
Home » Posts tagged 'PFIZER' (Page 2)
Tag Archives: PFIZER
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
FDA approves Mylotarg (gemtuzumab ozogamicin) for treatment of acute myeloid leukemia
The U.S. Food and Drug Administration today approved Mylotarg (gemtuzumab ozogamicin) for the treatment of adults with newly diagnosed acute myeloid leukemia whose tumors express the CD33 antigen (CD33-positive AML). The FDA also approved Mylotarg for the treatment of patients aged 2 years and older with CD33-positive AML who have experienced a relapse or who have not responded to initial treatment (refractory).
Mylotarg originally received accelerated approval in May 2000 as a stand-alone treatment for older patients with CD33-positive AML who had experienced a relapse. Mylotarg was voluntarily withdrawn from the market after subsequent confirmatory trials failed to verify clinical benefit and demonstrated safety concerns, including a high number of early deaths. Today’s approval includes a lower recommended dose, a different schedule in combination with chemotherapy or on its own, and a new patient population.
“We are approving Mylotarg after a careful review of the new dosing regimen, which has shown that the benefits of this treatment outweigh the risk,” said Richard Pazdur, M.D., director of the FDA’s Oncology Center of Excellence and acting director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “Mylotarg’s history underscores the importance of examining alternative dosing, scheduling, and administration of therapies for patients with cancer, especially in those who may be most vulnerable to the side effects of treatment.”
AML is a rapidly progressing cancer that forms in the bone marrow and results in an increased number of white blood cells in the bloodstream. The National Cancer Institute of the National Institutes of Health estimates that approximately 21,380 people will be diagnosed with AML this year and that 10,590 patients with AML will die of the disease.
Mylotarg is a targeted therapy that consists of an antibody connected to an anti-tumor agent that is toxic to cells. It is thought to work by taking the anti-tumor agent to the AML cells that express the CD33 antigen, blocking the growth of cancerous cells and causing cell death.
The safety and efficacy of Mylotarg in combination with chemotherapy for adults were studied in a trial of 271 patients with newly diagnosed CD33-positive AML who were randomized to receive Mylotarg in combination with daunorubicin and cytarabine or to receive daunorubicin and cytarabine without Mylotarg. The trial measured “event-free survival,” or how long patients went without certain complications, including failure to respond to treatment, disease relapse or death, from the date they started the trial. Patients who received Mylotarg in combination with chemotherapy went longer without complications than those who received chemotherapy alone (median, event-free survival 17.3 months vs. 9.5 months).
The safety and efficacy of Mylotarg as a stand-alone treatment were studied in two, separate trials. The first trial included 237 patients with newly diagnosed AML who could not tolerate or chose not to receive intensive chemotherapy. Patients were randomized to receive treatment with Mylotarg or best supportive care. The trial measured “overall survival,” or how long patients survived from the date they started the trial. Patients who received Mylotarg survived longer than those who received only best supportive care (median overall survival 4.9 months vs. 3.6 months). The second trial was a single-arm study that included 57 patients with CD33-positive AML who had experienced one relapse of disease. Patients received a single course of Mylotarg. The trial measured how many patients achieved a complete remission. Following treatment with Mylotarg, 26 percent of patients achieved a complete remission that lasted a median 11.6 months.
Common side effects of Mylotarg include fever (pyrexia), nausea, infection, vomiting, bleeding, low levels of platelets in the blood (thrombocytopenia), swelling and sores in the mouth (stomatitis), constipation, rash, headache, elevated liver function tests, and low levels of certain white blood cells (neutropenia). Severe side effects of Mylotarg include low blood counts, infections, liver damage, blockage of the veins in the liver (hepatic veno-occlusive disease), infusion-related reactions, and severe bleeding (hemorrhage). Women who are pregnant or breastfeeding should not take Mylotarg, because it may cause harm to a developing fetus or a newborn baby. Patients with hypersensitivity to Mylotarg or any component of its formulation should not use Mylotarg.
The prescribing information for Mylotarg includes a boxed warning that severe or fatal liver damage (hepatotoxicity), including blockage of veins in the liver (veno-occlusive disease or sinusoidal obstruction syndrome), occurred in some patients who took Mylotarg.
Mylotarg received Orphan Drug designation, which provides incentives to assist and encourage the development of drugs for rare diseases.
The FDA granted the approval of Mylotarg to Pfizer Inc.



| Monoclonal antibody | |
|---|---|
| Type | Whole antibody |
| Source | Humanized (from mouse) |
| Target | CD33 |
| Clinical data | |
| Trade names | Mylotarg |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a607075 |
| Pregnancy category |
|
| Routes of administration |
Intravenous |
| ATC code | |
| Legal status | |
| Legal status |
|
| Identifiers | |
| CAS Number | |
| DrugBank | |
| ChemSpider |
|
| KEGG | |
| ChEMBL | |
| Chemical and physical data | |
| Molar mass | 151–153 g/mol |
Gemtuzumab ozogamicin (marketed by Wyeth as Mylotarg) is a drug-linked monoclonal antibody (an antibody-drug conjugate) that was used to treat acute myelogenous leukemia from 2000 to 2010. It was withdrawn from market in June 2010 when a clinical trial showed the drug increased patient death and added no benefit over conventional cancer therapies.
Mechanism and side effects
Gemtuzumab is a monoclonal antibody to CD33 linked to a cytotoxic agent from the class of calicheamicins. CD33 is expressed in most leukemic blast cells but also in normal hematopoietic cells, the intensity diminishing with maturation of stem cells.
Common side effects of administration included shivering, fever, nausea and vomiting. Serious side effects included severe myelosuppression (suppressed activity of bone marrow, which is involved in formation of various blood cells [found in 98% of patients]), disorder of the respiratory system, tumor lysis syndrome, Type III hypersensitivity, venous occlusion, and death.
History
Gemtuzumab ozogamicin was created in a collaboration between Celltech and Wyeth that began in 1991.[1][2] The same collaboration later produced inotuzumab ozogamicin.[3] Celltech was acquired by UCB in 2004[4] and Wyeth was acquired by Pfizer in 2009.[5]
In the United States, it was approved under an accelerated-approval process by the FDA in 2000 for use in patients over the age of 60 with relapsed acute myelogenous leukemia (AML); or those who are not considered candidates for standard chemotherapy.[6] The accelerated approval was based on the surrogate endpoint of response rate.[7] It was the first antibody-drug conjugate to be approved.[8]
Within the first year after approval, the FDA required a black box warning be added to Gemtuzumab packaging. The drug was noted to increase the risk of veno-occlusive disease in the absence of bone marrow transplantation.[9] Later the onset of VOD was shown to occur at increased frequency in Gemtuzumab patients even following bone marrow transplantation.[10] The drug was discussed in a 2008 JAMA article, which criticized the inadequacy of postmarketing surveillance of biologic agents.[11]
A randomized phase 3 comparative controlled trial (SWOG S0106) was initiated in 2004 by Wyeth in accordance with the FDA accelerated-approval process. The study was stopped[when?] prior to completion due to worrisome outcomes. Among the patients evaluated for early toxicity, fatal toxicity rate was significantly higher in the gemtuzumab combination therapy group vs the standard therapy group. Mortality was 5.7% with gemtuzumab and 1.4% without the agent (16/283 = 5.7% vs 4/281 = 1.4%; P = .01).[7]
In June 2010, Pfizer withdrew Mylotarg from the market at the request of the US FDA.[12][13] However, some other regulatory authorities did not agree with the FDA decision, with Japan’s Pharmaceuticals and Medical Devices Agency stating in 2011 that the “risk-benefit balance of gemtuzumab ozogamicin has not changed from its state at the time of approval”.[14]
In early 2017 Pfizer reapplied for US and EU approval, based on a meta-analysis of prior trials and results of the ALFA-0701 clinical trial, an open-label Phase III trial in 280 older people with AML. [8]
References
- Jump up^ “Mylotarg”. Informa Biomedtracker. Retrieved 19 August 2017.
- Jump up^ Niculescu-Duvaz, I (December 2000). “Technology evaluation: gemtuzumab ozogamicin, Celltech Group.”. Current opinion in molecular therapeutics. 2 (6): 691–6. PMID 11249747.
- Jump up^ Damle, NK; Frost, P (August 2003). “Antibody-targeted chemotherapy with immunoconjugates of calicheamicin.”. Current opinion in pharmacology. 3 (4): 386–90. PMID 12901947. doi:10.1016/S1471-4892(03)00083-3.
- Jump up^ “Celltech sold to Belgian firm in £1.5bn deal”. The Guardian. 18 May 2004.
- Jump up^ Sorkin, Andrew Ross; Wilson, Duff (25 January 2009). “Pfizer Agrees to Pay $68 Billion for Rival Drug Maker Wyeth”. The New York Times.
- Jump up^ Bross PF, Beitz J, Chewn G, Chen XH, Duffy E, Kieffer L, Roy S, Sridhara R, Rahman A, Williams G, Pazdur R (2001). “Approval summary: gemtuzumab ozogamicin in relapsed acute myeloid leukemia.”. Clin Cancer Res. 7 (6): 1490–6. PMID 11410481.
- ^ Jump up to:a b Gemtuzumab Voluntarily Withdrawn From US Market. June 2010
- ^ Jump up to:a b Stanton, Dan (February 1, 2017). “Pfizer resubmits US and EU application for withdrawn ADC Mylotarg”. BioPharma Reporter.
- Jump up^ Giles FJ, Kantarjian HM, Kornblau SM, Thomas DA, Garcia-Manero G, Waddelow TA, David CL, Phan AT, Colburn DE, Rashid A, Estey EH (2001). “Mylotarg (gemtuzumab ozogamicin) therapy is associated with hepatic venoocclusive disease in patients who have not received stem cell transplantation.”. Cancer. 92 (2): 406–13. PMID 11466696. doi:10.1002/1097-0142(20010715)92:2<406::AID-CNCR1336>3.0.CO;2-U.
- Jump up^ Wadleigh M, Richardson PG, Zahrieh D, Lee SJ, Cutler C, Ho V, Alyea EP, Antin JH, Stone RM, Soiffer RJ, DeAngelo DJ (2003). “Prior gemtuzumab ozogamicin exposure significantly increases the risk of veno-occlusive disease in patients who undergo myeloablative allogeneic stem cell transplantation.”. Blood. 102 (5): 1578–82. PMID 12738663. doi:10.1182/blood-2003-01-0255.
- Jump up^ The Research on Adverse Drug Events and Reports (RADAR) Project, JAMA
- Jump up^ Mylotarg (gemtuzumab ozogamicin): Market Withdrawal, US FDA
- Jump up^ Pfizer pulls leukemia drug from U.S. market, Reuters
- Jump up^ Pharmaceuticals and Medical Devices Safety Information, No. 277, February 2011 (PDF) (Technical report). Pharmaceuticals and Medical Devices Agency of Japan. 2011.
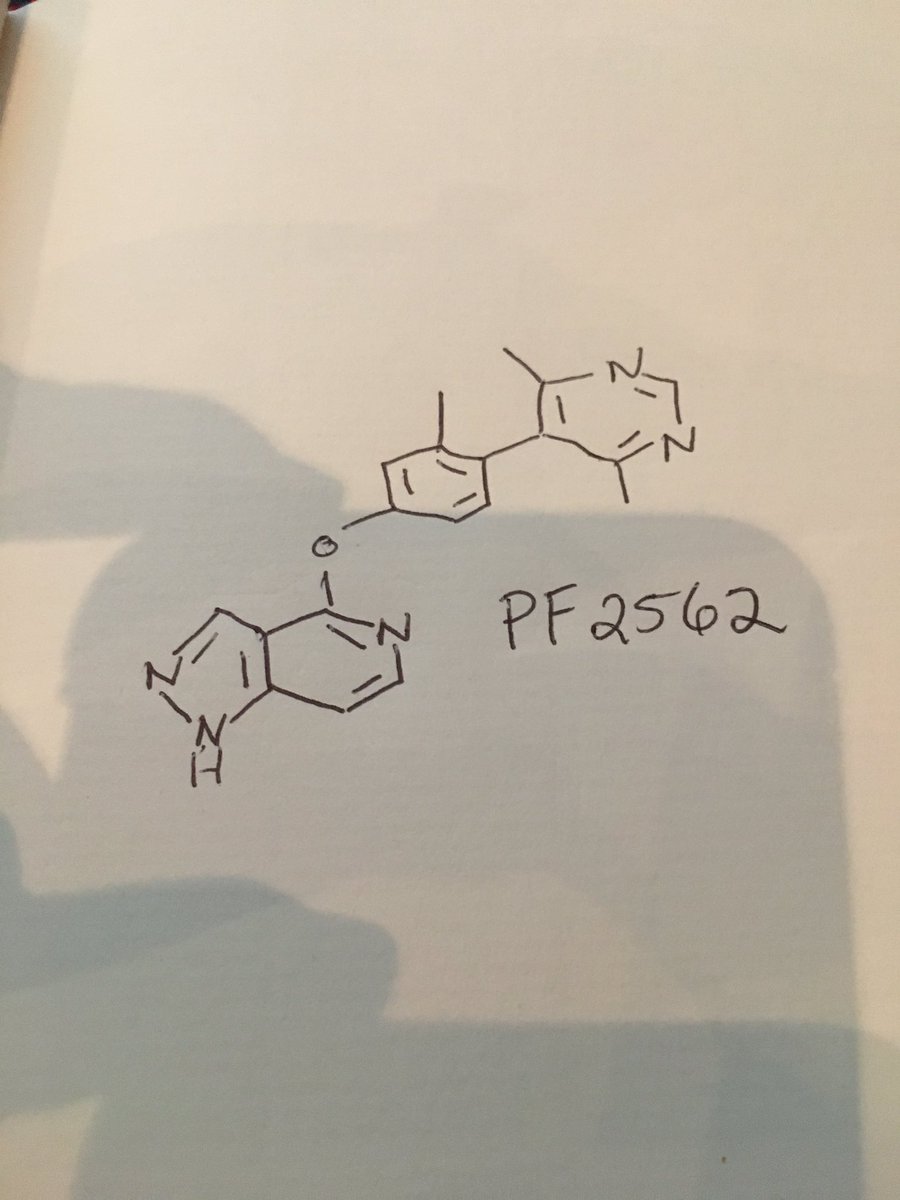
PF 2562
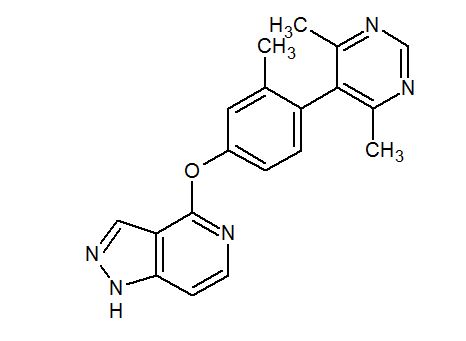
PF 2562
CAS 1609258-91-4
MF C19 H17 N5 O


Jennifer Elizabeth Davoren
Principal Scientist at Pfizer
SYNTHESIS


-
Dopamine acts upon neurons through two families of dopamine receptors, D1-like receptors (D1Rs) and D2-like receptors (D2Rs). The D1-like receptor family consists of D1 and D5 receptors which are expressed in many regions of the brain. D1 mRNA has been found, for example, in the striatum and nucleus accumbens. See e.g., Missale C, Nash S R, Robinson S W, Jaber M, Caron M G “Dopamine receptors: from structure to function”, Physiological Reviews 78:189-225 (1998). Pharmacological studies have reported that D1 and D5 receptors (D1/D5), namely D1-like receptors, are linked to stimulation of adenylyl cyclase, whereas D2, D3, and D4 receptors, namely D2-like receptors, are linked to inhibition of cAMP production.
-
Dopamine D1 receptors are implicated in numerous neuropharmacological and neurobiological functions. For example, D1 receptors are involved in different types of memory function and synaptic plasticity. See e.g., Goldman-Rakic P S et al., “Targeting the dopamine D1 receptor in schizophrenia: insights for cognitive dysfunction”, Psychopharmacology 174(1):3-16 (2004). Moreover, D1 receptors have been implicated in a variety of psychiatric, neurological, neurodevelopmental, neurodegenerative, mood, motivational, metabolic, cardiovascular, renal, ophthalmic, endocrine, and/or other disorders described herein including schizophrenia (e.g., cognitive and negative symptoms in schizophrenia), cognitive impairment associated with D2 antagonist therapy, ADHD, impulsivity, autism spectrum disorder, mild cognitive impairment (MCI), age-related cognitive decline, Alzheimer’s dementia, Parkinson’s disease (PD), Huntington’s chorea, depression, anxiety, treatment-resistant depression (TRD), bipolar disorder, chronic apathy, anhedonia, chronic fatigue, post-traumatic stress disorder, seasonal affective disorder, social anxiety disorder, post-partum depression, serotonin syndrome, substance abuse and drug dependence, Tourette’s syndrome, tardive dyskinesia, drowsiness, sexual dysfunction, migraine, systemic lupus erythematosus (SLE), hyperglycemia, dislipidemia, obesity, diabetes, sepsis, post-ischemic tubular necrosis, renal failure, resistant edema, narcolepsy, hypertension, congestive heart failure, postoperative ocular hypotonia, sleep disorders, pain, and other disorders in a mammal. See e.g., Goulet M, Madras B K “D(1) dopamine receptor agonists are more effective in alleviating advanced than mild parkinsonism in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-treated monkeys”, Journal of Pharmacology and Experimental Therapy 292(2):714-24 (2000); Surmeier D J et al., “The role of dopamine in modulating the structure and function of striatal circuits”, Prog. Brain Res. 183:149-67 (2010).New or improved agents that modulate (such as agonize or partially agonize) D1 are needed for developing new and more effective pharmaceuticals to treat diseases or conditions associated with dysregulated activation of D1, such as those described herein.
PATENT
Example 6
4-[4-(4,6-Dimethylpyrimidin-5-yl)-3-methylphenoxy]-1H-pyrazolo[4,3-c]pyridine (6)

Step 1. Synthesis of 4-[4-(4,6-dimethylpyrimidin-5-yl)-3-methylphenoxy]-1-(tetrahydro-2H-pyran-2-yl)-1H-pyrazolo[4,3-c]pyridine (C31)
Cesium carbonate (1.03 g, 3.16 mmol) and palladium(II) acetate (24 mg, 0.11 mmol) were added to a solution of C28 (225 mg, 1.05 mmol) and P3 (250 mg, 1.05 mmol) in 1,4-dioxane (10 mL) in a sealable reaction vessel, and the solution was purged with nitrogen for 10 minutes. Di-tert-butyl[3,4,5,6-tetramethyl-2′,4′,6-tri(propan-2-yl)biphenyl-2-yl]phosphane (97%, 104 mg, 0.210 mmol) was added, and the reaction mixture was briefly purged with nitrogen. The vessel was sealed and the reaction mixture was stirred at 100° C. for 3 hours. After cooling to room temperature, the mixture was filtered through Celite and the filter pad was washed with ethyl acetate; the combined filtrates were concentrated in vacuo and purified via silica gel chromatography (Eluents: 20%, then 50%, then 100% ethyl acetate in heptane). The product was obtained as an off-white solid. Yield: 272 mg, 0.655 mmol, 62%. LCMS m/z 416.5 [M+H+]. 1H NMR (400 MHz, CDCl3) δ 8.99 (s, 1H), 8.11 (d, J=0.6 Hz, 1H), 7.99 (d, J=6.0 Hz, 1H), 7.25-7.27 (m, 2H, assumed; partially obscured by solvent peak), 7.20-7.24 (m, 1H), 7.10 (d, J=8.4 Hz, 1H), 5.73 (dd, J=9.4, 2.5 Hz, 1H), 4.04-4.10 (m, 1H), 3.74-3.82 (m, 1H), 2.49-2.59 (m, 1H), 2.28 (s, 6H), 2.08-2.21 (m, 2H), 2.04 (s, 3H), 1.66-1.84 (s, 3H).
Step 2. Synthesis of 4-[4-(4,6-dimethylpyrimidin-5-yl)-3-methylphenoxy]-1H-pyrazolo[4,3-c]pyridine (6)
C31 (172 mg, 0.414 mmol) was dissolved in 1,4-dioxane (5 mL) and dichloromethane (5 mL), and cooled to 0° C. A solution of hydrogen chloride in 1,4-dioxane (4 M, 1.04 mL, 4.16 mmol) was added, and the reaction mixture was allowed to stir at room temperature for 45 hours. After removal of solvent in vacuo, the residue was partitioned between saturated aqueous sodium bicarbonate solution and dichloromethane. The aqueous layer was extracted twice with dichloromethane, and the combined organic layers were dried over sodium sulfate, filtered, and concentrated under reduced pressure, affording the product as an off-white solid. Yield: 130 mg, 0.392 mmol, 95%. LCMS m/z 332.3 [M+H+]. 1H NMR (400 MHz, CDCl3) δ 9.00 (s, 1H), 8.20 (br s, 1H), 7.99 (d, J=6.0 Hz, 1H), 7.28-7.30 (m, 1H), 7.23-7.27 (m, 1H), 7.16 (dd, J=6.0, 1.0 Hz, 1H), 7.11 (d, J=8.2 Hz, 1H), 2.28 (s, 6H), 2.05 (s, 3H).
Preparation P8
6-(4-Hydroxy-2-methylphenyl)-1,5-dimethylpyrazin-2(1H)-one (P8)

