
FLAGS AND HITS
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO FACEBOOK
...................................................................Join me on twitter
FACEBOOK
...................................................................Join me on twitter
 ..................................................................Join me on google plus
..................................................................Join me on google plus
 Googleplus
GoogleplusMYSELF

Alpha lipoic acid
Alpha lipoic acid
(+)-Thioctic acid
- Molecular FormulaC8H14O2S2
- Average mass206.326 Da
5-[3-(1,2-Dithiolanyl)]pentanoic Acid
5-19-07-00237[Beilstein]
62-46-4[RN](+)-Thioctic acid, (+)-α-Lipoic acid, (3R)-1,2-Dithiolane-3-pentanoic acid
(R)-(+)-1,2-Dithiolane-3-pentanoic acid, (R)-(+)-lipoic acid, (R)-(+)-α-Lipoic acid
(R)-6,8-Dithiooctanoic acid, (R)-6,8-thioctic acid, (R)-α-Lipoic Acid, (R)-α-Lipoic Acid
1,2-Dithiolane-3-pentanoic acid, (3R)-
5-[(3R)-1,2-Dithiolan-3-yl]pentanoic acidd-Thioctic acid, (R)-(+)-alpha-Lipoic acid, (R)-(+)-Thioctic acid, Dexlipotam
Thioctic Acid
CAS Registry Number: 62-46-4
CAS Name: 1,2-Dithiolane-3-pentanoic acid
Additional Names: 1,2-dithiolane-3-valeric acid; 6,8-thioctic acid; a-lipoic acid; 5-(1,2-dithiolan-3-yl)valeric acid; 5-[3-(1,2-dithiolanyl)]pentanoic acid; d-[3-(1,2-dithiacyclopentyl)]pentanoic acid; protogen A; acetate replacing factor; pyruvate oxidation factor
Trademarks: Biletan (Gador); Thioctacid (Viatris); Thioctan (Katwijk); Tioctan (Fujisawa)
Molecular Formula: C8H14O2S2, Molecular Weight: 206.33
Percent Composition: C 46.57%, H 6.84%, O 15.51%, S 31.08%
Literature References: Growth factor for many bacteria and protozoa; prosthetic group, coenzyme, or substrate in plants, microorganisms, and animal tissues. Isoln of naturally occurring d-form: L. J. Reed et al.,Science114, 93 (1951); eidem,J. Am. Chem. Soc.75, 1267 (1953); Patterson et al.,ibid.76, 1823 (1954). Syntheses of dl-form: Bullock et al.,ibid.74, 1868, 3455 (1952); Hornberger et al.,ibid. 2382; Reed, US2980716 and US3049549 (1961, 1962 to Res. Corp.); Lewis, Raphael, J. Chem. Soc.1962, 4263; Ose et al.,US3223712 (1965 to Yamanouchi); J. Tsuji et al.,J. Org. Chem.43, 3606 (1978). Biosynthesis via linoleic acid: J. P. Carreau Methods Enzymol.62, 152-158 (1974). Enantioselective synthesis of d-form: P. C. Bulmanpage et al.,Chem. Commun.1986, 1408. Clinical study in treatment of Wilson’s disease: S. F. Gomes da Costa, Arzneim.-Forsch.20, 1210 (1970). Use in treatment of mushroom poisoning: R. Plotzker et al.,Am. J. Med. Sci.283, 79 (1982); J. P. Hanrahan, M. A. Gordon, J. Am. Med. Assoc.251, 1057 (1984). Reviews: Wagner, Folkers, Vitamins and Coenzymes (Interscience, New York, 1964) pp 244-263; Schmidt et al.,Angew. Chem. Int. Ed.4, 846 (1965); Schmidt et al.,Adv. Enzymol. Relat. Areas Mol. Biol.32, 423 (1969).
Derivative Type: Sodium salt
CAS Registry Number: 2319-84-8
Molecular Formula: C8H13NaO2S2, Molecular Weight: 228.31
Percent Composition: C 42.09%, H 5.74%, Na 10.07%, O 14.02%, S 28.09%
Properties: White powder, sol in water. pH of aq solns about 7.4.
Derivative Type:d-Form
CAS Registry Number: 1200-22-2
Properties: Crystals by vacuum sublimation (at 85-90° and 25 microns). mp 46-48° (microblock). [a]D23 +104° (c = 0.88 in benzene). uv max (methanol): 333 nm (e 150). pKa 5.4. Practically insol in water. Sol in fat solvents.Melting point: mp 46-48° (microblock)
pKa: pKa 5.4
Optical Rotation: [a]D23 +104° (c = 0.88 in benzene)
Absorption maximum: uv max (methanol): 333 nm (e 150)
Derivative Type:dl-Form
CAS Registry Number: 1077-28-7
Properties: Yellow needles from cyclohexane, mp 60-61°. bp 160-165°. uv spectrum: Calvin, Fed. Proc.13, 703 (1954). Practically insol in water. Sol in fat solvents. Forms a water-soluble sodium salt.
Melting point: mp 60-61°
Boiling point: bp 160-165°
Derivative Type:l-Form
CAS Registry Number: 1077-27-6
Properties: Crystals from cyclohexane, mp 45-47.5° (microblock). [a]D23 -113° (c = 1.88 in benzene). uv max (methanol): 330 nm (e 140).
Melting point: mp 45-47.5° (microblock)
Optical Rotation: [a]D23 -113° (c = 1.88 in benzene)
Absorption maximum: uv max (methanol): 330 nm (e 140)
Derivative Type: Ethylenediamine
Trademarks: Tioctidasi (ISI)
Therap-Cat: Treatment of liver disease; antidote to poisonous mushrooms (Amanita species).
Keywords: Hepatoprotectant.
Lipoic acid (LA), also known as α-lipoic acid, alpha-lipoic acid (ALA) and thioctic acid, is an organosulfur compound derived from caprylic acid (octanoic acid).[3] ALA is made in animals normally, and is essential for aerobic metabolism. It is also manufactured and is available as a dietary supplement in some countries where it is marketed as an antioxidant, and is available as a pharmaceutical drug in other countries.[3]
Physical and chemical properties
Lipoic acid (LA), also known as α-lipoic acid,[3][4] alpha-lipoic acid (ALA), and thioctic acid[5] is an organosulfur compound derived from octanoic acid.[3] LA contains two sulfur atoms (at C6 and C8) connected by a disulfide bond and is thus considered to be oxidized although either sulfur atom can exist in higher oxidation states.[3]
The carbon atom at C6 is chiral and the molecule exists as two enantiomers (R)-(+)-lipoic acid (RLA) and (S)-(-)-lipoic acid (SLA) and as a racemic mixture (R/S)-lipoic acid (R/S-LA).
LA appears physically as a yellow solid and structurally contains a terminal carboxylic acid and a terminal dithiolane ring.
For use in dietary supplement materials and compounding pharmacies, the USP has established an official monograph for R/S-LA.[6][7]
Biological function
“Lipoate” is the conjugate base of lipoic acid, and the most prevalent form of LA under physiological conditions.[3] Most endogenously produced RLA are not “free” because octanoic acid, the precursor to RLA, is bound to the enzyme complexes prior to enzymatic insertion of the sulfur atoms. As a cofactor, RLA is covalently attached by an amide bond to a terminal lysine residue of the enzyme’s lipoyl domains. One of the most studied roles of RLA is as a cofactor of the pyruvate dehydrogenase complex (PDC or PDHC), though it is a cofactor in other enzymatic systems as well (described below).[3]
Only the (R)-(+)-enantiomer (RLA) exists in nature and is essential for aerobic metabolism because RLA is an essential cofactor of many enzyme complexes.[3]
Biosynthesis and attachment
The precursor to lipoic acid, octanoic acid, is made via fatty acid biosynthesis in the form of octanoyl-acyl carrier protein.[3] In eukaryotes, a second fatty acid biosynthetic pathway in mitochondria is used for this purpose.[3] The octanoate is transferred as a thioester of acyl carrier protein from fatty acid biosynthesis to an amide of the lipoyl domain protein by an enzyme called an octanoyltransferase.[3] Two hydrogens of octanoate are replaced with sulfur groups via a radical SAM mechanism, by lipoyl synthase.[3] As a result, lipoic acid is synthesized attached to proteins and no free lipoic acid is produced. Lipoic acid can be removed whenever proteins are degraded and by action of the enzyme lipoamidase.[8] Free lipoate can be used by some organisms as an enzyme called lipoate protein ligase that attaches it covalently to the correct protein. The ligase activity of this enzyme requires ATP.[9]
Cellular transport
Along with sodium and the vitamins biotin (B7) and pantothenic acid (B5), lipoic acid enters cells through the SMVT (sodium-dependent multivitamin transporter). Each of the compounds transported by the SMVT is competitive with the others. For example research has shown that increasing intake of lipoic acid[10] or pantothenic acid[11] reduces the uptake of biotin and/or the activities of biotin-dependent enzymes.
Enzymatic activity
Lipoic acid is a cofactor for at least five enzyme systems.[3] Two of these are in the citric acid cycle through which many organisms turn nutrients into energy. Lipoylated enzymes have lipoic acid attached to them covalently. The lipoyl group transfers acyl groups in 2-oxoacid dehydrogenase complexes, and methylamine group in the glycine cleavage complex or glycine dehydrogenase.[3]
2-Oxoacid dehydrogenase transfer reactions occur by a similar mechanism in:
- the pyruvate dehydrogenase complex
- the α-ketoglutarate dehydrogenase or 2-oxoglutarate dehydrogenase complex
- the branched-chain oxoacid dehydrogenase (BCDH) complex
- the acetoin dehydrogenase complex.
The most-studied of these is the pyruvate dehydrogenase complex.[3] These complexes have three central subunits: E1-3, which are the decarboxylase, lipoyl transferase, and dihydrolipoamide dehydrogenase, respectively. These complexes have a central E2 core and the other subunits surround this core to form the complex. In the gap between these two subunits, the lipoyl domain ferries intermediates between the active sites.[3] The lipoyl domain itself is attached by a flexible linker to the E2 core and the number of lipoyl domains varies from one to three for a given organism. The number of domains has been experimentally varied and seems to have little effect on growth until over nine are added, although more than three decreased activity of the complex.[12]
Lipoic acid serves as co-factor to the acetoin dehydrogenase complex catalyzing the conversion of acetoin (3-hydroxy-2-butanone) to acetaldehyde and acetyl coenzyme A.[3]
The glycine cleavage system differs from the other complexes, and has a different nomenclature.[3] In this system, the H protein is a free lipoyl domain with additional helices, the L protein is a dihydrolipoamide dehydrogenase, the P protein is the decarboxylase, and the T protein transfers the methylamine from lipoate to tetrahydrofolate (THF) yielding methylene-THF and ammonia. Methylene-THF is then used by serine hydroxymethyltransferase to synthesize serine from glycine. This system is part of plant photorespiration.[13]
Biological sources and degradation
Lipoic acid is present in many foods in which it is bound to lysine in proteins,[3] but slightly more so in kidney, heart, liver, spinach, broccoli, and yeast extract.[14] Naturally occurring lipoic acid is always covalently bound and not readily available from dietary sources.[3] In addition, the amount of lipoic acid present in dietary sources is low. For instance, the purification of lipoic acid to determine its structure used an estimated 10 tons of liver residue, which yielded 30 mg of lipoic acid.[15] As a result, all lipoic acid available as a supplement is chemically synthesized.
Baseline levels (prior to supplementation) of RLA and R-DHLA have not been detected in human plasma.[16] RLA has been detected at 12.3−43.1 ng/mL following acid hydrolysis, which releases protein-bound lipoic acid. Enzymatic hydrolysis of protein bound lipoic acid released 1.4−11.6 ng/mL and <1-38.2 ng/mL using subtilisin and alcalase, respectively.[17][18][19]
Digestive proteolytic enzymes cleave the R-lipoyllysine residue from the mitochondrial enzyme complexes derived from food but are unable to cleave the lipoic acid-L–lysine amide bond.[20] Both synthetic lipoamide and (R)-lipoyl-L-lysine are rapidly cleaved by serum lipoamidases, which release free (R)-lipoic acid and either L-lysine or ammonia.[3] Little is known about the degradation and utilization of aliphatic sulfides such as lipoic acid, except for cysteine.[3]
Lipoic acid is metabolized in a variety of ways when given as a dietary supplement in mammals.[3][21] Degradation to tetranorlipoic acid, oxidation of one or both of the sulfur atoms to the sulfoxide, and S-methylation of the sulfide were observed. Conjugation of unmodified lipoic acid to glycine was detected especially in mice.[21] Degradation of lipoic acid is similar in humans, although it is not clear if the sulfur atoms become significantly oxidized.[3][22] Apparently mammals are not capable of utilizing lipoic acid as a sulfur source.
Chemical synthesis
-Lipoic_acid_molecular_structure.png)
(R)-Lipoic acid (RLA, top) and (S)-lipoic acid (SLA, down). A 1:1 mixture (racemate) of (R)- and (S)-lipoic acid is called (RS)-lipoic acid or (±)-lipoic acid (R/S-LA).
SLA did not exist prior to chemical synthesis in 1952.[23][24] SLA is produced in equal amounts with RLA during achiral manufacturing processes. The racemic form was more widely used clinically in Europe and Japan in the 1950s to 1960s despite the early recognition that the various forms of LA are not bioequivalent.[25] The first synthetic procedures appeared for RLA and SLA in the mid-1950s.[26][27][28][29] Advances in chiral chemistry led to more efficient technologies for manufacturing the single enantiomers by both classical resolution and asymmetric synthesis and the demand for RLA also grew at this time. In the 21st century, R/S-LA, RLA and SLA with high chemical and/or optical purities are available in industrial quantities. At the current time, most of the world supply of R/S-LA and RLA is manufactured in China and smaller amounts in Italy, Germany, and Japan. RLA is produced by modifications of a process first described by Georg Lang in a Ph.D. thesis and later patented by DeGussa.[30][31] Although RLA is favored nutritionally due to its “vitamin-like” role in metabolism, both RLA and R/S-LA are widely available as dietary supplements. Both stereospecific and non-stereospecific reactions are known to occur in vivo and contribute to the mechanisms of action, but evidence to date indicates RLA may be the eutomer (the nutritionally and therapeutically preferred form).[32][33]
Pharmacology
Pharmacokinetics
A 2007 human pharmacokinetic study of sodium RLA demonstrated the maximum concentration in plasma and bioavailability are significantly greater than the free acid form, and rivals plasma levels achieved by intravenous administration of the free acid form.[34] Additionally, high plasma levels comparable to those in animal models where Nrf2 was activated were achieved.[34]
The various forms of LA are not bioequivalent.[25][non-primary source needed] Very few studies compare individual enantiomers with racemic lipoic acid. It is unclear if twice as much racemic lipoic acid can replace RLA.[34]
The toxic dose of LA in cats is much lower than that in humans or dogs and produces hepatocellular toxicity.[35]
Pharmacodynamics
The mechanism and action of lipoic acid when supplied externally to an organism is controversial. Lipoic acid in a cell seems primarily to induce the oxidative stress response rather than directly scavenge free radicals. This effect is specific for RLA.[4] Despite the strongly reducing milieu, LA has been detected intracellularly in both oxidized and reduced forms.[36] LA is able to scavenge reactive oxygen and reactive nitrogen species in a biochemical assay due to long incubation times, but there is little evidence this occurs within a cell or that radical scavenging contributes to the primary mechanisms of action of LA.[4][37] The relatively good scavenging activity of LA toward hypochlorous acid (a bactericidal produced by neutrophils that may produce inflammation and tissue damage) is due to the strained conformation of the 5-membered dithiolane ring, which is lost upon reduction to DHLA. In cells, LA is reduced to dihydrolipoic acid, which is generally regarded as the more bioactive form of LA and the form responsible for most of the antioxidant effects and for lowering the redox activities of unbound iron and copper.[38] This theory has been challenged due to the high level of reactivity of the two free sulfhydryls, low intracellular concentrations of DHLA as well as the rapid methylation of one or both sulfhydryls, rapid side-chain oxidation to shorter metabolites and rapid efflux from the cell. Although both DHLA and LA have been found inside cells after administration, most intracellular DHLA probably exists as mixed disulfides with various cysteine residues from cytosolic and mitochondrial proteins.[32] Recent findings suggest therapeutic and anti-aging effects are due to modulation of signal transduction and gene transcription, which improve the antioxidant status of the cell. However, this likely occurs via pro-oxidant mechanisms, not by radical scavenging or reducing effects.[4][37][39]
All the disulfide forms of LA (R/S-LA, RLA and SLA) can be reduced to DHLA although both tissue specific and stereoselective (preference for one enantiomer over the other) reductions have been reported in model systems. At least two cytosolic enzymes, glutathione reductase (GR) and thioredoxin reductase (Trx1), and two mitochondrial enzymes, lipoamide dehydrogenase and thioredoxin reductase (Trx2), reduce LA. SLA is stereoselectively reduced by cytosolic GR whereas Trx1, Trx2 and lipoamide dehydrogenase stereoselectively reduce RLA. (R)-(+)-lipoic acid is enzymatically or chemically reduced to (R)-(-)-dihydrolipoic acid whereas (S)-(-)-lipoic acid is reduced to (S)-(+)-dihydrolipoic acid.[40][41][42][43][44][45][46] Dihydrolipoic acid (DHLA) can also form intracellularly and extracellularly via non-enzymatic, thiol-disulfide exchange reactions.[47]
RLA may function in vivo like a B-vitamin and at higher doses like plant-derived nutrients, such as curcumin, sulforaphane, resveratrol, and other nutritional substances that induce phase II detoxification enzymes, thus acting as cytoprotective agents.[39][48] This stress response indirectly improves the antioxidant capacity of the cell.[4]
The (S)-enantiomer of LA was shown to be toxic when administered to thiamine-deficient rats.[49][50]
Several studies have demonstrated that SLA either has lower activity than RLA or interferes with the specific effects of RLA by competitive inhibition.[51][52][53][54][55]

