

7056500 United States
7662365 United States
8067431 United States
8617530 United States
9012469 United States
PROUD Indian WORLD RECORD VIEWS holder on THIS BLOG, ………live, by DR ANTHONY MELVIN CRASTO, Worldpeaceambassador, Worlddrugtracker, Helping millions, 100 million hits on google, pushing boundaries,2.5 lakh plus connections worldwide, 45 lakh plus VIEWS on this blog in 227 countries, 7 CONTINENTS ……A 90 % paralysed man in action for you, I am suffering from transverse mylitis and bound to a wheel chair, [THIS BLOG HOLDS WORLD RECORD VIEWS ] A PROUD INDIAN
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO FACEBOOK
...................................................................Join me on twitter
FACEBOOK
...................................................................Join me on twitter
 ..................................................................Join me on google plus
..................................................................Join me on google plus
 Googleplus
Googleplus
Mitiglinide (INN, trade name Glufast) is a drug for the treatment of type 2 diabetes.[1]
Mitiglinide belongs to the meglitinide class of blood glucose-lowering drugs and is currently co-marketed in Japan by Kissei and Takeda. The North America rights to mitiglinide are held by Elixir Pharmaceuticals. Mitiglinide has not yet gained FDA approval.
Mitiglinide calcium hydrate was approved by Pharmaceuticals and Medical Devices Agency of Japan (PMDA) on January 29, 2004. It was co-developed and co-marketed as Glufast® by Takeda and Kissei in Japan.
Mitiglinide is a rapid-acting insulin secretion-stimulating agent. It stimulates insulin secretion by closing the ATP-sensitive K+ (ATP) channels in pancreatic beta-cells. It is indicated for the treatment of type 2 diabetes mellitus.
Glufast® is available as tablet for oral use, containing 5 mg or 10 mg of Mitiglinide calcium hydrate. The recommended dose is 10 mg three times daily just before each meal (within 5 minutes).
China , Approved 2010-04-19, 快如妥/Glufast, Kissei
ミチグリニドカルシウム水和物

C38H48CaN2O6▪2H2O : 704.92
[207844-01-7]
Pharmacology
Mitiglinide is thought to stimulate insulin secretion by closing the ATP-sensitive K(+) K(ATP) channels in pancreatic beta-cells.

Dosage
Mitiglinide is delivered in tablet form.

| Molecular Weight | 333.42 |
| Formula | C19H27NO4 |
| CAS Number | 207844-01-7 |




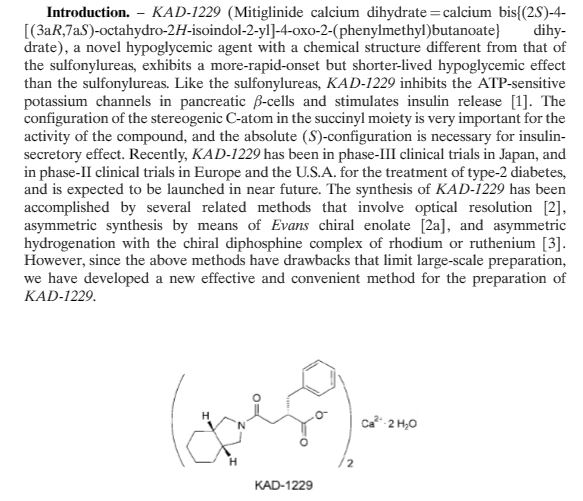
The condensation of dimethyl succinate (I) with benzaldehyde (II) by means of NaOMe in refluxing methanol followed by hydrolysis with NaOH in methanol/water gives 2-benzylidenesuccinic acid (III). Compound (III) is treated with refluxing Ac2O, yielding the corresponding anhydride (IV), which by reaction with cis-perhydroisoindole (V) in toluene affords the monoamide (VI). This amide is reduced with H2 over a chiral Rhodium catalyst and treated with (R)-1-phenylethylamine (VII) to provide the chiral salt (VIII) as a single diastereomer isolated by crystallization. Finally, this salt is treated first with aqueous NH4OH and then with aqueous CaCl2.

he optical resolution of racemic 2-benzylsuccinic acid (XV) using the chiral amines (R)-1-phenylethylamine (VII), (R)-1-(1-naphthyl)ethylamine (XIV) or (S)-1-phenyl-2-(4-tolyl)ethylamine (XVI) is carried out by fractional crystallization of the corresponding diastereomeric salts and treatment with 2N HCl, providing the desired enantiomer 2(S)-benzylsuccinic acid (XVII). Reaction of (XVII) with SOCl2 gives the corresponding acyl chloride (XVIII), which is treated with 4-nitrophenol (XIX) and TEA in dichloromethane to yield the activated diester (XX). The regioselective reaction of (XX) with cis-perhydroisoindole (V) in dichloromethane affords the monoamide (XXI), which by reaction with HCl and methanol provides the corresponding methyl ester (XXII). This ester is hydrolyzed with NaOH to the previously described chiral succinamic acid (XIII), which is finally converted into its calcium salt.

PATENT
https://www.google.com/patents/WO2009047797A2?cl=en
Perhydroisoindole derivative, (S)-mitiglinide of formula I is a potassium channel antagonist for the treatment of type 2 diabetes mellitus and is chemically known as (5)-2-benzyl-3-(cis-hexahydro-2- isoindolinylcarbonyl) propionic acid.
Formula I

It has potent oral hypoglycemic activity and is structurally different from the sulphonylureas, although it stimulates calcium influx by binding to the sulphonylurea receptor on pancreatic β-cells and closing K+ ATP channels. Perhydroisoindole derivatives including (S)-mitiglinide and salts thereof were first disclosed in US patent 5,202,335. This patent discloses preparation of (S)-mitiglinide by the reaction of (5)-3-benzyloxycarbonyl-4-phenylbutyric acid with cis-hexahydroisoindoline in the presence of N- methylmorpholine and isobutyl chloroformate followed by debenzylation with palladium on carbon in ethyl acetate to yield (5)-mitiglinide as viscous oil. (S)-Mitiglinide is isolated as its hemi calcium salt using calcium chloride in water which is further recrystallized with diisopropyl ether. Melting point of calcium salt of mitiglinide calcium dihydrate salt is herein reported as 179-185 0C. (S)-Mitiglinide prepared by the above process is obtained in low yields. Further, the synthetic method described in the patent does not enable the desired regioselectivity. Extensive purification steps are required to obtain the desired compound, which makes the process unattractive from industrial point of view. US patent 6,133,454 discloses a process for the preparation of (S)-mitiglinide by reacting dimethyl succinate with benzaldehyde in methanolic medium, to yield a diacid which is converted to corresponding anhydride and is further reacted with the perhydroisoindole to yield 2-[(cis- perhydroisomdol^-ytycarbonylmethyl^-phenylacrylic acid which is then subjected to catalytic hydrogenation using the complex rhodium/(2S,4S)-N-butoxycarbonyl-4-diphenylphosphino-2-diphenyl- phosphino-methylpyrrolidine (Rh/(S,S) BPPM) as asymmetric hydrogenation catalyst, followed by conversion to pharmaceutically acceptable salt of (S)-mitiglinide. The above patent utilizes ruthenium complex which is expensive, carcinogenic and toxicity, hence not recommended for industrial scale. European patent publication no. EP 0967204 discloses the preparation of mitiglinide by deprotecting benzyl-(S)-2-benzyl-3-(cis-hexahydro-2-isoindolinyl-carbonyl) propionate and converting the same to calcium dihydrate salt in crystalline form using calcium chloride, water and ethanol. The crystals of calcium salt are further recrystallized using ethanol and water. But the patent is silent about the crystalline form of mitiglinide calcium.
It will be appreciated by those skilled in the art that perhydroisoindole derivative, (S)-mitiglinide of formula I contains a chiral centre and therefore exists as enantiomers. Optically active compounds have increasingly gained importance since the technologies to develop optically active compounds in high purity have considerably improved. Obtaining asymmetric molecules has traditionally involved resolving the desired molecule from a racemic mixture using a chiral reagent, which is not profitable as it increases the cost and processing time. Alternatively, desired enantiomer can be obtained by selective recrystallization of one enantiomer. However such a process is considered inefficient, in that product recovery is often low, purity is uncertain and more than 50% of the material is lost. Enantiomers can also be resolved chromatographically, although the large amount of solvent required for conventional batch chromatography is cost prohibitive and results in the preparation of relatively dilute products. Limited throughput volumes also often make batch chromatography impractical for large-scale production. Even so, it is a common experience for those skilled in the art to find chiral separation of certain chiral mixtures to be inefficient or ineffective, thereby resulting in the efforts towards development of newer methodologies for asymmetric synthesis.
It would be of significant advantage to obtain (.S)-mitiglinide by development of reaction conditions necessary for productive manufacture of the required (5)-enantiomer, substantially free of the unwanted (R)-enantiomer, in large quantities that meet acceptable pharmaceutical standards. It is the property of the solid compounds to exist in different polymorphic form. By the term polymorphs mean to include different physical forms, crystal forms, crystalline/liquid crystalline/non-crystalline (amorphous) forms. This has especially become very interesting after observing that many antibiotics, antibacterials, tranquilizers etc, exhibit polymorphism and some/one of the polymorphic forms of a given drug exhibit superior bio-availability and consequently show much higher activity compared to other polymorphs. It has also been disclosed that the amorphous forms in a number of drugs exhibit different dissolution characteristics and in some cases different bioavailability patterns compared to the crystalline form [Konne T., Chem. Pharm. Bull. 38, 2003 (1990)]. The solubility of a material is also influenced by its solid-state properties, and it has been suggested that the solubility of an amorphous compound is 10 to 1600 times higher than that of its most stable crystalline structures (Bruno C. Hancock and Michael Parks, ‘What is the true solubility advantage for amorphous pharmaceuticals’, Pharmaceutical Research 2000, Apr; 17(4):397-404). Thus it can be concluded that amorphous products are in general more soluble and often show improved absorption in humans.
Thus, there is a widely recognized need for developing a stable polymorph, which would further offer advantages over crystalline forms in terms of better dissolution and the availability profiles. Also none of the prior art references disclose amorphous form of mitiglinide calcium. Thus present invention provides amorphous form of mitiglinide calcium.
It is also required that the final API like mitiglinide whether in the amorphous form or crystalline form must be free from the other impurities including the unwanted enantiomer, these can be side product and by product of the reaction, degradation products and starting materials. Impurities in final API are undesirable and in extreme cases, might even be harmful to a patient being treated with a dosage form containing the API. Therefore impurities introduced during commercial manufacturing processes must be limited to very small amounts and are preferably substantially absent. These limits are less than about 0.15 percent by weight of each identified impurity and 0.10 % by weight of unidentified and/or uncharacterized impurities. After the manufacture of APIs, the purity of the products, such as (S)- mitiglinide calcium dihydrate is required before commercialization, and in the manufacture of formulated pharmaceuticals. Therefore, pharmaceutical active compounds must be either free from these impurities or contain the impurities in acceptable limits. There is also a need for the isolation, characterization and identification of the impurities and their use as reference markers and reference standard. Thus, the present invention meets the need in the art for a novel, efficient and industrially advantageous process for providing optically pure perhydroisoindole derivatives, particularly (iS)-mitiglinide, which is unique with respect to its simplicity, scalability and involves controlling the steps of the reaction so that predominantly the desired (S)-enantiomer is produced in high yields and purity. The present invention also provides substantially pure (S)-mitiglinide and salts thereof having novel amide impurity in acceptable limit or free from this impurity.
Example 1: Preparation of (R) 4-benzyl-3-(3-phenylpropionv0-oxazolidin-2-one To a solution of (R)-4-benzyloxazolidin-2-one (50 g), 4-dimethylaminopyridine (4.85 g), 3-phenyl propionic acid (55.08 g) in dichloromethane (375 ml) under nitrogen atmosphere at 0-5 0C, dicyclohexylcarbodiimide (975.65 g) was added. The temperature was slowly raised to 25-30 0C and stirring was continued until no starting material was left as was confirmed by thin layer chromatography. Dicyclohexylurea formed during the reaction was filtered, washed with dichloromethane (200 ml) and the filtrate was washed with saturated solution of sodium bicarbonate (500 ml). The solution was dried over sodium sulphate and solvent was distilled off to obtained crude product which was purified from methanol (200 ml) at 10-15 °C and washed with methanol (50 ml) to obtain 81.0 g of the title compound. Example 2: Preparation of 3(5)-benzyl-4-(4-(J?)-benzyl-2-oxo-oxazolidin-3-yl)-4-oxo-butyrϊc acid tert-butyl ester
To a solution of (/?)-4-benzyl-3-(3-phenyl-propionyl)-oxazolidin-2-one (150 g) in anhydrous tetrahydrofuran (1.5 It) was added a solution of sodium hexamethyldisilazane (462 ml, 36-38% solution in tetrahydrofuran) with stirring at -85 to -95 0C for 60 minutes. Tert-butyl bromo acetate (137.5 g) in tetrahydrofuran (300 ml) was added to reaction mass and then stirred to 60 minutes at -85 to -95 0C. After completion of the reaction (monitored by TLC), the reaction mixture was poured into ammonium chloride solution (10%, 2.0 It) and extracted with ethyl acetate (2×750 ml). The combined organic layer was washed with demineralized water (1×750 ml) and dried over sodium sulphate. The solvent was evaporated under reduced pressure to obtain oily residue which was stirred with mixture of n-hexane (100 ml) and isopropyl alcohol (100 ml) at Oto -50C, filtered and dried under vacuum to obtain 153.12 g of title compound having chemical purity 99.41%, chiral purity 99.91% by HPLC, [α]D 20: (-)97.52° (c = 1, CHCl3) and M.P. : 117.1-118.20C.
Example 3: Preparation of 3(5)-benzyl-4-(4(i?)-benzyl-2-oxo-oxazolidin-3-yl)-4-oxobutyric acid Trifluoroacetic acid (100 g) was added to a solution of 3(5)-benzyl-4-(4-(/?)-benzyl-2-oxo-oxazolidin-3- yl)-4-oxobutyric acid tert-butyl ester (100 g) in dichloromethane (700 ml) at 25 0C and mixture was stirred further for about 12 hours ( when TLC indicated reaction to be complete). The reaction mixture was poured in to ammonium chloride solution (10%, 500 ml). The dichloromethane layer was separated and aqueous layer was extracted with dichloromethane (2 x 250 ml). The combined organic layer was dried over sodium sulphate and evaporated under reduced pressure to obtain title compound. The crude product was recrystallized from a mixture of ethyl acetate: n-hexane (1:4, 500 ml) to obtain 78.75g of the title compound having purity 99.56% by HPLC and M.P.: 145.9-146.40C.
Example 4: Preparation of (2S)-2-benzyl-l-((4R)-4-benzyl-2-oxo-oxazolidin-3-vI)-4-(hexahydro- isoindolin-2-yl)-butane-l,4-dione
To a solution of 3(5)-benzyl-4-(4-(/?)-benzyl-2-oxo-oxazolidin-3-yl)-4-oxo-butyric acid (50 g) in anhydrous dichloromethane (1.25 It) was added triethylamine (50 ml) with stirring at -20 to -30 0C and the stirred for 15 minutes. A solution of isobutylchloroformate (37.50g) in anhydrous dichloromethane (50 ml) was added at -20 to -30 0C and stirred for 60 minutes. Thereafter, a solution of cis- hexahydroisoindoline (32.50 g) in anhydrous dichloromethane (50 ml) was slowly added by maintaining temperature -20 to -300C. After the completion of the reaction (monitored by HPLC), the mixture was successively washed with 0.5N hydrochloric acid solution (500 ml), brine (300 ml) and dried over sodium sulphate. The solvent was evaporated under reduced pressure to obtain 102.0 g of the title compound having purity 94.39% by HPLC.
Example 5: Purification of r2S)-2-benzyl-l-((4R)-4-benzyl-2-oxo-oxazolidin-3-yl)-4-(hexahydro- isoindolin-2-vD-butane-l,4-dione
To the crude (2S)-2-benzyl-l-((4R)-4-benzyl-2-oxo-oxazolidin-3-yl)-4-(hexahydro-isoindolin-2-yl)- butane- 1,4-dione (51.0 g) was added methanol (150 ml) and the mixture was stirred for 5 hours at 0 to 5 0C. Solid that precipitated out was filtered, slurry washed with cold methanol (25 ml) and dried at 45 -50 0C under vacuum to obtain 28.80 g of pure title compound as a crystalline solid having purity of 99.71% by HPLC and M. P.: 104.1-105.70C.
Example 6: Preparation of calcium salt of (-SVmitiglinide. Step-1: Preparation of (-SVmitiglinide
(2S)-2-Benzyl- 1 -((4R)-4-benzyl-2-oxo-oxazolidin-3-yl)-4-(hexahydro-isoindolin-2-yl)-butane- 1 ,4-dione (28.0 g) was dissolved in tetrahydrofuran (196 ml) and a mixture of lithium hydroxide monohydrate (3.51 g) in demineralized water (56 ml) and hydrogen peroxide (40% solution, 5.5 ml) was added with stirring at 0 to 5 0C over a period of 30 minutes. The reaction mixture was further stirred at 0 to 5 0C till the completion of the reaction. After the completion of the reaction (monitored by TLC), the reaction was quenched with the addition of cooled sodium meta-bisulphate solution (25%, 168 ml) at 0 to 10 0C. The reaction mixture was extracted with ethyl acetate (2×112 ml), the layers were separated and the aqueous layer was discarded. The HPLC analysis of the aqueous layer shows 0.77% of amide impurity. The ethyl acetate layer was then extracted with aqueous ammonia solution (4%, 2×40 ml). The layers were separated and the aqueous layer was further extracted with ethyl acetate (2×280 ml). Combined ethyl acetate layer was discarded. This aqueous layer (280 ml) was used as such in the next stage. The aqueous layer display purity 96.19 % by HPLC and amide impurity 0.04% by HPLC. Step-2: Preparation of calcium salt of dSVmitiglinide
To the above stirred solution of (S)-mitiglinide in water and ammonia(280 ml), methanol (168 ml) was added, followed by calcium chloride (4.48 g) dissolved in demineralized water (56 ml) at ambient temperature and the mixture was stirred for 2 hours. The resulting precipitate was filtered, successively slurry washed with water (3 x 140 ml) and acetone (2 x 70 ml) and dried at 450C -500C under vacuum to obtain 16.1 g of title compound having purity 99.67% by HPLC and amide impurity 0.01% by HPLC. The title product was re-precipitated from a mixture of methanol and water and dried to obtain pure title compound.
Example 7: Preparation of (.SVmitiglinide
To a solution of (2S)-2-benzyl-l-((4R)-4-benzyl-2-oxo-oxazolidin-3-yl)-4-(hexahydro-isoindolin-2-yl)- butane- 1,4-dione (50 g) in tetrahydrofuran (350 ml) was added a solution of lithium hydroxide monohydrate (8.65 g) in demineralized water (100 ml) and hydrogen peroxide (30% w/w, 40 ml) with stirring at 5 to 10 0C over a period of 15 minutes. After the completion of reaction, sodium meta- bisulphate solution (40%, 500 ml) was added to the reaction mixture and the mixture was extracted with ethyl acetate (2 x 250 ml). The organic layer was dried over sodium sulphate and evaporated under vacuum to obtain 45.5 g of title compound having 35 % of R-benzyl oxozolidin-2-one as impurity. Example 8: Purification of (.S)-mitiglinide
Aqueous ammonia solution (4%, 300 ml) was added to the crude (5)-mitiglinide (30 g) and stirred. The reaction mixture was washed with ethyl acetate (3 x 300 ml). Thereafter the reaction mixture was acidified to pH 1 to 2 with IN hydrochloric acid solution (250 ml) and extracted with ethyl acetate (2 x 150 ml). The layers were separated and ethyl acetate layer was washed with demineralized water (2 x 150 ml), dried over sodium sulphate and then evaporated under reduced pressure to obtain 16.2 g of pure (5)-mitiglinide having purity 95.55% by HPLC Example 9: Preparation of calcium salt of (S)-mitiglinide
To a solution of (<S)-mitiglinide (15 g) in water (150 ml) and aqueous ammonia solution (25%, 15 ml) at 25 to 30 0C, a solution of calcium chloride (7.5 g) in demineralized water (37.5 ml) was added. The mixture was stirred for 1 hour to precipitate the calcium salt of (5)-mitiglinide dihydrate. The resulting precipitate was filtered, slurry washed with water (3 x 150ml) and dried at 45 to 50 0C to obtain 13.25 g of the title compound having purity of 98.84% by HPLC. Example 10: Purification of calcium salt of (5)-mitiglinide
(iS)-mitiglinide calcium (10 g) was dissolved in dimethylformamide (100 ml). This is followed by the addition of demineralized water (500 ml) at 25 to 30 0C. The mixture was stirred for 30 minutes. The precipitated solid was filtered, washed with water (10x 50ml) and dried at 45 to 50 0C under vacuum to obtain 8g of pure title compound as a crystalline solid having purity of 99.62% by HPLC. Example 11: Preparation of amorphous mitiglinide calcium
Crystalline mitiglinide calcium (2.0 g) was dissolved in tetrahydrofuran (20 ml) and filtered to remove undissolved and suspended particles. The solvent was then evaporated under vacuum to obtain a powder which was then dried under vacuum at 40-600C to obtain 1.70 g of the title compound. Example 12: Preparation of amorphous mitiglinide calcium
Crystalline mitiglinide calcium (2.0 g) was dissolved in dichloromethane (30 ml) and filtered to remove undissolved and suspended particles. The solvent was then evaporated under vacuum to obtain a powder which was then dried under vacuum at 40-600C to obtain 1.64 g of the title compound. Example 13: Preparation of amorphous mitiglinide calcium
Mitiglinide (2.0 g) was dissolved in methanol (20 ml) and methanolic ammonia (5.0 ml) solution was added to it. The solution was stirred at 25-30 0C and calcium chloride (1.5 g) dissolved in methanol was mixed with the solution of mitiglinide and ammonia in methanol and the solution was filtered to remove the suspended particles. The solvent was then evaporated under vacuum to obtain a powder which was then dried under vacuum at 40-600C to obtain 1.9 g of the title compound. Example 14: Preparation of amorphous mitiglinide calcium
Mitiglinide (2.0 g) was dissolved in dichloromethane (20 ml) and aqueous ammonia (3.6 ml, 25 % solution) was added to it. The solution was stirred at 25-300C and solid calcium chloride (1.5 g) was mixed with the solution of mitiglinide and ammonia in dichloromethane and the solution warmed at 30 – 35 0C. The solution was washed with water (2 xlO ml) and the clear solution was dried over sodium sulfate, filtered and evaporated under vacuum and finally dried at under vacuum at 40-60 0C to obtain 1.75 g of the title compound.
Example 15: Preparation of amorphous mitiglinide calcium
Crystalline mitiglinide calcium dihydrate (2.0 g) was dissolved in ethyl acetate (30 ml) and filtered to remove undissolved and suspended particles. Approimately. 60 % of the solvent was distilled off under vacuum to obtain a stirrable solution. The solution was then cooled to 15-2O0C, mixed with n-heptane (20 ml) and the mixture was stirred for 30 minutes. The resulting solid was filtered, washed with n-heptane and dried under vacuum at 45-600C to yield 1.72 g of the title compound. Example 16: Preparation of amorphous mitiglinide calcium
Crystalline mitiglinide calcium (2.Og) was dissolved in dichloromethane (30 ml) and filtered to remove undissolved and suspended particles. Approximately 60 % of the solvent was distilled off under vacuum to obtain a stirrable solution. The solution was then cooled to 15-200C and mixed with diisopropyl ether (20 ml). The mixture was stirred for 30 minutes and the resulting solid was filtered, washed with diisopropyl ether and dried under vacuum at 45-600C to obtain 1.70 g of the title compound. Example 17: Preparation of amorphous mitiglinide calcium
Mitiglinide (2.0 g) was dissolved in dichloromethane (20 ml) and aqueous ammonia (3.6 ml, 25 % solution) solution was added to it. The solution was stirred at 25-30 0C and mixed with solid calcium chloride (1.5 g) and the solution warmed at 30-35 0C and stirred for 30 minutes. The solution was washed with water (2 x 10 ml) and the clear solution was dried over sodium sulfate, and filtered. Approximately 60% of the solvent was distilled off under vacuum and the resulting viscous oil was cooled to 10-15 0C and mixed with diisopropyl ether (50 ml). The reaction mixture was stirred for 30-35 minutes and the resulting solid was filtered and dried at 40-600C to obtain 1.75 g of the title compound. Example 18: Conversion of amorphous mitiglinide calcium into crystalline mitiglinide calcium A suspension of amorphous mitiglinide calcium in diisopropyl ether (30 ml) was stirred for 2 hours at 25- 300C, filtered and dried under vacuum at 45-600C to obtain crystalline form of mitiglinide calcium. Example 19: Preparation of crystalline mitiglinide calcium
To a solution of mitiglinide (2.5 g) in water (2.5 ml), aqueous ammonia solution (approx 25%, 4.0 ml) and acetonitrile (2.5 ml) at 10-150C, calcium chloride (1.32 g) dissolved in demineralized water (15 ml) was added. The mixture was stirred for 2 hours. The resulting precipitate was filtered, slurry washed with water (3 x 25 ml) and acetone (2 x 5 ml) and dried at 45-500C under vacuum to obtain 2.12 g of title compound having purity: 99.72 % by HPLC.
Example 20: Preparation of crystalline mitiglinide calcium
To a solution of mitiglinide (2.5 g) in water (2.5 ml), aqueous ammonia solution (approx 25%, 4.0 ml) and tetrahydrofuran (2.5 ml) at 10-150C, calcium chloride (1.32 g) dissolved in demineralized water (15 ml) was added. The mixture was stirred for 2 hours. The resulting precipitate was filtered, slurry washed with water (3 x 25 ml) and acetone (2 x 5 ml) and dried at 45-500C under vacuum to obtain 1.95 g of title compound having purity: 99.52 % by HPLC.
Example 21; Preparation of crystalline mitiglinide calcium
To a solution of mitiglinide (30.0 g) in water (300 ml), aqueous ammonia solution (approx 25%, 48 ml) and acetone (300 ml) at 10-150C, calcium chloride (15.8 g) dissolved in demineralized water (180 ml) was added. The mixture was stirred for 2 hours. The resulting precipitate was filtered, slurry washed with water (3 x 300 ml) and acetone (2 x 60 ml) and dried at 45-500C under vacuum to obtain 24.32 g of title compound having purity: 99.42 % by HPLC.
Example 22: Preparation of crystalline mitiglinide calcium
To a solution of mitiglinide (3.0 g) in water (30 ml), aqueous ammonia solution (approx 25%, 4.8 ml) and isopropyl alcohol (300 ml) at 10-150C, calcium chloride (1.58 g) dissolved in demineralized water
(18 ml) was added. The mixture was stirred for 2 hours. The resulting precipitate was filtered, slurry washed with water (3 x 30 ml) and acetone (2 x 6 ml) and dried at 45-500C under vacuum to obtain 1.92 g of title compound having purity: 99.65 % by HPLC.
Example 23: Preparation of (2S)-2-benzyWV-((lR)-l-benzyl-2-hydroxy-ethyl)-4-(hexahvdro- isoindolin-2-yl)-4-oxo-buryramide
To a solution of (2S)-2-benzyl-l-((4R)-4-benzyl-2-oxo-oxazolidin-3-yl)-4-(hexahydro-isoindolin-2-yl)- butane-l,4-dione (20.0 g) in tetrahydrofuran (140 ml), a solution of lithium hydroxide monohydrate
(3.43 g,) in demineralized water (40 ml) was added and the reaction mixture was refluxed for 4 hours till the completion of the reactions (monitored by thin layer chromatography). After the completion of the reaction, the reaction mixture was poured into demineralized water (100 ml) and extracted with ethyl acetate (2 x 80 ml). The combined organic layer was washed with water (80 ml) and dried over sodium sulphate. The solvent was evaporated under reduced pressure to give residue which was stirred in isopropyl alcohol at 0-5 0C for 5 hours. The mixture was filtered and then dried at 40-45 0C under vacuum to obtain 12.48 g of title compound having purity 99.77 % by HPLC. Melting point = 77 – 800C.

PAPER
Helvetica Chimica ActaVolume 87, Issue 8, Version of Record online: 27 AUG 2004

PAPER
asian journal of chemistry asian journal of chemistry
(S)-Mitiglinide calcium dihydrate is designated chemically … Identification, Synthesis and Characterization of Impurities of (S)-Mitiglinide Calcium Dihydrate………http://www.asianjournalofchemistry.co.in/(X(1))/User/ViewFreeArticle.aspx?ArticleID=26_9_51
PATENT
CN 102382033

PATENT
https://www.google.com/patents/CN104311471A?cl=en
Mitiglinide calcium (mitiglinide calcium), the chemical name (2S) -2_ benzyl-3- (cis – hexahydro-2-isoindoline-carbonyl) propionic acid calcium salt dihydrate , for the treatment of type II diabetes. Kissei by Japanese pharmaceutical company research and development, and for the first time on sale in Japan in May 2004. Mitiglinide calcium is the second repaglinide, nateglinide after the first three columns MAG urea drugs, are ATP-dependent potassium channel blocker, is a derivative of phenylalanine, and its mechanism Similar sulfonylureas, but a faster onset of action and short half-life, is conducive to reducing postprandial blood glucose in diabetic patients, and avoid continuous glucose-induced low blood sugar, with the “in vitro pancreas” reputation.
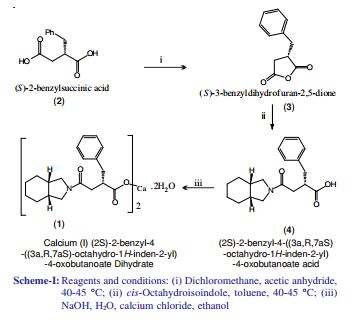
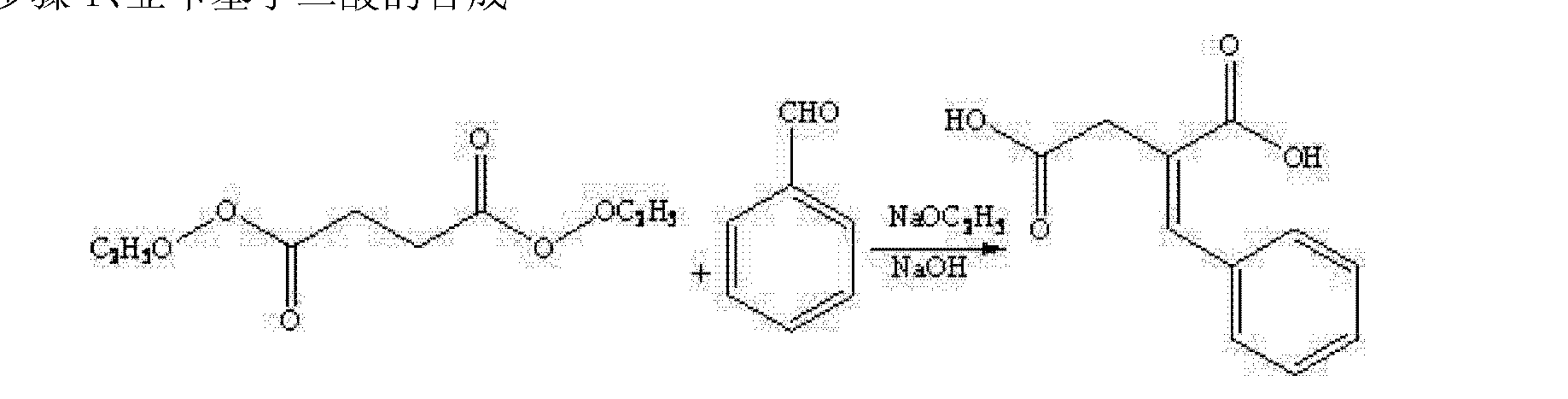
郑德强 etc. on “Food and Drug” magazine was first disclosed the synthesis of calcium Mitiglinide, this method dimethyl succinate and benzaldehyde for raw materials, Stobble condensation, hydrolysis, dehydration anhydride, cis – perhydro isoindole reduced to give racemic acid after condensation, and then split, and salt get Mitiglinide calcium. Specific synthetic route the following equation. The method is relatively complex, in the preparation process to generate half of the unwanted enantiomer, which will waste a lot of cis – perhydro isoindole, and in the preparation of cis – to use science as a whole hydride hydrogen isoindole time reducing agent, the operation is more complicated, the cost is relatively high, and the chiral amine as a resolving agent split, the yield is low.

