


















| Publication Number | Publication Date | IPCR Assignee/Applicant | Structure hits | Tools | |
|---|---|---|---|---|---|
|
1.
WO-2015178020-A1 |
2015-11-26 |
EN
|
|
||
|
2.
WO-2015174098-A1 |
2015-11-19 |
EN
|
|
||
|
3.
US-9187463-B2 |
2015-11-17 |
|
|||
|
4.
US-20150322055-A1 |
2015-11-12 |
|
|||
|
5.
EP-2922849-A1 |
2015-09-30 |
EN
|
|
||
|
6.
EP-2710002-A4 |
2014-10-01 |
EN
|
|
||
|
7.
US-8816090-B2 |
2014-08-26 |
|
|||
|
8.
EP-1856114-B1 |
2014-08-20 |
EN
|
|
||
|
9.
US-20140187583-A1 |
2014-07-03 |
|
|||
|
10.
WO-2014080633-A1 |
2014-05-30 |
EN
|
|
||
|
11.
EP-2710002-A1 |
2014-03-26 |
EN
|
|
||
|
12.
US-20140051726-A1 |
2014-02-20 |
|
|||
|
13.
EP-2688648-A1 |
2014-01-29 |
EN
|
|
||
|
14.
WO-2012157288-A1 |
2012-11-22 |
EN
|
|
||
|
15.
WO-2012127878-A1 |
2012-09-27 |
EN
|
|
||
|
16.
US-20080207690-A1 |
2008-08-28 |
|
|||
|
17.
EP-1856114-A1 |
2007-11-21 |
EN
|
|
||
|
18.
WO-2006090224-A1 |
2006-08-31 |
EN
|
|
Home » Posts tagged 'PFIZER' (Page 3)
Tag Archives: PFIZER
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on
ORGANIC SPECTROSCOPY INTERNATIONAL

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()




Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
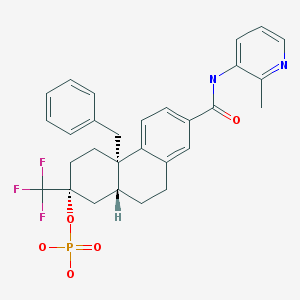
PF-06747775 (Pfizer) Third generation covalent EGFR inhibitors

 .
.

PF-06747775 (Pfizer)
PF06747775; PF06747775; PF 06747775; PF6747775; PF 6747775; PF6747775. PFE-X775
N-((3R,4R)-4-fluoro-1-(6-((3-methoxy-1-methyl-1H-pyrazol-4-yl)amino)-9-methyl-9H-purin-2-yl)pyrrolidin-3-yl)acrylamide
N-((3R,4R)-4-fluoro-1-(6-((3-methoxy-1-methyl-1H-pyrazol-4-yl)amino)-9-methyl-9H-purin-2-yl)pyrrolidin-3-yl)acrylamide
CAS 1776112-90-3
Chemical Formula: C18H22FN9O2
Exact Mass: 415.188
Recruiting, Phase I/II (NTC02349633)
Epidermal growth factor receptor antagonists
Antineoplastics
Non-small cell lung cancer
Dose escalation study to evaluate safety, PK, PD and efficacy in advanced EGFRm+ NSCLC
- 02 May 2015Phase-I clinical trials in Non-small cell lung cancer (Metastatic disease, Second-line therapy or greater) in USA (PO) (NCT02349633)
- 05 Feb 2015Pfizer plans a phase I trial for Non-small cell lung cancer (Second-line therapy or greater) in USA (NCT02349633)
- 05 Jan 2015Preclinical trials in Non-small cell lung cancer in USA (PO)
PF-06747775 is an orally available inhibitor of the epidermal growth factor receptor (EGFR) mutant form T790M, with potential antineoplastic activity. EGFR T790M inhibitor PF-06747775 specifically binds to and inhibits EGFR T790M, a secondarily acquired resistance mutation, which prevents EGFR-mediated signaling and leads to cell death in EGFR T790M-expressing tumor cells. Compared to some other EGFR inhibitors, PF-06747775 may have therapeutic benefits in tumors with T790M-mediated drug resistance.
for the oral treatment of patients with locally advanced or metastatic EGFR mutant (del19 or L858R) non-small cell lung cancer

Kinetic mechanism for two-step covalent inhibition of EGFR
PATENT
Example 7
(Scheme F): Preparation of N-((3R,4R)-4-fluoro-1-(6-((3-methoxy-1-methyl-1H-pyrazol-4-yl)amino)-9-methyl-9H-purin-2-yl)pyrrolidin-3-yl)acrylamide
Step 1: Preparation of 2-fluoro-N-(3-methoxy-1-methyl-1H-pyrazol-4-yl)-9H-purin -6-amine
Step 2: Preparation of 2-fluoro-N-(3-methoxy-1-methyl-1H-pyrazol-4-yl)-9-methyl -9H-purin-6-amine
Step 3: Preparation of N-((3R,4R)-4-fluoro-1-(6-((3-methoxy-1-methyl-1H-pyrazol -4-yl)amino)-9-methyl-9H-purin-2-yl)pyrrolidin-3-yl)acrylamide
Example 7A
(Scheme F): Preparation of N-((3R,4R)-4-fluoro-1-(6-((3-methoxy-1-methyl-1H-pyrazol-4-yl)amino)-9-methyl-9H-purin-2-yl)pyrrolidin-3-yl)acrylamide
Preparation Step 1A: Preparation of (3R,4R)-1-benzyl-3,4-dihydroxypyrrolidine-2,5-dione
Preparation Step 2A: Preparation of (3S,4S)-1-benzylpyrrolidine-3,4-diol
Preparation Step 3A: Preparation of (3aR,6aS)-5-benzyl-2,2-dioxo-tetrahydro-1-oxa-2λ6-thia-3-5-diaza-pentalene-3-carboxylic acid t-butyl ester
Preparation Step 4A: Preparation of (3R,4R)-1-benzyl-4-fluoropyrrolidin-3-amine bis-tosylate
Preparation Step 5A: N-((3R,4R)-1-benzyl-4-fluoropyrrolidin-3-yl)-3-(methylsulfonyl)propanamide
Preparation Step 6A: N-((3R,4R)-4-fluoropyrrolidin-3-yl)-3-(methylsulfonyl)propanamide
Step 1: Preparation of 2-fluoro-N-(3-methoxy-1-methyl-1H-pyrazol-4-yl)-9H-purin-6-amine
Step 2: Preparation of 2-fluoro-N-(3-methoxy-1-methyl-1H-pyrazol-4-yl)-9-methyl-9H-purin-6-amine
Step 3: Preparation of N-((3R,4R)-4-fluoro-1-(6-((3-methoxy-1-methyl-1H-pyrazol-4-yl)amino)-9-methyl-9H-purin-2-yl)pyrrolidin-3-yl)acrylamide

Summary of 1st generation and 2nd generation EGFR inhibitors

REFERENCES
Planken, S.; Murray, B. W.; Lafontaine, J.; Weinrich, S.; Hemkens, M.; Kath, J. C.; Nair, S. K.; Johnson, T. O.; Cheng, H.; Sutton, S. C.; Zientek, M.; Yin, M. -J.; Solowiej, J.; Nagata, A.; Gajiwala, K. Abstracts of Papers, 249th ACS National Meeting & Exposition, Denver, CO, United States, March 22–26, 2015; MEDI-248
//////Third generation, covalent EGFR inhibitors, PF-06747775, Pfizer, PFE-X775
Compound name AND SMILES string
Rociletinib COC(C=C(N1CCN(C(C)=O)CC1)C=C2)=C2NC3=NC=C(C(F)(F)F)C(NC4=CC=CC(NC(C=C)=O)=C4)=N3
Osimertinib CN(CCN(C)C)C(C(NC(C=C)=O)=C1)=CC(OC)=C1NC2=NC=CC(C3=CN(C)C4=C3C=CC=C4)=N2
EGF816 ClC1=C2C(N=C(NC(C3=CC(C)=NC=C3)=O)N2[C@H]4CN(C(/C=C/CN(C)C)=O)CCCC4)=CC=C1
PF-06747775 CN1C2=NC(N3C[C@@H](NC(C=C)=O)[C@H](F)C3)=NC(NC4=CN(C)N=C4OC)=C2N=C1
PF-06459988 CN(N=C1)C=C1NC2=NC3=C(C(Cl)=CN3)C(OC[C@H]4CN(C(C=C)=O)C[C@@H]4OC)=N2
WZ4002 ClC1=CN=C(NC2=C(OC)C=C(N3CCN(C)CC3)C=C2)N=C1OC4=CC=CC(NC(C=C)=O)=C4
Fosfluconazole
Fosfluconazole
Fosfluconazole; 194798-83-9; UNII-3JIJ299EWH; 3JIJ299EWH; NCGC00182029-01;
2-(2,4-difluorophenyl)-1,3-di(1h-1,2,4-triazol-1-yl)propan-2-yl dihydrogen phosphate;
2,4-difluoro-α,α-bis(1H-1,2,4-triazol-1-ylmethyl) benzyl alcohol, dihydrogen phosphate
| Molecular Formula: | C13H13F2N6O4P |
|---|---|
| Molecular Weight: | 386.250688 g/mol |
Research Code:UK-292663, UK 292663, F-FLCZ, F FLCZ
Trade Name:Prodif® PFIZER
MOA:Azole antifungal
Indication:Cryptococcus neoformans; Candidiasis
Status:Approved, Japan PMDA OCT 16 2003
Company:Pfizer (Originator)
Candidiasis,Cryptococcus neoformans, Injection, Solution, Eq. 100 mg/200 mg/400 mg fluconazole per vial
Fosfluconazole (INN) is a water-soluble phosphate prodrug of fluconazole – a triazole antifungal drug used in the treatment and prevention of superficial and systemic fungal infections. The phosphate ester bond is hydrolysed by the action of a phosphatase – an enzyme that removes a phosphate group from its substrate by hydrolysing phosphoric acid monoesters into a phosphate ion and a molecule with a free hydroxyl group (see dephosphorylation).
Fosfluconazole was approved by Pharmaceuticals and Medicals Devices Agency of Japan (PMDA) on Oct 16, 2003. It was developed and marketed as Prodif® by Pfizer in Japan.
Fosfluconazole is a water-soluble phosphate prodrug of fluconazole – a triazole antifungal drug. It is indicated for the treatment of candida and cryptococcus infections.
Prodif® is available as solution for intravenous use, containing 100, 200 or 400 mg of free Fosfluconazole per vial. The recommended dose is 50 to 100 mg administered intravenously once daily for candidiasis. Another dose is 50 to 200 mg fluconazole once daily for cryptococcosis.


Route 1


Reference:1. WO9728169A1 / US6977302B2.
2. Org. Process Res. Dev.2002, 6, 109-112.
http://pubs.acs.org/doi/pdf/10.1021/op010064%2B
2-(2,4-Difluorophenyl)-1,3-bis(1H-1,2,4-triazole-1-yl)- 2-propyl dihydrogen phosphate (2). A slurry of dibenzyl 2-(2,4-difluorophenyl)-1,3-bis(1H-1,2,4-triazole-1-yl)-2-propyl phosphate (30.1 kg, 53.13 mol), 5% palladium-on-carbon catalyst (50% wet, type 5R39, 1.5 kg), and sodium hydroxide (4.36 kg, 108.9 mol) in low-endotoxin water (75.7 L) was hydrogenated at ambient temperature and 414 kPa (60 psi) for 12 h. The slurry was filtered, and the catalyst was washed with low-endotoxin water (9.8 L). After separating the toluene by-product, the aqueous phase was slurried with carbon (3.1 kg) for 30 min. After the carbon was removed by filtration, the aqueous phase was acidified to pH 1.45 by that addition of sulfuric acid (6.69 kg) in low-endotoxin water (25 L) over 2 h. The resulting slurry was granulated at ambient temperature for 1 h and then filtered. The product was sequentially washed with filtered low-endotoxin water (103 L) and filtered acetone (103 L). The product was dried under vacuum at 50 °C for 12 h to give the title compound (18.1 kg, 88%) a white powder: mp 223-224 °C.
1H NMR (DMSO) δ 5.07 (2H, d), 5.24 (2H, d), 6.77-6.83 (1H, m), 7.00-7.18 (2H, m), 7.75 (2H, s), 8.53 (2H, s).
Found: C, 40.28; H, 3.39; N, 21.63;
[MH]+ 387.0786. C13H13F2N6O4P requires: C, 40.43; H, 3.39; N, 21.78; [MH]+ 387.0782.
US6977302
https://www.google.com/patents/US6977302
EXAMPLE 1 1-(2,4-Difluorophenyl)-1,3-bis(1H-1,2,4-triazol-1-yl)-2-propyl dihydrogen phosphate
(a) Dibenzyl 2-(2,4-difluorophenyl)-1,3-bis(1H-1,2,4-triazol-1-yl)-2-propyl phosphate
Method A

A solution of 2-(2,4-difluorophenyl)-1,3-bis(1H-1,2,4-triazol-1-yl)propan-2-ol (also known as fluconazole, 10.0 g, 32.6 mmol), 1H-tetrazole (6.85 g, 97.8 mmol), dibenzyl diisopropyl phosphoramidite (22.55 g, 65.2 mmol) in methylene chloride (100 ml) was stirred at room temperature under a nitrogen atmosphere for 2 hours. The mixture was then cooled to 0° C., and a solution of 3-chloroperoxybenzoic acid (13.5 g, 50-55% w/w, 39.1 mmol) in methylene chloride (50 ml) was added maintaining the temperature at 0° C. The resulting mixture was allowed to warm to room temperature for 1 hour before washing with aqueous sodium metabisulphite and sodium bicarbonate. After drying (MgSO4) the solvent was removed and replaced with methyl isobutyl ketone (37 ml) and tert-butyl methyl ether (74 ml). After granulating at −10° C. for 1 hour the product was filtered and washed with ice cold methyl isobutyl ketone and tert-butyl methyl ether (1:3, 15 ml) and dried at 50° C. under vacuum for 18 hours to give the subtitle compound (16.05 g, 87%), m.p. 93° C.
Found: C, 57.12; H, 4.46; N, 14.85. C27H25F2N6O4P requires C, 57.24; H, 4.46; N, 14.84%. m/z 567 (MH+) 1H NMR (300 MHz, CDCl3) δ=4.90 (d, 2H), 4.95 (d, 2H), 5.05 (d, 2H), 5.19 (d, 2H), 6.58-6.73 (m, 2H), 6.88-6.95 (m, 1H), 7.20-7.30 (m, 4H) 7.32-7.38 (m; 6H), 7.80 (s, 2H), 8.36 (s, 2H).
Method B

To stirred ethyl acetate (1530 ml) was added 2-(2,4-difluorophenyl)-1,3-bis(1H-1,2,4-triazol-1-yl)propan-2-ol (also known as fluconazole, 306 g, 1.00 mol) and pyridine (237.3 g, 3.00 mol) before cooling to 0° C. Phosphorus trichloride (137.4 g, 1.00 mol) was added dropwise to the reaction mixture maintaining the temperature between 0-5° C. before allowing the reaction mixture to warm to 15° C. over 30 minutes. Benzyl alcohol (216 g, 2.00 mol) was then added over 30 minutes at 15-20° C. After a further 30 minutes hydrogen peroxide (27.5% w/w in water, 373 g) was added maintaining the temperature at 15-20° C. After 30 minutes the aqueous phase was removed and the organic phase washed with aqueous sodium metabisulphite, dilute hydrochloric acid and water. The solvent was removed at reduced pressure and replaced with methyl isobutyl ketone (850 ml) and tert-butyl methyl ether (1132 ml). After granulating at 20° C. for 1 hour and at 0° C. for 1 hour, the product was filtered and washed with ice cold tert-butyl methyl ether (2×220 ml) and dried at 50° C. under vacuum for 18 hours to give the subtitle compound (358 g, 63%). The melting point and spectroscopic data was identical to that stated in method A.
(b) 2-(2,4-Difluorophenyl)-1,3-bis(1H-1,2,4-triazol-1-yl)-2-propyl dihydrogen phosphate

A slurry of the compound of step (a) (9.80 g, 17.3 mmol), 5% palladium on carbon catalyst (50% wet, 1.0 g) and sodium hydroxide (1.38 g, 34.6 mmol) in water (26 ml) was hydrogenated at room temperature and 414 kPa (60 p.s.i.) for 20 hours. The solution was filtered through a pad of celite (trade mark) and washed with water (5 ml). The toluene was separated and the aqueous phase cooled to 0° C. whereupon sulphuric acid (1.70 g, 17.3 mmol) was added. The resulting slurry was granulated at 0° C. for 1 hour and then filtered, washed with water (2×5 ml) and dried under vacuum at 50° C. to give the title compound (5.80 g, 87%). m.p. 223-224° C.
Found: C, 40.28; H, 3.39; N, 21.63. C13H13F2N6O4P requires C, 40.43; H, 3.39; N, 21.76%. 1H NMR (300 MHz, DMSO) δ=5.07 (d, 2H) 5.24 (d, 2H), 6.77-6.83 (m, 1H), 7.00-7.18 (m, 2H), 7.75 (s, 2H), 8.53 (s, 2H).
EXAMPLE 2 2-(2,4-Difluorophenyl)-1,3-bis(1H-1,2,4-triazol-1-yl)-2-propyl disodium phosphate

A solution of the compound of Example 1(a) (10.0 g, 17.7 mmol) and sodium acetate (2.90 g, 35.3 mmol) in ethanol (160 ml) and water (20 ml) was hydrogenated over Pearlman’s catalyst (1.00 g) at room temperature and at 345 kPa (50 p.s.i.) for 16 hours. The solution was filtered through a pad of celite (trade mark) and the solvents removed at reduced pressure to leave a thick syrup. This was dissolved in ethanol (100 ml) with the aid of sonication and warmed to reflux. The resulting solution was allowed to cool slowly and granulate for 1 hour at room temperature. The product was filtered, washed with ethanol (10 ml) and dried under vacuum at 50° C. to give the title compound (4.48 g, 59%). m.p. 160-162° C.
1H NMR (300 MHz, D2O) δ=5.01 (d, 2H), 5.40 (d, 2H), 6.60 (m, 1H), 6.79 (m, 1H), 7.11 (m, 1H), 7.63 (s, 2H), 8.68 (s, 2H).
Route 2

Reference:1. CN103864844A.
http://www.google.com/patents/CN103864844A?cl=en
TRANSLATED BY MACHINE…….TEXT MAY VARY
forskolin fluconazole (fosf Iuconazole, Formula I) is fluconazole (Formula IV) of monophosphate prodrugs, fluconazole in the tertiary alcohol into a phosphate ester, not only did not introduce a chiral center, also increased water solubility, because a long time to overcome the low water solubility of fluconazole resulting larger infusion volume defects. After intravenous administration in the role of phosphatases in vivo hydrolysis into fluconazole, pharmacological effect. Blessing from the Central Institute of the United States Secretary of fluconazole Fai end developed, launched in Japan in 2004 I May 15, for the treatment of candidiasis and cryptococcal infections caused deep as true bacteremia, respiratory fungal disease, fungal peritoneum
Inflammation, gastrointestinal fungal disease, fungal urinary tract infections, fungal meningitis.

Synthesis gas itraconazole on forskolin in W09728169, Organic Process Research & Development (200 2), 6 (2), 109-112, CN1789270, Art of Drug Synthesis (2007), 71-82, etc. have been reported in the literature . Which Organic Process Research & Development (2002) described in detail in the first blessing Secretary fluconazole and improved synthetic route for the route problems to adapt to industrial mass production of synthetic routes.
Document Organic Process Research & Development (2002), 6,109-112 discloses the following two synthetic routes.
Route One:

Route two:

The final step is a route to the removal of benzyl group in a methanol solvent by palladium on carbon catalyzed hydrogenation reaction yield was 65%. Since forskolin fluconazole final product insoluble in methanol, and therefore there is a route following shortcomings: a catalyst poisoning, the final product is easy to form methanol solvate, removing the catalyst in the loss of product, the final product are difficult to separate, low yield not suitable for industrial production.
Two routes still using palladium on carbon hydrogenation debenzylation, except that the solvent was changed to sodium hydroxide solution, the product of soluble and stable in aqueous sodium hydroxide solution, after filtering off the catalyst, forskolin fluoro itraconazole by acidification of sodium sulfate can be easily obtained blessing Secretary of fluconazole, the reaction yield of 85-90%.
In the prior art, the removal of benzyl preparation blessing Secretary of fluconazole, the use of a pressure hydrogenation, relatively harsh reaction conditions; and blessing Secretary of fluconazole in water and slightly soluble in methanol, for blessing Secretary fluconazole further refined and purified more difficult. The present invention aims to provide a new and suitable for industrial production methods blessing Secretary fluconazole.
Example 1
2- (2,4-gas-phenyl) -1,3-bis (1H-1, 2,4- two P sat 1-yl) -2-propyl-di-benzyl-pity Cool ( Preparation blessing Secretary fluconazole dibenzyl ester)
Step The method according to CN1210540A in Example 1 A or Method B of (a), was prepared to give the title compound, having 1H-NMR shown in Figure 1 (SOi) MHz, DMS0-D6) spectrum.
Example 2
2_ (2,4_ two gas-phenyl) -1, double 3_ (1H-1, 2,4_ two 1-yl) propyl pity _2_ di press (forskolin gas
Itraconazole ammonium salt) Preparation

Formula III blessing Secretary fluconazole two benzyl ester (566g, lmol), 120g of dry Pd / C (containing 5% palladium) and ammonium formate (315g, 5mol) in methanol (6L), and stirred under reflux for 5h , TLC monitoring completion of the reaction was filtered, 50 ° C the solvent was distilled off under reduced pressure, ethanol was added (566ml), stirred for beating, and filtered to give a solid forskolin fluconazole salt 415g, yield 98.8%.
] lH-Mffi (500MHz, DMS0-D6) δ: 4.87-4.90, 5.58-5.61,6.56-6.60, 6.94-7.03,7.52-7.61,8.96, having 1H-NMR shown in Figure 2 (500MHz, DMS0 -D6) spectrum.
Example 3
2- (2,4-gas-phenyl) -1,3-bis (1H-1, 2,4- two 1-yl) -2-propyl-pity acid dioxide Cool (forskolin
Fluconazole) Preparation of

[0052] Formula II forskolin fluconazole salt (420g, Imol), in water (IL) while stirring, filtered, 2mol / L sulfuric acid aqueous solution (500ml), 5 ° C under stirring for lh, filtered, cold water ( 200ml) wash, 50 ° C under dry blessed Division fluconazole 379g, yield 98%.
1H-Mffi (SOOMHz) DMSO-De) δ:. 5.09-5.12,5.25-5.28,6.80-6.84,7.05-7.16,7.77,8.55,10.32 [0054] Example 4
2_ (2,4_ two gas-phenyl) -1, double 3_ (1Η-1, 2,4_ two 1-yl) propyl pity _2_ di press (forskolin gas itraconazole salt) Preparation
Under nitrogen, forskolin fluconazole dibenzyl ester (566g, lmol), 84g of dry Pd / C (5% containing button) and ammonium formate (189g, 3mol) in anhydrous methanol (5L) in the mixture was stirred at reflux for 5h, TLC monitoring completion of the reaction was filtered, 50 ° C the solvent was distilled off under reduced pressure, ethanol was added (300ml), stirred for beating, and filtered to give a solid forskolin fluconazole salt 410g, yield 97.5%.
Example 5
2_ (2,4_ two gas-phenyl) -1, double 3_ (1H-1, 2,4_ two 1-yl) propyl pity _2_ di press (forskolin gas itraconazole salt) Preparation
Under nitrogen, forskolin fluconazole dibenzyl ester (566g, lmol), 30g of dry Pd / C (containing 10% palladium) and ammonium formate (315g, 5mol) in anhydrous methanol (5L) in the mixture was stirred at reflux for 5h, TLC monitoring completion of the reaction was filtered, 50 ° C the solvent was distilled off under reduced pressure, ethanol was added (300ml), stirred for beating, and filtered to give a solid forskolin fluconazole salt 405g, yield 96.4%.
Example 6
2_ (2,4_ two gas-phenyl) -1, double 3_ (1H-1, 2,4_ two 1-yl) propyl pity _2_ di press (forskolin gas itraconazole salt) Preparation
Under nitrogen, forskolin fluconazole dibenzyl ester (566g, lmol), 30g of dry Pd / C (containing 10% palladium) and ammonium formate (315g, 5mol) in ethanol (12L) and stirred was refluxed for 5h, TLC monitoring completion of the reaction, was filtered, 50 ° C the solvent was distilled off under reduced pressure, ethanol was added (300ml), stirred for beating, and filtered to give a solid forskolin fluconazole salt 395g, 94% yield.
Example 7
2_ (2,4_ two gas-phenyl) -1, double 3_ (1H-1, 2,4_ two 1-yl) propyl pity _2_ di press (forskolin gas itraconazole salt) Preparation
forskolin fluconazole dibenzyl ester (566g, lmol), 170g of dry Pd / C (containing 5% of palladium) and ammonium formate (315g, 5mol) in ethanol (16L) was stirred under reflux for 5h, TLC monitoring completion of the reaction was filtered, 50 ° C the solvent was distilled off under reduced pressure, ethanol was added (300ml), stirred for beating, and filtered to give a solid forskolin fluconazole salt 398g, yield 94.7%.
Example 8
2_ (2,4_ two gas-phenyl) -1, double 3_ (1H-1, 2,4_ two 1-yl) propyl pity _2_ di press (forskolin gas itraconazole salt) Preparation
Under nitrogen, forskolin fluconazole dibenzyl ester (566g, lmol), 120g of dry Pd / C (containing 5% palladium) and ammonium formate (315g, 5mol) in isopropanol (12L) in the mixture was stirred at reflux for 5h, TLC monitoring completion of the reaction was filtered, 50 ° C the solvent was distilled off under reduced pressure, ethanol was added (300ml), stirred for beating, and filtered to give a solid forskolin fluconazole salt 402g, a yield of 95.7%.
Example 9
2_ (2,4_ two gas-phenyl) -1, double 3_ (1H-1, 2,4_ two 1-yl) propyl pity _2_ di press (forskolin gas itraconazole salt) Preparation
[0071] under nitrogen blessing Secretary fluconazole dibenzyl ester (566g, lmol), 60g of dry Pd / C (containing 5% palladium) and ammonium formate (504g, 8mol) in methanol (8L) in, 50 ° C under stirring reaction 40h, TLC monitoring completion of the reaction, was filtered, 50 ° C the solvent was distilled off under reduced pressure, ethanol was added ^ OOml), stirred for beating, and filtered to give a solid forskolin fluconazole salt 398g, yield 94.8%.
Example 10
2_ (2,4_ two gas-phenyl) -1, double 3_ (1H-1, 2,4_ two 1-yl) propyl pity _2_ di press (forskolin gas itraconazole salt) Preparation
Under nitrogen, forskolin fluconazole dibenzyl ester (5668,111101), 8 (^ dry? (1 / (:( containing palladium 5%) and ammonium formate (315g, 5mol) for n-propyl alcohol (12L) in, 60 ° C the reaction was stirred 20h, TLC monitoring completion of the reaction was filtered, 50 ° C the solvent was distilled off under reduced pressure, ethanol was added (300ml), stirred for beating, and filtered to give a solid forskolin fluconazole salt 398g 95% yield.
Example 11
2- (2,4-gas-phenyl) -1,3-bis (1H-1, 2,4- sit two P-1-yl) -2-propyl-pity acid dioxide Cool (forskolin fluconazole) Preparation of [0077] under nitrogen blessing Secretary fluconazole dibenzyl ester 566 g (Imol) adding 56g of dry Pd / C (containing 5% palladium), methanol 6L, 315 g of ammonium formate, stirring boil under reflux for 5h, TLC after completion of the reaction was filtered, 50 ° C the solvent was distilled off under reduced pressure, addition of IL of water and dissolved with stirring, filtered, 2mol / L sulfuric acid 500mL, 5 ° C with stirring to precipitate lh, filtered, 200mL cold water, 50 ° C drying 365 g, 95% yield.
Example 12 forskolin fluconazole salt and HPLC detection methods blessing Secretary fluconazole:
High performance liquid chromatography (Chinese Pharmacopoeia 2010 edition two Appendix VD): octadecylsilane bonded silica as a filler, Column: Thermo BDS C18 (4.6 X 150mm, 3.5 μ m); methanol as mobile phase A, phosphate buffer (take potassium dihydrogen phosphate 0.68g, set 1000ml water, triethylamine 6ml, adjusted to pH 5.0 with phosphoric acid) as the mobile phase B, a flow rate of 1.0ml / min; column temperature 35 ° C; detection wavelength was 210nm, linear gradient.