Step 1. Synthesis of 1-(4-methoxy-2-methylphenyl)propan-2-one (C8)
Four batches of this experiment were carried out (4×250 g substrate). Tributyl(methoxy)stannane (400 g, 1.24 mol), 1-bromo-4-methoxy-2-methylbenzene (250 g, 1.24 mol), prop-1-en-2-yl acetate (187 g, 1.87 mol), palladium(II) acetate (7.5 g, 33 mmol) and tris(2-methylphenyl)phosphane (10 g, 33 mmol) were stirred together in toluene (2 L) at 100° C. for 18 hours. After cooling to room temperature, the reaction mixture was treated with aqueous potassium fluoride solution (4 M, 400 mL) and stirred for 2 hours at 40° C. The resulting mixture was diluted with toluene (500 mL) and filtered through Celite; the filter pad was thoroughly washed with ethyl acetate (2×1.5 L). The organic phase from the combined filtrates was dried over sodium sulfate, filtered, and concentrated in vacuo. Purification via silica gel chromatography (Gradient: 0% to 5% ethyl acetate in petroleum ether) provided the product as a yellow oil. Combined yield: 602 g, 3.38 mol, 68%. LCMS m/z 179.0 [M+H+]. 1H NMR (400 MHz, CDCl3) δ 7.05 (d, J=8.3 Hz, 1H), 6.70-6.77 (m, 2H), 3.79 (s, 3H), 3.65 (s, 2H), 2.22 (s, 3H), 2.14 (s, 3H).
Step 2. Synthesis of 1-(4-methoxy-2-methylphenyl)propane-1,2-dione (C9)
C8 (6.00 g, 33.7 mmol) and selenium dioxide (7.47 g, 67.3 mmol) were suspended in 1,4-dioxane (50 mL) and heated at 100° C. for 18 hours. The reaction mixture was cooled to room temperature and filtered through Celite; the filtrate was concentrated in vacuo. Silica gel chromatography (Eluent: 10% ethyl acetate in heptane) afforded the product as a bright yellow oil. Yield: 2.55 g, 13.3 mmol, 39%. LCMS m/z 193.1 [M+H+]. 1H NMR (400 MHz, CDCl3) δ 7.66 (d, J=8.6 Hz, 1H), 6.81 (br d, half of AB quartet, J=2.5 Hz, 1H), 6.78 (br dd, half of ABX pattern, J=8.7, 2.6 Hz, 1H), 3.87 (s, 3H), 2.60 (br s, 3H), 2.51 (s, 3H).
Step 3. Synthesis of 6-(4-methoxy-2-methylphenyl)-5-methylpyrazin-2(1H)-one (C10)
C9 (4.0 g, 21 mmol) and glycinamide acetate (2.79 g, 20.8 mmol) were dissolved in methanol (40 mL) and cooled to −10° C. Aqueous sodium hydroxide solution (12 N, 3.5 mL, 42 mmol) was added, and the resulting mixture was slowly warmed to room temperature. After stirring for 3 days, the reaction mixture was concentrated in vacuo. The residue was diluted with water, and 1 N aqueous hydrochloric acid was added until the pH was approximately 7. The aqueous phase was extracted with ethyl acetate, and the combined organic extracts were washed with saturated aqueous sodium chloride solution, dried over magnesium sulfate, filtered, and concentrated under reduced pressure. The resulting residue was slurried with 3:1 ethyl acetate/heptane, stirred for 5 minutes, filtered, and concentrated in vacuo. Silica gel chromatography (Eluent: ethyl acetate) provided the product as a tan solid that contained 15% of an undesired regioisomer; this material was used without further purification. Yield: 2.0 g. LCMS m/z 231.1 [M+H+]. 1H NMR (400 MHz, CDCl3) δ 8.09 (s, 1H), 7.14 (d, J=8.2 Hz, 1H), 6.82-6.87 (m, 2H), 3.86 (s, 3H), 2.20 (s, 3H), 2.11 (s, 3H).
Step 4. Synthesis of 6-(4-methoxy-2-methylphenyl)-1,5-dimethylpyrazin-2(1H)-one (C11)
C10 (from the previous step, 1.9 g) was dissolved in N,N-dimethylformamide (40 mL). Lithium bromide (0.86 g, 9.9 mmol) and sodium bis(trimethylsilyl)amide (95%, 1.91 g, 9.89 mmol) were added, and the resulting solution was stirred for 30 minutes. Methyl iodide (0.635 mL, 10.2 mmol) was added and stirring was continued at room temperature for 18 hours. The reaction mixture was then diluted with water and brought to a pH of approximately 7 by slow portion-wise addition of 1 N aqueous hydrochloric acid. The aqueous layer was extracted with ethyl acetate and the combined organic layers were washed several times with water, dried over magnesium sulfate, filtered, and concentrated. Silica gel chromatography (Gradient: 75% to 100% ethyl acetate in heptane) afforded the product as a viscous orange oil. Yield: 1.67 g, 6.84 mmol, 33% over two steps. LCMS m/z 245.1 [M+H+]. 1H NMR (400 MHz, CDCl3) δ 8.17 (s, 1H), 7.03 (br d, J=8 Hz, 1H), 6.85-6.90 (m, 2H), 3.86 (s, 3H), 3.18 (s, 3H), 2.08 (br s, 3H), 2.00 (s, 3H).
Step 5. Synthesis of P8
To a −78° C. solution of C11 (1.8 g, 7.37 mmol) in dichloromethane (40 mL) was added a solution of boron tribromide in dichloromethane (1 M, 22 mL, 22 mmol). The cooling bath was removed after 30 minutes, and the reaction mixture was allowed to warm to room temperature and stir for 18 hours. The reaction was cooled to −78° C., and methanol (10 mL) was slowly added; the resulting mixture was slowly warmed to room temperature. The reaction mixture was concentrated in vacuo, methanol (20 mL) was added, and the mixture was again concentrated under reduced pressure. The residue was diluted with ethyl acetate (300 mL) and water (200 mL) and the aqueous layer was brought to pH 7 via portion-wise addition of saturated aqueous sodium carbonate solution. The mixture was extracted with ethyl acetate (3×200 mL). The combined organic extracts were washed with water and with saturated aqueous sodium chloride solution, dried over magnesium sulfate, filtered, and concentrated in vacuo to afford the product as a light tan solid. Yield: 1.4 g, 6.0 mmol, 81%. LCMS m/z 231.1 [M+H+]. 1H NMR (400 MHz, CDCl3) δ 8.21 (s, 1H), 6.98 (d, J=8.2 Hz, 1H), 6.87-6.89 (m, 1H), 6.85 (br dd, J=8.2, 2.5 Hz, 1H), 3.22 (s, 3H), 2.06 (br s, 3H), 2.03 (s, 3H).
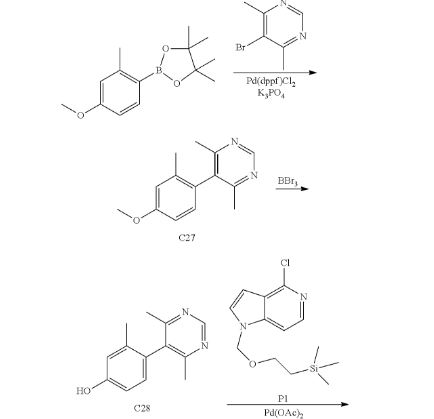
Step 1. Synthesis of 5-(4-methoxy-2-methylphenyl)-4,6-dimethylpyrimidine (C27)
1,1′-Bis(diphenylphosphino)ferrocene]dichloropalladium(II)-dichloromethane complex (5 g, 6 mmol) was added to a degassed mixture of 2-(4-methoxy-2-methylphenyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (30 g, 120 mmol), 5-bromo-4,6-dimethylpyrimidine (22.5 g, 120 mmol), and potassium phosphate (76.3 g, 359 mmol) in 1,4-dioxane (300 mL) and water (150 mL). The reaction mixture was heated at reflux for 4 hours, whereupon it was filtered and concentrated in vacuo. Purification via silica gel chromatography (Gradient: ethyl acetate in petroleum ether) provided the product as a brown solid. Yield: 25 g, 110 mmol, 92%. LCMS m/z 229.3 [M+H+]. 1H NMR (300 MHz, CDCl3) δ 8.95 (s, 1H), 6.94 (d, J=8.2 Hz, 1H), 6.87-6.89 (m, 1H), 6.84 (dd, J=8.3, 2.5 Hz, 1H), 3.86 (s, 3H), 2.21 (s, 6H), 1.99 (s, 3H).
Step 2. Synthesis of 4-(4,6-dimethylpyrimidin-5-yl)-3-methylphenol (C28)
Boron tribromide (3.8 mL, 40 mmol) was added drop-wise to a solution of C27 (3.0 g, 13 mmol) in dichloromethane (150 mL) at −70° C. The reaction mixture was stirred at room temperature for 16 hours, then adjusted to pH 8 with saturated aqueous sodium bicarbonate solution. The aqueous layer was extracted with dichloromethane (3×200 mL), and the combined organic layers were dried over sodium sulfate, filtered, and concentrated in vacuo. Silica gel chromatography (Gradient: 60% to 90% ethyl acetate in petroleum ether) afforded the product as a yellow solid. Yield: 1.2 g, 5.6 mmol, 43%. LCMS m/z 215.0 [M+H+]. 1H NMR (400 MHz, CDCl3) δ 8.98 (s, 1H), 6.89 (d, J=8.0 Hz, 1H), 6.86 (d, J=2.3 Hz, 1H), 6.80 (dd, J=8.3, 2.5 Hz, 1H), 2.24 (s, 6H), 1.96 (s, 3H).
//////////////PF 2562, non-catechol dopamine 1 receptor agonist, PFIZER, Jennifer Elizabeth Davoren, Amy Beth Dounay, Ivan Viktorovich Efremov, David Lawrence Firman Gray, Scot Richard Mente, Steven Victor O’Neil, Bruce Nelsen Rogers, Chakrapani Subramanyam, Lei Zhang, 1609258-91-4
Now at #MEDI 1st time disclosures David Gray of @pfizer on a non-catechol dopamine 1 receptor agonist #ACSSanFran
Cc1ncnc(C)c1c2ccc(cc2C)Oc4nccc3nncc34
PF 06821497
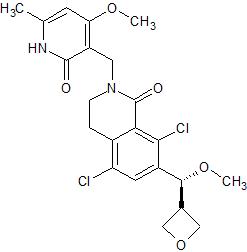
PF 06821497
Cas 1844849-11-1
Designed to treat lymphoma
1(2H)-Isoquinolinone, 5,8-dichloro-2-[(1,2-dihydro-4-methoxy-6-methyl-2-oxo-3-pyridinyl)methyl]-3,4-dihydro-7-[(S)-methoxy-3-oxetanylmethyl]-
MF C22 H24 Cl2 N2 O5,
MW 467.34
PF 06821497
5,8-Dichloro-2-[(4-methoxy-6-methyl-2-oxo-1,2-dihydro-3-pyridinyl)methyl]-7-[methoxy(3-oxetanyl)methyl]-3,4-dihydro-1(2H)-isoquinolinone
1(2H)-Isoquinolinone, 5,8-dichloro-2-[(1,2-dihydro-4-methoxy-6-methyl-2-oxo-3-pyridinyl)methyl]-3,4-dihydro-7-(methoxy-3-oxetanylmethyl)-
- Molecular Formula C22H24Cl2N2O5
- Average mass 467.342 Da
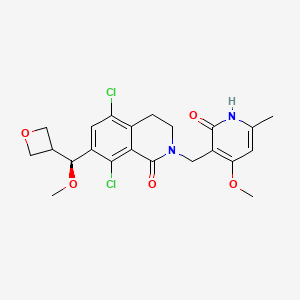
5,8-dichloro-2-[(4-methoxy-6-methyl-2-oxo-1H-pyridin-3-yl)methyl]-7-[(S)-methoxy(oxetan-3-yl)methyl]-3,4-dihydroisoquinolin-1-one
| Inventors | Michael Raymond Collins, Robert Steven Kania, Robert Arnold Kumpf, Pei-Pei Kung, Daniel Tyler Richter, Scott Channing Sutton, Martin James Wythes |
| Original Assignee | Pfizer Inc. |
-
Epigenetic alterations play an important role in the regulation of cellular processes, including cell proliferation, cell differentiation and cell survival. The epigenetic silencing of tumor suppressor genes and activation of oncogenes may occur through alteration of CpG island methylation patterns, histone modification, and dysregulation of DNA binding protein. Polycomb genes are a set of epigenetic effectors. EZH2 (enhancer of zeste homolog 2) is the catalytic component of the Polycomb Repressor Complex 2 (PRC2), a conserved multi-subunit complex that represses gene transcription by methylating lysine 27 on Histone H3 (H3K27). EZH2 plans a key role in regulating gene expression patterns that regulate cell fate decisions, such as differentiation and self-renewal. EZH2 is overexpressed in certain cancer cells, where it has been linked to cell proliferation, cell invasion, chemoresistance and metastasis.
-
High EZH2 expression has been correlated with poor prognosis, high grade, and high stage in several cancer types, including breast, colorectal, endometrial, gastric, liver, kidney, lung, melanoma, ovarian, pancreatic, prostate, and bladder cancers. See Crea et al., Crit. Rev. Oncol. Hematol. 2012, 83:184-193, and references cited therein; see also Kleer et al., Proc. Natl. Acad. Sci. USA 2003, 100:11606-11; Mimori et al., Eur. J. Surg. Oncol. 2005, 31:376-80; Bachmann et al., J. Clin. Oncol. 2006, 24:268-273; Matsukawa et al., Cancer Sci. 2006, 97:484-491; Sasaki et al. Lab. Invest. 2008, 88:873-882; Sudo et al., Br. J. Cancer 2005, 92(9):1754-1758; Breuer et al., Neoplasia 2004, 6:736-43; Lu et al., Cancer Res. 2007, 67:1757-1768; Ougolkov et al., Clin. Cancer Res. 2008, 14:6790-6796; Varambally et al., Nature 2002, 419:624-629; Wagener et al., Int. J. Cancer 2008, 123:1545-1550; and Weikert et al., Int. J. Mol. Med. 2005, 16:349-353.Recurring somatic mutations in EZH2 have been identified in diffuse large B-cell lymphoma (DLBCL) and follicular lymphomas (FL). Mutations altering EZH2 tyrosine 641 (e.g., Y641C, Y641F, Y641N, Y641S, and Y641H) were reportedly observed in up to 22% of germinal center B-cell DLBCL and 7% of FL. Morin et al. Nat. Genetics 2010 February; 42(2):181-185. Mutations of alanine 677 (A677) and alanine 687 (A687) have also been reported. McCabe et al., Proc. Natl. Acad. Sci. USA 2012, 109:2989-2994; Majer et al. FEBS Letters 2012, 586:3448-3451. EZH2 activating mutations have been suggested to alter substrate specificity resulting in elevated levels of trimethylated H3K27 (H3K27me3).Accordingly, compounds that inhibit the activity of wild type and/or mutant forms of EZH2 may be of interest for the treatment of cancer.
SYNTHESIS

Steps
1 COUPLING, Ag2CO3
2 Alkylation, K2CO3
3 LiAlH4 REDUCTION
4 THIONYL CHLORIDE
5 N-Alkylation of Amides, t-BuOK
6 A GRIGNARD REACTION
7 AN ALKYLATION , METHYL IODIDE, t-BuOK
8 HYDROGENATION, DE BENZYLATION, PLATINUM OXIDE
9 LAST STEP separation by chiral preparative, SFC on (R,R) Whelk O1 column, TO GET PF 06821497
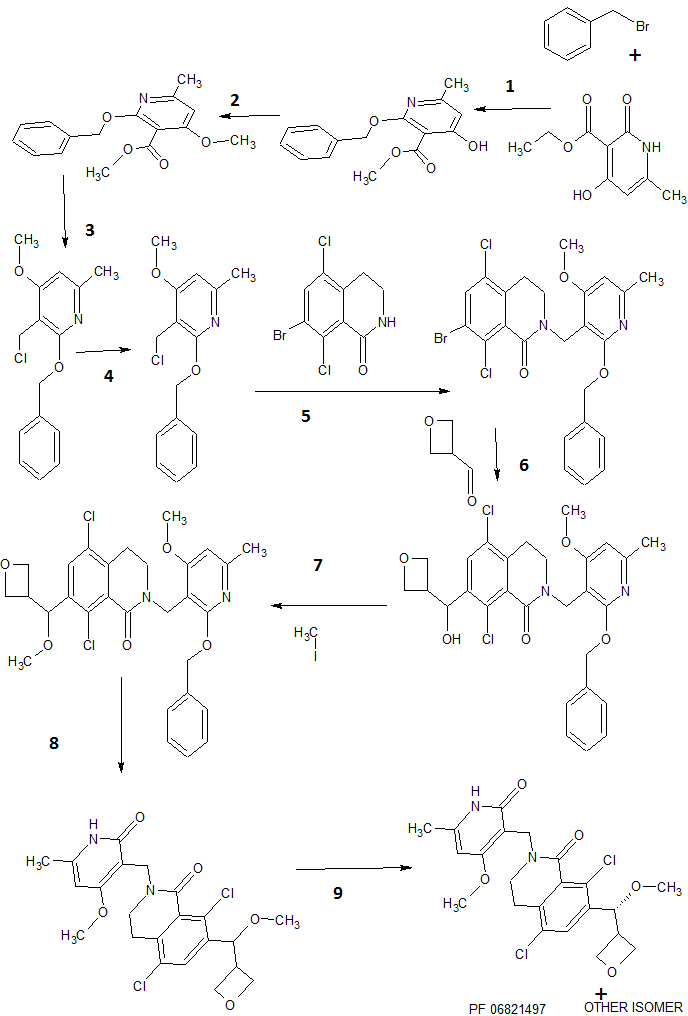
PATENT
US 20150361067

///////////////PF 06821497, 1844849-11-1, PFIZER, lymphoma, Pei-Pei Kung, @pfizer, #ACSSanFran, Michael Raymond Collins, Robert Steven Kania, Robert Arnold Kumpf, Pei-Pei Kung, Daniel Tyler Richter, Scott Channing Sutton, Martin James Wythes
Next up in #MEDI 1st time disclosures Pei-Pei Kung from @pfizer presenting a molecule designed to treat lymphoma #ACSSanFran
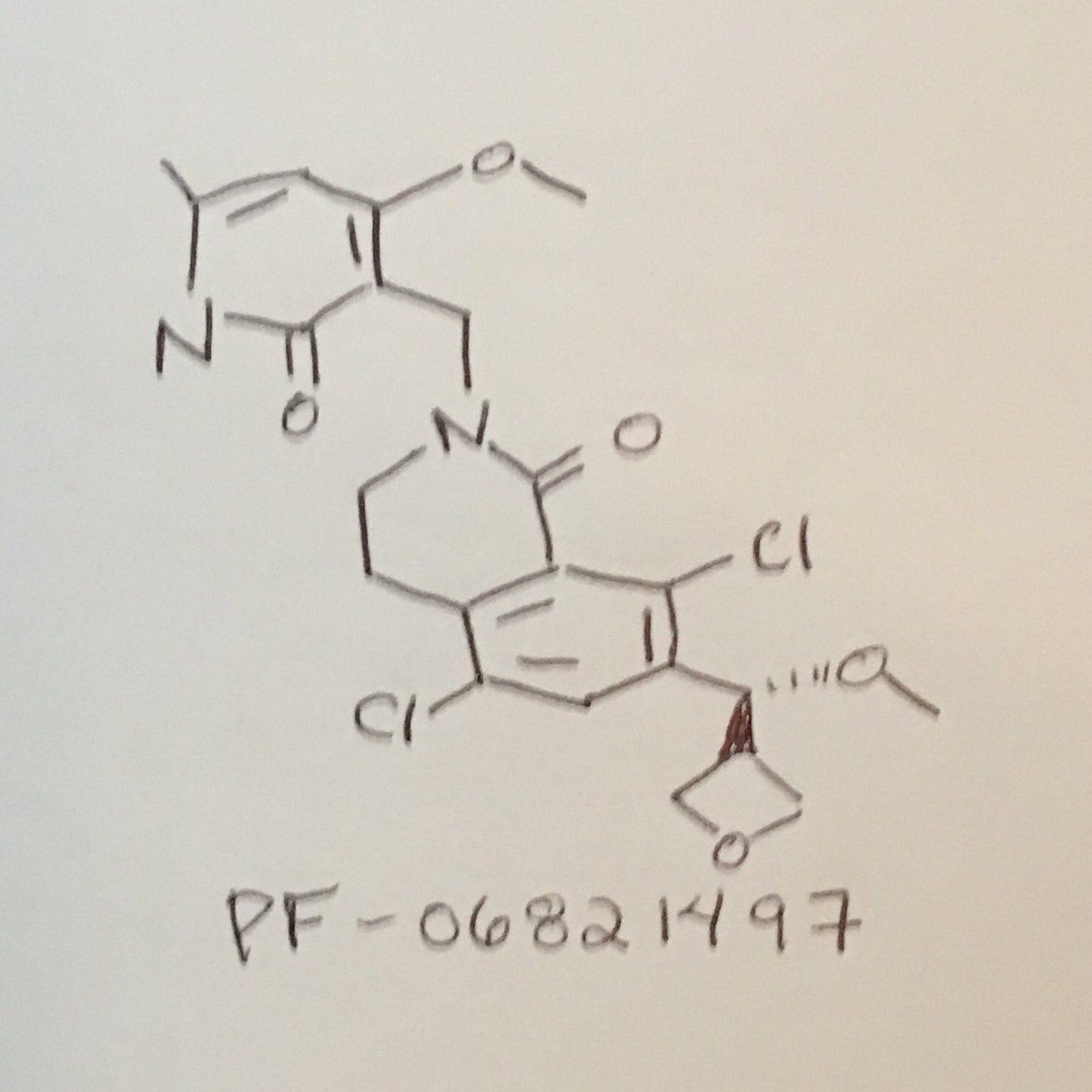
CO[C@H](c2cc(Cl)c3CCN(CC1=C(OC)C=C(C)NC1=O)C(=O)c3c2Cl)C4COC4
| CC1=CC(=C(C(=O)N1)CN2CCC3=C(C=C(C(=C3C2=O)Cl)C(C4COC4)OC)Cl)OC |
Lorlatinib, лорлатиниб , لورلاتينيب , 洛拉替尼 , PF-6463922

Lorlatinib, PF-6463922
For Cancer; Non-small-cell lung cancer
- Molecular Formula C21H19FN6O2
- Average mass 406.413 Da
Phase 2
WO 2013132376
| Andrew James Jensen, Suman Luthra, Paul Francis RICHARDSON | |
| Applicant | Pfizer Inc. |

Ros1 tyrosine kinase receptor inhibitor; Anaplastic lymphoma kinase receptor inhibitor
useful for treating cancer mediated by anaplastic lymphoma kinase (ALK) or c-ros oncogene 1 (ROS1) receptor tyrosine kinase, particularly NSCLC. an ATP-competitive inhibitor of ROS1/ALK, for treating NSCLC. In February 2017, lorlatinib was reported to be in phase 2 clinical development.
- Originator Pfizer
- Developer Pfizer; The Childrens Hospital of Philadelphia; Yale University
- Class Antineoplastics; Aza compounds; Benzoxazines; Pyrazoles; Pyrazolones; Small molecules
- Mechanism of Action Anaplastic lymphoma kinase inhibitors; ROS1-protein-inhibitors
- Orphan Drug Status Yes – Non-small cell lung cancer
Lorlatinib (PF-6463922) is an experimental anti-neoplastic drug in development by Pfizer. It is a orally-administered small molecule inhibitor of ROS1 and ALK.
In 2015, FDA granted Pfizer orphan drug status for lorlatinib for the treatment of non-small cell lung cancer.[1]
- 05 Oct 2016 Massachusetts General Hospital plans a phase II trial for Non-small cell lung cancer (Late-stage disease, Metastatic disease) in USA (PO, unspecified formulation) (NCT02927340)
- 01 Oct 2016 Pfizer completes a phase I trial in pharmacokinetic trial in Healthy volunteers in USA (NCT02804399)
- 01 Aug 2016 Pfizer initiates a phase I drug-drug interaction trial in Healthy volunteers in Belgium (PO, unspecified formulation) (NCT02838264)

Structures of ALK inhibitors marketed or currently in the clinic
Synthesis

NEED COLOUR

Clinical studies
Several clinical trials are ongoing. A phase II trial comparing avelumab alone and in combination with lorlatinib or crizotinib for non-small cell lung cancer is expected to be complete in late 2017. A phase II trial comparing lorlatinib with crizotinib is expected to be complete in mid-2018.[2] A phase II trial for treatment of ALK-positive or ROS1-positive non-small cell lung cancer with CNA metastases is not expected to be complete until 2023.[3] Preclinical studies are investigating lorlatinib for treatment of neuroblastoma.
Lorlatinib is an investigational medicine that inhibits the anaplastic lymphoma kinase (ALK) and ROS1 proto-oncogene. Due to tumor complexity and development of resistance to treatment, disease progression is a challenge in patients with ALK-positive metastatic non-small cell lung cancer (NSCLC). A common site for progression in metastatic NSCLC is the brain. Lorlatinib was specifically designed to inhibit tumor mutations that drive resistance to other ALK inhibitors and to penetrate the blood brain barrier.
ABOUT LORLATINIB
ALK in NSCLC ROS1 in NSCLC PRECLINICAL DATA CLINICAL STUDIES Originally discovered as an oncogenic driver in a type of lymphoma, ALK gene alterations were also found to be among key drivers of tumor development in cancers, such as NSCLC.1 In ALK-positive lung cancer, a normally inactive gene called ALK is fused with another gene. This genetic alteration creates the ALK fusion gene and ultimately, the production of an ALK fusion protein, which is responsible for tumor growth.1,2 This genetic alteration is present in 3-5% of NSCLC patients.3,4,5 Another gene that can fuse with other genes is called ROS1. Sometimes a ROS1 fusion protein can contribute to cancer-cell growth and tumor survival. This genetic alteration is present in approximately 1% of NSCLC patients.5 Preclinical data showed lorlatinib is capable of overcoming resistance to existing ALK inhibitors and penetrated the blood brain barrier in ALK-driven tumor models.2 Specifically, in these preclinical models, lorlatinib had activity against all tested clinical resistance mutations in ALK.
A Phase 1/2 clinical trial of lorlatinib in patients with ALK-positive or ROS1-positive advanced NSCLC is currently ongoing. • The primary objective of the Phase 1 portion was to assess safety and tolerability of single-agent lorlatinib at increasing dose levels in patients with ALK-positive or ROS1-positive advanced NSCLC.6 • Data from the Phase 1 study showed that lorlatinib had promising clinical activity in patients with ALK-positive or ROS1- positive advanced NSCLC. Most of these patients had developed CNS metastases and had received ≥1 prior tyrosine kinase inhibitor.7 o The most common treatment-related adverse events (AEs) were hypercholesterolemia (69%) and peripheral edema (37%). Hypercholesterolemia was the most common (11%) grade 3 or higher treatment-related AE and the most frequent reason for dose delay or reduction. No patients discontinued due to treatment-related AEs. At the recommended Phase 2 dose, 4 out of 17 patients (24%) experienced a treatment-related AE of any grade that led to a dose delay or hold.
PATENT
WO2014207606
This invention relates to crystalline forms of the macrocyclic kinase inhibitor, (10R)-7-amino-12-fluoro-2, 10,16-trimethyl-15-OXO-10,15, 16,17-tetrahydro-2H-8,4-(metheno)pyrazolo[4, 3-?][2,5,1 1 ]benzoxadiazacyclotetradecine-3-carbonitrile, including crystalline solvates thereof, that may be useful in the treatment of abnormal cell growth, such as cancer, in mammals. The invention also relates to compositions including such crystalline forms, and to methods of using such compositions in the treatment of abnormal cell growth in mammals, especially humans.
Background of the Invention
The compound (10R)-7-amino-12-fluoro-2,10,16-trimethyl-15-oxo-10,15,16,17-tetrahydro-2/-/-8,4-(metheno)pyrazolo[4,3- ?][2,5,1 1 ]benzoxadiazacyclotetradecine-3-carbonitrile, represented by the formula (I):

(I)
is a potent, macrocyclic inhibitor of both wild type and resistance mutant forms of anaplastic lymphoma kinase (ALK) and c-ros oncogene 1 (ROS1) receptor tyrosine kinase. Preparation of the free base compound of formula (I) as an amorphous solid is disclosed in International Patent Publication No. WO 2013/132376 and in United States Patent Publication No. 2013/0252961 , the contents of which are incorporated herein by reference in their entirety.
Human cancers comprise a diverse array of diseases that collectively are one of the leading causes of death in developed countries throughout the world (American Cancer Society, Cancer Facts and Figures 2005. Atlanta: American Cancer Society; 2005). The progression of cancers is caused by a complex series of multiple genetic and molecular events including gene mutations, chromosomal translocations, and karyotypic abnormalities (Hanahan & Weinberg, The hallmarks of cancer. Cell 2000; 100: 57-70). Although the underlying genetic causes of
cancer are both diverse and complex, each cancer type has been observed to exhibit common traits and acquired capabilities that facilitate its progression. These acquired capabilities include dysregulated cell growth, sustained ability to recruit blood vessels (i.e., angiogenesis), and ability of tumor cells to spread locally as well as metastasize to secondary organ sites (Hanahan & Weinberg 2000). Therefore, the ability to identify novel therapeutic agents that inhibit molecular targets that are altered during cancer progression or target multiple processes that are common to cancer progression in a variety of tumors presents a significant unmet need.
Receptor tyrosine kinases (RTKs) play fundamental roles in cellular processes, including cell proliferation, migration, metabolism, differentiation, and survival. RTK activity is tightly controlled in normal cells. The constitutively enhanced RTK activities from point mutation, amplification, and rearrangement of the corresponding genes have been implicated in the development and progression of many types of cancer. (Gschwind et al., The discovery of receptor tyrosine kinases: targets for cancer therapy. Nat. Rev. Cancer 2004; 4, 361-370; Krause & Van Etten, Tyrosine kinases as targets for cancer therapy. N. Engl. J. Med. 2005; 353: 172-187.)
Anaplastic lymphoma kinase (ALK) is a receptor tyrosine kinase, grouped together with leukocyte tyrosine kinase (LTK) to a subfamily within the insulin receptor (IR) superfamily. ALK was first discovered as a fusion protein with nucleophosmin (NPM) in anaplastic large cell lymphoma (ALCL) cell lines in 1994. (Morris et al., Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin’s lymphoma. Science 1994; 263:1281-1284.) NPM-ALK, which results from a chromosomal translocation, is implicated in the pathogenesis of human anaplastic large cell lymphoma (ALCL) (Pulford et al., Anaplastic lymphoma kinase proteins in growth control and cancer. J. Cell Physiol., 2004; 199: 330-58). The roles of aberrant expression of constitutively active ALK chimeric proteins in the pathogenesis of ALCL have been defined (Wan et. al., Anaplastic lymphoma kinase activity is essential for the proliferation and survival of anaplastic large cell lymphoma cells. Blood, 2006; 107:1617-1623). Other chromosomal rearrangements resulting in ALK fusions have been subsequently detected in ALCL (50-60%), inflammatory myofibroblastic tumors (27%), and non-small-cell lung cancer (NSCLC) (2-7%). (Palmer et al., Anaplastic lymphoma kinase: signaling in development and disease. Biochem. J. 2009; 420:345-361 .)
The EML4-ALK fusion gene, comprising portions of the echinoderm microtubule associated protein-like 4 (EML4) gene and the ALK gene, was first discovered in NSCLC archived clinical specimens and cell lines. (Soda et al., Identification of the transforming EML4-ALK fusion gene in non-small cell lung cancer. Nature 2007; 448:561-566; Rikova et al., Cell 2007; 131 :1 190-1203.) EML4-ALK fusion variants were demonstrated to transform NIH-3T3 fibroblasts and cause lung adenocarcinoma when expressed in transgenic mice, confirming the
potent oncogenic activity of the EML4-ALK fusion kinase. (Soda et al., A mouse model for EML4-ALK-positive lung cancer. Proc. Natl. Acad. Sci. U.S.A. 2008; 105:19893-19897.) Oncogenic mutations of ALK in both familial and sporadic cases of neuroblastoma have also been reported. (Caren et al., High incidence of DNA mutations and gene amplifications of the ALK gene in advanced sporadic neuroblastoma tumors. Biochem. J. 2008; 416:153-159.)
ROS1 is a proto-oncogene receptor tyrosine kinase that belongs to the insulin receptor subfamily, and is involved in cell proliferation and differentiation processes. (Nagarajan et al. Proc Natl Acad Sci 1986; 83:6568-6572). ROS is expressed, in humans, in epithelial cells of a variety of different tissues. Defects in ROS expression and/or activation have been found in glioblastoma, as well as tumors of the central nervous system (Charest et al., Genes Chromos. Can. 2003; 37(1): 58-71). Genetic alterations involving ROS that result in aberrant fusion proteins of ROS kinase have been described, including the FIG-ROS deletion translocation in glioblastoma (Charest et al. (2003); Birchmeier et al. Proc Natl Acad Sci 1987; 84:9270-9274; and NSCLC (Rimkunas et al., Analysis of Receptor Tyrosine Kinase ROS1 -Positive Tumors in Non-Small Cell Lung Cancer: Identification of FIG-ROS1 Fusion, Clin Cancer Res 2012; 18:4449-4457), the SLC34A2-ROS translocation in NSCLC (Rikova et al. Cell 2007;131 :1 190-1203), the CD74-ROS translocation in NSCLC (Rikova et al. (2007)) and cholangiocarcinoma (Gu et al. PLoS ONE 201 1 ; 6(1 ): e15640), and a truncated, active form of ROS known to drive tumor growth in mice (Birchmeier et al. Mol. Cell. Bio. 1986; 6(9):3109-31 15). Additional fusions, including TPM3-ROS1 , SDC4-ROS1 , EZR-ROS1 and LRIG3-ROS1 , have been reported in lung cancer patient tumor samples (Takeuchi et al., RET, ROS1 and ALK fusions in lung cancer, Nature Medicine 2012; 18(3):378-381).
The dual ALK/c-MET inhibitor crizotinib was approved in 201 1 for the treatment of patients with locally advanced or metastatic NSCLC that is ALK-positive as detected by an FDA-approved test. Crizotinib has also shown efficacy in treatment of NSCLC with ROS1 translocations. (Shaw et al. Clinical activity of crizotinib in advanced rson-smali cell lung cancer (NSCLC) harboring ROS1 gene rearrangement. Presented at the Annual Meeting of the American Society of Clinical Oncology, Chicago, June 1-5, 2012.) As observed clinically for other tyrosine kinase inhibitors, mutations in ALK and ROS1 that confer resistance to ALK inhibitors have been described (Choi et ai., EML4-ALK Mutations in Lung Cancer than Confer Resistance to ALK Inhibitors, N Engl J Med 2010; 363:1734-1739; Awad et ai., Acquired Resistance to Crizotinib from a Mutation in CD74-ROS1, Engl J Med 2013; 368:2395-2401 ).
Thus, ALK and ROS1 are attractive molecular targets for cancer therapeutic intervention. There remains a need to identify compounds having novel activity profiles against wild-type and mutant forms of ALK and ROS1 .
The present invention provides crystalline forms of the free base of (10R)-7-amino-12-fluoro-2, 10,16-trimethyl-15-OXO-10,15, 16,17-tetrahydro-2H-8,4-(metheno)pyrazolo[4,3- ?][2, 5,1 1 ]-benzoxadiazacyclotetradecine-3-carbonitrile having improved properties, such as improved crystallinity, dissolution properties, decreased hygroscopicity, improved mechanical properties, improved purity, and/or improved stability, while maintaining chemical and enantiomeric stability.
Comparative Example 1A
Preparation of (10f?)-7-amino-12-fluoro-2,10,16-trimethyl-15-oxo-10,15,16,17-tetrahydro-2H-8,4-(metheno)pyrazolo[4,3- ?l[2,5,1 Hbenzoxadiazacyclo-tetradecine-3-carbonitrile (amorphous)

