



Graphical abstract

PROUD Indian WORLD RECORD VIEWS holder on THIS BLOG, ………live, by DR ANTHONY MELVIN CRASTO, Worldpeaceambassador, Worlddrugtracker, Helping millions, 100 million hits on google, pushing boundaries,2.5 lakh plus connections worldwide, 45 lakh plus VIEWS on this blog in 227 countries, 7 CONTINENTS ……A 90 % paralysed man in action for you, I am suffering from transverse mylitis and bound to a wheel chair, [THIS BLOG HOLDS WORLD RECORD VIEWS ] A PROUD INDIAN
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO FACEBOOK
...................................................................Join me on twitter
FACEBOOK
...................................................................Join me on twitter
 ..................................................................Join me on google plus
..................................................................Join me on google plus
 Googleplus
GoogleplusHuman pluripotent stem cells (hPSCs) show great potential and versatility in regenerative medicine and new therapeutic approaches to fight disease. Patient-specific, individualized treatments using stem cells have even been generated for a number of diseases. Although further research into hPSCs is needed in order to harness their full potential, preserving the stem cells and storing them in the large numbers required for research has proved difficult.
Teruo Akuta and colleagues at the RIKEN Center for Developmental Biology, together with scientists from the Foundation for Biomedical Research and Innovation, have now developed a cost-effective, efficient and reliable slow-freezing method for preserving hPSCs in large numbers with a high survival rate.
Vitrification, which involves the use of cryoprotectants to chill cells to low temperatures without freezing, and conventional slow-freezing techniques are currently used for the cryopreservation of hPSCs. “Vitrification using liquid nitrogen is a highly skilled task,” notes Akuta, “and is not…
View original post 263 more words

Plazomicin
Plazomicin is a neoglycoside antibiotic with activity against a broad range of Gram-positive and Gram-negive pathogens. Plazomicin showed potent in vitro activity against multidrug-resistant Klebsiella pneumoniae and Escherichia coli.
| Synonyms: O-2-Amino-2,3,4,6-tetradeoxy-6-[(2-hydroxyethyl)amino]-α-D-glycero-hex-4-enopyranosyl-(1→4)-O-[3-deoxy-4-C-methyl-3-(methylamino)-β-L-arabinopyranosyl-(1→6)]-N1-[(2S)-4-amino-2-hydroxy-1-oxobutyl]-2-deoxy-D-streptamine; ACHN 490; |
| CAS Number: 1154757-24-0 |
| Achaogen (USA)Phase II completed |
| Mol. Formula: C25H48N6O10 |
| Aminoglycosides, Broad-spectrum, |
| Mol. Weight: 592.68 |
To continue the development of plazomicin, the company has received a contract option of US$ 60M from the Biomedical Advanced Research and Development Authority (BARDA) to support a global Phase III clinical study. The study will evaluate plazomicin in treating patients with serious Gram-negative bacterial infections due to carbapenem-resistant Enterobacteriaceae. The study is expected to start in the fourth quarter of 2013 [4].

Achaogen (a-KAY-o-jen) is developing plazomicin, its lead product candidate, for the treatment of serious bacterial infections due to MDR Enterobacteriaceae, including carbapenem-resistant Enterobacteriaceae, or CRE. In 2013, the Centers for Disease Control and Prevention identified CRE as a “nightmare bacteria” and an immediate public health threat that requires “urgent and aggressive action.” We expect to initiate a Phase 3 superiority trial of plazomicin in the first quarter of 2014.
CRE are one of many types of MDR gram-negative pathogens threatening patients. Bacteria such as Pseudomonas aeruginosa, Acinetobacter baumannii, and extended-spectrum beta-lactamase producing Enterobacteriaceae each pose “serious” resistance threats, according to the CDC, and also drive a great need for new, safe, and effective antibiotics. We have assembled the chemistry and microbiology expertise and capabilities required to develop new agents for the treatment of gram-negative infections. Plazomicin was the first clinical candidate from our gram-negative antibiotic discovery engine. In addition, our research and development pipeline includes two antipseudomonal programs targeting P. aeruginosa—a program to discover and develop small molecule inhibitors of LpxC, which is an enzyme essential for the synthesis of the outer membrane of gram-negative bacteria, and a therapeutic antibody program. We are also pursuing small molecule research programs targeting other essential gram-negative enzymes.
Achaogen has built an exceptional research and development team with deep expertise in the discovery and development of new drugs from research through commercialization. Our executive team has over 60 years of combined industry experience, and a proven track record of leadership, global registration, and lifecycle management for over 20 products. Our facility is located on the shores of the San Francisco Bay, ten minutes from the San Francisco International Airport, and only fifteen minutes from downtown San Francisco.
http://www.google.com/patents/US20100099661
Common Intermediates Sisomicin

Amberlite IRA-400 (OH form) (200 g) was washed with MeOH (3×200 m1). To a stirring suspension of the washed resin in MeOH (150 mL) was added sisomicin sulfate (20.0 g, 0.029 mol) and the mixture was stirred overnight. The resin was then filtered and washed with MeOH (100 mL) and the combined organic layers were concentrated to dryness to yield the desired sisomicin (11.57 g, 0.026 mol, 89.6% yield): MS m/e [M+H]+ calcd 448.3, found 448.1.
Example 1 6′-(2-Hydroxy-ethyl)-1-(4-amino-2(S)-hydroxy-butyryl)-sisomicin

6′-(2-tert-Butyldimethylsililoxy-ethyl)-2′,3,3″-triBoc-1-(N-Boc-4-amino-2(S)-hydroxy-butyryl)-sisomicin
2′,3,3″-triBoc-1-(N-Boc-4-amino-2(S)-hydroxy-butyryl)-sisomicin (0.10 g, 0.105 mmol) was treated with tert-butyldimethylsilyloxy acetaldehyde following Procedure 1-Method A to yield the desired 6′-(2-tert-butyldimethylsilyloxy-ethyl)-2′,3,3″-triBoc-1-(N-Boc-4-amino-2(S)-hydroxy-butyryl)-sisomicin (MS m/e [M+H]+ calcd 1107.6, found 1107.4), which was carried through to the next step without further purification.

6′-(2-Hydroxy-ethyl)-1-(4-amino-2(S)-hydroxy-butyryl)-sisomicin
6′ -(2-tert-butyldimethylsililoxy-ethyl)-2′,3,3″-triBoc-1-(N-Boc-4-amino-2(S)-hydroxy-butyryl)-sisomicin (0.105 mmol) was submitted to Procedure 3-Method B for Boc removal to yield a crude, which was purified by RP HPLC Method 1-Column A to yield 6′-(2-hydroxy-ethyl)-1-(4-amino-2(S)-hydroxy-butyryl)-sisomicin: MS m/e [M+H]+ calcd 593.3, found 593.2, [M+Na]+615.3 ; CLND 97.5% purity.
|
4-23-2010
|
ANTIBACTERIAL AMINOGLYCOSIDE ANALOGS
|

| US8318685 | Nov 14, 2011 | Nov 27, 2012 | Achaogen, Inc. | Antibacterial aminoglycoside analogs |
| US8367625 | Apr 7, 2011 | Feb 5, 2013 | Achaogen, Inc. | Antibacterial aminoglycoside analogs |
| US8372813 | Apr 7, 2011 | Feb 12, 2013 | Achaogen, Inc. | Antibacterial aminoglycoside analogs |
| US8377896 | Mar 9, 2011 | Feb 19, 2013 | Isis Pharmaceuticals, Inc | Antibacterial 4,6-substituted 6′, 6″ and 1 modified aminoglycoside analogs |
| US8399419 | Mar 9, 2011 | Mar 19, 2013 | Achaogen, Inc. | Antibacterial aminoglycoside analogs |
| US8481502 | Apr 6, 2012 | Jul 9, 2013 | Achaogen, Inc. | Antibacterial aminoglycoside analogs |
| US8492354 | Nov 14, 2011 | Jul 23, 2013 | Achaogen, Inc. | Antibacterial aminoglycoside analogs |
| US8524675 | Nov 14, 2011 | Sep 3, 2013 | Achaogen, Inc. | Antibacterial aminoglycoside analogs |
| US8524689 | Nov 14, 2011 | Sep 3, 2013 | Achaogen, Inc. | Antibacterial aminoglycoside analogs |
| US8569264 | Jan 5, 2012 | Oct 29, 2013 | Isis Pharmaceuticals, Inc. | Antibacterial 4,5-substituted aminoglycoside analogs having multiple substituents |
| US8653041 | Oct 15, 2012 | Feb 18, 2014 | Achaogen, Inc. | Antibacterial aminoglycoside analogs |
| US8653042 | Nov 14, 2011 | Feb 18, 2014 | Achaogen, Inc. | Antibacterial aminoglycoside analogs |
| US8658606 | Nov 14, 2011 | Feb 25, 2014 | Achaogen, Inc. | Antibacterial aminoglycoside analogs |

7-[(4S)-4-Amino-6-azaspiro[2.4]heptan-6-yl]-8-chloro-6-fluoro-1-[(2S)-2-fluorocyclopropyl]-4-oxoquinoline-3-carboxylic acid
(1R-(1a(S*),2a))-7-(7-Amino-5-azaspiro[2.4]hept-5-yl)-8-chloro-6-fluoro-1-(2-fluorocyclopropyl)-1,4-dihydro-4-oxo-3-quinolinecarboxylic Acid
SYNTHESIS……….http://www.drugfuture.com/synth/syndata.aspx?ID=176447
127254-10-8 [RN]
127254-10-8(ACETATE)
Sitafloxacin (INN; also called DU-6859a) is a fluoroquinolone antibiotic[1] that shows promise in the treatment of Buruli ulcer. The molecule was identified by Daiichi Sankyo Co., which brought ofloxacin and levofloxacin to the market. Sitafloxacin is currently marketed in Japan by Daiichi Sankyo under the tradename Gracevit.
Sitafloxacin is a new-generation, broad-spectrum oral fluoroquinolone antibiotic.It is very active against many Gram-positive, Gram-negative and anaerobic clinical isolates, including strains resistant to other fluoroquinolones, was recently approved in Japan for the treatment of respiratory and urinary tract infections. Sitafloxacin is active against methicillin-resistant staphylococci, Streptococcus pneumoniae and other streptococci with reduced susceptibility to levofloxacin and other quinolones and enterococci

AU 8933702; EP 0341493; JP 1990231475; JP 1995300416; JP 1999124367; JP 1999124380; US 5587386; US 5767127
The condensation of 3-chloro-2,4,5-trifluorobenzoylacetic acid ethyl ester (I) with (1R,2S)-N-(tert-butoxycarbonyl)-2-fluorocyclopropylamine (III) and ethyl orthoformate (II) in hot acetic anhydride gives (1R,2S)-2-(3-chloro-2,4,5-trifluorobenzoyl)-3-(2-fluorocyclopropylamino)acrylic acid ethyl ester (IV). The cyclization of (IV) by means of NaH yields the quinolone (V), which is hydrolyzed with HCl to the free acid (VI). The condensation of (VI) with 7(S)-(tert-butoxycarbonylamino)-5-azaspiro[2.4]heptane (VII) by means of triethylamine in refluxing acetonitrile affords the protected final product (VIII), which is finally deprotected with trifluoroacetic acid and anisole.