After the examination, according to the peak area calculation, purity prepared in Example 2-11 was the implementation of the target product of 99.5%.
| Patent | Submitted | Granted |
|---|---|---|
| Nanoparticulate Anidulafungin Compositions and Methods for Making the Same [US2009238867] | 2009-09-24 | |
| IMIDAZOPYRIDINE SUBSTITUTED TROPANE DERIVATIVES WITH CCR5 RECEPTOR ANTAGONIST ACTIVITY FOR THE TREATMENT OF HIV AND INFLAMMATION [US7790740] | 2008-02-21 | 2010-09-07 |
| Pharmaceutical formulations of cyclodextrins and antifungal azole compounds [US2007082870] | 2007-04-12 | |
| TRIAZOLE DERIVATIVES USEFUL IN THERAPY [EP0880533] | 1998-12-02 | 2002-06-12 |
| Triazole derivatives useful in therapy [US6790957] | 2003-07-31 | 2004-09-14 |
| Process for controlling the hydrate mix of a compound [US7323572] | 2004-01-15 | 2008-01-29 |
| TOPICAL TERBINAFINE FORMULATIONS AND METHODS OF ADMINISTERING SAME FOR THE TREATMENT OF FUNGAL INFECTIONS [US7820720] | 2010-04-29 | 2010-10-26 |
| PHARMACEUTICAL COMPOSITION COMPRISING PHENYLAMIDINE DERIVATIVE AND METHOD OF USING THE PHARMACEUTICAL COMPOSITION IN COMBINATION WITH ANTIFUNGAL AGENT [US8173157] | 2010-04-22 | 2012-05-08 |
| COMPOSITIONS COMPRISING POLYUNSATURATED FATTY ACID MONOGLYCERIDES OR DERIVATIVES THEREOF AND USES THEREOF [US8222295] | 2009-11-26 | 2012-07-17 |
| MASKED CARBOXYLATE NEOPENTYL SULFONYL ESTER CYCLIZATION RELEASE PRODRUGS OF ACAMPROSATE, COMPOSITIONS THEREOF, AND METHODS OF USE [US2009069419] | 2009-03-12 |
| Patent | Submitted | Granted |
|---|---|---|
| Triazole derivatives useful in therapy [US2005130940] | 2005-06-16 | |
| Chemical compounds [US7309790] | 2005-06-16 | 2007-12-18 |
| Combination of voriconazole and an antifungal CYP2C19 inhibitor [US2005182074] | 2005-08-18 | |
| Inhibitors of fungal invasion [US2004106663] | 2004-06-03 | |
| Triazole derivatives useful in therapy [US6977302] | 2004-11-25 | 2005-12-20 |
| Pharmaceuticals [US7691877] | 2007-08-23 | 2010-04-06 |
| SIMPLE PANTOIC ACID ESTER NEOPENTYL SULFONYL ESTER CYCLIZATION RELEASE PRODRUGS OF ACAMPROSATE, COMPOSITIONS THEREOF, AND METHODS OF USE [US7994218] | 2009-03-26 | 2011-08-09 |
| COMPLEX PANTOIC ACID ESTER NEOPENTYL SULFONYL ESTER CYCLIZATION RELEASE PRODRUGS OF ACAMPROSATE, COMPOSITIONS THEREOF, AND METHODS OF USE [US8168617] | 2009-03-19 | 2012-05-01 |
| Purine derivatives [US7642350] | 2006-11-23 | 2010-01-05 |
| IMIDAZOPYRIDINONES [US2009221631] | 2009-09-03 |
IMPURITIES
1









 |
|
| Systematic (IUPAC) name | |
|---|---|
|
{[2-(2,4-Difluorophenyl)-1,3-bis(1H-1,2,4-triazol-1-yl)propan-2-yl]oxy}phosphonic acid
|
|
| Clinical data | |
| AHFS/Drugs.com | International Drug Names |
| Legal status |
|
| Routes of administration |
IV |
| Identifiers | |
| CAS Number | 194798-83-9 |
| ATC code | None |
| PubChem | CID 214356 |
| ChemSpider | 185843 |
| UNII | 3JIJ299EWH |
| ChEMBL | CHEMBL1908301 |
| Chemical data | |
| Formula | C13H13F2N6O4P |
| Molar mass | 386.25 g/mol |
| CN1210540A * | Jan 27, 1997 | Mar 10, 1999 | 辉瑞研究开发公司 | Triazole derivatives useful in therapy |
| CN1789270A * | Dec 16, 2005 | Jun 21, 2006 | 西安新安医药科技有限公司 | Mycotic ingection-resisting fosfluconazole hydrate and preparation method thereof |
| CN101890028A * | Feb 22, 2007 | Nov 24, 2010 | 卫材R&D管理有限公司 | Stabilized pharmaceutical composition |
| CN102439018A * | Mar 3, 2010 | May 2, 2012 | 塞普斯制药有限公司 | Fosfluconazole derivatives, synthesis, and use in long acting formulations |
| US20040007689 * | Jun 23, 2003 | Jan 15, 2004 | Pfizer Inc. | Process for controlling the hydrate mix of a compound |
| Reference | ||||
|---|---|---|---|---|
| 1 | * | ARTHUR BENTLEY等: “The Discovery and Process Development of a Commercial Route to the Water Soluble Prodrug, Fosfluconazole“, 《ORGANIC PROCESS RESEARCH & DEVELOPMENT》, vol. 6, no. 2, 18 December 2001 (2001-12-18), XP002491526, DOI: doi:10.1021/op010064+ | ||
| 2 | * | 国大亮 等: “福司氟康唑“, 《齐鲁药事》, vol. 24, no. 1, 30 January 2005 (2005-01-30), pages 60 | ||
| 3 | * | 村上尚道: “fosfluconazole“, 《NEW DRUGS OF THE WORLD:2003》, vol. 33, no. 10, 15 September 2004 (2004-09-15), pages 56 | ||
//////UK-292663, UK 292663, F-FLCZ, F FLCZ, Fosfluconazole, 194798-83-9, UNII-3JIJ299EWH, 3JIJ299EWH, NCGC00182029-01
Fc1ccc(c(F)c1)C(OP(=O)(O)O)(Cn2ncnc2)Cn3ncnc3
Pfizer’s Fosdagrocorat, PF-04171327 for Rheumatoid Arthritis
Fosdagrocorat, PF-04171327,
CAS 1044535-58-1
(2R,4aS,10aR)-4a-Benzyl-7-((2-methylpyridin-3-yl)carbamoyl)-2-(trifluoromethyl)-1,2,3,4,4a,9,10,10a-octahydrophenanthren-2-yl dihydrogen phosphate
2-Phenanthrenecarboxamide, 4b,5,6,7,8,8a,9,10-octahydro-N-(2-methyl-3-pyridinyl)-4b-(phenylmethyl)-7-(phosphonooxy)-7-(trifluoromethyl)-, (4bS,7R,8aR)-
(2R,4aS,10aR)-4a-benzyl-7-((2-methylpyridin-3-yl)carbamoyl)-2-(trifluoromethyl)-1,2,3,4,4a,9,10,10a-octahydrophenanthren-2-yl dihydrogen phosphate
MF C29H30F3N2O5P
Exact Mass: 574.1844
- PF 04171327
- PF-04171327
- UNII-HPI19004QS
- Selective Glucocorticoid Receptor Modulator
phase 2 .Rheumatoid Arthritis
Glucocorticoid receptor modulators
Pfizer
- 03 Sep 2015Phase II development of fosdagrocorat is ongoing
- 01 Jun 2014Pfizer completes a phase II trial in Rheumatoid arthritis in US, Bulgaria, Colombia, the Czech Republic, Germany, Hungary, India, South Korea, Malaysia, Mexico, Poland, Romania, Russia, Serbia, Slovakia, South Africa, Spain and the Ukraine (NCT01393639)
- 30 Sep 2011Phase-II clinical trials in Rheumatoid arthritis in Bulgaria, Colombia, Germany, India, Malaysia, Mexico, Poland, Romania and South Africa (PO)
Fosdagrocorat, also known as PF-04171327, a dissociated agonist of the glucocorticoid receptor (DAGR), a selective high-affinity partial agonist of the GR with potent anti-inflammatory activity at exposures that provide less undesirable effects on bone and glucose metabolism compared with prednisone (pred).
Glucocorticoid receptor modulators are glucocorticoid receptor ligands that are used to treat a variety of conditions because of their powerful anti-inflammatory, antiproliferative and immunomodulatory activity. J. Miner, et al., Expert Opin. Investig. Drugs (2005) 14(12):1527-1545.
Examples of glucocorticoid receptor modulators include dexamethasone, prednisone, prednisolone, RU-486, and as described in WO 2000/66522 and WO 2004/005229.
Treatment with glucocorticoid receptor modulators is often associated with side effects, such as bone loss and osteoporosis.
Identifying a glucocorticoid receptor modulator that is efficacious, potent, and has mitigated side-effects fulfills a medical need.
SYNTHESIS COMING…………
PATENT
WO 2008093227/US 20100286214
https://www.google.com/patents/WO2008093227A1?cl=en
SCHEME A

The 1 (/?)-Benzyl-5-bromo-9(S)-hydro-10(R)-hydroxy-10(R)-methyl-tricyclo[7.3.1.02‘7]trideca-2,4,6-trien-13-one of Formula A-8 was prepared using the protocol described in Scheme A, which is generally disclosed in WO 00/66522. Ph depicts Phenyl. Bn depicts Benzyl. Compound A-1 can be purchased (for example, VOUS and Riverside; CAS No. 4133-35-1 ). Compound A-2 can be prepared as described in Org. Syn. 1971 , 51 , 109-112.
SCHEME B


The (4βS,7R,8αR)-4β-benzyl-7-hydroxy-Λ/-(2-methylpyridin-3-yl)-7-(trifluoromethyl)-4b,5,6,7,8α,9,10-octahydrophenanthrene-2-carboxamide was prepared as described in Scheme B.
SCHEME C
 The (2R,4αS, 10αR)-4α-benzyl-7-((2-methylpyridin-3-yl)carbamoyl)-2-(trifluoromethyl)-1 ,2,3,4,4α,9,10,10α-octahydrophenanthren-2-yl dihydrogen phosphate of C-3 was prepared as described in Scheme C. Bn depicts benzyl.
The (2R,4αS, 10αR)-4α-benzyl-7-((2-methylpyridin-3-yl)carbamoyl)-2-(trifluoromethyl)-1 ,2,3,4,4α,9,10,10α-octahydrophenanthren-2-yl dihydrogen phosphate of C-3 was prepared as described in Scheme C. Bn depicts benzyl.
SCHEME D


The (2R,4αS,10αR)-4α-benzyl-7-((2-methylpyridin-3-yl)carbamoyl)-2-(trifluoromethyl)-1 ,2,3,4,4α,9,10,10α-octahydrophenanthren-2-yl dihydrogen phosphate of C-3 was prepared as described in Scheme D. Bn depicts benzyl. Ph depicts phenyl.
SCHEME E


The (2R,4αS, 10αR)-4α-benzyl-7-((2-methylpyridin-3-yl)carbamoy[)-2-(trifluoromethyl)-1 ,2,3,4,4α,9,10,10α-octahydrophenanthren-2-yl dihydrogen phosphate of C-3 was prepared as described in Scheme E. Bn depicts benzyl. Ph depicts phenyl.
Starting Material A-8 is 1(R)~Benzyl-5-bromo-9(S)-hydro-10(R)-hydroxy-10(R)-methyl-tricyclo[7.3.1.02‘7]trideca-2,4,6-trien-13-one as depicted by the following formula:

Preparation 1 : (S)-4a-benzyl-7-bromo-2-ethoxy-3,4,4a,9-tetrahydrophenanthrene

Starting Material A-8 (450 g; 1.17 moles) was dissolved in ethanol (4.5 L) at ambient temperature. 21% sodium ethoxide in ethanol (44 mL; 0.12 moles) was added and the mixture was heated to reflux for three hours. Once the Starting Material A-8 was consumed, the reaction mixture was chilled to -250C. Acetyl chloride (250 mL; 3.51 moles) was slowly added to the mixture while the temperature was maintained near -25°C. After the addition was complete, the mixture was warmed to O0C and held there until the intermediate enone was consumed. The mixture was slurry at this point. 21 % sodium ethoxide in ethanol (1.31 L; 3.51 moles) was added to the mixture while the temperature was maintained between -5°C and 50C. If the mixture was not basic, more sodium ethoxide was added. The temperature of the mixture was increased to 25°C and then diluted with water (5.9 L). The mixture was filtered and the solid was washed with water (3 X). The title compound (440 g; 85 area %) was obtained as a beige solid. 1H NMR (DMSO) δ ppm: 1.27 (t, 3H), 1.65 (dt, 1 H), 2.06 (d, 1 H), 2.21 (dd, 1 H)1 2.49 (m, 1 H), 2.65 (m, 2H), 2.89 (m, 2H), 3.85 (q, 2H), 5.45 (m, 2H), 6.44 (d, 2H), 6.98 (t, 2H), 7.06 (m, 2H), 7.25 (d, 1 H), 7.33 (dd, 1 H).
Preparation 2: (S)-4a-benzyl-7-bromo-2,2-(1,2-ethylenedioxy)-1,2,3,4,4a,9-hexahydrophenanthrene

The (S)-4α-benzyl-7-bromo-2-ethoxy-3,4,4α,9-tetrahydrophenanthrene (1270 g; 3.2 moles; 85 area %, which may be prepared as described in Preparation 1 ) was dissolved in toluene (6.45 L). The ethylene glycol (898 mL; 16.1 moles) and p-toluenesulfonic acid (6.1 g; 0.03 moles) were added and the reaction heated to reflux. Solvent (1 L) was distilled from the mixture and replaced with fresh toluene (1 L). This distillation process was repeated twice more. More p-toluenesulfonic acid (6.1 g) was added each time fresh toluene was added. During the reaction, two intermediates (detected by LC) were formed as the substrate was converted into product. The end point of the reaction was an equilibrium point between the two intermediates and the product. Once the endpoint was reached, the mixture was cooled to ambient temperature. The mixture was washed with 0.5 M NaOH (2 L). The phases separated quickly and both were dark with a small rag layer. The mixture was washed with water (2 L). The phases
separated very slowly. The mixture was dried by azeotropic distillation. Methanol (4 L) was added to the mixture and solvent (4 L) was distilled from the mixture. The methanol addition and solvent distillation were repeated twice more. Methanol was added to the mixture and precipitation occurred a few minutes later. More methanol (4 L) was added to the mixture and then brought to reflux. After 30 minutes, the mixture was cooled to 00C. The mixture was filtered and the solid was washed with chilled methanol (2 X 2L). The solid was dried in a vacuum oven at 65°C. The title compound (882 g; 98 area %) was obtained as a beige solid. 1H NMR (DMSO) δ ppm: 1.71 (m, 2H), 2.06 (m, 2H), 2.31 (dd, 1 H), 2.39 (m, 1 H), 2.68 (d, 1 H), 2.77 (m, 1 H), 2.86 (dd, 1 H), 3.36 (d, 1 H), 3.86 (m, 4H), 5.45 (m, 1 H), 6.50 (m, 2H), 7.00 (m, 4H), 7.37 (dd, 1 H), 7.44 (d, 1 H).
Preparation 3: (S)-methyl 4β-benzyl-7,7-(1,2-ethylenedioxy)-4β,5,6,7,8,10-hexahydrophenanthrene-2-carboxylate

The (S)-4α-benzyl-7-bromo-2,2-(1 ,2-ethylenedioxy)-1 ,2,3,4,4α,9-hexahydrophenanthrene (719 g; 1.75 moles, which may be prepared as described in Preparation 2) was dissolved in tetrahydrofuran (7.19 L) and chilled to -7O0C. The 1.6 M n-butyl lithium in hexane (2270 mL; 2.27 moles) was added at a rate such that the temperature was maintained below -6O0C. The mixture held an additional 15 minutes after the addition. Carbon dioxide (108 g; 2.45 moles) was added while the temperature was maintained below -60°C. The mixture held an additional 15 minutes after the addition. The mixture was warmed to ambient temperature. Solvent (7 L) was distilled from the mixture at atmospheric pressure. DMF (7 L) was added to the mixture. The mixture was cooled to ambient temperature. Methyl iodide (152 mL; 2.45 moles) was added and the mixture was held until the reaction was completed (~1 hour). The mixture was heated to 7O0C and solvent was distilled by gradually reducing the pressure to 70 mmHg. Once distillation had ceased, the mixture was cooled to room
temperature. Water (6.5 L) was slowly added to the mixture to precipitate the product. The mixture was filtered and the solid washed with water (3 X). The solid was dried on the filter. The crude product (736 g; 74 area %) was obtained as a beige solid. The product was purified by chromatography. 463 g of product was recovered from the chromatography. This material was separated from n-heptane (6130 mL). 394 g of the title compound was recovered. Another 70 g of title compound was recovered from the mother liquor by chromatography. 1H NMR (DMSO) δ ppm: 1.74 (m, 2H), 2.10 (m, 2H)1 2.33 (dd, 1 H), 2.45 (m, 1 H), 2.72 (d, 1 H), 2.79 (m, 1 H), 2.94 (dd, 1 H), 3.40 (d, 1 H), 3.87 (m, 7H), 5.49 (m, 1 H), 6.47 (m, 2H), 6.93 (m, 2H), 7.01 (m, 1 H), 7.42 (d, 1 H), 7.64 (d, 1 H), 7.79 (dd, 1 H).
Preparation 4: (4βS,8α/?)-methyl 4β-benzyl-7,7-(1,2-ethylenedioxy)-4β,5,6,7,8,8α,9,10-octahydrophenanthrene-2-carboxylate

The (S)-methyl 4β-benzyl-7,7-(1 ,2-ethylenedioxy)-4β,5,6,7,8,10-hexahydrophenanthrene-2-carboxylate (201 g; 0.515 moles, which may be prepared as described in Preparation 3) and 50 ml of ethylene glycol was dissolved in toluene (2.0 L) in an autoclave. To this was added 10 grams of a 5% Pd/C (dry catalyst). The autoclave was then sealed and purged with nitrogen (three cycles) followed by hydrogen (three cycles). The reaction was run for 18 hours with a pressure of 80 psig and temperature of 50 0C. HPLC analysis for completion and selectivity (typical selectivity’s are: 95 to 5, Trans to Cis). The suspension was filtered through Celite® to remove the catalyst and the toluene solution is concentrated at 50 0C, under vacuum, to
approximately 200 ml. While still at 50 0C, 1 L of 1-butanol was added and the solution heated to 60 0C, until clear. Upon cooling, the resulting solid title compound was isolated by vacuum filtration (196 grams; 97%; Trans to Cis 95.75 to 4.24). 1H NMR (300 MHz, CDCI3) δ ppm: 7.79 (bs, 1 H1 Ar-H), 7.47 (d, J= 9 Hz, 1 H, Ar-H), 7.13-7.05 (cm, 3H, Ar-H), 6.56-6.53 (cm, 2H, Ar-H), 6.43 (d, J= 9 Hz, 1 H, Ar-H), 4.04-3.93 (cm, 4H, 2-CH2), 3.89 (s, 3H, CH3),3.08-3.03 (cm, 3H, CH2, CH-H), 2.63 (d, J= 15 Hz, CH-H), 2.22-1.72 (cm, 8H, 4-CH2), 1.57 (cm, 1 H, CH-H).; 13CNMR (CDCI3, δ): 167.7, 149.2, 137.7, 136.4, 131.1 , 130.5, 127.8, 127.7, 127.4, 126.3, 125.5, 108.9, 64.6, 64.5, 52.1 , 40.5, 39.8, 38.3, 35.8, 31.6, 30.3, 27.9, 24.6.
Preparation 5: (4βS,8α/?)-methyl 4β-benzyl-7-oxo-4β,5,6,7,8,8α,9,10-octahydrophenanthrene-2-carboxylate

ThΘ (4βS,8αR)-mΘthyl 4β-benzyl-7,7-(1 ,2-ethylenΘdioxy)-4β,5,6,7,8,8α,9,10-octahydrophenanthrene-2-carboxylate (150 g, 382 mmol, which may be prepared as described in Preparation 4) was dissolved in dichloromethane (630 ml). Water (270 ml) was added with stirring followed by trifluoroacetic acid (73 ml. 1150 mmol) via drop funnel over 30 minutes, maintaining the internal temperature below 3O0C. After the addition was complete, the reaction was heated at 4O0C for 2 hours. In process check indicated incomplete reaction with around 9% (area percent) starting material. The layers were separated and fresh water (270 ml) and trifluoroacetic acid (31 ml) was added. The reaction mixture was heated at 4O0C for 1 hour. This process was continued until the starting material was consumed. The organic phase was washed with 5% aqueous sodium bicarbonate (300 ml), water (300 ml) and dried over MgSO4 and concentrated to dryness to give 126.4 g of the title compound (representing a 95% yield). 1H NMR (DMSO) δ ppm: 7.70 (s, 1 H), 7.37 (d, J=8.4 Hz, 1 H), 7.11 (m, 3H), 6.6 (d, J= 5.70 Hz, 2H), 6.45 (d, J=8.4 Hz, 1H), 3.80 (s, 3H), 3.80 (m, 2H), 3.04-1.48 (m, 11 H).
Preparation 6: (4βS,7f?,8α/?)-methyl 4β-benzyl-7-hydroxy-7-(trifluoromethyl)-4β,5J6,7,8,8α,9,10-octahydrophenanthrene-2-carboxylate

The (4βS,8αf?)-methyl 4β-benzyl-7-oxo-4β,5,6,7,8I8α,9,10-octahydrophenanthrene-2-carboxylate (118g, 0.339 mole, which may be prepared as described in Preparation 5) dissolved in dichloromethane was chilled to -5O0C. The solution became turbid. 1.0 M Tetrabutylammonium fluoride a solution in THF (3.4 ml, 0.003 mol) was added with no appreciable temperature change. Trifluorotrimethylsilane (79 ml, 0.51 mol) was added over 20 minutes with a color change to bright orange to light red in color. The reaction mixture was held at -50 0C for about 2 hours and then allowed to warm to 0 0C.
Tetrabutylammonium fluoride (340 ml, 0.34 moles) was added very slowly at 0 0C, to the reaction mixture over 45 minutes. An exotherm was observed with gas evolution. The reaction mixture was stirred 10 minutes and HPLC analysis indicated complete desilylialation. Water (1 L) was added to the reaction mixture and with vigorous stirring and allowed to warm to room temperature. The organic layer was washed with water (1 L). The organic layer was concentrated and chromatographed to produce 72 g, 51 % of the title compound, with an additional 32 g of impure product. 1H NMR (DMSO) δ ppm: 7.70 (s, 1 H), 7.37 (d, J=8.1 Hz, 1 H)1 7.09 (m, 3H), 6.5 (dd, J=1.2, 6.6 Hz, 2H), 6.38 (d, J=8.4 Hz, 1 H), 3.80 (s, 3H), 3.80 (m, 2H), 3.09-1.21 (m, 13H).
Preparation 7: (4βS,7/?,8α/?)-methyl 4β-benzyl-7-(bis(benzyloxy)phosphoryloxy)-7-(trifluoromethyl)-4β,5,6,7,8,8α,9,10-octahydrophenanthrene-2-carboxylate

The (4βS,7R,8αf?)-methyl 4β-benzyl-7-hydroxy-7-(trifluoromethyl)-4β)5,6,7)8,8α,9,10-octahydrophenanthrene-2-carboxylate (5.0 g; 11.9 mmol, which may be prepared as in Preparation 6) and 5-methyltetrazole (3.6 g; 43.0 mmol) were mixed together in dichloromethane (50 mL) at ambient temperature. Dibenzylphosphoramidite (8.3 mL; 25.1 mmol) was added and the mixture was stirred until the reaction was completed (1 hour). The mixture was chilled to 00C and 30% hydrogen peroxide (10 mL) was added. The reaction was stirred until the oxidation was completed (30 minutes). The aqueous phase was separated from the organic phase. The organic phase was washed with 10% sodium meta-bisulfite (50 ml_). The organic phase was dried with anhydrous magnesium sulfate and concentrated. The crude product was purified by silica gel chromatography with 15% ethyl acetate in hexanes. The purified title compound (8.41 g; 94% yield) was obtained as a colorless oil that contained 6% ethyl acetate by weight. 1H NMR (DMSO): δ 1.31 (t, 1 H), 1.63-1.92 (m, 3H), 2.05-2.35 (m, 3H), 2.63 (d, 1 H), 2.75-3.16 (m, 4H), 3.80 (s, 3H), 5.13 (m, 4H), 6.43 (d, 1 H), 6.49 (m, 2H), 7.04-7.17 (m, 3H), 7.33-7.42 (m, 12H), 7.71 (d, 1 H).
Preparation 8: dibenzyl (2f?,4αS,10αR)-4α-benzyl-7-((2-methylpyridin-3-o yl)carbamoyl)-2-(trifluoromethyl)-1 ,2,3,4,4α,9,10,10α-octahydrophenanthren-2-yI phosphate

The (4βS,7R,8αf?)-methyl 4β-benzyl-7-(bis(benzyloxy)phosphoryloxy)-7- (trifluoromethyl)-4β,5,6,7,8,8α,9,10-octahydrophenanthrene-2-carboxylate (7.9 g; 11.6 5 mmol, which may be prepared as in Preparation 7) and 3-amino-2-picoline (1.3 g; 12.2 mmol) were mixed together in tetrahydrofuran (80 ml_) and chilled to 0°C. The 1 M solution of lithium bis(trimethylsilyl)amide in tetrahydrofuran (24 ml_; 24.4 mmol) was added while maintaining the temperature below 100C. The mixture was stirred for 30 minutes. Water (50 mL) was added to the reaction mixture. The mixture was extracted with ethyl acetate. The organic extract was washed with water. The organic phase was dried with anhydrous magnesium sulfate and concentrated. The crude product was purified by silica gel chromatography with 70% ethyl acetate in hexanes. The purified title compound (6.79 g; 68% yield) was obtained as a yellow gum that contained 6% ethyl acetate by weight. 1H NMR (DMSO): δ 1.33 (t, 1 H), 1.66-1.93 (m, 3H), 2.08-2.34 (m, 3H), 2.41 (s, 3H), 2.68 (d, 1 H), 2.76-3.19 (m, 4H), 5.14 (m, 4H), 6.47 (d, 1 H), 6.56 (m, 2H), 7.07-7.19 (m, 3H), 7.20-7.53 (m, 12H), 7.71 (d, 1 H), 7.76 (s, 1 H), 8.32 (d, 1 H), 9.93 (s, 1 H).
Example 1 : (4βS,7/?,8αR)-4β-benzyl-7-hydroxy-W-(2-methylpyridin-3-yl)-7-(trifluoromethyl)-4β,5,6,7,8,8α,9,10-octahydrophenanthrene-2-carboxamide

