WORLD RECORD VIEWS holder on THIS BLOG, ………live, by DR ANTHONY MELVIN CRASTO, Worldpeaceambassador, Worlddrugtracker, Helping millions, 100 million hits on google, pushing boundaries,2.5 lakh plus connections worldwide, 45 lakh plus VIEWS on this blog in 227 countries, 7 CONTINENTS ……A 90 % paralysed man in action for you, I am suffering from transverse mylitis and bound to a wheel chair, [THIS BLOG HOLDS WORLD RECORD VIEWS ]
DR ANTHONY MELVIN CRASTO, Born in Mumbai in 1964 and graduated from Mumbai University, Completed his Ph.D from ICT, 1991,Matunga, Mumbai, India, in Organic Chemistry, The thesis topic was Synthesis of Novel Pyrethroid Analogues, Currently he is working with AFRICURE PHARMA, ROW2TECH, NIPER-G, Department of Pharmaceuticals, Ministry of Chemicals and Fertilizers, Govt. of India as ADVISOR, earlier assignment was
with GLENMARK LIFE SCIENCES LTD, as CONSUlTANT, Retired from GLENMARK in Jan2022 Research Centre as Principal Scientist, Process Research (bulk actives) at Mahape, Navi Mumbai, India. Total Industry exp 32 plus yrs, Prior to joining Glenmark, he has worked with major multinationals like Hoechst Marion Roussel, now Sanofi, Searle India Ltd, now RPG lifesciences, etc. He has worked with notable scientists like Dr K Nagarajan, Dr Ralph Stapel, Prof S Seshadri, etc, He did custom synthesis for major multinationals in his career like BASF, Novartis, Sanofi, etc., He has worked in Discovery, Natural products, Bulk drugs, Generics, Intermediates, Fine chemicals, Neutraceuticals, GMP, Scaleups, etc, he is now helping millions, has 9 million plus hits on Google on all Organic chemistry websites. His friends call him Open superstar worlddrugtracker. His New Drug Approvals, Green Chemistry International, All about drugs, Eurekamoments, Organic spectroscopy international,
etc in organic chemistry are some most read blogs He has hands on experience in initiation and developing novel routes for drug molecules
and implementation them on commercial scale over a 32 PLUS year tenure till date Feb 2023, Around 35 plus products in his career. He has good knowledge of IPM, GMP, Regulatory aspects, he has several International patents published worldwide . He has good proficiency in Technology transfer, Spectroscopy, Stereochemistry, Synthesis, Polymorphism etc., He suffered a paralytic stroke/ Acute Transverse mylitis in Dec 2007 and is 90 %Paralysed, He is bound to a wheelchair, this seems to have injected feul in him to help chemists all around the world, he is more active than before and is pushing boundaries, He has 100 million plus hits on Google, 2.5 lakh plus connections on all networking sites, 100 Lakh plus views on dozen plus blogs, 227 countries, 7 continents, He makes himself available to all, contact him on +91 9323115463, email amcrasto@gmail.com, Twitter, @amcrasto , He lives and will die for his family, 90% paralysis cannot kill his soul., Notably he has 38 lakh plus views on New Drug Approvals Blog in 227 countries......https://newdrugapprovals.wordpress.com/ , He appreciates the help he gets from one and all, Friends, Family, Glenmark, Readers, Wellwishers, Doctors, Drug authorities, His Contacts, Physiotherapist, etc
He has total of 32 International and Indian awards
CAS Name: 1-[[(6R,7R)-7-[[(2Z)-(2-Amino-4-thiazolyl)(methoxyimino)acetyl]amino]-2-carboxy-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-en-3-yl]methyl]-1-methylpyrrolidinium inner salt
Additional Names: 1-[[(6R,7R)-7-[2-(2-amino-4-thiazolyl)glyoxylamido]-2-carboxy-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-en-3-yl]methyl]-1-methylpyrrolidinium hydroxide inner salt 72-(Z)-2-(O-methyloxime); 7-[(Z)-2-(2-aminothiazol-4-yl)-2-methoxyiminoacetamido]-3-(1-methylpyrrolidinio)methyl-3-cephem-4-carboxylate
Manufacturers’ Codes: BMY-28142
Molecular Formula: C19H24N6O5S2
Molecular Weight: 480.56
Percent Composition: C 47.49%, H 5.03%, N 17.49%, O 16.65%, S 13.34%
Literature References: Semisynthetic, fourth generation cephalosporin antibiotic. Prepn: S. Aburaki et al.,DE3307550; eidem,US4406899 (both 1983 to Bristol-Myers); and antibacterial activity: T. Naito et al.,J. Antibiot.39, 1092 (1986). In vitro comparative antimicrobial spectrum: N. J. Khan et al.,Antimicrob. Agents Chemother.26, 585 (1984); and b-lactamase stability: H. C. Neu et al.,J. Antimicrob. Chemother.17, 441 (1986). HPLC determn in plasma and urine: R. H. Barbhaiya et al.,Antimicrob. Agents Chemother.31, 55 (1987). Clinical evaluations in infection: N. Clynes et al.,Diagn. Microbiol. Infect. Dis.12, 257 (1989); S. Oster et al.,Antimicrob. Agents Chemother.34, 954 (1990). Review of clinical pharmacokinetics: M. P. Okamoto et al.,Clin. Pharmacokinet.25, 88-102 (1993).
Properties: Colorless powder, mp 150° (dec). uv max (pH 7 phosphate buffer): 235, 257 nm (e 16700, 16100).
Melting point: mp 150° (dec)
Absorption maximum: uv max (pH 7 phosphate buffer): 235, 257 nm (e 16700, 16100)
Derivative Type: Sulfate
Molecular Formula: C19H24N6O5S2.H2SO4
Molecular Weight: 578.64
Percent Composition: C 39.44%, H 4.53%, N 14.52%, O 24.89%, S 16.62%
Properties: mp 210° (dec). uv max (pH 7 phosphate buffer): 236, 258 nm (e 17200, 16900).
Melting point: mp 210° (dec)
Absorption maximum: uv max (pH 7 phosphate buffer): 236, 258 nm (e 17200, 16900)
Derivative Type: Hydrochloride monohydrate
CAS Registry Number: 123171-59-5
CAS Name: 1-[[(6R,7R)-7-[[(2Z)-(2-Amino-4-thiazolyl)(methoxyimino)acetyl]amino]-2-carboxy-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-en-3-yl]methyl]-1-methylpyrrolidinium chloride monohydrochloride monohydrate
FDA APPROVED 2/22/2024, To treat complicated urinary tract infections, Exblifep
BMY 28142
BMY-28142
Cefepime is a fourth-generation cephalosporinantibiotic. Cefepime has an extended spectrum of activity against Gram-positive and Gram-negativebacteria, with greater activity against both types of organism than third-generation agents. A 2007 meta-analysis suggested when data of trials were combined, mortality was increased in people treated with cefepime compared with other β-lactam antibiotics.[1] In response, the U.S. Food and Drug Administration (FDA) performed their own meta-analysis which found no mortality difference.[2]
Cefepime is a broad-spectrum cephalosporin antibiotic and has been used to treat bacteria responsible for causing pneumonia and infections of the skin and urinary tract. Some of these bacteria include Pseudomonas, Escherichia, and Streptococcus species. The following represents MIC susceptibility data for a few medically significant microorganisms:[7]
Escherichia coli: ≤0.007 – 128 μg/ml
Pseudomonas aeruginosa: 0.06 – >256 μg/ml
Streptococcus pneumoniae: ≤0.007 – >8 μg/ml
Chemistry
The combination of the syn-configuration of the methoxyiminomoiety and the aminothiazole moiety confers extra stability to β-lactamase enzymes produced by many bacteria. The N–methylpyrrolidine moiety increases penetration into Gram-negative bacteria. These factors increase the activity of cefepime against otherwise resistant organisms including Pseudomonas aeruginosa and Staphylococcus aureus.
Semisynthetic, fourth generation cephalosporin antibiotic. Prepn: S. Aburaki et al., DE 3307550; eidem, US 4406899 (both 1983 to Bristol-Myers); and antibacterial activity: T. Naito et al., J. Antibiot. 39, 1092 (1986).
Trade names
Following expiration of the Bristol-Myers Squibb patent,[] cefepime became available as a generic and is now] marketed by numerous companies worldwide under tradenames including Neopime (Neomed), Maxipime, Cepimax, Cepimex, and Axepim.
^ Yahav D, Paul M, Fraser A, Sarid N, Leibovici L (May 2007). “Efficacy and safety of cefepime: a systematic review and meta-analysis”. The Lancet. Infectious Diseases. 7 (5): 338–348. doi:10.1016/S1473-3099(07)70109-3. PMID17448937.
^ “Cefepime (maxipime), large spectrum 4th generation cephalosporin, resistant to beta-lactamases]”.
^World Health Organization (2019). Executive summary: the selection and use of essential medicines 2019: report of the 22nd WHO Expert Committee on the selection and use of essential medicines. Geneva: World Health Organization. hdl:10665/325773. WHO/MVP/EMP/IAU/2019.05. License: CC BY-NC-SA 3.0 IGO.
^ Chapman TM, Perry CM (2003). “Cefepime: a review of its use in the management of hospitalized patients with pneumonia”. American Journal of Respiratory Medicine. 2 (1): 75–107. doi:10.1007/bf03256641. PMID14720024.
Berdazimer sodium, sold under the brand name Zelsuvmi, is a medication used for the treatment for molluscum contagiosum.[1] Berdazimer sodium is a nitric oxide releasing agent.[1] It is a polymer formed from sodium 1-hydroxy-3-methyl-3-(3-(trimethoxysilyl)propyl)-1-triazene-2-oxide and tetraethyl silicate.[2]
Berdazimer sodium was approved for medical use in the United States in January 2024.[3][4][5]
Medical uses
Berdazimer sodium is indicated for the topical treatment of molluscum contagiosum.[1]
Pharmacology
Mechanism of action
Berdazimer sodium is a nitric oxide releasing agent.[1] The mechanism of action for the treatment of molluscum contagiosum is unknown.[1]
Pharmacodynamics
The pharmacodynamics of berdazimer sodium are unknown.[1]
Society and culture
Legal status
Berdazimer sodium was approved for medical use in the United States in January 2024.[4]
Berdazimer is a polymeric substance consisting of a polysiloxane backbone (Si-O-Si bonds) with covalently bound N-diazeniumdiolate nitric oxide (NO) donors. It releases NO through exposure to proton donors like water, which will degrade the N-diazeniumdiolate entity.2 Berdazimer was previously investigated as a potential treatment for molluscum contagiosum, a viral cutaneous infection mainly affecting children, sexually active adults, and immunocompromised patients. It is one of the 5 most prevalent skin diseases in the world and the third-most common viral skin infection in children.3 Previously, the first line treatment for molluscum contagiosum was surgical excision, although it poses challenges such as repeated doctor visits, post-surgical scarring and skin discoloration, and fear and anxiety in the pediatric population.3
On Jan 05, 2024, the FDA approved berdazimer under the brand name ZELSUVMI for the treatment of adult and pediatric molluscum contagiosum, and it is the first drug to be approved for this condition. This decision is based on positive results demonstrated in 2 Phase 3 trials, B-SIMPLE 4 and B-SIMPLE 2, where reduced lesion counts were observed with once-a-day uses of berdazimer.5
^World Health Organization (2018). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 79”. WHO Drug Information. 32 (1). hdl:10665/330941.
Further reading
Pera Calvi I, R Marques I, Cruz SA, Mesquita YL, Padrao EM, Souza RM, et al. (2023). “Safety and efficacy of topical nitric oxide-releasing berdazimer gel for molluscum contagiosum clearance: A systematic review and meta-analysis of randomized controlled trials”. Pediatric Dermatology. 40 (6): 1060–1063. doi:10.1111/pde.15419. PMID37721050. S2CID262045499.
Han H, Smythe C, Yousefian F, Berman B (February 2023). “Molluscum Contagiosum Virus Evasion of Immune Surveillance: A Review”. Journal of Drugs in Dermatology. 22 (2): 182–189. doi:10.36849/JDD.7230. PMID36745361. S2CID256613906.
Clinical trial number NCT04535531 for “A Phase 3 Molluscum Contagiosum Efficacy and Safety Study (B-SIMPLE4)” at ClinicalTrials.gov
Clinical trial number NCT03927703 for “A Phase 3 Efficacy & Safety of SB206 & Vehicle Gel for the Treatment of MC (B-SIMPLE2)” at ClinicalTrials.gov
Clinical trial number NCT03927716 for “A Phase 3 Randomized Parallel Group Study Comparing the Efficacy & Safety of SB206 & Vehicle Gel in the Treatment of MC (B-SIMPLE1)” at ClinicalTrials.gov
fda approved 4/3/2024, To treat certain bloodstream infections, bacterial skin and associated tissue infections, and community-acquired bacterial pneumonia Press Release zevtera
Ceftobiprole, sold under the brand name Zevtera among others, is a fifth-generation[5]cephalosporin antibacterial used for the treatment of hospital-acquired pneumonia (excluding ventilator-associated pneumonia) and community-acquired pneumonia. It is marketed by Basilea Pharmaceutica under the brand names Zevtera and Mabelio.[6][7][8][9][10][11] Like other cephalosporins, ceftobiprole exerts its antibacterial activity by binding to important penicillin-binding proteins and inhibiting their transpeptidase activity which is essential for the synthesis of bacterial cell walls. Ceftobiprole has high affinity for penicillin-binding protein 2a of methicillin-resistant Staphylococcus aureus strains and retains its activity against strains that express divergent mecA gene homologues (mecC or mecALGA251). Ceftobiprole also binds to penicillin-binding protein 2b in Streptococcus pneumoniae (penicillin-intermediate), to penicillin-binding protein 2x in Streptococcus pneumoniae (penicillin-resistant), and to penicillin-binding protein 5 in Enterococcus faecalis.[12]
Medical uses
In the US, ceftobiprole is indicated for the treatment of adults with Staphylococcus aureus bloodstream infections (bacteremia) including those with right-sided infective endocarditis;[4] adults with acute bacterial skin and skin structure infections;[4] and people with community-acquired bacterial pneumonia.[4]
Microbiology
Ceftobiprole has shown in vitro antimicrobial activity against a broad range of Gram-positive and Gram-negative pathogens. Among the Gram-positive pathogens, ceftobiprole has demonstrated good in vitro activity against methicillin-resistant Staphylococcus aureus, methicillin-susceptible Staphylococcus aureus and coagulase-negative staphylococci, as well as against strains of methicillin-resistant Staphylococcus aureus with reduced susceptibility to linezolid, daptomycin or vancomycin.[13] Ceftobiprole has also displayed potent activity against Streptococcus pneumoniae (including penicillin-sensitive, penicillin-resistant and ceftriaxone-resistant strains) and Enterococcus faecalis, but not against Enterococcus faecium. For Gram-negative pathogens, ceftobiprole has shown good in vitro activity against Haemophilus influenzae (including both ampicillin-susceptible and ampicillin-non-susceptible isolates), Pseudomonas aeruginosa and strains of Escherichia coli, Klebsiella pneumoniae and Proteus mirabilis that do not produce extended-spectrum β-lactamases (ESBL). Like all other cephalosporins, ceftobiprole was inactive against strains that produce extended-spectrum β-lactamases.[14]
The efficacy of ceftobiprole has been demonstrated in two large randomized, double-blind, phase 3 clinical trials in patients with hospital-acquired and community-acquired pneumonia. Ceftobiprole was non-inferior to ceftazidime plus linezolid in the treatment of hospital-acquired pneumonia (excluding ventilator-acquired pneumonia) and non-inferior to ceftriaxone with or without linezolid in the treatment of community-acquired pneumonia.[15][16]
Pharmacology
Ceftobiprole medocaril
Ceftobiprole is the active moiety of the prodrug ceftobiprole medocaril and is available for intravenous treatment only. It is mainly excreted via the kidney.[17]
Society and culture
Legal status
500 mg powder
Ceftobiprole has been approved for the treatment of adults with hospital acquired pneumonia (excluding ventilator-acquired pneumonia) and community-acquired pneumonia in twelve European countries, Canada, and Switzerland.[18]
In February 2010, the Committee for Medicinal Products for Human Use of the European Medicines Agency adopted a negative opinion, recommending the refusal of the marketing authorization for the medicinal product Zeftera, intended for treatment of complicated skin and soft-tissue infections in adults. The company that applied for authorization is Janssen-Cilag International N.V. The applicant requested a re-examination of the opinion. After considering the grounds for this request, the CHMP re-examined the opinion, and confirmed the refusal of the marketing authorization in June 2010.[19]
Processes for producing ceftobiprole medocaril are known per se. What the processes known from the prior art have in common is that, starting from 7-aminocephalosporanic acid, a large number of intermediates have to be produced, isolated and purified in order to obtain ceftobiprole medocaril of the general formula (1) in sufficient purity.
The compound of the general formula (1) is known per se and is described, for example, in WO 99/65920. It can be used for the treatment and prophylaxis of bacterial infectious diseases, especially infectious diseases caused by methicillin-resistant Staphylococcus Aureus strains.
WO 99/65920 describes, as the last step in the production process of ceftobiprole Medocaril, a reaction in which the Medocaril prodrug unit is introduced into a compound of the general formula (2).
The compound of the general formula (2) is also known per se and has been described, for example, in EP 0 849 269 A1. The compound of the general formula (2) is prepared according to EP 0 849 269 A1 starting from (2R,6R,7R)-te rt. B u toxyc abonylamin o-3-formyl-8-oxo-5-thia-1 -azabicyclo[4.2.0]oct-3-ene-2-carboxylic acid benzhydryl ester by Wittig reaction with (1 ‘-allyloxycarbonyl-2- oxo-[1,3’]bipyrrolidinyl-3-yl)-triphenylphosphonium bromide. The resulting Δ2 reaction product is reisomerized to the desired Δ3 isomer by sulfoxidation and subsequent reduction and then deprotected from the benzhydryl ester with trifluoroacetic acid. The acylation in position 7 occurs by reaction with (Z)-(5-amino-[1,2,4]-thiadiazol-3-yl)-trityloxyiminothioacetic acid S-benzothiazol-2-yl ester. The compound of the general formula (2) is then obtained by removing the protective groups.
In EP 1 067 131 A1 the formation of the ylide in toluene or a mixture of toluene and dichloromethane is tert by adding alkali. Butylate in tetrahydrofuran, which allows the base to be added as a solution. The reaction of the ylide with the corresponding aldehyde is described at a reaction temperature of -70 0 C.
EP 0 841 339 A1 relates to cephalosporin derivatives and processes for their production. WO 95/29182 also discloses intermediates for the production of cephalosporins.
WO 01/90111 describes a further production of ceftobiprole Medocaril in several stages starting from desacetyl-7-aminocephalosporanic acid by acylation with (Z)-(5-amino-[1,2,4]-thiadiazol-3-yl)- trityloxyiminothioacetic acid S-benzothiazol-2-yl ester in N,N-dimethylformamide, followed by in situ esterification with diphenyldiazomethane in dichloromethane to give the corresponding benzohydryl ester, which is precipitated and isolated by adding hexane. In the next step, this product is oxidized to the corresponding aldehyde using TEMPO/NaOCI in dichloromethane/water or with Braunstein in tetrahydrofuran/dichloromethane. The next reaction step involves the Wittig reaction to the 3-vinyl-substituted derivative, in which the reaction takes place in dichloromethane/toluene/tetrahydrofuran at -78°C. The crude product is stirred with ethanol and made from dichloromethane/tert. Butyl methyl ether recrystallized or purified chromatographically. According to the method disclosed in WO 01/901 11, the Wittig reaction is carried out at low temperatures of -80 to -70 0 C in a complex solvent mixture of dichloromethane, toluene and tetrahydrofuran. This leads to significant disadvantages when carrying out the reaction on a production scale, since regeneration of the process solvents is difficult.
5 , 1 4 g of 7-amino-3-formyl-ceph-3-em-4-carboxy I at was dissolved in 2 7 , 8 m bis(trimethylsilyl)acetamide and 50 ml propylene oxide. 16.8 g of (1 R/S,3’R)-(1′-tert-butyloxycarbonyl-2-oxo-[1,3′]bipyrrolidinyl-3-yl)-triphenylphosphonium bromide (EP1067131, WO02/14332) slowly added in portions. Stirring was continued at 1 ° C until the starting material had reacted and then the crystalline precipitate was added
Nitrogen atmosphere filtered off and washed with 50 ml cyclohexane/bis(tirmethylsilyl)acetamide 99.5/0.5. After drying under vacuum, the desired product was obtained in silylated form.
The material was dissolved in 100ml dichloromethane and at 0 0 C with 50ml 3%
NaHCC> 3 solution added. The phases were separated, the organic phase was washed with 30 ml of water and the combined water phases were adjusted to pH 3.5 with 3% H 3 PO 4 after activated carbon treatment. The crystalline precipitate was filtered, washed with water and dried under vacuum.
10.28 g of 7-amino-3-formyl-ceph-3-em-4-carboxylate were dissolved in 55.6 ml of bis(trimethylsilyl)acetamide and 100 ml of propylene oxide. 33.6 g of (1R/S, 3’R)-(1′-tert. Butyloxycarbonyl-2-oxo-[1,3′]bipyrrolidinyl-3-yl)-triphenylphosphonium bromide (EP1067131, WO02.) were then added at 0 0 C /14332) slowly added in portions over 22 hours. The mixture was stirred at 1° C. until the starting material had reacted and then the reaction mixture was cooled to -20 ° C. The crystalline precipitate was filtered off under a nitrogen atmosphere and washed in portions with 180 ml of cyclohexane/bis(trimethylsilyl)acetamide 99.5/0.5. After drying under vacuum, the bissilylated
1.0g (6R,7R)-7-Trimethylsilylamino-3[E-(R)-1′-(5-tert.butyloxycarbonyl)-2-oxo-[I.Slbipyrrolidinyl-S-ylidenemethyO-δ-oxo-δ -thia-i-aza-bicyclo^^.Oloct^-ene^- carboxylic acid trimethylsilyl esters were dissolved in 10ml dichloromethane and a solution of 300mg dicyclohexylamine in 1ml EtOH and 10ml ethyl acetate was added. The precipitate was filtered off, washed with ethyl acetate and dried in vacuo.
3.0g (6R,7R)-7-Amino-3[E-(R)-1′-(5-tert-butyloxycarbonyl)-2-oxo-[1,3′]bipyrrolidinyl-3-ylidenemethyl]-8 -oxo-5-thia-1-aza-bicyclo[4.2.0]oct-2-ene-2-carboxylic acid in silylated form was dissolved in 150 ml of dichloromethane at 0°. 600 μl of DMF/water 5/1 and 1.8 ml of bis(trimethylsilyl) acetamide and 2.29 g of 2-trityloxyimino-2-(5-amino-1,2,4-thiadiazol-3-yl) acetic acid chloride were then added Hydrochoride (J. Antibiotics 37:557 – 571, 1984) was added in portions.
After 3 hours at 0°, the mixture was poured into 30 ml MeOH/120 ml water and the methylene chloride phase was separated off. The organic phase was concentrated to 66g and 25ml of trifluoroacetic acid was added. After 10 minutes, 1.5 ml triethylsilane and 10 ml water were added and the mixture was cooled to -15 ° C. The organic phase was separated off and again with 6 ml
Washed trifluoroacetic acid/water 1/1. The combined aqueous phases were diluted to 150 ml with water and filtered through an adsorber resin column with XAD-1600. After washing out the column with water, elution was carried out with water/acetonitrile 85/15. The product-containing fractions were concentrated in vacuo and allowed to stand at 0° for post-crystallization. The crystalline
Product was filtered off, washed with water and dried under vacuum.
Auswaage: 2,66g
4.2 Variant B:
7.4g (6R,7R)-7-Amino-3[E-(R)-1′-(5-tert-butyloxycarbonyl)-2-oxo-[1,3′]bipyrrolidinyl-3-ylidenemethyl]-8 -oxo-5-thia-1-aza-bicyclo[4.2.0]oct-2-ene-2-carboxylic acid was dissolved at 0° in 781 ml of dichloromethane with the addition of 6.7 ml of triethylamine. 8.65 g of 2-trityloxyimino-2-(5-amino-1,2,4-thiadiazol-3-yl)-acetic acid chloride hydrochloride were then added in portions. After the starting material had reacted, the mixture was poured into 500 ml of water and the methylene chloride phase was separated off. The organic phase was dried over Na 2 SC> 4 and concentrated in vacuo.
The residue was dissolved in 148 ml of dichloromethane and 4.5 ml of triethylsilane and 74 ml of trifluoroacetic acid were added at room temperature. After 30 minutes, 222 ml of dichloromethane and 222 ml of water were added and the mixture was cooled to -20 0 C. The organic phase was separated off and washed again with a mixture of 37 ml of trifluoroacetic acid and 148 ml of water. The combined aqueous phases were diluted with water to 364 ml, filtered through an adsorber resin and eluted with acetonitrile/water 15/85.
The filtrate was concentrated to 35g on a Rotavapor, filtered and washed with water.
After drying in a vacuum, 4.5 g of the sample was obtained.
6.0g (6R,7R)-7-Amino-3[E-(R)-1′-(5-tert-butyloxycarbonyl)-2-oxo-[1,3′]bipyrrolidinyl-3-ylidenemethyl]-8 -oxo-5-thia-1-aza-bicyclo[4.2.0]oct-2-ene-2-carboxylic acid in silylated form was dissolved in 300 ml of dichloromethane at 0°. 1200 μl of DMF/water 5/1 and 8.1 ml of bis(tirmethylsilyl)acetamide were then added as well as 5.3g of 2-trityloxyimino-2-(5-amino-1,2,4-thiadiazol-3-yl)acetic acid chloride Hydrochoride (J. Antibiotics 37:557 – 571, 1984) added in portions. The mixture was then poured into 60 ml MeOH/240 ml water and the methylene chloride phase was separated off. The organic phase was concentrated to 48g and 1.5 ml of triethylsilane was added. After adding 50ml
Trifluoroacetic acid was stirred at room temperature for 60 min, 20 ml of water was added and the mixture was cooled to -15°C. The organic phase was separated off and washed again with 20 ml trifluoroacetic acid/water 1/1. The combined aqueous phases were diluted to 500 ml with water and treated with 2.0 g of activated carbon. After filtration, the solution was concentrated in vacuo.
The residue was diluted to 50 ml with water and adjusted to pH 6.9 with saturated NaHCO 3 solution. The mixture was stirred at 0 0 C for 2 hours, filtered and the precipitate washed with water.
0, 5 3 g ( 6 R , 7 R )-7-[(Z)-2-(5-amino-[1,2,4]thiadiazol-3-yl)-2-hydroxyimino-acetylamino]-8- oxo-3-[(E)-(R)-2-oxo-[1,3′]bipyrrolidinyl-3-ylidenemethyl]-5-thia-1 – aza-bicyclo[4.2.0]oct-2-ene- 2 carboxylic acid were dissolved in 5 ml of dimethyl sulfoxide and 0.27 g of carbonic acid (5-methyl-2-oxo-[1,3]dioxol-4-ylmethyl)-4-nitrophenyl ester were added and stirred at room temperature. A solution of sodium ethyl hexanoate in 30 ml of acetone was added for precipitation. The precipitate was filtered and washed with acetone.
