





- Young Chemists who have just started work in industry as development chemists
- Organic Chemists/Medicinal Chemists in Research and Development who would like to gain an appreciation of development and scale up and who are perhaps contemplating moving into chemical development.
- Development and Production Chemists in industry who would like to improve their efficiency and gain an insight into alternative approaches to chemical development.
- Chemical Engineers who wish to understand a chemist’s approach to chemical development of batch processes. (Engineers would, however, need a good grounding in organic chemistry)
- Students who are about to enter the industry and can obtain company sponsorship.
- Experienced Chemists looking to refresh and/or augment their knowledge of chemical development
- Analytical Chemists who wish to gain a broader appreciation of process chemistry
- Managers who might benefit from a comprehensive and up to date overview of chemical development
Home » Posts tagged 'INDIA' (Page 3)
Tag Archives: INDIA
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on
ORGANIC SPECTROSCOPY INTERNATIONAL

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()




Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Flow Chemistry India 2016, 21-22 January 2016, Mumbai, India

Flow Chemistry India 2016Date: Thursday, 21 January 2016 – Friday, 22 January 2016
|
SELECTBIO INDIA
http://selectbiosciences.com/conferences/index.aspx?conf=FCINDIA16&se=india
Register…………..http://selectbiosciences.com/conferences/registration.aspx?conf=FCINDIA16&se=india

venue
Hotel Ramada Powai and Convention Centre, Mumbai, India


Professor & Research Chair, Nelson Mandela Metropolitan University |

Professor, Synthetic Organic Chemistry , The University of Tokyo |

Deputy Director and Chair, National Chemical Laboratory |

Professor, Eindhoven University of Technology |

Scientific Director, University of Lyon |

Chairman, Flow Chemistry Society
Professor, University of Warsaw |


Maninderjit Singh Ahluwalia
Overview
SELECTBIO INDIA is delighted to welcome you all at the 4th International Conference Flow Chemistry India 2016 to be held in Mumbai on January 21-22, 2016 under the auspices of the Flow Chemistry Society. The society aims to unite and represent those who are actively working on this rapidly developing field. This meeting is dedicated to the integration of flow chemistry into everyday practice throughout the world by delivering the latest knowledge and making it available for the entire chemistry community.
Society members save 25% on the registration fee and non-members will receive their first year’s membership included in the fee.
Running alongside the conference will be an exhibition covering the latest technological advances in the area of flow chemistry.
Who Should Attend
• Scientists, Chemists, Chemical Engineers and Researchers working in Pharmaceutical and Fine Chemicals Research and Development including Drug Discovery, Medicinal Chemistry and Chemical Process Development
• Scientists, Chemists and Chemical Engineers working in Pharmaceutical and Fine Chemical Bulk Manufacturing Units
• Corporate Management, Scientists, Managers responsible for development of Pharmaceutical and Fine Chemicals R & D and Manufacturing activities
• Scientists, Chemists & Engineers belonging to the fields of Inorganic, Organic, Medicinal, Natural Products, Analytical, High-throughput and Process Chemistry in the Academic research as well as in Applied research and development in the area of Agrochemical, Petrochemical and Fragrance industry
• Scientists working in or interested in applications of Flow Chemistry in Material science, Green chemistry, Nanotechnology, Biotechnology, Theoretical Chemistry, Information technology and Flow synthesis instruments including Engineering & Automation
Conference Package – Includes Registration, 2 Nights Accommodation, Dinner & Airport Transfers (Valid up to January 5, 2016 only)
Call for Posters
You can also present your research on a poster while attending the meeting. Submit an abstract for consideration now!
Poster Submission Deadline: 30 November 2015
Agenda Topics
- Advances in Micro & Continuous Flow Reactors, Systems & Processes
- Applications in Pharmaceutical Industry & API Synthesis
- Engineering Aspects of Flow Chemistry
- Flow Reactor – Choosing the Right One
- Photochemistry & Multistep Synthesis in Flow
- Quality Issue and QbD in Flow Chemistry
- Scale up – From Micro to Commercial Scale
- Yield Improvement, Cost Cutting and Waste Reduction in Flow Chemistry
Sponsorship and Exhibition Opportunities
http://selectbiosciences.com/conferences/index.aspx?conf=FCINDIA16&se=india

Workshop Tutor
Charlotte Wiles
CEO CHEMTRIX
A Workshop on “Flow Chemistry Demonstrations (Lab & Plant Scale) for Chemical and Pharmaceutical Industry-” will be held one day prior to the training course i.e. on 20th January, 2016 from 10:00 am – 05:00 pm in Mumbai. This workshop is supported by Process Intensification will be jointly conducted by :
Dr. Dinesh Kudav (Mumbai University); Dr. Charlotte Wiles (Chemtrix BV-Neth); Mr. Wouter Stam (Flowid, NV-Neth); Mr. Manjinder Singh (CIPLA & VP-FCS-India Chapter); Dr. Viktor Gyollai, (AM Technology-UK); Dr. Prashant Kini (UPL Ltd.); Mr. Kumar Oza (Pi & TCPL); Mr. Madhav Sapre (Pi & Sharon Bio); et al .
This workshop is specially designed to demonstrate application/capabilities of Flow Chemistry running “live” reactions in Continuous Flow Reactors. The reactions likely to be demonstrated using Flow Chemistry includes :• Nitration • Organometallic reaction• Oxidation • Bi-phasic reaction• Nano-Particle preparation in Flow• Biocatalytic Reaction with enhanced enzyme life.
This workshop is free for the registered delegates of Flow Chemistry India 2016 Conference and Continuous Flow Reactors Training Course.
You can visit Mumbai city
Taj hotel, mumbai
Gateway of india
 Food in mumbai
Food in mumbai
 mumbai skyline
mumbai skyline

The Bandra-Worli Sea Link is a cable-stayed bridge that connects central Mumbai with its western suburbs

get in if you can
The Mumbai Suburban Railway system carries more than 6.99 million commuters on a daily basis. It has the highest passenger densities of any urban railway …

Chhatrapati shivaji in mumbai india
British-victoria terminus


VADA PAV


SELECTBIO CONFERENCES PICS










/////////
Pharma Regulations for Generic Drug Products in India and US: Case Studies and Future Prospectives

Dr. Suryakanta Swain
Introduction
The Indian pharmaceutical industry has come a long way from being non-existent before independence to a prominent provider of medicines and health care products in the current decade. The Indian pharmaceutical industry at present is the global leader of growing pharmaceutical manufacturing companies, providing wide range capabilities in the complex field of technology and drug manufacturing. Indian pharma market growing at a rapid pace currently providing Indian pharmaceutical industry third rank all over the world in terms of volume and fourteen ranks, according to market value [1]. The major strength of currently growing Indian pharmaceutical sector is its capability to manufacture wide range of simple analgesic pills to complicated antibiotics, cardiac compounds with peer quality and efficacy and altogether exporting them to developed world. The industry bulk profit comes from exporting generics and API to the developed market mainly US followed by UK, Germany, Brazil etc. The total share of generics accounts in export is 58% providing the major boost, the Indian commerce ministry has set an ambitious export target of $ 25 billion by 2013-14, which can be achieved only by major contribution from generics market [2]. The Indian generics market is growing day by day with Indian pharmaceutical companies seeking more Abbreviated New Drug Application approvals (ANDAs) in US in major segments such as cardiovascular, antibiotics and other groups. The major force for the development of generics market in US came in the form of enacting the Drug Price Competition and Patent Restoration Act of 1984, public law 98-417 better known as “The Hatch- Waxman Act” which created opportunities for developing and marketing generics or better called as abbreviated new drug applications for 180 days. Under ANDAs a pharmaceutical manufacturer can develop and market low price generic version of previously approved innovator drugs, thus providing the same product to patient in pregnable price with safety and efficacy. A generic or biosimilar drug product is one that is comparable to an innovators drug product in dosage form, strength and route of administration, quality, performance characteristics and intended use. All approved products, both innovator and generics, are enlisted in FDA’s orange book. Generic drug application are termed as “abbreviated” because they are generally not required to include preclinical (animal) and clinical (human) data to establish safety and efficacy instead, generics applicant must demonstrate that there product is bioequivalent (i.e., performs in similar manner to innovator products). India has its unique position all over the world generics market, providing drugs at low cost to the developed world, this is because of its rigid and flexible pharma regulations, patent act which is updated from time to time, thus Indian generics market is playing a major role in growth of Indian economy as it provides a major share in export, mainly exporting generics to US, therefore a proper set of rules and regulations is required in future for producing generics and exporting them, so that Indian pharmaceutical sector and economy maintains its growth and becomes leaders globally.
|
|||||||
| Table 1: Regulatory requirements for generic drugs. |
Pharma Regulations for Generic Product in India and US
Generics have an important role to play in public health as they are well known to medical community and usually more affordable due to competition. They are formulated when patent and other exclusivity rights expire. The key for generic medicines is their therapeutic interchangeability with originator products. To ensure the therapeutic efficacy generic products must be pharmaceutically interchangeable (contain the same amount of active ingredient and have the same dosage form) and bioequivalent to the originator product. Bioequivalence is usually established using comparative in-vivo pharmacokinetic studies with originator products. The detailed description how it is carried out is described in respective WHO document and national regulatory guidelines. Well resourced regulatory authorities require that a generic medicine must meet certain regulatory criteria [3,4]. The major regulatory requirement for generic drug is presented in Table 1. For applying the ANDA’s in US, application is submitted under any of the below subsections of 505(j) of Federal act, it is important to comply with rule and regulations of US because it’s the major export destination for Indian generics manufacturers [5], the various application which can be applied for ANDAs in US is depicted in Table 2. The ANDAs review process is most important for developing generics, the review by FDA and CDER is done for generic applicant to compare its therapeutic bioequivalent with brand drugs after its approval for equivalency generic version of drug can be marketed (Figure 1). The review for equivalency is done by taking into account the bioavailability of product with branded drug, its microbiology, chemistry and labeling of product, this are current regulation to follow for generic approvals given by respective FDA.
|
||||||||||
| Table 2: Different types of ANDA applications in US. |
 |
| Figure 1: Explain the ANDA reviews process for development of generic drugs. |
Future Generic Products in India and US
It is seen that there is an upward swing in the generic market. It has reached 100 billion dollars in the past and is estimated to be three times higher than the overall growth of drugs. The current trend exhibits that blockbuster drugs are scheduled to lose their patent protection, opening the doors to cheaper generic drugs between 2013 and 2015 with the total market value in billions. It is expected that the percentage of generic drugs in the US market will rise from 14 to 21. This growth will enhance the export prospect of India and it will be doubled every year. It will be due to increase in the number of low cost workers and degree of innovation. Recent success in track record in design operation of high tech manufacturing, testing, quality control, research, clinical testing and biotechnology also contribute to this higher growth. Indian pharmaceutical industries those who have USFDA (United States Food and Drug Administration) affiliations and approval of ANDA (Abbreviated New Drug Applications) will stand benefited. Now India’s global share in the field of generic market is stipulated at 35% which is very high [6,7]. Table 3 describes list of various drugs going to get off-patent in 2015. To make the situation more favorable the Indian government has also introduced scheme of providing generic drugs to patient in hospitals with various Jan-aushadhi Kendra (Facilitation Centre). Thus future prospects of generics in India and US are very high as they are the next big thing in health care scenario. Consistent with prior research, MEPs (Market Exclusivity Periods) for drugs experiencing initial generic entry in 2011-2012 was 12.6 years for New Molecular Entities (NMEs) with sales greater than $100 million in the year prior to generic entry, and 12.9 years for all NMEs. Further research may reveal variation by type of NME, whether defined by molecule type or other classification. Generic competition has intensified over the past 10-15 years, and the MEP has become an even more important indicator of the economics of brand-name drugs. The MEP is critical to manufacturers’ ability to earn profits on brand-name drugs to fund future research and development activities, and brand-name drug shares rapidly drop following initial generic entry. Over 80% of brandname drugs experiencing initial generic entry in 2012 had faced at least one Paragraph IV patent challenge from a generic manufacturer, up from only 9% for drugs experiencing initial generic entry in 1995. These challenges are filed relatively early in the brand drug life cycle, on average within 7 years of brand launch. Developments for the generic pharmaceutical industry are encouraging as more brand-name drugs come off patent and payers push for cost cuts in health care. In addition, due to increasing FDA budget and staffing should begin to cut the backlog of branded and generic drug applications and increase the ability of the FDA to inspect facilities here and overseas as generic biologics get to market in the next few years [8].
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Table 3: List of some important drugs going to be off-patents |
Upcoming Challenges for Indian Generics Manufacturers in Global Market
The generic drug companies in India have broad technological and diversified market capabilities. As more and more patents expire, the generic portion of the pharmaceutical market is expected to continue to have increased sales. The scientific capability for manufacturing and supplying generic drugs of these companies will give them an edge over others and make them major players in the international generics market. Fortunately India has the best subject skills to galvanize foreign investors. The encouraging scenario of basic research and drug discovery will also support the changed dynamics. But their future sustainable growth depends on sustaining in competitive markets of developed world. The major challenges for generic manufactures are strengthening the existing regulatory system especially for enabling more detailed and universal classification of drugs and chemicals between branded generic and generics. High R&D cost and investment in research is also a major stumbling block in this direction [9].
|
||||||||||||||||||||||||||||||||||||||||
| Table 4: Describes list of various new ANDAs approval in the year 2013. |
Amendments in the Pharma Regulations for Generic Products
The Hatch-Waxman Act enacted 1984 is a landmark act. It allows generic drugs to enter the market without repeating expensive clinical trials required for their branded drugs. The legislation is meant for balancing the world of generic and branded drug industries. It provides accessibility to lower-cost generic drugs while still encouraging innovation and development of new drugs. Nevertheless, the legislation created unintended legal barriers that have slowed the entry of generic drugs into the market due to significant legal loopholes. The generic drug companies are allowed to market the drug after the patent and certain exclusivities expire. It has led to the prolific growth of generic drugs in the market. Thus some changes are required so that the loopholes can be filled and the regulation can be strengthened and selling of low cost drugs can be achieved. The change in rule related to alleged abuse of the 30-months stay provision is to be taken care were the ANDA applicant informs the original patent holder about the generic version filing, where they have 45 days to file a patent infringement suit against the generic applicant. If an infringement suit is filed within the 45-days period, FDA approval to market the generic version is automatically postponed for 30 months. These stays are extremely advantageous to innovating companies, because they provide over 2 years of additional market sales. Company takes profit by utilizing this route and delays the entry of generic drug in market; many steps have been taken by amending act of Greater Access to Affordable Pharmaceuticals Act passed in 2003 by American government. Extending the extensions by alleged abuse of the 30-month stay provision is done by many companies that holds patent, the companies are able to further delay the market entry of generic drugs is through multiple patent listings in the Orange Book, which is the FDA’s official listing of all the approved products. There are instances in which brand-name companies listed related patents in the Orange Book after an ANDA had already been filed by a generic manufacturer. The effect of these “later-listings” is that the generic applicant is then required to re-certify that the laterlisted patent is also invalid or not infringed and notify the patent holder of the re-certification. Thus more delay occurs in generic drug to reach market [10]( Figure 2).
Recent Cases and Incidents of Generic Products Regulation in India and US
The future prospects of generic product regulation in India and US are of great importance as they will decide the direction of growth of Indian Pharmaceutical Industries. Based on the recent cases and incidents that have occurred in India and US related to the generic product utilization, the new crucial roles will be implemented. The list of a few recent cases and incidents that happened in connection with generics in India & US are discussed in detail below (Figure 3).
 |
| Figure 2: Schematic overview for the benefits of Hatch-Waxman Act. |
 |
| Figure 3: Steps for the launching of generic drugs |
The Karen L. Bartlett case
In December-2004, Physician of Karen L. Bartlett was prescribed Clinoril, the brand-name version of the Non-Steroidal Anti- Inflammatory Drug (NSAID) sulindac, for shoulder pain of Karen L. Bartlett. Her pharmacist dispensed a generic form of sulindac manufactured by petitioner Mutual Pharmaceutical. Karen L. Bartlett soon developed an acute case of toxic epidermal necrolysis. She is severely disfigured, has physical disabilities, and is nearly blind. At the time of the prescription, sulindacs label did not specifically refer to toxic epidermal necrolysis. By 2005, however, the FDA had recommended changing all NSAID labeling to contain a more explicit toxic epidermal necrolysis warning. Respondent sued Mutualin New Hampshire state court. A jury found Mutual liable on respondent’s design-defect claim and awarded her over $21 million. The First Circuit gets ratified. As relevant, it found that neither the FDCA nor the FDA’s regulations pre-empted respondent’s design-defect claim. It distinguished PLIVA, Inc. v. Mensing, 564 U.S in which the Court held that failure-to-warn claims against generic manufacturers are pre-empted by the FDCA’s prohibition on changes to generic drug labels by arguing that generic manufacturers facing design-defect claims could comply with both federal and state law simply by choosing not to make the drug at all. This case is being closely watched by pharmaceutical companies, federal regulators and others, the Supreme Court will decide on whether Mutual can be held responsible for Ms. Bartlett’s injuries. The outcome is likely to further clarify the legal recourse for patients who take generic drugs, which now account for 80 percent of all prescriptions in the US. The verdict on both the sides will be playing a crucial role in drafting future pharma regulations as if the court agrees with Mutual and rules that generic companies cannot be sued for defective products, trial lawyers warn that patients will be left with very few options if they are injured by a generic drug whereas manufacturers of generic drugs and other business groups have said that if the court sides with Ms. Bartlett, the decisions of individual juries could trump the authority of federal agencies like the Food and Drug Administration and potentially lead drug makers to remove valuable medicines from the market. Thus this case will be important for the future of generics drug market in US and India [11,12].
Pay to delay pharmaceutical case
The question of whether the manufacturer of a branded drug can pay another drug manufacturer to keep a generic version of the drug off the market was heard by the United States Supreme Court on 25th March, 2013. The court will decide whether “pay-to-delay” or reverse settlements arrangements, in which the manufacturer of a branded medication pays another company to keep a generic version off the market, are legal or not, the outcome of the case is very important because it will decide for how many patients pay for medications. Federal Trade Commission challenges the payments. It sees these arrangements as collusion, design to stop competition in the market place and is meant for violation of antitrust laws of the nations. The drug makers, in contrast, see the settlements as a routine way of settling a legal dispute, with each side getting something it wants. The Hatch- Waxman Act 1984 has some loophole. Payments are made possible by using these loopholes. Certain amendments are made in the last decade to encourage generic manufacturers to challenge patents held by branded manufacturers before they are set to expire. Typically, the generic manufacturer files for FDA approval to market a generic version of a branded medication that is still under patent protection, and the branded manufacturer sues the generic manufacturer for patent infringement. An increasing number of such cases end in “payto- delay” agreements according to which the generic manufacturer agrees to hold off on introducing the generic version in exchange for payment from the branded manufacturer. The case in point is Androgen (testosterone gel), produced by Solvay Pharmaceuticals whose patent is set to expire in 2020. The bone of contention between Actavis (formerly Watson Pharmaceuticals) and Solvay Pharmaceuticals was Andro Gel. Actavis filed for FDA approval to market a generic version of Andro Gel in 2003, and Solvay sued. In 2006, the FDA approved the generic version for marketing of Actavis, but the suit remained status quo. Later in 2006, the companies came to a settlement according to which Solvay would pay Actavis $20 to $30 million per year in exchange for help with marketing and an agreement to keep its generic version of Andro Gel off the market until 2015. The FTC (Federal Trade Commission) contends that the drug companies colluded to maintain Solvay’s monopoly on Andro Gel because, without the settlement, the generic version would have become available in 2006. A federal district court dismissed the FTC’s argument in this case, but another district court in a similar case decided the opposite way, so it is now up to the Supreme Court to decide and decision is expected. Moreover the best verdict according to many experienced federal judges that supreme court should not generalize the law, where as it should be implemented on case to case basis, thus this case should be great importance for Pharma regulators to draw guidelines for future regulations of generics in India and US and it will be important for patients to decide whether they will opt for cheaper or expensive medicines [13,14].
The Ranbaxy saga case
The criminal fraud that Ranbaxy has done with US FDA has let down many but it’s the fellow generics drug maker of India that will face the heat, this will be a very important incident which will decide fate of generics drug market of India in US and its regulation. Ranbaxy pharmaceutical of India is charged with producing low quality generic drugs in US and manipulating data’s required for filing NDA and ANDA approvals in US, thus cheating their counter parts in many ways to be first in the race of producing generic version. Ranbaxy pleaded guilty to seven federal criminal counts of selling adulterated drugs with intent to defraud, failing to report that its drugs did not meet specifications, and making intentionally false statements to the government. Ranbaxy agreed to pay $500 million in fines, forfeitures, and penalties-the most ever levied against a generic-drug company. The company, now majority owned by Japanese drug maker Daiichi Sankyo, sells its products in more than 150 countries and has 14,600 employees. It also came to light that even Ranbaxy scientist adulterated there generic testing drug with branded drugs for manipulating bioequivalence study.
Thus these serious allegations on one of the top India pharmaceutical company could be a major setback for generic manufactures and Indian Pharma regulator as they have failed to, therefore some strict regulations could be implemented by US FDA in future for Indian generics producers which could be a serious issue as it will lead to effect the generics drug market in India. Thus this will be the major factor which will decide the fate of future regulation of generics in India and US [15,16].
Miscellaneous cases and incidents
The study discusses the case of Swiss drug maker Novartis plea overruled recently by the Supreme Court was an attempt to win patent protection for its cancer drug Glivec. This was a serious blow to Western pharmaceutical firms who are increasingly focusing on India to drive sales and it also affects Indian and US generic market. Glivec (ß-polymorphic form of imatinib mesylate) is indicated for treatment of certain blood and stomach cancers. The Supreme Court decision implies that a clutch of Indian companies, including Cipla, Ranbaxy and Natco, could continue marketing generic versions of the drug at a fraction of the cost of Novartis’ product. While Novartis’ Glivec costs over one lakh a month, local companies sell versions of the drug at roughly ten thousand a month. Supreme Court’s ruling states that the drug has failed in “both the tests of invention and patentability” under Indian law. On the other hand, Glivec is widely recognized as one of the most important medical discoveries in decades, but it lost the battle on innovative quality grounds. The verdict can be interpreted as a battle between research and innovation on one side and public health and affordability on the other. It is true that the prospect of producing cheaper generic versions of lifesaving drugs in the country, thus sale of generics will increase and generic market will be boosted up. Thus the case study suggests that the future of generics in India is bright and this case will be a benchmark for it. The well documented Novartis case in the ‘Glivec’ matter has brought the Indian patent system into sharp focus, whereas Indian regulatory authority should reform new rules for granting patent so that bigger MNCs should be attracted to India in future for better business [18–20].
Recent patents
With expiration of patent branded drugs are applied for generics version, some of the new ANDAs approval in year 2013 [17] are described briefly in Table 4.
CONCLUSION
In situations where demand for medicines exceeds supply, and cost effective drug in demand with minimum expenditure, generic drug are best choice fulfilling this demand. The current and future prospective of generics in India and US is very bright as Indian government looking towards generic drugs for providing better health care to public. Indian pharmaceutical industries grow rapidly all over the world and one of largest generic exporter in world where as, US being the major destination for export. Thus, the proper validated regulation is required for manufacturing generic drugs in India and US which requires proper symbiotic relation between India and US. Some amendments are warranted in Hatch Waxman Act 1984 for developing generic drug in better way, where as re-election of Barack Obama in US provides positive increase in generic market as his government extending health care insurance for additional 30 million Americans in the health care ambit, creating increased demand for generics.
REFERENCES
- http://www.financial express.com/news/Indian-pharma-exports-may-grow-by-20 pct/8397241.
- Ramesh T, Saravanan V, Khullar D (2011) Regulatory perspective for entering global pharmamarkets. Pharma-time 43.
- Gattani; “Branded to generic drugs”. The Indian pharmacist, June 2012.
- Indian Pharma Industry: SWOT Analysis; Internet report, June, 2009.
- Yourlegalhelp.com/generic-drug…liability…/1879.
- NYTimes.com/pharmaceutical/justices-to-take-up-case-on-generic-drug-makers- liability.html.
- http://www.npr.org/Nina Totenberg/Supreme Court Hears ‘Pay to Delay’ Pharmaceutical Case.
- Katherine Eban /Dirty medicine-Fortune Features.htm.
Pharma Regulations for Generic Drug Products in India and US: Case Studies and Future Prospectives
Suryakanta Swain*, Ankita Dey, Chinam Niranjan Patra and Muddana Eswara Bhanoji Rao
Roland Institute of Pharmaceutical Sciences, Department of Pharmaceutics, Berhampur, Odisha, India
Suryakanta Swain
Assistant Professor
Roland Institute of Pharmaceutical Sciences
Department of Pharmaceutics
P.O.: Khodasingi, Berhampur-7600 10, Odisha, India
Tel: 91-943-803-8643; 909-037-4275
E-mail: swain_suryakant@yahoo.co.in
Citation: Swain S, Dey A, Patra CN, Bhanoji Rao ME (2014) Pharma Regulations for Generic Drug Products in India and US: Case Studies and Future Prospectives. Pharmaceut Reg Affairs 3:119. doi: 10.4172/2167-7689.1000119
|
Suryakanta Swain |
|
| Biography | |
| Dr. Suryakanta Swain was born on 8th June 1980 in Debendrapur, Balasore, Odisha (INDIA). After completing his B. Pharm with 79.37% from Berhampur University, Odisha, India and join in to M. Pharm (Pharmaceutics) by qualifying GATE and N.I.P.E.R with All India entrance examinations with C.G.P.A 8.89 from Biju Patnaik University of Technology, Rourkela, Odisha, India. He is completed his Ph.D in Pharmacy from Berhampur University on 09.12.2013. He started his career as a Research Trainee Executive in Formulation Research & Development in Medley Pharmaceuticals Pvt. Ltd, Daman, India. Presently he is working as Asst. Professor-cum-Placement Officer in Department of Pharmaceutics at Roland Institute of Pharmaceutical Sciences, Berhampur, Odisha, India. So far he has published thirty articles of reputed national & international journals with high indexing or impact factor. He has edited one book, authored four books & one book chapter an international level. He has filled One Indian patent. He has permanent Editor, Advisary, Editorial board members and reviewers in more than 15 national & international journals. | |
| Research Interest | |
| Mucoadhesive DDS, Transdermal DDS, Liposomal DDS, Selfemulsifying DDS, Micro and Nanoparticulate DDS, Gastro-Intestinal DDS, Colon Specific DDS and Controlled DDS. | |
| Publications | |
| Solid Lipid Nanoparticle: An Overview | |
| Suryakanta Swain and Sitty Manohar Babu | |
| Editorial: Pharmaceut Reg Affairs 2015, 4: e154 | |
| doi: 10.4172/2167-7689.1000e154 | |
| Pharmaceutical Impurities and Degradation Products: An Overview | |
| Prafulla Kumar Sahu, Suryakanta Swain and Manohar Babu S | |
| Editorial: Pharmaceut Reg Affairs 2015, 4: e146 | |
| doi: 10.4172/2167-7689.1000e146 | |
| Impact of Pharmacovigilance in Healthcare System: Regulatory Perspective | |
| Suryakanta Swain and Chinam Niranjan Patra | |
| Editorial: Pharmaceut Reg Affairs 2014, 3: e143 | |
| doi: 10.4172/2167-7689.1000e143 | |
| Bio-Relevant and Bioequivalence Studies: An Overview | |
| Suryakanta Swain and Nerella Nagadivya | |
| Editorial: Pharmaceut Reg Affairs 2014, 3: e140 | |
| doi: 10.4172/2167-7689.1000e140 | |
Roland Institute of Pharmaceutical Sciences, Department of Pharmaceutics, Berhampur, Odisha, India