The chiral intermediate (1R,2S)-N-(tert-butoxycarbonyl)-2-fluorocyclopropylamine (III) is obtained as follows: 1) The cyclization of butadiene (IX) with dibromofluoromethane by means of BuONa, followed by oxidation with KMnO4, esterification with ethanol – sulfuric acid and reduction with tributyltin hydride gives 2-fluorocyclopropanecarboxylic acid ethyl ester as a cis/trans mixture (X), which is separated by crystallization. The cis-racemic-isomer (XI) is hydrolyzed with NaOH to the corresponding acid (XII), which is condensed with (R)-alpha-methylbenzylamine (XIII) by means of diphenyl chlorophosphate to give the mixture of diastereomers (XIV). This mixture is separated by crystallization, yielding pure (1S,2S)-2-fluoro-N-[alpha(R)-methylbenzyl]cyclopropanecarboxamide (XV), which is hydrolyzed with HCl to the corresponding free acid (XVI). Finally, this compound is converted into (III) by treatment with diphenylphosphoryl azide in refluxing tert-butanol.

b) The intermediate 7(S)-(tert-Butoxycarbonylamino)-5-azaspiro[2.4]heptane (VII) can also be obtained as follows: 1) The cyclopropanation of ethyl acetoacetate (XXXI) with 1,2-dibromoethane (XXXII) by means of K2CO3 in DMF gives 1-acetylcyclopropane-1-carboxylic acid ethyl ester (XXXIII), which is brominated with Br2 in ethanol yielding the bromoacetyl derivative (XXXIV). The cyclization of (XXXI) with (R)-alpha-methylbenzylamine (XIII) by means of triethylamine affords 5-[1(R)-phenylethyl]-5-azaspiro[2.4]heptane-4,7-dione (XXXV), which by reaction with hydroxylamine is converted into the monooxime (XXXVI). The reduction of (XXXVI) with H2 over RaNi in methanol affords 7-amino-5-[1(R)-phenylethyl]-5-azaspiro[2.4]heptan-4-one as a diastereomeric mixture (XXXVII) + (XXXVIII), which is separated by column chromatography. The reduction of the (7S)-isomer (XXXVIII) with LiAlH4 in THF gives 7(S)-amino-5-[1(R)-phenylethyl]-5-azaspiro[2.4]heptane (XXXIX), which is protected in the usual way to the tert-butoxycarbonyl derivative (XL). Finally, this compound is debenzylated to (VII) by hydrogenation with H2 over Pd/C in ethanol.
The chiral intermediate (1R,2S)-N-(tert-butoxycarbonyl)-2-fluorocyclopropylamine (III) is obtained as follows: 1) The cyclization of butadiene (IX) with dibromofluoromethane by means of BuONa, followed by oxidation with KMnO4, esterification with ethanol – sulfuric acid and reduction with tributyltin hydride gives 2-fluorocyclopropanecarboxylic acid ethyl ester as a cis/trans mixture (X), which is separated by crystallization. The cis-racemic-isomer (XI) is hydrolyzed with NaOH to the corresponding acid (XII), which is condensed with (R)-alpha-methylbenzylamine (XIII) by means of diphenyl chlorophosphate to give the mixture of diastereomers (XIV). This mixture is separated by crystallization, yielding pure (1S,2S)-2-fluoro-N-[alpha(R)-methylbenzyl]cyclopropanecarboxamide (XV), which is hydrolyzed with HCl to the corresponding free acid (XVI). Finally, this compound is converted into (III) by treatment with diphenylphosphoryl azide in refluxing tert-butanol.

b) The intermediate 7(S)-(tert-Butoxycarbonylamino)-5-azaspiro[2.4]heptane (VII) can also be obtained as follows: 2) The reaction of 1-acetylcyclopropane-1-carboxylic acid ethyl ester (XXXIII) with (R)-alpha-methylbenzylamine (XIII) by means of NaOH and ethyl chloroformate gives the corresponding amide (XLI), which by reaction with ethylene glycol and p-toluenesulfonic acid is converted into the ethylene ketal (XLII). The bromination of (XLII) with Br2 in dioxane affords the bromomethyl dioxolane (XLIII), which is finally cyclized to 5-[1(R)-phenylethyl]-5-azaspiro[2.4]heptane-4,7-dione (XXXV), already obtained as an intermediate in the preceding synthesis.

The chiral intermediate (1R,2S)-N-(tert-butoxycarbonyl)-2-fluorocyclopropylamine (III) can also be obtained as follows: 3) A study of the influence of different substituents in the cis/trans ratio of the cyclopropanation process has been performed. The general method is as follows: the reaction of benzylamine (XXIII) with acetaldehyde and trichloromethyl chloroformate gives the N-benzyl-N-vinylcarbamoyl chloride (XXIV), which by treatment with alcohol yields the N-vinylcarbamate (XXV). The cyclopropanation of (XXV) with fluorodiiodomethane and diethyl zinc as before preferentially affords the cis-N-(2-fluorocyclopropyl)carbamate (XXVI), which is purified by crystallization. The hydrogenolysis of (XXVI) with H2 over Pd/C in acetic acid gives cis-racemic-2-fluorocyclopropylamine (XXVII), which is submitted to optical resolution with L-menthyl chloroformate to afford pure (1R,2S)-isomer (XXII). Finally, this compound is converted into (III) with tert-butoxycarbonyl anhydride as before.
|
3-7-2012
|
Method for Production of Quinolone-Containing Lyophilized Preparation
|
|
|
12-5-2007
|
Stabilized liquid preparation
|
|
|
8-24-2007
|
PHARMACEUTICAL COMPOSITION
|
|
|
6-29-2007
|
PHARMACEUTICAL COMPOSITION
|
|
|
7-15-2005
|
Pharmaceutical composition
|
|
|
3-2-2005
|
Highly absorptive solid preparation
|
|
|
7-9-2004
|
Highly absorbable solid preparation
|
|
|
2-6-2004
|
Medicinal composition
|
|
|
12-17-1999
|
NOVEL THERAPEUTIC AGENTS THAT MODULATE ENZYMATIC PROCESSES
|

Xing Ren is the mature seed of Prunus armeniaca L. var. ansu Maxim, P. armeniaca L., P. mandshurica (Maxim.) Koehne or P. sibirica L., family Rosaceae.
Main ingredients
Xing Ren contains 50% of fatty oils (including oleic, linoleic. palmitic, stearic and linolenic acids), as well as amygdalin, amygdalase and prunase. Hydrolysing amygdalin generates benzaldehyde and hydrocyanic acid, which is toxic

Bitter Apricot Seed ( Xingren ) 杏仁 , also known as xing ren 杏仁,ku xing ren 苦杏仁,kuang xing ren 光杏仁,ku he ren 杏核仁,炒杏仁,jian xing ren 尖杏仁,xing ren xiang 杏仁霜. It belong to the “Rosaceae” family.
Bitter Apricot Seed ( Xingren ) 杏仁 has a warm, bitter and slightly toxic. It is use for treating the lung and large intestine.

1. Arresting coughing and asthma.
2. Expelling phlegm.
3. Help bowel movements.
Bitter Apricot Seed ( Xingren ) 杏仁 contain hydrogen cyanide which is a strong toxin. Eating 20 to 30 piece may cause toxic reaction even death. The toxin can be hydrolyzed in cooking and can render it non toxic. It is not recommended for small children.
Properties
Taste: bitter; nature: slightly warm; slightly toxic.
Channels entered
Lung and Large Intestine.
Functions and indications
Stops coughing and calms wheezing, moistens the Intestines and frees the bowels. It is indicated for coughing and wheezing, sore throat andconstipation.
Common dosage
3-10g, decocted for a short time only.
Precautions and contraindications

Remarks
Ku Xing Ren (Semen Pruni Armeniacae Amarum), or bitter apricot kernel, is normally used for this herb.
In China, people who can afford to buy apricot as a fresh fruit generally throw the stones away. Servants and the less fortunate children gatherthe stones and sell them to collectors who use cheap labor to crack the shells (endocarp) and free the seeds (kernel) with the brown seed coat tightly covering the embryo (two cotyledons with the small radical and plumule).
In Chinese prescriptions, an apricot seed with the brown seed coat intact is called bei-xing-ren (northern apricot seed). All Chinese apothecaries keep a supply of apricot seeds in this state.
Detoxificated apricot seed: The major portion of the annual production of apricot seed is detoxified, processed by being first subjected to boiling water to loosen the seed coat, which can then be rubbed off by hand, followed by soaking the white cotyledons in cold water with several changes to eliminate the bitter element and to detoxify them. Then the detoxified white cotyledons are dried for the market. In Chinese prescriptions, this decoated and detoxified material is called nan-xing-ren (southern apricot seed) or tian-xing-ren (sweet apricot seed), which is also available in apothecaries. However, a greater portion of the detoxified material is used in pastry and for food.
Toxicity
The LD for intravenous injection of amygdalin in mice or rats is 25g/kg; for intraperitoneal injection, it is 8g/kg; and for oraladministration, it is O.6g/kg. Oral administration of 55 pieces (the equivalem of about 60g) of Ku Xing Ren, containing 1.8g of amygdalin, can cause death in humans. The main symptoms oftoxic reaction include a bitter taste in the mouth, dizziness, nausea, vomiting, pain in the abdomen, diarrhoea, agitation, vexation and restlessness, palpitations, and weakness of the limbs, and in severe cases, oppression in the chest, difficulty in breathing, loss of consciousness, a reduction in blood pressure and even coma. Ku Xing Ren should therefore not be used raw and overdosage must be avoided.
Modern Research


