
FLAGS AND HITS
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO FACEBOOK
...................................................................Join me on twitter
FACEBOOK
...................................................................Join me on twitter
 ..................................................................Join me on google plus
..................................................................Join me on google plus
 Googleplus
GoogleplusMYSELF

Asparaginase erwinia chrysanthemi (recombinant)-rywn
Sequence:
1ADKLPNIVIL ATGGTIAGSA ATGTQTTGYK AGALGVDTLI NAVPEVKKLA51NVKGEQFSNM ASENMTGDVV LKLSQRVNEL LARDDVDGVV ITHGTDTVEE101SAYFLHLTVK SDKPVVFVAA MRPATAISAD GPMNLLEAVR VAGDKQSRGR151GVMVVLNDRI GSARYITKTN ASTLDTFKAN EEGYLGVIIG NRIYYQNRID201KLHTTRSVFD VRGLTSLPKV DILYGYQDDP EYLYDAAIQH GVKGIVYAGM251GAGSVSVRGI AGMRKAMEKG VVVIRSTRTG NGIVPPDEEL PGLVSDSLNP301AHARILLMLA LTRTSDPKVI QEYFHTY
>Protein sequence for asparaginase (Erwinia chrysanthemi) monomer ADKLPNIVILATGGTIAGSAATGTQTTGYKAGALGVDTLINAVPEVKKLANVKGEQFSNM ASENMTGDVVLKLSQRVNELLARDDVDGVVITHGTDTVEESAYFLHLTVKSDKPVVFVAA MRPATAISADGPMNLLEAVRVAGDKQSRGRGVMVVLNDRIGSARYITKTNASTLDTFKAN EEGYLGVIIGNRIYYQNRIDKLHTTRSVFDVRGLTSLPKVDILYGYQDDPEYLYDAAIQH GVKGIVYAGMGAGSVSVRGIAGMRKAMEKGVVVIRSTRTGNGIVPPDEELPGLVSDSLNP AHARILLMLALTRTSDPKVIQEYFHTY
References:
- Therapeutic Targets Database: TTD Biologic drug sequences in fasta format [Link]
Asparaginase erwinia chrysanthemi (recombinant)-rywn
JZP458-201
JZP458
CAS Registry Number 1349719-22-7
Protein Chemical FormulaC1546H2510N432O476S9
Protein Average Weight 140000.0 Da
Rylaze, FDA APPROVED 6/30/2021, BLA 761179
L-Asparaginase (ec 3.5.1.1, L-asparagine amidohydrolase) erwinia chrysanthemi tetramer alpha4Asparaginase (Dickeya chrysanthemi subunit)
Other Names
- Asparaginase Erwinia chrysanthemi
- Crisantaspase
- Cristantaspase
- Erwinase
- Erwinaze
- L-Asparagine amidohydrolase (Erwinia chrysanthemi subunit)
Asparaginase erwinia chrysanthemi [USAN]
L-Asparaginase, erwinia chrysanthemi
Asparaginase (erwinia chrysanthemi)
Asparaginase erwinia chrysanthemi
L-Asparaginase (ec 3.5.1.1, L-asparagine amidohydrolase) erwinia chrysanthemi tetramer alpha4
Asparaginase erwinia chrysanthemi (recombinant) [USAN]
Asparaginase erwinia chrysanthemi (recombinant)
A hydrolase enzyme that converts L-asparagine and water to L-aspartate and NH3.
NCI: Asparaginase Erwinia chrysanthemi. An enzyme isolated from the bacterium Erwinia chrysanthemi (E. carotovora). Asparagine is critical to protein synthesis in leukemic cells, which cannot synthesize this amino acid due to the absence of the enzyme asparagine synthase. Asparaginase hydrolyzes L-asparagine to L-aspartic acid and ammonia, thereby depleting leukemic cells of asparagine and blocking protein synthesis and tumor cell proliferation, especially in the G1 phase of the cell cycle. This agent also induces apoptosis in tumor cells. The Erwinia-derived product is often used for those patients who have experienced a hypersensitivity reaction to the E. Coli formulation. (NCI Thesaurus)
- Treatment of Acute Lymphoblastic Leukemia (ALL)
- Antineoplastic Agents
| 10MG/0.5ML | INJECTABLE;INTRAMUSCULAR |
PATENT
WO 2011003633
https://patents.google.com/patent/WO2011003633A1/en
The present invention concerns a conjugate of a protein having substantial L-asparagine aminohydrolase activity and polyethylene glycol, particularly wherein the polyethylene glycol has a molecular weight less than or equal to about 5000 Da, particularly a conjugate wherein the protein is a L-asparaginase from Erwinia, and its use in therapy.Proteins with L-asparagine aminohydrolase activity, commonly known as L- asparaginases, have successfully been used for the treatment of Acute Lymphoblastic Leukemia(ALL) in children for many years. ALL is the most common childhood malignancy (Avramis and Panosyan, Clin. Pharmacokinet. (2005) 44:367-393).[0003] L-asparaginase has also been used to treat Hodgkin’s disease, acute myelocytic leukemia, acute myelomonocytic leukemia, chronic lymphocytic leukemia, lymphosarcoma, reticulosarcoma, and melanosarcoma (Kotzia and Labrou, J. Biotechnol. 127 (2007) 657-669).The anti-tumor activity of L-asparaginase is believed to be due to the inability or reduced ability of certain malignant cells to synthesize L-asparagine (Kotzia and Labrou, J. Biotechnol. 127 (2007) 657-669). These malignant cells rely on an extracellular supply of L-asparagine. However, the L-asparaginase enzyme catalyzes the hydrolysis of L-asparagine to aspartic acid and ammonia, thereby depleting circulating pools of L-asparagine and killing tumor cells which cannot perform protein synthesis without L-asparagine (Kotzia and Labrou, J. Biotechnol. 127 (2007) 657-669).[0004] L-asparaginase from E. coli was the first enzyme drug used in ALL therapy and has been marketed as Elspar® in the USA or as Kidrolase® and L-asparaginase Medac® in Europe. L- asparaginases have also been isolated from other microorganisms, e.g., an L-asparaginase protein from Erwinia chrysanthemi, named crisantaspase, that has been marketed as Erwinase® (Wriston Jr., J.C. (1985) “L-asparaginase” Meth. Enzymol. 113, 608-618; Goward, CR. et al. (1992) “Rapid large scale preparation of recombinant Erwinia chrysanthemi L-asparaginase”, Bioseparation 2, 335-341). L-asparaginases from other species of Erwinia have also been identified, including, for example, Erwinia chrysanthemi 3937 (Genbank Accession#AAS67028), Erwinia chrysanthemi NCPPB 1125 (Genbank Accession #CAA31239), Erwinia carotovora (Genbank Accession #AAP92666), and Erwinia carotovora subsp. Astroseptica (Genbank Accession #AAS67027). These Erwinia chrysanthemi L-asparaginases have about 91-98% amino acid sequence identity with each other, while the Erwinia carotovora L- asparaginases have approximately 75-77% amino acid sequence identity with the Erwinia chrysanthemi L-asparaginases (Kotzia and Labrou, J. Biotechnol. 127 (2007) 657-669).[0005] L-asparaginases of bacterial origin have a high immunogenic and antigenic potential and frequently provoke adverse reactions ranging from mild allergic reaction to anaphylactic shock in sensitized patients (Wang, B. et al. (2003) “Evaluation of immunologic cross reaction of anti- asparaginase antibodies in acute lymphoblastic leukemia (ALL and lymphoma patients),Leukemia 17, 1583-1588). E. coli L-asparaginase is particularly immunogenic, with reports of the presence of anti-asparaginase antibodies to E. coli L-asparaginase following i.v. or i.m. administration reaching as high as 78% in adults and 70% in children (Wang, B. et al. (2003) Leukemia 17, 1583-1588).[0006] L-asparaginases from Escherichia coli and Erwinia chrysanthemi differ in their pharmacokinetic properties and have distinct immunogenic profiles, respectively (Klug Albertsen, B. et al. (2001) “Comparison of intramuscular therapy with Erwinia asparaginase and asparaginase Medac: pharmacokinetics, pharmacodynamics, formation of antibodies and influence on the coagulation system” Brit. J. Haematol. 115, 983-990). Furthermore, it has been shown that antibodies that developed after a treatment with L-asparaginase from E. coli do not cross react with L-Asparaginase from Erwinia (Wang, B. et al., Leukemia 17 (2003) 1583-1588). Thus, L-asparaginase from Erwinia (crisantaspase) has been used as a second line treatment of ALL in patients that react to E. coli L-asparaginase (Duval, M. et al. (2002) “Comparison of Escherichia co/z-asparaginase with £Vwzm‘α-asparaginase in the treatment of childhood lymphoid malignancies: results of a randomized European Organisation for Research and Treatment ofCancer, Children’s Leukemia Group phase 3 trial” Blood 15, 2734-2739; Avramis and Panosyan,Clin. Pharmacokinet. (2005) 44:367-393).[0007] In another attempt to reduce immunogenicity associated with administration of microbial L-asparaginases, an E. coli L-asparaginase has been developed that is modified with methoxy- polyethyleneglycol (mPEG). This method is commonly known as “PEGylation” and has been shown to alter the immunological properties of proteins (Abuchowski, A. et al. (1977) “Alteration of Immunological Properties of Bovine Serum Albumin by Covalent Attachment of Polyethylene Glycol,” J.Biol.Chem. 252 (11), 3578-3581). This so-called mPEG-L- asparaginase, or pegaspargase, marketed as Oncaspar® (Enzon Inc., USA), was first approved in the U.S. for second line treatment of ALL in 1994, and has been approved for first- line therapy of ALL in children and adults since 2006. Oncaspar® has a prolonged in vivo half-life and a reduced immunogenicity/antigenicity.[0008] Oncaspar® is E. coli L-asparaginase that has been modified at multiple lysine residues using 5 kDa mPEG-succinimidyl succinate (SS-PEG) (U.S. Patent No. 4,179,337). SS-PEG is aPEG reagent of the first generation that contains an instable ester linkage that is sensitive to hydro lysis by enzymes or at slightly alkaline pH values (U.S. Patent No. 4,670,417; Makromol. Chem. 1986, 187, 1131-1144). These properties decrease both in vitro and in vivo stability and can impair drug safety.[0009] Furthermore, it has been demonstrated that antibodies developed against L-asparaginase from E. coli will cross react with Oncaspar® (Wang, B. et al. (2003) “Evaluation of immunologic cross-reaction of anti-asparaginase antibodies in acute lymphoblastic leukemia (ALL and lymphoma patients),” Leukemia 17, 1583-1588). Even though these antibodies were not neutralizing, this finding clearly demonstrated the high potential for cross-hypersensitivity or cross-inactivation in vivo. Indeed, in one report 30-41% of children who received pegaspargase had an allergic reaction (Wang, B. et al. (2003) Leukemia 17, 1583-1588).[0010] In addition to outward allergic reactions, the problem of “silent hypersensitivity” was recently reported, whereby patients develop anti-asparaginase antibodies without showing any clinical evidence of a hypersensitivity reaction (Wang, B. et al. (2003) Leukemia 17, 1583-1588). This reaction can result in the formation of neutralizing antibodies to E. coli L-asparaginase and pegaspargase; however, these patients are not switched to Erwinia L-asparaginase because there are not outward signs of hypersensitivity, and therefore they receive a shorter duration of effective treatment (Holcenberg, J., J. Pediatr. Hematol. Oncol. 26 (2004) 273-274).[0011] Erwinia chrysanthemi L-asparaginase treatment is often used in the event of hypersensitivity to E. co/z-derived L-asparaginases. However, it has been observed that as many as 30-50% of patients receiving Erwinia L-asparaginase are antibody-positive (Avramis andPanosyan, Clin. Pharmacokinet. (2005) 44:367-393). Moreover, because Erwinia chrysanthemi L-asparaginase has a significantly shorter elimination half-life than the E. coli L-asparaginases, it must be administered more frequently (Avramis and Panosyan, Clin. Pharmacokinet. (2005) 44:367-393). In a study by Avramis et al., Erwinia asparaginase was associated with inferior pharmacokinetic profiles (Avramis et al., J. Pediatr. Hematol. Oncol. 29 (2007) 239-247). E. coli L-asparaginase and pegaspargase therefore have been the preferred first-line therapies for ALL over Erwinia L-asparaginase.[0012] Numerous biopharmaceuticals have successfully been PEGylated and marketed for many years. In order to couple PEG to a protein, the PEG has to be activated at its OH terminus. The activation group is chosen based on the available reactive group on the protein that will bePEGylated. In the case of proteins, the most important amino acids are lysine, cysteine, glutamic acid, aspartic acid, C-terminal carboxylic acid and the N-terminal amino group. In view of the wide range of reactive groups in a protein nearly the entire peptide chemistry has been applied to activate the PEG moiety. Examples for this activated PEG-reagents are activated carbonates, e.g., p-nitrophenyl carbonate, succinimidyl carbonate; active esters, e.g., succinimidyl ester; and for site specific coupling aldehydes and maleimides have been developed (Harris, M., Adv. Drug – A -DeI. Rev. 54 (2002), 459-476). The availability of various chemical methods for PEG modification shows that each new development of a PEGylated protein will be a case by case study. In addition to the chemistry the molecular weight of the PEG that is attached to the protein has a strong impact on the pharmaceutical properties of the PEGylated protein. In most cases it is expected that, the higher the molecular weight of the PEG, the better the improvement of the pharmaceutical properties (Sherman, M. R., Adv. Drug Del. Rev. 60 (2008), 59-68; Holtsberg, F. W., Journal of Controlled Release 80 (2002), 259-271). For example, Holtsberg et al. found that, when PEG was conjugated to arginine deaminase, another amino acid degrading enzyme isolated from a microbial source, pharmacokinetic and pharmacodynamic function of the enzyme increased as the size of the PEG attachment increased from a molecular weight of 5000Da to 20,000 Da (Holtsberg, F.W., Journal of Controlled Release 80 (2002), 259-271).[0013] However, in many cases, PEGylated biopharmaceuticals show significantly reduced activity compared to the unmodified biopharmaceutical (Fishburn, CS. (2008) Review “The Pharmacology of PEGylation: Balancing PD with PK to Generate Novel Therapeutics” J. Pharm. Sd., 1-17). In the case of L-asparaginase from Erwinia carotovora, it has been observed that PEGylation reduced its in vitro activity to approximately 57% (Kuchumova, A.V. et al. (2007) “Modification of Recombinant asparaginase from Erwinia carotovora with Polyethylene Glycol 5000” Biochemistry (Moscow) Supplement Series B: Biomedical Chemistry, 1, 230-232). The L-asparaginase from Erwinia carotovora has only about 75% homology to the Erwinia chrysanthemi L-asparaginase (crisantaspase). For Oncaspar® it is also known that its in vitro activity is approximately 50% compared to the unmodified E. coli L-asparaginase.[0014] The currently available L-asparaginase preparations do not provide alternative or complementary therapies— particularly therapies to treat ALL— that are characterized by high catalytic activity and significantly improved pharmacological and pharmacokinetic properties, as well as reduced immunogenicity. L-asparaginase protein has at least about 80% homology or identity with the protein comprising the sequence of SEQ ID NO:1, more specifically at least about 85%, 86%, 87%, 88%, 89%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99%, or 100% homology or identity with the protein comprising the sequence of SEQ ID NO:1. SEQ ID NO:1 is as follows:ADKLPNIVILATGGTIAGSAATGTQTTGYKAGALGVDTLINAVPEVKKLANVKGE QFSNMASENMTGDVVLKLSQRVNELLARDDVDGVVITHGTDTVEESAYFLHLTV KSDKPVVFVAAMRPATAISADGPMNLLEAVRVAGDKQSRGRGVMVVLNDRIGSA RYITKTNASTLDTFKANEEGYLGVIIGNRIYYQNRIDKLHTTRSVFDVRGLTSLPKV DILYGYQDDPEYLYDAAIQHGVKGIVYAGMGAGSVSVRGIAGMRKAMEKGVVVIRSTRTGNGIVPPDEELPGLVSDSLNPAHARILLMLALTRTSDPKVIQEYFHTY (SEQ ID NO:1) [0048] The term “comprising the sequence of SEQ ID NO:1” means that the amino-acid sequence of the protein may not be strictly limited to SEQ ID NO:1 but may contain additional amino-acids.ExamplesExample 1 : Preparation of Recombinant Crisantaspase [0100] The recombinant bacterial strain used to manufacture the naked recombinant Erwinia chrysanthemi L-asparaginase protein (also referred to herein as “r-crisantaspase”) was an E. coli BL21 strain with a deleted ansB gene (the gene encoding the endogenous E. coli type II L- asparaginase) to avoid potential contamination of the recombinant Erwinia chrysanthemi L- asparaginase with this enzyme. The deletion of the ansB gene relies on homologous recombination methods and phage transduction performed according to the three following steps:1) a bacterial strain (NMI lOO) expressing a defective lambda phage which supplies functions that protect and recombine electroporated linear DNA substrate in the bacterial cell was transformed with a linear plasmid (kanamycin cassette) containing the kanamycin gene flanked by an FLP recognition target sequence (FRT). Recombination occurs to replace the ansB gene by the kanamycin cassette in the bacterial genome, resulting in a ΛansB strain; 2) phage transduction was used to integrate the integrated kanamycin cassette region from the ΛansB NMI lOO strain to the ansB locus in BL21 strain. This results in an E. coli BL21 strain with a deleted ansB gene and resistant to kanamycin; 3) this strain was transformed with a FLP -helper plasmid to remove the kanamycin gene by homologous recombination at the FRT sequence. The genome of the final strain (BL21 ΛansB strain) was sequenced, confirming full deletion of the endogenous ansB gene.[0101] The E. co/z‘-optimized DNA sequence encoding for the mature Erwinia chrysanthemi L- asparaginase fused with the ENX signal peptide from Bacillus subtilis was inserted into an expression vector. This vector allows expression of recombinant Erwinia chrysanthemi L- asparaginase under the control of hybrid T5/lac promoter induced by the addition of Isopropyl β- D-1-thiogalactopyranoside (IPTG) and confers resistance to kanamycin.[0102] BL21 ΛansB strain was transformed with this expression vector. The transformed cells were used for production of the r-crisantaspase by feed batch glucose fermentation in Reisenberg medium. The induction of the cell was done 16h at 23°C with IPTG as inducer. After cell harvest and lysis by homogenization in 1OmM sodium phosphate buffer pH6 5mM EDTA (Buffer A), the protein solution was clarified by centrifugation twice at 1500Og, followed by 0.45μm and 0.22μm filtration steps. The recombinant Erwinia chrysanthemi L-asparaginase was next purified using a sequence of chromatography and concentration steps. Briefly, the theoretical isoelectric point of the Erwinia chrysanthemi L-asparaginase (7.23) permits the recombinant enzyme to adsorb to cation exchange resins at pH6. Thus, the recombinant enzyme was captured on a Capto S column (cation exchange chromatography) and eluted with salt gradient in Buffer A. Fractions containing the recombinant enzyme were pooled. The pooled solution was next purified on Capto MMC column (cation exchange chromatography) in Buffer A with salt gradient. . The eluted fractions containing Erwinia chrysanthemi L-asparaginase were pooled and concentrated before protein separation on Superdex 200pg size exclusion chromatography as polishing step. Fractions containing recombinant enzymes were pooled, concentrated, and diafiltered against 10OmM sodium phosphate buffer pH8. The purity of the final Erwinia chrysanthemi L-asparaginase preparation was evaluated by SDS-PAGE (Figure 1) and RP-HPLC and was at least 90%. The integrity of the recombinant enzyme was verified byN-terminal sequencing and LC-MS. Enzyme activity was measured at 37°C using Nessler’s reagent. The specific activity of the purified recombinant Erwinia chrysanthemi L-asparaginase was around 600 U/mg. One unit of enzyme activity is defined as the amount of enzyme that liberates lμmol of ammonia from L-asparagine per minute at 37°C. Example 2: Preparation of 10 kDa mPEG-L- Asparaginase Conjugates[0103] A solution of L-asparaginase from Erwinia chrysanthemi was stirred in a 100 mM sodium phosphate buffer at pH 8.0, at a protein concentration between 2.5 and 4 mg/mL, in the presence of 150 mg/mL or 36 mg/mL 10 kDa mPEG-NHS, for 2 hours at 22°C. The resulting crude 10 kDa mPEG-L-asparaginase was purified by size exclusion chromatography using a Superdex 200 pg column on an Akta purifier UPC 100 system. Protein-containing fractions were pooled and concentrated to result in a protein concentration between 2 and 8 mg/mL. Two 10 kDa mPEG-L-asparaginase conjugates were prepared in this way, differing in their degree of PEGylation as determined by TNBS assay with unmodified L-asparaginase as a reference, one corresponding to full PEGylation (100% of accessible amino groups (e.g., lysine residues and/or the N-terminus) residues being conjugated corresponding to PEGylation of 78% of total amino groups (e.g., lysine residues and/or the N-terminus)); the second one corresponding to partial PEGylation (39% of total amino groups (e.g., lysine residues and/or the N-terminus) or about 50% of accessible amino groups (e.g., lysine residues and/or the N-terminus)) . SDS-PAGE analysis of the conjugates is shown in Figure 2. The resulting conjugates appeared as an essentially homogeneous band and contained no detectable unmodified r-crisantaspase.Example 3: Preparation of 5 kDa mPEG-L-Asparaginase Conjugates[0104] A solution of L-asparaginase from Erwinia chrysanthemi was stirred in a 100 mM sodium phosphate buffer at pH 8.0, at a protein concentration of 4 mg/mL, in the presence of 150 mg/mL or 22.5 mg/mL 5 kDa mPEG-NHS, for 2 hours at 22°C. The resulting crude 5 kDa mPEG-L-asparaginase was purified by size exclusion chromatography using a Superdex 200 pg column on an Akta purifier UPC 100 system. Protein-containing fractions were pooled and concentrated to result in a protein concentration between 2 and 8 mg/mL. Two 5 kDa mPEG-L- asparaginase conjugates were prepared in this way, differing in their degree of PEGylation as determined by TNBS assay with unmodified L-asparaginase as a reference, one corresponding to full PEGylation (100% of accessible amino groups (e.g., lysine residues and/or the N-terminus) being conjugated corresponding to PEGylation of 84% of total amino groups (e.g., lysine residues and/or the N-terminus)); the second one corresponding to partial PEGylation (36% of total amino groups (e.g., lysine residues and/or the N-terminus) or about 43% of accessible amino groups (e.g., lysine residues and/or the N-terminus)). SDS-PAGE analysis of the conjugates is shown in Figure 2. The resulting conjugates appeared as an essentially homogeneous band and contained no detectable unmodified r-crisantaspase.Example 4: Preparation of 2 kDa mPEG-L-Asparaginase Conjugates[0105] A solution of L-asparaginase from Erwinia chrysanthemi was stirred in a 100 mM sodium phosphate buffer pH 8.0 at a protein concentration of 4 mg/mL in the presence of150 mg/mL or 22.5 mg/mL 2 kDa mPEG-NHS for 2 hours at 22°C. The resulting crude 2 kDa mPEG-L-asparaginase was purified by size exclusion chromatography using a Superdex 200 pg column on an Akta purifier UPC 100 system. Protein containing fractions were pooled and concentrated to result in a protein concentration between 2 and 8 mg/mL. Two 2 kDa mPEG-L- asparaginase conjugates were prepared in this way, differing in their degree of PEGylation as determined by TNBS assay with unmodified L-asparaginase as reference, one corresponding to maximum PEGylation (100% of accessible amino groups (e.g., lysine residues and/or the N- terminus) being conjugated corresponding to PEGylation of 86% of total amino groups (e.g., lysine residues and/or the N-terminus)); the second one corresponding to partial PEGylation (47% of total amino groups (e.g., lysine residues and/or the N-terminus) or about 55% of accessible amino groups {e.g., lysine residues and/or the N-terminus)). SDS-PAGE analysis of the conjugates is shown in Figure 2. The resulting conjugates appeared as an essentially homogeneous band and contained no detectable unmodified r-crisantaspase.Example 5: Activity of mPEG-r-Crisantaspase Conjugates[0106] L-asparaginase aminohydrolase activity of each conjugate described in the proceeding examples was determined by Nesslerization of ammonia that is liberated from L-asparagine by enzymatic activity. Briefly, 50μL of enzyme solution were mixed with 2OmM of L-asparagine in a 50 mM Sodium borate buffer pH 8.6 and incubated for 10 min at 37°C. The reaction was stopped by addition of 200μL of Nessler reagent. Absorbance of this solution was measured at 450 nm. The activity was calculated from a calibration curve that was obtained from Ammonia sulfate as reference. The results are summarized in Table 2, below:Table 2: Activity of mPEG-r-crisantaspase conjugates