Example 1A
Step 1 :
Palladium (II) acetate (53 mg, 0.24 mmol) and cataCXium® A (180 mg, 0.5 mmol) were mixed together in toluene (1 .5 mL, de-gassed) and the resulting solution was added via pipette to a stirred solution of compound 7 (0.9 g, 2.4 mmol), compound 15 (1 .0 g, 3.0 mmol) bis-pinacolato diboron (0.9 g, 3.6 mmol) and CsF (1 .9 g, 12.6 mmol) in MeOH/H20 (9:1 , 12 mL, degassed) at 60 °C. The resulting mixture was then stirred at reflux for 3 hrs. A further portion of Palladium (II) acetate (26 mg, 0.12 mmol) and cataCXium® A (90 mg, 0.25 mmol) in toluene (1 .5 mL, de-gassed) was added, and the yellow reaction mixture stirred at 60 °C overnight. After cooling to room temperature, the mixture was diluted with EtOAc (150 mL) and filtered through CELITE®. The filtrate was washed with water (100 mL), then brine (100 mL), dried (Na2S04) and evaporated. The residue was purified by flash chromatography over silica gel, which was eluted with 1 :1 EtOAc/cyclohexane, to give compound 22 as a yellow oil (570 mg, 43% yield). TLC (Rf = 0.40, 1 :1 EtOAc/cyclohexane). 1H NMR (400 MHz, CDCI3) δ 8.03 (m, 1 H), 7.65 (s, 1 H), 7.27 (dd,1 H, J = 9.9, 2.7 Hz), 7.01 (m, 1 H), 6.68 (m, 1 H), 6.40 (m, 1 H), 4.90 (br s, 2 H), 4.20 – 4.30 (m, 2 H), 3.96 (s, 3 H), 3.94 (s, 3 H), 2.55 – 2.85 (m, 3 H), 1 .68 (d, 3 H, J = 6.6 Hz), 1 .24 (s, 9 H). LCMS ES m/z 539 [M+H]+.
Step 2:
To a solution of compound 22 (69% purity, 0.95 g, assumed 1 .05 mmol) in MeOH (20 mL) was added a solution NaOH (1 .0 g, 25 mmol) in water (2 mL). The mixture was stirred at 40 °C for 3.5 hours. The reaction was diluted with water (80 mL), concentrated by 20 mL to remove MeOH on the rotary evaporator, and washed with MTBE (100 mL). The aqueous layer was then acidified carefully with 1 M aq HCI to approx. pH 2 (pH paper). Sodium chloride (15 g) was added to the mixture and the mixture was extracted with EtOAc (100 mL). The organic layer was separated, dried (Na2S04) and evaporated to give compound 23 as a pale yellow solid (480 mg, 87% yield). 1H NMR (400 MHz, CD3OD) δ 8.05 (m, 1 H), 7.45 (s, 1 H), 7.37 (dd,1 H, J = 10.4, 2.8 Hz), 7.10 (dt, 1 H, J = 8.5, 2.4 Hz), 6.50 – 6.60 (m, 2 H), 4.05 – 4.30 (m, 2 H), 3.99 (s, 3 H), 2.60 – 2.80 (m, 3 H), 1 .72 (d, 3 H, J = 6.5 Hz). LCMS ES m/z 525 [M+H]+.
Step 3:
A solution of HCI in dioxane (4 M, 6.0 mL) was added to a solution of compound 23
(480 mg, 0.91 mmol) in MeOH (methanol) (6 mL) and the reaction was stirred at 40 °C for 2.5 hours. The reaction mixture was then concentrated to dryness under reduced pressure. The residue was taken-up in MeOH (50 mL) and acetonitrile (100 mL) was added and the mixture was then again evaporated to dryness, to give compound 24 as an off white solid (400 mg, 87% yield). 1H NMR (400 MHz, CD3OD) δ 8.07 (dd, 1 H, J = 8.9. 5.9 Hz), 7.51 (d, 1 H, J = 1 .7 Hz), 7.42 (dd, 1 H, J = 9.8, 2.6 Hz), 7.23 (d, 1 H, J = 1 .6 Hz), 7.16 (dt, 1 H, J = 8.5, 2.7 Hz), 6.73 (dd, 1 H, J = 1 1 .9, 6.9 Hz), 4.22 (d, 1 H, J = 14.7 Hz), 4.14 (d, 1 H, J = 14.7 Hz), 4.07 (s, 3 H), 2.75 (s, 3 H), 1 .75 (d, 3 H, J = 5.5 Hz). LCMS ES m/z 425 [M+H]+.
Step 4:
A solution of compound 24 (400 mg, assumed 0.91 mmol) as the HCI salt and DIPEA
(diisopropylethylamine) (1 .17 g, 9.1 mmol) in DMF (dimethylformamide) (5.0 mL) and THF (0.5 mL) was added drop-wise to a solution of HATU (2-(1 H-7-azabenzotriazol-1 -yl)-1 ,1 ,3,3-tetramethyl uronium hexafluorophosphate methanaminium) (482 mg, 1 .27 mmol) in DMF (10.0 mL) at 0 °C over 30 minutes. After complete addition, the mixture was stirred at 0 °C for a further 30 mins. Water (70 mL) was added and the mixture was extracted into EtOAc (2 x 60 mL). The combined organics were washed with saturated aqueous NaHC03 (2 x 100 mL), brine (100 mL), dried over Na2S04, and evaporated. The residue was purified by column chromatography over silica gel, which was eluted with 70% EtOAc/cyclohexane giving 205 mg of a pale yellow residue (semi-solid). The solids were dissolved in MTBE (7 mL) and cyclohexane (20 mL) was added slowly with good stirring to precipitate the product. After stirring for 30 minutes, the mixture was filtered, and Example 1A was collected as an
amorphous white solid (1 10 mg, 29% yield). TLC (Rf = 0.40, 70% EtOAc in cyclohexane). 1H NMR (400 MHz, CDCI3) δ 7.83 (d, 1 H, J = 2.0 Hz), 7.30 (dd, 1 H, J = 9.6, 2.4 Hz), 7.21 (dd, 1 H, J = 8.4, 5.6 Hz), 6.99 (dt, 1 H, J = 8.0, 2.8 Hz), 6.86 (d, 1 H, J = 1 .2 Hz), 5.75 – 5.71 (m, 1 H), 4.84 (s, 2 H), 4.45 (d, 1 H, J = 14.4 Hz), 4.35 (d ,1 H, J = 14.4 Hz), 4.07 (s, 3 H), 3.13 (s, 3 H), 1 .79 (d, 3 H, J = 6.4Hz). LCMS ES m/z 407 [M+H]+.
Example 1
Preparation of crystalline hydrate of (10 ?)-7-amino-12-fluoro-2,10,16-trimethyl-15-oxo- 10,15,16,17-tetrahvdro-2/-/-8,4-(metheno)pyrazolo[4,3- ?l[2,5,1 Hbenzoxa-diazacyclo-tetradecine-3-carbonitrile (Form 1)

Example 1A Example 1
(amorphous) (Form 1 }
Amorphous (10f?)-7-amino-12-fluoro-2,10,16-trimethyl-15-oxo-10,15,16,17-tetrahydro-2H-8,4-(metheno)pyrazolo[4,3- ?][2,5,11 ]benzoxa-diazacyclo-tetradecine-3-carbonitrile free base, prepared as described in Example 1A (and Example 2 of United States Patent Publication No. 2013/0252961), was dissolved in 1 .0 : 1 .1 (v:v) H20:MeOH at a concentration of 22 mg/mL at 50°C, then allowed to cool to room temperature . This slurry was granulated for approximately 72 hours. The solids were isolated by filtration and vacuum dried overnight at 60°C to produce crystalline hydrate Form 1 of (10R)-7-amino-12-fluoro-2,10,16-trimethyl-15-oxo-10,15,16,17-tetrahydro-2H-8,4-(metheno)pyrazolo[4,3-/?][2,5,1 1 ]benzoxadiazacyclotetradecine-3-carbonitrile.
Example 4
Alternative preparation of crystalline acetic acid solvate of (10 ?)-7-amino-12-fluoro-2, 10,16-trimethyl-15-OXO-10,15, 16,17-tetrahvdro-2H-8,4-(metheno)pyrazolo[4,3- ?U2,5, 1 1 lbenzoxa-diazacyclotetradecine-3-carbonitrile (Form 3)

Step 1 :
To a reaction vessel under N2 were charged compound 9 (9.97 kg, 17.95 mol), compound 21 (3.52 kg, 18.85 mol) and 2-methyltetrahydrofuran (97 L). Triethylamine (7.45 kg, 73.6 mol) was added while keeping the internal temperature below 35°C. The reaction mixture was held for 30 min and n-propylphosphonic anhydride (T3P), 50% solution in ethyl acetate (22.85 kg, 35.9 mol) was charged slowly, maintaining the internal temperature below 25°C. The reaction mixture was held at 20°C for at least 2 h until reaction was deemed complete. Ethyl acetate (35 L) and water (66 L) were added followed by 0.5N Hydrochloric acid solution (80 L). The aqueous layer was removed and the organic layer was washed with brine solution (80 L). The organic layer was concentrated and solvent exchanged with 2-methyl-2-butanol (80 L) give compound 25 (23 wt/wt%) solution in 2-methyl-2-butanol . This solution was carried forward to the next step directly in three batches, assuming 12.00 kg (100% yield) from this step.
Step 2:
2-Methyl-2-butanol (100 L) was combined with potassium acetate (1 .8 kg, 18.34 mol), palladium(ll) acetate (0.10 kg, 0.46 mol) and water (0.10 kg, 5.73 mol). The resulting mixture was purged with nitrogen. Di(1 -adamantyl)n-butylphosphine (0.23 kg, 0.43 mol) was added. An amount of 20% of compound 25 (3.97 kg active or 17.3 L of step 1 solution in 2-methyl-2-butanol) was added, and the resulting reaction mixture was heated at reflux for 2 h. The remaining solution of compound 25 in 2-methyl-2-butanol was subsequently added to the reaction over a period of 5 h. The resulting mixture was heated until the reaction was deemed complete (typically 16 – 20 h). This reaction step was processed in three batches, and the isolation was done in one single batch. Thus, the combined three batches were filtered through CELITE® to remove insoluble materials. The filtrate was concentrated to a low volume (approximately 20 L). Acetonitrile (60 L) was added. The resulting mixture was heated to reflux for 2 – 4 h, then cooled to RT for granulation. The resulting slurry was filtered to give compound 26 as a crude product. The crude product was combined with ethyl acetate (80 L) and Silicycle thiol (5 kg). The resulting mixture was heated for 2 h, cooled to RT and filtered. The filtrate was concentrated to approx. 20 L, and the resulting slurry was granulated and filtered. The filter cake was rinsed with ethyl acetate (4 L) and dried in a vacuum oven to give compound 26 as a pure product (4.74 kg, 43.5% overall last two steps). 1H NMR (CDCI3) δ 8.25 – 8.23 (m, 1 H), 7.28 (1 H, dd, 2.76 and 9.79 Hz), 7.22 (1 H, dd, 5.52 and 8.53 Hz), 7.18 (1 H, d, J = 1 .76 Hz), 7.01 (1 H, dt, J = 2.50 and 8.03 Hz), 5.78 – 5.70 (m, 1 H), 4.76 (1 H, d, J = 14.3 Hz), 4.13 (s, 3H), 3.16 (s, 3H), 1 .78 (d, 3H, J = 6.02 Hz), 1 .45 (s, 18H); 13C NMR (CDCI3) δ 167.0, 162.9, 160.4, 148.7, 146.3, 143.0, 140.7, 139.9, 135.5, 129.9, 129.8, 126.1 , 123.8, 123.5, 1 19.7, 1 13.8, 1 13.5, 1 1 1 .6, 108.1 , 81 .1 , 70.1 , 45.5, 37.0, 29.7, 26.0, 20.7; LCMS (M+1)+ 607.3, 507.1 , 451 .2.
Step 3:
To a reactor under N2 was added compound 26 (4.74 kg, 7.82 mol) and ethyl acetate (54 L). Hydrochloric acid 37% (5.19 L, 63.2 mol) was charged slowly while keeping the internal temperature below 25°C. The reaction mixture was stirred for 24 – 48 h until the reaction was complete. Ethyl acetate (54L) and water (54 L) were added. The reaction mixture was then treated with triethylamine until pH 8 – 9 was reached. The aqueous layer was removed and then the organic layer was washed water (2 x 54 L). The organic layer was concentrated under reduced pressure to approx. 54 L to give compound 27 (unisolated).
Step 4:
Acetic acid (1 .0 kg, 16.6 mol) was added to the organic layer containing compound 27. The reaction mixture was concentrated and then held for at least 3 h with stirring at RT. The resulted slurry was filtered. The filter cake was washed with ethyl acetate (2 L) and dried under vacuum to give 3.20 kg (87.8% yield) of Example 4 acetic acid solvate (Form 3). The spectroscopic data of this material was identical to that of an authentic sample of the crystalline acetic acid Form 3 of (10R)-7-amino-12-fluoro-2, 10, 16-trimethyl-15-oxo-10, 15,16, 17-tetrahydro-2/-/-8,4-(metheno)pyrazolo[4,3- ?][2,5,1 1 ]-benzoxadiazacyclo-tetradecine-3-carbonitrile prepared according to Example 3.
Preparation of Synthetic Intermediates

7 6 5
Step 1 :
A solution of (-)-DIPCI ((-)-B-chlorodiisopinocampheylborane) (57.1 g, 178 mmol) in THF
(tetrahydrofuran) (100 ml) was cooled to -20 to -30 °C. A solution of compound 1 (31 .3 g, 1 19 mmol) in THF (100 ml) was then added dropwise, via addition funnel (30 min addition). The reaction was left to warm up to room temperature (RT). After 2 h, the reaction was cooled to -30 °C and another portion of (-)-DIPCI (38.0 g, 1 19 mmol) was added. After 30 min, the reaction was allowed to warm to RT and after 1 h, the solvents were removed in vacuo and the residue re-dissolved in MTBE (methyl tertiary-butyl ether) (200 ml). A solution of diethanolamine (31 g, 296 mmol) in ethanol/THF (15 ml/30 ml) was added via addition funnel, to the reaction mixture under an ice bath. The formation of a white precipitate was observed. The suspension was heated at reflux for 2 hours then cooled to room temperature, filtered and the mother liquids concentrated in vacuo. The residue was suspended in heptane/EtOAc (7:3, 200 ml) and again
filtered. This procedure was repeated until no more solids could be observed after the liquids were concentrated. The final yellow oil was purified by column chromatography (eluent: cyclohexane/EtOAc 99:1 to 96:4). The resulting colorless oil was further purified by recrystallization from heptanes, to give alcohol compound 2 (25 g, 80% yield, 99% purity and 96% ee) as white crystals. 1H NMR (400 MHz, CDCI3) δ 7.73 (dd, 1 H), 7.32 (dd, 1 H), 6.74 (ddd, 1 H), 4.99 – 5.04 (m, 1 H), 2.01 (d, 1 H), 1 .44 (d, 3 H). LCMS-ES: No ionization, Purity 99%. Chiral GC (column CP-Chirasil-DexnCB): 96% ee; Rt (minor) 17.7 minutes and Rt (major) 19.4 minutes.
Step 2:
A solution of compound 2 (22 g, 83 mmol) in MTBE (350 mL) was cooled under an ice bath and triethylamine (23 mL, 166 mmol) followed by mesyl chloride (9.6 mL, 124 mmol) were added drop-wise. The reaction was then warmed to RT and stirred for 3 h. The reaction mixture was filtered and the solids washed with EtOAc. The mother liquids were concentrated in vacuo to give compound 3 (35 g, 80% yield) as a pale yellow oil. This material was taken into the following step without further purification. 1H NMR (400 MHz, CDCI3) δ 7.78 (dd, 1 H), 7.24 (dd, 1 H), 6.82 (ddd, 1 H), 2.92 (s, 3 H), 1 .64 (d, 3 H). LCMS-ES no ionization.
Step 3:
A suspension of Cs2C03 (65 g, 201 mmol) and compound 4 (13.3 g, 121 mmol) in 2-CH3-THF (2-methyitetrahydrofuran) (600 mL) and acetone (300 mL) was stirred at RT for 30 minutes then heated at 40 °C before drop-wise addition of a solution of compound 3 (34.4 g, 80 mmol) in 2-CH3-THF (300 mL) via addition funnel. The resulting mixture was left stirring at 75 -80 °C for 24 h. The reaction was then filtered through CELITE® with MTBE, the solvents removed in vacuo and the residue purified by column chromatography over silica gel which was eluted with cyclohexane/EtOAc (9:1 to 1 :1) to give compound 5 (14.3 g, 39 % yield, 90% ee) as a white solid. The solids were then re crystallized from heptane/EtOAc to give compound 5 (10.8 g, 37% yield, 95% ee). 1H NMR (400 MHz, CDCI3) 5 7.38 (dd, 1 H), 7.62 (dd, 1 H), 7.10 (dd, 1 H), 6.75 (ddd, 1 H), 6.44 – 6.51 (m, 2 H), 5.34 – 5.39 (m, 1 H), 4.73 (br s, 2 H), 1 .61 (d, 3 H). LCMS-ES m/z 359 [M+H]+. HPLC (Chiralpak IC 4.6 x 250 mm): 95% ee; Rt (minor) 10.4 minutes; Rt (major) 14.7 minutes; eluent: Heptane 80%/IPA 20% with 0.2% DEA, 0.7 mL/min. Step 4:
Compound 5 (20 g, 57 mmol) was dissolved in methanol (300 mL), and sequentially treated with triethylamine (TEA) (15.4 mL, 1 13 mmol) and PdCI2(dppf) (1 ,1 -bis(diphenylphosphino)ferrocene]dichloropalladium(ll) ) (4.1 g, 5.7 mmol). This mixture was heated at 100 °C for 16 hours, under a 100 psi carbon monoxide atmosphere. LCMS indicated consumption of starting material. The reaction mixture was filtered through a pad of CELITE®, and the filtrate evaporated to a brown oil. The crude product was purified by flash
chromatography over silica gel which was eluted with 50% to 75% ethyl acetate in cyclohexane, affording the pure product 6 as a brick-red solid (13.0 g, 79% yield). 1H NMR (400 MHz, CDCI3) δ 1 .65 (d, 3 H), 3.94 (s, 3 H), 4.75 (br s, 2 H), 6.32 (q, 1 H), 6.42 (dd, 1 H), 6.61 (dd, 1 H), 7.00 (ddd, 1 H), 7.28 (dd, 1 H), 7.60 (dd, 1 H), 8.03 (dd, 1 H). LCMS ES m/z 291 for [M+H]+.
Step 5:
Compound 6 (13.0 g, 45 mmol) was dissolved in acetonitrile (195 mL), and cooled to <10 °C in an ice water bath. NBS (N-bromosuccinimide) (7.9 g, 45 mmol) was added drop-wise to the cooled reaction mixture as a solution in acetonitrile (195 mL), monitoring the internal temperature to ensure it did not rise above 10 °C. After addition was complete, the mixture was stirred for 15 minutes. Thin layer chromatography (TLC) (1 :1 cyclohexane/ethyl acetate) showed consumption of starting material. The reaction mixture was evaporated, and the residue redissolved in ethyl acetate (400 mL), and washed with 2M aqueous NaOH (2 x 300 mL), and 10% aqueous sodium thiosulfate solution (300 mL). The organic extracts were dried over MgS04, and evaporated to a red oil (17.6 g). The crude product was purified over silica gel, which was eluted with 10% to 50% ethyl acetate in cyclohexane, which gave compound 7 (12.0 g, 73% yield). 1H NMR (400 MHz, CDCI3) δ 1 .65 (d, 3 H), 3.96 (s, 3 H), 4.74 – 4.81 (br s, 2 H), 6.33 (q, 1 H), 6.75 (d, 1 H), 7.03 (ddd, 1 H), 7.25 (dd, 1 H), 7.66 (d, 1 H), 8.06 (dd, 1 H). LCMS ES m/z 369/371 [M+H]+. A Chiralpak AD-H (4.6 x 100 mm, 5 micron) column was eluted with 10% MeOH (0.1 % DEA) in C02 at 120 bar. A flow rate of 5.0 mL/min gave the minor isomer Rt 0.6 minutes and the major isomer Rt 0.8 minutes (99% ee). Optical rotation: [ ]d20 = -92.4 deg (c=1 .5, MeOH).
Preparation of (/?)-methyl 2-(1 -((N,N-di-Boc-2-amino-5-bromopyridin-3-yl)oxy)ethyl)-4-fluorobenzoic acid (9)

7
Step 1 :
To a solution of compound 7 (2000 g, 5.4 mol) in dry DCM (dichloromethane) (32000 mL) was added DIPEA (N.N-dsisopropyleibylamine) (2100 g, 16.28 mol) and DMAP (4-dimethylaminopyridine) (132 g, 1 .08 mol). Then Boc20 (di-tert-butyl-dicarbonate) (3552 g, 16.28 mol) was added to the mixture in portions. The reaction was stirred at RT for overnight. TLC (petroleum ether/EtOAc =5:1) show the reaction was complete, the mixture was washed with sat. NH4CI (15 L) two times, then dried over Na2S04and concentrated to give a crude product which was purified by column (silica gel, petroleum ether/EtOAc from 20:1 to 10:1) to give compound 8 (2300 g, 75%) as a white solid.
Step 2:
Compound 8 (50 g, 87.81 mmol, 100 mass%) was charged to a round bottom flask (RBF) containing tetrahydrofuran (12.25 mol/L) in Water (5 mL/g, 3060 mmol, 12.25 mol/L) and sodium hydroxide (1 mol/L) in Water (1 .5 equiv., 131 .7 mmol, 1 mol/L). The biphasic mixture was stirred at RT for 14 hours. 1 N HCI was added to adjust pH to < 2. THF was then removed by vacuum distillation. The product precipitated out was collected by filtration. The filter cake was rinsed with water, pulled dried then dried in vacuum oven to constant weight (48 h, 55°C, 25 mbar). 48.3g isolated, 99% yield. 1H NMR (CDCI3, 400MHz) δ 8.24 (1 H, dd, 1 H, J = 5.76 and 3.0 Hz), 8.16 (1 H, d, J = 2.0 Hz), 7.37 (1 H, dd, J = 2.5 and 9.8 Hz), 7.19 (1 H, d, J = 2 Hz), 7.14 – 7.06 (1 H, m), 6.50 (1 H, q, J = 6.3 Hz), 1 .67 (3H, d, J = 8.4 Hz), 1 .48 (18H, s). 13C NMR (CDCI3, 100 MHz), δ 170.1 , 169.2, 167.6, 165.1 , 150.6, 149.2, 148.6, 141 .4, 140.7, 135.2, 135.1 , 124.2, 122.2,122.1 , 1 19.9, 1 15.4, 1 15.1 , 1 13.4, 1 13.2, 100.0, 83.4, 73.3, 27.9, 23.9. LCMS (M+ +1) 557.2, 555.3, 457.1 , 455.1 , 401 , 0, 399.0.