The patent discloses a CN201010573666 diethyl succinate and benzaldehyde, condensation occurs Stobble sodium ethoxide in ethanol and then hydrolyzed benzylidene succinic acid, succinic acid benzylidene get by catalytic hydrogenation DL-2-benzyl succinic acid, DL-2-benzyl succinic acid by (R) – a chiral amine resolving to give (S) -2- benzyl succinic acid, (S) -2- benzyl succinic acid anhydride to generate its role in the acetic anhydride, and the resulting acid anhydride and cis – hexahydro isoindole reaction of Mitiglinide acid, calcium chloride and ammonia most 后米格列奈 acid reacts with calcium Mitiglinide dihydrate. The synthesis route following formula. This method effectively avoids the expensive intermediate cis – perhydro isoindole waste, reduce costs, but still amounted to a six-step synthesis route much so that the reagent type, long cycle, low yield, and direct use in the synthesis process Sodium block protonated reagent preparation sodium methylate, generate a lot of flammable hydrogen gas, limiting the industrial application of the method.
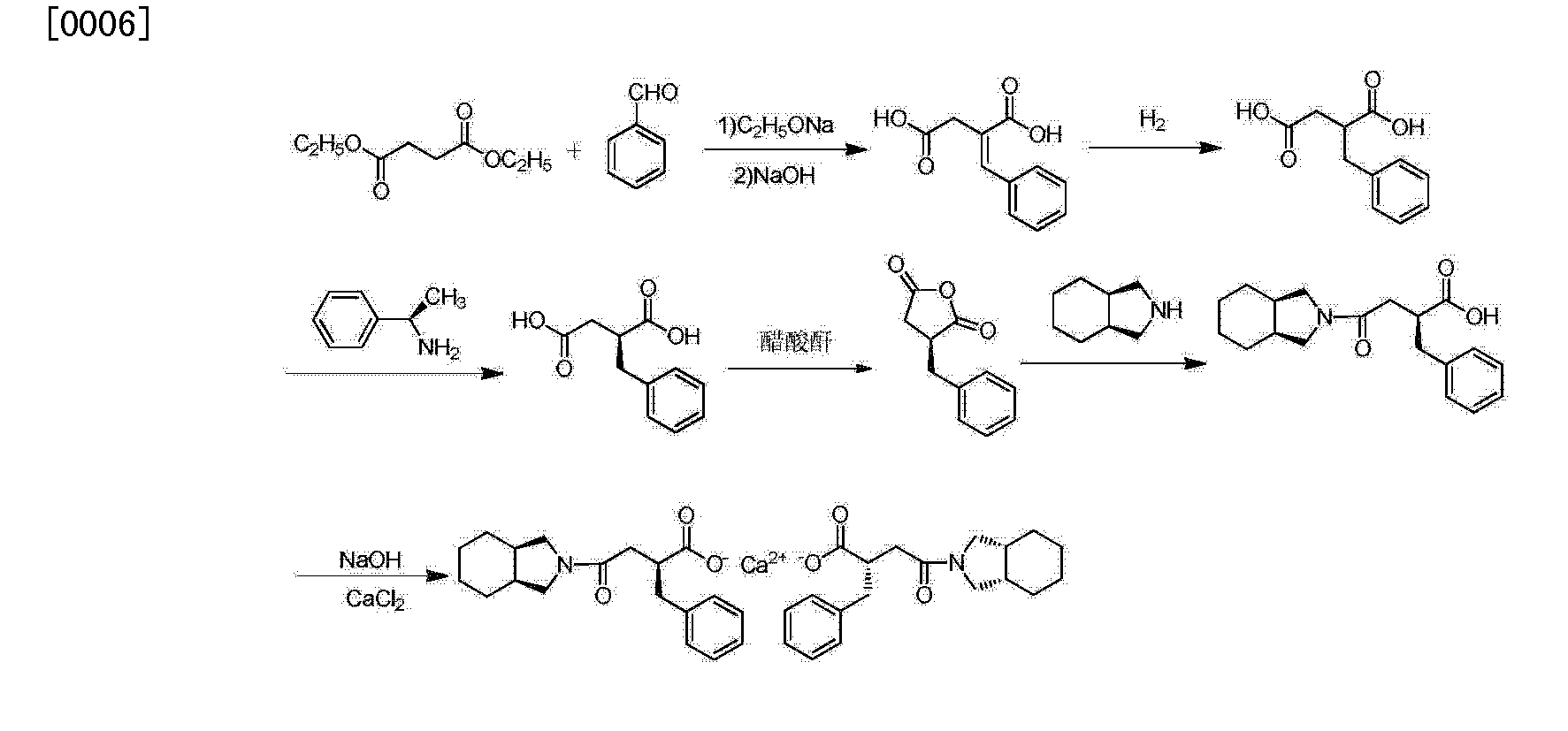
The present invention solves is to overcome the existing routes that exist in step lengthy reagent variety, low yield, long cycle, high cost, not suitable for industrial production shortcomings. The present invention provides the following formula preparation process route mitiglinide calcium, organic solvent for this preparation method uses less synthesis process is simple, high yield, good purity, suitable for industrial production.

An improved Mitiglinide calcium industrialized preparation method comprises the following steps: Step 1: Preparation of 2-benzylidene succinic acid; 2 steps: (S) prepared _2_ section succinic acid; Step 3: 2- (S) – section group _4_ oxo – (cis – perhydro isoindol-2-yl) butyric acid; Step 4: Preparation Mitiglinide calcium. Characterized in that: in step 1, using commercially available reagents protonated organic bases, protonation process using an organic alkali solution was slowly feeding methods. Step 2 chiral asymmetric reduction. Step 3 fails anhydride using direct selective amidation. Step 4 beating impurities using an aqueous solvent, prepared mitiglinide calcium dihydrate purification method.
The preparation step 1, using a commercially available organic bases as sodium methoxide or sodium ethoxide protonation agent. As optimization program, feeding method using sodium methoxide or sodium ethoxide solution formulated as the corresponding alcohol and the corresponding dialkyl succinate protonating a nucleophilic substitution reaction.
The preparation method described in Step 2, the use of Ru with BINAP homogeneous catalyst Ru (OAc) 2 [(S) -BINAP] as a chiral asymmetric synthesis of chiral reducing reagent.
The steps of the preparation method 3, using ethyl acetate as a reaction solvent, acid binding agent triethylamine do, imidazole and thionyl chloride selective amidation reagent, for cis – perhydro isoindole conduct Selective condensation title intermediate.
The step of preparing said 4, mitiglinide calcium crude product was slurried in 95% ethanol by suction, after simple preparation of high purity mitiglinide calcium dihydrate.
More specifically, the industrialized Mitiglinide calcium preparation, the following steps: Step 1: Preparation of succinate 2_ Benzylidene

Sodium methoxide (sodium ethoxide) was dissolved in methanol (ethanol), was added dropwise to dimethyl succinate (ethyl) ester, was heated at reflux for 30min, benzaldehyde was added dropwise under reflux, stirring at reflux completed the dropwise 3~5h, drops adding an aqueous solution of 4N NaOH dropwise Bi refluxed 4~6h, cooled to room temperature, adjusted with 6N HCl San PH 2, a solid precipitated, centrifuged, and dried to give the title intermediate 1. Step 2: Preparation of (S) -2- acid, benzyl butyl

Intermediate 1, methanol, and Ru (OAc) 2 [(S) -BINAP] into the reactor, the reactor with N2 the replacement air after heating to 50 ° C, a hydrogen pressure through 10h, cooled, filtered, The filtrate was concentrated to dryness to give the title intermediate 2. Step 3: 2- (S) – benzyl-4-oxo – (cis – perhydro isoindol-2-yl) butyric acid

Ethyl acetate was added to the reactor, triethylamine, imidazole and Intermediate 2, was stirred and cooled to -15~-5 ° C, was added dropwise thionyl chloride addition was complete, the -15 ° C~_5 ° C Under continued stirring 6h, a solution of cis – perhydro isoindole, drip completed, stirred at room temperature overnight, the reaction mixture was added IN hydrochloric acid, stirred Ih, separation, and the organic layer was washed with sodium hydroxide solution to extract IN The combined aqueous layer was washed with a small amount of ethyl acetate, the aqueous layer was adjusted with IN hydrochloric acid and the PH = 3, the aqueous layer was extracted with ethyl acetate, the organic layers combined, washed with water and saturated brine, and the organic layer was dried over anhydrous Na2SO4, filtered and the filtrate concentrated under reduced pressure to obtain the objective compound 3 billion Step 4: Preparation of calcium Mitiglinide

The 3 was dissolved in ethanol, was added 2N sodium hydroxide solution, after mixing the solution was added dropwise a 10% aqueous solution of calcium chloride, the reaction mixture was stirred vigorously 3~5h, ice-cooled, filtered, the filter cake with 95% ethanol beating crystallization, filtration, and dried in vacuo to give the title compound I.
Accordingly, the present invention is a method for preparing mitiglinide calcium has the following advantages:
1, Step 1, using commercially available sodium methylate (sodium ethanol) instead of sodium block as a proton agent, effectively avoid the risk of sodium block formed during the reaction a lot of flammable hydrogen gas, industrial production safer. Another use dropping protonated reagent feeding method can effectively avoid succinic acid alkyl ester of two methylene groups are protonated and reduce the incidence of side effects, so that the yield increased by nearly 20%.
2, Step 2, the selective reduction of chiral reagent (S) -BINAP instead of the original route after the first split reduction method, not only simplifies the reaction step, but low yield while avoiding split It leads to the risk of an increase in cost.
3, Step 3, the fixed selective amidation reaction conditions instead of the original first into anhydride after amidation reaction that simplifies the reaction steps to reduce the unit operations, shortening the production cycle, improve production efficiency.
4, Step 4, by using an aqueous solution of calcium Mitiglinide ethanol refining crude beating, then dried under reduced pressure to control the moisture content and reduce the difficulty of the operation, more conducive to industrial production.DETAILED DESCRIPTION The following examples further illustrate the invention, but the present invention is not limited thereto. Example One Step I: Preparation 2_ benzylidene succinic acid Sodium methoxide (9kg) and methanol (48L) into the 100L reactor, stirring to dissolve, into the high slot 50L. The dimethyl succinate (20kg) into the 200L reaction vessel, heated to reflux, methanol was added dropwise a solution of fast high tank of sodium methoxide, refluxed for reaction completion dropwise 30min, was added dropwise under reflux benzaldehyde (10. 9kg) dropwise with stirring at reflux completed 3~5h, HPLC detection benzaldehyde completion of the reaction, a solution of aqueous 4N NaOH (38L), Bi dropwise refluxed 4~6h, cooled to room temperature, 2, adjusted with 6N HCl and the precipitated solid was San PH, centrifugation, and dried in vacuo to give a pale yellow solid 19kg, i.e. an intermediate, yield 90%. Step 2: Preparation of (S) -2- butyric acid benzyl 200L detecting a high pressure hydrogenation reactor airtight, Intermediate I (19kg), methanol (95L) containing 5% Ru (0Ac) 2 [(S ) -BINAP] molecular sieve (SBA-15) supported catalyst (0. 95kg, homemade) into the reactor, purge the inside of the reactor with N2 atmosphere, followed by heating to 50 ° C, atmospheric pressure hydrogen-10h, cooled, filtered and the filtrate was concentrated to dryness under reduced pressure, the resulting solid was recrystallized from ethyl acetate and dried in vacuo to give an off-white solid 15. 5kg, i.e. intermediate 2, yield 81%, chiral purity 90. 5% θ. θ .. Step 3: 2- (S) – benzyl-4-oxo – (cis – perhydro isoindol-2-yl) butyric acid in 500L reaction vessel was charged with ethyl acetate (225L), triethylamine (1.8kg), imidazole (9. 8kg) and Intermediate 2 (15kg), stirred and cooled to -KTC, was added dropwise thionyl chloride (17. 2kg), the addition was complete, the -KTC~-5 ° C under Stirring was continued for 6h, a solution of cis – perhydro isoindole (9kg), drip completed, the reaction was stirred at room temperature for 18h, the reaction mixture was added IN HCl (150L) was stirred Ih, liquid separation, the organic layer was washed with IN sodium hydroxide solution (100LX3) extracted aqueous layers were combined, washed with ethyl acetate (50L) with, water layer was washed with IN of hydrochloric acid adjusted to PH = 3, the aqueous layer was extracted with ethyl acetate (IOOmLX 3), the combined organic layers , saturated brine (50LX 3) was washed, and the organic layer was dried over anhydrous Na2SO4, filtered, and the filtrate was concentrated under reduced pressure to give an oil 19. 8kg, i.e. Intermediate 3 Yield: 87%. Step 4: Preparation of mitiglinide calcium Intermediate 3 (. 19 8kg) and absolute ethanol (99L) into the 200L reactor, and stirred to dissolve, was added 2N sodium hydroxide solution (35L), minutes after mixing Batch into the high slot. The 500L reaction vessel was added 5% aqueous calcium chloride solution (155L), stirring was added dropwise a solution of the high slot, dropwise with vigorous stirring the reaction completion 3~5h, centrifuged, the cake was washed with 95% ethanol (99L) was recrystallized beating, centrifugation and dried in vacuo (50 ° C / 0. 09MPa), to give the title compound I 16. lkg, yield 73%.
PATENT
https://www.google.com/patents/CN102424664A?cl=en
Mitiglinide calcium Phenylalanine belong chiral compound synthesis routes according to different methods of constructing chiral center has the following three synthetic process:
① split method 😦 Document: CN 102101838A, CN 1844096, etc.)

In this method, diethyl succinate and benzaldehyde by Mobbe condensation, hydrolysis, dehydration anhydride, and after cis-hydrogenated isoindole condensation is reduced to give racemic acid, and then split, and salt to give Mitiglinide calcium. The first method step condensation reaction impurities, product separation and purification difficult, finally resolving the yield is low. This method is also a lack atom economy.
② asymmetric hydrogenation 😦 Document tetrahedron Letters, 1987,28 (17), 1905-1908; Tetrahedron Letters, 1989,30 (6), 735-738)

[0027] This method requires expensive rhodium complexes (Rh, (2S, 4Q-N_-butoxycarbonyl-4-diphenylphosphino _2_ diphenylphosphino-2-diphenylphosphino methylpyrrolidine alkyl), making the production cost is greatly improved, and the need for high-pressure hydrogenation reaction, is not conducive to industrial production.
③ chiral method 😦 Document: CN 1680321A)

The method uses phenylalanine as chiral starting materials, after diazotization, nucleophilic substitution, high temperature decarboxylation and condensation reaction product. Wherein the decarboxylation temperature is too low yield, making the overall process costs.
DISCLOSURE
The object of the present invention is to provide a simple, effective and easy-to-operate preparation Mitiglinide calcium.
The present invention provides a process for the preparation of calcium Mitiglinide, the synthesis route is as follows:

Step 1: D- phenylalanine in the acid hydrolysis of formula (¾ 2- hydroxy acid;
Step 2: formula (¾ 2- hydroxy acid under basic conditions to give protected hydroxyl sulfonate of formula (¾-hydroxyphenyl propionic acid ester;
Step 3: The formula (¾-hydroxyphenyl propionic acid ester in the acid-catalyzed carboxyl ester-protected formula (4) phenylalanine methyl sulfonate carboxylate;
Step 4: cis-hydrogen isoindole synthesis formula (6) perhydro isoindole halide;
Step 5: Under alkaline conditions, the formula ⑷ formula (6) nucleophilic substitution reaction formula (5) Mitiglinide acid
Step 6: Under alkaline conditions, the formula (¾ Mitiglinide ester hydrolysis to the calcium salt of formula (1) Mitiglinide calcium.
Preferably, the specific steps include:
Step 1: (D) – phenylalanine hydrolysis in a strong acid of formula (2) 2-hydroxyphenyl propionic acid
In (D) – phenylalanine as a starting material, in the presence of a strong acid such as sulfuric acid, _5 ° C _5 ° C hydrolysis, to give Formula (2) 2-hydroxyphenyl propionic acid White solid.
Step 2: The formula (¾ 2- hydroxy acid under basic conditions to protect the hydroxyl group sulfonic acid ester of formula (¾-hydroxyphenyl propionic acid ester
2-hydroxyphenyl propionic acid in an organic base such as triethylamine or pyridine, or an inorganic base such as sodium bicarbonate, sodium carbonate or potassium carbonate effect, p-hydroxybenzoic acid ester protecting performed, the protecting group used is an aliphatic or aromatic sulfonic acid group such as mesylate, tosylate or p-toluenesulfonic acid group, a sulfonic acid group is preferably methyl group or p-toluenesulfonic acid.
Step 3: Protect formula formula (¾-hydroxyphenyl propionic acid ester in the acid-catalyzed carboxyl ester group (4) benzenepropanoic
MitigIinide1 (I) carboxylic acid ester sulfonate
In the catalytic acid carboxyl benzenepropanoic acid ester group protection, the use of alcohol may be fatty alcohols or aromatic alcohols, preferably ethanol, t-butanol or benzyl alcohol.
Step 4: cis-hydrogen isoindole synthesis formula (6) perhydro isoindole halide
In the synthesis of perhydro isoindole halide in the haloacetyl halide can be used chloroacetyl chloride, bromoacetyl chloride or bromoacetyl bromide, chloroacetyl chloride is preferred.
Step 5: Under alkaline conditions, (4) and (6) a nucleophilic substitution reaction formula (¾ Mitiglinide acid
Under the conditions of a strong base, such as sodium alkoxide such as sodium ethoxide or sodium methylate, perhydro isoindole halide and phenylalanine sulfonate nucleophilic substitution reaction Mitiglinide ethyl reaction temperature of -10 ° C -25 ° c, preferably 0 ° C.
Step 6: Under alkaline conditions, the formula (¾ Mitiglinide ester hydrolysis to the calcium salt of formula (1) calcium Mitiglinide
Ethyl mitiglinide under basic conditions such as sodium hydroxide, potassium hydroxide, or an amine (ammonia) in the presence of an aqueous solution of calcium chloride, and hydrolyzed as calcium salt, in aqueous solution under conditions of heavy alcohol crystallization, high purity mitiglinide calcium.
The present invention and the prior art comparison, has the following advantages:
1, to find an innovative high-yield process for preparing calcium Mitiglinide route, a total yield of 47%;
2, with respect to the routing methods reported in the literature, the optical yield doubled, ee greater than 99%;
3. The process route of the raw materials are cheap, readily available, avoiding costly chiral resolving agents or the use of a catalyst;
4. The process route mild conditions, high temperature decarboxylation overcome the harsh reaction conditions.
In the present invention, (D) – phenylalanine as a starting material, after diazotization, a hydroxyl group and a carboxyl group protected, nucleophilic substitution, hydrolysis and other reactions prepared mitiglinide calcium, high yield. The present invention provides a process used by a wide range of raw materials, low prices, the total yield of 47%, optical purity greater than 99%, and mild reaction conditions, the reaction process is simple, avoid the literature, such as split, high-pressure hydrogenation method low yield, long reaction steps and other shortcomings, but also to overcome the harsh conditions of high temperature reaction deacidification, etc. for preparation and production of calcium Mitiglinide provides a new choice.
The process route mild conditions, high temperature decarboxylation overcome the harsh reaction conditions.
In the present invention, (D) – phenylalanine as a starting material, after diazotization, a hydroxyl group and a carboxyl group protected, nucleophilic substitution, hydrolysis and other reactions prepared mitiglinide calcium, high yield. The present invention provides a process used by a wide range of raw materials, low prices, the total yield of 47%, optical purity greater than 99%, and mild reaction conditions, the reaction process is simple, avoid the literature, such as split, high-pressure hydrogenation method low yield, long reaction steps and other shortcomings for Mitiglinide calcium preparation and production of a new choice.
Preferably, in the above embodiment, each step may be the following alternative, the embodiment can achieve the same advantageous effects to a third embodiment of embodiment:
Step 1: (D) – phenylalanine in the acid hydrolysis of formula (¾ 2- hydroxy acid
In (D) – phenylalanine as a starting material, in the presence of sulfuric acid, -50C _5 ° C hydrolysis, to give Formula O) 2-hydroxyphenyl propionic acid White solid.
Step 2: formula (¾ 2- hydroxy acid under basic conditions to give protected hydroxyl sulfonate of formula C3) hydroxyphenyl propionic acid ester
2-hydroxyphenyl propionic acid in an organic base such as triethylamine or pyridine, or an inorganic base such as sodium bicarbonate, sodium carbonate or potassium carbonate effect, p-hydroxybenzoic acid ester protecting performed, the protecting group used is an aliphatic or aromatic sulfonic acid group such as mesylate, tosylate or p-toluenesulfonic acid group, a sulfonic acid group is preferably methyl group or p-toluenesulfonic acid.
Step 3: Formula C3) hydroxyphenyl propionic acid ester in the acid-catalyzed carboxyl ester-protected formula (4) phenylalanine methyl sulfonate carboxylate [0118] In the acid-catalyzed, styrene-acrylic acid ester-protected carboxy, the use of alcohol may be fatty alcohols or aromatic alcohols, preferably ethanol, t-butanol or benzyl alcohol.
Step 4: cis-hydrogen isoindole synthesis formula (6) perhydro isoindole halide
In the synthesis of perhydro isoindole halide in the haloacetyl halide can be used chloroacetyl chloride, bromoacetyl chloride or bromoacetyl bromide, chloroacetyl chloride is preferred.
Step 5: Under alkaline conditions, the formula ⑷ formula (6) nucleophilic substitution reaction formula (5) Mitiglinide acid
Under the conditions of a strong base, such as sodium alkoxide such as sodium ethoxide or sodium methylate, perhydro isoindole halide and phenylalanine sulfonate nucleophilic substitution reaction Mitiglinide ethyl reaction temperature of -10 ° C -25 ° c, preferably 0 ° C.
Step 6: Under alkaline conditions, the formula (¾ Mitiglinide ester hydrolysis to the calcium salt of formula (1) calcium Mitiglinide
Ethyl mitiglinide under basic conditions such as sodium hydroxide, potassium hydroxide, or an amine (ammonia) in the presence of an aqueous solution of calcium chloride, and hydrolyzed as calcium salt, in aqueous solution under conditions of heavy alcohol crystallization, high purity mitiglinide calcium.

Patent
https://www.google.com/patents/CN103724253A?cl=en
bis [(2s) -2- benzyl-3- (cis – hexahydro isoindole-2-carbonyl) propionic acid] monocalcium dihydrate (mitiglinide calcium), the formula C38H48CaN206.2Η20 English called Mitiglinide Calcium Hydrate, structural formula (I) as

Mitiglinide Calcium is synthesized by Japan Orange Health Pharmaceutical Co., Ltd., in April 2004 in Japan, for through diet and exercise therapy can effectively control high blood sugar in type II diabetes patients.Mitiglinide calcium is the second repaglinide, nateglinide third after the United States and Glenn urea drugs belong phenylalanine derivatives. By closing ΑΤΡ Mitiglinide calcium-dependent pancreatic β cell membrane Κ channel, resulting in the Ca flow, increase intracellular Ca concentration of extracellular vesicles containing threshing leaving insulin, thereby stimulating the secretion of insulin.And only when the meal will be rapid and transient stimulates the pancreas to secrete insulin, sulphonylureas with the traditional Compared to the rapid onset and short duration of action, inhibition of postprandial hyperglycemia characteristic of type II diabetes, to avoid low blood sugar react, early first- and mild diabetes treatment, and well tolerated.
According to the literature and patent reports, prepared Mitiglinide calcium are the following methods.
Method I: 2_ (S) _ benzyl succinic acid as raw material, amides, reduction, calcium salt formation Mitiglinide this method, although fewer steps, but the chiral compound materials, expensive , the production cost is high, not suitable for industrial production. References: Sorbera LA, Leeson PA, Castaner RM, et al.Mitiglinidecalcium (KAD-1229) [J] .Drugs Future, 2000,25 (10):. 1034-1042 [0007] Method Two: succinate methyl ester with benzaldehyde for raw materials, Stobble condensation, hydrolysis, dehydration anhydride, cis – perhydro isoindole after condensation is reduced to give racemic acid, and then split into calcium salts and the like have Mitiglinide. This method is relatively complex and condensation reaction impurities, product separation and purification difficult, costly, and chiral separation time yield is low.[Reference: Zheng Dejiang, Liu Wentao, Wu Lihua synthetic calcium Mitiglinide [J] Food and Drug, 2007,9 (11): 13-15]
Method three: dimethyl succinate and benzaldehyde for raw materials, Stobbe condensation, reduction, split, with p-nitrophenol and dicyclohexyl carbodiimide activated calcium salt formation Mitiglinide This production cost is relatively high, and used column chromatography, suitable for industrial production. References: Synthesis Technology Zhang Hongmei Chen meritorious, Cao Xiaohui Mitiglinide of [J], modern chemicals, 2008,28 (8): 56-59.]
Example 1:
The cis – hexahydro-isoquinoline (250.4g, 2mol), anhydrous potassium carbonate (304.0g, 2.2mol), methylene burn (1000ml) was added to the reaction flask, keeping the temperature 0-5 ° C with vigorous stirring, dropwise acetyl chloride (271.0g, 2.4mol) in dichloromethane (500ml) solution, drip completed, room temperature 2.5h, point board monitoring, reaction complete, additional water 1000ml, organic layer was separated, water (1000ml), saturated brine (1000ml), dried over anhydrous sodium sulfate overnight, dichloromethane was distilled off under reduced pressure to give cis -N- chloroacetyl hexahydro isoindole (2) 357.4g oil close Rate: 88.6%.
The cis -N- chloroacetyl hexahydro isoindole (302.5g, 1.5mol), N_ within phenylpropionyl camphor sulfonamide (573.0g, 1.65mol), 70% sodium hydride (56.6g, 1.65 mol), Ν, Ν- dimethylformamide (900ml) was added to the reaction flask, at 50 ° C, the reaction was stirred vigorously 12h, to give the alkylated product, placed to room temperature before use.
100ml of water was slowly dropped to the above-mentioned system, drip complete, lithium hydroxide (39.5g, 1.65mol), tetrahydrofuran (600ml), at 0-5 ° C under a 30% solution of hydrogen peroxide solution 680ml, drop Albert, was transferred to the reaction was continued at room temperature for 18h, point board monitoring, reaction complete, additional water 1200ml, adjusting the pH to about 2_3, extracted with dichloromethane (900ml X 3), the combined organic phases with saturated brine (1500ml) wash, overnight over anhydrous sodium sulfate, the solvent was distilled off under reduced pressure to give a viscous liquid, to which was added ethyl acetate 250ml, stirred at room temperature, suction filtered, the filter cake with ethyl acetate (150ml) and dried to give (2s) – 2-benzyl-3- (cis – hexahydro isoindole-2-carbonyl) – propionic acid (6) as a white solid 231.8g, two steps yield: 49%. Compound 6 (230g, 0.73mol), water 1150ml, added to the reaction flask. After the whole solution, was added 2mol / L sodium hydroxide solution, 400ml, stirred at rt for 30min, was slowly added dropwise with vigorous stirring chloride (162.0g, 1.46mol) in water (320ml) solution dropwise was completed, the reaction was continued for 1.5h, filtration, water (200ml X 2) washing the filter cake to give a white solid, 60 ° C and dried under reduced pressure to 3h, the filter cake with 95% ethanol (2300ml) recrystallized Mitiglinide calcium (I) 430g, yield: 83.6%, mp: 178 ~ 183 ° C, FAB-MS: m / z316 [M + l] +; [α] D20 = + 5.45 ° (C = 1, methanol) [Document: m.ρ.: 179 ~ 185Ό, [α] d20 = + 5.64 ° (C = L 0, methanol)]; purity: 99.8% [HPLC normalization method : Column C18, mobile phase L OOmol / L potassium dihydrogen phosphate buffered saline – acetonitrile-water (20:35: 30) (adjusted pH = 2.10); detection wavelength 210nm]; iH-NMlUCDCldOOM), δ: 1.1 ~ 1.5 (16Η, m), 1.8 ~ 2.4 (6Η, m), 2.5 ~ 3.1 (14Η, m) 3.3 ~
3.8 (6H, m) 7.4 ~ 7.6 (10H, m); Elemental analysis (%):. C64.68, Η7.35, Ν3.94, Theory: C64.75, Η7.44, Ν3.97 yield : 36.05%, a purity of 99.8%.

