
Home » SPOTLIGHT (Page 5)
Category Archives: SPOTLIGHT
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Drug Spotlight: Zioptan, Tafluprost, Merck,

 |
|---|
isopropyl (5Z)-7-{(1R,2R,3R,5S)-2-[(1E)-3,3-difluoro-4-phenoxybut-1-en-1-yl]-3,5-dihydroxycyclopentyl}hept-5-enoate,
Drug: Zioptan
Generic molecule: tafluprost
Company: Merck
Approval date: Feb. 10, 2012
The scoop: Merck says this is the first (get ready for a mouthful) preservative-free prostaglandin analog ophthalmic solution and is for treating elevated eye pressure in some patients with the most common form of glaucoma. Merck sells the ointment in the U.S. and most of Europe, while it licensed it to Japanese drugmaker Santen in Japan, Germany and northern Europe.
Tafluprost (trade names Taflotan, marketed by Santen Pharmaceutical Co. and Zioptan, by Merck (U.S.)) is a prostaglandin analogue used topically (as eye drops) to control the progression of glaucoma and in the management of ocular hypertension. It reduces http://en.wikipedia.org/wiki/Intraocular_pressure”; rel=”nofollow”>intraocular pressure by increasing the outflow of aqueous fluid from the eyes.[1][2]
Taflotan contains 15 µg/ml Tafluprost. Taflotan sine is a preservative-free, single-dose formulation containing 0.3 ml per dose.[3]

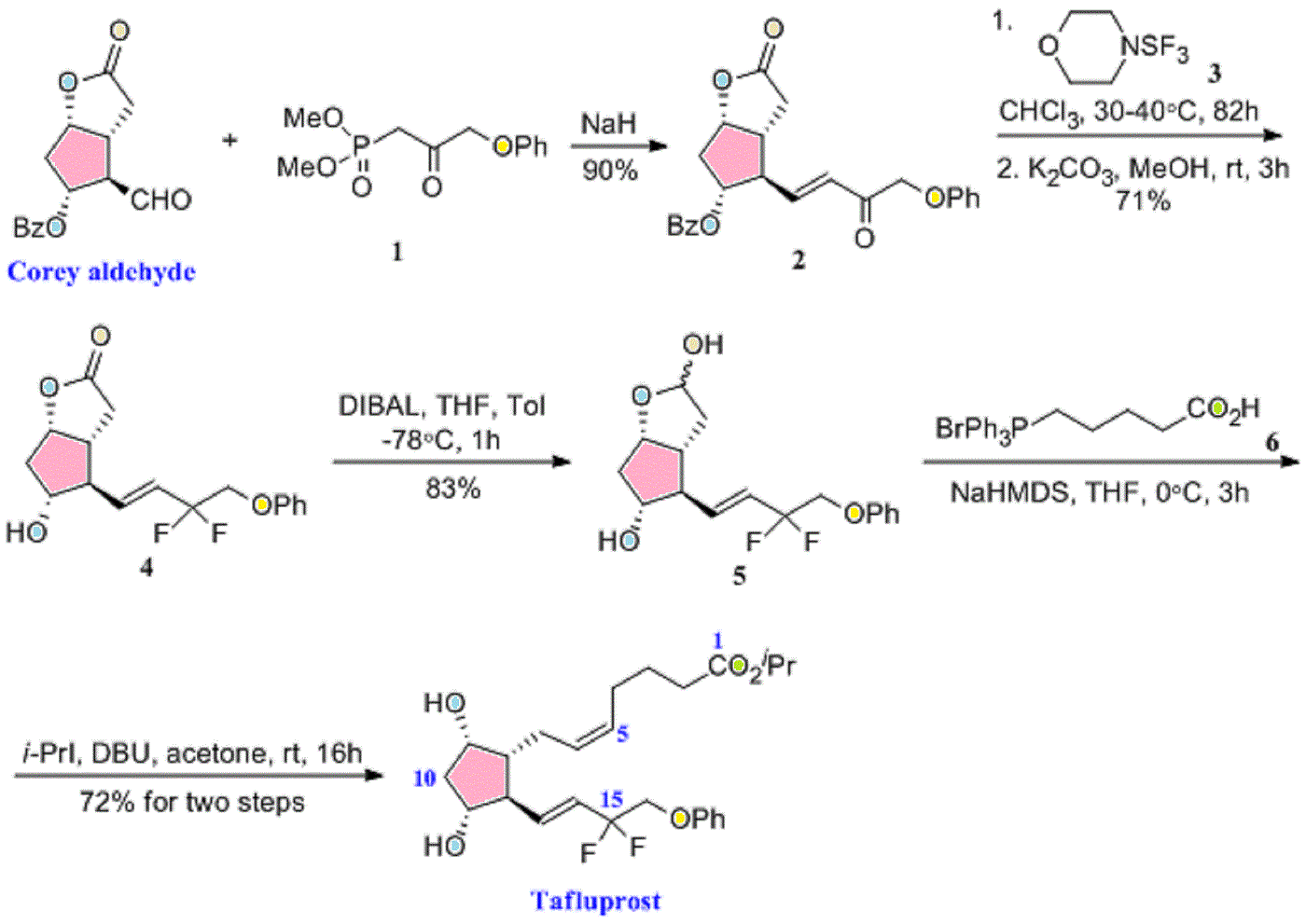
|
|
| Systematic (IUPAC) name | |
|---|---|
| isopropyl (5Z)-7-{(1R,2R,3R,5S)-2-[(1E)-3,3-difluoro-4-phenoxybut-1-en-1-yl]-3,5-dihydroxycyclopentyl}hept-5-enoate | |
| Clinical data | |
| Trade names | Saflutan, Taflotan, Tapros, Zioptan |
| AHFS/Drugs.com | International Drug Names |
| Pregnancy cat. | C (US) |
| Legal status | ℞-only (US) |
| Routes | Topical (eye drops) |
| Identifiers | |
| CAS number | 209860-87-7 |
| ATC code | S01EE05 |
| PubChem | CID 6433101 |
| ChemSpider | 8044182 |
| UNII | 1O6WQ6T7G3 |
| ChEBI | CHEBI:66899 |
| ChEMBL | CHEMBL1963683 |
| Chemical data | |
| Formula | C25H34F2O5 |
| Mol. mass | 452.531266 g/mol |


- Schubert-Zsilavecz, M, Wurglics, M, Neue Arzneimittel 2008/2009
- Santen Home Page
- Gelbe Liste (in German)

European Patent No. 8509621 discloses a process for the preparation of tafluprost. In the first step, (3afl,4fl,5fl,6aS)-4-formyl-2-oxohexahydro-2 — cyclopenta[b]furan-5-ylbenzoate (CTAF 1 (i)) is condensed with dimethyl (2-oxo-3- phenoxypropyl)-phosphonate in the presence of lithium chloride and triethylamine, to provide (3aft,4F?,5F?,6aS)-2-oxo-4-((£)-3-oxo-4-phenoxybut-1 -en-1 -yl)hexahydro-2H- cyclopenta[b]-furan-5-ylbenzoate (CTAF1 ). In the second step, CTAF 1 is reacted with morpholinosulfurtrifluoride to provide (3aH,4H,5H,6aS)-4-((£)-3,3-difluoro-4- phenoxybut-1 -en-1 -yl)-2-oxohexahydro-2 –cyclopenta-[b]furan-5-yl benzoate (CTAF2). CTAF 2 is debenzoylated by potassium carbonate in methanol, to provide (3aH,4H,5H,6aS)-4-((£)-3,3-difluoro-4-phenoxybut-1 -en-1 -yl)-5-hydroxyhexahydro-2H- cyclopenta[b]furan-2-one(CTAF 3), which is further reduced by diisobutyl aluminum hydride (DIBALH) to provide (3af?,4f?,5f?,6aS)-4-((£)-3,3-difluoro-4-phenoxybut-1 -en-1 – yl) hexahydro-2H-cyclopenta[b]furan-2,5-diol (CTAF 4). CTAF 4 is then treated with (4- carboxybutyl)triphenylphosphonium bromide, in the presence of potassium bis(trimethylsilyl)amide in THF, to provide (Z)-7-((1 f?,2f?,3f?,5S)-2-((£)-3,3-difluoro-4- phenoxybut-1 -en-1 -yl)-3,5-dihydroxycyclopentyl)hept-5-enoic acid (“tafluprost free acid,” CTAF5), which is reacted with isopropyl iodide in the presence of DBU to provide (Z)- isopropyl 7-((1 F?,2F?,3F?,5S)-2-((£)-3,3-difluoro-4-phenoxybut-1 -en-1 -yl)-3,5-dihydroxy- cyclopentyl)hept-5-enoate (“tafluprost,” CTAF 6). The reaction sequence is summarized in Scheme 1 .

CTAF 1(i)
CTAF 1 CTAF 2

U.S. Patent Application Publication No. 2010/0105775A1 discloses amino acid salts of prostaglandins. The application also discloses a process for the preparation of prostaglandins, comprising forming an amino acid salt of a prostaglandin and converting the amino acid salt to the prostaglandin.
EXAMPLE 1 : Preparation of CTAF 1

CTAF1(i)
CTAF1
To a stirred suspension of sodium hydride (60% dispersion in mineral oil, 0.217 g, 5.429 mmol) in THF (5 ml_) was added a solution of dimethyl (2-oxo-3- phenoxypropyl)phosphonate(1 .21 g, 4.705 mmol) in THF (2 ml_), over 15 minutes at 0- 5°C under a nitrogen atmosphere. The mixture was warmed to 25-35 , 0.5 M zinc chloride solution in THF (9.4 ml_, 4.705 mmol) was added over 10 minutes, and then the mixture was stirred for 15 minutes at 25-35<€. CTAF1 (i) (3af?,4F?,5F?,6aS)-4-formyl-2- oxohexahydro-2 –cyclopenta[b]furan-5-yl benzoate (1 g) in dichloromethane (10 ml_) was added over 5 minutes at 25-35 °C. The temperature was raised to 35-40 °C and the mixture was stirred for 2hours under a nitrogen atmosphere. The mixture was cooled to 15°C and the reaction was quenched by adding acetic acid (0.2 mL), followed by adding saturated ammonium chloride solution (10 mL), and further stirring for 15 minutes. The organic layer was separated and the aqueous layer was extracted with ethyl acetate (5 mL). The combined organic layers were evaporated under reduced pressure below 50°C. The crude product was purified by column chromatography on silica gel (100-200 mesh) with 30% ethyl acetate in hexane, to afford the title compound (0.9 g, 61 %yield).
EXAMPLE 2: Preparation of CTAF 2

CTAF1 CTAF2
To a stirred solution of CTAF1 (5 g, 0.0123 mol) in dichloromethane (100 mL) was added diethylaminosulfurtrifluoride (13 mL, 0.09841 mol) at 0-5 °C under a nitrogen atmosphere. The temperature was raised to 25-35 °C and maintained for 24 hours under a nitrogen atmosphere at the same temperature. The mass was slowly added into a saturated sodium bicarbonate solution (75 mL) at 0-5 °C. Temperature was raised to 25- 35 °C, the layers were separated, and the aqueous layer was extracted with dichloromethane (2×25 mL). The combined organic layer was washed with water (25mL) and dried over sodium sulfate (5 g). The organic layer was evaporated to dryness under reduced pressure below 40 °C. The crude product was purified by column chromatography on silica gel (100-200 mesh) with 30% ethyl acetate in hexane, to afford the title compound (4.2 g, 79% yield). EXAMPLE 3: Preparation of CTAF 4

CTAF 2 CTAF 4
CTAF 2 (2.30 g, 5.37 mmol) was dissolved in toluene (25 mL) and the solution was cooled to -65 °C under nitrogen. Diisobutyl aluminum hydride (1 .5 M in toluene, 1 1 .8 mL, 17.7mmol) was added over 15 minutes at -61 to -65 . The mixture was stirred for 3hours and then the reaction was quenched by adding methanol (1 .5 mL). Sulfuric acid (1 M, 25 mL) was added and the temperature rose to -20°C during the addition. Methyl t-butyl ether (MTBE) (10 mL) was added and the mixture was allowed to warm to room temperature. The organic phase was separated and the aqueous phase was extracted with MTBE (2x 10 mL). The combined organic phase was washed with water (10 mL), saturated aqueous sodium bicarbonate (10 mL), and then brine (10 mL). The washes were back-extracted with MTBE (10 mL). The combined organic phases were dried with magnesium sulfate, filtered, and evaporated to give a colourless oil (2.20 g). The crude product was chromatographed on silica (60 g), eluting with a mixture of ethyl acetate and heptane (2:1 by volume), and then with ethyl acetate, to give CTAF 4 as a colourless oil (1 .71 g, 97% yield).
EXAMPLE 4: Preparation of CTAF 2

CTAF1 CTAF2
To a stirred solution of CTAF1 (20 g, 0.0492 mol) in dichloromethane(400 mL) was added diethylaminosulfurtrifluoride (52 mL, 0.393 mol) at 0-10°C under a nitrogen atmosphere. The temperature was raised to 25-35 and maintained for 96hours under a nitrogen atmosphere at that temperature. The mass was slowly added to a saturated NaHCOs solution (600 mL) at 0-10°C. The mixture was heated to 25-35 <€ and filtered through aCelite bed. The layers were separated and the aqueous layer was extracted with DCM (2×100 mL). The combined organic layer was washed with 10% NaCI solution (100 mL) and evaporated to dryness under reduced pressure below 40°C. The residue was purified by column chromatography on silica gel (100-200 mesh) with 30% ethyl acetate in hexane.
Column purified material was dissolved in MTBE (80 mL) at 40°C and stirred for 30 minutes at that temperature. Diisopropyl ether (160 mL) was added at 35-40 and stirring continued for 30 minutes at 35-40 . Cooled the mass to 5-15°C and stirred for 30 minutes at that temperature. The solid was filtered, washed with a mixture of MTBE and diisopropyl ether (DIPE) (1 :2 by volume, 60 mL), and dried at 40°C under vacuum, to afford pure CTAF2 (12.0 g, 57% yield).
EXAMPLE 5: Preparation of CTAF 5

