

DASABUVIR, ABT 333,
CAS 1132935-63-7,
N-[6-[3-tert-butyl-5-(2,4-dioxopyrimidin-1-yl)-2-methoxyphenyl]naphthalen-2-yl]methanesulfonamide; Dasabuvir; N-(6-(3-tert-butyl-5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-2-methoxyphenyl)naphthalen-2-yl)methanesulfonamide
Non-nucleoside NS5B polymerase inhibitor
-
Methanesulfonamide, N-(6-(5-(3,4-dihydro-2,4-dioxo-1(2H)-pyrimidinyl)-3-(1,1-dimethylethyl)-2-methoxyphenyl)-2-naphthalenyl)-
-
N-(6-(3-tert-Butyl-5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-2-methoxyphenyl)naphthalen-2-yl)methanesulfonamide
- Methanesulfonamide, N-(6-(5-(3,4-dihydro-2,4-dioxo-1(2H)-pyrimidinyl)-3-(1,1-dimethylethyl)-2-methoxyphenyl)-2-naphthalenyl)-
N-(6-(3-tert-Butyl-5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-2-methoxyphenyl)naphthalen-2-yl)methanesulfonamide
- 493.5813
- UNII-DE54EQW8T1,
![ChemSpider 2D Image | Sodium {6-[5-(2,4-dioxo-3,4-dihydro-1(2H)-pyrimidinyl)-2-methoxy-3-(2-methyl-2-propanyl)phenyl]-2-naphthyl}(methylsulfonyl)azanide | C26H26N3NaO5S](http://www.chemspider.com/ImagesHandler.ashx?id=34999596&w=250&h=250)
Dasabuvir sodium anhydrous
RN: 1132940-11-4
UNII: R2M8F5TK9T
http://chem.sis.nlm.nih.gov/chemidplus/rn/1132940-11-4
Sodium {6-[5-(2,4-dioxo-3,4-dihydro-1(2H)-pyrimidinyl)-2-methoxy-3-(2-methyl-2-propanyl)phenyl]-2-naphthyl}(methylsulfonyl)azanide
Sodium {6-[5-(2,4-dioxo-3,4-dihydro-1(2H)-pyrimidinyl)-2-methoxy-3-(2-methyl-2-propanyl)phenyl]-2-naphthyl}(methylsulfonyl)azanide
ABT-333 sodium
dasabuvir sodium
DASABUVIR SODIUM ANHYDROUS
sodium {6-[3-tert-butyl-5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-2-methoxyphenyl]naphthalen-2-yl}(methanesulfonyl)azanide
- Molecular FormulaC26H26N3NaO5S
- Average mass515.557
Dasabuvir (ABT-333), an oral non-nucleoside NS5B polymerase inhibitor, is a component of an all-oral hepatitis C treatment regimen under FDA review for the chronic Hepatitis C treatment.
On April 22, 2014, AbbVie submitted a New Drug Application (NDA) to the U.S. Food and Drug Administration (FDA) seeking approval for its investigational, all-oral, interferon-free regimen for the treatment of adult patients with chronic genotype 1 (GT1) hepatitis C virus (HCV) infection.
Dasabuvir (trade name Exviera in Europe) is an antiviral drug for the treatment of hepatitis C virus (HCV) infection. In the United States, it is approved by the Food and Drug Administration for use in combination with ombitasvir, paritaprevir, and ritonavir in the product Viekira Pak.[1]
Dasabuvir acts as a NS5B (an RNA-directed RNA polymerase) inhibitor.[2]
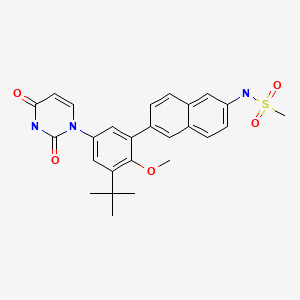
Patent
WO2009039127
http://www.google.com/patents/WO2009039127A1?cl=en