PAPER
WEI HUANG,等: “Novel Convenient Synthesis of Mitiglinide“, 《SYNTHETIC COMMUNICATIONS》, vol. 37, no. 13, 3 July 2007 (2007-07-03), pages 2153 – 2157, XP055079498, DOI: doi:10.1080/00397910701392590
http://www.tandfonline.com/doi/abs/10.1080/00397910701392590
Abstract: A novel convenient synthesis of the hypoglycemic agent mitiglinide was developed. (2S)-4-[(3aR,7aS)-Octahydro-2H-isoindol-2-yl]-4-oxo-2-benzyl-butanoic acid (6) was prepared by selective hydrolysis of ethyl 4-[(3aR,7aS)-octahydro-2Hisoindol-2-yl]-4-oxo-2-benzyl-butanoate (5) using a-chymotrypsin; the latter was prepared by a novel facile route from (3aR,7aS)-octahydro-2H-isoindole. The overall yield was 25.6%.
Keywords: a-chymotrypsin, mitiglinide, synthesis
Mitiglinide (calcium bis[(2S)-4-[(3aR,7aS)-octahydro-2H–isoindol-2-yl]-4oxo-2-benzylbutanoate]dihydrate) is a novel oral hypoglycemic agent. It inhibits the adenosine triphosphate (ATP)-sensitive potassium channels in pancreatic b-cells and stimulates insulin release like sulfonylureas,[1] but has a rapid onset and short-lasting hypoglycemic effect as compared with the latter.
Mitiglinide has been synthesized by several related methods that involve optical resolution,[2] asymmetric synthesis,[2a,3] and diasteroselective alkylation using chiral auxiliary.[4]
In a previous article,[2] two optical resolution methods of the key compound racemic acid 4 were reported. One of them involves esterification with optically active alcohols, which are separated into the diastereomers by column chromatogeaphy and hydrolyzed. Only the diastereomeric (S)-Nbenzyl mandelamide ester could be separated; the overall yield was 28%,
The alternative method was optical resolution by optically active bases. The best result was 30.8% yield and 97% ee when using (R)-1-(1-naphthyl)-ethylamine as a base. In this article, we have developed a new optical resolution method of racemic ester 5 by a-chymotrypsin in 45.3% yield; the optical purity of (S)-acid (6) determined by chiral-phase high performance liquid chromatography (HPLC) on Sumichiral
OA3300 was 99.2% ee, and, the method can be used for scale-up preparation.
The synthesis of free acid 6 is shown in Scheme 1. (3aR,7aS)-Octahydro2H-isoindole was chloroacetylated in the presence of Et3N to afford (3aR, 7aS)-2-(chloro-acetyl)-octahydro-2H-isoindole (2), which was condensed with diethyl benzylmalonate followed by hydrolysis and decarbonylation to obtain 4-[(3aR,7aS)-octahydro-2H-isoindol-2-yl]-4-oxo-2-benzyl-butanoic acid (4). The overall yield of the three-step synthesis was 62.9%. The racemic acid (4) was esterified with SOCl2/EtOH to give the corresponding racemic ester (5). The (R)-ester was selectively hydrolyzed by a-chymotrypsin to separate out the (S)-ester, which was subjected to hydrolysis, giving 6.
The overall yield was 28.5% [based on (3aR,7aS)-octahydro-2H-isoindole].
Compound 6 was treated with calcium chloride and 25% ammonium hydroxide to give mitiglinide; after recrystallization from 95% EtOH, the pure product was obtained in 90% yield.
Patent
https://www.google.com/patents/WO2009047797A2?cl=en
EXAMPLES
Example 1: Preparation of (R) 4-benzyl-3-(3-phenylpropionv0-oxazolidin-2-one To a solution of (R)-4-benzyloxazolidin-2-one (50 g), 4-dimethylaminopyridine (4.85 g), 3-phenyl propionic acid (55.08 g) in dichloromethane (375 ml) under nitrogen atmosphere at 0-5 0C, dicyclohexylcarbodiimide (975.65 g) was added. The temperature was slowly raised to 25-30 0C and stirring was continued until no starting material was left as was confirmed by thin layer chromatography. Dicyclohexylurea formed during the reaction was filtered, washed with dichloromethane (200 ml) and the filtrate was washed with saturated solution of sodium bicarbonate (500 ml). The solution was dried over sodium sulphate and solvent was distilled off to obtained crude product which was purified from methanol (200 ml) at 10-15 °C and washed with methanol (50 ml) to obtain 81.0 g of the title compound. Example 2: Preparation of 3(5)-benzyl-4-(4-(J?)-benzyl-2-oxo-oxazolidin-3-yl)-4-oxo-butyrϊc acid tert-butyl ester
To a solution of (/?)-4-benzyl-3-(3-phenyl-propionyl)-oxazolidin-2-one (150 g) in anhydrous tetrahydrofuran (1.5 It) was added a solution of sodium hexamethyldisilazane (462 ml, 36-38% solution in tetrahydrofuran) with stirring at -85 to -95 0C for 60 minutes. Tert-butyl bromo acetate (137.5 g) in tetrahydrofuran (300 ml) was added to reaction mass and then stirred to 60 minutes at -85 to -95 0C. After completion of the reaction (monitored by TLC), the reaction mixture was poured into ammonium chloride solution (10%, 2.0 It) and extracted with ethyl acetate (2×750 ml). The combined organic layer was washed with demineralized water (1×750 ml) and dried over sodium sulphate. The solvent was evaporated under reduced pressure to obtain oily residue which was stirred with mixture of n-hexane (100 ml) and isopropyl alcohol (100 ml) at Oto -50C, filtered and dried under vacuum to obtain 153.12 g of title compound having chemical purity 99.41%, chiral purity 99.91% by HPLC, [α]D 20: (-)97.52° (c = 1, CHCl3) and M.P. : 117.1-118.20C.
Example 3: Preparation of 3(5)-benzyl-4-(4(i?)-benzyl-2-oxo-oxazolidin-3-yl)-4-oxobutyric acid Trifluoroacetic acid (100 g) was added to a solution of 3(5)-benzyl-4-(4-(/?)-benzyl-2-oxo-oxazolidin-3- yl)-4-oxobutyric acid tert-butyl ester (100 g) in dichloromethane (700 ml) at 25 0C and mixture was stirred further for about 12 hours ( when TLC indicated reaction to be complete). The reaction mixture was poured in to ammonium chloride solution (10%, 500 ml). The dichloromethane layer was separated and aqueous layer was extracted with dichloromethane (2 x 250 ml). The combined organic layer was dried over sodium sulphate and evaporated under reduced pressure to obtain title compound. The crude product was recrystallized from a mixture of ethyl acetate: n-hexane (1:4, 500 ml) to obtain 78.75g of the title compound having purity 99.56% by HPLC and M.P.: 145.9-146.40C.
Example 4: Preparation of (2S)-2-benzyl-l-((4R)-4-benzyl-2-oxo-oxazolidin-3-vI)-4-(hexahydro- isoindolin-2-yl)-butane-l,4-dione
To a solution of 3(5)-benzyl-4-(4-(/?)-benzyl-2-oxo-oxazolidin-3-yl)-4-oxo-butyric acid (50 g) in anhydrous dichloromethane (1.25 It) was added triethylamine (50 ml) with stirring at -20 to -30 0C and the stirred for 15 minutes. A solution of isobutylchloroformate (37.50g) in anhydrous dichloromethane (50 ml) was added at -20 to -30 0C and stirred for 60 minutes. Thereafter, a solution of cis- hexahydroisoindoline (32.50 g) in anhydrous dichloromethane (50 ml) was slowly added by maintaining temperature -20 to -300C. After the completion of the reaction (monitored by HPLC), the mixture was successively washed with 0.5N hydrochloric acid solution (500 ml), brine (300 ml) and dried over sodium sulphate. The solvent was evaporated under reduced pressure to obtain 102.0 g of the title compound having purity 94.39% by HPLC.
Example 5: Purification of r2S)-2-benzyl-l-((4R)-4-benzyl-2-oxo-oxazolidin-3-yl)-4-(hexahydro- isoindolin-2-vD-butane-l,4-dione
To the crude (2S)-2-benzyl-l-((4R)-4-benzyl-2-oxo-oxazolidin-3-yl)-4-(hexahydro-isoindolin-2-yl)- butane- 1,4-dione (51.0 g) was added methanol (150 ml) and the mixture was stirred for 5 hours at 0 to 5 0C. Solid that precipitated out was filtered, slurry washed with cold methanol (25 ml) and dried at 45 -50 0C under vacuum to obtain 28.80 g of pure title compound as a crystalline solid having purity of 99.71% by HPLC and M. P.: 104.1-105.70C.
Example 6: Preparation of calcium salt of (-SVmitiglinide. Step-1: Preparation of (-SVmitiglinide
(2S)-2-Benzyl- 1 -((4R)-4-benzyl-2-oxo-oxazolidin-3-yl)-4-(hexahydro-isoindolin-2-yl)-butane- 1 ,4-dione (28.0 g) was dissolved in tetrahydrofuran (196 ml) and a mixture of lithium hydroxide monohydrate (3.51 g) in demineralized water (56 ml) and hydrogen peroxide (40% solution, 5.5 ml) was added with stirring at 0 to 5 0C over a period of 30 minutes. The reaction mixture was further stirred at 0 to 5 0C till the completion of the reaction. After the completion of the reaction (monitored by TLC), the reaction was quenched with the addition of cooled sodium meta-bisulphate solution (25%, 168 ml) at 0 to 10 0C. The reaction mixture was extracted with ethyl acetate (2×112 ml), the layers were separated and the aqueous layer was discarded. The HPLC analysis of the aqueous layer shows 0.77% of amide impurity. The ethyl acetate layer was then extracted with aqueous ammonia solution (4%, 2×40 ml). The layers were separated and the aqueous layer was further extracted with ethyl acetate (2×280 ml). Combined ethyl acetate layer was discarded. This aqueous layer (280 ml) was used as such in the next stage. The aqueous layer display purity 96.19 % by HPLC and amide impurity 0.04% by HPLC. Step-2: Preparation of calcium salt of dSVmitiglinide
To the above stirred solution of (S)-mitiglinide in water and ammonia(280 ml), methanol (168 ml) was added, followed by calcium chloride (4.48 g) dissolved in demineralized water (56 ml) at ambient temperature and the mixture was stirred for 2 hours. The resulting precipitate was filtered, successively slurry washed with water (3 x 140 ml) and acetone (2 x 70 ml) and dried at 450C -500C under vacuum to obtain 16.1 g of title compound having purity 99.67% by HPLC and amide impurity 0.01% by HPLC. The title product was re-precipitated from a mixture of methanol and water and dried to obtain pure title compound.
Example 7: Preparation of (.SVmitiglinide
To a solution of (2S)-2-benzyl-l-((4R)-4-benzyl-2-oxo-oxazolidin-3-yl)-4-(hexahydro-isoindolin-2-yl)- butane- 1,4-dione (50 g) in tetrahydrofuran (350 ml) was added a solution of lithium hydroxide monohydrate (8.65 g) in demineralized water (100 ml) and hydrogen peroxide (30% w/w, 40 ml) with stirring at 5 to 10 0C over a period of 15 minutes. After the completion of reaction, sodium meta- bisulphate solution (40%, 500 ml) was added to the reaction mixture and the mixture was extracted with ethyl acetate (2 x 250 ml). The organic layer was dried over sodium sulphate and evaporated under vacuum to obtain 45.5 g of title compound having 35 % of R-benzyl oxozolidin-2-one as impurity. Example 8: Purification of (.S)-mitiglinide
Aqueous ammonia solution (4%, 300 ml) was added to the crude (5)-mitiglinide (30 g) and stirred. The reaction mixture was washed with ethyl acetate (3 x 300 ml). Thereafter the reaction mixture was acidified to pH 1 to 2 with IN hydrochloric acid solution (250 ml) and extracted with ethyl acetate (2 x 150 ml). The layers were separated and ethyl acetate layer was washed with demineralized water (2 x 150 ml), dried over sodium sulphate and then evaporated under reduced pressure to obtain 16.2 g of pure (5)-mitiglinide having purity 95.55% by HPLC Example 9: Preparation of calcium salt of (S)-mitiglinide
To a solution of (<S)-mitiglinide (15 g) in water (150 ml) and aqueous ammonia solution (25%, 15 ml) at 25 to 30 0C, a solution of calcium chloride (7.5 g) in demineralized water (37.5 ml) was added. The mixture was stirred for 1 hour to precipitate the calcium salt of (5)-mitiglinide dihydrate. The resulting precipitate was filtered, slurry washed with water (3 x 150ml) and dried at 45 to 50 0C to obtain 13.25 g of the title compound having purity of 98.84% by HPLC. Example 10: Purification of calcium salt of (5)-mitiglinide
(iS)-mitiglinide calcium (10 g) was dissolved in dimethylformamide (100 ml). This is followed by the addition of demineralized water (500 ml) at 25 to 30 0C. The mixture was stirred for 30 minutes. The precipitated solid was filtered, washed with water (10x 50ml) and dried at 45 to 50 0C under vacuum to obtain 8g of pure title compound as a crystalline solid having purity of 99.62% by HPLC. Example 11: Preparation of amorphous mitiglinide calcium
Crystalline mitiglinide calcium (2.0 g) was dissolved in tetrahydrofuran (20 ml) and filtered to remove undissolved and suspended particles. The solvent was then evaporated under vacuum to obtain a powder which was then dried under vacuum at 40-600C to obtain 1.70 g of the title compound. Example 12: Preparation of amorphous mitiglinide calcium
Crystalline mitiglinide calcium (2.0 g) was dissolved in dichloromethane (30 ml) and filtered to remove undissolved and suspended particles. The solvent was then evaporated under vacuum to obtain a powder which was then dried under vacuum at 40-600C to obtain 1.64 g of the title compound. Example 13: Preparation of amorphous mitiglinide calcium
Mitiglinide (2.0 g) was dissolved in methanol (20 ml) and methanolic ammonia (5.0 ml) solution was added to it. The solution was stirred at 25-30 0C and calcium chloride (1.5 g) dissolved in methanol was mixed with the solution of mitiglinide and ammonia in methanol and the solution was filtered to remove the suspended particles. The solvent was then evaporated under vacuum to obtain a powder which was then dried under vacuum at 40-600C to obtain 1.9 g of the title compound. Example 14: Preparation of amorphous mitiglinide calcium
Mitiglinide (2.0 g) was dissolved in dichloromethane (20 ml) and aqueous ammonia (3.6 ml, 25 % solution) was added to it. The solution was stirred at 25-300C and solid calcium chloride (1.5 g) was mixed with the solution of mitiglinide and ammonia in dichloromethane and the solution warmed at 30 – 35 0C. The solution was washed with water (2 xlO ml) and the clear solution was dried over sodium sulfate, filtered and evaporated under vacuum and finally dried at under vacuum at 40-60 0C to obtain 1.75 g of the title compound.
Example 15: Preparation of amorphous mitiglinide calcium
Crystalline mitiglinide calcium dihydrate (2.0 g) was dissolved in ethyl acetate (30 ml) and filtered to remove undissolved and suspended particles. Approimately. 60 % of the solvent was distilled off under vacuum to obtain a stirrable solution. The solution was then cooled to 15-2O0C, mixed with n-heptane (20 ml) and the mixture was stirred for 30 minutes. The resulting solid was filtered, washed with n-heptane and dried under vacuum at 45-600C to yield 1.72 g of the title compound. Example 16: Preparation of amorphous mitiglinide calcium
Crystalline mitiglinide calcium (2.Og) was dissolved in dichloromethane (30 ml) and filtered to remove undissolved and suspended particles. Approximately 60 % of the solvent was distilled off under vacuum to obtain a stirrable solution. The solution was then cooled to 15-200C and mixed with diisopropyl ether (20 ml). The mixture was stirred for 30 minutes and the resulting solid was filtered, washed with diisopropyl ether and dried under vacuum at 45-600C to obtain 1.70 g of the title compound. Example 17: Preparation of amorphous mitiglinide calcium
Mitiglinide (2.0 g) was dissolved in dichloromethane (20 ml) and aqueous ammonia (3.6 ml, 25 % solution) solution was added to it. The solution was stirred at 25-30 0C and mixed with solid calcium chloride (1.5 g) and the solution warmed at 30-35 0C and stirred for 30 minutes. The solution was washed with water (2 x 10 ml) and the clear solution was dried over sodium sulfate, and filtered. Approximately 60% of the solvent was distilled off under vacuum and the resulting viscous oil was cooled to 10-15 0C and mixed with diisopropyl ether (50 ml). The reaction mixture was stirred for 30-35 minutes and the resulting solid was filtered and dried at 40-600C to obtain 1.75 g of the title compound. Example 18: Conversion of amorphous mitiglinide calcium into crystalline mitiglinide calcium A suspension of amorphous mitiglinide calcium in diisopropyl ether (30 ml) was stirred for 2 hours at 25- 300C, filtered and dried under vacuum at 45-600C to obtain crystalline form of mitiglinide calcium. Example 19: Preparation of crystalline mitiglinide calcium
To a solution of mitiglinide (2.5 g) in water (2.5 ml), aqueous ammonia solution (approx 25%, 4.0 ml) and acetonitrile (2.5 ml) at 10-150C, calcium chloride (1.32 g) dissolved in demineralized water (15 ml) was added. The mixture was stirred for 2 hours. The resulting precipitate was filtered, slurry washed with water (3 x 25 ml) and acetone (2 x 5 ml) and dried at 45-500C under vacuum to obtain 2.12 g of title compound having purity: 99.72 % by HPLC.
Example 20: Preparation of crystalline mitiglinide calcium
To a solution of mitiglinide (2.5 g) in water (2.5 ml), aqueous ammonia solution (approx 25%, 4.0 ml) and tetrahydrofuran (2.5 ml) at 10-150C, calcium chloride (1.32 g) dissolved in demineralized water (15 ml) was added. The mixture was stirred for 2 hours. The resulting precipitate was filtered, slurry washed with water (3 x 25 ml) and acetone (2 x 5 ml) and dried at 45-500C under vacuum to obtain 1.95 g of title compound having purity: 99.52 % by HPLC.
Example 21; Preparation of crystalline mitiglinide calcium
To a solution of mitiglinide (30.0 g) in water (300 ml), aqueous ammonia solution (approx 25%, 48 ml) and acetone (300 ml) at 10-150C, calcium chloride (15.8 g) dissolved in demineralized water (180 ml) was added. The mixture was stirred for 2 hours. The resulting precipitate was filtered, slurry washed with water (3 x 300 ml) and acetone (2 x 60 ml) and dried at 45-500C under vacuum to obtain 24.32 g of title compound having purity: 99.42 % by HPLC.
Example 22: Preparation of crystalline mitiglinide calcium
To a solution of mitiglinide (3.0 g) in water (30 ml), aqueous ammonia solution (approx 25%, 4.8 ml) and isopropyl alcohol (300 ml) at 10-150C, calcium chloride (1.58 g) dissolved in demineralized water
(18 ml) was added. The mixture was stirred for 2 hours. The resulting precipitate was filtered, slurry washed with water (3 x 30 ml) and acetone (2 x 6 ml) and dried at 45-500C under vacuum to obtain 1.92 g of title compound having purity: 99.65 % by HPLC.
Example 23: Preparation of (2S)-2-benzyWV-((lR)-l-benzyl-2-hydroxy-ethyl)-4-(hexahvdro- isoindolin-2-yl)-4-oxo-buryramide
To a solution of (2S)-2-benzyl-l-((4R)-4-benzyl-2-oxo-oxazolidin-3-yl)-4-(hexahydro-isoindolin-2-yl)- butane-l,4-dione (20.0 g) in tetrahydrofuran (140 ml), a solution of lithium hydroxide monohydrate
(3.43 g,) in demineralized water (40 ml) was added and the reaction mixture was refluxed for 4 hours till the completion of the reactions (monitored by thin layer chromatography). After the completion of the reaction, the reaction mixture was poured into demineralized water (100 ml) and extracted with ethyl acetate (2 x 80 ml). The combined organic layer was washed with water (80 ml) and dried over sodium sulphate. The solvent was evaporated under reduced pressure to give residue which was stirred in isopropyl alcohol at 0-5 0C for 5 hours. The mixture was filtered and then dried at 40-45 0C under vacuum to obtain 12.48 g of title compound having purity 99.77 % by HPLC. Melting point = 77 – 800C.
PATENT
https://www.google.com/patents/CN102101838A?cl=en
Mitiglinide calcium (mitiglinide calcium), the chemical name (2S) _2_ benzyl _3_ (cis – hexahydro _2_ isoindolinyl-carbonyl) propionate dihydrate by Japanese pharmaceutical company developed Kissei ATP-dependent potassium channel blockers, 2004 for the first time in Japan for the treatment of type II diabetes.
Mitiglinide calcium is the second repaglinide, nateglinide after the first three columns MAG urea drugs, is a derivative of phenylalanine, which acts like mechanism sulfonylurea, but faster onset and the short half-life, is conducive to reducing postprandial blood glucose in diabetic patients, but also to avoid low blood sugar caused by continuous glucose, with “in vitro pancreas” reputation.
In recent years, synthetic methods as described in patent application number: Patent 200510200127 9, the synthesis process first synthesized racemic (±) 2_-benzyl-3- (cis – hexahydro iso-indole-2. carbonyl) propionic acid, and then split to give (2S) -2- benzyl-3- (cis – hexahydro isoindole-2-carbonyl) propionic acid, not a lot of waste material along _ hexahydro isoindole, and Chiral separation is not high.
DISCLOSURE
The technical problem to be solved by the present invention is to provide a material savings along _ hexahydro isoindole, and preparation of a high degree of chiral separation.
To solve the above technical problem, the technical solution of the present invention is employed as a method for preparing mitiglinide calcium, comprising the steps of:
Step 1 Synthesis, benzylidene succinic acid
With stirring, was added sodium metal in absolute ethanol, under an inert gas, the solution was heated to reflux with stirring, reflux for 45 fly 0 minutes, under reflux before the dropwise addition of benzaldehyde, and then added dropwise diethyl succinate esters, reaction stirring was continued for 2 to 3 hours, slowly reducing the LC-Ms detection, the ratio of formaldehyde starting material benzene, cooled to room temperature, after use 5 (T55wt% aqueous solution of NaOH to adjust the PH San 13.0, and then heated at reflux;. Γ4 hours, cooled to at room temperature, keeping the reaction solution temperature <25 ° C, pH adjusted with concentrated hydrochloric San 2.0, filtration, recrystallization cold tetrahydrofuran, wherein the molar ratio of sodium metal with benzaldehyde and diethyl succinate is: 0.3 … ~ 0 5: 1 2~1 5: 1; Step 2 synthesis, benzyl butyl acid

The benzylidene succinic acid into the reactor, 10% Pd / C and ethanol, evacuated, and then replaced with hydrogen three times, introducing hydrogen, atmospheric hydrogenation reaction 12~15 hours, the reaction solution suction After the filtrate was evaporated to dryness under reduced pressure, the resulting solid was recrystallized from ethyl acetate, wherein the mass ratio of benzylidene succinic acid with 10% Pd / C is 1: 0 0 15 ^ 20.
3 Synthesis [0006] step, (S) -2- acid, benzyl butyl

Benzyl succinic acid dissolved in methanol was added dropwise with stirring (R) – a chiral amine, stirred at room temperature 2 hours wide, and the precipitated solid was filtered and the solid dispersed in water, under stirring 6 mol / mL hydrochloric acid adjusted ρΗ = 1 (Γ2.0, stirred for 30 minutes, the solid by suction filtration, dried, and wherein the benzyl succinic acid (R) – chiral amine molar ratio of 1: 0~2 5 2;…
Said (R) – a chiral amine (R) -I- phenylethylamine, (R) -I- naphthylethylamine or (R) -I- phenyl-2-p-amine;
4 (S) synthesis step, -2-benzyl succinic anhydride

Reactor, has added (S) -2- benzyl succinic acid and acetic anhydride, at 7 5,0 ° C reaction 1 to 2 hours, isopropyl ether low temperature crystallization after cooling, heavy with ethyl acetate crystallization, wherein (S) -2- molar ratio of benzyl succinic acid and acetic anhydride: 1: 7 · 0 to 7 · 5;
Step 5, (2S) -2- benzyl-3- (cis – hexahydro isoindole-2-carbonyl) propionic acid Synthesis

Stirring, S- benzyl succinic anhydride is dissolved in dichloromethane, control the internal temperature <0 V, a solution of cis _ hexahydro isoindole, dropping it, keeping the internal temperature at <0! : Continue stirring for 2 to 3 hours, the reaction in 2 (T25 ° C 10~15 hours, concentrated to give a pale yellow viscous material, wherein the (S) -2- benzyl succinic anhydride and cis – hexahydro isoindole molar ratio of 1: 2 (Γ2 5; step 6, mitiglinide calcium synthesis.

To the reactor was added (2S) -2- benzyl-3- (cis – hexahydro isoindole-2-carbonyl) propionic acid, water and concentrated ammonia, stir until completely dissolved, a solution of anhydrous calcium chloride aqueous solution, gradually precipitated white solid was added dropwise and then stirred at room temperature 12~ after 15 hours, suction filtered, the filter cake washed with water, dried to give a white solid, i.e. Mitiglinide calcium crude, obtained crude product with methanol and water (volume Than 0.5 4~0 5: 1) and recrystallized as a white solid Mitiglinide calcium;
Beneficial effects: The invention provides a method for preparing calcium Mitiglinide not only saves raw material cis – hexahydro isoindole, and chiral separation is high.
Embodiment 1
Step 1 Synthesis, benzylidene succinic acid
Under stirring, sodium metal (1.7 g, 0. 072 mol) was added absolute ethanol (50 mL), and under argon, the solution was heated to reflux with stirring, reflux for 50 minutes under reflux before the dropwise addition of benzene Formaldehyde (23 mL, 0. 183 mol), and then added dropwise diethyl succinate (50 mL, 0. 275 mol), stirring was continued for 2.5 hours the reaction, reducing the slow LC-Ms detection, the ratio of formaldehyde starting material benzene , was cooled to room temperature, with 55wt.% aqueous NaOH solution adjusting pH ≥ 13.0, and then heated at reflux for 3 hours, cooled to room temperature, the reaction solution temperature maintained <25 ° C, with concentrated hydrochloric pH≤2.0, leaching, cryogenic tetrahydrofuran recrystallization, yield = 81.3%;
Step 2 synthesis, benzyl butyl acid
The benzylidene succinic acid (23. 7 g, 0. 114 mol) into the reactor, then add 10% Pd / C (4. 7g) and anhydrous ethanol (300 mL), evacuated, then Hydrogen replacement three times, introducing hydrogen, hydrogenated at atmospheric pressure for 14 hours, the reaction solution after filtration, evaporated to dryness under reduced pressure, the resulting solid was recrystallized from ethyl acetate, yield: 98% 9; step 3, (S). -2-butyric acid benzyl
Benzyl succinic acid (31. 2 g, 0. 156 mol) was dissolved in methanol (500 mL), and added dropwise with stirring (R) -I- phenylethylamine (41.2 g, 0. 343 mol), room temperature stirred for 1.5 hours, the precipitated solid was filtered, the solid dispersion to water (100 mL) and stirred at with 6 mol / mL hydrochloric acid adjusted ρΗ = 1. (Γ2. 0, stirred for 30 minutes, the solid was suction filtered, and dried Yield 87. 3%; 4 (S) synthesis step, -2-benzyl succinic anhydride
Reactor, has added (S) -2- benzylbutyl acid (27. 8 g, 0. 132 mol) and acetic anhydride (88 mL, 0. 964 mol), at 7 5,0 ° C for 1 hours, cooled and added to isopropyl ether (150 mL) low temperature crystallization, after recrystallization from ethyl acetate, yield: 73% 9;.
Under – (hexahydro-isoindole-2-carbonyl cis) acid synthesis stirring S- benzyl succinic anhydride (12. 7 g, 0 Step 5, (2S) -2- benzyl-3. 067 mol) was dissolved in dichloromethane (250 mL), to control the internal temperature <0 ° C, a solution of cis – hexahydro isoindole (18. 5 g, 0 154 mol), the addition was complete, maintaining the internal temperature in <0! : Continue stirring for 2.5 hours, the reaction in 2 (T25 ° C 12 hours, concentrated to give a pale yellow viscous material, yield: 83 1%; Step 6 Synthesis Mitiglinide calcium.
To the reactor was added (2S) -2- benzyl-3- (cis – hexahydro isoindole-2-carbonyl) propionic acid (. 28. 7 g, 0 091 mol), water (150 mL), and concentrated aqueous ammonia (12 mL), stirring until completely dissolved, a solution of anhydrous calcium chloride (12. 1 g, 0.109 mol) water (100 mL) solution was gradually precipitated white solid was added dropwise at room temperature and then stirred for 13 End hours, filtration, washing the filter cake, and drying to give a white solid, crude Mitiglinide calcium, derived from crude methanol and water (volume ratio 0.5 4~0 5: 1) and recrystallized as a white solid MIG Chennai column calcium, yield: 87.3%.
Second Embodiment
Example A similar experimental method steps 1 through 6 was carried out except in step 3, using (R) -1- naphthyl-amine (61. 4 g, 0. 359 mol) substituted (R) -I- phenylethylamine Other operating homogeneous reaction similar to this step of the synthesis yield: 87.3%.
Third Embodiment
Example A similar experimental method steps 1 through 6 was carried out except in step 3, using (R) -I- phenyl-2-p-tolyl-ethylamine (90. 2 g, 0. 374 mol) substituent (R ) -I- phenethylamine, other homogeneous reaction procedure similar to the synthesis yield of this step:. 83 4% ο
PATENT
WO 199832736

CLIP
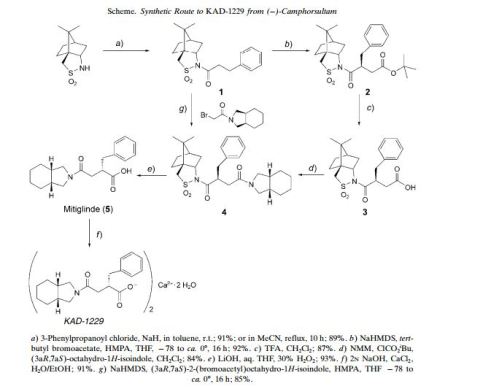
The process for the preparation of KAD-1229 starts from ()-camphorsultam ((3aS)-8,8-dimethylhexahydro-3a,6-methano-2,1-benzisothiazole 2,2-dioxide), readily available in 85% yield from ()- -camphor [4]. Treatment of ()-camphorsultam with excess 3-phenylpropionyl chloride in the presence of NaH in toluene at room temperature gave 1 in 91% yield (Scheme) [5]. An alternative procedure is to reflux camphorsultam with 1.1 to ca. 1.5 equiv. of 3-phenylpropionyl chloride in MeCN for 8 ± 10 h [6]. The crude product, acylsultam 1, purified by recrystallization from EtOH/H2O in 89% yield, was reacted with an equimolar amount of base to form the chiral enolate in dry ice/EtOH bath, followed by C()-re-alkylation [7] with tert-butyl bromoacetate to give 2. The choice of the organic base was very important: the reaction with BuLi, lithium diisopropylamide (LDA), or NaHMD (sodium hexamethyldisilazane) gave 2 in 30 ± 40%, 60%, or 90% yield, respectively, after recrystallization from MeOH. Alkylation promoted by these bases tends to give products with high diastereoselectivity, and the diastereoisomeric purity of crude product 2 was determined to be 93%. However, the reaction with NaHMDS as the base proceeded smoothly in high yield. The tert-butyl ester 2 was cleaved with TFA (CF3COOH) in CH2Cl2 to give the free acid 3 in 87% yield [8]. Acylation of (3aR,7aS)-octahydro-1H-isoindole with 3 by a mixed anhydride method afforded 4 in 84% yield [9]. Compound 4 can be also obtained in 85% yield via direct alkylation of 1 with (3aR,7aS)-2-(bromoacetyl)octahydro-1H-isoindole; however, the yield of the (2-bromoacetyl)octahydro-1H-isoindole prepared from 2-bromoacetyl bromide and cis-octahydro-1H-isoindole was only 40%. Nondestructive cleavage of 4 by hydroperoxide-assisted saponification (LiOH, aq. H2O2 , THF, r.t.) regenerated the camphorsultam (96% recovered yield) and gave mitiglinide (5) in 93% yield and high enantiomeric excess ( 99% by HPLC analysis of the corresponding methyl ester) [7]. Product 5 was treated with 2 NaOH, followed by treatment with CaCl2 . Recrystallization from aqueous EtOH gave KAD-1229 in 91% yield, with a melting point and specific rotation data identical to those in the literature [2b]. Co
(2S)-4-[(3aR,7aS)-Octahydro-2H-isoindol-2-yl]-4-oxo-2-(phenylmethyl)butanoic Acid ( Mitiglinide, 5). base
mitiglinide as a colorless viscous oil. The ee was determined to be 99.4% by HPLC analysis of the corresponding Me ester on a Chiralcel AS column (250 4.6 mm, flow rate 0.7 ml/min, UV 214 nm, n-hexane/i-PrOH 80 : 20 as the eluent).
20 Dalpha= -3.5 (c 1.0, MeOH).
1 H-NMR: 1.23 ± 1.63 (m, 8 H); 2.13 ± 2.22 (m, 2 H); 2.42 ± 2.52 (m, 2 H); 2.73 ± 3.32 (m, 7 H); 7.18 ± 7.32 (m, 5 H).
ESI-MS: 316.15 ( [M H]). Anal. calc. for C19H25NO3 (315.41): C 72.35, H 7.99, N 4.44; found: C 72.51, H 8.03, N 4.31.
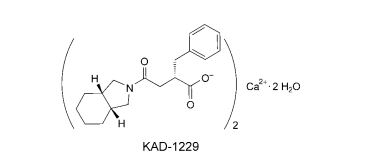
Calcium Bis{(2S)-4-[(3aR,7aS)-octahydro-2H-isoindol-2-yl]-4-oxo-2-(phenylmethyl)butanoate} Dihydrate (KAD-1229).
KAD-1229 as colorless crystals (0.82 g, 91%).
M.p. 179 ± 185 (lit. 179 ± 185 [2b]). 20 D 5.4 (c 0.6, MeOH) (lit. 20 D 5.7, c 1.0, MeOH [2b]).
1 H-NMR: 1.13 ± 1.39 (m, 16 H); 2.0 ± 2.3 (m, 6 H); 2.54 ± 2.83 (m, 14 H); 3.20 ± 3.22 (m, 6 H); 7.11 ± 7.28 (m, 10 H).
ESI-MS: 669.32 ( [M 2 H2O H]). Anal. calc. for C38H48CaN2O6 ¥2H2O (704.91): C 64.75, H 7.44, N 3.94; found: C 64.46, H 7.35, N 3.73.
REFERENCES for aboveclip
[1] H. Ohnota, T. Koizumi, N. Tsutsumi, M. Kobayashi, S. Inoue, S. J. Sato, Pharmacol. Exp. Ther. 1994, 269, 489; H. Ohnota, M. Kobayashi, T. Kiozumi, K. Katsuno, F. Sato, T. Azisawa, Biochem. Pharmacol. 1995, 49, 165; M. Kinukawa, H. Ohnota, T. Azisawa, Br. J. Pharmacol. 1996, 117, 17021.
[2] a) T. Yamaguchi, T. Yanagi, H. Hokari, Y. Mukaiyama, T. Kamijo, I. Yamamoto, Chem. Pharm. Bull. 1997, 45, 1518; b) T. Yamaguchi, T. Yanagi, H. Hokari, Y. Mukaiyama, T. Kamijo, I. Yamamoto, Chem. Pharm. Bull. 1998, 46, 337.
[3] J. P. Lecouve, C. Fugier, J. C. Souvie, Pat. WO9901430, 1999 (Chem. Abstr. 1999, 130, 110156r).
[4] M. Vandewalle, J. Van der Eycken, W. Oppolzer, C. Vullioud, Tetrahedron 1986, 42, 4035; F. A. Davis, J. C. Towson, M. C. Weismiller, S. Lal, P. J. Carroll, J. Am. Chem. Soc. 1988, 110, 8477.
[5] W. Oppolzer, O. Tamura, J. Deerberg, Helv. Chim. Acta 1992, 75, 1965.
[6] M. C. William, B. Corey, J. Org. Chem. 1998, 63, 6732.
[7] W. Oppolzer, R. Moretti, S. Thomi, Tetrahedron Lett. 1989, 30, 5603.
[8] H. Heitsch, R. Henning, H. W. Kleemann, W. Linz, W. U. Nicke, D. Ruppert, H. Urbach, A. Wagner, J. Med. Chem. 1993, 36, 2788.
[9] J. J. Plattner, P. A. Marcotte, H. D. Kleinert, H. H. Stein, J. Greer, G. Bolis, A. K. L. Fung, B. A. Bopp, J. R. Luly, J. Med. Chem. 1988, 31, 2277.
References
| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| EP0507534A1 * | Mar 30, 1992 | Oct 7, 1992 | Kissei Pharmaceutical Co., Ltd. | Succinic acid compounds |
| EP0967204A1 * | Jan 22, 1998 | Dec 29, 1999 | Kissei Pharmaceutical Co Ltd | Process for producing benzylsuccinic acid derivatives |
| US6133454 * | Jul 1, 1998 | Oct 17, 2000 | Adir Et Compagnie | Method for preparing a substituted perhydroisoindole |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| CN102898348A * | Sep 8, 2012 | Jan 30, 2013 | 迪沙药业集团有限公司 | Preparation method for Mitiglinide calcium |
| CN102898348B * | Sep 8, 2012 | Sep 2, 2015 | 迪沙药业集团有限公司 | 一种米格列奈钙的制备方法 |
| CN103450069A * | Jun 24, 2013 | Dec 18, 2013 | 山西大同大学 | Preparation method of mitiglinide calcium |
| CN103724253A * | Dec 11, 2013 | Apr 16, 2014 | 苑振亭 | Preparation method for Mitiglinide calcium hydrate |
| CN103724253B * | Dec 11, 2013 | Jun 15, 2016 | 苑振亭 | 一种米格列奈钙的制备方法 |
| CN102659562A * | May 9, 2012 | Sep 12, 2012 | 山东铂源药业有限公司 | Synthesis method of mitiglinide calcium intermediate |
| CN102898348A * | Sep 8, 2012 | Jan 30, 2013 | 迪沙药业集团有限公司 | Preparation method for Mitiglinide calcium |
| CN102898348B * | Sep 8, 2012 | Sep 2, 2015 | 迪沙药业集团有限公司 | 一种米格列奈钙的制备方法 |
| CN103450069A * | Jun 24, 2013 | Dec 18, 2013 | 山西大同大学 | Preparation method of mitiglinide calcium |
| CN103709092A * | Nov 4, 2013 | Apr 9, 2014 | 河北科技大学 | High purity mitiglinide calcium preparation method |
| CN103709092B * | Nov 4, 2013 | Jul 6, 2016 | 河北科技大学 | 米格列奈钙的制备方法 |
| CN104311471A * | Sep 23, 2014 | Jan 28, 2015 | 山东省药学科学院 | Improved mitiglinide calcium industrialized preparation method |
| CN1616427A * | Nov 13, 2003 | May 18, 2005 | 中国科学院上海药物研究所 | New method for preparing medicine mitiglinide for treating diabetes |
| CN101270074A * | Mar 21, 2007 | Sep 24, 2008 | 北京德众万全药物技术开发有限公司 | Method for preparing high purity mitiglinide calcium |
| CN101492411A * | Jan 22, 2008 | Jul 29, 2009 | 北京华禧联合科技发展有限公司 | Improved method for preparation of mitiglinide |
| WO2009047797A2 * | Oct 7, 2008 | Apr 16, 2009 | Ind-Swift Laboratories Limited | Process for the preparation of perhydroisoindole derivative |
| Reference | ||
|---|---|---|
| 1 | * | 张永亮,等: “米格列奈合成方法研究“, 《化工中间体》, no. 1, 31 December 2009 (2009-12-31), pages 16 – 22 |
| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| CN101492411A * | Jan 22, 2008 | Jul 29, 2009 | 北京华禧联合科技发展有限公司 | Improved method for preparation of mitiglinide |
| WO2005030719A1 * | Sep 24, 2004 | Apr 7, 2005 | Les Laboratoires Servier | Novel method for preparing cis-octahydro-isoindole |
| Reference | ||
|---|---|---|
| 1 | * | WEI HUANG,等: “Novel Convenient Synthesis of Mitiglinide“, 《SYNTHETIC COMMUNICATIONS》, vol. 37, no. 13, 3 July 2007 (2007-07-03), pages 2153 – 2157, XP055079498, DOI: doi:10.1080/00397910701392590 |
| 2 | * | 张永亮,等: “米格列奈合成方法研究“, 《化工中间体》, no. 1, 31 January 2009 (2009-01-31), pages 16 – 22 |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| CN103709092A * | Nov 4, 2013 | Apr 9, 2014 | 河北科技大学 | High purity mitiglinide calcium preparation method |
| CN103709092B * | Nov 4, 2013 | Jul 6, 2016 | 河北科技大学 | 米格列奈钙的制备方法 |
| EP0507534A1 * | Mar 30, 1992 | Oct 7, 1992 | Kissei Pharmaceutical Co., Ltd. | Succinic acid compounds |
| EP0967204A1 * | Jan 22, 1998 | Dec 29, 1999 | Kissei Pharmaceutical Co Ltd | Process for producing benzylsuccinic acid derivatives |
| US6133454 * | Jul 1, 1998 | Oct 17, 2000 | Adir Et Compagnie | Method for preparing a substituted perhydroisoindole |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| CN102898348A * | Sep 8, 2012 | Jan 30, 2013 | 迪沙药业集团有限公司 | Preparation method for Mitiglinide calcium |
| CN102898348B * | Sep 8, 2012 | Sep 2, 2015 | 迪沙药业集团有限公司 | 一种米格列奈钙的制备方法 |
| CN103450069A * | Jun 24, 2013 | Dec 18, 2013 | 山西大同大学 | Preparation method of mitiglinide calcium |
| CN103724253A * | Dec 11, 2013 | Apr 16, 2014 | 苑振亭 | Preparation method for Mitiglinide calcium hydrate |
| CN103724253B * | Dec 11, 2013 | Jun 15, 2016 | 苑振亭 | 一种米格列奈钙的制备方法 |
|
|
| Systematic (IUPAC) name | |
|---|---|
|
(2S)-2-benzyl-4-[(3aR,7aS)-octahydro-2H-isoindol- 2-yl]-4-oxobutanoic acid
|
|
| Clinical data | |
| AHFS/Drugs.com | International Drug Names |
| Routes of administration |
oral |
| Identifiers | |
| CAS Number | 145375-43-5 |
| ATC code | A10BX08 (WHO) |
| PubChem | CID 121891 |
| DrugBank | DB01252 |
| ChemSpider | 108739 |
| UNII | D86I0XLB13 |
| KEGG | D01854 |
| ChEMBL | CHEMBL471498 |
| Chemical data | |
| Formula | C19H25NO3 |
| Molar mass | 315.41 g/mol |
/////////207844-01-7, 145525-41-3, KAD-1229, S-21403, MITIGLINIDE, Glufast, Kissei, 145375-43-5
O=C(O)[C@@H](Cc1ccccc1)CC(=O)N3C[C@H]2CCCC[C@H]2C3
DRUG REGULATORY AFFAIRS INTERNATIONAL

