PROUD Indian WORLD RECORD VIEWS holder on THIS BLOG, ………live, by DR ANTHONY MELVIN CRASTO, Worldpeaceambassador, Worlddrugtracker, Helping millions, 100 million hits on google, pushing boundaries,2.5 lakh plus connections worldwide, 45 lakh plus VIEWS on this blog in 227 countries, 7 CONTINENTS ……A 90 % paralysed man in action for you, I am suffering from transverse mylitis and bound to a wheel chair, [THIS BLOG HOLDS WORLD RECORD VIEWS ] A PROUD INDIAN
Join me on Facebook
FACEBOOK
...................................................................Join me on twitter
..................................................................Join me on google plus
Googleplus
MYSELF
DR ANTHONY MELVIN CRASTO Ph.D ( ICT, Mumbai)
, INDIA
36Yrs Exp. in the feld of Organic Chemistry,Working for AFRICURE PHARMA as ADVISOR earlier with GLENMARK PHARMA at Navi Mumbai, INDIA. Serving chemists around the world. Helping them with websites on Chemistry.Million hits on google,
NO ADVERTISEMENTS , ACADEMIC , NON COMMERCIAL SITE, world acclamation from industry, academia, drug authorities for websites, blogs and educational contribution, ........amcrasto@gmail.com..........+91 9323115463, Skype amcrasto64
DR ANTHONY MELVIN CRASTO, Born in Mumbai in 1964 and graduated from Mumbai University, Completed his Ph.D from ICT, 1991,Matunga, Mumbai, India, in Organic Chemistry, The thesis topic was Synthesis of Novel Pyrethroid Analogues, Currently he is working with AFRICURE PHARMA, ROW2TECH, NIPER-G, Department of Pharmaceuticals, Ministry of Chemicals and Fertilizers, Govt. of India as ADVISOR, earlier assignment was
with GLENMARK LIFE SCIENCES LTD, as CONSUlTANT, Retired from GLENMARK in Jan2022 Research Centre as Principal Scientist, Process Research (bulk actives) at Mahape, Navi Mumbai, India. Total Industry exp 32 plus yrs, Prior to joining Glenmark, he has worked with major multinationals like Hoechst Marion Roussel, now Sanofi, Searle India Ltd, now RPG lifesciences, etc. He has worked with notable scientists like Dr K Nagarajan, Dr Ralph Stapel, Prof S Seshadri, etc, He did custom synthesis for major multinationals in his career like BASF, Novartis, Sanofi, etc., He has worked in Discovery, Natural products, Bulk drugs, Generics, Intermediates, Fine chemicals, Neutraceuticals, GMP, Scaleups, etc, he is now helping millions, has 9 million plus hits on Google on all Organic chemistry websites. His friends call him Open superstar worlddrugtracker. His New Drug Approvals, Green Chemistry International, All about drugs, Eurekamoments, Organic spectroscopy international,
etc in organic chemistry are some most read blogs He has hands on experience in initiation and developing novel routes for drug molecules
and implementation them on commercial scale over a 32 PLUS year tenure till date Feb 2023, Around 35 plus products in his career. He has good knowledge of IPM, GMP, Regulatory aspects, he has several International patents published worldwide . He has good proficiency in Technology transfer, Spectroscopy, Stereochemistry, Synthesis, Polymorphism etc., He suffered a paralytic stroke/ Acute Transverse mylitis in Dec 2007 and is 90 %Paralysed, He is bound to a wheelchair, this seems to have injected feul in him to help chemists all around the world, he is more active than before and is pushing boundaries, He has 100 million plus hits on Google, 2.5 lakh plus connections on all networking sites, 100 Lakh plus views on dozen plus blogs, 227 countries, 7 continents, He makes himself available to all, contact him on +91 9323115463, email amcrasto@gmail.com, Twitter, @amcrasto , He lives and will die for his family, 90% paralysis cannot kill his soul., Notably he has 38 lakh plus views on New Drug Approvals Blog in 227 countries......https://newdrugapprovals.wordpress.com/ , He appreciates the help he gets from one and all, Friends, Family, Glenmark, Readers, Wellwishers, Doctors, Drug authorities, His Contacts, Physiotherapist, etc
He has total of 32 International and Indian awards
The FDA has approved a new type of sleep drug. This new drug is an orexin receptor antagonist and is the first approved drug of this type. Orexins are chemicals that are involved in regulating the sleep-wake cycle and play a role in keeping people awake. Learn more here:http://go.usa.gov/EcEz
The U.S. Food and Drug Administration today approved Belsomra (suvorexant) tablets for use as needed to treat difficulty in falling and staying asleep (insomnia).
Belsomra is an orexin receptor antagonist and is the first approved drug of this type. Orexins are chemicals that are involved in regulating the sleep-wake cycle and play a role in keeping people awake. Belsomra alters the signaling (action) of orexin in the brain.
Insomnia is a common condition in which a person has trouble falling or staying asleep. It can range from mild to severe, depending on how often it occurs and for how long. Insomnia can cause daytime sleepiness and lack of energy. It also can make a person feel anxious, depressed, or irritable. People with insomnia may have trouble with attentiveness, learning, and memory.
“To assist health care professionals and patients in finding the best dose to treat each individual patient’s sleeplessness, the FDA has approved Belsomra in four different strengths – 5, 10, 15, and 20 milligrams,” said Ellis Unger, M.D., director of the Office of Drug Evaluation I in the FDA’s Center for Drug Evaluation and Research. “Using the lowest effective dose can reduce the risk of side effects, such as next-morning drowsiness.”
Belsomra should be taken no more than once per night, within 30 minutes of going to bed, with at least seven hours remaining before the planned time of waking. The total dose should not exceed 20 mg once daily.
The most commonly reported adverse reaction reported by clinical trial participants taking Belsomra was drowsiness. Medications that treat insomnia can cause next-day drowsiness and impair driving and other activities that require alertness. People can be impaired even when they feel fully awake.
The FDA asked the drug manufacturer, Merck, Sharpe & Dohme Corp., to study next-day driving performance in people who had taken Belsomra. The testing showed impaired driving performance in both male and female participants when the 20 mg strength was taken. Patients using the 20 mg strength should be cautioned against next-day driving or activities requiring full mental alertness. Patients taking lower doses should also be made aware of the potential for next-day driving impairment, because there is individual variation in sensitivity to the drug.
The effectiveness of Belsomra was studied in three clinical trials involving more than 500 participants. In the studies, patients taking the drug fell asleep faster and spent less time awake during the remainder of the night compared to people taking an inactive pill (placebo). Belsomra was not compared to other drugs approved to treat insomnia, so it is not known if there are differences in safety or effectiveness between Belsomra and other insomnia medications.
Like other sleep medicines, there is a risk from Belsomra of sleep-driving and other complex behaviors while not being fully awake, such as preparing and eating food, making phone calls, or having sex. Chances of such activity increase if a person has consumed alcohol or taken other medicines that make them sleepy. Patients or their families should call the prescribing health care professional if this type of activity occurs.
Belsomra will be dispensed with an FDA-approved patient Medication Guide that provides instructions for its use and important safety information. Belsomra is a controlled substance (Schedule-IV) because it can be abused or lead to dependence.
Belsomra is made by Merck, Sharpe & Dohme Corp. of Whitehouse Station, N.J.
A panel of experts at the US Food and Drug Administration has recommended Merck & Co’s insomnia drug suvorexant when given in lower dosages but rejected the higher dose that the company was seeking.———read more at
Suvorexant (MK-4305) is a dual orexin receptor antagonist in development by Merck & Co.[1][2][3] Suvorexant works by turning off wakefulness rather than by inducing sleep.[4] It is not currently approved for commercial use, but it has completed three Phase III trials.[5]The recent FDA review showed that the drug is associated with increased somnolence the next day and users of higher doses had an increased rate of suicidal ideation. [6] It is one of two such compounds currently in development, the other being GlaxoSmithKline‘s SB-649,868.
Ref:Org.Process Res.Dev-2011-15-367.
PAPER
Mangion IK, * Sherry BD, Yin J, Fleitz FJ. Merck & Co., Rahway, USA
Enantioselective Synthesis of a Dual Orexin Receptor Antagonist.Org. Lett. 2012; 14: 3458-3461
OREXINS A AND B ARE EXCITATORY NEUROPEPTIDES THAT STIMULATE WAKEFULNESS. SUVOREXANT IS A DUAL OREXIN RECEPTOR ANTAGONIST THAT IS IN PHASE III CLINICAL TRIALS FOR THE TREATMENT OF INSOMNIA. THE KEY STEP IN THE ASYMMETRIC SYNTHESIS DEPICTED IS A TANDEM ENZYMATIC TRANSAMINATION–ANNULATION SEQUENCE (F → G → H).
A previous synthesis of suvorexant (N. A. Strotman et al. J. Am. Chem. Soc. 2011, 133, 8362) involved an asymmetric Ru-catalyzed reductive amination in the construction of the diazepane ring. The present route benefits from the circumvention of transition-metal catalysis and dichloromethane as solvent.
To a solution of 22.3 g (78 mmol) of the hydrochloride salt of F-1, 15.9 g (78 mmol) A-2, 12.8 g (94 mmol) 1-hydroxy-7-azabenzotriazole, and 43.1 mL (392 mmol) N-methylmorpholine in 300 mL of DMF was added 22.5 g (118 mmol) EDC and the reaction was stirred overnight at room temperature. The reaction was partitioned between EtOAc and saturated aqueous NaHCO3, washed with water, brine, dried over MgSO4, and concentrated by rotary evaporation. The residue was purified by column chromatography on silica gel (EtOAc/hexanes) to provide G-1 as a colorless gum. Data for G-1: LC/MS: rt=2.22 min; m/z (M+H)=434.2 found; 434.2 required.
A round bottom flask containing a solution of 29.6 g (68.3 mmol) G-1 in 300 mL EtOAc and 200 ml MeOH was evacuated under reduced pressure and purged three times with an atmosphere of N2. To the flask was then added 2.4 g of 20% Pd(OH)2on carbon. The flask was again evacuated under reduced pressure and purged three times with an atmosphere of N2, and then three times with H2. The reaction was stirred under an atmosphere of H2 for three days, then filtered through a pad of celite, rinsing with EtOAc followed by MeOH. The filtrate was concentrated to provide G-2 as a white foam. Data for G-2: LC/MS: rt=0.96 & 1.13 min (see two conformers under these conditions); m/z (M+H)=300.0 found; 300.2 required.
To 21.0 g (70.1 mmol) G-2 in 250 mL DMF was added 29.3 mL (210 mmol) triethylamine and 13.2 g (70.1 mmol) D-1 and the mixture was heated in an oil bath at 75° C. for 2 h. After cooling to room temperature, the reaction was diluted with EtOAc, washed with saturated aqueous NaHCO3, water, brine and dried over MgSO4. Following concentration by rotary evaporation, the residue was purified by flash column chromatography (hexanes/EtOAc) to provide a gum. The gum was stirred in a mixture of 150 ml EtOAc and 300 ml hexanes overnight. Filtration provided G-3 as a white solid. Data for G-3: LC/MS: rt=2.29 min; m/z (M+H)=451.1 found; 451.2 required; HRMS (APCI) m/z (M+H) 451.1631 found; 451.1644 required.
(2) Org.Process Res.Dev.2011,15,367 – 375 reported a synthetic route is as follows:
the two lines above has the following disadvantages: starting materials using highly toxic compound methyl vinyl ketone, methyl vinyl ketone to the eyes, skin, mucous membranes and upper respiratory tract irritation strong, easy to operate when used; and finally to preparation suvorexant, the need to chiral separation, is not conducive to industrial production, but low yield.
(3) W02012148553 and J.Am.Chem.Soc.2011,133,8362 – Scheme 8371 report as follows:
The route disadvantages: starting materials using highly toxic compound methyl vinyl ketone, methyl vinyl ketone to the eyes, skin, mucous membranes and upper respiratory tract irritation strong, easy to operate when used; also use a heavy metal catalyst, high cost, and environmentally unfriendly.
(4) Org.Lett, synthetic route Vol.14, N0.13,2012,3458-3461 reported as follows:
The disadvantage of this route: starting materials using highly toxic compound methyl vinyl ketone, methyl vinyl ketone pairs of eyes, skin, mucous membranes and upper respiratory tract irritation strong.; Additional use of biocatalysis, high cost.
(5) Angew.Chem.1nt.Ed.2011,50,11511 – 11515 reported synthetic route is as follows:
The methyl-2- (benzylamino) ethyl ester (20mmol), (R) _3_ ((tert-butoxycarbonyl) amino) butyric acid (21mmol), 1- hydroxybenzotriazole (25mmol), Sodium hydride (24mmol) added to the flask, anhydrous acetone 50ml, was added with stirring 1 (Shu ^ (25 dirty 01), 301:! 411. The reaction was added 10% citric acid solution, extracted with ethyl acetate, 5% Na2CO3 The organic layer was washed with a solution, and saturated brine, MgSO4 dried, filtered and evaporated to dryness, the product obtained from ethyl acetate and petroleum ether (1: 2, volume ratio) was recrystallized to obtain (yield 97%, mp: 107 ° C, [a] 26D =
21.97 (103.76mg / 20ml, MeOH)).
Example 4: [0074] (R) -4- benzyl-7-methyl-1,4-diaza Synthesis heptane-2,5-dione
The 3g (8.2mmol) (R) – methyl _2_ (N- benzyl _3_ ((tert-butoxycarbonyl) amino) butanamide yl) acetate were added to the flask, and dissolved in ethyl acetate was added IOml added 30ml45% of acetate hydrochloride gas, 25 ° C reaction 4h.Evaporated to dryness, and saturated NaHC03 solution, methylene chloride and ethanol (2: 1, volume ratio) was extracted, MgSO4 organic layer was dried and evaporated to dryness to give a pale yellow oil.It was dissolved in 30ml MeOH and dried added 0.487g (9.02mmol) NaOMe, under nitrogen, 10 ° C reaction 4h.Quenched with saturated NH4Cl solution was added 5 ^ Na2CO3 solution, methylene chloride and ethanol (2: 1, volume ratio) was extracted organic layers were combined, MgSO4 dried, rotary evaporated to give a white solid (yield 98.93%, mp: 122_123 ° C , [a] 26D = 33.49 (112.87mg / 20ml, MeOH)).IH NMR (600MHz, DMS0_d6) δ ppm7.77-7.76 (bd, 1H), 7.33-7.25 (m, 5H), 4.59-4.53 (m, 2H), 4.10-4.02 (m, 2H), 3.65-3.62 ( m, 1H), 2.93-2.90 (m, 1H),
2.76-2.72 (m, 1H), 1.14-1.13 (d, 3H); (FIG. 2) MS (ESI) m / z233.10 ([M + H] +) ..
The 3g (8.2mmol) (R) – methyl _2_ (N_ _ _3 benzyl ((tert-butoxycarbonyl) amino) butanamide yl) acetate were added to the flask, dissolved in dichloromethane was added IOml adding 30ml methylene chloride solution containing 10% of CF3COOH of, 25 ° C reaction 4h.Evaporated to dryness and saturated NaHCO3 solution, methylene chloride and ethanol (2: 1, volume ratio) was extracted, MgSO4 organic layer was dried and evaporated to dryness to give a yellow oil.This was dissolved in 50ml of dry toluene, was added 0.156g (6.5mmol) of sodium hydride, 110 ° C reaction 4h.After cooling to room temperature, quenched with saturated NH4Cl solution, 5% Na2CO3 solution is added, methylene chloride and ethanol (2: 1, volume ratio) was extracted organic layers were combined, MgSO4 dried, rotary evaporated to give a white solid 1.83g (yield 90.34 %, mp: 122-123 ° C, [a J26D = 33.49 (112.87mg / 20ml, MeOH)).
The 3g (8.2mmol) (R) – methyl _2_ (N_ _ _3 benzyl ((tert-butoxycarbonyl) amino) butanamide yl) acetate were added to the flask, methanol was added IOml dissolved, 30ml36% methanol solution of hydrochloric acid gas, 25 ° C reaction 4h.Evaporated to dryness, and saturated NaHC03 solution, methylene chloride and ethanol (2: 1, volume ratio) was extracted, MgSO4 organic layer was dried and evaporated to dryness to give a yellow oil.This was dissolved in 50ml of dry toluene, was added 1.7g (12.3mmol) of potassium carbonate, 110 ° C reaction 8h.After cooling to room temperature, quenched with saturated NH4Cl solution, 5% Na2CO3 solution is added, methylene chloride and ethanol (2: 1, volume ratio) was extracted organic layers were combined, MgSO4 dried, rotary evaporated to give a white solid (yield 95.78%, mp: 122_123 ° C, [a] 26D = 33.49 (112.87mg / 20ml, MeOH)).
The 3g (8.2mmol) (R) – methyl _2_ (N_ _ _3 benzyl ((tert-butoxycarbonyl) amino) butanamide yl) acetate were added to the flask, methanol was added IOml dissolved, 30ml of 36% methanol containing hydrochloric acid gas solution, 25 ° C reaction 4h.Evaporated to dryness and saturated NaHCO3 solution, methylene chloride and ethanol (2: 1, volume ratio) was extracted, MgSO4 organic layer was dried and evaporated to dryness to give a yellow oil.Which was dissolved in 30ml of ethyl acetate and dried, was added 0.88g (16.4mmOl) sodium alkoxide, 10 ° C the reaction 6h.Quenched with saturated NH4Cl solution was added 5 ^ Na2CO3 solution, methylene chloride and ethanol (2: 1, volume ratio) was extracted organic layers were combined, MgSO4 dried, rotary evaporated to give a white solid (yield 93%, mp: 122_123 ° C , [a] 26D = 33.49 (112.87mg / 20ml, Me0H)) ο Example 8:
The 3g (8.2mmol) (R) – methyl _2_ (N- benzyl _3_ ((tert-butoxycarbonyl) amino) butanamide yl) acetate were added to the flask, methanol was added IOml dissolved, 30ml hydrochloric acid gas containing 36% methanol solution, 25 ° C reaction 4h.Evaporated to dryness, and saturated NaHC03 solution, methylene chloride and ethanol (2: 1, by volume) to extract, MgS04 organic layer was dried and evaporated to dryness to give a yellow oil.Which was dissolved in 30ml of dry methanol was added 2.07g (20.5mmol) of triethylamine, 60 ° C the reaction 8h.After cooling to room temperature, quenched with saturated NH4Cl solution, 5% Na2CO3 solution is added, methylene chloride and ethanol (2: 1, volume ratio) was extracted organic layers were combined, MgSO4 dried, rotary evaporated to give a white solid (yield 92.68%, mp: 122_123 ° C, [a] 26D = 33.49 (112.87mg / 20ml, MeOH)).
The 3g (8.2mmol) (R) – methyl _2_ (N_ _ _3 benzyl ((tert-butoxycarbonyl) amino) butanamide yl) acetate were added to the flask, methanol was added IOml dissolved, 30ml hydrochloric acid gas containing 36% methanol solution, 25 ° C reaction 4h.Evaporated to dryness, and saturated NaHC03 solution, methylene chloride and ethanol (2: 1, by volume) to extract, MgSO4 organic layer was dried and evaporated to dryness to give a yellow oil.Which was dissolved in 30ml of dry acetonitrile was added 1.38g (12.3mmol) of potassium t-butoxide, 30 ° C the reaction 8h.Quenched with saturated NH4Cl solution was added 5 ^ Na2CO3 solution, methylene chloride and ethanol (2: 1, volume ratio) was extracted organic layers were combined, MgSO4 dried, rotary evaporated to give a white solid (yield 89.86%, mp: 122_123 ° C , [a] 26D = 33.49 (112.87mg / 20ml, MeOH)).