^ Zhanel GG, Lam A, Schweizer F, Thomson K, Walkty A, Rubinstein E, et al. (2008). “Ceftobiprole: a review of a broad-spectrum and anti-MRSA cephalosporin”. American Journal of Clinical Dermatology. 9 (4): 245–254. doi:10.2165/00128071-200809040-00004. PMID18572975. S2CID24357533.
^ Farrell DJ, Flamm RK, Sader HS, Jones RN (April 2014). “Activity of ceftobiprole against methicillin-resistant Staphylococcus aureus strains with reduced susceptibility to daptomycin, linezolid or vancomycin, and strains with defined SCCmec types”. International Journal of Antimicrobial Agents. 43 (4): 323–327. doi:10.1016/j.ijantimicag.2013.11.005. PMID24411474.
^ Nicholson SC, Welte T, File TM, Strauss RS, Michiels B, Kaul P, et al. (March 2012). “A randomised, double-blind trial comparing ceftobiprole medocaril with ceftriaxone with or without linezolid for the treatment of patients with community-acquired pneumonia requiring hospitalisation”. International Journal of Antimicrobial Agents. 39 (3): 240–246. doi:10.1016/j.ijantimicag.2011.11.005. PMID22230331.
^“Zeftera (previously Zevtera) EPAR”. European Medicines Agency (EMA). 18 February 2010. Retrieved 6 April 2024. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
External links
Clinical trial number NCT03138733 for “Ceftobiprole in the Treatment of Patients With Staphylococcus Aureus Bacteremia” at ClinicalTrials.gov
Clinical trial number NCT03137173 for “Ceftobiprole in the Treatment of Patients With Acute Bacterial Skin and Skin Structure Infections” at ClinicalTrials.gov
Clinical trial number NCT00326287 for “Ceftobiprole in the Treatment of Patients With Community-Acquired Pneumonia” at ClinicalTrials.gov
Clinical trial number NCT03439124 for “Ceftobiprole in the Treatment of Pediatric Patients With Pneumonia” at ClinicalTrials.gov
////////Ceftobiprole, BAL-9141, BAL-9141-000, BAL-9141000, BAL9141-000, RO 63-9141, RO-63-9141, RO-639141, fda 2024, zevtera, approvals 2024, Ceftobiprole medocaril sodium salt
fda approved 4/26/2024, To treat WHIM syndrome (warts, hypogammaglobulinemia, infections and myelokathexis), Xolremdi
Mavorixafor (AMD-070) is a potent, selective and orally available CXCR4 antagonist, with an IC50 value of 13 nM against CXCR4125I-SDF binding, and also inhibits the replication of T-tropic HIV-1 (NL4.3 strain) in MT-4 cells and PBMCs with an IC50 of 1 and 9 nM, respectively.
AMD-070 is a small molecule drug candidate that belongs to a new investigational class of anti-HIV drugs known as entry (fusion) inhibitors. Approximately 76% of HIV-patients with measurable viral load are infected with a strain of virus that is resistant to one or more classes of antiretroviral agents, thus reducing treatment options. Unlike many existing HIV drugs that target the virus after it has infected a healthy cell, AMD-070 blocks the virus from entering a healthy cell, thus preventing the replication process. AMD-070 targets the CXCR4 receptor on HIV and prevents the virus from entering and infecting healthy cells. AMD-070 is specific for the CXCR4 receptor and does not interact with any other chemokine receptors in vitro. AMD-070 strongly inhibits viral infection by all CXCR4 using virus (including virus using CXCR4 alone and/or virus using CXCR4 and CCR5) in vitro. AMD-070 is orally bioavailable in animals, it has suitable PK and toxicity profile for oral dosing. AMD-070 shows additive or synergistic effects in vitro in combination with other known anti-HIV agents. AMD-070 is active against CXCR4 using HIV strains that are resistant to existing antiretroviral therapies in vitro, reveals potent anti-HIV activity against CXCR4-using laboratory strains and clinical isolates. MD-070 had been in phase II clinical trials by Genzyme for the treatment of HIV infection. However, this research has been discontinued. AMD-070 has been studied in Phase I/II clinical trials for the treatment of Renal cell carcinoma and Phase I clinical trials for the treatment of malignant melanoma and solid tumours.
A novel and practical synthesis of mavorixafor (1) is reported. The novelty of this synthetic route is the use of 8-chloro-5,6,7,8-tetrahydroquinoline (9) and 1,4-diaminobutane as the materials, instead of 8-amino-5,6,7,8-tetrahydroquinoline (4) and N,N-diprotected aminobutyraldehyde (6a or 6b). The preparation of (S)-8-(4-aminobutylamino)-5,6,7,8-tetrahydroquinoline (13) by resolution with N-acetyl-l-leucine was first achieved. Then the one-pot synthesis of 1 from 13 involving protection, condensation, and subsequent hydrolysis was successfully developed. In addition, the final product with a satisfactory purity (>99.5%, detected by both achiral and chiral HPLC) was obtained by a simple operation (salification) without column chromatographic purification.
Charge diethyl-4-aminobutyl acetal (E) (1.00 wt, 1.00 eq) to vessel A. Charge acetonitrile (10.0 vol, 7.8 wt) and adjust temperature to 20°C. Heat the mixture to 40°C. Concentrate the reaction mixture to 6.0 vol under reduced pressure at 35 to 45°C.
[0098] Acetonitrile filler (5.0 vol, 3.9wt) at 35 to 45°C. Concentrate the reaction mixture to 6.0 vol under reduced pressure 35 to 45°C. This step is repeated once as described below.
[0099] Acetonitrile filler (5.0 vol, 3.9wt) at 35 to 45°C. Concentrate the reaction mixture to 6.0 vol under reduced pressure at 35 to 45°C. Cool to 20°C.
[00100] Charge di-tert-butyl dicarbonate (1.1 eq, 1.5 wt) to a drum, followed by acetonitrile (0.4 vol, 0.3 wt) and agitate until fully dissolved. Concentrate the reaction mixture to 6.0 vol under reduced pressure at 35 to 45°C.
[00101] Charge this di-tert-butyl dicarbonate solution in acetonitrile to vessel A maintaining 20°C. Charge acetonitrile (1.5 vol, 1.1 wt) to the solution as a line rinse and stir at 20°C for 30 to 60 min..
[00102] Charge 4-dimethylaminopyridine (0.076 wt, 0.10 eq) to the vessel A at 20°C. Heat the solution to 40°C. Concentrate the reaction mixture to 5.0 vol under reduced pressure. Charge acetonitrile (5.0 vol, 3.9 wt) to the solution. Concentrate the reaction mixture to 5.0 vol under reduced pressure.
[00103] Take the resulting solution of Dl into next reaction without isolation.
Step IB: Preparation of Cl
[00104] Charge acetonitrile (2.0 vol, 1.6 wt) at 35 to 45°C to vessel A containing solution of D-1 from Step 1A.
[00105] Charge di-tert-butyl dicarbonate (1.4 eq, 1.9 wt) to a drum, followed by acetonitrile (10.0 vol, 7.8 wt) and agitate until fully dissolved. Charge this di-tert-butyl dicarbonate solution to vessel A, 2 to 6 h while distilling under vacuum at 35 to 45°C maintaining the volume of the reaction at 7.0 vol. Load acetonitrile (3.0 vol, 2.4 wt) over 20 to 40 min. as a line rinse while distilling under vacuum at 35 to 45°C, maintaining the volume of the reaction at 7.0 vol.
[00106] Charge di-tert-butyl dicarbonate, (0.14 eq, 0.19 wt) to a drum, followed by acetonitrile (1.0 vol, 0.74 wt) and agitate until fully dissolved. Charge this di-tert-butyl dicarbonate solution to vessel A over 20 to 40 min.. Charge acetonitrile (0.3 vol, 0.24 wt) over 10 to 20 min as a line rinse while distilling under vacuum at 35 to 45°C, maintaining the volume of the reaction at 7.0 vol.
[00107] Concentrate the reaction mixture to 5.0 vol distilling under vacuum at 35 to 45°C.
[00108] Charge n-heptane, (7.5 vol, 5.1 wt) to the reaction mixture, and concentrate the reaction mixture to 5.0 vol under reduced pressure at 40°C. This step is repeated once as described below.
[00109] Charge n-heptane, (7.5 vol, 5.1 wt) to the reaction mixture, and concentrate the reaction mixture to 5.0 vol under reduced pressure at 40°C.
[00110] Charge decolorizing, activated charcoal (0.2 wt) to the solution and stir for 1 to 2 h at 40°C. Filter the reaction mixture at 40°C. Charge n-heptane, (2.0 vol, 1.4 wt) to the reactor vessel and stir for 5 to 15 min. at 20°C before charging to the filter as a line rinse. Combine the filtrate and wash, and as required adjust to 20°C.
[00111] Take the resulting solution of Cl into next reaction without isolation.
Step 1C: Preparation of Bl
[00112] Charge 15% v/v acetic acid (2.0 vol) caution gas evolution, to vessel A containing solution of Cl from Step IB, maintaining the temperature at 20°C and stir for 10 min. at 20°C. Allow the phases to separate for 15 min. at 20°C. Discharge the aqueous phase to waste, retaining the organic phase in vessel A. This step is repeated once as described below.
[00113] Charge 15% v/v acetic acid (2.0 vol) maintaining 20°C and stir for 10 min. at 20°C. Allow the phases to separate for 15 min. at 20°C. Discharge the aqueous phase to waste, retaining the organic phase in vessel A.
[00114] Adjust the reaction to 30°C. Charge 4% w/w sodium chloride solution (2.1 vol) to the vessel maintaining the temperature at 30°C. Charge glacial acetic acid (4.1 vol, 4.3 wt) to the vessel maintaining 30°C. Stir the reaction mixture for 2 h maintaining the temperature at 30°C.
[00115] Charge purified water, (6.0 vol) at 30°C. Stir the contents for 5 to 10 min. at 30°C, and separate the phases, retaining the upper organic phase in vessel A. Charge the lower aqueous phase to vessel B.
[00116] Charge purified water (4.0 vol) at 30°C and stir for 5 to 10 min. maintaining the temperature at 30°C. Separate the phases at 30°C, retaining the upper organic phase in vessel A. Charge the lower aqueous phase to vessel B.
[00117] Adjust the temperature to 30°C of vessel B containing combined aqueous phases. Charge n-heptane, (2.0 vol, 1.4 wt) to vessel B and stir for 5 to 10 min. maintaining the temperature at 30°C. Separate the phases at 30°C, over 15 min.. Charge the upper organic phase to vessel A and recharge the lower aqueous phase to vessel B. This step is repeated two additional times as described below.
[00118] Charge n-heptane, (2.0 vol, 1.4 wt) to vessel B and stir for 5 to 10 min. maintaining the temperature at 30°C. Separate the phases at 30°C, over 15 min.. Charge the upper organic phase to vessel A and recharge the lower aqueous phase to vessel B.
[00119] Charge n-heptane, (2.0 vol, 1.4 wt) to vessel B and stir for 5 to 10 min. maintaining the temperature at 30°C. Separate the phases at 30°C, over 15 min., discharge the lower aqueous phase to waste and charge the upper organic layer to vessel A.
[00120] Concentrate the combined organic phases in vessel A to 3.0 vol at 10 to 20°C under reduced pressure. Offload the solution to new HDPE drum(s) and line rinse with n-heptane (0.5
vol, 0.4 wt) at 20°C. Homogenize the drum and store as “Bl solution in n-heptane,” and take into next reaction without isolation.
Step ID: Preparation of F-2d
[00121] Calculate a new 1.00 wt based on the above assay.
[00122] Charge “Bl solution in n-heptane” from Step 1C (1.00 wt, 1.00 eq, corrected for w/w assay, ca. 3.0 vol), into an appropriate vessel. THF load (3.0 vol, 2.7 wt). Heat the reaction mixture to 40°C.
[00123] Charge purified water, (0.02 vol, 0.02 wt) followed by sodium metabisulphite, (0.125 eq, 0.08 wt) as a solid via the charge hole at 40°C. Stir the resulting mixture for 30 to 35 min. at 40°C. This step was repeated four additional times to add the reagent in five portions total, as detailed below.
[00124] Charge purified water, (0.02 vol, 0.02 wt) followed by sodium metabisulphite, (0.125 eq, 0.08 wt) as a solid via the charge hole at 40°C. Stir the resulting mixture for 30 to 35 min. at 40°C.
[00125] Charge purified water, (0.02 vol, 0.02 wt) followed by sodium metabisulphite, (0.125 eq, 0.08 wt) as a solid via the charge hole at 40°C. Stir the resulting mixture for 30 to 35 min. at 40°C.
[00126] Charge purified water, (0.02 vol, 0.02 wt) followed by sodium metabisulphite, (0.125 eq, 0.08 wt) as a solid via the charge hole at 40°C. Stir the resulting mixture for 30 to 35 min. at 40°C.
[00127] Charge purified water, (0.02 vol, 0.02 wt) followed by sodium metabisulphite, (0.125 eq, 0.08 wt) as a solid via the charge hole at 40°C. Stir the resulting mixture for 36 hours at 40°C.
[00128] Cool the reaction mixture to 20°C over 3 to 4 h at a target constant rate. Filter the reaction mixture at 20°C on a 1-2 pm cloth.
[00129] Wash the solid with a pre-mixed mixture of THF (0.5 vol, 0.5 wt) and n-heptane (0.5 vol, 0.3 wt) maintaining the temperature at 20°C. This step was repeated an additional three times, as detailed below.
[00130] Wash the solid with n-heptane, (2.0 vol, 1.4 wt) as a line rinse and apply to the filtercake at 20°C.
[00131] Wash the solid with n-heptane, (2.0 vol, 1.4 wt) as a line rinse and apply to the filtercake at 20°C.
[00132] Wash the solid with acetonitrile, (2.0 vol, 1.6 wt) as a line rinse and apply to the filtercake at 20°C.
[00133] Dry the solid at 38°C under a flow of nitrogen for 12 h.
[00137] Charge J, (1.00 wt, 1.00 eq) to vessel A. Charge purified water, (1.0 vol, 1.0 wt) to vessel A and as necessary adjust the temperature to 20°C. Charge concentrated hydrochloric acid, (4.0 eq, 3.0 vol, 3.6 wt) to vessel A maintaining the temperature at 20°C. Line rinse with purified water, (0.5 vol, 0.5 wt) maintaining the contents of vessel A at 15 to 25°C.
[00138] Charge chloroacetic acid, (1.3 wt, 1.5 eq) and purified water, (1.0 vol, 1.0 wt) to vessel B and as necessary, adjust the temperature to 20°C. Stir until fully dissolved, expected 10 to 20 min.
[00139] Charge the chloroacetic acid solution to vessel A maintaining the temperature of vessel A at 20°C. Line rinse vessel A with purified water, (0.5 vol, 0.5 wt) at 15 to 25°C and charge to vessel B at 20°C. Heat the reaction mixture to 80°C. Stir the reaction mixture at 80°C for 20 h.
[00140] Cool the reaction mixture to 10°C over 1.5 h. Load 47% w/w potassium phosphate solution (6.0 vol) over 60 min. targeting a constant rate maintaining 10°C. Adjust the pH of the reaction mixture by charging 47% w/w potassium phosphate solution to pH 7.0 maintaining the reaction temperature at 10°C. Expected charge is 2.0 to 3.5 vol 47% w/w potassium phosphate solution.
[00141] Stir the slurry for >30 min. maintaining 10°C and rechecking the pH, pass criterion pH 7.0. Filter the reaction mixture through 20 pm cloth at 10°C. Wash the filter-cake with purified water, (1.0 vol, 1.0 wt) at 10°C. This step is repeated additional three times as described below.
[00142] Slurry wash the filter-cake in the reactor vessel with purified water, (10.0 vol, 10.0 wt) for 45 min. to 90 min. at 10°C. Filter the mixture through 20 pm cloth at 10°C.
[00143] Slurry wash the filter-cake in the reactor vessel with purified water, (10.0 vol, 10.0 wt) for 45 min. to 90 min. at 10°C. Filter the mixture through 20 pm cloth at 10°C.
[00144] Slurry wash the filter-cake in the reactor vessel with purified water, (10.0 vol, 10.0 wt) for 45 min. to 90 min. at 10°C. Filter the mixture through 20 pm cloth at 10°C.
[00145] Wash the filter-cake with purified water, (1.0 vol, 1.0 wt) at 10°C. The filter-cake was washed with purified water additional five times as described below.
[00146] Wash the filter-cake with purified water, (1.0 vol, 1.0 wt) at 10°C.
[00147] Wash the filter-cake with acetonitrile, (2×1.3 vol, 2×1.0 wt) at 10°C.
[00148] Dry the filter-cake on the filter under vacuum and strong nitrogen flow through the filter cake at 20°C until the water content is <15.0% w/w by Karl-Fisher analysis.
[00149] Dry the filter-cake on the filter under vacuum and strong nitrogen flow through the filter cake at 30°C until the water content is <5.0% w/w by Karl-Fisher analysis.
[00150] Dry the filter-cake on the filter under vacuum and strong nitrogen flow through the filter cake at 50°C until the water content is <1.0% w/w by Karl-Fisher analysis.
[00151] Yield of compound Gl: about 75%.
Step 2B: Preparation of F-3a
Charge di-/c/7-butyl dicarbonate, (1.85 wt, 1.4 eq) to vessel A followed by N,N-dimethylformamide, (2.6 wt, 2.7 vol) and stir at 20°C for 20 min. until dissolution achieved. Add A,A-diisopropylethylamine, (0.08 wt, 0.11 vol, 0.1 eq) to contents of vessel A at 20°C. Heat the contents of vessel A to 40°C.
[00153] Charge Gl, (1.00 wt) to vessel B followed by YW-di methyl form am ide, (5.2 wt, 5.5 vol) and adjust to 14°C.
[00154] Charge the Gl/DMF solution from vessel B to vessel A over 5 h at 40°C, at an approximately constant rate. Line rinse with Y,Y-di methyl form am ide, (0.4 wt, 0.4 vol), maintaining vessel A at 40°C. Stir the resulting reaction mixture at 40°C for 16 h.
[00155] Charge decolorizing charcoal activated, (0.20 wt). Adjust the mixture to 40°C and stir at 40°C for 60 to 90 min..
[00156] Clarify (filter) the reaction mixture into vessel B at 40°C. Charge N,N-dimethylformamide, (0.9 wt, 1.0 vol) via vessel A and filter at 40°C. Charge purified water, (3.5 vol) to the combined filtrates, over 60 min., maintaining the temperature at 40°C. As required, cool the mixture to 35°C over 30 to 60 min..
[00157] Filler F-3a, (0.02 wt) as seed material at 35°C. Stir at 34°C for 1.5 h then check for crystallization. Cool slurry to 30°C over 40 min.
[00158] Filler F-3a, (0.02 wt) as seed material at 30°C. Stir at 30°C for 1.5 h then check for crystallization.
[00159] Cool slurry at 20°C over 3.5 h at a targeted constant rate. Stir at 20°C for 3 hours. Charge purified water, (1.0 vol), maintaining the temperature at 20°C over 60 min..
Stir at 20°C for 3 hours.
[00160] Cool slurry to 2°C over 2.5 h. Stir at 2°C for 2.5 hours. Filter through 20 pm cloth and pull dry until no further filtrate passes. Wash the solid with pre-mixed Y,Y-di methyl form am ide / purified water, (2.0 vol, 1:2 v:v) at 2°C. Wash the solid with purified water, (2 x 3.0 vol) at 2°C. Dry under vacuum at 28°C until KF <0.2% w/w, and Y,Y-di methyl form am ide <0.4% w/w.
[00161] Yield of compound F-3a: 62-70%.
Example 3: Synthesis of Mavorixafor:
Scheme VI:
nce
Step 3A: Preparation of imine Q-1
[00162] To vessel A charge purified water, (8.7 vol, 8.7 wt) followed by potassium phosphate, (5.52 eq, 5.3 wt) portion-wise and cool to 15°C. Charge tetrahydrofuran, (4.3 vol, 3.8 wt) and n-heptane, (2.2 vol, 1.5 wt) to vessel A and cool the biphasic mixture to 0°C. Charge Fl, (1.00 eq, 1.00 wt) to the vessel in 2 portions maintaining 0°C.
[00163] Charge F-2d, (1.10 eq, 1.95 wt) to the vessel in 4 portions maintaining 0°C, ensuring portions are spaced by 10 min.. Stir the resulting biphasic mixture for 1.5 h at 0°C. Allow the layers to separate for 45 min. at 0°C before separating the layers. Retain the upper organic phase within vessel A.
[00164] Take the resulting solution of Ql into next reaction without isolation.
Step 3B: Preparation of amine P-1
[00165] To vessel B, charge tetrahydrofuran, (6.0 vol, 5.3 wt) and adjust to 15°C. Charge zinc chloride, (1.5 eq, 0.92 wt) to vessel B in 4 portions, maintaining 10 to 30°C. Adjust the reaction mixture in vessel B to 15°C. Stir the mixture at 15°C for 1 hour. Charge sodium borohydride,(1.0 eq, 0.17 wt) to vessel B in 2 portions maintaining 15°C. Cool the reaction mixture in vessel B to 15°C. Stir the mixture for 1 hour maintaining 15°C. Cool the reaction mixture in vessel B to -5°C.
[00166] Cool the retained organic solution of Ql in vessel A, from Step 3A, to -5°C.
[00167] Charge the organic solution in vessel A into vessel B over 1 to 2 h maintaining -5°C. Charge tetrahydrofuran, (1.0 vol, 0.9 wt) to vessel A as a line rinse and adjust to -5°C. Transfer the contents of vessel A to vessel B maintaining -5°C.
[00168] Stir the resulting reaction mixture in vessel B for 1.5 h maintaining -5°C.
[00169] Charge purified water, (4.5 vol, 4.5 wt) and glacial acetic acid, (1.0 eq, 0.27 wt, 0.26 vol) to the cleaned vessel A and cool to 0°C. Charge the contents of vessel B to vessel A over 1 to 2 h maintaining 0°C. Charge tetrahydrofuran, (1.0 vol, 0.9 wt) to vessel B as a vessel rinse, cool to 0°C and transfer to vessel A maintaining 0°C.
[00170] Warm the resulting mixture in vessel A to 30°C. Stir the resulting mixture in vessel A at 30°C for 1 h. Allow the layers to settle for 15 min. at 30°C before separating the layers. Retain the upper organic phase.
[00171] Cool the retained organic phase to 15°C. Charge to the vessel 25% w/w ammonia solution (3.0 vol) at 10 to 30°C. Cool the reaction mixture to 20°C. Charge to the vessel 25% w/w ammonium chloride solution (3.0 vol) at 20°C and stir for 1 h. Separate the layers for 15 min. at 20°C, retain the upper organic phase. Wash the retained organic phase with 10% w/w sodium chloride solution (3.0 vol) at 20°C for 10 min.. Allow the layers to settle for 10 min. at 20°C before separating and retaining the upper organic phase within the vessel.
[00172] Charge tert-butyl methyl ether, (0.5 vol, 0.4 wt) to the organic phase. Cool the mixture to 5°C. Adjust the pH of the reaction mixture to pH 5 with hydrochloric acid aqueous solution (expected ca. 9.0 vol) over 1 h at a targeted constant rate at 5°C. Stir the mixture at 5°C for 45 min.. Measure the pH of the aqueous phase to confirm the value is pH 5.
[00173] Charge sodium chloride, (2.1 wt) to the reaction mixture at 5°C and stir the mixture until everything is dissolved. Adjust the temperature of the reaction mixture to 20°C. Separate the layers at 20°C and retain the organic phase within the vessel. Tetrahydrofuran charge, (1.5 vol, 1.3 wt) maintaining 20°C.
[00174] Charge to the vessel 24% w/w sodium chloride solution (7.5 vol) at 20°C and stir for 10 min.. Separate the layers at 20°C and retain the organic phase in the vessel. This step is repeated additional one more time as described below.
[00175] Charge to the vessel 24% w/w sodium chloride solution (7.5 vol) at 20°C and stir for 10 min.. Separate the layers at 20°C and retain the organic phase in the vessel.
[00176] Heat the retained organic phase to 35°C and concentrate the mixture to 6.0 vol under reduced pressure maintaining 35°C.
[00177] Tetrahydrofuran charge, (15.0 vol, 13.2 wt) maintaining 35°C. Concentrate the mixture to 6.0 vol under reduced pressure maintaining 35°C.
[00178] Tetrahydrofuran charge, (15.0 vol, 13.2 wt) maintaining 35°C. Concentrate the mixture to 11.0 vol under reduced pressure maintaining 35°C.
[00179] Cool the mixture to -5°C. Load tert-butyl methyl ether, (10.0 vol, 7.4 wt) over 1 h maintaining -5°C. Stir the mixture at -5°C for 1.5 hours. Filter the solid on 1 to 2 pm filter cloth at -5°C. Wash the solid with pre-mixed tetrahydrofuran, (1.9 vol, 1.7 wt) and tert-butyl methyl ether, (3.1 vol, 1.9 wt) at -5°C as a displacement wash.
[00180] Wash the solid with tert-butyl methyl ether, (5.0 vol, 3.7 wt) at -5°C.
[00181] Dry the solid on the filter under a flow of nitrogen at 23°C.
[00182] Yield of compound P-1: 76-87%.
Step 3C: Preparation of compound 0-1
[00183] Charge purified water, (2.0 vol, 2.0 wt) followed by potassium phosphate, (3.3 eq, 1.54 wt), carefully portion-wise, maintaining <15°C, to vessel A. Charge toluene, (4.5 vol, 3.9 wt) to the vessel maintaining <15°C. As necessary, adjust the temperature to 10°C.
[00184] Charge P-1, (1.00 eq, 1.00 wt) to the vessel in two portions maintaining 10°C. Stir the reaction mixture at 10°C for 15 min..
[00185] Load F-3a, (1.1 eq, 0.64 wt) in 4 equal portions ensuring portions are spaced by 10 min. at 10°C.
[00186] Tetrabutylammonium iodide (TBAI) filler (0.20 eq, 0.16 wt). Heat the reaction mixture to 40°C. Stir the reaction mixture at 40°C for 30 h.
[00187] Charge pre-mixed 2-mercaptoacetic acid, (0.40 eq, 0.08 wt, 0.06 vol), and toluene, (0.5 vol, 0.4 wt) over 20 min. to Vessel A at 40°C. Line rinse with toluene, (0.5 vol, 0.4 wt) at 40°C. Adjust the temperature of the reaction mixture to 50°C. Stir the mixture at 50°C for 2.5 hours.
[00188] Adjust the temperature of Vessel A to 20°C. Charge purified water, (3.0 vol, 3.0 wt) maintaining 20°C. Stir the reaction mixture at 20°C for 15 min. and transfer to a new, clean HDPE container. Line/vessel rinse with toluene, (0.5 vol, 0.4 wt) at 20°C. Clarify (filter) the reaction mixture via a 1 pm filter at 20°C into clean Vessel A. Wash the vessel and the filter with toluene, (0.5 vol, 0.4 wt) at 20°C. Allow the layers to separate for 15 min. at 20°C, retaining the upper organic layer (organic layer 1).
[00189] Wash the aqueous layer with toluene, (2.5 vol, 2.2 wt) at 20°C for 15 min.. Allow the layers to separate for 15 min. at 20°C. Retain the upper organic layer (organic layer 2).
[00190] Combine the organic layer 1 and organic layer 2 and adjust the temperature to 20°C. Wash the combined organic layers with 10% w/w sodium chloride solution (5.0 vol) at 20°C for 15 min.. Allow the layers to settle for 15 min. at 20°C. Retain the upper organic layer.
[00191] Take the resulting solution of Ol into next reaction without isolation.
Step 3D: Preparation of compound Kl
[00192] Charge n-butanol, (2.4 wt, 3.0 vol) to vessel B and adjust to 5°C. Charge concentrated sulfuric acid, (1.1 wt, 5.0 eq, 0.6 vol) slowly to Vessel B maintaining <15°C. Line rinse with toluene, (0.4 wt, 0.5 vol) maintaining <15°C. Adjust the temperature of Vessel B to 25°C.
[00193] Heat the n-butanol/ sulfuric acid solution in Vessel B to 55°C. Charge the organic layer from Vessel A (from Step 3C) to the butanol/ sulfuric acid solution in Vessel B over 60 to 90 min. maintaining 55°C. Charge toluene, (1.3 wt, 1.5 vol) to Vessel A as a line rinse and transfer to Vessel B maintaining 55°C. Stir the contents of Vessel B at 55°C for 1.5 h.
[00194] Stir the mixture in Vessel B for 4.5 h at 55°C. Cool the contents of Vessel B to 20°C over 10 h. Filter the slurry over 1-2 pm filter cloth under nitrogen at 20°C. Wash the filter cake with pre-mixed toluene, (3.5 wt, 4.0 vol) and n-butanol, (1.0 vol, 0.8 wt) at 20°C. Wash the filter cake with toluene, (4.3 wt, 5.0 vol) at 20°C. Dry the solid at 30°C under vacuum.
[00195] Correct the output weight for assay. Expected 50-55% w/w.
[00196] Yield of compound K1: 89-92%.
Step 3E: Preparation of Mavorixafor Drug Substance