 /////////Abbreviated new drug application approvals, Cases and incidents, Pharma regulations, Recent patents, Roland Institute of Pharmaceutical Sciences, Department of Pharmaceutics, Berhampur, Odisha, India
/////////Abbreviated new drug application approvals, Cases and incidents, Pharma regulations, Recent patents, Roland Institute of Pharmaceutical Sciences, Department of Pharmaceutics, Berhampur, Odisha, India
Process Development for Low Cost Manufacturing

-
Process Development for Low Cost Manufacturing on 23-24 nov 2015 , Hyderabad, INDIA
- 23.11.2015 – 24.11.2015


- Hotel Green Park – Hyderabad, India

- View Brochure
- or click
- https://scientificupdate.co.uk/index.php/training/scheduled-training-courses/details/213-Low-Cost-Manufacturing.html?utm_source=Scientific+Update+News&utm_campaign=d31c03d4e3-India_Courses_Hyderabad7_22_2015&utm_medium=email&utm_term=0_08c5e1fb69-d31c03d4e3-78584097
Chemical process research and development is recognised as a key function during the commercialisation of a new product particularly in the generic and contract manufacturing arms of the chemical, agrochemical and pharmaceutical industries.
The synthesis and individual processes must be economic, safe and must generate product that meets the necessary quality requirements.
This 2-day course presented by highly experienced process chemists will concentrate on the development and optimisation of efficient processes to target molecules with an emphasis on raw material cost, solvent choice, yield improvement, process efficiency and work up, and waste minimisation.
Process robustness testing and reaction optimisation via stastical methods will also be covered.
A discussion of patent issues and areas where engineering and technology can help reduce operating costs.
The use of engineering and technology solutions to reduce costs will be discussed and throughout the course the emphasis will be on minimising costs and maximising returns.



///////////
Gnidia glauca

Introduction
Search of complementary and alternative medicine has gained a thrust in the recent decade due to the pronounced side effects and health hazards of the chemically synthesized drugs. Hereby, a comprehensive knowledge about the traditionally used medicinal plants is indispensable for exploration of its novel bioactive components. One of such comparatively less explored medicinal plant is Gnidia glauca. Although, it has folkloric, traditional phytomedicinal and agrochemical applications in various parts of the world, still there are no available scientific validations or evidences to support the fact. In African medicine it is used for treatment of abdominal pain, cancers, wounds, snake bites, sore throat and burns. It is also well known for its piscicidal, insecticidal, molluscicidal and even homicidal activity for its use as arrow poisons. Similarly, its antineoplastic activity is reported to be remarkably superior [1]. However, till date there is no comprehensive information on the plant.

In view of the background, herein we present the first commentary on complete research carried out till date on G. glauca and its promises as complementary and alternative medicine (Figure 1).
Antimicrobial Activity
Plant pathogenic fungi are major cause of heavy losses in the crop yield as well as the economic turnover of the farmers. Hereby, development of eco-friendly herbal and cheap antifungal agents is of utmost importance. Aqueous extracts of various parts of G. glaucaexhibited variable mycelia inhibition against Phytophthora parasitica, a plant pathogenic fungi causing heart rot in pineapple. At a concentration of 5% the G. glauca seeds, leaves and barks showed an inhibition upto 19.16, 15.90 and 23.46%, respectively. Similarly, an enhanced activity was observed with a higher concentration at 10%, equivalent to 28.47, 34.59, 33.60% for seed, leaves and bark respectively [2]. A significant anticariogenic activity against Streptococcus mutans by the methanolic extract of G. glauca leaves was reported recently. The active extracts showed a high total phenolic (126.25 ± 0.20 μg GAE/ mg) and flavonoid (25.75 ± 0.10 μg CE/mg) content [3]. G. glauca bark extract is reported to have superior antibacterial activity against urinary tract infection causing pathogens likeEscherichia coli, Klebsiella pneumoniae, Pseudomonas aeruginosa, Staphylococcus aureus and Enterococcus faecalisas compared to leaf and flower [4].

