
FLAGS AND HITS
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO FACEBOOK
...................................................................Join me on twitter
FACEBOOK
...................................................................Join me on twitter
 ..................................................................Join me on google plus
..................................................................Join me on google plus
 Googleplus
GoogleplusMYSELF

Ipragliflozin

Ipragliflozin
ASP-1941 , 1(S)-[3-(1-benzothien-2-ylmethyl)-4-fluorophenyl]-1-deoxy-beta-D-glucopyranose L-proline cocrystal

Kotobuki (Originator)
| (1S)-1,5-Anhydro-1-C-[3-[(1-benzothiophen-2-yl)methyl]-4-fluorophenyl]-D-glucitol |
| Molecular Formula | C21H21FO5S | |
| Molecular Weight | 404.45 | |
| CAS Registry Number | 761423-87-4 |
Ipragliflozin (formerly ASP1941) has been filed in Japan on the back of phase III trials which showed that it could provide significant reductions in glycated haemoglobin levels (HbA1c) levels – a marker of glucose control over time – compared to placebo
According to Astellas’ latest R&D pipeline update in February 2013, Astellas is developing ipragliflozin only in Japan. The same document in August 2012 indicated it was also carrying out phase II studies with the drug in the US and Japan.
Astellas Pharma Inc.: Submits Application for Marketing Approval of
Ipragliflozin (ASP1941), SGLT2 Inhibitor for Treatment of
Type 2 Diabetes, in Japan
TOKYO, March 13, 2013 – Astellas Pharma Inc. (“Astellas”; Tokyo:4503; President and CEO:
Yoshihiko Hatanaka) announced today that it has submitted a market authorization application for aSGLT2 inhibitoripragliflozin (generic name; development code: ASP1941) to the Ministry of Health, Labour and Welfare in Japan seeking an approval forthe indication of type 2 diabetes.
Ipragliflozin is a selective SGLT2 (sodium-glucose co-transporter 2)inhibitor discovered through research collaboration with Kotobuki Pharmaceutical Co., Ltd. SGLTs are membrane proteins that
exist on the cell surface and transfer glucose into cells. SGLT2 is a subtype of the sodium-glucose co-transporters and plays a key role in the reuptake of glucose in the proximal tubule of the kidneys.
Ipragliflozin reduces blood glucose levels by inhibiting the reuptake of glucose.
In the Phase III pivotal study in monotherapy for type 2 diabetesin Japan, ipragliflozin
demonstrated significant decreases of HbA1c, an index of glycemic control, in change from baseline compared to placebo. Based on the safety resultsin this study, ipragliflozin was safe and well tolerated. Patients with type 2 diabetes generally need combination therapy, so it is important
for a novel oral hypoglycemic agent to be safe to use with existing diabetes therapies. In this regard, Astellas has conducted six Phase III studies to investigate the safety and efficacy of ipragliflozin
used in combination with other hypoglycemic agentsfor a long term period. In these Phase IIIstudies, effectiveness and favorable safety of ipragliflozin was confirmed even in combination with
other hypoglycemic agents.
Astellas expects to provide an additional therapeutic option and further contribute to the treatment of type 2 diabetes by introducing ipragliflozin, an oral hypoglycemic agent with a novel mechanism
of action, into the Japanese market.
About Type 2 Diabetes
Diabetes (medically known as diabetes mellitus) is a disorder in which the body has difficulty regulating its blood glucose (sugar) level. There are two major types of diabetes: type 1 and type 2.
Type 2 diabetes (formerly called non-insulin-dependent diabetes mellitus or adult-onset diabetes) is a disorder that is characterized by high blood glucose in the context of insulin resistance and relative insulin deficiency. Patients are instructed to increase exercise and diet restrictions, but most
require treatment with an anti-diabetic agent to control blood glucose.
structure:

The compound and methods of its synthesis are described in WO 2004/080990, WO 2005/012326 and WO 2007/114475 for example.
The gluconolactone method: In 1988 and 1989 a general method was reported to prepare C-arylglucosides from tetra-6>-benzyl protected gluconolactone, which is an oxidized derivative of glucose (see J. Org. Chem. 1988, 53, 752-753 and J. Org. Chem. 1989, 54, 610- 612). The method comprises: 1) addition of an aryllithium derivative to the hydroxy-protected gluconolactone to form a hemiketal (a.k.ci., a lactol), and 2) reduction of the resultant hemiketal with triethylsilane in the presence of boron trifluoride etherate. Disadvantages of this classical, but very commonly applied method for β-C-arylglucoside synthesis include:
1) poor “redox economy” (see J. Am. Chem. Soc. 2008, 130, 17938-17954 and Anderson, N. G. Practical Process Research & Development, 1st Ed.; Academic Press, 2000 (ISBN- 10: 0120594757); pg 38)— that is, the oxidation state of the carbon atom at CI, with respect to glucose, is oxidized in the gluconolactone and then following the arylation step is reduced to provide the requisite oxidation state of the final product. 2) due to a lack of stereospecificity, the desired β-C-arylglucoside is formed along with the undesired a-C-arylglucoside stereoisomer (this has been partially addressed by the use of hindered trialkylsilane reducing agents (see Tetrahedron: Asymmetry 2003, 14, 3243-3247) or by conversion of the hemiketal to a methyl ketal prior to reduction (see J. Org. Chem. 2007, 72, 9746-9749 and U.S. Patent 7,375,213)).
Oxidation Reduction

Glucose Gluconoloctone Hemiketal a-anomer β-anomer
R = protecting group
The metalated glucal method: U.S. Patent 7,847,074 discloses preparation of SGLT2 inhibitors that involves the coupling of a hydroxy-protected glucal that is metalated at CI with an aryl halide in the presence of a transition metal catalyst. Following the coupling step, the requisite formal addition of water to the C-arylglucal double bond to provide the desired C-aryl glucoside is effected using i) hydroboration and oxidation, or ii) epoxidation and reduction, or iii) dihydroxylation and reduction. In each case, the metalated glucal method represents poor redox economy because oxidation and reduction reactions must be conducted to establish the requisite oxidation states of the individual CI and C2 carbon atoms.
U.S. Pat. Appl. 2005/0233988 discloses the utilization of a Suzuki reaction between a CI -boronic acid or boronic ester substituted hydroxy-protected glucal and an aryl halide in the presence of a palladium catalyst. The resulting 1- C-arylglucal is then formally hydrated to provide the desired 1- C-aryl glucoside skeleton by use of a reduction step followed by an oxidation step. The synthesis of the boronic acid and its subsequent Suzuki reaction, reduction and oxidation, together, comprise a relatively long synthetic approach to C-arylglucosides and exhibits poor redox economy. Moreover, the coupling catalyst comprises palladium which is toxic and therefore should be controlled to very low levels in the drug substance.

R = protecting group; R’ = H or alkyl
The glucal epoxide method: U.S. Patent 7,847,074 discloses a method that utilizes an organometallic (derived from the requisite aglycone moiety) addition to an electrophilic epoxide located at C1-C2 of a hydroxy-protected glucose ring to furnish intermediates useful for SGLT2 inhibitor synthesis. The epoxide intermediate is prepared by the oxidation of a hydroxy- protected glucal and is not particularly stable. In Tetrahedron 2002, 58, 1997-2009 it was taught that organometallic additions to a tri-6>-benzyl protected glucal-derived epoxide can provide either the a-C-arylglucoside, mixtures of the a- and β-C-arylglucoside or the β-C-arylglucoside by selection of the appropriate counterion of the carbanionic aryl nucleophile (i.e., the
organometallic reagent). For example, carbanionic aryl groups countered with copper (i.e., cuprate reagents) or zinc (i.e., organozinc reagents) ions provide the β-C-arylglucoside, magnesium ions provide the a- and β-C-arylglucosides, and aluminum (i.e., organoaluminum reagents) ions provide the a-C-arylglucoside.

or Zn
The glycosyl leaving group substitution method: U.S. Patent 7,847,074, also disclosed a method comprising the substitution of a leaving group located at CI of a hydroxy-protected glucosyl species, such as a glycosyl halide, with a metalated aryl compound to prepare SGLT2 inhibitors. U.S. Pat. Appl. 2011/0087017 disclosed a similar method to prepare the SGLT2 inhibitor canagliflozin and preferably diarylzinc complexes are used as nucleophiles along with tetra- >-pivaloyl protected glucosylbromide.

Glucose Glucosyl bromide β-anomer
Methodology for alkynylation of 1,6-anhydroglycosides reported in Helv. Chim. Acta. 1995, 78, 242-264 describes the preparation of l,4-dideoxy-l,4-diethynyl^-D-glucopyranoses (a. La., glucopyranosyl acetylenes), that are useful for preparing but-l,3-diyne-l,4-diyl linked polysaccharides, by the ethynylating opening (alkynylation) of partially protected 4-deoxy-4-C- ethynyl-l,6-anhydroglucopyranoses. The synthesis of β-C-arylglucosides, such as could be useful as precursors for SLGT2 inhibitors, was not disclosed. The ethynylation reaction was reported to proceed with retention of configuration at the anomeric center and was rationalized (see Helv. Chim. Acta 2002, 85, 2235-2257) by the C3-hydroxyl of the 1,6- anhydroglucopyranose being deprotonated to form a C3-0-aluminium species, that coordinated with the C6-oxygen allowing delivery of the ethyne group to the β-face of the an oxycarbenium cation derivative of the glucopyranose. Three molar equivalents of the ethynylaluminium reagent was used per 1 molar equivalent of the 1,6-anhydroglucopyranose. The
ethynylaluminium reagent was prepared by the reaction of equimolar (i.e., 1:1) amounts of aluminum chloride and an ethynyllithium reagent that itself was formed by the reaction of an acetylene compound with butyllithium. This retentive ethynylating opening method was also applied (see Helv. Chim. Acta. 1998, 81, 2157-2189) to 2,4-di-<9-triethylsilyl- 1,6- anhydroglucopyranose to provide l-deoxy-l-C-ethynyl- -D-glucopyranose. In this example, 4 molar equivalents of the ethynylaluminium reagent was used per 1 molar equivalent of the 1,6- anhydroglucopyranose. The ethynylaluminium regent was prepared by the reaction of equimolar (i.e., 1: 1) amounts of aluminum chloride and an ethynyl lithium reagent that itself was formed by reaction of an acetylene compound with butyllithium.
It can be seen from the peer-reviewed and patent literature that the conventional methods that can be used to provide C-arylglucosides possess several disadvantages. These include (1) a lack of stereoselectivity during formation of the desired anomer of the C- arylglucoside, (2) poor redox economy due to oxidation and reduction reaction steps being required to change the oxidation state of CI or of CI and C2 of the carbohydrate moiety, (3) some relatively long synthetic routes, (4) the use of toxic metals such as palladium, and/or (5) atom uneconomic protection of four free hydroxyl groups. With regard to the issue of redox economy, superfluous oxidation and reduction reactions that are inherently required to allow introduction of the aryl group into the carbohydrate moiety of the previously mention synthetic methods and the subsequent synthetic steps to establish the required oxidation state, besides adding synthetic steps to the process, are particular undesirable for manufacturing processes because reductants can be difficult and dangerous to operate on large scales due to their flammability or ability to produce flammable hydrogen gas during the reaction or during workup, and because oxidants are often corrosive and require specialized handling operations (see Anderson, N. G. Practical Process Research & Development, 1st Ed.; Academic Press, 2000 (ISBN-10: 0120594757); pg 38 for discussions on this issue).
-
The C-glycoside derivative represented by the formula (1) and its salt [hereinafter, they are referred to as “compound (1)” or “compound of formula (1)” in some cases] is known to be useful for treatment and prevention of diabetes such as insulin-dependent diabetes (type 1 diabetes), non-insulin-dependent diabetes (type 2 diabetes) and the like and various diabetes-related diseases including insulin-resistant diseases and obesity (Patent Literature 1).
-


-
The method for producing the C-glycoside derivative represented by the formula (1), described in the Patent Literature 1 is understood to be represented by the below-shown reaction formula (I), by referring to the Examples and Reference Examples, described in the Patent Literature 1. Roughly explaining, it is a method which comprises reacting [1-benzothien-2-yl(5-bromo-2-fluorophenyl)methoxy]tert-butyl)dimethylsilane (synthesized in accordance with Reference Example 37 of the Literature) in a manner shown in Example 65 of the Literature, to obtain (1S)-1,5-anhydro-1-[3-(1-benzothien-2-ylmethyl)-4-fluorophenyl]-2,3,4,6-tetra-O-benzyl-D-glucitol and then reacting the obtained compound in accordance with Example 100 of the Literature to synthesize intended (1S)-1,5-anhydro-1-C-[3-(1-benzothiophene-2-ylmethyl)-4-fluorophenyl]-D-glucitol.
-


-
However, the method for producing the C-glycoside derivative of the formula (1), disclosed in the Patent Literature 1 is not industrially satisfactory in yield and cost, as is seen in later-shown Reference Example 1 of the present Description.
-
For example, as described later, the method includes a step of low product yield (for example, a step of about 50% or lower yield) and the overall yield of the C-glycoside derivative (final product) represented by the formula (1) from the compound (8) (starting raw material) is below 7%; therefore, the method has problems in yield and cost from the standpoint of medicine production and has not been satisfactory industrially. In addition, the method includes an operation of purification by column chromatography which uses chloroform as part of purification solvents; use of such a solvent poses a problem in environmental protection and there are various restrictions in industrial application of such an operation; thus, the method has problems in providing an effective medicine.
-
Also, an improved method of conducting an addition reaction with trimethylsilyl carbohydrate instead of benzyl carbohydrate and then conducting deprotection for acetylation, is known for a compound which has a structure different from that of the compound of the formula (1) but has a structure common to that of the compound of the formula (1) (Patent Literature 2). It is described in the Patent Literature 2 that the improved method enhances the overall yield to 6.2% from 1.4%. Even in the improved method, however, the yield is low at 6.2% which is far from satisfaction in industrial production.
- Patent Literature 1: WO 2004/080990 Pamphlet
- Patent Literature 2: WO 2006/006496 Pamphlet