The USPTO continues to offer an online discussion tool for commenting on selected chapters of the Manual. To participate in the discussion and to contribute your ideas go to: http://uspto-mpep.ideascale.com.
Men suffering from sexual dysfunction can be successful at reversing their problem, by focusing on lifestyle factors and not just relying on medication, according to new research at the University of Adelaide.
In a new paper published in the Journal of Sexual Medicine, researchers highlight the incidence of erectile dysfunction and lack of sexual desire among Australian men aged 35-80 years.
Over a five-year period, 31% of the 810 men involved in the study developed some form of erectile dysfunction.
“Sexual relations are not only an important part of people’s wellbeing. From a clinical point of view, the inability of some men to perform sexually can also be linked to a range of other health problems, many of which can be debilitating or potentially fatal,” says Professor Gary Wittert, Head of the Discipline of Medicine at the University of Adelaide and Director of the University’s Freemasons Foundation Centre for…
View original post 255 more words

DS-8587
Daiichi Sankyo (Japan)
7-[3a(R)-Amino-6a(S)-fluoroperhydrocyclopenta[c]pyrrol-2-yl]-6-fluoro-1-[(1R,2S)-2-fluorocyclopropyl]-8-methyl-4-oxo-1,4-dihydroquinoline-3-carboxylic acid hydrochloride dihydrate
7-[(1S,6S)-1-amino-4-oxa-8-azabicyclo[4.3.0]nonan-8-yl]-6-fluoro-1-[(1R,2S)-2-fluorocyclopropan-1-yl]-1,4-dihydro-8-methoxy-4-oxoquinoline-3-carboxylic acid
| C21 H22 F3 N3 O3 . Cl H . 2 H2 O | |
| Mw | 493.904 |
DS-8587, a new broad-spectrum antibacterial agent, is in phase I clinical trials at Daiichi Sankyo for the treatment of bacterial infection.
DS-8587, from Daiichi Sankyo, is a fluoroquinolone with improved activity against both Gram-negative and Gram-positive bacteria. The compound is especially effective against Acinetobacter baumannii but also has improved activity against streptococci, staphylococci, enterococci, E. coli, and anaerobes . The compound is currently under Phase I of clinical development .
DS-8587, a new generation of fluoroquinolone, against Acinetobacter baumannii. The MICs against clinical isolates and inhibitory activity against target enzymes of DS-8587 was superior to ciprofloxacin and levofloxacin. Furthermore, the antibacterial activity of DS-8587 was less affected by adeA/adeB/adeC or abeM efflux pumps and frequency of single-step mutations with DS-8587 was lower as compared to those with ciprofloxacin. DS-8587 might be an effective agent against A. baumanniiinfection.
WO 2008082009 or
http://www.google.com/patents/EP2540715A1?cl=en
(3S)-3-[3-(tert-Butyldimethylsilyloxy)-1-propyl]-5-oxo-1-[(1R)-1-phenylethyl]pyrrolidine-3-carboxylic acid tert-butyl ester

[Reference Example 72]
(3S)-3-[3-(tert-Butyldimethylsilyloxy)-1-propyl]-4-fluoro-5-oxo-1-[(1R)-1-phenylethyl]pyrrolidine-3-carboxylic acid tert-butyl ester

[Reference Example 73]
(3S)-4-Fluoro-3-(3-hydroxy-1-propyl)-5-oxo-1-[(1R)-1-phenylethyl]pyrrolidine-3-carboxylic acid tert-butyl ester

[Reference Example 74]
(3S)-3-(3-benzenesulfonyloxy-1-propyl)-4-Fluoro-5-oxo-1-[(1R)-1-phenylethyl]pyrrolidine-3-carboxylic acid tert-butyl ester

[Reference Example 75]
(1S,5R)-5-Fluoro-4-oxo-3-[(1R)-1-phenylethyl]-3-azabicyclo[3.3.0]octan-1-ylcarboxylic acid tert-butyl ester

[Reference Example 76]
(1R,5R)-1-(tert-Butoxycarbonylamino)-5-fluoro-4-oxo-3-[(1R)-1-phenylethyl]-3-azabicyclo[3.3.0]octane

[Reference Example 77]
(1R,5S)-1-(tert-Butoxycarbonylamino)-5-fluoro-3-[(1R)-1-phenylethyl]-3-azabicyclo[3.3.0]octane

(1R,5S)-1-(tert-Butoxycarbonylamino)-5-fluoro-3-azabicyclo[3.3.0]octane

7-[(1R,5S)-1-Amino-5-fluoro-3-azabicyclo[3.3.0]octan-3-yl]-6-fluoro-1-[(1R,2S)-2-fluorocyclopropane]-1,4-dihydro-8-methyl-4-oxoquinoline-3-carboxylic acid

WO 2012018105
http://www.google.st/patents/WO2012018105A1?cl=en
The following structural formula

compound represented by, 7 – [(1R, 5S) -1 – amino-5 – fluoro-3 – azabicyclo [3.3.0] octan-3 – yl] -6 – fluoro -1 – [(1R, . the 2S) -2 – fluoro-cyclopropane-1 – yl] -1,4 – dihydro-8 – methyl-4 – oxo-3-quinoline -) is referred to as carboxylic acid (hereinafter referred to as Compound A multi-agent containing quinolone resistance including resistant Gram-positive cocci resistant pneumococcus, etc., widely against gram-negative bacteria from Gram-positive bacteria, and, in addition to show strong antibacterial activity, convulsions, which is known in the art as a side effect of the antimicrobial agent of the present system potential cardiotoxicity light and toxicity-inducing activity (photosensitivity), has been reported recently in clinical further (QT prolongation), blood sugar abnormalities, and to express the side effects of delayed-type drug 疹等 is excellent safety low, Then, it is excellent oral absorbability and organ migration properties become apparent, is expected as an antimicrobial agent superior (Patent Document 1).
WO 2008/082009 pamphlet
The compound A, was synthesized according to the method described in Patent Document 1.
Preparation 7 1 hydrochloride dihydrate Ratings (1) Compound A crystalline acid addition salt preparation of acid addition salts of (Example 1) Compound A, and Compound A – [(1R, 5S) – 1 – amino-5 – fluoro-3 – azabicyclo [3.3.0] octan-3 – yl] -6 – fluoro -1 – [(1R, 2S) -2 – fluoro-cyclo-1 – yl] -1 , 4 – dihydro-8 – methyl-4 – oxo-3-quinoline – was added 1mol / L hydrochloric acid (74μL) carboxylic acid (Compound A) (31.3mg,, 0.074mmol) in, and dried under reduced pressure at room temperature.10% aqueous 2 the residue – was added to (100μL) propanol was dissolved by heating at 60 ℃, and allowed to stand day out on the room temperature. Collected by filtration the precipitated crystals, and the 1st air dried, 19.9mg (yield: 54%) was obtained.
Elemental analysis: C 21 H 22 F 3 N 3 O 3 · HCl · 2H 2 O
Theoretical value: C; 51.07, H; 5.51, N; 8.51, F; 11.54, Cl; 7.18
Measured value: C; 50.93, H; 5.40, N; 8.49, F; 11.30, Cl; 7.47
The characteristic diffraction peaks in the powder X-ray diffraction: 2θ = 5.3,7.9,10.6,13.3,21.1,23.0,25.1,27.6 (°)
2 5% aqueous (1001.6mg, 0.746mmol) in the preparation of Compound A one hydrochloride monohydrate (2) Compound A – was added propanol (30mL), was dissolved by heating at 60 ℃. After stirring day out on the room temperature and stirred for 6 hours at 10 ℃. Collected by filtration the precipitated crystals, and the 1st dried air, 839.3 mg (yield: 87%) was obtained.
Elemental analysis: C 21 H 22 F 3 N 3 O 3 · HCl · 1H 2 O
Theoretical value: C; 53.00, H; 5.30, N; 8.83, F; 11.98, Cl; 7.45
Measured value: C; 53.25, H; 5.43, N; 8.51, F; 11.58, Cl; 7.18
The characteristic diffraction peaks in the powder X-ray diffraction: 2θ = 11.3,14.0,20.1,21.4,22.8,24.0,26.0,26.6 (°)
ref
| EP0343524A1 | May 19, 1989 | Nov 29, 1989 | Shionogi Seiyaku Kabushiki Kaisha | Pyridonecarboxylic acids and antibacterial agents |
| JPH0395176A | Title not available | |||
| JPH02231475A | Title not available | |||
| JPH08225567A | Title not available | |||
| JPS6345261A | Title not available | |||
| JPS6456673A | Title not available | |||
| JPS61282382A | Title not available | |||
| US5017708 * | Sep 8, 1989 | May 21, 1991 | Shionogi & Co., Ltd. | Azabicycloalkanes |
| WO1994014794A1 | Dec 28, 1993 | Jul 7, 1994 | Hideki Ao | 8-methoxyquinolonecarboxylic acid derivative |
| WO1995021163A1 | Feb 2, 1995 | Aug 10, 1995 | Katsumi Chiba | Pyridonecarboxylic acid derivative substituted by bicyclic amino group, ester thereof, salt thereof, and bicyclic amine as intermediate therefor |
| WO1996023782A1 | Feb 1, 1996 | Aug 8, 1996 | Daiichi Seiyaku Co | Heterocyclic compounds |

1, nemonoxacin; 2, delafloxacin; 3, finafloxacin; 4, zabofloxacin; 5, JNJ-Q2; 6, DS-8587; 7, KPI-10; 8, ozenoxacin; 9, chinfloxacin; 10, ACH-702.

