

| Patent ID | Date | Patent Title |
|---|---|---|
| US7378424 | 2008-05-27 | Derivatives of pyrimido[6, 1-A]isoquinolin-4-one |
| US7105663 | 2006-09-12 | Derivatives of pyrimido[6, 1-a]isoquinolin-4-one |
| US6794391 | 2004-09-21 | Derivatives of pyrimido[6.1-a]isoquinolin-4-one |
| US2004001895 | 2004-01-01 | Combination treatment for depression and anxiety |
| US2003235631 | 2003-12-25 | Combination treatment for depression and anxiety |
FLAGS AND HITS
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
Join me on Linkedin
Join me on Researchgate
Join me on Facebook
 FACEBOOK
...................................................................Join me on twitter
FACEBOOK
...................................................................Join me on twitter
 ..................................................................Join me on google plus
..................................................................Join me on google plus
 Googleplus
Googleplus
FACEBOOK
...................................................................Join me on twitter
..................................................................Join me on google plus
GoogleplusMYSELF
DR ANTHONY MELVIN CRASTO Ph.D ( ICT, Mumbai)
, INDIA
36Yrs Exp. in the feld of Organic Chemistry,Working for AFRICURE PHARMA as ADVISOR earlier with GLENMARK PHARMA at Navi Mumbai, INDIA. Serving chemists around the world. Helping them with websites on Chemistry.Million hits on google,
NO ADVERTISEMENTS , ACADEMIC , NON COMMERCIAL SITE, world acclamation from industry, academia, drug authorities for websites, blogs and educational contribution, ........amcrasto@gmail.com..........+91 9323115463, Skype amcrasto64


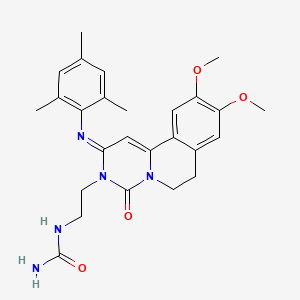
RPL 554, Ensifentrine


RPL-554, Ensifentrine
- Molecular FormulaC26H31N5O4
- Average mass477.555
FDA 6/26/2024, Ohtuvayre, To treat chronic obstructive pulmonary disease
Drug Trials Snapshot
RPL 554
Urea, N-[2-[(2E)-6,7-dihydro-9,10-dimethoxy-4-oxo-2-[(2,4,6-trimethylphenyl)imino]-2H-pyrimido[6,1-a]isoquinolin-3(4H)-yl]ethyl]-
(2-[(2E)-9,10-DIMETHOXY-4-OXO-2-[(2,4,6-TRIMETHYLPHENYL)IMINO]-2H,3H,4H,6H,7H-PYRIMIDO[4,3-A]ISOQUINOLIN-3-YL]ETHYL)UREA
2-[9,10-dimethoxy-4-oxo-2-(2,4,6-trimethylphenyl)imino-6,7-dihydropyrimido[6,1-a]isoquinolin-3-yl]ethylurea
{2-[(2E)-9,10-dimethoxy-4-oxo-2-[(2,4,6-trimethylphenyl)imino]-2H,3H,4H,6H,7H-pyrimido[4,3-a]isoquinolin-3-yl]ethyl}urea
2-[4-keto-9,10-dimethoxy-2-(2,4,6-trimethylphenyl)imino-6,7-dihydropyrimido[4,3-a]isoquinolin-3-yl]ethylurea
2-[9,10-dimethoxy-4-oxo-2-(2,4,6-trimethylphenyl)imino-6,7-dihydropyrimido[4,3-a]isoquinolin-3-yl]ethylurea
298680-25-8 CAS
UNII:3E3D8T1GIX
CFTR stimulator; PDE 3 inhibitor; PDE 4 inhibitor
RPL-554 is a mixed phosphodiesterase (PDE) III/IV inhibitor in phase II clinical development at Verona Pharma for the treatment of asthma, allergic rhinitis, chronic obstructive pulmonary disease (COPD) and inflammation.
RPL-554 is expected to have long duration of action and will be administered nasally thereby preventing gastrointestinal problems often resulting from orally administered PDE4 antiinflammatory drugs.
The company is now seeking licensing agreements or partnerships for the further development and commercialization of the drug.
RPL-554 (LS-193,855) is a drug candidate for respiratory diseases. It is an analog of trequinsin, and like trequinsin, is a dual inhibitor of the phosphodiesterase enzymes PDE-3 and PDE-4.[1] As of October 2015, inhaled RPL-554 delivered via a nebulizer was in development for COPD and had been studied in asthma.[2]
PDE3 inhibitors act as bronchodilators, while PDE4 inhibitors have an anti-inflammatory effect.[1][3]
RPL554 was part of a family of compounds invented by Sir David Jack, former head of R&D for GlaxoSmithKline, and Alexander Oxford, a medicinal chemist; the patents on their work were assigned to Vernalis plc.[4][5]:19-20
In 2005, Rhinopharma Ltd, acquired the rights to the intellectual property from Vernalis.[5]:19-20 Rhinopharma was a startup founded in Vancouver, Canada in 2004 by Michael Walker, Clive Page, and David Saint, to discover and develop drugs for chronic respiratory diseases,[5]:16 and intended to develop RPL-554, delivered with an inhaler, first for allergic rhinitis, then asthma, then forCOPD.[5]:16-17 RPL554 was synthesized at Tocris, a contract research organization, under the supervision of Oxford, and was studied in collaboration with Page’s lab at King’s College, London.[1] In 2006 Rhinopharma recapitalized and was renamed Verona Pharma plc.[5]
This was first seen in April 2015 when it was published as a France national. Verona Pharma (formerly Rhinopharma), under license from Kings College via Vernalis, is developing the long-acting bronchodilator, RPL-554 the lead in a series dual inhibitor of multidrug resistant protein-4 and PDE 3 and 4 inhibiting trequinsin analogs which included RPL-565, for treating inflammatory respiratory diseases, such as allergic rhinitis, asthma, and COPD.
RPL554
Verona Pharma’s lead drug, RPL554, is a “first-in-class” inhaled drug under development for chronic obstructive pulmonary disease (COPD), asthma and cystic fibrosis. The drug is an inhibitor of the phosphodiesterase 3 (PDE3) and phosphodiesterase 4 (PDE4) enzymes, two enzymes known to be of importance in the development and progression of immunological respiratory diseases. The drug has the potential to act as both a bronchodilator and an anti-inflammatory which would significantly differentiate it from existing drugs.
RPL554 was selected from a class of compounds co-invented by Sir David Jack, the former Director of Research at Glaxo who led the team that discovered many of the commercially successful drugs in the respiratory market.
Verona Pharma has successfully completed two double-blind placebo controlled randomised Phase 2b studies of RPL554: one in mild to moderate asthma and another in mild to moderate COPD. The drug was found to be well tolerated, free from drug-related adverse effects (especially cardiovascular and gastro-intestinal effects) and generated significant bronchodilation. Additionally, double-blind placebo controlled exploratory studies in healthy volunteers challenged with an inhaled irritant also generated consistent, clinically meaningful anti-inflammatory effects.
Verona Pharma is also carrying out exploratory studies to investigate the potential of RPL554 as a novel treatement for cystic fibrosis. In November 2014, the Company received a Venture and Innovation Award from the UK Cystic Fibrosis Trust to further such studies.
For further information on the potential of RPL554 for the treatment of respiratory diseases, refer to the peer-reviewed paper available on-line in the highly-respected medication journal, The Lancet Respiratory Medicine, entitled “Efficacy and safety of RPL554, a dual PDE3 and PDE4 inhibitor, in healthy volunteers and in patients with asthma or chronic obstructive pulmonary disease: findings from four clinical trials”.
The competitive advantages of RPL554 include the following:
- combining bronchodilator (PDE 3) and anti-inflammatory actions (PDE 4) in a single drug, something that is currently only achieved with a combination LABA and glucocorticosteroid inhaler,
- unique in not using steroids or beta agonists, which have known side effects,
- planned to be administered by nasal inhalation, thereby reducing the unwanted gastrointestinal side effects of many orally administered drugs.
History of Clinical Trials
- Following completion in May 2008 of toxicological studies of RPL554, the Company commenced in February 2009 a Phase I/IIa clinical trial of the drug at the Centre for Human Drug Research (CHDR) at Leiden in the Netherlands. In September 2009, the Company announced that it had successfully completed the trial, demonstrating that RPL554 has a good safety profile and has beneficial effects in terms of bronchodilation and bronchoprotection in asthmatics and a reduction in the numbers of inflammatory cells in the nasal passages of allergic rhinitis patients.
- In November 2010, the Company successfully completed a further trial that examined the safety and bronchodilator effectiveness of the drug administered at higher doses.
- In August 2011, the Company demonstrated that bronchodilation is maintained over a period of 6 days with daily dosing of RPL554 in asthmatics.
- In November 2011, the Company successfully demonstrated safety and bronchodilation of RPL554 in patients with mild to moderate forms of COPD.
- In March 2013, the Company demonstrated positive airway anti-inflammatory activity with respect to COPD at a clinical trial carried out at the Medicines Evaluation Unit (MEU) in Manchester, UK.
![]()
Synthesis
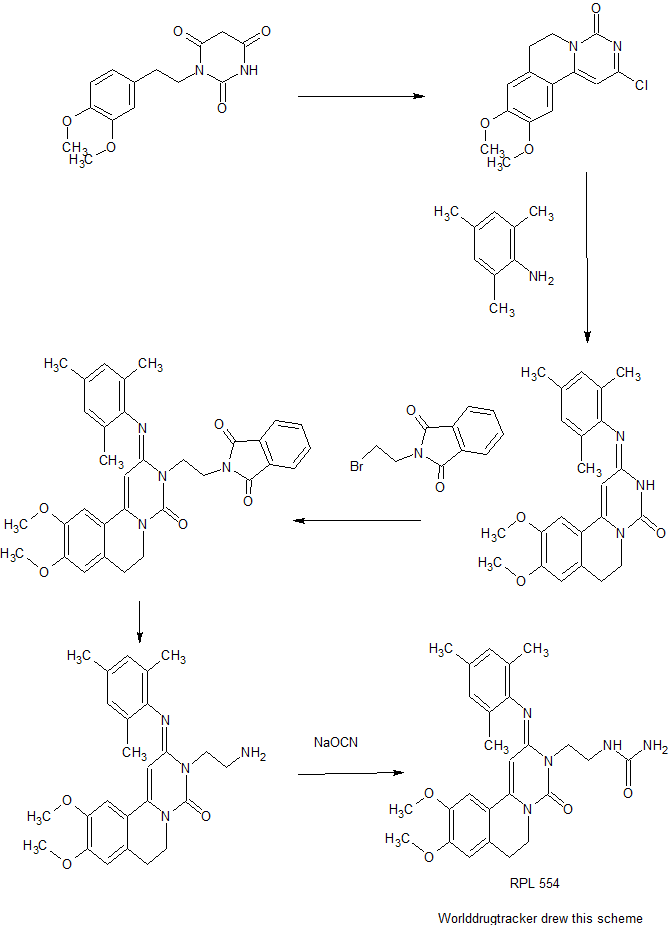
Cyclization of 1-(3,4-dimethoxyphenethyl)barbituric acid in refluxing POCl3 produces the pyrimidoisoquinolinone , which is further condensed with 2,4,6-trimethylaniline in boiling isopropanol to afford the trimethylphenylimino derivative . Subsequent alkylation of with N-(2-bromoethyl)phthalimide in the presence of K2CO3 and KI, followed by hydrazinolysis of the resulting phthalimidoethyl compound yields the primary amine . This is finally converted into the title urea RPL 554 by reaction with sodium cyanate in aqueous HCl.
Example 1 : 9 Λ 0-Dimethoxy-2-(2.4-6-trimethy-phen yliminoY-3-(N-carbamoyl-2- aminoethylV3.4.6.7-tetrahydro-2H-pyrimido[6.1-a]isoquinolin-4-one

Sodium cyanate (6.0g, 0.092 mol) in water (100 ml) was added dropwise to a stirred solution of 9,10-Dimethoxy-2-(2,4,6-trimethylphenylimino)-3-(2-aminoethyl)-3,4,6,7- tetrahydro-2H-pyrimido[6,l-a]isoquinolin-4-one, prepared according to Preparation 4 above (20.0g, 0.046 mol) in water (600 ml) and IN ΗC1 (92 ml) at 80°C. After stirring for 2h at 80°C the mixture was cooled in an ice-bath and basified with 2N NaOH. The mixture was extracted with dichloromethane (3 x 200 ml) and the combined extract was dried (MgSO- ) and evaporated in vacuo. The resulting yellow foam was purified by column chromatography on silica gel eluting with CH2CI2 / MeOH (97:3) and triturated with ether to obtain the title compound as a yellow solid, 11.9g, 54%.
M.p.: 234-236°C m/z: C26H31N5O4 requires M=477 found (M+l) = 478
HPLC: Area (%) 99.50 Column ODS (150 x 4.6 mm)
MP pH3 KH2PO4 / CH3CN (60/40)
FR (ml/min) 1.0 RT (min) 9.25 Detection 250 nm
lK NMR (300 MHz, CDCI3): δ 1.92 (1H, br s, NH), 2.06 (6H, s, 2xCH3), 2.29 (3H, s, CH3), 2.92 (2H, t, CH2), 3.53 (2H, m, CH2), 3.77 (3H, s, OCH3), 3.91 (3H, s, OCH3), 4.05 (2H, t, CH2), 4.40 (2H, t, CH2), 5.35 (2H, br s, NH2), 5.45 (1H, s, C=CH), 6.68 (1H, s, ArH), 6.70 (1H, s, ArH), 6.89 (2H, s, 2xArH).
Preparation 1 : Synthesis of 2-Chloro-6.7-d-hydro-9.10-Dimethoxy-4H-pyrimido- [6,l-a]isoquinoHn-4-one (shown as (1) in Figure 1

A mixture of l-(3,4-dimethoxyphenyl) barbituric acid (70g, 0.24mol), prepared according to the method described in B. Lai et al. J.Med.Chem. 27 1470-1480 (1984), and phosphorus oxychloride (300ml, 3.22mol) was refluxed for 2.5h. The excess phosphorous oxychloride was removed by distillation (20mmHg) on wa ming. After cooling the residue was slurried in dioxan (100ml) and cautiously added to a vigorously stirred ice/water solution (11). Chloroform (11) was added and the resulting mixture was basified with 30% sodium hydroxide solution. The organic layer was separated and the aqueous phase further extracted with chloroform (2x750ml). The combined organic extracts were washed with water (1.51), dried over magnesium sulphate and concentrated in vacuo to leave a gummy material (90g). This was stirred in methanol for a few minutes, filtered and washed with methanol (200ml), diethyl ether (2x200ml) and dried in vacuo at 40°C to yield the title compound as a yellow/orange solid. 47g, 62%
(300MHz, CDCI3) 2.96(2H, t, C(7) H2); 3.96(6H, s, 2xOCH3; 4.20(2H, t, C(6) H2); 6.61(1H, s, C(1) H); 6.76(1H, s, Ar-H); 7.10(1H, s, Ar-H). Preparation 2: 9.10-Dimethoxy-2-(2.4.6-trimethylphenyliminoV3.4.6.7- tetrahydro-2H-pyrimido[6.1-a]isoquinolin-4-one (shown as (2) in Figure 1
2-Chloro-9,10-dimethoxy-6,7-dihydro-4H-pyrimido[6,l-a]isoquinolin-4-one, prepared according to Preparation 1, (38.5g, 0.13 mol) and 2,4,6-trimethylaniline (52.7g, 0.39 mol) in propan-2-ol (3 1) was stirred and heated at reflux, under nitrogen, for 24h. After cooling to room temperature, the solution was evaporated in vacuo and the residue was purified by column chromatography on silica gel, eluting with CΗ2CI2 /
MeOH, initially 98:2, changing to 96:4 once the product began to elute from the column. The title compound was obtained with a slight impurity, (just above the product on tic). Yield 34.6g, 67%.
Preparation 3: 9.10-Dimethoxy-2-(2.4.6-trimethylphenyliminoV3-(2-N- phthalimidoethyπ-3.4.6.7-tetrahydro-2H-pyrimido[6.1-a]isoquinolin-4-one
(shown as (3 in Figure 1)
A mixture of 9,10-Dimethoxy-2-(2,4,6-trimethylphenylimino)-3,4,6,7-tetrahydro-2H- pyrimido[6,l-a]isoquinolin-4-one (which was prepared according to Preparation 2) (60.0g, 0.153 mol), potassium carbonate (191g, 1.38 mol), sodium iodide (137g, 0.92 mol) and N-(2-bromoethyl)phthalimide (234g, 0.92 mol) in 2-butanone (1500 ml) was stirred and heated at reflux, under nitrogen, for 4 days. After cooling to room temperature the mixture was filtered and the filtrate was evaporated in vacuo. The residue was treated with methanol (1000 ml) and the solid filtered off, washed with methanol and recrystallised from ethyl acetate to obtain the title compound as a pale yellow solid in yield 40. Og, 46%. Evaporation of the mother liquor and column chromatography of the residue on silica gel (CΗ2C-2 / MeOH 95:5) provided further product 11.7g, 13.5%. Preparation 4: 9.10-Dimethoxy-2-(2A6-trimethylphenylimino)-3-(2-arninoethyO- 3.4.6.7-tetrahydro-2H-pyrimido[6.1-a]isoquino-in-4-one (shown as (4) in Figure 1)
A mixture of 9,10-Dimethoxy-2-(2,4,6-trimethylphenylimino)-3-(2-N- phthalimidoethyl)-3,4,6,7-tetrahydro-2H-pyrimido[6,l-a]isoquinolin-4-one (22. Og, 0.039 mol), prepared according to Preparation 3, and hydrazine hydrate (11.3g, 0.195 mol) in chloroform (300 ml) and ethanol (460 ml) was stined at room temperature, under nitrogen, for 18h. Further hydrazine hydrate (2.9g, 0.05 mol) was added and the mixture was stirred a further 4h. After cooling in ice / water, the solid was removed by filtration and the filtrate evaporated in vacuo. The residue was dissolved in dichloromethane and the insoluble material was removed by filtration. The fitrate was dried (MgSO-i) and evaporated in vacuo to afford the title compound as a yellow foam in yield 16.2g, 96%.
PATENT
Novel crystalline acid addition salts forms of RPL-554 are claimed, wherein the salts, such as ethane- 1,2-disulfonic acid, ethanesulfonic acid, methanesulfonic acid, benzenesulfonic acid, p-toluenesulfonic acid, hydrochloric acid, hydrobromic acid, phosphoric acid or sulfuric acid. .
RPL554 (9, 10-dimethoxy-2-(2,4,6-trimethylphenylimino)-3-(/V-carbamoyl-2-aminoethyl)-3,4,6,7-tetrahydro-2H-pyrimido[6, l-a]isoquinolin-4-one) is a dual PDE3/PDE4 inhibitor and is described in WO 00/58308. As a combined PDE3/PDE4 inhibitor, RPL554 has both antiinflammatory and bronchodilatory activity and is useful in the treatment of respiratory disorders such as asthma and chronic obstructive pulmonary disease (COPD). The structure of RPL554 is shown below.

Owing to its applicability in the treatment of respiratory disorders, it is often preferable to administer RPL554 by inhalation. Franciosi et al. disclose a solution of RPL554 in a citrate-phosphate buffer at pH 3.2 (The Lancet: Respiratory Medicine 11/2013; l(9):714-27. DOI: 10.1016/S2213-2600(13)70187-5). The preparation of salts of RPL554 has not been described.
PATENT
http://www.google.ch/patents/WO2000058308A1?cl=en&hl=de
PATENT
http://www.google.ch/patents/WO2012020016A1?cl=en
U.S. Pat. No. 6,794,391, 7,378,424, and 7,105,663, which are each incorporated herein by reference, discloses compound RPL-554 (N-{2-[(2iT)-2-(mesityiimino)-9,10- dimethoxy-4-oxo-6,7-dihydro-2H-pyrimido[6,l-a]-isoquinolin-3 4H)-yl]ethyl}urea).