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
Uses
R/S-LA and RLA are widely available as over-the-counter nutritional supplements in the United States in the form of capsules, tablets, and aqueous liquids, and have been marketed as antioxidants.[3]
Although the body can synthesize LA, it can also be absorbed from the diet. Dietary supplementation in doses from 200–600 mg is likely to provide up to 1000 times the amount available from a regular diet. Gastrointestinal absorption is variable and decreases with the use of food. It is therefore recommended that dietary LA be taken 30–60 minutes before or at least 120 minutes after a meal. Maximum blood levels of LA are achieved 30–60 minutes after dietary supplementation, and it is thought to be largely metabolized in the liver.[56]
In Germany, LA is approved as a drug for the treatment of diabetic neuropathy since 1966 and is available as a non-prescription pharmaceutical.[57]
Clinical research
According to the American Cancer Society as of 2013, “there is no reliable scientific evidence at this time that lipoic acid prevents the development or spread of cancer”.[58] As of 2015, intravenously administered ALA is unapproved anywhere in the world except Germany for diabetic neuropathy, but has been proven reasonably safe and effective in four clinical trials; however another large trial over four years found no difference from placebo.[59] As of 2012, there was no good evidence alpha lipoic acid helps people with mitochondrial disorders.[60] A 2018 review recommended ALA as an anti-obesity supplement with low dosage (< 600 mg/day) for a short period of time (<10 weeks); however, it is too expensive to be practical as a complementary therapy for obesity.[61]
SYN
WO 0210151
DE 19709069; EP 0863125; US 6013833
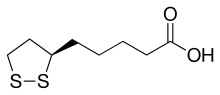
A synthetic route based on the asymmetric reduction of oxo diesters has been reported. Meldrum’s acid (LII) was acylated by methyl adipoyl chloride (LI) in the presence of pyridine to produce the intermediate (LIII) which, upon alcoholysis with isobutanol, led to oxo diester (LIV). Enantioselective reduction of (LIV) by means of baker’s yeast furnished the (S)-hydroxy diester (LV). Alternatively, the analogous oxo diester (LVI) was prepared by acylation of methyl acetoacetate with methyl adipoyl chloride (LI), followed by deacetylation in the presence of ammonium hydroxide. Then, asymmetric chemical reduction of (LVI) by hydrogenation in the presence of the chiral catalyst Ru2Cl4[(S)-BINAP]2 provided the (S)-hydroxy diester (LVII). Regioselective reduction of either diester (LV) or (LVII) by means of NaBH4 in refluxing THF furnished dihydroxy ester (XLVIII). After conversion of (XLVIII) to the dimesylate (XLIX), displacement with potassium thioacetate afforded the bis(acetylthio) derivative (LVIII), which was further hydrolyzed with KOH to provide dihydrolipoic acid (LIX). In a related procedure, dihydrolipoic acid (LIX) was prepared by reaction of dimesylate (XLIX) with sodium disulfide, followed by reductive treatment with NaBH4 and NaOH. The title cyclic disulfide was then obtained by oxidation of the dithiol (LIX) using oxygen in the presence of FeCl3.
SYN
DE 10036516; WO 0210113
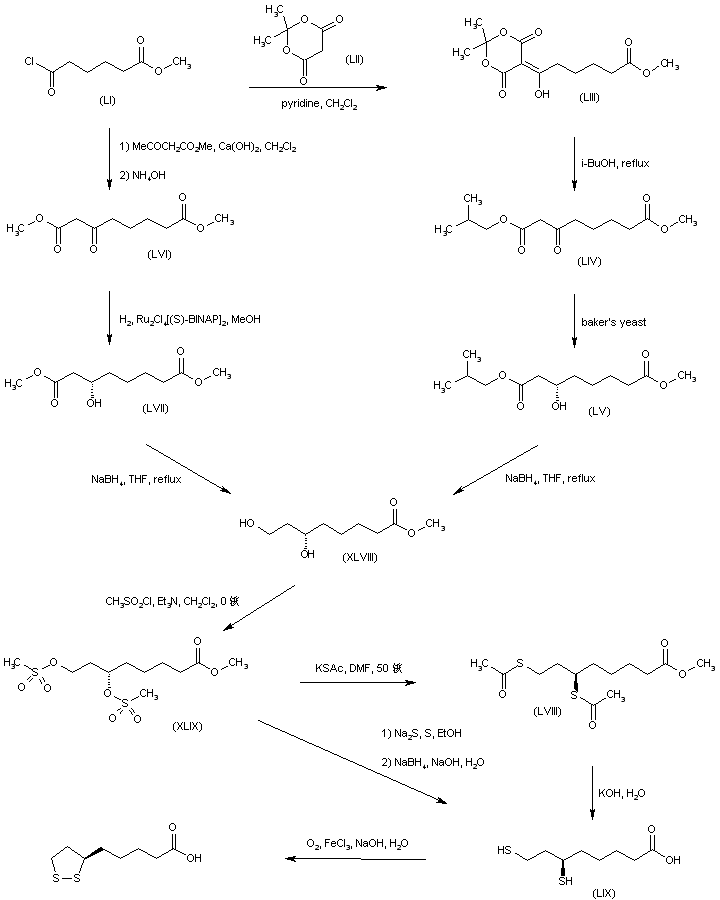
The key dihydroxy ester intermediate (XIII) was also obtained by asymmetric hydrogenation of hydroxy ketoester (XLIII) in the presence of (S)-BINAP-dichlororuthenium catalyst. The precursor hydroxy ketoester (XLIII) was prepared by two alternative procedures. In one method, the racemic dihydroxy ester (XLII) was selectively oxidized to (XLIII) by means of NaOCl. In another method, the unsaturated keto ester (XLIV) was epoxidized by means of sodium percarbonate, and the resultant epoxide (XLV) was then reduced to the hydroxy ketoester (XLIII) by catalytic hydrogenation over PtO2.
SYN
WO 0230919
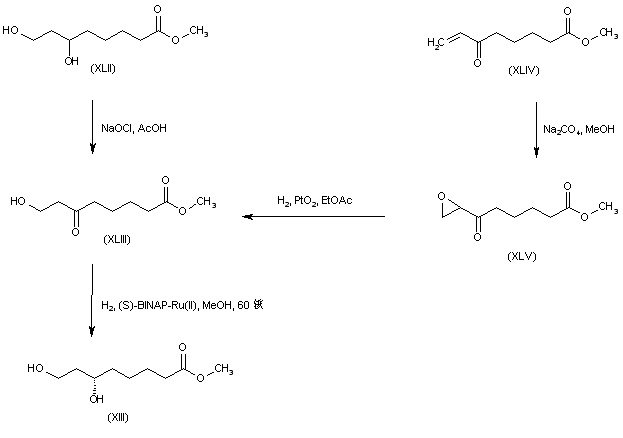
Both enantiomers of racemic 8-chloro-6-hydroxyoctanoic acid (LX) were separated employing either (+)- or (-)-alpha-methylbenzylamine. Esterification of the (R)-(-)-enantiomer with HCl-MeOH provided the chloro hydroxy ester (LXI). Further chlorination of (LXI) with SOCl2 and pyridine proceeded with inversion of configuration at C-6 to furnish the (S)-dichloro derivative (LXII). The cyclic disulfide (L) was then prepared by treatment of chloride (LXII) with sulfur and sodium sulfide in boiling EtOH. Basic hydrolysis of the methyl ester group of (LXII) then afforded (R) alpha lipoic acid. The title compound was also obtained from the (S)-(+)-acid (LXIII). Reaction of hydroxy acid (LXIII) with methanesulfonyl chloride produced the chloro mesylate (LXIV), which was then cyclized to the target disulfide in the presence of sulfur and Na2S.
SYN
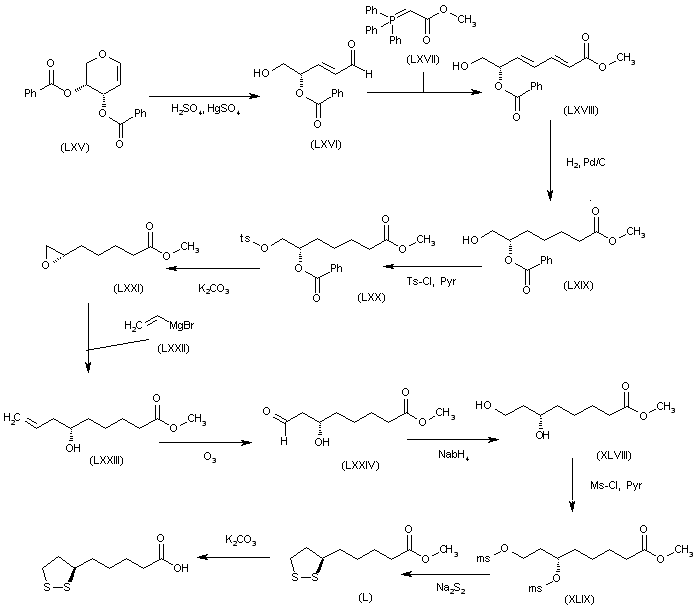
The reaction of the chiral dibenzoyloxy-dihydropyran (LXV) with H2SO4 and HgSO4 gives the unsaturated aldehyde (LXVI), which is condensed with the phosphorane (LXVII) to yield the hepatdienoic ester (LXVIII). The hydrogenation of (LXVIII) with H2 over Pd/C affords the heptanoic ester (LXIX), which is treated with Ts-Cl and pyridine to provide the tosyloxy derivative (LXX). The cyclization of (LXX) by means of K2CO3 gives the chiral epoxide (LXXI), which is condensed with vinylmagnesium bromide (LXXII) to yield 6(S)-hydroxy-8-nonenoic acid methyl ester (LXXIII). The oxidation of the terminal double bond of (LXXIII) with ozone affords the carbaldehyde (LXXIV), which is reduced with NaBH4 to provide 6(S),8-dihydroxyoctanoic acid methyl ester (XLVIII). The reaction of (XLVIII) with Ms-Cl and pyridine gives the dimesylate (XLIX), which is treated with Na2S2 to yield the lipoic acid methyl ester (L), which is hydrolyzed to the target acid with KOH in H2O.
SYN
DE 3629116; EP 0261336
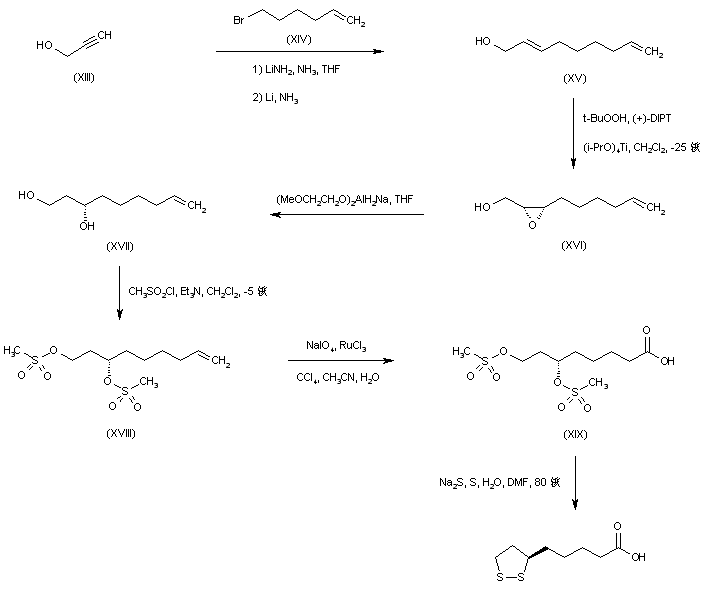
Alkylation of the lithio-dianion of propargyl alcohol (XIII) with 6-bromo-1-hexene (XIV), followed by in situ reduction of the resultant disubstituted acetylene with lithium metal gave the allylic alcohol (XV). Asymmetric Sharpless epoxidation of (XV) using tert-butyl hydroperoxide in the presence of L-(+)-diisopropyl tartrate afforded the (S,S)-epoxy alcohol (XVI). This was reduced to the chiral diol (XVII) employing Red-Al?in THF. After formation of the bis-mesylate (XVIII), oxidative cleavage of the terminal double bond by means of NaIO4 in the presence of ruthenium catalyst furnished the carboxylic acid (XIX). The mesylate groups were finally displaced by sodium disulfide to produce the desired cyclic disulfide compound.
SYN
Both enantiomers of racemic 8-chloro-6-hydroxyoctanoic acid (LX) were separated employing either (+)- or (-)-alpha-methylbenzylamine. Esterification of the (R)-(-)-enantiomer with HCl-MeOH provided the chloro hydroxy ester (LXI). Further chlorination of (LXI) with SOCl2 and pyridine proceeded with inversion of configuration at C-6 to furnish the (S)-dichloro derivative (LXII). The cyclic disulfide (L) was then prepared by treatment of chloride (LXII) with sulfur and sodium sulfide in boiling EtOH. Basic hydrolysis of the methyl ester group of (LXII) then afforded (R) alpha lipoic acid. The title compound was also obtained from the (S)-(+)-acid (LXIII). Reaction of hydroxy acid (LXIII) with methanesulfonyl chloride produced the chloro mesylate (LXIV), which was then cyclized to the target disulfide in the presence of sulfur and Na2S.
| DE 19533881; EP 0763533; US 5731448 |
SYN
WO 9638437
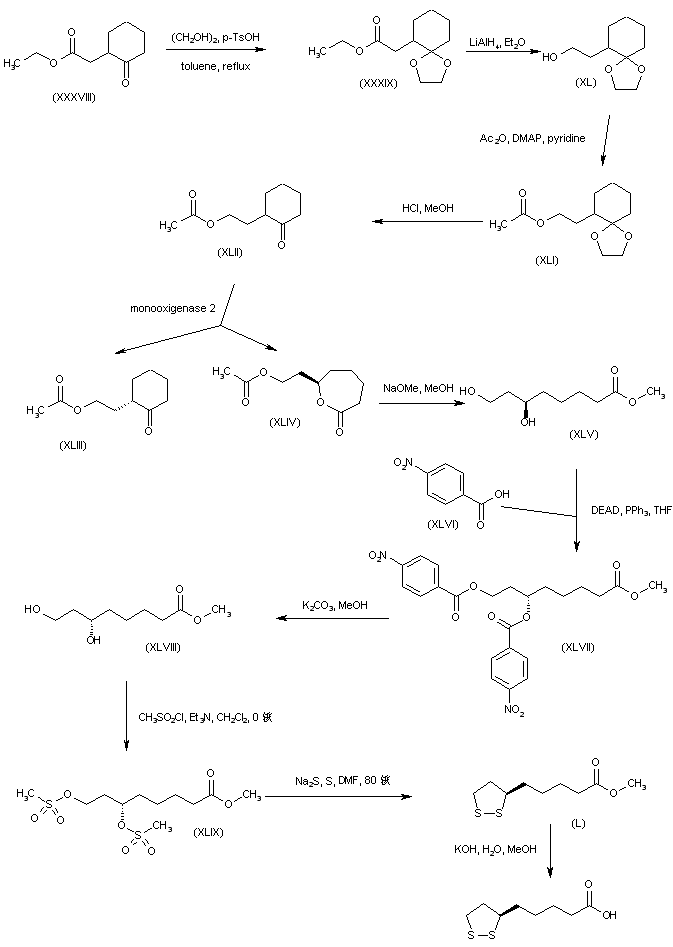
A different strategy was based on the enantioselective oxidation of a cyclohexanone derivative by enzymic Baeyer-Villiger reaction. Keto ester (XXXVIII) was protected as the ethylene ketal (XXXIX) and subsequently reduced to alcohol (XL) using LiAlH4. Acetylation of alcohol (XL) to acetate (XLI), followed by acidic ketal hydrolysis afforded cyclohexanone (XLII) (9,10). The racemic ketone (XLII) was then subjected to oxidative cleavage by monooxigenase 2 obtained from Pseudomonas putida to furnish the (R)-lactone (XLIV) along with unreacted (S)-cyclohexanone (XLIII) (9-11). The use of cyclohexanone monooxigenase from Acinetobacter NCIMB 9871 has also been reported for this reaction (12). Methanolysis of lactone (XLIV) in the presence of NaOMe gave rise to the (R)-dihydroxy ester (XLV). Inversion of the configuration of (XLV) was accomplished by Mitsunobu coupling with p-nitrobenzoic acid (XLVI) to produce the (S)-p-nitrobenzoate ester (XLVII). Smooth hydrolysis of ester (XLVII) provided methyl (S)-6,8-dihydroxyoctanoate (XLVIII), which was processed through intermediates (XLIX) and (L), as for the isopropyl (X) (Scheme 29605101a) and ethyl (XXIX) (Scheme 29605103a) homologues, to afford the title compound.
SYN
| Tetrahedron Lett 2001,42(29),4891 |
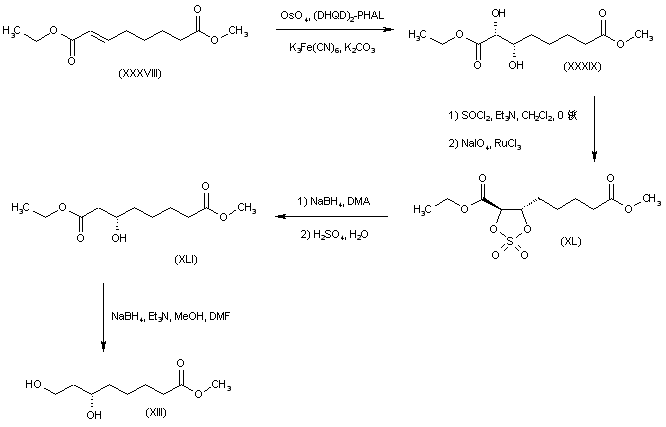
The olefinic diester (XXXVIII) was subjected to OsO4-catalyzed asymmetric dihydroxylation using hydroquinidine 1,4-phthalazinediyl diether [(DHQD)2-PHAL] as chiral ligand to afford diol (XXXIX). This was converted to the cyclic sulfate (XL) by treatment with SOCl2, followed by RuCl3-catalyzed NaIO4 oxidation of the intermediate sulfite. Regioselective reduction of sulfate (XL) at the alpha position with NaBH4 in DMA led to the (3S)-alcohol (XLI). Further selective reduction of the ethyl ester group of (XLI) was achieved by treatment with NaBH4-Et3N in MeOH-DMF, yielding the target intermediate dihydroxy ester (XIII).
SYN
1,6-Hexanediol (I) was protected as the mono-tetrahydropyranyl ether (II), and the free hydroxyl group was subsequently oxidized to aldehyde (III) under Swern conditions. Reformatskii reaction of aldehyde (III) with the organozinc reagent generated from ethyl bromoacetate yielded the racemic hydroxy ester (IV). The requisite (S)-enantiomer (VI) was obtained via oxidation of (IV) to oxo ester (V) using pyridinium chlorochromate, and then asymmetric hydrogenation in the presence of (S)-(-)-2,2′-bis(diphenylphosphino)-1,1′-binaphthyl dichlororuthenium complex. Oxo ester (V) was also prepared by SnCl2-catalyzed insertion of ethyl diazoacetate into aldehyde (III). The chiral hydroxy ester (VI) was then reduced to diol (VII) by means of NaBH4-CuSO4. After conversion of (VII) to the corresponding dimesylate (VIII), removal of the tetrahydropyranyl protecting group under acidic conditions gave alcohol (IX). This was sequentially oxidized with PCC to aldehyde, and then with Ag2O to furnish the target dimesylate acid intermediate (X).
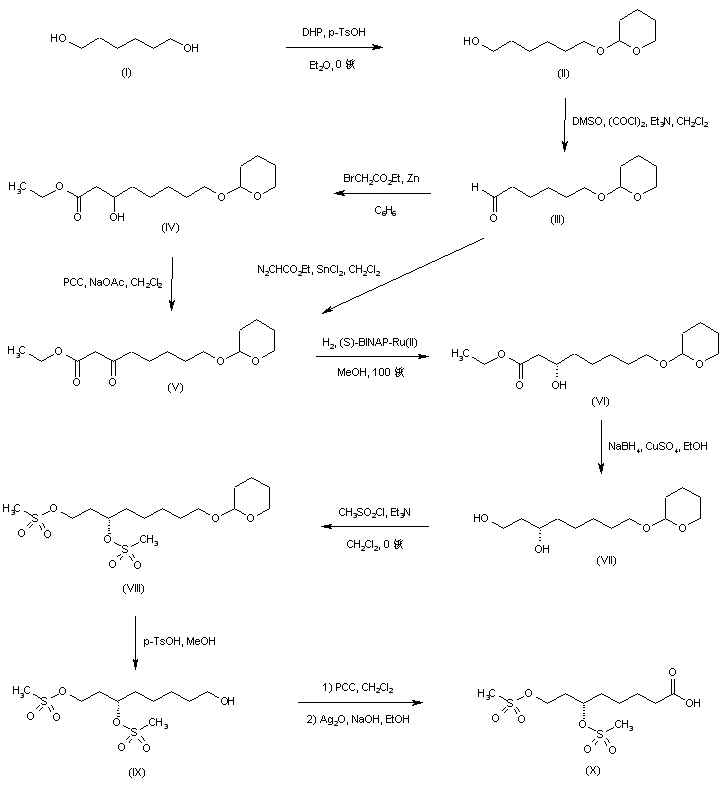
SYN
Tetrahedron Asymmetry 2000,11(4),879
The intermediate 6(S)-hydroxy-8-nonenoic acid methyl ester (III) has been obtained by enantioselective allylation of 6-oxohexanoic acid methyl ester (I) with allyltributylstannane (II) catalyzed by the chiral catalyst (R)-BINOL/Ti(O-iPr)4 in refluxing dichloromethane (other BINOL/metal catalysts have also been studied).
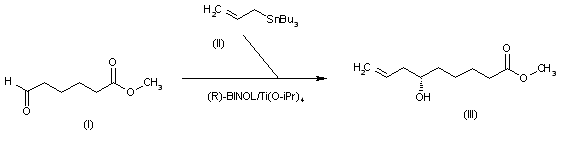
SYN
Tetrahedron Lett 1985,26(21),2535
Aldehyde (II), prepared by ozonolysis of cyclohexene (I), was ketalized with (S,S)-2,4-pentanediol (III) to afford dioxane (IV). Titanium chloride-mediated coupling of acetal (IV) with the ketene acetal (V) afforded diastereoselectively adduct (VI), which was subsequently hydrolyzed to carboxylic acid (VII) by means of trifluoroacetic acid. Removal of the pentanediol moiety to furnish the (R)-alcohol (IX) was accomplished via Jones oxidation of the secondary alcohol (VII) to ketone (VIII), followed by beta-elimination in the presence of piperidinium acetate. Reduction of the free carboxyl group by borane-tetrahydrofuran complex gave diol (X), which was further converted to dimesylate (XI). Disulfide displacement of the mesylate groups provided (+)-lipoic acid isopropyl ester (XII), which was finally hydrolyzed to the title acid using K2CO3 in MeOH/H2O.
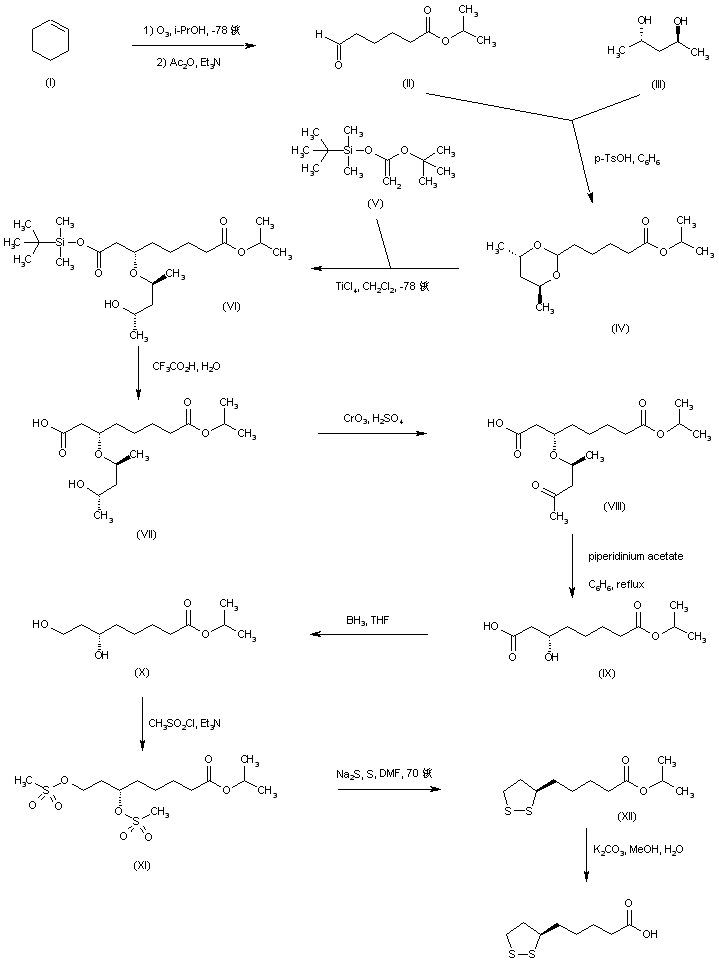
SYN
Tetrahedron Lett 1987,28(44),5313
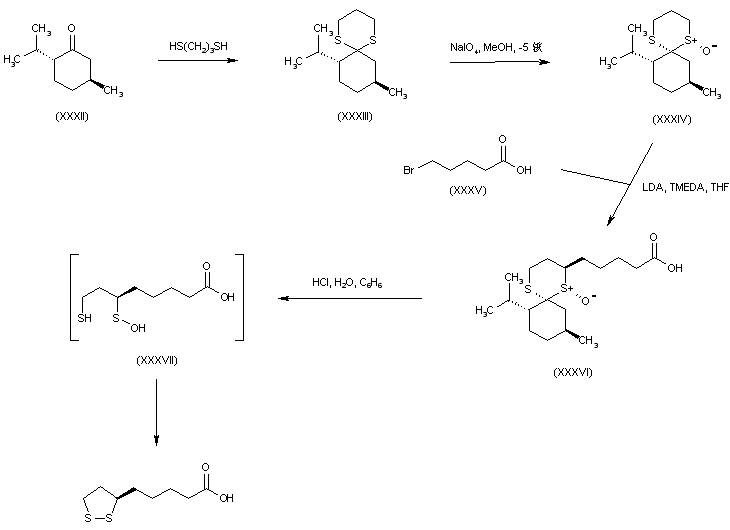
A short synthetic strategy utilized the cyclic thioketal (XXXIII), derived from d-menthone (XXXII) and 1,3-propanedithiol, as the chiral template. Stereospecific oxidation of dithiane (XXXIII) employing NaIO4 produced sulfoxide (XXXIV). The carbanion generated from sulfoxide (XXXIV) was stereoselectively alkylated by 5-bromopentanoic acid (XXXV) in the presence of TMEDA to furnish the trans alkylated compound (XXXVI). Finally, acidic hydrolysis of (XXXVI) formed the intermediate mercapto sulfinic acid (XXXVII) which spontaneously cyclized to the desired dithiolane derivative.
SYN
Tetrahedron Lett 1987,28(19),2183
Diisopropylidene mannitol (I) was first converted into the dibutyltin derivative (II), which was subsequently mono-benzylated to (III). Acetylation of (III) with acetic anhydride in pyridine gave (IV). After acidic hydrolysis of the isopropylidene ketals of (IV), the resultant tetraol (V) was converted into tetramesylate (VI). Reductive elimination in (VI) with Zn and NaI produced diene (VII). The acetate group of (VII) was then hydrolyzed to (VIII) using NaOMe. Intermediate (VIII) was reacted with triethyl orthoacetate in the presence of propionic acid to generate the allyl vinyl ether (IX), which underwent a Claisen rearrangement to the diene-ester (X). Selective hydroboration-oxidation of the terminal double bond of (X) yielded the primary alcohol (XI). Subsequent benzyl group hydrogenolysis in (XI) furnished the target intermediate diol (XII).
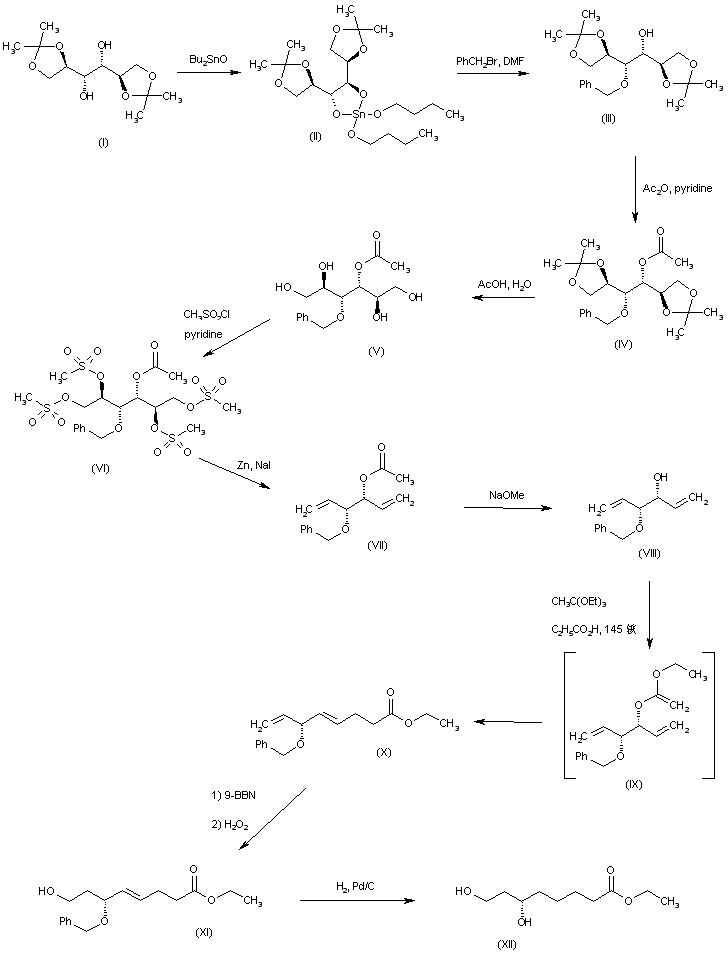
SYN
Esterification of diisopropylidene mannitol (I) with benzoyl chloride in pyridine afforded dibenzoate (II). Hydrolysis of the isopropylidene ketals of (II) with aqueous HOAc gave tetraol (III), which was further converted to tetramesylate (IV) on treatment with methanesulfonyl chloride and pyridine. Reductive elimination of the mesylate groups of (IV) using Zn dust and NaI yielded diene (V). The benzoate esters of (V) were then removed by treatment with sodium methoxide. The resultant divinylglycol (VI) was reacted with dibutyltin oxide to produce the tin derivative (VII), which was converted to the target intermediate, themono-benzyl ether (VIII), by treatment with benzyl bromide in hot DMF.
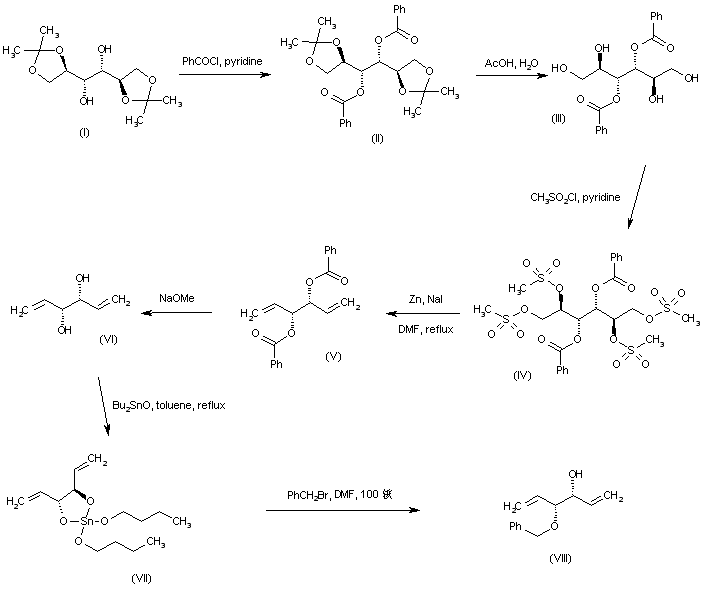
SYN
Tetrahedron Lett 1989,30(42),5705
Alkylation of the dianion of octyl acetoacetate (XIII) with 4-iodobutyronitrile (XIV) provided the cyano keto ester (XV). Enantiospecific reduction of (XV) utilizing baker’s yeast gave rise to the desired (S)-hydroxy ester (XVI) in high enantiomeric excess. Subsequent ester group reduction in (XVI) by means of LiBH4 provided diol (XVII). The target dihydroxy ester (XII) was then obtained by alcoholysis of nitrile (XVII) under acidic conditions.
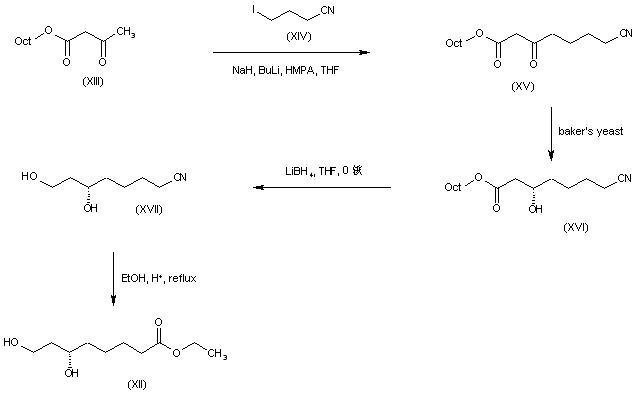
SYN
J Chem Soc Chem Commun 1995,(15),1563
A different strategy was based on the enantioselective oxidation of a cyclohexanone derivative by enzymic Baeyer-Villiger reaction. Keto ester (XXXVIII) was protected as the ethylene ketal (XXXIX) and subsequently reduced to alcohol (XL) using LiAlH4. Acetylation of alcohol (XL) to acetate (XLI), followed by acidic ketal hydrolysis afforded cyclohexanone (XLII) (9,10). The racemic ketone (XLII) was then subjected to oxidative cleavage by monooxigenase 2 obtained from Pseudomonas putida to furnish the (R)-lactone (XLIV) along with unreacted (S)-cyclohexanone (XLIII) (9-11). The use of cyclohexanone monooxigenase from Acinetobacter NCIMB 9871 has also been reported for this reaction (12). Methanolysis of lactone (XLIV) in the presence of NaOMe gave rise to the (R)-dihydroxy ester (XLV). Inversion of the configuration of (XLV) was accomplished by Mitsunobu coupling with p-nitrobenzoic acid (XLVI) to produce the (S)-p-nitrobenzoate ester (XLVII). Smooth hydrolysis of ester (XLVII) provided methyl (S)-6,8-dihydroxyoctanoate (XLVIII), which was processed through intermediates (XLIX) and (L), as for the isopropyl (X) (Scheme 29605101a) and ethyl (XXIX) (Scheme 29605103a) homologues, to afford the title compound.
SYN
Synthesis (Stuttgart) 1996,(5),594
Racemic tetrahydro-2-furylmethanol (I) was converted to tosylate (II), which was further displaced by KCN to yield nitrile (III). Basic hydrolysis of nitrile (III), followed by Fischer esterification of the resultant carboxylic acid (IV) provided ethyl ester (V). Enzymatic resolution of racemic ester (V) by means of the lipase from Candida cylindracea generated a mixture of the (R)-acid (VI) and the unreacted (S)-ester (VII), which were separated by column chromatography. The desired (S) ester (VII) was then reduced to alcohol (VIII) with LiAlH4 in cold Et2O. Regioselective opening of the cyclic ether (VIII) with iodotrimethylsilane in acetone furnished the acetonide of 6-iodo-1,3-hexanediol (IX). Alkylation of benzyl methyl malonate (X) with iodide (IX) provided malonate (XI). Hydrogenolysis of the benzyl ester group of (XI), followed by thermal decarboxylation led to ester (XII). The target dihydroxy ester precursor (XIII) was then obtained by acid-catalyzed hydrolysis of the acetonide function.
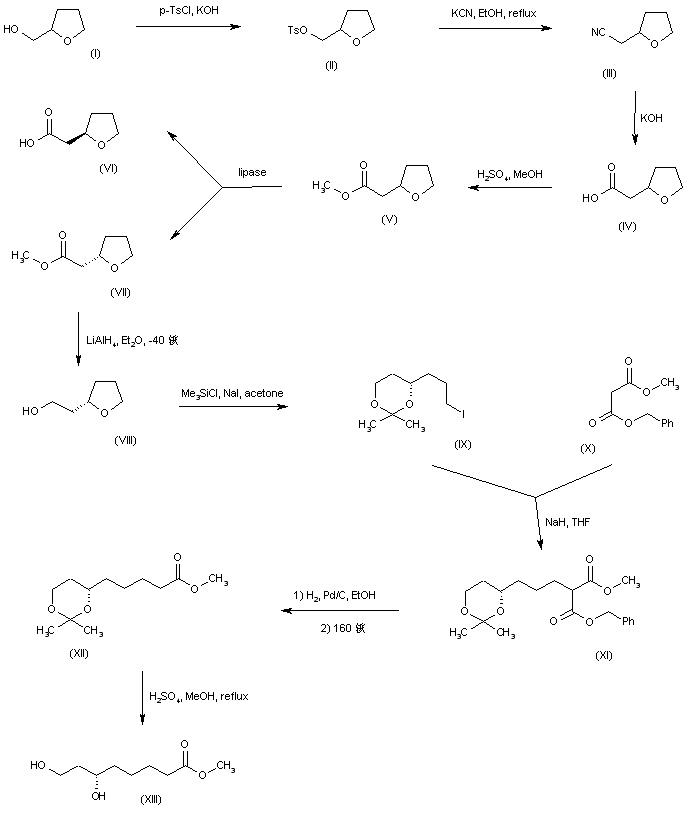
SYN
Synthesis (Stuttgart) 1996,(11),1289
Addition of vinylmagnesium bromide to 2-nitrocyclohexanone (XIV) afforded the nitro alcohol (XV). Ring cleavage of (XVI) in the presence of anhydrous CuSO4 absorbed on silica gel gave the nitro ketone (XVI). Nitro group hydrolysis in (XVI) by successive treatment with NaOMe and H2SO4 in MeOH furnished oxo ester (XVII) as the main product. This was enantiospecifically reduced with baker’s yeast to yield the (S)-alcohol (XVIII). Selective methyl ether cleavage with tetrabutylammonium iodide and BF3 provided the dihydroxy ester precursor (XIII).
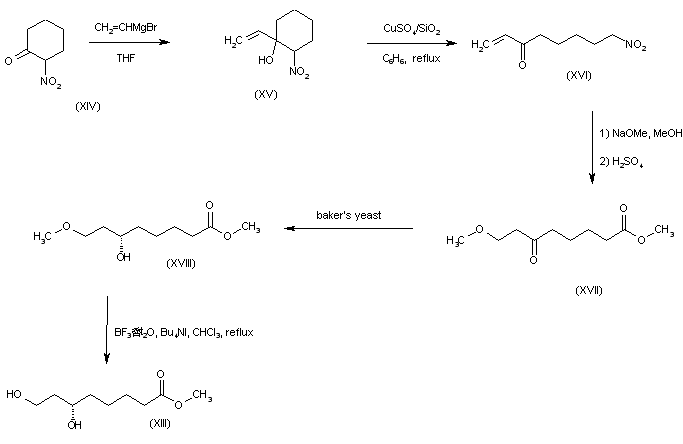
SYN
An alternative route to (+)-lipoic acid used ethyl 4,6-di-O-acetyl-2,3-dideoxy-alpha-D-erythro-hexopyranoside (XX), prepared from triacetyl-D-glucal, as the chiral starting point. Deacetylation of (XX) with sodium methoxide under Zemplen conditions gave diol (XXI) which, after conventional benzylation, led to the 4,6-di-O-benzyl derivative (XXII). Ring opening of the cyclic acetal (XXII) with propanediol in the presence of boron trifluoride afforded the dithiane derivative (XXIII). The free hydroxyl group of (XXIII) was converted into xanthate (XXIV) by reaction with NaH and CS2, followed by methyl iodide. Reductive cleavage of the xanthate group by means of Bu3SnH and AIBN provided (XXV). Hydrolysis of the thioacetal function with HgO and BF3 provided aldehyde (XXVI). Chain homologation was performed by Wittig reaction of aldehyde (XXVI) with phosphorane (XXVII) to afford the unsaturated ester (XXVIII). Simultaneous double bond hydrogenation and benzyl ether cleavage in the presence of Raney nickel led to dihydroxy ester (XXIX). This was converted to the corresponding dimesylate (XXX), which was further cyclized to disulfide (XXXI) using the in situ generated sodium disulfide as in the precedent Schemes. Finally, basic hydrolysis of the ethyl ester (XXXI) yielded the title carboxylic acid.
| Carbohydr Res 1986,148(1),51 |
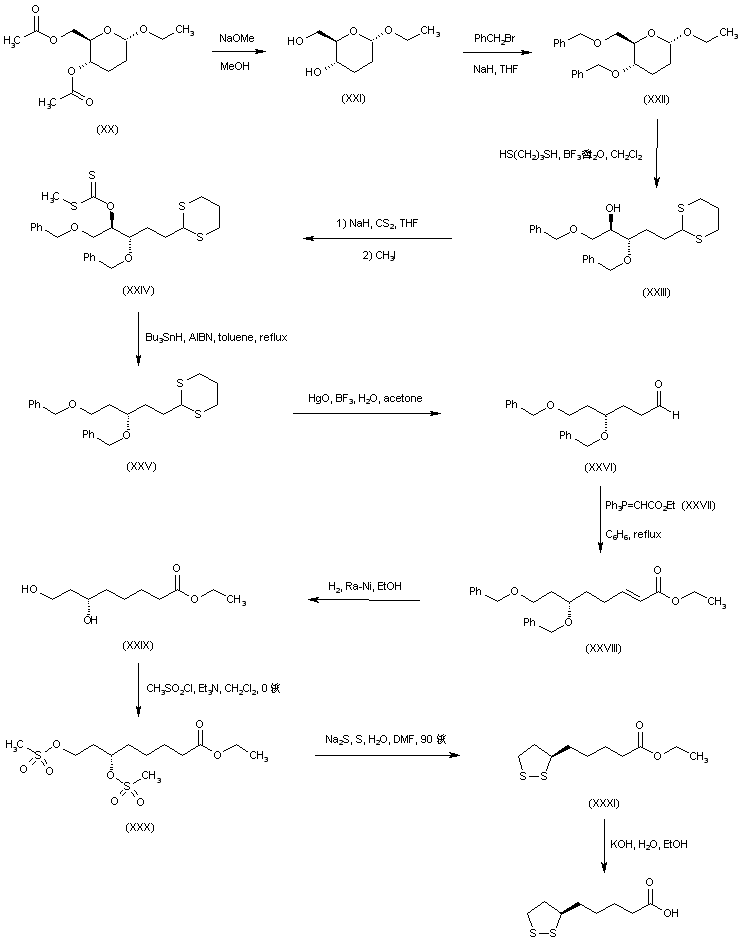
SYN
Diisopropylidene mannitol (I) was first converted into the dibutyltin derivative (II), which was subsequently mono-benzylated to (III). Acetylation of (III) with acetic anhydride in pyridine gave (IV). After acidic hydrolysis of the isopropylidene ketals of (IV), the resultant tetraol (V) was converted into tetramesylate (VI). Reductive elimination in (VI) with Zn and NaI produced diene (VII). The acetate group of (VII) was then hydrolyzed to (VIII) using NaOMe. Intermediate (VIII) was reacted with triethyl orthoacetate in the presence of propionic acid to generate the allyl vinyl ether (IX), which underwent a Claisen rearrangement to the diene-ester (X). Selective hydroboration-oxidation of the terminal double bond of (X) yielded the primary alcohol (XI). Subsequent benzyl group hydrogenolysis in (XI) furnished the target intermediate diol (XII).
| J Carbohydr Chem 1990,9(2-3),307 |
SYN
J Chem Soc Chem Commun 1986,(18),1408
SYN
https://www.sciencedirect.com/science/article/abs/pii/S1381117713003342