Example 10:
(R) -1- benzyl-5-methyl-1,4-Synthesis diazepan the
A 1.4g (R) -4- benzyl-7-methyl-diaza heptane _2,5_ _1,4_ dione (6mmol) was dissolved in 60ml dry THF, was added portionwise under ice- 1.35g LiAlH4 (36mmol), 25 ° C was stirred for 4h.Cooled to -10 ° C, was added 1.5mlH2O quenched and then 1.5mll5% NaOH, 4.5ml H20, part MgSO4, stirring lh, filtration, spin dried to give 1.2g oil (yield 97.56%, [a] 29D = -5.87 (200.86mg / 20ml, CHCl 3)).ee> 99%, Chrom Techchiral-AGP150 * 4mm Mobile phase: Ammonium dihydrogen sulfate (IM): acetonitrile = 99: 1, column temperature: 30 ° C, flow rate: 0.5ml / Hiin0 IH NMR (600MHz, DMS0_d6) δ ppm7.32-7.20 (m, 5Η), 3.57 (s, 2Η), 3.48 (bs, 1Η), 2.99-2.95 (m, 1Η), 2.86-2.82 (m, 1Η), 2.72-2.68 (m, 1Η ), 2.65-2.61 (m, 1Η), 2.58-2.49 (m, 3Η), 1.75-1.70 (m, 1Η), 1.46-1.41 (m, 1Η), 1.01-1.00 (d, 3Η); (Figure 3 .) MS (ESI) m / z205.10 ([M + H] +) [0095] Example 11:
(R) -1- benzyl-5-methyl-1,4-Synthesis diazepan the
A 1.4g (R) -4- benzyl-7-methyl-diaza heptane _2,5_ _1,4_ dione (6mmol) was dissolved in 60mlTHF TEMPERATURE dropwise 2 equivalents of borane ( 12mm0l), reflux 8h.Cooled to _10 ° C, quenched by addition of methanol, adjusted pH = 3, stirred for 2h, sodium carbonate adjusted to pH = 10, extracted with methylene chloride three times, the combined organic layer, MgSO4 drying, rotary evaporation.(Yield 95.32%, [a] 29D = -5.87 (200.86mg / 20ml, CHCl 3)).[0098] Example 12:
(R) -1- benzyl-5-methyl-1,4-Synthesis diazepan the
The (R) -4- benzyl-7-methyl-1,4-diaza heptane-2,5-dione (5mmol) was dissolved in 15ml dry THF, was added under ice-cooling to a solution of Ig sodium boron (27mmol) in 15ml dry THF hydride was added dropwise a solution of iodine in 20ml THF 12mmol dried under nitrogen, at reflux for 6h.Cooled to (TC, quenched 5ml3N HCl was added, followed by addition of 8ml3NNaOH, liquid separation, the aqueous layer was extracted three times with ether, the combined organic layer was washed with saturated brine, MgSO4 drying, filtration, spin dry (yield 90.34%, [a ] 29D = -5.87 (200.86mg / 20ml, CHC13)).
Example 13:
(R) – (4_-Benzyl-7-methyl-1,4-diazepan-1-yl) (5-methyl _2_ (2H-1,2,3_ three
Synthesis of 2-yl) phenyl) methyl ketone
The 3g (R) -1- benzyl-5-methyl-1,4-diazepane (14.7mmol), 3.66g5_ methyl -2- (2Η-1, 2,3- triazol-2-yl) benzoic acid (18.03mmol) was dissolved in DMF, 2.43gHOBt (18.55mmol), 6ml TEA (42.75mmol), 3.45g EDC (17.99mmol), warmed to 50 ° C, the reaction 2h.Was added a saturated NaHCO3 solution and EA, the aqueous layer was washed three times with EA, the combined organic layers.The organic layer was washed with citric acid solution, the product salified fully into the aqueous phase, the aqueous phase was washed with EA after the addition of sodium carbonate to adjust the pH> 9, EA and washed three times, the organic layers combined, washed with water and saturated brine, MgSO4 dried, rotary dried, PE and EA (4: 1) and recrystallized (yield 98.36%, mp: 108-109 ° C, [α] 31D = -58.37 (202.5mg / 20ml, MeOH)).IH NMR (600MHz, DMS0_d6) δ ppm8.00-7.76 (m, 3H), 7.37-7.17 (m, 7H), 4.40-4.09 (m, 1H), 3.63-3.48 (m, 2H), 3.44-3.02 ( m, 3H), 2.82-2.75 (m, 1H), 2.63-2.47 (m, 1H), 2.63-2.14 (m, 5H), 2.02-1.63 (m, 2H), 1.17-0.99 (m, 3H); (Figure 4) MS (ESI) m / z390.30 ([M + H] +) [0105] Example 14:
The 3g (R) -1- benzyl-5-methyl-1,4-diazepane (14.7mmol), 2.98g5_ methyl -2- (2H-1, 2,3- triazol-2-yl) benzoic acid (14.7mmol) was dissolved in methylene chloride, was added 18.55mmolHOAt, 6ml TEA (42.75mmol), 2.86g CDI (17.64mmol), 30 ° C reaction 4h.Was added a saturated NaHC03 solution and EA, the aqueous layer was washed three times with EA, the combined organic layers.The organic layer was washed with citric acid solution, the product salified fully into the aqueous phase, the aqueous phase was washed with EA after the addition of sodium carbonate to adjust the pH> 9, EA and washed three times, the combined organic layer was washed with saturated brine paint, MgS04 drying, spin dry, PE and EA (4: 1) and recrystallized (yield 96.45%, mp = 108-109 ° C, [a J31D = -58.37 (202.5mg / 20ml, MeOH)).
Example 15:
(R) – (4_-Benzyl-7-methyl-1,4-diazepan-1-yl) (5-methyl _2_ (2H-1,2,3_ triazol – 2- yl) phenyl) -methanone [0110] The 3g (R) -1- benzyl-5-methyl-1,4-diazepane (14.7mmol), 3.28g5_ methyl – 2- (2Η-1,2,3- triazol-2-yl) benzoic acid (16.17mmol) was dissolved in acetone was added 2.43gHOBt (18.55mmol), 6ml TEA (42.75mmol), 3.33gDCC (16.17mmol) After the addition of sodium carbonate, 3 (TC reaction 4h. Saturated NaHCO3 solution was added and EA, the aqueous layer was washed three times with EA, the combined organic layers. The organic layer was washed with citric acid solution, the product salified fully into the aqueous phase, the aqueous phase was washed with EA adjust pH> 9, EA and washed three times, the organic layers combined, washed with water and saturated brine, MgSO4 dried, rotary dried, PE and EA (4: 1) and recrystallized (yield 92.43%, m.ρ .: 108-109 .. , [a J31D = -58.37 (202.5mg / 20ml, MeOH)).
A 2.08g (R) – (4_ _1,4_ Benzyl-7-methyl-diazepan-1-yl) (5_-methyl -2- (2Η-1, 2, 3- triazol-2-yl) phenyl) methyl ketone (7.2mmol) was dissolved in 20ml THF, 10% of the PdC12,50 ° C through the H2 reaction 2h.Filtration, rotary evaporation to give the product (yield 93.24%, [a] 26D = -14.36 (199.12mg / 20ml, MeOH)).
A 2.08g (R) – (4_ _1,4_ Benzyl-7-methyl-diazepan-1-yl) (5_-methyl -2- (2H-1, 2, 3- triazol-2-yl) phenyl) methanone (7.2mmol) was dissolved in 20ml of methanol was added 10% Pd / C, was added ammonium formate (21.6_ο1), the reaction was refluxed for 6h.Filtration, rotary evaporation to give the product (yield 92.68%, [a] 26D = -14.36 (199.12mg / 20ml, MeOH)).
Example 19
Synthesis Suvorexant of
To 0.9g (R) – (7- methyl-1,4-diazepan-1-yl) (methyl 5_ _2_ (2H-1,2,3_ triazol-2 yl) phenyl) methanone (3.0lmmol) of IOml DMF was added 0.57g2, 5- dichlorobenzene and oxazole (3.03mmol), 0.91g TEA (9mmol), heated to 75 ° C, the reaction 2h.Cooled to room temperature, EA dispersion, washed with a saturated NaHCO3 solution, saturated brine, MgSO4 dried, rotary evaporated to give a white solid (yield 93.02%, mp: 128-129 ° C, [a] 3C1.9D = -11.7 (199.99 mg / 20ml, MeOH)).IH NMR (600MHz, DMS0_d6) δ ρρm8.05-7.88 (m, 2Η), 7.82-7.78 (m, 1Η), 7.42-7.25 (m, 2Η), ζ, 06-7.00 (m, IH), 4.29- 4.06 (m, 1Η), 4.01-3.72 (m, 2Η), 3.66-3.49 (m, 2Η), 2.10 (s, 3Η), 2.06-2.01 (m, IH), 1.50 (m, 1Η), 1.78- 1.50 (m, 1Η), 1.14-1.13 (d, 3Η); (FIG. 6) MS (ESI) m / z451.20 ([Μ + Η] +).
The compound of the formula I is disclosed as an antagonist of orexin receptors in US Patent 7,951,797, US Patent Application Publication US 2008/0132490, PCT PatentPublication WO 2008/069997, Cox et al, J. Med. Chem. 2010, 53, 5320-5332, Strotman et al, JACS, 2011, 133(21), 8362-8371, and Baxter et al, Org. Process Res. & Dev., 201 1, 15(2) 367- 375.
This compound is disclosed as having activity in antagonizing the human orexin-1 (OX1) receptor with a Ki of 0.55 nM and in antagonizing the human orexin-2 (0X2) receptor with a Ki of 0.35 nM. The processes disclosed in US Patent 7,951,797, US Patent Application Publication US 2008/0132490, PCT Patent Publication WO 2008/069997, Cox et al, J. Med. Chem. 2010, 53, 5320-5332, Strotman et al, JACS, 201 1, 133(21), 8362-8371, and Baxter et al, Org. Process Res. & Dev., 2011, 15(2) 367-375 are lengthy, suffer from low yields, necessitate multiple protecting groups, rely on chiral chromatography to prepare a single isomer and require microwave technology to prepare the acid intermediate. Relative to the processes disclosed in US Patent 7,951,797, US Patent Application Publication US 2008/0132490, PCT Patent Publication WO 2008/069997, Cox et al, J. Med. Chem. 2010, 53, 5320-5332, Strotman et al, JACS, 2011, 133(21), 8362-8371, and Baxter et al, Org. Process Res. & Dev., 201 1, 15(2) 367- 375, the present invention may provide improved processes for the efficient, scalable, chromatography-free and cost-effective preparation of the formula I, to give higher isolated yield of the subject compound.
EXAMPLE 1
2. DMF
5-Chloro-l,3-benzoxazole-2-thiol (9a)
2-Amino-4-chlorophenol (2.50 kg, 17.4 mol) was charged to a vessel and suspended in water (52 L) and methanol (10.4 L). High dilution was required to prevent slow and difficult filtration of the product. The mixture was stirred, cooled to 0 °C, then thiophosgene (2.00 kg, 17.4 mol) was added to the suspension ensuring that the internal temperature remained at 5 °C throughout the addition. Water (8 L) and methanol (2 L) were added to aid stirring and the slurry was warmed to 13 °C for 1 h, followed by aging at 20 °C for a further 1 h. The slurry was then filtered and the solid washed with water (5 L). The batch was repeated and combined to dry in a vacuum oven (T = 40 °C) for 15 h to give 9-a (5.81 kg, 31.3 mol). The data corresponds to the commercially available material. XH NMR (400 MHz, d6-DMSO): δ 7.51 (d, 1 H, J = 9.2 Hz), 7.307.26 (m, 2 H). 13C NMR (100.6 MHz, d6-DMSO): δ 181.2, 147.4, 133.1, 129.7, 123.9, 1 11.6, 110.8. HRMS (ESI): m/z [M+ + H] calcd for C7H4CINOS: 185.9780; found: 185.9785.
Thiol 9a (10.5 kg, 54.6 mol) was added to a vessel and suspended in DCM (141 kg). Oxalyl chloride (10.4 kg, 82.3 mol) was added (slightly endothermic) followed by DMF (40.0 kg, 547 mol) over 1.25 h, such that the batch temperature was≤ 25 °C. The batch was aged at 20 °C for approximately 30 min, HPLC analysis showed reaction to be complete. The batch was cooled to 10 °C then triethylamine (16.64 kg, 164.4 mol) was added via a sub-surface sample line at such a rate as to maintain a batch temperature of≤ 10 °C. A sub-surface addition protocol was required to prevent build up of triethylamine hydrochloride solid on the walls of the vessel. The batch was cooled to 0 °C, then a solution of N-Boc-ethylenediamine (10.5 kg, 61.2 mol) in DCM (10 kg) was added such that the batch temperature was≤ 10 °C. The reaction was warmed to 20 °C and stirred for 2.5 h, HPLC analysis showed the reaction to be complete. Water (63.6 kg) was charged to the batch and the mixture stirred for 5 min. The layers were separated and the aqueous phase re-extracted with DCM (42.2 kg). The organic solutions were then combined and approximately half of the total DCM volume was distilled from the batch under vacuum whilst maintaining a temperature of≤ 40 °C. MeCN (83.3 kg) was then added and the remaining DCM removed by distillation (0.5 mol % DCM left by XH NMR wrt MeCN). MVK (4.61 kg, 65.8 mol) was added to the batch followed by DBU (4.17 kg, 27.4 mol) such that the temperature was≤ 20 °C. The batch was aged for 10 h at 20 °C then analyzed by HPLC. The reaction was then diluted with water (42.4 kg) and aged for a further 30 min. The mixture was filtered and the slurry washed with MeCN (33.3 kg). The solid was washed with MeCN (-10 L) then dried in a vacuum oven (T = 60 °C) for 22 h. MVK adduct 10 (15.5 kg) was isolated as an off-white solid, mp 145-148 °C. ¾ NMR (400 MHz, CDC13): δ 7.24 (d, 1 H, J = 2.3 Hz), 7.09 (d, 1 H, J = 8.5 Hz), 6.91 (dd, 1 H, J = 8.5, 2.3 Hz), 5.06 (s, 1 H, br), 3.73 (t, 2 H, J = 6.7 Hz), 3.63 (t, 2 H, J = 6.1 Hz), 3.37 (d, 2 H, br), 2.89 (t, 2 H, J = 6.7 Hz), 2.14 (s, 3H), 1.33 (s, 9 H). 13C NMR (100.6 MHz, CDC13): 8 206.7, 163.0, 156.0, 147.4, 144.6, 129.2, 120.3, 116.6, 109.2, 79.4, 49.3, 44.3, 41.9, 39.1, 30.2, 28.3. HRMS (ESI): m/z [M+ + H] calcd for
382.1534; found: 382.1544.
EXAMPLE 2
□ HMDS, THF/hexane (3.6:1.0), -25 to -15 °C; NBS
5-Chlorobenzoxazole (3-2)
To a 250 mL 3-neck round bottom flask equipped with a distillation head, glass stopper, septum, thermocouple and magnetic stir bar was charged 2-amino-4-chlorophenol (20.00 g, 0.139 mol). The solid was dissolved in THF (60 mL) and p-TsOH (0.265 g, 1.39 mmol) was added. The brown solution was warmed to 60 °C over 10 min and aged for 90 min. HPLC assay of the reaction mixture showed 1 LCAP unreacted starting material. The temperature was increased from 60 °C to 74 °C, and at 63 °C solvent distillation began. A total of 58 mL was collected during the first distillation. The mixture was diluted with THF (60 mL) and a total of 67 mL of solvent was removed between 71 and 84 °C. The mixture was again diluted with THF (60 mL) and 61 mL of solvent was removed between 74 and 1 14 °C. The dark brown solution was cooled to room temperature. The final mass of the solution was 27.96 g. Analysis of the crude stream by XH NMR showed 0.1 wt% MeOH present in the sample. XH NMR (500 MHz, CDC13): δ = 8.10 (s, 1H), 7.76 (d, J= 1.5 Hz, 1H), 7.50 (d, J= 8.7 Hz, 1H), 7.36 ppm (dd, J= 8.7, 1.7 Hz, 1H).
A 500 mL 3-neck round bottom flask equipped with a septum, thermocouple, 125 mL addition funnel, inert gas inlet and magnetic stir bar was purged with nitrogen for 10 min. Hexamethyldisilazane (42 mL, 0.20 mol) and THF (78 mL) were charged against positive nitrogen pressure. The addition funnel was charged with a hexane solution of n-butyllithium (78.0 mL, 195 mmol). The amine solution was cooled to -52 °C and n-butyllithium was added over 84 min, resulting in a temperature increase to 12.5 °C over the course of the addition. The resulting lithium hexamethyldisilazide solution was removed from the cooling bath and aged for 30 minutes. To a 500 mL 3 -neck round bottom flask equipped with a septum, thermocouple, inert gas inlet and magnetic stir bar was charged 5-chlorobenzoxazole (20.00 g, 130 mmol). The gray solid was dissolved in THF (100 mL) and the resulting colorless solution was cooled to -25 °C. The freshly prepared lithium hexamethyldisilazide solution was added via cannula over 80 minutes. The temperature of the anion solution was maintained between -25 and -15 °C during the addition. The resulting dark brown solution was aged for 90 minutes between -25 and -15 °C. To a 1000 mL 3-neck round bottom flask equipped with a Claisen adapter, septum,
thermocouple, inert gas inlet, stir rod bearing, and blade was charged THF (100 mL) and N- bromosuccinimide (34.8 g, 195 mmol). The resulting slurry was cooled to -20 °C and the anion solution was added via cannula over 150 minutes. During the addition the anion solution and reaction mixture were maintained between -25 and -15 °C. The resulting brown slurry was removed from the cooling bath and aged for 50 minutes while warming to room temperature. To the resulting bromide slurry was added a solution of ethanolamine (12.6 mL, 208 mmol) in MeCN (38 mL) via syringe pump over 5 hours. During the addition the reaction temperature was maintained between 20 and 27 °C. The resulting brown slurry was aged at room temperature overnight. The reaction mixture was cooled in an ice water bath and the septum replaced with a 50 mL addition funnel charged with concentrated HC1 (32 mL, 390 mmol). The acid solution was added over 10 min, during which time the addition the temperature increased from 10 to 20 °C. The reaction mixture was removed from the ice water bath and aged for 5 min. A 20% (w/w) solution of K2HPO4 in water (170 mL) was added and the resulting biphasic mixture was transferred to a seperatory funnel. The flask was washed with THF (3x, 10 mL) and the washings were added. The aqueous phase was cut; the organic phase was washed with 20% (w/w) K2HPO4 in water (200 mL), separated and analyzed. The crude reaction stream had a total mass of 396.47 g. By quantitative HPLC assayed 25.81 g of 3-3 in the organic phase. XH NMR (500 MHz, DMSO-i¾): δ = 8.17 (t, J= 5.6 Hz, 1H), 7.34 (d, J= 8.4 Hz, 1H), 7.25 (d, J= 1.8 Hz, 1H), 6.97 (dd, J= 8.4, 1.8 Hz, 1H), 4.81 (t, J= 5.4 Hz, 1H), 3.56 (q, J= 5.7 Hz, 2H), 3.35 pm (q, J= 5.8 Hz, 2H).
To a 1000 mL 3-neck round bottom flask equipped with a septum, thermocouple, inert gas inlet and magnetic stir bar was charged 3-3 (25.2 g, 119 mmol). To this flask was added 126 mL DMF, 12.2 mL methyl vinyl ketone (148 mmol) and 0.119 mL 10M NaOH (1.19 mmol). The reaction was then aged for 6 hours, at which time conversion was judged to be complete by HPLC. The solution was diluted with 252 mL iPAc and cooled to 0 °C, then 23.1 mL Et3 (166 mmol) followed by dropwise addition of 12.0 mL methanesulfonyl chloride (154 mmol) over 45 minutes, maintaining internal temperature less than 10 °C. After a further 30 minutes, conversion was judged to be complete by HPLC. The solution was washed with 3x 63 mL 5 w/w% aqueous aHC03 solution, then 66 mL water. After cutting the aqueous layer, the organics were reduced to approximately two volumes or 50 mL iPAc. The organics were then agitated by an overhead stirrer during slow addition of 151 mL n-Heptane over 4 hours. Over this time a crystalline white precipitate developed, and was allowed to stir overnight. At this time there was a thick slurry, which was filtered and washed with 2x 50 mL 90: 10 n- HeptaneTPAc, and after drying with a nitrogen stream over the filter pad, 3-4 was obtained as a white crystalline solid (34.6 g., 96 mmol). ‘H NMR (500 MHz, CDC13): δ = 7.29 (s, 1H), 7.16 (d, J= 8.2 Hz, 1H), 6.97 (d, J= 7.8 Hz, 1H), 4.46 (s, 2H), 3.92 (s, 2H), 3.81 (t, J= 5.9 Hz, 2H), 2.98-2.92 (m, 5H), 2.16 (s, 3H).
EXAMPLE 3
5-Chloro-2-((R)-5-methyl-[l,4]diazepan-l-yl)-benzooxazole hydrochloride (R-11) To a 1000 mL 3 -necked flask was charged isopropylamine hydrochloride (25.8 g., 270 mmol) and 525 mL 0.1 M aqueous triethanolamine solution. To this was added 750 mg pyridoxal 5′-phosphate hydrate (PLP) and 3.0 g of the transaminase polypeptide having the amino acid sequence SEQ ID NO: l and the suspension was stirred until all components dissolved. The transaminase polypeptide having the amino acid sequence SEQ ID NO: 1 was obtained as disclosed in US Patent Publication US 2010/0285541 for the identical sequence “SEQ ID NO: 1 10” therein. The solution was heated to 40 °C and the pH of the solution was adjusted to pH 8.5 with an aqueous 4M solution of isopropylamine. Mesylate 3-4 was added as a 225 mL DMSO solution via syringe over 6 hours, and the resulting mixture stirred for a further 5 hours. At this time, the solution was poured into a 3L separatory funnel and extracted with 1.5 L of 1 : 1 iPAc:IPA. The aqueous layer was cut then extracted again with 750 mL 4: 1 iPAc:IPA. The organics were combined, then washed with 750 mL brine. Then the organics were concentrated with IPA flushing to establish a 45 mL solution in IPA which was then treated with 4.6M HC1 in IPA (9.94 mL, 45.7 mmol) via dropwise addition. The resulting solution was stirred vigorously while 52 mL IP Ac was added slowly over 5 hours, creating a slurry of HQ salt 6. The slurry was then slowly cooled to 0 °C and allowed to stir overnight. At this time the slurry was filtered and dried with a nitrogen stream over the filter pad, providing R-11 as a white crystalline solid (7.80 g., 25.8 mmol). ¾ NMR (500 MHz, CD3OD): δ = 7.13-7.10 (m, 2H), 6.97 (dd, J= 8.2, 1.8 Hz, 1H), 3.99-3.79 (m, 3H), 3.67-3.57 (m, 3H), 3.39-3.33 (m, 1H), 2.24 (s,
1H), 2.12-2.07 (m, 1H), 1.42 (d, J= 6.7 Hz, 3H).