Back Ache and Joint Ache
According to ethnobotanical information, roots of G. glauca are widely used as a traditional medicine in Embu and Mbeere districts, Eastern Province of Kenya for treatment of back ache and joint ache [5].
Insecticidal and Larvicidal Activity
Leaves of G. glauca are used in Kenya as insecticidal agent [5]. Sequentially extracted hexane and chloroform extracts of dried bark ofG. glauca exhibited moderate mosquito larvicidal activity, whereas hexane, choloroform and MeOH extracts of fresh bark of the plant showed superior larvicidal activity against second instar larvae of Aedes aegypti. Maximum activity upto 100 % mortality was exhibited by the chloroform extract of fresh bark within a few minutes. Bioassay guided fractionation confirmed that compounds like bicoumarin and Pimelea factor P2 are mostly responsible for larvicidal activity [6]. Aqueous extract of G. glauca leaf and bark showed a notable ovicidal activity against the eggs of teak defoliator, Hyblaea puera Cramer upto 44.4 and 45.7 %, respectively [7]. In order to check the antileukemic and piscicidal activity of G. glauca, dried ground roots were extracted at room temperature with 95% ethanol under stirring condition for 24 h. The extract was further partitioned in various proportion of chloroform – water mixture to yield the gold fish piscitoxic fraction identified as gnidiglaucin (C32H46O10 ). However, the isolated compound failed to show inhibitory activity in in-vivo assay for antileukemic activity (P- 388) [8].
Antiviral
A recent ethnobotanical study on medicinal plants used by people in Zegie, Peninsula, Northwestern Ethiopia revealed that the root powder of G. glauca mixed with skimmed milk is taken orally for seven days for treatment of rabies [9].
Antioxidant Activity
The methanolic extract of G. glauca leaf with high antioxidant activity showed major phenolic content of 203.3 GAE/g. It could scavenge both ABTS (IC50 = 16.3 μg/mL) and nitric oxide (IC50 = 360.8 μg/mL) radicals. Further, FRAP value of 993.7 μm TE/mg was recorded at 30 min and 142.5 mg AAE/g of total antioxidant activity was evaluated [1]. In our previous report as well, we observed similar trend where the alcoholic extracts of G. glauca leaf showed high phenolic and flavonoid content. In case of pulse radiolysis generated hydroxyl radical scavenging second order rate constants of ethanolic extracts of G. glauca flower (4×106) was found to be very high indicating superior activity, followed by its leaf (3.73×106) and stem (3.66×106). Methanol extract of leaf showed efficient scavenging activity against DPPH radical, super oxide and nitric oxide radicals [10].
Antidiabetic Activity
Metabolic enzymes, like α-amylase and α-glucosidase are considered as key targets for discovery of antidiabetic drugs. Ethanolic, methanolic and ethyl acetate extracts of G. glauca flowers showed an excellent inhibitory potential (~70 % and above) against α-amylase while only methanol extract of leaf showed high inhibition against α-glucosidase [11].
Nanobiotechnology
The higher content of phenolics and flavonoids is responsible for the synthesis of gold nanoparticles by G. glauca flower extract. It showed one of the most rapid routes for synthesis to be completed entirely within just 20 min. The resulting AuNPs were small spheres with a diameter of 10 nm in majority. Exotic shapes like nanotriangles were also observed employing high resolution transmission electron microscopy along with other characterization tools. These AuNPs exhibited excellent catalytic properties in a reaction where 4-nitrophenol is reduced to 4-aminophenol by NaBH4 [12].
Toxicology Study
Toxicology studies to establish the safety of methanolic extract of G. glauca barks and roots involved the evaluation of acute oral toxicity in female rats. Neither mortality, nor morbidity was observed at administered dosages of 175, 550 and 2000 mg/kg body wt., which reveal the safety of these extracts in the doses up to 2000 mg/kg body weight. This study establishing that an LD50 value of G. glauca bark and root extracts, higher than 2000 mg/kg body weight is definitely advantageous for its clinical studies [13]. Thus it provided the scientific rationale supporting the wide usage of G. glauca for diverse therapeutic purposes [14].
Conclusion
G. glauca being one of the very important ethnomedicinal plant, will continue to be explored by researchers from various disciplines. In near future scientific discoveries, adding newer attributes to its therapeutic spectrum will surely enable it to emerge as one of the very vital model system, pivotal to many field of research like, pharmacognosy, pharmacy, phytochemistry, drug discovery and nanobiotechnology.
References
- Rao SB, Jayanthi M, Yogeetha R, Ramakrishnaiah H, Nataraj J (2013) Free radical scavenging activity and reducing power of Gnidia glauca (Fresen.) Gilg. J App Pharm Sci 3: 203-207.
- Naik ST, Maheswarappa V (2007) Prospects of using plant extracts in management of pineapple heart rot. Karnataka J AgricSci 20: 180-182.
- Junaid S, Dileep N, Rakesh KN, Pavithra GM, Vinayaka KS, et al. (2013) Anticariogenic activity of Gnidia glauca (Fresen.) Gilg, Pothosscandens L. and ElaegnuskologaSchlecht. J App Pharm Sci 3: 020-023.
- Prashith KTR, Vivek MN, Junaid S, Rakesh KN, Dileep N, et al. (2014) Inhibitory activity of Polyalthialongifolia, Anaphalislawii andGnidia glauca against Colletotrichumcapsici and urinary tract pathogens. SciTechnol Arts Res J 3: 26-30.
- Kareru PG, Kenji GM, Gachanja AN, Keriko JM, Mungai G (2006) Traditional medicines among the Embu and Mbeere peoples of Kenya. Afr J Tradit Complement Altern Med 4: 75-86.
- Amarajeewa BWRC, Mudalige AP, Kumar V (2007) Chemistry and mosquito larvicidal activity of Gnidia glauca. Proceedings of the Peradeniya University Research Sessions, Sri Lanka, 12: 101-102.
- Kupchan SM, Shizuri Y, Sumner WC Jr, Haynes HR, Leighton AP, et al. (1976) Isolation and structural elucidation of new potent antileukemicditerpenoid esters from Gnidia species. J Org Chem 41: 3850-3853.
- Ghosh S,Derle A,Ahire M, More P,Jagtap S, et al. (2013) Phytochemical analysis and free radical scavenging activity of medicinal plants Gnidia glauca andDioscoreabulbifera. PLoS One 8: e82529.
- Ghosh S,Ahire M, Patil S, Jabgunde A, BhatDusane M, et al. (2012) Antidiabetic Activity of Gnidia glauca and Dioscoreabulbifera: Potent Amylase and Glucosidase Inhibitors. Evid Based Complement Alternat Med 2012: 929051.
- Ghosh S, Patil S, Ahire M, Kitture R, Gurav DD, et al. (2012) Gnidia glauca flower extract mediated synthesis of gold nanoparticles and evaluation of its chemocatalytic potential. J Nanobiotechnology 10: 17.
- Khadke SS, Pachauri DR, Mahajan SD.(2011) An acute oral toxicity study of Gnidia glauca (Fresen.) Gilg. in albino rats as per OECD guideline 425. Int J PharmTech Res 3: 787-791.




Professor B.A. Chopade
M.Sc., Ph.D. Nottingham University, England
Fogarty Fellow Illinois University, Chicago, USAVice- Chancellor
Fogarty Fellow Illinois University, Chicago, USAVice- Chancellor
Dr. Babasaheb Ambedkar Marathwada University
Aurangabad-431004
Maharashtra State, India
Ph.No. : (office) (0240)-2403111 Fax No. (0240)-2403113/335
Aurangabad-431004
Maharashtra State, India
Ph.No. : (office) (0240)-2403111 Fax No. (0240)-2403113/335
E-mail : vc@bamu.ac.in

Balu A Chopade
Professor B.A. Chopade has been working as a Vice-Chancellor of Dr. Babasaheb Ambedkar Marathwada University, Aurangabad, Maharashtra from 04/06/2014. He has been working as Professor of Microbiology and Coordinator of University of Potential Excellence Programme (UPE Phase I & II) of UGC in Biotechnology at University of Pune. He was Director of Institute of Bioinformatics and Biotechnology (IBB), University of Pune from 2006 to 2012. He has established and developed IBB as a unique national institute and as a centre of excellence in research, innovation and teaching in biotechnology in India. He has successfully established an innovative benchmarking of publications in peer reviewed international journals of repute by undergraduate students at IBB. He was Head of the Department of Microbiology, University of Pune from 1994, 1996-2000 and 2003-2006. He has 35 years of experience in research, innovation, teaching and administration at the University of Pune.
Professor Chopade has several national and international academic honors and professional distinctions to his credits. He was the Government of India Scholar at the University of Nottingham, England and obtained a Ph.D. degree in microbiology and molecular genetics (1983-1986). He was also the recipient of the most prestigious Fogarty International NIH Research Fellowship Award from Govt. of USA for Post-Doctoral Research at the University of Illinois at Chicago (1994-1996) in genetic engineering. He is also recipient of International Award in Microbiology from International Union of Microbiological Societies (IUMS) in 1986. He has had very distinguished academic career and has carved his career entirely on the basis of merit and academic excellence.He was also coordinator of ALIS link programme between British Council London and Department of Microbiology, University of Pune (1994-1997).
He has published more than 100 research papers in peer reviewed international and national journals with high impact factor. The total impact factor of his research is more than 260, with h-index 26 and i10-index 52. His work is cited more than 2002 times (www.scholar.google.com). He has obtained 2 USA and 8 Indian patents. His research work has been cited by Nobel Laureate Professor Arthur Kornberg from University of Stanford, California, USA. His pioneering work on e-DNA and Acinetobacter vesicles is also cited by “Nature” journal from England. His work also has been cited in 3 textbooks of microbiology published from USA and Europe. He has presented more than 150 papers in International and National Conferences and has given large number of plenary lectures. He has successfully supervised 27 Ph.D., 4 M.Phil. and 10 Post Doctoral scholars for their research. Currently 2 Post-Doctoral Fellows and 4 Ph.D. students are working with him. His 3 students had obtained Young Scientist Awards in 1993 at Stockholm, from International Congress of Chemotherapy (ICC), Europe. His research area includes microbial and molecular genetics, biotechnology and nanomedicine. He is on editorial board of Wealth of India Publication series, from CSIR New Delhi, as well as number of research journals. He has obtained research grants and funding of more than rupees 10 crores from national and international funding agencies. He has successfully completed 32 major research projects from various National and International funding agencies. He has developed a new herbal medicine “Infex” which is manufactured by Shrushti Herbal Pharma Ltd., Bangalore. He is a pioneer in the area of Industry-Academia interactions and entrepreneurship in biotechnology and microbiology at IBB, University of Pune.
He was a visiting scientist at the Pasteur Institute, Paris, France and King’s College, University of London in 1990. He has received number of awards and most notable are: Pradnya Bhushan Dr. Babasaheb Ambedkar Award(2014) Aurangabad. Bronze Medal, International Genetically Engineered Machines (iGEM), Massachusetts Institute of Technology (MIT), USA (2009), Pradnyavant Award (2011) by Undalkar Foundation, Karad. Maharashtra, Best teacher award by Pune Municipal Corporation (1993); Best research paper awards in microbial and molecular genetics (1988 & 2002) by Association of Microbiologist of India; He was recipient of Wadia Oration award (2008) by Institution of Engineers, India. Best research paper award in Bioinformatics (2009) by SBC, India. Summer Fellowship of Indian Academy of Sciences, Bangalore (2001). His biography is published by American Biographical Institute, USA (2000) and International Biographical Centre Cambridge (1991). He is member of American Society for Microbiology, USA and Society for General Microbiology, England since 1984. He is also a life member of number of national organizations like Association of Microbiologist of India (AMI) and Biotechnology Society of India (BSI), Society of Biological Chemists of India (SBC) Indian Science Congress (ISC). He is recipient of Marcus’s Who’s Who in Science and Engineering U.S.A. (2001), Marcus’s Who’s Who of the World, U.S.A. (2000), Marcus’s Who’s Who in Medicine, U.S.A. (2002), Marcus’s Who’s Who in Education, U.S.A. (2002).
He has been working on various authorities of University of Pune, as well as many State and Central Universities in India. Such as, Chairman, Board of Studies in Microbiology from 1997-2000 & 2005-2007. Member, BOS in Biotechnology (2005-2006, 2012-2017), Member of Academic Council (1997-2000 & 2000-2005) and Board of College and University Development (BCUD) of University of Pune from 1997-2000 & 2000-2005. Member, Faculty of Science, University of Pune (1997-2000, 2003-2005) and Member, Board of Teaching and Research (BUTR), (1997-2002). Member, Board of studies in Biochemistry and Molecular Biology, Central University Pondichery (2001-2003). Member, Board of Studies in Biochemistry and Molecular Biology, Shivaji University Kolhapur (2009-2014). Member, Board of Studies in Life Sciences, North Maharashtra University, Jalgaon (1994-1999). Member, Faculty of Science, Bharti Vidyapeeth Pune (2013-2018). Member, Faculty of Science, North Maharashtra University, Jalgaon (1990-1999).
He was chairman of large number of committees of UGC, New Delhi such as 11th Plan Research Committee, Research Projects and Deemed University Status since 2008. Chairman, International Travel Grants, (2008-2013). He was Chairman of State Eligibility Test (SET) in Microbiology for Govt. of Maharashtra and Goa from (1997-2000). He has active an involvement in the national and international scientific organizations. He has been involved in University administration in the various capacities for more than 33 years, as a chairman and member of large number of development, finance, examination and administration committees of University of Pune.
He is member of research and recognition committees of numerous state and central universities in India. He also worked as a coordinator of DBT Potential Excellence Programme at the Department of Microbiology, University of Pune (1994-1998). He is nominee of Department of Biotechnology, Government of India for Reliance Industries limited Mumbai, Biorefinery of Somaiya Group of Industries in Karnataka and Agharkar Research Institute (ARI) Pune.
His vision for Dr.Babasaheb Ambedkar Marathwada University (BAMU), Aurangabad is to transform it as one of the best research and innovation Universities in India and subsequently develop as a world class University.
///////Gnidia glauca, Phytochemistry, Ethnomedicine, Dr. Babasaheb Ambedkar Marathwada University, Aurangabad, Maharashtra, india, Balu A Chopade
Indian pharma’s struggle to tighten standards paves way for M&A deals

People walk past a chemist shop at a market in Mumbai. Photo: Reuters
MUMBAI – India’s smaller generic drugmakers, struggling to cope with a bruised reputation and tougher regulation in the United States, are under pressure to consider branching out to new, less-profitable markets or sell out to larger rivals.
Two years after its most high-profile regulatory setback to date in the United States – Ranbaxy’s $500 million U.S. fine for drug safety violations – India’s $15 billion a year generic drug industry is still rebuilding its image in its biggest market.
Many of its top firms are facing sanctions at some of their factories, as the U.S. Food and Drug Administration (FDA) tightens checks and its approvals process.
Combined with government-mandated price controls on drugs at home, that is piling pressure on smaller players.
“If they want to have a presence globally, they have to make investments. If they can’t, then they’ll have to focus on other markets or scale back their ambition outside of India, and that’s probably what will happen,” said Subhanu Saxena, CEO of Cipla , India’s fourth-largest drugmaker by revenue.
Ashok Anand, president of Hikal Ltd , a Mumbai-based drugmaker with a market value of $167 million, said some peers were putting themselves on the block.
“If they cannot deal with the stricter regulations, they might just prefer to sell out,” he said.
Pressure on U.S. sales has been felt across the Indian industry, with all drugmakers hit by delays in FDA approvals as the U.S. safety body overhauls its review process. Growth in U.S. revenue for drugmakers slowed to 14 percent in the year to March 2015, less than half what it was in the year to March 2012, according to brokerage Edelweiss.

But for larger players who want to plug gaps or, for the likes of Glenmark and Aurobindo who aim to grow in the United States, this pressure has lowered prices and could pave the way for attractive deals, bankers said.
“Now that some of the smaller companies are reeling under intensive regulatory scrutiny and want to cash out on their investments, valuations would be much more realistic,” said the head of India M&A at a large European bank in Mumbai.
SPENDING SPREE
Indian manufacturers say they have spent millions in high-end testing equipment, improved training and have hired larger teams in quality control since Ranbaxy was fined for manipulating clinical data.
Some consultants estimate spending on compliance has more than doubled to reach about 6 to 7 percent of sales for the larger companies.
But while the number of U.S. export bans issued to Indian companies fell to eight in 2014 from 21 in 2013, according to FDA data, the agency continues to find manufacturing violations at the plants of some of the biggest drugmakers in the country, an indication of the pervasiveness of the problem.
Sun Pharmaceutical Industries , Wockhardt , Dr Reddy’s Laboratories and Cadila Healthcarehave all faced FDA rebukes over the past year.
Smaller firms Ipca and Aarti Drugs faced FDA bans on their plants this year.
These failures – which executives blame on India’s “quick fix” culture and consultants blame on a failure to prioritize compliance – have clouded short-term growth prospects and added to pressure on smaller players, pushing some to look elsewhere.
“They can choose to be in lesser-regulated markets, such as Latin America, where there is a lot of demand. But they will have to live with much thinner margins,” said the finance director of a small Indian drugmaker, who did not want to be named. “It’s survival of the fittest.” REUTERS
http://m.todayonline.com/business/indian-pharmas-struggle-tighten-standards-paves-way-ma-deals
///////
AN IMPROVED PROCESS FOR THE PREPARATION OF DOLUTEGRAVIR

 Aurobindo Pharma MD and CEO N. Govindarajan at a company research centre. “It [the transition] is purely driven by the need to get more into areas where there is scope for better profit margins,
Aurobindo Pharma MD and CEO N. Govindarajan at a company research centre. “It [the transition] is purely driven by the need to get more into areas where there is scope for better profit margins,
Dolutegravir (I) is chemically known as (4/?,12aS)-N-[(2,4-difluorophenyl)methyl]-3,4,6,8,12,12a-hexahydro-7-hydroxy-4-methyl-6,8-dioxo-2//-pyrido[r,2′:4,5]pyrazino[2,l-b][l,3]oxazine-9-carboxamide. Dolutegravir is a human immunodeficiency virus type 1 (HIV-1) integrase strand transfer inhibitor (INSTI) indicated in combination with other antiretroviral agents for the treatment of HIV-1 infection. Dolutegravir is being marketed under the trade name Tivicay®. US 8,129,385 disclosed Dolutegravir or its pharmaceutically acceptable salts thereof. US ‘385 also discloses a process for the preparation of Dolutegravir (I). The process involves the condensation of 5-benzyloxy-4-hydroxy-6-hydroxymethyl nicotinic acid (II) with 2,4-difluorobenzylamine (III) to produce 5-benzyloxy-N-(2,4-difluorobenzyl)-4-hydroxy-6-hydroxymethyl nicotinic acid amide (IV), which is further under goes oxidation using manganese dioxide (Mn02) to produce 5-benzyloxy-N-(2,4-difluorobenzyl)-6-formyl-4-hydroxy-nicotinic acid amide (V). This amide compound (V) is reacted with sodium chlorite (NaClCh) to produce 3-benzyloxy-5-(2,4-difluorobenzylcarbamoyl)-4- hydroxy-pyridine-2-carboxylic acid (VI), which is further treated with methanol (MeOH) to produce 3-benzyloxy-5-(2,4-difluorobenzyl)-4-hydroxy-pyridine-2-carboxylic acid methyl ester (VII).

