














| Patent | Submitted | Granted |
|---|---|---|
| BENZIMIDAZOLE MODULATORS OF VR1 [US2011190344] | 2011-08-04 | |
| BENZIMIDAZOLE MODULATORS OF VR1 [US2011190364] | 2011-08-04 | |
| Benzimidazole Modulators of VR1 [US7951829] | 2007-11-08 | 2011-05-31 |
FLAGS AND HITS
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
Join me on Linkedin
Join me on Researchgate
Join me on Facebook
 FACEBOOK
...................................................................Join me on twitter
FACEBOOK
...................................................................Join me on twitter
 ..................................................................Join me on google plus
..................................................................Join me on google plus
 Googleplus
Googleplus
FACEBOOK
...................................................................Join me on twitter
..................................................................Join me on google plus
GoogleplusMYSELF
DR ANTHONY MELVIN CRASTO Ph.D ( ICT, Mumbai)
, INDIA
36Yrs Exp. in the feld of Organic Chemistry,Working for AFRICURE PHARMA as ADVISOR earlier with GLENMARK PHARMA at Navi Mumbai, INDIA. Serving chemists around the world. Helping them with websites on Chemistry.Million hits on google,
NO ADVERTISEMENTS , ACADEMIC , NON COMMERCIAL SITE, world acclamation from industry, academia, drug authorities for websites, blogs and educational contribution, ........amcrasto@gmail.com..........+91 9323115463, Skype amcrasto64


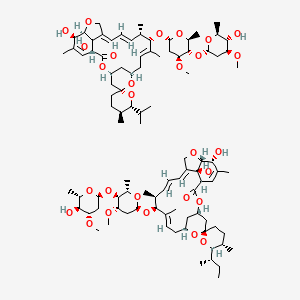
Ivermectin
IVERMECTIN
MK933
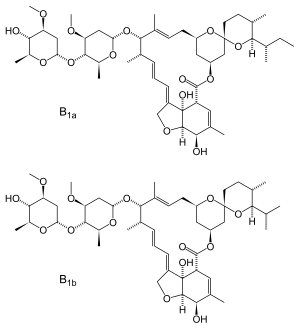
22,23-dihydroavermectin B1a + 22,23-dihydroavermectin B1b
70288-86-7 ![]() 71827-03-7
71827-03-7
| C95H146O28 | |
| Molecular Weight: | 1736.15894 g/mol |
|---|
 C48H74O14, 875.09
C48H74O14, 875.09
Ivermectin is a macrocyclic lactone derived from Streptomyces avermitilis with antiparasitic activity. Ivermectin exerts its anthelmintic effect via activating glutamate-gated chloridechannels expressed on nematode neurons and pharyngeal muscle cells. Distinct from the channel opening induced by endogenous glutamate transmitter, ivermectin-activated channels open very slowly but essentially irreversibly. As a result, neurons or muscle cells remain at either hyperpolarisation or depolarization state, thereby resulting in paralysis and death of the parasites. Ivermectin does not readily pass the mammal blood-brain barrier to the central nervous system where glutamate-gated chloride channels locate, hence the hosts are relatively resistant to the effects of this agent.
This drug, ivermectin, was developed by William C. Campbell of Drew University and Satoshi Ōmura of Japan’s Kitasato University. They were awarded the Nobel Prize in Physiology or Medicine. Originally, the drug was used to treat parasites in livestock and pets before becoming the mainstay of the global campaigns to combat lymphatic filariasis and onchocerciasis.
A workhorse of a drug that a few weeks ago earned its developers a Nobel prize for its success in treating multiple tropical diseases is showing early promise as a novel and desperately needed tool for interrupting malaria transmission, according to new findings presented today at the American Society of Tropical Medicine and Hygiene (ASTMH) Annual Meeting.
At ASTMH annual meeting, new studies explore advances in using ivermectin in ‘mass drug administration’ campaigns to reduce infections in Africa and slow spread of drug resistance in Asia…http://www.pharmpro.com/news/2015/10/nobel-prize-winning-drug-could-also-fight-malaria?et_cid=4908183&et_rid=577220619&type=cta
This new finding was presented today at the American Society of Tropical Medicine and Hygiene (ASTMH) Annual Meeting by researchers from Colorado State University.
Ivermectin has been used for decades, given once per year as a part of Mass Drug Administration (MDA) programs, to reduce the disabling worm infections onchocerciasis, which causes river blindness, and filariasis, the cause of the hugely swollen legs (elephantiasis). Merck has generously donated the entire supply of drug; other companies have followed suit with different drugs for other neglected tropical diseases.
Ivermectin (22,23-dihydroavermectin B1a + 22,23-dihydroavermectin B1b) is a broad-spectrum antiparasitic drug in theavermectin family. It is sold under brand names Heartgard, Sklice[1] and Stromectol[2] in the United States, Ivomecworldwide by Merial Animal Health, Mectizan in Canada by Merck, Iver-DT[3] in Nepal by Alive Pharmaceutical and Ivextermin Mexico by Valeant Pharmaceuticals International. In Southeast Asian countries, it is marketed by Delta Pharma Ltd. under the trade name Scabo 6. While in development, it was assigned the code MK-933 by Merck.[4]
It is taken internally or used topically, depending on the treated condition.
It is on the World Health Organization’s List of Essential Medicines, a list of the most important medication needed in a basichealth system.[5]
It is a drug for the treatment of Onchocerciasis.
The disease is also known as river blindness. It is sometimes called Robles’ disease, after the Guatemalan doctor Rodolfo Robles, who first linked the blindness with an insect a century ago (1915).
Medical uses
Ivermectin is a broad-spectrum antiparasitic agent, traditionally against parasitic worms. It is mainly used in humans in the treatment of onchocerciasis (river blindness), but is also effective against other worm infestations (such as strongyloidiasis,ascariasis, trichuriasis, filariasis and enterobiasis), and some epidermal parasitic skin diseases, including scabies.
Ivermectin is currently being used to help eliminate river blindness (onchocerciasis) in the Americas, and to stop transmissionof lymphatic filariasis and onchocerciasis around the world in programs sponsored by the Carter Center using ivermectin donated by Merck.[6][7][8] The disease is endemic in 30 African countries, six Latin American countries, and Yemen, according to studies conducted by the World Health Organization.[9] The drug rapidly kills microfilariae, but not the adult worms. A single oral dose of ivermectin, taken annually for the 10- to 15-year lifespan of the adult worms, is all that is needed to protect the individual from onchocerciasis.[10]
An Ivermectin cream called Soolantra has been approved by the FDA for treatment of rosacea.[11][12]
SOOLANTRA (ivermectin) cream, 1% is a white to pale yellow hydrophilic cream. Each gram of SOOLANTRA cream contains 10 mg of ivermectin. It is intended for topical use.
Ivermectin is a semi-synthetic derivative isolated from the fermentation of Streptomyces avermitilis that belongs to the avermectin family of macrocyclic lactones.
Ivermectin is a mixture containing not less than 95.0 % and not more than 102.0 % of 5-O-demethyl-22,23-dihydroavermectin A1a plus 5-O-demethyl-25-de(1-methylpropyl)-25-(1-methylethyl)-22,23-dihydroavermectin A1a, generally referred to as 22,23-dihydroavermectin B1a and B1b or H2B1a and H2B1b, respectively; and the ratio (calculated by area percentage) of component H2B1a/(H2B1a + H2B1b)) is not less than 90.0 %.
The respective empirical formulas of H2B1a and H2B1b are C48H74O14and C47H72O14 with molecular weights of 875.10 and 861.07 respectively.
The structural formulas are:
 |
Component H2B1a: R = C2H5, Component H2B1b: R = CH3.SOOLANTRA cream contains the following inactive ingredients: carbomer copolymer type B, cetyl alcohol, citric acid monohydrate, dimethicone, edetate disodium, glycerin, isopropyl palmitate, methylparaben, oleyl alcohol, phenoxyethanol, polyoxyl 20 cetostearyl ether, propylene glycol, propylparaben, purified water, sodium hydroxide, sorbitan monostearate, and stearyl alcohol.
River blindness?
The disease is also known as river blindness. It is sometimes called Robles’ disease, after the Guatemalan doctor Rodolfo Robles, who first linked the blindness with an insect a century ago (1915).

The infection is associated with a nematode worm Onchocerca volvulus, which are transmitted by Simulium blackflies which live and breed near fast-flowing streams and rivers. The worms carry parasitic Wolbachia bacteria. The bite of the flies enables the worm larvae to enter the human’s body; after maturing into adults, followed by breeding, the larvae (microfilariae) formed move towards the skin, and release the bacteria when they die. The bacteria trigger an immune response which leads to lesions on the eye and possible blindness (the “river blindness”).


Left: the Simulium fly (from http://flipper.diff.org/app/items/6730). Right: Simplified life cycle of Onchocerciasis volvulus, modified from the original at: http://emedicine.medscape.com/article/224309-overview#a0104.
Arthropod
More recent evidence supports its use against parasitic arthropods and insects:
- Mites such as scabies:[13][14][15] It is usually limited to cases that prove to be resistant to topical treatments or that present in an advanced state (such as Norwegian scabies).[15]
- Lice:[16][17] Ivermectin lotion (0.5%) is FDA-approved for patients six months of age and older.[18] After a single, 10-minute application of this formulation on dry hair, 78% of subjects were found to be free of lice after two weeks.[19] This level of effectiveness is equivalent to other pediculicide treatments requiring two applications.[20]
- Bed bugs:[21] Early research shows that the drug kills bed bugs when taken by humans at normal doses. The drug enters the human bloodstream and if the bedbugs bite during that time, they will die in a few days.
Contraindications
Ivermectin is contraindicated in children under the age of five, or those who weigh less than 15 kg (33 lb);[22] and those who are breastfeeding, and have a hepatic or renal disease.[23]
Side effects
The main concern is neurotoxicity, which in most mammalian species may manifest as central nervous system depression, and consequent ataxia, as might be expected from potentiation of inhibitory GABA-ergic synapses.
Dogs with defects in the P-glycoprotein gene (MDR1), often collie-like herding dogs, can be severely poisoned by ivermectin.
Since drugs that inhibit CYP3A4 enzymes often also inhibit P-glycoprotein transport, the risk of increased absorption past the blood-brain barrier exists when ivermectin is administered along with other CYP3A4 inhibitors. These drugs include statins, HIV protease inhibitors, many calcium channel blockers, and glucocorticoids such as dexamethasone, lidocaine, and the benzodiazepines.[24]
For dogs, the insecticide spinosad may have the effect of increasing the potency of ivermectin.[25]
Pharmacology
Pharmacodynamics
Ivermectin and other avermectins (insecticides most frequently used in home-use ant baits) are macrocyclic lactones derived from the bacterium Streptomyces avermitilis. Ivermectin kills by interfering with nervous system and muscle function, in particular by enhancing inhibitory neurotransmission.
The drug binds and activates glutamate-gated chloride channels (GluCls).[26] GluCls are invertebrate-specific members of the Cys-loop family of ligand-gated ion channelspresent in neurons and myocytes.
Pharmacokinetics
Ivermectin can be given either by mouth or injection. It does not readily cross the blood–brain barrier of mammals due to the presence of P-glycoprotein,[27] (the MDR1 gene mutation affects function of this protein). Crossing may still become significant if ivermectin is given at high doses (in which case, brain levels peak 2–5 hr after administration). In contrast to mammals, ivermectin can cross the blood–brain barrier in tortoises, often with fatal consequences.
Ecotoxicity
Field studies have demonstrated the dung of animals treated with ivermectin supports a significantly reduced diversity of invertebrates, and the dung persists longer.[28]
History
The discovery of the avermectin family of compounds, from which ivermectin is chemically derived, was made by Satoshi Ōmura of Kitasato University, Tokyo and William C. Campbell of the Merck Institute for Therapeutic research. Ōmura identified avermectin from the bacterium Streptomyces avermitilis. Campbell purified avermectin from cultures obtained from Ōmura and led efforts leading to the discovery of ivermectin, a derivative of greater potency and lower toxicity.[29] Ivermectin was introduced in 1981.[30] Half of the 2015 Nobel Prize in Physiology or Medicine was awarded jointly to Campbell and Ōmura for discovering avermectin, “the derivatives of which have radically lowered the incidence of river blindness and lymphatic filariasis, as well as showing efficacy against an expanding number of other parasitic diseases”.[31]
It started with the avermectins. In 1974, a group of researchers headed by Professor Satoshi Ōmura of the Kitasato Institute, isolated an organism with promising antimicrobial properties in a soil sample (sample OS-3153) picked up near a golf course at Kawana, Ito City, Shizuoka Prefecture, Japan. This was passed on to researchers at the Merck, Sharpe and Dohme (MSD) research laboratories in the USA, who isolated a small family of natural products that became known as avermectins. For many years, scientists have looked in soil samples for the source of potential medicines, like the tetracyclines or streptomycin . There are 8 avermectins, molecules with closely related structures. They are made by fermentation from the bacterium Streptomyces avermitilis.
 Professor Satoshi Ōmura
Professor Satoshi Ōmura

The 8 different avermectins, with the differences between them shown in the table below.
| Name | R1 | R2 | X-Y |
|---|---|---|---|
| Avermectin A1a | Me | Et | CH=CH |
| Avermectin A1b | Me | Me | CH=CH |
| Avermectin A2a | Me | Et | CH2CH(OH) |
| Avermectin A2b | Me | Me | CH2CH(OH) |
| Avermectin B1a | H | Et | CH=CH |
| Avermectin B1b | H | Me | CH=CH |
| Avermectin B2a | H | Et | CH2CH(OH) |
| Avermectin B2b | H | Me | CH2CH(OH) |
The avermectins proved to be have biocidal activity against a wide range of parasites – such as roundworms, lungworms, mites, lice and arachnids; one of these parasites is the tick Rhipicephalus (Boophilus) microplus, one of the most important cattle parasites in tropical regions. Those with the -CH=CH- function are the more active; the most potent was Avermectin B1, occurring as an 80:20 mixture of the similar molecules B1a and B1b, particularly the B1a component. Commercially it is known as Abamectin.
Veterinary use
In veterinary medicine ivermectin is used against many intestinal worms (but not tapeworms), most mites, and some lice. Despite this, it is not effective for eliminating ticks, flies, flukes, or fleas. It is effective against larval heartworms, but not against adult heartworms, though it may shorten their lives. The dose of the medicine must be very accurately measured as it is very toxic in over-dosage. It is sometimes administered in combination with other medications to treat a broad spectrum of animal parasites. Some dog breeds (especially the Rough Collie, the Smooth Collie, the Shetland Sheepdog, and the Australian Shepherd), though, have a high incidence of a certain mutation within the MDR1 gene (coding for P-glycoprotein); affected animals are particularly sensitive to the toxic effects of ivermectin.[32][33] Clinical evidence suggests kittens are susceptible to ivermectin toxicity.[34] A 0.01% ivermectin topical preparation for treating ear mites in cats (Acarexx) is available.
Ivermectin is sometimes used as an acaricide in reptiles, both by injection and as a diluted spray. While this works well in some cases, care must be taken, as several species of reptiles are very sensitive to ivermectin. Use in turtles is particularly contraindicated.
 IVERMECTIN
IVERMECTIN
Chlorotris(triphenylphosphine)rhodium(I), [RhCl(PPh3)3]
http://www.chm.bris.ac.uk/motm/wilcat/wilcath.htm
Such selectivity found an important application in the synthesis of Ivermectin (MectizanTM). Avermectin is a naturally-occurring molecule with anthelmintic and insecticidal properties; selectively reducing one double bond using Wilkinson’s catalyst afforded Ivermectin. The resultant small change in molecular shape makes Ivermectin a much more effective drug to combat onchocerciasis (river blindness), a disease which affects many millions of people, mainly in poor African communities.