Step 1 :
Ethyl 1 ,3-dimethylpyrazole-5-carboxylate (5.0 g, 30 mmol) was dissolved in 1 ,2-dichloroethane (200 mL), followed by addition of NBS (5.3 g, 30 mmol) and dibenzoyi peroxide (727 mg, 3.0 mmol), in small portions and stirred at 85 °C for 2 hours. The mixture was allowed to cool, diluted to 400 mL with dichloromethane, and washed with water (2 x 200 mL). The organic layer was dried over MgS04, and evaporated to give compound 10 (4.1 g, 42% yield). TLC (EtOAc/Cyclohexane; 1 :10; KMn04): Rf~0.3. 1H NMR (400 MHz, CDCI3) δ 4.47 (s, 2 H), 4.41 (q, 2 H), 4.15 (s, 3 H), 1 .42 (t, 3 H). LCMS ES m/z 324/326/328 [M+H]+.
Step 2:
Compound 10 (3.0 g, 9.2 mmol) was dissolved in methylamine solution (33% solution in ethanol, 70 mL), and stirred at RT for 16 hours. The mixture was evaporated to give compound 11 (1 .8 g, 71 % yield). 1H NMR (400 MHz, CDCI3) δ 4.39 (q, 2 H), 4.14 (s, 3 H), 4.05 (s, 2 H), 2.62 (d, 3 H), 1 .41 (t, 3 H). LCMS ES m/z 276/278 [M+H]+.
Step 3:
Compound 11 (1 .8 g, 6.5 mmol) was dissolved in dichloromethane (20 mL), and the mixture cooled to 0 °C. A solution of di(fe/?-butyl) dicarbonate (1 .75 g, 8 mmol) in dichloromethane (17.5 mL) was added dropwise. The ice bath was removed and the mixture stirred for 18 hours at room temperature. The mixture was diluted to 100 mL with dichloromethane, and washed with water (2 x 50 mL). Organic extracts were dried over magnesium sulfate, and evaporated to give compound 12 (1 .8 g, 72% yield). 1H NMR (400 MHz, CDCI3) δ 4.48 – 4.44 (m, 2 H), 4.41 (q, 2 H), 4.12 (s, 3 H), 2.82 – 2.79 (m, 3 H), 1 .47 (s, 9 H), 1 .41 (t, 3 H). LCMS ES m/z 376/378 [M+H]+ and 276/278 [M-BOC]+.
Step 4:
Compound 12 (4 g, 1 1 mmol) was dissolved in dioxane (43 mL). Sodium amide (1 g, 27 mmol) was added in one portion. The reaction mixture was stirred at 100 °C for 24 h. After this time, the solvent was removed under reduced pressure to give a white solid. The material was suspended in EtOAc (100 mL) and washed with 5% citric acid solution (100 mL). The organic phase was separated and washed with water (100 mL), dried over MgS04, filtered and the solvent removed in vacuo to give compound 13 as a yellow gum (3.1 g, 84% yield). 1H NMR (400 MHz, DMSO-c/6) δ 4.27 (s, 2 H), 3.92 (s, 3 H), 2.70 (s, 3 H), 1 .40 (s, 9 H). LCMS ES m/z 348/350 [M+H]+ and 248/250 [M-BOC]+.
Step 5:
Compound 13 (3 g, 8.6 mmol) was dissolved in DMF (43 mL, 0.2 M). HOBt (1 .2 g, 8.6 mmol) was added, followed by ammonium chloride (0.9 g, 17.2 mmol). EDCI (2.5 g, 13 mmol) was then added, followed by TEA (2.4 mL, 17 mmol). The reaction mixture was stirred at room temperature. After 18h, the solvent was removed under reduced pressure to give a yellow oil
(8.0 g). The residue was dissolved in EtOAc (75ml_). The organic phase was washed with NaHC03 (sat. solution, 70 ml_) and then brine (100 ml_). The combined organic layers were dried over MgS04 and the solvent removed in vacuo to give compound 14 as a dark yellow oil (2.7 g, 91 % yield). This material was used directly in the next step without further purification. 1H NMR (400 MHz, CDCI3) δ 6.74 (br s, 1 H), 5.95 (br s, 1 H), 4.49 (br s, 2 H), 4.16 (s, 3 H), 2.81 (br s, 3 H), 1 .47 (s, 9 H). LCMS ES m/z 347/349 [M+H]+ and 247/249 [M-BOC]+.
Step 6:
Compound 14 (2.7 g, 7.9 mmol) was dissolved in DCM (80 ml_, 0.1 M). TEA (3.3 ml_, 23.8 mmol) was then added and the reaction mixture cooled down to -5 °C. Trifluoroacetic anhydride (2.2 ml_, 15.8 mmol) in DCM (15 ml_) was added dropwise over 30 min. After addition, the reaction mixture was stirred at 0 °C for 1 h. After this time, the solvents were removed under reduced pressure to give a dark yellow oil. This residue was diluted in DCM (100 ml_), washed with 5% citric acid, sat. NaHC03and brine, dried over MgS04, filtered and the solvents removed in vacuo to give a dark yellow oil (2.6 g). The crude product was purified by reverse phase chromatography to give compound 15 as a yellow oil (2.3 g, 87% yield). 1H NMR (400 MHz, CDCI3) δ 4.46 (br s, 2 H), 4.01 (s, 3 H), 2.83 (br s, 3 H), 1 .47 (s, 9 H). LCMS ES m/z 331 /329 [M+H]+ and 229/231 [M-BOC]+ as the base ion.
Preparation o/: 1 -methyl-3-((methylamino)methyl)-1 H-pyrazole-5-carbonitrile (21)

Step 1 :
To /V-benzylmethylamine (2.40 kg, 19.8 mol) and ethyldiisopropylamine (2.61 kg, 20.2 mol) in acetonitrile (6 L) at 16°C was added chloroacetone (1 .96 kg, 21 .2 mol) over 60 mins [exothermic, temp kept <30°C]. The mixture was stirred at 22°C for 18 hours then concentrated to an oily solid. The residue was triturated with MTBE (5 L), and then filtered through a pad of CELITE® (600 g, top) and silica (1 .5 kg, bottom), washing with MTBE (8 L). The filtrate was evaporated to afford compound 16 (3.35 kg, 18.9 mol, 95%) as a brown oil.
Step 2:
Compound 16 (1 .68 kg, 9.45 mol), Boc-anhydride (2.1 kg, 9.6 mol) and 20wt% Pd/C (50% H20, 56 g) in ethanol (5 L) were hydrogenated in an 1 1 -L autoclave at 50 psi [exotherm to 40°C with 20°C jacket]. The atmosphere became saturated with carbon dioxide during the reaction and so needed to be vented and de-gassed twice to ensure sufficient hydrogen uptake and completion of the reaction. The total reaction time was ~1 .5 hours. Two runs (for a total of 18.9 mol) were combined and filtered through a pad of SOLKA-FLOC®, washing with methanol. The filtrate was treated with DMAP (45 g, 0.37 mol) and stirred at room temperature overnight to destroy the excess Boc-anhydride. The mixture was then concentrated to dryness, dissolved in MTBE (6 L) and filtered through a pad of magnesol (1 kg), washing with MTBE (4 L). The filtrate was evaporated to afford compound 17 (3.68 kg, ~95 wt%, 18.7 mol, 99%) as an orange-brown oil.
Step 3:
To compound 17 (3.25 kg, -95 wt%, 16.5 mol) and diethyl oxalate (4.71 kg, 32.2 mol) in methanol (12 L) at 15°C was added 25 wt% sodium methoxide in methanol (6.94 kg, 32.1 mol) over 25 mins [temp kept <25°C]. The mixture was stirred at 20°C for 16 hours then cooled to -37°C and 37% hydrochloric acid (3.1 kg, 31 mol) was added over 5 mins [temp kept <-10°C]. The mixture was cooled to -40°C and methylhydrazine (1 .42 kg, 30.8 mol) was added over 7 mins [temp kept <-17°C]. The mixture was warmed to 5°C over 90 minutes, then re-cooled to 0°C and quenched by addition of 2.4M KHS04 (6.75 L, 16.2 mol) in one portion [exotherm to 27°C]. The mixture was diluted with water (25 L) and MTBE (15 L), and the layers separated. The organic layer was washed with brine (7 L) and the aqueous layers then sequentially re-extracted with MTBE (8 L). The combined organics were evaporated and azeotroped with toluene (2 L) to afford crude compound 18. Chromatography (20 kg silica, 10-40% EtOAc in hexane) afforded compound 18 (3.4 kg, ~95 wt%, 11 .4 mol, 69%) as an orange oil.
Step 4:
Ammonia (3 kg, 167 mol) was bubbled in to cooled methanol (24 L) [temp kept <18°C]. A solution of compound 18 (4.8 kg, ~95 wt%, 16.1 mol) in methanol (1 .5 L) was added over 30 minutes and the mixture stirred at 25°C for 68 hours and then at 30°C for 24 hours. Two runs (from a total of 9.68 kg of ~95 wt% Step 3) were combined and concentrated to ~13 L volume. Water (30 L) was slowly added over 80 minutes, keeping the temperature 30 to 40°C. The resulting slurry was cooled to 20°C, filtered, washed with water (12 L) and pulled dry on the filter overnight. The solids were triturated in MTBE (8 L) and hexane (8 L) at 45°C then re-cooled to 15°C, filtered, washed with hexane (4 L) and dried under vacuum to afford compound 19 (7.95 kg, 29.6 mol, 90%) as an off-white solid.
Step 5:
To compound 19 (7.0 kg, 26.1 mol) in DCM (30 L) at 0°C was added triethylamine (5.85 kg, 57.8 mol). The mixture was further cooled to -6°C then trifluoroacetic anhydride (5.85 kg, 27.8 mol) added over 90 minutes [temp kept 0 to 5°C]. TLC assay showed the reaction was incomplete. Additional triethylamine (4.1 kg, 40.5 mol) and trifluoroacetic acid (4.1 kg, 19.5 mol) were added over 2 hours until TLC showed complete reaction. The reaction mixture was quenched in to water (40 L) [temp to 23°C]. The layers were separated and the aqueous re-extracted with DCM (8 L). The organic layers were sequentially washed with brine (7 L), filtered through a pad of silica (3 kg) and eluted with DCM (10 L). The filtrate was evaporated and chromatographed (9 kg silica, eluent 10-30% EtOAc in hexane). Product fractions were evaporated and azeotroped with IPA to afford compound 20 (6.86 kg, -94 wt%, 25.8 mol, 99%) as an orange oil.
Step 6:
To compound 20 (6.86 kg, -94 wt%, 25.8 mol) in IPA (35 L) at 17°C was added 37% hydrochloric acid (6.4 L, 77.4 mol). The mixture was heated to 35°C overnight then concentrated to a moist solid and residual water azeotroped with additional IPA (8 L). The resulting moist solid was triturated with MTBE (12 L) at 45°C for 30 minutes then cooled to 20°C and filtered, washing with MTBE (5 L). The solids were dried under vacuum at 45°C to afford compound 21 (4.52 kg, 24.2 mol, 94%) as a white solid. 1H-NMR was consistent with desired product; mp 203-205°C; HPLC 99.3%. 1H NMR (CD3OD, 400 MHz) δ 7.12 (1 H, s), 4.28 (2H, s), 4.09 (3H, s), 2.77 (3H, s). 13C NMR (CD3OD, 100 MHz) δ 144.5, 177.8, 1 14.9, 110.9, 45.9, 39.0, 33.2. LCMS (M++1) 151 .1 , 138.0, 120.0.
PATENT
PATENT
PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2017021823&redirectedID=true
Preparation of the free base of lorlatinib as an amorphous solid is disclosed in
International Patent Publication No. WO 2013/132376 and in United States Patent No. 8,680,1 1 1 . Solvated forms of lorlatinib free base are disclosed in International Patent Publication No. WO 2014/207606.
Example 1
Lab Scale Preparation of Form 7 of (10 ?‘)-7-amino-12-fluoro-2,10,16-trimethyl-15-oxo-10,15,16,17-tetrahydro-2/-/-8,4-(metheno)pyrazolo[4,3- ?l[2,5,1 l lbenzoxadiazacyclotetra-decine- -carbonitrile (lorlatinib) Free Base
[AcOH solvate]
Form 7 of lorlatinib free base was prepared by de-solvation of the acetic acid solvate of lorlatinib (Form 3), prepared as described in International Patent Publication No. WO 2014/207606, via an intermediate methanol solvate hydrate form of lorlatinib (Form 2).
The acetic acid solvate of lorlatinib (Form 3) (5 g, 10.72 mmol) was slurried in methanol
(10 mL/g, 1235.9 mmol) at room temperature in an Easymax flask with magnetic stirring to which triethylamine (1 .2 equiv., 12.86 mmol) was added over 10 minutes. The resulting solution was heated to 60°C and water (12.5 mL/g, 3469.3 mmol) was added over 10 minutes, while maintaining a temperature of 60°C. Crystallization was initiated by scratching the inside of the glass vessel to form a rapidly precipitating suspension which was triturated to make the system mobile. The suspension was then cooled to 25°C over 1 hour, then cooled to 5°C and granulated for 4 hours. The white slurry was filtered and washed with 1 mL/g chilled
water/methanol (1 :1) then dried under vacuum at 50°C overnight to provide the methanol solvate hydrate Form 2 of lorlatinib.
Form 7 was then prepared via a re-slurry of the methanol solvate hydrate Form 2 of lorlatinib in heptane. 100 mg of lorlatinib Form 2 was weighed into a 4-dram vial and 3 mL of heptane was added. The mixture was slurried at room temperature on a roller mixer for 2 hours. Form conversion was confirmed by PXRD revealing complete form change to Form 7 of lorlatinib free base.
Paper
http://pubs.acs.org/doi/abs/10.1021/jm500261q
*E-mail: ted.w.johnson@pfizer.com. Phone: (858) 526-4683., *E-mail: paul.f.richardson@pfizer.com. Phone: (858) 526-4290.

Although crizotinib demonstrates robust efficacy in anaplastic lymphoma kinase (ALK)-positive non-small-cell lung carcinoma patients, progression during treatment eventually develops. Resistant patient samples revealed a variety of point mutations in the kinase domain of ALK, including the L1196M gatekeeper mutation. In addition, some patients progress due to cancer metastasis in the brain. Using structure-based drug design, lipophilic efficiency, and physical-property-based optimization, highly potent macrocyclic ALK inhibitors were prepared with good absorption, distribution, metabolism, and excretion (ADME), low propensity for p-glycoprotein 1-mediated efflux, and good passive permeability. These structurally unusual macrocyclic inhibitors were potent against wild-type ALK and clinically reported ALK kinase domain mutations. Significant synthetic challenges were overcome, utilizing novel transformations to enable the use of these macrocycles in drug discovery paradigms. This work led to the discovery of 8k (PF-06463922), combining broad-spectrum potency, central nervous system ADME, and a high degree of kinase selectivity.
Discovery of (10R)-7-Amino-12-fluoro-2,10,16-trimethyl-15-oxo-10,15,16,17-tetrahydro-2H-8,4-(metheno)pyrazolo[4,3-h][2,5,11]-benzoxadiazacyclotetradecine-3-carbonitrile (PF-06463922), a Macrocyclic Inhibitor of Anaplastic Lymphoma Kinase (ALK) and c-ros Oncogene 1 (ROS1) with Preclinical Brain Exposure and Broad-Spectrum Potency against ALK-Resistant Mutations
References
1H NMR PREDICT


13C NMR PREDICT


 |
|
| Clinical data | |
|---|---|
| Routes of administration |
PO |
| Legal status | |
| Legal status |
|
| Identifiers | |
| CAS Number | 1454846-35-5 |
| ChemSpider | 32813339 |
| Chemical and physical data | |
| Formula | C22H20FN5O2 |
| Molar mass | 405.43 g·mol−1 |
| 3D model (Jmol) | Interactive image |
///////////////////Lorlatinib, PF-6463922, anti-neoplastic, Pfizer, ROS1, ALK, phase 2, UNII:OSP71S83EU, лорлатиниб , لورلاتينيب , 洛拉替尼 , Orphan Drug, PF 6463922
Fc2ccc3C(=O)N(C)Cc1nn(C)c(C#N)c1c4cc(O[C@H](C)c3c2)c(N)nc4
PF-04136309

PF 4136309
PF4136309; PF 4136309; PF-4136309; PF04136309; PF4136309; PF-04136309; INCB8761; INCB 8761; INCB-8761
(S)-N-(2-(3-((4-hydroxy-4-(5-(pyrimidin-2-yl)pyridin-2-yl)cyclohexyl)amino)pyrrolidin-1-yl)-2-oxoethyl)-3-(trifluoromethyl)benzamide
N-[2-[(3S)-3-[[trans-4-Hydroxy-4-[5-(2-pyrimidinyl)-2-pyridinyl]cyclohexyl]amino]-1-pyrrolidinyl]-2-oxoethyl]-3-(trifluoromethyl)benzamide
N-[2-((3S)-3-[4-hydroxy-4-(4-pyrimidin-2-ylphenyl)cyclohexyl]aminopyrrolidin-1-yl)-2- oxoethyl]-3-(trifluoromethyl)benzamide
1341224-83-6
MF: C29H31F3N6O3
MW: 568.24097
CC chemokine receptor 2 (CCR2) antagonist


PF-4136309, also known as INCB8761, is an orally available human chemokine receptor 2 (CCR2) antagonist with potential immunomodulating and antineoplastic activities. Upon oral administration, CCR2 antagonist PF-04136309 specifically binds to CCR2 and prevents binding of the endothelium-derived chemokine ligand CLL2 (monocyte chemoattractant protein-1 or MCP1) to its receptor CCR2, which may result in inhibition of CCR2 activation and signal transduction. This may inhibit inflammatory processes as well as angiogenesis, tumor cell migration, and tumor cell proliferation. The G-protein coupled receptor CCR2 is expressed on the surface of monocytes and macrophages, stimulates the migration and infiltration of these cell types, and plays an important role in inflammation, angiogenesis, and tumor cell migration and proliferation.
- Originator Pfizer
- Class Analgesics
- Mechanism of Action CCR2 receptor antagonists
Highest Development Phases
- Phase I/II Pancreatic cancer
- Discontinued Hepatic fibrosis; Pain
Most Recent Events
- 01 Apr 2016 Phase-I/II clinical trials in Pancreatic cancer (Combination therapy, First-line therapy, Metastatic disease) in USA (PO) (NCT02732938)
- 01 Dec 2015 Phase-I clinical trials in Pancreatic cancer (In volunteers) in Belgium (PO) (NCT02598206)
- 09 Nov 2015 Pfizer plans a phase I trial in Healthy volunteers in Belgium and USA (NCT02598206)


(S)-N-[2-(3-{trans-4-Hydroxy-4-[5-(pyrimidin-2-yl)pyridin-2-
yl]cyclohexylamino}pyrrolidin-1-yl)-2-oxoethyl]-3-(trifluoromethyl)benzamide
MS (M+H)+:569.2.
1H NMR (400 MHz, CD3OD): δ 9.57 – 9.45 (m, 1H), 8.94-8.84 (m, 2H), 8.82 –
8.72 (m, 1H), 8.27 – 8.19 (m, 1H), 8.15 (d, J = 7.8 Hz, 1H), 7.91 – 7.84 (m, 2H), 7.69
(dd, J = 7.8, 7.8 Hz, 1H), 7.46-7.39 (m, 1H), 4.29 – 4.12 (m, 2H), 3.87 (dd, J = 10.1, 6.4
Hz, 0.5H), 3.83 – 3.39 (m, 3.5H), 3.38 – 3.32 (m, 1H), 3.02 – 2.91 (m, 1H), 2.51 – 2.35
(m, 2H), 2.34 – 2.14 (m, 1H), 2.13 – 1.88 (m, 2.5H), 1.88 – 1.76 (m, 0.5H), 1.74 – 1.56
(m, 4H).
Anal. (C29H31F3N6O3): calcd C 61.24, H 5.50, N 14.79; found C 61.18, H 5.59,
N 14.87.
INTERMEDIATES
8-(5-Bromopyridin-2-yl)-1,4-dioxaspiro[4.5]decan-8-ol

LC-MS (M+H)+: 316.1/314.1. 1H NMR (300 MHz,CDCl3): δ 8.60 (s, 1 H), 7.82 (d, 1 H), 7.38 (d, 1 H), 4.6 (s, 1 H), 4.0 (m, 4 H), 2.2 (m, 4
H), 1.7 (m, 4 H).
8-(5-Pyrimidin-2-ylpyridin-2-yl)-1,4-dioxaspiro[4.5]decan-8-ol
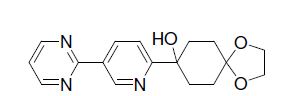
LC-MS (M+H)+: 314.2.
4-Hydroxy-4-(5-pyrimidin-2-ylpyridin-2-yl)cyclohexanone

MS
(M+H)+: 270.2.
tert-Butyl [(S)-1-({[3-(Trifluoromethyl)benzoyl]amino}acetyl)
pyrrolidin-3-yl]carbamate.
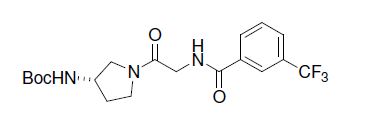
MS (M-Boc+H)+: 316.
(S)-N-{2-[3-Aminopyrrolidin-1-yl]-2-oxoethyl}-3-(trifluoromethyl)
benzamide hydrochloride
MS
(M+H)+: 316.
PATENT
WO 2012114223
https://www.google.com/patents/WO2012114223A1?cl=en
Example 35
Step A
8-(4-lodo-phenyl)-1 ,4-dioxa-spiro[4.5]decan-8-ol. To a solution of 1 ,4-diiodobenzene (16.5 g, 50 mmol) in THF (350 mL) at -78°C was added n-BuLi (2.5 M, 24 mL) over 1 hour. After stirred additional 30 minutes, a solution of 1 ,4-dioxa-spiro[4.5]decan-8-one (7.8 g, 50 mmol) in THF (30 mL) was added in and the resulting mixture was stirred for 3 hours. To the mixture was added TMSCI (5.4 g, 50 mmol) and the resulting mixture was allowed to warm to rt and stirred at rt for 18 hours. The reaction mixture was neutralized to pH 6.0, and extracted with ethyl acetate (3X 50 mL). The organic extracts were combined, washed with saline solution (2X 50 mL), dried over sodium sulfate, concentrated in vacuo. The residue was chromatographed on silica gel, eluting with hexane/ethyl acetate (95/5 to 100/0). The appropriate fractions were combined to give 8-(4-lodo-phenyl)-1 ,4-dioxa-spiro[4.5]decan-8-ol (12 g, 66.6%) with LCMS: 361 .2 (M+H+, 100%) and {[8-(4-iodophenyl)-1 ,4- dioxaspiro[4.5]dec-8-yl]oxy}(trimethyl)silane (6 g, 27%) with LCMS: 433.1 (M+H+, 100%). Step B

8-(4-pyrimidin-2-ylphenyl)-1 ,4-dioxaspiro[4.5]decan-8-ol. To a solution of 8-(4-iodo- phenyl)-1 ,4-dioxa-spiro[4.5]decan-8-ol (450.0 mg, 1.249 mmol) in THF (1.0 mL) at room temperature was added dropwise isopropylmagnesium chloride (2.0 M in THF, 1 .37 mL) and the reaction mixture was stirred at room temperature for 30 mins. To another flask charged with nickel acetylacetonate (20 mg, 0.06 mmol) and 1 ,3-bis(diphenylphosphino)-propane (26 mg, 0.062 mmol) suspened in THF (3 mL) under N2 was added 2-bromopyrimidine (199 mg, 1.25 mmol). The resulting mixture was stirred at room temperature until it is clear. The second mixture was transferred into the degassed Grignard solution prepared in step 1. The resulting mixture was stirred at room temperature overnight. The reaction mixture was diluted with EtOAc, quenched with water, washed with brine, dried overNa2S04, and concentrated. The residue was columned on silica gel, eluted with hexane/EtOAc (2/1 ), to gave the desired compound (270 mg, 69%) as white solid. LCMS: 313.1 , (M+H, 100%). 1H
NMR (CDCIs): δ 8.86 (d, 2H), 8.46 (dd, 2H), 7.71 (dd, 2H), 7.24 (t, 1 H), 4.05 (d, 4H), 2.30 (dt, 2H), 2.18 (dt, 2H), 1 .90 (m, 2H), 1 .78 (m, 2H).
Step C

4-Hydroxy-4-(4-pyrimidin-2-ylphenyl)cyclohexanone. The title compound was prepared by treating the ketal of step B with HCI in water following the procedure described in step B of Example 2. MS (M+H)+ 269.
Step D

N-[2-((3S)-3-[4-hydroxy-4-(4-pyrimidin-2-ylphenyl)cyclohexyl]aminopyrrolidin-1-yl)-2- oxoethyl]-3-(trifluoromethyl)benzamide bis(trifluoroacetate) (salt). To a 1-neck round-bottom flask charged with methylene chloride (1 ml.) was added 4-hydroxy-4-(4-pyrimidin-2- ylphenyl)cyclohexanone (50.0 mg, 0.186 mmol), N-2-[(3S)-3-aminopyrrolidin-1-yl]-2- oxoethyl-3-(trifluoromethyl)benzamide hydrochloride (65.5 mg, 0.186 mmol), and triethylamine (85.7 uL, 0.615 mmol). The resulting mixture was stirred at 25°C for 30 minutes, and to it was added sodium triacetoxyborohydride (62.4 mg, 0.28 mmol) in portion. The reaction mixture was stirring at rt overnight. The reaction was concentrated, and the residue was chromatographed on Si02, eluted with acetone/methanol (100% to 90%/10%) to give two fractions, which were further purified on prep-LCMS separately to afford F1 (24.2 mg ) and F2 (25.9 mg) as white powder in total 34% of the yield. LCMS: 568.2 (M+H, 100%)
Paper
Discovery of INCB8761/PF-4136309, a Potent, Selective, and Orally Bioavailable CCR2 Antagonist

We report the discovery of a new (S)-3-aminopyrrolidine series of CCR2 antagonists. Structure–activity relationship studies on this new series led to the identification of 17 (INCB8761/PF-4136309) that exhibited potent CCR2 antagonistic activity, high selectivity, weak hERG activity, and an excellent in vitro and in vivo ADMET profile. INCB8761/PF-4136309 has entered human clinical trials.
HPLC
http://link.springer.com/article/10.1007/s10337-015-2860-8
A precise and sensitive LC method was developed and further validated for the determination of enantiomeric purity of (S)-N-[2-(3-{trans-4-hydroxy-4-[5-(pyrimidin-2-yl)pyridin-2-yl] cyclohexylamino} pyrrolidin-1-yl)-2-oxoethyl]-3-(trifluoromethyl) benzamide (PF-04136309). Baseline separation with a resolution higher than 1.8 was accomplished within 40 min using a CHIRALPAK AD (250 × 4.6 mm; particle size 5 μm) column, with n-hexane:2-propanol (70:30v/v) as mobile phase at a flow rate of 1 mL min−1. The eluted analytes were subsequently detected with a UV detector at 260 nm. The effects of mobile phase components and temperature on enantiomeric selectivity as well as the resolution of enantiomers were thoroughly investigated. The calibration curves were plotted within a concentration range between 0.01 and 1 mg mL−1 (n = 9), and recoveries between 98.17 and 101.28 % were obtained, with relative standard deviation (RSD) lower than 1.44 %. The LOD and LOQ for PF-04136309 were 3.59 and 11.54 μg mL−1 and for its enantiomer were 3.39 and 11.28 μg mL−1, respectively. The developed method was demonstrated to be accurate, robust and sensitive for the determination of enantiomeric purity of PF-04136309, especially for the analysis of bulk samples.
REFERENCES
1: Xue CB, Wang A, Han Q, Zhang Y, Cao G, Feng H, Huang T, Zheng C, Xia M, Zhang K, Kong L, Glenn J, Anand R, Meloni D, Robinson DJ, Shao L, Storace L, Li M, Hughes RO, Devraj R, Morton PA, Rogier DJ, Covington M, Scherle P, Diamond S, Emm T, Yeleswaram S, Contel N, Vaddi K, Newton R, Hollis G, Metcalf B. Discovery of INCB8761/PF-4136309, a Potent, Selective, and Orally Bioavailable CCR2 Antagonist. ACS Med Chem Lett. 2011 Oct 5;2(12):913-8. doi: 10.1021/ml200199c. eCollection 2011 Dec 8. PubMed PMID: 24900280; PubMed Central PMCID: PMC4018168.
http://www.pfizer.com/files/news/asco/ASCO2016_PipelineFactSheet_CCR2.pdf
//////1341224-83-6, PF 4136309, PF4136309, PF 4136309, PF-4136309, PF04136309, PF4136309, PF-04136309, INCB8761, INCB 8761, INCB-8761, PFIZER, PHASE 2
O=C(NCC(N1C[C@@H](NC2CCC(C3=NC=C(C4=NC=CC=N4)C=C3)(O)CC2)CC1)=O)C5=CC=CC(C(F)(F)F)=C5
ANIDULAFUNGIN