It would be beneficial to provide a composition of a stable polymorph of RPL-554, that has advanrtages over less stable polymorphs or amorphous forms, including
stability, compressibility, density, dissolution rates, increased potency or. lack toxicity.
| WO2000058308A1 * | Mar 29, 2000 | Oct 5, 2000 | Vernalis Limited | DERIVATIVES OF PYRIMIDO[6,1-a]ISOQUINOLIN-4-ONE |
| US6794391 | Sep 26, 2001 | Sep 21, 2004 | Vernalis Limited | Derivatives of pyrimido[6.1-a]isoquinolin-4-one |
| US7105663 | Feb 24, 2004 | Sep 12, 2006 | Rhinopharma Limited | Derivatives of pyrimido[6,1-a]isoquinolin-4-one |
| US7378424 | Feb 24, 2004 | May 27, 2008 | Verona Pharma Plc | Derivatives of pyrimido[6, 1-A]isoquinolin-4-one |
| WO2012020016A1 * | 9. Aug. 2011 | 16. Febr. 2012 | Verona Pharma Plc | Crystalline form of pyrimidio[6,1-a]isoquinolin-4-one compound |
| WO2014140647A1 | 17. März 2014 | 18. Sept. 2014 | Verona Pharma Plc | Drug combination |
| WO2014140648A1 | 17. März 2014 | 18. Sept. 2014 | Verona Pharma Plc | Drug combination |
| WO2015173551A1 * | 11. Mai 2015 | 19. Nov. 2015 | Verona Pharma Plc | New treatment |
| US8883857 | 8. März 2013 | 11. Nov. 2014 | Baylor College Of Medicine | Small molecule xanthine oxidase inhibitors and methods of use |
| US8883858 | 23. Juli 2014 | 11. Nov. 2014 | Baylor College Of Medicine | Small molecule xanthine oxidase inhibitors and methods of use |
| US8895626 | 23. Juli 2014 | 25. Nov. 2014 | Baylor College Of Medicine | Small molecule xanthine oxidase inhibitors and methods of use |
| US8987337 | 23. Juli 2014 | 24. März 2015 | Baylor College Of Medicine | Small molecule xanthine oxidase inhibitors and methods of use |
| US9061983 | 23. Juli 2014 | 23. Juni 2015 | Baylor College Of Medicine | Methods of inhibiting xanthine oxidase activity in a cell |
| US9062047 | 9. Aug. 2011 | 23. Juni 2015 | Verona Pharma Plc | Crystalline form of pyrimido[6,1-A] isoquinolin-4-one compound |
References
- Boswell-Smith V et al. The pharmacology of two novel long-acting phosphodiesterase 3/4 inhibitors, RPL554 [9,10-dimethoxy-2(2,4,6-trimethylphenylimino)-3-(n-carbamoyl-2-aminoethyl)-3,4,6,7-tetrahydro-2H-pyrimido[6,1-a]isoquinolin-4-one] and RPL565 [6,7-dihydro-2-(2,6-diisopropylphenoxy)-9,10-dimethoxy-4H-pyrimido[6,1-a]isoquinolin-4-one]. J Pharmacol Exp Ther. 2006 Aug;318(2):840-8. PMID 16682455
- Nick Paul Taylor for FierceBiotech. October 1, 2015 Verona sets sights on PhIIb after COPD drug comes through early trial
- Turner MJ et al. The dual phosphodiesterase 3 and 4 inhibitor RPL554 stimulates CFTR and ciliary beating in primary cultures of bronchial epithelia. Am J Physiol Lung Cell Mol Physiol. 2016 Jan 1;310(1):L59-70. PMID 26545902
- Jump up^ see US20040171828, identified in the citations of PMID 16682455
- ISIS Resources, PLC. August 23, 2006 Proposed Acquisition of Rhinopharma
REFERENCES
1: Calzetta L, Cazzola M, Page CP, Rogliani P, Facciolo F, Matera MG. Pharmacological characterization of the interaction between the dual phosphodiesterase (PDE) 3/4 inhibitor RPL554 and glycopyrronium on human isolated bronchi and small airways. Pulm Pharmacol Ther. 2015 Jun;32:15-23. doi: 10.1016/j.pupt.2015.03.007. Epub 2015 Apr 18. PubMed PMID: 25899618.
2: Franciosi LG, Diamant Z, Banner KH, Zuiker R, Morelli N, Kamerling IM, de Kam ML, Burggraaf J, Cohen AF, Cazzola M, Calzetta L, Singh D, Spina D, Walker MJ, Page CP. Efficacy and safety of RPL554, a dual PDE3 and PDE4 inhibitor, in healthy volunteers and in patients with asthma or chronic obstructive pulmonary disease: findings from four clinical trials. Lancet Respir Med. 2013 Nov;1(9):714-27. doi: 10.1016/S2213-2600(13)70187-5. Epub 2013 Oct 25. PubMed PMID: 24429275.
3: Wedzicha JA. Dual PDE 3/4 inhibition: a novel approach to airway disease? Lancet Respir Med. 2013 Nov;1(9):669-70. doi: 10.1016/S2213-2600(13)70211-X. Epub 2013 Oct 25. PubMed PMID: 24429260.
4: Calzetta L, Page CP, Spina D, Cazzola M, Rogliani P, Facciolo F, Matera MG. Effect of the mixed phosphodiesterase 3/4 inhibitor RPL554 on human isolated bronchial smooth muscle tone. J Pharmacol Exp Ther. 2013 Sep;346(3):414-23. doi: 10.1124/jpet.113.204644. Epub 2013 Jun 13. PubMed PMID: 23766543.
5: Gross N. The COPD pipeline XX. COPD. 2013 Feb;10(1):104-6. doi: 10.3109/15412555.2013.766103. PubMed PMID: 23413896.
6: Gross NJ. The COPD Pipeline XIV. COPD. 2012 Feb;9(1):81-3. doi: 10.3109/15412555.2012.646587. PubMed PMID: 22292600.
7: Boswell-Smith V, Spina D, Oxford AW, Comer MB, Seeds EA, Page CP. The pharmacology of two novel long-acting phosphodiesterase 3/4 inhibitors, RPL554 [9,10-dimethoxy-2(2,4,6-trimethylphenylimino)-3-(n-carbamoyl-2-aminoethyl)-3,4,6, 7-tetrahydro-2H-pyrimido[6,1-a]isoquinolin-4-one] and RPL565 [6,7-dihydro-2-(2,6-diisopropylphenoxy)-9,10-dimethoxy-4H-pyrimido[6,1-a]isoquino lin-4-one]. J Pharmacol Exp Ther. 2006 Aug;318(2):840-8. Epub 2006 May 8. PubMed PMID: 16682455.
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
N-{2-[(2E)-2-(mesitylimino)-9,10-dimethoxy-4-oxo-6,7-dihydro-2H-pyrimido[6,1-a]-isoquinolin-3(4H)-yl]ethyl}urea
|
|
| Identifiers | |
| PubChem | CID 9934746 |
| ChemSpider | 8110374 |
| Synonyms | 9,10-Dimethoxy-2-(2,4,6-trimethylphenylimino)-3-(N-carbamoyl-2-aminoethyl)-3,4,6,7-tetrahydro-2H-pyrimido[6,1-a]isoquinolin-4-one |
| Chemical data | |
| Formula | C26H31N5O4 |
| Molar mass | 477.554 g/mol |
///////////RPL-554, LS-193,855, 298680-25-8, UNII:3E3D8T1GIX, RPL554, RPL 554, phase 2, Chronic Obstructive Pulmonary Diseases , COPD, Allergic Rhinitis, Asthma Therapy, Cystic Fibrosis, Inflammation, Bronchodilators
Cc3cc(C)cc(C)c3N=c2cc1-c(cc4OC)c(cc4OC)CCn1c(=O)n2CCNC(N)=O
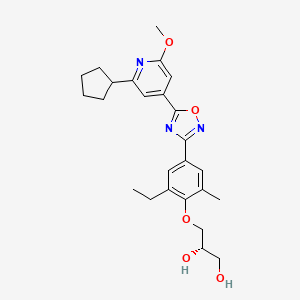
ACT-334441, Cenerimod an S1P receptor 1 agonist

ACT-334441
Cenerimod
UNII-Y333RS1786; Y333RS1786
S1P receptor 1 agonist
CAS 1262414-04-9
Chemical Formula: C25H31N3O5
Exact Mass: 453.22637
Martin Bolli, Cyrille Lescop, Boris Mathys,Keith Morrison, Claus Mueller, Oliver Nayler,Beat Steiner,
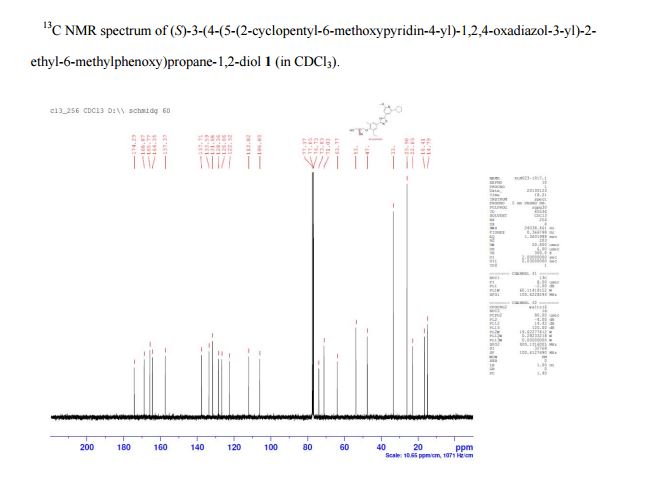
(S)-3-(4-(5-(2-cyclopentyl-6-methoxypyridin-4-yl)-1,2,4-oxadiazol-3-yl)-2-ethyl-6-methylphenoxy)propane-1,2-diol
(2S)-3-[4-[5-(2-cyclopentyl-6-methoxypyridin-4-yl)-1,2,4-oxadiazol-3-yl]-2-ethyl-6-methylphenoxy]propane-1,2-diol
(S)-3-{4-[5-(2-Cyclopentyl-6-methoxy-pyridin-4-yl)-[1,2,4]oxadiazol-3-yl]-2-ethyl-6-methyl-phenoxy}-propane-1,2-diol
| Mechanism Of Action | Sphingosine 1 phosphate receptor modulator |
|---|---|
| Who Atc Codes | L03A-X (Other immunostimulants) |
| Ephmra Codes | L3A (Immunostimulating Agents Excluding Interferons) |
| Indication | Systemic Lupus Erythematosus |
Cenerimod is a potent and orally active immunomodulator, exhibited EC50 value of 2.7 nM. Cenerimod is an agonist for the G protein-coupled receptor S1 P1/EDG1 and has a powerful and long-lasting immunomodulating effect which is achieved by reducing the number of circulating and infiltrating T- and B-lymphocytes, without affecting their maturation, memory, or expansion. Cenerimod may be useful for prevention or treatment of diseases associated with an activated immune system

CENERIMOD
ACT-334441; lysosphingolipid receptor agonist – Actelion; S1P1 receptor modulator – Actelion; Second selective S1P1 receptor agonist – Actelion; Sphingosine 1 phosphate receptor modulators – Actelion; Sphingosine 1-phosphate receptor 1 agonists – Actelion
- Mechanism of Action Lysosphingolipid receptor agonists
- Highest Development Phases
- Phase I/II Systemic lupus erythematosus
Most Recent Events
- 09 Jun 2016 Actelion terminates a phase I drug interaction trial for Systemic lupus erythematosus (In volunteers) in France (NCT02479204)
- 22 Dec 2015 Phase-I/II clinical trials in Systemic lupus erythematosus in Ukraine, Belarus (PO) (NCT02472795)
- 24 Sep 2015 Phase-I/II clinical trials in Systemic lupus erythematosus in USA (PO) (NCT02472795)
| # | Nct Number | Title | Recruitment | Conditions | Interventions | Phase | |
|---|---|---|---|---|---|---|---|
| 1 | NCT02472795 | Clinical Study to Investigate the Biological Activity, Safety, Tolerability, and Pharmacokinetics of ACT-334441 in Subjects With Systemic Lupus Erythematosus | Recruiting | Systemic Lupus Erythematosus | Drug: ACT-334441|Drug: Placebo | Phase 2 Actelion | |
| 2 | NCT02479204 | Drug Interaction Study of ACT-334441 With Cardiovascular Medications in Healthy Subjects | Suspended | Healthy Subjects | Drug: ACT-334441 2 mg|Drug: ACT-334441 4 mg|Drug: placebo|Drug: atenolol|Drug: diltiazem ER | Phase 1 Actelion |

The human immune system is designed to defend the body against foreign micro-organisms and substances that cause infection or disease. Complex regulatory mechanisms ensure that the immune response is targeted against the intruding substance or organism and not against the host. In some cases, these control mechanisms are unregulated and autoimmune responses can develop. A consequence of the uncontrolled inflammatory response is severe organ, cell, tissue or joint damage. With current treatment, the whole immune system is usually suppressed and the body’s ability to react to infections is also severely compromised. Typical drugs in this class include azathioprine, chlorambucil, cyclophosphamide, cyclosporin, or methotrexate. Corticosteroids which reduce inflammation and suppress the immune response, may cause side effects when used in long term treatment. Nonsteroidal anti-inflammatory drugs (NSAIDs) can reduce pain and inflammation, however, they exhibit considerable side effects. Alternative treatments include agents that activate or block cytokine signaling.
Orally active compounds with immunomodulating properties, without compromising immune responses and with reduced side effects would significantly improve current treatments of uncontrolled inflammatory diseases.
In the field of organ transplantation the host immune response must be suppressed to prevent organ rejection. Organ transplant recipients can experience some rejection even when they are taking immunosuppressive drugs. Rejection occurs most frequently in the first few weeks after transplantation, but rejection episodes can also happen months or even years after transplantation. Combinations of up to three or four medications are commonly used to give maximum protection against rejection while minimizing side effects. Current standard drugs used to treat the rejection of transplanted organs interfere with discrete intracellular pathways in the activation of T-type or B-type white blood cells. Examples of such drugs are cyclosporin, daclizumab, basiliximab, everolimus, or FK506, which interfere with cytokine release or signaling; azathioprine or leflunomide, which inhibit nucleotide synthesis; or 15-deoxyspergualin, an inhibitor of leukocyte differentiation.
The beneficial effects of broad immunosuppressive therapies relate to their effects; however, the generalized immunosuppression which these drugs produce diminishes the immune system’s defense against infection and malignancies. Furthermore, standard immunosuppressive drugs are often used at high dosages and can cause or accelerate organ damage.
SYNTHESIS
PATENT
https://www.google.com/patents/WO2011007324A1?cl=zh
The human immune system is designed to defend the body against foreign microorganisms and substances that cause infection or disease. Complex regulatory mechanisms ensure that the immune response is targeted against the intruding substance or organism and not against the host. In some cases, these control mechanisms are unregulated and autoimmune responses can develop. A consequence of the uncontrolled inflammatory response is severe organ, cell, tissue or joint damage. With current treatment, the whole immune system is usually suppressed and the body’s ability to react to infections is also severely compromised. Typical drugs in this class include azathioprine, chlorambucil, cyclophosphamide, cyclosporin, or methotrexate. Corticosteroids which reduce inflammation and suppress the immune response, may cause side effects when used in long term treatment. Nonsteroidal anti-inflammatory drugs (NSAIDs) can reduce pain and inflammation, however, they exhibit considerable side effects. Alternative treatments include agents that activate or block cytokine signaling.
Orally active compounds with immunomodulating properties, without compromising immune responses and with reduced side effects would significantly improve current treatments of uncontrolled inflammatory diseases.
In the field of organ transplantation the host immune response must be suppressed to prevent organ rejection. Organ transplant recipients can experience some rejection even when they are taking immunosuppressive drugs. Rejection occurs most frequently in the first few weeks after transplantation, but rejection episodes can also happen months or even years after transplantation. Combinations of up to three or four medications are commonly used to give maximum protection against rejection while minimizing side effects. Current standard drugs used to treat the rejection of transplanted organs interfere with discrete intracellular pathways in the activation of T-type or B-type white blood cells. Examples of such drugs are cyclosporin, daclizumab, basiliximab, everolimus, or FK506, which interfere with cytokine release or signaling; azathioprine or leflunomide, which inhibit nucleotide synthesis; or 15-deoxyspergualin, an inhibitor of leukocyte differentiation.
The beneficial effects of broad immunosuppressive therapies relate to their effects; however, the generalized immunosuppression which these drugs produce diminishes the immune system’s defense against infection and malignancies. Furthermore, standard immunosuppressive drugs are often used at high dosages and can cause or accelerate organ damage.
Description of the invention
The present invention provides novel compounds of Formula (I) that are agonists for the G protein-coupled receptor S1 P1/EDG1 and have a powerful and long-lasting immunomodulating effect which is achieved by reducing the number of circulating and infiltrating T- and B-lymphocytes, without affecting their maturation, memory, or expansion. The reduction of circulating T- / B-lymphocytes as a result of S1 P1/EDG1 agonism, possibly in combination with the observed improvement of endothelial cell layer function associated with S1 P1/EDG1 activation, makes such compounds useful to treat uncontrolled inflammatory diseases and to improve vascular functionality. Prior art document WO 2008/029371 discloses compounds that act as S1 P1/EDG1 receptor agonists and show an immunomodulating effect as described above. Unexpectedly, it has been found that the compounds of the present invention have a reduced potential to constrict airway tissue/vessels when compared to compounds of the prior art document WO 2008/029371. The compounds of the present invention therefore demonstrate superiority with respect to their safety profile, e.g. a lower risk of bronchoconstriction.
Examples of WO 2008/029371 , which are considered closest prior art analogues are shown in Figure 1.

Figure 1 : Structure of Examples of prior art document WO 2008/029371 , which are considered closest analogues to the compounds of the present invention.
The data on the constriction of rat trachea rings compiled in Table 1 illustrate the superiority of the compounds of the present invention as compared to compounds of prior art document WO 2008/029371.
For instance, the compounds of Example 1 and 6 of the present invention show a significantly reduced potential to constrict rat trachea rings when compared to the compounds of prior art Examples 222 and 226 of WO 2008/029371 , respectively. Furthermore, the compounds of Example 1 and 6 of the present invention also show a reduced potential to constrict rat trachea rings when compared to the compounds of prior art Examples 196 and 204 of WO 2008/029371 , respectively. These data demonstrate that compounds wherein R1 represents 3-pentyl and R2 represents methoxy are superior compared to the closest prior art compounds of WO 2008/029371 , i.e. the compounds wherein R1 represents an isobutyl and R2 represents methoxy or wherein R1represents methyl and R2 represents 3-pentyl. Moreover, also the compound of Example 16 of the present invention, wherein R1 is 3-methyl-but-1-yl and R2 is methoxy, exhibits a markedly reduced potential to constrict rat trachea rings when compared to its closest analogue prior art Example 226 of WO 2008/029371 wherein R1 is isobutyl and R2 is methoxy.
The unexpected superiority of the compounds of the present invention is also evident from the observation that the compounds of Example 2 and 7 of the present invention show a markedly reduced potential to constrict rat trachea rings when compared to the compounds of prior art Examples 229 and 233 of WO 2008/029371 , respectively. This proves that compounds wherein R1represents cyclopentyl and R2 represents methoxy are superior compared to the closest prior art compounds of WO 2008/029371 , i.e. the compounds wherein R1 represents methyl and R2 represents cyclopentyl.
Also, the compound of Example 3 of the present invention exhibits the same low potential to constrict rat trachea rings as its S-enantiomer, i.e. the compound of Example 2 of the present invention, indicating that the configuration at this position has no significant effect on trachea constriction. Furthermore, also Example 21 of the present invention exhibits the same low potential to constrict rat trachea rings as present Example 2, which differs from Example 21 only by the linker A (forming a 5-pyridin-4-yl-[1 ,2,4]oxadiazole instead of a 3- pyridin-4-yl-[1 ,2,4]oxadiazole). This indicates that also the nature of the oxadiazole is not critical regarding trachea constriction.
Table 1 : Rat trachea constriction in % of the constriction induced by 50 mM KCI. n.d. = not determined. For experimental details and further data see Example 33.


result obtained at a compound concentration of 300 nM.
The compounds of the present invention can be utilized alone or in combination with standard drugs inhibiting T-cell activation, to provide a new immunomodulating therapy with a reduced propensity for infections when compared to standard immunosuppressive therapy. Furthermore, the compounds of the present invention can be used in combination with reduced dosages of traditional immunosuppressant therapies, to provide on the one hand effective immunomodulating activity, while on the other hand reducing end organ damage associated with higher doses of standard immunosuppressive drugs. The observation of improved endothelial cell layer function associated with S1 P1/EDG1 activation provides additional benefits of compounds to improve vascular function.
The nucleotide sequence and the amino acid sequence for the human S1 P1/EDG1 receptor are known in the art and are published in e.g.: HIa, T., and Maciag, T., J. Biol
Chem. 265 (1990), 9308-9313; WO 91/15583 published 17 October 1991 ; WO 99/46277 published 16 September 1999. The potency and efficacy of the compounds of Formula (I) are assessed using a GTPγS assay to determine EC5O values and by measuring the circulating lymphocytes in the rat after oral administration, respectively (see in experimental part). i) In a first embodiment, the invention relates to pyridine compounds of the Formula (I),

Formula (I)
PATENT
WO 2013175397
https://www.google.com/patents/WO2013175397A1?cl=en
Pyridine-4-yl derivatives of formula (PD),

Formula (PD) A represents

(the asterisks indicate the bond that is linked to the pyridine group of Formula (PD));
Ra represents 3-pentyl, 3-methyl-but-1-yl, cyclopentyl, or cyclohexyl;
Rb represents methoxy;
Rc represents 2,3-dihydroxypropoxy, -OCH2-CH(OH)-CH2-NHCO-CH2OH,
-OCH2-CH(OH)-CH2N(CH3)-CO-CH2OH, -NHS02CH3, or -NHS02CH2CH3; and
Rd represents ethyl or chloro.)
disclosed in WO201 1007324, have immunomodulating activity through their S1 P1/EDG1 receptor agonistic activity. Therefore, those pyridine-4-yl derivatives are useful for prevention and / or treatment of diseases or disorders associated with an activated immune system, including rejection of transplanted organs such as kidney, liver, heart, lung, pancreas, cornea, and skin; graft-versus-host diseases brought about by stem cell transplantation; autoimmune syndromes including rheumatoid arthritis, multiple sclerosis, inflammatory bowel diseases such as Crohn’s disease and ulcerative colitis, psoriasis, psoriatic arthritis, thyroiditis such as Hashimoto’s thyroiditis, uveo-retinitis; atopic diseases such as rhinitis, conjunctivitis, dermatitis; asthma; type I diabetes; post-infectious autoimmune diseases including rheumatic fever and post-infectious glomerulonephritis; solid cancers and tumor metastasis. 2-Cyclopentyl-6-methoxy-isonicotinic acid, which is also disclosed in WO201 1007324, is a useful intermediate for the synthesis of the pyridine-4-yl derivatives of formula (PD), wherein Ra is a cyclopentyl group.
In the process described in WO201 1007324, 2-cyclopentyl-6-methoxy-isonicotinic acid was prepared according to the following reaction scheme 1 :

Compound D Compound E
Rieke Zinc: cyclopentylzinc bromide;
PdCI2(dppf)dcm: 1 ,1 ‘-Bis(diphenylphosphino)ferrocene-palladium(ll)dichloride
dichloromethane complex
However, the abovementioned process has drawbacks for larger scale, i.e. industrial scale synthesis of 2-cyclopentyl-6-methoxy-isonicotinic acid, for the following reasons:
a) The commercially available starting material, 2,6-dichloro-isonicotinic acid (Compound A) is expensive.
b) The conversion of Compound C to Compound D is cost-intensive. The reaction has to be performed under protective atmosphere with expensive palladium catalysts and highly reactive and expensive Rieke zinc complex. Such synthesis steps are expensive to scale up and it was therefore highly desired to find alternative synthesis methods.
Even though Goldsworthy, J. Chem. Soc. 1934, 377-378 discloses the preparation of 1 -cyclopentylethanone, which is a key building block in the new process of the present invention, by using ethyl 1 -acetoacetate as a starting material, this synthesis was far from being suitable in an industrial process. The reported yield was low (see also under “Referential Examples” below). Scheme 2

ethyl 1 -acetylcyclo- 1-cyclopentyl- pentanecarboxylate ethanone
Besides the early work by Goldsworthy there are several recent examples for the preparation of 1 -cyclopentylethanone described in the literature. Such examples include:
1 ) Addition of methyl lithium to a N-cyclopentanecarbonyl-N,0-dimethylhydroxylamine at -78°C in a yield of 77%. US2006/199853 A1 , 2006 and US2006/223884 A1 , 2006.
2) Addition of methyl lithium to a cyclopentyl carboxylic acid in diethylether at -78°C in a yield of 81 %. J. Am. Chem. Soc, 1983, 105, 4008-4017.
3) Addition of methylmagnesiumbromide to cyclopentanecarbonitrile.
Bull. Soc. Chim. Fr., 1967, 3722-3729.
4) Oxidation of 1 -cyclopentylethanol with chromtrioxide. US5001 140 A1 , 1991.
WO2009/71707 A1 , 2009.
5) Addition of cyclopentylmagnesium bromide to acetic anhydride at -78 °C with a yield of 54%. WO2004/74270 A2, 2004.
6) Synthesis of 1-cyclopentylethanone in 5 steps from cyclopentanone. Zhang, Pang; Li, Lian-chu, Synth. Commun., 1986, 16, 957-966.
However, the processes described in the above-listed publications are not efficient for scale-up since they require cryogenic temperatures, expensive starting materials, toxic reagents or many steps. The lack of an efficient process to manufacture 1 -cyclopentylethanone is further also mirrored by the difficulty in sourcing this compound on kilogram scale for a reasonable price and delivery time. Therefore, the purpose of the present invention is to provide a new, efficient and cost effective process for the preparation of 2-cyclopentyl-6-methoxy-isonicotinic acid, which is suitable for industrial scale synthesis.
Patent
Disclosed in WO2011007324, have immunomodulating activity through their S1P1/EDG1 receptor agonistic activity. Therefore, those pyridine-4-yl derivatives are useful for prevention and/or treatment of diseases or disorders associated with an activated immune system, including rejection of transplanted organs such as kidney, liver, heart, lung, pancreas, cornea, and skin; graft-versus-host diseases brought about by stem cell transplantation; autoimmune syndromes including rheumatoid arthritis, multiple sclerosis, inflammatory bowel diseases such as Crohn’s disease and ulcerative colitis, psoriasis, psoriatic arthritis, thyroiditis such as Hashimoto’s thyroiditis, uveo-retinitis; atopic diseases such as rhinitis, conjunctivitis, dermatitis; asthma; type I diabetes; post-infectious autoimmune diseases including rheumatic fever and post-infectious glomerulonephritis; solid cancers and tumor metastasis. 2-Cyclopentyl-6-methoxy-isonicotinic acid, which is also disclosed in WO2011007324, is a useful intermediate for the synthesis of the pyridine-4-yl derivatives of formula (PD), wherein Ra is a cyclopentyl group.
 Rieke Zinc: cyclopentylzinc bromide;
Rieke Zinc: cyclopentylzinc bromide;
PdCl2(dppf)dcm: 1,1′-Bis(diphenylphosphino)ferrocene-palladium(II)dichloride dichloromethane complex


EXAMPLES
Example 1a
1-Cyclopentylethanone

Example 1 b
1-Cyclopentylethanone
Example 1c
1-Cyclopentylethanone
Example 1d
1-Cyclopentylethanone
Example 1e
1-Cyclopentylethanone
Example 1f
Tert-butyl 1-acetylcyclopentanecarboxylate

Example 1 g
Tert-butyl 1-acetylcyclopentanecarboxylate
Example 2
2-Cyclopentyl-6-hydroxyisonicotinic acid

Example 3
Methyl 2-cyclopentyl-6-hydroxyisonicotinate

Example 4a
Methyl 2-chloro-6-cyclopentylisonicotinate

Example 4b
Methyl 2-chloro-6-cyclopentylisonicotinate
Example 5
2-Cyclopentyl-6-methoxyisonicotinic acid

Example 6
Ethyl 4-cyclopentyl-2,4-dioxobutanoate

Example 7
Ethyl 3-cyano-6-cyclopentyl-2-hydroxyisonicotinate

REFERENTIAL EXAMPLES

Referential Example 1
Referential Example 2
PATENT
https://www.google.com/patents/US8658675
Martin Bolli, Cyrille Lescop, Boris Mathys,Keith Morrison, Claus Mueller, Oliver Nayler,Beat Steiner,
novel compounds of Formula (I) that are agonists for the G protein-coupled receptor S1P1/EDG1 and have a powerful and long-lasting immunomodulating effect which is achieved by reducing the number of circulating and infiltrating T- and B-lymphocytes, without affecting their maturation, memory, or expansion. The reduction of circulating T-/B-lymphocytes as a result of S1P1/EDG1 agonism, possibly in combination with the observed improvement of endothelial cell layer function associated with S1P1/EDG1 activation, makes such compounds useful to treat uncontrolled inflammatory diseases and to improve vascular functionality. Prior art document WO 2008/029371 discloses compounds that act as S1P1/EDG1 receptor agonists and show an immunomodulating effect as described above. Unexpectedly, it has been found that the compounds of the present invention have a reduced potential to constrict airway tissue/vessels when compared to compounds of the prior art document WO 2008/029371. The compounds of the present invention therefore demonstrate superiority with respect to their safety profile, e.g. a lower risk of bronchoconstriction.
Examples of WO 2008/029371, which are considered closest prior art analogues are shown in FIG. 1.


