 (m, 2H), 1.94 – 1.84 (m, 1H), 1.78 – 1.66 (m, 1H), 1.65 – 1.51 (m, 1H).
Example 1 was identified by a method described hereinafter as (^-S-iS-iS^-dimethyl-l pyrazol-l-yl)phenyl)-4-((>?)-3-(2-(5,6,7,8-tetrahydro-l,8-naphthyridin-2-yl)ethyl)pyrrolidin-l- yl)butanoic acid.

PAPER
Organic & Biomolecular Chemistry (2016), 14(25), 5992-6009
http://pubs.rsc.org/en/content/articlelanding/2016/ob/c6ob00496b#!divAbstract
Synthesis and determination of absolute configuration of a non-peptidic αvβ6 integrin antagonist for the treatment of idiopathic pulmonary fibrosis
Abstract
A diastereoselective synthesis of (S)-3-(3-(3,5-dimethyl-1H-pyrazol-1-yl)phenyl)-4-((R)-3-(2-(5,6,7,8-tetrahydro-1,8-naphthyridin-2-yl)ethyl)pyrrolidin-1-yl)butanoic acid (1), a potential therapeutic agent for the treatment of Idiopathic Pulmonary Fibrosis, which is currently undergoing Phase I clinical trials is reported. The key steps in the synthesis involved alkylation of 2-methylnaphthyridine with (R)-N-Boc-3-(iodomethyl)-pyrrolidine, and an asymmetric Rh-catalysed addition of an arylboronic acid to a 4-(N-pyrrolidinyl)crotonate ester. The overall yield of the seven linear step synthesis was 8% and the product was obtained in >99.5% ee proceeding with 80% de. The absolute configuration of 1 was established by an alternative asymmetric synthesis involving alkylation of an arylacetic acid using Evans oxazolidinone chemistry, acylation using the resulting 2-arylsuccinic acid, and reduction. The absolute configuration of the benzylic asymmetric centre was established as (S).

naphthyridin-2-yl)ethyl)pyrrolidin-1-yl)butanoic acid (1a) FREE FORM
isopropylamine), flow-rate = 1 mL/min, detecting at 235 nm, RT=12.5 min, 100% (other
diastereoisomer not present RT=9.6 min);
J=5.4 Hz, 2H), 3.27 (t, J=10.6 Hz, 1H), 3.09 (br. s.,1H), 2.93 – 2.86 (m, 1H), 2.82 (d, J=10.1Hz, 1H), 2.86 – 2.75 (m, 2H), 2.72 (t, J=6.2 Hz, 1H), 2.74 – 2.67 (m, 2H), 2.75 (d, J=9.0 Hz,1H), 2.61 – 2.50 (m, 1H), 2.31 (s, 3H), 2.29 (s, 3H), 2.33 – 2.26 (m, 1H), 2.24 – 2.11 (m, 1H),1.94 – 1.86 (m, 2H), 1.94 – 1.84 (m, 1H), 1.78 – 1.66 (m, 1H), 1.65 – 1.51 (m, 1H);
126.2, 123.7, 123.2, 117.4, 109.7, 107.0, 63.3, 56.7 , 54.5, 44.1, 40.9, 40.0, 36.9, 35.5, 32.8,
30.3, 25.8, 19.9, 13.5, 12.5;