The methyl ester compound (VII) is reacted with 3-bromopropene to produce l-allyl-3-benzyloxy-5-(2,4-difluorobenzyl)-4-oxo-l,4-dihydro-pyridine-2- carboxylic acid methyl ester (VIII), which is further reacted with potassium osmate dihydrate (K2OSO4.2H2O) to produce 3-benzyloxy-5-(2,4-difluorobenzylcarbamoyl)-4-oxo-l-(2-oxo-ethyl)-l,4-dihydropyridine-2-carboxylic acid methyl ester (IX). The compound (IX) is reacted with (R)-3-amino-l-butanol (X) to produce benzyloxy Dolutegravir (XI), which is deprotected by treating with TFA to produce Dolutegravir (I). The process is as shown in scheme-I below:
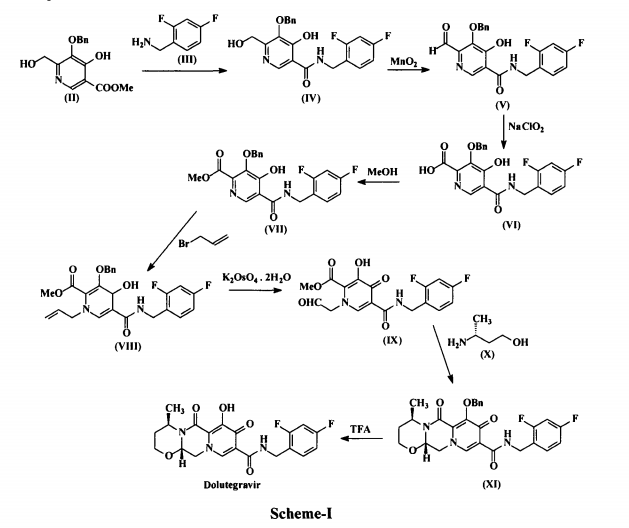
The major disadvantage with the above prior-art process is that it involves large no of steps and tedious work-up procedures to isolate the required product. This results a longer period of time cycle is required to produce Dolutegravir (I), which in turn renders the process more costly and less eco friendly. Further the above processes are low yielding and with less purity. US 8,217,034 discloses variant process for the preparation of Dolutegravir.
This process involves the reaction of methyl l-(2,2-dihydroxyethyl)-4-oxo-3-[(phenylmethyl)oxy]-l,4-dihydro-2-pyridine carboxylate (XII) with (R)-3-amino-l-butanol (X) to produce (4R, 12o5)-4-methyl-7-[(phenylmethyl)oxy]-3,4,12,12a-tetrahydro-2//-pyrido[ 1 \2′,4,5] pyrazino[2,l-b][l,3]oxazine-6,8-dione (XIII), which is further undergoes bromination using NBS to produce (4R,12aS)-9-bromo-4-methyl-7-[(phenylmethyl)oxy]-3,4,12,12a-tetrahydro-2H-pyrido[r,2′:4,5]pyrazino[2,l-b][l,3]oxazine-6,8-dione (XIV). The bromo Compound (XIV) is condensed with 2,4-difluorobenzylamine (III) in the presence of Tetrakis(triphenylphosphine)palladium (Pd(PPh3)4) to produce benzyloxy Dolutegravir (XI), which is hydrogenated in the presence of Pd/C to produce Dolutegravir (I). The process is as shown in Scheme-II below:

The major disadvantage with the above prior art process of preparing Dolutegravir is the use of expensive reagent tetrakis(triphenylphosphine)palladium (Pd(PPh3)4> in coupling step. Use of this reagent on industrial scale is not preferred, which makes the process more expensive. WO 2011/119566 discloses another variant process for the preparation of Dolutegravir.
This process involves the reaction of l-(2,2-dimethoxyethyl)-5-methoxy-6-(methoxycarbonyl)-4-oxo-l,4-dihydropyridine-3-carboxylic acid (XV) with acetic acid in presence of methane sulfonic acid to produce 5-methoxy-6-(methoxycarbonyl)-4-oxo-l-(2-oxoethyl)-l,4-dihydropyridine-3-carboxylic acid (XVI), which is further condensed with (R)-3-amino-l-butanol (X) to produce (4R,12aS)-7-methoxy-4-methyl-6,8-dioxo-3,4,6,8,12,12a-hexahydro-2//-pyrido[ 1 ‘,2’:4,5]pyrazino[2,1 -b] [ 1,3]-oxazine-9-carboxylic acid (XVII). This acid Compound XVII is acylated with 2,4-difluorobenzylamine (III) in the presence of carbonyldiimidazole (CDI) to produce methoxy Dolutegravir (XVIII), which is demethylated in the presence of lithium bromide (LiBr) to produce Dolutegravir (I).
The process is as shown in Scheme-3 below:
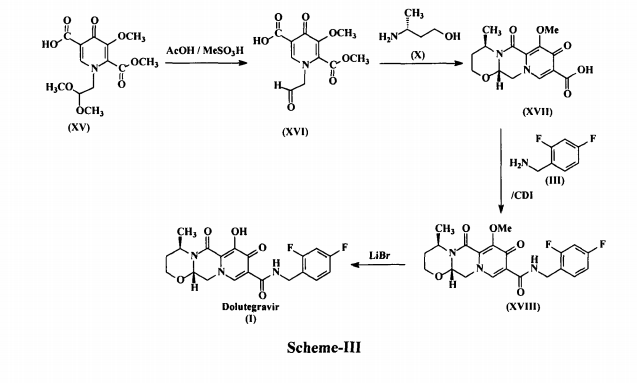
The major disadvantage of the above prior art process of preparing Dolutegravir is the use of expensive and highly moisture sensitive reagent, 1,1-carbonyldiimidazole (CDI), during acylation. Use of this reagent on industrial scale is not preferred due to anhydrous conditions required in the process. However, there is always a need for alternative preparative routes, which for example, involve fewer steps, use reagents that are less expensive and/or easier to handle, consume smaller amounts of reagents, provide a higher yield of product, have smaller and/or more eco-friendly waste products, and/or provide a product of higher purity. Hence, there is a need to develop cost effective and commercially viable process for the preparation of Dolutegravir of formula (I). The present invention is related to a process for the preparation of pure Dolutegravir of formula (I), wherein optically active acid addition salt of (R)-3-amino-l-butanol (X) is directly condensed with 5-methoxy-6-(methoxycarbonyl)-4-oxo-l-(2-oxoethyl)-l,4-dihydropyridine-3-carboxylic acid (XVI) instead of condensing with free base of (R)-3-amino-1-butanol (X). The present invention is also related to a process for the preparation of pure Dolutegravir of formula (I), wherein, inexpensive and easily handling condensing reagents in the condensation of (4R, 12aS)-7-methoxy-4-methyl-6,8-dioxo-3,4,6,8,12,12a-hexahydro-2//-pyrido[l’,2′:4,5]pyrazino [2,l-b][l,3]oxazine-9-carboxylic acid (XVII) with 2,4-difluorobenzylamine (III).


AN IMPROVED PROCESS FOR THE PREPARATION OF DOLUTEGRAVIR
| APPLICATION NUMBER | 1361/CHE/2013 |
| APPLICANT NAME | AUROBINDO PHARMA LTD |
| DATE OF FILING | 27/03/2013 |
| PUBLICATION DATE (U/S 11A) | 16/01/2015 |
In another embodiment, 5-methoxy-6-(methoxycarbonyl)-4-oxo-l-(2-oxoethyl)-l,4- dihydropyridine-3-carboxylic acid (XVI) used in the present invention is prepared by reacting 4-methoxyacetoacetate (XIX) with N,N-dimethyl-l,l- bis(methyloxy)methanamine (DMF-DMA) (XX) to produce methyl-2- (dimethylaminomethylene)-4-methoxy-3-oxo-butanoate(methyl-3-(dimethylamino)-2 [(methyloxy)acetyl]-2-propenoate) (XXI), which is reacted with aminoacetaldehyde dimethyl acetal (XXII) to produce methyl-2-(2,2-dimethoxyethylaminomethylene)-4-methoxy-3-oxo-butanoate(methyl-3-{[2,2-bis(methyloxy)ethyl]amino}-2-[(methyloxy) acetyl]-2-propenoate) (XXIII).
The compound (XXIII) is contacted with dimethyl ethanedioate in presence of alkali metal alkoxide to produce dimethyl-1-(2,2-dimethoxyethyl)-3-methoxy-4-oxo-l ,4-dihydropyridine-2,5-dicarboxylate (XXIV), which is selectively hydrolyzed with a base to produce l-[2,2-bis(methyloxy)ethyl]-5-(methyloxy)-6-[(methyloxy)carbonyl]-4-oxo-l ,4-dihydro-3-pyridinecarboxylic acid (XV). The compound (XV) is treated with a catalytic amount of a strong protic acid in the presence of acetic acid in an organic solvent to produce a reaction mixture containing 5- methoxy-6-(methoxycarbonyl)-4-oxo-l-(2-oxoethyl)-l,4-dihydropyridine-3-carboxylic acid (XVI), The process is as shown in Scheme-IV below:

The following examples illustrate the nature of the invention and are provided for illustrative purposes only and should not be construed to limit the scope of the invention.
Example-1:
EXAMPLES: Example-1: Process for the preparation of Dolutegravir
Step-i: Preparation of (/?)-3-amino-l-butanol tartarate salt: D-(+) Tartaric acid (12.7 g, 0.085 mol) was added in to a solution of (i?,5)-3-amino-l-butnaol (7.5 g, 0.084 mol) in methanol (100 ml) at 40 °C. The reaction mixture was stirred for about 1 hour at 35-40 °C and the reaction mass was cooled to 0-5°C and maintained for 30-40 minutes. The obtained solid was filtered and washed with chilled methanol (10 ml) at 0-5 °C. The solid was dried to get (i?)-3-amino-l-butanol tartarate salt (8.0 g, 40%).
Step-ii: Preparation of (4rt,12a£)-7-methoxy-4-methyl-6,8-dioxo-3,4,6,8,12,12a-hexahydro-2H-pyrido[l’,2′;4,5]pyrazino[2,l-b][l,3]oxazine-9-carboxylic acid (XVII): l-[2,2-Bis(methyloxy)ethyl]-5-(methyloxy)-6-[(methyloxy)carbonyl]-4-oxo-l,4-dihydro-3-pyridinecarboxylic acid (XV) (lOOg; 0.3175 moles) was suspended in acetonitrile (800 ml) and heated to 80-82°C. A mixture of acetic acid (95.25 g), methanesulfonic acid (9.14 g; 0.09525 moles) and acetonitrile (200 ml) were added to the slurry at 80-82°C. The reaction mass was continued at 80-82°C to complete the reaction. After completion of the reaction, anhydrous sodium acetate (65 g) and (/?)-3-amino-l-butanol tartrate salt (79.68g; 0.3334 moles) were added at 20-25°C and stirred at 60-65°C to complete the reaction. The reaction mass was concentrated and acidified with IN aqueous hydrochloric acid (750 ml) and extracted with methylene chloride (1500 ml) at ice cold temperature. The organic layer was separated, concentrated, treated with hot methanol (350 ml) for 2 h, filtered, washed with methanol and dried to yield (4R,12aS)-7-methoxy-4-methyl-6,8-dioxo-3,4,6,8,12,12a-hexahydro-2H-pyrido[ 1′ ,2′ :4,5]pyrazino[2,1 -b] [ 1,3]oxazine-9-carboxylic acid (XVII) (72 g; HPLC purity: 99.07%).
Step-iii: Process for the preparation of Dolutegravir (I). Method A: Triethylamine (3.61 g; 0.0357 moles) was added to the suspension of (4R,12aS)-7- methoxy-4-methyl-6,8-dioxo-3,4,6,8,12,12a-hexahydro-2H-pyrido[ 1′ ,2′ :4,5]pyrazino[2,1 – b][l,3]oxazine-9-carboxylic acid (XVII) (10 g; 0.0325 moles) in methylene chloride (50 ml), and cooled to 10-15°C. Pivaloyl chloride (4.3 g; 0.0357 moles) was added to the reaction mass, and stirred at 10-15°C for 1 h. Thereafter, 2,4-difiuorobenzylamine (5.58 g; 0.0389 moles) was added at 10-15°C and then warmed to 20-25°C to complete the reaction. After completion of the reaction, IN aqueous hydrochloric acid (20 ml) was added, organic layer was separated, washed with 5% w/w aqueous sodium bicarbonate solution (10 ml) followed by 15% w/w aqueous sodium chloride solution (10 ml) and concentrated. To the concentrated mass, acetonitrile (100 ml) and Lithium bromide (5.08 g; 0.0584 moles) were added and heated to 65-70°C for 3 h to complete the reaction. After completion of the reaction, the reaction mass was acidified with 5N aqueous hydrochloric acid (40 ml), concentrated to about 50 ml and DM water was added to crystallize the product at 20-25°C. The slurry was stirred for 2 h, filtered, washed with DM water and dried to yield (4R,12aS)-N-(2,4-difluorobenzyl)-7-hydroxy-4-methyl-6,8-dioxo-3,4,6,8,12,12a,-hexahydro-2H-pyrido[ 1′ ,2′ :4,5]pyrazino[2,1 -b] [ 1,3]oxazine-9-carboxamide (I) (11.5 g, HPLC purity: 99.63%).
Method B: Isobutyl chloroformate (4.65 gm, 0.03404 moles) in methylene chloride (10 ml) was added to the solution of N-methylmorpholine (3.45 gm, 0.03410 moles) and (4R,12aS)-7-methoxy-4-methyl-6,8-dioxo-3,4,6,8,12,12a-hexahydro-2H-pyrido[ 1′ ,2′ :4,5]pyrazino-[2,1 -b][l,3]oxazine-9-carboxy!ic acid (XVII) (10.0 gm, 0.03245 moles) in methylene chloride (60 ml) at -10 to 0°C in about 1 h. 2,4-Difloro benzyl amine (4.88 gm, 0.03409 moles) in methylene chloride (10 ml) was added to the cold reaction mass, and stirred at 20-30°C for completion of reaction. After completion of reaction, the reaction mass was washed with 5%w/w aqueous sodium bicarbonate solution (20 ml), IN hydrochloric acid (20 ml), DM water (20 ml) and concentrated. Acetonitrile (120 ml) and lithium bromide (4.8 gm, 0.05516 moles) were added to the concentrated mass, and stirred at 70-80°C for 3 h to complete the reaction. After completion of reaction, the reaction mass was acidified with 5N aqueous hydrochloric acid (40 ml) and concentrated to about 50 ml. DM Water (100 ml) was added to the concentrated reaction mass and stirred for 2 h at 25-30°C to crystallize the product. The product was filtered, washed with DM Water (50 ml) and dried to yield Dolutegravir (I) (10.7 gm, HPLC purity: 99.60%).
Example-2: Process for the preparation of Dolutegravir (I) (4R, 12aS)-N-(2,4-difluorobenzyl)-7-methoxy-4-methyl-6,8-dioxo-3,4,6,8,12,12a,-hexahydro-2H-pyrido[r,2′:4,5]pyrazino[2,l-b][l,3]oxazine-9-carboxamide (XVIII) (2 g, 0.0046 moles) was suspended in isopropyl alcohol (20 ml) and lithium bromide (0.8 g, 0.00924 moles) was added and stirred at 70-80°C for 15 h to complete the reaction. After completion of reaction the reaction mass was acidified with 5N aqueous hydrochloric acid (5 ml) and concentrated. DM Water (20 ml) was added to the concentrated mass and stirred at 25-30°C to crystallize the product. The product was filtered, washed with DM Water and dried to yield Dolutegravir (I) (1.5 g, HPLC purity: 97.93%).


/////
Indian Generics 2016
The generic APIs market is expected to continue to rise faster than the branded/innovative APIs, by 7.7%/year to reach $30.3 billion in 2016. Asia-Pacific is expected to show the fastest growth rates (10.8%/year). The 24 fastest growing markets will include 11 in Asia-Pacific, seven in Eastern Europe and CIS, four in Africa-Middle East and two in Latin America (Figure ).

Figure – Top growth markets for generic APIs to 2016
By 2016, China will account for 27.7% of the global generic API merchant market, while the US will have fallen to 23.8%; the mature markets as a whole will see their share fall from 41.8% in 2012 to 36.9%. India will be the third largest, with a 7.2% share.
सुकून उतना ही देना प्रभू, जितने से जिंदगी चल जाये।औकात बस इतनी देना,कि औरों का भला हो जाये।………..P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
सुकून उतना ही देना प्रभू, जितने से जिंदगी चल जाये। औकात बस इतनी देना, कि औरों का भला हो जाये।
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
Join me on Linkedin

Join me on Facebook 
Join me on twitter
Join me on google plus  Googleplus
Googleplus
Googleplus amcrasto@gmail.com
amcrasto@gmail.com
 LIONEL MY SON
LIONEL MY SONHe was only in first standard in school when I was hit by a deadly one in a million spine stroke called acute transverse mylitis, it made me 90% paralysed and bound to a wheel chair, Now I keep him as my source of inspiration and helping millions, thanks to millions of my readers who keep me going and help me to keep my son happy

सुकून उतना ही देना प्रभू, जितने से
जिंदगी चल जाये।
औकात बस इतनी देना,
कि औरों का भला हो जाये।
जिंदगी चल जाये।
औकात बस इतनी देना,
कि औरों का भला हो जाये।
Gatifloxacin

GATIFLOXACIN
BMS-206584, CG-5501, AM-1155, Zymar, Bonoq, Gatiflo, AM-1155
(±)-1-Cyclopropyl-6-fluoro-8-methoxy-7-(3-methyl-1-piperazinyl)-4-oxo-1,4-dihydroquinoline-3-carboxylic acid
Gatifloxacin sold under the brand names Gatiflo, Tequin and Zymar, is an antibiotic of the fourth-generation fluoroquinolonefamily,[1] that like other members of that family, inhibits the bacterial enzymes DNA gyrase and topoisomerase IV. Bristol-Myers Squibb introduced Gatifloxacin in 1999 under the proprietary name Tequin for the treatment of respiratory tract infections, having licensed the medication from Kyorin Pharmaceutical Company of Japan. Allergan produces it in eye-drop formulation under the names Zymar and Zymaxid. In many countries, gatifloxacin is also available as tablets and in various aqueous solutions forintravenous therapy.
Originally developed at Kyorin, gatifloxacin was first licensed to Gruenenthal in Europe, and that company still maintains rights to the oral and injectable formulations of the product. In October 1996, Kyorin licensed gatifloxacin to BMS, granting the company development and marketing rights in the U.S., Canada, Australia, Mexico, Brazil and certain other markets. In 2006, rights to the compound were returned by BMS. Subsequently, Senju and Kyorin signed a licensing agreement regarding the development of ethical eye drops containing the fluoroquinolone. In April 2000, Sumitomo Dainippon Pharma agreed to comarket the oral formulation in Japan. In August of that year, Allergan in-licensed gatifloxacin from Kyorin, gaining development and commercialization rights to the drug in all territories except Japan, Korea, China and Taiwan. The India-based Lupin Pharmaceuticals signed an agreement in June 2004 with Allergan to promote the ophthalmic solution of gatifloxacin in the pediatric specialty area in the U.S. PediaMed Pharmaceuticals also holds rights to the drug. In 2009, Kyorin licensed the drug candidate to Senju in China.
Gatifloxacin is the common name for (±)-1-cyclopropyl-6-fluoro-1,4-dihydro-8-methoxy-7-(3-methyl-1-piperazinyl)-4-oxo-3-quinolinecarboxylic acid (1), one of the most important broad-spectrum antibacterial agents and a member of the fourth-generation fluoroquinolone family.(1)Fluoroquinolones inhibit the enzyme DNA gyrase (topoisomerase II), which is responsible for the supercoiling of the DNA double helix, preventing the replication and repair of bacterial DNA and RNA.(2) Gatifloxacin (1) reached the market in 1999 under the brand name Tequin for the treatment of respiratory tract infections. The drug is available as tablets and aqueous solutions for intravenous therapy as well as eye drop formulation (Zymar).
To date, there are several processes described for the preparation of gatifloxacin, which can be grouped into two main categories: direct substitution of the 7-position fluorine atom of 1-cyclopropyl-6,7-difluoro-1,4-dihydro-8-methoxy-4-oxo-3-quinolinecarboxylic acid (2) by 2-methylpiperazine (Scheme 1),(3-5) and through boron chelate-type intermediates to overcome the diminished reactivity induced by the 8-methoxy group, which uses as starting material the ethyl ester derivative 3 (Scheme 2).(6-9)
SCHEME1