(4-Carboxybutyl)triphenylphosphonium bromide (10.32 g, 23.3 mmol, 4 eq) was suspended in THF (20 mL) under a nitrogen atmosphere and cooled to 5°C. NaHMDS solution (1 M in THF, 46.6 mL, 46.6 mmol, 8 eq) was added over 10 minutes. The red/orange mixture was stirred for 30 minutes. A solution of CTAF 4 (1 .90 g, 5.82 mmol) in THF (10 mL) was added over 30 minutes at 0-3 . The mixture was stirred for 1 .5hours and then the reaction was quenched by adding water (30 mL) and the masswas warmed to room temperature. The aqueous phase was separated and the organic phase was washed with water (20 mL). The combined aqueous phases were washed with MTBE (30 mL). The organic phases up to this point were discarded. The aqueous phase was acidified with 2M hydrochloric acid (14 mL, to pH 3-4) and extracted with ethyl acetate (2×30 mL). The combined ethyl acetate layers were washed with brine (20 mL), dried with magnesium sulfate, filtered, and evaporated under reduced pressure to give CTAF 5 asa yellow oil (8.60 g).
A 2.96 g sample was removed and the remainder (5.64 g) was chromatographed on silica (30 g) eluting with ethyl acetate to give purified CTAF 5 (1 .41 g) asa yellow oil. NMR analysis showed approximately 90% purity, remainder triphenyl phosphine oxide.
EXAMPLE 6: Preparation of CTAF 5 DCHA salt

CTAF 5 CTAF 5 DCHA sa t
CTAF5 (1 1 .72 g, 90% purity, 25.7 mmol, containing 1 .4% trans isomer) was dissolved in acetone (60 mL). Dicyclohexylamine (4.66 g, 25.7 mmol) was added and the mixture was stirred at room temperature overnight. The solid was filtered and washed with acetone (6 mL), then dried to give the DCHA salt (12.93 g, 85% yield, 0.29% trans-isomer).
A sample (7.03 g) was further purified by recrystallisation. It was dissolved in hot acetone (30 mL) and cooled to room temperature with stirring. The mixture was stirred for 3 hours, filtered and the solid was washed with acetone (3 mL) and dried to give a white solid (6.41 g, 91 % recovery, 0.1 1 % trans-isomer).
A PXRD pattern of the product is shown as Fig. 1 , obtained using copper Ka radiation. In the drawing, the y-axis is intensity units and the x-axis is the 2-theta angle, in degrees. EXAMPLE 7: Pre aration of CTAF 6

CTAF 5 DCH A sa l ^ I AI- O
CTAF 5 DCHA salt (5.80 g, 9.80 mmol) was suspended in ethyl acetate (20 mL). Sulfuric acid (1 M, 20 mL) was added and the mixture was stirred until a clear solution was obtained. The organic phase was separated and the aqueous phase was extracted with ethyl acetate (2×20 mL). The combined organic layers were washed with water (15 mL) and brine (15 mL), dried with magnesium sulfate, filtered, and evaporated. The residue was dissolved in acetone (40 mL) and charged into a jacketed vessel at 30°C. 1 ,8-Diazabicyclo[5.4.0]undec-7-ene (DBU) (8.95 g, 58.8 mmol) was added, then 2- iodopropane (10.0 g, 58.8 mmol) was added, and the mixture was stirred for 20hours. The mixture was concentrated under reduced pressure and the residue was partitioned between ethyl acetate (30 mL) and aqueous potassium dihydrogen orthophosphate (8 g) in water (50 mL). The organic phase was separated and the aqueous was extracted with ethyl acetate (30 mL). The combined organic phases were washed with brine (20 mL), dried with magnesium sulfate, filtered and evaporated to give a yellow oil (4.83 g). The crude product was chromatographed on silica (130 g), eluting with a mixture of ethyl acetate and heptane (2:1 by volume), to give CTAF 6 (3.98 g, 90% yield) as a colorless oil.
DRUG SPOTLIGHT-Afinitor (everolimus) , Novartis:

Afinitor (everolimus)
40-O-(2-hydroxyethyl)-rapamycin
Company: Novartis
Approval Status: Approved July 2012
Treatment Area: hormone receptor-positive, HER2-negative breast cancer
Everolimus is a derivative of Rapamycin (sirolimus), and works similarly to Rapamycin as an mTOR (mammalian target of rapamycin) inhibitor. It is currently used as an immunosuppressant to prevent rejection of organ transplants. In a similar fashion to other mTOR inhibitors Everolimus’ effect is solely on the mTORC1 protein and not on the mTORC2 protein.
159351-69-6 CAS NO
BRANDS
| Afinitor | Novartis |
| Certican | Novartis |
| VOTUBIA | Novartis |
| Zortress | Novartis |
Afinitor (everolimus), an inhibitor of mTOR (mammalian target of rapamycin), is an antineoplastic agent.
Afinitor is specifically approved for the treatment of postmenopausal women with advanced hormone receptor-positive, HER2-negative breast cancer (advanced HR+ BC) in combination with exemestane, after failure of treatment with letrozole or anastrozole.
Afinitor is supplied as a tablet for oral administration. The recommended dose of Afinitor for breast cancer is 10 mg, to be taken once daily, at the same time every day, either consistently with food or consistently without food.
FDA Approval
The FDA approval of Afinitor for the treatment of advanced hormone receptor-positive, HER2-negative breast cancer was based on a randomized, double-blind, multicenter study in 724 postmenopausal women with estrogen receptor-positive, HER 2/neu-negative advanced breast cancer with recurrence or progression following prior therapy with letrozole or anastrozole.
Everolimus is indicated for the treatment of postmenopausal women with advanced hormone receptor-positive, HER2-negative breast cancer (advanced HR+ BC) in combination with exemestane, after failure of treatment with letrozole or anastrozole. Indicated for the treatment of adult patients with progressive neuroendocrine tumors of pancreatic origin (PNET) with unresectable, locally advanced or metastatic disease. Indicated for the treatment of adult patients with advanced renal cell carcinoma (RCC) after failure of treatment with sunitinib or sorafenib. Indicated for the treatment of adult patients with renal angiomyolipoma and tuberous sclerosis complex (TSC), not requiring immediate surgery. Indicated in pediatric and adult patients with tuberous sclerosis complex (TSC) for the treatment of subependymal giant cell astrocytoma (SEGA) that requires therapeutic intervention but cannot be curatively resected.
Everolimus (RAD-001) is the 40-O-(2-hydroxyethyl) derivative of sirolimus and works similarly to sirolimus as an inhibitor of mammalian target of rapamycin (mTOR).
It is currently used as an immunosuppressant to prevent rejection of organ transplants and treatment of renal cell cancer and other tumours. Much research has also been conducted on everolimus and other mTOR inhibitors for use in a number of cancers.
It is marketed by Novartis under the tradenames Zortress (USA) and Certican (Europe and other countries) in transplantation medicine, and Afinitor in oncology.
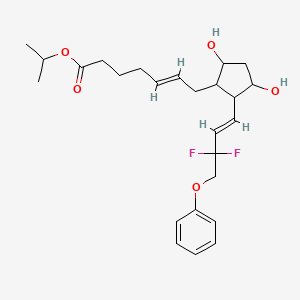
AFINITOR (everolimus), an inhibitor of mTOR, is an antineoplastic agent.
The chemical name of everolimus is (1R,9S,12S,15R,16E,18R,19R,21R,23S,24E,26E,28E,30S,32S,35R)-1,18- dihydroxy-12-{(1R)-2-[(1S,3R,4R)-4-(2-hydroxyethoxy)-3-methoxycyclohexyl]-1-methylethyl}-19,30-dimethoxy15,17,21,23,29,35-hexamethyl-11,36-dioxa-4-aza-tricyclo[30.3.1.04,9]hexatriaconta-16,24,26,28-tetraene-2,3,10,14,20pentaone.
The molecular formula is C53H83NO14 and the molecular weight is 958.2. The structural formula is:
 |
AFINITOR Tablets are supplied for oral administration and contain 2.5 mg, 5 mg, 7.5 mg, or 10 mg of everolimus. The tablets also contain anhydrous lactose, butylated hydroxytoluene, crospovidone, hypromellose, lactose monohydrate, and magnesium stearate as inactive ingredients.
AFINITOR DISPERZ (everolimus tablets for oral suspension) is supplied for oral administration and contains 2 mg, 3 mg, or 5 mg of everolimus. The tablets for oral suspension also contain butylated hydroxytoluene, colloidal silicon dioxide, crospovidone, hypromellose, lactose monohydrate, magnesium stearate, mannitol, and microcrystalline cellulose as inactive ingredients.
- R.N Formica Jra, K.M Lorberb, A.L Friedmanb, M.J Biaa, F Lakkisa, J.D Smitha, M.I Lorber (March 2004). “The evolving experience using everolimus in clinical transplantation”. Elsevier 36 (2): S495–S499.
- “Afinitor approved in US as first treatment for patients with advanced kidney cancer after failure of either sunitinib or sorafenib” (Press release). Novartis. 2009-03-30. Retrieved April 6, 2009.
- “Novartis receives US FDA approval for Zortress (everolimus) to prevent organ rejection in adult kidney transplant recipients” (Press release). Novartis. 2010-04-22. Retrieved April 26, 2010.
- “Novartis’ Afinitor Cleared by FDA for Treating SEGA Tumors in Tuberous Sclerosis”. 1 Nov 2010.
- http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm254350.htm
- “US FDA approves Novartis drug Afinitor for breast cancer”. 20 Jul 2012.
PATENTS
|
Country
|
Patent Number
|
Approved
|
Expires (estimated)
|
|---|---|---|---|
| United States | 6440990 | 1993-09-24 | 2013-09-24 |
| Canada | 2145383 | 2004-11-16 | 2013-09-24 |
| Canada | 2225960 | 2004-05-11 | 2016-07-12 |
| United States | 7297703 | 1999-12-06 | 2019-12-06 |
|
10-28-2011
|
METHODS OF TREATMENT
|
|
|
1-21-2011
|
ANTI-IGF1R
|
|
|
1-14-2011
|
HISTONE H2AX (HH2AX) BIOMARKER FOR FTI SENSITIVITY
|
|
|
3-24-2010
|
Thermal treatment of a drug eluting implantable medical device
|
|
|
1-13-2010
|
Therapeutic phosphonate compounds
|
|
|
10-21-2009
|
Processes for preparing water-soluble polyethylene glycol conjugates of macrolide immunosuppressants
|
|
|
10-16-2009
|
Heparin Prodrugs and Drug Delivery Stents Formed Therefrom
|
|
|
9-11-2009
|
PHOSPHONATE COMPOUNDS HAVING IMMUNO-MODULATORY ACTIVITY
|
|
|
12-31-2008
|
Phosphonate compounds having immuno-modulatory activity
|
|
|
10-8-2008
|
Anti-inflammatory phosphonate compounds
|
|
6-27-2008
|
Genes Involved in Neurodegenerative Conditions
|
|
|
10-24-2007
|
Fluid treatment of a polymeric coating on an implantable medical device
|
|
|
7-11-2007
|
Oxepane isomer of 42-O-(2-hydroxy)ethyl-rapamycin
|
|
|
2-9-2007
|
40-O-(2-hydroxy)ethyl-rapamycin coated stent
|
|
|
1-5-2007
|
Methods for treating neurofibromatosis 1
|
|
|
9-8-2006
|
Anti-inflammatory phosphonate compounds
|
| WO1994009010A1 | Sep 24, 1993 | Apr 28, 1994 | Sandoz Ag | O-alkylated rapamycin derivatives and their use, particularly as immunosuppressants |
| WO2007135397A1 * | May 18, 2007 | Nov 29, 2007 | Christoph Beckmann | 36 -des (3 -methoxy-4 -hydroxycyclohexyl) 36 – (3 -hydroxycycloheptyl) derivatives of rapamycin for the treatment of cancer and other disorders |
| EP0663916A1 | Sep 24, 1993 | Jul 26, 1995 | Novartis AG | O-alkylated rapamycin derivatives and their use, particularly as immunosuppressants |
| US5665772 | Sep 24, 1993 | Sep 9, 1997 | Sandoz Ltd. | O-alkylated rapamycin derivatives and their use, particularly as immunosuppressants |
| US20030125800 | Apr 24, 2002 | Jul 3, 2003 | Shulze John E. | Drug-delivery endovascular stent and method for treating restenosis |
…………………………………….
Rapamycin is a known macrolide antibiotic produced by Streptomvces hvgroscopicus. having the structure depicted in Formula A:

See, e.g., McAlpine, J.B., et al., J. Antibiotics (1991) 44: 688; Schreiber, S.L., et al., J. Am. Chem. Soc. (1991) J_13: 7433‘- US Patent No. 3 929 992. Rapamycin is an extremely potent immunosuppressant and has also been shown to have antitumor and antifungal activity. Its utility as a pharmaceutical, however, is restricted by its very low and variable bioavailabiiity as well as its high toxicity. Moreover, rapamycin is highly insoluble, making it difficult to formulate stable galenic compositions.
Everolimus, 40-O-(2-hydroxyethyl)-rapamycin of formula (1) is a synthetic derivative of rapamycin (sirolimus) of formula (2), which is produced by a certain bacteria strain and is also pharmaceutically active.