The (4βS,7ft,8αR)-methyl 4β-benzyl-7-hydroxy-7-(trifluoromethyl)-4β,5,6,7,8,8α,9,10-octahydrophenanthrene-2-carboxylate (10 g; 23.9 mmol, which may be prepared as described in Preparation 6), and 3-amino-2-picoline (2.71 g; 25.1 mmol) were dissolved in toluene (200 ml_). The 1 M lithium bis(trimethylsilyl)amide in tetrahydrofuran (74.1 mL; 74.1 mmol) was added at a rate such that the temperature was maintained below 350C. There was a mild exotherm and a solid precipitated during the addition. The mixture was held an additional 30 minutes after the addition. Water (250 mL) was added to the mixture. There was a mild exotherm and the solid dissolved. Ethyl acetate (50 mL) was added to the mixture to ensure the product did not precipitate. Stirring was stopped to allow the phases to separate. The aqueous phase was removed. The organic phase was washed with water (250 mL). Solvent (230 mL) was distilled at atmospheric pressure from the organic phase. The mixture was cooled to ambient temperature. The mixture was filtered and the solid was washed with toluene (2 times) followed by heptane (2 times). The solid was dried in a vacuum oven at 700C. The title compound of the present example (10 g) was obtained as a beige solid. 1H NMR (DMSO) δ ppm: 1.32 (m, 1 H), 1.82 (m, 4H), 2.10 (m, 4H), 2.41 (s, 3H), 2.68 (d, 1 H), 3.08 (m, 3H), 6.00 (s, 1H), 6.43 (d, 1 H), 6.59 (m, 2H), 7.12 (m, 3H), 7.25 (dd, 1H), 7.44 (dd, 1H), 7.71 (dd, 1 H), 7.75 (d, 1 H), 8.31 (dd, 1 H), 9.91 (s, 1 H).
Example 2: (2f?,4αS,10αR)-4α-benzyl-7-((2-methylpyridin-3-yl)carbamoyl)-2-(trifluoromethyl)-i ,2,3,4,4α,9,10,1 Oα-octahydrophenanthren-2-yl dihydrogen phosphate

The dibenzyl (2R,4αS, 10αR)-4α-bθnzyl-7-((2-methylpyridin-3-yl)carbamoyl)-2-(trifluoromethyl)-1 ,2,3,4,4a,9,10,10a-octahydrophenanthren-2-yl phosphate (6 g; 7.9 mmol, which may be prepared as described in Preparation 8) was dissolved in methanol (120 ml_). 5% palladium on carbon (63% water) (1.3 g; 0.4 mmol) was added to the mixture. The mixture was treated with hydrogen (50 psi) at room temperature. The reaction stalled with 12% of the monobenzylic intermediate remaining. The mixture was filtered through a pad of Celite®. Fresh catalyst (1.3 g) was added to the solution and resubmitted to the hydrogenation conditions. Once the reaction was completed, the mixture was filtered through a pad of Celite®. The solution was concentrated to about 60 ml_ by distillation and not by using a rotary evaporator. During the distillation a white solid precipitated. The mixture was cooled to ambient temperature. The mixture was filtered and the solid washed with methanol. The solid was dried in a vacuum oven at 700C. The compound of the present example (3.36 g; 75% yield) was obtained as a white solid and had an LC purity of 98 area %. 1H NMR (DMSO): δ 1.33 (t, 1 H)1 1.69-1.98 (m, 3H), 2.07-2.29 (m, 3H)1 2.42 (s, 3H), 2.61-2.80 (m, 2H)1 2.93-3.19 (m, 3H)1 3.30 (d, 1 H), 6.50 (d, 1 H), 6.64 (m, 2H), 7.08-7.20 (m, 3H), 7.29 (dd, 1 H), 7.48 (dd, 1 H), 7.75 (dd, 2H), 8.33 (dd, 1 H), 9.96 (s, 1 H).
PATENT
WO 2008093236
http://www.google.co.in/patents/WO2008093236A1?cl=en
Example 1 : (4βS,7/?,8α/?)-4β-benzyl-7-hydroxy-N-(2-methylpyridin-3-yl)-7- (trifluoromethyl)-4β,5,6,7,8,8α,9,10-octahydrophenanthrene-2-carboxamide

The (4βS,7R,8α/?)-methyl 4β-benzyl-7-hydroxy-7-(trifluoromethyl)-4β,5,6J7,8,δα,9, 10- octahydrophenanthrene-2-carboxylate (10 g; 23.9 mmol, which may be prepared as described in Preparation 6), and 3-amino-2-picoline (2.71 g; 25.1 mmol) were dissolved in toluene (200 ml_). The 1 M lithium bis(trimethylsilyl)amide in tetrahydrofuran (74.1 ml_; 74.1 mmol) was added at a rate such that the temperature was maintained below 350C. There was a mild exotherm and a solid precipitated during the addition. The mixture was held an additional 30 minutes after the addition. Water (250 ml_) was added to the mixture. There was a mild exotherm and the solid dissolved. Ethyl acetate (50 ml_) was added to the mixture to ensure the product did not precipitate. Stirring was stopped to allow the phases to separate. The aqueous phase was removed. The organic phase was washed with water (250 ml_). Solvent (230 ml_) was distilled at atmospheric pressure from the organic phase. The mixture was cooled to ambient temperature. The mixture was filtered and the solid was washed with toluene (2 times) followed by heptane (2 times). The solid was dried in a vacuum oven at 700C. The title compound of the present example (10 g) was obtained as a beige solid. 1H NMR (DMSO) δ ppm: 1.32 (m, 1H), 1.82 (m, 4H), 2.10 (m, 4H), 2.41 (s, 3H), 2.68 (d, 1 H), 3.08 (m, 3H), 6.00 (s, 1 H), 6.43 (d, 1 H), 6.59 (m, 2H), 7.12 (m, 3H), 7.25 (dd, 1 H), 7.44 (dd, 1 H), 7.71 (dd, 1 H), 7.75 (d, 1 H), 8.31 (dd, 1 H), 9.91 (s, 1 H).
Example 2: (2f?,4αS,10α/?)-4α-benzyl-7-((2-methylpyridin-3-yl)carbamoyl)-2- (trifluoromethyl)-1,2,3,4,4α,9,10,10α-octahydrophenanthren-2-yl dihydrogen phosphate

The dibenzyl (2R,4αS,10αR)-4α-benzyl-7-((2-methylpyridin-3-yl)carbamoyl)-2- (trifluoromethyl)-1 ,2,3,4,4a,9,10,10a-octahydrophenanthren-2-yl phosphate (6 g; 7.9 mmol, which may be prepared as described in Preparation 8) was dissolved in methanol (120 ml_). 5% palladium on carbon (63% water) (1.3 g; 0.4 mmol) was added to the mixture. The mixture was treated with hydrogen (50 psi) at room temperature. The reaction stalled with 12% of the monobenzylic intermediate remaining. The mixture was filtered through a pad of Celite®. Fresh catalyst (1.3 g) was added to the solution and resubmitted to the hydrogenation conditions. Once the reaction was completed, the mixture was filtered through a pad of Celite®. The solution was concentrated to about 60 ml_ by distillation and not by using a rotary evaporator. During the distillation a white solid precipitated. The mixture was cooled to ambient temperature. The mixture was filtered and the solid washed with methanol. The solid was dried in a vacuum oven at 7O0C. The compound of the present example (3.36 g; 75% yield) was obtained as a white solid and had an LC purity of 98 area %. 1H NMR (DMSO): δ 1 .33 (t, 1 H), 1 .69- 1.98 (m, 3H), 2.07-2.29 (m, 3H), 2.42 (s, 3H), 2.61 -2.80 (m, 2H), 2.93-3.19 (m, 3H), 3.30 (d, 1 H), 6.50 (d, 1 H), 6.64 (m, 2H), 7.08-7.20 (m, 3H), 7.29 (dd, 1 H), 7.48 (dd, 1 H), 7.75 (dd, 2H), 8.33 (dd, 1 H), 9.96 (s, 1 H).
REFERENCES
https://www.pfizer.com/sites/default/files/product-pipeline/July%2028%202015%20Pipeline%20Update.pdf
https://clinicaltrials.gov/ct2/show/NCT00938587
////////
Cc1c(cccn1)NC(=O)c2ccc3c(c2)CC[C@H]4[C@]3(CC[C@@](C4)(C(F)(F)F)OP(=O)(O)O)Cc5ccccc5
O=P(O)(O[C@@]1(C(F)(F)F)C[C@@]2([H])CCC3=C(C=CC(C(NC4=CC=CN=C4C)=O)=C3)[C@]2(CC5=CC=CC=C5)CC1)O
PF 04995274, a 5-HT4Partial Agonist
PF-04995274,
(R)-4-((4-(((4-(Tetrahydrofuran-3-yloxy)-1,2-benzisoxazol-3-yl)oxy)methyl)piperidin-1-yl)methyl)tetrahydro-2H-pyran-4-ol
4-(4-{4-[(R)-(Tetrahydro-furan-3-yl)oxy]-benzo[d]isoxazol-3-yloxymethyl}-piperidin-1-ylmethyl)-tetrahydro-pyran-4-ol
CAS 1331782-27-4
UNII: XI179PG9LV
MF C23-H32-N2-O6
MW 432.5138
a 5-HT4Partial Agonist
PHASE 1 Alzheimer’s type dementia.
Pfizer Inc. INNOVATOR
5-HT4 agonists have attracted attention for therapeutic value in the treatment of Alzheimer’s Disease (AD) and cognitive impairment.Acting to increase levels of acetylcholine and soluble APP alpha, 5-HT4 agonists have the potential to demonstrate both ameliorative and disease modifying effects
(R)-4-((4-((4-(tetrahydrofuran-3-yloxy)benzo[d]isoxazol-3-yloxy)methyl)piperidin-1-yl)methyl)tetrahydro-2/-/-pyran-4-ol and pharmaceutically acceptable salts thereof. This invention also is directed, in part, to a method for treating a 5-HT4 mediated disorder in a mammal. Such disorders include acute neurological and psychiatric disorders, stroke, cerebral ischemia, spinal cord trauma, head trauma, perinatal hypoxia, cardiac arrest, hypoglycemic neuronal damage, dementia, Alzheimer’s disease, Huntington’s Chorea, amyotrophic lateral sclerosis, ocular damage, retinopathy, cognitive disorders, idiopathic and drug- induced Parkinson’s disease, muscular spasms and disorders associated with muscular spasticity including tremors, depression, epilepsy, convulsions, migraine, urinary incontinence, substance tolerance, substance withdrawal, psychosis, schizophrenia, anxiety, mood disorders, trigeminal neuralgia, hearing loss, tinnitus, macular degeneration of the eye, gastroesophageal reflux disease, gastrointestinal disease, gastric motility disorder, non-ulcer dyspepsia, functional dyspepsia, irritable bowel syndrome, constipation, dyspepsia, esophagitis, gastroesophageral disease, nausea, emesis, brain edema, pain, tardive dyskinesia, sleep disorders, attention deficit/hyperactivity disorder, attention deficit disorder, disorders that comprise as a symptom a deficiency in attention and/or cognition, and conduct disorder

a(a) SOCl2, DMAP, acetone, DME, RT, 81%;
(b) DEAD, PPh3, THF, RT, 65%;
(c) K2CO3, MeOH, RT, 92%;
(d) K2CO3, water, MeOH, 50 °C, 76%;
(e) CDI, THF, 50 °C, 43%;
(f) DEAD, PPh3, THF, reflux, 51%;
(g) HCl, Et2O, RT, 81%;
(h) TEA, MeOH, reflux, 50%.

PAPER
Journal of Medicinal Chemistry (2012), 55(21), 9240-9254
http://pubs.acs.org/doi/abs/10.1021/jm300953p

The cognitive impairments observed in Alzheimer’s disease (AD) are in part a consequence of reduced acetylcholine (ACh) levels resulting from a loss of cholinergic neurons. Preclinically, serotonin 4 receptor (5-HT4) agonists are reported to modulate cholinergic function and therefore may provide a new mechanistic approach for treating cognitive deficits associated with AD. Herein we communicate the design and synthesis of potent, selective, and brain penetrant 5-HT4 agonists. The overall goal of the medicinal chemistry strategy was identification of structurally diverse clinical candidates with varying intrinsic activities. The exposure–response relationships between binding affinity, intrinsic activity, receptor occupancy, drug exposure, and pharmacodynamic activity in relevant preclinical models of AD were utilized as key selection criteria for advancing compounds. On the basis of their excellent balance of pharmacokinetic attributes and safety, two lead 5-HT4 partial agonist candidates 2d and 3 were chosen for clinical development.
PATENT
https://www.google.co.in/patents/WO2011101774A1?cl=en
(R)-4-((4-((4-(tetrahydrofuran-3-yloxy)benzo[d]isoxazol-3-yloxy)methyl)piperidin-1-yl)methyl)tetrahydro-2H-pyran-4-ol , hereinafter referred to as “Compound X,” and having the following structure:

Compound X
Example 1 : Synthesis of iR)-4-ii4-i(4-itetrahvdrofuran-3-yloxy)benzord1isoxazol-3-yloxy)methyl)piperidin-1 -yl)methyl)tetrahvdro- 2 -pyran-4-ol

Methyl 2-fluoro-6-hydroxybenzoate (2): To a 20L jacketed reactor were charged 2-fluoro-6-hydroxybenzoic acid (Oakwood Products; 0.972 kg, 6.31 mol), methanol (7.60 L) and sulfuric acid (0.710 kg, 7.24 mol, 1 .15 eq). The jacket temperature was heated to 60°C and the reaction mixture was stirred for 45 h. The reaction mixture was concentrated under vacuum and approximately 7.5 L of methanol distillates were collected. The resulting thin oil was cooled to 20°C. Water (7.60 L) and ethyl acetate (7.60 L) were charged to the reactor, and the product extracted into the organic layer. The EtOAc solution was washed with a solution of sodium bicarbonate (1.52 Kg) in water (6.92 L) followed by a brine solution of sodium chloride (1.74 kg) in water (4.08 L). The resulting EtOAc solution was concentrated to dryness. A light orange oil was isolated; the oil slowly crystallized upon standing to give the title compound (2) (0.952 Kg, 5.60 mol, 89% yield). 1 H NMR (400 MHz, CDCI3) δ ppm 3.97 (s, 3H), 6.59 (ddd, J=10.9, 8.2,1 .2, 1 H), 6.76 (dt, J=8.2, 1 .1 , 1 H), 7.35 (td, J=8.6, 6.3, 1 H), 1 1.24 (s, 1 H); 13C NMR (400 MHz, CDCI3) δ ppm 52.65, 102.56 (d, J=13), 106.90 (d, J=23), 1 13.31 (d, J=3.1 ), 135.34 (d, J=1 1 .5), 161 .02, 163.31 (d, J=62.2), 169.87 (d, 3.8); MS 171.045 (m+1 ). 2-Fluoro-N,6-dihydroxybenzamide (3): To a 50L reactor was charged water (4.47 L) and hydroxylamine sulfate (6.430 kg, 39.17 mol), the mixture was stirred at 25°C. A solution of potassium carbonate (3.87 Kg, 27.98 mol) in water (5.05 L) was slowly added to the reaction mixture to form a thick white mixture that was stirred at 20°C. A solution of methyl 2-fluoro-6-hydroxybenzoate (2) (0.952 Kg, 5.60 mol) in methanol (9.52 L) was slowly added to the reactor resulting in mild off gassing. The reaction mixture was then heated to 35°C and stirred for 20 h. The reaction mixture was cooled to 15°C and stirred for 1 h. The mixture was filtered to remove inorganic material. The reactor was rinsed with methanol (2.86 L) and the tank rinse was used to wash the inorganic cake.
Analysis of the cake indicated that it contained product. To a 20L reactor was charged methanol (10 L) and the inorganic cake and the mixture was stirred at 25°C for 30 min. The mixture was filtered and the cake washed with methanol (3 L).
The combined filtrates were charged back into the reactor and concentrated under vacuum with the jacket temperature set at 40°C until approximately 10 L remained. The mixture was held at 25°C and cone. HCI (5.51 L) was added. The reactor was cooled to 15°C and stirred for 2 h. The white slurry was filtered and the resulting product cake was washed with water (4.76L), blown dry with nitrogen and then dried in a vacuum oven at 40°C for 12 h. The desired product (3) (747 g, 4.36 mol), was isolated in 78% yield. 1 H NMR (400 MHz, CD3OD) δ ppm 4.91 (s, 3H), 6.63 (ddd, J=10.9, 8.5, 0.8, 1 H), 6.72 (dt, J=8.2, 0.8, 1 H), 7.31 (td, J=8.2, 6.6, 1 H); MS 172.040 (m+1 ).
4-Fluorobenzo[d]isoxazol-3-ol (4): To a 20L jacketed reactor were charged tetrahydrofuran (2.23 L) and 1 ,1 ‘-carbonyldiimidazole (0.910 Kg, 5.64 mol). The resulting mixture was stirred at 20°C. Then a solution of 2-fluoro-N,6-dihydroxybenzamide (3) (744 g, 4.34 mol) in tetrahydrofuran (4.45 L) was slowly charged to the reactor maintaining the temperature below 30°C and stirred at 25°C for 30 min during which some off gassing was observed. The reaction mixture was heated to 60°C over 30 min and stirred for 6 h. The reactor was cooled to 20°C followed by the addition of 1 N aqueous hydrogen chloride (7.48L) over 15 min to adjust the pH to 1. The jacket temperature was set to 35°C and the reaction mixture concentrated under vacuum to remove approximately 6.68L of THF. The reactor was cooled to 15°C and stirred for 1 h. The resulting white slurry was filtered, the cake was washed with water (3.71 L) and dried in a vacuum oven at 40°C for 12 h. The desired product, (4) (597 g, 3.90 mol), was isolated in 90% yield. 1 H NMR (400 MHz, CD3OD) δ ppm 4.93 (b, 1 H), 6.95 (dd, J=10.1 , 8.6, 1 H), (d, J=8.6, 1 H), 7.52-7.57 (m, 1 H); LRMS 154.029 (m+1 ).
Tert-butyl 4-(tosyloxymethyl)piperidine-1-carboxylate (5): To a 20L jacketed reactor were charged dichloromethane (8 L), N-boc-4-piperdine methanol (0.982 Kg, 4.56 mol) and p-toluenesulfonyl chloride (0.970 Kg, 5.09 mol) and the resulting mixture was stirred at 20°C for 5 min. Triethylamine (0.94 Kg, 9.29 mol) was added to the reactor via an addition funnel and the resulting deep red solution was stirred at 25°C for 16 h. A solution of sodium carbonate (0.96 Kg, 9.06 mol) in water (7.04 L) was charged to the reaction mixture and stirred for 1 h at 20°C. The phases were split and the organic layer washed with brine (6 L) and concentrated at 40°C to a low stir volume. Dimethylacetamide (2 L) was charged to the reactor and concentration continued under full vacuum at 40°C for 1 h. The solution of tert-butyl 4-(tosyloxymethyl)piperidine-l -carboxylate (5) in dimethyl acetamide was held for further processing. Yield was assumed to be 100% with approximately
90% potency. A sample was pulled and concentrated to dryness for purity analysis. 1 H NMR (400 MHz, CDCI3) δ ppm 1 .02-1 .12 (m, 2H), 1.14 (s, 9H), 1 .59-1.64 (m, 2H), 1.75-1.87 (m, 1 H), 2.43 (s, 3H), 2.55-2.75 (m, 2H), 3.83 (d, J=6.7, 2H), 3.95-4.20 (b, 2H), 7.33 (d, 8.6, 2H), 7.76 (d, 8.2, 2H); 13C NMR (400 MHz, CDCI3) δ ppm 21 .64, 28.15, 28.39, 35.74, 73.97, 79.50, 126.99, 127.84, 129.86, 132.84, 144.84, 154.63; LRMS 739.329 (2m+1 ).
Tert-butyl 4-((4-fluorobenzo[d]isoxazol-3-yloxy)methyl)piperidine-1-carboxylate (6): To a 20L jacketed reactor were charged dimethylacetamide (4.28 L), tert-butyl 4-(tosyloxymethyl)piperidine-1 -carboxylate (5) (1.68 Kg, 4.56 mol), 4-fluorobenzo[d]isoxazol-3-ol (4) (540 g, 3.51 mol), and potassium carbonate (960 g, 6.98 mol) resulting in a thick beige slurry. The reaction mixture was heated to 50°C and stirred for 20 h and then cooled to 20°C, followed by the addition of water (7.5 L) and ethyl acetate (5.37 L). After mixing for 15 min, the phases were settled and split. The organic layer was washed with water (5.37 L), sending the aqueous wash to waste. The organic mixture was distilled under vacuum with a maximum jacket temperature of 40°C until approximately 5 L remained in the reactor. Methanol (2.68 L) was added and the resulting solution concentrated under vacuum to about 3 L of a yellow oil. Methanol (2.68 L) was charged to the reactor and the resulting solution was stirred at 25°C for 15 min. Water (0.54 L) was added over 15 min resulting in a white slurry. The mixture was cooled to 15°C, stirred for 1 h and then filtered. The filter cake was washed with a solution of water (0.54 L) in methanol (2.14 L), then air dried for 30 min, transferred to a vacuum oven and dried at 40°C for 12 h. The desired product, (6) (746 g, 2.13 mol), was isolated in 61 % yield. 1 H NMR (400 MHz, CDCI3) δ ppm 1.23-1 .37 (m, 2H), 1 .45 (s, 9H), 1 .78-1 .88 (m, 2H), 2.04-2.17 (m, 1 H), 2.67-2.83 (m, 2H), 4.02-4.26 (m, 2H), 4.28 (d, 6.6, 2H), 6.89 (dd, J=8.6, 7.5, 1 H), 7.21 (d, J=9, 1 H), (td, 8.6, 4.9); LRMS 351.171 (m+1 ).
(R)-Tert-butyl 4-((4-(tetrahydrofuran-3-yloxy)benzo[d]isoxazol-3-yloxy)methyl)piperidine-1-carboxylate (8): To a 20 L glass reactor with the jacket set to 20°C were charged (R)-tetrahydrofuran-3-ol (7) (297 g, 3.37 mol) and dimethylacetamide (5.1 L). 2.0 M sodium bis(trimethylsilyl)amide in THF (1.37 L, 2.74 mol) was slowly added via an addition funnel while maintaining a pot temperature less than 30°C. The resulting orange/red solution was stirred at 25°C for 30 min. Then, tert-butyl 4-((4-fluorobenzo[d]isoxazol-3-yloxy)methyl)piperidine-1 -carboxylate (6) (640.15 g, 1.83 mol) was charged and the reaction mixture was stirred at 25°C for 16 h. The reaction mixture was cooled to 20°C and water (6.4 L) was slowly added over 45 min maintaining a pot temperature of less than 35°C. Ethyl acetate (6 L) was added and the biphasic mixture was stirred for 15 min and then separated. The aqueous layer was back extracted with additional ethyl acetate (4 L). The combined organics were then washed with water (5 L) and a 20% brine solution (5 L). The organic mixture was concentrated under vacuum with the jacket temperature set to 40°C to approximately 3 L and held for further processing. Quantitative yield of the desired product, (8) (0.76 Kg, 1 .82 mol), in ethyl acetate was assumed. A sample was pulled and concentrated to dryness for purity analysis. 1 H NMR (400 MHz, CDCI3) δ ppm 1 .25-1.38 (m, 2H), 1 .44 (s, 9H), 1.76-1 .84 (m, 2H), 1 .89-1.97 (b, 1 H), 1 .99-2.12 (m, 1 H), 2.14-2.28 (m, 2H), 2.63-2.84 (m, 2H), 3.90-4.21 (m, 6H), 4.24 (d, J=6.3, 2H), 5.00-5.05 (m, 1 H), 6.48 (d, J=8.2, 1 H), 6.98 (d, J=8.6, 1 H), 7.37 (t, J=8.2, 1 H); LRMS 419.216 (m+1 ).
(R)-3-(Piperidin-4-ylmethoxy)-4-(tetrahydrofuran-3-yloxy)benzo[d]isoxazole 4-methylbenzenesulfonate (9): To a 20L jacketed reactor charged ethyl acetate (6.1 L), (R)-tert-butyl 4-((4-(tetrahydrofuran-3-yloxy)benzo[d]isoxazol-3-yloxy)methyl)piperidine-1 -carboxylate (8) (0.76 kg, 1 .82 mol) and p-toluenesulfonic acid monohydrate (0.413 kg, 2.17 mol) and stirred at 20°C for 30 min. The reactor jacket was heated from 20 to 65°C over
1 h and then held at 65°C for 16 h. The reactor was cooled to 15°C over 1 h and granulated for 2 h. The resulting slurry was filtered, the cake was washed with EtOAc (3 L) and then air dried on the filter for 30 min. The cake was transferred to a vacuum oven and dried at 40°C for 12 h. The desired product, (9) (854 g, 1.74 mol), was isolated in 96% yield (two steps). 1 H NMR (400
MHz, CD3OD) δ ppm 1.54-1 .67 (m, 2H), 2.04-2.18 (m, 3H), 2.19-2.36 (m, 2H), 2.33 (s, 3H), 3.01 -3.12 (m, 2H), 3.41-3.50 (m, 2H), 3.86-4.01 (m, 4H), 4.26 (d, J=6.3, 2H), 4.90 (s, 2H), 5.14-5.19 (m, 1 H), 6.72 (d, J=8.2, 1 H), 7.02 (d, J=8.6, 1 H), 7.21 (d, J=7.8, 2H), 7.48 (t, J=8.6, 1 H), 7.70 (d, J=8.2, 2H); LRMS 319.165 (m+1 ).
(R)-4-((4-((4-(Tetrahydrofuran-3-yloxy)benzo[d]isoxazol-3-yloxy)methyl)piperidin-1-yl)methyl)tetrahydro-2H-pyran-4-ol (11): To a
20L jacketed reactor were charged water (7.5 L) and sodium carbonate (0.98 kg); the mixture was stirred at 20°C until all solids had dissolved. Then (R)-3-(piperidin-4-ylmethoxy)-4-(tetrahydrofuran-3-yloxy)benzo[d]isoxazole 4-methylbenzenesulfonate (9) (750 g, 1 .53 mol) and ethyl acetate (6.0 L) were added to the reactor and stirred at 20°C for 30 min. The phases were split and the lower aqueous layer was back extracted twice with ethyl acetate (6.0 L and then 3.75 L). The organic layers were combined in the 20L reactor and washed twice with brine (3.0 L). The ethyl acetate solution was concentrated to under vacuum at 45°C to a low stir volume. Isopropyl alcohol (3.75 L) was added and concentration continued until 2 L remained in the reactor.
Additional isopropyl alcohol (2.75 L) was added and the mixture cooled to 25°C. To the reactor was charged 1 ,6-dioxaspiro[2.5]octane (10) (260 g, 2.29 mol) and the resulting solution heated to 50°C and stirred for 16 h. The reaction mixture was cooled to 30°C and water (15 L) was added over 60 min. Product crystallized from solution and the resulting slurry was cooled to 15°C over 1 h and then granulated for 4 h. The product was filtered and washed with water (3.75 L). The cake was blown dry with nitrogen for 30 min and then transferred to a vacuum oven and dried at 40°C for 12 h. The desired product, (11 ) (588 g, 1 .36 mol), was isolated in 89% yield.
1 H NMR (400 MHz, CDCI3) δ ppm 1 .41-1 .63 (m, 6H), 1.71 -1.81 (m, 2H), 1.81 -1.94 (m, 1 H), 2.17-2.26 (m, 2H), 2.33 (s, 2H), 2.4 (td, J=1 1.7, 2.3, 2H), 2.92 (d, J=1 1 .8, 2H), 3.46 (s, 1 H), 3.71-3.84 (m, 4H), 3.91 -4.10 (m, 4H), 4.24 (d, J=5.9, 2H), 5.03-5.08 (m, 1 H), 6.50 (d, J=8.2, 1 H), 7.00 (d, J=8.2, 1 H), 7.38 (t, J=8.2, 1 H);
13C NMR (400 MHz, CDCI3) δ ppm 29.1 1 , 33.10, 35.20, 36.92, 36.96, 56.15, 63.93, 67.14, 67.46, 68.27, 72.94, 74.06, 78.37, 103.17, 105.15, 131.71 , 152.71 , 166.02, 166.28;
LRMS 433.232 (m+1 ).
Example 2: Synthesis of iR)-4-ii4-i(4-itetrahvdrofuran-3-yloxy)benzord1isoxazol-3-yloxy)methyl)piperidin-1 -yl)methyl)tetrahvdro- 2H-pyran-4-ol