[00197] Charge Kl, (1.00 eq, 1.00 wt, corrected for HPLC assay) in vessel A followed by nitrogen-purged purified water, (2.0 wt, 2.0 vol) and if necessary, adjust the temperature to 20°C. Charge nitrogen-purged toluene, (12.0 wt, 14.0 vol) to the solution maintaining 20°C. Charge nitrogen-purged n-butanol, (0.8 wt, 1.0 vol) to the solution maintaining 20°C. Heat the biphasic mixture to 30°C. Charge nitrogen-purged 3.0 M aqueous sodium hydroxide solution (6.2 eq, 5.9 vol) maintaining 30°C. Check the pH (expected 12 to 13). Adjust the pH of the aqueous layer to pH 10.0 with nitrogen-purged 0.3 M sulfuric acid solution (expected up to 2.5 vol) maintaining 30°C. Stir the mixture at 30°C for 45 min..
[00198] Measure the pH to confirm the value is pH 10.0.
[00199] Allow the layers to settle at 30°C for 30 min. and separate the layers retaining the organic phase in the vessel, and discharge the aqueous layer into a separate container (container C).
[00200] Charge pre-mixed toluene, (4.1 wt, 4.7 vol) and n-butanol, (0.24 wt, 0.3 vol) to a separate vessel; heat the contents to 30°C and charge the aqueous layer from container C. As required adjust the temperature to 30°C and stir for 5 to 10 min. at 30°C. Allow the phases to separate for 10 to 15 min. at 30°C. Discharge the aqueous phase to waste and combine the organic phase to the organic phase in vessel A.
[00201] Charge nitrogen-purged purified water, (2.0 wt, 2.0 vol) to the organic layer maintaining the temperature at 30°C and stir for 5 to 10 min. at 30°C. Allow the phases to separate for 10 to 15 min. at 30°C. Discharge the aqueous phase to waste retaining the organic phase in the vessel. Heat the retained organic solution to 40°C. Concentrate the resulting organic phase to 7.0 vol by vacuum distillation at 40°C.
[00202] Charge nitrogen -purged toluene, (13.0 wt, 15.0 vol) to the mixture and concentrate the solution 7.0 vol by vacuum distillation at 40°C. This step is repeated additional one time as described below.
[00203] Charge nitrogen -purged toluene, (13.0 wt, 15.0 vol) to the mixture and concentrate the solution 7.0 vol by vacuum distillation at 40°C.
[00204] Charge nitrogen-purged toluene, (7.0 wt, 8.0 vol) to the mixture at 40°C, heat to 55°C and clarify the hot reaction mixture under nitrogen via a 1 pm filter.
[00205] Charge clarified nitrogen-purged toluene, (1.7 wt, 2.0 vol) to the mixture as a line and vessel rinse at 40°C. Concentrate the solution to 7.0 vol by vacuum distillation at 40°C. At the end of the distillation the product is expected to have precipitated. Heat the mixture to 63°C.
[00206] Adjust the temperature to 60.5°C. This batch will be referred to as the main batch.
[00207] Load seed material, (0.02 wt) to a new clean container. Charge clarified nitrogen-purged toluene, (0.09 wt, 0.10 vol) to this seed material and gently shake.
[00208] Seed the main batch with the slurry maintaining the temperature at 60.5 ± 2°C. Stir the reaction at the 60.5± 2°C for 1 hour.
[00209] Cool to 40°C for 2.5 h. Stir the reaction at 40°C for 1 hour.
[00210] Cool to 30°C over 2 h.. Stir the reaction at 30°C for 1 h.
[00211] Cool to 25°C 50 min. Stir the reaction at 25°C over 2 hours.
[00212] Cool to 2°C over 4 h. Stir the mixture for 12 hours at 2°C.
[00213] Filter the mixture at 2°C over 1 to 2 pm cloth. Wash the filter cake with clarified nitrogen-purged toluene, (2.0 vol, 1.7 wt) at 2°C. Dry the filter cake under vacuum and a flow of nitrogen for 1.5 h.
[00214] Dry the solid at 40°C under vacuum and a flow of nitrogen until drying specification is achieved.
[00215] Yield of the final compound mavorixafor: 72%.
[00216] When toluene is used as the recrystallization solvent, optionally with a dissolution aid such butanol or methanol, for maxorixa for recrystallization, advantages were found compared to using dichloromethane and isopropyl acetate. We have found that these solvents do not react with the API, and accordingly we believe that this change has caused the significant reduction of impurities A (imine), B (N-formyl) and C (acetamide) that we have observed.
[00217] In some embodiments, the mavorixafor composition included 7000, 6000, 5000, 4500, 4450, 4000, 3500, 3000, 2500, 2000, 1750, 1700, 1650, 1600, 1550, 1500, 1450, 1400, 1400, 1400, 1400 gold 50 ppm of toluene or less. In some embodiments, the mavorixafor composition comprises a detectable amount of toluene. In some embodiments, the mavorixafor composition comprises from a detectable amount of toluene to 1350 ppm of toluene.
////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
Mavorixafor (AMD-070) is a potent, selective and orally available CXCR4 antagonist, with an IC50 value of 13 nM against CXCR4125I-SDF binding, and also inhibits the replication of T-tropic HIV-1 (NL4.3 strain) in MT-4 cells and PBMCs with an IC50 of 1 and 9 nM, respectively.
125I-SDF-CXCR413 nM (IC50)HIV-1 (NL4.3 strain)1 nM (IC50, in MT-4 cells)HIV-1 (NL4.3 strain)9 nM (IC50, in PBMCs)HIV-1 (NL4.3 strain)3 nM (IC90, in MT-4 cells)HIV-1 (NL4.3 strain)26 nM (IC90, in PBMCs)
In Vitro
Mavorixafor (AMD-070) is a potent and orally available CXCR4 antagonist, with an IC50 value of 13 nM against CXCR4 125I-SDF binding, and also inhibits the replication of T-tropic HIV-1 (NL4.3 strain) in MT-4 cells and PBMCs with an IC50 of 1 and 9 nM, respectively. Mavorixafor (AMD-070) shows no effect on other chemokine receptors (CCR1, CCR2b, CCR4, CCR5, CXCR1, and CXCR2)[1]. Mavorixafor (AMD-070) (6.6 µM) significantly suppresses the anchorage-dependent growth, the migration and matrigel invasion of the B88-SDF-1 cells[2].MCE has not independently confirmed the accuracy of these methods. They are for reference only.
In Vivo
Mavorixafor (AMD-070) (2 mg/kg, p.o.) significantly reduces the number of metastatic lung nodules in mice, and lowers the expression of human Alu DNA in mice, without body weight loss[2].MCE has not independently confirmed the accuracy of these methods. They are for reference only.
Arimoclomol maleate is in a phase III clinical trials by Orphazyme for the treatment of Niemann-Pick disease type C (NP-C). It is also in phase II clinical studies for the treatment of amyotrophic lateral sclerosis (ALS).
Arimoclomol (INN; originally codenamed BRX-345, which is a citrate salt formulation of BRX-220) is an experimental drug developed by CytRx Corporation, a biopharmaceutical company based in Los Angeles, California. In 2011 the worldwide rights to arimoclomol were bought by Danish biotech company Orphazyme ApS.[1] The European Medicines Agency (EMA) and U.S. Food & Drug Administration (FDA) granted orphan drug designation to arimoclomol as a potential treatment for Niemann-Pick type C in 2014 and 2015 respectively.[2][3]
Fig. 1 Structures of (±)-bimoclomol (1) and (R)-(+)-arimoclomol (2).
The present disclosure provides an optimized four-step process for preparing an ultra-pure composition comprising arimoclomol citrate, i.e. N-{[(2R)-2-hydroxy-3-piperidin-l-ylpropyl]oxy}pyridine-3-carboximidoyl chloride 1-oxide citrate. The optimized process comprises a plurality of optimized sub-steps, each contributing to an overall improved process, providing the ultra-pure composition comprising arimoclomol citrate. The ultra-pure composition comprising arimoclomol citrate meets the medicines agencies’ high regulatory requirements. An overview of the four-steps process is outlined below:
Step 1: Overview of process for preparing ORZY-01
Step 2: Overview of process for preparing ORZY-03
Step 4: Overview of process for preparing BRX-345 (ORZY-05)
The previously reported two-step synthesis of ORZY-01 as shown below includes a 2 hour reflux in step 1A, followed by purification of intermediate compound (V) to increase the batch quality.
(R,Z)-3-(N’-(2-hydroxy-3-(piperidin-1-yl)propoxy)carboximidoyl chloride)pyridine-1-oxide1 – (R)-(+)-Arimoclomol – 2 A solution of (R,Z)-3-(N’-(2-hydroxy-3-(piperidin-1-yl)propoxy)carbamimidoyl)pyridine-1-oxide 12 (205 mg, 0.70 mmol) in conc. hydrochloric acid (1.1 mL, 13.9 mmol) and water (3 mL) was cooled to -5 °C for 15 minutes. Sodium nitrite (63 mg, 0.91 mmol) in water (0.5 mL) was then added dropwise to the reaction mixture and the reaction was stirred at -5 °C for 2.5 hours. The reaction mixture was made alkaline with NaOH (7 M, 3 mL). An additional 10 mL of water was added followed by DCM (30 mL) containing EtOAc (5 mL) and the organics were dried over MgSO4, filtered and concentrated in vacuo. The residue was purified by FCC on Biotage Isolera using Biotage SNAP 10 g Si cartridge eluting with gradient elution 0-30% MeOH:DCM both containing 0.1% Et3N to afford the title compound (160 mg, 73% yield) as a colourless semi-solid. Analytical data was consistent with literature values. See ESI section SFC traces for specific enantiomeric ratios of 2 synthesised under the various methodologies quoted in the text. Optical rotation was not determined as it was determined in the ultimate product of this 2·citrate and comparative run times on SFC. 1H NMR (600 MHz, CDCl3) δ: 8.63 (t, J = 1.4 Hz, 1H), 8.16 (ddd, J = 6.4, 1.6, 0.9 Hz, 1H), 7.66 – 7.62 (m, 1H), 7.25 (dd, J = 8.0, 6.6 Hz, 1H), 4.26 (qd, J = 11.3, 5.2 Hz, 2H), 4.07 (dd, J = 9.2, 4.7 Hz, 1H), 2.62 (s, 2H), 2.47 – 2.31 (m, 4H), 1.65 – 1.51 (m, 4H), 1.42 (s, 2H); 13C NMR (151 MHz, CDCl3) δ: 140.3, 137.7, 133.1, 132.5, 125.7, 123.9, 78.7, 64.9, 60.9, 54.8, 25.8, 24.0.
(R)-(+)- Arimoclomol citrate – 2·citrate (R,Z)-3-(N’-(2-hydroxy-3-(piperidin-1-yl)propoxy)carboximidoyl chloride)pyridine-1-oxide (159 mg, 0.51 mmol) was dissolved in acetone (3 mL) and citric acid (97 mg, 0.51 mmol) was added. The reaction mixture was left to stir at room temperature for 18 hours. After this time the mixture was sonicated and the precipitate was filtered, rinsed with cold acetone (1 mL) and dried under vacuum to afford the title compound (165 mg, 64% yield) as a white amorphous solid. Analytical data was consistent with literature values. m.p. 161-162 °C, Acetone (lit. 163-165 °C, EtOH); [α]D 20 +8.0 (c=1, H2O); IR νmax (neat): 3423, 3228, 2949, 2868, 1722, 1589, 1483, 1433, 1307, 1128, 972, 829 cm-1; 1H NMR (600 MHz, d6-DMSO) δ: 8.54 (t, J = 1.5 Hz, 1H), 8.39 – 8.35 (m, 1H), 7.72 – 7.68 (m, 1H), 7.55 (dd, J = 8.0, 6.5 Hz, 1H), 4.28 (ddd, J = 17.6, 13.3, 7.4 Hz, 3H), 3.35 (br. s, 2H), 3.13 – 2.74 (m, 6H), 2.59 (d, J = 15.2 Hz, 2H), 2.56 – 2.51 (m, 2H), 1.77 – 1.61 (m, 4H), 1.48 (s, 2H); 13C NMR (151 MHz, d6-DMSO) δ: 176.6, 171.3, 140.5, 136.4, 132.7, 131.5, 126.8, 123.3, 77.8, 71.4, 63.8, 58.7, 53.1, 44.0, 30.7, 23.0, 21.9; HRMS (m/z TOF MS ES+) for C14H20ClN3O3 [M+H]+ calc. 314.1271, observed 314.1263; SFC er purity R:S >99:1
Procedure for the conversion of (R)-(+)-Bimoclomol 1 into (R)-(+)-Arimoclomol 2 To a solution of (R)-(+)-bimoclomol (61 mg, 0.21 mmol) in acetone (2 mL) was added benzenesulfonic acid (33 mg, 0.21 mmol). The reaction mixture was stirred at room temperature for 1.5 hours. The reaction mixture was concentrated in vacuo. Separately to a suspension of hydrogen peroxide-urea adduct (39 mg, 0.41 mmol) in acetonitrile (6 mL) at -5°C (ice-salt bath) was added trifluoroacetic anhydride (58 μL, 0.41 mmol) dropwise. A suspension of (R)-(+)-bimoclomol, 1, benzenesulfonic acid salt, as made above, in acetonitrile (3 mL) was then added dropwise to this solution. The reaction mixture was stirred for 18 hours, whilst slowly warming to room temperature. Aqueous Na2S2O5 solution (0.5 M, 1 mL) was added and the reaction mixture stirred for 1 hour. The reaction mixture was made alkaline with NaOH (7 M) and extracted with DCM (2 x 30 mL). The combined organics were washed with brine, dried over MgSO4, filtered and concentrated in vacuo. The residue was purified by FCC on a Biotage Isolera using Biotage SNAP 10g Si cartridge eluting with gradient elution 0-35% MeOH in DCM to afford the title compound (35 mg, 55% yield) as a colourless semi-solid. Analytical data of the products was consistent with literature and/or previous samples synthesised above.
Arimoclomol is believed to function by stimulating a normal cellular protein repair pathway through the activation of molecular chaperones. Since damaged proteins, called aggregates, are thought to play a role in many diseases, CytRx believes that arimoclomol could treat a broad range of diseases.
Arimoclomol has been shown to extend life in an animal model of ALS[11] and was well tolerated in healthy human volunteers in a Phase I study. CytRx is currently conducting a Phase II clinical trial.[12]
Arimoclomol also has been shown to be an effective treatment in an animal model of Spinal Bulbar Muscular Atrophy (SBMA, also known as Kennedy’s Disease).[13]
Arimoclomol was discovered by Hungarian researchers, as a drug candidate to treat insulin resistance[14][15] and diabetic complications such as retinopathy, neuropathy and nephropathy. Later, the compound, along with other small molecules, was screened for further development by Hungarian firm Biorex, which was sold to CytRx Corporation, who developed it toward a different direction from 2003.
^ Kieran D, Kalmar B, Dick JR, Riddoch-Contreras J, Burnstock G, Greensmith L (April 2004). “Treatment with arimoclomol, a coinducer of heat shock proteins, delays disease progression in ALS mice”. Nat. Med. 10 (4): 402–5. doi:10.1038/nm1021. PMID15034571. S2CID2311751.
^ Kalmar B, Greensmith L, Malcangio M, McMahon SB, Csermely P, Burnstock G (December 2003). “The effect of treatment with BRX-220, a co-inducer of heat shock proteins, on sensory fibers of the rat following peripheral nerve injury”. Exp. Neurol. 184 (2): 636–47. doi:10.1016/S0014-4886(03)00343-1. PMID14769355. S2CID5316222.
^ Rakonczay Z, Iványi B, Varga I, et al. (June 2002). “Nontoxic heat shock protein coinducer BRX-220 protects against acute pancreatitis in rats”. Free Radic. Biol. Med. 32 (12): 1283–92. doi:10.1016/S0891-5849(02)00833-X. PMID12057766.
^ Kalmar B, Burnstock G, Vrbová G, Urbanics R, Csermely P, Greensmith L (July 2002). “Upregulation of heat shock proteins rescues motoneurones from axotomy-induced cell death in neonatal rats”. Exp. Neurol. 176 (1): 87–97. doi:10.1006/exnr.2002.7945. PMID12093085. S2CID16071543.
1H-1,2,3-Triazolium, 3-(((2S,3S,5R)-2-carboxy-3-methyl-4,4-dioxido-7-oxo-4-thia-1-azabicyclo(3.2.0)hept-3-yl)methyl)-1-methyl-, inner salt
Enmetazobactam
The Board of directors of Orchid Pharma Ltd has announced that the company had developed a new molecule known as OCID-5090, which was licensed to a company named Allecra Therapeutics, this molecule was undergoing the clinical trials and the company is happy to announce that the molecule has cleared the Phase 3 clinical trials.
Allecra Therapeutics would now either directly or through out license file for NDA of this molecule. Allecra has already out licensed the product to Haini Pharmaceuticals, China for the Chinese Territory at a value of $78mn plus royalties.
As per the IP Agreement between Orchid Pharma Limited and Allecra Therapeutics, Orchid is entitled to receive a Royalty of 6-8% on the worldwide sales of the product. Therefore, once the molecule is commercialised, Orchid can expect a regular stream of Royalty from Allecra. Further, the rights to develop and commercialise the molecule in India (which is under patent protection) remain with Orchid Pharma Limited, and the company is evaluating the various options to commercialise the product.
Orchid had developed a new molecule known as OCID-5090, which was licensed to a company named Allecra Therapeutics, this molecule was undergoing the clinical trials and the molecule has cleared the Phase 3 clinical trials.
Allecra Therapeutics would now either directly or through out license file for NDA of this molecule. Allecra has already out licensed the product to Haini Pharmaceuticals, China for the Chinese Territory at a value of $78mn plus royalties.
As per the IP Agreement between Orchid Pharma Limited and Allecra Therapeutics, Orchid is entitled to receive a Royalty of 6-8% on the worldwide sales of the product. Therefore, once the molecule is commercialised, Orchid can expect a regular stream of Royalty from Allecra. Further, the rights to develop and commercialise the molecule in India (which is under patent protection) remain with Orchid Pharma Limited, and the company is evaluating the various options to commercialise the product.
INVENTOR
Senthilkumar U P
ORCHID
Summary of Profile of Dr. U. P. Senthilkumar, R&D Centre, Orchid Pharma Ltd. Dr. U. P. Senthilkumar Ph.D., the principal inventor of novel beta-lactamase inhibitor, OCID5090, is currently serving as the senior vice-president at Orchid’s Research and Development Centre, Chennai. With illustrious credentials — top ranks in B.Sc. and M.Sc. degrees, first rank in the Graduate Aptitude Test in Engineering (GATE), UGC-CSIR Junior Research Fellowship (JRF) and the prestigious Dr. K.S. Krishnan Fellowship from the Department of Atomic Energy (DAE) and publication of M.Sc. project work in the Indian Journal of Chemistry in 1987 — Mr. Senthilkumar chose to pursue his doctoral research in synthetic organic chemistry with his mentor Prof. Ramasubbu Jeyaraman at Bharathidasan University, Tiruchirapalli. His research focus on the conformational preferences of sterically challenged novel N-Nitroso heterocycles and their conformation dependent anti-cancer properties, led to the publication of 9 articles in reputed peer-reviewed international journals – a commendable accomplishment in the 90s. After a brief post-doctoral stint on fluorescent dicyclopentapyridines, Dr. U. P. Senthilkumar joined Torrent Research Centre at Ahmedabad and started his new endeavor of drug discovery on ACE inhibitors. At the process research and development laboratory, he was actively involved in asymmetric and stereo-selective synthesis of Active Pharmaceutical Ingredients (APIs), and exploited the full potential of chiral prep-HPLC to realize the target molecules. After joining Orchid Pharma Ltd., Chennai, Dr. Senthilkumar led the efforts in the development of differentiated and patentable manufacturing processes for APIs related to both non-antibiotics and beta-lactam antibiotics. He played a significant role in successfully implementing the manufacturing processes overcoming several challenging problems. In addition, his scientific insights and breath of understanding on the patent landscape were invaluable and impactful in creating significant value to the organization and growth of the company in realizing the mission to become a leader in the pharmaceutical generic business. One finds more than 100 articles/patents/publications to his credit, which include inventions on new drugs, drug-intermediates, products, processes, new synthetic routes, rearrangements and novel polymorphs. As a Leadership Persona of the IP management team, he had exhibited a thoroughness of the science/invention and meticulously executed the task of prosecution of few hundred patents in many countries from both New Drug Discovery and Process Chemistry space. All the successful effort earned Orchid Pharma Ltd the National Intellectual Property Award from the Department of Industrial Policy and Promotion, Ministry of Commerce and Industry,Government of India. Through his executive and decision-making skills combined with scientific rationale and clarity, Dr. Senthilkumar played significant role in the selection of products and creation of generic product portfolio for Orchid, with unique IP strategies, analysis of patents, patent mapping, designing & developing invalidation/non-infringing positions, and early launch opportunity, including first-to-file (FTF) positioning. His appearance in the US courts, for deposition in couple of patent litigations, and successful accomplishment of the same are testimony to his depth, thoroughness of science and the ability to defend the invention with grit and professionalism. Additionally, his effectual role in the first-to-launch of one of the large volume sterile penicillins with regulatory exclusivity, achieved successfully by overcoming the citizen petition process in the regulatory pathway, is another shining example of his leadership and scientific strength. To support in-house projects as well as multinational pharma majors, Dr. Senthilkumar has taken up CRAMS (Contract Research and Manufacturing Services) and CMC (Chemistry, Manufacturing and Control) for new chemical entities. Besides, he passionately focused on novel beta-lactamase inhibitors and their antibiotic combinations that were envisaged by him to exhibit potent activity against multi-drug resistant bacteria. His dedicated effort brought a novel extended spectrum beta-lactamase inhibitor, OCID5090, which was out-licensed to Allecra Inc. OCID5090/cefepime combination has completed successfully the Phase III clinical trials for treating complicated urinary-track-infections (cUTI), including acute pyelonephritis (AP), and rightfully, OCID5090 has gotten the US FDA fast track designation as a Qualified Infectious Disease product (QIDP) that provides a five-year additional market exclusivity and priority review. His never-ending passion for research is infectious and roped him with academic institutions to explore novel technologies including electron-beam irradiated heterogeneous catalysis. His commendable knowledge on intellectual property is being utilized by the IP Cells of various institutions as well as the Tamil Nadu State Technology Development and Promotion Centre. A sincere student he is, Dr. Senthilkumar is also a founder-member of Prof. Ramasubbu Jeyaraman Science Foundation (RJSF). Since 2011, he has been playing a significant role in rganizing several academic events (seminars, work-shops, invited lectures, state-level proficiency tests, and research-orientation programs) for post-graduate chemistry students to create passion for research. His concern and help for poor and rural students show his human face.
[0051]To a suspension of (2S,3S,5R)-3-methyl-7-oxo-3-(1H-1,2,3-triazol-1-ylmethyl)-4-thia-1-azabicyclo-[3.2.0]heptane-2-carboxylic acid 4,4-dioxide (25 g) in acetone (100 mL) at 25-30° C. was added slowly N,O-bis(silylacetamide) (18.6 g) with stirring. The reaction mixture was stirred at this temperature (25-30° C.) for 15-20 min. To the clear solution obtained, methyl iodide (100 mL) was added over a period of 15 min. and stirred at 25-30 min. for 24 h. The precipitated solid was separated by filtration and washed with acetone (25 mL). Wet weight of the solid obtained was 30 g.
[0052]The above wet solid was stirred with purified water (300 mL) at 10-15° C. for 2.5 h. To the resulted reaction mixture was added sodium thiosulfate (0.1 g) and stirred at 10-15° C. for 10-15 min. To the reaction mixture, dichloromethane (300 mL) was added, stirred and the organic layer separated. The aqueous layer was washed with a solution of Amberlite LA-2 resin (5% solution in dichloromethane twice, followed by dichloromethane twice. To the aqueous solution, activated carbon (1 g) was added, stirred for 15 min, filtered and washed with purified water (25 mL). The solution was filtered and lyophilized to get the title compound in pure form (10 g). 1H NMR (400 MHz, DMSO) δ ppm: 1.39 (s, 3H), 3.14 (dd, J=16.0, 1.3 Hz, 1H), 3.55 (dd, J=16.0, 4.2 Hz, 1H), 3.97 (s, 1H), 4.34 (s, 3H), 5.05 (dd, J=4.2, 1.3 Hz, 1H), 5.29 (d, J=14.7 Hz, 1H), 5.42 (d, J=14.7 Hz, 1H), 8.91 (d, J=1.3 Hz, 1H), 8.99 (d, J=1.3 Hz, 1H). Mass m/z: M+1 peak at 315. Alternatively the solution could be subjected to spray-drying to yield the title compound.
Synthesis of (2535.5R)-3-methyl-3-((3-methyl-lH-1.2 -triazol-3-ium-l-yl)methvn-7-oxo-4-thia-l-azabicyclor3.2.01heptane-2-carboxylate 4,4-dioxide (4),
Compound (4) was prepared according to Scheme 2.
Scheme 2
i) Ν,Ο-bis-trimethylsilylacetamide, CH2CI2; ii) CH3OTf; iii) Na 2-ethylhexanoate
In a round bottom flask under nitrogen flow 100 g of Tazobactam acid (1) and 500 mL of Dichloromethane are loaded. The temperature is adjusted to +30/35°C then 37 g of Ν,Ο-Bis(trimethylsilyl) acetamide are loaded in 15-20 minutes maintaining the temperature to +35/42°C. The mixture is heated to reflux (+40/42°C) for 60 minutes. If the solution is not clear, N,0-Bis(trimethylsilyl) acetamide is loaded in small portions (0,5-1.0 g each) waiting 15 minutes every time till a clear solution containing intermediate (2) is obtained. 0.55 moles of N,0-Bis(trimethylsilyl) acetamide is used, with further 0.1-0.2 equivalents being added if the reaction is not complete.
Then the temperature is cooled down to 0/+5°C and 70 g of Methyl trifluoromethanesulfonate are loaded in 60-90 minutes maintaining the temperature at 0/+5°C. After 30 minutes the reaction is monitored by HPLC to control the disappearance of intermediate (2) and formation of intermediate (3). The reaction is monitored every 30 minutes until completion.
In a round bottom flask, under nitrogen, are loaded 500 mL of Ethanol and 55 g of Sodium 2-Ethylhexanoate and the temperature is adjusted to +20/25°C, then the reaction solution containing intermediate (3) is added in 60-90 minutes maintaining the temperature of +20/25 °C under vigorous stirring. The suspension is stirred for 30 minutes then is filtered and washed with 300 mL of Ethanol followed by 500 mL of Dichloromethane under nitrogen. The crude product (4) is dried under nitrogen flow till constant weight (150 g) is obtained. The crude product compound (4) was isolated as a solid product (HPLC assay = 70%, yield = 80%).
Purification of (2tS’,3^5^)-3-methyl-3-((3-methyl-lH-l,2,3-triazol-3-ium-l-yl)methyl)-7-oxo-4-thia-l-azabicyclor3.2.01heptane-2-carboxylate 4,4-dioxide (4)
In a round bottom flask 800 mL of Dimethylformamide are loaded, the temperature is adjusted to +20/25°C then crude Compound 4 (150g) obtained above is loaded using 100 mL of Dimethylformamide to facilitate the transfer. The mixture is stirred for 5 minutes and a solution is obtained, then and after a few minutes crystallization takes place. The suspension is stirred for about 3 hours, then is cooled to 0/+5°C and stirred for another 3 hours.