http://www.google.com/patents/EP2105442A1
-
Into a tetrahydrofuran (20 ml) solution of benzo[b]thiophene (5.0 g) was dropwise added a n-hexane solution (25 ml) of n-butyl lithium (1.58 M) at -78°C in an argon atmosphere, followed by stirring at -78°C for 10 minutes. Into this solution was dropwise added a tetrahydrofuran (80 ml) solution of 5-bromo-2-fluorobenzaldehyde (8.0 g), followed by stirring at -78°C for 2.5 hours. The temperature of the reaction mixture was elevated to room temperature. Water was added thereto, followed by extraction with ethyl acetate. The organic layer was washed with a saturated aqueous sodium chloride solution, dried over anhydrous magnesium sulfate, filtered, and concentrated. The residue was purified by silica gel column chromatography (n-hexane/ethyl acetate) to obtain 1-benzothien-2-yl(5-bromo-2-fluorophenyl)methanol (10.5 g, yield: 83.6%).
1H-NMR (CDCl3): δ
2.74 (1H, d), 6.35 (1H, d), 6.93 (1H, dd), 7.14 (1H, s), 7.27-7.38 (2H, m), 7.39 (1H, m), 7.68 (1H, dd), 7.74 (2H, m)
- First step: synthesis of 1-benzothien-2-yl(5-bromo-2-fluorophenyl)methanol
Second step: synthesis of [1-benzothien-2-yl(5-bromo-2-fluorophenyl)methoxy](tert-butyl)dimethylsilane
-
To a dimethylformamide (20 ml) solution of 1-benzothien-2-yl(5-bromo-2-fluorophenyl)methanol (5.0 g) were added imidazole (1.3 g), a catalytic amount of 4-(dimethylamino)pyridine and tert-butyldimethylchlorosilane (2.7 g), followed by stirring at room temperature for 7 hours. To the reaction mixture was added a saturated aqueous ammonium chloride solution, followed by extraction with ethyl acetate. The organic layer was washed with a saturated aqueous ammonium chloride solution and a saturated aqueous sodium chloride solution, dried over anhydrous magnesium sulfate, filtered and concentrated. The residue was purified by silica gel column chromatography (n-hexane/ethyl acetate) to obtain [1-benzothien-2-yl(5-bromo-2-fluorophenyl)methoxy](tert-butyl)dimethylsilane (5.22 g, yield: 78.0%).
MS: 451 (M+)
1H-NMR (CDCl3): δ
0.05 (3H, s), 0.11 (3H, s), 0.95 (9H, s), 6.34 (1H, s), 6.91 (1H, t), 7.08 (1H, d), 7.23-7.38 (2H, m), 7.64-7.68 (1H, m), 7.75-7.28 (2H, m)
Third step: Synthesis of 1-C-[3-(1-benzothien-2-yl{[tert-butyl-(dimethyl)silyloxy}methyl)4-fluorophenyl]-2,3,4,6-tetra-O-benzyl-D-glucopyranose
-
Into a tetrahydrofuran (15 ml) solution of [1-benzothien-2-yl(5-bromo-2-fluorophenyl)methoxy](tert-butyl)dimethylsilane (1.5 g) was dropwise added a n-hexane solution (2.2 ml) of n-butyl lithium (1.58 M) in an argon atmosphere at -78°C, followed by stirring at -78°C for 30 minutes. Into the solution was dropwise added a tetrahydrofuran (20 ml) solution of 2,3,4,6-tetra-O-benzyl-D-glucono-1,5-lactone (1.9 g), followed by stirring at -78°C for 15 minutes and then at 0°C for 1.5 hours. To the reaction mixture was added a saturated aqueous ammonium chloride solution, followed by extraction with ethyl acetate. The organic layer was washed with a saturated aqueous ammonium chloride solution and a saturated aqueous sodium chloride solution, dried over anhydrous magnesium sulfate, filtered and concentrated. The residue was purified by silica gel column chromatography (n-hexane/chloroform/acetone) to obtain 1-C-[3-(1-benzothien-2-yl{[tert-butyl-(dimethyl)silyloxy}methyl)-4-fluorophenyl]-2,3,4,6-tetra-O-benzyl-D-glucopyranose (1.52 g, yield: 50.2%). MS: 933 (M+Na)
Fourth step: Synthesis of 1-C-{3-[1-benzothien-2-yl(hydroxy)methyl]-4-fluorophenyl}-2,3,4,6-tetra-O-benzyl-D-glucopyranose
-
To a tetrahydrofuran (15 ml) solution of 1-C-[3-(1-benzothien-2-yl{[tert-butyl-(dimethyl)silyloxy}methyl)-4-fluorophenyl]-2,3,4,6-tetra-O-benzyl-D-glucopyranose (1.52 g) was added a tetrahydrofuran solution (2.0 ml) of tetrabutylammonium fluoride (1.0 M), followed by stirring at room temperature for 1 hour. The reaction mixture was concentrated per se. The residue was purified by silica gel column chromatography (n-hexane/ethyl acetate) to obtain 1-C-{3-[1-benzothien-2-yl(hydroxy)methyl]-4-fluorophenyl}-2,3,4,6-tetra-O-benzyl-D-glucopyranose (0.99 g, yield: 74.7%). MS: 819 (M+Na), 779 (M+H-H2O)
Fifth step: Synthesis of (1S)-1,5-anhydro-1-[3-(1-benzothien-2-ylmethyl)-4-fluorophenyl]-2,3,4,6-tetra-O-benzyl-D-glucitol
-
To an acetonitrile (5.0 ml) solution of 1-C-{3-[1-benzothien-2-yl(hydroxy)methyl]-4-fluorophenyl}-2,3,4,6-tetra-O-benzyl-D-glucopyranose (500 mg) were added triethylsilane (175 mg) and boron trifluoride-diethyl ether complex (196 mg) in an argon atmosphere at -20°C, followed by stirring at -20°C for 5 hours. To the reaction mixture was added a saturated aqueous sodium bicarbonate solution, followed by extraction with chloroform. The organic layer was washed with a saturated aqueous sodium bicarbonate solution and a saturated aqueous sodium chloride solution, dried over anhydrous magnesium sulfate, filtered and concentrated. The residue was purified by silica gel column chromatography (n-hexane/ethyl acetate) to obtain (1S)-1,5-anhydro-1-[3-(1-benzothien-2-ylmethyl)-4-fluorophenyl]-2,3,4,6-tetra-O-benzyl-D-glucitol (150 mg, yield: 30.2%) MS: 787 (M+Na)
1H-NMR (CDCl3): δ
3.42-3.48 (1H, m), 3.55-3.58 (1H, m), 3.72-3.78 (4H, m), 3.83 (1H, d), 4.14-4.30 (3H, m), 4.39 (1H, d), 4.51-4.67 (4H, m), 4.83-4.94 (2H, m), 6.86-6.90 (1H, m), 6.98 (1H, brs), 7.06-7.37 (24H, m), 7.57-7.60 (1H, m), 7.66-7.69 (1H, m)
Sixth step: Synthesis of (1S)-1,5-anhydro-1-C-[3-(1-benzothiophene-2-ylmethyl)-4-fluorophenyl]-D-glucitol
-
To a dichloromethane (10 ml) solution of (1S)-1,5-anhydro-1-[3-(1-benzothien-2-ylmethyl)-4-fluorophenyl]-2,3,4,6-tetra-O-benzyl-D-glucitol (137 mg) were added pentamethylbenzene (382 mg) and a n-heptane solution (0.75 ml) of boron trichloride (1.0 M) in an argon atmosphere at -78°C, followed by stirring at -78°C for 3 hours. Methanol was added to the reaction mixture, the temperature of the resulting mixture was elevated to room temperature, and the mixture was concentrated per se. The residue was purified by silica gel column chromatography (chloroform/methanol) to obtain (1S)-1,5-anhydro-1-C-[3-(1-benzothiophene-2-ylmethyl)-4-fluorohenyl]-D-glucitol OR IPRAGLIFLOZIN (63 mg, yield: 87.8%).
1H-NMR (CD3OD): δ
3.29-3.48 (4H, m), 3.68 (1H, dd), 3.87 (1H, dd), 4.11 (1H, d), 4.20-4.29 (2H, m), 7.03 (1H, s), 7.08 (1H, dd), 7.19-7.29 (2H, m), 7.35 (1H, m), 7.42 (1H, dd), 7.64 (1H, d), 7.72 (1H, d)
(1S)-1,5-anhydro-1-C-[3-(1-benzothiophene-2-ylmethyl)-4-fluorohenyl]-D-glucitol OR IPRAGLIFLOZIN
Empagliflozin

Empagliflozin
BI-10773
(2S,3R,4R,5S,6R)-2-[4-chloro-3-[[4-[(3S)-oxolan-3-yl]oxyphenyl]methyl]phenyl]-6-(hydroxymethyl)oxane-3,4,5-triol
M.Wt: 450.91
: C23H27ClO7
Sponsor/Developer: Eli Lilly and Boehringer Ingelheim
Mechanism of action: SGLT 2 inhibitor
Indication (Phase): Oral treatment of adults with type 2 diabetes (Phase III, expected to conclude by year’s end); Oral treatment of adults with type 2 diabetes plus high blood pressure (Phase IIb; trial results released Oct. 2)
NDA, MAA filings planned for 2013
Empagliflozin is a potent, selective sodium glucose co-transporter-2 inhibitor that is in development for the treatment of type 2 diabetes. Empagliflozin is an inhibitor of the sodium glucose co-transporter-2 (SGLT-2), which is found almost exclusively in the proximal tubules of nephronic components in the kidneys. SGLT-2 accounts for about 90 percent of glucose reabsorption into the blood. Blocking SGLT-2 causes blood glucose to be eliminated through the urine via the urethra. The Empagliflozin phase III clinical trial program will include about 14,500 patients. The program consists of twelve ongoing international phase III clinical trials, including a large cardiovascular outcomes trial.
Empagliflozin is a novel SGLT2 inhibitor that is described for the treatment or improvement in glycemic control in patients with type 2 diabetes mellitus, for example in WO 05/092877, WO 06/117359, WO 06/120208, WO 2010/092126, WO 2010/092123, WO 2011/039107, WO 2011/039108. The use of a SGLT2 inhibitor in a method for treating obesity is described in WO 08/116,195
WO2005/092877
WO2006/117359 MP 149 DEG CENT
Empagliflozin is drug which is being investigated in clinical trials for the oral treatment oftype 2 diabetes by Boehringer Ingelheim and Eli Lilly and Company.[1][2] It is an inhibitor of the sodium glucose co-transporter-2 (SGLT-2), which is found almost exclusively in theproximal tubules of nephronic components in the kidneys. SGLT-2 accounts for about 90 percent of glucose reabsorption into the blood. Blocking SGLT-2 causes blood glucose to be eliminated through the urine via the urethra.[3][4]
SGLT-2 inhibitors such as empagliflozin reduce blood glucose by blocking glucose reabsorption in the kidney and thereby excreting glucose (i.e., blood sugar) via the urine.[5]
As of December 2013, empagliflozin is in phase III clinical trials.[2]

When taken in dosages of 10 or 25 mg once a day, the incidence of adverse events was similar to placebo. However, there was a higher frequency of genital infections at both the 10 mg and the 25 mg dosages.
1-chloro-4-(β-D-glucopyranos-1-yl)-2-[4-((S)-tetrahydrofuran-3-yloxy)-benzyl]-benzene of the formula

as described for example in WO 2005/092877. Methods of synthesis are described in the literature, for example WO 06/120208 and WO 2011/039108. According to this invention, it is to be understood that the definition of empagliflozin also comprises its hydrates, solvates and polymorphic forms thereof, and prodrugs thereof. An advantageous crystalline form of empagliflozin is described in WO 2006/117359 and WO 2011/039107 which hereby are incorporated herein in their entirety. This crystalline form possesses good solubility properties which enables a good bioavailability of the SGLT2 inhibitor. Furthermore, the crystalline form is physico-chemically stable and thus provides a good shelf-life stability of the pharmaceutical composition. Preferred pharmaceutical compositions, such as solid formulations for oral administration, for example tablets, are described in WO 2010/092126,