REFERENCES
| MacDonald, S.; Pritchard, J.; Anderson, N. Discovery of a small molecule alphavbeta6 inhibitor for idiopathic pulmonary fibrosis 253rd Am Chem Soc (ACS) Natl Meet (April 2-6, San Francisco) 2017, Abst MEDI 362 |
///////////////GSK 3008348, phase 1, idiopathic pulmonary fibrosis, GSK, Niall Andrew ANDERSON, Brendan John FALLON, John Martin Pritchard, Integrin alphaV antagonists
Next talk in #MEDI 1st time disclosures is Simon MacDonald of @GSK on a treatment for idiopathic pulmonary fibrosis
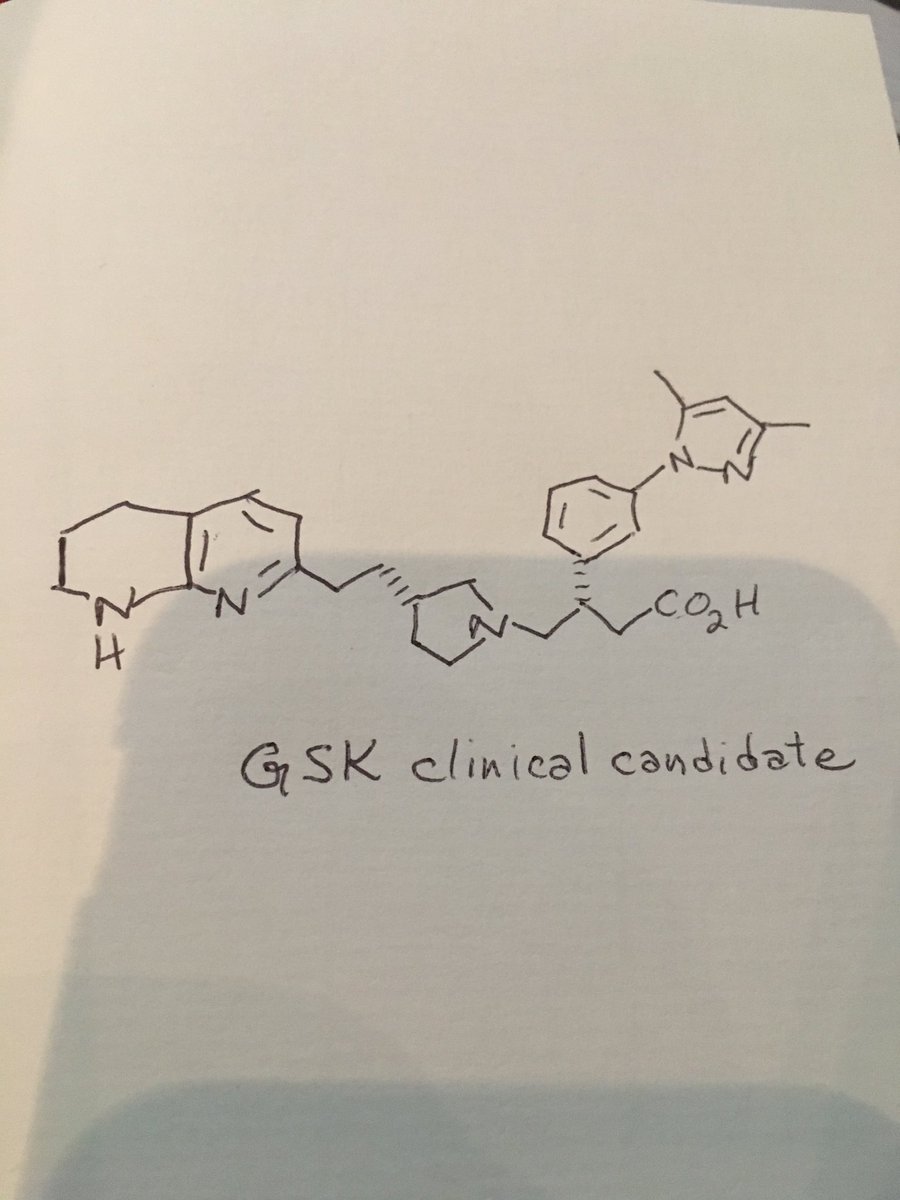
AZD 9567
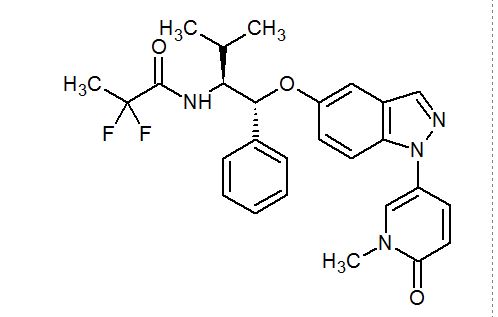
AZD 9567
CAS 1893415-00-3
1893415-64-9 as MONOHYDRATE
2,2-Difluoro-N-[(1R,2S)-3-methyl-1-[[1-(1-methyl-6-oxo-1,6-dihydropyridin-3-yl)-1H-indazol-5-yl]oxy]-1-phenylbutan-2-yl]propanamide
Propanamide, N-[(1S)-1-[(R)-[[1-(1,6-dihydro-1-methyl-6-oxo-3-pyridinyl)-1H-indazol-5-yl]oxy]phenylmethyl]-2-methylpropyl]-2,2-difluoro-
2,2-difluoro-N-[(1R,2S)-3-methyl-1-[1-(1-methyl-6-oxopyridin-3-yl)indazol-5-yl]oxy-1-phenylbutan-2-yl]propanamide
2,2-difluoro- V-[(lR,25)-3-methyl-l-{[l-(l-methyl-6-oxo-l,6-dihydropyridin-3-yl)-lH-indazol-5-yl]oxy}-l-phenylbutan-2-yl]propanamide
MF C27 H28 F2 N4 O3, MF 494.533
AstraZeneca INNOVATOR
AZD-9567, a glucocorticoid receptor modulator, is in early clinical development at AstraZeneca in healthy male volunteers.
Phase I Rheumatoid arthritis
- Originator AstraZeneca
- Class Antirheumatics
- Mechanism of Action Glucocorticoid receptor modulators
-
- 01 Sep 2016 AstraZeneca completes a phase I trial (In volunteers) in Germany (NCT02512575)
- 24 May 2016 Phase-I clinical trials in Rheumatoid arthritis (In volunteers) in United Kingdom (PO) (NCT02760316)
- 24 May 2016 AstraZeneca initiates a phase I trial in Rheumatoid arthritis (In volunteers) in Germany (PO) (NCT02760316)
| Inventors | Lena Elisabeth RIPA, Karolina Lawitz, Matti Juhani Lepistö, Martin Hemmerling, Karl Edman, Antonio Llinas |
| Applicant | Astrazeneca |