The solid is filtered and washed with 300 mL of Dimethylformamide pre-cooled to 0/+5°C. Compound 4 is then suspended in 700 mL of Ethyl acetate and the temperature is adjusted to +40/45°C. The suspension is stirred for 30 minutes then the solid is filtered and washed with 150 mL of Ethyl acetate pre-heated to +40/45°C. The suspension with
Ethyl acetate is repeated twice. Finally Compound 4 is dried under vacuum at +40°C till constant weight is achieved (66 g, HPLC assay = 99%, yield = 76%).
Compound 4 Sterile filtration and recrystallization Procedure
In a round bottom flask 350 mL of Methanol are loaded, the temperature is adjusted to +30/35°C then 100 g of Compound 4 are loaded and finally the flask is washed with 60 mL of Methanol. After 5-10 minutes a solution is obtained. The solution is diluted with 330 mL of acetone adjusting the temperature to +20/+25°C. The obtained solution is treated with 2,2 g of charcoal for 20 minutes then filtered using a 0.22microM filter and the filter is washed with a mixture of 13 mL of Methanol and 110 mL of Acetone. The temperature of the solution is adjusted to +30/35°C and under vigorous stirring 830 mL of Acetone are loaded in about 15-20 minutes. After stirring for 60 minutes at temperature of +30/35°C 1170 mL of Acetone are loaded in 45-60 minutes. Then the temperature is adjusted to +20/25 °C in about 30-60 minutes and maintained for 30 minutes. The obtained crystalline solid is filtered and washed with 430 mL of Acetone. Finally the product is dried under vacuum at +40°C till constant weight is achieved (83 g of Compound 4) are obtained with an HPLC assay = 98-99%, yield =t 80%).
Mr. Ram Gopal Agarwal
Chairman and Non-Executive Director
Mr. Ram Gopal Agarwal is Founder Chairman of Dhanuka Group.
He is a decisive and action oriented visionary who took over a sick pesticide Company named Northern Mineral Pvt. Ltd. in 1980 and transformed it today into a Rs 1000 Crore organization called Dhanuka Agritech Ltd.
His deep commitment and inspiring leadership in initial turbulent days is an example worth inculcating and his passion to contribute to Indian Agriculture is commendable.
His ability to prioritize and deal effectively with a number of tasks simultaneously reinforced with the skills to make effective decisions, has metamorphosed the business venture into one of the fastest growing Agrochemical Company in India which has thrice been rated as ‘Best under a Billion Company’ by Forbes Magazine.
In order to achieve his aspiration of “Transforming India through Agriculture” he has dedicated himself to bring changes in Agrochemicals Industry and the farming community. His contribution for adopting newer farming techniques at the grass root level, judicious use of agro chemicals in farming and imparting knowledge through his nationwide network of distributors and Dhanuka Doctors in field has resulted in the overall prosperity of farmers.
Mr. Ram Gopal Agarwal has been the past Chairman of CCFI, (Crop Care Federation of India) the apex Chamber of all Indian Agrochemical majors. He is also Chairman of Advisory Committee of AGRO Chemicals Federation of India.
Mr. Ram Gopal Agarwal, Group Chairman, has been bestowed with many Awards for his tremendous contribution in Agro Industry like “Life Time Achievement Award” by Agri Business Summit and Agri Awards 2019, “Distinguished Contribution to Indian Agrochemicals Industry” during India Chem 2016 International Conference organised by FICCI etc.
Mr. Manish Dhanuka
Managing Director
Mr. Manish Dhanuka is the Director of Orchid Pharma Limited; he has the vision to rejuvenate Orchid Pharma Ltd. and take it on a fruitful path. His wide-ranging experience of handling operations, commercial, marketing and finance in the manufacturing industry provides for his analytical and decision-making skills facilitating the restoration of the company to its glorious past and to achieve even greater heights.
He excels in creating economical Pharmaceutical technologies and accelerated evaluation process for improving healthcare. Experience of 25 years in research, evaluation, and teaching in the pharmaceutical industry equips him with the expertise in innovative pharmaceutical technologies…
He holds a B.Tech in Chemical Engineering from IIT, New Delhi, and M.S in Chemical Engineering from the University of Akron, USA.
Before establishing Dhanuka Laboratories Ltd. in 1993, he began his career at Ranbaxy Labs Ltd. in New Delhi and worked there for 5 years. His vision and strategy to grow the Pharmaceutical industry in the Indian sub-continent, have helped the Dhanuka Group of companies enhance its Bulk Drugs manufacturing arm exponentially. He spearheaded the acquisition of Synmedic Laboratories in the year 2013 which is involved in pharmaceutical formulations. This entrepreneurial vigor enabled him to take over the operations of Orchid Pharma Ltd. in March 2020.
Outside of work, he likes to travel for wildlife adventures.
Mr. Mridul Dhanuka
Whole-Time Director
He is associated with Dhanuka Group Ltd. since 2005. He was responsible in successfully realigning the entire supply chain vertical from procurement to sales. At Orchid, he hopes to replicate the Group’s success and put another feather in Dhanuka cap.
CLIP
Orchid Chemicals & Pharmaceuticals, or Orchid Pharma since its recent name change in 2015, was established in 1992 in Chennai to manufacture antibiotics, and entered drug discovery in 2001 with projects in the areas of anti-infectives and treatments for pain.32, 197 In 2002, the company engaged in a joint venture to develop US-based firm Bexel Biotechnology’s BLX-1002, an oral, non-PPAR AMPK activator for the treatment of diabetes,198 later repositioned for NASH (2012), but no further progress has been reported recently.197 In 2008, Orchid invested in Diakron Pharmaceuticals, a US-based company that had an exclusive license to MSD′s investigational oral anticoagulant drug, a direct thrombin inhibitor later known as DPOC-4088 (or DP-4088),199 which reached Phase 1 clinical studies in Europe in 2012 (Supporting Information Table 6b, entries 5–6).200 The company’s own internal discovery efforts had a broad therapeutic focus, covering infectious diseases, inflammation, pain, oncology, metabolic disorders, and CNS diseases. OCID-2987,197, 201 a PDE4 inhibitor for the treatment of inflammatory disorders such as COPD, completed successfully Phase 1 studies in Europe in 2012, and OCID-4681 29,202, 203 a histone deacetylase (HDAC) inhibitor for cancer had received approval in 2011 for Phase 1 studies for solid tumors in India, but we assume both have been abandoned, as cancer and inflammation are not mentioned in the company’s latest annual reports.197 Two additional compounds were abandoned at the preclinical stage: OCID-5005, a STAT-3/IL-6 inhibitor for oncology, and a unnamed Th1/Th2 cytokine synthesis inhibitor for inflammation (Supporting Information Table 2a, entries 134–138).197 Financial issues led Orchid, as of 2009, to sell parts of its business to Hospira (now part of Pfizer). As a consequence, no progress has been reported on its discovery programs since 2010, and no further NCE patent application has been published since 2012. However, in 2013 Orchid licensed its broad-spectrum β-lactamase inhibitor OCID-5090, a zwitterionic N-methylated tazobactam derivative, to the German Allecra Therapeutics for a 20 % stake in the company, for use in combination with antibiotics to treat multidrug-resistant gram negative bacteria.204–207 Allecra’s lead compound AAI202, a combination of cefepime and AAI101/OCID-5090 30, is currently in Phase 1 studies in France.208, 209
NEW DRUG APPROVALS
ONE TIME
$10.00
Dr. B. Gopalan
Scientific Advisor
Dr. Gopalan is a synthetic organic chemist with extensive experience in the field of drug discovery and development. After completing his PhD from University of Madras, he went to Harvard University where he worked with the Nobel Laureate, Prof. E.J. Corey, as a post-doctoral fellow. Subsequent to this he joined Syntex Research Inc. in California to work on the synthesis of unnatural amino acids. After a year, he moved to Bristol-Meyers Squibb, Princeton, New Jersey, to contribute to their program on novel antibiotics and ACE inhibitors. Dr. Gopalan then moved back to India in 1982 to join the Drug Discovery Research Division of Boots Pharmaceuticals (India) Ltd. in Mumbai. Over his decade long stint there he contributed extensively to their drug discovery program, and one of the product candidates that he developed went up to Phase-2 clinical trials in both USA and UK. He then moved to Sun Pharma Advanced Research Center as Vice-President and, after a year, took up the position as General Manager at Glaxo (India) Ltd. in 1993. Here, he worked in a broad range of areas that included process development, synthesis of impurities of APIs, and generation of small molecule libraries to support drug discovery efforts to Glaxo, France. In 1999 he took over as Senior Vice President of the Drug Discovery Chemistry Division of Glenmark Pharmaceuticals Ltd. where he was involved in the design and development of inhibitors for PDE IV and DPP IV, as well as agonists for CB2. After a 6-year stint at Glenmark, Dr. Gopalan joined Matrix Laboratories Ltd. as CSO and Executive Vice-President, where he successfully helped to develop novel and selective inhibitors for PDE4 and DPP4. Five years later he became CSO and Executive Director of Orchid Pharmaceuticals Ltd in Chennai. He served in this capacity for close to a decade, contributing extensively to drug design and development in the broad segments of oncology, anti-infectives, and anti-inflammatory & metabolic disorders. Since 2017, Dr. Gopalan has been associated with CSIR-Indian Institute of Chemical Technology as a Scientific Advisor.
Dr. Gopalan’s illustrious career is endowed with numerous successes. He has been inventor, or co-inventor, of several drugs or candidate drugs. These include the novel potassium channel blockers BTS-67582 (BTI-2927) for tpe-2 diabetes, the PDE IV inhibitors Oglemilast (COPD) and Revamilast (RA); DPP IV inhibitor Melogliptin; a selective Cannaboid-2 agonist Tedalinib (Neuropathic pain); a Beta lactamase inhibitor Enmetazobactum (OCID-5090); OCID-18034 (an inhibitor of KPC enzyme); and OCID-18174 (an inhibitor of P. arugenosa). Most of these compounds were out-licensed to major international pharmaceutical companies such as Forest Laboratories Inc. USA, Teijin of Japan, Merck KGaA of Germany, Allecra of Switzerland, and Merck & Co. USA. Dr.Gopalan has 34 publications in National and International Journals, has contributed a Chapter,Co-authored with Professor K.K.Balasubramanian (IITM) on Applications of Click Chemistry in Drug Discovery and Development in a Book on Click reaction in Organic Synthesis, published by Wiley-VCH VERLAG GmbH &Co,KGaA, Weinheim,Germany,Chapter 2, p 25-70,2016, edited by Prof. S. Chandrasekharan (IISc,Bangalore) & 51 Patents.
Commensurate with his achievements, Dr. Gopalan has also received many awards. The more prominent of these include Inventor’s award by Glenmark (2004), Ranbaxy Science Foundation Award in Pharmaceutical Sciences (2005), and the Lifetime Achievement Award in the Field of Chemistry from Vels University (2011).
Madrigal Pharmaceuticals , following the merger between Synta and Madrigal Pharmaceuticals (pre-merger) (following the acquisition of VIA Pharmaceuticals ‘ assets (originally under license from Roche )), is developing resmetirom (MGL-3196, VIA-3196), the lead from oral capsule formulation thyroid hormone receptor (THR) beta agonists, cholesterol and triglyceride modulators, for the use in the treatment of metabolic disorders including hypercholesterolemia and other dyslipidemias, and non-alcoholic steatohepatitis.
MGL-3196 is a first-in-class, orally administered, small-molecule, liver-directed, THR β-selective agonist. Preclinical, toxicology and Phase 1 clinical data suggest MGL-3196 has an attractive, differentiated profile as a potential treatment for non-alcoholic steatohepatitis (NASH) and dyslipidemias. THR-β selectivity also enhances the safety profile of MGL-3196, compared to non-selective agents. MGL-3196 has shown no suppression of the central thyroid axis, no THR-α effects on heart rate or bone, and no elevation of liver enzymes. These characteristics make MGL-3196 among the most promising molecules in development in this therapeutic area. MGL-3196 is in a Phase 2 clinical trial for the treatment of non-alcoholic steatohepatitis (NASH).
PATENT
WO-2020010068
Novel crystalline salt of resmetirom as thyroid hormone receptor agonists useful for treating obesity, hyperlipidemia, hypercholesterolemia and diabetes. Appears to be the first filing from the assignee and the inventors on this compound,
Thyroid hormones are critical for normal growth and development and for maintaining metabolic homeostasis (Paul M. Yen, Physiological reviews, Vol. 81(3): pp. 1097-1126 (2001)). Circulating levels of thyroid hormones are tightly regulated by feedback mechanisms in the hypothalamus/pituitary/thyroid (HPT) axis. Thyroid dysfunction leading to hypothyroidism or hyperthyroidism clearly demonstrates that thyroid hormones exert profound effects on cardiac function, body weight, metabolism, metabolic rate, body temperature, cholesterol, bone, muscle and behavior.
[0005] The biological activity of thyroid hormones is mediated by thyroid hormone receptors (TRs or THRs) (M. A. Lazar, Endocrine Reviews, Vol. 14: pp. 348-399 (1993)). TRs belong to the superfamily known as nuclear receptors. TRs form heterodimers with the retinoid receptor that act as ligand-inducible transcription factors. TRs have a ligand binding domain, a DNA binding domain, and an amino terminal domain, and regulate gene expression through interactions with DNA response elements and with various nuclear co-activators and co repressors. The thyroid hormone receptors are derived from two separate genes, a and b. These distinct gene products produce multiple forms of their respective receptors through differential RNA processing. The major thyroid receptor isoforms are aΐ, a2, bΐ, and b2. Thyroid hormone receptors aΐ, bΐ, and b2 bind thyroid hormone. It has been shown that the thyroid hormone receptor subtypes can differ in their contribution to particular biological responses. Recent studies suggest that TIIb 1 plays an important role in regulating TRH (thyrotropin releasing hormone) and on regulating thyroid hormone actions in the liver. T11b2 plays an important role in the regulation of TSH (thyroid stimulating hormone) (Abel et. al, J. Clin. Invest., Vol 104: pp. 291-300 (1999)). TIIb 1 plays an important role in regulating heart rate (B. Gloss et. al. Endocrinology, Vol. 142: pp. 544-550 (2001); C. Johansson et. al, Am. J. Physiol., Vol. 275: pp. R640-R646 (1998)).
[0006] Efforts have been made to synthesize thyroid hormone analogs which exhibit increased thyroid hormone receptor beta selectivity and/or tissue selective action. Such thyroid hormone mimetics may yield desirable reductions in body weight, lipids, cholesterol, and lipoproteins, with reduced impact on cardiovascular function or normal function of the hypothalamus/pituitary/thyroid axis (see, e.g., Joharapurkar et al, J. Med. Chem, 2012, 55 (12), pp 5649-5675). The development of thyroid hormone analogs which avoid the undesirable effects of hyperthyroidism and hypothyroidism while maintaining the beneficial effects of thyroid hormones would open new avenues of treatment for patients with metabolic disease such as obesity, hyperlipidemia, hypercholesterolemia, diabetes and other disorders and diseases such as liver steatosis and NASH, atherosclerosis, cardiovascular diseases, hypothyroidism, thyroid cancer, thyroid diseases, a resistance to thyroid hormone (RTH) syndrome, and related disorders and diseases.
Example 3: Preparation of (Z)-ethyl (2-cyano-2-(2-(3,5-dichloro-4-((5-isopropyl-6- oxo- l,6-dihydropyridazin-3-yl)oxy)phenyl)hydrazono)acetyl)carbamate (Int. 8)
A 2 L, three-neck, round-bottom flask equipped with overhead stirring, a thermocouple, N2 inlet/outlet was charged with Int. 7 (75.0 g, 0.239 mol, 1 wt), acetic acid (600 mL, 8 vol), water (150 mL, 2 vol), and concentrated HC1 (71.3 mL, 0.95 vol). The resulting thin slurry was cooled to 6 °C and a solution of NaN02 (16.8 g, 0.243 mol, 1.02 equiv) in water (37.5 mL, 0.5 vol) was added over a period of 10 min while maintaining a batch temperature below 10 °C. After an additional 10 min of agitation between 5-10 °C, HPLC analysis showed complete conversion of Int. 7 to the diazonium intermediate. A solution of NaOAc (54.5 g, 0.664 mol, 2.78 equiv) in water (225 mL, 3 vol) was added over a period of 6 min while maintaining a batch temperature below 10 °C. N-cyanoacetylurethane (37.9 g, 0.243 mol, 1.02 equiv) was immediately added, the cooling was removed, and the batch naturally warmed to 8 °C over 35 min. HPLC analysis showed complete consumption of the diazonium intermediate and the reaction was deemed complete. The batch warmed naturally to 21 °C and was filtered through Sharkskin filter paper. The reactor and cake were washed sequentially with water (375 mL, 5 vol) twice. The collected orange solid was dried in a 35 °C vacuum oven for 64 h to provide crude Int. 8 (104.8 g, 91%).
A I L, three-neck, round-bottom flask equipped with overhead stirring, a
thermocouple, and N2 inlet/outlet was charged with crude Int. 8 (104.4 g, 1 wt) and acetic acid (522 mL, 5 vol). The resulting slurry was heated to 50 °C and held at that temperature for 1.5 h. The batch cooled naturally to 25 °C over 2 h and was filtered through Sharkskin filter paper. The reactor and cake were washed sequentially with water (522 mL, 5 vol) and the cake conditioned under vacuum for 1.75 h. The light orange solid was dried to constant weight in a 40 °C vacuum oven to provide 89.9 g (78% from Int. 7) of the desired product. 1H NMR (DMSO) was consistent with the assigned structure.
Example 4: Preparation of 2-(3,5-dichloro-4-((5-isopropyl-6-oxo-l,6- dihydropyridazin-3-yl)oxy)phenyl)-3,5-dioxo-2,3,4,5-tetrahydro-l,2,4-triazine-6-carbonitrile (Compound A)
A 2 L, three-neck, round-bottom flask equipped with overhead stirring, a
thermocouple, N2 inlet/outlet, and reflux condenser was charged with Int. 8 (89.3 g, 0.185 mol, 1 wt), DMAC (446 mL, 5 vol), and KOAc (20.0 g, 0.204 mol, 1.1 equiv). The mixture was heated to 120 °C and held at that temperature for 2 h. HPLC analysis showed complete conversion to Compound A. The batch temperature was adjusted to 18 °C over 1 h, and acetic acid (22.3 mL, 0.25 vol) was added. The batch temperature was adjusted to 8 °C, and water (714 mL, 8 vol) was added over 1 h; an orange slurry formed. The batch was filtered through Sharkskin filter paper and the cake was allowed to condition overnight under N2 without vacuum for convenience. A premixed solution of 1 : 1 acetone/water (445 mL, 5 vol) was charged to the flask and added to the cake as a rinse with vacuum applied. After 2 h of conditioning the cake under vacuum, it was transferred to a clean 1 L, three-neck, round- bottom flask equipped with overhead stirring, a thermocouple, and N2inlet/outlet. Ethanol (357 mL, 4 vol) and acetone (357 mL, 4 vol) were charged and the resulting slurry was heated to 60 °C; dissolution occurred. Water (890 mL, 10 vol) was added over a period of 90 min while maintaining a batch temperature between 55-60 °C. The resulting slurry was allowed to cool to 25 °C and filtered through Sharkskin filter paper. The reactor and cake were washed sequentially with a solution of 1:1 EtOH/water (446 mL, 5 vol). The cake was conditioned overnight under N2 without vacuum for convenience. The cracks in the cake were smoothed and vacuum applied. The cake was washed with water (179 mL, 2 vol) and dried in a 45 °C vacuum oven to a constant weight of 70.5 g (87%, crude Compound A). HPLC analysis showed a purity of 94.8%.
A 500 mL, three-neck, round-bottom flask equipped with overhead stirring, a thermocouple, N2 inlet/outlet, and reflux condenser was charged with crude Compound A (70.0 g) and MIBK (350 mL, 5 vol). The orange slurry was heated to 50 °C and held at that temperature for 2 h. The batch cooled naturally to 23 °C and was filtered through Sharkskin filter paper. The reactor and cake were washed sequentially with MIBK (35 mL, 0.5 vol) twice. The collected solids were dried in a 45 °C vacuum oven to a constant weight of 58.5 g (84%). This solid was charged to a 500 mL, three-neck, round-bottom flask equipped with overhead stirring, a thermocouple, N2 inlet/outlet, and reflux condenser. Ethanol (290 mL, 5 vol) was added and the slurry was heated to reflux. After 3.5 h at reflux, XRPD showed the solid was consistent with Form I, and heating was removed. Upon reaching 25 °C, the batch was filtered through filter paper, and the reactor and cake were washed sequentially with EtOH (174 mL, 3 vol). The tan solid Compound A was dried in a 40 °C vacuum oven to a constant weight of 50.4 g (87%, 64% from Int. 8). HPLC analysis showed a purity of 99.1%. 1H NMR (DMSO) was consistent with the assigned structure.
Example 5: Scaled up preparation of 2-(3,5-dichloro-4-((5-isopropyl-6-oxo-l,6- dihydropyridazin-3-yl)oxy)phenyl)-3,5-dioxo-2,3,4,5-tetrahydro-l,2,4-triazine-6-carbonitrile (Compound A)
A larger scale batch of Compound A was synthesized according to the scheme below. The conditions in the scheme below are similar to those described in Examples 1-4 above.
6A
Compound A
Synthesis of 4: A 50 L jacketed glass vessel (purged with N2) was charged with 3,6- dichloropyridazine (2.00 kg), 4-amino-2,6-dichlorophenol (2.44 kg) and N,N- dimethylacetamide (10.0 L). The batch was vacuum (26 inHg) / nitrogen (1 PSIG) purged 3 times. Cesium carbonate (5.03 kg) was added and the batch temperature was adjusted from 22.3 °C to 65.0 °C over 3.5 hours. The batch was held at 65.0 °C for 20 hours. At this point,
NMR analysis indicated 3.34% 3.6-dichloropyridazine relative to 2. The batch temperature was adjusted to 21.5 °C and ethyl acetate (4.00 L) was added to the batch. The batch was agitated for 10 minutes and then filtered through a 18″ Nutsche filter equipped with polypropylene filter cloth. The filtration took 15 minutes. Ethyl acetate (5.34 L) was charged to the vessel and transferred to the filter as a rinse. The batch was then manually re- suspended in the filter before re-applying vacuum. This process was repeated 2 more times and the filter cake was conditioned for 10 minutes. The filtrate was charged to a 100-L vessel that contained (16.0 L) of a previously prepared 15% sodium chloride in H20. The batch was agitated for 5 minutes and then allowed to separate for 35 minutes. The interface was not visible, so the calculated 23 L of the lower aqueous phase was removed. 16.0 L of 15% Sodium chloride in H20 was added to the batch. The batch was agitated for 6 minutes and then allowed to separate for 7 minutes. The interface was visible at -19 L and the lower aqueous phase was removed. 17.0 L of 15% Sodium chloride in H20 was added to the batch. The batch was agitated for 7 minutes and then allowed to separate for 11 minutes. The lower aqueous phase was removed. The vessel was set up for vacuum distillation and the batch was concentrated from 17.0 L to 8.0 L over 2 hours 20 minutes with the batch temperature kept around 21 °C. Benzoic anhydride (3.19 kg) and acetic acid (18.0 L) were charged to the vessel. The vessel was set up for vacuum distillation and the batch was concentrated from 28.0 L to 12.0 L over 2 days (overnight hold at 20 °C) with the batch temperature kept between 20 and 55 °C. At this point, JH NMR analysis indicated a mol ratio of acetic acid to ethyl acetate of 1.0:0.015. Acetic acid (4.0 L) was charged to the batch and the batch was distilled to 12 L. JH NMR analysis indicated a mol ratio of acetic acid to ethyl acetate of 1.0:0.0036. Acetic acid (20.0 L) was charged to the batch and the batch temperature was adjusted to 70.0 °C. The batch was sampled for HPLC analysis and 2 was 0.16%. Sodium acetate (2,20 kg) was added to the batch and the batch temperature was adjusted from 72.4 °C to 110.0 °C. After 18.5 hours, HPLC analysis indicated no Int. B detected. The batch temperature was adjusted from 111.3 to 74.7 °C and DI water (30.0 L) was added to the batch over 2 hours. The batch temperature was adjusted to 20 .5 °C and then filtered using a 24″ Haselloy Nutsche filter equipped with polypropylene filter cloth. A previously prepared solution of 1:1 acetic acid in DI H20 (10.0 L) was charged to the vessel and agitated for 5 minutes. The wash was transferred to the filter and the batch was then manually re- suspended in the filter before re-applying vacuum. DI H20 (10.0 L) was charged to the vessel and then transferred to the filter. The batch was manually re-suspended in the filter before re-applying vacuum. DI H20 (10.0 L) was charged directly to the filter and the batch was then manually re-suspended in the filter before re-applying vacuum. The filter cake was allowed to condition for 18 hours to give 14.4 kg of 4. HPLC analysis indicated a purity of 93.7%. This wet cake was carried forward into the purification. A 100 L jacketed glass vessel (purged with N2) was charged with crude 4 (wet cake 14.42 kg), acetic acid (48.8 L) and the agitator was started. DI H20 (1.74 L) was charged. The batch (a slurry) temperature was adjusted from 18.1 to 100.1 °C over 4.25 hours. The batch was held at 100.1 to 106.1 °C for 1 hour and then adjusted to 73.1 °C. DI H20 (28.0 L) was added to the batch over 1 hour keeping the batch temperature between 73.1 and 70.3 °C. The batch temperature was adjusted further from 70.3 °C to 25.0 °C overnight. The batch was filtered using a 24″ Hastelloy Nutsche filter equipped with polypropylene filter cloth. The filtration took 13 minutes. A solution of DI H20 (9.00 L) and acetic acid (11.0 L) was prepared and added to the 100 L vessel. The mixture was agitated for 5 minutes and then transferred to the filter cake. DI H20 (20.0 L) was charged to the vessel, agitated for 6 minutes and then transferred to the filter cake. DI H20 (20.0 L) was charged to the vessel, agitated for 9 minutes and then transferred to the filter cake. The batch was allowed to condition for 3 days and then transferred to drying trays for vacuum oven drying. After 3 days at 50 °C and 28’7Hg, the batch gave a 74% yield (3.7 kg) of4 as an off-white solid. The JH NMR spectrum was consistent with the assigned structure, HPLC analysis indicated a purity of 98.87% and KF analysis indicated 0.14% H20. Synthesis of Int. 7: A 100-L jacketed glass vessel (purged with N2) was charged with tetrahydrofuran (44.4 L). The agitator was started (125 RPM) and 4 (3.67 kg) was charged followed by lithium chloride (1.26 kg). The batch temperature was observed to be 26.7 ° C and was an amber solution. Isopropenylmagnesium bromide 1.64 molar solution in 2-methyl THF (21.29 kg) was added over 2 ½ hours keeping the batch between 24.3 and 33.6 °C. The batch was agitated at 24.5 °C for 17 hours at which point HPLC analysis indicated 9% 4. A 2nd 100-L jacketed glass vessel (purged with N2) was charged with 3N hydrogen chloride (18.3 L). The batch was transferred to the vessel containing the 3N HC1 over 25 minutes keeping the batch temperature between 20 and 46 °C. A bi-phasic solution was observed. The quenched batch was transferred back to the 1st 100-L vessel to quench the small amount of residue left behind. THF (2.00 L) was used as a rinse. The batch temperature was observed to be 40.9 ° C and was agitated at 318 RPM for 45 minutes. The batch temperature was adjusted to 21.8 ° C and the layers were allowed to separate. The separation took 10 minutes. The lower aqueous phase was removed (-26.0 L). A solution of sodium chloride (1.56 kg) in DI water (14.0 L) was prepared and added to the batch. This was agitated at 318 RPM for 10 minutes and agitator was stopped. The separation took 3 minutes. The lower aqueous phase was removed (-16.0 L). The batch was vacuum distilled from 58.0 L to 18.4 L using ~24’7Hg and a jacket temperature of 50 to 55 °C. A solution of potassium hydroxide (2.30 kg) in DI water (20.7 L) was prepared in a 72-L round bottom flask. The vessel was set up for atmospheric distillation using 2 distillation heads and the batch was transferred to the 72-L vessel. THF (0.75 L) was used as a rinse. The batch volume was -41.0 L, the temperature was adjusted to 64.1 °C and distillation started with the aid of a N2 sweep. Heating was continued to drive the batch temperature to 85.4 °C while distilling at which point the 72-L vessel was set up for reflux (batch volume was about 28.0 L at the end of the distillation). The batch was held at 85 °C for 13 hours at which point HPLC analysis indicated 0.3% compound 6A. Heating was stopped and the batch was transferred to a 100-L jacketed glass vessel. Solids were observed. The batch temperature was adjusted from 70.6 °C to 56.7 °C. A previously prepared solution of sodium hydrogen carbonate (2.82 kg) in DI water (35.0 L) was added over 80 minutes keeping the batch temperature between 56.7 and 46.7 °C. The batch pH at the end of the addition was 9.8. The batch was held at