http://www.google.com/patents/WO2011039108A2
Example 1 : Synthesis of the fluoride VIII.1
Oxalylchloride (176kg; 1386mol; 1 ,14eq) is added to a mixture of 2-chloro-5-iodo benzoic acid (343kg; 1214mol) (compound IX.1 ), fluorobenzene (858kg) and N,N-dimethylformamide (2kg) within 3 hours at a temperature in the range from about 25 to 30°C (gas formation). After completion of the addition, the reaction mixture is stirred for additional 2 hours at a temperature of about 25 to 30°C. The solvent (291 kg) is distilled off at a temperature between 40 and 45°C (p=200mbar). Then the reaction solution (91 1 kg) is added to aluminiumchloride AICI3 (181 kg) and fluorobenzene (192kg) at a temperature between about 25 and 30°C within 2 hours. The reaction solution is stirred at the same temperature for about an additional hour. Then the reaction mixture is added to an amount of 570 kg of water within about 2 hours at a temperature between about 20 and 30°C and stirred for an additional hour. After phase separation the organic phase (1200kg) is separated into two halves (600kg each). From the first half of the organic phase solvent (172kg) is distilled off at a temperature of about 40 to 50°C (p=200mbar). Then 2-propanole (640kg) is added. The solution is heated to about 50°C and then filtered through a charcoal cartouche (clear filtration). The cartouche may be exchanged during filtration and washed with a
fluorobenzene/2-propanole mixture (1 :4; 40kg) after filtration. Solvent (721 kg) is distilled off at a temperature of about 40 to 50°C and p=200mbar. Then 2-propanole (240kg) is added at a temperature in the range between about 40 to 50°C. If the content of fluorobenzene is greater than 1 % as determined via GC, another 140kg of solvent are distilled off and 2- propanole (140kg) is added. Then the solution is cooled from about 50°C to 40°C within one hour and seeding crystals (50g) are added. The solution is further cooled from about 40°C to 20°C within 2 hours. Water (450kg) is added at about 20°C within 1 hour and the suspension is stirred at about 20°C for an additional hour before the suspension is filtered. The filter cake is washed with 2-propanole/water (1 :1 ; 800kg). The product is dried until a water level of <0.06%w/w is obtained. The second half of the organic phase is processed identically. A total of 410kg (94%yield) of product which has a white to off-white crystalline appearance, is obtained. The identity of the product is determined via infrared spectrometry.
Example 2: Synthesis of the ketone VII.1
To a solution of the fluoride VIII.1 (208kg), tetrahydrofuran (407kg) and (S)-3- hydroxytetrahydrofuran (56kg) is added potassium-ie f-butanolate solution (20%) in tetrahydrofuran (388kg) within 3 hrs at 16 to 25°C temperature. After completion of the addition, the mixture is stirred for 60min at 20°C temperature. Then the conversion is determined via HPLC analysis. Water (355kg) is added within 20 min at a temperature of 21 °C (aqueous quench). The reaction mixture is stirred for 30 min (temperature: 20°C). The stirrer is switched off and the mixture is left stand for 60 min (temperature: 20°C). The phases are separated and solvent is distilled off from the organic phase at 19 to 45°C temperature under reduced pressure. 2-Propanol (703kg) is added to the residue at 40 to 46°C temperature and solvent is distilled off at 41 to 50°C temperature under reduced pressure. 2-Propanol (162kg) is added to the residue at 47°C temperature and solvent is distilled off at 40 to 47°C temperature under reduced pressure. Then the mixture is cooled to 0°C within 1 hr 55 min. The product is collected on a centrifuge, washed with a mixture of 2- propanol (158kg) and subsequently with ie f.-butylmethylether (88kg) and dried at 19 to 43°C under reduced pressure. 227kg (91 ,8%) of product are obtained as colourless solid. The identity of the product is determined via infrared spectrometry.
Example 3: Synthesis of the iodide V.1
To a solution of ketone VII.1 (217,4kg) and aluminium chloride (AICI3; 81 ,5kg) in toluene (366,8kg) is added 1 ,1 ,3,3-tetramethyldisiloxane (TMDS, 82,5kg) within 1 hr 30 min
(temperature: 18-26°C). After completion of the addition, the mixture is stirred for additional 1 hr at a temperature of 24°C. Then the conversion is determined via HPLC analysis.
Subsequently the reaction mixture is treated with acetone (15,0kg), stirred for 1 hr 5 min at 27°C temperature and the residual TMDS content is analyzed via GC. Then a mixture of water (573kg) and concentrated HCI (34kg) is added to the reaction mixture at a temperature of 20 to 51 °C (aqueous quench). The reaction mixture is stirred for 30 min (temperature:
51 °C). The stirrer is switched off and the mixture is left stand for 20 min (temperature: 52°C). The phases are separated and solvent is distilled off from the organic phase at 53-73°C temperature under reduced pressure. Toluene (52,8kg) and ethanol (435,7kg) are added to the residue at 61 to 70°C temperature. The reaction mixture is cooled to 36°C temperature and seeding crystals (0,25kg) are added. Stirring is continued at this temperature for 35 min. Then the mixture is cooled to 0 to 5°C and stirred for additional 30 min. The product is collected on a centrifuge, washed with ethanol (157kg) and dried at 15 to 37°C under reduced pressure. 181 kg (82,6%) of product are obtained as colourless solid. The identity of the product is determined via the HPLC retention time.
Example 4: Synthesis of the lactone IV.1
A suspension of the D-(+)-gluconic acid-delta-lactone IVa.1 (42,0kg), tetrahydrofuran (277,2kg), 4-methylmorpholine (NMM; 152,4kg) and 4-dimethylaminopyridine (DMAP;
1 ,44kg) is treated with chlorotrimethylsilane (TMSCI; 130,8kg) within 50 min at 13 to 19°C. After completion of the addition stirring is continued for 1 hr 30 min at 20 to 22°C and the conversion is determined via HPLC analysis. Then n-heptane (216,4kg) is added and the mixture is cooled to 5°C. Water (143kg) is added at 3 to 5°C within 15 min. After completion of the addition the mixture is heated to 15°C and stirred for 15 min. The stirrer is switched off and the mixture is left stand for 15 min. Then the phases are separated and the organic layer is washed in succession two times with water (143kg each). Then solvent is distilled off at 38°C under reduced pressure and n-heptane (130kg) is added to the residue. The resulting solution is filtered and the filter is rinsed with n-heptane (63kg) (filter solution and product solution are combined). Then solvent is distilled off at 39 to 40°C under reduced pressure. The water content of the residue is determined via Karl-Fischer analysis (result: 0,0%).
1 12,4kg of the product is obtained as an oil (containing residual n-heptane, which explains the yield of >100%). The identity of the product is determined via infrared spectrometry.
Example 5a: Synthesis of the glucoside 11.1
To a solution of the iodide V.1 (267kg) in tetrahydrofuran (429kg) is added Turbogrignard solution (isopropylmagnesium chloride/lithium chloride solution, 14 weight-% iPrMgCI in THF, molar ratio LiCI : iPrMgCI = 0,9 – 1 .1 mol/mol) (472kg) at -21 to -15°C temperature within 1 hr 50 min. On completion of the addition the conversion is determined via HPLC analysis. The reaction is regarded as completed when the area of the peak corresponding to the iodide V.1 is smaller than 5,0% of the total area of both peaks, iodide V.1 and the corresponding desiodo compound of iodide V.1 . If the reaction is not completed, additional Turbogrignard solution is added until the criterion is met. In this particular case the result is 3,45%. Then the lactone IV.1 (320kg) is added at -25 to -18°C temperature within 1 hr 25 min. The resulting mixture is stirred for further 1 hr 30 min at -13 to -18°C. On completion the conversion is determined via HPLC analysis (for information). On completion, a solution of citric acid in water (938L; concentration: 10 %-weight) is added to the reaction mixture of a volume of about 2500L at -13 to 19°C within 1 hr 25 min.
The solvent is partially distilled off from the reaction mixture (residual volume: 1816-1905L) at 20 to 30°C under reduced pressure and 2-methyltetrahydrofuran (532kg) is added. Then the stirrer is switched off and the phases are separated at 29°C. After phase separation the pH value of the organic phase is measured with a pH electrode (Mettler Toledo MT HA 405 DPA SC) or alternatively with pH indicator paper (such as pH-Fix 0-14, Macherey and Nagel). The measured pH value is 2 to 3. Then solvent is distilled off from the organic phase at 30 to 33°C under reduced pressure and methanol (1202kg) is added followed by the addition of a solution of 1 ,25N HCI in methanol (75kg) at 20°C (pH = 0). Full conversion to the acetale 111.1 is achieved by subsequent distillation at 20 to 32°C under reduced pressure and addition of methanol (409kg).
Completion of the reaction is obtained when two criteria are fulfilled:
1 ) The ratio of the sum of the HPLC-area of the alpha-form + beta-form of intermediate 111.1 relative to the area of intermediate llla.1 is greater or equal to 96,0% : 4,0%. 2) The ratio of the HPLC-area of the alpha-form of intermediate 111.1 to the beta-form of 111.1 is greater or equal to 97,0% to 3,0%.
In this particular case both criteria are met. Triethylamin (14kg) is added (pH = 7,4) and solvent is distilled off under reduced pressure, acetonitrile (835kg) is added and further distilled under reduced pressure. This procedure is repeated (addition of acetonitrile: 694kg) and methylene chloride (640kg) is added to the resulting mixture to yield a mixture of the acetale 111.1 in acetonitrile and methylene chloride. The water content of the mixture is determined via Karl Fischer titration (result: 0,27%).
The reaction mixture is then added within 1 hr 40 min at 10 to 19°C to a preformed mixture of AICI3 (176kg), methylene chloride (474kg), acetonitrile (340kg), and triethylsilane (205kg). The resulting mixture is stirred at 18 to 20°C for 70 min. After completion of the reaction, water (1263L) is added at 20 to 30°C within 1 hr 30 min and the mixture is partially distilled at 30 to 53°C under atmospheric pressure and the phases are separated. Toluene (698kg) is added to the organic phase and solvent is distilled off under reduced pressure at 22 to 33°C. The product is then crystallized by addition of seeding crystals (0,5kg) at 31 °C and water (267kg) added after cooling to 20°C. The reaction mixture is cooled to 5°C within 55 min and stirred at 3 to 5°C for 12 hrs. Finally the product is collected on a centrifuge as colourless, crystalline solid, washed with toluene (348kg) and dried at 22 to 58°C. 21 1 kg (73%) of product are obtained. The identity of the product is determined via the HPLC retention time.
Example 5b: Synthesis of the glucoside 11.1
To a solution of the iodide V.1 (30g) in tetrahydrofuran (55ml_) is added Turbogrignard solution (isopropylmagnesium chloride/lithium chloride solution, 14 weight-% iPrMgCI in THF, molar ratio LiCI : iPrMgCI = 0,9 – 1 .1 mol/mol) (53g) at -14 to -13°C temperature within 35 min. On completion of the addition the conversion is determined via HPLC analysis. The reaction is regarded as completed when the area of the peak corresponding to the iodide V.1 is smaller than 5,0% of the total area of both peaks, iodide V.1 and the corresponding desiodo compound of iodide V.1 . If the reaction is not completed, additional Turbogrignard solution is added until the criterion is met. In this particular case the result is 0,35%. Then the lactone IV.1 (36g) is added at -15 to -6°C temperature within 15 min. The resulting mixture is stirred for further 1 hr at -6 to -7°C. On completion, the conversion is determined via HPLC analysis (for information). On completion, a solution of citric acid in water (105mL;
concentration: 10 %-weight) is added to the reaction mixture at -15 to 10°C within 30 min. The solvent is partially distilled off from the reaction mixture (residual volume: 200mL) at 20 to 35°C under reduced pressure and 2-methyltetrahydrofuran (71 mL) is added. Then the mixture is stirred for 25min at 30°C. Then the stirrer is switched off and the phases are separated at 30°C. After phase separation the pH value of the organic phase is measured with a pH electrode (Mettler Toledo MT HA 405 DPA SC) or alternatively with pH indicator paper (such as pH-Fix 0-14, Macherey and Nagel). The measured pH value is 3. Then solvent is distilled off from the organic phase at 35°C under reduced pressure and methanol (126ml_) is added followed by the addition of a solution of 1 ,25N HCI in methanol (10,1 ml_) at 25°C (pH = 1 -2). Full conversion to the acetale 111.1 is achieved by subsequent distillation at 35°C under reduced pressure and addition of methanol (47ml_).
Completion of the reaction is obtained when two criteria are fulfilled:
1 ) The ratio of the sum of the HPLC-area of the alpha-form + beta-form of intermediate 111.1 relative to the area of intermediate llla.1 is greater or equal to 96,0% : 4,0%. In this particular case the ratio is 99,6% : 0,43%.
2) The ratio of the HPLC-area of the alpha-form of intermediate 111.1 to the beta-form of III.1 is greater or equal to 97,0% to 3,0%. In this particular case the ratio is 98,7% : 1 ,3%.
Triethylamin (2,1 mL) is added (pH = 9) and solvent is distilled off at 35°C under reduced pressure, acetonitrile (120ml_) is added and further distilled under reduced pressure at 30 to 35°C. This procedure is repeated (addition of acetonitrile: 102ml_) and methylene chloride (55ml_) is added to the resulting mixture to yield a mixture of the acetale 111.1 in acetonitrile and methylene chloride. The water content of the mixture is determined via Karl Fischer titration (result: 0,04%).
The reaction mixture is then added within 1 hr 5 min at 20°C to a preformed mixture of AICI3 (19,8g), methylene chloride (49ml_), acetonitrile (51 mL), and triethylsilane (23g). The resulting mixture is stirred at 20 to 30°C for 60 min. After completion of the reaction, water (156mL) is added at 20°C within 25 min and the mixture is partially distilled at 55°C under atmospheric pressure and the phases are separated at 33°C. The mixture is heated to 43°C and toluene (90mL) is added and solvent is distilled off under reduced pressure at 41 to 43°C. Then acetonitrile (1 OmL) is added at 41 °C and the percentage of acetonitrile is determined via GC measurement. In this particular case, the acetonitrile percentage is 27%- weight. The product is then crystallized by addition of seeding crystals (0,1 g) at 44°C and the mixture is further stirred at 44°C for 15min. The mixture is then cooled to 20°C within 60min and water (142mL) is added at 20°C within 30min. The reaction mixture is cooled to 0 to 5°C within 60 min and stirred at 3°C for 16 hrs. Finally the product is collected on a filter as colourless, crystalline solid, washed with toluene (80mL) and dried at 20 to 70°C. 20, 4g (62,6%) of product are obtained. The identity of the product is determined via the HPLC retention time.
- Grempler R, Thomas L, Eckhardt M, Himmelsbach F, Sauer A, Sharp DE, Bakker RA, Mark M, Klein T, Eickelmann P (January 2012). “Empagliflozin, a novel selective sodium glucose cotransporter-2 (SGLT-2) inhibitor: characterisation and comparison with other SGLT-2 inhibitors”. Diabetes Obes Metab 14 (1): 83–90. doi:10.1111/j.1463-1326.2011.01517.x. PMID 21985634.
- “Empagliflozin”. clinicaltrials.gov. U.S. National Institutes of Health. Retrieved 22 September 2012.
- Nair S, Wilding JP (January 2010). “Sodium glucose cotransporter 2 inhibitors as a new treatment for diabetes mellitus”. J. Clin. Endocrinol. Metab. 95 (1): 34–42. doi:10.1210/jc.2009-0473. PMID 19892839.
- Bays H (March 2009). “From victim to ally: the kidney as an emerging target for the treatment of diabetes mellitus”. Curr Med Res Opin25 (3): 671–81. doi:10.1185/03007990802710422. PMID 19232040.
- Abdul-Ghani MA, DeFronzo RA (September 2008). “Inhibition of renal glucose reabsorption: a novel strategy for achieving glucose control in type 2 diabetes mellitus”. Endocr Pract 14 (6): 782–90. PMID 18996802.
[1]. Grempler R, Thomas L, Eckhardt M et al. Empagliflozin, a novel selective sodium glucose cotransporter-2 (SGLT-2) inhibitor: characterisation and comparison with other SGLT-2 inhibitors. Diabetes Obes Metab. 2012 Jan;14(1):83-90.
Abstract
AIMS: Empagliflozin is a selective sodium glucose cotransporter-2 (SGLT-2) inhibitor in clinical development for the treatment of type 2 diabetes mellitus. This study assessed pharmacological properties of empagliflozin in vitro and pharmacokinetic properties in vivo and compared its potency and selectivity with other SGLT-2 inhibitors. METHODS: [(14)C]-alpha-methyl glucopyranoside (AMG) uptake experiments were performed with stable cell lines over-expressing human (h) SGLT-1, 2 and 4. Two new cell lines over-expressing hSGLT-5 and hSGLT-6 were established and [(14)C]-mannose and [(14)C]-myo-inositol uptake assays developed. Binding kinetics were analysed using a radioligand binding assay with [(3)H]-labelled empagliflozin and HEK293-hSGLT-2 cell membranes. Acute in vivo assessment of pharmacokinetics was performed with normoglycaemic beagle dogs and Zucker diabetic fatty (ZDF) rats. RESULTS: Empagliflozin has an IC(50) of 3.1 nM for hSGLT-2. Its binding to SGLT-2 is competitive with glucose (half-life approximately 1 h). Compared with other SGLT-2 inhibitors, empagliflozin has a high degree of selectivity over SGLT-1, 4, 5 and 6. Species differences in SGLT-1 selectivity were identified. Empagliflozin pharmacokinetics in ZDF rats were characterised by moderate total plasma clearance (CL) and bioavailability (BA), while in beagle dogs CL was low and BA was high. CONCLUSIONS: Empagliflozin is a potent and competitive SGLT-2 inhibitor with an excellent selectivity profile and the highest selectivity window of the tested SGLT-2 inhibitors over hSGLT-1. Empagliflozin represents an innovative therapeutic approach to treat diabetes.
[2]. Thomas L, Grempler R, Eckhardt M et al. Long-term treatment with empagliflozin, a novel, potent and selective SGLT-2 inhibitor, improves glycaemic control and features of metabolic syndrome in diabetic rats. Diabetes Obes Metab. 2012 Jan;14(1):94-6.
Abstract
Empagliflozin is a potent, selective sodium glucose co-transporter-2 inhibitor that is in development for the treatment of type 2 diabetes. This series of studies was conducted to assess the in vivo pharmacological effects of single or multiple doses of empagliflozin in Zucker diabetic fatty rats. Single doses of empagliflozin resulted in dose-dependent increases in urinary glucose excretion and reductions in blood glucose levels. After multiple doses (5 weeks), fasting blood glucose levels were reduced by 26 and 39% with 1 and 3 mg/kg empagliflozin, respectively, relative to vehicle. After 5 weeks, HbA1c levels were reduced (from a baseline of 7.9%) by 0.3 and 1.1% with 1 and 3 mg/kg empagliflozin, respectively, versus an increase of 1.1% with vehicle. Hyperinsulinaemic-euglycaemic clamp indicated improved insulin sensitivity with empagliflozin after multiple doses versus vehicle. These findings support the development of empagliflozin for the treatment of type 2 diabetes.
[3]. Luippold G, Klein T, Mark M, Grempler R. Empagliflozin, a novel potent and selective SGLT-2 inhibitor, improves glycaemic control alone and in combination with insulin in streptozotocin-induced diabetic rats, a model of type 1 diabetes mellitus. Diabetes Obes Metab. 2012 Jul;14(7):601-7.
Abstract
AIM: Sodium glucose cotransporter-2 (SGLT-2) is key to reabsorption of glucose in the kidney. SGLT-2 inhibitors are in clinical development for treatment of type 2 diabetes mellitus (T2DM). The mechanism may be of value also in the treatment of type 1 diabetes mellitus (T1DM). This study investigated effects of the SGLT-2 inhibitor, empagliflozin, alone and in combination with insulin, on glucose homeostasis in an animal model of T1DM. METHODS: Sprague-Dawley rats were administered a single intraperitoneal injection of streptozotocin (STZ; 60 mg/kg). Acutely, STZ rats received two doses of insulin glargine with or without empagliflozin, and blood glucose was measured. In a subchronic study, STZ rats received empagliflozin alone, one or two insulin-releasing implants or a combination of one implant and empagliflozin over 28 days; blood glucose and HbA(1c) were measured. RESULTS: In the acute setting, empagliflozin in combination with 1.5 IU insulin induced a similar glucose-lowering effect as 6 IU insulin. Both interventions were more efficacious than monotherapy with 1.5 IU insulin. In the subchronic study, 12-h blood glucose profile on day 28 in the combination group was lower than with one implant, and similar to two implants. Plasma HbA(1c) was improved in the combination group and in animals with two implants. CONCLUSIONS: Empagliflozin reduced blood glucose levels in a T1DM animal model. Empagliflozin combined with low-dose insulin showed comparable glucose-lowering efficacy to treatment with high-dose insulin. Our data suggest that empagliflozin is an efficacious adjunctive-to-insulin therapy with the clinical potential for the treatment of T1DM.
[4]. Macha S, Rose P, Mattheus M et al. Lack of drug-drug interaction between empagliflozin, a sodium glucose cotransporter-2 inhibitor, and warfarin in healthy volunteers. Diabetes Obes Metab. 2012 Oct 24. doi: 10.1111/dom.12028. [Epub ahead of print]
Abstract
AIM: To investigate potential drug-drug interactions between empagliflozin and warfarin. MATERIALS AND METHODS: Healthy subjects (n=18) received empagliflozin 25 mg qd for 5 days (treatment A), followed by empagliflozin 25 mg qd for 7 days (days 6-12) with a single 25 mg dose of warfarin on day 6 (B), and a single 25 mg dose of warfarin alone (C), in an open-label, crossover study. Subjects received treatments in sequence AB_C or C_AB with a washout period of ≥14 days between AB and C or C and AB. RESULTS: Warfarin had no effect on empagliflozin area under concentration-time curve or maximum plasma concentration at steady-state (AUC(τ) (,ss) or C(max,ss) ): geometric mean ratios (GMRs) (90% confidence intervals [CI]) were 100.89% (96.86, 105.10) and 100.64% (89.79, 112.80), respectively. Empagliflozin had no effect on AUC from 0 hours to infinity (AUC(0) (-∞) ) or C(max) for R-warfarin or S-warfarin (GMRs [90% CI] for AUC(0) (-∞) : 98.49% [95.29, 101.80] and 95.88% [93.40, 98.43], respectively; C(max) : 97.89% [91.12, 105.15] and 98.88% [91.84, 106.47], respectively). Empagliflozin had no clinically relevant effects on warfarin’s anticoagulant activity (international normalised ratio [INR]) (GMR [95% CI] for peak INR: 0.87 [0.73, 1.04]; area under the effect-time curve from 0 to 168 hours: 0.88 [0.79, 0.98]. No drug-related adverse events were reported for empagliflozin after monotherapy or combined administration. The combination of empagliflozin and warfarin was well tolerated. CONCLUSIONS: No drug-drug interactions were observed between empagliflozin and warfarin, indicating that empagliflozin and warfarin can be co-administered without dosage adjustments of either drug.
[5]. Sarashina A, Koiwai K, Seman LJ et al. Safety, Tolerability, Pharmacokinetics and Pharmacodynamics of Single Doses of Empagliflozin, a Sodium Glucose Cotransporter-2 (SGLT-2) Inhibitor, in Healthy Japanese Subjects. Drug Metab Pharmacokinet. 2012 Nov 13. [Epub ahead of print]
Abstract
This randomized, placebo-controlled within dose groups, double-blind, single rising dose study investigated the safety, tolerability, pharmacokinetics and pharmacodynamics of 1 mg to 100 mg doses of empagliflozin in 48 healthy Japanese male subjects. Empagliflozin was rapidly absorbed, reaching peak levels in 1.25 to 2.50 hours; thereafter, plasma concentrations declined in a biphasic fashion, with mean terminal elimination half-life ranging from 7.76 to 11.7 hours. Increase in empagliflozin exposure was proportional to dose. Oral clearance was dose independent and ranged from 140 to 172 mL/min. In the 24 hours following 100 mg empagliflozin administration, the mean (%CV) amount of glucose excreted in urine was 74.3 (17.1) g. The amount and the maximum rate of glucose excreted via urine increased with dose of empagliflozin. Nine adverse events, all of mild intensity, were reported by 8 subjects (7 with empagliflozin and 1 with placebo). No hypoglycemia was reported. In conclusion, 1 mg to 100 mg doses of empagliflozin had a good safety and tolerability profile in healthy Japanese male subjects. Exposure to empagliflozin was dose-proportional. The amount and rate of urinary glucose excretion were higher with empagliflozin than with placebo, and increased with empagliflozin dose.
…………………………….
formula A

Example I

(5-bromo-2-chloro-phenyl)-(4-methoxy-phenyl)-methanone
38.3 ml oxalyl chloride and 0.8 ml of dimethylformamide are added to a mixture of
100 g of 5-bromo-2-chloro-benzoic acid in 500 ml dichloromethane. The reaction mixture is stirred for 14 h, then filtered and separated from all volatile constituents in the rotary evaporator. The residue is dissolved in 150 ml dichloromethane, the solution is cooled to -5 0C, and 46.5 g of anisole are added. Then 51.5 g of aluminum trichloride are added batchwise so that the temperature does not exceed 5 0C. The solution is stirred for another 1 h at 1 to 5 0C and then poured onto crushed ice. The organic phase is separated, and the aqueous phase is extracted another three times with dichloromethane. The combined organic phases are washed with aqueous 1 M hydrochloric acid, twice with aqueous 1 M sodium hydroxide solution and with brine. Then the organic phase is dried, the solvent is removed and the residue is recrystallised in ethanol. Yield: 86.3 g (64% of theory)
Mass spectrum (ESI+): m/z = 325/327/329 (Br+CI) [M+H]+
Example Il
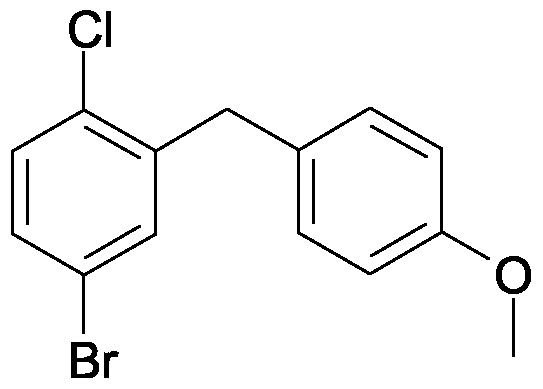
4-bromo-1-chloro-2-(4-methoxy-benzyl)-benzene
A solution of 86.2 g (5-bromo-2-chloro-phenyl)-(4-methoxy-phenyl)-methanone and 101.5 ml triethylsilane in 75 ml dichloromethane and 150 ml acetonitrile is cooled to 1O0C. Then with stirring 50.8 ml of boron trifluoride etherate are added so that the temperature does not exceed 2O0C. The solution is stirred for 14 h at ambient temperature, before another 9 ml triethylsilane and 4.4 ml boron trifluoride etherate are added. The solution is stirred for a further 3 h at 45 to 5O0C and then cooled to ambient temperature. A solution of 28 g potassium hydroxide in 70 ml of water is added, and the resulting mixture is stirred for 2 h. Then the organic phase is separated off and the aqueous phase is extracted another three times with diisopropylether. The combined organic phases are washed twice with aqueous 2 M potassium hydroxide solution and once with brine and then dried over sodium sulfate. After the solvent has been removed the residue is washed in ethanol, separated again and dried at 6O0C. Yield: 50.0 g (61 % of theory)
Mass spectrum (ESI+): m/z = 310/312/314 (Br+CI) [M+H]+
Example III

