
FLAGS AND HITS
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO FACEBOOK
...................................................................Join me on twitter
FACEBOOK
...................................................................Join me on twitter
 ..................................................................Join me on google plus
..................................................................Join me on google plus
 Googleplus
GoogleplusMYSELF

LEUCODERMA CASE ; PROGRESSING FOR CURE ; सफेद दाग , लियूकोडर्मा का एक केस जिसे ई०टी०जी० आयुर्वेदास्कैन तकनीक आधारित आयुर्वेदिक इलाज से फायदा
दिनान्क २८ अगस्त २०१३ को एक २७ साल के लड़्के ने सफेद दाग के इलाज के लिये मेरे OUT DOOR HOSPITAL मे consultation के लिये समपर्क किया था / इस लड़्के के सारे शरीर पर छोटे बड़े सैकड़ों की सन्ख्या मे LUECODERMA यानी सफेद दाग के चकत्ते पड़े हुये थे, जो उसको पिछले १५ साल पहले हुये थे / इसके पिता एक होम्योपैथी के डाक्टर है जो प्रैक्टिस करते है / वे ही इसे लेकर इलाज के लिये मेरे OUT-DOOR HOSPITAL मे लेकर आये थे /
मैने उनको बताया कि बिना ई०टी०जी० आयुर्वेदास्कैन और आयुर्वेद के रकत और पेशाब के परीक्शन के इलाज कराना बेकार है / LEUCODERMA के ईलाज के लिये परीक्षण कराना सबसे पहली आवश्यकता है /
दिनाक २८ अगस्त २०१३ को इस लड़्के का शरीर के दो हिस्सो का PHOTOGRAPH लिया गया था / नीचे दिया गया photograph इसी दिन का है /
यह PHOTOGRAPH मरीज के…
View original post 1,012 more words
CLAZOSENTAN

Clazosentan
READ ALL AT
http://www.allfordrugs.com/2014/01/22/clazosentan/

READ MORE ON SNTAN SERIES……http://medcheminternational.blogspot.in/p/sentan-series.html
Tezosentan Disodium for pulmonary hypertension

TEZOSENTAN
180384-57-0 CAS OF FREE ACID
N-[6-(2-Hydroxyethoxy)-5-(2-methoxyphenoxy)-2-[2-(2H-tetrazol-5-yl)pyridin-4-yl]pyrimidin-4-yl]-5-propan-2-ylpyridine-2-sulfonamide
5-isopropyl-pyridine-2-sulphonic acid 6-(2-hydroxy-ethoxy)-5- (2-methoxy-phenoxy)-2-(2-1 H-tetrazol-5-yl-pyridin-4-yl)- pyrimidin-4-ylamide
| Formula | C27H27N9O6S |
|---|---|
| Mol. mass | 605.624 |
…………………………………………………………………………….

Tezosentan disodium, Ro-61-0612, Veletri
5-isopropyl-pyridine-2-sulfonic acid [6-(2-hydroxy-ethoxy)-5-(2-methoxy-phenoxy)-2-[2-(1H-tetrazole-5-yl)-pyridine-4-yl]-pyrimidine-4-yl]-amide sodium salt (1:2)
180384-58-1 of disodium salt, 180384-57-0 (free acid)

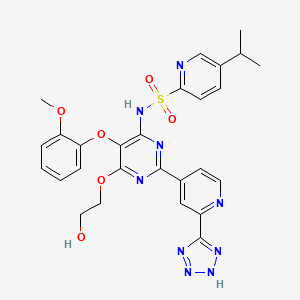
Tezosentan is a non-selective ETA and ETB receptor antagonist.[1] It acts as a vasodilator and was designed as a therapy for patients with acuteheart failure. Recent studies have shown however, that tezosentan does not improve dyspnea or reduce the risk of fatal or nonfatal cardiovascular events.[2]
Pulmonary disease (COPD), which may possibly be associated with pulmonary hypertension, as well as allergic and non-allergic rhinitis, provided that treatment with endothelin from a therapeutic standpoint is not contraindicated.
Tezosentan disodium is an endothelin ETB receptor antagonist in phase II clinical development for the treatment of stable, chronic pulmonary arterial hypertension. The drug was previously being evaluated for heart failure, but trials in that indication have been discontinued. The compound is being developed by Actelion.
………………………………..
SYNTHESIS