Example 4A. Preparation of N-(6-(3-tert-butyl-5-(2,4-dioxo-3,4-dihydropyrimidin-l(2H)-yl)-2- methoxyphenyl)naphthalen-2-yl)methanesulfonamide (compound IB-LO-2.3).
[00768] Part A. Preparation of N-(6-bromonaphthalen-2-yl)methanesulfonamide. [00769] A solution of the product from Example 3, Part B (4.48g, 20.17mmol) in pyridine (10OmL) was treated drop wise with methanesulfonyl chloride (1.97mL, 2.89 g, 25.2mmol) followed by stirring at room temperature for Ih. Diluted with toluene and concentrated under vacuum twice. The residue was extracted with EtOAc and washed with water, IM citric acid and brine. Treated with Darco G-60, dried over Na2SO4, filtered through celite and concentrated under vacuum. Solid was triturated with ether- hexane, collected by filtration and dried under vacuum to give the title compound as a faint pink solid (3.32g, 55 %).
[00770] Part B. Preparation of N-(6-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)naphthalen-2-yl) methanesulfonamide .
[00771] A mixture of the product from Part A (1.0Og, 3.33mmol), bis(pincolato)diboron (1.27g,
5.00mmol), potassium acetate (0.98 g, 9.99mmol) and Combiphos Pd6 (84mg, 0.17mmol) in toluene
(22mL) was heated at reflux for 3h. Cooled and diluted with ethyl acetate and water. The mixture was treated with Darco G-60 and filtered through celite. The filtrate was washed with water and brine. Dried over Na2SO4, filtered and concentrated under vacuum. Oil was dissolved in ether and precipitated by addition of hexanes. The product was collected by filtration and washed with hexanes. Evaporation of the filtrate and purification by silica gel column chromatography eluting with EtOAc/hexanes. The title compound from crystallization and chromatography was obtained as a white solid (927mg, 80%).
Part C. Preparation of tert-butyl 3-tert-butyl-4-methoxy-5-(6-(methylsulfonamido) naphthalen-
2-yl)phenylcarbamate.
Combined the product from Example 3, Part H (87mg, 0.243mmol), the product from Part B
(169mg, 0.486mmol), toluene (1.0ml), ethanol (1.0ml) and sodium carbonate (0.243ml, 0.243mmol) in a sealed tube and de-gassed with N2 gas for 20min. Tetrakis(triphenylphosphine)palladium(0) (5.61mg,
4.86μmol) was added and de-gassing was continued another 5-10 min. Heated at 90-950C for 16h.
Cooled and concentrated under vacuum. Purification by silica gel column chromatography eluting with
EtOAc/hexanes gave the title compound (92.2mg, 76 %).
[00774] Part D. Preparation of N-(6-(3-tert-butyl-5-(2,4-dioxo-3,4-dihydropyrimidin-l(2H)-yl)-2- methoxyphenyl)naphthalen-2-yl)methanesulfonamide.
[00775] A solution of the product from Part C (90mg, 0.180mmol) in CH2Cl2 (2.0ml) was treated with trifluoroacetic acid (1.0ml, 12.98mmol) at room temperature for Ih. Concentrated under vacuum, dissolved residue in EtOAc, washed with 10% NaHCO3, and brine. Dried over Na2SO4, filtered and concentrated under vacuum. Dissolved in DMF (1.4ml) and cooled to -250C and added (E)-3-methoxy- acryloyl isocyanate (0.633ml, 0.361mmol) drop wise while maintaining the temperature below -1O0C. Warmed to room temperature and stirred for 2h. Poured into ether, washed with water, and brine. Dried over Na2SO4, filtered and concentrated under vacuum. Added a mixture OfH2SO4 (0.1ml, 1.876mmol), water (1.0ml) and EtOH (1.0ml) and stirred at 1000C 16h. Cooled and concentrated under vacuum. Poured into water, extracted with EtOAc, combined extracts and washed with brine. Dried over Na2SO4, filtered and concentrated under vacuum. Purification by silica gel column chromatography eluting with MeOH/CHCl3 gave the title compound (53mg, 59%). 1H NMR (300 MHz DMSO-J6) δ 1.42 (s, 9 H) 3.08 (s, 3 H) 3.25 (s, 3 H) 5.65 (d, J=7.72 Hz, 1 H) 7.34 (dd, J=15.81, 2.57 Hz, 2 H) 7.42 (dd, J=8.82, 1.84 Hz, 1 H) 7.65 – 7.76 (m, 2 H) 7.80 (d, J=8.09 Hz, 1 H) 7.96 (t, J= 8.27 Hz, 2 H) 8.02 (s, 1 H) 10.04 (s, 1 H) 11.41 (s, 1 H); MS (ESI+) m/z 494 (M+H)+; (ESI-) m/z 492 (M-H)“.
Example 4B. Preparation of N-(6-(3-tert-butyl-5-(2,4-dioxo-3,4-dihydropyrimidin-l(2H)-yl)-2- methoxyphenyl)naphthalen-2-yl)methanesulfonamide (compound IB-LO-2.3).
Part A. Preparation of 2-tert-butyl-6-iodo-4-nitrophenol.
To the product from Example 3, Part E (4.5g, 23.05mmol) dissolved in MeOH (120ml) and water (3OmL) was added iodine monochloride (1.155ml, 23.05mmol) drop wise over a period of lOmin.
The mixture was stirred for 2h and diluted into IL of water and allowed to stand overnight. The solid material was collected by filtration and washed 3 x 5OmL with water and dried under vacuum overnight to give a tan solid (7.14g, 96%).
Part B. Preparation of l-tert-butyl-3-iodo-2-methoxy-5-nitrobenzene.
To an ice bath cooled solution of the product from Part A (5.5g, 17.13mmol) in MTBE (15ml) in a 5OmL pressure vessel was added 2.0M trimethylsilyl diazomethane (12.85ml, 25.7mmol) followed by drop-wise addition of methanol (1.OmL) resulting in calm bubbling. The vessel was sealed and stirred at room temperature for 16h, cooled and the pressure was released. The solution was partitioned between
EtOAc and water. The organic layer was washed with 1.0M HCl, saturated potassium carbonate solution, and saturated NaCl. The organic layer was dried over sodium sulfate, filtered and concentrated to give a red oil that was used without purification (5.4g, 84%).
Part C. Preparation of 3-tert-butyl-5-iodo-4-methoxyaniline.
[00782] A mixture of the product from Part B (5.80g, 17.31mmol), ammonium chloride (1.389g,
26.0mmol), and iron (4.83g, 87mmol) in THF/MeOH/water (20OmL total, 2/2/1) was refluxed for 2h, cooled and filtered through Celite. The filtrate was evaporated and the residue was partitioned between water and EtOAc. The organic layer was washed with saturated brine, dried with sodium sulfate, filtered and evaporated to give a brown oil (5.28g, 100% yield).
[00783] Part D. Preparation of (E)-N-(3-tert-butyl-5-iodo-4-methoxyphenylcarbamoyl)-3-methoxy acrylamide.
[00784] To a solution of the product from Part C (3.05g, lOmmol) in DMF (50ml) at -20 0C under N2 was added at a fast drip a 0.4M solution in benzene of (E)-3-methoxyacryloyl isocyanate (50.0ml,
20.00mmol, prepared by the method of Santana et al., J. Heterocyclic. Chem. 36:293 (1999). The solution was stirred for 15min at -20 0C, warmed to room temperature for 45min and diluted with EtOAc. The organic was washed with water and brine. Dried over Na2SO4, filtered and concentrated to a brown solid. The residue was triturated in Et2θ/hexane to give a fine powder that was collected by filtration and dried under vacuum to give the title compound as a tan powder (2.46g, 57%).
[00785] Part E. Preparation of l-(3-tert-butyl-5-iodo-4-methoxyphenyl)dihydropyrimidine-2,4(lH,3H)- dione.
[00786] To a suspension of the product from Part D (2.46g, 5.69mmol) in ethanol (50ml) was added a solution of 5.5mL OfH2SO4 in 5OmL water and the mixture was heated at 1100C for 2.5h to give a clear solution. Cooled and diluted with 5OmL of water while stirring to give an off-white solid that was collected by filtration, washed with water and dried under vacuum to give the title compound (2.06g,
90%).
[00787]Part F. Preparation of N-(6-(3-tert-butyl-5-(2,4-dioxo-3,4-dihydropyrimidin-l(2H)-yl)-2- methoxyphenyl)naphthalen-2-yl)methanesulfonamide.
[00788] In a microwave tube, the product from Part E (104mg, 0.26mmol), the product from Example
4A, Part B (108mg, 0.31mmol), and 1.0M sodium carbonate solution (312μL, 0.31mmol) in 1: 1 ethanol- toluene (1.7mL) was degassed by nitrogen sparge for 15min. l,l’-Bis(diphenylphosphino) ferrocene palladium (II) chloride dichloromethane complex (9mg, 0.01 lmmol) was added, and degassing was continued for another 5min. The tube was sealed and heated in the microwave at 1000C for Ih. Diluted with dichloromethane and washed with IM citric acid solution and brine. The organic layer was then stirred with (3-mercaptopropyl) silica gel for Ih. Filtered through celite and concentrated under vacuum.
Triturated with ether, methanol, and then again with ether to give the title compound as a nearly white solid (32mg, 25 %). 1H NMR (300 MHz, DMSO-J6): δ 11.41 (d, J=I.84 Hz, 1 H) 10.04 (s, 1 H) 8.03 (s,
1 H) 7.96 (t, J=8.09 Hz, 2 H) 7.80 (d, J=8.09 Hz, 1 H) 7.63 – 7.79 (m, 2 H) 7.35 – 7.45 (m, 1 H) 7.37 (d,
J=2.57 Hz, 1 H) 7.32 (d, J=2.57 Hz, 1 H) 5.65 (dd, J=8.09, 2.21 Hz, 1 H) 3.25 (s, 3 H) 3.09 (s, 3 H) 1.43
(s, 9 H). MS (+ESI)m/z (rel abundance): 494 (100,M+H), 511 (90, M+NH4), 987 (20, 2M+H), 1009
(8, 2M+Na).
PATENT
http://www.google.com/patents/WO2009039134A1?cl=en
Example 2A. Preparation of N-(6-(3-tert-butyl-5-(2,4-dioxo-3,4-dihydropyrimidin-l(2H)-yl)-2- methoxyphenyl)naphthalen-2-yl)methanesulfonamide (compound IB-LO-2.3).
[00511] Part A. Preparation of N-(6-bromonaphthalen-2-yl)methanesulfonamide. [00512] A solution of the product from Example 1, Part B (4.48g, 20.17mmol) in pyridine (10OmL) was treated drop wise with methanesulfonyl chloride (1.97mL, 2.89 g, 25.2mmol) followed by stirring at room temperature for Ih. Diluted with toluene and concentrated under vacuum twice. The residue was extracted with EtOAc and washed with water, IM citric acid and brine. Treated with Darco G-60, dried over Na2SO4, filtered through celite and concentrated under vacuum. Solid was triturated with ether- hexane, collected by filtration and dried under vacuum to give the title compound as a faint pink solid (3.32g, 55 %).
Part B. Preparation of N-(6-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)naphthalen-2-yl) methanesulfonamide .
[00514] A mixture of the product from Part A (1.0Og, 3.33mmol), bis(pincolato)diboron (1.27g,
5.00mmol), potassium acetate (0.98 g, 9.99mmol) and Combiphos Pd6 (84mg, 0.17mmol) in toluene
(22mL) was heated at reflux for 3h. Cooled and diluted with ethyl acetate and water. The mixture was treated with Darco G-60 and filtered through celite. The filtrate was washed with water and brine. Dried over Na2SO4, filtered and concentrated under vacuum. Oil was dissolved in ether and precipitated by addition of hexanes. The product was collected by filtration and washed with hexanes. Evaporation of the filtrate and purification by silica gel column chromatography eluting with EtOAc/hexanes. The title compound from crystallization and chromatography was obtained as a white solid (927mg, 80%).
[00515] Part C. Preparation of tert-butyl 3-tert-butyl-4-methoxy-5-(6-(methylsulfonamido) naphthalen-
2-yl)phenylcarbamate.
[00516] Combined the product from Example 1, Part H (87mg, 0.243mmol), the product from Part B
(169mg, 0.486mmol), toluene (1.0ml), ethanol (1.0ml) and sodium carbonate (0.243ml, 0.243mmol) in a sealed tube and de-gassed with N2 gas for 20min. Tetrakis(triphenylphosphine)palladium(0) (5.61mg,
4.86μmol) was added and de-gassing was continued another 5-10 min. Heated at 90-950C for 16h.
Cooled and concentrated under vacuum. Purification by silica gel column chromatography eluting with
EtOAc/hexanes gave the title compound (92.2mg, 76 %).
[00517]Part D. Preparation of N-(6-(3-tert-butyl-5-(2,4-dioxo-3,4-dihydropyrimidin-l(2H)-yl)-2- methoxyphenyl)naphthalen-2-yl)methanesulfonamide.
[00518] A solution of the product from Part C (90mg, 0.180mmol) in CH2Cl2 (2.0ml) was treated with trifluoroacetic acid (1.0ml, 12.98mmol) at room temperature for Ih. Concentrated under vacuum, dissolved residue in EtOAc, washed with 10% NaHCO3, and brine. Dried over Na2SO4, filtered and concentrated under vacuum. Dissolved in DMF (1.4ml) and cooled to -250C and added (E)-3-methoxy- acryloyl isocyanate (0.633ml, 0.361mmol) drop wise while maintaining the temperature below -1O0C. Warmed to room temperature and stirred for 2h. Poured into ether, washed with water, and brine. Dried over Na2SO4, filtered and concentrated under vacuum. Added a mixture Of H2SO4 (0.1ml, 1.876mmol), water (1.0ml) and EtOH (1.0ml) and stirred at 1000C 16h. Cooled and concentrated under vacuum. Poured into water, extracted with EtOAc, combined extracts and washed with brine. Dried over Na2SO4, filtered and concentrated under vacuum. Purification by silica gel column chromatography eluting with MeOH/CHCl3 gave the title compound (53mg, 59%). 1H NMR (300 MHz OMSO-d6) δ 1.42 (s, 9 H) 3.08 (s, 3 H) 3.25 (s, 3 H) 5.65 (d, J=7.72 Hz, 1 H) 7.34 (dd, J=15.81, 2.57 Hz, 2 H) 7.42 (dd, J=8.82, 1.84 Hz, 1 H) 7.65 – 7.76 (m, 2 H) 7.80 (d, J=8.09 Hz, 1 H) 7.96 (t, J= 8.27 Hz, 2 H) 8.02 (s, 1 H) 10.04 (s, 1 H) 11.41 (s, 1 H); MS (ESI+) m/z 494 (M+H)+; (ESI-) m/z 492 (M-H)“.
[00519] Example 2B. Preparation of N-(6-(3-tert-butyl-5-(2,4-dioxo-3,4-dihydropyrimidin-l(2H)-yl)-2- methoxyphenyl)naphthalen-2-yl)methanesulfonamide (compound IB-LO-2.3).
[00520] Part A. Preparation of 2-tert-butyl-6-iodo-4-nitrophenol.
[00521] To the product from Example 1, Part E (4.5g, 23.05mmol) dissolved in MeOH (120ml) and water (3OmL) was added iodine monochloride (1.155ml, 23.05mmol) drop wise over a period of lOmin.
The mixture was stirred for 2h and diluted into IL of water and allowed to stand overnight. The solid material was collected by filtration and washed 3 x 5OmL with water and dried under vacuum overnight to give a tan solid (7.14g, 96%).
[00522]Part B. Preparation of l-tert-butyl-3-iodo-2-methoxy-5-nitrobenzene.
[00523] To an ice bath cooled solution of the product from Part A (5.5g, 17.13mmol) in MTBE (15ml) in a 5OmL pressure vessel was added 2.0M trimethylsilyl diazomethane (12.85ml, 25.7mmol) followed by drop-wise addition of methanol (1.OmL) resulting in calm bubbling. The vessel was sealed and stirred at room temperature for 16h, cooled and the pressure was released. The solution was partitioned between
EtOAc and water. The organic layer was washed with 1.0M HCl, saturated potassium carbonate solution, and saturated NaCl. The organic layer was dried over sodium sulfate, filtered and concentrated to give a red oil that was used without purification (5.4g, 84%).
[00524] Part C. Preparation of 3-tert-butyl-5-iodo-4-methoxyaniline.
[00525] A mixture of the product from Part B (5.8Og, 17.31mmol), ammonium chloride (1.389g,
26.0mmol), and iron (4.83g, 87mmol) in THF/MeOH/water (20OmL total, 2/2/1) was refluxed for 2h, cooled and filtered through Celite. The filtrate was evaporated and the residue was partitioned between water and EtOAc. The organic layer was washed with saturated brine, dried with sodium sulfate, filtered and evaporated to give a brown oil (5.28g, 100% yield).
[00526] Part D. Preparation of (E)-N-(3-tert-butyl-5-iodo-4-methoxyphenylcarbamoyl)-3-methoxy acrylamide.
[00527] To a solution of the product from Part C (3.05g, lOmmol) in DMF (50ml) at -20 0C under N2 was added at a fast drip a 0.4M solution in benzene of (E)-3-methoxyacryloyl isocyanate (50.0ml,
20.00mmol, prepared by the method of Santana et al., J. Heterocyclic. Chem. 36:293 (1999). The solution was stirred for 15min at -20 0C, warmed to room temperature for 45min and diluted with EtOAc. The organic was washed with water and brine. Dried over Na2SO4, filtered and concentrated to a brown solid. The residue was triturated in Et2O/hexane to give a fine powder that was collected by filtration and dried under vacuum to give the title compound as a tan powder (2.46g, 57%).
[00528] Part E. Preparation of l-(3-tert-butyl-5-iodo-4-methoxyphenyl)dihydropyrimidine-2,4(lH,3H)- dione.
[00529] To a suspension of the product from Part D (2.46g, 5.69mmol) in ethanol (50ml) was added a solution of 5.5mL OfH2SO4 in 5OmL water and the mixture was heated at 110°C for 2.5h to give a clear solution. Cooled and diluted with 5OmL of water while stirring to give an off-white solid that was collected by filtration, washed with water and dried under vacuum to give the title compound (2.06g,
[00530] Part F. Preparation of N-(6-(3-tert-butyl-5-(2,4-dioxo-3,4-dihydropyrimidin-l(2H)-yl)-2- methoxyphenyl)naphthalen-2-yl)methanesulfonamide.
[0053I]In a microwave tube, the product from Part E (104mg, 0.26mmol), the product from Example 2A, Part B (108mg, OJ lmmol), and 1.0M sodium carbonate solution (312μL, 0.31mmol) in 1:1 ethanol- toluene ( 1.7mL) was degassed by nitrogen sparge for 15min. 1 , 1 ‘-Bis(diphenylphosphino) ferrocene palladium (II) chloride dichloromethane complex (9mg, O.Ol lmmol) was added, and degassing was continued for another 5min. The tube was sealed and heated in the microwave at 1000C for Ih. Diluted with dichloromethane and washed with IM citric acid solution and brine. The organic layer was then stirred with (3-mercaptopropyl) silica gel for Ih. Filtered through celite and concentrated under vacuum. Triturated with ether, methanol, and then again with ether to give the title compound as a nearly white solid (32mg, 25 %). 1H NMR (300 MHz, OMSO-d6): δ 11.41 (d, J=1.84 Hz, 1 H) 10.04 (s, 1 H) 8.03 (s, 1 H) 7.96 (t, J=8.09 Hz, 2 H) 7.80 (d, J=8.09 Hz, 1 H) 7.63 – 7.79 (m, 2 H) 7.35 – 7.45 (m, 1 H) 7.37 (d, J=2.57 Hz, 1 H) 7.32 (d, J=2.57 Hz, 1 H) 5.65 (dd, J=8.09, 2.21 Hz, 1 H) 3.25 (s, 3 H) 3.09 (s, 3 H) 1.43 (s, 9 H). MS (+ESI) m/z (rel abundance): 494 (100, M+H), 511 (90, M+NH4), 987 (20, 2M+H), 1009 (8, 2M+Na).
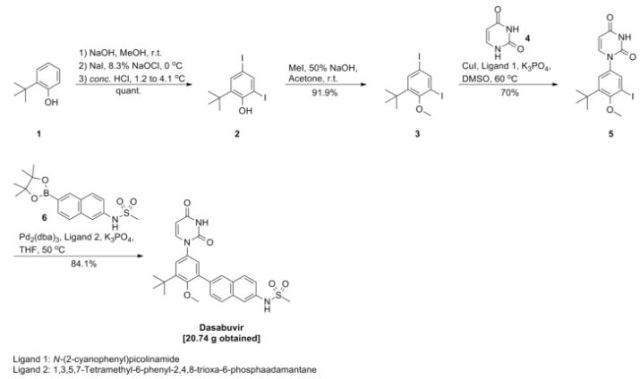
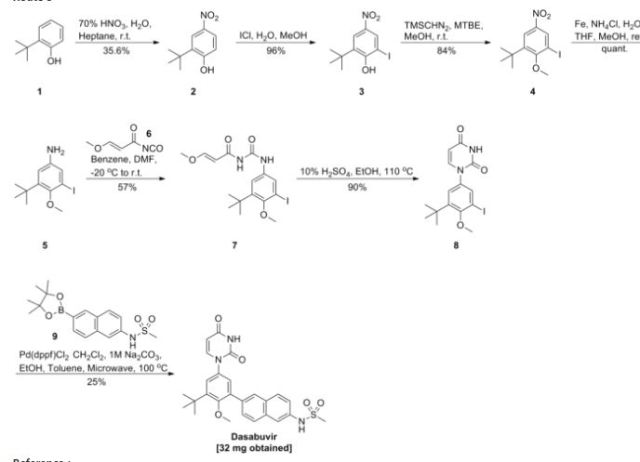
PATENT
WO 2014031791
https://www.google.com/patents/WO2014031791A1?cl=en
Example 4. Preparation of ^-(6-(3-?eri-butyl-5-(2,4-dioxo-3,4-dihydropyrimidin-l(2H)-yl)-2- methoxyphenyl)naphthalen-2-yl)methanesulfonamide (compound (A-l)).