* the numbers “40%” and “100%” indicate an approximate degree of PEGylation of respectively 40-55% and 100% of accessible amino groups (see Examples 2-4, supra).** the ratio mol PEG / mol monomer was extrapolated from data using TNBS assay, that makes the assumption that all amino groups from the protein (e.g., lysine residues and the N-terminus) are accessible.[0107] Residual activity of mPEG-r-crisantaspase conjugates ranged between 483 and 543 Units/mg. This corresponds to 78-87% of L-asparagine aminohydrolase activity of the unmodified enzyme. Example 6: L-Asparagine-Depleting Effect of Unmodified Crisantaspase
PAPER
Biotechnology and Applied Biochemistry (2019), 66(3), 281-289. |
https://iubmb.onlinelibrary.wiley.com/doi/10.1002/bab.1723
Crisantaspase is an asparaginase enzyme produced by Erwinia chrysanthemi and used to treat acute lymphoblastic leukemia (ALL) in case of hypersensitivity to Escherichia coli l-asparaginase (ASNase). The main disadvantages of crisantaspase are the short half-life (10 H) and immunogenicity. In this sense, its PEGylated form (PEG-crisantaspase) could not only reduce immunogenicity but also improve plasma half-life. In this work, we developed a process to obtain a site-specific N-terminal PEGylated crisantaspase (PEG-crisantaspase). Crisantaspase was recombinantly expressed in E. coli BL21(DE3) strain cultivated in a shaker and in a 2-L bioreactor. Volumetric productivity in bioreactor increased 37% compared to shaker conditions (460 and 335 U L−1 H−1, respectively). Crisantaspase was extracted by osmotic shock and purified by cation exchange chromatography, presenting specific activity of 694 U mg−1, 21.7 purification fold, and yield of 69%. Purified crisantaspase was PEGylated with 10 kDa methoxy polyethylene glycol-N-hydroxysuccinimidyl (mPEG-NHS) at different pH values (6.5–9.0). The highest N-terminal pegylation yield (50%) was at pH 7.5 with the lowest poly-PEGylation ratio (7%). PEG-crisantaspase was purified by size exclusion chromatography and presented a KM value three times higher than crisantaspase (150 and 48.5 µM, respectively). Nonetheless, PEG-crisantaspase was found to be more stable at high temperatures and over longer periods of time. In 2 weeks, crisantaspase lost 93% of its specific activity, whereas PEG-crisantaspase was stable for 20 days. Therefore, the novel PEG-crisantaspase enzyme represents a promising biobetter alternative for the treatment of ALL.
ADKLPNIVILATGGTIAGSAATGTQTTGYKAGALGVDTLINAVPEVKKLANVKGEQFSN
MASENMTGDVVLKLSQRVNELLARDDVDGVVITHGTDTVEESAYFLHLTVKSDKPVV
FVAAMRPATAISADGPMNLLEAVRVAGDKQSRGRGVMVVLNDRIGSARYITKTNAST
LDTFKANEEGYLGVIIGNRIYYQNRIDKLHTTRSVFDVRGLTSLPKVDILYGYQDDPEY
LYDAAIQHGVKGIVYAGMGAGSVSVRGIAGMRKAMEKGVVVIRSTRTGNGIVPPDEE
LPGLVSDSLNPAHARILLMLALTRTSDPKVIQEYFHTY
Figure S1 – Amino acid sequence of the enzyme crisantaspase without the signal peptide and with the lysines highlighted in red (Swiss-Prot/TrEMBL accession number: P06608|22-348 AA).
……………………………………………………………………………………………………………………………..
As a component of a chemotherapy regimen to treat acute lymphoblastic leukemia and lymphoblastic lymphoma in patients who are allergic to E. coli-derived asparaginase products
Press ReleaseFor Immediate Release:June 30, 2021
FDA Approves Component of Treatment Regimen for Most Common Childhood Cancer
Alternative Has Been in Global Shortage Since 2016
Today, the U.S. Food and Drug Administration approved Rylaze (asparaginase erwinia chrysanthemi (recombinant)-rywn) as a component of a chemotherapy regimen to treat acute lymphoblastic leukemia and lymphoblastic lymphoma in adult and pediatric patients who are allergic to the E. coli-derived asparaginase products used most commonly for treatment. The only other FDA-approved drug for such patients with allergic reactions has been in global shortage for years.
“It is extremely disconcerting to patients, families and providers when there is a lack of access to critical drugs for treatment of a life-threatening, but often curable cancer, due to supply issues,” said Gregory Reaman, M.D., associate director for pediatric oncology in the FDA’s Oncology Center of Excellence. “Today’s approval may provide a consistently sourced alternative to a pivotal component of potentially curative therapy for children and adults with this type of leukemia.”
Acute lymphoblastic leukemia occurs in approximately 5,700 patients annually, about half of whom are children. It is the most common type of childhood cancer. One component of the chemotherapy regimen is an enzyme called asparaginase that kills cancer cells by depriving them of substances needed to survive. An estimated 20% of patients are allergic to the standard E. coli-derived asparaginase and need an alternative their bodies can tolerate.
Rylaze’s efficacy was evaluated in a study of 102 patients who either had a hypersensitivity to E. coli-derived asparaginases or experienced silent inactivation. The main measurement was whether patients achieved and maintained a certain level of asparaginase activity. The study found that the recommended dosage would provide the target level of asparaginase activity in 94% of patients.
The most common side effects of Rylaze include hypersensitivity reactions, pancreatic toxicity, blood clots, hemorrhage and liver toxicity.
This review was conducted under Project Orbis, an initiative of the FDA Oncology Center of Excellence. Project Orbis provides a framework for concurrent submission and review of oncology drugs among international partners. For this review, FDA collaborated with Health Canada, where the application review is pending.
Rylaze received Fast Track and Orphan Drug designations for this indication. Fast Track is a process designed to facilitate the development and expedite the review of drugs to treat serious conditions and fulfill an unmet medical need. Orphan Drug designation provides incentives to assist and encourage drug development for rare diseases.
The FDA granted approval of Rylaze to Jazz Pharmaceuticals.
REF
DUBLIN, June 30, 2021 /PRNewswire/ — Jazz Pharmaceuticals plc (Nasdaq: JAZZ) today announced the U.S. Food and Drug Administration (FDA) approval of Rylaze™ (asparaginase erwinia chrysanthemi (recombinant)-rywn) for use as a component of a multi-agent chemotherapeutic regimen for the treatment of acute lymphoblastic leukemia (ALL) or lymphoblastic lymphoma (LBL) in pediatric and adult patients one month and older who have developed hypersensitivity to E. coli-derived asparaginase.1 Rylaze is the only recombinant erwinia asparaginase manufactured product that maintains a clinically meaningful level of asparaginase activity throughout the entire duration of treatment, and it was developed by Jazz to address the needs of patients and healthcare providers with an innovative, high-quality erwinia-derived asparaginase with reliable supply.
“We are excited to bring this important new treatment to patients who are in critical need, and we are grateful to FDA for the approval of Rylaze based on its established safety and efficacy profile. We are pleased Rylaze was approved before the trial is complete and are diligently working to advance additional clinical trial data. We are committed to quickly engaging with FDA to evolve the Rylaze product profile with additional dosing options and an IV route of administration,” said Bruce Cozadd, chairman and CEO of Jazz Pharmaceuticals. “Thank you to our collaborators within the Children’s Oncology Group, the clinical trial investigators, patients and their families, and all of the other stakeholders who helped us achieve this significant milestone.”
Rylaze was granted orphan drug designation for the treatment of ALL/LBL by FDA in June 2021. The Biologics Licensing Application (BLA) approval followed review under the Real-Time Oncology Review (RTOR) program, an initiative of FDA’s Oncology Center of Excellence designed for efficient delivery of safe and effective cancer treatments to patients.
The company expects Rylaze will be commercially available in mid-July.
“The accelerated development and approval of Rylaze marks an important step in bringing a meaningful new treatment option for many ALL patients – most of whom are children – who cannot tolerate E. coli-derived asparaginase medicine,” said Dr. Luke Maese, assistant professor at the University of Utah, Primary Children’s Hospital and Huntsman Cancer Institute. “Before the approval of Rylaze, there was a significant need for an effective asparaginase medicine that would allow patients to start and complete their prescribed treatment program with confidence in supply.”
Recent data from a Children’s Oncology Group retrospective analysis of over 8,000 patients found that patients who did not receive a full course of asparaginase treatment due to associated toxicity had significantly lower survival outcomes – regardless of whether those patients were high risk or standard risk, slow early responders.2
About Study JZP458-201
The FDA approval of Rylaze, also known as JZP458, is based on clinical data from an ongoing pivotal Phase 2/3 single-arm, open-label, multicenter, dose confirmation study evaluating pediatric and adult patients with ALL or LBL who have had an allergic reaction to E. coli-derived asparaginases and have not previously received asparaginase erwinia chrysanthemi. The study was designed to assess the safety, tolerability and efficacy of JZP458. The determination of efficacy was measured by serum asparaginase activity (SAA) levels. The Phase 2/3 study is being conducted in two parts. The first part is investigating the intramuscular (IM) route of administration, including a Monday-Wednesday-Friday dosing schedule. The second part remains active to further confirm the dose and schedule for the intravenous (IV) route of administration.
The FDA approval of Rylaze was based on data from the first of three IM cohorts, which demonstrated the achievement and maintenance of nadir serum asparaginase activity (NSAA) greater than or equal to the level of 0.1 U/mL at 48 hours using IM doses of Rylaze 25 mg/m2. The results of modeling and simulations showed that for a dosage of 25 mg/m2 administered intramuscularly every 48 hours, the proportion of patients maintaining NSAA ≥ 0.1 U/mL at 48 hours after a dose of Rylaze was 93.6% (95% CI: 92.6%, 94.6%).1
The most common adverse reactions (incidence >15%) were abnormal liver test, nausea, musculoskeletal pain, fatigue, infection, headache, pyrexia, drug hypersensitivity, febrile neutropenia, decreased appetite, stomatitis, bleeding and hyperglycemia. In patients treated with the Rylaze, a fatal adverse reaction (infection) occurred in one patient and serious adverse reactions occurred in 55% of patients. The most frequent serious adverse reactions (in ≥5% of patients) were febrile neutropenia, dehydration, pyrexia, stomatitis, diarrhea, drug hypersensitivity, infection, nausea and viral infection. Permanent discontinuation due to an adverse reaction occurred in 9% of patients who received Rylaze. Adverse reactions resulting in permanent discontinuation included hypersensitivity (6%) and infection (3%).1
The company will continue to work with FDA and plans to submit additional data from a completed cohort of patients evaluating 25mg/m2 IM given on Monday and Wednesday, and 50 mg/m2 given on Friday in support of a M/W/F dosing schedule. Part 2 of the study is evaluating IV administration and is ongoing. The company also plans to submit these data for presentation at a future medical meeting.
Investor Webcast
The company will host an investor webcast on the Rylaze approval in July. Details will be announced separately.
About Rylaze™ (asparaginase erwinia chrysanthemi (recombinant)-rywn)
Rylaze, also known as JZP458, is approved in the U.S. for use as a component of a multi-agent chemotherapeutic regimen for the treatment of acute lymphoblastic leukemia (ALL) or lymphoblastic lymphoma (LBL) in pediatric and adult patients one month and older who have developed hypersensitivity to E. coli-derived asparaginase. Rylaze has orphan drug designation for the treatment of ALL/LBL in the United States. Rylaze is a recombinant erwinia asparaginase that uses a novel Pseudomonas fluorescens expression platform. JZP458 was granted Fast Track designation by the U.S. Food and Drug Administration (FDA) in October 2019 for the treatment of this patient population. Rylaze was approved as part of the Real-Time Oncology Review program, an initiative of the FDA’s Oncology Center of Excellence designed for efficient delivery of safe and effective cancer treatments to patients.
The full U.S. Prescribing Information for Rylaze is available at: <http://pp.jazzpharma.com/pi/rylaze.en.USPI.pdf>
Important Safety Information
RYLAZE should not be given to people who have had:
- Serious allergic reactions to RYLAZE
- Serious swelling of the pancreas (stomach pain), serious blood clots, or serious bleeding during previous asparaginase treatment
RYLAZE may cause serious side effects, including:
- Allergic reactions (a feeling of tightness in your throat, unusual swelling/redness in your throat and/or tongue, or trouble breathing), some of which may be life-threatening
- Swelling of the pancreas (stomach pain)
- Blood clots (may have a headache or pain in leg, arm, or chest)
- Bleeding
- Liver problems
Contact your doctor immediately if any of these side effects occur.
Some of the most common side effects with RYLAZE include: liver problems, nausea, bone and muscle pain, tiredness, infection, headache, fever, allergic reactions, fever with low white blood cell count, decreased appetite, mouth swelling (sometimes with sores), bleeding, and too much sugar in the blood.
RYLAZE can harm your unborn baby. Inform your doctor if you are pregnant, planning to become pregnant, or nursing. Females of reproductive potential should use effective contraception (other than oral contraceptives) during treatment and for 3 months following the final dose. Do not breastfeed while receiving RYLAZE and for 1 week after the final dose.
Tell your healthcare provider if there are any side effects that are bothersome or that do not go away.
These are not all the possible side effects of RYLAZE. For more information, ask your healthcare provider.
You are encouraged to report negative side effects of prescription drugs to the FDA. Visit www.fda.gov/medwatch, or call 1-800-FDA-1088 (1-800-332-1088).
About ALL
ALL is a cancer of the blood and bone marrow that can progress quickly if not treated.3 Leukemia is the most common cancer in children, and about three out of four of these cases are ALL.4 Although it is one of the most common cancers in children, ALL is among the most curable of the pediatric malignancies due to recent advancements in treatment.5,6 Adults can also develop ALL, and about four of every 10 cases of ALL diagnosed are in adults.7 The American Cancer Society estimates that almost 6,000 new cases of ALL will be diagnosed in the United States in 2021.7 Asparaginase is a core component of multi-agent chemotherapeutic regimens in ALL.8 However, asparaginase treatments derived from E. coli are associated with the potential for development of hypersensitivity reactions.9
About Lymphoblastic Lymphoma
LBL is a rare, fast-growing, aggressive subtype of Non-Hodgkin’s lymphoma, most often seen in teenagers and young adults.8 LBL is a very aggressive lymphoma – also called high-grade lymphoma – which means the lymphoma grows quickly with early spread to different parts of the body.10,11
About Jazz Pharmaceuticals plc
Jazz Pharmaceuticals plc (NASDAQ: JAZZ) is a global biopharmaceutical company whose purpose is to innovate to transform the lives of patients and their families. We are dedicated to developing life-changing medicines for people with serious diseases – often with limited or no therapeutic options. We have a diverse portfolio of marketed medicines and novel product candidates, from early- to late-stage development, in neuroscience and oncology. We actively explore new options for patients including novel compounds, small molecules and biologics, and through cannabinoid science and innovative delivery technologies. Jazz is headquartered in Dublin, Ireland and has employees around the globe, serving patients in nearly 75 countries. For more information, please visit www.jazzpharmaceuticals.com and follow @JazzPharma on Twitter.
About The Children’s Oncology Group (COG)
COG (childrensoncologygroup.org), a member of the NCI National Clinical Trials Network (NCTN), is the world’s largest organization devoted exclusively to childhood and adolescent cancer research. COG unites over 10,000 experts in childhood cancer at more than 200 leading children’s hospitals, universities, and cancer centers across North America, Australia, and New Zealand in the fight against childhood cancer. Today, more than 90% of the 14,000 children and adolescents diagnosed with cancer each year in the United States are cared for at COG member institutions. Research performed by COG institutions over the past 50 years has transformed childhood cancer from a virtually incurable disease to one with a combined 5-year survival rate of 80%. COG’s mission is to improve the cure rate and outcomes for all children with cancer.
Caution Concerning Forward-Looking Statements
This press release contains forward-looking statements, including, but not limited to, statements related to Jazz Pharmaceuticals’ belief in the potential of Rylaze to provide a reliable therapeutic option for adult and pediatric patients to maximize their chance for a cure, plans for a mid-July 2021 launch of Rylaze, the availability of a reliable supply of Rylaze and other statements that are not historical facts. These forward-looking statements are based on Jazz Pharmaceuticals’ current plans, objectives, estimates, expectations and intentions and inherently involve significant risks and uncertainties. Actual results and the timing of events could differ materially from those anticipated in such forward-looking statements as a result of these risks and uncertainties, which include, without limitation, effectively launching and commercializing new products; obtaining and maintaining adequate coverage and reimbursement for the company’s products; delays or problems in the supply or manufacture of the company’s products and other risks and uncertainties affecting the company, including those described from time to time under the caption “Risk Factors” and elsewhere in Jazz Pharmaceuticals’ Securities and Exchange Commission filings and reports (Commission File No. 001-33500), including Jazz Pharmaceuticals’ Annual Report on Form 10-K for the year ended December 31, 2020 and future filings and reports by Jazz Pharmaceuticals. Other risks and uncertainties of which Jazz Pharmaceuticals is not currently aware may also affect Jazz Pharmaceuticals’ forward-looking statements and may cause actual results and the timing of events to differ materially from those anticipated. The forward-looking statements herein are made only as of the date hereof or as of the dates indicated in the forward-looking statements, even if they are subsequently made available by Jazz Pharmaceuticals on its website or otherwise. Jazz Pharmaceuticals undertakes no obligation to update or supplement any forward-looking statements to reflect actual results, new information, future events, changes in its expectations or other circumstances that exist after the date as of which the forward-looking statements were made.
Jazz Media Contact:
Jacqueline Kirby
Vice President, Corporate Affairs
Jazz Pharmaceuticals plc
CorporateAffairsMediaInfo@jazzpharma.com
Ireland, +353 1 697 2141
U.S. +1 215 867 4910
Jazz Investor Contact:
Andrea N. Flynn, Ph.D.
Vice President, Head, Investor Relations
Jazz Pharmaceuticals plc
investorinfo@jazzpharma.com
Ireland, +353 1 634 3211
References
- Rylaze (asparaginase erwinia chrysanthemi (recombinant)-rywn) injection, for intramuscular use Prescribing Information. Palo Alto, CA: Jazz Pharmaceuticals, Inc.
- Gupta S, Wang C, Raetz EA et al. Impact of Asparaginase Discontinuation on Outcome in Childhood Acute Lymphoblastic Leukemia: A Report From the Children’s Oncology Group. J Clin Oncol. 2020 Jun 10;38(17):1897-1905. doi: 10.1200/JCO.19.03024
- National Cancer Institute. Adult Acute Lymphoblastic Leukemia Treatment (PDQ®)–Patient Version. Available at www.cancer.gov/types/leukemia/patient/adult-all-treatment-pdq. Accessed June 29, 2021
- American Cancer Society. Key Statistics for Childhood Leukemia. Available at https://www.cancer.org/cancer/leukemia-in-children/about/key-statistics.html. Accessed June 29, 2021.
- American Cancer Society. Cancer Facts & Figures 2019. www.cancer.org/research/cancer-facts-statistics/all-cancer-facts-figures/cancer-facts-figures-2019.html. Accessed June 29, 2021.
- Pui C, Evans W. A 50-Year Journey to Cure Childhood Acute Lymphoblastic Leukemia. Seminars in Hematology. 2013;50(3), 185-196.
- American Cancer Society. Key Statistics for Acute Lymphocytic Leukemia (ALL). Available at https://cancerstatisticscenter.cancer.org/?_ga=2.8163506.1018157754.1621008457-1989786785.1621008457#!/data-analysis/NewCaseEstimates. Accessed June 29, 2021.
- Salzer W, Bostrom B, Messinger Y et al. 2018. Asparaginase activity levels and monitoring in patients with acute lymphoblastic leukemia. Leukemia & Lymphoma. 59:8, 1797-1806, DOI: 10.1080/10428194.2017.1386305.
- Hijiya N, van der Sluis IM. Asparaginase-associated toxicity in children with acute lymphoblastic leukemia. Leuk Lymphoma. 2016;57(4):748–757. DOI: 10.3109/10428194.2015.1101098.
- Leukemia Foundation. Lymphoblastic Lymphoma. Available at https://www.leukaemia.org.au/disease-information/lymphomas/non-hodgkin-lymphoma/other-non-hodgkin-lymphomas/lymphoblastic-lymphoma/. Accessed June 29, 2021.
- Mayo Clinic. Acute Lymphocytic Leukemia Diagnosis. Available at https://www.mayoclinic.org/diseases-conditions/acute-lymphocytic-leukemia/diagnosis-treatment/drc-20369083. Accessed June 29, 2021.
SOURCE Jazz Pharmaceuticals plc
Related Links
CLIP
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4776285/

/////////////asparaginase erwinia chrysanthemi (recombinant)-rywn, Rylaze, Jazz Pharmaceuticals, JZP458-201, JZP458, FDA 2021, APPROVALS 2021, ORPHAN, Fast Track, Acute Lymphoblastic Leukemia, ALL, Antineoplastic Agents
https://chem.nlm.nih.gov/chemidplus/id/1349719227
https://go.drugbank.com/drugs/DB08886

NEW DRUG APPROVALS
ONE TIME
$10.00
Soberana 02, FINLAY-FR-2

Soberana 02
FINLAY-FR-2
cas 2543416-58-4
A SARS-CoV-2 vaccine comprising a conjugate of the spike protein RBD domain with tetanus toxoid (Finlay Vaccine Institute of Cuba)
Soberana 02, is a conjugate vaccine developed by Instituto Finlay de Vacunas.[517]
Cuba[518]
Iran[517]
517 Zimmer, Carl; Corum, Jonathan; Wee, Sui-Lee. “Coronavirus Vaccine Tracker”. The New York Times. Retrieved 30 June 2021.
518 Sesin, Carmen (14 May 2021). “Cuba begins mass Covid-19 vaccine inoculation before concluding trials”. NBC News. Retrieved 2 July 2021.
Soberana 02, technical name FINLAY-FR-2, is a COVID-19 vaccine produced by the Finlay Institute, a Cuban epidemiological research institute. It is a conjugate vaccine. This candidate followed a previous one called SOBERANA-01 (FINLAY-FR-1).[2] Professor Ihosvany Castellanos Santos said that the antigen is safe because it contains parts instead of the whole live virus, and therefore it does not require extra refrigeration, like other candidates in the world.[3] According to the WHO candidate landscape vaccine document, this vaccine requires two doses, the second one being administered 28 days after the first shot.[4]
The name of the vaccine, Soberana, is a Spanish word that means “sovereign”.[5]