46.7 to 49.0 °C for 40 minutes and then cooled to 25.0 °C. The batch was filtered using a 18″ stainless steel Nutsche filter. DI water (18.4 L) was charged to the vessel and transferred to the filter. The filter cake was manually re-suspended in the filter and then the liquors were removed. This process was repeated once more and the filter cake was 3″ thick. The filter cake was conditioned on the filter for 3 days, was transferred to drying trays and dried in a vacuum oven at 45 °C to provide 2.93 kg Int. 7 (95% yield) with an HPLC purity of 87.6%.
Synthesis of Int. 8: A 100 L jacketed glass vessel (purged with N2 and plumbed to a caustic scrubber) was charged with acidic acid (13.0 L). Int. 7 (2.85 kg) was charged to the vessel and the agitator was started. N-Cyanoacetylurethane (1.56 kg) and DI water (5.70 L) were charged to the vessel. The batch temperature was adjusted from 17.0 °C to 5.5 °C and a thin slurry was observed. At this point 37% hydrogen chloride (2.70 L) was added over 10 minutes keeping the batch temperature between 4.8 °C and 8.8 °C. A previously prepared solution of sodium nitrite (638 g) in DI water (1.42 L) was added over 26 minutes keeping the batch temperature between 5.8 °C and 8.7 °C. A brown gas was observed in the vessel head space during the addition. HPLC analysis indicated no Int. 7 detected. At this point a previously prepared solution of sodium acetate (2.07 kg) in DI water (8.50 L) was added over 47 minutes keeping the batch temperature between 5.5 °C and 9.5 °C. After the addition, a thin layer of orange residue was observed on the vessel wall just above the level of the batch. The batch temperature was adjusted from 9.4 °C to 24.5 °C and held at 25 °C (+ 5 °C) for 12 hours. The batch was filtered using a 24″ Hastelloy Nutsche filter equipped with tight-weave polypropylene filter cloth. The filtration took 30 minutes. The vessel was rinsed with 14.3 L of a 1 : 1 acidic acid / DI water. The orange residue on the reactor washed away with the rinse. The rinse was transferred to the filter where the batch was manually re-suspended. Vacuum was re-applied to remove the wash. A 2nd 1 : 1 acidic acid / DI water wash was performed as above and the batch was conditioned on the filter for 26 hours. HPLC analysis of the wet filter cake indicated purity was 90.4%. The batch was dried to a constant weight of 3.97 kg (91% yield) in a vacuum oven at 45 °C and 287Hg. Preparation of Compound A DMAC Solvate
A 100 L, jacketed, glass vessel purged with N2 was charged with Int. 8 (3.90 kg) and potassium acetate (875 g). N,N-dimethylacetamide (DMAC, 18.3 L) was charged to the vessel and the agitator was started. The batch temperature was adjusted to 115 °C over 2 h. After 2 h at 115 °C, the batch was sampled and HPLC analysis indicated 0.27% Int. 8 remained. The batch temperature was adjusted to 25.0 °C overnight. Acetic acid (975 mL) was added to the batch and the batch was agitated further for 3 h. The batch was transferred to a carboy and the vessel was rinsed clean with 800 mL of DMAC. The batch was transferred back to the 100 L vessel using vacuum through a 10 μιη in-line filter and a DMAC rinse (1.15 L) was used. The filtration was fast at the beginning but slow at the end, plugging up the filter. The batch temperature was adjusted to 11.1 °C and DI water (35.1 L) was added over 2 h 20 min, keeping the batch temperature between 5-15 °C. The batch was held for 1 h and filtered, using an 18″ Nutsche filter equipped with tight-weave
polypropylene cloth. The filtration took 15 h. A 1: 1 ethanol/DI water wash (19.5 L) was charged to the vessel, cooled to 10 °C, and transferred to the filter cake. The cake was allowed to condition under N2 and vacuum for 8 h and transferred to drying trays. The batch was dried in a vacuum oven at 45 °C and 28’7Hg to give 89% yield (3.77 kg) of Compound A DMAC solvate as an orange/tan solid. The 1H NMR spectrum was consistent with the assigned structure and Karl Fischer analysis indicated 0.49% H20. XRPD indicated the expected form, i.e., Compound A DMAC solvate. Thermogravimetric analysis (TGA) indicated 16% weight loss. HPLC analysis indicated a purity of 93.67%.
Preparation of Crude Compound A
A 100 L, jacketed, glass vessel purged with N2 was charged with Compound A
DMAC solvate (3.75 kg) and ethanol (15.0 L). The agitator was started and acetone (15.0 L) was added. The batch temperature was adjusted from 10.6 °C to 60.0 °C over 1 h. At this point, the batch was in solution. DI water was added to the batch over 1.5 h, keeping the batch temperature at 60 + 5 °C. The batch was held at 60 + 5 °C for 1 h and cooled to 23.5 °C. An 18″ Nutsche filter equipped with tight-weave (0.67 CFM) polypropylene cloth was set up and the batch was filtered. The filtration took 15 h. A 1: 1 ethanol/DI water wash (19.5 L) was charged to the vessel and transferred to the filter cake. The cake was allowed to condition under N2 and vacuum for 8 h and transferred to drying trays. The batch was dried in a vacuum oven at 45 °C and 28’7Hg for five days to give a 94% yield (2.90 kg) of Compound A as a powdery tan solid. The NMR spectrum is consistent with the assigned structure and Karl Fischer analysis indicated 6.6% H20. XRPD indicated the expected form of dihydrate. TGA indicated 6.7% weight loss. HPLC analysis indicated a purity of 96.4% (AUC).
Purification of Crude Compound A
A 50 L, jacketed, glass vessel purged with N2 was charged with Compound A crude
(2.90 kg) and methyl isobutyl ketone (14.5 L). The agitator was started and the batch temperature was adjusted from 20.2 °C to 50.4 °C over 1.5 h. The batch was held at 50 °C (+ 5 °C) for 1 h and cooled to 20-25 °C. The batch was held at 20-25 °C for 2.5 h. An 18″ Nutsche filter equipped with tight- weave (0.67 CFM) polypropylene cloth was set up and the batch was filtered. The filtration took 20 min. Methyl isobutyl ketone (MIBK, 1.45 L) was charged to the vessel and transferred to the filter cake. The cake was manually resuspended and the liquors were pulled through with vacuum. Methyl isobutyl ketone (2.90 L) was charged to the filter cake and the cake was manually resuspended. The liquors were pulled through with vacuum and the cake was conditioned with vacuum and nitrogen for 15 h. The filter cake dried into a tan, hard 18″ x 1 ½” disc. This was manually broken up and run through coffee grinders to give a 76% yield (2.72 kg) of MGL-3196 MIBK solvate as a tan, powdery solid. No oven drying was necessary. The NMR spectrum was consistent with the assigned structure and Karl Fischer analysis indicated <0.1 % H20. XRPD indicated the expected form MIBK solvate. TGA indicated 17.3% weight loss. HPLC analysis indicated a purity of 98.5%.
Example 6: Conversion of Compound A to Form I
Purified Compound A (4802 g) as a 1:1 MIBK solvate which was obtained from Int. 8 as described in Example 5 above was added into a jacketed, 100 L reactor along with 24 liters of ethanol. The resulting slurry was heated to 80 + 5 °C (reflux) over 1 h 25 min; the mixture was stirred at that temperature for 4 h 25 min. Analysis of the filtered solids at 2 h 55 min indicated that the form conversion was complete, with the XRPD spectra conforming to Form I. The mixture was cooled to 20 + 5 °C over 45 min and stirred at that temperature for 15 min. The slurry was filtered and the filter cake was washed twice with prefiltered ethanol (2 x 4.8 L). The wet cake (4.28 kg) was dried under vacuum at 40 + 5 °C for 118 h to afford 3390 g of Compound A form I.
PAPER
Journal of Medicinal Chemistry (2014), 57(10), 3912-3923
The beneficial effects of thyroid hormone (TH) on lipid levels are primarily due to its action at the thyroid hormone receptor β (THR-β) in the liver, while adverse effects, including cardiac effects, are mediated by thyroid hormone receptor α (THR-α). A pyridazinone series has been identified that is significantly more THR-β selective than earlier analogues. Optimization of this series by the addition of a cyanoazauracil substituent improved both the potency and selectivity and led to MGL-3196 (53), which is 28-fold selective for THR-β over THR-α in a functional assay. Compound 53 showed outstanding safety in a rat heart model and was efficacious in a preclinical model at doses that showed no impact on the central thyroid axis. In reported studies in healthy volunteers, 53 exhibited an excellent safety profile and decreased LDL cholesterol (LDL-C) and triglycerides (TG) at once daily oral doses of 50 mg or higher given for 2 weeks.
//////////////RESMETIROM , MGL-3196, VIA-3196, UNII-RE0V0T1ES0, Phase III
Xanomeline(LY246708) is a selective M1 muscarinic receptor agonist. in vitro: Xanomeline had high affinity for muscarinic receptors in brain homogenates, but had substantially less or no affinity for a number of other neurotransmitter receptors and uptake sites. In cells stably expressing genetic m1 receptors, xanomeline increased phospholipid hydrolysis in CHO, BHK and A9 L cells to 100, 72 and 55% of the nonselective agonist carbachol. In isolated tissues, xanomeline had high affinity for M1 receptors in the rabbit vas deferens (IC50 = 0.006 nM), low affinity for M2 receptors in guinea pig atria (EC50 = 3 microM), was a weak partial agonist in guinea pig ileum and was neither an agonist nor antagonist in guinea pig bladder. Xanomeline produced small increases in striatal acetylcholine levels and did not antagonize the large increases in acetylcholine produced by the nonselective muscarinic agonist oxotremorine, indicating that xanomeline did not block M2 autoreceptors. in vivo: Xanomeline increased striatal levels of dopamine metabolites, presumably by acting at M1 heteroreceptors on dopamine neurons to increase dopamine release. In contrast, xanomeline had only a relatively small effect on acetylcholine levels in brain, indicating that it is devoid of actions at muscarinic autoreceptors. The effects of xanomeline on ex vivo binding and DOPAC levels lasted for about 3 hr and were evident after oral administration. An analog of xanomeline with similar in vivo effects did not inhibit acetylcholinesterase or choline acetyltransferase and inhibited choline uptake only at concentrations much higher than those required to inhibit binding. These data indicate xanomeline is selective agonist for M1 over M2 and M3 receptors in vivo in rat.
CAS Name: 3-[4-(Hexyloxy)-1,2,5-thiadiazol-3-yl]-1,2,5,6-tetrahydro-1-methylpyridine
Molecular Formula: C14H23N3OS
Molecular Weight: 281.42
Percent Composition: C 59.75%, H 8.24%, N 14.93%, O 5.69%, S 11.39%
Literature References: Selective muscarinic M1-receptor agonist.
Prepn: P. Sauerberg, P. H. Olesen, EP384288 (1990 to Ferrosan); eidem,US5043345 (1991 to Novo Nordisk); eidemet al.,J. Med. Chem.35, 2274 (1992).
Prepn of crystalline tartrate: L. M. Osborne et al.,WO9429303 (1994 to Novo Nordisk).
Muscarinic receptor binding study: H. E. Shannon et al.,J. Pharmacol. Exp. Ther.269, 271 (1994). Pharmacology: F. P. Bymaster et al.,ibid. 282.
HPLC determn in plasma: C. L. Hamilton et al.,J. Chromatogr.613, 365 (1993).
Derivative Type: Oxalate
CAS Registry Number: 141064-23-5
Molecular Formula: C14H23N3OS.C2H2O4
Molecular Weight: 371.45
Percent Composition: C 51.74%, H 6.78%, N 11.31%, O 21.54%, S 8.63%
Properties: Crystals from acetone, mp 148°.
Melting point: mp 148°
Derivative Type: (+)-L-Hydrogen tartrate
CAS Registry Number: 152854-19-8
Additional Names: Xanomeline tartrate
Manufacturers’ Codes: LY-246708; NNC-11-0232
Trademarks: Lomeron (Lilly); Memcor (Lilly)
Molecular Formula: C14H23N3OS.C4H6O6
Molecular Weight: 431.50
Percent Composition: C 50.10%, H 6.77%, N 9.74%, O 25.95%, S 7.43%
Properties: Crystals from 2-propanol, mp 95.5°.
Melting point: mp 95.5°
Therap-Cat: Cholinergic; nootropic.
Keywords: Cholinergic; Nootropic.
SYNTHESIS WILL BE UPDATED
EP 0384288; US 5260311; US 5264444; US 5328925, US 5834495; WO 9429303, EP 0687265; JP 1996507298; WO 9420495
The reaction of pyridine-3-carbaldehyde (I) with KCN in acetic acid, followed by a treatment with NH4Cl in aqueous NH4OH yields 2-amino-2-(3-pyridyl)acetonitrile (II), which is cyclized to 3-chloro-4-(3-pyridyl)-1,2,5-thiadiazole (III) by a treatment with S2Cl2 in DMF. The reaction of (III) with sodium hexyloxide in hexanol yields 3-(hexyloxy)-4-(3-pyridyl)-1,2,5-thiadiazole (IV), which is treated with methyl iodide in acetone to afford the corresponding N-methylpyridinium salt (V). Finally, this compound is hydrogenated with NaBH4 in ethanol and salified with oxalic or L-tartaric acid in acetone or isopropanol.
Xanomeline (39) has emerged as one of the most potent unbridged arecoline derivatives. It has higher potency and efficacy for m1 and m4 than for m2, m3 and m5 receptor subtypes [73], binds to the m1receptor subtype uniquely tightly [74,75] and stimulates phosphoinositide hydrolysis in the brain. In cells containing human m1 receptors which are stably expressing amyloid precursor protein (APP), xanomeline (39) stimulates APP release with a potency 1000 greater than carbachol and reduces the secretion of Aβ by 46% [76] (cf 2.6 Central nervous system). In patients with Alzheimer’s disease, it halted cognitive decline and reduced behavioural symptoms such as hallucinations, delusions and vocal outbursts [77,78]. As might be expected there have been numerous attempts to prepare analogues with comparable potency and efficacy. Transplanting the thiadiazole ring of xanomeline to a range of bicyclic amines reduced selectivity [79,80] as did the use of pyrazine analogues (40) [81].
Xanomeline (1) is an orthosteric muscarinic acetylcholine receptor (mAChR) agonist, often referred to as M1/M4-preferring, that received widespread attention for its clinical efficacy in schizophrenia and Alzheimer’s disease (AD) patients. Despite the compound’s promising initial clinical results, dose-limiting side effects limited further clinical development. While xanomeline, and related orthosteric muscarinic agonists, have yet to receive approval from the FDA for the treatment of these CNS disorders, interest in the compound’s unique M1/M4-preferring mechanism of action is ongoing in the field of chemical neuroscience. Specifically, the promising cognitive and behavioral effects of xanomeline in both schizophrenia and AD have spurred a renewed interest in the development of safer muscarinic ligands with improved subtype selectivity for either M1 or M4. This Review will address xanomeline’s overall importance in the field of neuroscience, with a specific focus on its chemical structure and synthesis, pharmacology, drug metabolism and pharmacokinetics (DMPK), and adverse effects.
PAPER
References
Jump up^Farde L, Suhara T, Halldin C, et al. (1996). “PET study of the M1-agonists [11C]xanomeline and [11C]butylthio-TZTP in monkey and man”. Dementia (Basel, Switzerland). 7 (4): 187–95. PMID8835881.
Jump up^Messer WS (2002). “The utility of muscarinic agonists in the treatment of Alzheimer’s disease”. Journal of Molecular Neuroscience : MN. 19 (1-2): 187–93. PMID12212779. doi:10.1007/s12031-002-0031-5.
Jump up^Mirza NR, Peters D, Sparks RG (2003). “Xanomeline and the antipsychotic potential of muscarinic receptor subtype selective agonists”. CNS Drug Reviews. 9 (2): 159–86. PMID12847557. doi:10.1111/j.1527-3458.2003.tb00247.x.
Vadadustat, also known as AKB-6548 and PG-1016548, is a potent Hypoxia-inducible factor-proline dioxygenase inhibitor. AKB-6548 works by inhibiting hypoxia inducible factor-prolyl hydroxylase (HIP-PH), leading to stabilization and increased levels of HIFα. In turn HIFα improves production of hemoglobin and red blood cells (RBCs), while maintaining normal levels of erythropoietin (EPO) in patients. We believe this differentiated mechanism of action has the potential to be safer than that of injectable recombinant erythropoietin stimulating agents (rESAs), avoiding supra-physiological levels of EPO and saturation of EPO receptors for prolonged periods of time.
Akebia Therapeutics, under license from Procter & Gamble, and sublicensee Mitsubishi Tanabe Pharma are developing vadadustat, an orally active small-molecule hypoxia-inducible factor prolyl hydroxylase (HIF-PH) inhibitor that stabilize HIF2-α, as a once-daily formulation, for treating anemia. Also the company is investigating AKB-6899, an oral HIF-PH inhibitor, for treating cancer and ocular diseases. In March 2016, the IND application was opened. Aerpio Therapeutics, a spinoff of Akebia, is investigating AKB-4924, a HIF2-α stabilizer, which inhibits HIF prolyl hydroxylase-2, for treating inflammatory bowel disease and wound healing
Hypoxia-inducible factor (HIF) is a transcription factor that is a key regulator of responses to hypoxia. In response to hypoxic conditions, i.e., reduced oxygen levels in the cellular environment, HIF upregulates transcription of several target genes, including those encoding erythropoietin. HIF is a heteroduplex comprising an alpha and beta subunit. While the beta subunit is normally present in excess and is not dependent on oxygen tension, the HIF-alpha subunit is only detectable in cells under hypoxic conditions. In this regard, the accumulation of HIF-alpha is regulated primarily by hydroxylation at two proline residues by a family of prolyl hydroxylases known as HIF prolyl hydroxylases, wherein hydroxylation of one or both of the proline residues leads to the rapid degradation of HIF-alpha. Accordingly, inhibition of HIF prolyl hydroxylase results in stabilization and accumulation of HIF-alpha {i.e., the degradation of HIF-alpha is reduced), thereby leading to an increase in the amount of HIF-alpha available for formation of the HIF heterodimer and upregulation of target genes, such as the Erythropoietin gene. Conversely, activation of HIF prolyl hydroxylase results in destabilization of HIF-alpha {i.e., the degradation of HIF-alpha is increased), thereby leading to a decrease in the amount of HIF-alpha available for formation of the HIF heterodimer and downregulation of target genes, such as VEGF.
The family of hypoxia inducible factors includes HIF- 1 -alpha, HIF-2-alpha, and HIF-3 -alpha.
A new class of prolyl hydroxylase inhibitors and their use to treat or prevent diseases ameliorated by modulation of hypoxia-inducible factor (HIF) prolyl hydroxylase are described in U.S. Patent No. 7,811,595, which is incorporated herein by reference in its entirety. The synthesis of such prolyl hydroxylase inhibitors is described in U.S. Patent Publication No.2012/0309977, which is incorporated herein by reference in its entirety. Such compounds inhibit HIF prolyl hydroxylase, thereby stabilizing HIF-alpha. As a consequence of stabilizing HIF-alpha, endogenous erythropoietin (EPO) production is increased. As with all drugs, proper doses and dosing regimens for treating patients having diseases such as anemia are essential for achieving a desired or optimal therapeutic effect without adverse effects or unwanted side-effects. Indeed, many active compounds fail in clinical trials because an effective and safe dosing regimen cannot be found.
Vadadustat (also known as AKB-6548) in anemia secondary to chronic kidney disease (CKD)
We are developing our lead product candidate, vadadustat, to be the potential best-in-class hypoxia inducible factor–prolyl hydroxylase inhibitor for the treatment of anemia secondary to CKD.
HIF inhibitor Vadadustat (Code AKB-6548) The chemical name N- [5- (3- chlorophenyl) -3-hydroxypyridine-2-carbonyl] glycine,
Vadadustat is a treatment for anemia associated with chronic kidney disease oral HIF inhibitor, is an American biopharmaceutical company Akebia Therapeutics invention in the research of new drugs, has completed Phase II pivotal clinical trial treatment studies, successfully met the researchers set given the level of hemoglobin in vivo target and good security, a significant effect, and phase III clinical trials.
U.S. Patent Publication US20120309977 synthetic route for preparing a Vadadustat: A 3-chlorophenyl boronic acid and 3,5_-dichloro-2-cyanopyridine as starting materials, by-catalyzed coupling methoxy substituted, cyano hydrolysis and condensation and ester hydrolysis reaction Vadadustat, process route is as follows:
Since the entire synthetic route 12 steps long, complicated operation, high cost.U.S. Patent No. 1 2 ^ ¥ disclosed 20070299086 & (^ (Scheme 3 1118 seven seven to 3,5-dichloro-2-cyanopyridine starting material, first-dichloro substituted with benzyloxy, then cyano hydrolysis, condensation, hydrogenation and deprotection trifluorosulfonyl, to give N- [5- trifluoromethanesulfonyloxy-3-hydroxypyridine-2-carbonyl) glycine methyl ester, 3-chlorophenyl and then boronic acid catalyzed coupling reactions, the final ester hydrolysis reaction Vadadustat, process route is as follows:
The synthesis steps long, intermediate products and final products contain more impurities and byproducts, thus purified requires the use of large amounts of solvents, complicated operation, low yield, and because the hydrogenation reaction is a security risk on the production, not conducive to the promotion of industrial production, it is necessary to explore a short process, simple operation, low cost synthetic method whereby industrial production Vadadus tat fit.
Example 1
A) Preparation of N- (3,5_-dichloro-2-carbonyl) glycine methyl ester:
3,5-dichloro-2-pyridinecarboxylic acid (19.2g, 0.10mol) and N, N’_ carbonyldiimidazole (24.3g, 0.15mol) was dissolved in N, N- dimethylformamide (100 mL ), was added glycine methyl ester hydrochloride (15.18,0.12111〇1), 11 was added dropwise diisopropylethylamine (51.7g, 0.40mol), the reaction mixture was stirred 35 ° C for 8 hours, TLC determined the completion of reaction gussets The reaction solution was concentrated by rotary evaporation to dryness, dilute hydrochloric acid was adjusted to neutral by adding ethyl acetate, dried over magnesium sulfate, and concentrated by rotary evaporation to dryness, and recrystallized from methanol to give N- (3,5- dichloro-pyridin-2 – carbonyl) glycine methyl ester, an off-white solid (21.6g), a yield of 82.0%, this reaction step is as follows:
1234567 B) Preparation of N- [5- (3- chlorophenyl) -3-chloropyridine-2-carbonyl] glycine methyl ester: 2