SCHEME2

- 1.
Mather, R.; Karenchak, L. M.; Romanowski, E. G.; Kowalski, R. P. Am. J. Ophthalmol.2002, 133 ( 4) 463
- 2.
Corey, E. J.; Czakó, B.; Kürti, L. Molecules and Medicine; Wiley: NJ, 2007; p 135.
- 3.
Masuzawa, K.; Suzue, S.; Hirai, K.; Ishizaki, T. 8-Alkoxyquinolonecarboxylic acid and salts thereof excellent in the selective toxicity and process of preparing the same EP 0 230 295 A3, 1987.
- 4.
Niddam-Hildesheim, V.; Dolitzky, B.-Z.; Pilarsky, G.; Steribaum, G. Synthesis of Gatifloxacin WO 2004/069825 A1, 2004.
- 5.
Ruzic, M; Relic, M; Tomsic, Z; Mirtek, M. Process for the preparation of Gatifloxacin and regeneration of degradation products WO 2006/004561 A1, 2006.
- 6.
Iwata, M.; Kimura, T.; Fujiwara, Y.; Katsube, T. Quinoline-3-carboxylic acid derivatives, their preparation and use EP 0 241 206 A2, 1987.
- 7.
Sanchez, J. P.; Gogliotti, R. D.; Domagala, J. M.; Garcheck, S. J.; Huband, M. D.; Sesnie,J. A.; Cohen, M. A.; Shapiro, M. A. J. Med. Chem. 1995, 38, 4478
- 8.
Satyanarayana, C.; Ramanjaneyulu, G. S.; Kumar, I. V. S. Novel crystalline forms of Gatifloxacin WO 2005/009970 A1 2005.
- 9.
Takagi, N.; Fubasami, H.; Matsukobo, H.; (6,7-Substituted-8-alkoxy-1-cyclopropyl-1,4-dihydro-4-oxo-3-quinolinecarboxylic acid-O3,O4)bis(acyloxy-O)borates and the salts thereof, and methods for their manufacture EP 0 464 823 A1, 1991.
………………………….
WO 2005009970
http://www.google.com/patents/WO2005009970A1?cl=en
preparation of Gatifloxacin hemihydrate from Ethyl-1- Cyclopropyl-6, 7-difluoro-8-methoxy-4-oxo-l, 4-dihydro-3-quinoline carboxylate through boron difluoride chelate. Ethyl-1-cyclopropyl- 6, 7-difluoro-8-methoxy-4-oxo-l, 4-dihydro-3-quinoline carboxylate is reacted with aqueous hydrofluoroboric acid followed by condensation with 2-methyl piperazine in polar organic solvent resulting in an intermediate l-Cyclopropyl-7- (3-methyl piperazin-1- yl). -6-fluoro-8-methoxy-4-oxo-l, 4-dihydro-3-quinoline carboxylic acid boron difluoride chelate. This intermediate may be further hydrolyzed to yield Gatifloxacin. Gatifloxacin so obtained may needs purification to yield high purity product. However to obtain directly high purity Gatifloxacin it is desirable to isolate the intermediate by cooling to low temperatures . Treating with an alcohol or mixture of alcohols purifies this intermediate. The purified condensed chelate in aqueous ethanol on hydrolysis with triethylamine followed by crystallization in ethanol gives Gatifloxacin hemihydrate with high purity.
STAGE – I:

Ethyl l-cyclopropyl-6,7-difluoro-8-met oxy l-Cycloproρyl-6, 7-difluoro-8-methoxy -4-oxo-l, -dihydro-3-quinoline -4-oxo-l, 4-dihydro-3-quinoline carboxylate carboxylic acid boron difluoride chelate
STAGE – II :

l-Cycloprop l-7- ( 3-methylpiperazin-l-yl.
6-fluoro~8-methoxy-4-oxo-l , 4-dihydro-3- carboxylicacid borondifluoride chelate quinoline carboxylicacid borondifluoride chelate
STAGE -III :

l-Cyclopropyl-7- (3- ethylpiperaz.in-l-yl . GATIFLOXACIN
-6-fluoro-8-methoxy-4-oxo-l , 4-dihydro-3- quinoline carboxylicacid borondifluoride chelate
Example-I: Preparation of Gatifloxacin • with isolation of intermediate (boron difluoride chelate derivative)
Stage-1: Preparation of l-cyclopropyl-6, 7-di luoro-8-methoxy-4-oxo- 1, 4-dihydro-3-quinoline carboxylic acid boron difluoride chelate. Ethyl-l-cyclopropyl-6, 7-difluoro-8-methoxy-4-oxo-l, -dihydro-3- quinόline carboxylate (100g)is suspended in ,40%aq..hydrofluoroboric acid -(1000 ml). Temperature of • the reaction mass is raised and maintained at 95°C to 100°C for 5hrs followed by cooling to 30°C – 35°C. Water (400 ml) is added and maintained at 25°C – 30°C for 2hrs . Product is filtered, washed with water (500 ml) and dried at 40°C – 45°C to constant weight. Dry weight of the product: 101.6 g (Yield: 95.8 %)
Stage-2: Preparation of 1- Cyclopropyl-7- (3-methylpiperazin-l-yl) – 6-fluoro-8-methoxy-4-oxo-l, -dihydro-3-quinoline carboxylic acid boron difluoride chelate
100 g of Boron difluoride chelate derivative prepared as above in stage-1 is suspended in acetonitrile (800 ml) , to that 2-methyl piperazine (44.0 g, 1.5 mole equiv.) is added and mixed for 15 min to obtain a clear solution. The reaction mass is maintained at 30°C – 35°C for 12 hrs followed by cooling to -10°C to -5°C. The reaction mass is maintained at -10°C to -5°C for 1 hr. The product is filtered and dried at 45°C – 50°C to constant weight. Dry weight of the product: 116.0 g (Yield: 93.9 %) .
The condensed chelate (100 g) prepared as above is suspended in methanol (1500 ml), maintained at 40°C – 45°C for 30 min. The reaction mass is gradually cooled, maintained for 1 hr at -5°C to 0°C. The product is filtered, washed with methanol (50 ml) and dried at 45°C – 50°C to constant weight. Dry weight of the product: 80.0 g (Yield: 80.0 %)
Stage -3: Preparation of Gatifloxacin (Crude)
The pure condensed chelate (100.0 g) prepared as above in stage-2 is suspended in 20% aq. ethanol (1000 ml) , the temperature is raised and maintained at 75°C to 80°C for 2 hrs. The reaction mass is cooled, filtered to remove insolubles, distilled under vacuum to remove solvent. Fresh ethanol (200 ml) is added and solvent is removed under vacuum at temperature below 50°C. Ethanol (200 ml) is added to the residue and gradually cooled to -10°C to -5°C. The reaction mass is mixed at -10°C to -5°C for 1 hr and then filtered. The wet cake is washed with ethanol (25 ml) and dried at 45°C – 50°C to constant weight.
The dry weight of the Gatifloxacin is 83.3 g (Yield: 91.7 %)
Stage- 4: Purification of crude Gatifloxacin
Crude Gatifloxacin (100.0 g) prepared as above in stage-3 is suspended in methanol (4000 ml), the temperature is raised and maintained at 60°C to 65°C for 20 min. to get a clear solution. Activated carbon (5 g) is added, maintained for 30 min and the solution is filtered. The filtrate is concentrated to one third of its original volume under vacuum at temperature below 40°C. The reaction mass is gradually cooled and maintained at -10°C to -5°C for 2 hrs. The product is filtered, washed with methanol (50 ml) and dried at 45°C – 50°C to constant weight. The dry weight of the pure Gatifloxacin is 76.0 g (Yield: 76.0 %)
Example-II: Preparation of Gatifloxacin without isolation of intermediate (boron difluoride chelate derivative)
Stage-1: Preparation of l-cyclopropyl-6, 7-difluoro-8-methoxy-4- oxo-1, 4-dihydro-3-quinoline carboxylic acid boron difluoride chelate.
Ethyll-cyclopropyl-6, 7-difluoro-8-methoxy-4-oxo-l, 4-dihydro-3- quinoline carboxylate (lOOg) is suspended in 40% aq. hydrofluoroboric acid (1000 ml) . Temperature of the reaction mass is raised and maintained at 95°C to 100°C for 5 hrs followed by cooling to 30°C – 35°C. 400 ml DM water is added, maintained at 25°C – 30°C for 2hrs . The product is filtered, washed with DM water (500 ml) and dried at 40°C – 45°C to constant weight. The dry wt is 102.5 g (Yield: 96.6 %)
Stage – 2: Preparation of Gatifloxacin (Crude)
The boron difluoride chelate derivative (100 g) prepared as above in stage-1 is suspended in acetonitrile (800 ml) , 2-methyl piperazine (44 g, 1.5 mole equiv.) is added and mixed for 15 min to obtain a clear solution. The reaction mass is maintained at 30°C – 35°C for 12 hrs. Removed the solvent by vacuum distillation. 20% Aq. ethanol (1000 ml) is added, raised the temperature and maintained at 75°C to 80°C for 2 hrs. The reaction mass is cooled, filtered to remove insolubles. The filtrate is distilled under vacuum to remove solvent completely. Fresh ethanol (250 ml) is added and distilled under vacuum at temperature below 50°C. Fresh Ethanol (250 ml) is added to the residue and gradually cooled to -10°C to -5°C. The reaction mass is maintained at -10°C to -5°C for 1 hr and filtered. The wet cake is washed with ethanol (30 ml) and dried at 45°C – 50°C to constant weight.
The dry weight of the Gatifloxacin is 73.5 g (Yield: 65.4 %)
Stage -3: Purification of crude Gatifloxacin
Crude Gatifloxacin (80.0 g) prepared as above in stage-2 is suspended in methanol (2000 ml) , the temperature is raised and maintained at 60°C to 65°C for 20 min. to get a clear solution. The reaction mixture is filtered. The filtrate is gradually cooled and maintained at -10°C to -5°C for 2 hrs. The product is filtered, washed with methanol (50 ml) and dried at 45°C – 50°C to constant weight.
The dry weight of the pure Gatifloxacin is 56.0 g (Yield: 70.0 %)
……………………….
WO 2005047260
http://www.google.co.in/patents/WO2005047260A1?cl=en
Gatifloxacin is the international common name of l-cyclopropyl-6-fluoro-l, 4-dihydro-8-methoxy- 1- (3-methyl-l-piperazinyl) -4-oxo-3-guinolin-carboxylic acid of formula (I) , with application in medicine and known for its antibiotic activity:

European patent application EP-A-230295 discloses a process for obtaining gatifloxacin that consists on the reaction of compound (II) with 2-

In this process the gatifloxacin is isolated in the form of a hemihydrate after a laborious process of column chromatography and recrystallisation in methanol, which contributes towards making the final yield lower than 20% by weight. Moreover, in said process an undesired by-product is formed, resulting from demethylation at position 8 of the ring. European patent application EP-A-241206 discloses a process for preparing gatifloxacin, whose final steps are as follows:

(III) H ft N Me H DMSO
Gatifloxacin (I)

(IV) This process uses the intermediate compound (III) , which has been prepared and isolated in a separate operation, while the intermediate compound (IV) is also isolated before proceeding to its conversion into gatifloxacin by treatment with ethanol in the presence of triethylamine. The overall yield from these three steps is lower than 40%. These disadvantages — a synthesis involving several steps, low yields, and the need to isolate the intermediate products — hinder the production of gatifloxacin on an industrial scale. There is therefore a need to provide a process for preparing gatifloxacin with a good chemical yield, without the need to isolate the intermediate compounds and that substantially avoids demethylation in position 8 of the ring. The processes termed in English “one pot” are characterised in that the synthesis is carried out in the same reaction vessel, without isolating the intermediate compounds, and by means of successive addition of the reacting compounds. The authors of the present invention have discovered a simplified process for preparing gatifloxacin which does not require isolation of the intermediate compounds .
Example 1: Preparing gatifloxacin from compound (II) 10 g (0.0339 moles, 1 equivalent) of compound
(II) is placed in a flask, 30 ml of acetonitryl (3 volumes) is added and this is heated to a temperature of 76-80° C.

Once reflux has been attained, and being the temperature maintained, 3.28 g (0.0203 moles, 0.6 equivalents) of hexamethyldisilazane (HMDS) is added with a compensated adding funnel. Once addition is completed, the reaction is maintained with stirring for 1 hour at a temperature of 76-80° C. Once this period has elapsed, the reaction mixture is cooled to a temperature ranging between 0 and 15° C, and 5.78 g (0.0407 moles, 1.2 equivalents) of boron trifluoride ethyletherate is added while keeping the temperature below 15° C. Once addition is completed, the temperature is allowed to rise to 15- 25° C and it is kept under these conditions for approximately 2 hours. The pH of the mixture is then adjusted to an approximate value of 9 with triethylamine (approximately 2 ml) . To the resulting suspension is added a solution of 10.19 g (0.1017 moles, 3 equivalents) of 2-methylpiperazine in 28 ml of acetonitryl, while maintaining the temperature between 15 and 25° C. The resulting amber solution is kept with stirring under these conditions for approximately 3 hours . Once the reaction has been completed, the solution is distilled at low pressure until a stirrable paste is obtained. At this point 50 ml of methanol is added, the resulting suspension is raised to a temperature of 63-67° C and is kept under these conditions for approximately 5 hours . Once the reaction has been completed, the mixture is cooled to a temperature of 25-35° C in a water bath, and then at a temperature of 0-5° C in a water/ice bath for a further 1 hour. The resulting precipitate is filtered, washed with cold methanol (2 x 10 ml) and dried at 40° C in a vacuum oven to constant weight. 10.70 g of crude gatifloxacin is obtained, having a water content of 2.95% by weight. The yield of the process is 81.8%.
The crude product is crystallised in methanol by dissolving 20 g of crude gatifloxacin in 1 1 of methanol (50 volumes) at a temperature of 63-67° C. Once all the product has been dissolved, the solution is left to cool to a temperature of 30-40° C, and then to a temperature of 0-5° C in a water/ice bath, maintaining it under these conditions for 1 hour. The resulting suspension is filtered and the solid retained is washed with 20 ml (1 volume) of cold methanol. The solid obtained is dried at 40° C in a vacuum oven to provide 18.65 g of gatifloxacin with a water content of 2.36% by weight.
The overall yield from the compound (II) is 77.7%, with a purity exceeding 99.8% as determined by HPLC chromatography. The content of by-product resulting from demethylation in position 8 of the ring is lower than 0.1% as determined by HPLC chromatography.
 |
|
| Systematic (IUPAC) name | |
|---|---|
| 1-cyclopropyl-6-fluoro- 8-methoxy-7-(3-methylpiperazin-1-yl)- 4-oxo-quinoline-3-carboxylic acid | |
| Clinical data | |
| Trade names | Zymar |
| AHFS/Drugs.com | monograph |
| MedlinePlus | a605012 |
|
|
| Oral (discontinued), Intravenous(discontinued) ophthalmic |
|
| Pharmacokinetic data | |
| Protein binding | 20% |
| Half-life | 7 to 14 hours |
| Identifiers | |
| 112811-59-3 |
|
| J01MA16 S01AE06 | |
| PubChem | CID: 5379 |
| DrugBank | DB01044 |
| ChemSpider | 5186 |
| UNII | 81485Y3A9A |
| KEGG | D08011 |
| ChEBI | CHEBI:5280 |
| ChEMBL | CHEMBL31 |
| NIAID ChemDB | 044913 |
| Chemical data | |
| Formula | C19H22FN3O4 |
| 375.394 g/mol | |
PAPER