………………………………..
SYNTHESIS

Reaction of 4-cyano-pyridine (I) with Na in methanol followed by treatment with ammonium chloride provides 4-amidino-pyridine hydrochloride (II), which is then converted into 5-(2-methoxyphenoxy)-2-(pyridin-4-yl)-pyrimidine-4,6-diol (IV) by condensation with diethyl malonate derivative (III) by means of Na in MeOH. By heating compound (IV) with phosphorus oxychloride (POCl3), 4,6-dichloro-5-(2-methoxyphenoxy)-2-pyridin-4-yl)pyrimidine (V) is obtained, which in turn is oxidized with peracetic acid in refluxing acetonitrile to afford N-oxide derivative (VI). Condensation of (VI) with 5-isopropylpyridine-2-sulfonamide potassium (VII) furnishes 5-isopropylpyridine-2-sulfonic acid 6-chloro-5-(2-methoxyphenoxy)-2-(1-oxy-pyridin-4-yl)-pyrimidin-4-yl amide (VIII), which is then dissolved in dimethoxyethane and subjected to reaction with Na in hot ethylene glycol (IX) to provide N-[6-(2-hydroxyethoxy)-5-(2-methoxyphenoxy)-2-(1-oxy-pyridin-4-yl)-pyrimidin-4-yl]-5-isopropylpyridine-2-sulfonamide (X). Refluxing of (X) with trimethylsilylcyanide and Et3N in acetonitrile yields cyano derivative (XI), which is then converted into the tetrazole derivative (XII) by reaction with sodium azide and NH4Cl in DMF at 70 C. Finally, the disodium salt of tezosentan is obtained by treatment of (XII) with Na/MeOH in THF. refEP 0799209; JP 1998509182; WO 9619459
…………………………………..
SYNTHESIS PROCEDURE as in EP0979822A1
Examples
- 1360 ml of formamide were added to 136 g (437 mmol) of 5-(2-methoxy-phenoxy)-2-pyridine-4-yl-pyrimidine-4,6-diole. Then, at a temperature of 0°C, 11.7 ml (219 mmol) of concentrated sulfuric acid and thereafter 36.5 g (130 mmol) of iron(II)sulfate heptahydrate were added to the suspension. After that, 89 ml (874 mmol) of 30% hydrogen peroxide were added dropwise within 1 hr at a temperature of 0°C to 5°C. The viscous yellow-brownish suspension was stirred at 0°C for 1.5 hr. Subsequently, a solution of 83 g (437 mmol) of sodium pyrosulfite in 680 ml of de-ionized water was added dropwise to the reaction mixture within 30 min. at 0°C to 5°C and the reaction mixture was stirred at 0°C to 5°C for 30 min. The suspension was then filtered under reduced pressure. The filtrate was first washed with 1750 ml of de-ionized water and thereafter with 700 ml of ethanol. Then the solid was dried at 80°C, 2000 Pa for 16 hr. There were obtained 132.4 g (91% of theory) of 4-[4,6-dihydroxy-5-(2-methoxy-phenoxy)-pyrimidine-2-yl]-pyridine-2-carboxylic acid amide with a HPLC purity of 91.4% (w/w).
-
Preparation of starting material:
- a) 53.1 g of 4-cyano-pyridine (98%) are added all at once to a solution of 1.15 g of sodium in 200 ml of abs. MeOH. After 6 hr 29.5 g of NH4Cl are added while stirring vigorously. The mixture is stirred at room temperature overnight. 600 ml of ether are added thereto, whereupon the precipitate is filtered off under suction and thereafter dried at 50°C under reduced pressure. There is thus obtained 4-amidino-pyridine hydrochloride (decomposition point 245-247°C).
- b) 112.9 g of diethyl (2-methoxyphenoxy)malonate are added dropwise within 30 min. to a solution of 27.60 g of sodium in 400 ml of MeOH. Thereafter, 74.86 g of the amidine hydrochloride obtained in a) are added all at once. The mixture is stirred at room temperature overnight and evaporated at 50°C under reduced pressure. The residue is treated with 500 ml of ether and filtered off under suction. The filter cake is dissolved in 1000 ml of H2O and treated little by little with 50 ml of CH3COOH. The precipitate is filtered off under suction, washed with 400 ml of H2O and dried at 80°C under reduced pressure. There is thus obtained 5-(2-methoxy-phenoxy)-2-(pyridine-4-yl)-pyrimidine-4,6-diole (or tautomer), melting point above 250°C.
- Example 1
Example 2
- Within 20 min. 61 ml (633 mmol) of POCl3 were added dropwise to 34 ml (200 mmol) of diisopropyl ethylamine at 5°C to 10°C followed by stirring at 5°C to 10°C for 15 min. Then 23.5 g (66 mmol) of 4-[4,6-dihydroxy-5-(2-methoxy-phenoxy)-pyrimidine-2-yl]-pyridine-2-carboxylic acid amide were added in four portions under cooling followed by stirring at 90°C for 25 hr. The reaction mixture was cooled down to 20°C and transferred to a new flask together with 50 ml of dichloromethane. Volatile components (i.e. excess of POCl3) was removed by evaporation from 20°C to 70°C followed by re-distillation with 100 ml of toluene. After adding 250 ml of dichloromethane to the residue (88 g of a black oil) the solution was heated to 35°C to 40°C and 80 ml of de-ionized water were added dropwise within 30 min. whereby the pH was kept constant by the subsequent addition of 28% NaOH solution (60 ml) within 5 to 6 hr. The mixture was stirred at 35°C to 40°C for 30 min. followed by removal of dichloromethane by distillation. The resulting suspension was allowed to cool down to 20°C and was stirred for additional 2 hr. The solid was filtered off under suction, washed with 500 ml of water and dried at 70°C, 2000 Pa for 16 hr. There were obtained 21.3 g (86% of theory) of 4-[4,6-dichloro-5-(2-methoxy-phenoxy)-pyrimidine-2-yl]-pyridine-2-carbonitrile with a HPLC purity of 94.3% (w/w).
- 8.95 g (24 mmol) of 4-[4,6-dichloro-5-(2-methoxy-phenoxy)-pyrimidine-2-yl]-pyridine-2-carbonitrile were suspended in 100 ml of acetone. At a temperature of 20°C, 5.04 g (25 mmol) of 5-isopropyl-pyridine-2-sulfonamide, 1 ml of de-ionized water, 10.6 g (77 mmol) of potassium carbonate and 135 mg (1.2 mmol) 1,4-diazobicyclo[2.2.2]octane were added. The mixture was stirred at 40°C for 20 hr. Thereafter, another 240 mg (1.2 mmol) of 5-isopropyl-pyridine-2-sulfonamide and 80 mg (0.7 mmol) of 1,4-diazobicyclo[2.2.2]octane were added. The reaction mixture was stirred for 24 hr at 40°C followed by cooling to 20°C. Then 50 ml of de-ionized water and 45 ml of 3 N aqueous hydrochloric acid were added slowly until pH = 1. The acetone was removed by distillation and the resulting suspension was stirred at 20°C for 1.5 hr. The solid was filtered off under suction, washed first with 100 ml of de-ionized water and thereafter with 50 ml of t-butylmethylether. Then the solid was dried at 70°C, 2000 Pa for 20 hr. There were obtained 13.2 g (102% of theory) of 5-isopropyl-pyridine-2-sulfonic acid [6-chloro-2-(2-cyano-pyridine-4-yl)-5-(2-methoxy-phenoxy)-pyrimidine-4-yl]-amide with a HPLC purity of 87.8% (w/w).
- Example 4
- 122 g (233 mmol) of 5-isopropyl-pyridine-2-sulfonic acid [6-chloro-2-(2-cyano-pyridine-4-yl)-5-(2-methoxy-phenoxy)-pyrimidine-4-yl]-amide was suspended in 450 ml of N,N-dimethyl formamide and the mixture was cooled down to 15°C. At this temperature, 35 ml of hydrazine hydrate were added dropwise within 1 hr. The resulting solution was stirred at 15°C to 20°C for 16 hr and thereafter diluted with 600 ml of de-ionized water. Then 50 ml of glacial acetic acid were added dropwise at 0°C to 5°C until pH = 5.5. 600 g of ice were added and the suspension was stirred for 1 hr. The solid was filtered off under suction, washed with 3000 ml of water and dried at 40°C, 2000 Pa for 24 hr. There were obtained 126 g (97% of theory) of 5-isopropyl-pyridine-2-sulfonic acid [6-chloro-2-[2-(hydrazino-imino-methyl)-pyridine-4-yl]-5-(2-methoxy-phenoxy)-pyrimidine-4-yl]-amide with a HPLC purity of 91.8% (w/w).
- Example 6
- 20 g (35 mmol) of 5-isopropyl-pyridine-2-sulfonic acid [6-chloro-2-[2-(hydrazino-imino-methyl)-pyridine-4-yl]-5-(2-methoxy-phenoxy)-pyrimidine-4-yl]-amide were added to 160 ml of N,N-dimethyl formamide. The solution was kept at 15°C to 20°C and 23 ml of 6 N aqueous hydrochloric acid were added, followed by addition of a solution containing 4.8 g (7 mmol) of sodium nitrite in 20 ml de-ionized water within 10 min. The mixture was stirred at 20°C for 1 hr, then 140 ml of de-ionized water were added and the suspension was stirred at 0°C for 1 hr. The solid was filtered, firstly washed with 80 ml of de-ionized water and thereafter with 80 ml of t-butylmethylether. Then the solid was dried at 70°C and 2000 Pa for 16 hr. The crude product (23.4 g) was taken up with 117 ml of tetrahydrofuran for 1 hr. After filtration at 0°C the crystallized product was washed with 25 ml of t-butylmethylether and was then dried at 70°C, 2000 Pa for 16 hr. There were obtained 17.3 g (84% of theory) of 5-isopropyl-pyridine-2-sulfonic acid [6-chloro-5-(2-methoxy-phenoxy)-2-[2-(1H-tetrazole-5-yl)-pyridine-4-yl]-pyrimidine-4-yl]-amide with a HPLC purity of 91.1% (w/w).
- Example 8
- Example 10
- 6.2 g of sodium hydroxide were added to 15 g (26 mmol) of 5-isopropyl-pyridine-2-sulfonic acid [6-chloro-5-(2-methoxy-phenoxy)-2-[2-(1H-tetrazole-5-yl)-pyridine-4-yl]-pyrimidine-4-yl]-amid and 75 ml of ethylene glycol. The mixture was heated to 85°C for 5 hr. Then 55 ml of de-ionized water were added and thereafter 55 ml of 3 N hydrochloric acid were added dropwise. The mixture was allowed to cool down to 20°C and was stirred for 1 hr. The solid was filtered off and dried at 70°C, 2000 Pa for 18 hr. There were obtained 16.2 g (103%) of 5-isopropyl-pyridine-2-sulfonic acid 16-(2-hydroxy-ethoxy)-5-(2-methoxy-phenoxy)-2-[2-(1H-tetrazole-5-yl)-pyridine-4-yl]-pyrimidine-4-yl]-amide with a HPLC purity of 92% (w/w). 80 ml of dioxane and 80 ml of ethanol were added to this solid. At a temperature of 60°C, gaseous ammonia was introduced into the liquid until pH = 9 to 10. The resulting suspension was allowed to cool down to 20°C and was stirred at 20°C for 20 hr and thereafter at 0°C for 2.5 hr. Then the solid was filtered off and dried at 70°C, 2000 Pa for 18 hr. There were obtained 14.2 g of mono ammonium salt with a HPLC purity of 96.2% (w/w). The solid was heated (reflux) in 70 ml of methanol, cooled down slowly to 20°C and stirred at 20°C for 19 hr and thereafter at 0°C for 2 hr. Then the solid was filtered off and dried at 70°C, 2000 Pa for 19 hr. There were obtained 11.5 g (66% of theory) of 5-isopropyl-pyridine-2-sulfonic acid [6-(2-hydroxy-ethoxy)-5-(2-methoxy-phenoxy)-2-[2-(1H-tetrazole-5-yl)-pyridine-4-yl]-pyrimidine-4-yl]-amide sodium salt (1:2) with a HPLC purity of 98.6% (w/w).