1-cyclopropyl-8-methyl-7-[5-methyl-6-(methylamino)-3-pyridinyl]-4-oxo-1 ,4-dihydro-3- quinolinecarboxylic acid
1-cyclopropyl-8-methyl-7-{5-methyl-6-[(methylamino)methyl]-3-pyridyl}-4-oxo-1,4-dihydro-3-quinolinecarboxylic acid.
Ferrer Internacional (Spain), phase 3 Gram-positive
Ferrer Internacional has completed one Phase III clinical trial to evaluate the topical formulation of ozenoxacin in the treatment of impetigo [
|
poster……http://landing.quotientbioresearch.com/blog/bid/50380/Ozenoxacin-Activity-against-Atypical-Bacteria
Ozenoxacin is active against a great number of pathogens, such as Propionibacterium acnes, Staphylococcus aureus, methicillin-susceptible Staphylococcus aureus (MSSA), methicillin-resistant Staphylococcus aureus (MRSA) including ciprofloxacin-resistant strains, methicillin-susceptible Staphylococcus epidermidis (MSSE), methicillin-resistant Staphylococcus epidermidis (MRSE), Streptococcus pyogenes, Group G Streptococci, penicillin-resistant Streptococcus pneumoniae, Beta-lactamase positive Haemophilus influenzae, non-typeable strains of Haemophilus influenzae, Beta-lactamase positive Moraxella catarrhalis, Neisseria meningitides, Legionella pneumophila, Mycoplasma pneumoniae, Legionella pneumophila, Mycobacterium tuberculosis, Streptococcus agalactiae group B, Neisseria gonorrhoeae, Chlamydia trachomatis, Mycoplasma hominis, Ureaplasma urealyticum Helicobacter pylori, Bacteroides fragilis, Clostridium perfringens, Escherichia coli, quinolone-resistant Escherichia coli, Salmonella spp., Shigella spp., ciprofloxacin-susceptible Pseudomonas aeruginosa, Clostridium difficile, and Listeria monocytogenes.
Ozenoxacin is a novel non-fluorinated quinolone antibacterial agent. It is currently in late stage phase 3 trials for the topical treatment of impetigo. The bacterial action of ozenoxacin is through the dual inhibition of DNA gyrase and topoisomerase IV. Excellent in vitro and in vivo antibacterial activity has been demonstrated in pre-clinical and clinical studies against a broad range of bacterial organisms. This includes organisms with emerging resistance to quinolones. Phase I and II clinical trials have also shown that ozenoxacin is a safe and effective antibacterial agent. No evidence of adverse effects as linked to topically formulated halogenated quinolones has been shown.
Ozenoxacin (I) was firstly disclosed in US6335447, and equivalent patents. Its chemical name is 1-cyclopropyl-8-methyl-7-[5-methyl-6-(methylamino)-3-pyridinyl]-4-oxo-1 ,4-dihydro-3- quinolinecarboxylic acid. Its chemical formula is: H

Ozenoxacin (I)
Topical application of antimicrobial agents is a useful tool for therapy of skin and skin structures infections, sexually transmitted diseases and genital tract infections and some systemic infections susceptible to topical treatment. Topical antimicrobial therapy has several potential advantages compared with systemic therapy.
Firstly, it can avoid an unnecessary exposure of the gut flora which may exert selection for resistance. Secondly, it is expected that the high local drug concentration in topical application and the negligible systemic absorption should overwhelm many mutational resistances. Thirdly, topical applications are less likely than systemic therapy to cause side effects. Accordingly, some topical compositions comprising ozenoxacin have been reported in the art.
JP2002356426A discloses ointments and gels for skin. An ointment comprising ozenoxacin 1%, N-methyl-2-pyrrolidone 8%, propylene glycol 14.9%, oleic acid 0.9%, diisopropanolamine 2.3%, polyethylene glycol 400 20.2%, polyethylene glycol 4000 50.2%, and water 3.2% is reported in Example 2.
JP2003226643A discloses aqueous solutions comprising ozenoxacin, cyclodextrin, and a viscous agent.
EP1731138A1 discloses fine particle dispersion liquid comprising ozenoxacin to be used in the manufacture of pharmaceutical compositions.
WO2007015453A1 discloses lotions comprising ozenoxacin.
JP2007119456A discloses aqueous suspensions containing nanoparticles and solution granules of ozenoxacin to be used in the manufacture of pharmaceutical compositions. Ophthalmic solutions are mentioned preferably. A combined use of ozenoxacin, magnesium ions, and hydroxypropyl-β-cyclodextrin specially for ophthalmic use is disclosed in Yamakawa, T. et al., Journal of Controlled Release (2003), 86(1 ), 101-103.
Semisolid topical compositions are useful alternatives to liquid compositions, because of their better manipulation and consequent patient preferences. However, in spite of the great diversity of components present in the semisolid compositions disclosed in the art, no quantitative stability studies are available for them.
Thus, there is a need of proved stable semisolid topical compositions comprising ozenoxacin as active ingredient, wherein microbiological and therapeutic activities are warranted because of demonstrated durable and prolonged pharmaceutical stability.
Synthesis
US6335447
http://www.google.co.in/patents/US6335447
EXAMPLE 5
To a solution of 0.80 g of 7-[6-({[(benzyloxy)-carbonyl] (methyl)amino}methyl)-5-methyl-3-pyrdyl]-1-cyclo-propyl-8-methyl-4-oxo-1,4-dihydro-3-quinoline-carboxylic acid in 16 ml of acetic acid was added 0.20 g of 5% (w/w) palladium-carbon and the mixture was stirred at ambient temperature and atmospheric pressure for 2 hours under a hydrogen atmosphere. The reaction mixture was filtered and the solvent was evaporated under reduced pressure. The obtained residue was dissolved in a mixed solvent consisting of 3.8 ml of ethanol and 3.8 ml of water. After adding 3.8 ml of an aqueous 1 mol/l sodium hydroxide solution thereto and adjusting the solution to pH 5.5with 1 mol/l hydrochloric acid, 10 ml of chloroform was added thereto. An organic layer was separated and dried over anhydrous magnesium sulfate and the solvent was evaporated under reduced pressure. Addition of diethyl ether to the obtained residue and filtration of crystals afforded 0.25 g of colorless crystals of 1-cyclopropyl-8-methyl-7-{5-methyl-6-[(methylamino)methyl]-3-pyridyl}-4-oxo-1,4-dihydro-3-quinolinecarboxylic acid.
IR (KBr) cm−1: 3322, 1721; NMR(d1-TFA) δ: 1.2-1.9 (4H, m), 2.94 (3H, s), 3.05 (3H, s), 3.29 (3H, s), 4.6-5.0 (1H, m), 5.12 (2H, s), 7.91 (1H, d, J=8.5 Hz), 8.6-9.0 (2H, m), 9.0-9.3 (1H, brs), 9.75 (1H, s). Melting point: 199° C.

FINAFLOXACIN
(S-cyano-1-cyclopropyl-ό-fluoro-T-^aS, 7aS)-hexahydropyrrolo [3,4- b]-1,4-oxazin-6(2H)-yl]-1,4-dihydro-4-oxo-3-quinolinecarboxylic acid)
7-[(4aS,7aS)-3,4,4a,5,7,7a-hexahydro-2H-pyrrolo[3,4-b][1,4]oxazin-6-yl]-8-cyano-1-cyclopropyl-6-fluoro-4-oxoquinoline-3-carboxylic acid |
BAY-35-3377
BY-377
CAS Registry Number: 209342-40-5
HYD SALT
(-)-(4aS,7aS)-8-Cyano-1-cyclopropyl-6-fluoro-4-oxo-7-(perhydropyrrolo[3,4-b]-1,4-oxazin-6-yl)-1,4-dihydroquinoline-3-carboxylic acid hydrochloride
209342-41-6,
| C20 H19 F N4 O4 . Cl H | |
| MW | 434.849 |
Synonyms: Finafloxacin, UNII-D26OSN9Q4R,
MerLion Pharmaceuticals (Singapore)…POSTER…….http://www.merlionpharma.com/sites/default/files/file/PPS/F1-2036_Wohlert.pdf
H. pylori, Broad-Spectrum
Finafloxacin is a novel fluoroquinolone being developed by MerLion Pharmaceuticals. Under neutral pH conditions (pH 7.2–7.4), the compound has shown in vitro activity equivalent to that of ciprofloxacin. However, under slightly acidic pH5.8 the compound shows enhanced potency.
Other marketed fluoroquinolones, such as ciprofloxacin, levofloxacin and moxifloxacin, exhibit reduced activity at slightly acidic pH 5.0–6.5. This feature of finafloxacin makes the compound suitable for use in the treatment of infections in acidic foci of infections such as urinary tract infections
Finafloxacin hydrochloride, a novel highly potent antibiotic, is in phase III clinical trials at Alcon for the treatment of ear infections. MerLion Pharmaceuticals is evaluating the product in phase II clinical trials at for the treatment of Helicobacter pylori infection and for the treatment of lower uncomplicated urinary tract infections in females.
A quinolone, finafloxacin holds potential for the treatment of Helicobacter pylori infection and urinary tract infection. Unlike existing antibiotics, finafloxacin demonstrates a unique acid activated activity whereby it becomes increasingly active under acidic conditions.
In 2009, a codevelopment agreement was signed between Chaperone Technologies and MerLion Pharmaceuticals. In 2011, finafloxacin hydrochloride was licensed to Alcon by MerLion Pharmaceuticals in North America for the treatment of ear infections.
MerLion Pharmaceuticals has announced that the FDA has granted a Qualified Infectious Disease Product Designation and Fast Track Status for finafloxacin. The company is currently recruiting patients for the Phase II clinical trial of the compound for the treatment of complicated urinary tract infections (cUTI) and/or acute pyelonephritis compared to ciprofloxacin
Finafloxacin and derivatives thereof can be synthesized according to the methods described in U.S. Patent No. 6,133,260 to Matzke et al., the contents of which are herein incorporated by reference in their entirety. The compositions of the invention are particularly directed toward treating mammalian and human subjects having or at risk of having a microbial tissue infection. Microbial tissue infections that may be treated or prevented in accord with the method of the present invention are referred to in J. P. Sanford et al., “The Sanford Guide to Antimicrobial Therapy 2007” 37 Edition (Antimicrobial Therapy, Inc.). Particular microbial tissue infections that may be treatable by embodiments of the present invention include those infections caused by bacteria, protozoa, fungi, yeast, spores, and parasites.
SYNTHESIS
WO1998026779A1
http://www.google.sc/patents/WO1998026779A1 COPY PASTE ON BROWSER
8-cyano-l-cyclopropyl-6-fluoro-7-((lS, 6S)-2-oxa-5 ,8-di-azabicyclo [4.3.0] non-8-yl)-l, 4-dihydro-4-oxo-3-quinolinecarboxylic acid.
The compounds, which are suitable for use in the invention are known already to some extent in EP-A-0350733, EP-A-0550903 as well as from DE-A-4329600 or can be prepared according to the processes described in .
If, for example 9,10-difluoro-3 ,8-dimethyl-7-oxo-2 ,3-dihydro-7H-pyrido [l ,2,3-d, e] [l, 3,4] benzoxadiazine-6 -carboxylic acid and 2-oxa-5 ,8-diazabicyclo [4.3.0] nonane, the reaction can be represented by the following equation:

The 7-halo-quinolonecarboxylic acid derivatives used for preparing the compounds of Fomel (I) of the invention are known or can be prepared by known methods. Thus, the 7-chloro-8-cyano-l-cyclopropyl-6-fluoro-1 ,4-dihydro-4-oxo-3-quinolinecarboxylic acid, or of the 7-chloro-8-cyano-l-cyclopropyl-6-fluoro- l been ,4-dihydro-4-oxo-3-quinolinecarboxylic acid ethyl ester described in EP-A-0 276 700th The corresponding 7-fluoro derivatives can be, for example, via the following reaction sequence to build:

An alternative process for preparing the intermediate compound 2,4-dichloro-3-cyano-5-fluoro-benzoyl chloride as the starting material for the preparation of 7-chloro-
8-cyano-1-cyclopropyl-6-fluoro-1 ,4-dihydro-4-oxo-3-quinolinecarboxylic acid is used (EP-A-0276700) and in the 3-cyano-2 ,4,5-trifluoro- benzoyl can be converted, is based on 5-fluoro-l ,3-xylene, 5-fluoro-l ,3-xylene, in the presence of a catalyst under ionic conditions in the nucleus disubstituted to 2,4-dichloro-5-fluoro-l ,3-dimethylbenzene, and this is subsequently chlorinated chlorinated under free radical conditions in the side chains of 2,4-dichloro-5-fluoro-3-dichloromethyl-l-trichloro-methylbenzene. This is the 2,4-dichloro-5-fluoro-3-dichloromethyl-benzoic acid to give 2,4-dichloro-5-fluoro-3-formyl-benzoic acid, and then hydrolyzed to 2,4-dichloro-5-fluoro-3 N-hydroxyiminomethyl acid implemented. By treatment with thionyl chloride, 2,4-dichloro-3-cyano-5-fluoro-benzoyl chloride is obtained, which can still be ,4,5-trifluoro-ben-zoylfluorid converted by a chlorine / fluorine exchange on-3-cyano-2 .



The amines used for the preparation of compounds of formula (I) according to the invention are known from EP-A-0550903, EP-A-0551653 as well as from DE-A-4 309 964th
An alternative to the synthesis of lS, 6S-2-oxa-5 ,8-diazabicyclo [4.3.0] nonane-dihydro-drobromid or the free base 1 S, 6S-2-oxa-5 ,8-diazabicyclo [4.3.0 ] nonane and the corresponding IR, 6R enantiomer provides the following path represents:
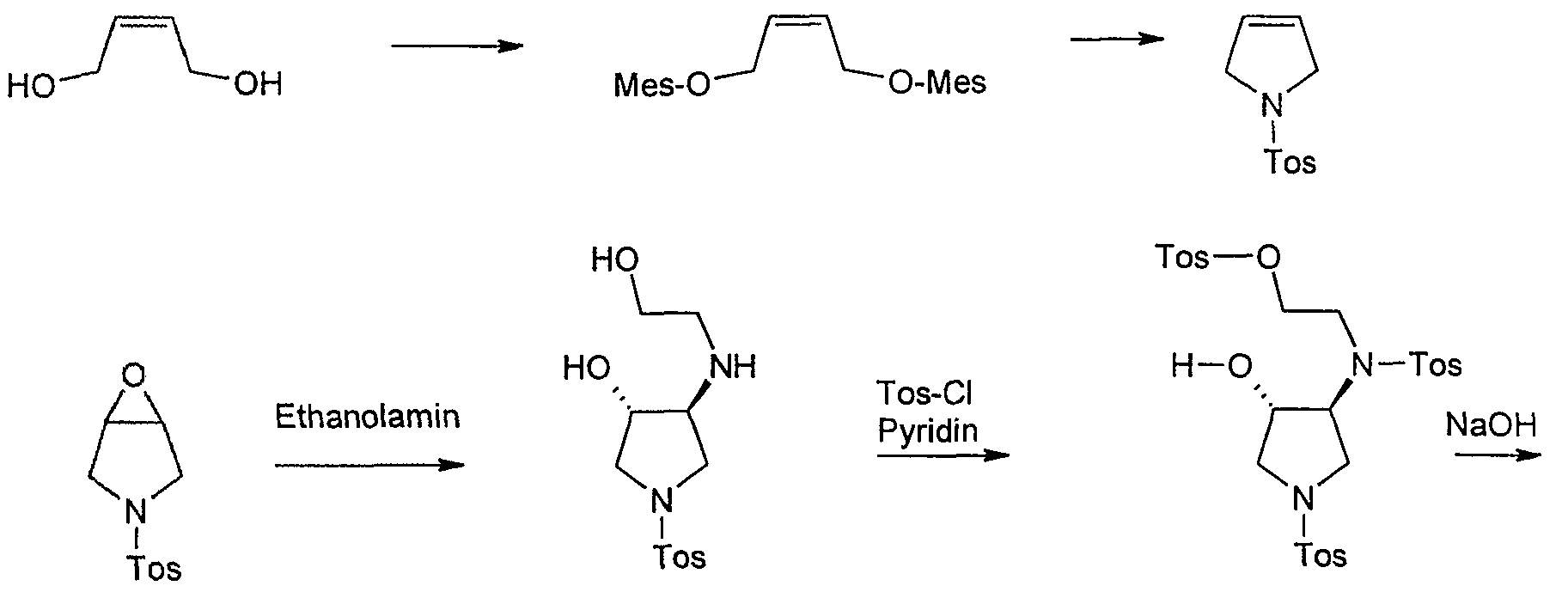
Starting material for this synthesis is the cis-l ,4-dihydroxy-2-butene, which is converted to the bis-mesylate with mesylation tosylamide for 1-tosylpyrrolidine. This is converted into the epoxide m-chloroperbenzoic. The ring opening of the epoxide by heating in isopropanol with ethanolamine to trans-3-hydroxy-4 – (2-hydroxy-ethylamino)-l-(toluene-4-sulfonyl)-pyrrolidine in 80% yield. Tetrahydrofuran is then in pyridine / reacted with tosyl chloride, with cooling to Tris-tosylate, which as a crude product in a mixture with some tetra-tosyl derivative with basichen reaction conditions to give the racemic trans-5 ,8-bis-tosyl-2-oxa-5, 6 – diazabicyclo [4.3.0] nonane is cylisiert. At this stage occurs with high selectivity of a chromatographic resolution kieselgelgebundenem poly (N-methacryloyl-L-leucine-d menthylamide) as the stationary phase. The desired enantiomer, (lS, 6S) -5,8-bis-tosyl-2-oxa-5 ,6-diazabicyclo [4.3.0] nonane, is of a purity of
> 99% ee. Cleavage of the p-tosyl protecting groups is carried out with HBr-acetic acid to the lS, 6S-2-Oxa-5 ,8-diazabicyclo [4.3.0] nonane dihydrobromide, the one with a base such as sodium or potassium hydroxide or with the aid of ion exchanger can be converted into the free base. The analogous sequence may be used for the preparation of lR, 6R-2-Oxa-5 ,8-diazabicyclo [4.3.0] nonane dihydrobromide.

HBr / AcOH

Synthesis of lS, 6S-2-oxa-5 ,8-diazabicyclo [4.3.0] nonane
Examples of compounds of the invention are mentioned in addition to the compounds listed in the preparation examples, the compounds listed in Table 1 below, which can be used both in racemic form as well as enantiomerically pure or diastereomerically pure compounds. Table 1:


Example 1 Z
8-cyano-1-cyclopropyl-6 ,7-difluoro-1 ,4-dihydro-4-oxo-3-quinoline-carboxylic acid ethyl ester

a 3-bromo-2 ,4,5-trifluoro-benzoate
To a mixture of 1460 ml of methanol and 340 g of triethylamine, 772 g of 3-bromo-2 ,4,5-trifluoro-benzoyl fluoride was added dropwise under ice cooling. There is one
Stirred for an hour at room temperature. The Reaktionsgemsich is concentrated, the residue dissolved in water and methylene chloride, and the aqueous phase was extracted with methylene chloride. After drying the organic phase over sodium sulfate, concentrated, and the residue was distilled in vacuum. This gives 752.4 g of 3-bromo-2 ,4,5-trifluoro-benzoic acid methyl ester of boiling point 122 ° C/20 mbar.
b. 3-Cyano-2 ,4,5-trifluoro-benzoic acid methyl ester:
269 g of 3-bromo-2 ,4,5-trifluoro-benzoic acid methyl ester and 108 g of copper cyanide are heated to reflux in 400 ml of dimethylformamide for 5 hours. , All volatile components of the reaction mixture are then distilled off in vacuo. The distillate was then fractionated on a column. This gives 133 g of 3-cyano-2 ,4,5-trifluoro-benzoate of boiling point 88-89 ° C / 0.01 mbar.
c. 3-Cyano-2 ,4,5-trifluoro-benzoic acid
A solution of 156 g of 3-cyano-2 ,4,5-trifluoro-benzoate in 960 ml of glacial acetic acid, 140 ml of water and 69 ml concentrated sulfuric acid is heated for 8 hours under reflux. Then the acetic acid is distilled off under vacuum and the residue treated with water. Of failed-ne solid is filtered off, washed with water and dried. Obtained
118.6 g of 3-cyano-2 ,4,5-trifluoro-benzoic acid as a white solid, mp 187-190 ° C.
d 3-cyano-2 ,4,5-trifluoro-benzoyl chloride:
111 g of 3-cyano-2 ,4,5-trifluoro-benzoic acid and 84 g of oxalyl chloride are stirred in 930 ml of dry methylene chloride with the addition of a few drops of dimethylformamide for 5 hours at room temperature. The methylene chloride is evaporated and the residue distilled in vacuo. This gives 117.6 g of 3-cyano-2 ,4,5-trifluoro-benzoyl chloride as a yellow oil.
e 2 – (3-cyano-2 ,4,5-trifluoro-benzoyl)-3-dimethylamino-acrylic acid ethyl ester:
To a solution of 36.5 g of 3-dimethylamino-acrylate and 26.5 g of triethylamine in 140 ml toluene, a solution of 55 g 3-cyano-2, 4,5 – trifluoro-benzoyl chloride are added dropwise in 50 ml of toluene so that the temperature 50-55 ° C remains. Then stirred for 2 hours at 50 ° C.
The reaction mixture is concentrated in vacuo and used without further
Processing used in the next step. f 2 – (3-cyano-2 ,4,5-trifluoro-benzoyl)-3-cyclopropylamino-acrylic acid ethyl ester:
To the reaction product of step e 30 g of glacial acetic acid are added dropwise at 20 ° C. A solution of 15.75 g of cyclopropyl amine in 30 ml of toluene is added dropwise. The mixture is stirred at 30 ° C for 1 hour. Are then added 200 ml of water, stirred 15 minutes, the organic phase is separated off and shakes it again with 100 ml of water. The organic phase is dried over sodium sulfate and concentrated in vacuo. The crude product thus obtained is a set-without further purification in the next step.
g 8-cyano-l-cyclopropyl-6 ,7-difluoro-l ,4-dihydro-4-oxo-3-quinolinecarboxylic acid ethyl ester:
The reaction product from stage f and 27.6 g of potassium carbonate are stirred in 80 ml dimethylformamide for 16 hours at room temperature. The reaction mixture is then poured into 750 ml ice water, the solid filtered off with suction and washed with 80 ml cold methanol. After drying, 47 g of 8 – cyano-l-cyclopropyl-6 ,7-difluoro-l ,4-dihydro-4-oxo-3-quinoline carboxylic acid ethyl ester, mp 209-211 ° C.
Example 2 Z
2,4-dichloro-5-fluoro-l ,3-dimethylbenzene

