
Home » Posts tagged 'FDA 2014' (Page 2)
Tag Archives: FDA 2014
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on
ORGANIC SPECTROSCOPY INTERNATIONAL

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
FDA gives green light to Novartis acromegaly drug Pasireotide

Pasireotide, Signifor; SOM 320; HY-16381; 396091-73-9
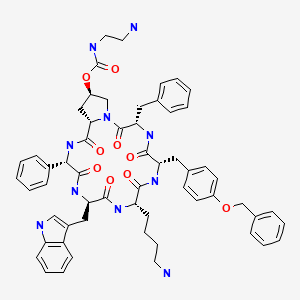
Cyclo[4(R)-[N-(2-aminoethyl)carbamoyloxy]-L-prolyl-L-phenyl-glycyl-D-tryptophyl-L-lysyl-(4-O-benzyl)-L-tyrosyl-L-phenylalanyl]bis(L-aspartic acid)
Regulators in the USA has approved a long-acting release of Novartis’ Signifor as a treatment for acromegaly.
The Food and Drug Administration has approved Signifor LAR (pasireotide) for the treatment of patients with acromegaly who have had an inadequate response to surgery or for whom the latter is not an option. The thumbs-up comes a month after the European Medicines Agency approved the drug, a next-generation somatostatin analogue administered intramuscularly once-monthly.
Read more at: http://www.pharmatimes.com/Article/14-12-16/FDA_gives_green_light_to_Novartis_acromegaly_drug.aspx#ixzz3M8Ibn14Q
clinical…..https://clinicaltrials.gov/search/intervention=Pasireotide+OR+SOM-230
Pasireotide (SOM230, trade name Signifor[1]) is an orphan drug approved in the U.S. and Europe for the treatment of Cushing’s disease in patients who fail or are ineligible for surgical therapy.[2][3] It was developed by Novartis. Pasireotide is a somatostatinanalog which has a 40-fold increased affinity to somatostatin receptor 5 than other somatostatin analogs.
The drug showed therapeutical potential in a recent study (PASPORT-CUSHINGS B2305) where 162 patients were treated with either 2x 600 µg or 2x 900 µg pasireotide s.c. daily.[4] The effectiveness of the treatment was checked by the UFC-value (urinary free cortisol) after six months of treatment. The mean reduction of UFC after six months was 47.9%, which also lead to amelioration of clinical symptoms such as blood pressure, cholesterol value, and weight loss.[5]
Pasireotide was approved by the EMEA in October 2009[6] and by the FDA in December 2012.[7]
At present, it is in phase III clinical trials at Novartis for the treatment of carcinoid tumors and symptoms that are not adequately controlled by somatostatin analogues (Sandostatin). Phase II clinical development is also under way at the company for the treatment of gastric dumping syndrome, metastatic carcinoid tumors, meningioma and pituitary adenoma and for the treatment of hepatocellular carcinoma in combination with everolimus. Early clinical trials are also ongoing for the treatment of patients with metastatic melanoma or Merkel cell carcinoma. A phase I clinical trial for the treatment of alcoholic cirrhosis has been completed. The company intends to file for approval in 2007 for these indications. Novartis and Thomas Jefferson University are conducting phase II clinical trials for the treatment of prostate cancer, alone or in combination with everolimus. The Mayo Clinic is conducting phase II clinical trials for the treatment of polycystic liver disease. Phase III clinical trials had been ongoing for the reduction of post-pancreatectomy fistula, leak, and abscess; however, in 2010 these trials were suspended. In 2004, orphan drug designation was assigned in the E.U. for the treatment of functional gastroenteropancreatic endocrine tumors. In 2009, orphan drug designation was received in the U.S. and the E.U. for the treatment of Cushing’s disease and acromegaly. The designation for the treatment of Cushing’s disease was assigned in Australia in 2011 and in Japan in 2012. In 2013, orphan drug designation was assigned in Australia for the treatment of acromegaly.

SIGNIFOR (pasireotide diaspartate) injection is prepared as a sterile solution of pasireotide diaspartate in a tartaric acid buffer for administration by subcutaneous injection. SIGNIFOR is a somatostatin analog. Pasireotide diaspartate, chemically known as (2-Aminoethyl) carbamic acid (2R,5S,8S,11S,14R,17S,19aS)-11-(4-aminobutyl)-5-benzyl-8-(4-benzyloxybenzyl)-14-(1H-indol-3ylmethyl)-4,7,10,13,16,19-hexaoxo-17-phenyloctadecahydro-3a,6,9,12,15,18hexaazacyclopentacyclooctadecen-2-yl ester, di[(S)-2-aminosuccinic acid] salt, is a cyclohexapeptide with pharmacologic properties mimicking those of the natural hormone somatostatin.
The molecular formula of pasireotide diaspartate is C58H66N10O9 • 2C4H7NO4 and the molecular weight is 1313.41. The structural formula is:
 SIGNIFOR is supplied as a sterile solution in a single-dose, 1 mL colorless glass ampule containing pasireotide in 0.3 mg/mL, 0.6 mg/mL, or 0.9 mg/mL strengths for subcutaneous injection.
SIGNIFOR is supplied as a sterile solution in a single-dose, 1 mL colorless glass ampule containing pasireotide in 0.3 mg/mL, 0.6 mg/mL, or 0.9 mg/mL strengths for subcutaneous injection.
Each glass ampule contains:
| 0.3 MG | 0.6 MG | 0.9 MG | |
| Pasireotide diaspartate | 0.3762* | 0.7524* | 1.1286* |
| Mannitol | 49.5 | 49.5 | 49.5 |
| Tartaric acid | 1.501 | 1.501 | 1.501 |
| Sodium hydroxide | ad pH 4.2 | ad pH 4.2 | ad pH 4.2 |
| Water for injection | ad 1ml | ad 1ml | ad 1ml |
| * corresponds to 0.3/0.6/0.9 mg pasireotide base Note: Each ampule contains an overfill of 0.1ml to allow accurate administration of 1 ml from the ampule. |
|||
 |
|
| Systematic (IUPAC) name | |
|---|---|
| [(3S,6S,9S,12R,15S,18S,20R)-9-(4-aminobutyl)-3-benzyl-12-(1H-indol-3-ylmethyl)-2,5,8,11,14,17-hexaoxo-15-phenyl-6-[(4-phenylmethoxyphenyl)methyl]-1,4,7,10,13,16-hexazabicyclo[16.3.0]henicosan-20-yl] N-(2-aminoethyl)carbamate | |
| Clinical data | |
| Trade names | Signifor |
| Licence data | EMA:Link |
| Legal status |
|
| Routes | Subcutaneous |
| Identifiers | |
| CAS number | 396091-73-9 |
| ATC code | H01CB05 |
| PubChem | CID 9941444 |
| UNII | 98H1T17066 |
| Synonyms | SOM230 |
| Chemical data | |
| Formula | C58H66N10O9 |
| Mol. mass | 1107.26 g/mol |
Pasireotide is a multiligand somatostatin analogue with high binding affinity to somatostatin receptors sst1, sst2, sst3 and sst5. Novartis Oncology, a division of Novartis, filed for approval in the E.U. for the treatment of Cushing’s syndrome in 2010. A positive opinion was granted in 2011 and final approval was obtained in 2012. The E.U.’s first launch took place in Germany in June 2012. Also in 2011, Novartis filed an NDA in the U.S. seeking approval of the compound for the treatment of Cushing’s syndrome; however, the application was withdrawn the same year due to an issue related to chemistry, manufacturing and controls. In November 2012, the product was recommended for approval in the U.S. for Cushing’s syndrome. In December 2012, final FDA approval was granted. Phase III clinical trials are ongoing in Japan for this indication. In 2014, the product was approved in the E.U and the U.S. for the treatment of adult patients with acromegaly for whom surgery is not an option or has not been curative and who are inadequately controlled on treatment with a first-generation somatostatin analogue (SSA).
- http://www.google.com/patents/EP2310042B1?cl=en
-
The present invention relates to a new use of Somatostatin (SRIF) peptidomimetics (also referred to as Somatostatin- or SRIF-analogs).
-
Somatostatin is a tetradecapeptide having the structure

-
The somatostatin class is a known class of small peptides comprising the naturally occurring somatostatin-14 and analogues having somatostatin related activity, e.g. as disclosed by A.S. Dutta in Small Peptides, Vol.19, Elsevier (1993). By “somatostatin analog” as used herein is meant any straight-chain or cyclic polypeptide having a structure based on that of the naturally occurring somatostatin-14 wherein one or more amino acid units have been omitted and/or replaced by one or more other amino radical(s) and/or wherein one or more functional groups have been replaced by one or more other functional groups and/or one or more groups have been replaced by one or several other isosteric groups. In general, the term covers all modified derivatives of the native somatostatin-14 which exhibit a somatostatin related activity, e.g. they bind to at least one of the five somatostatin receptor (SSTR), preferably in the nMolar range.
-
Natural somatostatin binds and activates all 5 somatostatin receptors (SSTR1-5) with nmol efficacy and thus causes its multiple physiological effects.
-
Synthetically available somatostatin analogs differ in their binding affinity to the different somatostatin receptor subtypes and often bind selectively to one or few subtypes with significantly higher affinity.
-
Somatostatin analogs of particular interest according to the present invention have a high binding affinity to human SSTR1,2,3,5 and have been described e.g. in WO 97/01579 , the contents of which being incorporated herein by reference. Said somatostatin analogs comprise the amino acid sequence of formula I-(D/L)Trp-Lys-X1 -X2 - Iwherein X1 is a radical of formula (a) or (b)

wherein R1 is optionally substituted phenyl, wherein the substituent may be halogen, methyl, ethyl, methoxy or ethoxy,
R2 is -Z1-CH2-R1, -CH2-CO-O-CH2-R1,
wherein Z1 is O or S, and
X2 is an α-amino acid having an aromatic residue on the Cα side chain, or an amino acid unit selected from Dab, Dpr, Dpm, His,(Bzl)HyPro, thienyl-Ala, cyclohexyl-Ala and t-butyl-Ala, the residue Lys of said sequence corresponding to the residue Lys9 of the native somatostatin-14. -
Somatostatin analogs of particular interest which have a high binding affinity to human SSTR1,2,3,5 have also been described e.g. inWO02/10192. Said somatostatin analogs comprise the compound of formula

also called cyclo[{4-(NH2-C2H4-NH-CO-O-)Pro}-Phg-DTrp-Lys-Tyr(4-Bzl)-Phe] or pasireotide, as well as diastereoisomers and mixtures thereof, in free form, in salt or complex form or in protected form. Phg means -HN-CH(C6H5)-CO- and Bzl means benzyl.




…………………
http://www.google.com/patents/WO2002010192A2?cl=en
Example 1 : Cyclo[{4-(NH2-C2H4-NH-CO-O-
a) Synthesis of Fmoc-Pro(4-OCO-NH-CH2-CH2-NH-Boc)-OH
L-hydroxyproline methylester hydrochloride is reacted with Fmoc-OSu in aqueous 1.0 N sodium carbonate/THF at room temperature. After completion of the reaction, Fmoc-Pro(4- OH)-OMe is isolated by precipitation. Fmoc-Pro(4-OH)-OMe is then added dropwise into a solution of trisphosgene (0.6 eq.) in THF to give a chlorocarbonate intermediate. After 1 h dimethylaminopyridine (1.0 eq.) and N-Boc-diaminoethane (6.0 eq.) are added and the reaction is stirred at room temperature. After completion of the reaction, the solvent is removed in vacuo and the resulting Fmoc-Pro(4-OCO-NH-CH2-CH2-NH-Boc)-OMe is extracted from a two phase system of ethyl acetate/0.1 M HCI to give crude product (MH+ = 554) which is purified by crystallization from ethyl acetate. The methyl ester is then cleaved to the free acid by treatment with 1 N NaOH in dioxane/water and the product Fmoc-Pro(4-OCO-NH-CH2-CH2-NH-Boc)-OH is purified on silica gel, [(M+Na)]+= 562).
b) H-Phe-Pro(4-OCO-NH-CH2-CH2-NH-Boc)-Phg-DTrp(Boc)-Lys(Boc)-Tyr(Bzl)-OH Commercially available Fmoc-Tyr(Bzl)-O-CH2-Ph(3-OCH3)-O-CH2-Polystyrene resin (SASRIN-resin, 2.4 mM) is used as starting material and carried through a standard protocol consisting of repetitive cycles of Nα-deprotection (Piperidine/DMF, 2:8), repeated washings with DMF and coupling (DIPCI: 4.8 mM/HOBT: 6mM, DMF). The following amino acid- derivatives are sequentially coupled: Fmoc-Lys(Boc)-OH, Fmoc-DTrp(Boc)-OH, Fmoc-Phg- OH, Fmoc-Pro(4-OCO-NH-CH2-CH2-NH-Boc)-OH, Fmoc-Phe-OH. Couplings (2 eq. amino acids) are continued or repeated until completion, i.e. until complete disappearance of residual amino groups which is monitored by a negative ‘Kaiser* Ninhydrin test. Before cleavage of the completely assembled protected linear peptide from its resin support the Nα-Fmoc protection from the last residue is removed.
c) H-Phe-Pro(4-OCO-NH-CH2-CH2-NH-Boc)-Phg-DTrp(Boc)-Lys(Boc)-Tyr(Bzl)-OH After washings with CH2CI2) the peptide-resin is transferred into a column or a stirred suction filter and the peptide fragment is cleaved and eluted with a short treatment with 2% TFA in CH2CI2 within 1 h. The eluate is immediately neutralized with a saturated NaHCO3 solution. The organic solution is separated and evaporated and the side chain protected precursor (MH+ = 1366) is cyclized without further purification.
d) cyclo[-Pro(4-OCO-NH-CH2-CH2-NH2)-Phg-DT -Lys-Tyr(Bzl)-Phe-], trifluoroacetate The above linear fragment is dissolved in DMF (4 mM), cooled to minus 5°C and treated with 2 eq. DIPEA then 1.5 eq. of DPPA and stirred until completion (ca. 20h) at 0-4°C. The solvent was almost completely removed in vacuo; the concentrate is diluted with ethyl acetate, washed with NaHCO3, water, dried and evaporated in vacuo.
For deprotection the residue is dissolved at 0°C in TFA H2O 95:5 (ca.50 mM) and stirred in the cold for 30 min. The product is then precipitated with ether containing ca. 10 eq. HCI, filtered, washed with ether and dried. In order to completely decompose remaining Indole-N carbaminic acid the product is dissolved in 5% AcOH and lyophilized after 15 h at ca. 5°C. Preparative RP-HPLC is carried out on a C-18 10 μm STAGROMA column (5-25 cm) using a gradient of 0.5% TFA to 0.5% TFA in 70% acetonitrile. Fractions containing the pure title compound are combined, diluted with water and lyophilized. The lyophilisate is dissolved in water followed by precipitation with 10% Na2CO3 in water. The solid free base is filtered of, washed with water and dried in vacuum at room temperature. The resulting white powder is directly used for the different salts.
Example 2: Cyclo[{4-(NH2-C2H4-NH-CO-O-)Pro}-Phg-DTrp-Lys-Tyr(4-Bzl)-Phe] in salt form a. Acetate
Conversion to the acetate salt form is carried out using an ion-exchange resin (e.g. AG 3- X4). MS (ESI): m/z 524.5 [M+2H]2+ [α]D 20= -42°, c=0.26 in AcOH 95%
b. Aspartate
Conversion to the mono- or di-aspartate is obtained by reacting 1 equivalent of the compound of Example 1 with 1 or 2 equivalent of aspartic acid in a mixture of acetonitrile/water 1 :3. The resulting mixture is frozen and lyophilized. The di-aspartate may also be obtained by dissolving the compound of Example 1 in water/acetonitrile 4:1, filtering, loading on a an ion-exchange resin, e.g. BioRad AG4X4 column, and eluting with water/acetonitrile 4:1. The eluate is concentrated, frozen and lyophilized. [ ]D 20= -47.5°, c= 2.5mg/ml in methanol
……………..
WO2013/174978 A1
http://www.google.im/patents/WO2013174978A1?cl=ru
………………………..
WO2013/131879 A1,
………………………..
WO2005/53732 A1,
http://www.google.com/patents/WO2005053732A1?cl=en
……………………………
Journal of Medicinal Chemistry, 2003 , vol. 46, 12 pg. 2334 – 2344
http://pubs.acs.org/doi/abs/10.1021/jm021093t