[00254] A 3-L, 3 -neck, round-bottom flask was equipped with an overhead stirrer, a thermocouple, a Claisen condenser and a reflux condenser. Tris(dibenzylideneacetone)dipalladium(0) (0.330 g, 0.360 mmol), di-?er?-butyl(2′,4′,6′-triisopropyl-3,4,5,6-tetramethylbiphenyl-2-yl)phosphine (0.416 g, 0.864 mmol) and milled potassium phosphate tribasic (21.0 g, 99.0 mmol) were charged to the 3-L flask. The flask was purged with argon for not less than 90 minutes with constant stirring of the solids. i-Amyl alcohol (250 ml) was charged to a separate 500-mL round-bottom flask and was purged with argon for not less than 30 minutes and was transferred to the 3-L flask using a cannula under argon atmosphere. The contents of the 3-L flask were heated to 80 °C and stirred at this temperature for 30 minutes. A 1-L round-bottom flask equipped with a magnetic stir bar was charged with 6-(3-teri-butyl-5-(2,4-dioxo-3,4- dihydropyrimidin- 1 (2H)-yl)-2-methoxyphenyl)naphthalen-2-yl 1 , 1 ,2,2,3,3,4,4,4-nonafluorobutane- 1 – sulfonate (62.9 g, 90 mmol), methanesulfonamide (12.85 g, 135 mmol) and i-amyl alcohol (505 mL), purged with argon and heated to 60 °C. The reagent mixture was stirred under argon for not less than 30 minutes. A clear yellow solution was observed. This solution was transferred to the 3-L flask using a cannula under argon atmosphere. The temperature of the 3-L flask was raised to 85 °C and the contents were stirred for 14 hours under a positive pressure of argon. The temperature was then raised to 95 °C and the contents were stirred for an additional 4 hours under a positive pressure of argon. The reaction mixture was allowed to cool down to room temperature, diluted with tetrahydrofuran (2200 mL) and water (800 mL) and was transferred to a 6-L separatory funnel. The organic layer was washed thrice with water (2000 mL) containing L-cysteine (17.3 g) and NaCl (235 g). The organic layer was collected, filtered through a pad of diatomaceous earth and was concentrated in vacuo to approximately 250 mL. Ethyl acetate (775 mL) was added over 7 hours with stirring, and the mixture was allowed to stir for an additional 14 hours. White solid was isolated by filtration, and the solid was washed with ethyl acetate (1000 mL). The solid was then dissolved in tetrahydrofuran (1500 mL) and filtered through a pad of diatomaceous earth to obtain a clear solution. The diatomaceous earth was washed with tetrahydrofuran (300 mL). The combined tetrahydrofuran solution was concentrated in vacuo to approximately 250 mL, and then ethyl acetate (775 mL) was added over 7 hours with stirring. The product solution was allowed to stir for an additional 14 hours. White solid was isolated by filtration. The solid was washed with ethyl acetate (1000 mL) and dried in a vacuum oven at 60 °C for 24 hours. The solid was slurried in 308 mL of 200 proof ethanol for 1.5 hours, then isolated by filtration. The solid was washed with 132 mL of 200 proof ethanol and dried in a vacuum oven at 50 °C for 18 hours. The title compound was isolated as a white solid (32.6 g, 100% potency vs. standard, 73% yield). !H NMR (400 MHz, DMSO-i¾) δ ppm 1 1.41 (d, J= 2.1 Hz, 1H), 10.04 (s, 1H), 8.02 (d, J= 0.9 Hz, 1H), 7.98 – 7.91 (m, 2H), 7.79 (d, J = 7.9 Hz, 1H), 7.72 (d, J= 2.0 Hz, 1H), 7.69 (dd, J = 8.5, 1.7 Hz, 1H), 7.41 (dd, J = 8.8, 2.2 Hz, 1H), 7.36 (d, J= 2.7 Hz, 1H), 7.31 (d, J= 2.7 Hz, 1H), 5.65 (dd, J = 7.9, 2.2 Hz, 1H), 3.24 (s, 3H), 3.08 (s, 3H), 1.42 (s, 9H). 13C NMR (101 MHz, DMSO-i¾) δ ppm 163.1 (C), 156.0 (C), 150.0 (C), 145.3 (CH), 142.9 (C), 136.0 (C), 134.3 (C), 134.2 C(), 133.5 (C), 132.2 (C), 129.5 (C), 129.0 (CH), 127.6 (CH), 127.1 (CH), 127.0 (CH), 126.5 (CH), 124.3 (CH), 120.2 (CH), 1 14.5 (CH), 101.1 (CH), 60.3 (CH3), 39.4 (CH3), 35.1(C), 30.5 (CH3).
Example 5. Preparation of the sodium salt of /V-(6-(3-teri-butyl-5-(2,4-dioxo-3,4- dihydropyrimidin- 1 (2H)-yl)-2-methoxyphenyl)naphthalen-2-yl)methanesulfonamide (compound (A-sl)).
[00286] A solution of 2-propanol and water was prepared by combining 18.5 g of water and 512 g of 2- propanol. Hereafter, this solution is referred to as the “antisolvent solution.”
[00287] A solution of 2-propanol and water was prepared by combining 23.94 g of water and 564 g of 2- propanol. This solution was cooled in a refrigerator prior to use. Hereafter, this solution is referred to as the “chilled wash solution.”
[00288] A jacketed reactor was equipped with an overhead stirrer and charged with 32.0 g (64.8 mmol) of A^-(6-(3-?er^butyl-5-(2,4-dioxo-3,4-dihydropyrimidin-l(2H)-yl)-2-methoxyphenyl)naphthalen-2- yl)methanesulfonamide and 105.9 g of dimethyl sulfoxide. With stirring the mixture was heated to an internal temperature of 68 °C. A solution of 2.66 g of sodium hydroxide (66.5 mmol, 1.026 equiv) in 16 g of water was added to the reactor over several minutes, followed by 12.4 g of 2-propanol while maintaining the internal temperature at 68 °C. Antisolvent solution (24.5 g) was added to the reactor while maintaining the internal temperature at 68 °C. A slurry of 0.32 g of seed crystals of the final product in 22.8 g of antisolvent solution was added to the reactor, followed by a 2.6 g rinse of the flask with antisolvent solution. The reaction mixture was stirred for 1.5 hours while maintaining the internal temperature at 68 °C. Antisolvent solution (354 g) was added to the reactor over 7 hours while maintaining the internal temperature at 68 °C. The contents of the reactor were cooled to an internal temperature of 0 °C over 7 hours and then mixed at 0 °C for 7 hours. The solids were isolated by filtration and washed with 252 g of the chilled wash solution. The isolated solids were dried in a vacuum oven at 50 °C for 19 hours. The title compound was isolated as a white solid (30.7 g, 92% potency vs. free acid standard, 57.2 mmol free acid equivalent, 88% yield). !H NMR (400 MHz, DMSO-i¾) δ ppm 7.75 (s, 1H), 7.72 (d, J= 7.8 Hz, 1H), 7.59 (dd, J = 8.8, 2.2 Hz, 2H), 7.45 (dd, J = 8.5, 1.8 Hz, 1H), 7.27 (d, J = 2.6 Hz, 2H), 7.21 (d, J = 2.7 Hz, 1H), 7.06 (dd, J = 8.8, 2.2 Hz, 1H), 5.62 (d, J = 7.8 Hz, 1H), 3.24 (s, 3H), 2.68 (s, 3H), 1.40 (s, 9H).
PATENT
http://www.google.com/patents/EP2593439A2?cl=en
Example 2. Preparation of l -(3-teri-butyl-5-(6-hydroxynaphthalen-2-yl)-4- methoxyphenyl)pyrimidine-2,4(l f,3H)-dione (compound (4)).
[00165] This reaction is sensitive to oxygen, and so all vessels were sealed with rubber septa. All solution transfers were accomplished by cannula technique using nitrogen as the inert gas. Anhydrous tetrahydrofuran was sparged with nitrogen gas for 2 hours prior to use to render it anaerobic. Hereafter this is referred to as degassed tetrahydrofuran. [00166] A 100-mL round-bottom flask was charged with 12.9 g of potassium phosphate tribasic (60.8 mmol, 2.0 equivalents), a magnetic stir bar, and 60 mL of water. The mixture was stirred to dissolve the solids, and the aqueous solution was sparged with nitrogen gas for 2 hours prior to use. Hereafter this is referred to as the phosphate solution.
[00167] A 100-mL round-bottom flask was purged with nitrogen gas and charged with 282 mg of tris(dibenzylideneacetone)dipalladium(0) (0.31 mmol, 0.02 equivalents Pd), 413 mg ofphosphine ligand, l ,3,5,7-tetramethyl-8-phenyl-2,4,6-trioxa-8-phosphatricyclo[3.3.1.1 ‘7]decane (1.4 mmol, 2.3 equivalents relative to Pd) and a magnetic stir bar. The flask was sealed with a septum and the atmosphere above the solids was purged with nitrogen gas. Sixty mL of degassed tetrahydrofuran was added to the flask and the mixture was stirred under a nitrogen atmosphere. This solution was sparged for 15 minutes prior to use and is hereafter referred to as the catalyst solution.
[00168] A 500-mL jacketed reactor was equipped with an overhead stirrer and reflux condenser and the atmosphere was purged with nitrogen gas. The reactor was charged with 12.1 g of l -(3-?er?-butyl-5-iodo- 4-methoxyphenyl)pyrimidine-2,4(l f,3 /)-dione, (30.3 mmol, 1.0 equivalent) and 5.98 g of 6- hydroxynaphthalen-2-ylboronic acid (31.8 mmol, 1.05 equivalents). The atmosphere was purged with nitrogen gas with stirring of the solid reagents for 20 minutes. The reactor was charged with 120 mL of degassed tetrahydrofuran, and the mixture was stirred to dissolve the solids. The solution was sparged with nitrogen gas for 10 minutes. The phosphate solution was added to the reactor by cannula, followed by the catalyst solution. The resulting biphasic mixture was stirred aggressively to ensure adequate phase mixing, and the jacket was warmed to 65 °C. The reaction jacket was cooled to room temperature prior to quench.
[00169] After 2.5 hours, the reaction jacket was cooled to room temperature prior to quench.
[00170] The workup of the reaction was also conducted under anaerobic conditions. Fifty-seven grams of sodium chloride and 4.2 g of cysteine (15 weight equivalents relative to palladium catalyst) were dissolved in 300 mL of water, and the resulting solution was sparged for 2 hours prior to use. To quench the reaction, approximately 1/3 of this solution was transferred to the reaction mixture by cannula under nitrogen gas and the resulting biphasic mixture was stirred vigorously for 2 hours. The mechanical agitation was halted, the two solutions were allowed to separate, and the aqueous solution was drained out of the reactor through the bottom valve. Approximately 1/3 of the quench solution was transferred to the reaction mixture by cannula under nitrogen gas and the resulting biphasic mixture was stirred vigorously for 45 minutes. The mechanical agitation was halted, the two solutions were allowed to separate, and the aqueous solution was drained out of the reactor through the bottom valve. The final portion of the quench solution was transferred to the reaction mixture by cannula, the resulting biphasic mixture was stirred vigorously for 45 minutes and the aqueous solution was drained out of the reactor through the bottom valve. [00171] The remainder of the workup was not conducted under anaerobic conditions. The pale yellow organic solution was drained from the reactor through the bottom valve and filtered over a pad of grade 4 Filtrol® (1 cm deep by 4.5 cm diameter). The reactor and filter cake were rinsed with 70 mL of tetrahydrofuran. The bulk of the solvent was distilled in vacuo (ca 90-130 torr) at ca 40 °C with good agitation from an overhead stirrer. The solution was concentrated to approximately 50 mL volume, during which time the product began to precipitate out. Ethyl acetate (100 mL, 8 volume/weight relative to product) was added to the mixture, and the resultant slurry was stirred overnight at room temperature. The crystalline material was isolated by filtration and the filter cake was washed twice with 20 mL portions of ethyl acetate. The wet-cake was air-dried on the filter and dried in a vacuum oven at 50 °C at approximately 250 torr with a gentle nitrogen sweep overnight.
[00172] The desired product was isolated as a white solid (11.6 g, 96.4% potency vs. standard, 88% potency-adjusted yield).!H NMR (400 MHz, DMSO-4) δ ppm δ 1 1.39 (d, J = 2.1 Hz, 1H), 9.82 (s, 1H), 7.91 (d, J = 0.8 Hz, 1H), 7.80 (d, J= 8.9 Hz, 1H), 7.77 – 7.74 (m, 2H), 7.58 (dd, J = 8.5, 1.7 Hz, 1H), 7.32 (d, J = 2.7 Hz, 1H), 7.27 (d, J= 2.7 Hz, 1H), 7.16 (d, J = 2.3 Hz, 1H), 7.10 (dd, J = 8.8, 2.4 Hz, 1H), 5.64 (dd, J = 7.9, 2.2 Hz, 1H), 3.23 (s, 3H), 1.41 (s, 9H).
Example 3. Preparation of 6-(3-?eri-butyl-5-(2,4-dioxo-3,4-dihydropyrimidin-l (2H)-yl)-2- methoxyphenyl)naphthalen-2-yl 1 ,1 ,2,2,3,3,4,4,4-nonafluorobutane-l -sulfonate (compound (5a)).
[00174] A reactor was equipped with an overhead stirrer in the central neck and charged with 45.0 g of 1- (3-?eri-butyl-5-(6-hydroxynaphthalen-2-yl)-4-methoxyphenyl)pyrimidine-2,4(l f,3H)-dione (97.8 weight%>, 106 mmol, 1.0 equivalent) and 21.9 g of 325 mesh potassium carbonate (159 mmol, 1.5 equivalents). The atmosphere was purged with nitrogen gas while the solids were stirred. The flask was charged with 445 mL of Λ^Λ^-dimethylformamide, and the slurry was stirred to dissolve the l-(3-?eri- butyl-5-(6-hydroxynaphthalen-2-yl)-4-methoxyphenyl)pyrimidine-2,4(l f,3H)-dione. The purge was stopped and the reaction was conducted under a slight positive pressure of nitrogen gas.
Perfluorobutanesulfonyl fluoride (35.2 g, 117 mmol, 1.1 equivalents) was added in one portion, and the mixture was stirred vigorously to mix the immiscible liquids overnight.
[00175] The inorganic solids were separated by filtration, and the flask and filter cake were rinsed with approximately 30 mL of Λ^,Λ^-dimethylformamide. The Λ^,Λ^-dimethylformamide solution was filtered directly into a second flask with an overhead stirrer. With stirring, 1 12 g of water (25 weight% of total Λ^,Λ^-dimethylformamide employed) was added to the Λ^,Λ^-dimethylformamide solution of product over approximately 0.5 hour to induce precipitation of the desired product, and the mixture was allowed to stir for 5 hours. The wet-cake was isolated by filtration with recirculation of the liquors to recover all the solids. The wet-cake was washed with 60 mL of 25% (v/v) water yV-dimethylformamide, then 85 mL water.
[00176] The solids were dissolved in 760 mL of isopropyl acetate. The resultant organic solution was washed once with 200 mL of water, twice with 270 mL portions of water and once with 200 mL of water to remove residual AyV-dimethylformamide. Solvent was removed by distillation at approximately 130 torr with heating to 55 °C until the total volume was approximately 200 mL. With efficient stirring, heptane (450 mL) was added to the warm (55 °C) slurry. The slurry was allowed to cool to room temperature overnight with stirring. The desired product was isolated by filtration, with recycling of the liquors to isolate all of the solids material. The wet-cake was washed twice with 100 mL portions of 20% (v/v) isopropyl acetate/heptane. The wet-cake was air-dried on the filter and dried in a vacuum oven at 50 °C at approximately 250 torr with a gentle nitrogen sweep overnight. The title compound was isolated as a white solid (64.0 g, 100% potency vs. standard, 87% yield). !H NMR (600 MHz, DMSO- d6) δ ppm 1 1.42 (s, 1H), 8.21 – 8.15 (m, 4H), 7.84 (dd, J = 8.6, 1.7 Hz, 1H), 7.77 (d, J = 7.9 Hz, 1H), 7.60 (dd, J = 9.0, 2.5 Hz, 1H), 7.39 (d, J = 2.7 Hz, 1H), 7.35 (d, J = 2.7 Hz, 1H), 5.66 (d, J = 7.9 Hz, 1H), 3.21 (s, 3H), 1.41 (s, 9H).
[00177] Example 3-1. Alternative Preparation of 6-(3-teri-butyl-5-(2,4-dioxo-3,4-dihydropyrimidin- 1 (2//)-yl)-2-methoxyphenyl)naphthalen-2-yl 1 , 1 ,2,2,3 ,3 ,4,4,4-nonafluorobutane- 1 -sulfonate (compound (5a)).
[00178] A 250-L, 3-neck round-bottom flask equipped with an overhead stirrer was charged with 10 g of 1 -(3-ier?-butyl-5-(6-hydroxynaphthalen-2-yl)-4-methoxyphenyl)pyrimidine-2,4( l//,3//)-dione (98 wt%>, 23.5 mmol, 1.0 equiv) and 6.5 g of milled potassium carbonate (325 mesh, 47.1 mmol, 2.0 equiv). Acetonitrile (MeCN, 60 mL, 6 volumes with respect to naphthol) and dimethylformamide (dimethylformide, 40 mL, 4 volumes with respect to naphthol) was charged to the reactor and the slurry was stirred. Perfluorobutanesulfonyl fluoride (96 wt%>, 8.3 g, 26 mmol, 1.1 equiv) was charged to the well-stirred mixture over 60 minutes by syringe pump. A trace (<0.1 area%) of starting material was detected by HPLC analysis of an aliquot at 20 minutes reaction time. The
acetonitrile/dimethylformamide solution was filtered over a coarse fritted funnel to separate the inorganic solids, and the flask and filter was rinsed with 15 mL of 3 :2 (v/v)
acetonitrile/dimethylformamide. The total mass of solvents employed was approximately 92 g.
[00179] First crystallization: The acetonitrile/dimethylformamide solution was transferred to a 3- neck flask equipped with an overhead stirrer. Water (50 g, 54 wt%> with respect to total solution charged) was added to the well-stirred solution over 100 minutes. This adjusts the solvent
composition to 35 wt% water. During the addition of water the mixture self-seeded, and the solution was held for approximately 1 hour after complete addition of water. The solids were isolated by filtration, and the wetcake was washed with two 30 mL portions of a rinse solution of 40 wt%
water/27 wt% dimethylformamide/33 wt% acetonitrile and then once with 40 mL of water.
[00180] Aqueous washing: A 500-L jacketed cylindrical reactor equipped with an overhead stirrer and Teflon baffle to aid in vertical mixing was charged with the wetcake and 133 g of ethyl acetate (8X theoretical mass of product, 150 mL). The mixture was stirred to dissolve the substrate and the solution was washed twice with 40 mL portions of water.
[00181] Concentration and crystallization: A constant-volume distillation was conducted with heptanes, in vacuo (ca 100 mmHg, jacket temperature of 50 °C), to adjust the solvent composition to approximately 12 wt% ethyl acetate/88 wt% heptanes. During the distillation, solids begin to crystallize out of the solution. Once the distillation was complete, the solution was cooled to ambient temperature (23 °C). The solids were isolated by filtration and the wet cake was washed with a 50-mL portions heptane. The wet cake was dried to give the final product (14.0 g). The solids were 98.1% pure by HPLC analysis and 100% potent vs. reference standard, for an isolated yield of 85%o.
[00182] Example 4. Preparation of ^-(6-(3-ier?-butyl-5-(2,4-dioxo-3,4-dihydropyrimidin-l(2H)-yl)-2- methoxyphenyl)naphthalen-2-yl)methanesulfonamide (compound (A)).