Efficacy
It has shown an efficacy of 62% after only two doses, according to BioCubaFarma, though a pre-print or details of the study have not been released.[6][7][8]
Pharmacology
FINLAY-FR-2 is a conjugate vaccine. It consists of the receptor binding domain of the SARS-CoV-2 spike protein conjugated chemically to tetanus toxoid.[2]
Manufacturing
The spike protein subunit is produced in Chinese hamster ovary cell culture.[2] In a pre-print article scientists from Cuba explain details of the vaccines technology and production.[9][non-primary source needed]
| Production Deliveries Planned Production Potential Production |
Deliveries (0)Effective production (implies deliveries) (1)
Planned production
- Iran
Potential Production
- Ghana
- Argentina
In Cuba
The Cuban government says it is planning to produce 100 million doses of its vaccine to respond to its own demand and that of other countries.[12][13] Cuba has also suggested that, once it’s approved, it will offer the vaccine to tourists visiting the country.[14][15][16]
The production of the first batch of about 100,000 doses will start in April.[17] José Moya, representative of the World Health Organization and the Pan American Health Organization (PAHO) in Cuba, suggested that after the vaccine passes all clinical stages, it could be included as part of PAHO’s Revolving Fund.[18]
The roll-out began with an “Interventional Trial”[19] that consisted of inoculating 150,000 at-risk participants which seems to be defined as health-care workers.[20][21] On April 11, 2021, the Ministry of Public Health of Cuba announced that 75,000 health-care workers were inoculated with their first dose of either of the two Cuba’s Phase III vaccines (the other being Abdala).[22][23]
Outside Cuba
Vietnam, Iran, Venezuela, Argentina,[24][25][26] Pakistan, India, the African Union, Jamaica and Suriname[27] have expressed interest in purchasing the vaccine, although they are waiting on Phase 3 results.[28][29]
Iran has signed an agreement to manufacture the vaccine[30] and Argentina is negotiating one.[24][25][26] Additionally, the Cuban government offered a “transfer of technology” to Ghana and will also supply “active materials” needed to make the vaccine.[31][32][33]
While the price is currently unknown, the commercialization strategy of the vaccine will be a combination of the “impact on health” and the capability of Cuba’s system to financially support “the production of vaccines and drugs for the country”, per the director of the Finlay Institute, Vicente Vérez.[34]
Clinical trials
Phase I
FINLAY-FR-2, which started being developed in October 2020, had 40 volunteers for its Phase I, according to the Cuban Public Registry of Clinical Trials, with an open, sequential and adaptive study to assess safety, reactogenicity and explore immunogenicity of the vaccine.[35]
Phase II
Phase IIa involved 100 Cubans, and phase IIb of the vaccine will have 900 volunteers between 19 and 80 years.[36][37] Vicente Vérez, director general of the Finlay Vaccine Institute, said that the vaccine has shown to give an immune response after 14 days.[38] The second phase has been supervised by Iranian officials from the Pasteur Institute.[5]
Phase III
Phase III commenced at the beginning of March as originally scheduled,[39][15] and “ready to publish” results are expected by June.[40][41][42] The trial volunteers are divided into three groups: some will receive two doses of the vaccine 28 days apart, another group will get two doses plus a third immune booster (Soberana Plus[43][44][45]), and the third a placebo.[39]
Although the trials involve thousands of adult volunteers recruited in Havana,[46] Cuba’s public health officials have said that they will also need to conduct phase III trials abroad because the island doesn’t have an outbreak of sufficient scale to produce meaningful statistics on vaccine protection.[5][14]
On March 13, 2021, the Cuban Biotechnology and Pharmaceutical Industries Business Group (BioCubaFarma) announced on social media that it had sent 100,000 doses of its Soberana 02 coronavirus vaccine candidate to the Pasteur Institute of Iran for clinical testing, “as part of the collaboration with other countries in the development of COVID-19 vaccines.” [47]
On April 26, 2021, it was reported that a Phase III conducted by the Pasteur Institute of Iran was approved to be started in Iran[48][49][50] It was previously reported that the Institute will host Phase 3 but the pre-requisites were “technology transfer and joint production”.[51][5]
Mexico plans to host a phase 3 trial.[52]
Interventional Study
The “Interventional Study” is set both in Havana,[53] Cuba’s capital and Santiago de Cuba, Cuba’s second most populous city [54][55] and in other provinces.[56] On May 6, 2021, the Finlay Institute of Vaccines announced on social media that the following adverse events have been observed: injection site pain (20%), inflammation at the injection site (5%), and general discomfort (5%).[57][58]
Authorizations
| Full authorization Emergency authorization |
See also: List of COVID-19 vaccine authorizations § Soberana 02
References
- ^ “Cuba’s Soberana Plus against Covid-19 is showing good results”. Prensa Latina. Retrieved 10 May 2021.
- ^ Jump up to:a b c Malik JA, Mulla AH, Farooqi T, Pottoo FH, Anwar S, Rengasamy KR (January 2021). “Targets and strategies for vaccine development against SARS-CoV-2”. Biomedicine & Pharmacotherapy. 137: 111254. doi:10.1016/j.biopha.2021.111254. PMC 7843096. PMID 33550049.
- ^ Santos IC (January 2021). “Rapid response to: Covid 19: Hope is being eclipsed by deep frustration”. BMJ. 372: n171. doi:10.1136/bmj.n171.
- ^ “Draft landscape and tracker of COVID-19 candidate vaccines”. http://www.who.int. World Health Organization. Retrieved 2021-02-04.
- ^ Jump up to:a b c d Rasmussen SE, Eqbali A (12 January 2021). “Iran, Cuba, Under U.S. Sanctions, Team Up for Covid-19 Vaccine Trials”. The Wall Street Journal.
- ^ “Cuba’s homegrown Covid vaccine shows promise”. http://www.ft.com. Retrieved 2021-06-20.
- ^ “Cuba encouraged by early efficacy results of homegrown COVID-19 vaccine”. http://www.zawya.com. Retrieved 2021-06-20.
- ^ Acosta, Nelson (2021-06-20). “Cuba encouraged by early results of homegrown COVID-19 vaccine amid worst outbreak”. The Age. Retrieved 2021-06-20.
- ^ Valdes-Balbin, Yury; Santana-Mederos, Darielys; Quintero, Lauren; Fernández, Sonsire; Rodriguez, Laura; Ramirez, Belinda Sanchez; Perez, Rocmira; Acosta, Claudia; Méndez, Yanira; Ricardo, Manuel G.; Hernandez, Tays (2021-02-09). “SARS-CoV-2 RBD-Tetanus toxoid conjugate vaccine induces a strong neutralizing immunity in preclinical studies”. doi:10.1101/2021.02.08.430146.
- ^ Melimopoulos, Elizabeth. “Is Cuba closing in on COVID vaccine sovereignty?”. http://www.aljazeera.com. Retrieved 2021-05-07.
- ^ “Optimism as Cuba set to test its own Covid vaccine”. BBC News. 2021-02-16. Retrieved 2021-05-07.
- ^ “Cuba espera fabricar 100 millones de dosis de su candidato vacunal Soberana 02”. Nodal (in Spanish). 21 January 2021.
- ^ “Vaccino, Cuba pronta a produrre 100 milioni di dosi di ‘Soberana 02′”. Dire (in Italian). 21 January 2021.
- ^ Jump up to:a b Ribeiro G (4 February 2021). “Cuba to offer coronavirus vaccines to tourists”. Brazilian Report.
- ^ Jump up to:a b “Coronavirus: Vacuna cubana Soberana 02 alista fase 3 y ensayos”. Deutsche Welle (in Spanish). 5 February 2021.
- ^ Meredith S (23 February 2021). “‘Sun, sea, sand and Soberana 02’: Cuba open to inoculating tourists with homegrown Covid vaccine”. CNBC.
- ^ “Coronavirus: Vacuna cubana Soberana 02 alista fase 3 y ensayos”. Deutsche Welle (in Spanish). 5 February 2021.
Las expectativas sobre Soberana 02 son tales que el titular del organismo estatal que desarrolló la vacuna, Vicente Vérez, confirmó que mientras se aguarden los resultados de la Fase 3 solo en La Habana, en abril se dará inicio a la producción del primer lote, de alrededor de 100 mil dosis.
- ^ “Cuba anuncia fase 3 de la vacuna Soberana 02”. La Jornada(in Spanish). 7 February 2021.
Una vez que superen las etapas clínicas, la OMS podría contar con el fármaco cubano, afirmó Moya, y “pasar a ser parte del grupo de vacunas que se oferten a través del Fondo Rotatorio”, un mecanismo que desde hace cuatro décadas permite gestionar antígenos e insumos a los países de las Américas.
- ^ “SOBERANA – INTERVENTION | Registro Público Cubano de Ensayos Clínicos”. rpcec.sld.cu. Retrieved 2021-04-11.
- ^ “Cuba says it’s ‘betting it safe’ with its own Covid vaccine”. NBC News. Retrieved 2021-04-11.
- ^ “Cuba begins testing 2nd COVID-19 vaccine on health care workers”. medicalxpress.com. Retrieved 2021-04-11.
- ^ Ministry of Public Health of Cuba (11 April 2021). “[Translated] “The administration of the 1st dose of the Cuban vaccine candidates #Soberana02 and #Abdala to the 75 thousand health workers and Biocubafarma who are part of the intervention study taking place in #LaHabana has concluded.””. Twitter. Retrieved 2021-04-11.
- ^ “Cuban scientists, health workers received first anti-Covid-19 dose”. http://www.plenglish.com/index.php?o=rn&id=66247&SEO=cuban-scientists-health-workers-received-first-anti-covid-19-dose (in Spanish). Retrieved 2021-04-11.
- ^ Jump up to:a b “ILARREGUI (EMBAJADOR EN CUBA): “DURANTE ESTE AÑO PODREMOS TENER VACUNAS CUBANAS EN ARGENTINA””. RadioCut. Retrieved 2021-05-07.
- ^ Jump up to:a b Argentina, Cadena 3. “Argentina comenzó a negociar con Cuba la vacuna Soberana”. Cadena 3 Argentina (in Spanish). Retrieved 2021-05-07.
- ^ Jump up to:a b de 2021, 6 de Mayo. “Sin definiciones sobre cuándo podrían llegar, el Gobierno avanza para conseguir las vacunas Soberana y Abdala de Cuba”. infobae (in Spanish). Retrieved 2021-05-07.
- ^ admin (2021-04-09). “Cuba’s COVID-19 Vaccines Being Sought After by CARICOM Countries”. Caribbean News. Retrieved 2021-05-07.
- ^ Guenot, Marianne (2021-02-15). “Cuba is working on a homegrown COVID-19 vaccine program. It has a history of fighting disease without help from the West”. Business Insider France (in French). Retrieved 2021-05-07.
- ^ Página12 (2021-01-22). “Soberana 02: Cuba prepara cien millones de dosis de la vacuna contra el coronavirus | “No somos una multinacional. Nuestro fin es crear salud”, dijo el director del Instituto Finlay de Vacunas”. PAGINA12. Retrieved 2021-05-07.
- ^ “Cuban coronavirus vaccine to start third clinical trial phase in Iran”. Tehran Times. 2021-04-18. Retrieved 2021-05-07.
- ^ Banini | 0542440286, Awofisoye Richard. “CEO OF FDA DISCUSSES PRODUCTION OF COVID-19 VACCINE WITH CUBAN AMBASSADOR”. http://www.fdaghana.gov.gh. Retrieved 2021-05-05.
- ^ “Cuba To Transfer COVID-19 Vaccine Technology To Ghana”. http://www.gnbcc.net. Retrieved 2021-05-05.
- ^ “Cuban government offers to transfer COVID-19 Soberana 02 vaccine technology to Ghana”. Rio Times Online. 16 February 2021.
- ^ “Coronavirus: Cuba will produce 100 million doses of its Soberana 02 vaccine”. OnCubaNews English. 2021-01-21. Retrieved 2021-05-07.
- ^ “SOBERANA 02 | Registro Público Cubano de Ensayos Clínicos”. Cuban Registry of Clinical Trials (in Spanish). Retrieved 24 January 2021.
- ^ Cuba inicia nova fase de testes com vacina que desenvolve contra covid-19 (in Portuguese), Universo Online, 19 January 2021, Wikidata Q105047566
- ^ “Cuba apuesta por crear primera vacuna de América Latina contra el covid-19”. France 24 (in Spanish). 2021-01-21. Retrieved 24 January 2021.
- ^ “Cuba negotiates with other countries to develop phase 3 of Soberana 02 vaccine”. OnCubaNews English. 2020-12-30. Retrieved 24 January 2021.
- ^ Jump up to:a b “Cuban-developed vaccine enters Phase III trial”. ABS CBN. 5 March 2021.
- ^ Mega, Emiliano Rodríguez (2021-04-29). “Can Cuba beat COVID with its homegrown vaccines?”. Nature. doi:10.1038/d41586-021-01126-4. PMID 33927405.
- ^ “Cuban Vaccine Ready in July. Interview with the Cuban Ambassador to the Czech Republic”. Pressenza. 2021-03-23. Retrieved 2021-04-29.
- ^ Augustin, Ed (2021-05-12). “Cuba deploys unproven homegrown vaccines, hoping to slow an exploding virus outbreak”. The New York Times. ISSN 0362-4331. Retrieved 2021-05-14.
- ^ “L’esempio cubano sui vaccini”. http://www.ilfoglio.it (in Italian). Retrieved 2021-05-07.
- ^ Avances de las vacunas cubanas contra la COVID-19, retrieved 2021-05-07
- ^ Mega, Emiliano Rodríguez (2021-04-29). “Can Cuba beat COVID with its homegrown vaccines?”. Nature. doi:10.1038/d41586-021-01126-4. PMID 33927405.
- ^ Yaffe, Helen. “Cuba’s five COVID-19 vaccines: the full story on Soberana 01/02/Plus, Abdala, and Mambisa”. LSE Latin America and Caribbean blog. Retrieved 2021-03-31.
- ^ “Cuba sends 100,000 doses of the Soberana 02 vaccine candidate to Iran” oncubanews.com. Retrieved 19 March 2021.
- ^ “Iran-Cuba vaccine enters phase three clinical trials”. Tehran Times. 2021-04-26. Retrieved 2021-04-28.
- ^ “Cuban coronavirus vaccine to start third clinical trial phase in Iran”. Tehran Times. 2021-04-18. Retrieved 2021-04-28.
- ^ “América Latina apura una vacuna propia. Cuba, adelante; México avanza. Pero no son los únicos”. http://www.poresto.net (in Spanish). Retrieved 2021-04-28.
- ^ Marsh S (2021-01-09). “Cuba to collaborate with Iran on coronavirus vaccine”. Reuters. Retrieved 2021-01-24.
- ^ “Mexico Hopes to Work With Cuba on Covid Vaccine Phase 3 Trial”. Bloomberg.com. 2021-02-14. Retrieved 2021-05-07.
- ^ Marsh, Sarah (2021-03-24). “Nearly all Havana to receive experimental Cuban COVID-19 vaccines”. Reuters. Retrieved 2021-04-28.
- ^ BioCubaFarma (April 6, 2021). “[Translated] Updating the vaccination process with vaccine candidates #Soberana02 and #Abdala during ongoing clinical trials.#VacunasCubanasCovid19”. Twitter (in Spanish). Retrieved 2021-04-11.
- ^ “Intervention study with Covid-19 vaccine candidate Abdala begins”. Radio Cadena Agramonte. Retrieved 2021-04-28.
- ^ “Cuba administers over 62,000 doses in intervention trials”. http://www.plenglish.com/index.php?o=rn&id=66012&SEO=cuba-administers-over-62000-doses-in-intervention-trials (in Spanish). Retrieved 2021-04-28.
- ^ “[Trnslated] In more than 62 thousand applied doses of #Soberana02 the safety of the vaccine has been demonstrated. Adverse effects have been: 👉 Pain at the injection site (20%). 👉 Redness at the injection site (5%). 👉 Feeling of general malaise (5%)”. Twitter. Retrieved 2021-05-07.
- ^ “[Translated]In more than 62 thousand applied doses of #Soberana02 the safety of the vaccine has been demonstrated. Adverse effects have been: 👉 Pain at the injection site (20%). 👉 Redness at the injection site (5%). 👉 Feeling of general malaise (5%)”. Facebook. Retrieved 2021-05-07.
External links
| Scholia has a profile for SOBERANA 02 (Q105047585). |
| Vaccine description | |
|---|---|
| Target | SARS-CoV-2 |
| Vaccine type | Conjugate |
| Clinical data | |
| Other names | FINLAY-FR-2, SOBERANA PLUS[1] |
| Routes of administration | Intramuscular |
| Legal status | |
| Legal status | Full and Emergency Authorizations: List of Soberana 02 authorizations |
| Part of a series on the |
| COVID-19 pandemic |
|---|
| COVID-19 (disease)SARS-CoV-2 (virus)CasesDeaths |
| showTimeline |
| showLocations |
| showInternational response |
| showMedical response |
| showEconomic impact and recession |
| showImpacts |
| COVID-19 portal |
/////////////////SARS-CoV-2, covid 19, corona virus, vaccine, iran, cuba, Soberana 02, FINLAY-FR-2
Nature (London, United Kingdom) (2021),

NEW DRUG APPROVALS
one time
$10.00
Rezivertinib
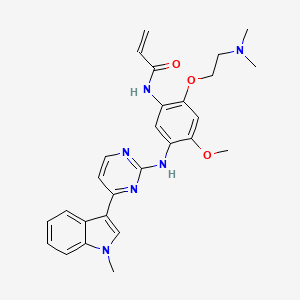
BPI-7711, Rezivertinib
1835667-12-3
C27H30N6O3, 486.576
N-[2-[2-(dimethylamino)ethoxy]-4-methoxy-5-[[4-(1-methylindol-3-yl)pyrimidin-2-yl]amino]phenyl]prop-2-enamide
Beta Pharma in collaboration Chinese licensee CSPC Pharmaceuticals Group , is developing BPI-7711
In June 2021, this drug was reported to be in phase 3 clinical development.
APPROVALS 2024, CHINA 2024
- OriginatorBeta Pharma
- ClassAmides; Amines; Antineoplastics; Indoles; Phenyl ethers; Pyrimidines; Small molecules
- Mechanism of ActionEpidermal growth factor receptor antagonists
- Phase IIINon-small cell lung cancer
- 30 Dec 2020Chemical structure information added
- 09 Apr 2020Beta Pharma initiates a phase I trial for Non-small cell lung cancer (In volunteers) in China (PO) (NCT04135833)
- 25 Mar 2020Beta Pharma completes a phase I pharmacokinetic trial for Non-small cell lung cancer (In volunteers) in China (NCT04135820)
N-(2-(2-(Dimethylamino)ethoxy)-4-methoxy-5-((4-(1-methyl-1H-indol-3-yl)-2-pyrimidinyl)amino)phenyl)-2-propenamideThe epidermal growth factor receptor (EGFR, Herl, ErbB l) is a principal member of the ErbB family of four structurally-related cell surface receptors with the other members being Her2 (Neu, ErbB2), Her3 (ErbB3) and Her4 (ErbB4). EGFR exerts its primary cellular functions though its intrinsic catalytic tyrosine protein kinase activity. The receptor is activated by binding with growth factor ligands, such as epidermal growth factor (EGF) and transforming growth factor-alpha (TGF-a), which transform the catalytically inactive EGFR monomer into catalytically active homo- and hetero- dimers. These catalytically active dimers then initiate intracellular tyrosine kinase activity, which leads to the autophosphorylation of specific EGFR tyrosine residues and elicits the downstream activation of signaling proteins. Subsequently, the signaling proteins initiate multiple signal transduction cascades (MAPK, Akt and JNK), which ultimately mediate the essential biological processes of cell growth, proliferation, motility and survival.EGFR is found at abnormally high levels on the surface of many types of cancer cells and increased levels of EGFR have been associated with advanced disease, cancer spread and poor clinical prognosis. Mutations in EGFR can lead to receptor overexpression, perpetual activation or sustained hyperactivity and result in uncontrolled cell growth, i.e. cancer. Consequently, EGFR mutations have been identified in several types of malignant tumors, including metastatic lung, head and neck, colorectal and pancreatic cancers. In lung cancer, mutations mainly occur in exons 18 to 21, which encode the adenosine triphosphate (ATP)-binding pocket of the kinase domain. The most clinically relevant drug- sensitive EGFR mutations are deletions in exon 19 that eliminate a common amino acid motif (LREA) and point mutations in exon 21, which lead to a substitution of arginine for leucine at position 858 (L858R). Together, these two mutations account for nearly 85% of the EGFR mutations observed in lung cancer. Both mutations have perpetual tyrosine kinase activity and as a result they are oncogenic. Biochemical studies have demonstrated that these mutated EGFRs bind preferentially to tyrosine kinase inhibitor drugs such as erlotinib and gefitinib over adenosine triphosphate (ATP).Erlotinib and gefitinib are oral EGFR tyrosine kinase inhibitors that are first line monotherapies for non-small cell lung cancer (NSCLC) patients having activating mutations in EGFR. Around 70% of these patients respond initially, but unfortunately they develop resistance with a median time to progression of 10-16 months. In at least 50% of these initially responsive patients, disease progression is associated with the development of a secondary mutation, T790M in exon 20 of EGFR (referred to as the gatekeeper mutation). The additional T790M mutation increases the affinity of the EGFR kinase domain for ATP, thereby reducing the inhibitory activity of ATP- competitive inhibitors like gefitinib and erlotinib.Recently, irreversible EGFR tyrosine kinase inhibitors have been developed that effectively inhibit the kinase domain of the T790M double mutant and therefore overcome the resistance observed with reversible inhibitors in the clinic. These inhibitors possess reactive electrophilic functional groups that react with the nucleophilic thiol of an active-site cysteine. Highly selective irreversible inhibitors can be achieved by exploiting the inherent non-covalent selectivity of a given scaffold along with the location of a particular cysteine residue within the ATP binding site. The acrylamide moieties of these inhibitors both undergo a Michael reaction with Cys797 in the ATP binding site of EGFRT790M to form a covalent bond. This covalent mechanism is thought to overcome the increase in ATP affinity of the T790M EGRF double mutant and give rise to effective inhibition. However, these inhibitors may cause various undesired toxicities. Therefore, development of new inhibitors for treatment of various EGFR-related cancers is still in high demand.
PatentCN201580067776) N-(2-(2-(dimethylamino)ethoxy)-4-methoxy-5-((4-(1-methyl-1H- Indol-3-yl)pyrimidin-2-yl)amino)phenyl)acrylamide (compound of formula I) can be prepared by the following synthetic route:
PATENT
https://patents.google.com/patent/WO2016094821A2/enExample 1N-(2-(2-(Dimethylamino)ethoxy)-4-methoxy-5-((4-(l-methyl-lH-indol-3- yl)pyrimidin-2-yl)amino)phenyl)acrylamide (1) Sche