5-Hydroxy-2,2-dimethyl-benzo[1,3]dioxin-4-one: Thionyl chloride (83.8 g, 0.71 mol) was slowly added to a solution of 2,6-dihydroxy-benzoic acid (77 g, 0.5 mol), acetone (37.7 g, 0.65 mol) and DMAP (3.1 g, 0.025 mol) in dimethoxyethane (375 mL). The mixture was stirred at RT for 7 h. The residue obtained after concentration under reduced pressure was dissolved in ethyl
acetate and washed with water and aqueous saturated sodium bicarbonate solution. The organic layer was dried (Na2S04) and concentrated to afford 79 g desired product as a red solid (81 % yield). 1 H NMR (400 MHz, CDCI3) δ ppm 1 .68 (s, 6H), 6.37 (dd, J=8, 0.8, 11-1) 6.56 (dd, J=8, 0.8, 1 H), 7.34 (t, J=8, 1 H), 10.27( brs, 1 H).
2,2-Dimethyl-5-[(R)-(tetrahydro-furan-3-yl)oxy]-benzo[1,3]dioxin-4-one:
Diethyl azodicarboxylate (130.5 g, 0.75 mol) was added in a dropwise fashion to a mixture of 5-hydroxy-2,2-dimethyl-benzo[1 ,3]dioxin-4-one (100 g, 0.51 mol), triphenylphosphine (196.5 g, 0.75 mol), and (S)-tetrahydro-furan-3-ol (44 g, 0.5 mol) in 600 ml. of anhydrous THF. The resulting mixture was stirred at RT for 18 h. The solvent was removed under reduced pressure and the crude material was purified on a silica gel flash column, eluting with petroleum ether/ ethyl acetate (15:1 -> 3:1 ). 86 g (65% yield) of product was isolated as a colorless oil. 1 H NMR (400 MHz, CDCI3) δ ppm 1.67 (s, 6H), 2.30 (m, 2H), 4.2 (m, 4H) 4.97 (m, 1 H), 6.49 (d, J=8.4, 1 H) 6.51 (d, J=8.4, 1 H), 7.39 (t,
J=8.4, 1 H).
2-Hydroxy-6-[(R)-(tetrahydro-furan-3-yl)oxy]-benzoic acid methyl ester: Potassium carbonate (134.8 g, 0.98 mol) was added to a solution of 2,2-dimethyl-5-[(R)-(tetrahydro-furan-3-yl)oxy]-benzo[1 ,3]dioxin-4-one (86 g, 0.33 mol) in 1 L methanol. The mixture was stirred at RT for 2 h, then concentrated in vacuo. The residue was dissolved in ethyl acetate and washed with aqueous ammonium chloride solution. The organic layer was dried (Na2S04) and concentrated to afford 72 g of the product as a yellow solid (92% yield). 1 H NMR (400 MHz, CDCI3) δ ppm 2.20 (m, 2H), 3.99 (s, 3H), 4.80(m, 4H). 4.94 (m, 1 H), 6.31 (dd, J=8.4, 0.8, 1 H), 6.59 (dd, J=8.4, 0.8, 1 H), 7.30 (t, J=8.4, 1 H).
2,N-Dihydroxy-6-[(R)-(tetrahydro-furan-3-yl)oxy]-benzamide: Potassium carbonate (121 g. 0.867mmol) was added portionwise to a solution of hydroxylamine sulfate (120 g, 0.732 mol) in 360 ml. of water at 0°C. After stirring for 30 min, sodium sulfite (3.74 g, 0.029 mol) and a solution of 2-hydroxy-6-[(R)-(tetrahydro-furan-3-yl)oxy]-benzoic acid methyl ester (35 g, 0.146 mol) in 360 ml. of methanol were added and the mixture was stirred at 50°C for 30 h. Methanol was removed from the cooled reaction mixture under reduced pressure and the resulting aqueous layer was acidified with 2N HCI. The aqueous layer was extracted with ethyl acetate and the organic layer was dried (Na2S04) and concentrated to afford 25 g (76% yield ) of the product as a yellow solid. 1 H NMR (400 MHz, CDCI3) δ ppm 2.00 (m, 1 H), 2.15 (m, 1 H), 3.80 (m, 4H), 5.05 (m, 1 H), 6.48 (d, J=8, 1 H), 6.49 (d, J=8, 1 H), 7.19 (t, J=8, 1 H), 10.41 (brs, 1 H), 1 1.49 (brs, 1 H); LRMS m/z 239 (m+1 ).
4-[(R)-(Tetrahydro-furan-3-yl)oxy]-benzo[d]isoxazol-3-ol: A solution of 2, N-dihydroxy-6-[(R)-(tetrahydro-furan-3-yl)oxy]-benzamide (25 g, 0.105 mol) in 250 ml. of THF was heated to 50°C. Carbonyl diimidazole was added portionwise and the resulting mixture was stirred at 50°C for 14 h. After cooling to RT, 100 ml. of 2N HCI was added and the aqueous layer was extracted with ethyl acetate. The combined organic layers were then extracted three times with 10% aqueous potassium carbonate. The potassium carbonate aqueous extracts were washed with ethyl acetate and then acidified to pH 2 – 3 with 2N HCI. The acidified aqueous layer was extracted with ethyl acetate. The ethyl acetate extracts were washed with brine, dried (Na2S04) and concentrated to afford 20 g of product as a yellow solid (43% yield). 1 H NMR (400 MHz, CDCI3) δ ppm 2.20 (m, 2H), 3.89 (m, 1 H), 4.01 (m, 3H), 5.05 (m, 1 H), 6.48 (d, J=7.6, 1 H). 6.92 (d, J=7.6, 1 H), 7.37 (t, J=7.6, 1 H); LRMS m/z 222 (m+1 ).
4-{4-[(R)-(Tetrahydro-furan-3-yl)oxy]-benzo[d]isoxazol-3-yloxymethyl}-piperidine-1-carboxylic acid tert-butyl ester: Diethyl azodicarboxylate (15.6 g, 0.09 mol) was added to a mixture of 4-[(R)-(tetrahydro-furan-3-yl)oxy]-benzo[d]isoxazol-3-ol (10 g, 0.045 mol), 4-hydroxymethyl-piperidine-1 -carboxylic acid tert-butyl ester (1 1.6 g, 0.054 mol) and triphenylphosphine (23.5 g, 0.09 mol) in 300 mL THF. After the addition was complete the mixture was heated at reflux for 18 h. After concentration in vacuo, the crude product was purified on a silica gel flash column, eluting with petroleum ether/ ethyl acetate (15:1 -» 5:1 ) to afford 22 g of the product as an oil (51 % yield). 1 H NMR (400 MHz, CDCI3) δ ppm 1.25 (m, 2H), 1.39 (s, 9H), 1.76 (m, 2H), 1.99 (m, 1 H). 2.15 (m, 2H), 2.70 (bt, J=1 1.6, 2H), 3.95 (m, 4H). 4.13 (m, 2H). 4.34 (d J=6.4, 2H), 4.98 (m, 1 H), 6.43 (d, J=8, 1 H), 6.93 (d, J=8, 1 H), 7.31 (t, J=8, 1 H).
3-(Piperidin-4-ylmethoxy)-4-[(R)-(tetrahydro-furan-3-yl)oxy]-benzo[d]isoxazole: A 0°C solution of 4-{4-[(R)-(tetrahydro-furan-3-yl)oxy]-benzo[d]isoxazol-3-yloxymethyl}-piperidine-1 -carboxylic acid tert-butyl ester in 500 mL ether was treated with a saturated solution of HCI (g) in 200 mL ether. After addition was complete, the mixture was warmed to RT and stirred for 16 h. The reaction mixture was filtered. The white solid was washed with ethyl acetate followed by ether and dried to yield 15 g (81 % yield) of the desired product as a white solid. 1 H NMR (400 MHz, CD3OD) 5 ppm 1 .51 – 1.69 (m, 2 H) 2.04 – 2.19 (m, 3 H) 2.22 – 2.37 (m, 2 H) 2.99 – 3.14 (m, 2 H) 3.40 – 3.51 (m, 2 H) 3.85 – 4.02 (m, 4 H) 4.25 – 4.31 (m, 2 H) 5.17 (td, J= >1^ , 1 .56 Hz, 1 H) 6.72 (d, J=8.00 Hz, 1 H) 7.01 (d, J=8.59 Hz, 1 H) 7.47 (t, J=8.20 Hz, 1 H); LRMS m/z 319 (m+1 ).
4-(4-{4-[(R)-(Tetrahydro-furan-3-yl)oxy]-benzo[d]isoxazol-3-yloxymethyl}-piperidin-1-ylmethyl)-tetrahydro-pyran-4-ol: 1 ,6-Dioxa-spiro[2.5]octane (Focus Synthesis; 9.7 g, 0.084 mol) and triethylamine (8.6 g, 0.084 mol) were added to a solution of 3-(piperidin-4-ylmethoxy)-4-[(R)-(tetrahydro-furan-3-yl)oxy]-benzo[d]isoxazole (15 g, 0.042 mol) in 200 mL methanol. The resulting solution was heated at reflux for 18 h. The cooled mixture was concentrated and ethyl acetate and water were added to the residue. The layers were separated and the organic extracts were washed with brine, dried (Na2S04) and concentrated to provide 17 g crude product as a yellow oil. The crude material was purified by prep HPLC to afford 10 g of the desired product as a white solid. (50% yield).
1 H NMR (400 MHz, CDCI3) δ ppm 1.41 -1.63 (m, 6H), 1.71-1.81 (m, 2H), 1 .81 -1 .94 (m, 1 H), 2.17-2.26 (m, 2H), 2.33 (s, 2H), 2.4 (td, J=1 1 .7, 2.3, 2H), 2.92 (d, J=1 1.8, 2H), 3.46 (s, 1 H), 3.71-3.84 (m, 4H), 3.91-4.10 (m, 4H), 4.24 (d, J=5.9, 2H), 5.03-5.08 (m, 1 H), 6.50 (d, J=8.2, 1 H), 7.00 (d, J=8.2, 1 H), 7.38 (t, J=8.2, 1 H);
13C NMR (101 MHz, CDCI3) δ ppm 29.1 1 , 33.10, 35.20, 36.92, 36.96, 56.15, 63.93, 67.14, 67.46, 68.27, 72.94, 74.06, 78.37, 103.17, 105.15, 131.71 , 152.71 , 166.02, 166.28.
PAPER
Two Routes to 4-Fluorobenzisoxazol-3-one in the Synthesis of a 5-HT4Partial Agonist
† Groton Laboratories, Worldwide Research & Development, Pfizer Inc., Eastern Point Road, Groton, Connecticut 06340,United States
‡ Porton Fine Chemical, 1 Fine Chemical Zone, Chongqing Chemical Industrial Park, Changshou, Chongqing 401221China
Org. Process Res. Dev., Article ASAP
DOI: 10.1021/acs.oprd.5b00389
Publication Date (Web): February 2, 2016
Copyright © 2016 American Chemical Society
*E-mail: daniel.w.widlicka@pfizer.com.
http://pubs.acs.org/doi/abs/10.1021/acs.oprd.5b00389

A potent 5-HT4 partial agonist, 1 (PF-04995274), targeted for the treatment of Alzheimer’s disease and cognitive impairment, has been prepared on a multi-kilogram scale. The initial synthetic route, that proceeded through a 4-substituted 3-hydroxybenzisoxazole core, gave an undesired benzoxazolinone through a Lossen-type rearrangement. Route scouting led to two new robust routes to the desired 4-substituted core. Process development led to the efficient assembly of the API on a pilot plant scale under process-friendly conditions with enhanced throughput. In addition, crystallization of a hemicitrate salt of the API with pharmaceutically beneficial properties was developed to enable progression of clinical studies.
REFERNCES
Noguchi, H.; Waizumi, N. Preparation of benzisoxazole derivatives for treatment of 5-HT4 mediated disorders. PCT Int. Appl. WO/2011/101774 A1, 20110825
////////PF-04995274, PF 04995274, PFIZER, Alzheimer’s type dementia, PHASE 1
c1cc2c(c(c1)O[C@@H]3CCOC3)c(no2)OCC4CCN(CC4)CC5(CCOCC5)O
RQ 00000010 for the treatment of GERD, functional dyspepsia and chronic constipation.

RQ 00000010
CAS 907607-22-1
| Molecular Formula: | C22H27F3N2O6 |
|---|---|
| Molecular Weight: | 472.45479 g/mol |
HSMMHNBGQLGCBY-UHFFFAOYSA-N;
RaQualia Pharma Inc
PFIZER INNOVATOR
RQ-00000010; RQ-10
4-{[4-({[4-(2,2,2-trifluoroethoxy)-1,2-benzisoxazol-3-yl]oxy}methyl)piperidin-1-yl]methyl}tetrahydro-2H-pyran-4-carboxylic acid
4-{[4-({[4-(2,2,2-trifluoroethoxy)-1,2-benzisoxazol-3-yl]oxy}methyl)piperidin-1-yl]methyl}tetrahydro-2H-pyran-4-carboxylic acid
ΦirΦfff^fΣ^^-TrifluoroethoxyVi.a-benzisoxazol-S-vnoxylmethvπpiperidin-i-vπmethylltetrahydro-2H-pyran-4-carboxylic acid
4-[[4-[[4-(2,2,2-trifluoroethoxy)-1,2-benzoxazol-3-yl]oxymethyl]piperidin-1-yl]methyl]oxane-4-carboxylic acid
PHASE 1 for the treatment of GERD, functional dyspepsia and chronic constipation.
Useful for treating diseases mediated by 5-HT4 receptor activity eg such as gastroesophageal reflux disease (GERD), gastric motility disorder, dyspepsia, constipation, esophagitis, diabetes, CNS and cardiovascular diseases.
RaQualia, following its spin-out from Pfizer, is developing RQ-00000010, a 5-HT4 receptor partial agonist, for the treatment of gastric motility disorders, including gastroparesis associated with Parkinson’s disease.
In November 2015, the drug was reported to be in phase 1 clinical development. RaQualia and licensee CJ CheilJedang are investigating the drug for the treatment of GERD, functional dyspepsia and chronic constipation.
4-{[4-({[4-(2,2,2-trifluoroethoxy)-1,2-benzisoxazol-3-yl]oxy}methyl)piperidin-1-yl]methyl}tetrahydro-2H-pyran-4-carboxylic acid is disclosed in PL1 as a 5-HT4 receptor agonist, which is useful in the treatment or alleviation of disease conditions mediated by 5-HT4 receptor activity; in particular 5-HT4 receptor agonistic activity, such as gastroesophageal reflux disease (GERD), gastrointestinal disease, gastric motility disorder, non-ulcer dyspepsia, functional dyspepsia (FD), irritable bowel syndrome (IBS), constipation, dyspepsia, esophagitis, gastroesophageal disease, gastritis, nausea, central nervous system disease, Alzheimer’s disease, cognitive disorder, emesis, migraine, neurological disease, pain, cardiovascular disorders, cardiac failure, heart arrhythmia, diabetes, and apnea syndrome (See NPL 1 to 13 and PL 2 to 7).
Simply an white solid has been produced in the previously known methods of preparing 4-{[4-({[4-(2,2,2-trifluoroethoxy)-1,2-benzisoxazol-3-yl]oxy}methyl)piperidin-1-yl]methyl}tetrahydro-2H-pyran-4-carboxylic acid, described in PL 1. A generic disclosure of pharmaceutically-acceptable salts of 4-{[4-({[4-(2,2,2-trifluoroethoxy)-1,2-benzisoxazol-3-yl]oxy}methyl)piperidin-1-yl]methyl}tetrahydro-2H-pyran-4-carboxylic acid of the instant application is disclosed, and the free base of the compound of the instant invention is disclosed and claimed, in PL 1 having an international filing date of December 6, 2006, assigned to the assignee hereof. Thus any salts of the compound have been neither pacifically described nor synthesized in prior art.
It has been found that HCl-salt, HBr-salt, pTSA-salt and EDSA-salt of 4-{[4-({[4-(2,2,2-trifluoroethoxy)-1,2-benzisoxazol-3-yl]oxy}methyl)piperidin-1-yl]methyl}tetrahydro-2H-pyran-4-carboxylic acid shown below, can be isolated as a crystalline form which has advantageous properties such as ease of making a formulation, high solubility, and good stability. In addition the salts of the present invention are more easily purified than a non-crystalline form disclosed in PL 1 (WO2006/090224) and crystalline form disclosed in PL 3 (WO2012/157288).
Patent Literature
{PL 1} WO2006/090224.
{PL 2} US Patent No. 6,106,864.
{PL 3} WO2012/157288
{PL 4} WO00/35298.
{PL 5} WO91/11172.
{PL 6} WO94/02518.
{PL 7} WO98/55148.
Non Patent Literature
{NPL 1} Bockaert J. et al., TiPs 13; 141-145, 1992.
{NPL 2} Ford A. P et al., Med. Res. Rev. 13: 633-662, 1993.
{NPL 3} Gullikson G. W. et al., Drug Dev. Res. 26; 405-417, 1992.
{NPL 4} Richard M. Eglen et al., TiPs 16; 391-398, 1995.
{NPL 5} Bockaert J. et al., CNS Drugs 1; 6-15, 1994.
{NPL 6} Romanelli M. N. et al., Arzheim Forsch./Drug Res., 43; 913-918, 1993.
{NPL 7} Kaumann A. J. et al., Naunyn-Schmiedebergs Arch Pharmacol., 344; 150-159, 1991.
{NPL 8} Remington’s Pharmaceutical Sciences, 19th Edition (Mack Publishing Company, 1995).
{NPL 9} Expert Opinion in Therapeutic Patents, H (6), 981-986, by Liang and Chen (2001).
{NPL 10} Tablets, Vol. 1, by H. Lieberman and L. Lachman (Marcel Dekker, New York, 1980).
{NPL 11} Pharmaceutical Technology On-line, 25(2), 1-14, by Verma et al. (2001).
{NPL 12} J Pharm Sci, 88 (10), 955-958, by Finnin and Morgan (October 1999).
{NPL 13} Evrard, B., et al., Journal of Controlled Release 96 (3), pp. 403-410, 2004.
{NPL 14} Byrn S. R. et al., Solid-State Chemistry of Drugs 2nd ed., pp 3-43 and 461-503, 1999, SSCI, Inc.