Reaction of 2-chloro-5-ispropylpyridine (VII) with thiourea (A) in aqueous HCl gives 5-isopropyl- pyridine-2-thiol (VIII), which is chlorinated with chlorine in acetic acid to yield 5-isopropylpyridine-2-sulfochloride (IX). This compound is converted into 5-isopropylpyridine-2-sulfonamide potassium salt (X).
…………………………
synthesis
. Example 1
a) 200 ml of dimethoxyethane and 1 10.9 g of 4-[4-(4-tert- butyl-phenyl-sulphonylamino)-6-chloro-5-(2-methoxy-phenoxy)- pyrimidin-2-yl]-pyridine 1 -oxide are added all at once to a solution of 23.80 g of sodium in 660 ml of ethylene glycol. The solution is heated at 90°C for 20 hours while stirring, thereafter cooled, poured into 2500 ml of H2O and thereafter treated with CH3COOH to pH 5. The mixture is extracted three times with EtOAc, the organic phase is washed with H2O, dried with Na2Sθ4 and evaporated under reduced pressure. The residue is recrystall- ized from CH3CN and thereafter twice from a mixture of acetone and CH3CN. There is thus obtained 4-[4-(4-tert-butyl-phenyl- sulphonylamino)-6-(2-hydroxy-ethoxy)-5-(2-methoxy-phenoxy)- pyrimidin-2-yl]-pyridine 1 -oxide.
Preparation of the starting material:
b) 53.1 g of 4-cyano-pyridine (98%) are added all at once to a solution of 1.15 g of sodium in 200 ml of abs. MeOH. After
6 hours 29.5 g of NH4CI are added while stirring vigorously. The mixture is stirred at room temperature overnight. 600 ml of ether are added thereto, whereupon the precipitate is filtered off under suction and thereafter dried at 50°C under reduced pressure. There is thus obtained 4-amidino-pyridine hydro- chloride (decomposition point 245-247°C).
c) 1 12.9 g of diethyl (2-methoxyphenoxy)malonate are added dropwise within 30 minutes to a solution of 27.60 g of sodium in 400 ml of MeOH. Thereafter, 74.86 g of the amidine hydro- chloride obtained in b) are added all at once. The mixture is stirred at room temperature overnight and evaporated at 50°C under reduced pressure. The residue is treated with 500 ml of ether and filtered off under suction. The filter cake is dissolved in 1000 ml of H2O and treated little by little with 50 ml of CH3COOH. The precipitate is filtered off under suction, washed with 400 ml of H2O and dried at 80°C under reduced pressure. There is thus obtained 5-(2-methoxy-phenoxy)-2-(pyridin-4-yl)- pyrimidine-4,6-diol (or tautomer), melting point above 250°C.
d) A suspension of 1 54.6 g of 5-(2-methoxy-phenoxy)-2- (pyridin-4-yl)-pyrimidine-4,6-diol (or tautomer) in 280 ml of POCI3 is heated at 120°C in an oil bath for 24 hours while stirring vigorously. The reaction mixture changes gradually into a dark brown liquid which is evaporated under reduced pressure and thereafter taken up three times with 500 ml of toluene and evaporated. The residue is dissolved in 1000 ml of CH2CI2, treated with ice and H2O and thereafter adjusted with 3N NaOH until the aqueous phase has pH 8. The organic phase is separated and the aqueous phase is extracted twice with CH2CI2. The combined CH2CI2 extracts are dried with MgSθ4, evaporated to half of the volume, treated with 1000 ml of acetone and the CH2CI2 remaining is distilled off at normal pressure. After standing in a refrigerator for 2 hours the crystals are filtered off under suction and dried at 50°C overnight. There is thus obtained 4,6-dichloro-5-(2-methoxy-phenoxy)-2-pyridin-4-yl)- pyrimidine, melting point 1 78-1 80°C.
e) A solution of 1 7.4 g of 4,6-dichloro-5-(2-methoxy- phenoxy)-2-pyridin-4-yl)-pyrimidine in 100 ml of CH3CN is boiled at reflux for 3 hours with 1 5 ml of a 32% peracetic acid solution, thereafter cooled and stored in a refrigerator overnight. The crystals are filtered off under suction and dried at 50°C under reduced pressure. There is thus obtained 4-[4,6-dichloro- 5-(2-methoxy-phenoxy)-pyrimidin-2-yl]-pyridine 1 -oxide, melting point 189-1 90°C.
f) A solution of 36.4 g of 4-[4,6-dichloro-5-(2-methoxy- phenoxy)-pyrimidin-2-yl]-pyridine 1 -oxide and 52.8 g of p-tert- butylphenyl-sulphonamide potassium in 1 50 ml of abs. DMF is stirred at room temperature for 24 hours. Thereafter, it is poured into a mixture of 1 500 ml of H2O and 1000 ml of ether while stirring mechanically, whereby a precipitate forms. The suspension is adjusted to pH 5 with CH3COOH, suction filtered, the crystals are washed with cold water and thereafter with ether and dried at 50°C. There is thus obtained 4-[4-(4-tert- butyl-phenylsulphonylamino)-6-chloro-5-(2-methoxy-phenoxy)- pyrimidin-2-yl]-pyridine 1 -oxide as a colourless material of melting point 247-249°C.
Example 2
A solution of 78.45 g of 4-[4-(4-tert-butyl-phenyl- sulphonylamino)-6-(2-hydroxy-ethoxy)-5-(2-methoxy-phenoxy)- pyrimidin-2-yl]-pyridine 1 -oxide, 122.5 g of trimethylsilyl cyanide, 127.8 g of triethylamine and 1200 ml of CH3CN is boiled at reflux for 20 hours and thereafter evaporated under reduced pressure. The oily residue is taken up in 1000 ml of EtOAc and the solution is washed with CH3COOH:H2θ 9:1 and then with H2O. The EtOAc extracts are dried with Na2SO4. After evaporation of the solvent the residue is taken up in a mixture of CH3CN and CF3COOH (20:1 ), whereby a crystalline precipitate separates. There is thus obtained 4-tert-butyl-N-[2-(2-cyano-pyridin-4- yl)-6-(2-hydroxy-ethoxy)-5-(2-methoxy-phenoxy)-pyrimidin-4- yl]-benzenesulphonamide of melting point 176-1 79°C.
Example 3 for analogy only compd is different
A suspension of 50.0 g of 4-tert-butyl-N-[2-(2-cyano- pyridin-4-yl)-6-(2-hydroxy-ethoxy)-5-(2-methoxy-phenoxy)- pyrimidin-4-yl]-benzenesulphonamide, 46.33 g of NH4CI and 56.47 g of NaN3 in 1600 ml of DMF is heated to 70°C for 24 hours while stirring vigorously. The majority of the solvent is distilled off under reduced pressure, the residue is dissolved in H2O, the solution is extracted four times at pH 6.5 with ether, thereafter treated with CH3COOH to pH = 4.5 and extracted with EtOAc. After working up there is obtained a residue which is treated with ether and filtered off under suction therefrom. There is thus obtained 4-tert-butyl-N-[6-(2-hydroxy-ethoxy)-5-(2- methoxy-phenoxy)-2-(2-1 H-tetrazol-5-yl-pyridin-4-yl)- pyrimidin-4-yl]-benzenesulphonamide, melting point 225-227°C.
Example 30 final product
In analogy to Example 3, from 5-isopropyl-pyridine-2- sulphonic acid 2-(2-cyano-pyridin-4-yl)-6-(2-hydroxy-ethoxy)- 5-(2-methoxy-phenoxy)-pyrimidin-4-ylamide there is obtained 5-isopropyl-pyridine-2-sulphonic acid 6-(2-hydroxy-ethoxy)-5- (2-methoxy-phenoxy)-2-(2-1 H-tetrazol-5-yl-pyridin-4-yl)- pyrimidin-4-ylamide (tezosantan free base) as a white substance of melting point 1 98- 200°C from acetonitrile.
The corresponding disodium salt (tezosantan di sodium salt) is obtained as a white powder from this product using sodium methylate in analogy to Example 5
Example 5 for analogy only, compd is different
A solution of 47.8 g of 2-[6-(4-tert-butyl-phenylsulphonyl- amino)-5-(2-methoxy-phenoxy)-2-(2-1 H-tetrazol-5-yl-pyridin- 4-yl)-pyrimidin-4-yloxy]-ethyl pyridin-2-ylcarbamate in 500 ml of abs. THF is treated dropwise with a cold solution of 2.8 g of sodium in 50 ml of methanol, whereby there forms gradually a solid precipitate which, after stirring at room temperature for 1 hour, is filtered off under suction, dried under greatly reduced pressure at 35°C for 3 days and thereafter at 50°C for 2 days. There is thus obtained the bis-sodium salt, decomposition point above 250°C.
References
- Urbanowicz, W; Sogni, P, Moreau, R, Tazi, K A, Barriere, E, Poirel, O, Martin, A, Guimont, M C, Cazals-Hatem, D, Lebrec, D (2004). “Tezosentan, an endothelin receptor antagonist, limits liver injury in endotoxin challenged cirrhotic rats”. Gut (BMJ Publishing Group Ltd & British Society of Gastroenterology) 53 (12): 1844–1849. doi:10.1136/gut.2003.036517. PMC 1774327. PMID 15542526.
- “Tezosentan does not appear to improve symptoms for patients with acute heart failure”. Medical Studies/Trials. news-medical.net. 7 Nov 2007. Retrieved 2007-11-24.
4 US2003/100507 A1
5 Drugs Fut 2003,28(8),754
6 WO 1996019459……
7 EP 0897914
8 WO 2011163085
9 WO 2004082637
| 10 WO 2002074034 |
11…
| 15055997 | 4-8-2004 | Discovery, modeling, and human pharmacokinetics of N-(2-acetyl-4,6-dimethylphenyl)-3-(3,4-dimethylisoxazol-5-ylsulfamoyl)thiophene-2-carboxamide (TBC3711), a second generation, ETA selective, and orally bioavailable endothelin antagonist. | Journal of medicinal chemistry |
12 ..
| 10610277 | 7-1-1999 | RO 610612 . | Drugs in R&D |
13….
| 3-27-2003 | Aqueous pharmaceutical composition comprising Tezosentan | |
| US6103902 | 8-16-2000 | Carbamoylation process |
| WO0036918 | 6-30-2000 | METHODS AND COMPOSITIONS FOR TREATMENT OF CELL PROLIFERATIVE DISORDERS METHODS AND COMPOSITIONS FOR TREATMENT OF CELL PROLIFERATIVE DISORDERS |
| US6063911 | 5-17-2000 | Methods and compositions for treatment of cell proliferative disorders |
READ MORE ON SNTAN SERIES……http://medcheminternational.blogspot.in/p/sentan-series.html
Ciprostene calcium
Ciprostene calcium
(5Z)-9β-Methyl-6a-carbaprostaglandin I2, calcium salt, 9-β-methylcarbacyclin,
Restenosis Treatment of Antiplatelet Therapy
81703-55-1 (anhydrous ca salt)
81845-44-5 (free base, anhydrous)
| Chemical Name: | 6,9ALPHA-METHYLENE-9BETA-METHYL-11ALPHA,15S-DIHYDROXY-PROSTA-5Z,13E-DIEN-1-OIC ACID, CALCIUM SALT |
| Synonyms: | U-61431F;CIPROSTENE CALCIUM;CIPROSTENE CALCIUM SALT;9-beta-methylcarbacyclin;pentalenylidene)-,calciumsalt(2:1),(3as-(2z,3a-alpha,5-beta,6-alpha(1e,3r*;5-(hexahydro-5-hydroxy-6-(3-hydroxy-1-octenyl)-3a-methyl-2(1h)-pentanoicaci;6,9ALPHA-METHYLENE-9BETA-METHYL-11ALPHA,15S-DIHYDROXY-PROSTA-5Z,13E-DIEN-1-OIC ACID, CALCIUM SALTPentanoicacid,5-[hexahydro-5-hydroxy-6-(3-hydroxy-1-octenyl)-3a-methyl-2(1H)-pentalenylidene]-,calcium salt (2:1), [3aS-[2Z,3aa,5b,6a(1E,3R*),6aa]]-; Ciprostene calcium; U 61431F |
| Molecular Formula: | C44H70CaO8 |
| Formula Weight: | 767.1 |
U-61431F (anhydrous)
- 9-beta-Methylcarbacyclin
- Ciprostene calcium
- U 61431F
- U-61,431F
- UNII-A85Y5Y98EJ
Pfizer (Originator)
 CIPROSTENE Ca
CIPROSTENE Ca