4-(5-bromo-2-chloro-benzyl)-phenol
A solution of 14.8 g 4-bromo-1-chloro-2-(4-methoxy-benzyl)-benzene in 150 ml dichloromethane is cooled in an ice bath. Then 50 ml of a 1 M solution of boron tribromide in dichloromethane are added, and the solution is stirred for 2 h at ambient temperature. The solution is then cooled in an ice bath again, and saturated aqueous potassium carbonate solution is added dropwise. At ambient temperature the mixture is adjusted with aqueous 1 M hydrochloric acid to a pH of 1 , the organic phase is separated, and the aqueous phase is extracted another three times with ethyl acetate. The combined organic phases are dried over sodium sulphate, and the solvent is removed completely. Yield: 13.9 g (98% of theory) Mass spectrum (ESI ): m/z = 295/297/299 (Br+CI) [M-HV
Example IV

r4-(5-bromo-2-chloro-benzyl)-phenoxyl-tert-butyl-dimethyl-silane
A solution of 13.9 g 4-(5-bromo-2-chloro-benzyl)-phenol in 140 ml dichloromethane is cooled in an ice bath. Then 7.54 g tert-butyldimethylsilylchlorid in 20 ml dichloromethane are added followed by 9.8 ml triethylamine and 0.5 g 4- dimethylaminopyridine. The solution is stirred for 16 h at ambient temperature and then diluted with 100 ml dichloromethane. The organic phase is washed twice with aqueous 1 M hydrochloric acid and once with aqueous sodium hydrogen carbonate solution and then dried over sodium sulfate. After the solvent has been removed the residue is filtered through silica gel (cyclohexane/ethyl acetate 100:1 ). Yield: 16.8 g (87% of theory) Mass spectrum (El): m/z = 410/412/414 (Br+CI) [M]+
Example V

2.3.4.6-tetrakis-O-(trimethylsilyl)-D-glucopyranone
A solution of 20 g D-glucono-1 ,5-lactone and 98.5 ml Λ/-methylmorpholine in 200 ml of tetrahydrofuran is cooled to -5 0C. Then 85 ml trimethylsilylchloride are added dropwise so that the temperature does not exceed 5 0C. The solution is then stirred for 1 h at ambient temperature, 5 h at 35 0C and again for 14 h at ambient temperature. After the addition of 300 ml of toluene the solution is cooled in an ice bath, and 500 ml of water are added so that the temperature does not exceed 100C. The organic phase is then separated and washed in each case once with aqueous sodium dihydrogen phosphate solution, water and brine. The solvent is removed, the residue is taken up in 250 ml of toluene, and the solvent is again removed completely. Yield: 52.5 g (approx. 90% pure)
Mass spectrum (ESI+): m/z = 467 [M+H]+
Example Vl

1-chloro-4-(β-D-qlucopyranos-1-yl)-2-(4-hvdroxybenzyl)-benzene
A solution of 4.0 g [4-(5-bromo-2-chloro-benzyl)-phenoxy]-te/Tf-butyl-dimethyl-silane in 42 ml dry diethyl ether is cooled to -800C under argon. 11.6 ml of a 1.7 M solution of te/if-butyllithium in pentane are slowly added dropwise to the cooled solution, and then the solution is stirred for 30 min at -80 0C. This solution is then added dropwise through a transfer needle, which is cooled with dry ice, to a solution of 4.78 g
2,3,4,6-tetrakis-O-(trimethylsilyl)-D-glucopyranone in 38 ml diethyl ether chilled to – 80 0C. The resulting solution is stirred for 3 h at -78 0C. Then a solution of 1.1 ml methanesulphonic acid in 35 ml of methanol is added and the solution is stirred for 16 h at ambient temperature. The solution is then neutralised with solid sodium hydrogen carbonate, ethyl acetate is added and the methanol is removed together with the ether. Aqueous sodium hydrogen carbonate solution is added to the remaining solution, and the resulting mixture is extracted four times with ethyl acetate. The organic phases are dried over sodium sulphate and evaporated down. The residue is dissolved in 30 ml acetonitrile and 30 ml dichloromethane and the solution is cooled to -10 0C. After the addition of 4.4 ml triethylsilane 2.6 ml boron trifluoride etherate are added dropwise so that the temperature does not exceed -5 0C. After the addition is complete the solution is stirred for another 5 h at -5 to -10 0C and then quenched by the addition of aqueous sodium hydrogen carbonate solution. The organic phase is separated, and the aqueous phase is extracted four times with ethyl acetate. The combined organic phases are dried over sodium sulfate, the solvent is removed, and the residue is purified by chromatography on silica gel (dichoromethane/methanol 1 :0->3:1 ). The product then obtained is an approx. 6:1 mixture of β/α which can be converted into the pure β-anomer by global acetylation of the hydroxy groups with acetic anhydride and pyridine in dichloromethane and recrystallization of the product from ethanol. The product thus obtained is converted into the title compound by deacetylation in methanol with aqueous 4 M potassium hydroxide solution. Yield: 1.6 g (46% of theory)
Mass spectrum (ESI+): m/z = 398/400 (Cl) [M+H]+
Preparation of the compound A:

1-chloro-4-(β-D-qlucopyranos-1-yl)-2-r4-(<fS)-tetrahvdrofuran-3-yloxy)-benzyll- benzene
0.19 g (f?)-3-(4-methylphenylsulfonyloxy)-tetrahydrofuran are added to a mixture of 0.20 g 1-chloro-4-(β-D-glucopyranos-1-yl)-2-(4-hydroxybenzyl)-benzene and 0.29 g cesium carbonate in 2.5 ml dimethylformamide. The mixture is stirred at 75 0C for 4 h, before another 0.29 g caesium carbonate and 0.19 g (f?)-3-(4-methylphenyl- sulfonyloxy)-tetrahydrofuran are added. After an additional 14 h stirring at 75 0C the mixture is cooled to ambient temperature and brine is added. The resulting mixture is extracted with ethyl acetate, the combined organic extracts are dried over sodium sulfate, and the solvent is removed. The residue is purified by chromatography on silica gel (dichloromethane/methanol 1 :0 -> 5:1 ). Yield: 0.12 g (49% of theory) Mass spectrum (ESI+): m/z = 451/453 (Cl) [M+H] +
TOFOGLIFLOZIN » All About Drugs
TOFOGLIFLOZIN » All About Drugs
CLICK ABOVE for full article
ALSO same article at
SEE……..http://apisynthesisint.blogspot.in/2015/12/tofogliflozin.html
SEE ALL FLOZINS AT
EG, Dapagliflozin, canagliflozin and all
http://medcheminternational.blogspot.in/p/flozin-series.html
DAPAGLIFLOZIN SEES LIGHT

DAPAGLIFLOZIN, BMS-512148
(2S,3R,4R,5S,6R)-2-[4-chloro-3-(4-ethoxybenzyl)phenyl]-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol,
cas 461432-26-8
| Molecular Formula: C21H25ClO6 |
| Molecular Weight: 408.87 |
Bristol-Myers Squibb (Originator)
AstraZeneca
TYPE 2 DIABETES,SGLT-2 Inhibitors
launched 2012, as forxiga in EU

Dapagliflozin propanediol is a solvate containing 1:1:1 ratio of the dapagliflozin, (S)-(+)-1,2-propanediol, and water.
US——-In 2011, the product was not recommended for approval by the FDA’s Endocrinologic and Metabolic Drugs Advisory Committee. In 2011, the FDA assigned a complete response letter to the application. A new application was resubmitted in 2013 by Bristol-Myers Squibb and AstraZeneca in the U.S
WILMINGTON, Del. & PRINCETON, N.J.--(BUSINESS WIRE)--December 12, 2013--
AstraZeneca (NYSE:AZN) and Bristol-Myers Squibb Company (NYSE:BMY) today announced the U.S. Food and Drug Administration’s (FDA) Endocrinologic and Metabolic Drugs Advisory Committee (EMDAC) voted 13-1 that the benefits of dapagliflozin use outweigh identified risks and support marketing of dapagliflozin as an adjunct to diet and exercise to improve glycemic control in adults with type 2 diabetes mellitus. The Advisory Committee also voted 10-4 that the data provided sufficient evidence that dapagliflozin, relative to comparators, has an acceptable cardiovascular risk profile.
The FDA is not bound by the Advisory Committee’s recommendation but takes its advice into consideration when reviewing the application for an investigational agent. The Prescription Drug User Fee Act (PDUFA) goal date for dapagliflozin is Jan. 11, 2014.

Dapagliflozin is being reviewed by the FDA for use as monotherapy, and in combination with other antidiabetic agents, as an adjunct to diet and exercise to improve glycemic control in adults with type 2 diabetes. It is a selective and reversible inhibitor of sodium-glucose cotransporter 2 (SGLT2) that works independently of insulin to help remove excess glucose from the body. Dapagliflozin, an investigational compound in the U.S., was the first SGLT2 inhibitor to be approved anywhere in the world. Dapagliflozin is currently approved under the trade name [Forxiga](TM) for the treatment of adults with type 2 diabetes, along with diet and exercise, in 38 countries, including the European Union and Australia.
http://online.wsj.com/article/PR-CO-20131212-910828.html?dsk=y
………………………………………………………………..
PATENTS
WO 2010138535
WO 2011060256
WO 2012041898
WO 2012163990
WO 2013068850
WO 2012163546
WO 2013068850
WO 2013079501

Dapagliflozin (INN/USAN,[1] trade name Forxiga) is a drug used to treat type 2 diabetes. It was developed by Bristol-Myers Squibb in partnership with AstraZeneca. Although dapagliflozin’s method of action would operate on both types of diabetes[1] and other conditions resulting inhyperglycemia, the current clinical trials specifically exclude participants with type 1 diabetes.[2][3]
In July 2011 an US Food and Drug Administration (FDA) committee recommended against approval until more data was available.[4] The Prescription Drug User Fee Act (PDUFA) date for dapagliflozin for the treatment of Type 2 diabetes was extended three months by the FDA to January 28, 2012.
In April 2012, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency issued a positive opinion on the drug. It is now marketed in a number of European countries including the UK and Germany.
Dapagliflozin inhibits subtype 2 of the sodium-glucose transport proteins (SGLT2), which is responsible for at least 90% of the glucose reabsorption in the kidney. Blocking this transporter causes blood glucose to be eliminated through the urine.[5] The efficacy of the this medication class has yet to be determined, but in initial clinical trials, dapagliflozin lowers HbA1c by 0.90 percentage points when added to metformin.[6]
Type II diabetes is the most common form of diabetes accounting for 90% of diabetes cases. Over 100 million people worldwide have type-2 diabetes (nearly 17 million in the U.S.) and the prevalence is increasing dramatically in both the developed and developing worlds. Type-II diabetes is a lifelong illness, which generally starts in middle age or later part of life, but can start at any age. Patients with type-2 diabetes do not respond properly to insulin, the hormone that normally allows the body to convert blood glucose into energy or store it in cells to be used later. The problem in type-2 diabetes is a condition called insulin resistance where the body produces insulin, in normal or even high amounts, but certain mechanisms prevent insulin from moving glucose into cells. Because the body does not use insulin properly, glucose rises to unsafe levels in the blood, the condition known as hyperglycemia.
Hyperglycemia, that is, elevated plasma glucose, is a hallmark of diabetes. Plasma glucose is normally filtered in the kidney in the glomerulus but is actively reabsorbed in the proximal tubule (kidney). Sodium-dependent glucose co-transporter SGLT2 appears to be the major transporter responsible for the reuptake of glucose at this site. The SGLT inhibitor phlorizin, and closely related analogs, inhibit this reuptake process in diabetic rodents and dogs, resulting in normalization of plasma glucose levels by promoting glucose excretion without hypoglycemic side effects. Long term (6 month) treatment of Zucker diabetic rats with an SGLT2 inhibitor has been reported to improve insulin response to glycemia, improve insulin sensitivity, and delay the onset of nephropathy and neuropathy in these animals, with no detectable pathology in the kidney and no electrolyte imbalance in plasma. Selective inhibition of SGLT2 in diabetic patients would be expected to normalize plasma glucose by enhancing the excretion of glucose in the urine, thereby improving insulin sensitivity and delaying the development of diabetic complications.
The treatment of diabetes is an important health concern and despite a wide range of available therapies, the epidemic continues. Type 2 diabetes (T2DM) is a progressive disease caused by insulin resistance and decreased pancreatic β-cell function. Insulin is produced by the pancreatic β-cell and mediates cellular glucose uptake and clearance. Insulin resistance is characterized by the lack of response to the actions of this hormone which results in decreased cellular clearance of glucose from the circulation and overproduction of glucose by the liver.
The currently available therapies to treat type 2 diabetes augment the action or delivery of insulin to lower blood glucose. However, despite therapy, many patients do not achieve control of their type 2 diabetes. According to the National Health and Nutrition Examination Survey (NHANES) III, only 36% of type 2 diabetics achieve glycemic control defined as a A1C<7.0% with current therapies. In an effort to treat type 2 diabetes, aggressive therapy with multiple pharmacologic agents may be prescribed. The use of insulin plus oral agents has increased from approximately 3 to 11% from NHANES II to III.
Thus, treatment of hyperglycemia in type 2 diabetes (T2DM) remains a major challenge, particularly in patients who require insulin as the disease progresses. Various combinations of insulin with oral anti-diabetic agents (OADs) have been investigated in recent years, and an increasing number of patients have been placed on these regimens. Poulsen, M. K. et al., “The combined effect of triple therapy with rosiglitazone, metformin, and insulin in type 2 diabetic patients”,Diabetes Care, 26 (12):3273-3279 (2003); Buse, J., “Combining insulin and oral agents”, Am. J. Med., 108 (Supp. 6a):23S-32S (2000). Often, these combination therapies become less effective in controlling hyperglycemia over time, particularly as weight gain and worsening insulin resistance impair insulin response pathways.
Hypoglycemia, weight gain, and subsequent increased insulin resistance are significant factors that limit optimal titration and effectiveness of insulin. (Holman, R. R. et al., “Addition of biphasic, prandial, or basal insulin to oral therapy in type 2 diabetes”, N. Engl. J. Med., 357 (17):1716-1730 (2007)). Weight gain with insulin therapy is predominantly a consequence of the reduction of glucosuria, and is thought to be proportional to the correction of glycemia. (Makimattila, S. et al., “Causes of weight gain during insulin therapy with and without metformin in patients with Type II diabetes mellitus”, Diabetologia, 42 (4):406-412 (1999)). Insulin drives weight gain when used alone or with OADs. (Buse, J., supra). In some cases, intensive insulin therapy may worsen lipid overload and complicate progression of the disease through a spiral of caloric surplus, hyperinsulinemia, increased lipogenesis, increased adipocity, increased insulin resistance, beta-cell toxicity, and hyperglycemia. (Unger, R. H., “Reinventing type 2 diabetes: pathogenesis, treatment, and prevention”, JAMA, 299 (10):1185-1187 (2008)). Among commonly used OADs, thiazolidinediones (TZDs) and sulfonylureas intrinsically contribute to weight gain as glucosuria dissipates with improved glycemic control. Weight gain is less prominent with metformin, acting through suppression of hepatic glucose output, or with incretin-based DPP-4 inhibitors. Overall, there is a pressing need for novel agents that can be safely added to insulin-dependent therapies to help achieve glycemic targets without increasing the risks of weight gain or hypoglycemia.
A novel approach to treating hyperglycemia involves targeting transporters for glucose reabsorption in the kidney. (Kanai, Y. et al., “The human kidney low affinity Na+/glucose cotransporter SGLT2. Delineation of the major renal reabsorptive mechanism for D-glucose”, J. Clin. Invest., 93 (1):397-404 (1994)). Agents that selectively block the sodium-glucose cotransporter 2 (SGLT2) located in the proximal tubule of the kidney can inhibit reabsorption of glucose and induce its elimination through urinary excretion. (Brown, G. K., “Glucose transporters: structure, function and consequences of deficiency”, J. Inherit. Metab. Dis., 23 (3):237-246 (2000)). SGLT2 inhibition has been shown in pre-clinical models to lower blood glucose independently of insulin. (Han, S. et al., “Dapagliflozin, a selective SGLT2 inhibitor, improves glucose homeostasis in normal and diabetic rats”, Diabetes, 57 (6):1723-1729 (2008); Katsuno, K. et al., “Sergliflozin, a novel selective inhibitor of low-affinity sodium glucose cotransporter (SGLT2), validates the critical role of SGLT2 in renal glucose reabsorption and modulates plasma glucose level”, J. Pharmacol. Exp. Ther., 320 (1):323-330 (2007)).
Dapagliflozin(BMS-512148) is a potent sodium-glucose transport proteins inhibitor with IC50 of 1.1 nM and 1.4uM for SGLT2 and SGLT1, respectively. Dapagliflozin (BMS-512148) inhibits subtype 2 of the sodium-glucose transport proteins (SGLT2), which is responsible for at least 90% of the glucose reabsorption in the kidney. Blocking this transporter causes blood glucose to be eliminated through the urine. Symptoms of hypoglycaemia occurred in similar proportions of patients in the dapagliflozin (2~4%) and placebo groups (3%). Signs, symptoms, and other reports suggestive of genital infections were more frequent in the dapagliflozin groups (2•5 mg, [8%]; 5 mg, [13%]; 10 mg, [9%]) than in the placebo group ( [5%]).
Dapagliflozin (which is disclosed in U.S. Pat. No. 6,515,117) is an inhibitor of sodium-glucose reabsorption by the kidney, by inhibiting SGLT2, which results in an increased excretion of glucose in the urine. This effect lowers plasma glucose in an insulin-independent manner.
Dapagliflozin is currently undergoing clinical development for treatment of type 2 diabetes. (Han, S. et al., supra; Meng, W. et al., “Discovery of dapagliflozin: a potent, selective renal sodium-dependent glucose cotransporter 2 (SGLT2) inhibitor for the treatment of type 2 diabetes”, J. Med. Chem., 51 (5):1145-1149 (2008)). Phase 2a and 2b studies with dapagliflozin have demonstrated efficacy in reducing hyperglycemia either alone or in combination with metformin in patients with T2DM. (Komoroski, B. et al., “Dapagliflozin, a novel, selective SGLT2 inhibitor, improved glycemic control over 2 weeks in patients with type 2 diabetes mellitus”, Clin. Pharmacol. Ther., 85 (5):513-519 (2009); List, J. F. et al., “Dapagliflozin-induced glucosuria is accompanied by weight loss in type 2 diabetic patients”, 68th Scientific Sessions of the American Diabetes Association, San Francisco, Calif., Jun. 6-10, 2008, Presentation No. 0461P).
It has been found that dapagliflozin does not act through insulin signaling pathways and is effective in controlling blood sugar in patients whose insulin signaling pathways do not work well. This applies to extremes of insulin resistance, in type 2 diabetes as well as in insulin resistance syndromes, caused by, for example, mutations in the insulin receptor.
Since dapagliflozin leads to heavy glycosuria (sometimes up to about 70 grams per day) it can lead to rapid weight loss and tiredness. The glucose acts as an osmotic diuretic (this effect is the cause of polyuria in diabetes) which can lead to dehydration. The increased amount of glucose in the urine can also worsen the infections already associated with diabetes, particularly urinary tract infections and thrush (candidiasis). Dapagliflozin is also associated with hypotensive reactions.