N-(4-(2-(Dimethylamino)ethoxy)-2-methoxy-5-nitrophenyl)-4-(l-methyl-lH- indol-3-yl)pyrimidin-2-amine (Scheme 1, Intermediate B). To a slurry of NaH (30 mmol, 60% oil dispersion prewashed with hexanes) and 50 mL of 1,4-dioxane was added 2-dimethylaminoethanol (27 mmol, 2.7 mL) dropwise with stirring under N2. After stirring for 1 h, a slurry of A (5.4 mmol) in 50 mL of 1,4-dioxane was added portion-wise over 15 min under a stream of N2. The resulting mixture was stirred overnight, then poured into water and the solid was collected, rinsed with water, and dried under vacuum to yield 2.6 g of product as a yellow solid. A purified sample was obtained from chromatography (silica gel; CH2C12-CH30H gradient). 1H NMR (300 MHz, DMSO) δ 2.26 (s, 6H), 2.70 (t, 2H, J = 6 Hz), 3.87 (s, 3H), 4.01 (s, 3H), 4.32 (t, 2H, J = 6 Hz), 7.00-7.53 (m, 5H), 8.18-8.78 (m, 5H); C24H26N604 m/z MH+ 463.4-(2-(Dimethylamino)ethoxy)-6-methoxy-Nl-(4-(l-methyl-lH-indol-3- yl)pyrimidin-2-yl)benzene-l,3-diamine (Scheme 1, Intermediate C). A suspension of 2.6 g of Intermediate B, 1.6 g of Fe°, 30 mL of ethanol, 15 mL of water, and 20 mL of cone. HC1 was heated to 78 °C for 3 h. The solution was cooled to room temperature, adjusted to pH 10 with 10% NaOH (aq) and diluted with CH2C12. The mixture was filtered through Dicalite, and the filtrate layers were separated. The aqueous phase was extracted with CH2C12 twice, and the combined organic extracts were dried over Na2S04 and concentrated. Column chromatography (silica gel, CH2Cl2-MeOH gradient) afforded 1.2 g of Intermediate C as a solid. C24H28N602 m/z MH+ 433.N-(2-(2-(Dimethylamino)ethoxy)-4-methoxy-5-((4-(l-methyl-lH-indol-3- yl)pyrimidin-2-yl)amino)phenyl)acrylamide (1). To a solution of Intermediate C (2.8 mmol) in 50 mL of THF and 10 mL of water was added 3-chloropropionychloride (2.8 mmol) dropwise with stirring. After 5 h of stirring, NaOH (28 mmol) was added and the mixture was heated at 65°C for 18 h. After cooling to room temperature, THF was partially removed under reduced pressure, and the mixture was extracted with CH2C12, dried over Na2S04, and concentrated. Chromatography of the crude product (silica gel, CH2Cl2-MeOH) afforded 0.583 g of Example 1 as a beige solid. 1H NMR (300 MHz, DMSO) δ 2.28 (s, 6H), 2.50-2.60 (m, 2H), 3.86 (s, 3H), 3.90 (s, 3H), 4.19 (t, 2H, = 5.5 Hz), 5.73-5.77 (m, IH), 6.21-6.27 (m, IH), 6.44-6.50 (m, IH), 6.95 (s, IH), 7.11-7.53 (overlapping m, 3H), 7.90 (s, IH), 8.27-8.30 (overlapping m, 3H), 8.55 (s, IH), 8.84 (s, IH), 9.84 (s, IH) ppm; C27H30N6O3 m/z MH+ 487
PATENT WO2021115425
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2021115425&tab=FULLTEXT&_cid=P20-KQN9F3-73566-1Epidermal growth factor receptors (EGFR, Her1, ErbB1) are the main members of the ErbB family of four structurally related cell surface receptors, and the other members are Her2 (Neu, ErbB2), Her3 (ErbB3) and Her4 (ErbB4). EGFR exerts its main cellular functions through its inherent catalytic tyrosine protein kinase activity. The receptor is activated by binding to growth factor ligands, such as epidermal growth factor (EGF) and transforming growth factor-α (TGF-α). The catalytically inactive EGFR monomer is transformed into a catalytically active homopolymer and Heterodimer. These catalytically active dimers then initiate intracellular tyrosine kinase activity, which leads to autophosphorylation of specific EGFR tyrosine residues and elicits downstream activation of signaling proteins. Subsequently, the signal protein initiates multiple signal transduction cascades (MAPK, Akt, and JNK), which ultimately regulate the basic biological processes of cell growth, proliferation, motility, and survival.
EGFR has been found to have abnormally high levels on the surface of many types of cancer cells, and elevated EGFR levels have been associated with advanced disease, cancer spread, and poor clinical prognosis. Mutations in EGFR can lead to overexpression of the receptor, permanent activation or continuous hyperactivity, leading to uncontrolled cell growth, which is cancer. Therefore, EGFR mutations have been identified in several types of malignant tumors, including metastatic lung cancer, head and neck cancer, colorectal cancer, and pancreatic cancer. In brain cancer, mutations mainly occur in exons 18-21, which encode the adenosine triphosphate (ATP)-binding pocket of the kinase domain. The most clinically relevant drug-sensitive EGFR mutations are deletions in exon 19 and point mutations in exon 21. The former eliminates a common amino acid motif (LREA), and the latter results in position 858 (L858R). The arginine is replaced by leucine. Together, these two mutations account for nearly 85% of the EGFR mutations observed in lung cancer. Both mutations have permanent tyrosine kinase activity, so they are carcinogenic. In at least 50% of patients who initially responded to current therapies, the progression of the disease is related to the development of a secondary mutation, T790M (also known as the goalkeeper mutation) in exon 20 of EGFR.
BPI-7711 is a third-generation EGFR-TKI compound developed by Beida Pharmaceuticals and disclosed in International Patent No. WO2017/218892. It is the N-(2-(2-(dimethylamino) )Ethoxy)-4-methoxy-5-((4-(1-methyl-1H-indol-3-yl)pyrimidin-2-yl)amino)phenyl)acrylamide methanesulfonic acid salt:
Need to develop improved properties containing N-(2-(2-(dimethylamino)ethoxy)-4-methoxy-5-((4-(1-methyl-1H-indole-3 -Yl)pyrimidin-2-yl)amino)phenyl)acrylamide pharmaceutically acceptable salt, in particular the pharmaceutical composition of BPI-7711 and its use, and the preparation of said pharmaceutical composition suitable for large-scale production method.
PATENT
WO2021061695 , for another filing, assigned to Beta Pharma, claiming a combination of an EGFR inhibitor (eg BPI-7711) and a CDK4/6 inhibitor, useful for treating cancer.
PATENT
WO-2021121146
Novel crystalline polymorphic form A of rezivertinib – presumed to be BPI-7711 – useful for treating diseases mediated by EGFR mutations eg lung cancer, preferably non-small cell lung cancer (NSCLC).Epidermal growth factor receptor (EGFR) is a type of transmembrane receptor tyrosine kinase in the human body. The activation (ie phosphorylation) of this kinase is of great significance to the inhibition of tumor cell proliferation, angiogenesis, tumor invasion, metastasis and apoptosis. EGFR kinase is involved in the disease process of most cancers, and these receptors are overexpressed in many major human tumors. Overexpression, mutations, or high expression of ligands associated with these family members can lead to some tumor diseases, such as non-small cell lung cancer, colorectal cancer, breast cancer, head and neck cancer, cervical cancer, bladder cancer, and thyroid. Cancer, stomach cancer, kidney cancer, etc.
In recent years, epidermal growth factor receptor tyrosine kinase has become one of the most attractive targets in current anti-tumor drug research. In 2003, the US FDA approved the first epidermal growth receptor tyrosine kinase inhibitor (EGFR-TKI) drug (gefitinib) for the treatment of advanced non-small cell lung cancer (NSCLC). Development of a generation of EGFR inhibitors. Numerous clinical trials have confirmed that for patients with EGFR-positive non-small cell lung cancer, the therapeutic effect of molecular targeted drugs is significantly better than traditional chemotherapy.
Although the first-generation EGFR-inhibiting targeted drugs responded well to the initial treatment of many non-small cell lung cancer (NSCLC) patients, most patients will eventually develop disease progression due to drug resistance (such as EGFR secondary T790M mutation). The emergence of drug resistance is caused by various mechanisms based on the mutations in the original EGFR pathway activity. In the drug resistance research on the first generation of EGFR inhibitors, the research frontier is the irreversible third generation EFGR inhibitor.
But so far, the third-generation EGFR inhibitors worldwide, in addition to AstraZeneca O’Higgins imatinib developed, there is no other effective against T790M resistance mutations in patients with drug approved for clinical use; Several drug candidates for the T790M mutation are in clinical development. The chemical structure of this third-generation EGFR inhibitor is completely different from that of the first-generation. The main difference from the first-generation EGFR inhibitors is that they both use a highly selective core structure to replace the low-selective aminoquinoline core structure of the first and second-generation EGFR-TKIs. Compared with wild-type EGFR, these third-generation compounds are highly specific and selective for the T790M mutation after EGFR positive resistance.
Chinese Patent Application No. CN201580067776.8 discloses a compound of the following formula I, which also belongs to the third-generation EGFR-TKI class of small molecule targeted drugs. The compound has a high inhibitory effect on non-small cell lung cancer (NSCLC) cells with single-activity mutation and T790M double-mutant EGFR, and its effective inhibitory concentration is significantly lower than the concentration required to inhibit the activity of wild-type EGFR tyrosine kinase. It has good properties, low side effects and good safety.
Chinese Patent Application No. CN201780050034.3 also discloses various salts and corresponding crystal forms of the compound of the above formula I. Example 2 discloses two crystal forms of the methanesulfonate of the compound of formula I, 2A and 2B, respectively.In the following examples, the “room temperature” can be 15-25°C.[0041](1) N-(2-(2-(Dimethylamino)ethoxy)-4-methoxy-5-((4-(1-methyl-1H-indol-3-yl)pyrimidine -2-yl)amino)phenyl)acrylamide (compound of formula I)[0042]
[0043]Known (for example, see CN201580067776.8) N-(2-(2-(dimethylamino)ethoxy)-4-methoxy-5-((4-(1-methyl-1H- Indol-3-yl)pyrimidin-2-yl)amino)phenyl)acrylamide (compound of formula I) can be prepared by the following synthetic route:[0044]
[0045]Step 1-Preparation of Intermediate J:[0046]
[0047]Preparation: In a 10L reaction flask, add 6L of anhydrous tetrahydrofuran solvent, protected by nitrogen, and cool to 0°C. While stirring, slowly add 101 g of sodium hydride (101 g, 2.52 mol), and the internal temperature does not exceed 10° C., and add 234 g of dimethylaminoethanol (234 g, 2.62 mol). After the addition, the temperature is adjusted to room temperature to prepare a sodium alkoxide solution.[0048]In a 30L reaction flask, add N-(4-fluoro-2-methoxy-5-nitrophenyl)-4-(1-methyl-1H-indol-3-yl)-2-pyrimidinamine ( Starting material B) (430g, 1.10mol), then add 9L of tetrahydrofuran, start stirring, dissolve it, control the temperature at 10±10°C, slowly add the prepared sodium alkoxide solution dropwise. Control the temperature at 10±10℃ and keep it for 5.0h. When the raw material content is ≤0.5%, the reaction ends. Control the temperature at 10±10°C, slowly add 3% hydrochloric acid solution dropwise, adjust the pH of the solution to 6-7, stir for 1.5h and then stand for stratification, separate the organic phase, and concentrate to 15-20L. After cooling to 20±5°C, 4.3 kg of water was slowly added dropwise, filtered, and dried to obtain 497 g of yellow powder intermediate J with a yield of 98.0% and an HPLC purity of 99.3%. MS m/z: 463.2 [M+1].[0049]Nuclear magnetic data: 1 HNMR (d 6 -DMSO): δ ppm: 8.78 (s, 1H); 8.42-8.28 (m, 3H); 8.16 (s, 1H); 7.53 (d, 1H, J = 8.28); 7.29- 7.20 (m, 2H); 7.13-7.07 (m, 1H); 7.01 (s, 1H); 4.33 (t, 2H, J = 5.65); 4.02 (s, 3H); 3.88 (s, 3H); 2.71 ( t, 2H, J = 5.77); 2.27 (s, 6H).[0050]Step 2-Preparation of Intermediate K:[0051]
[0052]Preparation: Add 5L of tetrahydrofuran and Intermediate J (350g, 108mmol) to a 10L hydrogenation reactor, add 17.5g of wet palladium charcoal, replace the hydrogenation reactor with hydrogen, adjust the pressure value to 0.2MPa, control the temperature at 25°C, and keep the temperature for reaction. At 9h, HPLC monitors the progress of the reaction, and stops the reaction when the substrate is ≤0.5%. Filter, concentrate the filtrate under reduced pressure until the solvent volume is about 2L, adjust the internal temperature to room temperature, slowly add 4L n-heptane dropwise within 4-7 hours, filter and dry the solid under reduced pressure to obtain 285g of white powder intermediate K The yield was 86%, and the HPLC purity was 99.60%. MS m/z: 433.3 [M+1].
Nuclear magnetic data: 1 HNMR (CDCl 3 ): δ ppm: 8.42 (d, 1H, J = 7.78), 8.28 (s, 1H), 8.26-8.23 (m, 1H), 7.78 (s, 1H), 7.51 (d, 1H,J=8.28),7.41(s,1H),7.26-7.23(m,1H),7.19- 7.11(m,2H),6.72(s,1H), 4.38(br,2H),4.06(t, 2H,J=5.77), 3.88(s,3H), 3.75(s,3H), 2.63(t,2H,J=5.77), 2.26(s,6H).
Step 3-Preparation of compound of formula I:
Add 250 mL of anhydrous tetrahydrofuran solvent and Intermediate K (14 g, 32 mmol) to the reaction flask and stir, cool to 0-5° C., add 10% hydrochloric acid (12 ml), and stir for 20 minutes. At 0-5°C, slowly drop 3-chloropropionyl chloride (5.6 g, 45 mmol) into the reaction flask. Stir for 3 hours, after sampling test (K/(U+K)≤0.5%) is qualified, add 36% potassium hydroxide aqueous solution (75ml, 480mmol), heat to 23-25°C, and stir for 12 hours. Raise the temperature to 50-60°C and stir for 4 hours. After the sampling test (U/(U+L)≤0.1%) is qualified, stand still for liquid separation. Separate the organic phase, wash with 10% brine three times, dry, filter, and concentrate the organic phase to 150 ml. The temperature was raised to 40° C., 150 ml of n-heptane was slowly added dropwise, and the temperature was lowered to room temperature to precipitate crystals. Filtered and dried to obtain 10.71 g of light brown solid (compound of formula I), yield 68%, HPLC purity: 99.8% (all single impurities do not exceed 0.15%). MS m/z: 487.3 [M+1].[0057]Nuclear magnetic data (Figure 1): 1 HNMR (d 6 -DMSO): δppm: 9.84 (s, 1H), 8.90 ~ 8.82 (m, 1H), 8.32-8.25 (m, 2H), 7.89 (s, 1H) ,7.51(d,1H,J=8.25), 7.27~7.10(m,1H), 6.94(s,1H), 6.49(dd,1H,J=16.88,10.13), 6.25(dd,1H,J=16.95 ,1.81),5.80~5.75(m,1H),4.19(t,2H,J=5.57),3.88(d,6H,J=14.63,6H),3.34(s,3H),2.58(d,2H, J=5.5), 2.28 (s, 6H).
(2) N-(2-(2-(Dimethylamino)ethoxy)-4-methoxy-5-((4-(1-methyl-1H-indol-3-yl)pyrimidine -2-yl)amino)phenyl)acrylamide methanesulfonate (Form A) preparation
Example 1
The compound of formula I (3 g, 6.1 mmol) was dissolved in 24 ml of dimethyl sulfoxide DMSO solvent, the temperature was raised to 65° C., and the mixture was stirred and dissolved. Add an equivalent amount of methanesulfonic acid (0.59 g, 6.1 mmol) to the system. The temperature was lowered to 50°C, and 12ml of isopropyl acetate IPAc was slowly added. Stir at 50°C for 1 hour, then lower the temperature to 15°C. 21ml IPAc was added in 4 hours. The solution was stirred and crystallized at 15°C, filtered under reduced pressure, the filter cake was washed with isopropyl acetate, and washed with acetone to reduce the residual DMSO solvent. Blow drying at 50°C (or vacuum drying at 50°C) to obtain 3.16 g of a pale yellow solid (crystal form A). HPLC purity is 100%, yield is 88%, DMSO: <100ppm; IPAc: <100ppm. MS m/z: 487.2 [M+1-MsOH]. Melting point: 242-244°C.
Nuclear magnetic data (figure 2): 1 HNMR(d 6 -DMSO): δppm: 9.57(brs,1H), 9.40(s,1H), 8.71(s,1H), 8.48(s,1H), 8.32(d ,1H,J=7.9),8.29(d,1H,J=5.3),7.96(s,1H),7.51(d,1H,J=8.2),7.23(ddd,1H,J=7.9,7.1,0.8 ), 7.19 (d, 1H, J = 5.4), 7.15 (ddd, 1H, J = 7.8, 7.3, 0.5), 6.94 (s, 1H), 6.67 (dd, 1H, J = 16.9, 10.2), 6.27 ( dd, 1H, J = 16.9, 1.8), 5.57 (dd, 1H, J = 16.9, 1.7), 4.44 (t, 2H, J = 4.6), 3.89 (s, 3H), 3.88 (s, 3H), 3.58 (t, 2H, J=4.6), 2.93 (s, 6H), 2.39 (s, 3H).
After testing, the powder X-ray diffraction pattern of crystal form A obtained in this example has diffraction angle 2θ values of 11.06±0.2°, 12.57±0.2°, 13.74±0.2°, 14.65±0.2°, 15.48±0.2°, 16.58±0.2°, 17.83±0.2°, 19.20±0.2°, 19.79±0.2°, 20.88±0.2°, 22.05±0.2°, 23.06±0.2°, 24.23±0.2°, 25.10±0.2°, 25.71±0.2°, 26.15±0.2°, 27.37±0.2°, 27.42±0.2° has a characteristic peak; its XRPD spectrum is shown in Figure 3 and the attached table, DSC diagram is shown in Figure 4, TGA diagram is shown in Figure 5, and infrared spectrum IR diagram is shown in Figure 6. Show.
Example 2
[0066]The compound of formula I (28.25 g, 58.1 mmol) was dissolved in 224 ml of dimethyl sulfoxide DMSO solvent, the temperature was raised to 15-35° C., and the mixture was stirred to clear. 0.97 equivalents of methanesulfonic acid (5.4 g, 0.97 mmol) were added to the system in batches. Slowly add 448 ml of methyl isobutyl ketone (MIBK). Stir for 1 hour, then lower the temperature to 10-15°C. The solution was reacted with salt formation at 10-15°C, sampled, and HPLC detected the residue of the compound of formula I in the mother liquor (≤0.4%). After the reaction was completed, vacuum filtration was performed to obtain 32 g of the crude methanesulfonate of the compound of formula I.Add 3g of the crude methanesulfonate of the compound of formula I into 24ml of dimethyl sulfoxide DMSO solvent, stir to clear at 65°C, cool down, slowly add 48ml of methyl isobutyl ketone (MIBK) dropwise, stir and crystallize 6-8 After hours, vacuum filtration, drying at 60° C. (or 60° C. vacuum drying) to obtain the target crystal form A. Melting point: 242-244°C. The XRPD pattern of the crystal form is consistent with Figure 3 (Figure 7), and all characteristic peaks are within the error range.
SYN
European Journal of Medicinal Chemistry 291 (2025) 117643
Rezivertinib, also known as BPI-7711, is a third-generation epidermal growth factor receptor (EGFR) TKI, developed by Beta Pharm. Rezivertinib selectively targets both EGFR-sensitizing mutations
and the T790 M resistance mutation, thereby addressing resistance mechanisms associated with first- and second-generation EGFR-tyrosine kinase inhibitors. In 2024, the NMPA approved Rezivertinib mesylate capsules (trade name: Ruibida) for the treatment of adult patients with locally advanced or metastatic NSCLC who have progressed during or after EGFR-TKI therapy and have confirmed EGFR T790 M mutation-positive status. Rezivertinib exerts its antitumor activity by forming covalent bonds with mutant EGFR, particularly the T790 M mutation, which effectively blocks the downstream signaling pathways responsible for promoting tumor cell proliferation and survival [21]. The mechanism of Rezivertinib effectively inhibits tumor growth in patients harboring T790M-mediated resistance to first- and second-generation EGFR-TKIs. In a Phase IIb clinical trial (NCT03812809), Rezivertinib demonstrated significant clinical efficacy among patients with EGFR T790 M mutation-positive NSCLC who had experienced disease progression following prior EGFR-TKI therapy. The trial reported an ORR of
64.6 % and a median PFS of 12.2 months, highlighting its potent antitumor activity in this specific patient cohort. In terms of safety, Rezivertinib exhibited a favorable tolerability profile [22]. The most
frequently observed treatment-related adverse events were rash, diarrhea, and elevated liver enzymes, predominantly of mild to moderate severity (grade 1 or 2). No dose-limiting toxicities were noted, and its safety profile aligned with those of other third-generation EGFR-TKIs.
The synthesis of Rezivertinib, illustrated in Scheme 5, initiates with nucleophilic substitution reaction between Rezi-001 and Rezi-002,affording Rezi-003 [23]. Fe-mediated reduction of Rezi-003 yields
Rezi-004, followed by amidation with Rezi-005 to deliver Rezivertinib [20] J.J. Cui, E.W. Rogers, Preparation of Fluorodimethyltetrahydroethenopyrazolobenzoxatriazacyclotridecinone
Derivatives for Use as Antitumor Agents, 2017. US20180194777A1.
[21] Y. Shi, Y. Zhao, S. Yang, J. Zhou, L. Zhang, G. Chen, J. Fang, B. Zhu, X. Li, Y. Shu,
J. Shi, R. Zheng, D. Wang, H. Yu, J. Huang, Z. Zhuang, G. Wu, L. Zhang, Z. Guo,
M. Greco, X. Li, Y. Zhang, Safety, efficacy, and pharmacokinetics of rezivertinib
(BPI-7711) in patients with advanced NSCLC with EGFR T790M mutation: a phase
1 dose-escalation and dose-expansion study, J. Thorac. Oncol. 17 (2022) 708–717.

//////////// BPI-7711, BPI 7711, rezivertinib, phase 3, CHINA 2024, APPROVALS 2024



AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

……
CN1C=C(C2=CC=CC=C21)C3=NC(=NC=C3)NC4=CC(=C(C=C4OC)OCCN(C)C)NC(=O)C=C
NEW DRUG APPROVALS
one time
$10.00
Tralokinumab
(Heavy chain)
QVQLVQSGAE VKKPGASVKV SCKASGYTFT NYGLSWVRQA PGQGLEWMGW ISANNGDTNY
GQEFQGRVTM TTDTSTSTAY MELRSLRSDD TAVYYCARDS SSSWARWFFD LWGRGTLVTV
SSASTKGPSV FPLAPCSRST SESTAALGCL VKDYFPEPVT VSWNSGALTS GVHTFPAVLQ
SSGLYSLSSV VTVPSSSLGT KTYTCNVDHK PSNTKVDKRV ESKYGPPCPS CPAPEFLGGP
SVFLFPPKPK DTLMISRTPE VTCVVVDVSQ EDPEVQFNWY VDGVEVHNAK TKPREEQFNS
TYRVVSVLTV LHQDWLNGKE YKCKVSNKGL PSSIEKTISK AKGQPREPQV YTLPPSQEEM
TKNQVSLTCL VKGFYPSDIA VEWESNGQPE NNYKTTPPVL DSDGSFFLYS RLTVDKSRWQ
EGNVFSCSVM HEALHNHYTQ KSLSLSLGK
(Light chain)
SYVLTQPPSV SVAPGKTARI TCGGNIIGSK LVHWYQQKPG QAPVLVIYDD GDRPSGIPER
FSGSNSGNTA TLTISRVEAG DEADYYCQVW DTGSDPVVFG GGTKLTVLGQ PKAAPSVTLF
PPSSEELQAN KATLVCLISD FYPGAVTVAW KADSSPVKAG VETTTPSKQS NNKYAASSYL
SLTPEQWKSH RSYSCQVTHE GSTVEKTVAP TECS
(Disulfide bridge: H22-H96, H149-H205, H263-H323, H369-H427, H228-H’228, H231-H’231, L22-L87, L136-L195, H136-L213)
Tralokinumab
トラロキヌマブ (遺伝子組換え)
| Formula | C6374H9822N1698O2014S44 |
|---|---|
| CAS | 1044515-88-9 |
| Mol weight | 143873.2167 |
EU APPROVED, Adtralza, 2021/6/17
Antiasthmatic, Anti-inflammatory, Anti-IL-13 antibody
Tralokinumab is a human monoclonal antibody which targets the cytokine interleukin 13,[1] and is designed for the treatment of asthma and other inflammatory diseases.[2] Tralokinumab was discovered by Cambridge Antibody Technology scientists, using Ribosome Display, as CAT-354[3] and taken through pre-clinical and early clinical development.[4] After 2007 it has been developed by MedImmune, a member of the AstraZeneca group, where it is currently in Ph3 testing for asthma and Ph2b testing for atopic dermatitis.[5][6] This makes it one of the few fully internally discovered and developed drug candidates in AstraZeneca’s late stage development pipeline.
Discovery and development
Tralokinumab (CAT-354) was discovered by Cambridge Antibody Technology scientists[7] using protein optimization based on Ribosome Display.[8] They used the extensive data sets from ribosome display to patent protect CAT-354 in a world-first of sequence-activity-relationship claims.[7] In 2004, clinical development of CAT-354 was initiated with this first study completing in 2005.[9] On 21 July 2011, MedImmune LLC initiated a Ph2b, randomized, double-blind study to evaluate the efficacy of tralokinumab in adults with asthma.[10]
In 2016, MedImmune and AstraZeneca were developing tralokinumab for asthma (Ph3) and atopic dermatitis (Ph2b) while clinical development for moderate-to-severe ulcerative colitis and idiopathic pulmonary fibrosis (IPF) have been discontinued.[9] In July of that year AstraZeneca licensed Tralokinumab to LEO Pharma for skin diseases.[11]
A phase IIb study of Tralokinumab found that treatment was associated with early and sustained improvements in atopic dermatitis symptoms and tralokinumab had an acceptable safety and tolerability profile, thereby providing evidence for targeting IL-13 in patients with atopic dermatitis.[12]
On 15 June 2017, Leo Pharma announced that they were starting phase III clinical trials with tralokinumab in atopic dermatitis.[13]
Society and culture
Legal status
On 22 April 2021, the Committee for Medicinal Products for Human Use (CHMP) adopted a positive opinion, recommending the granting of a marketing authorization for the medicinal product Adtralza, intended for the treatment of moderate‑to‑severe atopic dermatitis.[14]
The applicant for this medicinal product is LEO Pharma A/S.
References
- ^ Kopf M, Bachmann MF, Marsland BJ (September 2010). “Averting inflammation by targeting the cytokine environment”. Nature Reviews. Drug Discovery. 9 (9): 703–18. doi:10.1038/nrd2805. PMID 20811382. S2CID 23769909.
- ^ “Statement On A Nonproprietary Name Adopted By The USAN Council: Tralokinumab” (PDF). American Medical Association.
- ^ Thom G, Cockroft AC, Buchanan AG, Candotti CJ, Cohen ES, Lowne D, et al. (May 2006). “Probing a protein-protein interaction by in vitro evolution” [P]. Proceedings of the National Academy of Sciences of the United States of America. 103 (20): 7619–24. Bibcode:2006PNAS..103.7619T. doi:10.1073/pnas.0602341103. PMC 1458619. PMID 16684878.
- ^ May RD, Monk PD, Cohen ES, Manuel D, Dempsey F, Davis NH, et al. (May 2012). “Preclinical development of CAT-354, an IL-13 neutralizing antibody, for the treatment of severe uncontrolled asthma”. British Journal of Pharmacology. 166 (1): 177–93. doi:10.1111/j.1476-5381.2011.01659.x. PMC 3415647. PMID 21895629.
- ^ “Pipeline”. MedImmune. Retrieved 11 June 2013.
- ^ “Studies found for CAT-354”. ClinicalTrials.gov. Retrieved 11 June 2013.
- ^ Jump up to:a b Human Antibody Molecules for Il-13, retrieved 2015-07-26
- ^ Jermutus L, Honegger A, Schwesinger F, Hanes J, Plückthun A (January 2001). “Tailoring in vitro evolution for protein affinity or stability”. Proceedings of the National Academy of Sciences of the United States of America. 98 (1): 75–80. Bibcode:2001PNAS…98…75J. doi:10.1073/pnas.98.1.75. PMC 14547. PMID 11134506.
- ^ Jump up to:a b “Tralokinumab”. Adis Insight. Springer Nature Switzerland AG.
- ^ Clinical trial number NCT01402986 for “A Phase 2b, Randomized, Double-blind Study to Evaluate the Efficacy of Tralokinumab in Adults With Asthma” at ClinicalTrials.gov
- ^ “AstraZeneca enters licensing agreements with LEO Pharma in skin diseases”.
- ^ Wollenberg A, Howell MD, Guttman-Yassky E, Silverberg JI, Kell C, Ranade K, et al. (January 2019). “Treatment of atopic dermatitis with tralokinumab, an anti-IL-13 mAb”. The Journal of Allergy and Clinical Immunology. 143 (1): 135–141. doi:10.1016/j.jaci.2018.05.029. PMID 29906525.
- ^ “LEO Pharma starts phase 3 clinical study for tralokinumab in atopic dermatitis”. leo-pharma.com. AstraZeneca. 1 July 2016.
- ^ “Adtralza: Pending EC decision”. European Medicines Agency. 23 April 2021. Retrieved 23 April 2021.
| Tralokinumab Fab fragment bound to IL-13. From PDB 5L6Y. | |
| Monoclonal antibody | |
|---|---|
| Type | Whole antibody |
| Source | Human |
| Target | IL-13 |
| Clinical data | |
| ATC code | D11AH07 (WHO) |
| Identifiers | |
| CAS Number | 1044515-88-9 |
| ChemSpider | none |
| UNII | GK1LYB375A |
| KEGG | D09979 |
| Chemical and physical data | |
| Formula | C6374H9822N1698O2014S44 |
| Molar mass | 143875.20 g·mol−1 |
| (what is this?) (verify) |
/////////Tralokinumab, Adtralza, EU 2021, APPROVALS 2021, Antiasthmatic, Anti-inflammatory, Anti-IL-13 antibody, MONOCLONAL ANTIBODY, PEPTIDE, トラロキヌマブ (遺伝子組換え) ,
NEW DRUG APPROVALS
ONE TIME
$10.00
Upacicalcet sodium hydrate
Upacicalcet sodium hydrate, ウパシカルセトナトリウム水和物
CAS 2052969-18-1
1333218-50-0 free
PMDA JAPAN APPROVED 2021/6/23, Upasita
Calcium sensing receptor agonist
(2S)-2-amino-3-[(3-chloro-2-methyl-5-sulfophenyl)carbamoylamino]propanoic acid
| Formula | C11H13ClN3O6S. Na. xH2O |
|---|
- OriginatorAjinomoto Pharma
- DeveloperSanwa Kagaku Kenkyusho
- ClassAmines; Chlorobenzenes; Propionic acids; Small molecules; Sulfonic acids; Toluenes
- Mechanism of ActionCalcium-sensing receptor agonists
- RegisteredSecondary hyperparathyroidism
- 25 Jun 2021Chemical structure information added
- 23 Jun 2021Sanwa Kagaku Kenkyusho and Kissei Pharmaceutical agree to co-promote upacicalcet in Japan for Secondary hyperparathyroidism
- 23 Jun 2021Registered for Secondary hyperparathyroidism in Japan (IV) – First global approval
| Upacicalcet Sodium HydrateMonosodium 3-({[(2S)-2-amino-2-carboxyethyl]carbamoyl}amino)-5-chloro-4-methylbenzenesulfonate hydrateC11H13ClN3NaO6S▪xH2O [2052969-18-1 , anhydride] |
Announcement of Marketing Authorization Approval in Japan and Co-promotion Agreement of UPASITA® IV Injection Syringe for the Treatment of Secondary Hyperparathyroidism in Dialysis Patients
SANWA KAGAKU KENKYUSHO Co., Ltd. (Head Office: Nagoya, President and CEO : Shusaku Isono, Suzuken Group, ; “SANWA KAGAKU”) has received Marketing Authorization approval today for UPASITA® IV Injection Syringes (generic name: Upacicalcet Sodium Hydrate; “UPASITA®”) for the treatment of secondary hyperparathyroidism in patients on hemodialysis.
UPASITA® was created by Ajinomoto Pharmaceuticals Co., Ltd. (currently EA Phama Co., Ltd.) and developed by SANWA KAGAKU for the treatment of secondary hyperparathyroidism under a licensing agreement with EA Pharma. UPASITA® acts on calcium sensing receptor in the parathyroid and suppresses excessive secretions of parathyroid hormones (PTH). UPASITA® is administered by intravenous injection to dialysis patients through dialysis circuit by physicians or medical staffs upon completion of dialysis and such administration is expected to reduce the burden of patients with many oral medications whose drinking water volume is severely restricted.
Regarding provision of medical and drug information, SANWA KAGAKU entered into a co-promotion agreement in Japan with Kissei Pharmaceutical Co., Ltd. (Head Office: Matsumoto, Nagano; Chairman and CEO: Mutsuo Kanzawa ; “Kissei”). SANWA KAGAKU will handle the production, marketing, and distribution of the Product while SANWA KAGAKU and Kissei collaboratively promote it to medical institutions in the field in accordance with the agreement. Through the co-promotion activity in the field, SANWA KAGAKU and Kissei will contribute to the treatment of dialysis patients suffering from secondary hyperparathyroidism.
《Reference》
About secondary hyperparathyroidism (SHPT)
SHTP is one of complications that occur as chronic kidney disease (chronic kidney failure) progresses and is a pathological condition where excessive PTH is secreted by the parathyroid gland. It has been reported that excessive secretion of parathyroid hormone promotes efflux of phosphorus and calcium from the bone into the blood, thereby increasing the risk of developing bone fractures and arteriosclerosis due to calcification of the cardiovascular system and affecting the vital prognosis.
Product Summary of UPASITA® IV Injection Syringe for Dialysis
Brand name:
UPASITA® IV Injection Syringe for Dialysis 25μg
UPASITA® IV Injection Syringe for Dialysis 50μg
UPASITA® IV Injection Syringe for Dialysis 100μg
UPASITA® IV Injection Syringe for Dialysis 150μg
UPASITA® IV Injection Syringe for Dialysis 200μg
UPASITA® IV Injection Syringe for Dialysis 250μg
UPASITA® IV Injection Syringe for Dialysis 300μg
Generic Name (JAN):
Upacicalcet Sodium Hydrate
Date of Marketing Approval:
June 23, 2021
Indications:
Secondary hyperparathyroidism in patients on hemodialysis
Dosage and Administration:
In adults, UPASITA® is usually administered into venous line of the dialysis circuit at the end of dialysis session during rinse back at a dose of 25 μg sodium upacicalcet 3 times a week as a starting dose.
The starting dose can be 50 μg depending on the concentration of serum calcium. Thereafter, the dose may be adjusted in a range from 25 to 300 μg while parathyroid hormone (PTH) and serum calcium level should be carefully monitored in patients.
SYN
WO 2020204117