[00183] A 3-L, 3-neck, round-bottom flask was equipped with an overhead stirrer, a thermocouple, a Claisen condenser and a reflux condenser. Tris(dibenzylideneacetone)dipalladium(0) (0.330 g, 0.360 mmol), di-ier?-butyl(2′,4′,6′-triisopropyl-3,4,5,6-tetramethylbiphenyl-2-yl)phosphine (0.416 g, 0.864 mmol) and milled potassium phosphate tribasic (21.0 g, 99.0 mmol) were charged to the 3-L flask. The flask was purged with argon for not less than 90 minutes with constant stirring of the solids. i-Amyl alcohol (250 ml) was purged with argon for not less than 30 minutes and was transferred to the 3-L flask using a cannula under argon atmosphere. The contents of the 3-L flask were heated to 80 °C and stirred at this temperature for 30 minutes. A 1-L round bottom flask equipped with a magnetic stir bar was charged with 6-(3-ier?-butyl-5-(2,4-dioxo-3,4-dihydropyrimidin-l(2H)-yl)-2-methoxyphenyl)naphthalen-2-yl 1,1,2,2,3,3,4,4,4-nonafluorobutane-l-sulfonate (62.9 g, 90 mmol), methanesulfonamide (12.85 g, 135 mmol) and i-amyl alcohol (505 mL), purged with argon and heated to 60 °C. The reaction mixture was stirred under argon for not less than 30 minutes. A clear yellow solution was observed. This solution was transferred to the 3-L flask using a cannula under argon atmosphere. The temperature of the 3-L flask was raised to 85 °C and the contents were stirred for 14 hours under a positive pressure of argon. The temperature was then raised to 95 °C and the contents were stirred for an additional 4 hours under a positive pressure of argon. The reaction mixture was allowed to cool down to room temperature, diluted with tetrahydrofuran (2200 mL) and water (800 mL) and was transferred to a 6-L separatory funnel. The organic layer was washed thrice with water (2000 mL) containing L-cysteine (17.3 g) and NaCl (235 g). The organic layer was collected, filtered through a pad of diatomaceous earth and was concentrated in vacuo to approximately 250 mL. Ethyl acetate (775 mL) was added over 7 hours with stirring, and the mixture was allowed to stir for an additional 14 hours. White solid was isolated by filtration, and the solid was washed with ethyl acetate (1000 mL). The solid was dissolved in tetrahydrofuran (1500 mL) and filtered through a pad of diatomaceous earth to obtain a clear solution. The diatomaceous earth was washed with tetrahydrofuran (300 mL). The combined tetrahydrofuran solution was concentrated in vacuo to approximately 250 mL, and then ethyl acetate (775 mL) was added over 7 hours with stirring. The product solution was allowed to stir for an additional 14 hours. White solid was isolated by filtration. The solid was washed with ethyl acetate (1000 mL) and dried in a vacuum oven at 60 °C for 24 hours. The solid was slurried in 308 mL of 200 proof ethanol for 1.5 hours, then isolated by filtration. The solid was washed with 132 mL of 200 proof ethanol and dried in a vacuum oven at 50 °C for 18 hours. The title compound was isolated as a white solid (32.6 g, 100% potency vs. standard, 73% yield). !H NMR (400 MHz, DMSO-i/6) δ ppm 11.41 (d, J = 2.1 Hz, 1H), 10.04 (s, 1H), 8.02 (d, J = 0.9 Hz, 1H), 7.98 – 7.91 (m, 2H), 7.79 (d, J = 7.9 Hz, 1H), 7.72 (d, J = 2.0 Hz, 1H), 7.69 (dd, J = 8.5, 1.7 Hz, 1H), 7.41 (dd, J = 8.8, 2.2 Hz, 1H), 7.36 (d, J = 2.7 Hz, 1H), 7.31 (d, J = 2.7 Hz, 1H), 5.65 (dd, J = 7.9, 2.2 Hz, 1H), 3.24 (s, 3H), 3.08 (s, 3H), 1.42 (s, 9H).
[00184] Other ligands such as 2,2,7,7-tetramethyl-l-(2′,4′,6′-triisopropylbiphenyl-2-yl)phosphepane; 7,7,9,9-tetramethyl-8-(2′,4′,6′-triisopropyl-3,6-dimethoxybiphenyl-2-yl)-l,4-dioxa-8- phosphaspiro[4.5]decane; and 8-(2-(2-methoxynaphthalen- 1 -yl)phenyl)-7,7,9,9-tetramethyl- 1 ,4-dioxa-8- phosphaspiro[4.5]decane were tested under the conditions described above and produced favorable yields of greater than 50% of the sulfonamidated product.
PREPN OF SODIUM SALT
Example 5. Preparation of the sodium salt of V-(6-(3-teri-butyl-5-(2,4-dioxo-3,4- dihydropyrimidin- 1 (2H)-yl)-2-methoxyphenyl)naphthalen-2-yl)methanesulfonamide (compound (As)).
[00192] A solution of 2-propanol and water was prepared by combining 18.5 g of water and 512 g of 2- propanol. Hereafter, this solution is referred to as the “antisolvent solution.”
[00193] A solution of 2-propanol and water was prepared by combining 23.94 g of water and 564 g of 2- propanol. This solution was cooled in a refrigerator prior to use. Hereafter, this solution is referred to as the “chilled wash solution.”
[00194] A jacketed reactor was equipped with an overhead stirrer and charged with 32.0 g (64.8 mmol) of A^-(6-(3-?er^butyl-5-(2,4-dioxo-3,4-dihydropyrimidin-l(2H)-yl)-2-methoxyphenyl)naphthalen-2- yl)methanesulfonamide and 105.9 g of dimethyl sulfoxide. With stirring the mixture was heated to an internal temperature of 68 °C. A solution of 2.66 g of sodium hydroxide (66.5 mmol, 1.026 equiv) in 16 g of water was added to the reactor over several minutes, followed by 12.4 g of 2-propanol while maintaining the internal temperature at 68 °C. Antisolvent solution (24.5 g) was added to the reactor while maintaining the internal temperature at 68 °C. A slurry of 0.32 g of seed crystals of the final product in 22.8 g of antisolvent solution was added to the reactor, followed by a 2.6 g rinse of the flask with antisolvent solution. The reaction mixture was stirred for 1.5 hours while maintaining the internal temperature at 68 °C. Antisolvent solution (354 g) was added to the reactor over 7 hours while maintaining the internal temperature at 68 °C. The contents of the reactor were cooled to an internal temperature of 0 °C over 7 hours and then mixed at 0 °C for 7 hours. The solids were isolated by filtration and washed with 252 g of the chilled wash solution. The isolated solids were dried in a vacuum oven at 50 °C for 19 hours. The title compound was isolated as a white solid (30.7 g, 92% potency vs. free acid standard, 57.2 mmol free acid equivalent, 88% yield). !H NMR (400 MHz, DMSO-i¾) δ ppm 7.75 (s, 1H), 7.72 (d, J= 7.8 Hz, 1H), 7.59 (dd, J= 8.8, 2.2 Hz, 2H), 7.45 (dd, J= 8.5, 1.8 Hz, 1H), 7.27 (d, J = 2.6 Hz, 2H), 7.21 (d, J= 2.7 Hz, 1H), 7.06 (dd, J= 8.8, 2.2 Hz, 1H), 5.62 (d, J= 7.8 Hz, 1H), 3.24 (s, 3H), 2.68 (s, 3H), 1.40 (s, 9H).
CLIP
DASABUVIR, ABT 333,