(1) (2)
Everolimus is marketed under the brand name Certican for the prevention of rejection episodes following heart and kidney transplantation, and under the brand name Afinitor for treatment of advanced kidney cancer.
Due to its complicated macrolide chemical structure, everolimus is, similarly as the parent rapamycin, an extremely unstable compound. It is sensitive, in particular, towards oxidation, including aerial oxidation. It is also unstable at temperatures higher than 25°C and at alkaline pH.
Everolimus and a process of making it have been disclosed in WO 94/09010
Synthesis

Alkylation of rapamycin (I) with 2-(tert-butyldimethylsilyloxy)ethyl triflate (II) by means of 2,6-lutidine in hot toluene gives the silylated target compound (III), which is deprotected by means of 1N HCl in methanol (1). (Scheme 21042401a) Manufacturer Novartis AG (CH). References 1. Cottens, S., Sedrani, R. (Sandoz-Refindungen VmbH; Sandoz-Patent GmbH; Sandoz Ltd.). O-Alkylated rapamycin derivatives and their use, particularly as immunosuppressants. EP 663916, EP 867438, JP 96502266, US 5665772, WO 9409010.EP 0663916; EP 0867438; JP 1996502266; JP 1999240884; US 5665772; WO 9409010
…………..
SYNTHESIS
https://www.google.com/patents/WO2012103960A1
(US 5,665,772, EP 663916). The process principle is shown in the scheme below, wherein the abbreviation RAP-OH has been used as an abbreviation for the rapamycin structure of formula (2) above, L is a leaving group and P is a trisubstituted silyl group serving as a OH- protective group.
RAP-OH + L-CH2-CH2-0-P — –> RAP-O-CH2-CH2-O-P — – > RAP-O-CH2-CH2-OH
(2) (4) (1)
Specifically, the L- group is a trifluoromethanesulfonate (triflate) group and the protective group P- is typically a tert-butyldimethylsilyloxy- group. Accordingly, the known useful reagent within the above general formula (3) for making everolimus from rapamycin is 2-(tert-butyldimethylsilyloxy)ethyl triflate of formula (3 A):

According to a known synthetic procedure disclosed in Example 8 of WO 94/09010 and in Example 1 of US application 2003/0125800, rapamycin (2) reacts in hot toluene and in the presence of 2,6-lutidine with a molar excess of the compound (3 A), which is charged in several portions, to form the t-butyldimethylsilyl-protected everolimus (4A). This compound is isolated and deprotected by means of IN aqueous HC1 in methanol. Crude everolimus is then purified by column chromatography. Yields were not reported.

(2) (3A) (4A) (1)
In an article of Moenius et al. (J. Labelled Cpd. Radiopharm. 43, 113-120 (2000)), which used the above process for making C14-labelled and tritiated everolimus, a diphenyl- tert.butylsilyloxy -protective group was used as the alkylation agent of formula (3B).

Only 8% yield of the corresponding compound (4B)

and 21% yield of the compound (1) have been reported.
Little is known about the compounds of the general formula (3) and methods of their preparation. The synthesis of the compound (3 A) was disclosed in Example 1 of US application 2003/0125800. It should be noted that specification of the reaction solvent in the key step B of this synthesis was omitted in the disclosure; however, the data about isolation of the product allow for estimation that such solvent is dichloromethane. Similarly also a second article of Moenius et al. (J. Labelled Cpd. Radiopharm.42, 29-41 (1999)) teaches that dichloromethane is the solvent in the reaction.
It appears that the compounds of formula (3) are very reactive, and thus also very unstable compounds. This is reflected by the fact that the yields of the reaction with rapamycine are very low and the compound (3) is charged in high molar extent. Methods how to monitor the reactivity and/or improve the stability of compounds of general formula (3), however, do not exist.
Thus, it would be useful to improve both processes of making compounds of formula (3) and, as well, processes of their application in chemical synthesis.
xample 6: 40-O-[2-((2,3-dimethylbut-2-yl)dimethylsilyloxy)ethyl]rapamycin
In a 100 mL flask, Rapamycin (6 g, 6.56 mmol) was dissolved in dimethoxyethane (4.2 ml) and toluene (24 ml) to give a white suspension and the temperature was raised to 70°C. After 20 min, N,N-diisopropylethylamine (4.56 ml, 27.6 mmol) and 2-((2,3-dimethylbutan-2- yl)dimethylsilyloxy)ethyl trifluoromethanesulfonate (8.83 g, 26.3 mmol) were added in 2 portions with a 2 hr interval at 70°C. The mixture was stirred overnight at room temperature, then diluted with EtOAc (40 ml) and washed with sat. NaHC03 (30 ml) and brine (30 ml). The organic layer was dried with Na2S04, filtered and concentrated. The cmde product was chromatographed on a silica gel column (EtOAc/heptane 1/1 ; yield 4.47 g).
Example 7: 40-O-(2-hydroxyethyl)-rapamycin [everolimus]
In a 100 mL flask, 40-O-[2-((2,3-dimethylbut-2-yl)dimethylsilyloxy)ethyl]rapamycin (4.47 g, 4.06 mmol) was dissolved in methanol (20 ml) to give a colorless solution. At 0°C, IN aqueous hydrochloric acid (2.0 ml, 2.0 mmol) was added and the mixture was stirred for 90 min. The reaction was followed by TLC (ethyl acetate/n-heptane 3 :2) and HPLC. Then 20 ml of saturated aqueous NaHC03 were added, followed by 20 ml of brine and 80 ml of ethyl acetate. The phases were separated and the organic layer was washed with saturated aqueous NaCl until pH 6/7. The organic layer was dried by Na2S04, filtered and concentrated to yield 3.3 g of the product.
……………………….
SYNTHESIS
https://www.google.co.in/patents/WO1994009010A1
Example 8: 40-O-(2-Hydroxy)ethyl-rapamycin
a) 40-O-[2-(t-Butyldimethylsilyl)oxy]ethyl-rapamycin
A solution of 9.14 g (10 mmol) of rapamycin and 4.70 mL (40 mmol) of 2,6-lutidine in 30 mL of toluene is warmed to 60°C and a solution of 6.17 g (20 mmol) of 2-(t-butyldimethylsilyl)oxyethyl triflate and 2.35 mL (20 mmol) of 2,6-lutidine in 20 mL of toluene is added. This mixture is stirred for 1.5h. Then two batches of a solution of 3.08 g (10 mmol) of triflate and 1.2 mL (10 mmol) of 2,6-lutidine in 10 mL of toluene are added in a 1.5h interval. After addition of the last batch, stirring is continued at 60°C for 2h and the resulting brown suspension is filtered. The filtrate is diluted with ethyl acetate and washed with aq. sodium bicarbonate and brine. The organic solution is dried over anhydrous sodium sulfate, filtered and concentrated. The residue is purified by column chromatography on silica gel (40:60 hexane-ethyl acetate) to afford 40-O-[2-(t-butyldimethylsilyl)oxy]ethyl-rapamycin as a white solid: 1H NMR (CDCl3) δ 0.06 (6H, s), 0.72 (1H, dd), 0.90 (9H, s), 1.65 (3H, s), 1.75 (3H, s), 3.02 (1H, m), 3.63 (3H, m), 3.72 (3H, m); MS (FAB) m/z 1094 ([M+Na]+), 1022 ([M-(OCH3+H2O)]+).
b) 40-O-(2-Hydroxy)ethyl-rapamycin
To a stirred, cooled (0°C) solution of 4.5 g (4.2 mmol) of 40-O-[2-(t-butyldimethylsilyl)oxy]ethyl-rapamycin in 20 mL of methanol is added 2 mL of IN HCl. This solution is stirred for 2h and neutralized with aq. sodium bicarbonate. The mixture is extracted with three portions of ethyl acetate. The organic solution is washed with aq.
sodium bicarbonate and brine, dried over anhydrous sodium sulfate, filtered and
concentrated. Purification by column chromatography on silica gel (ethyl acetate) gave the title compound as a white solid:1H NMR (CDCl3) δ 0.72 (1H, dd), 1.65 (3H, s), 1.75 (3H, s), 3.13 (5H, s and m), 3.52-3.91 (8H, m); MS (FAB) m/z 980 ([M+Na]+), 926 ([M-OCH3]+), 908 ([M-(OCH3+H2O)]+), 890 ([M-(OCH3+2H2O)]+), 876 ([M-(2CH3OH+OH)]+), 858 ([M-(OCH3+CH3OH+2H2O)]+).
MBA (rel. IC50) 2.2
IL-6 dep. prol. (rel. IC50) 2.8
MLR (rel. IC50) 3.4
…………………..
synthesis
Everolimus (Everolimus) was synthesized by the Sirolimus (sirolimus, also known as rapamycin Rapamycin) ether from. Sirolimus is from the soil bacterium Streptomyces hygroscopicus isolated metabolites. Activation end sirolimus (triflate, Tf) the other end of the protection (t-butyldimethylsilyl, TBS) of ethylene glycol 1 reaction of 2 , because the hydroxyl group 42 hydroxyl site over the 31-bit resistance is small, so the reaction only occurs in 42. Compound 2under acidic conditions TBS protection is removed everolimus.
DRUG SPOTLIGHT –CUBICIN, DAPTOMYCIN
Daptomycin

N-decanoyl-L-tryptophyl-L-asparaginyl-L-aspartyl-L-threonylglycyl-
L-ornithyl-L-aspartyl-D-alanyl-L-aspartylglycyl-D-seryl-threo-3-methyl-L-glutamyl-3-anthraniloyl-L-alanine[egr]1-lactone
Daptomycin is a lipopeptide antibiotic used in the treatment of systemic and life-threatening infections caused by Gram-positive organisms. It is a naturally occurring compound found in the soil saprotroph Streptomyces roseosporus. Its distinct mechanism of action makes it useful in treating infections caused by multi-resistant bacteria. It is marketed in the United States under the trade name Cubicin by Cubist Pharmaceuticals.

The compound LY 146032 was discovered by researchers at Eli Lilly and Company in the late 1980s.LY 146032 showed promise in Phase I/II clinical trials for treatment of infection caused by Gram-positive organisms. Lilly ceased development because high-dose therapy was associated with adverse effects on skeletal muscle, including myalgia and potential myositis.
The rights to LY 146032 were acquired by Cubist Pharmaceuticals in 1997, which following U.S. Food and Drug Administration (FDA) approval in September 2003 for use in people older than 18 years began marketing the drug under the trade name CUBICIN. Cubicin is marketed in the EU and in several other countries by Novartis following its purchase of Chiron Corporation, previous licensee.

Daptomycin has a distinct mechanism of action, disrupting multiple aspects of bacterial cell membrane function. It appears to bind to the membrane and cause rapid depolarization, resulting in a loss of membrane potential leading to inhibition of protein, DNA and RNA synthesis, which results in bacterial cell death
DRUG SPOTLIGHT- EXENATIDE, BYETTA

EXENATIDE

ACETATE CAS NO 141732-76-5
Exenatide, derived from a compound found in the saliva of the Gila monster, a large lizard native to the southwestern US, is a functional analog of Glucagon-Like Peptide-1 (GLP-1), a naturally occuring peptide.
Exenatide (INN, marketed as Byetta, Bydureon) is a glucagon-like peptide-1 agonist(GLP-1 agonist) medication approved in April 2005 for the treatment of diabetes mellitus type 2. It belongs to the group of incretin mimetics and is manufactured by Amylin Pharmaceuticals. Exenatide in its Byetta form is administered as a subcutaneous injection (under the skin) of the abdomen, thigh, or arm, any time within the 60 minutes before the first and last meal of the day. A once-weekly injection has been approved as of January 27, 2012 under the trademark Bydureon

Exenatide is a synthetic version of exendin-4, a hormone found in the saliva of the Gila monster that was first isolated by Dr. John Eng in 1992 while working at the Veterans Administration Medical Center in the Bronx, New York. It displays biological properties similar to human glucagon-like peptide-1 (GLP-1), a regulator of glucose metabolism andinsulin secretion. According to the package insert, exenatide enhances glucose-dependent insulin secretion by the pancreatic beta-cell, suppresses inappropriately elevated glucagon secretion, and slows gastric emptying, although the mechanism of action is still under study.

Exenatide is a 39-amino-acid peptide, an insulin secretagogue, with glucoregulatory effects. Exenatide was approved by the FDA on April 28, 2005 for patients whose diabetes was not well-controlled on other oral medication. The medication is injected subcutaneously twice per day using a filled pen-like device. The abdomen is a common injection site, after the area is cleaned with an alcohol pad. A new pen must first be tested to see if the medicine is flowing.

GILA MONSTER
| Indication | Indicated as adjunctive therapy to improve glycemic control in patients with Type 2 diabetes mellitus who are taking metformin, a sulfonylurea, or a combination of both, but have not achieved adequate glycemic control. |
| Pharmacodynamics | Exenatide is an incretin mimetic, which has glucoregulatory effects. While it is has blood-sugar lowering actions alone, it can also be combined with other medications such as pioglitazone, metformin, sulfonylureas, and/or insulin to improve glucose control. The approved use of exenatide is with either sulfonylureas, metformin and thiazolinediones. The medication is injected twice per day using a pre-filled pen device. Typical human responses to exenatide plus eating include improvements in the initial rapid release of endogenous insulin, suppression of glucagon release by the pancreas, regulation of gastric empyting and reduced appetite; all behaviors more typical of individuals without blood sugar control problems. Exenatide is self-regulating in that in lowers blood sugar when levels are elevated but does not continue to lower blood sugar when levels return to normal, unlike with sulfonylureas or insulins. |
| Mechanism of action | Exenatide is a functional analog of the human incretin Glucagon-Like Peptide-1 (GLP-1). Incretins enhance glucose-dependent insulin secretion and exhibit other antihyperglycemic actions following their release into the circulation from the gut. The GLP-1 system increases insulin secretion only in the presence of elevated plasma glucose levels, avoiding inappropriately high insulin levels during fasting. The drug also moderates peak serum glucagon levels during hyperglycemic periods following meals, but does not interfere with glucagon release in response to hypoglycemia. Secondary effects of drug administration reduces the rate of gastric emptying and decreases food intake, mitigating the potential severity of hyperglycemic events after meal |
| Following subcutaneous administration to patients with type 2 diabetes, exenatide reaches median peak plasma concentrations in 2.1 hours. |
DRUG SPOTLIGHT-Cobicistat