EXAMPLE 4
19 5
5-Methyl-2-[l,2,3]triazol-2-yl-benzoic acid (5) The iodide 19 (6.04 kg, 23.0 mol), THF (45 L) and DMF (9.0 L) were charged to a vessel. Copper iodide (218 g, 1.15 mol) and potassium carbonate (7.94 kg, 57.4 mol) were added and the mixture heated to an internal temperature of 40 °C. 1,2,3-Triazole (3.16 kg, 46.0 mol) was added as a solution in THF (6.0 L) over half an hour (no exotherm) and heating continued to 65 °C (again no exotherm observed) and the reaction monitored by HPLC. Once complete N,N-dimethylethylenediamine (244 mL, 2.30 mol) was added and mixture cooled to RT. Aqueous 3.6 M HC1 (36 L) was added (exotherm) and the mixture extracted twice with ethyl acetate (2 x 30 L). The combined organics were washed with LiCl solution (2 x 20 L). The acid solution assayed for 3.79 kg of 5 (81%) and 4.64 kg of 5 and 20 combined (99%). A solution of acids 5 and 20 (approx. 4.64 kg, 22.9 mol) in THF and EtOAc (approx. 1 10 L) was concentrated to low volume. THF (90 L) was added and the solvent composition checked by XH NMR to ensure most ethyl acetate had been removed. Sodium tert-butoxide (2.42 kg, 25.2 mol) was added slowly as a solid over 1-2 h (slight exotherm), allowing the sodium salt to form and stirred overnight at RT. The liquors showed a 45:55 ratio of product: starting material and the solid was collected by filtration, washed with THF (2 x 20 L) and dried in a vacuum oven (T = 40 °C) for 15 h to afford 4.22 kg of crude sodium salt. The crude sodium salt (4.22 kg, 14.9 mol) was charged to a 50 L vessel and 3.6 M HC1 (21.2 L) was added with cooling. The slurry was then stirred at room temperature for 16 h and the off-white solid isolated by filtration. The cake was washed with water (11 L) and iP Ac/Heptane (2 x 5L), then dried in a vacuum oven (T = 35 °C) for 15 h to give 3.10 kg of crude acid 5 (97.9 LCAP, 92 wt%, corrected weight 2.85 kg, 61% yield from 19). The acid 5 (2.85 kg corrected, 14.0 mol) was charged to a 50 L vessel and EtOAc (28 L) and dilute 0.22 M HC1 (14 L) were added and the mixture stirred until two clear phases resulted. The aqueous layer was removed and the organic layer filtered to remove any particulate matter. The ethyl acetate was reduced to about 8 L and then heptane (15.6 L) was added over 1 h and the liquors sampled to check for appropriate losses. The solid was isolated by filtration, washed with heptane:ethyl acetate (3 : 1 , 4 L) and dried on the filter under nitrogen to give 2.81 kg of acid 5. m.p. 167.5 °C. XH NMR (400 MHz, d6-DMSO): δ 12.09 (br s, 1H), 8.04 (s, 1H), 7.62 (d, 1H, J = 8.4 Hz), 7.58 (d, 1H, J = 1.2 Hz), 7.49 (dd, 1H, J = 8.4, 1.2 Hz), 2.41 (s, 3H). 13C NMR (100.6 MHz, d6-DMSO): δ 168.0, 139.2, 136.4, 135.8, 132.5, 130.3, 128.7, 124.8, 20.9. HRMS (ESI): m/z [M+ + H] calcd for C10H9N3O2: 204.0773; found: 204.0781. EXAMPLE 5
A round bottom flask was charged 6.86 g of 5-methyl-2-[l,2,3]triazol-2-yl- benzoic acid (5) along with 7.0 vol or 70 mis of dry iPAc (KF < 200 ppm) forming a slurry. To this was charged 0.73 g of DMF then the system was purged thoroughly with nitrogen and temperature was set at 20°C-25°C. 5.04 g of oxalyl chloride was added while maintaining 20°C- 25°C and controlling off-gassing since it is extremely vigorous. With the feed of oxalyl chloride the previous slurry dissolved. The batch was aged for 1 hr, sampled for acid chloride formation (< 1 LCAP) and allowed to proceed to amidation. In a separate vessel a solution of potassium carbonate was prepared in 5.0 vol or 50 mL water (note: exotherm). The solution was cooled to 0 °C. When acid chloride (above) was prepared, added 2.5 vol or 25 mL iPAc to the aqueous solution with overhead stirring, then added 10.0 g. amine hydrochloride salt (R-ll) to solution, and stirred for 15 minutes. Then using a cannula, the acid chloride solution was transferred over from separate vessel over the course of 1 hour, maintaining less than 5°C internal temperature. The vessel was flushed with 2.5 vol or 25 mL iPAc and sampled to determine completion. The slurry was heated to 40 °C. Upon reaching 40 °C, 1.5 vol or 15 mL Acetonitrile was and agitated for 5 minutes, and all material went into solution (98% AY observed). Agitation was stopped. After phase separation, the aqueous layer was cut, the organics were stirred with DARCO (10 wt% 6 basis) at 40°C for 3 hours, then filtered hot and taken through to
crystallization. Additional product was recovered from the carbon with an iPAc flush.
The batch was concentrated in iPAc and flushed to 7.5 vol (L/Kg of 1) and heated to 80-85C until complete dissolution. The solution was cooled to 65 °C linearly over 2 hrs, and the agitation speed was adjusted to high. At 65 °C, the solution was charged with 0.3 wt% seed in n-Heptane and aged for 1 hour. After the age and confirmation of the seed bed, the batch was cooled to 45 °C over 2.5 hrs. At this time a solvent switch was conducted at constant volume to a ratio of 90: 10 n-Heptane: iP Ac. The material was filtered hot at 45 °C, the cake was washed with 3 vol (L/Kg of 1) of 90: 10 n-Heptane :iP Ac twice, followed by 3 vol (L/Kg of 1) of n- Heptane twice. The cake was dried at 70 °C under vacuum to give 14.4 g. 1 (31.8 mmol,) as a crystalline white powder.
A reaction vessel was charged with 213.4 g of triazole acid (5) along with 7.4 vol or 2236 mis of dry iPAc (KF < 200 ppm) forming a slurry. To this charge was added 21.93 g of DMF then the system was purged thoroughly with nitrogen and temperature was maintained at 20- 25C. Charged 152.3 g of oxalyl chloride while maintaining 20-25C and control of off-gassing since it is extremely vigorous. With the feed of oxalyl chloride the previous slurry all dissolved. The batch was aged for 1 hr. The reaction was sampled for Acid Chloride formation (< 1 LCAP) and proceeded to distillation. Distillation was conducted down to 11 18 ml or constant volume distillation using 7.4 vol of fresh iPAc under vacuum maintaining less than 30°C.
In a separate vessel prepared a solution of 302.2 g of amine hydrochloride salt (R-ll) in 15.3 vol or 4624 mis of dry iPAc (KF < 200 ppm) to form a slurry. Then transferred the acid chloride solution using a cannula over from a separate vessel followed by flushing the vessel with 6.9 vol or 2085 mis of iPAc. With the amine and acid chloride in the same vessel began addition of 404.8 g of triethylamine. This charge was made over 1 to 4 hrs at a temperature between 20-40C with a desired control of the temperature between 20-30C. Once feed of the TEA was complete, the batch was aged for lhr and then sampled to determine completion.
Once the batch was complete, charged 7.4 vol of water or 2236 mis and then heated the solution to 40C. Once at 40C, the mixture was aged 5 minutes then agitation was stopped. The phases separated but there was an appreciable rag layer so it was allowed to settle and the rag was cut along with the aqueous layer. The aqueous rag was filtered then the aqueous layer was back extracted with 3.5 vol or 1058 ml of iPAc and all iPAc layers were combined.
The batch was recycled in iPAc (~60 g per kg of iPAc) via a Cuno filter (1 bundle per 39 Kg Amine HC1 Salt) for several hours at 40°C. The batch was drummed off through a sparkler filter and additional material was recovered from the carbon with an iPAc flush.
The batch was concentrated in iPAc and flushed to 7.5 vol (L/Kg of product) and heated to 80-85°C until complete dissolution. The mixture was cooled to 65°C linearly over 2 hrs, and agitation speed was adjusted to high from this point forward. At 65°C, the mixture was charged with 0.3 wt% of [(R)-4-(5-chloro-benzooxazol-2-yl)-7-methyl-[l,4]diazepan-l-yl]-(5- methyl-2-[l,2,3]triazol-2-yl-phenyl)-methanone seed in n-Heptane and aged for 1-3 hour. After the age and confirmation of the seed bed, the batch was cooled to 45°C over 2.5 hrs. A solvent switch was conducted at constant volume to a ratio of 90: 10 n-Heptane :iP Ac.
The batch was wet milled to a uniform particle size and filter hot at 45C. The cake was washed with 3 vol (L/Kg of product) of 90: 10 n-Heptane :iP Ac twice, followed by 3 vol (L/Kg of product) of n-heptane twice. The cake was dried at 70°C under vacuum.
Suvorexant (MK-4305) is a potent dual Orexin antagonist under development for the treatment of sleep disorders at Merck. The key transformation is an asymmetric Ru-catalyzed transfer hydrogenation (using a modified Noyori RuCl(p-cymene)(DPEN) complex) of an in-situ generated cyclic imine resulting in the formation of the desired chiral diazepane in 97% yield and 94.5% ee. Mechanistic studies have revealed that CO2 (derived from the formic acid) has pronounced effect on reaction outcome. Studies have determined that the efficiency of the Ru-catalyst, the composition of the resulting amine (via carbamate formation), and the reaction kinetics are mediated by the amount of CO2 generated during the reaction. The efficiency of the reductive-amination can be enhanced by either purging the CO2 or by trapping the newly formed nucleophilic secondary amine.
A new synthetic route to drug candidate 1, a potent and selective dual orexin antagonist for the treatment of sleep disorders, has been developed. The key acyclic precursor 10 was prepared in a one-step process in 75% isolated yield from commercially available starting materials using novel chemistry to synthesize 2-substituted benzoxazoles. A reductive amination was followed by a classical resolution to afford chiral diazepane (R)-11. Finally, coupling of (R)-11 with acid 5 furnished the desired drug candidate 1.
The amine DBT salt 16 (5.67 kg, 9.09 mol) was charged to a vessel and inerted. DCM (28 L) was added, followed by 4 N sodium hydroxide solution (prepared from 10 N NaOH [22.4 L] and water [36 L]). The slurry was then stirred at ambient temperature for 1 h until a solution was obtained. The layers were separated, and the aqueous phase was treated with sodium chloride solution (10.1 kg in 20 L water). DCM (5 L) was then added and the biphasic mixture stirred for 10 min before separating the layers. The combined organic layers were then concentrated under reduced pressure to a 10 L volume. The solution of the free amine was used directly in the next reaction
The triazole acid 5 (13.25 kg, 65.2 mol), DCM (88 L), and DMF (1.35 L, 17.4 mol) were charged to a vessel, and the resulting suspension was cooled to 0 °C. Oxalyl chloride (8.28 kg, 65.2 mol) was added portionwise, keeping the internal temperature between 5 and 10 °C (the anhydride formed above 10 °C), and then the reaction was aged for 30 min at this temperature. HPLC analysis showed acid 5 remained; an additional charge of oxalyl chloride (160 g, 1.26 mol) was made, and the solution stirred at 5 °C for 30 min. A solution of the amine (R)-11 (16.5 kg, 62.1 mol) and triethylamine (13.19 kg, 130.0 mol) in DCM (∼8 L) was added to the acid chloride over 30 min, keeping the internal temperature less than 15 °C. The resulting slurry was aged for 30 min and then quenched by the addition of water (167 L) over 10 min, keeping the internal temperature <15 °C. The lower organic layer was removed and then concentrated under atmospheric pressure to a volume of 100 L. Assay at this stage showed 27.3 kg 1, 98%. The solution was solvent switched to MeCN (∼560 L, 20 mL/g) by distillation under reduced pressure at <50 °C. The MeCN solution was treated with Ecosorb C-941 (2.8 kg) slurried in MeCN (10 L). The resulting slurry was aged for 30 min and then filtered through a Solka Flok pad and a 0.1 um cartridge filter, washing with MeCN (2 × 30 L). The MeCN filtrate was concentrated under reduced pressure at <50 °C to a final volume of ∼112 L. The slurry was cooled to 25 °C and water (280 L) added over 40 min. The resulting slurry was aged at 20 °C for 1 h and then filtered, washing the cake with 5:1 water/MeCN (60 L) followed by water (40 L). The solid was dried in the vacuum oven with nitrogen purge overnight at 50 °C. The final target 1 was isolated as a white solid, 26.72 kg, 95%, 98.5% ee, 99.6 LCAP, mp 153.1 °C.
The 1H NMR data for this compound was extremely complicated due to its existence as four rotamers. These rotamers did not coalesce during high-temperature experiments.(4)
[α]25D −11.8 (c 1.0, MeOH) for a sample of 97.8% ee. HRMS (ESI): m/z [M+ + H] calcd for C23H23ClN6O2: 451.1649; found: 451.1640.
A highly regioselective halogenation of 2-substituted-1,2,3-triazoles was developed via sp2 C–H activation. This method is compatible with halogen atoms, as well as electron-donating and electron-withdrawing groups. Meanwhile, the strategy is also efficient for the synthesis of a key intermediate of Suvorexant.
PAPER
2.1. Synthesis of (R)-methyl 2-(N-benzyl-3-((tert-butoxycarbonyl)amino)butanamido)acetate (3)
To a solution of methyl 2-(benzylamino)acetate (compound 10, 50.14 g,0.28 mol),(R)-3-((tert-butoxycarbonyl)amino)butanoic acid (50.75 g,0.25 mol),1-hydroxy-1H-benzotriazole (41.88 g, 0.31 mol),and dry triethylamine (37.95 g,0.38 mol) in 320 mL of DMF was added EDC hydrochloride (57.51 g,0.30 mol),and the reaction was stirred for 5 h at room temperature. The reaction was partitioned between EtOAc and 10% aqueous citric acid,the layers were separated and the organic was washed with 5% aqueous Na2CO3,then with brine,dried over MgSO4 and concentrated by rotary evaporation. The residue was recrystallized from a mixture solvent (PE:EtOAc = 2:1) to provide compound 3 as a white solid, 83.01 g in 91% yield. Mp: 107 ℃,[α]D 25 22.0 (c0.52,MeOH). 1H NMR (600 MHz,DMSO-d6): δ 7.38-7.23 (m,5H),6.73-6.72 (d,1H, J = 6 Hz),4.75-4.43 (m,2H),4.31-3.95 (m,2H),3.89-3.87 (t,1H, J = 12 Hz),3.64-3.62 (d,3H,J = 12 Hz),2.64-2.50 (m,1H),2.37- 2.23 (m,1H),1.38-1.37 (d,9H,J = 6 Hz),1.08-1.06 (m,3H); MS (ESI) m/z: 365.20 [M+H]+. HR-MS(ESI): m/z [M+H] calcd. for C19H28N2O5: 365.2071; found: 365.2066.
2.2. Synthesis of (R)-4-benzyl-7-methyl-1,4-diazepane-2,5-dione (4)
A solution of compound 3 (15.93 g,43.74 mmol) in 10 mL EtOAc was added 150 mL 45% HCl/EtOAc and the reaction was stirred for 4 h. The solvents were removed by rotary evaporation,and the residue was basified with saturated aqueous NaHCO3,and extracted with CH2Cl2. The organic extracts were concentrated. The residue was dissolved in 150 mL of dehydrated MeOH, treated with CH3ONa (2.84 g,52.49 mmol),and stirred at room temperature overnight (N2 protected,slightly exothermic). The reaction was cooled to room temperature and quenched with aqueous NH4Cl. Most of the solvent was removed and the reaction was then dumped into a separatory funnel containing 5% aqueous Na2CO3 and extracted with CH2Cl2 three times. The organic layers were combined,dried over MgSO4,and concentrated to provide compound 4 as a white solid 9.50 g in 94% yield. Analytical HPLC analysis carried out on Chiralpak AD column (4.6 mm × 250 mm) with 60% EtOH in hexanes (containing 0.1% diethylamine as a modifier),flow rate of 1 mL/min,indicated that intermediate (R)-4 was of >99% ee. Mp: 122-123 ℃. [α]D2533.5 (c 0.56,MeOH). 1H NMR (600 MHz,DMSO-d6): δ 7.77-7.76 (bd,1H,J = 6 Hz),7.33-7.25 (m,5H),4.59-4.53 (m,2H),4.10- 4.02 (m,2H),3.65-3.62 (m,1H),2.93-2.90 (m,1H),2.76-2.72 (m,1H), 1.14-1.13 (d,3H,J = 6 Hz); 13C NMR (150 MHz,DMSO-d6): δ 171.1, 168.4,138.1,128.9,128.0,127.7,53.1,50.6,46.5,40.5,23.3. MS (ESI) m/z: 233.10 [M+H]+. HR-MS(ESI): m/z [M+H] calcd. for C13H16N2O2: 233.1285; found: 233.1289.
2.3. Synthesis of (R)-1-benzyl-5-methyl-1,4-diazepane (6)
A solution of compound 4 (1.40 g,6.0 mmol) in 60 mL THF at 0 ℃ was treated with LiAlH4 (1.36 g,36.0 mmol) in batches. The reaction was slowly warmed to room temperature and stirred for another 4 h. The reaction was then cooled to -10 ℃ and was carefully quenched with 1.5 mL water,then NaOH (1.5 mL,15%) followed by an additional 4.5 mL of water. A portion of MgSO4was added and the mixture was stirred for 1 h before filtered. The filtrate was concentrated to provide light yellow oil 1.10 g in 88% yield. [α]D25 -5.9 (c 1.00,CHCl3),ee >99%,Analytical analysis was performed on Chrom Tech chiral-AGP column (150 mm × 4 mm) with 99% 1 mol/L ammonium dihydrogen phosphate and 1% acetonitrile,at flow rate of 0.5 mL/min with column temperature of 40 ℃. 1H NMR (600 MHz,DMSO-d6): δ 7.32-7.20 (m,5H),3.57 (s, 2H),3.48 (bs,1H),2.99-2.95 (m,1H),2.86-2.82 (m,1H),2.72-2.68 (m,1H),2.65-2.61 (m,1H),2.58-2.49 (m,3H),1.75-1.70 (m,1H), 1.46-1.41 (m,1H),1.01-1.00 (d,3H,J = 6 Hz); 13C NMR (150 MHz, DMSO-d6): δ 140.1,128.9,128.5,127.1,62.5,58.8,52.7,52.6,47.0, 37.5,23.9. MS (ESI) m/z: 205.10 [M+H]+. HR-MS(ESI): m/z [M+H] calcd. for C13H20N2: 205.1699; found: 205.1692.
2.4. Synthesis of (R)-(4-benzyl-7-methyl-1,4-diazepan-1-yl)(5-methyl-2-(2H-1,2,3-triazol-2-yl)phenyl)methanone (7)
To a solution of compound 6 (2.40 g,11.76 mmol),compound 5 (2.86 g,14.11 mmol),1-hydroxy-1H-benzotriazole (1.90 g, 14.11 mmol),and dry triethylamine (3.56 g,35.28 mmol) in 18 mL of dry DMF was added EDC hydrochloride (2.70 g, 14.11 mmol),and the reaction was stirred 2 h at room temperature. The reaction was partitioned between EtOAc and saturated aqueous NaHCO3,the layers were separated and the organic was added to aqueous citric acid stirring for 1 h. Water was added and the mixture was partitioned. Combined the water layers and added saturated aqueous Na2CO3 to regulate pH > 9,then extracted with three portions of EtOAc. The organic layers were combined,dried over MgSO4 and concentrated by rotary evaporation to provide compound 7 as a white power 4.30 g in 93% yield. Mp: 108-109 ℃, [α]D25-58.4 (c 1.01,MeOH). 1HNMR(600 MHz,DMSO-d6): δ 8.00- 7.76 (m,3H),7.37-7.17 (m,7H),4.40-4.09 (m,1H),3.63-3.48 (m, 2H),3.44-3.02 (m,3H),2.82-2.75 (m,1H),2.63-2.47 (m,1H), 2.63-2.14 (m,5H),2.02- 1.63 (m,2H),1.17-0.99 (m,3H); MS (ESI) m/z: 390.30 [M+H]+. HR-MS(ESI): m/z [M+H] calcd. for C23H27N5O: 390.2288; found: 390.2281.