CLIP
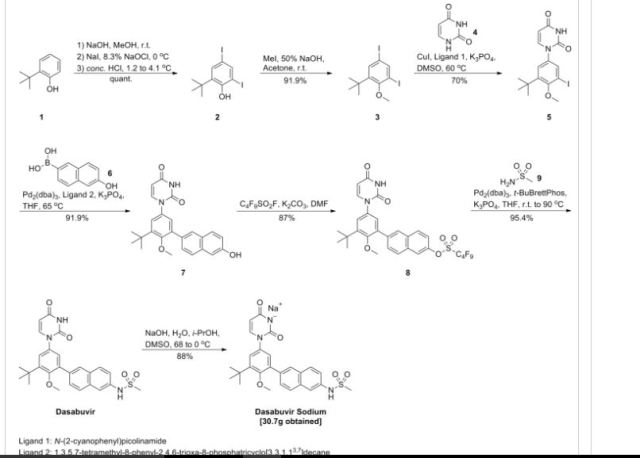
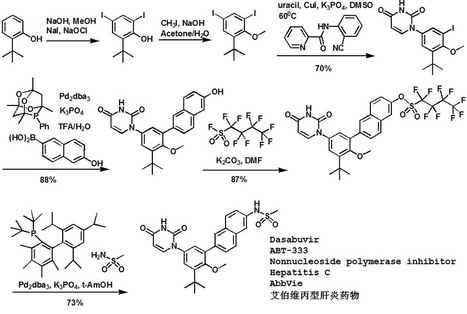
Dasabuvir sodium (Exviera) Dasabuvir sodium (Exviera), an oral non-nucleoside NS5B polymerase inhibitor discovered and developed by Abbvie, is a component of an all-oral hepatitis C treatment regimen Viekira Pak approved by the US FDA in December 2014 for the treatment of adult patients with chronic genotype 1 (GT1) hepatitis C virus (HCV) infection.84 The investigational regimen consists of the fixed-dose combination of paritaprevir (XXVII) (veruprevir, ABT- 450, vide infra) with ritonavir booster (150/100 mg) co-formulated with the NS5A inhibitor ombitasvir (XXV) (ABT-267, vide infra)25 mg, dosed once daily, and nonnucleoside NS5B polymerase inhibitor dasabuvir (X) (ABT-333) 250 mg with or without ribavirin (weight-based), dosed twice daily.85,86 The drug was granted breakthrough therapy designation by the US FDA in May 2013. AbbVie’s application is supported by the data from six Phase III studies covering over 2300 patients in 25 countries representing one of the largest clinical programs in hepatitis C research and development.87 Across six studies, the 12-week therapeutic regimen achieved impressive cure rates, notching a 99% sustained virologic response mark in some populations.88 Although several syntheses of dasabuvir sodium (X) have been disclosed,89–91 the most likely scale approach is outlined in Scheme 12.91 Commercially available 2-tert-butyl phenol (65) was polyiodinated to furnish diiodophenol 66 in 93% yield. Thiswas followed by methylation of the phenol to provide methyl phenyl ether 67 in 99% yield. Next, sequential couplings were employed to install the periphery about the central phenyl core. First, Goldberg coupling of 67 with pyrimidine-2,4-(1H,3H)-dione 68 in presence of CuI (10 mol %) and 69 provided compound 70 in 70% yield. Subsequently, the remaining iodide underwent Suzuki coupling with boronic acid 71 in the presence of Pd2(dba)3 and 72 to yield the naphthol 73 in high yield. Naphthol 73 was then converted to the corresponding polyfluorinated naphthol sulfonate 75, which was subsequently converted to dasabuvir through a palladium- mediated installation of methyl sulfonamide 76. Dasabuvir sodium (X) was then crystallized upon treatment with aq NaOHin i-PrOH and DMSO in 88% yield.


84. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm427530.htm.
85. Poordad, F.; Lawitz, E.; Kowdley, K. V.; Cohen, D. E.; Podsadecki, T.; Siggelkow,S.; Heckaman, M.; Larsen, L.; Menon, R.; Koev, G.; Tripathi, R.; Pilot-Matias, T.;
Bernstein, B. N. Engl. J. Med. 2013, 368, 45.
86. Kowdley, K. V.; Lawitz, E.; Poordad, F.; Cohen, D. E.; Nelson, D. R.; Zeuzem, S.;Everson, G. T.; Kwo, P.; Foster, G. R.; Sulkowski, M. S.; Xie, W.; Pilot-Matias, T.;Liossis, G.; Larsen, L.; Khatri, A.; Podsadecki, T.; Bernstein, B. N. Engl. J. Med.2014, 370, 222.
87. http://www.marketwatch.com/story/abbvie-submits-new-drug-applicationto-us-fda-for-its-investigational-all-oral-interferon-free-therapy-for-thetreatment-of-hepatitis-c-2014-04-22.
88. Feld, J. J.; Kowdley, K. V.; Coakley, E.; Sigal, S.; Nelson, D. R.; Crawford, D.;Weiland, O.; Aguilar, H.; Xiong, J.; Pilot-Matias, T.; DaSilva-Tillmann, B.;Larsen, L.; Podsadecki, T.; Bernstein, B. N. Engl. J. Med. 2014, 370, 1594.
89. Flentge, C., A.;; Hutchinson, D. K.; Betebenner, D. A.; Degoey, D. A.; Donner, P.L.; Kati, W. M.; Krueger, A. C.; Liu, D. C.; Liu, Y.; Longenecker, K. L.; Maring, C.J.; Motter, C. E.; Pratt, J. K.;
PATENT
http://www.google.com/patents/US20130224149
Example 2 Preparation of 1-(3-tert-butyl-5-(6-hydroxynaphthalen-2-yl)-4-methoxyphenyl)pyrimidine-2,4(1H,3H)-dione (compound (4a))
This reaction is sensitive to oxygen, and care was taken to establish and maintain an inert atmosphere in the handling and use of air-sensitive materials or mixtures. All solution transfers were accomplished by cannula technique using nitrogen as the inert gas. Anhydrous tetrahydrofuran was sparged with nitrogen gas for 2 hours prior to use to render it anaerobic. Hereafter this is referred to as degassed tetrahydrofuran.
A 100-mL round-bottom flask was charged with 12.9 g of potassium phosphate tribasic (60.8 mmol, 2.0 equivalents), a magnetic stir bar, and 60 mL of water. The mixture was stirred to dissolve the solids, and the aqueous solution was sparged with nitrogen gas for 2 hours prior to use. Hereafter this is referred to as the phosphate solution.
A 100-mL round-bottom flask was purged with nitrogen gas and charged with 282 mg of tris(dibenzylideneacetone)dipalladium(0) (0.31 mmol, 0.02 equivalents Pd), 413 mg of phosphine ligand, 1,3,5,7-tetramethyl-8-phenyl-2,4,6-trioxa-8-phosphatricyclo[3.3.1.13,7]decane (1.4 mmol, 2.3 equivalents relative to Pd) and a magnetic stir bar. The flask was sealed with a septum and the atmosphere above the solids was purged with nitrogen gas. Sixty mL of degassed tetrahydrofuran was added to the flask and the mixture was stirred under a nitrogen atmosphere. This solution was sparged with nitrogen for 15 minutes prior to use and is hereafter referred to as the catalyst solution.
A 500-mL jacketed reactor was equipped with an overhead stirrer and reflux condenser and the atmosphere was purged with nitrogen gas. The reactor was charged with 12.1 g of 1-(3-tert-butyl-5-iodo-4-methoxyphenyl)pyrimidine-2,4(1H,3H)-dione, (30.3 mmol, 1.0 equivalent) and 5.98 g of 6-hydroxynaphthalen-2-ylboronic acid (31.8 mmol, 1.05 equivalents). The atmosphere was purged with nitrogen gas with stirring of the solid reagents for 20 minutes. The reactor was charged with 120 mL of degassed tetrahydrofuran, and the mixture was stirred to dissolve the solids. The solution was sparged with nitrogen gas for 10 minutes. The phosphate solution was added to the reactor by cannula, followed by the catalyst solution. The resulting biphasic mixture was stirred aggressively to ensure adequate phase mixing, and the jacket was warmed to 65° C. The reaction jacket was cooled to room temperature prior to quench.
After 2.5 hours, the reaction jacket was cooled to room temperature prior to quench.
The workup of the reaction was also conducted under anaerobic conditions. Fifty-seven grams of sodium chloride and 4.2 g of cysteine (15 weight equivalents relative to palladium catalyst) were dissolved in 300 mL of water, and the resulting solution was sparged with inert gas for 2 hours prior to use. To quench the reaction, approximately ⅓ of this solution was transferred to the reaction mixture by cannula under nitrogen gas and the resulting biphasic mixture was stirred vigorously for 2 hours. The mechanical agitation was halted, the two solutions were allowed to separate, and the aqueous solution was drained out of the reactor through the bottom valve. Approximately ⅓ of the quench solution was transferred to the reaction mixture by cannula under nitrogen gas and the resulting biphasic mixture was stirred vigorously for 45 minutes. The mechanical agitation was halted, the two solutions were allowed to separate, and the aqueous solution was drained out of the reactor through the bottom valve. The final portion of the quench solution was transferred to the reaction mixture by cannula, the resulting biphasic mixture was stirred vigorously for 45 minutes and the aqueous solution was drained out of the reactor through the bottom valve.
The remainder of the workup was not conducted under anaerobic conditions. The pale yellow organic solution was drained from the reactor through the bottom valve and filtered over a pad of grade 4 Filtrol® (1 cm deep by 4.5 cm diameter). The reactor and filter cake were rinsed with 70 mL of tetrahydrofuran. The bulk of the solvent was distilled in vacuo (ca 90-130 torr) at ca 40° C. with good agitation from an overhead stirrer. The solution was concentrated to approximately 50 mL volume, during which time the product began to precipitate out. Ethyl acetate (100 mL, about 8 mL of solvent per gram of the product) was added to the mixture, and the resultant slurry was stirred overnight at room temperature. The crystalline material was isolated by filtration and the filter cake was washed twice with 20 mL portions of ethyl acetate. The wet cake was air-dried on the filter and dried in a vacuum oven at 50° C. at approximately 250 torr with a gentle nitrogen sweep overnight.
The desired product was isolated as a white solid (11.6 g, 96.4% potency vs. standard, 88% potency-adjusted yield). 1H NMR (400 MHz, DMSO-d6) δ ppm 11.39 (d, J=2.1 Hz, 1H), 9.82 (s, 1H), 7.91 (d, J=0.8 Hz, 1H), 7.80 (d, J=8.9 Hz, 1H), 7.77-7.74 (m, 2H), 7.58 (dd, J=8.5, 1.7 Hz, 1H), 7.32 (d, J=2.7 Hz, 1H), 7.27 (d, J=2.7 Hz, 1H), 7.16 (d, J=2.3 Hz, 1H), 7.10 (dd, J=8.8, 2.4 Hz, 1H), 5.64 (dd, J=7.9, 2.2 Hz, 1H), 3.23 (s, 3H), 1.41 (s, 9H).
Example 2-1 Alternative preparation of 1-(3-tert-butyl-5-(6-hydroxynaphthalen-2-yl)-4-methoxyphenyl)pyrimidine-2,4(1H,3H)-dione (compound (4a))
This reaction is air-sensitive and the reaction was conducted under anaerobic conditions. A 100-mL round-bottom flask was purged with nitrogen gas and charged with 229 mg of tris(dibenzylideneacetone)dipalladium(0) (0.25 mmol, 0.02 equivalents Pd), 323 mg of 1,3,5,7-tetramethyl-8-phenyl-2,4,6-trioxa-8-phosphatricyclo[3.3.1.13,7]decane (1.13 mmol, 0.045 equivalents) and a magnetic stir bar. The flask was sealed with a septum and the atmosphere above the solids was purged with nitrogen gas. Sixty mL of degassed tetrahydrofuran was added to the flask and the mixture was stirred under a nitrogen atmosphere for 20 minutes. This solution is hereafter referred to as the catalyst solution.
A 500-mL jacketed reactor was equipped with an overhead stirrer and reflux condenser and the atmosphere was purged with nitrogen gas. The reactor was charged with 10.0 g of 1-(3-tert-butyl-5-iodo-4-methoxyphenyl)pyrimidine-2,4(1H,3H)-dione, (25.1 mmol, 1.0 equivalent), 4.98 g of 6-hydroxynaphthalen-2-ylboronic acid (26.6 mmol, 1.06 equivalents) and 10.3 g of potassium phosphate tribasic (48.7 mmol, 2.0 equivalents). The atmosphere was purged with nitrogen gas with stirring of the solid reagents for 20 minutes. The reactor was charged with 100 mL of tetrahydrofuran, 50 mL of water, and the mixture was stirred to dissolve the solids. The biphasic mixture was sparged with nitrogen gas for 30 minutes. The catalyst solution was transferred to the main reactor by positive nitrogen pressure through a cannula. The resulting biphasic mixture was stirred aggressively and warmed to an internal temperature between 60 and 65° C. under nitrogen for 2 hours. The reaction mixture was cooled to an internal temperature between 50 and 55° C. before quench.
The workup of the reaction was conducted under anaerobic conditions at an internal temperature between 50 and 55° C. Fifteen grams of sodium chloride and 1.0 g of cysteine were dissolved in 80 mL of water, and the resulting solution was sparged for 1 hour. This solution was transferred to the reaction mixture by cannula with nitrogen gas pressure and the resulting biphasic mixture was stirred vigorously for 45 minutes. The mechanical agitation was halted, the two solutions were allowed to separate, and the aqueous solution was drained out of the reactor through the bottom valve. Fifteen grams of sodium chloride and 1.0 g of cysteine were dissolved in 80 mL of water, and the resulting solution was sparged for 1 hour. This solution was transferred to the reaction mixture by cannula with nitrogen gas pressure and the resulting biphasic mixture was stirred vigorously for 45 minutes. The mechanical agitation was halted, the two solutions were allowed to separate, and the aqueous solution was drained out of the reactor through the bottom valve.
The pale yellow organic solution was drained from the reactor through the bottom valve and filtered over a polypropylene filter to remove palladium black. The reactor and filter cake were rinsed with 22 mL of tetrahydrofuran and 50 mL of ethyl acetate was added to the organic solution. The solution was distilled at atmospheric pressure (approximately 66° C. internal temperature) with continuous addition of 110 mL of ethyl acetate, keeping the volume of the solution roughly constant during the distillation. During the constant-volume distillation, solids began to precipitate in the reactor. After the ethyl acetate was charged, the distillation was continued at atmospheric pressure, concentrating the slurry to approximately 60 mL total volume. The solution was cooled to an internal temperature of approximately 30° C. and held for 3 hours with stirring. The crystalline material was isolated by filtration and the filter cake was washed twice with 20 mL portions of ethyl acetate. The wet cake was dried in a vacuum oven at 50° C. with a gentle nitrogen sweep overnight. The desired product was isolated as an off-white solid (8.33 g, 80% yield). 1H NMR (400 MHz, DMSO-d6) δ ppm δ 11.39 (d, J=2.1 Hz, 1H), 9.82 (s, 1H), 7.91 (d, J=0.8 Hz, 1H), 7.80 (d, J=8.9 Hz, 1H), 7.77-7.74 (m, 2H), 7.58 (dd, J=8.5, 1.7 Hz, 1H), 7.32 (d, J=2.7 Hz, 1H), 7.27 (d, J=2.7 Hz, 1H), 7.16 (d, J=2.3 Hz, 1H), 7.10 (dd, J=8.8, 2.4 Hz, 1H), 5.64 (dd, J=7.9, 2.2 Hz, 1H), 3.23 (s, 3H), 1.41 (s, 9H).
Example 2-2 Alternative preparation of 1-(3-tert-butyl-5-(6-hydroxynaphthalen-2-yl)-4-methoxyphenyl)pyrimidine-2,4(1H,3H)-dione (compound (4a))
This reaction is air-sensitive and the reaction was conducted under nitrogen atmosphere. A 100-mL round-bottom flask was purged with nitrogen gas and charged with 303 mg of tris(dibenzylideneacetone)dipalladium(0) (0.33 mmol, 0.02 equivalents Pd), 411 mg of 1,3,5,7-tetramethyl-8-phenyl-2,4,6-trioxa-8-phosphatricyclo[3.3.1.13,7]decane (1.40 mmol, 0.045 equivalents) and a magnetic stir bar. The flask was sealed with a septum and the atmosphere above the solids was purged with nitrogen gas. Seventy-five (75) mL of degassed tetrahydrofuran was added to the flask and the mixture was stirred under a nitrogen atmosphere for 25 minutes. This solution is hereafter referred to as the catalyst solution.
A 500-mL jacketed reactor was equipped with an overhead stirrer and reflux condenser and the atmosphere was purged with nitrogen gas. The reactor was charged with 12.5 g of 1-(3-tert-butyl-5-iodo-4-methoxyphenyl)pyrimidine-2,4(1H,3H)-dione, (31.2 mmol, 1.0 equivalent), 6.20 g of 6-hydroxynaphthalen-2-ylboronic acid (33.0 mmol, 1.06 equivalents) and 13.0 g of potassium phosphate tribasic (61.2 mmol, 2.0 equivalents). The reactor was charged with 130 mL of tetrahydrofuran, 65 mL of water, and the mixture was stirred to dissolve the solids. The biphasic mixture was sparged with nitrogen gas for 30 minutes. The catalyst solution was transferred to the main reactor by positive nitrogen pressure through a cannula. The resulting biphasic mixture was stirred aggressively and warmed to an internal temperature between 60 and 65° C. under nitrogen for 2.5 hours. The reaction mixture was cooled to an internal temperature between 50 and 55° C. before quench.
The workup of the reaction was conducted under anaerobic conditions at an internal temperature between 50 and 55° C. Sodium chloride (18.8 g) and cysteine (1.25 g) were dissolved in 100 mL of water, and the resulting solution was sparged with nitrogen for 40 minutes. This solution was transferred to the reaction mixture by cannula with nitrogen gas pressure and the resulting biphasic mixture was stirred vigorously for 45 minutes. The mechanical agitation was halted, the two solutions were allowed to separate, and the aqueous solution was drained out of the reactor through the bottom valve. Sixty-three (63) mL of degassed tetrahydrofuran were added to the reactor by cannula with positive nitrogen pressure. Sodium chloride (18.9 g) and cysteine (1.333 g) were dissolved in 100 mL of water, and the resulting solution was sparged with nitrogen for 40 minutes. This solution was transferred to the reaction mixture by cannula with nitrogen gas pressure and the resulting biphasic mixture was stirred vigorously for 45 minutes. The mechanical agitation was halted, the two solutions were allowed to separate, and the aqueous solution was drained out of the reactor through the bottom valve.
The pale yellow organic solution was drained from the reactor through the bottom valve and filtered through a thin pad of filter aid on a polyethylene filter while warm. The reactor and filter cake were rinsed with 32 mL of tetrahydrofuran, and 65 mL of ethyl acetate was added to the organic solution. The solution was distilled at atmospheric pressure (approximately 66° C. internal temperature) with continuous addition of 190 mL of ethyl acetate, keeping the volume of the solution roughly constant during the distillation. During the constant-volume distillation, solids began to precipitate in the reactor. After the ethyl acetate was charged, the distillation was continued at atmospheric pressure, concentrating the slurry to approximately 90 mL total volume. The slurry was cooled to an internal temperature of approximately 40° C. and was concentrated further in vacuo to a total volume of approximately 50 mL. The slurry was cooled to an internal temperature of 30° C. and held for 16 hours with stirring. The crystalline material was isolated by filtration, and the filter cake was washed twice with 25 mL portions of ethyl acetate. The wet cake was dried in a vacuum oven at 50° C. with a gentle nitrogen sweep overnight. The desired product was isolated as an off-white solid (11.4 g, 99.5% potent vs. standard, 87% potency-adjusted yield).
Example 4 Preparation of N-(6-(3-tert-butyl-5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-2-methoxyphenyl)naphthalen-2-yl)methanesulfonamide (compound (A-1))