The data on the constriction of rat trachea rings compiled in Table 1 illustrate the superiority of the compounds of the present invention as compared to compounds of prior art document WO 2008/029371.
For instance, the compounds of Example 1 and 6 of the present invention show a significantly reduced potential to constrict rat trachea rings when compared to the compounds of prior art Examples 222 and 226 of WO 2008/029371, respectively. Furthermore, the compounds of Example 1 and 6 of the present invention also show a reduced potential to constrict rat trachea rings when compared to the compounds of prior art Examples 196 and 204 of WO 2008/029371, respectively. These data demonstrate that compounds wherein R1 represents 3-pentyl and R2represents methoxy are superior compared to the closest prior art compounds of WO 2008/029371, i.e. the compounds wherein R1 represents an isobutyl and R2represents methoxy or wherein R1 represents methyl and R2 represents 3-pentyl. Moreover, also the compound of Example 16 of the present invention, wherein R1is 3-methyl-but-1-yl and R2 is methoxy, exhibits a markedly reduced potential to constrict rat trachea rings when compared to its closest analogue prior art Example 226 of WO 2008/029371 wherein R1 is isobutyl and R2 is methoxy.
The unexpected superiority of the compounds of the present invention is also evident from the observation that the compounds of Example 2 and 7 of the present invention show a markedly reduced potential to constrict rat trachea rings when compared to the compounds of prior art Examples 229 and 233 of WO 2008/029371, respectively. This proves that compounds wherein R1 represents cyclopentyl and R2 represents methoxy are superior compared to the closest prior art compounds of WO 2008/029371, i.e. the compounds wherein R1represents methyl and R2 represents cyclopentyl.
Preparation of Intermediates2-Chloro-6-methyl-isonicotinic acid
The title compound and its ethyl ester are commercially available.
2-(1-Ethyl-propyl)-6-methoxy-isonicotinic acid
a) To a solution of 2,6-dichloroisonicotinic acid (200 g, 1.04 mol) in methanol (3 L), 32% aq. NaOH (770 mL) is added. The stirred mixture becomes warm (34° C.) and is then heated to 70° C. for 4 h before it is cooled to rt. The mixture is neutralised by adding 32% aq. HCl (100 mL) and 25% aq. HCl (700 mL). The mixture is stirred at rt overnight. The white precipitate that forms is collected, washed with methanol and dried. The filtrate is evaporated and the residue is suspended in water (200 mL). The resulting mixture is heated to 60° C. Solid material is collected, washed with water and dried. The combined crops give 2-chloro-6-methoxy-isonicotinic acid (183 g) as a white solid; LC-MS: tR=0.80 min, [M+1]+=187.93.
b) To a suspension of 2-chloro-6-methoxy-isonicotinic acid (244 g, 1.30 mol) in methanol (2.5 L), H2SO4 (20 mL) is added. The mixture is stirred at reflux for 24 h before it is cooled to 0° C. The solid material is collected, washed with methanol (200 mL) and water (500 mL) and dried under HV to give 2-chloro-6-methoxy-isonicotinic acid methyl ester (165 g) as a white solid; LC-MS: tR=0.94 min, [M+1]+=201.89.
c) Under argon, Pd(dppf) (3.04 g, 4 mmol) is added to a solution of 2-chloro-6-methoxy-isonicotinic acid methyl ester (50 g, 0.248 mol) in THF (100 mL). A 0.5 M solution of 3-pentylzincbromide in THF (550 mL) is added via dropping funnel. Upon complete addition, the mixture is heated to 85° C. for 18 h before it is cooled to rt. Water (5 mL) is added and the mixture is concentrated. The crude product is purified by filtration over silica gel (350 g) using heptane:EA 7:3 to give 2-(1-ethyl-propyl)-6-methoxy-isonicotinic acid methyl ester (53 g) as a pale yellow oil; 1H NMR (CDCl3): δ0.79 (t, J=7.5 Hz, 6H), 1.63-1.81 (m, 4H), 2.47-2.56 (m, 1H), 3.94 (s, 3H), 3.96 (s, 3H), 7.12 (d, J=1.0 Hz, 1H), 7.23 (d, J=1.0 Hz, 1H).
d) A solution of 2-(1-ethyl-propyl)-6-methoxy-isonicotinic acid methyl ester (50 g, 0.211 mol) in ethanol (250 mL), water (50 mL) and 32% aq. NaOH (50 mL) is stirred at 80° C. for 1 h. The mixture is concentrated and the residue is dissolved in water (200 mL) and extracted with TBME. The org. phase is separated and washed once with water (200 mL). The TBME phase is discarded. The combined aq. phases are acidified by adding 25% aq. HCl and then extracted with EA (400+200 mL). The combined org. extracts are concentrated. Water (550 mL) is added to the remaining residue. The mixture is heated to 70° C., cooled to rt and the precipitate that forms is collected and dried to give the title compound (40.2 g) as a white solid; LC-MS: tR=0.95 min, [M+1]+=224.04; 1H NMR (D6-DMSO): δ 0.73 (t, J=7.3 Hz, 6H), 1.59-1.72 (m, 4H), 2.52-2.58 (m, 1H), 3.88 (s, 3H), 7.00 (d, J=1.0 Hz, 1H), 7.20 (d, J=1.0 Hz, 1H).
2-Methoxy-6-(3-methyl-butyl)-isonicotinic acid
The title compound is prepared in analogy to 2-(1-ethyl-propyl)-6-methoxy-isonicotinic acid; LC-MS: tR=0.94 min, [M+1]+=224.05; 1H NMR (D6-DMSO): δ 0.92 (d, J=5.8 Hz, 6H), 1.54-1.62 (m, 3H), 2.70-2.76 (m, 2H), 3.88 (s, 3H), 6.99 (s, 1H), 7.25 (s, 1H), 13.52 (s).
2-Cyclopentyl-6-methoxy-isonicotinic acid
The title compound is prepared in analogy to 2-(1-ethyl-propyl)-6-methoxy-isonicotinic acid; LC-MS: tR=0.93 min, [M+1]+=222.02; 1H NMR (CDCl3): δ 1.68-1.77 (m, 2H), 1.81-1.90 (m, 4H), 2.03-2.12 (m, 2H), 3.15-3.25 (m, 1H), 3.99 (s, 3H), 7.18 (d, J=1.0 Hz, 1H), 7.35 (d, J=0.8 Hz, 1H).
2-Cyclohexyl-6-methoxy-isonicotinic acid
The title compound is prepared in analogy to 2-(1-ethyl-propyl)-6-methoxy-isonicotinic acid; LC-MS: tR=0.98 min, [M+1]+=236.01; 1H NMR (D6-DMSO): δ 1.17-1.29 (m, 1H), 1.31-1.43 (m, 2H), 1.44-1.55 (m, 2H), 1.67-1.73 (m, 1H), 1.76-1.83 (m, 2H), 1.84-1.92 (m, 2H), 2.66 (tt, J=11.3, 3.3 Hz, 1H), 3.88 (s, 3H), 7.00 (d, J=1.0 Hz, 1H), 7.23 (d, J=1.0 Hz, 1H).
2-Cyclopentyl-N-hydroxy-6-methoxy-isonicotinamidine
a) A solution of 2-cyclopentyl-6-methoxy-isonicotinic acid methyl ester (3.19 g, 13.6 mmol) in 7 N NH3 in methanol (50 mL) is stirred at 60° C. for 18 h. The solvent is removed in vacuo and the residue is dried under HV to give crude 2-cyclopentyl-6-methoxy-isonicotinamide (3.35 g) as a pale yellow solid; LC-MS**: tR=0.57 min, [M+1]+=221.38.
b) Pyridine (8.86 g, 91.3 mmol) is added to a solution of 2-cyclopentyl-6-methoxy-isonicotinamide (3.35 g, 15.2 mmol) in DCM (100 mL). The mixture is cooled to 0° C. before trifluoroacetic acid anhydride (9.58 g, 45.6 mmol) is added portionwise. The mixture is stirred at 0° C. for 1 h before it is diluted with DCM (100 mL) and washed with sat. aq. NaHCO3 solution (100 mL) and brine (100 mL). The separated org. phase is dried over MgSO4, filtered and concentrated. The crude product is purified by CC on silica gel eluting with heptane:EA 9:1 to give 2-cyclopentyl-6-methoxy-isonicotinonitrile (2.09 g) as pale yellow oil; LC-MS**: tR=0.80 min, [M+1]+=not detectable; 1H NMR (D6-DMSO): δ 1.61-1.82 (m, 6H), 1.94-2.03 (m, 2H), 3.16 (quint, J=7.8 Hz, 1H), 3.89 (s, 3H), 7.15 (s, 1H), 7.28 (s, 1H).
c) To a solution of 2-cyclopentyl-6-methoxy-isonicotinonitrile (2.09 g, 10.3 mmol) in methanol (100 mL), hydroxylamine hydrochloride (2.15 g, 31.0 mmol) and NaHCO3 (3.04 g, 36.2 mmol) are added. The mixture is stirred at 60° C. for 18 h before it is filtered and the filtrate is concentrated. The residue is dissolved in EA (300 mL) and washed with water (30 mL). The washings are extracted back with EA (4×100 mL) and DCM (4×100 mL). The combined org. extracts are dried over MgSO4, filtered, concentrated and dried under HV to give the title compound (2.74 g) as a white solid; LC-MS**: tR=0.47 min, [M+1]+=236.24; 1H NMR (D6-DMSO): δ 1.61-1.82 (m, 6H), 1.92-2.01 (m, 2H), 3.04-3.13 (m, 1H), 3.84 (s, 3H), 5.90 (s, 2H), 6.86 (s, 1H), 7.13 (s, 1H), 9.91 (s, 1H).
2-Cyclopentyl-6-methoxy-isonicotinic acid hydrazide
a) To a solution of 2-cyclopentyl-6-methoxy-isonicotinic acid (2.00 g, 9.04 mmol), hydrazinecarboxylic acid benzyl ester (1.50 g, 9.04 mmol) and DIPEA (2.34 g, 18.1 mmol) in DCM (40 mL), TBTU (3.19 g, 9.94 mmol) is added. The mixture is stirred at rt for 2 h before it is diluted with EA (250 mL), washed twice with sat. aq. NaHCO3 solution (150 mL) followed by brine (100 mL), dried over MgSO4, filtered and concentrated. The crude product is purified by CC on silica gel eluting with heptane:EA 4:1 to give N′-(2-cyclopentyl-6-methoxy-pyridine-4-carbonyl)-hydrazinecarboxylic acid benzyl ester (2.74 g) as pale yellow oil; LC-MS**: tR=0.74 min, [M+1]+=369.69; 1H NMR (D6-DMSO): δ 1.62-1.83 (m, 6H), 1.95-2.05 (m, 2H), 3.10-3.21 (m, 1H), 3.88 (s, 3H), 5.13 (s, 2H), 6.97 (s, 1H), 7.23 (s, 1H), 7.28-7.40 (m, 5H), 9.45 (s, 1H), 10.52 (s, 1H).
b) Pd/C (500 mg, 10% Pd) is added to a solution of N′-(2-cyclopentyl-6-methoxy-pyridine-4-carbonyl)-hydrazinecarboxylic acid benzyl ester (2.74 g, 7.42 mmol) in THF (50 mL) and methanol (50 mL). The mixture is stirred at rt under 1 bar of H2 for 25 h. The catalyst is removed by filtration and the filtrate is concentrated and dried under HV to give the title compound (1.58 g) as an off-white solid; LC-MS**: tR=0.51 min, [M+1]+=236.20; 1H NMR (D6-DMSO): δ 1.60-1.82 (m, 6H), 1.94-2.03 (m, 2H), 3.08-3.19 (m, 1H), 3.86 (s, 3H), 4.56 (s br, 2H), 6.93 (d, J=1.0 Hz, 1H), 7.20 (d, J=1.0 Hz, 1H), 9.94 (s, 1H).
3-Ethyl-4-hydroxy-5-methyl-benzonitrile
The title compound is prepared from 3-ethyl-4-hydroxy-5-methyl-benzaldehyde following literature procedures (A. K. Chakraborti, G. Kaur, Tetrahedron 55 (1999) 13265-13268); LC-MS: tR=0.90 min; 1H NMR (CDCl3): δ1.24 (t, J=7.6 Hz, 3H), 2.26 (s, 3H), 2.63 (q, J=7.6 Hz, 2H), 5.19 (s, 1H), 7.30 (s, 2H).
3-Chloro-4-hydroxy-5-methyl-benzonitrile
The title compound is prepared from commercially available 2-chloro-6-methyl-phenol in analogy to literature procedures (see 3-ethyl-4-hydroxy-5-methyl-benzonitrile); LC-MS: tR=0.85 min. 1H NMR (CDCl3): δ2.33 (s, 3H), 6.10 (s, 1H), 7.38 (s, 1H), 7.53 (d, J=1.8 Hz, 1H).
3-Ethyl-4,N-dihydroxy-5-methyl-benzamidine
The title compound is prepared from 3-ethyl-4-hydroxy-5-methyl-benzonitrile or from commercially available 2-ethyl-6-methyl-phenol following literature procedures (G. Trapani, A. Latrofa, M. Franco, C. Altomare, E. Sanna, M. Usala, G. Biggio, G. Liso, J. Med. Chem. 41 (1998) 1846-1854; A. K. Chakraborti, G. Kaur, Tetrahedron 55 (1999) 13265-13268; E. Meyer, A. C. Joussef, H. Gallardo, Synthesis 2003, 899-905); LC-MS: tR=0.55 min; 1H NMR (D6-DMSO): δ 9.25 (s br, 1H), 7.21 (s, 2H), 5.56 (s, 2H), 2.55 (q, J=7.6 Hz, 2H), 2.15 (s, 3H), 1.10 (t, J=7.6 Hz, 3H).
3-Chloro-4,N-dihydroxy-5-methyl-benzamidine
The title compound is prepared from commercially available 2-chloro-6-methyl-phenol in analogy to literature procedures (e.g. B. Roth et al. J. Med. Chem. 31 (1988) 122-129; and literature cited for 3-ethyl-4,N-dihydroxy-5-methyl-benzamidine); 3-chloro-4-hydroxy-5-methyl-benzaldehyde: LC-MS: tR=0.49 min, [M+1]+=201.00; 1H NMR 82.24 (s, 2H), 2.35 (s, 4H), 5.98 (s br, 1H), 7.59 (d, J=1.8 Hz, 1H), 7.73 (d, J=1.8 Hz, 1H), 9.80 (s, 1H); 3-chloro-4,N-dihydroxy-5-methyl-benzamidine: 1H NMR (D6-DMSO): δ 2.21 (s, 3H), 5.72 (s br, 2H), 7.40 (s, 1H), 7.48 (s, 1H), 9.29 (s br, 1H), 9.48 (s br, 1H).
(R)-4-(2,2-Dimethyl-[1,3]dioxolan-4-ylmethoxy)-3-ethyl-N-hydroxy-5-methyl-benzamidine
a) To a solution of 3-ethyl-4-hydroxy-5-methyl-benzonitrile (2.89 g, 17.9 mmol) in THF (80 mL), (R)-(2,2-dimethyl-[1,3]dioxolan-4-yl)methanol (2.84 g, 21.5 mmol) followed by triphenylphosphine (5.81 g, 21.5 mmol) is added. The mixture is cooled with an ice-bath before DEAD (9.36 g, 21.5 mmol) is added dropwise. The mixture is stirred at rt for 1 h, the solvent is removed in vacuo and the residue is purified by CC on silica gel eluting with heptane:EA 85:15 to give (R)-4-(2,2-dimethyl-[1,3]dioxolan-4-ylmethoxy)-3-ethyl-5-methyl-benzonitrile (4.45 g) as a pale yellow oil; LC-MS**: tR=0.75 min, [M+1]+=not detected; 1H NMR (CDCl3): δ1.25 (t, J=7.5 Hz, 3H), 1.44 (s, 3H), 1.49 (s, 3H), 2.34 (s, 3H), 2.65-2.77 (m, 2H), 3.80-3.90 (m, 2H), 3.94-4.00 (m, 1H), 4.21 (t, J=7.3 Hz, 1H), 4.52 (quint, J=5.8 Hz, 1H), 7.35 (s, 1H), 7.38 (s, 1H).
b) To a mixture of (R)-4-(2,2-dimethyl-[1,3]dioxolan-4-ylmethoxy)-3-ethyl-5-methyl-benzonitrile (4.45 g, 16.2 mmol) and NaHCO3 (4.75 g, 56.6 mmol) in methanol (30 mL), hydroxylamine hydrochloride (3.37 g, 48.5 mmol) is added. The mixture is stirred at 60° C. for 18 h before it is filtered and the solvent of the filtrate is removed in vacuo. The residue is dissolved in EA and washed with a small amount of water and brine. The org. phase is separated, dried over MgSO4, filtered, concentrated and dried to give the title compound (5.38 g) as a white solid; LC-MS**: tR=0.46 min, [M+1]+=309.23; 1H NMR (D6-DMSO): δ 1.17 (t, J=7.5 Hz, 3H), 1.33 (s, 3H), 1.38 (s, 3H), 2.25 (s, 3H), 2.57-2.69 (m, 2H), 3.73-3.84 (m, 3H), 4.12 (t, J=7.0 Hz, 1H), 4.39-4.45 (m, 1H), 5.76 (s br, 2H), 7.34 (s, 1H), 7.36 (s, 1H), 9.47 (s, 1H).
(R)-3-Chloro-4-(2,2-dimethyl-[1,3]dioxolan-4-ylmethoxy)-N-hydroxy-5-methyl-benzamidine
The title compound is obtained as a colorless oil (1.39 g) in analogy to (R)-4-(2,2-dimethyl-[1,3]dioxolan-4-ylmethoxy)-3-ethyl-N-hydroxy-5-methyl-benzamidine starting from 3-chloro-4-hydroxy-5-methyl-benzonitrile and L-α,β-isopropyliden glycerol; LC-MS: tR=0.66 min, [M+H]+=314.96.
(S)-4-(3-Amino-2-hydroxypropoxy)-3-ethyl-5-methylbenzonitrile
a) To a solution of 3-ethyl-4-hydroxy-5-methyl-benzonitrile (5.06 g, 31.4 mmol) in THF (80 mL), PPh3 (9.06 g, 34.5 mmol) and (R)-glycidol (2.29 mL, 34.5 mmol) are added. The mixture is cooled to 0° C. before DEAD in toluene (15.8 mL, 34.5 mmol) is added. The mixture is stirred for 18 h while warming up to rt. The solvent is evaporated and the crude product is purified by CC on silica gel eluting with heptane:EA 7:3 to give 3-ethyl-5-methyl-4-oxiranylmethoxy-benzonitrile (5.85 g) as a yellow oil; LC-MS: tR=0.96 min; [M+42]+=259.08.
b) The above epoxide is dissolved in 7 N NH3 in methanol (250 mL) and the solution is stirred at 65° C. for 18 h. The solvent is evaporated to give crude (S)-4-(3-amino-2-hydroxypropoxy)-3-ethyl-5-methylbenzonitrile (6.23 g) as a yellow oil; LC-MS: tR=0.66 min; [M+1]+=235.11.
N—((S)-3-[2-Ethyl-4-(N-hydroxycarbamimidoyl)-6-methyl-phenoxy]-2-hydroxy-propyl)-2-hydroxy-acetamide
a) To a solution of (S)-4-(3-amino-2-hydroxypropoxy)-3-ethyl-5-methylbenzonitrile (6.23 g, 26.59 mmol) in THF (150 mL), glycolic acid (2.43 g, 31.9 mmol), HOBt (4.31 g, 31.9 mmol), and EDC hydrochloride (6.12 g, 31.9 mmol) are added. The mixture is stirred at rt for 18 h before it is diluted with sat. aq. NaHCO3 and extracted twice with EA. The combined org. extracts are dried over MgSO4, filtered and concentrated. The crude product is purified by CC with DCM containing 8% of methanol to give (S)—N-[3-(4-cyano-2-ethyl-6-methyl-phenoxy)-2-hydroxy-propyl]-2-hydroxy-acetamide (7.03 g) as a yellow oil; LC-MS: tR=0.74 min, [M+1]+=293.10; 1H NMR (CDCl3): δ 1.25 (t, J=7.5 Hz, 3H), 2.32 (s, 3H), 2.69 (q, J=7.5 Hz, 2H), 3.48-3.56 (m, 3H), 3.70-3.90 (m, 3H), 4.19 (s, br, 3H), 7.06 (m, 1H), 7.36 (s, 1H), 7.38 (s, 1H).
b) The above nitrile is converted to the N-hydroxy-benzamidine according to literature procedures (e.g. E. Meyer, A. C. Joussef, H. Gallardo, Synthesis 2003, 899-905); LC-MS: tR=0.51 min, [M+1]+=326.13; 1H NMR (D6-DMSO): δ 1.17 (t, J=7.4 Hz, 3H), 2.24 (s, 3H), 2.62 (q, J=7.4 Hz, 2H), 3.23 (m, 1H), 3.43 (m, 1H), 3.67 (m, 2H), 3.83 (s, 2H), 3.93 (m, 1H), 5.27 (s br, 1H), 5.58 (s br, 1H), 5.70 (s, 2H), 7.34 (s, 1H), 7.36 (s, 1H), 7.67 (m, 1H), 9.46 (s br, 1H).
(S)—N-(3-[2-Chloro-4-(N-hydroxycarbamimidoyl)-6-methyl-phenoxy]-2-hydroxy-propyl)-2-hydroxy-acetamide
The title compound is obtained as a beige wax (1.1 g) in analogy to N—((S)-3-[2-ethyl-4-(N-hydroxycarbamimidoyl)-6-methyl-phenoxy]-2-hydroxy-propyl)-2-hydroxy-acetamide starting from 3-chloro-4-hydroxy-5-methyl-benzonitrile; LC-MS: tR=0.48 min, [M+H]+=331.94.
3-Chloro-N-hydroxy-4-methanesulfonylamino-5-methyl-benzamidine
a) A mixture of 4-amino-3-chloro-5-methylbenzonitrile (155 mg, 930 μmol) and methanesulfonylchloride (2.13 g, 18.6 mmol, 1.44 mL) is heated under microwave conditions to 150° C. for 7 h. The mixture is cooled to rt, diluted with water and extracted with EA. The org. extract is dried over MgSO4, filtered and concentrated. The crude product is purified on prep. TLC using heptane:EA 1:1 to give N-(2-chloro-4-cyano-6-methyl-phenyl)-methanesulfonamide (105 mg) as an orange solid; LC-MS**: tR=0.48 min; 1H NMR (CDCl3): δ2.59 (s, 3H), 3.18 (s, 3H), 6.27 (s, 1H), 7.55 (d, J=1.3 Hz, 1H), 7.65 (d, J=1.5 Hz, 1H).
b) Hydroxylamine hydrochloride (60 mg, 858 μmol) and NaHCO3 (72 mg, 858 μmol) is added to a solution of N-(2-chloro-4-cyano-6-methyl-phenyl)-methanesulfonamide (105 mg, 429 μmol) in methanol (10 mL). The mixture is stirred at 65° C. for 18 h. The solvent is removed in vacuo and the residue is dissolved in a small volume of water (2 mL) and extracted three times with EA (15 mL). The combined org. extracts are dried over MgSO4, filtered, concentrated and dried to give the title compound (118 mg) as a white solid; LC-MS**: tR=0.19 min, [M+1]+=277.94; 1H NMR (CDCl3): δ2.57 (s, 3H), 3.13 (s, 3H), 6.21 (s, 1H), 7.49 (d, J=1.5 Hz, 1H), 7.63 (d, J=1.5 Hz).
3-Ethyl-N-hydroxy-4-methanesulfonylamino-5-methyl-benzamidine
a) In a 2.5 L three-necked round-bottom flask 2-ethyl-6-methyl aniline (250 g, 1.85 mol) is dissolved in DCM (900 mL) and cooled to 5-10° C. Bromine (310.3 g, 1.94 mol) is added over a period of 105 min such as to keep the temperature at 5-15° C. An aq. 32% NaOH solution (275 mL) is added over a period of 10 min to the greenish-grey suspension while keeping the temperature of the reaction mixture below 25° C. DCM (70 mL) and water (100 mL) are added and the phases are separated. The aq. phase is extracted with DCM (250 mL). The combined org. phases are washed with water (300 mL) and concentrated at 50° C. to afford the 4-bromo-2-ethyl-6-methyl-aniline (389 g) as a brown oil; 1H NMR (CDCl3): δ 1.27 (t, J=7.3 Hz, 3H), 2.18 (s, 3H), 2.51 (q, J=7.3 Hz, 2H), 3.61 (s br, 1H), 7.09 (s, 2H).
b) A double-jacketed 4 L-flask is charged with 4-bromo-2-ethyl-6-methyl-aniline (324 g, 1.51 mol), sodium cyanide (100.3 g, 1.97 mol), potassium iodide (50.2 g, 0.302 mol) and copper(I)iodide (28.7 g, 0.151 mol). The flask is evacuated three times and refilled with nitrogen. A solution of N,N′-dimethylethylenediamine (191.5 mL, 1.51 mol) in toluene (750 mL) is added. The mixture is heated to 118° C. and stirred at this temperature for 21 h. The mixture is cooled to 93° C. and water (1250 mL) is added to obtain a solution. Ethyl acetate (1250 mL) is added at 22-45° C. and the layers are separated. The org. phase is washed with 10% aq. citric acid (2×500 mL) and water (500 mL). The separated org. phase is evaporated to dryness to afford 4-amino-3-ethyl-5-methyl-benzonitrile (240 g) as a metallic black solid; 1H NMR (CDCl3): δ1.29 (t, J=7.5 Hz, 3H), 2.19 (s, 3H), 2.52 (q, J=7.3 Hz, 2H), 4.10 (s br, 1H), 7.25 (s, 2H).
c) The title compound is then prepared from the above 4-amino-3-ethyl-5-methyl-benzonitrile in analogy to 3-chloro-N-hydroxy-4-methanesulfonylamino-5-methyl-benzamidine; LC-MS**: tR=0.26 min, [M+1]+=272.32.
3-Chloro-4-ethanesulfonylamino N-hydroxy-5-methyl-benzamidine
The title compound is prepared in analogy to 3-chloro-N-hydroxy-4-methanesulfonylamino-5-methyl-benzamidine using ethanesulfonylchloride; LC-MS**: tR=0.27 min, [M+1]+=292.13; 1H NMR (D6-DMSO): δ 1.36 (t, J=7.5 Hz, 3H), 2.40 (s, 3H), 3.22 (q, J=7.5 Hz), 5.88 (s, 2H), 7.57 (d, J=1.5 Hz, 1H), 7.63 (d, J=1.5 Hz, 1H), 9.18 (s, 1H), 9.78 (s, 1H).
4-Benzyloxy-3-ethyl-5-methyl-benzoic acid
a) To a solution of 3-ethyl-4-hydroxy-5-methyl-benzaldehyde (34.9 g, 0.213 mol, prepared from 2-ethyl-6-methyl-phenol according to the literature cited for 3-ethyl-4,N-dihydroxy-5-methyl-benzamidine) in MeCN (350 mL), K2CO3 (58.7 g, 0.425 mol) and benzylbromide (36.4 g, 0.213 mol) are added. The mixture is stirred at 60° C. for 2 h before it is cooled to rt, diluted with water and extracted twice with EA. The org. extracts are washed with water and concentrated to give crude 4-benzyloxy-3-ethyl-5-methyl-benzaldehyde (45 g) as an orange oil. 1H NMR (CDCl3): δ1.29 (t, J=7.5 Hz, 3H), 2.40 (s, 3H), 2.77 (q, J=7.8 Hz, 2H), 4.90 (s, 2H), 7.31-7.52 (m, 5H), 7.62 (d, J=1.5 Hz, 1H), 7.66 (d, J=1.8 Hz, 1H), 9.94 (s, 1H).
b) To a mixture of 4-benzyloxy-3-ethyl-5-methyl-benzaldehyde (132 g, 0.519 mol) and 2-methyl-2-butene (364 g, 5.19 mol) in tert.-butanol (1500 mL), a solution of NaH2PO4 dihydrate (249 g, 2.08 mol) in water (1500 mL) is added. To this mixture, NaClO2 (187.8 g, 2.08 mol) is added in portions. The temperature of the reaction mixture is kept below 30° C., and evolution of gas is observed. Upon completion of the addition, the orange bi-phasic mixture is stirred well for 3 h before it is diluted with TBME (1500 mL). The org. layer is separated and washed with 20% aq. NaHS solution (1500 mL) and water (500 mL). The org. phase is then extracted three times with 0.5 N aq. NaOH (1000 mL), the aq. phase is acidified with 25% aq. HCl (500 mL) and extracted twice with TBME (1000 mL). These org. extracts are combined and evaporated to dryness to give the title compound; 1H NMR (D6-DMSO): δ 1.17 (t, J=7.5 Hz, 3H), 2.31 (s, 3H), 2.67 (q, J=7.5 Hz, 2H), 4.86 (s, 2H), 7.34-7.53 (m, 5H), 7.68 (s, 2H), 12.70 (s, 1H).
Example 1 (S)-3-(2-Ethyl-4-{5-[2-(1-ethyl-propyl)-6-methoxy-pyridin-4-yl]-[1,2,4]oxadiazol-3-yl}-6-methyl-phenoxy)-propane-1,2-diol
a) To a solution of 2-(1-ethyl-propyl)-6-methoxy-isonicotinic acid (190 mg, 732 μmol) in THF (10 mL) and DMF (2 mL), DIPEA (190 mg, 1.46 mmol) followed by TBTU (235 mg, 732 μmol) is added. The mixture is stirred at rt for 10 min before (R)-4-(2,2-dimethyl-[1,3]dioxolan-4-ylmethoxy)-3-ethyl-N-hydroxy-5-methyl-benzamidine 226 mg, 732 μmol) is added. The mixture is stirred at rt for 1 h before it is diluted with EA and washed with water. The org. phase is separated and concentrated. The remaining residue is dissolved in dioxane (10 mL) and heated to 105° C. for 18 h. The mixture is cooled to rt, concentrated and the crude product is purified on prep. TLC plates using DCM containing 10% of methanol to give 4-{3-[4-((R)-2,2-dimethyl-[1,3]dioxolan-4-ylmethoxy)-3-ethyl-5-methyl-phenyl]-[1,2,4]oxadiazol-5-yl}-2-(1-ethyl-propyl)-6-methoxy-pyridine (256 mg) as a yellow oil; LC-MS: tR=1.28 min, [M+H]+=496.23.
b) A solution of 4-{3-[4-((R)-2,2-dimethyl-[1,3]dioxolan-4-ylmethoxy)-3-ethyl-5-methyl-phenyl]-[1,2,4]oxadiazol-5-yl}-2-(1-ethyl-propyl)-6-methoxy-pyridine (250 mg, 504 μmol) in 4 M HCl in dioxane (10 mL) is stirred at rt for 90 min before it is concentrated. The crude product is purified on prep. TLC plates using DCM containing 10% of methanol to give the title compound (76 mg) as a pale brownish solid; LC-MS: tR=1.12 min, [M+H]+=456.12; 1H NMR (CDCl3): δ0.85 (t, J=7.0 Hz, 6H), 1.33 (t, J=7.0 Hz, 3H), 1.70-1.89 (m, 4H), 2.42 (s, 3H), 2.61-2.71 (m, 1H), 2.78 (q, J=7.3 Hz, 2H), 3.82-4.00 (m, 4H), 4.04 (s, 3H), 4.14-4.21 (m, 1H), 7.34 (s, 1H), 7.46 (s, 1H), 7.86-7.91 (m, 2H).
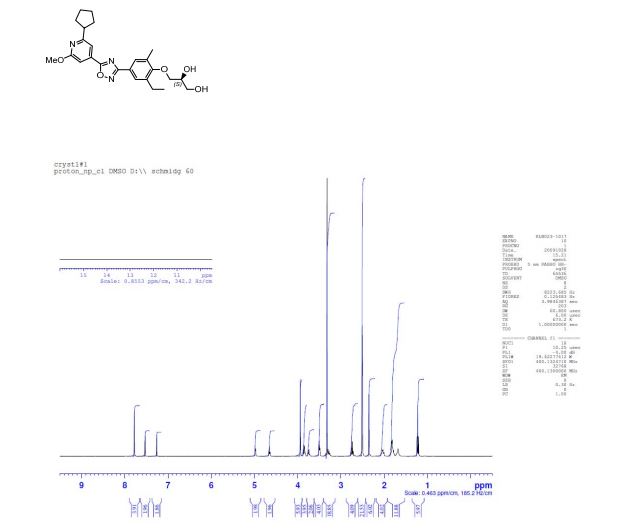
Example 2 (S)-3-{4-[5-(2-Cyclopentyl-6-methoxy-pyridin-4-yl)-[1,2,4]oxadiazol-3-yl]-2-ethyl-6-methyl-phenoxy}-propane-1,2-diol
The title compound is prepared in analogy to Example 1 starting from 2-cyclopentyl-6-methoxy-isonicotinic acid; LC-MS: tR=1.14 min, [M+H]+=454.16; 1H NMR (CDCl3): δ1.33 (t, J=7.5 Hz, 3H), 1.72-1.78 (m, 2H), 1.85-1.94 (m, 4H), 2.03-2.15 (m, 2H), 2.41 (s, 3H), 2.72 (d, J=5.3 Hz, 1H), 2.77 (q, J=7.5 Hz, 2H), 3.19-3.28 (m, 1H), 3.81-3.94 (m, 2 H), 3.95-3.98 (m, 2H), 4.02 (s, 3H), 4.14-4.21 (m, 1H), 7.31 (d, J=1.3 Hz, 1H), 7.51 (d, J=1.0 Hz, 1H), 7.88 (d, J=1.8 Hz), 7.89 (d, J=2.0 Hz, 1H).
PAPER