Carbacyclin and closely related compounds are known in the art. See Japanese Kokia 63,059 and 63,060, also abstracted respectively as Derwent Farmdoc CPI Numbers 48154B/26 and 48155B/26. See also British published specifications 2,012,265 and German Offenlegungsschrift 2,900,352, abstracted as Derwent Farmdoc CPI Number 54825B/30. See also British published applications 2,017,699, 2,014,143 and 2,013,661.
The synthesis of carbacyclin and related compounds is also reported in the chemical literature, as follows: Morton, D. R., et al., J. Organic Chemistry, 44:2880 (1979); Shibasaki, M., et al. Tetrahedron Letters, 433-436 (1979); Kojima, K., et al., Tetrahedron Letters, 3743-3746 (1978); Nicolaou, K. C., et al., J. Chem. Soc., Chemical Communications, 1067-1068 (1978); Sugie A., et al., Tetrahedron Letters 2607-2610 (1979); Shibasaki, M., Chemistry Letters, 1299-1300 (1979), and Hayashi, M., Chem. Lett. 1437-40 (1979); and Li, Tsung-tee, “A Facial Synthesis of 9(0)-Methano-prostacyclin”, Abstract No. 378, (Organic Chemistry), and P. A. Aristoff, “Synthesis of 6a-Carbaprostacyclin I.sub.2 “, Abstract No. 236 (Organic Chemistry) both at Abstract of Papers (Part II) Second Congress of the North American Continent, San Francisco, Calif. (Las Vegas, Nev.), USA, 24-29 August 1980.
7-Oxo and 7-hydroxy-CBA.sub.2 compounds are apparently disclosed in U.S. Pat. No. 4,192,891. 19-Hydroxy-CBA.sub.2 compounds are disclosed in U.S. Ser. No. 054,811, filed July 5, 1979. CBA.sub.2 aromatic esters are disclosed in U.S. Pat. No. 4,180,657. 11-Deoxy-Δ.sup.10 – or Δ.sup.11 -CBA.sub.2 compounds are described in Japanese Kokai 77/24,865, published Feb. 24, 1979.
Prostaglandin E.sub.1 (3-hydroxy-2-(3-hydroxy-1-octenyl)-5-oxocyclopentaneheptanoic acid) is a naturally occurring prostaglandin and was one of the first to be isolated and characterised. It is available commercially for the treatment of peripheral vascular disease.
Prostacyclin (otherwise known as epoprostenol and PGI.sub.2) is also a natural prostaglandin occurring within the arterial wall of mammals. It has potent vasodilatory and antiplatelet properties and is available commercially as its sodium salt, sodium epoprostenol, for use in extracorporeal circuits during cardiopulmonary bypass, renal dialysis, and charcoal haemoperfusion. A number of recent publications in the literature have suggested that prostacyclin may also have fibrinolytic activity (J. Pharmac. Exp. Therap. 1982, 222(3), 544 to 549 and Thrombos, Res., 1983, 29, 655 to 660). Similar reports have also occurred for the prostacyclin analogue, iloprost (Brit. J. Pharmac., 1985, 86, 8138 and Thromb. Haemost., 1983, 50, 893). It has also been suggested that prostacyclin augments the thrombolytic activity of streptokinase (J. Cardiovasc. Pharmac., 1985, 7, 739 to 746).
A number of prostacyclin analogues have also been synthesised and evaluated as antithrombotic or antiplatelet agents (Circulation, 1985, 72(6), 1219 to 1225 and Progress in Medicinal Chemistry, 1984, 21, 237 to 279).
………………………………………………………..