Cobicistat
Thiazol-5-ylmethyl N-[1-benzyl-4-[[2-[[(2-isopropylthiazol-4-yl)methyl-methyl-carbamoyl]amino]-4-morpholino-butanoyl]amino]-5-phenyl-pentyl]carbamate
1004316-88-4 CAS NO
- Cobicistat, formerly known as GS-9350, is a pharmacokinetic enhancer (a drug used to boost other medications in the blood to make them more effective) by Gilead Sciences. It is a component of the approved fixed-dose combination tablet Stribild.
- Cobicistat is not active against HIV. It works by inhibiting an enzyme called CYP3A4 that is responsible for breaking down (or metabolism) of certain medications, including several HIV drugs. This helps boost the effectiveness of these drugs, while allowing fewer pills or doses on a daily basis.
Cobicistat is Gilead’s proprietary potent mechanism-based inhibitor of cytochrome P450 3A (CYP3A), an enzyme that metabolizes drugs in the body. Unlike ritonavir, cobicistat acts only as a pharmacoenhancer and has no antiviral activity. Pharmacokinetic studies have demonstrated that cobicistat boosts the widely prescribed protease inhibitors atazanavir and darunavir.
Cobicistat is a licensed drug for use in the treatment of infection with the human immunodeficiency virus (HIV).
Like ritonavir (Norvir), cobicistat is of interest not for its anti-HIV properties, but rather its ability to inhibit liver enzymes that metabolize other medications used to treat HIV, notablyelvitegravir, an HIV integrase inhibitor currently under investigation itself. By combining cobicistat with elvitegravir, higher concentrations of elvitgravir are achieved in the body with lower dosing, theoretically enhancing elvitgravir’s viral suppression while diminishing its adverse side-effects. In contrast with ritonavir, the only currently approved booster, cobicistat has no anti-HIV activity of its own.[1]
Cobicistat is a component of the four-drug, fixed-dose combination HIV treatmentelvitegravir/cobicistat/emtricitabine/tenofovir (known as the “Quad Pill” or Stribild).[1][2] The Quad Pill/Stribild was approved by the FDA in August 2012 for use in the United States and is owned by Gilead Sciences.
Cobicistat is a potent inhibitor of cytochrome P450 3A enzymes, including the importantCYP3A4 subtype. It also inhibits intestinal transport proteins, increasing the overall absorption of several HIV medications, including atazanavir, darunavir and tenofovir alafenamide fumarate.[3]
- Highleyman, L. Elvitegravir “Quad” Single-tablet Regimen Shows Continued HIV Suppression at 48 Weeks. HIV and Hepatitis.com
- R Elion, J Gathe, B Rashbaum, and others. The Single-Tablet Regimen of Elvitegravir/Cobicistat/Emtricitabine/Tenofovir Disoproxil Fumarate (EVG/COBI/FTC/TDF; Quad) Maintains a High Rate of Virologic Suppression, and Cobicistat (COBI) is an Effective Pharmacoenhancer Through 48 Weeks. 50th Interscience Conference on Antimicrobial Agents and Chemotherapy (ICAAC 2010). Boston, September 12–15, 2010.
- Lepist, E. -I.; Phan, T. K.; Roy, A.; Tong, L.; MacLennan, K.; Murray, B.; Ray, A. S. (2012). “Cobicistat Boosts the Intestinal Absorption of Transport Substrates, Including HIV Protease Inhibitors and GS-7340, in Vitro”. Antimicrobial Agents and Chemotherapy 56 (10): 5409–5413. doi:10.1128/AAC.01089-12. PMC 3457391. PMID 22850510.
SPOTLIGHT-Linaclotide, Linzess, Ironwood Pharmaceuticals,

Drug: Linzess
Generic molecule: linaclotide
Company: Ironwood Pharmaceuticals
Approval date: Aug. 30,2012
851199-59-2 CAS NO
L-Cysteinyl-L-cysteinyl-L-glutamyl-L-tyrosyl-L-cysteinyl-L-cysteinyl-L-asparaginyl-L-prolyl-L-alanyl-L-cysteinyl-L-threonylglycyl-L-cysteinyl-L-tyrosine cyclo(1-6),(2-10),(5-13)-tris(disulfide)
Linaclotide is a peptide consisting of 14 amino acids. The sequence is
H–Cys1–Cys2–Glu3–Tyr4–Cys5–Cys6–Asn7–Pro8–Ala9–Cys10–Thr11–Gly12–Cys13–Tyr14–OH
There are three disulfide bonds: Between Cys1 and Cys6, between Cys2 and Cys10, and between Cys5 and Cys13.[8]
Linaclotide (marketed under the trade name Linzess) is an experimentalpeptide agonist of guanylate cyclase 2C that is undergoing clinical trials for use in treating abdominal pain in patients with irritable bowel syndrome (IBS) accompanied by constipation. The drug also looks promising in the treatment of gastroparesis, chronic intestinal pseudo-obstruction (CIPO), andinertia coli as well.[1] The drug was developed by Ironwood Pharmaceuticals, based in Cambridge, Massachusetts.
Linaclotide was approved by the FDA on August 30, 2012 for the treatment of chronic idiopathic constipation and to treat irritable bowel syndrome with constipation (IBS-C) in adults.[2] It became available in US pharmacies on December 17, 2012. [3] That same month, it was forecast by market research firm Decision Resources to achieve blockbuster status by 2021.[4]
The National Institutes of Health (NIH) estimates that as many as 20% of Americans may experience signs of irritable bowel syndrome, with approximately one-third of those affected experiencing constipation often accompanied by abdominal pain, affecting as many as 10 million Americans.Laxatives can assist with constipation but don’t treat pain, while use ofopiates to treat pain can aggravate constipation. While low-cost laxatives and pain killers would likely be tried first, linaclotide targets both associated conditions in a once-daily pill and could be used if standard treatments are unsuccessful in treating symptoms, though it would likely cost as much as several dollars per day.[5]
The approval of partner Ironwood’s linaclotide in late August is one of many reasons Forest Labs has been oft-cited as a takeover target in biopharma. Forest markets the drug, which is OK’d for chronic idiopathic constipation and to treat irritable bowel syndrome with constipation. Morgan Stanley has estimated potential peak sales at $2 billion.
- Tadataka Yamada, ed. (2011). Textbook of Gastroenterology. John Wiley & Sons. ISBN 9781444359411.
- “FDA approves Linzess to treat certain cases of irritable bowel syndrome and constipation”. 30 Aug 2012.
- “Ironwood and Forest Announce U.S. Availability of LINZESS”. 17 Dec 2012.
- “Constella/Linzess Will Achieve Blockbuster Sales of $1.2 Billion in 2021 in the Irritable Bowel Syndrome Drug Market”. 19 Dec 2012.
- Pollack, Andrew. “Drug for Irritable Bowel Achieves Goals in Trial”, The New York Times, September 13, 2010. Accessed September 14, 2010.
- Jeffrey M Johnston , Caroline B Kurtz , Douglas A Drossman , Anthony J Lembo , Brenda I Jeglinski , James E MacDougall , Stephen M Antonelli & Mark G Currie . “Pilot Study on the Effect of Linaclotide in Patients With Chronic Constipation”, The American Journal of Gastroenterology 104, 125–132 (1 January 2009) | doi:10.1038/ajg.2008.59. Accessed September 15, 2010.
- Staff. “Daily International Pharma Alert”, FDANews, September 17, 2007, Vol. 4 No. 182. Accessed September 15, 2010.
- Albericio, F; Giraud, M; Gongora, M; Paradis, M; Tulla-Puche, J; Werbitzky, O. Solid-Phase Synthesis of the Cys-rich Peptide Linaclotide.
DRUG SPOTLIGHT-Ambrisentan

Ambrisentan
(2S)-2-[(4,6-dimethylpyrimidin-2-yl)oxy]-3-methoxy- 3,3-diphenylpropanoic acid
177036-94-1 cas no
Ambrisentan (U.S. trade name Letairis; E.U. trade name Volibris; India trade namepulmonext by MSN labs ) is a drug indicated for use in the treatment of pulmonary hypertension.
It functions as an endothelin receptor antagonist, and is selective for the type A endothelin receptor (ETA).[1] Once daily oral ambrisentan 2.5 to 10 mg/day significantly improved exercise capacity (6-minute walk distance) compared with placebo in two double-blind, multicenter trials (ARIES-1 & ARIES-2).[2]
Ambrisentan was approved for sale by the U.S. Food and Drug Administration (FDA) on June 15, 2007 for the once-daily treatment of pulmonary arterial hypertension.[3][4][5] It was later approved by the European Medicines Agency for use in the EU on April 2008.[6]Ambrisentan had previously been designated an orphan drug by both the FDA and the European Commission, in August 2004 and May 2005 respectively.[7]
Ambrisentan is indicated for the treatment of pulmonary arterial hypertension (WHO Group 1) in patients with WHO class II or III symptoms to improve exercise capacity and delay clinical worsening.
The LETAIRIS Education and Access Program (LEAP) is a program to help physicians and patients learn about the risks of LETAIRIS, including the serious risks of liver injury and birth defects.
LEAP works by:
- Providing information to prescribers on the risks of LETAIRIS
- Providing comprehensive education to patients and assistance with obtaining LETAIRIS
- Requiring enrollment of both prescriber and patient in LEAP
- Controlling dispensing through a specialized distribution network (specialty pharmacies)
- Letairis website run by Gilead Sciences
- Prescribing information
- Information on the LETAIRIS Education and Access Program (LEAP)
- Vatter H, Seifert V (2006). “Ambrisentan, a non-peptide endothelin receptor antagonist”. Cardiovasc Drug Rev 24 (1): 63–76.doi:10.1111/j.1527-3466.2006.00063.x. PMID 16939634.
- Frampton JE. Ambrisentan. American Journal of Cardiovascular Drugs August 1, 2011; 11 (4): 215-226.Link text
- Pollack, Andrew (2007-06-16). “Gilead’s Drug Is Approved to Treat a Rare Disease”. New York Times. Archived from the original on 20 June 2007. Retrieved 2007-05-25.
- “U.S. Food and Drug Administration Approves Gilead’s Letairis Treatment of Pulmonary Arterial Hypertension” (Press release).Gilead Sciences. 2007-06-15. Retrieved 2007-06-16.
- “FDA Approves New Orphan Drug for Treatment of Pulmonary Arterial Hypertension” (Press release). Food and Drug Administration. 2007-06-15. Archived from the original on 23 June 2007. Retrieved 2007-06-22.
- “GlaxoSmithKline’s Volibris (ambrisentan) receives authorisation from the European Commission for the treatment of Functional Class II and III Pulmonary Arterial Hypertension” (Press release). GlaxoSmithKline. 2008-04-25. Archived from the original on 30 April 2008. Retrieved 2008-04-29.
- Waknine, Yael (2005-05-09). “International Approvals: Ambrisentan, Oral-lyn, Risperdal”. Medscape. Retrieved 2007-06-16.


Patent EP2547663A1


READ MORE ON SNTAN SERIES……http://medcheminternational.blogspot.in/p/sentan-series.html
DRUG SPOTLIGHT–Treprostinil

Treprostinil (marketed under the trade names Remodulin for infusion and Tyvaso for inhalation) is a synthetic analog of prostacyclin (PGI2).
Treprostinil sodium, Uniprost, LRX-15, U-62840, UT-15, BW-15AU, 15AU81, Remodulin
289480-64-4, 81846-19-7 (free acid

During the 1960s a U.K. research team, headed by Professor John Vane began to explore the role of prostaglandins in anaphylaxis and respiratory diseases. Working with a team from the Royal College of Surgeons, Vane discovered that aspirin and other oral anti-inflammatory drugs worked by inhibiting the synthesis of prostaglandins. This finding opened the door to a broader understanding of the role of prostaglandins in the body.

Vane and a team from the Wellcome Foundation had identified a lipid mediator they called “PG-X,” which inhibited platelet aggregation. PG-X, which later would become known as prostacyclin, was 30 times more potent than any other known anti-aggregatory agent.
By 1976, Vane and fellow researcher Salvador Moncada published the first paper on prostacyclin, in the scientific journal Nature. The collaboration produced a synthetic molecule which was given the name epoprostenol. But like native prostacyclin, the structure of the epoprostenol molecule proved to be unstable in solution, prone to rapid degradation. This presented a challenge for both in vitro experiments and clinical applications. To overcome this challenge, the research team that discovered prostacyclin was determined to continue the research in an attempt to build upon the success they had seen with the prototype molecule. The research team synthesized nearly 1,000 analogs.
Treprostinil has demonstrated a unique effect on PPAR gamma, a transcription factor important in vascular pathogenesis as a mediator of proliferation, inflammation and apoptosis. Through a complementary, yet cyclic AMP-independent pathway, treprostinil activates PPARs, another mechanism that contributes to the anti-growth benefits of the prostacyclin class.
Treprostinil is indicated for the treatment of pulmonary arterial hypertension in patients with NYHA Class II-IV symptoms to diminish symptoms associated with exercise.[1] It may be administered as a continuous subcutaneous infusion or continuous intravenous infusion; however, because of the risks associated with chronic indwelling central venous catheters, including serious blood stream infections, continuous intravenous infusion should be reserved for patients who are intolerant of the subcutaneous route, or in whom these risks are considered warranted.
In patients with pulmonary arterial hypertension requiring transition from epoprostenol sodium (Flolan), treprostinil is indicated to diminish the rate of clinical deterioration. The risks and benefits of each drug should be carefully considered prior to transition.
The pharmacokinetics of continuous subcutaneous treprostinil are linear over the dose range of 1.25 to 125 ng/kg/min (corresponding to plasma concentrations of about 15 pg/mL to 18,250 pg/m) and can be described by a two-compartment model. Dose proportionality at infusion rates greater than 125 ng/kg/min has not been studied.
The major effects of treprostinil are vasodilation of arteries in the pulmonary (lung) and body. Treprostinil also inhibits platelet aggregation.
Treprostinil may be administered as a continuous subcutaneous infusion or continuous intravenous infusion via a small infusion pumpthat the patient must wear at all times. Treprostinil can be given subcutaneously by continuous infusion using an infusion set connected to an infusion pump, but also may be given intravenously via a central venous catheter if the patient is unable to tolerate subcutaneous administration because of severe site pain or reaction.
1. Remodulin Full Prescribing Information US Patent No. 5,153,222
2. UT_OpinVEvid_FEB09v.1
3. Cost-minimization analysis of treprostinil vs. epoprostenol as an alternate to oral therapy nonresponders for the treatment of pulmonary arterial hypertension L. Narine, L. K. Hague, J. H. Walker, C. Vicente, R. Schilz, O. Desjardins, T. R. Einarson and M. Iskedjian

…..
(+)-Treprostinil (also known as UT-15) is the active ingredient in Remodulin®, a commercial drug approved by FDA for the treatment of pulmonary arterial hypertension (PAH). It was first described in U.S. Pat. No. 4,306,075. Treprostinil is a stable analog of prostacyclin (PGI2) belonging to a class of compounds known as benzindene prostacyclins, which are useful pharmaceutical compounds possessing activities such as platelet aggregation inhibition, gastric secretion reduction, lesion inhibition, and bronchodilation.