The current work outlines the application of an up-to-date and regulatory-based pharmaceutical quality management method, applied as a new development concept in the process of formulating dry powder inhalation systems (DPIs). According to the Quality by Design (QbD) methodology and Risk Assessment (RA) thinking, a mannitol based co-spray dried formula was produced as a model dosage form with meloxicam as the model active agent.
The concept and the elements of the QbD approach (regarding its systemic, scientific, risk-based, holistic, and proactive nature with defined steps for pharmaceutical development), as well as the experimental drug formulation (including the technological parameters assessed and the methods and processes applied) are described in the current paper.
Findings of the QbD based theoretical prediction and the results of the experimental development are compared and presented. Characteristics of the developed end-product were in correlation with the predictions, and all data were confirmed by the relevant results…
View original post 223 more words
DRUG REGULATORY AFFAIRS INTERNATIONAL

Data Integrity has become one of the most frequently observed GMP deviations at FDA and EU Inspections. For that reason the ECA Foundation decided to set up a Task Force on Data Integrity in December 2015 – with the goal to provide Guidance for the implementation in practice. Read more about the ECA Guidance on Data Integrity.
http://www.gmp-compliance.org/eca_mitt_05545_15488_n.html
Data Integrity has become one of the most frequently observed GMP deviations at FDA and EU Inspections. This is why the topic is currently in the centre of attention of both regulators and industry. And for that reason the ECA Foundation decided to set up a Task Force on Data Integrity in December 2015 – with the goal to provide Guidance for the implementation in practice.
The ECA Task Force will be comprised of members from both the IT Compliance Group and the Analytical QC Group. Current Members are:
– Dr. Wolfgang Schumacher…
View original post 177 more words

(±)-SIPI 6360
D2/5-HT2A receptor dual antagonist
7-[3-[4-(6-fluoro-1,2-benzoxazol-3-yl)piperidin-1-yl]propoxy]-3-methyl-3,4-dihydro-1H-quinolin-2-one
2(1H)-Quinolinone, 7-[3-[4-(6-fluoro-1,2-benzisoxazol-3-yl)-1-piperidinyl]propoxy]-3,4-dihydro-3-methyl-
| Molecular Formula: | C25H28FN3O3 |
|---|---|
| Molecular Weight: | 437.506523 g/mol |
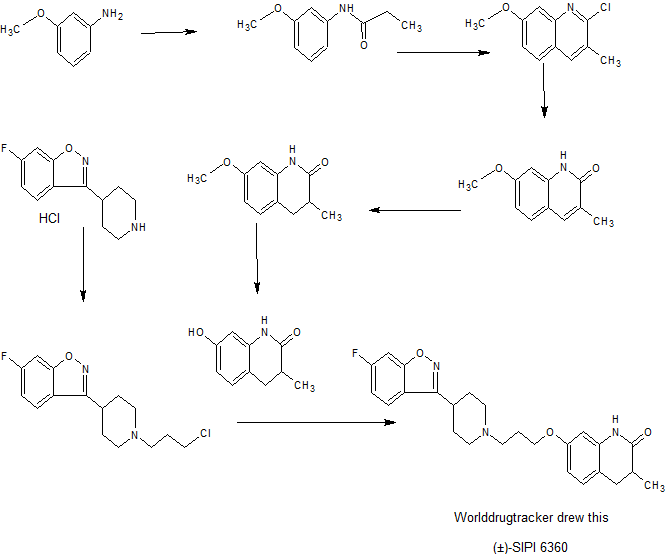
Schizophrenia is a common severe mental patients, mental illness is the most serious of all, the most dangerous kind, the worldwide incidence of about I%, with the accelerate pace of social life, The incidence was significantly increased. Most schizophrenic patients due to the long treatment period, high cost, side effects and give up the treatment, often lead to more serious social consequences.
Numerous studies show that the brain monoamine neurotransmitters, especially dopamine and 5-hydroxytryptamine system is closely related to the body’s normal mental activity, these two types of system disorder can lead to a variety of neuropsychiatric diseases such as schizophrenia , neuropathic pain, mania, anxiety disorders, all kinds of depression, Parkinson’s disease and the like.
The drugs currently used clinically primarily for conventional antipsychotics (such as dopamine D2 receptor antagonists) and atypical antipsychotics (such as D2 / 5-HT2a dual antagonist), where conventional antipsychotics because it is easy leads to extrapyramidal symptoms (EPS) and gradually phased out, atypical antipsychotics variety, but no one medication to improve the overall spectrum of schizophrenia has the absolute advantage, most of the positive or negative symptoms of a a symptom improvement, or reduced side effects. So look for low toxicity, rapid onset, treatment spectral width of new anti-schizophrenia drug has been a hot topic in the world pharmaceutical industry.
In recent years, scientists have found that the dopamine D2 partial agonist can over time reduce dopamine activity in the transfer of dopamine, but not all block; the other hand, when the low dopaminergic activity is caused by stimulating effect on both positive and negative symptoms of mental illness have a significant effect. 5-HT2a receptor antagonists can improve negative symptoms, while synergies D2 EPS side effects can be reduced to about 1% level (classical antipsychotic drugs EPS incidence is about 30%), part of the 5-HTla agonism and 5-HT2a and synergy can make in therapeutic doses under observation EPS decreased to undetectable levels, therefore, has D2 ,5-HT2a, 5HTla synergy targets three new anti-drugs are currently developed Jingshenfenlie focus and an important development direction.
The present invention relates to a quinoline derivative can stabilize the brain dopaminergic, serotonergic energy system, may for a variety of neurological and psychiatric diseases have improved and treatment can be used for neuropathic pain, mania, schizophrenia, anxiety disorders, all kinds of depression, Parkinson’s disease, especially in the treatment of schizophrenia.
PATENT

PATENT

Example 1
1-1
7- (3- (4- (6-fluorophenyl and [d] different dumb-3-yl) piperidin-1-yl) propoxy) -3,4-dihydro-3-carboxylic acid -one – yl quinolin -2 (1H)
1) N- (3- methoxyphenyl) propionamide
3-methoxy-aniline (0.1mol), methylene chloride (30 mL), triethylamine (0.2mol), was added to the flask lOOmL three, propionyl chloride was added dropwise under ice (0.12mol) in methylene chloride 30 mL, temperature does not exceed 5 ° C, the addition was complete, the ice bath was removed and stirred at room temperature 0.5h, the system was washed with water, dilute hydrochloric acid, saturated brine, dried over anhydrous magnesium sulfate, and evaporated to dryness to give a white powdery solid 17.01g yield 95%.
2) 2-chloro-7-methoxy-3-methylquinoline
The DMF (20mL) was added to the three 250mL flask, was added dropwise under ice-salt bath of POCl 3 (100 mL), temperature does not exceed 0 ° C, the addition was completed stirring 0.5h, was added portionwise N- (3- methoxyphenoxy yl) propanamide powder (31.0g), was slowly warmed to 50 ° C, violent reaction, to be exothermic easing slowly warmed to reflux, the reaction was kept 2h, cooled to room temperature, the system was poured into 800 g of crushed ice to sodium carbonate to adjust the pH to 7 to precipitate a yellow solid with petroleum ether – ethyl acetate to give pure product 20.86g, yield 58%.
3) 3-methyl-7-methoxy-quinolin -2 (1H) – one
2-Chloro-7-methoxy-3-methyl-quinoline (20.76g), acetic acid (150 mL) placed in 250mL one-neck flask, heated at reflux for 24h, acetic acid recovery, and the residue was recrystallized from ethanol to 95%, white needle crystalline 16.08g, yield 85%.
4) 7-methoxy-3,4-dihydro-3-methyl-quinolin -2 (1H) – one
7-Methoxy-3-methyl-quinolin -2 (1H) – one (18.92g), acetic acid (150mL), 10% Pd / C (lg) was added to the three 250mL flask, the system was replaced with nitrogen air, and then the nitrogen was replaced with hydrogen, and then the reaction was heated to 80 ° C overnight, cooled to room temperature, filtered and the filtrate evaporated to dryness to give a white powder, washed with water once, 50 ° C and dried in vacuo 4h, as a white powdery solid 18.91g yield of 98.95%.
5) 7-hydroxy-3,4-dihydro-3-methyl-quinolin -2 (1H) – one
7-Methoxy-3,4-dihydro-3-methyl-quinolin -2 (1H) – one (19.12g), 40% hydrobromic acid (150 mL) placed in 250mL one-neck flask was heated at reflux for 12h cooled to room temperature, the precipitated solid was filtered, the filter cake successively with hydrobromic acid, washed with water, 50 ° C and dried in vacuo 4h, 14.60 g as a white powdery solid, yield 82.4%.
6) 3- (1- (3-chloropropyl) piperidin-4-yl) -6-fluorophenyl and [d] oxazole different dumb
6-fluoro-3- (piperidin-4-yl) benzo [d] isoxazol dummy oxazole (22.00g), 1- bromo-3-chloropropane (40mL), anhydrous potassium carbonate (40g), acetone ( 250mL) was added to a 500mL one-neck flask was refluxed overnight, cooled to room temperature, filtered, the filter cake was washed twice with hot acetone and the combined filtrate was added dropwise a solution of anhydrous hydrogen chloride in ethanol, the precipitated white solid was filtered, the filter cake washed with acetone after washing once, it was dissolved in 200mL of water, adjusted with sodium carbonate to pH 9, and filtered to obtain a white powdery solid 15.94 g, yield 48.0%
7) 7- (3- (4- (6-fluorobenzo [d] isoxazol-3-yl dummy) piperidin-1-yl) propoxy) -3,4-dihydro-3-methyl quinolin -2 (1H) – one
3- (1- (3-chloropropyl) piperidin-4-yl) -6-fluorophenyl and [d] oxazole different dumb (lmmol), 7- hydroxy-3,4-dihydro-3-carboxylic acid yl quinolin -2 (1H) – one (1.0 mmol), anhydrous potassium carbonate (3.0mmol) were added to the lOmLDMF, 60 ° C overnight the reaction, potassium carbonate was filtered off, the mother liquor evaporated to dryness to give a pale yellow solid, the filter cake recrystallized with 95% ethanol, 50 ° C and dried in vacuo 4h, as a white powdery solid 0.30g, 69% yield.
NMR IH (of DMSO-D . 6 ): L up to .27 (D, 3H, J = 9.2Hz), 2.06-2.32 (m, 9H), 2.67-2.69 (T, 2H), 2.95 (D * D, lH, J = 3.2Hz, 12.8Hz), 3.15-3.17 ( m, 2H), 4.05 (t, 2H, J = 6Hz), 6. 37 (d, lH, J = 2.4Hz), 6.56 (d * d, lH, J = 2.4Hz, 8.0Hz), 7.05-7.11 (m, 2H), 7.25-7.29 (m, lH), 7.73-7.76 (m, lH), 7.98 (s, lH), 11.43 (brs, lH)
ESI-MS: 438 (M + 1)
Example 2
Preparation 1-1 hydrochloride
7- (3- (4- (6-fluorophenyl and [d] different dumb-3-yl) piperidin-1-yl) propoxy) -3,4-dihydro-3-methyl-quinoline morpholine -2 (1H) – one (lmmol) was dissolved with ethyl acetate (50 mL) was slowly added dropwise a solution of anhydrous hydrogen chloride in ethyl acetate (lmol / L, 5mL), stirred for 2h, the precipitated solid was filtered, the filter cake washed with ethyl acetate, 50 ° C and dried in vacuo 4h, as a white powdery solid 0.436g, yield 92%.
ESI-MS: 438 (M + 1)
Elemental analysis results:
Calcd: C, 63.35%; H, 6.17%; Cl, 7.48%; F, 4.01%; N, 8.87%; O, 10.13%
Found: C, 63.29%; H, 6.24%; CI, 7.43%; F, 4.05%; N, 8.82%; O, 10.17%
Example 3
Preparation 1-1 methanesulfonate
The 1-1 (lmmol) was dissolved with ethyl acetate (50 mL) was slowly added dropwise a solution of methanesulfonic acid in ethyl acetate (lmol / L, 5mL), stirred for 2h, the precipitated solid was filtered, the filter cake with ethyl acetate wash, 50 ° C and dried in vacuo 4h, as a white powdery solid 0.487g, yield 91.3%.
ESI-MS: 438 (M + 1, positive mode), 95 (CH 3 the SO 3 -, negative mode) Elemental analysis:
Calcd: C, 58.52%; H, 6.04%; F, 3.56%; N, 7.87%; 0, 17.99%; S, 6.01%
Found: C, 58.49%; H, 6.09%; F, 3.50%; N, 7.81%; 0, 18.02%; S, 6.09%
PATENT
Paper
Development and Kilogram-Scale Synthesis of a D2/5-HT2A Receptor Dual Antagonist (±)-SIPI 6360

The kilogram-scale synthesis of a D2/5-HT2A receptor dual antagonist (±)-SIPI 6360 was developed as an alternative treatment for schizophrenia. Specifically, three conditions were modified and optimized, including the Vilsmeier conditions, to prepare quinoline 3. In addition, the palladium-catalyzed hydrogenation was modified to synthesize dihydroquinolin-2(1H)-one 5, and the reduction of β-chloroamide was altered to form 3-chloropropanamine 8. Ultimately these improvements led to the preparation of a 1.5 kg of (±)-SIPI 6360 batch in eight steps with an overall yield of 34% and purity of 99.8%.
//////// D2/5-HT2A receptor dual antagonist (±)-SIPI 6360, 1401333-14-9
c21CC(C(Nc1cc(cc2)OCCCN3CCC(CC3)c4c5ccc(cc5on4)F)=O)C

Olanexidine Gluconate
OPB-2045G, Gluconate olanexidin, Olanedine, OPB-2045, OPB 2045G,
(Olanedine®)Approved in Japan PMDA 2015-07-03, Olanedine® by Otsuka

A disinfectant uesd to prevent of postoperative bacterial infections.


CAS .146510-36-3(Olanexidine free form),
Imidodicarbonimidic diamide, N-((3,4-dichlorophenyl)methyl)-N’-octyl
| C17H27Cl2N5 | |
| Formula Weight: | 372.341 |
CAS 799787-53-4(Olanexidine Gluconate)
| 568.49 | |
| Formula | C17H27Cl2N5 ● C6H12O7 |
1-(3,4-Dichlorobenzyl)-5-octylbiguanide mono-D-gluconate
| オラネキシジングルコン酸塩 Olanexidine Gluconate  C17H27Cl2N5▪C6H12O7 : 568.49 [799787-53-4] |
Indication:Bacterial infection
Otsuka (Originator)



SEE ALSO
Olanexidine hydrochloride [USAN]
146509-94-6 HCL
RN: 218282-71-4 HCL HYDRATE
UNII: R296398ALN
Molecular Formula, C17-H27-Cl2-N5.Cl-H.1/2H2-O
Molecular Weight, 835.6192
Imidodicarbonimidic diamide, N-((3,4-dichlorophenyl)methyl)-N’-octyl-, monohydrochloride, hydrate (2:1)
INTRODUCTION
Olanexidine gluconate was approved by Pharmaceuticals and Medical Devices Agency of Japan (PMDA) on Jul 03, 2015. It was developed and marketed as Olanedine® by Otsuka in Japan.
Olanexidine gluconate is an antiseptic/disinfectant compound with potent bactericidal activity against Gram-negative and Gram-positive bacteria, for use in preparing patients for surgery and preventing of postoperative bacterial infections.
Olanedine® is available as topical solution (1.5%), containing 3 g/200 mL, 0.15 g/10 mL and 0.375 g/25 mL, and the recommendation is applying appropriate amount of the drug.
PRODUCT PATENT
Kazuyoshi Miyata, Yasuhide Inoue, Akifumi Hagi, Motoya Kikuchi, Hitoshi Ohno, Kinji Hashimoto, Kinue Ohguro, Tetsuya Sato,Hidetsugu Tsubouchi, Hiroshi Ishikawa,Takashi Okamura, Koushi Iwata,
| Otsuka Pharmaceutical Co., Ltd., Otsuka Pharmaceutical Factory, Inc. |

CN1065453A
http://www.google.com.au/patents/CN1065453A?cl=en
WO2008026757A1
https://google.com/patents/WO2008026757A1?cl=en
Example 1: l-cyano-3-n-octylguanidine
A 7.00-kg quantity of Compound (4) (54.16 mol) was dissolved in 105 liters of ethyl acetate, and the resulting mixture was cooled to 5°C or below. A 2.66-kg quantity of concentrated sulfuric acid (27.12 mol) was added thereto dropwise at a temperature of 4O0C or below while stirring. To the thus- obtained suspension of 1/2 sulfate of Compound (4) was added 5.06 kg of sodium dicyanamide (56.83 mol), and the resulting suspension was heated under reflux for 7 hours. The reaction solution was cooled to 400C or below, and 70 liters of water was added thereto. Subsequently, the resulting solution was heated to 80 to 900C (internal temperature) to distill the ethyl acetate off. The remaining liquid was cooled to 400C or below, and 70 liters of toluene was then added thereto, followed by the extraction of 1-cyano — 3-n-octyl guanidine at about 500C. The extracted toluene layer was washed with 35 liters of water at about 500C and cooled to 100C or below, followed by stirring for about 30 minutes. The resulting precipitated crystals were separated and washed with 7 liters of toluene. The resulting crystals were dried at 400C for 7.5 hours, yielding l-cyano-3-n- octylguanidine. 2007/067107
-16-
Yield: 9.11 kg (The yield was 85.7% based on the Compound(4).) White crystals having a melting point of 69 to 740C (no clear melting point was observed)
IR(KBr) spectrum: 3439, 3296, 2916, 2164, 1659, 1556, 1160, 718, and 572 cm“1
Thermogravimetric measurement/differential thermal analysis: 73.5°C (weak), an endothermic peak at 77.50C
1H-NMR(CDCl3) spectrum: 0.88 ppm (t, J = 6.6 Hz, 3H), 1.20-1.38 ppm (m, 10H), 1.43-1.62 ppm (m, 2H), 3.17 ppm (dd, J = 6.9 Hz, J = 6.0 Hz, 2H), 5.60-5.70 ppm (bs, 2H), 5.80-5.95 ppm (bs, IH)
Reference Example 2: Acidolysis of 1- (3,4-dichlorobenzyl) -5- octylbiguanide dihydrochloride
A 1-g quantity of 1- (3, 4-dichlorobenzyl) -5-octyl biguanide dihydrochloride was dissolved in 15 ml of 10% ethanol, followed by refluxing for 5 hours. HPLC analysis was conducted under the conditions described below.
The yield of 1-[N- (3,4-dichlorobenzyl) carbamoyl-3- octyl]guanidine (holding time: 9.84 minutes) was 0.91%, and the yield of 1- (N-octyl-carbamoyl) -3- (3, 4-dichlorobenzyl) guanidine
(holding time: 10.54 minutes) was 0.22%.
HPLC analysis conditions:
Column: YMC AM302 4.6 mm I. D. x 150 mm
Eluate: MeCN/0.05 M aqueous solution of sodium 1- octanesulfonate/acetic acid = 700/300/1
Detector: UV 254 nm
The physical property values of the resulting 1-[N- (3,4- dichlorobenzyl) carbamoyl-3-octyl] guanidine were as follows: NMR (DMSO-de) δ: 0.86 (3H, t, J = 6.0 Hz), 1.07-1.35 (1OH, m) , 1.35-1.49 (2H, m) , 2.95-3.15 (2H, m) , 4.12 (2H, d, J = 6.3 Hz), 6.78-7.40 (4H, m) , 7.23 (IH, dd, J = 2.1 Hz, J = 8.4 Hz), 7.46 (IH, d, J = 2.1 Hz), 7.54 (IH, d, J = 8.4 Hz)
The physical property values of the resulting 1- (N-octyl- carbamoyl) -3- (3, 4-dichlorobenzyl) guanidine were as follows: NMR (DMSO-d6) δ: 0.85 (3H, t, J = 6.6 Hz), 1.02-1.40 (12H, m) , 2.89-2.95 (2H, m) , 4.33 (2H, bs) , 5.76-7.00 (4H, m) , 7.28 (IH, dd, J = 2.1 Hz, J = 8.1 Hz), 7.52 (IH, d, J = 2.1 Hz), 7.58 (IH, d, J = 8.1 Hz)
Example 1: 1- (3, 4-dichlorobenzyl) -5-octylbiguanide monohydrochloride 1/2 hydrate
A 9.82-g quantity of Compound (2) (0.05 mol) and 10.63 g of 3, 4-dichlorobenzylamine (0.05 mol) were added to 49 ml of butyl acetate, followed by refluxing for 6 hours. The reaction solution was concentrated under reduced pressure, and a mixture of 12 ml of water and 47 ml of isopropyl alcohol was added and dissolved into the remainder. To the thus-obtained solution was added, dropwise, 10.13 g of concentrated hydrochloric acid. The resulting mixture was stirred at 28 to 300C for 30 minutes, and the precipitated crystals were then filtered out. The thus- obtained crystals were washed with a small amount of isopropyl alcohol, yielding 23.42 g of (non-dried) 1- (3, 4-dichlorobenzyl) – 5-octylbiguanide dihydrochloride. The resulting crystals were suspended in 167 ml of water without drying, the suspension was then stirred at 25 to 27°C for 2 hours, followed by separation of the crystals by filtration. The thus-obtained crystals were washed with a small amount of water and dried at 400C for 20 hours, yielding 17.05 g of 1- (3, 4-dichlorobenzyl) -5-octyl biguanide monohydrochloride 1/2 hydrate having a purity of 99.9% at a yield of 81.6%.
Example 2 : 1- (3, 4-dichlorobenzyl) -5-octylbiguanide dihydrochloride
A 100-g quantity of Compound (4) (0.774 mol) was dissolved in 1 liter of n-butyl acetate, and 37.6 g of concentrated sulfuric acid (0.383 mol) was added thereto while stirring. To the thus-obtained suspension of 1/2 sulfate of Compound (4) was added 68.9 g of sodium dicyanamide (0.774 mol), 7107
-18- and the resulting suspension was heated under reflux for 3 hours. The reaction solution was cooled to about 200C, and the organic layer thereof was sequentially washed with about 500 ml each of (i) 5% hydrochloric acid, (ii) 5% aqueous caustic soda solution, (iii) 5% aqueous sodium bicarbonate solution, and (iv) water.
To the thus-obtained n-butyl acetate solution of Compound (2) were added 118.5 g of Compound (3) (0.673 mol) and then 58.4 ml of concentrated hydrochloric acid while stirring. The reaction solution was heated, and about 800 ml of n-butyl acetate was distilled off under atmospheric pressure (ordinary pressure) , followed by heating the reaction solution under reflux for 3.5 hours . Subsequently, the reaction solution was cooled to about 400C, and 900 ml of isopropanol, 100 ml of water, and 134 ml of concentrated hydrochloric acid were added thereto. The mixture was stirred at 60 to 70°C for 1 hour and cooled to 100C or below and the precipitated crystals were then separated. The resulting crystals were washed with 200 ml of isopropanol and dried at 6O0C, yielding 1- (3, 4-dichlorobenzyl) -5-octylbiguanide dihydrochloride. Yield: 243.8 g (The yield was 81.3% based on the Compound (3).) Melting point: 228.90C IR(KBr) spectrum: 2920, 1682, 1634, 1337, 1035, 820, and 640 cm“1
PATENT
WO2004105745A1
PATENT
WO2009142715A1
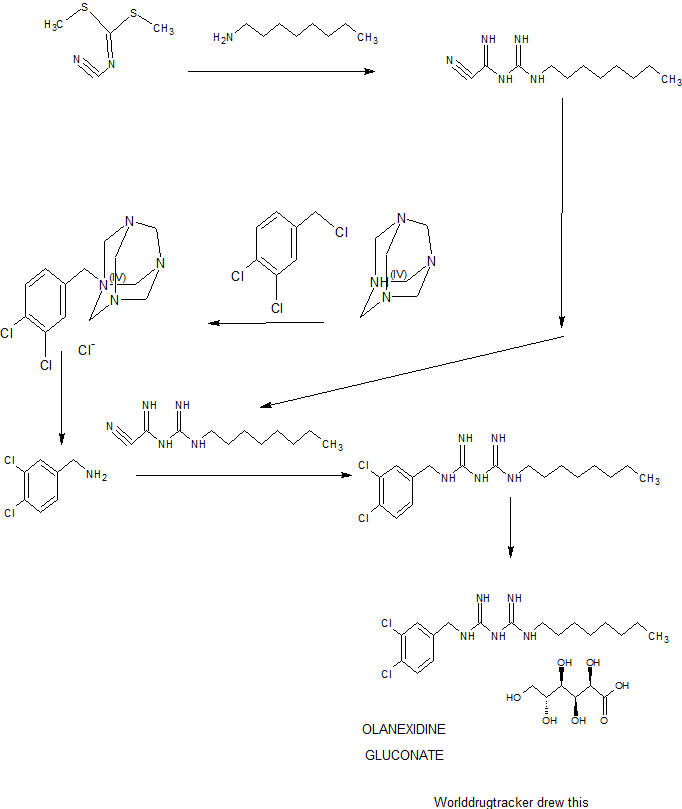
PATENT
https://www.google.com/patents/US8334248
Olanexidine is a compound with high bactericidal activity having the chemical name 1-(3,4-dichlorobenzyl)-5-octylbiguanide. Research has been carried out into bactericides containing, olanexidine hydrochloride as an active ingredient (see Japanese Patent No. 2662343, etc.).
Olanexidine has very poor solubility in water, and hitherto known salts of olanexidine are also poorly soluble in water. For example, the solubility at 0° C. of olanexidine hydrochloride in water has been measured to be less than 0.05% (W/V), and the solubility of free olanexidine is a further order of magnitude less than this. Consequently, sufficient bactericidal activity cannot be expected of an aqueous solution merely having olanexidine dissolved therein, and moreover, depending on the conditions the olanexidine may precipitate out.
In the case of making an aqueous preparation of olanexidine in particular, to make the concentration of the olanexidine sufficient for exhibiting effective bactericidal activity, and to reduce the possibility of the olanexidine precipitating out, it has thus been considered necessary to use a dissolution aid such as a surfactant.
EXAMPLE 1 Preparation of an Aqueous Solution Aqueous Solution 1
20.9 g (50 mmol) of olanexidine hydrochloride hemihydrate was added to 250 mL of a 1 N aqueous sodium hydroxide solution, and the suspension was stirred for 1.5 hours at room temperature (25° C.). The solid was filtered off, and washed with water. The solid obtained was further suspended in 250 mL of purified water, the suspension was stirred for 5 minutes at room temperature, and the solid was filtered off, and washed with water. This operation was carried out once more to remove sodium chloride formed. The solid obtained (free olanexidine) was put into purified water in which 8.9 g (50 mmol) of gluconolactone had been dissolved, and the mixture was stirred at room temperature until the solid dissolved, and then purified water was further added to give a total volume of 300 mL. The concentration of olanexidine in the aqueous solution obtained was measured by using high performance liquid chromatography to be 6% in terms of free olanexidine.
This aqueous solution was still transparent and colorless even after being left for several months at room temperature.
CLIP
http://dmd.aspetjournals.org/content/28/12/1417/F9.expansion.html


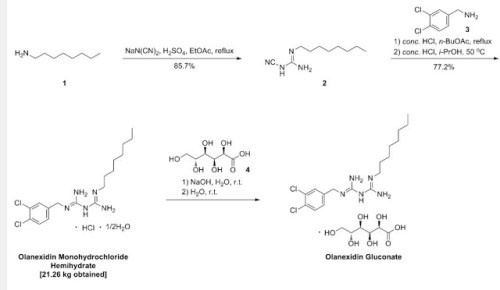
REFERENCES
http://www.otsukakj.jp/en/news/photo/photo-14423716650.pdf
| Patent ID | Date | Patent Title |
|---|---|---|
| US8979785 | 2015-03-17 | Fluid application device and method |
| US8911771 | 2014-12-16 | Fluid application device and method |
| US8858484 | 2014-10-14 | Fluid application device and method |
| US2013330114 | 2013-12-12 | FLUID APPLICATION DEVICE AND METHOD |
| US2012095254 | 2012-04-19 | METHOD AND APPARATUS FOR PREPARING A SOLUTION OF A SHEAR SENSITIVE MATERIAL |
| US7868207 | 2011-01-11 | PROCESS FOR PRODUCING 1-(3, 4-DICHLOROBENZYL)-5-OCTYLBIGUANIDE OR A SALT THEREOF |
| US2010331421 | 2010-12-30 | DISINFECTANT AND/OR BACTERICIDAL AQUEOUS COMPOSITIONS |
| US2010331423 | 2010-12-30 | AQUEOUS SOLUTION OF OLANEXIDINE, METHOD OF PREPARING THE AQUEOUS SOLUTION, AND DISINFECTANT |
| US7829518 | 2010-11-09 | Aqueous solution of olanexidine, method of preparing the aqueous solution, and disinfectant |
| US7825080 | 2010-11-02 | Aqueous solution of olanexidine, method of preparing the aqueous solution, and disinfectant |
| Patent ID | Date | Patent Title |
|---|---|---|
| US7622469 | 2009-11-24 | 2, 4-diamino-1, 3, 5-triazine derivatives |
| US2009287021 | 2009-11-19 | METHOD AND APPARATUS FOR PREPARING A SOLUTION OF A SHEAR SENSITIVE MATERIAL |
| US2007053942 | 2007-03-08 | Disinfectant and/or bactericidal aqueous compositions |
| EP0507317 | 1997-01-15 | BIGUANIDE DERIVATIVES, MANUFACTURING METHOD THEREOF, AND DISINFECTANTS CONTAINING THE DERIVATIVES |
| EP0507317A2 * | Apr 3, 1992 | Oct 7, 1992 | Otsuka Pharmaceutical Co., Ltd. | Biguanide derivatives, manufacturing method thereof, and disinfectants containing the derivatives |
| EP1634589A1 * | May 25, 2004 | Mar 15, 2006 | Otsuka Pharmaceutical Co., Ltd. | Aqueous olanexidine solution, method of preparing the same, and disinfectant |
| Reference | ||
|---|---|---|
| 1 | * | TSUBOUCHI H ET AL: “Synthesis and Structure-Activity Relationships of Novel Antiseptics” BIOORGANIC & MEDICINAL CHEMISTRY LETTERS, OXFORD, GB, vol. 7, no. 13, 8 July 1997 (1997-07-08), pages 1721-1724, XP004136287 ISSN: 0960-894X |
//////////Olanexidine Gluconate, OPB-2045G, (Olanedine®, Approved, japan 2015-07-03, Olanedine, Otsuka, PMDA, Olanexidine, オラネキシジングルコン酸塩 , Gluconate olanexidin, Olanedine, OPB-2045, OPB 2045G, JAPAN 2015
CCCCCCCCN=C(N)NC(=NCC1=CC(=C(C=C1)Cl)Cl)N
Clc1ccc(CNC(=N)NC(=N)NCCCCCCCC)cc1Cl.O=C(O)[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO

Fortune India has published list of 500 mid-size companies who ranked them on various parameters based on the results of 2013-2014. Ajanta pharma features very prominently in the lists. Ajanta’s ranking on various pararmeters is given below:
Ranked 3’d largest Wealth Creator on 5 year CAGR 93.14%
Ranked 10th on Capital Employed (ROCE)
Ranked 21st in Net profit .
Ranked 182nd in Sales
On 17’th August 2015, Fortune India organized an award function to present the awards to Top 10 largest weatth creator companies and Ajanta is one of those elite companies.
The awards were presented by Mr. Piyush Goyal, Minister of State-Power,Coal& New and Renewable Energy, Govt. of India to Mr. yogesh Agrawal, Managing Director and Mr.Rajesh Agrawal,Jt.Managing Director of the company.