Macclesfield AstraZeneca site in Cheshire

Glucocorticoids (GCs) have been used for decades to treat acute and chronic inflammatory and immune conditions, including rheumatoid arthritis, asthma, chronic obstructive pulmonary disease (“COPD”), osteoarthritis, rheumatic fever, allergic rhinitis, systemic lupus erythematosus, Crohn’s disease, inflammatory bowel disease, and ulcerative colitis. Examples of GCs include dexamethasone, prednisone, and
prednisolone. Unfortunately, GCs are often associated with severe and sometimes irreversible side effects, such as osteoporosis, hyperglycemia, effects on glucose metabolism (diabetes mellitus). skin thinning, hypertension, glaucoma, muscle atrophy. Cushing’s syndrome, fluid homeostasis, and psychosis (depression ). These side effects can particularly limit the use of GCs in a chronic setting. Thus, a need continues to exist for alternative therapies that possess the beneficial effects of GCs, but with a reduced likel ihood of side effects.
GCs form a complex with the GC receptor ( GR ) to regulate gene transcription. The GC-GR complex translocates to the cell nucleus, and then binds to GC response elements (GREs) in the promoter regions of various genes. The resulting GC-GR- GRE complex, in turn, activates or inhibits transcription of proximally located genes. The GC-GR complex also (or alternatively) may negatively regulate gene transcription by a process that does not involve DNA binding. In this process, termed transrepression, the GC-GR complex enters the nucleus and directly interacts (via protein-protein interaction) with other transcription factors, repressing their ability to induce gene transcription and thus protein expression.
Some of the side effects of GCs are believed to be the result of cross-reactivity with other steroid receptors (e.g., progesterone, androgen, mineralocorticoid, and estrogen receptors), which have somewhat homologous ligand binding domains; and/or the inability to selectively modulate gene expression and downstream signaling. Consequently, it is believed that an efficacious selective GR modulator (SGRM), which binds to GR with greater affinity relative to other steroid hormone receptors, would provide an alternative therapy to address the unmet need for a therapy that possesses the beneficial, effects of GCs, while, at the same time, having fewer side effects.
A range of compounds have been reported to have SGRM activity. See, e.g., WO2007/0467747, WO2007/114763, WO2008/006627, WO2008/055709, WO2008/055710, WO2008/052808, WO2008/063116, WO2008/076048,
WO2008/079073, WO2008/098798, WO2009/065503, WO2009/142569,
WO2009/142571, WO2010/009814, WO2013/001294, and EP2072509. Still, there continues to be a need for new SGRMs that exhibit, for example, an improved potency, efficacy, effectiveness in steroid-insensitive patients, selectivity, solubility allowing for oral administration, pharmacokinetic profile allowing for a desirable dosing regimen, stability on the shelf {e.g., hydro lytic, thermal, chemical, or photochemical stability), crystallinity, tolerability for a range of patients, side effect profile and/or safety profile.
PATENT
Scheme 1 below illustrates a general protocol for making compounds described in this specification, using either an Ullman route or an aziridine route.
Scheme 1