PATENT
WO2006090224
| PFIZER JAPAN INC. |
EXAMPLE 1 :
ΦirΦfff^fΣ^^-TrifluoroethoxyVi.a-benzisoxazol-S-vnoxylmethvπpiperidin-i-vπmethylltetrahydro-2H-pyran-4-carboxylic acid
Step 1. Methyl 2-hvdroxy-6-(2,2,2-trifluoroethoxy)benzoate
A mixture of 5-hydroxy-2,2-dimethyl-4tø-1 ,3-benzodioxin-4-one (123 g, 633 mmol, Synth. Commun.
1994, 24t 1025), potassium carbonate (262 g, 1.9 mol) and 2,2,2-trifluoroethyl trifluoromethanesulfonate (95.8 mL, 665 mmol) in Λ/,Λ/-dimethylformamide (600 mL) was stirred at 50 0C for 30 min. Then methanol (300 ml_) was added to the mixture, and stirring was continued for 5 h at that temperature. After cooling to room temperature, the mixture was diluted with water (500 ml_) and neutralized with 2Λ/ hydrochloric acid. Product was extracted with a mixture of ethyl acetate-hexane (5:1 , 500 mL x 3). Combined organic layers were washed with water (500 mL), dried over magnesium sulfate and concentrated under reduced pressure. The residual solid was recrystallized from methanol-water to afford 125 g (79%) of the desired product as colorless crystals.
1H-NMR (CDCI3) δ: 11.47 (1 H, s), 7.36 (1 H, t, J = 8.4 Hz), 6.72 (1 H, dd, J = 1.1 , 8.4 Hz), 6.38 (1 H, q, J = 8.1 Hz), 4.36 (2 H, q, J= 8.0 Hz), 3.96 (3 H, s).
MS (ESI) m/z: 251 (M+H) +, 249 (M-H) \
Step 2. 4-(2,2,2-Trifluoroethoxy)-1 ,2-benzisoxazol-3-ol
To a solution of hydroxylamine sulfate (120 g, 732 mmol) in water (360 mL) was added potassium carbonate (121 g, 875 mmol) at 0 0C. After 30 min of stirring, sodium sulfite (3.74 g, 29.7 mmol) and a methanolic solution of methyl 2-hydroxyl-6-(2,2,2-trifluoroethoxy)benzoate (36.4 g, 146 mmol, EXAMPLE 1 , step 1 , in 360 mL of methanol) were added to the mixture. Then the mixture was warmed to 50 °C and stirred for 30 h. After cooling to room temperature, reaction mixture was partially concentrated to approx. 2/3 volume and acidified with 2Λ/ hydrochloric acid. Product was extracted three times with ethyl acetate. Combined organic layer was washed with brine, dried over sodium sulfate and concentrated under reduced pressure to afford the desired product as a crystalline solid. Crude product (36.3 g) was used for the next step without further purification.
The described above crude product (5.56 g, 22.14 mmol) was suspended in tetrahydrofuran (22.0 mL) and heated at 50 °C. 1 ,1 ‘-carbonyldiimidazole (7.54 g, 46.48 mmol) was added to the suspension at 50 °C. After addition, the mixture was stirred at 50 0C for 14 h, the mixture was cooled to room temperature. 2Λ/ hydrochloric acid was added to the mixture and extracted with ethyl acetate. The organic layer was extracted with 10% aq. potassium carbonate (100 mL x 5). The water layers were acidified with 2Λ/ hydrochloric acid and extracted with ethyl acetate (200 mL x 2). The extracts were combined and dried over sodium sulfate and concentrated in vacuo to give brown solid. The residual solid was recrystallized from ethyl acetate/hexane to give 3.21 g (61 %) of the title compound as colorless needles.
1H-NMR (CDCl3) δ: 7.53 (1 H1 1, J = 8.5 Hz), 7.14 (1 H, d, J= 8.5 Hz), 6.73 (1 H, d, J = 7.9 Hz), 4.63 (2 H, q, J= 8.0 Hz), 3.83 (1 H, br).
MS (ESI) m/z: 234 (M+H) +, 232 (M-H) “.
Step 3. rMethoxy(tetrahydro-4H-pyran-4-ylidene)methoxyKtrimethyl)silane
To a stirred solution of diisopropylamine (5.2 mL, 37 mmol) in tetrahydrofuran (15 mL) was added dropwise n-butyllithium (1.6 M in hexane, 21 mL, 34 mmol) at 0 0C and stirred for 20 min. A mixture of methyl tetrahydro-2W-pyran-4-carboxylate (4.5 g, 31 mmol) and trimethylsilyl chloride (4.3 mL, 34 mmol) was added to the mixture at -40 0C, then trimethylsilyl chloride (0.4 mL, 0.3 mmol) was added to the mixture. The mixture was stirred at room temperature for 2 h. The volatile components were removed by evaporation and the residual mixture was filtered through a pad of celite washing with hexane. The filtrate was evaporated to give 6.9 g (quant.) of the title compound as a clear yellow oil.
1H-NMR (CDCI3) δ: 3.64-3.59 (4 H, m), 3.52 (3 H, s), 2.24 (2 H, t, J = 5.6 Hz), 2.15 (2 H, t, J = 5.4 Hz), 0.22 (9 H, s).
Step 4. Methyl 4-{f4-(hvdroxymeth’vDpiperidin-1 -yllmethylltetrahvdro^rt-pyran^-carboxylate
To a stirred mixture of piperidin-4-ylmethanol (5.0 g, 43.4 mmol), f-butyldimethylsilylchloride (7.2 g, 47.8 mmol), and triethylamine (7.3 ml_, 52.1 mmol) in dichloromethane (50 mL) was added 4-dimethylaminopyridine (530 mg, 4.3 mmol) at 0 0C. After being stirred at 0 0C for 2 h, 50 mL of water was added to the mixture. The mixture was extracted with dichloromethane (50 mL x 3) and the extracts were combined, dried over sodium sulfate, and concentrated in vacuo to give 10.2 g of a crude oil. The residual oil was dissolved with 86 mL of ethanol, and potassium carbonate (7.2 g, 52.1 mmol) and paraformaldehyde (1.56 g, 52.1 mmol) were added to the solution. After being stirred at room temperature for 2 days, the mixture was filtered and the filtrate was concentrated in vacuo to give a yellow oil. The residual oil was dissolved with 45 mL of acetonitrile and magnesium chloride (414 mg, 4.3 mmol) was added to the solution. [methoxy(tetrahydro-4H-pyran-4-ylidene)methoxy](trimethyl)silane (11.3 g, 52.1 mmol, EXAMPLE 1 , step 3) was added to the mixture at 0 0C. After being stirred at 0 0C for 20 h, 100 mL of 2Λ/ hydrochloric acid was added to the mixture. The mixture was stirred for 30 min and washed with diethyl ether (100 mL x 2). The water layer was neutralized with aq. ammonia and extracted with ethyl acetate (100 mL x 2). The extracts were combined and dried over sodium sulfate and concentrated in vacuo to give a yellow oil. The residual oil was purified by silica gel column chromatography (dichloromethane/methanol/aq. ammonia 400: 10: 1 ) to give 6.8 g (41%) of the title compound as a colorless waxy solid.
1H-NMR (CDCI3) δ: 3.75-3.90 (2 H, m), 3.71 (3 H, s), 3.40-3.55 (4 H, m), 2.73 (2 H, m), 2.49 (2 H, m), 2.10-2.25 (2 H, m), 1.95-2.10 (2 H, m), 1.50-1.70 (4 H, m), 1.30-1.50 (2 H, m), 1.10-1.30 (2 H, m).
MS (ESI) m/z: 272 (M+H) +.
Step 5. Methyl 4-{r4-((r4-(2,2,2-trifluoroethoxy)-1 ,2-benzisoxazol-3-vπoxy)methyl)piperidin-1 -yllmethyll-tetrahydro-2H-pyran-4-carboxylate
A mixture of 4-(2,2,2-trifluoroethoxy)-1 ,2-benzisoxazol-3-ol (230 mg, 1 mmol, EXAMPLE 1 , step
2), methyl 4-{[4-(hydroxymethyl)piperidin-1 -yl]methyl}tetrahydro-2/-/-pyran-4-carboxylate (270 mg, 1 mmol, EXAMPLE 1 , step 4), and cyanomethyltributylphosphorane (400 mg, 1.5 mmol) in toluene (1.0 mL) was stirred at 100 0C for 16 h. After cooling, the mixture was concentrated in vacuo to give a dark brown oil. The residual oil was purified by silica gel column chromatography (hexane/ethyl acetate 2 : 1 ) to give 250 mg (51 %) of the title compound as a white solid.
1H-NMR (CDCl3) δ: 7.44 (1 H, dd, J= 7.9, 8.4 Hz), 7.12 (1 H, d, J= 8.4 Hz), 6.61 (1 H, d, J= 7.9 Hz), 4.49 (2 H, q, J= 8.1 Hz), 4.24 (2 H, d, J= 6.4 Hz), 3.88-3.78 (2 H, m), 3.72 (3 H, s), 3.54-3.41 (2 H, m), 2.83-2.71 (2 H, m), 2.52 (2 H, s), 2.35-1.29 (11 H, m).
MS (ESI) m/z: 487 (M+H) +.
Step 6. 4-(r4-(ir4-(2,2,2-Trifluoroethoxy)-1 ,2-benzisoxazol-3-vπoxy)methyl)piperidin-1 -ylimethylltetrahydro-2H-pyran-4-carboxylic acid
A mixture of methyl 4-{[4-({[4-(2,2,2-trifluoroethoxy)-1 ,2-benzisoxazol-3-yl]oxy}methyl)piperidin-1-yl]methyl}tetrahydro-2/+pyran-4-carboxylate (89 mg, 0.18 mmol, EXAMPLE 1 , Step 5) in tetrahydrofuran (1 mL), methanol (1 ml_) and 2 Λ/ aq. sodium hydroxide (1 ml_) was stirred at 70 °C for 17 h. The mixture was neutralized with 2 N hydrochloric acid (1 mL) and formed precipitate was filtered.
The precipitate was triturated with diethylether to give 50 mg (58%) of the title compound as a white solid.
1H-NMR (DMSO-d6) δ: 7.59 (1 H1 dd, J= 8.1 , 8.4 Hz), 7.25 (1 H, d, J = 8.4 Hz), 6.94 (1 H, d, J = 8.1 Hz), 4.93 (2 H, q, J= 8.7 Hz), 4.19 (2 H, d, J= 5.9 Hz), 3.75-3.62 (2 H, m), 3.48-3.30 (2 H, m), 2.90-2.74 (2 H, m), 2.50 (2 H, s), 2.29-2.13 (2 H, m), 1.94-1.23 (9 H, m).
A signal due to CO2H was not observed.
MS (ESI) m/z: 473 (M+H) +, 471 (M-H) “.
m.p.: 171.7 °C.
IR (KBr) v: 2950, 1617, 1527, 1188, 1113 cm”1.
Anal. Calcd for C22H27N2O6F3: C, 55.93; H, 5.76; N, 5.93. Found: C, 55.72; H, 5.78; N, 5.80.
PATENT
WO2015174098
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015174098
PATENT
WO2014080633
http://www.google.com/patents/WO2014080633A1?cl=en
PATENT
WO 2015178020
The present invention relates to novel salts of 4-{[4-({[4-(2,2,2-trifluoroethoxy)-1,2-benzisoxazol-3-yl]oxy}methyl)piperidin-1-yl]methyl}tetrahydro-2H-pyran-4-carboxylic acid. More particularly, the invention relates to salt forms (HCl-salt, HBr-salt, p-toluenesulfonate salt and ethanedisulfonate salt), and to processes for the preparation of, compositions containing and to uses of, such salt forms.
EXAMPLE 1
Preparation of
4-{[4-({[4-(2,2,2-trifluoroethoxy)-1,2-benzisoxazol-3-yl]oxy}methyl)piperidin-1-yl]methyl}tetrahydro-2H-pyran-4-carboxylic acid according to the conventional process
A slurry of 4-{[4-({[4-(2,2,2-trifluoroethoxy)-1,2-benzisoxazol-3-yl]oxy}methyl)piperidin-1-yl]-methyl}tetrahydro-2H-pyran-4-carboxylic acid (1.326 kg, 2.807 mol, a white solid) in ethyl acetate (18.564 L) is dissolved at 70 oC. The solution is cooled to 64 oC during 35 min and 200 mg of seed crystal (0.423 mmol) is seeded to the mixture. The mixture is cooled to 40 oC over 5 h period and stirred at this temperature for 14.5 h. The slurry is gradually cooled to 19 oC during 6 h period and the mixture is stirred at this temperature for 46 h. The formed precipitate is collected by filtration and the filter cake is washed with 2.0 L of ethyl acetate. The filter cake is dried under reduced pressure at 50 oC to afford 1.140 kg of the desired crystalline form of 4-{[4-({[4-(2,2,2-trifluoroethoxy)-1,2-benzisoxazol-3-yl]oxy}methyl)piperidin-1-yl]-
methyl}tetrahydro-2H-pyran-4-carboxylic acid (86%).
1H-NMR (DMSO-d6) delta: 7.59 (1 H, dd, J = 8.1, 8.4 Hz), 7.25 (1 H, d, J = 8.4 Hz), 6.94 (1 H, d, J = 8.1 Hz), 4.93 (2 H, q, J = 8.7 Hz), 4.19 (2 H, d, J = 5.9 Hz), 3.75-3.62 (2 H, m), 3.48-3.30 (2 H, m), 2.90-2.74 (2 H, m), 2.50 (2 H, s), 2.29-2.13 (2 H, m), 1.94-1.23 (9 H, m).
A signal due to CO2H is not observed.
m.p. (DSC onset): 169 oC.
The temperature has a margin of error of +/- 1 oC.
Crystallinity by PXRD: Crystal (Figure 1): Main peaks at 2-Theta: 5.9, 9.3, 9.8, 11.9, 13.7, 14.3, 15.0, 17.8, 18.2-19.3, 19.7, 22.6, 23.4-24.5 and 24.9 (o ). Each peak has a margin of error of +/- 0.2.
IR nu (diffuse reflection) (Figure 6): 4389-4383, 3426, 2943-2937, 2120, 1904, 1724, 1614, 1535, 1508, 1437, 1420, 1287, 1261, 1221, 1180, 1121, 1094, 1059, 1022, 991, 974, 957, 934, 918, 868, 827, 783, 746, 731, 654, 638, 615, 588, 554, 542 and 507 cm-1. Each peak has a margin of error of +/- 2 cm-1.
Anal. Calcd for C22H27N2O6F3: C, 55.93; H, 5.76; N, 5.93. Found: C, 55.76; H, 5.74; N, 5.85.
PATENT
WO2012/157288
http://www.google.co.in/patents/WO2012157288A1?cl=pt-PT
EXAMPLE 1
Preparation of
4-{[4-({[4-(2,2,2-Trifluoroethoxy)-1,2-benzisoxazol-3-yl]oxy}methyl)piperidin-1-yl]methyl}-tetrahydro-2H-pyran-4-carboxylic acid according to the conventional process
A mixture of methyl 4-{[4-({[4-(2,2,2-trifluoroethoxy)-1,2-benzisoxazol-3-yl]oxy}methyl)piperidin-1-yl]methyl}-tetrahydro-2H-pyran-4-carboxylate (89 mg, 0.18 mmol, PCT WO2006090224 EXAMPLE 1, Step 5) in tetrahydrofuran (1 mL), methanol (1 mL) and 2 N aq. sodium hydroxide (1 mL) is stirred at 70 oC for 17 h. The mixture is neutralized with 2 N hydrochloric acid (1 mL) and formed precipitate is filtered. The precipitate is triturated with diethylether to give 50 mg (58%) of the title compound as a white solid.
1H-NMR (DMSO-d6) delta: 7.59 (1 H, dd, J = 8.1, 8.4 Hz), 7.25 (1 H, d, J = 8.4 Hz), 6.94 (1 H, d, J = 8.1 Hz), 4.93 (2 H, q, J = 8.7 Hz), 4.19 (2 H, d, J = 5.9 Hz), 3.75-3.62 (2 H, m), 3.48-3.30 (2 H, m), 2.90-2.74 (2 H, m), 2.50 (2 H, s), 2.29-2.13 (2 H, m), 1.94-1.23 (9 H, m).
A signal due to CO2H is not observed.
MS (ESI) m/z: 473 (M+H) +, 471 (M-H) –.
Anal. Calcd for C22H27N2O6F3: C, 55.93; H, 5.76; N, 5.93. Found: C, 55.72; H, 5.78; N, 5.80.
| Patent | Submitted | Granted |
|---|---|---|
| Benzisoxazole Derivatives [US2008207690] | 2008-08-28 | |
| 5-HT4 Receptor Agonist as a Prokinetic Agent [US2014051726] | 2012-03-23 | 2014-02-20 |
| Polymorph Form of 4-methyl)piperidin-1-yl]methyl}-tetrahydro-2H-pyran-4-carboxylic acid [US2014187583] | 2012-05-18 | 2014-07-03 |
see……….http://apisynthesisint.blogspot.in/2015/12/rq-00000010-for-treatment-of-gerd.html
/////c12c(cccc1onc2OCC3CCN(CC3)CC4(CCOCC4)C(=O)O)OCC(F)(F)F
C1CN(CCC1COC2=NOC3=C2C(=CC=C3)OCC(F)(F)F)CC4(CCOCC4)C(=O)O
Pfizer’s PF 04991532 a Hepatoselective Glucokinase Activator Clinical Candidate for Treating Type 2 Diabetes Mellitus

PF 04991532
GKA PF-04991532
(S)-6-{3-cyclopentyl-2-[4-(trifluoromethyl)-1H-imidazol-1-yl]propanamido}nicotinic acid
(S)-6-(3-Cyclopentyl-2-(4-(trifluoromethyl)-1H-imidazol-1-yl)propanamido)nicotinic Acid
(S)-6-(3-cyclopentyl-2-(4-(trifluoromethyl)-1H-imidazol-1-yl)propanamido)nicotinic acid
MW 396.36, MF C18 H19 F3 N4 O3
CAS 1215197-37-7
3-Pyridinecarboxylic acid, 6-[[(2S)-3-cyclopentyl-1-oxo-2-[4-(trifluoromethyl)-1H-imidazol-1-yl]propyl]amino]-
http://www.biochemj.org/content/441/3/881
Type 2 diabetes mellitus (T2DM) is a rapidly expanding public epidemic affecting over 300 million people worldwide. This disease is characterized by elevated fasting plasma glucose (FPG), insulin resistance, abnormally elevated hepatic glucose production (HGP), and reduced glucose-stimulated insulin secretion (GSIS). Moreover, long-term lack of glycemic control increases risk of complications from neuropathic, microvascular, and macrovascular diseases.
The standard of care for T2DM is metformin followed by sulfonylureas, dipeptidyl peptidase-4 (DPP-IV) inhibitors, and thiazolidinediones (TZD) as second line oral therapies. As disease progression continues, patients typically require injectable agents such as glucagon-like peptide-1 (GLP-1) analogues and, ultimately, insulin to help maintain glycemic control. Despite these current therapies, many patients still remain unable to safely achieve and maintain tight glycemic control, placing them at risk of diabetic complications and highlighting the need for novel therapeutic options.

Glucokinase (hexokinase IV) continues to be a compelling target for the treatment of type 2 diabetes given the wealth of supporting human genetics data and numerous reports of robust clinical glucose lowering in patients treated with small molecule allosteric activators. Recent work has demonstrated the ability of hepatoselective activators to deliver glucose lowering efficacy with minimal risk of hypoglycemia.
While orally administered agents require a considerable degree of passive permeability to promote suitable exposures, there is no such restriction on intravenously delivered drugs. Therefore, minimization of membrane diffusion in the context of an intravenously agent should ensure optimal hepatic targeting and therapeutic index.
Diabetes is a major public health concern because of its increasing prevalence and associated health risks. The disease is characterized by metabolic defects in the production and utilization of carbohydrates which result in the failure to maintain appropriate blood glucose levels. Two major forms of diabetes are recognized. Type I diabetes, or insulin-dependent diabetes mellitus (IDDM), is the result of an absolute deficiency of insulin. Type II diabetes, or non-insulin dependent diabetes mellitus (NIDDM), often occurs with normal, or even elevated levels of insulin and appears to be the result of the inability of tissues and cells to respond appropriately to insulin. Aggressive control of NIDDM with medication is essential; otherwise it can progress into IDDM.
As blood glucose increases, it is transported into pancreatic beta cells via a glucose transporter. Intracellular mammalian glucokinase (GK) senses the rise in glucose and activates cellular glycolysis, i.e. the conversion of glucose to glucose-6-phosphate, and subsequent insulin release. Glucokinase is found principally in pancreatic β-cells and liver parenchymal cells. Because transfer of glucose from the blood into muscle and fatty tissue is insulin dependent, diabetics lack the ability to utilize glucose adequately which leads to undesired accumulation of blood glucose (hyperglycemia). Chronic hyperglycemia leads to decreases in insulin secretion and contributes to increased insulin resistance. Glucokinase also acts as a sensor in hepatic parenchymal cells which induces glycogen synthesis, thus preventing the release of glucose into the blood. The GK processes are thus critical for the maintenance of whole body glucose homeostasis.
It is expected that an agent that activates cellular GK will facilitate glucose-dependent secretion from pancreatic beta cells, correct postprandial hyperglycemia, increase hepatic glucose utilization and potentially inhibit hepatic glucose release. Consequently, a GK activator may provide therapeutic treatment for NIDDM and associated complications, inter alia, hyperglycemia, dyslipidemia, insulin resistance syndrome, hyperinsulinemia, hypertension, and obesity.
Several drugs in five major categories, each acting by different mechanisms, are available for treating hyperglycemia and subsequently, NIDDM (Moller, D. E., “New drug targets for Type II diabetes and the metabolic syndrome” Nature 414; 821-827, (2001)): (A) Insulin secretogogues, including sulphonyl-ureas (e.g., glipizide, glimepiride, glyburide) and meglitinides (e.g., nateglidine and repaglinide) enhance secretion of insulin by acting on the pancreatic beta-cells. While this therapy can decrease blood glucose level, it has limited efficacy and tolerability, causes weight gain and often induces hypoglycemia. (B) Biguanides (e.g., metformin) are thought to act primarily by decreasing hepatic glucose production. Biguanides often cause gastrointestinal disturbances and lactic acidosis, further limiting their use. (C) Inhibitors of alpha-glucosidase (e.g., acarbose) decrease intestinal glucose absorption. These agents often cause gastrointestinal disturbances. (D) Thiazolidinediones (e.g., pioglitazone, rosiglitazone) act on a specific receptor (peroxisome proliferator-activated receptor-gamma) in the liver, muscle and fat tissues. They regulate lipid metabolism subsequently enhancing the response of these tissues to the actions of insulin. Frequent use of these drugs may lead to weight gain and may induce edema and anemia. (E) Insulin is used in more severe cases, either alone or in combination with the above agents.
Ideally, an effective new treatment for NIDDM would meet the following criteria: (a) it would not have significant side effects including induction of hypoglycemia; (b) it would not cause weight gain; (c) it would at least partially replace insulin by acting via mechanism(s) that are independent from the actions of insulin; (d) it would desirably be metabolically stable to allow less frequent usage; and (e) it would be usable in combination with tolerable amounts of any of the categories of drugs listed herein.
Substituted heteroaryls, particularly pyridones, have been implicated in mediating GK and may play a significant role in the treatment of NIDDM. For example, U.S. Patent publication No. 2006/0058353 and PCT publication Nos. WO2007/043638, WO2007/043638, and WO2007/117995 recite certain heterocyclic derivatives with utility for the treatment of diabetes. Although investigations are on-going, there still exists a need for a more effective and safe therapeutic treatment for diabetes, particularly NIDDM.

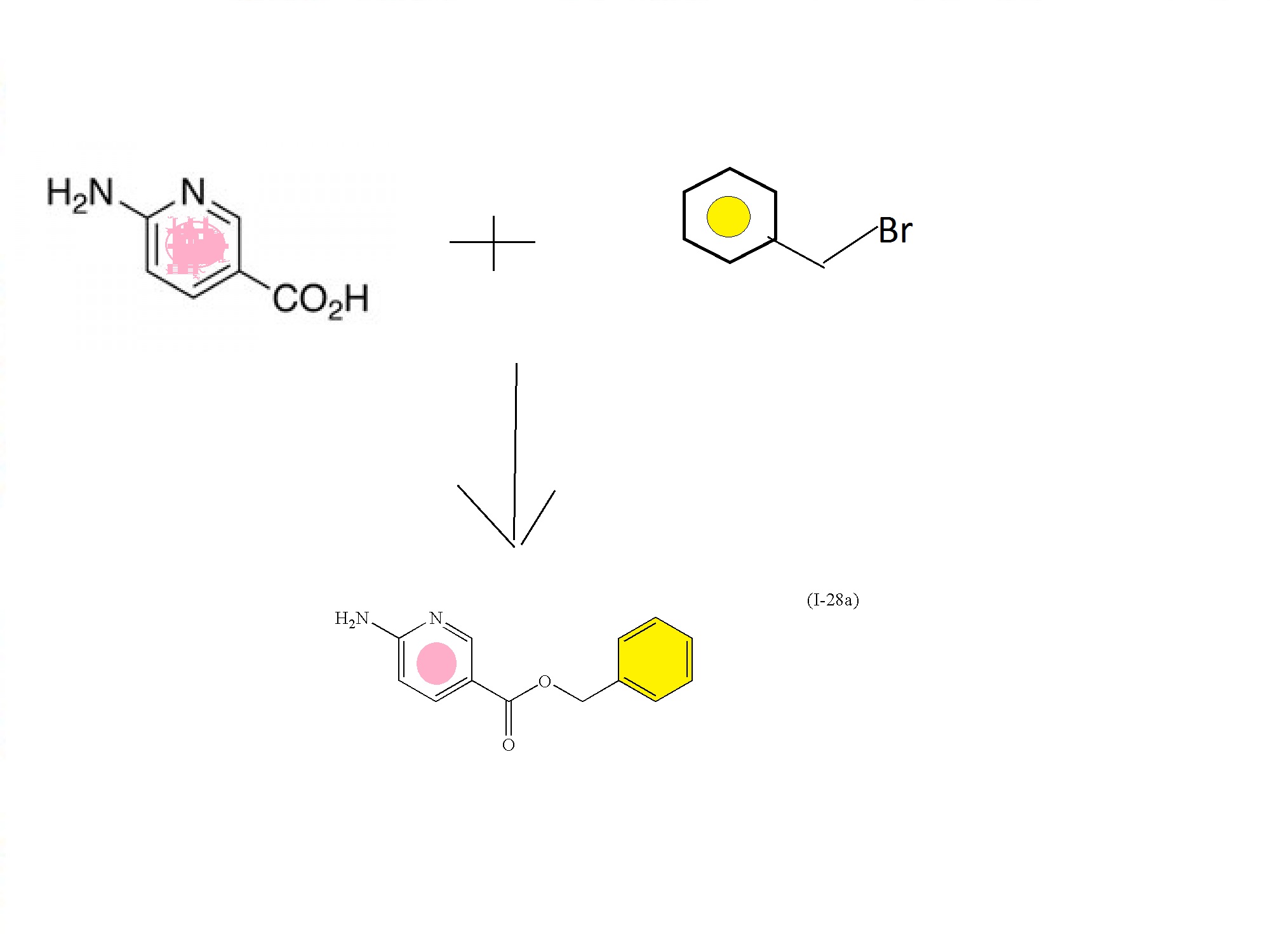
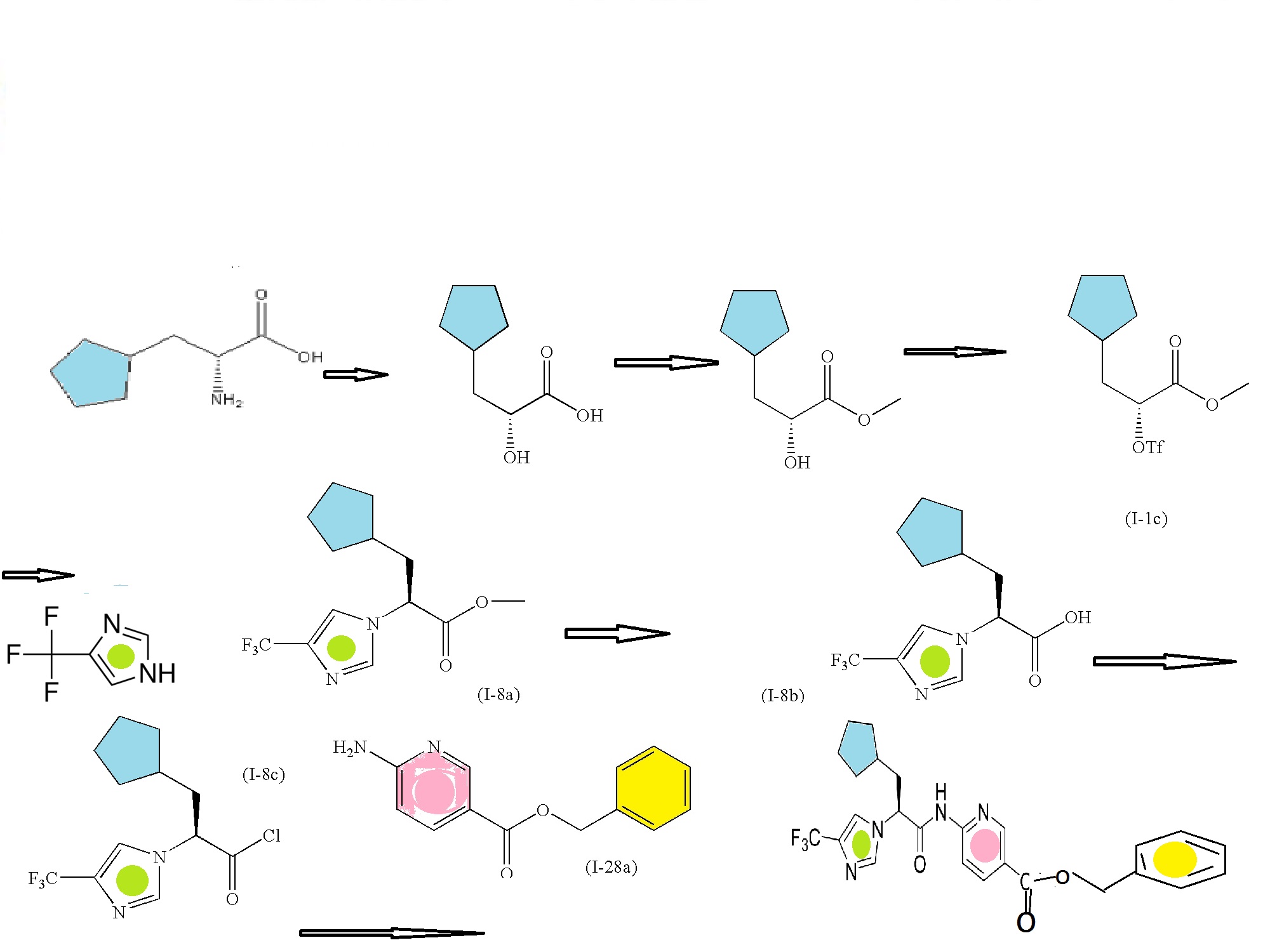
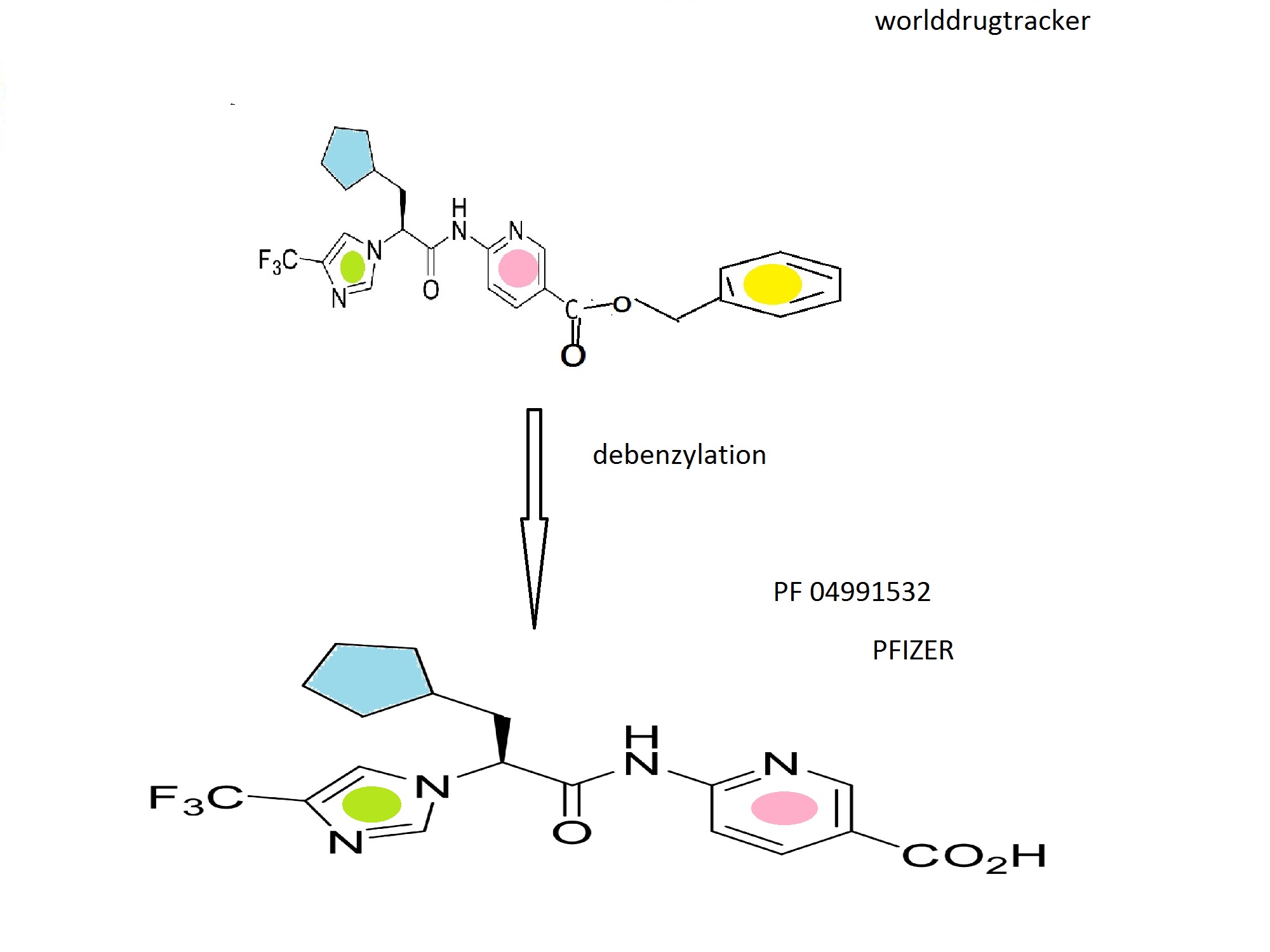
PATENT
US 20100063063
http://www.google.com/patents/US20100063063
SYNTHESIS CONSTRUCTION

6-aminonicotinic acid

BENZYL BROMIDE

FIRST KEY INTERMEDIATE
SECOND SERIES FOR NEXT INTERMEDIATE

(R)-2-amino-3-cyclopentylpropanoic acid

(R)-methyl 3-cyclopentyl-2-hydroxypropanoic acid (I-1a)

(R)-methyl 3-cyclopentyl-2-hydroxypropanoate (I-1b)
Trifluoromethanesulfonic acid anhydride

(R)-methyl 3-cyclopentyl-2-(trifluoromethylsulfonyloxy)propanoate (I-1c)
CONDENSED WITH

4-Trifluoromethyl-1H-imidazole
TO GIVE PRODUCT SHOWN BELOW

(S)-methyl 3-cyclopentyl-2-(4-(trifluoromethyl)-1H-imidazol-1-yl)propanoate (I-8a)