2.5. Synthesis of (R)-(7-methyl-1,4-diazepan-1-yl)(5-methyl-2-(2H-1,2,3-triazol-2-yl)phenyl)methanone (9)
Compound 7 (5.86 g,15.05 mmol) was dissolved in 58 mL MeOH. After a portion of 10% Pd/C was added,the reaction was stirred for 4 h under H2 atmosphere at room temperature. The reaction was filtered through a pad of celite and the filtrate was concentrated to provide compound 9 as a white solid 4.01 g in 89% yield. Mp: 119-121 ℃,[a]D 26 -14.4 (c 1.00,MeOH)). 1H NMR (600 MHz,DMSO-d6): δ 8.24-8.02 (m,2H),7.88-7.29 (m,3H), 4.42-2.50 (m,7H),2.41 (s,3H),2.24-1.98 (m,2H),1.17-0.99 (m, 3H); 13C NMR (150 MHz,DMSO-d6): δ 168.6,138.3,136.9,134.1, 131.1,129.2,128.3,122.5,52.6,49.1,44.4,43.1,37.8,20.8,20.6. MS (ESI)m/z: 300.20 [M+H]+. HR-MS(ESI): m/z [M+H] calcd. for C16H21N5O: 300.1819; found: 300.1812.
2.6. Synthesis of suvorexant
To compound 8 (0.56 g,3 mmol) in 10 mL dry DMF was added TEA (0.91 g,9 mmol) and compound 9 (0.89 g,3 mmol),the mixture was stirred at 75 ℃ for 2 h. After cooling to room temperature,the reaction was diluted with EtOAc,washed with saturated aqueous NaHCO3,water,brine and dried over MgSO4. The residue was recrystallized from i-PrOH/EtOAc to provide a white solid 1.20 g in 90% yield. Mp: 149-150 ℃,[α]D25 -11.6 (c 1.00,MeOH). Analytical HPLC analysis carried out on a Chiralpak AD column (4.6 mm × 250 mm) with 60% EtOH in hexanes (containing 0.1% diethylamine as a modifier) at a flow rate of 1 mL/min,indicated that intermediate (R)-4 was of >99% ee. Mp: 153 ℃,[α]D25 -11.7 (c 1.00,MeOH) [ OPRD REF ],
Baxter, C. A.; Cleator, E.; Brands, K. M. J.; Edwards, J. S.; Reamer, R. A.; Sheen, F. J.; Stewart, G. W.; Strotman, N. A.; Wallace, D. J. (2011). “The First Large-Scale Synthesis of MK-4305: A Dual Orexin Receptor Antagonist for the Treatment of Sleep Disorder”.Organic Process Research & Development15 (2): 367–375. doi:10.1021/op1002853.
“Suvorexant: A Dual Orexin Receptor Antagonist for the Treatment of Sleep Onset and Sleep Maintenance Insomnia.”. Ann Pharmacother49: 477–483. Feb 9, 2015.doi:10.1177/1060028015570467. PMID25667197.
Label: BELSOMRA- Suvorexant Tablet, Film Coated”Label: BELSOMRA- Suvorexant Tablet, Film Coated.” DailyMed. Merck Sharp & Dohme Corp. & the U.S. National Library of Medicine, 01 Aug. 2014. Web. 29 Oct. 2014.
Jacobson, LH; Callander, GE; Hoyer, D (Nov 2014). “Suvorexant for the treatment of insomnia.”. Expert review of clinical pharmacology7 (6): 711–30.doi:10.1586/17512433.2014.966813. PMID25318834.
“Belsomra”. drugs.com. Retrieved 20 February 2015.
“U.S. Food and Drug Administration.” Drug Development and Drug Interactions: Table of Substrates, Inhibitors and Inducers. U.S. Food and Drug Administration, 27 Oct. 2014. Web. 30 Oct. 2014.
Suvorexant synthesis There are several ways, the following is a scaled-up process (OPRD, 2011, 15, 367). A compound with sulfur phosgene in ring closure to give 2,2 thiol group with oxalyl chloride to chlorine after conversion to give the intermediate 4 with a primary amine 3 attack, followed by Michael addition occurred with 5 6.6 mesylate de Boc protected After the reductive amination get 7, this is the racemic product. 7 8 after two crystallization with tartaric acid split to give 9 (> 97% ee).Triazole carboxylic acid 10 with 11 to give 12, 12 coupled after conversion to the acid chloride under basic conditions with pH 9 condensation Suvorexant.
Pantoprazole is a proton pump inhibitor drug used for short-term treatment of erosion and ulceration of the esophagus caused by gastroesophageal reflux disease.
Use
Pantoprazole is used for short-term treatment of erosion and ulceration of the oesophagus caused by gastroesophageal reflux disease. Initial treatment is generally of eight weeks’ duration, after which another eight week course of treatment may be considered if necessary. It can be used as a maintenance therapy for long term use after initial response is obtained.
Adverse effects
Antacid preparations such as pantoprazole work by suppressing the acid-mediated breakdown of proteins. This leads to an elevated risk of developing food and drug allergies due to undigested proteins passing into the gastrointestinal tract where sensitisation occurs. It is unclear whether this risk occurs with short-term or only long-term use.[1]
Nutrition: May reduce the absorption of important nutrients, vitamins and minerals, as well as medications, leaving users at increased risk for pneumonia.[2]
Cardiovascular: Increase in a chemical that suppresses the production of nitric oxide by 25% in humans, which have proven to relax and protect arteries and veins. Causes blood vessels to constrict, a development that could lead to a number of cardiovascular problems if continued for a prolonged period of time.[2]
Pharmacology
Wyeth pantoprazole 20mg.
Pantoprazole is metabolized in the liver by the cytochrome P450 system.[3] Metabolism mainly consists of demethylation by CYP2C19followed by sulfation. Another metabolic pathway is oxidation by CYP3A4. Pantoprazole metabolites are not thought to have any pharmacological significance. Pantoprazole is relatively free of drug interactions;[4] however, it may alter the absorption of other medications that depend on the amount of acid in the stomach, such as ketoconazole or digoxin. Generally inactive at acidic pH of stomach, thus it is usually given with a pro kinetic drug. Pantoprazole binds irreversibly to H+K+ATPase (proton pumps) and suppresses the secretion of acid. As it binds irreversibly to the pumps, new pumps have to be made before acid production can be resumed. The drug’s plasma half-life is about 2 hours.[5]
Pharmacokinetics
Absorption
Bioavailability: (oral, delayed release tablets), approximately 77%
Effect of food: (oral, delayed-release tablets), AUC and Cmax no effect, Tmax variable, absorption delayed, no net effect
Effect of food: (oral, for-delayed-release suspension), administer 30 minutes before a meal
Tmax, Oral, delayed-release suspension: 2 to 2.5 h
Tmax, Oral, delayed-release tablets: 2.5 h
Tmax, Oral, delayed-release tablets: 1.5 to 2 hours (pediatrics)
Distribution
Protein binding: about 98% to primarily albumin
Vd, extensive metabolizers (IV): approximately 11 L to 23.6 L
Vd, pediatrics (oral): 0.21 to 0.43 L/kg.
Metabolism
Hepatic; cytochrome P450 CYP2C19; minor metabolism from CYP3A4, 2D6, and 2C9
Excretion
Fecal: (oral or IV, normal metabolizers), 18%
Renal: (oral or IV, normal metabolizers), approximately 71%, none as unchanged
Dialyzable: no (hemodialysis)
Total body clearance: (IV) 7.6 to 14 L/hour.
Total body clearance: (oral, pediatrics) 0.18 to 2.08 L/h/kg
Elimination Half Life
Oral or IV, 1 hour
Oral or IV, slow metabolizers, 3.5 to 10 hours
Pediatrics, 0.7 to 5.34 hours
Availability
Pantoprazole was developed by Altana (owned by Nycomed) and was licensed in the USA to Wyeth (which was taken over by Pfizer). It was initially marketed under the brand name Protonix by Wyeth-Ayerst Laboratories and now is available as a generic. It is available by prescription in delayed-release tablets. It is also available for intravenous use.
On 24 December 2007, Teva Pharmaceutical released an AB-rated generic alternative to Protonix.[6] This was followed by generic equivalents from Sun Pharma and Kudco Pharma. Wyeth sued all three for patent infringement and launched its own generic version of Protonix with Nycomed.[7][8]
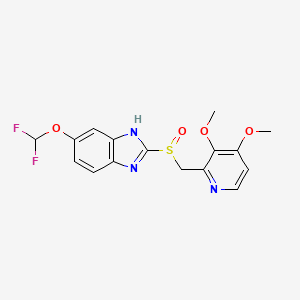
Pantoprazole is the international non-proprietary name of the chemical product 5-(difluoromethoxy)-2-[[(3,4-dimethoxy-2- pyridinyl)methyl]sulfmyl]-lH-benzimidazole of formula
Pantoprazole This product is an active ingredient used in the treatment of gastric ulcers, usually in the form of its sodium salt.
The product was described for the first time in European patent application EP-A-0166287 that also describes several processes for the preparation of products assignable to a general formula among which pantoprazole is to be found. The reaction sequences of these processes, applied precisely to the preparation of pantoprazole, are given in Scheme 1.
Scheme 1
In Scheme 1, the variables Y, Z, Z’ and Z” are leaving groups, for example atoms of halogen, and the variables M and M’ are atoms of alkali metals.
Austrian patent AT-B-394368 discloses another process based on a different route of synthetis, the reaction sequence of which is given in Scheme 2.
Pantoprazole Scheme 2
Nevertheless, this process has obvious drawbacks, since the methylation can take place not only in OH in the 4-position of the pyridine ring, but also in the nitrogen linked to a hydrogen of the benzimidazole ring, which can give place to mixtures of the desired product with the two possible methylated isomers of the benzimidazole compounds obtained, 3- methyl or 1 -methyl, which means that additional chromatographic purification steps are needed and the yields obtained are low.
PCT application WO97/29103 discloses another process for the preparation of pantoprazole, the reaction sequence of which is given in Scheme 3.
Scheme 3 As may be seen, different synthesis strategies have been proposed for the preparation of pantoprazole, some of them recently, which is an indication that the preparation of the product is still not considered to be sufficiently well developed, whereby there is still a need for developing alternative processes that allow pantoprazole to be prepared by means of simpler techniques and more accessible intermediate compounds and with good chemical yields.
EXAMPLES
Example 1. – Preparation of compound (IX)
47.5 ml (0.502 mol) of acetic anhydride were mixed with 1.65 g (0.0135 mol) of 4-dimethylaminopyridine, giving a transparent yellow solution which was heated to 65° – 70°C. This temperature was held by cooling since the reaction is exothermic. 25 g (0.1441 mol) of 2-methyl-3- methoxy-4-chloropyridine N-oxide (X) were added over a period of about 70 minutes. Once the addition was completed, the reaction was held at 65° – 70°C for a further 2h 20 minutes and after this time it was allowed to cool down to below 65°C and 90 ml of methanol were added gradually, while holding the temperature below 65°C. The resulting reaction mass was distilled at reduced pressure in a rotavap to remove the volatile components and the residue containing compound (IX) was used as such for the following reaction. Thin layer chromatography on silica gel 60 F254, eluting with CHCl3/MeOH (15: 1), showed a single spot at Rf – 0.82, indicating that the reaction has been completed.
Example 2. – Preparation of compound fVIII
(IX) (VIII)
11.5 ml methanol and 11.5 ml of water were added over the crude residue from Example 1 containing compound (IX), and thereafter, while holding the temperature to between 25° and 30°C with a water bath, the residual acetic acid contained in the crude residue was neutralized by the addition of 33% aqueous NaOH. Once the residual acid had been neutralized, 19 ml (0.2136 mol) of the 33% aqueous NaOH were added over 20 minutes, while holding the temperature to between 25° and 30°C, and, on completion of the addition, the hydrolysis reaction at pH 11.7 – 11.8 was held for 2h 30 minutes, to between 25° and 30°C. On completion of the reaction, the pH was adjusted to 7.0 – 7.5 by the addition of HC1 35%, while holding the temperature to 25°C. Thereafter, 50 ml of methylene chloride were added and, after stirring and allowing to rest, the phases were decanted. A further five extractions were carried out with 30 ml methylene chloride each and the pooled organic phases were dried with anhydrous sodium sulfate, were filtered and washed, and were evaporated at reduced pressure in a rotavap, providing a solid residue having a melting point around 73°C and containing compound (VIII). Thin layer chromatography on silica gel 60 F254, eluting with CHCl3/MeOH (15: 1), gave a main spot at Rf = 0.55, showing that the reaction was complete. The thus obtained crude residue was used as such in the following reaction.
Example 3. – Preparation of compound (VI)
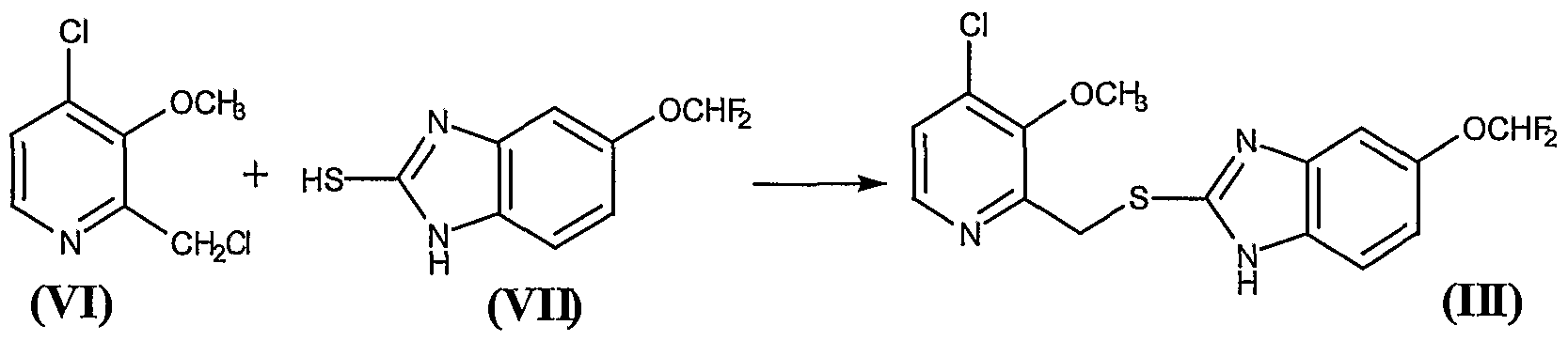
24.5 g of the residue obtained in Example 2, containing approximately 0.142 mol of the compound 2-hydroxymethyl-3-methoxy-4-chloropyridine (VIII), were mixed with 0.5 ml of DMF and 300 ml of anhydrous methylene chloride, to give a brown solution which was cooled to 0° – 5°C in an ice water bath. Thereafter, a solution of 11.5 ml (0.1585 mol) of thionyl chloride in 50 ml of anhydrous methylene chloride was added over 20 minutes, while holding the above-mentioned temperature,. Once the addition was complete, the reaction was held at 0° – 5°C for a further 90 minutes and then 120 ml of water and NaOH 33% were added to pH 5 – 6, requiring approximately 29 ml of NaOH. The phases were then decanted and separated. The organic phase was extracted with a further 120 ml of water and the pooled aqueous phases were extracted with a further 4×25 ml of methylene chloride, in order to recover the greatest possible amount of product. The pooled organic phases were dried over anhydrous sodium sulfate, filtered and washed, and evaporated at reduced pressure in a rotavap, to give a residue containing the compound 2-chloromethyl-3- methoxy-4-chloropyridine (VI). Thin layer chromatography on silica gel 60 F254, eluting with CHCl3/MeOH (15:1), showed a main spot at Rf = 0.83, indicating that the reaction was complete. The thus obtained crude residue was used as such in the following reaction. Example 4. – Preparation of compound (III)
26.11 g of the residue obtained in the Example 3 containing approximately 0.136 mol of the compound 2-chloromethyl-3-methoxy-4- chloropyridine (VI) were mixed with 370 ml of methylene chloride, to give a brown solution over which were added, at 20° – 25°C, 29.3 g (0.136 mol) of 5-difluoromethoxy-2-mercaptobenzimidazole (VII) and 17.10 ml (0.136 mol) of tetramethylguanidine (TMGH). The mixture was stirred at this temperature for 2 hours, after which 450 ml of water were added, with the pH being held to between 9.5 and 10. Thereafter the phases were decanted and the organic phase was washed 5×50 ml of a IN NaOH aqueous solution and, thereafter, with 2×50 ml of water. The organic phase was treated with 50 ml of water and an amount of HC1 30% sufficient to adjust the pH to between 5 and 6. Thereafter, the phases were decanted, and the organic phase was dried over anhydrous sodium sulfate, was filtered and washed, and evaporated at reduced pressure in a rotavap, to give a solid residue of melting point 64° – 73 °C that contains the compound (III). Thin layer chromatography on silica gel 60 F254, eluting with CHCl3/MeOH (15: 1), presented a main spot at Rf = 0.52. Yield 82%. The thus obtained compound 5-(difluoromethoxy)-2-[[(3-methoxy-4-chlorine-2 pyridinyl)methyl]mercapto]- lH-benzimidazole (III) was used as such in the following reaction Example 5. – Preparation of compound (IV)
25.8 g (0.0694 mol) of the compound (III) obtained in the Example 4 were mixed with 88 ml of methanol, to give a brown solution to which 3.7 ml of water, 0.99 g of ammonium molybdate and 0.78 g of sodium carbonate were added. The system was cooled to 0°C – 5°C, 3.4 ml (0.0756 mol) of 60% hydrogen peroxide were added, and the reaction mixture was held at 0°C – 5°C for 1 – 2 days, the end point of the reaction being checked by thin layer chromatography on silica gel 60 F254, eluting with CHCl3/MeOH (15: l).
During the reaction the presence of hydrogen peroxide in the reaction medium was controlled by testing with potassium iodide, water and starch. When effected on a sample containing hydrogen peroxide, it provides a brown-black colour. If the assay is negative before the chromatographic control indicates completion of the reaction, more hydrogen peroxide is added.
On completion of the reaction, 260 ml of water were added, the system was cooled to 0°C – 5°C again and the mixture was stirred for 2 hours at this temperature. The solid precipitate was filtered, washed with abundant water, and dried at a temperature below 60°C, to give 5-(difluoromethoxy)-2-[[(3- methoxy-4-chlorine-2-pyridinyl)methyl]sulfinyl]-lH-benzimidazole (IV), melting point 130° – 136°C, with an 83.5% yield. Thin layer chromatography on silica gel 60 F254, eluting with CHCl3/MeOH (15: 1), gave a main spot at Rf = 0.5.
Compound (IV) can be purified, if desired, by the following crystallization method:
5 g of crude product was suspended in 16 ml of acetone and was heated to boiling until a dark brown solution was obtained. Thereafter the thus obtained solution was allowed to cool down to room temperature and then was then chilled again to -20°C, at which temperature the mixture was held for 23 hours without stirring. Thereafter the solid was filtered and washed with 6×4 ml of acetone chilled to -20°C. Once dry, the resulting white solid weighed 2.73 g, had a point of melting of 142°C and gave a single spot in thin layer chromatography. The IR spectrum of the compound on KBr is given in Figure 1.
The acetonic solution comprising the mother liquors of filtration and the washes was concentrated to a volume of 20 ml and a further 5 g of crude compound were added. The above described crystallization process was repeated to obtain a further 4.11 g of purified product of characteristics similar to the previous one.
The acetonic solution from the previous crystallization was concentrated to a volume of 17 ml and a further 4 g of crude compound were added. The above described crystallization process was repeated to obtain a further 2.91 g of purified product of similar characteristics to the previous ones.
The acetonic solution from the previous crystallization was concentrated to a volume of 15 ml and a further 4 g of crude compound were added. The above described crystallization process was repeated to obtain a further 3.3 g of purified product of similar characteristics to the previous ones.
The acetonic solution from the previous crystallization was concentrated to a volume of 16 ml and a further 4.36 g of crude compound were added. The above described crystallization process was repeated to obtain a further 3.62 g of purified product of similar characteristics to the previous ones.
Finally, the acetonic solution from the previous crystallization was concentrated to a volume of 10 – 12 ml and held at -20°C for two days without stirring. Thereafter, the solid was filtered and washed with 5×3 ml of acetone chilled to -20°C. Once dry, the solid weighed 1.26 g and had similar characteristics to the previous ones.
The total yield of all the crystallizations was 80%.