A practical synthesis of S1P receptor 1 agonist ACT-334441 (1) through late-stage convergent coupling of two key intermediates is described. The first intermediate is 2-cyclopentyl-6-methoxyisonicotinic acid whose skeleton was built from 1-cyclopentylethanone, ethyl oxalate, and cyanoacetate in a Guareschi–Thorpe reaction in 42% yield over five steps. The second, chiral intermediate, is a phenol ether derived from enantiomerically pure (R)-isopropylidene glycerol ((R)-solketal) and 3-ethyl-4-hydroxy-5-methylbenzonitrile in 71% yield in a one-pot reaction. The overall sequence entails 18 chemical steps with 10 isolated intermediates. All raw materials are cheap and readily available in bulk quantities, the reaction conditions match with standard pilot plant equipment, and the route reproducibly afforded 3–20 kg of 1 in excellent purity and yield for clinical studies.
Practical Synthesis of a S1P Receptor 1 Agonist via a Guareschi–Thorpe Reaction
Chemistry Process R&D, Actelion Pharmaceuticals Ltd., Gewerbestrasse 16, CH-4123 Allschwil, Switzerland
Org. Process Res. Dev., Article ASAP
DOI: 10.1021/acs.oprd.6b00210
*E-mail: stefan.abele@actelion.com. Telephone: +41 61 565 67 59.
(1H NMR): 99.40% w/w; er (HPLC method 2): (S):(R) = 99.7:0.3, tR 10.70 min (S-isomer), 14.5 min (R-isomer);
mp 80 °C (DSC);
1H NMR (d6-DMSO): δ 7.78 (s, 2 H), 7.53 (s, 1 H), 7.26 (s, 1 H), 4.98 (d, J = 4.6 Hz, 1 H), 4.65 (s, 1 H), 3.94 (s, 3 H), 3.86 (m, 2 H), 3.75 (m, 1 H), 3.50 (t, J = 5.4 Hz, 2 H), 3.28 (m, 1 H), 2.75 (d, J = 7.5 Hz, 2 H), 2.35 (s, 3 H), 2.03 (m, 2 H), 1.81 (m, 4 H), 1.69 (m, 2 H), 1.22 (t, J = 7.5 Hz, 3 H).
13C NMR (CDCl3): δ 174.3, 168.9, 165.8, 164.4, 157.4, 137.7, 133.6, 131.7, 128.4, 126.7, 122.5, 112.0, 106.0, 73.9, 71.1, 63.8, 53.7, 47.5, 33.3, 25.9, 22.9, 16.4, 14.8.
| Patent ID | Date | Patent Title |
|---|---|---|
| US2015133669 | 2015-05-14 | NEW PROCESS FOR THE PREPARATION OF 2-CYCLOPENTYL-6-METHOXY-ISONICOTINIC ACID |
| US8658675 | 2014-02-25 | Pyridin-4-yl derivatives |
//////////ACT-334441, ACT 334441, ACT334441, CENERIMOD, S1P receptor 1 agonist, Systemic lupus erythematosus, UNII-Y333RS1786 Y333RS1786, phase 2, Actelion Pharmaceuticals Ltd., Martin Bolli, Cyrille Lescop, Boris Mathys,Keith Morrison, Claus Mueller, Oliver Nayler,Beat Steiner,
OC[C@H](O)COC1=C(C)C=C(C2=NOC(C3=CC(C4CCCC4)=NC(OC)=C3)=N2)C=C1CC
Is AQL Testing required within the 100% Visual Inspection?
DRUG REGULATORY AFFAIRS INTERNATIONAL

Is AQL Testing required within the 100% Visual Inspection?
One of the most frequently asked questions is whether an additional testing based on samples is required after the 100% visual inspection of parenterals. The answer is: basically, “yes”.
One of the most frequently asked questions is whether an additional AQL testing based on samples is required after the 100% visual inspection of parenterals. The background for that question is the probabilistic nature of visual inspection. It is known that the discovery of defects (like for example particulates) is a matter of detection probability. In other words, visual inspection cannot exclude that defective containers may still be in the batch which hasn’t been sorted out. This applies to manual, semi-automatic and also automatic visual inspection.
The American Pharmacopoeia has reacted to that and has integrated AQL testing in the monograph Visible Particulates in Injections. Here, the value 0.65 has been…
View original post 203 more words
Two new FDA Warning Letters for API Manufacturers in China
DRUG REGULATORY AFFAIRS INTERNATIONAL


Two new FDA Warning Letters for API Manufacturers in China
In June 2016, two API manufacturers in China received a Warning Letter from the FDA. Both companies had major deficiencies regarding data integrity. For instance, manipulations were found in HPLC analyses as well as in GC analyses. You will find more information on the current FDA Warning Letters for Chongqing Lummy and Shanghai Desano here. http://www.gmp-compliance.org/enews_05496_Two-new-FDA-Warning-Letters-for-API-Manufacturers-in-China_15488,15484,Z-QCM_n.html
The Chinese Company Chongqing Lummy Pharmaceutical Co., Ltd. received a Warning Letter from the FDA on June 21, 2016. This Warning Letter referred to both the FDA inspection from March 14-16, 2016 and the response which the API manufacturer had sent to the FDA on March 31, 2016.
It was claimed that Chongqing Lummy Pharmaceuticals had no adequate control in place to prevent data manipulation or deletion. The FDA investigator’s review of the audit trail revealed that an analyst had manipulated the computerized gas…
View original post 425 more words
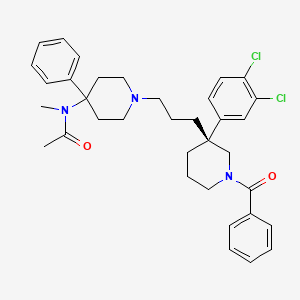
Osanetant , SR-142,801

Osanetant (SR-142,801)
160492-56-8 CAS
| : MW 605.257582985 | |
| Chemical Formula | C35H41Cl2N3O2 |
|---|
(R)-(+)-N-[[3-[1-benzoyl-3-(3,4-dichlorophenyl)piperidin-3-yl]prop-1-yl]-4-phenylpiperidin-4-yl]-N-methylacetamide
Osanetant (SR-142,801) was a neurokinin 3 receptor antagonist developed by Sanofi-Synthélabo, which was being researched for the treatment of schizophrenia, but was discontinued.[1][2] It was the first non-peptide NK3 antagonist developed in the mid-1990s,[3][4] Other potential applications for osanetant is in the treatment of drug addiction, as it has been found to block the effects ofcocaine in animal models.[5][6]
Developed by Sanofi-Aventis (formerly Sanofi-Synthelabo), osanetant (SR-142801) is an NK3 receptor antagonist which was under development for the treatment of schizophrenia and other Central Nervous System (CNS) disorders. In a review of its R&D portfolio, the company announced in August 2005 that it would cease any further development ofosanetant. This follows an earlier decision to discontinue development of eplivanserin for schizophrenia
![]()
(R)-(+)-N-[[3-[1-benzoyl-3-(3,4-dichlorophenyl)piperidin-3-yl]prop-1-yl]-4-phenylpiperidin-4-yl]-N-methylacetamide and to a process for their preparation. (R)-(+)-N-[[3-[1-Benzoyl-3-(3,4-dichlorophenyl)piperidin-3-yl]prop-1-yl]-4-phenylpiperidin-4-yl]-N-methylacetamide, hereinafter denoted by its International Non-proprietary Name “osanetant”, is the first antagonist of the NK-3 receptor described in the literature, the preparation of which, in particular in the hydrochloride form, is illustrated in EP-A-673 928.
According to this document, osanetant is prepared by reacting N-methyl-N-(4-phenylpiperidin-4-yl)acetamide with 1-benzoyl-3-(3,4-dichlorophenyl)-3-(methanesulfonyloxyprop-1-yl)piperidine and by converting the osanetant thus obtained to its hydrochloride. It has been found that osanetant hydrochloride is isolated in the form of an amorphous solid which is difficult to purify. This product comprises impurities originating from the preceding synthetic stages.
Preparative chromatography starting from osanetant base can be used to obtain osenetant in the pure form.
Osanetant is a neurokinin (NK3) receptor antagonist under development by Sanofi-Synthélabo (formerly Sanofi) as a potential treatment for schizophrenia . Sanofi was originally investigating its potential use as a treatment for psychosis and anxiety . Following phase IIa clinical trials , osanetant entered phase IIb development in February 2001 . Osanetant was the first potent and selective non-peptide antagonist described for the NK3 tachykinin receptor . It has a higher affinity for human and guinea pig NK3 receptors than for rat NK3 receptors . In October 1999, Lehman Brothers predicted that the probability of the product reaching the market was 10%, with a possible launch in 2003 and potential peak sales of US $200 million in 2011 .