A rational drug design approach, capitalizing on structure−activity relationships and involving transposition of functional groups from somatotropin release inhibitory factor (SRIF) into a reduced size cyclohexapeptide template, has led to the discovery of SOM230 (25), a novel, stable cyclohexapeptide somatostatin mimic that exhibits unique high-affinity binding to human somatostatin receptors (subtypes sst1−sst5). SOM230 has potent, long-lasting inhibitory effects on growth hormone and insulin-like growth factor-1 release and is a promising development candidate currently under evaluation in phase I clinical trials.
5.1.3.2. Cyclization, Deprotection, and Purification of Cyclo[(diaminoethylcarbamoyl)-HyPro-Phg-d-Trp-Lys-Tyr(Bzl)-Phe] (25). For cyclization, the above linear fragment was dissolved in DMF to a concentration of 4 mM, cooled to −5 °C, treated with 2 equiv of DIPEA and then 1.5 equiv of DPPA, and stirred at 0−4 °C until completion (ca. 20 h). The solvent was almost completely removed in vacuo. The concentrate was diluted with ethyl acetate, washed with NaHCO3 and water, dried, and evaporated in vacuo. The protected cyclized product was obtained in good yield.
For complete deprotection, the residue was dissolved at 0 °C in TFA/H2O, 95:5 (ca. 50 mM), and the mixture was stirred in the cold for 30 min. The product was then precipitated with ether containing ca. 10 equiv of HCl, filtered, washed with ether, and dried. To completely decompose the remaining indole-N carbaminic acid, the product was dissolved in 5% AcOH and lyophilized after 15 h at ca. 5 °C. Analytical RP-HPLC indicated a purity of 75% for the crude product.Preparative HPLC purification afforded 25: 3.1 g, 20% yield, purity 98%, RtI = 10.70, RtII = 10.20, RtIV = 3.90, HRMS 1047.51 (calcd 1047.5014).
Table 2. 1H and 13C NMR Assignments of SOM230, Using Numbering Scheme in NMR Assignment
| residue | group | δ 1H [ppm] | δ 13C [ppm] | residue | group | δ 1H [ppm] | δ 13C [ppm] |
| 1 | l-phenylglycine |
| 1 | NH | 9.73 | 1 | α-CH | 6.47 | 59.3 | |||
| 1 | 2/6-CH | 8.02 | 127.3 | 1 | CO | 169.6 | |||
| 1 | 3/5-CH | 7.41 | 129.1 | 1 | 1-C | 141.0 |
| 1 | 4-CH | 7.21 | 128.0 | |
| 2 | d-tryptophane |
| 2 | 1‘-NH | 12.20 | 2 | α-CH | 5.28 | 55.6 | |||
| 2 | NH | 10.34 | 2 | β-CH2 | 3.72 | 3.30 | 28.5 | ||
| 2 | 7-CH | 7.65 | 112.0 | 2 | CO | 173.9 | |||
| 2 | 4-CH | 7.43 | 119.2 | 2 | 8-C | 137.5 | |||
| 2 | 2-CH | 7.28 | 124.7 | 2 | 9-C | 128.3 | |||
| 2 | 6-CH | 7.23 | 121.6 | 2 | 3-C | 110.3 |
| 2 | 5-CH | 6.96 | 119.2 | |
| 3 | l-lysine |
| 3 | NH | 10.10 | 3 | δ-CH2 | 1.41 | 1.32 | 31.5 | ||
| 3 | α-CH | 4.62 | 55.2 | 3 | γ-CH2 | 0.89 | 23.5 | ||
| 3 | ε-CH2 | 2.80 | 41.0 | 3 | CO | 171.9 | |||
| 3 | β-CH2 | 1.87 | 1.32 | 31.6 | 3 | NH3+ | a |
| 4 | (4-O-benzyl)-l-tyrosine | |||
| 4 | NH | 7.99 | 4 | 7-CH2 | 4.92 | 69.9 | |||
| 4 | 2‘/6‘-CH | 7.46 | 128.0 | 4 | β-CH2 | 3.46 | 3.10 | 39.7 | |
| 4 | 3‘/5‘-CH | 7.37 | 128.9 | 4 | CO | 171.8 | |||
| 4 | 4‘-CH | 7.30 | 128.2 | 4 | 4-C | 157.9 | |||
| 4 | 2/6-CH | 7.21 | 131.5 | 4 | 1‘C | 137.9 | |||
| 4 | 3/5-CH | 6.85 | 114.7 | 4 | 1-C | 129.8 |
| 4 | α-CH | 5.23 | 53.1 | |
| 5 | l-phenylalanine |
| 5 | NH | 9.82 | 5 | α-CH | 4.42 | 53.9 | |||
| 5 | 2/6-CH | 7.38 | 130.0 | 5 | β-CH2 | 3.23 | 3.06 | 37.8 | |
| 5 | 3/5-CH | 7.27 | 129.3 | 5 | CO | 171.2 | |||
| 5 | 4-CH | 7.16 | 127.6 | 5 | 1-C | 136.3 |
| 6 | (γ-O-diaminoethylcarbamate)-l-hydroxyproline | ||||||
| 6 | 2-NH | 8.04 | 6 | 4-CH2 | 2.95 | 42.4 | |||
| 6 | γ-CH | 5.23 | 70.9 | 6 | β-CH2 | 2.63 | 1.25 | 37.0 | |
| 6 | α-CH | 4.22 | 60.6 | 6 | CO | 170.7 | |||
| 6 | δ-CH2 | 4.12 | 51.4 | 6 | 1-CO | 156.7 | |||
| 6 | 3-CH2 | 3.42 | 44.5 | 6 | 4-NH3+ | a |
| A | acetate |
| A | CH3 | 2.20 | 22.1 | A | CO | 174.3 |
a The NH3+ protons are part of the water peak at 5.82 ppm.
References
- Signifor® (pasireotide) Official Website for healthcare professionals outside the US http://www.signifor.com/
- “Novartis drug Signifor® approved in the EU as the first medication to treat patients with Cushing’s disease”. Retrieved 2012-07-08.
- Mancini et al. Therapeutics and Clinical Risk Management 2010;6:505-516
- Colao et al. Pasireotide (SOM230) provides clinical benefit in patients with Cushing’s disease: results from a large, 12-month, randomized-dose, double-blind, Phase III study, Abstract OC1.7. European Neuroendocrine Association (ENEA) 14th Congress, 2010:62-63
- U.S. National Library of Medicine: Treatment of pituitary-dependent Cushing’s disease with the multireceptor ligand somatostatin analog pasireotide (SOM230): a multicenter, phase II trial. http://www.ncbi.nlm.nih.gov/pubmed/18957506?dopt=Abstract
- EMEA Approval for Pasireotide
- “FDA Approves Pasireotide for Cushing’s Disease”.
| WO2005117830A1 | 6 Jun 2005 | 15 Dec 2005 | Camurus Ab | Liquid depot formulations |
| WO2006075124A1 * | 9 Dec 2005 | 20 Jul 2006 | Camurus Ab | Somatostatin analogue formulations |
| WO2006131730A1 | 6 Jun 2006 | 14 Dec 2006 | Camurus Ab | Glp-1 analogue formulations |
| WO2007096055A1 * | 7 Feb 2007 | 30 Aug 2007 | Novartis Ag | Combination of somatostatin-analogs with different selectivity for human somatostatin receptor subtypes |
| WO2010003939A1 * | 7 Jul 2009 | 14 Jan 2010 | Novartis Ag | Use of pasireotide for the treatment of endogenous hyperinsulinemic hypoglycemia |
| US20090155193 * | 9 Dec 2005 | 18 Jun 2009 | Fredrik Joabsson | Topical Bioadhesive Formulations |
FDA approves Gardasil 9 for prevention of certain cancers caused by five additional types of HPV
12/10/2014 01:39 PM EST
The U.S. Food and Drug Administration today approved Gardasil 9 (Human Papillomavirus 9-valent Vaccine, Recombinant) for the prevention of certain diseases caused by nine types of Human Papillomavirus (HPV). Covering nine HPV types, five more HPV types than Gardasil (previously approved by the FDA), Gardasil 9 has the potential to prevent approximately 90 percent of cervical, vulvar, vaginal and anal cancers.

GARDASIL is the only human papillomavirus (HPV) vaccine that helps protect against 4 types of HPV. In girls and young women ages 9 to 26, GARDASIL helps protect against 2 types of HPV that cause about 75% of cervical cancer cases, and 2 more types that cause about 90% of genital warts cases. In boys and young men ages 9 to 26, GARDASIL helps protect against approximately 90% of genital warts cases.
GARDASIL also helps protect girls and young women ages 9 to 26 against approximately 70% of vaginal cancer cases and up to 50% of vulvar cancer cases.
GARDASIL may not fully protect everyone, nor will it protect against diseases caused by other HPV types or against diseases not caused by HPV. GARDASIL does not prevent all types of cervical cancer, so it’s important for women to continue routine cervical cancer screenings. GARDASIL does not treat cancer or genital warts. GARDASIL is given as 3 injections over 6 months.
尼达尼布 ニンテダニブ NINTEDANIB For Idiopathic pulmonary fibrosis

NINTEDANIB, BBIF 1120, Intedanib
Boehringer Ingelheim Corp
As a potential treatment for a range of different solid tumour types
CAS 656247-17-5
CAS 1377321-64-6 (nintedanib bisethanesulfonate)
CAS [656247-18-6] mono ethane sulfonate
3(Z)-[1-[4-[N-Methyl-N-[2-(4-methylpiperazin-1-yl)acetyl]amino]phenylamino]-1-phenylmethylene]-2-oxo-2,3-dihydro-1H-indole-6-carboxylic acid methyl ester
MW 539.62, MF C31 H33 N5 O4
Launched 2014 USA….Idiopathic pulmonary fibrosis
| chinese, japanese | 尼达尼布 ニンテダニブ |
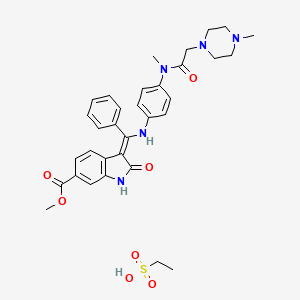
Ethanesulfonic acid – methyl (3Z)-3-{[(4-{methyl[(4-methyl-1-piperazinyl)acetyl]amino}phenyl)amino](phenyl)methylene}-2-oxo-6-indolinecarboxylate (1:1)
Nintedanib esylate
Cas 656247-18-6 [RN]
Methyl (3Z)-3-[({4-[N-methyl-2-(4-methylpiperazin-1-yl)acetamido]phenyl}amino)(phenyl)methylidene]-2-oxo-2,3-dihydro-1H-indole-6-carboxylate ethanesulfonate
Nintedanib esylate [USAN]
(3Z)-2,3-Dihydro-3-[[[4-[methyl[2-(4-methyl-1-piperazinyl)acetyl]amino]phenyl]amino]phenylmethylene]-2-oxo-1H-indole-6-carboxylic acid methyl ester ethanesulfonate
1H-Indole-6-carboxylic acid, 2,3dihydro-3-[[[4-[methyl[(4-methyl-1-piperazinyl)acetyl]amino]phenyl]amino]phenylmethylene]-2-oxo-,methyl ester, (3Z)-, ethanesulfonate (1:1)
Nintedanib esylate, 656247-18-6, UNII-42F62RTZ4G, , NSC753000, NSC-753000, KB-62821
Molecular Formula: C33H39N5O7S Molecular Weight: 649.75706
ニンテダニブエタンスルホン酸塩
Highly crystalline (mp = 305 °C) and exhibits a log P of 3.0 and good aqueous solubility (>20 mg/mL in water)…..J. Med. Chem., 2015, 58 (3), pp 1053–1063
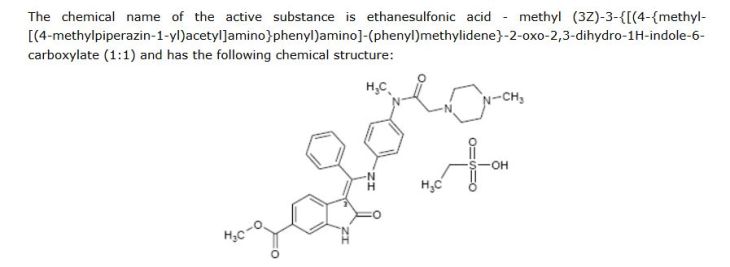
Nintedanib esilate is a bright yellow powder soluble in water. The solubility increases at lower pH and decrease at higher pH due to the non-protonated free base which has a low solubility in water.At room temperature, the active substance exists only in one single crystalline form . The active substance contains no chiral centres. The double bond at C
-3 of the indole moiety allows forE/Zisomerism, but the activesubstance is the Z

Trade Name:Ofev® / Vargatef®
MOA:Tyrosine kinase inhibitor
Indication:Idiopathic pulmonary fibrosis (IPF); Non small cell lung cancer (NSCLC)
In 2011, orphan drug designation was assigned in the U.S. and Japan for the treatment of idiopathic pulmonary fibrosis. In 2013, orphan drug designation was also assigned for the same indication in the E.U. In 2014, a Breakthrough Therapy Designation was assigned to the compound for the treatment of idiopathic pulmonary fibrosis.
Nintedanib (formerly BIBF 1120) is a small molecule tyrosine-kinase inhibitor, targeting vascular endothelial growth factor receptor (VEGFR), fibroblast growth factor receptor (FGFR) and platelet derived growth factor receptor (PDGFR) being developed by Boehringer Ingelheim as an anti-angiogenesis anti-cancer agent under the trade name Vargatef, and recently approved for treatment of idiopathic pulmonary fibrosis as Ofev.
The use of nintedanib or its salts, particularly its esylate salt is claimed for treating non-small cell lung cancer (NSCLC) in a patient who has received prior treatment with an anti-tumor therapy other than with nintedanib, wherein the patient to be treated is selected for treatment on the basis showing progression of the cancer within a period of 9 months or less after the initiation of said prior treatment. It is also claimed that the compound may be administered in combination with an anti-cancer drug, eg docetaxel. Nintedanib is known to be an antagonist of FGF-1, FGF-2, FGF-3, VEGF-1, VEGF-2, VEGF-3, PDGF-α and PDGF-β receptors.
Use of nintedanib for the treatment of non-small cell lung cancer in a patient who has received prior anti-tumour therapy other than with nintedanib. Boehringer Ingelheim has developed and launched Ofev, an oral capsule formulation of nintedanib, for the treatment of idiopathic pulmonary fibrosis (IPF), hepatic insufficiency and cancer, including metastatic NSCLC, ovarian, prostate and colorectal cancer. In October 2014, the US FDA approved the drug and an NDA was filed in Japan for IPF. Picks up from WO2014049099, claiming pharmaceutical combinations comprising nintedanib and sunitinib.
Nintedanib is an indolinone derivative angiogenesis inhibitor, originated at Boehringer Ingelheim. In 2014, the product candidate was approved and launched in the U.S. for the treatment of idiopathic pulmonary fibrosis, and a positive opinion was received by the EMA for the same indication. Also in 2014, Nintedanib was approved in the E.U. for the oral treatment of locally advanced, metastatic or locally recurrent non-small cell lung cancer (NSCLC) of adenocarcinoma tumour histology after first-line chemotherapy, in combination with docetaxel.The drug candidate is a small-molecule triple kinase inhibitor targeting the angiogenesis kinases (angiokinases) vascular endothelial growth factor receptor (VEGFR), fibroblast growth factor receptor (FGFR) and platelet-derived growth factor receptor (PDGFR). By allowing the vascularization necessary for the nourishment of tumors, these angiokinases have been implicated in tumor growth, proliferation and metastasis. In previous studies, intedanib potently and selectively inhibited human endothelial cell proliferation and induced apoptosis in human umbilical vein endothelial cells (HUVEC). It showed good oral bioavailability and tolerance, and significant antitumor activity was observed in a number of human tumor xenograft models.
Mechanism of action
Nintedanib is an indolinone-derived drug that inhibits the process of blood vessel formation (angiogenesis). Angiogenesis inhibitors stop the formation and reshaping of blood vessels in and around tumours, which reduces the tumour’s blood supply, starving tumour cells of oxygen and nutrients leading to cell death and tumour shrinkage. Unlike conventional anti-cancer chemotherapy which has a direct cell killing effect on cancer cells, angiogenesis inhibitors starve the tumour cells of oxygen and nutrients which results in tumour cell death. One of the advantages of this method of anti-cancer therapy is that it is more specific than conventional chemotherapy agents, therefore results in fewer and less severe side effects than conventional chemotherapy.
The process of new blood vessel formation (angiogenesis) is essential for the growth and spread of cancers. It is mediated by signaling molecules (growth factors) released from cancer cells in response to low oxygen levels. The growth factors cause the cells of the tumour’s blood vessel to divide and reorganize resulting in the sprouting of new vessels in and around the tumour, improving its blood supply.
Angiogenesis is a process that is essential for the growth and spread of all solid tumours, blocking it prevents the tumour from growing and may result in tumour shrinkage as well as a reduction in the spread of the cancer to other parts of the body. Nintedanib exerts its anti-cancer effect by binding to and blocking the activation of cell receptors involved in blood vessel formation and reshaping (i.e. VEGFR 1-3, FGFR 1-3 AND PDGFRα and β). Inhibition of these receptors in the cells that make up blood vessels (endothelial cells, smooth muscle cells and pericytes) by Nintedanib leads to programmed cell death, destruction of tumor blood vessels and a reduction in blood flow to the tumour. Reduced tumour blood flow inhibits tumor cell proliferation and migration hence slowing the growth and spread of the cancer.[1]
Adverse effects
Preclinical studies have shown that nintedanib binds in a highly selective manner to the ATP binding pocked of its three target receptor families, without binding to similarly shaped ATP domains in other proteins, which reduces the potential for undesirable side effects.[2]
The most common side effects observed with nintedanib were reversible elevation in liver enzymes (10-28% of patients) and gastrointestinal disturbance (up to 50%). Side effects observed with nintedanib were worse with the higher 250 mg dose, for this reason subsequent trials have used the equally clinically effective 200 mg dose.[1][2][3][4][5][6][7][8][9]
Nintedanib inhibits the growth and reshaping of blood vessels which is also an essential process in normal wound healing and tissue repair. Therefore a theoretical side effect of nintedanib is reduced wound healing however, unlike other anti-angiogenic agents, this side effect has not been observed in patients receiving nintedanib.
Studies
Preclinical studies have demonstrated that nintedanib selectively binds to and blocks the VEGF, FGF and PDGF receptors, inhibiting the growth of cells that constitute the walls of blood vessels (endothelial and smooth muscle cells and pericytes) in vitro. Nintedanib reduces the number and density of blood vessels in tumours in vivo, resulting in tumour shrinkage.[1][2] Nintedanib also inhibits the growth of cells that are resistant to existing chemotherapy agents in vitro, which suggests a potential role for the agent in patients with solid tumours that are unresponsive to or relapse following current first line therapy.[10]
Early clinical trials of nintedanib have been carried out in patients with non-small cell lung, colorectal, uterine, endometrial, ovarian and cervical cancer and multiple myeloma.[4][5][7][8][9] These studies reported that the drug is active in patients, safe to administer and is stable in the bloodstream. They identified that the maximum tolerated dose of nintedanib is 20 0 mg when taken once a day.
Clinical studies
In the first human trials, nintedanib halted the growth of tumours in up to 50% of patients with non-small cell lung cancer and 76% of patients with advanced colorectal cancer and other solid tumours.[4][8] A complete response was observed in 1/26 patients with non-small cell lung and 1/7 patients with ovarian cancer treated with nintedanib. A further 2 patients with ovarian cancer had partial responses to nintedanib.[8][9]
Two phase II trials have been carried out assessing the efficacy, dosing and side effects of nintedanib in non-small cell lung and ovarian cancer. These trials found that nintedanib delayed relapse in patients with ovarian cancer by two months[6] and that overall survival of patients with non-small cell lung who received nintedanib was similar to that observed with the FDA approved VEGFR inhibitor sorafenib. These trials also concluded that increasing the dose of the nintedanib has no effect on survival.[3]
SYNTHESIS
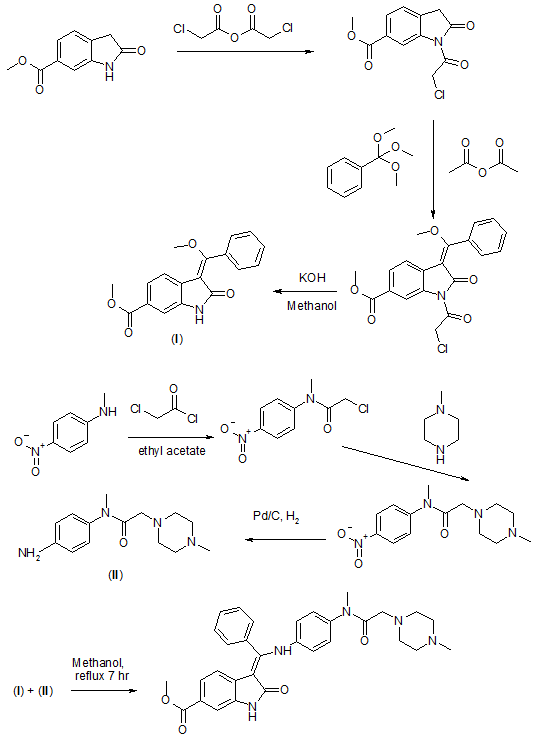
WO2009071523A1
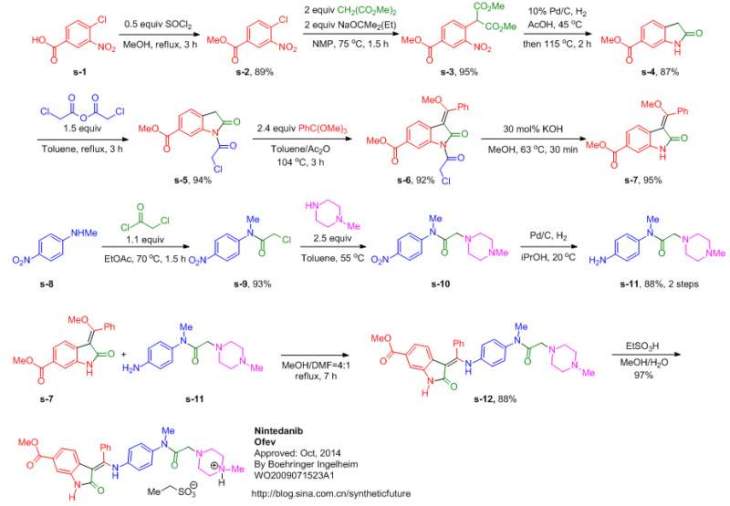
MORE SYNTHESIS
Route 1


Reference:1. WO0127081A1.
2. US6762180B1.
3. J. Med. Chem. 2009, 52, 4466-4480.
Route 2