(S)-3-cyclopentyl-2-(4-(trifluoromethyl)-1H-imidazol-1-yl)propanoic acid (I-8b)

CONVERTED TO ACID CHLORIDE, (S)-3-cyclopentyl-2-(4-(trifluoromethyl)-1H-imidazol-1-yl)propanoyl chloride (I-8c)
AND CONDENSED WITH
WILL GIVE BENZYL DERIVATIVE AS BELOW

THEN DEBENZYLATION TO FINAL PRODUCT
Intermediate: (R)-methyl 3-cyclopentyl-2-hydroxypropanoic acid (I-1a)
To a stirred solution of (R)-2-amino-3-cyclopentylpropanoic acid (5.0 grams; Chem-Impex International, Inc., Wood Dale, Ill.) and 1 M H2SO4 (45.1 mL) at 0° C., was added a solution of NaNO2 (3.12 g) in H2O (15.6 mL) drop wise over 10 minutes. The reaction mixture was stirred for 3 hours at 0° C., then for 2 hours at room temperature. The solution was then extracted (3 times) with diethyl ether. The combined organic extracts were dried over MgSO4, filtered, and the filtrate concentrated to afford 2.36 g of (I-1a). 1H NMR (400 MHz, CDCl3) δ 4.26-4.28 (1H), 1.99-2.07 (1H), 1.76-1.81 (4H), 1.60-1.62 (4H), 1.12-1.16 (2H); LCMS for C8H14O3 m/z 157.1 (M−H)−.
Intermediate: (R)-methyl 3-cyclopentyl-2-hydroxypropanoate (I-1b)
To a stirred solution of 2.36 g of (I-1a) in anhydrous methanol (15 mL) at room temperature was added SOCl2(1.64 mL). The resulting mixture was heated at reflux for 2 hours. It was then cooled and concentrated under reduced pressure. The residue was partitioned between ethyl acetate and aqueous saturated NaHCO3 solution. The biphasic mixture was separated and the aqueous portion was extracted with ethyl acetate. The combined extracts were dried over MgSO4, filtered, and the filtrate concentrated under reduced pressure. The resulting residue was purified by flash column chromatography (silica gel, heptanes/ethyl acetate) to afford 1.5 g of (I-1b) as a clear oil.1H NMR (400 MHz, CDCl3) δ 4.15-4.20 (1H), 3.77 (3H), 2.62-2.63 (1H), 1.97-2.05 (1H), 1.49-1.86 (8H), 1.06-1.17 (2H); LCMS for C9H16O3 m/z 171.6 (M)+. Intermediate (I-1b) can alternatively be prepared by the method described below.
A 0.2M solution of Li2CuCl4 was prepared as follows: Anhydrous CUCl2 (26.9 g, 200 mol) and anhydrous LiCl (17.0 g, 400 mmol) were dissolved in THF (1000 mL). The mixture required gentle heating to completely dissolve the solids. After cooling the solution is ready for use.
A solution of Li2CuCl4 (0.2 M in THF, 125 mL, 25.0 mmol) was added slowly to a suspension of cyclopentylmagnesium bromide (2 M in diethyl ether, 135 mL, 270 mmol; Aldrich Chemical Company, Inc., Milwaukee, Wis.) and THF (500 mL) at −50° C. over 2-3 mins. The pale grey/brown suspension was then allowed to warm slowly to −10° C. over 30 mins, by which time the color had developed to a dark grey. The mixture was re-cooled to −78° C. and (R)-methyl oxirane-2-carboxylate (25.0 g, 245 mmol; Aldrich Chemical Company, Inc., Milwaukee, Wis.) was added neat via syringe over 90 seconds. The reaction was then stirred at −78° C. for 20 mins, before removing the ice-bath and allowing to warm to approximately −50° C. over 30 mins. Saturated NH4Cl (aq, 700 mL) was then added and the mixture stirred for 30 mins. The organic layer was collected and the aqueous layer extracted with diethyl ether (2×250 mL). The combined organics were washed with saturated NH4Cl (aq, 350 mL), dried over MgSO4, and evaporated. Distillation of the crude residue (68-70° C. at 0.8 mbar) yielded 65-70% of (I-1b) as a pale yellow oil. A small amount of less volatile material remained in the still pot. 1H NMR (400 MHz; CDCl3): δ 4.17(1H), 3.76 (3H), 2.67 (1H), 2.01 (1H), 1.48-1.88 (8H), 1.11 (2H).
Intermediate: (R)-methyl 3-cyclopentyl-2-(trifluoromethylsulfonyloxy)propanoate (I-1c)
Intermediate: (R)-methyl 3-cyclopentyl-2-(trifluoromethylsulfonyloxy)propanoate (I-1c
Intermediate (I-1b) (6.37 g, 37.0 mmol) was dissolved in dry dichloromethane (260 mL) and stirred under nitrogen in an ice bath. 2,6-Lutidine (9.0 mL, 77 mmol) was added. Trifluoromethanesulfonic acid anhydride (11 mL, 65 mmol) in dry dichloromethane (75 mL) was added dropwise. The reaction was stirred in the ice bath for 60 minutes, concentrated under reduced pressure, and taken up in 1N HCl and methyl t-butyl ether. The aqueous layer was separated, and the organic layer was washed with additional 1N HCl to insure the removal of all the lutidine. The combined organic layer was then washed with brine, dried over sodium sulfate, filtered, concentrated under reduced pressure, and dried under high vacuum to afford (I-1c) (11.3 g, 37 mmol, 100%), which was used immediately without further purification; 1H NMR (400 MHz, CDCl3) δ 5.10-5.14 (1H), 3.82 (3H), 2.02-2.12 (1H), 1.79-1.98 (4H), 1.51-1.66 (4H), 1.08-1.18 (2H).
Intermediate (I-1b) (6.37 g, 37.0 mmol) was dissolved in dry dichloromethane (260 mL) and stirred under nitrogen in an ice bath. 2,6-Lutidine (9.0 mL, 77 mmol) was added. Trifluoromethanesulfonic acid anhydride (11 mL, 65 mmol) in dry dichloromethane (75 mL) was added dropwise. The reaction was stirred in the ice bath for 60 minutes, concentrated under reduced pressure, and taken up in 1N HCl and methyl t-butyl ether. The aqueous layer was separated, and the organic layer was washed with additional 1N HCl to insure the removal of all the lutidine. The combined organic layer was then washed with brine, dried over sodium sulfate, filtered, concentrated under reduced pressure, and dried under high vacuum to afford (I-1c) (11.3 g, 37 mmol, 100%), which was used immediately without further purification; 1H NMR (400 MHz, CDCl3) δ 5.10-5.14 (1H), 3.82 (3H), 2.02-2.12 (1H), 1.79-1.98 (4H), 1.51-1.66 (4H), 1.08-1.18 (2H)
Intermediate: (S)-methyl 3-cyclopentyl-2-(4-(trifluoromethyl)-1H-imidazol-1-yl)propanoate (I-8a)
4-Trifluoromethyl-1H-imidazole (5.0 g, 37.0 mmol; Apollo Scientific Ltd., Bredbury, Cheshire, UK) was stirred in dry THF (180 mL) under nitrogen at room temperature. Lithium hexamethyldisilazide (1M in THF, 33.4 mL, 33.4 mmol) was added dropwise via addition funnel. The mixture was stirred at room temperature for 50 minutes and then chilled in an ice bath. A solution of (I-1c) (11.3 g, 37 mmol) in dry THF (45 mL), which had been chilled in an ice bath, was added in one portion. The reaction was allowed to warm to room temperature, stirred for 2 hours, quenched with saturated aqueous ammonium chloride solution (20 mL) and allowed to stir overnight. The aqueous layer was separated, and the organic layer was concentrated and then diluted with water and ethyl acetate. The organic layer was washed in series with dilute aqueous phosphoric acid, aqueous 10% potassium carbonate, and brine. The organic layer was then dried over sodium sulfate, filtered, and concentrated under reduced pressure to a brown oil. The crude material, containing the undesired regioisomer as a small impurity, was purified by chromatography on a 330 g pre-packed silica gel column, eluting with 10% ethyl acetate/heptane, linear gradient to 70% ethyl acetate/heptane. The product fractions were located by spotting on a silica TLC plate and visualizing with KMnO4 stain. TLC (1:1 ethyl acetate/heptane, developed in potassium permanganate) located the pure and mixed fractions. The clean product fractions were combined, evaporated, and dried under high vacuum to afford (I-8a) as a clear oil (6.61 g, 22.4 mmol, 67%). 1H NMR (400 MHz, CDCl3) δ 7.57 (1H), 7.38 (1H), 4.71-4.74 (1H), 3.76 (3H), 2.01-2.14 (2H), 1.45-1.79 (7H), 1.03-1.18 (2H); m/z 291.4 (M+H)+.
Intermediate: (S)-3-cyclopentyl-2-(4-(trifluoromethyl)-1H-imidazol-1-yl)propanoic acid (I-8b
6N HCl (140 mL) was added to (I-8a) (6.61 g, 22.4 mmol) and the mixture was warmed to 95° C. for 16 hours and then allowed to cool. Solid potassium carbonate (58 g) was added in portions to bring the pH to about 4. A precipitate crashed out. Ethyl acetate was added, and the mixture was stirred until everything dissolved. The aqueous layer was extracted once with ethyl acetate. The combined organics were washed with brine, dried over sodium sulfate, filtered, concentrated under reduced pressure, and dried under high vacuum to afford (I-8b) as a clear glass (6.15 g, 21.9 mmol, 98%). 1H NMR (400 MHz, CDCl3) δ 7.73 (1H), 7.34 (1H), 6.85-7.15 (1H), 4.66-4.70 (1H), 1.98-2.17 (2H), 1.41-1.75 (7H), 1.01-1.19 (2H); m/z 277.4 (M+H)+.
Intermediate: (S)-3-cyclopentyl-2-(4-(trifluoromethyl)-1H-imidazol-1-yl)propanoyl chloride (I-8c)
To a suspension of intermediate (I-8b) (0.25 g, 0.9 mmol) in dichloromethane (5 mL) was added oxalyl chloride (0.35 g, 2.7 mmol) and N,N-dimethylformamide (1 drop) at room temperature. The mixture was stirred for 2 hours at room temperature. The reaction mixture was concentrated in vacuo, and the residue was chased with dichloromethane two times and concentrated in vacuo to afford (I-8c) (0.27 g, 100%) as an oil, which was used in the next step directly.
Intermediate: (S)-6-(3-cyclopentyl-2-(4-(trifluoromethyl)-1H-imidazol-1-yl)propanamido) nicotinoyl chloride (I-21a)
-
 Thionyl chloride (225 mg, 1.89 mmol) was added to a solution of the compound of Example 48 (150 mg, 0.387 mmol) in dichloromethane (1.5 mL) and the reaction stirred at room temperature for 1 hour. LCMS of an aliquot in methanol showed ˜67% methyl ester. To the reaction mixture was added another 25 uL of thionyl chloride and this was stirred at room temp for another 30 minutes. Solvents were evaporated to afford 157 mg (100%) of (I-21a) as a grayish-white solid. LCMS in methanol to generate the methyl ester gave m/z 395.9 (M+H)+.
Thionyl chloride (225 mg, 1.89 mmol) was added to a solution of the compound of Example 48 (150 mg, 0.387 mmol) in dichloromethane (1.5 mL) and the reaction stirred at room temperature for 1 hour. LCMS of an aliquot in methanol showed ˜67% methyl ester. To the reaction mixture was added another 25 uL of thionyl chloride and this was stirred at room temp for another 30 minutes. Solvents were evaporated to afford 157 mg (100%) of (I-21a) as a grayish-white solid. LCMS in methanol to generate the methyl ester gave m/z 395.9 (M+H)+.

(I-8b
Intermediate: (S)-3-cyclopentyl-2-(4-(trifluoromethyl)-1H-imidazol-1-yl)propanoic acid (I-8b)
6N HCl (140 mL) was added to (I-8a) (6.61 g, 22.4 mmol) and the mixture was warmed to 95° C. for 16 hours and then allowed to cool. Solid potassium carbonate (58 g) was added in portions to bring the pH to about 4. A precipitate crashed out. Ethyl acetate was added, and the mixture was stirred until everything dissolved. The aqueous layer was extracted once with ethyl acetate. The combined organics were washed with brine, dried over sodium sulfate, filtered, concentrated under reduced pressure, and dried under high vacuum to afford (I-8b) as a clear glass (6.15 g, 21.9 mmol, 98%). 1H NMR (400 MHz, CDCl3) δ 7.73 (1H), 7.34 (1H), 6.85-7.15 (1H), 4.66-4.70 (1H), 1.98-2.17 (2H), 1.41-1.75 (7H), 1.01-1.19 (2H); m/z 277.4 (M+H)+.
(I-28a
Intermediate: benzyl 6-aminonicotinate (I-28a)
To a stirred suspension of 6-aminonicotinic acid (100 g, 0.72 mol; Aldrich Chemical Company, Inc., Milwaukee, Wis.) in N,N-dimethylformamide (700 mL) with brisk mechanical stirring was added potassium carbonate (150 g, 1.08 mol) and the reaction was stirred for 10 min before the portionwise addition of benzyl bromide (95 mL, 0.80 mol). The reaction was stirred at room temperature overnight, then the solids were filtered off and washed thoroughly with ethyl acetate, and the solvent was removed under vacuum. The filter cake was dissolved in water and extracted with ethyl acetate. The residue after evaporation of N,N-dimethylformamide was combined with the ethyl acetate extracts (total volume 2 L of ethyl acetate) and the combined organic extracts washed with brine (5×500 mL), dried (MgSO4) and the solvent removed under reduced pressure. The crude product was refluxed with 1:1 diethyl ether:hexane for 30 min then the solids filtered off (warm), washed with diethyl ether:hexane (1:1), and dried. This solid was precipitated from hot toluene (hot filtration required to remove dibenzylated material) and dried to afford (I-28a) (107.2 g, 65%) as an off-white solid; 1H NMR (DMSO-d6): δ 8.50 (1H), 7.82 (1H), 7.34-7.29 (5H), 6.84 (2H), 6.43 (1H), 5.23 (2H); m/z 229.4 (M+H)+.
Example 47
(S)-benzyl 6-(3-cyclopentyl-2-(4-(trifluoromethyl)-1H-imidazol-1-yl)propanamido)nicotinate
Formula (1A-4) wherein R4 is
To Intermediate (I-8b) (16.28 g, 59.8 mmol) stirring in dry dichloromethane (400 mL) at room temperature under nitrogen was added 2 drops of DMF. Oxalyl chloride (11 mL, 130 mmol) was added dropwise. After the bubbling subsided the reaction was left stirring for 90 minutes and then concentrated under reduced pressure. Two successive portions of 1,2-dichloroethane were added and evaporated to remove all excess oxalyl chloride. The crude acid chloride was taken up in dichloromethane (150 mL) and stirred at room temperature. Intermediate (I-28a) (14.3 g, 62.5 mmol) and pyridine (10 mL, 130 mmol) were stirred in 400 mL dry dichloromethane. This was added to the acid chloride solution, using another 50 mL dry dichloromethane to complete the transfer. The mixture was left stirring at room temperature under nitrogen for 18 hours. The reaction was diluted with dichloromethane and water, and 1M aqueous phosphoric acid was added. The organic layer was separated and washed sequentially with dilute aqueous potassium carbonate, and brine. This was then dried over sodium sulfate, filtered, and concentrated under reduced pressure to a glass, which was taken up in hot ethyl acetate and stirred at room temperature. A precipitate appeared at about 30 minutes. The mixture was stirred for 16 hours and then filtered. The precipitate was washed with ethyl acetate and then diethyl ether and dried under high vacuum at 60° C. to afford the title compound as a white solid (17.8 g, 36.6 mmol, 61%). The mother liquor was evaporated and purified by silica gel chromatography on a 120 g pre-packed column, eluting with 40% ethyl acetate/heptane. The product fractions were combined, concentrated under reduced pressure, dried under high vacuum to a glass, and converted as previously described to additional product (3.5 g, 7.2 mmol, 12%, total yield 73%). 1H NMR (400 MHz, DMSO-d6) δ 11.50 (1H), 8.87-8.88 (1H), 8.29-8.32 (1H), 8.12-8.14 (1H), 7.93-7.94 (2H), 7.39-7.46 (2H), 7.30-7.37 (3H), 5.32 (2H), 5.21-5.25 (1H), 2.06-2.19 (2H), 1.26-1.63 (8H), 1.01-1.06 (1H); m/z 487.5 (M+H)+.
Example 48
(S)-6-(3-cyclopentyl-2-(4-(trifluoromethyl)-1H-imidazol-1-yl)propanamido)nicotinic acid
Formula (1A-4) wherein R4 is

The compound of Example 47 (4.07 g, 8.35 mmol) was added to a 500 mL Parr bottle, followed by ethyl acetate (50 mL) and ethanol (100 mL). The mixture was warmed until all of the solid dissolved, and then cooled to room temperature. 10% Pd/C (450 mg) was added, and the mixture was shaken under 50 psi hydrogen for 90 minutes. The reaction was filtered through a microfiber filter. The filtrate was concentrated under reduced pressure and dried under high vacuum at 50° C. to afford product as a glassy solid (3.0 g, 7.75 mmol, 90.6%). The glassy solid was stirred overnight in diethyl ether. The white solid precipitate was filtered, washed with diethyl ether, suction dried, and dried under high vacuum at 50° C. to afford the title compound as a white solid.
1H NMR (400 MHz, DMSO-d6) δ 13.10-13.25 (1H), 11.44 (1H), 8.83 (1H), 8.23-8.26 (1H), 8.09-8.12 (1H), 7.94-7.95 (2H), 5.22-5.26 (1H), 2.06-2.17 (2H), 1.29-1.64 (8H), 1.04-1.07 (1H);
m/z 397.3 (M+H)+.
THIS NMR IS FROM SUPPORTING INFO OF A JOURNAL
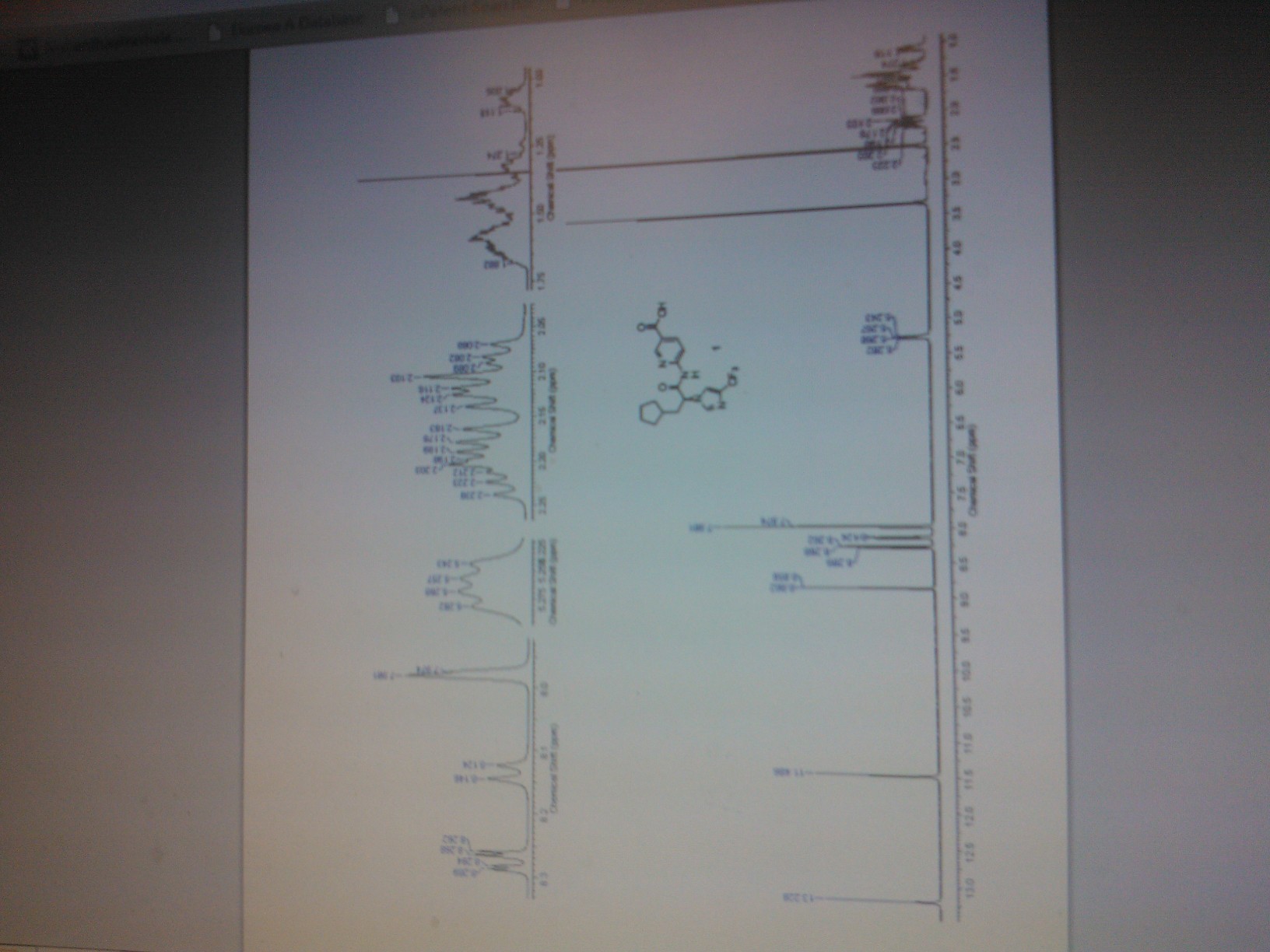
PAPER
Organic Process Research & Development (2012), 16(10), 1635-1645
http://pubs.acs.org/doi/abs/10.1021/op300194c

This work describes the process development and manufacture of early-stage clinical supplies of a hepatoselective glucokinase activator, a potential therapy for type 2 diabetes mellitus. Critical issues centered on challenges associated with the synthesis of intermediates and API bearing a particularly racemization-prone α-aryl carboxylate functionality. In particular, a T3P-mediated amidation process was optimized for the coupling of a racemization-prone acid substrate and a relatively non-nucleophilic amine. Furthermore, an unusually hydrolytically-labile amide in the API also complicated the synthesis and isolation of drug substance. The evolution of the process over multiple campaigns is presented, resulting in the preparation of over 110 kg of glucokinase activator.
(S)-6-(3-Cyclopentyl-2-(4-(trifluoromethyl)-1H-imidazol-1-yl)propanamido)nicotinic Acid (1)
Pressure Hydrogenation
1 (89% yield) as a white solid:
mp 187–189 °C;
1H NMR (400 MHz, d6-DMSO) δ 13.23 (s, 1H), 11.49 (s, 1H), 8.86 (dd, J = 0.4, 2.4 Hz, 1H), 8.27 (dd, J = 2.4, 8.8 Hz, 1H), 8.13 (d, J = 8.8 Hz, 1H), 7.97–7.99 (m, 2H), 5.27 (dd, J = 5.6, 10.0 Hz, 1H), 2.20 (ddd, J = 6.0, 10.0, 14.0, 1H), 2.10 (ddd, J = 5.6, 8.4, 14.0, 1H), 1.27–1.69 (m, 8H), 1.03–1.12 (m, 1H);
13C NMR (100 MHz, d6-DMSO) δ 168.8, 165.7, 154.3, 149.7, 139.6, 138.8, 129.9 (q, JCF = 38 Hz), 122.6, 122.0 (q, JCF = 265 Hz), 120.0 (q, JCF = 4 Hz), 112.8, 60.0, 37.6, 36.2, 32.0, 30.8, 24.6, 24.4;
19F NMR (376 MHz, d6-DMSO) δ −60.7.
HRMS-ESI m/z: [M + H]+ calcd for C18H19F3N4O3, 397.1482; found, 397.1481.
Achiral HPLC: rt 4.6 min. Chiral SFC: rt 4.1 min (1), 3.1 min (ent-1).
PAPER
Journal of Medicinal Chemistry (2012), 55(3), 1318-1333
http://pubs.acs.org/doi/abs/10.1021/jm2014887

Glucokinase is a key regulator of glucose homeostasis, and small molecule allosteric activators of this enzyme represent a promising opportunity for the treatment of type 2 diabetes. Systemically acting glucokinase activators (liver and pancreas) have been reported to be efficacious but in many cases present hypoglycaemia risk due to activation of the enzyme at low glucose levels in the pancreas, leading to inappropriately excessive insulin secretion. It was therefore postulated that a liver selective activator may offer effective glycemic control with reduced hypoglycemia risk. Herein, we report structure–activity studies on a carboxylic acid containing series of glucokinase activators with preferential activity in hepatocytes versus pancreatic β-cells. These activators were designed to have low passive permeability thereby minimizing distribution into extrahepatic tissues; concurrently, they were also optimized as substrates for active liver uptake via members of the organic anion transporting polypeptide (OATP) family. These studies lead to the identification of 19 as a potent glucokinase activator with a greater than 50-fold liver-to-pancreas ratio of tissue distribution in rodent and non-rodent species. In preclinical diabetic animals, 19 was found to robustly lower fasting and postprandial glucose with no hypoglycemia, leading to its selection as a clinical development candidate for treating type 2 diabetes.
(S)-6-(3-Cyclopentyl-2-(4-(trifluoromethyl)-1H-imidazol-1-yl)propanamido)nicotinic Acid (19)
afford 19 as a white solid (3.22 g, 71%).
1H NMR (400 MHz, DMSO-d6) δ 11.47 (s, 1H), 8.86 (d, J = 1.95 Hz, 1H), 8.27 (dd, J = 2.24, 8.68 Hz, 1H), 8.13 (d, J = 8.78 Hz, 1H), 7.97 (d, J = 4.88 Hz, 2H), 5.27 (dd, J = 5.37, 9.66 Hz, 1H), 2.04–2.26 (m, 2H), 1.38–1.72 (m, 7H), 1.26–1.37 (m, 1H), 1.08 (td, J = 7.88, 11.75 Hz, 1H);
LCMS m/z 397.5 (M + H)+.
HPLC purity (method A): tR = 7.690 min, 100%.