Example 6. – Preparation of pantoprazole
12.95 g (0.0334 mol) of compound (IV) purified by crystallization of Example 5 were mixed with 38 ml of N,N-dimethylacetamide and thereafter 7.03 g (0.1003 mol) of potassium methoxide were added, while holding the temperature to between 20°C and 30°C, whereby a dark brown mixture was obtained. The system was held at approximately 25°C for about 23 hours, after which, once the reaction was complete, the pH was adjusted to 7 with the addition of 3.82 ml of acetic acid. The N,N-dimethylacetamide was removed at reduced pressure at an internal temperature of not more than 75°C. 65 ml of water and 50 ml of methylene chloride were added over the thus obtained residue, followed by decantation of the phases. Once the phases were decanted, the aqueous phase was extracted a with further 3×25 ml of methylene chloride, the organic phases were pooled and the resulting solution dried over anhydrous sodium sulfate, was filtered and washed, and evaporated at reduced pressure in a rotavap, to give a crude residue over which 55 ml of water were added, to give a suspension (if the product does not solidify at this point the water is decanted and a further 55 ml of water are added to remove remains of N,N-dimethylacetamide that hinder the solidification of the product). The solid was filtered and, after drying, 11.61 g of crude pantoprazole of reddish brown colour were obtained (Yield 90%). The thus obtained crude product was decoloured by dissolving the crude product in 150 ml of methanol, whereby a dark brown solution was obtained. 7.5 g of active carbon were added, while maintaining stirring for 45 minutes at 25°C – 30°C, after which the carbon was filtered out and the filter was washed. The methanol was then removed in the rotavap at reduced pressure, a temperature below 40°C. 10.33 g of a solid residue were obtained and were mixed with 14.9 ml of methylethylketone, and the suspension was heated to 45°C for about 10 minutes, after which it was cooled, first to room temperature and then to -20°C. This temperature was held over night and thereafter the solid was filtered, washed with 6×5 ml of methylethylketone chilled to -20°C. Once dry, 7.75 g of a white solid, melting point 140°C – 141 °C, were obtained. Thin layer chromatography on silica gel F254, eluting with CHCl3/MeOH (15: 1), gave a single spot at Rf =
0.41 and a IR spectrum corresponding identically with that of pantoprazole.
The ketonic solution comprising the mother liquors of filtration and the washes, was concentrated to 9.7 ml, was heated to 40°C, was held at this temperature for about five minutes and was then cooled, first to room temperature and then to -20°C, this temperature being held for 4 hours. At the end of this time, the solid was filtered and was washed with 4×2 ml of methylethylketone chilled to -20°C. Once dry, 0.42 g of a white solid of similar characteristics to the previous one was obtained.
The ketone solution from the previous treatment was concentrated to 3.1 ml, was heated to 40°C, was held to this temperature for about five minutes and then was cooled, first to room temperature and then to -20°C, this temperature being held for 4 hours. At the end of this time, the solid was filtered and was washed with 5×3 ml of methylethylketone chilled to – 20°C. Once dry, 0.41 g of a white-beige solid of similar characteristics to the previous one was obtained. The total yield, including purifications, was 67%.
If a whiter solid is desired, one or several washes can be carried with isopropyl acetate as follows: 6.6 g of pantoprazole from the methylethylketone treatment were suspended in 50 ml of isopropyl acetate. The system (white suspension) was stirred for about 30 minutes at 25°C, was then cooled to 0°C – 5°C, was stirred for about 15 minutes at this temperature and the solid was then filtered, was washed with 3×15 ml of isopropyl acetate. Once dry, 6.26 g of a pure white solid were obtained.
Trade Names
Country
Trade name
Manufacturer
Germany
Pantozol
Nycomed
Rifun
– “-
France
Eupantol
Altana
Inipomp
Sanofi-Aventis
United Kingdom
Protium
ALTANA
Italy
Pantekta
Abbott
Pantopan
Pharmacia
Pantork
Altana
USA
Protonix
Wyeth
Ukraine
Kontrolok
Nycomed Oranienburg GmbH, Germany
Nolpaza
Krka
Pultset
Nobel Ilach Sanayi ve Ticaret AS, Turkey
Proksium
JSC “Lubnyfarm”, Ukraine
various generic drugs
Formulations
ampoule 40 mg;
Tablets 40 mg
UV – spectrum
Conditions : Concentration – 1 mg / 100 ml
Solvent designation schedule
Methanol
Water
0.1 M HCl
0.1M NaOH
The absorption maximum
289 nm
291nm
Observed
decay
295 nm
391
346
–
418
ε
16600
14700
–
17700
IR – spectrum
Wavelength (μm)
Wavenumber (cm -1 )
NMR Spectrum
will be added
Links
EP 134 400 (Byk Gulden Lomberg; appl. 1.5.1984; CH-prior. 3.5.1983).
US 4,555,518 (Byk Gulden Lomberg; 26.11.1985; appl. 1.5.1984; CH-prior. 3.5.1983).
US 4,758,579 (Byk Gulden Lomberg; 19.7.1988; appl. 28.4.1987; CH-prior. 16.6.1984).
UV and IR Spectra. H.-W. Dibbern, RM Muller, E. Wirbitzki, 2002 ECV
NIST / EPA / NIH Mass Spectral Library 2008
Handbook of Organic Compounds. NIR, IR, Raman, and UV-Vis Spectra Featuring Polymers and Surfactants, Jr., Jerry Workman.Academic Press, 2000.
Handbook of ultraviolet and visible absorption spectra of organic compounds, K. Hirayama. Plenum Press Data Division, 1967.
[Dr. John Cooke, chair of Methodist Hospital’s cardiovascular services] [Houston Chronicle Health Zone dated Thursday, July 11, 2013 chron.com/refluxmeds] (Journal: Circulation)
Jump up^ Meyer, U A (1996). “Metabolic interactions of the proton-pump inhibitors lansoprazole, omeprazole and pantoprazole with other drugs”. European journal of gastroenterology & hepatology8 (Suppl 1): S21–25. doi:10.1097/00042737-199610001-00005.
Steinijans, V. W.; Huber, R.; Hartmann, M.; Zech, K.; Bliesath, H.; Wurst, W.; Radtke, H. W. (1996). “Lack of pantoprazole drug interactions in man: An updated review”. International Journal of Clinical Pharmacology and Therapeutics34 (6): 243–262. PMID8793611.
Terconazole is an anti-fungal medication, primarily used to treat vaginal fungal infections.
The synthesis of racemic terconazole [J. Heeres et al., J. Med . Chem . , 26 , 611 11983)] is similar. differing in the introduction of a 1 H- 1 , 2,4-triazol-1-yl substituent in place of 1H-imidazol-1-yl and in the nature of the phenol used in the last step of the synthetic sequence, which phenol is 1-methylethyl-4-(4- hydroxyphenyl)piperazme instead of 1-acetyl-4-(4-nydroxyphenyl)piperazine.
Example 20: (2S,4R) -(-)-1-[4-[[2-(2,4-dichlorophenyl)-2-[(1H-1,2,4-triazol-1-yl]methyl-1,3-dioxolane-4-yl]methoxy]phenyl]-4-(1-methylethyl)piperazine, (2S,4R) – (-)-terconazole.
This compound is prepared following the process described for (+)-torconazole, starting from (2S,4S)-(-)-IV (Ar = 2,4-dichlorophenyl, Y = N, R = CH3) (224 mg, 0.55 mmol), 4-(4-hydroxyphenyl)-1-(1-methylethyl)-piperazine (121 mg, 0.55 mmol), NaH (22.4 mg, 0.56 mmol) in 8 ml of DMSO. (2S,4R) -(-(-terconazole ((2S,4R)-V, Ar
= 2,4-dichlorophenyl, Y = N, Z = CH(CH3)2) is obtained as a white solid, m.p. 76-78ºC, [α]D20= -12.0 (c = 0.4.
CHCl3).
Example 17 : (2R,4S)-(+)-1-[4-[[2-(2,4-dichlorophenyl)- 2-[(1H-1,2,4-triazol-1-yl]methyl-1,3-dioxolane-4-yl]methyl]phenyl]-4-(1-methylethyl)piperazine, (2R,4S)-(+)-terconazole.
To a suspension of NaH (60-65% dispersion in paraffin, 36 mg, 0.90 mmol) in anhydrous DMSO (8 ml), 4-(4-hydroxyphenyl) -1 – ( 1-methyle thyl ) p iper az ine ( 193 mg , 0 . 88 mmol ) is added and the mixture is stirred for 1 hour at room temperature. Then, (2R,4R)-(+)-IV (Ar = 2,4-dichlorophenyl, Y = N, R = CH3 ) is added (180 mg, 0.44 mmol) and the mixture is heated at 80°C for 4 hours. The reaction mixture is allowed to cool to room temperature, diluted with water (20 ml) and extraoteo with CH2Cl2 (3 × 25 ml). The combined organic phases are washed with 5N NaOH (3 × 25 ml) and water (3 × 25 ml dried with Na2SO4 and the solvent is evaporated of: under vacuum. The oily residue thus obtained is crystallized from diisopropyl ether to give (2R,4S)-(+)-terconazole ((2R,4S)-V, Ar = 2,4-cichlorophenyl, Y = N, Z = CH(CH3)2) (140 mg, 59 % yield) as a white solid, m.p. 72-74’C, [α]D20 = + 11,05 (c = 0.4, CHCl3).
The U.S. Food and Drug Administration today approved Cologuard, the first stool-based colorectal screening test that detects the presence of red blood cells and DNA mutations that may indicate the presence of certain kinds of abnormal growths that may be cancers such as colon cancer or precursors to cancer.
Colorectal cancer primarily affects people age 50 and older, and among cancers that affect both men and women, it is the third most common cancer and the second leading cause of cancer-related death in the United States, according to the Centers for Disease Control and Prevention (CDC). Colorectal cancer screening is effective at reducing illness and death related to colon cancer. The CDC estimates that if everyone age 50 or older had regular screening tests as recommended, at least 60 percent of colorectal cancer deaths could be avoided.
Colorectal cancer occurs in the colon (large intestine) or rectum (the passageway that connects the colon to the anus). Most colorectal cancers start as abnormal raised or flat tissue growths on the wall of the large intestine or rectum (polyps). Some very large polyps are called advanced adenomas and are more likely than smaller polyps to progress to cancer.
Using a stool sample, Cologuard detects hemoglobin, a protein molecule that is a component of blood. Cologuard also detects certain mutations associated with colorectal cancer in the DNA of cells shed by advanced adenomas as stool moves through the large intestine and rectum. Patients with positive test results are advised to undergo a diagnostic colonoscopy.
“This approval offers patients and physicians another option to screen for colorectal cancer,” said Alberto Gutierrez, Ph.D., director of the Office of In Vitro Diagnostics and Radiological Health at the FDA’s Center for Devices and Radiological Health. “Fecal blood testing is a well-established screening tool and the clinical data showed that the test detected more cancers than a commonly used fecal occult test.”
Today’s approval of the Cologuard does not change current practice guidelines for colorectal cancer screening. Stool DNA testing (also called “fecal DNA testing”) is not currently recommended as a method to screen for colorectal cancer by the United States Preventive Services Task Force (USPSTF). Among other guidelines, the USPSTF recommends adults age 50 to 75, at average risk for colon cancer, be screened using fecal occult blood testing, sigmoidoscopy, or colonoscopy.
The safety and effectiveness of Cologuard was established in a clinical trial that screened 10,023 subjects. The trial compared the performance of Cologuard to the fecal immunochemical test (FIT), a commonly used non-invasive screening test that detects blood in the stool. Cologuard accurately detected cancers and advanced adenomas more often than the FIT test. Cologuard detected 92 percent of colorectal cancers and 42 percent of advanced adenomas in the study population, while the FIT screening test detected 74 percent of cancers and 24 percent of advanced adenomas. Cologuard was less accurate than FIT at correctly identifying subjects negative for colorectal cancer or advanced adenomas. Cologuard correctly gave a negative screening result for 87 percent of the study subjects, while FIT provided accurate negative screening results for 95 percent of the study population.
Today the Centers for Medicare & Medicaid Services (CMS) issued a proposed national coverage determination for Cologuard. Cologuard is the first product reviewed through a joint FDA-CMS pilot program known as parallel review where the agencies concurrently review medical devices to help reduce the time between the FDA’s approval of a device and Medicare coverage. This voluntary pilot program is open to certain premarket approval applications for devices with new technologies and to medical devices that fall within the scope of a Part A or Part B Medicare benefit category and have not been subject to a national coverage determination.
“Parallel review allows the last part of the FDA process to run at the same time as the CMS process, cutting as many as six months from the time from study initiation to coverage,” said Nancy Stade, CDRH’s deputy director for policy. “The pilot program is ongoing, but we will apply what we have learned to improve the efficiency of the medical device approval pathway for devices that address an important public health need.”
“This is the first time in history that FDA has approved a technology and CMS has proposed national coverage on the same day,” said Patrick Conway, chief medical officer and deputy administrator for innovation and quality for CMS. “This parallel review represents unprecedented collaboration between the two agencies and industry and most importantly will provide timely access for Medicare beneficiaries to an innovative screening test to help in the early detection of colorectal cancer.”
CMS proposes to cover the Cologuard test once every three years for Medicare beneficiaries who meet all of the following criteria:
age 50 to 85 years,
asymptomatic (no signs or symptoms of colorectal disease including but not limited to lower gastrointestinal pain, blood in stool, positive guaiac fecal occult blood test or fecal immunochemical test), and
average risk of developing colorectal cancer (no personal history of adenomatous polyps, of colorectal cancer, or inflammatory bowel disease, including Crohn’s Disease and ulcerative colitis; no family history of colorectal cancers or an adenomatous polyp, familial adenomatous polyposis, or hereditary nonpolyposis colorectal cancer).
Cologuard is manufactured by Exact Sciences in Madison, Wisconsin.
The European Medicines Agency relies on the results of clinical trials carried out by pharmaceutical companies to reach its opinions on the authorisation of medicines. Although the authorisation of clinical trials occurs at Member State level, the Agency plays a key role in ensuring that the standards of good clinical practice (GCP) are applied across the European Economic Area in cooperation with the Member States. It also manages a database of clinical trials carried out in the European Union.
Clinical trials are studies that are intended to discover or verify the effects of one or more investigational medicines. The regulation of clinical trials aims to ensure that the rights, safety and well-being of trial subjects are protected and the results of clinical trials are credible.
Regardless of where they are conducted, all clinical trials included in applications for marketing authorisation for human medicines in the European Economic Area (EEA) must have been carried out in accordance with the requirements set out in Annex 1 ofDirective 2001/83/EC. This means that:
clinical trials conducted in the EEA have to comply with European Union (EU) clinical-trial legislation (Directive 2001/20/EC);
In the EEA, approximately 4,000 clinical trials are authorised each year. This equals approximately 8,000 clinical-trial applications, with each trial involving two Member States on average. Approximately 61% of clinical trials are sponsored by the pharmaceutical industry and 39% by non-commercial sponsors, mainly academia.
Role of the Agency
Clinical-trial data is included in clinical-study reports that form a large part of the application dossiers submitted by pharmaceutical companies applying for a marketing authorisation via the Agency.
The Agency’s Committee for Medicinal Products for Human Use (CHMP) is responsible for conducting the assessment of a human medicine for which an EU-wide marketing authorisation is sought. As part of its scientific evaluation work, the CHMP reviews the clinical-trial data included in the application.
Assessments are based on purely scientific criteria and determine whether or not the medicines concerned meet the necessary quality, safety and efficacy requirements in accordance with EU legislation, particularly Directive 2001/83/EC.
Good clinical practice
The Agency plays a central role in ensuring application of good clinical practice (GCP). GCP is the international ethical and scientific quality standard for designing, recording and reporting clinical trials that involve the participation of human subjects.
The Agency works in cooperation with GCP inspectors from medicines regulatory authorities (‘national competent authorities’) in EEA Member States on the harmonisation and coordination of GCP-related activity at an EEA level.
The Agency does not have a role in the approval of clinical-trial applications in the EEA. The approval of clinical-trial applications is the responsibility of the national competent authorities.
EudraCT database and the EU Clinical Trials Register
The Agency is responsible for the development, maintenance and coordination of the EudraCT database. This is a database used by national competent authorities to enter clinical-trial data from clinical trial sponsors and paediatric-investigation-plan (PIP) addressees.
A subset of this data is made available through the European Union Clinical Trials Register, which the Agency manages on behalf of EU Member States and forms part ofEudraPharm, the EU database of medicines.
Users are able to view:
the description of phase-II to phase-IV adult clinical trials where the investigator sites are in the EEA;
the description of any clinical trials in children with investigator sites in the EU and any trials that form part of a PIP including those where the investigator sites are outside the EU.
As of 21 July 2014, it will be mandatory for sponsors to post clinical trial results in the EudraCT database. A subset of the data included in EudraCT is made available to the public in the European Union Clinical Trials Register. The content and level of detail of these summary results is set out in a European Commission guideline and in its technical guidance. A typical set of summary results provides information on the objectives of a given study, explains how it was designed and gives its main results and conclusions.
The Agency is also working towards the proactive publication of data from clinical trials carried out on the medicines that it authorises. For more information, see release of data from clinical trials.
Clinical trials conducted in countries outside the EU
Clinical trials conducted outside the EU but submitted in an application for marketing authorisation in the EU have to follow the principles which are equivalent to the provisions of the Directive 2001/20/EC.
In April 2012, the Agency published the final version of this paper:
This paper aims to strengthen existing processes to provide assurance that clinical trials meet the required ethical and GCP standards, no matter where in the world they have been conducted.
The number of clinical trials and clinical-trial subjects outside Western Europe and North America has been increasing for a number of years. More information is available in this document:
Linnaeus named the genus Aesculus after the Roman name for an edible acorn. Common names for these trees include “buckeye” and “horse chestnut”. Some are also called white chestnut or red chestnut (as in some of the Bach flower remedies). In Britain, they are sometimes called conker trees because of their link with the game of conkers, played with the seeds, also called conkers. Aesculus seeds were traditionally eaten, after leaching, by the Jōmon people of Japan over about four millennia, until 300 AD.[6]
Aesculus species have stout shoots with resinous, often sticky, buds; opposite, palmately divided leaves, often very large—to 65 cm (26 in) across in the Japanese horse chestnut Aesculus turbinata. The seeds of the Aesculus are traditionally used in a game called conkers in Europe. Species are deciduous or evergreen. Flowers are showy, insect- or bird-pollinated, with four or five petals fused into a lobedcorolla tube, arranged in a panicle inflorescence. Flowering starts after 80–110 growing degree days. The fruit matures to a capsule, 2–5 cm (25⁄32–1 31⁄32 in) diameter, usually globose, containing one to three seeds (often erroneously called a nut) per capsule. Capsules containing more than one seed result in flatness on one side of the seeds. The point of attachment of the seed in the capsule (hilum) shows as a large circular whitish scar. The capsule epidermis has “spines” (botanically: prickles) in some species, while other capsules are warty or smooth. At maturity, the capsule splits into three sections to release the seeds.[7][8][9]
The most familiar member of the genus worldwide is the common horse chestnut Aesculus hippocastanum. The yellow buckeye Aesculus flava (syn. A. octandra) is also a valuable ornamental tree with yellow flowers, but is less widely planted. Among the smaller species, the bottlebrush buckeye Aesculus parviflora also makes a very interesting and unusual flowering shrub. Several other members of the genus are used as ornamentals, and several horticultural hybrids have also been developed, most notably the red horse chestnut Aesculus × carnea, a hybrid between A. hippocastanum and A. pavia.
Use in alternative medicine
Aesculus has been listed as one of the 38 substances used to prepare Bach flower remedies,[10] a kind of alternative medicine promoted for its effect on health. However according to Cancer Research UK, “there is no scientific evidence to prove that flower remedies can control, cure or prevent any type of disease, including cancer”.[11]
Jump up^ Ogg, James G.; Gradstein, F. M; Gradstein, Felix M. (2004). A geologic time scale 2004. Cambridge, UK: Cambridge University Press.ISBN0-521-78142-6.
Jump up^ Hardin, JW. 1957. A revision of the American Hippocastanaceae I. Brittonia 9:145-171.
Jump up^ Judd, WS, RW Sanders, MJ Donoghue. 1994. Angiosperm family pairs. Harvard Papers in Botany. 1:1-51.
Jump up^ Harrington, Mark G.; Edwards, Karen J.; Johnson, Sheila A.; Chase, Mark W.; Gadek, Paul A. (Apr–Jun 2005). “Phylogenetic inference in Sapindaceae sensu lato using plastid matK and rbcL DNA sequences”. Systematic Botany30 (2): 366–382. doi:10.1600/0363644054223549. JSTOR25064067.
Jump up^ Harlan, Jack R. (1995). The Living Fields: Our Agricultural Heritage (1. publ. ed.). Cambridge [u.a.]: Cambridge Univ. Press. p. 15. ISBN0-521-40112-7.Harlan cites Akazawa, T & Aikens, CM, Prehistoric Hunter-Gathers in Japan (1986), Univ. Tokyo Press; and cites Aikens, CM & Higachi, T, Prehistory of Japan (1982), NY Academic Press.