References
- ^ “Lipoic Acid”. Pubmed. NCBI. Retrieved October 18, 2018.
- ^ Teichert, J; Hermann, R; Ruus, P; Preiss, R (November 2003). “Plasma kinetics, metabolism, and urinary excretion of alpha-lipoic acid following oral administration in healthy volunteers”. The Journal of Clinical Pharmacology. 43 (11): 1257–67. doi:10.1177/0091270003258654. PMID 14551180. S2CID 30589232.
- ^ Jump up to:a b c d e f g h i j k l m n o p q r s t u v w x y “Lipoic acid”. Micronutrient Information Center, Linus Pauling Institute, Oregon State University, Corvallis. 1 January 2019. Retrieved 5 November 2019.
- ^ Jump up to:a b c d e Shay, KP; Moreau, RF; Smith, EJ; Hagen, TM (June 2008). “Is alpha-lipoic acid a scavenger of reactive oxygen species in vivo? Evidence for its initiation of stress signaling pathways that promote endogenous antioxidant capacity”. IUBMB Life. 60 (6): 362–7. doi:10.1002/iub.40. PMID 18409172. S2CID 33008376.
- ^ Reljanovic, M; Reichel, G; Rett, K; Lobisch, M; et al. (September 1999). “Treatment of diabetic polyneuropathy with the antioxidant thioctic acid (alpha-lipoic acid): A two year multicenter randomized double-blind placebo-controlled trial (ALADIN II). Alpha Lipoic Acid in Diabetic Neuropathy”. Free Radical Research. 31 (3): 171–9. doi:10.1080/10715769900300721. PMID 10499773.
- ^ USP32-NF27. p. 1042.
- ^ “Pharmacopeial Forum”. 34 (5): 1209.
- ^ Jiang, Y; Cronan, JE (2005). “Expression cloning and demonstration of Enterococcus faecalis lipoamidase (pyruvate dehydrogenase inactivase) as a Ser-Ser-Lys triad amidohydrolase”. Journal of Biological Chemistry. 280 (3): 2244–56. doi:10.1074/jbc.M408612200. PMID 15528186.
- ^ Cronan, JE; Zhao, X; Jiang, Y (2005). Poole, RK (ed.). Function, attachment and synthesis of lipoic acid in Escherichia coli. Advances in Microbial Physiology. 50. pp. 103–46. doi:10.1016/S0065-2911(05)50003-1. ISBN 9780120277506. PMID 16221579.
- ^ Zempleni, J.; Trusty, T. A.; Mock, D. M. (1997). “Lipoic acid reduces the activities of biotin-dependent carboxylases in rat liver”. The Journal of Nutrition. 127 (9): 1776–81. doi:10.1093/jn/127.9.1776. PMID 9278559.
- ^ Chirapu, S. R.; Rotter, C. J.; Miller, E. L.; Varma, M. V.; Dow, R. L.; Finn, M. G. (2013). “High specificity in response of the sodium-dependent multivitamin transporter to derivatives of pantothenic acid”. Current Topics in Medicinal Chemistry. 13 (7): 837–42. doi:10.2174/1568026611313070006. PMID 23578027.
- ^ Machado, RS; Clark, DP; Guest, JR (1992). “Construction and properties of pyruvate dehydrogenase complexes with up to nine lipoyl domains per lipoate acetyltransferase chain”. FEMS Microbiology Letters. 79 (1–3): 243–8. doi:10.1111/j.1574-6968.1992.tb14047.x. PMID 1478460.
- ^ Douce, R; Bourguignon, J; Neuburger, M; Rebeille, F (2001). “The glycine decarboxylase system: A fascinating complex”. Trends in Plant Science. 6 (4): 167–76. doi:10.1016/S1360-1385(01)01892-1. PMID 11286922.
- ^ Durrani, AI; Schwartz, H; Nagl, M; Sontag, G (October 2010). “Determination of free [alpha]-lipoic acid in foodstuffs by HPLC coupled with CEAD and ESI-MS”. Food Chemistry. 120 (4): 38329–36. doi:10.1016/j.foodchem.2009.11.045.
- ^ Reed, LJ (October 2001). “A trail of research from lipoic acid to alpha-keto acid dehydrogenase complexes”. Journal of Biological Chemistry. 276 (42): 38329–36. doi:10.1074/jbc.R100026200. PMID 11477096.
- ^ Hermann, R; Niebch, G; Borbe, HO; Fieger, H; et al. (1996). “Enantioselective pharmacokinetics and bioavailability of different racemic formulations in healthy volunteers”. European Journal of Pharmaceutical Sciences. 4 (3): 167–74. doi:10.1016/0928-0987(95)00045-3.
- ^ Teichert, J; Preiss, R (1997). High-performance Liquid Chromatography Methods for Determination of Lipoic and Dihydrolipoic Acid in Human Plasma. Methods in Enzymology. 279. pp. 159–66. doi:10.1016/S0076-6879(97)79019-0. ISBN 9780121821807. PMID 9211267.
- ^ Teichert, J; Preiss, R (October 1995). “Determination of lipoic acid in human plasma by high-performance liquid chromatography with electrochemical detection”. Journal of Chromatography B. 672 (2): 277–81. doi:10.1016/0378-4347(95)00225-8. PMID 8581134.
- ^ Teichert, J; Preiss, R (November 1992). “HPLC-methods for determination of lipoic acid and its reduced form in human plasma”. International Journal of Clinical Pharmacology, Therapy, and Toxicology. 30 (11): 511–2. PMID 1490813.
- ^ Biewenga, GP; Haenen, GR; Bast, A (September 1997). “The pharmacology of the antioxidant lipoic acid”. General Pharmacology. 29 (3): 315–31. doi:10.1016/S0306-3623(96)00474-0. PMID 9378235.
- ^ Jump up to:a b Schupke, H; Hempel, R; Peter, G; Hermann, R; et al. (June 2001). “New metabolic pathways of alpha-lipoic acid”. Drug Metabolism and Disposition. 29 (6): 855–62. PMID 11353754.
- ^ Teichert, J; Hermann, R; Ruus, P; Preiss, R (November 2003). “Plasma kinetics, metabolism, and urinary excretion of alpha-lipoic acid following oral administration in healthy volunteers”. Journal of Clinical Pharmacology. 43 (11): 1257–67. doi:10.1177/0091270003258654. PMID 14551180. S2CID 30589232.
- ^ Hornberger, CS; Heitmiller, RF; Gunsalus, IC; Schnakenberg, GHF; et al. (1953). “Synthesis of DL—lipoic acid”. Journal of the American Chemical Society. 75 (6): 1273–7. doi:10.1021/ja01102a003.
- ^ Hornberger, CS; Heitmiller, RF; Gunsalus, IC; Schnakenberg, GHF; et al. (1952). “Synthetic preparation of lipoic acid”. Journal of the American Chemical Society. 74 (9): 2382. doi:10.1021/ja01129a511.
- ^ Jump up to:a b Kleeman, A; Borbe, HO; Ulrich, H (1991). “Thioctic Acid-Lipoic Acid”. In Borbe, HO; Ulrich, H (eds.). Thioctsäure: Neue Biochemische, Pharmakologische und Klinische Erkenntnisse zur Thioctsäure [Thioctic Acid. New Biochemistry, Pharmacology and Findings from Clinical Practice with Thioctic Acid]. Symposium at Wiesbaden, DE, 16–18 February 1989. Frankfurt, DE: Verlag. pp. 11–26. ISBN 9783891191255.
- ^ Fontanella, L (1955). “Preparation of optical antipodes of alpha-lipoic acid”. Il Farmaco; Edizione Scientifica. 10 (12): 1043–5. PMID 13294188.
- ^ Walton, E; Wagner, AF; Bachelor, FW; Peterson, LH; et al. (1955). “Synthesis of (+)-lipoic acid and its optical antipode”. Journal of the American Chemical Society. 77 (19): 5144–9. doi:10.1021/ja01624a057.
- ^ Acker, DS; Wayne, WJ (1957). “Optically active and radioactive α-lipoic acids”. Journal of the American Chemical Society. 79 (24): 6483–6487. doi:10.1021/ja01581a033.
- ^ Deguchi, Y; Miura, K (June 1964). “Studies on the synthesis of thioctic acid and its related compounds. XIV. Synthesis of (+)-thioctamide”. Yakugaku Zasshi. 84 (6): 562–3. doi:10.1248/yakushi1947.84.6_562. PMID 14207116.
- ^ Lang, G (1992). In Vitro Metabolism of a-Lipoic Acid Especially Taking Enantioselective Bio-transformation into Account (Ph.D. thesis). Münster, DE: University of Münster.
- ^ US patent 5281722, Blaschke, G; Scheidmantel, U & Bethge, H et al., “Preparation and use of salts of the pure enantiomers of alpha-lipoic acid”, issued 1994-01-25, assigned to DeGussa.
- ^ Jump up to:a b Carlson, DA; Young, KL; Fischer, SJ; Ulrich, H. “Ch. 10: An Evaluation of the Stability and Pharmacokinetics of R-lipoic Acid and R-Dihydrolipoic Acid Dosage Forms in Plasma from Healthy Human Subjects”. Lipoic Acid: Energy Production, Antioxidant Activity and Health Effects. pp. 235–70. In Packer & Patel 2008.
- ^ Packer, L; Kraemer, K; Rimbach, G (October 2001). “Molecular aspects of lipoic acid in the prevention of diabetes complications”. Nutrition. 17 (10): 888–95. doi:10.1016/S0899-9007(01)00658-X. PMID 11684397.
- ^ Jump up to:a b c Carlson, DA; Smith, AR; Fischer, SJ; Young, KL; et al. (December 2007). “The plasma pharmacokinetics of R-(+)-lipoic acid administered as sodium R-(+)-lipoate to healthy human subjects” (PDF). Alternative Medicine Review. 12 (4): 343–51. PMID 18069903.
- ^ Hill, AS; Werner, JA; Rogers, QR; O’Neill, SL; et al. (April 2004). “Lipoic acid is 10 times more toxic in cats than reported in humans, dogs or rats”. Journal of Animal Physiology and Animal Nutrition. 88 (3–4): 150–6. doi:10.1111/j.1439-0396.2003.00472.x. PMID 15059240.
- ^ Packer, L; Witt, EH; Tritschler, HJ (August 1995). “Alpha-lipoic acid as a biological antioxidant”. Free Radical Biology and Medicine. 19 (2): 227–50. doi:10.1016/0891-5849(95)00017-R. PMID 7649494.
- ^ Jump up to:a b Shay, KP; Moreau, RF; Smith, EJ; Smith, AR; et al. (October 2009). “Alpha-lipoic acid as a dietary supplement: Molecular mechanisms and therapeutic potential”. Biochimica et Biophysica Acta (BBA) – General Subjects. 1790 (10): 1149–60. doi:10.1016/j.bbagen.2009.07.026. PMC 2756298. PMID 19664690.
- ^ Haenen, GRMM; Bast, A (1991). “Scavenging of hypochlorous acid by lipoic acid”. Biochemical Pharmacology. 42 (11): 2244–6. doi:10.1016/0006-2952(91)90363-A. PMID 1659823.
- ^ Jump up to:a b Shay, KP; Shenvi, S; Hagen, TM. “Ch. 14 Lipoic Acid as an Inducer of Phase II Detoxification Enzymes Through Activation of Nr-f2 Dependent Gene Expression”. Lipoic Acid: Energy Production, Antioxidant Activity and Health Effects. pp. 349–71. In Packer & Patel 2008.
- ^ Arnér, ES; Nordberg, J; Holmgren, A (August 1996). “Efficient reduction of lipoamide and lipoic acid by mammalian thioredoxin reductase”. Biochemical and Biophysical Research Communications. 225 (1): 268–74. doi:10.1006/bbrc.1996.1165. PMID 8769129.
- ^ Biaglow, JE; Ayene, IS; Koch, CJ; Donahue, J; et al. (April 2003). “Radiation response of cells during altered protein thiol redox”. Radiation Research. 159 (4): 484–94. Bibcode:2003RadR..159..484B. doi:10.1667/0033-7587(2003)159[0484:RROCDA]2.0.CO;2. PMID 12643793.
- ^ Haramaki, N; Han, D; Handelman, GJ; Tritschler, HJ; et al. (1997). “Cytosolic and mitochondrial systems for NADH- and NADPH-dependent reduction of alpha-lipoic acid”. Free Radical Biology and Medicine. 22 (3): 535–42. doi:10.1016/S0891-5849(96)00400-5. PMID 8981046.
- ^ Constantinescu, A; Pick, U; Handelman, GJ; Haramaki, N; et al. (July 1995). “Reduction and transport of lipoic acid by human erythrocytes”. Biochemical Pharmacology. 50 (2): 253–61. doi:10.1016/0006-2952(95)00084-D. PMID 7632170.
- ^ May, JM; Qu, ZC; Nelson, DJ (June 2006). “Cellular disulfide-reducing capacity: An integrated measure of cell redox capacity”. Biochemical and Biophysical Research Communications. 344 (4): 1352–9. doi:10.1016/j.bbrc.2006.04.065. PMID 16650819.
- ^ Jones, W; Li, X; Qu, ZC; Perriott, L; et al. (July 2002). “Uptake, recycling, and antioxidant actions of alpha-lipoic acid in endothelial cells”. Free Radical Biology and Medicine. 33 (1): 83–93. doi:10.1016/S0891-5849(02)00862-6. PMID 12086686.
- ^ Schempp, H; Ulrich, H; Elstner, EF (1994). “Stereospecific reduction of R(+)-thioctic acid by porcine heart lipoamide dehydrogenase/diaphorase”. Zeitschrift für Naturforschung C. 49 (9–10): 691–2. doi:10.1515/znc-1994-9-1023. PMID 7945680.
- ^ Biewenga, GP; Haenen, GRMM; Bast, A (1997). “Ch. 1: An Overview of Lipoate Chemistry”. In Fuchs, J; Packer, L; Zimmer, G (eds.). Lipoic Acid In Health & Disease. CRC Press. pp. 1–32. ISBN 9780824700935.
- ^ Lii, CK; Liu, KL; Cheng, YP; Lin, AH; et al. (May 2010). “Sulforaphane and alpha-lipoic acid upregulate the expression of the pi class of glutathione S-transferase through c-jun and Nrf2 activation”. Journal of Nutrition. 140 (5): 885–92. doi:10.3945/jn.110.121418. PMID 20237067.
- ^ Gal, EM; Razevska, DE (August 1960). “Studies on the in vivo metabolism of lipoic acid. 1. The fate of DL-lipoic acid-S35 in normal and thiamine-deficient rats”. Archives of Biochemistry and Biophysics. 89 (2): 253–61. doi:10.1016/0003-9861(60)90051-5. PMID 13825981.
- ^ Gal, EM (July 1965). “Reversal of selective toxicity of (-)-alpha-lipoic acid by thiamine in thiamine-deficient rats”. Nature. 207 (996): 535. Bibcode:1965Natur.207..535G. doi:10.1038/207535a0. PMID 5328673. S2CID 4146866.
- ^ US patent 6271254, Ulrich, H; Weischer, CH & Engel, J et al., “Pharmaceutical compositions containing R-alpha-lipoic acid or S-alpha.-lipoic acid as active ingredient”, issued 2001-08-07, assigned to ASTA Pharma.
- ^ Kilic, F; Handelman, GJ; Serbinova, E; Packer, L; et al. (October 1995). “Modelling cortical cataractogenesis 17: In vitro effect of a-lipoic acid on glucose-induced lens membrane damage, a model of diabetic cataractogenesis”. Biochemistry and Molecular Biology International. 37 (2): 361–70. PMID 8673020.
- ^ Artwohl, M; Schmetterer, L; Rainer, G; et al. (September 2000). Modulation by antioxidants of endothelial apoptosis, proliferation, & associated gene/protein expression. 36th Annual Meeting of the European Association for the Study of Diabetes, 17–21 September 2000, Jerusalem, Israel. Diabetologia. 43 (Suppl 1) (published August 2000). Abs 274. PMID 11008622.
- ^ Streeper, RS; Henriksen, EJ; Jacob, S; Hokama, JY; et al. (July 1997). “Differential effects of lipoic acid stereoisomers on glucose metabolism in insulin-resistant skeletal muscle”. AJP: Endocrinology and Metabolism. 273 (1 Pt 1): E185–91. doi:10.1152/ajpendo.1997.273.1.E185. PMID 9252495.
- ^ Frölich, L; Götz, ME; Weinmüller, M; Youdim, MB; et al. (March 2004). “(r)-, but not (s)-alpha lipoic acid stimulates deficient brain pyruvate dehydrogenase complex in vascular dementia, but not in Alzheimer dementia”. Journal of Neural Transmission. 111 (3): 295–310. doi:10.1007/s00702-003-0043-5. PMID 14991456. S2CID 20214857.
- ^ McIlduff, Courtney E; Rutkove, Seward B (2011-01-01). “Critical appraisal of the use of alpha lipoic acid (thioctic acid) in the treatment of symptomatic diabetic polyneuropathy”. Therapeutics and Clinical Risk Management. 7: 377–385. doi:10.2147/TCRM.S11325. ISSN 1176-6336. PMC 3176171. PMID 21941444.
- ^ Ziegle, D.; Reljanovic, M; Mehnert, H; Gries, F. A. (1999). “α-Lipoic acid in the treatment of diabetic polyneuropathy in Germany”. Experimental and Clinical Endocrinology & Diabetes. 107 (7): 421–30. doi:10.1055/s-0029-1212132. PMID 10595592.
- ^ “Lipoic Acid”. American Cancer Society. November 2008. Retrieved 5 October 2013.
- ^ Javed, S; Petropoulos, IN; Alam, U; Malik, RA (January 2015). “Treatment of painful diabetic neuropathy”. Therapeutic Advances in Chronic Disease. 6 (1): 15–28. doi:10.1177/2040622314552071. PMC 4269610. PMID 25553239.
- ^ Pfeffer G, Majamaa K, Turnbull DM, Thorburn D, Chinnery PF (April 2012). “Treatment for mitochondrial disorders”. Cochrane Database Syst Rev (4): CD004426. doi:10.1002/14651858.CD004426.pub3. PMC 7201312. PMID 22513923.
- ^ Namazi, Nazli; Larijani, Bagher; Azadbakht, Leila (2018). “Alpha-lipoic acid supplement in obesity treatment: A systematic review and meta-analysis of clinical trials”. Clinical Nutrition. 37 (2): 419–428. doi:10.1016/j.clnu.2017.06.002. ISSN 0261-5614. PMID 28629898
| Names | |
|---|---|
| IUPAC name(R)-5-(1,2-Dithiolan-3-yl)pentanoic acid | |
| Other namesα-Lipoic acid; Alpha lipoic acid; Thioctic acid; 6,8-Dithiooctanoic acid | |
| Identifiers | |
| CAS Number | 1077-28-7 (racemate) 1200-22-2 (R) |
| 3D model (JSmol) | Interactive image |
| ChEBI | CHEBI:30314 |
| ChEMBL | ChEMBL134342 |
| ChemSpider | 5886 |
| DrugBank | DB00166 |
| ECHA InfoCard | 100.012.793 |
| IUPHAR/BPS | 4822 |
| KEGG | C16241 |
| MeSH | Lipoic+acid |
| PubChem CID | 6112 |
| UNII | 73Y7P0K73Y (racemate) VLL71EBS9Z (R) |
| CompTox Dashboard (EPA) | DTXSID7025508 |
| showInChI | |
| showSMILES | |
| Properties | |
| Chemical formula | C8H14O2S2 |
| Molar mass | 206.32 g·mol−1 |
| Appearance | Yellow needle-like crystals |
| Melting point | 60–62 °C (140–144 °F; 333–335 K) |
| Solubility in water | Very Slightly Soluble(0.24 g/L)[1] |
| Solubility in ethanol 50 mg/mL | Soluble |
| Pharmacology | |
| ATC code | A16AX01 (WHO) |
| Pharmacokinetics: | |
| Bioavailability | 30% (oral)[2] |
| Related compounds | |
| Related compounds | Lipoamide Asparagusic acid |
| Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa). | |
| Infobox references |
//////////Alpha lipoic acid, d-Thioctic acid, (R)-(+)-alpha-Lipoic acid, (R)-(+)-Thioctic acid, Dexlipotam,

NEW DRUG APPROVALS
one time
$10.00
Maribavir

Maribavir
- Molecular FormulaC15H19Cl2N3O4
- Average mass376.235 Da
FDA APROVED 11/23/2021, Livtencity1263 W94, 1263W94
176161-24-3[RN]
1H-Benzimidazol-2-amine, 5,6-dichloro-N-(1-methylethyl)-1-β-L-ribofuranosyl-
UNII-PTB4X93HE1, марибавир , ماريبافير ,马立巴韦 , BW-1263W94
Camvia, D04859, G1263, GW257406X
1263W94; BW-1263W94; GW-1263; GW-257406X; SHP-620; VP-41263
Company:GlaxoSmithKline (Originator) , Shire
MOA:UL97 kinase inhibitorIndication:CMV prophylaxis
To treat post-transplant cytomegalovirus (CMV) infection/disease that does not respond (with or without genetic mutations that cause resistance) to available antiviral treatment for CMV
Press Release
Reference:1. WO9601833A1.
Syn
US 6204249
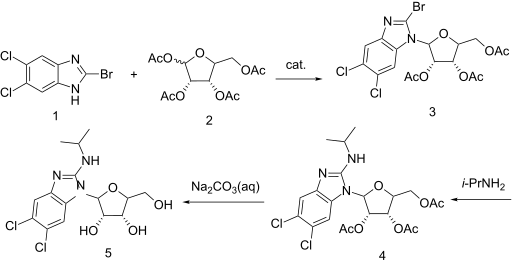

https://patents.google.com/patent/WO2001077083A1/enExample 7: 5,6-Dichloro-2-(isoproylamino)-1-(β-L-ribofuranosyl)-1 H-benzimidazolesoprylamino (10 mL) and 2-bromo-5,6-dichloro-1-(2,3,5-tri-0-acetyl-β-L- ribofuranosyl)-1 H-benzimidazole (1.0 g, 1.9 mmol) were combined with absolute ethanol (20 mL) and stirred at 75°C for 48 h. The reaction mixture was concentrated and purified on a silica gel column (2.5 vm x 16 cm, 230-400 mesh) with 1 :20 methanol: dichloromethane to give product contaminated with a small amount of higher Rf material. This was repurified on a chromatotron, fitted with a 2 mm silica gel rotor, with 1 :25 methanol.dichloromethane to give a white solid (0.43 g, 1.15 mmol, 60o/o); [a]20D=(-)22.4 (c=0.5 DMF); UVλ™* (E): pH 7.0:304 nm (95,00), 275 (1 ,800) 260 (8,300); 0.1 NaOH: 304 nm (9,900), 275 (19,00), 260 (8,100); MS (Cl): m/z (re/, intensity) 376 (100, M+1); ‘H NMR (DMSO-de) d 7.59 (s, 1 H, Ar-H), 7.35 (s, 1 H, Ar- H), 6.90 (d, 1 H, NH, J=7.8 Hz), 5.73 (d, 1 H, H-1′, J=6.5 Hz), 5.62 (t, 1 H, OH, J=4.2 Hz), 5.27-5.23 (m, 2H, OH), 4.27 (apparent dd, 1 H, J=13.4 Hz, J=7.6 Hz), 4.11 -3.99 (m, 2H), 3.97 (br. s, 1 H), 3.72-3.61 (m, 2H, H-5’), 1.18 (d, 6H, CH(CH3)2, J=6.6 Hz).Anal. Calcd. for

H2O: C, 45.70; H, 5.37; N, 10.66. Found: C, 45.75; H, 4.98; N, 10.50.
Maribavir was in phase II clinical trials for the treatment of cytomegalovirus (CMV) infection. It was granted orphan drug designation by the FDA for the indication.
The drug was originally developed by the University of Michigan and was licensed to GlaxoSmithKline. ViroPharma (now subsidiary of Shire) acquired worldwide rights to the drug from GlaxoSmithKline in 2003.
Maribavir, sold under the brand name Livtencity, is an antiviral medication that is used to treat post-transplant cytomegalovirus (CMV).[1][2]
The most common side effects include taste disturbance, nausea, diarrhea, vomiting and fatigue.[2]
Maribavir is a cytomegalovirus pUL97 kinase inhibitor that works by preventing the activity of human cytomegalovirus enzyme pUL97, thus blocking virus replication.[2]
Maribavir was approved for medical use in the United States in November 2021.[2][3]
Medical uses
Maribavir is indicated to treat people twelve years of age and older and weighing at least 35 kilograms (77 lb) with post-transplant cytomegalovirus infection/disease that does not respond (with or without genetic mutations that cause resistance) to available antiviral treatment for cytomegalovirus.[2]
Contraindications
Maribavir may reduce the antiviral activity of ganciclovir and valganciclovir, so coadministration with these medications is not recommended.[2]
History
Maribavir is licensed by ViroPharma from GlaxoSmithKline in 2003, for the prevention and treatment of human cytomegalovirus (HCMV) disease in hematopoietic stem cell/bone marrow transplant patients. The mechanism by which maribavir inhibits HCMV replication is by inhibition of an HCMV encoded protein kinase enzyme called UL97 or pUL97.[4] Maribavir showed promise in Phase II clinical trials and was granted fast track status, but failed to meet study goals in a Phase III trial.[5] However, the dosage used in the Phase III trial may have been too low to be efficacious.[6]
A Phase II study with maribavir demonstrated that prophylaxis with maribavir displayed strong antiviral activity, as measured by statistically significant reduction in the rate of reactivation of CMV in recipients of hematopoietic stem cell/bone marrow transplants.[7] In an intent-to-treat analysis of the first 100 days after the transplant, the number of subjects who required pre-emptive anti-CMV therapy was statistically significantly reduced with maribavir compared to placebo.
ViroPharma conducted a Phase III clinical study to evaluate the prophylactic use for the prevention of cytomegalovirus disease in recipients of allogeneic stem cell transplant patients. In February 2009, ViroPharma announced that the Phase III study failed to achieve its goal, showing no significant difference between maribavir and a placebo at reducing the rate at which CMV DNA levels were detected in patients.[8]
The safety and efficacy of maribavir were evaluated in a Phase III, multicenter, open-label, active-controlled trial that compared maribavir with a treatment assigned by a researcher running the study, which could include one or two of the following antivirals used to treat cytomegalovirus: ganciclovir, valganciclovir, foscarnet, or cidofovir.[2] In the study, 352 transplant recipients with cytomegalovirus infections who did not respond (with or without resistance) to treatment randomly received maribavir or treatment assigned by a researcher for up to eight weeks.[2] The study compared the two groups’ plasma cytomegalovirus DNA concentration levels at the end of the study’s eighth week, with efficacy defined as having a level below what is measurable.[2] Of the 235 participants who received maribavir, 56% had levels of cytomegalovirus DNA below what was measurable versus 24% of the 117 participants who received an investigator-assigned treatment.[2]
The U.S. Food and Drug Administration (FDA) granted the application for maribavir orphan drug, breakthrough therapy and priority review designations.[2][3][9][10] The FDA granted the approval of Livtencity to Takeda Pharmaceuticals Company Limited.[2][3]
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
FDA Approves First Treatment for Common Type of Post-Transplant Infection that is Resistant to Other Drugs
Approval is for Cytomegalovirus, a Type of Herpes Virus
https://www.fda.gov/news-events/press-announcements/fda-approves-first-treatment-common-type-post-transplant-infection-resistant-other-drugsFor Immediate Release:November 23, 2021
Today, the U.S. Food and Drug Administration approved Livtencity (maribavir) as the first drug for treating adults and pediatric patients (12 years of age and older and weighing at least 35 kilograms) with post-transplant cytomegalovirus (CMV) infection/disease that does not respond (with or without genetic mutations that cause resistance) to available antiviral treatment for CMV. Livtencity works by preventing the activity of human cytomegalovirus enzyme pUL97, thus blocking virus replication.
“Transplant recipients are at a much greater risk for complications and death when faced with a cytomegalovirus infection,” said John Farley, M.D., M.P.H., director of the Office of Infectious Diseases in the FDA’s Center for Drug Evaluation and Research. “Cytomegalovirus infections that are resistant or do not respond to available drugs are of even greater concern. Today’s approval helps meet a significant unmet medical need by providing a treatment option for this patient population.”
CMV is a type of herpes virus that commonly causes infection in patients after a stem cell or organ transplant. CMV infection can lead to CMV disease and have a major negative impact on transplant recipients, including loss of the transplanted organ and death.
Livtencity’s safety and efficacy were evaluated in a Phase 3, multicenter, open-label, active-controlled trial that compared Livtencity with a treatment assigned by a researcher running the study, which could include one or two of the following antivirals used to treat CMV: ganciclovir, valganciclovir, foscarnet or cidofovir. In the study, 352 transplant recipients with CMV infections who did not respond (with or without resistance) to treatment randomly received Livtencity or treatment assigned by a researcher for up to eight weeks.
The study compared the two groups’ plasma CMV DNA concentration levels at the end of the study’s eighth week, with efficacy defined as having a level below what is measurable. Of the 235 patients who received Livtencity, 56% had levels of CMV DNA below what was measurable versus 24% of the 117 patients who received an investigator-assigned treatment.
The most common side effects of Livtencity include taste disturbance, nausea, diarrhea, vomiting and fatigue. Livtencity may reduce the antiviral activity of ganciclovir and valganciclovir, so coadministration with these drugs is not recommended. Virologic failure due to resistance can occur during and after treatment with Livtencity, therefore CMV DNA levels should be monitored and Livtencity resistance should be checked if the patient is not responding to treatment or relapses.
Livtencity received Breakthrough Therapy and Priority Review designations for this indication. Breakthrough Therapy designation is a process designed to expedite the development and review of drugs that are intended to treat a serious condition and preliminary clinical evidence indicates that the drug may demonstrate substantial improvement over available therapy on a clinically significant endpoint(s). Priority Review designation directs overall attention and resources to the evaluation of applications for drugs that, if approved, would be significant improvements in the safety or effectiveness of the treatment, diagnosis or prevention of serious conditions when compared to standard applications.
The FDA granted the approval of Livtencity to Takeda Pharmaceuticals Company Limited.
Related Information
References
- ^ Jump up to:a b https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/215596lbl.pdf
- ^ Jump up to:a b c d e f g h i j k l m “FDA Approves First Treatment for Common Type of Post-Transplant Infection that is Resistant to Other Drugs”. U.S. Food and Drug Administration (FDA) (Press release). 23 November 2021. Retrieved 23 November 2021.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ Jump up to:a b c “Takeda’s Livtencity (maribavir) Approved by U.S. FDA as the First and Only Treatment for People Ages 12 and Older with Post-Transplant Cytomegalovirus (CMV), Refractory (With or Without Genotypic Resistance) to Conventional Antiviral Therapies”. Takeda (Press release). 23 November 2021. Retrieved 26 November 2021.
- ^ Biron KK, Harvey RJ, Chamberlain SC, Good SS, Smith AA, Davis MG, et al. (August 2002). “Potent and selective inhibition of human cytomegalovirus replication by 1263W94, a benzimidazole L-riboside with a unique mode of action”. Antimicrobial Agents and Chemotherapy. 46 (8): 2365–72. doi:10.1128/aac.46.8.2365-2372.2002. PMC 127361. PMID 12121906.
- ^ Marty FM, Ljungman P, Papanicolaou GA, Winston DJ, Chemaly RF, Strasfeld L, et al. (April 2011). “Maribavir prophylaxis for prevention of cytomegalovirus disease in recipients of allogeneic stem-cell transplants: a phase 3, double-blind, placebo-controlled, randomised trial”. The Lancet. Infectious Diseases. 11 (4): 284–92. doi:10.1016/S1473-3099(11)70024-X. PMID 21414843.
- ^ Snydman DR (April 2011). “Why did maribavir fail in stem-cell transplants?”. The Lancet. Infectious Diseases. 11 (4): 255–7. doi:10.1016/S1473-3099(11)70033-0. PMID 21414844.
- ^ Phase 2 Data Shows Maribavir Markedly Reduced Rate Of Cytomegalovirus Infection And Disease In Bone Marrow Transplant Patients, Medical News Today, Jun 2, 2008
- ^ ViroPharma:Maribavir Phase III Study Missed Goal;Shares Plunge, CNN Money, February 09, 2009
- ^ “Maribavir Orphan Drug Designations and Approvals”. U.S. Food and Drug Administration (FDA). 1 February 2007. Retrieved 26 November 2021.
- ^ “Maribavir Orphan Drug Designations and Approvals”. U.S. Food and Drug Administration (FDA). 7 June 2011. Retrieved 26 November 2021.
External links
- “Maribavir”. Drug Information Portal. U.S. National Library of Medicine.
- Clinical trial number NCT02931539 for “Efficacy and Safety Study of Maribavir Treatment Compared to Investigator-assigned Treatment in Transplant Recipients With Cytomegalovirus (CMV) Infections That Are Refractory or Resistant to Treatment With Ganciclovir, Valganciclovir, Foscarnet, or Cidofovir” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | Livtencity |
| Other names | 1263W94 |
| License data | USDailyMed: Maribavir |
| Routes of administration | By mouth |
| ATC code | J05AX10 (WHO) |
| Legal status | |
| Legal status | US:℞-only[1][2] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 176161-24-3 |
| PubChemCID | 471161 |
| DrugBank | DB06234 |
| ChemSpider | 413807 |
| UNII | PTB4X93HE1 |
| ChEMBL | ChEMBL515408 |
| NIAID ChemDB | 070966 |
| CompTox Dashboard (EPA) | DTXSID60170091 |
| Chemical and physical data | |
| Formula | C15H19Cl2N3O4 |
| Molar mass | 376.23 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
| (what is this?) (verify) |
/////////Maribavir, APPROVALS 2021, FDA 2021, Livtencity, Takeda, Breakthrough Therapy, Priority Review , ORPHAN, UNII-PTB4X93HE1, марибавир , ماريبافير ,马立巴韦 , BW-1263W94, Camvia, D04859, G1263, GW257406X, 1263W94, BW-1263W94, GW-1263, GW-257406X, SHP-620, VP-41263,

NEW DRUG APPROVALS
ONE TIME
$10.00
Pafolacianine

Pafolacianine
OTL-38
- Molecular FormulaC61H67N9O17S4
- Average mass1326.495 Da
FDA APPROVED NOV 2021
2-{(E)-2-[(3E)-2-(4-{2-[(4-{[(2-Amino-4-oxo-3,4-dihydro-6-pteridinyl)methyl]amino}benzoyl)amino]-2-carboxyethyl}phenoxy)-3-{(2E)-2-[3,3-dimethyl-5-sulfo-1-(4-sulfobutyl)-1,3-dihydro-2H-indol-2-ylidene ]ethylidene}-1-cyclohexen-1-yl]vinyl}-3,3-dimethyl-1-(4-sulfobutyl)-3H-indolium-5-sulfonate OTL-38Tyrosine, N-[4-[[(2-amino-3,4-dihydro-4-oxo-6-pteridinyl)methyl]amino]benzoyl]-O-[(6E)-6-[(2E)-2-[1,3-dihydro-3,3-dimethyl-5-sulfo-1-(4-sulfobutyl)-2H-indol-2-ylidene]ethylidene]-2-[(E)-2-[3,3-dimethy l-5-sulfo-1-(4-sulfobutyl)-3H-indolium-2-yl]ethenyl]-1-cyclohexen-1-yl]-, inner salt
2-(2-(2-(4-((2S)-2-(4-(((2-amino-4-oxo-3,4-dihydropteridin-6-yl)methyl)amino)benzamido)-2-carboxyethyl)phenoxy)-3-(2-(3,3-dimethyl-5-sulfo-1-(4-sulfobutyl)-1,3-dihydro-2H-indol-2-ylidene)ethylidene)cyclohex-1-en-1-yl)ethenyl)-3,3-dimethyl-5-sulfo-1-(4-sulfobutyl)-3H-indolium inner salt,sodium salt (1:4)
- 3H-Indolium, 2-(2-(2-(4-((2S)-2-((4-(((2-amino-3,4-dihydro-4-oxo-6-pteridinyl)methyl)amino)benzoyl)amino)-2-carboxyethyl)phenoxy)-3-(2-(1,3-dihydro-3,3-dimethyl-5-sulfo-1-(4-sulfobutyl)-2H-indol-2-ylidene)ethylidene)-1-cyclohexen-1-yl)ethenyl)-3,3-dimethyl-5-sulfo-1 (4-sulfobutyl)-, inner salt,sodium salt (1:4)
1628423-76-6 [RN]
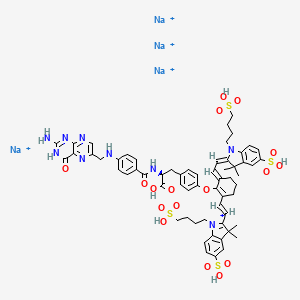
Pafolacianine sodium [USAN]
RN: 1628858-03-6
UNII: 4HUF3V875C
C61H68N9Na4O17S4+5
- Intraoperative Imaging and Detection of Folate Receptor Positive Malignant Lesions
Pafolacianine, sold under the brand name Cytalux, is an optical imaging agent.[1][2]
The most common side effects of pafolacianine include infusion-related reactions, including nausea, vomiting, abdominal pain, flushing, dyspepsia, chest discomfort, itching and hypersensitivity.[2]
It was approved for medical use in the United States in November 2021.[2][3]
Pafolacianine is a fluorescent drug that targets folate receptor (FR).[1]
Medical uses
Pafolacianine is indicated as an adjunct for intraoperative identification of malignant lesions in people with ovarian cancer.[1][2]
History
The safety and effectiveness of pafolacianine was evaluated in a randomized, multi-center, open-label study of women diagnosed with ovarian cancer or with high clinical suspicion of ovarian cancer who were scheduled to undergo surgery.[2] Of the 134 women (ages 33 to 81 years) who received a dose of pafolacianine and were evaluated under both normal and fluorescent light during surgery, 26.9% had at least one cancerous lesion detected that was not observed by standard visual or tactile inspection.[2]
The U.S. Food and Drug Administration (FDA) granted the application for pafolacianine orphan drug, priority review, and fast track designations.[2][4] The FDA granted the approval of Cytalux to On Target Laboratories, LLC.[2]
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////////////////////////////
SYN

WO 2014149073
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2014149073
In another aspect of the invention, this disclosure provides a method of synthesizing a compound having the formula
[0029] In a fourth embodiment of the invention, this disclosure provides a method of synthesizing a compound having the formula
[0030]
[0032] wherein C is any carbon isotope. In this embodiment, the amino acid linker is selected from a group consisting of methyl 2-di-tert-butyl dicarbonate-amino-3-(4-phenyl)propanoate, 3-(4-hydroxyphenyl)-2-(di-tert-butyl-dicarbonate methylamino)propanoic acid, 2-amino-4-(4-hydroxyphenyl)butanoic acid, and Tert-butyl (2-di-tert-butyl dicarbonate- amino)-3-(4-hydroxyphenyl)propanoate . In a particular embodiment, the aqueous base is potassium hydroxide (KOH). The method of this embodiment may also further include purifying the compound by preparatory HPLC.
EXAMPLE 1 : General synthesis of Pte – L Tyrosine – S0456 (OTL-0038)
[0088] Scheme:
C33H37CIF3N
Reactants for Step I:
[0089] A 500 mL round bottom flask was charged with a stirring bar, pteroic acid
(12.0 g, 29.40 mmol, 1 equiv), (L)-Tyr(-OfBu)-OfBu- HCI (1 1 .63 g, 35.28 mmol, 1 .2
equiv) and HATU (13.45 g, 35.28 mmol, 1 .2 equiv) then DMF (147 mL) was added to give a brown suspension [suspension A]. DIPEA (20.48 mL, 1 17.62 mmol, 4.0 equiv) was added slowly to suspension A at 23 °C, over 5 minutes. The suspension turned in to a clear brown solution within 10 minutes of addition of DIPEA. The reaction was stirred at 23 °C for 2.5 h. Reaction was essentially complete in 30 minutes as judged by LC/MS but was stirred further for 2.5 h. The formation of Pte_N10(TFA)_L_Tyr(-OfBu)-OfBu HCI (Figure 12) was confirmed by LC/MS showing m/z 409→m/z 684. LC/MS method: 0-50% acetonitrile in 20 mM aqueous NH4OAc for 5 min using Aquity UPLC-BEH C18, 1 .7μιη 2.1 * 50 mm column . The reaction mixture was cannulated as a steady stream to a stirred solution of aq. HCI (2.0 L, 0.28 M) over the period of 30 minutes to give light yellow precipitate of Pte_N10(TFA)_L_Tyr(-OfBu)-OfBu HCI. The precipitated Pte_N 10(TFA)_L_Tyr(- OfBu)-OfBu HCI was filtered using sintered funnel under aspirator vacuum, washed with water (8 * 300 mL) until the pH of the filtrate is between 3 and 4. The wet solid was allowed to dry under high vacuum for 12 hours on the sintered funnel. In a separate batch, where this wet solid (3) was dried under vacuum for 48 hours and then this solid was stored at -20 0 C for 48 h. However, this brief storage led to partial decomposition of 3. The wet cake (58 g) was transferred to a 500 mL round bottom flask and was submitted to the next step without further drying or purification.
Reactants for Step II:
The wet solid (58 g) was assumed to contain 29.40 mmol of the desired compound (3) (i. e. quantitative yield for the step I ).
[0090] A 500 mL round bottom flask was charged with a stirring bar, Pte_N10(TFA)_L_Tyr(-OfBu)-OfBu HCI as a wet cake (58 g, 29.40 mmol, 1 equiv). A solution of TFA:TIPS:H20 (95:2.5:2.5, 200 mL) was added at once to give a light brown suspension. The reaction content was stirred at 23°C for 1 .5 hours and was monitored by LC/MS. The suspension became clear dull brown solution after stirring for 5 minutes. LC/MS method: 0-50% acetonitrile in 20 mM aqueous NH4OAc for 5 min using Aquity UPLC-BEH C18, 1 .7μιη 2.1 * 50 mm column. The formation of Pte_TFA_L_Tyr (Figure 12) was confirmed by showing m/z 684→m/z 572. Reaction time varies from 30 min to 1 .5 hours depending on the water content of Pte_N10(TFA)_L_Tyr(-OfBu)-OfBu HCI. The reaction mixture was cannulated as a steady stream to a stirred MTBE (1 .8 L) at 23 °C or 100 °C to give light yellow precipitate of Pte_TFA_L_Tyr. The precipitated Pte_TFA_L_Tyr was filtered using sintered funnel under aspirator vacuum, washed with MTBE (6 * 300 mL) and dried under high vacuum for 8 hours to obtain Pte_TFA_L_Tyr (14.98 g, 83.98% over two steps) as a pale yellow solid. The MTBE washing was tested for absence of residual TFA utilizing wet pH paper (pH between 3-4). The yield of the reaction was between 80-85% in different batches. The deacylated side product was detected in 3.6% as judged by LC/MS. For the different batches this impurity was never more than 5%.
Reactants for Step III:
[0091] A 200 mL round bottom flask was charged with a stirring bar and Pte_TFA_L_Tyr (13.85 g, 22.78 mmol, 1 equiv), then water (95 mL) was added to give a yellow suspension [suspension B]. A freshly prepared solution of aqueous 3.75 M NaOH (26.12 mL, 97.96 mmol, 4.30 equiv), or an equivalent base at a corresponding temperature using dimethylsulfoxide (DMSO) as a solvent (as shown in Table 1 ), was added dropwise to suspension B at 23 °C, giving a clear dull yellow solution over 15 minutes [solution B]. The equivalence of NaOH varied from 3.3 to 5.0 depending on the source of 4 (solid or liquid phase synthesis) and the residual TFA. Trianion 5 (Figure 12) formation was confirmed by LC/MS showing m/z 572→m/z 476 while the solution pH was 9-10 utilizing wet pH paper. The pH of the reaction mixture was in the range of 9-10. This pH is crucial for the overall reaction completion. Notably, pH more than 10 leads to hydrolysis of S0456. Excess base will efficiently drive reaction forward with potential hydrolysis of S0456. The presence of hydrolysis by product can be visibly detected by the persistent opaque purple/blue to red/brown color.
TABLE 1 : Separate TFA deprotection via trianion formation; S0456
[0092] The precipitated OTL-0038 product could also be crashed out by adding the reaction solution steady dropwise to acetone, acetonitrile, isopropanol or ethyl acetate/acetone mixture. Acetone yields optimal results. However, viscous reactions could be slower due to partial insolubility and/or crashing out of S0456. In this reaction, the equivalence of the aqueous base is significant. Excess base will efficiently drive reaction forward with potential hydrolysis of S0456. This solution phase synthesis provides Pte_N10(TFA)_Tyr-OH »HCI salt and desires approximately 4.1 to approximately 4.8 equiv base as a source to hydrolyze the product. Particularly, precipitation of Pte_Tyr_S0456 was best achieved when 1 mL of reaction mixture is added dropwise to the stirred acetone (20 mL). Filtration of the precipitate and washing with acetone (3 x10 mL) gave the highest purity as judged from LC/MS chromatogram.
[0093] During experimentation of this solution-phase synthesis of Pte – L Tyrosine -S0456 (OTL-0038) at different stages, some optimized conditions were observed:
Mode of addition: Separate TFA deprotection via trianion formation; S0456 @ 23 °C; reflux.
Stability data of Pte – L Tyrosine – S0456 (OTL-0038):
Liquid analysis: At 40 °C the liquid lost 8.6% at 270 nm and 1 % at 774 nm. At room temperature the liquid lost about 1 .4% at 270 nm and .5% at 774 nm. At 5 °C the
270 nm seems stable and the 774 nm reasonably stable with a small degradation purity.
Source Purity Linker S0456 Base Solvent Duration % Conversion
4.3-4.6
Solution 0.95
95% 1 equiv equiv H20 15 min 100% phase equiv
K2C03
PATENT
US 20140271482
FDA approves pafolacianine for identifying malignant ovarian cancer lesions
On November 29, 2021, the Food and Drug Administration approved pafolacianine (Cytalux, On Target Laboratories, LLC), an optical imaging agent, for adult patients with ovarian cancer as an adjunct for interoperative identification of malignant lesions. Pafolacianine is a fluorescent drug that targets folate receptor which may be overexpressed in ovarian cancer. It is used with a Near-Infrared (NIR) fluorescence imaging system cleared by the FDA for specific use with pafolacianine.
Efficacy was evaluated in a single arm, multicenter, open-label study (NCT03180307) of 178 women diagnosed with ovarian cancer or with high clinical suspicion of ovarian cancer scheduled to undergo primary surgical cytoreduction, interval debulking, or recurrent ovarian cancer surgery. All patients received pafolacianine. One hundred and thirty-four patients received fluorescence imaging evaluation in addition to standard of care evaluation which includes pre-surgical imaging, intraoperative palpation and normal light evaluation of lesions. Among these patients, 36 (26.9%) had at least one evaluable ovarian cancer lesion detected with pafolacianine that was not observed by standard visual or tactile inspection. The patient-level false positive rate of pafolacianine with NIR fluorescent light with respect to the detection of ovarian cancer lesions confirmed by central pathology was 20.2% (95% CI 13.7%, 28.0%).
The most common adverse reactions (≥1%) occurring in patients were nausea, vomiting, abdominal pain, flushing, dyspepsia, chest discomfort, pruritus, and hypersensitivity.
The recommended pafolacianine dose is 0.025 mg/kg administered intravenously over 60 minutes, 1 to 9 hours before surgery. The use of folate, folic acid, or folate-containing supplements should be avoided within 48 hours before administration of pafolacianine.
View full prescribing information for Cytalux.
This application was granted priority review, fast track designation, and orphan drug designation. A description of FDA expedited programs is in the Guidance for Industry: Expedited Programs for Serious Conditions-Drugs and Biologics.
USFDA approves new drug to help identify cancer lesions
This drug is indicated for use in adult patients with ovarian cancer to help identify cancerous lesions during surgery.By The Health Master -December 2, 2021
The U.S. Food and Drug Administration (USFDA) has approved Cytalux (pafolacianine), an imaging drug intended to assist surgeons in identifying ovarian cancer lesions. The drug is designed to improve the ability to locate additional ovarian cancerous tissue that is normally difficult to detect during surgery.
Cytalux is indicated for use in adult patients with ovarian cancer to help identify cancerous lesions during surgery. The drug is a diagnostic agent that is administered in the form of an intravenous injection prior to surgery.
Alex Gorovets, M.D., deputy director of the Office of Specialty Medicine in the FDA’s Center for Drug Evaluation and Research said, “The FDA’s approval of Cytalux can help enhance the ability of surgeons to identify deadly ovarian tumors that may otherwise go undetected.
By supplementing current methods of detecting ovarian cancer during surgery, Cytalux offers health care professionals an additional imaging approach for patients with ovarian cancer.”
The American Cancer Society estimates there will be more than 21,000 new cases of ovarian cancer and more than 13,000 deaths from this disease in 2021, making it the deadliest of all female reproductive system cancers.
Conventional treatment for ovarian cancer includes surgery to remove as many of the tumors as possible, chemotherapy to stop the growth of malignant cells or other targeted therapy to identify and attack specific cancer cells.
Ovarian cancer often causes the body to overproduce a specific protein in cell membranes called a folate receptor. Following administration via injection, Cytalux binds to these proteins and illuminates under fluorescent light, boosting surgeons’ ability to identify the cancerous tissue.
Currently, surgeons rely on preoperative imaging, visual inspection of tumors under normal light or examination by touch to identify cancer lesions. Cytalux is used with a Near-Infrared fluorescence imaging system cleared by the FDA for specific use with pafolacianine.
The safety and effectiveness of Cytalux was evaluated in a randomized, multi-center, open-label study of women diagnosed with ovarian cancer or with high clinical suspicion of ovarian cancer who were scheduled to undergo surgery.
Of the 134 women (ages 33 to 81 years) who received a dose of Cytalux and were evaluated under both normal and fluorescent light during surgery, 26.9% had at least one cancerous lesion detected that was not observed by standard visual or tactile inspection.
The most common side effects of Cytalux were infusion-related reactions, including nausea, vomiting, abdominal pain, flushing, dyspepsia, chest discomfort, itching and hypersensitivity. Cytalux may cause fetal harm when administered to a pregnant woman.
The use of folate, folic acid, or folate-containing supplements should be avoided within 48 hours before administration of Cytalux. There is a risk of image interpretation errors with the use of Cytalux to detect ovarian cancer during surgery, including false negatives and false positives.
References
- ^ Jump up to:a b c d https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/214907s000lbl.pdf
- ^ Jump up to:a b c d e f g h i “FDA Approves New Imaging Drug to Help Identify Ovarian Cancer Lesions”. U.S. Food and Drug Administration (FDA) (Press release). 29 November 2021. Retrieved 30 November 2021. This article incorporates text from this source, which is in the public domain.
- ^ “On Target Laboratories Announces FDA Approval of Cytalux (pafolacianine) injection for Identification of Ovarian Cancer During Surgery”. On Target Laboratories. 29 November 2021. Retrieved 30 November 2021 – via PR Newswire.
- ^ “Pafolacianine Orphan Drug Designations and Approvals”. U.S. Food and Drug Administration (FDA). 23 December 2014. Retrieved 30 November 2021.
External links
- “Pafolacianine”. Drug Information Portal. U.S. National Library of Medicine.
| Clinical data | |
|---|---|
| Trade names | Cytalux |
| Other names | OTL-0038 |
| License data | US DailyMed: Pafolacianine |
| Pregnancy category | Not recommended |
| Routes of administration | Intravenous |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only [1][2] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 1628423-76-6 |
| PubChem CID | 135565623 |
| DrugBank | DB15413 |
| ChemSpider | 64880249 |
| UNII | F7BD3Z4X8L |
| ChEMBL | ChEMBL4297412 |
| Chemical and physical data | |
| Formula | C61H67N9O17S4 |
| Molar mass | 1326.49 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI |
////////////Pafolacianine, FDA 2021, APPROVALS 2021, Cytalux, OVARIAN CANCER, OTL 38,
[Na+].[Na+].[Na+].[Na+].CC1(C)\C(=C/C=C/2\CCCC(=C2Oc3ccc(C[C@H](NC(=O)c4ccc(NCc5cnc6N=C(N)NC(=O)c6n5)cc4)C(=O)O)cc3)\C=C\C7=[N](CCCCS(=O)(=O)O)c8ccc(cc8C7(C)C)S(=O)(=O)O)\N(CCCCS(=O)(=O)O)c9ccc(cc19)S(=O)(=O)O