Reference:1. WO2009071523A1 / US8304541B2.
Route 3


Reference:1. CN104262232A.
Route 4


Reference:1. CN104844499A.
Current clinical trials
Nintedanib is being tested in several phase I to III clinical trials for cancer. Angiogenesis inhibitors such as nintedanib may be effective in a range of solid tumour types including; lung, ovarian, metastatic bowel, liver and brain cancer. Patients are also being recruited for three phase III clinical trials that will evaluate the potential benefit of nintedanib when added to existing 1st line treatments in patients with ovarian.[11] and 2nd line treatment in non-small cell lung cancer [12][13] The phase III trials of nintedanib in lung cancer have been named LUME-Lung 1 and LUME-Lung 2.
Current phase II trials are investigating the effect of nintedanib in patients with metastatic bowel cancer, liver cancer and the brain tumour: glioblastoma multiforme.[14]
Phase III trials are investigating the use of nintedanib in combination with the existing chemotherapy agents permexetred and docetaxel in patients with non-small cell lung cancer,[15] and in combination with carboplatin and paclitaxel as a first line treatment for patients with ovarian cancer.[16]
A phase III clinical trial was underway examining the safety and efficacy of nintedanib on patients with the non-cancerous lung condition idiopathic pulmonary fibrosis.[17] Nintedanib, under the brand name Ofev, was approved by the FDA for treatment of idiopathic pulmonary fibrosis on 15 Oct 2014. [18]
In terms of clinical development, additional phase III clinical trials are ongoing for the treatment of epithelial ovarian cancer, fallopian tube or primary peritoneal cancer, in combination with chemotherapy, and for the treatment of refractory metastatic colorectal cancer. Phase II clinical trials are also ongoing at the company for the treatment of glioblastoma multiforme, previously untreated patients with renal cell cancer, and for the treatment of patients with unresectable malignant pleural mesothelioma. The National Cancer Center of Korea (NCC) is evaluating the compound in phase II studies as second line treatment for small cell lung cancer (SCLC). The Centre Oscar Lambret is also conducting phase II clinical trials for the treatment of breast cancer in combination with docetaxel. Phase II trials are under way at EORTC as second line therapy for patients with either differentiated or medullary thyroid cancer progressing after first line therapy. The compound is also in early clinical development for the treatment of cancer of the peritoneal cavity, hepatocellular carcinoma, acute myeloid leukemia and ovarian cancer. Clinical trials have been completed for the treatment of prostate cancer and for the treatment of colorectal cancer. Boehringer Ingelheim is also conducting phase I/II clinical trials for the treatment of NSCLC and acute myeloid leukemia in addition to low-dose cytarabine. Phase I clinical studies are ongoing at the company for the treatment of epithelial ovary cancer and for the treatment of patients with mild and moderate hepatic impairment. The company had been evaluating the compound in early clinical trials for the treatment of prostate cancer in combination with docetaxel, but recent progress reports for this indication are not available at present.
In 2011, orphan drug designation was assigned in the U.S. and Japan for the treatment of idiopathic pulmonary fibrosis. In 2013, orphan drug designation was also assigned for the same indication in the E.U. In 2014, a Breakthrough Therapy Designation was assigned to the compound for the treatment of idiopathic pulmonary fibrosis.
PAPER
http://pubs.acs.org/doi/full/10.1021/jm501562a
Nintedanib: From Discovery to the Clinic
†Department of Medicinal Chemistry; §Department of Drug Metabolism and Pharmacokinetics; ‡Department of Non-Clinical Drug Safety; ∥Department of Translational Medicine and Clinical Pharmacology; ⊥Department of Respiratory Diseases Research; and #Corporate Division Medicine, TA Oncology, Boehringer Ingelheim Pharma GmbH & Co. KG, 88397 Biberach an der Riss, Germany
○ Clinical Development and Medical Affairs, Respiratory, Boehringer Ingelheim Inc., Ridgefield, Connecticut 06877, United States
∇ Boehringer Ingelheim RCV GmbH & Co. KG, A-1121 Vienna, Austria
J. Med. Chem., 2015, 58 (3), pp 1053–1063
DOI: 10.1021/jm501562a

Nintedanib (BIBF1120) is a potent, oral, small-molecule tyrosine kinase inhibitor, also known as a triple angiokinase inhibitor, inhibiting three major signaling pathways involved in angiogenesis. Nintedanib targets proangiogenic and pro-fibrotic pathways mediated by the VEGFR family, the fibroblast growth factor receptor (FGFR) family, the platelet-derived growth factor receptor (PDGFR) family, as well as Src and Flt-3 kinases. The compound was identified during a lead optimization program for small-molecule inhibitors of angiogenesis and has since undergone extensive clinical investigation for the treatment of various solid tumors, and in patients with the debilitating lung disease idiopathic pulmonary fibrosis (IPF). Recent clinical evidence from phase III studies has shown that nintedanib has significant efficacy in the treatment of NSCLC, ovarian cancer, and IPF. This review article provides a comprehensive summary of the preclinical and clinical research and development of nintedanib from the initial drug discovery process to the latest available clinical trial data.
-
Roth, G. J.; Heckel, A.; Colbatzky, F.; Handschuh, S.; Kley, J.; Lehmann-Lintz, T.; Lotz, R.; Tontsch-Grunt,U.; Walter, R.; Hilberg, F.Design, synthesis, and evaluation of indolinones as triple angiokinase inhibitors and the discovery of a highly specific 6-methoxycarbonyl-substituted indolinone (BIBF 1120) J. Med. Chem.2009, 52, 4466– 4480
-
2.Roth, G. J.; Sieger, P.; Linz, G.; Rall, W.; Hilberg, F.; Bock, T. 3-Z-[1-(4-(N-((4-Methyl-piperazin-1-yl)-methylcarbonyl)-N-methyl-amino)-anilino)-1-phenyl-methylene]-6-methoxycarbonyl-2-indolinone monoethanesulphonate and the use thereof as a pharmaceutical composition. WO2004/013099. 2004.
-
3.Merten, J.; Linz, G.; Schnaubelt, J.; Schmid, R.; Rall, W.; Renner, S.; Reichel, C.; Schiffers, R. Process for the manufacture of an indolinone derivative. WO2009/071523. 2009
PAPER
http://pubs.acs.org/doi/abs/10.1021/jm900431g
J. Med. Chem., 2009, 52 (14), pp 4466–4480
DOI: 10.1021/jm900431g

Inhibition of tumor angiogenesis through blockade of the vascular endothelial growth factor (VEGF) signaling pathway is a new treatment modality in oncology. Preclinical findings suggest that blockade of additional pro-angiogenic kinases, such as fibroblast and platelet-derived growth factor receptors (FGFR and PDGFR), may improve the efficacy of pharmacological cancer treatment. Indolinones substituted in position 6 were identified as selective inhibitors of VEGF-, PDGF-, and FGF-receptor kinases. In particular, 6-methoxycarbonyl-substituted indolinones showed a highly favorable selectivity profile. Optimization identified potent inhibitors of VEGF-related endothelial cell proliferation with additional efficacy on pericyctes and smooth muscle cells. In contrast, no direct inhibition of tumor cell proliferation was observed. Compounds 2 (BIBF 1000) and 3 (BIBF 1120) are orally available and display encouraging efficacy in in vivo tumor models while being well tolerated. The triple angiokinase inhibitor 3 is currently in phase III clinical trials for the treatment of nonsmall cell lung cancer.
PATENT
The present invention relates to a beneficial treatment of tumours in patients suffering from NSCLC, and to a clinical marker useful as predictive variable of the responsiveness of tumours in patients suffering from NSCLC. The present invention further relates to a method for selecting patients likely to respond to a given therapy, wherein said method optionally comprises the use of a specific clinical marker. The present invention further relates to a method for delaying disease progression and/or prolonging patient survival of NSCLC patients, wherein said method comprises the use of a specific clinical marker.
The monoethanesulphonate salt form of this compound presents properties which makes this salt form especially suitable for development as medicament. The chemical structure of 3-Z-[l-(4-(N-((4-methyl-piperazin-l-yl)-methylcarbonyl)-N-methyl-amino)-anilino)- 1 -phenyl-methylene] -6-methoxycarbonyl-2-indolinone-monoethanesulphonate (ΓΝΝ name nintedanib esylate) is depicted below as Formula Al .
Formula Al

This compound is thus for example suitable for the treatment of diseases in which angiogenesis or the proliferation of cells is involved. The use of this compound for the treatment of immunologic diseases or pathological conditions involving an
immunologic component is being described in WO 2004/017948, the use for the treatment of, amongst others, oncological diseases, alone or in combination, is being described in WO 2004/096224 and WO 2009/147218, and the use for the treatment of fibrotic diseases is being described in WO 2006/067165.
A method using biomarkers for monitoring the treatment of an individual with the compound 3-Z-[l-(4-(N-((4-methyl-piperazin-l-yl)-methylcarbonyl)-N-methyl-amino)-anilino)-l -phenyl-methylene] -6-methoxycarbonyl-2-indolinone or a pharmaceutically acceptable salt thereof, wherein it is determined if a sample from said individual comprises a biomarker in an amount that is indicative for said treatment, is disclosed in WO 2010/103058.
PATENT
http://www.google.com/patents/US20110201812
The present invention relates to a process for the manufacture of a specific indolinone derivative and a pharmaceutically acceptable salt thereof, namely 3-Z-[1-(4-(N-((4-methyl-piperazin-1-yl)-methylcarbonyl)-N-methyl-amino)-anilino)-1-phenyl-methylene]-6-methoxycarbonyl-2-indolinone and its monoethanesulfonate, to new manufacturing steps and to new intermediates of this process.
The indolinone derivative 3-Z-[1-(4-(N-((4-methyl-piperazin-1-yl)-methylcarbonyl)-N-methyl-amino)-anilino)-1-phenyl-methylene]-6-methoxycarbonyl-2-indolinone and its monoethanesulfonate are known from the following patent applications: WO 01/027081, WO 04/013099, WO 04/017948, WO 04/096224 and WO 06/067165. These patent applications disclose the compound, a process for its manufacture, a specific salt form of this compound and the use of the compound or its salt in a pharmaceutical composition to treat oncological or non-oncological diseases via inhibition of the proliferation of target cells, alone or in combination with further therapeutic agents. The mechanism of action by which the proliferation of the target cells occurs is essentially a mechanism of inhibition of several tyrosine kinase receptors, and especially an inhibition of the vascular endothelial growth factor receptor (VEGFR).




EXAMPLE 1Synthesis of the 6-methoxycarbonyl-2-oxindole in accordance with the process shown in synthesis scheme CSynthesis of benzoic acid, 4-chloro-3-nitro-, methylester
-
- 20 kg of 4-chloro-3-nitro-benzoic acid (99.22 mol) is suspended in 76 L methanol. 5.9 kg thionylchloride (49.62 mol) is added within 15 minutes and refluxed for about 3 hours. After cooling to about 5° C., the product is isolated by centrifugation and drying at 45° C.
- Yield: 19.0 kg (88.8% of theoretical amount)
- Purity (HPLC): 99.8%
Synthesis of propanedioic acid, [4-(methoxycarbonyl)-2-nitrophenyl]-, dimethylester
-
- 12.87 kg of malonic acid, dimethylester (97.41 mol) is added to a hot solution (75° C.) of 10.73 kg sodium-tert.amylate (97.41 mol) in 35 L 1-methyl-2-pyrrolidinone (NMP). A solution of 10 kg benzoic acid, 4-chloro-3-nitro-, methylester (46.38 mol) in 25 L 1-methyl-2-pyrrolidinone is added at 75° C. After stirring for 1.5 hours at about 75° C. and cooling to 20° C., the mixture is acidified with 100 L diluted hydrochloric acid to pH 1. After stirring for 1.5 hours at about 5° C., the product is isolated by centrifugation and drying at 40° C.
- Yield: 13.78 kg (95.4% of theoretical amount)
- Purity (HPLC): 99.9%
- Alternatively, propanedioic acid, [4-(methoxycarbonyl)-2-nitrophenyl]-, dimethylester can be synthesized as follows:
- 33.1 kg of malonic acid, dimethylester (250.6 mol) and 27.0 kg benzoic acid, 4-chloro-3-nitro-, methylester (125.3 mol) are subsequently added to a solution of 45.1 kg sodium-methylate (250.6 mol) in 172 kg 1-methyl-2-pyrrolidinone (NMP) at 20° C. After stirring for 1.5 hours at about 45° C. and cooling to 30° C., the mixture is acidified with 249 L diluted hydrochloric acid. At the same temperature, the mixture is seeded, then cooled to 0° C. and stirred for an additional hour. The resulting crystals are isolated by centrifugation, washed and dryed at 40° C.
- Yield: 37.5 kg (86% of theoretical amount)
- Purity (HPLC): 99.7%
Synthesis of 6-methoxycarbonyl-2-oxindole
A solution of 13 kg propanedioic acid, [4-(methoxycarbonyl)-2-nitrophenyl]-, dimethylester (41.77 mol) in 88 L acetic acid is hydrogenated at 45° C. and under 40-50 psi in the presence of 1.3 kg Pd/C 10%. After standstill of the hydrogenation, the reaction is heated up to 115° C. for 2 hours. The catalyst is filtered off and 180 L water is added at about 50° C. The product is isolated after cooling to 5° C., centrifugation and drying at 50° C.
-
- Yield: 6.96 kg (87.2% of theoretical amount)
- Purity (HPLC): 99.8%
EXAMPLE 2Synthesis of the “chlorimide” (methyl-1-(chloroacetyl)-2-oxoindoline-6-carboxylate)

Method 1
6-methoxycarbonyl-2-oxindole (400 g; 2.071 mol) is suspended in toluene (1200 ml) at room temperature. Chloroacetic anhydride (540 g; 3.095 mol) is added to this suspension. The mixture is heated to reflux for 3 h, then cooled to 80° C. and methyl cyclohexane (600 ml) is added within 30 min. The resulting suspension is further cooled down to room temperature within 60 min. The mother liquor is separated and the solid is washed with ice cold methanol (400 ml). The crystals are dried to afford 515.5 g (93.5%) of the “chlorimide” compound as a white solid. 1H-NMR (500 MHz, DMSO-d6) δ: 8.66 (s, 1H, 6-H); 7.86 (d, J=8.3 Hz, 1H, 8-H); 7.52 (d, J=8.3 Hz, 1H, 9-H); 4.98 (s, 2H, 15-H2); 3.95 (s, 3H, 18-H3); 3.88 (s, 2H, 3-H2). 13C-NMR (126 MHz, DMSO-d6) δ: 174.7 (C-2); 36.0 (C-3); 131.0 (C-4); 140.8 (C-5); 115.7 (C-6); 128.9 (C-7); 126.1 (C-8); 124.6 (C-9); 166.6 (C-10); 165.8 (C-13); 46.1 (C-15); 52.3 (C-18). MS: m/z 268 (M+H)+. Anal. calcd. for C12H10ClNO4: C, 53.85; H, 3.77; Cl, 13.25; N, 5.23. Found: C, 52.18; H, 3.64; Cl, 12.89; N, 5.00.
Method 2
6-Methoxycarbonyl-2-oxindole (10 g; 0.052 mol) is suspended in n-butyl acetate (25 ml) at room temperature. To this suspension a solution of chloroacetic anhydride (12.8 g; 0.037 mol) in n-butyl acetate (25 ml) is added within 3 min. The mixture is heated to reflux for 2 h, then cooled to 85° C. and methyl cyclohexane (20 ml) is added. The resulting suspension is further cooled down to room temperature and stirred for 2 h. The mother liquor is separated and the solid is washed with methanol (400 ml) at ambient temperature. The crystals are dried to afford 12.7 g (91.5%) of the “chlorimide” compound as a slightly yellow solid.
EXAMPLE 3Synthesis of the “chlorenol” (methyl-1-(chloroacetyl)-3-[methoxy(phenyl)methylene]-2-oxoindoline-6-carboxylate)

Method 1
Methyl-1-(chloroacetyl)-2-oxoindoline-6-carboxylate (12.0 g; 0.045 mol) is suspended in toluene (60 ml) at ambient temperature. Acetic anhydride (16.2 g; 0.157 mol) is added to this suspension. The mixture is heated to not less than 104° C. and trimethyl orthobenzoate (20.0 g; 0.108 mol) is added within 60 min. During the addition period and subsequent stirring at the same temperature for 3 h, volatile parts of the reaction mixture are distilled off. The concentration of the reaction mixture is kept constant by replacement of the distilled part by toluene (40 ml). The mixture is cooled down to 5° C., stirred for 1 h and filtrated. The solid is subsequently washed with toluene (14 ml) and with a mixture of toluene (8 ml) and ethyl acetate (8 ml). After drying, 16.3 g (91.7%) of the “chlorenol” compound are isolated as slightly yellow crystals. 1H-NMR (500 MHz, DMSO-d6) δ: 8.73 (d, J=1.5 Hz, 1H, 6-H); 8.09 (d, J=8.0 Hz, 1H, 9-H); 7.90 (dd, J=8.1; 1.5 Hz, 1H, 8-H); 7.61-7.48 (m, 5H, 21-H, 22-H, 23-H, 24-H, 25-H); 4.85 (s, 2H, 18-H2); 3.89 (s, 3H, 27-H3); 3.78 (s, 3H, 15-H3). 13C-NMR (126 MHz, DMSO-d6) δ: 165.9 (C-2+C16); 103.9 (C-3); 127.4; 128.6; 130.0; 135.4 (C-4+C-5+C-7+C-20); 115.1 (C-6); 126.1 (C-8); 122.5 (C-9); 166.7 (C-10); 173.4 (C-13); 58.4 (C-15); 46.4 (C-18); 128.6 (C-21+C-22+C-24+C-25); 130.5 (C-23); 52.2 (C-27). MS: m/z 386 (M+H)+. Anal. calcd. for C20H16ClNO5: C, 62.27; H, 4.18; Cl, 9.19; N, 3.63. Found: C, 62.21; H, 4.03; Cl, 8.99; N, 3.52.
Method 2
Methyl-1-(chloroacetyl)-2-oxoindoline-6-carboxylate (12.0 g; 0.045 mol) is suspended in xylene (60 ml) at ambient temperature. Acetic anhydride (16.2 g; 0.157 mol) is added to this suspension. The mixture is heated to reflux, trimethyl orthobenzoate (20.0 g; 0.108 mol) is added within 40 min and heating is maintained for 4 h. The mixture is cooled down to 0° C. and the mother liquor is separated. The solid is subsequently washed with xylene (14 ml) and a mixture of xylene (8 ml) and ethyl acetate (8 ml). After drying 14.3 g (81.0%) of the “chlorenol” compound are isolated as yellow crystals.
Method 3
Methyl-1-(chloroacetyl)-2-oxoindoline-6-carboxylate (12.0 g; 0.045 mol) is suspended in toluene (60 ml) at ambient temperature. Acetic anhydride (16.2 g; 0.157 mol) is added to this suspension. The mixture is heated to reflux, trimethyl orthobenzoate (20.0 g; 0.108 mol) is added within 40 min and heating is maintained for 3 h. The mixture is cooled down to 0° C. and the mother liquor is separated. The solid is subsequently washed with toluene (14 ml) and a mixture of toluene (8 ml) and ethyl acetate (8 ml). After drying 15.3 g (87.3%) of the “chlorenol” compound are isolated as fawn crystals.
EXAMPLE 4Synthesis of the “enolindole” (methyl-3-[methoxy(phenyl)methylene]-2-oxoindoline-6-carboxylate)

Method 1
A solution of potassium hydroxide (0.41 g, 0.006 mol) in methanol (4 ml) is added at 63° C. to a suspension of methyl-1-(chloroacetyl)-3-[methoxy(phenyl)methylene]-2-oxoindoline-6-carboxylate (8.0 g; 0.020 mol) in methanol (32 ml). The mixture is then stirred for 30 min, cooled to 0° C. and stirring is maintained for 2 h. After filtration, the solid is washed with methanol (24 ml) and dried to afford 6.0 g (94.6%) of the “enolindole” compound as yellow crystals. 1H-NMR (500 MHz, CDCl3) δ: 8.08 (s, 1H, 1-H); 7.88 (d, J=7.8 Hz, 1H, 9-H); 7.75 (m, 1H, 8-H); 7.52-7.56 (m, 3H, 18-H, 19-H, 20-H); 7.40-7.45 (m, 3H, 6-H, 17-H, 21-H); 3.92 (s, 3H, 23-H3); 3.74 (s, 3H, 13-H3). 13C-NMR (126 MHz, CDCl3) δ: 168.8 (C-2); 107.4 (C-3); 130.8 (C-4); 138.2 (C-5); 109.4 (C-6); 128.2 and 128.3 (C-7, C-16); 123.5 (C-8); 123.1 (C-9); 170.1 (C-11); 57.6 (C-13); 167.2 (C-14); 128.7 and 128.9 (C-17, C-18, C-20, C-21); 130.5 (C-19); 52.1 (C-23). MS (m/z): 310 (M+H)+. Anal. calcd. for C18H15NO4: C, 69.89; H, 4.89; N, 4.53. Found: C, 69.34; H, 4.92; N, 4.56.
Method 2
A suspension of methyl-1-(chloroacetyl)-3-[methoxy(phenyl)methylene]-2-oxoindoline-6-carboxylate (7.0 g; 0.018 mol) in methanol (28 ml) is heated to reflux. Within 3 min, a solution of sodium methoxide in methanol (0.24 g, 30 (w/w), 0.001 mol) is added to this suspension. The mixture is then stirred for 30 min, cooled to 5° C. and stirring is maintained for 2 h. After filtration, the solid is washed with methanol (9 ml) and dried to afford 5.4 g (89.7%) of the “enolindole” compound as yellow crystals.
Method 3
A suspension of methyl-1-(chloroacetyl)-3-[methoxy(phenyl)methylene]-2-oxoindoline-6-carboxylate (8.0 g; 0.021 mol) in methanol (32 ml) is heated to reflux. A solution of sodium methoxide in methanol (0.74 g, 30% (w/w), 0.004 mol), further diluted with methanol (4 ml), is added dropwise to this suspension. The mixture is then stirred for 90 min, cooled to 0° C. and stirring is maintained for 2 h. After filtration, the solid is washed with methanol (24 ml) and dried to afford 5.9 g (91.2%) of the “enolindole” compound as yellow crystals.
EXAMPLE 5Synthesis of the “chloroacetyl” (N-(4-nitroanilino)-N-methyl-2-chloro-acetamide)