Jump up^ Hardin, JW. 1957. A revision of the American Hippocastanaceae I. Brittonia 9:145-171
Jump up^ Hardin, JW. 1957. A revision of the American Hippocastanaceae II. Brittonia 9:173-195
Jump up^ Hardin, JW. 1960. A revision of the American Hippocastanaceae V, Species of the Old World. Brittonia 12:26-38
Forest, F., Drouin, J. N., Charest, R., Brouillet, L., & Bruneau A. (2001). A morphological phylogenetic analysis of Aesculus L. and Billia Peyr. (Sapindaceae). Canad. J. Botany79 (2): 154-169. Abstract.
A. hippocastanum grows to 36 metres (118 ft) tall, with a domed crown of stout branches; on old trees the outer branches often pendulous with curled-up tips. The leaves are opposite and palmately compound, with 5–7 leaflets; each leaflet is 13–30 cm long, making the whole leaf up to 60 cm across, with a 7–20 cm petiole. The leaf scars left on twigs after the leaves have fallen have a distinctive horseshoe shape, complete with seven “nails”. The flowers are usually white with a small red spot; they are produced in spring in erect panicles 10–30 cm tall with about 20–50 flowers on each panicle. Usually only 1–5 fruit develop on each panicle; the shell is a green, spiky capsule containing one (rarely two or three) nut-like seeds called conkers or horse-chestnuts. Each conker is 2–4 cm diameter, glossy nut-brown with a whitish scar at the base.[2]
Etymology
The common name “horse-chestnut” (often unhyphenated) is reported as having originated from the erroneous belief that the tree was a kind of chestnut (though in fact only distantly related), together with the observation that eating the fruit cured horses of chest complaints[3] despite this plant being poisonous to horses.
In Britain and Ireland, the nuts are used for the popular children’s game conkers. During the First World War, there was a campaign to ask for everyone (including children) to collect horse-chestnuts and donate them to the government. The conkers were used as a source of starch for the fermentation via the Clostridium acetobutylicum method devised by Chaim Weizmann to produce acetone. Any starch plant would have done, but they chose to ask for conkers to avoid causing starvation by using food. Weizmann’s process could use any source of starch, but it was never particularly efficient and the factory only produced acetone for three months. The aim was to produce acetone for use as solvent which aided in the production of cordite, which was then used in military armaments.
A selection of fresh conkers from a horse-chestnut
The nuts, especially those that are young and fresh, are slightly poisonous, containing alkaloidsaponins and glucosides. Although not dangerous to touch, they cause sickness when eaten; consumed by horses, they can cause tremors and lack of coordination.[6] Somemammals, notably deer, are able to break down the toxins and eat them safely.[citation needed]
Though the seeds are said to repel spiders there is little evidence to support these claims. The presence of saponin may repel insects but it is not clear whether this is effective on spiders.[7]
Horse-chestnuts have been threatened by the leaf-mining moth Cameraria ohridella, whose larvae feed on horse chestnut leaves. The moth was described from Macedonia where the species was discovered in 1984 but took 18 years to reach Britain.[8]
The flower is the symbol of the city of Kiev, capital of Ukraine.[9] Although the horse-chestnut is sometimes known as the buckeye, this name is generally reserved for the New World members of the Aesculus genus.
Medical uses
The seed extract standardized to around 20 percent aescin (escin) is used for its venotonic effect, vascular protection, anti-inflammatory and free radical scavenging properties.[10][11] Primary indication is chronic venous insufficiency.[11][12] A recent Cochrane Review found the evidence suggests that Horse Chestnut Seed Extract is an efficacious and safe short-term treatment for chronic venous insufficiency.[13]
Aescin reduces fluid leaks to surrounding tissue by reducing both the number and size of membrane pores in the veins.
Safety in medical use
Two preparations are considered; whole horsechestnut extract (whole HCE) and purified β-aescin. Historically, whole HCE has been used both for oral and IV routes (as of year 2001). The rate of adverse effects are low, in a large German study, 0.6%, consisting mainly of gastrointestinal symptoms. Dizziness, headache and itching have been reported. One serious safety issue is rare cases of acute anaphylactic reactions, presumably in a context of whole HCE. Purified β-aescin would be expected to have a better safety profile.
Another is the risk of acute renal failure, “when patients, who had undergone cardiac surgery were given high doses of horse chestnut extract i.v. for postoperative oedema. The phenomenon was dose dependent as no alteration in renal function was recorded with 340 μg kg−1, mild renal function impairment developed with 360 μg kg−1 and acute renal failure with 510 μg kg−1″.[14] This almost certainly took place in a context of whole HCE.
Three clinical trials were since performed to assess the effects of aescin on renal function. A total of 83 subjects were studied; 18 healthy volunteers given 10 or 20 mg iv. for 6 days, 40 in-patients with normal renal function given 10 mg iv. two times per day (except two children given 0.2 mg/kg), 12 patients with cerebral oedema and normal renal function given a massive iv. dose on the day of surgery (49.2 ± 19.3 mg) and 15.4 ± 9.4 mg daily for the following 10 days and 13 patients with impaired renal function due to glomerulonephritis or pyelonephritis, who were given 20–25 mg iv. daily for 6 days. “In all studies renal function was monitored daily resorting to the usual tests of renal function: BUN, serum creatinine, creatinine clearance, urinalysis. In a selected number of cases paraaminohippurate and labelled EDTA clearance were also measured. No signs of development of renal impairment in the patients with normal renal function or of worsening of renal function in the patients with renal impairment were recorded.” It is concluded that aescin has excellent tolerability in a clinical setting.[15]
Raw Horse Chestnut seed, leaf, bark and flower are toxic due to the presence of esculin and should not be ingested. Horse chestnut seed is classified by the FDA as an unsafe herb.[11] The glycoside and saponin constituents are considered toxic.[11]
Aesculus hippocastanum is used in Bach flower remedies. When the buds are used it is referred to as “chestnut bud” and when the flowers are used it is referred to as “white chestnut”.
A famous specimen of the horse-chestnut was the Anne Frank Tree in the centre of Amsterdam, which she mentioned in her diary and which survived until August 2010, when a heavy wind blew it over.[17][18] Eleven young specimens, sprouted from seeds from this tree, were transported to the United States. After a long quarantine in Indianapolis, each tree was shipped off to a new home at a notable museum or institution in the United States, such as the 9/11 Memorial Park, Central H.S. in Little Rock, and two Holocaust Centers. One of them was planted outdoors in March 2013 in front of the Children’s Museum of Indianapolis, where they were originally quarantined. [1]
Bonsai
The horse-chestnut is a favourite subject for bonsai.[19]
Diseases
Bleeding Canker. Half of all horse-chestnuts in Great Britain are now showing symptoms to some degree of this potentially lethal bacterial infection.[20][21]
Guignardia leaf blotch, caused by the fungus Guignardia aesculi
Wood rotting fungi, e.g. such as Armillaria and Ganoderma
An antimicrobial agent that destroys fungi by suppressing their ability to grow or reproduce. Antifungal agents differ from industrial fungicides in that they defend against fungi present in human or animal tissues.
An antimicrobial agent that destroys fungi by suppressing their ability to grow or reproduce. Antifungal agents differ from industrial fungicides in that they defend against fungi present in human or animal tissues.
Ketoconazole, 1-acetyl-4-[4-[[2-(2,4-dichlorophenyl)-2-[(1H-imidazol-1-yl)-methyl]-1,3– dioxolan-4-yl]methoxy]phenyl]piperazine, is a racemic mixture of the cis enantiomers (-)-(2S,4R) and (+)-(2R,4S) marketed as an anti-fungal agent. Ketoconazole inhibits fungal growth through the inhibition of ergosterol synthesis.(-)-Ketoconazole, the (2S,4R) enantiomer contained in the racemate of ketoconazole, is in phase III clinical trials at Cortendo for the treatment of endogenous Cushing’s syndrome. The company and licensee DiObex had also been developing the drug candidate for the treatment of type 2 diabetes; however, no recent development has been reported for this research.Preclinical studies have demonstrated the drug candidate’s ability to inhibit the synthesis of cortisol, resulting in substantial clinical benefits including lowering both blood pressure and cholesterol in addition to controlling glucose levels. It has also been shown that (-)-ketoconazole is responsible for virtually all of the cortisol synthesis inhibitory activity present in the racemate. Rights to the compound are shared with Cortendo.In 2012, orphan drug designation was assigned in the U.S. for the treatment of endogenous Cushing’s syndrome.
August 12, 2014 02:30 AM Eastern Daylight Time
GÖTEBORG, Sweden.–(BUSINESS WIRE)–Cortendo AB (OSE:CORT) today announced that the first patient has been enrolled into the Phase 3 SONICS trial, i.e., “Study Of NormoCort In Cushing’s Syndrome.”
“The enrollment of the first patient into the SONICS trial represents a significant milestone for Cortendo”
The patient was enrolled by one of the trial’s lead principal investigators at a Pituitary Center from a prestigious institution in Baltimore, Maryland. “The enrollment of the first patient into the SONICS trial represents a significant milestone for Cortendo”, said Dr. Theodore R Koziol. ”The SONICS clinical trial team is acutely focused on the implementation of the trial following a successful EU Investigator’s meeting in Barcelona in July, which we believe further solidified the foundation for the trial.”
Cortendo successfully completed its European Investigator meeting supporting SONICS held in Barcelona, Spain on July 17-18. More than 35 investigators/study coordinators, including many of the world’s leading Cushing’s experts from 24 study sites, were in attendance and received training for the trial. Based on the positive feedback from the meeting, Cortendo has gained further confidence that NormoCort (COR-003) has the potential to be an important future treatment option for patients afflicted with Cushing’s Syndrome. A second US Investigator meeting is also being planned for later this year.
”It was gratifying to participate in the NormoCort SONICS trial investigator meeting in my home town of Barcelona with so many esteemed colleagues dedicated to treating patients with Cushing’s Syndrome”, said Susan Webb M.D. Ph.D. Professor of Medicine Universitat Autonoma de Barcelona. ”There remains a significant unmet medical need for patients, and I am delighted to be part of the development of this new therapy”.
Cortendo has also further strengthened its internal as well as external teams to support the study and to position the trial for an increased recruitment rate. In July, Cortendo added both an experienced physician and internal Clinical Operations Director to the NormoCort development team. Cortendo, working in concert with its CROs supporting the SONICS trial, now has a team of approximately 20 personnel on the NormoCort development program.
Cortendo has previously communicated its plan to meet the recruitment goal by increasing the number of study sites from 38 to 45 worldwide. The company is at various levels of activation with more than 30 study sites to date. Therein, Cortendo expects a large proportion of the sites to be activated by the end of the third quarter this year and remains confident that essentially all sites will be open by the end of 2014.
Risk and uncertainty
The development of pharmaceuticals carries significant risk. Failure may occur at any stage during development and commercialization due to safety or clinical efficacy issues. Delays may occur due to requirements from regulatory authorities not anticipated by the company.
About Cortendo
Cortendo AB is a biopharmaceutical company headquartered in Göteborg, Sweden. Its stock is publicly traded on the NOTC-A-list (OTC) in Norway. Cortendo is a pioneer in the field of cortisol inhibition and has completed early clinical trials in patients with Type 2 diabetes. The lead drug candidate NormoCort, the 2S, 4R-enantiomer of ketoconazole, has been re-focused to Cushing’s Syndrome, and has entered Phase 3 development. The company’s strategy is to primarily focus its resources within orphan drugs and metabolic diseases and to seek opportunities where the path to commercialization or partnership is clear and relatively near-term. Cortendo’s business model is to commercialize orphan and specialist product opportunities in key markets, and to partner non-specialist product opportunities such as diabetes at relevant development stages.
Alexander Lindström Chief Financial Officer Office +1 610 254 9200 Mobile : +1 917 349 7210 E-mail : alindstrom@cortendo.com
Ketoconazole, 1-acetyl-4- [4-[[2-(2,4-dichlorophenyl)-2-[(1H-imidazol-1-yl)-methyl]-1,3-dioxolan-4-yl] methoxy] phenyl] piperazine, is a racemic mixture of the cis enantiomers (-)-(2S, 4R) and (+)-(2R, 4S) marketed as an anti-fungal agent. Ketoconazole inhibits fungal growth through the inhibition of ergosterol synthesis. Ergosterol is a key component of fungal cell walls.
More recently, ketoconazole was found to decrease plasma cortisol and to be useful, alone and in combination with other agents, in the treatment of a variety of diseases and conditions, including type 2 diabetes, Metabolic Syndrome (also known as the Insulin Resistance Syndrome, Dysmetabolic Syndrome or Syndrome X), and other medical conditions that are associated with elevated cortisol levels. SeeU.S. Patent Nos. 5,584,790 ; 6,166,017 ; and 6,642,236 , each of which is incorporated herein by reference. Cortisol is a stress-related hormone secreted from the cortex of the adrenal glands. ACTH (adenocorticotropic hormone) increases cortisol secretion. ACTH is secreted by the pituitary gland, a process activated by secretion of corticotropin releasing hormone (CRH) from the hypothalamus.
Cortisol circulates in the bloodstream and activates specific intracellular receptors, such as the glucocorticoid receptor (GR). Disturbances in cortisol levels, synthetic rates or activity have been shown to be associated with numerous metabolic complications, including insulin resistance, obesity, diabetes and Metabolic Syndrome. Additionally, these metabolic abnormalities are associated with substantially increased risk of cardiovascular disease, a major cause of death in industrialized countries. See Mårin P et al., “Cortisol secretion in relation to body fat distribution in obese premenopausal women.” Metabolism 1992; 41:882-886, Bjorntorp, “Neuroendocrine perturbations as a cause of insulin resistance.” Diabetes Metab Res Rev 1999; 15(6): 427-41, and Rosmond, “Role of stress in the pathogenesis of the metabolic syndrome.” Psychoneuroendocrinology 2005; 30(1): 1-10, each of which is incorporated herein by reference.
While ketoconazole is known to inhibit some of the enzymatic steps in cortisol synthesis, such as, for example, 17α hydroxylase (Wachall et al., “Imidazole substituted biphenyls: a new class of highly potent and in vivo active inhibitors of P450 17 as potential therapeutics for treatment of prostate cancer.” Bioorg Med Chem 1999; 7(9): 1913-24, incorporated herein by reference) and 11b-hydroxylase (Rotstein et al., “Stereoisomers of ketoconazole: preparation and biological activity.” J Med Chem 1992; 35(15): 2818-25) and 11β-hydroxy steroid dehydrogenase (11β-HSD) (Diederich et al., “In the search for specific inhibitors of human 11β-hydroxysteroid-dehydrogenases (11β-HSDs): chenodeoxycholic acid selectively inhibits 11β-HSD-L” Eur J Endocrinol 2000; 142(2): 200-7, incorporated herein by reference) the mechanisms by which ketoconazole decreases cortisol levels in the plasma have not been reported. For example, there is uncertainty regarding the effect of ketoconazole on the 11β-hydroxy steroid dehydrogenase (11β-HSD) enzymes. There are two 11β-HSD enzymes. One of these, 11β-HSD-I, is primarily a reductase that is highly expressed in the liver and can convert the inactive 11-keto glucocorticoid to the active glucocorticoid (cortisol in humans and corticosterone in rats). In contrast, the other, 11β-HSD-II, is primarily expressed in the kidney and acts primarily as an oxidase that converts active glucocorticoid (cortisol in humans and corticosterone in rats) to inactive 11-keto glucocorticoids. Thus, the plasma concentration of active glucocorticoid is influenced by the rate of synthesis, controlled in part by the activity of adrenal 11β-hydroxylase and by the rate of interconversion, controlled in part by the relative activities of the two 11β-HSD enzymes. Ketoconazole is known to inhibit these three enzymes (Diederich et al., supra) and the 2S,4R enantiomer is more active against the adrenal 11β-hydroxylase enzyme than is the 2R,4S enantiomer (Rotstein et al., supra). However, there are no reports describing the effect of the two ketoconazole enantiomers on either of 11β-HSD-I or 11β-HSD-II, so it is not possible to predict what effects, if any, the two different ketoconazole enantiomers will each have on plasma levels of the active glucocorticoid levels in a mammal.
Ketoconazole has also been reported to lower cholesterol levels in humans (Sonino et al. (1991). “Ketoconazole treatment in Cushing’s syndrome: experience in 34 patients.” Clin Endocrinol (Oxf). 35(4): 347-52; Gylling et al. (1993). “Effects of ketoconazole on cholesterol precursors and low density lipoprotein kinetics in hypercholesterolemia.” J Lipid Res. 34(1): 59-67) each of which is incorporated herein by reference). The 2S,4R enantiomer is more active against the cholesterol synthetic enzyme 14 αlanosterol demethylase than is the other (2R,4S) enantiomer (Rotstein et al infra). However, because cholesterol level in a human patient is controlled by the rate of metabolism and excretion as well as by the rate of synthesis it is not possible to predict from this whether the 2S,4R enantiomer of ketoconazole will be more effective at lowering cholesterol levels.
The use of ketoconazole as a therapeutic is complicated by the effect of ketoconazole on the P450 enzymes responsible for drug metabolism. Several of these P450 enzymes are inhibited by ketoconazole (Rotsteinet al., supra). This inhibition leads to an alteration in the clearance of ketoconazole itself (Brass et al., “Disposition of ketoconazole, an oral antifungal, in humans.” Antimicrob Agents Chemother 1982; 21(1): 151-8, incorporated herein by reference) and several other important drugs such as Glivec (Dutreix et al., “Pharmacokinetic interaction between ketoconazole and imatinib mesylate (Glivec) in healthy subjects.” Cancer Chemother Pharmacol 2004; 54(4): 290-4) and methylprednisolone (Glynn et al., “Effects of ketoconazole on methylprednisolone pharmacokinetics and cortisol secretion.” Clin Pharmacol Ther 1986; 39(6): 654-9). As a result, the exposure of a patient to ketoconazole increases with repeated dosing, despite no increase in the amount of drug administered to the patient. This exposure and increase in exposure can be measured and demonstrated using the “Area under the Curve” (AUC) or the product of the concentration of the drug found in the plasma and the time period over which the measurements are made. The AUC for ketoconazole following the first exposure is significantly less than the AUC for ketoconazole after repeated exposures. This increase in drug exposure means that it is difficult to provide an accurate and consistent dose of the drug to a patient. Further, the increase in drug exposure increases the likelihood of adverse side effects associated with ketoconazole use.
[0008]
Rotstein et al. (Rotstein et al., supra) have examined the effects of the two ketoconazole cis enantiomers on the principal P450 enzymes responsible for drug metabolism and reported “…almost no selectivity was observed for the ketoconazole isomers” and, referring to drug metabolizing P450 enzymes: “[t]he IC50 values for the cis enantiomers were similar to those previously reported for racemic ketoconazole”. This report indicated that both of the cis enantiomers could contribute significantly to the AUC problem observed with the ketoconazole racemate.
One of the adverse side effects of ketoconazole administration exacerbated by this AUC problem is liver reactions. Asymptomatic liver reactions can be measured by an increase in the level of liver specific enzymes found in the serum and an increase in these enzymes has been noted in ketoconazole treated patients (Sohn, “Evaluation of ketoconazole.” Clin Pharm 1982; 1(3): 217-24, and Janssen and Symoens, “Hepatic reactions during ketoconazole treatment.” Am J Med 1983; 74(1B): 80-5, each of which is incorporated herein by reference). In addition 1:12,000 patients will have more severe liver failure (Smith and Henry, “Ketoconazole: an orally effective antifungal agent. Mechanism of action, pharmacology, clinical efficacy and adverse effects.” Pharmacotherapy 1984; 4(4): 199-204, incorporated herein by reference). As noted above, the amount of ketoconazole that a patient is exposed to increases with repeated dosing even though the amount of drug taken per day does not increase (the “AUC problem”). The AUC correlates with liver damage in rabbits (Ma et al., “Hepatotoxicity and toxicokinetics of ketoconazole in rabbits.” Acta Pharmacol Sin 2003; 24(8): 778-782 incorporated herein by reference) and increased exposure to the drug is believed to increase the frequency of liver damage reported in ketoconazole treated patients.
Additionally, U.S. Patent No. 6,040,307 , incorporated herein by reference, reports that the 2S,4R enantiomer is efficacious in treating fungal infections. This same patent application also reports studies on isolated guinea pig hearts that show that the administration of racemic ketoconazole may be associated with an increased risk of cardiac arrhythmia, but provides no data in support of that assertion. However, as disclosed in that patent, arrhythmia had not been previously reported as a side effect of systemic racemic ketoconazole, although a particular subtype of arrhythmia, torsades de pointes, has been reported when racemic ketoconazole was administered concurrently with terfenadine. Furthermore several published reports (for example, Morganroth et al. (1997). “Lack of effect of azelastine and ketoconazole coadministration on electrocardiographic parameters in healthy volunteers.” J Clin Pharmacol. 37(11): 1065-72) have demonstrated that ketoconazole does not increase the QTc interval. This interval is used as a surrogate marker to determine whether drugs have the potential for inducing arrhythmia. US Patent Number 6,040,307 also makes reference to diminished hepatoxicity associated with the 2S,4R enantiomer but provides no data in support of that assertion. The method provided in US Patent Number 6,040,307 does not allow for the assessment of hepatoxicity as the method uses microsomes isolated from frozen tissue.