The IC50 for SGLT2 is less than one thousandth of the IC50 for SGLT1 (1.1 versus 1390 nmol/l), so that the drug does not interfere with the intestinal glucose absorption.[7]
- Statement on a nonproprietory name adopted by the USAN council
- Efficacy and Safety of Dapagliflozin, Added to Therapy of Patients With Type 2 Diabetes With Inadequate Glycemic Control on Insulin, ClinicalTrials.gov, April 2009
- Trial Details for Trial MB102-020, Bristol-Myers Squibb, May 2009
- “FDA panel advises against approval of dapagliflozin”. 19 July 2011.
- Prous Science: Molecule of the Month November 2007
- UEndocrine: Internet Endocrinology Community
- Schubert-Zsilavecz, M, Wurglics, M, Neue Arzneimittel 2008/2009
- more1) Pal, Manojit et al; Improved Process for the preparation of SGLT2 inhibitor dapagliflozin via glycosylation of 5-bromo-2-Chloro-4′-ethoxydiphenylmethane with Gluconolactone ;. Indian Pat Appl,. 2010CH03942 , 19 Oct 20122) Lemaire, Sebastien et al; Stereoselective C-Glycosylation Reactions with Arylzinc Reagents ;Organic Letters , 2012, 14 (6), 1480-1483;3) Zhuo, Biqin and Xing, Xijuan; Process for preparation of Dapagliflozin amino acid cocrystals ;Faming Zhuanli Shenqing , 102 167 715, 31 Aug 20114) Shao, Hua et al; Total synthesis of SGLT2 inhibitor Dapagliflozin ; Hecheng Huaxue , 18 (3), 389-392; 2010
5) Liou, Jason et al; Processes for the preparation of C-Aryl glycoside amino acid complexes as potential SGLT2 Inhibitors ;. PCT Int Appl,. WO2010022313
6) Seed, Brian et al; Preparation of Deuterated benzyl-benzene glycosides having an inhibitory Effect on sodium-dependent glucose co-transporter; . PCT Int Appl,. WO2010009243
7) Song, Yanli et al; Preparation of benzylbenzene glycoside Derivatives as antidiabetic Agents ;. PCT Int Appl,. WO2009026537
8) Meng, Wei et al; D iscovery of Dapagliflozin: A Potent, Selective Renal Sodium-Dependent Glucose cotransporter 2 (SGLT2) Inhibitor for the Treatment of Type 2 Diabetes ; Journal of Medicinal chemistr y, 2008, 51 (5), 1145 -1149;
9) Gougoutas, Jack Z. et al; Solvates Crystalline complexes of amino acid with (1S)-1 ,5-anhydro-LC (3 – ((phenyl) methyl) phenyl)-D-glucitol were prepared as for SGLT2 Inhibitors the treatment of Diabetes ;. PCT Int Appl,. WO2008002824
10) Deshpande, Prashant P. et al; Methods of producing C-Aryl glucoside SGLT2 Inhibitors ;.. U.S. Pat Appl Publ,. 20,040,138,439

dapagliflozin being an inhibitor of sodiumdependent glucose transporters found in the intestine and kidney (SGLT2) and to a method for treating diabetes, especially type II diabetes, as well as hyperglycemia, hyperinsulinemia, obesity, hypertriglyceridemia, Syndrome X, diabetic
complications, atherosclerosis and related diseases, employing such C-aryl glucosides alone or in combination with one, two or more other type antidiabetic agent and/or one, two or more other type therapeutic agents such as hypolipidemic agents.
Approximately 100 million people worldwide suffer from type II diabetes (NIDDM – non-insulin-dependent diabetes mellitus), which is characterized by hyperglycemia due to excessive hepatic glucose production and peripheral insulin resistance, the root causes for which are as yet unknown. Hyperglycemia is considered to be the major risk factor for the development of diabetic complications, and is likely to contribute directly to the impairment of insulin secretion seen in advanced NIDDM. Normalization of plasma glucose in NIDDM patients would be predicted to improve insulin action, and to offset the development of diabetic complications. An inhibitor of the sodium-dependent glucose transporter SGLT2 in the kidney would be expected to aid in the normalization of plasma glucose levels, and perhaps body weight, by enhancing glucose excretion.
Dapagliflozin can be prepared using similar procedures as described in U.S. Pat. No. 6,515,117 or international published applications no. WO 03/099836 and WO 2008/116179
WO 03/099836 A1 refers to dapagliflozin having the structure according to formula 1 .

formula 1
WO 03/099836 A1 discloses a route of synthesis on pages 8-10, whereby one major step is the purification of a compound of formula 2

formula 2
The compound of formula 2 provides a means of purification for providing a compound of formula 1 since it crystallizes. Subsequently the crystalline form of the compound of formula 2 can be deprotected and converted to dapagliflozin. Using this process, dapagliflozin is obtained as an amorphous glassy off-white solid containing 0.1 1 mol% of EtOAc. Crystallization of a pharmaceutical drug is usually advantageous as it provides means for purification also suitable for industrial scale preparation. However, for providing an active pharmaceutical drug a very high purity is required. In particular, organic impurities such as EtOAc either need to be avoided or further purification steps are needed to provide the drug in a
pharmaceutically acceptable form, i.e. substantially free of organic solvents. Thus, there is the need in the art to obtain pure and crystalline dapagliflozinwhich is substantially free of organic solvents.
WO 2008/002824 A1 discloses several alternative solid forms of dapagliflozin, such as e.g. solvates containing organic alcohols or co-crystals with amino acids such as proline and phenylalanine. For instance, the document discloses crystalline
dapagliflozin solvates which additionally contain water molecules (see e.g.
Examples 3-6), but is silent about solid forms of dapagliflozin which do not contain impurities such as organic alcohols. As described above, it is desirable to provide the pharmaceutical active drug in a substantially pure form, otherwise triggering further expensive and time-consuming purification steps. In contrast, the document relates to dapagliflozin solvates where an alcohol and water are both incorporated into the crystal lattice. Hence, there is the need in the art to obtain pure and crystalline dapagliflozin suitable for pharmaceutical production.
WO 2008/1 16179 A1 refers to an immediate release pharmaceutical composition comprising dapagliflozin and propylene glycol. Propylene glycol is a chiral
substance and (S)-propylene glycol used is very expensive. Consequently, also the immediate release pharmaceutical composition is more expensive.
Crystalline forms (in comparision to the amorphous form) often show desired different physical and/or biological characteristics which may assist in the manufacture or formulation of the active compound, to the purity levels and uniformity required for regulatory approval. As described above, it is desirable to provide the pharmaceutical active drug in a substantially pure form, otherwise triggering further expensive and time-consuming purification steps.
…..
WO 2008/ 1 16179 Al seems to disclose an immediate release formulation comprising dapagliflozin and propylene glycol hydrate. WO 2008/ 116195 A2 refers to the use of an SLGT2 inhibitor in the treatment of obesity
http://www.google.com/patents/US20120282336
http://www.tga.gov.au/pdf/auspar/auspar-dapagliflozin-propanediol-monohydrate-130114.pdf
Example 2 Dapagliflozin (S) PGS—(2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (S)-propane-1,2-diol hydrate (1:1:1)
Dapagliflozin (S) propylene glycol hydrate (1:1:1) can be prepared using similar procedures as described in published applications WO 08/002824 and WO 2008/116179, the disclosures of which are herein incorporated by reference in their entirety for any purpose. SGLT2 EC50=1.1 nM.
Example 3 Dapagliflozin (R) PGS—(2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (R)-propane-1,2-diol hydrate (1:1:1)
Dapagliflozin (R) propylene glycol hydrate (1:1:1) can be prepared using similar procedures as described in WO 08/002824 and WO 2008/116179, the disclosures of which are herein incorporated by reference in their entirety for any purpose. SGLT2 EC50=1.1 nM.
WO 2008/002824 A1 discloses several alternative solid forms of dapagliflozin, such as e.g. solvates containing organic alcohols or co-crystals with amino acids such as proline and phenylalanine. For instance, the document discloses crystalline
dapagliflozin solvates which additionally contain water molecules (see e.g.
Examples 3-6), but is silent about solid forms of dapagliflozin which do not contain impurities such as organic alcohols. As described above, it is desirable to provide the pharmaceutical active drug in a substantially pure form, otherwise triggering further expensive and time-consuming purification steps. In contrast, the document relates to dapagliflozin solvates where an alcohol and water are both incorporated into the crystal lattice. Hence, there is the need in the art to obtain pure and crystalline dapagliflozin suitable for pharmaceutical production.
WO 2008/1 16179 A1 refers to an immediate release pharmaceutical composition comprising dapagliflozin and propylene glycol. Propylene glycol is a chiral
substance and (S)-propylene glycol used is very expensive. Consequently, also the immediate release pharmaceutical composition is more expensive.
Surprisingly, amorphous dapagliflozin can be purified with the process of the present invention. For instance amorphous dapagliflozin having a purity of 99,0% can be converted to crystalline dapagliflozin hydrate having a purity of 100% (see examples of the present application). Moreover, said crystalline dapagliflozin hydrate does not contain any additional solvent which is desirable. Thus, the process of purifying dapagliflozin according to the present invention is superior compared with the process of WO 03/099836 A1 .
Additionally, the dapagliflozin hydrate obtained is crystalline which is advantageous with respect to the formulation of a pharmaceutical composition. The use of expensive diols such as (S)-propanediol for obtaining an immediate release pharmaceutical composition as disclosed in WO 2008/1 16179 A1 can be avoided
………………………………
In Vitro Characterization and Pharmacokinetics of Dapagliflozin …
dmd.aspetjournals.org/content/…/DMD29165_supplemental_data_.doc

Dapagliflozin (BMS-512148), (2S,3R,4R,5S,6R)-2-(3-(4-Ethoxybenzyl)-4-chlorophenyl)
-6-hydroxymethyl-tetrahydro-2H-pyran-3,4,5-triol. 1H NMR (500 MHz, CD3OD) δ 7.33
(d, J = 6.0, 1H), 7.31 (d, J = 2.2, 1H), 7.31 (dd, J = 2.2, 6.0, 1H), 7.07 (d, J = 8.8, 2H),
6.78 (d, J = 8.8, 2H), 4.07-3.90 (m, 7H), 3.85 (d, J = 10.6, 1H), 3.69 (dd, J = 5.3, 10.6,
1H), 3.42-3.25 (m, 4H), 1.34 (t, J = 7.0, 3H). 13C NMR (125 MHz, CD3OD) δ 158.8,
140.0, 139.9, 134.4, 132.9, 131.9, 130.8, 130.1, 128.2, 115.5, 82.9, 82.2, 79.7, 76.4, 71.9,
64.5, 63.1, 39.2, 15.2.
HRMS calculated for C21H25ClNaO6 (M+Na)+
For C21H25ClO6: C, 61.68; H, 6.16. Found: C, 61.16; H, 6.58.
: 431.1237; found 431.1234. Anal. Calcd
SECOND SET
1H NMR (500 MHz, CD3OD) δ 7.33 (d, J = 6.0, 1H), 7.31 (d, J = 2.2, 1H), 7.31 (dd, J = 2.2, 6.0, 1H), 7.07 (d, J = 8.8, 2H), 6.78 (d, J = 8.8, 2H), 4.07–3.90 (m, 7H), 3.85 (d, J = 10.6, 1H), 3.69 (dd, J = 5.3, 10.6, 1H), 3.42–3.25 (m, 4H), 1.34 (t, J = 7.0, 3H);
13C NMR (125 MHz, CD3OD) δ 158.8, 140.0, 139.9, 134.4, 132.9, 131.9, 130.8, 130.1, 128.2, 115.5, 82.9, 82.2, 79.7, 76.4, 71.9, 64.5, 63.1, 39.2, 15.2;
HRMS calcd for C21H25ClNaO6 (M + Na)+ 431.1237, found 431.1234. Anal. Calcd for C21H25ClO6: C, 61.68; H, 6.16. Found: C, 61.16; H, 6.58.
………………………
HPLC
-
HPLC measurements were performed with an Agilent 1100 series instrument equipped with a UV-vis detector set to 240 nm according to the following method:
Column: Ascentis Express RP-Amide 4.6 x 150 mm, 2.7 mm;
Column temperature: 25 °C
– Eluent A: 0.1 % formic acid in water
– Eluent B: 0.1 % formic acid in acetonitrile
– Injection volume: 3 mL
– Flow: 0.7 mL/min
– Gradient:Time [min] [%] B 0.0 25 25.0 65 26.0 70 29.0 70 29.5 25 35.0 25 ……………………..

……..
http://www.google.com/patents/WO2013068850A2?cl=en
EXAMPLE 24 – Synthesis of 2,4-di-6>-ieri-butyldiphenylsilyl-l-C-(4-chloro-3-(4- ethoxybenzyl)phenyl)- -D-glucopyranoside 2,4-di-6>-TBDPS-dapagliflozin; (IVj”))

[0229] l-(5-Bromo-2-chlorobenzyl)-4-ethoxybenzene (1.5 g, 4.6 mmol) and magnesium powder (0.54 g, 22.2 mmol) were placed in a suitable reactor, followed by THF (12 mL) and 1,2- dibromoethane (0.16 mL). The mixture was heated to reflux. After the reaction had initiated, a solution of l-(5-bromo-2-chlorobenzyl)-4-ethoxybenzene (4.5 g, 13.8 mmol) in THF (28 mL) was added dropwise. The mixture was allowed to stir for another hour under reflux, and was then cooled to ambient temperature, and then titrated to determine the concentration. The above prepared 4-chloro-3-[(4-ethoxyphenyl)methyl]phenyl magnesium bromide (31 mL, 10 mmol, 0.32 M in THF) and A1C13 (0.5 M in THF, 8.0 mL, 4.0 mmol) were mixed at ambient temperature to give a black solution, which was stirred at ambient temperature for 1 hour. To a solution of
I, 6-anhydro-2,4-di-6>-ieri-butyldiphenylsilyl- -D-glucopyranose (0.64 g, 1.0 mmol) in PhOMe (3.0 mL) at ambient temperature was added phenylmagnesium bromide (0.38 mL, 1.0 mmol, 2.6 M solution in Et20). After stirring for about 5 min the solution was then added into the above prepared aluminum mixture via syringe, followed by additional PhOMe (1.0 mL) to rinse the flask. The mixture was concentrated under reduced pressure (50 torr) at 60 °C (external bath temperature) to remove low-boiling point ethereal solvents and then PhOMe (6mL) was added. The reaction mixture was heated at 130 °C (external bath temperature) for 8 hours at which time HPLC assay analysis indicated a 51% yield of 2,4-di-6>-ieri-butyldiphenylsilyl-l-C-(4-chloro-3- (4-ethoxybenzyl)phenyl)- -D-glucopyranoside. After cooling to ambient temperature, the reaction was treated with 10% aqueous NaOH (1 mL), THF (10 mL) and diatomaceous earth at ambient temperature, then the mixture was filtered and the filter cake was washed with THF. The combined filtrates were concentrated and the crude product was purified by silica gel column chromatography (eluting with 1:30 EtOAc/77-heptane) affording the product 2,4-di-6>- ieri-butyldiphenylsilyl- 1 – -(4-chloro-3 -(4-ethoxybenzyl)phenyl)- β-D-glucopyranoside (0.30 g, 34%) as a white powder.
1H NMR (400 MHz, CDC13) δ 7.56-7.54 (m, 2H), 7.43-7.31 (m, 13H), 7.29-7.22 (m, 6H), 7.07- 7.04 (m, 2H), 7.00 (d, J= 2.0 Hz, IH), 6.87 (dd, J= 8.4, 2.0 Hz, IH), 6.83-6.81 (m, 2H), 4.18 (d, J= 9.6 Hz, IH), 4.02 (q, J= 6.9 Hz, 2H), 3.96 (d, J= 10.8 Hz, 2H), 3.86 (ddd, J= 11.3, 7.7, 1.1 Hz, IH), 3.76 (ddd, J= 8.4, 8.4, 4.8 Hz, IH), 3.56 (ddd, J= 9.0, 6.4, 2.4 Hz, IH), 3.50 (dd, J=
I I.4, 5.4 Hz, IH), 3.44 (dd, J= 9.4, 8.6 Hz, IH), 3.38 (dd, J= 8.8, 8.8 Hz, IH), 1.70 (dd, J= 7.8, 5.4 Hz, IH, OH), 1.42 (t, J= 6.8 Hz, 3H), 1.21 (d, J= 5.2 Hz, IH, OH), 1.00 (s, 9H), 0.64 (s, 9H); 13C NMR (100 MHz, CDC13) δ 157.4 (C), 138.8 (C), 137.4 (C), 136.3 (CH x2), 136.1 (CH x2), 135.2 (CH x2), 135.0 (C), 134.9 (CH x2), 134.8 (C), 134.2 (C), 132.8 (C), 132.0 (C), 131.6 (CH), 131.1 (C), 129.9 (CH x2), 129.7 (CH), 129.6 (CH), 129.5 (CH), 129.4 (CH), 129.2 (CH), 127.58 (CH x2), 127.57 (CH x2), 127.54 (CH x2), 127.31 (CH), 127.28 (CH x2), 114.4 (CH x2), 82.2 (CH), 80.5 (CH), 79.3 (CH), 76.3 (CH), 72.7 (CH), 63.4 (CH2), 62.7 (CH2), 38.2 (CH2), 27.2 (CH3 x3), 26.6 (CH3 x3), 19.6 (C), 19.2 (C), 14.9 (CH3). EXAMPLE 25 -Synthesis of dapagliflozin ((25,3R,4R,55,6/?)-2-[4-chloro-3-(4- ethoxybenzyl)phenyl]-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol; (Ij))