Sanofi-Aventis CEO, Chris Vihebacher,
PATENT

EP 0673928; FR 2717477; FR 2717478; FR 2719311; JP 1996048669; US 5741910; US 5942523; US 6124316
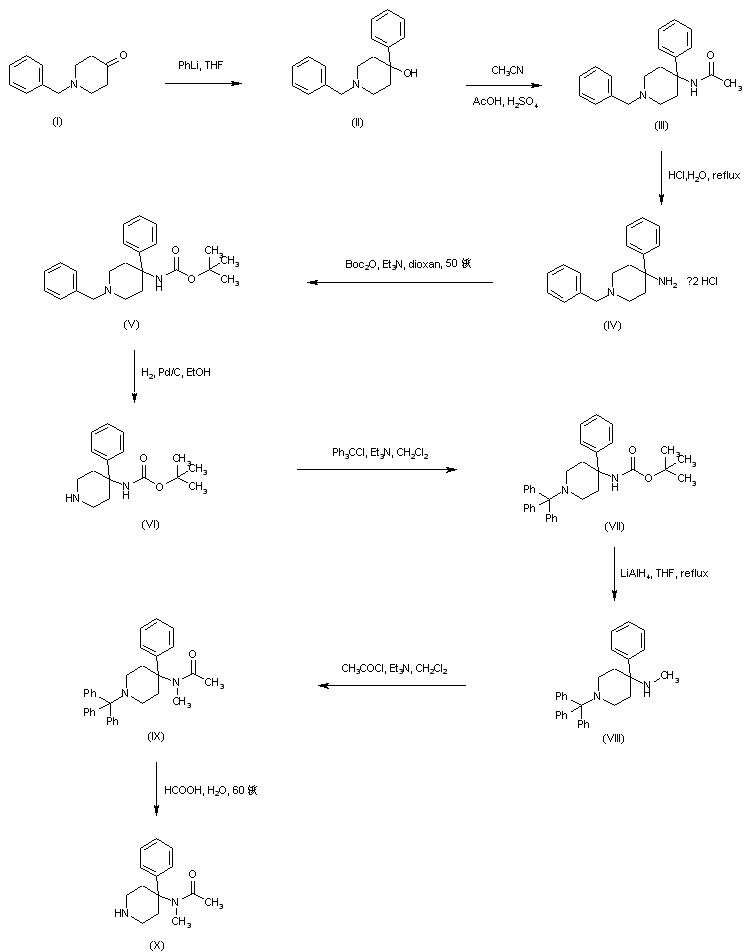
N-Benzyl-4-hydroxy-4-phenylpiperidine (II) was prepared by addition of phenyllithium to N-benzyl-4-piperidone (I). Carbinol (II) was then converted to acetamide (III) by acid-catalyzed Ritter reaction with acetonitrile. Replacement of the acetamido for an N-Boc group in (III) was effected by acidic hydrolysis of amide (III) to give (IV), followed by treatment with di-tert-butyl dicarbonate. The resultant 1-benzyl-4-(Boc-amino)-4-phenylpiperidine (V) was subjected to catalytic hydrogenolysis in the presence of Pd/C, and the N-debenzylated piperidine (VI) was reprotected as the N-trityl derivative (VII) by treatment with triphenylmethyl chloride and triethylamine. Reduction of the N-Boc group of (VII) by LiAlH4, yielded the N-methyl amine (VIII). After acylation of (VIII) with acetyl chloride to acetamide (IX), its N-trityl group was cleaved by treatment with hot aqueous formic acid to produce the intermediate piperidine (X).


Michael addition of methyl acrylate (XII) to (3,4-dichlorophenyl)acetonitrile (XI) produced the cyano diester adduct (XIII). Catalytic hydrogenation of the cyano group of (XIII) over Raney nickel with concomitant intramolecular cyclization gave rise to the piperidinone (XIV). After basic hydrolysis of the methyl ester function of (XIV), the resultant piperidone propionic acid (XV) was reduced to piperidino alcohol (XVI) by means of borane in THF. Resolution of the racemic piperidine (XVI) employing (+)-camphorsulfonic acid provided the dextro enantiomer (XVII). After N-protection of (XVII) as the Boc derivative (XVIII), its primary alcohol was activated as the corresponding mesylate (XIX) with methanesulfonyl chloride and Et3N. Condensation between mesylate (XIX) and intermediate piperidine (X) in acetonitrile at 60 C, produced (XX). The title benzamido derivative was then obtained by acid-promoted Boc group cleavage in (XX), followed by acylation with benzoyl chloride.
WO 9805640
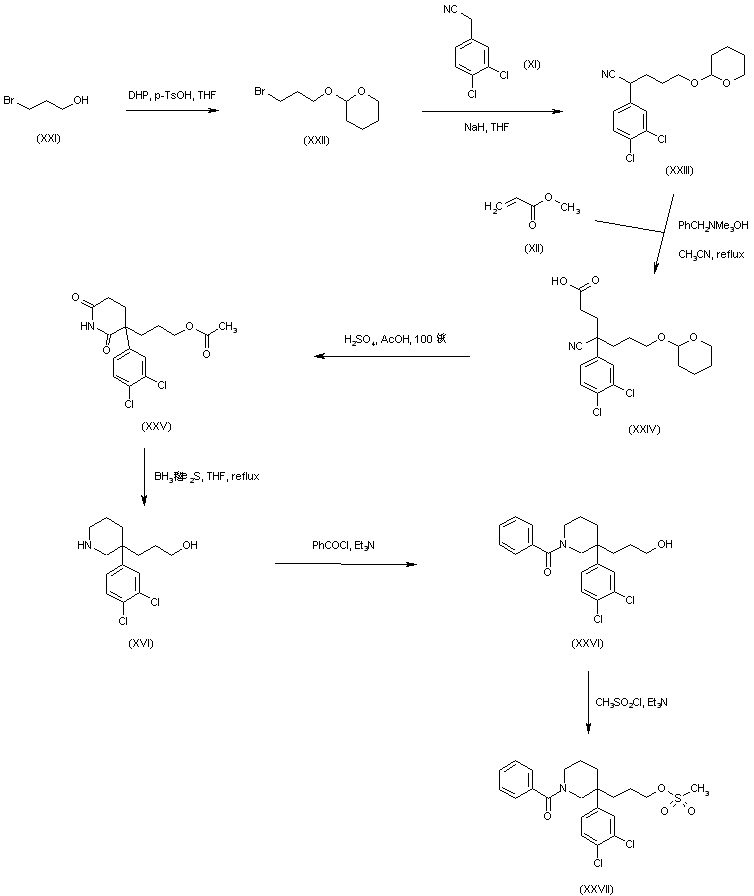
Bioorg Med Chem Lett 1996,6(19),2307
In a related synthesis, (3,4-dichlorophenyl)acetonitrile (XI) was alkylated with bromide (XXII) –prepared by protection of 3-bromopropanol (XXI) with dihydropyran– to afford (XXIII). Subsequent Michael addition of methyl acrylate (XII) to (XXIII) in the presence of Triton B?gave the cyanoacid (XXIV). This was cyclized to the glutarimide (XXV) by refluxing in HOAc in the presence of H2SO4. Reduction of (XXV) using borane-dimethylsulfide complex produced the already reported racemic piperidinoalcohol (XVI). After acylation of the amine group of (XVI) with benzoyl chloride to yield (XXVI), its hydroxyl group was converted into the target mesylate precursor (XXVII) with methanesulfonyl chloride and Et3N.

An alternative preparation of the precursor 4-(N-methyl-N-acetyl)amino-4-phenylpiperidine (XXXIX) has been reported. The N-benzyl protecting group of piperidine (III) was replaced with an N-Boc group by catalytic hydrogenolysis to (XXXVI), followed by treatment with Boc2O to yield (XXXVII). Amide (XXXVII) alkylation with iodomethane under phase-transfer conditions gave the N-methyl derivative (XXXVIII). Subsequent N-Boc group cleavage in (XXXVIII) was accomplished by using zinc chloride in CH2Cl2 to afford the piperidine-ZnCl2 complex (XXXIX). This was then alkylated with mesylate (XXVII), and the title compound was finally isolated from the racemic mixture by means of preparative chiral HPLC.


In a further method, aminopiperidine (IV) was converted to the formamide (XL) by heating in ethyl formate. Formyl group reduction in (XL) with LiAlH4 provided the N-metyl amine (XLI). The N-benzyl group of (XLI) was then removed by catalytic hydrogenation over Pd/C. Alkylation of the resultant piperidine (XLII) with mesylate (XXVII) gave adduct (XLIII). After acetylation of (XLIII) in neat Ac2O, the racemic mixture was separated by chiral HPLC.

In a further procedure, nitrile (XXIII) was alkylated with ethyl 3-bromopropionate (XXVIII) to give cyano ester (XXIX). Catalytic hydrogenation of the cyano group of (XXIX) gave rise to the piperidinone (XXX), which was further reduced to piperidine (XXXI) with LiAlH4 in THF. Acid deprotection of the tetrahydropyranyl group of (XXXI), followed by resolution with (+)-camphorsulfonic acid, furnished the desired (S)-piperidinoalcohol camphorsulfonate salt (XXXII). Treatment of piperidine (XXXII) with benzoyl chloride in the presence of DIEA yielded benzamide (XXXIII). Conversion of the primary alcohol of (XXXIII) into the desired alkyl iodide (XXXV) was achieved via formation of the mesylate ester (XXXIV), followed by displacement of the mesylate group with KI in refluxing acetone.
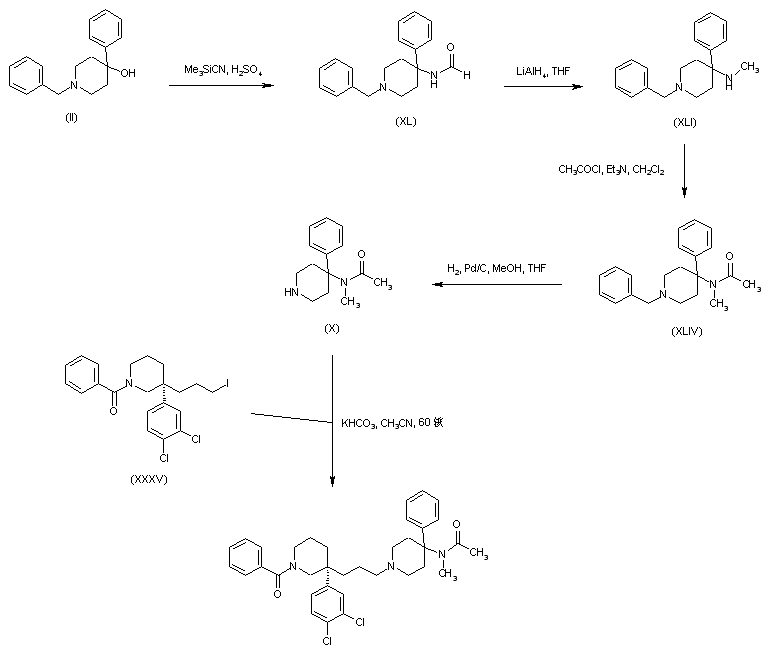
Bioorg Med Chem Lett 1997,7(5),555
A new method has been reported. Formamide (XL) was prepared form carbinol (II) by a modified Ritter reaction with cyanotrimethylsilane. Subsequent reduction of (XL) with LiAlH4 gave the N-methyl amine (XLI), which was converted to acetamide (XLIV) by treatment with acetyl chloride. Benzyl group hydrogenolysis in (XLIV) afforded the piperidine (X). Finally, alkylation of piperidine (X) with the chiral alkyl iodide (XXXV) provided the title compound.
| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| US5741910 * | Feb 29, 1996 | Apr 21, 1998 | Sanofi | Compounds which are selective antagonists of the human NK3 receptor and their use as medicinal products and diagnostic tools |
| US5942523 * | Feb 29, 1996 | Aug 24, 1999 | Sanofi | Compounds which are selective antagonists of the human NK3 receptor and their use as medicinal products and diagnostic tools |
| US6040316 * | Sep 2, 1997 | Mar 21, 2000 | Warner-Lambert Company | 3-alkyl-3-phenyl-piperidines |
| US6124316 * | May 7, 1999 | Sep 26, 2000 | Sanofi | Compounds which are specific antagonists of the human NK3 receptor and their use as medicinal products and diagnostic tools |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| US7648992 | Jul 4, 2005 | Jan 19, 2010 | Astrazeneca Ab | Hydantoin derivatives for the treatment of obstructive airway diseases |
| US7655664 | Dec 14, 2005 | Feb 2, 2010 | Astrazeneca Ab | Hydantoin derivatives as metalloproteinase inhibitors |
| US7662845 | Aug 7, 2006 | Feb 16, 2010 | Astrazeneca Ab | 2,5-Dioxoimidazolidin-4-yl acetamides and analogues as inhibitors of metalloproteinase MMP12 |
| US7666892 | May 5, 2008 | Feb 23, 2010 | Astrazeneca Ab | Metalloproteinase inhibitors |
| US7700604 | Dec 14, 2005 | Apr 20, 2010 | Astrazeneca Ab | Hydantoin derivatives as metalloproteinase inhibitors |
| US7754750 | Jul 13, 2010 | Astrazeneca Ab | Metalloproteinase inhibitors | |
| US7989620 | Aug 2, 2011 | Astrazeneca Ab | Hydantoin derivatives for the treatment of obstructive airway diseases | |
| US8153673 | Jan 26, 2010 | Apr 10, 2012 | Astrazeneca Ab | Metalloproteinase inhibitors |
| US8183251 | Nov 28, 2007 | May 22, 2012 | Astrazeneca Ab | Hydantoin compounds and pharmaceutical compositions thereof |
| US20080032997 * | Dec 14, 2005 | Feb 7, 2008 | Astrazeneca Ab | Novel Hydantoin Derivatives as Metalloproteinase Inhibitors |
| US20080064710 * | Jul 4, 2005 | Mar 13, 2008 | Astrazeneca Ab | Novel Hydantoin Derivatives for the Treatment of Obstructive Airway Diseases |
| US20080221139 * | Nov 28, 2007 | Sep 11, 2008 | David Chapman | Novel Compounds |
| US20080262045 * | May 5, 2008 | Oct 23, 2008 | Anders Eriksson | Metalloproteinase Inhibitors |
| US20080293743 * | Dec 14, 2005 | Nov 27, 2008 | Astrazeneca Ab | Novel Hydantoin Derivatives as Metalloproteinase Inhibitors |
| US20080306065 * | May 6, 2008 | Dec 11, 2008 | Anders Eriksson | Metalloproteinase Inhibitors |
| US20100144771 * | Dec 2, 2009 | Jun 10, 2010 | Balint Gabos | Novel Hydantoin Derivatives for the Treatment of Obstructive Airway Diseases |
| WO2007106022A2 * | Mar 15, 2007 | Sep 20, 2007 | Astrazeneca Ab | A new crystalline form g of (5s) -5- [4- (5-chloro-pyridin-2- yloxy) -piperidine-1-sulfonylmethyl] – 5 -methyl -imidazolidine – 2,4-dione (i) and intermediates thereof. |
| WO2007106022A3 * | Mar 15, 2007 | Nov 1, 2007 | Astrazeneca Ab | A new crystalline form g of (5s) -5- [4- (5-chloro-pyridin-2- yloxy) -piperidine-1-sulfonylmethyl] – 5 -methyl -imidazolidine – 2,4-dione (i) and intermediates thereof. |

References
- “osanetant Sanofi-Aventis discontinued, France.”. Highbeam.
- Kamali, F (July 2001). “Osanetant Sanofi-Synthélabo”. Current opinion in investigational drugs (London, England : 2000). 2 (7): 950–6.PMID 11757797.
- Emonds-Alt, X; Bichon, D; Ducoux, JP; Heaulme, M; Miloux, B; Poncelet, M; Proietto, V; Van Broeck, D; et al. (1995). “SR 142801, the first potent non-peptide antagonist of the tachykinin NK3 receptor”. Life Sciences. 56 (1): PL27–32. doi:10.1016/0024-3205(94)00413-M.PMID 7830490.
- Quartara L, Altamura M (August 2006). “Tachykinin receptors antagonists: from research to clinic”. Current Drug Targets. 7 (8): 975–92.doi:10.2174/138945006778019381. PMID 16918326. Retrieved 2011-04-14.
- Desouzasilva, M; Mellojr, E; Muller, C; Jocham, G; Maior, R; Huston, J; Tomaz, C; Barros, M (May 2006). “The tachykinin NK3 receptor antagonist SR142801 blocks the behavioral effects of cocaine in marmoset monkeys”. European Journal of Pharmacology. 536 (3): 269–78.doi:10.1016/j.ejphar.2006.03.010. PMID 16603151.
- Jocham, Gerhard; Lezoch, Katharina; Müller, Christian P.; Kart-Teke, Emriye; Huston, Joseph P.; De Souza Silva, M. AngéLica (September 2006). “Neurokinin receptor antagonism attenuates cocaine’s behavioural activating effects yet potentiates its dopamine-enhancing action in the nucleus accumbens core”. European Journal of Neuroscience. 24 (6): 1721–32. doi:10.1111/j.1460-9568.2006.05041.x.PMID 17004936.
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
N-(1-{3-[(3R)-1-benzoyl-3-(3,4-dichlorophenyl)piperidin-3-yl]propyl}-4-phenylpiperidin-4-yl]-N-methylacetamide
|
|
| Identifiers | |
| CAS Number | 160492-56-8 |
| ATC code | none |
| PubChem | CID 219077 |
| IUPHAR/BPS | 2110 |
| ChemSpider | 189901 |
| UNII | K7G81N94DT |
| ChEMBL | CHEMBL346178 |
| Chemical data | |
| Formula | C35H41Cl2N3O2 |
| Molar mass | 606.625 g/mol |
///////Osanetant , SR-142,801,
CC(=O)N(C)C1(CCN(CC1)CCC[C@@]2(CCCN(C2)C(=O)C3=CC=CC=C3)C4=CC(=C(C=C4)Cl)Cl)C5=CC=CC=C5

AZD-1236 Revisited
AZD1236
CAS 459814-89-2,
MF C15 H19 Cl N4 O5 S. MW402.85
2,4-Imidazolidinedione, 5-[[[4-[(5-chloro-2-pyridinyl)oxy]-1-piperidinyl]sulfonyl]methyl]-5-methyl-, (5S)-
(5S)-5-[4-(5-chloro-pyridin-2-yloxy)-piperidine-1-sulfonylmethyl]-5-methyl-imidazolidine-2,4-dione
(S)-5-[4-(5-ChIoro-pyridin-2-yloxy)-piperidine-l-suIfonylmethyl]-5-methyl- imidazoIidine-2,4-dione
UNII-B4OQY51WZS; B4OQY51WZS; (S)-5-(((4-((5-Chloropyridin-2-yl)oxy)piperidin-1-yl)sulfonyl)methyl)-5-methylimidazolidine-2,4-dione; AZD1236; AZD-1236;
Piperidine, 4-[(5-chloro-2-pyridinyl)oxy]-1-[[[(4S)-4-methyl-2,5-dioxo-4-imidazolidinyl]methyl]sulfonyl]- (9CI)(5S)-5-[[[4-[(5-Chloro-2-pyridinyl)oxy]-1-piperidinyl]sulfonyl]methyl]-5-methyl-2,4-imidazolidinedione
Mechanism of Action: Matrix metalloproteinase 9 & 12 (MMP9,12) inhibitor MMP9 MMP12i
Anders Eriksson, Matti Lepistö, Michael Lundkvist, af Rosenschöld Magnus Munck,Pavol Zlatoidsky,
Astrazeneca Ab INNOVATOR
- OriginatorAstraZeneca
- Class
- Mechanism of ActionMatrix metalloproteinase inhibitors
- Highest Development Phases
- DiscontinuedChronic obstructive pulmonary disease
Most Recent Events
- 29 Jul 2010Discontinued – Phase-II for Chronic obstructive pulmonary disease in Europe (PO)
- 29 Jul 2010Discontinued – Phase-I for Chronic obstructive pulmonary disease in Japan (PO)
- 29 Jul 2010Discontinued – Phase-I for Chronic obstructive pulmonary disease in Japan (PO)
AZD1236 is a selective MMP-9 and MMP-12 inhibitor (IC50 4.5 and 6.1nM) from Astrazeneca that, since it failed biomarker endpoints for COPD is included in the AZ Open Innovation 2014 set for repurposing. Pending any published link the structure identification is tenatative but seems likely to be the structure crystalised in WO2007106022.
Matrix metallopeptidase 9 and 12 (MMP9|MMP12) inhibitor http://www.ncbi.nlm.nih.gov/gene/4318; http://www.ncbi.nlm.nih.gov/gene/4321 Preclinical Pharmacology AZD1236 is a potent and reversible inhibitor of human MMP9 and MMP12 (IC50’s = 4.5 and 6.1nM, respectively), with 10 – 15-fold selectivity to MMP2 and MMP13 and >350-fold selectivity to other members of the enzyme family. Its activity is approximately 20- to 50-fold lower at the rat, mouse, and guinea pig orthologues. In acute models of lung injury, AZD1236 inhibited the hemorrhage and inflammation induced by instillation of human MMP12 into rat lungs by ~80% at 0.81 mg/kg, and also abolished macrophage infiltration into BAL fluid induced by tobacco smoke inhalation in the mouse. Safety and Tolerability In healthy human volunteers, AZD1236 was well tolerated in single doses from 2 to 1500 mg and in multiple doses of 15, 75 and 500 mg for periods of up to 13 days. AZD1236 was also well tolerated in COPD patients with moderate to severe disease when given at 75 mg BID for 6 weeks. Pre-clinical toxicology studies of up to 12 month duration have been performed. Toxicologically important findings mainly relate to chronic treatment and included: diffuse eye lens opacities after 6 months administration to rats and fibrodysplasia in the subcutis after 12 months to dogs. Clinical Pharmacology Target coverage data to date have been mixed. In healthy subjects, single dose of 30 or 75 mg inhibited ex vivo zymosanstimmulated whole blood MMP activity (the 75 mg dose yielding plasma compound levels at Cmax steady state of ~120 x IC50). In contrast, 75 mg BID for 6 wks in COPD patients compared to placebo did not identify any significant change in whole blood MMP activity.
![]()
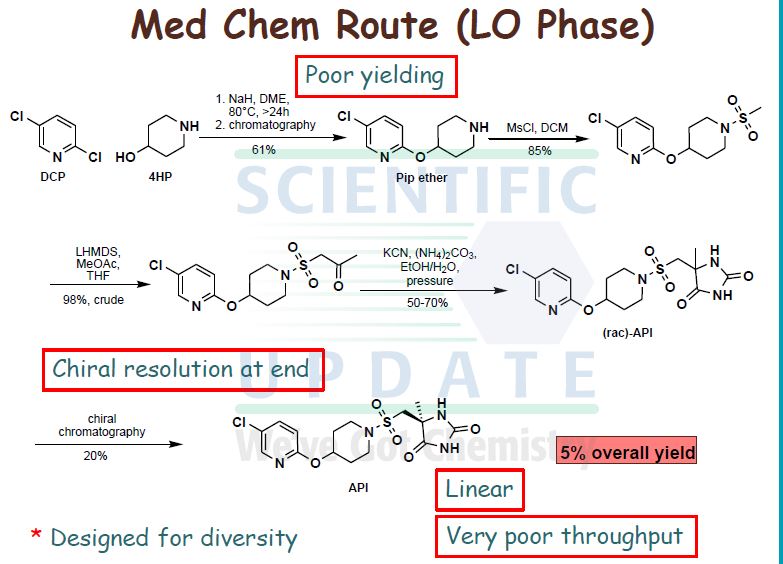
PATENT
WO 02/074767 further discloses a specific metalloproteinase inhibitor compound identified therein as (5S)-5-[4-(5-chloro-pyridin-2-yloxy)-piperidine-l-sulfonylmethyl]-5- methyl-imidazolidine-2,4-dione (page 65, lines 15 to 27; and page 120, lines 23 to 29). This compound is designated herein as compound (I).