You need to add just two hydrogen atoms to reduce a C=C bond in avermectin.
Notes and references
- “SKLICE- ivermectin lotion (NDC Code(s): 49281-183-71)”. DailyMed. February 2012. Retrieved 2015-09-09.
- “STROMECTOL- ivermectin tablet (NDC Code(s): 0006-0032-20)”. DailyMed. May 2010. Retrieved 2015-09-09.
- Adhikari, Santosh (2014-05-27). “ALIVE PHARMACEUTICAL (P) LTD.: Iver-DT”. ALIVE PHARMACEUTICAL (P) LTD. Retrieved 2015-10-07.
- Pampiglione S, Majori G, Petrangeli G, Romi R (1985). “Avermectins, MK-933 and MK-936, for mosquito control”. Trans R Soc Trop Med Hyg 79 (6): 797–9. doi:10.1016/0035-9203(85)90121-X. PMID 3832491.
- “WHO Model List of Essential Medicines” (PDF). World Health Organization. October 2013. Retrieved 22 April 2014.
- The Carter Center. “River Blindness (Onchocerciasis) Program”. Retrieved2008-07-17..
- The Carter Center. “Lymphatic Filariasis Elimination Program”. Retrieved 2008-07-17..
- WHO. “African Programme for Onchocerciasis Control”. Retrieved 2009-11-12..
- United Front Against Riverblindness. “Onchocerciasis or Riverblindness”..
- United Front Against Riverblindness. “Control of Riverblindness”..
- Galderma Receives FDA Approval of Soolantra (Ivermectin) Cream for Rosacea“
- “SOOLANTRA- ivermectin cream (NDC Code(s): 0299-3823-30, 0299-3823-45, 0299-3823-60)”. DailyMed. December 2014. Retrieved 2015-09-09.
- Brooks PA, Grace RF (August 2002). “Ivermectin is better than benzyl benzoate for childhood scabies in developing countries”. J Paediatr Child Health 38 (4): 401–4.doi:10.1046/j.1440-1754.2002.00015.x. PMID 12174005.
- Victoria J, Trujillo R (2001). “Topical ivermectin: a new successful treatment for scabies”. Pediatr Dermatol 18 (1): 63–5. doi:10.1046/j.1525-1470.2001.018001063.x.PMID 11207977.
- ^ Jump up to:a b Strong M, Johnstone PW (2007). Strong, Mark, ed. “Interventions for treating scabies”. Cochrane Database of Systematic Reviews (Online) (3): CD000320.doi:10.1002/14651858.CD000320.pub2. PMID 17636630.
- Dourmishev AL, Dourmishev LA, Schwartz RA (December 2005). “Ivermectin: pharmacology and application in dermatology”. International Journal of Dermatology 44(12): 981–8. doi:10.1111/j.1365-4632.2004.02253.x. PMID 16409259.
- Strycharz JP, Yoon KS, Clark JM (January 2008). “A new ivermectin formulation topically kills permethrin-resistant human head lice (Anoplura: Pediculidae)”. Journal of Medical Entomology 45 (1): 75–81. doi:10.1603/0022-2585(2008)45[75:ANIFTK]2.0.CO;2.ISSN 0022-2585. PMID 18283945.
- “Sklice lotion”.
- David M. Pariser, M.D., Terri Lynn Meinking, Ph.D., Margie Bell, M.S., and William G. Ryan, B.V.Sc. (November 1, 2012). “Topical 0.5% Ivermectin Lotion for Treatment of Head Lice”. New England Journal of Medicine 367: 1687–1693.doi:10.1056/NEJMoa1200107.
- Study shows ivermectin ending lice problem in one treatment, Los Angeles Times, Nov 5, 2012
- DONALD G. MCNEIL JR. (2012-12-31). “Pill Could Join Arsenal Against Bedbugs”. The New York Times. Retrieved 2013-04-05.
- Jump up^ Dourmishev AL, Dourmishev LA, Schwartz RA (December 2005). “Ivermectin: pharmacology and application in dermatology”. International Journal of Dermatology 44(12): 981–988. doi:10.1111/j.1365-4632.2004.02253.x. PMID 16409259.
- Huukelbach J, Winter B, Wilcke T, et al. (August 2004). “Tratmient masivo selectivo con ivermectina contra las helmintiasis intestinales y parasitos cutáneas en una población gravemente afectada”. Bull World Health Organ 82 (7): 563–571. doi:10.1590/S0042-96862004000800005.
- Goodman and Gilman’s Pharmacological Basis of Therapeutics, 11th edition, pages 122, 1084-1087.
- Jump up^ “COMFORTIS® and ivermectin interaction Safety Warning Notification”. U.S. Food and Drug Administration (FDA) Center for Veterinary Medicine (CVM).
- Yates DM, Wolstenholme AJ (August 2004). “An ivermectin-sensitive glutamate-gated chloride channel subunit from Dirofilaria immitis”. Int. J. Parasitol. 34 (9): 1075–81.doi:10.1016/j.ijpara.2004.04.010. PMID 15313134.
- Borst P, Schinkel AH (June 1996). “What have we learnt thus far from mice with disrupted P-glycoprotein genes?”. European Journal of Cancer 32 (6): 985–990.doi:10.1016/0959-8049(96)00063-9.
- Iglesias LE, Saumell CA, Fernández AS, et al. (December 2006). “Environmental impact of ivermectin excreted by cattle treated in autumn on dung fauna and degradation of faeces on pasture”. Parasitology Research 100 (1): 93–102. doi:10.1007/s00436-006-0240-x. PMID 16821034.
- Fisher MH, Mrozik H (1992). “The chemistry and pharmacology of avermectins”. Annu. Rev. Pharmacol. Toxicol. 32: 537–53. doi:10.1146/annurev.pa.32.040192.002541.PMID 1605577.
- W. C. CAMPBELL; R. W. BURG, , M. H. FISHER, and , R. A. DYBAS (June 26, 1984).“The Discovery of Ivermectin and Other Avermectins”. American Chemical Society. pp. 5–20. ISBN 9780841210837.
|chapter=ignored (help) - “The Nobel Prize in Physiology or Medicine 2015” (PDF). Nobel Foundation. Retrieved7 October 2015.
- “MDR1 FAQs”, Australian Shepherd Health & Genetics Institute, Inc.
- “Multidrug Sensitivity in Dogs”, Washington State University’s College of Veterinary Medicine
- Frischke H, Hunt L (April 1991). “Suspected ivermectin toxicity”. Canadian Veterinary Journal 32 (4): 245. PMC 1481314. PMID 17423775.
External links
- Stromectol
- The Carter Center River Blindness (Onchocerciasis) Control Program
- Mectizan Donation Program
- American NGDO Treating River Blindness
- MERCK. 25 Years: The MECTIZAN® Donation Program
- Trinity College Dublin. Prof William Campbell – The Story of Ivermectin
- “IVERMECTIN- ivermectin tablet (NDC Code(s): 42799-806-01)”. DailyMed. November 2014. Retrieved 2015-09-09
Bibliography
- Chapman and Hall Combined Chemical Dictionary compound code number CMD99-Y (Ivermectin); CLF13-X (Avermectin).
- The World Health Organisation page on Onchocerciasis
- “Organic Chemists Fighting Blindness” – the Mectizan story from the RSC Organic Division
- A. Crump and K. Otoguro, Trends in Parasitology, 2005, 21, 126-132. (Ōmura’s work)
- Nobel committee’s announcement.
Avermectin
- R. W. Burg, B. M. Miller, E. E. Baker, J. Birnbaum, S. A. Currie, R. Hartman, Y.-L. Kong, R. L. Monaghan, G. Olson, I. Putter, J. B. Tunac, H. Wallick, E. O. Stapley, R. Oiwa, and S. Ōmura, Antimicrob. Agents Chemother., 1979, 15, 361-367 (production of avermectins)
- T. W. Miller, L. Chaiet, D. J. Cole, L. J. Cole, J. E. Flor, R. T. Goegelman, V. P. Gullo, H. Joshua, A. J. Kempf, W. R. Krellwitz, R. L. Monaghan, R. E. Ormond, K. E. Wilson, G. Albers-Schönberg and I. Putter., Antimicrob. Agents Chemother., 1979, 15, 368-371 (isolation of avermectins)
- J. R. Egerton, D. A. Ostlind, L. S. Blair, C. H. Eary, D. Suhayda, S. Cifelli, R. F. Riek and W. C. Campbell, Antimicrob. Agents Chemother., 1979, 15, 372-378 (efficacy of avermectins)
- M. H. Fisher, Pure Appl. Chem., 1990, 62, 1231-1240 (avermectin review)
- Y. J. Yoon, E.-S. Kim, Y.-S. Hwang and C.-Y. Choi, Appl. Microbiol. Biotechnol., 2004, 63, 626–634 (biosynthesis)
Ivermectin
- J. C. Chabala, H. Mrozik, R. L. Tolman, P. Eskola, A. Lusi, L. H. Peterson, M. F. Woods, M. H. Fisher and W. C. Campbell, J. Med. Chem., 1980, 23, 1134-1136 (synth)
- W. C. Campbell, M. H. Fisher, E. O. Stapley, G. Albers-Schönberg and T. A. Jacob, Science, 1983, 221, 823–828 (ivermectin as a new antiparasitic agent)
- S. Ōmura and A. Crump, Nat. Rev. Microbiol., 2004, 2, 984-989. (“The life and times of ivermectin – a success story”).
- K. Collins, Perspect. Biol. Med., 2004, 47, 100-109. (History of the Merck Mectizan donation program)
- A. D. Hopkins, Eye, 2005, 19, 1057–1066 (improvements upon ivermectin treatment)
- T. G. Geary, Trends in Parasitology, 2005, 21, 530–532 (20 years of ivermectin)
- S. Ōmura, Int. J. Antimicrob. Ag., 2008, 31, 91–98 (25 years of Ivermectin)
- A. G. Canga, A. M. S. Prieto, M. J. D. Liébana, N. F. Martínez, M. S.Vega and J. J. G. Vieitez, Vet. J., 2009, 179, 25–37 (pharmacokinetics and metabolism of ivermectin in domestic animal species)
- A. Crump and S. Ōmura, Proc. Jpn. Acad., Ser. B., 2011, 87, 13-28 (ivermectin review)
- I. Farrell, Education in Chemistry, November 2013. (“One in the eye for river blindness”), online.
- The Mectizan donation program
- Colombia eliminates river blindness
Doramectin
- K. Stutzman-Engwall, S. Conlon, R. Fedechko, H. McArthur, K. Pekrun, Y. Chen, S. Jenne, C. La, N. Trinh, S. Kim, Y.-X. Zhang, R. Fox, C. Gustafsson and A. Krebber, Metabolic Engineering, 2005, 7, 27–37 (synth.)
- J.-B. Wang, H.-X. Pan and G.-L. Tang, Bioorg. Med. Chem. Lett., 2011, 21, 3320–3323 (synth.)
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
22,23-dihydroavermectin B1a + 22,23-dihydroavermectin B1b
|
|
| Clinical data | |
| Trade names | Stromectol |
| AHFS/Drugs.com | monograph |
| MedlinePlus | a607069 |
| Pregnancy category |
|
| Legal status |
|
| Routes of administration |
Oral, topical |
| Pharmacokinetic data | |
| Protein binding | 93% |
| Metabolism | Liver (CYP450) |
| Biological half-life | 18 hours |
| Excretion | Feces; <1% urine |
| Identifiers | |
| CAS Registry Number | 70288-86-7 |
| ATC code | D11AX22 P02CF01 QP54AA01QS02QA03 |
| PubChem | CID: 9812710 |
| DrugBank | DB00602 |
| ChemSpider | 7988461 |
| UNII | 8883YP2R6D |
| KEGG | D00804 |
| ChEMBL | CHEMBL341047 |
| PDB ligand ID | IVM (PDBe, RCSB PDB) |
| Chemical data | |
| Formula | C 48H 74O 14 (22,23-dihydroavermectin B1a) C 47H 72O 14 (22,23-dihydroavermectin B1b) |
| Molecular mass | 875.10 g/mol |
SIMILAR
Doramectin is a similar molecule, used to treat parasites in animals, such as cattle, horses, sheep and pigs.

/////////////////ivermectin, MALARIA
Study Demonstrates Efficacy of New Tumor Treatment

Study Demonstrates Efficacy of New Tumor Treatment |
||||
|
The results of a study demonstrate the efficacy of this drug in treating nonfunctional neuroendocrine tumors of lung or gastrointestinal origin.
|
What are “complex manufacturing processes”? A recent reply from the EMA
DRUG REGULATORY AFFAIRS INTERNATIONAL
Sometimes a clear definition of terms is crucial in the communication between authorities and pharmaceutical companies. Find out what the European Medicines Agency EMA defines as “complex manufacturing steps” and what authorisation holders providing a variation application need to consider.
The Variations Regulation (EC) no. 1234/2008 of the European Commission defines the procedure for variations of existing marketing authorisations. The “detailed guidelines for the various categories of variations“, which were published in the consolidated version in August 2013 in the European Official Journal, explain the interpretation and application of this Variations Regulation.
Although the “detailed guidelines” describe a number of scenarios of possible variations in some detail, there are formulations in the Guideline text which require clarification due to their blur. The EMA adopted such a case in a recent update of itsquestions and answers collection “Quality of Medicines Questions and Answers: Part 1”…
View original post 258 more words
Voxtalisib, SAR-245409, XL-765

Voxtalisib
SAR-245409, XL-765
2-amino-8-ethyl-4-methyl-6-(1H-pyrazol-3-yl)pyrido[2,3-d]pyrimidin-7(8H)-one
2-Amino-8-ethyl-4-methyl-6-(1H-pyrazol-5-yl)pyrido[2,3-d]pyrimidin-7(8H)-one hydrochloride
C13 H14 N6 O . Cl H, 306.751
934493-76-2
INNOVATOR Exelixis Inc,, LICENSE SANOFI
PHASE 2, Malignant neoplasms
0.2H2O
- Mol. Formula:C13H14N6O∙0.2H2O, MW:273.9
- NMR………http://www.chemietek.com/Files/Line2/CHEMIETEK,%20XL765,%20Lot%2001,%20NMR%20in%20CD3OD.pdf
- Mechanism of Action:selective oral inhibitor of PI3K and mTOR
Indication:Cancer Treatment
Stage of Development: phase ll study in chronic lymphocytic leukemia (CLL) and non-Hodgkin’s lymphoma (NHL). A phase I/II trial is assessing SAR245409 in combination with letrozole in ER/PR+ HER2- breast cancer. 
SAR245409 (XL765)
SAR245409 (XL765) is an orally available inhibitor of PI3K and the mammalian target of rapamycin (mTOR), which are frequently activated in human tumors and play central roles in tumor cell proliferation. Exelixis discovered SAR245409 internally and out-licensed the compound to Sanofi. SAR245409 is being evaluated by Sanofi as a single agent and in multiple combination regimens in a variety of cancer indications. Clinical trials have included a single agent phase 2 trial in Non-Hodgkin’s lymphoma, combination phase 1b/2 trials with temozolomide in patients with glioblastoma, with letrozole in hormone receptor positive breast cancer, with bendamustine and/or rituximab in lymphoma or leukemia, and a phase 1 trial in combination with a MEK inhibitor.
SAR-245409 is an investigational drug originated by Exelixis that dually inhibits mammalian target of rapamycin (mTOR) and phosphatidylinositol 3-kinase (PI3K).

Sanofi is also evaluating the compound in phase I/II clinical trials for the treatment of malignant neoplasm as monotherpay or in combination regimen. It has also completed phase I clinical trials as an oral treatment for brain cancer.
In 2009, the drug candidate was licensed to Sanofi (formerly known as sanofi-aventis) by Exelixis worldwide for the treatment of solid tumors.
XL765 (Voxtalisib, SAR245409, Sanofi)*, a PYRIDOPYRIMIDINONE-derivative, is a highly selective, potent and reversible ATP-competitive inhibitor of pan-Class I PI3K (α, β, γ, and δ) and mTORC1/mTORC2. It is orally active, highly selective over 130 other protein kinases. In cellular assays, XL765 inhibits the formation of PIP3 in the membrane, and inhibits phosphorylation of AKT, p70S6K, and S6 phosphorylation in multiple tumor cell lines with different genetic alterations affecting the PI3K pathway.

In mouse xenograft models, oral administration of XL-765 results in dose-dependent inhibition of phosphorylation of AKT, p70S6K, and S6 with a duration of action of approximately 24 hours. Repeat dose administration of XL765 results in significant tumor growth inhibition in multiple human xenograft models in nude mice that is associated with antiproliferative, antiangiogenic, and proapoptotic effects
PATENT
WO 2014058947
http://www.google.co.in/patents/WO2014058947A1?cl=en
Example 1. Synthesis of Compound (1)
Compound (1) can be synthesized as described in WO 07/044813, which is hereby incorporated in its entirety.

Briefly, a base and an intermediate, compound (a), are added to solution of commercially available 2-metfiyl-2-thiopseudourea sulfate in a solvent such as water and stirred overnight at room temperature. After neutralization, compound (b) is collected by filtration and dried under vacuum. Treatment of compound (b) with POCI3 and heating at reflux for 2 hours yields compound (c) which can be concentrated under vacuum to dryness. Compound (c) can be used directly in the following reaction with ethylamine carried out in a solvent such as water with heating to give compound (d). Compound (d) is then treated with iodine monochloride in a solvent such as methanol to form compound (e). Compound (e) is then dissolved in DMA, to which ethyl acrylate, Pd(OAc)2 and a base are added. This reaction mixture is heated and reacted overnight until completion of the reaction to give compound (f), which can be purified via column chromatography.
Compound (f) is then be treated with DBU in the presence of a base, such as DIEA, and heated at reflux for 15 hours. Upon completion of the reaction, the solvent is evaporated and the residue triturated with acetone to yield compound (g). Bromination of compound (g) can be achieved through drop-wise addition of Br2 to compound (g) in CH2C12, followed by stirring overnight at room temperature. Next, filtration is carried out, and triethylamine is added so that, upon washing and drying, the product, compound (h) is obtained. A Suzuki coupling between compound (h) and lH-pyrazol-5-yl boronic acid is carried out using a Pd- catalyst such as [1,1 -bis(diphenylphosphino)ferrocene]dichloropalladium(II) in the presence of a base to yield compound (i). Finally, compound (i) can be converted to compound (1) of the instant invention through 1) oxidation of the methylthio group with m-CPBA, carried out at room temperature with stirring and 2) treatment of the resulting product dissolved in dioxane, with liquid ammonia. Stirring at room temperature overnight followed by purification by column chromatography gives the desired product, 2-amino-8-ethyl-4-methyl- 6-(lH-pyrazol-5-yl)pyrido[2,3-d]pyrimidin-7(8H)-one, compound (1).
PATENT
WO 2007044813
http://www.google.co.in/patents/WO2007044813A1?cl=en
Example 1 2-amino-8-ethyl-4-methyl-6-(lJΪ-pyrazol-5-yl)pyrido[2,3-</]pyrimidin-7(8J?)-one

To a solution of 2-methyl-2-thiopseudourea sulfate (Aldrich, 58.74 g, 0.422 mol) in water (1000 mL) were added sodium carbonate (81.44 g, 0.768 mol) and ethyl acetoacetate (50 g, 0.384 mol) at room temperature. The reaction mixture was stirred overnight. After neutralizing to pH = 8, the solid was collected through filtration followed by drying under vacuum overnight to afford 6-methyl-2-(methylthio)pyrimidin-4(3H)-one (57.2 g, 95% yield) of product. 1H NMR (400 MHz, DMSO-d6): δ 12.47 (bs, IH), 5.96 (bs, lH), 2.47(s, 3H), 2.17 (s, 3H).

To the round bottom flask containing 6-methyl-2-(methylthio)pyrimidin-4(3H)- one (19 g, 121.6 mmol) was added POCl3 (30 mL). The reaction mixture was heated to reflux for 2 h and then concentrated on a rotary evaporator to dryness. The crude 4-chloro- 6-methyl-2-(methylthio)pyrimidine was used directly in the next reaction without further purification.

To the 4-chloro-6-methyl-2-(methylthio)pyrimidine from above was added 30 mL of a solution of 70% ethylamine in water. The reaction mixture was heated to 50 0C for 3 h. After completion, excess ethylamine was evaporated on rotary evaporator under vacuum. The solid was filtered and dried under vacuum to afford 7V-ethyl-6-methyl-2- (methylthio)pyrimidin-4-amine (20 g, 90% yield).

To the solution of N-emyl-6-methyl-2-(methylthio)pyrimidin-4-amine (20 g, 121.6 mmol) in methanol was added iodine monochloride (26.58 g, 163.7 mmol) in small portions at 0 °C. Then the reaction mixture was stirred overnight. After evaporation of solvent, the residue was triturated with acetone. The product iV-ethyl-5-iodo-6-methyl-2- (methylthio)pyrimin-4-amine (25.2 g, 75% yield) was collected by filtration. 1H NMR (400 MHz, CDCl3): δ 5.37 (bs, IH), 3.52 (q, J = 7.2 Hz, IH), 2.50 (s, 3H), 1.26 (t, J = 7.2 Hz, 3H).

To the solution of N-ethyl-5-iodo-6-methyl-2-(methylthio)pyrimin-4-amine (25.2 g, 81.48 mmol) in DMA (260 mL) were added ethyl acrylate (12.23 g, 122.2 mmol), Pd(OAc)2 (3.65 g, 16.25 mmol), (+)BINAP and triethyl amine (24.68 g, 244.4 mmol). Then the reaction mixture was heated to 100 0C and reacted overnight. After evaporation of solvent, the residue was diluted with water and the aqueous layer was extracted with ethyl acetate. The product (E)-ethyl-3-(4-(ethylamino)-6-methyl-2-(methylthio)pyrimidin-5- yl)acrylate (16.8 g, 73% yield) was isolated by silica gel column chromatography with 6-8% ethyl acetate in hexane as eluent. 1H NMR (400 MHz, CDCl3): δ 7.65 (d, J = 16.4Hz, IH), 6.20 (d, J = 16.4Hz, IH), 5.15 (bs, IH), 4.28(q, J = 7.2 Hz, 2H), 3.54 (q, J = 7.2 Hz, 2H), 2.53 (s, 3H), 2.37 (s, 3H), 1.35 (t, J = 7.2 Hz, 3H), 1.24 (t, J = 7.2 Hz, 3H).