a solvent-free
In 124 g of 3,5-dimethyl-fluorobenzene 1 g of anhydrous iron (III) chloride are pre-loaded and launched with the speed of chlorine (about 4 h), with which the reaction. This is initially slightly exothermic (temperature increase from 24 to 32 ° C) and is maintained by cooling below 30 ° C. After addition of 120 g of chlorine, the mixture is determined. According to GC analysis are 33.4% monochloro compound, formed 58.4% desired product and 5%> overchlorinated connections. The hydrogen chloride is removed and the reaction mixture is then distilled in a column in a water jet vacuum:
In the run 49 g of 2-chloro-5-fluoro-l ,3-dimethylbenzene obtained at 72-74 ° C/22 mbar. After 5 g of an intermediate fraction proceed at 105 ° C/22 mbar 75 g of 2,4 – dichloro-5-fluoro-l ,3-dimethylbenzene via, Melting range: 64 – 65 ° C.
b in 1,2-dichloroethane
1 kg of 3,5-dimethyl-fluorobenzene and 15 g of anhydrous iron (III) chloride are placed in 1 1 1 ,2-dichloroethane and chlorine is introduced in the same extent as the reaction proceeds (about 4 h). The reaction is initially exothermic (temperature rise from 24 to 32 ° C) and is kept below 30 ° C by cooling. After the introduction of 1200 g of chlorine are according to GC analysis 4% monochloro compound, 81.1% and 13.3% desired product overchlorinated connections emerged. After distilling off the solvent and the hydrogen chloride is distilled in a column in a water jet vacuum:
In the run 40 g of 2-chloro-5-fluoro-l ,3-dimethylbenzene receive. After some intermediate run going at 127-128 ° C/50 mbar 1115 g of 2,4-dichloro-5-fTuor-l ,3-dimethyl-ethylbenzene over.
Example 3 Z
2,4-dichloro-5-fluoro-3-dichloromethyl-l-trichloromethylbenzene

In a photochlorination using chlorine inlet and outlet for the hydrogen chloride to a scrubber and a light source in the vicinity of the chlorine inlet tube, 1890 g of 2,4-dichloro-5-fluoro-l ,3-dimethylbenzene pre-loaded and at 140 to 150 ° C. Chlorine metered. Within 30 hours 3850 g of chlorine are introduced. The content of the desired product according to GC analysis is 71.1% and the proportion of connections minderchlorierten 27.7%. The DestiUaton a 60 cm column with Wilson spirals provides a flow of 1142 g, which can be reused in the chlorination. The main fraction at 160-168 ° C / 0.2 mbar gives 2200 g of 2,4-dichloro-5-fluoro-3-dichloromethyl-l-trichloro-methyl benzene having a melting range of 74-76 ° C. After one recrystallization
Sample from methanol, the melting point 81-82 ° C.
Example Z 4
2,4-dichloro-5-fluoro-3-formyl-benzoic acid

In a 2500 ml stirred apparatus with gas discharge are presented 95% sulfuric acid at 70 ° C. and under stirring, 500 g of molten added dropwise 2,4-dichloro-5-fluoro-3-dichloromethyl-1 trichloromethylbenzene. It is after a short while hydrochloric development. Is metered during a 2 h and stirred until the evolution of gas after. After cooling to 20 ° C., the mixture is discharged ice to 4 kg and the precipitated solid is filtered off with suction. The product is after-washed with water and dried.
Yield: 310 g, melting range: 172-174 ° C
Example Z 5
2,4-dichloro-5-fluoro-3-N-hydroxyiminomethyl-benzoic acid

In a stirred reactor 80 g of hydroxylamine hydrochloride in 500 ml of ethanol are charged and added dropwise 200 ml of 45% strength sodium hydroxide solution and then with 40 – 200 g of 2,4-dichloro-5-fluoro-3-formyl-benzoic acid added 45.degree.The reaction is slightly exothermic and it is stirred for 5 h at 60 ° C. After cooling to
Room temperature is provided by the dropwise addition of hydrochloric acid to pH <3, the product taken up in tert-butyl methyl ether, the organic phase separated and the solvent distilled off. The residue obtained 185 g of 2,4-dichloro-5-fluoro-3-N-hydroxyiminomethyl benzoic acid, melting range: 190 – 194 ° C.
Example No. 6
2,4-dichloro-3-cyano-5-benzoyl-fιuor

In a stirred vessel with metering and gas outlet via a reflux condenser to a scrubber 600 ml of thionyl chloride are introduced and registered at 20 ° C. 210 g of 2,4-dichloro-5-fluoro-3-N-hydroxyiminomethyl benzoic acid in the proportion as hydrochloric developed and sulfur dioxide. After the addition the mixture is heated until the gas evolution under reflux. Mixture is then distilled, and boiling in the range of 142-145 ° C/10 mbar, 149 g of 2,4-dichloro-3-cyano-5-fluoro-benzoyl chloride (98.1% purity by GC) Melting range: 73-75 ° C.
Example No. 7
3-Cyano-2 ,4,5-trifluoro-benzoyl

50 g of potassium fluoride are suspended in 120 ml of tetramethylene sulfone and at 15 mbar for drying distilled (ca. 20 mL).Then, 50.4 g of 2,4 – dichloro-3-cyano-5-fluoro-benzoyl chloride was added and stirred at an internal temperature with exclusion of moisture for 12 hours at 180 ° C. Are removed by vacuum distillation to 32.9 g of 3-cyano-2 ,4,5-trifluoro-benzoyl fluoride in the boiling range of 98 –
Obtain 100 ° C/12 mbar.
Example No. 8
3-Cyano-2 ,4,5-trifluoro-benzoyl chloride

76.6 g of 3-cyano-2 ,4,5-trifluoro-benzoyl fluoride together with 1 g of anhydrous
Aluminum chloride introduced at 60-65 ° C and then added dropwise 25 g of silicon tetrachloride gas in the course of development. After the evolution of gas at 65 ° C is distilled in a vacuum. Boiling range 120-122 ° C/14 mbar, 73.2 g of 3 – cyano-2 ,4,5-trifluoro-benzoyl chloride over.
Example No. 9
1 – (toluene-4-sulfonyl-pyrroline

In a 20 1 HC4-HWS boilers are 2.016 kg (17.6 mol)
Submitted methanesulfonyl chloride in dichloromethane and 12 1 at -10 ° C internal temperature under strong cooling (-34 ° C) solution of 705 g (8.0 mol) of 2-butene-l ,4-diol in 1.944 kg (2.68 1 , 19.2 mol) of triethylamine was added dropwise over 30 minutes. A yellow suspension stirred for 1 hour at -10 ° C and then treated with 4 1 of water, the temperature rises to 0 ° C.The suspension is warmed to room temperature, stirred for 10 minutes at room temperature and then fed in a 30 1 separating funnel. The phases are stirred separately (good phase separation) and the aqueous phase extracted with 2 1 of dichloromethane. The combined dichloromethane phases are presented in a pre-cooled 20 1 HC4 vessel and kept at 0 ° C.
In another 20-1 HC4 boiler distillation 1.37 kg (8.0 mol) toluenesulfonamide be submitted in 6 1 toluene. It is mixed with 3.2 kg of 45% sodium hydroxide solution, 0.8 1 of water and 130.5 g Tetrabutylammomiimhydrogensulfat, heated to 40 ° C maximum temperature inside and creates a vacuum. Then, the previously obtained
Dichloromethane (15.2 1) was added dropwise over 1.5 hours while the dichloromethane was removed by distillation at 450 mbar (bath temperature: 60 ° C). During the distillation is foaming. In the end, a solution is available at an internal temperature of 33-40 ° C. After the addition of dichloromethane is distilled off, until barely distillate is (duration: about 85 minutes; internal temperature 40 ° C at 60 ° C bath temperature at the end). The vessel contents will be warm transferred to a separating funnel and rinsed the tank with water and 5 1 2 1 toluene at 50 ° C. Before phase separation, the solids are extracted in the intermediate phase and washed with 0.5 1 of toluene. The organic phase is extracted with 2.4 1 of water, separated and evaporated to dryness on a rotary evaporator. The solid residue (1758 g) is suspended in 50 ° C bath temperature in 1.6 1 of methanol, the suspension is transferred into a 10 1-flanged flask and the flask rinsed with diisopropyl 2,4 1. The mixture is heated to reflux temperature (59 ° C) and stirred for 30 minutes under reflux. The suspension is cooled to 0 ° C., stirred at 0 ° C for 1 hour and extracted with 0.8 1 of a cold mixture of ether Methanol/Diisopropyl-: washed (1 1.5). The crystals are dried under a nitrogen atmosphere at 50 ° C/400 mbar.
Yield: 1456 g (81.5% of theory)
Example Z 10
3 – (toluene-4-sulfonylV6-oxa-3-aza-bicvclo [3.1.0] hexane
o “|” h “CH3
334.5 g (1.5 mol) of l-(toluene-4-sulphonyl)-pyrroline are dissolved in 1.5 1 of dichloromethane at room temperature and over 15 minutes with a suspension of 408 g (approx. 1.65 to 1, 77 mol) of 70-75% m-chloroperbenzoic acid in 900 ml of dichloromethane (cools added in manufacturing from). The mixture is heated under reflux for 16 hr (test for
Peroxide with KI / starch paper shows yet to peroxide), the suspension was cooled to 5 ° C, sucks the precipitated m-chlorobenzoic acid and washed with 300 ml of dichloromethane (peroxide with Precipitation: negative; precipitate was discarded). The filtrate is to destroy excess peroxide with 300 ml of 10% sodium sulfite solution, washed twice (test for peroxide runs now negative), extracted with 300 ml of saturated sodium bicarbonate solution, washed with water, dried with sodium sulfate and about a quarter of the volume evaporated. Again on test peroxide: negative. The mixture is concentrated and the solid residue is stirred with ice cooling, 400 ml of isopropanol, the precipitate filtered off and dried at 70 ° C in vacuum.
Yield: 295 g (82.3%),
Mp: 136-139 ° C,
TLC (dichloromethane methanol 98:2): 1 HK (Jodkammer)
Example CLOSED
trans-3-Hydroxy-4-(2-hydroxy-ethylamino-l-(‘toluene-4-sulfonyl’) pyrrolidine