Method 1
A suspension of N-methyl-4-nitroaniline (140 g; 0.920 mol) in ethyl acetate (400 ml) is heated to 70° C. Within 90 min, chloro acetylchloride (114 g; 1.009 mol) is added to this suspension. The resulting solution is then refluxed for 1 h, cooled to 60° C. and methyl cyclohexane (245 ml) is added. The suspension is further cooled down to 0° C. and stirred for 1 h. The reaction mixture is filtrated, washed with methyl cyclohexane (285 ml) and the precipitate is dried to afford 210.4 g (92.7%) of the “chloroacetyl” compound as white crystals. 1H-NMR (500 MHz, DMSO-d6) δ: 8.29 (d, J=8.5 Hz, 2H, 1-H+3-H); 7.69 (d, J=8.5 Hz, 2H, 4-H+6-H); 4.35 (s, 2H, 9-H2); 3.33 (s, 3H, 12-H3). 13C-NMR (126 MHz, DMSO-d6) δ: 124.6 (C-1+C-3); 145.6 (C-2); 127.4 (C-4+C-6); 148.6 (C-5); 165.6 (C-8); 42.7 (C-9); 37.2 (C-12). MS (m/z): 229 (M+H)+. Anal. calcd. for C9H9ClN2O3: C, 47.28; H, 3.97; N, 12.25. Found: C, 47.26; H, 3.99; Cl, 15.73; N, 12.29.
Method 2
A suspension of N-methyl-4-nitroaniline (20.0 g; 0.131 mol) in ethyl acetate (20 ml) is heated to 60° C. Within 15 min, a solution of chloro acetic anhydride (26.0 g; 0.151 mol) in ethyl acetate (60 ml) is added to this suspension. The resulting solution is then refluxed for 1 h, cooled to 75° C. ° C. and methyl cyclohexane (80 ml) is added. After seeding at 60° C., the suspension is further cooled down to 0° C. and stirred for 1 h. The reaction mixture is filtrated, washed with methyl cyclohexane (40 ml) and the precipitate is dried to afford 25.9 g (83.3%) of the “chloroacetyl” compound as grey crystals.
EXAMPLE 6Synthesis of the “nitroaniline” (N-(4-nitrophenyl)-N-methyl-2-(4-methylpiperazin-1-yl)acetamide) and of the “aniline” (N-(4-aminophenyl)-N-methyl-2-(4-methylpiperazin-1-yl)acetamide)

Method 1
A suspension of N-(4-nitroanilino)-N-methyl-2-chloro-acetamide (20.0 g; 0.087 mol) in toluene (110 ml) is heated to 40° C. Within 30 min, 1-methylpiperazine (21.9 g; 0.216 mol) is added dropwise. After purging of the dropping funnel with toluene (5 ml) the reaction mixture is stirred for 2 h at 55° C., cooled to ambient temperature and washed with water (15 ml). The organic layer is diluted with isopropanol (100 ml) and Pd/C (10%; 1.0 g) is added. After subsequent hydrogenation (H2, 4 bar) at 20° C. the catalyst is removed. Approximately ⅘ of the volume of the resulting solution is evaporated at 50° C. The remaining residue is dissolved in ethyl acetate (20 ml) and toluene (147 ml) heated to 80° C., then cooled to 55° C. and seeded. The reaction mixture is further cooled to 0° C. and stirred for 3 h at the same temperature. After filtration, the solid is washed with ice cold toluene (40 ml) and dried to afford 20.2 g (88.0%) of the “aniline” compound as white crystals. 1H-NMR (500 MHz, DMSO-d6) δ: 6.90 (d, J=8.5 Hz, 2H, 4-H+6-H); 6.65 (d, J=8.5 Hz, 2H, 1-H+3-H); 5.22 (2H, 19-H2); 3.04 (s, 3H, 9-H3); 2.79 (s, 2H, 11-H2); 2.32 (m, 4H, 13-H2+17-H2); 2.23 (m, 4H, 14-H2+16-H2); 2.10 (s, 3H, 18-H3). 13C-NMR (126 MHz, DMSO-d6) δ: 114.0 (C-1+C-3); 148.0 (C-2); 127.6 (C-4+C-6); 131.5 (C-5); 168.9 (C-8); 36.9 (C-9); 58.5 (C-11); 52.4 (C-13+C-17); 54.6 (C-14+C-16); 45.7 (C-18). MS (m/z): 263 (M+H)+. Anal. calcd. for C14H22N4O: C, 64.09; H, 8.45; N, 21.36. Found: C, 64.05; H, 8.43; N, 21.39.
Method 2
A suspension of N-(4-nitroanilino)-N-methyl-2-chloro-acetamide (14.5 g; 0.063 mol) in ethyl acetate (65 ml) is heated to 40° C. Within 30 min, 1-methylpiperazine (15.8 g; 0.156 mol) is added dropwise. After purging of the dropping funnel with ethyl acetate (7 ml) the reaction mixture is stirred at 50° C. for 90 min, cooled to ambient temperature and washed with water (7 ml). The organic layer is diluted with isopropanol (75 ml) and dried over sodium sulphate. After separation of the solid, Pd/C (10%; 2.0 g) is added and the solution is hydrogenated (H2, 5 bar) at ambient temperature without cooling. Subsequently the catalyst is removed by filtration and the solvent is evaporated at 60° C. The remaining residue is dissolved in ethyl acetate (250 ml) and recrystallized. After filtration and drying 10.4 g (60.4%) of the “aniline” compound are isolated as white crystals.
EXAMPLE 7Synthesis of the “anilino” (3-Z-[1-(4-(N-((4-methyl-piperazin-1-yl)-methylcarbonyl)-N-methyl-amino)-anilino)-1-phenyl-methylene]-6-methoxycarbonyl-2-indolinone)

Method 1
A suspension of methyl-3-[methoxy(phenyl)methylene]-2-oxoindoline-6-carboxylate (10.0 g; 0.032 mol) and N-(4-aminophenyl)-N-methyl-2-(4-methylpiperazin-1-yl)acetamide (8.6 g; 0.032 mol) in a mixture of methanol (72 ml) and N,N-dimethylformamide (18 ml) is heated to reflux. After 7 h of refluxing the suspension is cooled down to 0° C. and stirring is maintained for additional 2 h. The solid is filtered, washed with methanol (40 ml) and dried to afford 15.4 g (88.1%) of the “anilino” compound as yellow crystals. 1H-NMR (500 MHz, DMSO-d6) δ: 11.00 (s, 1H, 23-H); 12.23 (s, 19-H); 7.61 (t; J=7.1 Hz, 1H, 33-H); 7.57 (t, J=7.5 Hz, 2H, 32-H+34-H); 7.50 (d, J=7.7 Hz, 2H, 31-H+35-H); 7.43 (d, J=1.6 Hz, 1H, 29-H); 7.20 (dd, J=8.3; 1.6 Hz, 1H, 27-H); 7.13 (d, J=8.3 Hz, 2H, 14-H+18-H); 6.89 (d, 8.3 Hz, 2H, 15-H+17-H); 5.84 (d, J=8.3 Hz, 1H, 26-H); 3.77 (s, 3H, 40-H3); 3.06 (m, 3H, 12-H3); 2.70 (m, 2 H, 8-H2); 2.19 (m, 8H, 2-H2, 3-H2, 5-H2, 6-H2); 2.11 (s, 3H, 7-H3). 13C-NMR (126 MHz, DMSO-d6) δ: 54.5 (C-2+C-6); 52.2 (C-3+C-5); 45.6 (C-7); 59.1 (C-8); 168.5 (C-9); 36.6 (C-12); 140.1 (C-13); 127.6 (C-14+C-18); 123.8 (C-17+C-15); 137.0 (C-16); 158.3 (C-20); 97.5 (C-21); 170.1 (C-22); 136.2 (C-24); 128.9 (C-25); 117.2 (C-26); 121.4 (C-27); 124.0 (C-28); 109.4 (C-29); 131.9 (C-30); 128.4 (C-31+C-35); 129.4 (C-32+C-34); 130.4 (C-33); 166.3 (C-37); 51.7 (C-40). MS (m/z): 540 (M+H)+. Anal. calcd. for C31H33N5O4: C, 69.00; H, 6.16; N, 12.98. Found: C, 68.05; H, 6.21; N, 12.81.
Method 2
A suspension of methyl-3-[methoxy(phenyl)methylene]-2-oxoindoline-6-carboxylate (20.0 g; 0.064 mol) and N-(4-aminophenyl)-N-methyl-2-(4-methylpiperazin-1-yl)acetamide (17.1 g; 0.065 mol) in methanol (180 ml) is heated to reflux for 7.5 h. The resulting suspension is cooled down to 10° C. within 1 h and stirring is maintained for 1 h. After filtration, the solid is washed with ice cold methanol (80 ml) and dried to afford 31.0 g (89.0%) of the “anilino” compound as yellow crystals.
EXAMPLE 8Synthesis of the 3-Z-[1-(4-(N-((4-methyl-piperazin-1-yl)-methylcarbonyl)-N-methyl-amino)-anilino)-1-phenyl-methylene]-6-methoxycarbonyl-2-indolinone, monoethanesulfonate

A suspension of 3-Z-[1-(4-(N-((4-methyl-piperazin-1-yl)-methylcarbonyl)-N-methyl-amino)-anilino)-1-phenyl-methylene]-6-methoxycarbonyl-2-indolinone (30.0 g; 0.055 mol) in methanol (200 ml) and water (2.4 ml) is heated to 60° C. Aqueous ethanesulfonic acid (70% (w/w); 8.75 g; 0.056 mol) is added to the reaction mixture. The resulting solution is cooled to 50° C., seeded and then diluted with isopropanol (200 ml). The mixture is further cooled to 0° C. and stirred for 2 h at this temperature. The precipitate is isolated, washed with isopropanol (120 ml) and dried to furnish 35.1 g (97.3%) of the monoethanesulfonate salt of the compound as yellow crystals. 1H-NMR (400 MHz, DMSO-d6) δ: 12.26 (s, 11-H); 10.79 (s, 1H, 1-H); 9.44 (s, 1H, 24-H); 7.64 (m, 1H, 32-H); 7.59 (m, 2H, 31-H+33-H); 7.52 (m, 2H, 30-H+34-H); 7.45 (d, J=1.6 Hz, 1H, 7-H); 7.20 (dd, J=8.2; 1.6 Hz, 1H, 5-H); 7.16 (m, 2H, 14-H+16-H); 6.90 (m, 2H, 13-H+17-H); 5.85 (d, J=8.2 Hz, 1H, 4-H); 3.78 (s, 3H, 37-H3); 3.45-2.80 (broad m, 4H, 23-H2+25-H2); 3.08 (s, 3H, 28-H3); 2.88 (s, 2H, 20-H2); 2.85-2.30 (broad m, 4H, 22-H2+26-H2); 2.75 (s, 3H, 27-H3); 2.44 (q, J=7.4 Hz, 2H, 39-H2); 1.09 (t, J=7.4 Hz, 3H, 38-H3). 13C-NMR (126 MHz, DMSO-d6) δ: 9.8 (C-38); 36.6 (C-28); 42.3 (C-27); 45.1 (C-39); 51.7 (C-37); 48.9 (C-22+C-26); 52.6 (C-23+C-25); 57.5 (C-20); 97.7 (C-3); 109.5 (C-7); 117.3 (C-4); 121.4 (C-5); 123.8 (C-13+C-17); 124.1 (C-6); 127.7 (C-14+C-16); 128.4 (C-30+C-34); 128.8 (C-9); 129.5 (C-31+C-33); 130.5 (C-32); 132.0 (C-29); 168.5 (C-9); 136.3 (C-8); 137.3 (C-12); 139.5 (C-15); 158.1 (C-10); 166.3 (C-35); 168.0 (C-19); 170.1 (C-2). MS (m/z): 540 (M(base)+H)+. Anal. calcd. for C33H39N5O7S: C, 60.17; H, 6.12; N, 10.63; S, 4.87. Found: C, 60.40; H, 6.15; N, 10.70; S, 4.84.
CLIPS

After a classical malonic ester addition to arene 3, the resulting nitro benzene (4) is hydrogenated under acidic conditions, furnishing the 6-methoxycarbonyl-substituted oxindole 5 via decarboxylative cyclization. Condensation of 5 with trimethyl orthobenzoate in acetic anhydride leads to compound 6, one of the two key building blocks of the synthesis. The concomitant N-acetylation of the oxindole activates the scaffold for the condensation reaction.
The aniline side chain (9) can be prepared by a one-pot bromo-acetylation/amination of the para-nitro-phenylamine (7) using bromoacetyl bromide and N-methylpiperazine and a subsequent hydrogenation furnishing 9 as the second key building block. Condensation of both building blocks in an addition–elimination sequence and subsequent acetyl removal with piperidine furnishes 2 as free base (pKa = 7.9), which subsequently is converted into its monoethanesulfonate salt (1). Compound 1 is highly crystalline (mp = 305 °C) and exhibits a log P of 3.0 and good aqueous solubility (>20 mg/mL in water).
CLIPS
see
http://www.yaopha.com/2014/07/09/synthesis-of-vargatef-nintedanib-boehringer-ingelheim-idiopathic-pulmonary-fibrosis-drug/

CLICK ON PIC
Updates………..
“J.Med.Chem” 2009 Vol. 52, page 4466-4480 and the “Chinese Journal of Pharmaceuticals” 2012, Vol. 43, No. 9, page 726-729 reported a further intermediate A and B synthesis, and optimized from the reaction conditions, the reaction sequence, the feed ratio and catalyst selection, etc., so that the above-described synthetic routes can be simplified and reasonable.
PATENT
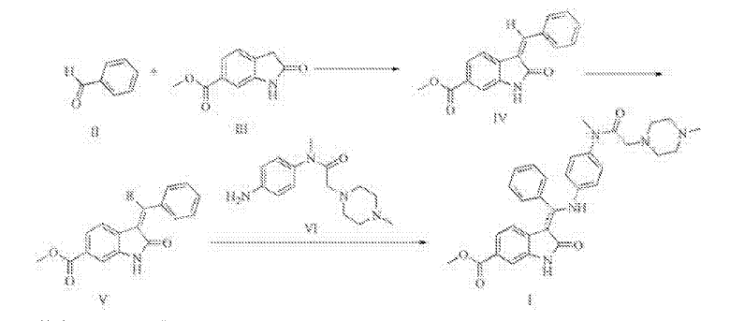
MACHINE TRANSLATED FROM CHINESE
Synthesis of Trinidad Neeb (I),
A 500ml reaction flask was charged 30g of compound V, 22.5g compound of the VI, ethanol 300ml, sodium bicarbonate and 15g, the reaction was heated to reflux for 2 hours, the reaction mixture was added to 600ml of water, there are large amount of solid precipitated, was filtered, the cake washed with 100ml washed once with methanol, a yellow solid 41.9g refined Trinidad Neeb (I). Yield 92.7%.
4 bandit R (400MHz, dmso) δ11 · 97 (s, 1H), 8.38 (s, 1H), 7.97 (dd, J = 11.9, 5.0Hz, 2H), 7.67 (d, J = 8.1Hz, 1H), 7.16 (ddd, J = 26.9, 22.1, 7.0Hz, 5H), 6.85 (d, J = 8.6Hz, 2H), 6.63 (d, J = 8.7Hz, 2H), 3.90 (s, 3H), 2.99 (s, 3H), 2.69 (s, 2H), 2.51-2.24 (m, 8H), 2.20 (s, 3H) MS:. m / z540 (m + 1) + 2 Example: Preparation of compound IV 250ml reaction flask was added 28.7g of 2- oxindole-6-carboxylate, 130ml ethanol, stirred open, then added 30.3ml (31.8g) benzaldehyde, 2.97 mL piperidine was heated to 70 ° C-80 after ° C for 2 hours, allowed to cool to 20 ° C- 30 ° C, the precipitate was filtered, the filter cake was washed with absolute ethanol, 50 ° C 5 hours and dried in vacuo give a yellow solid 38.7g (IV of), yield: 92.4% Preparation of compound V square in 500ml reaction flask was added 30g compound IV, dichloromethane 360ml, cooled with ice water to 0-5 ° C, 71/92 bromine 3.lml (9.7g), drop finished warmed to 20- 30 ° C, 3 hours after the reaction, the reaction solution was washed once with 150ml dichloromethane layer was concentrated oil was done by adding 200ml ethanol crystallization, filtration, 60 ° C and dried under vacuum 36.lg white solid (V ), yield: 93 · 8%.
After Trinidad Technip (I) are synthesized in the reaction flask was added 500ml of 30g compound V, 33.0g compound of the VI, ethanol 300ml, sodium bicarbonate, 15g, was heated to reflux for 2 hours, the reaction mixture was added to 600ml water, there are large amount of solid precipitated, was filtered, the filter cake washed once with 100ml methanol obtained 42.3g of yellow solid was purified by Technip Trinidad (I). Yield 93.6%.
ΧΗNMR (400MHz, dmso) δ11.94 (s, 1Η), 8.36 (s, 1H), 7.96 (dd, J = 11.9, 5.0Hz, 2H), 7.67 (d, J = 8.1Hz, 1H) , 7.16 (ddd, J = 26.9, 22.1, 7.0Hz, 5H), 6.85 (d, J = 8.6Hz, 2H), 6.61 (d, J = 8.7Hz, 2H), 3.90 (s, 3H), 2.99 ( s, 3H), 2.65 (s, 2H), 2.50-2.30 (m, 8H), 2.20 (s, 3H) MS:. m / z540 (m + 1) + square
PATENT
(I) 2.30g, yield 85.3%. Melting point 241 ~ 243 ℃, Mass spectrum (the EI): m / Z 540 (the M + the H), 1 the H NMR (of DMSO D . 6 ): 2.27 (S, 3H), 2.43 (m, 8H), 2.78 (S, 2H) , 3.15 (s, 3H), 3.82 (s, 3H), 5.97 (d, J = 8.3Hz, 1H), 6.77 (d, J = 8.7Hz, 1H), 6.96 (d, J = 8.6Hz, 2H) , 7.32-7.62 (m, 8H), 8.15 (s, 1H), 12.15 (s, 1H).
CLIPS
http://pubs.rsc.org/en/content/articlelanding/2015/ay/c5ay01207d#!divAbstract