U.S. Pat. No. 5,153,222 describes use of treprostinil for treatment of pulmonary hypertension. Treprostinil is approved for the intravenous as well as subcutaneous route, the latter avoiding potential septic events associated with continuous intravenous catheters. U.S. Pat. Nos. 6,521,212 and 6,756,033 describe administration of treprostinil by inhalation for treatment of pulmonary hypertension, peripheral vascular disease and other diseases and conditions. U.S. Pat. No. 6,803,386 discloses administration of treprostinil for treating cancer such lung, liver, brain, pancreatic, kidney, prostate, breast, colon and head-neck cancer. U.S. patent application publication No. 2005/0165111 discloses treprostinil treatment of ischemic lesions. U.S. Pat. No. 7,199,157 discloses that treprostinil treatment improves kidney functions. U.S. Pat. No. 7,879,909 discloses treprostinil treatment of neuropathic foot ulcers. U.S. publication No. 2008/0280986 discloses treprostinil treatment of pulmonary fibrosis, interstitial lung disease with treprostinil and asthma. U.S. Pat. No. 6,054,486 discloses treatment of peripheral vascular disease with treprostinil. U.S. patent application publication No. 2009/0036465 discloses combination therapies comprising treprostinil. U.S. publication No. 2008/0200449 discloses delivery of treprostinil using a metered dose inhaler. U.S. Pat. Nos. 7,417,070, 7,384,978 and 7,544,713 as well as U.S. publications Nos. 2007/0078095, 2005/0282901, and 2008/0249167 describe oral formulations of treprostinil and other prostacyclin analogs as well as their use for treatment of a variety of conditions. U.S. provisional application No. 61/354,949 filed Jun. 15, 2010 discloses the use of orally administered treprostinil for treatment of Raynaud’s phenomenon, systemic sclerosis and digital ischemic lesions.
Treprostinil and other prostacyclin derivatives have been prepared as described in Moriarty, et al in J. Org. Chem. 2004, 69, 1890-1902, Drug of the Future, 2001, 26(4), 364-374, U.S. Pat. Nos. 4,306,075, 6,441,245, 6,528,688, 6,700,025, 6,765,117, 6,809,223 and US Publication No. 2009/0163738. The entire teaching of these documents are incorporated herein by reference in their entirety. The methods described in these patent documents, however, do not describe a feasible production method for producing stereochemically pure treprostinil because, for example, the methods require the use of expensive reagents and tedious chromatographic purification techniques. Therefore, there is a need in the art for an economical, efficient and simplified method for preparing treprostinil and its synthetic intermediates.

NMR
The 1HNMR and HPLC of the samples were compared with reference UT-15 and were identical; 1H NMR (CDCl3, 300 MHz) δ 0.90 (t, 3H, 6 Hz), 1.05-1.78 (m, 13H), 2.85-2.85-2.98 (m, 1H), 2.03 2.12 (m, 1H), 2.21-2.32 (m, 1H), 2.45-2.53 (m, 1H), 2.61-2.81 (m, 3H), 3.52 (br s, 1H), 3.58-3.69 (m, 1H), 4.62 (s, 2H), 6.69 (d, 1H, J=8 Hz), 6.78 (d, 1H, J=8 Hz), 7.04 (dd, 1H, J=8 Hz).
J. Org. Chem. 2004, 69, 1890-1902
mp 126−127 °C;
[α]25D +52.6 (c 0.453, MeOH), [α]25D + 34.0° (c 0.457, EtOH).
IR 3385, 2928, 2856, 1739, 1713, 1585, and 779 cm–1;
1H NMR (CDCl3, 300 MHz) δ 0.87 (t, 3 H, J = 6 Hz), 1.21−1.86 (m, 13H), 2.02−2.44 (m, 4H), 3.42−3.76 (m, 3H), 3.81 (s, 2H), 3.82−3.94 (m, 1H), 4.63−4.68 (m, 1H), 4.88−4.92 (m, 1H), 4.94−4.98 (m, 1H), 4.99−5.02 (m, 1H), 5.60 (s, 1H), 5.92−6.06 (m, 1H), 6.85 (d, 1H, J = 6 Hz), 7.20−7.27 (m, 1H), 7.31−7.37 (m, 1H);
13C NMR (MeOH, 75 MHz) δ 13.1, 22.4, 25.1, 25.3, 28.3, 31.8, 32.7, 33.2, 34.7, 36.9, 40.7, 41.0, 51.3, 65.2, 71.6, 76.3, 109.5, 121.1, 125.8, 127.4, 140.8, 155.2, 171.5; UV, λmax MeOH, 217 nm;
HPLC, Hypersil ODS column (4.6 × 250 mm2), 5 μm; flow rate 2.0 mL/min; mobile phase A, water (60%):acetonitrile (40%):trifluoroacetic acid (0.1%), and mobile phase B, water (22%):acetonitrile (78%):trifluoroacetic acid (0.1%); retention time, 15 min (purity 99.7%). Anal. Calcd for C23H34O5: C, 70.74; H, 8.78. Found: C, 70.41; H, 8.83.
DRUG SPOTLIGHT – HALOPERIDOL

HALOPERIDOL
CAS No:- [52-86-8]
IUPAC Name:- 4-[4-(4-Chlorophenyl)-4-hydroxy-1-piperidinyl]-1-(4-fluorophenyl)-1-butanone
MW: 375.86, C21H23ClFNO2
Drug information:- Haloperidol is an anti-psychotic drug. This butyrophenone compound is used for the treatment of schizophrenia and other psychotic disorders in adults and childrens.
Haloperidol was discovered by Paul Janssen. It was developed in 1958 at the Belgian company Janssen Pharmaceutica and submitted to the first of clinical trials in Belgium later that year.
Haloperidol was approved by the U.S. Food and Drug Administration (FDA) on April 12, 1967; it was later marketed in the U.S. and other countries under the brand name Haldol by McNeil Laboratories.
Haloperidol is a dopamine inverse agonist of the typical antipsychotic class of medications. It is a butyrophenone derivative and has pharmacological effects similar to the phenothiazines.
Haloperidol is an older antipsychotic used in the treatment of schizophrenia and acutepsychotic states and delirium. A long-acting decanoate ester is used as an injection given every four weeks to people with schizophrenia or related illnesses who have poor adherence to medication regimens and suffer frequent relapses of illness, or to overcome the drawbacks inherent to its orally administered counterpart that burst dosage increases risk or intensity of side effects. In some countries, such as the United States of America, injections of antipsychotics such as haloperidol can be ordered by a court at the request of a psychiatrist.
Haloperidol is sold under the tradenames Aloperidin, Bioperidolo, Brotopon, Dozic,Duraperidol (Germany), Einalon S, Eukystol, Haldol (common tradename in the US and UK), Halosten, Keselan, Linton, Peluces, Serenace, Serenase, andSigaperidol


Conditions:-
i. Fluorobenzene, aluminum chloride, carbon disulfide, room temperature, 2 h,
ii. 4-(p-chlorophenyl)piperadine-4-ol, potassium iodide, toluene, 100 – 110 ºC
Drug spotlight, Celecoxib from G. D. Searle Company


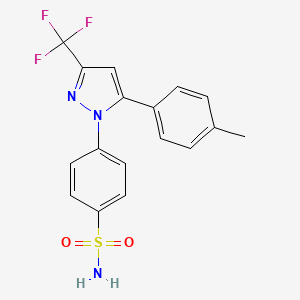
CELECOXIB
4-[5-(4- methylphenyl)-3-(trifluoromethyl)-lH-pyrazol-l-yl]benzenesulfonamide
169590-42-5
mp…157-159 deg C
Celecoxib is a sulfonamide non-steroidal anti-inflammatory drug (NSAID) and selective COX-2 inhibitor used in the treatment of osteoarthritis, rheumatoid arthritis, acute pain, painful menstruation and menstrual symptoms, and to reduce numbers of colon and rectum polyps in patients with familial adenomatous polyposis. It is marketed by Pfizer. It is known under the brand name Celebrex or Celebra for arthritis and Onsenal for polyps. Celecoxib is available by prescription in capsule form.
Celecoxib was discovered developed by G. D. Searle & Company and was approved by the FDA on December 31, 1998. It was co-promoted by Monsanto Company (parent company of Searle) and Pfizer under the brand name Celebrex. Monsanto merged with Pharmacia, from which the Medical Research Division was acquired by Pfizer, giving Pfizer ownership of Celebrex. The drug was at the core of a major patent dispute that was resolved in Searle’s favor (later Pfizer) in 2004. In University of Rochester v. G.D. Searle & Co., 358 F.3d 916 (Fed. Cir. 2004), the University of Rochester claimed that United States Pat. No. 6,048,850 (which claimed a method of inhibiting COX-2 in humans using a compound, without actually disclosing what that compound might be) covered drugs such as celecoxib. The court ruled in favor of Searle, holding in essence that the University had claimed a method requiring, yet provided no written description of, a compound that could inhibit COX-2 and therefore the patent was invalid.

After the withdrawal of rofecoxib (Vioxx) from the market in September 2004, Celebrex enjoyed a robust increase in sales. However, the results of the APC trial in December of that year raised concerns that Celebrex might carry risks similar to those of Vioxx, and Pfizer announced a moratorium on direct-to-consumer advertising of Celebrex soon afterwards. After a significant drop, sales of Celebrex have recovered, and reached $2 billion in 2006.[6] Pfizer resumed advertising Celebrex in magazines in 2006, and resumed television advertising in April 2007 with an unorthodox, 2 1⁄2-minute advertisement which extensively discussed the adverse effects of Celebrex in comparison with other anti-inflammatory drugs. The ad drew criticism from the consumer advocacy group Public Citizen, which called the ad’s comparisons misleading. Pfizer has responded to Public Citizen’s concerns with assurances that they are truthfully advertising the risk and benefits of Celebrex as set forth by the FDA.
In late 2007, Pfizer released another U.S. television ad for Celebrex, which also discussed celecoxib’s adverse effects in comparison with those of other anti-inflammatory drugs.
Daniel L. Simmons of Brigham Young University, who discovered the COX-2 enzyme, is suing Pfizer to be credited with discovery of the technique in 1989 that eventually led to the drug, and for $1 billion USD. The company has made about $30 billion from the drug as of 2006. A settlement was finally reached in April 2012.
Celecoxib is licensed for use in osteoarthritis, rheumatoid arthritis, acute pain, painful menstruation and menstrual symptoms, ankylosing spondylitis and to reduce the number of colon and rectal polyps in patients with familial adenomatous polyposis. It was originally intended to relieve pain while minimizing the gastrointestinal adverse effects usually seen with conventional NSAIDs. In practice, its primary indication is in patients who need regular and long term pain relief; there is probably no advantage to using celecoxib for short term or acute pain relief over conventional NSAIDs, except in the situation where non-selective NSAIDs or aspirin cause cutaneous reactions (urticaria or “hives”). In addition, the pain relief offered by celecoxib is similar to that offered by paracetamol (acetaminophen).
Synthesis
https://www.google.com/patents/WO2010095024A2?cl=en
US 5,466,823, also discloses a process for the preparation of Celecoxib, which comprises reacting 4-methylacetophenone (II) with 1-ethyltrifluoroacetate (III) in the presence of methyl t-butyl ether and sodium methoxide, followed by recrystallisation from isooctane to produce l-(4-methylphenyl)-4,4,4-trifluorobutane-l ,3-dione (IV), which is further condensed with 4-hydrazinophenylsulfonamide hydrochloride (V) in the presence of ethanol to produce crude Celecoxib, which is recrystallised from ethyl acetate and isooctane to give Celecoxib (I),
The process is as shown in Scheme -I below:

HI rv

The synthesis of celecoxib was first described in 1997 by a team of researchers at Searle Research and Development. Celecoxib is synthesized by a Claisen condensation reaction of an acetophenone with N-(trifluoroacetyl)imidazole catalyzed by the strong base, sodium bis(trimethylsilyl)amide to produce a 1,3-dicarbonyl adduct. Condensation of the diketone with (4-sulfamoylaphenyl)hydrazine produces the 1,5-diarylpyrazole drug moiety.
Scheme-I The above process involves isolation of the intermediate l-(4-methylphenyl)-4,4,4- tiϊfluorobutane-l ,3-dione (IV) by crystallization, before condensing with 4- sulphonamido-phenylhydrazine, which adds to the cost and complexity of the synthesis.
Further, the above process proceeds with less selectivity to Celecoxib, which is having about 4 wt. % of regioisomer (VI) by-product under commercial conditions.
US 6, 150,534 discloses a process for the preparation of Celecoxib, which comprises, condensing l-(4-methylphenyl)-4,4,4-trifluorobutane-l ,3-dione (IV) with 4- sulphonamido-phenylhydrazine in presence of an amide solvent at controlled temperature to produce amide solvate of Celecoxib, which is further desolvated by recrystallization from isopropanol and water.
The above process also involves isolation of the intermediate l-(4-methylphenyl)- 4,4,4-trifluorobutane-l ,3-dione (IV) by crystallization, before condensing with 4- sulphonamido-phenylhydrazine,
US 5,892,053 discloses a process for the preparation of Celecoxib by condensing 4- methylacetophenone (II) with 1-ethyltrifluoro acetate (III) to produce l-(4- methylphenyl)-4,4,4-trifluoiObutane-l ,3-dione (IV), which is further reacted with 4- hydrazinophenylsulfonamide (V) in presence of aqueous mixture of alcohol and acid to produce Celecoxib.
US 6,579,988 discloses a preparation of Celecoxib via novel intermediate compound of formula VII. Formula VII