643.7 g (2.65 mol) 3 – (Toluoι-4-sulfonyl)-6-oxa-3-aza-bicyclo [3.1.0] hexane to 318.5 ml with ethanolamine in 4 1 of isopropanol at reflux for 16 hours cooked. After TLC monitoring, further 35.1 ml (total 5.86 mol) of ethanolamine added to the mixture and boiled again until the next morning. The mixture is filtered hot with suction and the filtrate concentrated on a rotary evaporator to 3.5 ltr. After seeding and stirring at room temperature for 3.5 1 diisopropyl ether are added, and stirred at 0 ° C for 6 hours. The precipitated crystals are filtered off, with 250 ml of a mixture of isopropanol / diisopropyl ether (1: 1) and washed 2 times with 300 ml of diisopropyl ether and dried overnight under high vacuum.
Yield: 663.7 g (83% of theory), content: 96.1% (area% by HPLC). Example Z 12
trans-toluene-4-sulfonic acid {2 – [[4-hydroxy-l-(toluene-4-sulfonyl)-pyrrolidin-3-yl] – ftoluol-4-sulfonyl)-amino]-ethyl ester)

552 g (1.837 mol) of trans-3-hydroxy-4-(2-hydroxy-ethylamino)-l-(toluene-4-sulfonyl) – pyrrolidine are dissolved under argon in 1.65 1 tetrahydrofuran and 0.8 1 of pyridine dissolved and at -10 ° C in portions 700 g (3.675 mol) p-toluenesulfonyl chloride are added thereto. The mixture is then stirred at this temperature for 16 hours. The work is done by adding 4.3 18.5 1% aqueous hydrochloric acid, extraction twice with dichloromethane (3 1, 2 1), washing the combined organic phases with saturated Natriurnhydrogencarbonatlösung (3 1, 2 1), drying over sodium sulfate, extracting and distilling off the solvent in vacuo. The residue is dried overnight at the oil pump and crude in the next reaction. There were 1093 g as a hard foam (content [area% by HPLC]: 80% Tris-tosyl-product and 13% tetra-tosyl-product, yield see next step). Example Z 13
rac. trans-5 ,8-bis-tosyl-2-oxa-5 .6-diazabicyclor4 .3.01 nonane

1092 g of crude trans-toluene-4-sulfonic acid {2 – [[4-hydroxy-l-(toluene-4-sulfonyl) – pyrrolidin-3-yl] – (toluene-4-sulfonyl)-amino]-ethyl} were dissolved in tetrahydrofuran and 9.4 1 at 0-3 ° C with 1.4 1 of a 1.43 molar solution of sodium hydroxide in
Methanol reacted. After half an hour at this temperature, 2.1 1 of water and 430 ml of diluted (2:1) was added to the mixture and acetic acid with previously isolated crystals of trans-toluene-4-sulfonic acid {2 – [[4-hydroxy-l – (toluene-4-sulfo-phenyl)-pyrrolidin-3-yl] – (toluene-4-sulfonyl)-amino] ethyl}-seeded. The suspension is stirred overnight at 0 to -4 ° C. The next morning, the crystals are filtered off, washed twice with 400 ml of cold mixture of tetrahydrofuran / water (4:1) and dried at 3 mbar at 50 ° C overnight.
Yield: 503 g of white crystals (62.7%> of theory over 2 steps), content: 99.7% (area% by HPLC). Example Z 14
Preparative chromatographic resolution of racemic rac. trans-5.8-bis-tosyl-2-oxa-5.6-diazabicyclor4.3.0] nonane
The chromatography of the racemate at room temperature in a column (inner diameter 75 mm), which with 870 g of a chiral stationary phase (kie-selgelgebundenes poly (N-methacryloyl-L-leucine-d menthylamide) based on the mer captomodifizierten silica Polygosil 100 , 10 microns; see EP-A 0 379 917) is filled (bed height: 38 cm). Detection is carried out using a UV detector at 254 nm
For the sample application using a solution of a concentration of 100 g of rac. trans-5 ,8-bis-tosyl-2-oxa-5 ,6-diazabicyclo [4.3.0] nonane in 3000 ml of tetrahydrofuran. A Trenncyclus is carried out under the following conditions: with the aid of a pump is required for 2 min at a flow of 50 ml / min, a part of the sample solution and the same time at a flow rate of 50 ml / min, pure n-heptane to the column.
Thereafter eluted at a flow rate of 100 ml / min 18 minutes with a mixture of n-Heptan/Tetrahydrofuran (3/2 vol / vol). This is followed for 3 minutes at a flow of 100 ml / min elution with pure tetrahydrofuran. Thereafter, further eluted with n-Heptan/Tetrahydro-furan (3/2 vol / vol). This cycle is repeated several times.
The first eluted enantiomer is the (lS, 6R) -5,8-bis-tosyl-2-oxa-5 ,6-diazabicyclo-[4.3.0] nonane, which is isolated by concentration. The eluate of the more retarding enantiomers is largely evaporated in vacuo, and the precipitated crystals are filtered off with suction and dried. From the separation of 179 g of racemate in this
As 86.1 g (96.2% of theory) of the enantiomer (lS, 6S) -5,8-bis-tosyl-2-oxa-5, 6 – diazabicyclo [4.3.0] nonane having a purity of> 99 % ee. Example Z 15
(LR, 6R-2-oxa-5.6-diazabicvclo [4.3.0] nonane dihydrobromide

38.3 g (87 mmol) of (lS, 6R) -5,8-bis-tosyl-2-oxa-5 ,6-diazabicyclo [4.3.0] nonane in 500 ml of 33 -% HBr / glacial acetic acid 10 g added anisole and heated for 4 hours at 60 ° C (bath). After standing overnight, the suspension is cooled, the precipitate filtered, with
100 ml of abs. Ethanol and dried at 70 ° C under high vacuum.
Yield: 23.5 g (93%) of white solid product, mp 309-310 ° C (dec.), DC (dichloromethane/methanol/17% aq ammonia 30:8:1.): 1 HK
[Α] D: + 0.6 ° (c = 0.53, H 2 O) (fluctuating).
Example Z 16
(LS.6S-2-oxa-5.6-diazabicvclor4.3.01nonan-Dihvdrobromid

Z is analogous to Example 15 from (lS, 6S) -5,8-bis-tosyl-2-oxa-5 ,6-diazabicyclo [4.3.0] no-nan (1S, 6S)-2-oxa-5, 6-diazabicyclo [4.3.0] nonane dihydrobromide receive. Example Z 17
(1 R.6R-2-oxa-5.8-diazabicvclo [4.3.Olnonan

1 Method: 5,8 g (20 mmol) of (lS, 6R)-2-oxa-5 ,8-diazabicyclo [4.3.0] nonane dihydro-drobromid are suspended in 100 ml of isopropanol at room temperature with 2.4 g ( 42.9 mmol) and powdered potassium hydroxide while leaving about 1 hour in an ultrasonic bath. The suspension is cooled in an ice bath, filtered, washed with isopropanol and the undissolved salt, the filtrate was concentrated and distilled in a Kugelrohr oven at 150-230 ° C oven temperature and 0.7 mbar. Obtained 2.25 g (87.9% of theory) of a viscous oil which crystallizes. [Α] D -21.3 ° (c = 0.92, CHC1 3) Accordingly, this reaction can be carried out in ethanol.
2 Method: A homosexual genie catalyzed mixture of (lR, 6R)-2-oxa-5 ,8-diazabicyclo [4.3.0] nonane dihydrobromide and 620 mg (11 mmol) of powdered potassium hydroxide is dry in a Kugelrohr apparatus at 0.2 mbar and increasing oven temperature to 250 ° C distilled. Obtained 490 mg (76.6% of theory) of (lR, 6R) -2 – oxa-5 ,8-diazabicyclo [4.3.0] nonane as a viscous oil which slowly crystallized.
3 Method: 100 g of moist, pretreated cation exchanger (Dowex 50WX, H + – form, 100-200 mesh, capacity: 5.1 meq / g of dry or 1.7 meq / mL) are charged into a column with about 200 ml 1 N HC1 activated and washed neutral with water 3 1. A solution of 2.9 g (10 mmol) of (lS, 6R)-2-oxa-5 ,8-diazabicyclo [4.3.0] nonane
Dihydrobromide in 15 ml of water is added to the ion exchanger, and then washed with 2 1 water, and eluted with approximately 1 1 1 N ammonia solution. The eluate is evaporated. concentrated. Yield: 1.3 g of a viscous oil (quantitative), DC (dichloromethane/methanol/17% NH 3 30:8:1): 1 HK, GC: 99.6% (area).
Example Z 18
(LS.6SV2-oxa-5.8-diazabicvclor4.3.01nonan

Z is analogous to Example 17 from (lS, 6S)-2-oxa-5 ,8-diazabicyclo [4.3.0] nonane-di-hydrobromide the free base (lS, 6S)-2-oxa-5 ,8-diazabicyclo [ 4.3.0] nonane made.
Example Z 19
2 – (2,4-dichloro-3-cyano-5-fluoro-benzoyl)-3-dimethylamino-acrylic acid ethyl ester

To a solution of 626 g (4.372 mol) of 3-dimethylamino-acrylate and 591 g (4.572 mol) of ethyl-diisopropyl-amine (Hunigs base) in 1060 ml of dichloromethane, a solution of 1075 g starting at room temperature 2,4-dichloro -3-cyano-5-fluoro-benzoyl chloride (94% pure, corresponding to 1010.5 g = 4.00 mol) was dropped in 850 ml of dichloromethane. The temperature rises to 50-55 ° C (dropwise addition about 90 minutes). Then stirred for 2 hours at 50 ° C and the reaction mixture was used without further purification in the next step.
Example Z 20
2 – (2,4-dichloro-3-Cyano-5-fluoro-benzoyl-3-cvclopropylamino-acrylate

To the reaction mixture from the above step 306 g (5.1 mol) of glacial acetic acid are added dropwise under cooling at about 15 ° C. Then, with further cooling at 10-15 ° C. 267.3 g (4.68 mol) of cyclopropyl amine is added dropwise. Immediately after which the reaction mixture is mixed with 1300 ml of water under ice-cooling and 15 minutes stirred well. The dichloromethane layer was separated and used in the next step.
Example 21 Z
7-chloro-8-cyano-1-cyclopropyl-6-fluoro-1.4-dihydro-4-oxo-3-chinolincarbonsäureethyl ester

To a heated to 60-70 ° C suspension of 353 g (2.554 mol) of potassium carbonate in 850 ml of N-methylpyrrolidone, the dichloromethane phase is dropped from the precursor (about 90 minutes). During the addition of the dichloromethane at the same time
Reaction mixture was distilled off. Then the reaction mixture for 5 Vz hours at 60-70 ° C is well stirred. The mixture is cooled to about 50 ° C. and distilled under a vacuum of about 250 mbar residual dichloromethane from. At room temperature is added dropwise 107 ml 30% hydrochloric acid under ice cooling, then to obtain a pH of 5-6 is set. Then, 2,200 ml of water are added under ice cooling. The reaction mixture is thoroughly stirred for 15 minutes, the solid was then filtered off and washed on the filter twice with 1000 ml of water and extracted three times with 1000 ml of ethanol and then dried in a vacuum oven at 60 ° C.
Yield: 1200 g (89.6% of theory).
This product can be purified, if desired by, the solid is stirred in 2000 ml of ethanol for 30 minutes at reflux. You filtered hot with suction, washed with 500 ml of ethanol and dried at 60 ° C in vacuum. Melting point: 180-182 ° C.
Η-NMR (400 MHz, CDC1 3): d = 1.2 to 1.27 (m, 2H), 1.41 (t, 3H), 1.5-1.56 (m, 2H), 4, 1 to 4.8 (m, 1H), 4.40 (q, 2H), 8.44 (d, J = 8.2 Hz, H), 8.64 (s, 1H) ppm.
Example Z 22
7-chloro-8-cyano-1-cvclopropyl-6-fluoro-1 ,4-dihydro-4-oxo-3-quinolinecarboxylic acid

33.8 g (0.1 mol) of 7-chloro-8-cyano-l-cyclopropyl-6-fluoro-l ,4-dihydro-4-oxo-3-quinolinecarboxylate dissolved in a mixture of 100 ml of acetic acid, 20 ml water and 10 ml concentrated sulfuric acid was heated for 3 hours under reflux. After cooling, the mixture is poured onto 100 ml of ice water, the precipitate filtered off, washed with water and ethanol and dried at 60 ° C in vacuum.
Yield: 29.6 g (96% of theory),
Mp 216-21 C. (with decomposition)
Example 1

A 8-Cyano-l-cvclopropyl-6-fluoro-7-((lS.6S-2-oxa-5.8-diazabicvclo [4.3.0] non-8-yl – 1 ,4-dihydro-4-oxo-3 -quinoline carboxylic acid
1.00 g (3.26 mmol) of 7-chloro-8-cyano-l-cyclopropyl-6-fluoro-l ,4-dihydro-4-oxo-3-quinolinecarboxylic acid are heated with 501 mg (3.91 mmol) of ( lS, 6S)-2-oxa-5 ,8-diazabicyclo [4.3.0] nonane and 0.9 ml of triethylamine in 30 ml of acetonitrile was stirred at 40-45 ° C under argon for 25 hours. All volatile components in vacuo. removed and the residue recrystallized from ethanol. Yield: 1.22 g (94%)
Melting point: 294 ° C. (with decomposition)
B) 8-Cyano-l-cyclopropyl-6-fluoro-7-(‘(lS.6S-2-oxa-5 ,8-diazabicvclo [4.3.01nonan-8-YLV 1.4-dihydro-4-oxo-3- quinoline carboxylic acid Hvdrochlorid
200 mg (0.63 mmol) of 8-cyano-l-cyclopropyl-6 ,7-difluoro-l ,4-dihydro-4-oxo-3-quinolinecarboxylic acid ethyl ester to be 97 mg (0.75 mmol) of (lS, 6S)-2-oxa-5, 8 – diazabicyclo [4.3.0] nonane and 0.17 ml of triethylamine in 3 ml of acetonitrile was stirred at 40-45 ° C for 2 hours under argon. All volatile components in vacuo. removed, the residue treated with water, insolubles filtered off and the filtrate was extracted with dichloromethane. The organic phase is dried over sodium sulfate and then concentrated under reduced pressure. a. The resulting residue is dissolved in 6 ml of tetrahydrofuran and 2 ml of water and 30 mg (0.72 mmol) of lithium hydroxide monohydrate was added. After 16 hours of stirring at room temperature, acidified with dilute hydrochloric acid and the resulting precipitate was filtered off with suction and dried. Yield: 155 mg (57%) Melting point:> 300 ° C
C) 8-Cyano-l-cvclopropyl-6-fluoro-7-((lS, 6S-2-oxa-5.8-diazabicvclo [4.3.01non-8 yiyi.4-dihydro-4-oxo-3-quinolinecarboxylic acid hydrochloride
1 g (2.5 mmol) of 8-cyano-l-cyclopropyl-6-fluoro-7-((lS, 6S)-2-oxa-5 ,8-diazabicyclo [4.3.0] non-8-yl )-l ,4-dihydro-4-oxo-3-quinolinecarboxylic acid is suspended in 20 ml of water was added to the suspension, 10 ml hydrochloric acid and stirred for In at room temperature for 3 hours. The resulting precipitate is filtered off, washed with ethanol and dried at 80 ° C under high vacuum.
Yield: 987 mg (90.6% of theory), Melting point: 314-316 ° C. (with decomposition).
D) 8-Cyano-l-cvclopropyl-6-fluoro-7-(iS, 6S)-2-oxa-5.8-diazabicyclo [4.3.0] non-8-YLV 1 ,4-dihydro-4-oxo-3 -quinoline carboxylic acid hydrochloride
86.4 g (217 mmol) of 8-cyano-l-cyclopropyl-6-fluoro-7-((lS, 6S)-2-oxa-5, 8 – diazabicyclo [4.3.0] non-8-yl) – l ,4-dihydro-4-oxo-3-quinolinecarboxylic acid are dissolved at room temperature in 963 ml of water and 239 ml of 1 N aqueous sodium hydroxide solution. After filtration and washing with 200 ml of water is added to 477 ml in aqueous hydrochloric acid and the precipitated crystals placed at 95 ° C to 100 ° C in solution. The solution is cooled overnight, the precipitated crystals are filtered off with suction and washed three times with 500 ml of water and dried in vacuum.
Yield 90 g (94.7% of theory), content:> 99% (area% by HPLC) 99.6% ee. [] D 23: -112 ° (c = 0.29, N NaOH).
……………….
Tetrahedron Lett 2009, 50(21): 2525
A novel approach to Finafloxacin hydrochloride (BAY35-3377)Pages 2525-2528 |
||
Finafloxacin hydrochloride, an important clinical compound was synthesized by a novel synthetic approach. An active intermediate ethyl 7-chloro-8-cyano-1-cyclopropyl-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylate 19 was prepared by a new route. The chiral (S,S′)-N-Boc 10 was derived from protected pyrrolidine and the absolute stereochemistry was established by X-ray analysis.
http://www.sciencedirect.com/science/article/pii/S0040403909005875
……………….



| WO2011003091A1 * | 2 Jul 2010 | 6 Jan 2011 | Alcon Research, Ltd. | Compositions comprising finafloxacin and methods for treating ophthalmic, otic, or nasal infections |
| US7723524 | 29 Sep 2004 | 25 May 2010 | Daiichi Pharmaceutical Co., Ltd. | 8-cyanoquinolonecarboxylic acid derivative |
| US8536167 | 2 Jul 2010 | 17 Sep 2013 | Alcon Research, Ltd. | Methods for treating ophthalmic, otic, or nasal infections |
| DE4329600A1 * | 2 Sep 1993 | 9 Mar 1995 | Bayer Ag | Pyrido [1,2,3-d,e] [1,3,4] benzoxadiazinderivate |
| EP0276700A1 * | 15 Jan 1988 | 3 Aug 1988 | Bayer Ag | 8-Cyano-1-cyclopropyl-1,4-dihydro-4-oxo-3-quinolinecarboxylic acids, process for their preparation, and antibacterial agents containing them |
| EP0350733A2 * | 30 Jun 1989 | 17 Jan 1990 | Bayer Ag | 7-(1-Pyrrolidinyl)-3-quinolone- and -naphthyridone-carboxylic-acid derivatives, method for their preparation and for substituted mono- and bi-cyclic pyrrolidine intermediates, and their antibacterial and feed additive compositions |
| EP0550903A1 * | 28 Dec 1992 | 14 Jul 1993 | Bayer Ag | Quinolone- and naphthyridone carboxylic acid derivatives as antibacterial agents |
| EP0603887A2 * | 23 Dec 1993 | 29 Jun 1994 | Daiichi Pharmaceutical Co., Ltd. | Bicyclic amine derivatives |
| EP0676199A1 * | 23 Mar 1995 | 11 Oct 1995 | Pfizer Inc. | Use of trovafloxacin or derivatives thereof for the manufacture of a medicament for the treatment of H. pylori infections |
| GB2289674A * | Title not available |