To a solution of (E)-ethyl-3-(4-(ethylamino)-6-methyl-2-(methylthio)pyrimidin- 5-yl)acrylate (16.8 g, 59.8 mmol) in DIPEA was added l,8-diazabicyclo[5.4.0]undec-7-ene (DBU, 18.21 g, 119.6 mmol) at room temperature. Then the reaction mixture was heated to reflux and reacted for 15 h. After evaporation of solvent, the residue was triturated with acetone. The product 8-ethyl-4-methyl-2-(methylthio)pyrido[2,3-d]pyrimidin-7(8H)-one (10.77 g, 77% yield) was collected by filtration. 1H NMR (400 MHz, CDCl3): δ 7.78 (d, J = 9.6 Hz, IH), 6.63 (d, J = 9.6 Hz5 IH), 4.5(q, J = 7.2 Hz, 2H), 2.67 (s, 3H), 2.62 (s, 3H), 1.33 (t, J = 7.2 Hz, 3H).

[00187] To a solution of 8-ethyl-4-methyl-2-(methylthio)pyrido[2,3-d]pyrimidin-7(8H)- one (6.31 g, 26.84 mmol) in DCM was added Br2 (4.79 g, 29.52 mmol) dropwise at room temperature. Then the reaction mixture was stirred at room temperature overnight. After filtration the solid was suspended in DCM (100 mL), and triethylamine (20 mL) was added. The mixture was washed with water and dried with Na2SO4, and the product 6-bromo-8- ethyl-4-methyl-2-(methylthio)pyrido[2,3-d]pyrimidin-7(8H)-one (6.96 g, 83 % yield) was obtained after evaporation of DCM. 1H NMR (400 MHz, CDCl3): δ 8.22 (s, IH), 4.56 (q, J = 7.2 Hz, 2H), 2.68 (s, 3H), 2.62 (s, 3H), 1.34 (t, J = 7.2Hz, 3H).

To a solution of 6-bromo-8-ethyl-4-methyl-2-(methylthio)ρyrido[2,3- d]pyrimidin-7(8H)-one (0.765 g, 2.43 mmol) in DME-H2O (10:1 11 mL) was added IH- pyrazol-5-ylboronic acid (Frontier, 0.408 g, 3.65 mmol), [1,1′- bis(diphenylphosphino)ferrocene]dichloropalladium(II) complex with CH2Cl2 (Pd(dρρρf),0.198 g, 0.243 mmol) and triethylamine (0.736 g, 7.29 mmol) at room temperature. Then the reaction mixture was heated to reflux and reacted for 4 h. After cooling down to room temperature, the reaction mixture was partitioned with water and ethyl acetate. After separation, the. organic layer was dried with Na2SO4, and the product 8- ethyl-4-methyl-2-(methylthio)-6-(lH-pyrazol-5-yl)pyrido[2,3-d]pyrimidin-7(8H)-one (0.567 g, 77% yield) was obtained by silica gel column chromatography. 1H NMR (400 MHz, CDCl3): δ 13.3 (bs, IH), 8.54 (s, IH), 7.82-7.07 (m, 2H), 4.45 (q, J = 7.2 Hz, 2H), 2.71 (s, 3H), 2.60 (s, 3H), 1.26 (t, J = 7.2Hz, 3H).

To the solution of 8-ethyl-4-methyl-2-(methylthio)-6-(lH-pyrazol-5- yl)pyrido[2,3-d]pyrimidin-7(8H)-one (0.123 g, 0.41mmol) in DCM (2 mL) was added MCPBA (0.176 g, 77%, 0.785 mmol) in a small portion at room temperature. Then the reaction mixture was stirred for 4 h. After evaporation of DCM, dioxane (1 mL) and liquid ammonia (1 mL) were introduced. The reaction was stirred at room temperature overnight. The product 2-amino-8-ethyl-4-methyl-6-(lH-pyrazol-5-yl)pyrido[2,3-(/lpyrimidin-7(8H)- one (50.4 mg) was obtained by silica gel column chromatography. 1H NMR (400 MHz, CD3OD): δ 8.41 (s, IH), 7.62 (d, J – 2.0 Hz, IH), 6.96 (d, J = 2.0Hz5 IH), 4.51 (q, J = 7.2Hz, 2H), 2.64 (s, 3H), 1.29 (t, J = 7.2Hz, 3H); MS (EI) for C13H14N6O: 271.3 (MH+)
References:
1. P. W. Yu, et al., Characterization of the Activity of the PI3K/mTOR Inhibitor XL765 (SAR245409) in Tumor Models with Diverse Genetic Alterations Affecting the PI3K Pathway, Mol Cancer Ther, May 2014 13; 1078-91
2. K. P. Papadopoulos, et al., Phase I Safety, Pharmacokinetic, and Pharmacodynamic Study of SAR245409 (XL765), a Novel, Orally Administered PI3K/mTOR Inhibitor in Patients with Advanced Solid Tumors, Clin Cancer Res, May 1, 2014 20; 2445
3 WO 2014058947
4 WO 2013040337
5 WO 2012065019
6 WO 2009017838
7 WO 2008127678
8 WO 2008124161
9 WO 2007044698
10 WO 2007044813
| WO2007044813A1 | 9 Oct 2006 | 19 Apr 2007 | Exelixis Inc | PYRIDOPYRIMIDINONE INHIBITORS OF PI3Kα |
| WO2012054748A2 * | 20 Oct 2011 | 26 Apr 2012 | Seattle Genetics, Inc. | Synergistic effects between auristatin-based antibody drug conjugates and inhibitors of the pi3k-akt mtor pathway |
| WO2012065019A2 * | 11 Nov 2011 | 18 May 2012 | Exelixis, Inc. | Pyridopyrimidinone inhibitors of p13k alpha |
| US7811572 | 14 Aug 2006 | 12 Oct 2010 | Immunogen, Inc. | Process for preparing purified drug conjugates |
| US20040235840 | 20 May 2004 | 25 Nov 2004 | Immunogen, Inc. | Cytotoxic agents comprising new maytansinoids |
Exelixis, Inc.
210 East Grand Avenue
So. San Francisco, CA 94080
(650) 837-7000 phone
(650) 837-8300 fax

////////////Voxtalisib hydrochloride, Exelixis, SANOFI, PHASE 2, Malignant neoplasms, SAR-245409, XL-765


New “mTOR” inhibitor from Exelixis, Inc., XL 388

XL 388
A Novel Class of Highly Potent, Selective, ATP-Competitive, and Orally Bioavailable Inhibitors of the Mammalian Target of Rapamycin (mTOR)
Benzoxazepine-Containing Kinase Inhibitor

[7-(6-Aminopyridin-3-yl)-2,3-dihydro-1,4-benzoxazepin-4(5H)-yl][3-fluoro-2-methyl-4-(methylsulfonyl)phenyl]methanone
[7-(6-amino-3-pyridinyl)-2,3-dihydro-1,4-benzoxazepin-4(5H)-yl][3-fluoro-2-methyl-4-(methylsulfonyl)phenyl]-Methanone,
(7-(6-Aminopyridin-3-yl)-2,3-dihydrobenz[f][1,4]oxazepin-4(5H)-yl)(3-fluoro-2-methyl-4-(methylsulfonyl)phenyl)methanone
MW 455.50, CAS 1251156-08-7, MF C23 H22 F N3 O4 S
Exelixis, Inc. INNOVATOR, IND Filed
½H2O
C23H22FN3O4S.½H2O , Molecular Weight: 464.51
MONO HYDROCHLORIDE…..CAS 1777807-51-8, [7-(6-Aminopyridin-3-yl)-2,3-dihydro-1,4-benzoxazepin-4(5H)-yl][3-fluoro-2-methyl-4-(methylsulfonyl)phenyl]methanone Hydrochloride (1·HCl)
TLC Rf = 0.33 (Dichloromethane:Methanol [95:5])
Potent and selective mTOR inhibitor (IC50 = 9.9 nM). Inhibits mTOR activity in an ATP-competitive manner. Exhibits >300-fold selectivity for mTOR over PI 3-K and a range of other kinases. Displays antitumor activity in athymic nude mice implanted with tumor xenografts.
SYNTHESIS
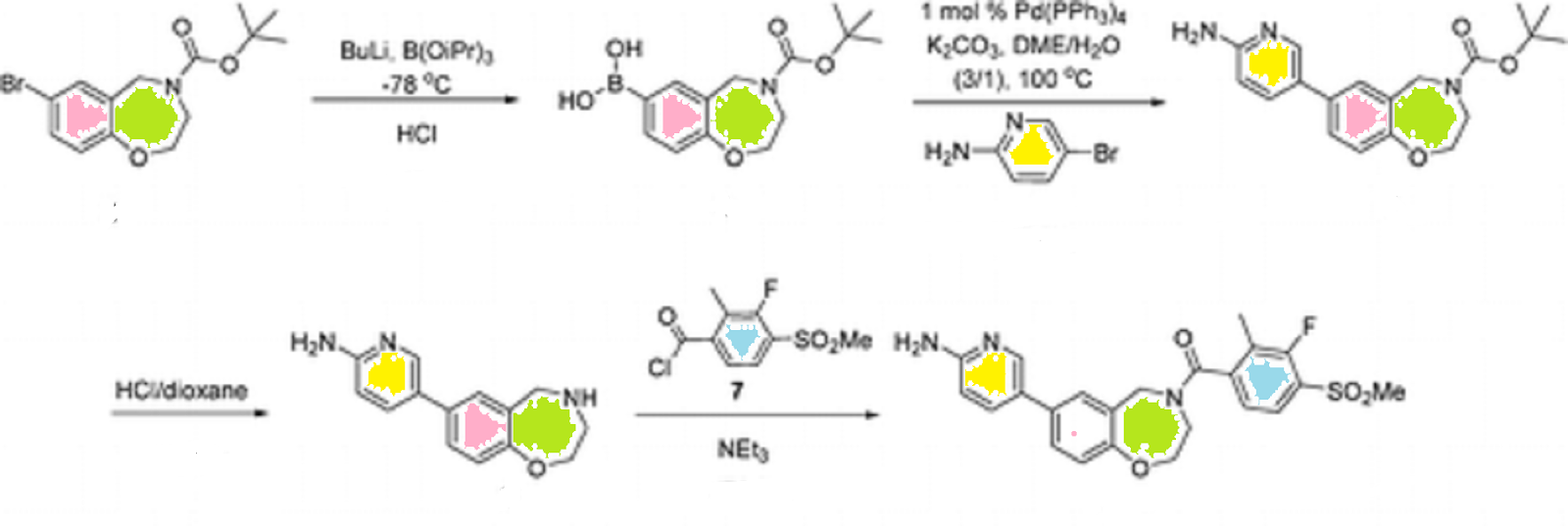
CLICK ON IMAGE FOR CLEAR VIEW……………..
Tyrosine kinases are important enzymes for signal transduction in cells. Therefore, they are often targets for the treatment of diseases that are caused by dysregulation of cellular processes, such as cancers. Mammalian target of rapamycin (mTOR) is a kinase in the phosphatidylinositol-3-kinase (PI3K) family of enzymes and is implicated in the regulation of cell growth and proliferation. Various inhibitors of mTOR have been explored as possible agents for treatment of various cancers
The mammalian target of rapamycin (mTOR) is a large protein kinase that integrates both extracellular and intracellular signals of cellular growth, proliferation, and survival. Both extracellular mitogenic growth factor signaling from cell surface receptors and intracellular signals that convey hypoxic stress, energy, and nutrient status converge at mTOR. mTOR exists in two distinct multiprotein complexes: mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2).
mTORC1 is a key mediator of translation and cell growth, via its substrates p70S6 kinase (p70S6K) and eIF4E binding protein 1 (4E-BP1), and promotes cell survival via the serum and glucocorticoid-activated kinase (SGK), whereas mTORC2 promotes activation of prosurvival kinase AKT. mTORC1, but not mTORC2, can be inhibited by an intracellular complex between rapamycin and FK506 binding protein (FKBP). However, rapamycin–FKBP may indirectly inhibit mTORC2 in some cells by sequestering mTOR protein and thereby inhibiting assembly of mTORC2.
Given the role of mTOR signaling in cellular growth, proliferation, and survival as well as its frequent deregulation in cancers, several rapamycin analogues (rapalogues) that are selective allosteric mTORC1 inhibitors have been extensively evaluated in a number of cancer clinical trials.
Demonstrated clinical efficacy for rapalogues is currently limited to patients with advanced, metastatic renal cell carcinoma (RCC) despite extensive development efforts.
This result is likely attributed not only to a lack of inhibition of mTORC2 by rapalogues that leads to upregulation of Akt through a negative feedback loop, but also to only partial inhibition of mTORC1.Therefore, ATP-competitive mTOR inhibitors that should simultaneously inhibit both mTORC1 and mTORC2 may offer a clinical advantage over rapalogues.
As a key component of the phosphoinositide 3-kinase-related kinase (PIKK) family, which is comprised of phosphoinositide 3-kinases (PI3Ks), DNA-PK, ATM, and ATR, mTOR shares the highly conserved ATP binding pockets of the PI3K family with sequence similarity of 25% in the kinase catalytic domain.
In light of this fact, it is not surprising that many of the first reported ATP-competitive mTOR inhibitors such as BEZ235 and GDC-0980 also inhibited PI3Ks. PI3Ks are responsible for the production of 3-phosphoinositide lipid second messengers such as phosphatidylinositol 3,4,5-triphosphate (PIP3), which are involved in a number of critical cellular processes, including cell proliferation, cell survival, angiogenesis, cell adhesion, and insulin signaling.
Therefore, the development of ATP-competitive mTOR inhibitors that are selective over PI3Ks may offer an improved therapeutic potential relative to rapalogues as well as dual PI3K/mTOR inhibitors. Recently, several selective ATP-competitive mTOR inhibitors such as Torin 2 and AZD8055 have been reported with sufficient promise to warrant clinical trials.
PATENT
WO 2010118208
Example 2:
[7-(6-Aminopyridin-3-yl)-2,3-dihydro-l,4-benzoxazepin-4(5H)-yl] [3-fluoro- 2-methyl-4-(methylsulfonyl)phenyl]methanone

tørt-Butyl 7-(6-aminopyridin-3-yl)-2,3-dihydrobenzo[/] [l,4]oxazepine-4(5H)- carboxylate. To a mixture of 4-(te/t-butoxycarbonyl)-2,3,4,5- tetrahydrobenzo[/][l,4]oxazepin-7-ylboronic acid (1.52 g, 5.2 mmol), prepared as described in Reference Example 5, 2-amino-5-bromopyridine (900 mg, 5.2 mmol), and potassium carbonate (1.73 g, 12.5 mmol) in 1 ,2-dimethoxyethane/water (30 mL/10 mL) was added tetrakis(triphenylphosphine)palladium(0) (90 mg, 1.5 mol%) and the reaction mixture was purged with nitrogen and stirred at reflux for 3 h. The reaction was cooled to rt, diluted with water/ethyl acetate (50 mL/50 mL), and the separated aqueous layer was extracted with ethyl acetate. The resulting emulsion was removed by filtration. The combined organic layer was washed with brine, dried with sodium sulfate, filtered and concentrated under reduced pressure, and the residue was triturated with toluene for 1 h. The resulting off-white solid was isolated by filtration to give the desired product (1.37 g, 77 %) as an off-white solid. MS (EI) for Ci9H23N3O3: 342 (MH+).
5-(2,3,4,5-Tetrahydrobenzo[/] [l,4]oxazepin-7-yl)pyridine-2-amine. To a stirred solution of tert-butyl 7-(6-aminopyridin-3-yl)-2,3-dihydrobenzo[/][l,4]oxazepine- 4(5H)-carboxylate (1.36 g, 3.98 mmol) in 1,4-dioxane (5 mL) was added 4 N hydrogen chloride in 1 ,4-dioxane (5 mL) and the reaction mixture was stirred at rt overnight. The reaction was concentrated on a rotary evaporator and the residue was triturated with ether. The solid was isolated by filtration. This solid was dissolved in water (5 mL) and made basic with 5 N sodium hydroxide to pH 11-12. The brownish sticky oil that aggregated at the bottom was isolated and the aqueous layer was extracted with 5 % methanol in ethyl acetate. The extracts were dried with sodium sulfate and concentrated on a rotary evaporator. The brownish sticky oil was dissolved with a mixture of methanol/ethyl acetate, combined with the isolated organic residue and concentrated under reduced pressure to give a yellow solid. This solid was triturated with dichloromethane (10 mL) for 1 h and a yellow solid was isolated by filtration and dried under high vacuum to give amine the desired product (920 mg, 96 %). MS (EI) for Ci4Hi5N3O: 242 (MH+).
[7-(6-Aminopyridin-3-yl)-2,3-dihydro-l,4-benzoxazepin-4(5H)-yl][3-fluoro-2- methyl-4-(methylsulfonyl)phenyl]methanone.
To a stirred suspension of 5-(2, 3,4,5- tetrahydrobenzo[/][l,4]oxazepin-7-yl)pyridine-2-amine (85 mg, 352 μmol) and triethylamine (54 μL, 387 μmol) in dichloromethane (10 mL) was added 3-fluoro-2-methyl-4- (methylsulfonyl)benzoyl chloride (91 mg, in 3 mL of dichloromethane), prepared as described in Reference Example 1, at 0 0C for 2 h. After stirring for an additional 1 h at rt, the reaction mixture was diluted with water (5 mL) and the separated aqueous layer was extracted with dichloromethane. The combined extracts were dried with sodium sulfate, filtered and concentrated under reduced pressure to give a light-yellow solid that was purified via silica gel chromatography to give the desired product (113 mg, 70%) as a white solid.
1H NMR (400 MHz, DMSO-d6): δ 8.24-8.03 (dd, IH), 7.79-7.71 (m, IH), 7.71-7.69 (dd, 0.5H), 7.57-7.57 (d, 0.5H), 7.44-7.40 (m, 1.5H), 7.29-7.19 (dd, IH), 7.05-7.01 (dd, IH), 6.64-6.63 (d, 0.5H), 6.54-6.45 (dd, IH), 6.06 (s, 2H), 4.93-4.31 (m, 2H), 4.31-3.54 (m, 4H), 3.37-3.36(d, 3H), 2.12-1.77 (d, 3H).
MS (EI) C23H22FN3O4S: 456 (MH+).
PAPER
Journal of Medicinal Chemistry (2013), 56(6), 2218-2234.
J. Med. Chem., 2013, 56 (6), pp 2218–2234
DOI: 10.1021/jm3007933