US 2007/0004924 Al discloses a process for the preparation of Celecoxib by condensing l-(4-methylphenyl)-4,4,4-trifluorobutane-1.3-dione (IV) with 4- hydrazinophenylsulfonamide (V) in presence of a solvent system containing an organic solvent, the salt of the 4-sulphonamidophenylhydrazine having a solubility in the organic solvent at least 0.05 M. ‘
US 2008/0234491 Al discloses the condensation of l-(4-methylphenyl)-4,4,4- trifluorobutane-l,3-dione (IV) with 4-hydrazinophenylsulfonamide (V) or its acid addition salts in the presence of a solvent medium comprising an alkyl ester, water or mixtures thereof to produce Celecoxib. Further, crystallization of crude Celecoxib is carried out in toluene alone.
l-(4-Methylphenyl)-4,4,4-trifluorobutane-l,3-dione (IV) is condensed with 4- hydrazinophenylsulfonamide (V) or its acid addition salt in a solvent selected from water, inert organic solvent to produce 4-[5-(4-methylphenyl)-3-(trifluoiOmethyl)-lH- pyrazol-l-yl]benzenesulfonamide (Celecoxib) of Formula I. The acid addition salts of compound of the formula IV includes, but are not limited to, hydrochloride, hydrobromide, sulfate, nitrate, oxalate, mesylate, methane sulfonate, and tartrate, preferably, hydrochloride salt. The suitable inert organic solvents for the above reaction include but are not limited to ketone solvents, such as acetone, methyl ethyl ketone, methyl isobutyl ketone, n-butanone, and tertiary-butyl ketone; nitrile solvents, such as acetonitrile. and propionitrile; halogenated solvents, such as dichloromethane, ethylene dichloride, and chloroform; esters, such as ethyl acetate, n-propylacetate, isopropyl acetate, and tertiary-butyl acetate; aprotic polar solvents, such as N,N- dimethylformamide, dimethylsulfoxide, and N,N-dimethylacetamide; ethers, such as diisopropyl ether, tetrahydrofuran and 1,4-dioxane; hydrocarbon solvents, such as cyclohexane, toluene and xylene; and mixtures thereof. The preferred solvent is water. The reaction may be performed at a temperature ranging from about 25°C to about reflux temperature of the solvent or mixture of solvents used for the reaction. The above reaction is conducted in presence of an acid selected from aqueous hydrochloric acid, aqueous sulfuric acid, p-toluene sulfonic acid, trifluoroacetic acid, and acetic acid to maintain the pH of the reaction mixture is below 7. More preferably, aqueous HCl is added. Crude Celecoxib (I) produced may be isolated by precipitation of compound from the reaction mixture, which may be performed by cooling the reaction mixture, followed by addition of an organic solvent selected from alcohols such as methanol, ethanol, isopropanol or aromatic hydrocarbons such as toluene, xylene, ethyl benzene and mixtures thereof solvents. The preferred solvent is mixture of methanol and toluene.
It has been observed that preparation of Celecoxib (I) using above reaction conditions results in regioisomer of compound (VI) to less than 2.5% by HPLC analysis.
EXAMPLE 1
Stage-1:
Preparation of l-(4-methylphenyl)-4,4,4-trifluorobutane-l,3-dione (IV)
4-Methylacetophenone (50 g, 0.373 mol) was dissolved in toluene (250 ml) and 30% methanolic sodium methoxide solution (80.6 g, 0.447 mol), followed by 1- ethyltrifluoro acetate (63.58 g, 0.447 mol) were added at 25-3O0C. Temperature of the reaction mass was raised to 55-600C and stirred ~ 4 hr to complete the reaction. The reaction mass was cooled to 20-250C and washed with 10% aqueous hydrochloric acid (200 ml). The layers were separated and concentrated the organic layer at 50-550C under reduced pressure to produce 80 g of l-(4-methylphenyl)-4,4,4-trifluoiObutane- 1,3-dione (IV) as an oily mass.
Stage-2:
Preparation of 4-[5-(4-methylphenyl)-3-(trifluorornethyl)-lh-pyrazol-l- yljbenzenesulfonamide (Celecoxib) (I) l-(4-Methylphenyl)-4,4,4-trifluorobutane-l,3-dione (IV) (80 g, 0.348 mol), 4- hydrazinophenylsulfonamide (V) (77.74 g, 0.348 mol) and concentrated hydrochloric acid (18.6 g) were added to DM water (500 ml) and heated to 98-1000C. The mass was stirred for 4 hr to complete the reaction. The reaction mass was cooled to 70-75 C and a mixture of toluene (600 ml) and methanol (10 ml) was added to the reaction mass. After 1 hr stirring at 70-750C, the reaction mass was cooled to 20-250C, the product was filtered and λvashed with toluene (100 ml) followed by DM water (200 ml). The product obtained was dried at 55-600C under reduced pressure to produce 1 15 g of Celecoxib crude. Chromatographic purity: 99%(by PTPLC, by area normalization)
……………………..

The synthesis of celecoxib was first described in 1997 by researchers at Searle Research and Development. It is synthesized by a Claisen condensation reaction of an acetophenone with N-(trifluoroacetyl)imidazole catalyzed by the strong base, sodium bis(trimethylsilyl)amide to produce a 1,3-dicarbonyl adduct. Condensation of the diketone with (4-sulfamoylaphenyl)hydrazine produces the 1,5-diarylpyrazole drug moiety.
Penning TD, Talley JJ, Bertenshaw SR, Carter JS, Collins PW, Docter S, Graneto MJ, Lee LF, Malecha JW, Miyashiro JM, Rogers RS, Rogier DJ, Yu SS, Burton EG, Cogburn JN, Gregory SA, Koboldt CM, Perkins WE, Seibert K, Veenhuizen AW, Zhang YY, Isakson PC (1997). “Synthesis and Biological Evaluation of the 1.5 Diarylpyrazole Class of Cyclooxygenase-2 Inhibitors: Identification of 4-[5-(4-Methylphenyl)-3-(trifluoromethyl)-1H-pyrazole-1-yl]benzenesulfonamide (SC-58634, Celecoxib)”. Journal of Medicinal Chemistry40 (9): 1347–1365. doi:10.1021/jm960803q. PMID 9135032.

……………………………….


- The condensation of 4-methylacetophenone (I) with ethyl trifluoroacetate (II) by means of NaOMe in refluxing methanol gives 4,4,4-trifluoro-1-(4-methylphenyl)butane-1,3-dione, which is cyclized with 4-hydrazinophenylsulfonamide (III) in refluxing ethanol.
…………………
http://www.google.com/patents/US7759497
In U.S. Pat. Nos. 5,892,053 and 5,910,597, Zhi et al. describe a scalable two step process for the preparation of pyrazoles from the condensation of diketones and hydrazines. In the first step, a diketone is formed by the treatment of a ketone with base and ester in a suitable solvent. In the second step, the diketone is solubilized in an aqueous alcohol and condensed with a hydrazine to form the pyrazole product. This two step process has been used on a commercial scale for the preparation of celecoxib (4-[5-(4-methylphenyl)-3-(trifluoromethyl)-1H-pyrazole-1-yl]benzenesulfonamide) sold under the trademark CELEBREX® by Pharmacia Corporation as shown in the following reaction:

While this synthetic approach proceeds with high selectivity to celecoxib, about 2-5 wt. % of regioisomer and hydroxyregioisomer by-products are formed under commercial conditions.

The regioisomer and hydroxyregioisomer by-products must be separated from celecoxib in a purification step to enable the celecoxib to meet purity requirements. The separation is typically done through a crystallization step in which celecoxib preferentially crystallizes while the regioisomer and hydroxyregioisomer by-products predominantly remain in solution. The celecoxib crystals are then removed from the resultant slurry and separated from impurities by solid-liquid separation techniques known to those skilled in the art, such as centrifugation or filtration.
Under commercial conditions used to date, of the two by-products, regioisomer is selectively formed over hydroxyregioisomer. This is problematic, however, since the regioisomer is generally more difficult to separate through crystallization from celecoxib than is the hydroxyregioisomer, and regioisomer concentrations of greater than about 1% typically require two crystallizations to achieve desired celecoxib purity. The second crystallization adds time to the manufacturing process and thus negatively impacts product throughput. Additionally, a second crystallization reduces yield as some celecoxib remains uncrystallized and is not recovered from the liquid phase.

Example 7Preparation of Celecoxib with Hydrazine Reactant Containing Water
To a 250 mL reactor which had been purged with nitrogen and which had been fitted with a mechanical stirrer and a chilled condenser was charged while stirring, isopropyl alcohol (50.75 g), ethyltrifluoroacetate (37.95 g), sodium methoxide (25% in methanol, 53.28 g) and 4′-methylacetophenone (27.43 g). The reaction mixture was heated to 50-55° C. and held for at least 2 hours. To a separate 1 L reactor which had been purged with nitrogen and fitted with a mechanical stirrer and a chilled condenser, was charged 4-SAPH•HCl (45.96 g), isopropyl alcohol (101.2 g), water (74 g) and trifluoroacetic acid (23.43 g). The 4-SAPH•HCl was heated to 50° C. with agitation. At the completion of the 2 hour reaction period, the contents of the first reactor was transferred to the second reactor containing the 4-SAPH•HCl over a period of at least five minutes and the reaction mixture was then brought to 55° C. and maintained at that temperature for at least 30 minutes. The pH of the reaction mixture was then adjusted to be within the range of 3 to 9 followed by the addition of water (95 g). The contents were then heated to 65° C. and the pH was again adjusted to be within the range of 3 to 9. Water (90 g) was then added to the mixture over a time period of about 20 minutes while maintaining the temperature at about 65° C. The reaction mixture was then cooled to about 20° C. over a period of 12 to 14 hours to produce celecoxib (62-65 g) with less than 0.05% regio-isomer and undetectable regioisomer.
Example 8Preparation of Celecoxib with Anhydrous Hydrazine Reactant
To a 250 mL reactor which had been purged with nitrogen and which had been fitted with a mechanical stirrer and a chilled condenser was charged while stirring, isopropyl alcohol (50.75 g), ethyltrifluoroacetate (37.95 g), sodium methoxide (25% in methanol, 53.28 g) and 4′-methylacetophenone (27.43 g). The reaction mixture was heated to 50-55° C. and held for at least 2 hours. To a separate 1 L reactor which had been purged with nitrogen and fitted with a mechanical stirrer and a chilled condenser, was charged 4-SAPH•HCl (45.96 g), isopropyl alcohol (101.2 g) and trifluoroacetic acid (23.43 g). The 4-SAPH•HCl was heated to 50° C. with agitation. At the completion of the 2 hour reaction period, the contents of the first reactor was transferred to the second reactor containing the 4-SAPH•HCl over a period of at least five minutes and the reaction mixture was then brought to 55° C. and maintained at that temperature for at least 30 minutes. The pH of the reaction mixture was then adjusted to be within the range of 3 to 9 followed by the addition of water (95 g). The contents were then heated to 65° C. and the pH was again adjusted to be within the range of 3 to 9. Water (90 g) was then added to the mixture over a time period of about 20 minutes while maintaining the temperature at about 65° C. The reaction mixture was then cooled to about 20° C. over a period of 12 to 14 hours to produce celecoxib (62-65 g) with less than 0.05% regio-isomer. Analysis of the reaction mixture prior to initiation of crystallization indicated that the regio-isomer content was less than 0.5 mole percent of the reaction products.
Example 9Preparation of Celecoxib by Addition of Diketone Salt to 4-SAPH-HCl
To a 250 mL reactor, fitted with a mechanical stirrer and maintained under a nitrogen atmosphere, was added isopropyl alcohol (54.8 g, 0.912 moles), ethyl trifluoroacetate (38.0 g, 0.267 moles) and 25% sodium methoxide in methanol (53.3 g, 0.246 moles). To the agitated reactor was added 4-methylacetophenone (27.6 g, 0.206 moles). The reaction mixture was heated to 50° C. and maintained for 2 hours. To a second (1 liter) reactor was added 4-sulphamidophenyl hydrazine hydrochloride (46.0 g, 0.206 moles), isopropyl alcohol (101.3 g, 1.685 moles) and trifluoroacetic acid (11.7 g, 0.103 moles) with stirring. The reaction mixture was heated to approximately 45° C. Upon completion of the 2-hour reaction period in the 250 mL reactor, the contents was added to the second reactor over approximately 10 minutes. The reaction mixture maintained at 55° C. for 30 minutes. The pH was adjusted with 50% aqueous sodium hydroxide to a pH of 5-6. The reaction mixture was heated to 65° C. and water was added (95 g, 5.3 moles). The pH was again adjusted with 50% aqueous sodium hydroxide to a value of 5-6. Water (90 g, 5.0 moles) was added over 20 minutes while maintaining the temperature at 65° C. The reaction mixture was then cooled over 9 hours to 20° C. The reaction mixture was filtered, washed twice with 50% aqueous isopropyl alcohol and dried in a vacuum over for 16 hours to yield celecoxib (65.6 g) whose HPLC retention time was identical to that of authentic celecoxib. Regio-isomer was not detected by HPLC.
Example 10Preparation of Celecoxib by the Addition of 4-SAPH-HCl to Diketone
To a 1 L reactor fitted with a mechanical stirrer and maintained under a nitrogen atmosphere, was added isopropyl alcohol (54.7 g, 0.912 moles), ethyl trifluoroacetate (37.7 g, 0.267 moles), and 25% sodium methoxide in methanol (53.3 g, 0.247 moles). To the agitated reactor was added 4-methylacetophenone (27.32 g, 0.205 moles). The reaction mixture was heated to 50° C. and maintained for 2 hours. Trifluoroacetic acid (36.69, 0.321 moles) was added to the reaction mixture over a period of five minutes. 4-SAPH-HCl (46.0 g, 0.205 moles) was added through a power addition funnel over a period of 10 minutes. The reaction mixture was brought to 55° C. and maintained for one hour. Isopropyl alcohol (81.5 g, 1.36 moles) was added followed by the addition of 50% sodium hydroxide (18.5 g, 0.231 moles) to achieve a pH of 7. Water (87.8 g, 4.88 moles) was added and the reaction mixture heated to 65° C. Water (90.0 g 5.00 moles) was added over ten minutes. The reaction mixture was cooled to 20° C. over nine hours. The slurry was filtered and washed twice with 50% (weight) aqueous isopropyl alcohol (100 g). The solid was dried in a vacuum oven for 16 hours to yield celecoxib (67.2 g) whose HPLC retention time was identical to that of authentic material. Regio-isomer was not detected by HPLC.
4-[5-(4-Methylphenyl)-3-(trifluoromethyl)-1H-pyrazol-1-yl]benzenesulfonamide (Celecoxib)
E-mail: prataprp@ drreddys.com. Fax: 914044346285. Telephone: 9989997176