(I)
WO 02/074767 further discloses processes for the preparation of compound (I). Thus, in one embodiment, compound (I) is prepared by a route analogous to that shown in the following Scheme (WO 02/074767; pages 87, 113 and 120) but substituting the appropriate amine in step (d):
Scheme 1


Reagents and conditions for Scheme 1: a) KCN, (NHLj)2CO3, EtOHTH2O, +900C, 3h;. b) Chiral separation, CHIRALPAK AD, methanol as eluent;. c) Cl2 (g), AcOH/H2O, <+15 0C, 25min; d) Diisopropylethylamine, THF. -20 0C, 30 min.
The obtained compound (I) is then purified either by precipitation and washing with ethanol/water or by preparative HPLC. In a second embodiment, the racemate of compound (I), (5RS)-5-[4-(5-chloro-pyridin-2- yloxy)-piperidine-l-sulfonylmethyl]-5-methyl-imidazolidine-2,4-dione, was prepared by reacting l-[4-(5-chloro-pyridin-2-yloxy)-piperidine-l-sulfonyl]-propan-2-one with an excess of potassium cyanide and ammonium carbonate in ethanol, and isolating the product by precipitation. Compound (I), the (5S)-enantiomer, was then obtained by chiral HPLC (WO 02/074767; pages 55 and 65).
No crystalline forms of compound (I) are disclosed in WO 02/074767.
Compound (I) is a potent metalloproteinase inhibitor, particularly a potent inhibitor of
MMP 12, and as such is useful in therapy. However, when made according to the processes described in WO 02/074767, compound (I) exhibits unpredictable solid state properties with respect to thermodynamic stability. To prepare pharmaceutical formulations containing compound (I) for administration to humans in accordance with the requirements of U.S. and other international health registration authorities, there is a need to produce compound (I) in a stable form, such as a stable crystalline form, having constant physical properties.


PATENT
WO 02/074767 further discloses a specific metalloproteinase inhibitor compound identified therein as (5S)-5-[4-(5-chloro-pyridin-2-yloxy)-piperidine-l-sulfonylmethyl]-5- methyl-imidazolidine-2,4-dione (page 65, lines 15 to 27; and page 120, lines 23 to 29). This compound is designated herein as compound (I).
(I)
WO 02/074767 further discloses processes for the preparation of compound (I). Thus, in one embodiment, compound (I) is prepared by a route analogous to that shown in the following Scheme (WO 02/074767; pages 87, 113 and 120) but substituting the appropriate amine in step (d):
Scheme 1
Reagents and conditions for Scheme 1: a) KCN, (NHLj)2CO3, EtOHTH2O, +900C, 3h;. b) Chiral separation, CHIRALPAK AD, methanol as eluent;. c) Cl2 (g), AcOH/H2O, <+15 0C, 25min; d) Diisopropylethylamine, THF. -20 0C, 30 min.
The obtained compound (I) is then purified either by precipitation and washing with ethanol/water or by preparative HPLC. In a second embodiment, the racemate of compound (I), (5RS)-5-[4-(5-chloro-pyridin-2- yloxy)-piperidine-l-sulfonylmethyl]-5-methyl-imidazolidine-2,4-dione, was prepared by reacting l-[4-(5-chloro-pyridin-2-yloxy)-piperidine-l-sulfonyl]-propan-2-one with an excess of potassium cyanide and ammonium carbonate in ethanol, and isolating the product by precipitation. Compound (I), the (5S)-enantiomer, was then obtained by chiral HPLC (WO 02/074767; pages 55 and 65).
No crystalline forms of compound (I) are disclosed in WO 02/074767.
Compound (I) is a potent metalloproteinase inhibitor, particularly a potent inhibitor of
MMP 12, and as such is useful in therapy. However, when made according to the processes described in WO 02/074767, compound (I) exhibits unpredictable solid state properties with respect to thermodynamic stability. To prepare pharmaceutical formulations containing compound (I) for administration to humans in accordance with the requirements of U.S. and other international health registration authorities, there is a need to produce compound (I) in a stable form, such as a stable crystalline form, having constant physical properties.
A preferred process for the synthesis of compound (I) is shown in Scheme 2.
Scheme 2
KCN, (NH4)2CO3
(H) 2-propanol

Chromatography KOBu’

Cl2
AcOH AcOH, H2O

Compound (I)

Recrystallisation EtOH, H2O
Compound (I) Form G

Example 5
(S)-5-[4-(5-ChIoro-pyridin-2-yloxy)-piperidine-l-suIfonylmethyl]-5-methyl- imidazoIidine-2,4-dione Process 1
I5 a) 5-Chloro-2-(piperidin-4-yloxy)-pyridine
5-Chloro-2-(piperidin-4-yloxy)-pyridine acetate (40 g, 0.146 mol) was slurried in iso- PrOAc (664 mL) at 300C. To this slurry was added Na2CO3 (1.5 mol per litre; 196 mL, 2 mol eq.). The slurry was then rapidly stirred at 30 0C for 15 minutes. The biphasic mixture was allowed to settle, and the bottom aqueous phase was separated and discarded.
20 The above base washing procedure was repeated twice more. The organic phase was then washed once with water (200 mL). The resulting iso-VxOAc solution was reduced in volume to approximately 300 mL by distillation under reduced pressure. The solution was then diluted with zsø-PrOAc (400 mL) and again distilled down to approximately 300 mL. This procedure was repeated once more. A sample was removed for analysis of 5-chloro-
25 2-(piperidm-4-yloxy)-pyridine content and water content. The weight or the volume of the solution was measured in order to calculate the concentration of 5-chloro-2-(piperidin-4- yloxy)-pyridme in the Z-PrOAc solution.
fr) rSV5-r4-(5-Chloro-pyridin-2-yloxyVpiperidine-l-sulfonylmethvn-5-methyl- 30 imidazolidine-2 ,4-dione Diisopropylethylamine (24.3 mL, 0.139 mol, 1 mol eq.) was added to the iso-PrOAc solution prepared in part (a) [ca. 300 mL; equivalent to 31.2 g, 0.146 mol, 1.05 mol eq. of 5-chloro-2-(piperidin-4-yloxy)-pyridine] in one portion at RT. The solution was then cooled to -15 °C.
((S)-4-Methyl-2,5-dioxo-imidazolidin-4-yl)-methanesulfonyl chloride (31.65 g, 0.139 mol, 1 mol eq.) was dissolved in dry THF (285 mL) at RT with stirring. The resulting solution was then added to the iso-PrOAc solution of 5-chloro-2-(piperidin-4-yloxy)- pyridine dropwise at -15 0C over about 1.5 h. A precipitate was seen on addition of the ((S)-4-methyl-2,5-dioxo-imidazolidin-4-yl)-methanesulfonyl chloride. At the end of the addition, dry THF (32 mL) was added to the reaction mixture to wash the line and the mixture was stirred for 1 h at — 15 0C. It was then warmed to 20 °C over 1 h and stirred at 20 °C for 1 h further. The reaction was quenched with 10 wt% NaHSO4 (157 mL) with rapid stirring. After about 15 minutes, the biphasic mixture was allowed to settle, and the bottom aqueous phase was separated and discarded. This acid wash procedure was repeated once more. The organic phase was then washed with water (157 mL) using rapid stirring and allowing complete phase separation before partitioning. The reaction solution was then warmed to 40 °C and washed again with water (157 mL). THF (95 mL) was added to the organic layer that was then warmed to 40 0C and filtered at 400C to remove any particulate matter. The solvent volume was then reduced to about 157 mL by reduced pressure distillation with the jacket temperature at 55 °C. zso-PrOAc (317 mL) was then added and the volume was again reduced to about 157 mL. Two more put-and-takes of zsø-PrOAc (317 mL) were carried out. Solids began to precipitate out during the distillations and a suspension resulted. The volume was reduced to about 157 mL each time and after the final distillation a small sample of solvent was then removed from the reaction mixture for residual THF analysis. The 1H NMR showed no THF peaks. The contents of the reaction were then cooled to 0 °C and the product was collected by filtration. The reaction vessel was washed with zsø-PrOAc (63 mL) and this rinse was used to wash the product on the filter. The product was dried overnight in a vacuum oven at 40 °C. The required (S)-5-[4- (5-chloro-pyridin-2-yloxy)-piperidine-l-sulfonyhnethyl]-5-methyl-imidazolidine-2,4-dione was isolated as a white solid in 71% yield (41.8 g).
1H NMR (300MHz, d6-DMSO) δ 10.74 (IH, s), 8.20 (IH, d), 8.01 (IH, s), 7.81 (IH, dd), 6.87 (IH, d), 5.09 (IH, m), 3.52-3.35 (4H, m), 3.13 (2H, m), 2.02 (2H, m), 1.72 (2H, m), 1.33 (3H, s).
Example 6
(S)-5-[4-(5-Chloro-pyridin-2-yloxy)-piperidine-l-sulfonylmethyl]-5-methyl- imidazolidine-2,4-dione Process 2 a) 5-Chloro-2-(piperidm-4-yloxy)-pyridine
5-Chloro-2-(piperidm-4-yloxy)-pyridine acetate (70 g, 257 mmol) was slurried in toluene
(560 mL) at RT. IM NaOH (420 mL) was added and the slurry was then rapidly stirred at RT for 15 min. The biphasic mixture was allowed to settle, and the bottom aqueous phase was separated and discarded. The organic phase was then washed with water (2 x 420 mL). A sample was removed from the organic phase and assayed for 5-chloro-2-(piperidin-
4-yloxy)-pyridine.
The resulting toluene solution was then reduced in volume by distillation at reduced pressure, down to approximately 168 mL (2.4 vol eq. with respect to 5-chloro-2-(piperidin-
4-yloxy)-pyridine acetate charge). The solution was then diluted with toluene (420 mL) and again distilled down to approx 168 mL (2.4 vol eq.). A sample was removed for analysis of water content.
b*) (S)-5-r4-r5-Chloro-pyridm-2-yloxy)-piperidine-l-sulfonylmethvH-5-methyl- imidazolidine-2 ,4-dione
Diisopropylethylamine (38.4 mL, 220 mmol) was added to the toluene solution of 5-chloro-2-(piperidin-4-yloxy)-pyridine obtained in step (a) (containing 236 mmol) in one portion followed by dry THF (151 mL) as a line wash. ((S)-4-Methyl-2,5-dioxo- imidazolidin-4-yl)-methanesulfonyl chloride (48.7 g, 215 mmol) was dissolved in dry THF (352 mL) at RT with stirring. The resulting solution of the sulfonyl chloride was then added dropwise to the toluene/THF solution of 5-chloro-2-(piperidin-4-yloxy)-pyridine and diisopropylethylamine at RT over 1 to 2 h. A precipitate was seen on addition of the sulfonyl chloride. At the end of the addition, dry THF (50 mL) was added to the reaction 5 mixture as a line wash. After the addition was complete, the reaction was stirred for about 30 min at RT.
The reaction was quenched with 10 wt% NaHSO4 (251 mL) with rapid stirring for approx 15 min. The biphasic mixture was allowed to settle, when the bottom aqueous phase was io separated and discarded. This acid wash procedure was repeated once more. The solvent volume was then reduced to about 220 mL by reduced pressure distillation. Toluene (300 mL) was then added and the volume was reduced to about 245 mL Solids begin to precipitate during the distillations and a suspension resulted. After the final distillation, a small sample of solvent was then removed from the reaction mixture for residual THF i5 analysis.
The contents of the reaction mixture were then cooled to 0 °C, stirred for about 30 minutes at this temperature and the product was collected by filtration. The reaction vessel was washed with toluene (100 mL) and this rinse was used to wash the product on the filter. 20 The product was dried in a vacuum oven at 40 0C to constant weight. (S)-5-[4-(5-Chloro- pyridin-2-yloxy)-piperidine-l-sulfonylmethyl]-5-methyl-imidazolidine-2,4-dione was isolated as a white solid in typically 85 to 88% yield over the two steps.

Aerial view of Mölndal
Patent
WO 2003106689

Paul Hudson, President, AstraZeneca U.S. and Executive Vice President, North America, joined by AstraZeneca volunteers to celebrate the AstraZeneca Hope Lodge’s fifth birthday.
CLIPS
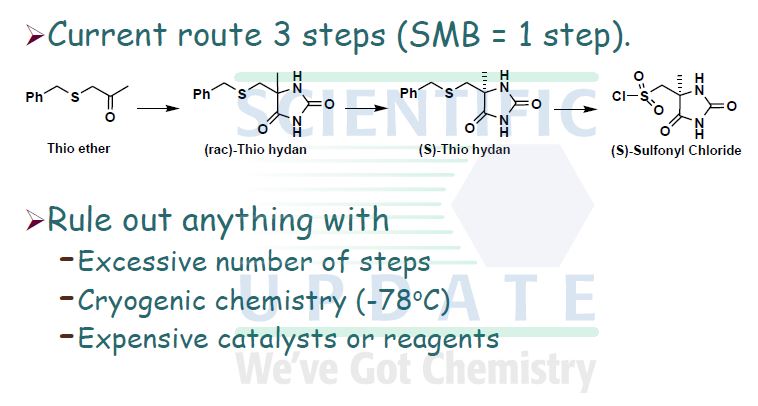
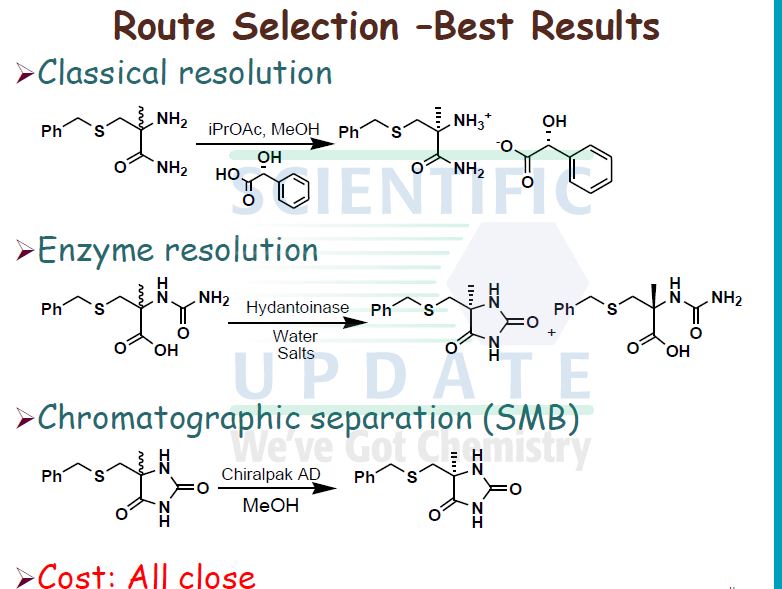

Astra boss Pascal Soriot
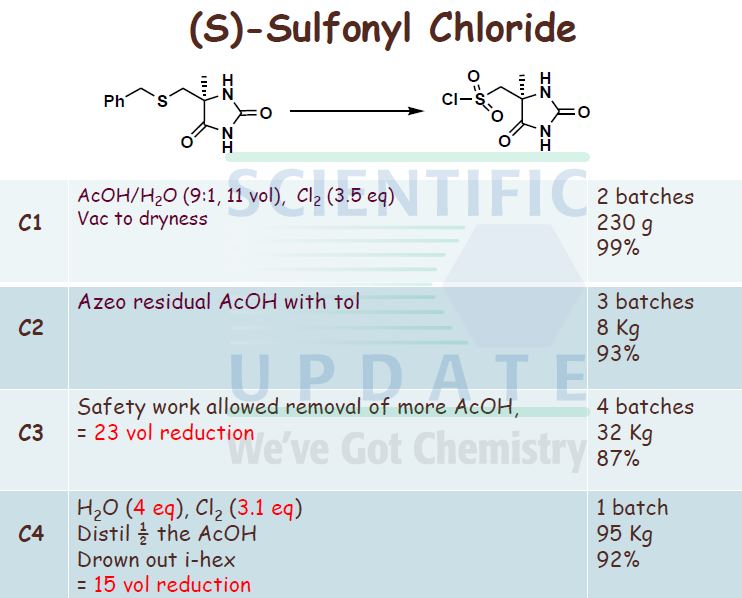
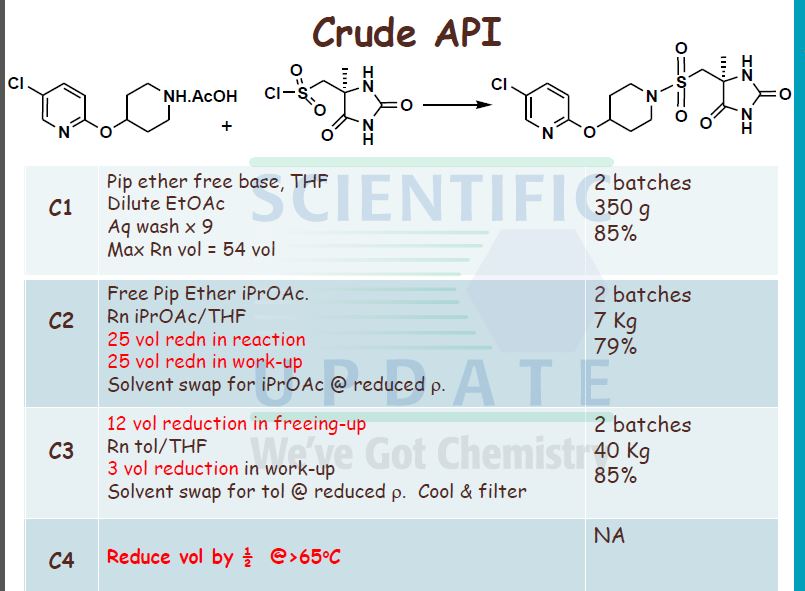

Massachusetts Economic Development Secretary Jay Ash (left) congratulates Kumar Srinivasan, Head of AstraZeneca R&D Boston (right), at a ceremony to launch AstraZeneca’s Gatehouse Park BioHub.
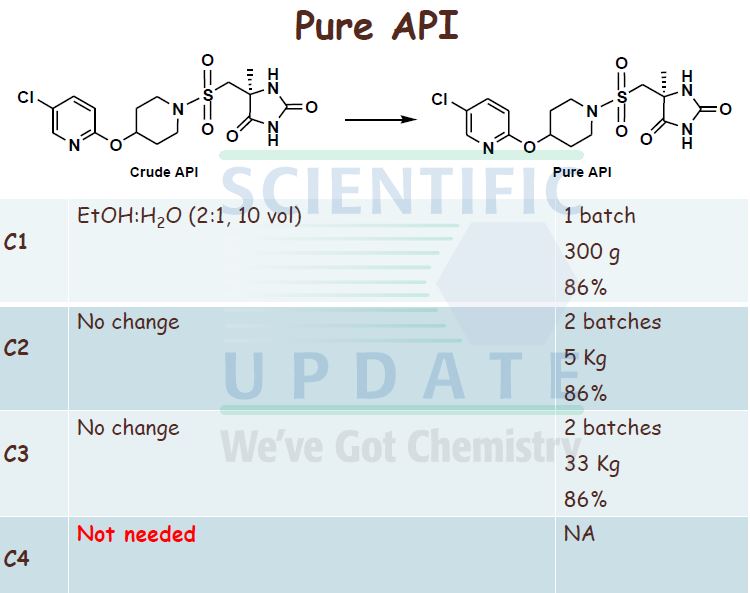
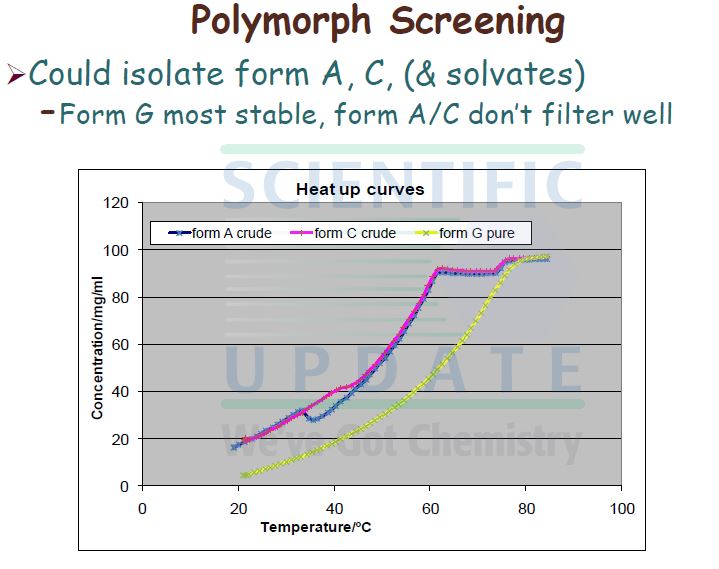


| References |
| 1. AstraZeneca. AZD1236. Accessed on 31/10/2014. Modified on 31/10/2014. Open Innovation, http://openinnovation.astrazeneca.com/what-we-offer/compound/azd1236/ |
| 2. Dahl R, Titlestad I, Lindqvist A, Wielders P, Wray H, Wang M, Samuelsson V, Mo J, Holt A. (2012) Effects of an oral MMP-9 and -12 inhibitor, AZD1236, on biomarkers in moderate/severe COPD: a randomised controlled trial. Pulm Pharmacol Ther, 25 (2): 169-77. [PMID:22306193] |
https://ncats.nih.gov/files/AZD1236.pdf
http://openinnovation.astrazeneca.com/what-we-offer/compound/azd1236/
| WO1992001062A1 * | Jul 4, 1991 | Jan 23, 1992 | Novo Nordisk A/S | Process for producing enantiomers of 2-aryl-alkanoic acids |
| WO1996021640A1 * | Jan 16, 1996 | Jul 18, 1996 | Teva Pharmaceutical Industries, Ltd. | Optically active aminoindane derivatives and preparation thereof |
| WO2002074767A1 * | Mar 13, 2002 | Sep 26, 2002 | Astrazeneca Ab | Metalloproteinase inhibitors |
| WO2003093260A1 * | Apr 29, 2003 | Nov 13, 2003 | Biogal Gyogyszergyar Rt. | Novel crystal forms of ondansetron, processes for their preparation, pharmaceutical compositions containing the novel forms and methods for treating nausea using them |
| WO2003094919A2 * | May 12, 2003 | Nov 20, 2003 | Teva Pharmaceutical Industries Ltd. | Novel crystalline forms of gatifloxacin |
| EP0175312A2 * | Sep 14, 1985 | Mar 26, 1986 | Kanegafuchi Kagaku Kogyo Kabushiki Kaisha | Process for preparing optically active hydantoins |
| EP0255390A2 * | Jul 30, 1987 | Feb 3, 1988 | MediSense, Inc. | Rhodococcus bacterium for the production of aryl acylamidase |
| EP0442584A1 * | Feb 14, 1991 | Aug 21, 1991 | Dsm N.V. | Process for the preparation of an optically active amino acid amide |
| EP0580210A1 * | Jul 6, 1993 | Jan 26, 1994 | Dsm N.V. | Process for the preparation of optically active methionine amide |
| EP0909754A1 * | Oct 13, 1998 | Apr 21, 1999 | Eli Lilly And Company | Process to make chiral compounds |
| EP1550725A1 * | Jun 5, 2003 | Jul 6, 2005 | Kaneka Corporation | PROCESS FOR PRODUCING OPTICALLY ACTIVE alpha-METHYLCYSTEINE DERIVATIVE |
| US4983771 * | Sep 18, 1989 | Jan 8, 1991 | Hexcel Corporation | Method for resolution of D,L-alpha-phenethylamine with D(-)mandelic acid |
| US20040044215 * | Aug 28, 2003 | Mar 4, 2004 | Alain Alcade | Crystalline forms of osanetant |
| US20040266832 * | Jun 24, 2004 | Dec 30, 2004 | Li Zheng J. | Crystal forms of 2-(3-difluoromethyl-5-phenyl-pyrazol-1-yl)-5-methanesulfonyl pyridine |
| Reference | ||
|---|---|---|
| 1 | * | HIRRLINGER B. ET AL.: ‘Purification and properties of an amidase from Rhodococcus erythropolis MP50 which enantioselectively hydrolyzes 2-arylpropionamides‘ JOURNAL OF BACTERIOLOGY vol. 178, no. 12, June 1996, pages 3501 – 3507, XP001174103 |
| 2 | * | See also references of EP2064202A2 |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| US7625934 | Dec 1, 2009 | Astrazeneca Ab | Metalloproteinase inhibitors | |
| US7772403 | Mar 15, 2007 | Aug 10, 2010 | Astrazeneca Ab | Process to prepare sulfonyl chloride derivatives |
///////AZD1236, AZD-1236, AZD 1236,
O=S(=O)(C[C@@]1(C)NC(=O)NC1=O)N3CCC(Oc2ccc(Cl)cn2)CC3
CDMO Ash Stevens to Be Acquired by Piramal Enterprises