Nintedanib
|
|
| Systematic (IUPAC) name | |
|---|---|
| Methyl (3Z)-3-{[(4-{methyl[(4-methylpiperazin-1-yl)acetyl]amino}phenyl)amino](phenyl)methylidene}-2-oxo-2,3-dihydro-1H-indole-6-carboxylate | |
| Clinical data | |
| Trade names | Vargatef, Ofev |
| AHFS/Drugs.com | Consumer Drug Information |
| Pregnancy cat. |
|
| Legal status | |
| Routes | Oral and intravenous |
| Identifiers | |
| CAS number | 656247-17-5 |
| ATC code | None |
| Chemical data | |
| Formula | C31H33N5O4 |
| Mol. mass | 539.6248 g/mol |
References
- Hilberg, F.; G. J. Roth, M. Krssak, S. Kautschitsch, W. Sommergruber, U. Tontsch-Grunt, P. Garin-Chesa, G. Bader, A. Zoephel, J. Quant, A. Heckel, W. J. Rettig (2008). “BIBF 1120: triple angiokinase inhibitor with sustained receptor blockade and good antitumor efficacy”. Cancer Res 68 (12): 4774–82. doi:10.1158/0008-5472.CAN-07-6307. ISSN 1538-7445. PMID 18559524.
- Hilberg, F.; U. Tontsch-Grunt, F. Colbatzky, A. Heckel, R. Lotz, J.C.A. van Meel, G.J. Roth (2004). “BIBF1120 a novel, small molecule triple angiokinase inhibitor: profiling as a clinical candidate for cancer therapy”. European Journal of Cancer Supplements 2 (50).
- Reck, M.; R. Kaiser; C. Eschbach; M. Stefanic; J. Love; U. Gatzemeier; P. Stopfer; J. von Pawel (2011). “A phase II double-blind study to investigate efficacy and safety of two doses of the triple angiokinase inhibitor BIBF 1120 in patients with relapsed advanced non-small-cell lung cancer”. Ann Oncol. ISSN 1569-8041.
- Okamoto, I.; H. Kaneda, T. Satoh, W. Okamoto, M. Miyazaki, R. Morinaga, S. Ueda, M. Terashima, A. Tsuya, A. Sarashina, K. Konishi, T. Arao, K. Nishio, R. Kaiser, K. Nakagawa (2010). “Phase I safety, pharmacokinetic, and biomarker study of BIBF 1120, an oral triple tyrosine kinase inhibitor in patients with advanced solid tumors”. Mol Cancer Ther 9 (10): 2825–33. doi:10.1158/1535-7163.MCT-10-0379. ISSN 1538-8514. PMID 20688946.
- Mross, K.; M. Stefanic, D. Gmehling, A. Frost, F. Baas, C. Unger, R. Strecker, J. Henning, B. Gaschler-Markefski, P. Stopfer, L. de Rossi, R. Kaiser (2010). “Phase I study of the angiogenesis inhibitor BIBF 1120 in patients with advanced solid tumors”. Clin Cancer Res 16 (1): 311–9. doi:10.1158/1078-0432.CCR-09-0694. ISSN 1078-0432. PMID 20028771.
- Ledermann, J.A. (2009). “A randomised phase II placebo-controlled trial using maintenance therapy to evaluate the vascular targeting agent BIBF 1120 following treatment of relapsed ovarian cancer (OC)”. J Clin Oncol 27 (15s): (suppl; abstr 5501).
- Kropff, M.; J. Kienast; G. Bisping; W. E. Berdel; B. Gaschler-Markefski; P. Stopfer; M. Stefanic; G. Munzert (2009). “An open-label dose-escalation study of BIBF 1120 in patients with relapsed or refractory multiple myeloma”. Anticancer Res 29 (10): 4233–8. ISSN 1791-7530. PMID 19846979.
- Ellis, P. M.; R. Kaiser; Y. Zhao; P. Stopfer; S. Gyorffy; N. Hanna (2010). “Phase I open-label study of continuous treatment with BIBF 1120, a triple angiokinase inhibitor, and pemetrexed in pretreated non-small cell lung cancer patients”. Clin Cancer Res 16 (10): 2881–9. doi:10.1158/1078-0432.CCR-09-2944. ISSN 1078-0432. PMID 20460487.
- du Bois, A.; J. Huober; P. Stopfer; J. Pfisterer; P. Wimberger; S. Loibl; V. L. Reichardt; P. Harter (2010). “A phase I open-label dose-escalation study of oral BIBF 1120 combined with standard paclitaxel and carboplatin in patients with advanced gynecological malignancies”. Ann Oncol 21 (2): 370–5. doi:10.1093/annonc/mdp506. ISSN 1569-8041. PMID 19889612.
- Xiang, Q. F.; F. Wang; X. D. Su; Y. J. Liang; L. S. Zheng; Y. J. Mi; W. Q. Chen; L. W. Fu (2011). “Effect of BIBF 1120 on reversal of ABCB1-mediated multidrug resistance”. Cell Oncol (Dordr) 34 (1): 33–44. doi:10.1007/s13402-010-0003-7. ISSN 2211-3436.
- “Boehringer Ingelheim – AGO-OVAR 12 / LUME-Ovar 1 Trial Information”. 2011.
- “Boehringer Ingelheim – LUME-Lung 2 Trial Information”. 2011.
- “Boehringer Ingelheim – LUME-Lung 1 Trial Information”. 2011.
- http://clinicaltrials.gov/ct2/results?term=++%09+BIBF+1120&phase=1
- http://clinicaltrials.gov/ct2/show/NCT00805194 Phase III LUME-Lung 1: BIBF 1120 Plus Docetaxel as Compared to Placebo Plus Docetaxel in 2nd Line Non Small Cell Lung Cancer
- http://clinicaltrials.gov/ct2/show/NCT01015118 Phase III BIBF 1120 or Placebo in Combination With Paclitaxel and Carboplatin in First Line Treatment of Ovarian Cancer
- http://clinicaltrials.gov/ct2/show/NCT01335477 Safety and Efficacy of BIBF 1120 at High Dose in Idiopathic Pulmonary Fibrosis Patients II
- “FDA approves Ofev to treat idiopathic pulmonary fibrosis”. 2014.
-
F. Hilberg et al. Cancer Res. 2008, 68, 4774
2. M. Reck et al. Ann. Oncol. 2011, 22, 1374
3. M. Reck et al. J. Clin. Oncol. 2013 (suppl.), Abst LBA8011
4. N. H. Hanna et al. J. Clin. Oncol. 2013, 2013 (suppl.), Abst 8034
5. J.A. Ledermann et al. J. Clin Oncol. 2011, 29, 3798
6. Glioblastoma: A. Muhac et al. J. Neurooncol. 2013, 111, 205
7. O. Bouche et al. Anticancer Res. 2011, 31, 2271
8. T. Eisen et al. J. Clin. Oncol. 2013 (suppl.), Abst. 4506
MORE…………….
Reference:
[6]. Japan PMDA.
[7]. Drug@FDA, NDA205832 Pharmacology Review(s).
[8]. Med. Chem. 2015, 58, 1053-1063.
[9]. Drug@EMA, EMEA/H/C/002569 Vargatef: EPAR-Assessment Report.
[10]. Drug Des. Devel. Ther. 2015, 9, 6407-6419.
[11]. Cancer Res. 2008, 68, 4774-4782.
[12]. J. Med. Chem. 2009, 52, 4466-4480.
Merten, J.; et. al. Process for the manufacture of an indolinone derivative. US20110201812A1
2. Roth, G. J.; et. al. 3-z-[1-(4-(n-((4-methyl-piperazin-1-yl)-methylcarbonyl)-n-methyl-amino)-anilino)-1-phenyl-methylene]-6-methoxycarbonyl-2-indolinone-monoethanesulphonate and the use thereof as a pharmaceutical composition. WO2004013099A1
3. Roth, G. J.; et. al. Design, Synthesis, and Evaluation of Indolinones as Triple Angiokinase Inhibitors and the Discovery of a Highly Specific 6-Methoxycarbonyl-Substituted Indolinone (BIBF 1120). J Med Chem, 2009, 52(14), 4466-4480. -

ニンテダニブエタンスルホン酸塩
Nintedanib Ethanesulfonate
C31H33N5O4.C2H6O3S : 649.76
[656247-18-6]US7119093 * Jul 21, 2003 Oct 10, 2006 Boehringer Ingelheim Pharma Gmbh & Co. Kg 3-Z-[1-(4-(N-((4-Methyl-piperazin-1-yl)-methylcarbonyl)-N-methyl-amino)-anilino)-1-phenyl-methylene]-6-methoxycarbonyl-2-indolinone-monoethanesulphonate and the use thereof as a pharmaceutical composition ///////////////
FDA Approves Blincyto (blinatumomab) for Precursor B-Cell Acute Lymphoblastic Leukemia
 Blinatumomab linking a T cell to a malignant B cell.
Blinatumomab linking a T cell to a malignant B cell.
FDA Approves Blincyto (blinatumomab) for Precursor B-Cell Acute Lymphoblastic Leukemia
December 3, 2014 — The U.S. Food and Drug Administration today
approved Blincyto (blinatumomab) to treat patients with Philadelphia
chromosome-negative precursor B-cell acute lymphoblastic leukemia
(B-cell ALL), an uncommon form of ALL.

Blinatumomab (AMG103) is a drug that has anti-cancer properties. It belongs to a new class of constructed monoclonal antibodies,bi-specific T-cell engagers (BiTEs), that exert action selectively and direct the human immune system to act against tumor cells. Blinatumomab specifically targets the CD19 antigen present on B cells.[1]
The drug was developed by a German-American company Micromet, Inc. in cooperation with Lonza; Micromet was later purchases by Amgen, which has furthered the drug’s clinical trials. In July 2014, the FDA granted breakthrough therapy status to blinatumomab for the treatment of acute lymphoblastic leukemia (ALL).[2] In October 2014, Amgen’s Biologics License Application for blinatumomab was granted priority review designation by the FDA, thus establishing a deadline of May 19, 2015 for completion of the FDA review process.[3]
Structure and mechanism of action
Blinatumomab linking a T cell to a malignant B cell.
Blinatumomab enables a patient’s T cells to recognize malignant B cells. A molecule of blinatumomab combines two binding sites: a CD3site for T cells and a CD19 site for the target B cells. CD3 is part of the T cell receptor. The drug works by linking these two cell types andactivating the T cell to exert cytotoxic activity on the target cell.[4] CD3 and CD19 are expressed in both pediatric and adult patients, making blinatumomab a potential therapeutic option for both pediatric and adult populations.[5]
Therapeutic use
Clinical trials
In a phase 1 clinical study with blinatumomab, patients with non-Hodgkin’s lymphoma showed tumor regression, and in some cases complete remission.[6] There are ongoing phase 1 and phase 2 clinical trials of blinatumomab in patients with acute lymphoblastic leukemia (ALL).[7] One phase II trial for ALL reported good results in 2010 and another is starting.[8]
Adverse effects
Common side effects observed in Phase 2 trials are listed below; they were temporary and typically occurred during the first treatment cycle:[5]
- Flu-like symptoms (i.e. fever, headache, and fatigue)
- Tremor
- Weight increase
- Hypokalemia
- Decrease of blood immunoglobulin
CNS effects were also observed during clinical trials and were treated via a lower dose of blinatumomab, administration of dexamethasone, or treatment discontinuation. Because the side effects were reversible, early monitoring for the CNS symptoms listed below is important:[5]
- Seizure
- Encephalopathy
- Tremor
- Apraxia
- Speech disorders
- Disorientation
Less common side effects include cytokine release syndrome and immunogenicity.[5]
References
- Statement on a Nonproprietary Name adopted by the USAN Council: Blinatumomab
- Amgen Receives FDA Breakthrough Therapy Designation For Investigational BiTE® Antibody Blinatumomab In Acute Lymphoblastic Leukemia
- Amgen’s BiTE® Immunotherapy Blinatumomab Receives FDA Priority Review Designation In Acute Lymphoblastic Leukemia
- Mølhøj, M; Crommer, S; Brischwein, K; Rau, D; Sriskandarajah, M; Hoffmann, P; Kufer, P; Hofmeister, R; Baeuerle, PA (March 2007). “CD19-/CD3-bispecific antibody of the BiTE class is far superior to tandem diabody with respect to redirected tumor cell lysis”. Mol Immunol 44 (8): 1935–43. doi:10.1016/j.molimm.2006.09.032. PMID 17083975.
- Background Information for the Pediatric Subcommittee of the Oncologic Drugs Advisory Committee Meeting 04 December 2012
- Bargou, R; et al. (2008). “Tumor regression in cancer patients by very low doses of a T cell-engaging antibody”. Science 321 (5891): 974–977. doi:10.1126/science.1158545.PMID 18703743.
- ClinicalTrials.gov NCT00560794 Phase II Study of the BiTE Blinatumomab (MT103) in Patients With Minimal Residual Disease of B-precursor Acute ALL
- “Micromet initiates MT103 phase 2 trial in adult ALL patients”. 20 Sep 2010.
External links
| Monoclonal antibody | |
|---|---|
| Type | Bi-specific T-cell engager |
| Source | Mouse |
| Target | CD19, CD3 |
| Clinical data | |
| Legal status |
?
|
| Identifiers | |
| CAS number | 853426-35-4 |
| ATC code | None |
| UNII | 4FR53SIF3A |
| Chemical data | |
| Formula | C2367H3577N649O772S19 |
| Mol. mass | 54.1 kDa |
The U.S. Food and Drug Administration approved Ofev (nintedanib) for the treatment of idiopathic pulmonary fibrosis (IPF).
Nintedanib
FDA approves Ofev to treat idiopathic pulmonary fibrosis
10/15/2014
The U.S. Food and Drug Administration today approved Ofev (nintedanib) for the treatment of idiopathic pulmonary fibrosis (IPF).
read at
see synthesis
https://newdrugapprovals.org/2014/05/21/in-battle-of-ipf-drugs-bis-nintedanib-impresses/

FDA approves Esbriet (pirfenidone ピルフェニドン 吡非尼酮) to treat idiopathic pulmonary fibrosis

The U.S. Food and Drug Administration today approved Esbriet (pirfenidone)
ピルフェニドン 吡非尼酮
for the treatment of idiopathic pulmonary fibrosis (IPF).
read at
SYNTHESIS
Click for synthesis



//////////
FDA Approves Spiriva Respimat (tiotropium) for the Maintenance Treatment of COPD

Ridgefield, Conn., September 25, 2014 – Boehringer Ingelheim Pharmaceuticals, Inc. announced today that the U.S. Food and Drug Administration (FDA) approved Spiriva Respimat (tiotropium bromide) inhalation spray for the long-term, once-daily maintenance treatment of bronchospasm associated with chronic obstructive pulmonary disease (COPD), including chronic bronchitis and emphysema and to reduce exacerbations in COPD patients. Boehringer Ingelheim anticipates Spiriva Respimat to be available in January 2015.

Spiriva Respimat provides a pre-measured amount of medicine in a slow-moving mist that helps patients inhale the medicine. Spiriva Respimat was developed to actively deliver medication in a way that does not depend of how fast air is breathed in from the inhaler.
READ AT




MAKE IN INDIA
FDA Approves Tybost (cobicistat) for use in the treatment of HIV-1 Infection

Cobicistat, GS-9350
1004316-88-4
| C 40 H 53 N 7 O 5 S 2 |
N-[1(R)-Benzyl-4(R)-[2(S)-[3-(2-isopropylthiazol-4-ylmethyl)-3-methyl]ureido]-4-(4-morpholinyl)butyramido]-5-phenylpentyl]carbamic acid thiazol-5-ylmethyl ester
(1,3-thiazol-5-yl) methyl (5S, 8R, 11R) -8,11-dibenzyl-2-methyl-5-[2 – (morpholin-4-yl) ethyl] -1 – [2 – (propan-2-yl) -1,3-thiazol-4-yl] -3,6-dioxo-2 ,4,7,12-tetraazatridecan-13-oate
cytochrome P450 3A4 (CYP3A4) inhibitor
FDA Approves Tybost (cobicistat) for use in the treatment of HIV-1 Infection
September 24, 2014 — The U.S. Food and Drug Administration (FDA) has approved Tybost (cobicistat), a CYP3A inhibitor used in combination with atazanavir or darunavir for the treatment of human immunodeficiency virus type 1 (HIV-1) infection
Cobicistat is a pharmacokinetic enhancer that works by inhibiting the enzyme (CYP3A) that metabolizes atazanavir and darunavir. It increases the systemic exposure of these drugs and prolongs their effect. Cobicistat is also one of the ingredients in the combination HIV drug Stribild, which was approved by the FDA in August, 2012.
Tybost comes in 150 mg tablets and is administered once daily in combination with the protease inhibitors atazanavir (Reyataz), or darunavir (Prezista).
Because Tybost inhibits CYP3A, other medications metabolized by CYP3A may result in increased plasma concentrations and potentially severe side effects, which may be life-threatening or even fatal. Extra care should be exercised by healthcare professionals to ensure than other medications are reviewed and their concentrations monitored, especially when initiating new medicines or changing doses.
The approval of Tybost was based on the following clinical trials:
•The data to support the use of atazanavir and Tybost were from a phase 2 and 3 trial in treatment-naïve adults comparing atazanavir/cobicistat 300/150 mg and atazanavir/ritonavir 300/100 mg once daily each in combination with Truvada. The atazanavir/cobicistat based regimen was non-inferior to the atazanavir/ritonavir based regimen.
•The data to support the use of cobicistat with darunavir is from a multiple dose trial in healthy subjects comparing the relative bioavailability of darunavir/cobicistat 800/150 mg to darunavir/ritonavir 800/100 mg.
The most common adverse drug reactions observed with Tybost (in combination with atazanavir) in clinical trials were jaundice, ocular icterus, and nausea.
Tybost is a product of Gilead Sciences, Foster City, CA.

Cobicistat (formerly GS-9350) is a licensed drug for use in the treatment of infection with the human immunodeficiency virus (HIV).
Like ritonavir (Norvir), cobicistat is of interest not for its anti-HIV properties, but rather its ability to inhibit liver enzymes that metabolize other medications used to treat HIV, notablyelvitegravir, an HIV integrase inhibitor currently under investigation itself. By combining cobicistat with elvitegravir, higher concentrations of elvitgravir are achieved in the body with lower dosing, theoretically enhancing elvitgravir’s viral suppression while diminishing its adverse side-effects. In contrast with ritonavir, the only currently approved booster, cobicistat has no anti-HIV activity of its own.[1]
Cobicistat, a cytochrome P450 CYP3A4 inhibitor, was approved in the E.U. in 2013 as a pharmacokinetic enhancer of the HIV-1 protease inhibitors atazanavir and darunavir in adults. First launch took place in 2014 in United Kingdom. In 2012, Gilead filed a New Drug Application in the U.S. for the same indication. In April 2013, the FDA issued a Complete Response Letter from the FDA. In 2014 the FDA accepted Gilead’s resubmission.
Cobicistat is a component of the four-drug, fixed-dose combination HIV treatmentelvitegravir/cobicistat/emtricitabine/tenofovir (known as the “Quad Pill” or Stribild).[1][2] The Quad Pill/Stribild was approved by the FDA in August 2012 for use in the United States and is owned by Gilead Sciences.
Cobicistat is a potent inhibitor of cytochrome P450 3A enzymes, including the importantCYP3A4 subtype. It also inhibits intestinal transport proteins, increasing the overall absorption of several HIV medications, including atazanavir, darunavir and tenofovir alafenamide fumarate.[3]
The drug candidate acts as a pharmaco-enhancer to boost exposure of HIV protease inhibitors. In 2011, cobicistat was licensed to Japan Tobacco by Gilead for development and commercialization in Japan as a stand-alone product for the treatment of HIV infection. In 2012, orphan drug designation was assigned in Japan for the pharmacokinetic enhancement of anti-HIV agent.
Oxidative metabolism by cytochrome P450 enzymes is one of the primary mechanisms of drug metabolism.. It can be difficult to maintain therapeutically effective blood plasma levels of drugs which are rapidly metabolized by cytochrome P450 enzymes. Accordingly, the blood plasma levels of drugs which are susceptible to cytochrome P450 enzyme degradation can be maintained or enhanced by co-administration of cytochrome P450 inhibitors, thereby improving the pharmacokinetics of the drug.
While certain drugs are known to inhibit cytochrome P450 enzymes, more and/or improved inhibitors for cytochrome P450 monooxygenase are desirable. Particularly, it would be desirable to have cytochrome P450 monooxygenase inhibitors which do not have appreciable biological activity other than cytochrome P450 inhibition. Such inhibitors can be useful for minimizing undesirable biological activity, e.g., side effects. In addition, it would be desirable to have P450 monooxygenase inhibitors that lack significant or have a reduced level of protease inhibitor activity. Such inhibitors could be useful for enhancing the effectiveness of antiretroviral drugs, while minimizing the possibility of eliciting viral resistance, especially against protease inhibitors.
…………………………….
Cobicistat (GS-9350): A potent and selective inhibitor of human CYP3A as a novel pharmacoenhancer
ACS Med Chem Lett 2010, 1(5): 209
http://pubs.acs.org/doi/abs/10.1021/ml1000257
http://pubs.acs.org/doi/suppl/10.1021/ml1000257/suppl_file/ml1000257_si_001.pdf

Cobicistat (3, GS-9350) is a newly discovered, potent, and selective inhibitor of human cytochrome P450 3A (CYP3A) enzymes. In contrast to ritonavir, 3 is devoid of anti-HIV activity and is thus more suitable for use in boosting anti-HIV drugs without risking selection of potential drug-resistant HIV variants. Compound 3 shows reduced liability for drug interactions and may have potential improvements in tolerability over ritonavir. In addition, 3 has high aqueous solubility and can be readily coformulated with other agents.
1-Benzyl-4-{2-[3-(2-isopropyl-thiazol-4-ylmethyl)-3-methyl-ureido]-4-morpholin-4-yl-butyrylamino}-5-phenyl-pentyl)-carbamic acid thiazol-5-ylmethyl ester (GS-9350)
HPLC (Chiral CelROD-H, Chiral Technologies Inc;heptane/iPrOH = 70/30).
1H NMR (CD3OD)
δ8.98 (1 H, s), 7.82 (1 H, s), 7.25-7.05
(11 H, m), 5.25-5.10 (2 H, m), 4.60-4.50 (2 H, m), 4.21-4.03 (2 H, m), 3.82-3.72 (1
H, m), 3.65-3.65 (4 H, m), 3.35-3.25 (1 H, m), 2.98 (3 H, s), 2.8-2.6 (4 H, m), 2.4-2.2
(6 H, m), 1.95-1.8 (1 H, m), 1.8-1.6 (1 H, m), 1.6-1.4 (4 H, m), 1.42-1.32 (6 H, m).
MS (ESI) m/z: 776.2 (M+H)+.
HRMS calc. for C40H53N7O5S2: 775.355, found: 775.353.
…………………………………
http://www.google.com/patents/CN103694196A?cl=en
CN 103694196
oxidative metabolism by cytochrome P450 enzymes is one of the main mechanisms of drug metabolism, generally by administration of cytochrome P450 inhibitors to maintain or increase the degradation of cytochrome P450 enzymes are sensitive to the drug plasma levels, in order to improve the pharmacokinetics of drugs dynamics, can be used to enhance the effectiveness of anti-retroviral drugs. For example W02008010921 discloses compounds of formula I as a cytochrome P450 monooxygenase specific compounds (Cobicistat):

W02008010921 discloses the synthesis of compounds of formula I with a variety of, as one of the methods of the following routes
Shows:

The reagents used in the method is expensive, and more difficult to remove by-products, long reaction time, high cost, is not conducive to industrial
Production.
W02010115000 on these routes has been improved:

The first step in the route used for the ring-opening reaction reagent trimethylsilyl iodide, trimethylsilyl iodide expensive. W02010115000 reports this step and the subsequent ring-opening reaction of morpholine substitution reaction yield of two steps is not high, only 71%, so that only iodotrimethylsilane a high cost of raw material is not suitable for industrial production.