HPLC Conditions:
 29.2 min30.9 min
29.2 min30.9 minhttps://newdrugapprovals.org/2013/04/07/drug-spotlight-celecoxib-from-g-d-searle-company/
Celecoxib extraction
Celecoxib was extracted from Celebra™ 100
mg and 200 mg capsules. Ten capsules were
crushed to fine powder, using agate mortar and
pestle, transferred to a 50 ml volumetric flask
and diluted to volume with methanol. The solution
was shaked for 5 min and filtered. The
residue was recrystallized from acetonitrile after
evaporation of the solvent in a water bath at 50
°C, under a stream of nitrogen.
Thin layer Chromatography (TLC) In this procedure it was used silica gel 60 F254 plates (20 x 20 cm) with a thickness of 0.25 mm. All plates used were commercially prepared by MERCK (lot # 040422153). The mobile phase used to develop the system consisted of chloroform-ethyl acetate-ether (10:5:1, v/v/v). Just in order to verify the selectivity of the proposed system, another COX-2 inhibitor (rofecoxib) with similar properties was used. 20 µl of celecoxib reference substance and extracted from tablets and rofecoxib solutions (50 µg.ml–1) were spotted on the TLC plates, and transferred to a developing tank containing the mobile phase. The plate was then examined under UV light (254 and 365 nm).
Thin-layer chromatography One of the most effective screening methods is the thin-layer chromatography (TLC), which is the simplest of all the widely used chromatographic methods to perform. In the determination of this method, different chromatography systems were tested and analyzed according to the classification proposed by Moffat 13, which divide the drugs in three categories: acid, basic and neutrals based on their polarity and acid characteristics. This procedure is important in
order to increase the information obtained only by changing the mobile phase, and thus resulting in a significant change in selectivity. These chromatographic systems consisted of mixtures of the following solvents: chloroformacetonitrile, ethyl acetate, acetone and methanol, in different concentrations. Nevertheless, all the mobile phases tested were not adequate for the proper identification of the drug. With the objective to develop more reliable chromatographic method, a modification in the system tested was proposed, in order to improve the chromatographic resolution. The mobile phase used consisted of chloroform-ethyl acetate-ether (10:5:1 v/v/v). The system was chosen due to its sensitivity, simplicity and efficacy
The preference to use commercially prepared silica gel plates was due to its durability and homogeneity of the absorbent layer. A sample solution of the working standard and the drug sample (50 (µg ml–1) were spotted onto a silica gel plate with a micropipette and the chromatogram was developed by placing the plate in a tank containing the mobile phase. Following development the individual solute spots were identified under UV lamp (254 and 365 nm). The spots in the drug sample and the working standard presented similar Rf values. The Rf value obtained for drug sample and the working standard were 0.45 and for rofecoxib 0.32.
NMR
The spectrum shown in Figure 3 possesses two characteristic sharp singlet peaks at 2.3 and3.3 ppm that belong to the methyl and sulfonamide protons of celecoxib. The spectrum also reveals peaks from 7.3 to 7.9, which is due to the protons of aromatic groups. The characteristics peaks from the working standard agree well with those observed in the samples, considering the fact that a constant shift is observed in all peaks 11, 12.
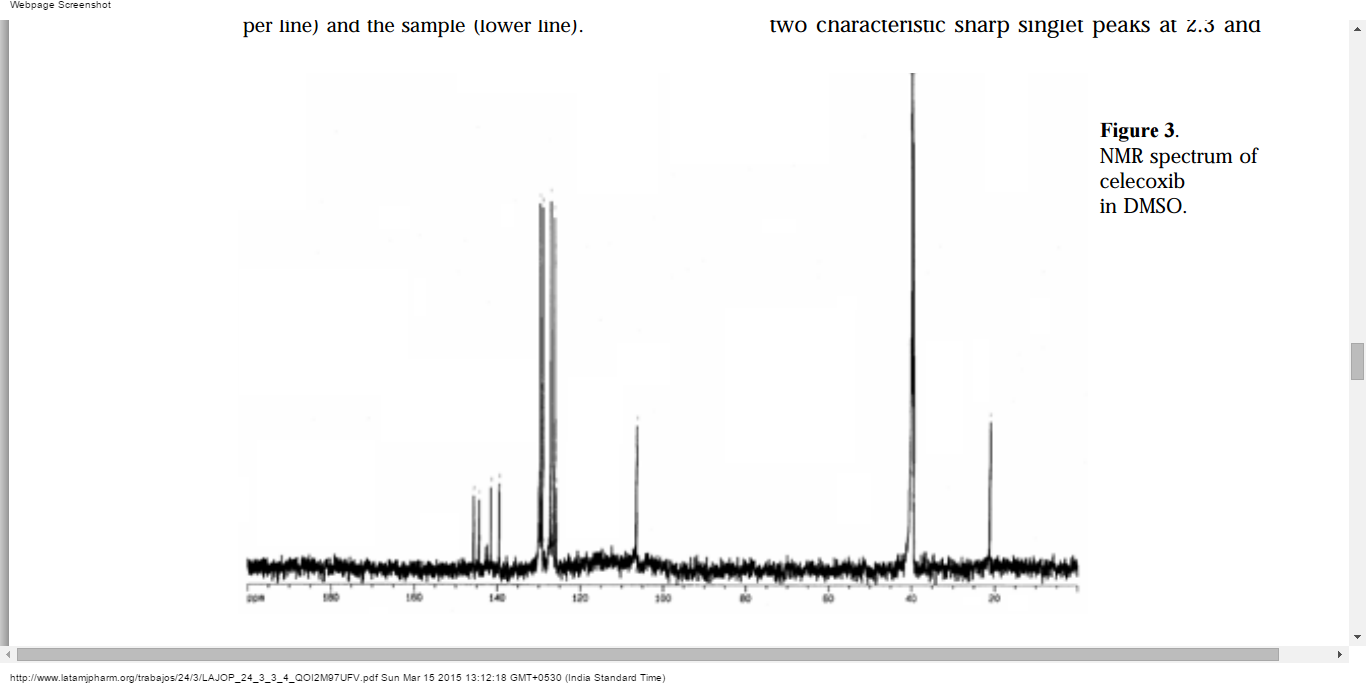
UV

IR
1150 – 1350 S = O stretching (sulfonamide group)
1550 – 1600 N – H stretching
3300 – 3500 NH2 stretching


1H NMR PREDICT

13 C NMR

(4) (a) Matsuo, M.; Tsuji, K.; Konishi, N.; Nakamura, K. EP patent 0,418,845, A1, 1990. (b) Matsuo, M.; Tsuji, K.; Konishi, N.; Ogino, T. EP patent 0,554,829, A1, 1993. (c) Nishiwaki, T. Bull. Chem. Soc. Jpn. 1969, 42, 3024. (d) Soliman, R.; Feid-allah, H. J. Pharm. Sci. 1980, 70, 602. (e) Wright, J. B.; Dulin, W. E.; Markillie, J. H. J. Med. Chem. 1963, 7, 102. (f) Habeeb, A. G.; Rao, P. N. P.; Knaus, E. E. J. Med. Chem. 2001, 44, 3039. (g) Szabo, G.; Fischer, J.; Kis-Varga, A.; Gyires, K. J. Med. Chem. 2008, 51, 142. (h) Oh, L. M. Tetrehedron Lett. 2006, 47, 7943.
(5) Talley, J. J.; Penning, T. D.; Collins, P. W.; Rogier, D. J.; Malecha, J. W.; Miyashiro, J. M.; Bertenshaw, S. R.; Khanna, I. K.; Graneto, M. J.; Rogers, R. S.; Carter, J. S. US patent 5,466,823, 1995.
(6) Penning, T. D.; Talley, J. J.; Bertenshaw, S. R.; Carter, J. S.; Collins, P. W.; Docter, S.; Graneto, M. J.; Lee, L. F.; Malecha, J. W.; Miyashiro, J. M.; Rogers, R. S.; Rogier, D. J.; Yu, S. S.; Anderson, G. D.; Burton, E. G.; Cogburn, J. N.; Gregory, S. A.; Koboldt, C. M.; Perkins, W. E.; Seibert, K.; Veenhuizen, A. W.; Zhang, Y. Y.; Isakson, P. C. J. Med. Chem. 1997, 40, 1347.
7 Talley, J. J.; Penning, T. D.; Collins, P. W.; Rogier, D. J.; Malecha, J. W.; Miyashiro, J. M.; Bertenshaw, S. R.; Khanna, I. K.; Graneto, M. J.; Rogers, R. S.; Carter, J. S.; Docter, S. H.; Yu, S. S. US patent 6,586,603, B1, 2003.
(8) (a) O′ Shea, P. ; Tillyer, R. D. ; Wang, X. ; Clas, S. D. ; Dalton, C. US patent 6,150,534, 2000. (b) O′ Shea, P. ; Tillyer, R. D. ; Wang, X. ; Clas, S. D. ; Dalton, C. US patent 6,232,472, 2001.
(9) (a) Zhi, B. ; Newaz, M. US patent 5,,892,053, 1999. (b) Zhi, B. ; Newaz, M. US patent 5,910,597, 1999.
10(a) Letendre, L. J.; McGhee, W. D.; Snoddy, C.; Klemm, G.; Graud, H. T. US patent 7,141,678, 2006. (b) Letendre, L. J.; McGhee, W. D.; Snoddy, C.; Klemm, G.; Graud, H. T. US patent 2007/0,004, 924 A1, 2007.
(11) ICH harmonized tripartite guideline, Impurities in New Drug Substances Q3A (R2), current step 4 version dated 25 October2006.
(12) Ahlstrom, M. M.; Ridderstrom, M.; Zamora, I.; Luthman, K. J. Med. Chem. 2007, 50, 4444. (13) Soliman, R. J. Med. Chem. 1979, 22, 321.
SYNTHESIS
 +
+
![]() CELECOXIB
CELECOXIB
EUCLISES PHARMACEUTICALS, INC.; MARTINEZ, Eduardo, J.; TALLEY, John, J.; JEROME, Kevin, D.; BOEHM, Terri, L. Patent: WO2014/12074 A2, 2014 ; Location in patent: Paragraph 00209; 00210
2
+
![]()
 +
+
Reddy, Anumula Raghupathi; Sampath, Alla; Goverdhan, Gilla; Yakambaram, Bojja; Mukkanti, Kagga; Reddy, Padi Pratap Organic Process Research and Development, 2009 , vol. 13, # 1 p. 98 – 101
3
Prabhakaran, Jaya; Underwood, Mark D.; Parsey, Ramin V.; Arango, Victoria; Majo, Vattoly J.; Simpson, Norman R.; Van Heertum, Ronald; Mann, J. John; Kumar, J.S. Dileep Bioorganic and Medicinal Chemistry, 2007 , vol. 15, # 4 p. 1802 –
4
Li, Feng; Nie, Jing; Sun, Long; Zheng, Yan; Ma, Jun-An Angewandte Chemie – International Edition, 2013 , vol. 52, # 24 p. 6255 – 6258
5
Synlett, , vol. 1997, # 4 p. 375 – 377
6
Tetrahedron Letters, , vol. 52, # 45 p. 6000 – 6002
7
Bioorganic and Medicinal Chemistry Letters, , vol. 21, # 22 p. 6636 – 6640
8

![]() celecoxib
celecoxib
Angewandte Chemie – International Edition, , vol. 52, # 24 p. 6255 – 6258
9
Tetrahedron Letters, , vol. 52, # 45 p. 6000 – 6002

![]()
celecoxib
10Angewandte Chemie – International Edition, , vol. 52, # 24 p. 6255 – 6258

![]()
11
Bioorganic and Medicinal Chemistry, , vol. 15, # 4 p. 1802 – 1807

![]()
..celecoxib
………
http://www.google.com/patents/WO2014012074A2?cl=en
4,4,4-Trifluoro-l-(p-tolyl)butane-l,3-dione (C01)

25% sodium methoxide in methanol (51.3 ml, 223.5 mmol) and ethyl
trifluoroacetate (24.4 ml, 204.9 mmol) were dissolved in 110 mL methyl tert-butyl ether under N2, at room temperature. 4′-methyl acetophenone (25.0 ml, 186.3 mmol) was added and stirred at room temperature overnight. The reaction was washed with 3M HC1 and dried over magnesium sulfate. The solution was then evaporated and the resulting oil dried under vacuum overnight. The resulting light orange crystalline solid was washed with cold isooctane and dried under vacuum to yield an off white crystalline solid (37.3 g, 87% yield). LC tr=3.49 minutes (C- 18 column, 5 to 95% acetonitrile/water over 6 minutes at 1.7 mL/min with detection 254 nm, at 23 °C).
4-(5-(p-Tolyl)-3-(trifluoromethyl)-lH-pyrazol-l-yl)benzenesulfonamide (C02)

] C01 (23.55 g, 102.3 mmol) was refluxed with 4-sulphonamidophenyl
hydrazine HCl (23.95 g, 127.9 mmol) in 700 mL ethanol overnight. The reaction was evaporated, dissolved in 700 mL ethyl acetate, washed with water and brine, dried over magnesium sulfate and evaporated to -100 mL ethyl acetate. The product was crystalized by the addition of ~ 400 mL isooctane. After 15 minutes, the white crystalline solid was broken up, washed with isooctane and dried under vacuum (35.15 g, 90% yield). 1H NMR (400 MHz, CDC13) δ 7.94-7.91 (m, 2H), 7.51-7.49 (m, 2H), 7.21-7.20 (m, 2H), 7.15-7.13 (m, 2H), 6.77 (s, 1H), 2.41 (s, 3H). LC tr=4.27 minutes (C-18 column, 5 to 95% acetonitrile/water over 6 minutes at 1.7 mL/min with detection 254 nm, at 23° C). ES(neg)MS m/z 380 (M-H calcd for C17H14F3N302S requires 380).