A series of novel, highly potent, selective, and ATP-competitive mammalian target of rapamycin (mTOR) inhibitors based on a benzoxazepine scaffold have been identified. Lead optimization resulted in the discovery of inhibitors with low nanomolar activity and greater than 1000-fold selectivity over the closely related PI3K kinases. Compound 28 (XL388) inhibited cellular phosphorylation of mTOR complex 1 (p-p70S6K, pS6, and p-4E-BP1) and mTOR complex 2 (pAKT (S473)) substrates. Furthermore, this compound displayed good pharmacokinetics and oral exposure in multiple species with moderate bioavailability. Oral administration of compound 28 to athymic nude mice implanted with human tumor xenografts afforded significant and dose-dependent antitumor activity.
(7-(6-Aminopyridin-3-yl)-2,3-dihydrobenz[f][1,4]oxazepin-4(5H)-yl)(3-fluoro-2-methyl-4-(methylsulfonyl)phenyl)methanone (28)
1H NMR (400 MHz, DMSO-d6): δ (rotamers are observed) 8.24 and 8.03 (d, J = 2.4 Hz, 1H), 7.77 and 7.72 (t, J = 7.6 Hz, 1H), 7.71–7.39 (m, 2H), 7.57 and 6.63 (d, J = 2.4 Hz, 1H), 7.28 and 7.19 (d, J = 7.6 Hz, 1H), 7.04 and 7.02 (d, J = 8.0 Hz, 1H), 6.52 and 6.46 (d, J = 8.8 Hz, 1H), 6.05 (br s, 2H), 4.93–4.31 (m, 2H), 4.28–3.56 (m, 4H), 3.37 and 3.34 (s, 3H), 2.12 and 1.77 (d,J = 1.6 Hz, 3H). 13C NMR (100 MHz, DMSO-d6): δ 167.3, 167.2, 166.6, 166.6, 158.9, 158.9, 158.4, 158.4, 157.4, 157.2, 155.9, 155.8, 145.4, 145.1, 145.1, 144.0, 143.9, 135.0, 134.7, 132.9, 132.8, 129.4, 129.2, 128.2, 128.2, 128.1, 128.0, 127.0, 126.9, 126.8, 125.9, 125.6, 125.4, 123.6, 123.5, 123.3, 123.1, 122.8, 122.0, 122.0, 121.9, 121.9, 121.2, 120.7, 107.8, 107.8, 70.9, 70.8, 51.1, 51.1, 47.4, 46.5, 43.5, 43.5, 43.5, 43.4, 11.0, 10.9, 10.7, 10.6. IR (KBr pellet): 1623, 1487, 1457, 1423, 1385, 1314, 1269, 1226, 1193, 1144, 1133, 1054, 1031, 962, 821, 768 cm–1. Mp: 204–205 °C. MS (EI): m/z for C23H22FN3O4S, 456.0 (MH+). High-resolution MS (FAB MS using glycerol as the matrix): m/z calcd for C23H22FN3O4S 456.13878, found 456.13943.
PATENT
- SYNTHETIC EXAMPLES
-

-
1-Bromo-3,4-difluoro-2-methylbenzene. To a stirred mixture of 2,3-difluorotoluene (1.9 g, 14.8 mmol) and iron (82.7 mg, 1.48 mmol) in chloroform (10 mL) at rt was added bromine (76 μL, 14.8 mmol) over 2 h. The resulting mixture was stirred at rt overnight. Excess water (10 mL) was added and the reaction mixture was diluted with ether (20 mL). The separated organic layer was washed with aqueous sodium thiosulfate, brine, dried over sodium sulfate and concentrated on a rotary evaporator. The residue was distilled to give the desired product (2.49 g, 81%) as a colorless oil.
-
3,4-Difluoro-2-methylbenzoic acid. To a stirred solution of 1-bromo-3,4-difluoro-2-methylbenzene (940 mg, 4.54 mmol) in tetrahydrofuran (5 mL) was added isopropylmagnesium bromide (3.0 mL, 6.0 mmol) over 1 h at 0° C. The resulting mixture was stirred at rt for 24 h. Carbon dioxide (CO2), generated from dry ice, was introduced to the reaction mixture over 2 h and the resulting mixture was stirred for an additional 30 min. The reaction mixture was quenched with addition of an excess amount of water (5 mL) and the tetrahydrofuran was removed on a rotary evaporator. The resulting aqueous layer was diluted with water (5 mL) and acidified with concentrated hydrochloric acid to pH 1-2. The white precipitate was filtered and washed with water and cold hexanes and dried under high vacuum to give the desired product (630 mg, 81%) as a white powder. MS (EI) for C8H6F2O2: 171 (MH−).
-
3-Fluoro-2-methyl-4-(thiomethyl)benzoic acid. To a stirred solution of acid 3,4-difluoro-2-methylbenzoic acid (700 mg, 4.1 mmol) in dimethylsulfoxide (5 mL) was added powdered potassium hydroxide (274 mg, 4.9 mmol) and the mixture was stirred at rt for 30 min. Sodium thiomethoxide (342 mg, 4.9 mmol) was added to the mixture and the resulting mixture was stirred at 55-60° C. for 4 h. Additional powdered potassium hydroxide (70 mg, 1.2 mmol), sodium thiomethoxide (60 mg, 0.8 mmol), and dimethylsulfoxide (2 mL) were added to the reaction mixture. After stirring for 4 h, the mixture was cooled to 0° C. and quenched with excess water (10 mL). The resulting suspension was acidified at 0° C. with concentrated hydrochloric acid to pH 1-2. The white precipitate was collected by suction filtration, washed with water and dried under vacuum overnight to give the desired product (870 mg, 100%). The intermediate sulfide was used in the next step without further purification. MS (EI) for C9H9FO2S: 199.1 (MH−).
-
3-Fluoro-2-methyl-4-(methylsulfonyl)benzoic acid. To a stirred suspension of 3-fluoro-2-methyl-4-(thiomethyl)benzoic acid in an acetone/water (1 mL/10 mL) mixture was added sodium hydroxide (330 mg, 8.25 mmol) and sodium bicarbonate (680 mg, 8.1 mmol). Oxone (˜4 g) was added portionwise to the reaction mixture at 0° C. over 2 h. The reaction was monitored by LC/MS. Concentrated hydrochloric acid was added to adjust the pH 2-3 and the white precipitate was collected by suction filtration, washed with water, and dried under vacuum. Dried precipitate was suspended in water (10 mL), stirred vigorously at rt for 1 h, filtered, washed with water, and hexanes and dried under vacuum to give the desired product (886 mg, 94%) as a white powder. MS (EI) for C9H9FO4S: 231 (MH−).
-
3-Fluoro-2-methyl-4-(methylsulfonyl)benzoyl chloride. A mixture of 3-fluoro-2-methyl-4-(methylsulfonyl)benzoic acid (860 mg, 3.7 mmol) in thionyl chloride (10 mL) was heated to reflux for 3 h. (the reaction mixture became homogenous). The reaction mixture was concentrated on a rotary evaporator to give the crude acid chloride. This acid chloride was triturated with dichloromethane (2 mL) and concentrated under reduced pressure. The trituration process was repeated 3 times until the product (900 mg, 98%) was obtained as a white powder.
- Reference Example 13-Fluoro-2-methyl-4-(methylsulfonyl)benzoyl chloride
Reference Example 2Ethyl 4-(2,3,4,5-tetrahydro-1,4-benzoxazepin-7-yl)benzoate hydrochloride salt
-

-
4-(ethoxycarbonyl)phenylboronic acid (22.16 g, 114 mmol), tert-butyl 7-bromo-2,3-dihydrobenzo[f][1,4]oxazepin-4(5H)-carboxylate (34.08 g, 104 mmol), prepared as described in Reference Example 4, Pd(dppf)Cl2 and TEA (21 g, 208 mmol) were combined in a mixture of dioxane (200 mL) and water (20 mL). The reaction mixture was heated to 90° C. for 2 h, then cooled and the solvent removed. Purification of the residue by silica chromatography gave the desired product ester (31.3 g, 69% yield).
-
To the solution of tert-butyl 7-(4-(ethoxycarbonyl)phenyl)-2,3-dihydrobenzo[f][1,4]oxazepine-4(5H)-carboxylate (10.3 g, 25.93 mmol) in MeOH (120 mL) was added a solution of 4 N HCl in dioxane (50 mL). The reaction mixture was heated to 50° C. for 3 h (monitored by LC/MS). The reaction mixture was allowed to cool to rt. Ethyl 4-(2,3,4,5-tetrahydro-1,4-benzoxazepin-7-yl)benzoate as the hydrochloride salt (8.8 g, 99% yield) was collected by suction filtration.
-

-
tert-Butyl-5-bromo-2-hydroxybenzyl(2-hydroxyethyl)carbamate. Commercially-available 5-bromo-2-hydroxybenzaldehyde (4.0 g, 10 mmol) and 2-aminoethanol were combined in THF/MeOH (100 mL, 10:1) and sodium borohydride (0.76 g, 2.0 mmol) was added with stirring. The resulting reaction mixture was stirred at 40° C. for 4 h, concentrated on a rotary evaporator then diluted with EtOAc (50 mL) and saturated NaHCO3 (30 mL). To this suspension was added di-tert-butyl dicarbonate (2.83 g, 13 mmol). The mixture was stirred at rt overnight. The organic layer was washed with water, dried over anhydrous magnesium sulfate, filtered, and concentrated on a rotary evaporator. Hexane was subsequently added to the crude reaction product which resulted in the formation of a white solid. This slurry was filtered to obtain the desired product (6.8 g, 98%) as a white solid. MS (EI) for C14H20BrNO4, found 346 (MH+).
-
tert-Butyl-7-bromo-2,3-dihydrobenzo[f][1,4]oxazepine-4(5H)-carboxylate. tert-Butyl-5-bromo-2-hydroxybenzyl(2-hydroxyethyl)carbamate (3.46 g, 10 mmol) and triphenylphosphine (3.96 g, 15 mmol) were combined in DCM (100 mL) and diisopropyl azodicarboxylate (3.03 g, 15 mmol) was added. The resulting reaction mixture was stirred at rt for 12 h. The reaction mixture was washed with water, dried, filtered, and concentrated on a rotary evaporator. The resulting crude product was purified via silica gel chromatography eluting with 8:2 hexane/ethyl acetate to give the desired product (1.74 g, 53%) as a white solid. MS (EI) for C14H18BrNO3, found 328 (MH+).
- Reference Example 4tert-Butyl-7-bromo-2,3-dihydrobenzo[f][1,4]oxazepine-4(5H)-carboxylate
Reference Example 54-(tert-Butoxycarbonyl)-2,3,4,5-tetrahydrobenzo[f][1,4]oxazepin-7-ylboronic acid
-

-
To a stirred solution of tert-butyl-7-bromo-2,3-dihydrobenzo[f][1,4]oxazepine-4(5H)-carboxylate (10 g, 30.5 mmol), prepared as described in Reference Example 4, and triisopropylborate (9.1 mL, 40 mmol) in dry tetrahydrofuran (100 mL) was added dropwise n-butyllithium in tetrahydrofuran (1.6 M, 25 mL, 40 mmol) while maintaining the temperature below −60° C. Upon completion of addition, the reaction mixture was stirred for 30 min, then quenched with 1 N aqueous hydrochloric acid (35 mL) and allowed to warm to rt. The reaction mixture was extracted with ethyl acetate, dried over anhydrous magnesium sulfate, filtered and concentrated on a rotary evaporator. Hexane was subsequently added to the crude reaction product which resulted in the formation of a white solid. This slurry was stirred for 1 h and filtered to obtain 4-(tert-butoxycarbonyl)-2,3,4,5-tetrahydrobenzo[f][1,4]oxazepin-7-ylboronic acid (8.6 g, 95%) as a white solid. MS (EI) for C14H20BNO5: 194 (M-Boc).
- Example 2[7-(6-Aminopyridin-3-yl)-2,3-dihydro-1,4-benzoxazepin-4(5H)-yl][3-fluoro-2-methyl-4-(methylsulfonyl)phenyl]methanone
-

-
tert-Butyl 7-(6-aminopyridin-3-yl)-2,3-dihydrobenzo[f][1,4]oxazepine-4(5H)-carboxylate. To a mixture of 4-(tert-butoxycarbonyl)-2,3,4,5-tetrahydrobenzo[f][1,4]oxazepin-7-ylboronic acid (1.52 g, 5.2 mmol), prepared as described in Reference Example 5, 2-amino-5-bromopyridine (900 mg, 5.2 mmol), and potassium carbonate (1.73 g, 12.5 mmol) in 1,2-dimethoxyethane/water (30 mL/10 mL) was added tetrakis(triphenylphosphine)palladium(0) (90 mg, 1.5 mol %) and the reaction mixture was purged with nitrogen and stirred at reflux for 3 h. The reaction was cooled to rt, diluted with water/ethyl acetate (50 mL/50 mL), and the separated aqueous layer was extracted with ethyl acetate. The resulting emulsion was removed by filtration. The combined organic layer was washed with brine, dried with sodium sulfate, filtered and concentrated under reduced pressure, and the residue was triturated with toluene for 1 h. The resulting off-white solid was isolated by filtration to give the desired product (1.37 g, 77%) as an off-white solid. MS (EI) for C19H23N3O3: 342 (MH+).
-
5-(2,3,4,5-Tetrahydrobenzo[f][1,4]oxazepin-7-yl)pyridine-2-amine. To a stirred solution of tert-butyl 7-(6-aminopyridin-3-yl)-2,3-dihydrobenzo[f][1,4]oxazepine-4(5H)-carboxylate (1.36 g, 3.98 mmol) in 1,4-dioxane (5 mL) was added 4 N hydrogen chloride in 1,4-dioxane (5 mL) and the reaction mixture was stirred at rt overnight. The reaction was concentrated on a rotary evaporator and the residue was triturated with ether. The solid was isolated by filtration. This solid was dissolved in water (5 mL) and made basic with 5 N sodium hydroxide to pH 11-12. The brownish sticky oil that aggregated at the bottom was isolated and the aqueous layer was extracted with 5% methanol in ethyl acetate. The extracts were dried with sodium sulfate and concentrated on a rotary evaporator. The brownish sticky oil was dissolved with a mixture of methanol/ethyl acetate, combined with the isolated organic residue and concentrated under reduced pressure to give a yellow solid. This solid was triturated with dichloromethane (10 mL) for 1 h and a yellow solid was isolated by filtration and dried under high vacuum to give amine the desired product (920 mg, 96%). MS (EI) for C14H15N3O: 242 (MH+).
-
[7-(6-Aminopyridin-3-yl)-2,3-dihydro-1,4-benzoxazepin-4(5H)-yl][3-fluoro-2-methyl-4-(methylsulfonyl)phenyl]methanone. To a stirred suspension of 5-(2,3,4,5-tetrahydrobenzo[f][1,4]oxazepin-7-yl)pyridine-2-amine (85 mg, 352 μmol) and triethylamine (54 μL, 387 μmol) in dichloromethane (10 mL) was added 3-fluoro-2-methyl-4-(methylsulfonyl)benzoyl chloride (91 mg, in 3 mL of dichloromethane), prepared as described in Reference Example 1, at 0° C. for 2 h. After stirring for an additional 1 h at rt, the reaction mixture was diluted with water (5 mL) and the separated aqueous layer was extracted with dichloromethane. The combined extracts were dried with sodium sulfate, filtered and concentrated under reduced pressure to give a light-yellow solid that was purified via silica gel chromatography to give the desired product (113 mg, 70%) as a white solid. 1H NMR (400 MHz, DMSO-d6): δ 8.24-8.03 (dd, 1H), 7.79-7.71 (m, 1H), 7.71-7.69 (dd, 0.5H), 7.57-7.57 (d, 0.5H), 7.44-7.40 (m, 1.5H), 7.29-7.19 (dd, 1H), 7.05-7.01 (dd, 1H), 6.64-6.63 (d, 0.5H), 6.54-6.45 (dd, 1H), 6.06 (s, 2H), 4.93-4.31 (m, 2H), 4.31-3.54 (m, 4H), 3.37-3.36 (d, 3H), 2.12-1.77 (d, 3H). MS (EI) C23H22FN3O4S: 456 (MH+).
PAPER
Org. Process Res. Dev., 2015, 19 (7), pp 721–734
DOI: 10.1021/acs.oprd.5b00037
http://pubs.acs.org/doi/abs/10.1021/acs.oprd.5b00037

The benzoxazepine core is present in several kinase inhibitors, including the mTOR inhibitor 1. The process development for a scalable synthesis of 7-bromobenzoxazepine and the telescoped synthesis of 1 are reported. Compound 1 consists of three chemically rich, distinct fragments: the tetrahydrobenzo[f][1,4]oxazepine core, the aminopyridyl fragment, and the substituted (methylsulfonyl)benzoyl fragment. Routes were developed for the preparation of 3-fluoro-2-methyl-4-(methylsulfonyl)benzoic acid (17) and tert-butyl 7-bromo-2,3-dihydrobenzo[f][1,4]oxazepine-4(5H)-carboxylate (2). The processes for the two compounds were scaled up, and over 15 kg of each starting material was prepared in overall yields of 42% and 58%, respectively.
A telescoped sequence beginning with compound 2 afforded 7.5 kg of the elaborated intermediate 5-(2,3,4,5-tetrahydrobenzo[f][1,4]oxazepin-2-amine dihydrochloride (6) in 63% yield. Subsequent coupling with benzoic acid 17 gave 7.6 kg of the target compound 1 in 84% yield. The preferred hydrochloride salt was eventually prepared. The overall yield for the synthesis of inhibitor 1 was 21% over eight isolated synthetic steps, and the final salt was obtained with 99.7% HPLC purity.
[7-(6-Aminopyridin-3-yl)-2,3-dihydro-1,4-benzoxazepin-4(5H)-yl][3-fluoro-2-methyl-4-(methylsulfonyl)phenyl]methanone (1)
Compound 1 was observed as a mixture of two rotational isomers in the 1H and 13C NMR spectra.
1H NMR (400 MHz, DMSO-d6): δ 8.24–8.03 (dd, 1H), 7.79–7.71 (m, 1H), 7.71–7.69 (dd, 0.5H), 7.57–7.57 (d, 0.5H), 7.44–7.40 (m, 1.5H), 7.29–7.19 (dd, 1H), 7.05–7.01 (dd, 1H), 6.64–6.63 (d, 0.5H), 6.54–6.45 (dd, 1H), 6.06 (s, 2H), 4.93–4.31 (m, 2H), 4.31–3.54 (m, 4H), 3.37–3.36 (d, 3H), 2.12–1.77 (d, 3H). 13C NMR (100 MHz, DMSO-d6): δ 167.3, 167.2, 166.6, 166.6, 158.9, 158.9, 158.4, 158.4, 157.4, 157.2, 155.9. 155.8, 145.4, 145.1, 145.1, 144.0, 143.9, 135.0, 134.7, 132.9, 132.8, 129.4, 129.2, 128.2, 128.2, 128.1, 128.0, 127.0, 126.9, 126.8, 125.9, 125.6, 125.4, 123.6, 123.5, 123.3, 123.1, 122.8, 122.0, 122.0, 121.9, 121.9, 121.2, 120.7, 107.8, 107.8, 70.9, 70.8, 51.1, 51.1, 47.4, 46.5, 43.5, 43.5, 43.5, 43.4, 11.0, 10.9, 10.7, 10.6. IR (KBr pellet): 1623, 1487, 1457, 1423, 1385, 1314, 1269, 1226, 1193, 1144, 1133, 1054, 1031, 962, 821, 768 cm–1. MS (EI) C23H22FN3O4S: found 456.2 ([M + H]+). High-resolution MS (FAB-MS using glycerol as a matrix) for C23H22FN3O4S: found 456.13943 ([M + H]+), calcd 456.13878.
[7-(6-Aminopyridin-3-yl)-2,3-dihydro-1,4-benzoxazepin-4(5H)-yl][3-fluoro-2-methyl-4-(methylsulfonyl)phenyl]methanone Hydrochloride (1·HCl)
1·HCl as a white solid (7.81 kg, 95%, 99.7% purity by AN-HPLC).
Analyses: OVI: DMF < 100 ppm, DMC < 100 ppm, acetone = 3081 ppm, MTBE < 100 ppm, iPAc < 100 ppm, THF < 100 ppm. Heavy metals: Pd ≤ 0.2 ppm, others < 20 ppm (USP ⟨231⟩). 1H NMR (400 MHz, DMSO-d6), equimolar amounts of two rotamers: δ 8.20–8.40 (br s, 2H), 8.33 (s, 0.5H), 8.31 (d, J = 2.8 Hz, 0.5H), 8.15 (d, J = 2.0 Hz, 0.5H), 7.96 (dd, J = 9.7, 2.0 Hz, 0.5H), 7.70–7.78 (m, 1.5H), 7.55–7.57 (m, 0.5H), 7.51–7.55 (m, 0.5H), 7.28 (d, J = 8.6 Hz, 0.5H), 7.17 (d, J = 3.1 Hz, 0.5H), 7.15 (d, J = 5.1 Hz, 0.5H), 7.05–7.11 (m, 1.5H), 6.83 (d, J = 2.7 Hz, 0.5H), 4.86–4.99 (m, 1H), 4.29–4.56 (m, 1H), 4.10–4.27 (m, 2H), 3.93–4.04 (m, 0.5H), 3.45–3.65 (m, 1.5H), 3.37 (s, 1.5 H), 3.35 (s, 1.5H), 2.12 (d, J = 2.0 Hz, 1.5H), 1.76 (d, J = 2.0 Hz, 1.5H). 13C NMR (100 MHz, DMSO-d6), equimolar amounts of two rotamers: δ 168.1, 167.5, 159.4, 159.2, 159.1, 156.6, 153.9, 153.8, 144.6, 142.9, 142.3, 133.0, 132.7, 130.0, 129.9, 129.7, 129.5, 129.1, 129.0, 128.9, 128.8, 128.5, 127.7, 127.6, 127.5, 127.1, 126.9, 124.4, 124.3, 124.1, 122.7, 122.1, 121.6, 114.4, 71.2, 51.7, 51.3, 47.9, 46.9, 44.3, 44.2, 11.7, 11.4.
REFERENCES
Anand, N.; Benzoxazepines as Inhibitors of PI3K/mTOR and Methods of their Use and Manufacture. U.S. Patent 8,648,066, Feb 11, 2014.
Aay, N.; Benzoxazepines as Inhibitors of PI3K/mTOR and Methods of their Use and Manufacture. U.S. Patent 8,637,499, Jan 28,2014.
| US8637499 * | May 25, 2010 | Jan 28, 2014 | Exelixis, Inc. | Benzoxazepines as inhibitors of PI3K/mTOR and methods of their use and manufacture |
| US20120258953 * | May 25, 2010 | Oct 11, 2012 | Exelixis, Inc. | Benzoxazepines as Inhibitors of PI3K/mTOR and Methods of Their Use and Manufacture |
PROFILE