Preparation of compounds of formula I
Example [0126] Implementation
[0127] I1-a (20g) was dissolved in dichloromethane, was added 50% K0H (5.5g) solution, control the internal temperature does not exceed 25 ° C, TLC analysis ΙΙ-a disappears. Was cooled to O ~ 10 ° C, was added (2R, 5R) -5 – amino-1 ,6 – diphenyl-2 – hexyl-carbamic acid 5 – methyl-thiazole ester hydrochloride (14.8g), stirred for I ~ 2 h, 1 – hydroxybenzotriazole triazole (5.5g), stirred for I h, 1 – ethyl – (3 – dimethylaminopropyl) carbodiimide hydrochloride (15g), and incubated for 5 ~ 10 hours, TLC analysis of the starting material disappeared, the reaction was completed. The reaction was quenched with aqueous acetic acid, methylene chloride layer was separated, washed with saturated aqueous NaHCO3, washed with water, dried and concentrated. By HPLC purity of 99.1%. Adding ethanol, the ethanol was evaporated to give the product compound of part I of a solution in ethanol. Molar yield 88%, LC-MS: M +1 = 777.1 [0128] All publications mentioned in the present invention are incorporated by reference as if each reference was individually incorporated by reference, as cited in the present application. It should also be understood that, after reading the foregoing teachings of the present invention, those skilled in the art that various modifications of the present invention or modifications, and these equivalents falling as defined by the appended claims scope of claims of the present application.
…………………………
US 2014088304
http://www.google.com/patents/US20140088304
International Patent Application Publication Number WO 2008/010921 and International Patent Application Publication Number WO 2008/103949 disclose certain compounds that are reported to be useful to modify the pharmacokinetics of a co-administered drug, e.g. by inhibiting cytochrome P450 monooxygenase. One specific compound identified therein is a compound of the following formula I:

There is currently a need for improved synthetic methods and intermediates that can be used to prepare the compound of formula I and its salts
Schemes 1-4 below.




Preparation of a Compound of Formula IV

Scheme V.
Example 14Preparation of Compound I

To the solution of L-thiazole morpholine ethyl ester oxalate salt XIVa (35.6 kg) in water (66.0 kg) was charged dichloromethane (264 kg), followed by a slow addition of 15 wt % KHCO3 solution (184.8 kg). The resulting mixture was agitated for about 1 hour. The layers were separated and the organic layer was washed with water (132 kg). The organic layer was concentrated under vacuum to dryness. Water (26.5 kg) was charged and the content temperature was adjusted to about 10° C., followed by slow addition of 45% KOH solution (9.8 kg) while maintaining the content temperature at less than or equal to 20° C. The mixture was agitated at less than or equal to 20° C. until the reaction was judged complete by HPLC. The reaction mixture was concentrated under vacuum to dryness and co-evaporated five times with dichloromethane (132 kg each time) under reduced pressure to dryness. Co-evaporation with dichloromethane (132 kg) was continued until the water content was <4% by Karl Fischer titration. Additional dichloromethane (264 kg) was charged and the content temperature was adjusted to −18° C. to −20° C., followed by addition of monocarbamate.HCl salt IXa (26.4 kg). The resulting mixture was agitated at −18° C. to −20° C. for about 1 hour. HOBt (11.4 kg) was charged and the reaction mixture was again agitated at −18° C. to −20° C. for about 1 hour. A pre-cooled solution (−20° C.) of EDC.HCl (21.4 kg) in dichloromethane (396 kg) was added to the reaction mixture while the content temperature was maintained at less than or equal to −20° C. The reaction mixture was agitated at −18° C. to −20° C. until the reaction was judged complete. The content temperature was adjusted to about 3° C. and the reaction mixture quenched with a 10 wt % aqueous citric acid solution (290 kg). The layers were separated and the organic layer was washed once with 15 wt % potassium bicarbonate solution (467 kg) and water (132 kg). The organic layer was concentrated under reduced pressure and then co-evaporated with absolute ethanol.
The product I was isolated as the stock solution in ethanol (35.0 kg product, 76.1% yield).
1H NMR (dDMSO) δ□ 9.05 (s, 1H), 7.85 (s, 1H), 7.52 (d, 1H), 7.25-7.02 (m, 12H), 6.60 (d, 1H), 5.16 (s, 2H), 4.45 (s, 2H), 4.12-4.05 (m, 1H), 3.97-3.85 (m, 1H), 3.68-3.59 (m, 1H), 3.57-3.45 (m, 4H), 3.22 (septets, 1H), 2.88 (s, 3H), 2.70-2.55 (m, 4H), 2.35-2.10 (m, 6H), 1.75 (m, 1H), 1.62 (m, 1H), 1.50-1.30 (m, 4H), 1.32 (d, 6H).
13C NMR (CD3OD) δ 180.54, 174., 160.1, 157.7, 156.9, 153.8, 143.8, 140.1, 140.0, 136.0, 130.53, 130.49, 129.4, 127.4, 127.3, 115.5, 67.7, 58.8, 56.9, 55.9, 54.9, 53.9, 51.6, 49.8, 42.7, 42.0, 35.4, 34.5, 32.4, 32.1, 29.1, 23.7.
Example 13Preparation of L-Thiazole Morpholine Ethyl Ester Oxalate Salt XIVa

To a solution of (L)-thiazole amino lactone XII (33.4 kg) in dichloromethane (89.5 kg) was charged dichloromethane (150 kg) and absolute ethanol (33.4 kg). The content temperature was then adjusted to about 10° C., followed by slow addition of TMSI (78.8 kg) while the content temperature was maintained at less than or equal to 22° C. and agitated until the reaction was judged complete. The content temperature was adjusted to about 10° C., followed by a slow addition of morpholine (49.1 kg) while the content temperature was maintained at less than or equal to 22° C. Once complete, the reaction mixture was filtered to remove morpholine.HI salt and the filter cake was rinsed with two portions of dichloromethane (33.4 kg). The filtrate was washed twice with water (100 kg). The organic layer was concentrated under vacuum to dryness. Acetone (100 kg) was then charged to the concentrate and the solution was concentrated under reduced pressure to dryness. Acetone (233.8 kg) was charged to the concentrate, followed by a slow addition of the solution of oxalic acid (10 kg) in acetone (100 kg). The resulting slurry was refluxed for about 1 hour before cooling down to about 3° C. for isolation. The product XIVa was filtered and rinsed with acetone (66.8 kg) and dried under vacuum at 40° C. to afford a white to off-white solid (40 kg, 71% yield). 1H NMR (CDCl3) δ □7.00 (s, 1H), 6.35 (broad s, 1H), 4.60-4.40 (m, 3H), 4.19 (quartets, 2H), 4.00-3.90 (m, 4H), 3.35-3.10 (m, 7H), 3.00 (s, 3H), 2.40-2.30 (m, 1H), 2.15-2.05 (m, 1H), 1.38 (d, 6H), 1.25 (triplets, 3H).
……………………………………..
W02008010921
http://www.google.co.in/patents/WO2008010921A2?cl=en
Preparation of Example A
Scheme 1

Example A Compound 2
To a solution of Compound 1 (ritonavir) (1.8 g, 2.5 mmol) in 1,2- dichloroethane (15 mL) was added l,l’-thiocarbonyldiimidazole (890 mg, 5.0 mmol). The mixture was heated at 75 SC for 6 hours and cooled to 25 SC. Evaporation under reduced pressure gave a white solid. Purification by flash column chromatography (stationary phase: silica gel; eluent: EtOAc) gave Compound 2 (1.6 g). m/z: 831.1 (M+H)+. Example A
To the refluxing solution of tributyltin hydride (0.78 mL, 2.9 mmol) in toluene (130 mL) was added a solution of Compound 2 (1.6 g, 1.9 mmol) and 2,2′- azobisisobutyronitrile (31 mg, 0.19 mmol) in toluene (30 mL) over 30 minutes. The mixture was heated at 1152C for 6 hours and cooled to 25 BC. Toluene was removed under reduced pressure. Purification by flash column chromatography (stationary phase: silica gel; eluent: hexane/EtOAc = 1/10) gave Example A (560 mg). m/z: 705.2 (M+H)+. 1H-NMR (CDCl3) δ 8.79 (1 H, s), 7.82 (1 H, s), 7.26-7.05 (10 H, m), 6.98 (1 H, s), 6.28 (1 H, m), 6.03 (1 H, m), 5.27 (1 H7 m), 5.23 (2 H, s), 4.45-4.22 (2 H, m), 4.17 (1 H, m), 3.98 (1 H, m), 3.75 (1 H, m), 3.25 (1 H7 m), 2.91 (3 H, s), 2.67 (4 H, m), 2.36 (1 H, m), 1.6-1.2 (10 H, m), 0.85 (6 H, m).
| EP1183026A2 * | 25 May 2000 | 6 Mar 2002 | Abbott Laboratories | Improved pharmaceutical formulations |
| US20060199851 * | 2 Mar 2006 | 7 Sep 2006 | Kempf Dale J | Novel compounds that are useful for improving pharmacokinetics |
| Thiazol-5-ylmethyl N-[1-benzyl-4-[[2-[[(2-isopropylthiazol-4-yl)methyl-methyl-carbamoyl]amino]-4-morpholino-butanoyl]amino]-5-phenyl-pentyl]carbamate | |
| Clinical data | |
|---|---|
| Legal status |
fda approved sept 2014
|
| Identifiers | |
| CAS number | 1004316-88-4 |
| ATC code | V03AX03 |
| PubChem | CID 25151504 |
| ChemSpider | 25084912 |
| UNII | LW2E03M5PG |
| Chemical data | |
| Formula | C40H53N7O5S2 |
| Mol. mass | 776.023 g/mol |
| US7939553 * | Jul 6, 2007 | May 10, 2011 | Gilead Sciences, Inc. | co-administered drug (as HIV protease inhibiting compound, an HIV (non)nucleoside/nucleotide inhibitor of reverse transcriptase, capsid polymerization inhibitor, interferon, ribavirin analog) by inhibiting cytochrome P450 monooxygenase; ureido- or amido-amine derivatives; side effect reduction |
- Highleyman, L.
Elvitegravir “Quad” Single-tablet Regimen Shows Continued HIV Suppression at 48 Weeks
- R Elion, J Gathe, B Rashbaum, and others. The Single-Tablet Regimen of Elvitegravir/Cobicistat/Emtricitabine/Tenofovir Disoproxil Fumarate (EVG/COBI/FTC/TDF; Quad) Maintains a High Rate of Virologic Suppression, and Cobicistat (COBI) is an Effective Pharmacoenhancer Through 48 Weeks. 50th Interscience Conference on Antimicrobial Agents and Chemotherapy (ICAAC 2010). Boston, September 12–15, 2010.
- Lepist, E. -I.; Phan, T. K.; Roy, A.; Tong, L.; MacLennan, K.; Murray, B.; Ray, A. S. (2012). “Cobicistat Boosts the Intestinal Absorption of Transport Substrates, Including HIV Protease Inhibitors and GS-7340, in Vitro”. Antimicrobial Agents and Chemotherapy 56 (10): 5409–5413. doi:10.1128/AAC.01089-12. PMC 3457391. PMID 22850510.
-
Patent No all US
Expiry 5814639 Sep 29, 2015 5814639*PED Mar 29, 2016 5914331 Jul 2, 2017 5914331*PED Jan 2, 2018 5922695 Jul 25, 2017 5922695*PED Jan 25, 2018 5935946 Jul 25, 2017 5935946*PED Jan 25, 2018 5977089 Jul 25, 2017 5977089*PED Jan 25, 2018 6043230 Jul 25, 2017 6043230*PED Jan 25, 2018 6642245 Nov 4, 2020 6642245*PED May 4, 2021 6703396 Mar 9, 2021 6703396*PED Sep 9, 2021 7176220 Nov 20, 2023 7635704 Oct 26, 2026 8148374 Sep 3, 2029
FDA approves AstraZeneca’s constipation drug Movantik
September 16, 2014
The US Food and Drug Administration has approved AstraZeneca’s Movantik for opioid-induced constipation in adults with chronic non-cancer pain.
Movantik (naloxegol), an oral once-a-day treatment licensed from Nektar Therapeutics, belongs to a class of drugs called peripherally-acting mu-opioid receptor antagonists, which are used to decrease the constipating effects of opioids. The drug’s safety and effectiveness were established in two trials of 1,352 participants who had taken opioids for at least four weeks for non-cancer related pain and had opioid-induced constipation.
Read more at: http://www.pharmatimes.com/Article/14-09-16/FDA_approves_AstraZeneca_s_constipation_drug_Movantik.aspx#ixzz3DdGiFse8
Eliquis, Apixaban for the Treatment of Deep Vein Thrombosis and Pulmonary Embolism
Apixaban
CAS 503612-47-3
APPROVALS
EMA————MAY 18, 2011
FDA…………………DEC28, 2012
PMDA………….. DEC25, 2012
CFDA………………JAN 22, 2013

Apixaban, sold under the tradename Eliquis, is a anticoagulant for the treatment of venous thromboembolic events. It is taken by mouth. It is a direct factor Xa inhibitor.
Apixaban was approved in Europe in 2012.[1] It was approved in the U.S. in 2014 for treatment and secondary prophylaxis of deep vein thrombosis (DVT) and pulmonary embolism (PE).[2] It is being developed in a joint venture by Pfizer and Bristol-Myers Squibb.[3][4]


INVENTIVE
Donald Pinto (left) and Michael Orwat (right) work on developing new products for BMS.
Credit: Bristol-Myers Squibb
Ruth R. Wexler, executive director of cardiovascular diseases chemistry at Bristol-Myers Squibb, who led the group that designed and synthesized Eliquis (apixaban) to reduce the risk of stroke in patients with an abnormal heart rhythm called atrial fibrillation, recalls hearing about the drug’s success in late-stage clinical trials for the first time.
“I was at the European Society of Cardiology meeting when the results of ARISTOTLE, our large Phase 3 trial, were announced,” she says. “I was sitting in the audience, and it was just amazing to see the data released for the first time. It blew my mind that the data was that spectacular.”
In the trial, which compared apixaban with the workhorse anticoagulant Coumadin (warfarin), apixaban reduced the risk of stroke in patients with atrial fibrillation by 21%, major bleeding by 31%, and mortality by 11%. Unlike Coumadin, apixaban doesn’t require regular monitoring of the blood.
Medical uses
Apixaban is indicated for the following:[5]
- To lower the risk of stroke and embolism in patients with nonvalvular atrial fibrillation.
- Deep vein thrombosis (DVT) prophylaxis. DVT’s may lead to pulmonary embolism (PE) in knee or hip replacement surgery patients.
- Treatment of both DVT and PE.
- To reduce the risk of recurring DVT and PE after initial therapy.
Atrial fibrillation
Apixaban is recommended by the National Institute for Health and Clinical Excellence for the prevention of stroke and systemic embolism in people with non-valvular atrial fibrillation and at least one of the following risk factors: prior stroke or transient ischemic attack, age 75 years or older, diabetes mellitus, or symptomatic heart failure.[6]
Apixaban and other newer anticoagulants (dabigatran and rivaroxaban) appear equally effective as warfarin in preventing non-hemorrhagic stroke in people with atrial fibrillation and are associated with lower risk of intracranial bleeding.[7]
Mechanism of action
Apixaban is a highly selective, orally bioavailable, and reversible direct inhibitor of free and clot-bound factor Xa. Factor Xa catalyzes the conversion of prothrombin to thrombin, the final enzyme in the coagulation cascade that is responsible for fibrin clot formation.[10] Apixaban has no direct effect on platelet aggregation, but by inhibiting factor Xa, it indirectly decreases clot formation induced by thrombin.[5]
FDA approval
A new drug application (NDA) for the approval of apixaban was submitted to the FDA by Bristol-Myers Squibb and Pfizer jointly after conclusion of the ARISTOTLE clinical trial in 2011.[11]
Apixaban was approved for the prevention of stroke in people with atrial fibrillation on December 28, 2012.[12] On March 14, 2014, it was approved for the additional use of preventing deep vein thrombosis and pulmonary embolism in people that had recently undergone knee or hip replacement.[13] On August 21, 2014, the FDA approved apixaban for the treatment of recurring deep vein thrombosis and pulmonary embolism.[2]
During development it was known as BMS-562247-01.
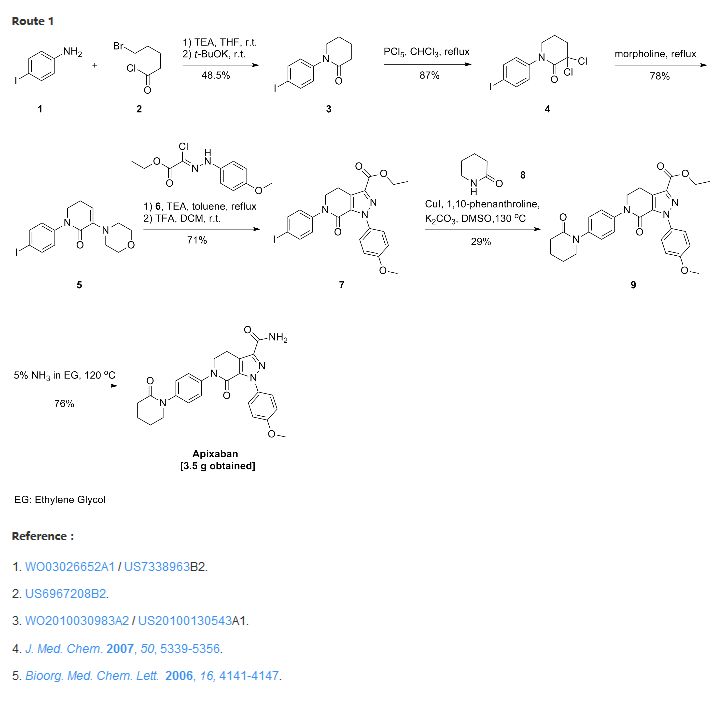