In Scheme 1, Ar is 
[182] The amino alcohol reagent used in Scheme 1 may be made using the below Scheme 2.
Scheme 2

The Grignard reagent (ArMgBr) used in Scheme 2 can be obtained commercially, or, if not, can generally be prepared from the corresponding aryl bromide and Mg and/or iPrMgCl using published methods.
[183] The iodo and hydroxy pyridone indazole reagents used in Scheme 1 may be made using the below Scheme 3A or 3B, respectively.
Scheme 3A



[184] Scheme 4 below provides an alternative protocol for making compounds described in this specification.
Scheme 4

Example 1. Preparation of 2,2-difluoro- V-[(lR,2S)-3-methyl-l-{[l-(l-methyl-6-oxo-l,6-dihydropyridin-3-yl)-lH-indazol-5-yl]oxy}-l-phenylbutan-2-yl]propanamide.

[199] Step A. Preparation of 5-[5-[(te^butyldimethylsilyl)oxy]-lH-indazol-l-yl]-l-methyl-l,2-dihydropyridin-2-one.

Into a 2 L 4-necked, round-bottom flask, purged and maintained with an inert atmosphere of N2, was placed a solution of 5-[(tert-butyldimethylsilyl)oxy]-lH-indazole (805 g, 3.2 mol) in toluene (8 L), 5 -iodo-1 -methyl- 1 ,2-dihydropyridin-2-one (800 g, 3.4 mol) and
K3PO4 (1.2 kg, 5.8 mol). Cyclohexane-l,2-diamine (63 g, 0.5 mol) was added followed by the addition of Cul (1.3 g, 6.8 mmol) in several batches. The resulting solution was stirred overnight at 102°C. The resulting mixture was concentrated under vacuum to yield 3.0 kg of the title compound as a crude black solid. LC/MS: m/z 356 [M+H]+.
[200] Step B. Preparation of 5-(5-hydroxy-lH-indazol-l-yl)-l-methylpyridin-2(lH)-one.

Into a 2 L 4-necked, round-bottom flask was placed 5-[5-[(fert-butyldimethylsilyl)oxy]-lH-indazol-l-yl]-l-methyl-l,2-dihydropyridin-2-one (3.0 kg, crude) and a solution of HCl (2 L, 24 mol, 36%) in water (2 L) and MeOH (5 L). The resulting solution was stirred for 1 hr at 40°C and then evaporated to dryness. The resulting solid was washed with water (4 x 5 L) and ethyl acetate (2 x 0.5 L) to afford 480 g (61%, two steps) of the title product as a brown solid. LC/MS: m/z 242 [M+H]+. 1HNMR (300 MHz, DMSO-d6): δ 3.52 (3H, s),6.61 (lH,m),7.06 (2H,m),7.54 (lH,m), 7.77 (lH,m), 8.19 (2H, m) 9.35 (lH,s).
[201] Ste C. Preparation of tert-butyl((lR,25)-l-hydroxy-3-methyl-l-phenylbutan-2-yl)carbamate.

(S)-tert-butyl 3 -methyl- l-oxo-l-phenylbutan-2-ylcarbamate (1.0 kg, 3.5 mol) was dissolved in toluene (4 L). Afterward, 2-propanol (2 L) was added, followed by triisopropoxyaluminum (0.145 L, 0.73 mol). The reaction mixture was heated at 54-58°C for 1 hr under reduced pressure (300-350 mbar) to start azeothropic distillation. After the collection of 0.75 L condensate, 2-propanol (2 L) was added, and the reaction mixture was stirred overnight at reduced pressure to afford 4 L condensate in total. Toluene (3 L) was added at 20°C, followed by 2M HC1 (2 L) over 15 min to keep the temperature below 28°C. The layers were separated (pH of aqueous phase 0-1) and the organic layer was washed successively with water (3 L), 4% NaHCCte (2 L) and water (250 mL). The volume of the organic layer was reduced from 6 L at 50°C and 70 mbar to 2.5 L. The resulting mixture was heated to 50°C and heptane (6.5 L) was added at 47-53°C to maintain the material in solution. The temperature of the mixture was slowly decreased to 20°C, seeded with the crystals of the title compound at 37°C (seed crystals were prepared in an earlier batch made by the same method and then evaporating the reaction mixture to dryness, slurring the residue in heptane, and isolating the crystals by filtration), and allowed to stand overnight. The product was filtered off, washed with heptane (2 x 1 L) and dried under vacuum to afford 806 g (81%) of the title compound as a white solid. 1HNMR (500 MHz, DMSO-d6): δ 0.81 (dd, 6H), 1.16 (s, 8H), 2.19 (m, 1H), 3.51 (m, 1H), 4.32 (d, 1H), 5.26 (s, 1H), 6.30 (d, 1H), 7.13 – 7.2 (m, 1H), 7.24 (t, 2H), 7.3 – 7.36 (m, 3H).
[202] Step D. Preparation of (lR,2S)-2-amino-3-methyl-l-phenylbutan-l-ol hydrochloride salt.