OR

Anidulafungin
V-Echinocandin
| CAS Number | 166663-25-8 |
|---|---|
N-[(3S,6S,9S,11R,15S,18S,20R,21R,24S,25S,26S)-6-[(1S,2R)-1,2-dihydroxy-2-(4-hydroxyphenyl)ethyl]-11,20,21,25-tetrahydroxy-3,15-bis[(1R)-1-hydroxyethyl]-26-methyl-2,5,8,14,17,23-hexaoxo-1,4,7,13,16,22-hexaazatricyclo[22.3.0.09,13]heptacosan-18-yl]- 4-{4-[4-(pentyloxy)phenyl]phenyl}benzamide
- LY-307853
- LY-329960
- LY-333006
- LY303366
- VEC
- VER-002
1H NMR (700 MHz, d6-DMSO) δ 0.91 (t, 3H), 1.12 (d, 3H), 1.36 (m, 2H), 1.41 (m, 2H), 1.74 (p, 2H), 1.88 and 1.97 (overlapped, 2H), 3.85 (overlapped, 1H), 4.01 (t, 2H), 4.35 (overlapped, 1H), 4.44 (m, 1H), 4.76 (m, 1H), 4.80 (m, 1H), 5.02 (m, 1H), 5.07 (d, 1H), 5.52 (d, 1H), 7.04 (d, 1H), 7.66 (d, 1H), 7.74 (d, 1H), 7.80 (d, 1H), 7.82 (d, 1H), 7.97 (d, 1H), 8.01 (d, 1H), 8.14 (broad s, 1H), 8.60 (d, 1H). IR (cm−1)
KBr νmax; 3450 (O−H), 2932 (C−H), 2871 (C−H), 1632 (C═O), 1517 (Ar), 1488 (Ar), 1248 (C−O), 821 (C−H out-of-plane bending Ar 2 adj H’s).
Anidulafungin (brand names: Eraxis (in U.S. and Russia), Ecalta (in Europe)) is a semisynthetic echinocandin used as anantifungal drug. Anidulafungin was originally manufactured and submitted for FDA approval by Vicuron Pharmaceuticals.[1] Pfizeracquired the drug upon its acquisition of Vicuron in the fall of 2005.[2] Pfizer gained approval by the Food and Drug Administration(FDA) on February 21, 2006;[3] it was previously known as LY303366. Preliminary evidence indicates it has a similar safety profile tocaspofungin. Anidulafungin has proven efficacy against esophageal candidiasis, but its main use will probably be in invasive Candidainfection;[4][5][6] it may also have application in treating invasive Aspergillus infection. It is a member of the class of antifungal drugs known as the echinocandins; its mechanism of action is by inhibition of (1→3)-β-D-glucan synthase, an enzyme important to the synthesis of the fungal cell wall.
Pharmacodynamics and pharmacokinetics
Anidulafungin significantly differs from other antifungals in that it undergoes chemical degradation to inactive forms at body pH and temperature. Because it does not rely on enzymatic degradation or hepatic or renal excretion, the drug is safe to use in patients with any degree of hepatic or renal impairment.[7]
Distribution: 30–50 L. Protein binding: 84%.
Anidulafungin is not evidently metabolized by the liver. This specific drug undergoes slow chemical hydrolysis to an open-ring peptide which lacks antifungal activity. The half-life of the drug is 27 hours. Thirty percent is excreted in the feces (10% as unchanged drug). Less than 1% is excreted in the urine.[8][9][10]
Mechanism of action
Anidulafungin inhibits glucan synthase, an enzyme important in the formation of (1→3)-β-D-glucan, a major fungal cell wall component. Glucan synthase is not present in mammalian cells, so it is an attractive target for antifungal activity.[11]
Semisynthesis
Anidulafungin is manufactured via semisynthesis. The starting material is echinocandin B (a lipopeptide fermentation product ofAspergillus nidulans or the closely related species, A. rugulosus), which undergoes deacylation (cleavage of the linoleoyl side chain) by the action of a deacylase enzyme from the bacterium Actinoplanes utahensis;[12] in three subsequent synthetic steps, including a chemical reacylation, the antifungal drug anidulafungin[11][13] is synthesized.
Aspergillus nidulans. Anidulafungin is an echinocandin, a class of antifungal drugs that inhibits the synthesis of 1,3-β-D-glucan, an essential component of fungal cell walls.
ERAXIS (anidulafungin) is 1-[(4R,5R)-4,5-dihydroxy-N -[[4“-(pentyloxy)[1,1′:4′,1”-terphenyl]-4-yl]carbonyl]-L-ornithine]echinocandin B. Anidulafungin is a white to off-white powder that is practically insoluble in water and slightly soluble in ethanol. In addition to the active ingredient, anidulafungin, ERAXIS for Injection contains the following inactive ingredients:
50 mg/vial – fructose (50 mg), mannitol (250 mg), polysorbate 80 (125 mg), tartaric acid (5.6 mg), and sodium hydroxide and/or hydrochloric acid for pH adjustment.
100 mg/vial – fructose (100 mg), mannitol (500 mg), polysorbate 80 (250 mg), tartaric acid (11.2 mg), and sodium hydroxide and/or hydrochloric acid for pH adjustment.
The empirical formula of anidulafungin is C58H73N7O17 and the formula weight is 1140.3. The structural formula is
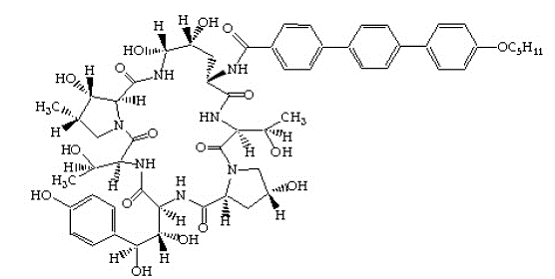
Prior to administration, ERAXIS for Injection requires reconstitution with sterile Water for Injection and subsequent dilution with either 5% DextroseInjection, USP or 0.9% Sodium Chloride Injection, USP (normal saline).
SYNTHESIS
J MED CHEM 1995, 38 3271-3281
Semisynthetic Chemical Modification of the Antifungal Lipopeptide …
pubs.acs.org/doi/abs/10.1021/jm00017a012
Aug 1, 1995 – J. Med. Chem. , 1995, 38 (17), pp 3271–3281. DOI: 10.1021/jm00017a012 … Journal ofMedicinal Chemistry 2001 44 (16), 2671-2674
Echinocandin B (ECB) is a lipopeptide composed of a complex cyclic peptide acylated at the N-terminus by linoleic acid. Enzymatic deacylation of ECB provided the peptide “nucleus” as a biologically inactive substrate from which novel ECB analogs were generated by chemical reacylation at the N-terminus. Varying the acyl group revealed that the structure and physical properties of the side chain, particularly its geometry and lipophilicity, played a pivotal role in determining the antifungal potency properties of the analog. Using CLOGP values to describe and compare the lipophilicities of the side chain fragments, it was shown that values of > 3.5 were required for expression of antifungal activity. Secondly, a linearly rigid geometry of the side chain was the most effective shape in enhancing the antifungal potency. Using these parameters as a guide, a variety of novel ECB analogs were synthesized which included arylacyl groups that incorporated biphenyl, terphenyl, tetraphenyl, and arylethynyl groups. Generally the glucan synthase inhibition by these analogs correlated well with in vitro and in vivo activities and was likewise influenced by the structure of the side chain. These structural variations resulted in enhancement of antifungal activity in both in vitro and in vivo assays. Some of these analogs, including LY303366 (14a), were effective by the oral route of administration.
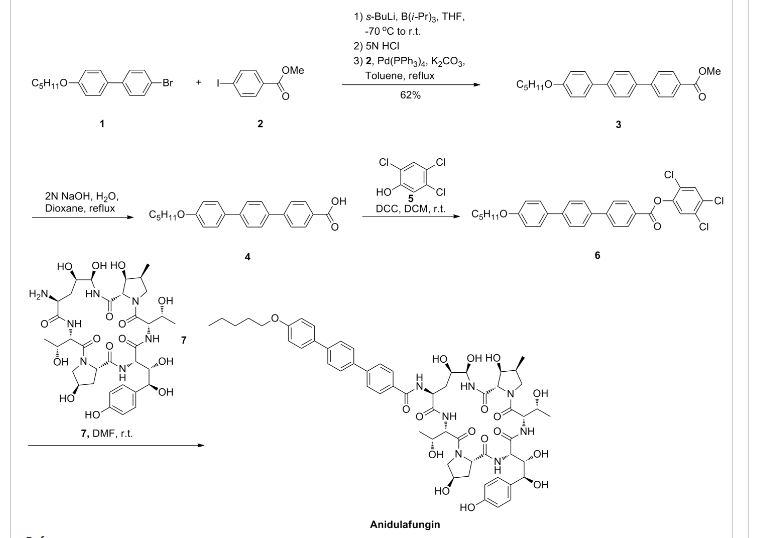
PATENT
US 5965525
http://www.google.co.in/patents/US5965525
PATENT
US 4293482
http://www.google.co.in/patents/US4293482
Paper
Commercialization and Late-Stage Development of a Semisynthetic Antifungal API: Anidulafungin/d-Fructose (Eraxis)
http://pubs.acs.org/doi/abs/10.1021/op800055h

Many years ago anidulafungin 1 was identified as a potentially useful medicine for the treatment of fungal infections. Its chemical and physical properties as a relatively high molecular weight semisynthetic derived from echinocandin B proved to be a significant hurdle to its final presentation as a useful medicine. It has recently been approved as an intravenous treatment for invasive candidaisis, an increasingly common health hazard that is potentially life-threatening. The development and commercialization of this API, which is presented as a molecular mixture of anidulafungin and d-fructose is described. This includes, single crystal X-ray structures of the starting materials, the echinocandin B cyclic-peptide nucleus (ECBN·HCl) and the active ester 1-({[4′′-(pentyloxy)-1,1′:4′,1′′-terphenyl-4-yl]carbonyl}oxy)-1H-1,2,3-benzotriazole (TOBt). Details of the structure and properties of starting materials, scale-up chemistry and unusual crystallization phenomena associated with the API formation are discussed.
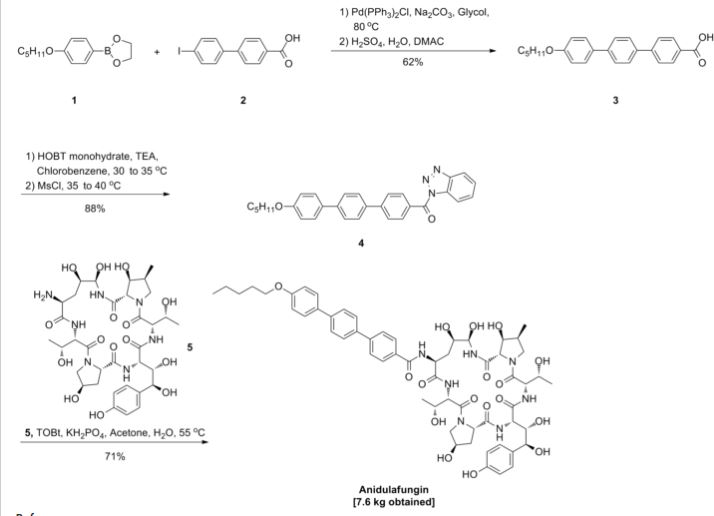
References
- PRNewswire. Vicuron Pharmaceuticals Files New Drug Application (NDA) for Anidulafungin for Treatment of Invasive Candidiasis/Candidemia 08-18-2005.
- Jump up^ PRNewswire. Vicuron Pharmaceuticals Stockholders Approve Merger With Pfizer 08-15-2005
- “FDA Approves New Treatment for Fungal Infections”. FDA News Release. Food and Drug Administration. 2006-02-21. Archived from the original on 10 July 2009. Retrieved 2009-08-01.
- Krause DS, Reinhardt J, Vazquez JA, Reboli A, Goldstein BP, Wible M, Henkel T (2004). “Phase 2, randomized, dose-ranging study evaluating the safety and efficacy of anidulafungin in invasive candidiasis and candidemia”. Antimicrob Agents Chemother 48 (6): 2021–4.doi:10.1128/AAC.48.6.2021-2024.2004. PMC 415613. PMID 15155194.
- Jump up^ Pfaller MA, Boyken L, Hollis RJ, Messer SA, Tendolkar S, Diekema DJ (2005). “In Vitro Activities of Anidulafungin against More than 2,500 Clinical Isolates of Candida spp., Including 315 Isolates Resistant to Fluconazole”. J Clin Microbiol 43 (11): 5425–7.doi:10.1128/JCM.43.11.5425-5427.2005. PMC 1287823. PMID 16272464.
- J Pfaller MA, Diekema DJ, Boyken L, Messer SA, Tendolkar S, Hollis RJ, Goldstein BP (2005). “Effectiveness of anidulafungin in eradicating Candida species in invasive candidiasis”. Antimicrob Agents Chemother 49 (11): 4795–7. doi:10.1128/AAC.49.11.4795-4797.2005.PMC 1280139. PMID 16251335.
- Jump up^ “Eraxis at RxList”. 2009-06-24. Retrieved 2009-08-01.
- Trissel LA and Ogundele AB, “Compatibility of Anidulafungin With Other Drugs During Simulated Y-Site Administration,”Am J Health-Sys Pharm, 2005, 62:834-7.
- Vazquez JA, “Anidulafungin: A New Echinocandin With a Novel Profile,” Clin Ther, 2005, 27(6):657-73.
- Jump up^ Walsh TJ, Anaissie EJ, Denning DW, et al., “Treatment of Aspergillosis: Clinical Practice Guidelines of the Infectious Diseases Society of America,” Clin Infect Dis, 2008, 46(3):327-60
- Denning DW (1997). “Echinocandins and pneumocandins – a new antifungal class with a novel mode of action”. J Antimicrob Chemother 40 (5): 611–614. doi:10.1093/jac/dkf045.PMID 9421307.
- Lei Shao; Jian Li; Aijuan Liu; Qing Chang; Huimin Lin; Daijie Chen (2013). “Efficient Bioconversion of Echinocandin B to Its Nucleus by Overexpression of Deacylase Genes in Different Host Strains”. Applied and Environmental Microbiology 79 (4): 1126–1133. doi:10.1128/AEM.02792-12. PMC 3568618. PMID 23220968.
- “Anidulafungin EMA Europa” (PDF).
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
N-[(3S,6S,9S,11R,15S,18S,20R,21R,24S,25S,26S)-6-[(1S,2R)-1,2-dihydroxy-2-(4-hydroxyphenyl)ethyl]-11,20,21,25-tetrahydroxy-3,15-bis[(1R)-1-hydroxyethyl]-26-methyl-2,5,8,14,17,23-hexaoxo-1,4,7,13,16,22-hexaazatricyclo[22.3.0.09,13]heptacosan-18-yl]- 4-{4-[4-(pentyloxy)phenyl]phenyl}benzamide
|
|
| Clinical data | |
| Trade names | Eraxis |
| AHFS/Drugs.com | Monograph |
| Pharmacokinetic data | |
| Protein binding | 84 % |
| Biological half-life | 40–50 hours |
| Identifiers | |
| CAS Number | 166663-25-8 |
| ATC code | J02AX06 (WHO) |
| PubChem | CID 166548 |
| DrugBank | DB00362 |
| ChemSpider | 21106258 |
| UNII | 9HLM53094I |
| KEGG | D03211 |
| ChEBI | CHEBI:55346 |
| ChEMBL | CHEMBL1630215 |
| Chemical data | |
| Formula | C58H73N7O17 |
| Molar mass | 1140.24 g/mol |
//////////FUNGIN, ANIDULAFUNGIN, Eraxis , Ecalta, semisynthetic echinocandin, anantifungal drug, FDA 2006, PFIZER, LY-307853, LY-329960, LY-333006, LY303366, VEC, VER-002, 166663-25-8, Eli Lilly and Company Inc.

CCCCCOc1ccc(cc1)c2ccc(cc2)c3ccc(cc3)C(=O)N[C@H]6C[C@@H](O)[C@@H](O)NC(=O)C4[C@@H](O)[C@@H](C)CN4C(=O)C(NC(=O)C(NC(=O)C5C[C@@H](O)CN5C(=O)C(NC6=O)[C@@H](C)O)[C@@H](O)[C@H](O)c7ccc(O)cc7)[C@@H](C)O
Varenicline (Chantix™) バレニクリン酒石酸塩

Varenicline (Chantix™)
Varenicline
- MF C13H13N3
- MW 211.26
Varenicline (trade name Chantix and Champix usually in the form of varenicline tartrate), is a prescription medication used to treatnicotine addiction. Varenicline is a nicotinic receptor partial agonist—it stimulates nicotine receptors more weakly than nicotine itself does. In this respect it is similar to cytisine and different from the nicotinic antagonist, bupropion, and nicotine replacement therapies(NRTs) like nicotine patches and nicotine gum. As a partial agonist it both reduces cravings for and decreases the pleasurable effects of cigarettes and other tobacco products. Through these mechanisms it can assist some patients to quit smoking.



Varenicline Tartrate

C13H13N3
 C4H6O6 : 361.35
C4H6O6 : 361.35[375815-87-5]
Medical uses
Varenicline is used for smoking cessation. In a 2009 meta-analysis varenicline was found to be more effective than bupropion (odds ratio 1.40) and NRTs (odds ratio 1.56).[1]
A 2013 Cochrane overview and network meta-analysis concluded that varenicline is the most effective medication for tobacco cessation and that smokers were nearly three times more likely to quit on varenicline than with placebo treatment. Varenicline was more efficacious than bupropion or NRT and as effective as combination NRT for tobacco smoking cessation.[2][3]
The United States’ Food and Drug Administration (US FDA) has approved the use of varenicline for up to twelve weeks. If smoking cessation has been achieved it may be continued for another twelve weeks.[4]
Varenicline has not been tested in those under 18 years old or pregnant women and therefore is not recommended for use by these groups. Varenicline is considered a class C pregnancy drug, as animal studies have shown no increased risk of congenital anomalies, however, no data from human studies is available.[5] An observational study is currently being conducted assessing for malformations related to varenicline exposure, but has no results yet.[6] An alternate drug is preferred for smoking cessation during breastfeeding due to lack of information and based on the animal studies on nicotine.[7]

Varenicline L-tartrate (Compound I) is the international commonly accepted name for 7,8,9,10- tetrahydro-6, 10-methano-6i7-pyrazino [2, 3- h] [3 ] benzazepme, (2R, 3R) -2 , 3-dihydroxybutanedioate (1:1) (which is also known as 5,8,14- tπazatetracyclo [10.3.1. O2‘11. O4‘9] -hexadeca-2 (11) , 3, 5, 7, 9-pentaene, (2R, 3R)-2,3- dihydroxybutanedioate (1:1)) and has an empirical formula of C13H13N3 • C4H6O6 and a molecular weight of 361.35. Varenicline L-tartrate is a commercially marketed pharmaceutically active substance known to be useful for the treatment of smoking addiction.

(D
Varenicline L-tartrate is a partial agonist selective for (X4β2 nicotinic acetylcholine receptor subtypes. In the United States, varenicline L-tartrate is marketed under the name Chantix™ for the treatment of smoking cessation. Varenicline base and its pharmaceutically acceptable acid addition salts are described in U.S. Patent No. 6,410,550. In particular, Example 26 of U.S. Patent No. 6,410,550 describes the preparation of varenicline hydrochloride salt using 1- (4 , 5-dinitro-10- aza-tπcyclo [6.3.1.O2‘7] dodeca-2, 4, 6-trien-10-yl) -2,2,2- tπfluoroethanone (compound of formula (III)) as starting compound. On the other hand, Example HA) of U.S. Patent No. 6,410,550 illustrates the preparation of compound of formula (III) via nitration of compound of formula (II) using an excess of nitronium triflate (>4 equiv) as a nitrating agent. The process disclosed in U.S. Patent No. 6,410,550 is depicted in Scheme 1.

VareniclineΗCl
Scheme 1
However, Coe et al., J. Med. Chem., 48, 3474 (2005), describes the same process and examples as U.S. Patent No. 6,410,550, and it also reveals that this process affords intermediate ortho-4 , 5-dinitrocompound of formula (III) together with the meta-3, 5-dinitro- isomer (i.e. the meta-dinitrocompound) in a ratio 9:1. The presence of the meta-dinitrocompound may affect not only the purity of the intermediate compound of formula III but it may also have an effect on the purity of the final varenicline tartrate, given that it can be carried along the synthetic pathway and/or it can also give rise to other derivative impurities. Thereby, as well as in U.S. Patent No. 6,410,550, in order to isolate pure compound of formula (III) , the raw product is triturated with ethyl acetate/hexane to afford compound of formula (III) with 77% yield. Additionally, the mother liquor is purified by chromatography on silica gel to improve the yield to a total of 82.8%. However, this process is not desirable for industrial implementation since it requires extensive and complicated purification procedures, i.e. trituration of the solid product along with column chromatography purification of the mother liquor, which is not very efficient or suitable for industrial scale-up.
Several improved processes for the synthesis of varenicline or its salts have been reported in the literature (e.g. WO2006/090236) . However, none of these processes tackle the optimization of the purification step of compound of formula (III).
There is therefore the need for providing an improved process for the preparation of varenicline L- tartrate which involves simple experimental procedures well suited to industrial production, which avoids the use of column chromatography purifications, and which affords high pure varenicline L-tartrate which hence can be used directly as a starting product for the preparation of the marketed pharmaceutical speciality.
Additionally, it has been observed that varenicline L-tartrate is usually obtained as a yellow solid under – A –
standard synthetic conditions. In this regard, colour must be attributed to the presence of some specific impurities that may or may not be detectable by conventional methods such as HPLC. The presence of impurities may adversely affect the safety and shelf life of formulations. In this connection, International application No. WO2006/090236 describes the isolation of vareniclme L- tartrate as a white solid. However, in order to remove coloured impurities, the varenicline L-tartrate obtained in WO2006/090236 is treated with a particular activated carbon having a specific grade (i.e. Darco KB-B™) . In fact, Example 5 of WO2006/090236 describes a large reprocessing step which comprises: dissolving varenicline L-tartrate in water, adding toluene, basifying with NaOH aqueous solution, collecting the toluene phase containing varenicline free base, distilling, adding methanol, azeotropically distilling the mixture, and adding more methanol to obtain a methanolic solution containing varenicline free base, adding Darco KB-B™ (10% w/w) , stirring for one hour, filtering through a pad of celite, and treating with L-tartaric acid to give varenicline L- tartrate salt as a white solid. Further, WO2006/090236 provides the absorbance at 430 nm of a varenicline L- tartrate salt solution, either in dichloromethane or in toluene, with or without using Darco KB-B™ activated carbon. However, this measure cannot be used to corroborate the whiteness of the solid varenicline L- tartrate. In addition, Example 3 of International application No. WO2002/092089, also disclose the preparation of varenicline L-tartrate polymorphic form C (i.e. a hydrate polymorph) as a white precipitate. Therefore, there is also a need for a simple and efficient method for preparing varenicline L-tartrate with enhanced whiteness and having a high purity.
SYNTHESIS



Synthesis of Intermediate VIII
Paper
J. Med. Chem. 48, 3474 (2005).
http://pubs.acs.org/doi/pdf/10.1021/jm050069n
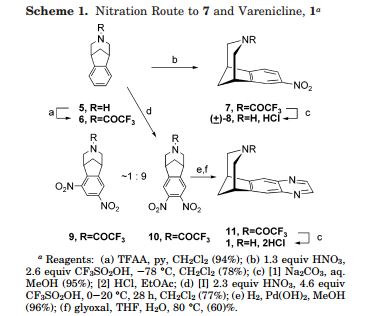
PATENT
https://www.google.com/patents/WO2001062736A1?cl=en
CLIP
Profiles of Drug Substances, Excipients and Related Methodology, Volume 37
edited by Harry G. Brittain

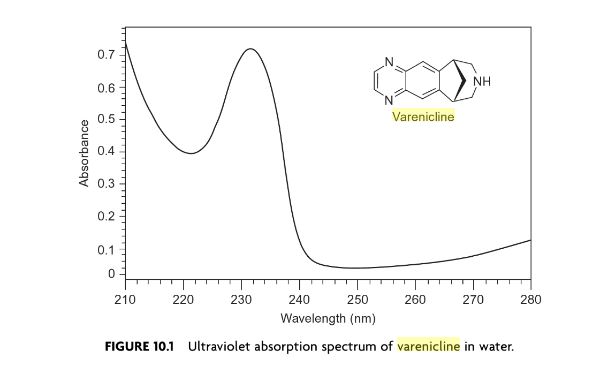

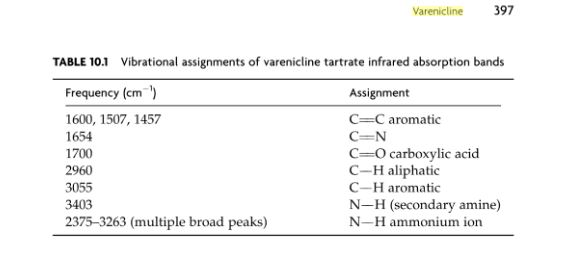
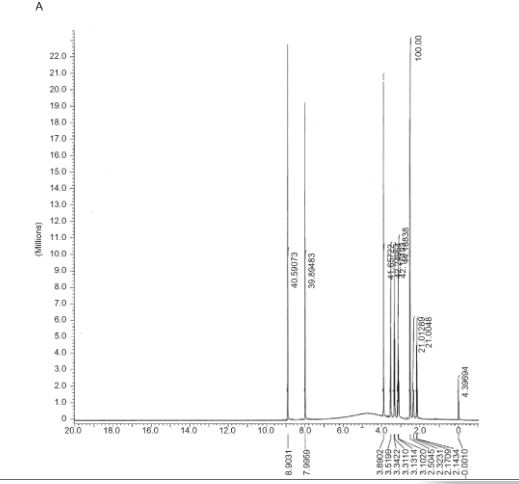

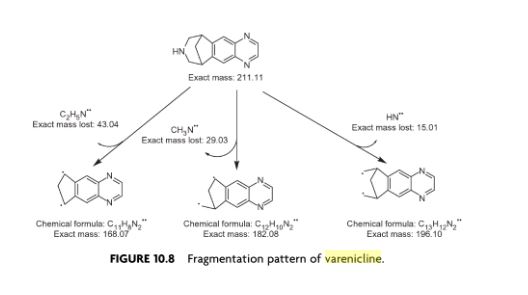
SYNTHESIS

|
DOI: 10.1021/jm00190a020
|
|
| DOI: 10.1021/jm050069n |
CLIP
Scheme (I) compound patent US6410550B1 is provided adjacent difluorobromobenzene as raw materials by DA reaction, oxidation, cyclization, debenzylation get varenicline intermediate (II). The synthesis route is as follows:

Patent CN101693712A mainly given varenicline intermediate (II) The preparation process is different from the compound patented. After the five-step method patents cited compounds. The entire route is longer, while using a large number of precious metal catalysts and reaction conditions need very strict control, inappropriate EVAL industry production.