1 (3,5-dichloro-2-carbonyl) glycine methyl ester (20 (^, 〇1 76111111), 3-chlorophenyl boronic acid (13.18, 3 83.7mmol), [l, l’- bis (diphenylphosphino) ferrocene] dichloropalladium (2.8g, 3.8mmol), potassium carbonate (14.2g, 4 0. lmo 1) and N, N- dimethylformamide (75mL) was added The reaction flask, the reaction mixture was heated to 60 ° C for 20 hours the reaction was stirred for 5:00, point TLC plates to determine completion of the reaction, the reaction solution was cooled to room temperature, was concentrated by rotary evaporation to dryness, extracted with ethyl acetate, washed with brine, sulfuric acid 6 magnesium dried and concentrated by rotary evaporation to dryness, a mixed solvent of ethyl acetate and n-hexane was recrystallized to give N- [5- (3- chlorophenyl) -3-7-chloro-2-carbonyl] glycine methyl ester, white solid (19.7g), yield 76.4%, this reaction step is as follows:
C) Preparation of N_ [5- (3- chlorophenyl) -3-methoxy-pyridine-2-carbonyl] glycine:
N- [5- (3- chlorophenyl) -3-chloropyridine-2-carbonyl] glycine methyl ester (19 (^, 56111 111〇1) and sodium methoxide (7.6g, 0.14mol) was dissolved in methanol (150 mL), the reaction mixture was heated to 65 ° C, the reaction was stirred at reflux for 24 hours, TLC determined gussets completion of the reaction the reaction solution was cooled to room temperature, water (300mL) was stirred for 3h, cooled to 0 ° C, stirred for 2h, precipitated solid was filtered, the filter cake was dried to give N- [5- (3- chlorophenyl) -3-methoxy-pyridine-2-carbonyl] glycine, off-white solid (17.4 g of), a yield of 96.5%, of the reaction steps are as follows:
D) Preparation Vadadustat:
N- [5- (3- chlorophenyl) -3-methoxy-pyridine-2-carbonyl] glycine (16.68,51.7111111〇1) and 48% hydrobromic acid solution (52mL, 0.46mol) added to the reaction bottle, the reaction mixture was heated to 100 ° C, the reaction was stirred at reflux for 24 hours, TLC determined gussets completion of the reaction the reaction solution cooled square ~ 5 ° C, was slowly added 50% sodium hydroxide solution was adjusted to pH 2 at 0 -5 ° C under crystallization 3h, the filter cake washed with ethyl acetate and n-hexane mixed solvent of recrystallization, in finished Vadadustat, off-white solid (15.6g), a yield of 98.0%, this reaction step is as follow
Lanthier et al. (U.S. Patent Application 2012/0309977) described a procedure for synthesizing a compound of Formula (II) starting from 3-chloroboronic acid and 3,5-dichloropicolinonitrile, as shown in the scheme below:
which has an X-ray powder diffraction pattern as shown in FIG. 1. In certain embodiments, Form A of Compound (I) has an X-ray powder diffraction pattern comprising one, two, three, four, or five peaks at approximately 18.1 , 20.3, 22.9, 24.0, and 26.3 °2Θ; and wherein the crystalline Compound (I) is substantially free of any other crystalline form of Compound (I).
Compound (I) as prepared according to e.g., U.S. 7,811,595 and/or U.S. Patent Application No. 13/488,554 and then subjecting the resulting Compound (I)
(I),
to a procedure comprising
a) preparing a solution of Compound (I) in 2-methyltetrahydrofuran;
b) adding n-heptane;
c) heating the suspension {e.g., to about 40-50 °C);
d) cooling the suspension {e.g., to about 0-10 °C); and
c) isolating the crystals.
SYNTHESIS
US 2015361043
Synthesis of vadadustat and its intermediates is described. The process involves Suzuki coupling of 3,5-dichloropyridine-2-carbonitrile with (3-chlorophenyl)boronic acid, selective chloride displacement, simultaneous hydrolysis of nitrile and methyl ether, activation with CDI, condensation with methyl glycinate hydrochloride and finally ester hydrolysis. The process is simple and provides high product yield with high quality. Vadadustat is expected to be useful for the treatment of renal failure anemia (1). Suzuki coupling of 3,5-dichloropyridine-2-carbonitrile (I) with (3-chlorophenyl)boronic acid (II) in the presence of PdCl2(dppf) and K2CO3 in DMF yields 3-chloro-5-(3-chlorophenyl)pyridine-2-carbonitrile (III), which upon selective chloride displacement with NaOMe in refluxing MeOH affords methyl ether (IV). Hydrolysis of nitrile and methyl ether in intermediate (IV) with HBr or HCl at 100 °C furnishes 5-(3-chlorophenyl)-3-hydroxypyridine-2-carboxylic acid (V). After activation of carboxylic acid (V) with CDI or pivaloyl chloride and DIEA in DMSO, condensation with methyl glycinate hydrochloride (VI) in the presence of DIEA provides vadadustat methyl ester (VII). Finally, hydrolysis of ester (VII) with NaOH in H2O/THF produces the target vadadustat (1).
FIG. 1 depicts an outline of one embodiment for preparing the disclosed prolyl hydroxylase inhibitors.
FIG. 2 depicts an outline of one embodiment for preparing the disclosed prolyl hydroxylase inhibitor ester prodrugs.
FIG. 3 depicts an outline of one embodiment for preparing the disclosed prolyl hydroxylase inhibitor amide prodrugs.
Example 1 describes a non-limiting example of the disclosed process for the preparation of a prolyl hydroxylase ester pro-drug
EXAMPLE 1Methyl {[5-(3-chlorophenyl)-3-hydroxypyridin-2-yl]amino}acetate (4)
Preparation of 5-(3-chlorophenyl)-3-chloro-2-cyanopyridine (1): To a 100 mL round bottom flask adapted for magnetic stirring and equipped with a nitrogen inlet was charged (3-chlorophenyl)boronic acid (5 g, 32 mmol), 3,5-dichloro-2-cyanopyridine (5.8 g, 34 mmol), K2CO3 (5.5 g, 40 mmol), [1,1′-bis(diphenyphosphino)ferrocene]dichloro-palladium(II) [PdCl2(dppf)] (0.1 g, 0.13 mmol), dimethylformamide (50 mL) and water (5 mL). The reaction solution was agitated and heated to 45° C. and held at that temperature for 18 hours after which the reaction was determined to be complete due to the disappearance of 3,5-dichloro-2-cyanopyridine as measured by TLC analysis using ethyl acetate/methanol (4:1) as the mobile phase and UV 435 nm to visualize the reaction components. The reaction solution was then cooled to room temperature and the contents partitioned between ethyl acetate (250 mL) and saturated aqueous NaCl (100 mL). The organic phase was isolated and washed a second time with saturated aqueous NaCl (100 mL). The organic phase was dried for 4 hours over MgSO4, the MgSO4 removed by filtration and the solvent removed under reduced pressure. The residue that remained was then slurried in methanol (50 mL) at room temperature for 20 hours. The resulting solid was collected by filtration and washed with cold methanol (50 mL) then hexanes (60 mL) and dried to afford 5.8 g (73% yield) of an admixture containing a 96:4 ratio of the desired regioisomer. 1H NMR (DMSO-d6) δ 9.12 (d, 1H), 8.70 (d, 1H), 8.03 (t, 1H) 7.88 (m, 1H), and 7.58 (m, 2H)
Preparation of 5-(3-chlorophenyl)-3-methoxy-2-cyanopyridine (2): To a 500 mL round bottom flask adapted for magnetic stirring and fitted with a reflux condenser and nitrogen inlet was charged with 5-(3-chlorophenyl)-3-chloro-2-cyanopyridine, 1, (10 g, 40 mmol), sodium methoxide (13.8 mL, 60 mmol) and methanol (200 mL). With stirring, the reaction solution was heated to reflux for 20 hours. The reaction was determined to be complete due to the disappearance of 5-(3-chlorophenyl)-3-chloro-2-cyanopyridine as measured by TLC analysis using hexane/ethyl acetate (6:3) as the mobile phase and UV 435 nm to visualize the reaction components. The reaction mixture was cooled to room temperature and combined with water (500 mL). A solid began to form. The mixture was cooled to 0° C. to 5° C. and stirred for 3 hours. The resulting solid was collected by filtration and washed with water, then hexane. The resulting cake was dried in vacuo at 40° C. to afford 9.4 g (96% yield) of the desired product as an off-white solid. 1H NMR (DMSO-d6) δ 8.68 (d, 1H), 8.05 (d, 1H), 8.01 (s, 1H) 7.86 (m, 1H), 7.59 (s, 1H), 7.57 (s, 1H) and 4.09 (s, 3H).
Preparation of 5-(3-chlorophenyl)-3-hydroxypyridine-2-carboxylic acid (3): To a 50 mL round bottom flask adapted for magnetic stirring and fitted with a reflux condenser was charged 5-(3-chlorophenyl)-3-methoxy-2-cyanopyridine, 2, (1 g, 4 mmol) and a 48% aqueous solution of HBr (10 mL). While being stirred, the reaction solution was heated to reflux for 20 hours. The reaction was determined to be complete due to the disappearance of 5-(3-chlorophenyl)-3-methoxy-2-cyanopyridine as measured by TLC analysis using hexane/ethyl acetate (6:3) as the mobile phase and UV 435 nm to visualize the reaction components. The reaction contents was then cooled to 0° C. to 5° C. with stirring and the pH was adjusted to approximately 2 by the slow addition of 50% aqueous NaOH. Stirring was then continued at 0° C. to 5° C. for 3 hours. The resulting solid was collected by filtration and washed with water, then hexane. The resulting cake was dried in vacuo at 40° C. to afford 1.03 g (quantitative yield) of the desired product as an off-white solid. 1H NMR (DMSO-d6) δ 8.52 (d, 1H), 7.99 (d, 1H), 7.95 (s, 1H) 7.81 (t, 1H), 7.57 (s, 1H), and 7.55 (s, 1H).
Preparation of methyl {[5-(3-chlorophenyl)-3-hydroxypyridin-2-yl]amino}acetate (4): To a 50 mL round bottom flask adapted for magnetic stirring and fitted with a nitrogen inlet tube was charged 5-(3-chlorophenyl)-3-hydroxypyridine-2-carboxylic acid, 3, (1 gm, 4 mmol), N,N′-carbonyldiimidazole (CDI) (0.97 g, 6 mmol) and dimethyl sulfoxide (5 mL). The reaction mixture was stirred at 45° C. for about 1 hour then cooled to room temperature. Glycine methyl ester hydrochloride (1.15 g, 12 mmol) is added followed by the dropwise addition of diisopropylethylamine (3.2 mL, 19 mmol). The mixture was then stirred for 2.5 hours at room temperature after which water (70 mL) was added. The contents of the reaction flask was cooled to 0° C. to 5° C. and 1N HCl was added until the solution pH is approximately 2. The solution was extracted with dichloromethane (100 mL) and the organic layer was dried over MgSO4 for 16 hours. Silica gel (3 g) is added and the solution slurried for 2 hours after which the solids are removed by filtration. The filtrate is concentrated to dryness under reduced pressure and the resulting residue was slurried in methanol (10 mL) for two hours. The resulting solid was collected by filtration and washed with cold methanol (20 mL) then hexane and the resulting cake is dried to afford 0.85 g of the desired product as an off-white solid. The filtrate was treated to afford 0.026 g of the desired product as a second crop. The combined crops afford 0.88 g (68% yield) of the desired product. 1H NMR (DMSO-d6) δ 12.3 (s, 1H), 9.52 (t, 1H), 8.56 (d, 1H), 7.93 (s, 1H), 7.80 (q, 2H), 7.55 (t, 2H), 4.12 (d, 2H), and 3.69 (s, 3H).
The formulator can readily scale up the above disclosed synthesis. Disclosed herein below is a synthesis wherein the disclosed process is scaled up for commercial use
EXAMPLE 2Methyl {[5-(3-chlorophenyl)-3-hydroxypyridin-2-yl]amino}acetate (4)
Preparation of 5-(3-chlorophenyl)-3-chloro-2-cyanopyridine (1): A 20 L reactor equipped with a mechanical stirrer, dip tube, thermometer and nitrogen inlet was charged with (3-chlorophenyl)boronic acid (550 g, 3.52 mol), 3,5-dichloro-2-cyanopyridine (639 g, 3.69 mol), K2CO3 (5.5 g, 40 mmol), [1,1′-bis(diphenyphosphino)ferrocene]dichloro-palladium(II) [PdCl2(dppf)] (11.5 g, 140 mmol), and dimethylformamide (3894 g, 4.125 L). The reaction solution was agitated and purged with nitrogen through the dip-tube for 30 minutes. Degassed water (413 g) was then charged to the reaction mixture while maintaining a temperature of less than 50° C. 25 hours. The reaction was determined to be complete due to the disappearance of 3,5-dichloro-2-cyanopyridine as measured by TLC analysis using ethyl acetate/methanol (4:1) as the mobile phase and UV 435 nm to visualize the reaction components. The reaction solution was then cooled to 5° C. and charged with heptane (940 g, 1.375 L) and agitated for 30 minutes. Water (5.5 L) was charged and the mixture was further agitated for 1 hour as the temperature was allowed to rise to 15° C. The solid product was isolated by filtration and washed with water (5.5 L) followed by heptane (18881 g, 2750 ML). The resulting cake was air dried under vacuum for 18 hours and then triturated with a mixture of 2-propanol (6908 g, 8800 mL0 and heptane (1 g, 2200 mL0 at 50° C. for 4 hours, cooled to ambient temperature and then agitated at ambient temperature for 1 hour. The product was then isolated by filtration and washed with cold 2-propanol (3450 g, 4395 mL) followed by heptane (3010 g, 4400 mL). The resulting solid was dried under high vacuum at 40° C. for 64 hours to afford 565.9 g (65% yield) of the desired product as a beige solid. Purity by HPLC was 98.3. 1H NMR (DMSO-d6) δ 9.12 (d, 1H), 8.70 (d, 1H), 8.03 (t, 1H) 7.88 (m, 1H), and 7.58 (m, 2H).
Preparation of 5-(3-chlorophenyl)-3-methoxy-2-cyanopyridine (2): A 20 L reactor equipped with a mechanical stirred, condenser, thermometer and nitrogen inlet was charged with 5-(3-chlorophenyl)-3-chloro-2-cyanopyridine, 1, (558 g, 2.24 mol) and sodium methoxide (25% solution in methanol, 726.0 g, 3.36 mol). With agitation, the reaction solution was heated to reflux for 24 hours, resulting in a beige-colored suspension. The reaction was determined to be complete due to the disappearance of 5-(3-chlorophenyl)-3-chloro-2-cyanopyridine as measured by TLC analysis using hexane/ethyl acetate (6:3) as the mobile phase and UV 435 nm to visualize the reaction components. The reaction mixture was cooled to 5° C. and then charged with water (5580 mL). The resulting slurry was agitated for 3 hours at 5° C. The solid product was isolated by filtration and washed with water (5580 mL) until the filtrate had a pH of 7. The filter cake was air dried under vacuum for 16 hours. The filter cake was then charged back to the reactor and triturated in MeOH (2210 g, 2794 mL) for 1 hour at ambient temperature. The solid was collected by filtration and washed with MeOH (882 g, 1116 mL, 5° C.) followed by heptane (205 mL, 300 mL), and dried under high vacuum at 45° C. for 72 hours to afford 448 g (82% yield) of the desired product as an off-white solid. Purity by HPLC was 97.9%. 1H NMR (DMSO-d6) δ 8.68 (d, 1H), 8.05 (d, 1H), 8.01 (s, 1H) 7.86 (m, 1H), 7.59 (s, 1H), 7.57 (s, 1H) and 4.09 (s, 3H).
Preparation of 5-(3-chlorophenyl)-3-hydroxypyridine-2-carboxylic acid (3): A 20 L reactor equipped with a mechanical stirrer, condenser, thermometer, nitrogen inlet and 25% aqueous NaOH trap was charged 5-(3-chlorophenyl)-3-methoxy-2-cyanopyridine, 2, (440.6 g, 1.8 mol) and 37% aqueous solution of HCl (5302 g). While being agitated, the reaction solution was heated to 102° C. for 24 hours. Additional 37% aqueous HCl (2653 g) was added followed by agitation for 18 hours at 104° C. The reaction contents was then cooled to 5° C., charged with water (4410 g) and then agitated at 0° C. for 16 hours. The resulting precipitated product was isolated by filtration and washed with water until the filtrate had a pH of 6 (about 8,000 L of water). The filter cake was pulled dry under reduced pressure for 2 hours. The cake was then transferred back into the reactor and triturated in THF (1958 g, 2201 mL) at ambient temperature for 2 hours. The solid product was then isolated by filtration and washed with THF (778 g, 875 mL) and dried under reduced pressure at 5° C. for 48 hours to afford 385 g (89% yield) of the desired product as an off-white solid. HPLC purity was 96.2%. 1H NMR (DMSO-d6) δ 8.52 (d, 1H), 7.99 (d, 1H), 7.95 (s, 1H) 7.81 (t, 1H), 7.57 (s, 1H), and 7.55 (s, 1H).
Preparation of methyl {[5-(3-chlorophenyl)-3-hydroxypyridin-2-yl]amino}acetate (4): A 20 L reactor equipped with a mechanical stirrer, condenser, thermometer and nitrogen inlet was charged with 5-(3-chlorophenyl)-3-hydroxypyridine-2-carboxylic acid, 3, (380 g, 1.52 mol) and diisopropylethylamine (DIPEA) (295 g, 2.28 mol). With agitation, the solution was cooled to 3° C. and charged with trimethylacetyl chloride (275.7 g, 2.29 mol) while maintaining a temperature of less than 11° C., The mixture was then agitated at ambient temperature for 2 hours. The mixture was then cooled to 10° C. and charged with a slurry of glycine methyl ester HCl (573.3 g, 4. 57 mol) and THF (1689 g, 1900 mL), then charged with DIPEA (590.2 g, 4.57 mol) and agitated at ambient temperature for 16 hours. The mixture was then charged with EtOH (1500 g, 1900 mL) and concentrated under reduced pressure to a reaction volume of about 5.8 L. The EtOH addition and concentration was repeated twice more. Water (3800 g) was then added and the mixture was agitated for 16 hours at ambient temperature. The resulting solid product was isolated by filtration and washed with a mixture of EtOH (300 g, 380 mL) and water (380 g), followed by water (3800 g), dried under reduced pressure for 18 hours at 50° C. to afforded 443 g (91% yield) of the desired product as an off-white solid. Purity by HPLC was 98.9%. 1H NMR (DMSO-d6) δ 12.3 (s, 1H), 9.52 (t, 1H), 8.56 (d, 1H), 7.93 (s, 1H), 7.80 (q, 2H), 7.55 (t, 2H), 4.12 (d, 2H), and 3.69 (s, 3H).
Scheme II herein below outlines and Example 2 describes a non-limiting example of the disclosed process for preparing a prolyl hydroxylase inhibitor from an ester prodrug.
EXAMPLE 3{[5-(3-Chlorophenyl)-3-hydroxypyridin-2-yl]amino}acetic acid (5)
Preparation of {[5-(3 -chlorophenyl)-3-hydroxypyridin-2-yl]amino}acetic acid (5): To a 50 mL flask is charged methyl {[5-(3-chlorophenyl)-3-hydroxypyridin-2-yl]amino}-acetate, 4, (0.45 g, 1.4 mmol), tetrahydrofuran (4.5 mL) and 1 M NaOH (4.5 mL, 4.5 mmol). The mixture was stirred for 2 hours at room temperature after which it was determined by TLC analysis using hexane/ethyl acetate (6:3) as the mobile phase and UV 435 nm to visualize the reaction components that the reaction was complete. The reaction solution was adjusted to pH 1 with concentrated HCl and the solution was heated at 35° C. under vacuum until all of the tetrahydrofuran had been removed. A slurry forms as the solution is concentrated. With efficient stirring the pH is adjusted to ˜2 with the slow addition of 1 M NaOH. The solid which forms was collected by filtration, washed with water, followed by hexane, then dried under vacuum to afford 0.38 g (88% yield) of the desired product as a white solid. 1H NMR (DMSO-d6) δ 12.84 (s, 1H), 12.39 (s, 1H), 9.39 (t, 1H), 8.56 (d, 1H), 7.94 (s, 1H), 7.81 (m, 2H), 7.55 (q, 2H), and 4.02 (d, 2H).
The formulator can readily scale up the above disclosed synthesis. Disclosed herein below is a synthesis wherein the disclosed process is scaled up for commercial use.
EXAMPLE 4{[5-(3-Chlorophenyl)-3-hydroxypyridin-2-yl]amino}acetic acid (5)
Preparation of {[5-(3-chlorophenyl)-3-hydroxypyridin-2-yl]amino}acetic acid (5): To a 20 L reactor equipped with a mechanical stirrer, condenser, thermometer and nitrogen inlet was charged methyl {[5-(3-chlorophenyl)-3-hydroxypyridin-2-yl]amino}-acetate, 4, (440 g, 1.42 mol), tetrahydrofuran (3912 g, 4400 mL) and 1 M NaOH (4400 mL). The mixture was stirred for 2 hours at room temperature after which it was determined by TLC analysis using hexane/ethyl acetate (6:3) as the mobile phase and UV 435 nm to visualize the reaction components that the reaction was complete. The reaction solution was acidified to a pH of 2 with slow addition of 2M HCl (2359 g). The resulting mixture was concentrated under reduced pressure to a volume of about 7.5 L. Ware (2210 g) was added and the solution cooled to ambient temperature and agitated for 18 hours. The solid product was isolated by filtration and washed with water (6 L). the crude product was transferred back into the reactor and triturated with 2215 g o deionized water at 70° C. for 16 hours. The mixture was cooled to ambient temperature, The solid product was isolated by filtration and washed with water (500 mL) and dried under reduced pressure at 70° C. for 20 hours to afford 368 g (87% yield) of the desired product as an off-white solid. Purity by HPLC was 99.3%. 1H NMR (DMSO-d6) δ 12.84 (s, 1H), 12.39 (s, 1H), 9.39 (t, 1H), 8.56 (d, 1H), 7.94 (s, 1H), 7.81 (m, 2H), 7.55 (q, 2H), and 4.02 (d, 2H).
Scheme III herein below outlines and Example 3 describes a non-limiting example of the disclosed process for preparing a prolyl hydroxylase amide prodrug.
EXAMPLE 55-(3-Chlorophenyl)-N-(2-amino-2-oxoethyl)-3-hydroxylpyridin-2-yl amide
Preparation of 5-(3-chlorophenyl)-N-(2-amino-2-oxoethyl)-3-hydroxylpyridin-2-yl amide (6): To a solution of 5-(3-chlorophenyl)-3-hydroxypyridine-2-carboxylic acid, 3, (749 mg, 3 mmol) in DMF (20 mL) at room temperature under N2 is added 1-(3-dimethyl-aminopropyl)-3-ethylcarbodiimide (EDCI) (0.925 g, 5.97 mmol) and 1-hydroxybenzo-triazole (HOBt) (0.806 g, 5.97 mmol). The resulting solution is stirred for 15 minutes then 2-aminoacetamide hydrochloride (0.66 g, 5.97 mmol) and diisopropylethylamine (1.56 ml, 8.96 mmol) are added. The reaction is monitored by TLC and when the reaction is complete the reaction mixture is concentrated under reduced pressure and H2O added. The product can be isolated by normal work-up: The following data have been reported for compound (6). 1H NMR (250 MHz, DMSO-d6) δ ppm 12.46 (1H, s), 9.17 (1H, t, J=5.9 Hz), 8.55 (1H, d, J=2.0 Hz), 7.93 (1H, d, J=0.9 Hz), 7.75-7.84 (2H, m), 7.49-7.60 (3H, m), 7.18 (1H, s), 3.91 (2H, d, J=5.9 Hz). HPLC-MS: m/z 306 [M+H]+.
Scheme IV herein below depicts a non-limiting example the hydrolysis of an amide pro-drug to a prolyl hydroxylase inhibitor after removal of a R10 protecting group
Beuck S, Schänzer W, Thevis M. Hypoxia-inducible factor stabilizers and other small-molecule erythropoiesis-stimulating agents in current and preventive doping analysis. Drug Test Anal. 2012 Nov;4(11):830-45. doi: 10.1002/dta.390. Epub 2012 Feb 24. Review. PubMed PMID: 22362605.
Solid forms of 2-(5-(3-fluorophenyl)-3-hydroxypicolinamido)acetic acid, compositions, and uses thereof
clip
Oct 6, 2015
Akebia Reaches Agreement with FDA and EMA on Vadadustat Global Phase 3 Program
Plans to Initiate Phase 3 PRO2TECT™ Clinical Program by Year-End
CAMBRIDGE, Mass.–(BUSINESS WIRE)– Akebia Therapeutics, Inc. (NASDAQ: AKBA), a biopharmaceutical company focused on delivering innovative therapies to patients with kidney disease through the biology of hypoxia inducible factor (HIF), today announced the successful completion of the End-of-Phase 2 Meeting process with the United States Food and Drug Administration (FDA) and the Scientific Advice Process with the European Medicines Agency (EMA) for its lead product, vadadustat (formerly AKB-6548), for patients with anemia related to non-dialysis dependent chronic kidney disease (NDD-CKD). The company has reached agreement with both the FDA and EMA regarding key elements of the Phase 3 program, known as the PRO2TECT™ program, and expects to launch the program later this year.
The PRO2TECT™program includes two separate studies and will collectively enroll approximately 3,100 NDD-CKD patients across 500 sites globally. The correction study will address anemia patients not currently being treated with recombinant erythropoiesis stimulating agents (rESAs). The conversion study includes patients currently receiving rESA who will be converted to either vadadustat or the active control with the goal of maintaining their baseline hemoglobin levels. Both studies will include a 1:1 randomization and an open label, active-control, non-inferiority design. Primary endpoints include an efficacy assessment of the hemoglobin response and an assessment of cardiovascular safety measured by major adverse cardiovascular events.
“Akebia’s Phase 3 program is designed to provide the medical community and regulators with a clear understanding of vadadustat’s potential benefit and safety advantages over rESAs, the current standard of care worldwide and, with a positive outcome, to establish vadadustat as the best-in-class treatment option for patients with renal anemia,” stated John P. Butler, President and Chief Executive Officer of Akebia. “We are pleased that the regulators are in agreement regarding the importance of an active-control trial as this design is the most clinically relevant and commercially valuable, and will allow us the quickest path to full enrollment. We are now moving rapidly to launch these studies and advance our goal of bringing forward new treatment options for patients suffering from renal anemia.”
“This Phase 3 program builds on the positive data from our Phase 2 program in NDD-CKD patients which demonstrated that once-daily vadadustat can control and maintain hemoglobin levels in a clinically relevant range while minimizing fluctuations in hemoglobin levels that are associated with increased cardiovascular safety risks,” stated Brad Maroni, M.D., Chief Medical Officer at Akebia. “These two Phase 3 event-driven studies are designed to establish the safety and efficacy of vadadustat in the setting of contemporary clinical practice patterns, and support regulatory approvals globally.”
In addition, Akebia discussed with the FDA and EMA a parallel Phase 3 program, known as the INNO2VATE™ program, for vadadustat in patients with anemia related to chronic kidney disease who are undergoing dialysis (DD-CKD). Akebia expects to formalize its Phase 3 program in DD-CKD patients after presenting the results from its recently completed Phase 2 study to both regulatory agencies.
About Vadadustat (Formerly AKB-6548)
Vadadustat is an oral therapy currently in development for the treatment of anemia related to chronic kidney disease (CKD). Vadadustat is designed to stabilize HIF, a transcription factor that regulates the expression of genes involved with red blood cell (RBC) production in response to changes in oxygen levels, by inhibiting the hypoxia-inducible factor prolyl hydroxylase (HIF-PH) enzyme. Vadadustat exploits the same mechanism of action used by the body to naturally adapt to lower oxygen availability associated with a moderate increase in altitude. At higher altitudes, the body responds to lower oxygen availability with increased production of HIF, which coordinates the interdependent processes of iron mobilization and erythropoietin (EPO) production to increase RBC production and, ultimately, improve oxygen delivery.