To a solution of HC1 in propan-2-ol (5-6 N, 3.1 L, 16 mol) at 20°C was added tert-butyl((li?,25)-l-hydroxy-3-methyl-l-phenylbutan-2-yl)carbamate (605 g, 2.2 mol) in portions over 70 min followed by the addition of MTBE (2 L) over 30 min. The reaction mixture was cooled to 5°C and stirred for 18 hr. The product was isolated by filtration and dried to afford 286 g of the title compound as an HC1 salt (61% yield). The mother liquor was concentrated to 300 mL. MTBE (300 mL) was then added, and the resulting precipitation was isolated by filtration to afford additional 84 g of the title compound as a HC1 salt (18% yield). Total 370 g (79%). 1HNMR (400 MHz, DMSO-d6): δ 0.91 (dd, 6H), 1.61 – 1.81 (m, 1H), 3.11 (s, 1H), 4.99 (s, 1H), 6.08 (d, 1H), 7.30 (t, 1H), 7.40 (dt, 4H), 7.97 (s, 2H).
[203] Step E. Preparation of (2S,35)-2-isopropyl-l-(4-nitrophenylsulfonyl)-3-phenylaziridine.

(li?,25)-2-Amino-3-methyl-l-phenylbutan-l-ol hydrochloride (430 g, 2.0 mol) was mixed with DCM (5 L) at 20°C. 4-Nitrobenzenesulfonyl chloride (460 g, 2.0 mol) was then added over 5 min. Afterward, the mixture was cooled to -27°C. Triethylamine (1.0 kg, 10 mol) was slowly added while maintaining the temperature at -18°C. The reaction mixture was cooled to -30°C, and methanesulfonyl chloride (460 g, 4.0 mol) was added slowly while maintaining the temperature at -25 °C. The reaction mixture was then stirred at 0°C for 16 hr before adding triethylamine (40 mL, 0.3 mol; 20 mL ,0.14 mol and 10 mL, 0.074 mol) w at 0°C in portions over 4 hr. Water (5 L) was subsequently added at 20°C, and the resulting layers were separated. The organic layer was washed with water (5 L) and the volume reduced to 1 L under vacuum. MTBE (1.5 L) was added, and the mixture was stirred on a rotavap at 20°C over night and filtered to afford 500 g (70%) of the title product as a solid. 1HNMR (400 MHz, CDCls): δ 1.12 (d, 3H), 1.25 (d, 3H), 2.23 (ddt, 1H), 2.89 (dd, 1H), 3.84 (d, 1H), 7.08 – 7.2 (m, 1H), 7.22 – 7.35 (m, 4H), 8.01 – 8.13 (m, 2H), 8.22 – 8.35 (m, 2H)
[204] Step F. Preparation of V-((lR,2S)-3-methyl-l-(l-(l-methyl-6-oxo-l,6-dihydropyridin-3-yl)-lH-indazol-5-yloxy)-l-phenylbutan-2-yl)-4-nitrobenzenesulfonamide.