Piramal Enterprises Limited announced that its wholly owned subsidiary in the U.S. has entered into an agreement to acquire 100 percent stake in Ash Stevens Inc., a U.S.-based contract development and manufacturing organization (CDMO), in an all cash deal for a consideration of USD $42.95 million plus an earn-out consideration capped at $10 million. This potential transaction is expected to be completed by the end of August.
Located in Riverview, Michigan, Ash Stevens has over 50 years of experience in contract manufacturing, and serves several biotech, mid-size pharma, and large pharmaceutical clients worldwide.
With over 60,000 sq. ft. of facilities, eight chemical drug development and production laboratories, and six full-scale production areas, Ash Stevens has built a stellar reputation, led by science, driven by operational excellence, and one that emphasizes quality as a culture. As one of the leaders in HPAPI manufacture, Ash Stevens has an impeccable safety record of working with high potency anti-cancer agents and other highly-potent therapeutics. The state-of-the-art manufacturing facility in Michigan features all necessary engineering and containment controls for the safe handling and cGMP manufacture of small and large-scale HPAPIs, with Occupational Exposure Limits (OELs) ≤ 0.1µg/m3. The facility has approvals from U.S., EU, Australia, Japan, Korea, and Mexico regulatory agencies.
“The acquisition of Ash Stevens fits well with our strategy to build an asset platform that offers value to our partners and collaborators. Currently, around 25 percent of the molecules in clinical development are potent. Our clients are looking for reliable partners that can assist them in advancing these programs forward,” said Vivek Sharma, CEO of Piramal Pharma Solutions. He further adds, “North America is a key market that we can now service with our three local facilities – the Coldstream Labs in Kentucky for fill finish needs, the Torcan facility in Toronto for complex high value APIs and now, Ash Stevens in Michigan for HPAPIs. Having facilities with a differentiated platform and geographical proximity to clients are keys towards building strategic partnerships. We expect this acquisition to also be synergistic with our Antibody Drug Conjugates (ADCs) and injectable business. We can now fulfill client requirements for a single source of supply for both high potent APIs and drug products.”
“With its rich history of scientific excellence, a track record of 12 product launches, Ash Stevens is well poised to become the partner of choice for clients looking to advance programs from early development through launch. In addition to the business benefits that the combined entity will bring to our clients, I am also pleased that the firms share common core values: both were founded by successful entrepreneurs, value integrity, and are committed to a customer-first approach,” said Dr. Mark Cassidy, President of the API Business at Piramal Pharma Solutions. “I am pleased to welcome the Ash Stevens team into the Piramal group. We expect them to be an integral part of our future growth plans.”
Added Dr. Stephen Munk, CEO of Ash Stevens, “We look forward to working with the Piramal leadership and management team, to develop API solutions that benefit customers and improve the lives of patients. The commitment that Piramal has shown towards growing its healthcare businesses, coupled with the complementary capabilities that our two firms have, makes this an exciting time for Ash Stevens and our employees. We have already identified areas where we can create significant value together, and will be moving forward rapidly to achieve those objectives.”
The transaction is not subject to any regulatory approvals. No related party of PEL has any interest in Ash Stevens.
Wells Fargo Securities, LLC served as exclusive financial advisor to Ash Stevens, with legal counsel provided by Morrison & Foerster LLP.
For further information on the financials, please visit our website: www.piramal.com.

Dr. Stephen A. Munk, President and CEO of Ash Stevens Inc.

Large scale reactor train with 2000, 3000, and 4000 L glass-lined reactors equipped with split butterfly valves.


Ajay Piramal

From left: Anand Piramal, executive director, Piramal Group; Swati Piramal, vice-chairperson, Piramal Group; Ajay Piramal, chairman, Piramal Group; Nandini Piramal, executive director, Piramal Enterprises; and Peter DeYoung, president, Piramal Enterprises
////////////CDMO, Ash Stevens, Piramal Enterprises, Stephen A. Munk
ULIXERTINIB, уликсертиниб , أوليكسيرتينيب , 优立替尼 ,
OR
ULIXERTINIB
4-(5-chloro-2-isopropylaminopyridin-4-yl)-1H-pyrrole-2-carboxylic acid[1-(3-chlorophenyl)-2-hydroxyethyl]amide
| Molecular Formula: | C21H22Cl2N4O2 |
|---|---|
| Molecular Weight: | 433.33098 g/mol |
BVD-523; BVD-ERK; BVD-ERK/HM; BVD-ERK/ST; VRT-0752271; VRT-752271; VX-271, V
уликсертиниб , أوليكسيرتينيب , 优立替尼 ,
4-[5-chloro-2-(isopropylamino)-4-pyridyl]-N-[(1S)-1-(3-chlorophenyl)-2-hydroxy-ethyl]-1H-pyrrole-2-carboxamide
CAS 869886-67-9
Vertex Pharmaceuticals, Incorporated innovators
ULIXERTINIB HCl

| Molecular Weight | 469.79 |
| Formula | C21H22Cl2N4O2●HCl |
| CAS | 1956366-10-1 |
| Chemical Name | 1H-Pyrrole-2-carboxamide, 4-[5-chloro-2-[(1-methylethyl)amino]-4-pyridinyl]-N-[(1S)-1-(3-chlorophenyl)-2-hydroxyethyl]-,hydrochloride(1:1) |
Ulixertinib malonate
4-(5-chloro-2-isopropylaminopyridin-4-yl)-1H-pyrrole-2-carboxylic acid[1-(3-chlorophenyl)-2-hydroxyethyl]amide (referred to as ulixertinib malonate)
- Originator Vertex Pharmaceuticals
- Developer BioMed Valley Discoveries
- Class Aminopyridines; Antineoplastics; Pyrroles; Small molecules
- Mechanism of Action Mitogen activated protein kinase 3 inhibitors; Mitogen-activated protein kinase 1 inhibitor
Highest Development Phases
- Phase I/II Acute myeloid leukaemia; Cancer; Myelodysplastic syndromes
- Phase I Pancreatic cancer
Most Recent Events
- 01 Mar 2016 Phase-I clinical trials in Pancreatic cancer (Combination therapy, First-line therapy, Metastatic disease) in USA (PO) (NCT02608229)
- 23 Nov 2015 BioMed Valley Discoveries and Washington University School of Medicine plan a phase Ib trial for Pancreatic cancer (First-line therapy, Metastatic disease, Combination therapy) (PO) (NCT02608229)
- 01 Nov 2014 Phase-I/II clinical trials in Acute myeloid leukaemia (Second-line therapy or greater) and Myelodysplastic syndromes (Second-line therapy or greater) in USA (NCT02296242) (PO)
![]()
INTRODUCTION
Ulixertinib is in phase I/II clinical trials for the treatment of acute myelogenous leukemia (AML), myelodysplasia and advanced solid tumors.
Members of the family of B-cell CLL/lymphoma 2 proteins (BCL-2) are apoptosis regulators. These proteins control mitochondrial outer
membrane permeabilization (MOMP). Expression of BCL-2 protein blocks cell death in response to various cellular injuries. A number of cancers, including melanoma, breast, prostate, chronic lymphocytic leukemia, and lung cancer, may be caused by damage to the BCL-2 gene. Mutations in BCL-2 may also be a cause of resistance to cancer treatments. Unfortunately, resistance can quickly develop using conventional BCL-2 inhibitor therapies to treat cancer.
Extracellular-signal-regulated kinases (ERKs) are protein kinases that are involved in cell cycle regulation, including the regulation of meiosis, mitosis, and postmitotic functions in differentiated cells. Disruption of the ERK pathway is common in cancers. However, to date, little progress has been made developing effective ERK inhibitors for the treatment of cancer.
As the understanding of the molecular basis of cancer grows, there is an increased emphasis on developing drugs that specifically target particular nodes in pathways that lead to cancer. In view of the deficiencies noted above, there is, inter alia, a need for effective molecularly targeted cancer treatments, including combination therapies. The present invention is directed to meeting these and other needs.

Mitogen-activated protein kinase (MAPK) pathways mediate signals which control diverse cellular processes including growth, differentiation, migration, proliferation and apoptosis. One MAPK pathway, the extracellular signal-regulated kinase (ERK) signaling pathway, is often found to be up-regulated in tumors. Pathway members, therefore, represent attractive blockade targets in the development of cancer therapies (Kohno and Pouyssegur, 2006). For example, U.S. Patent No. 7,354,939 B2 discloses, inter alia, compounds effective as inhibitors of ERK protein kinase. One of these compounds, 4-(5-Chloro-2-isopropylaminopyridin-4-yl)-1 H-pyrrole-2-carboxylic acid [1 -(3-chlorophenyl)-2-hydroxyethyl]amide, is a compound according to formula (I):

Pharmaceutical compositions are often formulated with a crystalline solid of the active pharmaceutical ingredient (API). The specific crystalline form of the API can have significant effects on properties such as stability and solubility / bioavailability. Instability and solubility characteristics can limit the ability to formulate a composition with an adequate shelf life or to effectively deliver a desired amount of a drug over a given time frame (Peterson et al., 2006).
Synergistic combination comprising an ERK1/2 inhibitor (such as ulixertinib) and a BCL-2 family inhibitor (such as navitoclax), assigned to BioMed Valley Discoveries (BVD), naming Decrescenzo and Welsch. BVD, presumably under license from Vertex, is developing ulixertinib (phase 2 trial), a small-molecule ERK 1/2 inhibitor for treating cancers including acute myelogenous leukemia and myelodysplastic syndrome. In June 2015, clinical data were presented at the 51st ASCO meeting in Chicago, IL.
BIOMED VALLEY DISCOVERIES

PATENT
WO2005113541 PDT PATENT
I-9 COMPD
SEE BELOW
PATENT
Novel crystalline forms of 4-(5-chloro-2-isopropylaminopyridin-4-yl)-1H-pyrrole-2-carboxylic acid[1-(3-chlorophenyl)-2-hydroxyethyl]amide (referred to as ulixertinib) can be prepared which exhibit improved properties, eg surprisingly improved stability and solubility characteristics. Also claimed is their use for treating cancer.
EXAMPLE 2
Preparation of Crystaline Free Base 4-(5-Chloro-2-isopropylaminopyridin-4-yl)-1 H-pyrrole-2-carboxylic acid [1 -(3-chlorophenyl)-2-hydroxyethyl]amide
4-(5-Chloro-2-isopropylaminopyridin-4-yl)-1 H-pyrrole-2-carboxylic acid [1 -(3-chlorophenyl)-2-hydroxyethyl]amide free base was prepared according to the following synthesis scheme.
Stepl

C5H2CIFIN
257.43 C8H10CIIN2
ASYM-11 1606 296.54
ASYM-1 12060

ASYM-111938 ASYM-112393

ASYM-1 11935
In Step 1 , a clean and dry 200 L glass-lined reactor was evacuated to <-0.08 MPa, and then filled with nitrogen to normal pressure three times. Anhydrous ethanol (49.90 kg) was charged into the 200 L glass-lined reactor. ASYM-1 1 1606 (Asymchem) (12.70 kg) and isopropylamine (29.00 kg) were added into the mixture in turn. The mixture was heated to 65-75°C for refluxing. The mixture reacted at 65-75°C. After 20 h, the reaction was sampled and analyzed by HPLC every 4-6 h until the content of ASYM-1 1 1606 was <1 %. The mixture was cooled to 40-45°C and was concentrated at <45°C under reduced pressure (<-0.08 MPa) until 13-26 Lremained. The organic phase was washed with a sodium chloride solution and was stirred for 20-30 min and settled for 20-30 min before separation. The organic phase was concentrated at <30°C under reduced pressure (<-0.06 MPa) until 13-20 L remained. Petroleum ether (8.55 kg) was added into the concentrated mixture. The mixture was transferred into a 20 L rotary evaporator and continued concentrating at <30°C under reduced pressure (<-0.06 MPa) until 13-20 L remained. Then petroleum ether (8.55 kg) was added into the concentrated mixture. The mixture was cooled to 0-5°C and stirred for crystallization. After 1 h, the mixture was sampled and analyzed by wt% every 1 -2 h until the wt% of the mother liquor was <1 1 % or the change of the wt% between consecutive samples was <1 %. The mixture was filtered with a 10 L filter flask. The filter cake was sampled and analyzed for purity by HPLC. 10.50 kg of product was recovered as a brownish yellow solid at 99.39% purity.
In Step 2, a clean and dry 300 L glass-lined reactor was evacuated to <-0.08 MPa, and then filled with nitrogen to normal pressure three times. Glycol dimethyl ether (73.10 kg) was charged into the 300 L glass-lined reactor at 20-30°C. ASYM-1 12060 (Asymchem) (10.46 kg) and ASYM-1 1 1938 (Asymchem) (12.34 kg, 1 1 .64 kg after corrected) were added into the mixture in turn under the protection of nitrogen. Maintaining the temperature at 20-30°C, purified water (10.50 kg) and anhydrous sodium carbonate (5.67 kg) were added into the mixture. Palladium acetate (0.239 kg) and tricyclohexylphosphonium tetrafluoroborate (0.522 kg) were added into the mixture under the protection of nitrogen. After addition, the mixture was evacuated to <-0.06 MPa, and then filled with nitrogen to normal pressure. This was repeated for ten times until residual oxygen was <300 ppm. The mixture was heated to 75-85°C for refluxing. The mixture reacted at 75-85°C. After 4 h, the mixture was sampled and analyzed by HPLC every 2-3 h for content of ASYM-
1 12060. The content of AS YM-1 12060 was 6.18%, so additional ASYM-1 1 1938 (0.72 kg) was added and continued reaction until the content of ASYM-1 12060 was <3%. The mixture was cooled to 25-35°C and filtered with a 30 L stainless steel vacuum filter. The filter cake was soaked and washed twice with THF (14.10kg). The filtrate and washing liquor were combined and concentrated at <50°C under reduced pressure (<-0.08 MPa) until 10-15 L remained. The mixture was cooled to 15-25°C. Methanol (1 1 .05 kg) was added into the concentrated mixture. Then the mixture was stirred for crystallization. After 2 h, the mixture was sampled and analyzed by HPLC every 2-4 h until the wt% of the mother liquor was <2%. The mixture was filtered with a 30 L stainless steel vacuum filter. The filter cake was soaked and washed twice with methanol (8.30 kg). The filter cake was transferred into a 50 L plastic drum. Then ethyl acetate (7.10 kg) and petroleum ether (46.30 kg) were added into the drum. The mixture was stirred for 1.5-2 h and then filtered with a nutsche filter. The filter cake was soaked and washed with petroleum ether (20.50 kg). The filter cake was dried in the nutsche filter under nitrogen at 30-40°C. After 8 h, the solid was sampled and Karl Fischer (KF) analysis was performed in intervals of 4-8 h to monitor the drying process. Drying was completed when the KF result was <1 .0% water. During drying, the solid was turned over and mixed every 4-6 h. 12.15 kg of product was recovered as a brownish yellow solid at 98.32% purity.
In Step 3, a clean and dry 300 L glass-lined reactor was evacuated to <-0.08 MPa, and then filled with nitrogen to normal pressure three times. THF (62.58 kg) was charged into the 300 L glass-lined reactor at 15-30°C. Then the stirrer was started. ASYM-1 12393 (12.00 kg, 1 1 .70 kg after corrected) was added into the mixture. The mixture was stirred until the solid dissolved completely. Maintaining the temperature at 15-30°C, a lithium hydroxide solution which was
prepared with lithium hydroxide monohydrate (5.50 kg) in purified water (70.28 kg) was added into the mixture. Then diethylamine (3.86 kg) was added. The mixture was heated to 60-70°C for refluxing. The mixture reacted at 60-70°C. After 30 h, the reaction was sampled and analyzed by HPLC every 4-6 h until the content of intermediate at relative retention time (RRT)=1 .39-1 .44 was <1 % and the content of ASYM-1 12393 was <1 %. HPLC conditions for this analysis are set forth in Table 1 .
Table 1 : HPLC Parameters

The mixture was cooled to 25-35°C and MTBE (25.97 kg) was added into the mixture. The mixture was stirred for 20-30 min and filtered via an in-line fluid filter. The filtrate was transferred into a 300 L glass-lined reactor and settled for 20-30 min before separation. The pH of the obtained aqueous phase was adjusted with a 6 N hydrochloric acid solution which was prepared from concentrated hydrochloric acid (14.86 kg) in purified water (10.88 kg) at the rate of 5-8 kg/h at 15-25°C until the pH was 1 -2. The pH of the mixture was adjusted again with a saturated sodium carbonate solution which was prepared from sodium carbonate (5.03 kg) in purified water (23.56 kg) at the rate of 3-5 kg/h at 15-25°C until the pH was 6.4-6.7. Then the pH of the mixture was adjusted with a hydrochloric acid solution which was prepared from concentrated hydrochloric acid (1 .09 kg) in purified water (0.80 kg) until the pH was 6.2-6.4. The mixture was filtered with a nutsche filter. The filter cake was transferred into a 300 L glass-lined reactor and purified water (1 17.00 kg) was added. The mixture was stirred and sampled and analyzed by HPLC until the p-toluenesulfonic acid residue of the filter cake was <0.5%. Then the mixture was filtered. The filter cake was dried in the tray drier under nitrogen at 55-65°C until KF<10%. The solid and MTBE (8.81 kg) were charged into a 50 L stainless steel drum. The mixture was stirred for 1 -2 h. The mixture was filtered with a 30 L stainless steel vacuum filter. The filter cake was dried in the nutsche filter at 50-60°C. After 8 h, the solid was sampled and analyzed by KF every 4-8 h until KF<5%. During drying, the solid was turned over and mixed every 4-6 h. 6.3 kg of product was recovered as an off-white solid at 98.07% purity.
In Step 4, a dry and clean 50 L flask was purged with nitrogen for 20 min. DMF (30.20 kg) was charged into the 50 L flask reactor. Then the stirrer was started. Maintaining the temperature at 15-25°C, ASYM-1 12394 (3.22 kg, 2.76 kg after corrected) was added into the mixture. The mixture was stirred until the solid dissolved completely. The mixture was cooled to -10 to -20°C and 1 -hydroxybenzotriazole hydrate (2.10 kg) was added into the mixture at -10 to -20°C. Then EDCI (2.41 kg) was added into the mixture in five portions at an interval of about 5-10 min. The mixture was cooled to -20 to -30°C and ASYM-1 1 1888 (Asymchem) (1 .96 kg) was added into the mixture at -20 to -30°C. Then DIEA (1 .77 kg) was added into the mixture at the rate of 3-4 kg/h. The mixture was heated to 15-25°C at the rate of 5-10°C/h. The mixture was reacted at 15-25°C. After 6-8 h, the mixture was sampled and analyzed by HPLC every 2-4 h until the content of ASYM-1 12394 was <2%. The mixture was cooled to 0-10°C and the reaction mixture was quenched with a solution which was prepared from ethyl acetate (28.80 kg) in purified water (12.80 kg) at 0-10°C. The mixture was extracted three times with ethyl acetate (28.80 kg). For each extraction the mixture was stirred for 20-30 min and settled for 20-30 min before separation. The organic phases were combined and washed twice with purified water (12.80 kg). The mixture was stirred for 20-30 min and settled for 20-30 min before separation for each time. Then the obtained organic phase was filtered through an in-line fluid filter. The filtrate was transferred into a 300 L glass-lined reactor. The mixture was washed twice with a 5% acetic acid solution, which was prepared from acetic acid (2.24 kg) in purified water (42.50 kg). The solution was added at the rate of 10-20 kg/h. The organic phase was washed twice with a sodium carbonate solution, which was prepared from sodium carbonate (9.41 kg) in purified water (48.00 kg). The organic phase was washed twice with a sodium chloride solution, which was prepared from sodium chloride (16.00 kg) in purified water (44.80 kg). The organic phase was transferred into a 300 L glass-lined reactor. Anhydrous sodium sulfate (9.70 kg) was added into the mixture and the mixture was stirred for 2-4 h at 15-30°C. The mixture was filtered with a nutsche filter, which was pre-loaded with about 1 cm thick silica gel (7.50 kg). The filter cake was soaked and washed with ethyl acetate (14.40 kg) before filtration. The filtrates were combined and the combined filtrate was added into a 72 L flask through an in-line fluid filter. The mixture was concentrated at T≤40°C under reduced pressure (P<-0.08 MPa) until 3-4 L remained. Then MTBE (4.78 kg) was added into the mixture. The mixture was cooled to 0-10°C for crystallization with stirring. After 1 h, the mixture was sampled and analyzed by wt% every 1-2 h until the wt% of the mother liquor was <5% or the change of wt% between consecutive samples was <1%. The mixture was filtered with a vacuum filter flask and the filter cake was dried in the tray drier under nitrogen at 30-40°C until KF<0.5%. 3.55 kg of product was recovered as an off-white solid at 100% purity.
EXAMPLE 3A
Preparation of 4-(5-Chloro-2-isopropylaminopyridin-4-yl)-1 H-pyrrole-2-carboxylic acid [1 -(3-chlorophenyl)-2-hydroxyethyl]amide Form C

4-(5-Chloro-2-isopropylaminopyridin-4-yl)-1 H-pyrrole-2-carboxylic acid [1 -(3-chlorophenyl)-2-hydroxyethyl]amide Form C was prepared from 4-(5-Chloro-2-isopropylaminopyridin-4-yl)-1 H-pyrrole-2-carboxylic acid [1 -(3-chlorophenyl)-2-hydroxyethyl]amide free base as follows. ASYM-1 1 1935 (10.4 kg) was added to a stirred mixture of anhydrous ethanol (73.9 kg), methanol (4.1 kg) and isopropanol (4.1 kg). The mixture was heated to 70-75°C and stirred until all the solids dissolved. Anhydrous HCI (37 wt%, 1 .1 eq) in a mixture of ethanol/methanol/isopropanol (90:5:5) was added and the mixture maintained at 70-75°C for 2 hours after the addition was completed. The mixture was then cooled to 15-25°C at a rate of 5-15°C per hour and stirred at this temperature until the desired polymorphic purity was reached. The end point of the crystallization/polymorph conversion was
determined by the absence of an XRPD peak at about 10.5° 2Θ in three successive samples.
The mixture was then filtered and washed successively with a pre-prepared solution of anhydrous ethanol (14.8 kg), methanol (0.8 kg) and isopropanol (0.8 kg), followed by MTBE (2 x 21 kg). Avoidance of delay in the washing of the filter cake is preferable because the polymorph may be unstable in the wet filter cake in the presence of reagent alcohol and improved stability was observed after the MTBE wash has been performed. The wet filter cake was then dried in a heated filter funnel or a tray drier at 40-50°C until dry. Typical yields were about 85-90%.
EXAMPLE 3B
Alternative Preparation of 4-(5-Chloro-2-isopropylaminopyridin-4-yl)-1 H-pyrrole-2-carboxylic acid [1 -(3-chlorophenyl)-2-hydroxyethyl]amide Form C