IVj’ U
[0230] A solution of the 2,4-di-6>-ieri-butyldiphenylsilyl-l-C-(4-chloro-3-(4- ethoxybenzyl)phenyl)- -D-glucopyranoside (60 mg, 0.068 mmol) in THF (3.0 mL) and TBAF (3.0 mL, 3.0 mmol, 1.0 M in THF) was stirred at ambient temperature for 15 hours. CaC03 (0.62 g), Dowex^ 50WX8-400 ion exchange resin (1.86 g) and MeOH (5mL) were added to the product mixture and the suspension was stirred at ambient temperature for 1 hour and then the mixture was filtrated through a pad of diatomaceous earth. The filter cake was rinsed with MeOH and the combined filtrates was evaporated under vacuum and the resulting residue was purified by column chromatography (eluting with 1 : 10 MeOH/DCM) affording dapagliflozin (30 mg).
1H NMR (400 MHz, CD3OD) δ 7.37-7.34 (m, 2H), 7.29 (dd, J= 8.2, 2.2 Hz, 1H), 7.12-7.10 (m, 2H), 6.82-6.80 (m, 2H), 4.10 (d, J= 9.6 Hz, 2H), 4.04 (d, J= 9.2 Hz, 2H), 4.00 (q, J= 7.1 Hz, 2H), 3.91-3.87 (m, 1H), 3.73-3.67(m, 1H), 3.47-3.40 (m, 3H), 3.31-3.23 (m, 2H), 1.37 (t, J= 7.0 Hz, 3H);
13C NMR (100 MHz, CD3OD) δ 157.4 (C), 138.6 (C), 138.5 (C), 133.1 (C), 131.5 (C), 130.5 (CH), 129.4 (CH x2), 128.7 (CH), 126.8 (CH), 114.0 (CH x2), 80.5 (CH), 80.8 (CH), 78.3 (CH), 75.0 (CH), 70.4 (CH), 63.0 (CH2), 61.7 (CH2), 37.8 (CH2), 13.8 (CH3);
LCMS (ESI) m/z 426 (100, [M+NH4]+), 428 (36, [M+NH4+2]+), 447 (33, [M+K]+).
Example 1 – Synthesis of l,6-anhydro-2,4-di-6>-ieri-butyldiphenylsilyl- -D-glucopyranose (II”)

III II”
[0206] To a suspension solution of l,6-anhydro- -D-glucopyranose (1.83 g, 11.3 mmol) and imidazole (3.07 g, 45.2 mmol) in THF (10 mL) at 0 °C was added dropwise a solution of TBDPSC1 (11.6 mL, 45.2 mmol) in THF (10 mL). After the l,6-anhydro-P-D-gJucopyranose was consumed, water (10 mL) was added and the mixture was extracted twice with EtOAc (20 mL each), washed with brine (10 mL), dried (Na2S04) and concentrated. Column
chromatography (eluting with 1 :20 EtOAc/rc-heptane) afforded 2,4-di-6>-ieri-butyldiphenylsilyl- l,6-anhydro- “D-glucopyranose (5.89 g, 81%).
1H NMR (400 MHz, CDC13) δ 7.82-7.70 (m, 8H), 7.49-7.36 (m, 12H), 5.17 (s, IH), 4.22 (d, J= 4.8 Hz, IH), 3.88-3.85 (m, IH), 3.583-3.579 (m, IH), 3.492-3.486 (m, IH), 3.47-3.45 (m, IH), 3.30 (dd, J= 7.4, 5.4 Hz, IH), 1.71 (d, J= 6.0 Hz, IH), 1.142 (s, 9H), 1.139 (s, 9H); 13C NMR (100 MHz, CDCI3) δ 135.89 (CH x2), 135.87 (CH x2), 135.85 (CH x2), 135.83 (CH x2), 133.8 (C), 133.5 (C), 133.3 (C), 133.2 (C), 129.94 (CH), 129.92 (CH), 129.90 (CH), 129.88 (CH), 127.84 (CH2 x2), 127.82 (CH2 x2), 127.77 (CH2 x4), 102.4 (CH), 76.9 (CH), 75.3 (CH), 73.9 (CH), 73.5 (CH), 65.4 (CH2), 27.0 (CH3 x6), 19.3 (C x2).
Perampanel

Perampanel
5′-(2-cyanophenyl)-1′-phenyl-2,3′-bipyridinyl-6′(1’H)-one
cas no 380917-97-5
FDA-approved drug to treat epilepsy. Trade name Fycompa, Eisai (Eisai) research and development.
FYCOMPA tablets contain perampanel, a non-competitive AMPA receptorantagonist. Perampanel is described chemically as 2-(2-oxo-1-phenyl-5-pyridin-2-yl-1,2-dihydropyridin-3-yl) benzonitrile hydrate (4:3).
The molecular formula is C23H15N3O •3/4H2O and the molecular weight is 362.90 (3/4 hydrate). The chemical structure of perampanel is:
 |
Perampanel is a white to yellowish white powder. It is freely soluble in N-methylpyrrolidone, sparingly soluble in acetonitrile and acetone, slightly soluble in methanol, ethanol and ethyl acetate, very slightly soluble in 1-octanol and diethyl ether and practically insoluble in heptane and water.
Perampanel (INN/USAN, trade name Fycompa) is an antiepileptic drug developed by Eisai Co. that acts as a selective noncompetitive antagonist of AMPA receptors, the major subtype of ionotropic glutamate receptors.[1][2]
Perampanel was found to be effective in the treatment of refractory partial-onset seizures in three pivotal (Phase 3) clinical trials[3][4] and has been approved for marketing under the brand name Fycompa by the European Medicines Agency.[5] The minimum effective dose is 4 mg once daily; doses of 8 mg and 12 mg daily provide a greater therapeutic benefit with a corresponding increase in adverse events. Dizziness and somnolence/sedation/fatigue are the most frequent dose-related adverse events. The drug is currently approved, for the control of partial-onset seizures, in those of both sexes who suffer from epilepsy and who are 12 years of age and older, by the Food and Drug Administration, and is considered to be a scheduled drug (an agent with the potential for addiction). Perampanel has been studied in other clinical indications includingParkinson’s disease.[6][7]
It has high potency (IC50 in vitro in functional studies of about 100-250 nM) and a prolonged terminal half-life in humans of approximately 105 hours. The drug is 95% bound to plasma protein. Its primary route of metabolism is by CYP3A4. It does not induce or inhibit P450 enzymes. About 70% of the dose is excreted in the feces and 30% in the urine; less than 2% of the dose is excreted unchanged into the urine.
In clinical trials, perampanel was generally well tolerated although the incidence of adverse events increased in a dose-dependent fashion. There was no increase in serious adverse events compared with placebo. According to the Food and Drug Administration, most common adverse reactions reported by patients receiving Fycompa in clinical trials include dizziness, drowsiness, fatigue, irritability, falls, upper respiratory tract infection,weight increase, vertigo, loss of muscle coordination (ataxia), gait disturbance, balance disorder, anxiety, blurred vision, stuttering (dysarthria), weakness (asthenia), aggression, and excessive sleep (hypersomnia).[8]
Fycompa’s label has a boxed warning to alert prescribers and patients about the risk of serious neuropsychiatric events. Some of these events were reported as serious and life-threatening. Violent thoughts or threatening behavior (including homicidal ideation) was also observed in a few patients. Patients and caregivers should alert a health care professional immediately if changes in mood or behavior that are not typical for the patient are observed. Health care professionals should closely monitor patients during the titration period when higher doses are used.[9]
- Rogawski, M. A. (2011). “Revisiting AMPA Receptors as an Antiepileptic Drug Target”. Epilepsy Currents 11 (2): 56–63. doi:10.5698/1535-7511-11.2.56. PMC 3117497. PMID 21686307. edit
- Rogawski MA, Hanada T. Preclinical pharmacology of perampanel, a selective non-competitive AMPA receptor antagonist. Acta Neurol Scand 2013;127 (Suppl. 197): 19–24.Rogawski, M. A.; Kaukinen, T.; Collin, P.; Krekelä, I.; Patrikainen, H.; Tillonen, J.; Nyrke, T.; Laurila, K.; Haimila, K.; Partanen, J.; Valve, R.; Mäki, M.; Luostarinen, L. (2013). “Preclinical pharmacology of perampanel, a selective non-competitive AMPA receptor antagonist”. Acta Neurologica Scandinavica 127 (1): 19–25. doi:10.1111/ane.12100. PMID 22494246. edit
- Krauss, G. L.; Serratosa, J. M.; Villanueva, V.; Endziniene, M.; Hong, Z.; French, J.; Yang, H.; Squillacote, D.; Edwards, H. B.; Zhu, J.; Laurenza, A. (2012). “Randomized phase III study 306: Adjunctive perampanel for refractory partial-onset seizures”. Neurology 78 (18): 1408–1415.doi:10.1212/WNL.0b013e318254473a. PMID 22517103. edit
- French, J. A.; Krauss, G. L.; Biton, V.; Squillacote, D.; Yang, H.; Laurenza, A.; Kumar, D.; Rogawski, M. A.; Campanille, V.; Floridia, J.; Ilari, R.; Consalvo, D. E.; Thomson, A.; Sfaello, I.; Pociecha, J.; Nieto, F.; Firstenfeld, A.; Zuin, D.; Mesri, J.; Silva, W.; Nofal, P.; Cristalli, D.; Clement, J. F.; Hwang, P.; McLachlan, R.; Pillay, N.; Lasso, J.; Peralta, B. L.; Hernandez, M. L.; Tenhamm, E. (2012). “Adjunctive perampanel for refractory partial-onset seizures: Randomized phase III study 304”. Neurology 79 (6): 589–596. doi:10.1212/WNL.0b013e3182635735. PMC 3413761. PMID 22843280. edit
- “European Medicines Agency Report on Perampanel”.
- Gottwald MD, Aminoff MJ (July 2008). “New frontiers in the pharmacological management of Parkinson’s disease”. Drugs Today 44 (7): 531–45.doi:10.1358/dot.2008.44.7.1217105. PMID 18806903.
- http://www.webmd.com/epilepsy/news/20121024/epilepsy-drug-fycompa-approved
- http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm325038.htm
- http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm325038.htm
Perampanel structure is formed by the coupling of an aromatic ring . Pyridone centrally located, surrounded by connecting two benzene rings and a pyridine ring. The synthesis of 2,5 – dibromopyridine (1) Start with sodium methoxide to produce 2-substituted, and an organic tin compound occurs Stille Coupling 3 4 4 HBr generated after acid hydrolysis and coupling of benzyl bromide with NBS to give 5,5. After 6 coupling of boronic ester and get Perampanel.
Perampanel is a pharmaceutically active agent, currently in clinical phase 3. It can be used to treat Parkinson’s disease, epilepsy and multiple sclerosis.
Perampanel, having the following chemical formula

is also known as E 2007, ER 155055-90 and 3-(2-cyanophenyl)-1-phenyl-5-(2-pyridil)-1,2-dihydropyridin-2-one
Various methods of synthesis of such molecules are known, such as those reported in EP1300396, EP 1465626, EP 1772450, EP 1764361 and EP 1970370.
Many of the methods of synthesis of such active substances reported by the prior art use the key intermediate 5-(2-pyridil)-1,2-dihydropyridin-2-one also known as 2,3′-bipyridin-6′(1′H)-one having the following chemical formula:

Other methods use the synthetic precursor of this intermediate known as 2-methoxy-5-(pyridin-2-yl)pyridine or 6′-methoxy-2,3′-bipyridine having the formula:

2,3′-bipyridin-6′(1′H)-one. it is in fact prepared by simple acid-catalysed demethylation of the 6′-methoxy-2,3′-bipyridine as is reported in the prior art.
Various ways of synthesising 2-methoxy-5-(pyridin-2-yl)pyridine are known. The process summarised in Diagram (I) below is described in WO 2001096308:

Such process highlights clear disadvantages such as the need to operate in cryogenic conditions (T=−78° C.) using special equipment and the need to isolate boronic acid via work-up. In addition the use of 2-Bromopyridine is required, which exacerbates the production of waste compared to 2-chloropyridine.
Another process described in WO 2004009553 is summarised in Diagram (II):

Disadvantages of this process include the use of high molecular weight benzene-sulfonyl pyridine entailing a scarce atom-economy of the process and the need to operate at low temperature T (−78° C.) using special equipment.
Lastly, a completely different process is described in WO20087093392 for the preparation of 2,3′-bipyridin-6′(1′H)-one (Diagram (III)) which however does not include the preparation of the intermediate precursor 2-methoxy-5-(pyridin-2-yl)pyridine:

Perampanel and other 1 ,2-dihydropyridine compounds which possess antagonistic action against AMPA receptor and/or inhibitory action against kainate receptor are described in WO 01/96308. Example 7 in WO 01/96308 discloses a process for producing perampanel by reacting 3-(2-cyanophenyl)-5-(2-pyridyl)-2(lH)-pyridone with phenyl boronic acid, copper acetate and triethylamine in methylene chloride, followed by addition of concentrated aqueous ammonia, water and ethyl acetate. After work-up (phase separation, washing the organic phase and drying over magnesium sulfate), the solvent was concentrated in vacuo and the residue was purified by a silica gel column chromatography (ethyl acetate:hexane=l :2) to give the title product as pale yellow powder. There is no disclosure regarding the polymorphic nature of the product.
A new crystalline or amorphous form of a compound may possess physical properties that differ from, and are advantageous over, those of other crystalline or amorphous forms. These include, packing properties such as molar volume, density and hygroscopicity; thermodynamic properties such as melting temperature, vapor pressure and solubility; kinetic properties such as dissolution rate and stability under various storage conditions; surface properties such as surface area, wettability, interfacial tension and shape; mechanical properties such as hardness, tensile strength, compactibility, handling, flow and blend; and filtration properties. Variations in any one of these properties may affect the chemical and pharmaceutical processing of a compound as well as its bioavailability and may often render the new form advantageous for pharmaceutical and medical use.
EP 1764361 (US 2010/324297) discloses three anhydrous crystalline forms ofperampanel, designated Form I, Form III and Form V and a hydrate form ofperampanel. Anhydrous Form I is prepared in accordance with Example Dl by dissolving perampanel in ethyl acetate (EtOAc) under reflux, cooling the solution, seeding with anhydrous perampanel crystals, continued cooling and collecting the precipitated crystals. Anhydrous Form V is prepared in accordance with Example CI, by dissolving perampanel in acetone, heating to reflux and concentrating the solution to solidification, dissolving the solids in acetone-water, refluxing then cooling and collecting the precipitate. The hydrate form is prepared in accordance with Example Bl by dissolving perampanel in acetone-water, heating, cooling the solution, seeding with perampanel hydrate crystals, continued cooling and collecting the precipitated crystals. US 2009/0088574 discloses a crystalline form of perampanel designated Form IV, which is prepared by slurring perampanel in an acetone/water mixture.
US 7,803,818 discloses an amorphous form of perampanel which is prepared by spray drying perampanel from an acetone solution.
US 7,718,807 discloses acid addition salts of perampanel or a hydrate thereof, wherein the acid is selected from the group consisting of benzenesulfonic acid, p- toluenesulfonic acid, hydrochloric acid, hydrobromic acid, sulfuric acid, methanesulfonic acid, fumaric acid, tartaric acid, succinic acid and benzoic acid.
…………………………………………………………………
Perampanel aromatic ring structure is made of highly coupled. Pyridone centrally located, surrounded by connecting two benzene ring and a pyridine ring. The synthesis of 2,5 – dibromo pyridine ( 1) Start (Synthesis, 2012, 57), sodium methoxide instead of generating 2 , and organotin compounds 3 Stille Coupling occurs to generate 4 . 4 in HBr phenylboronic acid after hydrolysis and coupling to get 5 , 5 after bromination with NBS and borate 6 coupled to get Perampanel.
…………………………….
nmr
A Practical, Laboratory-Scale Synthesis of Perampanel
……………………
updated info
-
Perampanel is a pharmaceutical active substance, currently in clinical phase 3, used to treat Parkinson’s disease, epilepsy and multiple sclerosis.
-
[0003]Perampanel, having the following chemical formula
is also known as E 2007, ER 155055-90 and 3-(2-cyanophenyl)-1-phenyl-5-(2-pyridil)-1,2-dihydropyridin-2-one
-
[0004]Various methods of synthesis of such molecule are known, such as those reported in the patent publications EP1300396 , EP1465626 ,EP1772450 , EP1764361 and EP 1970370 .
-
[0005]Many of the methods of synthesis of such active substance reported by the prior art use the key intermediate 5-(2-pyridil)-1,2-dihydropyridin-2-one also known as 2,3′-bipyridin-6′(1’H)-one having the following chemical formula:
or use the synthetic precursor thereof named 2-methoxy-5-(pyridin-2-yl)pyridine or 6′-methoxy-2,3′-bipyridine having the formula:

2,3′-bipyridin-6′(1’H)-one is in fact prepared by simple acid-catalysed demethylation of the 6′-methoxy-2,3′-bipyridine as thoroughly reported in the prior art.
-
[0006]Various ways of synthesising 2-methoxy-5-(pyridin-2-yl)pyridine are known. The process summarised in the diagram (I) below is described in the publication WO 2001096308 :
Diagram (I)
-
[0007]
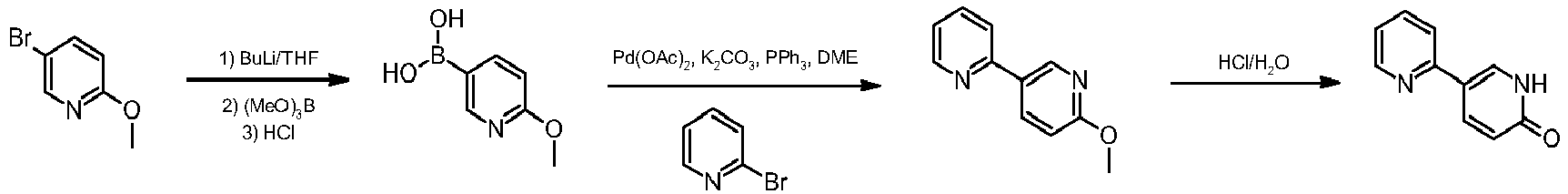
-
[0008]Such process highlights clear disadvantages such as the need to operate in cryogenic conditions (T=-78°C) using special equipment and the need to isolate the boronic acid via work-up; in addition the use of 2-Bromopyridine is envisaged, which is less convenient as regards the production of waste compared to 2-chloropyridine.
-
[0009]Another process described in WO 2004009553 is summarised in the diagram (II) :
Diagram (II)
-
[0010]
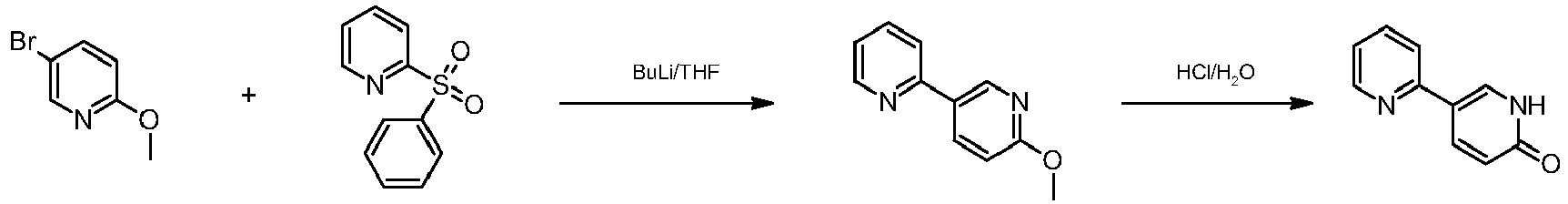
-
[0011]It presents clear disadvantages such as the use of high molecular weight benzenesulfonyl pyridine entailing a scarce atom-economy of the process and the need to operate at low temperature T (-78°C) using special equipment.
-
[0012]Lastly, a completely different process is described in WO20087093392for the preparation of 2,3′-bipyridin-6′(1’H)-one which however does not include the preparation of the intermediate precursor named 2-methoxy-5-(pyridin-2-yl)pyridine, process shown in the diagram (III) :
diagram (III)
-
[0013]

LOSARTAN

E-3340
L-158086
MK-0954
MK-954
Ex-89 (free acid)
COZAAR (losartan potassium, cas 124750-99-8) is an angiotensin II receptor (type AT1)antagonist. Losartan potassium, a nonpeptide molecule, is chemically described as 2-butyl-4-chloro-1-[p-(o-1H-tetrazol-5-ylphenyl)benzyl]imidazole-5-methanol monopotassium salt. Its empirical formula is C22H22ClKN6O, and its structural formula is:
 |
Losartan potassium is a white to off-white free-flowing crystalline powder with a molecular weight of 461.01. It is freely soluble in water, soluble in alcohols, and slightly soluble in common organic solvents, such as acetonitrile and methyl ethyl ketone. Oxidation of the 5-hydroxymethyl group on the imidazole ring results in the active metabolite of losartan.
COZAAR is available as tablets for oral administration containing either 25 mg, 50 mg or 100 mg of losartan potassium and the following inactive ingredients: microcrystalline cellulose, lactose hydrous, pregelatinized starch, magnesium stearate, hydroxypropyl cellulose, hypromellose, and titanium dioxide.
COZAAR 25 mg, 50 mg and 100 mg tablets contain potassium in the following amounts: 2.12 mg (0.054 mEq), 4.24 mg (0.108 mEq) and 8.48 mg (0.216 mEq), respectively. COZAAR 25 mg, COZAAR 50 mg, and COZAAR 100 mg may also contain carnauba wax.
|
Losartan (rINN) /loʊˈsɑrtən/ is an angiotensin II receptor antagonist drug used mainly to treat high blood pressure (hypertension). Losartan was the first angiotensin II antagonist to be marketed. Losartan potassium is marketed by Merck & Co. Inc. under the trade nameCozaar. Losartan is available in generic form.
As with all angiotensin II type 1 receptor (AT1) antagonists, losartan is indicated for the treatment of hypertension. It may also delay progression of diabetic nephropathy, and is also indicated for the reduction of renal disease progression in patients with type 2 diabetes, hypertension and microalbuminuria (>30 mg/24 hours) or proteinuria (>900 mg/24 hours).
Although clinical evidence shows calcium channel blockers and thiazide-type diuretics are preferred first-line treatments for most patients (from both efficacy and cost points of view), an angiotensin II receptor antagonist such as losartan is recommended as first-line treatment in patients under the age of 55 who cannot tolerate an ACE inhibitor.The LIFE study demonstrated losartan was significantly superior to atenolol in the primary prevention of adverse cardiovascular events (myocardial infarction or stroke), with a significant reduction in cardiovascular morbidity and mortality for a comparable reduction in blood pressure. A study hints that losartan has a beneficial effect on mitochondria by reversing age related dysfunction in maintaining normal blood pressure and cellular energy usage. The maximal effects on blood pressure usually occur within 3–6 weeks upon starting losartan.
Losartan is also available as hydrochlorothiazide/losartan, a combination drug with a low dose thiazide diuretic to achieve an additive antihypertensive effect.

-
Activation of AT1 receptors in the outer membrane of vascular smooth muscle cells of the heart and arteries causes those tissues to constrict. Blocking of vasoconstriction mediated by AT1 receptors has been found to be beneficial to patients with hypertension.
-
[0003]AT1 receptors are activated by an octa-peptide, angiotensin II. Angiotensin II helps to maintain constant blood pressure despite fluctuations in a person’s state of hydration, sodium intake and other physiological variables. Angiotensin II also performs the regulatory tasks of inhibiting excretion of sodium by the kidneys, inhibiting norephedrin reuptake and stimulating aldosterone biosynthesis.
-
[0004]Inhibiting angiotensin II binding to AT1 receptors with an AT1 receptor antagonist disrupts the vasoconstriction mediated by AT1 receptors that contributes to hypertension.
-
[0005]In the early 1970s, it was discovered that certain oligopeptides competitively inhibited angiotensin receptors (at that time the existence of two receptor subtypes, AT1 and AT2, was unknown). This discovery spurred interest in development of therapeutic oligopeptides with increased potency, but interest in peptide analogs waned due in part to their poor oral bioavailability.
-
[0006]In 1982, Furukawa. Kishimoto and Nishikawa of Taketa Chemical Indus. discovered a class of non-peptide-containing imidazoles that also inhibited the vasoconstriction effect of angiotensin II. See U.S. Patents Nos. 4,340,598 and 4,355,040. Later, U.S. Patent No. 5,138,069 was obtained by Carini, Denucia and Pancras of E.I. DuPont de Nemours on another class of imidazoles, which encompasses the compound losartan. In 1995, losartan (CA Index: 2-butyl-4-chloro-1-[[2′-(1H-tetrazol-5-yl) [1,1′-biphenyl] -4-yl]methyl]-1H-imidazole-5-methanol) (formula I):

became the first nonpeptide AT1 antagonist approved by the U.S. Food and Drug Administration for clinical use. Losartan can be administered orally as its monopotassium salt. Losartan potassium is available by prescription in tablet form as a sole active ingredient (Cozaar®: Merck) and as a co-active ingredient with hydrochlorothiazide (Hyzaar®: Merck).
-
[0007]Losartan has been prepared by a variety of synthetic pathways. In several of these synthetic pathways, the penultimate product is 2-butyl-4-chloro-1-[[2′-(2-triphenylmethyl-2H-tetrazol-5-yl) [1,1′-biphenyl] -4-yl]methyl]-1H-imidazole-5-methanol (“trityl losartan”). Trityl losartan is an intermediate in processes described in U.S. Patents Nos. 5,138,069; 5,962,500 and 5,206,374.
-
[0008]In a process described in Example 316 of U.S. Patent No. 5,138,069, the tetrazole ring of losartan is formed by reacting 1-[(2′-cyanobiphenyl-4-yl)methyl]-2-butyl-4-chloro-5-hydroxymethylimidazole with trimethyltin azide. The reaction gives a trimethylstannyl substituted tetrazole compound directly. The trimethylstannyl group is cleaved from the product by reacting with trityl chloride. This reaction results in attachment of the trityl group to the tetrazole ring. In the last step, the trityl group is cleaved with acid to give losartan (Scheme 1).

-
[0009]In the last step, trityl losartan was suspended in methanol and cooled to ~10°C. 3.4 N Hydrochloric acid was added to the slurry. After a period of time, the pH of the reaction mixture was raised to 13 with 10 N NaOH. Methanol was then distilled off while makeup water was added. After distillation, additional water and toluene were added. The toluene phase was separated and the aqueous phase was extracted once more with toluene. Ethyl acetate and acetic acid were then added to the aqueous phase. Losartan was recovered from the aqueous phase as a solid and further purified by slurrying in ethyl acetate. Losartan was obtained in 88.5% yield and 98.8% purity as determined by HPLC. This process is also described in U.S. Patents Nos. 5,128,355 and 5,155,188.
-
[0010]U.S. Patent No. 5,962,500, Examples 3-5, describe a process for preparing losartan in which the tetrazole ring of losartan is present in the starting material, 5-phenyltetrazole. The ‘500 patent process, depicted in Scheme 2, is convergent and uses a Suzuki coupling reaction (Miyaura, N.; Suzuki, A. Chem. Rev., 1995, 95, 2457) in the convergent step. On one branch of the synthesis, 5-phenyltetrazole is converted into the boronic acid coupling partner for the Suzuki reaction by ortho metalation with n-butyl lithium, followed by reaction with trisopropylborate. The tetrazole ring is protected from reacting with the strong allcyl lithium base with a trityl group. The trityl group is conventionally attached by reacting the tetrazole with trityl chloride in the presence of a non-nucleophilic base. On the other branch of the convergent synthesis, 2-n-butyl-4-chloro-1H-imidazole-5-carboxaldehyde is alkylated with 4-bromobenzylbromide, followed by reduction of the aldehyde with sodium borohydride to yield the other Suzuki coupling partner.

-
[0011]The direct product of Suzuki coupling is trityl losartan. In the next and last step, the tetrazole ring of trityl losartan is deprotected with 4N H2SO4 in THF. In that step, the acidic solution was aged overnight at 20 to 25°C. The solution was then extracted with isopropyl acetate and residual organic solvent was removed from the aqueous phase under vacuum. The solution was then carried forward to from the potassium salt without intermediate isolation of losartan. This process is also described in U.S.Patents Nos, 5,206,374, Example 21, and 5,310,928, Example 21.
-
[0012]Larsen, R.D et al. [J. Org. Chem. (1994), 59, 6391-6394] discloses a similar convergent synthesis of lasartan, whereby the trityl lasartan, generated by Suzuki coupling, is deprotected using 0.7 M H2SO4 in a 50 : 50 mixture of acetonitrile /water.
-
[0013]U.S. Patent No. 5,206,374 Examples 1 and 4-8 describe another process for making Iosartan that also involves a Suzuki coupling reaction. However, unlike the ‘500 patent process, the ‘374 patent process is not convergent. The ‘374 patent process is depicted in Scheme 3.

-
[0014]In the ‘374 patent process, as in the `500 patent process, the tetrazole ring of 5-phenyltetrazole is protected with a trityl group before orthometallation of the phenyl moiety with n-butyl lithium in preparation for making the boronic acid Suzuki coupling partner. In the Suzuki coupling step, the boronic acid is reacted with 4-bromotoluene. The methyl group attached to one of the phenyl rings of the Suzuki product is then halogenated with N-bromosuccinamide and the benzylic bromine atom of that product is displaced with 2-n-butyl-4-chloro-1H-imidazole-5-carboxaldehyde. Reduction of the aldehyde group with sodium borohydride yields trityl losartan. The tetrazole group of trityl losartan was deprotected with 12% aqueous HCl in THF. After 12 hours, the pH of the reaction mixture was raised to 12.5 with 30% NaOH. The THF was then distilled off while make-up water was added to the mixture. After distillation, the mixture was cooled and the triphenyl methanol byproduct of deprotection, which had precipitated, was removed by filtration. The filtrate and rinsate, with which it was combined, were extracted with toluene. Then, ethyl acetate was added and 36% HCI was added until the pH of the reaction mixture was lowered to 3.8. The mixture was cooled, causing losartan to precipitate from the solution. Losartan was obtained in 83% theoretical yield starting from trityl losartan.




EP 253310 discloses a process, wherein 2-n-butyl-4-chloro-1H-imidazolyl-5-methanol (III) is coupled with 5-(4′-bromomethyl-1,1′-biphenyl-2-yl)-2-triphenylmethyl-2H-tetrazole (IV) in N,N-dimethylformamide as solvent in presence of sodium methoxide as the base to furnish trityl losartan. The other bases that have been claimed are sodium hydride, alkali metal carbonates such as sodium carbonate and potassium carbonate and amine bases such as triethyl amine and pyridine.

The coupling reaction results in a mixture of trityl losartan and its regio isomer (V). These are separated by column chromatography.
U.S. Pat. Nos. 5,130,439 and 5,310,928 disclose a method for coupling (IV) and (VI) in N,N-dimethylacetamide solvent in the presence of anhydrous potassium carbonate as base. The imidazole aldehyde (VI) gives predominantly the desired regio isomer (VII). The intermediate VII is then reduced with sodium borohydride to furnish the trityl losartan. The product is isolated by extraction into toluene from aqueous N,N-dimethylacetamide, concentration of the toluene solution and crystallization using ethyl acetate or ethanol as solvent. The synthesis steps are depicted as follows.

In a process published in J. Med. Chem. (1991), 34, 2525-2547, Losartan is prepared by coupling (III) and (IV) in N,N-dimethylformamide in the presence of sodium methoxide. The desired compound is isolated after vacuum distillation of solvent followed by extractive work-up. The resultant product mixture is purified by chronmatography.
The U.S. Pat. Nos. 5,138,069, 5,128,355 and 5,155,118 describe a process for the preparation of losartan, wherein the tetrazole ring of losartan is formed by reacting 1-((2′-cyanobiphenyl-4-yl)methyl)-2-butyl-4-chloro-5-hydroxymethylimidazole with trimethyltin azide. The reaction results in trimethylstannyl substituted tetrazole compound, which is then reacted with trityl chloride and sodium hydroxide.

The trityl losartan thus formed is treated with 3.4N hydrochloric acid in methanol at about 10° C. to give losartan.
The U.S. Pat. Nos. 5,138,069, 5,128,355 and 5,155,118 also disclose another process for making trityl losartan, where in the coupling between IV and VI is carried out in a biphasic solvent system comprising of chlorinated solvent and water. The reaction is carried out at room temperature in presence of sodium hydroxide as the base and aliquat 336 as the phase transfer catalyst. The resulting intermediate VII is then reduced in situ with sodium borohydride to furnish trityl losartan.

U.S. Pat. No. 5,206,374, 5,310,928 and 5,962,500 disclose another process for preparing losartan in which 5-phenyltetrazole (X) is converted into the boronic acid coupling partner (XII) for the Suzuki reaction by tritylation of phenyltetrazole with trityl chloride in presence of a non-nucleophilic base, ortho metalation with n-butyl lithium, followed by reaction with triisopropylborate. 2-n-butyl-4-chloro-1H-imidazole-5-carboxaldehyde (VI) is alkylated with 4-bromobenzylbromide, followed by reduction of the aldehyde with sodium borohydride to yield the other Suzuki coupling partner (XIII). The product of Suzuki coupling is trityl losartan. This process is published in J. Org. Chem. (1994), 59, 6391-6394.


European patents EP 470,794 and EP 470,795 describe a method for the manufacture of biphenyl carbonitriles (XVI). These patents also describe a method of preparation of trityl losartan by coupling of intermediates (III) and (IV) employing the procedure described in EP 253,310.