[205] (25′,35)-2-Isopropyl-l-(4-nitrophenylsulfonyl)-3-phenylaziridine (490 g, 1.3 mol) was mixed with 5-(5-hydroxy-lH-indazol-l-yl)-l-methylpyridin-2(lH)-one (360 g, 1.4 mol) in acetonitrile (5 L) at 20°C. Cesium carbonate (850 g, 2.6 mol) was added in portions over 5 min. The reaction mixture was then stirred at 50°C overnight. Water (5 L) was added at 20°C, and the resulting mixture was extracted with 2-methyltetrahydrofuran (5L and 2.5 L). The combined organic layer was washed successively with 0.5 M HC1 (5 L), water (3 x 5L) and brine (5L). The remaining organic layer was concentrated to a thick oil, and then MTBE (2 L) was added. The resulting precipitate was filtered to afford 780 g (purity 71% w/w) of the crude title product as a yellow solid, which was used in the next step without further purification. 1HNMR (400 MHz, DMSO-d6): δ 0.93 (dd, 6H), 2.01 -2.19 (m, 1H), 3.50 (s, 3H), 3.74 (s, 1H), 5.00 (d, 1H), 6.54 (d, 1H), 6.78 (d, 1H), 6.95 -7.15 (m, 4H), 7.23 (d, 2H), 7.49 (d, 1H), 7.69 (dd, 1H), 7.74 (d, 2H), 8.00 (s, 1H), 8.08 (d, 2H), 8.13 (d, 2H).
[206] Step G. Preparation of 2,2-difluoro- V-[(lR,25)-3-methyl-l-{[l-(l-methyl-6-oxo-l,6-dihydropyridin-3-yl)-lH-indazol-5-yl]oxy}-l-phenylbutan-2-yl]propanamide.