An improved process to obtain gatifloxacin (1) through use of boron chelate intermediates has been developed. The methodology involves an initial activation step which accelerates the formation of the first chelate under low-temperature conditions and prevents demethylation of the starting material. To increase the overall yield and to avoid the isolation and manipulation of the resulting intermediates, the process has been designed to be carried out in one pot. As a result, we present here an easy, scaleable and substantially impurity-free process to obtain gatifloxacin (1) in high yield.
A High-Throughput Impurity-Free Process for Gatifloxacin
Department of Research & Development, Química Sintética S.A., c/ Dulcinea s/n, 28805 Alcalá de Henares, and Department of Organic Chemistry, University of Alcalá, 28871 Madrid, Alcalá de Henares, Spain
Org. Process Res. Dev., 2008, 12 (5), pp 900–903
DOI: 10.1021/op800042a
gatifloxacin (1) as white crystals. Yield 32.3 kg, (93%); purity by HPLC 99.87%; Assay by HPLC 100.8%; mp 167−168 °C(18) (Lit. (J. Med. Chem. 1995, 38, 4478)159−162 °C).
18
DSC analysis showed two endothermic peaks at 166.2 °C (T onset = 164.3 °C) and 190.0 °C (T onset = 188.2 °C) and an exothermic one at 168.1 °C. The shape of this DSC curve is characteristic of a monotropic transition between crystalline forms
Water content by Karl Fischer 3.0%(19) MS m/z 376 (M+ + H);
19
Although there are several hydrates described for gatifloxacin such as, among others, the hemimydrate, sesquihydrate, and pentahydrate(Raghavan, K. S.; Ranadive, S. A.;Gougoutas, J. Z.; Dimarco, J. D.; Parker, W. L.; Dovich, M.; Neuman, A.Gatifloxacin pentahydrate. WO 2002/22126 A1, 2002) , the Gatifloxacin obtained by the present procedure does not seem to form a stoichometric hydrate, but instead it retains moisture.
Thus, the product is usually obtained with a Karl-Fischer value below 1% after drying, but it can absorb moisture until a final content of about 3%. This water content can vary between 2.0% and 3.5%, depending on the relative humidity of the environment. DSC analysis revealed a broad endothermic signal with minimum at 76 °C, while TGA analysis showed that the product loses all the water below 80 °C.
No loss of weight is registered when the product melts, and the weight is constant until the decomposition of the material at about 200 °C. On the basis of these results, it can be said that the water content of the gatifloxacin obtained by the present process is retained moisture instead of water belonging to the lattice. The shape of the derivative of the weight curve at the beginning of the analysis shows that the sample has already lost part of the moisture when the register starts. This is probably due to the sample starting to lose weight when makes contact with the dry atmosphere of the TGA oven that could explain the different values obtained for water content of the analyzed sample by TGA (1.90%) and Karl-Fischer (2.64%) methods.
1H NMR (DMSO-d6) δ 0.97 (d, J = 6.1 Hz, 3H), 1.04 (m, 2H), 1.15 (m, 2H), 2.75−2.94 (m, 4H) 3.14 (m, 1H), 3.30 (m, 2H), 3.74 (s, 3H), 4.15 (m, 1H), 7.70 (d, JH−F = 12.2 Hz, 1H), 8.67 (s, 1H).
13C NMR (DMSO-d6) δ 8.40, 8.42, 18.66, 40.28, 45.46, 50.17, 50.29 (d, JC−F = 3.44 Hz), 57.36 (d, JC−F = 3.74 Hz), 62.15, 106.0 (d, JC−F = 22.7 Hz), 106.04, 120.05 (d, JC−F = 8.6 Hz), 133.6 (d, JC−F = 1.1 Hz), 138.9 (d, JC−F = 11.9 Hz), 145.2 (d, JC−F = 5.87 Hz), 149.88, 155.06 (d, JC−F = 249.2 Hz), 165.56, 175.56 (d, JC−F = 3.3 Hz).
19F NMR (DMSO-d6) δ −120.4 (d, J = 12.2 Hz).
Anal. Calcd for C19H22N3O4F + 3.0% H2O; C, 58.95; H, 6.07; N, 10.85. Found: C, 58.90; H, 5.82; N, 10.90.
Boron content 170 ppm.(The respective boron contents found for batches 2 and 3 were 160 and 300 ppm)
Side-effects and removal from the market
A Canadian study published in the New England Journal of Medicine in March 2006 claims Tequin can have significant side effectsincluding dysglycemia.[2] An editorial by Dr. Jerry Gurwitz in the same issue called for the Food and Drug Administration (FDA) to consider giving Tequin a black box warning.[3] This editorial followed distribution of a letter dated February 15 by Bristol-Myers Squibb to health care providers indicating action taken with the FDA to strengthen warnings for the medication.[4] Subsequently it was reported on May 1, 2006 that Bristol-Myers Squibb would stop manufacture of Tequin, end sales of the drug after existing stockpiles were exhausted, and return all rights to Kyorin.[5]
Union Health and Family Welfare Ministry of India on 18 March 2011 banned the manufacture, sale and distribution of Gatifloxacin as it caused certain adverse side effects[6]
Contraindications
Availability
Gatifloxacin is currently available only in the US and Canada as an ophthalmic solution.
In China it is sold in tablet as well as in eye drop formulations.
Ophthalmic anti-infectives are generally well tolerated. The concentration of the drug observed following oral administration of 400 mg gatifloxacin systemically is approximately 800 times higher than that of the 0.5% Gatifloxacin eye drop. Given as an eye drop, Gatifloxacin Ophthalmic Solution 0.3% & 0.5% cause very low systemic exposures. Therefore, the systemic exposures resulting from the gatifloxacin ophthalmic solution are not likely to pose any risk for systemic toxicities.

- The reaction of 1-bromo-2,4,5-trifluoro-3-methoxybenzene (I) with CuCN and N-methyl-2-pyrrolidone at 150 C gives 2,4,5-trifluoro-3-methoxybenzonitrile (II), which by treatment with concentrated H2SO4 yields the benzamide (III) The hydrolysis of (III) with H2SO4 -. water at 110 C affords 2,4,5-trifluoro-2-methoxybenzoic acid (IV), which by reaction with SOCl2 is converted into the acyl chloride (V). The condensation of (V) with diethyl malonate by means of magnesium ethoxide in toluene affords diethyl 2- (2,4,5-trifluoro-3-methoxybenzoyl) malonate (VI), which by treatment with p-toluenesulfonic acid in refluxing water gives ethyl 2- (2,4,5-trifluoro-3-methoxybenzoyl) acetate (VII). The condensation of (VII) with triethyl orthoformate in refluxing acetic anhydride yields 3-ethoxy -2- (2,4,5-trifluoro-3-methoxybenzoyl) acrylic acid ethyl ester (VIII), which is treated with cyclopropylamine (IX) to afford the corresponding cyclopropylamino derivative (X). The cyclization of (X) by means of NaF in refluxing DMF gives 1-cyclopropyl-6,7-difluoro-8-methoxy-4-oxo-1,4-dihydroquinoline-3-carboxylic acid ethyl ester (XI), which is hydrolyzed with H2SO4 in acetic acid to yield the corresponding free acid (XII). Finally, this compound is condensed with 2-methylpiperazine (XIII) in hot DMSO.
 |
|
Title: Gatifloxacin
CAS Registry Number: 112811-59-3
CAS Name: 1-Cyclopropyl-6-fluoro-1,4-dihydro-8-methoxy-7-(3-methyl-1-piperazinyl)-4-oxo-3-quinolinecarboxylic acid
Trademarks: Tequin (BMS); Zymar (Allergan)
Molecular Formula: C19H22FN3O4
Molecular Weight: 375.39
Percent Composition: C 60.79%, H 5.91%, F 5.06%, N 11.19%, O 17.05%
Literature References: Fluorinated quinolone antibacterial. Prepn: K. Masuzawa et al., EP 230295; eidem, US 4980470 (1987, 1990 both to Kyorin); J. P. Sanchez et al., J. Med. Chem. 38, 4478 (1995); of the sesquihydrate: T. Matsumoto et al., US5880283 (1999 to Kyorin). In vitro antibacterial activity: A. Bauernfeind, J. Antimicrob. Chemother. 40, 639 (1997); H. Fukuda et al., Antimicrob. Agents Chemother. 42, 1917 (1998). Clinical pharmacokinetics: M. Nakashima et al., ibid. 39, 2635 (1995). Clinical study in urinary tract infection: H. Nito, 10th Mediterranean Congr. Chemother. 1996, 327; in respiratory tract infection: S. Sethi, Expert Opin. Pharmacother. 4, 1847 (2003).
Properties: Pale yellow prisms from methanol as hemihydrate, mp 162°.
Melting point: mp 162°
Derivative Type: Sesquihydrate
CAS Registry Number: 180200-66-2
Manufacturers’ Codes: AM-1155
Molecular Formula: C19H22FN3O4.1½H2O
Molecular Weight: 384.40
Percent Composition: C 59.37%, H 6.03%, F 4.94%, N 10.93%, O 18.73%
Therap-Cat: Antibacterial.
Keywords: Antibacterial (Synthetic); Quinolones and Analogs
|
References
- Burka JM, Bower KS, Vanroekel RC, Stutzman RD, Kuzmowych CP, Howard RS (July 2005). “The effect of fourth-generation fluoroquinolones gatifloxacin and moxifloxacin on epithelial healing following photorefractive keratectomy”. Am. J. Ophthalmol. 140 (1): 83–7. doi:10.1016/j.ajo.2005.02.037.PMID 15953577.
- Park-Wyllie, Laura Y.; David N. Juurlink; Alexander Kopp; Baiju R. Shah; Therese A. Stukel; Carmine Stumpo; Linda Dresser; Donald E. Low; Muhammad M. Mamdani (March 2006).“Outpatient Gatifloxacin Therapy and Dysglycemia in Older Adults”. The New England Journal of Medicine 354 (13): 1352–1361. doi:10.1056/NEJMoa055191. PMID 16510739. Retrieved 2006-05-01. Note: publication date 30 March; available on-line 1 March
- Gurwitz, Jerry H. (March 2006). “Serious Adverse Drug Effects — Seeing the Trees through the Forest”. The New England Journal of Medicine 354 (13): 1413–1415.doi:10.1056/NEJMe068051. PMID 16510740. Retrieved2006-05-01.
- Lewis-Hall, Freda (February 15, 2006). “Dear Healthcare Provider:” (PDF). Bristol-Myers Squibb. Retrieved May 1, 2006.
- Schmid, Randolph E. (May 1, 2006). “Drug Company Taking Tequin Off Market”. Associated Press. Archived from the original on November 25, 2007. Retrieved 2006-05-01.[dead link]
- “Two drugs banned”. The Hindu (Chennai, India). 19 March 2011.
- Peggy Peck (2 May 2006). “Bristol-Myers Squibb Hangs No Sale Sign on Tequin”. Med Page Today. Retrieved 24 February2009.
| EP0610958A2 * | 20 Jul 1989 | 17 Aug 1994 | Ube Industries, Ltd. | Intermediates in the preparation of 4-oxoquinoline-3-carboxylic acid derivatives |
| ES2077490A1 * | Title not available |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2008126384A1 | 31 Mar 2008 | 23 Oct 2008 | Daiichi Sankyo Co Ltd | Method for producing quinolone carboxylic acid derivative |
| CN101659654B | 28 Aug 2008 | 6 Nov 2013 | 四川科伦药物研究有限公司 | 2-Methylpiperazine fluoroquinolone compound and preparation method and application thereof |
| CN102351843A * | 18 Aug 2011 | 15 Feb 2012 | 张家口市格瑞高新技术有限公司 | Synthesis method of 2-methyl piperazine lomefloxacin |
| EP1832587A1 * | 2 Mar 2007 | 12 Sep 2007 | Quimica Sintetica, S.A. | Method for preparing moxifloxacin and moxifloxacin hydrochloride |
| US7365201 | 2 Mar 2006 | 29 Apr 2008 | Apotex Pharmachem Inc. | Process for the preparation of the boron difluoride chelate of quinolone-3-carboxylic acid |
| US7875722 | 30 Sep 2009 | 25 Jan 2011 | Daiichi Sankyo Company, Limited | Method for producing quinolone carboxylic acid derivative |
| EP0464823A1 * | Jul 4, 1991 | Jan 8, 1992 | Kyorin Pharmaceutical Co., Ltd. | (6,7-Substituted-8-alkoxy-1-cyclopropyl-1,4-dihydro-4-oxo-3-quinolinecarboxylic acid-O3,O4)bis(acyloxy-O)borates and the salts thereof, and methods for their manufacture |
| US4997943 * | Mar 31, 1987 | Mar 5, 1991 | Sankyo Company Limited | Quinoline-3-carboxylic acid derivatives |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| CN101659654B | Aug 28, 2008 | Nov 6, 2013 | 四川科伦药物研究有限公司 | 2-Methylpiperazine fluoroquinolone compound and preparation method and application thereof |
| CN102351843A * | Aug 18, 2011 | Feb 15, 2012 | 张家口市格瑞高新技术有限公司 | Synthesis method of 2-methyl piperazine lomefloxacin |
* Cited by examiner
TAKE A TOUR
TAKE A TOUR
Amritsar, punjab, India
-
Amritsar – Wikipedia, the free encyclopedia
https://en.wikipedia.org/?title=AmritsarAmritsar is one of the largest cities of the Punjab state in India. The city origin lies in the village of Tung, and was named after the lake founded by the fourth Sikh …
 GOLDEN TEMPLE
GOLDEN TEMPLE

 .
.






 Tandoori chicken at Surjit Food Plaza. amritsar
Tandoori chicken at Surjit Food Plaza. amritsar
Bullet marks on the walls of the park premises







The Jallianwalla Bagh in 1919, months after the massacre
Mealtime at the Golden Temple Amritsar
 Golden Temple – Harmandir Sahib: Free food for everyone
Golden Temple – Harmandir Sahib: Free food for everyone
Sri Guru Ram Dass Jee International Airport in Amritsar
Amritsar – Wagah Border – Street food stall | Explore bernic… |

-

Golden Temple
-

Maharaja Ranjit Singh’s Ram Bagh Gardens
-

Golden Temple
-

Durgiana Temple
-

The holy water
-

Jallianwala Bagh
-

Jallianwala Bagh
-

The holy water
-

Golden Temple
-

Golden Temple
-

Sikh Gurdwara
-

The holy water
 Night view of the Harmandir Sahib///////////
Night view of the Harmandir Sahib/////////// -



%27,_oil_on_canvas_painting_by_Charles_W._Bartlett,_1920,_Honolulu_Academy_of_Arts.jpg)
 tandoori chicken
tandoori chicken













FDA grants orphan drug status for Lipocine’s LPCN 1107 to prevent preterm birth

17-Hydroxyprogesterone caproate
[(8R,9S,10R,13S,14S,17R)-17-Acetyl-10,13-dimethyl-3-oxo-2,6,7,8,9,11,12,14,15,16-decahydro-1H-cyclopenta[a]phenanthren-17-yl] hexanoate
Molecular Weight: 428.6041 g/mol
Molecular Formula: C27H40O4
Pregn-4-ene-3,20-dione,17-hydroxy-, hexanoate (7CI,8CI);Progesterone, 17-hydroxy-, hexanoate (6CI);Hexanoic acid, ester with 17-hydroxypregn-4-ene-3,20-dione (8CI);17a-Caproyloxypregn-4-ene-3,20-dione;17a-Hydroxypregn-4-ene-3,20-dionecaproate;17a-Hydroxypregn-4-ene-3,20-dionehexanoate;17a-Hydroxyprogesteronecaproate;17a-Hydroxyprogesteronen-caproate;Delalutin;Depo-proluton;Hormofort;Pregn-4-ene-3,20-dione,17-[(1-oxohexyl)oxy]-;NSC 17592;Neolutin;Primolut Depot;Procyte Depo;Proge;Syngynon;Teralutil;
FDA grants orphan drug status for Lipocine’s LPCN 1107 to prevent preterm birth
Specialty pharmaceutical firm Lipocine has received orphan drug designation from the US Food and Drug Administration (FDA) for its LPCN 1107 to prevent preterm birth (PTB).

LPCN 1107 is an oral product candidate of 17-alpha hydroxyprogesterone caproate under development for the indication of prevention of recurrent preterm birth. LPCN 1107 has the potential to become the first oral HPC product for the prevention of preterm birth in women with a prior history of at least one preterm birth. Potential benefits of our oral product candidate relative to current injectable products include the elimination of pain and site reactions associated with weekly injections, elimination of weekly doctor visits or visits from the nurse, and elimination of interference/disruption of personal, family or professional activities associated with weekly visits.
Preterm Birth (PTB) is defined as delivery of less than 37 weeks of gestation. PTB occurs in ~12% of all US births. PTB remains the leading cause of perinatal mortality and morbidity, accounting for as many as 75% of perinatal deaths.
The expense associated with PTB involves not only the immediate cost of the preterm baby being treated in the hospital ICU setting, but includes the long term treatment costs for disabilities for the life of the child. Current total PTB related economic impact on the US health system far exceeds $26 billion, an estimated cost in 2006.

Behrman RE et al. in: Behrman RE, Butler AS, eds. Preterm Birth: Causes, Consequences, and Prevention. Washington, DC: The National Academies Press; 2006:329-354.
There is a significant unmet need for a ‘patient friendly’ product for the prevention of PTB. The only FDA approved product for the prevention of PTB must be given by an intra-muscular injection each week for a total of 18-22 injections.
LPCN 1107 Product Attributes:
- Designed for oral administration twice daily of hydroxyprogesterone caproate (same active as in the only FDA approvd injectable product for the prevention of recurrent PTB).
- Eliminates site reaction and pain at the site of injection
- Eliminates regular doctor office visits or visits from the nurse (weekly visits for 16 – 20 weeks)
- Significant absorption upon oral dosing of LPCN 1107 in healthy non-pregnant women
- Good dose response demonstrated in healthy non-pregnant women
- LPCN 1107 was well tolerated in single dose study
- LPCN 1107 may be eligible for orphan drug designation
LPCN 1107, Lipocine’s oral hydroxyprogesterone caproate (HPC) product candidate has the potential to become the first oral HPC product for the prevention of preterm birth in women with a prior history of at least one preterm birth. Potential benefits of our oral product candidate relative to current once-a-week intramuscular (IM) injectable product include the elimination of pain and site reactions associated with weekly injections, elimination of weekly doctor visits or visits from the nurse, and elimination of interference/disruption of personal, family or professional activities associated with weekly visits. Lipocine has successfully completed a Phase 1 study under a US IND designed to determine the pharmacokinetics and bioavailability of LPCN 1107 relative to an IM HPC, as well as safety and tolerability, in healthy non-pregnant female volunteers.
17α-Hydroxyprogesterone caproate is a synthetic, steroidalprogestin that is similar to medroxyprogesterone acetate andmegestrol acetate. It is an ester derivative of 17α-hydroxyprogesterone formed from caproic acid (hexanoic acid).
17α-Hydroxyprogesterone caproate was previously marketed under the trade name Delalutin by Squibb, which was approved by the U.S. Food and Drug Administration (FDA) in 1956 and withdrawn from marketing in 1999.
The US FDA approved Makena from KV Pharmaceutical (previously named as Gestiva) on February 4, 2011 for prevention ofpreterm delivery in women with a history of preterm delivery, sparking a pricing controversy.
Synthesis
Hydroxyprogesterone caproate can be prepared by the following sequence:[13]
It is made from 16-dehydropregnenolone acetate (16-DPA),[14] product of the Marker degradation.