Losartan potassium exhibits polymorphism. Several polymorphic forms have been prepared and characterized. The following paragraphs briefly describe various polymorphs.
U.S. Pat. No. 5,608,075 discloses the polymorphic forms of losartan, wherein the trityl losartan is deprotected with H2SO4 in 50:50 acetonitrile:water and the free acid is treated with KOH solution. The aqueous solution containing losartan potassium is added slowly to a refluxing azeotropic mixture of cyclohexane/iso propanol and the ternary azeotrope cyclohexane/iso propanol/water is distilled till the water content of the pot is less than 0.05%. The white crystalline solid thus obtained is polymorphic form-I, which is characterized by DSC, XRD and IR. Polymorphic form-II is prepared by heating form-I in a DSC cell. This process is also described in U.S. Pat. No. 5,859,258.
U.S. Pat. No. 6,710,183 discloses the synthesis of losartan potassium starting from trityl losartan, wherein trityl losartan is reacted in an alcohol of formula R—OH (where R is C1 to C4 straight chain alkyl group) with 0.1 to 1 equivalent KOH. Losartan potassium thus formed is isolated after crystallizing out by changing the solvent to an aprotic or weakly protic solvent. The alcohol used is preferably methanol and the protic dipolar solvent used for the crystallization of the final product is preferably acetonitrile or straight or branched chain or cyclic aliphatic hydrocarbons.
EP 1294712 (WO 02/094816) discloses the process to manufacture losartan potassium form-I, wherein trityl losartan or losartan is suspended in a solvent and KOH is added to obtain a clear solution, which is then concentrated under reduced pressure to remove most of the solvent. An anti solvent is added to crystallize losartan potassium. The solvents to prepare losartan potassium include methanol, ethanol, and butanol but preferably the salt formation is carried out in methanol. Anti solvent is selected from common solvents such as ethyl acetate, acetonitrile, toluene and acetone, but the preferred anti solvent is acetone.
US application 2004/0006237 (WO 03/048135) relates to novel amorphous and novel crystalline forms III, IV, V of losartan potassium and the processes for their preparation. The patent also discloses novel processes for preparing losartan potassium forms I and II. The preparation of amorphous losartan includes the step of dissolving losartan potassium in a solvent to form a solution and distilling the solvent form the solution to dryness. Losartan form III (hydrated) is obtained by exposing losartan potassium amorphous or form I to an atmosphere having high relative humidity. Losartan potassium form IV is obtained by treating a saturated solution of losartan potassium in ethanol with methylene chloride. Losartan form V is obtained by treating a saturated solution of losartan potassium in ethanol with hexane. Losartan potassium form II is obtained by adding a saturated solution of losartan potassium in ethanol to xylene to form a mixture and evaporating ethanol from the mixture. Losartan form I is obtained by treating a saturated solution of losartan potassium in ethanol or iso propanol, with less soluble solvent like ethyl acetate, toluene, acetone, methyl ethyl ketone, methylene chloride, acetonitrile, dimethyl carbonate or hexane.
US application 2004/0034077 (WO 03/093262) discloses a process for preparing losartan and losartan potassium, wherein trityl losartan is treated with an acid in a diluent comprising a ketone. Especially preferred liquid ketones are acetone, methyl ethyl ketone and methyl isobutyl ketone, and acetone being the most preferred. Acids, which have been found suitable, include hydrochloric acid, sulphuric acid, acetic acid, trifluoroacetic acid, hydrobromic acid and formic acid. After the trityl losartan has been substantially converted to losartan, reaction mixture is basified. Preferred bases are alkali metal hydroxides and alkoxides. After addition of the base, the liquid ketone is evaporated under vacuum. After separation of triaryl methyl alcohol the residue is acidified to yield losartan. Free losartan is suspended in an alcohol and treated with a solution of potassium ions. Finally losartan potassium is precipitated from the alcohol. The alcohol is selected from the group consisting of isopropyl alcohol, butyl alcohol and isobutyl alcohol. The potassium ion solution is prepared by dissolving potassium iso propoxide, potassium butoxide and potassium iso butoxide or potassium hydroxide in the diluent.
US application 2004/0097568 discloses a process for preparing form III of losartan potassium, wherein trityl losartan is treated with aqueous solution of potassium hydroxide in methanol to obtain losartan potassium. The solvent is evaporated under vacuum and traces of water are removed as an azeotrope with toluene. Methanol and carbon are added to the resulting mixture. The carbon is filtered and the methanol is distilled. The resulting mixture is cooled to 20-25° C. to obtain crystalline form III losartan potassium.
US 5,138,069 and
WO 93/10106. The advantages provided by pharmaceutical products in the crystalline form in terms of easiness of processes for the preparation of related medicaments are well known. Crystalline compounds are in fact known to be more suited to the formulation of galenic forms, thanks both to their flowability in the form of powders or granulates, and to the surface properties of the crystals which promote adhesion, for example during the preparation of tablets. Furthermore, the solubility of crystalline compounds in aqueous solutions, in particular in the gastric juices, can also be significantly different than that of the corresponding amorphous compounds. There is therefore the need to discriminate between the crystalline and the amorphous forms of biologically active compounds, so as to fulfil the various pharmaceutical requirements.
A number of crystalline and amorphous forms of losartan potassium are known from
WO 95/17396 and
WO 03/048135. According to
WO 95/17396, crystalline losartan potassium is prepared by salification of acid losartan with an alkali hydroxide. The losartan potassium aqueous solution is then added to a isopropanol-cyclohexane azeotropic mixture under reflux. Water is then removed by azeotropic distillation of the resulting water-isopropanol-cyclohexane ternary mixture, which boils at 64°C. When the solution is anhydrous, the head temperature raises to 69°C and losartan potassium crystallizes.
US 5,859,258 discloses another crystallization process which comprises dissolution of losartan potassium in isopropanol-water, distillation of the binary azeotrope to an approx. 2.6% water content, precipitation by addition of a losartan potassium suspension in cyclohexane, subsequent distillation of the ternary azeotrope to a water content ranging from 0.02 to 0.11 %, and finally drying crystalline losartan potassium under vacuum at a temperature of approx. 45-50°C.

 , Ian R. Baxendale, Steven V. Ley and Nikzad Nikbin Corresponding author email
, Ian R. Baxendale, Steven V. Ley and Nikzad Nikbin Corresponding author emailThe imidazole ring of losartan, an antihypertensive and angiotensin II blocker is formed in a condensation reaction between valeroamidine 160 and dihydroxyacetone [50]. It was found that direct chlorination of the imidazole 162also forms the dichlorination product 164 (as shown in Scheme 33) with formaldehyde as a by-product which proved difficult to suppress and made purification of the reaction mixture problematic. Hence, a sequence involving silyl protection, chlorination and deprotection was established which gave the desired product in 90% overall yield (Scheme 33).
![[1860-5397-7-57-i33]](https://i0.wp.com/beilstein-journals.org/bjoc/content/inline/1860-5397-7-57-i33.png)
Alternatively, glycine can be reacted with methyl pentanimidate 169 to form the corresponding amidine 171 in high yield. Cyclisation, followed by a Vilsmeier-type reaction then furnishes the key chloroimidazolyl building block 172in good yield (Scheme 34) [51].
![[1860-5397-7-57-i34]](https://i0.wp.com/beilstein-journals.org/bjoc/content/inline/1860-5397-7-57-i34.png)
- 50———Shi, Y.-J.; Frey, L. F.; Tschaen, D. M.; Verhoeven, T. R. Synth. Commun. 1993, 23, 2623–2630.doi:10.1080/00397919308012598
- 51—-Griffiths, G. J.; Hauck, M. B.; Imwinkelried, R.; Kohr, J.; Roten, C. A.; Stucky, G. C.; Gosteli, J. J. Org. Chem. 1999,64, 8084–8089. doi:10.1021/jo9824910
- 52–Zhong, Y.-L.; Lee, J.; Reamer, R. A.; Askin, D. Org. Lett. 2004, 6, 929–931. doi:10.1021/ol036423y
NMR: (1H, DMSO, 300 mHz): δ 0.80 (3H, t, J=10. CH3), 1.25 (2H, sext, J=10. CH3CH2), 1.45 (2H, quin, J=10. CH3CH2CH2), 2.45-2.55 (2H, m, CH3CH2CH2CH2), 4.25 (2H, d, J= 3, CH2OH), 5.15-5.25 (3H, m, CH2Ar and OH), 6.88 (d, 2H, J=12, ArH), 7.08 (d, 2H, J=12, ArH), 7.23-7.36 (3H, m, ArH), 7.50-7.55 (1H, ArH).
SEACOND SET
http://www.google.co.in/patents/US7915425
IR v max (KBR): 3201.01, 1580.73, 1460.18, 764.81, 540.09
1H NMR (MeOD) δ, 0.87 (t, 3H), 1.33 (sext, 2H), 1.53 (quint, 2H), 2.56 (t, 2H), 4.43 (s, 2H), 5.24 (s, 2H), 6.89-7.53 (m, 8H).
13C NMR (MeOD) δ, 14.07, 23.24, 27.40, 30.92, 126.71, 126.86, 127.35, 128.21, 130, 130.8, 131, 131.19, 131.81, 136.09, 142.21, 149.97, 162.72
MS (m/z)=423.3 (M+1).
……………………………..
Melting point: 179-180.2
IR, v max (KBR): 3376.27, 1579.77, 1468.86, 762.88, 556.4
1H NMR (CDCl3) δ, 0.87 (t, 3H), 1.31 (sext, 2H), 1.54 (quint, 2H), 2.57 (t, 2H), 4.45 (s, 2H), 5.30 (s, 2H), 7.01-7.68 (m, 8H).
13C NMR (CDCl3) δ, 14.07, 23.24, 27.40, 30.92, 126.71, 126.86, 127.35, 128.21, 130, 130.8, 131, 131.19, 131.81, 136.09, 142.21, 149.97, 162.72
MS (m/z)=423.5 (M+1).
……………………………..
ADDITIONAL WRITEUP FOR READERS, NUMBERINGS ARE ALL NEW
Losartan and its potassium salt, having the formulae (1) & (2) respectively are angiotensin-II receptor (Type AT1) antagonists.

In adults Losartan is currently indicated for the treatment of hypertension (in hypertensive patients with left ventricular hypertrophy, it is also indicated to reduce the risk of stroke).
Losartan Potassium having the formula 2 and its principle active metabolite block the vasoconstrictor and aldosterone. Secreting effects of angiotensin II by selectively blocking the binding of angiotensin II to the AT1 receptor found in many tissues (e.g., vasicular smooth muscle, adrenal gland) otherwise called as angiotensin receptor blockers (ARBs).
The present invention relates to a short, simple and practical process for the preparation of Losartan 1 which belongs to a novel class of tetrazole-imidazole compounds.
There are many processes recorded in literature. The latest prior art information for the preparation of Losartan is the disclosure made in the patent application of Novartis in their PCT WO 2005/014602 dated 17 Feb. 2005.
The process described in the application comprises the reaction of 4′-(Bromomethyl)-2-cyanobiphenyl (BromoOTBN) of the formula 3 with 2-n-butyl-4-chloro-5-formyl imidazole (BCFI) of the formula of 4 in the presence of Potassium carbonate and acetonitrile to give ‘cyano aldehyde’ of the formula 5. The Cyano aldehyde of the formula 5 is reduced with sodium borohydride to get ‘cyano alcohol’ of the formula 6. The Cyano alcohol is reacted with diethyl aluminium azide in the presence of triethyl aluminium to give Losartan of the formula 1.
The reaction scheme of the process is shown in the Scheme 1

Even though the process is simple, handling of triethyl aluminium used needs special attention like very anhydrous conditions, reactions are to be performed under nitrogen or argon and transferring of triethyl aluminium from the containers needs anhydrous systems. The neat liquid and dense solutions of triethyl aluminium are known to ignite very easily at room temperature in presence of air (Pyrophoric). So handling of both triethyl aluminium and diethyl aluminium needs special attention like anhydrous conditions, nitrogen atmosphere etc.,
In EP 0578125A1 of Takeda Chemical Industries dated 12 Jan. 1994, yet another method for the preparation of Losartan has been disclosed in which Trioctadecyl or Trioctyl tin azide has been used as a tetrazole-forming agent. This method also uses the Cyano alcohol of the formula (6). The process comprises reacting the cyano alcohol of the formula (6) with tri-n-octyl tin azide in presence of toluene to give tri-n-octyl tetrazole derivative, which was treated with nitrous acid to give Losartan of the formula (1) in 94.7% yield. The process is shown in the reaction scheme 2

Even though the yields are better (94.7%) in this process again handling of tri-n-octyl tin azide is involved.
Dupont/Merck in their patents and papers always described that trityl Losartan of the formula 7 is detritylated to get Losartan 1 For example they described in J. Med. Chem., 1991, 34, 2525-2547, the preparation of Losartan of the formula 1, from trityl Losartan of the formula 7 using mineral acids such as Hydrochloric acid and sulfuric acid in 93% yield. The reaction scheme of the process is shown in the scheme 3

In this paper ‘Aldehyde Tetrazole’ of the formula 8 is isolated from trityl tetrazole aldehyde of the formula 21 and were further used for preparing derivatives of aldehyde such as benzene sulfonyl hydrazones of the formula 9 but not for Losartan. This process is shown in the scheme 4

In J. Org. Chem 1994, 59, 6391-6394 again by Merck team reported Trityl Losartan and Losartan synthesis by coupling of boronic acid derivative 11 with 3-(4-bromobenzyl) derivative of BCBMI of the formula 10. The formed trityl Losartan of the formula 7 is converted to Losartan of the formula 1 with acid. The whole process is described in Scheme 5

The Compound of the formula 10 is prepared from the reaction of BCFI of the formula 4 with p-bromo benzyl bromide of the formula 12 in potassium carbonate and Dimethyl formamide followed by reduction with sodium borohydride (NaBH4). The details are given in the Scheme 6

The Compound of the formula 11 is prepared from 5-phenyl tetrazole of the formula 14 by reacting with trityl chloride to get N-trityl-5-phenyl tetrazole of the formula 13, which on reaction with butyl lithium and triisopropyl borate followed by hydrolysis to give compound of the formula 11. This process is shown in the Scheme 7

In one of the first patent filed by Dupont/Merck (date of filing 9 Jul. 1987, priority 11 January 1986 EP0253310) reported a procedure for the preparation of Losartan. Bromo OTBN of the formula 3 is reacted with BCHMI of the formula 15 in the presence of a base to give cyano alcohol of the formula 6, and its regioisomer of the formula 14. Separation of the isomer needs column chromatography. The cyano alcohol 6 is reacted with sodium/ammonium azide in DMF for 13 days to get Losartan 1 in 21% yield. The process is shown in the Scheme 8

The drawbacks of the above process are
- 1). Separation of the regioisomer using column chromatography which is industrially not feasible for the preparation of large scale (ton) material/product
- 2). The tetrazole formation takes 13 days with 21% yield, which is unproductive.
- 3). Dupont/Merck uses BCHMI 15 as the starting material for preparing cyano alcohol of the formula 6. BCHMI 15 is an expensive intermediate compared to BCFI 4, and also the formation of unwanted regio isomer 14 is higher. The process is schematically described in scheme 8. Even though the process looks simple it has two problems.
First: Cyano alcohol is produced as a mixture of regioisomers and needs column chromatography for purification.
Second: Tetrazole formation. This takes 13 days with 21% yield, which limits commercialization of the process.
In U.S. Pat. No. 4,820,843 and U.S. Pat. No. 4,879,186, Dupont prepares Losartan by reaction of BCFI of the formula 4 and N-Triphenylmethyl-5-[2-(4′-bromomethyl biphenyl)]tetrazole of the formula 16 in the presence of base, followed by reduction with sodium borohydride to give Trityl Losartan of the formula 7, which is treated with mineral acid to give Losartan 1.
The process is shown in scheme 9

In U.S. Pat. No. 4,874,867 of Dupont/Merck, a process for the preparation of N-Triphenylmethyl-5-[2-(4′-bromomethyl biphenyl)]tetrazole of the formula 16 is described by the reaction of OTBN of the formula 20 with trimethyl tin azide to give the compound 17, which is treated with Hydrochloric acid to give tetrazole derivative of OTBN of the formula 18. The tetrazole derivative of OTBN of the formula 18 is protected with trityl chloride to give compound of the formula 19, followed by bromination with N-bromosuccinimide to give N-Triphenylmethyl-5-[2-(4′-bromomethyl biphenyl)]tetrazole of the formula 16.
The process is shown in the scheme 10.

In all the above papers and patents by Dupont/Merck, the process yields in many steps are good 75-95% and in some steps are less to moderate 21-49%. The drawbacks, or the problems in all these processes is, the number of unit operations.
For example:
- 1). In J. Med. Chem 1991, 34, 2525-2547 the number of steps are six (6) to prepare Losartan of the formula 1 from the readily available intermediates.
- 2). In J. Org. Chem 1994, 59, 6391-6394 the number of steps are nine (9) to prepare Losartan of the formula 1 from the readily available intermediates.
- 3). In EP 0253310 patent the number of operations are two (2) but the problem is time & yields i.e., 13 days and poor yield (21%), also the uneconomical column chromatographic separation of regioisomer.
- 4). In U.S. Pat. Nos. 4,820,843 and 4,879,186 the number of steps are six (6).
- 5). In U.S. Pat. No. 4,874,867 the number of steps are seven (7).
………………………..
INTERMEDIATES
(1-(2′-Cyano biphenyl-4-methyl)-2-butyl-4-chloro-5-formyl imidazole) of the formula 5.

Melting point: 107-108° C.
HPLC Purity: >98%
IR. v max (KBR): 2218 (—CN), 1662.40 (—CHO)
1H NMR (CDCl3) δ, 0.91 (t, 3H), 1.38 (sext, 2H), 1.73 (quint, 2H), 2.67 (t, 2H), 5.61 (s, 2H), 7.16-7.77 (m, 8H), 9.77 (s, 1H).
13C NMR (CDCl3) δ, 13.51, 22.18, 26.33, 29.04, 47.74, 110.05, 118.36, 124.11, 126.59, 127.65, 129.16, 129.81, 132.76, 133.61, 136.01, 137.69, 142.96, 144.33, 154.46, 177.73
DR ANTHONY MELVIN CRASTO

ORGANIC SPECTROSCOPY