PAPER
Bioorganic & Medicinal Chemistry Letters (2013), 23(24), 6588-6592
http://www.sciencedirect.com/science/article/pii/S0960894X13012638


Figure 1.
Structure of Hepatoselective GKA PF-04991532 (1).
References
Drug Metabolism & Disposition (2015), 43(2), 190-198
PLoS One (2014), 9(5), e97139/1-e97139/9,
Journal of Biological Chemistry (2012), 287(17), 13598-13610
Drug Discovery Today (2012), 17(9-10), 528-529
Biochemical Journal (2012), 441(3), 881-887.
///////////

Figure 1. Representative structures of glucokinase activators.
Pfizer’s PF 04937319 glucokinase activators for the treatment of Type 2 diabetes

PF 04937319
N,N-dimethyl-5-(2-methyl-6-((5-methylpyrazin-2-yl)-carbamoyl)benzofuran-4-yloxy)pyrimidine-2-carboxamide
MW 432.43
MF C22 H20 N6 O4
CAS 1245603-92-2
2-Pyrimidinecarboxamide, N,N-dimethyl-5-[[2-methyl-6-[[(5-methyl-2-pyrazinyl)amino]carbonyl]-4-benzofuranyl]oxy]-
N,N-Dimethyl-5-(2-methyl-6-((5-methylpyrazin-2-yl)carbamoyl)-benzofuran-4- yloxy)pyrimidine-2-carboxamide
Pfizer Inc. clinical candidate currently in Phase 2 development.
CLINICAL TRIALS
A trial to assess the safety, tolerability, pharmacokinetics, and pharmacodynamics of single doses of PF-04937319 in subjects with type 2 diabetes mellitus (NCT01044537)
Multiple dose study of PF-04937319 in patients with type 2 diabetes (NCT01272804)
Phase 2 study to evaluate safety and efficacy of investigational drug – PF04937319 in patients with type 2 diabetes (NCT01475461)
SYNTHESIS

Glucokinase is a key regulator of glucose homeostasis and small molecule activators of this enzyme represent a promising opportunity for the treatment of Type 2 diabetes. Several glucokinase activators have advanced to clinical studies and demonstrated promising efficacy; however, many of these early candidates also revealed hypoglycemia as a key risk. In an effort to mitigate this hypoglycemia risk while maintaining the promising efficacy of this mechanism, we have investigated a series of substituted 2-methylbenzofurans as “partial activators” of the glucokinase enzyme leading to the identification of N,N-dimethyl-5-(2-methyl-6-((5-methylpyrazin-2-yl)-carbamoyl)benzofuran-4-yloxy)pyrimidine-2-carboxamide as an early development candidate.
Diabetes is a major public health concern because of its increasing prevalence and associated health risks. The disease is characterized by metabolic defects in the production and utilization of carbohydrates which result in the failure to maintain appropriate blood glucose levels. Two major forms of diabetes are recognized. Type I diabetes, or insulin-dependent diabetes mellitus (IDDM), is the result of an absolute deficiency of insulin. Type Il diabetes, or non-insulin dependent diabetes mellitus (NIDDM), often occurs with normal, or even elevated levels of insulin and appears to be the result of the inability of tissues and cells to respond appropriately to insulin. Aggressive control of NIDDM with medication is essential; otherwise it can progress into IDDM. As blood glucose increases, it is transported into pancreatic beta cells via a glucose transporter. Intracellular mammalian glucokinase (GK) senses the rise in glucose and activates cellular glycolysis, i.e. the conversion of glucose to glucose-6-phosphate, and subsequent insulin release. Glucokinase is found principally in pancreatic β-cells and liver parenchymal cells. Because transfer of glucose from the blood into muscle and fatty tissue is insulin dependent, diabetics lack the ability to utilize glucose adequately which leads to undesired accumulation of blood glucose (hyperglycemia). Chronic hyperglycemia leads to decreases in insulin secretion and contributes to increased insulin resistance. Glucokinase also acts as a sensor in hepatic parenchymal cells which induces glycogen synthesis, thus preventing the release of glucose into the blood. The GK processes are thus critical for the maintenance of whole body glucose homeostasis.
It is expected that an agent that activates cellular GK will facilitate glucose-dependent secretion from pancreatic beta cells, correct postprandial hyperglycemia, increase hepatic glucose utilization and potentially inhibit hepatic glucose release. Consequently, a GK activator may provide therapeutic treatment for NIDDM and associated complications, inter alia, hyperglycemia, dyslipidemia, insulin resistance syndrome, hyperinsulinemia, hypertension, and obesity. Several drugs in five major categories, each acting by different mechanisms, are available for treating hyperglycemia and subsequently, NIDDM (Moller, D. E., “New drug targets for Type 2 diabetes and the metabolic syndrome” Nature 414; 821 -827, (2001 )): (A) Insulin secretogogues, including sulphonyl-ureas (e.g., glipizide, glimepiride, glyburide) and meglitinides (e.g., nateglidine and repaglinide) enhance secretion of insulin by acting on the pancreatic beta-cells. While this therapy can decrease blood glucose level, it has limited efficacy and tolerability, causes weight gain and often induces hypoglycemia. (B) Biguanides (e.g., metformin) are thought to act primarily by decreasing hepatic glucose production. Biguanides often cause gastrointestinal disturbances and lactic acidosis, further limiting their use. (C) Inhibitors of alpha-glucosidase (e.g., acarbose) decrease intestinal glucose absorption. These agents often cause gastrointestinal disturbances. (D) Thiazolidinediones (e.g., pioglitazone, rosiglitazone) act on a specific receptor (peroxisome proliferator-activated receptor-gamma) in the liver, muscle and fat tissues. They regulate lipid metabolism subsequently enhancing the response of these tissues to the actions of insulin. Frequent use of these drugs may lead to weight gain and may induce edema and anemia. (E) Insulin is used in more severe cases, either alone or in combination with the above agents. Ideally, an effective new treatment for NIDDM would meet the following criteria: (a) it would not have significant side effects including induction of hypoglycemia; (b) it would not cause weight gain; (c) it would at least partially replace insulin by acting via mechanism(s) that are independent from the actions of insulin; (d) it would desirably be metabolically stable to allow less frequent usage; and (e) it would be usable in combination with tolerable amounts of any of the categories of drugs listed herein.
Substituted heteroaryls, particularly pyridones, have been implicated in mediating GK and may play a significant role in the treatment of NIDDM. For example, U.S. Patent publication No. 2006/0058353 and PCT publication No’s. WO2007/043638, WO2007/043638, and WO2007/117995 recite certain heterocyclic derivatives with utility for the treatment of diabetes. Although investigations are on-going, there still exists a need for a more effective and safe therapeutic treatment for diabetes, particularly NIDDM.
Designing glucokinase activators with reduced hypoglycemia risk: discovery of N,N-dimethyl-5-(2-methyl-6-((5-methylpyrazin-2-yl)-carbamoyl)benzofuran-4-yloxy)pyrimidine-2-carboxamide as a clinical candidate for the treatment of type 2 diabetes mellitus
*Corresponding authors
aPfizer Worldwide Research & Development, Eastern Point Road, Groton
E-mail: jeffrey.a.pfefferkorn@pfizer.com
Tel: +860 686 3421
E-mail: jeffrey.a.pfefferkorn@pfizer.com
Tel: +860 686 3421
Med. Chem. Commun., 2011,2, 828-839
DOI: 10.1039/C1MD00116G
http://pubs.rsc.org/en/content/articlelanding/2011/md/c1md00116g/unauth#!divAbstract
http://www.rsc.org/suppdata/md/c1/c1md00116g/c1md00116g.pdf
N,N-Dimethyl-5-(2-methyl-6-((5-methylpyrazin-2-yl)carbamoyl)-benzofuran-4- yloxy)pyrimidine-2-carboxamide (28). To a solution of the 5-methyl-2-aminopyrazine (38.9 g, 356 mmol) in dimethoxyethane (315 mL) in a 3-neck flask equipped with overhead stirring and a condenser at 0 o C was added Me2AlCl (1 M solution in hexanes) (715 mL). The mixture was warmed to room temperature and stirred for 1.5 h. In a separate flask, 26 (52.6 g, 142.5 mmol) was dissolved in dimethoxyethane (210 mL). This mixture was then added to the amine mixture. A gum precipitated and upon scratching the flask it dissipated into a solid. The reaction was refluxed for 3.5 h. Aq. Rochelle’s salt (5 L) and 2-MeTHF (2 L) was added to the mixture and this was allowed to stir with overhead stirring for 14 h, after which time, a yellow solid precipitated. The solid was collected by filtration, washing with 2-MeTHF. The resulting solid was dried in a vacuum oven overnight to afford the desired material (50.0g) in 81% yield.
1 H NMR (400MHz, CDCl3) δ 9.54 (d, J = 1.56 Hz, 1H), 8.50 (s, 2H), 8.37 (s, 1H), 8.14 (d, J = 0.78 Hz, 1H), 7.88 – 7.92 (m, 1H), 7.52 (d, J = 1.37 Hz, 1H), 6.28 (t, J = 0.98 Hz, 1H), 3.14 (s, 3H), 2.98 (s, 3H), 2.55 (s, 3H), 2.49 (d, J = 1.17 Hz, 3H);
MS(ES+ ): m/z 433.4 (M+1), MS(ES- ): m/z 431.3 (M-1).
PAPER

http://pubs.rsc.org/en/content/articlelanding/2013/md/c2md20317k#!divAbstract
PAPER
Bioorganic & Medicinal Chemistry Letters (2013), 23(16), 4571-4578
http://www.sciencedirect.com/science/article/pii/S0960894X13007452

Figure 1.
Glucokinase activators 1 and 2.
PATENT
WO 2010103437
https://www.google.co.in/patents/WO2010103437A1?cl=en
Scheme I outlines the general procedures one could use to provide compounds of the present invention having Formula (I).


Preparations of Starting Materials and Key Intermediates
Preparation of Intermediate (E)-3-(ethoxycarbonyl)-4-(5-methylfuran-2-yl)but- 3-enoic acid (I- 1a):

(Ma) To a vigorously stirred solution of 5-methyl-2-furaldehyde (264 ml_, 2650 mmol) and diethyl succinate (840 ml_, 5050 mmol) in ethanol (1.820 L) at room temperature was added sodium ethoxide (0.93 L of a 21 weight % solution in ethanol) in one portion. The reaction mixture was then heated at reflux for 13 hours. After cooling to room temperature, the mixture was concentrated in vacuo (all batches were combined at this point). The resulting residue was partitioned between ethyl acetate (1 L) and hydrochloric acid (1 L of a 2M aqueous solution). After separation, the aqueous layer was extracted with ethyl acetate (2 x 1 L). The combined organic extracts were then extracted with sodium hydrogen carbonate (2 x 1 L of a saturated aqueous solution). These aqueous extracts were combined and adjusted to pH 2 with hydrochloric acid (2M aqueous solution) then extracted with ethyl acetate (2 x 1 L). These organic extracts were combined and concentrated in vacuo to give desired (E)-3-(ethoxycarbonyl)-4-(5-methylfuran-2-yl)but-3-enoic acid (J1 Ia: 34.34 g, 5%). The original organic extract was extracted with sodium hydroxide (2 L of a 2M aqueous solution). This aqueous extract was adjusted to pH 2 with hydrochloric acid (2M aqueous solution) then extracted with ethyl acetate (2 x 1 L). These organic extracts were combined and concentrated in vacuo to give additional desired materials (395.2 gram, 63%) as red liquid. 1H NMR (CDCI3, 300 MHz) δ ppm 7.48 (s, 1 H), 6.57 (d, 1 H), 6.09 (d, 1 H), 4.24 (q, 2H), 3.87 (s, 2H), 2.32 (s, 3H), 1.31 (t, 3H).
Preparation of Intermediate ethyl 4-acetoxy-2-methylbenzofuran-6- carboxylate (1-1 b):

(M b) To a vigorously stirred solution of (E)-3-(ethoxycarbonyl)-4-(5- methylfuran-2-yl)but-3-enoic acid (1-1 a: 326.6 g, 1 .371 mol) in acetic anhydride (1 .77 L, 18.72 mol) at room temperature was added sodium acetate (193 g, 2350 mmol) in one portion. The reaction mixture was then heated at reflux for 2.5 hours. After cooling to room temperature, the mixture was concentrated in vacuo (all batches were combined at this point). The resulting residue was suspended in dichloromethane (1 .5 L) and filtered, washing the solids with dichloromethane (3 x 500 ml_). The combined filtrate and washings were then washed with sodium hydrogencarbonate (2 x 1 L of a saturated aqueous solution) and brine (2 L), then concentrated in vacuo to give desired ethyl 4-acetoxy-2-methylbenzofuran-6-carboxylate (H b: 549.03 g, quantitative). 1H NMR (CDCI3, 300 MHz) δ ppm 8.00-7.99 (m, 1 H), 7.64 (d, 1 H), 6.32-6.32 (m, 1 H), 4.38 (q, 2H), 2.47 (d, 3H), 2.37 (s, 3H), 1 .39 (t, 3H).
Preparation of Intermediate ethyl 4-hydroxy-2-methylbenzofuran-6- carboxylate (1- 1 c):

(He) To a stirred solution of ethyl 4-acetoxy-2-methylbenzofuran-6- carboxylate (Hb: 549.03 g, 1 .37 mol) in ethanol (4.00 L) at room temperature was added potassium carbonate (266 g, 1 .92 mol) in one portion. The reaction mixture was then heated at 600C for 3 hours. Potassium carbonate (100 g, 0.720 mol) was then added in one portion and the reaction mixture was heated at 600C for a further 3 hours. After cooling to room temperature the mixture was diluted with dichloromethane (2 L) and the suspension filtered, washing the solids with dichloromethane (2 x 1 L) (all batches were combined at this point). The combined filtrate and washings were then washed with citric acid (2.5 L of a 1 M aqueous solution), then concentrated in vacuo and the resulting residue purified by dry flash chromatography (hexane then 2:1 hexane:ethyl acetate). All fractions containing the desired product were combined and concentrated in vacuo. The resulting residue, which solidified on standing, was slurried with cold toluene and filtered. The solids were then stirred with hot toluene and decolourising charcoal for 1 hour, followed by filtration of the hot mixture through a pad of celite. The filtrate was allowed to cool and the resulting precipitate isolated by filtration to give desired ethyl 4-hydroxy-2- methylbenzofuran-6-carboxylate (1-1 c: 360 g, 90%) as orange powder.
1H NMR (CDCI3, 300 MHz) δ ppm 7.73-7.73 (m, 1 H), 7.45 (d, 1 H), 6.51 -6.50 (m, 1 H), 5.85 (s, 1 H), 4.39 (q, 2H), 2.48 (d, 3H), 1.40 (t, 3H). LCMS (liquid chromatography mass spectrometry): m/z 221.06 (96.39 % purity).
Preparation of SM-25-bromo-N,N-dimethylpyrimidine-2-carboxamide (SM-
£1:

(SM-2) Oxalyl chloride (47.4g, 369mmol) was added to a suspension of 5-
Bromo-pyrimidine-2-carboxylic acid (5Og, 250mmol) in dichloromethane (821 ml) at room temperature followed by 1 -2 drop of dimethylformamide. The reaction mixture was stirred under nitrogen for 2 hours LCMS in methanol indicated the presence of the methyl ester and some acid. Dimethylformamide (0.2ml) was added to the reaction mixture. The acid dissolved after 30 minutess. LCMS showed corresponding methyl ester and no starting material peak was observed. The solvent was removed and dried in vacuo to afford the crude 5-Bromo-pyrimidine-2-carbonyl chloride (55g, 100%). The 5-Bromo-pyrimidine-2-carbonyl chloride (55g, 250mmol) was dissolved in tetrahydrofuran (828ml) and dimethyl-amine (2M solution in tetrahydrofuran) (373ml, 745mmol) was added portionwise at room temperature. The reaction was stirred at room temperature under nitrogen for 16 hours, after which time, LCMS indicated completion. The mixture was diluted with ethyl acetate (500ml) and washed with H2O (500ml). The water layer was further extracted with CH2CI2 (5x500ml), all organics combined, and dried over magnesium sulfate. The filtrate was concentrated in vacuo and then suspended in methyl-/-butylether (650ml). The solution was then heated to reflux. The hot solution was allowed to cool overnight to afford pink crystals. The crystals were filtered and washed with cold methyl-t-butylether (100ml) the solid was dried in a vacuum oven at 550C for 12 hourrs to afford the title compound 5-bromo-N,N-dimethylpyhmidine-2-carboxamide (SM-2: 44g, 77%) as a pink solid.
1H NMR (400 MHz, CHLOROFORM-d) δ ppm 2.94 (s, 3 H) 3.13 (s, 3 H) 8.85 (s, 2 H) m/z (M+1 ) = 232.
Preparation of Intermediate Ethyl 4-(2-(dimethylcarbamoyl)Dyrimidin-5- yloxy)-2-methylbenzofuran-6-carboxylate (l-2a):

A mixture of Cs2CO3 (62.1 g, 191 mmol), 5-bromo-N,N- dimethylpyrimidine-2-carboxamide (SM-2: 24g, 104mmol) and ethyl 4- hydroxy-2-methylbenzofuran-6-carboxylate (1-1 c: 2Og, 91 mmol); 1 ,10- phenanthroline (1.64g, 9.07mmol) and copper iodide (864mg, 4.54mmol) in dimethylformamide (200ml) was purged with N2 gas and then heated to 90°C using a mechanical stirrer. The heterogeneous reaction mixture was stirred at this temperature for 18 hours. HPLC indicated near completion. The reaction mixture was cooled to 350C and diluted with ethyl acetate (300ml). The mixture was filtered to remove any cesium carbonate. The filtrate was then partitioned between water (500ml) and ethyl acetate (500ml); however, no separation was observed. Concentrated HCL (20ml) was added to the mixture. When the aqueous phase was about pH1 , the phases separated. The organics were separated and the aqueous layer reextracted with ethyl acetate (2x500ml). All organics were combined and back extracted with water (200ml) and brine (500ml). The organics were separated and treated with activated charcoal (10g) and magnesium sulfate. The mixture was allowed to stir for 10 minutes and then filtered through a plug of celite to afford a crude yellow solution. The filter cake was washed with ethyl acetate (100 ml_). The organics were concentrated in vacuo to afford a crude solid this was dried under high vacuum for 4 days. The dry crude solid was triturated using methanol (80 ml_). The solids were dispersed into a fine light orange crystalline powder with a red liquor. The solids were isolated by filtration and rinsed with methanol (20 ml_). The solid was dried in the vacuum oven at 550C for 12 hours to afford ethyl 4-(2- (dimethylcarbamoyl)pyrimidin-5-yloxy)-2-methylbenzofuran-6-carboxylate (J1 2a) as a yellow solid (18.2g, 54%)
1H NMR (400 MHz, CHLOROFORM-d) δ ppm 1.41 (t, J=7.12 Hz, 3 H) 2.50 (d, J=0.98 Hz, 3 H) 3.00 (s, 3 H) 3.17 (s, 3 H) 4.41 (d, J=7.22 Hz, 2 H) 6.29 (s, 1 H) 7.62 (d, J=1.17 Hz, 1 H) 8.06 (s, 1 H) 8.50 (s, 2 H). m/z (M+1 ) = 370.5
Preparation of Starting material 5-bromo-N-ethyl-N-methylpyrimidine-2- carboxamide (SM-3):

(SM-3) Oxalyl chloride (1 .45g, 1 1 .1 mmol) was added to a suspension of 5-
Bromo-pyrimidine-2-carboxylic acid (1 .5g, 7.4mmol) in dichloromethane (50ml) at room temperature followed by 1 -2 drop of dimethylformamide. The reaction mixture was stirred under nitrogen for 2 hours LCMS in methanol indicated the presence of the methyl ester and some acid. Dimethylformamide (0.2ml) was added to the reaction mixture and all of the acid dissolved after 30 minutes. LCMS showed corresponding methyl ester and no starting material peak was observed. The solvent was removed and dried in vacuo to afford the crude 5-Bromo-pyrimidine-2-carbonyl chloride (1 -6g). 5-Bromo-pyrinnidine-2-carbonyl chloride (1600mg, 7.225mnnol) was dissolved in dichloromethane (25ml) and triethylamine (4.03ml, 28.9mmol) was added followed by ethyl-methyl-amine (0.68 mL, 7.92 mmol). The reaction was stirred at room temperature under nitrogen for 16 ours, after which time, LCMS indicated completion. The mixture was diluted with dichloromethane (50ml) and washed with water (50ml) followed by 10% citric acid (50ml) and brine (50ml). The organic layer was separated and dried over MgSO4, the residue was filtered and the solvent was removed in vacuo to afford the title compound 5-bromo-N-ethyl-N-methylpyrimidine-2- carboxamide (SM-3): (1.4g, 79.4%) as a brown oil.
1H NMR (400 MHz, CHLOROFORM-d) δ ppm 1.08 – 1.31 (m, 3 H) 2.99 (d, J=79.05 Hz, 3 H) 3.19 (q, J=7.22 Hz, 1 H) 3.59 (q, J=7.22 Hz, 1 H) 8.84 (d, J=3.12 Hz, 2 H)
Example 2
Preparation of N,N-dimethyl-5-(2-methyl-6-((5-methylpyrazin-2- yl)carbamoyl)-benzofuran-4-yloxy)Dyrimidine-2-carboxamide (2):

(2)
To a solution of the 5-methyl-2-aminopyrazine (38.9 g, 356 mmol) in dimethylether (315 ml_) in a 3-neck flask equipped with overhead stirring and a condensor at O0C was added Me2AICI (1 M solution in hexanes) (715 ml_). The mixture was warmed at room temperature and stirred for 1.5 hours. In a separate flask, ethyl 4-(2-(dimethylcarbamoyl)pyrimidin-5-yloxy)-2- methylbenzofuran-6-carboxylate (l-2a: 52.6g, 142.5mmol) was dissolved in dimethylether (210 ml_). This mixture was then added to the complexed amine. A gum precipitated upon scratching the flask and dissipated into a solid. The resultant reaction was refluxed for 3.5 hours HPLC indicated 93% complete. Five liters of Rochelles salt made up in water and 2 liters of 2- methyltetrahydrofuran was added to the mixture. The reaction mixture was then poured into the biphasic system. The mixture was allowed to stir with overhead stirring for 14 hours, after which time, a yellow solid precipitated. The solid was collected through filteration. The solid retained was washed with 2-methyltetrahydrofuran. The resultant solid was dried in vacuo oven overnight to afford the title compound N,N-dimethyl-5-(2-methyl-6-((5- methylpyrazin-2-yl)carbamoyl)benzofuran-4-yloxy)pyhmidine-2-carboxamide (2): (49.98g, 81 %)
1H NMR (400 MHz, CHLOROFORM-d) d ppm 2.49 (d, J=1 .17 Hz, 3H) 2.55 (s, 3H) 2.98 (s, 3 H) 3.14 (s, 3 H) 6.28 (t, J=0.98 Hz, 1 H) 7.52 (d, J=1 .37 Hz, 1 H) 7.88 – 7.92 (m, 1 H) 8.14 (d, J=0.78 Hz, 1 H) 8.37 (s, 1 H) 8.50 (s, 2 H) 9.54 (d, J=1 .56 Hz, 1 H).
m/z (M+1 ) = 433.4, m/z (M-1 )= 431 .5
REFERENCES
Beebe, D.A.; Ross, T.T.; Rolph, T.P.; Pfefferkorn, J.A.; Esler, W.P.
The glucokinase activator PF-04937319 improves glycemic control in combination with exercise without causing hypoglycemia in diabetic rats
74th Annu Meet Sci Sess Am Diabetes Assoc (ADA) (June 13-17, San Francisco) 2014, Abst 1113-P
Amin, N.B.; Aggarwal, N.; Pall, D.; Paragh, G.; Denney, W.S.; Le, V.; Riggs, M.; Calle, R.A.
Two dose-ranging studies with PF-04937319, a systemic partial activator of glucokinase, as add-on therapy to metformin in adults with type 2 diabetes
Diabetes Obes Metab 2015, 17(8): 751
Study to compare single dose of three modified release formulations of PF-04937319 with immediate release material-sparing-tablet (IR MST) formulation previously studied in adults with type 2 diabetes mellitus (NCT02206607)
OTHERS

///////////Pfizer , PF 04937319, glucokinase activators, Type 2 diabetes
FDA grants breakthrough status for Pfizer’s leukaemia drug inotuzumab ozogamicin


Inotuzumab ozogamicin
RN: 635715-01-4
UNII: P93RUU11P7
Pfizer Inc., Oncology Institute Of Southern Switzerland INNOVATOR
http://chem.sis.nlm.nih.gov/chemidplus/rn/635715-01-4
- MF 1680.6764
-
Oncological Treatment
FDA grants breakthrough status for Pfizer’s leukaemia drug inotuzumab ozogamicin
The US Food and Drug Administration (FDA) has granted breakthrough therapy designation for Pfizer’s investigational antibody-drug conjugate (ADC) inotuzumab ozogamicin to treat acute lymphoblastic leukaemia (ALL).
The US Food and Drug Administration (FDA) has granted breakthrough therapy designation for Pfizer’s investigational antibody-drug conjugate (ADC) inotuzumab ozogamicin to treat acute lymphoblastic leukaemia (ALL).
The breakthrough status was based on data from the Phase III INO-VATE ALL trial, which enrolled 326 adult patients with relapsed or refractory CD22-positive ALL and compared inotuzumab ozogamicin to standard of care chemotherapy………….http://www.pharmaceutical-technology.com/news/newsfda-grants-breakthrough-status-pfizer-leukaemia-drug-inotuzumab-ozogamicin-4697877?WT.mc_id=DN_News

EVER SINCE POST WAS WRITTEN…..FGD APPROVAL Inotuzumab ozogamicin
PFIZER

![]()

| Besponsa | FDA
8/17/2017 |
To treat adults with relapsed or refractory acute lymphoblastic leukemia Press Release Drug Trials Snapshot |
Inotuzumab ozogamicin (CMC-544) is an antibody-drug conjugate for the treatment of cancers.[1] It consists of the humanized monoclonal antibody inotuzumab (for CD22), linked to a cytotoxic agent from the class of calicheamicins (which is reflected by ‘ozogamicin‘ in the drug’s name).[2]
This drug is being developed by Pfizer and UCB.
It is undergoing numerous clinical trials,[3] including two phase II trials for Non-Hodgkin lymphoma (NHL).
A phase III trial in patients with follicular b-cell NHL has been terminated due to poor enrollment.[4] A Phase III trial in patients with relapsed or refractory CD22+ aggressive non-Hodgkin lymphoma (NHL) who were not candidates for intensive high-dose chemotherapy was terminated for futility.[5]
Monoclonal antibodies (mAbs) and derivatives are currently the fastest growing class of therapeutic molecules. More than 30 G-type immunoglobulins (IgG) and related agents have been approved over the past 25 years mainly for cancers and inflammatory diseases. In oncology, mAbs are often combined with cytotoxic drugs to enhance their therapeutic efficacy. Alternatively, small anti-neoplastic molecules can be chemically conjugated to mAbs, used both as carriers (increased half-life) and as targeting agents (selectivity). Potential benefits of antibody-drug conjugates (ADCs), strategies, and development challenges are discussed in this review. Several examples of ADCs are presented with emphasis on three major molecules currently in late clinical development as well as next generation thio-mAbs conjugates with improved therapeutic index.

PATENT
http://www.google.com/patents/WO2013088304A1?cl=en
Inotuzumab ozogamicin:

is described in U.S. Patent Application No. 10/428894
U.S. Patent Application No. 10/428894


References
- Statement On A Nonproprietary Name Adopted By The Usan Council – Inotuzumab ozogamicin, American Medical Association.
- Takeshita, A; Shinjo, K; Yamakage, N; Ono, T; Hirano, I; Matsui, H; Shigeno, K; Nakamura, S; Tobita, T; Maekawa, M (2009). “CMC-544 (inotuzumab ozogamicin) shows less effect on multidrug resistant cells: analyses in cell lines and cells from patients with B-cell chronic lymphocytic leukaemia and lymphoma.”. British journal of haematology 146 (1): 34–43.doi:10.1111/j.1365-2141.2009.07701.x. PMID 19388933.
- http://clinicaltrials.gov/ct2/results?term=Inotuzumab+ozogamicin
- http://clinicaltrials.gov/ct2/show/NCT00562965
- http://pfizer.newshq.businesswire.com/press-release/pfizer-discontinues-phase-3-study-inotuzumab-ozogamicin-relapsed-or-refractory-aggress
- http://pubs.rsc.org/en/content/articlelanding/2008/np/b514294f#!divAbstract
Structure of inotuzumab ozogamicin. ABOVE
| Monoclonal antibody | |
|---|---|
| Type | Whole antibody |
| Source | Humanized (from mouse) |
| Target | CD22 |
| Identifiers | |
| CAS Registry Number | 635715-01-4 |
| ATC code | None |
| UNII | P93RUU11P7 |
| KEGG | D08933 |
| Chemical data | |
| Formula | C6518H10002N1738O2036S42 |
| Molecular mass | 150,000 Daltons |
//////////
Sandoz, Pfizer closing Mumbai API plant

Sandoz closing Mumbai API plant but says it remains committed to India

Sandoz will shutter an Indian API facility in 2016 as part of a manufacturing refocus in the region.
From online sources
Drug major Sandoz will discontinue operations at its Turbhe site (Maharashtra) by end December 2016, as part of global plans to optimise its manufacturing network.