[207] N-((lR,2S)-3-Methyl- 1 -(1 -(1 -methyl-6-oxo- 1 ,6-dihydropyridin-3-yl)- \H-indazol-5-yloxy)-l-phenylbutan-2-yl)-4-nitrobenzenesulfonamide (780 g, 71%w/w) was mixed with DMF (4 L). DBU (860 g, 5.6 mol) was then added at 20°C over 10 min. 2-Mercaptoacetic acid (170 g, 1.9 mol) was added slowly over 30 min, keeping the temperature at 20°C. After 1 hr, ethyl 2,2-difluoropropanoate (635 g, 4.60 mol) was added over 10 min at 20°C. The reaction mixture was stirred for 18 hr. Subsequently, additional ethyl 2,2-difluoropropanoate (254 g, 1.8 mol) was added, and the reaction mixture was stirred for an additional 4 hr at 20°C. Water (5 L) was then slowly added over 40 min, maintaining the temperature at 20°C. The water layer was extracted with isopropyl acetate (4 L and 2 x 2 L). The combined organic layer was washed with 0.5M HC1 (4 L) and brine (2 L). The organic layer was then combined with the organic layer from a parallel reaction starting from 96 g of N-((li?,25)-3-methyl-l-((l-(l-methyl-6-oxo-l,6-dihydropyridin-3-yl)- lH-indazol-5-yl)oxy)- 1 -phenylbutan-2-yl)-4-nitrobenzenesulfonamide, and concentrated to approximate 1.5 L. The resulting brown solution was filtered. The filter was washed twice with isopropyl acetate (2 x 0.5 L). The filtrate was evaporated until a solid formed. The solid was then co evaporated with 99.5% ethanol (1 L), affording 493 g (77%, two steps) of an amorphous solid.
[208] The solid (464 g, 0.94 mol) was dissolved in ethanol/water 2: 1 (3.7 L) at 50°C. The reaction mixture was then seeded with crystals () of the title compound (0.5 g) at 47°C, and a slight opaque mixture was formed. The mixture was held at that temperature for 1 hr. Afterward, the temperature was decreased to 20°C over 7 hr, and kept at 20°C for 40 hr. The solid was filtrated off, washed with cold (5°C) ethanol/water 1 :2 (0.8 L), and dried in vacuum at 37°C overnight to afford 356 g (0.70 mol, 74%, 99.9 % ee) of the title compound as a monohydrate. LC/MS: m/z 495 [M+H]+. ‘HNMR (600 MHz, DMSO-d6) δ 0.91 (dd, 6H), 1.38 (t, 3H), 2.42 (m, 1H), 3.50 (s, 3H), 4.21 (m, 1H), 5.29 (d, 1H), 6.53 (d, 1H), 7.09 (d, 1H), 7.13 (dd, 1H), 7.22 (t, 1H), 7.29 (t, 2H), 7.47 (d, 2H), 7.56 (d, 1H), 7.70 (dd, 1H), 8.13 (d, 1H), 8.16 (d, 1H), 8.27 (d, 1H).
[209] The seed crystals may be prepared from amorphous compound prepared according to Example 2 using 2,2-difluoropropanoic acid, followed by purification on HPLC. The compound (401 mg) was weighed into a glass vial. Ethanol (0.4 mL) was added, and the vial was shaken and heated to 40°C to afford a clear, slightly yellow solution. Ethanol/Water (0.4 mL, 50/50% vol/vol) was added. Crystallization started to
occur within 5 min, and, after 10 min, a white thick suspension formed. The crystals were collected by filtration
/////////////AZD 9567, AstraZeneca, lucocorticoid receptor modulator, Rheumatoid arthritis, phase 1, Lena Elisabeth RIPA, Karolina Lawitz, Matti Juhani Lepistö, Martin Hemmerling, Karl Edman, Antonio Llinas
3rd speaker this afternoon in 1st time disclosures is Lena Ripa of @AstraZeneca on a glucocorticoid receptor modulator #ACSSanFran
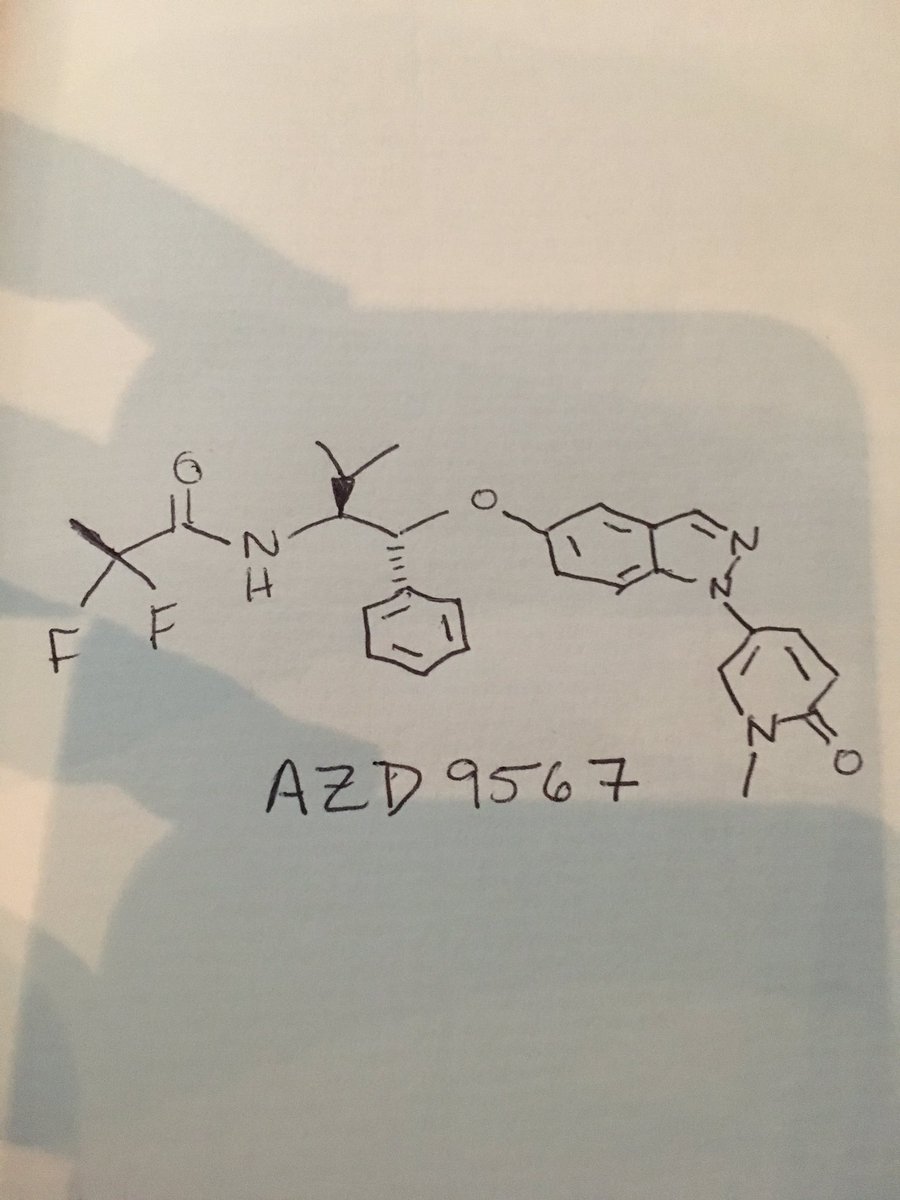
CC(F)(F)C(=O)N[C@@H](C(C)C)[C@H](Oc1cc2cnn(c2cc1)C=3C=CC(=O)N(C)C=3)c4ccccc4