PATENT
WO 2011108724
WO 2011108690
JP 2013063971
WO 2016194881
JP 6510136
PATENT
WO 2016194881
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2016194881&tab=FULLTEXT(Example 1) Synthesis of
(2S) -2-amino-3-{[(5-chloro-2-hydroxy-3-sulfophenyl) carbamoyl] amino} propanoic acid (Compound 1 )
[Chemical formula 14]CDI 150. 2 g (926.6Mmol, 1.1 eq. vs Boc-DAP-O t Bu) to and stirred at 5 ° C. acetone was added 750mL (3.0L / kg). 250 g (842.6 mmol) of Boc-DAP-OtBu was added in two portions, and the mixture was washed with 125 mL (0.5 L / kg) of acetone. After stirring for 30 minutes, completion of the IC (imidazolylcarbonylation) reaction was confirmed by HPLC. 282.6 g (1263.8 mmol, 1.5 eq.) Of ACHB was added in 3 portions, and the mixture was washed with 125 mL (0.5 L / kg) of acetone. After raising the temperature to 30 ° C. and stirring for 18 hours, the completion of the urea conversion reaction was confirmed by HPLC. After cooling to 5 ° C., 124.5 mL (1432.4 mmol, 1.7 eq.) Of concentrated hydrochloric acid was added, and the mixture was stirred for 1 hour. The precipitated unwanted material was filtered and washed with 1000 mL (4.0 L / kg) of acetone. The filtrate was concentrated to 1018 g (4.1 kg / kg), the temperature was raised to 50 ° C., and 625.0 mL (7187 mmol, 8.5 eq.) Of concentrated hydrochloric acid was added dropwise. After stirring for 30 minutes and confirming the completion of deprotection by HPLC, 750 mL of water was added (3.0 L / kg). This liquid was concentrated under reduced pressure to 1730 g (6.9 kg / kg) to precipitate a solid. After stirring at 20 ° C. for 14 hours, vacuum filtration was performed. The filtered solid was washed with 500 mL (2.0 L / kg) of acetone and then dried under reduced pressure at 60 ° C. for 6 hours to obtain 201.4 g of the target product (64.5%).
1H-NMR (400MHz, DMSO-d6): δ 8.3 (s, 1H), 8.2 (bs, 3H), 8.1 (d, 1H, J = 2.6Hz), 7.3 (t, 1H, J = 6.0Hz), 7.0 (d, 1H, J = 2.6Hz), 4.0-4.1 (m, 1H), 3.6-3.7 (m, 1H), 3.4-3.5 (m, 1H)[0026](Example 2) Synthesis of
(2S) -2-amino-3-{[(3-sulfophenyl) carbamoyl] amino} propanoic acid (Compound 2 )
[Chemicalformula 15] CDI 120.2 g (741.2 mmol, 1. 600 mL (3.0 L / kg) of acetone was added to 1 eq. Vs Boc-DAP-OtBu), and the mixture was stirred at 5 ° C. 200 g (673.9 mmol) of Boc-DAP-OtBu was added in two portions, and the mixture was washed with 100 mL (0.5 L / kg) of acetone. After stirring for 30 minutes, the completion of the IC reaction was confirmed by HPLC. 175.0 g (1010.8 mmol, 1.5 eq.) Of ABS was added in 3 portions and washed with 100 mL (0.5 L / kg) of acetone. After raising the temperature to 30 ° C. and stirring for 18 hours, the completion of the urea conversion reaction was confirmed by HPLC. After cooling to 5 ° C., 99.6 mL (1145.4 mmol, 1.7 eq.) Of concentrated hydrochloric acid was added, and the mixture was stirred for 1 hour. The precipitated unwanted material was filtered and washed with 1400 mL (7.0 L / kg) of acetone. The filtrate was concentrated to 800.1 g (4.0 kg / kg), heated to 50 ° C., and then 500.0 mL (5750.0 mmol, 8.5 eq.) Of concentrated hydrochloric acid was added dropwise. After stirring for 30 minutes and confirming the completion of deprotection by HPLC, 600 mL of water was added (3.0 L / kg). This liquid was concentrated under reduced pressure to 1653.7 g to precipitate a solid. After aging at 20 ° C. for 15 hours, vacuum filtration was performed. The filtered solid was washed with 400 mL (2.0 L / kg) of acetone and then dried under reduced pressure at room temperature for 6 hours to obtain 140.3 g of the desired product (net 132.2 g, 64.7%).
1H-NMR (400MHz, DMSO-d6): δ 8.8 (s, 1H), 8.2 (bs, 3H), 7.7 (s, 1H), 7.3-7.4 (m, 1H), 7.1-7.2 (m, 2H) , 6.3-6.4 (bs, 1H), 4.0-4.1 (bs, 1H), 3.6-3.7 (bs, 1H), 3.5-3.6 (bs, 1H)[0027](Example 3) Synthesis of
(2S) -2-amino-3-{[(3-chloro-2-methyl-5-sulfophenyl) carbamoyl] amino} propanoic acid (Compound 3 )
[Chemical formula 16]CDI 14. To 4 g (88.8 mmol, 1.05 eq. Vs Boc-DAP-OtBu), 75 mL (3.0 L / kg vs DAP-OtBu) of acetone was added and stirred at 5 ° C. After adding 25 g (84.3 mmol) of Boc-DAP-OtBu in two portions and stirring for 30 minutes, the completion of the IC reaction was confirmed by HPLC. 26.1 g (118.0 mmol, 1.4 eq.) Of ACTS was added in 3 portions and washed with 25 mL (1.0 L / kg) of acetone. After the temperature was raised to 30 ° C., the mixture was stirred overnight, and the completion of the urea conversion reaction was confirmed by HPLC. After concentrating under reduced pressure at 10 kPa and 40 ° C. until the solvent was completely removed, 37.5 mL (1.5 L / kg) of water and 22.8 mL (257.6 mmol) of concentrated hydrochloric acid were added to perform deprotection for 2 hours. After confirming the completion of the reaction by HPLC, the mixture was cooled to 5 ° C., 60 mL (2.4 L / kg) of MeCN was added, and the mixture was stirred overnight. Further, when 120 mL (4.8 L / kg) of MeCN was added, stratification occurred, so 10 mL (0.4 L / kg) of water and 2.5 mL (0.1 L / kg) of MeCN were added. The precipitated solid was filtered under reduced pressure, washed with 60 mL of MeCN / water (1/2), and then dried under reduced pressure at 60 ° C. for 14 hours to obtain 20.1 g of the desired product as a white solid (net18.3 g, yield 61). 0.8%).
1H-NMR (400MHz, DMSO-d6): δ 14.70-13.30 (bs, 1H), 8.27 (bs, 3H), 8.15 (s, 1H), 7.98 (d, 1H, J = 1.6Hz), 7.27 (d , 1H, J = 1.6Hz), 6.82 (t, 1H, J = 6.0Hz), 4.04 (bs, 1H), 3.70-3.60 (m, 1H), 3.60-3.50 (m, 1H), 2.22 (s, 3H)[0028](Example 4) Synthesis of
compound 3 using phenylchloroformate as a carbonyl group-introducing reagent
(Step 1)
[Chemicalformula 17] MeCN 375 mL (7.5 L / kg vs ACTS), Py for 50 g (225.6 mmol) of ACTS. 38.1 mL (473.7 mmol, 2.1 eq.) Was added and stirred at 25 ° C. 29.9 mL (236.8 mmol, 1.05 eq.) Of ClCO 2 Ph (phenyl chloroformate) was added dropwise, and after stirring for 30 minutes, completion of the CM (carbamate) reaction was confirmed by HPLC. 68.9 g (232.4 mmol) of Boc-DAP-OtBu was added, 97.5 mL (699.3 mmol, 3.1 eq.) Of TEA was added dropwise, and the mixture was stirred at 25 ° C. for 3 hours. The completion of the urea conversion reaction was confirmed by HPLC. Here, 103.5 g of the total amount of 517.43 g was used to move to the next step (down to ACTS 10 g scale).
30 mL of water was added and concentrated to 77.0 g at 40 ° C. and 5 kPa. After 100 mL (10 L / kg) of AcOEt was added and the liquid separation operation was performed, 30 mL of water was added to the organic layer and the liquid separation operation was performed again. The organic layer was concentrated to 47.6 g at 40 ° C. and 10 kPa, and then 15 mL (1.5 L / kg) of AcOEt and 100 mL (10 L / kg) of THF were added. Again, it was concentrated to 50.7 g and THF was added up to 146 g. When it was concentrated again to 35.5 g and added to AcOEt 30 mL (3 L / kg) and THF 100 mL (10 L / kg), a solid was precipitated. It was cooled to 5 ° C. and aged overnight. The precipitated solid was filtered under reduced pressure, washed with 20 mL (2.0 L / kg) of THF, and then dried under reduced pressure at 40 ° C. for 3 hours overnight at 30 ° C. to obtain 24.9 g of the desired product as a white solid (net). 23.0 g, 83.6%).
1 H-NMR (400MHz, DMSO-d6): δ 8.86 (bs, 1H), 8.09 (s, 1H), 7.88 (s, 1H), 7.25 (d, 1H, J = 1.6Hz), 7.14 (d, 1H, J = 7.6Hz), 6.60 (t, 1H, J = 5.6Hz), 4.00-3.90 (m, 1H), 3.60-3.50 (m, 1H), 3.30-3.20 (m, 1H), 3.15-3.05 (m, 6H), 2.19 (s, 3H), 1.50-1.30 (m, 18H), 1.20-1.10 (m, 9H)
(Step 2)
[Chemical
formula 18] Compound 4 21.64 g (net. 20.0 g, 68 mL of water (3.4 L / kg vs. compound 4) vs. 32.8 mmol) ) Was added, the mixture was stirred at 50 ° C., and 12 mL (135.6 mmol, 4.1 eq.) Of concentrated hydrochloric acid was added dropwise. After stirring for 1 hour, the temperature was raised to 70 ° C. to dissolve the precipitated solid. After confirming the completion of the reaction by HPLC, the mixture was cooled to 50 ° C. and aged for 1 hour, and then cooled to 5 ° C. over 4 hours. The precipitated solid was filtered under reduced pressure, washed with 40 mL (2.0 L / kg) of MeCN / water (2/1), and then dried under reduced pressure at 60 ° C. for 3 hours to obtain 11.2 g of the desired product as a white solid (11.2 g). net 10.5 g, 91.1%).[0029](Example 5)
[Chemicalformula 19] MeCN 10.0 mL (10.0 L / kg vs ACSS), Py 0.75 mL (9.25 mmol, 2.05 eq.) For 1.00 g (4.51 mmol) of ACTS. , And stirred at 8 ° C. After dropping 0.59 mL (4.74 mmol, 1.05 eq.) Of ClCO 2 Ph, raising the temperature to room temperature and stirring for 1 hour, completion of the CM conversion reaction was confirmed by HPLC. 1.33 g (4.51 mmol, 1.0 eq.) Of Boc-DAP-OtBu was added, 1.92 mL (13.76 mmol, 3.05 eq.) Of TEA was added dropwise, and the mixture was stirred at 40 ° C. for 1 hour. After confirming the completion of the urea conversion reaction by HPLC, the mixture was concentrated until the solvent was completely removed. 1.0 mL of water and 2.0 mL of concentrated hydrochloric acid (22.6 mmol, 5.0 eq.) Were added, and the mixture was stirred at 50 ° C. for 4 hours. After confirming the completion of deprotection by HPLC, MeCN 7.5 mL (7.5 L / kg), 1 M HCl aq. After adding 4.5 mL, the mixture was stirred at 5 ° C. overnight. The precipitated solid was filtered under reduced pressure, washed with 3.0 mL (3.0 L / kg) of MeCN, and then dried at 60 ° C. overnight to obtain 1.28 g of the desired product as a white solid (net 1.18 g, 77). .0%).[0030](Example 6)
(Step 1)
3-({[(2S) -2-amino-3-methoxy-3-oxopropyl] carbamoyl} amino) -5-chloro-4-methylbenzene-1-sulfonic acid ( Synthesis of Compound 5 )
[Chemical formula 20] To5 g (22.56 mmol) of ACTS, 37.5 mL (7.5 L / kg vs ACTS) of MeCN and 3.81 mL (47.38 mmol, 2.1 eq.) Of Py were added. The mixture was stirred at 25 ° C. 2.99 mL (23.68 mmol, 1.05 eq.) Of ClCO 2 Ph was added dropwise, and after stirring for 30 minutes, the completion of the CM reaction was confirmed by HPLC. 5.92 g (23.23 mmol, 1.03 eq.) Of Boc-DAP-OMe was added, 9.75 mL (69.93 mmol, 3.1 eq.) Of TEA was added dropwise, and the mixture was stirred at 25 ° C. for 3 hours. 0.4 g (1.58 mmol, 0.07 eq.) Of Boc-DAP-OMe and 0.22 mL (1.58 mmol, 0.07 eq.) Of TEA were added, and the completion of the ureaization reaction was confirmed by HPLC. 7.32 mL (112.8 mmol, 5.0 eq.) Of MsOH was added, the temperature was raised to 50 ° C., and the mixture was stirred for 4 hours. After confirming the completion of deprotection by HPLC, the mixture was cooled to 25 ° C. and 37.5 mL (7.5 L / kg) of MeCN and 7.5 mL (1.5 L / kg) of water were added to precipitate a solid. It was cooled to 5 ° C. and aged for 16 hours. The precipitated solid was filtered under reduced pressure, washed with 20 mL (4.0 L / kg) of water / MeCN (1/2), and then dried under reduced pressure at 40 ° C. for 5 hours to obtain 7.72 g of the target product as a white solid (772 g of the target product). net 7.20 g, 87.3%).
1H-NMR (400MHz, DMSO-d6): δ 8.39 (bs, 3H), 8.16 (d, 1H, J = 1.2Hz), 7.90 (d, 1H, J = 1.6Hz), 7.28 (d, 1H, J = 1.6Hz), 6.78 (t, 1H, J = 5.6Hz), 4.20-4.10 (m, 1H), 3.77 (s, 3H), 3.70-3.60 (m, 1H), 3.55-3.45 (m, 1H) , 2.21 (s, 3H)
HRMS (FAB – ): calcd for m / z 364.0369 (MH), found The m / z 364.0395 (MH)
(step 2)
[Formula 21]
compound 5 10.64 g (net Non 10.0 g, To 27.34 mmol), 18 mL of water (1.8 L / kg vs. compound 5 ) was added and stirred at 8 ° C. 3.42 mL (57.41 mmol, 2.1 eq.) Of a 48% aqueous sodium hydroxide solution was added dropwise, and the mixture was washed with 1.0 mL (1.0 L / kg) of water and then stirred at 8 ° C. for 15 minutes. After confirming the completion of hydrolysis by HPLC, the temperature was raised to 25 ° C. and 48% HBr aq. The pH was adjusted to 5.8 by adding about 3.55 mL. After confirming the precipitation of the target product by dropping 65 mL (6.5 L / kg) of IPA, the mixture was aged for 1 hour. 81 mL (8.1 L / kg) of IPA was added dropwise and aged at 8 ° C. overnight. The precipitated solid was filtered under reduced pressure, washed with 20 mL (2.0 L / kg) of IPA, and then dried under reduced pressure at 40 ° C. for 4 hours to obtain 10.7 g of the desired product as a white solid (net 9.46 g, 92. 6%).
1 H-NMR (400MHz, DMSO-d6): δ8.76 (s, 1H), 7.91 (d, 1H, J = 1.6Hz), 8.00-7.50 (bs, 2H), 7.24 (d, 1H, J = 1.6Hz), 7.20 (t, 1H, J = 5.6Hz), 3.58-3.54 (m, 1H), 3.47-3.43 (m, 1H), 3.42-3.37 (m, 1H), 2.23 (s, 3H)[0031](Example 7)
(Step 1)
[Chemicalformula 22] For 10.0 g (45.1 mmol) of ACTS, 50 mL (5.0 L / kg vs ACTS) of MeCN, 7.46 mL (92.5 mmol, 2.05 eq. ) Was added, and the mixture was stirred at 8 ° C. 5.98 mL (47.4 mmol, 1.05 eq.) Of ClCO 2 Ph was added dropwise, the temperature was raised to 25 ° C., and the mixture was stirred for 1 hour, and then the completion of the CM reaction was confirmed by HPLC. 100 ml of acetone (10.0 L / kg vs ACTS) was added, the mixture was cooled to 8 ° C., and aged for 1 hour. The precipitated solid was filtered under reduced pressure, washed with 30 mL of acetone (3.0 L / kg vs ACTS), and then dried under reduced pressure at 60 ° C. for 2 hours to obtain 17.8 g of the target product (net 14.4 g as a free form). Quant).
1 H-NMR (400MHz, DMSO-d6): δ 9.76 (bs, 1H), 8.93-8.90 (m, 2H), 8.60-8.50 (m, 1H), 8.10-8.00 (m, 2H), 7.60 (s , 1H), 7.50-7.40 (m, 3H), 7.30-7.20 (m, 3H), 2.30 (s, 3H)
(Step 2)
[Chemical 23]
Compound 6 To 5.0 g (11.9 mmol), 50 ml of acetonitrile and 3.53 g (11.9 mmol) of Boc-DAP-OtBu were added, and the mixture was stirred at 8 ° C. 3.5 ml (25 mmol) of triethylamine was added dropwise, and the mixture was stirred overnight at room temperature. The solvent was distilled off under reduced pressure, and 25 ml of ethyl acetate and 5 ml of water were added for extraction. The organic layer was washed with 5 ml of water, the solvent was distilled off, 50 ml of tetrahydrofuran was added, the mixture was cooled to 8 ° C., and aged for 1 hour. The precipitated solid was filtered under reduced pressure, washed with 10 ml of tetrahydrofuran, and dried under reduced pressure at 60 ° C. overnight to obtain 6.3 g of the desired product as a white solid.[0032](Example 8)
[Chemicalformula 24] For 1.08 g (4.89 mmol) of ACTS, 8.1 mL (7.5 L / kg vs ACTS) of MeCN and 827 μL (10.27 mmol, 2.1 eq.) Of Py were added. In addition, it was stirred at room temperature. ClCO 2 Ph 649 μL (5.14 mmol, 1.05 eq.) Was added dropwise, and the mixture was stirred for 30 minutes, and then the completion of the CM conversion reaction was confirmed by HPLC. 1.48 g (5.04 mmol, 1.03 eq.) Of Cbz-DAP-OMe HCl was added, 2.1 mL (15.17 mmol, 3.1 eq.) Of TEA was added dropwise, and the mixture was stirred at room temperature for about 5 hours. After confirming the completion of the urea conversion reaction by HPLC, the mixture was concentrated until the solvent was completely removed. 15.0 mL of 30% HBr / AcOH was added, and the mixture was stirred at room temperature for 70 minutes, and the completion of deprotection was confirmed by HPLC. After concentration to dryness, 10 mL of water and 4 mL of AcOEt were added to carry out an extraction operation, and then the aqueous layer was stirred at room temperature overnight. The precipitated solid was filtered under reduced pressure, washed with 15 mL of water and 10 mL of AcOEt, and then dried at 40 ° C. for 3 hours to obtain 1.45 g of the desired product as a white solid (58.8%).[0033](Example 9) Synthesis of compound 7 ( methyl ester of compound 1 )
using phenyl chloroformate as a carbonyl group introduction reagent [Chemical formula 25] MeCN 73 mL (14.6 L) with respect to 5.00 g (22.4 mmol) of ACHB. / Kg vs ACHB), Py 3.8 mL (47 mmol, 2.1 eq.), Was added and stirred at 40 ° C. After adding 3.0 mL (24 mmol, 1.05 eq.) Of ClCO 2 Ph and stirring for 30 minutes, the completion of the CM conversion reaction was confirmed by HPLC. 5.87 g (23 mmol, 1.0 eq.) Of Boc-DAP-OMe was added, washed with a small amount of MeCN, 9.7 mL (70 mmol, 3.1 eq.) Of TEA was added dropwise, and the mixture was stirred at 40 ° C. for 3 hours. After confirming the completion of the urea conversion reaction by HPLC, the mixture was cooled to room temperature. 7.3 mL (112 mmol, 5.0 eq.) Of MsOH was added, the temperature was raised to 50 ° C., and the mixture was stirred for 7 hours. Further, 1.5 mL (23 mmol, 1.0 eq.) Of MsOH was added, and the reaction was carried out at 50 ° C. overnight. After confirming the completion of deprotection by HPLC, 90 mL of acetone was added to the reaction solution, and the mixture was cooled to room temperature. The precipitated solid was obtained and dried under reduced pressure at 60 ° C. to obtain the desired product. 1 H-NMR (400MHz, DMSO-d6): δ 7.22 (m, 1H), 7.14 (m, 1H), 4.36 (m, 1H), 3.80 (s, 3H), 3.20-3.40 (m, 2H).[0034](Example 10) Synthesis of
compound 5 using 4-chlorophenylchloroformate as a carbonyl group-introducing reagent
[Chemical formula 26] For5.00 g (22.6 mmol) of ACTS, 73 mL (14.6 L / kg vs ACTS) of MeCN, 3.8 mL (47 mmol, 2.1 eq.) Of Py was added and stirred at 40 ° C. After adding 3.25 mL (23.7 mmol, 1.05 eq.) Of 4-chloroformic acid 4-chlorophenylate and stirring at 40 ° C. for 1.5 hours, completion of the CM conversion reaction was confirmed by HPLC. Add 5.92 g (23.2 mol, 1.0 eq.) Of Boc-DAP-OMe, wash with a small amount of MeCN, add 9.7 mL (70 mmol, 3.1 eq.) Of TEA, and stir at 40 ° C. for 2 hours. did. After confirming the completion of the urea conversion reaction by HPLC, the mixture was cooled to room temperature. 7.3 mL (113 mmol, 5.0 eq.) Of MsOH was added, the temperature was raised to 50 ° C., and the mixture was stirred for 3.5 hours. After confirming the completion of deprotection by HPLC, the reaction solution was cooled to room temperature, 7.5 mL of water was added, the mixture was cooled to 8 ° C., and the mixture was stirred overnight. The precipitated solid was filtered, washed with a small amount of MeCN water, and dried at 60 ° C. overnight to obtain 6.94 g of the desired product as a white solid (84.1%).[0035](Example 11) Synthesis of
compound 5 using 4-nitrophenyl chloroformate as a carbonyl group-introducing reagent
[Chemical formula 27]73 mL (14.6 L / kg vs. ACTS) of MeCN with respect to 5.00 g (22.6 mmol) of ACTS. , Py 3.8 mL (47 mmol, 2.1 eq.), And stirred at 40 ° C. 4.77 mL (23.7 mmol, 1.05 eq.) Of 4-nitrophenyl chloroformate was added dropwise, and the mixture was stirred at 40 ° C. for 3.5 hours, and then the completion of the CM reaction was confirmed by HPLC. Add 5.92 g (23.2 mmol, 1.0 eq.) Of Boc-DAP-OMe, wash with a small amount of MeCN, add 9.7 mL (70 mmol, 3.1 eq.) Of TEA, and stir at 40 ° C. for 2 hours. did. After confirming the completion of the urea conversion reaction by HPLC, the mixture was cooled to room temperature. 7.3 mL (113 mmol, 5.0 eq.) Of MsOH was added, the temperature was raised to 50 ° C., and the mixture was stirred for 3.5 hours. After confirming the completion of deprotection by HPLC, the reaction solution was cooled to room temperature, 7.5 mL of water was added, the mixture was cooled to 8 ° C., and the mixture was stirred overnight. The precipitated solid was filtered, washed with a small amount of MeCN water, and dried at 60 ° C. overnight to obtain 5.96 g of the desired product as a white solid (72.2%).[0036](Example 12) Synthesis of
compound 3 using Boc-DAP-OH
[Chemical 28]MeCN 73 mL (14.6 L / kg vs ACTS), Py 3.8 mL, relative to 5.00 g (22.6 mmol) of ACTS. (47 mmol, 2.1 eq.) Was added and stirred at 40 ° C. After adding 3.00 mL (23.8 mmol, 1.05 eq.) Of phenylchloroformate and stirring at 40 ° C. for 0.5 hours, the completion of the CM conversion reaction was confirmed by HPLC (CM conversion reaction product: 4.37 minutes). , ACTS: N.D.). Add 4.75 g (23.2 mmol, 1.0 eq.) Of Boc-DAP-OH, wash with a small amount of MeCN, add 9.7 mL (70 mmol, 3.1 eq.) Of TEA, and stir at 40 ° C. for 2 hours. did. After confirming the completion of the urea-forming reaction by HPLC (urea-forming reaction product: 3.81 minutes, CM-forming reaction product: 0.02 area% vs. urea-forming reaction product), the mixture was cooled to room temperature. By adding 7.3 mL (113 mmol, 5.0 eq.) Of MsOH, raising the temperature to 50 ° C., stirring for 4.5 hours, and further adding 1.5 mL (23 mmol, 1.0 eq.) Of MsOH, stirring for 1 hour. , The formation of the target product was confirmed by HPLC (Compound 3: 2.49 minutes, urea conversion reaction product: 0.50 area vs. compound 3, area of compound 3 with respect to the total area excluding pyridine: 71.0 area).
PATENT
JP 6510136
PATENT
WO 2020204117
Reference Example 1
Synthesis of 3-{[(2S) -2-amino-2-carboxyethyl] carbamoylamino} -5-chloro-4-methylbenzenesulfonate sodium (Compound A1)
(Step 1)
Synthesis of
3 -({[(2S) -2-amino-3-methoxy-3-oxopropyl] carbamoyl} amino) -5-chloro-4-methylbenzene-1-sulfonic acid 3-amino- 37.5 mL (7.5 L / kg vs ACTS) of acetonitrile and 3.81 mL (47.38 mmol, 2.1 eq.) Of pyridine against 5 g (22.56 mmol) of 5-chloro-4-methylbenzenesulfonic acid (ACTS). Was added and stirred at 25 ° C. 2.99 mL (23.68 mmol, 1.05 eq.) Of ClCO 2 Ph was added dropwise, and after stirring for 30 minutes, the completion of the carbamate reaction was confirmed by HPLC. Add 5.92 g (23.23 mmol, 1.03 eq.) Of 3-amino-N- (tert-butoxycarbonyl) -L-alanine methyl ester hydrochloride and 9.75 mL (69.93 mmol, 3.1 eq.) Triethylamine. Was added dropwise, and the mixture was stirred at 25 ° C. for 3 hours. Add 0.4 g (1.58 mmol, 0.07 eq.) Of 3-amino-N- (tert-butoxycarbonyl) -L-alanine methyl ester hydrochloride and 0.22 mL (1.58 mmol, 0.07 eq.) Of triethylamine. Then, the completion of the urea conversion reaction was confirmed by HPLC. 7.32 mL (112.8 mmol, 5.0 eq.) Of methanesulfonic acid was added, the temperature was raised to 50 ° C., and the mixture was stirred for 4 hours. After confirming the completion of deprotection by HPLC, the mixture was cooled to 25 ° C. and 37.5 mL (7.5 L / kg) of acetonitrile and 7.5 mL (1.5 L / kg) of water were added to precipitate a solid. It was cooled to 5 ° C. and aged for 16 hours. The precipitated solid was filtered under reduced pressure, washed with 20 mL (4.0 L / kg) of water / acetonitrile (1/2), and then dried under reduced pressure at 40 ° C. for 5 hours to obtain 7.72 g of the desired product as a white solid (. net 7.20 g, 87.3%).
1 H-NMR (400MHz, DMSO-d6): δ 8.39 (bs, 3H), 8.16 (d, 1H, J = 1.2Hz), 7.90 (d, 1H, J = 1.6Hz), 7.28 (d, 1H, J = 1.6Hz), 6.78 (t, 1H, J = 5.6Hz), 4.20-4.10 (m, 1H), 3.77 (s, 3H), 3.70-3.60 (m, 1H), 3.55-3.45 (m, 1H) ), 2.21 (S, 3H)HRMS (FAB – ): Calcd For M / Z 364.0369 (MH & lt;), Found M / Z 364.0395 (MH & lt;)
(Step 2)
(2)
Compound obtained in step 1 of synthesis of 3-{[(2S) -2-amino-2-carboxyethyl] carbamoylamino} -5-chloro-4-methylbenzenesulfonate . To 64 g (net 10.0 g, 27.34 mmol), 18 mL of water (1.8 L / kg vs. the compound of Step 1) was added, and the mixture was stirred at 8 ° C. 3.42 mL (57.41 mmol, 2.1 eq.) Of a 48% aqueous sodium hydroxide solution was added dropwise, and the mixture was washed with 1.0 mL (1.0 L / kg) of water and then stirred at 8 ° C. for 15 minutes. After confirming the completion of hydrolysis by HPLC, the temperature was raised to 25 ° C. and 48% HBr aq. About 3.55 mL was added to adjust the pH to 5.8. After confirming the precipitation of the desired product by dropping 65 mL (6.5 L / kg) of isopropyl alcohol, the mixture was aged for 1 hour. 81 mL (8.1 L / kg) of isopropyl alcohol was added dropwise and the mixture was aged at 8 ° C. overnight. The precipitated solid was filtered under reduced pressure, washed with 20 mL (2.0 L / kg) of isopropyl alcohol, and then dried under reduced pressure at 40 ° C. for 4 hours to obtain 10.7 g of the desired product as a white solid (net 9.46 g, 92). .6%).
1 H-NMR (400MHz, DMSO-d6): δ8.76 (s, 1H), 7.91 (d, 1H, J = 1.6Hz), 8.00-7.50 (bs, 2H), 7.24 (d, 1H, J = 1.6Hz), 7.20 (t, 1H, J = 5.6Hz), 3.58-3.54 (m, 1H), 3.47-3.43 (m, 1H), 3.42-3.37 (m, 1H), 2.23 (s, 3H)
///////////Upacicalcet sodium hydrate, Upasita, ウパシカルセトナトリウム水和物 , APPROVALS 2021, JAPAN 2021, Upacicalcet
NEW DRUG APPROVALS
ONE TIME
$10.00
Meglimin hydrochloride