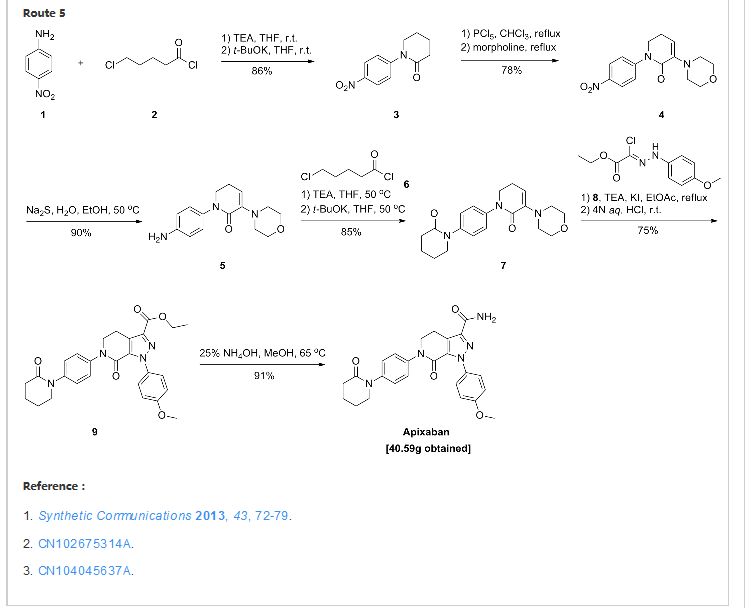
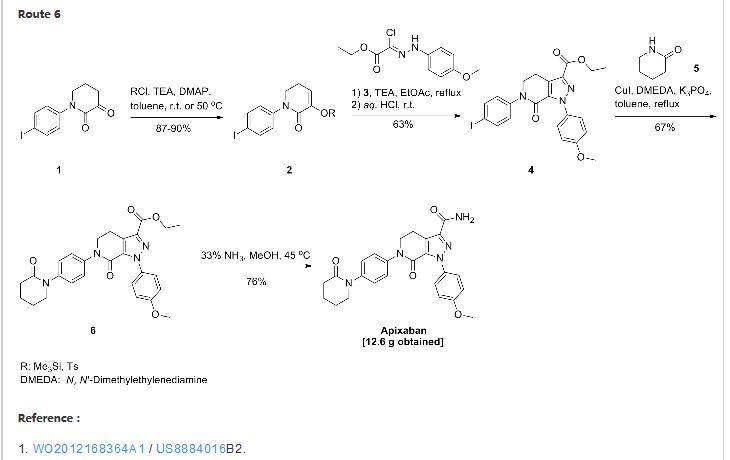
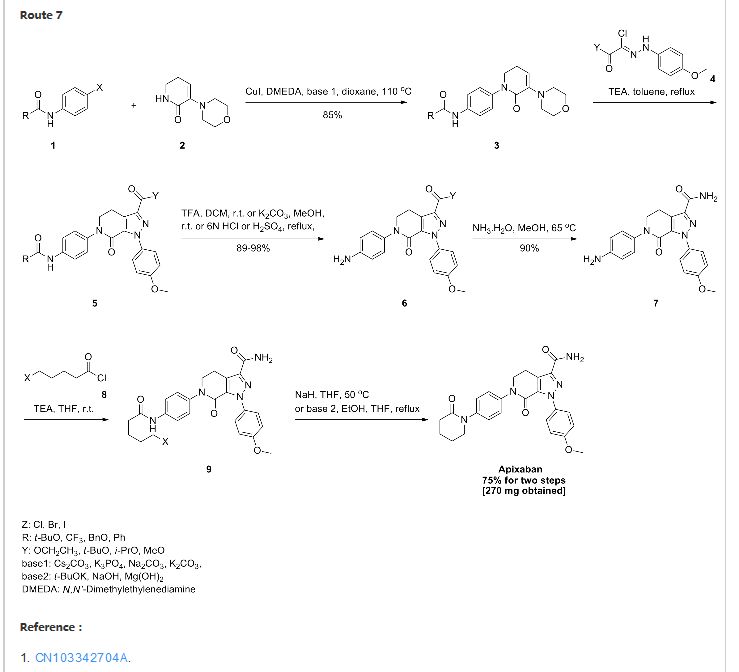

Thursday, August 21, 2014 – Bristol-Myers Squibb Company (NYSE: BMY) and Pfizer Inc. (NYSE: PFE) today announced the U.S. Food and Drug Administration (FDA) has approved a Supplemental New Drug Application (sNDA) for Eliquis for the treatment of DVT and PE, and for the reduction in the risk of recurrent DVT and PE following initial therapy. Combined, DVT and PE are known as VTE. It is estimated that every year, approximately 900,000 Americans are affected by DVT and PE.
See more at: http://worlddrugtracker.blogspot.in/2014/08/fda-approves-eliquis-apixaban-for.html


![1-(4-methoxyphenyl)-7-oxo-6-[4-(2-oxopiperidin-1-yl)phenyl]-4,5-dihydropyrazolo[3,4-c]pyridine-3-carboxamide](https://i0.wp.com/pic8.molbase.net/molpic/ae/b8/543077.png) APIXABAN
APIXABAN
PREDICTIONS
1H NMR