Ringold, H. J.; Loken, B.; Rosenkraz, G.; Sondheimer, F.; J. Amer. Chem. Soc. 1956, 78, 816.
……………………………….
PATENT
http://www.google.com/patents/CN104017041A?cl=en
method of synthesizing progesterone caproate, comprising the steps of:
[0006] Step one to 17 α- hydroxy progesterone as a raw material, and n-hexyl acid in pyridine and p-toluene sulfonic acid catalysis by esterification reaction mixture esterified, the reaction is as follows

Step two, to the mixture of step one described esterified in an alcohol solution of acid catalysis to give progesterone caproate
Ketone crude reaction is as follows:

Step one to obtain a mixture containing progesterone caproate ester compound of step two the mixture is esterified in an alcohol solution of acid catalysis to give progesterone caproate crude. The reaction process of the present invention avoids the costly esterification agent n-hexyl anhydride used materials costs and recovery costs are significantly reduced.
Example 1
17 a – hydroxy progesterone 20g, n-caproic acid 40ml, topiramate 唳 16ml, p-toluenesulfonic acid 1.6g, toluene 300ml, 500ml three-necked flask were put, the reaction temperature was raised to between 110 ~ 120 ° C 3 hours TLC sampling The reaction was monitored. The reaction is as follows:

[0016] 17 a – hydroxy progesterone concentration treatment made after completion of the reaction, as a method for the enrichment process concentrated under reduced pressure and toluene, pyridine, and the unfinished batch reaction of n-hexanoic acid.
After the end of the [0017] concentrated in the three-necked flask was added 100mL ethanol, 3ml of concentrated hydrochloric acid was heated to reflux alcohol solution 2 hours, the reaction was monitored sampling TLC, complete hydrolysis of the diester into progesterone caproate stop the reaction. The reaction is as follows:

cooled to below 5 ° C, filtered and dried to obtain crude progesterone caproate 24g, crude yield of 120%.Progesterone caproate crude was purified with ethanol to give progesterone caproate boutique 19.Sg, progesterone caproate Collectibles yield based on the crude progesterone caproate 82.5% of the total yield of 99.0% o
Example 2
17 α – hydroxy progesterone 20g, n-caproic acid 50ml, topiramate 唳 30ml, p-toluenesulfonic acid 3g, toluene 300ml, 500ml three-necked flask were put, the reaction temperature was raised to between 110 ~ 120 ° C 3 hours TLC monitoring sampling reaction. 17 α – hydroxy progesterone concentration treatment made after completion of the reaction, as the concentration treatment method evaporated toluene, pyridine and n-hexyl Unreacted acid.
After the end of the [0022] concentrated in the three-necked flask was added 100mL ethanol, 5ml of concentrated hydrochloric acid was heated to reflux alcohol solution I hour, the reaction was monitored sampling TLC, complete hydrolysis of the diester into progesterone caproate stop the reaction. Cooled to below 5 ° C, filtered and dried to obtain crude progesterone caproate 23.5g, crude yield of 117.5%. Progesterone caproate crude was purified with ethanol to give progesterone caproate boutique 19.2g, progesterone caproate Collectibles yield based on the crude progesterone caproate 81.7%, the total yield was 96.0%.
Example 3
17 α – hydroxy progesterone 20g, n-caproic acid 60ml, topiramate 唳 40ml, p-toluenesulfonic acid 4g, toluene 300ml, 500ml three-necked flask were put, the reaction temperature was raised to between 110 ~ 120 ° C 2.5 hours TLC monitoring sampling reaction. 17 α – hydroxy progesterone concentration treatment made after completion of the reaction, as the concentration treatment method evaporated toluene, pyridine and n-hexyl Unreacted acid.
After the end of the [0025] concentrated in the three-necked flask was added 100mL ethanol, 8ml of concentrated hydrochloric acid was heated to reflux alcohol solution 40 minutes, the reaction was monitored sampling TLC, complete hydrolysis of the diester into progesterone caproate stop the reaction. Cooled to below 5 ° C, filtered and dried to obtain crude progesterone caproate 23g, crude yield of 115%. Progesterone caproate crude was purified with ethanol to give progesterone caproate fine 19g, progesterone caproate Collectibles yield based on the crude progesterone caproate 82.6% of the total yield of 95.0%.
Example 4
17 α – hydroxy progesterone 20g, n-caproic acid 60ml, topiramate 唳 40ml, p-toluenesulfonic acid 4g, benzene, 300ml, 500ml three-necked flask were put, the reaction temperature was raised to between 110 ~ 120 ° C 2.5 hours TLC monitoring sampling reaction. 17 α- hydroxy progesterone concentration treatment made after completion of the reaction, as a method for the enrichment process concentrated under reduced pressure benzene, pyridine and non-completion of the reaction of n-hexanoic acid.
After the end of the [0028] concentrated in the three-necked flask was added 100mL of methanol, 8ml of concentrated sulfuric acid was heated to reflux alcohol solution 40 minutes, the reaction was monitored sampling TLC, complete hydrolysis of the diester into progesterone caproate stop the reaction. Cooled to below 5 ° C, filtered and dried to obtain crude progesterone caproate 23g, crude yield of 115%. Progesterone caproate crude was purified with ethanol to give progesterone caproate fine 19g, progesterone caproate Collectibles yield based on the crude progesterone caproate 82.6% of the total yield of 95.0%.
Notes
- SMFM Clinical Guideline: Progesterone and preterm birth prevention: translating clinical trials data into clinical practice, AJOG May 2012
- Meirs et al. NEJM 2003
- Dodd JM, Flenady V, Cincotta R, Crowther CA; The Cochrane Database of Systematic Reviews 2006 Issue 1
- Keirse, MJNC; Progesterone (2004). “déjà vu” or “still to be seen”?.”. Birth 31: 3.
- Johnson, JWC; Austin, KL; Jones, GS; Davis, GH; King, TM (1975). “Efficacy of 17 alpha-hydroxyprogesterone caproate in the prevention of premature labor”. NEJM 293 (14): 675.doi:10.1056/nejm197510022931401.
- Yemini, M; Borenstein, R; Dreazen et al. (1985). “Prevention of premature labor by 17 alpha-hydroxyprogesterone caproate”. Am J Obstet Gynecol 151 (5): 574–7. doi:10.1016/0002-9378(85)90141-3.
- Meis PJ et al. Prevention of Recurrent Preterm Delivery by 17 Alpha-hydroxyprogesterone Caproate. NEJM, 2003: vol 348, no 24, pg 2379-2385.
- Keirse MJNC, Progestogen administration in pregnancy may prevent preterm delivery. Br J Obstet Gynecol 1990 February; 97:149.
- Advisory Committees: CDER 2006 Meeting Documents
- Hendrix AG, et al. Embriotoxicity of sex steroidal hormones in nonhuman primates: II. Hydroxyprogesterone caproate, estradiol valerate. Teratology 1987 February. 35 (1): 129.
- Duke University Medical Center, New England Journal of Medicine, correspondence, vol 349.
- Hauth, JC; Gilstrap, LC; Brekken, AL; Hauth, JM (1983). “The effect of 17 alpha-hydroxyprogesterone caproate on pregnancy outcome in an active-duty military population”. Am J Obstet Gynecol 146 (2): 187.
- Ringold, H. J.; Loken, B.; Rosenkraz, G.; Sondheimer, F. (1956). “Steroids. LXXIII. The Direct Oppenauer Oxidation of Steroidal Formate Esters. A New Synthesis of 17α-Hydroxyprogesterone”. J. Amer. Chem. Soc. 78 (4): 816. doi:10.1021/ja01585a030.
- Goswami, A.; Kotoky, R.; Rastogi, R. C.; Ghosh, A. C. (2003). “A One-Pot Efficient Process for 16-Dehydropregnenolone Acetate”. Organic Process Research & Development 7 (3): 306.doi:10.1021/op0200625.
- Armstrong J (May 2011). “Unintended consequences — the cost of preventing preterm births after FDA approval of a branded version of 17OHP”. N. Engl. J. Med. 364 (18): 1689–91.doi:10.1056/NEJMp1102796. PMID 21410391.
Sources
- FDA Reproductive Health Drugs Advisory Committee. August 29, 2006 Meeting to discuss NDA 21-945 Gestiva (Adeza Biomedical)
17α-hydroxyprogesterone caproate injection, 250 mg/mL, for the proposed indication: prevention of preterm delivery in women with a history of a prior preterm delivery. - Adeza Biomedical (October 23, 2006) Receives FDA Approvable Letter For Gestiva
- Adeza gets orphan drug designation for Gestiva (January 31, 2007)
- Cytyc Acquires Adeza Biomedical Corporation (April 3, 2007)
Adeza’s name changed to Cytyc Prenatal Products Corp.

TAKE A TOUR
KODAIKANAL, TAMILNADU, INDIA
-
Kodaikanal – Wikipedia, the free encyclopedia
en.wikipedia.org/wiki/Kodaikanal
Kodaikanal is a city in the hills of the Dindigul district in the state of Tamil Nadu, India. Its name in the Tamil language means “The Gift of the Forest”. Kodaikanal …
.

 .
.
 .
.

 .
.

.
 .
.




 .
.

CUISINE OF TAMILNADU
Cuisine of Tamil Nadu

Like all other South Indian states, Tamil Nadu is also known for a wide variety of delicious food both for the vegetarians as well as the non-vegetarians. Grains, lentils, rice and vegetables are the main ingredients of the traditional foods of Tamil Nadu. Spices add flavor and give a distinctive taste to the Tamil cuisines. Some of the most common and popular dishes of the region are idly, dosai, vada, pongal and Uppuma. Coconut chutney and sambhar invariably form a part of most of the Tamil dishes.
The typical Tamil breakfast includes dosai, which is a pancake made from a batter of rice, idly (steamed rice cakes) and lentils (crisp fried on a pan), vada (deep fried doughnuts prepared from a batter of lentils), pongal (a mash of rice and lentils boiled together and seasoned with cashew nuts, ghee, pepper and cummin seed), uppuma (cooked semolina seasoned in oil with mustard, pepper, cummin seed and dry lentils). These are the main local dishes but there are several variations that are eaten with coconut chutney and mulaga podi.For lunch and the main course, the food consists of boiled rice, which is served with an assortment of vegetable dishes, sambar, chutneys, rasam (a hot broth prepared from tamarind juice and pepper) and curd. On the other hand, the non-vegetarian lunch and dinner include curries and dishes cooked with chicken, mutton or fish. Crispy Papad/Papar and appalam form an important part of a typical Tamil meal.Filter coffee is a famous and popular beverage of the people of Tamil Nadu in general and Chennai in particular. It is interesting to note that making of filter coffee is like a ritual as the coffee beans are first roasted and then powdered. After the grinding work is over, the powder is put into a filter set and then boiling water is added to prepare the decoction, which is allowed to set for about 15-18 minutes. The decoction is ready and can be added to milk with sugar according to taste. The coffee is poured from one container to another in quick succession so that the ideal frothy cup of filter coffee is ready.

Chettinad Cuisine
Chettinad cuisine is one of the spiciest and most aromatic in India. The name Chettinad cuisine comes from the place of its origin, Chettinad. Chettinad cuisine and delicacy is a specialty of Tamil Nadu and is a delight for non-vegetarian food lovers. The Chettinad cuisine consists of several variations of mutton, fish, and chicken items. The Chettinad Pepper Chicken is a specialty of all the non-vegetarian dishes. Dishes like biryani and paya are popular Tamil style of Mughali food. Paya is a type of spiced trotters broth and is usually eaten with either parathas or appam.






Tapioca Masala

 .
.


…………….

THANKS AND REGARD’S
DR ANTHONY MELVIN CRASTO Ph.D
DR ANTHONY MELVIN CRASTO Ph.D
MOBILE-+91 9323115463
GLENMARK SCIENTIST , INDIA
web link
web link
http://anthonycrasto.jimdo.com/
blogs are


Congratulations! Your presentation titled “Anthony Crasto Glenmark scientist, helping millions with websites” has just crossed MILLION views.
アンソニー 安东尼 Энтони 안토니 أنتوني
join my process development group on google
you can post articles and will be administered by me on the google group which is very popular across the world
LinkedIn group
blogs are
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
Join me on Linkedin
Join me on Facebook
Join me on twitter
Join me on google plus Googleplus
Googleplus amcrasto@gmail.com
BMS 564929, Androgen receptor (AR) modulator

BMS-564929; BMS564929; 627530-84-1; 2-Chloro-4-[(7r,7as)-7-Hydroxy-1,3-Dioxotetrahydro-1h-Pyrrolo[1,2-C]imidazol-2(3h)-Yl]-3-Methylbenzonitrile; hydantoin,
CAS 627530-84-1, Squibb Bristol Myers Co
| Molecular Formula: | C14H12ClN3O3 |
|---|---|
| Molecular Weight: | 305.71638 g/mol |
4-[(7R,7aS)-7-Hydroxy-1,3-dioxoperhydropyrrolo[1,2-c]imidazol-2-yl]-2-chloro-3-methylbenzonitrile
7-K,7aS)-2-Chloro-4-(7-hydroxy-l,3-dioxotetrahydropyrrolo[l,2- c]imidazol-2-yl)-3-methylbenzonitrile
4-[(7R,7aS)-7-hydroxy-1,3-dioxo-5,6,7,7a-tetrahydropyrrolo[1,2-c]imidazol-2-yl]-2-chloro-3-methylbenzonitrile
BMS-564929 is a highly potent, orally active and nonsteroidal tissue selective modulator of androgen receptor (AR) with Ki value of 2.11 nM.
BMS-564929 is a selective androgen receptor (AR) modulator with Ki value of 2.11 ± 0.16 nM [1].
The AR is a type of nuclear receptor that is activated by the androgenic hormones, testosterone, or dihydrotestosterone. The important function is regulating gene expression.
BMS-564929 is a muscle-tissue specific agonist for AR with a bicyclic hydantoin structure [2]. BMS-564929 is about 400-fold selective for AR vs. PR and more than 1000-fold selective for AR vs. GR, MR and ERα and β. In the C2C12 myoblast cell line, BMS-564929 shows a potency of 0.44 ± 0.03 nM compared with 2.81 ± 0.48 nM measured for testosterone
In castrated male rats, BMS-564929 is substantially more potent than testosterone (T) in promoting the growth of the levator ani muscle, and is highly selective for muscle vs. Prostate. Because of its potent oral activity and tissue selectivity, BMS-564929 is expected to yield beneficial clinical effects in muscle and other tissues with a more favorable safety way
BMS-564,929 is an investigational selective androgen receptor modulator, which is being developed by Bristol-Myers Squibb for treatment of the symptoms of age-related decline in androgen levels in men (“andropause“). These symptoms may includedepression, loss of muscle mass and strength, reduction in libido and osteoporosis. Treatment with exogenous testosterone is effective in counteracting these symptoms but is associated with a range of side effects, the most serious of which is enlargement of the prostate gland, which can lead to benign prostatic hypertrophy and even prostate cancer. This means there is a clinical need for selective androgen receptor modulators, which produce anabolic effects in some tissues such as muscle and bone, but without stimulating androgen receptors in the prostate.[1]
BMS-564,929 is one such compound currently in early human clinical trials, which is an orally active, potent and selective agonist for androgen receptors (Ki 2.1nM, 20x functional selectivity for muscle tissue over prostate) and in studies on castrated rats it was shown to counteract decrease in muscle mass over time, and at higher doses even increased muscle mass, without significantly affecting prostate tissue.[2] It does however vastly reduce luteinizing hormone levels, it being an astonishing 33x more suppressive compound than testosterone,[3] which may be a problem in human clinical use.[4]
Selective androgen receptor modulators may also be used by athletes to assist in training and increase physical stamina and fitness, potentially producing effects similar to anabolic steroids but with significantly fewer side effects. For this reason, SARMs have already been banned by the World Anti-Doping Agency since January 2008 despite no drugs from this class yet being in clinical use, and blood tests for all known SARMs are currently being developed.[5][6]
| Patent | Submitted | Granted |
|---|---|---|
| Bicyclic modulators of androgen receptor function [US2004019063] | 2004-01-29 | |
| BICYCLIC MODULATORS OF ANDROGEN RECEPTOR FUNCTION [US7772267] | 2008-05-08 | 2010-08-10 |
| Bicyclic modulators of androgen receptor function [US7405234] | 2004-09-16 | 2008-07-29 |
WO 2003096980
http://www.google.com/patents/WO2003096980A2?cl=en
Example 23
(7-K,7aS)-2-Chloro-4-(7-hydroxy-l,3-dioxotetrahydropyrrolo[l,2- c]imidazol-2-yl)-3-methylbenzonitrile