Meglimin hydrochloride
Imeglimin
hydrochloride
Twymeeg
| Formula | C6H13N5. HCl |
|---|---|
| CAS | 775351-61-6 (HCl). , C6H14ClN5 191.66CAS 775351-65-0, FREEFORM 155.20 |
| Mol weight | 191.6619 |
AntidiabeticAPPROVED PMDA JAPAN2021/6/23, イメグリミン塩酸塩
(4R)-6-N,6-N,4-trimethyl-1,4-dihydro-1,3,5-triazine-2,6-diamine
1,3,5-Triazine-2,4-diamine,1,6-dihydro-N,N,6-trimethyl-,(+)-(9CI)
(4R)-6-N,6-N,4-trimethyl-1,4-dihydro-1,3,5-triazine-2,6-diamine
JAPAN
Twymeeg Tablets 500 mg
(Sumitomo Dainippon Pharma Co., Ltd.)

Imeglimin is an experimental drug being developed as an oral anti-diabetic.[1][2] It is an oxidative phosphoryl
Imeglimin (brand name Twymeeg) is an oral anti-diabetic medication.[1][2] It was approved for use in Japan in June 2021.[3]
It is an oxidative phosphorylation blocker that acts to inhibit hepatic gluconeogenesis, increase muscle glucose uptake, and restore normal insulin secretion. It is the first approved drug of this class of anti-diabetic medication.


PATENT
https://patents.google.com/patent/WO2012072663A1/enEXAMPLESExample 1 : Synthesis and isolation of (+)-2-amino-3,6-dihydro-4-dimethylamino-6- methyl-l,3,5-triazine hydrochloride by the process according to the invention
Preliminary step: Synthesis of racemic 2-amino-3,6-dihydro-4-dimethylamino- 6-methyl-l,3,5-triazine hydrochloride:
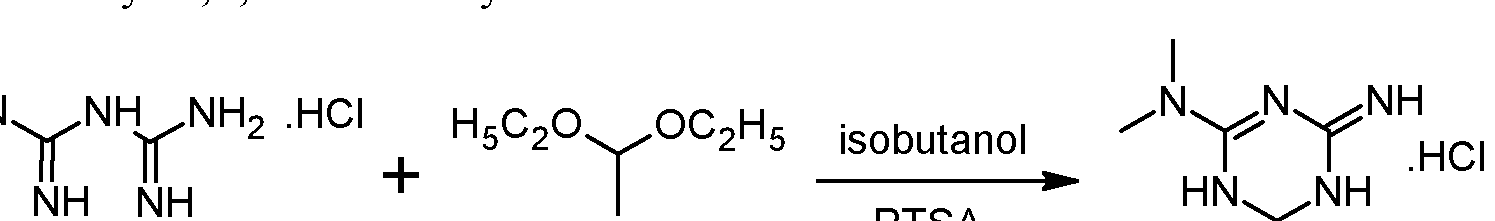
Metformin hydrochloride is suspended in 4 volumes of isobutanol. Acetaldehyde diethylacetal (1.2 eq.) and para-toluenesulfonic acid (PTSA) (0.05 eq) are added and the resulting suspension is heated to reflux until a clear solution is obtained. Then 2 volumes of the solvent are removed via distillation and the resulting suspension is cooled to 20°C. The formed crystals are isolated on a filter dryer and washed with isobutanol (0.55 volumes). Drying is not necessary and the wet product can be directly used for the next step.Acetaldehyde diethylacetal can be replaced with 2,4,6-trimethyl-l,3,5-trioxane (paraldehyde).- Steps 1 and 2: formation of the diastereoisomeric salt and isolation of the desired diastereoisomer

Racemic 2-amino-3,6-dihydro-4-dimethylamino-6-methyl-l,3,5-triazine hydrochloride wet with isobutanol (obtained as crude product from preliminary step without drying) and L-(+)-Tartaric acid (1 eq.) are dissolved in 2.2 volumes of methanol at 20-40°C. The obtained clear solution is filtered and then 1 equivalent of triethylamine (TEA) is added while keeping the temperature below 30°C. The suspension is heated to reflux, stirred at that temperature for 10 minutes and then cooled down to 55°C. The temperature is maintained at 55°C for 2 hours and the suspension is then cooled to 5- 10°C. After additional stirring for 2 hours at 5-10°C the white crystals are isolated on a filter dryer, washed with methanol (2 x 0.5 Vol) and dried under vacuum at 50°C. The yield after drying is typically in the range of 40-45%
– Steps 3 and 4: transformation of the isolated diastereoisomer of the tartrate salt into the hydrochloride salt and recovery of the salt

γ ethanol HN^NH(+) 2-amino-3,6-dihydro-4-dimethylamino-6-methyl-l,3,5-triazine tartrate salt is suspended in 2 volumes of ethanol and 1.02 equivalents of HCl-gas are added under vacuum (-500 mbar). The suspension is heated to reflux under atmospheric pressure (N2) and 5% of the solvent is removed via distillation. Subsequent filtration of the clear colourless solution into a second reactor is followed by a cooling crystallization, the temperature is lowered to 2°C. The obtained suspension is stirred at 2°C for 3 hours and afterwards the white crystals are isolated with a horizontal centrifuge. The crystal cake is washed with ethanol and dried under vacuum at 40°C. The typical yield is 50-55% and the mother liquors can be used for the recovery of about 25-30%) of (+)-2-amino- 3,6-dihydro-4-dimethylamino-6-methyl-l,3,5-triazine tartrate.Example 2: Modification of the solvent of steps 3 and 4
– Steps 3 and 4: transformation of the isolated diastereoisomer of the tartrate salt into the hydrochloride salt and recovery of the salt
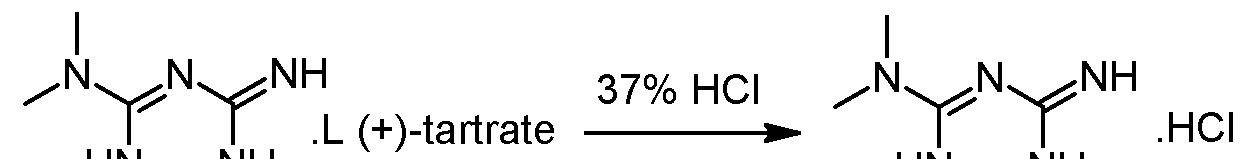
HN^NH acetone HN^NH(+) 2-amino-3,6-dihydro-4-dimethylamino-6-methyl-l,3,5-triazine tartrate salt synthesized according to steps 1 and 2 of example 1 is suspended in 1 volume (based on total amount of (+) 2-amino-3,6-dihydro-4-dimethylamino-6-methyl-l,3,5-triazine tartrate salt) of acetone at 20°C. To this suspension 1.01 equivalents of 37% Hydrochloric acid are added. The suspension is heated to reflux under atmospheric pressure (N2) and water is added until a clear solution is obtained. 1.5 vol of acetone are added at reflux temperature. The compound starts crystallising and the obtained suspension is kept at reflux for 2 hours followed by a cooling crystallization to 0°C. The obtained suspension is stirred at 0°C for 2 hours and the white crystals are isolated by centrifugation. The crystal cake is washed with isopropanol and dried under vacuum at 40°C in a continuous drying oven.
References
- ^ Vuylsteke V, Chastain LM, Maggu GA, Brown C (September 2015). “Imeglimin: A Potential New Multi-Target Drug for Type 2 Diabetes”. Drugs in R&D. 15 (3): 227–32. doi:10.1007/s40268-015-0099-3. PMC 4561051. PMID 26254210.
- ^ Dubourg J, Fouqueray P, Thang C, Grouin JM, Ueki K (April 2021). “Efficacy and Safety of Imeglimin Monotherapy Versus Placebo in Japanese Patients With Type 2 Diabetes (TIMES 1): A Double-Blind, Randomized, Placebo-Controlled, Parallel-Group, Multicenter Phase 3 Trial”. Diabetes Care. 44 (4): 952–959. doi:10.2337/dc20-0763. PMID 33574125.
- ^ Poxel SA (June 23, 2021). “Poxel and Sumitomo Dainippon Pharma Announce the Approval of TWYMEEG® (Imeglimin hydrochloride) for the Treatment of Type 2 Diabetes in Japan” (Press release).
| Clinical data | |
|---|---|
| Trade names | Twymeeg |
| Legal status | |
| Legal status | Rx-only in Japan |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 775351-65-0 |
| PubChem CID | 24812808 |
| ChemSpider | 26232690 |
| UNII | UU226QGU97 |
| CompTox Dashboard (EPA) | DTXSID50228237 |
| Chemical and physical data | |
| Formula | C6H13N5 |
| Molar mass | 155.205 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI |
/////////Imeglimin hydrochloride, Twymeeg, JAPAN 2021, APPROVALS 2021, Antidiabetic, イメグリミン塩酸塩, ATI DIABETES, DIABETES, Imeglimin
CC1N=C(NC(=N1)N(C)C)N.Cl
NEW DRUG APPROVALS
ONE TIME
$10.00
CVnCoV, zorecimeran, CureVac COVID-19 vaccine
CVnCoV
cas 2541470-90-8
An optimized, non-chemical modified mRNA encoding the prefusion-stabilized full-length spike protein of SARS-CoV-2 virus (Curevac)
zorecimeran, CureVac COVID-19 vaccine
CureVac/Bayer
GSK
NCT04674189 NCT04449276 NCT04515147 NCT04652102
EudraCT-2020-004066-19
mRNA-based vaccine
PHASE 3
| CVnCoV | Humoral and cellular responses | CD4+ T-cells, CD8+ T-cells | N/A | N/A | Rhesus macaque | [124] |
124. Rauch S, Gooch K, Hall Y, Salguero FJ, Dennis MJ, Gleeson FV. et al. mRNA vaccine CVnCoV protects non-human primates from SARS-CoV-2 challenge infection. bioRxiv. 2020. 2020 12.23.424138
The CureVac COVID-19 vaccine is a COVID-19 vaccine candidate developed by CureVac N.V. and the Coalition for Epidemic Preparedness Innovations (CEPI).[1] The vaccine showed inadequate results in its Phase III trials with only 47% efficacy.[2] The European Medicines Agency stated that: “(…) medicine developers should design studies to demonstrate a rate of efficacy of at least 50%.”[3].
The CVnCov Vaccine (or CV07050101) is in development by CureVac AG. The vaccine uses mRNA technology to create a protein associated with SARS-CoV2, and upon administration and replication, to initiate subsequent immune responses in the body. As of June 2020, the company received regulatory approval from German and Belgian Authorities to commence Phase 1 clinical trials of this vaccine (NCT04449276).
Efficacy
On 16 June 2021,[4] CureVac said its vaccine showed 47% efficacy from its Phase III trial. This was based on interim analysis of 134 COVID cases in its Phase III study conducted in Europe and Latin America. The final analysis for the trials requires a minimum of 80 additional cases.[2]
Pharmacology
CVnCoV is an mRNA vaccine that encodes the full-length, pre-fusion stabilized coronavirus spike protein, and activates the immune system against it.[5][6][7] CVnCoV technology does not interact with the human genome.[6] CVnCoV uses unmodified RNA,[8] unlike the Pfizer–BioNTech COVID-19 vaccine and Moderna COVID-19 vaccine, which both use nucleoside-modified RNA.[9]
Manufacturing
Manufacturing of mRNA vaccines can be performed rapidly in high volume,[10] including use of portable, automated printers (“RNA microfactories”) for which CureVac has a joint development partnership with Tesla.[11]
mRNA vaccines require stringent cold chain refrigeration throughout manufacturing, distribution and storage.[12][13] The CureVac technology for CVnCoV uses a non-modified, more natural mRNA less affected by hydrolysis, enabling storage at 5 °C (41 °F) and relatively simplified cold chain requirements that facilitate up to three months of storage and distribution to world regions that do not have specialized ultracold equipment.[6][10]
CureVac has a European-based network to accelerate manufacturing of CVnCoV, if proven safe and effective, for production of up to 300 million doses in 2021 and 600 million doses in 2022.[10][14] An estimated 405 million doses will be provided to EU states.[14]
Clinical trials
In November 2020, CureVac reported results of a Phase I-II clinical trial that CVnCoV (active ingredient zorecimeran) was well-tolerated, safe, and produced a robust immune response.[15][16]
In December 2020, CureVac began a Phase III clinical trial of CVnCoV with 36,500 participants.[17][18] Bayer will provide clinical trial support and international logistics for the Phase III trial, and may be involved in eventual manufacturing should the vaccine prove to be safe and effective.[19][20] In February 2021, the EU’s CHMP started a rolling review of CVnCoV.[21][22] In April 2021, the same procedure began in Switzerland.[23]
Brand names
The manufacturer currently markets the vaccine under the name CVnCoV.[24] Zorecimeran is the proposed international nonproprietary name (pINN).[25]
References
- ^ “CureVac focuses on the development of mRNA-based coronavirus vaccine to protect people worldwide”. CureVac(Press release). 15 March 2020. Retrieved 17 February 2021.
- ^ Jump up to:a b Burger, Ludwig (16 June 2021). “CureVac fails in pivotal COVID-19 vaccine trial with 47% efficacy”. Reuters. Retrieved 17 June 2021.
- ^ https://www.ema.europa.eu/en/human-regulatory/overview/public-health-threats/coronavirus-disease-covid-19/treatments-vaccines/vaccines-covid-19/covid-19-vaccines-studies-approval#what-is-the-level-of-efficacy-that-can-be-accepted-for-approval?-section
- ^ “CureVac Provides Update on Phase 2b/3 Trial of First-Generation COVID-19 Vaccine Candidate, CVnCoV”. 16 June 2021.
- ^ https://www.curevac.com/wp-content/uploads/2020/10/20201023-CureVac-Manuscript-draft-preclinical-data.pdf
- ^ Jump up to:a b c Schlake T, Thess A, Fotin-Mleczek M, Kallen KJ (November 2012). “Developing mRNA-vaccine technologies”. RNA Biology. 9(11): 1319–30. doi:10.4161/rna.22269. PMC 3597572. PMID 23064118.
- ^ “Understanding mRNA COVID-19 vaccines”. US Centers for Disease Control and Prevention. 18 December 2020. Retrieved 5 January 2021.
- ^ “COVID-19”. CureVac. Retrieved 21 December 2020.
- ^ Dolgin, Elie (25 November 2020). “COVID-19 vaccines poised for launch, but impact on pandemic unclear”. Nature Biotechnology: d41587–020–00022-y. doi:10.1038/d41587-020-00022-y. PMID 33239758. S2CID 227176634.
- ^ Jump up to:a b c Nawrat A (3 December 2020). “Q&A with CureVac: resolving the ultra-cold chain logistics of Covid-19 mRNA vaccines”. Pharmaceutical Technology. Retrieved 5 January 2021.
- ^ “Tesla to make molecule printers for German COVID-19 vaccine developer CureVac”. Reuters. 2 July 2020. Retrieved 19 December 2020.
- ^ Kartoglu U, Milstien J (July 2014). “Tools and approaches to ensure quality of vaccines throughout the cold chain”. Expert Review of Vaccines. 13 (7): 843–54. doi:10.1586/14760584.2014.923761. PMC 4743593. PMID 24865112.
- ^ Hanson CM, George AM, Sawadogo A, Schreiber B (April 2017). “Is freezing in the vaccine cold chain an ongoing issue? A literature review”. Vaccine. 35 (17): 2127–2133. doi:10.1016/j.vaccine.2016.09.070. PMID 28364920.
- ^ Jump up to:a b Kansteiner F (17 November 2020). “CureVac, armed with COVID-19 vaccine deal, plots ‘pandemic-scale’ Euro manufacturing expansion”. FiercePharma, Questex LLC. Retrieved 5 January2021.
- ^ “CureVac’s Covid-19 vaccine induces immune response in study”. Clinical Trials Arena. 3 November 2020. Retrieved 5 January 2021.
- ^ “CureVac’s COVID-19 vaccine triggers immune response in Phase I trial”. Reuters. 2 November 2020. Retrieved 5 January2021.
- ^ “Multicenter Clinical Study Evaluating the Efficacy and Safety of Investigational SARS-CoV-2 mRNA Vaccine CVnCoV in Adults 18 Years of Age and Older”. EU Clinical Trials Register. 19 November 2020. Retrieved 5 January 2021.
Proposed INN: zorecimeran
- ^ “A Study to Determine the Safety and Efficacy of SARS-CoV-2 mRNA Vaccine CVnCoV in Adults”. ClinicalTrials.gov. 8 December 2020. NCT04652102. Retrieved 19 December 2020.
- ^ Burger L (7 January 2021). “CureVac strikes COVID-19 vaccine alliance with Bayer”. Reuters. Retrieved 17 February 2021.
- ^ “CureVac and Bayer join forces on COVID-19 vaccine candidate CVnCoV”. CureVac (Press release). 7 January 2021. Retrieved 17 February 2021.
- ^ “EMA starts rolling review of CureVac’s COVID-19 vaccine (CVnCoV)”. European Medicines Agency (EMA) (Press release). 11 February 2021. Retrieved 12 February 2021.
- ^ “CureVac Initiates Rolling Submission With European Medicines Agency for COVID-19 Vaccine Candidate, CVnCoV”. CureVac(Press release).
- ^ “CureVac starts review process in Switzerland for COVID-19 vaccine hopeful”. Reuters. 19 April 2021. Retrieved 19 April 2021.
- ^ “Celonic and CureVac Announce Agreement to Manufacture over 100 Million Doses of CureVac’s COVID-19 Vaccine Candidate, CVnCoV”. CureVac (Press release). 30 March 2021. Retrieved 14 April 2021.
- ^ World Health Organization (October 2020). “International Nonproprietary Names for Pharmaceutical Substances (INN). Proposed INN: List 124 – COVID-19 (special edition)” (PDF). WHO Drug Information. 34 (3): 668–69. Archived (PDF) from the original on 27 November 2020.
External links
| Scholia has a profile for zorecimeran (Q97154239). |
- “Zorecimeran”. Drug Information Portal. U.S. National Library of Medicine.
| Vaccine description | |
|---|---|
| Target | SARS-CoV-2 |
| Vaccine type | mRNA |
| Clinical data | |
| Other names | CVnCoV, CV07050101 |
| Routes of administration | Intramuscular |
| ATC code | None |
| Identifiers | |
| DrugBank | DB15844 |
| UNII | 5TP24STD1S |
| Part of a series on the |
| COVID-19 pandemic |
|---|
| COVID-19 (disease)SARS-CoV-2 (virus) |
| showTimeline |
| showLocations |
| showInternational response |
| showMedical response |
| showImpact |
| COVID-19 portal |
- Rego GNA, Nucci MP, Alves AH, Oliveira FA, Marti LC, Nucci LP, Mamani JB, Gamarra LF: Current Clinical Trials Protocols and the Global Effort for Immunization against SARS-CoV-2. Vaccines (Basel). 2020 Aug 25;8(3). pii: vaccines8030474. doi: 10.3390/vaccines8030474. [Article]
- Speiser DE, Bachmann MF: COVID-19: Mechanisms of Vaccination and Immunity. Vaccines (Basel). 2020 Jul 22;8(3). pii: vaccines8030404. doi: 10.3390/vaccines8030404. [Article]
- CureVac & Covid-19 [Link]
- Smart Patients [Link]
- Regulatory News [Link]
////////////zorecimeran, CVnCoV, CV07050101, CORONA VACCINE, COVID 19, VACCINE, CUREVAC, SARS-CoV-2, CV07050101, SARS-CoV-2 mRNA vaccine
NEW DRUG APPROVALS
one time
$10.00
Tucidinostat, Chidamide
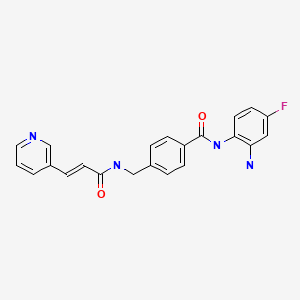