As a HIF stabilizer with best-in-class potential, vadadustat raises hemoglobin levels predictably and sustainably, with a dosing regimen that allows for a gradual and controlled titration. Vadadustat has been shown to improve iron mobilization, potentially eliminating the need for intravenous iron administration and reducing the overall need for iron supplementation.
About Anemia Related to CKD
Approximately 30 million people in the United States have CKD, with an estimated 1.8 million of these patients suffering from anemia. Anemia results from the body’s inability to coordinate RBC production in response to lower oxygen levels due to the progressive loss of kidney function, which occurs in patients with CKD. Left untreated, anemia significantly accelerates patients’ overall deterioration of health with increased morbidity and mortality. Renal anemia is currently treated with injectable rESAs, which are associated with inconsistent hemoglobin responses and well-documented safety risks.
About Akebia Therapeutics
Akebia Therapeutics, Inc. is a biopharmaceutical company headquartered in Cambridge, Massachusetts, focused on delivering innovative therapies to patients with kidney disease through HIF biology. The company has completed Phase 2 development of its lead product candidate, vadadustat, an oral therapy for the treatment of anemia related to CKD in both non-dialysis and dialysis patients.
clip
Akebia Announces Positive Top-Line Results from its Phase 2 Study of Vadadustat in Dialysis Patients with Anemia Related to Chronic Kidney Disease
-Treatment with Vadadustat Successfully Maintained Mean Hemoglobin Levels Following Conversion from rESA Therapy-
-Vadadustat Demonstrated a Favorable Safety Profile with Once Daily and Three Times per Week Dosing-
CAMBRIDGE, Mass.–(BUSINESS WIRE)–Akebia Therapeutics, Inc. (NASDAQ:AKBA), a biopharmaceutical company focused on delivering innovative therapies to patients with kidney disease through the biology of hypoxia inducible factor (HIF), today announced positive top-line results from its Phase 2 study of vadadustat (formerly AKB-6548) in dialysis patients with anemia related to chronic kidney disease (CKD). The study achieved its primary objective, indicating that vadadustat maintained stable hemoglobin (HGB) levels throughout the 16-week treatment period following conversion from recombinant erythropoiesis-stimulating agent (rESA) therapy. Vadadustat demonstrated a favorable safety profile with no drug-related serious adverse events and no deaths. The results highlight the potential of vadadustat, dosed either once daily or three times per week, to safely and predictably manage and sustain HGB levels in CKD patients undergoing dialysis.
“This study was a clear success, demonstrating the potential of vadadustat to effectively and safely treat anemia in dialysis patients switching from injectable rESA therapy”
The open-label, multi-center, 94 patient study was designed to evaluate the ability of vadadustat to maintain hemoglobin levels in patients undergoing hemodialysis who were previously being treated with rESAs. Patients were assigned to one of three dose cohorts: once daily vadadustat at a starting dose of 300mg, once daily vadadustat at a starting dose of 450mg, or vadadustat three times per week in conjunction with the patient’s hemodialysis schedule at a starting dose of 450mg. The study achieved its primary endpoints of maintaining stable hemoglobin levels over 16 weeks of treatment in all three cohorts of patients converting from rESAs to vadadustat.
Vadadustat was well tolerated among patients in all three dose cohorts. Treatment-emergent adverse events (TEAEs) with vadadustat were balanced across the cohorts. Serious adverse events (SAEs) were reported in 13 subjects (13.8%), well within the expected range for this patient population. There were no drug-related SAEs and no deaths reported in the study.
“This study was a clear success, demonstrating the potential of vadadustat to effectively and safely treat anemia in dialysis patients switching from injectable rESA therapy,” said Brad Maroni, M.D., Chief Medical Officer at Akebia. “We are impressed with the consistency in hemoglobin levels across the duration of the study, which highlights the ability of vadadustat to control and maintain hemoglobin levels in this patient population. Furthermore, the results indicate that daily and three times per week dosing regimens are both viable options for patients on dialysis.”
John P. Butler, President and Chief Executive Officer of Akebia, stated, “These results further confirm vadadustat as a potential best-in-class anemia treatment for CKD patients, and reinforce our confidence in this product candidate as we advance toward our Phase 3 program. Adding these results to the 12 other clinical studies we have completed, we are confident in the potential for vadadustat to treat anemia in a broad array of patients with CKD. We are pleased to have successfully completed this stage of our drug development and look forward to initiating Phase 3 studies.”
Complete efficacy and safety data from this Phase 2 study will be presented at an upcoming medical meeting.
About the Phase 2 Study Design of Vadadustat in Dialysis Patients with Anemia Related to CKD
The Phase 2 multi-center, open-label study evaluated 94 patients over 16 weeks of treatment, at 20 dialysis centers in the United States, including an assessment of HGB response to the starting dose of vadadustat during the first 8 weeks, followed by an assessment of HGB response to algorithm-guided dose adjustments of vadadustat during the subsequent 8 weeks of treatment. The study enrolled three cohorts, each consisting of approximately 30 CKD patients with anemia undergoing dialysis who were switched from injectable rESA therapy to vadadustat. Patients in the first two cohorts received once daily doses of vadadustat, while patients in the third cohort received vadadustat three times per week in conjunction with their hemodialysis schedule.
Jump up^Gupta N, Wish JB. Hypoxia-Inducible Factor Prolyl Hydroxylase Inhibitors: A Potential New Treatment for Anemia in Patients With CKD. Am J Kidney Dis. 2017 Jun;69(6):815-826. . doi:10.1053/j.ajkd.2016.12.011. PMID28242135. Missing or empty |title= (help)
Jump up^Martin ER, Smith MT, Maroni BJ, Zuraw QC, deGoma EM. Clinical Trial of Vadadustat in Patients with Anemia Secondary to Stage 3 or 4 Chronic Kidney Disease. Am J Nephrol. 2017;45(5):380-388. . doi:10.1159/000464476. PMID28343225. Missing or empty |title= (help)
Vadadustat (Vafseo). Vadadustat (28) is a hypoxia inducible factor prolyl hydroxylase (HIF-PH) inhibitor developed by Akebia Therapeutics. It was approved by the European Commission in April 2023, and recently also by the USFDA, for the treatment of symptomatic anemia associated with chronic kidney disease in adults receiving chronic maintenance dialysis. Vadadustat acts by inhibiting HIFPH, 214 which results in increases of endogenous erythropoietin production, red blood cell synthesis, and iron mobilization. 215 While a number of syntheses of vadadustat (28) have been published in previous patents 216−228 and a journal article, 229 Akebia Therapeutics has published two patents regarding the large-scale preparation of vadadustat (Scheme 52). 218,226 The key intermediate nitrile 28.3 could be accessed in two steps: the neat SNAr reaction between commercially available 2,3,5trichloropyridine (28.1) and 4-DMAP to generate pyridinium salt 28.2, followed by a second SNAr reaction of 28.2 with NaCN. The Suzuki coupling between 28.3 and 3-chlorophenyl boronic acid (28.4) gave the biaryl 28.5, and the subsequent SNAr reaction of 28.5 with NaOMe replaced the 3-chloro substitution on the pyridine ring with a methoxy group, generating intermediate 28.6. Global acidic hydrolysis of both methyl ether and nitrile group in 28.6 gave the 3 hydroxypicolinic acid 28.7. Treatment of 28.7 with DIPEA and excess pivaloyl chloride (PivCl) resulted in the formation of mixed anhydride 28.8 with concomitant acylation of the 3 hydroxy group. Without isolation of 28.8, glycine methyl ester hydrochloride (28.9) was then charged with additional DIPEA to generate the corresponding amide 28.10. The residual amount (∼0.5%) of 28.7 in 28.10 was hard to remove, but this impurity could be effectively rejected with an extra amount of DIPEA during workup and solvent switch. Finally, the Opivaloyl group and methyl ester were both removed via basic hydrolysis, giving vadadustat (28) in about 90% yield from 28.7.
REF
(215) Pergola, P. E.; Spinowitz, B. S.; Hartman, C. S.; Maroni, B. J.; Haase, V. H. Vadadustat, a novel oral HIF stabilizer, provides effective anemia treatment in nondialysis-dependent chronic kidney disease. Kidney Int. 2016, 90, 1115−1122. (216) Lanthier, C. M.; Gorin, B.; Oudenes, J.; Dixon, C. E.; Lu, A. Q.; Copp, J. D.; Janusz, J. M. Preparation of [(3-hydroxypyridine-2carbonyl)amino]alkanoic acids, esters and amides as prolyl hydroxylase inhibitors. US 20120309977, 2012. (217) Li, X.; Chen, J. Process for the preparation of vadadustat. CN105837502, 2016. (218) Gorin, B. I.; Lanthier, C. M.; Luong, A. B. C.; Copp, J. D.; Gonzalez, J. Process for preparing 2-[[5-(3-chlorophenyl)-3-hydroxypyridine-2-carbonyl]amino]acetic acid. WO 2019217550, 2019. (219) Kou, J.; Li, Y.; Xiao, Q.; Lin, B.; Sun, J.; Wang, Z.; Luo, Z.;Huang, F. Preparation method of vadadustat. CN 110903238, 2020. (220) Machida, K.; Yasukouchi, H.; Nishiyama, A. Method for producing vadadustat intermediate. WO 2020217733, 2020. (221) Xiao, Q.; Lin, B.; Kou, J.; Sun, J.; Qiu, X.; Wang, Z.; Luo, Z.;Huang, F. Preparation of vadadustat intermediate. CN 111848505,2020
(222) Xiao, Q.; Lin, B.; Wang, Z.; Kou, J.; Li, Y.; Sun, J.; Jin, L.; Luo, Z.; Huang, F. Preparation of vadadustat and intermediate thereof. CN 111205222, 2020. (223) Xiao, Q.; Lin, B.; Wang, Z.; Kou, J.; Luo, Z.; Huang, F.; Li, Y. Preparation of vadadustat and intermediate thereof. CN 111423367, 2020. (224) Xiao, Q.; Qiu, X.; Lin, B.; Kou, J.; Li, Y.; Sun, J.; Wang, Z.; Luo, Z.; Huang, F. Preparation of vadadustat. CN 111320577, 2020. (225) Xiao, Q.; Lin, B.; Wang, Z.; Kou, J.; Qiu, X.; Cai, X.; Li, Y.; Luo, Z.; Huang, F. Method for preparing vadadustat and intermediate thereof. WO 2021179540, 2021. (226) Jurkauskas, V.; Jung, Y. C.; Kwon, T.; Kannan, A.; Gondi, V. B. Manufacturing process for 3,5-dichloropicolinonitrile for synthesis of vadadustat. WO 2022006427, 2022. (227) Chen, Z.; Zheng, Y.; Zhang, L.; Yu, C.; Liu, L.; He, B. Preparation of a pyridine compound used for the preparation of vadadustat. CN 117843565, 2024. (228) Patel, K. R.; Thakrar, V. H.; Mehta, T. B.; Wagh, A. G.; Patel, J. A.; Patil, R. R.; Solanki, Y. U.; Ladumor, C. B. A process for the preparation of Vadadustat or salts thereof. WO 2024079708, 2024. (229) Lin, B. Y.; Kou, J. P.; Wu, S. M.; Cai, X. R.; Xiao, Q. B.; Li, Y. L.; Hu, J.; Li, J. B.; Wang, Z. Q. Development of a robust and scalable process for the large-scale preparation of Vadadustat. Org. Process Res. Dev. 2021, 25, 960−968.
.
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
RPL-554 is a mixed phosphodiesterase (PDE) III/IV inhibitor in phase II clinical development at Verona Pharma for the treatment of asthma, allergic rhinitis, chronic obstructive pulmonary disease (COPD) and inflammation.
RPL-554 is expected to have long duration of action and will be administered nasally thereby preventing gastrointestinal problems often resulting from orally administered PDE4 antiinflammatory drugs.
The company is now seeking licensing agreements or partnerships for the further development and commercialization of the drug.
RPL-554 (LS-193,855) is a drug candidate for respiratory diseases. It is an analog of trequinsin, and like trequinsin, is a dual inhibitor of the phosphodiesterase enzymes PDE-3 and PDE-4.[1] As of October 2015, inhaled RPL-554 delivered via a nebulizer was in development for COPD and had been studied in asthma.[2]
RPL554 was part of a family of compounds invented by Sir David Jack, former head of R&D for GlaxoSmithKline, and Alexander Oxford, a medicinal chemist; the patents on their work were assigned to Vernalis plc.[4][5]:19-20
In 2005, Rhinopharma Ltd, acquired the rights to the intellectual property from Vernalis.[5]:19-20 Rhinopharma was a startup founded in Vancouver, Canada in 2004 by Michael Walker, Clive Page, and David Saint, to discover and develop drugs for chronic respiratory diseases,[5]:16 and intended to develop RPL-554, delivered with an inhaler, first for allergic rhinitis, then asthma, then forCOPD.[5]:16-17 RPL554 was synthesized at Tocris, a contract research organization, under the supervision of Oxford, and was studied in collaboration with Page’s lab at King’s College, London.[1] In 2006 Rhinopharma recapitalized and was renamed Verona Pharma plc.[5]
This was first seen in April 2015 when it was published as a France national. Verona Pharma (formerly Rhinopharma), under license from Kings College via Vernalis, is developing the long-acting bronchodilator, RPL-554 the lead in a series dual inhibitor of multidrug resistant protein-4 and PDE 3 and 4 inhibiting trequinsin analogs which included RPL-565, for treating inflammatory respiratory diseases, such as allergic rhinitis, asthma, and COPD.
RPL554
Verona Pharma’s lead drug, RPL554, is a “first-in-class” inhaled drug under development for chronic obstructive pulmonary disease (COPD), asthma and cystic fibrosis. The drug is an inhibitor of the phosphodiesterase 3 (PDE3) and phosphodiesterase 4 (PDE4) enzymes, two enzymes known to be of importance in the development and progression of immunological respiratory diseases. The drug has the potential to act as both a bronchodilator and an anti-inflammatory which would significantly differentiate it from existing drugs.
RPL554 was selected from a class of compounds co-invented by Sir David Jack, the former Director of Research at Glaxo who led the team that discovered many of the commercially successful drugs in the respiratory market.
Verona Pharma has successfully completed two double-blind placebo controlled randomised Phase 2b studies of RPL554: one in mild to moderate asthma and another in mild to moderate COPD. The drug was found to be well tolerated, free from drug-related adverse effects (especially cardiovascular and gastro-intestinal effects) and generated significant bronchodilation. Additionally, double-blind placebo controlled exploratory studies in healthy volunteers challenged with an inhaled irritant also generated consistent, clinically meaningful anti-inflammatory effects.
Verona Pharma is also carrying out exploratory studies to investigate the potential of RPL554 as a novel treatement for cystic fibrosis. In November 2014, the Company received a Venture and Innovation Award from the UK Cystic Fibrosis Trust to further such studies.
The competitive advantages of RPL554 include the following:
combining bronchodilator (PDE 3) and anti-inflammatory actions (PDE 4) in a single drug, something that is currently only achieved with a combination LABA and glucocorticosteroid inhaler,
unique in not using steroids or beta agonists, which have known side effects,
planned to be administered by nasal inhalation, thereby reducing the unwanted gastrointestinal side effects of many orally administered drugs.
History of Clinical Trials
Following completion in May 2008 of toxicological studies of RPL554, the Company commenced in February 2009 a Phase I/IIa clinical trial of the drug at the Centre for Human Drug Research (CHDR) at Leiden in the Netherlands. In September 2009, the Company announced that it had successfully completed the trial, demonstrating that RPL554 has a good safety profile and has beneficial effects in terms of bronchodilation and bronchoprotection in asthmatics and a reduction in the numbers of inflammatory cells in the nasal passages of allergic rhinitis patients.
In November 2010, the Company successfully completed a further trial that examined the safety and bronchodilator effectiveness of the drug administered at higher doses.
In August 2011, the Company demonstrated that bronchodilation is maintained over a period of 6 days with daily dosing of RPL554 in asthmatics.
In November 2011, the Company successfully demonstrated safety and bronchodilation of RPL554 in patients with mild to moderate forms of COPD.
In March 2013, the Company demonstrated positive airway anti-inflammatory activity with respect to COPD at a clinical trial carried out at the Medicines Evaluation Unit (MEU) in Manchester, UK.
Cyclization of 1-(3,4-dimethoxyphenethyl)barbituric acid in refluxing POCl3 produces the pyrimidoisoquinolinone , which is further condensed with 2,4,6-trimethylaniline in boiling isopropanol to afford the trimethylphenylimino derivative . Subsequent alkylation of with N-(2-bromoethyl)phthalimide in the presence of K2CO3 and KI, followed by hydrazinolysis of the resulting phthalimidoethyl compound yields the primary amine . This is finally converted into the title urea RPL 554 by reaction with sodium cyanate in aqueous HCl.
Example 1 : 9 Λ 0-Dimethoxy-2-(2.4-6-trimethy-phen yliminoY-3-(N-carbamoyl-2- aminoethylV3.4.6.7-tetrahydro-2H-pyrimido[6.1-a]isoquinolin-4-one
Sodium cyanate (6.0g, 0.092 mol) in water (100 ml) was added dropwise to a stirred solution of 9,10-Dimethoxy-2-(2,4,6-trimethylphenylimino)-3-(2-aminoethyl)-3,4,6,7- tetrahydro-2H-pyrimido[6,l-a]isoquinolin-4-one, prepared according to Preparation 4 above (20.0g, 0.046 mol) in water (600 ml) and IN ΗC1 (92 ml) at 80°C. After stirring for 2h at 80°C the mixture was cooled in an ice-bath and basified with 2N NaOH. The mixture was extracted with dichloromethane (3 x 200 ml) and the combined extract was dried (MgSO- ) and evaporated in vacuo. The resulting yellow foam was purified by column chromatography on silica gel eluting with CH2CI2 / MeOH (97:3) and triturated with ether to obtain the title compound as a yellow solid, 11.9g, 54%.
M.p.: 234-236°C m/z: C26H31N5O4 requires M=477 found (M+l) = 478
Preparation 1 : Synthesis of 2-Chloro-6.7-d-hydro-9.10-Dimethoxy-4H-pyrimido- [6,l-a]isoquinoHn-4-one (shown as (1) in Figure 1
A mixture of l-(3,4-dimethoxyphenyl) barbituric acid (70g, 0.24mol), prepared according to the method described in B. Lai et al. J.Med.Chem. 27 1470-1480 (1984), and phosphorus oxychloride (300ml, 3.22mol) was refluxed for 2.5h. The excess phosphorous oxychloride was removed by distillation (20mmHg) on wa ming. After cooling the residue was slurried in dioxan (100ml) and cautiously added to a vigorously stirred ice/water solution (11). Chloroform (11) was added and the resulting mixture was basified with 30% sodium hydroxide solution. The organic layer was separated and the aqueous phase further extracted with chloroform (2x750ml). The combined organic extracts were washed with water (1.51), dried over magnesium sulphate and concentrated in vacuo to leave a gummy material (90g). This was stirred in methanol for a few minutes, filtered and washed with methanol (200ml), diethyl ether (2x200ml) and dried in vacuo at 40°C to yield the title compound as a yellow/orange solid. 47g, 62%
2-Chloro-9,10-dimethoxy-6,7-dihydro-4H-pyrimido[6,l-a]isoquinolin-4-one, prepared according to Preparation 1, (38.5g, 0.13 mol) and 2,4,6-trimethylaniline (52.7g, 0.39 mol) in propan-2-ol (3 1) was stirred and heated at reflux, under nitrogen, for 24h. After cooling to room temperature, the solution was evaporated in vacuo and the residue was purified by column chromatography on silica gel, eluting with CΗ2CI2 /
MeOH, initially 98:2, changing to 96:4 once the product began to elute from the column. The title compound was obtained with a slight impurity, (just above the product on tic). Yield 34.6g, 67%.
A mixture of 9,10-Dimethoxy-2-(2,4,6-trimethylphenylimino)-3,4,6,7-tetrahydro-2H- pyrimido[6,l-a]isoquinolin-4-one (which was prepared according to Preparation 2) (60.0g, 0.153 mol), potassium carbonate (191g, 1.38 mol), sodium iodide (137g, 0.92 mol) and N-(2-bromoethyl)phthalimide (234g, 0.92 mol) in 2-butanone (1500 ml) was stirred and heated at reflux, under nitrogen, for 4 days. After cooling to room temperature the mixture was filtered and the filtrate was evaporated in vacuo. The residue was treated with methanol (1000 ml) and the solid filtered off, washed with methanol and recrystallised from ethyl acetate to obtain the title compound as a pale yellow solid in yield 40. Og, 46%. Evaporation of the mother liquor and column chromatography of the residue on silica gel (CΗ2C-2 / MeOH 95:5) provided further product 11.7g, 13.5%. Preparation 4: 9.10-Dimethoxy-2-(2A6-trimethylphenylimino)-3-(2-arninoethyO- 3.4.6.7-tetrahydro-2H-pyrimido[6.1-a]isoquino-in-4-one (shown as (4) in Figure 1)
A mixture of 9,10-Dimethoxy-2-(2,4,6-trimethylphenylimino)-3-(2-N- phthalimidoethyl)-3,4,6,7-tetrahydro-2H-pyrimido[6,l-a]isoquinolin-4-one (22. Og, 0.039 mol), prepared according to Preparation 3, and hydrazine hydrate (11.3g, 0.195 mol) in chloroform (300 ml) and ethanol (460 ml) was stined at room temperature, under nitrogen, for 18h. Further hydrazine hydrate (2.9g, 0.05 mol) was added and the mixture was stirred a further 4h. After cooling in ice / water, the solid was removed by filtration and the filtrate evaporated in vacuo. The residue was dissolved in dichloromethane and the insoluble material was removed by filtration. The fitrate was dried (MgSO-i) and evaporated in vacuo to afford the title compound as a yellow foam in yield 16.2g, 96%.
Novel crystalline acid addition salts forms of RPL-554 are claimed, wherein the salts, such as ethane- 1,2-disulfonic acid, ethanesulfonic acid, methanesulfonic acid, benzenesulfonic acid, p-toluenesulfonic acid, hydrochloric acid, hydrobromic acid, phosphoric acid or sulfuric acid. .
RPL554 (9, 10-dimethoxy-2-(2,4,6-trimethylphenylimino)-3-(/V-carbamoyl-2-aminoethyl)-3,4,6,7-tetrahydro-2H-pyrimido[6, l-a]isoquinolin-4-one) is a dual PDE3/PDE4 inhibitor and is described in WO 00/58308. As a combined PDE3/PDE4 inhibitor, RPL554 has both antiinflammatory and bronchodilatory activity and is useful in the treatment of respiratory disorders such as asthma and chronic obstructive pulmonary disease (COPD). The structure of RPL554 is shown below.
Owing to its applicability in the treatment of respiratory disorders, it is often preferable to administer RPL554 by inhalation. Franciosi et al. disclose a solution of RPL554 in a citrate-phosphate buffer at pH 3.2 (The Lancet: Respiratory Medicine 11/2013; l(9):714-27. DOI: 10.1016/S2213-2600(13)70187-5). The preparation of salts of RPL554 has not been described.
U.S. Pat. No. 6,794,391, 7,378,424, and 7,105,663, which are each incorporated herein by reference, discloses compound RPL-554 (N-{2-[(2iT)-2-(mesityiimino)-9,10- dimethoxy-4-oxo-6,7-dihydro-2H-pyrimido[6,l-a]-isoquinolin-3 4H)-yl]ethyl}urea).
It would be beneficial to provide a composition of a stable polymorph of RPL-554, that has advanrtages over less stable polymorphs or amorphous forms, including
Crystalline form of pyrimido[6,1-A] isoquinolin-4-one compound
References
Boswell-Smith V et al. The pharmacology of two novel long-acting phosphodiesterase 3/4 inhibitors, RPL554 [9,10-dimethoxy-2(2,4,6-trimethylphenylimino)-3-(n-carbamoyl-2-aminoethyl)-3,4,6,7-tetrahydro-2H-pyrimido[6,1-a]isoquinolin-4-one] and RPL565 [6,7-dihydro-2-(2,6-diisopropylphenoxy)-9,10-dimethoxy-4H-pyrimido[6,1-a]isoquinolin-4-one]. J Pharmacol Exp Ther. 2006 Aug;318(2):840-8. PMID 16682455
Turner MJ et al. The dual phosphodiesterase 3 and 4 inhibitor RPL554 stimulates CFTR and ciliary beating in primary cultures of bronchial epithelia. Am J Physiol Lung Cell Mol Physiol. 2016 Jan 1;310(1):L59-70. PMID 26545902
1: Calzetta L, Cazzola M, Page CP, Rogliani P, Facciolo F, Matera MG. Pharmacological characterization of the interaction between the dual phosphodiesterase (PDE) 3/4 inhibitor RPL554 and glycopyrronium on human isolated bronchi and small airways. Pulm Pharmacol Ther. 2015 Jun;32:15-23. doi: 10.1016/j.pupt.2015.03.007. Epub 2015 Apr 18. PubMed PMID: 25899618.
2: Franciosi LG, Diamant Z, Banner KH, Zuiker R, Morelli N, Kamerling IM, de Kam ML, Burggraaf J, Cohen AF, Cazzola M, Calzetta L, Singh D, Spina D, Walker MJ, Page CP. Efficacy and safety of RPL554, a dual PDE3 and PDE4 inhibitor, in healthy volunteers and in patients with asthma or chronic obstructive pulmonary disease: findings from four clinical trials. Lancet Respir Med. 2013 Nov;1(9):714-27. doi: 10.1016/S2213-2600(13)70187-5. Epub 2013 Oct 25. PubMed PMID: 24429275.
3: Wedzicha JA. Dual PDE 3/4 inhibition: a novel approach to airway disease? Lancet Respir Med. 2013 Nov;1(9):669-70. doi: 10.1016/S2213-2600(13)70211-X. Epub 2013 Oct 25. PubMed PMID: 24429260.
4: Calzetta L, Page CP, Spina D, Cazzola M, Rogliani P, Facciolo F, Matera MG. Effect of the mixed phosphodiesterase 3/4 inhibitor RPL554 on human isolated bronchial smooth muscle tone. J Pharmacol Exp Ther. 2013 Sep;346(3):414-23. doi: 10.1124/jpet.113.204644. Epub 2013 Jun 13. PubMed PMID: 23766543.
5: Gross N. The COPD pipeline XX. COPD. 2013 Feb;10(1):104-6. doi: 10.3109/15412555.2013.766103. PubMed PMID: 23413896.
6: Gross NJ. The COPD Pipeline XIV. COPD. 2012 Feb;9(1):81-3. doi: 10.3109/15412555.2012.646587. PubMed PMID: 22292600.
7: Boswell-Smith V, Spina D, Oxford AW, Comer MB, Seeds EA, Page CP. The pharmacology of two novel long-acting phosphodiesterase 3/4 inhibitors, RPL554 [9,10-dimethoxy-2(2,4,6-trimethylphenylimino)-3-(n-carbamoyl-2-aminoethyl)-3,4,6, 7-tetrahydro-2H-pyrimido[6,1-a]isoquinolin-4-one] and RPL565 [6,7-dihydro-2-(2,6-diisopropylphenoxy)-9,10-dimethoxy-4H-pyrimido[6,1-a]isoquino lin-4-one]. J Pharmacol Exp Ther. 2006 Aug;318(2):840-8. Epub 2006 May 8. PubMed PMID: 16682455.

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....














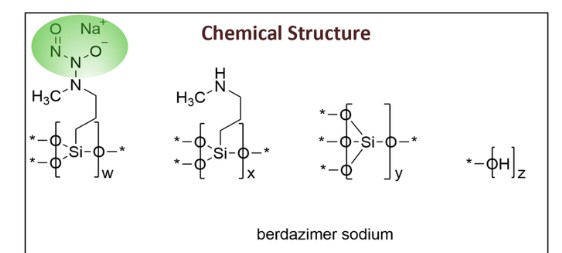


 References
References

(1)_(30204271666).jpg)
(1)_(29608654394).jpg)

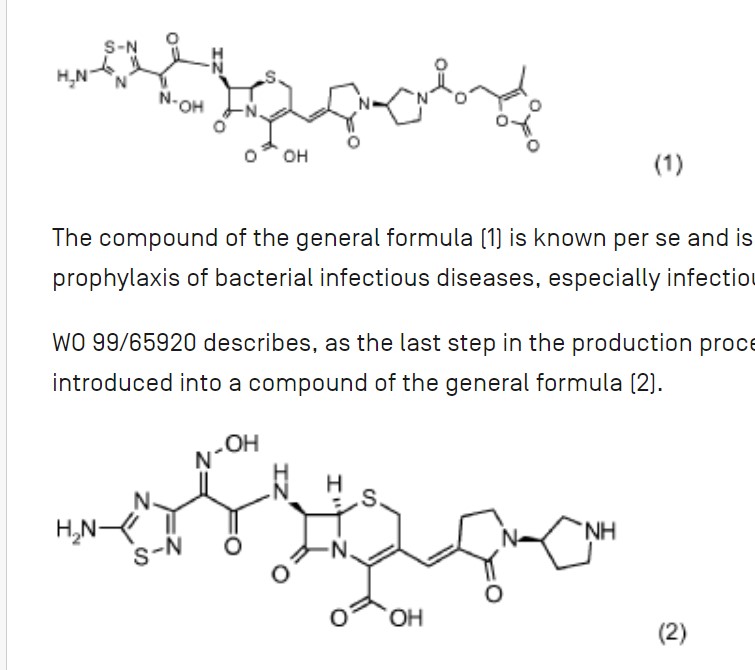





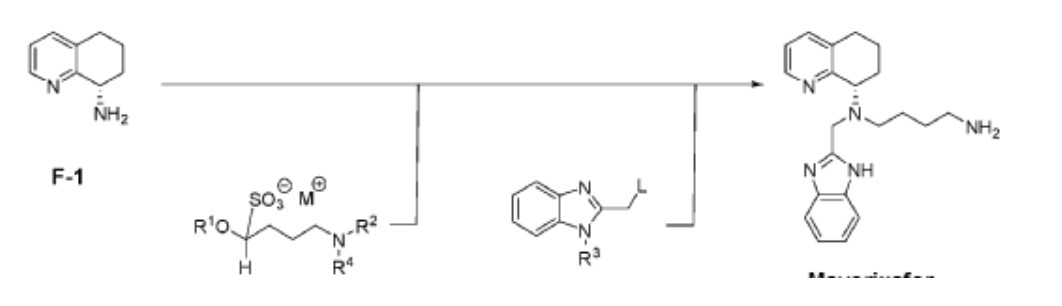

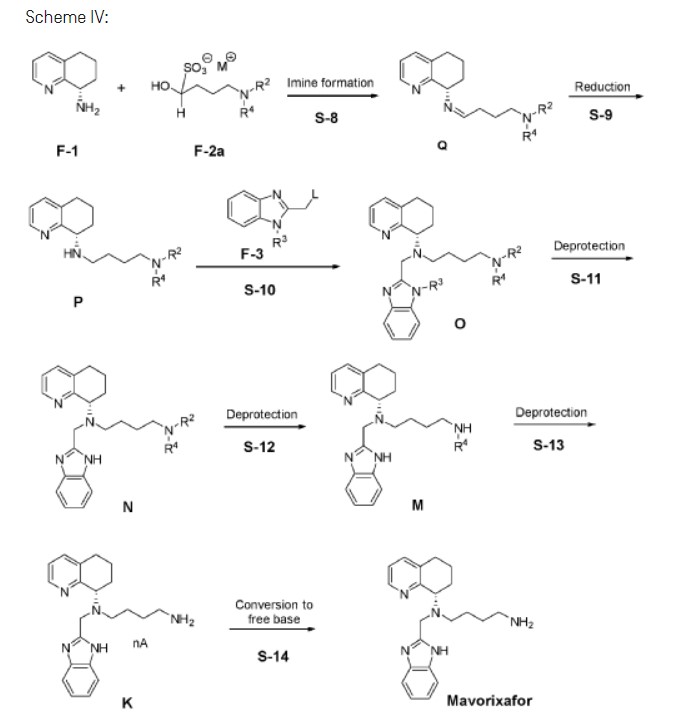

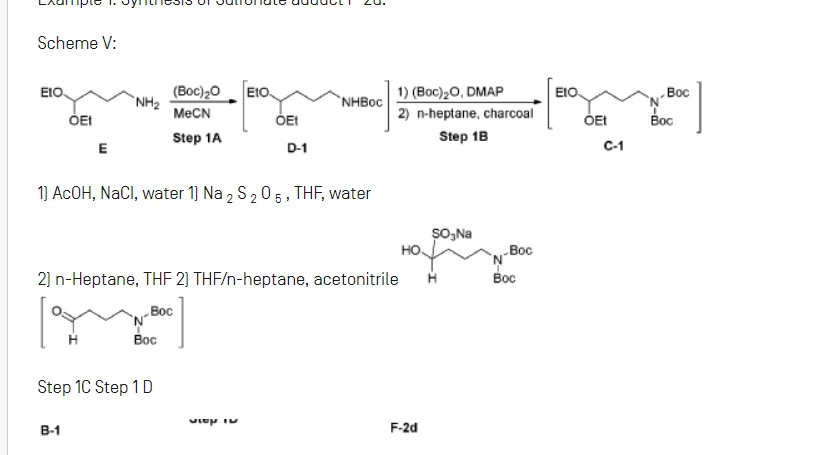

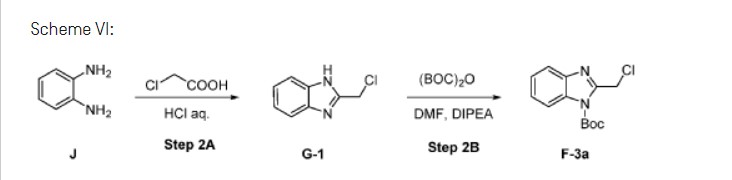

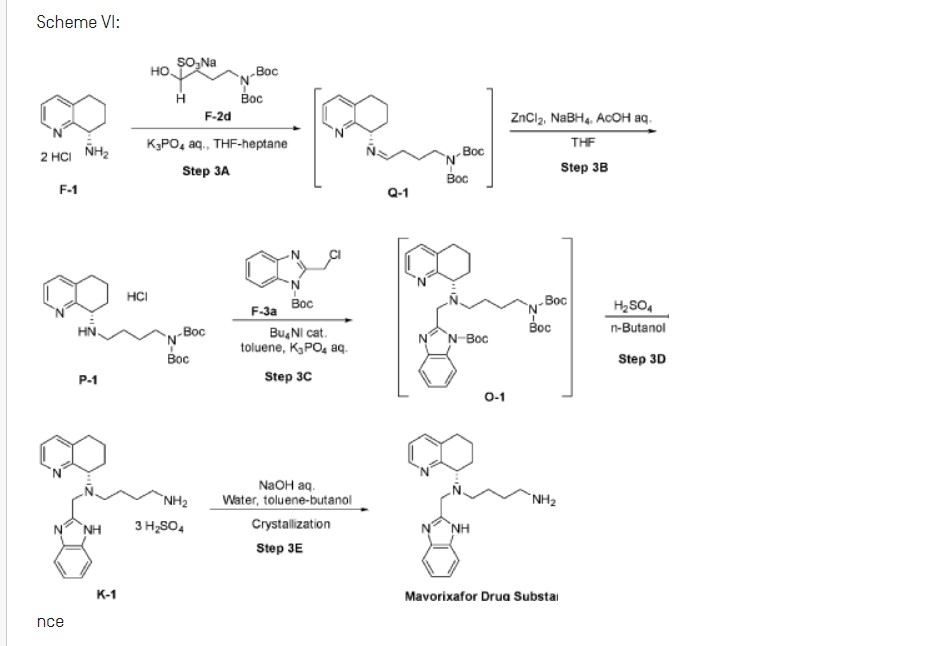







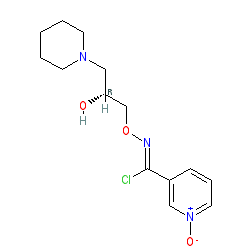
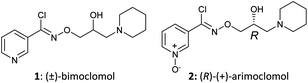




![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif)
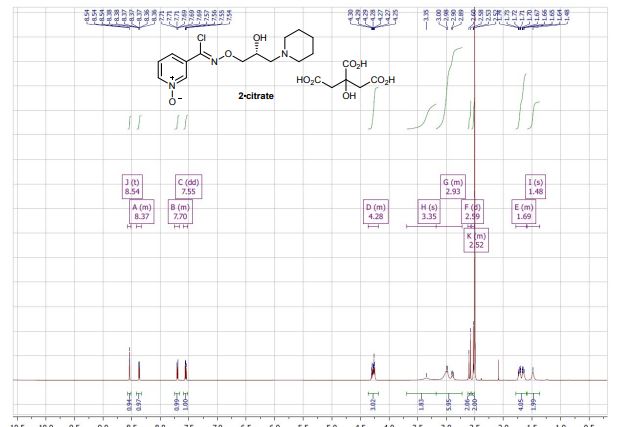





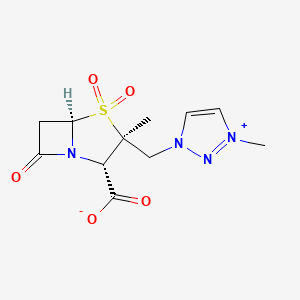













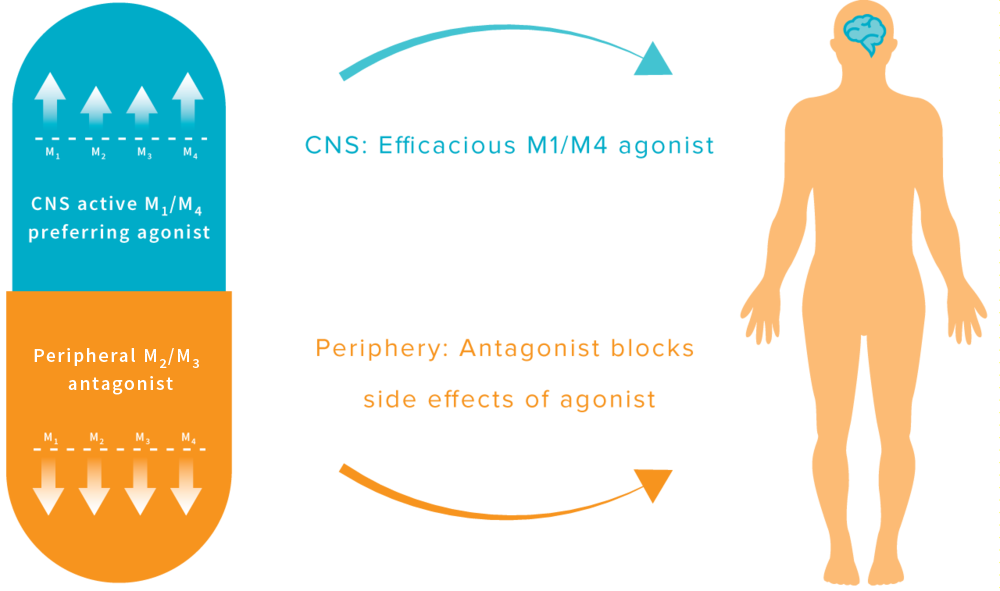






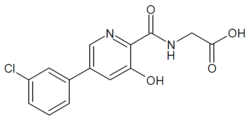
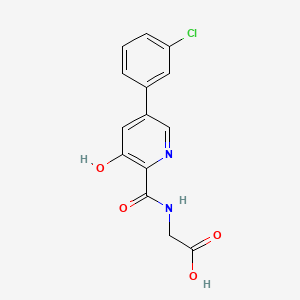



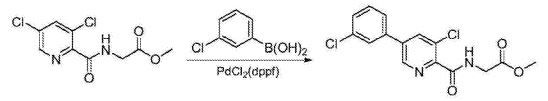
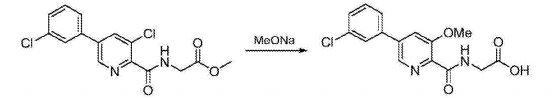
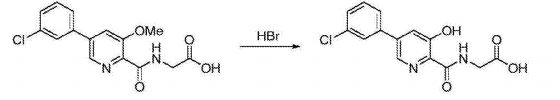


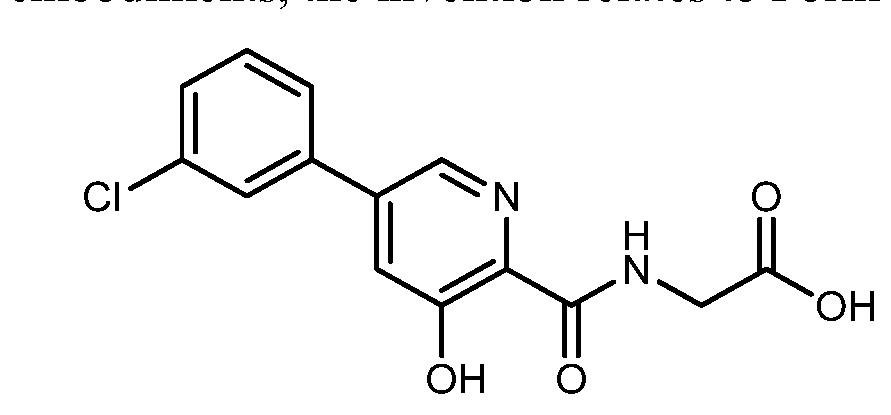
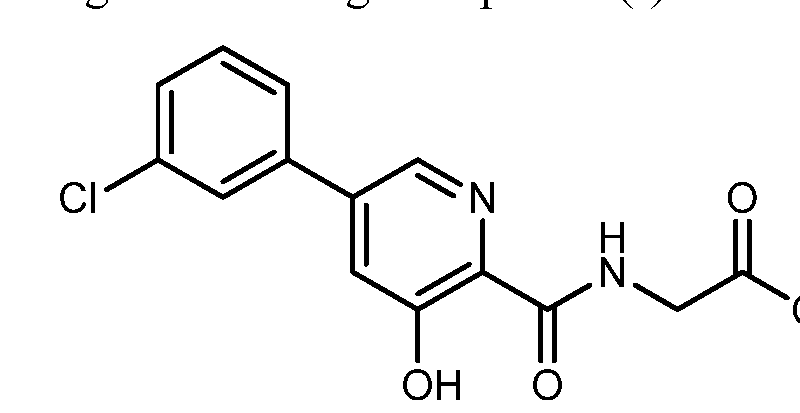







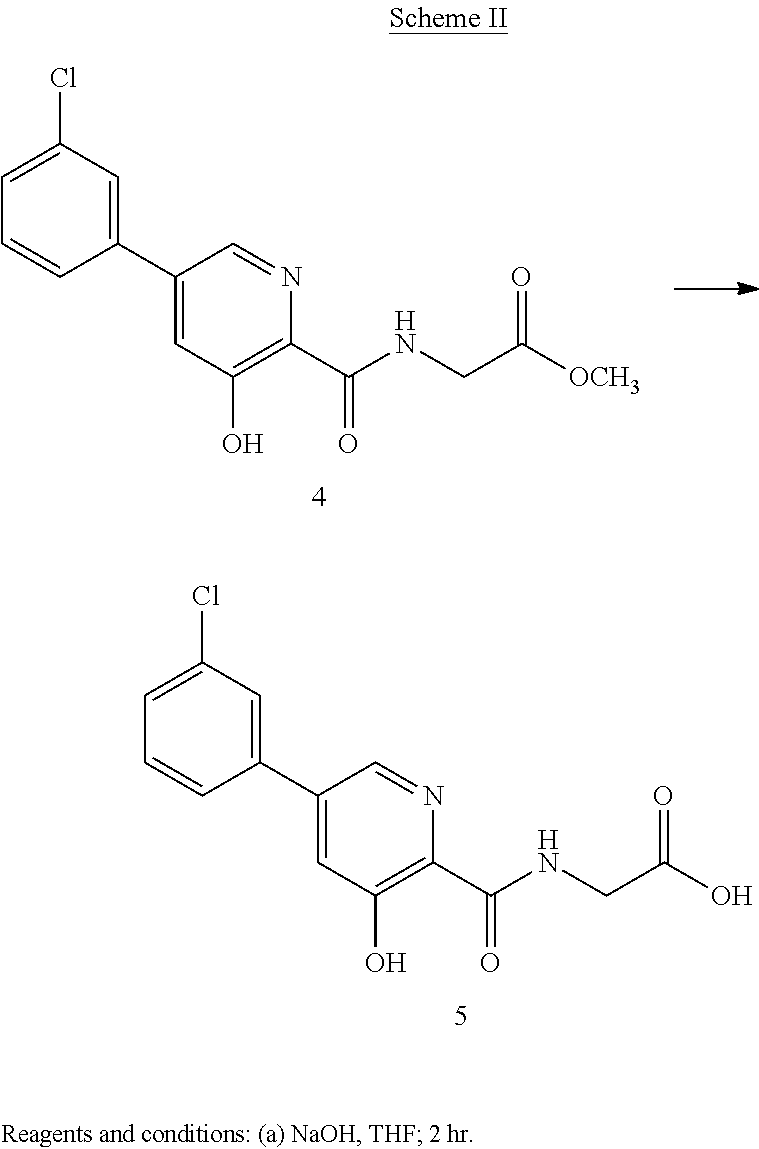
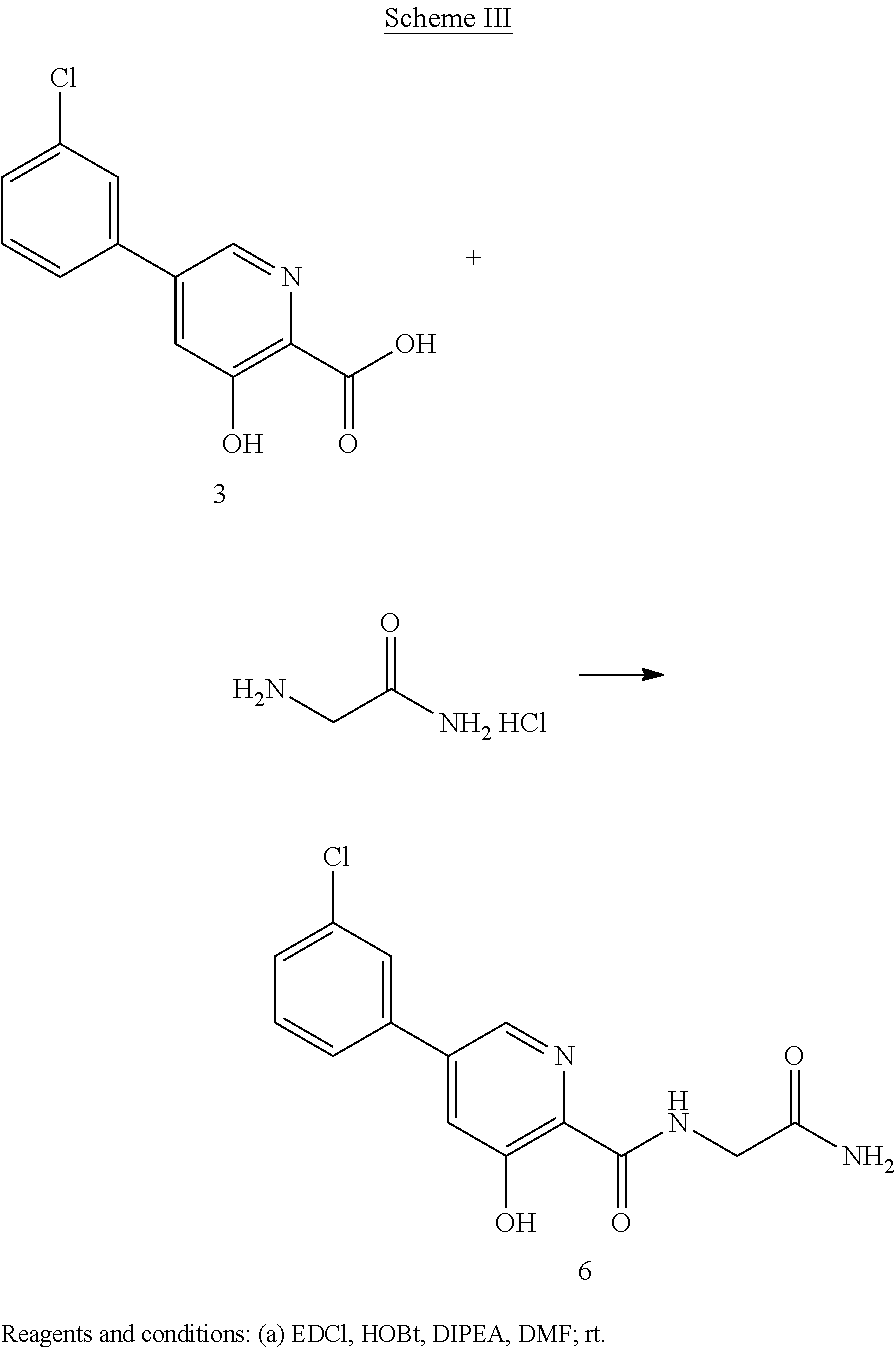




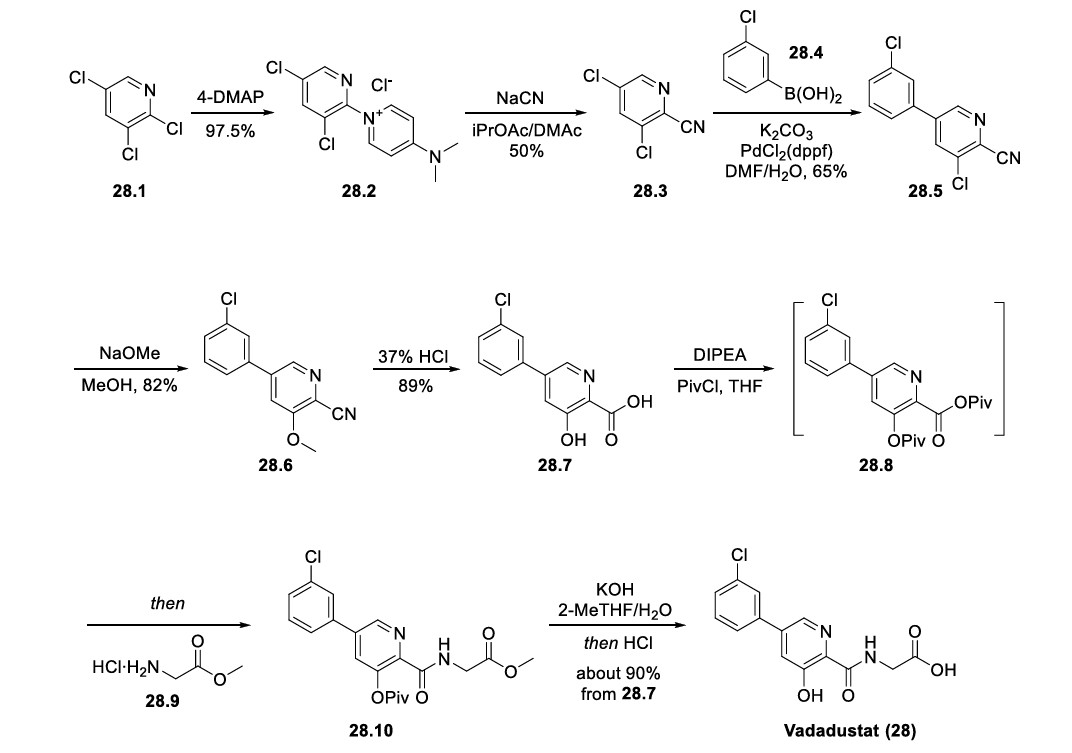





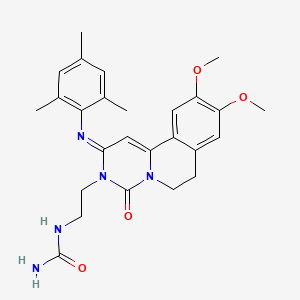
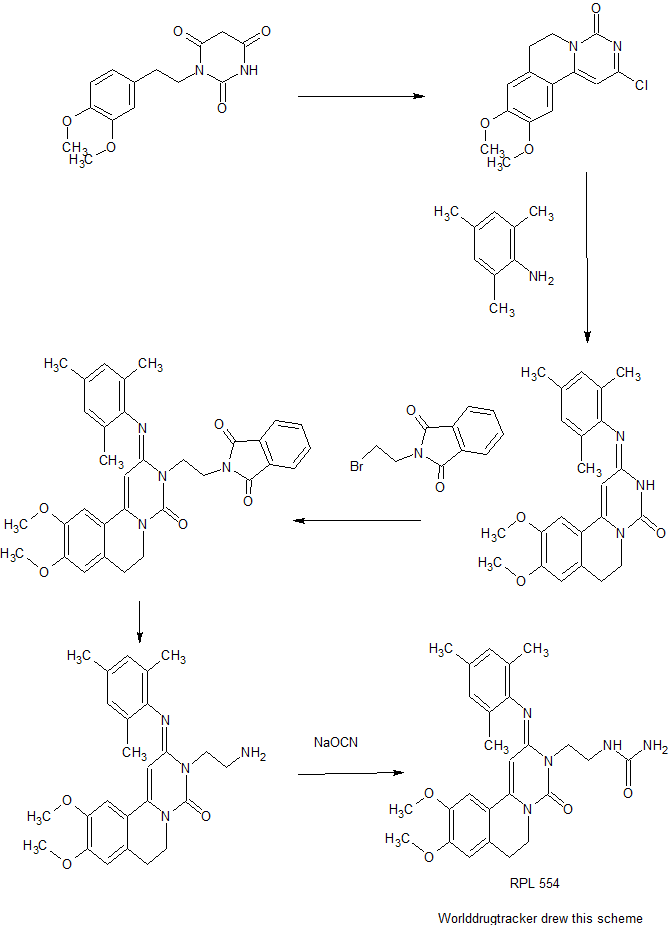






{kind=link}
{kind=link}
{kind=link}
{kind=link}
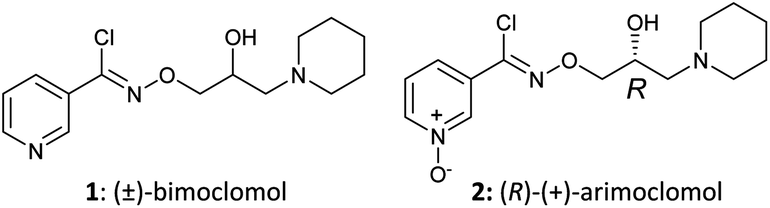{kind=link}
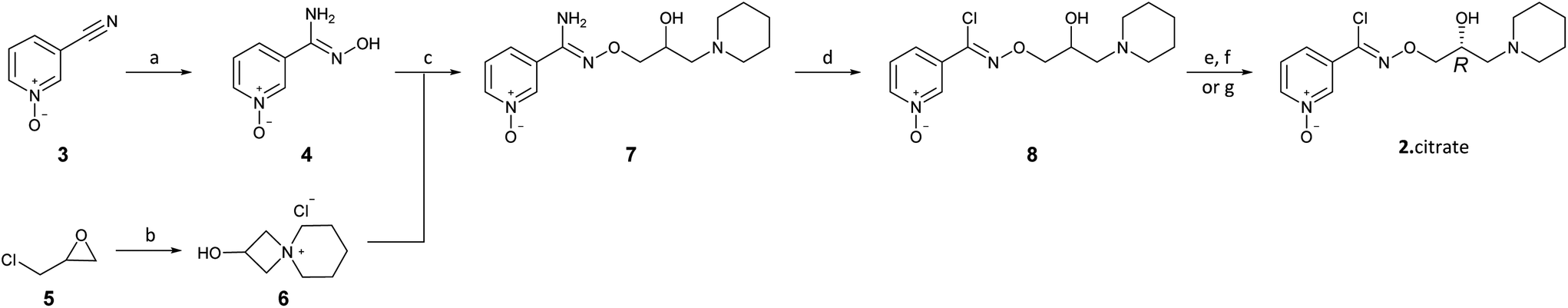{kind=link}
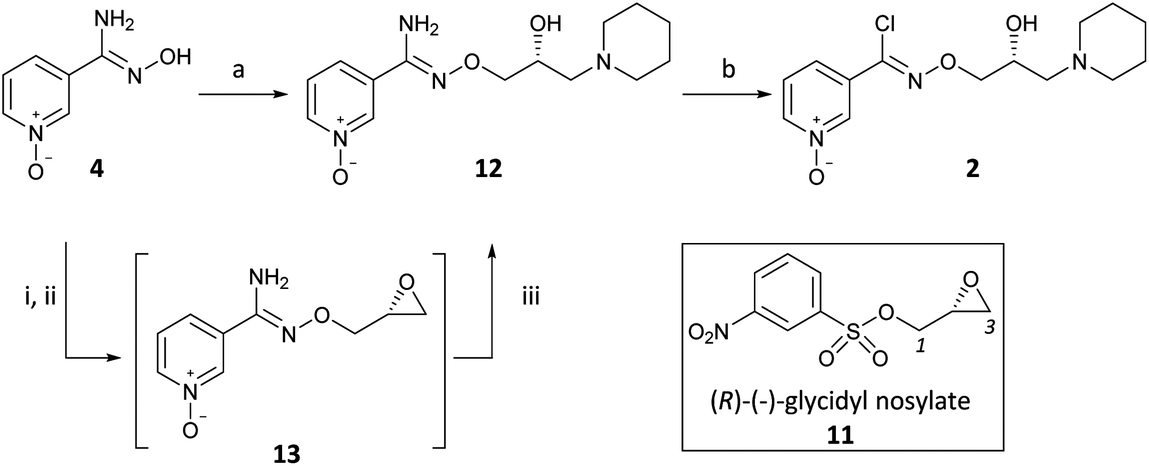{kind=link}
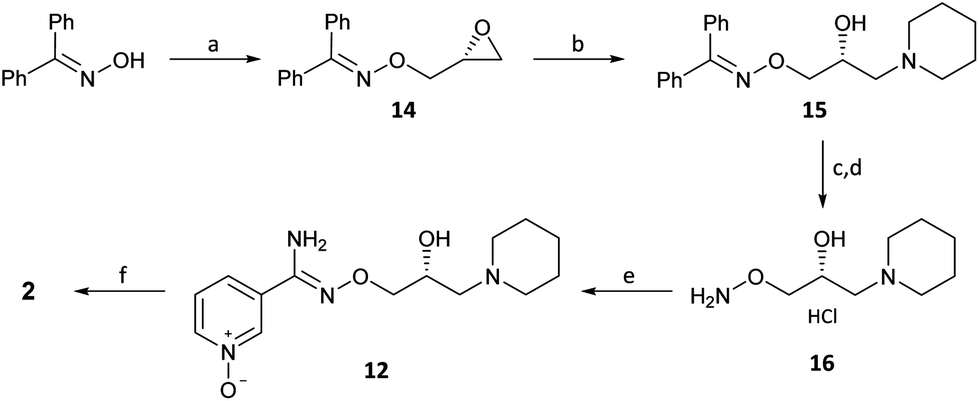{kind=link}
{kind=link}
{kind=link}