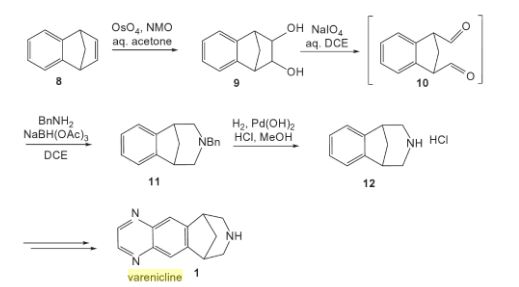
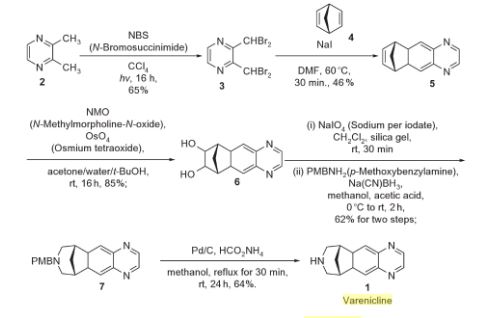
PATENT
A varenicline intermediate 2,3, 4, 5-tetrahydro-1,5-methylene bridge synthesis -1H-3- benzazepine hydrochloride, which comprises the following Step: (1) 2-indanone of formula 3 and the compound and paraformaldehyde under alkaline or acidic conditions Mannich reaction, as shown in general formula 2 intermediate; (2) the step (I) obtained through reaction of Formula 2 intermediate under basic or acidic conditions by reducing the role of the carbonyl group is reduced to a methylene group, and get varenicline intermediate (II) by debenzylation, the reaction is:

Wherein, R groups are selected from _H, _Me, _Et, _iPr> _t_Bu.
Figure 2;

Wherein, R group is -H, -Me, -Et, -iPr or -t_Bu.
(2) Step (I) obtained by the reaction intermediates of formula under basic or acidic conditions by reducing the role of the carbonyl group is reduced 2 methylene, and get by debenzylation cutting Lenk Lin intermediate (II);

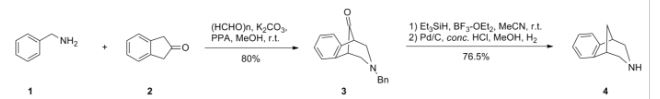
CLIP
Varenicline, a nicotinic 42 partial agonist, was approved in the US for the treatment of smoking cessation in May of 2006. It was developed and marketed by Pfizer as a treatment for cigarette smokers who want to quit. Varenicline partially activates the nicotinic receptors and thus reduces the craving for cigarette that smokers feel when they try to quit smoking. By mitigating this craving and antagonizing nicotine activity without other symptoms, this novel drug helps quitting this dangerous addiction easier on the patients [6,52]. Several modifications [54,55] to the original synthesis [53,56] have been reported in the literature, including an improved process scale synthesis of the last few steps (Scheme 15) [57]. The Grignard reaction was initiated on a small scale by addition of 2-bromo fluorobenzene 113 to a slurry of Magnesium turnings and catalytic 1,2-dibromoethane in THF and heating the mixture until refluxing in maintained. To this refluxing mixture was added a mixture of the 2-bromo fluorobenzene 113 and cyclopentadiene 114 over a period of 1.5 h. After complete addition, the reaction was allowed to reflux for additional 1.5 h to give the Diels- Alder product 115 in 64% yield. Dihydroxylation of the olefin 115 by reacting with catalytic osmium tetraoxide in the presence of N-methylmorpholine N-oxide (NMO) in acetone: water mixture at room temperature provided the diol 116 in 89% yield. Oxidative cleavage of diol 116 with sodium periodate in biphasic mixture of water: DCE at 10ºC provided di-aldehyde 117 which was immediately reacted with benzyl amine in the presence of sodium acetoxyborohydride to give benzyl amine 118 in 85.7% yield. The removal of the benzyl group was effected by hydrogenation of the HCl salt in 40-50 psi hydrogen pressure with 20% Pd(OH)2 in methanol to give amine hydrochloride 119 in 88% yield. Treatment of amine 119 with trifluoroacetic anhydride and pyridine in dichloromethane at 0ºC gave trifluoroacetamide 120 in 94% yield. Dinitro compound 121 was prepared by addition of trifluoroacetamide 120 to a mixture of trifluoromethane sulfonic acid and nitric acid, which was premixed, in dichloromethane at 0ºC. Reduction of the dinitro compound 121 by hydrogenation at 40-50 psi hydrogen in the presence of catalytic 5%Pd/C in isopropanol:water mixture provided the diamine intermediate 122 which was quickly reacted with glyoxal in water at room temperature for 18h to give compound 123 in 85% overall yield. The trifluoroacetamide 123 was then hydrolyzed with 2 M sodium hydroxide in toluene at 37-40ºC for 2-3h followed by preparation of tartrate salt in methanol to furnish varenicline tartrate (XV).
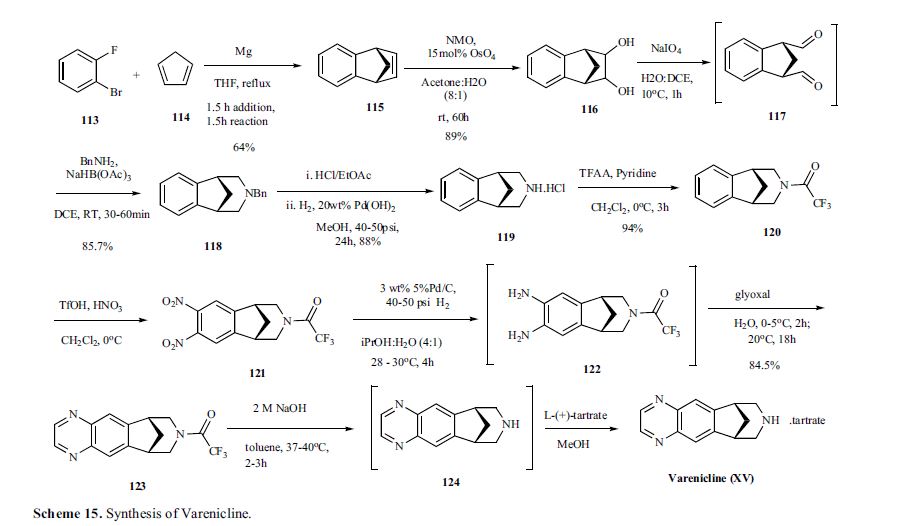
[52]Keating, G.; Siddiqui, M. A. A. CNSdrugs, 2006, 11, 946.
[53] Coe, J. W.; Brooks, P. R.; Vetelino, M. G.; Wirtz, M. C.; Arnold,E. P. ; Huang, J.; Sands, S. B.; Davis, T. I.; Lebel, L. A.; Fox, C.
B.; Shrikhande, A.; Heym, J. H.; Schaeffer, E.; Rollema, H.; Lu,Y.; Mansbach, R. S.; Chambers, L. K.; Rovetti, C. C.; Schulz, D.
W.; Tingley, III, F. D.; O’Neill, B. T. J. Med. Chem., 2005, 48,3474.
[54] Brooks, P. R.; Caron, S.; Coe, J. W.; Ng, K. K.; Singer, R. A.;Vazquez, E.; Vetelino, M. G.; Watson, Jr. H. H.; Whritenour, D.
C.; Wirtz, M. C. Synthesis, 2004, 11, 1755.
[55] Singer, R. A.; McKinley, J. D.; Barbe, G.; Farlow, R. A. Org. Lett.,2004, 6, 2357.
[56] Coe, J. W.; Brooks, P. R. P. US-6410550 B1, 2002.
[57] Busch, F. R.; Hawkins, J. M.; Mustakis, L. G.; Sinay, T. G., Jr.;Watson, T. J. N.; Withbroe, G. J. WO-2006090236 A1, 2006.
PATENT
WO 2002085843
https://google.com/patents/WO2002085843A2?cl=en
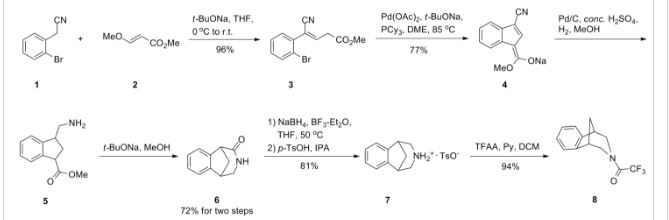
PATENT
https://www.google.com/patents/EP2204369A1?cl=en
Varenicline (a compound I of formula I) is the international commonly accepted non-proprietary name for 7,8,9,10-tetrahydro-6,10-methano-6H-pyrazino[2,3-h][3]benzazepine (which is also known as 5,8,14-triazatetracyclo[10.3.1.02,11.04,9]-hexadeca-2(11),3,5,7,9-pentaene), and has an empirical formula of C13H13N3 and a molecular weight of 211.26.

The L-tartrate salt of varenicline is known to be therapeutically useful and is commercially marketed for the treatment of smoking addiction. Varenicline L-tartrate is a partial agonist selective for α4β2 nicotinic acetylcholine receptor subtypes. In the United States, varenicline L-tartrate is marketed under the trade mark Chantix and is indicated as an aid to smoking cessation treatment.
Varenicline base and its pharmaceutically acceptable acid addition salts are described in U.S. Patent No. 6,410,550 . In particular, the preparation of varenicline provided in this reference makes use of 10-aza-tricyclo[6.3.1.02,7]-dodeca-2(7),3,5-triene (a compound of Formula VI), as a key intermediate compound (see Scheme 1 below). Specifically, Example 1 of U.S. Patent No. 6,410,550 describes the synthetic preparation of key intermediate compound of Formula VI as depicted in Scheme 1.

1,2,3,4-tetrahydro-1,4-methano-naphthalene-cis-2,3-diol (a compound of Formula III), and / or indane-1,3-dicarbaldehyde (a compound of Formula IV).
Example 1: Preparation of 1,2,3,4-tetrahydro-1,4-methano-naphthalene-cis-2,3-diol (a compound of Formula III)
A 10mL round bottom flask was charged with a compound of formula II (142mg, 1mmol), N-methylmorpholine-N-oxide (120mg, 1.03mmol), tert-butanol (3mL) and water (1mL). FibreCat™ 3003 (OsO4 anchored onto a polymeric support) (11.6mg, 0.0025mmol) was added to this solution and the mixture was heated to reflux. Complete conversion to a compound of formula III was detected by GC, method A, after 48h.
Example 2: Preparation of 1,2,3,4-tetrahydro-1,4-methano-naphthalene-cis-2,3-diol (a compound of Formula III)Step A) Preparation of hexadecyl-trimethylammoniumpermanganate (HTAP):
HTAP was prepared from ion exchange reaction between hexadecyltrimethylammoniumbromide and potassium permanganate.
Potassium permanganate (17.38g, 0.11mol, 1equiv.) was dissolved in 500mL water. A solution of hexadecyltrimethylammoniumbromide (40.10g, 0.11mol, 1equiv) in 500mL water was added drop-wise over 45 min at 20-22°C, and the mixture stirred for 30 minutes at this temperature. The precipitated solid was collected by filtration, washed with water (3 x 100mL) and dried under vacuum at 35°C for 24 hours to give 34.38g of HTAP as a light purple solid.
Step B) Preparation of a compound of formula III:
Compound II (3.52g, 24.8mmol, 1equiv.) was dissolved in anhydrous tetrahydrofuran (80mL) and a solution of HTAP (10g, 24.8mmol, 1.0equiv.) in anhydrous tetrahydrofuran (125mL) was added drop-wise at 23-30°C over 45min. The reaction was monitored by TLC (hexane-ethyl acetate = 1:1). After complete reaction the mixture was cooled to below 10°C, and methyl tert-butyl ether (50mL) and 5% aqueous NaOH solution (50mL) were added and the mixture stirred for 30min. The solid was removed by filtration, and washed with methyl tert-butyl ether (2 x 30mL). The combined layers of the filtrate were separated and the aqueous phase extracted with methyl tert-butyl ether (2 x 30mL). The organic layers were combined and washed with 5% aqueous NaOH solution (50mL), water (2 x 50mL), dried over MgSO4, filtered and concentrated to obtain a dark green solid. This residue was suspended in acetone (15mL) and collected by filtration, washing with additional acetone (3 x 5mL). The product was dried under vacuum at 40°C to give 2.215g (50.7% yield) as a white crystalline solid.
Analytical data: m.p. = 178.8-179.3°C; 1H-NMR: See Figure 1; 13C-NMR: See Figure 2.
Example 3: Preparation of indane-1,3-dicarbaldehyde (a compound of Formula IV)
A 25 mL round bottom flask was charged with a compound of formula I (142mg, 1mmol), Ruthenium (III) chloride hydrate (Aldrich, Reagent Plus™) (7.2mg, 0.035mmol), acetonitrile (8.5mL) and water (1.1mL). The solution was heated to 45°C and sodium periodate (449mg, 2.1mmol) was added portionwise over 25 minutes. After 1h, the reaction was cooled to ambient temperature and filtered. The solids were washed with ethyl acetate (3 x 2mL) and water (3mL). The filtrate was concentrated under vacuum and 5mL of water were added to the obtained residue. The mixture was extracted with ethyl acetate (2 x 5mL) and the combination of the organic layers was washed with water (3 x 5mL), dried with MgSO4 and concentrated under vacuum to obtain a compound of formula IV (118mg) in 68% yield, 70.9% purity (analyzed by GC, method A).
PATENT
WO 199935131, WO 2002092089, US 2013030179
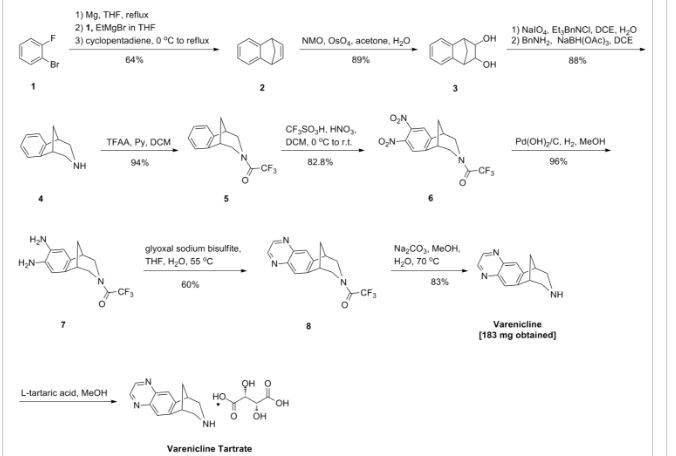
PATENT
https://www.google.com/patents/WO2009065872A2?cl=en
Example 1: Preparation of 7,8,9,10- tetrahydro-6, 10-methano-6H-pyrazino [2, 3-h] [3] benzazepine L-tartrate (i.e. varenicline L-tartrate)
A) Preparation of compound of formula (III)
This example is based on U.S. Patent No. 6,410,550.
A 250 mL round bottom flask with thermometer, condenser, addition funnel and magnetic stirring was charged with 10-aza-tricyclo [ 6.3.1. O2‘7] dodeca-2, 4, 6- triene para-toluene sulfonic acid salt (12.4g, 37.5 mmol) and 44 mL of CH2Cl2. Triethylamine (8.3 g, 82.5 mmol) was added to the slurry and the resulting solution was cooled to 0-5 0C. The addition funnel was charged with a solution of (CF3CO)2O (8.1q, 41.25 mmol) in 19 mL of CH2Cl2. This solution was slowly added to the reaction mixture, maintaining the temperature < 15 0C. The resulting mixture was stirred for 1 hour, and the complete conversion was monitored by GC. The crude reaction mixture was washed with water (2 * 40 mL) and brine (40 mL) . The organic phase was used in the next step without further purification.
On the other hand, a 500 mL round bottom flask with thermometer, condenser, addition funnel and magnetic stirring was charged with CF3SO3H (25.9 g, 172.5 mmol), CH2Cl2 (110 mL) and cooled to 0-5 0C. At this temperature, fuming nitric acid (5.4 g, 86.25 mmol) was added slowly. To the resulting slurry at 0-5 0C, the solution obtained in the previous step was slowly added, maintaining the temperature < 15 0C. After the addition, the reaction mixture was stirred overnight. The complete dinitration was confirmed by GC. The crude reaction mixture was poured into water (60 mL) an ice (80 g) and stirred. The phases were separated and the aqueous phase was extracted with CH2Cl2 (3 x 50 mL) . The mixture of the organic phases was washed with aqueous saturated NaHCO3, dried over Na2SO4 and volatiles evaporated under vacuum to obtain 11.9 g of a solid that was suspended and stirred for 2 hours in AcOEt (12 mL) and hexanes (24 mL) . The solid was filtered and washed with hexanes to obtain the compound of formula (III), 9.1g with a purity of 88.9% by GC (9.8% of meta-dimtrocompound impurity) .
B) Preparation of compound of formula (IV)
This example is based on International Patent No. WO/2006/090236.
A 200 mL autoclave was charged with (III) (9.1 g, 26.3 mmol), damp 5% Pd/C 50% and 180 mL of a 2- propanol/water (80/20 wt/wt) . The reaction was stirred under 50 psi of hydrogen for 18 hours. The complete hydrogenation was confirmed by GC analysis. The reaction was filtered through Celite and washed with 2-propanol (40 mL) . To this solution, K2HPO4(458 mg, 2.63 mmol) was added. The mixture was cooled at 0-5 0C and a solution of 4.07 g of 40% aqueous glyoxal diluted with water (14.5 mL) was added slowly. The resulting solution was stirred 2 hours at this temperature and overnight at room temperature. The complete conversion was confirmed by GC analysis. The reaction was concentrated under vacuum to a volume of 68 mL and water (128 mL) was added drop- wise. The resulting suspension was stirred for 2 hours at room temperature, 1 hour in a ice/water bath, filtered, washed with water (20 mL) and dried m a oven at 50 0C to obtain the compound of formula (IV), 6.78 g.
C) Preparation of vareniclme L-tartrate (compound of formula (I) )
This example is based on International Patent No. WO/2006/090236.
A 250 mL round bottom flask with thermometer, condenser, and magnetic stirring was charged with compound of formula (IV) (6.78 g, 22 mmol) and toluene
(47 mL) . To this solution was added a solution of NaOH (2.7 g, 68.2 mmol) in water (34 mL) . The mixture was heated to 400C and stirred for 4 hours. The complete hydrolysis was confirmed by GC analysis. Toluene (68 mL) was added and the reaction was cooled. The phases were separated and the aqueous phase was extracted with toluene (30 mL) . The organic phases were evaporated under vacuum. The residue was dissolved in MeOH (90 mL) and evaporated again. The final residue was dissolved in 156 mL of MeOH. 1.3 g of activated carbon “Darco G-60 100 mesh” were added and the mixture was stirred for 30 min and filtered through Celite to obtain an intense yellow solution. The process with activated carbon was repeated without any improvement in the colour. This solution was added drop-wise over a solution of L- tartaric acid (3.63 g, 24.2 mmol) in MeOH (47 mL) . The slurry was stirred for 72 hours at room temperature, filtered, washed with MeOH and dried in an oven at 50 0C for 8 hours, to obtain 5.05 g of varenicline L-tartrate as a yellow solid with a 95.5% purity by HPLC (4.4% of unknown impurity A). Colour L: 92.75, a*: -7.19, b*:43.08.
Comparative Example 2: Preparation of 7,8,9,10- tetrahydro-6, 10-methano-6H-pyrazmo [2, 3-h] [3 ] benzazepine L-tartrate (i.e. varenicline L-tartrate) A) Preparation of compound of formula (IV)
This example is based on International Patent No. WO/2006/090236.
A 200 mL autoclave was charged with (III) prepared according to Comparative Example 1.A) (4.1 g) , 123 mg of damp 5% Pd/C 50% and 81 mL of a 2-propanol/water (80/20 wt/wt) . The reaction was stirred under 50 psi of hydrogen for 24 hours. The complete hydrogenation was confirmed by GC analysis. The reaction was filtered through Celite and washed with 2-propanol (16 mL) . To this solution, K2HPO4 (207 mg, 1.19 mmol) was added. The mixture was cooled at 0-5 0C and a solution of 1.84 g of 40% aqueous glyoxal diluted with water (6.6 mL) was added slowly. The resulting solution was stirred 2 hours at this temperature and overnight at room temperature. The complete conversion was confirmed by GC analysis. The reaction was concentrated under vacuum to a volume of 30 mL and water (56 mL) was added drop-wise. The resulting suspension was stirred for 2 hours at room temperature, 1 hour in a ice/water bath, filtered, washed with water and dried in a oven at 50 0C to obtain 3.15 g of compound of formula (IV) .
B) Preparation of vareniclme L-tartrate (compound of formula (I) )
This example is based on International application No. WO/2006/090236. A 100 mL round bottom flask with thermometer, condenser, and magnetic stirring was charged with
7, 8, 9, 10-tetrahydro-8- (tπfluoroacetyl) -6, 10-methano-6H- pyrazino [2 , 3-h] [3] benzazepine, i.e. compound of formula
(IV) (3.14 g, 10.2 mmol) and toluene (22 mL) . To this solution was added a solution of NaOH (1.3 g, 31.6 mmol) in water (16 mL) . The mixture was heated to 40 0C and stirred for 2.5 hours. The complete hydrolysis was confirmed by GC analysis. Toluene (30 mL) was added and the reaction was cooled. The phases were separated and the aqueous phase was extracted with toluene (15 mL) . The organic phases were evaporated under vacuum. The residue was dissolved in MeOH (45 mL) and evaporated again. The final residue was dissolved m 70 mL of MeOH. 314 mg of activated carbon “Darco G-60 100 mesh” were added and the mixture was stirred for 30 mm and filtered through Celite to obtain a yellow solution. This solution was added drop-wise over a solution of L- tartaπc acid (1.68 g, 11.22 mmol) m MeOH (22 mL) . The slurry was stirred for 1 hour at room temperature, filtered, washed with MeOH (2 x 5 mL) and dried under vacuum, to obtain vareniclme L-tartrate (2.48 g) as a yellow solid with a 95.6% purity by HPLC (4.4% of unknown impurity A). Colour L: 99.50, a*: -4.98, b*:43.02
Comparative Example 3: Preparation of 7,8,9,10- tetrahydro-6, 10-methano-6H-pyrazino [2, 3-h] [3 ] benzazepine L-tartrate (i.e. vareniclme L-tartrate)
This example is based on International application No. WO/2002/092089.
2 g of vareniclme L-tartrate as obtained from Comparative Example 1 were dissolved in 3 mL of water.
To this solution, 100 mL of CH3CN were added, and the resulting slurry was stirred for 10 mm and filtered.
After drying the product was analysed to be a 98.2% purity by HPLC (1.7% of unknown impurity A) . Colour L: 91.44, a*: -3.24, b* : 33.47
Example 1: Preparation of 7, 8, 9, lO-tetrahydro-6, 10- methano-6H-pyrazmo [2, 3-h] [3] benzazepine L-tartrate
(i.e. vareniclme L-tartrate)
A) Preparation of compound of formula (III) This example is based on U.S. Patent No. 6,410,550, except for the purification step, which is the object of the present invention (i.e. crystallization in toluene) .
A 500 mL round bottom flask with thermometer, condenser, addition funnel and magnetic stirring was charged with 10-aza-tricyclo [ 6.3.1. O2‘7] dodeca-2, 4, 6- tπene para-toluene sulfonic acid salt (32.5g, 98.2 mmol) and 115 mL of CH2Cl2. Triethylamine (21.8 g, 216 mmol) was added to the slurry and the resulting solution was cooled to 0-5 0C. The addition funnel was charged with a solution of (CF3CO)2O (22.7 g, 108 mmol) in 50 mL of CH2Cl2. This solution was slowly added to the reaction mixture, maintaining the temperature < 15 0C. The resulting mixture was stirred for 1 hour, and the complete conversion was monitored by GC. The crude reaction mixture was washed with water (2 x 100 mL) and brine (100 mL) . The organic phase was used in the next step without further purification.
A l L round bottom flask with thermometer, condenser, addition funnel and magnetic stirring was charged with CF3SO3H (67.8 g, 452 mmol), CH2Cl2 (280 mL) and cooled to 0-5 0C. At this temperature, fuming nitric acid (14.2 g, 226 mmol) was slowly added. To the resulting slurry at 0-5 0C, the solution obtained in the previous step was slowly added, maintaining the temperature < 15 0C. After the addition, the reaction mixture was stirred overnight. The complete dinitration was confirmed by GC. The crude reaction mixture was poured into water (150 mL) an ice (200 g) and stirred. The phases were separated and the aqueous phase was extracted with CH2Cl2 (100 mL) . The mixture of the organic phases was washed with aqueous saturated NaHCO3 (2×100 mL) , water (100 mL) , dried over Na2SO4 and volatiles evaporated under vacuum to obtain 30.5 g of a solid with a 83.6% purity by GC (12.5% of meta- dinitrocompound impurity) . 20 g of this solid were crystallized in toluene (100 mL) to obtain the compound of formula (III), 15 g of a pale brown solid with a 98.5 % purity by GC (meta-dinitrocompound impurity not detected) .
B) Preparation of compound of formula (IV) This example is based on International Patent No. WO/2006/090236.
A 200 mL autoclave was charged with (III) (9.1 g, 26.3 mmol, crystals from toluene), damp 5% Pd/C 50% and 180 mL of a 2-propanol/water (80/20 wt/wt) . The reaction was stirred under 50 psi of hydrogen for 18 hours. The complete hydrogenation was confirmed by GC analysis. The reaction was filtered over Celite and washed with 2- propanol (40 mL) . To this solution, K2HPO4 (458 mg, 2.63 mmol) was added. The mixture was cooled at 0-5 0C and a solution of 4.07 g of 40% aqueous glyoxal diluted with water (14.5 mL) was added slowly. The resulting solution was stirred 2 hours at this temperature and overnight at room temperature. The complete conversion was confirmed by GC analysis. The reaction was concentrated under vacuum to a volume of 68 mL and water (128 mL) was added drop-wise. The resulting suspension was stirred for 2 hours at room temperature, 1 hour in a ice/water bath, filtered, washed with water (20 mL) and dried m a oven at 50 0C to obtain the product, 7.16 g of compound of formula (IV) with a 99.9% purity by HPLC. C) Preparation of varenicline L-tartrate (compound of formula ( I) )
Thrs example rs based on International Patent No. WO/2006/090236. A 250 mL round bottom flask with thermometer, condenser, and magnetic stirring was charged with a solution of NaOH (2.89 g, 72.23 mmol) in water (36 mL) , compound of formula (IV) (7.15 g, 23.3 mmol) and toluene (50 mL) . The mixture was heated to 40 0C and stirred for 4 hours. The complete hydrolysis was confirmed by GC analysis. Toluene (71 mL) was added and the reaction was cooled. The phases were separated and the aqueous phase was extracted with toluene (36 mL) . The organic phases were evaporated under vacuum. The residue was dissolved in MeOH (110 mL) and evaporated again. The final residue was dissolved in 164 mL of MeOH. 750 mg of activated carbon “Darco G-60 100 mesh” were added and the mixture was stirred for 30 min and filtered through Celite to obtain a yellow solution. This solution was added drop- wise over a solution of L-tartaric acid (3.84 g, 25.6 mmol) in MeOH (50 mL) . The slurry was stirred for 14 hours at room temperature, filtered, washed with MeOH and dried under vacuum, to obtain varenicline L-tartrate
(7.04 g) as an off-white solid with a >99.9% purity by HPLC (unknown impurity A not detected) . Colour L: 94.39, a*: 2.27, b*:9.02.