http://www.slideshare.net/kennytirorx/celebrex-ms-report
………………………………….
http://www.google.co.in/patents/WO2011055233A2?cl=en
The preparation and use as COX-2 inhibitors of benzenesulfonamide derivatives such as celecoxib is described in US Patent No. 5,466,823. Processes for the preparation of celecoxib are also described in U.S. Patent Nos. 5,760,068 and 5,910,597.
As per the process exemplified in the U.S. Patent No. 5,760,068 (hereinafter referred to as the Ό68 patent), celecoxib is prepared by the reaction of l-(4-methylphenyl)- 4,4,4-trifluorobutane-l,3-dione with 4-sulphonamidophenyl hydrazine hydrochloride in absolute ethanol at reflux temperature under argon for 24 hours. The resulting mass is cooled to room temperature, followed by filtering and concentrating the reaction mixture to afford an orange solid, which is then recrystallized from a solvent system containing methylene chloride/hexane to produce the product as a pale yellow solid (melting point: 157° – 159°C).
The recrystallization process for preparing celecoxib described in the ‘608 patent suffers from disadvantages since the recrystallization process requires large volumes of solvents (more than 20 volumes each of methylene chloride and hexane solvents per gram of celecoxib), which is not commercially and environmentally, advisable for scale up operations. Moreover, the use of methylene chloride is hazardous to the environment and human health. The use of n-hexane is not advisable because it causes an ignition and fire risk due to its electrostatic charge accumulation property.
PCT Publication No. WO 01/42222 (hereinafter referred to as the ‘222 application) discloses three polymorphic forms (Form I, Form II and Form III) of celecoxib, pharmaceutical compositions, and methods of use thereof. The crystalline forms are characterized by powder X-ray diffraction (P-XRD), differential scanning calorimetry (DSC) and Infrared (IR) spectroscopy. The ‘222 application further teaches that the crystalline Form III of celecoxib is more thermodynamically stable than Form I and Form II. The ‘222 application also teaches that crystalline Form III of celecoxib is produced by crystallization of celecoxib from a solvent comprising isopropanol and water, for example, as described in U.S. Patent No. 5,910,597.
According to the ‘222 application, the polymorphic Form I is characterized by an X-ray powder diffraction pattern having peaks expressed as 2-theta at about 5.5, 5.7, 7.2 and 16.6 degrees; a melting point of about 162.5°C to about 163°C; a differential scanning calorimetry (DSC) endotherm maximum at about 163.3°C; and an Infra Red (IR) spectrum with peaks at about 3256 and 3356 cm-1. The polymorphic Form II is characterized by an X- ray powder diffraction pattern having peaks expressed as 2-theta at about 10.3, 13.8 and 17.7 degrees, a melting point of about 161°C to about 162°C; a differential scanning calorimetry (DSC) endotherm maximum at about 162°C. The polymorphic Form III is characterized by a melting point of about 160.8°C.
However, it has been observed by the present inventors that the celecoxib obtained after crystallization from isopropanol and water is fluffier resulting in low bulk density and poor flow properties. Moreover, it has also been observed that the particles of the crystalline form III of celecoxib obtained by the aforementioned crystallization processes are static or cohesive thereby increasing the difficulties of formulation scientists.
PCT Publication No. WO 01/42221 discloses an amorphous form of celecoxib, and processes for preparing amorphous celecoxib using crystallization inhibitors. Amorphous celecoxib exhibits an apparent glass transition at 111.4°C (onset).
EP Patent No. 1167355 (hereinafter referred to as the ‘355 patent) discloses a crystalline form, designated as Form I, of celecoxib, processes for the preparation, and pharmaceutical compositions thereof. The crystalline form is characterized by powder X-ray diffraction (P-XRD) and scanning electron microscopy (SEM). According to the ‘355 patent, the crystalline Form I is characterized by an X-ray powder diffraction pattern having peaks expressed as 2-theta at about 14.8, 16.05, 17.875, 19.615, 21.455, 22.080, 22.385, 23.425, 25.33 and 29.355 degrees. The crystalline Form I is further characterized by an X-ray powder diffraction pattern having additional peaks expressed as 2-theta at about 10.67, 10.97, 12.985, 13.855, 18.340, 18.685, 20.425, 20.67, 23.185, 24.51, 24.93, 25.73, 26.915, 27.63, 28.185, 29.955, 30.375, 31.405, 34.915, 35.585, 37.895, 44.070 and 45.250 degrees. The ‘355 patent teaches that the crystalline Form I has improved properties over prior art crystal form which is used for formulating celecoxib as disclosed in International Application No. WO 95/15316. The ‘355 patent teaches that the prior art crystal form (designated as Form II) has several disadvantages, caused by its crystal structure, since it has low bulk density and a crystal morphology that tends to form long cohesive needles.
According to the ‘355 patent, the celecoxib crystalline Form I is prepared by dissolving celecoxib in a solvent system comprising at least one amide solvent selected from the group consisting of N,N-dimethylformamide, Ν,Ν-dimethylacetamide, and mixtures thereof; and isolating the crystals of Form I by adding a non-solvent, especially water, to the solution.
The process for the preparation of the celecoxib crystalline Form I described in the ‘355 patent also suffers from drawbacks since the use of amide solvents in the purification/crystallization of celecoxib leads to the formation of solvates (for example, the formation of solvates of celecoxib with amide solvents such as dimethylacetamide and dimethylformamide can be found in the preparative examples of the WO 01/42222). It is well known that the removal of these residual amide solvents from the celecoxib crystalline form is very difficult and requires high temperatures.
Formation of solvates of celecoxib with amide solvents such as dimethylacetamide and dimethylformamide is also described in PCT Publication No. WO2005/014546. Moreover, celecoxib obtained by the crystallization process using amide solvents described in the ‘355 patent does not have satisfactory purity for pharmaceutical use.
The solvated forms of celexicob are not acceptable from regulatory point of view since they include substantial amounts of organic solvents, and thus are not acceptable for clinical use. It is well known that impurities and residual solvents in celecoxib or any active pharmaceutical ingredient (API) are undesirable and might be harmful. Purity standards are set by regulatory authorities with the intention of ensuring that an API is as free of impurities and residual solvents as possible, and, thus, are as safe as possible for clinical use. Furthermore, it is required to control the levels of these impurities in the final drug compound obtained by the manufacturing process and to ensure that the impurities and/or residual solvents are present in the lowest possible levels.
celecoxib characterized by a powder X-ray diffraction (P-XRD) pattern having peaks (designated as d-values) at about 16.0, 15.3, 12.3, 10.6 ± 0.2 A. According to the ‘340 application, the crystalline Form N of celecoxib is prepared by suspending celecoxib Form III in a hydrocarbon solvent selected from the group consisting of n-tetradecane, and n-decane, heating the suspension at high temperatures (about 165°C) while stirring, stirring the resulting emulsion at the same high temperature, followed by cooling to 145°C. The resulting mass is then reheated to about 165°C, followed by cooling to about 110°C, filtering the separated crystals, and drying at 100°C under the vacuo for 12 hours to produce celecoxib Form N.
The crystallization process for preparing celecoxib described in the ‘340 application also suffers from disadvantages since the processes involve tedious and cumbersome procedures such as the use of high boiling point solvents, large amounts of solvents (about 20 volumes of high boiling point solvents per gram of celecoxib), high temperatures (about 165°C), high drying temperatures (about 100°C), and prolonged periods of drying at a high temperature, resulting in formation of unwanted impurities, thereby making the process industrially unfeasible.
PCT Publication No. WO 05/089511 discloses a hydrate of celecoxib sodium salt characterized by a powder X-ray diffraction (P-XRD) pattern having peaks at 3.05, 8.91 and 10.77 degrees 2-theta.
PCT Publication No. WO 2006079923A1 discloses a crystalline Form IV of celecoxib characterized by a powder X-ray diffraction (P-XRD) pattern having peaks at about 4.46, 13.13, 18.29, 20.21, 21.83 and 26.24 degrees 2-theta. [0020] Based on the aforementioned drawbacks, the prior art crystallization processes may be unsuitable for the preparation of celecoxib, especially in crystalline Form III, in commercial scale operations.
| Patent | Submitted | Granted |
|---|---|---|
| 5-oxo-pyrrolidine-2-carboxylic acid hydroxamide derivatives [EP1004578] | 2000-05-31 | 2004-02-25 |
| ANALGESIC COMPOSITIONS COMPRISING ANTI-EPILEPTIC COMPOUNDS AND METHODS OF USING SAME [EP1011658] | 2000-06-28 | 2005-12-07 |
| SYNERGISTIC ANALGESIC COMBINATION OF OPIOID ANALGESIC AND CYCLOOXYGENASE-2 INHIBITOR [EP1014886] | 2000-07-05 | 2004-11-24 |
| BIARYLALKANOIC ACIDS AS CELL ADHESION INHIBITORS [EP1017382] | 2000-07-12 | 2006-03-01 |
| BIOADHESIVE COMPOSITIONS AND METHODS FOR TOPICAL ADMINISTRATION OF ACTIVE AGENTS [EP1021204] | 2000-07-26 | 2005-12-28 |
| Implantable medical device with enhanced biocompatibility and biostability [EP1023879] | 2000-08-02 | 2005-04-06 |
| NOVEL COMBINATION [EP1027052] | 2000-08-16 | |
| CYCLIC AMINO ACID DERIVATIVES AS CELL ADHESION INHIBITORS [EP1033983] | 2000-09-13 | |
| SUBSTITUTED BETA-ALANINE DERIVATIVES AS CELL ADHESION INHIBITORS [EP1034164] | 2000-09-13 | 2004-05-19 |
| Delayed-release oral formulation of dihydroxy open acid simvastatin and salts and esters thereof [EP1036563] | 2000-09-20 |
| Patent | Submitted | Granted |
|---|---|---|
| Crystalline hydrated dihydroxy open-acid simvastatin calcium salt [EP1036783] | 2000-09-20 | 2003-05-21 |
| CYCLOOXYGENASE-2 INHIBITION CYCLOOXYGENASE-2 INHIBITION [EP1039914] | 2000-10-04 | |
| Dioxocyclopentyl hydroxamic acids [EP1041072] | 2000-10-04 | 2003-07-16 |
| PHARMACEUTICAL ORAL DOSAGE FORM COMPRISING A COMBINATION OF AN OPIOID AGONIST AND NALTREXONE [EP1041987] | 2000-10-11 | 2006-04-19 |
| Method and compositions for the treatment and prevention of pain and inflammation [US2005101563] | 2005-05-12 | |
| Compositions of a cyclooxygenase-2 selective inhibitor and a neurotrophic factor-modulating agent for the treatment of central nervous system mediated disorders [US2005148589] | 2005-07-07 | |
| Compositions and methods of treatment involving peroxisome proliferator-activated receptor-gamma agonists and cyclooxygenase-2 selective inhibitors [US2003220374] | 2003-11-27 | |
| Methods for treating carbonic anhydrase mediated disorders [US2003220376] | 2003-11-27 | |
| Method of using a Cox-2 inhibitor and a 5-HT1A receptor modulator as a combination therapy [US2004147581] | 2004-07-29 | |
| Tyrosine kinase inhibitors [US2002137755] | 2002-09-26 |
| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO1995015316A1 | 14 Nov 1994 | 8 Jun 1995 | Stephen R Bertenshaw | Substituted pyrazolyl benzenesulfonamides for the treatment of inflammation |
| WO2001042221A1 | 6 Dec 2000 | 14 Jun 2001 | Michael J Hageman | Solid-state form of celecoxib having enhanced bioavailability |
| WO2001042222A1 | 1 Dec 2000 | 14 Jun 2001 | Leonard J Ferro | Polymorphic crystalline forms of celecoxib |
| WO2002000627A1 * | 26 Jun 2001 | 3 Jan 2002 | Mehmet Bahar | A crystalline form of celecoxib |
| WO2005014546A1 | 8 Aug 2003 | 17 Feb 2005 | Hetero Drugs Ltd | Novel crystalline forms of celecoxib |
| WO2005089511A2 | 17 Mar 2005 | 29 Sep 2005 | Transform Pharmaceuticals Inc | Novel pharmaceutical forms, and methods of making and using the same |
| WO2006051340A1 | 21 Jul 2005 | 18 May 2006 | Pliva Istrazivanje I Razvoj D | Novel form of celecoxib |
| WO2006079923A2 | 19 Jan 2006 | 3 Aug 2006 | Pharmacia & Upjohn Co Llc | Form iv crystalline celecoxib |
| EP1167355A1 | 15 Mar 2001 | 2 Jan 2002 | Fako Ilaclari A.S. | A crystalline form of celecoxib |
| US5466823 | 30 Nov 1993 | 14 Nov 1995 | G.D. Searle & Co. | Antiinflammatory agents; FDA Orange book listed patent for Celebrex |
| US5760068 | 14 Nov 1994 | 2 Jun 1998 | G.D. Searle & Co. | Substituted pyrazolyl benzenesulfonamides for the treatment of inflammation |
| US5910597 | 14 Oct 1998 | 8 Jun 1999 | G.D. Searle & Co. | Process for preparing 3-haloalkyl-1H-pyrazoles |
………..

Dr Raghupathi Reddy Anumula
Managing Director- Chemikos Laboratories Private Limited
-
Managing Director
Chemikos Laboratories Private Limited
August 2014 – Present (Hyderabad Area, India
Director- API development
Dr. Reddy’s Laboratories
July 2012 – July 2014 (2 years 1 month)Hyderabad Area, India
Managing the three teams consisting of 50 members and guiding them to deliver the projects in time without any major surprises.

Associate Director- API development
Dr. Reddy’s Laboratories
October 2006 – June 2012 (5 years 9 months)Hyderabad Area, India
New product initiation, development ,Regulatory filing and launch support for API’s.
New product initiation, development ,Regulatory filing and launch support for API’s.
1994 – 2006