The Turbhe sites employs 170 people and manufactures antibiotics and active pharmaceutical ingredients (API), a note from the company said. Sandoz is the generic drugs arm of pharmaceutical company Novartis.
“Sandoz will refocus its manufacturing set up in India as part of its strategy to optimise its global manufacturing network, while continuing to serve patients in India,” the company said. As part of the plan, Sandoz will focus its manufacturing at other sites which employ over 1,300 employees and produce over three billion tablets and 180 tonnes of API annually, it added. The company has two manufacturing facilities at Kalwe and Mahad.
“We made the announcement today to ensure our associates are informed as soon as possible about our decisions and to ensure a transparent process,” Vivek Devaraj, Sandoz Country Head in India, said in the statement. “We are committed to managing the process with care, sensitivity and respect for all impacted associates at Turbhe, to supporting our customers through the transition and to meeting patient needs for access to important medicines,” he added.
In 2012, the company had shut down its formulations and API development centres, respectively. Drug companies have in the past shut down plants in India as a fallout of global strategies, mergers and acquisitions. At present, Pfizer’s plant in Thane faces an uncertain future.

……………………
ABOUT PFIZER
 There has been practically no production at Pfizer’s Thane plant (pic above) since 2013.
There has been practically no production at Pfizer’s Thane plant (pic above) since 2013.

Sandoz India’s Turbhe plant to down shutters by December 2016
Less than a week after Sandoz, the generic division of Novartis, announced that it would discontinue operations at its Turbhe site by end December 2016, another MNC, Pfizer India announced the closure of its manufacturing facility at Thane, two months from today, from September 16, 2015.
According to Pfizer India spokesperson, the Thane plant was commissioned in the 1960s, manufacturing medicines for both domestic and international markets but there has been ‘practically been no production activity at this plant since 2013′. Hence closure of the site would not impact supply of Pfizer India’s medicines.
Both plant closures are a consolidation of manufacturing facilities, with the shutting down of older facilities and re-direction to more modern facilities, with Pfizer India’s statement attributing the decision to ‘an assessment of its long term viability and its ability to achieve the needed production.’
132 of the 212 Pfizer India workmen at the Thane plant had already taken up the voluntary retirement scheme (VRS) offered by the company and the statement indicated that the remaining 80 workmen who continued to receive full wages despite plant inactivity, would also receive requisite compensation as mandated by law.
While the close down process is in the final stages at Pfizer India’s Thane facility, Sandoz’ July 10 announcement is the beginning of the process at its Turbhe plant, which employs 170 associates and manufactures antibiotics and APIs.
“We made the announcement (on July 10) to ensure our associates are informed as soon as possible about our decisions and to ensure a transparent process,” said Vivek Devaraj, Sandoz Country Head in India. He said the company was “committed to managing the process with the utmost care, sensitivity and respect for all impacted associates at Turbhe, to supporting our customers through the transition and to meeting patient needs for access to important medicines.” Manufacturing would now focus at its other sites which employ over 1,300 associates and produce over three billion tablets and 180 tonnes of API annually.”
/////////Sandoz, shutter, Indian API facility, Pfizer
GOSOGLIPTIN
GOSOGLIPTIN
CAS 869490-23-3 FREE BASE
DIHYDROCHLORIDE..869490-47-1
GOSOGLIPTIN; UNII-GI718UO477; PF-00734200; PF-734200;
(3,3-difluoropyrrolidin-1-yl)-[(2S,4S)-4-(4-pyrimidin-2-ylpiperazin-1-yl)pyrrolidin-2-yl]methanone
| Molecular Formula: | C17H24F2N6O |
|---|---|
| Molecular Weight: | 366.408866 g/mol |
| Company | Pfizer Inc. |
| Description | Dipeptidyl peptidase-4 (DPP-4) inhibitor |
| Molecular Target | Dipeptidyl peptidase-4 (DPP-4) (CD26) |
| Mechanism of Action | Dipeptidyl peptidase-4 (DPP-4) inhibitor |
| Latest Stage of Development | Phase II |
| Standard Indication | Diabetes |
| Indication Details | Treat Type II diabetes |
Type 2 diabetes mellitus is a chronic disorder characterized by hyperglycemia coupled with a gradual decline in insulin sensitivity and insulin secretion. The incretin hormone glucagon-like peptide-1 (GLP-1), which is released post-prandially from the L-cells of the intestine, stimulates the release of insulin from pancreatic β-cells. However, GLP-1 is rapidly degraded in vivo by peptidases, including dipeptidyl peptidase IV (DPP-4), which is a widely distributed serine protease that specifically cleaves N-terminal dipeptides from polypeptides with proline or alanine at the penultimate position.
In vivo administration of DPP-4 inhibitors to human subjects results in higher circulating concentrations of endogenous GLP-1 and subsequent decrease in plasma glucose. Long term treatment with a DPP-4 inhibitor leads to a reduction in circulating HbA1c (glycosylated hemoglobin). DPP-4 inhibition also offers the potential to improve the insulin producing function of the pancreas through either β-cell preservation or regeneration. Therefore, DPP-4 inhibition has emerged as a promising new treatment of Type 2 diabetes
PF-734200 is a potent, selective, orally active dipeptidyl peptidase IV inhibitor. It had been in phase II clinical development at Pfizer for the treatment of type 2 diabetes; however, in 2010 the company discontinued these trials. In 2012, the product was licensed to SatRx, a spin-off of the ChemRar High Tech Center, by Pfizer on an exclusive worldwide basis (with the exception of China) for the development and commercialization as monotherapy or in combination with other therapies for the treatment of type 2 diabetes. SatRx is conducting phase II clinical trials for the treatment of type 2 diabetes.
……………………….
PAPER
New synthetic route to a dipeptidyl peptidase-4 inhibitor
Org Process Res Dev 2012, 16(3): 409
http://pubs.acs.org/doi/abs/10.1021/op200309z

A new synthetic route to a dipeptidyl peptidase-4 (DPP4) inhibitor was developed and demonstrated on a multigram scale. This approach takes advantage of the cheap and readily available Boc-trans-4-hydroxy-l-proline methyl ester as starting material which was derivatized through an SN2 reaction. Several leaving groups were studied, and the nosylate group showed superiority over other derivatives. Formation of an amide using the most costly starting material, 3,3-difluoropyrrolidine, was performed late in the synthesis to minimize its economical impact on the overall cost of the API.
(3,3-Difluoropyrrolidin-1-yl)-(2S,4S)-4-(4-(pyrimidin-2-yl)piperazin-1-yl)pyrrolidin-2-yl)methanone.FREE BASE
Mp 149 °C (decomp).
[α]d = −31.1 (T = 24 °C, c = 1, CHCl3). Specific rotation of product 4 prepared using the initial route: [α]d = −31.5 (T = 24 °C, c = 1, CHCl3).
1H NMR (400 MHz; CDCl3) δ 8.30 (d, J = 4 Hz, 2H), 6.48 (t, J = 4 Hz, 1H), 3.95–3.6 (m, 9H), 3.25–2.85 (m, 4H), 2.6–2.25 (m, 7H), 1.75–1.6 (m, 1H).
13C NMR (100 MHz; CDCl3) δ 172.28; 161.55; 157.70; 127.22 (t, 1J C–F = 248 Hz), 126.22 (t, 1J C–F = 246 Hz), 109.95; 66.54; 58.87; 57.99; 52.71 (t, 2 J C–F = 32 Hz); 52.00; 50.41; 43.03; 34.46, 34.37, 34.25; 19F NMR (377 MHz, CDCl3) δ −102.1 (m, 2F).
IR (neat): 2951w, 2864w, 2799w, 2759w, 1630s, 1585vs, 1547m, 1449m, 1172m, 1254m, 1129m, 982w, 923m, 796m, 638w.
HRMS (ES, N2) Calcd for C17H24F2N6O: 367.20524, found: 367.20592.
……………………….
PAPER
(3,3-difluoro-pyrrolidin-1-yl)-((2S,4S)-(4-(4-pyrimidin-2-yl-piperazin-1-yl)-pyrrolidin-2-yl)-methanone: A potent, selective, orally active dipeptidyl peptidase IV inhibitor
Bioorg Med Chem Lett 2009, 19(7): 1991
http://www.sciencedirect.com/science/article/pii/S0960894X09001966?np=y
- Pfizer Global Research & Development, Groton/New London Laboratories, Pfizer Inc, Groton, CT 06340, United States
A series of 4-substituted proline amides was evaluated as inhibitors of dipeptidyl pepdidase IV for the treatment of type 2 diabetes. (3,3-Difluoro-pyrrolidin-1-yl)-[(2S,4S)-(4-(4-pyrimidin-2-yl-piperazin-1-yl)-pyrrolidin-2-yl]-methanone (5) emerged as a potent (IC50 = 13 nM) and selective compound, with high oral bioavailability in preclinical species.

………………….
PATENT
WO 2005116014
http://www.google.co.in/patents/WO2005116014A1?cl=en
Example 113 (3.3-Difluoropyrrolidin-1-yl)-((2S,4S)-4-(4-(pyrimidin-2-yl)piperazin-1-yl)pyrrolidin-2-yl)-methanone

Step 1 – (S)-2-(3.3-Difluoro-pyrrolidine-1-carbonyl)-4-oxo-pyrrolidine-1 -carboxylic acid tert-butyl ester
(S)-4-Oxo-pyrrolidine-1 ,2-dicarboxylic acid 1-tert-butyl ester (6.6 kg, 1.0 equivalent) was charged to a reactor, followed by addition of dichloromethane (15 volumes). The reaction mixture was cooled to 0°C. Triethylamine (4.82 liters, 1.2 equiv) was added over 30 minutes. The mixture turned from suspension to a clear solution at the end of triethylamine addition. The mixture was held at 0°C to 5°C for 10 minutes. Pivaloyl chloride (3.65 kg, 1.05 equivalents) was added slowly while keeping the reaction temperature at 0°C to 5°C. The reaction mixture turned back to aslurry. The reaction mixture was sampled for completion by HPLC (using diethylamine to derivatize) after held for 1 hour at 0°C to 5°C.
3,3-Difluoro- pyrrolidine hydrochloride (4.13 kg, 1.0 equivalent) was charged to the above mixture over 10 minutes at – 10°C to 0°C. Triethylamine (4.0 liters, 1.0 equiv) was introduced slowly over 70 minutes at -10°C to 0°C. Upon completion of triethylamine addition, the mixture was stirred for 1h at 0 to 5°C. The reaction was complete by HPLC assay (-1% starting material). The reaction was quenched with water (10 volumes) at 0°C to 5 °C. The mixture was heated to 20°C to 25 °C. The layers were separated, and the organic layer was washed with 0.5 M HCI (5 volumes). The organic layer was again washed with combined 5% NaHC03 (2 volumes) and half saturated brine solution (1.64 M, 3 volumes). The organic solution was concentrated atmospherically to a low stirrable volume (approximately 20 liters). Ethyl acetate (12.6 volumes, 82.8 liters) was added, the solution was concentrated atmospherically to -6 volumes. The mixture was held at 60°C to 65 °C for 2 hours and cooled to room temperature over 3 hours. The mixture was held at 20°C to 25 °C for 8 hours. Heptane (8 volumes) was added, and the mixture was granulated for a minimum of 2 hours. The solid was filtered, rinsed with 2:1 heptane/ethyl acetate (1 volume), and dried in a tray dryer at 25°C to 35°C for a minimum of 12 h. Yield: 7.26 kg, 79%. HPLC purity: 99.7%. The mother liquor (86 liters) was concentrated to 12 liters under partial vacuum at 65°C to 70°C. The mixture was cooled to 60°C to 65 °C. Ethyl acetate (4.0 liters) was added slowly over 15 minutes. The mixture was cooled to 20°C to 25 °C over 2 hours and was held at that temperature for at least 2 hours. The solid was filtered and rinsed with heptane/ethyl acetate (3:1 v/v, 1.7 liters). Drying in a tray dryer for 12 hours at 35°C to 45 °C yielded 435 grams of product. HPLC purity: 96.4%.
Step 2 – (2S.4S)-2-(3.3-Dif luoro-pyrrolidine-1 -carbonyl)-4-(4-pyrimidin-2-yl-piperazin-1 -yl)-pyrrolidine-1 – carboxylic acid tert-butyl ester A reactor was charged with THF (20 volumes), 2-piperazin-1-yl-pyrimidine (2.17 kg, 1.05 equivalents) and the product from Step 1 (4.00 kg, 1.0 equivalent). The mixture was held at 20°C to 25°C until all material was dissolved over 30 minutes. Acetic acid (0.792 kg, 1.05 equivalents) as added. The mixture was stirred for 1 hour during which the reaction mixture turned to cloudy. The reaction mixture was refluxed for 30 minutes and then concentrated at 60°C to 70°C until a steady temperature of 66.9°C was observed in the overheads indicating complete removal of water from the system. More THF was added as necessary. At the end, THF was added to bring the total volume in the reactor to 15 volumes of the limit reagent. The reaction mixture was cooled to -3°C to 7°C and sampled for complete formation of imine by HPLC (using sodium triacetoxyborohydride to reduce imine). Sodium triacetoxyborohydride (5.33 kg, 2.0 equivalents) was added portion-wise to the suspension at -5°C to 15°C. The reaction mixture was heated to 20°C to 25°C and held for 12 hours. HPLC results confirmed the reaction was complete by 99.8%. Sodium bicarbonate aqueous solution (10% w/w, 10 volumes) was added. The slurry was concentrated to remove 10 volumes of THF under partial vacuum at 30°C to 60°C. Ethyl acetate (10 volumes) was added to the suspension after it cooled to 20°C to 25CC. The organic phase was separated and the aqueous phase was checked by HPLC. It contained less than 2% of the product. The organic phase was washed with water (5 volumes), saturated brine solution (5 volumes) and concentrated to a small volume (2 volumes) under partial vacuum at 45°C to 50°C. To the slurry was added heptane (10 volumes) at 45°C to 50°C over 30 minutes. The mixture was cooled to 20°C to 25°C and granulated for 2 hours. Solid was collected by filtration, rinsed with heptane (2 volumes). Drying in a tray dryer for 12 hours at 35°C to 45°C yield 5.35 kg (91.3%) of the product. Step 3 – (3.3-Dif luoro-pyrrolidin-1 -yl)-f(2S.4S)-4-(4-pyrimidin-2-yl-piperazin-1 -yl)-pyrrolidin-2-yll- methanone Water (19 liters, 2 volumes) was charged to a reactor followed by the product from Step 2 (9.57 kg,
1.0 equivalent). To the slurry was added concentrated HCI (37 wt% in water, 19.1 liters, 2 volumes) slowly at 20°C to 30°C over 4 hours. The slurry went into solution after 12 liters of HCI was added. After the addition completion, the reaction was complete by HPLC assay. The reaction mixture was cooled to 5°C to 15°C. To the mixture was added 50% NaOH aqueous solution slowly with agitation to pH 10 to pH 11. The pH was monitored with a pH meter closely during the neutralization. The total volume of 50% NaOH added was 12.45 liters. The mixture was warmed to 20°C to 25°C and extracted with ethyl acetate twice (115 liters, 12 volumes and 57 liters, 6 volumes, respectively). The sample from aqueous layer after second extraction was analyzed by HPLC and showed only 1% of the product in that aqueous solution.
The organic layers were combined and treated with magnesium sulfate (5 kg) for 1 hour. The mixture was filtered. The filter cake was rinsed with ethyl acetate (10 liters). The filtrate was charged back to the reactor via a 0.2 micron in-line filter for speck free operation. (The following operations were performed under speck free conditions.) The solution was concentrated to 20 liters (2 volumes) under partial vacuum at 50°C to 60°C. The mixture was cooled to 20°C to 25°C over 30 minutes. Upon cooling to room temperature, crystallization occurred. The mixture was held for 30 minutes. Hexanes (20 liters, 2 volumes) was added slowly over 1 hour. The mixture was granulated for 2 hours. The solid product was collected by filtration and rinsed with hexanes/ethyl acetate (10 liters, 1 :1 v/v). The filter was blown dry with nitrogen for a minimum of 2 hours. The product was dried in a tray dryer at 44°C for 12 hours.
Yield: 5.7 kg, 75.9%.
m.p. 156°C. MS m/z 367 (MH+).
FREE BASE
1H NMR (400 MHz, D20): δ 8.15 (d, 2H, J = 5.0 Hz, CH of pyrimidine), 6.55 (t, 1 H, J = 4.8 Hz, CH of pyrimidine), 3.87-3.81 (dd, 1 H, H2b of proline, rotomeric), 3.78-3.50 (m, 4H, N-CH2 of pyrrolidide), 3.55-3.40 (m, 4H, N-CH2 of piperazine), 2.97 (dd, 1 H, J = 10.2, 6.6 Hz, H5a of proline), 2.85-2.75 (m, 1 H, H4b of proline), 2.69 (dd, 1 H, J = 10.0, 9.1 Hz, H5b of proline), 2.55-2.20 (m, 7H, overlapping N-CH2 of piperazine, CH2 of pyrrolidide and H3b of proline), 1.47-1.38 (m, 1 H, H3a of proline).
Alternatively, the dihydrochloride salt of the titled compound was prepared according to the method of Example 1.
………………
US 2005/0256310
http://www.google.com/patents/US20050256310

This approach begins with N–t-Boc-4-oxo-l-proline (1) that undergoes a mixed anhydride activation with pivaloyl chloride at 0 °C, followed by amidation with 3,3-difluoropyrrolidine to yield the intermediate 2. Reductive amination with 1-(2-pyrimidyl)piperazine using sodium triacetoxyborohydride in THF/AcOH provided the desired stereoisomer 3 in high yield and selectivity, the undesired diastereomer being completely removed by crystallization. Deprotection of 3 with 6 N HCl, followed by neutralization with 50% NaOH and extraction provided PF-734200 (4) in good yield.
EXAMPLE 113 (3,3-Difluoropyrrolidin-1-yl)-((2S,4S)-4-(4-(pyrimidin-2-yl)piperazin-1-yl)pyrrolidin-2-yl)-methanone

Step 1—(S)-2-(3,3-Difluoro-pyrrolidine-1-carbonyl)-4-oxo-pyrrolidine-1-carboxylic acid tert-butyl
(S)-4-Oxo-pyrrolidine-1,2-dicarboxylic acid 1-tert-butyl ester (6.6 kg, 1.0 equivalent) was charged to a reactor, followed by addition of dichloromethane (15 volumes). The reaction mixture was cooled to 0° C. Triethylamine (4.82 liters, 1.2 equiv) was added over 30 minutes. The mixture turned from suspension to a clear solution at the end of triethylamine addition. The mixture was held at 0° C. to 5° C. for 10 minutes. Pivaloyl chloride (3.65 kg, 1.05 equivalents) was added slowly while keeping the reaction temperature at 0° C. to 5° C. The reaction mixture turned back to a slurry. The reaction mixture was sampled for completion by HPLC (using diethylamine to derivatize) after held for 1 hour at 0° C. to 5° C. 3,3-Difluoro-pyrrolidine hydrochloride (4.13 kg, 1.0 equivalent) was charged to the above mixture over 10 minutes at −10° C. to 0° C. Triethylamine (4.0 liters, 1.0 equiv) was introduced slowly over 70 minutes at −10° C. to 0° C. Upon completion of triethylamine addition, the mixture was stirred for 1 h at 0 to 5° C. The reaction was complete by HPLC assay (˜1% starting material). The reaction was quenched with water (10 volumes) at 0° C. to 5 ° C. The mixture was heated to 20° C. to 25 ° C. The layers were separated, organic layer was washed with 0.5 M HCl (5 volumes). The organic layer was again washed with combined 5% NaHCO3 (2 volumes) and half saturated brine solution (1.64 M, 3 volumes). The organic solution was concentrated atmospherically to a low stirrable volume (approximately 20 liters). Ethyl acetate (12.6 volumes, 82.8 liters) was added, the solution was concentrated atmospherically to ˜6 volumes. The mixture was held at 60° C. to 65° C. for 2 hours and cooled to room temperature over 3 hours. The mixture was held at 20° C. to 25 ° C. for 8 hours. Heptane (8 volumes) was added, and the mixture was granulated for a minimum of 2 hours. The solid was filtered, rinsed with 2:1 heptane/ethyl acetate (1 volume), and dried in a tray dryer at 25° C. to 35° C. for a minimum of 12 h. Yield: 7.26 kg, 79%. HPLC purity: 99.7%. The mother liquor (86 liters) was concentrated to 12 liters under partial vacuum at 65° C. to 70° C. The mixture was cooled to 60° C. to 65° C. Ethyl acetate (4.0 liters) was added slowly over 15 minutes. The mixture was cooled to 20° C. to 25° C. over 2 hours and was held at that temperature for at least 2 hours. The solid was filtered and rinsed with heptane/ethyl acetate (3:1 v/v, 1.7 liters). Drying in a tray dryer for 12 hours at 35° C. to 45° C. yielded 435 grams of product. HPLC purity: 96.4%.
Step 2—(2S,4S)-2-(3,3-Difluoro-pyrrolidine-1-carbonyl)-4-(4-pyrimidin-2-yl-piperazin-1-yl)-pyrrolidine-1-carboxylic acid tert-butyl ester
A reactor was charged with THF (20 volumes), 2-piperazin-1-yl-pyrimidine (2.17 kg, 1.05 equivalents) and the product from Step 1 (4.00 kg, 1.0 equivalent). The mixture was held at 20° C. to 25° C. until all material was dissolved over 30 minutes. Acetic acid (0.792 kg, 1.05 equivalents) as added. The mixture was stirred for 1 hour during which the reaction mixture turned to cloudy. The reaction mixture was refluxed for 30 minutes and then concentrated at 60° C. to 70° C. until a steady temperature of 66.9° C. was observed in the overheads indicating complete removal of water from the system. More THF was added as necessary. At the end, THF was added to bring the total volume in the reactor to 15 volumes of the limit reagent. The reaction mixture was cooled to −3° C. to 7° C. and sampled for complete formation of imine by HPLC (using sodium triacetoxyborohydride to reduce imine). Sodium triacetoxyborohydride (5.33 kg, 2.0 equivalents) was added portion-wise to the suspension at −5° C. to 15° C. The reaction mixture was heated to 20° C. to 25° C. and held for 12 hours. HPLC results confirmed the reaction was complete by 99.8%. Sodium bicarbonate aqueous solution (10% w/w, 10 volumes) was added. The slurry was concentrated to remove 10 volumes of THF under partial vacuum at 30° C. to 60° C. Ethyl acetate (10 volumes) was added to the suspension after it cooled to 20° C. to 25° C. The organic phase was separated and the aqueous phase was checked by HPLC. It contained less than 2% of the product. The organic phase was washed with water (5 volumes), saturated brine solution (5 volumes) and concentrated to a small volume (2 volumes) under partial vacuum at 45° C. to 50° C. To the slurry was added heptane (10 volumes) at 45° C. to 50° C. over 30 minutes. The mixture was cooled to 20° C. to 25° C. and granulated for 2 hours. Solid was collected by filtration, rinsed with heptane (2 volumes). Drying in a tray dryer for 12 hours at 35° C. to 45° C. yield 5.35 kg (91.3%) of the product.
Step 3—(3,3-Difluoro-pyrrolidin-1-yl)-[(2S,4S)-4-(4-pyrimidin-2-yl-piperazin-1-yl)-pyrrolidin-2-yl]-methanone
Water (19 liters, 2 volumes) was charged to a reactor followed by the product from Step 2 (9.57 kg, 1.0 equivalent). To the slurry was added concentrated HCl (37 wt % in water, 19.1 liters, 2 volumes) slowly at 20° C. to 30° C. over 4 hours. The slurry went into solution after 12 liters of HCl was added. After the addition completion, the reaction was complete by HPLC assay. The reaction mixture was cooled to 5° C. to 15° C. To the mixture was added 50% NaOH aqueous solution slowly with agitation to pH 10 to pH 11. The pH was monitored with a pH meter closely during the neutralization. The total volume of 50% NaOH added was 12.45 liters. The mixture was warmed to 20° C. to 25° C. and extracted with ethyl acetate twice (115 liters, 12 volumes and 57 liters, 6 volumes, respectively). The sample from aqueous layer after second extraction was analyzed by HPLC and showed only 1% of the product in that aqueous solution. The organic layers were combined and treated with magnesium sulfate (5 kg) for 1 hour. The mixture was filtered. The filter cake was rinsed with ethyl acetate (10 liters). The filtrate was charged back to the reactor via a 0.2 micron in-line filter for speck free operation. (The following operations were performed under speck free conditions.) The solution was concentrated to 20 liters (2 volumes) under partial vacuum at 50° C. to 60° C. The mixture was cooled to 20° C. to 25° C. over 30 minutes. Upon cooling to room temperature, crystallization occurred. The mixture was held for 30 minutes. Hexanes (20 liters, 2 volumes) was added slowly over 1 hour. The mixture was granulated for 2 hours. The solid product was collected by filtration and rinsed with hexanes/ethyl acetate (10 liters, 1:1 v/v). The filter was blown dry with nitrogen for a minimum of 2 hours. The product was dried in a tray dryer at 44° C. for 12 hours.
Yield: 5.7 kg, 75.9%. m.p. 156° C. MS m/z 367 (MH+).
1H NMR (400 MHz, D2O): δ 8.15 (d, 2H, J=5.0 Hz, CH of pyrimidine), 6.55 (t, 1H, J=4.8 Hz, CH of pyrimidine), 3.87-3.81 (dd, 1H, H2b of proline, rotomeric), 3.78-3.50 (m, 4H, N—CH2 of pyrrolidide), 3.55-3.40 (m, 4H, N—CH2 of piperazine), 2.97 (dd, 1H, J=10.2, 6.6 Hz, H5a of proline), 2.85-2.75 (m, 1H, H4b of proline), 2.69 (dd, 1H, J=10.0, 9.1 Hz, H5b of proline), 2.55-2.20 (m, 7H, overlapping N—CH2 of piperazine, CH2 of pyrrolidide and H3b of proline), 1.47-1.38 (m, 1H, H3a of proline).
Alternatively, the dihydrochloride salt of the titled compound was prepared according to the method of Example 1.
……………..
PAPER

Scheme 1.
Reagents and conditions: (a) 3,3-difluoropyrrolidine hydrochloride, EDC, HOBt, TEA, DCM, rt; (b) NaBH4, MeOH, (c) (1) trifluoromethane-sulphonyl chloride, DIPEA, DCM; (2) 2-(1-piperazinyl)pyrimidine, DCM, −10 °C; (d) 4 N HCl in dioxane, rt; (e) 2-(1-piperazinyl)pyrimidine, NaBH(OAc)3, AcOH, DCE; (f) R1R2NH hydrochloride, EDC, HOBt TEA, DCM, 0–rt; (g) N-heterocyclic piperazine, NaBH(OAc)3, AcOH, DCE.
……………………….
if image is not clear see at………..http://www.allfordrugs.com/2015/07/03/gosogliptin/
| Patent | Submitted | Granted |
|---|---|---|
| Therapeutic compounds [US7291618] | 2005-11-17 | 2007-11-06 |
| (2S,4S)-4-(piperazin-1-yl)pyrrolidine-2-methanone derivatives [US7465732] | 2007-05-03 | 2008-12-16 |
| THERAPEUTIC COMPOUNDS [US2007161664] | 2007-07-12 | |
| Therapeutic compounds [US2006079498] | 2006-04-13 |
//////////
see gliptins at…………http://drugsynthesisint.blogspot.in/p/gliptin-series.html

{kind=link}