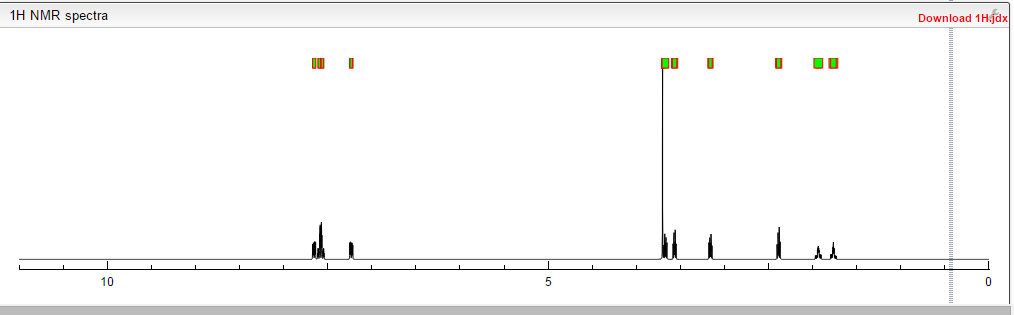
13C NMR

COSY
1H NMR PREDICT


13 C NMR PREDICT


|
|
![1-(4-methoxyphenyl)-7-oxo-6-[4-(2-oxopiperidin-1-yl)phenyl]-4,5-dihydropyrazolo[3,4-c]pyridine-3-carboxamide NMR spectra analysis, Chemical CAS NO. 503612-47-3 NMR spectral analysis, 1-(4-methoxyphenyl)-7-oxo-6-[4-(2-oxopiperidin-1-yl)phenyl]-4,5-dihydropyrazolo[3,4-c]pyridine-3-carboxamide H-NMR spectrum](https://i0.wp.com/pic11.molbase.net/nmr/nmr_image/2014-08-08/000/543/077/503612-47-3-1h.png)
![1-(4-methoxyphenyl)-7-oxo-6-[4-(2-oxopiperidin-1-yl)phenyl]-4,5-dihydropyrazolo[3,4-c]pyridine-3-carboxamide NMR spectra analysis, Chemical CAS NO. 503612-47-3 NMR spectral analysis, 1-(4-methoxyphenyl)-7-oxo-6-[4-(2-oxopiperidin-1-yl)phenyl]-4,5-dihydropyrazolo[3,4-c]pyridine-3-carboxamide C-NMR spectrum](https://i0.wp.com/pic11.molbase.net/nmr/nmr_image/2014-08-08/000/543/077/503612-47-3-13c.png)
C-NMR spectral analysis
|
CAS NO. 503612-47-3, 1-(4-methoxyphenyl)-7-oxo-6-[4-(2-oxopiperidin-1-yl)phenyl]-4,5-dihydropyrazolo[3,4-c]pyridine-3-carboxamide C-NMR spectral analysis
l-(4-Methoxyphenyl)-7-oxo-6-[4-(2-oxopiperidin-l -yl)phenyl]-4, 5,6,7- tetrahydro- lH-pyrazolo[3,4-c]pyridine-3-carboxyamide of formula (I), also known come apixaban, is a powerful inhibitor of coagulation factor Xa disclosed in US 6,967,208. Said compound is used in the prevention and treatment of thromboembolic disorders.
 (I)
US 7, 153,960 discloses a process for the preparation of apixaban wherein the key step is the formation of intermediate (A) by 1 ,3 dipolar cycloaddition reaction between the compounds of formula (B) and (C) and its subsequent conversion to the compound of formula (D) by treatment with an acid. The compound of formula (D), after simple manipulations of functional groups, is converted to apixaban
 B C A D
Said patent discloses the preparation of the compounds of formula (B) and (C). While the synthesis of the hydrazone of formula (B) has been known for some time, the preparation of the key intermediate of formula (C) is complex and uses reagents which are expensive and potentially hazardous, such as phosphorus pentachloride (PC15), and drastic reaction conditions.
US 7, 153,960, for example, discloses as preferred the preparation of an enamine intermediate of formula (C) wherein the amine residue NRbRc is a morpholine. The conditions used for the success of the reaction actually involve the use of morpholine as solvent at high temperatures, such as reflux temperature (about 130- 135°C).
The complexity of the known processes for the preparation of the intermediate of formula C, the expense and danger of the reagents and the drastic reaction conditions used make said processes difficult to apply and scale up industrially, especially for the purpose of preparing the intermediates of formula A and D and apixaban.
Example 6. Synthesis of compound of formula (I): l-(4- Methoxyphenyl)-6-[4-(2-oxo-piperidinyl)phenyl]-7-oxo-4,5,6,7-tetrahydro- l//-pyrazolo[3,4-c]pyridine-3-carboxyamide: Apixaban (I)
 The compound of formula II, prepared as in Example 5 (17.50 g, 35.82 mmol), is suspended in 100 ml of 33% NH3 and 200 ml of MeOH in a 1L 4-necked flask equipped with coolant, thermometer and magnetic stirrer, in nitrogen atmosphere, and heated to 45°. MeOH (250 ml) is added until completely dissolved, and the solution is left under stirring for 2h. Another addition of 33% NH3 (50 ml) is performed, and the progress of the reaction is monitored by TLC (AcOEt/MeOH 9: 1) and HPLC. After 18h the solvent is evaporated under low pressure, and the solid residue obtained is suspended in 200 ml of H2O and left under stirring for 2h. The white solid is filtered through a Buchner funnel, and washed with H2O (50 ml). The product of formula (I) is stove-dried at 50°C to a constant weight (12.60 g, yield 76%). The HPLC purity of the product exceeds 99%
 .
1H NMR (300 MHz, CDC13): DELTA
7.47 (2H, dd, J0=8.7 Hz, Ar-H),
7.31(2H, dd, J0=8.7 Hz, Ar-H),
7.23 (2H, dd, J0=8.7 Hz, Ar-H),
6.93 (2H, dd, J0=8.7 Hz, Ar-H),
6.83 (1H, s, N-H),
5.53 (1H, s, N-H),
4.1 1 (2H, t, J=6.6 Hz, CH2CH2N),
3.81 (3H, s, Ar-OCH3),
3.59 (2H, m, NCH2CH2CH2CH2CO)
3.37 (2H, t, J=6.6 Hz, CH2CH2N),
2.55 (2H, m, NCH2CH2CH2CH2CO),
1.93 (4H, m, NCH2CH^CH2CH2CO).
SEE
NMR For apixaban (in CDCl3) 1  Zhou , J. C. ; Oh , L. M. ; Ma , P. ; Li , H. Y. Synthesis of 4,5-dihydro-pyrazolo[3,4-c]pyrid-2-ones. WO Patent 2003/0 49681, June 19 , 2003 .
  J. Med. Chem. 2007 , 50 , 5339 – 5356 .
1-(4-Methoxyphenyl)-7-oxo-6-(4-(2-oxopiperidin-1-yl)phenyl)-4,5,6,7-tetrahydro-1H-pyrazolo[3,4-c]pyridine-3-carboxamide (1)[ref ]J. Med. Chem. 2007 , 50 , 5339 – 5356 .To the advanced intermediate 2 (2.44 g, 5.0 mmol) was added 25% ammonia water (1.5 mL, 20 mmol) in methanol (20 mL), and the mixture was heated to 65 °C for 5 h in an autoclave of 50 mL. The resulting mixture was cooled to room temperature, poured into water (30 mL), and crystalized below 0°C. The precipitate was filtrated and dried in vacuo at 50°C to afford the desired product 1 as a pale white solid. Yield: 2.09 g, 91%; mp 171–173 °C; IR (KBr, cm−1): 3448 and 3298 (N-H stretching), 2940 (C-H aliphatic), 1669 (C˭N stretching), 1614 (C˭O stretching), 1544 (aliphatic C˭C), 1513, 1463 and 1441 (aromatic C˭C), 1334, 1300 and 1254 (C-N stretching), 1146, 1111, 1090 and 1024 (C-O stretching), 835, 816, 794 and 758 (Ar-H aromatic bending); 1H NMR (500 MHz, CDCl3, ppm), δ: 7.48 (d, J = 8.0 Hz, 2H), 7.35 (d, J = 8.0 Hz, 2H), 7.27 (d, J = 8.0 Hz, 2H), 6.95 (d, J = 8.0 Hz, 2H), 5.66 (brs, 2H), 4.12 (t, J = 5.6 Hz, 2H), 3.84 (s, 3H), 3.55–3.65 (m, 2H), 3.39 (t, J = 5.6 Hz, 2H), 2.57 (t, J = 6.2 Hz, 2H), 1.91–2.01 (m, 4H); 13C NMR (125 MHz, CDCl3, ppm), δ: 170.9, 164.4, 160.5, 158.0, 142.1, 140.6 (2C), 134.0, 133.2, 127.4 (4C), 126.9 (2C), 126.5, 114.4 (2C), 56.2, 52.3, 51.8, 33.5, 24.2, 22.1, 21.9; MS/EI m/z = 459.2 (M+).
SEE
http://www.google.com/patents/WO2014056434A1?cl=en ………………….. http://www.google.com/patents/WO2014108919A2?cl=en HPLC method of Analysis:
Apixaban compound of formula- 1 of the present invention is analyzed by HPLC using the following conditions:
Apparatus: A liquid chromatographic system is to be equipped with variable wavelength UV- detector; Column: Zorbax Bonus RP, 250 x 4.6 mm, 5μιη or equivalent; Flow rate: 1.2 ml/min; wavelength: 270 nm; column temperature: 40°C; Injection volume; 5 uL; Run time: 35 minutes; Needle wash: diluent; Diluent: Acetonitrile: water (90: 10 v/v); Elution: Gradient; Mobile phase-A: Buffer; Mobile phase-B: acetonitrile:water (90:10 v/v); Buffer: Weigh accurately about 1.36 g of potassium dihydrogen ortho phosphate in 1000 10 ml of milli-Q water and adjust pH 6.0 with dil KOH solution, then filter through 0.22 μιη nylon membrane filter paper. The following impurities have been observed during the preparation of Apixaban.
 methyl esterImpurity Chloro Impurity Dehydro Impurity
Scheme-I:
Apixaban
 Scheme-II:
 Pure Apixaban Formula-1 [Apixaban]
Example-1: Preparation of 3-chloro-l-(4-iodophenyI)-5,6-dihydropyridin-2(lH)-one (Formula-6)
Lithium carbonate (4.08 gm) followed by lithium chloride (2.28 gm) were added to a mixture of 3,3-dichloro-l-(4-iodophenyl)piperidin-2-one compound of formula-5 (30 gm) and dimethylformamide (60 ml) at 25-30°C and stirred for 5 min at the same temperature. Heated the reaction mixture to 110-115°C and stirred for 4 hrs at the same temperature. Cooled the reaction mixture to 25-30°C. Water was added to the reaction mixture at 25-30°C and stirred for 1 hr at the same temperature. Filtered the precipitated solid and then dried to get the title compound. Yield: 25.0 gm; MR: 120-130°C.
Example-2: Preparation of 3-chIoro-l-(4-iodophenyl)-5,6-dihydropyridin-2(lH)-one (Formula-6)
Lithium carbonate (2.99 gm) followed by sodium chloride (2.76 gm) were added to a mixture of 3,3-dichloro-l-(4-iodophenyl)piperidin-2-one compound of formula-5 (50 gm) and dimethylformamide (150 ml) at 30-35°C and stirred for 10 min at the same temperature. Heated the reaction mixture to 110-115°C and stirred for 6 hrs at the same temperature. Cooled the reaction mixture to 25-30°C. Water was added to the reaction mixture at 25-30°C and stirred for 1 hr at the same temperature. Filtered the precipitated solid and then dried to get the title compound.
Yield: 42.0 gm; M.R: 120-130°C.
Example-3: Preparation of l-(4-iodophenyl)-3-morpholino-5,6-dihydropyridin-2(lH)-one (Formula-7)
Morpholine (5.09 gm) was added to a mixture of 3-chloro-l-(4-iodophenyl)-5,6-dihydro pyridin-2(lH)-one compound of formula-6 (5 gm) and toluene (5 ml) at 25-30°C and stirred for 5 min at the same temperature. Heated the reaction mixture to 115-120°C and stirred for 3 hrs at the same temperature. Cooled the reaction mixture to 25-30°C. Water was added to the reaction mixture at 25-30°C and stirred for 15 hrs at the same temperature. Filtered the precipitated solid and then dried to get the title compound. Yield: 3.8 gm.
Example-4: Preparation of l-(4-iodophenyl)-3-morpholino-5,6-dihydropyridin-2(lH)-one (Formula-7)
Morpholine (28.73 gm) was added to a mixture of 3-chloro-l-(4-iodophenyl)-5,6- dihydropyridin-2(lH)-one compound of formula-6 (50 gm) and toluene (50 ml) at 30-35°C. Heated the reaction mixture to 115-120°C and stirred for 8 hrs at 115-120°C. After completion of the reaction, cooled the reaction mixture to 25-30°C. Methyl tert-butyl ether (100 ml) followed by water were slowly added to the reaction mixture at 25-30°C. Cooled the reaction mixture to 5- 10°C and stirred for 2 hours at 5-10°C. Filtered the precipitated solid and then dried to get the title compound. Yield: 45 gm.
Example-5: Preparation of ethyl 6-(4-iodophenyl)-l-(4-methoxyphenyI)-7-oxo-4,5,6,7-tetra hydro-lH-pyrazoIo[3,4-c]pyridine-3-carboxyIate (FormuIa-13)
A mixture of 3-chloro-l-(4-iodophenyl)-5,6-dihydropyridin-2(lH)-one compound of formula-6 (79.2 gm), (Z)-ethyl 2-chloro-2-(2-(4-methoxyphenyl)hydrazono)acetate compound of formula-9 (65 gm) and toluene (450 ml) was heated to 90-100°C and stirred for 5 min at the same temperature. Triethyl amine (72 gm) was slowly added to the reaction mixture at 95-100°C and stirred for 2½ hrs at the same temperature. Cooled the reaction mixture to 25-30°C. Water (110 ml) was added to the reaction mixture at 25-30°C and stirred for 8 hrs at the same temperature. Filtered the solid, washed with water and then dried to get the title compound.
Yield: 78.5 gm.
Example-6: Preparation of 5-bromo-N-(4-iodophenyl)pentanamide (Formula-3)
A mixture of 5-bromopentanoic acid (54 g), thionyl chloride (41 g), dimethylformamide (2 ml) and toluene (100 ml) was heated to 40-45°C and stirred for 2 hours at the same temperature. Distilled off the reaction mixture to remove the un-reacted thionyl chloride under reduced pressure at a temperature below 40°C. Toluene (50 ml) was added to the reaction mixture and stirred for 15 minutes. The reaction mixture was cooled to 25-30°C under nitrogen atmosphere and it slowly added to a pre-cooled mixture of 4-iodoaniline compound of formula-2 (50 g) and toluene (350 ml) at 0-5°C. Triethyl amine (29 g) was added to it at 0-5°C. The above reaction mixture containing acid chloride was slowly added to the reaction mixture containing 4- iodoaniline under nitrogen atmosphere and stirred for 2 hours at 0-5°C. Water (250 ml) was added to the reaction mixture and stirred for 2 hours at 0-5°C. Filtered the precipitated solid and then dried to get title compound. Yield: 83 gm; MR: 135-140°C; HPLC purity: 99%.
Example-7: Preparation of 3-chloro-l-(4-iodophenyl)-5,6-dihydropyridin-2(lH)-one (Formula-6)
Step-a) Preparation of l-(4-iodophenyl)piperidin-2-one (Formula-4)
Sodium tert-butoxide (18.86 g) was added to a mixture of 5-bromo-N-(4- iodophenyl)pentanamide compound of formula-3 (50 g) and toluene (250 ml) at 0-5°C and stirred for 2 hours at 0-5°C. Water (100 ml) followed by aqueous hydrochloric acid solution (50 ml) were added to the reaction mixture and stirred for 10 minutes at 5-10°C. Both the organic and aqueous layers were separated; the organic layer was washed with water. Distilled off the solvent from the organic layer under reduced pressure at a temperature below 60°C to get title compound as a solid.
Step-b) Preparation of 3,3-dichIoro-l-(4-iodophenyI)piperidin-2-one (Formula-5)
The compound obtained in step-a) was dissolved in dichloromethane (100 ml) and slowly added to a mixture of phosphorous pentachloride (95 g) and dichloromethane (150 ml) at 25- 30°C. The reaction mixture was heated to 35-40°C and stirred for 4 hours at the same temperature. Cooled the reaction mixture to 5-10°C. Chilled water (150 ml) was added to the reaction mixture and stirred for 1.5 hours at 10-15°C. Both the organic and aqueous layers were separated; the organic layer was washed with water followed by 10% aqueous sodium carbonate solution. Distilled off the solvent completely from the organic layer to get title compound as a solid.
Step-c) Preparation of 3-chloro-l-(4-iodophenyl)-5,6-dihydropyridin-2(lH)-one (Formula- 6)
To the obtained compound in step-b), dimethylformamide (100 ml), followed by lithium carbonate (2.2 g) and sodium chloride (2.0 g) were added at 25-30°C. The reaction mixture was heated to 115-120°C and stirred for 6 hours at the same temperature. Cooled the reaction mixture to 30-35°C, water (350 ml) was added to it and stirred for 2 hours at 25-30°C. Filtered the precipitated solid and washed with water. Methanol (360 ml) was added to the obtained solid and the reaction mixture was heated to 65-70°C. Stirred the reaction mixture for 20 minutes at the same temperature. Carbon (3.0 g) was added to the reaction mixture and stirred for 20 minutes at 65-70°C. Filtered the reaction mixture through hyflow bed and washed with methanol. Distilled off the solvent from the filtrate under reduced pressure and methanol (300 ml) was added to the residue and stirred for 20 minutes at 25-30°C. Cooled the reaction mixture to -5 to 0°C and stirred for 60 minutes at the same temperature. Filtered the precipitated solid, washed with methanol and then dried to get title compound.
Yield: 25 gm; MR: 115- 120°C: HPLC purity: 98%.
Example-8: Preparation of 3-morpholino-l-(4-(2-oxopiperidin-l-yl)phenyl)-5,6-dihydro pyridin-2(lH)-one (Formula-8)
A mixture of l-(4-iodophenyl)-3-mo holino-5,6-dihydropyridin-2(lH)-one compound of formula-7 (50 g), piperidin-2-one (32.25 g) and o-xylene (75 ml) was stirred for 10 minutes at 25-30°C. Potassium carbonate (27.0 g), followed by copper iodide (7.43 g) were added to the reaction mixture. The reaction mixture was heated to 140-145°C under azeotropic distillation condition and stirred for 6 hours at the same temperature. Cooled the reaction mixture to 35- 40°C, water (175 ml) was slowly added to the reaction mixture at 35-40°C. Cooled the reaction mixture to 10-15°C and ammonia (125 ml) was added to the reaction mixture at 10-15°C. The temperature of the reaction mixture was raised to 25-30°C and stirred for 2 hours at the same temperature. Filtered the precipitated solid, washed with water and then dried to get title compound.
Yield: 35 gm; MR: 195-200°C; HPLC purity: 95%.
Example-9: Preparation of (Z)-ethyl 2-chloro-2-(2-(4-nlethoxyphenyl)hydrazono)acetate (FormuIa-9)
A mixture of 4-methoxyaniline compound of formula- 12 (50 g) and water (150 ml) was cooled to 5-10°C. Hydrochloric acid (100 ml), followed by a solution of sodium nitrite (30.81 g) in water (50 ml) were slowly added to the reaction mixture at 5-10°C and stirred for 2 hours at 5- 10°C to provide diazotized compound. Ethyl acetate (250 ml) was added to the reaction mixture. Ethyl 2-chloro acetoacetate (76.84 g) was slowly added to a mixture of sodium acetate (76.6 g), ethyl acetate (250 ml) and water (150 ml) at 25-30°C and the reaction mixture was stirred for 2 hours at 25-30°C. The reaction mixture was slowly added to the reaction mixture containing diazotized compound at a temperature below 10°C. The temperature of the reaction mixture was raised to 25-30°C and stirred for 16 hours at the same temperature. Both the organic and aqueous layers were separated and the organic layer was washed with 10% aqueous sodium bicarbonate solution followed by 10% aqueous sodium chloride solution. Distilled off the solvent completely from the organic layer under reduced pressure and then co-distilled with toluene. Toluene was added to the obtained compound and stirred for 15 minutes at 25-30°C. Silica-gel was added to the reaction mixture and stirred for 30 minutes at 25-30°C. Filtered the reaction mixture and the solvent from the filtrate was distilled off completely under reduced pressure. Cyclohexane (400 ml) was added to the obtained compound and the reaction mixture was stirred for 60 minutes at 25-30°C. Filtered the precipitated solid, washed with cyclohexane and then dried to get title compound. Yield: 60 gm; MR: 95-100°C; HPLC purity: 99%.
ExampIe-10: Preparation of ethyl l-(4-methoxyphenyl)-7-oxo-6-(4-(2-oxopiperidin-l-yl) phenyl)-4,5,6,7-tetrahydro-lH-pyrazolo[3,4-c]pyridine-3-carboxylate (Formula-11)
A mixture of 3-morpholino-l-(4-(2-oxopiperidin-l-yl)phenyl)-5,6-dihydropyridin-2(lH)- one compound of formula-8 (30 g), sodium carbonate (26.83 g) and acetone (150 ml) was heated to 45-50°C. (Z)-ethyl 2-chloro-2-(2-(4-methoxyphenyl)hydrazono)acetate compound of formula- 9 (32.5 g) was added to the reaction mixture at 45-50°C and stirred for 3 hours at the same temperature. Cooled the reaction mixture to 25-30°C and aqueous hydrochloric acid (50 ml) in 50 ml of water was added to it at 25-30°C. Stirred the reaction mixture for 2 hours at 25-30°C. Water was slowly added to the reaction mixture and stirred for 45 minutes at 25-30°C. Filtered the obtained solid and washed with water. The obtained solid was recrystallized from toluene (150 ml) to get the title compound. Yield: 35 gm; MR: 155-160°C; HPLC purity: 97%.
Example- 11: Preparation of l-(4-methoxyphenyl)-7-oxo-6-[4-(2-oxo piperidin-l-yl)phenyl]- 4,5,6,7-tetrahydro-lH-pyrazolo[3,4-c]pyridine-3-carboxamide (Formula-1)
A mixture of ethyl l-(4-methoxyphenyl)-7-oxo-6-(4-(2-oxopiperidin-l-yl)phenyl)- 4,5,6,7-tetrahydro-lH-pyrazolo[3,4-c]pyridine-3-carboxylate compound of formula-11 (50 g), formamide (150 ml), sodium methoxide (30 ml) and isopropanol (300 ml) was heated to 65-70°C and stirred for 2 hours at 65-70°C. Cooled the reaction mixture to 0-5°C and stirred for 30 minutes at 0-5°C. Filtered the precipitated solid and washed with isopropanol. Methanol (150 ml) was added to the obtained solid, the reaction mixture was heated to 65-70°C and stirred for 15 minutes at 65-70°C. Cooled the reaction mixture to 0-5°C and stirred for 30 minutes at 0-5°C. Filtered the precipitated solid, washed with methanol and then dried to get title compound. Yield: 35 g. MR: 230-235°C; HPLC purity: 98%.
The PXRD of the crystalline solid obtained from the above example is matches with the PXRD of crystalline form-M of the present invention.
Example-12: Purification of l-(4-methoxyphenyl)-7-oxo-6-[4-(2-oxo piperidin-l-yl)phenyl]- 4,5,6, 7-tetrahydro-lH-pyrazolo[3,4-c]pyridine-3-carboxamide (Formula-1)
1 -(4-methoxyphenyl)-7-oxo-6-[4-(2-oxopiperidin- 1 -yl)phenyl]-4,5,6,7-tetrahydro- 1 H- pyrazolo[3,4-c]pyridine-3-carboxamide compound of formula-1 (100 g) was dissolved in a mixture of dichloromethane (1200 ml) and methanol (200 ml) at 25-30°C. 10% aqueous sodium carbonate solution (200 ml) was added to the reaction mixture and stirred for 15 minutes at 25- 30°C. Both the organic and aqueous layers were separated, methanol (100 ml) was added to the organic layer and again 200 ml of 10% aqueous sodium carbonate solution was added to the reaction mixture. The reaction mixture was stirred for 15 minutes at 25-30°C and separated the organic and aqueous layers. To the organic layer methanol (100 ml) followed by water (200 ml) were added. Both the organic and aqueous layers were separated. The solvent from organic layer was distilled under reduced pressure at a temperature below 40°C. 3000 ml of a mixture of dichloromethane and methanol (in the ratio of 3:7) was added to the crude compound and the reaction mixture was heated to reflux temperature and stirred for 10 minutes. Carbon (10 g) was added to the reaction mixture and stirred for 15 minutes at the reflux temperature. Filtered the reaction mixture through hyflow bed, washed with a mixture of dichloromethane and methanol. The filtrate was cooled to 0-5°C and stirred for 2 hours at 0-5°C. Filtered the precipitated solid and washed with a mixture of dichloromethane and methanol. Isopropanol (1000 ml) was added to the reaction mixture. Heated the reaction mixture to 80-85°C and stirred for 15 minutes. Cooled the reaction mixture to 25-30°C and stirred for 2 hours at 35-30°C. Filtered the precipitated solid, washed with isopropanol and then dried to get title compound.
Yield: 80 gm; MR: 235-240°C.
The PXRD pattern of crystalline solid obtained from the above example is matches with PXRD of crystalline form-M of the present invention.
Example-13: Preparation of crystalline form-M of l-(4-methoxyphenyl)-7-oxo-6-[4-(2-oxo piperidin-l-yl)phenyl]-4,5,6,7-tetrahydro-lH-pyrazolo[3,4-c]pyridine-3-carboxamide (Formula-1)
l-(4-methoxyphenyl)-7-oxo-6-[4-(2-oxopiperidin-l-yl)phenyl]-4,5,6,7-tetrahydro-lH- pyrazolo[3,4-c] pyridine-3-carboxamide compound of formula-1 (6.25 gm) was added to isopropanol (400 ml) at 25-30°C. Heated the reaction mixture to reflux temperature and stirred for 15 min at the same temperature. Cooled the reaction mixture to 0-5°C and stirred for 60 min the same temperature. Filtered the solid, washed with isopropanol and then dried to get the title compound. Yield: 4.5 gm; Water content: 0.30% w/w. HPLC purity: 99.8%; Acid impurity: 0.02%; Amino acid impurity: Not detected; Chloro impurity: 0.01%; Methyl ester impurity: 0.05%; Ethyl ester impurity: 0.01%; Dehydro impurity: 0.07%.
Particle size distribution: D(0.1): 9.183 μπι; D(0.5): 25.991 um; D(0.9): 60.749 μιη; D[4,3]: 31.066 μπι.
The PXRD and DSC of the obtained compound are illustrated in figure- 1 and figure-2 respectively.
Example-14: Preparation of crystalline form-M of l-(4-methoxyphenyl)-7-oxo-6-[4-(2-oxo piperidin-l-yI)phenyl]-4,5,6,7-tetrahydro-lH-pyrazolo[3,4-c]pyridine-3-carboxamide (Formula-1)
1 -(4-methoxyphenyl)-7-oxo-6-[4-(2-oxopiperidin- 1 -yl)phenyl]-4,5,6,7-tetrahydro- 1 H- pyrazolo[3,4-c]pyridine-3-carboxamide compound of formula-1 (6.25 gm) was added to 50% aqueous isopropanol (60 ml) at 25-30°C. Heated the reaction mixture to 50-60°C and stirred for 4 hrs at the same temperature. Cooled the reaction mixture to 25-30°C and stirred for 60 min at the same temperature. Filtered the solid and then dried to get the title compound.
Yield: 4.1 gm; Water content: 0.35% w/w.
The PXRD and DSC of the obtained compound are illustrated in figure- 1 and figure-2 respectively.
Example-15: Preparation of crystalline form-S of l-(4-methoxyphenyl)-7-oxo-6-[4-(2-oxo piperidin-l-yl)phenyl]-4,5,6,7-tetrahydro-lH-pyrazolo[3,4-c]pyridine-3-carboxamide (Formula-1)
l-(4-methoxyphenyl)-7-oxo-6-[4-(2-oxopiperidin-l-yl)phenyl]-4,5,6,7-tetrahydro-lH- pyrazolo[3,4-c]pyridine-3-carboxamide compound of formula-1 (34 gm) was added to a mixture of dichloromethane and methanol at 25-30°C. Heated the reaction mixture to reflux temperature and stirred for 15 min at the same temperature. Filtered the reaction mixture and washed with a mixture of dichloromethane and methanol. Cooled the filtrate to 0-5°C and stirred for 60 min at the same temperature. Filtered the precipitated solid and then dried to get the title compound. Yield: 24.0 gm; M.R: 235-245°C; Water content: 7.38% w/w.
The PXRD and DSC of the obtained compound are illustrated in figure-3 and figure-4 respectively.
Example-16: Preparation of crystalline form-N of l-(4-methoxyphenyl)-7-oxo-6-[4-(2-oxo piperidin-l-yl)phenyl]-4,5,6,7-tetrahydro-lH-pyrazolo[3,4-c]pyridine-3- carboxamide(Formula-l)
A mixture of dichloromethane and ethyl acetate (625 ml, in 3:7 ratio) was added to l-(4- methoxyphenyl)-7-oxo-6-[4-(2-oxopiperidin-l-yl)phenyl]-4,5,6,7-tetrahydro-lH-pyrazolo[3,4-c] pyridine-3-carboxamide compound of formula- 1 (6.25 gm) at 25-30°C. Heated the reaction mixture to reflux temperature and stirred for 15 min at the same temperature. Cooled the reaction mixture to 0-5°C and stirred for 60 min at the same temperature. Filtered the solid and then dried to get title compound. Yield: 3.9 g; Water content: 5.21% w/w.
The PXRD and DSC of the obtained compound are illustrated in figure-5 and figure-6 respectively.
Example-17: Preparation of crystalline form-M of l-(4-methoxyphenyl)-7-oxo-6-[4-(2-oxo piperidin-l-yl)phenyl]-4,5,6,7-tetrahydro-lH-pyrazolo[3,4-c]pyridine-3-carboxamide (Formula-1)
1 -(4-methoxyphenyl)-7-oxo-6-[4-(2-oxopiperidin- 1 -yl)phenyl]-4,5,6,7-tetrahydro- 1 H- pyrazolo[3,4-c]pyridine-3-carboxamide compound of formula-1 (34 gm) was added to a mixture of dichloromethane and methanol (1020 ml, in 3:7 ratio) at 25-30°C. Heated the reaction mixture to reflux temperature and stirred for 15 min at the same temperature. Filtered the reaction mixture and washed with a mixture of dichloromethane and methanol. Cooled the filtrate to 0- 5°C and stirred for 60 min at the same temperature. Filtered the precipitated solid and added to isopropanol (510 ml). Heated the reaction mixture to reflux temperature and stirred for 15 Minutes at the same temperature. The reaction mixture was cooled to 0-5°C and stirred for 60 minutes at the same temperature. Filtered the solid and then dried to get crystalline form-M of compound of formula-1. Yield: 23 g; Water content: 0.30%w/w.
The PXRD and DSC of the obtained compound are illustrated in figure- 1 and figure-2 respectively.
Example-18: Preparation of crystalline form-M of l-(4-methoxyphenyl)-7-oxo-6-[4-(2-oxo piperidin-l-yl)phenyl]-4,5,6,7-tetrahydro-lH-pyrazolo[3,4-c]pyridine-3-carboxamide (Formula-1)
l-(4-methoxyphenyl)-7-oxo-6-[4-(2-oxopiperidin-l-yl)phenyl]-4,5,6,7-tetrahydro-lH- pyrazolo[3,4-c]pyridine-3-carboxamide compound of formula-1 (34 gm) was added to a mixture of dichloromethane and methanol (1020 ml, in 3:7 ratio) at 25-30°C. Heated the reaction mixture to reflux temperature and stirred for 15 min at the same temperature. Filtered the reaction mixture and washed with a mixture of dichloromethane and methanol. Cooled the filtrate to 0- 5°C and stirred for 60 min at the same temperature. Filtered the precipitated solid and added to aq.isopropanol (340 ml). Heated the reaction mixture to 50-60°C and stirred for 15 minutes at the same temperature. The reaction mixture was cooled to 25-35°C and stirred for 60 minutes at the same temperature. Filtered the solid and then dried to get crystalline form-M of compound of formula-1.
Yield: 23 g; Water content: 0.35%w/w.
The PXRD and DSC of the obtained compound are illustrated in figure- 1 and figure-2 respectively
/…………………
|
References
- “ELIQUIS® (apixaban) Approved In Europe For Preventing Venous Thromboembolism After Elective Hip Or Knee Replacement” (Press release). Pfizer. April 20, 2012. Retrieved 2012-05-29.
- http://www.pfizer.com/news/press-release/press-release-detail/u_s_fda_approves_eliquis_apixaban_for_the_treatment_of_deep_vein_thrombosis_dvt_and_pulmonary_embolism_pe_and_for_the_reduction_in_the_risk_of_recurrent_dvt_and_pe_following_initial_therapy
- “Bristol-Myers Squibb News Release 26 April 2007”. Archived from the original on 11 September 2007. Retrieved 2007-09-15.
- Nainggolan, Lisa. “Apixaban better than European enoxaparin regimen for preventing VTE”. Retrieved 2011-04-01.
- “Eliquis (apixaban) [prescribing information]” (PDF). Princeton, NJ: Bristol-Myers Squibb. March 2014. Retrieved 2014-10-29.
- “www.nice.org.uk”.
- Gómez-Outes, A; Terleira-Fernández, AI; Calvo-Rojas, G; Suárez-Gea, ML; Vargas-Castrillón, E (2013). “Dabigatran, Rivaroxaban, or Apixaban versus Warfarin in Patients with Nonvalvular Atrial Fibrillation: A Systematic Review and Meta-Analysis of Subgroups.”. Thrombosis 2013: 640723. PMID 24455237.
- “ELIQUIS® (apixaban) tablets Factor Xa Inhibitor” (PDF). FDA. August 2014. Retrieved 2014-11-02.
- Enriquez A, Lip GY, Baranchuk A (2015). “Anticoagulation reversal in the era of the non-vitamin K oral anticoagulants”. Europace. doi:10.1093/europace/euv030. PMID 25816811.
- Frost C, Wang J, Nepal S; et al. (February 2013). “Apixaban, an oral, direct factor Xa inhibitor: single dose safety, pharmacokinetics, pharmacodynamics and food effect in healthy subjects”. Br J Clin Pharmacol 75 (2): 476–87. doi:10.1111/j.1365-2125.2012.04369.x. PMID 22759198.
- Granger, M.D. et. al., Christopher (September 15, 2011). “Apixaban versus Warfarin in Patients with Atrial Fibrillation”. New England Journal of Medicine (365): 981–992. doi:10.1056/NEJMoa1107039. Retrieved 17 September 2015.
- FDA approves Eliquis to reduce the risk of stroke, blood clots in patients with non-valvular atrial fibrillation
Neale, Todd (March 14, 2014). “FDA OKs Apixaban for DVT Prevention”. MedPage Today. Retrieved 17 September 2015.
//////////
SEE ABAN SERIES……http://organicsynthesisinternational.blogspot.in/p/aban-series.html
सुकून उतना ही देना प्रभू, जितने से
जिंदगी चल जाये।
औकात बस इतनी देना,
कि औरों का भला हो जाये।
 COCK WILL TEACH YOU
COCK WILL TEACH YOU
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE


 amcrasto@gmail.com
amcrasto@gmail.com

{kind=link}