23 A. 3-Chloro-2-methylphenyIacetamide

To a solution of 3-chloro-2-methylaniline (3.00 g, 21.2 mmol) in 25 mL of EtOH at rt was added acetic anhydride (2.40 mL, 25.4 mmol), and the solution was stirred at rt for 2 h. The mixture was concentrated under reduced pressure to give 3.89 g (100%) of the desired acetamide. 1H NMR (DMSO- ) δ 2.05 (s, 3H), 2.20 (s, 3H), 7.16 (t, J = 1.1, 8.3, 1H), 7.25 (d, J = 8.3, 1H), 7.31 (d, J = 8.3, 1H), 9.55 (s, 1H); 13C NMR (DMSO- ) δ 15.1, 23.1, 124.4, 125.8, 126.7, 130.3, 133.7, 138.0, 168.3; HPLC a) column: Phenominex ODS C18 4.6 x 50 mm, 4 min gradient, 10% MeOH/90% H2O/0.1% TFA to 90% MeOH/10% H2O/0.1% TFA; 1 min hold, 4 mL/min UV detection at 220 nm, 2.32 min retention time; HPLC b) column: Shimadzu Shim-Pack VP-ODS CI 8 4.6 x 50 mm, 4 min gradient, 10% MeOH/90% H2O/0.1% TFA to 90% MeOH/10% H2O/0.1% TFA, 1 min hold; 4 mL/min, UV detection at 220 nm, 2.20 min retention time (99%); MS (ES) m/z 184 [M+H]+.
no 23B. 4-Bromo-3-chloro-2-methylphenylacetamide

To a suspension of acetamide 23A (2.00 g, 10.9 mmol) in 15 mL of glacial AcOH cooled to approximately 15 °C was added bromine (1.67 mL, 32.7 mmol) over 20 min. The ice bath was removed and the solution was stirred for
2 h, poured into ice water with stirring, and the solid was then filtered and dried to give 2.75 g (96%) of the desired bromide. 1H NMR (DMSO-_i6) δ 2.05 (s,
3H), 2.28 (s, 3H), 7.29 (d, J = 8.3, 1H), 7.56 (d, J = 8.8, 1H), 9.60 (s, 1H); 13C NMR (DMSO–i6) δ 16.7, 23.1, 118.1, 125.5, 130.4, 132.7, 133.4, 137.1, 168.4;
HPLC a) column: Phenominex ODS C18 4.6 x 50 mm, 4 min gradient, 10%
MeOH/90% H2O/0.1% TFA to 90% MeOH/10% H2O/0.1 % TFA, 1 min hold,
4 mL/min, UV detection at 220 nm, 2.95 min retention time; HPLC b) column:
Shimadzu Shim-Pack VP-ODS C18 4.6 x 50 mm, 4 min gradient, 10% MeOH/90% H2O/0.1% TFA to 90% MeOH/10% H2O/0.1% TFA, 1 min hold,
4 mL/min, UV detection at 220 nm, 2.87 min retention time (98%); MS (ES) m/z 263 [M+H]+.
23C. 3-Chloro-4-cyano-2-methylphenylacetamide

A suspension of bromide 23B (2.70 g, 10.3 mmol) and copper cyanide (0.92 g,
10.3 mmol) in DMF (30 mL) was heated to 150 °C for 4 h. The suspension was cooled, poured into water with stirring, and the solid was filtered and dried to give 1.44 g (67%) of the desired nitrile. 1H NMR (DMSO-d6) δ 2.12 (s, 3H),
i n 2.29 (s, 3H), 7.72 (d, J = 8.8, 1H), 7.75 (d, J = 8.2, 1H), 9.73 (s, 1H); 13C NMR (DMSO- ) δ 15.3, 23.5, 107.7, 116.5, 123.0, 130.1, 131.5, 135.7, 142.3, 168.8; HPLC a) column: Phenominex ODS C18 4.6 x 50 mm, 4 min gradient, 10% MeOH/90% H2O/0.1% TFA to 90% MeOH/10% H2O/0.1% TFA, 1 min hold, 4 mL/min, UV detection at 220 nm, 2.23 min retention time; HPLC b) column: Shimadzu Shim-Pack VP-ODS C18 4.6 x 50 mm, 4 min gradient, 10% MeOH/90% H2O/0.1% TFA to 90% MeOH/10% H2O/0.1% TFA, 1 min hold, 4 mL/min, UV detection at 220 nm, 2.13 min retention time (95%); MS (ES) m/z 209 [M+H]+.
23D. 3-Chloro-4-cyano-2-methylphenylaniline

A solution of cyanoacetamide 23C (9.90 g, 47.4 mmol) in 100 mL of concentrated HCl EtOH (1 :1) was refluxed 30 min. The solution was then concentrated and dried under reduced pressure to give 9.41 g (98%) of the desired aniline as the hydrochloride salt. The free base of the aniline was obtained by suspending the salt in EtOAc and washing with saturated aqueous NaHC03 solution. The organic layer was then dried (MgS04), filtered and concentrated under reduced pressure. Η NMR (OMSO-dβ) δ 2.12 (s, 3H), 6.30 (s, 2H), 6.61 (d, J = 8.23, 1H), 7.36 (d, J = 8.23, 1H); 13C NMR DMSO-d6) δ 13.8, 96.9, 112.1, 118.3, 118.85, 132.2, 135.6, 152.5; HPLC a) column: Phenominex ODS C18 4.6 x 50 mm, 4 min gradient, 10% MeOH/90% H2O/0.1% TFA to 90% MeOH/10% H2O/0.1% TFA, 1 min hold, 4 mL/min, UV detection at 220 nm, 2.43 min retention time; HPLC b):column: Shimadzu Shim-Pack VP-ODS C18 4.6 x 50 mm, 4 min gradient, 10% MeOH/90% H2O/0.1% TFA to 90% MeOH/10% H2O/0.1% TFA, 1 min hold, 4 mL/min, UV detection at 220 nm, 2.31 min retention time (99%); MS (ES) m/z 167 [M+H]+.
23E. 2-Chloro-4-isocyanato-3-methylbenzonitriIe
5

The title compound was prepared from compound 23D in a manner similar to that described in Experiments 2D to 2E.
l o 23F. (2S,3-R)-l-(3-Chloro-4-cyano-2-methylphenylcarbamoyl)-3-hydroxy- pyrrolidine-2-carboxylic acid methyl ester

To a solution of hydroxyproline compound IF (493 mg, 3.40 mmol) in CH2C12
15 (15 mL) was added 4 A molecular sieves (~ 3.0 g), followed by isocyanate 23E (725 mg, 3.22 mmol), and the resulting mixture was stirred at rt overnight, filtered, and concentrated under reduced pressure. The residue was purified by flash chromatography (silica gel, 0.5% MeOH in EtOAc/hexane, 1: 1) to afford the title compound (736 mg) as an off-white solid. HPLC column: YMC S-5 0 C18 (4.6 x 50 mm), 0% to 100% B, 4 min gradient, 1 min hold (A = 90% H20 – 10% CH3CN – 0.1% TFA and B = 10% H20 – 90% CH3CN – 0.1% TFA), flow rate at 4 mL/min, UV detection at 220 nm, 1.57 min retention time (100%); MS (ES) m/z 338 [M+H]+. 23G. (7-R,7a5)-2-Chloro-4-(7-hydroxy-l,3-dioxotetrahydropyrroIo[l,2- c]imidazoI-2-yl)-3-methyIbenzonitrile.

To a suspension of cz‘s-3-hydroxyproline methyl ester, HCl salt (4.91 g, 27 mmol) in CH2C12 (100 mL) cooled to 0 °C was added -Pr2NEt (4.79 mL, 27.5 mmol). After stirring at rt for 15 min, isocyanate 23E was added as a solid in one portion through a powder addition funnel, rinsing with 50 mL CH2C12. The resulting light brown solution was stirred at rt until urea formation was complete (~ 2 h). To the mixture was then added DBU (4.6 mL, 30 mmol), and the resulting brown colored solution was stirred at rt until hydantoin formation was complete (~ 15 h). The product (4.72 g, 62%) was collected by filtration and washing with CH2C12 (2x). The mother liquor was then diluted with CH2C12 and washed with H20 (2x), 1 N HCl (2x) and brine. After removal of most of the solvent under reduced pressure, further product (1.2 g, 16%) was collected by filtration and washing with CH2C12 (2x). Recrystallization of the 4.72 g of crude product from hot THF and hexane gave 4.5 g of analytically pure product.
1H NMR (DMSO- ) δ 2.05-2.11 (m, 1H), 2.15-2.22 (m, 1H), 2.20, 2.24 (s, 3H), 3.29-3.33 (m, 1H), 3.59-3.68 (m, 1H), 4.42-4.50 (m, 2H), 5.64, 5.72 (d, J = 3.9, 3.3, 1H), 7.22, 7.51 (d, J = 8.3, 1H), 7.96 (d, I = 8.2,1H);
13C NMR (OMSO-d6) δ 15.4, 15.6, 35.5, 35.6, 43.3, 43.4, 68.8, 69.3, 69.8, 112.9, 113.1, 115.8, 128.1, 128.7, 132.1, 136.3, 136.4, 136.9, 137.1, 158.6, 169.1, 169.6;
HPLC a) column: Phenominex ODS C18 4.6 x 50 mm, 4 min gradient, 10% MeOH/90% H2O/0.1% TFA to 90% MeOH 10% H2O/0.1% TFA; 1 min hold; 4 mL/min, UV detection at 254 nm, 2.07 and 2.32 min retention time; HPLC b) column: Shimadzu Shim-Pack VP-ODS C18 (4.6 x 50 mm), 4 min gradient, 10% MeOH/90% H2O/0.1% TFA to 90% MeOH/10% H2O/0.1% TFA, 1 min hold, 4 mL/min, UV detection at 254 nm, 1.93 and 2.23 min retention time; Chiral HPLC column: Daicel Chiralcel OD 4.6 x 250 mm, isocratic, 30 min, 25% isopropanol/hexanes, 1 mL/min, UV detection at 254 nm; Shimadzu HPLC: 17.99 min retention time (>99%): Column: Hypercarb 5μ, 4.6 x 100 mm, 25 °C, isocratic, 30 min ACN/Η20 (35:65); 1 mL/min,
10.99 min retention time; MS (ES) m/z 306 [M+H]+. Alternatively, compound 23G can also be prepared by the following procedure: A solution of 22C (0.10 g, 0.28 mmol) and copper cyanide (0.03 g, 0.34 mmol) in DMF (1 mL) was refluxed for 3 h, cooled to rt, and diluted with water. The resulting solid was filtered, washed with water, dried and purified using preparative HPLC to afford the title compound (27 mg).
Alternatively, compound 23G can also be prepared by the following procedures: A solution of 22C (0.10 g, 0.278 mmol) and copper cyanide (0.03g, 0.334 mmol) in DMF (1 mL) was refluxed for 3 h, cooled to rt and diluted with water. The resulting solid was filtered, washed with water, dried and purified using preparative HPLC to afford the title compound (27 mg). HPLC: 99% at 2.06, 2.34 min (retention time) (Conditions: Phenom. Lura C18 (4.6 x 50 mm); Eluted with 0% to 100% B, 4 min gradient (A = 90% H20 – 10% MeOH – 0.1% TFA and B = 10% H20 – 90% MeOH – 0.1% TFA); Flow rate at 4.0 mL/min. UV detection at 220 nm). Chiral HPLC: retention time = 11.04 min (99%); Conditions: OD (4.6 x 250 mm); Eluted with 25% isopropanol in hexane for 30 min at 1 mL/min. MS (ES) m/z 306 [M+l]+.
References
- Gao, W; Dalton, JT (2007). “Expanding the therapeutic use of androgens via selective androgen receptor modulators (SARMs)”. Drug Discovery Today 12 (5–6): 241–8. doi:10.1016/j.drudis.2007.01.003. PMC 2072879. PMID 17331889.
- Ostrowski, J; Kuhns, JE; Lupisella, JA; Manfredi, MC; Beehler, BC; Krystek Jr, SR; Bi, Y; Sun, C et al. (2007). “Pharmacological and x-ray structural characterization of a novel selective androgen receptor modulator: potent hyperanabolic stimulation of skeletal muscle with hypostimulation of prostate in rats”. Endocrinology 148 (1): 4–12. doi:10.1210/en.2006-0843. PMID 17008401.
- http://antaeuslabs.blogspot.com/2011/01/hydantoin-derivative-sarms-bms-564929.html
- Gao, W; Dalton, JT (2007). “Ockham’s Razor and Selective Androgen Receptor Modulators (SARMs): Are We Overlooking the Role of 5α-Reductase?”. Molecular interventions 7 (1): 10–3.doi:10.1124/mi.7.1.3. PMC 2040232. PMID 17339601.
- Thevis, M; Kohler, M; Schlörer, N; Kamber, M; Kühn, A; Linscheid, MW; Schänzer, W (2008). “Mass spectrometry of hydantoin-derived selective androgen receptor modulators”. Journal of mass spectrometry : JMS 43 (5): 639–50. doi:10.1002/jms.1364. PMID 18095383.
- Thevis, M; Kohler, M; Thomas, A; Maurer, J; Schlörer, N; Kamber, M; Schänzer, W (2008). “Determination of benzimidazole- and bicyclic hydantoin-derived selective androgen receptor antagonists and agonists in human urine using LC-MS/MS”. Analytical and Bioanalytical Chemistry 391 (1): 251–61. doi:10.1007/s00216-008-1882-6. PMID 18270691.
External links
- Crystal Structure Of The Rat Androgen Receptor Ligand Binding Domain Complex With Bms-564929 at Protein Data Bank
 |
|
| Systematic (IUPAC) name | |
|---|---|
| (7R,7aS)-2-Chloro-4-(7-hydroxy-1,3-dioxotetrahydropyrrolo[1,2-c]imidazol-2-yl)-3-methylbenzonitrile | |
| Clinical data | |
|
|
| Identifiers | |
| PubChem | CID 9882972 |
| DrugBank | DB07286 |
| ChemSpider | 8058647 |
| Chemical data | |
| Formula | C14H12ClN3O3 |
| 305.716 g/mol | |
//////////
TAKE A TOUR
TAKE A TOUR
TAKE A TOUR
Jejuri, Pune district, Maharashtra, INDIA
-
Jejuri – Wikipedia, the free encyclopedia
en.wikipedia.org/wiki/JejuriJejuri is a city and a municipal council in Pune district in the Western Indian state of Maharashtra. It is famous for the main temple of Lord Khandoba.
















Jejuri is situated 48 km from Pune in Maharashtra State. Jejuri can be reached is by Road or Rail from Pune. Number of State Transport buses ply from Pune. It can be reached by Express trains from Pune Railway Station. GKP LTT Express Train no.15018 departure 0450 hrs from Pune PN arrival Jejuri JJR 0548 hrs, Maharshtra Express Train no.11040 departure 0450 hrs from Pune PN arrival Jejuri JJR 0549 hrs Koyana Express Train no.11029 departure 0045 hrs from Pune PN arrival Jejuri JJR 0148 hrs Sahyadri Express Train no.11023 departure 2205 hrs from Pune PN arrival Jejuri JJR 2308 hrs.These trains runs all days.
Jejuri is one of the most famous religious places in Maharashtra. The Village Jejuri is popularly known as Khanderayachi Jejuri.
Jejuri’s Khandoba Temple is built on a hill, which is approximately 51 kilometers away from Pune Railway Station. As the Temple is on the hill, one has to ascend more than 200 steps. But the ascending is not so tough and the wonderful view of Jejuri village is superb. If weather permits, One can easily see the spectacular view of Dive and Saswad Ghat. One can enjoy number of `Deep Mala’ (lamp post) while climbing the hill. Jejuri is really popular for its old Deep Malas.
The Jejuri temple was constructed in 1608. The Sabhamandap (Audience Hall) and other parts of the structure were completed subsequently. In 1742, Holkars constructed pillars and completed battlements and tank. The devotees added gateways, stairways, lamp pillars, cloisters etc.
The Idol of Lord khandoba in the Temple is beautiful.
The shepherd community considers Khandoba as their family deity.
One must visit Jejuri to look the Crystal Stands. Jejuri is one of the important temples in Maharashtra with historical significance.
Khandobacha Yelkot, Yelkot Yelkot Jay Malhar, Sadanandacha Yelkot, Kadepathar Maharaj Ki Jay are some of the popular terms here.
One can find many idols in and nearby the Jejuri Temple.
Temple Festivals :-
Chaitra Pournima, Dussehra, Champa Shashthi, Paush Pournima, Magh Pournima, Mahashivratri, Somvati Amavasya, Guru Pournima
Pandit/Brahmin for Pooja in Jejuri :-
Upadhye Guruji-9850150797, 02115-253152
Accommodation :-
Shree Siddhi Lodge and Hotel # 02115-253090
Best time to visit Jejuri :-
Throughout the year
Nearest Railway Station :-
Jejuri Railway Station
Distance :-
Mumbai – Jejuri – 206 Kilometers (By Road) Via Mumbai Pune Express Way
Nearby Attractions / Holy Places :-
Shree Mayureshwar Temple, Morgaon (Ashta Vinayak)
Pandeshwar
Bhuleshwar
Saswad
Kanifnath Temple
Balaji Temple


A painting depicts Khandoba riding a white horse with Mhalsa, accompanied with a dog and attendants including a Waghya dancing before him.
Khandoba and Mhalsa killing demons Mani-Malla — a popularoleograph, c.1880.
| Khandoba | |
|---|---|

|