Ajanta Pharma, “One of the Giants of Tomorrow” – Fortune India
We are pleased to share with you that Ajanta Pharma has been honoured as “ONE OF THE GIANTS OF TOMORROW” by prestigious Fortune India magazine on 19th August 2016 at New Delhi.
The honour was conferred to our Managing Director, Mr. Yogesh Agrawal and our Jt. Managing Director, Mr. Rajesh Agrawal at the hands of Hon. Mr. Nitin Gadkari, Union Minister for Road Transport and Highways and Shipping, Govt of India. This is the 2nd year in row where Ajanta has received the recognition from Fortune India.
Fortune India (June 2016 Issue) published the list of mid-size companies based on the financial year 2014-15 results and we are pleased to share with you that Ajanta has been ranked 3rd Top Wealth creator over last 5 years.


Ajanta Pharma Limited (APL) is a pharmaceutical company headquartered in Mumbai, India. It has strong presence in Branded Generic business in India & Emerging markets; and Generic business in USA. In India, company operates in selected therapeutic areas of Cardiology, Dermatology, Ophthalmology and Pain management. Its brands in each of sub-therapeutic areas or molecules hold leadership positions. In Emerging Markets, company has presences in Africa, Asia, Middle East, and CIS on broader therapeutic segments such as anti-malarial, gastro, antibiotics, cardiology, dermatology, pain management, etc. In USA, company has already no. of approved ANDA’s which are either commercialized or in process of being commercialized and large no. of ANDA’s are awaiting US FDA approval. We have state-of-the-art research facilities for formulation (finished product) and API development located at Mumbai, India. Our R&D capabilities are evident from number of products launched 1st to market by the company providing patients most needed compliance and convenience. A dedicated and focused team of more than 750 Ajantaites work for R&D, which is growing continuously. Ajanta has four formulations manufacturing facilities located in India and 1 in Mauritius. Besides that, we also have an API manufacturing facility located at Waluj, India. Ajanta’s flagship formulation facility at Paithan (Maharashtra, India) has approval of USFDA, WHO- Geneva (prequalification), UNICEF and many regulatory authorities from different parts of the world. We continuously invest in enhancing our existing manufacturing facilities to meet current cGMP requirements and also construct new facilities to meet the company’s growth requirements. We are in process of setting up 1 more formulations manufacturing facility for domestic and emerging markets at Guwahati, Assam. Please visit http://ajantapharma.com/ for more information. Contact: careers@ajantapharma.com
Speciality Branded Generics, Generics, Complex Formulations
Pharmaceuticals
Public Company
98 Ajanta House Charkop, Kandivili West Mumbai,Maharashtra 400067 India
5001-10,000 employees
1973
Rajesh Agrawal (left), Ajanta Pharma’s joint managing director, with brother Yogesh, who is also managing director of the company, at their Kandivli facility
Ajanta Pharma needed a shot of its own medicine, an energiser like 30-Plus. It found its antidote in the new generation of Agrawals: Mannalal’s sons, Yogesh and Rajesh.

“When I joined Ajanta (in 2000), and realised what was going on, I wanted to run away. I thought to myself, ‘Why did I return from the US? I could have had a job there,’” says Rajesh, 39, Ajanta’s joint managing director, who has a management degree from Bentley College, Massachusetts. “It was tough in the beginning, especially the situation with creditors and debtors.”
Together, Rajesh and his older brother Yogesh, 43, who is managing director, changed Ajanta’s trajectory by focusing on the ‘specialty’ generic drug market and putting an end to the company’s legacy businesses, which included OTC drug sales and supplying drugs to government health agencies in India and other countries.
This was a risky move, but it has paid off. Ajanta Pharma closed FY15 with a consolidated net sales of Rs 1,481 crore and a net profit of Rs 310 crore (this is a compound annual growth rate, or CAGR, of 57 percent for four years since 2011). In terms of net sales, it recorded a CAGR of 31 percent for the same period. This growth has come on a low base, but the signs are encouraging. Its market value currently stands at around Rs 13,500 crore; this is a 65-fold growth in 15 years.
References
https://www.linkedin.com/company/263285
/////////Ajanta Pharma, “One of the Giants of Tomorrow” , Fortune India, AWARD, Fortune India, RAJESH AGRAWAL
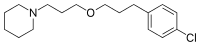
Pitolisant
CAS 362665-56-3
FDA APPROVED 2019 AUG
1-(3-(3-(4-Chlorophenyl)propoxy)propyl)piperidine
MF C17H26ClNO
MW 295.1703
(Wakix®)Approved EU 31/3/2016, Narcolepsy
A histamine H3 receptor antagonist/inverse agonist used to treat narcolepsy.
![]()
BF-2649; BF-2.649; FUB-649, Ciproxidine, Tiprolisant
CAS 362665-56-3, 362665-57-4 (oxalate)
CAS 903576-44-3(Pitolisant Hydrochloride)
APPROVED IN EU, European Medicine Agency (EMA) on Mar 31, 2016.
1-{3-[3-(4-Chlorophenyl)propoxy]propyl}piperidine hydrochloride (1:1)
| Molecular Weight | 332.31 |
| Formula | C17H26ClNO ● HCl |
![]()
Bioprojet INNOVATOR
Jean-Charles Schwartz, Jeanne-Marie Lecomte
Pitolisant (INN) or tiprolisant (USAN) is a histamine receptor inverse agonist/antagonist selective for the H3 subtype.[1] It hasstimulant and nootropic effects in animal studies,[2] and may have several medical applications, having been researched for the treatment of narcolepsy, for which it has been granted orphan drug status in the EU and US.[3][4] It is currently in clinical trials forschizophrenia and Parkinson’s disease.[4][5][6]
Pitolisant hydrochloride was approved by European Medicine Agency (EMA) on Mar 31, 2016. It was developed and marketed as Wakix® by Bioprojet in EU.
Pitolisant is being developed by Bioprojet for the oral treatment of central nervous system disorders. Pitolisant is a selective histamine H3-receptor antagonist/inverse agonist which enhances the activity of histaminergic neurons. Pitolisant has been launched in several countries for the treatment of narcolepsy, and is approved in the US, EU, Iceland and Liechtenstein. Clinical development is underway for type-1 diabetes, hypersomnia and drug abuse in countries worldwide.
Phase III development was also conducted for the treatment of hypersomnia in Switzerland. Phase II development for attention-deficit hyperactivity disorder was conducted in France. However, there were no recent reports on development identified. Development in epilepsy and obesity has been discontinued.
Ferrer and Bioprojet appeared to have a co-development agreement for pitolisant that allowed the mutual use of both companies’ technical and scientific resources; however, as per Ferrer’s communication dated June 2016, the drug is no longer in its portfolio.

Pitolisant hydrochloride is an antagonist/inverse agonist of the histamine H3 receptor, which is indicated in adults for the treatment of narcolepsy with or without cataplexy.
Wakix® is available as tablet for oral use, containing 4.5 mg and 18 mg of Pitolisant hydrochloride. The initial dose of 9 mg (two 4.5 mg, tablets) per day, and it should be used at the lowest effective dose, depending on individual patient response and tolerance, according to an up-titration scheme, without exceeding the dose of 36 mg/day.
Pitolisant was developed by Jean-Charles Schwartz, Walter Schunack and colleagues after the former discovered H3 receptors.[7]Pitolisant was the first clinically used H3 receptor inverse agonist.
Pitolisant, also known as Tiprolisant, is a histamine receptor inverse agonist/antagonist selective for the H3 subtype. It has stimulant and nootropic effects in animal studies, and may have several medical applications, having been researched for the treatment of narcolepsy, for which it has been granted orphan drug status in the EU and US. It is currently in clinical trials for schizophrenia and Parkinson’s disease. Pitolisant was the first clinically used H3 receptor inverse agonist.

The European Medicines Agency (EMA) has recommended granting marketing authorization for pitolisant (Wakix, Bioprojet Pharma) for narcolepsy with or without cataplexy, the agency announced today.
Narcolepsy is a rare sleep disorder that affects the brain’s ability to regulate the normal sleep-wake cycle, leading to excessive daytime sleepiness, including the sudden urge to sleep, and disturbed night-time sleep. Some patients also experience sudden episodes of cataplexy, potentially causing dangerous falls and increasing the risks for accidents, including car accidents. Symptoms of narcolepsy can be severe and significantly reduce quality of life.
Pitolisant “will add to the available treatment options for narcolepsy. It is a first-in-class medicine that acts on histamine H3 receptors in the brain. This leads to increased histamine release in the brain, thereby enhancing wakefulness and alertness,” the EMA notes in a news release.
The EMA recommendation for approval of pitolisant is based on an evaluation of all available safety and efficacy data conducted by the Committee for Medicinal Products for Human Use (CHMP). The data include two pivotal placebo-controlled trials involving 259 patients, as well as one uncontrolled, open-label study involving 102 patients with narcolepsy and one supportive study in 105 patients.
The studies showed that pitolisant was effective in reducing excessive daytime sleepiness in patients with narcolepsy. The beneficial effect of the drug on cataplexy was demonstrated in one of the pivotal studies as well as in the supportive study.
No major safety concerns with pitolisant emerged in testing. Insomnia, headache, and nausea were among the most common adverse effects observed in the clinical trials, and the CHMP decided on measures to mitigate these risks, the EMA said. The CHMP also requested the company conduct a long-term safety study to further investigate the safety of the drug when used over long periods.
Pitolisant for narcolepsy received orphan designation from the Committee for Orphan Medicinal Products in 2007. Orphan designation provides medicine developers access to incentives, such as fee reductions for scientific advice, with the aim of encouraging the development of treatments for rare disorders.
The CHMP opinion will now be sent to the European Commission for the adoption of a decision on a European Union–wide marketing authorization. Once that has been granted, each member state will decide on price and reimbursement based on the potential role/use of this medicine in the context of its national health system.

Narcolepsy-cataplexy.
Narcolepsy-cataplexy, or Gelineau syndrome, is a rare but serious disorder characterized by excessive daytime sleepiness which can be an extreme hindrance to normal professional and social activities, and which is accompanied by more or less frequent attacks of cataplexy (a sudden loss of muscle tone triggered by emotions as varied as laughter or fear) and erratic episodes of REM sleep (during wakefulness and during sleep), sometimes associated with hypnagogic hallucinations. Moreover, individuals with narcolepsy have various degrees of cognitive impairment and tend to be obese (reviewed by Dauvilliers et al., Clin. Neurophysiol., 2003, 114, 2000; Baumann and Bassetti, Sleep Med. Rev., 2005, 9, 253).
The disorder is caused by the loss of a group of neurons in the brain which produce two peptides, orexins, also known as hypocretins, located in the anterior hypothalamus and projecting to the main groups of aminergic neurons which regulate wakefulness and sleep. Patients with the disorder generally have very low levels of orexins in cerebrospinal fluid. Orexin knock-out mice display many of the symptoms seen in narcoleptic subjects, confirming the role of these peptides and thereby providing an excellent animal model of the disease (Chemelli et al., Cell, 1999, 98, 437).
Several types of treatments which can improve the symptoms of narcolepsy already exist, although they do not completely relieve symptoms and, furthermore, can cause significant side effects limiting their usefulness.
For instance, amphetamines or analogues such as methylphenidate which release catecholamines are used to treated daytime sleepiness, but these agents induce a state of excessive excitation as well as cardiovascular disturbances and also carry a potential for drug addiction.
Modafinil, a drug whose mechanism of action is unclear, also improves daytime sleepiness without causing as many side effects as amphetamines. Nonetheless, its efficacy is limited and it can cause headaches and nausea, particularly at high doses. Moreover amphetamines and/or modafinil do not appear to improve some of the most disabling symptoms of the disease, particularly cataplexy attacks, cognitive deficits and weight gain. With regard to cataplexy, treatments include antidepressants and oxybate. Effectiveness of the former has not been demonstrated (Cochrane Database Syst. Rev., 2005, 20, 3), and the latter is a drug of illegal abuse and its use is restricted.
It has also been shown that histamine H3 receptor antagonists induce the activation of histaminergic neurons in the brain which release histamine, a neurotransmitter with a crucial role in maintaining wakefulness (Schwartz et al., Physiol. Rev. 1991, 71, 1).
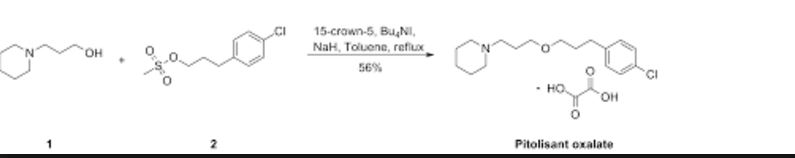
PATENT
Pharmaceutical products with histamine H3 receptor ligand properties and 0 subsequent pharmacological activities thereof are described in EP-980300. An especially important product among those disclosed is 1-[3-[3-(4- chlorophenyl)propoxy] propyl]-piperidine. This compound is disclosed as the free base and as the oxalate salt.
5 The use of 1-[3-[3-(4-chlorophenyl)propoxy]propyl]-piperidine as the free base is limited because of its oily nature. On the contrary, 1-[3-[3-(4- chlorophenyl)propoxy]propyl]-piperidine oxalate is a crystalline substance but its low aqueous solubility (0.025 g/ml at 230C) also limits its use as a
pharmaceutical ingredient.
0
Subsequent patents EP-1100503 and EP-1428820 mention certain salts of 1- [3-[3-(4-chlorophenyl)propoxy]propyl]-piperidine. However, the only one specifically described is the oxalate salt. The crystalline monohydrochloride salt is not described.
Example 1 : 1-[3-[3-(4-chlorophenyl)propoxy]propyl]-piperidine
According to the method disclosed in EP-982300, Example 78, sodium 3-piperidinopropanolate (2.127 kg; 12.88 mol), 3-(4-chlorophenyl)propyl mesylate (1.121 kg; 4.51 mol) and 0.322 mol of 15-crown-5 in 4.5 kg of dry toluene were refluxed for 4 hours. The solvent was evaporated and the residue purified by column chromatography on silica gel (eluent: methylene chloride/methanol (90/10)). The obtained oil was distilled in a fractionating equipment at reduced pressure (0.3-0.7 mmHg) and with a heating jacket at 207-2100C. The head fractions and the distilled fraction at 0.001-0.010 mmHg with a jacket temperature of 180-2000C were collected. The obtained oil (1.0 kg; 3.38 mol) corresponds to 1-[3-[3-(4-chlorophenyl)propoxy] propyl]-piperidine. Yield 75%.
Example 2: 1-[3-[3-(4-chlorophenyl)propoxy]propyl]-piperidine
monohydrochloride
Preparation
Distilled 1-[3-[3-(4-chlorophenyl)propoxy]propyl]-piperidine (1.0 kg) and anhydrous ethyl acetate (4.5 kg) are transferred to a 10-L glass vessel fitted with a cooling bath and a gas inlet. A stream of gaseous hydrogen chloride is bubbled in the reaction mixture at 20-250C.
The pH of the solution is checked by taking a 0.5 mL sample of the reaction mixture and diluting it with 5 mL of deionized water. The final pH must be about 3-4.
The mixture is cooled to -10°C-(-12°C) and stirred at this temperature for 1 h. The precipitate is filtered by using a sintered glass filter and washed with 0.5 L of anhydrous ethyl acetate previously cooled to 0-50C. The product is dried in a vacuum oven at 5O0C for a minimum period of 12 hours. The resulting crude 1 -[3-[3-(4-chlorophenyl)propoxy]propyl]-piperidine monohydrochloride weighs 1.10 kg.
Purification
A mixture of the above-described crude, 3.98 kg of anhydrous ethyl acetate and 0.35 kg of /-propanol is heated slowly at 55-6O0C in a 10-L glass vessel fitted with a heating and cooling system. When the solution has been completed, it is filtered through a heat-isolated sintered glass filter, keeping the temperature at 55-6O0C. The solution is transferred to a 10 L glass vessel and the mass is slowly cooled to 0-50C for about 1 hour. The mixture is stirred at this temperature for 1 hour and the precipitate is filtered through a sintered glass filter. The solid is washed with a mixture of 1.6 kg of anhydrous ethyl acetate and 0.14 kg of /-propanol cooled at 0-50C. The solid is dried in a vacuum oven at 5O0C for a minimum period of 12 hours. M. p. 117-1190C. Yield 80%.
IR spectrum (KBr): bands at 1112 and 1101 (C-O Ether/ St. asym), 2936 and 2868 (Alkane CH(CH2)) / St.), 1455 (Alkane CH(CH2)) / Deform.), 2647 and 2551 (Amine Salt / St.), 1492 (Amine / St.), 802 (Aromatic / Deform.) cm“1.
SEE
Eur. J. Pharm. Sci. 2001, 13, 249–259.
1: Leu-Semenescu S, Nittur N, Golmard JL, Arnulf I. Effects of pitolisant, a histamine H3 inverse agonist, in drug-resistant idiopathic and symptomatic hypersomnia: a chart review. Sleep Med. 2014 Jun;15(6):681-7. doi: 10.1016/j.sleep.2014.01.021. Epub 2014 Mar 18. PubMed PMID: 24854887.
2: Dauvilliers Y, Bassetti C, Lammers GJ, Arnulf I, Mayer G, Rodenbeck A, Lehert P, Ding CL, Lecomte JM, Schwartz JC; HARMONY I study group. Pitolisant versus placebo or modafinil in patients with narcolepsy: a double-blind, randomised trial. Lancet Neurol. 2013 Nov;12(11):1068-75. doi: 10.1016/S1474-4422(13)70225-4. Epub 2013 Oct 7. PubMed PMID: 24107292.
3: Nirogi R, Ajjala DR, Kandikere V, Pantangi HR, Jonnala MR, Bhyrapuneni G, Muddana NR, Vurimindi H. LC-MS/MS method for the determination of pitolisant: application to rat pharmacokinetic and brain penetration studies. Biomed Chromatogr. 2013 Nov;27(11):1431-7. doi: 10.1002/bmc.2939. Epub 2013 Jun 13. PubMed PMID: 23760876.
4: Kasteleijn-Nolst Trenité D, Parain D, Genton P, Masnou P, Schwartz JC, Hirsch E. Efficacy of the histamine 3 receptor (H3R) antagonist pitolisant (formerly known as tiprolisant; BF2.649) in epilepsy: dose-dependent effects in the human photosensitivity model. Epilepsy Behav. 2013 Jul;28(1):66-70. doi: 10.1016/j.yebeh.2013.03.018. Epub 2013 May 8. PubMed PMID: 23665640.
5: Uguen M, Perrin D, Belliard S, Ligneau X, Beardsley PM, Lecomte JM, Schwartz JC. Preclinical evaluation of the abuse potential of Pitolisant, a histamine H₃ receptor inverse agonist/antagonist compared with Modafinil. Br J Pharmacol. 2013 Jun;169(3):632-44. doi: 10.1111/bph.12149. PubMed PMID: 23472741; PubMed Central PMCID: PMC3682710.
6: Brabant C, Charlier Y, Tirelli E. The histamine H₃-receptor inverse agonist pitolisant improves fear memory in mice. Behav Brain Res. 2013 Apr 15;243:199-204. doi: 10.1016/j.bbr.2012.12.063. Epub 2013 Jan 14. PubMed PMID: 23327739.
7: Zhang DD, Sisignano M, Schuh CD, Sander K, Stark H, Scholich K. Overdose of the histamine H₃ inverse agonist pitolisant increases thermal pain thresholds. Inflamm Res. 2012 Nov;61(11):1283-91. doi: 10.1007/s00011-012-0528-5. Epub 2012 Jul 21. PubMed PMID: 22820944.
8: Inocente C, Arnulf I, Bastuji H, Thibault-Stoll A, Raoux A, Reimão R, Lin JS, Franco P. Pitolisant, an inverse agonist of the histamine H3 receptor: an alternative stimulant for narcolepsy-cataplexy in teenagers with refractory sleepiness. Clin Neuropharmacol. 2012 Mar-Apr;35(2):55-60. doi: 10.1097/WNF.0b013e318246879d. PubMed PMID: 22356925.
9: Schwartz JC. The histamine H3 receptor: from discovery to clinical trials with pitolisant. Br J Pharmacol. 2011 Jun;163(4):713-21. doi: 10.1111/j.1476-5381.2011.01286.x. Review. PubMed PMID: 21615387; PubMed Central PMCID: PMC3111674.
 |
|
| Clinical data | |
|---|---|
| Trade names | Wakix |
| Synonyms | Tiprolisant; Ciproxidine; BF2.649 |
| License data | |
| Routes of administration |
Oral |
| Drug class | Histamine H3 receptor inverse agonists |
| ATC code | |
| Identifiers | |
| CAS Number |
|
| PubChem CID | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEBI | |
| ChEMBL | |
| Chemical and physical data | |
| Formula | C17H26ClNO |
| Molar mass | 295.851 g/mol g·mol−1 |
| 3D model (JSmol) | |
//////////Pitolisant Hydrochloride, Wakix, histamine H3 receptor antagonist/inverse agonist, narcolepsy, orphan drug, tiprolisant, EU 2016, FDA 2019

![Skeletal formula of (1S,4S,7Z,10S,16E,21R)-7-ethylidene-4,21-diisopropyl-2-oxa-12,13-dithia-5,8,20,23-tetrazabicyclo[8.7.6]tricos-16-ene-3,6,9,19,22-pentone](https://upload.wikimedia.org/wikipedia/commons/thumb/7/71/Romidepsin_structure_%282%29.svg/200px-Romidepsin_structure_%282%29.svg.png)
| Romidepsin; Depsipeptide; FK228; Chromadax; FR901228; Istodax; | |
| Molecular Formula: | C24H36N4O6S2 |
|---|---|
| Molecular Weight: | 540.69584 g/mol |
CAS 128517-07-7
(1S,4S,7Z,10S,16E,21R)-7-ethylidene-4,21-di(propan-2-yl)-2-oxa-12,13-dithia-5,8,20,23-tetrazabicyclo[8.7.6]tricos-16-ene-3,6,9,19,22-pentone
(E)-N-(2-amino-4-fluorophenyl)-4-((3-(pyridin-3-yl)acrylamido)methyl)benzamide
Romidepsin, also known as Istodax, is an anticancer agent used in cutaneous T-cell lymphoma (CTCL) and other peripheral T-cell lymphomas (PTCLs). Romidepsin is a natural product obtained from the bacteria Chromobacterium violaceum, and works by blocking enzymes known as histone deacetylases, thus inducing apoptosis.[1] It is sometimes referred to as depsipeptide, after the class of molecules to which it belongs. Romidepsin is branded and owned by Gloucester Pharmaceuticals, now a part of Celgene.[2]
Romidepsin, a histone deacetylase inhibitor, originally developed by Fujisawa (now Astellas Pharma), causes cell cycle arrest,
differentiation, and apoptosis in various cancer cells.
In 2004, the FDA granted fast-track designation for romidepsin as monotherapy for the treatment of cutaneous T-cell lymphoma (CTCL) in patients who have relapsed following, or become refractory to, other systemic therapies. The FDA designated romidepsin as an orphan drug and it was approved in 2009 for this indication and it was commercialized in 2010. In 2007, another fast-track designation was granted for the product as monotherapy of previously treated peripheral T-cell lymphoma.
Romidepsin (FR901228) was originally discovered and isolated from the fermentation broth of Chromobacterium violaceum No. 968. It was identified through efforts in the search for novel agents which selectively reverse the morphological phenotype of Ras oncogene-transformed cells since the Ras signaling pathway plays a critical role in cancer development. Therefore, the drug could also have multiple molecular targets for its anticancer activity besides HDAC.
FR901228 is a bicyclic depsipeptide which is structurally unrelated to any known class of cyclic peptides with an unusual disulfide bond connecting a thiol and D-cysteine.
This drug is commercially produced by fermentation; however its interesting and novel structure warrants examination of its synthesis within the context of this review
Romidepsin is a histone deacetylase (HDAC) inhibitor.HDACs catalyze the removal of acetyl groups from acetylated lysine residues in histone and non-histone proteins, resulting in the modulation of gene expression.
Romidepsin is indicated for treatment of cutaneous T-cell lymphoma (CTCL) in patients who have received at least
one prior systemic therapy; treatment of peripheral T-cell lymphoma (PTCL) in patients who have received at least
one prior therapy.
Available as an injection, containing 10 mg of romidepsin and recommended dose is 14 mg/m2 administered intravenously over a 4-hour period on days 1, 8, and 15 of a 28-day cycle until disease progression or unacceptable toxicity.