A 3-L, 3-neck, round-bottom flask was equipped with an overhead stirrer, a thermocouple, a Claisen condenser and a reflux condenser. Tris(dibenzylideneacetone)dipalladium(0) (0.330 g, 0.360 mmol), di-tert-butyl(2′,4′,6′-triisopropyl-3,4,5,6-tetramethylbiphenyl-2-yl)phosphine (0.416 g, 0.864 mmol) and milled potassium phosphate tribasic (21.0 g, 99.0 mmol) were charged to the 3-L flask. The flask was purged with argon for not less than 90 minutes with constant stirring of the solids. t-Amyl alcohol (250 ml) was charged to a separate 500-mL round-bottom flask and was purged with argon for not less than 30 minutes and was transferred to the 3-L flask using a cannula under argon atmosphere. The contents of the 3-L flask were heated to 80° C. and stirred at this temperature for 30 minutes. A 1-L round-bottom flask equipped with a magnetic stir bar was charged with 6-(3-tert-butyl-5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-2-methoxyphenyl)naphthalen-2-yl-1,1,2,2,3,3,4,4,4-nonafluorobutane-1-sulfonate (62.9 g, 90 mmol), methanesulfonamide (12.85 g, 135 mmol) and t-amyl alcohol (505 mL), purged with argon and heated to 60° C. The reagent mixture was stirred under argon for not less than 30 minutes. A clear yellow solution was observed. This solution was transferred to the 3-L flask using a cannula under argon atmosphere. The temperature of the 3-L flask was raised to 85° C. and the contents were stirred for 14 hours under a positive pressure of argon. The temperature was then raised to 95° C. and the contents were stirred for an additional 4 hours under a positive pressure of argon. The reaction mixture was allowed to cool down to room temperature, diluted with tetrahydrofuran (2200 mL) and water (800 mL) and was transferred to a 6-L separatory funnel. The organic layer was washed thrice with water (2000 mL) containing L-cysteine (17.3 g) and NaCl (235 g). The organic layer was collected, filtered through a pad of diatomaceous earth and was concentrated in vacuo to approximately 250 mL. Ethyl acetate (775 mL) was added over 7 hours with stirring, and the mixture was allowed to stir for an additional 14 hours. White solid was isolated by filtration, and the solid was washed with ethyl acetate (1000 mL). The solid was then dissolved in tetrahydrofuran (1500 mL) and filtered through a pad of diatomaceous earth to obtain a clear solution. The diatomaceous earth was washed with tetrahydrofuran (300 mL). The combined tetrahydrofuran solution was concentrated in vacuo to approximately 250 mL, and then ethyl acetate (775 mL) was added over 7 hours with stirring. The product solution was allowed to stir for an additional 14 hours. White solid was isolated by filtration. The solid was washed with ethyl acetate (1000 mL) and dried in a vacuum oven at 60° C. for 24 hours. The solid was slurried in 308 mL of 200 proof ethanol for 1.5 hours, then isolated by filtration. The solid was washed with 132 mL of 200 proof ethanol and dried in a vacuum oven at 50° C. for 18 hours. The title compound was isolated as a white solid (32.6 g, 100% potency vs. standard, 73% yield). 1H NMR (400 MHz, DMSO-d6) δ ppm 11.41 (d, J=2.1 Hz, 1H), 10.04 (s, 1H), 8.02 (d, J=0.9 Hz, 1H), 7.98-7.91 (m, 2H), 7.79 (d, J=7.9 Hz, 1H), 7.72 (d, J=2.0 Hz, 1H), 7.69 (dd, J=8.5, 1.7 Hz, 1H), 7.41 (dd, J=8.8, 2.2 Hz, 1H), 7.36 (d, J=2.7 Hz, 1H), 7.31 (d, J=2.7 Hz, 1H), 5.65 (dd, J=7.9, 2.2 Hz, 1H), 3.24 (s, 3H), 3.08 (s, 3H), 1.42 (s, 9H). 13C NMR (101 MHz, DMSO-d6) δ ppm 163.1 (C), 156.0 (C), 150.0 (C), 145.3 (CH), 142.9 (C), 136.0 (C), 134.3 (C), 134.2 CO, 133.5 (C), 132.2 (C), 129.5 (C), 129.0 (CH), 127.6 (CH), 127.1 (CH), 127.0 (CH), 126.5 (CH), 124.3 (CH), 120.2 (CH), 114.5 (CH), 101.1 (CH), 60.3 (CH3), 39.4 (CH3), 35.1 (C), 30.5 (CH3).
Other ligands such as 2,2,7,7-tetramethyl-1-(2′,4′,6′-triisopropylbiphenyl-2-yl)phosphepane; 7,7,9,9-tetramethyl-8-(2′,4′,6′-triisopropyl-3,6-dimethoxybiphenyl-2-yl)-1,4-dioxa-8-phosphaspiro[4.5]decane; and 8-(2-(2-methoxynaphthalen-1-yl)phenyl)-7,7,9,9-tetramethyl-1,4-dioxa-8-phosphaspiro[4.5]decane were tested under the conditions described above and produced favorable yields of greater than 50% of the sulfonamidated product.
Example 4-1 Alternative Preparation of N-(6-(3-tert-butyl-5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-2-methoxyphenyl)naphthalen-2-yl)methanesulfonamide (compound (A-1))
A 450-mL, stainless steel Parr® pressure reactor equipped with an overhead stirrer was charged with tris(dibenzylideneacetone)dipalladium(0) (0.131 g, 0.143 mmol), di-tert-butyl(2′,4′,6′-triisopropyl-3,6-dimethoxybiphenyl-2-yl)phosphine (0.167 g, 0.344 mmol) and milled potassium phosphate tribasic (6.69 g, 31.5 mmol). The flask was purged with argon for not less than 90 minutes. Tetrahydrofuran (90 mL) was taken in a 100-mL round bottom flask, purged with argon for not less than 30 minutes and was transferred to the 450-mL reactor using a cannula under argon atmosphere. The contents of the 450-mL reactor were heated to 80° C. and stirred at this temperature for 30 minutes. A 250-mL, round-bottom flask equipped with a magnetic stir bar was charged with 6-(3-tert-butyl-5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-2-methoxyphenyl)naphthalen-2-yl-1,1,2,2,3,3,4,4,4-nonafluorobutane-1-sulfonate (20.0 g, 28.6 mmol), methanesulfonamide (3.27 g, 34.4 mmol) and tetrahydrofuran (160 mL), purged with argon for not less than 45 minutes. A clear yellow solution was observed. This solution was transferred to the 450-mL reactor that has been cooled to the room temperature using a cannula under argon atmosphere. The temperature of the 450-mL reactor was raised to 90° C. and the contents were stirred for 20 hours. The reaction mixture was allowed to cool down to 50° C., diluted with tetrahydrofuran (70 mL) and water (70 mL) containing L-cysteine (0.875 g) and sodium chloride (7.7 g). The contents were stirred for 2 hours at 50° C. The aqueous layer was discarded and the organic layer was filtered through an approximately 2-inch pad of diatomaceous earth and rinsed with tetrahydrofuran (45 mL) to obtain a clear, light yellow solution. The total weight of reaction mixture was 363.43 g. HPLC analysis of the reaction mixture revealed 13.71 g (97%) of the title compound was present in the reaction mixture. A portion of the reaction mixture (50 g) was concentrated to a final volume of 12-14 mL under vacuum. Ethyl acetate (45 mL) was added slowly and the reaction mixture was stirred over night at room temperature to obtain white slurry. Product was collected by filtration, washed with ethyl acetate (7 mL) and dried overnight in a vacuum oven at 50-60° C. to obtain 2.02 g of white solid. Ethanol (14 mL) was added to the solid and stirred overnight at the room temperature. The product was collected by filtration, washed with ethanol (4 mL) and dried overnight in a vacuum oven at 50-60° C. to obtain the title compound (1.79 g, 95.4%).
Example 4-2 Alternative preparation of N-(6-(3-tert-butyl-5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-2-methoxyphenyl)naphthalen-2-yl)methanesulfonamide (compound (A-1))
A 450-mL, stainless steel Parr® pressure reactor equipped with an overhead stirrer was charged with tris(dibenzylideneacetone)dipalladium(0) (0.105 g, 0.115 mmol), di-tert-butyl(2′,4′,6′-triisopropyl-3,6-dimethoxybiphenyl-2-yl)phosphine (0.133 g, 0.275 mmol) and milled potassium phosphate tribasic (5.35 g, 25.2 mmol). The flask was purged with argon for not less than 90 minutes. 2-Methyltetrahydrofuran (70 mL) was taken in a 100-mL round bottom flask, purged with argon for not less than 30 minutes and was transferred to the 450-mL reactor using a cannula under argon atmosphere. The contents of the 450-mL reactor were heated to 80° C. and stirred at this temperature for 30 minutes. A 250-mL, round bottom flask equipped with a magnetic stir bar was charged with 6-(3-tert-butyl-5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-2-methoxyphenyl)naphthalen-2-yl-1,1,2,2,3,3,4,4,4-nonafluorobutane-1-sulfonate (16.0 g, 22.9 mmol), methanesulfonamide (2.61 g, 27.5 mmol) and 2-methyltetrahydrofuran (155 mL), purged with argon for not less than 60 minutes. This solution was transferred to the 450-mL reactor that has been cooled to the room temperature using a cannula under argon atmosphere. The temperature of the 450-mL flask was raised to 90° C. and the contents were stirred for 14 hours. The reaction mixture was allowed to cool down to 70° C., diluted with ethyl acetate (190 mL) and stirred for 3 hours at 70° C., cooled to the room temperature, stirred for an additional 4 hours, filtered through a fine frit filter funnel and rinsed with ethyl acetate (90 mL) to obtain 29.4 g of light brown solid. A portion of this solid (13.04 g) was transferred to a 500-mL, 3-neck round bottom flask equipped with an overhead stirrer and a thermocouple. Tetrahydrofuran (175 mL) was added, followed by the addition of water 50 mL containing L-cysteine (0.63 g) and sodium chloride (5.5 g). The reaction mixture was stirred for 2 hours at 50° C. under a slight positive pressure of argon. The reaction mixture was transferred to a 500-mL separatory funnel and the aqueous layer was discarded. The organic layer was filtered through an approximately 2-inch pad of diatomaceous earth and rinsed with tetrahydrofuran (45 mL) to obtain a clear, light yellow solution. The organic layer was concentrated to a total weight of 45.59 g. A portion of this organic solution (41.58 g) was charged to a 250-mL, 3-neck round bottom flask fitted with an overhead stirrer. Ethyl acetate (80 mL) was added over 6 hours by a pump with constant stirring at room temperature. The product was collected by filtration, rinsed with ethyl acetate (20 mL) and dried in a vacuum oven for 2 hours to obtain 3.17 g of the title compound (>99.8 pure and 94.6% potent vs. standard).
Example 4-3 Alternative preparation of N-(6-(3-tert-butyl-5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-2-methoxyphenyl)naphthalen-2-yl)methanesulfonamide (compound (A-1))
A 600-mL, stainless steel Parr® pressure reactor equipped with an overhead stirrer was charged with tris(dibenzylideneacetone)dipalladium(0) (0.229 g, 0.251 mmol), di-tert-butyl(2′,4′,6′-triisopropyl-3,6-dimethoxybiphenyl-2-yl)phosphine (0.291 g, 0.601 mmol) and milled potassium phosphate tribasic (11.70 g, 55.1 mmol). The flask was purged with argon for not less than 90 minutes. Ethyl acetate (140 mL) was taken in a 250-mL, round bottom flask, purged with argon for not less than 30 minutes and was transferred to the 600-mL reactor using a cannula under argon atmosphere. The contents of the 600-mL reactor were heated to 80° C. and stirred at this temperature for 30 minutes. A 500-mL round bottom flask equipped with a magnetic stir bar was charged with 6-(3-tert-butyl-5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-2-methoxyphenyl)naphthalen-2-yl-1,1,2,2,3,3,4,4,4-nonafluorobutane-1-sulfonate (35.0 g, 50.1 mmol), methanesulfonamide (5.72 g, 60.1 mmol) and ethyl acetate (280 mL), purged with argon for not less than 60 minutes while stirring at 50° C. This solution was transferred to the 600-mL reactor that had been cooled to room temperature using a cannula under argon atmosphere. The temperature of the 600-mL flask was raised to 90° C., and the contents were stirred for 18 hours. The reaction mixture was allowed to cool down to 40° C., filtered and rinsed with ethyl acetate (140 mL). Solid (41.50 g) was obtained after drying for 2 hours on high vacuum. This solid contained the titled product (23.06 g, 93%).
Example 4-4 Alternative preparation of N-(6-(3-tert-butyl-5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-2-methoxyphenyl)naphthalen-2-yl)methanesulfonamide (compound (A-1))
Tris(dibenzylideneacetone)dipalladium(0) (0.0066 g, 7.16 μmol), di-tert-butyl(2′,4′,6′-triisopropyl-3,6-dimethoxybiphenyl-2-yl)phosphine (0.0083 g, 17 μmol) and milled potassium phosphate tribasic (0.334 g, 1.58 mmol) were charged to a 40-mL reaction vial inside an inert atmosphere glove box. t-Amyl alcohol (4 mL) was added, the vial was capped, and the contents were heated to 80° C. and stirred at this temperature for 30 minutes. The reaction mixture was cooled down to the room temperature. 6-(3-tert-Butyl-5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-2-methoxyphenyl)naphthalen-2-yl-1,1,2,2,3,3,4,4,4-nonafluorobutane-1-sulfonate (1.0 g, 1.43 mmol), methanesulfonamide (0.163 g, 1.72 mmol) and t-amyl alcohol (8 mL) were added to the 40-mL reaction vial, and the vial was capped. The reaction temperature was raised to 90° C. and the contents were stirred for 5 hours. HPLC analysis of the reaction mixture showed that the product was formed in 94 area % at 210 nm.
Example 4-5 Alternative preparation of N-(6-(3-tert-butyl-5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-2-methoxyphenyl)naphthalen-2-yl)methanesulfonamide (compound (A-1))
A 600-mL, stainless steel, Parr® reactor was equipped with an overhead stirrer, thermocouple and a heating mantle. Tris(dibenzylideneacetone)dipalladium(0) (0.164 g, 0.179 mmol), di-tert-butyl(2′,4′,6′-triisopropyl-3,6-dimethoxybiphenyl-2-yl)phosphine (0.208 g, 0.429 mmol) and milled potassium phosphate tribasic (8.36 g, 39.4 mmol) were charged to the 600-mL reactor. The reactor was purged with argon for not less than 90 minutes. 2-Methyltetrahydrofuran (100 mL) was purged with argon for not less than 30 minutes and was transferred to the 600-mL reactor using a cannula under argon atmosphere. The reactor was tightly sealed, the contents were heated to 80° C. and stirred at this temperature for 30 minutes. A 500-mL round bottom flask equipped with a magnetic stir bar was charged with 6-(3-tert-butyl-5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-2-methoxyphenyl)naphthalen-2-yl 1,1,2,2,3,3,4,4,4-nonafluorobutane-1-sulfonate (25 g, 35.8 mmol), methanesulfonamide (4.09 g, 42.9 mmol) and ethyl acetate (200 mL), purged with argon for not less than 30 minutes with stirring and heated to 60° C. A clear solution was observed. This solution was transferred to the 600-mL reactor using a cannula under argon atmosphere. The reactor was tightly sealed, the contents were heated to 90° C. and stirred at this temperature for 14 hours. The reaction mixture was cooled to 35° C., solids were collected by filtration, washed with ethyl acetate (300 mL) and dried under high vacuum for 2-4 hours. The solids were then transferred to a 1-L, three-neck, round-bottom flask equipped with an overhead stirrer and a thermocouple. N-Acetyl-L-cysteine (0.58 g, 3.5 mmol), dimethylformamide (DMF) (100 mL) and glacial acetic acid (0.85 g) were charged to the 1-L flask; the contents were heated to 60° C. and mixed for 1 hour. The mixture was filtered through approximately 2-inch pad of diatomaceous earth and washed with DMF (50 mL). The dark-brown/black-colored solid collected on diatomaceous earth was discarded and the light yellow/clear filtrate was charged to a separate 1-L, three-neck, round-bottom flask equipped with an overhead stirrer, a thermocouple and a syringe pump. The DMF solution was mixed and methanol (300 mL) was added over 8 hours, while maintaining the internal temperature at 25±5° C. The white solid was collected by filtration washed with methanol (150 mL) and dried in a vacuum oven at 50° C. for not less than 8 hours. The title compound was isolated as a white solid (15.8 g, 89% yield).
Example 4-6 Alternative preparation of N-(6-(3-tert-butyl-5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-2-methoxyphenyl)naphthalen-2-yl)methanesulfonamide (compound (A-1))
A 600-mL, stainless steel, Parr® reactor was equipped with an overhead stirrer, thermocouple and a heating mantle. Tris(dibenzylideneacetone)dipalladium(0) (0.164 g, 0.179 mmol), 7,7,9,9-tetramethyl-8-(2′,4′,6′-triisopropyl-3,6-dimethoxybiphenyl-2-yl)-1,4-dioxa-8-phosphaspiro[4.5]decane (0.238 g, 0.429 mmol) and milled potassium phosphate tribasic (8.36 g, 39.4 mmol) were charged to the 600-mL reactor. The reactor was purged with argon for not less than 90 minutes. 2-Methyltetrahydrofuran (100 mL) was purged with argon for not less than 30 minutes and was transferred to the 600-mL reactor using a cannula under argon atmosphere. The reactor was tightly sealed, the contents were heated to 80° C. and stirred at this temperature for 30 minutes. A 500-mL round bottom flask equipped with a magnetic stir bar was charged with 6-(3-tert-butyl-5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-2-methoxyphenyl)naphthalen-2-yl 1,1,2,2,3,3,4,4,4-nonafluorobutane-1-sulfonate (25 g, 35.8 mmol), methanesulfonamide (4.09 g, 42.9 mmol) and ethyl acetate (200 mL), purged with argon for not less than 30 minutes with stirring and heated to 60° C. A clear solution was observed. This solution was transferred to the 600-mL reactor using a cannula under argon atmosphere. The reactor was tightly sealed, the contents were heated to 90° C. and stirred at this temperature for 14 hours. The reaction mixture was cooled to 35° C., 5% aqueous N-acetyl-L-cysteine solution (100 mL) was added and the contents were mixed for 1 hour at 35° C. Solids were collected by filtration, washed with water (2×25 mL) and ethyl acetate (3×80 mL) and were dried under high vacuum for 2-4 hours. The solids were then transferred to a 1-L, three-neck, round-bottom flask equipped with an overhead stirrer and a thermocouple. N-Acetyl-L-cysteine (0.58 g, 3.5 mmol), dimethylformamide (DMF) (100 mL) and glacial acetic acid (0.85 g) were charged to the 1-L flask; the contents were heated to 60° C. and mixed for 1 hour. The mixture was filtered through an approximately 2-inch pad of diatomaceous earth and washed with DMF (50 mL). The dark-brown/black-colored solid collected on the diatomaceous earth was discarded and the light yellow/clear filtrate was charged to a separate 1-L, three-neck, round-bottom flask equipped with an overhead stirrer, a thermocouple and a syringe pump. The DMF solution was mixed and methanol (300 mL) was added over 8 hours, while maintaining the internal temperature at 25±5° C. The white solid was collected by filtration washed with methanol (150 mL) and dried in a vacuum oven at 50° C. for not less than 8 hours. The title compound was isolated as a white solid (15.6 g, 88% yield).
Example 4-7 Alternative preparation of N-(6-(3-tert-butyl-5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-2-methoxyphenyl)naphthalen-2-yl)methanesulfonamide (compound (A-1))