Tucidinostat, Chidamide
ツシジノスタット
2021/6/23 PMDA JAPAN APPROVED,
| Hiyasta |
| Formula | C22H19FN4O2 |
|---|---|
| CAS | 1616493-44-7 |
| Mol weight | 390.4103 |
Antineoplastic, Histone deacetylase inhibitor
Chidamide (Epidaza) is a histone deacetylase inhibitor (HDI) developed in China.[1] It was also known as HBI-8000.[2] It is a benzamide HDI and inhibits Class I HDAC1, HDAC2, HDAC3, as well as Class IIb HDAC10.[3]
Chidamide is approved by the Chinese FDA for relapsed or refractory peripheral T-cell lymphoma (PTCL), and has orphan drug status in Japan.[2][better source needed] As of April 2015 it is only approved in China.[1]
Chidamide is being researched as a treatment for pancreatic cancer.[4][5][6] However, it is not US FDA approved for the treatment of pancreatic cancer.
Chidamide (Epidaza®), a class I HDAC inhibitor, was discovered and developed by ChipScreen and approved by the CFDA in December 2014 for the treatment of recurrent of refractory peripheral T-cell lymphoma. Chidamide, also known as CS055 and HBI- 8000, is an orally bioavailable benzamide type inhibitor of HDAC isoenzymes class I 1–3, as well as class IIb 10, with potential antineoplastic activity. It selectively binds to and inhibits HDAC, leading to an increase in acetylation levels of histone protein H3.74 This agent also inhibits the expression of signaling kinases in the PI3K/ Akt and MAPK/Ras pathways and may result in cell cycle arrest and the induction of tumor cell apoptosis. Currently, phases I and II clinical trials are underway for the treatment of non-small cell lung cancer and for the treatment of breast cancer, respectively.
Chemical Synthesis
The scalable synthetic approach to chidamide very closely follows the discovery route. The sequence began with the condensation of commercial nicotinaldehyde (52) and malonic acid (53) in a mixture of pyridine and piperidine. Next, activation of acid 54 with N,N0-carbonyldiimidazole (CDI) and subsequent reaction with 4-aminomethyl benzoic acid (55) under basic conditions afforded amide 56 in 82% yield. Finally, activation of 56 with CDI prior to treatment with 4-fluorobenzene- 1,2-diamine (57) and subsequent treatment with TFA and THF yielded chidamide (VIII) in 38% overall yield from 52. However, no publication reported that mono-N-Boc-protected bis-aniline was used to approach Chidamide.
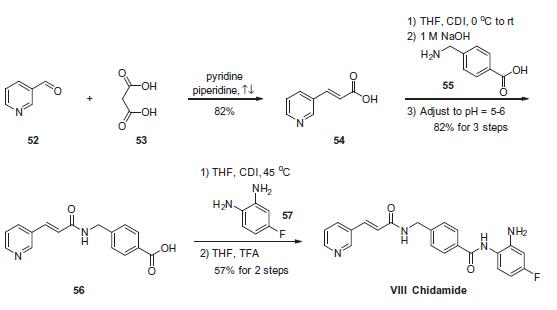
References
- ^ Jump up to:a b Lowe D (April 2015). “China’s First Homegrown Pharma”. Seeking Alpha.
- ^ Jump up to:a b “Chipscreen Biosciences Announces CFDA Approval of Chidamide (Epidaza) for PTCLs in China”. PR Newswire Association LLC.
- ^ “HUYA Bioscience International Grants An Exclusive License For HBI-8000 In Japan And Other Asian Countries To Eisai”. PR Newswire Association LLC. February 2016.
- ^ Qiao Z, Ren S, Li W, Wang X, He M, Guo Y, et al. (April 2013). “Chidamide, a novel histone deacetylase inhibitor, synergistically enhances gemcitabine cytotoxicity in pancreatic cancer cells”. Biochemical and Biophysical Research Communications. 434 (1): 95–101. doi:10.1016/j.bbrc.2013.03.059. PMID 23541946.
- ^ Guha M (April 2015). “HDAC inhibitors still need a home run, despite recent approval”. Nature Reviews. Drug Discovery. 14 (4): 225–6. doi:10.1038/nrd4583. PMID 25829268. S2CID 36758974.
- ^ Wang SS (2015-04-02). “A New Cancer Drug, Made in China”. The Wall Street Journal. Retrieved 13 April 2015.
| Clinical data | |
|---|---|
| Trade names | Epidaza |
| Other names | Tucidinostat |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 1616493-44-7 |
| PubChem CID | 9800555 |
| ChemSpider | 7976319 |
| UNII | 87CIC980Y0 |
| Chemical and physical data | |
| Formula | C22H19FN4O2 |
| Molar mass | 390.418 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI |
/////Tucidinostat, Antineoplastic, Histone deacetylase inhibitor, ツシジノスタット , Epidaza, Chidamide, APPROVALS 2021, JAPAN 2021
NEW DRUG APPROVALS
one time
$10.00
Isotretinoin

Isotretinoin
Title: Isotretinoin
CAS Registry Number: 4759-48-2
CAS Name: 13-cis-Retinoic acid
Additional Names: 2-cis-vitamin A acid; neovitamin A acid
Manufacturers’ Codes: Ro-4-3780Trademarks: Accutane (Roche); Isotrex (Stiefel); Oratane (Douglas); Roaccutane (Roche)
Molecular Formula: C20H28O2Molecular Weight: 300.44Percent Composition: C 79.95%, H 9.39%, O 10.65%
Literature References: Naturally occurring metabolite of vitamin A, q.v.; inhibits sebum production. Prepn: C. D. Robeson et al.,J. Am. Chem. Soc.77, 4111 (1955). Stereoselective process: R. Lucci, EP111325; idem,US4556518 (1984, 1985 both to Hoffmann-La Roche). Toxicology and teratogenicity study: J. J. Kamm, J. Am. Acad. Dermatol.6, 652 (1982). Identification as endogenous metabolite of all-trans-retinoic acid: M. E. Cullum, M. H. Zile, J. Biol. Chem.260, 10590 (1985). HPLC determn in serum: G. Tang, R. M. Russell, J. Lipid Res.31, 175 (1990). Review of pharmacology and clinical efficacy in acne: A. R. Shalita et al.,Cutis42, Suppl. 6A, 1-19 (1988). Symposium on clinical experience: Dermatology195, Suppl. 1, 1-37 (1997).
Properties: Reddish-orange plates from isopropyl alcohol, mp 174-175°. uv max: 354 nm (e 39800). LD50 (20 day) in mice, rats (mg/kg): 904, 901 i.p.; 3389, >4000 orally (Kamm).
Melting point: mp 174-175°Absorption maximum: uv max: 354 nm (e 39800)Toxicity data: LD50 (20 day) in mice, rats (mg/kg): 904, 901 i.p.; 3389, >4000 orally (Kamm)Therap-Cat: Antiacne.Keywords: Antiacne.
Isotretinoin, also known as 13-cis-retinoic acid and sold under the brand name Accutane among others, is a medication primarily used to treat severe acne. It is also used to prevent certain skin cancers (squamous-cell carcinoma), and in the treatment of other cancers. It is used to treat harlequin-type ichthyosis, a usually lethal skin disease, and lamellar ichthyosis. It is a retinoid, meaning it is related to vitamin A, and is found in small quantities naturally in the body. Its isomer, tretinoin, is also an acne drug.
The most common adverse effects are a transient worsening of acne (lasting 1–4 months), dry lips (cheilitis), dry and fragile skin, and an increased susceptibility to sunburn. Uncommon and rare side effects include muscle aches and pains (myalgias), and headaches. Isotretinoin is known to cause birth defects due to in-utero exposure because of the molecule’s close resemblance to retinoic acid, a natural vitamin A derivative which controls normal embryonic development. It is also associated with psychiatric side effects, most commonly depression but also, more rarely, psychosis and unusual behaviours. Other rare side effects include hyperostosis, and premature epiphyseal closure, have been reported to be persistent.
In the United States, a special procedure is required to obtain the pharmaceutical. In most other countries, a consent form is required which explains these risks. In other countries, such as Israel, it is prescribed like any other medicine from a dermatologist (after proper blood tests).
Women taking isotretinoin must not get pregnant during and for one month after the discontinuation of isotretinoin therapy. Sexual abstinence or effective contraception is mandatory during this period. Barrier methods by themselves (e.g., condoms) are not considered adequate due to the unacceptable failure rates of approximately 3%. Women who become pregnant while taking isotretinoin therapy are generally counseled to have an abortion.
It was patented in 1969 and approved for medical use in 1982.[2] It sold well, but in 2009, Roche decided to discontinue manufacturing due to diminishing market share due to the availability of the many generic versions and the settling of multiple lawsuits over side effects. It continues to be manufactured as of 2019 by Absorica, Amnesteem, Claravis, Myorisan, Sotret, and Zenatane.[3]
Medical uses
Isotretinoin is used primarily for severe cystic acne and acne that has not responded to other treatments.[4][5][6][7] Many dermatologists also support its use for treatment of lesser degrees of acne that prove resistant to other treatments, or that produce physical or psychological scarring.[8] Isotretinoin is not indicated for treatment of prepubertal acne and is not recommended in children less than 12 years of age.[9]
It is also somewhat effective for hidradenitis suppurativa and some cases of severe rosacea.[10] It can also be used to help treat harlequin ichthyosis, lamellar ichthyosis and is used in xeroderma pigmentosum cases to relieve keratoses. Isotretinoin has been used to treat the extremely rare condition fibrodysplasia ossificans progressiva. It is also used for treatment of neuroblastoma, a form of nerve cancer.
Isotretinoin therapy has furthermore proven effective against genital warts in experimental use, but is rarely used for this indication as there are more effective treatments. Isotretinoin may represent an efficacious and safe alternative systemic form of therapy for recalcitrant condylomata acuminata (RCA) of the cervix. In most countries this therapy is currently unapproved and only used if other therapies failed.[11][12]
Prescribing restrictions
Isotretinoin is a teratogen; there is about a 20–35% risk for congenital defects in infants exposed to the drug in utero, and about 30–60% of children exposed to isotretinoin prenatally have been reported to show neurocognitive impairment.[13] Because of this, there are strict controls on prescribing isotretinoin to women who may become pregnant and women who become pregnant while taking isotretinoin are strongly advised to terminate their pregnancies.[13]
In most countries, isotretinoin can only be prescribed by dermatologists or specialist physicians; some countries also allow limited prescription by general practitioners and family doctors. In the United Kingdom[14] and Australia,[15][16] isotretinoin may be prescribed only by or under the supervision of a consultant dermatologist. Because severe cystic acne has the potential to cause permanent scarring over a short period, restrictions on its more immediate availability have proved contentious.[17] In New Zealand, isotretinoin can be prescribed by any doctor but subsidised only when prescribed by a vocationally-registered general practitioner, dermatologist or nurse practitioner.[18]
In the United States, since March 2006 the dispensing of isotretinoin is run through a website called iPLEDGE. The FDA required the companies marketing the drug in the US, which at the time that iPLEDGE was launched were Roche, Mylan, Barr, and Ranbaxy, to put this website in place as a risk evaluation and mitigation strategy. These companies formed a group called the Isotretinoin Products Manufacturing Group, and it hired Covance to run the website.[19][20] Prescribers, pharmacists, and all people to whom the drug is prescribed need to register on the site and log information into it. Women with child-bearing potential must commit to using two forms of effective contraception simultaneously for the duration of isotretinoin therapy and for a month immediately preceding and a month immediately following therapy. Additionally they must have two negative pregnancy tests 30 days apart and have negative pregnancy tests before each prescription is written.[21][22]
History[edit]
The compound 13-cis retinoic acid was first studied in the 1960s at Roche Laboratories in Switzerland by Werner Bollag as a treatment for skin cancer. Experiments completed in 1971 showed that the compound was likely to be ineffective for cancer and, surprisingly, that it could be useful to treat acne. However, they also showed that the compound was likely to cause birth defects, so in light of the events around thalidomide, Roche abandoned the product. In 1975, Gary Peck and Frank Yoder independently rediscovered the drug’s use as a treatment of cystic acne while studying it as a treatment for lamellar ichthyosis, and published that work. Roche resumed work on the drug. In clinical trials, subjects were carefully screened to avoid including women who were or might become pregnant. Roche’s New Drug Application for isotretinoin for the treatment of acne included data showing that the drug caused birth defects in rabbits. The FDA approved the application in 1982.
Scientists involved in the clinical trials published articles warning of birth defects at the same time the drug was launched in the US, but nonetheless isotretinoin was taken up quickly and widely, both among dermatologists and general practitioners. Cases of birth defects showed up in the first year, leading the FDA to begin publishing case reports and to Roche sending warning letters to doctors and placing warning stickers on drug bottles, and including stronger warnings on the label. Lawsuits against Roche started to be filed. In 1983 the FDA’s advisory committee was convened and recommended stronger measures, which the FDA took and were that time unprecedented: warning blood banks not to accept blood from people taking the drug, and adding a warning to the label advising women to start taking contraceptives a month before starting the drug. However use of the drug continued to grow, as did the number of babies born with birth defects. In 1985 the label was updated to include a boxed warning. In early 1988 the FDA called for another advisory committee, and FDA employees prepared an internal memo estimating that around 1,000 babies had been born with birth defects due to isotretinoin, that up to around 1,000 miscarriages had been caused, and that between 5,000 and 7,000 women had had abortions due to isotretinoin. The memo was leaked to the New York Times[77] a few days before the meeting, leading to a storm of media attention. In the committee meeting, dermatologists and Roche each argued to keep the drug on the market but to increase education efforts; pediatricians and the CDC argued to withdraw the drug from the market. The committee recommended to restrict physicians who could prescribe the drug and to require a second opinion before it could be prescribed. The FDA, believing it did not have authority under the law to restrict who had the right to prescribe the drug, kept the drug on the market but took further unprecedented measures: it required to Roche to make warnings yet more visible and graphic, provide doctors with informed consent forms to be used when prescribing the drug, and to conduct follow up studies to test whether the measures were reducing exposure of pregnant women to the drug. Roche implemented those measures, and offered to pay for contraception counseling and pregnancy testing for women prescribed the drug; the program was called the “Pregnancy Prevention Program”.
A CDC report published in 2000[78] showed problems with the Pregnancy Prevention Program and showed that the increase in prescriptions was from off-label use, and prompted Roche to revamp its program, renaming it the “Targeted Pregnancy Prevention Program” and adding label changes like requirements for two pregnancy tests, two kinds of contraception, and for doctors to provide pharmacists with prescriptions directly; providing additional educational materials, and providing free pregnancy tests. The FDA had another advisory meeting in late 2000 that again debated how to prevent pregnant women from being exposed to the drug; dermatologists testified about the remarkable efficacy of the drug, the psychological impact of acne, and demanded autonomy to prescribe the drug; others argued that the drug be withdrawn or much stricter measures be taken. In 2001 the FDA announced a new regulatory scheme called SMART (the System to Manage Accutane Related Teratogenicity) that required Roche to provide defined training materials to doctors, and for doctors to sign and return a letter to Roche acknowledging that they had reviewed the training materials, for Roche to then send stickers to doctors, which doctors would have to place on prescriptions they give people after they have confirmed a negative pregnancy test; prescriptions could only be written for 30 days and could not be renewed, thus requiring a new pregnancy test for each prescription.[citation needed]
In February 2002, Roche’s patents for isotretinoin expired, and there are now many other companies selling cheaper generic versions of the drug. On June 29, 2009, Roche Pharmaceuticals, the original creator and distributor of isotretinoin, officially discontinued both the manufacture and distribution of their Accutane brand in the United States due to what the company described as business reasons related to low market share (below 5%), coupled with the high cost of defending personal-injury lawsuits brought by some people who took the drug.[79] Generic isotretinoin will remain available in the United States through various manufacturers. Roche USA continues to defend Accutane and claims to have treated over 13 million people since its introduction in 1982. F. Hoffmann-La Roche Ltd. apparently will continue to manufacture and distribute Roaccutane outside of the United States.[80]
Among others, actor James Marshall sued Roche over allegedly Accutane-related disease that resulted in removal of his colon.[81] The jury, however, decided that James Marshall had a pre-existing bowel disease.[82]
Several trials over inflammatory bowel disease claims have been held in the United States thus far, with many of them resulting in multimillion-dollar judgments against the makers of isotretinoin.[83]
Society and culture
Brands
As of 2017 isotretinoin was marketed under many brand names worldwide: A-Cnotren, Absorica, Accuran, Accutane, Accutin, Acne Free, Acnecutan, Acnegen, Acnemin, Acneone, Acneral, Acnestar, Acnetane, Acnetin A, Acnetrait, Acnetrex, Acnogen, Acnotin, Acnotren, Acretin, Actaven, Acugen, Acutret, Acutrex, Ai Si Jie, Aisoskin, Aknal, Aknefug Iso, Aknenormin, Aknesil, Aknetrent, Amnesteem, Atlacne, Atretin, Axotret, Casius, Ciscutan, Claravis, Contracné, Curacne, Curacné, Curakne, Curatane, Cuticilin, Decutan, Dercutane, Effederm, Epuris, Eudyna, Farmacne, Flexresan, Flitrion, I-Ret, Inerta, Inflader, Inotrin, Isac, Isdiben, Isoacne, Isobest, Isocural, Isoderm, Isoface, IsoGalen, Isogeril, Isolve, Isoprotil, Isoriac, Isosupra, Isosupra Lidose, Isotane, Isotina, Isotinon, Isotren, Isotret, Isotretinoin, Isotretinoina, Isotretinoína, Isotretinoine, Isotretinoïne, Isotrétinoïne, Isotretinoinum, Isotrex, Isotrin, Isotroin, Izotek, Izotziaja, Lisacne, Locatret, Mayesta, Myorisan, Neotrex, Netlook, Nimegen, Noitron, Noroseptan, Novacne, Oralne, Oraret, Oratane, Piplex, Policano, Procuta, Reducar, Retin A, Roaccutan, Roaccutane, Roacnetan, Roacta, Roacutan, Rocne, Rocta, Sotret, Stiefotrex, Tai Er Si, Teweisi, Tretin, Tretinac, Tretinex, Tretiva, Tufacne, Zenatane, Zerocutan, Zonatian ME, and Zoretanin.[1]
As of 2017 it was marketed as a topical combination drug with erythromycin under the brand names Isotrex Eritromicina, Isotrexin, and Munderm.[1]
Research
While excessive bone growth has been raised a possible side effect, a 2006 review found little evidence for this.[84]
syn