ASYM-1 15985
4-(5-Chloro-2-isopropylaminopyridin-4-yl)-1 H-pyrrole-2-carboxylic acid [1 -(3-chlorophenyl)-2-hydroxyethyl]amide Form C was also prepared from 4-(5-Chloro-2-isopropylaminopyridin-4-yl)-1 H-pyrrole-2-carboxylic acid [1 -(3-chlorophenyl)-2-hydroxyethyl]amide free base as follows. A dry and clean 72 L flask was purged with nitrogen for 20 min. Anhydrous ethanol (21 .35 kg) methanol (1 .17 kg) and isopropanol (1 .19 kg) were charged into the 72 L flask at 15-25°C and the mixture was stirred for 20-30 min. ASYM-1 1 1935 (3.01 kg) was added into the mixture and heated to 70-75°C at the rate of 15-25°C/h and stirred until the solid dissolved completely.
An alcohol / HCI solution was prepared as follows. Anhydrous ethanol (1.500 kg) methanol (0.088 kg) and isopropanol (0.087 kg) were charged into a 5 L flask at 15-25°C and the mixture was stirred for 20-30 min. The mixture was bubbled with hydrogen chloride through a dip tube under stirring at 10-25°C. After 2 h, the mixture was sampled and analyzed every 2-4 h until the wt% of hydrogen chloride was > 35%.
The alcohol / HCI solution (0.519 kg) prepared above was added dropwise into the mixture at the rate of 0.5-1.0 kg/h at 70-75°C. Seed crystal (0.009 kg) was added into the mixture and the alcohol / HCI solution (0.173 kg) prepared above was added into the mixture at the rate of 0.5-1 .0 kg/h at 70-75°C. After addition, the mixture was stirred for 1 -2 h at 70-75°C. The mixture was cooled to 15-25°C at the rate of 5-15°C/h and stirred for 4-6 h. The mixture was heated to 70-75°C at the rate of 15-25°C/h and stirred for 8-10 h at 70-75°C. The mixture was cooled to 15-25°C at the rate of 5-15°C/h and stirred for 4-6 h. The mixture was filtered with a vacuum filter flask. The filter cake was soaked and rinsed with a solution which was prepared from anhydrous ethanol (4.25 kg) and methanol (0.24 kg) and isopropanol (0.24 kg) before filtration. The filter cake was dried in a drying room under nitrogen at 40-50°C until the ethanol residue was <0.5% and methanol residue was <0.3% and isopropanol residue was <0.3%. 2.89 kg of product was recovered as a white solid at 99.97% purity.
PATENT
WO-2016123581
Novel crystalline malonate salt forms of 4-(5-chloro-2-isopropylaminopyridin-4-yl)-1H-pyrrole-2-carboxylic acid[1-(3-chlorophenyl)-2-hydroxyethyl]amide (referred to as ulixertinib malonate) and composition comprising them. Also claimed is their use for treating cancer.
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2016123581&redirectedID=true
EXAMPLE 6
Aqueous Disolution of 4-(5-Chloro-2-isopropylaminopyridin-4-yl)-1 H-pyrrole-2-carboxylic acid [1-(3-chlorophenyl)-2-hydroxyethyl]amide Malonate Form A
Samples of 4-(5-Chloro-2-isopropylaminopyridin-4-yl)-1 H-pyrrole-2-carboxylic acid [1 -(3-chlorophenyl)-2-hydroxyethyl]amide Form C and 4-(5-Chloro-2-isopropylaminopyridin-4-yl)-1 H-pyrrole-2 -carboxylic acid [1 -(3-chlorophenyl)-2-hydroxyethyl]amide malonate Form A were each shaken at ambient temperature in fasting state simulated gastric fluid (FaSSGF) pH 1.6 for 30 minutes. Concentration of 4-(5-Chloro-2-isopropylaminopyridin-4-yl)-1 H-pyrrole-2-carboxylic acid [1 -(3-chlorophenyl)-2-hydroxyethyl]amide was measured at 5, 15 and 30 minutes.
After 30 minutes, the samples were removed from FaSSGF, placed in fasting state simulated intestinal fluid (FaSSIF) pH 6.5, with shaking, for an additional 5 hours. Concentration of 4-(5-Chloro-2-isopropylaminopyridin-4-yl)-1 H-pyrrole-2-carboxylic acid [1 -(3-chlorophenyl)-2-hydroxyethyl]amide was measured at 10, 30, 60 90, 120, 180, 270, and 300 minutes. Results are summarized in Table 13 and shown in FIG. 10A (FaSSGF) and FIG. 10B (FaSSIF).
Table 13: Solubility of 4-(5-Chloro-2-isopropylaminopyridin-4-yl)-1 H-pyrrole-2-carboxylic acid [1 -(3-chlorophenyl)-2-hydroxyethyl]amide Form C and Malonate Form A.

PATENT
PATENT
WO2015095834
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015095834&redirectedID=true
PATENT
![]()
Example 1 Compound 1-9 was prepared as follows:

1-9
2,2,2-TrichIoro-l-(4-iodo-lH-pyrrol-2-yl)ethanone: To a stirred solution of 50 g (235 mmol, 1.0 equiv.) of 2,2,2-trichloro-l-(lH-pyrrol-2-yl)-ethanone in dry dichloromethane (400 mL) under nitrogen, a solution of iodine monochloride (39 g, 240 mmol, 1.02 equivalents) in of dichloromethane (200 mL) was added dropwise. The resulting mixture was stirred at room temperature for 2 hours. The solution was washed with 10% potassium carbonate, water, 1.0 M sodium thiosulfate, saturated sodium chloride, dried over sodium sulfate, filtered, and concentrated under reduced pressure. The solid was recrystallized from hexanes/methyl acetate to afford the title compound (68.5g, 86%) as a colorless solid (86%). MS FIA: 335.8, 337.8 ES-.
4-Iodo-lH-pyrrole-2-carboxyIic acid methyl ester: To a stirred solution of 2,2,2- trichloro-l-(4-iodo-lH-pyrrol-2-yl)ethanone (68g, 201 mmol, 1.0 equivalent) in dry methanol (400 mL) under nitrogen, was added a solution of sodium methoxide in methanol (4.37 M, 54 mL, 235 mmol, 1.2 equivalents) over 10 minutes. The resulting mixture was stirred at room temperature for 1 hour. The volatiles were removed under reduced pressure and the crude was then partitioned between water and tert- butylmethyl ether. The organic phase was separated, washed two times with water, saturated sodium chloride, dried over sodium sulfate, filtered and concentrated under vacuum to afford the title compound (48g, 96%) as a colorless solid, that was used directly without further purification.
4-Iodo-l-(toluene-4-sulfonyl)-lH-pyrrole-2-carboxylic acid methyl ester: 4-Iodo- lH-pyrrole-2-carboxylic acid methyl ester (24.6 g, 98 mmol, 1.0 equivalent) was dissolved in dichloromethane (150 mL) and triethylamine (30 mL, 215.6 mmol, 2.2 equivalents). 4-(Dimethylamino)pyridine (1.2 g, 9.8 mmol, 0.1 equivalent) and p- toluenesulfonylchloride (20.6 g, 107.8 mmol, 1.1 equivalents) were added and the reaction mixture was stirred for 16 hours at room temperature. The reaction was quenched with 1 M ΗC1 and the organic layer was washed with aqueous sodium bicarbonate and brine. After drying over magnesium sulfate, the solvent was removed under reduced pressure and the residue was crystallized from tert-butylmethyl ether, yielding the title compound as a pale yellow solid (30 g, 75%). Rt(min) 8.259 minutes.
4-(4,4,5,5-Tetramethyl-[l,3,2]dioxaborolan-2-yI)-l-(toluene-4-sulfonyl)-lH- pyrrole-2-carboxylic acid methyl ester: To a degassed solution of 4-iodo-l- (toluene-4-sulfonyl)-lH-pyrrole-2-carboxylic acid methyl ester (20 g, 49.4 mmol, 1.0 equivalent) and bis(pinacolato)diborane (15 g, 65 mmol, 1.3 equivalents) in DMF (200 mL) under nitrogen, was added dichloro[l,l ‘- bis(diphenylphosphino)ferrocene]palladium (II) dichloromethane adduct (3.6 g, 4.9 mmol, 0.1 equivalent). The reaction mixture was then stirred at 80 °C for 18 hours. After removing the DMF under reduced pressure, the resulting thick oil residue was suspended in diethyl ether (500 mL) and a solid precipitated immediately. This solid was removed by filtration and the filtrate was washed with IM HCl, water, brine and dried over MgS0 . Concentration afforded the title compound as a white solid and used without further purification (10 g, 50%). LC/MS: Rt(min) 4.6; 406.4 ES+. MS FIA: 406.2 ES+. ‘pfNMR δ 1.2 (s, 12H), 2.35 (s, 3H), 3.8 (s, 3H), 7.2 (m, 3H), 7.8 (d, 2H), 8.0 (s, IH).
N,N’-2-(5-Chloro-4-iodo-pyridyI)-isopropyIarnine:
Method A. (Microwave)
In a 10 mL microwave tube, 5-chloro-2-fluoro-4-iodopyridine (1.0 g, 3.9 mmol, 1.0 equivalent) was dissolved in DMSO (4.0 mL) and then ispropylamine (0.99 mL, 11.7 mmol, 3.0 equivalents) was added. The tube was sealed and placed under microwave irradiation for 600 sec at 150 °C. This reaction was repeated six times. The reaction mixtures were combined, then diluted in ethyl acetate and washed with water. After drying over sodium sulfate, the solvent was evaporated to afford the title compound as a thick brown oil (5.6 g, 80% ) which was used directly without further purification. Rt(min) 4.614; MS FIA: 296.9 ES+. ‘pfNMRsssssss δ 1.25 (d, 6H), 3.65 (m, IH), 7.15 (s, IH), 7.75 (s, IH).
Method B: (Thennal)
5-Chloro-2-fluoro-4-iodopyridine (400 mg, 1.55 mmol, 1.0 equivalent) was dissolved in ethanol (5.0 mL) and then isopropylamine (0.66 mL, 7.8 mmol, 5.0 equivalents) was added. The resulting solution was stirred at 80 °C for 48 hours. The reaction mixture was then diluted in ethyl acetate and washed with water. After drying over sodium sulfate, the solvent was evaporated and a thick brown oil was obtained, which was then purified by flash chromatography on silica gel eluting with mixtures of hexanes/ethyl acetate (from 99:1 to 80:20) to afford the title compound as a pale yellow solid (96 mg, 21%).
4-(5-Chloro-2-isopropylaminopyridin-4-yl)-l-(toluene-4-suIfonyl)-lH-pyrrole-2- carboxylic acid methyl ester: To a solution of N,N’-2-(5-chloro-4-iodo-pyridyl)- isopropylamine (0.53 g, 1.8 mmol, 1.0 equivalent) and 4-(4,4,5,5-tetramethyl- [l,3,2]dioxaborolan-2-yl)-l-(toluene-4-sulfonyl)-lH-pyrrole-2-carboxylic acid methyl ester (0.78 g, 1.8 mmol, 1.0 equivalent) in DME (4.0 mL) was added a solution of aqueous 2 M sodium carbonate (1.0 mL) followed by Pd(PPh3)4 (0.21 mg, 0.18 mmol, 0.1 equivalent). The microwave tube was sealed and the reaction mixture was irradiated by microwave for 1800 sec. at 170 °C. The cmde of six reactions were combined and diluted in ethyl acetate and washed with water. After drying the organic layer with sodium sulfate, the solvent was removed and the resulting thick oil was adsorbed on silica gel. The crude was then purified by flash chromatography on silica, eluting with hexanes/ethyl acetate mixtures (from 99:1 to 70:30) to afford the title compound as a yellow solid (3.1 g, 61% over two steps). Rt(min) 6.556. MS FIA: 448.1 ES+. ‘HNMR δ 1.45 (d, 6H), 2.5 (s, 3H), 3.81 (s, 3H), 6.8 (s, IH), 7.35 (s, IH),
7.4 (d, 2H), 8.0 (m ,3H), 8.3 (s, IH).
4-(5-Chloro-2-isopropylaminopyridin-4-yl)-l-(2,4,6-trimethylbenzenesulfonyl)- lH-pyrrole-2-carboxylic acid methyl ester: To a solution of N,N’-2-(5-chloro-4- iodo-pyridyl)-isopropylamine (96 mg, 0.32 mmol, 1.0 equivalent) and 4-(4,4,5,5- tetramethyl-[ 1 ,3,2]dioxaborolan-2-yl)- 1 -(2,4,6-trimethylbenzenesulfonyl)- lH-pyrrole- 2-carboxylic acid methyl ester (152 mg, 0.35 mmol, 1.1 equivalents) in DME (2 mL), was added a solution of aqueous 2 M sodium carbonate (0.2 mL) followed by Pd(PPh ) (37 mg, 0.032 mmol, 0.1 equivalent). The reaction mixture was stirred at 80 °C for 16 hours. The crude was diluted in ethyl acetate and washed with water. After drying the organic layer with sodium sulfate, the solvent was removed and the resulting thick oil was adsorbed on silica gel. The cmde was then purified by flash chromatography on silica, eluting with hexanes/ethyl acetate mixtures (from 99:1 to 80:20) to afford the title compound as a yellow solid (65 mg, 43%). Rt(min) 7.290. MS FIA:476.1 ES+.
4-(5-Chloro-2-isopropylaminopyridin-4-yl)-lH-pyrrole-2-carboxyIic acid:
Method A. (Microwave)
A solution of 4-(5-chloro-2-isopropylaminopyridin-4-yl)-l-(toluene-4-sulfonyl)-lH- pyrrole-2-carboxylic acid methyl ester (3.1 g, 6.9 mmol, 1.0 equivalent) in TΗF (2.0 mL) was added to a solution of lithium hydroxide monohydrated (710 mg, 17.3 mmol,
2.5 equivalents) in water (3.0 mL). The microwave tube was sealed and the reaction mixture was irradiated by microwave for 1200 sec. at 150 °C. The cmde solution was acidified with aqueous 6Ν ΗC1. The solvent was evaporated off to afford the title compound which was used directly without further purification. Rt(min): 3.574. FIA MS: 279.9 ES+; 278.2 ES-.
Method B: (Thermal)
A solution of 4-(5-chloro-2-isopropylaminoρyridin-4-yl)-l-(2,4,6- trimethylbenzenesulfonyl)-lH-pyrrole-2-carboxylic acid methyl ester (0.69 g, 1.4 mmol, 1.0 equivalent) in TΗF (3.0 mL) was added to a solution of lithium hydroxide monohydrated (1.19 g, 29 mmol, 20.0 equivalents) in water (3.0 mL). The mixture was then refluxed for 8 hours. The cmde solution was acidified with aqueous 6N ΗC1 until cloudy, the organic solvent was partially removed and the product precipitated. The title compound was isolated by filtration and washed with water and diethyl ether, yielding a white solid (0.38 g, 96%).
4-(5-Chloro-2-isopropylaminopyridin-4-yl)-lH-pyrrole-2-carboxyIic acid [l-(3- ch!orophenyl)-2-hydroxyethyl] amide: To a suspension of 4-(5-chloro-2- isopropylaminopyridin-4-yl)-lH-pyrrole-2-carboxylic acid (1.93 g, 6.9 mmol, 1.0 equivalent) in DMF (5.0 mL) was added EDCI (1.45 g, 7.6 mmol, 1.1 equivalents), ΗOBt (0.94 g, 6.9 mmol, 1.0 equivalent) and (5)-3-chlorophenylglycynol (1.58 g, 7.6 mmol, 1.1 equivalents). Dusopropylethylamme (2.7 mL) was then added and the resulting mixture was stirred a room temperature overnight. The mixture was then poured into water and extracted with ethyl acetate. After drying over sodium sulfate, the solvent was removed and the crude was adsorbed on silica gel. Purification was effected by flash chromatography on silica, eluting with mixtures of hexanes/acetone (from 80:20 to 60:40) to afford the title compound as white solid (1.9 g, 64%). Rt(min) 4.981s. FIA MS: 433.1 ES+; 431.2 ES-. 1ΗNMR (CD3OD) δ 1.31 (d, 6H), 3.85 (m, 3H), 5.15 (t, IH), 7.01 (s, IH), 7.25 (m, 3H), 7.4 (s, IH), 7.45 (s, IH), 7.7 (s, IH), 7.95 (s, IH).
Example 2 Compound 1-9 was also prepared according to following alternate method:

2,5-DichIoro-4-nitropyridine N-oxide: To a suspension of 2-chloro-5-chloropyridine (10 g, 0.067 mol) in acetic anhydride (25 mL) was added hydrogen peroxide 30% (25 mL) in small portions. This mixture was stirred at room temperature for 24 hours and then heated at 60 °C for 30 hours. After removing the excess of acetic acid under reduced pressure, the residue was added in small portions to concentrated sulfuric acid (15 mL). The resulting solution was added to a mixture of concentrated sulfuric acid (15 mL) and fuming nitric acid (25 mL) and then heated at 100 °C for 90 minutes. The reaction mixture was poured on ice, neutralized with solid ammonium carbonate and finally with aqueous ammonia until a basic pH was obtained and. A precipitate formed. The precipitate was collected by filtration to afford the title compound as a pale yellow solid (3.1 g), Rt(min) 3.75. MS FIA shows no peak. ‘pfΝMR (DMSO-de) δ 8.78 (s, IH), 9.15 (s, IH).
4-Bromo-2-chloro-5-N-isopropylpyridin-2-amine N-oxide: To 2,5-dichloro-4- nitropyridine Ν-oxide (400 mg, 1.9 mmol) was added acetyl bromide (2 mL) very slowly. The reaction mixture was then heated at 80 °C for 10 minutes. The solvent was removed under a stream of nitrogen and the cmde product was dried under high vacuum. The cmde material (165 mg, 0.62 mmol) was dissolved in ethanol (2 mL), z‘so-propylamine (0.53 mL) added and the resulting mixture was heated at 80 °C for 2 hours. The cmde solution was then purified by reversed phase HPLC (acetonitrile/water/TFA 1%) to afford the title compound as a pale yellow solid (60 mg, 36.6%). Rt(min) 5.275. MS FIA264.8, 266.9 ES+.
4-(5-chloro-2-isopropylaminopyridin-4-yl)-lH-pyrrole-2-carboxylic acid [l-(3- chlorophenyl)-2-hydroxyethyl] amide (1-9): 4-Bromo-2-chloro-5-N- isopropylpyridin-2-amine N-oxide (25 mg, 0.094 mmol, 1.0 equivalent) and 4- (4,4,5, 5-tetramethyl-[l,3,2]dioxaborolan-2-yl)-l-(2,4,6-trimethylbenzensulfonyl)-lH- pyrrole-2-carboxylic acid methyl ester (39 mg, 0.094 mmol, 1.0 equivalent) were dissolved in benzene (5 mL) then aqueous 2M Νa2C03 (1 mL) and Pd(PPh3)4 (115.6 mg, 0.1 mmol, 0.2 equivalent) were added and the resulting suspension was heated at reflux at 80 °C for 16 hours. The reaction mixture was diluted in ethyl acetate, washed with water and dried over anhydrous sodium sulfate to afford 4-(5-chloro-2- isopropylamino-pyridin-4-yl)- 1 -(2,4,6-trimethyl-benzenesulfonyl)- lH-pyrrole-2- carboxylic acid methyl ester N-oxide (R (min) 6.859. MS FIA: 492.0 ES+) which was then treated with a 2 M solution of PC13 in dichloromethane (1 mL) at room temperature. After 10 minutes, the solvent was removed under a stream of nitrogen and the cmde oil was dissolved in methanol (1 mL) and aqueous 1 M ΝaOΗ (1 mL). The resulting mixture was heated at reflux for 16 hours then the cmde solution was acidified using aqueous 1 M ΗC1 and the solvent was removed. The resulting 4-(5- chloro-2-isopropylamino-pyridin-4-yl)-lΗ-pyrrole-2-carboxylic acid (R (min) 3.527. MS FIA: 279.4 ES+; 278.2 Es-) was suspended in DMF (3 mL) together with EDCI (36 mg, 0.19 mmol, 2 equivalents), HOBt (26 mg, 0.19 mmol, 2 equivalents), (S)-3- chlorophenylglycinol HCl salt (59 mg, 0.28 mmol, 3 equivalents) and DIEA (0.12 mL, 0.75 mmol, 8 equivalents). The resulting mixture was stirred at room temperature for 16 hours. The reaction mixture was diluted in ethyl acetate, washed with water and dried over sodium sulfate. After removing the solvent under reduced pressure, the cmde product was purified by reversed phase HPLC (acetonitrile/water/TFA 1%) to afford the title compound as a white solid (4.8 mg, 8.1%).
PATENT
US20150512092015-02-19COMPOUNDS AND COMPOSITIONS AS INHIBITORS OF MEK
US73549392008-04-08Pyrrole inhibitors of ERK protein kinase, synthesis thereof and intermediates thereto
Research scientist Tony Huang works in a laboratory at Vertex Pharmaceuticals Inc. in San Diego
REFERENCES
1 . Kohno M, Pouyssegur J (2006) Targeting the ERK signaling pathway in cancer therapy. Ann Med 38: 200-21 1 .
2. Kuby, J., Immunology, 3rd Ed., W.H. Freeman & Co., New York.
3. Lee DC, Webb ML(2003) Pharmaceutical Analysis. John Wiley & Sons, Inc., New York: 255-257.
4. Peterson ML, Hickey MB, Zaworotko MJ and Almarsson O (2006) Expanding the Scope of Crystal Form Evaluation in Pharmaceutical Science. J Pharm Pharmaceut Sci 9(3):317-326.
5. Pierce Catalog and Handbook, 1994-1995; Pierce Chemical Co., Rockford, III.
6. Remington, The Science and Practice of Pharmacy (21 st Edition, Lippincott Williams and Wilkins, Philadelphia, PA.
7. The United States Pharmacopeia-National Formulary, The United States Pharmacopeial Convention, Rockville, MD.

Gabriel Martinez-Botella
Director, Chemistry at Sage Therapeutics
Experience

Director, Chemistry
Sage Therapeutics
July 2012 – Present (4 years 2 months)

Principal Scientist, Team Leader
AstraZeneca
March 2008 – July 2012 (4 years 5 months)

Sr Scientist
Vertex Pharmaceuticals
2002 – 2008 (6 years)
Education


PIC NOT AVAILABLE
///////////ULIXERTINIB, BVD-523; BVD-ERK, BVD-ERK/HM, BVD-ERK/ST, VRT-0752271, VRT-752271, VX-271, уликсертиниб ,أوليكسيرتينيب ,优立替尼 , PHASE 2, Vertex Pharmaceuticals, BioMed Valley Discoveries, UNII:16ZDH50O1U, 869886-67-9 , Gabriel Martinez-Botella
CC(C)NC1=NC=C(C(=C1)C2=CNC(=C2)C(=O)NC(CO)C3=CC(=CC=C3)Cl)Cl

DR ANTHONY MELVIN CRASTO

ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on
ORGANIC SPECTROSCOPY INTERNATIONAL