Romidepsin was first reported in the scientific literature in 1994, by a team of researchers from Fujisawa Pharmaceutical Company (now Astellas Pharma) in Tsukuba, Japan, who isolated it in a culture of Chromobacterium violaceum from a soil sample obtained inYamagata Prefecture.[3] It was found to have little to no antibacterial activity, but was potently cytotoxic against several human cancercell lines, with no effect on normal cells; studies on mice later found it to have antitumor activity in vivo as well.[3]
The first total synthesis of romidepsin was accomplished by Harvard researchers and published in 1996.[4] Its mechanism of actionwas elucidated in 1998, when researchers from Fujisawa and the University of Tokyo found it to be a histone deacetylase inhibitorwith effects similar to those of trichostatin A.[5]

Phase I studies of romidepsin, initially codenamed FK228 and FR901228, began in 1997.[6] Phase II and phase III trials were conducted for a variety of indications. The most significant results were found in the treatment of cutaneous T-cell lymphoma (CTCL) and other peripheral T-cell lymphomas (PTCLs).[6]
In 2004, romidepsin received Fast Track designation from the FDA for the treatment of cutaneous T-cell lymphoma, and orphan drugstatus from the FDA and the European Medicines Agency for the same indication.[6] The FDA approved romidepsin for CTCL in November 2009[7] and approved romidepsin for other peripheral T-cell lymphomas (PTCLs) in June 2011.[8]
Mechanism of action
Romidepsin acts as a prodrug with the disulfide bond undergoing reduction within the cell to release a zinc-binding thiol.[3][9][10] The thiol reversibly interacts with a zinc atom in the binding pocket of Zn-dependent histone deacetylase to block its activity. Thus it is anHDAC inhibitor. Many HDAC inhibitors are potential treatments for cancer through the ability to epigenetically restore normal expression of tumor suppressor genes, which may result in cell cycle arrest, differentiation, and apoptosis.[11]

The use of romidepsin is uniformly associated with adverse effects.[12] In clinical trials, the most common were nausea and vomiting,fatigue, infection, loss of appetite, and blood disorders (including anemia, thrombocytopenia, and leukopenia). It has also been associated with infections, and with metabolic disturbances (such as abnormal electrolyte levels), skin reactions, altered taste perception, and changes in cardiac electrical conduction.[12]
CLIP
http://www.scielo.br/scielo.php?script=sci_arttext&pid=S0103-50532012001200003

Romidepsin was first isolated from the fermentation broth of Chromobacterium Violaceum WB968 in a nutrient
medium. Sterilized of 1% glucose and 1% bouillon solution were incubated with Chromobacterium Violaceum WB968, followed by further incubation with 1% glucose solution, 1% bouillon solution and adekanol gave the target romidepsin after extraction, silica gel chromatography and recrystallization.[Okuhara, M.; Goto, T.; Hori, Y. et al. US4977138A, 1990.]
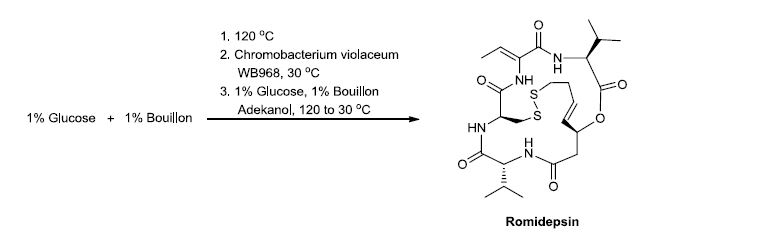
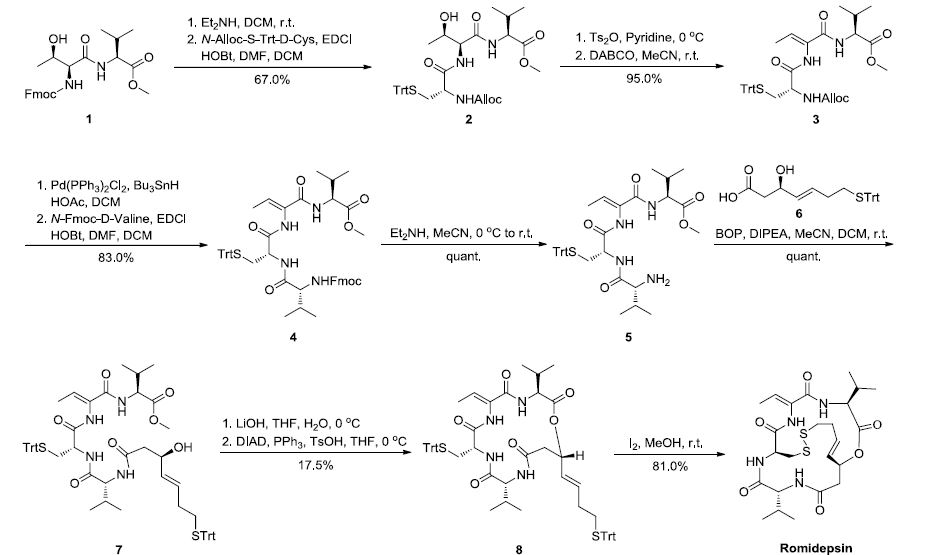

The synthetic route was initiated by the deprotection L-(Fmoc)Thr-L-Val-OMe 1, subsequently coupled with
N-Alloc-S-Trt-D-Cys, followed by tosylation and then elimination to produce tripeptide 3 in the yield of 63.7% over four steps. The N-Alloc deprotection of 3 and then coupling with N-Fmoc-D-Valine were proceeded to provide tetrapeptide 4, which was subsequently removed Fmoc group to afford relative tetrapeptide 5 in 83.0% yield from compound 3. Condensation of 5 with β-hydroxy mercapto acid 6 was carried out by treating with benzotriazol-1-yl-oxy-tris-(dimethylamino)-phosphonium hexafluorphosphate (BOP) to give relative amide 7, and sequential hydrolysis yielded corresponding acid, which was performed by Mitsunobu macrolactonization
to produce depsipeptide 8 in 17.5% yield over three steps. Finally, romidepsin was obtained in the presence of iodine
in 81.0% yield and the overall yield of 7.5%.
The synthesis of intermediate β-hydroxy mercapto acid 6 commenced with the commercially available methyl 3,3-dimethoxypropionate 9. Nucleophilic addition of 9 with N,O-dimethylhydroxylamine provided Weinreb amide 10, followed by addition with lithium acetylide to give propargylic ketone 12 in the yield of 50.2% over two steps. Noyori’s asymmetric hydrogenation of ketone 12 provided (E)-alkene 14, which was removed the silyl group and then substituted with paratoluensulfonyl chloride to yield tosylate 15 in 40.6% yield across three steps. The dimethyl acetal of 15 was hydrolyzed to corresponding aldehyde by using lithium tetrafluoroborate,
which was immediately oxidized to relative carboxylic acid by applying Pinnick oxidation conditions. The trityl mercaptan was introduced by tosylate displacement to provide 6 in 65.0% yield over three steps and the overall yield of 13.3%.[2]
REF Greshock, T. J.; Johns, D. M.; Noguchi, Y., et al. Org. Lett. 2008, 10 (4), 613-616.
CLIP
Romidepsin (Istodax)
Romidepsin, a histone deacetylase inhibitor, originally developed by Fujisawa (now Astellas Pharma), causes cell cycle arrest,
differentiation, and apoptosis in various cancer cells.111 In 2004, the FDA granted fast-track designation for romidepsin as monotherapy for the treatment of cutaneous T-cell lymphoma (CTCL) in patients who have relapsed following, or become refractory
to, other systemic therapies. The FDA designated romidepsin as an orphan drug and it was approved in 2009 for this indication
and it was commercialized in 2010. In 2007, another fast-track designation was granted for the product as monotherapy of
previously treated peripheral T-cell lymphoma. Romidepsin (FR901228) was originally discovered and isolated from the fermentation
broth of Chromobacterium violaceum No. 968. It was identified through efforts in the search for novel agents which
selectively reverse the morphological phenotype of Ras oncogene-transformed cells since the Ras signaling pathway plays a
critical role in cancer development. Therefore, the drug could also have multiple molecular targets for its anticancer activity besides
HDAC.112 FR901228 is a bicyclic depsipeptide which is structurally unrelated to any known class of cyclic peptides with an unusual
disulfide bond connecting a thiol and D-cysteine. This drug is commercially produced by fermentation; however its interesting
and novel structure warrants examination of its synthesis within the context of this review.113,114 The synthesis of romidepsin
described is based on the total synthesis reported by the Williams115 and Simon groups (Scheme 20).116
L-Valine methyl ester (134) was coupled to N-Fmoc-L-threonine in the presence of the BOP reagent in 95% yield. The N-Fmoc protecting group was removed with Et2NH and the corresponding free amine was coupled to N-alloc-(S-triphenylmethyl)-D-cysteine with 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDCI) and HOBT in DMF and CH2Cl2 to yield the tripeptide 135 in good yield. The threonine residue of tripeptide 135 was then subjected to dehydrating conditions to give alkene 136 in 95% yield. The N-alloc protecting group of the dehydrated tripeptide 136 was removed with palladium and tin reagents and the corresponding free amine was subsequently coupled with N-Fmoc-D-valine to give tetrapeptide 137 in 83% yield. After removal of the N-Fmoc protecting group of compound 137 with Et2NH amine 138 was obtained in quantitative yield. The acid coupling partner 145 for
amine 138 was prepared as follows: methyl 3,3-dimethoxypropionate (139) was converted to its corresponding Weinreb amide by standard conditions and reacted with lithium acetylide 140 to give propargylic ketone 141 in 75% yield. Noyori’s asymmetric reduction of ketone 141 using ruthenium catalyst 142 gave the (R)-propargylic alcohol in 98% ee. This was followed by Red-Al reduction of the alkyne to selectively yield (E)-alkene 143 in 58% yield for the two steps. Liberation of the primary alcohol
with tetrabutylammonium fluoride (TBAF) followed by selective tosylation gave 144 in 70% yield in two steps. Hydrolysis of the dimethyl acetal of 144 with LiBF4 was followed by a Pinnick oxidation to give the corresponding carboxylic acid. The tosylate was displaced with trityl mercaptan in the presence of tert-butyl alcohol to give allylic alcohol 145 in 65% yield for the three steps.
Aminoamide 138 was then coupled to acid 145 using BOP to give peptide 146 in quantitative yield. The methyl ester of compound 146 was hydrolyzed with lithium hydroxide to provide the free carboxylic acid which underwent macrolactonization under Mitsunobu conditions in the presence of diisopropyl azodicarboxylate (DIAD) and triphenylphosine to give macrocycle 147 in 24% yield.
Finally, the disulfide linkage was formed by treating bis-tritylsulfane 147 with iodine in methanol at room temperature to give romidepsin (XIII) in 81% yield.
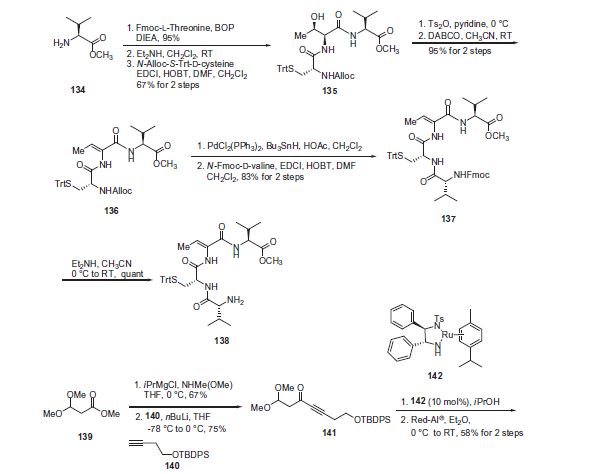
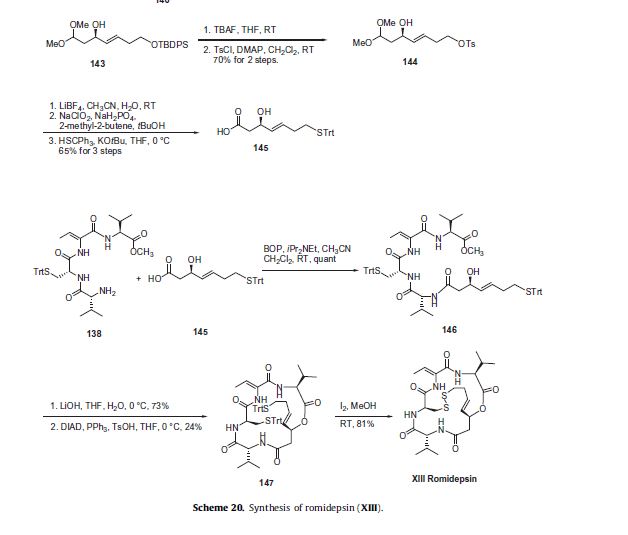
111 Bertino, E. M.; Otterson, G. A. Expert Opin. Invest. Drugs 2011, 20, 1151.
112. Furumai, R.; Matsuyama, A.; Kobashi, N.; Lee, K.-H.; Nishiyama, M.; Nakajima,
H.; Tanaka, A.; Komatsu, Y.; Nishino, N.; Yoshida, M.; Horinouchi, S. Cancer
Res. 2002, 62, 4916.
113. Verdine, G. L.; Vrolijk, N. H.; Bertel, S. WO 2008083288 A2, 2008.
114. Verdine, G. L.; Vrolijk, N. H. WO 2008083290 A1, 2008.
115. Greshock, T. J.; Johns, D. M.; Noguchi, Y.; Williams, R. M. Org. Lett. 2008, 10,
613.
116. Li, K. W.; Wu, J.; Xing, W.; Simon, J. A. J. Am. Chem. Soc. 1996, 118, 7237.
http://pubs.rsc.org/en/content/articlelanding/2009/np/b817886k#!divAbstract

Williams’ improved synthesis of FK228.


Williams’ synthesis of the FK228 amide isostere (74).
![Skeletal formula of (1S,4S,7Z,10S,16E,21R)-7-ethylidene-4,21-diisopropyl-2-oxa-12,13-dithia-5,8,20,23-tetrazabicyclo[8.7.6]tricos-16-ene-3,6,9,19,22-pentone](https://en.wikipedia.org/wiki/File:Romidepsin_structure_(2).svg) |
|
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
(1S,4S,7Z,10S,16E,21R)-7-ethylidene-4,21-diisopropyl-2-oxa-12,13-dithia-5,8,20,23-tetrazabicyclo[8.7.6]tricos-16-ene-3,6,9,19,22-pentone
|
|
| Clinical data | |
| Trade names | Istodax |
| MedlinePlus | a610005 |
| License data |
|
| Pregnancy category |
|
| Routes of administration |
Intravenous infusion |
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Bioavailability | Not applicable (IV only) |
| Protein binding | 92–94% |
| Metabolism | Hepatic (mostly CYP3A4-mediated) |
| Biological half-life | 3 hours |
| Identifiers | |
| CAS Number | 128517-07-7 |
| ATC code | none |
| PubChem | CID 5352062 |
| IUPHAR/BPS | 7006 |
| UNII | CX3T89XQBK |
| ChEBI | CHEBI:61080 |
| ChEMBL | CHEMBL1213490 |
| Synonyms | FK228; FR901228; Istodax |
| Chemical data | |
| Formula | C24H36N4O6S2 |
| Molar mass | 540.695 g/mol |
//////////fast-track designation, Romidepsin, Depsipeptide, FK228, Chromadax, FR901228, Istodax, FDA 2009, Fujisawa, Astellas Pharma, 128517-07-7
CC=C1C(=O)NC(C(=O)OC2CC(=O)NC(C(=O)NC(CSSCCC=C2)C(=O)N1)C(C)C)C(C)C
Movantik; NKTR-118; NKTR118; UNII-44T7335BKE; NKTR 118
854601-70-0 cas
1354744-91-4 (Naloxegol Oxalate)
(4R,4aS,7S,7aR,12bS)-7-[2-[2-[2-[2-[2-[2-(2-methoxyethoxy)ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]-3-prop-2-enyl-1,2,4,5,6,7,7a,13-octahydro-4,12-methanobenzofuro[3,2-e]isoquinoline-4a,9-diol
| MF | C34H53NO11 |
|---|---|
| MW | 651.78472 g/mol |
Naloxegol oxalate (MovantikTM, Moventig)
Naloxegol (INN; PEGylated naloxol;[1] trade names Movantik and Moventig) is a peripherally–selective opioid antagonistdeveloped by AstraZeneca, licensed from Nektar Therapeutics, for the treatment of opioid-induced constipation.[2] It was approved in 2014 in adult patients with chronic, non-cancer pain.[3] Doses of 25 mg were found safe and well tolerated for 52 weeks.[4] When given concomitantly with opioid analgesics, naloxegol reduced constipation-related side effects, while maintaining comparable levels of analgesia.[5]

Naloxegol Oxalate was approved by the U.S. Food and Drug Administration (FDA) on Sept 16, 2014, then approved by European Medicine Agency (EMA) on Dec 8, 2014. It was developed and marketed as Movantik®(in the US)/Moventig®(in EU) by AstraZeneca.
Naloxegol oxalate is an antagonist of opioid binding at the mu-opioid receptor. It is indicated for the treatment of opioid-induced constipation (OIC) in adult patients with chronic non-cancer pain.
Movantik® is available as tablets for oral use, containing 12.5 mg or 25 mg of free Naloxegol. The recommended dose is 25 mg once daily (reduce to 12.5 mg if not tolerated).
Chemically, naloxegol is a pegylated (polyethylene glycol-modified) derivative of α-naloxol. Specifically, the 5-α-hydroxyl group of α-naloxol is connected via an ether linkage to the free hydroxyl group of a monomethoxy-terminated n=7 oligomer of PEG, shown extending at the lower left of the molecule image at right. The “n=7” defines the number of two-carbon ethylenes, and so the chain length, of the attached PEG chain, and the “monomethoxy” indicates that the terminal hydroxyl group of the PEG is “capped” with amethyl group.[6] The pegylation of the 5-α-hydroxyl side chain of naloxol prevents the drug from crossing the blood-brain barrier(BBB).[5] As such, it can be considered the antithesis of the peripherally-acting opiate loperamide which is utilized as an opiate-targeting anti-diarrheal agent that does not cause traditional opiate side-effects due to its inability to accumulate in the central nervous system in normal subjects.
Naloxegol was previously a Schedule II drug in the United States because of its chemical similarity to opium alkaloids, but was recently reclassified as a prescription drug after the FDA concluded that the impermeability of the blood-brain barrier to this compound made it non-habit-forming, and so without the potential for abuse — specifically, naloxegol was officially decontrolled on 23. January 2015. [7]

As an opiate antagonist, it is not expected to be capable of inducing the euphoria and anxiolytic effects which are generally cited as the desirable effects of commonly abused opiates (all of which are opiate agonists) if it were to cross the BBB; it would in fact reverse the effects of opiate drugs of abuse if it entered the central nervous system.
Naloxegol is an oral polyethylene glycol (PEG) derivative of naloxone, a peripherally acting µ-opioid receptor antagonist (PAMORA) with limited potential for interfering with centrally mediated opioid analgesia. The incorporation of a polyethylene glycol moiety aims at inhibiting naloxone’s capacity to cross the blood-brain barrier, while preserving the affinity for the µ-opioid receptor [1].

Opioid-induced bowel dysfunction (OIBD) represents a broad spectrum of symptoms that result from the actions of opioids on the CNS as well as the gastrointestinal tract. The majority of gastrointestinal effects seem to be mediated by the high number of µ-receptors that are expressed in the enteric nervous system. Naloxegol was more effective than placebo in increasing the number of spontaneous bowel movements in patients with opioid-induced constipation, including those with an inadequate response to laxatives.
Recognition of Naloxegol as a useful option in the treatment of opioid-induced constipation resulted in its approval by US-FDA for adult patients with chronic, non-cancer pain in 2014.
Naloxegol oxalate (XXI) is a peripherally acting l-opioid receptor antagonist that was approved in the USA and EU for the treatment of opioid-induced constipation in adults with chronic non-cancer pain. The drug, a pegylated version of naloxone, has significantly reduced central nervous system (CNS) penetration and works by inhibiting the binding of opioids in the gastrointestinal tract.152–154 Naloxegol oxalate was developed by Nektar and licensed to AstraZeneca. Although we were unable to find a single report in the primary or patent literature that describes the exact experimental procedures to prepare naloxegol oxalate, there havebeen reports on the preparation of closely related analogs155 with specific reports on improving the selectivity of the reduction step156 and the salt formation of the final drug substance.157 Taken together, the likely synthesis of naloxegol oxalate (XXI) is
described in Scheme 28. Naloxone (180) was treated with methoxyethyl chloride in the presence of Hunig’s base to give the protected ketone 181. Reduction of the ketone with potassium trisec-butylborohydride exclusively provided the a-alcohol 182 in 85% yield. Alternatively, sodium trialkylborohydrides could also be used to provide similar a-selective reduction in high yield.
Deprotonation of the alcohol with sodium hydride followed by alkylation with CH3(OCH2CH2)7Br (183) provided the pegylated intermediate 184 in 88% yield. Acidic removal of the methoxyethyl ether protecting group followed by treatment with oxalic acid and crystallization provided naloxegol oxalate (XXI) in good yield.
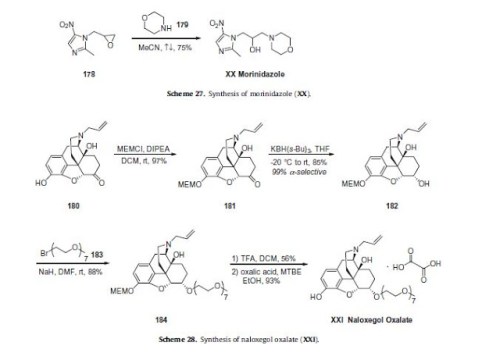
152. Corsetti, M.; Tack, J. Expert Opin. Pharmacol. 2015, 16, 399.
153. Garnock-Jones, K. P. Drugs 2015, 75, 419.
154. Leonard, J.; Baker, D. E. Ann. Pharmacother. 2015, 49, 360.
155. Bentley, M. D.; Viegas, T. X.; Goodin, R. R.; Cheng, L.; Zhao, X. US Patent
2005136031A1, 2005.
156. Cheng, L.; Bentley, M. D. WO Patent 2007124114A2, 2007.
157. Aaslund, B. L.; Aurell, C.-J.; Bohlin, M. H.; Sebhatu, T.; Ymen, B. I.; Healy, E. T.;
Jensen, D. R.; Jonaitis, D. T.; Parent, S. WO Patent 2012044243A1, 2012.
158. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm4183
Naloxegol Synthesis
CREDIT
https://ayurajan.blogspot.in/2016/08/naloxegol.html
US20050136031A1: The patent reports detailed synthetic procedures to manufacture gram quantities of Naloxegol. The synthesis starts with Naloxone which was treated with methoxyethyl chloride in the presence of Hunig’s base to give the protected ketone. Reduction of the ketone with potassium tri-sec-butylborohydride exclusively provided the α-alcohol in 85% yield. Deprotonation of the alcohol with sodium hydride followed by alkylation with CH3(OCH2CH2)7Br provided the pegylated Naloxone in 88% yield.
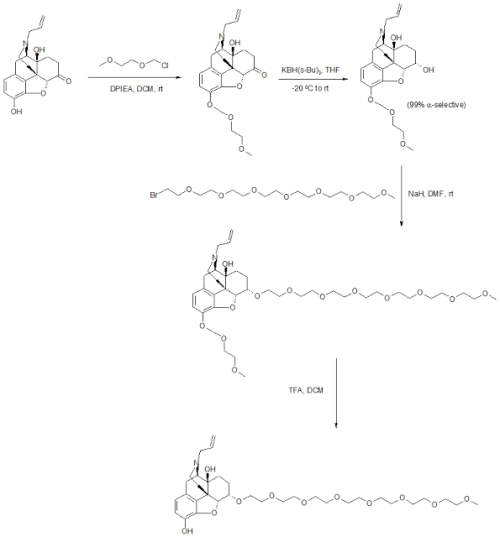
Identifications:

| 1H NMR (Estimated) for Naloxegol |


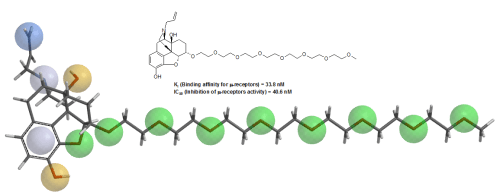

PATENT

PATENT
http://www.google.co.in/patents/WO2005058367A2?cl=en
EXAMPLE 4 SYNTHESIS OF PEG 3-NALθxoL [0211] The structure of the naloxol, an exemplary small molecule drug, is shown below.

Naloxol [0212] This molecule was prepared (having a protected hydroxyl group) as part of a larger synthetic scheme as described in Example 5.
EXAMPLE 5
[0213] α,β-PEGι-naloxol was prepared. The overview of the synthesis is provided below.

(3)

(4)
5.A. Synthesis of 3-MEM-naloxone
[0214] Diisopropylethylamine (390 mg, 3.0 mmole) was added to a solution of naloxone ■ HCl • 2H2O (200 mg, 0.50 mmole) in CH2C12 (10 mL) with stining. Methoxyethyl chloride (“MEMCl,” 250 mg, 2.0 mmole) was then added dropwise to the above solution. The solution was stined at room temperature under N2 overnight.
[0215] The crude product was analyzed by HPLC, which indicated that 3-
MEM-O-naloxone (1) was formed in 97% yield. Solvents were removed by rotary evaporation to yield a sticky oil.
5.B. Synthesis of α and β epimer mixture of 3-MEM-naloxoI (2)
[0216] 3 mL of 0.2 N NaOH was added to a solution of 3-MEM-naloxone
(1) (obtained from 5.A. above, and used without further purification) in 5mL of ethanol. To this was added a solution of NaBHLt (76 mg, 2.0 mmole) in water (1 mL) dropwise. The resulting solution was stined at room temperature for 5 hours. The ethanol was removed by rotary evaporation followed by addition of a solution of 0.1 N HCl solution to destroy excess NaBKj and adjust the pH to a value of 1. The solution was washed with CHC13 to remove excess methoxyethyl chloride and its derivatives (3 x 50 mL), followed by addition of K2OO3 to raise the pH of the solution to 8.0. The product was then extracted with CHC13 (3 x 50 mL) and dried over Na2SO4. The solvent was removed by evaporation to yield a colorless sticky solid (192 mg, 0.46 mmole, 92% isolated yield based on naloxone • HCl • 2H2O).
[0217] HPLC indicated that the product was an α and β epimer mixture of
3-MEM-naloxol (2).
5.C. Synthesis of α and β epimer mixture of 6-CH3-OCH2CH2-O-3-MEM- naloxol (3a).
[0218] NaH (60% in mineral oil, 55 mg, 1.38 mmole) was added into a solution of 6-hydroxyl-3-MEM-naloxol (2) (192 mg, 0.46 mmole) in dimethylformamide (“DMF,” 6 mL). The mixture was stined at room temperature under N2 for 15 minutes, followed by addition of 2-bromoethyl methyl ether (320 mg, 2.30 mmole) in DMF (1 mL). The solution was then stirred at room temperature under N2 for 3 hours.
[0219] HPLC analysis revealed formation of a mixture of α- and β-6-CH3-OCH2CH2-0-3-MEM-naloxol (3) in about 88% yield. DMF was removed by a rotary evaporation to yield a sticky white solid. The product was used for subsequent transformation without further purification.
5.D. Synthesis of α and β epimer mixture of 6-CH3-OCH2CH2-naloxoI (4)
[0220] Crude α- and β-6-CH3-OCH2CH2-O-3-MEM-naloxol (3) was dissolved in 5 mL of CH2C12 to form a cloudy solution, to which was added 5 mL of trifluoroacetic acid (“TFA”). The resultant solution was stined at room temperature for 4 hours. The reaction was determined to be complete based upon HPLC assay. CH2C12 was removed by a rotary evaporator, followed by addition of 10 mL of water. To this solution was added sufficient K2OO3 to destroy excess TFA and to adjust the pH to 8. The solution was then extracted with CHC13 (3 x 50 mL), and the extracts were combined and further extracted with 0.1 N HCl solution (3 x 50 mL). The pH of the recovered water phase was adjusted to a pH of 8 by addition of K2CO3>followed by further extraction with CHC13 (3 x 50 mL). The combined organic layer was then dried with Na2SO4. The solvents were removed to yield a colorless sticky solid.
[0221] The solid was purified by passage two times through a silica gel column (2 cm x 30 cm) using CHCl3/CH3OH (30:1) as the eluent to yield a sticky solid. The purified product was determined by 1H NMR to be a mixture of α- and β epimers of 6-CH3-OCH2CH2-naloxol (4) containing ca. 30% α epimer and ca. 70% β epimer [100 mg, 0.26 mmole, 56% isolated yield based on 6-hydroxyl-3-MEM- naloxol (2)].
[0222] 1H NMR (δ, ppm, CDC13): 6.50-6.73 (2 H, multiplet, aromatic proton of naloxol), 5.78 (1 H, multiplet, olefinic proton of naloxone), 5.17 (2 H, multiplet, olefinic protons of naloxol), 4.73 (1 H, doublet, C5 proton of α naloxol), 4.57 (1 H, doublet, C5 proton of β naloxol), 3.91 (1H, multiplet, C6 proton of naloxol), 3.51-3.75 (4 H, multiplet, PEG), 3.39 (3 H, singlet, methoxy protons of PEG, α epimer), 3.36 (3 H, singlet, methoxy protons of PEG, β epimer), 3.23 (1 H, multiplet, C6 proton of β naloxol), 1.46-3.22 (14 H, multiplet, protons of naloxol).
SYN 1

PATENT
https://www.google.com/patents/WO2012044243A1?cl=en
Naloxol-polyethylene glycol conjugates are provided herein in solid phosphate and oxalate salt forms. Methods of preparing the salt forms, and pharmaceutical compositions comprising the salt forms are also provided herein. ACKGROUND
Effective pain management therapy often calls for an opioid analgesic. In addition to the desired analgesic effect, however, certain undesirable side effects, such as bowel dysfunction, nausea, constipation, among others, can accompany the use of an opioid analgesic. Such side effects may be due to opioid receptors being present outside of the central nervous system, principally in the gastrointestinal tract. Clinical and preclinical studies support the use of mPEG7-0-naloxol, a conjugate of the opioid antagonist naloxol and polyethylene glycol, to counteract undesirable side effects associated with use of opioid analgesics. When administered orally to a patient mPEG7-0-naloxol largely does not cross the blood brain barrier into the central nervous system, and has minimal impact on opioid- induced analgesia. See, e.g., WO 2005/058367; WO 2008/057579; Webster et al., “NKTR-118 Significantly Reverses Opioid-Induced Constipation,” Poster 39, 20th AAPM Annual Clinical Meeting (Phoenix, AZ), October 10, 2009.
To move a drug candidate such as mPEG7-O-naloxol to a viable pharmaceutical product, it is important to understand whether the drug candidate has polymorphic forms, as well as the relative stability and interconversions of these forms under conditions likely to be encountered upon large-scale production, transportation, storage and pre-usage preparation. Solid forms of a drug substance are often desired for their convenience in formulating a drug product. No solid form of mPEG7-O-naloxol drug substance has been made available to date, which is currently manufactured and isolated as an oil in a free base form. Exactly how to accomplish this is often not obvious. For example the number of pharmaceutical products that are oxalate salts is limited. The free base form of mPEG7-0-naloxol has not been observed to form a crystalline phase even when cooled to -60 °C and has been observed to exist as a glass with a transition temperature of
approximately -45 °C. Furthermore, mPEG7-0-naloxol in its free base form can undergo oxidative degradation upon exposure to air. Care can be taken in handling the free base, for example, storing it under inert gas, to avoid its degradation. However, a solid form of mPEG7-0-naloxol, preferably one that is stable when kept exposed to air, is desired
a naloxol-polyethlyene glycol conjugate oxalate salt, the salt comprising ionic species of mPEG7-0-naloxol and oxalic acid. The formulas of mPEG7-0-naloxol and oxalic acid are as follows:

In certain embodiments, the methods provided comprise dissolving mPEG7-0- naloxol free base in ethanol; adding methyl t-butyl ether to the dissolved mPEG?–
O-naloxol solution; adding oxalic acid in methyl t-butyl ether to the dissolved mPEG7-0-naloxol over a period of at least 2 hours to produce a slurry; and filtering the slurry to yield the naloxol-polyethlyene glycol conjugate oxalate salt in solid form.
In certain embodiments, the methods provided comprise dissolving mPEG7-0- naloxol free base in acetonitrile; adding water to the dissolved mPEG7-0-naloxol solution; adding oxalic acid in ethyl acetate to the dissolved mPEG7-0-naloxol over a period of at least 2 hours to produce a slurry; and filtering the slurry to yield the naloxol-polyethlyene glycol conjugate oxalate salt in solid form.
In some embodiments, the solid salt form of mPEG7-0-naloxol is a crystalline form.
In certain embodiments a solid crystalline salt provided herein is substantially pure, having a purity of at least about 80%, at least about 85%, at least about 90%, at least about 92%, at least about 95%, at least about 96%, at least about 97%, at least about 98%, or at least about 99%.
In certain embodiments, the solid salt form of mPEG7-0-naloxol is a phosphate salt.
In other embodiments, the solid mPEG7-0-naloxol salt form is an oxalate salt. For instance, in some embodiments of solid oxalate salt forms provided herein, the solid mPEG7-0-naloxol oxalate salt form is in Form A, as described herein. As another example, in some embodiments of solid oxalate salt forms provided herein, the solid mPEG7-0-naloxol oxalate salt form is in Form B, as described herein. In yet other embodiments, an oxalate salt of mPEG7-0-naloxol in solid form prepared according to the methods described herein is provided.
In yet other embodiments, an dihydrogenphosphate salt of mPEG7-0-naloxol in solid form prepared according to the methods described herein is provided.
In certain embodiments of a solid mPEG7-0-naloxol oxalate salt Form B provided herein, the salt form exhibits a single endothermic peak on differential scanning calorimetry between room temperature and about 150 °C. The single endothermic peak can occur, for instance, between about 91 °C to about 94 °C. For example, in some embodiments the endothermic peak is at about 92 °C, about 92.5 °C, or about93 °C.
PATENT
http://www.google.co.in/patents/WO2005058367A2?cl=en
PATENT
PATENT
https://www.google.com/patents/CN102174049A?cl=en
1. WO2012044243A / US12015038524A1.
2. WO2005058367A2 / US7786133B2.
3. US20060182692A1 / US8067431B2.
4. CN101033228A.
5. Fudan Univ. J. Med. Sci. 2007, 34, 888-890.
| WO2008057579A2 * | Nov 7, 2007 | May 15, 2008 | Nektar Therapeutics Al, Corporation | Dosage forms and co-administration of an opioid agonist and an opioid antagonist |
| WO2009137086A1 * | May 7, 2009 | Nov 12, 2009 | Nektar Therapeutics | Oral administration of peripherally-acting opioid antagonists |
| US20050136031 * | Dec 16, 2004 | Jun 23, 2005 | Bentley Michael D. | Chemically modified small molecules |
| FDA Orange Book Patents: 1 of 6 | |
|---|---|
| Patent | 7056500 |
| Expiration | Jun 29, 2024 |
| Applicant | ASTRAZENECA PHARMS |
| Drug Application | N204760 (Prescription Drug: MOVANTIK. Ingredients: NALOXEGOL OXALATE) |
| FDA Orange Book Patents: 2 of 6 | |
|---|---|
| Patent | 7662365 |
| Expiration | Oct 18, 2022 |
| Applicant | ASTRAZENECA PHARMS |
| Drug Application | N204760 (Prescription Drug: MOVANTIK. Ingredients: NALOXEGOL OXALATE) |
| FDA Orange Book Patents: 3 of 6 | |
|---|---|
| Patent | 8617530 |
| Expiration | Oct 18, 2022 |
| Applicant | ASTRAZENECA PHARMS |
| Drug Application | N204760 (Prescription Drug: MOVANTIK. Ingredients: NALOXEGOL OXALATE) |
| FDA Orange Book Patents: 4 of 6 | |
|---|---|
| Patent | 9012469 |
| Expiration | Apr 2, 2032 |
| Applicant | ASTRAZENECA PHARMS |
| Drug Application | N204760 (Prescription Drug: MOVANTIK. Ingredients: NALOXEGOL OXALATE) |
| FDA Orange Book Patents: 5 of 6 | |
|---|---|
| Patent | 7786133 |
| Expiration | Dec 19, 2027 |
| Applicant | ASTRAZENECA PHARMS |
| Drug Application | N204760 (Prescription Drug: MOVANTIK. Ingredients: NALOXEGOL OXALATE) |
| FDA Orange Book Patents: 6 of 6 | |
|---|---|
| Patent | 8067431 |
| Expiration | Dec 16, 2024 |
| Applicant | ASTRAZENECA PHARMS |
| Drug Application | N204760 (Prescription Drug: MOVANTIK. Ingredients: NALOXEGOL OXALATE) |
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
(5α,6α)-4,5-epoxy-6-(3,6,9,12,15,18,21-heptaoxadocos-1-yloxy)-17-(2-propen-1-yl)morphinan-3,14-diol
|
|
| Clinical data | |
| Trade names | Movantik, Moventig |
| AHFS/Drugs.com | movantik |
| License data | |
| Pregnancy category |
|
| Routes of administration |
Oral |
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Protein binding | ~4.2% |
| Metabolism | Hepatic (CYP3A) |
| Biological half-life | 6–11 h |
| Excretion | Feces (68%), urine (16%) |
| Identifiers | |
| CAS Number | 854601-70-0 |
| ATC code | A06AH03 (WHO) |
| PubChem | CID 56959087 |
| ChemSpider | 28651656 |
| ChEBI | CHEBI:82975 |
| Synonyms | NKTR-118 |
| Chemical data | |
| Formula | C34H53NO11 |
| Molar mass | 651.785 g/mol |
//////////////Naloxegol, Movantik, NKTR-118, NKTR118, UNII-44T7335BKE, NKTR 118, 854601-70-0, EU 2014, FDA 2014
COCCOCCOCCOCCOCCOCCOCCO[C@H]1CC[C@]2([C@H]3Cc4ccc(c5c4[C@]2([C@H]1O5)CCN3CC=C)O)O
Naloxegol oxalate (MovantikTM, Moventig)
Naloxegol oxalate (XXI) is a peripherally acting l-opioid receptor antagonist that was approved in the USA and EU for the treatment of opioid-induced constipation in adults with chronic non-cancer pain. The drug, a pegylated version of naloxone, has significantly reduced central nervous system (CNS) penetration and works by inhibiting the binding of opioids in the gastrointestinal tract.152–154 Naloxegol oxalate was developed by Nektar and licensed to AstraZeneca. Although we were unable to find a single report in the primary or patent literature that describes the exact experimental procedures to prepare naloxegol oxalate, there have been reports on the preparation of closely related analogs155 with specific reports on improving the selectivity of the reduction step156 and the salt formation of the final drug substance.157 Taken together, the likely synthesis of naloxegol oxalate (XXI) is described in Scheme 28. Naloxone (180) was treated with methoxyethyl chloride in the presence of Hunig’s base to give the protected ketone 181. Reduction of the ketone with potassium trisec-butylborohydride exclusively provided the a-alcohol 182 in 85% yield. Alternatively, sodium trialkylborohydrides could also be used to provide similar a-selective reduction in high yield.
Deprotonation of the alcohol with sodium hydride followed by alkylation with CH3(OCH2CH2)7Br (183) provided the pegylated intermediate 184 in 88% yield. Acidic removal of the methoxyethyl ether protecting group followed by treatment with oxalic acid and crystallization provided naloxegol oxalate (XXI) in good yield.