Tris(dibenzylideneacetone)dipalladium(0) (0.0026 g, 2.80 μmol), di-tert-butyl(2′,4′,6′-triisopropyl-3,6-dimethoxybiphenyl-2-yl)phosphine (0.0033 g, 6.72 μmol) and milled potassium phosphate tribasic (0.131 g, 0.616 mmol) were charged to a 40-mL reaction vial inside an inert atmosphere glove box. 2-Methyltetrahydrofuran (1.5 mL) was added, the vial was capped, and the contents were heated to 80° C. and stirred at this temperature for 30 minutes. The reaction mixture was cooled down to room temperature. 6-(3-tert-Butyl-5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-2-methoxyphenyl)naphthalen-2-yl 1,1,2,2-tetrafluoro-2-(perfluoroethoxy)ethanesulfonate (0.4 g, 0.560 mmol, Example 3-7, compound (5f)), methanesulfonamide (0.064 g, 0.672 mmol) and ethyl acetate (3 mL) were added to the 40-mL reaction vial. The temperature of the closed vial was raised to 90° C. and the contents were magnetically stirred for 16 hours. HPLC analysis of the reaction mixture showed that the product was formed in 97 area % at 210 nm.
Example 4-8 Alternative preparation of N-(6-(3-tert-butyl-5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-2-methoxyphenyl)naphthalen-2-yl)methanesulfonamide (compound (A-1))

Tris(dibenzylideneacetone)dipalladium(0) (0.0071 g, 7.71 μmol), di-tert-butyl(2′,4′,6′-triisopropyl-3,6-dimethoxybiphenyl-2-yl)phosphine (0.0089 g, 19.0 μmol) and milled potassium phosphate tribasic (0.360 g, 1.696 mmol) were charged to a 40-mL reaction vial inside an inert atmosphere glove box. 2-Methyltetrahydrofuran (4 mL) was added, and the closed vial and its contents were heated to 80° C. with magnetic stirring for 30 minutes. The reaction mixture was cooled down to room temperature. 6-(3-tert-Butyl-5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-2-methoxyphenyl)naphthalen-2-yl 1,1,1,2,3,3,3-heptafluoropropane-2-sulfonate (1.0 g, 1.542 mmol, Example 3-4, compound (5c)), methanesulfonamide (0.176 g, 1.850 mmol) and ethyl acetate (8 mL) were added to the 40-mL reaction vial. The temperature of the closed vial and its contents was raised to 90° C. and stirred for 20 hours. HPLC analysis of the reaction mixture showed that the product was formed in 95 area % at 210 nm.
Example 4-9 Alternative preparation of N-(6-(3-tert-butyl-5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-2-methoxyphenyl)naphthalen-2-yl)methanesulfonamide (compound (A-1))

Tris(dibenzylideneacetone)dipalladium(0) (0.0055 g, 6.02 μmol), di-tert-butyl(2′,4′,6′-triisopropyl-3,6-dimethoxybiphenyl-2-yl)phosphine (0.0070 g, 14.0 μmol) and milled potassium phosphate tribasic (0.281 g, 1.324 mmol) were charged to a 40-mL reaction vial inside an inert atmosphere glove box. 2-Methyltetrahydrofuran (3.4 mL) was added, and the closed vial and its contents were heated to 80° C. with magnetic stirring for 30 minutes. The reaction mixture was cooled down to room temperature. 6-(3-tert-Butyl-5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-2-methoxyphenyl)naphthalen-2-yl sulfofluoridate (0.6 g, 1.204 mmol, Example 3-8, compound (5g)), methanesulfonamide (0.137 g, 1.444 mmol) and ethyl acetate (6.7 mL) were added to the 40-mL reaction vial. The temperature of the closed reaction vial and its contents was raised to 90° C. and the contents were stirred for 20 hours. HPLC analysis of the reaction mixture showed that the product was formed in 79 area % at 210 nm.
Example 4-10 Alternative preparation of N-(6-(3-tert-butyl-5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-2-methoxyphenyl)naphthalen-2-yl)methanesulfonamide (compound (A-1))

Tris(dibenzylideneacetone)dipalladium(0) (0.0042 g, 4.56 μmol), di-tert-butyl(2′,4′,6′-triisopropyl-3,6-dimethoxybiphenyl-2-yl)phosphine (0.0053 g, 12.0 μmol) and milled potassium phosphate tribasic (0.213 g, 1.003 mmol) were charged to a 40-mL reaction vial inside an inert atmosphere glove box. 2-Methyltetrahydrofuran (1.9 mL) was added, and the closed vial and its contents were heated to 80° C. with magnetic stirring for 30 minutes. The reaction mixture was cooled down to room temperature. 6-(3-tert-Butyl-5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-2-methoxyphenyl)naphthalen-2-yl trifluoromethanesulfonate (0.5 g, 0.912 mmol, Example 3-6, compound (5e)), methanesulfonamide (0.104 g, 1.094 mmol) and ethyl acetate (5.7 mL) were added to the 40-mL reaction vial. The temperature of the closed vial and its contents was raised to 90° C. and stirred for 14 hours. HPLC analysis of the reaction mixture showed that the product was formed in 91 area % at 210 nm.
Example 4-11 Alternative preparation of N-(6-(3-tert-butyl-5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-2-methoxyphenyl)naphthalen-2-yl)methanesulfonamide (compound (A-1))

Tris(dibenzylideneacetone)dipalladium(0) (0.0037 g, 4.04 μmol), di-tert-butyl(2′,4′,6′-triisopropyl-3,6-dimethoxybiphenyl-2-yl)phosphine (0.0047 g, 9.7 μmol) and milled potassium phosphate tribasic (0.094 g, 0.445 mmol) were charged to a 40-mL reaction vial inside an inert atmosphere glove box. tert-Amyl alcohol (1.0 mL) was added, the contents were heated to 80° C. and stirred at this temperature for 30 minutes. The reaction mixture was cooled down to room temperature. 6-(3-tert-Butyl-5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-2-methoxyphenyl)naphthalen-2-yl methanesulfonate (0.2 g, 0.404 mmol), methanesulfonamide (0.046 g, 0.485 mmol) and tert-amyl alcohol (1.5 mL) were added to a 40-mL reaction vial. The reaction temperature was raised to 110° C., and the contents were stirred for 14 hours. HPLC analysis of the reaction mixture showed that the titled compound was formed in 7 area % at 210 nm.
Example 4-12 Alternative preparation of N-(6-(3-tert-butyl-5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-2-methoxyphenyl)naphthalen-2-yl)methanesulfonamide (compound (A-1))

Palladium acetate (0.0018 g, 8.09 μmol), di-tert-butyl(2′,4′,6′-triisopropyl-3,6-dimethoxybiphenyl-2-yl)phosphine (0.0086 g, 0.018 mmol) and water (0.6 μL, 0.032 mmol) were charged to a 40-mL reaction vial inside an inert atmosphere glove box. tert-Amyl alcohol (1.0 mL) was added, and the contents were heated to 80° C. and stirred at this temperature for 15 minutes. The reaction mixture was cooled down to room temperature. Potassium phosphate tribasic (0.094 g, 0.445 mmol), 6-(3-tert-butyl-5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-2-methoxyphenyl)naphthalen-2-yl methanesulfonate (0.2 g, 0.404 mmol), methanesulfonamide (0.046 g, 0.485 mmol) and tert-amyl alcohol (1.5 mL) were added to the 40-mL reaction vial. The reaction temperature was raised to 110° C., and the contents were stirred for 14 hours. HPLC analysis of the reaction mixture showed that the titled compound was formed in 5 area % at 210 nm.
REF…
Wagner, Rolf et al, Uracil or thymine derivative for treating hepatitis C and their preparation, PCT Int. Appl., WO2009039127, 26 Mar 2009
Flentge, Charles A. et al, Preparation of anti-infective pyrimidines for treating hepatitis C,PCT Int. Appl., WO2009039134, 26 Mar 2009
Shekhar, Shashank et al,N-(6-(3-tert-Butyl-5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-2-methoxyphenyl)naphthalen-2-yl)methanesulfonamide as HCV polymerase inhibitor and its preparation, pharmaceutical compositions and use in the treatment of hepatitis C,PCT Int. Appl., WO2012009699, 19 Jan 2012
Shekhar, Shashank et al,Process for preparing antiviral pyrimidinylphenylnaphthalenyl sulfonamide compounds,PCT Int. Appl.,US20130224149, 29 Aug 2013
Shekhar, Shashank et al,Preparation and use of phosphine ligands for catalytic reactions,U.S. Pat. Appl. Publ., US20130217876, 22 Aug 2013
Want to know everything on vir series
click
http://drugsynthesisint.blogspot.in/p/vir-series-hep-c-virus-22.html
AND
http://medcheminternational.blogspot.in/p/vir-series-hep-c-virus.html
Reference
///////////////////Dasabuvir sodium, Exviera, FDA 2014, Hepatitis C treatment, Dasabuvir
[Na+].COc1c(cc(cc1C(C)(C)C)N2C=CC(=O)[N-]C2=O)c3ccc4cc(NS(=O)(=O)C)ccc4c3

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....














 BELINOSTAT
BELINOSTAT












































 38 nos
38 nos


 disodium salt is TR 701
disodium salt is TR 701


















 GADOBUTROL
GADOBUTROL




 WORLDCUP FOOTBALL WEEK 2014 BRAZIL
WORLDCUP FOOTBALL WEEK 2014 BRAZIL













 …………………..
…………………..
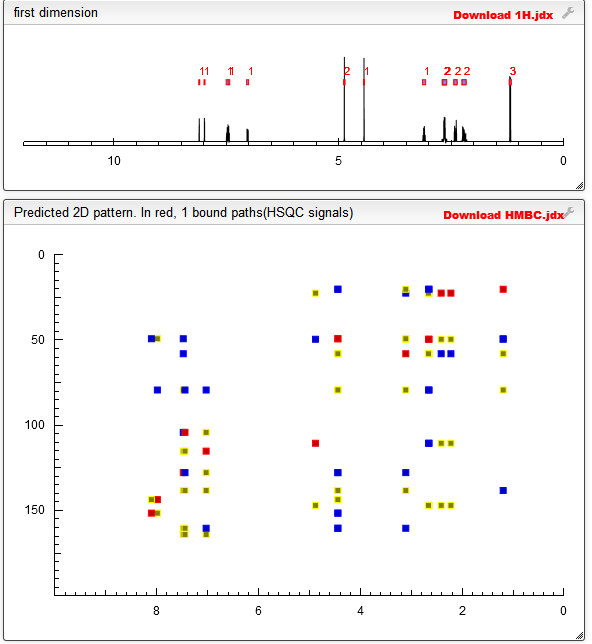

 efinaconazole
efinaconazole








 COCK WILL TEACH YOU NMR
COCK WILL TEACH YOU NMR COCK SAYS MOM CAN TEACH YOU NMR
COCK SAYS MOM CAN TEACH YOU NMR









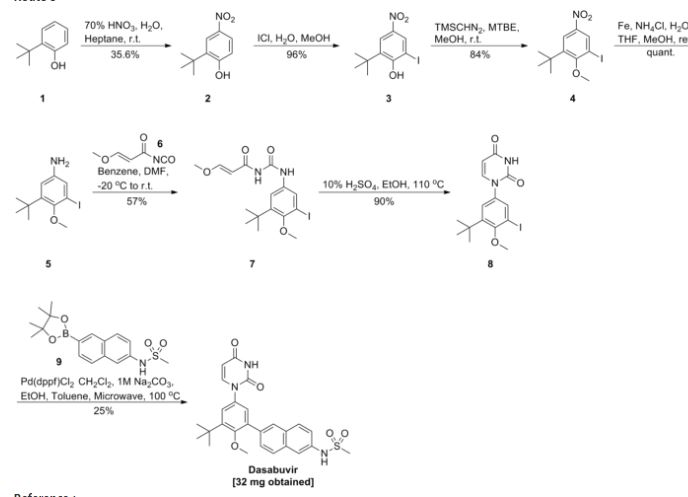
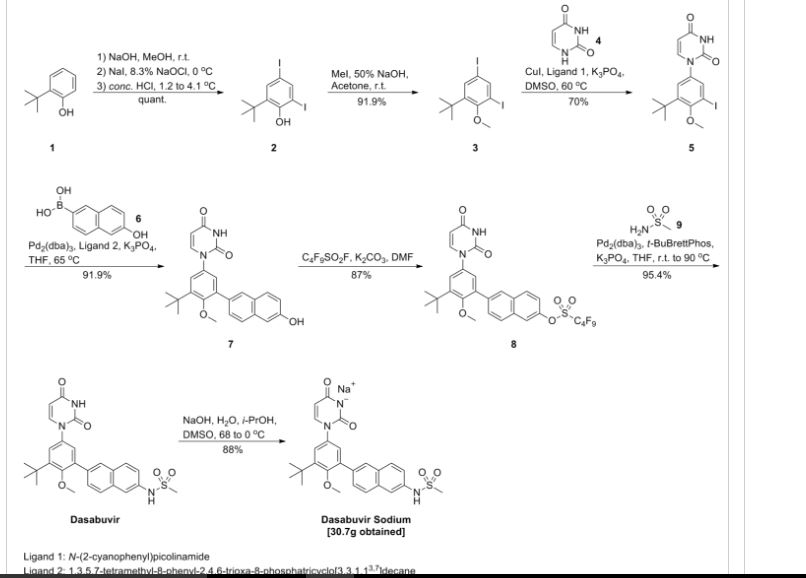






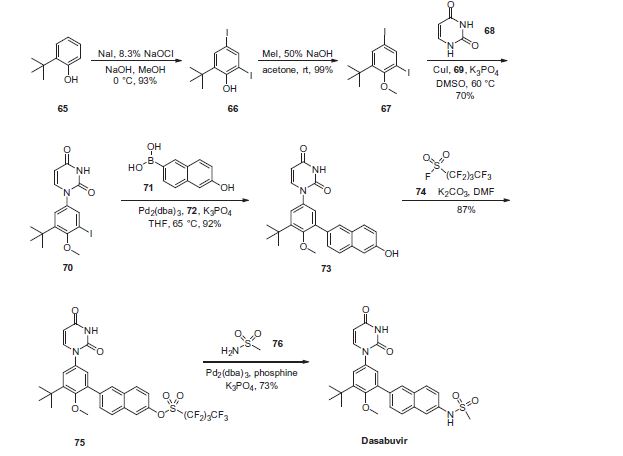



















 66
66