Senior Director
Chemical Development at Dermira, Inc.
Lives San jose caifornia
E-mail: sriram.naganathan@dermira.com.

LINKS
https://www.linkedin.com/pub/sriram-naganathan/3/50a/5b6
https://www.facebook.com/sriram.naganathan.5
snaganat@exelixis.com, sriramrevathi@yahoo.com
Summary
Chemical process-development and CMC professional offering 20 years of experience from preclinical development through commercialization of small molecules and peptides.
Hands-on experience in multi-step synthesis, route-scouting, process development, scale-up, tech transfer to CRO/CMO, including manufacture under cGMP and process validation.
Extensive knowledge of CMC regulatory landscape (FDA, EMEA) including preparation of CMC sections of IND, IMPD, NDA and MAA
Experience
Senior Director, Chemical Development
Dermira, Inc.
January 2015 – Present (10 months)Menlo Park, CA
Consultant
Intarcia Therapeutics
December 2014 – January 2015 (2 months)
Senior Director
Exelixis
March 2013 – November 2014 (1 year 9 months)South San Francisco, CA
210 E. Grand Ave
South San Francisco , California 94080
United States
United States
Company Description: Exelixis, Inc. (Exelixis) is developing therapies for cancer and other serious diseases. Through its drug discovery and development activities, the Company is… more
Senior Scientist II
Exelixis
August 2004 – January 2008 (3 years 6 months)
Associate Director
CellGate, Inc.
2000 – 2004 (4 years)
Research Scientist
Roche Bioscience
1997 – 2000 (3 years)
Research Scientist
Cultor
1995 – 1997 (2 years)
Research Scientist
Pfizer
1994 – 1997 (3 years)
Research Assistant Professor
University of Pittsburgh
April 1992 – October 1994 (2 years 7 months)
Worked on Vitamin K mechanism in the labs of (Late) Prof Paul Dowd
Education
Vivekananda College (University of Madras), India
Bachelor of Science (B.Sc.), Chemistry
1980 – 1983
![]()

(Above) Former Group members join Professor Block at the National ACS Meeting in San Francisco, March 2010: from left, Dr. Shuhai Zhao, Dr. Sherida Johnson, Professor Block, Dr. Sriram Naganathan.
Sriram Naganathan, Ph.D. 1992, snaganat@exelixis.com, sriramrevathi@yahoo.com

As many things change, many things remain constant. One such constant is the frequent reminder that “You can take the boy out of sulfur chemistry but you cannot take sulfur chemistry out of the boy”. At every stage of my professional career organic chemistry of sulfur and sulfur-containing compounds have followed me (or is it the other way around?). Not many can point to the cover of an Angewandte Chemie issue as a synopsis of his/her thesis work – I will be forever grateful for that opportunity received in the Block Group.
As a post-doc in the late Prof. Paul Dowd’s lab at the University of Pittsburgh we used sulfur-containing analogs of vitamin K to probe the mechanism of action. I was then hired at Pfizer Central Research in Groton, CT in the Specialty Chemicals Division to investigate possible decomposition pathways of sulfur-containing high-intensity artificial sweeteners.
At Roche Bioscience (Palo Alto, CA) and Exelixis (South San Francisco, CA – my current job………CHANGED……Dermira) I was involved in process development for the preparation of therapeutic agents, several of them sulfur-containing molecules. Between those two positions I was a Senior Scientist at CellGate (Sunnyvale, CA).
We attempted to exploit the chemistry of sulfur-containing linkers to target the delivery active pharmaceutical agents, using the transport properties of polyarginines. Although I thought I was only training to become a synthetic organic chemist, I did not realize that my passion was really organic reaction mechanisms until I arrived in the Block lab – the two arms of the science are truly inseparable.
I realize after many years that the seed was really sown and nurtured during the many friendly and sometimes-fiery discussions in the lab, and further solidified in my post-doc years. I learned that every “blip-in-the-baseline” cannot to be ignored, and is part of the whole story.
As a process chemist in the pharma industry, I can attribute much of my success to lessons about careful and critical evaluation of primary data and thorough knowledge of reaction mechanisms. I am currently Director, Chemical Development, at Exelixis.NOW DERMIRA.
My primary responsibility involves the manufacture and potential commercialization of our primary product, cabozantinib. It was only natural that I developed a strong interest in the science of cooking and food. I have been pursuing this avenue since moving to Northern California.
I am also an avid gardener, experimenting with growing interesting varieties of chilies, tomatoes and then combining those with all sorts of alliums. It does help that I live close enough to Gilroy, CA, that I can often smell what they are famous for as I walk out of the front door!! I have shared my knowledge in several lectures at the Tech Museum (San Jose, CA) where I was a volunteer exhibit explainer.
My family (my wife Revathi and our two high-school-age daughters Swetha and Sandhya) like to travel and also enjoy the outdoor recreation so abundant in Northern California. We try to take in a new country each year and accomplish personal challenges. After many interesting years in the tech-industry, Revathi is a full-time mom. She is also a fitness instructor at the Y. Swetha and Sandhya are part of the water polo and swim teams at their school.
Swetha is very active in a leadership role for the robotics team, and Sandhya belongs to the quiz team. Revathi and I climbed Half Dome (Yosemite) a few years ago and I just completed a 100-mile bicycle ride around Lake Tahoe.
I remain a highly-opinionated baseball and college basketball fan (favorite teams: in order, Kansas, North Carolina and whoever happens to be playing Missouri and Duke). I am still an avid photographer, although I spend no money on film (I thought I was going to be the last guy on the planet still shooting film!!). I greatly value the many friendships developed during my stay in Albany and keep in touch with many.
In fact, one of my roommates from the SUNY days was instrumental in me getting my present position. Of course, this also means that I have lost touch with several friends during the past decades. If you are reading this and haven’t contacted me in a few years, please do, via e-mail.
We enjoy entertaining guests who drop by – so now you have no excuse not to contact us, especially when you visit the SF Bay Area.

OLD PROFLE……Dr Sriram Naganathan received his Ph.D. from SUNY-Albany where he studied organosulfur chemistry. He is currently an Associate Director at CellGate, Inc. located in Sunnyvale, California. CellGate is involved in the commercialization of novel medicines by utilizing proprietary transporter technology, based on oligomers of arginine, to enhance the therapeutic potential of existing drugs. His responsibilities include process development, scale-up and GMP production of clinical candidates, as well some basic research. He previously held positions at Pfizer Central Research and Roche Bioscience.
Dermira


Thomas G. Wiggans | Founder & Chief Executive Officer……..http://dermira.com/about-us/management-team/

CEO TOM WIGGANS, LEFT AND CMO GENE GAUER, RIGHT


Exelixis, Inc.
![]()

210 East Grand Avenue
So. San Francisco, CA 94080
(650) 837-7000 phone
(650) 837-8300 fax
Directions to Exelixis, Inc.
101 Northbound from San Francisco Airport:
- Take 101 North toward San Francisco.
- Take the Grand Avenue exit, exit 425A, toward So San Francisco.
- Turn right onto East Grand Ave.
- 210 East Grand Ave is on your right-hand side.
101 Southbound from San Francisco:
- Take 101 South.
- Take the Grand Avenue exit. Turn left at the first light.
- Immediately turn left at the first light onto Grand Avenue (which will become East Grand Avenue)
- 210 East Grand Ave is on your right-hand side.
////////////mTOR inhibitor, Exelixis, Inc., PI3K, phosphatidylinositol-3-kinase, XL 388, XL388, IND Filed
IPI 504, Retaspamycin, Retaspimycin

IPI 504, Retaspamycin, Retaspimycin
CAS 857402-63-2
Cas 857402-23-4 ( Retaspimycin); 857402-63-2 ( Retaspimycin HCl).
MF C31H45N3O8 BASE
MW: 587.32067 BASE
Infinity Pharmaceuticals Inc, INNOVATOR
[(3R,5S,6R,7S,8E,10S,11S,12Z,14E)-6,20,22-trihydroxy-5,11-dimethoxy-3,7,9,15-tetramethyl-16-oxo-21-(prop-2-enylamino)-17-azabicyclo[16.3.1]docosa-1(22),8,12,14,18,20-hexaen-10-yl] carbamate;hydrochloride
17-Allylamino-17-demethoxygeldanamycin Hydroquinone Hydrochloride
| Retaspimycin hydrochloride; 8,21-didehydro-17-demethoxy-18,21-dideoxo-18,21-dihydroxy-17-(2-propenylamino)-geldanamycin monohydrochloride | |
| Application: | A novel, water-soluble, potent inhibitor of heat-shock protein 90 (Hsp90) |
| Molecular Weight: | 624.17 ……….HCl salt |
| Molecular Formula: | C31H46ClN3O8……….HCl salt |
Introduction
IPI-504 is a novel, water-soluble, potent inhibitor of heat-shock protein 90 (Hsp90).
Orphan drug designation was assigned to the compound by the FDA for the treatment of gastrointestinal stromal cancer (GIST).

Retaspimycin Hydrochloride is the hydrochloride salt of a small-molecule inhibitor of heat shock protein 90 (HSP90) with antiproliferative and antineoplastic activities. Retaspimycinbinds to and inhibits the cytosolic chaperone functions of HSP90, which maintains the stability and functional shape of many oncogenic signaling proteins and may be overexpressed or overactive in tumor cells. Retaspimycin-mediated inhibition of HSP90 promotes the proteasomal degradation of oncogenic signaling proteins in susceptible tumor cell populations, which may result in the induction of apoptosis.
Phase I study of Retaspimycin: A phase 1 study of IPI-504 (retaspimycin hydrochloride) administered intravenously twice weekly for 2 weeks at 22.5, 45, 90, 150, 225, 300 or 400 mg/m(2) followed by 10 days off-treatment was conducted to determine the safety and maximum tolerated dose (MTD) of IPI-504 in patients with relapsed or relapsed/refractory multiple myeloma (MM). Anti-tumor activity and pharmacokinetics were also evaluated. Eighteen patients (mean age 60.5 years; median 9 prior therapies) were enrolled. No dose-limiting toxicities (DLTs) were reported for IPI-504 doses up to 400 mg/m(2).
The most common treatment-related adverse event was grade 1 infusion site pain (four patients). All other treatment-related events were assessed as grade 1 or 2 in severity. The area under the curve (AUC) increased with increasing dose, and the mean half-life was approximately 2-4 h for IPI-504 and its metabolites. Four patients had stable disease, demonstrating modest single-agent activity in relapsed or relapsed/refractory MM. (source: Leuk Lymphoma. 2011 Dec;52(12):2308-15.)

Figure Hsp90 protein partners and clients destabilized by Hsp90 inhibition (Jackson et al., 2004).
In a different approach, Infinity Pharmaceuticals has developed IPI504 (retaspimycin or 17-AAG hydroquinone, Figure 4) (Adams et al., 2005; Sydor et al., 2006), a new GA analogue, in which the quinone moiety was replaced by a dihydroquinone one. Indeed, the preclinical data suggested that the hepatotoxicity of 17-AAG was attributable to the ansamycin benzoquinone moiety, prone to nucleophilic attack.
Furthermore, it was recently reported that the hydroquinone form binds Hsp90 with more efficiency than the corresponding quinone form (Maroney et al., 2006). In biological conditions, the hydroquinone form can interconvert with GA, depending on redox equilibrium existing in cell. It has been recently proposed, that NQ01 (NAD(P)H: quinone oxidoreductase) can produce the active hydroquinone from the quinone form of IPI504 (Chiosis, 2006).
However, Infinity Pharmaceuticals showed that if the overexpression of NQ01 increased the level of hydroquinone and cell sensitivity to IPI504, it has no significant effect on its growth inhibitory activity. These results suggest that NQ01 is not a determinant of IPI504 activity in vivo (Douglas et al., 2009).

Figure 4: GA, 17-AAG, 17-DMAG and IPI504.
PATENT
http://www.google.com/patents/EP2321645A1?cl=en
Geldanamycin (IUPAC name ([18S-(4E,5Z,8R*.9R*.10E,12R*.13S*,14R*,l6S*)]- 9- [(aminocarbonyl)oxy]- 13- hydroxy- 8,14,19- trimetoxy- 4,10,12,16- tetramethyl- 2- azabicyclo[16.3.1.]docosa- 4,6.10,18,21- pentan- 3.20,22trion) is a benzoquinone ansamycin antibiotic which may be produced by the bacterium Streptorayces hygroscopicus. Geldanamycin binds specifically to HSP90 (Heat Shock Protein 90) and alters its function.
While Hsp90 generally stabilizes folding of proteins and, in particular in tumor cells, folding of overexpressed/mutant proteins such as v-Src. Bcr-Abl and p53. the Hsp90 inhibitor Geldanamycin induces degradation of such proteins.
The respectiv e formula of geldanamycin is given herein below:

E\en though geldanamycin is a potent antitumor agent, the use of geldanamycin also shows some negathe side-effects (e.g. hepatotoxicity) which led to the dev elopment of geldanamycin analogues/derivatives, in particular analogues/deriv atives containing a derivatisation at the 17 position. Without being bound by theory , modification at the 17 position of geldanamycin may lead to decreases hepatotoxicity.
Accordingly geldanamycin analogues/derivatives which are modified at the 17 position, such as 17-AAG (17-N-Allylamino-17-demethoxygeldanamycin), are preferred in context of the present invention. Also preferred herein are geldanamycin derivatives to be used in accordance with the present invention which are water-soluble or which can be dissoh ed in water completely (at least 90 %. more preferably 95 %. 96 %. 97 %, 98 % and most preferably 99 %). 17-AAG ([QS.5S,6RJS$EΛ0R,l \SΛ2E,14E)-2\- (allylamino)-6-hydroxy-5.11-diraethoxy-
3.7.9,15-tetramethyl-16.20.22-trioxo-17-azabicyclo[16.3.1]docosa-8,12.14,18,21-pentaen-10- yl] carbamate) is. as mentioned above a preferred derivative of geldanamycin. 17- AAG is commercially available under the trade name “Tanespimycin“ (also known as KOS-953) for example from Kosan Biosciences Incorporated (Acquired by Bristol-Myers Squibb Company). Tanespimycin is presently studied in phase II clinical trials for multiple myeloma and breast cancer and is usually administered intravenously.
The respective formula of 17- AAG is given herein below:

Preferred geldanamycin-derh ative (HSP90 inhibitor) to be used in context of the present invention are IPI-504 (also known as retaspiimcin or Mcdi-561 : lnfinin Pharmaceuticals (Medlmmunc/ Astra Zeneca)). Clinical trials on the use of IPI-504 (which is usually administered intravenously) in the treatment of non-small cell lung cancer (NSCLC) and breast cancer are performed. Also alvespimycin hy drochloride (Kosan Biosciences Incorporated Acquired By : Bristol-Myers Squibb Company) is a highly potent, water-soluble and orally acti\e derivative of geldanamycin preferably used in context of the present invention.