NEW DRUG APPROVALS
ONE TIME
$10.00
AMOROLFINE
AMOROLFINE(2R,6S)-2,6-Dimethyl-4-{2-methyl-3-[4-(2-methyl-2-butanyl)phenyl]propyl}morpholine
(2R,6S)-2,6-Dimethyl-4-{2-methyl-3-[4-(2-methylbutan-2-yl)phenyl]propyl}morpholine
78613-35-1[RN]
(±)-cis-2,6-Dimethyl-4-(2-methyl-3-(p-tert-pentylphenyl)propyl)morpholine
Ro 14-4767-002
аморолфин , أمورولفين ,阿莫罗芬 ,
Title: Amorolfine
CAS Registry Number: 78613-35-1
CAS Name:cis-4-[3-[4-(1,1-Dimethylpropyl)phenyl]-2-methylpropyl]-2,6-dimethylmorpholine
Additional Names:cis-4-[3-(4-tert-amylphenyl)-2-methylpropyl]-2,6-dimethylmorpholine; (±)-cis-2,6-dimethyl-4-[2-methyl-3-(p-tert-pentylphenyl)propyl]morpholine
Manufacturers’ Codes: Ro-14-4767/000
Molecular Formula: C21H35NO
Molecular Weight: 317.51
Percent Composition: C 79.44%, H 11.11%, N 4.41%, O 5.04%
Literature References: Antimycotic morpholine derivative; inhibits fungal ergosterol biosynthesis. Prepn (unspec stereochem): A. Pfiffner, K. Bohnen, DE2752096; A. Pfiffner, US4202894 (1978, 1980 both to Hoffmann-La Roche); of cis-form: NL8004537 (1980 to Hoffmann-La Roche). In vitro comparative antifungal spectrum: S. Shadomy et al.,Sabouraudia22, 7 (1984). Mechanism of action: A. Polak-Wyss et al.,ibid.23, 433 (1985); A. Polak, Ann. N.Y. Acad. Sci.544, 221 (1988). LC determn in pharmaceutical formulations: M. A. Czech et al.,J. Pharm. Biomed. Anal.9, 1019 (1991). Series of articles on mode of action and clinical trials: Clin. Exp. Dermatol.17, Suppl. 1, 1-70 (1992). Review of pharmacology and clinical efficacy: M. Haria, H. M. Bryson, Drugs49, 103-120 (1995).
Properties: bp0.1 120°.
Boiling point: bp0.1 120°
Amorolfine hydrochloride (Ro 14-4767/002) is a antifungal reagent.
Derivative Type: Hydrochloride
CAS Registry Number: 78613-38-4
Manufacturers’ Codes: Ro-14-4767/002
Trademarks: Loceryl (Roche)
Molecular Formula: C21H35NO.HCl
Molecular Weight: 353.97
Percent Composition: C 71.26%, H 10.25%, N 3.96%, O 4.52%, Cl 10.02%
Therap-Cat: Antifungal (topical).
Amorolfine hydrochloride (Ro 14-4767/002) is a antifungal reagent. Target: Antifungal Amorolfine is an antifungal showing activity against fungi pathogenic to plants, animals and humans. Amorolfine possesses a broad antifungal spectrum including dermatophytes, yeasts, dimorphic fungi and moulds and is not only fungistatic but fungicidal against most species [1]. At 0.2, 2 and 5 micrograms/ml amorolfine did not have any significant inhibitory or enhancing effect on phagocytosis whether following simultaneous addition of blastospores and drug to the neutrophils, prior treatment of neutrophils for 2 h before addition of blastospores or prior treatment of blastospores for 2 h. Simultaneous addition of amorolfine resulted in a significant increase in killing at all concentrations. This increase was not significantly enhanced by either preincubation of neutrophils or blastospores for 2 h with the drug [2].
Amorolfine (or amorolfin), is a morpholineantifungal drug that inhibits Δ14-sterol reductase and cholestenol Δ-isomerase, which depletes ergosterol and causes ignosterol to accumulate in the fungal cytoplasmiccell membranes. Marketed as Curanail, Loceryl, Locetar, and Odenil, amorolfine is commonly available in the form of a nail lacquer, containing 5% amorolfine hydrochloride as the active ingredient. It is used to treat onychomycosis (fungal infection of the toe- and fingernails). Amorolfine 5% nail lacquer in once-weekly or twice-weekly applications has been shown in two studies to be between 60% and 71% effective in treating toenail onychomycosis; complete cure rates three months after stopping treatment (after six months of treatment) were 38% and 46%. However, full experimental details of these trials were not available and since they were first reported in 1992 there have been no subsequent trials.[1]
It is a topical solution for the treatment of toenail infections.[2][3] Systemic treatments may be considered more effective.[1]
It is approved for sale over-the-counter in Australia, Brazil, Russia, Germany and the UK, and is approved for the treatment of toenail fungus by prescription in other countries. It is not approved for the treatment of onychomycosis in the United States or Canada, but can be ordered from there by mail from other countries.[4]
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////////////////////////////
SYN
Indian Pat. Appl., 2010MU01980,







SYN
https://pubs.rsc.org/en/content/articlelanding/2017/ob/c6ob02765b/unauth
The acid-promoted crystallization-induced diastereoisomer transformation (CIDT) of naphthoxazines derived from racemic O-protected 2-substituted 4-hydroxybutyraldehydes and enantiopure Betti’s base allows the deracemization of the starting aldehydes with ee up to 96%. As an alternative, reduction with lithium aluminum hydride of the diastereoisomerically enriched naphthoxazines leads to enantioenriched primary amines. The utility of the latter strategy was demonstrated by applying it to the synthesis of enantioenriched fenpropimorph and to the first synthesis of enantiopure amorolfine, with ee up to 99.5%.
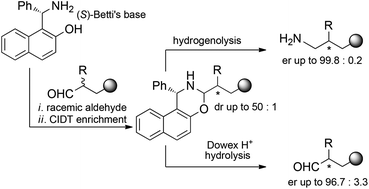
PATENT
https://patents.google.com/patent/WO2013097629A1/en
Amorolfine hydrochloride, chemical name is cis-4-[3-[4-(1,1-dimethyl-propyl)phenyl]-2-mercaptopropyl]-2 , 6-diamidino-morpholine hydrochloride, CAS registration number is 78613-38-4, the chemical knot is as follows:

Amoxifen hydrochloride is an antifungal drug developed by Roche and launched in 1991 under the trade name Leceryl. Regarding the synthesis process of amorolfine hydrochloride, the prior art has been described:


US7795425B2 synthetic route: (1) 2-nonyl cinnamaldehyde is condensed with cis-2,6-dimethylmorpholine to give cis-4-(3-phenyl-2-methylpropyl)-2,6- Dimercapto-morpholine hydrochloride, (2) cis-4-(3-phenyl-2-methylpropyl)-2,6-dimethyl-morpholine hydrochloride followed by 2-methyl – 2-chlorobutane, with acid Catalytic, Heck reaction occurs, and amorolfine is obtained. In step (1), palladium carbon catalytic hydrogenation is required, so the cost is high; in addition, there may be multiple rearrangement reactions in step (2), many by-products, difficult product purification, low quality of finished product and low yield. And it requires a low temperature reaction equipment of -40 ~ -65 °C, which consumes a lot of energy. International patent application WO2007113218A1 improves the synthesis method of amorolfine hydrochloride, the first step of Heck reaction, 4-iodo-t-amylbenzene and 2-methylallyl alcohol are reacted in the presence of a palladium catalyst and a base to obtain 3-un Butyl phenyl-2-methylpropanal, the reaction solvent is selected from N,N-dimercaptocarboxamide (abbreviated as DMF), polar protic solvent or non-polar solvent; second step reductive amination reaction, 3 – tert-amylphenyl-2-mercaptopropanal is reacted with cis-2,6-dimercaptomorpholine to give amorolfine, the reducing agent is selected from palladium

The WO2007113218A1 process still has defects: (1) The first step of the Heck reaction, the reaction solvent DMF is moderately toxic, and the International Agency for Research on Cancer (IARC) considers it to be a carcinogen. DMF is chemically stable and can exist for a long time in wastewater. It is highly polluted by water and difficult to biodegrade. Its BOD5/COD value is 0.065 ( BOD5/COD is an indicator of biodegradability of wastewater, and 0.3 is the lower limit of biodegradable degradation of wastewater). value). Wastewater treatment costs are high during large production. Although the boiling point of DMF is 154 ° C, it is unstable under alkaline conditions, especially at high temperatures, and decomposition starts at 100 ° C or higher. The polar protic solvent, such as the lower alcohol described in the patent, cannot meet the high temperature reaction requirements, and the high boiling polar protic solvent has poor solubility to the catalyst and is difficult to react. The non-polar solvent does not substantially dissolve the palladium catalyst, so the application value is not large. (2) The second step of reductive amination reaction, using expensive The cost of catalytic hydrogenation of heavy metal palladium is high, and the high pressure reaction equipment is unsafe; the reduction of metal borohydride is easy to generate a large amount of hydrogen, which poses a safety hazard, and also reduces 3-tert-pentylphenyl-2-methylpropanal to The corresponding alcohol increases the impurities; the reduction by-product of the metal cyanoborohydride is highly toxic. (3) The product yield was low, and the total yield of the product of the example was about 50%. None of the purity of the products and intermediates has been disclosed.The chemical reaction equation of the present invention is expressed as follows:

(la) (lb)In a 10L clean reaction kettle, add 2600 mL of acetic anhydride, 5200 mL of glacial acetic acid, 350 g of sodium periodate, break 1236 g, cool to 5 ° C, add 810 mL of sulfuric acid, control the dropwise addition within 1 hour, and then add 1130 g of t-amyl. The benzene was stirred at room temperature for more than 16 hours, and the reaction of the raw materials was confirmed by thin layer chromatography. The reaction mixture was poured into a mixture of 8 L of water and 4 L of dichloromethane, and the mixture was separated. The organic layer was washed with 4L of 25% aqueous sodium sulfite, and the organic layer was dried over anhydrous sodium sulfate. It was 4-iodo-t-amylbenzene 2013 g, yield: 96%, and the GC purity was 94.2%. NMR spectral data: (400 MHz, CDC1 3 ): 0.73 (3H, t, J = 7.4 Hz), 1.31 (6H, s), 1.67 (2H, q, J – 7.4 Hz), 7.13 (2H, d, J = 8.56 Hz), 7.66 (2H, d, J = 8.56 Hz) 0 Example 22 kg of 4-iodo-t-amylbenzene prepared according to the method of Example 1 and 6 L of N-methylpyrrolidone were added to a 10 L clean reaction vessel, and the mixture was stirred under nitrogen, stirring was carried out, and 300 g of palladium acetate and 1.7 kg of sodium hydrogencarbonate were added. Finally, 2.5 kg of 2-mercaptopropanol was added, the temperature was raised to 105 C, and the GC content of 4-iodo-t-amylbenzene was measured to monitor the progress of the reaction, and the reaction was completed for 2 hours. Cool to room temperature, filter, concentrate the filtrate, add the residue to 12 L of ethyl acetate, wash with 20 L of water, rectify the organic phase, collect 125-128 ° C fraction (vacuum degree ≤ -0.099)\3⁄4^), and obtain 3- Tert-amylphenyl-2-mercaptopropanal L41 kg, yield: 88.6%, GC purity: 93.5%. NMR spectral data: (400 MHz, CDC1 3 ): 0.69 (3H, t, J = 7.45 Hz), 1.11 (3H, d, J = 6.87 Hz), 1.29 (6H, s), 1.65 (2H, q, J =7.43 Hz), 2.60 (13⁄4 dd, J=13.52 Hz), 2.69 (1H, J=7.06 Hz), 3.08 (1H, dd, J = 13.54 Hz), 7.12 (2H, d, J = 8.27 Hz), 7.27 (2H, d, J = 8.27 Hz), 9,75 (1H, s).The above 3-tert-pentylphenyl-2-methylpropanal lkg, 5 L of ethyl acetate was added to a 10 L reactor, protected with nitrogen, cooled to 10 ° C, and 600 g of 2,6-dimethylmorpholine was added dropwise. , add about 30 minutes. Then, 300 mL of glacial acetic acid was added dropwise, the temperature was kept at 15 C, the addition was completed, and the temperature was raised to 18 ° C for 30 minutes. After cooling to 10 Torr, 1,3 kg of sodium triacetoxyborohydride was added. After the addition, the temperature was maintained at 18 ° C, and the GC content of 3-tert-amylphenyl-2-methylpropanal was detected to monitor the progress of the reaction. Ended in 2 hours. After cooling to 10 ° C or lower, the pH was adjusted to 10 with a sodium hydroxide solution, and the layers were allowed to stand, and the organic layer was washed with 4 L of water. The organic phase was added with concentrated hydrochloric acid, adjusted to pH 2, filtered, and the filter cake was dried under reduced pressure at 65 V for 14 hours to obtain 1.59 kg of amorolamine hydrochloride, yield: 85.6%, HPLC purity: 99.6%. R spectrum data: 3⁄4 NMR (400MHz, CD 3 OD) 5: 0.64 (3H, t, J=7, 2Hz), 1.03 (3H, d, J=6.8Hz), 1.15(6H, d, J=6 , 0 Hz), 1.25 (63⁄4 s), 1.64 (2H, m, J = 7.6 Hz), 2.34 (1H, d, J = 6.8 Hz), 2.48 (23⁄4 d, J = 6.8 Hz), 2.75 (2H, d , J=6.0Hz), 3.1(2H, d, J=8.8Hz) 5 3.4(2H, d, J=11.2Hz), 3,9(2H, m), 7.16(2H, dd, J=8.4Hz ), 7.27 (2H, dd, J = 8.4 Hz). Example 3 In a 10 L clean reaction kettle, 2 kg of 4-substituted tert-amylbenzene prepared according to the method of Example 1 and 6 L of N-mercaptopyrrolidone were protected by nitrogen, stirring was started, and 150 g of palladium acetate and 2.5 kg of dipotassium hydrogen phosphate were added. Finally, 1.8 kg of 2-methylallyl alcohol was added, and the temperature was raised to 130. C reaction, the GC content of 4-deuterated tert-amylbenzene was measured to control the progress of the reaction, and the reaction was completed for 10 hours. Cool to room temperature, filter, concentrate the filtrate, add the residue to 12 L of ethyl acetate, dissolve 20 L of water, concentrate the organic phase, recover ethyl acetate, and add the residue to 10 L of saturated sodium hydrogen sulfite solution at room temperature to precipitate solid. The mixture was stirred for 6 hours, filtered, and filtered, washed with EtOAc EtOAc EtOAc EtOAc. The filtrate was concentrated to dry ethyl acetate to give 1. <RTI ID=0.0>#</RTI><RTIgt;</RTI><RTIgt;</RTI><RTIgt; -NMR spectral data: (400 MHz, CDC1 3 ): 0.69 (3H, t, J-7.45 Hz), 1.11 (3H, d, J-6.87 Hz), 1.29 (6H, s), 1.65 (2H, q, J=7.43 Hz), 2.60 (1H, dd, J=13.52 Hz), 2.69 (1H, J=7.06 Hz), 3.08 (1H, dd, J = 13.54 Hz), 7.12 (2H, d, J = 8.27 Hz) ), 7.27 (2H, d, J = 8.27 Hz), 9.75 (1H, s).Add 1 kg of the above 3-tert-pentylphenyl-2-methylpropanal, 5 L of ethyl acetate in a 10 L reactor, protect with nitrogen, cool to 10 C, and add 1.2 kg of 2,6-dimethylmorpholine dropwise. , 40 minutes added. Then, 780 mL of glacial acetic acid was added dropwise, the temperature was kept at 15 ° C, the addition was completed, and the temperature was raised to 20 ° C for 60 minutes. After cooling to 10 ° C, 2.3 kg of sodium triacetoxyborohydride was added. After the addition, the temperature was checked at 25 ° C, and the GC content of 3-tert-amylpyridyl-2-methylpropanal was detected to monitor the progress of the reaction. The reaction was completed in 2 hours. Cool to below 10 ,, adjust the pH to 11 with sodium hydroxide solution, let stand for stratification, wash the organic layer with 4 L of water, add concentrated hydrochloric acid to the organic phase, adjust pH to 2, filter, filter cake at 70 ° C decompression After drying for 14 hours, 1.75 kg of amorolfine hydrochloride was obtained, yield: 84.6%, HPLC purity: 99.7%. R spectrum data: 3⁄4 NMR (400MHz, CD 3 OD) 5: 0.64 (3H, t, J = 7.2Hz), 1.03 (3H, d, J = 6.8Hz), L15(6H, d, J=6.0Hz ), 1.25(6H, s), L64(2H 5 m, J=7.6Hz), 2.34(1H, d, J=6.8Hz), 2.48(2H, d, J=6.8Hz), 2.75(2H, d , J=6.0Hz), 3.1(2H, d, J=8.8Hz), 3.4(2H 5 d, J=11.2Hz), 3·9(2Η, m), 7.16(2H, dd, J=8.4Hz ), 7.27 (2H ; dd, J = 8.4 Hz). Example 4In a 10 L clean reaction kettle, 2 kg of 4-iodo-t-amylbenzene prepared according to the method of Example 1, 2 N of N-methylpyrrolidone, protected by nitrogen, stirring was started, and palladium nitrate 6 g, acetic acid was added. Sodium 627 g, and finally 592 g of 2-methylallyl alcohol was added thereto, and the temperature was raised to 140 ° C to carry out a reaction. The GC content of 4-deactivated t-amylbenzene was examined to monitor the progress of the reaction, and the reaction was terminated for 24 hours. Cool to room temperature, filter, concentrate the filtrate, add the residue to 8 L of ethyl acetate, dissolve in 16 L of water, rectify the organic phase, collect 125-128 C fraction (vacuum degree ≤ -0.0991 ^ & ) to give 3-tert-pentylphenyl 2-mercaptopropanal 1.37 kg, yield: 86%, GC purity: 93.0%. MR spectrum data: (400 MHz, CDC1 3 ): 0.69 (3H, t, J = 7.45 Hz), 1 , 11 (3H, d, J = 6.87 Hz), 1.29 (6H, s), 1.65 (2H, q , 3=1 A3 Hz), 2.60 (IH, dd, J=13.52 Hz), 2.69 (IH, J=7.06 Hz), 3.08 (IH, dd, J = 13.54 Hz), 7.12 (2H, d, J = 8.27 Hz), 7.27 (2H, d, J = 8.27 Hz), 9.75 (IH, s).The above 3-tert-pentylphenyl-2-mercaptopropanal lkg, 5 L of dichloromethane was added to a 10 L reactor, protected with nitrogen, cooled to 10 ° C, and 1.6 kg of 2,6-dimethyl was added dropwise. Morpholine, added in 45 minutes. Then, 300 mL of glacial acetic acid was added dropwise, the temperature was kept at 15 ° C, the addition was completed, and the temperature was raised to 23 Torr for 60 minutes. After cooling to 10 ° C, 1.6 kg of sodium triacetoxyborohydride was added. After the addition, the temperature was checked at 23 ° C, and the GC content of 3-tert-pentylphenyl-2-methylpropanal was detected to monitor the progress of the reaction. The end of the hour. Cool to below 10 °C, adjust the pH to 10 with sodium hydroxide solution, let stand for layering, wash the organic layer with 4L water, add concentrated hydrochloric acid to the organic phase, adjust the pH to 1, filter, filter cake at 70 °C After drying under reduced pressure for 14 hours, 1.59 kg of amorolamine hydrochloride was obtained, yield: 83.6%, HPLC purity: 99.6%. iH-NMR spectral data: ! H NM (400 MHz, CD 3 OD) 5: 0.64 (3H, t, J = 7.2 Hz), 1.03 (3H, d, J = 6.8 Hz), 1.15 (6H, d, J= 6.0Hz), 1.25(6H, s), 1.64(2H, m, J=7.6Hz), 2.34(1H, d, J=6.8Hz), 2.48(2H, d, J=6.8Hz), 2.75(2H , d, J=6.0Hz), 3.1(2H, d, J=8.8Hz), 3.4(2H, d, J=11.2Hz), 3.9(2H, m), 7.16(2H, dd, J=8.4Hz ), 7.27 (2H, dd, J = 8.4 Hz). Example 52 kg of 4-iodo-t-amylbenzene prepared according to the method of Example 1 and 4 L of N-methylpyrrolidone were added to a 10 L clean reaction vessel, and the mixture was stirred under nitrogen, stirring was carried out, 30 g of palladium chloride and 750 g of sodium hydrogencarbonate were added. Finally, 1.3 kg of 2-methylallyl alcohol was added, and the mixture was heated to 120 ° C to measure the GC content of 4-iodo-t-amylbenzene to control the progress of the reaction, and the reaction was completed for 13 hours. It was cooled to room temperature, filtered, and the filtrate was concentrated. The residue was dissolved in 8 L of chloroform, washed with 16 L of water, and the organic phase was concentrated. The ethyl acetate was recovered. The residue was added dropwise to 10 L of saturated sodium hydrogensulfite solution at room temperature to precipitate a solid. Hour, filter, filter cake washed with 5 L of ethyl acetate, solid dispersed in 3 L 3 mol / liter The mixture was stirred at room temperature for 5 hours, and the reaction mixture was dried over EtOAcjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjj Yield: 91,7%, GC purity: 98.8%. – Spectrum data: (400 MHz, CDC1 3 ): 0.69 (3H, t, J=7.45 Hz), 1.11 (3H, d, J-6.87 Hz), 1.29 (6H, s), 1.65 (2H, q, J=7.43 Hz), 2.60 (IH, dd, J=13.52 Hz), 2.69 (IH, J=7.06 Hz), 3.08 (IH, dd, J = 13.54 Hz), 7.12 (2H, d, J = 8.27 Hz) ), 7.27 (2H, d, J = 8.27 Hz), 9.75 (IH, s).Add 1 kg of the above 3-tert-pentylphenyl-2-methylpropanal, 5 L of absolute ethanol in a 10 L reactor, protect with nitrogen, cool to 10 ° C, and add 600 g of 2,6-dimercaptomorpholine. , added in 30 minutes. Then, 500 mL of glacial acetic acid was added dropwise, the temperature was kept at 15 ° C, the addition was completed, and the temperature was raised to 23 ° C for 60 minutes. After cooling to 10 ° C, 1.2 kg of sodium triacetoxyborohydride was added. After the addition, the temperature was monitored at 10 Torr, and the GC content of 3-tert-pentylphenyl-2-nonylpropionaldehyde was detected to monitor the progress of the reaction. The end of the hour. 10. Under C, adjust the pH value to 11 with sodium hydroxide solution, add 3 L of dichloromethane, let stand for layering, wash the organic layer with 4 L of water, add concentrated hydrochloric acid to the organic phase, adjust pH to 2, filter, filter cake at 7 CTC minus After drying for 14 hours, 1.45% of amorolfine hydrochloride was obtained, yield: 87.0%, HPLC purity: 99.7% – NMR spectral data: J H NMR (400 MHz 5 CD 3 OD) 6: 0.64 (3H, t, J= 7,2Hz), 1.03(3H, d, J=6.8Hz), 1.15(6H, d, J=6.0Hz), 1.25(6H, s), 1.64(2H, m, J=7.6Hz), 2.34( 1H ? d, J = 6.8 Hz), 2.48 (2H, d, J = 6.8 Hz), 2.75 (23⁄4 d, J = 6.0 Hz), 3.1 (2H, d, J = 8.8 Hz), 3.4 (2H, d , J = 11.2 Hz) 5 3.9 (2H, m), 7.16 (2H, dd, J = 8.4 Hz), 7.27 (2H, dd, J = 8.4 Hz). Example 62 kg of 4-iodo-t-amylbenzene prepared in accordance with the method of Example 1 and 4 L of N-methylpyrrolidone were added to a 10 L clean reaction vessel. The mixture was stirred under nitrogen, stirring was started, 10 g of palladium acetate was added, and 800 g of carbonic acid was added. 1.1 kg of 2-mercaptopropanol was heated to 80 ° C, and the GC content of 4-deactivated t-amylbenzene was measured to control the progress of the reaction, and the reaction was terminated for 24 hours. Cool to room temperature, filter, concentrate the filtrate, add 8 L of chloroform to dissolve, 16 L of water, rectify the organic phase, collect 125-128 ° C 真空 (vacuum degree ≤ -0.099 ^ ^ & ), to obtain 3-tert-amylbenzene Base-2-mercaptopropanal 1.42 kg, yield: 89.2%, GC purity: 92.5%. ^- MR Spectral Data: (400 MHz, CDC1 3 ): 0.69 (33⁄4 t, J=7.45 Hz), 1.11 (3H, d, J=6.87 Hz), 1.29 (6H, s), 1.65 (2H, q, J=7.43 Hz), 2.60 (IH, dd, J=13.52 Hz), 2.69 (IH, J=7.06 Hz), 3.08 (IH, dd, J = 13.54 Hz), 7.12 (2H, d, J = 8.27 Hz), 7.27 (2H, d, J = 8.27 Hz), 9.75 (1H, s).The above 3-tert-pentylphenyl-2-methylpropanal lkg, 5 L of decyl alcohol was added to a 10 L reactor, protected with nitrogen, cooled to 10 C, and 600 g of 2,6-dimethylmorpholine was added dropwise for 30 minutes. Plus finished. Then, 500 mL of water acetic acid was added dropwise, the temperature was kept at 10 ° C, the addition was completed, and the temperature was raised to 20 ° C for 60 minutes. After cooling to 10 C, 1.2 kg of sodium triacetoxyborohydride was added. After the addition, the temperature was maintained at 23 ° C, and the GC content of 3-tert-pentylphenyl-2-methylpropanal was detected to monitor the progress of the reaction. End of 2 hours. Cool to 10 ° C, adjust the pH to 10 with sodium hydroxide solution, add 3 L of dichloromethane, let stand for layering, wash the organic layer with 4 L of water, add concentrated hydrochloric acid to the organic phase, adjust pH to 1.5, filter, filter The cake was dried under reduced pressure at 65 C for 15 hours to obtain 1.46 kg of amorolfine hydrochloride, yield: 90.1%, HPLC purity: 99,8%. ^-NMR spectral data: l R NMR (400 MHz, CD 3 OD) 5: 0.64 (3H, t, J = 7.2 Hz), 1.03 (3H, d, J = 6.8 Hz), U5 (6H, d, J = 6.0Hz), 1.25(6H, s), 1.64(23⁄4 m, J=7.6Hz), 2.34(1H, d, J=6.8Hz), 2.48(2H, d, J=6.8Hz), 2.75(2H, d, J=6.0Hz), 3.1(2H, d, J=8.8Hz), 3.4(2H, d, J=l 1.2Hz), 3.9(2H, m), 7.16(2H, dd, J=8.4Hz ), 7.27 (2H, dd, J = 8.4 Hz). Example 72 kg of 4-iodo-t-amylbenzene prepared according to the method of Example 1 and 6 L of N-decylpyrrolidone were added to a 10 L clean reaction kettle, protected by nitrogen, stirring was started, and 75 g of palladium acetate and 2.0 kg of disodium hydrogen phosphate were added. Finally, 780 g of 2-methylallyl alcohol was added, and the temperature was raised to 125 Torr. The GC content of 4-iodo-t-amylbenzene was measured to control the progress of the reaction, and the reaction was terminated for 8 hours. The mixture was cooled to room temperature, filtered, and the filtrate was concentrated. The residue was evaporated, evaporated, evaporated, evaporated, evaporated. The solid was precipitated, stirred for 6 hours, filtered, and the filter cake was washed with 5 L of ethyl acetate. The solid was dispersed in 10 L 2 mol/L hydrochloric acid, stirred at room temperature for 5 hours, and the reaction mixture was extracted with 10 L of ethyl acetate. The mixture was dried, filtered, and the filtrate was evaporated to ethyl acetate to ethylamine (ethyldiethyldithioacetate). 3⁄4-NMR spectral data: (400 MHz, CDC1 3 ): 0.69 (3H, t, J = 7.45 Hz), 1.11 (3H, d, J = 6.87 Hz), 1.29 (6H, s), 1.65 (2H, q , J=7.43 Hz), 2.60 (1H, dd, J=13.52 Hz), 2.69 (1H, J=7.06 Hz), 3.08 (1H, dd, J = 13.54 Hz), 7.12 (2H, d, J = 8.27 Hz), 7.27 (2H, d, J = 8.27 Hz), 9.75 (1H, s).Add the above 3-tert-pentylphenyl-2-mercaptopropanal lkg, 5L hydrazine, in a 10L reactor Under nitrogen atmosphere, cooled to 10 Torr, 700 g of 2,6-dimercaptomorpholine was added dropwise, then 280 mL of glacial acetic acid was added, the temperature was maintained at 15 C, and then the temperature was raised to 23 ° C for 60 minutes. After cooling to 10 ° C, 1.0 kg of sodium triacetoxyborohydride was added, and 20 was added. The temperature was maintained under C, and the GC content of 3-tert-amylphenyl-2-methylpropanal was examined to monitor the progress of the reaction, and the reaction was completed for 3 hours. Cool to below 10 ° C, adjust the pH to 11 with sodium hydroxide solution, let stand for layering, wash the organic layer with 4 L of water, add concentrated hydrochloric acid to the organic phase, adjust the pH to 1, filter, filter cake at 70 ° C After drying under reduced pressure for 14 hours, 1.59 kg of amorolamine hydrochloride was obtained, yield: 83.8%, HPLC purity: 99.6%. ^-NMR spectral data: 3⁄4 NMR (400MHz, CD 3 OD) 5: 0.64 (3H, t, J- 7.2 Hz), 1.03 (3H, d, J = 6.8 Hz), 1.15 (6H ; d, J = 6.0 Hz), 1.25(6H, s), 1.64(2H, m, J=7.6Hz), 2.34(1H, d, J=6.8Hz), 2.48(2H, d, J=6.8Hz) } 2.75(2H, d, J=6.0Hz), 3.1(2H, d, J=8.8Hz), 3.4(2H, d, J=11.2Hz), 3.9(2H, m), 7.16(2H, dd, J=8.4Hz) , 7.27 (2H, dd, J = 8.4 Hz). Example 83-tert-pentylphenyl-2-mercaptopropanol lkg, 5 L of dichloromethane prepared by the method of Example 5 was added to a 10 L reactor, and was purged with nitrogen and cooled to 10. C, 1000 g of 2,6-dimethylmorpholine was added dropwise, then 400 mL of water acetic acid was added, the temperature was maintained at 15 ° C, and then the temperature was raised to 20 ° C for 60 minutes. After cooling to 0 C, 1.5 kg of sodium triacetoxyborohydride was added, and 6 C was added after the addition, and the GC content of 3-tert-pentylphenyl-2-mercaptopropanal was detected to monitor the progress of the reaction for 5 hours. End. Adjust the pH to 10 with sodium hydroxide solution at 6 °C, let stand for layering, wash the organic layer with 4L of water, add concentrated hydrochloric acid to the organic phase, adjust the pH to 2, filter, filter cake and dry at 65 Ό for 14 hours under reduced pressure. , Amofufen hydrochloride 1.48kg, yield: 91.2%, HPLC purity: 99.7%. ^- MR spectral data: NMR (400MHz, CD 3 OD) 5: 0·64 (3Η, ΐ, J=7, 2Hz), 1.03(3Η, d, J=6.8Hz), 1.15(6H, d, J =6.0Hz), 1.25(6H, s), 1.64(2H, m, J=7.6Hz), 2.34(1H, d, J=6.8Hz), 2.48(2H, d, J=6.8Hz), 2.75( 2H, d, J=6.0Hz), 3,1(2H, d, J=8.8Hz), 3.4(2H, d, J=11.2Hz), 3.9(23⁄4 m), 7.16(2H, dd, J- 8.4 Hz), 7.27 (2H, dd, J = 8.4 Hz). Example 9Add 3-tert-pentylphenyl-2-mercaptopropanol lkg prepared in the same manner as in Example 2, 4 L of tetrahydrofuran, protect with nitrogen, cool to 10 ° C, add 820 g of 2,6-two Mercaptomorpholine, Then, 380 mL of glacial acetic acid was added, the temperature was maintained at 15 ° C, and then kept at room temperature for 60 minutes. After cooling to 10 ° C, 1.8 kg of sodium triacetoxyborohydride was added, and after 10 liters of the addition, the GC content of 3-tert-amylphenyl-2-nonylpropionaldehyde was detected to monitor the progress of the reaction for 5 hours. End. The pH was adjusted to 10 with sodium hydroxide solution at 10 ° C, and the layers were allowed to stand. The organic layer was washed with 4 L of water, and the organic phase was added with concentrated hydrochloric acid, adjusted to pH 2, filtered, and the filter cake was dried under reduced pressure at 65 Torr for 14 hours. , amlofol hydrochloride 1.41 kg, yield: 87.1%, HPLC purity: 99.8%. NMR spectral data: J H NMR (400 MHz, CD 3 OD) 5: 0.64 (3H, t, J- 7.2 Hz), L03 (3H, d, J = 6.8 Hz), 1.15 (6H, d, J = 6.0 Hz) ), 1.25(6H, s), 1.64(2H, m, J=7.6Hz), 2.34(1H, d, J-6.8Hz), 2.48(2H, d, J=6.8Hz), 2.75(2H, d , J=6.0Hz), 3.1(2H, d, J=8.8Hz), 3.4(2H, d, J=11.2Hz), 3.9(2H, m), 7.16(2H, dd, J=8.4Hz), 7.27 (2H, dd, J = 8.4 Hz). Comparative example 1In a 1000 mL four-necked flask, 137 g of 4-deuterated tert-amylbenzene prepared according to the method of Example 1, 1.12 g of palladium acetate, 50.4 g of sodium hydrogencarbonate, N,N-dimethylformamide 500 mL, nitrogen gas, added 54 g of 2-mercaptopropanol, warmed to 10 (TC for 10 hours, cooled to room temperature, filtered, filter cake washed with hydrazine, hydrazine-dimethylformamide 300 mL, combined filtrate, poured into 2000 mL of saturated brine and 1000 mL The mixture was extracted with ethyl acetate, and the organic phase was washed with water, dried over anhydrous magnesium sulfate, filtered, and concentrated, dried, and evaporated, and the residue was distilled in vacuo to collect fractions of 125-128 ° C (vacuum degree <-0.099 MPa) to obtain 3-un Amyl phenyl-2-mercaptopropanal 84 g, Yield: 77%, GC purity: 88.0% – R spectrum data: (400 MHz, CDC1 3 ): 0.69 (3H, t, J = 7.45 Hz) , 1.11 (3H : d, J=6.87 Hz), 1.29 (6H, s), 1.65 (2H, q, J=7.43 Hz), 2,60 (1H, dd, J=13.52 Hz), 2.69 (1H, J-7.06 Hz), 3.08 (1H, dd, J = 13.54 Hz), 7.12 (2H, d, J = 8.27 Hz), 7.27 (2H, d, J = 8.27 Hz), 9.75 (1H, s).109 g of the above 3-tert-amylphenyl-2-mercaptopropanal and 500 mL of ethanol were placed in a 1000 mL four-necked flask, cooled to 0 ° C, and 30 mL of glacial acetic acid and 69 g of 2,6-dimethylmorpholine were added. Stir at room temperature for 30 minutes, cool to -15 ° C, add 15.93 g of sodium borohydride in 1 hour. After the addition, warm to 0 C for 2 hours, adjust the pH to 12 with 25% sodium hydroxide solution. The mixture was extracted with 2000 mL of saturated brine and 1000 mL of ethyl acetate. The organic phase was washed with water and concentrated to dryness. The obtained residue was added to 500 mL of isopropyl ether, hydrogen chloride gas to pH 2, stirred at room temperature for 2 hours, filtered, and washed with isopropyl ether. , the filter cake is dried under reduced pressure at 70 ° C for 14 hours to obtain hydrochloric acid. Morofen 119 g, yield: 67%, HPLC purity: 97.1%. 3⁄4-NMR spectral data: ‘H NMR (400 MHz, CD 3 OD) 5: 0, 64 (3H, t, J = 7, 2 Hz), 1.03 (3H, d, J = 6.8 Hz), 1.15 (6H, d , J=6.0Hz), 1.25(6H, s), 1.64(2H, m, J=7.6Hz), 2.34(1H, d, J=6.8Hz), 2.48(2H, d, J=6.8Hz), 2.75(2H, d, J=6.0Hz), 3.1(2H, d, J=8.8Hz), 3.4(2H, d, J=11.2Hz), 3,9(2H, m), 7.16(2H, dd , J = 8.4 Hz), 7.27 (2H, dd, J = 8.4 Hz). Comparative example 2109 g of 3-tert-pentylphenyl-2-methylpropanal prepared according to the method of Comparative Example 1 and 500 mL of methanol were added to a 1000 mL four-necked flask, cooled to 0 ° C, and 30 mL of glacial acetic acid and 69 g of 2, 6 were added. – dimethylmorpholine, stirred at room temperature for 30 minutes, cooled to -15 ° C, replaced with nitrogen, added 5 g of 0% palladium on carbon, passed through hydrogen, reduced at 40 ° C, 4 atm, until the hydrogen pressure did not decrease, The reaction is complete. Cool to room temperature, replace with nitrogen, filter, adjust the pH of the filtrate with 25% sodium hydroxide solution, add 2000 mL of saturated brine and 1000 mL of ethyl acetate for extraction, wash the organic phase, concentrate and dry, add the residue to 500 mL Isopropyl ether, hydrogen chloride gas to pH 2, stirred at room temperature for 2 hours, filtered, washed with isopropyl ether, and the filter cake was dried under reduced pressure at 70 ° C for 14 hours to obtain amolofol hydrochloride 113 g, yield: 64%. HPLC purity: 97.8%. NMR spectral data: 3⁄4 NMR (400MHz, CD 3 OD) 5: 0.64 (3H, t, J = 7.2 Hz), 1.03 (3H, d, J = 6.8 Hz), 1.15 (6H, d, J = 6.0 Hz) , 1.25(6H, s), 1.64(2H, m, J-7.6Hz), 2.34(1H, d, J=6.8Hz), 2.48(2H, d, J=6.8Hz) ? 2.75(2H, d, J=6.0Hz), 3.1(2H, d, J=8.8Hz), 3.4(2H, d, J-11.2Hz), 3.9(2H, m), 7.16(2H, dd, J=8.4Hz), 7.27 (2H, dd, J=8, 4Hz).
Patent
Publication numberPriority datePublication dateAssigneeTitleEP0447947A1 *1990-03-231991-09-25BASF AktiengesellschaftN-(3-Phenyl-2-methylpropyl and -methyl-prop-2-enyl)-azaheterocyclesWO2007113218A1 *2006-04-032007-10-11Galderma S.A.Process for producing 3-[4-(1,1-dimethyl-propyl)-phenyl]-2-methyl-propionaldehyde and cis-4-{3-[4-(1,1-dimethyl-propyl)-phenyl]-2-methyl-propyl}-2,6-dimethyl-morpholine (amorolfine)Family To Family CitationsEP1749826A1 *2005-07-282007-02-07Galderma S.A.Process of producing bepromolineCN101485625B *2009-02-192010-09-22中国药科大学Amoluofen emulsifiable paste
Publication numberPriority datePublication dateAssigneeTitle
CN105130808A *2015-08-132015-12-09上海瑞博化学有限公司High purity 2,5-dimethyl-3,4-dihydroxy methylbenzoate synthesis methodFamily To Family CitationsCN103288768B *2013-06-182015-02-18中国人民解放军第四军医大学Asymmetric synthetic method of optical pure amorolfine hydrochlorideCN104926629B *2015-05-302016-06-22江苏科本医药化学有限公司Domino reaction is utilized to prepare the green method of 3,3-diaryl acrylic aldehydeCN108997246B *2017-06-062021-08-31江苏礼华生物技术有限公司Preparation method of amorolfine hydrochlorideCN110498729A *2019-09-092019-11-26武汉诺安药业有限公司A kind of clean method for preparing of hydrochloric acid Amorolfine intermediate
Notes
- ^ Jump up to:a b Williams HC (2003). Evidence-Based Dermatology. Blackwell. ISBN 9781444300178.
- ^ Flagothier C, Piérard-Franchimont C, Piérard GE (March 2005). “New insights into the effect of amorolfine nail lacquer”. Mycoses. 48 (2): 91–4. doi:10.1111/j.1439-0507.2004.01090.x. PMID 15743424.
- ^ Feng X, Xiong X, Ran Y (May 2017). “Efficacy and tolerability of amorolfine 5% nail lacquer in combination with systemic antifungal agents for onychomycosis: A meta-analysis and systematic review”. Dermatologic Therapy. 30 (3): e12457. doi:10.1111/dth.12457. PMID 28097731.
- ^ It can readily be verified that Curanail is advertised on websites such as US Amazon.com, shipped from abroad.
| Clinical data | |
|---|---|
| AHFS/Drugs.com | International Drug Names |
| ATC code | D01AE16 (WHO) |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 78613-35-1 |
| PubChem CID | 54260 |
| ChemSpider | 49010 |
| UNII | AB0BHP2FH0 |
| KEGG | D02923 |
| ChEBI | CHEBI:599440 |
| ChEMBL | ChEMBL489411 |
| CompTox Dashboard (EPA) | DTXSID0046690 |
| Chemical and physical data | |
| Formula | C21H35NO |
| Molar mass | 317.517 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
| (verify) |
/////////////AMOROLFINE, Ro 14-4767-002, аморолфин ,أمورولفين ,阿莫罗芬 , antifungal

NEW DRUG APPROVALS
one time
$10.00
Acetaminosalol

Acetaminosalol
- Molecular FormulaC15H13NO4
- Average mass271.268 Da
- ацетаминосалол [Russian] [INN], أسيتامينوسالول [Arabic] [INN], 醋氨沙洛 [Chinese] [INN]
(1E)-N-{4-[(2-Hydroxybenzoyl)oxy]phenyl}ethanimidic acid118-57-0[RN]
204-261-3[EINECS]
CAS Registry Number: 118-57-0
CAS Name: 2-Hydroxybenzoic acid 4-(acetylamino)phenyl ester
Additional Names:p-acetamidophenyl salicylate; acetylaminophenyl salicylate; acetyl-p-aminosalol; p-acetylaminophenol salicylic acid ester; phenetsal
Trademarks: Salophen (Bayer); Phenosal
Molecular Formula: C15H13NO4
Molecular Weight: 271.27
Percent Composition: C 66.41%, H 4.83%, N 5.16%, O 23.59%
Literature References: Prepn: Brewster, J. Am. Chem. Soc.40, 1136 (1918).
Properties: Crystals from hot ethanol, mp 187°. Practically insol in petr ether, cold water, more sol in warm water. Sol in alcohol, ether, benzene. Incompatible with alkalies and alkaline solns which dissolve it with decompn. The alkaline soln gradually becomes blue when boiled, the blue color being discharged upon continued boiling and again produced upon cooling and exposure to air.
Melting point: mp 187°
Therap-Cat: Analgesic; antipyretic; anti-inflammatory.
Therap-Cat-Vet: Analgesic; antipyretic.
Keywords: Analgesic (Non-Narcotic); Anti-inflammatory (Nonsteroidal); Salicylic Acid Derivatives; Antipyretic.
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////////////////////////////
Acetaminosalol is an organic compound with the chemical formula C15H13NO4.
It is an esterification product of salicylic acid and paracetamol. It was marketed by Bayer under the brand name Salophen as an analgesic in the late 19th and early 20th centuries.
Action and uses
In a warm alkaline solution acetaminosalol is broken up into salicylic acid and paracetamol. It is decomposed in the intestines, even when given as an injection. It was used as a substitute for salicylic acid in acute rheumatism, and as an intestinal antiseptic. It was similarly effective and much safer than salol, another intestinal antiseptic commonly used at the time. The fact that it is tasteless renders it easy to administer.Advertisement for early 20th century Bayer products, including Salophen
SYNJournal of Organic Chemistry, 86(5), 4254-4261; 2021

| Names | |
|---|---|
| Preferred IUPAC name4-Acetamidophenyl 2-hydroxybenzoate | |
| Identifiers | |
| CAS Number | 118-57-0 |
| 3D model (JSmol) | Interactive imageInteractive image |
| ChEBI | CHEBI:250620 |
| ChEMBL | ChEMBL92590 |
| ChemSpider | 1907 |
| ECHA InfoCard | 100.003.875 |
| EC Number | 204-261-3 |
| MeSH | Salophen |
| PubChem CID | 1984 |
| UNII | O3J7H54KMD |
| CompTox Dashboard (EPA) | DTXSID7045865 |
| showInChI | |
| showSMILES | |
| Properties | |
| Chemical formula | C15H13NO4 |
| Molar mass | 271.272 g·mol−1 |
| Density | 1.327 g cm−3 |
| log P | 2.562 |
| Acidity (pKa) | 7.874 |
| Basicity (pKb) | 6.123 |
| Hazards | |
| Flash point | 241.9 °C (467.4 °F; 515.0 K) |
| Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa). | |
| Infobox references |
///////////////Acetaminosalol, nalgesic , Anti-inflammatory, Salicylic Acid Derivatives, Antipyretic, ацетаминосалол , أسيتامينوسالول , 醋氨沙洛 ,

NEW DRUG APPROVALS
ONE TIME
$10.00
LINZAGOLIX
LINZAGOLIX
CAS 935283-04-8
C22H15F3N2O7S
- Hormone Antagonists
3-[5-[(2,3-difluoro-6-methoxyphenyl)methoxy]-2-fluoro-4-methoxyphenyl]-2,4-dioxo-1H-thieno[3,4-d]pyrimidine-5-carboxylic acid
- WHO 10711
- Treatment of Endometriosis Pain and Uterine Myoma-Associated Heavy Menstrual Bleeding
- OriginatorKissei Pharmaceutical
- DeveloperKissei Pharmaceutical; ObsEva
- Class2 ring heterocyclic compounds; Antihormones; Antineoplastics; Carboxylic acids; Fluorinated hydrocarbons; Ketones; Pyrimidines; Small molecules; Thiophenes
- Mechanism of ActionLHRH receptor antagonists
- PreregistrationUterine leiomyoma
- Phase IIIEndometriosis
- Phase IIAdenomyosis
- 22 Nov 2021FDA assigns PDUFA action date of (13/09/2022) for linzagolix for Uterine leiomyoma
- 22 Nov 2021The US FDA accepts NDA for linzagolix for Uterine leiomyoma for review
- 20 Oct 2021Efficacy and adverse events data from a phase II trial in Adenomyosis presented at the American Society for Reproductive Medicine (ASRM) 2021 Scientific Congress & Expo


Linzagolix choline
CAS#: 1321816-57-2 (choline)
Chemical Formula: C27H28F3N3O8S
Exact Mass: 611.1549
Molecular Weight: 611.58
Linzagolix is an orally bioavailable gonadotropin-releasing hormone (GnRH or LHRH) receptor antagonist, with potential hormone production inhibitory activity. Upon oral administration of linzagolix, this agent competes with GnRH for receptor binding and inhibits GnRH receptor signaling in the anterior pituitary gland, thereby inhibiting the secretion and release of luteinizing hormone (LH) and follicle stimulating hormone (FSH). In males, the inhibition of LH secretion prevents the release of testosterone. As a result, this may relieve symptoms associated with hormonally dependent disease states such as hormone-dependent prostate cancer. In women, this prevents the production of estrogen by the ovaries and may relieve symptoms from sex-hormone dependent diseases, such as pain associated with endometriosis, heavy menstrual bleeding or uterine fibroids.
Linzagolix (INN; developmental code names KLH-2109, OBE-2109; tentative brand name Yselty) is a small-molecule, non-peptide, orally active gonadotropin-releasing hormone antagonist (GnRH antagonist) which is under development by Kissei Pharmaceutical and ObsEva for the treatment of uterine fibroids, endometriosis, and adenomyosis.[1][3][2] As of December 2020, it is under review for approval for uterine fibroids, is in phase III clinical trials for endometriosis, and is in phase II clinical studies for adenomyosis.[1]
Estrogen-dependent disorders represent a challenging class of diseases that have a high incidence in the general population and are often associated with particularly severe symptomology. Uterine fibroids, for example, also referred to as leiomyomata, are among the most common benign tumors in women. Symptoms associated with uterine fibroids commonly include heavy or prolonged menstrual bleeding, pelvic pressure and pelvic organ compression, back pain, and adverse reproductive outcomes. Heavy menstrual bleeding may lead to iron deficiency anemia, a key symptom of uterine fibroids and the leading cause of surgical interventions that may include hysterectomy. Endometriosis is another estrogen-dependent gynecological condition, characterized by the presence of endometrial-like tissue outside the uterus.
Additional examples of estrogen-dependent diseases include adenomyosis and rectovaginal endometriosis, which are particularly severe endometrial growth disorders characterized by the invasion of endometrial tissue into the uterine myometrium and rectovaginal zones, respectively. The term adenomyosis or uterine adenomyosis is used to describe the presence of both endometrial glands and stroma deep within the myometrium. This condition is associated with hypertrophy and hyperplasia of the subjacent muscle cells, which may ultimately result in an altered size and globulous morphology of the uterus. Due to the severity of this disorder, one of the key symptoms is strong menstrual and even non-menstrual pelvic pain with abnormal uterine bleeding. Like adenomyosis, rectovaginal endometriosis patients present with a variety of pain symptoms including dysmenorrhea, dyspareunia, chronic pelvic pain, dysuria, and dyschezia. Treatment options for rectovaginal endometriosis are limited. Since medical therapies are either ineffective or have considerable side effects, rectovaginal endometriosis patients often undergo surgical procedures to reduce the endometrial node, and may even be subject to resection of the bowel if the node infiltrates the rectal or sigmoidal wall.
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////////////////////////////
Obseva Announces U.S. FDA Acceptance of New Drug Application for Linzagolix
November 22, 2021 01:05 ET | Source: ObsEva SA………. https://www.globenewswire.com/news-release/2021/11/22/2338610/0/en/Obseva-Announces-U-S-FDA-Acceptance-of-New-Drug-Application-for-Linzagolix.html
FDA Accepts NDA for Linzagolix for the Management of Heavy Menstrual Bleeding Associated with Uterine Fibroids
GENEVA, Switzerland November 22, 2021 – Obseva SA (NASDAQ: OBSV; SIX: OBSN), a biopharmaceutical company developing and commercializing novel therapies to improve women’s reproductive health, today announced that the New Drug Application (NDA) for linzagolix for the management of heavy menstrual bleeding associated with uterine fibroids in premenopausal women has been accepted for review by the United States Food and Drug Administration (FDA). The submission is based on data from the two Phase 3 PRIMROSE trials. Linzagolix has a differentiated profile and if approved, would be the first and only GnRH receptor antagonist with flexible dosing options for uterine fibroids, including a low dose option to address the needs of women who cannot or do not want to take hormones.1,4 The FDA set a target action date of September 13, 2022 for this NDA under the Prescription Drug User Fee Act (PDUFA).
“Today marks an important milestone not only in the linzagolix clinical development process, but for Obseva as a company, and most importantly, the millions of women living with uterine fibroids throughout the US. Linzagolix is a significant innovation in the field of women’s health – an area that is consistently underinvested in – and we are incredibly excited about the potential of bringing this important treatment to market” said Brian O’Callaghan, CEO of Obseva. “We are encouraged by our positive Phase 3 PRIMROSE results. If approved, we believe linzagolix will address a significant unmet need in offering a more individualized treatment option for a broader range of women.”
The Phase 3 PRIMROSE trials of linzagolix (PRIMROSE 1: US; n=574 and PRIMROSE 2: Europe and US; n=535) investigated the efficacy and safety of two dosing regimens, 100mg once daily and 200mg once daily, alone or in combination with hormonal ABT (1 mg estradiol and 0.5 mg norethisterone acetate) for the treatment of heavy menstrual bleeding associated with uterine fibroids. The NDA submission comprises positive 24-week treatment results from both studies, as well as supportive results from Week 52 and the 76-week post-treatment follow-up.
“Uterine fibroids can have a devastating impact on women’s day-to-day life. With its unique dosing options, linzagolix has the potential to significantly advance medical options for women,” stated Elizabeth Garner, MD, MPH, Chief Medical Officer of Obseva. “A dosing option without hormonal ABT would be welcomed by the significant number of women who either have contraindications to or a personal preference to avoid the use of estrogen-based therapies, while also providing a dosing option for women in whom hormonal ABT is indicated.”
The linzagolix marketing authorization application (MAA) was validated by the European Medicine Agency (EMA) with an approval recommendation from the Committee for Medicinal Products for Human Use (CHMP) expected in Q4 2021. Obseva announced previously that the company has entered into a partnership with Syneos Health to support commercialization of linzagolix in the US and EU.
About Linzagolix
Linzagolix is a novel, once daily, oral GnRH receptor antagonist with a potentially best-in-class profile1,2,3. Linzagolix is the subject of submitted marketing authorization applications for the treatment of heavy menstrual bleeding associated with uterine fibroids and is currently in late-stage clinical development for the treatment of pain associated with endometriosis. Obseva licensed linzagolix from Kissei in late 2015 and retains worldwide commercial rights, excluding Asia, for the product. Linzagolix is not currently approved anywhere in the world.
About the Phase 3 PRIMROSE Program in Uterine Fibroids
PRIMROSE 1 & 2 were prospective, randomized, parallel group, double-blind, placebo-controlled Phase 3 studies that investigated the efficacy and safety of two dosing regimens of linzagolix, 100 mg and 200 mg once daily, alone and in combination with hormonal ABT (1 mg estradiol and 0.5 mg norethisterone acetate) for the treatment of heavy menstrual bleeding associated with uterine fibroids. PRIMROSE 1 was conducted in the United States and enrolled 574 women. PRIMROSE 2 was conducted in Europe and the United States and enrolled 535 women. Both trials comprised a 52-week treatment period followed by a 6-month post treatment follow-up period. Additional information can be found here.
About Uterine Fibroids
Uterine fibroids are common benign tumors of the muscular tissue of the uterus which affect women of childbearing age and can vary in size from undetectable to large bulky masses. Few long-term medical treatments are available, and as a result, approximately 300,000 hysterectomies are performed for uterine fibroids every year in the US.
The symptoms of uterine fibroids are wide-ranging and include heavy menstrual bleeding, anemia, pelvic pressure and bloating, urinary frequency and pain that can be extremely debilitating with a significant impact on quality of life. These symptoms can also have an impact on mental health, creating the additional burden of anxiety and distress.
About Obseva
Obseva is a biopharmaceutical company built to address some of the most challenging unmet needs in women’s health – an under-researched, under-invested field of medicine. With deep expertise in clinical development, Obseva is passionate about the pursuit of advances that benefit women and their health and the importance of delivering truly meaningful innovation in this space. Through strategic in-licensing and disciplined drug development, Obseva has established a late-stage clinical pipeline with development programs focused on new therapies for the treatment of uterine fibroids, endometriosis, and preterm labor. Obseva is listed on the Nasdaq Global Select Market and is traded under the ticker symbol “OBSV” and on the SIX Swiss Exchange where it is traded under the ticker symbol “OBSN”. For more information, please visit http://www.ObsEva.com.
About Kissei
Kissei is a Japanese pharmaceutical company with approximately 70 years of history, specialized in the field of urology, kidney-dialysis and unmet medical needs. Silodosin is a Kissei product for the treatment of the signs and symptoms of benign prostatic hyperplasia which is sold worldwide through its licensees. KLH-2109/OBE2109 is a new chemical entity discovered by Kissei R&D.

……………………………

PATENT
WO 2007046392
https://patents.google.com/patent/WO2007046392A1/en
PATENT
WO 2014042176
https://patents.google.com/patent/WO2014042176A1/en

(Process 1)
Compound (D) can be produced by reacting compound (B) or a salt thereof with compound (C) in the presence of a base in a solvent. Examples of the solvent include halogen solvents such as dichloromethane, cyclic ethers such as tetrahydrofuran, 2-methyltetrahydrofuran, and tetrahydropyran, amide solvents such as N, N-dimethylformamide, aromatic hydrocarbon solvents such as toluene, A nitrile solvent such as acetonitrile, an ester solvent such as ethyl acetate, or a mixed solvent thereof and a mixed solvent thereof and water are preferable, and a mixed solvent of tetrahydrofuran and water is preferable. Examples of the base include organic bases such as triethylamine and pyridine, and inorganic bases such as sodium hydrogen carbonate, potassium hydrogen carbonate, cesium carbonate, sodium carbonate, and potassium carbonate, preferably triethylamine, sodium hydrogen carbonate, or potassium carbonate Is mentioned. The equivalent of the base may be an equivalent amount capable of neutralizing the salt and neutralizing the acid generated by the reaction. The equivalent of (C) can be used in an amount of 0.8 to 1.1 equivalents relative to (B), preferably 1.0 equivalent. The reaction temperature is usually 0 to 30 ° C., and the reaction time is usually 0.5 to 3 hours, although it varies depending on the raw material used, the solvent, the reaction temperature and the like. Examples of the salt of the compound (B) include a salt with an inorganic acid, a salt with an organic acid, a salt with an acidic amino acid, and the like. Examples of the salt with an inorganic acid include salts with hydrochloric acid, hydrobromic acid, nitric acid, sulfuric acid, phosphoric acid and the like. Examples of salts with organic acids include formic acid, acetic acid, trifluoroacetic acid, fumaric acid, oxalic acid, tartaric acid, maleic acid, citric acid, succinic acid, malic acid, methanesulfonic acid, benzenesulfonic acid, p-toluene And salts with sulfonic acid and the like. Examples of salts with acidic amino acids include salts with aspartic acid, glutamic acid and the like. Among these salts, salts with hydrochloric acid and methanesulfonic acid are preferable. Compound (C) used in Scheme 1 may be a commercially available product, or can be produced according to a known method or a method analogous thereto. Compound (D) may be isolated before the next step, but it can also be used in the next step without isolation.(Process 2)
Compound (F) can be produced by reacting compound (D) with compound (E) or a salt thereof in a solvent in the presence or absence of a base. Examples of the solvent include cyclic ethers such as tetrahydrofuran, 2-methyltetrahydrofuran, tetrahydropyran, amide solvents such as N, N-dimethylformamide, aromatic hydrocarbon solvents such as toluene, nitrile solvents such as acetonitrile, An ester solvent such as ethyl acetate or a mixed solvent thereof and a mixed solvent thereof with water, and the like are preferable, and a mixed solvent of tetrahydrofuran and water is preferable. Examples of the base include organic bases such as N, N-dimethylaminopyridine, triethylamine, N-methylpyrrolidine, N-methylmorpholine, diisopropylethylamine, and preferably N, N-dimethylaminopyridine, triethylamine and the like. . The equivalent of the base can be used in an amount of 0.1 to 2.0 equivalents relative to the compound (E), preferably 0.1 to 0.5 equivalents (provided that when a salt of the compound (E) is used, Further base necessary for neutralization is required). The reaction temperature is from room temperature to 60 ° C., and the reaction time is usually from 1 to 24 hours, although it varies depending on the raw material used, the solvent, the reaction temperature, and the like. Examples of the salt of compound (E) include a salt with an inorganic acid, a salt with an organic acid, a salt with an acidic amino acid, and the like. Examples of the salt with an inorganic acid include salts with hydrochloric acid, hydrobromic acid, nitric acid, sulfuric acid, phosphoric acid and the like. Examples of salts with organic acids include formic acid, acetic acid, trifluoroacetic acid, fumaric acid, oxalic acid, tartaric acid, maleic acid, citric acid, succinic acid, malic acid, methanesulfonic acid, benzenesulfonic acid, p-toluene And salts with sulfonic acid and the like. Examples of salts with acidic amino acids include salts with aspartic acid, glutamic acid and the like. Compound (F) may be isolated before the next step, but it can also be used in the next step without isolation.(Process 3)
The intramolecular cyclization and hydrolysis reaction in this step can be performed simultaneously or separately.
(Step 3-1)
Compound (A) can be produced by subjecting compound (F) to intramolecular cyclization and hydrolysis in the presence of a base in a solvent. Examples of the solvent include cyclic ethers such as tetrahydrofuran, 2-methyltetrahydrofuran and tetrahydropyran, lower alcohols such as methanol, ethanol and 2-propanol, amide solvents such as N, N-dimethylformamide, and nitriles such as acetonitrile. Examples thereof include a solvent and the like or a mixed solvent of a mixed solvent thereof and water, and a mixed solvent of tetrahydrofuran / methanol / water is preferable. Examples of the base include inorganic bases such as sodium hydroxide, potassium hydroxide, lithium hydroxide and sodium hydride, and metal alkoxides such as sodium methoxide and potassium tert-butoxide, preferably lithium hydroxide and sodium And methoxide. The base can be used in an amount of 3.0 to 6.0 equivalents, preferably 4.0 to 4.5 equivalents, relative to compound (F). The reaction temperature is usually from 0 to 20 ° C., and the reaction time is usually from 1 to 10 hours, although it varies depending on the raw material used, solvent, reaction temperature and the like.
(Step 3-2)
When isolating compound (G), compound (G) can be produced by subjecting compound (F) to an intramolecular cyclization reaction in a solvent in the presence of a base. Examples of the solvent include cyclic ethers such as tetrahydrofuran, 2-methyltetrahydrofuran and tetrahydropyran, lower alcohols such as methanol, ethanol and 2-propanol, amide solvents such as N, N-dimethylformamide, and nitriles such as acetonitrile. Examples thereof include a solvent and the like or a mixed solvent thereof, and a mixed solvent of tetrahydrofuran / methanol is preferable. Examples of the base include inorganic bases such as sodium hydroxide, potassium hydroxide, lithium hydroxide or sodium hydride, metal alkoxides such as sodium methoxide and potassium tert-butoxide, and lithium hydroxide, sodium methoxide and the like. preferable. The base can be used in an amount of 0.1 to 1.5 equivalents, preferably 1.0 to 1.1 equivalents, relative to compound (F). The reaction temperature is usually from 0 to 20 ° C., and the reaction time is usually from 1 to 10 hours, although it varies depending on the raw material used, solvent, reaction temperature and the like.
(Step 3-3)
The hydrolysis reaction in this step can be performed by the same method as in step 3-1 or a method analogous thereto.(Process 4)
Compound (A) can be converted to a salt thereof by a conventional method. Examples of such salts include inorganic salts such as sodium salt, potassium salt, calcium salt, magnesium salt, triethylamine, diisopropylamine, N, N′-dibenzylethylenediamine, ethanolamine, (2-hydroxyethyl) trimethylammonium. (Hereinafter referred to as choline), addition salts with organic bases such as N-methylglucamine, arginine, lysine and the like, and choline salts are preferred. Examples of the reagent used for conversion to the choline salt include choline hydroxide, choline bicarbonate, choline chloride and choline acetate.Here, the compound (B) and the salt thereof used in the above-mentioned scheme 1 are commercially available, or manufactured by the method described in a) to c), the method described in the reference examples, or a method analogous thereto. Can do.
a) JP-A 64-29373
b) Synthetic Communications, 32, 2565 (2002)
c) Synthesis, 200 (1977)Further, the compound (E) or a salt thereof used in the scheme 1 can be produced by the method described in Patent Document 1, the method described in Reference Examples, or a method analogous thereto.The compound obtained in the production process in the present specification includes hydrates or solvates thereof, and any of them can be used. Furthermore, the compound obtained in the production process in the present specification may have tautomers and / or geometric isomers, any of which can be used, and also a mixture thereof. be able to.By the production method of the present invention, the compound (A) useful as a pharmaceutical product or a salt thereof can be obtained in high yield and high purity through the compound (D) which is a production intermediate.The content of the present invention will be described in more detail by the following examples, but the present invention is not limited to the content.Reference example 1
Dimethyl 4-oxothiolane-2,3-dicarboxylate methylthioglycolate (15.0 g), tetrahydrofuran (45 g), piperidine (0.361 g) in a reaction mixture at room temperature with dimethyl maleate (21.4 g) in tetrahydrofuran (30 g) The solution was added. To the reaction mixture was added 20% sodium methoxide in methanol (43 g) at 55 ° C. under a nitrogen atmosphere. The reaction mixture was stirred at reflux for 3 hours. Diisopropyl ether (105 g) and acetic acid (0.85 g) were added to the reaction mixture at 45-50 ° C., and then cooled. The suspension was filtered to obtain wet crystals (43.3 g) of sodium salt of dimethyl 4-oxothiolane-2,3-dicarboxylate. The wet crystals were added to a mixture of 85% phosphoric acid (9.8 g), water (20 g) and ethyl acetate (150 g) at room temperature, and the aqueous layer was removed. The obtained organic layer was washed with 10% brine and then dried over anhydrous magnesium sulfate. The drying agent was removed by filtration, and the filtrate was concentrated under reduced pressure to obtain the title compound (22.7 g).Reference example 2
Dimethyl 4- (hydroxyimino) thiolane-2,3-dicarboxylate Dimethyl 4-oxothiolane-2,3-dicarboxylate (10.0 g), pyridine (5.44 g), hydroxylamine hydrochloride (3.34 g) Was stirred at 50 ° C. for 1 hour. Ethyl acetate and 7% aqueous phosphoric acid solution were added to the reaction mixture at room temperature, and the aqueous layer was removed. The obtained organic layer was washed with 5% sodium bicarbonate water and 10% brine. The organic layer was dried over anhydrous sodium sulfate. After removing the desiccant by filtration, the filtrate was concentrated under reduced pressure to obtain the title compound (10.4 g).Reference example 3
4-Aminothiophene-2,3-dicarboxylic acid dimethyl hydrochloride 4- (hydroxyimino) thiolane-2,3-dicarboxylate (10.4 g) in acetic acid (32 g) solution in 4N-hydrogen chloride / ethyl acetate solution ( 120 g) was added at room temperature. The reaction mixture was stirred at room temperature for 8 hours. After filtering the suspension, the obtained solid was dried to obtain the title compound (9.42 g).Reference example 4
4-Aminothiophene-2,3-dicarboxylic acid dimethyl methanesulfonate To a solution of methanesulfonic acid (80.0 g) in ethyl acetate (900 g), dimethyl 4- (hydroxyimino) thiolane-2,3-dicarboxylate (97. 1 g) of ethyl acetate (500 g) was added at 65-75 ° C. The reaction mixture was stirred at the same temperature for 2 hours. Methyl isobutyl ketone (100 g) was added at 45-50 ° C. and cooled to room temperature. After filtering the suspension, the obtained solid was dried to obtain the title compound (102 g).Reference Example 5
1,2-difluoro-3-[(4-fluoro-2-methoxyphenoxy) methyl] -4-methoxybenzene sodium borohydride in a solution of 2,3-difluoro-6-methoxybenzaldehyde (150 g) in toluene (900 g) (13.2 g) of 0.1N sodium hydroxide aqueous solution (180 g) was added at 35 to 39 ° C. The reaction mixture was stirred at the same temperature for 5 hours. After cooling the reaction mixture to room temperature, the aqueous layer was removed. The obtained organic layer was washed with 20% brine to obtain a toluene solution of 2,3-difluoro-6-methoxybenzyl alcohol. To this solution was added concentrated hydrochloric acid (610 g) at room temperature. The reaction mixture was stirred at 38-43 ° C. for 5 hours. After cooling the reaction mixture to room temperature, the aqueous layer was removed. The obtained organic layer was washed with water and 20% brine to obtain a toluene solution of 3- (chloromethyl) -1,2-difluoro-4-methoxybenzene. To this solution, 4-fluoro-2-methoxyphenol (125 g) and tetrabutylammonium bromide (56.2 g) were added at room temperature. A 25% aqueous sodium hydroxide solution (170 g) was added to the reaction mixture at 60 to 63 ° C., and the mixture was stirred at the same temperature for 4 hours. Water was added to the reaction mixture and the aqueous layer was removed. The obtained organic layer was washed with water and concentrated under reduced pressure. The residue was dissolved in 2-propanol and water was added. After filtering the suspension, the obtained solid was dried to obtain the title compound (232 g).Reference Example 6
1,2-difluoro-3-[(4-fluoro-2-methoxy-5-nitrophenoxy) methyl] -4-methoxybenzene 1,2-difluoro-3-[(4-fluoro-2-methoxyphenoxy) methyl ] To a solution of 4-methoxybenzene (158 g) in acetic acid (1200 g) was added 60% nitric acid (72.2 g) at 59-62 ° C., and the mixture was stirred at the same temperature for 2 hours. Water (1200 g) was added to the suspension at 15 to 19 ° C., and the mixture was stirred at the same temperature for 1 hour. After filtering the suspension, the obtained solid was washed with water to obtain wet crystals of the title compound (190 g, Net amount 168 g).Reference Example 7
2-Fluoro-5-[(2,3-difluoro-6-methoxyphenyl) methoxy] -4-methoxyaniline Raney nickel (2.5 g), ethyl acetate (180 g), 1,2-difluoro-3-[(4 -Fluoro-2-methoxy-5-nitrophenoxy) methyl] -4-methoxybenzene wet crystal (10.9 g, Net amount 10.0 g) was stirred at room temperature under a hydrogen atmosphere for 4 hours. The catalyst was removed by filtration, and the filtrate was concentrated under reduced pressure. The residue was dissolved with methanol and water was added. After filtering the suspension, the obtained solid was dried to obtain the title compound (7.97 g).Example 1
4- (phenoxycarbonylamino) thiophene-2,3-dicarboxylic acid dimethyl potassium carbonate (17.1 g), water (90 g), tetrahydrofuran (150 g) and 4-aminothiophene-2,3-dicarboxylic acid dimethyl hydrochloride (30 0.06) was added phenyl chloroformate (18.6 g) at 6-13 ° C. The reaction mixture was stirred at 12-13 ° C. for 30 minutes, and then the aqueous layer was removed. To the obtained organic layer, tert-butyl methyl ether was added and washed with 20% brine. The obtained organic layer was concentrated under reduced pressure. The residue was dissolved with diisopropyl ether and n-hexane was added. After filtering the suspension, the obtained solid was dried to obtain the title compound (37.0 g).
1 H-NMR (DMSO-d 6 ) δ ppm: 3.82 (3H, s), 3.82 (3H, s), 7.13-7.30 (3H, m), 7.40-7.46 (2H, m), 7.80 (1H, s ), 10.24 (1H, s)Example 2
4- {3- [2-Fluoro-5- (2,3-difluoro-6-methoxybenzyloxy) -4-methoxyphenyl] ureido} dimethyl thiophene-2,3-dicarboxylate 2-fluoro-5-[( 2,3-difluoro-6-methoxyphenyl) methoxy] -4-methoxyaniline (7.70 g), dimethyl 4- (phenoxycarbonylamino) thiophene-2,3-dicarboxylate (8.65 g), triethylamine (0. 37 g) and tetrahydrofuran (80 mL) were stirred at room temperature for 24 hours. The reaction mixture was concentrated under reduced pressure. Ethyl acetate and methanol were added to the residue. After filtering the suspension, the obtained solid was dried to obtain the title compound (12.0 g).
1 H-NMR (DMSO-d 6 ) δ ppm: 3.71 (3H, s), 3.82 (3H, s), 3.83 (3H, s), 3.89 (3H, s), 5.00 (2H, d, J = 1.6 Hz), 6.87-6.93 (1H, m), 7.00 (1H, d, J = 12.8Hz), 7.41-7.50 (1H, m), 7.75 (1H, d, J = 8.0Hz), 7.94 (1H, s ), 8.82 (1H, s), 8.95 (1H, s)Example 3
3- [2-Fluoro-5- (2,3-difluoro-6-methoxybenzyloxy) -4-methoxyphenyl] -2,4-dioxo-1,2,3,4-tetrahydrothieno [3,4 d] methyl pyrimidine-5-carboxylate 4- {3- [2-fluoro-5- (2,3-difluoro-6-methoxybenzyloxy) -4-methoxyphenyl] ureido} thiophene-2,3-dicarboxylic acid A methanol solution (3.48 g) of 28% sodium methoxide was added to a suspension of dimethyl (10.0 g) in tetrahydrofuran (40 g), stirred at room temperature for 3 hours, and acetic acid (1.30 g) was added. The reaction mixture was concentrated under reduced pressure. Methanol was added to the residue, and water was further added. After filtering the suspension, the obtained solid was dried to obtain the title compound (8.58 g).
1 H-NMR (DMSO-d 6 ) δ ppm: 3.79 (3H, s), 3.81 (3H, s), 3.84 (3H, s), 4.95 (2H, s), 6.88-6.94 (1H, m), 7.08 (1H, d, J = 11.6Hz), 7.19-7.23 (2H, m), 7.44-7.53 (1H, m), 11.62 (1H, s)Example 4
4- (phenoxycarbonylamino) thiophene-2,3-dicarboxylate potassium carbonate (9.38 kg), water (49 kg), tetrahydrofuran (82 kg), dimethyl 4-aminothiophene-2,3-dicarboxylate hydrochloride (16 4 kg) of the reaction mixture was stirred for 40 minutes, and then phenyl chloroformate (10.1 kg) was added at 11-21 ° C. The reaction mixture was stirred for 30 minutes, and then the aqueous layer was removed to obtain a tetrahydrofuran solution of the title compound.Example 5
4- {3- [2-Fluoro-5- (2,3-difluoro-6-methoxybenzyloxy) -4-methoxyphenyl] ureido} dimethyl thiophene-2,3-dicarboxylate 4-obtained in Example 4 To a tetrahydrofuran solution of dimethyl (phenoxycarbonylamino) thiophene-2,3-dicarboxylate, 2-fluoro-5-[(2,3-difluoro-6-methoxyphenyl) methoxy] -4-methoxyaniline (17.0 kg), Tetrahydrofuran (8.5 kg) and triethylamine (1.1 kg) were added, and the mixture was stirred at 50 ° C. for 3.5 hours to obtain a tetrahydrofuran solution of the title compound.Example 6
3- [2-Fluoro-5- (2,3-difluoro-6-methoxybenzyloxy) -4-methoxyphenyl] -2,4-dioxo-1,2,3,4-tetrahydrothieno [3,4 d] pyrimidine-5-carboxylic acid tetrahydrofuranate 4- {3- [2-fluoro-5- (2,3-difluoro-6-methoxybenzyloxy) -4-methoxyphenyl] ureido} obtained in Example 5 Methanol (41 kg) and water (47 kg) are added to a tetrahydrofuran solution of dimethyl thiophene-2,3-dicarboxylate, a 7.3% lithium hydroxide aqueous solution (80.1 kg) is added at 11 to 13 ° C., and 90 ° C. at 11 ° C. Stir for minutes. Acetic acid (11.4 kg) was added to the reaction mixture at 9 to 16 ° C., and acetic acid (13.0 kg) was further added at 29 to 31 ° C. Seed crystals were added to the reaction mixture, and the mixture was stirred at the same temperature for 30 minutes. Water (34 kg) was added to the suspension and stirred at 30 ° C. for 40 minutes. The suspension was stirred at 4-9 ° C. for 90 minutes. After the suspension was filtered, the obtained solid was washed with a mixed solution of methanol (54 kg) and water (68 kg) to give wet crystals of the title compound (31.64 kg, Net amount (compound (A) free form equivalent)) 26 0.7 kg) was obtained.
A part of the wet crystals of the title compound was dried under reduced pressure at an external temperature of 60 ° C., and 1 H-NMR, HPLC and powder X-ray diffraction were measured on the obtained dried crystals of the title compound.
1 H-NMR (DMSO-d 6 ) δ ppm: 1.68-1.82 (3H, m), 3.53-3.65 (3H, m), 3.80 (3H, s), 3.81 (3H, s), 4.94-4.98 (2H , m), 6.87-6.94 (1H, m), 7.13 (1H, d, J = 11.2Hz), 7.25 (1H, d, J = 7.2Hz), 7.39 (1H, s), 7.43-7.52 (1H, m), 11,99 (1H, s), 14.53 (1H, s)
PATENT
WO 2020089190
https://patents.google.com/patent/WO2020089190A2/enFor example, the GnRH antagonist may be 3-[2-fluoro-5-(2,3-difluoro-6-methoxybenzyloxy)4- methoxyphenyl]-2,4-dioxo-1 ,2,3,4- tetrahydrothieno [3,4d]pyrimidine-5-carboxylic acid, or a pharmaceutically acceptable salt thereof. The salt may be, for instance, the choline salt thereof, represented by formula (Via), below.
Compound (VI) and pharmaceutically acceptable salts thereof, such as the choline salt thereof (compound (Via)), can be synthesized, for example, using the methodology described in WO 2014/042176, the disclosure of which is incorporated herein by reference in its entirety. An exemplary synthetic scheme that may be used for the preparation of compound (VI) and the choline salt thereof is shown in Scheme 1 , below.Scheme 1 . Exemplary preparation of compound (VI) and the choline salt thereof




wherein Ri and R are each independently C alkoxy groups; LG is a nucleofugal leaving group, such as chlorine or bromine, among others; R represents an optional substituent, such as halogen, acyl group, C alkyl group, or a nitro substituent; DMAP denotes A/-dimethylaminopyridine; and TEA denotes trimethylamine.Crystalline compound (Via) has been characterized spectroscopically, for instance, in US Patent No. 9,169,266, the disclosure of which is incorporated herein by reference in its entirety. The foregoing crystalline form has been shown to exhibit characteristic X-ray powder diffraction peaks at about 7.10 2Q, about 11 .5° 2Q, about 19.4° 2Q, about 21 .5° 2Q, about 22.0° 2Q, about 22.6° 2Q, about 23.5° 2Q, and about 26.2° 2Q. Additionally, this crystalline form exhibits 13C solid-state nuclear magnetic resonance (NMR) peaks centered at about 55.5 ppm, about 57.1 ppm, about 58.7 ppm, about 69.8 ppm, about 98.1 ppm, about 110.3 ppm, about 1 1 1 .6 ppm, about 113.7 ppm, about 1 18.0 ppm, about 145.3 ppm, about 149.8 ppm, and about 155.8 ppm. This crystalline form further exhibits 19F solid-state NMR peaks centered at about -151.8 ppm, -145.2 ppm, and -131 .6 ppm.Compound (VI), as well as pharmaceutically acceptable salts thereof, such as the choline salt thereof, exhibit a high affinity for human GnRH receptor (27.4 nM). Using the compositions and methods described herein, a patient that is presenting with or has been diagnosed as having, adenomyosis or rectovaginal endometriosis may be administered a compound of formula (VI), or a pharmaceutically acceptable salt thereof, such as the choline salt thereof, to treat the disease or ameliorate one or more symptoms of the disease. Exemplary doses of compound (VI) and pharmaceutically acceptable salts thereof, such as the choline salt thereof, include doses of from 25 mg to 500 mg daily, such as doses of 100 mg per day and 200 mg per day. Additional dosing information is provided below.3-Aminoalkyl pyrimidine-2, 4(1 H,3H)-dionesAdditional GnRH antagonists that may be used in conjunction with the compositions and methods described herein include optionally substituted 3-aminoalkyl pyrimidine-2, 4(1 H,3H)-dione derivatives, such as compounds represented by formula (VII)

PATENTWO 2021023876https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2021023876&_cid=P11-KWFRM2-91270-1
In some embodiments, the compound is the choline salt of the compound represented by formula (VI), choline 3- [2-fluoro-5- (2,3-difluoro-6-methoxybenzyloxy) 4-methoxyphenyI] -2,4- dioxo-1,2,3,4-
tetrahydrothieno [3,4d] pyrimidine-5-carboxylate. It is to be understood that references herein to a compound represented by formula (VI) specifically include the choline salt of compound (VI), which is represented by formula (VIa), below.
In some embodiments, the choline 3- [2-fluoro-5- (2,3-difluoro-6-methoxybenzyloxy) 4-methoxyphenyI] -2,4-dioxo-1,2,3,4- tetrahydrothieno [3,4d ] pyrimidine-5-carboxylate is in a crystalline state.
PATENT
WO 2021023877
References
- ^ Jump up to:a b c “Linzagolix – Kissei Pharmaceutical/ObsEva – AdisInsight”.
- ^ Jump up to:a b Ezzati M, Carr BR (2015). “Elagolix, a novel, orally bioavailable GnRH antagonist under investigation for the treatment of endometriosis-related pain”. Womens Health (Lond). 11 (1): 19–28. doi:10.2217/whe.14.68. PMID 25581052.
- ^ Chodankar, Rohan; Allison, Jennifer (2018). “New Horizons in Fibroid Management”. Current Obstetrics and Gynecology Reports. 7 (2): 106–115. doi:10.1007/s13669-018-0242-6. ISSN 2161-3303.
External links
| Clinical data | |
|---|---|
| Trade names | Yselty |
| Other names | KLH-2109; OBE-2109 |
| Routes of administration | By mouth[1][2] |
| Drug class | GnRH modulator; GnRH antagonist; Antigonadotropin |
| ATC code | None |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 935283-04-8 |
| PubChem CID | 16656889 |
| ChemSpider | 17590169 |
| UNII | 7CDW97HUEX |
| KEGG | D11608 |
| ChEMBL | ChEMBL3668014 |
| Chemical and physical data | |
| Formula | C22H15F3N2O7S |
| Molar mass | 508.42 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI |
////////LINZAGOLIX, Hormone Antagonists, WHO 10711, KLH-2109, KLH 2109, OBE-2109, OBE 2109

NEW DRUG APPROVALS
ONE TIME
$10.00
Ropeginterferon alfa-2b
PCDLPQTHSL GSRRTLMLLA QMRRISLFSC LKDRHDFGFP QEEFGNQFQK AETIPVLHEM
IQQIFNLFST KDSSAAWDET LLDKFYTELY QQLNDLEACV IQGVGVTETP LMKEDSILAV
RKYFQRITLY LKEKKYSPCA WEVVRAEIMR SFSLSTNLQE SLRSKE
(Disulfide bridge: 2-99, 30-139)
Ropeginterferon alfa-2b
- AOP2014
CAS 1335098-50-4
UNII981TME683S
FDA APPROVED, 2021/11/12, BESREMI
PEPTIDE, Antineoplastic, Antiviral
Polycythemia vera (PV) is the most common Philadelphia chromosome-negative myeloproliferative neoplasm (MPN), characterized by increased hematocrit and platelet/leukocyte counts, an increased risk for hemorrhage and thromboembolic events, and a long-term propensity for myelofibrosis and leukemia.1,2 Interferon alfa-2b has been used for decades to treat PV but requires frequent dosing and is not tolerated by all patients.2 Ropeginterferon alfa-2b is a next-generation mono-pegylated type I interferon produced from proline-IFN-α-2b in Escherichia coli that has high tolerability and a long half-life.4,6 Ropeginterferon alfa-2b has shown efficacy in PV in in vitro and in vivo models and clinical trials.3,4
Ropeginterferon alfa-2b was approved by the FDA on November 12, 2021, and is currently marketed under the trademark BESREMi by PharmaEssentia Corporation.6
Ropeginterferon alfa-2b, sold under the brand name Besremi, is a medication used to treat polycythemia vera.[1][2][3][4] It is an interferon.[1][3] It is given by injection.[1][3]
The most common side effects include low levels of white blood cells and platelets (blood components that help the blood to clot), muscle and joint pain, tiredness, flu-like symptoms and increased blood levels of gamma-glutamyl transferase (a sign of liver problems).[3] Ropeginterferon alfa-2b can cause liver enzyme elevations, low levels of white blood cells, low levels of platelets, joint pain, fatigue, itching, upper airway infection, muscle pain and flu-like illness.[2] Side effects may also include urinary tract infection, depression and transient ischemic attacks (stroke-like attacks).[2]
It was approved for medical use in the European Union in February 2019,[3] and in the United States in November 2021.[2][5] Ropeginterferon alfa-2b is the first medication approved by the U.S. Food and Drug Administration (FDA) to treat polycythemia vera that people can take regardless of their treatment history, and the first interferon therapy specifically approved for polycythemia vera.[2]
https://www.fda.gov/news-events/press-announcements/fda-approves-treatment-rare-blood-disease#:~:text=FDA%20NEWS%20RELEASE-,FDA%20Approves%20Treatment%20for%20Rare%20Blood%20Disease,FDA%2DApproved%20Option%20Patients%20Can%20Take%20Regardless%20of%20Previous%20Therapies,-ShareFor Immediate Release:November 12, 2021
Today, the U.S. Food and Drug Administration approved Besremi (ropeginterferon alfa-2b-njft) injection to treat adults with polycythemia vera, a blood disease that causes the overproduction of red blood cells. The excess cells thicken the blood, slowing blood flow and increasing the chance of blood clots.
“Over 7,000 rare diseases affect more than 30 million people in the United States. Polycythemia vera affects approximately 6,200 Americans each year,” said Ann Farrell, M.D., director of the Division of Non-Malignant Hematology in the FDA’s Center for Drug Evaluation and Research. “This action highlights the FDA’s commitment to helping make new treatments available to patients with rare diseases.”
Besremi is the first FDA-approved medication for polycythemia vera that patients can take regardless of their treatment history, and the first interferon therapy specifically approved for polycythemia vera.
Treatment for polycythemia vera includes phlebotomies (a procedure that removes excess blood cells though a needle in a vein) as well as medicines to reduce the number of blood cells; Besremi is one of these medicines. Besremi is believed to work by attaching to certain receptors in the body, setting off a chain reaction that makes the bone marrow reduce blood cell production. Besremi is a long-acting drug that patients take by injection under the skin once every two weeks. If Besremi can reduce excess blood cells and maintain normal levels for at least one year, then dosing frequency may be reduced to once every four weeks.
The effectiveness and safety of Besremi were evaluated in a multicenter, single-arm trial that lasted 7.5 years. In this trial, 51 adults with polycythemia vera received Besremi for an average of about five years. Besremi’s effectiveness was assessed by looking at how many patients achieved complete hematological response, which meant that patients had a red blood cell volume of less than 45% without a recent phlebotomy, normal white cell counts and platelet counts, a normal spleen size, and no blood clots. Overall, 61% of patients had a complete hematological response.
Besremi can cause liver enzyme elevations, low levels of white blood cells, low levels of platelets, joint pain, fatigue, itching, upper airway infection, muscle pain and flu-like illness. Side effects may also include urinary tract infection, depression and transient ischemic attacks (stroke-like attacks).
Interferon alfa products like Besremi may cause or worsen neuropsychiatric, autoimmune, ischemic (not enough blood flow to a part of the body) and infectious diseases, which could lead to life-threatening or fatal complications. Patients who must not take Besremi include those who are allergic to the drug, those with a severe psychiatric disorder or a history of a severe psychiatric disorder, immunosuppressed transplant recipients, certain patients with autoimmune disease or a history of autoimmune disease, and patients with liver disease.
People who could be pregnant should be tested for pregnancy before using Besremi due to the risk of fetal harm.
Besremi received orphan drug designation for this indication. Orphan drug designation provides incentives to assist and encourage drug development for rare diseases.
The FDA granted the approval of Besremi to PharmaEssentia Corporation.
Medical uses
In the European Union, ropeginterferon alfa-2b is indicated as monotherapy in adults for the treatment of polycythemia vera without symptomatic splenomegaly.[3] In the United States it is indicated for the treatment of polycythemia vera.[1][2][5]
History
The effectiveness and safety of ropeginterferon alfa-2b were evaluated in a multicenter, single-arm trial that lasted 7.5 years.[2] In this trial, 51 adults with polycythemia vera received ropeginterferon alfa-2b for an average of about five years.[2] The effectiveness of ropeginterferon alfa-2b was assessed by looking at how many participants achieved complete hematological response, which meant that participants had a red blood cell volume of less than 45% without a recent phlebotomy, normal white cell counts and platelet counts, a normal spleen size, and no blood clots.[2] Overall, 61% of participants had a complete hematological response.[2] The U.S. Food and Drug Administration (FDA) granted the application for Ropeginterferon_alfa-2b orphan drug designation and granted the approval of Besremi to PharmaEssentia Corporation[2]
REF
- Bartalucci N, Guglielmelli P, Vannucchi AM: Polycythemia vera: the current status of preclinical models and therapeutic targets. Expert Opin Ther Targets. 2020 Jul;24(7):615-628. doi: 10.1080/14728222.2020.1762176. Epub 2020 May 18. [Article]
- How J, Hobbs G: Use of Interferon Alfa in the Treatment of Myeloproliferative Neoplasms: Perspectives and Review of the Literature. Cancers (Basel). 2020 Jul 18;12(7). pii: cancers12071954. doi: 10.3390/cancers12071954. [Article]
- Verger E, Soret-Dulphy J, Maslah N, Roy L, Rey J, Ghrieb Z, Kralovics R, Gisslinger H, Grohmann-Izay B, Klade C, Chomienne C, Giraudier S, Cassinat B, Kiladjian JJ: Ropeginterferon alpha-2b targets JAK2V617F-positive polycythemia vera cells in vitro and in vivo. Blood Cancer J. 2018 Oct 4;8(10):94. doi: 10.1038/s41408-018-0133-0. [Article]
- Gisslinger H, Zagrijtschuk O, Buxhofer-Ausch V, Thaler J, Schloegl E, Gastl GA, Wolf D, Kralovics R, Gisslinger B, Strecker K, Egle A, Melchardt T, Burgstaller S, Willenbacher E, Schalling M, Them NC, Kadlecova P, Klade C, Greil R: Ropeginterferon alfa-2b, a novel IFNalpha-2b, induces high response rates with low toxicity in patients with polycythemia vera. Blood. 2015 Oct 8;126(15):1762-9. doi: 10.1182/blood-2015-04-637280. Epub 2015 Aug 10. [Article]
- EMA Approved Products: Besremi (ropeginterferon alfa-2b ) solution for injection [Link]
- FDA Approved Drug Products: BESREMi (ropeginterferon alfa-2b-njft) injection [Link]
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////////////////////////////
References
- ^ Jump up to:a b c d e https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/761166s000lbl.pdf
- ^ Jump up to:a b c d e f g h i j k l “FDA Approves Treatment for Rare Blood Disease”. U.S. Food and Drug Administration (FDA) (Press release). 12 November 2021. Retrieved 12 November 2021. This article incorporates text from this source, which is in the public domain.
- ^ Jump up to:a b c d e f g “Besremi EPAR”. European Medicines Agency (EMA). Retrieved 14 November 2021. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ Wagner SM, Melchardt T, Greil R (March 2020). “Ropeginterferon alfa-2b for the treatment of patients with polycythemia vera”. Drugs of Today. Barcelona, Spain. 56 (3): 195–202. doi:10.1358/dot.2020.56.3.3107706. PMID 32282866. S2CID 215758794.
- ^ Jump up to:a b “U.S. FDA Approves Besremi (ropeginterferon alfa-2b-njft) as the Only Interferon for Adults With Polycythemia Vera” (Press release). PharmaEssentia. 12 November 2021. Retrieved 14 November 2021 – via Business Wire.
External links
- “Ropeginterferon alfa-2b”. Drug Information Portal. U.S. National Library of Medicine.
- Clinical trial number NCT01193699 for “Safety Study of Pegylated Interferon Alpha 2b to Treat Polycythemia Vera (PEGINVERA)” at ClinicalTrials.gov
- Clinical trial number NCT02218047 for “AOP2014 vs. BAT in Patients With Polycythemia Vera Who Previously Participated in the PROUD-PV Study. (CONTI-PV)” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | Besremi |
| Other names | AOP2014, ropeginterferon alfa-2b-njft |
| License data | EU EMA: by INNUS DailyMed: Ropeginterferon_alfa |
| Pregnancy category | Contraindicated |
| Routes of administration | Subcutaneous |
| Drug class | Interferon |
| ATC code | L03AB15 (WHO) |
| Legal status | |
| Legal status | US: ℞-only [1][2]EU: Rx-only [3] |
| Identifiers | |
| CAS Number | 1335098-50-4 |
| DrugBank | DB15119 |
| UNII | 981TME683S |
| KEGG | D11027 |
/////////Ropeginterferon alfa-2b, FDA 2021, APPROVALS 2021, BESREMI, PEPTIDE, Antineoplastic, Antiviral, AOP 2014, PharmaEssentia

NEW DRUG APPROVALS
ONE TIME
$10.00
SULCONAZOLE

SULCONAZOLEсульконазол , سولكونازول , 硫康唑
- Molecular FormulaC18H15Cl3N2S
- Average mass397.749 Da
1H-Imidazole, 1-[2-[[(4-chlorophenyl)methyl]thio]-2-(2,4-dichlorophenyl)ethyl]- [ACD/Index Name]
4332
5D9HAA5Q5S
61318-90-9[RN]
(±)-1-[2,4-Dichloro-b-[(p-chlorobenzyl)thio]phenethyl]imidazole
1-[2-[[(4-Chlorophenyl)methyl]thio]-2-(2,4-dichlorophenyl)ethyl]-1H-imidazole: SulconazoleCAS Registry Number: 61318-90-9
CAS Name: 1-[2-[[(4-Chlorophenyl)methyl]thio]-2-(2,4-dichlorophenyl)ethyl]-1H-imidazole
Additional Names: (±)-1-[2,4-dichloro-b-[(p-chlorobenzyl)thio]phenethyl]imidazole
Molecular Formula: C18H15Cl3N2S
Molecular Weight: 397.75
Percent Composition: C 54.35%, H 3.80%, Cl 26.74%, N 7.04%, S 8.06%
Literature References: Prepn: K. A. M. Walker, DE2541833; idem,US4055652 (1976, 1977 both to Syntex). HPLC determn in plasma: M. Fass et al.,J. Pharm. Sci.70, 1338 (1981). Mechanism of action study: W. H. Beggs, Biochem. Arch.10, 117 (1994). Clinical trial in tinea pedis: W. A. Akers et al.,J. Am. Acad. Dermatol.21, 686 (1989). Review of pharmacology and clinical efficacy: P. Benfield, S. P. Clissold, Drugs35, 143-153 (1988).
Derivative Type: Nitrate
CAS Registry Number: 61318-91-0
Manufacturers’ Codes: RS-44872
Trademarks: Exelderm (Syntex); Myk (Cassenne); Sulcosyn (Syntex)
Molecular Formula: C18H15Cl3N2S.HNO3
Molecular Weight: 460.76
Percent Composition: C 46.92%, H 3.50%, Cl 23.08%, N 9.12%, S 6.96%, O 10.42%
Properties: Colorless crystals from acetone, mp 130.5-132°.
Melting point: mp 130.5-132°
Therap-Cat: Antifungal.
Keywords: Antifungal (Synthetic); Imidazoles.
Sulconazole (trade name Exelderm) is an antifungal medication of the imidazole class. It is available as a cream or solution to treat skin infections such as athlete’s foot, ringworm, jock itch, and sun fungus.[1][2] Although not used commercially for insect control, sulconazole nitrate exhibits a strong anti-feeding effect on the keratin-digesting Australian carpet beetle larvae Anthrenocerus australis.[3]
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////////////////////////////
SYN

DE 2541833 US 4038409
(Read example 5 and 9 in US patent.)
https://patents.google.com/patent/US4038409A/en
EXAMPLE 5Alternative Route to 1-[β-(R-carbonylthio)phenethyl]imidazolesA. Preparation of 1-[2,4-dichloro-β-(methylcarbonylthio)-phenethyl]imidazole, oxalate.1-(β,2,4-Trichlorophenethylimidazole (1.19g) in 5 ml of dry tetrahydrofuran was added to preformed sodium thioacetate, generated in situ from 720 mg thioacetic acid and sodium hydride (480 mg 57% dispersion in mineral oil) in 20 ml. tetrahydrofuran and the mixture stirred and refluxed under nitrogen for 18 hours. The solvent was removed under reduced pressure, water (20 ml) added and the product extracted with ether. The extracts were washed with water, dried (MgSO4), evaporated and the residue chromatographed on silica gel eluting with 10-20% acetone in dichloromethane. The pure product in ether was treated dropwise with ethereal oxalic acid until precipitation was complete, and the thus obtained oxalate salt of 1-[2,4-dichloro-β-(methylcarbonylthio)phenethyl]imidazole recrystallized from acetone/ethyl acetate with mpBy substituting other available sodium thioacids for sodium thioacetate, other compounds of this invention may be prepared.
EXAMPLE 9A. Preparation of 1-[2,4-dichloro-β-(4-chlorobenzylthio)-phenethyl]imidazoleTo a stirred solution of 330 mg sodium hydroxide in 30 ml methanol under nitrogen is added 810 mg of 1-[2,4-dichloro-β-(methylcarbonylthio)phenethyl]imidazole oxalate and the mixture is stirred at room temperature for ca. 30 minutes (until thin layer chromatography shows the disappearance of the ester). α,p-dichlorotoluene (350 mg) is then added, the solution stirred a further 15 minutes and the solvent removed under reduced pressure. Ether and water are then added to the residue and the ether extract washed with water, dried (MgSO4) and concentrated. Dropwise addition of nitric acid (d = 1.42) until precipitation is complete gives the nitrate salt of 1-[2,4-dichloro-β-(4-chlorobenzylthio)phenethyl]imidazole, recrystallized from acetone, mp 130.5°-132° C.B. By using other compounds of this invention exemplified by those set forth in Examples 2 and 4 and other suitable (substituted) hydrocarbyl halides (or mesylates, tosylates), other compounds may be prepared.
SYN
https://www.sciencedirect.com/science/article/pii/S2095177917301405
SYN
Synthesis Path
Substances Referenced in Synthesis Path
| CAS-RN | Formula | Chemical Name | CAS Index Name |
|---|---|---|---|
| 6258-66-8 | C7H7ClS | 4-chlorobenzyl mercaptan | Benzenemethanethiol, 4-chloro- |
| 24155-42-8 | C11H10Cl2N2O | 1-(2,4-dichlorophenyl)-2-(1H-imidazol-1-yl)ethanol | 1H-Imidazole-1-ethanol, α-(2,4-dichlorophenyl)- |
References
- ^ Drugs.com: sulconazole topical
- ^ Fromtling RA (April 1988). “Overview of medically important antifungal azole derivatives”. Clinical Microbiology Reviews. 1 (2): 187–217. doi:10.1128/CMR.1.2.187. PMC 358042. PMID 3069196.
- ^ Sunderland MR, Cruickshank RH, Leighs SJ (2014). “The efficacy of antifungal azole and antiprotozoal compounds in protection of wool from keratin-digesting insect larvae”. Textile Research Journal. 84 (9): 924–931. doi:10.1177/0040517513515312.
| Clinical data | |
|---|---|
| Trade names | Exelderm |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a698018 |
| Routes of administration | Topical |
| ATC code | D01AC09 (WHO) |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 61318-90-9 |
| PubChem CID | 5318 |
| ChemSpider | 5127 |
| UNII | 5D9HAA5Q5S |
| KEGG | D08535 |
| ChEBI | CHEBI:9325 |
| ChEMBL | ChEMBL1221 |
| CompTox Dashboard (EPA) | DTXSID8044129 |
| Chemical and physical data | |
| Formula | C18H15Cl3N2S |
| Molar mass | 397.74 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
| (what is this?) (verify) |
/////////SULCONAZOLE, сульконазол , سولكونازول , 硫康唑 , Antifungal,

NEW DRUG APPROVALS
ONE TIME
$10.00
TNO 155
TNO 155
2-Oxa-8-azaspiro[4.5]decan-4-amine, 8-[6-amino-5-[(2-amino-3-chloro-4-pyridinyl)thio]-2-pyrazinyl]-3-methyl-, (3S,4S)-
- (3S,4S)-8-[6-Amino-5-[(2-amino-3-chloro-4-pyridinyl)thio]-2-pyrazinyl]-3-methyl-2-oxa-8-azaspiro[4.5]decan-4-amine
- (3S,4S)-8-(6-amino-5-((2-amino-3-chloropyridin-4-yl)thio)pyrazin-2-yl)-3-methyl-2-oxa-8-azaspiro[4.5]decan-4-amine
| Molecular Weight |
421.95 |
|---|---|
| Formula |
C₁₈H₂₄ClN₇OS |
| CAS No. |
- PTPN11 inhibitor TNO155
- SHP2 inhibitor TNO155
- TNO-155
- TNO155
- UNII-FPJWORQEGI
TNO155 is a potent selective and orally active allosteric inhibitor of wild-type SHP2 (IC50=0.011 µM). TNO155 has the potential for the study of RTK-dependent malignancies, especially advanced solid tumors.
- Originator Novartis
- Developer Mirati Therapeutics; Novartis
- Class Antineoplastics
- Mechanism of ActionProtein tyrosine phosphatase non receptor antagonists
- Phase I/IISolid tumours
- Phase IColorectal cancer
- 11 Jul 2021Phase I trial in Solid tumours is still ongoing in USA, Canada, Japan, South Korea, Netherlands, Singapore, Spain, Taiwan (NCT03114319)
- 04 Jun 2021Efficacy, safety and pharmacokinetics data from phase I trial in Solid tumours presented at 57th Annual Meeting of the American Society of Clinical Oncology (ASCO-2021)
- 08 Jan 2021Novartis plans a phase Ib/II trial for Solid tumours (Combination therapy, Inoperable/Unresectable, Late-stage disease, Metastatic disease, Second-line therapy or greater) in February 2021 (NCT04699188)
CLIP
Combinations with Allosteric SHP2 Inhibitor TNO155 to Block Receptor Tyrosine Kinase Signaling
Results: In EGFR-mutant lung cancer models, combination benefit of TNO155 and the EGFRi nazartinib was observed, coincident with sustained ERK inhibition. In BRAFV600E colorectal cancer models, TNO155 synergized with BRAF plus MEK inhibitors by blocking ERK feedback activation by different RTKs. In KRASG12C cancer cells, TNO155 effectively blocked the feedback activation of wild-type KRAS or other RAS isoforms induced by KRASG12Ci and greatly enhanced efficacy. In addition, TNO155 and the CDK4/6 inhibitor ribociclib showed combination benefit in a large panel of lung and colorectal cancer patient–derived xenografts, including those with KRAS mutations. Finally, TNO155 effectively inhibited RAS activation by colony-stimulating factor 1 receptor, which is critical for the maturation of immunosuppressive tumor-associated macrophages, and showed combination activity with anti–PD-1 antibody.
Conclusions: Our findings suggest TNO155 is an effective agent for blocking both tumor-promoting and immune-suppressive RTK signaling in RTK- and MAPK-driven cancers and their tumor microenvironment. Our data provide the rationale for evaluating these combinations clinically.
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////////////////////////////
PATENT
WO 2015107495
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015107495
PATENT
WO 2020065453
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2020065453
(3S,4S)-8-(6-amino-5-((2-amino-3-chloropyridin-4-yl)thio)pyrazin-2-yl)-3-methyl-2-oxa-8-azaspiro[4.5]decan-4-amine, which has the formula I,
WO/2015/107495 A1 describes a method for the manufacture of the compound of the formula I which can be characterized by the following reaction scheme 1:
[0008] The last compound resulting from step g above was then reacted as in the following scheme 2:
Scheme 2:
[0009] Thus the compound of formula I is obtained (last compound in the scheme 2, above). The synthesis requires at least the 9 steps shown and is rather appropriate for synthesis in laboratory amounts.
Scheme 1A:
[0016] Therefore, the process, though readily feasible on a laboratory scale, is not ideal for manufacture at a large scale.
[0017] The compound added in reaction b in Scheme 2 is obtained in WO
2015/107495 A1 as “Intermediate 10” follows:
Scheme 3:
[0018] An issue here is the relatively low yield of the amine resulting from reaction a in
Scheme 3.
[0019] In addition, while WO 2015/107495 A1 generically mentions that pharmaceutically acceptable salts of the compound of the formula I may be obtainable, no concrete reason for obtaining such salts and no specific examples of salts are described.
[0020] In addition, given the many potentially salt forming groups in formula I, it is not clear whether any salts with a clear stoichiometry can be formed at all.
Example 1
Method of synthesis of the compound of the formula I ((3S,4S)-8-(6-amino-5-((2-amino-3- chloropyridin-4-yl)thio)pyrazin-2-yl)-3-methyl-2-oxa-8-azaspiro[4.5]decan-4-amine):
The overall synthesis can be described by the following Reaction Scheme A:
Scheme A:
Step a
[00293] To a solution of A1 (10.4 kg, 100 mol, 1.0 Eq) in CH2Cl2 (50 L) was added imidazole (8.16 kg, 120 mol, 1.2eq) and TBSCl (18 kg, 120 mol, 1.2 Eq) at 0 °C. After addition, the mixture was stirred at 0°C for 4 h . GC showed the reaction was finished. (A1/ (A1 + A2) < 1%). The reaction mixture was quenched with saturated NaHCO3 (14L) at 0-5°C. Phases were separated. The organic phase was washed with brine (14L). The organic layer was dried over Na2SO4, concentrated under vacuum at 40-45°C to afford A2 (23.3 kg, assay 88%, yield 94%) which was used for the next step directly. 1H NMR (400 MHz, CDC13) δ = 4.35 (d, J= 8.8 Hz, 1H), 3.74 (s, 3H), 2.48 (s, J= 8.8
Hz, 3H), 0.93 (s, 9H), 0.09 (s, 6H).
Step b
[00294] To a solution of A2 (7.5 kg, 34.3 mol, 1.0 Eq) and N,O-dimethylhydroxylamine hydrochloride (6.69 kg, 68.6mol, 2.0 Eq) in THF (20 L) was added drop-wise a solution
of chloro(isopropyl)magnesium (2 M, 51.45 L, 3.5 Eq) at 0 °C under N2 over 5-6 h. After addition, the reaction mixture was stirred at 0 °C for 1h, GC showed the reaction was finished (A2/(A2+A3) < 2 %). The mixture was quenched with NH4Cl (25 L) slowly by keeping the temperature at 0-5°C. After addition, the reaction mixture was stirred for 30min. Phase was separated. The aqueous layer was extracted with EA(2 x 20 L). The combined organic phase was washed with brine (25L), dried over Na2SO4, concentrated to give A3(9.4 kg, assay 86%, yield 95%) which was used for the next step directly. 1HNMR (400 MHz, CDCl3) δ = 4.67 (m, J= 6.6 Hz, 1H), 3.70 (s, 3H), 3.21 (s, 3H), 3.17 (d, 3H)2.48 (s , J= 6.6 Hz, 3H), 0.90 (s, 9H), 0.10 (s, 3H), 0.08 (s, 3H).
[00295] To a solution of A3 (7.1 kg, assay 86%, 24.65 mol, 1.0 Eq) in DCM (30 L) was added dropwise a solution of LiAlH4 (2.4 M, 11.3 L, 1.1 Eq) at -70 °C under N2. Then the reaction mixture was stirred at -70 °C for 3h, and TLC showed the reaction was finished (PSC-1). The mixture was warmed to 0 °C, and then quenched with sat. potassium sodium tartrate (35 L) at 0 °C. After addition, DCM (20L) was added and stirred for 2h at 20-25°C. Phases were separated. The aqueous layer was extracted with DCM (25 L). The combined organic phase was charged with sat. citric acid (45L) and stirred at 0°C for 8h. Phase was separated. The organic phase was washed with NaHCO3 (25L), brine (25 L), dried over Na2SO4, and the solvent was removed under vacuum at 25-30°C. n-Heptane (10 L) was added to the residue and concentrated under vacuum at 30-35°C. n-Heptane (10 L) was added to the residue again and concentrated under vacuum at 30-35°C to give A4 (4.2 kg, assay
60%, yield 54%) which was used for the next step directly.
Step d
[00296] To a solution of diisopropylamine (3.06 kg, 30.3 mol, 1.5 eq) in THF (20 L) cooled to approximately -10°C was added 2.5 M n-BuLi (12.12 L, 30.3 mol, 1.5 eq) under N2. The resulting mixture was stirred at approximately -10 °C for 30min, then a solution of A5 (5.2 kg, 20.20 mol, 1.0eq) in THF (10 L) was added slowly. After addition, the reaction mixture was stirred at -10°C for 30 min, and then cooled to -50°C. A4 (4.18 kg, 22.22 mol, 1.1eq) was added dropwise. After addition, the reaction mixture was stirred at -50°C for 30 min. The mixture was quenched with saturated aqueous NH4Cl (30L) and water (10L) at -50°C. The reaction mixture was warmed to 20-25°C. Phase was separated. The aqueous phase was extracted with EA (3 x 20 L). All organic phases were combined and washed with brine(20L), then concentrated to a yellow oil which was purified by column (silica gel, 100-200 mesh, eluted with n-heptane:EA from 50:1 to 10:1) to give A6 (5.5 kg, assay 90 %, yield 55%) as pale yellow oil. 1H NMR (400 MHz, CDCl3) δ = 4.35-4.15 (m, 2H), 3.95-3.74 (m, 3H), 3.52 (m, 2H), 2.67(m, 2H), 2.12-1.98 (m, 2H), 1.75-1.52 (m, 4H), 1.49 (s, 9H), 1.35-1.10 (m, 6H), 0.98 (s,
9H), 0.02 (s, 6H).
Step e
[00297] To a solution of A6 (11.4 kg, 25.58 mol, 1.0eq) in THF (60 L) was added LiBH4
(836 g, 38.37 mol, 1.5eq) in portions at 5-10 °C, and the reaction mixture was stirred at 20-25 °C for 18 h. HPLC showed the reaction was finished (A6/(A6+A7)<2%). The mixture was cooled to l0°C and slowly quenched with saturated NaHCO3 solution (15 L) and water (25L) with vigorously stirring. After gas formation stopped, vacuum filtration was applied to remove solids. The solid was washed with EA (2 x 15 L). Phase was separated; the aqueous phase was extracted with EA (3 x15L). All organic phases were combined and washed with brine (15L), and concentrated to obtain crude A7 (13.8 kg, assay 58%, yield 77%) which was used for the next step directly.
[00298] To a solution of A7 (8 kg, 19.82 mol, 1.0 eq) in THF (40 L) under nitrogen atmosphere was added TsCl (5.28 kg, 27.75 mol, 1.4 eq) at 10-15°C. After addition, the mixture was cooled to 0 °C, and 1M LiHMDS (29.7 L, 29.73 mol, 1.5 eq) was added dropwise during 2h. After addition, the mixture was stirred at 0°C for 3h. HPLC showed the reaction was finished (PSC-1 A7/ (A7+A8)<7%). TBAF (20.72 kg, 65.67 mol, 3.3 eq) was added into the mixture at 0 °C and the reaction mixture was stirred at 25-30 °C for 48h. HPLC showed the reaction was finished ( PSC-2, A9-intermedaite/(A9-intermediate+A9) < 2%). The mixture was quenched with saturated aqueous sodium bicarbonate solution (32L) and stirred for 30min at 0 °C. Phase was separated, and the aqueous phase was extracted with EA (3 x 20 L). The combined organic phase was washed with brine(20 L), dried over Na2SO4, and concentrated to a yellow oil which was purified by column (eluted with n-heptane:EA from 10:1 to 1:1) to give A9 (4.42 kg, assay 90%, yield 74 %) as pale yellow solid.
Step g
[00299] To a solution of A9 (4.0 kg, 14.74 mol, 1.0 eq) in DCM (40 L) cooled on an ice-bath was added DMP (9.36 kg, 23.58mol, 1.6eq) in portions, and it resulted in a suspension. After addition, the mixture stirred for 4 hours at 20-25°C. HPLC showed the reaction was finished (A9/(A9+A10)<2%). DCM (30L) was added at 0°C. After addition, the mixture was quenched with saturated aqueous Na2SO3 (20 L). The mixture was stirred for 30min at 0 °C, filtered and the white solid was washed with DCM (2 x15L). Phase was separated, and the organic phase was cooled to 0°C, to which was added saturated aqueous NaHCO3 (20L) and stirred for 1h. Phase was separated, and the organic phase was washed with brine(25L), dried over Na2SO4, and concentrated to a yellow oil which was purified by column (eluted with n-heptane:EA from 50:1 to 10:1) to give A10 (3.70 kg, assay 88%, ee value 95.3%, yield 82%) as white solid. 1H NMR (400 MHz, DMSO-d6) δ = 4.20 (d, J = 8.0 Hz,
1H), 3.98-3.67 (m, 4H), 3.08-2.90 (m, 2H), 1.54-1.39(m, 13H), 1.18 (d, J = 8.0 Hz, 3H).
Step h
[00300] To a solution of A10 (4.60 kg, 17.08 mol, 1.0 eq) in THF (40 L) was added
Ti(OEt)4 (15.58 kg, 68.32 mol, 4.0 eq) and (R)-t-Butyl sulfmamide (4.14 kg, 34.16 mol, 2.0 eq) at 25 °C. After addition, the mixture was heated to 70°C and stirred for 20h. HPLC showed the reaction was finished (PSC-l, A10/(A10+A12)<4%). The mixture was cooled to -30— 40°C, and MeOH (4 L) was added dropwise within 30 min and stirred for 1 h. 2M L1BH4 (8.1 L) solution was added dropwise to the reaction mixture at -40- -50°C and stirred for 1h. HPLC indicated all of imine was consumed (PSC-2, A12/(A12+A13)<1%). The mixture was warmed to -30 °C and stirred for 1h, then warmed to 0 °C within 2 h and stirred for 1h, then warmed to 20-25 °C and stirred for 30min. IP AC ( 25L) was added to above mixture, NaHCO3(5L) was added dropwise in about 1h at 25 °C and stirred for 30 min. The mixture was filtered under vacuum and the cake was washed with IP AC (8 x15L). The combined organic phase was washed with brine (25L), then evaporated under vacuum to get a solution of A13
(about 28kg) which was used for next step.
[00301] To a mixture of A13 in IPAC (about 28 kg, 17.08 mol, 1.0 eq) was added dropwise
4M HCl/IPA (8.54 L, 34.16 mol, 2.0 eq) at -5 °C and stirred for 5h at -5 °C. HPLC showed that A13 was consumed completely (A13/(A14+A13)<1%). MTBE (25 L) was added to above mixture within
30 min and stirred for 30 min at -5 °C .The solid was collected by vacuum filtration. The cake was washed with MTBE (2 x 2.5 L). The wet cake was used for next step directly.
Step j
[00302] The wet solid A14 (from 9.2 kg A10) was stirred in MTBE(76 L) at 25°C, then the
16% NaOH (9.84 kg) solution was added dropwise to the MTBE suspension while maintaining IT<10ºC. After addition, the mixture was stirred for 15 min and all solids were dissolved at 0°C. The organic phase was separated, and the aqueous phase was extracted with MTBE (2 x 20L). The combined organic phase was washed with brine (10 L) and evaporated under vacuum to remove all MTBE. ACN (24 L) was added to above residue, and the mixture was evaporated under vacuum to remove the organic solvents and yielded a crude A15 (5.42 kg, qnmr 90%, 18.04 mol, 1.0 eq). ACN (34.68 kg) was added to above residue and stirred for 10 min at 65°C. A solution of (-)-O-acetyl-D-mandelic acid (3.15kg,16.2 mol, 0.9 eq) in ACN(11.6 kg) was added drop-wise to the mixture (firstly added 1/3, stirred for 0.5 h, then added the others) over 3h. The mixture was stirred for 1 h at 65°C, then cooled to 25°C over 4h and stirred for l2h at 25°C . The solid was collected by vacuum filtration, and the cake was washed with pre-cooled ACN (2 x15kg) (PSC-1) and dried under vacuum to give
A16 (7.36 kg, yield 46% from A10 to A16). 1H NMR (400 MHz, DMSO-d6) δ = 7.43-7.29 (m, 5H),
5.58 (s, 2H), 4.12-4.07 (m, 1H), 3.75-3.65 (m, 3H), 3.51-3.49 (m, 1H), 3.18-3.17 (m, 1H), 2.84 (bs,
2H), 2.05 (s, 3H), 1.60-1.40 (m, 13H), 1.14-1.12 (d, J= 8.0 Hz, 3H).
Step k
[00303] To a solution of A16 (15 g) in MeOH (90 mL) was added dropwise 5N HC1/IPA
(45 mL) at room temperature within 15 minutes. After the addition, the mixture was stirred for 6 hours.
IP AC (180 mL) was added dropwise to above mixture within 1h at room temperature. The resulting mixture was stirred for another 30 minutes before it was cooled to 0-5 °C. The mixture was stirred at 0- 5 °C for another 2h and the precipitants were collected by filtration. The cake was washed with (45*2 mL) IP AC, dried under vacuum at 60 °C overnight to afford the product as a white solid. 1H NMR (400
MHz, DMSO-d6) δ = 9.37 (br s, 1H), 9.25 (br s, 1H), 8.42 (br s, 3H), 4.26 – 4.17 (m, 1H), 3.72 (ABq, J
= 9.1 Hz, 2H), 3.50 – 3.41 (m, 1H), 3.28 – 3.18 (m, 1H), 3.18 – 3.09 (m, 1H), 2.99 – 2.74 (m, 2H), 2.07 – 1.63 (m, 4H), 1.22 (d, J= 6.5 Hz, 3H).
Step l
[00304] To a mixture of A17 (10 g) and Z17a (9.5 g) in DMAC (60 mL) was added K2CO3
(22.5 g) and H2O (40 mL) at room temperature. The mixture was degassed with nitrogen and stirred at
90 °C overnight. The mixture was cooled to room temperature, diluted with Me-THF (500 mL) and
H2O (280 mL). The organic phase was separated and the aqueous phase was extracted with Me-THF
(300 mL*2). The combined organic phases were washed with brine (200 mL*3), concentrated under
vacuum to remove most of the solvent. The residue was diluted with IPA (60 mL) and H2O (20 mL), stirred at 50 °C for 1h, cooled to 5 °C within 3h, stirred at this temperature for 1h. The solid was collected by vacuum filtration, dried under vacuum to afford the product as a yellow solid (l2g,
87.4%). 1H NMR (400 MHz, DMSO-d6)δ = 7.64 (d, J= 6.2 Hz, 1H), 7.62 (s, 1H), 6.26 (s, 2H), 6.13 (s, 2H), 5.74 (d, J= 5.3 Hz, 1H), 4.12 – 4.02 (m, 1H), 3.90 – 3.78 (m, 2H), 3.67 (d, J= 8.4 Hz, 1H), 3.49 (d, J= 8.4 Hz, 1H), 3.33 (s, 2H), 2.91 (d, J= 5.1 Hz, 1H), 1.78 – 1.68 (m, 1H), 1.67 – 1.57 (m, 1H), 1.56 – 1.41 (m, 2H), 1.08 (d, J= 6.5 Hz, 3H).
Example 2
Formation of the succinate salt of the compound of the formula I:
[00305] The reaction is summarized by the following Reaction Scheme:
[00306] To a mixture of A18 (10 g) in MeOH (76 g) and H2O (24 g) was added succinic acid (2.94 g) at room temperature. The mixture was heated to 50 °C and stirred for 30 minutes to dissolve all solid. The solution was added to IPA (190 mL) at 60-65 °C. The resulting mixture was stirred at 60 °C >5 hours, cooled to -15 °C within 5 hours and stirred at this temperature >4 hours. The solid was collected by vacuum filtration, dried under vacuum to afford the product as an off-white solid(l0.8 g, 82.8%). 1H NMR (400 MHz, DMSO-d6)δ = 7.64 (d, J= 6.2 Hz, 1H), 7.63 (s, 1H), 6.26 (s, 2H), 6.16 (s, 2H), 5.74 (d, J= 5.3 Hz, 1H), 4.12 – 4.02 (m, 1H), 3.90 – 3.78 (m, 2H), 3.67 (d, J= 8.4 Hz, 1H), 3.49 (d, J= 8.4 Hz, 1H), 3.33 (s, 2H), 2.91 (d, J= 5.1 Hz, 1H), 2.34 (s, 4H), 1.71 – 1.60 (m, 4H), 1.13 (d, J = 6.5 Hz, 3H).
[00307] In a special variant, the reaction follows the following Reaction Scheme, also including an optional milling to yield the final product:
Example 3
Formation of the intermediate Z17a (3-((2-amino-3-chloropyridin-4-yl)thio)-6-chloropyrazin-2- amine). Variant 1:
[00308] The compound Z17a was obtained by reaction according to the following Reaction
Scheme:
[00309] In detail, the synthesis of Compound Z17a was carried out as follows:
Step a
[00310] Under nitrogen atmosphere, n-BuLi (2.5M, 7.6 L) was added dropwise to a solution of 3-chloro-2-fluoropyridine (2 kg) in THF (15 L) at -78°C. Then the resultant mixture was stirred for 1h. Then a solution of I2 (4.82 kg) in THF (6 L) was added dropwise. After addition, the reaction mixture was stirred for 30 min, and then quenched with sat. Na2SO3 (10 L), and warmed to 20- 25°C. Phase was separated. The aqueous phase was extracted with EA (2 x 10 L). The combined organic phase was washed with sat.Na2SO3 (2 x 8 L), brine (8 L), and dried over Na2SO4. The organic phase was concentrated under vacuum. The residue was slurried in MeOH (4 L), filtered, and dried to offer 3-chloro-2-fluoro-4-iodopyridine 1c (2.2 kg, yield 68%).
Step b
[00311] Into a solution of Compound 1c (8 kg) in DMSO (48 L) was passed through NH3
(gas) at 80 °C overnight. TLC showed the reaction was finished. The reaction mixture was cooled to RT. The reaction mixture was added to water (140 L). The solid was collected and washed with water (25 L), dried to afford Z17b (6.91 kg, yield 87%). 1H NMR (400 MHz, CDC13) δ = 7.61 (d, J= 6.8 Hz,
1H), 7.14 (s , J= 6.8 Hz, 1H), 5.09 (bs, 2H).
Step c
[00312] A solution of 2-amino-6-chloro-pyrazine la (1 kg, 7.69 mol) in DCM (15 L) was heated to reflux, to which was charged NBS (4l7g) in portions during 1 h. The reaction was cooled to room temperature. The reaction mixture was washed with water (3 L) and brine (3 L). The organic phase was evaporated, and the residue was purified by column chromatography to give product Z17f
(3-bromo-6-chloropyrazin-2-amine) (180 g, 11% yield).
[00313] To a solution of 3-bromo-6-chloropyrazin-2-amine Z17f (6.0 kg, 28.78 mol) in 1,4- Dioxane (40 L) was added Pd(OAc)2 (64.56 g, 287.6 mmol), Xantphos (333 g, 575.6 mmol), and DIPEA (7.44 kg, 57.56 mol) at room temperature under nitrogen. After another 30 minutes purging with nitrogen, methyl 3-mercaptopropanoate (3.81 kg, 31.70 mol) was added, resulting in darkening of the orange mixture. The mixture was heated to 90°C. HPLC showed complete conversion of the starting material. The mixture was allowed to cool to about room temperature, then diluted with EtOAc (40L). After aging for 30 min with stirring, the entire mixture was filtered and solids were washed with EtOAc (3 x 15L). The combined orange filtrate was concentrated to dryness and the solid residue was suspended in DCM (45 L). The mixture was heated to 35-40 °C and stirred for 1h until all solids were dissolved. Then n-heptane (45L) was added dropwise. Upon complete addition, the mixture was cooled to 15-20 °C with stirring for 1h. The solids were collected by vacuum filtration and solids were washed with cold 1:1 DCM/heptane (25 L), then heptane (25 L) (PSC-2). The solids were dried over the weekend to give Z17d (5.32 kg, yield 75%). 1H NMR (400 MHz, CDCl3) δ = 7.83 (s, 1H), 4.88 (bs,
2H), 3.73 (s, 3H), 3.47 (t, J= 9.2 Hz, 2H), 2.79 (t, J= 9.2 Hz, 2H).
Step e
[00314] To a solution of Z17d (8.0 kg, assay 95%, 30.68 mol) in THF (70 L) was added
EtONa (prepared from 776 g Na and 13.6 L EtOH) at room temperature and the mixture was stirred at
ambient temperature for 1 hour. The mixture was then concentrated to a wet yellow solid by rotary evaporation and the residue was suspended in DCM (40L). The mixture stirred under N2 for l6h. The solids were collected by vacuum filtration and the cake was washed with DCM (about 15 L) until the filtrate was colorless (PSC-2). The solids were then dried under vacuum to give Z17c (6.93 kg, qNMR
72 %, yield 88%). 1H NMR (400 MHz, D2O) δ = 7.37 (s, 1H).
Step f
[00315] To a mixture of Z17c (6.95 kg, assay 72%, 27.23 mol) in l,4-dioxane (72 L) was added Xantphos (233 g, 411 mmol, 0.015 eq), Pd2(dba)3 (186 g, 206 mmol, 0.0075 eq), Z17b (7.13 kg, 28.02 mol) and DIPEA (7.02 kg, 54.46 mol). The system was vacuated and purged with nitrogen gas three times. The mixture was stirred at 65 °C for 16 h under N2. The mixture was cooled to RT and water (50 L) was added, filtered. The cake was washed with EA (25 L). The filtrate was extracted with EA (4 x 20 L). The organic phase was concentrated in vacuum to offer the crude product which was combined with the cake. Then DCM (60 L) was added to the crude product and stirred at 25-30°C for l8h and then filtered. The filter cake was slurried with CH2Cl2 (30 L) for 4 hrs and filtered. The filter cake was slurred in CH2Cl2 (30 L) for 16 hrs and filtered. Then the filter cake was dried in vacuum to give Z17a (3-((2-amino-3-chloropyridin-4-yl)thio)-6-chloropyrazin-2-amine; 9.1 kg, 84 %) as light yellow solid. 1H NMR (400 MHz, DMSO-d6) δ = 7.89 (s, 1H), 7.7 (d, J= 7.6 Hz, 1H), 7.18 (bs, 2H), 6.40 (bs, 2H), 5.97 (d, J= 7.6 Hz, 1H).
Example 4
Alternative formation of the intermediate Z17a (here also named Y7a)
[00316] By way of alternative and according to a preferred reaction method, the compound of the formula Z17a was obtained according to the following Reaction Scheme:
In detail, the synthesis of the compound of the formula Y7a = Z17a was carried out as follows:
Step a
[00317] 2, 3, 5-trichloropyrazine (70.50 g, 384.36 mmol, 1 equiv) and ammonia solution
(25% wt, 364.00 g, 400 mL, 2.68 mol, 6.14 equiv) were added to a 1-L sealed reactor. The mixture was heated to 80 °C and stirred for 24 h, and the reaction was completed. The reaction mixture was cooled to 30 °C and filtered to give a brown filter cake. The brown filter cake was dissolved in acetone
(50 mL), and filtered. To the filtrate was added petroleum ether (300 mL). The suspension was stirred for 4 h, and filtered to give the crude product. The crude product was slurried in combined solvents of petroleum ether and acetone (10/1, 200 mL) and filtered to give the product Y7d (51.00 g, 307.91 mmol, 80% yield) as a light yellow solid. 1H NMR (400 MHz, DMSO-d6) δ = 7.63 (s, 1H).
[00318] To a 200 mL round bottom flask was added Na2S (10.816 g, 44wt% containing crystalline water, 60.978mmol) and toluene (100 mL). The mixture was heated to reflux, and water was removed with a Dean-Stark trap (about 5~6 mL water was distilled out). After cooling, the mixture was concentrated to dryness.
[00319] To above round bottom flask was added Y7d (5.000 g, 30.489mmol) and 2-methylbutan-2-ol (50 mL), the reaction was heated to reflux and stirred for 36 h. After cooling to 25 °C, the mixture was filtered. The solvent of the filtrate was exchanged with n-heptane (5 V, 3 times, based on Y7d), and finally concentrated to IV residue. THF (25 mL) was charged to the residue at 25 °C and stirred. The suspension was filtered and washed with THF/n-heptane (5 mL/5 mL) to give a brown solid (6.200 g).
[00320] To another 200 mL round bottom flask was added above brown solid (6.200 g),
10% brine (25 mL), Me-THF (30 mL) and n-Bu4NBr (9.829 g, 30.489 mmol). The mixture was stirred for 0.5 h at room temperature, and the phases were separated. The organic phase was washed with 20% brine (25 mL), and exchanged the solvent with iso-propanol (5 V *3 times, based on Y7d) to give the iso-propanol solution of Y7c (27.000g, 99.2% purity by HPLC area, 58.08% assay yield). 1H NMR (400 MHz, DMSO-d6) δ = 6.88 (s, 1H), 2.97 – 2.92 (m, 14H), 1.38 – 1.31 (m, 14H), 1.13 – 1.04 (m,
14H), 0.73 – 0.69 (t, 21H).
Step c
[00321] To a 25-mL round-bottom flask was added Y7c (4.7g, 23.27wt%, IPA solution from Step b, 2.723 mmol, 1.0 equiv), Y7b (1.052 g, 4.085 mmol, 1.5 equiv), l,lO-Phenanthroline (0.05 g, 0.272 mmol) and water (8 mL). The mixture was purged with nitrogen gas three times, and Cul (0.026 g, 0.136 mmol) was added under nitrogen atmosphere. The mixture was heated up to 65 °C and stirred for 3 h, and the reaction was completed. The reaction was cooled to room temperature and filtered, and the filter cake was washed with water (4 mL*3). The filter cake was slurried in MTBE (6 mL) for 30 min and filtered. The filter cake was washed with MTBE (6 mL) and dried to afford Y7a which is Z17a (565 mg, 72% yield).
[00322] Z17b is synthesized as described in Example 3 Step a and Step b.
Example 5
Alternative Synthesis of the intermediate Z17a:
[00323] According to another preferred method, the compound of the formula Z17a was obtained in accordance with the following Reaction Scheme:
[00324] The reactions were carried out as follows:
Step a
Y7d was synthesised as described in Example 4 step a.
[00325] To a three-necked round-bottle flask was added Y7d (200 mg, 1.22 mmol, 1 equiv), dioxane (4 mL). The solution was vacuated and purged with nitrogen gas three times. Xantphos (14mg, 0.024 mmol, 0.02 equiv), PdCl2(dppf) (8.9 mg, 0.012 mmol, 0.1 equiv), and DIPEA (0.32 g, 2.44 mmol, 2.0 equiv) were added under nitrogen atmosphere. The solution was heated to 85 °C for overnight. The reaction was cooled and evaporated. The residue was purified by column chromatography (eluent/ethyl acetate/heptane = 1/1) to give Z17d (259 mg, 0.99 mmol, 81%). 1H NMR (400 MHz, CDCl3) δ = 7.83 (s, 1H), 4.88 (bs, 2H), 3.73 (s, 3H), 3.47 (t, J= 9.2 Hz, 2H), 2.79 (t, J= 9.2 Hz, 2H).
[00326] The remaining steps were carried out as described in Example 4, Steps e and f, to yield Z17a. Z17b was synthesized as described in Example 3 Step a and Step b.
Example 6
(3S,4S)-8-(6-amino-5-((2-amino-3-chloropyridin-4-yl)thio)pyrazin-2-yl)-3-methyl-2-oxa-8- azaspiro[4.5]decan-4-amine. succinate (1:1) hemihydrate. modification (form) HA:Variant a)
[00327] 50 ml ethanol and 2.5 ml water were added to a 100ml flask containing 3.0 g of free base of 3S,4S)-8-(6-amino-5-((2-amino-3-chloropyridin-4-yl)thio)pyrazin-2-yl)-3-methyl-2-oxa-8-azaspiro[4.5]decan-4-amine (obtained as A18 for example as described in Example 1) and 848.0 mg of succinic acid. The mixture was heated to 50°C to generate a clear solution. The temperature was lowered to 15°C during a period of 3 hours. The solution was kept stirring at 15°C overnight.
Precipitated solid was separated via suction filtration and 50 ml of acetone was added to produce a suspension. The suspension was stirred at 50°C for 3 hours. The solid was separated with suction filtration and dried at room temperature under vacuum for 3 hours. Yield was about 60%.
[00328] The succinate appeared as a highly crystalline solid, with a melting point onset of
94.4°C and an accompanying enthalpy of 96 J/g. The succinate salt crystals showed aggregates of broken drusy tabular particles.
[00329] Variant b)
[00330] 14.34 g of 3S,4S)-8-(6-amino-5-((2-amino-3-chloropyridin-4-yl)thio)pyrazin-2-yl)- 3-methyl-2-oxa-8-azaspiro[4.5]decan-4-amine free form (obtained as A18 for example as described in Example 1) and 4.053 g of succinic acid were equilibrated in 100 mL 95% EtOH at 50°C. Add 5 mL of water into the system and heat to 70-75 °C. Add 95 mL of pure EtOH and heat for 30 min more. Stir over night at 25 oC. Filter the mixture wash with EtOH and dry under vacuum in an oven at room temperature. Yield is 87.5%.
PATENT
WO 2020065452
PATENT
PHARMACEUTICAL COMBINATION COMPRISING TNO155 AND NAZARTINIB
PAPER
Journal of Medicinal Chemistry (2020), 63(22), 13578-13594.
https://pubs.acs.org/doi/10.1021/acs.jmedchem.0c01170
SHP2 is a nonreceptor protein tyrosine phosphatase encoded by the PTPN11 gene and is involved in cell growth and differentiation via the MAPK signaling pathway. SHP2 also plays an important role in the programed cell death pathway (PD-1/PD-L1). As an oncoprotein as well as a potential immunomodulator, controlling SHP2 activity is of high therapeutic interest. As part of our comprehensive program targeting SHP2, we identified multiple allosteric binding modes of inhibition and optimized numerous chemical scaffolds in parallel. In this drug annotation report, we detail the identification and optimization of the pyrazine class of allosteric SHP2 inhibitors. Structure and property based drug design enabled the identification of protein–ligand interactions, potent cellular inhibition, control of physicochemical, pharmaceutical and selectivity properties, and potent in vivo antitumor activity. These studies culminated in the discovery of TNO155, (3S,4S)-8-(6-amino-5-((2-amino-3-chloropyridin-4-yl)thio)pyrazin-2-yl)-3-methyl-2-oxa-8-azaspiro[4.5]decan-4-amine (1), a highly potent, selective, orally efficacious, and first-in-class SHP2 inhibitor currently in clinical trials for cancer.

file:///C:/Users/Inspiron/Downloads/jm0c01170_si_001.pdf
(3S,4S)-8-(6-amino-5-((2-amino-3-chloropyridin-4-yl)thio)pyrazin-2-yl)-3-methyl-2-oxa-8- azaspiro[4.5]decan-4-amine (1):

Step a: A mixture of (3S,4S)-tert-butyl 4-((R)-1,1-dimethylethylsulfinamido)-3-methyl-2-oxa-8- azaspiro[4.5]decane-8-carboxylate (51 mg, 0.136 mmol) and HCl (4 M in dioxane, 340 L, 1.362 mmol) in MeOH (5 mL) was stirred for 1 h at 40 °C. After cooling to RT, the volatiles were removed under reduced pressure to give (3S,4S)-3-methyl-2-oxa-8-azaspiro[4.5]decane-4-amine which was used in next step without further purification. MS m/z 171.1 (M+H)+. Step b: A mixture of (3S,4S)-3-methyl-2-oxa-8-azaspiro[4.5]decane-4-amine crude, 3-((2-amino3-chloropyridin-4-yl)thio)-6-chloropyrazin-2-amine (35.5 mg, 0.123 mmol), and DIPEA (193 L, 1.11 mmol) in DMSO (600 L) was stirred for 16 h at 100 °C. After cooling to RT, the volatiles were removed under reduced pressure and the resulting residue was purified by HPLC (gradient elution 15-40% acetonitrile in water, 5 mM NH4OH modifier) to give (3S,4S)-8-(6-amino-5-((2-amino-3-chloropyridin-4-yl)thio)pyrazin-2-yl)-3-methyl-2-oxa-8-azaspiro[4.5]decan-4-amine (11 mg, 0.026 mmol). 1 H NMR (400 MHz, METHANOL-d4) δ ppm 7.67-7.47 (m, 2 H), 5.91 (d, J=5.5 Hz, 1 H), 4.22 (qd, J=6.4, 4.8 Hz, 1 H), 4.03 (ddt, J=13.5, 8.9, 4.7 Hz, 2 H), 3.86 (d, J=8.7 Hz, 1 H), 3.71 (d, J=8.7 Hz, 1 H), 3.37 (td, J=9.9, 4.9 Hz, 1 H), 3.29-3.23 (m, 1 H), 3.00 (d, J=5.0 Hz, 1H) 1.91-1.56 (m, 4 H), 1.21 (d, J=6.4 Hz, 3 H). HRMS calcd for C18H25ClN7OS (M+H)+ 422.1530, found 422.1514.
//////////TNO 155, CANCER
CILENGITIDE
| IUPAC Condensed | cyclo[Arg-Gly-Asp-D-Phe-N(Me)Val] |
|---|---|
| HELM | PEPTIDE1{R.G.D.[dF].[meV]}$PEPTIDE1,PEPTIDE1,5:R2-1:R1$$$ |
| IUPAC | cyclo[L-arginyl-glycyl-L-alpha-aspartyl-D-phenylalanyl-N-methyl-L-valyl] |
CILENGITIDE
- Molecular FormulaC27H40N8O7
- Average mass588.656 Da
2-[(2S,5R,8S,11S)-5-benzyl-11-[3-(diaminomethylideneamino)propyl]-7-methyl-3,6,9,12,15-pentaoxo-8-propan-2-yl-1,4,7,10,13-pentazacyclopentadec-2-yl]acetic acid188968-51-6[RN]
4EDF46E4GI
7823
циленгитид
سيلانجيتيد
西仑吉肽
EMD 121974, EMD-121974, UNII-4EDF46E4GI
Cilengitide has been in phase III clinical trials by Merck Serono and NCI for the treatment of glioblastoma multiforme. However, this research has been discontinued.
Cilengitide was originally developed by Merck KGaA in collaboration with the Technical University of Munich, then received orphan drug designation from FDA for the treatment of glioma in 2005.
Cilengitide (EMD 121974) is a molecule designed and synthesized at the Technical University Munich in collaboration with Merck KGaA in Darmstadt. It is based on the cyclic peptide cyclo(-RGDfV-), which is selective for αv integrins, which are important in angiogenesis (forming new blood vessels), and other aspects of tumor biology. Hence, it is under investigation for the treatment of glioblastoma, where it may act by inhibiting angiogenesis, and influencing tumor invasion and proliferation.[1][2]
The European Medicines Agency has granted cilengitide orphan drug status.[3]
Cilengitide seems to function by inhibiting the FAK/src/AKT pathway and inducing apoptosis in endothelial cells.[4] Preclinical studies in mice of cilengitide were able to demonstrate efficacious tumor regression.[4]
In a rat xenograft model, cilengitide was able to potentiate the cytotoxic effects of radiation when cilengitide was administered prior to radiation therapy.[5] When combined with radiation, inhibition of integrin expression by cilengitide synergistically improves the cytotoxic effects of ionizing radiation for glioblastoma.[5]
Clinical trials
Phase II studies were able to demonstrate that cilengitide as a potential monotherapy in patients with recurrent glioblastoma[6] with high intratumor drug levels when 2000 mg of cilengitide is given twice weekly.[7]
Cilengitide is well tolerated, in combination with radiation and temozolomide, at a dose of 2000 mg in patients with newly diagnosed glioblastoma, regardless of MGMT promoter status.[8] In a phase I/IIa study, the addition of cilengitide to the standard of care for newly diagnosed glioblastoma (surgical resection followed by temozolomide and radiation therapy) improves progression-free survival and overall survival in patients with MGMT promoter methylation.[9]
However, in a subsequent study, cilengitide does not seem to alter the pattern of glioblastoma progression,[10]
and in an EORTC phase III randomized, controlled, multicenter clinical trial, consisting of over 500 patients in 23 countries, the addition of cilengitide to the standard of care did not improve overall survival in patients with newly diagnosed glioblastoma and methylated MGMT promoter status [11] A phase II study, the CORE trial, is currently being conducted in patients with newly diagnosed glioblastoma and unmethylated MGMT promoter status.[12]
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////////////////////////////
SYN
Angewandte Chemie, International Edition, 55(4), 1540-1543; 2016

SYN
Chemistry – A European Journal, 16(18), 5385-5390, S5385/1-S5385/36; 2010
Reference:1. WO0047228A1 / US7115261B1.
2. US6001961A.Route 2
Reference:1. CN102731627A.PATENTWO/2021/224234ANTIVIRAL USE OF CILENGITIDEhttps://patentscope.wipo.int/search/en/detail.jsf?docId=WO2021224234&_cid=P20-KW0M52-85135-1
PATENThttps://patents.google.com/patent/CN102731627A/enEMD121974 (Cilengitide), the Chinese another name: ring (L-arginyl glycyl-L-aspartoyl-D-phenylalanyl-N-methyl-L-valyl) is an a kind of new classification cancer therapy drug of synthetic.Merkel company discovers that EMD121974 amalgamation radiotherapy (merging to reach assists TM to add radiotherapy) possibly prolong lifetime; Simultaneously integrate plain supressor antitumor drug as first; Got into the III clinical trial phase, its important mechanism is to grow targeting that the blood supply structure of nutrition, the growth of promotion cancer cell is provided in tumour and for tumour through line artery.The EMD121974 molecular formula is: C 27H 40N 8O 7, have following structure:
The preparation method of cyclic peptide mainly contains liquid phase synthesis process, solid phase synthesis precursor peptide cyclization process, process for solid phase synthesis in liquid phase at present; Wherein preceding two kinds of synthesis techniques all are the cyclisation in liquid phase of synthetic precursor peptide, and this method needs reactant in extremely rare solvent, to react (10 -3~10 -4Mol/L), and intermolecular be prone to react generation line style or cyclic polymer, greatly reduced the cyclisation yield, bring trouble for follow-up purifying, and in large-scale production, produce a large amount of waste liquids, be unfavorable for suitability for industrialized production.In conjunction with the structure of EMD121974, utilize the false rare principle of benefit of solid phase, developed a kind of efficient cyclization reaction, the cyclisation time shortens to 20%~30% of liquid phase cyclisation, and the 2%-8% of solvent as liquid phase used in reaction.Embodiment 1The preparation of Fmoc-L-Asp (OtBu)-Wang ResinThe Wang Resin that takes by weighing the 10g substitution degree and be 0.5mmol/g joins in the reactor drum, adds an amount of DCM, and swelling 30min takes out DCM; 6.17g Fmoc-L-Asp-OtBu, DIC 2.40ml, HOBT2.1g are dissolved among the 30ml DMF; At 0-5 ℃ of activation 15min, activation solution is joined in the reactor drum that contains Wang Resin, behind the reaction 10min; Add DMAP 0.18g again, at 0~30 ℃ of reaction 1~5h.After reaction finishes, add sealing Wang Resin unreacted hydroxylation reagent diacetyl oxide 1ml and pyridine 0.5ml, behind the capping 1h, DMF, DCM, the CH of 80ml used in washing successively 3OH, DMF washing 2,1,1,2 times, each 1min.Through detecting, obtain the Fmoc-L-Asp that substitution degree is 0.47mmol/g (OtBu)-Wang Resin.Embodiment 2The EMD121974 precursor:The preparation of A-Wang Resin (Fmoc-D-Phe-N-Me-L-Val-L-Arg (Mtr)-Gly-L-Asp (OtBu)-Wang Resin)Fmoc-L-Asp (OtBu)-Wang Resin is joined in the reactor drum, behind DMF swelling 30min, take out solvent, the piperidines-DMF that adds 80ml 25% reacts 5min, and 80ml DMF washs 1 time (3min), and the piperidines-DMF that adds 80ml 25% reacts 15min; DMF, DCM, the CH of 80ml used in washing successively 3OH, DMF washing 2,1,1,2 times, each 1min; With 4.45g Fmoc-Gly-OH, 5.68g HBTU, 2.03g HOBt, be dissolved among the DMF of 30ml, dissolve the back and added DIEA 2.45ml; 0~5 ℃ of activation 15min; Activation solution is joined in the above-mentioned reactor drum, and behind reaction 1-3h under 0~30 ℃, reaction end detects with ninhydrin method.Adopt aforesaid method coupling Fmoc-L-Arg (Mtr)-OH, Fmoc-N-Me-L-Val, Fmoc-D-Phe-OH successively, finally obtain Fmoc-D-Phe-N-Me-L-Val-L-Arg (Mtr)-Gly-L-Asp (OtBu)-Wang Resin.Embodiment 3EMD121974 precursor peptide: the preparation of B-Wang Resin (D-Phe-N-Me-L-Val-L-Arg (Mtr)-Gly-L-Asp-Wang Resin)With volume ratio is that piperidines-DMF of 25% is the Fmoc deprotection agent of Fmoc-D-Phe-N-Me-L-Val-L-Arg (Mtr)-Gly-L-Asp (OtBu)-Wang Resin; Add piperidines-DMF 80ml of 25% first time; Reaction 5min, 80ml DMF washs 1 time (3min), adds piperidines-DMF 80ml of 25% for the second time; Behind the reaction 15min, DMF, DCM, the CH of 80ml used in washing successively 3OH, DMF washing 2,1,1,2 times, each 1min gets D-Phe-N-Me-L-Val-L-Arg (Mtr)-Gly-L-Asp (OtBu)-Wang Resin after washing finishes.80% the PhOH-DCM solution that adds volume ratio and be 100ml takes off OtBu with the TFA of catalytic amount, reacts 8h; DMF, DCM, the CH of 80ml used in washing successively 3OH, DMF washing 2,1,1,2 times, each 1min gets D-Phe-N-Me-L-Val-L-Arg (Mtr)-Gly-L-Asp-Wang Resin.Embodiment 4The preparation of EMD121974-Wang Resin (Cyclo (D-Phe-N-Me-L-Val-L-Arg-Gly-L-Asp)-Wang Rsin)In above-mentioned reactor drum, add cyclization reagent 3.9g DPPA, 2.5ml DIEA (reactant cyclization reagent amount of substance ratio is 1: 3), at 10~40 ℃ of reaction 3h, the multiple cyclization reagent reaction 3~5h (reaction end detects with ninhydrin method) that throws once above-mentioned equivalent; DMF, DCM, the CH of 80ml used in washing successively 3OH washing 2,1,3 times, each 3min gets Cyclo (D-Phe-N-Me-L-Val-L-Arg-Gly-L-Asp)-Wang Rsin.Embodiment 5The preparation of EMD121974 (Cyclo (D-Phe-N-Me-L-Val-L-Arg-Gly-L-Asp))In above-mentioned reactor drum, add the TFA/H of lytic reagent 120ml again 2Behind O/TlS (volume ratio is 95: 2.5: 2.5) the reaction 3h, suction filtration is removed resin, and filtrating slowly joins in the no water-ice ether; Static 2-5h, high speed centrifugation obtain thick peptide, prepare through high-pressure liquid phase; Lyophilize gets smart EMD121974; Its purity>99.5%, single impurity<0.2%, total recovery reaches 63%.Choosing substitution degree in the present embodiment is the Wang Resin of 0.5mmol/g, and can also choose substitution degree is the arbitrary Wang Resin and Fmoc-L-Asp-OtBu prepared in reaction Fmoc-L-Asp (the OtBu)-Wang Resin of 0.4~0.9mmol/g scope.All can realize technical scheme of the present invention, and obtain technique effect of the present invention.Above content is an EMD121974 and become one of best preferred version of route; And to further explain that the present invention did; But can not assert that practical implementation of the present invention is only limited to these explanations; Under the prerequisite that does not break away from the present invention’s design, can also make some simple deductions and replacement, all should be regarded as protection domain of the present invention.
CLIPhttps://www.eurekaselect.net/article/2607Cilengitide, a cyclic RGD pentapeptide, is currently in clinical phase III for treatment of glioblastomas and in phase II for several other tumors. This drug is the first anti-angiogenic small molecule targeting the integrins αvβ3, αvβ5 and α5β1. It was developed by us in the early 90s by a novel procedure, the spatial screening. This strategy resulted in c(RGDfV), the first superactive αvβ3 inhibitor (100 to 1000 times increased activity over the linear reference peptides), which in addition exhibited high selectivity against the platelet receptor αIIbβ3. This cyclic peptide was later modified by N-methylation of one peptide bond to yield an even greater antagonistic activity in c(RGDf(NMe)V). This peptide was then dubbed Cilengitide and is currently developed as drug by the company Merck-Serono (Germany). This article describes the chemical development of Cilengitide, the biochemical background of its activity and a short review about the present clinical trials. The positive anti-angiogenic effects in cancer treatment can be further increased by combination with “classical” anti-cancer therapies. Several clinical trials in this direction are under investigation.
CLIPJournal of Protein Chemistry

Schematic of the one-step chemoenzymatic synthesis of cilengitide using wild-type Mcy TE. (1) The chemically synthesised (SPPS, solid-phase peptide synthesis) mimetic substrate was condensed with benzyl mercaptane to produce pentapeptide thioester (pentapeptide-BMT). (2) Models of the substrate-O-TE acyl enzyme intermediate are marked with brackets (protein data bank, 1JMK). (3) Mechanism of TE domain catalysis: a pentapeptide -O-TE acyl-enzyme intermediate is formed by transfer of the peptidyl chain from the phosphopantethiene of the terminal peptidyl carrier protein (PCP), which was substituted by benzyl mercaptane, to the active site serine of the TE domain. For hydrolyzing TE domains, the intermediate is captured by water, generating the linear peptide; for cyclizing TE domains, an intramolecular nucleophile captures the intermediate, resulting in “cilengitide”
PATENTWO 9745447
WO 9745137
DE 19534177
WO 2000053627
WO 2000047228
US 20040063790
WO 2009124754
WO 2011079015
WO 2011069629
WO 2011144756WO 2016059622
PATENTWO 2012062777https://patents.google.com/patent/WO2012062777A1/enSynthesis of cyclic peptidesCyclo[-Arg-Gly-Asp- 6 or 7 -Phe-Val-Ala-] (1 and 2). Resin loading. 2- chlorotrityl chloride-resin ( 1 50 m g , 1 .5m m ol/g ) was p laced i n a 20 m l polypropylene syringe fitted with a polyethylene filter disk. The resin was then washed with CH2CI2 (5 χ 0.5 min), and a solution of Fmoc-L-Gly-OH (334 mg, 1 .125 mmol, 5 equiv) and DIEA (239 μΙ_, 6.25 equiv) in CH2CI2 (2.5 ml_) was added. The mixture was then stirred for 15 min. Extra DIEA (239 μΙ_, total 12.5 mmol) was added, and the mixture was stirred for an additional 45 min. The reaction was stopped by adding 3 χ DCM/ MeOH/ DIEA (85: 10:5) and stirring for 1 0 m in. The Fmoc-L-Gly-O-resin product was subjected to the following washings/treatments with CH2CI2 (3 χ 0.5 min), DMF (3 χ 0.5 min), piperidine and DMF (5 χ 0.5 min). The loading was 0.50 mmol/g, as calculated by Fmoc determination.Peptide coupling. Fmoc-L-Arg(Pbf)-OH (243 mg, 0.375 mmol, 5 equiv), Fmoc- L-Ala-OH (1 17 mg, 0.375 mmol, 5 equiv), Fmoc-L-Val-OH ( 127 mg, 0.375 mmol, 5 equiv) and Fmoc- L-Phe-OH ( 145 mg, 0.375 mmol, 5 equiv) were added sequentially to the above obtained H-L-Gly-O-resin using HCTU (155 mg, 0.375 mmol, 5 equiv), HOBt (50 mg, 0.375 mmol, 5 equiv) and DIEA (127 μΙ_, 0.75 mmol, 10 equiv) in DMF (2.5 ml_). In all cases, after 90 min of coupling, the ninhydrin test was negative. Removal of Fmoc group and washings were performed as described in general procedures. /V-Alloc-thiazole 6 or 7 (92 mg, 0.375 mmol, 5 equiv) was coupled with HATU (143 mg, 0.375 mmol, 5 equiv), HOAt (51 mg, 0.375 mmol, 5 equiv) and DIEA (127 μΙ_, 0.75 mmol, 10 equiv) for 90 min. This coupling was repeated twice in the same conditions. The Alloc group of the peptide resin was removed with Pd (PPh3)4 (9 mg, 0.0075 mmol, 0.1 equiv) in the presence of PhSiH3 (92.5 μΙ_, 0.75 mmol, 10 equiv) in DCM for 20 min. This deprotection was repeated three times in the same conditions. After washing, the resin was treated with dry THF (2ml_) for 15 min. Meanwhile, Fmoc-L-Asp(tBu)-OH (154 mg, 0.375 mmol, 5 equiv) was added to a 68 mM solution of triphosgene in dry THF (1 .15 equiv). Sym-collidine (99.5 μΙ_, 0.75 mmol, 10 equiv) was added to the clear solution, upon which a precipitate of collidinium chloride was formed. DIEA (102 μΙ_, 0.6 mmol, 8 equiv) was added to the resin, immediately followed by addition of the suspension. This coupling was repeated four times in the same conditions. The reaction mixture was stirred at 50 °C during 48 h.Peptide cleavage. Following Fmoc deprotection, the peptidyl-resin was treated with TFA-CH2CI2 (1 :99) (5 χ 30 s). The filtrate was collected on H20 (4 ml_) and the H20 was partially removed under reduced pressure. MeCN was then added to dissolve solid that formed during the removal of H20, and the solution was lyophilized to give 12 mg and 10 mg of the linear compounds 28 and 29 respectively with a purity of > 91 % as checked by HPLC (Column A, Rt 7.43 min and Rt 7.38 min respectively, linear gradient 35%-40% ACN in 15 min.)], which was used without further purification. MALDI-TOF-MS calculated for C50H71 N11 O13S2 1098.29; found mlz 1099.29 [M + H]+, 1 121 .28 [M + Na]+, 1 137.39 [M + K]+.Synthesis in solution. Cyclization. The protected linear peptides 28 and 29 were dissolved in DMF (1 L, 10“4 M), and HOAt (9.6 mg, 0.07 mmol, 5 equiv), DIPEA (24 μΙ_, 0.14 mmol, 10 equiv), and PyAOP (36.6 mg, 0.07 mmol, 5 equiv) were added. The mixture was stirred for 24 h at room temperature, and the course of the cyclization step was then checked by HPLC (Column A, Rt 1 1 -67 min and Rt 10.70 min respectively, linear gradient 45%-55% ACN in 15 min.). The solvent was removed by evaporation under reduced pressure and the protected cycle 30 and 31 were used in the next step without further purification. MALDI-TOF-MS calculated for C50H69N11 O12S2 1080.28; found mlz 1081 .28 [M + H]+, 1 103.27 [M + Na]+, 1 1 19.38 [M + K]+.Side chain deprotection. The protected cyclopeptides 30 and 31 (14.7 mg, 19.04 pmol) were treated with TFA-H20 (95: 5) during 1 h. The solvent was removed by evaporation under reduced pressure.Peptide purification. The crude product was purified by HPLC (Symmetry C8 5 μη-Ί, 30 mm x 100 mm), gradient of MeCN (30% to 75% in 15 min) MeCN (+0.05% TFA) in water (+0.05% TFA), 20 mL/min, detection at 220 nm, to give the cyclopeptides 1 and 2 (4.5 mg, 5.8 pmol and 6.5 mg, 8.37 pmol, 7.7% and 12% yield respectively). The products were characterized by HPLC (Rt 8.99 min, and Rt 8.02 min Column A, respectively, linear gradient 0%-100% ACN in 1 5 min. ) and by MALDI-TOF-MS: calculated for C33H45N11 O9S 771 .84; found mlz 772.84 [M + H]+, 794.83 [M + Na]+, 810.94 [M + K]+.Cyc/o-[Arg-Gly-Asp-Thz1X-] (3). General procedure for cyclopeptide synthesis. Solid phase synthesis: The synthesis of the linear peptide H- Asp(tBu)-XX-Arg(Pbf)-Gly-OH was performed using Fmoc-based solid phase peptide synthesis with 2-chlorotrityl chloride resin (2.0 g, 3.2 mmol).Resin loading: Fmoc-Gly-OH (594 mg, 2.0 mmol) was attached to the resin with DIPEA in DCM at room temperature for 1 .5 h. The remaining trityl groups were capped adding 0.5 mL of MeOH for 30 min. After that, the resin was filtered and washed with DCM (2x), DMF (2x). The loading of the resin was determined by titration of the Fmoc group (Chan WC and White PD. Fmoc Solid Phase Peptide Synthesis. Oxford University Press: New York, 2000). The final loading was 2.0 mmol/g. The Fmoc group was eliminated by treatment with 20% piperidine in DMF (2X10 min). The resin was washed with DMF (3x), DCM (3x). Peptide coupling: Fmoc-Arg(Pbf)-OH (5.19 g, 8.0 mmol), DIPCDI (1.23 mL, 8.0 mmol) and HOBt (1.08 g, 8.0 mmol) were dissolved in DMF and added to the resin for 1 .5 h. The end of the coupling was monitored by ninhydrin test (free amine group) (Kaiser E et al. Anal Biochem 1970, 34:595-598). The resin was filtered and washed with DMF (3X) and DCM (3X). The Fmoc group was eliminated with 20 % piperidine in DMF (2X10 min).The coupling of the thiazole module was carried out with 8 (1 .14 g, 3.0 mmol), PyAOP (1 .56 g, 3.0 mmol) and DIPEA (1 .02 mL, 6.0 mmol) in DMF for 1 .5 h. The completion of the reaction was checked with the ninhydrin test. Finally the deprotection of the amine and coupling of the Fmoc-Asp(‘Bu)-OH were carried out under the same conditions of the second amino acid.Peptide cleavage: The resin bound peptide was treated with 2% TFA in DCM (6 x 30 sec.) The resin was washed with DCM and the combined solution was evaporated under vacuum with Et20 several times, furnishing the linear peptide 32 as a white solid. The peptide was used for the next step without purification.H PLC (gradient 20 to 80% of CH3CN in 1 5 m in): tR= 8.33 min. HPLC-MS (ES(+)): m/z 795.3.Synthesis in solution. Cyclization: The product 32 (200 mg, 0.251 mmol) was dissolved in anhydrous DMF (50 mL, 5 mM), PyAOP (262 mg, 0.503 mmol) and DIPEA (213 μί, 1 .255 mmol) were added. The reaction was monitored by HPLC. Once the reaction was finished, the DMF was evaporated under vacuum. The crude was dissolved in AcOEt and the solution was washed with NH4CISat and Na2CO3 sat. The organic layer was collected, dried over Na2SO4, filtered and concentrated under vacuum. The peptide was purified by flash chromatography (CHCIs/MeOH 8:2) furnishing the protected cyclic peptide 33 as a white solid (1 56 mg, XX%). HPLC (gradient 40 to 90% of CH3CN in 1 5 min): tR= 8.86 min. HPLC-MS (ES(+)): m/z 778.2Side chain deprotection: The protected peptide 33 (125 mg, XX mmol), was treated with 25 mL of a solution of TFA H2O (95:5). After 3 h, the solvent was evaporated under vacuum and the residue was precipitated with Et2O (4X). The Et2O solution was discarded and the white solid was lyophilized to afford 3 55 mg (XX%).
Peptide purification. The end product 3 was dissolved in 5 ml MilliQ water and it was filtered through a 0.2 pm filter. The cyclic peptide was purified by semipreparative RP-HPLC using acetronitrile (0.05% TFA)/water (0.1 % TFA). The HPLC sample was vacuum concentred and transformed into the hydrochloride salt lyophilized with water with 0.05% HCI.1H-NMR (500 MHz, H20:D20-d2 9: 1 , 278 K): δ = 9.29 (t, NH Gly), 9.20 (d, J = 7.24 Hz, NH Asp), 8.90 (t, J = 5.89/5.89 Hz, NH Thz), 8.46 (d, J = 8.93 Hz, NH Arg), 7.79 (s, CH Thz), 7.22 (t, J = 5.39/5.39 Hz, ΝΗε Arg), 4.75 (m, CHa Arg), 4.63 (m, CHa Asp), 4.04 (dd, J = 3.35/14.90 Hz, CHa Gly), 3.82 (dd, J = 6.69/14.96 Hz, CHa Gly), 3.17 (m, CH25 Arg), 2.89 (m, CH2p Asp), 1 .92 (m, CH p Arg), 1 .82 (m, CHP Arg), 1 .63 (m, CH2 Arg). HPLC (gradient 0 to 20% of CH3CN in 15 min): tR= 10.52 m in. HRMS (E IS) m/z calculated 468.1540

found 469.16099 (M+H)+.Cyc/o-[Arg-Gly-Asp-Thz2X-] (4). The cyclopeptide 4 was prepared according to the process followed for 3 and using bithiazole 9 (XX mg, YY mmol) instead of 8. The linear peptide 34: HPLC (gradient 0 to 100% CH3CN in 15 min.): tR = 10.34 min, HPLC-MS (ES(+)): m/z 877.81 . The protected peptide 35: HPLC (gradient 0 to 100% CH3CN in 15 min.): tR = 13.91 min, HPLC-MS (ES(+)): m/z 860.54. The final peptide 4: 1H-NMR (500 MHz, H20:D20-d2 9: 1 , 298 K): δ = 8.93 (sbroad, NH Gly), 8.82 (d, J = 7.62 Hz, NH Asp), 8.75 (t, J = 5.69/5.69 Hz, NH Thz), 8.51 (d, J = 7.62 Hz, NH Arg), 8.05 (s, CH Thz1), 7.50 (s, CH Thz2), 7.19 (t, J = 5.38/5.38 Hz, ΝΗε Arg), 4.13 (dd, J = 5.82/14.24 Hz, CH Gly), 3.87 (dd, J = 5.96/15.69 Hz, CH Gly), 3.21 (m , CH25 Arg), 2.94 (m, CH2p Asp), 1 .95 (m , CHP Arg), 1 .87 (m , CHP Arg), 1 .68 (m , CH2y Arg). HPLC (gradient 1 0 to 25% of CH3CN in 1 5 m in): tR = 8.73 min. HRMS (EIS) m/z calculated 551 .1369 (C2oH25N906S2) found 552.14392 (2M+2H)+.Cyc/o-[Arg-Gly-Asp-Thz3X-] (5). The cyclopeptide 5 was prepared according to the process for 3 and using trithiazole 10 (XX mg, YY mmol) instead of 8. The linear peptide 36: HPLC (gradient 20 to 80% of CH3CN in 15 min.): tR = 7.60 min, HPLC-MS (ES(+)): m/z 961 .23. The protected peptide 37: HPLC (gradient 20 to 80% of CH3CN in 15 m in. ): tR = 1 3.13 min, HPLC-MS (ES(+)): m/z 944.3. The final peptide 5: HPLC (gradient 10 to 30% CH3CN in 15 m in): tR = 8.26 m in. HRMS (E IS) m/z calculated 634.1 1 99 (C23H26N10O6S3) found 635.12683 (2M+2H)+. 1H-NMR (500 MHz, DMSO-d6 298 K): δ = 9.21 (t, J = 5.4, NH Gly), 8.72 (m, NH Asp + NH Thz), 8.37 (s, CH Thz1), 7.96 (d, J = 9.2, NHa Arg), 7.77 (s, CH Thz2), 7.68 (t, J = 6.0, ΝΗε Arg), 7.23 (s, CH Thz3), 4.83 (dd, J = 14.3, 8.5, CHa Arg), 4.72 (dd, J = 16.3, 6.6, CH Thz), 4.59 (m, CH Thz + CHa Asp), 3.89 (d, J = 1 1 .5, CH Gly), 3.59 (d, J = 9.7, CH Gly), 3.13 (dd, J = 12.6, 6.3, CH25 Arg), 2.81 (dd, J = 16.3, 4.3, CHP Asp), 2.58 (dd, J = 16.5, 8.7, CHP Asp), 1 .82 (m, CHP Arg), 1 .71 (m, CHP Arg), 1 .49 (m, CH2y Arg).Cilengitide. The cilengitide was prepared according to the method described in Dechantsreiter MA et al. (J Med Chem 1999, 42:3033-3040). 1H- NMR (500 MHz, H20:D20-d2 9: 1 , 298 K): δ = 8.55 (d, J = 8.06 Hz, NH Asp), 8.37 (d, J = 7.28 Hz, NH Arg), 8.13 ( d, J = 9.19 Hz, NH Phe), 7.97 (m, NH Gly), 7.34 (m, 2H, C6H5 Phe), 7.26 (m, 3H, C6H5 Phe), 7.22 (t, J = 5.53/5.53 Hz, ΝΗε Arg), 5.19 (dd, J = 8.58/16.02 Hz, CHa Phe), 4.56 (dd, J = 7.45/- Hz, CHa Asp), 4.34 (d, J = 10.89 Hz, CHa MeVal), 4.12 (dd, J = 7.80/14.63 Hz, CH Gly), 3.95 (dd, J = 6.84/15.33 Hz, CHa Arg), 3.54 (dd, J = 3.37/14.60 Hz, CH Gly), 3.20 (m , CH25 Arg), 3.02 (m, CH2p Phe), 2.88 (s, CH3 MeVal), 2.84 (dd, J = 7.26/16.68 Hz, CHP Asp), 2.63 (dd, J = 7.60/16.54 Hz, CHP Asp), 2.06 (m, CHP Val), 1 .91 (m, CH2p Arg), 1 .57 (m, CH2 Asp), 0.88 (d, J = 6.55 Hz, CH3 Val1), 0.56 (d, J = 6.49 Hz, CH3 Val2).
PAPERJournal of medicinal chemistry (1999), 42(16), 3033-40.Peptide Science (2001), Volume Date2000, 37th, 249-250. Current opinion in investigational drugs (London, England : 2000) (2003), 4(6), 741-5. Journal of medicinal chemistry (2005), 48(24), 7675-87.Peptide Science (2006), 43rd, 215-216Angewandte Chemie, International Edition (2010), 49(15), 2732-2737, S2732/1-S2732/53.Accounts of Chemical Research (2017), 50(7), 1541-1556.
References
- ^ Burke PA, DeNardo SJ, Miers LA, Lamborn KR, Matzku S, DeNardo GL (August 2002). “Cilengitide targeting of alpha(v)beta(3) integrin receptor synergizes with radioimmunotherapy to increase efficacy and apoptosis in breast cancer xenografts”. Cancer Research. 62 (15): 4263–72. PMID 12154028.
- ^ Goodman SL, Hölzemann G, Sulyok GA, Kessler H (February 2002). “Nanomolar small molecule inhibitors for alphav(beta)6, alphav(beta)5, and alphav(beta)3 integrins”. Journal of Medicinal Chemistry. 45 (5): 1045–51. doi:10.1021/jm0102598. PMID 11855984.
- ^ Spreitzer H (October 27, 2008). “Neue Wirkstoffe – Cilengitide”. Österreichische Apothekerzeitung (in German) (22/2008): 1136–7.
- ^ Jump up to:a b Yamada S, Bu XY, Khankaldyyan V, Gonzales-Gomez I, McComb JG, Laug WE (December 2006). “Effect of the angiogenesis inhibitor Cilengitide (EMD 121974) on glioblastoma growth in nude mice”. Neurosurgery. 59 (6): 1304–12, discussion 1312. doi:10.1227/01.NEU.0000245622.70344.BE. PMID 17277694. S2CID 19861713.
- ^ Jump up to:a b Mikkelsen T, Brodie C, Finniss S, Berens ME, Rennert JL, Nelson K, Lemke N, Brown SL, Hahn D, Neuteboom B, Goodman SL (June 2009). “Radiation sensitization of glioblastoma by cilengitide has unanticipated schedule-dependency”. International Journal of Cancer. 124 (11): 2719–27. doi:10.1002/ijc.24240. PMID 19199360.
- ^ Reardon DA, Fink KL, Mikkelsen T, Cloughesy TF, O’Neill A, Plotkin S, et al. (December 2008). “Randomized phase II study of cilengitide, an integrin-targeting arginine-glycine-aspartic acid peptide, in recurrent glioblastoma multiforme”. Journal of Clinical Oncology. 26 (34): 5610–7. CiteSeerX 10.1.1.688.8987. doi:10.1200/JCO.2008.16.7510. PMID 18981465.
- ^ Gilbert MR, Kuhn J, Lamborn KR, Lieberman F, Wen PY, Mehta M, Cloughesy T, Lassman AB, Deangelis LM, Chang S, Prados M (January 2012). “Cilengitide in patients with recurrent glioblastoma: the results of NABTC 03-02, a phase II trial with measures of treatment delivery”. Journal of Neuro-Oncology. 106 (1): 147–53. doi:10.1007/s11060-011-0650-1. PMC 4351869. PMID 21739168.
- ^ Nabors LB, Mikkelsen T, Hegi ME, Ye X, Batchelor T, Lesser G, Peereboom D, Rosenfeld MR, Olsen J, Brem S, Fisher JD, Grossman SA (November 2012). “A safety run-in and randomized phase 2 study of cilengitide combined with chemoradiation for newly diagnosed glioblastoma (NABTT 0306)”. Cancer. 118 (22): 5601–7. doi:10.1002/cncr.27585. PMC 3423527. PMID 22517399.
- ^ Stupp R, Hegi ME, Neyns B, Goldbrunner R, Schlegel U, Clement PM, et al. (June 2010). “Phase I/IIa study of cilengitide and temozolomide with concomitant radiotherapy followed by cilengitide and temozolomide maintenance therapy in patients with newly diagnosed glioblastoma” (PDF). Journal of Clinical Oncology. 28(16): 2712–8. doi:10.1200/JCO.2009.26.6650. PMID 20439646.
- ^ Eisele G, Wick A, Eisele AC, Clément PM, Tonn J, Tabatabai G, et al. (March 2014). “Cilengitide treatment of newly diagnosed glioblastoma patients does not alter patterns of progression”(PDF). Journal of Neuro-Oncology. 117 (1): 141–5. doi:10.1007/s11060-014-1365-x. PMID 24442484. S2CID 21636884.
- ^ Merck Group. “Phase III Trial of Cilengitide Did Not Meet Primary Endpoint in Patients With Newly Diagnosed Glioblastoma, Date accessed: 3/24/2014.”
- ^ ASCO Meeting Library. [1] “Cilengitide combined with standard treatment for patients with newly diagnosed glioblastoma and methylated O6-methylguanine-DNA methyltransferase (MGMT) gene promoter: Key results of the multicenter, randomized, open-label, controlled, phase III CENTRIC study, Date accessed: 3/24/2014
| Names | |
|---|---|
| IUPAC name2-[(2S,5R,8S,11S)-5-benzyl-11-{3-[(diaminomethylidene)amino]propyl}-7-methyl-3,6,9,12,15-pentaoxo-8-(propan-2-yl)-1,4,7,10,13-pentaazacyclopentadecan-2-yl]acetic acid | |
| Identifiers | |
| CAS Number | 188968-51-6 |
| 3D model (JSmol) | Interactive image |
| ChEMBL | ChEMBL429876 |
| ChemSpider | 154046 |
| IUPHAR/BPS | 6597 |
| KEGG | D03497 |
| MeSH | Cilengitide |
| PubChem CID | 176873 |
| UNII | 4EDF46E4GI |
| CompTox Dashboard (EPA) | DTXSID9044035 |
| showInChI | |
| showSMILES | |
| Properties | |
| Chemical formula | C27H40N8O7 |
| Molar mass | 588.656 g/mol |
| Density | 1.417 g/mL |
| Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa). | |
| Infobox references |
/////////CILENGITIDE, циленгитид , سيلانجيتيد ,西仑吉肽 , PHASE 3, EMD 121974, EMD-121974, UNII-4EDF46E4GI, orphan drug , MERCK, glioblastoma,
CC(C)C1C(=O)NC(C(=O)NCC(=O)NC(C(=O)NC(C(=O)N1C)CC2=CC=CC=C2)CC(=O)O)CCCN=C(N)N

NEW DRUG APPROVALS
ONE TIME
$10.00
DR ANTHONY MELVIN CRASTO

ORGANIC SPECTROSCOPY


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}