Post-marketing surveillance
No evidence for increased risks of cardiovascular events, depression, or self-harm with varenicline versus nicotine replacement therapy has been found in one post-marketing surveillance study.[23]
Mechanism of action
Varenicline displays full agonism on α7 nicotinic acetylcholine receptors.[24][25] And it is a partial agonist on the α4β2, α3β4, and α6β2 subtypes.[26] In addition, it is a weak agonist on the α3β2 containing receptors.
Varenicline’s partial agonism on the α4β2 receptors rather than nicotine’s full agonism produces less effect of dopamine release than nicotine’s. This α4β2 competitive binding, reduces the ability of nicotine to bind and stimulate the mesolimbic dopamine system – similar to the method of action of buprenorphine in the treatment of opioid addiction.[3]
Pharmacokinetics
Most of the active compound is excreted by the kidneys (92–93%). A small proportion is glucuronidated, oxidised, N-formylated or conjugated to a hexose.[27] The elimination half-life is about 24 hours.
History
Use of Cytisus plant as a smoking substitute during World War II[28] led to use as a cessation aid in eastern Europe and extraction of cytisine.[29] Cytisine analogs led to varenicline at Pfizer.[30][31][32]
Varenicline received a “priority review” by the US FDA in February 2006, shortening the usual 10-month review period to 6 months because of its demonstrated effectiveness inclinical trials and perceived lack of safety issues.[33] The agency’s approval of the drug came on May 11, 2006.[4] On August 1, 2006, varenicline was made available for sale in the United States and on September 29, 2006, was approved for sale in the European Union.[34]
SEE

| US6410550 | Nov 13, 1998 | Jun 25, 2002 | Pfizer Inc | Aryl fused azapolycyclic compounds |
| WO2009155403A2 * | Jun 18, 2009 | Dec 23, 2009 | Teva Pharmaceutical Industries Ltd. | Processes for the preparation of varenicline and intermediates thereof |
| Reference | ||
|---|---|---|
| 1 | * | BHUSHAN, VIDYA; RATHORE, RAJENDRA; CHANDRASEKARAN, S.: “A Simple and Mild Method for the cis-Hydroxylation of Alkenes with Cetyltrimethylammonium Permanganate” SYNTHESIS, no. 5, 1984, pages 431-433, XP002581198 |
| 2 | * | BROOKS P R ET AL: “Synthesis of 2,3,4,5-tetrahydro-1,5-methano-1H-3-benzaz epine via oxidative cleavage and reductive amination strategies” SYNTHESIS 20040803 DE, no. 11, 3 August 2004 (2004-08-03), pages 1755-1758, XP002581197 ISSN: 0039-7881 |
| 3 | * | SORBERA L A ET AL: “Varenicline tartrate: Aid to smoking cessation nicotinic [alpha]4[beta]2 partial agonist” DRUGS OF THE FUTURE 200602 ES LNKD- DOI:10.1358/DOF.2006.031.02.964028, vol. 31, no. 2, February 2006 (2006-02), pages 117-122, XP002581199 ISSN: 0377-8282 DOI: 10.1358/dof.2006.031.02.964028 |
| WO2001062736A1 * | Feb 8, 2001 | Aug 30, 2001 | Pfizer Products Inc. | Aryl fused azapolycyclic compounds |
| WO2002085843A2 * | Mar 4, 2002 | Oct 31, 2002 | Pfizer Products Inc. | Process for the preparation of 1,3-substituted indenes and aryl-fused azapolycyclic compounds |
| WO2006090236A1 * | Feb 21, 2006 | Aug 31, 2006 | Pfizer Products Inc. | Preparation of high purity substituted quinoxaline |
| WO2008060487A2 * | Nov 9, 2007 | May 22, 2008 | Pfizer Products Inc. | Polymorphs of nicotinic intermediates |
| Reference | ||
|---|---|---|
| 1 | * | COE J W ET AL: “Varenicline: an alpha4beta2 Nicotinic Receptor Partial Agonist for Smoking Cessation” JOURNAL OF MEDICINAL CHEMISTRY, AMERICAN CHEMICAL SOCIETY, WASHINGTON., US, vol. 48, no. 10, 1 January 2005 (2005-01-01), pages 3474-3477, XP002474642 ISSN: 0022-2623 cited in the application |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2010005643A1 * | May 28, 2009 | Jan 14, 2010 | Teva Pharmaceutical Industries Ltd. | Processes for purifying varenicline l-tartrate salt and preparing crystalline forms of varenicline l-tartrate salt |
| WO2011110954A1 * | Mar 8, 2011 | Sep 15, 2011 | Actavis Group Ptc Ehf | Highly pure varenicline or a pharmaceutically acceptable salt thereof substantially free of methylvarenicline impurity |
| WO2011154586A3 * | Jun 13, 2011 | Mar 22, 2012 | Medichem, S. A. | Improved methods for the preparation of quinoxaline derivatives |
| EP2581375A2 * | Jun 13, 2011 | Apr 17, 2013 | Medichem, S.A. | Improved methods for the preparation of quinoxaline derivatives |
| US8039620 | May 21, 2009 | Oct 18, 2011 | Teva Pharmaceutical Industries Ltd. | Varenicline tosylate, an intermediate in the preparation process of varenicline L-tartrate |
| US8178537 | Jun 22, 2010 | May 15, 2012 | Teva Pharmaceutical Industries Ltd. | Solid state forms of varenicline salts and processes for preparation thereof |
References
- Jump up^ Mills EJ, Wu P, Spurden D, Ebbert JO, Wilson K (2009). “Efficacy of pharmacotherapies for short-term smoking abstinance: a systematic review and meta-analysis” (PDF). Harm Reduct J 6: 25. doi:10.1186/1477-7517-6-25. PMC 2760513. PMID 19761618.
- ^ Jump up to:a b Cahill K, Stevens S, Perera R, Lancaster T (May 2013). “Pharmacological interventions for smoking cessation: an overview and network meta-analysis”. Cochrane Database Syst Rev (Systematic Review & Meta-Analysis) 5: CD009329.doi:10.1002/14651858.CD009329.pub2. PMID 23728690.
- ^ Jump up to:a b c d Elrashidi MY, Ebbert JO (June 2014). “Emerging drugs for the treatment of tobacco dependence: 2014 update”. Expert Opin Emerg Drug (Review) 19 (2): 243–60.doi:10.1517/14728214.2014.899580. PMID 24654737.
- ^ Jump up to:a b U.S. Food and Drug Administration.FDA Approves Novel Medication for Smoking Cessation. Press release, 11 May 2006.
- Jump up^ Cressman, AM; Pupco, A; Kim, E; Koren, G; Bozzo, P (May 2012). “Smoking cessation therapy during pregnancy.”. Canadian Family Physician 58 (5): 525–7. PMC 3352787.PMID 22586193.
- Jump up^ “Varenicline Pregnancy Cohort Study”. clinicaltrials.gov.
- Jump up^ “LactMed”. nih.gov.
- Jump up^ Leung, LK; Patafio, FM; Rosser, WW (September 28, 2011). “Gastrointestinal adverse effects of varenicline at maintenance dose: a meta-analysis”. BMC clinical pharmacology11 (1): 15. doi:10.1186/1472-6904-11-15. PMC 3192741. PMID 21955317.
- American Cancer Society. “Cancer Drug Guide: Varenicline”. Retrieved 2008-01-19.
- Jump up^ “DailyMed – CHANTIX- varenicline tartrate”. nih.gov.
- FDA. “Public Health Advisory: FDA Requires New Boxed Warnings for the Smoking Cessation Drugs Chantix and Zyban”. Retrieved 2009-07-01.
- ^ Jump up to:a b “www.accessdata.fda.gov” (PDF).
- Hughes, JR (8 January 2015). “Varenicline as a Cause of Suicidal Outcomes.”. Nicotine & tobacco research : official journal of the Society for Research on Nicotine and Tobacco.doi:10.1093/ntr/ntu275. PMID 25572451.
- “FDA Drug Safety Communication: Chantix (varenicline) may increase the risk of certain cardiovascular adverse events in patients with cardiovascular disease”. 2011-06-16.
- Jump up^ Singh, S; Loke, YK, Spangler, JG, Furberg, CD (Sep 6, 2011). “Risk of serious adverse cardiovascular events associated with varenicline: a systematic review and meta-analysis” (PDF). CMAJ : Canadian Medical Association 183 (12): 1359–66.doi:10.1503/cmaj.110218. PMC 3168618. PMID 21727225.
- Takagi, H; Umemoto, T (Sep 6, 2011). “Varenicline: quantifying the risk”. CMAJ : Canadian Medical Association 183 (12): 1404. doi:10.1503/cmaj.111-2063.PMC 3168634. PMID 21896705.
- Jump up^ Samuels, L (Sep 6, 2011). “Varenicline: cardiovascular safety”. CMAJ : Canadian Medical Association 183 (12): 1407–08. doi:10.1503/cmaj.111-2073. PMC 3168639.PMID 21896709.
- “European Medicine Agency confirms positive benefit-risk balance for Champix.”. 2011-07-21.
- ^ Jump up to:a b Prochaska JJ, Hilton JF (2012). “Risk of cardiovascular serious adverse events associated with varenicline use for tobacco cessation: systematic review and meta-analysis”. BMJ (Systematic Review & Meta-Analysis) 344: e2856.doi:10.1136/bmj.e2856. PMC 3344735. PMID 22563098.
- Mills EJ, Thorlund K, Eapen S, Wu P, Prochaska JJ (January 2014). “Cardiovascular events associated with smoking cessation pharmacotherapies: a network meta-analysis”.Circulation (Network Meta-Analysis) 129 (1): 28–41.doi:10.1161/CIRCULATIONAHA.113.003961. PMID 24323793.
- cessation in cardiovascular patients”. Evidence-Based Medicine (Review & Commentary) 19 (5): 193. doi:10.1136/eb-2014-110030.PMID 24917603.
- Rowland K (April 2014). “ACP Journal Club. Review: Nicotine replacement therapy increases CVD events; bupropion and varenicline do not”. Annals of Internal Medicine(Review & Commentary) 160 (8): JC2. doi:10.7326/0003-4819-160-8-201404150-02002.PMID 24733219.
- Jump up^ Kotz D, Viechtbauer W, Simpson C, van Schayck OC, West R, Sheikh A (2015).“Cardiovascular and neuropsychiatric risks of varenicline: a retrospective cohort study”.Lancet Respir Med (retrospective cohort) 3: 761–768. doi:10.1016/S2213-2600(15)00320-3. PMC 4593936. PMID 26355008.
- Jump up^ Mihalak KB, Carroll FI, Luetje CW; Carroll; Luetje (2006). “Varenicline is a partial agonist at alpha4beta2 and a full agonist at alpha7 neuronal nicotinic receptors”. Mol. Pharmacol.70 (3): 801–805. doi:10.1124/mol.106.025130. PMID 16766716.
- Jump up^ Mineur YS, Picciotto MR; Picciotto (December 2010). “Nicotine receptors and depression: revisiting and revising the cholinergic hypothesis”. Trends Pharmacol. Sci. 31 (12): 580–6. doi:10.1016/j.tips.2010.09.004. PMC 2991594. PMID 20965579.
- Tanuja Bordia. “Varenicline Is a Potent Partial Agonist at α6β2* Nicotinic Acetylcholine Receptors in Rat and Monkey Striatum”. aspetjournals.org.
- Obach, RS; Reed-Hagen, AE; Krueger, SS; Obach, BJ; O’Connell, TN; Zandi, KS; Miller, S; Coe, JW (2006). “Metabolism and disposition of varenicline, a selective alpha4beta2 acetylcholine receptor partial agonist, in vivo and in vitro”. Drug metabolism and disposition: the biological fate of chemicals 34 (1): 121–130.doi:10.1124/dmd.105.006767. PMID 16221753.
- “[Cytisine as an aid for smoking cessation].”. Med Monatsschr Pharm 15 (1): 20–1. Jan 1992. PMID 1542278.
- Prochaska, BMJ 347:f5198 2013 http://www.bmj.com/content/347/bmj.f5198
- Coe JW, Brooks PR, Vetelino MG, Wirtz MC, Arnold EP, Huang J, Sands SB, Davis TI, Lebel LA, Fox CB, Shrikhande A, Heym JH, Schaeffer E, Rollema H, Lu Y, Mansbach RS, Chambers LK, Rovetti CC, Schulz DW, Tingley FD 3rd, O’Neill BT (2005). “Varenicline: an alpha4beta2 nicotinic receptor partial agonist for smoking cessation”. J. Med. Chem. 48(10): 3474–3477. doi:10.1021/jm050069n. PMID 15887955.
- Schwartz JL (1979). “Review and evaluation of methods of smoking cessation, 1969–77. Summary of a monograph”. Public Health Rep 94 (6): 558–63. PMC 1431736.PMID 515342.
- Etter JF (2006). “Cytisine for smoking cessation: a literature review and a meta-analysis”. Arch. Intern. Med. 166 (15): 1553–1559. doi:10.1001/archinte.166.15.1553.PMID 16908787.
- Kuehn BM (2006). “FDA speeds smoking cessation drug review”. JAMA 295 (6): 614–614.doi:10.1001/jama.295.6.614. PMID 16467225.
- European Medicines Agency (2011-01-28). “EPAR summary for the public. Champix varenicline”. London. Retrieved 2011-02-14.
External links
Manufacturer’s website USA

|
|
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
7,8,9,10-Tetrahydro-6,10-methano-6H-pyrazino[2,3-h] [3]benzazepine
|
|
| Clinical data | |
| Trade names | Chantix |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a606024 |
| License data |
|
| Pregnancy category |
|
| Routes of administration |
Oral |
| Legal status | |
| Legal status | |
| Pharmacokinetic data | |
| Protein binding | <20% |
| Metabolism | Limited (<10%) |
| Biological half-life | 24 hours |
| Excretion | Renal (81–92%) |
| Identifiers | |
| CAS Number | 249296-44-4 |
| ATC code | N07BA03 (WHO) |
| PubChem | CID 5310966 |
| IUPHAR/BPS | 5459 |
| DrugBank | DB01273 |
| ChemSpider | 4470510 |
| UNII | W6HS99O8ZO |
| KEGG | D08669 |
| ChEBI | CHEBI:84500 |
| ChEMBL | CHEMBL1076903 |
| Chemical data | |
| Formula | C13H13N3 |
| Molar mass | 211.267 g/mol |
////////////Varenicline, Chantix™, FDA 2006, 249296-44-4, 375815-87-5, Champix , Pfizer, バレニクリン酒石酸塩
n1c2cc3c(cc2ncc1)[C@@H]4CNC[C@H]3C4
PF-06282999

PF 6282999
Alternative Names: PF-06282999; PF-6282999, PF-06282999
Cas 1435467-37-0
[2-(6-(5-chloro-2-methoxyphenyl)-4-oxo-2-thioxo-3,4-dihydropyrimidin-1(2H)-yl)acetamide]
2-(6-(5-chloro-2-methoxyphenyl)-4-oxo-2-thioxo-3,4-dihydropyrimidin-1(2H)-yl)acetamide
MF C13H12ClN3O3S
Molecular Weight: 325.767
Elemental Analysis: C, 47.93; H, 3.71; Cl, 10.88; N, 12.90; O, 14.73; S, 9.84
Irreversible inactivator of myeloperoxidase
Currently in clinical trials for the potential treatment of cardiovascular diseases.
Phase I
- Phase I Acute coronary syndromes
Most Recent Events
- 01 Mar 2015 Pfizer terminates phase I trial in Healthy volunteers in USA (NCT01965600)
- 10 Sep 2014 Pfizer completes enrolment in its phase I trial in Healthy volunteers in USA (NCT01965600)
- 01 Feb 2014 Phase-I clinical trials in volunteers in USA (PO)
A drug potentially for the treatment of acute coronary syndrome (ACS).

![]()
![]()
PF-06282999 is a potent and selective myeloperoxidase Inhibitor which is potential useful for the Treatment of Cardiovascular Diseases. PF-06282999 displayed excellent oral pharmacokinetics in preclinical species and robust irreversible inhibition of plasma MPO activity both in human blood stimulated exogenously and in plasma collected after oral (po) administration to lipopolysaccharide (LPS)-treated cynomolgus monkeys.
PF-06282999 has been advanced into first-in-human pharmacokinetics and safety studies. Myeloperoxidase (MPO) is a heme peroxidase that catalyzes the production of hypochlorous acid. Clinical evidence suggests a causal role for MPO in various autoimmune and inflammatory disorders including vasculitis and cardiovascular and Parkinson’s diseases, implying that MPO inhibitors may represent a therapeutic treatment option
The thiouracil derivative PF-06282999 [2-(6-(5-chloro-2-methoxyphenyl)-4-oxo-2-thioxo-3,4-dihydropyrimidin-1(2H)-yl)acetamide] is an irreversible inactivator of myeloperoxidase and is currently in clinical trials for the potential treatment of cardiovascular diseases. Concerns over idiosyncratic toxicity arising from bioactivation of the thiouracil motif to reactive species in the liver have been largely mitigated through the physicochemical (molecular weight, lipophilicity, and topological polar surface area) characteristics of PF-06282999, which generally favor elimination via nonmetabolic routes.

To test this hypothesis, pharmacokinetics and disposition studies were initiated with PF-06282999 using animals and in vitro assays, with the ultimate goal of predicting human pharmacokinetics and elimination mechanisms. Consistent with its physicochemical properties, PF-06282999 was resistant to metabolic turnover from liver microsomes and hepatocytes from animals and humans and was devoid of cytochrome P450 inhibition. In vitro transport studies suggested moderate intestinal permeability and minimal transporter-mediated hepatobiliary disposition. PF-06282999 demonstrated moderate plasma protein binding across all of the species.
Pharmacokinetics in preclinical species characterized by low to moderate plasma clearances, good oral bioavailability at 3- to 5-mg/kg doses, and renal clearance as the projected major clearance mechanism in humans. Human pharmacokinetic predictions using single-species scaling of dog and/or monkey pharmacokinetics were consistent with the parameters observed in the first-in-human study, conducted in healthy volunteers at a dose range of 20-200 mg PF-06282999.
In summary, disposition characteristics of PF-06282999 were relatively similar across preclinical species and humans, with renal excretion of the unchanged parent emerging as the principal clearance mechanism in humans, which was anticipated based on its physicochemical properties and supported by preclinical studies.
PAPER
Journal of Medicinal Chemistry (2015), 58(21), 8513-8528.
Discovery of 2-(6-(5-Chloro-2-methoxyphenyl)-4-oxo-2-thioxo-3,4-dihydropyrimidin-1(2H)-yl)acetamide (PF-06282999): A Highly Selective Mechanism-Based Myeloperoxidase Inhibitor for the Treatment of Cardiovascular Diseases

Myeloperoxidase (MPO) is a heme peroxidase that catalyzes the production of hypochlorous acid. Clinical evidence suggests a causal role for MPO in various autoimmune and inflammatory disorders including vasculitis and cardiovascular and Parkinson’s diseases, implying that MPO inhibitors may represent a therapeutic treatment option. Herein, we present the design, synthesis, and preclinical evaluation of N1-substituted-6-arylthiouracils as potent and selective inhibitors of MPO. Inhibition proceeded in a time-dependent manner by a covalent, irreversible mechanism, which was dependent upon MPO catalysis, consistent with mechanism-based inactivation. N1-Substituted-6-arylthiouracils exhibited low partition ratios and high selectivity for MPO over thyroid peroxidase and cytochrome P450 isoforms. N1-Substituted-6-arylthiouracils also demonstrated inhibition of MPO activity in lipopolysaccharide-stimulated human whole blood. Robust inhibition of plasma MPO activity was demonstrated with the lead compound 2-(6-(5-chloro-2-methoxyphenyl)-4-oxo-2-thioxo-3,4-dihydropyrimidin-1(2H)-yl)acetamide (PF-06282999, 8) upon oral administration to lipopolysaccharide-treated cynomolgus monkeys. On the basis of its pharmacological and pharmacokinetic profile, PF-06282999 has been advanced to first-in-human pharmacokinetic and safety studies.
tan solid (mp = 165.3 °C).
1H NMR (500 MHz, DMSO-d6) δ 12.85 (s, 1 H), 7.57 (dd, J = 9.03, 2.68 Hz, 1 H), 7.33 (s, 1 H), 7.17–7.23 (m, 2 H), 7.10 (s, 1 H), 5.89 (d, J = 1.71 Hz, 1 H), 5.41 (br s, 1 H), 3.89 (br s, 1 H), 3.84 (s, 3 H).
MS (ES+) m/z: 326.0 [M + H]+. HRMS: m/z calcd for C13H13ClN3O3S [M + H]+ 326.0366, found 326.0361.
Anal. Calcd for C13H12ClN3O3S: C, 47.93; H, 3.71; N, 12.90; S, 9.84. Found: C, 47.81; H, 3.70; N, 12.83; S, 9.83. HPLC purity: >95%.
PATENT
WO 2013068875
http://www.google.co.in/patents/WO2013068875A1?cl=en
Beta Keto Ester Route Section
A. Carboxylic Acid Route Section
Preparation 1

Ethyl 3-(5-chloro-2-methoxyphenyl)-3-oxopropanoate
A 3000 mL 3-necked round-bottomed flask flushed with nitrogen was charged with magnesium ethoxide (67.46 g, 589.51 mmoles) and THF (1 100 mL), and the resulting mixture was stirred as ethyl hydrogen malonate (162.26 g, 1 .18 moles; 145.00 mL diluted in 100 ml of THF) was added and the mixture was heated at 45 °C for 4 hours. Meanwhile, a 2000 mL 3-necked round-bottomed flask flushed with nitrogen was charged with 5-chloro-2-methoxybenzoic acid (100 g, 536 mmoles) and THF (600 mL). To this mixture stirring at room temperature was added 1 , 1 ‘-carbonyldiimidazole (95.59 g, 589.5 mmoles) in portions to avoid excess foaming. After stirring for 3 hours at room temperature the second solution was added gradually to the first solution. After addition the reaction mixture was heated to 45 °C. After 20 hours, the reaction mixture was concentrated under reduced pressure before adding ethyl acetate (1 L) followed by 2 N HCI (500 mL). After mixing, the layers were separated and the organic phase was washed sequentially with 2 N HCI (500 mL), saturated sodium bicarbonate (500 mL), and water (500 mL). The organic phase was concentrated under reduced pressure, the residue taken up in ethyl acetate (1000 mL) and concentrated again to afford the title compound (104.94 g).
MS (ES+) 257.2 [M+1 ]+. 1 H NMR showed product as a 7.5:1 keto:enol mixture. For the keto tautomer: 1 H NMR (500 MHz, CDCI3) δ ppm 7.85 (d, J=2.93 Hz, 1 H) 7.45 (dd, J=8.90, 2.81 Hz, 1 H) 6.92 (d, J=8.78 Hz, 1 H) 4.18 (q, J=7.16 Hz, 2 H) 3.95 (s, 2 H) 3.90 (s, 3 H) 1 .24 (t, J=7.07 Hz, 3 H). Preparation 2

(Z)-Ethyl 3-((2-amino-2-oxoethyl)amino)-3-(5-chloro-2-methoxyphenyl)acrylate A 5-L reaction vessel was charged with methanol (3.3 L), sodium methoxide (102.4 g, 1.8 moles), and glycinamide hydrochloride (202 g, 1.8 moles). The mixture was heated at 65 °C for 1 hour before cooling to 50 °C and adding acetic acid (514.25 mmoles, 30.88 g, 29.47 ml.) and ethyl 3-(5-chloro-2-methoxyphenyl)-3-oxopropanoate (300 g, 1.03 mole). After heating to reflux for 16 hours, the reaction mixture was stirred as it was cooled to 10 °C. After 30 min the resulting solid was collected by vacuum filtration, pulling dry to form a cake that was dried in a vacuum oven (20 mm Hg, 65 °C) for 14 hours to afford the title compound (339.4 g).
MS (ES+) 313.2 [M+1]+. 1H NMR (500 MHz, DMSO-d6) δ ppm 8.80 (t, J=5.00 Hz, 1 H) 7.47 (dd, J=8.90, 2.81 Hz, 1 H) 7.27 (br. s., 1 H) 7.22 (d, J=2.68 Hz, 1 H) 7.14 (d, J=8.78 Hz, 1 H) 7.09 (br. s., 1 H) 4.30 (s, 1 H) 4.03 (q, J=7.07 Hz, 2 H) 3.80 (s, 3 H) 3.56 (br. s., 1 H) 3.45 (br. s., 1 H) 1.18 (t, J=7.07 Hz, 3 H).
Example 1

2-( 6-(5-Chloro-2-methoxyphenyl)-4-oxo-2-thioxo-3, 4-dihydropyrimidin
acetamide
A reaction vessel equipped with an efficient stirrer was charged with (Z)-ethyl 3-((2- amino-2-oxoethyl)amino)-3-(5-chloro-2-methoxyphenyl)acrylate (15 g, 50.2 mmol), butyl acetate (150 ml.) and trimethylsilyl isothiocyanate (160.7 mmole, 21 .1 g, 22.7 ml.) and the mixture was heated to reflux. After 15 hours, the mixture was cooled to 30 °C and treated with 1 N aqueous sodium hydroxide (1 12.5 ml_, 1 12.5 mmoles). After 30 min, the organic layer was separated and extracted with another portion of 1 N sodium hydroxide (37.5 ml_, 37.5 mmoles). The combined aqueous phases were extracted twice with dichloromethane (2 x 45 mL), filtered, and treated with 6N HCI until a pH of 2.5 was achieved. After stirring for 1 hour, the resulting solid was isolated by vacuum filtration, resuspended in 100 mL of a 1 :1 methanol-water solution, heated with stirring at 50 °C for 2 hours, and cooled to room temperature before collecting the solid by vacuum filtration, pulling dry and drying in a vacuum oven (20 mm Hg, 50 °C) for 12 hours to afford 8.7 g of the desired product as a tan solid.
MS (ES+) 326.0 [M+1]+. 1H NMR (500 MHz, DMSO-d6) δ ppm 12.85 (s, 1 H) 7.57 (dd, J=9.03, 2.68 Hz, 1 H) 7.33 (s, 1 H) 7.17 – 7.23 (m, 2 H) 7.10 (s, 1 H) 5.89 (d, J=1.71 Hz, 1 H) 5.41 (br. s, 1 H) 3.89 (br. s, 1 H) 3.84 (s, 3 H).
Alternative Preparation of Example 1

2-( 6-( 5-Chloro-2-methoxyphenyl)-4-oxo-2-thioxo-3, 4-dihydropyrimidin- 1 ( 2H)-yl) acetamide A slurry of (Z)-ethyl 3-((2-amino-2-oxoethyl)amino)-3-(5-chloro-2- methoxyphenyl)acrylate (20 g, 63 mmol) in a mixture of butyl acetate (140 mL) and DMF (38 mL) was treated with trimethylsilyl isothiocyanate (16.8 g, 125 mmol) and the mixture was heated at 1 15-120 °C for 5-6 hours. The mixture was cooled to 0-5 °C, butyl acetate (100 mL) was added and the mixture was slurried for 8 hours. The formed solids were filtered, and the filter cake was washed with butyl acetate (2 x 100 mL). The solid was dried in a vacuum oven at 50 °C for 12 hours to a tan solid. The solid was dissolved in a 5:1 mixture of DMF and water at room temperature and additional water was added slowly to crystallize the material. The slurry was cooled to 10 °C and stirred for 8 hours, followed by filtration and washing with water. The filter cake was dried in a vacuum oven at 50 °C for 8 hours. The solid was dissolved in a 1 :1 mixture of methanol and water and the slurry was heated to 50 °C and held at this temperature for 2 hours. After cooling to 10 °C over 30 minutes, the slurry was held at this temperature for 1 hour, filtered and washed with water and dried in a vacuum oven at 50 °C for 8 hours to give the title compound as a white solid. MS (ES+) 326.0 [M+1]+.1H NMR (500 MHz, DMSO-d6) δ ppm 12.85 (s, 1 H) 7.57 (dd, J=9.03, 2.68 Hz, 1 H) 7.33 (s, 1 H) 7.17 – 7.23 (m, 2 H) 7.10 (s, 1 H) 5.89 (d, J=1.71 Hz, 1 H) 5.41 (br. s, 1 H) 3.89 (br. s, 1 H) 3.84 (s, 3 H).
REFERENCES
1: Ruggeri RB, Buckbinder L, Bagley SW, Carpino PA, Conn EL, Dowling MS, Fernando DP, Jiao W, Kung DW, Orr ST, Qi Y, Rocke BN, Smith A, Warmus JS, Zhang Y, Bowles D, Widlicka DW, Eng H, Ryder T, Sharma R, Wolford A, Okerberg C, Walters K, Maurer TS, Zhang Y, Bonin PD, Spath SN, Xing G, Hepworth D, Ahn K, Kalgutkar AS. Discovery of 2-(6-(5-Chloro-2-methoxyphenyl)-4-oxo-2-thioxo-3,4-dihydropyrimidin-1(2H)-yl)acetamide (PF-06282999): A Highly Selective Mechanism-Based Myeloperoxidase Inhibitor for the Treatment of Cardiovascular Diseases. J Med Chem. 2015 Oct 28. [Epubahead of print] PubMed PMID: 26509551.
////////////PF 06282999, 1435467-37-0, PFIZER, PHASE 1, PF-06282999; PF-6282999, PF06282999, ACUTE CORONARY SYNDROME
O=C(N)CN(C(N1)=S)C(C2=CC(Cl)=CC=C2OC)=CC1=O
PF 06650808
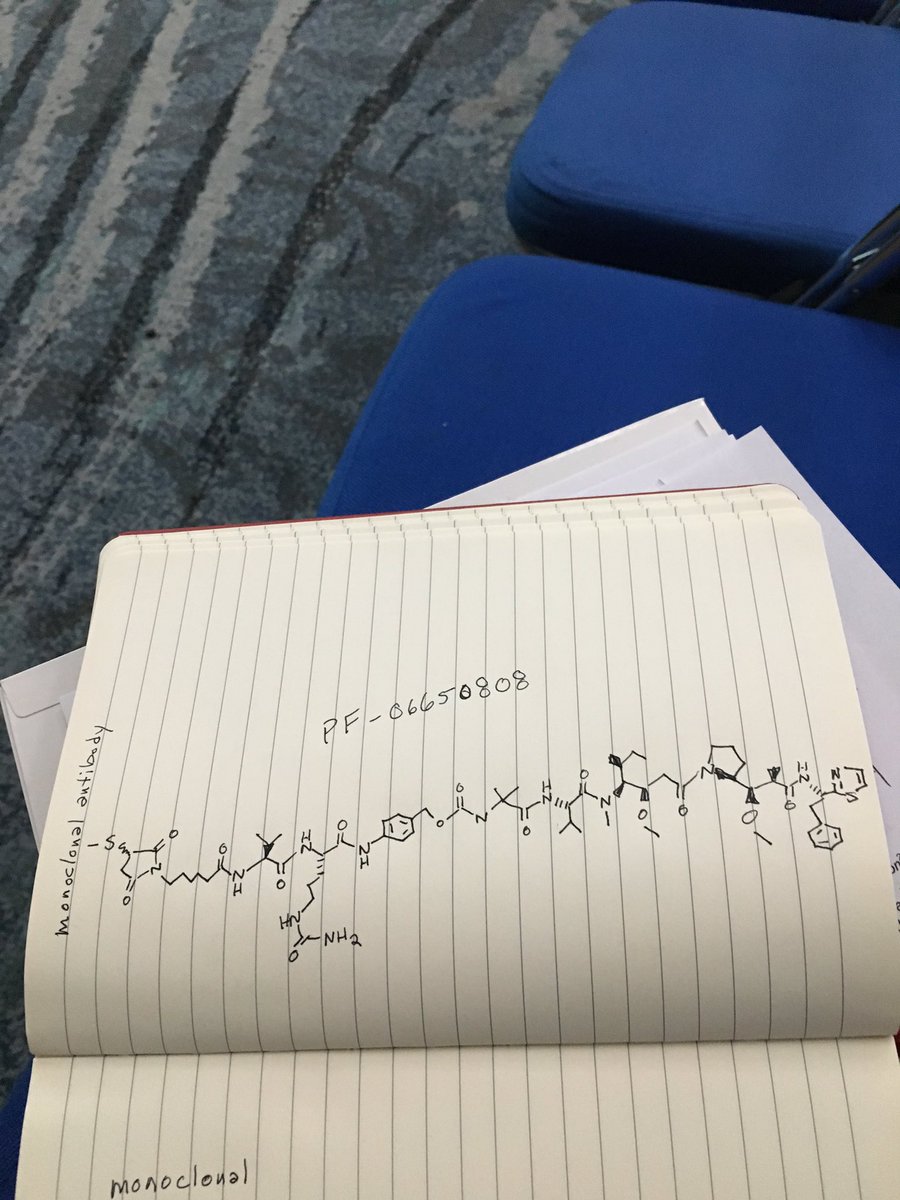 .
.
Picture credit….Bethany Halford

PF 06650808
CAS 1822383-80-1
A biologic for cancer treatment (Pfizer Inc.)
- Originator Pfizer
- Class Antineoplastics
- Mechanism of Action Notch-3 receptor antagonists
- No development reported Solid tumours
- 24 Jun 2018 Biomarkers information updated
- 28 Apr 2018 No recent reports of development identified for phase-I development in Solid-tumours(Late-stage disease) in USA (IV)
- 01 Jul 2017 Pfizer completes a phase I trial in Solid tumours (Late-stage disease) in USA (IV) (NCT02129205)
Company: Pfizer
Target: Neurogenic locus notch homolog protein 3 (NOTCH3): Activation and mutation of the NOTCH signaling pathway can lead to cancer.
Disease: Cancer
Notes: PF06650808 is an antibody-drug conjugate that delivers a cytotoxic payload molecule directly to tumor cells, explained Andreas Maderna, an associate research fellow at Pfizer. The payload molecule in PF06650808 was inspired by the marine natural product dolostatin 10, which is produced by cyanobacteria consumed by a type of sea slug.
https://cen.acs.org/articles/94/i15/New-drug-candidates-shine-San-Diego.html
PATENT
WO 2015171907
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015171907
The present invention relates to stable isotopic identification of biologic products, methods of stable isotopic identification of such biologic products, and stable isotopic methods and systems for correlating biologic products to the processes by which they are made.
PATENT
WO 2018045058
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2018045058&tab=PCTDESCRIPTION&maxRec=1000
CLIP
Rosen, L.S.; Wesolowski, R.; Gibson, B.; et al.
A Phase 1 dose escalation, safety, and pharmacokinetic study of PF-06650808, an anti-Notch3 antibody drug conjugate, in adult patients with advanced solid tumors
Eur Cancer Congr (September 25-29, Vienna) 2015, Abst 3OLBA
Maderna, A.
Therapeutic targeting the NOTCH3 receptor with antibody drug conjugates
251st Am Chem Soc (ACS) Natl Meet (March 13-17, San Diego) 2016, Abst MEDI 262
Hurvitz, S.A.; von Euw, E.; O’Brien, N.; et al.
Preclinical evaluation of targeting Notch-3 in breast cancer
107th Annu Meet Am Assoc Cancer Res (AACR) (April 16-20, New Orleans) 2016, Abst 1206
Chen, J.; Geles, K.; Silva, M.; Waterhouse, R.; Ma, D.; Charati, M.; Sapra, P.; Mccarthy, T.
Evaluate the impact of conjugation on targeting capacity, pharmacokinetics and tissue distribution of antibody drug conjugate, PF-06650808, in tumor bearing mice
22nd Int Symp Radiopharm Sci (ISRS) (May 14-19, Dresden) 2017, Abst P 052
///////////
PF 06650808
| Phase 1 |
$PFE compound inspired by auristatins
https://clinicaltrials.gov/ct2/show/NCT02129205
http://www.pfizer.com/sites/default/files/product-pipeline/8_7_2014_Pipeline_Update.pdf
ALL DATA COMING………
Notch-3 receptor antagonists
Neoplasms
Breast
| Pfizer |
Cancer
PF-06650808, is currently being examined in a Ph1 clinical trial (Protocol B7501001).
Notch3
Researchers are also exploring the use of Notch3 targeting. “The Notch pathway plays an important role in the growth of several solid tumours, including breast and ovarian cancer and melanoma,” explained Joerger. “In particular, Notch3 alterations such as gene amplification and upregulation are associated with poor patient survival. Research using Notch3 targeting as an innovative approach to treat solid malignancies included 27 patients unselected for Notch3 who received increasing doses of the anti-Notch3 antibody-drug conjugate PF-06650808. Responses were seen in two breast cancer patients (LBA 30). While preliminary, targeting Notch3 may become a new treatment approach in patients with selected solid tumours.”
The anti-Notch3 antibody-drug conjugate PF-06650808 is being developed by Pfizer.
- 31 Jul 2014 Phase-I clinical trials in Solid tumours (Late-stage disease) in USA (Parenteral)
- 30 Apr 2014 Preclinical trials in Solid tumours in USA (Parenteral)
- 30 Apr 2014 Pfizer plans a phase I trial for Solid tumours (late-stage disease, second-line therapy or greater) in USA (NCT02129205)
251st Am Chem Soc (ACS) Natl Meet (March 13-17, San Diego) 2016, Abst MEDI 262


/////////PF 06650808, PF-06650808, PF-6650808, monoclonal antibody, pfizer, phase 1, Solid tumours , Notch-3 receptor antagonists
C1(C(N(C(C1)=O)CCCCCC(=O)NC([C@H](C)C)C(=O)NC(C(=O)Nc2ccc(cc2)COC(=O)NC(C)(C)C(=O)N[C@@H](C(C)C)C(=O)[N@](C)C(C(CC)C)[C@@H](OC)CC(=O)N3CCC[C@H]3C(OO)C(C)C(=O)N[C@H](c4nccs4)CC)CCCNC(=O)N)=O)SC
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE



 Googleplus
Googleplus amcrasto@gmail.com
amcrasto@gmail.com
P.S
THE VIEWS EXPRESSED ARE MY PERSONAL AND IN NO-WAY SUGGEST THE VIEWS OF THE PROFESSIONAL BODY OR THE COMPANY THAT I REPRESENT, amcrasto@gmail.com, +91 9323115463 India.
I , Dr A.M.Crasto is writing this blog to share the knowledge/views, after reading Scientific Journals/Articles/News Articles/Wikipedia. My views/comments are based on the results /conclusions by the authors(researchers). I do mention either the link or reference of the article(s) in my blog and hope those interested can read for details. I am briefly summarising the remarks or conclusions of the authors (researchers). If one believe that their intellectual property right /copyright is infringed by any content on this blog, please contact or leave message at below email address amcrasto@gmail.com. It will be removed ASAP
PF 06650833
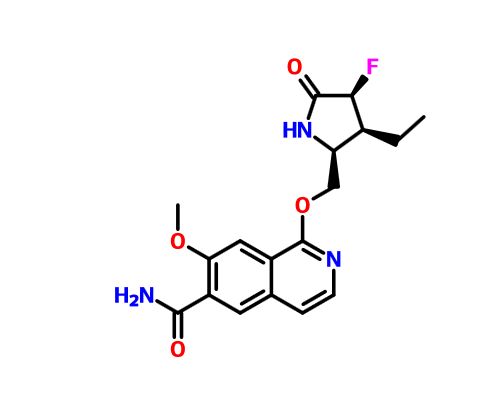
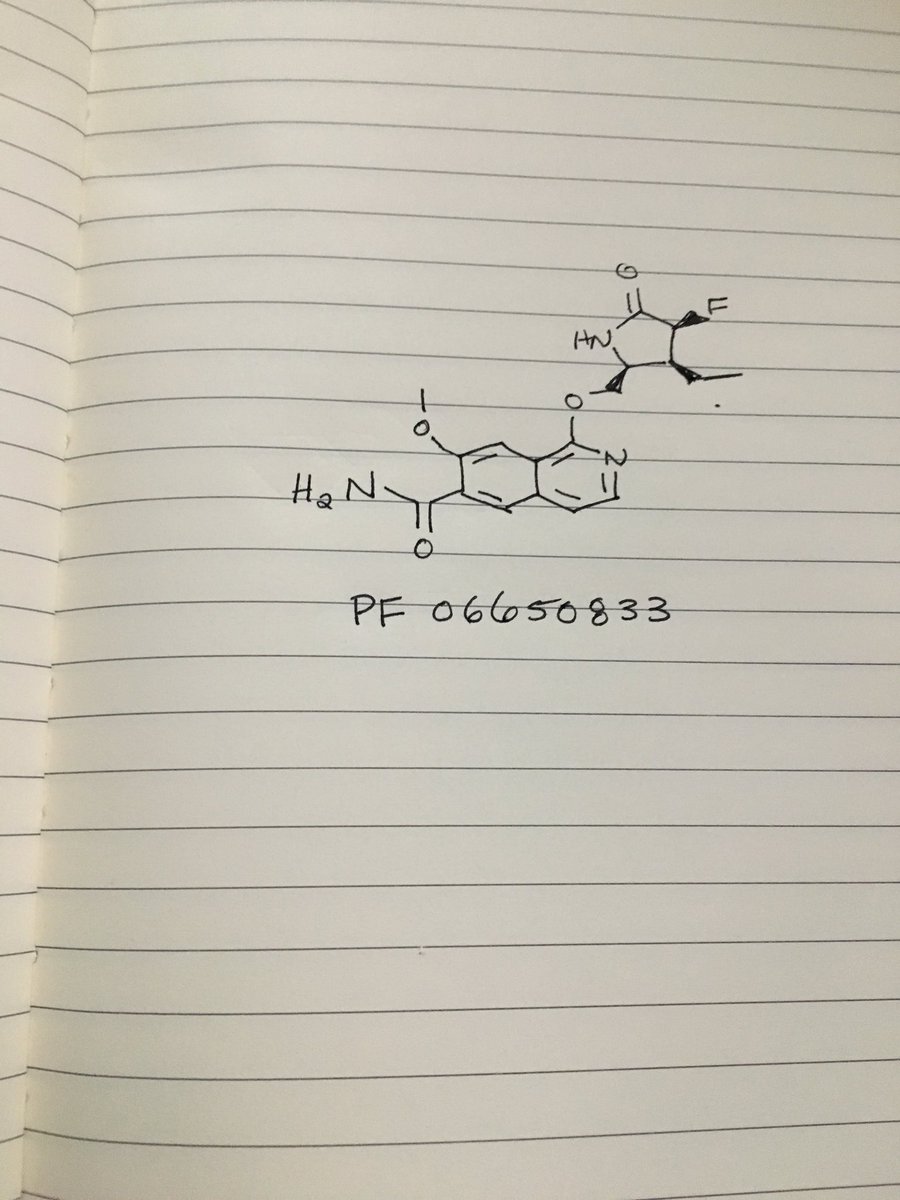 .
.
Picture credit….Bethany Halford
PF 06650833
MFC18H20FN3O4, MW361.37
1-{[(2S,3S,4S)-3-ethyl-4-fluoro-5-oxopyrrolidin-2-yl]methoxy}-7-methoxyisoquinoline-6-carboxamide
6-Isoquinolinecarboxamide, 1-[[(2S,3S,4S)-3-ethyl-4-fluoro-5-oxo-2-pyrrolidinyl]methoxy]-7-methoxy-
CAS 1817626-54-2
WO 2015150995
1st disclosures is @pfizer‘s on inflammatory disease treatment targeting IRAK4
$PFE IRAK4 inhibitor
Phase I Lupus vulgaris
- 01 Feb 2016 Pfizer completes a phase I pharmacokinetics trial in Healthy volunteers in USA (PO) (NCT02609139)
- 01 Nov 2015 Pfizer initiates a phase I pharmacokinetics trial in Healthy volunteers in USA (PO) (NCT02609139)
- 01 Jun 2015 Pfizer completes a phase I trial for Lupus (In volunteers) in USA (PO) (NCT02224651)
Compounds useful for the treatment of autoimmune and inflammatory diseases associated with lnterleukin-1 Receptor Associated Kinase (IRAK) and more particularly compounds that modulate the function of IRAK4.
Protein kinases are families of enzymes that catalyze the phosphorylation of specific residues in proteins, broadly classified in tyrosine and serine/threonine kinases. Inappropriate activity arising from dysregulation of certain kinases by a variety of mechanisms is believed to underlie the causes of many diseases, including but not limited to, cancer, cardiovascular diseases, allergies, asthma, respiratory diseases, autoimmune diseases, inflammatory diseases, bone diseases, metabolic disorders, and neurological and neurodegenerative diseases. As such, potent and selective inhibitors of kinases are sought as potential treatments for a variety of human diseases.
There is considerable interest in targeting the innate immune system in the treatment of autoimmune diseases and sterile inflammation. Receptors of the innate immune system provide the first line of defense against bacterial and viral insults. These receptors recognize bacterial and viral products as well as pro-inflammatory cytokines and thereby initiate a signaling cascade that ultimately results in the up-regulation of inflammatory cytokines such as TNFa, IL6, and interferons. Recently it has become apparent that self-generated ligands such as nucleic acids and products of inflammation such as high-mobility group protein B1 (HMGB1) and Advanced Glycated End-products (AGE) are ligands for Toll-like receptors (TLRs) which are key receptors of the innate immune system (O’Neill 2003, Kanzler et al 2007, Wagner 2006). This demonstrates the role of TLRs in the initiation and perpetuation of inflammation due to autoimmunity.
lnterleukin-1 receptor associated kinase 4 (I RAK4) is a ubiquitously expressed serine/threonine kinase involved in the regulation of innate immunity (Suzuki & Saito 2006). IRAK4 is responsible for initiating signaling from TLRs and members of the I L- 1/18 receptor family. Kinase-inactive knock-ins and targeted deletions of IRAK4 in mice were reported to cause reductions in TLR and IL-1 induced pro-inflammatory cytokines (Kawagoe et al 2007; Fraczek et al. 2008; Kim et al. 2007). IRAK4 kinase-dead knock-in mice have also been shown to be resistant to induced joint inflammation in the antigen-induced-arthritis (AIA) and serum transfer-induced (K/BxN) arthritis models (Koziczak-Holbro 2009). Likewise, humans deficient in IRAK4 also appear to display the inability to respond to challenge by Toll ligands and IL-1 (Hernandez & Bastian 2006). However, the immunodeficient phenotype of IRAK4-null individuals is narrowly restricted to challenge by gram positive bacteria, but not gram negative bacteria, viruses or fungi. This gram positive sensitivity also lessens with age, implying redundant or compensating mechanisms for innate immunity in the absence of IRAK4 (Lavine et al 2007).
These data indicate that inhibitors of IRAK4 kinase activity should have therapeutic value in treating cytokine driven autoimmune diseases while having minimal immunosuppressive side effects. Additional recent studies suggest that targeting IRAK4 may be useful in other inflammatory pathologies such as atherosclerosis and diffuse large B-cell lymphoma (Rekhter et al 2008; Ngo et al 2011). Therefore, inhibitors of IRAK4 kinase activity are potential therapeutics for a wide variety of diseases including but not limited to autoimmunity, inflammation, cardiovascular diseases, cancer, and metabolic diseases. See the following references for additional information: N. Suzuki and T. Saito, Trends in Immunology, 2006, 27, 566. T. Kawagoe, S. Sato, A. Jung, M. Yamamoto, K. Matsui, H. Kato, S. Uematsu, O. Takeuchi and S. Akira, Journal of Experimental Medicine, 2007, 204, 1013. J. Fraczek, T. W. Kim, H. Xiao, J. Yao, Q. Wen, Y. Li, J.-L. Casanova, J. Pryjma and X. Li, Journal of Biological Chemistry, 2008, 283, 31697. T. W. Kim, K. Staschke, K. Bulek, J. Yao, K. Peters, K.-H. Oh, Y. Vandenburg, H. Xiao, W. Qian, T. Hamilton, B. Min, G. Sen, R. Gilmour and X. Li, Journal of Experimental Medicine, 2007, 204, 1025. M. Koziczak-Holbro, A. Littlewood- Evans,
B. Pollinger, J. Kovarik, J. Dawson, G. Zenke, C. Burkhart, M. Muller and H. Gram, Arthritis & Rheumatism, 2009, 60, 1661. M. Hernandez and J. F. Bastian, Current Allergy and Asthma Reports, 2006, 6, 468. E. Lavine, R. Somech, J. Y. Zhang, A. Puel, X. Bossuyt, C. Picard, J. L. Casanova and C. M. Roifman, Journal of Allergy and Clinical Immunology, 2007, 120, 948. M. Rekhter, K. Staschke, T. Estridge, P. Rutherford, N. Jackson, D. Gifford-Moore, P. Foxworthy,
C. Reidy, X.-d. Huang, M. Kalbfleisch, K. Hui, M.S. Kuo, R. Gilmour and C. J. Vlahos, Biochemical and Biophysical Research Communications, 2008, 367, 642. O’Neill, L. A. (2003). “Therapeutic targeting of Toll-like receptors for inflammatory and infectious diseases.” Curr Opin Pharmacol 3(4): 396. Kanzler, H et al. (2007) “Therapeutic targeting of innate immunity with toll-like receptor agonists and antagonists.” Nature Medicine 13:552. Wagner, H. (2006) “Endogenous TLR ligands and autoimmunity” /Advances in Immunol 91 : 159. Ngo, V. N. et al. (2011) “Oncogenically active MyD88 mutations in human lymphoma” Nature 470: 115.
PATENT
Preparation 1 : 1-chloro-7-methoxyisoquinoline-6-carbonitrile (P1) Step 1. Synthesis of methyl 4-iodo-3-methoxybenzoate (CAS 35387-92-9. CD.
To a solution of 3-hydroxy-4-iodobenzoic acid (CAS 58123-77-6, C12) (10800 g, 40.9 moles) in DMF (65 L) was added K2C03 (25398 g, 184 moles), followed by the slow addition of dimethyl sulfate (11352 g, 90 moles). This mixture was heated to about 50 °C for over night. The reaction mixture was cooled to about 25 °C, diluted with EtOAc (50 L) and filtered through a plug of Celite®. The solid was thoroughly washed with EtOAc (10 L X 3). The combined EtOAc filtrates were poured into water. After stirring for about 30 min, the EtOAc layer was separated and it was further washed sequentially with water, 1 M NaOH and brine. The EtOAc layer was separated, dried over Na2S04, filtered and concentrated to provide the title compound C1. Yield: 11750 g (98%).
Step 2. Synthesis of (4-iodo-3-methoxyphenyl)methanol (CAS 244257-61-2, C2).
To a solution of compound C1 (11750 g, 40.2 moles) in THF (35 L) was added NaBH4 (7645 g, 201.09 moles) and refluxed. While refluxing, MeOH (25 L) was slowly added into the reaction mixture at a rate of about 1 L per hour. After completion of the reaction, it was poured into a solution of cold dilute HCI. Once the excess of NaBH4was quenched, the solution was filtered and extracted with EtOAc (2.5 L X 3). The combined EtOAc extracts were washed sequentially with water, brine and dried over Na2S04. The solvent was evaporated under reduced pressure and the resulting crude material was treated with MTBE. The resulting solid was filtered and filtrate was washed with water, brine, dried over Na2S0 , and filtered. The solvent was evaporated under reduced pressure to provide the title compound C2. Yield: 9900 g (93%).
Step 3. Synthesis of 4-iodo-3-methoxybenzaldehyde (CAS 121404-83-9, C3).
To a solution of compound C2 (9900 g, 34.5 moles) in CHCI3 (186 L), was added manganese dioxide (18000 g, 207 moles) and the resulting mixture was refluxed for about 16 h. The mixture was cooled to about 25 °C and filtered through a Celite pad, which was then washed thoroughly with CHCI3. The CHCI3 was evaporated under reduced pressure to provide the title compound C3. Yield: 9330 g (95%). 1 H NMR (400 MHz, CDCI3): δ 9.95 (s, 1 H), 7.99 (d, 1 H), 7.14 (dd, 1 H), 3.95 (s, 3 H).
Step 3. Synthesis of 6-iodo-7-methoxyisoquinoline (CAS 244257-63-4. C4).
To a solution of compound C3 (9300 g, 35 moles) in toluene (60 L) was added amino acetaldehyde dimethyl acetal (5590 g, 53 moles) and the mixture was refluxed for about 4 h, while removing the liberated water by the use of a Dean – Stark water separator. The reaction mixture was cooled to about 0 °C, after which trifluoroacetic anhydride (22305 g, 106 moles) followed by BF3-Et20 (15080 g, 106 moles) were added, keeping internal temperature below 5 °C. The reaction mixture was stirred at about 25 °C for about 16 h and quenched by pouring into a mixture of ice and ammonium hydroxide. The product was extracted with EtOAc (10 L X 3), and the combined EtOAc extracts were washed sequentially with water and brine. The combined EtOAc extracts were dried over Na2S04, filtered, and concentrated to afford a dark tan colored residue. This was treated with a mixture of MTBE and hexane (1 :1 v/v, 30 L), followed by 6 M HCI (9 L), with stirring. The precipitated solid was filtered and washed with MTBE. The solid was suspended in EtOAc (5 L) and made alkaline with ammonium hydroxide. The EtOAc layer was separated, washed with brine, dried over Na2S04, filtered, and concentrated to afford crude compound C4 as a brown solid. HPLC (230 nm) showed it to be about 83% pure.
The crude material (1000 g) was taken in AcOH (2.5 L) and stirred for about 90 min at about 25 °C. The solid was filtered and washed with AcOH (500 ml_). The filtrate was neutralized with saturated aqueous Na2C03 solution. The resulting precipitated solid was filtered, washed with water (4 L), and oven dried at about 70 – 75 °C for about 5 h to afford about 780 g of pure C4. Similarly, the remaining crude C4 (4 kg) was purified to provide the title compound C4. Yield: 4300 g (42%). 1H NMR (400 MHz, CDCI3): δ 9.15 (s, 1 H), 8.45 (d, 1 H), 8.35 (s, 1 H), 7.45 (d, 1 H), 7.15 (s, 1 H) 4.00 (s, 3 H).
Step 4. Synthesis of 7-methoxyisoquinoline-6-carbonitrile (C5).
To a solution of compound C4 (4300 g , 15 moles) in DMSO (39 L) was added copper(l) cyanide (2954 g, 33 moles) and the mixture was heated to about 120 °C for about 3 h. The reaction mixture was quenched by pouring into a mixture of ice and ammonium hydroxide (40 L) and filtered. The filtrate was extracted with EtOAc (10 L X 2). While stirring, the solid residue was again treated with ammonium hydroxide solution (10 L) and EtOAc (10 L). After filtration, the precipitated material was repeatedly washed with a mixture of MeOH and CHCI3 (1 :9, v/v) several times and the combined extracts were washed with brine. The extracts were dried over Na2S04, filtered, and concentrated under reduced pressure. The resulting crude material was triturated with hexane to provide the title compound C5. Yield: 2250 g (87%). 1H NMR (400 MHz, CDCI3): δ 9.25 (br. s, 1 H), 8.55 (br. s, 1 H), 8.15 (s, 1 H), 7.60 (d, 1 H), 7.30 (s, 1 H), 4.05 (s, 3 H).

A solution of a reactant such as 1-(((2S,3S,4S)-3-ethyl-4-fluoro-5-oxopyrrolidin-2-yl)methoxy)-7-methoxyisoquinoline-6-carbonitrile (200 mg, 0.5 mmol) in concentrated H2SO4 (1.5 ml.) was warmed to about 55 °C for about two hours, then cooled to about 20 °C. The reaction mixture was added dropwise with vigorous stirring to 7.3 ml_ of ice cold concentrated ammonium hydroxide with cooling in ice. The precipitated solid was filtered and washed with water, heptane, ether, and dried under vacuum. The residue may be used directly for subsequent work, or it may be purified by chromatography or HPLC.
ABSTRACTS
251st Am Chem Soc (ACS) Natl Meet (March 13-17, San Diego) 2016, Abst MEDI 261




//////////PF 06650833, IRAK4 inhibitor, inflammatory disease treatment , PFIZER, 1817626-54-2
N1C([C@H](C([C@H]1COc3c2cc(c(cc2ccn3)C(=O)N)OC)CC)F)=O
NC(=O)c2cc3ccnc(OC[C@H]1NC(=O)[C@@H](F)[C@H]1CC)c3cc2OC
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
Googleplus amcrasto@gmail.com
P.S
THE VIEWS EXPRESSED ARE MY PERSONAL AND IN NO-WAY SUGGEST THE VIEWS OF THE PROFESSIONAL BODY OR THE COMPANY THAT I REPRESENT, amcrasto@gmail.com, +91 9323115463 India.
I , Dr A.M.Crasto is writing this blog to share the knowledge/views, after reading Scientific Journals/Articles/News Articles/Wikipedia. My views/comments are based on the results /conclusions by the authors(researchers). I do mention either the link or reference of the article(s) in my blog and hope those interested can read for details. I am briefly summarising the remarks or conclusions of the authors (researchers). If one believe that their intellectual property right /copyright is infringed by any content on this blog, please contact or leave message at below email address amcrasto@gmail.com. It will be removed ASAP

{kind=link}