IPI-504
PATENT
WO 2005063714
http://www.google.co.ug/patents/WO2005063714A1?cl=en
Example 24
Preparation of Air-stable Hydroquinone Derivatives of the Geldanamycin Family of Molecules
,

17-Allylamino-17-Demethoxygeldanamycin (10.0 g, 17.1 mmol) in ethyl acetate
(200 mL) was stirred vigorously with a freshly prepared solution of 10% aqueous sodium hydrosulfite (200 mL) for 2 h at ambient temperature. The color changed from dark purple to bright yellow, indicating a complete reaction. The layers were separated and the organic phase was dried with magnesium sulfate (15 g). The drying agent was rinsed with ethyl acetate (50 mL). The combined filtrate was acidified with 1.5 M hydrogen chloride in ethyl acetate (12 mL) to pH 2 over 20 min. The resulting slurry was stirred for 1.5 h at ambient temperature. The solids were isolated by filtration, rinsed with ethyl acetate (50 mL) and dried at 40 °C, 1 mm Hg, for 16 h to afford 9.9 g (91%) of off-white solid. Crude hydroquinone hydrochloride (2.5 g) was added to a stirred solution of 5% 0.01 N aq. hydrochloric acid in methanol (5 mL). The resulting solution was clarified by filtration then diluted with acetone (70 mL). Solids appeared after 2-3 min. The resulting slurry was stirred for 3 h at ambient temperature, then for 1 h at 0-5 °C. The solids were isolated by filtration, rinsed with acetone (15 mL) and dried
PAPER
J. Med. Chem., 2006, 49 (15), pp 4606–4615
DOI: 10.1021/jm0603116

17-Allylamino-17-demethoxygeldanamycin (17-AAG)1 is a semisynthetic inhibitor of the 90 kDa heat shock protein (Hsp90) currently in clinical trials for the treatment of cancer. However, 17-AAG faces challenging formulation issues due to its poor solubility. Here we report the synthesis and evaluation of a highly soluble hydroquinone hydrochloride derivative of 17-AAG, 1a (IPI-504), and several of the physiological metabolites. These compounds show comparable binding affinity to human Hsp90 and its endoplasmic reticulum (ER) homologue, the 94 kDa glucose regulated protein (Grp94). Furthermore, the compounds inhibit the growth of the human cancer cell lines SKBR3 and SKOV3, which overexpress Hsp90 client protein Her2, and cause down-regulation of Her2 as well as induction of Hsp70 consistent with Hsp90 inhibition. There is a clear correlation between the measured binding affinity of the compounds and their cellular activities. Upon the basis of its potent activity against Hsp90 and a significant improvement in solubility, 1a is currently under evaluation in Phase I clinical trials for cancer.
17-Allylamino-17-demethoxygeldanamycin Hydroquinone Hydrochloride Ia
17-AAG hydroquinone hydrochloride (1a) as an off-white solid (11 g, 18 mmol, 80% yield). HPLC purity: 99.6%;
IR (neat): 3175, 2972, 1728, 1651, 1581, 1546, 1456, 1392, 1316, 1224, 1099, 1036 cm-1;
1H NMR (CDCl3:d6-DMSO, 6:1, 400 MHz):
δ 10.20 (1H, br), 9.62 (2H, br), 8.53 (1H, s), 8.47 (1H, s), 7.74 (1H, s), 6.72 (1H, d, J= 11.6 Hz), 6.28 (1H, dd, J = 11.6, 11.2 Hz), 5.73 (1H, dddd, J = 17.2, 10.0, 3.2, 2.4 Hz), 5.53 (1H, d, J = 10.4 Hz), 5.49 (1H, dd, J = 10.8, 10.0 Hz), 5.32 (2H, s), 5.04 (1H, d, J = 4.8 Hz), 5.02 (1H, d, J = 16.0 Hz), 4.81 (1H, s), 4.07 (1H, d, J = 9.6 Hz), 3.67 (2H, d, J = 6.4 Hz), 3.31 (1H, d,J = 8.8 Hz), 3.07 (3H, s), 3.07−3.04 (1H, m), 2.99 (3H, s), 2.64 (1H, m), 2.52−2.49 (1H, m), 1.76 (3H, s), 1.61−1.39 (3H, m), 0.78 (3H, d, J = 6.4 Hz), 0.64 (3H, d, J = 7.2 Hz);
13C NMR (CDCl3:d6-DMSO, 6:1, 100 MHz): δ 167.3, 155.8, 143.3, 136.3, 135.0, 134.2, 132.9, 132.1, 128.8, 127.6, 125.9, 125.3, 123.7, 123.0, 115.1, 104.5, 80.9, 80.7, 80.1, 72.5, 56.2, 56.2, 52.4, 34.6, 33.2, 31.1, 27.2, 21.6, 12.1, 12.1, 11.7;
HRMS calculated for C31H45N3O8 (M+ + H): 588.3285, Found 588.3273.
POSTER
Synthesis and biological evaluation of IPI-504, an aqueous soluble analog of 17-AAG and potent inhibitor of Hsp90
MEDI 210 |
| IPI-504 is the hydroquinone hydrochloride salt of 17-allylamino-17-demethoxy-geldanamycin (17-AAG), an Hsp90 inhibitor that is currently in clinical trials for the treatment of cancer.
IPI-504 demonstrates high aqueous solubility (>200 mg/mL). Interestingly, in vitro and in vivo IPI-504 interconverts with 17-AAG and exists in a pH and enzyme-mediated redox equilibrium. This occurs due to oxidation of the hydroquinone (IPI-504) to the quinone (17-AAG) at physiological pH and the reduction of 17-AAG by quinone reductases such as NQO1 to IPI-504. Here we report the design and synthesis of the stabilized hydroquinone IPI-504 and its inhibitory effect against Hsp90 and Grp94. Although IPI-504 was originally designed to be a soluble prodrug of 17-AAG, the hydroquinone is more potent than the quinone in the biochemical Hsp90 binding assay. Various hydroquinone analogs have been prepared to investigate the structure activity relationship of hydroquinone binding to Hsp90. Hydroquinone and quinone forms of 17-AAG metabolites show comparable binding affinities for Hsp90 and in cancer cell lines, hydroquinone analogs elicit specific responses consistent with Hsp90 inhibition. The desirable pharmacological properties as well as in vitro and in vivo activity of our lead compound, IPI-504, has led to the initiation of Phase I clinical trials in multiple myeloma. |
| http://oasys2.confex.com/acs/231nm/techprogram/P945016.HTM |
|
Geldanamycin and Beyond: Progress Toward the Development of HSP90 Inhibitors
8:30 AM-12:05 PM, Wednesday, 29 March 2006 Georgia World Congress Center — Georgia Ballroom 1, OralDivision of Medicinal ChemistryThe 231st ACS National Meeting, Atlanta, GA, March 26-30, 2006 |
References
Synthesis and biological evaluation of IPI-504, an aqueous soluble analog of 17-AAG and potent inhibitor of Hsp90
231st Am Chem Soc (ACS) Natl Meet (March 26-30, Atlanta) 2006, Abst MEDI 210
Design, synthesis, and biological evaluation of hydroquinone derivatives of 17-amino-17-demethoxygeldanamycin as potent, water-soluble inhibitors of Hsp90
J Med Chem 2006, 49(15): 4606
http://www.biotechduediligence.com/retaspamycin-hcl-ipi-504.html
///////////////////Hsp90, IPI-504, infinity pharma, Retaspamycin, Retaspimycin
MARIZEV® (Omarigliptin), Merck’s Once-Weekly DPP-4 Inhibitor for Type 2 Diabetes, Approved in Japan

MARIZEV® (Omarigliptin), Merck’s Once-Weekly DPP-4 Inhibitor for Type 2 Diabetes, Approved in Japan
KENILWORTH, N.J.–(BUSINESS WIRE)–Merck (NYSE:MRK), known as MSD outside the United States and Canada, today announced that the Japanese Pharmaceuticals and Medical Devices Agency (PMDA) has approved MARIZEV® (omarigliptin) 25 mg and 12.5 mg tablets, an oral, once-weekly DPP-4 inhibitor indicated for the treatment of adults with type 2 diabetes. Japan is the first country to have approved omarigliptin……….http://www.mercknewsroom.com/news-release/prescription-medicine-news/marizev-omarigliptin-mercks-once-weekly-dpp-4-inhibitor-type
syn…….https://newdrugapprovals.org/2014/04/18/omarigliptin-mk-3102-in-phase-3-for-type-2-diabetes/

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HEREJoin me on Linkedin
Join me on Facebook
Join me on twitter
Join me on google plus Googleplus
Googleplus amcrasto@gmail.com
amcrasto@gmail.com
/////////////MARIZEV, (Omarigliptin), Merck’s, Once-Weekly, DPP-4 Inhibitor, Type 2 Diabetes, Approved, Japan
Mavatrep, JNJ 39439335,

Mavatrep; UNII-F197218T99; Mavatrep (USAN); JNJ-39439335; 956274-94-5;
2-(2-(2-(2-(4-trifluoromethylphenyl)vinyl)-1H-benzimidazol-5-yl)phenyl)propan-2-ol
(E)-2-(2-{2-[2-(4-trifluoromethyl-phenyl)-vinyl]-1H-benzimidazol-5-yl}-phenyl)-propan-2-ol
(E)-2-(2-(2-(4-(Trifluoromethyl)styryl)-1H-benzo[d]imidazol-5-yl)phenyl)-propan-2-ol Hydrochloride
Phase I Musculoskeletal pain; Pain
- 01 Mar 2013 Janssen Research and Development completes a phase I trial in Japanese and Caucasian adult male volunteers in the US (NCT01631487)
- 01 Mar 2013 Janssen completes enrolment in its phase I trial for Pain (in volunteers) in the USA (NCT01631487)
- 05 Feb 2013 Janssen Research and Development initiates enrolment in a phase I trial for Pain (Japanese and Caucasian volunteers) in USA (NCT01631487)
- Originator Johnson & Johnson Pharmaceutical Research & Development
- Developer Janssen Research & Development
- Class Analgesics; Benzimidazoles; Small molecules
- Mechanism of Action TRPV1 receptor antagonists
| PHASE 1 Johnson & Johnson Pharmaceutical Research & Development, L.L.C. |
|
| Public title: | A Clinical Study to Investigate the Effect on Pain Relief of a Single Dose of JNJ-39439335 in Patients With Chronic Osteoarthritis Pain of the Knee |
http://clinicaltrials.gov/ct2/show/NCT01006304
http://apps.who.int/trialsearch/trial.aspx?trialid=NCT00933582
http://www.ama-assn.org/resources/doc/usan/mavatrep.pdf SEE STRUCTURE IN THIS FILE
MAVATREP IS JNJ-39439335
—
(E)-2-(2-(2-(4-(trifluoromethyl)styryl)-1H-benzo[d]imidazol-6-yl)phenyl)propan-2-ol hydrochloride
956282-89-6 CAS NO OF HCl SALT
Org. Process Res. Dev., Article ASAP
DOI: 10.1021/acs.oprd.5b00271, http://pubs.acs.org/doi/abs/10.1021/acs.oprd.5b00271

The process development of Mavatrep (1), a potent transient receptor potential vanilloid-1 (TRPV1) antagonist, is described. The two key synthetic transformations are the synthesis of (E)-6-bromo-2-(4-(trifluoromethyl)styryl)1H-benzo[d]imidazole (4) and the Suzuki coupling of 4 with 3,3-dimethyl-3H-benzo[c][1,2]oxaborol-1-ol (5). Compound 1a was prepared in four chemical steps in 63% overall yield.
HCl SALT
1a as an off-white solid. 1H NMR (DMSO-d6, 500 MHz) δ 8.35 (d, J = 16.6 Hz, 1H), 7.96 (d, J = 8.2 Hz, 2H), 7.89 (d, J = 8.3 Hz, 2H), 7.80 (dd, J = 8.1, 1.3 Hz, 1H), 7.77 (d, J = 8.4 Hz, 1H), 7.65 (d, J = 1.4 Hz, 1H), 7.53 (d, J = 6.6 Hz, 1H), 7.43 (dd,J = 8.4, 1.5 Hz, 1H), 7.39 (ddd, J = 8.1, 7.4, 1.5 Hz, 1H), 7.27 (ddd, J = 7.4, 7.4, 1.3 Hz, 1H), 7.04 (dd, J = 7.5, 1.5, 1H), 1.29 (s, 6H); 13C NMR (DMSO-d6, 125 MHz) δ 147.7 (C), 147.4 (C), 142.3 (C), 140.3 (CH), 139.0 (C), 138.0 (C), 131.7 (CH), 131.1 (C), 130.5 (C), 130.2 (C), 128.7 (2CH), 128.3 (CH), 127.4 (CH), 126.2 (CH), 126.2 (CH), 125.8 (2CH), 124.0 (CF3), 114.3 (CH), 113.2 (CH), 112.4 (CH), 71.6 (C), 32.4 (2CH3); Anal. Calcd for C25H22F3ClN2O·1.2H2O: C, 62.49; H, 5.12; Cl, 7.38; F, 11.86; N, 5.83. Found: C, 62.34; H, 4.93; Cl, 7.24; F, 11.61; N, 5.78. Water wt % calcd, 4.50%; found, 4.52% (determined by KF analysis).
Patent




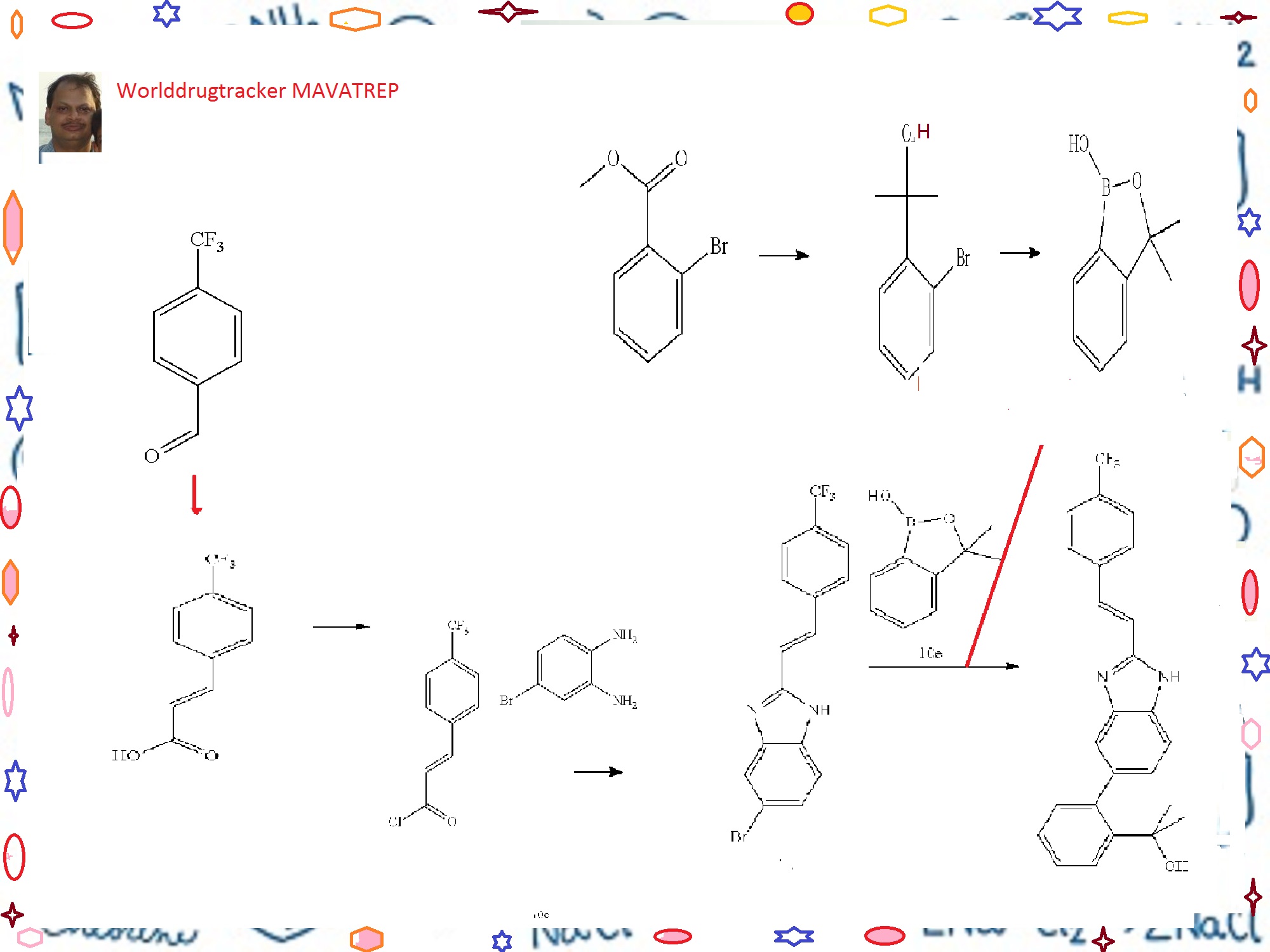
CLICK ON IMAGE FOR CLEAR VIEW
Example 10 (E)-2-(2-{2-[2-(4-trifluoromethyl-phenyl)-vinyl]-1H-benzimidazol-5-yl}-phenyl)-propan-2-ol(Cpd 18)
Step A. 3-(4-trifluoromethyl-phenyl)-acrylic acid
-
[0278]A solution of 4-trifluoromethylbenzaldehyde (7.7 mL, 57.7 mmol), malonic acid (12.0 g, 115.4 mmol), 0.567 μL piperidine (5.75 mmol) in 30 mL of pyridine was stirred at 70° C. for 18 h. The reaction solution was cooled to room temperature. Water (300 mL) was added and the resulting mixture was acidified to pH 4 (litmus) using concentrated hydrochloric acid to give a precipitate. The solid was filtered, and washed with water until the filtrate was neutral. The solid product was dried in vacuo to give the title Compound 10a as a white powder (11.2 g, 90%). 1HNMR (400 MHz, DMSO-d6) δ (ppm): 12.60 (bs, 1H), 7.92 (d, 2H, J=8.2 Hz), 7.77 (d, 2H, J=8.2 Hz), 7.66 (d, 1H, J=16.0 Hz), 6.70 (d, 1H, J=16.0 Hz).
-
[0000]

Step B. (E)-5-bromo-2-[2-(4-trifluoromethyl-phenyl)-vinyl]-1H-benzimidazole
-
[0279]A solution of Compound 10a (20.6 g, 95.4 mmol) in anhydrous methylene chloride (200 mL) was treated with oxalyl chloride (16.6 mL, 190 mmol) and “3 drops” of anhydrous dimethylformamide. The resulting solution was stirred at room temperature under an argon atmosphere for 18 h. The solvent was concentrated to give 3-(4-trifluoromethyl-phenyl)-acryloyl chloride Compound 10b as a solid, which was used without further purification in the next step.
-
[0280]To a solution of 4-bromo-benzene-1,2-diamine (16.1 g, 86.7 mmol) in acetic acid (100 mL) was added dropwise a solution of Compound 10b (assumed 95.4 mmol) in acetic acid (100 mL). The reaction mixture was stirred at 100° C. for 18 h. The reaction mixture was cooled to room temperature, and a mixture of ethyl acetate and hexanes 3:7 (500 mL) was added. The mixture was triturated at room temperature for 3 h to give a precipitate. The solid was filtered, and dried in vacuo to give the title Compound 10c (23.2 g, 73%). 1H NMR (400 MHz, DMSO-d6/CDCl3) δ (ppm): 8.45 (d, 1H, J=16.7 Hz), 7.84-7.90 (m, 1H), 7.74 (d, 2H, J=8.3
-
[0281]Hz), 7.56-7.62 (m, 3H), 7.50-7.52 (m, 1H), 7.34 (d, 1H, 16.7 Hz).
-
[0000]

Step C. 2-(2-bromo-phenyl)-propan-2-ol
-
[0282]To a solution of methyl 2-bromobenzoate (20.76 g, 96 mmol) in 120 mL of anhydrous ether under Argon at 0° C. was slowly added methylmagnesium bromide (77 mL, 3.26 M) at a rate that the internal temperature of the mixture was below 20° C. A white suspension resulted, and the mixture was stirred at room temperature for 2 h. The mixture was cooled in an ice-water bath. To the reaction mixture was very slowly added hydrochloric acid (400 mL, 0.5 M). The pH of the final mixture was adjusted to less than about 6 with few drops of 2M hydrochloric acid. The layers were separated, and the aqueous layer was extracted twice with ether. The organic layers were combined and dried over magnesium sulfate. The organic fraction was filtered, and the filtrate was concentrated to yield the title compound as a pale yellow liquid, which was distilled under vacuum to afford the title Compound 10d as a colorless liquid (16.9 g, 82%, b.p. about 65-70° C./0.3 mmHg). 1H NMR (400 MHz, CDCl3) δ (ppm): 7.67 (dd, 1H, J=1.7, 7.9 Hz), 7.58 (dd, 1H, J=1.3, 7.9 Hz), 7.30 (ddd, 1H, J=1.4, 7.4, 7.9 Hz), 7.10 (ddd, 1H, J=1.7, 7.4, 7.8 Hz), 2.77 (br s, 1H), 1.76 (s, 6H).
-
[0000]

Step D. 3,3-dimethyl-3H-benzo[c][1,2]oxaborol-1-ol
-
[0283]To a solution of n-BuLi (166 mL, 2.6 M, 432 mmol) in 200 mL of THF at −78° C. under argon was slowly added a solution of Compound 10d (42.2 g, 196 mmol) in 60 mL of THF at a rate that the internal temperature remained below −70° C. The mixture was stirred at −75° C. for 2 h. To the reaction mixture was then added triisopropylborate (59 mL, 255 mmol) in three portions. The mixture was allowed to warm slowly to room temperature overnight. The mixture was then cooled to 0° C., and was carefully quenched with dilute hydrochloric acid (250 mL, 2N). The mixture was then stirred at room temperature for 1 h. The pH of the mixture was checked and adjusted to acidic using additional 2N HCl if prophetic. The two layers were separated, and the aqueous layer was extracted twice with ether. The organic layers were combined, and dried with magnesium sulfate and filtered. The filtrate was concentrated under reduced pressure to yield a pale yellow oil. The residue was then diluted with ethyl acetate (400 mL) and, washed with 1N sodium hydroxide solution (150 mL×3). The basic aqueous layers were combined and acidified with 2N HCl. The clear solution turned cloudy when the acid was added. The mixture was extracted with ether (150 mL×3). The organic layers were combined and dried with magnesium sulfate. The solution was filtered, and the filtrate was concentrated under reduced pressure to yield the title Compound 10e as a colorless oil (26.2 g, 82%) which was used without further purification in the next step. 1H NMR (400 MHz, DMSO-d6) δ (ppm): 9.00 (s, 1H), 7.66 (dm, 1H, J=7.3 Hz), 7.45 (dt, 1H, J=1.1, 7.7 Hz), 7.40 (dm, 1H, J=7.6 Hz), 7.31 (dt, 1H, J=1.2, 7.1 Hz), 1.44 (s, 6H).
-
[0000]

Step E. (E)-2-(2-{2-[2-(4-trifluoromethyl-phenyl)-vinyl]-1H-benzimidazol-5-yl}-phenyl)-propan-2-ol
-
[0284]To a mixture of Compound 10e (11.7 g, 71 mmol), Compound 10c (19.9 g, 54 mmol), sodium carbonate (46 g, 435 mmol) and PdCl2(dppf).CH2Cl2 (8.9 g, 11 mmol) in a 1 L round bottom flask equipped with water condenser was added 400 mL of anhydrous DME and 200 mL of water. The mixture was evacuated and filled with Argon three times. The mixture was heated to 100° C. for 20 h. The mixture was then cooled to room temperature. The biphasic system was transferred to a 1 L separatory funnel and the two layers were separated. The organic layer was washed with brine (2×300 mL). The aqueous layers were combined and extracted with ethyl acetate once (about 300 mL). The organic layers were combined, dried with sodium sulfate, and filtered. The volume of the filtrate was reduced to about 170 mL under reduced pressure. The mixture was then filtered through a pad of silica gel and the pad was washed with ethyl acetate until the filtrate did not contain any product. After concentration, a light pink/beige solid was obtained. The solid was triturated with 50 mL ethyl acetate, and the mixture was heated to 85° C. for 5 min. The mixture was slowly cooled to r.t., then cooled at 0° C. for 0.5 h. The mixture was filtered, and the solid was washed with cold ethyl acetate twice, and dried under vacuum at 40° C. to yield the title Compound 18 as a light beige solid (7.58 g, 33%). RP-HPLC 95% pure.
-
1H NMR (400 MHz, DMSO-d6) δ (ppm): 12.73 (m, 1H,), 7.90 (d, 2H, J=8.2 Hz), 7.85 (dd, 1H, J=8.0, 0.6 Hz), 7.78 (d, 2H, J=8.4 Hz), 7.74 (d, 1H, J=16.8 Hz), 7.59-7.47 (m, 1H), 7.41 (s, 1H), 7.37-7.32 (m, 2H), 7.21 (dt, 1H, J=1.2, 7.4 Hz), 7.06 (s, 1H), 7.02 (d, 1H, J=7.4 Hz), 4.85 (s, 1H), 1.21 (s, 6H).
-
Mass Spectrum (LCMS, APCI pos.) Calcd. For C25H21F3N2O: 423.2 (M+H). Found 423.3.
-
m.p. (uncorr.) 250-251° C.
Example 10.1 Scale Up Preparation of (E)-2-(2-{2-[2-(4-trifluoromethyl-phenyl)-vinyl]-1H-benzimidazol-5-yl}-phenyl)-propan-2-ol (Cpd 18) Step A. 3-(4-trifluoromethyl-phenyl)-acrylic acid
-
[0286]A 2-L 4-neck round bottom flask equipped with an air condenser/argon inlet, mechanical stirrer, thermocouple and a stopper was charged with 4-(trifluoromethyl)benzaldehyde (250 g, 196.2 mL, 1.44 mol), malonic acid (302.6 g, 2.87 mol), and pyridine (750 mL). An exotherm developed (about 38-40° C.), which was maintained for 30 min. Piperidine (14.202 mL, 143.58 mmol) was then added to the reaction and a second exotherm developed (Tmax about 42° C. after about 10 min.). The reaction was stirred for 30 min and then heated to 60° C. for 18 h (overnight). The reaction appeared to be complete by TLC, and was cooled to about 40° C., diluted into water (2 L; done to prevent reaction freezing), cooled to room temperature, and further diluted with water (4 L, 6 L total). The slurry was acidified to pH=2.0-3.0 with concentrated hydrochloric acid (about 675-700 mL). The material was stirred for 30 min., and a white solid was collected by filtration. The filter cake was washed with water until the filtrate was neutral (pH about 5.5-6, 2.5 L), air-dried in a Buchner funnel for 2 h, and then further dried in a vacuum oven at 60° C. overnight to provide 300.5 g (96%) of the title Compound 10a as a white solid.
Step B. (E)-5-bromo-2-[2-(4-trifluoromethyl-phenyl)-vinyl]-1H-benzimidazole
-
[0287]To a 5-L 4-neck round bottom flask equipped with a magnetic stirrer, argon inlet-argon outlet to a carbonate scrub, two stoppers, and a room temperature water bath was charged with 4-(trifluoromethyl)cinnamic acid (315 g, 1.46 mol) and dichloromethane (3.15 L) to give a slurry. To the slurry was added oxalyl chloride (151.71 mL, 1.75 mol) and DMF (1.13 mL, 14.57 mmol). Upon addition of DMF, gas evolution commenced, and the reaction was continued for about 3 h during which time a solution developed. When the reaction was complete (LC-MS), it was concentrated to dryness to give 342.4 g of 3-(4-trifluoromethyl-phenyl)-acryloyl chloride Compound 10b (>100%) as a yellow oily solid.
-
[0288]A 5-L 4-neck round bottom flask equipped with mechanical stirrer, thermocouple, air condenser with argon inlet, and a stopper was charged with 4-bromo-benzene-1,2-diamine (244 g, 1.27 mol) and acetic acid (2.13 L). To this solution was added a solution of Compound 10b (327 g, 1.39 mol) in toluene (237 mL). After this addition, the temperature spiked to 45° C. in about 30 seconds and then subsided. The reaction was then heated to 90° C. for 16 h (overnight). The reaction was cooled to 40° C., and poured into a mixed solution of EtOAc and heptane (about 1:3, 5.75 L) and a precipitate occurred. The resulting slurry was stirred for 3 h, and the solid was collected by filtration, washed with EtOAc:heptane (1:3, 3 L), and then dried in a vacuum oven (60° C.) to give 324.3 g (65%) of the title Compound 10c as a partial acetate salt.
Step C. 2-(2-bromo-phenyl)-propan-2-ol
-
[0289]A 12-Liter 4-neck flask equipped with a thermocouple, condenser, septum, addition funnel and overhead mechanical stirrer under argon was charged with methyl-2-bromobenzoate (226.5 g, 1.05 mol) and THF (1.6 L, 19.66 mol). The mixture was cooled to a temperature between 2 and 5° C. with stirring and held for 30 min. To the solution was slowly added methyl magnesium bromide in diethyl ether (3M, 1.05 L; 3.15 mol) via the addition funnel at a rate to maintain the reaction temperature below 15° C. An exotherm was observed during the addition, the reaction temperature warmed from 3 to 15° C. The addition of 1.05 L Grignard was complete in 4 h (approximate feed rate was 4.17 mL/min). The reaction mixture appeared to be off-white/yellow slurry. The reaction was allowed to warm to room temperature and stirred overnight (15 h). The reaction was sampled by HPLC/TLC and showed no starting material present. The ice bath was again applied to the reaction flask and a 0.5 M HCl solution (4.5 L; 2.25 mol) was slowly added over a period of 2 h. The temperature increased dramatically from 0 to 15° C. After the quench was complete, the reaction was stirred at room temperature for 30 min. Additional 2 N HCl (500 mL; 1.00 mol) was slowly added to maintain a pH less than 6. MTBE (1 L) was added to help with the phase split. The reaction was stirred at room temperature for 1 to 2 h to dissolve the solid material into the aqueous phase (most likely Mg(OH)2 which is very basic). The pH must be checked and adjusted with additional acid when necessary. The phases were separated and the aqueous layer was washed with an additional 1 L MTBE (2×500 mL). The organic phases were combined, washed with NaHCO3 solution (2×300 mL), dried over MgSO4, filtered and the filtrate was concentrated under vacuum to yield the title Compound 10d (220.83 g, 97.48% yield) as a clear yellow oil.
Step D. 3,3-dimethyl-3H-benzo[c][1,2]oxaborol-1-ol
-
[0290]A 12-Liter 4-neck round bottom flask equipped with a thermocouple, condenser, addition funnel and overhead mechanical stirrer under dry Argon was charged with anhydrous THF, (3 L) and chilled to −70 to −78° C. via a dry ice/acetone bath. n-Butyl lithium (2.5N in hexanes, 860 mL, 2.15 mol) was slowly added via addition funnel. An exotherm was observed as the temperature rose from −78 to −70° C. To the addition funnel was added a solution of Compound 10d (220 g, 979.97 mmol) in anhydrous THF (1 L). The 2-(2-bromophenyl)propan-2-ol solution was slowly added to the n-BuLi solution. The addition took 90 min in order to maintain a reaction temperature below −70° C. After the addition was complete, the reaction mixture was stirred at −70 to −75° C. for 30 min. The triethylborate (230 mL, 1.35 mol) was quickly added in 3 portions at −70° C. An exotherm was observed, the batch temperature rose from −70 to −64° C. The reaction was stirred at −70° C. and slowly warmed to room temperature over night. After the reaction was cooled to 0-5° C., the reaction was slowly quenched with 2 M HCl (1 L, 2.00 mol) added via the addition funnel while maintaining the batch temperature 0-5° C. The reaction mixture was stirred for 1 h. The aqueous phase pH was 9-10. The pH was then adjusted to acidic (4-5) with 2 M HCl (200 mL). The two phases were separated and the aqueous layer was extracted with MTBE (2×500 mL). The combined organic phases were dried with anhydrous magnesium sulfate. The solution was filtered and concentrated to yield a yellow oil. The yellow oil was diluted with MTBE (1.5 L) and washed with 1M NaOH (3×500 mL). The product containing basic aqueous phases were combined and acidified with 2 M HCl (800 mL) (the clear solution turns turbid with the addition of acid). After stirring the turbid solution for 15 min (pH=4-5) (Note 1), it was extracted with MTBE (2×500 mL). The organic phases were combined and dried over MgSO4. The solution was filtered and the filtrate was concentrated to yield the title Compound 10e as a clear yellow oil (121.78 grams, 77% yield).
Step E. (E)-2-(2-{2-[2-(4-Trifluoromethyl-phenyl)-vinyl]-1H-benzimidazol-5-yl}-phenyl)-propan-2-ol
-
A 5-L 4-neck flask equipped with a thermocouple controller, condenser, overhead mechanical stirrer, Firestone Valve® and a nitrogen inlet/outlet was charged with dimethoxyethane (2 L), DI water (1 L) and sodium carbonate (230.9 g, 2.18 mol). The solution was degassed and purged with N2 three times. Compound 10e (71.7 g, 0.35 mol) and Compound 10c (100.0 g, 0.27 mol) were added to the degassed solution. The solution was degassed and purged with N2 three times. PdCl2(dppf) (44.48 g, 54.4 mmol) was added to the solution, and the solution was degassed and purged with N2 three times. The resulting two-phase suspension was heated to reflux for 18 h, and then cooled to room temperature. The reaction mixture was transferred to a 12-L separatory funnel, and the layers were separated. The organic layer was washed with brine (1 L). The two aqueous layers were combined and extracted with EtOAc (1 L). The combined organic layers were dried (Na2SO4), filtered, and the filtrate was concentrated to an oil. Two separate 100 g coupling reactions were combined and purified by chromatography in 10 successive chromatography runs on an ISCO preparative chromatography system (10×1.5 Kg SiO2, 5 column volumes of EtOAc, 250 mL/min flow rate). The combined fractions were transferred to two 22 L 4-neck round bottom flasks, and Silicycle Si-thiol functionalized silica gel (2 g) was added to each solution. The solutions were warmed to 40° C. and aged for 1 h. The solutions were filtered thru a medium glass funnel and washed with EtOAc (4 L) and combined. The filtrate was evaporated to a semi solid, which was transferred to a 2 L round bottom flask, to which EtOAc (0.4 L) was added. The resulting white precipitate slurry was cooled to −5° C. and stirred for 1 h. The slurry was filtered and washed twice with cold EtOAc (100 mL). The solids were dried in a vacuum oven at 40° C. for 40 h to afford 84.0 g (36.5% yield, 98.8 area % purity) of the title Compound 18 as a white solid. Anal. Calcd for C25H21N2OF3.0.04% H2O.0.15 mol MeOH: C, 70.48; H, 5.14: N, 6.42; F, 13.06 Found: C, 70.54; H, 4.83: N, 6.18; F, 13.33
Example 10.2 (E)-2-(2-{2-[2-(4-trifluoromethyl-phenyl)-vinyl]-1H-benzimidazol-5-yl}-phenyl)-propan-2-ol monosodium salt (Cpd 18)
-
A 5-L 4-neck flask equipped with a thermocouple controller, an overhead mechanical stirrer, and a nitrogen inlet/outlet was charged with (E)-2-(2-{2-[2-(4-trifluoromethyl-phenyl)-vinyl]-1H-benzimidazol-5-yl}-phenyl)-propan-2-ol. Compound 18 (125.0 g, 0.510 mol) and MeOH (1.25 L). A solution of sodium methoxide in methanol (0.5 M, 592 mL, 0.3 mol) was added. The reaction was heated to 65° C. for 30 min and all solids dissolved. The solution was cooled and evaporated to dryness. The foam was collected by scraping it out of the flask. The solids were placed in vacuum oven for 24 h at 40° C. to afford 139 g (about 100% isolated yield) of the title Compound 18 monosodium salt as a yellowish solid. 1H NMR (400 MHz, DMSO-d6) δ 7.80-7.84 (m, 3H), 7.74 (d, 2H, J=8.59 Hz), 7.65 (d, 1H, J=16.4 Hz), 7.40-7.44 (m, 2H), 7.25-7.37 (m, 2H), 7.16-7.20 (m, 1H), 7.01-7.05 (m, 1H), 6.84-6.87 (m, 1H), 1.23 (s, 6H). Mass Spectrum (LCMS, APCI pos.) Calcd. For C25H21F3N2O: 423.2 (M+H). Found 423.3. m.p. (uncorr.) 258-259° C.
Example 10.3 (E)-2-(2-{2-[2-(4-trifluoromethyl-phenyl)-vinyl]-1H-benzimidazol-5-yl}-phenyl)-propan-2-ol hydrochloride salt (Cpd 18)
-
A 250-mL separatory funnel was charged with (E)-2-(2-{2-[2-(4-trifluoromethyl-phenyl)-vinyl]-1H-benzimidazol-5-yl}-phenyl)-propan-2-ol. Compound 18 (1.0 g, 2.4 mmol) and EtOAc (20 mL). Aqueous HCl (1M, 20 mL) was added to the white slurry, and the separatory funnel was shaken. The solid product quickly dissolved, and a white precipitate started to form. The organic layer was transferred to a 100 mL round bottom flask equipped with a magnetic stir bar, and was stirred for 2 h. The thick slurry was filtered, rinsed with EtOAc (2×5 mL), and put into a vacuum oven at 40° C. for 36 h to afford 0.95 g (87.5%) of the title Compound 18 hydrochloride salt.
////////////Phase I, Musculoskeletal pain, Pain, Mavatrep, JNJ 39439335,
FDA grants breakthrough status for Pfizer’s leukaemia drug inotuzumab ozogamicin


Inotuzumab ozogamicin
RN: 635715-01-4
UNII: P93RUU11P7
Pfizer Inc., Oncology Institute Of Southern Switzerland INNOVATOR
http://chem.sis.nlm.nih.gov/chemidplus/rn/635715-01-4
- MF 1680.6764
-
Oncological Treatment
FDA grants breakthrough status for Pfizer’s leukaemia drug inotuzumab ozogamicin
The US Food and Drug Administration (FDA) has granted breakthrough therapy designation for Pfizer’s investigational antibody-drug conjugate (ADC) inotuzumab ozogamicin to treat acute lymphoblastic leukaemia (ALL).
The US Food and Drug Administration (FDA) has granted breakthrough therapy designation for Pfizer’s investigational antibody-drug conjugate (ADC) inotuzumab ozogamicin to treat acute lymphoblastic leukaemia (ALL).
The breakthrough status was based on data from the Phase III INO-VATE ALL trial, which enrolled 326 adult patients with relapsed or refractory CD22-positive ALL and compared inotuzumab ozogamicin to standard of care chemotherapy………….http://www.pharmaceutical-technology.com/news/newsfda-grants-breakthrough-status-pfizer-leukaemia-drug-inotuzumab-ozogamicin-4697877?WT.mc_id=DN_News

EVER SINCE POST WAS WRITTEN…..FGD APPROVAL Inotuzumab ozogamicin
PFIZER

![]()

| Besponsa | FDA
8/17/2017 |
To treat adults with relapsed or refractory acute lymphoblastic leukemia Press Release Drug Trials Snapshot |
Inotuzumab ozogamicin (CMC-544) is an antibody-drug conjugate for the treatment of cancers.[1] It consists of the humanized monoclonal antibody inotuzumab (for CD22), linked to a cytotoxic agent from the class of calicheamicins (which is reflected by ‘ozogamicin‘ in the drug’s name).[2]
This drug is being developed by Pfizer and UCB.
It is undergoing numerous clinical trials,[3] including two phase II trials for Non-Hodgkin lymphoma (NHL).
A phase III trial in patients with follicular b-cell NHL has been terminated due to poor enrollment.[4] A Phase III trial in patients with relapsed or refractory CD22+ aggressive non-Hodgkin lymphoma (NHL) who were not candidates for intensive high-dose chemotherapy was terminated for futility.[5]
Monoclonal antibodies (mAbs) and derivatives are currently the fastest growing class of therapeutic molecules. More than 30 G-type immunoglobulins (IgG) and related agents have been approved over the past 25 years mainly for cancers and inflammatory diseases. In oncology, mAbs are often combined with cytotoxic drugs to enhance their therapeutic efficacy. Alternatively, small anti-neoplastic molecules can be chemically conjugated to mAbs, used both as carriers (increased half-life) and as targeting agents (selectivity). Potential benefits of antibody-drug conjugates (ADCs), strategies, and development challenges are discussed in this review. Several examples of ADCs are presented with emphasis on three major molecules currently in late clinical development as well as next generation thio-mAbs conjugates with improved therapeutic index.

PATENT
http://www.google.com/patents/WO2013088304A1?cl=en
Inotuzumab ozogamicin:

is described in U.S. Patent Application No. 10/428894
U.S. Patent Application No. 10/428894


References
- Statement On A Nonproprietary Name Adopted By The Usan Council – Inotuzumab ozogamicin, American Medical Association.
- Takeshita, A; Shinjo, K; Yamakage, N; Ono, T; Hirano, I; Matsui, H; Shigeno, K; Nakamura, S; Tobita, T; Maekawa, M (2009). “CMC-544 (inotuzumab ozogamicin) shows less effect on multidrug resistant cells: analyses in cell lines and cells from patients with B-cell chronic lymphocytic leukaemia and lymphoma.”. British journal of haematology 146 (1): 34–43.doi:10.1111/j.1365-2141.2009.07701.x. PMID 19388933.
- http://clinicaltrials.gov/ct2/results?term=Inotuzumab+ozogamicin
- http://clinicaltrials.gov/ct2/show/NCT00562965
- http://pfizer.newshq.businesswire.com/press-release/pfizer-discontinues-phase-3-study-inotuzumab-ozogamicin-relapsed-or-refractory-aggress
- http://pubs.rsc.org/en/content/articlelanding/2008/np/b514294f#!divAbstract
Structure of inotuzumab ozogamicin. ABOVE
| Monoclonal antibody | |
|---|---|
| Type | Whole antibody |
| Source | Humanized (from mouse) |
| Target | CD22 |
| Identifiers | |
| CAS Registry Number | 635715-01-4 |
| ATC code | None |
| UNII | P93RUU11P7 |
| KEGG | D08933 |
| Chemical data | |
| Formula | C6518H10002N1738O2036S42 |
| Molecular mass | 150,000 Daltons |
//////////
Regulation of Herbal (Traditional) Medicinal Products in the European Union

Regulation of Herbal (Traditional) Medicinal Products in the European Union
| Introduction | |
| The European Union (EU) regulatory framework for medicinal products is complex and is based on the need of a marketing authorization before placing medicines in the market. The main objective is to protect public health by assuring quality, efficacy and safety. The requirements and procedures to obtain a marketing authorization are laid down in regulations, directives and scientific guidelines which are contained in the “Rules Governing Medicinal Products in the European Union”. Several volumes are included which are supported by other publications with complementary information such as scientific or Good Manufacturing Practice (GMP) guidelines, between others [1]. | |
 |
|
| Medicinal plants have been used since Ancient times in all parts of the world. Nonetheless, regulation of herbal medicines in a legal environment was introduced in the 20th century. The EU regulatory framework includes specific requirements for herbal medicinal products (HMP) which are independent from their legal status: traditional herbal medicinal product (THMP) or products based on clinical evidence – well established use (WEU). | |
| Before a HMP is placed in the market, it must be approved by a MS or by the European Commission by one of the existing types of application: full marketing authorization application, well-established use marketing authorization application or Traditional use marketing registration (Table 1).
|
|
| The applicant has to submit adequate quality, non-clinical and clinical documentation of the product, irrespectively of the procedure used. Quality requirements of the pharmaceutical product are the same, regardless of the type of application, while efficacy documentation differs between them. The full marketing application is chosen for new medicinal products (new chemical entity) and it has to be completed with the results of pharmaceutical tests (quality documentation), nonclinical (toxicological and pharmacological) studies and clinical trials. Safety data have to be of sufficient size according the existing guidelines; efficacy is demonstrated by results from the clinical trials which have to be in conformity with the guidelines of the corresponding therapeutic area. This type of application is open for HMP, but only a few examples of herbal products have obtained a marketing authorization in the EU in this way. | |
| EU Pharmaceutical Legislation for Herbal Medicinal Products for Human Use | |
 |
|
| Quality requirements | |
| The principles to assure quality of medicinal products are defined mainly in two Directives of volume 1: Directive 2001/83/EC (which was emended by Directive 2004/24/EC) and Directive 2003/63/EC. | |
| The basic legislation lay down in Directive 2001/83/EC describes the general requirements and provides legal definitions of herbal substances, herbal preparations and herbal medicinal products (Table 2). These concepts are essential for setting quality standards for HMP, as they are by definition complex in nature and so quality requirements set for purified compounds are not suitable for herbal products. | |
| According to the Directive 2001/83/EC, monographs in the European Pharmacopoeia (Eur. Ph.) are legally binding and applicable to all substances which are included in it. For substances which do not have a Eur. Ph. monograph, each Member State (MS) may apply its own national pharmacopoeia. Constituents which are not given in any pharmacopoeia shall be described in the form of a monograph under the same headings included in any monograph in the Eur. Ph., i.e., the name of the substance supplemented by any trade or scientific synonyms; the definition of the substance, set down in a form similar to that used in the European Pharmacopoeia; methods of identification and purity tests. | |
| Moreover, all medicinal products have to be manufactured according to the principles and guidelines of GMP for medicinal products. GMP are applicable to both finished HMP and active substances and, according to Article 46 (f) of Directive 2001/83/EC as amended, marketing authorization holders are required to use as starting materials only active substances which have been manufactured in accordance with the guidelines on the GMP for starting materials as adopted by the Community and distributed in accordance with good distribution practices for active substances. | |
| |
|
| Additional requirements are found in the Directive 2003/63/EC, as Herbal medicinal products differ substantially from conventional medicinal products in so far as they are intrinsically associated with the very particular notion of herbal substances and herbal preparations. It is therefore appropriate to determine specific requirements in respect of these products with regards to the standardized marketing authorization requirements. Then, detailed information on the herbal medicinal product, herbal substances and herbal preparations has to be included, such as the name, address and responsibility of each herbal substance supplier or description of the plant production process, geographical source or drying and storage conditions. The application dossier of a HMP should include specifications and details of all the analytical methods used for testing herbal substances and herbal preparations, results of batch analyses and analytical validation, together with the justification for the specifications. | |
| Most of the quality requirements for HMP are laid down in soft laws (considered as EU measures such as scientific guidelines) which do not have legal force but provide practical harmonization between the MS and the European Medicines Agency (EMA). | |
| The guideline on quality of HMP/THMP covers the general quality aspects of HMP for human and veterinary use, including THMP for human use (EMA, 2014) and indicates which information has to be included in the application dossier. It provides definitions to be taken in account such as genuine (native) herbal preparations, markers, drug to extract ratio (DER) and specifications. Which is more important, it states that the herbal substance or herbal preparation is considered as the whole active substance. In consequence, the quality control of these products has to include appropriate fingerprint analysis to cover not only the content of markers or constituents with known therapeutic activity but also a wider range of chemical constituents. | |
| |
|
| Efficacy requirements | |
| Before a HMP is placed in the market, it must be approved by a MS or by the European Commission by one of the existing types of application: full authorization application, well-established use authorization application or Traditional use registration (Table 3). | |
| The applicant has to submit adequate quality, non-clinical and clinical documentation of the product, irrespectively of the procedure used. Quality requirements of the pharmaceutical product are the same, regardless of the type of application, while efficacy documentation differs between them. The full marketing application is chosen for new medicinal products (new chemical entity) and it has to be completed with the results of pharmaceutical tests (quality documentation), nonclinical (toxicological and pharmacological) studies and clinical trials. Safety data have to be of sufficient size according the existing guidelines; efficacy is demonstrated by results from the clinical trials which have to be in conformity with the guidelines of the corresponding therapeutic area. This type of application is open for HMP, but only a few examples of herbal products have obtained a marketing authorization in the EU in this way. | |
| The well-established medicinal use (WEU) in the EU can be applied to medicinal products for which there exists a wide clinical experience within the EU (not only HMP). The assessment may be based in published controlled clinical trials, non-clinical studies and epidemiological studies. In this type of application, there are no limitations to the therapeutic indication, as this will be derived from the available documentation. | |
| |
|
| (Traditional) Herbal Medicinal Products | |
| Under the Traditional use registration for herbal medicinal product (article 16e), there exist some herbal products that not fulfill the efficacy requirements for a marketing authorization but are endorsed with a long tradition of use. In this case, no clinical trials on these products have been conducted and the efficacy is based on the long-standing use and experience. This simplified registration procedure is limited to products which are intended for use without medical supervision, with a specified strength and posology, to be used by oral, external or inhalation ways, and which can demonstrate a period of use equal or superior to 30 years, including at least 15 years within the EU. In this case, therapeutic indications are limited to those which can be considered safe for use without the supervision of a physician such as minor disorders or symptoms that are benign or self- limiting. In case the applicant should consider another kind of indication, the product must be documented with results of clinical and non-clinical studies, so a full application would be necessary. | |
| Simplified registration of THMP is described in Chapter 2a of Directive 2004/24/EC with three main objectives: a) to protect public health by allowing access to safe and high-quality HMP; b) to allow European citizens the access to medicines of their choice, even those HMP with a long tradition of use and which efficacy hasn’t been proved by clinical trials performed according the modern standards; c) to facilitate movement of medicinal products on the European market. | |
| Directive 2004/24/EC has two different dimensions: the evaluation by National Competent Authorities (NCA) of applications submitted by companies at any MS in the EU and at the EMA, and the establishment of advisory scientific opinions on the medicinal use of herbal substances or preparations. The directive on THMP also established a new scientific committee, the Herbal Medicinal Products Committee (HMPC) at the EMA in London, in 2004, to replace the previous Working Party on Herbal Medicinal Products (CPMP) with the following aims: to elaborate Community monographs and List entries for herbal substances/preparations; to publish scientific guidelines useful for the application of European legal framework; to publish its scientific opinion on questions related to herbal medicinal products and coordinate its work with the European Quality group. The HMPC is made up by 33 members, one member (and one alternate) nominated by each MS of the EU and by Iceland and Norway (the EFAEFTA states). Among them, also five experts are included, representing specific fields of expertise as clinical and non-clinical pharmacology, toxicology or pediatrician medicine. | |
| The guidelines and the monographs developed and approved by the HMPC are accepted by both companies and NCAs and are used for TUR and WEU marketing authorizations. This committee plays a key role in the harmonization of the regulation of HMP whereby Community herbal monographs have a fundamental role. | |
| Usage of Community herbal monographs in the EU regulation of traditional HMP | |
| These documents are established for HMP with regards to bibliographic applications (art. 10 a Directive 2001/83/EC) as well as THPMs. Community monographs reflect the scientific opinion of the HMPC on safety and efficacy data concerning a herbal substance. Any single plant or herbal preparation is assessed individually, according to the available information and includes qualitative and quantitative composition, pharmaceutical form(s), therapeutic indication(s), posology and method of administration, contraindications, special warnings and precautions of use, interactions, use in special population (pregnancy, lactation), effects on ability to drive and use machines, undesirable effects, overdose, pharmacological,pharmacodynamics, pharmacokinetic properties and preclinical safety data. | |
| Community list entry | |
| In the EU, a community list of herbal substances, preparations and combinations thereof for use in THMPs has been established. This list is based in the proposals form HMPC and is gradually developed. Substances or preparations which are included in the list have the main advantage that applicants do not need to provide evidence on the safe or traditional use for its registration at the NCA in the intended use and indication. | |
| A Public statement for one herbal substance/preparation is published because of safety reasons or lack of data to comply with the conditions in the Directive 2004/24/EC (the assessment work didn’t allow a monograph to be published) [2]. | |
| Community monographs are published by the EMA while list entries are approved and published by the European Commission because they are endorsed with a wider legal status: list entries are legally binding and NCAs should not request additional data on safety and traditional use. | |
| The establishment of monographs and list entries is based on the assessment of the published scientific data, together with the existing products in the market. Most of the assessment work is developed by the Monograph and List Working Party (MLWP) at the HMPC, which was established in 2006. In this working group, a member is designed as rapporteur and is responsible of drafting a monograph and/or list entry which will be later on considered and approved by the HPC and then, by the EMA. The documents are published on the EMA website: Community monographs have to be taken in account by the MS when assessing the application of any company. Monographs are note legally binding and MS are not obliged to follow the monographs. | |
| More than 100 species are included in the priority list with the following data: a) scientific data being assessed (R- Rapporteur assigned); b) evaluation report in progress and discussion in the MLWP (D- Draft under discussion); c) scientific opinion under public consultation (PDraft Published); d) comments after public consultation period being evaluated (PF- Assessment close to finalization – pre-final); e) final opinion adopted (F- Final opinion adopted). | |
| MLWP is also responsible of developing guidelines related to legal requirements for TU and WEU, as well as evaluating hazards and problems related to HMP. For the latter, coordination is established with the Safety Working Party (SWP) from the Committee for Medicinal Products for Human Use (CHMP). | |
| Community herbal monographs to support HMPC authorization | |
| A community monograph reflects the scientific opinion from the HMPC in relation to safety and efficacy of one herbal substance/ preparation for medicinal use. AS stated before, a community monograph may be used by a company for a TU or WEU application. That’s the reason why monographs are divided in to two columns: Well Established Use and Traditional Use (simplified application) (Figure 1). WEU is based in the existence of safety data of sufficient size and efficacy data derived from good-quality clinical trials. Traditional use is accepted for those applications which fulfill the criteria shown in the Directive 2004/24/EC. | |
| Each herbal substance/preparation is assessed individually, as the available information may be different for each one. As a result, some substances/preparations may be included in the WEU side, while others will be included in the TU side. If no enough data are available for the substance/preparation, it won’t be included in the monograph. | |
| The approved draft art he HMPC is published for public consultation for 3 month at the EMA website. Comments received are discussed and taken in account when necessary to achieve the final version of the monograph which will be finally published at the MA website. | |
| By the end of 2014, 126 monographs have been adopted and published by the EMA: 104 of them for TU only; 9 of the monographs refer only to WEU (Aloe vera, Cimicifuga racemosa, Rhamnus frangula, Plantago ovata – seed and tegumentum-, Plantago afra, Rheum palmatum, Cassia senna – leaves and fruits-.among them 13 monograph include both TU and WEU. | |
| The main application of a community monograph is to serve as a reference material for the marketing application, both for TU or WEU. Simplified registration is carried on at a national level, so the company gives the dossier to the NCA. With the aim of improving harmonization, the other MSs should recognize the first authorization granted in the first MS, considering that this is based in the European list. | |
| Directive 2004/24/CE established an adaptation period for those herbal products which were on the European market at the moment the Directive was approved. This seven-year period finalized last April 30th, 2011 and implies that nowadays those herbal preparations that not fulfill the actual legislation will not be marketed any more. | |
| In the public report form the EMA last June 2014, the status of updating the medicines registration in the EU was shown. The number Traditional use registrations (TUR) and Well-established use marketing authorizations (WEU) grouped for mono component and combination products has increased in the last years (Figure 2). | |
| The European market for HMP is increasing during the last years and even exceeds prescription medicines. The indications approved cover a wide range of therapeutic areas, most of them characteristic of self-medication diseases: the main therapeutic areas are respiratory tract disorders (cough and cold), mental stress and mood disorders, urinary tract and gynecology disorders, sleep disorders and temporary insomnia (Figure 3). Most of the approved THMP until now were updates of existing authorizations and were based on Community monographs. The Summary of Product Characteristics (SoPC) reflects the items in the corresponding monograph [3]. | |
| A good correlation between the HMPC work and the evaluation of the dossiers from the companies was detected. The relevance of these documents (as shown by the accepted dossiers) is reflected in the HMPC working plan; as an example, last December 2012, 54 among the 56 species with more than 3 marketing authorizations were listed in the priority list. | |
| Conclusión | |
| European legal framework for medicinal products does also include herbal medicinal products to assure their quality, efficacy and safety. The specific characteristics of these products led to the development of a simplified procedure to assure pharmaceutical quality, while keeping safety and efficacy criteria according to marketing authorization granted. | |
| Although the starting point was quite different for the MS, nowadays there exist Community monographs for most of the herbal substances/ preparations that are used in the European market and which form the basis for a harmonization scenario. Moreover, HMPC acts as an International Regulatory Body for herbal medicinal products in order to achieve global standards for this type of medicines, according to other International organization such as the International Conference on Harmonization (ICH). The main tasks the HMPC has to face are those related to herbal medicinal products which have been previously marketed abroad the EU and the increasing existence of combination products within the MS. | |
| References | |
|
|
|
|
||||||||||||
| Table 1: Types of applications for marketing authorization for a HMP in the EU according the Directive 2001/83/EC. |
|
||||||
| Table 2: Definitions applicable to herbal medicinal products (Directive 2001/83/EC). |
|
||||||
| Table 3: Definitions applicable to herbal medicinal products (Directive 2001/83/EC). |
 |
| Figure 1: European Community Monograph for Valeriana officinalis L., radix, for WEU and TU |
Ruiz-Poveda OMP*
Department of Pharmacology, Faculty of Pharmacy, Universidad Complutense de Madrid, 28040 Madrid, Spain
Ruiz-Poveda OMP
Department of Pharmacology, Faculty of Pharmacy
Universidad Complutense de Madrid, Madrid
Ciudad Universitaria s/n. 28040 Madrid, Spain
Tel: 913 941 767
Fax: 913 941 726
E-mail: olgapalomino@farm.ucm.es
Citation: Ruiz-Poveda OMP (2015) Regulation of Herbal (Traditional) Medicinal Products in the European Union. Pharmaceut Reg Affairs 4:142. doi: 10.4172/2167-7689.1000142
/////////Herbal medicines, Good manufacturing practices, Traditional uses
EMA: European Medicines Agency; DER: Drug to Extract Ratio; HMP: Herbal Medicinal Products; THMP: Traditional Herbal Medicinal Products
DR ANTHONY MELVIN CRASTO

ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on
ORGANIC SPECTROSCOPY INTERNATIONAL


{kind=link}