152. Corsetti, M.; Tack, J. Expert Opin. Pharmacol. 2015, 16, 399.
153. Garnock-Jones, K. P. Drugs 2015, 75, 419.
154. Leonard, J.; Baker, D. E. Ann. Pharmacother. 2015, 49, 360.
155. Bentley, M. D.; Viegas, T. X.; Goodin, R. R.; Cheng, L.; Zhao, X. US Patent
2005136031A1, 2005.
156. Cheng, L.; Bentley, M. D. WO Patent 2007124114A2, 2007.
157. Aaslund, B. L.; Aurell, C.-J.; Bohlin, M. H.; Sebhatu, T.; Ymen, B. I.; Healy, E. T.;
Jensen, D. R.; Jonaitis, D. T.; Parent, S. WO Patent 2012044243A1, 2012.

Opicapone
2,5-dichloro-3-(5-(3,4-dihydroxy-5-nitrophenyl)-1,2,4-oxadiazol-3-yl)-4,6-dimethylpyridine-1-oxide
BIA-9-1067; ONO-2370; BIA-91067
CAS No.923287-50-7
MF C15H10Cl2N4O6
MW: 411.9977
TRADE NAME (Ongentys®)
Approved EU 2016-06-24 BIAL PORTELA
| PORTELA & CA. S.A. [PT/PT]; Av. Da Siderurgia Nacional, P-4745-457 S. Mamede do Coronado (PT) |
LEARMONTH, David Alexander; (PT).
KISS, Laszlo Erno; (PT).
LEAL PALMA, Pedro Nuno; (PT).
DOS SANTOS FERREIRA, Humberto; (PT).
ARAÚJO SOARES DA SILVA, Patrício Manuel Vieira; (PT)
MOA:Catechol-O-methyl transferase (COMT) inhibitor
Indication:Parkinson’s disease (PD)
A COMT inhibitor used as adjunctive therapy for parkinson’s disease.

Opicapone, also known as BIA 9-1067, is a novel potent and selective catechol-O-methyltransferase inhibitor (COMT inhibitor ) under clinical evaluation as an adjunct to L-Dopa therapy of Parkinson’s disease. Opicapone, a novel third generation COMT inhibitor, when compared to entacapone, provides a superior response upon the bioavailability of levodopa associated to more pronounced, long-lasting, and sustained COMT inhibition
Opicapone was approved by European Medicine Agency (EMA) on Jun 24, 2016. It was developed and marketed as Ongentys® by Bial – Portela in EU.
Opicapone is a selective and reversible COMT inhibitor, used as adjunctive therapy for Parkinson’s disease.
Ongentys® is available as hard capsules, containing 25 mg and 50 mg of opicapone. The recommended dose is 50 mg, taken once a day at bedtime, at least one hour before or after levodopa combination medicines.
Catechol-O-methyltransferase (COMTa) catalyzes the transfer of a methyl group from S-adenosyl-l-methionine to catecholic substrates such as endogenous catechol neurotransmitters(2)and xenobiotics including (S)-2-amino-3-(3,4-dihydroxyphenyl)propanoic acid (l-Dopa), the gold standard drug for treatment of Parkinson’s disease (PD). Coadministration of a peripheral amino acid decarboxylase (AADC) inhibitor prevents breakdown of l-Dopa in the periphery by blocking enzymatic decarboxylation, and inhibition of COMT further improves its bioavailability by reducing the formation of 3-O-methyl-l-Dopa (3-OMD).
Abbreviations: COMT, catechol-O-methyltransferase; PD, Parkinson’s disease; AADC, amino acid decarboxylase; SAR, structure−activity relationship; ADMET, absorption, distribution, metabolism, excretion, toxicity; l-Dopa, (S)-2-amino-3-(3,4-dihydroxyphenyl)propanoic acid; 3-OMD, 3-O-methyl-l-Dopa.
![]()

Chemical structures of tolcapone 1, entacapone 2, and nebicapone 3.
Opicapone
A preferred method of treatment of Parkinson’s disease is the administration of a combination of levodopa and a peripherally selective aromatic amino acid decarboxylase inhibitor (AADCI) together with a catechol-O-methyltransferase (COMT) inhibitor. The currently employed COMT inhibitors are tolcapone and entacapone. However, some authorities believe that each of these COMT inhibitors have residual problems relating to pharmacokinetic or pharmacodynamic properties, or to clinical efficiency or safety. Hence, not all patients get most benefit from their levodopa/AADCI/COMT inhibitor therapy.
Favoured new COMT inhibitors were disclosed in L. E. Kiss et al, J. Med. Chem., 2010, 53, 3396-3411 (D1), WO 2007/013830 (D2) and WO 2007/117165 (D3) which are believed to have particularly desirable properties so that patients can benefit from enhanced therapy.
D1, D2 and D3 also disclosed methods of preparing the new COMT inhibitors. Those processes, although effective, would benefit from an increase in yields. Other benefits which would be appropriate include those selected from reduction in number of process steps, reduction in number of unit operations, reduction of cycle-times, increased space yield, increased safety, easier to handle reagents/reactants and/or increase in purity of the COMT inhibitor, especially when manufacture of larger quantities are envisaged. A process has now been discovered that proceeds via a new intermediate which is suitable for manufacture of commercially useful quantities of a particularly apt COMT inhibitor in good yield. Additional benefits occur such as those selected from a reduced number of process steps and number of unit operations, reduced cycle-times, increased space yield, increased safety, with easier to handle reagents/reactants, improved impurity profile and/or good purity.
https://clinicaltrials.gov/show/NCT01851850




PATENT
LEARMONTH, David Alexander; (PT).
KISS, Laszlo Erno; (PT).
LEAL PALMA, Pedro Nuno; (PT).
DOS SANTOS FERREIRA, Humberto; (PT).
ARAÚJO SOARES DA SILVA, Patrício Manuel Vieira; (PT)
The present invention in one aspect provides 5-[3-(2,5-dichloro-4,6-dimethyl-1-oxy-pyridin-3-yl)-[1,2,4,oxadiazol-5-yl]-2-hydroxy-3-methoxy-1-nitrobenzene and salts thereof, that is the compound of the formula (I):

and salts thereof.
Most aptly the compound of formula (I) is unsalted. However, salts of the hydroxy group with metal ions such as the alkali or alkaline earth metals, particularly the sodium and potassium salts are provided as well as those of highly basic organic compounds such as guanidine or the like.
Particularly suitably the compound of formula (I) or its salt is provided in a form suitable for use as a chemical intermediate. This may be, for example, in a form at least 50% pure, in crystalline form, in solid form or in an organic solvent or the like.
The compound of formula (I) is useful as an intermediate in the preparation of 5-[3-(2,5-dichloro-4,6-dimethyl-1-oxy-pyridin-3-yl)-[1,2,4]oxadiazol-5-yl]-3-nitrobenzene-1,2-diol i.e. the compound of formula II):

The compound of formula (II) may also be referred to as opicapone or 2,5-dichloro-3-(5-(3,4-dihydroxy-5-nitrophenyl)-[1,2,4]-oxadiazole-3-yl)-4,6-dimethylpyridine-1-oxide. Opicapone has been found to be more potent than tolcapone in inhibiting liver COMT both at 3 hours and 6 hours post oral administration to rats [ED50 in mg/kg, opicapone 0.87 at 3 hours and 1.12 at 6 hours as compared to tolcapone 1.28 at 3 hours and 2.08 at 6 hours]. Opicapone at a dose of 3 mg/kg was found to be more effective at inhibiting rat liver COMT with nearly complete inhibition occurring 2 to 6 hours post oral administration with only about 90% of enzyme activity recovered after 72 hours while tolcapone provided shorter duration of activity with about 84% recovery after only 9 hours. Both opicapone and tolcapone inhibit human recombinant S-COMT but opicapone has an inhibitory constant of 16pM being 10 fold lower than that for tolcapone. With respect to the desirable property of avoiding inhibition of COMT in the brain, opicapone following oral administration to the rat was found to be devoid of effect whereas tolcapone inhibited about 50% of enzyme activity over a period of 8 hours post administration.
Preparation 1

Cyanoacetamide (280g) was reacted with acetyl acetone (352.9g) in methanol (1015g) and morpholine (14.9g). The reaction was stirred under reflux at 65 °C until the reaction appeared complete. The resulting product suspension was filtered, washed with methanol and dried to provide the desired product about 97% yield.
Preparation 2

The product of Preparation 1 (159g) was suspended in acetonitrile (749.5g) and cooled to 0-5°C. Sulfuryl chloride (178.9g) was added and the reaction mixture warmed to room temperature and stirred until the reaction appeared complete.
The resulting suspension is cooled to 0-5°C and filtered. The solid was washed with acetonitrile, ethyl acetate and heptane. The product was then dried under vacuum at 50°C to yield the desired product (82%).
Preparation 3

Phosphoryl chloride (973.2g), tetramethylammonium chloride (67.3g) and compound of Preparation 2 (227.1g) were added to dichloromethane (500g). The suspension was heated to 85°C and stirred for 5 hours. Excess of phosphoryl chloride was removed by distillation in vacuo. The reaction mixture was cooled below 30°C and diluted with dichloromethane. The resulting solution was added to water (1350g) at room temperature and stirred for 30 minutes. The lower organic phase was separate and the aqueous phase extracted with dichloromethane. The organic phases were combined, washed with water and then treated with charcoal. The charcoal was filtered and a solvent swap to heptane was performed by distillation at atmospheric pressure. The solution was filtered at 50°C and then cooled to 30°C. On further cooling to 0°C
crystals were obtained. These were isolated by filtration, washed twice with heptane. After drying at 50°C the desired product was obtained typically at 88-91 % .
The above process was repeated with a reduction in dichloromethane during crystallisation and adding some methanol. The resulting plate-like crystals were more easily transferred for subsequent use.
Preparation 4a

Product of Preparation 3 (68.6g) and 1,10-phenanthroline monohydrate (0.9g) were suspended in methanol (240g) at room temperature. Water (518g) and a hydroxylamine solution (50% in water, 80.9g), were added and the mixture heated to 70-80°C and stirred for 5-6 hours. Water was added at 70-80 °C and the solution held for 1 hour to induce crystallization. Crystallization was completed by cooling to 15°C over 8 hours. The product was filtered off and washed twice with water and dried at 50°C under vacuum. The product was an off white to light yellow and the yield was 87.9% .
Preparation 4b

A suspension of 2,5-Dichloro-4,6-dimethyl-nicotinonitrile (45.0 kg) and 50% hydroxylamine (59.2 kg) in the presence of catalytic amount of 1,10-phenanthroline monohydrate (0.680 kg) in methanol / water (214 kg/362 kg) is heated to 70-80°C. The mixture is agitated at 70-80°C. Water (353 kg) is added slowly into the resulting solution while the temperature is maintained at > 79°C. The solution is cooled to 75 °C with stirring resulting in crystallization of (Z)-2,5-dichloro-N’-hydroxy-4,6-
dimethylnicotinimidamide. The suspension is further cooled to 20 °C, the solid is filtered off and the wet cake is washed with water (160 kg). (Z)-2,5-dichloro-N’-hydroxy-4,6-dimethylnicotinimidamide is dried under vacuum at max. 60°C until residual water level is max 0.15% (KF).
Example 1a
Preparation of 4-hydroxy-5-methoxy-3 nitrobenzoic acid

Vanillic acid (75g) was suspended in acetic acid (788g). The suspension was cooled to 10°C to 15°C and nitric acid (49g or 65% solution) was added over three hours at a rate which kept temperature between 10°C and 20°C. The resulting yellow orange was stirred for a further one hour at 18°C to 23°C. The suspension was filtered off, washed with acetic acid, then a mixture of acetic acid and water (1/2) and then water. Yield of 53% of a 87.9% pure product was obtained.
The above crude product was suspended in acetic acid and warmed to 105°C to 110°C until an orange brown solution is obtained. The solution was transferred to the crystallization vessel via a charcoal filter (or polish filtration) at a temperature above 85°C (optional step). The solution was then cooled to 80°C to 85°C. The mixture was stirred for one hour at 70°C to 80°C (optionally at 75°C) during which crystallization occurred. The product suspension was cooled to 20°C to 25°C for 17 hours or stirred for at least 12h at 20°C to 25 °C. The product suspension was filtered and washed with acetic acid, then acetic acid/ water (1/2) and finally water. The product was dried under vacuum at 50°C to 55°C. The yield of 70% corresponds to an overall yield of 44% for both parts of this preparation. The purity of the product assayed at 99.7% .
The preceding crystallization step is optional and the solution may be transferred to the crystallization vessel via polish filtration instead of via a charcoal filter.
The post crystallization suspension may be stirred for at least 12 hours at 20° C to 25 °C as an alternative to 17 hours.
Example 1b
Preparation of 4-hydroxy-5-methoxy-3 nitrobenzoic acid
A reactor was charged with 525 kg of glacial acetic acid and 50 kg vanillic acid. The mixture was heated with warm water gradually to 50°C in around 75 minutes. Temperature was set to 16°C. Nitric acid, 31.4 kg was then added gradually over a period of 3 hrs. When the administration was complete the mixture was allowed to stir for additional 3.5-4.5 hours.
The suspension was centrifuged whilst washed with 25 kg of acetic acid, 50 liter deionised water and 25 kg of acetic acid again. The wet crystalline material was suspended in 165 kg of acetic acid and heated at 91°C until complete dissolution. The solution was then cooled to 19.8°C and the mixture was allowed to stir for 1 hr. Centrifugation and washing with 15.2 kg acetic and 40 liter of deionised water was performed. The wet material was then dried in tray vacuum drier between 40-50°C until constant weight, for 72 hours. The dry material weight was 28.7 kg. The calculated yield was 45.4%.
Example 1c
Preparation of 4-h droxy-5-methoxy-3 nitrobenzoic acid

A suspension of vanillic acid (68.8 kg) in acetic acid (720 kg) is cooled to 17°C before an excess of a 65% nitric acid (44.0 kg) is added. After complete dosage of nitric acid the suspension is stirred for 2 hours. The suspension is filtered off and the wet cake is successively washed with acetic acid (80.0 kg), acetic acid/water (1:2 w/w – 105 kg) and finally water (80 kg – if necessary repeat). The solid is dried at 52°C for NMT 12 hours prior going to next step.
A suspension of the crude solid (650 kg) in acetic acid is warmed to 105 °C and stirred until complete dissolution of the crude solid. After polish filtration, the solution is cooled to 20°C over 3h resulting in crystallization and the suspension is stirred for 2h at 20°C. The solid is filtered off and the wet cake is successively washed with acetic acid (80 kg), acetic acid/water (1:2 w/w – 105 kg) and finally water (193 kg – if necessary repeat). 4-hydroxy-5-methoxy-3 nitrobenzoic acid pure is dried under vacuum at max. 55 °C until max 0.5% w/w residual acetic acid and max 0.2% w/w water is reached.
Example 2a
Preparation of 4-hydroxy-5-methoxy-3-nitrobenzoic acid
The process of Example la was scaled up to employ vanillic acid (375g) in acetic acid (3940g) to which was added nitric acid (65%, 245g) at 12°C over 3 hours followed by stirring for one hour. The overall yield was 40% of a 99.9% pure product.
Example 2b
Preparation of 4-hydroxy-5-methoxy-3-nitrobenzoic acid

Vanillic acid methyl ester (33g) and sodium nitrite (0.625g) are charged. Water (158mL) and 1,4-dioxane (158mL) are added at room temperature. The reaction mixture is heated to 40 °C. Nitric acid (65%) (15.75g) is added in the course of three hours and the resulting mixture is stirred for 4h after addition. The reaction mixture is sampled for completion.
The water/nitric-acid/dioxane azeotrope is distilled off in vacuum at 40 °C. The resulting product suspension is quenched by addition of sodium hydroxide solution (50% , 33.2 mL) and then stirred for 16h. The quench mixture is sampled for completion.
Then, HCl (18,5%, 70.2mL.) is added until the pH is below 1. The product is filtered off and washed with water (27.9mL). The product is then dried in vacuum at 50 °C. The overall yield was 81 % of a 97.3 % pure product.
Example 3a
Preparation of 4-hydroxy-5-methoxy-3-nitrobenzoyl chloride

A suspension of compound of Example la (1.0 eq) in dioxane (approx 4.5 vol) was treated with thionyl chloride (1.5 eq) and heated to 80°C. A clear solution formed at approximately 75 °C. The mixture was stirred for 3 hours at 80°C. Unreacted thionyl chloride was distilled off and after distillation the residue was cooled to 10°C.
Example 3 b
Preparation of 4-hydroxy-5-methoxy-3-nitrobenzoyl chloride
A suspension of compound of Example la (1.0 eq) in DCM (approx 3.4 vol) is treated with thionyl chloride (1.0 – 1.2 eq, for example 1.1 eq) and catalytic amount (0.011 eq) of DMF and the mixture is stirred for 16 h at 40°C. DCM is distilled off (approx 2.7 vol) and the residue is diluted with THF (approx 1.8 vol). The excess of thionylchloride is distilled off with THF/DCM and the residue after distillation is cooled to 10°C.
Example 3c
Preparation of 4-hydroxy-5-methoxy-3-nitrobenzoyl chloride
A suspension of compound of Example la (1.0 eq) in DCM (approx 4.5 vol) is treated with thionyl chloride (1.0 – 1.2 eq, for example 1.1 eq) and catalytic amount (0.0055 eq) of DMF and the mixture is stirred for 16 h at reflux. Unreacted thionylchloride is distilled off with DCM and the residue after distillation is diluted with THF (approx 1.8 vol) and cooled to 10°C.
The amount of DCM may be approx 3.4 as an alternative to approx 4.5 vol.
The catalytic amount of DMF may be about 0.011 eq as an alternative to 0.0055 eq.
Example 3d
Preparation of 4-hydroxy-5-methoxy-3-nitrobenzoyl chloride
In a reactor 68 kg dichloromethane, 20 kg 5-nitro- vanillic acid of example 1b, 76 gram of N,N-dimethylformamide and 13.4 kg (8 L) thionyl chloride, was charged at 20.2°C.
The mixture was heated to 40°C until all the starting material dissolved and the evolution of HCl and SO2 stopped. When all the starting material was consumed 5-10 L dichloromethane was distilled off at normal pressure at 40°C then the mixture was cooled to 20-25 °C and the distillation was continued until dry under vacuum at 40°C.
The evaporation residue was dissolved in 36 kg dry THF. The THF solution was used in
Example 4d.
Example 3e
Preparation of 4-hydroxy-5-methoxy-3-nitrobenzoyl chloride
A suspension of product of example 1C (4-hydroxy-5-methoxy-3 nitrobenzoic acid -160g, 1eq) in 1,4-dioxane (720mL, 4.5vol) is treated with thionyl chloride (169.8g, 103.7mL,1.5eq) and heated to 80°C. A clear solution is formed at approx. 75 °C. The mixture is stirred at 80 °C (3 hours). Unreacted thionyl chloride is distilled off and the residue after distillation is cooled to 10°C.
Example 4a
Preparation of 5-[3-(2,5-dichloro-4,6-dimethyl-pyridin-3-yl)-[1,2,4]oxadiazol-5-yl]-2-hydroxy-3-methoxy-1-nitrobenzene
In this example the compound of formula (IV) is reacted with the compound of formula (V) to produce the compound of the formula (III).



Compound of formula (V) (1.24 eq) was suspended in 1,4-dioxane (approximately 4.5 vol) and the suspension cooled to 10°C. The acyl chloride (compound of formula (IV)) solution of Example 3a in 1,4-dioxane was added slowly maintaining the temperature below 20°C. A clear orange solution was formed. After complete addition, the reaction mixture was stirred at 20°C for one hour. Pyridine (approximately 8eq) was added and the reaction mixture heated slowly to 115°C. The mixture was stirred for 6 hours at 115°C and then cooled to 20°C.
The dioxane/pyridine was distilled off under vacuum at 70°C. The residue was kept at 80°C and ethanol (approx 8 vol) added to induce crystallization. The resulting yellow suspension was cooled to 0°C and stirred for two hours. The product was filtered off and washed with ethanol (2.5 vol) water (3.8 vol) and ethanol 2.5 vol). The product was dried under vacuum at 50 °C. Typical yields for this process are 82 to 85%.
In an optional variant, methanol was employed in place of ethanol to induce crystallization.
Example 4b
Preparation of 5-[3-(2,5-dichloro-4,6-dimethyl-pyridin-3-yl)-[1,2,4]oxadiazol-5-yl]-2-hydroxy-3-methoxy-1-nitrobenzene
In a different reactor, compound of formula (V) (1.1 eq) is dissolved in DM Ac (approx 5.8 vol) and the solution is cooled to 5°C. The benzoyl chloride solution of Example 3b in THF/DCM is then added slowly maintaining the temperature below 10°C. After complete addition, the reaction mixture is stirred at 20 ±5°C. Pyridine (1.3 to 1.6 eq, for example 1.5 eq) is charged and the reaction mixture is heated slowly to 110±5°C removing low boiling components by distillation. The mixture is stirred for additional 3 h at 110±5°C.
In a further reactor, concentrated HCl (23.8 eq) is diluted with water (approx. 8.5 vol) and cooled to 10 °C. The reaction mixture in pyridine is dosed slowly to diluted hydrochloric acid. After complete addition, the resulting suspension is stirred for additional 2 h and the solid is filtered off. The crude solid is washed once with water and pre-dried on funnel.
The crude solid is suspended in DCM (approx. 28.6 vol) and the suspension is heated to 40°C to reach a clear solution. Resulting solution is cooled to 20°C and extracted with water. After phase separation, the aqueous phase is re-extracted with DCM and combined organic phase are washed once with water. DCM is distilled off under vacuum followed by addition of ethanol. Resulting suspension is further distilled to reduce the amount of DCM, then cooled to 5°C and stirred for additional 2 h. Finally, the product is filtered off, washed once with cold ethanol and dried under vacuum at 45°C.
Example 4c
Preparation of 5-[3-(2,5-dichloro-4,6-dimethyl-pyridin-3-yl)-[1,2,4]oxadiazol-5-yl]-2-hydroxy-3-methoxy-1-nitrobenzene
In a second reactor, compound of formula (V) (1.1 eq) is dissolved in DMAc (approx. 7 vol) and the solution is cooled to 5°C. The benzoyl chloride solution of Example 3c in THF/DCM is added slowly maintaining the temperature below 10 °C. After complete addition, the reaction mixture is stirred at 20 ± 5°C for 30 min. Pyridine (6.9 to 7.3 eq, for example 7.14 eq) is charged and the reaction mixture is heated slowly to 110°C removing low boiling components by distillation. The mixture is stirred for additional 4 h at 110°C and cooled to 20°C.
In a third reactor an emulsion of diluted hydrochloric acid (prepared from cone. HCl (19.6 eq) and approx. 7.6 vol distilled water) and DCM (approx. 25.5 vol) is cooled to about 15 °C before the reaction mixture in pyridine is dosed slowly to the emulsion. After complete addition, the organic phase is separated and washed with water before DCM is distilled off under vacuum followed by addition of ethanol. The resulting suspension is further distilled to reduce the amount of DCM, then cooled to 5°C and stirred for additional 2 h.
Finally, the product is filtered off, washed once with cold ethanol and dried under vacuum at 45 °C.
Example 4d
Preparation of 5-[3-(2,5-dichloro-4,6-dimethyl-pyridin-3-yl)-[1,2,4]oxadiazol-5-yl]-2-hydroxy-3-methoxy-1-nitrobenzene
140 kg Ν,Ν-dimethyl acetamide was charged into the reactor. 24.2 kg of amidoxime of Preparation 4 was dissolved in N,N-dimethyl acetamide while stirring at 21°C. The solution was cooled to 5-10°C. The THF solution of Example 3d was introduced slowly into the reaction mixture, 1.5-2 hrs, while the internal temperature was maintained at max. 9.5°C by external cooling. When the addition was complete the external cooling
was stopped. The internal temperature was allowed to raise to 21 °C in an hour. After stirring for 30 minutes, pyridine 53.0 kg was added to the mixture, while the temperature was in the range of 22.4°C – 20.6°C. Heating was started and the internal temperature raised to 105-115°C. The mixture started to reflux for 3h while the internal temperature managed to 113°C by partial distillation of some THF. The reaction mixture was then cooled and introduced to a mixture of 220 kg concentrated HCl and 170 kg of deionised water while the internal temperature was maintained between 14-16°C. The reactor was rinsed with 10 kg of Ν,Ν-dimethylacetamide and 20 kg deionised water. The rinse liquid was run to the mixture. The suspension was then further cooled to 5-10°C and stirred for 1.5-2.0 hours. The product was centrifuged and was washed 80 kg deionised water. Crude wet weight of the product was 88.6 kg.
The crude wet product, was dissolved in 460 kg (340 L) dichloromethane at max 40°C. When dissolved the temperature was set to 20-30°C and 120 kg deionised water was added. The organic phase was separated, the inorganic phase was extracted with 80 kg dichloromethane. The organic phase of 460 kg, was then washed with 200 kg deionised water and the phases were separated. The inorganic phase was extracted with the 80 kg dichloromethane and the organic phases were unified. The organic phase obtained so was concentrated in vacuum at 35°C to 200-240 Liter, then 260 kg ethanol 96% was continuously added and the evaporation was continued to a final 200-240 liter volume. Then the mixture was cooled to 5-10°C and was allowed to stir for 3 hrs. Centrifuging, washing with 20 kg ethanol resulted in 35.4 kg wet product. Vacuum drying for 16 hours at 45°C gave 34.09 kg dry product. The yield was 79.9%.
Example 4e
Preparation of 5-[3-(2,5-dichloro-4,6-dimethyl-pyridin-3-yl)-[1,2,4]oxadiazol-5-yl]-2-hydroxy-3-methoxy-1-nitrobenzene
In a second vessel, (Z)-2,5-dichloro-N’-hydroxy-4,6-dimethylnicotiriimidamide (201.2g, 1.24eq) is suspended in 1,4-dioxane (720mL, 4.5vol) and the suspension is cooled to 10°C. The residue of example 3e in 1,4-dioxane is added slowly maintaining the temperature below 20°C. A clear orange solution is formed. After complete addition, the reaction mixture is stirred at 20°C for 1 hour. Pyridine (483.7mL, 8eq) is then charged and the reaction mixture is heated slowly to 115°C. The mixture is stirred at 115°C for 6 hours. The solution is then cooled to 20°C. Dioxane/pyridine is distilled off.
After distillation, the pit is kept at 80 °C and ethanol (1.28L, 8vol) is added at this temperature to induce crystallization. The resulting yellow suspension is cooled to 75 °C and stirred for 1h at this temperature to allow crystal growth. The product suspension is then cooled to 0 °C and stirred for 2h at this temperature. The product is filtered off and washed subsequently with ethanol (400mL, 2.5vol), water (608mL, 3.8vol) and ethanol (400mL, 2.5vol). The product is dried under vacuum at 50°C until LOD is max 1% w/w.
Example 4f
Preparation of 5-[3-(2,5-dichloro-4,6-dimethyl-pyridin-3-yl)-[1,2,4]oxadiazol-5-yl]-2-hydroxy-3-methoxy-1-nitrobenzene
A mixture of compound of formula (V) (11.7g, 50 mmol, 1.25eq), methyl 4-hydroxy-3-methoxy-5-nitrobenzoate (10g, 40 mmol, leq) and a catalytic amount of p-toluenesulfonic acid (0.76g, 4mmol, 0.1eq) in dimethyl acetamide was heated to 80°C. The reaction was followed by HPLC. After 23h, 6% of conversion was obtained.
Example 4g
Preparation of 5-[3-(2,5-dichloro-4,6-dimethyl-pyridin-3-yl)-[1,2,4]oxadiazol-5-y1]-2-hydroxy-3-methoxy-1-nitrobenzene
A mixture of compound of formula (V) (11.7g, 50 mmol, 1.25eq), methyl 4-hydroxy-3-methoxy-5-nitrobenzoate (10g, 40 mmol, 1eq) and a catalytic amount of aluminum chloride (0.53g, 4mmol, 0.1eq) in dimethyl acetamide was heated to 80°C. The reaction was followed by HPLC. After 20h, 10% of conversion was obtained.
Example 5a
Preparation of 5-[3-(2,5-dichloro-4,6-dimethyl-1-oxy-pyridin-3-yl)-[1,2,4]oxadiazol-5-yl]-2-hydroxy-3-methoxy-1-nitrobenzene

A solution of the product of Example 4a (24g) was dissolved in dichloromethane (388g) at 20-40°C. The yellow solution was cooled to 5°C and urea hydrogen-peroxide (UHP) (17.6g) and trifluoroacetic acid anhydride (37g) added and stirring continued for 12hr at 5°C. The reaction mixture was warmed to room temperature over one hour and stirring continued for a further five hours. The precipitate that formed was filtered off and washed with dichloromethane. The combined filtrates were diluted further with dichloromethane, all washed and concentrated at atmospheric pressure. Toluene was added and the resulting suspension concentrated under vacuum, to remove residual dichloromethane. Further toluene was added and the mixture checked to ensure less than 0.5% dichloromethane and less than 0.1% water was present. Formic acid was added to provide a 10-12% formic acid in toluene mixture. The resulting suspension was warmed to 90°C and stirred until complete dissolution of solid. Crude product was obtained by cooling the solution to 5-10°C until crystallization commenced. The suspension was agitated at 5-10°C until crystallization appeared complete. The solid was filtered off, washed with toluene and dried under a stream of nitrogen.
The crude product was suspended in 10-12% wt/wt solution of formic acid in toluene and warmed to 90°C until dissolution of the solid. The solution was cooled to 5°C and stirred at 5°C until crystallisation occurred. The solid was obtained by filtration and washed with toluene. This recrystallization was repeated until the product tested as containing less than 0.1 % of starting material. The pure product was dried under vacuum at 50°C.
Example 5b
Preparation of 5-[3-(2,5-dichloro-4,6-dimethyl-1-oxy-pyridin-3-yl)-[1,2,4]oxadiazol-5-yl]-2-hydroxy-3-methoxy-1-nitrobenzene
After dissolution of the product of Example 4b (24g) in DCM (388g) at 20-40°C the yellow solution is cooled to 5°C before the temperature controlled addition of urea hydrogen peroxide complex (UHP)(17.6) and trifluoroacetic anhydride (TFAA) (37g). After addition of TFAA is complete stirring is continued for 12h at 5°C before the reaction mixture is warmed to room temperature (RT) within 1 h and stirring is continued for additional 5 h. The precipitate formed during the reaction is filtered and washed with DCM on the funnel filter. The combined filtrates are diluted with DCM (325g) and then repeatedly washed with water before concentrated at atmospheric pressure. DCM is replaced by toluene (170g) and the resulting suspension is concentrated again under vacuum to remove surplus DCM. Distillates are replaced by fresh toluene as before and the mixture is analyzed for residual water and DCM (Residual DCM after solvent switch max. 0.5%; residual water after solvent switch max. 0.1 %). Formic acid (24g) is charged resulting in an approx. 10-12 % w/w formic acid in toluene solvent mixture The resulting suspension is warmed to 90°C and stirred until compete dissolution of the solid is achieved. The crude product is crystallized by cooling of this solution to 5-10°C and subsequent agitation of the resulting suspension at 5-10°C. The solid is filtered of washed with toluene and then dried in a stream of nitrogen gas.
The crude product so obtained is suspended in an approx. 10-12 %w/w solution (176g) of formic acid in toluene. The suspension is warmed to 90°C and stirred until all product is dissolved. After cooling of this solution to 5°C and subsequent stirring at 5°C, crude product is isolated by filtration and subsequent washing of the wet product with toluene.
The re-crystallization of crude product is repeated (2 or more times). The pure product (11.8g) is dried at 50°C under vacuum.
Example 5c
Preparation of 5-[3-(2,5-dichloro-4,6-dimethyl-1-oxy-pyridin-3-yl)-[1,2,4]oxadiazol-5-yl]-2-hydroxy-3-methoxy-1-nitrobenzene
After dissolution of the product of Example 4c (24g) in DCM (388g) at 20-40°C the yellow solution is cooled to 5°C prior to the temperature controlled addition of urea hydrogen peroxide complex (UHP) (17.6g) and trifluoroacetic acid anhydride (TFAA) (37g). After addition of TFAA is complete stirring is continued for 12h at 5°C before the reaction mixture is warmed to RT within 1 h and stirring is continued for additional 5 h. The precipitate formed during the reaction is filtered and the filter cake is washed with DCM. The combined filtrates are diluted with DCM (325g) and then repeatedly washed with water before concentrated at atmospheric pressure. DCM is replaced by toluene (170g) and the resulting suspension is concentrated again in vacuum in order to remove surplus DCM and water. Distillates are replaced by fresh toluene followed by addition of formic acid (24g). The resulting suspension is warmed to 80°C and stirring is continued in order to dissolve the solid. The product is crystallized by cooling of this solution to 5°C and subsequent agitation of the resulting suspension at 5°C. The solid is filtered, washed with toluene and then dried in a stream of nitrogen gas.
The product is suspended in a formic acid / toluene (18g/158g) mixture followed by warming of the reaction mixture to 80°C. After dissolution of the product the solution is cooled to 5°C whereby the product precipitates. After additional stirring at 5°C the suspension is filtered and the filter cake is washed with toluene.
The re-crystallization of the product is repeated. The product is used as a wet material in the next process step (12.1g product obtained if dried at max. 60°C).
Example 5d
Preparation of 5-[3-(2,5-dichloro-4,6-dimethyl-1-oxy-pyridin-3-yl)-[1,2,4]oxadiazol-5-yI]-2-hydroxy-3-mefhoxy-1-nitrobenzene
550 kg (420 L) Dichloromethane was charged into a reactor. 34 kg of product of example 4d was added to in a short period at 20°C internal temperature. The solution was cooled to 6.5°C then 24.9 kg urea hydrogen peroxide complex (UHP) was added over a period of 20-40 minutes between 5-10°C. Stirring was continued for additional 20 minutes between 6.5-7.5°C. Trifluoroacetic anhydride, 53 kg, was administered into the reaction mixture, starting and maintaining the temperature at 6-7°C over a period of 2-3 hours. When the administration was complete the mixture was stirred for additional 30 minutes. Then the internal temperature was allowed to rise to a maximum of 25°C over a period of 1.5 hours. The internal temperature was maintained between 20-25°C and the mixture was allowed to react for additional 18-20 hrs. The reaction mixture was centrifuged and the fuge was washed with 45 kg dichloromethane. To the separated dichloromethane solution 460 kg (350 L) dichloromethane and 190 kg deionised water was added. The mixture was stirred for 10 minutes and the phases were separated for 30 minutes. The organic phase was washed again with 2×190 kg deionised water and separated as previously. Evaporation of the unified organic solution at max 35 °C under vacuum was done to a final volume of 100-120 L. Then a total of 105 kg acetonitrile was administered into the system while the distillation was continued to keep the volume at 100-120 L. When complete an additional 170 kg (220 L) acetonitrile was added to the mixture at normal pressure. This suspension was heated to 70-80°C at normal pressure while dichloromethane was distilled off continuously. The mixture was then kept stirred for an hour. The suspension was cooled to 20-25°C and was stirred for an additional 30 minutes. The suspension was then centrifuged and was washed with 30 kg acetonitrile. The wet material, 29.7 kg, was vacuum dried for 16 hrs at 30°C. Dried product yield was 81.5%.
27.7 kg product, 240 kg toluene and 29.2 kg formic acid was charged into reactor then heated to 90°C for complete dissolution for 1 hour. Then the solution was cooled to 7°C and then the suspension was kept at 7°C for additional 2 hrs. If necessary seeding was applied with 3-5 grams of pure product. The suspension was then centrifuged for 1 hour whilst washing with 28 kg cold toluene. The product was suspended in 225 kg toluene and 27.2 kg formic acid was charged. The mixture then was heated to 90°C for complete dissolution for 1 hour. Then the solution was cooled to 20-25 °C, then the suspension was kept between 15-25°C for additional 2 hrs, seeded if necessary. The suspension then was centrifuged for 60 minutes whilst washed with 28 kg cold toluene. The recrystallization process may be repeated 2-3 more times.
Drying for 24 hrs at 38-41°C under vacuum was conducted until constant weight. This resulted in 16.34 kg (58.8%) dry material.
Example 5e
Preparation of 5-[3-(2,5-dichloro-4,6-dimethyl-1-oxy-pyridin-3-yl)-[1,2,4]oxadiazol-5-yl]-2-hydroxy-3-methoxy-1-nitrobenzene
After dissolution of the product of Example 4e (150g) in DCM (2.43kg) at reflux, the yellow solution is cooled to 5°C prior to the temperature controlled addition of carbamide peroxide (UHP – urea hydrogen peroxide) ( 110g) and trifluoroacetic acid anhydride (TFAA) (155.1 ml in 4 portions within 2 hours). The mixture is stirred for 12h at 5°C then the reaction mixture is warmed to 25 °C over 1.5 hours and stirred for 5 hours. The precipitate formed during the reaction is filtered and the filter cake is washed with DCM (0.36 kg). The combined filtrates are warmed to 30°C and diluted with water (300g). 10% sodium hydroxide is added until pH= 4 is reached. The biphasic system is stirred for 10 minutes at 30°C and the mixture is then allowed to separate. The organic layer is then successively washed with a mixture water (750g) and 10% sodium hydroxide (7.5g) (until pH=4), 3.2% HCl solution (300g). DCM is distilled at atmospheric pressure and then replaced by toluene (1035g) applying vacuum (150mbar) and keeping internal temperature at 45°C. Formic acid (300g) and toluene (900g) are added keeping the internal temperature above 40°C. The resulting solution is distilled under vacuum (150 mbar, 45°C internal temperature) until distillation ceases. After seeding at 45°C, the slurry is stirred for 1 hour at 45°C then is cooled to 5°C over 2 hours. The suspension is stirred for at least 2 hours at 5°C and then filtered. The wet cake is washed with toluene (195g) and dried in a stream of nitrogen gas (Chemical purity of crude product min. 92 % area).
A suspension of crude product in formic acid (388g, 2wt) is warmed to 55°C and stirred until complete dissolution of the crude product. Toluene (1242g, 6.4wt) is added maintaining the internal temperature above 50 °C. The reaction is stirred at 150mBar and internal temperature 45 °C until distillation ceases. The vacuum and distillation is stopped and then seed is added at 45°C. The slurry is stirred for 1 hour at 45°C and cooled to 5°C in 2 hours. The resulting suspension is stirred for at least 2 hours at 5°C then filtered. The wet cake is washed with toluene (260g, 1.34wt). The wet cake is collected and charged into the reactor. This procedure is repeated at least twice until 5-[3-(2,5-dichloro-4,6-dimethyl-pyridin-3-yl)-[1,2,4]oxadiazol-5-y1]-2-hydroxy-3-methoxy-1-nitrobenzene level max is 0.1 % (a/a) prior to dry at 25°C max under vacuum.
Example 6
Example 5a was repeated on a larger scale employing product of Example 3 (82g), dichloromethane (1325g), urea peroxide (60.1g) and trifuoroacetic acid anhydride
(128g).
Example 7a
Preparation of 5-[3-(2,5-dichloro-4,6-dimethyl-1-oxy-pyridin-3-yl)-[1,2,4]oxadiazol-5-yl]-3-nitrobenzene-1,2-diol.

(Π)
Product of Example 6 (15g) was suspended in N-methyl pyrrolidone (NMP) (131.5g) and cooled to 5°C. Aluminium chloride (6.2g) and pyridine (12g) were added while maintaining the temperature at 5°C. After the addition of pyridine was complete the reaction mixture was warmed to 60 °C and maintained for 2 hours. After confirmation that less than 0.5 starting material remained, the reaction mixture was cooled, and aqueous HCl (water 233g, HCl 123g, 37%) added. The resulting yellow solid was isolated by suction filtration. The resulting wet product was washed with water and propan-2-ol (67g) and dried under vacuum.
Optionally, the crude product was suspended in ethanol (492g) and warmed to reflux. The suspension was stirred for 1 hour under reflux and then cooled to room temperature. The solid was obtained by filtration, washed with ethanol and dried under vacuum at 50°C. A typical yield of 85% was achieved.
If desired either the final ethanol crystallised material or the initially produced product after washing with propan-2-ol may be employed in preparation of micronized material for use in pharmaceutical compositions.
Example 7b
Preparation of 5-[3-(2,5-dichloro-4,6-dimethyl-1-oxy-pyridin-3-y1)-[1,2,4]oxadiazol-5-yl]-3-nitrobenzene-1,2-diol.
An approx. 11 % w/w suspension of the product of example 5b (20g) in NMP (150g) is cooled to 5°C followed by a consecutive temperature controlled addition of aluminium chloride (8g) and pyridine (15.3g). After addition of pyridine is complete the reaction mixture is warmed to 60°C followed by additional 2 h reaction time. After complete conversion of the product of example 5b the reaction mixture is cooled before an aqueous diluted hydrochloric acid (water 293g, HCl 177g, 34%) is dosed. By addition of the hydrochloric acid, crude product precipitates from the NMP/water matrix as a yellow solid which is isolated by suction filtration. The resulting wet product is washed with water and 2-propanol in a replacement wash followed by drying of the wet crude product under vacuum.
The crude product is suspended in ethanol (282g) followed by warming of the mixture to reflux. The suspension is stirred for 1 h at reflux conditions followed by cooling to room temperature. The product is isolated by filtration of the suspension. The wet product is washed with ethanol and subsequently dried in vacuo at approx 50°C (typically weight corrected yield was 85%).
The product (20g) is suspended in formic acid (725g) before the resulting suspension is warmed to max. 67°C. Stirring is continued until complete dissolution of the product is achieved. The hot solution is filtered and the filtrate is cooled to 40 – 45°C before the product is precipitated first by concentration of the solution to approx. 40% (v/v) of its original volume followed by addition of the anti solvent 2-propanol (390g). After addition of 2-propanol is finished the resulting suspension is kept at 55-60°C for crystal ripening followed by cooling to RT and filtration. The filter cake is washed with 2-propanol followed by drying of the material at max. 58°C until loss on drying (LOD) max. 0.5% . Typically, a yield of 97-98% was obtained.
If desired the product may be employed in preparation of micronized material for use in pharmaceutical compositions.
Example 7c
Preparation of 5-[3-(2,5-dichloro-4,6-dimethyl-1-oxy-pyridin-3-yl)-[1,2,4]oxadiazol-5-yl]-3-nitrobenzene-1,2-diol.
A suspension of the product of example 5c (20g) or of example 6 (20g) in NMP (153g) is cooled to 5°C followed by a consecutive temperature controlled addition of aluminium chloride (8.2g) and pyridine (15.4g). After addition of pyridine is complete the reaction mixture is warmed to 60°C followed by additional 3 h reaction time. After complete conversion of the product of example 5c or of example 6 the crude product is
precipitated by a temperature controlled addition of an aqueous hydrochloric acid solution (water 296g, HCl 179g, 34%). Filtration of the solid followed by washing of the wet filter cake with water and 2-propanol yields a crude product wet material which is immediately dissolved in formic acid (536g). After polish filtration the filtrate is concentrated under vacuum followed by addition of the anti-solvent 2-propanol (318g). After aging of the resulting suspension at 55-60°C the suspension is cooled to RT and filtered. The wet filter cake is washed with 2-propanol. The wet product is dried under vacuum at max. 58°C until LOD max. 0.5%. The yield was in the range of 70-95%
If desired the product may be employed in preparation of micronized material for use in pharmaceutical compositions.
Example 7d
Preparation of 5-[3-(2,5-dichloro-4,6-dimethyl-1-oxy-pyridin-3-yl)-[1,2,4]oxadiazol-5-yl]-3-nitrobenzene-1,2-diol.
132 kg (147 L) N-methylpyrrolidone was charged into a 1000 L reactor. 16.3 kg of product of example 5d was then added. The suspension was cooled to 5-7°C and 6.5 kg of sublimed aluminium chloride was added in portions keeping the internal temperature between 5-10°C. The mixture was stirred for 10 minutes then 12.6 kg pyridine was added maintaining the internal temperature between 5-10°C. The mixture was warmed with water in the jacket to 20-25°C and the mixture was stirred for 30 minutes. Then the mixture is heated to 58-62 °C and reacted for around 2 hours. In a separate reactor a mixture of 240.5 kg deionised water and 146.4 kg concentrated HCl was mixed. This was cooled to 15-20°C. The reaction mixture from the demethylation was introduced into the diluted hydrochloric acid between 20-25°C. Optionally, 51.2 kg dichloromethane was added to the suspension, stirred for 30 minutes and was centrifuged, washed with 60 kg deionised water and 20 kg isopropanol. Drying gave 15.9 kg of product.
The product was suspended in 185.3 kg of ethanol. The mixture was then stirred at 78°C for an hour, then cooled to 20-25°C and stirred for 1 hour. The suspension was then centrifuged and the filtercake was washed with 44.5 kg ethanol, 96% . The solid material was dried at 50°C in vacuum in a stainless steel tray drier. 14.35 kg (90.3% yield) dry product was obtained.
A reactor was charged with 317.2 kg formic acid and dry product. The mixture was heated to 65 °C until all the solid dissolves. The hot solution was then filtered to an empty 1000 L reactor, was rinsed with 20 kg formic acid, then the formic acid solution was distilled partially off under vacuum to around 80-100L. 260 kg isopropanol was then introduced at 50-60°C and stirred for 30-35 minutes. The mixture was then cooled to 20-25°C with water in the jacket and was allowed to stir min 2 hours. The suspension was then centrifuged and was washed with 25 kg isopropanol. The wet material was removed from the fuge and was transferred into vacuum tray drier and was dried until constant weight under vacuum at 45-50°C resulting in 13.6 kg product, with a yield of 95.3% .
If desired the product may be employed in preparation of micronized material for use in pharmaceutical compositions.
Example 7e
Preparation of 5-[3-(2,5-dichloro-4,6-dimethyl-1-oxy-pyridin-3-yl)-[1,2,4]oxadiazol-5-yl]-3-nitrobenzene-1,2-diol.
A suspension of product of Example 5e (34.1kg) in N-Methyl pyrrolidone (NMP) (182kg) is warmed to 50 °C until dissolution and then cooled to 5°C followed by a consecutive temperature controlled addition of aluminium chloride (9.8 kg) and pyridine (18.2kg). After addition of pyridine is complete the reaction mixture is warmed to 60°C and stirred for at least 2 hours. The reaction mixture is cooled to 10-16°C (e.g. 11, 13, 15°C) before an aqueous diluted hydrochloric acid (4M solution, 283L) is dosed maintaining the temperature below 25 °C. During the addition of the hydrochloric acid the crude product is precipitated from the NMP/water matrix as a yellow solid. The yellow solid is filtered and subsequently washed with water (179kg), 2-propanol (105kg). The wet solid is dried under vacuum at 55°C.
A suspension of wet product (25.1kg) in formic acid (813kg) is warmed to max. 67°C. The mixture is stirred at 67°C until complete dissolution of the product is achieved. The hot solution is filtered and the filtrate is cooled to 40 – 45°C before the product is precipitated first by concentration of the solution to approx. 40% (v/v) of its original volume followed by addition of the anti solvent 2-propanol (380kg). After addition of 2-propanol the resulting suspension is stirred at 55-60°C for crystal ripening followed by cooling to RT and filtration. The filter cake is washed with 2-propanol (38kg) and then dried at max. 58°C until LOD max. 0.5%). The product may be milled (for example using the method of Example 8).
Example 8
Micronization of 5-[3-(2,5-dichloro-4,6-dimethyl-1-oxy-pyridin-3-yl)-[1,2,4]oxadiazol-5-yl]-3-nitrobenzene-1,2-diol with MC JETMILL® type 200 milling equipment (micronization through spiral jet mills)
Equipment:
Mill: MC JETMILL® 200
Dosing unit: K-Tron T 35
Cyclone: type 600
Each micronization trial was performed on at least 2 kg of 5-(3-(2,5-dichloro-4,6-dimethyl-1-oxy-pyridin-3-yl)-[1,2,4]oxadiazol-5-yl]-3-nitrobenzene-1,2-diol.
The following working parameters have been defined for the micronization:
Feed rate range: 24.0-48.0 kg/h (200-400 g/30sec.)
Mill pressure range: 3.0-4.0 bar
Venturi pressure range: 3.0-4.0 bar; preferably the Venturi pressure is the same as the mill pressure
Using the above equipment and working parameters the microparticles of 5-(3-(2,5-dichloro-4,6-dimethyl-1-oxy-pyridin-3-yl)-[1,2,4]oxadiazol-5-yl]-3-nitrobenzene-1,2-diol comply with the following particle size specification (particle size determined by optical microscopy): D10 (EDC) is not less than 4 or 5 μm (for example not less than 5 μm), the D50 (EDC) is 10-45 or 15-30 μm (for example 15-30 μm) and the D95 (EDC) is not more than 60 or 70 μm (for example not more than 60 μm).
Example 9 (Figure 5)
2,5-Dichloro-4,6-dimethyl-nicotinonitrile is reacted with hydroxylamine in the presence of catalytic amounts of 1,10-phenanthroline monohydrate to yield the aldoxime (Z)-2,5-dichloro-N’-hydroxy-4,6-dimethylnicotinimidamide which represents the first coupling partner towards the synthesis of 5-[3-(2,5-dichloro-4,6-dimethyl-pyridin-3-yl)-[1,2,4]oxadiazol-5-yl]-2-hydroxy-3-methoxy-1-nitrobenzene. The second coupling partner 5-nitro-vanillic acid pure is synthesized from vanillic acid by nitration with 65 % nitric acid followed by re-crystallization of the crude 5-nitro-vanillic acid intermediate from acetic acid. The convergent assembly of the oxadiazole moiety in 5-[3-(2,5-dichloro-4,6-dimethyl-pyridin-3-yl)-[1,2,4]oxadiazol-5-yl]-2-hydroxy-3-methoxy-1-nitrobenzene is achieved by first activation of 5-nitro-vanillic acid as its acid chloride and subsequent coupling with the aldoxime (Z)-2,5-dichloro-N’-hydroxy-4,6-dimethylnicotinimidamide. Cyclisation of the initially formed coupling product is achieved thermally to give the oxadiazole moiety by elimination of water. The reaction
mixture of 5-[3-(2,5-dichloro-4,6-dimethyl-pyridin-3-yl)-[1,2,4]oxadiazol-5-yl]-2-hydroxy-3-methoxy-1-nitrobenzene, after ring closure reaction, is concentrated and product isolated from 1,4-dioxane/ethanol mixture in one step. Oxidation of the pyridine ring to the corresponding aryl-N-oxide (5-[3-(2,5-dichloro-4,6-dimethyl-1-oxy-pyridin-3-yl)-[1,2,4]oxadiazol-5-yl]-2-hydroxy-3-methoxy-1-nitrobenzene) is achieved with trifluoroperoxoacetic acid which is formed in situ from UHP (Urea hydrogen peroxide complex) and trifluoroacetic acid anhydride. Unreacted 5-[3-(2,5-dichloro-4,6-dimethyl-pyridin-3-yl)-[1,2,4]oxadiazol-5-yl]-2-hydroxy-3-methoxy-1-nitrobenzene is subsequently removed from 5-[3-(2,5-dichloro-4,6-dimethyl-1-oxy-pyridin-3-yl)-[1,2,4]oxadiazol-5-yl]-2-hydroxy-3-methoxy-1-nitrobenzene by repeated re-crystallisation from formic acid/toluene. The analogue intermediate 5-[3-(2,5-dichloro-4,6-dimethyl-1-oxy-pyridin-3-yl)-[1,2,4]oxadiazol-5-yl]-2-hydroxy-3-methoxy-1-nitrobenzene pure with a level of 5-[3-(2,5-dichloro-4,6-dimethyl-pyridin-3-yl)-[1,2,4]oxadiazol-5-yl]-2-hydroxy-3-methoxy-1-nitrobenzene below 0.10 %area is converted to 5-[3-(2,5-dichloro-4,6-dimethyl-1-oxy-pyridin-3-yl)-[1,2,4]oxadiazol-5-yl]-3-nitrobenzene-1,2-diol crude analogue by ether cleavage in the presence of a stoichiometric amount of aluminium chloride and pyridine. After completion of the reaction, the crude product is isolated by precipitation with an aqueous hydrochloric acid followed by dissolution of the precipitate in formic acid. After polish filtration of the resulting solution and partial solvent switch from formic acid to isopropanol, 5-[3-(2,5-dichloro-4,6-dimethyl-1-oxy-pyridin-3-yl)-[1,2,4]oxadiazol-5-yl]-3-nitrobenzene-1,2-diol is crystallized from the resulting formic acid/IPA crystallization matrix and finally optionally milled to the desired particle size.

PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2012107708
PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2007117165
scheme 1 depicts example how to produce a compound of the general formula IIB from a compound of the general formula IVB:

iii.

HB IVB
Scheme 1. Reagents: i. Piperidine, ethanol, reflux; ii. SO2Cl2, CCl4, reflux; iii. POCI3, 120 0C, 18 h; iv. 50% H2NOH, MeOH-H20, 1.25 mol % 1,10-phenanthroline hydrate.
The following reaction scheme 2 depicts an example how to produce certain compounds of general formula III:

I, R8 = methyl III, R8 = R9 = H
iv.
R9 = H

III, R8 = R9 = benzyl
Scheme 2. Reagents: i. 65 % HNO3, AcOH; ii. 48 % HBr (aq), 140 0C; iii. MeOH, HCl(g); iv. BnBr, K2CO3, CH3CN, reflux; v. 3N NaOH, MeOH/H2O.
The following reaction scheme 3 depicts an example how to produce the compound A, by activation of a compound according to general formula III followed by cyclisation involving condensation with a compound according to formula HB, dehydration and deprotection of the methyl residue protecting the hydroxyl group;
0C

compound A
| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2007013830A1 | Jul 26, 2006 | Feb 1, 2007 | Portela & Ca. S.A. | Nitrocatechol derivatives as comt inhibitors |
| WO2007117165A1 | Apr 10, 2007 | Oct 18, 2007 | Bial – Portela & Ca, S.A. | New pharmaceutical compounds |
| WO2008094053A1 * | Oct 10, 2007 | Aug 7, 2008 | Bial-Portela & Ca, S.A. | Dosage regimen for comt inhibitors |
| WO2012107708A1 * | Oct 21, 2011 | Aug 16, 2012 | Bial – Portela & Ca, S.A. | Administration regime for nitrocatechols |
| US20100168113 * | Apr 10, 2007 | Jul 1, 2010 | David Alexander Learmonth | Pharmaceutical Compounds |
| Reference | ||
|---|---|---|
| 1 | * | “[1,2,4]-oxadiazolyl nitrocatechol derivatives“, IP.COM JOURNAL, IP.COM INC., WEST HENRIETTA, NY, US, 3 May 2012 (2012-05-03), XP013150541, ISSN: 1533-0001 |
| 2 | * | KISS L E ET AL: “Discovery of a long-acting, peripherally selective inhibitor of catechol-O-methyltransferase“, JOURNAL OF MEDICINAL CHEMISTRY, AMERICAN CHEMICAL SOCIETY, US, vol. 53, no. 8, 22 April 2010 (2010-04-22), pages 3396 – 3411, XP002594266, ISSN: 0022-2623, [retrieved on 20100324], DOI: 10.1021/JM1001524 |
| 3 | L. E. KISS ET AL., J. MED. CHEM., vol. 53, 2010, pages 3396 – 3411 | |
| 4 | * | RASENACK N ET AL: “MICRON-SIZE DRUG PARTICLES: COMMON AND NOVEL MICRONIZATION TECHNIQUES“, PHARMACEUTICAL DEVELOPMENT AND TECHNOLOGY, NEW YORK, NY, US, vol. 9, no. 1, 1 January 2004 (2004-01-01), pages 1 – 13, XP009055393, ISSN: 1083-7450, DOI: 10.1081/PDT-120027417 |
1: Bicker J, Alves G, Fortuna A, Soares-da-Silva P, Falcão A. A new PAMPA model using an in-house brain lipid extract for screening the blood-brain barrier permeability of drug candidates. Int J Pharm. 2016 Jan 30. pii: S0378-5173(16)30072-2. doi: 10.1016/j.ijpharm.2016.01.074. [Epub ahead of print] PubMed PMID: 26836708.
2: Devos D, Moreau C. Opicapone for motor fluctuations in Parkinson’s disease. Lancet Neurol. 2015 Dec 22. pii: S1474-4422(15)00346-4. doi: 10.1016/S1474-4422(15)00346-4. [Epub ahead of print] PubMed PMID: 26725545.
3: Ferreira JJ, Lees A, Rocha JF, Poewe W, Rascol O, Soares-da-Silva P; Bi-Park 1 investigators. Opicapone as an adjunct to levodopa in patients with Parkinson’s disease and end-of-dose motor fluctuations: a randomised, double-blind, controlled trial. Lancet Neurol. 2015 Dec 22. pii: S1474-4422(15)00336-1. doi: 10.1016/S1474-4422(15)00336-1. [Epub ahead of print] PubMed PMID: 26725544.
4: Rascol O, Perez-Lloret S, Ferreira JJ. New treatments for levodopa-induced motor complications. Mov Disord. 2015 Sep 15;30(11):1451-60. doi: 10.1002/mds.26362. Epub 2015 Aug 21. Review. PubMed PMID: 26293004.
5: Gonçalves D, Alves G, Fortuna A, Soares-da-Silva P, Falcão A. Development of a liquid chromatography assay for the determination of opicapone and BIA 9-1079 in rat matrices. Biomed Chromatogr. 2016 Mar;30(3):312-22. doi: 10.1002/bmc.3550. Epub 2015 Aug 17. PubMed PMID: 26147707.
6: Ferreira JJ, Rocha JF, Falcão A, Santos A, Pinto R, Nunes T, Soares-da-Silva P. Effect of opicapone on levodopa pharmacokinetics, catechol-O-methyltransferase activity and motor fluctuations in patients with Parkinson’s disease. Eur J Neurol. 2015 May;22(5):815-25, e56. doi: 10.1111/ene.12666. Epub 2015 Feb 4. PubMed PMID: 25649051.
7: Bonifácio MJ, Torrão L, Loureiro AI, Palma PN, Wright LC, Soares-da-Silva P. Pharmacological profile of opicapone, a third-generation nitrocatechol catechol-O-methyl transferase inhibitor, in the rat. Br J Pharmacol. 2015 Apr;172(7):1739-52. doi: 10.1111/bph.13020. Epub 2015 Jan 20. PubMed PMID: 25409768; PubMed Central PMCID: PMC4376453.
8: Kiss LE, Soares-da-Silva P. Medicinal chemistry of catechol O-methyltransferase (COMT) inhibitors and their therapeutic utility. J Med Chem. 2014 Nov 13;57(21):8692-717. doi: 10.1021/jm500572b. Epub 2014 Sep 2. PubMed PMID: 25080080.
9: Rocha JF, Falcão A, Santos A, Pinto R, Lopes N, Nunes T, Wright LC, Vaz-da-Silva M, Soares-da-Silva P. Effect of opicapone and entacapone upon levodopa pharmacokinetics during three daily levodopa administrations. Eur J Clin Pharmacol. 2014 Sep;70(9):1059-71. doi: 10.1007/s00228-014-1701-2. Epub 2014 Jun 14. PubMed PMID: 24925090.
10: Rocha JF, Santos A, Falcão A, Lopes N, Nunes T, Pinto R, Soares-da-Silva P. Effect of moderate liver impairment on the pharmacokinetics of opicapone. Eur J Clin Pharmacol. 2014 Mar;70(3):279-86. doi: 10.1007/s00228-013-1602-9. Epub 2013 Nov 24. PubMed PMID: 24271646.
11: Bonifácio MJ, Sutcliffe JS, Torrão L, Wright LC, Soares-da-Silva P. Brain and peripheral pharmacokinetics of levodopa in the cynomolgus monkey following administration of opicapone, a third generation nitrocatechol COMT inhibitor. Neuropharmacology. 2014 Feb;77:334-41. doi: 10.1016/j.neuropharm.2013.10.014. Epub 2013 Oct 19. PubMed PMID: 24148813.
12: Gonçalves D, Alves G, Fortuna A, Soares-da-Silva P, Falcão A. An HPLC-DAD method for the simultaneous quantification of opicapone (BIA 9-1067) and its active metabolite in human plasma. Analyst. 2013 Apr 21;138(8):2463-9. doi: 10.1039/c3an36671e. PubMed PMID: 23476919.
13: Rocha JF, Almeida L, Falcão A, Palma PN, Loureiro AI, Pinto R, Bonifácio MJ, Wright LC, Nunes T, Soares-da-Silva P. Opicapone: a short lived and very long acting novel catechol-O-methyltransferase inhibitor following multiple dose administration in healthy subjects. Br J Clin Pharmacol. 2013 Nov;76(5):763-75. doi: 10.1111/bcp.12081. PubMed PMID: 23336248; PubMed Central PMCID: PMC3853535.
14: Almeida L, Rocha JF, Falcão A, Palma PN, Loureiro AI, Pinto R, Bonifácio MJ, Wright LC, Nunes T, Soares-da-Silva P. Pharmacokinetics, pharmacodynamics and tolerability of opicapone, a novel catechol-O-methyltransferase inhibitor, in healthy subjects: prediction of slow enzyme-inhibitor complex dissociation of a short-living and very long-acting inhibitor. Clin Pharmacokinet. 2013 Feb;52(2):139-51. doi: 10.1007/s40262-012-0024-7. PubMed PMID: 23248072.
15: Palma PN, Bonifácio MJ, Loureiro AI, Soares-da-Silva P. Computation of the binding affinities of catechol-O-methyltransferase inhibitors: multisubstate relative free energy calculations. J Comput Chem. 2012 Apr 5;33(9):970-86. doi: 10.1002/jcc.22926. Epub 2012 Jan 25. PubMed PMID: 22278964.
16: Gonçalves D, Alves G, Soares-da-Silva P, Falcão A. Bioanalytical chromatographic methods for the determination of catechol-O-methyltransferase inhibitors in rodents and human samples: a review. Anal Chim Acta. 2012 Jan 13;710:17-32. doi: 10.1016/j.aca.2011.10.026. Epub 2011 Oct 20. Review. PubMed PMID: 22123108.
17: Kiss LE, Ferreira HS, Torrão L, Bonifácio MJ, Palma PN, Soares-da-Silva P, Learmonth DA. Discovery of a long-acting, peripherally selective inhibitor of catechol-O-methyltransferase. J Med Chem. 2010 Apr 22;53(8):3396-411. doi: 10.1021/jm1001524. PubMed PMID: 20334432.
////////BIA-9-1067, ONO-2370, BIA-91067, 923287-50-7, Opicapone, Catechol-O-methyl transferase, COMT inhibitor, Parkinson’s disease, PD, BIA 9-1067, BIA 91067, BIA-91067, BIA91067, EU 2016
OC1=CC(C2=NC(C3=C(Cl)[N+]([O-])=C(C)C(Cl)=C3C)=NO2)=CC([N+]([O-])=O)=C1O