DIO-902 is the single enantiomer 2S,4R ketoconazole and is derived from racemic ketoconazole. It is formulated using cellulose, lactose, cornstarch, colloidal silicon dioxide and magnesium stearate as an immediate release 200 mg strength tablet. The chemical name is 2S,4R cis-1-acetyl-4-[4-[[2-(2,4-dichlorophenyl)-2-(1H-imidazol-1-ylmethyl)-1,3-dioxolan-4-yl] methoxyl]phenyl] piperazine, the formula is C26H28Cl2N4O4, and the molecular weight is 531.44. The CAS number is 65277-42-1, and the structural formula is provided below. The chiral centers are at the carbon atoms 2 and 4 as marked.
[0132]
Ketoconazole is an imidazole-containing fungistatic compound. DIO-902 is an immediate release tablet to be taken orally and formulated as shown in the table below.
Component
Percentage
2S,4R ketoconazole; DIO-902
50%
Silicified Microcrystalline Cellulose, NF (Prosolv HD 90)
16.5
Lactose Monohydrate, NF (316 Fast-Flo)
22.4
Corn Starch, NF (STA-Rx)
10
Colloidal Silicon Dioxide, NF (Cab-O-Sil M5P)
0.5
Magnesium Stearate, NF
0.6
The drug product may be stored at room temperature and is anticipated to be stable for at least 2 years at 25° C and 50% RH. The drug is packaged in blister packs.
A new process has been developed to separate ketoconazole (KTZ) enantiomers by membrane extraction, with the oppositely preferential recognition of hydrophobic and hydrophilic chiral selectors in organic and aqueous phases, respectively. This system is established by adding hydrophobic l-isopentyl tartrate (l-IPT) in organic strip phase (shell side) and hydrophilic sulfobutylether-β-cyclodextrin (SBE-β-CD) in aqueous feed phase (lumen side), which preferentially recognizes (+)-2R,4S-ketoconazole and (−)-2S,4R-ketoconazole, respectively. The studies performed involve two enantioselective extractions in a biphasic system, where KTZ enantiomers form four complexes with SBE-β-CD in aqueous phase and l-IPT in organic phase, respectively. The membrane is permeable to the KTZ enantiomers but non-permeable to the chiral selector molecules. Fractional chiral extraction theory, mass transfer performance of hollow fiber membrane, enantioselectivity and some experimental conditions are investigated to optimize the separation system. Mathematical model of I/II = 0.893e0.039NTU for racemic KTZ separation by hollow fiber extraction, is established. The optical purity for KTZ enantiomers is up to 90% when 9 hollow fiber membrane modules of 30 cm in length in series are used.
I, (−)-2S,4R-ketoconazole;
II, (+)-2R,4S-ketoconazole;
CDs, cyclodextrin derivatives;
l-IPT, l-isopentyl tartrate;
d-IPT, d-isopentyl tartrate;
HP-β-CD, hydroxypropyl-β-cyclodextrin;
Me-β-CD, methyl-β-cyclodextrin;
β-CD, β-cyclodextrin;
NTU, number of transfer units;
HTU, height of a transfer unit;
PVDF,polyvinylidene fluoride
…………………….
Stereoselective synthesis of both enantiomers of ketoconazole from (R)- and (S)-
Pelayo Camps, Xavier Farrés, Ma Luisa García, Joan Ginesta, Jaume Pascual, David Mauleón, Germano Carganico
Bromobenzoates (2R,4R)- and (2S,4S)-18, prepared stereoselectively from (R)- and (S)-epichlorohydrin, were transformed into (2R,4S)-(+)- and (2S,4R)-(−)-Ketoconazole, respectively, following the known synthetic protocols for the racemic mixture.
Tetrahedron Asymmetry 1995, 6(6): 1283
Stereoselective syntheses of both enantiomers of ketoconazole (1) from commercially available (R)- or (S)-epichlorohydrin has been developed. The key-step of these syntheses involves the selective substitution of the methylene chlorine atom by benzoate on a mixture of and or of their enantiomers, followed by crystallization of the corresponding cis-benzoates, (2S,4R)-18 or(2S,4S)-18, from which (+)- or (−)-1 were obtained as described for (±)-1. The ee’s of (+)- and (−)-ketoconazole were determined by HPLC on the CSP Chiralcel OD-H.
The incidence of fungal infections has considerably increased over the last decades. Notwithstanding the utility of the antifungal compounds commercialized in the last 15 years, the investigation in this field is however very extensive. During this time, compounds belonging to the azole class have beer, commercialized for both the topical and oral administrations, such a class including imidazoles as well as 1,2,4-triazoles. Some of these compounds car. show m some degree a low gastrointestinal tolerance as well as hepatotoxycity.
A large number of pharmaceutically active compounds are commercialized as stereoisomeric mixtures. On the other hand, the case in which only one of said stereoisomers is pharmaceutically active is frequent.
The undesired enantiomer has a lower activity and it sometimes may cause undesired side-effects.
Ketoconazole (1-acetyl-4-[4-[[2-(2,4-dichlorophenyl)-2-[(1H-imidazol-1-yl)methyl]-1,3-dioxolane-4-yl]methoxy]phenyl]piperazine), terconazole (1-[4-[[2(2,4-dichlorophenyl)-2-[(1H-1 , 2 ,4-triazol-1-yl)methyl]-1,3-dioxolane-4-yl]methoxy]phenyl]-4-(1-methylethyl)piperazine) and other related azole antifungal drugs contain in their structure a substituted 1,3-dioxolane ring, in which carbon atoms C2 and C4 are stereogenic centres, therefore four possible stereoisomers are possible. These compounds are commercialized in the form or cis racemates which show a higher antifungal activity than the corresponding trans racemates.
The cis homochiral compounds of the present invention, which are intermediates for the preparation of enantiomerically pure antifungal drugs, have been prepared previously in the racemic form and transformed into the different azole antifungal drugs in the racemic form [J. Heeres et al., J . Med . Chem . , 22 , 1003 (1979). J . Med . Chem . , 26, 611 (1983), J . Med . Chem . , 27 , 894 (1984) and US 4,144,346, 4,223,036, 4,358,449 and 4,335,125].
Scheme 1 shows the synthesis described for racemic ketoconazole [J. Heeres et al., J . Med . Chem . , 22 , 1003 (1979)]. Scheme 1
)
The synthesis of racemic terconazole [J. Heeres et al., J. Med . Chem . , 26 , 611 11983)] is similar. differing in the introduction of a 1 H- 1 , 2,4-triazol-1-yl substituent in place of 1H-imidazol-1-yl and in the nature of the phenol used in the last step of the synthetic sequence, which phenol is 1-methylethyl-4-(4- hydroxyphenyl)piperazme instead of 1-acetyl-4-(4-nydroxyphenyl)piperazine.
The preparation of racemic itraconazole [J. Heeres et al., J. Med . Chem. , 27 , 894 (1984)] is similar to that of terconazole, differing only in the nature of the phenol used in the last step of the synthetic sequence.
In the class of azoles containing a 1,3-dioxolane ring and a piperazine ring and moreover they are pure enantiomers, only the preparation of (+)- and (-)-ketoconazole has been described [D. M. Rotstein et al., J. Med . Chem . , 35, 2818 (1992)] (Scheme 2) starting from the tosylate of (+)- and (-) 2,2-dimethyl-1,3-dioxolane-4-methanol.
Scheme 2
This synthesis suffers from a series of drawbacks, namely: a) the use of expensive, high molecular weight starting products which are available only on a laboratory scale, and b) the need for several chromatographies during the process in order to obtain products of suitable purity, which maKes said synthesis economically unattractive and difficult to apply industrially.
Recently (N. M. Gray, WO 94/14447 and WO 94/14446) the use of (-)-ketoconazole and (+)-ketoconazole as antifungal drugs causing less side-effects than (±)-ketoconazole has been claimed.
The industrial preparation of enantiomerically pure antifungal drugs with a high antifungal activity and less side-effects is however a problem in therapy. The present invention provides novel homochiral compounds which are intermediates for the industrial preparation of already known, enantiomerically pure antifungal drugs such as ketoconazole enantiomers, or of others which have not yet been reported in literature, which are described first in the present invention, such as (+)-terconazole and (-)-terconazoie, which show the cited antifungal action, allowing to attain the same therapeutical effectiveness using lower dosages than those required for racemic terconazole
Example 14 : (2S,4R)-(-)-1-acetyl-4-[4-[ [2-(2,4-dichlorophenyl)-2-[(1H-imidazol-1-yl)-methyl]-1,3-dioxolane-4-yl]methoxy]phenyl]piperazine, (2S,4R) -(- )-ketoconazole.
This compound is prepared following the process described above for (2R,4S)-(+)-ketoconazole. Starting from HNa (60-65% dispersion in paraffin, 32 mg, 0.80 mmol), 1-acetyl-4-(4-hydroxyphenyl)piperazine (153 mg, 0.69 mol) and (2S,4S)-(-)-IV (Ar = 2,4-dichlorophenyl, Y = CH, R = CH3) (250 mg, 0.61 mmol), upon crystallization from an acetone:ethyl acetate mixture, (2S,4R) -(-)-ketoconazole is obtained [(2S,4R)-V Ar = 2,4-dichlorophenyl, Y = CH, Z = COCH3] (196 mg, 61% yield) as a solid, m.p. 153-155ºC (lit. 155-157ºC); [α]D20 = -10.50 (c = 0.4, CHCl3) (lit. [α]D25 = -10.58. c = 0.4, CHCl3) with e.e. > 99% (determined by HPLC using the chiral stationary phase CHIRALCEL OD-H and ethanol:hexane 1:1 mixtures containing 0.1 % diethylamine as the eluent).
+ KETOCONAZOLE…. UNDESIRED
Example 7: (2 R ,4S)-(+)-1-acetyl-4-[4-[[2-(2,4-dichlorophenyl)-2-[(1H-imidazol-1-yl)methyl]-1,3-dioxolane-4-yl]methoxy]phenyl]piperazine (22, 4 S)-(+)-ketoconazole.
To a suspension of NaH (dispersed in 60-65% paraffin, 19.2 mg, 0.48 mmol) in anhydrous DMSO (3 ml),
1-acetyl-4-(hydroxyphenyl)piperazine (102 mg, 0.46 mmol) is added and the mixture is stirred for 1 hour at room temperature. Then, a solution of (2R,4R) – (+)-IV (Ar = 2,4-dichlorophenyl, Y = CH, R = CH3) (160 mg, 0.39 mmol) in anhydrous DMSO (5 ml) is added, and the mixture is heated at 80ºC for 4 hours. The reaction mixture is allowed to cool to room temperature, diluted with water
(20 ml) and extracted with CH2Cl2 (3 × 25 ml). The combined organic phases are washed with water (3 × 25), dried with Na2SO4 and the solvent is evaporated off under vacuum. The oily residue thus obtained is crystallized from an acetone:ethyl acetate mixture to give (2R,4S)-(+)-ketoconazole ( (2R, 4 S) -V , Ar 2,4-dichlorophenyl, Y = CH , Z = COCH3 ) ( 110 mg , 5 3 % yie ld ) as a white solid, m.p. 155-156°C (lit. 154-156ºC), [α]D20 = + 8.99 (c = 0.4, CHCl3) (lit. [α]D25 = + 8.22, c = 0.4, CHCl3), with e.e. > 99% (determined by HPLC using the chirai stationary phase CHIRALCEL OD-H and ethanol:hexane 1:1 mixtures containing 0.1% of diethylamine, as the eluent; (+)-Ketoconazole retention time 73,28 min. (-)-Ketoconazole, retention time 79.06 min).
Experimental and theoretical analysis of the interaction of (+/-)-cis-ketoconazole with beta-cyclodextrin in the presence of (+)-L-tartaric acid J Pharm Sci 1999, 88(6): 599
1H NMR spectroscopy was used for determining the optical purity of cis-ketoconazole enantiomers obtained by fractional crystallization. The chiral analysis was carried out using β-cyclodextrin in the presence of (+)-l-tartaric acid. The mechanism of the chiral discrimination process, the stability of the complexes formed, and their structure in aqueous solution were also investigated by 1H and 13C chemical shift analysis, two-dimensional NOE experiments, relaxation time measurements, and mass spectrometry experiments. Theoretical models of the three-component interaction were built up on the basis of the available NMR data, by performing a conformational analysis on the relevant fragments on ketoconazole and docking studies on the components of the complex. The model derived from a folded conformation of ketoconazole turned out to be fully consistent with the molecular assembly found in aqueous solution, as inferred from NOE experiments. An explanation of the different association constants for the complexes of the two enantiomers is also provided on the basis of the interaction energies.
To start with the simplest one is Quantitative structure-activity relationship (QSAR) which is also referred to as 2D-QSAR sometimes. 3D-QSAR involving Comparative Molecular Field Analysis (CoMFA) and Comparative molecular similarity index analysis (CoMSIA) are extension of QSAR. QSAR is not able to take the three dimensional structure of a molecule into consideration due to absence of three-dimensional parameterization of structures. 3D-QSAR scores over QSAR in this respect. Docking studies throw more light on the binding modes of drugs with their target proteins but it is feasible only when the crystal structure of the target enzyme/protein is known with good resolution. Docking studies are also used for virtual screening of databases. But the ideal technique for virtual screening of compounds is through pharmacophore mapping and screening, especially when the structure of the target is not known. Very large databases can be first screened by pharmacophorebecause the technique is quite fast followed by screening of the positive hits using docking studies. Insilico designing of novel compounds can also be performed using deNovodesigning techniques subject to the condition that the target structure in known.
Yadav M R. New drug discovery: Where are we heading to?. J Adv Pharm Technol Res 2013;4:2-3
URL:
Yadav M R. New drug discovery: Where are we heading to?. J Adv Pharm Technol Res [serial online] 2013 [cited 2014 Aug 12];4:2-3. Available from: http://www.japtr.org/text.asp?2013/4/1/2/107493
SAN DIEGO,Aug. 11, 2014/PRNewswire/ — Mirati Therapeutics, Inc. (NASDAQ:MRTX) today announced that the U.S. FDA has granted Orphan Drug Designation to mocetinostat, a spectrum selective HDAC inhibitor, for diffuse large B-cell lymphoma (DLBCL). In June, mocetinostat was granted Orphan Drug Designation as a treatment for myelodysplastic syndrome (MDS). Orphan drug designation is also being sought for bladder cancer patients with specific genetic alterations.
Mocetinostat is an orally-bioavailable, spectrum-selective HDAC inhibitor. Mocetinostat is enrolling patients in a Phase 2 dose confirmation study in combination with Vidaza as treatment for intermediate and high-risk MDS. Mirati also plans to initiate Phase 2 studies of mocetinostat as a single agent in patients with mutations in histone acetyl transferases in bladder cancer and DLBCL. Initial data from the Phase 2 studies is expected by the end of 2014. In addition to the ongoing Phase 2 clinical trials, mocetinostat has completed 13 clinical trials in more than 400 patients with a variety of hematologic malignancies and solid tumors.
About Mirati Therapeutics
Mirati Therapeutics is a targeted oncology company developing an advanced pipeline of breakthrough medicines for precisely defined patient populations. Mirati’s approach combines the three most important factors in oncology drug development – drug candidates with complementary and compelling targets, creative and agile clinical development, and a highly accomplished precision medicine leadership team. The Mirati team is using a proven blueprint for developing targeted oncology medicines to advance and maximize the value of its pipeline of drug candidates, including MGCD265 and MGCD516, which are orally bioavailable, multi-targeted kinase inhibitors with distinct target profiles, and mocetinostat, an orally bioavailable, spectrum-selective histone deacetylase inhibitor. More information is available atwww.mirati.com.
In eukaryotic cells, nuclear DNA associates with histones to form a compact complex called chromatin. The histones constitute a family of basic proteins which are generally highly conserved across eukaryotic species. The core histones, termed H2A, H2B, H3, and H4, associate to form a protein core. DNA winds around this protein core, with the basic amino acids of the histones interacting with the negatively charged phosphate groups of the DNA. Approximately 146 base pairs of DNA wrap around a histone core to make up a nucleosome particle, the repeating structural motif of chromatin.
Csordas, Biochem. J., 286: 23-38 (1990) teaches that histones are subject to posttranslational acetylation of the α,ε-amino groups of N-terminal lysine residues, a reaction that is catalyzed by histone acetyl transferase (HAT1). Acetylation neutralizes the positive charge of the lysine side chain, and is thought to impact chromatin structure. Indeed, Taunton et al., Science, 272: 408-411 (1996), teaches that access of transcription factors to chromatin templates is enhanced by histone hyperacetylation. Taunton et al. further teaches that an enrichment in underacetylated histone H4 has been found in transcriptionally silent regions of the genome.
Histone acetylation is a reversible modification, with deacetylation being catalyzed by a family of enzymes termed histone deacetylases (HDACs). Grozinger et al., Proc. Natl. Acad. Sci. USA, 96: 4868-4873 (1999), teaches that HDACs are divided into two classes, the first represented by yeast Rpd3-like proteins, and the second represented by yeast Hda1-like proteins. Grozinger et al. also teaches that the human HDAC1, HDAC2, and HDAC3 proteins are members of the first class of HDACs, and discloses new proteins, named HDAC4, HDAC5, and HDAC6, which are members of the second class of HDACs. Kao et al., Genes & Dev., 14: 55-66 (2000), discloses HDAC7, a new member of the second class of HDACs. More recently, Hu et al. J. Bio. Chem. 275:15254-13264 (2000) and Van den Wyngaert, FEBS, 478: 77-83 (2000) disclose HDAC8, a new member of the first class of HDACs.
Richon et al., Proc. Natl. Acad. Sci. USA, 95: 3003-3007 (1998), discloses that HDAC activity is inhibited by trichostatin A (TSA), a natural product isolated from Streptomyces hygroscopicus, and by a synthetic compound, suberoylanilide hydroxamic acid (SAHA). Yoshida and Beppu, Exper. Cell Res., 177: 122-131 (1988), teaches that TSA causes arrest of rat fibroblasts at the G1 and G2 phases of the cell cycle, implicating HDAC in cell cycle regulation. Indeed, Finnin et al., Nature, 401: 188-193 (1999), teaches that TSA and SAHA inhibit cell growth, induce terminal differentiation, and prevent the formation of tumors in mice. Suzuki et al., U.S. Pat. No. 6,174,905, EP 0847992, JP 258863/96, and Japanese Application No. 10138957, disclose benzamide derivatives that induce cell differentiation and inhibit HDAC. Delorme et al., WO 01/38322 and PCT/IB01/00683, disclose additional compounds that serve as HDAC inhibitors.
The molecular cloning of gene sequences encoding proteins with HDAC activity has established the existence of a set of discrete HDAC enzyme isoforms. Some isoforms have been shown to possess specific functions, for example, it has been shown that HDAC-6 is involved in modulation of microtubule activity. However, the role of the other individual HDAC enzymes has remained unclear.
These findings suggest that inhibition of HDAC activity represents a novel approach for intervening in cell cycle regulation and that HDAC inhibitors have great therapeutic potential in the treatment of cell proliferative diseases or conditions. To date, few inhibitors of histone deacetylase are known in the art.
Example 426Synthesis of N-(2-Amino-phenyl)-4-[(4-pyridin-3-pyrimidin-2-ylamino)-methyl]-benzamide
Step 1: Synthesis of 4-Guanidinomethyl-benzoic acid methyl ester Intermediate 1
The mixture of 4-Aminomethyl-benzoic acid methyl ester HCl (15.7 g, 77.8 mmol) in DMF (85.6 mL) and DIPEA (29.5 mL, 171.2 mmol) was stirred at rt for 10 min. Pyrazole-1-carboxamidine HCl (12.55 g, 85.6 mmol) was added to the reaction mixture and then stirred at rt for 4 h to give clear solution. The reaction mixture was evaporated to dryness under vacuum. Saturated NaHCO3solution (35 mL) was added to give nice suspension. The suspension was filtered and the filter cake was washed with cold water. The mother liquid was evaporated to dryness and then filtered. The two solids were combined and re-suspended over distilled H2O (50 ml). The filter cake was then washed with minimum quantities of cold H2O and ether to give 12.32 g white crystalline solid intermediate 1 (77% yield, M+1: 208 on MS).
Step 2: Synthesis of 3-Dimethylamino-1-pyridin-3-yl-propenone Intermediate 2
3-Acetyl-pyridine (30.0 g, 247.6 mmol) and DMF dimethyl acetal (65.8 mL, 495.2 mmol) were mixed together and then heated to reflux for 4 h. The reaction mixture was evaporated to dryness and then 50 mL diethyl ether was added to give brown suspension. The suspension was filtered to give 36.97 g orange color crystalline product (85% yield, M+1: 177 on MS).