Treatment of the optically pure lactone (I) with lithium dimethyl methylphosphonate in tetrahydrofuran gives hemiacetal (II), which is oxidized to the diketone (III) using Jones’ reagent in acetone. Then in the key step, compound (III) cyclizes to enone (IV) using potassium carbonate and 18-crown-6 in warm toluene. Lithium dimethyl cuprate addition to enone (IV) in ether gives ketone (V), which is converted to acid (VI) (a 1:1 mixture of E and Z olefins at C-5) using (4-carboxybutyl)triphenylphosphorane in dimethyl sulfoxide. Cleavage of the alcohol-protecting groups in (VI) with an acetic acid-water-tetrahydrofuran mixture followed by chromatography to remove the 5-E isomer affords 9-methylcarbacyclin (VII). Finally, treatment of (VII) with calcium oxide in tetrahydrofuran gives U-61431F (ciprostene calcium).
…………………………..

J Org Chem 1983,v 48, 26, pg 5341 as label 10, mp , ir given
http://pubs.acs.org/doi/pdf/10.1021/jo00174a035 pdf dowload
Ciprostene calcium Calcium salt 10
5Z -9BETA-Methyl-6alpha-carbaprostaglandin I2, Calcium Salt (10). A suspension of 350 mg (0.96 mmol) of acid 8b, 23.6 mg (0.42 mmol) of calcium oxide, 5 mL of water, and 4 mL of THF was heated for 20 min at 50 “C and filtered, and the solvents were removed under reduced pressure. The resulting foam was dissolved in 4 mL of THF and then added dropwise to 50 mL of ether. The resulting suspension was stirred for 15 min, then filtered (rinsing with ether) to give 265 mg (82%) of calcium salt
10 as a white solid: mp 101-108 OC;
IR (mull) 3330,1670,1555, 1455, 1345, 1310, 1270, 1075, 1020, 970 cm-‘.
Anal. Calcd for C4H,,08Ca: C, 68.89; H, 9.20; Ca, 5.23. Found: C, 68.55; H, 8.94; Ca, 5.29
Ciprostene calcium FREE BASE 8b
(5Z)-9BETA-Methyl-6ALPHA-carbaprostaglandin I2 (8b) and (5E)-9BETA-Methyl-6a-carbaprostaglandin I2 (9b).
A solution of 17 mmol of sodium methylsulfinylmethide (prepared from 0.81 g of a 50% sodium hydride dispersion and 66 mL of Me2SO) was cooled to 15 “C, treated with 4.20 g (9.60 mmol) of (4-carboxybuty1)triphenylphosphonium bromide, stirred for 20 min, treated with 0.80 g (1.78 mmol) of ketone 6b in 12 mL of THF, stirred for 5 hat 45 “C, cooled to 0 “C, treated with 6 mL of water, stirred for 1 h, acidified with a solution of 5 mL of HZSO, in 100 mL of 1:1 water-brine, and extracted with ether. The ether extracts were washed several times with water and then with brine and were dried (Na2S04). The solvents were removed under reduced pressure and the residue was chromatographed on acid-washed silica gel eluted with 20% ethyl acetate in hexane to give 0.932 g (98%) of acid mixture 7b as an oil (Rf 0.38 in 65:34:1 hexane ethyl acetate-acetic acid). Without further purification, 0.75 g (1.41 mmol) of acid 7b was heated at 45 “C in a solution of 5 mL of THF, 7.5 mL of water, and 15 mL of glacial acetic acid. After 3 h the solution was cooled and partitioned between brine and 32 ethyl acetatehexme. The organic portion was dried (Na2S04) and the solvent removed under reduced pressure (using a toluene azeotrope to remove any remaining acetic acid). The crude product was chromatographed on HPLC silica gel eluted with 1000:405 chloroform-methanol-acetic acid to give 0.24 g (47%) of acid 8b as a colorless oil (Rf 0.25) and 0.23 g (45%) of acid 9b as a colorless oil (Rf 0.27). 8b:
NMR 6 0.89 (t, J = 5 Hz, 3 H), 1.02-2.8 (m including 3 H singlet at 6 1.08, 25 H), 3.5-4.35 (m, 2 H), 5.0-5.7 (m, 3 H), 6.05
(br s, 3 H);
IR (fh) 3340,2660,1710,1240,1205,1175,1130,1075, 1055,1020,970 cm-*;
mass spectrum, calcd for C30H5704Si3 [M’ – CH3 of tris(trimethylsily1) derivative],
m/e 565.3564; found, m/e 565.3552
DATA OF 9b ……….NOT DESIRED COMPD…please note
9b: NMR 6 0.90 (t, J = 5 Hz, 3 H), 1.06 (s, 3 H), 1.1-2.6 (m,22 H), 3.5-4.3 (m, 2 H), 5.0-5.7 (m, 3 H), 5.93 (br s, 3 H); IR (film) 3340, 2660, 1710, 1300, 1240, 1175, 1130, 1075, 1055, 1020, 970
cm-‘; mass spectrum, calcd for C30H5704Si3 [M+ – CH3 of tris-(trimethylsilyl) derivative], m/e 565.3564; found, m/e 565.3541
References
- Drugs Fut 1985, 10(11): 900
- Journal of Organic Chemistry, 1983 , vol. 48, 26 pg. 5341 – 5348 entry 10, mp,101 – 108 °CU-61,431F, a stable prostacyclin analogue, inhibits the proliferation of bovine vascular smooth muscle cells with little antiproliferative effect on endothelial cells.Shirotani M, Yui Y, Hattori R, Kawai C.Prostaglandins. 1991 Feb;41(2):97-110.
- J Org Chem 1983,v 48, 26, pg 5341 as label 10, mp , ir givenhttp://pubs.acs.org/doi/abs/10.1021/jo00174a035
- US 4420632
- EP257859 B1…
- US2002/147184 A1…
- J Org Chem 1981,46, 1954
| US4158667 * | 28 Jul 1977 | 19 Jun 1979 | The Upjohn Company | 6-Keto PGF analogs |
| US4338323 * | 10 Nov 1980 | 6 Jul 1982 | Science Union Et Cie | Piperidylbenzimidazolinone derivatives |
| US4539333 * | 10 May 1977 | 3 Sep 1985 | Burroughs Wellcome Co. | Prostacyclin, methods of using and method of making |
| US4632919 * | 27 Sep 1984 | 30 Dec 1986 | University Of Medicine & Dentistry Of N.J. | Process for prolonging recalcification, prothrombin and thrombin times of plasma |
| EP0112122A2 * | 8 Dec 1983 | 27 Jun 1984 | South African Inventions Development Corporation | Plasminogen activator |
| WO1987003488A1 * | 15 Dec 1986 | 18 Jun 1987 | Schering Ag | Treatment of thrombosis with fibrinolytic agents and prostacyclines |
| US4158667 * | 28 Jul 1977 | 19 Jun 1979 | The Upjohn Company | 6-Keto PGF analogs |
| US4338325 * | 27 Oct 1980 | 6 Jul 1982 | The Upjohn Company | PGI.sub.2 Pharmacologically acceptable salts |

Aegerion Pharmaceuticals: Juxtapid Sales Continue To Climb
.
.
Updated 01/20/14: With information on the approval of Juxtapid in Mexico for HoFH.
Aegerion Pharmaceuticals, a Cambridge, Massachusetts biopharmaceutical company focusing on development and commercialization of treatments for rare diseases, launches in the United States in January 2013, orphan drug Juxtapid (Lomitapide). Juxtapid is an oral once-a-day treatment for rare disease Homozygous Familial Hypercholesterolemia (HoFH). HoFH is caused by genetic defects inherited from both parents that affects the function of the LDL receptor, that is responsible for removing bad cholesterol (LDL-C) from the body.
Background Information On Juxtapid For HoFH
• Receives FDA Orphan Drug Designation (ODD) in October 2007
• Receives FDA approval in December 2012; Lojuxta (Juxtapid name in EU) receives EU approval in July 2013
• Launches in US in January 2013
• US price of $235,000 – 295,000/year
• Boxed warning of potential for liver toxicity
• Restricted distribution through Risk Evaluation &…
View original post 652 more words
Gilead’s HCV drug Sovaldi gets Europe OK
Gilead Sciences’ closely-watched hepatitis C drug Sovaldi has been given the green light in Europe.
The European Commission has granted marketing authorisation for Sovaldi (sofosbuvir) 400mg tablets
which, as part of HCV combination therapy with peg-interferon and ribavirin, offers cure rates of around 90% in previously-untreated adults. However, most significant is that the once-daily nucleotide analogue polymerase inhibitor is the first all-oral treatment option for up to 24 weeks for patients unsuitable for interferon.
Read more at: http://www.pharmatimes.com/Article/14-01-20/Gilead_s_HCV_drug_Sovaldi_gets_Europe_OK.aspx#ixzz2qwHI3iJi
SYNTHESIS
-
sofosbuvir » All About Drugs
http://www.allfordrugs.com/tag/sofosbuvir/ALL ABOUT DRUGS BY DR ANTHONY MELVIN CRASTO, WORLD DRUG TRACKER HELPING … US Approves Breakthrough Hepatitis C Drug,Sofosbuvir.
PSC 833 ( Valspodar )

Valspodar, SDZ-PSC-833, PSC-833, Amdray
P-Glycoprotein (MDR-1; ABCB1) Inhibitors , Multidrug Resistance Modulators
Valspodar is a cyclosporine derivative and a P-glycoprotein inhibitor currently in phase III clinical trials at the National Cancer Institute (NCI) in combination with chemotherapy for the treatment of leukemia. The drug was also being developed in combination with chemotherapy for the treatment of various other types of cancers, however, no recent developments on these trials have been reported.
P-glycoprotein is an ABC-transporter protein that has been implicated in conferring multidrug resistance to tumor cells. In previous trials, valspodar was associated with greater disease-free and overall survival in younger patients (45 years or below), and was shown to significantly increase the cellular uptake of daunorubicin in leukemic blast cells in vivo. However, in a phase III trial examining the drug candidate’s effects on AML in patients at least 60 years of age, valspodar was associated with excessive mortality and complete remission rates were higher in groups not treated with the compound.
Nonimmunosuppressive cyclosporin analog which is a potent multidrug resistance modifier; 7-10 fold more potent than cyclosporin A; a potent P glycoprotein inhibitor; MW 1215.
M.Wt: 1214.62
Formula: C63H111N11O12
CAS : 121584-18-7
IUPAC/Chemical name:
(3S,6S,9S,12R,15S,18S,21S,24S,30S,33S)-6,9,18,24-tetraisobutyl-3,21,30-triisopropyl-1,4,7,10,12,15,19,25,28-nonamethyl-33-((R,E)-2-methylhex-4-enoyl)-1,4,7,10,13,16,19,22,25,28,31-undecaazacyclotritriacontan-2,5,8,11,14,17,20,23,26,29,32-undecaone
6 – [(2S, 4R, 6E)-4-Methyl-2-(methylamino)-3-oxo-6-octenoic acid]-7-L-valine-cyclosporin A; Cyclo [[(2S, 4R, 6E) -4-methyl-2-(methylamino)-3-oxo-6-octenoyl]-L-valyl-N-methylglycyl-N-methyl-L-leucyl-L-valyl-N-methyl-L-leucyl-L- alanyl-D-alanyl-N-methyl-L-leucyl-Nm
[3′-oxo-4-butenyl-4-methyl-Thr1]-[Val2]-cyclosporine
Clinical trials
http://clinicaltrials.gov/search/intervention=psc+833
Synonyms
- 3′-Keto-bmt(1)-val(2)-cyclosporin A
- Amdray
- Psc 833
- PSC-833
- PSC833
- SDZ PSC 833
- Sdz-psc-833
- UNII-Q7ZP55KF3X
- Valspodar
Valspodar or PSC833 is an experimental cancer treatment and chemosensitizer drug.[1] It is a derivative of ciclosporin D.
Its primary use is that of a p-glycoprotein inhibitor. Previous studies in animal models have found it to be effective at preventing cancer cell resistance to chemotherapeutics, but these findings did not translate to clinical success.[2]
Valspodar, also known as PSC-833 is an analogue of cyclosporin-A. Valspodar inhibits p-glycoprotein, the multidrug resistance efflux pump, thereby restoring the retention and activity of some drugs in some drug-resistant tumor cells. This agent also induces caspase-mediated apoptosis.
PSC-833 is a non-immunosuppressive cyclosporin derivative that potently and specifically inhibits P-gp. In vitro experiments indicate that PSC-833interacts directly with P-gp with high affinity and probably interferes with the ATPase activity of P-gp. Studies in multidrug resistant tumor models confirm P-gp as the in vivo target of PSC-833 and demonstrate the ability of PSC-833 to reverse MDR leukemias and solid tumors in mice. Presently,PSC-833 is being evaluated in the clinic.
Valspodar can cause nerve damage.[1]
Valspodar

Synthesis By oxidation of cyclosporin D (I) with N-chlorosuccinimide and dimethylsulfide in toluene (1) Scheme 1 Description alpha (20, D) -..?. 255.1 (c 0.5, CHCl3) Manufacturer Sandoz Pharmaceuticals Corp (US).. . References 1 Bollinger, P., B flounder sterli, JJ, Borel, J.-F., Krieger, M., Payne, TG, Traber, RP, Wenger, R. (Sandoz AG; Sandoz Patent GmbH; Sandoz Erfindungen VmbH ). Cyclosporins and their use as pharmaceuticals.
AU 8817679, EP 296122, JP 89045396. AU 8817679; EP 0296122; JP 1989045396; JP 1996048696; US 5525590
……………………………..
- The cyclosporins comprise a class of structurally distinctive, cyclic, poly-N-methylated undecapeptides, generally possessing pharmacological, in particular immunosuppressive, anti-inflammatory and/or anti-parasitic activity, each to a greater or lesser degree. The first of the cyclosproins to be isolated was the naturally occurring fungal metabolite Ciclosporin or Cyclosporine, also known as cyclosporin A and now commercially available under the Registered Trade Mark SANDIMMUN®. Ciclosporin is the cyclosporin of formula A

wherein -MeBmt- represents the N-methyl-(4R)-4-but-2E-en-1-yl-4-methyl-(L)threonyl residue of formula B

in which -x-y- is trans -CH=CH- and the positive 2′, 3′ and 4′ have the configuration S, R and R respectively.
-
Since the original discovery of Ciclosporin, a wide variety of naturally occurring cyclosporins have been isolated and identified and many further non-natural cyclosporins have been prepared by total- or semi-synthetic means or by the application of modified culture techniques. The class comprised by the cyclosporins is thus now substantial and includes, for example, the naturally occurring cyclosporins A through Z [c.f. Traber et al. 1, Helv. Chim. Acta, 60, 1247-1255 (1977); Traber et al. 2, Helv. Chim. Acta, 65, 1655-1667 (1982); Kobel et al., Europ. J. Applied Microbiology and Biotechnology 14, 273-240 (1982); and von Wartburg et al. Progress in Allergy, 38, 28-45 (1986)], as well as various non-natural cyclosporin derivatives and artificial or synthetic cyclosporins including the dihydro- and iso-cyclosporins [in which the moiety -x-y- of the -MeBmt- residue (Formula B above) is saturated to give -x-y- = -CH₂-CH₂- / the linkage of the residue -MeBmt- to the residue at the 11-position of the cyclosporin molecule (Formula A above) is via the 3′-O-atom rather than the α-N-atom]; derivatised cyclosporins (e.g. in which the 3′-O-atom of the -MeBmt- residue is acylated or a further substituent is introduced at the α-carbon atom of the sarcosyl residue at the 3-position); cyclosporins in which the -MeBmt- residue is present in isomeric form (e.g. in which the configuration across positions 6′ and 7′ of the -MeBmt- residue is cis rather than trans); and cyclosporins wherein variant amino acids are incorporated at specific positions within the peptide sequence employing e.g. the total synthetic method for the production of cyclosporins developed by R. Wenger – see e.g. Traber et al. 1, Traber et al. 2 and Kobel et al. loc. cit.; U.S. Patents Nos 4 108 985, 4 210 581, 4 220 641, 4 288 431, 4 554 351 and 4 396 542; European Patent Publications Nos. 0 034 567 and 0 056 782; International Patent Publication No. WO 86/02080; Wenger 1, Transpl. Proc. 15, Suppl. 1:2230 (1983); Wenger 2, Angew. Chem. Int. Ed., 24, 77 (1985); and Wenger 3, Progress in the Chemistry of Organic Natural Products 50, 123 (1986).
-
The class comprised by the cyclosporins is thus now very large indeed and includes, for example [Thr]²-, [Val]²-, [Nva]²- and [Nva]²-[Nva]⁵-Ciclosporin (also known as cyclosporins C, D, G and M respectively), [3-O-acetyl-MeBmt]¹-Ciclosporin (also known as cyclosporin A acetate), [Dihydro-MeBmt]¹-[Val]²-Ciclosporin (also known as dihydro-cyclosporin D), [Iso-MeBmt]¹-[Nva]²-Ciclosporin (also known as isocyclosporin G), [(D)Ser]⁸-Ciclosporin, [MeIle]¹¹-Ciclosporin, [(D)MeVal]¹¹-Ciclosporin (also known as cyclosporin H), [MeAla]⁶-Ciclosporin, [(D)Pro]³-Ciclosporin and so on.
-
[In accordance with conventional nomenclature for cyclosporins, these are defined throughout the present specification and claims by reference to the structure of Ciclosporin (i.e. Cyclosporin A). This is done by first indicating the amino acid residues present which differ from those present in Ciclosporin (e.g. “[(D)Pro]³” to indicate that the cyclosporin in question has a -(D)Pro- rather than -Sar- residue at the 3-position) and then applying the term “Ciclosporin” to characterise remaining residues which are identical to those present in Ciclosporin.
-
The residue -MeBmt- at position 1 in Ciclosporin was unknown before the discovery of the cyclosporins. This residue and variants or modifications of it, e.g. as described below, are thus generally characteristic of the cyclosporins. In general, variants or alternatives to [MeBmt]¹ are defined by reference to the -MeBmt- structure. Thus for dihydrocyclosporins in which the moiety -x-y- (see formula B above) is reduced to -CH₂-CH₂-, the residue at the 1-position is defined as “-dihydro-MeBmt-“. Where the configuration across the moiety -x-y- is cis rather than trans, the resulting residue is defined as “-cis-MeBmt-“.
-
Where portions of the -MeBmt- residue are deleted, this is indicated by defining the position of the deletion, employing the qualifier “des” to indicate deletion, and then defining the group or atom omitted, prior to the determinant “-MeBmt-“, “-dihydro-MeBmt-“, “-cis-MeBmt-” etc.. Thus “-N-desmethyl-MeBmt-“, “-3′-desoxy-MeBmt-“, and “-3′-desoxy-4′-desmethyl-MeBmt-” are the residues of Formula B¹, B² and B³ respectively:

B¹ – X = CH₃, Y = OH, Z = H.
B² – X = CH₃, Y = H, Z = CH₃.
B³ – X = H, Y = H, Z = CH₃. -
Where positions or groups, e.g. in -MeBmt-, are substituted this is represented in conventional manner by defining the position and nature of the substitution. Thus -3′-O-acetyl-MeBmt- is the residue of formula B in which the 3′-OH group is acetylated (3′-O-COCH₃). Where substituents of groups, in e.g. -MeBmt-, are replaced, this is done by i) indicating the position of the replaced group by “des-terminology” as described above and ii) defining the replacing group. Thus -7′-desmethyl-7′-phenyl-MeBmt- is the residue of formula B above in which the terminal (8′) methyl group is replaced by phenyl. 3′-Desoxy-3′-oxo-MeBmt- is the residue of formula B above in which the 3′-OH group is replaced by =O.
-
In addition, amino acid residues referred to by abbreviation, e.g. -Ala-, -MeVal-, -αAbu- etc… are, in accordance with conventional practice, to be understood as having the (L)-configuration unless otherwise indicated, e.g. as in the case of “-(D)Ala-“. Residue abbreviations preceded by “Me” as in the case of “-MeLeu-“, represent α-N-methylated residues. Individual residues of the cyclosporin molecule are numbered, as in the art, clockwise and starting with the residue -MeBmt-, -dihydro-MeBmt- etc. … in position 1. The same numerical sequence is employed throughout the present specification and claims.]
-
[0010]Because of their unique pharmaceutical potential, the cyclosporins have attracted very considerable attention, not only in medical and academic circles, but also in the lay press. Cyclosporin itself is now commonly employed in the prevention of rejection following allogenic organ, e.g. heart, heart-lung, kidney and bone-marrow transplant, as well as, more recently, in the treatment of various auto-immune and related diseases and conditions. Extensive work has also been performed to investigate potential utility in the treatment of various parasitic diseases and infections, for example coccidiomycosis, malaria and schistosomiasis. Reports of investigative work into the potential utility of the very many other known cyclosporins in these or related indications now abound in the literature.



………………………………
References
- Wilkes, Gail; Ades, Terri B. (2004). Consumers Guide to Cancer Drugs. Jones & Bartlett Learning. p. 226. ISBN 9780763722548. Retrieved 29 May 2013.
- Tao, Jian’guo; Sotomayor, Eduardo. (2012). Hematologic Cancers: From Molecular Pathobiology to Targeted Therapeutics. Springer. p. 335. ISBN 9789400750289.
- PSC-833Drugs Fut 1995, 20(10): 1010
- US 5525590
- Synthesis of [S-[1-14C]Val(7)]VALSPODAR application of (+)/(-)-[13,14Cn]BABS and (+)/(-)-[13,14Cn]DPMGBS, part 4J Label Compd Radiopharm 2000, 43(3): 205
- WO 2006013094
- WO 2005013947
- WO 2002098418
- WO 1999017757
- Pharmaceutical Research, 2001 , vol. 18, 2 pg. 183 – 190
- US2003/158097 A1
- Valspodar; EP-B1 0 296 122:
- WO 94/07858
A New Class Of Antibiotics To Replace The Ones That Are No Longer Effective

As concerns about bacterial resistance to antibiotics grow, researchers are racing to find new kinds of drugs to replace ones that are no longer effective. One promising new class of molecules called acyldepsipeptides—ADEPs—kills bacteria in a way that no marketed antibacterial drug does—by altering the pathway through which cells rid themselves of harmful proteins.
Now, researchers from Brown Univ. and the Massachusetts Institute of Technology have shown that giving the ADEPs more backbone can dramatically increase their biological potency. By modifying the structure of the ADEPs in ways that make them more rigid, the team prepared new ADEP analogs that are up to 1,200 times more potent than the naturally occurring molecule.
A paper describing the research was released online by the Journal of the American Chemical Society.
“The work is significant because we have outlined and validated a strategy for the enhancing the potency of this promising class of antibacterial drug…
View original post 788 more words
Boehringer-Ingelheim …A Well-Balanced Pipeline
Promising Drugs in Boehringer-Ingelheim Pipeline
| Compound* | Clinical phase | Indication | Therapeutic principle | Mode of action |
|---|---|---|---|---|
| Olodaterol | Submitted | Chronic obstructive pulmonary Disease (COPD) | Long-acting beta-agonist | Bronchodilation |
| Tiotropium | Submitted | Cystic fibrosis (CF) | Bronchodilatator | Long Acting Muscarinic Antagonist |
| Afatinib | Phase III | Breast cancer | Signal transduction inhibition | Novel irreversible ErbB Family blocker |
| Afatinib | Phase III | Head and neck cancer | Signal transduction inhibition | Novel irreversible ErbB Family blocker |
| Deleobuvir (BI 207127) |
Phase III | Hepatitis C | Direct acting antiviral small molecule | Oral NS5B RNA-dependent polymerase inhibitor |
| Empagliflozin | Phase III | Diabetes mellitus type II |
SGLT-2-inhibitor | Inhibition of glucose transporter-2 |
| Faldaprevir (BI 201335) |
Phase III | Hepatitis C | Direct acting antiviral small molecule | Oral HCV NS3/4A protease inhibitor |
| Nintedanib | Phase III | Non-small cell lung cancer (NSCLC) | Angiogenesis inhibition | Triple angiokinase inhibitor, simultaneously blocks VEGFR, FGFR, PDGFR |
| Nintedanib | Phase III | Ovarian cancer | Angiogenesis inhibition | Triple angiokinase inhibitor, simultaneously blocks VEGFR, FGFR, PDGFR |
| Nintedanib | Phase III | Idiopathic pulmonary fibrosis (IPF) | Anti-fibrotic kinase inhibition | Anti-fibrotic kinase inhibitor |
| Tiotropium | Phase III | Asthma | Bronchodilatator | Long Acting Muscarinic Antagonist |
| Volasertib | Phase III | Various cancer types | Cell-cycle kinase inhibition | PLK-1 antagonist |
* These are investigational agents; their safety and efficacy have not yet been established.
Status: April 2013
Successful Products from our Boehringer-Ingelheim Research & Development
| Product name | First launch | Active ingredient | Indication |
|---|---|---|---|
| Gilotrif™ | 2013 | Afatinib | Non-small cell lung cancer (NSCLC) |
| Trajenta® | 2011 | Linagliptin | Diabetes mellitus type II |
| Pradaxa® | 2010 2008 |
Dabigatran etexilate | Stroke prevention in atrial fibrillationPrevention of venous thromboembolic events (VTE) in adults |
| Spiriva® Respimat Soft Mist™ InhalerSpiriva® |
2007 2002 |
Tiotropium | COPD |
| Micardis® | 1998 | Telmisartan | Essential hypertension |
| Sifrol® / Mirapex® /Mirapexin® | 20061997 | Pramipexole | Restless legs syndrome (RLS) Parkinson’s disease (PD) |
| Viramune® | 1996 | Nevirapine | HIV/AIDS |


Oncology Websites
No flash player detected. For optimized usage of this website your browser should support shockwave flash. For downloading see Macromedia Flash Player
Alexion Pharmaceuticals: The Power Of Soliris Continues
Alexion Pharmaceuticals is a global biopharmaceutical company focusing on developing therapies for patients with ultra-rare diseases. The company’s first and only marketed product, orphan drug Soliris (Eculizumab), generates blockbuster profits from two approved indications :
• Paroxysmal Nocturnal Hemoglobinuria (PNH), a rare genetic blood disorder
• Atypical Hemolytic Uremic Syndrome (aHUS), an ultra-rare genetic disorder.
Multiple FDA Orphan Drug Designation Indications
Soliris has FDA Orphan Drug Designation (ODD) for the following indications:
Num | Designation Date | Orphan Designation |
1 | 08-20-2003 | PNH |
2 | 04-29-2009 | aHUS |
3 | 10-18-2011 | Shiga-Toxin producing Escherichia Coli Hemolytic Uremic Syndrome (STEC-HUS) |
4 | 06-24-2013 | NeuroMyelitis Optica (NMO) |
5 | 01-10-2014 | Prevention of Delayed Graft Function (DGF) after Renal Transplantation |
.
On January 10, Soliris receives FDA ODD for the prevention of Delayed Graft Function (DGF) after renal transplantation.
J.P. Morgan Healthcare Conference
Leonard Bell, CEO of Alexion Pharmaceuticals, presents on January 15 at the J.P. Morgan Healthcare Conference
View original post 453 more words
DR ANTHONY MELVIN CRASTO

ORGANIC SPECTROSCOPY