C. D. Robeson et al., J. Am. Chem. Soc. 77, 4111 (1955). Stereoselective process: R. Lucci, EP 111325; idem, US 4556518 (1984, 1985 both to Hoffmann-La Roche). doi:10.1021/jo00349a001.
syn
J Chem Soc 1968,(16),1982-83
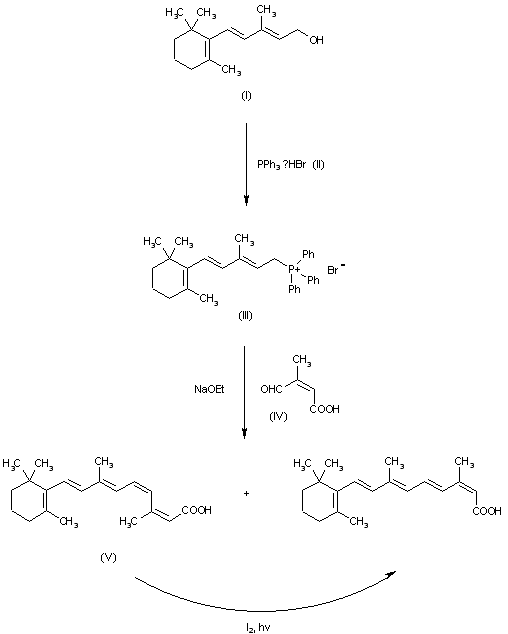
The reaction of vinyl-beta-ionol (I) with triphenylphosphonium bromide (II) in ethanol gives the corresponding phosphonium salt (III), which is condensed through a Wittig reaction with cis-beta-formylcrotonic acid (IV) by means of sodium ethoxide in ethanol to afford a mixture of cis-2-cis-4-vitamin A acid (V) and the desired product. Finally, compound (V) is isomerized bv irradiation with diffuse light in ether in the presence of iodine.
syn
Tetrahedron 2000,56(37),7211
The formylation of the beta-ionone (I) with methyl formate and NaOMe gives the enol (II), which by reaction with methanol and H2SO4 yields the dimethylacetal (III). The reaction of (III) with methylenetriphenylphosphorane (IV) affords the methylene compound (V), which is treated with formic acid to provide the aldehyde (VI). The condensation of (VI) with isopropylidenemalonic acid dimethyl ester (VII) by means of NaOH gives the polyenic malonic acid (VIII) as a mixture of isomers that is separated by crystallization in ethyl ether to yield the desired all-trans-isomer (IX). Finally, this malonic acid is selectively monodecarboxylated by means of refluxing 2,6-dimethylpyridine to afford the target (E,E,E,Z)-isomer.
References
- ^ Jump up to:a b c “Isotretinoin international brands”. Drugs.com. Retrieved 1 June 2017.
- ^ Fischer J, Ganellin CR (2006). Analogue-based Drug Discovery. John Wiley & Sons. p. 476. ISBN 978-3-527-60749-5.
- ^ “Isotretinoin (Oral Route) Description and Brand Names – Mayo Clinic”.
- ^ Merritt B, Burkhart CN, Morrell DS (June 2009). “Use of isotretinoin for acne vulgaris”. Pediatric Annals. 38 (6): 311–20. doi:10.3928/00904481-20090512-01. PMID 19588674.
- ^ Jump up to:a b Layton A (May 2009). “The use of isotretinoin in acne”. Dermato-Endocrinology. 1(3): 162–9. doi:10.4161/derm.1.3.9364. PMC 2835909. PMID 20436884.
- ^ Jump up to:a b c “Roaccutane 20mg Soft Capsules – Summary of Product Characteristics”. UK Electronic Medicines Compendium. 1 July 2015.
- ^ Jump up to:a b c US Label (PDF) (Report). FDA. 22 October 2010 [January 2010]. Retrieved 1 June2017. See FDA Index page for NDA 018662 for updates
- ^ Strauss JS, Krowchuk DP, Leyden JJ, Lucky AW, Shalita AR, Siegfried EC, Thiboutot DM, Van Voorhees AS, Beutner KA, Sieck CK, Bhushan R (April 2007). “Guidelines of care for acne vulgaris management”. Journal of the American Academy of Dermatology. 56 (4): 651–63. doi:10.1016/j.jaad.2006.08.048. PMID 17276540.
- ^ Jump up to:a b c d “Isotretinoin (oral formulations): CMDH scientific conclusions – Scientific conclusions and grounds for the variation to the terms of the Marketing Authorisation(s)”(PDF). European Medicines Agency. August 2017. Retrieved 17 May 2019.
- ^ Jump up to:a b Klasco RK, editor. Drugdex system, vol. 128. Greenwood Village (CO): Thomson Micromedex; 2006.[page needed]
- ^ Georgala S, Katoulis AC, Georgala C, Bozi E, Mortakis A (June 2004). “Oral isotretinoin in the treatment of recalcitrant condylomata acuminata of the cervix: a randomised placebo controlled trial”. Sexually Transmitted Infections. 80 (3): 216–8. doi:10.1136/sti.2003.006841. PMC 1744851. PMID 15170007.
- ^ Sehgal VN, Srivastava G, Sardana K (June 2006). “Isotretinoin–unapproved indications/uses and dosage: a physician’s reference”. International Journal of Dermatology. 45 (6): 772–7. doi:10.1111/j.1365-4632.2006.02830.x. PMID 16796650.
- ^ Jump up to:a b Choi JS, Koren G, Nulman I (March 2013). “Pregnancy and isotretinoin therapy”. Canadian Medical Association Journal. 185 (5): 411–3. doi:10.1503/cmaj.120729. PMC 3602257. PMID 23296582.
- ^ Joint Formulary Committee. British National Formulary (47th ed.). London: British Medical Association and Royal Pharmaceutical Society of Great Britain. ISBN 978-0-85369-584-4.[page needed]
- ^ “Fresh call for GPs to prescribe Roaccutane”. AustralianDoctor. 19 June 2012.
- ^ Specifically, doctors who are fellows of the Australasian College of Dermatologists (FACD); cf. Pharmaceutical Services Branch, Guide to poisons and therapeutic goods legislation for medical practitioners and dentists, Sydney: NSW Department of Health; 2006.[page needed]
- ^ James M (June 1996). “Isotretinoin for severe acne”. Lancet. 347 (9017): 1749–50. doi:10.1016/S0140-6736(96)90814-4. PMID 8656912. S2CID 28756302.
- ^ “Acne, Isotretinoin, and Depression”. MEDSAFE (New Zealand Ministry of Health). June 2013 [June 2005]. Retrieved 7 February 2014.
- ^ Thiboutot, D. M.; Cockerell, C. J. (1 August 2006). “iPLEDGE: A Report from the Front Lines of Dermatologic Practice”. AMA Journal of Ethics. 8 (8): 524–528. doi:10.1001/virtualmentor.2006.8.8.pfor1-0608. ISSN 1937-7010. PMID 23234692.
- ^ Darves, Bonnie (March 9, 2006). “Dermatologists Frustrated With Problematic iPledge Program”. Medscape.
- ^ “iPledge (About iPledge)”.
- ^ “Isotretinoin (marketed as Accutane) Capsule Information”. U.S. Food and Drug Administration (FDA). 2018-11-03.
- ^ Jump up to:a b c “Isotretinoin 20mg capsules – – (eMC)”. http://www.medicines.org.uk. Retrieved 2017-12-27.
- ^ “Isotretinoin 20mg capsules – – (eMC)”. http://www.medicines.org.uk. Retrieved 2018-01-10.
- ^ David M, Hodak E, Lowe NJ (1988). “Adverse effects of retinoids”. Medical Toxicology and Adverse Drug Experience. 3 (4): 273–88. doi:10.1007/bf03259940. PMID 3054426. S2CID 12432684.
- ^ DiGiovanna JJ (November 2001). “Isotretinoin effects on bone”. Journal of the American Academy of Dermatology. 45 (5): S176-82. doi:10.1067/mjd.2001.113721. PMID 11606950.
- ^ Ellis CN, Madison KC, Pennes DR, Martel W, Voorhees JJ (1984). “Isotretinoin therapy is associated with early skeletal radiographic changes”. Journal of the American Academy of Dermatology. 10 (6): 1024–9. doi:10.1016/S0190-9622(84)80329-1. PMID 6588057.
- ^ “Isotretinoin risks in acne treatment: Page 3 of 4”. October 2014.
- ^ Jump up to:a b Moy A, McNamara NA, Lin MC (September 2015). “Effects of Isotretinoin on Meibomian Glands”. Optometry and Vision Science. 92 (9): 925–30. doi:10.1097/OPX.0000000000000656. PMID 26154692. S2CID 205905994.
- ^ Jump up to:a b Lambert RW, Smith RE (March 1989). “Effects of 13-cis-retinoic acid on the hamster meibomian gland”. The Journal of Investigative Dermatology. 92 (3): 321–5. doi:10.1111/1523-1747.ep12277122. PMID 2918239.
- ^ Fraunfelder FT, Fraunfelder FW, Edwards R (September 2001). “Ocular side effects possibly associated with isotretinoin usage”. American Journal of Ophthalmology. 132 (3): 299–305. doi:10.1016/S0002-9394(01)01024-8. PMID 11530040.
- ^ Jump up to:a b c d e f g h Brelsford M, Beute TC (September 2008). “Preventing and managing the side effects of isotretinoin”. Seminars in Cutaneous Medicine and Surgery. 27 (3): 197–206. doi:10.1016/j.sder.2008.07.002. PMID 18786498.
- ^ Scheinfeld N, Bangalore S (May 2006). “Facial edema induced by isotretinoin use: a case and a review of the side effects of isotretinoin”. Journal of Drugs in Dermatology. 5 (5): 467–8. PMID 16703787.
- ^ Jump up to:a b “Updated measures for pregnancy prevention during retinoid use”. European Medicines Agency. 21 June 2018.
- ^ Roche Products Pty Ltd. Roaccutane (Australian Approved Product Information). Dee Why (NSW): Roche; 2005.[page needed]
- ^ Leyden JJ, Del Rosso JQ, Baum EW (February 2014). “The use of isotretinoin in the treatment of acne vulgaris: clinical considerations and future directions”. The Journal of Clinical and Aesthetic Dermatology. 7 (2 Suppl): S3–S21. PMC 3970835. PMID 24688620.
- ^ BNF, edition 57[page needed]
- ^ Jump up to:a b c d e f g h i j k l m Bremner JD, Shearer KD, McCaffery PJ (January 2012). “Retinoic acid and affective disorders: the evidence for an association”. The Journal of Clinical Psychiatry (Systematic Review). 73 (1): 37–50. doi:10.4088/JCP.10r05993. PMC 3276716. PMID 21903028.
- ^ Jump up to:a b c Kontaxakis VP, Skourides D, Ferentinos P, Havaki-Kontaxaki BJ, Papadimitriou GN (January 2009). “Isotretinoin and psychopathology: a review”. Annals of General Psychiatry. 8: 2. doi:10.1186/1744-859X-8-2. PMC 2637283. PMID 19154613.
- ^ Jump up to:a b c d Borovaya A, Olisova O, Ruzicka T, Sárdy M (September 2013). “Does isotretinoin therapy of acne cure or cause depression?”. International Journal of Dermatology. 52 (9): 1040–52. doi:10.1111/ijd.12169. PMID 23962262.
- ^ Jump up to:a b “Interactive Drug Analysis Profile – Isotretinoin”. mhra.gov.uk. Medicines & Healthcare Products Regulatory Agency. 31 March 2017.
- ^ Jump up to:a b Goodfield MJ, Cox NH, Bowser A, McMillan JC, Millard LG, Simpson NB, Ormerod AD (June 2010). “Advice on the safe introduction and continued use of isotretinoin in acne in the U.K. 2010”. The British Journal of Dermatology. 162 (6): 1172–9. doi:10.1111/j.1365-2133.2010.09836.x. PMID 21250961.
- ^ Jump up to:a b Ludot M, Mouchabac S, Ferreri F (June 2015). “Inter-relationships between isotretinoin treatment and psychiatric disorders: Depression, bipolar disorder, anxiety, psychosis and suicide risks”. World Journal of Psychiatry. 5 (2): 222–7. doi:10.5498/wjp.v5.i2.222. PMC 4473493. PMID 26110123.
- ^ Wysowski DK, Pitts M, Beitz J (October 2001). “An analysis of reports of depression and suicide in patients treated with isotretinoin”. Journal of the American Academy of Dermatology. 45 (4): 515–9. doi:10.1067/mjd.2001.117730. PMID 11568740.
- ^ Jump up to:a b Rowe C, Spelman L, Oziemski M, Ryan A, Manoharan S, Wilson P, Daubney M, Scott J (May 2014). “Isotretinoin and mental health in adolescents: Australian consensus”. The Australasian Journal of Dermatology (Review). 55 (2): 162–7. doi:10.1111/ajd.12117. PMID 24283385. S2CID 29178483.
- ^ Palha JA, Goodman AB (June 2006). “Thyroid hormones and retinoids: a possible link between genes and environment in schizophrenia” (PDF). Brain Research Reviews. 51(1): 61–71. doi:10.1016/j.brainresrev.2005.10.001. hdl:1822/3943. PMID 16325258. S2CID 30773986.
- ^ Jump up to:a b c d Goodman AB (March 1994). “Retinoid dysregulation as a cause of schizophrenia”. The American Journal of Psychiatry. 151 (3): 452–3. doi:10.1176/ajp.151.3.452b. PMID 8109664.
- ^ Goodman AB (May 1996). “Congenital anomalies in relatives of schizophrenic probands may indicate a retinoid pathology”. Schizophrenia Research. 19 (2–3): 163–70. doi:10.1016/0920-9964(96)88523-9. PMID 8789914. S2CID 12089905.
- ^ Goodman AB (July 2005). “Microarray results suggest altered transport and lowered synthesis of retinoic acid in schizophrenia”. Molecular Psychiatry. 10 (7): 620–1. doi:10.1038/sj.mp.4001668. PMID 15838536.
- ^ Samad TA, Krezel W, Chambon P, Borrelli E (December 1997). “Regulation of dopaminergic pathways by retinoids: activation of the D2 receptor promoter by members of the retinoic acid receptor-retinoid X receptor family”. Proceedings of the National Academy of Sciences of the United States of America. 94 (26): 14349–54. Bibcode:1997PNAS…9414349S. doi:10.1073/pnas.94.26.14349. PMC 24972. PMID 9405615.
- ^ Crockett SD, Porter CQ, Martin CF, Sandler RS, Kappelman MD (September 2010). “Isotretinoin use and the risk of inflammatory bowel disease: a case-control study”. The American Journal of Gastroenterology. 105 (9): 1986–93. doi:10.1038/ajg.2010.124. PMC 3073620. PMID 20354506.
- ^ Lowenstein EB, Lowenstein EJ (2011). “Isotretinoin systemic therapy and the shadow cast upon dermatology’s downtrodden hero”. Clinics in Dermatology. 29 (6): 652–61. doi:10.1016/j.clindermatol.2011.08.026. PMID 22014987.
- ^ “Drug Safety Update – Latest advice for medicines users – October 2017” (PDF). Medicines and Healthcare products Regulatory Agency. 3 October 2017. Retrieved 17 May2019.
- ^ “Pharmacovigilance Risk Assessment Committee (PRAC) – Minutes for the meeting on 3–6 July 2017” (PDF). European Medicines Agency. 1 September 2017. p. 44. Retrieved 17 May 2019.
- ^ Kremer I, Gaton DD, David M, Gaton E, Shapiro A (1994). “Toxic effects of systemic retinoids on meibomian glands”. Ophthalmic Research. 26 (2): 124–8. doi:10.1159/000267402. PMID 8196934.
- ^ Griffin JN, Pinali D, Olds K, Lu N, Appleby L, Doan L, Lane MA (November 2010). “13-Cis-retinoic acid decreases hypothalamic cell number in vitro”. Neuroscience Research. 68 (3): 185–90. doi:10.1016/j.neures.2010.08.003. PMID 20708044. S2CID 207152111.
- ^ Crandall J, Sakai Y, Zhang J, Koul O, Mineur Y, Crusio WE, McCaffery P (April 2004). “13-cis-retinoic acid suppresses hippocampal cell division and hippocampal-dependent learning in mice”. Proceedings of the National Academy of Sciences of the United States of America. 101 (14): 5111–6. Bibcode:2004PNAS..101.5111C. doi:10.1073/pnas.0306336101. JSTOR 3371827. PMC 387382. PMID 15051884.
- ^ Sakai Y, Crandall JE, Brodsky J, McCaffery P (June 2004). “13-cis Retinoic acid (accutane) suppresses hippocampal cell survival in mice”. Annals of the New York Academy of Sciences. 1021 (1): 436–40. Bibcode:2004NYASA1021..436S. doi:10.1196/annals.1308.059. PMID 15251924.
- ^ Nelson AM, Cong Z, Gilliland KL, Thiboutot DM (September 2011). “TRAIL contributes to the apoptotic effect of 13-cis retinoic acid in human sebaceous gland cells”. The British Journal of Dermatology. 165 (3): 526–33. doi:10.1111/j.1365-2133.2011.10392.x. PMC 3166444. PMID 21564055.
- ^ Nelson AM, Gilliland KL, Cong Z, Thiboutot DM (October 2006). “13-cis Retinoic acid induces apoptosis and cell cycle arrest in human SEB-1 sebocytes”. The Journal of Investigative Dermatology. 126 (10): 2178–89. doi:10.1038/sj.jid.5700289. PMID 16575387.
- ^ Wachter K (2009). “Isotretinoin’s Mechanism of Action Explored”. Skin & Allergy News. 40(11): 32. doi:10.1016/S0037-6337(09)70553-4.
- ^ Isotretinoin’s Mechanism of Action Elucidated Archived 2010-04-04 at the Wayback Machine. Medconnect (2009-08-28). Retrieved on 2010-11-13.
- ^ Nelson AM, Zhao W, Gilliland KL, Zaenglein AL, Liu W, Thiboutot DM (April 2008). “Neutrophil gelatinase-associated lipocalin mediates 13-cis retinoic acid-induced apoptosis of human sebaceous gland cells”. The Journal of Clinical Investigation. 118 (4): 1468–78. doi:10.1172/JCI33869. PMC 2262030. PMID 18317594.
- ^ Jump up to:a b Peck GL, Olsen TG, Yoder FW, Strauss JS, Downing DT, Pandya M, Butkus D, Arnaud-Battandier J (February 1979). “Prolonged remissions of cystic and conglobate acne with 13-cis-retinoic acid”. The New England Journal of Medicine. 300 (7): 329–33. doi:10.1056/NEJM197902153000701. PMID 153472.
- ^ Shalita A (2001). “The integral role of topical and oral retinoids in the early treatment of acne”. Journal of the European Academy of Dermatology and Venereology. 15: 43–9. doi:10.1046/j.0926-9959.2001.00012.x. PMID 11843233.
- ^ [unreliable medical source?]Farrell LN, Strauss JS, Stranieri AM (December 1980). “The treatment of severe cystic acne with 13-cis-retinoic acid. Evaluation of sebum production and the clinical response in a multiple-dose trial”. Journal of the American Academy of Dermatology. 3 (6): 602–11. doi:10.1016/S0190-9622(80)80074-0. PMID 6451637.
- ^ [unreliable medical source?]Jones H, Blanc D, Cunliffe WJ (November 1980). “13-cis retinoic acid and acne”. Lancet. 2 (8203): 1048–9. doi:10.1016/S0140-6736(80)92273-4. PMID 6107678. S2CID 40877032.
- ^ Pendino F, Flexor M, Delhommeau F, Buet D, Lanotte M, Segal-Bendirdjian E (June 2001). “Retinoids down-regulate telomerase and telomere length in a pathway distinct from leukemia cell differentiation”. Proceedings of the National Academy of Sciences of the United States of America. 98 (12): 6662–7. Bibcode:2001PNAS…98.6662P. doi:10.1073/pnas.111464998. JSTOR 3055868. PMC 34517. PMID 11371621.
- ^ Φαχαντίδης, Παναγιώτης Ε. (2007). Η επίδραση της ισοτρετινοϊνης και των αναστολέων της 5α-αναγωγάσης στις μεταλλοπρωτεάσες του συνδετικού ιστού σε ασθενείς με ακμή[The influence of isotretinoin and 5-a reductase inhibitors in metaloproteases of connective tissue in patients with ance] (in Greek). Aristotle University of Thessaloniki.[unreliable medical source?]
- ^ Toyoda M, Nakamura M, Makino T, Kagoura M, Morohashi M (June 2002). “Sebaceous glands in acne patients express high levels of neutral endopeptidase”. Experimental Dermatology. 11 (3): 241–7. doi:10.1034/j.1600-0625.2002.110307.x. PMID 12102663. S2CID 23468315.
- ^ Wysowski DK, Swartz L (May 2005). “Relationship between headache and depression in users of isotretinoin”. Archives of Dermatology. 141 (5): 640–1. doi:10.1001/archderm.141.5.640. PMID 15897395.
- ^ Magin P, Pond D, Smith W (February 2005). “Isotretinoin, depression and suicide: a review of the evidence”. The British Journal of General Practice. 55 (511): 134–8. PMC 1463189. PMID 15720936.
- ^ Ng CH, Schweitzer I (February 2003). “The association between depression and isotretinoin use in acne”. The Australian and New Zealand Journal of Psychiatry. 37 (1): 78–84. doi:10.1046/j.1440-1614.2003.01111.x. PMID 12534661. S2CID 8475675.
- ^ Jump up to:a b c d e “FDA information, side effects, and uses / Accutane (isotretinoin)”. U. S. Food and Drug Administration (FDA). Retrieved 20 January 2014.
- ^ “FDA information, side effects, and uses / Accutane (isotretinoin) : Table 2 Pharmacokinetic Parameters of Isotretinoin Mean (%CV), N=74“. U. S. Food and Drug Administration (FDA). Retrieved 20 January 2014.
- ^ “FDA information, side effects, and uses / Accutane (isotretinoin) : Drug Interactions“. U. S. Food and Drug Administration (FDA). Retrieved 20 January 2014.
- ^ Gina Kolata for the New York Times. April 22, 1988 Anti-Acne Drug Faulted in Birth
- ^ CDC. January 21, 2000 Accutane®-Exposed Pregnancies — California, 1999 MMWR Weekly 49(02);28-31
- ^ Shari Roan (7 November 2009). “New study may deal final blow to acne drug Accutane”. LA Times.
- ^ “Roche Discontinues and Plans to Delist Accutane in the U.S.” (Press release). Genentech. 2009-06-29. Archived from the original on 2009-11-08. Retrieved 2010-11-12.
- ^ Feeley J (2011-03-11). “Roche Accutane Acne Drug Caused ‘Tragedy’ for Actor, Brian Dennehy Says”. Bloomberg.
- ^ Silverman E (2011-11-04). “It’s Curtains On Actor’s Accutane Lawsuit”. Pharmalot. UBM Canon.
- ^ Voreacos D (May 30, 2007). “Roche Found Liable in First Of 400 Suits Over Accutane”. The Washington Post. Bloomberg News. Retrieved April 30, 2012.
- ^ Halverstam CP, Zeichner J, Lebwohl M (2006). “Lack of significant skeletal changes after long-term, low-dose retinoid therapy: case report and review of the literature”. Journal of Cutaneous Medicine and Surgery. 10 (6): 291–9. doi:10.2310/7750.2006.00065. PMID 17241599. S2CID 36785828.
External links
////////////Antiacne, 13-cis-Retinoic acid, 2-cis-vitamin A acid, neovitamin A acid, Isotretinoin
NEW DRUG APPROVALS
ONE TIME
$10.00
COVAX-19
Vaxine Pty Ltd company logo

Vaxine’s promising new COVID-19 vaccine candidate
A new multivalent COVID-19 vaccine developed by Australian company Vaxine to tackle the new virus variants could be game-changer in the fight against COVID-19
The world desperately needs a vaccine that blocks virus transmission and protects against all the variants. Covax-19 vaccine may soon change history”— Sharen Pringle, Vaxine Business Mananager
ADELAIDE, SA, AUSTRALIA, May 16, 2021 /EINPresswire.com/ — Professor Petrovsky, who is the Chairman and Research Director of Australian-based Vaxine Pty Ltd, explains that the two biggest challenges to tackling the COVID-19 pandemic are to develop a vaccine that completely prevents virus transmission something other COVID-19 have not been completely successful in achieving, and the second being to find a vaccine that protects equally against all the evolving immune-escape variants.
Professor Petrovsky has been researching coronavirus vaccines for the last 17 years, having previously published scientific papers on vaccines against both the SARS and MERS coronaviruses, which were highly protective in relevant animal models. He also recently published data from a collaboration with the US Army on development of a promising Ebola vaccine that protected mice against this most lethal disease after just a single vaccine dose. He has now successfully taken the same approach to design a protein-based vaccine against COVID-19.
Studies in a broad range of animal models including mice, hamsters, ferrets and monkeys, have recently revealed the high potential of this vaccine that is currently known as Covax-19(TM), but which likely will be soon rebranded as in its latest iteration it moves into late stage human trials in a number of countries.
Recent breakthrough data generated by Vaxine’s partner, Professor Kaissar Tabynov who leads the International Center for Vaccinology at the Kazakh National Agrarian University has shown that Vaxine’s unique spike protein antigen which is produced using insect cells in culture, was unique in that it not only totally protected hamsters from infection themselves but also prevented them from transmitting the virus to unvaccinated animals that were placed in the same cage two days after the vaccinated animals had been challenged with virus. Protection against transmission was not seen in hamsters given other vaccines making this finding unique to Vaxine’s spike protein antigen.
This hamster data reinforced findings in hamster, ferret and monkey challenge study performed by collaborating US Universities, who showed that two doses of Vaxine’s Covax-19 vaccine provided complete clearance of recoverable virus from the lungs and nose of animals when sampled just days after an infectious challenge.
“COVAX-19 vaccine has now been shown to be highly protective against the original Wuhan strain of the virus in hamster, ferret and monkey infection models performed by independent academic institutions in multiple countries, attesting to the strength of our protein-based vaccine approach”, says Prof. Petrovsky.
“A key element in the success of Covax-19 vaccine is the inclusion of Vaxine’s Advax adjuvant technology which acts as a turbocharger to drive an optimal immune response against the virus” explains Prof. Petrovsky who has been working on this promising vaccine adjuvant technology for the last 20 years with funding support from the US National Institutes of Health.
“We have now shown that our COVAX-19 vaccine can provide effective immunity including an ability to block nasal virus replication and this in turn successfully prevents transmission of the virus to vaccine-naïve animals,” he explains.
Follow on studies to confirm and expand upon these initial findings are currently underway at several US universities as well as Kazakh National Agrarian University, with a manuscript describing some of the initial animal data currently under review at a leading vaccine journal.
In another major breakthrough the team has now developed the vaccine into a multivariant format designed to protect against all the recently described variant strains of COVID-19, with work also underway on the most recently described Indian strains.
While the data is still preliminary says Prof. Petrovsky, the immune responses to the multivalent vaccine in mice are generating equally strong antibody binding activity against all the major virus variants. “This is extremely exciting as the world desperately needs vaccines able to protect against all the new strains of the virus including the UK, South African and Brazilian strains. By contrast , the currently available vaccines are clearly not as strong against some of these variants as they are against the original Wuhan strain” he explains.
Already there have been multiple confirmed cases of vaccine breakthrough where otherwise healthy individuals who have received mRNA, adenovirus or inactivated whole virus vaccines have become infected generally with either the South African or Brazilian variants.
This problem of immune-escape will only get worse over time as more complex variants emerge which is why Vaxine has been putting all its energy into finding a robust solution to this issue before proceeding with Phase 3 clinical trials of its Covax-19 vaccine.
Dr. Petrovsky went on to conclude “Now we have a multivalent formulation of Covax-19 vaccine that is showing high promise in animal studies, we plan to work as fast as we can to advance this new vaccine formulation in human trials, while expanding manufacturing capacity to ensure we are able to produce enough vaccine to meet the enormous global demand that will be attracted by such a successful vaccine.”
“To help us in this task Vaxine is looking to assemble a global network of partner organisations in countries around the world to assist Vaxine with vaccine development, clinical trials, manufacturing, distribution and sales. This is going to be a mammoth effort as we go to war against this insidious virus that continues to wreak havoc around the globe, with WHO recently predicting that the second year of the pandemic is likely to be much worse even than the first, an ominous warning for many countries that still remain poorly prepared and lacking in local vaccine manufacturing capability.
Vaxine wishes to help developing countries to establish their own local state-of-the-art vaccine manufacturing facilities, providing advice on appropriate facility design and undertaking technology transfer of its state of the art protein production technology to such facilities.
Countries in the developing world can no longer afford to sit and wait for outside organisations like COVAX to solve their vaccine supply problems, instead Vaxine proposes to help such countries find their own local solutions to the vaccine supply bottleneck for this.
Sharen Pringle
Vaxine Pty Ltd
437 033 400
email us here……..https://www.einnews.com/pr_news/541113168/covid-19-vaccine-breakthrough
Currently, the Australian influenza vaccine and adjuvant specialist and the Polish protein drug maker have just inked a memorandum of understanding, so the terms of a future contract remain to be defined. However, the technology behind is interesting.
The partners intent to utilize an insect cell-based recombinant spike protein of SARS-CoV–2 in combination with Vaxine’s proprietary Advax™ adjuvant and have already started Phase I testing in Australia with first result expected later this month. The company announced it will use artificial intelligence to evalutate clinical data in real time and announced the ambition to complete Phase II and III trials at the end of this year. “Supported by Microsoft technology, we aim to collect and analyse the COVAX-19™ trial data in real time, rather than waiting until the end of the trial before seeing if the vaccine is working, which is the traditional process,” said Vaxine’s Research Director Professor Nikolai Petrovsky from Flinders University in Adelaide.
Preclinically, Vaxine Pty Ltd’s syntetic spike protein with the company’s non-inflammatory Advax™ adjuvant, induced antibody and T-cell immune responses against the co-administered antigen. In various animal models, Covax-19 vaccination provided robust protection against an infection with the novel coronavirus.
The Phase I of Vaxine Pty Ltd in running since July in 40 healthy volunteers. If results are positive, the Australian vaccine maker is to expand studies and manufacturing to Europe. Under a future agreement Mabion SA would lead clinical development, manufacturing, regulatory negotiations and could exclusively market the vaccine in the EU and – optionally – in the US……..https://european-biotechnology.com/up-to-date/latest-news/news/mabion-to-licence-covid-19-jab-from-vaxine-pty-ltd.html
////////////////COVAX-19, corona virus, covid 19, Vaxine, australia, vaccine
NEW DRUG APPROVALS
ONE TIME
$10.00
DR ANTHONY MELVIN CRASTO

ORGANIC SPECTROSCOPY


{kind=link}
.svg){kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}