Step 3: Synthesis of 4-[(4Pyridin-3-pyrimidin-2-ylamino)-methyl]benzoic acid methyl ester Intermediate 3
Intermediate 1 (0.394 g, 1.9 mmol) and intermediate 2 (0.402 g, 2.3 mmol) and molecular sieves (0.2 g, 4A, powder, >5 micron) were mixed with isopropyl alcohol (3.8 mL). The reaction mixture was heated to reflux for 5 h. MeOH (50 mL) was added and then heated to reflux. The cloudy solution was filtrated over a pad of celite. The mother liquid was evaporated to dryness and the residue was triturated with 3 mL EtOAc. The suspension was filtrated to give 0.317 g white crystalline solid Intermediate 3 (52%, M+1: 321 on MS).
Step 4: Synthesis of N-(2-Amino-phenyl)-4-[(4-pyrymidin-2-ylamino)-methyl]-benzamide
Intermediate 3 (3.68 g, 11.5 mmol) was mixed with THF (23 mL), MeOH (23 mL) and H2O (11.5 mL) at rt. LiOH (1.06 g, 25.3 mmol) was added to reaction mixture. The resulting reaction mixture was warmed up to 40° C. overnight. HCl solution (12.8 mL, 2N) was added to adjust pH=3 when the mixture was cooled down to rt. The mixture was evaporated to dryness and then the solid was washed with minimum quantity of H2O upon filtration. The filter cake was dried over freeze dryer to give 3.44 g acid of the title compound (95%, M+1: 307 on MS).
Acid (3.39 g, 11.1 mmol) of the title compound, BOP (5.679 g, 12.84 mmol) and o-Ph(NH2)2(2.314 g, 21.4 mmol) were dissolved in the mixture of DMF (107 mL) and Et3N (2.98 mL, 21.4 mmol). The reaction mixture was stirred at rt for 5 h and then evaporated to dryness. The residue was purified by flash column (pure EtOAc to 5% MeOH/EtOAc) and then interested fractions were concentrated. The final product was triturated with EtOAc to give 2.80 g of title product
Pfefferli, Catherine; Müller, Fritz; Ja¿wi¿ska, Anna; Wicky, Chantal (2014). “Specific NuRD components are required for fin regeneration in zebrafish”.BMC Biol.12(30).doi:10.1186/1741-7007-12-30.PMID24779377.
Alnylam Pharmaceuticals, Inc., a leading RNAi therapeutics company, announced today positive top-line results from its ongoing Phase 1 trial of ALN-AT3, a subcutaneously administered RNAi therapeutic targeting antithrombin (AT) in development for the treatment of hemophilia and rare bleeding disorders (RBD). These top-line results are being presented at the World Federation of Hemophilia (WFH) 2014 World Congress being held May 11 – 15, 2014 in Melbourne, Australia. In Part A of the Phase 1 study, human volunteer subjects received a single subcutaneous dose of ALN-AT3 and, per protocol, the maximum allowable level of AT knockdown was set at 40%. Initial results show that a single, low subcutaneous dose of ALN-AT3 at 0.03 mg/kg resulted in an up to 28-32% knockdown of AT at nadir that was statistically significant relative to placebo (p < 0.01 by ANOVA). This led to a statistically significant (p < 0.01) increase in peak thrombin generation, that was temporally associated and consistent with the degree of AT knockdown. ALN-AT3 was found to be well tolerated with no significant adverse events reported. With these data, the company has transitioned to the Multiple Ascending Dose (MAD) Part B of the study in moderate-to-severe hemophilia subjects. Consistent with previous guidance, the company plans to present initial clinical results from the Phase 1 study, including results in hemophilia subjects, by the end of the year. These human study results are the first to be reported for Alnylam’s Enhanced Stabilization Chemistry (ESC)-GalNAc conjugate technology, which enables subcutaneous dosing with increased potency, durability, and a wide therapeutic index. Further, these initial clinical results demonstrate a greater than 50-fold potency improvement with ESC-GalNAc conjugates relative to standard template chemistry conjugates.
“We are excited by these initial positive results for ALN-AT3 in the human volunteer ‘Part A’ of our Phase 1 study. Indeed, within the protocol-defined boundaries of single doses that provide no more than a 40% knockdown of AT in normal subjects, we were able to demonstrate a statistically-significant knockdown of AT of up to 28-32% and an associated increase in thrombin generation. Remarkably, this result was achieved at the lowest dose tested of 0.03 mg/kg, demonstrating a high and better than expected level of potency for ALN-AT3, our first ESC-GalNAc conjugate to enter clinical development,” said Akshay Vaishnaw, M.D., Ph.D., Executive Vice President and Chief Medical Officer of Alnylam. “With these results in hand, we are now proceeding to ‘Part B’ of the study, where we will administer multiple ascending doses to up to 18 patients with moderate-to-severe hemophilia A or B. Patients will receive three weekly doses, and we fully expect to achieve robust levels of AT knockdown as we dose escalate. In addition, we will aim to evaluate a once-monthly dosing regimen in future clinical studies, as we believe this could provide a highly attractive prophylactic regimen for patients. We look forward to sharing our detailed Phase 1 results, including data in hemophilia subjects, later this year, consistent with our original guidance.”
“There are several notable implications of these exciting initial results with ALN-AT3. First, ALN-AT3 now becomes the fourth program in our ‘Alnylam 5×15’ pipeline to demonstrate clinical activity. As such, these results increase our confidence level yet further across the entirety of our pipeline efforts, where we remain focused on genetically defined, liver-expressed disease targets with a modular and reproducible delivery platform. Moreover, these results with ALN-AT3 establish human proof of concept for our ESC-GalNAc conjugate technology, extending and broadening the human results we have previously shown with ALN-TTRsc which employs our standard template chemistry. Our ESC-GalNAc conjugate technology enables subcutaneous dosing with increased potency and durability and a wide therapeutic index, and has now become our primary approach for the delivery of RNAi therapeutics,” said John Maraganore, Ph.D., Chief Executive Officer of Alnylam. “Finally, the achievement of target knockdown at such a low dose of 0.03 mg/kg is unprecedented. Based on our evaluation of datasets from non-human primate (NHP) and human studies, these results demonstrate a 10-fold improved potency for ALN-AT3 as compared with NHP and a 50-fold improved potency in humans as compared with ALN-TTRsc. Based on data we announced earlier this week at TIDES, we believe that this increased potency is the combined result of enhanced stability for ESC-GalNAc conjugates and an attenuated nuclease environment in human tissue compared with other species. If these results extend to other ESC-GalNAc-siRNA conjugates, such as those in our complement C5 and PCSK9 programs, we believe we can expect highly potent clinical activities with very durable target knockdown effects.”
The ongoing Phase 1 trial of ALN-AT3 is being conducted in the U.K. as a single- and multi-dose, dose-escalation study comprised of two parts. Part A – which has now been completed – was a randomized, single-blind, placebo-controlled, single-dose, dose-escalation study, intended to enroll up to 24 healthy volunteer subjects. The primary objective of this part of the study was to evaluate the safety and tolerability of a single dose of ALN-AT3, with the potential secondarily to show changes in AT plasma levels at sub-pharmacologic doses. This part of the study evaluated only low doses of ALN-AT3, with a dose-escalation stopping rule at no more than a 40% level of AT knockdown. Based on the pharmacologic response achieved in this part of the study, only the lowest dose cohort (n=4; 3:1 randomization of ALN-AT3:placebo) was enrolled. Part B of the study is an open-label, multi-dose, dose-escalation study enrolling up to 18 people with moderate-to-severe hemophilia A or B. The primary objective of this part of the study is to evaluate the safety and tolerability of multiple doses, specifically three doses, of subcutaneously administered ALN-AT3 in hemophilia subjects. Secondary objectives include assessment of clinical activity as determined by knockdown of circulating AT levels and increase in thrombin generation at pharmacologic doses of ALN-AT3; thrombin generation is known to be a biomarker for bleeding frequency and severity in people with hemophilia (Dargaud, et al., Thromb Haemost; 93, 475-480 (2005)). In this part of the study, dose-escalation will be allowed to proceed beyond the 40% AT knockdown level.
In addition to reporting positive top-line results from the Phase 1 trial with ALN-AT3, Alnylam presented new pre-clinical data with ALN-AT3. First, in a saphenous vein bleeding model performed in hemophilia A (HA) mice, a single subcutaneous dose of ALN-AT3 that resulted in an approximately 70% AT knockdown led to a statistically significant (p < 0.0001) improvement in hemostasis compared to saline-treated HA mice. The improved hemostasis was comparable to that observed in HA mice receiving recombinant factor VIII. These are the first results in what can be considered a genuine bleeding model showing that AT knockdown with ALN-AT3 can control bleeding. Second, a number of in vitro studies were performed in plasma from hemophilia donors. Stepwise AT depletion in these plasma samples was shown to achieve stepwise increases in thrombin generation. Furthermore, it was shown that a 40-60% reduction of AT resulted in peak thrombin levels equivalent to those achieved with 10-15% levels of factor VIII in HA plasma and factor IX in hemophilia B (HB) plasma. These levels of factor VIII or IX are known to significantly reduce bleeding in hemophilia subjects. As such, these results support the hypothesis that a 40-60% knockdown of AT with ALN-AT3 could be fully prophylactic. Finally, a modified Activated Partial Thromboplastin Time (APTT) assay – an ex vivomeasure of blood coagulation that is significantly prolonged in hemophilia – was developed, demonstrating sensitivity to AT levels. Specifically, depletion of AT in HA plasma led to a shortening of modified APTT. This modified APTT assay can be used to routinely and simply monitor functional activity of AT knockdown in further ALN-AT3 clinical studies.
“The unmet need for new therapeutic options to treat hemophilia patients remains very high, particularly in those patients who experience multiple annual bleeds such as patients receiving replacement factor ‘on demand’ or patients who have developed inhibitory antibodies. Indeed, I believe the availability of a safe and effective subcutaneously administered therapeutic with a long duration of action would represent a marked improvement over currently available approaches for prophylaxis,” said Claude Negrier, M.D., head of the Hematology Department and director of the Haemophilia Comprehensive Care Centre at Edouard Herriot University Hospital in Lyon. “I continue to be encouraged by Alnylam’s progress to date with ALN-AT3, including these initial data reported from the Phase 1 trial showing statistically significant knockdown of antithrombin and increased thrombin generation, which has been shown to correlate with bleeding frequency and severity in hemophilia. I look forward to the advancement of this innovative therapeutic candidate in hemophilia subjects.”
About Hemophilia and Rare Bleeding Disorders
Hemophilias are hereditary disorders caused by genetic deficiencies of various blood clotting factors, resulting in recurrent bleeds into joints, muscles, and other major internal organs. Hemophilia A is defined by loss-of-function mutations in Factor VIII, and there are greater than 40,000 registered patients in the U.S. and E.U. Hemophilia B, defined by loss-of-function mutations in Factor IX, affects greater than 9,500 registered patients in the U.S. and E.U. Other Rare Bleeding Disorders (RBD) are defined by congenital deficiencies of other blood coagulation factors, including Factors II, V, VII, X, and XI, and there are about 1,000 patients worldwide with a severe bleeding phenotype. Standard treatment for hemophilia patients involves replacement of the missing clotting factor either as prophylaxis or on-demand therapy. However, as many as one third of people with severe hemophilia A will develop an antibody to their replacement factor – a very serious complication; these ‘inhibitor’ patients become refractory to standard replacement therapy. There exists a small subset of hemophilia patients who have co-inherited a prothrombotic mutation, such as Factor V Leiden, antithrombin deficiency, protein C deficiency, and prothrombin G20210A. Hemophilia patients that have co-inherited these prothrombotic mutations are characterized as having a later onset of disease, lower risk of bleeding, and reduced requirements for Factor VIII or Factor IX treatment as part of their disease management. There exists a significant need for novel therapeutics to treat hemophilia patients.
About Antithrombin (AT)
Antithrombin (AT, also known as “antithrombin III” and “SERPINC1″) is a liver expressed plasma protein and member of the “serpin” family of proteins that acts as an important endogenous anticoagulant by inactivating Factor Xa and thrombin. AT plays a key role in normal hemostasis, which has evolved to balance the need to control blood loss through clotting with the need to prevent pathologic thrombosis through anticoagulation. In hemophilia, the loss of certain procoagulant factors (Factor VIII and Factor IX, in the case of hemophilia A and B, respectively) results in an imbalance of the hemostatic system toward a bleeding phenotype. In contrast, in thrombophilia (e.g., Factor V Leiden, protein C deficiency, antithrombin deficiency, amongst others), certain mutations result in an imbalance in the hemostatic system toward a thrombotic phenotype. Since co-inheritance of prothrombotic mutations may ameliorate the clinical phenotype in hemophilia, inhibition of AT defines a novel strategy for improving hemostasis.
About GalNAc Conjugates and Enhanced Stabilization Chemistry (ESC)-GalNAc Conjugates
GalNAc-siRNA conjugates are a proprietary Alnylam delivery platform and are designed to achieve targeted delivery of RNAi therapeutics to hepatocytes through uptake by the asialoglycoprotein receptor. Alnylam’s Enhanced Stabilization Chemistry (ESC)-GalNAc-conjugate technology enables subcutaneous dosing with increased potency and durability, and a wide therapeutic index. This delivery platform is being employed in several of Alnylam’s genetic medicine programs, including programs in clinical development.
About RNAi
RNAi (RNA interference) is a revolution in biology, representing a breakthrough in understanding how genes are turned on and off in cells, and a completely new approach to drug discovery and development. Its discovery has been heralded as “a major scientific breakthrough that happens once every decade or so,” and represents one of the most promising and rapidly advancing frontiers in biology and drug discovery today which was awarded the 2006 Nobel Prize for Physiology or Medicine. RNAi is a natural process of gene silencing that occurs in organisms ranging from plants to mammals. By harnessing the natural biological process of RNAi occurring in our cells, the creation of a major new class of medicines, known as RNAi therapeutics, is on the horizon. Small interfering RNA (siRNA), the molecules that mediate RNAi and comprise Alnylam’s RNAi therapeutic platform, target the cause of diseases by potently silencing specific mRNAs, thereby preventing disease-causing proteins from being made. RNAi therapeutics have the potential to treat disease and help patients in a fundamentally new way.
About Alnylam Pharmaceuticals
Alnylam is a biopharmaceutical company developing novel therapeutics based on RNA interference, or RNAi. The company is leading the translation of RNAi as a new class of innovative medicines with a core focus on RNAi therapeutics as genetic medicines, including programs as part of the company’s “Alnylam 5x15TM” product strategy. Alnylam’s genetic medicine programs are RNAi therapeutics directed toward genetically defined targets for the treatment of serious, life-threatening diseases with limited treatment options for patients and their caregivers. These include: patisiran (ALN-TTR02), an intravenously delivered RNAi therapeutic targeting transthyretin (TTR) for the treatment of TTR-mediated amyloidosis (ATTR) in patients with familial amyloidotic polyneuropathy (FAP); ALN-TTRsc, a subcutaneously delivered RNAi therapeutic targeting TTR for the treatment of ATTR in patients with TTR cardiac amyloidosis, including familial amyloidotic cardiomyopathy (FAC) and senile systemic amyloidosis (SSA); ALN-AT3, an RNAi therapeutic targeting antithrombin (AT) for the treatment of hemophilia and rare bleeding disorders (RBD); ALN-CC5, an RNAi therapeutic targeting complement component C5 for the treatment of complement-mediated diseases; ALN-AS1, an RNAi therapeutic targeting aminolevulinate synthase-1 (ALAS-1) for the treatment of hepatic porphyrias including acute intermittent porphyria (AIP); ALN-PCS, an RNAi therapeutic targeting PCSK9 for the treatment of hypercholesterolemia; ALN-AAT, an RNAi therapeutic targeting alpha-1 antitrypsin (AAT) for the treatment of AAT deficiency-associated liver disease; ALN-TMP, an RNAi therapeutic targeting TMPRSS6 for the treatment of beta-thalassemia and iron-overload disorders; ALN-ANG, an RNAi therapeutic targeting angiopoietin-like 3 (ANGPTL3) for the treatment of genetic forms of mixed hyperlipidemia and severe hypertriglyceridemia; ALN-AC3, an RNAi therapeutic targeting apolipoprotein C-III (apoCIII) for the treatment of hypertriglyceridemia; and other programs yet to be disclosed. As part of its “Alnylam 5×15” strategy, as updated in early 2014, the company expects to have six to seven genetic medicine product candidates in clinical development – including at least two programs in Phase 3 and five to six programs with human proof of concept – by the end of 2015. Alnylam is also developing ALN-HBV, an RNAi therapeutic targeting the hepatitis B virus (HBV) genome for the treatment of HBV infection. The company’s demonstrated commitment to RNAi therapeutics has enabled it to form major alliances with leading companies including Merck, Medtronic, Novartis, Biogen Idec, Roche, Takeda, Kyowa Hakko Kirin, Cubist, GlaxoSmithKline, Ascletis, Monsanto, The Medicines Company, and Genzyme, a Sanofi company. In March 2014, Alnylam acquired Sirna Therapeutics, a wholly owned subsidiary of Merck. In addition, Alnylam holds an equity position in Regulus Therapeutics Inc., a company focused on discovery, development, and commercialization of microRNA therapeutics. Alnylam scientists and collaborators have published their research on RNAi therapeutics in over 200 peer-reviewed papers, including many in the world’s top scientific journals such as Nature, Nature Medicine, Nature Biotechnology, Cell, the New England Journal of Medicine, and The Lancet. Founded in 2002, Alnylam maintains headquarters in Cambridge, Massachusetts. For more information, please visit www.alnylam.com.
Alnodesertib CAS 2267316-76-5 MF C18H24N6O2S MW388.49 4-[4-[(cyclopropyl-methyl-oxo-λ6-sulfanylidene)amino]-6-[(3R)-3-methylmorpholin-4-yl]pyrimidin-2-yl]pyridin-2-amine 4-[4-[(cyclopropyl-methyl-oxo-lambda6-sulfanylidene)amino]-6-[(3R)-3-methylmorpholin-4-yl]pyrimidin-2-yl]pyridin-2-amine (S)-({2-(2-aminopyridin-4-yl)-6-[(3R)-3-methylmorpholin-4-yl]pyrimidin-4-yl}imino)(cyclopropyl)(methyl)-λ6-sulfanoneserine/threonine kinase inhibitor, antineoplastic, ART 0380, EX-A9085 Alnodesertib (formerly known as ART0380) is an investigational, orally administered drug designed to…
Zelebrudomide CAS 2416131-46-7 MF C39H45N9O5 MW 719.8 g/mol 3-[4-[1-[[(3S)-1-[2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindol-5-yl]pyrrolidin-3-yl]methyl]piperidin-4-yl]anilino]-5-piperidin-1-ylpyrazine-2-carboxamide 3-[[4-[1-[[(3S)-1-[2-(2,6-Dioxo-3-piperidyl)-1,3-dioxo-5-isoindolinyl]-3-pyrrolidinyl]methyl]-4-piperidyl]phenyl]amino]-5-(1-piperidyl)pyrazine-2-carboxamide protein degrader, antineoplastic, NX 2127, LSC67HA8DE, NX-2127, BTK Degrader NX-2127 Zelebrudomide (NX-2127) is an investigational new drug that is…
Vormatrigine CAS 2392951-18-5 MF C16H12F6N4O2 MW406.28 g/mol 3-(ethoxydifluoromethyl)-6-(5-fluoro-6-(2,2,2-trifluoroethoxy)pyridin-3-yl)-[1,2,4]triazolo[4,3-a]pyridine 3-[ethoxydi(fluoro)methyl]-6-[5-fluoro-6-(2,2,2-trifluoroethoxy)pyridin-3-yl][1,2,4]triazolo[4,3-a]pyridinesodium channel blocker, PRAX-628, PRAX 628, QU3C48T4NV, Vormatrigine is a small molecule drug. The usage of the INN…
Ulacamten CAS 2830607-59-3 MF C21H25F2N3O3 MW405.4 g/mol 5-[(3,4-difluorophenyl)methyl]-8-(4-methylcyclohexyl)-6,9-dioxo-2,5,8-triazaspiro[3.5]nonane-2-carbaldehyde 5-[(3,4-difluorophenyl)methyl]-8-[(1r,4r)-4-methylcyclohexyl]-6,9-dioxo-2,5,8-triazaspiro[3.5]nonane-2-carbaldehydecardiac myosin inhibitor, CK-586, CK-4021586, CK 586, CK 4021586, X325G97HZJ Ulacamten (also known as CK-586 or CK-4021586) is an investigational drug developed by Cytokinetics that…
I , Dr A.M.Crasto is writing this blog to share the knowledge/views, after reading Scientific Journals/Articles/News Articles/Wikipedia. My views/comments are based on the results /conclusions by the authors(researchers). I do mention either the link or reference of the article(s) in my blog and hope those interested can read for details. I am briefly summarising the remarks or conclusions of the authors (researchers). If one believe that their intellectual property right /copyright is infringed by any content on this blog, please contact or leave message at below email address amcrasto@gmail.com. It will be removed ASAP

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO