WORLD RECORD VIEWS holder on THIS BLOG, ………live, by DR ANTHONY MELVIN CRASTO, Worldpeaceambassador, Worlddrugtracker, Helping millions, 100 million hits on google, pushing boundaries,2.5 lakh plus connections worldwide, 45 lakh plus VIEWS on this blog in 227 countries, 7 CONTINENTS ……A 90 % paralysed man in action for you, I am suffering from transverse mylitis and bound to a wheel chair, [THIS BLOG HOLDS WORLD RECORD VIEWS ]
DR ANTHONY MELVIN CRASTO, Born in Mumbai in 1964 and graduated from Mumbai University, Completed his Ph.D from ICT, 1991,Matunga, Mumbai, India, in Organic Chemistry, The thesis topic was Synthesis of Novel Pyrethroid Analogues, Currently he is working with AFRICURE PHARMA, ROW2TECH, NIPER-G, Department of Pharmaceuticals, Ministry of Chemicals and Fertilizers, Govt. of India as ADVISOR, earlier assignment was
with GLENMARK LIFE SCIENCES LTD, as CONSUlTANT, Retired from GLENMARK in Jan2022 Research Centre as Principal Scientist, Process Research (bulk actives) at Mahape, Navi Mumbai, India. Total Industry exp 32 plus yrs, Prior to joining Glenmark, he has worked with major multinationals like Hoechst Marion Roussel, now Sanofi, Searle India Ltd, now RPG lifesciences, etc. He has worked with notable scientists like Dr K Nagarajan, Dr Ralph Stapel, Prof S Seshadri, etc, He did custom synthesis for major multinationals in his career like BASF, Novartis, Sanofi, etc., He has worked in Discovery, Natural products, Bulk drugs, Generics, Intermediates, Fine chemicals, Neutraceuticals, GMP, Scaleups, etc, he is now helping millions, has 9 million plus hits on Google on all Organic chemistry websites. His friends call him Open superstar worlddrugtracker. His New Drug Approvals, Green Chemistry International, All about drugs, Eurekamoments, Organic spectroscopy international,
etc in organic chemistry are some most read blogs He has hands on experience in initiation and developing novel routes for drug molecules
and implementation them on commercial scale over a 32 PLUS year tenure till date Feb 2023, Around 35 plus products in his career. He has good knowledge of IPM, GMP, Regulatory aspects, he has several International patents published worldwide . He has good proficiency in Technology transfer, Spectroscopy, Stereochemistry, Synthesis, Polymorphism etc., He suffered a paralytic stroke/ Acute Transverse mylitis in Dec 2007 and is 90 %Paralysed, He is bound to a wheelchair, this seems to have injected feul in him to help chemists all around the world, he is more active than before and is pushing boundaries, He has 100 million plus hits on Google, 2.5 lakh plus connections on all networking sites, 100 Lakh plus views on dozen plus blogs, 227 countries, 7 continents, He makes himself available to all, contact him on +91 9323115463, email amcrasto@gmail.com, Twitter, @amcrasto , He lives and will die for his family, 90% paralysis cannot kill his soul., Notably he has 38 lakh plus views on New Drug Approvals Blog in 227 countries......https://newdrugapprovals.wordpress.com/ , He appreciates the help he gets from one and all, Friends, Family, Glenmark, Readers, Wellwishers, Doctors, Drug authorities, His Contacts, Physiotherapist, etc
He has total of 32 International and Indian awards
(4R–cis)-1-[[4-[[4-[3,3-Dibutyl-7-(dimethylamino)-2,3,4,5-tetrahydro-4-hydroxy-1,1-dioxido-1-benzothiepin-5-yl]phenoxy]methyl]phenyl]methyl]-4-aza-1-azoniabicyclo[2.2.2]octane Chloride Salt
It is well established that agents which inhibit the 20 transport of bile acids across the ileum can also cause a decrease in the level of cholesterol in blood serum. Stedronski, in “Interaction of bile acids and cholesterol with nonsystemic agents having hypocholesterolemic properties,” Biochimica et Biophysica Acta, 1210 (1994) 255- 25287, discusses biochemistry, physiology, and known active agents affecting bile acids and cholesterol.
A class of ileal bile acid transport-inhibiting compounds which was recently discovered to be useful for influencing the level of blood serum cholesterol is 30 tetrahydrobenzothiepine-l,l-dioxides (THBDO compounds). (U.S. Patent Application No. 08/816,065)
Some classes of compounds show enhanced potency as pharmaceutical therapeutics after they have been enantiomerically-enriched (see, for example, Richard B. Silverman, The Organic Chemistry of Drug Design and Drug Action, Academic Press, 1992, pp. 76-82) . Therefore, THBDO compounds that have been enantiomerically-enriched are of particular interest.
A class of chemistry useful as intermediates in the preparation of racemic THBDO compounds is tetrahydrobenzothiepine-1-oxides (THBO compounds) . THBDO compounds and THBO compounds possess chemical structures in which a phenyl ring is fused to a seven-member ring. A method of preparing enantiomerically-enriched samples of another phenyl/seven-member fused ring system, the benzothiazepines, is described by Higashikawa (JP 59144777) , where racemic benzothiazepine derivatives are optically resolved on a chromatographic column containing chiral crown ethers as a stationary phase. Although optical resolution is achieved, the Higashikawa method is limited to producing only small quantities of the enantiomerically-enriched benzothiazepine derivatives. Giordano (CA 2068231) reports the cyclization of (2S, 3S) -aminophenylthiopropionates in the presence of a phosphonic acid to produce (2S, 3S) -benzothiazepin-4-ones . However, that preparation is constrained by the need to use enantiomerically-enriched starting materials rather than racemic starting materials. In addition, the Giordano method controls the stereochemistry of the seven-member ring of the benzothiazepin-4-one only at the 2- and 3 -positions. The 4- and 5-positions of the seven-member ring of the benzothiazepin-4-one are not asymmetric centers, and the stereochemistry at these sites therefore cannot be controlled by the Giordano method. A method by which enantiomerically-enriched 1,5- benzothiazepin-3-hydroxy-4 (5H) -one compounds have been produced is through the asymmetric reduction of 1,5- benzothiazepin-3,4 (2H, 5H) -dione compounds, reported by Yamada, et al . (J“. Org. Chem. 1996, 61 (24), 8586-8590). The product is obtained by treating the racemic 1,5- benzothiazepin-3,4 (2H, 5H) -dione with the reaction product of an optically active alpha-amino acid and a reducing agent, for example sodium borohydride. Although a product with high optical purity was achieved, the method is limited by the use of a relatively expensive chemical reduction step.
The microbial reduction of racemic 1, 5-benzothiazepin- 3 , 4 (2H, 5H) -dione compounds to produce enantiomerically- enriched 1, 5-benzothiazepin-3-hydroxy-4 (5H) -one compounds is reported by Patel et al . , U.S. Patent 5,559,017. This method is limited by the inherent problems of maintaining a viable and pure bacterial culture of the appropriate species and variety. In addition, that method is limited in scale, producing only microgram quantities of the desired product. Until now, there have been no reported processes for preparing enantiomerically-enriched THBDO compounds or enantiomerically-enriched THBO compounds. Furthermore, there have been no reported processes for controlling the stereochemistry at the 4- and 5-positions of the seven- member rings of THBDO compounds or THBO compounds
FDA Grants Breakthrough Designation to Shire’s Rare GI Therapies
Tue, 06/14/2016
Shire announced that the U.S. Food and Drug Administration (FDA) has granted Breakthrough Therapy Designation for two investigational products for rare diseases: SHP621 (budesonide oral suspension, or BOS) for eosinophilic esophagitis (EoE), and SHP625 (maralixibat) for progressive familial intrahepatic cholestasis type 2 (PFIC2).
“Receiving Breakthrough Therapy Designation on two pipeline products this past week reflects the potential of our strong and innovative pipeline of more than 60 programs,” said Flemming Ornskov, M.D., MPH, and CEO, Shire. “Shire is committed to bringing innovation to the rare and specialty areas we focus on. We persevere to see compounds through the many stages of development through their challenges and successes, and always keep patients with unmet needs top of mind.”
EoE is a serious, chronic and rare disease that stems from an elevated number of eosinophils, a type of white blood cell, that infiltrate the walls of the esophagus. EoE is characterized by an inflammation of the esophagus that may lead to difficulty swallowing (dysphagia). The diagnosed prevalence of EoE ranges from approximately 15-55 cases per 100,000 persons, with high-end estimates reported by studies in Western regions.
PFIC refers to a group of autosomal-recessive liver disorders of childhood that disrupt bile formation and present with cholestasis. The symptoms of PFIC include severe itching of the skin (pruritus), and jaundice. PFIC is estimated to affect 1 in 50,000 to 1 in 100,000 births. PFIC2 is the most common type of PFIC, accounting for around half of cases.
According to the FDA, Breakthrough Therapy Designation is granted to a therapy that is intended to treat a serious or life-threatening disease or condition and preliminary clinical evidence indicates that the drug may demonstrate substantial improvement on one or more clinically significant endpoints over current standard of care. Under the designation, the FDA provides intensive guidance, organizational commitment involving senior managers, and eligibility for rolling and priority review of the application; this process helps ensure patients have access to therapies as soon as possible, pending approval. Breakthrough Therapy Designation does not guarantee that FDA will ultimately approve BOS for EoE or maralixibat for PFIC2, and the timing of any such approval is uncertain.
“On behalf of patients in the United States with EoE and PFIC2, we are so pleased that the FDA has granted Breakthrough Therapy Designation to BOS and maralixibat,” said Philip J. Vickers, Ph.D., Head of R&D, Shire. “We look forward to working with the agency to continue their development and, pending FDA approval, deliver these therapeutic options to the patients who need them most.”
It is well established that agents which inhibit the transport of bile acids across the tissue of the ileum can also cause a decrease in the levels of cholesterol in blood serum. Stedronski, in “Interaction of bile acids and cholesterol with nonsystemic agents having hypocholesterolemic properties,” Biochimica et Biophysica Acta, 1210 (1994) 255-287 discusses biochemistry, physiology, and known active agents surrounding bile acids and cholesterol. Bile acids are actively transported across the tissue of the ileum by an apical sodium co-dependent bile acid transporter (ASBT), alternatively known as an ileal bile acid transporter (IBAT). A class of ASBT-inhibiting compounds that was recently discovered to be useful for influencing the level of blood serum cholesterol comprises tetrahydrobenzothiepine oxides (THBO compounds, PCT Patent Application No. WO 96/08484). Further THBO compounds useful as ASBT inhibitors are described in PCT Patent Application No. WO 97/33882. Additional THBO compounds useful as ASBT inhibitors are described in U.S. Patent No. 5,994,391. Still further THBO compounds useful as ASBT inhibitors are described in PCT Patent Application No. WO 99/64409. Included in the THBO class are tetrahydrobenzo-thiepine-l -oxides and tetrahydrobenzothiepine- 1,1 -dioxides. THBO compounds possess chemical structures in which a phenyl ring is fused to a seven-member ring.
Published methods for the preparation of THBO compounds include the synthesis through an aromatic sulfone aldehyde intermediate. For example l-(2,2-dibutyl-3-oxopropylsulfonyl)-2-((4-methoxyphenyl)methyl)benzene (29) was cyclized with potassium t-butoxide to form tetrahydrobenzothiepine- 1,1 -dioxide (svn-24) as shown in Eq. 1.
Compound 29 was prepared by reacting 2-chloro-5-nitrobenzoic acid chloride with anisole in the presence of aluminum trichloride to produce a chlorobenzophenone compound; the chlorobenzophenone compound was reduced in the presence of trifluoromethanesulfonic acid and triethylsilane to produce a chlorodiphenylmethane compound; the chlorodiphenylmethane compound was treated with lithium sulfide and 2,2-dibutyl-3-(methanesulfonato)propanal to produce l-(2,2-dibutyl-3-oxopropylthio)-2-((4-methoxyphenyl)methyl)-4-dimethylaminobenzene (40); and 40 was oxidized with m-chloroperbenzoic acid to produce 29. The first step of that method of preparing compound 29 requires the use of a corrosive and reactive carboxylic acid chloride that was prepared by the reaction of the corresponding carboxylic acid with phosphorus pentachloride. Phosphorus pentachloride readily hydrolyzes to produce volatile and hazardous hydrogen chloride. The reaction of 2,2-dibutyl-3-(methanesulfonato)propanal with the lithium sulfide and the chlorodiphenylmethane compound required the intermediacy of a cyclic tin compound to make the of 2,2-dibutyl-3-(methanesulfonato)propanal. The tin compound is expensive and creates a toxic waste stream. In WO 97/33882 compound syn-24 was dealkylated using boron tribromide to produce the phenol compound 28. Boron tribromide is a corrosive and hazardous material that generates hydrogen bromide gas and requires special handling. Upon hydrolysis, boron tribromide also produces borate salts that are costly and time-consuming to separate and dispose of.
An alternative method of preparing THBO compounds was described in WO 97/33882, wherein a 1,3-propanediol was reacted with thionyl chloride to form a cyclic sulfite compound. The cyclic sulfite compound was oxidized to produce a cyclic sulfate compound. The cyclic sulfate was condensed with a 2-methylthiophenol that had been deprotonated with sodium hydride. The product of the condensation was a (2-methylphenyl) (3′-hydroxypropyl)thioether compound. The thioether compound was oxidized to form an thioether aldehyde compound. The thioether aldehyde compound was further oxidized to form an aldehyde sulfone compound which in turn was cyclized in the presence of potassium t-butoxide to form a 4-hydroxytetrahydrobenzothiepine 1,1 -dioxide compound. This cyclic sulfate route to THBO compounds requires an expensive catalyst. Additionally it requires the use of SOCI2, which in turn requires special equipment to handle. PCT Patent Application No. WO 97/33882 describes a method by which the phenol compound 28 was reacted at its phenol hydroxyl group to attach a variety of functional groups to the molecule, such as a quaternary ammonium group. For example, (4R,5R)-28 was reacted with l,4-bis(chloromethyl)benzene (?,??’-dichloro-p-xylene) to produce the chloromethyl benzyl- ether (4R,5R)-27. Compound (4R,5R)-27 was treated with diazabicyclo[2.2.2]octane (DABCO) to produce (4R,5R)-l-((4-(4-(3,3-dibutyl-7-(dimemylamino)-2,3,4,5-tetrahydro-4-hydroxy-l , 1 -dioxido-1 -benzothiepin-5-yl)phenoxy)methyl)phenyl)methyl-4-aza-l-azomabicyclo[2.2.2]octane chloride (41). This method suffers from low yields because of a propensity for two molecules of compound (4R,5R)-28 to react with one molecule of l,4-bis(chloromethyl)benzene to form a bis(benzothiepine) adduct. Once the bis-adduct forms, the reactive chloromethyl group of compound (4R,5R)-27 is not available to react with an amine to form the quaternary ammonium product.
A method of preparing enantiomerically enriched tetrahydrobenzothiepine oxides is described in PCT Patent Application No. WO 99/32478. In that method, an aryl-3- hydroxypropylsulfide compound was oxidized with an asymmetric oxidizing agent, for example (lR (->(8,9-dichloro-10-camphorsulfonyl)oxaziridine, to yield a chiral aryl-3-hydroxypropylsulfoxide. Reaction of the aryl-3-hydroxypropylsulfoxide with an oxidizing agent such as sulfur trioxide pyridine complex yielded an aryl-3-propanalsulfoxide. The aryl- 3-propanalsulfoxide was cyclized with a base such as potassium t-butoxide to enantioselectively produce a tetrahydrobenzothiepine- 1 -oxide. The tetrahydrobenzothiepine- 1 -oxide was further oxidized to produce a tetrahydrobenzothiepine- 1 , 1 -dioxide. Although this method could produce tetrahydrobenzothiepine- 1,1 -dioxide compounds of high enantiomeric purity, it requires the use of an expensive asymmetric oxidizing agent. Some 5-amidobenzothiepine compounds and methods to make them are described in
PCT Patent Application Number WO 92/18462. In Svnlett. 9, 943-944(1995) 2-bromophenyl 3-benzoyloxy-l-buten-4-yl sulfone was treated with tributyl tin hydride and AIBN to produce 3-benzoyloxytetrahydrobenzothiepine-1,1 -dioxide. In addition to forming the desired ASBT inhibitors, it is also desirable to form such
ASBT inhibitors of higher purity and having lower levels of residual solvent impurities. This is especially so with respect to ASBT inhibitors having a positively charged substituent, for example, the compounds designated as 41 (supra) and 60 (infra). It is further desirable to provide methods for making such high purity ASBT inhibitors.
( 4R, 5R) -26 A 1000 mL 4 neck jacketed Ace reactor flask was fitted with a mechanical stirrer, a nitrogen inlet, an addition funnel or condenser or distilling head with receiver, a thermocouple, four internal baffles and a 28 mm Teflon turbine agitator. The flask was purged with nitrogen gas and charged with 25.0 grams of (4R,5R)-28 and 125 mL of N,N-dimethylacetamide (DMAC). To this was added 4.2 grams of 50% sodium hydroxide. The mixture was heated to 50°C and stiπed for 15 minutes. To the flask was added 8.3 grams of 55 dissolved in 10 mL of DMAC, all at once. The temperature was held at 50°C for 24 hrs. To the flask was added 250 mL of toluene followed by 125 mL of dilution water. The mixture was stiπed for 15 minutes and the layers were then allowed to separate at 50°C. The flask was then charged with 125 mL of saturated sodium chloride solution and stiπed 15 minutes. Layers separated cleanly in 30 seconds at 50°C. Approximately half of the solvent was distilled off under vacuum at 50°C. The residual reaction mixture contained (4R,5R)-26.
Step 2. Preparation of (4R.5RV27.
( 4R, 5R) -27 Toluene was charged back to the reaction mixture of Step 1 and the mixture was cooled to 35°C. To the mixture was then added 7.0 grams of thionyl chloride over 5 minutes. The reaction was exothermic and reached 39°C. The reaction turned cloudy on first addition of thionyl chloride, partially cleared then finally remained cloudy. The mixture was stirred for 0.5 hr and was then washed with 0.25N NaOH. The mixture appeared to form a small amount of solids that diminished on stirring, and the layers cleanly separated. The solvent was distilled to a minimum stir volume under vacuum at 50°C. The residual reaction mixture contained (4R,5R)-27.
Step 3. Preparation of 41. To the reaction mixture of Step 2 was charged with 350 mL of methyl ethyl ketone (MEK) followed by 10.5 mL water and 6.4 grams of diazabicyclo[2.2.2]octane (DABCO) dissolved in 10 mL of MEK. The mixture was heated to reflux, and HPLC showed <0.5% of (4R,5R)-27. The reaction remained homogenous initially then crystallized at the completion of the reaction. An additional 5.3 mL of water was charged to the flask to redissolve product. Approximately 160 mL of solvent was then distilled off at atmospheric pressure. The mixture started to form crystals after 70 mL of solvent was distilled. Water separated out of distillate indicating a ternary azeotrope between toluene, water and methyl ethyl ketone (MEK). The mixture was then cooled to 25°C. The solids were filtered and washed with 150 mL MEK, and let dry under vacuum at 60°C. Isolated 29.8.0 g of off-white crystalline 4 Example 11a. Alternate Preparation of (4R,5R)-l-((4-(4-(3,3-dibutyl-7-(dimemylamino)-2,3,4,5-tetrahydro- 4-hydroxy- 1 , 1 -dioxido- 1 -benzithiepin-5-yl)phenoxy)methyl)phenyl)methyl-4-aza- 1 – azoniabicyclo[2.2.2]octane chloride, Form II of 41
A 1000 mL 4 neck jacketed Ace reactor flask is fitted with a mechanical stiπer, a nitrogen inlet, an addition funnel or condenser or distilling head with receiver, a thermocouple, four internal baffles and a 28 mm Teflon turbine agitator. The flask is purged with nitrogen gas and charged with 25.0 grams of (4R,5R)-28 and 100 mL of N,N-dimethylacetamide (DMAC). The mixture is heated to 50°C and to it is added 4.02 grams of 50% sodium hydroxide. The mixture is stiπed for 30 minutes. To the flask is added 8.7 grams of 55 dissolved in 12.5 mL of DMAC, all at once. The charge vessel is washed with 12.5 mL DMAC and the wash is added to the reactor. The reactor is stiπed for 3 hours. To the reactor is added 0.19 mL of 49.4% aq. NaOH and the mixture is stirred for 2 hours. To the mixture is added 0.9 g DABCO dissolved in 12.5 mL DMAC. The mixture is stiπed 30 to 60 minutes at 50°C. To the flask is added 225 mL of toluene followed by 125 mL of dilution water. The mixture is stiπed for 15 minutes and the layers are then allowed to separate at 50°C. The bottom aqueous layer is removed but any rag layer is retained. The flask is then charged with 175 mL of 5% hydrochloric acid solution and stiπed 15 minutes. Layers are separated at 50°C to remove the bottom aqueous layer, discarding any rag layer with the aqueous layer. Approximately half of the solvent is distilled off under vacuum at a maximum pot temperature of 80°C. The residual reaction mixture contains (4R,5R)-26.
Step 2. Preparation of (4R.5RV27.
Toluene (225 mL) is charged back to the reaction mixture of Step 1 and the mixture is cooled to 30°C. To the mixture is then added 6.7 grams of thionyl chloride over 30 to 45 minutes. The temperature is maintained below 35°C. The reaction turns cloudy on first addition of thionyl chloride, then at about 30 minutes the layers go back together and form a clear mixture. The mixture is stiπed for 0.5 hr and is then charged with 156.6 mL of 4% NaOH wash over a 30 minute period. The addition of the wash is stopped when the pH of the mixture reaches’ 8.0 to 10.0. The bottom aqueous layer is removed at 30°C and any rag layer is retained with the organic layer. To the mixture is charged 175 mL of saturated NaCl wash with agitation. The layers are separated at 30°C and the bottom aqueous layer is removed, discarding any rag layer with the aqueous layer. The solvent is distilled to a minimum stir volume under vacuum at 80°C. The residual reaction mixture contains (4R,5R)-27.
Step 3. Preparation of 41. To the reaction mixture of Step 2 is charged 325 mL of methyl ethyl ketone (MEK) and 13 mL water. Next, the reactor is charged 6.2 grams of diazabicyclo[2.2.2]octane (DABCO) dissolved in 25 mL of MEK. The mixture is heated to reflux and held for 30 minutes. Approximately 10% of solvent volume is then distilled off. The mixture starts to form crystals during distillation. The mixture is then cooled to 20°C for 1 hour. The off-white crystalline 41 (Form U) is filtered and washed with 50 mL MEK, and let dry under vacuum at 100°C.
Example lib. Alternate Preparation of (4R,5R)-1 -((4-(4-(3,3-dibutyl-7-(dimethylamino)-2,3,4,5-tetrahydro- 4-hydroxy- 1 , 1 -dioxido- 1 -benzithiepin-5-yl)phenoxy)methyl)phenyl)methyl-4-aza- 1 – azoniabicyclo[2.2.2]octane chloride, Form II of 41
A 1000 mL 4 neck jacketed Ace reactor flask is fitted with a mechanical stiπer, a nitrogen inlet, an addition funnel or condenser or distilling head with receiver, a thermocouple, four internal baffles and a Teflon turbine agitator. The flask is purged with nitrogen gas and charged with 25.0 grams of (4R,5R)-28 and 125 mL of N,N-dimethylacetamide (DMAC). The mixture is heated to 50°C and to it is added 7.11 grams of 30% sodium hydroxide over a period of 15 to 30 minutes with agitation. The mixture is stiπed for 30 minutes. To the flask is added 9.5 grams of solid 55. The reactor is stiπed for 3 hours. To the mixture is added 1.2 g of solid DABCO. The mixture is stiπed 30 to 60 minutes at 50°C. To the flask is added 225 mL of toluene followed by 125 mL of water. The mixture is stirred for 15 minutes and the layers are then allowed to separate at 50°C. The bottom aqueous layer is removed but any rag layer is retained with the organic layer. The flask is then charged with 175 mL of 5% hydrochloric acid solution and stirred 15 minutes. Layers are separated at 50°C to remove the bottom aqueous layer, discarding any rag layer with the aqueous layer. The flask is then charged with 225 mL of water and stirred 15 minutes. The layers are allowed to separate at 50°C. The bottom aqueous layer is removed, discarding any rag layer with the aqueous layer. Approximately half of the solvent is distilled off under vacuum at a maximum pot temperature of 80°C. The residual reaction mixture contains (4R,5R)-26.
Step 2. Preparation of (4R.5RV27.
Toluene (112.5 mL) is charged back to the reaction mixture of Step 1 and the mixture is cooled to 25°C. To the mixture is then added 7.3 grams of thionyl chloride over 15 to 45 minutes. The temperature of the mixture is maintained above 20°C and below 40°C. The reaction turns cloudy on first addition of thionyl chloride, then at about 30 minutes the layers go back together and form a clear mixture. The mixture is then charged with 179.5 mL of 4% NaOH wash over a 30 minute period. The mixture is maintained above 20°C and below 40°C during this time. The addition of the wash is stopped when the pH of the mixture reaches 8.0 to 10.0. The mixture is then allowed to separate at 40°C for at least one hour.
The bottom aqueous layer is removed and any rag layer is retained with the organic layer. To the mixture is charged 200 mL of dilution water. The mixture is stiπed for 15 minutes and then allowed to separate at 40°C for at least one hour. The bottom aqueous layer is removed, discarding any rag layer with the aqueous layer. The solvent is distilled to a minimum stir volume under vacuum at 80°C. The residual reaction mixture contains (4R,5R)-2 .
Step 3. Preparation of 41. To the reaction mixture of Step 2 is charged 350 mL of methyl ethyl ketone (MEK) and 7 mL water. The mixture is stiπed for 15 minutes and the temperature of the mixture is adjusted to 25°C. Next, the reactor is charged with 6.7 grams of solid diazabicyclo[2.2.2]octane (DABCO). The mixture is maintained at 25°C for three to four hours. It is then heated to 65°C and maintained at that temperature for 30 minutes. The mixture is then cooled to 25°C for 1 hour. The off-white crystalline 41 (Form II) is filtered and washed with 50 mL MEK, and let dry under vacuum at 100°C.
Example 12. Alternate preparation of (4R,5R)-1 -((4-(4-(3,3-dibutyl-7-(dimethylamino)-2,3,4,5-tetrahydro- 4-hydroxy- 1 , 1 -dioxido- 1 -benzithiepin-5-yl)phenoxy)methyl)phenyl)methyl-4-aza- 1 – azoniabicyclo[2.2.2]octane chloride, Form I of 41
(4R,5R)-27 (2.82 kg dry basis, 4.7 mol) was dissolved in MTBE (9.4 L). The solution of (4R,5R)-22 was passed through a 0.2 mm filter cartridge into the feeding vessel. The flask and was rinsed with MTBE (2 x 2.5 L). The obtained solution as passed through the cartridge filter and added to the solution of (4R,5R)-2 in the feeding vessel. DABCO (diazabicyclo[2.2.2]octane, 0.784 kg, 7.0 mol) was dissolved in MeOH (14.2 L). The DABCO solution was passed through the filter cartridge into the 100 L nitrogen-flushed reactor. The Pyrex bottle and the cartridge filter were rinsed with MeOH (7.5 L) and the solution was added to the reactor. The (4R,5R)-22 solution was added from the feeding vessel into the reactor at 37°C over a period of 10 min, while stirring. Methanol (6.5 L) was added to the Pyrex bottle and via the cartridge filter added to the feeding vessel to rinse the remaining (4R,5R)-2 into the reactor. The reaction mixture was brought to 50-60°C over 10-20 min and stiπed at that temperature for about 1 h. The mixture was cooled to 20-25°C over a period of 1 h. To the reaction mixture, methyl t-butyl ether (MTBE) (42 L) was added over a period of 1 h and stiπed for a minimum of 1 h at 20 – 25°C. The suspension was filtered through a Buchner funnel. The reactor and the filter cake were washed with MTBE (2 x 14 L). The solids were dried on a rotary evaporator in a 20 L flask at 400 – 12 mbar, 40°C, for 22 h. A white crystalline solid was obtained. The yield of 4 . (Form I) was 3.08 kg (2.97 kg dry, 93.8 %) and the purity 99.7 area % (HPLC; Kromasil C 4, 250 x 4.6 mm column; 0.05% TFA in H2O/0.05% TFA in ACN gradient, UV detection at 215 nm).
Example 12a. Conversion of Form I of Compound 41 into Form II of Compound 41.
To 10.0 grams of Form I of 4 . in a 400 mL jacketed reactor is added 140 mL of MEK. The reactor is stirred (358 φm) for 10 minutes at 23 °C for 10 minutes and the stirring rate is then changed to 178 φm. The suspension is heated to reflux over 1 hour using a programmed temperature ramp (0.95°C/minute) using batch temperature control (cascade mode). The delta Tmaχ is set to 5°C. The mixture is held at reflux for 1 hour. The mixture is cooled to
25°C. After 3 hours at 25°C, a sample of the mixture is collected by filtration. Filtration is rapid (seconds) and the filtrate is clear and colorless. The white solid is dried in a vacuum oven (80°C, 25 in. Hg) to give a white solid. The remainder of the suspension is stirred at 25°C for 18 hours. The mixture is filtered and the cake starts to shrink as the mother liquor reaches the top of the cake. The filtration is stopped and the reactor is rinsed with 14 mL of MEK. The reactor stirrer speed is increased from 100 to 300 φm to rinse the reactor. The rinse is added to the filter and the solid is dried with a rapid air flow for 5 minutes. The solid is dried in a vacuum oven at 25 in. Hg for 84 hours to give Form II of 4
Department of Discovery Chemistry and Department of Cardiovascular Disease, Pharmacia, 700 Chesterfield Parkway W, Chesterfield, Missouri 63017, Office of Science and Technology, Chemical Science Division, Pharmacia, 800 Lindbergh Boulevard, Creve Coeur, Missouri 63167, Department of Pharmaceutical Sciences, Pharmacia, Skokie, Illinois, and Department of Chemistry, University of Missouri, St. Louis, Missouri
In the preceding paper several compounds were reported as potent apical sodium-codependent bile acid transporter (ASBT) inhibitors. Since the primary site for active bile acid reabsorption is via ASBT, which is localized on the luminal surface of the distal ileum, we reasoned that a nonsystemic inhibitor would be desirable to minimize or eliminate potential systemic side effects of an absorbed drug. To ensure bioequivalency and product stability, it was also essential that we identify a nonhygroscopic inhibitor in its most stable crystalline form. A series of benzothiepines were prepared to refine the structure−activity relationship of the substituted phenyl ring at the 5-position of benzothiepine ring and to identify potent, crystalline, nonhygroscopic, and efficacious ASBT inhibitors with low systemic exposure.
compd
R
IC50 (nM)b
hygroscp I wt gain (%)c
hygroscp II % wt gain (%)d
crystallinitye
74
OCH2C6H4(p)CH2(N+)DB
0.28
1.59
2.1
yes
(4R–cis)-1-[[4-[[4-[3,3-Dibutyl-7-(dimethylamino)-2,3,4,5-tetrahydro-4-hydroxy-1,1-dioxido-1-benzothiepin-5-yl]phenoxy]methyl]phenyl]methyl]-4-aza-1-azoniabicyclo[2.2.2]octane Chloride Salt(74). Following a similar procedure as in General Method B, the title compound 74 was prepared from the corresponding chloromethyl benzyl ether and DABCO as a white solid, mp 223−230 °C (dec); 1H NMR (CDCl3) δ 0.89 (m, 6H), 1.27−1.52 (br m, 10H), 1.63 (m, 1H), 2.20 (m, 1H), 2.81 (s, 6H), 3.06 (ABq, JAB = 15.1 Hz, J = 43.3 Hz, 2H), 3.16 (s, 6H), 3.76 (s, 6H), 4.11 (d, J = 7.7 Hz, 1H), 5.09 (s, 2H), 5.14 (s, 2H), 5.48 (s, 1H), 5.96 (s, 1H), 6.49 (d, J = 8.9 Hz, 1H), 6.99 (d, J = 8.0 Hz, 2H), 7.26 (m, 1H), 7.44 (d, J = 8.0 Hz, 2H), 7.52 (d, J = 7.4 Hz, 2H), 7.68 (d, J = 7.4 Hz, 2H), 7.87 (d, J = 8.9 Hz, 1H). HRMS calcd for C40H56N3O4S: 674.3992; found, 674.4005. Anal. Calcd for C40H56N3O4S: ‘ C, 67.62; H, 7.95; N, 5.92; S, 4.51. Found: C, 67.48; H, 8.32; N, 5.85; S, 4.60.
a All compounds were prepared using method B in Scheme 3.b Taurocholate is transported across the baby hamster kidney cell membrane.c % weight gain in a 25 °C, 57% humidity chamber for 2 weeks.d % weight gain in a 40 °C, 80% humidity chamber for 2 weeks.e Crystallinity as determined by X-ray powder diffraction analysis.f (N+)DB is a DABCO terminal group with the quaternary ammonium attached to the linke
Example 10. Preparation of enantiomerically-enriched (4R.5R)-1- r.4- r _4- .3.3 -Dibutyl-7- (dimethylamino) -2.3 ,4.5- tetrahydro-4-hydroxy-1, l-dioxido-l-benzothiepin-5- yl] henoxy] ethyl] phenyl1methyl] -4-aza-l- azoniabicyclo [2.2.2] octane chloride ( (4R,5R) -XXVII) ♦
( (4R,5R) -XXVII) * = chiral center
Step 1. Preparation of 4-flUoro-2- ( (4- methoxyphenyl) methyl) -phenol To a stirred solution of 23.66 g of 95% sodium hydride (0.94 mol) in 600 mL of dry toluene was added 100.0 g of 4- fluorophenol (0.89 mol) at 0°C. The mixture was stirred at 90°C for 1 hour until gas evolution stopped. The mixture was cooled down to room temperature and a solution of 139.71 g of 3 -methoxybenzyl chloride (0.89 mol) in 400 mL of dry toluene was added. After refluxing for 24 hours, the mixture was cooled to room temperature and quenched with 500 mL of water. The organic layer was separated, dried over MgS04, and concentrated under high vacuum. The remaining starting materials were removed by distillation. The crude dark red oil was filtered through a layer of 1 L of silica gel with neat hexane to yield 53.00 g (25.6%) of the product as a pink solid: *H NMR (CDC13) d 3.79 (s, 3H) , 3.90 (s, 2H) , 4.58 (s, IH) , 6.70-6.74 (m, IH) , 6.79-6.88 (m, 4H) , 7.11-7.16 (m, 2H) .
Step 2. Preparation of 4-fluoro-2- ( (4- methoxyphenyl) methyl) -thiophenol
Step 2a. Preparation of thiocarbamate To a stirred solution of 50.00 g (215.30 mmol) of 4- fluoro-2- ( ( -methoxyphenyl) methyl) -phenol in 500 mL of dry DMF was added 11.20 g of 60% sodium hydride dispersion in mineral oil (279.90 mmol) at 2°C. The mixture was allowed to warm to room temperature and 26.61 g of dimethylthiocarbamoyl chloride (215.30 mmol) was added. The reaction mixture was stirred at room temperature overnight. The mixture was quenched with 100 mL of water in an ice bath. The solution was extracted with 500 mL of diethyl ether. The ether solution was washed with 500 mL of water and 500 mL of brine. The ether solution was dried over MgS04 and stripped to dryness. The crude product was filtered through a plug of 500 mL silica gel using 5% ethyl acetate/hexane to yield 48.00 g (69.8%) of the product as a pale white solid: XH NMR (CDC13) d 3.21 (s, 3H) , 3.46 (s, 3H) , 3.80 (s, 3H) , 3.82 (s, 2H) , 6.78-6.86 (m, 3H) , 6.90- 7.00 (m, 2H) , 7.09 (d, J = 8.7 Hz, 2H) .
Step 2b. Rearrangement and hydrolysis of thiocarbamate to 4-fluoro-2- ( (4 -methoxyphenyl) methyl) -thiophenol A stirred solution of 48.00 g (150.29 mmol) of thiocarbamate (obtained from Step 2a) in 200 mL of diphenyl ether was refluxed at 270°C overnight. The solution was cooled down to room temperature and filtered through 1 L of silica gel with 2 L of hexane to remove phenyl ether. The rearrangement product was washed with 5% ethyl acetate/hexane to give 46.00 g (95.8%) of the product as a pale yellow solid: XH NMR (CDC13) d 3.02 (s, 3H) , 3.10 (s, 3H) , 3.80 (s, 3H) , 4.07 (s, 2H) , 6.82-6.86 (m, 3H) , 6.93 (dt, J = 8.4 Hz, 2.7 Hz, IH) , 7.08 (d, J = 8.7 Hz, 2H) , 7.49 (dd, J = 6.0 Hz, 8.7 Hz, IH) . To a solution of 46.00 g (144.02 mmol) of the rearrangement product (above) in 200 mL of methanol and 200 mL of THF was added 17.28 g of NaOH (432.06 mmol) . The mixture was refluxed under nitrogen overnight . The solvents were evaporated off and 200 mL of water was added. The aqueous solution was washed with 200 mL of diethyl ether twice and placed in an ice bath. The aqueous mixture was acidified to pH 6 with concentrated HCl solution. The solution was extracted with 300 mL of diethyl ether twice. The ether layers were combined, dried over MgS04 and stripped to dryness to afford 27.00 g (75.5%) of the product as a brown oil: XH NMR (CDC13) d 3.24 (s, IH) , 3.80 (s, 3H) , 3.99 (s, 2H) , 6.81-6.87 (m, 4H) , 7.09 (d, J = 8.7 Hz, 2H) , 7.27- 7.33 (m, IH) .
Step 3. Preparation of dibutyl cyclic sulfate
Step 3a. Preparation of 2 , 2-dibutyl-l, 3-propanediol . To a stirred solution of di-butyl-diethylmalonate (Aldrich) (150g, 0.55 mol in dry THF (700ml) in an acetone/dry ice bath was added LAH (1 M THF) 662 ml (1.2 eq. , 0.66 mol) dropwise maintaining the temperature between -20 to 0°C. The reaction was stirred at RT overnight. The reaction was cooled to -20°C and 40 ml of water, and 80 mL of 10% NaOH and 80 ml of water were added dropwise. The resulting suspension was filtered. The filtrate was dried over sodium sulphate and concentrated in vacuo to give diol 598.4 g (yield 95%) as an oil. MS spectra and proton and carbon NMR spectra were consistent with the product.
Step 3b. Preparation of dibutyl cyclic sulfite
A solution of 2 , 2-dibutyl-l, 3-propanediol (103 g, 0.548 0 mol, obtained from Step 3a) and triethylamine (221 g, 2.19 mol) in anhydrous methylene chloride (500 ml) was stirred at 0°C under nitrogen. To the mixture, thionyl chloride (97.8* g, 0.‘82 mol) was added dropwise and within 5 min the solution turned yellow and then black when the addition was 5 completed within half an hour. The reaction mixture was stirred for 3 hrs. at 0°C. GC showed that there was no starting material left. The mixture was washed with ice water twice then with brine twice . The organic phase was dried over magnesium sulfate and concentrated under vacuum 0 to give 128 g (100%) of the dibutyl cyclic sulfite as a black oil. Mass spectrum (MS) was consistent with the product .
Step 3c. Oxidation of dibutyl cyclic sulfite to 5 dibutyl cyclic sulfate
To a solution of the dibutyl cyclic sulfite (127.5 g , 0.54 mol, obtained from Step 3b) in 600 ml acetonitrile and 500 ml of water cooled in an ice bath under nitrogen was added ruthenium (III) chloride (1 g) and sodium periodate 0 (233 g, 1.08 mol) . The reaction was stirred overnight and the color of the solution turned black. GC showed that there was no starting material left. The mixture was extracted with 300 ml of ether and the ether extract was washed three times with brine. The organic phase was dried over magnesium sulfate and passed through celite. The filtrate was 5 concentrated under vacuum and to give 133 g (97.8%) of the dibutyl cyclic sulfate as an oil. Proton and carbon NMR and MS were consistent with the product.
Step 4. Preparation of aryl-3-hydroxypropylsulfide
10 To a stirred solution of 27.00 g (108.73 mmol) of 4- fluoro-2- ( (4-methoxyphenyl) methyl) thiophenol (obtained from Step 2) in 270 mL of diglyme was added 4.35 g of 60% sodium-, hydride dispersion in mineral oil (108.73 mmol) at 0°C. After gas evolution ceased, 29.94 g (119.60 mmol) of the
15 dibutyl cyclic sulfate (obtained from Step 3c) was added at 0°C and stirred for 10 minutes. The mixture was allowed to warm up to room temperature and stirred overnight. The solvent was evaporated and 200 mL of water was added. The solution was washed with 200 mL of diethyl ether and added
2025 mL of concentrated sulfuric acid to make a 2.0 M solution that was refluxed overnight. The solution was extracted with ethyl acetate and the organic solution was dried over MgS04 and concentrated in vacuo. The crude aryl-3 – hydroxypropylsulfide was purified by silica gel
25 chromatography (Waters Prep 500) using 8% ethyl acetate/hexane to yield 33.00 g (72.5%) of the product as a light brown oil: E NMR (CDC13) d 0.90 (t, J = 7.1 Hz, 6H) , 1.14-1.34 (m, 12H) , 2.82 (s, 2H) , 3.48 (s, 2H) , 3.79 (s, 3H) , 4.10 (s, 2H) , 6.77-6.92 (m, 4H) , 7.09 (d, J = 8.7 Hz,
To a stirred solution of 20.00 g (47.78 mmol) of aryl- 53 -hydroxypropylsulfide (obtained from Step 4) in 1 L of methylene chloride was added 31.50 g of 96% (12?) – ( -) – (8 , 8- dichloro-10-camphor-sulfonyl) oxaziridine (100.34 mmol, Aldrich) at 2°C. After all the oxaziridine dissolved the mixture was placed into a -30 °C freezer for 72 hours. The
10 solvent was evaporated and the crude solid was washed with 1 L of hexane. The white solid was filtered off and the hexane solution was concentrated in vacuo. The crude oil was purified on a silica gel column (Waters Prep 500) using 15% ethyl acetate/hexane to afford 19.00 g (95%) of the
207.00 (d, J = 8.1 Hz, 2H) , 7.18-7.23 (m, IH) , 7.99-8.04 (m, IH) . Enantiomeric excess was determined by chiral HPLC on a (2?,2?) -Whelk-0 column using 5% ethanol/hexane as the eluent. It showed to be 78% e.e. with the first eluting peak as the major product.
25
Step 6. Preparation of enantiomerically-enriched aryl-3- propanalsulfoxide
To a stirred solution of 13.27 g of triethylamine (131.16 mmol, Aldrich) in 200 mL dimethyl sulfoxide were
30 added 19.00 g (43.72 mmol) of enantiomerically-enriched aryl-3 -hydroxypropylsulfoxide (obtained from Step 5) and 20.96 g of sulfur trioxide-pyridine (131.16 mmol, Aldrich) at room temperature. After the mixture was stirred at room temperature for 48 hours, 500 mL of water was added to the mixture and stirred vigorously. The mixture was then 5 extracted with 500 mL of ethyl acetate twice. The ethyl acetate layer was separated, dried over MgS04, and concentrated in vacuo. The crude oil was filtered through 500 mL of silica gel using 15% ethyl acetate/hexane to give 17.30 g (91%) of the enantiomerically-enriched aryl-3-
Step 7. Preparation of the enantiomerically-enriched tetrahydrobenzothiepine-1-oxide (4R, 5R)
20 To a stirred solution of 17.30 g (39.99 mmol) of enantiomerically-enriched aryl-3 -propanalsulfoxide (obtained from Step 6) in 300 mL of dry THF at -15°C was added 48 mL of 1.0 M potassium t-butoxide in THF (1.2 equivalents) under nitrogen. The solution was stirred at -15°C for 4 hours.
25 The solution was then quenched with 100 mL of water and neutralized with 4 mL of concentrated HCl solution at 0°C. The THF layer was separated, dried over MgS04, and concentrated in vacuo. The enantiomerically-enriched tetrahydrobenzothiepine-1-oxide (4R,5R) was purified by
To a stirred solution of 13.44 g (31.07 mmol) of enantiomerically-enriched tetrahydrobenzothiepine-1-oxide (obtained from Step 7) in 150 mL of methylene chloride was added 9.46 g of 68% m-chloroperoxybenzoic acid (37.28 mmol,
15 Sigma) at 0 °C. After stirring at 0 °C for 2 hours, the mixture was allowed to warm up to room temperature and stirred for 4 hours. 50 mL of saturated Na2S03 was added into the mixture and stirred for 30 minutes. The solution was then neutralized with 50 mL of saturated NaHC03 solution.
20 The methylene chloride layer was separated, dried over MgS04, and concentrated in vacuo to give 13.00 g (97.5%) of the enantiomerically-enriched tetrahydrobenzothiepine-1, 1- dioxide (4R,5R) as a light yellow solid: ‘H NMR (CDC13) d 0.89-0.95 (m, 6H) , 1.09-1.42 (m, 12H) , 2.16-2.26 (m, IH) ,
30 Step 9. Preparation of enantiomerically-enriched 7-
(dimethylamino) tetrahydrobenzothiepine-1 , 1-dioxide (4R.5R) – To a solution of 13.00 g (28.98 mmol) of enantiomerically-enriched tetrahydrobenzothiepine-1, 1- dioxide (obtained from Step 8) in 73 mL of dimethylamine (2.0 M in THF, 146 mmol) in a Parr Reactor was added ca . 20 5 mL of neat dimethylamine . The mixture was sealed and stirred at 110 °C overnight, and cooled to ambient temperature. The excess dimethylamine was evaporated. The crude oil was dissolved in 200 mL of ethyl acetate and washed with 100 mL of water, dried over MgS04 and
10 concentrated in vacuo. Purification on a silica gel column (Waters Prep 500) using 20% ethyl acetate/hexane gave 12.43 g (90.5%) of the enantiomerically- enriched 7- (dimethylamino) tetrahydrobenzothiepine-1, 1-dioxide (4R, 5R) as a colorless solid: *H NMR (CDC13) d 0.87-0.93 (m, 6H) ,
20 IH) . The product was determined to have 78% e.e. by chiral HPLC on a Chiralpak AD column using 5% ethanol/hexane as the eluent. Recrystallization of this solid from ethyl acetate/hexane gave 1.70 g of the racemic product. The remaining solution was concentrated and recrystallized to
25 give 9.8 g of colorless solid. Enantiomeric excess of this solid was determined by chiral HPLC on a Chiralpak AD column using 5% ethanol/hexane as the eluent. It showed to have 96% e.e with the first eluting peak as the major product.
30 Step 10: Demethylation of 5- (4 ‘ -methoxyphenyl) -7-
(dimethylamino) tetrahydrobenzothiepine-1.1-dioxide (4R, 5R) To a solution of 47 g (99 mmol) of enantiomeric- enriched (dimethylamino) tetrahydrobenzothiepine-1, 1-dioxide (obtained from Step 9) in 500 mL of methylene chloride at -10 °C was added dropwise a solution of boron tribromide (297 mL, 1M in methylene chloride, 297 mmol), and the resulting solution was stirred cold (-5 °C to 0 °C) for 1 hour or until the reaction was complete. The reaction was cooled in an acetone-dry ice bath at -10 °C, and slowly quenched with 300 mL of water. The mixture was warmed to 10 °C, and further diluted with 300 mL of saturated sodium bicarbonate solution to neutralize the mixture. The aqueous layer was separated and extracted with 300 mL of methylene chloride, and the combined extracts were washed with 200 mL of water, brine, dried over MgS04 and concentrated in vacuo. The residue was dissolved in 500 mL of ethyl acetate and stirred with 50 mL of glacial acetic acid for 30 minutes at ambient temperature. The mixture was washed twice with 200 mL of water, 200 mL of brine, dried over MgS04 and concentrated in vacuo to give the crude 4-hydroxyphenyl intermediate. The solid residue was recrystallized from methylene chloride to give 37.5 g (82%) of the desired (4R, 5R) -5- (4′ – hydoxyphenyl) -7- (dimethylamino) tetrahydrobenzothiepine-1, 1- dioxide as a white solid: *H NMR (CDC13) d 0.84-0.97 (m, 6H) , 1.1-1.5 (m, 10H) , 1.57-1.72 (m, IH) , 2.14-2.28 (m, IH) , 2.83 (s, 6H) , 3.00 (d, J = 15.3 Hz, IH) , 3.16 (d, J – 15.3 Hz, IH) , 4.11 (s, 2H) , 5.48 (s, IH) , 6.02 (d, J – 2.4 Hz, IH) , 6.55 (dd, J“ = 9, 2.4 Hz, IH) , 6.88 (d, 8 , 7 Hz , 2H) , 7.38 (d, J – 8.7 Hz, 2H) , 7.91 (d, J“ = 9 Hz, 2H) .
Step 11: Preparation of enantiomerically-enriched chlorobenzyl intermediate Treat a solution of enantiomerically-enriched (4R,5R)- 5- (4′ -hydoxypheny1) -7- (dimethylamino) tetrahydrobenzothiepine-1, 1-dioxide (5.0 g, 10.9 mmol, obtained from Step 10) in acetone (100 mL) at 25 °C under N2 with powdered 5 K2C03 (2.3 g, 16.3 mmol, 1.5 eq.) and a, a’ -dichloro-p-xylene (6.7 g, 38.1 mmol, 3.5 eq.) . Stir the resulting solution at 65 °C for about 48 hours. Cool the reaction mixture to 25 °C and concentrate to 1/5 of original volume. Dissolve the residue in EtOAc (150 mL) and wash with water (2 x 150 mL) .
10 Extract the aqueous layer with EtOAc (2 x 150 mL) and wash the combined organic extracts with saturated aqueous NaCI (2 x 150 mL. Dry the combined extracts with MgS04 and concentrate in vacuo to provide the crude product . Purification by flash chromatography (5.4 x 45 cm silica,
1525-40% EtOAc/hexane) will afford the enantiomerically- enriched chlorobenzyl intermediate .
Step 12: Preparation of enantiomerically-enriched (4R.5R)- 1- r [4- [ [4- [3 , 3-Dibutyl-7- (dimethylamino) -2,3 , 4 , 5-tetrahvdro-
Treat a solution of the enantiomerically-enriched chlorobenzyl intermediate (4.6 g, 7.7 mmol, obtained from
25 above in Step 11) in acetonitrile (100 mL) at 25 °C under N2 with diazabicyclo [2.2.2] -octane (DABCO, 0.95 g, 8.5 mmol, 1.1 eq.) and stir at 35 °C for 2 hours. Collect the precipitated solid and wash with CH3CN. Recrystallization from CH3OH/Et20 will give the desired title compound (XXVII) .
ANY ERROR, EMAIL amcrasto@gmail.com, +919323115463
Ponesimod (INN, codenamed ACT-128800) is an experimental drug for the treatment of multiple sclerosis (MS) and psoriasis. It is being developed by Actelion.
The first oral treatment for relapsing multiple sclerosis, the nonselective sphingosine-1-phosphate receptor (S1PR) modulator fingolimod, led to identification of a pivotal role of sphingosine-1-phosphate and one of its five known receptors, S1P1R, in regulation of lymphocyte trafficking in multiple sclerosis. Modulation of S1P3R, initially thought to cause some of fingolimod’s side effects, prompted the search for novel compounds with high selectivity for S1P1R. Ponesimod is an orally active, selective S1P1R modulator that causes dose-dependent sequestration of lymphocytes in lymphoid organs. In contrast to the long half-life/slow elimination of fingolimod, ponesimod is eliminated within 1 week of discontinuation and its pharmacological effects are rapidly reversible. Clinical data in multiple sclerosis have shown a dose-dependent therapeutic effect of ponesimod and defined 20 mg as a daily dose with desired efficacy, and acceptable safety and tolerability. Phase II clinical data have also shown therapeutic efficacy of ponesimod in psoriasis. These findings have increased our understanding of psoriasis pathogenesis and suggest clinical utility of S1P1R modulation for treatment of various immune-mediated disorders. A gradual dose titration regimen was found to minimize the cardiac effects associated with initiation of ponesimod treatment. Selectivity for S1P1R, rapid onset and reversibility of pharmacological effects, and an optimized titration regimen differentiate ponesimod from fingolimod, and may lead to better safety and tolerability. Ponesimod is currently in phase III clinical development to assess efficacy and safety in relapsing multiple sclerosis. A phase II study is also ongoing to investigate the potential utility of ponesimod in chronic graft versus host disease.http://www.ncbi.nlm.nih.gov/pmc/articles/PMC4707431/
Biology and pharmacology of sphingosine-1-phosphate receptor 1
The past decades have witnessed major advances in the treatment of autoimmune and chronic inflammatory diseases. A plethora of novel therapies targeting specific molecules involved in the inflammatory or immune system activation cascades have become available. These have significantly increased our understanding of disease pathogenesis and improved the management of immune-mediated disorders. However, most of the targeted therapies are biological drugs which need to be injected, are eliminated slowly (e.g. over several weeks) and can lose efficacy or tolerability due to their potential immunogenicity. In an attempt to overcome these hurdles, pharmaceutical research has made considerable efforts to develop novel oral targeted therapies for autoimmune and chronic inflammatory diseases.
Sphingosine-1-phosphate receptor 1 (S1P1R) is one of five known G protein-coupled receptors with nanomolar affinity for the lysophospholipid sphingosine-1-phosphate (S1P), which is generated through physiologic metabolism of the cell membrane constituent sphingomyelin by all cells [Brinkmann, 2007]. S1P receptors, including S1P1R, are widely expressed in many tissues [Chun et al. 2010]. S1P1R expression on lymphocytes controls their egress from thymus and secondary lymphoid organs [Cyster and Schwab, 2012]. Lymphocyte egress requires a gradient of S1P concentration, which is established by a high S1P concentration in blood and lymph compared with a low concentration in the interstitial fluid of lymphoid organs [Grigorova et al. 2009].
Synthetic S1P1 receptor modulators disrupt the interaction of the physiologic S1P ligand with S1P1R by promoting initial activation followed by sustained internalization and desensitization of S1P1R [Hla and Brinkmann, 2011; Pinschewer et al. 2011]. Experiments conducted in animal models of transplant rejection, multiple sclerosis, lupus erythematosus, arthritis and inflammatory bowel disease with the first-generation, nonselective S1P receptor modulator, fingolimod, have demonstrated the potential efficacy of this mode of action across several immune-mediated chronic inflammatory conditions [Brinkmann, 2007]. Fingolimod is a structural analog of sphingosine that is phosphorylated in the body by a sphingosine kinase to generate the bioactive form of the drug, fingolimod phosphate, which binds to multiple S1P receptors [Brinkmann, 2007]. Clinical trials in multiple sclerosis (MS) have confirmed the efficacy of fingolimod in relapsing MS, but not in primary progressive disease, and led to the approval of the first oral medication for the treatment of relapsing forms of MS in 2010 [Kappos et al. 2010].
The mechanism of action of fingolimod has increased our understanding of MS pathogenesis. T and B cells, but not natural killer (NK) cells, express functional S1P1R and are affected by fingolimod [Cyster and Schwab, 2012]. Furthermore, S1P1R is differentially expressed and regulated in functionally distinct subsets of lymphocytes and fingolimod has been shown to predominantly affect naïve T cells and central memory T cells (TCM) while sparing effector memory T cells (TEM), and terminally differentiated effector T cells (TE) in patients with relapsing MS [Mehling et al. 2008, 2011]. This has raised the possibility that, at least in MS, retention of TCM cells, which include pro-inflammatory T helper 17 (Th17) cells, by fingolimod may prevent their accumulation in the cerebrospinal fluid (CSF) and subsequent differentiation to TE cells in the central nervous system (CNS) [Hla and Brinkmann, 2011]. The effects of S1P1R modulation on B cells are less well defined. Recent data from patients with relapsing MS have shown predominant reduction of memory B cells and recently activated memory B cells (CD38int-high) in peripheral blood after treatment with fingolimod [Claes et al. 2014; Nakamura et al. 2014]. As memory B cells are implicated in the pathogenesis of MS and other autoimmune diseases, these observations suggest another potential mechanism underlying the therapeutic effects of S1P1R modulators.
Astrocytes, microglia, oligodendrocytes and neurons express various S1P receptors including S1P1R, S1P3R and S1P5R. Fingolimod has been shown to penetrate the CNS tissues and in vitro studies have shown activation of astrocytes and oligodendrocytes by fingolimod [Foster et al. 2007]. Conditional deletion of S1P1R on neural cells in mice reduced the severity of experimental autoimmune encephalomyelitis (EAE) and reductions in the clinical scores were paralleled by decreased demyelination, axonal loss and astrogliosis [Choi et al. 2011]. Unfortunately, there was no beneficial effect in a recently completed, large study of fingolimod in patients with primary progressive MS [Lublin et al. 2015], suggesting that the direct effect on CNS cells alone may not be sufficient. Taken together, these data suggest the possibility of a direct beneficial effect of S1P1R modulation in the brain of patients with relapsing MS [Dev et al. 2008]; however, its contribution to efficacy relative to the immunological effects remains unclear.
Initial studies in rodents suggested that modulation of S1P3R on cardiac myocytes by fingolimod was associated with a reduction of heart rate (HR) by activation of G-protein-coupled inwardly rectifying potassium channels (GIRK) that regulate pacemaker frequency, and the shape and duration of action potentials [Koyrakh et al. 2005; Camm et al. 2014]. Modulation of S1P2R and S1P3R on myofibroblasts by fingolimod was also shown to stimulate extracellular matrix synthesis [Sobel et al. 2013]. Modulation of these receptors on vascular smooth muscle cells appeared to be associated with vasoconstriction, leading to the slight increase in blood pressure observed with fingolimod treatment [Salomone et al. 2003; Watterson et al. 2005; Hu et al. 2006; Lorenz et al. 2007; Kappos et al. 2010]. These observations raised the possibility that some side effects associated with fingolimod treatment could be avoided by more selective S1P1R modulators, thus triggering the search for novel compounds.
Ponesimod, a selective, rapidly reversible, orally active, sphingosine-1-phosphate receptor modulator
Ponesimod (ACT-128800 (Z,Z)-5-[3-chloro-4-(2R)-2,3-dihydroxy-propoxy)-benzylidene]-2-propylimino-3-o-tolylthiazolidin-4-one) is a selective, rapidly reversible, orally active, S1P1R modulator. Ponesimod emerged from the discovery of a novel class of S1P1R agonists based on the 2-imino-thiazolidin-4-one scaffold (Figure 1) [Bolli et al. 2010]. Ponesimod activates S1P1R with high potency [half maximal effective concentration (EC50) of 5.7 nM] and selectivity. Relative to the potency of S1P, the potency of ponesimod is 4.4 higher for S1P1R and 150-fold lower for S1P3R, resulting in an approximately 650-fold higher S1P1R selectivity compared with the natural ligand.
In a 2009–2011 Phase II clinical trial including 464 MS patients, ponesimod treatment resulted in fewer new active brain lesions thanplacebo, measured during the course of 24 weeks.[3][4]
In a 2010–2012 Phase II clinical trial including 326 patients with psoriasis, 46 or 48% of patients (depending on dosage) had a reduction of at least 75% Psoriasis Area and Severity Index (PASI) score compared to placebo in 16 weeks.[3][5]
Common adverse effects in studies were temporary bradycardia (slow heartbeat), usually at the beginning of the treatment,dyspnoea (breathing difficulties), and increased liver enzymes (without symptoms). No significant increase of infections was observed under ponesimod therapy.[3]QT prolongation is detectable but was considered to be too low to be of clinical importance in a study.[6]
Sphingosine-1-phosphate (S1P) is a widespread lysophospholipid which displays a wealth of biological effects. Extracellular S1P conveys its activity through five specific G-protein coupled receptors numbered S1P1 through S1P5. Agonists of the S1P1 receptor block the egress of T-lymphocytes from thymus and lymphoid organs and hold promise for the oral treatment of autoimmune disorders. Here, we report on the discovery and detailed structure−activity relationships of a novel class of S1P1 receptor agonists based on the 2-imino-thiazolidin-4-one scaffold. Compound 8bo (ACT-128800) emerged from this series and is a potent, selective, and orally active S1P1 receptor agonist selected for clinical development. In the rat, maximal reduction of circulating lymphocytes was reached at a dose of 3 mg/kg. The duration of lymphocyte sequestration was dose dependent. At a dose of 100 mg/kg, the effect on lymphocyte counts was fully reversible within less than 36 h. Pharmacokinetic investigation of8bo in beagle dogs suggests that the compound is suitable for once daily dosing in humans.
…………..DELETED…………… column chromatography on silica gel eluting with heptane:ethyl acetate 1:4 to give the title compound (1.34 g, 37%) as a pale-yellow foam.
The present invention relates inter alia to a new process for the preparation of (2Z,5Z)-5-(3-chloro-4-((R)-2,3-dihydroxypropoxy)benzylidene)-2-(propylimino)-3-(o-tolyl)thiazolidin-4-one (hereinafter also referred to as the “COMPOUND” or “compound (2)”), especially in crystalline form C which form is described in WO 2010/046835. The preparation of COMPOUND and its activity as immunosuppressive agent is described in WO 2005/054215. Furthermore, WO 2008/062376 describes a new process for the preparation of (2Z,5Z)-5-(3-chloro-4-hydroxy-benzylidene)-2-propylimino-3-o-tolyl-thiazolidin-4-one which can be used as an intermediate in the preparation of COMPOUND.
Example 1 a) below describes such a process of preparing (2Z,5Z)-5-(3-chloro-4-hydroxy-benzylidene)-2-propylimino-3-o-tolyl-thiazolidin-4-one according to WO 2008/062376. According to WO 2008/062376 the obtained (2Z,5Z)-5-(3-chloro-4-hydroxy-benzylidene)-2-propylimino-3-o-tolyl-thiazolidin-4-one can then be transformed into COMPOUND by using standard methods for the alkylation of phenols. Such an alkylation is described in Example 1 b) below. Unfortunately, this process leads to the impurity (2Z,5Z)-5-(3-chloro-4-((1 ,3-dihydroxypropan-2-yl)oxy)benzylidene)-2-(propylimino)-3-(o-tolyl)thiazolidin-4-one which is present in about 2% w/w in the crude product (see Table 1 ) and up to 6 recrystallisations are necessary in order to get this impurity below 0.4% w/w (see Tables 1 and 2) which is the specified limit based on its toxicological qualification.
the obtained (R)-3-chloro-4-(2,3-dihydroxypropoxy)-benzaldehyde (1 ) with 2-[(Z)-propylimino]-3-o-tolyl-thiazolidin-4-one to form (2Z,5Z)-5-(3-chloro-4-((R)-2,3-dihydroxypropoxy)benzylidene)-2-(propylimino)-3-(o-tolyl)thiazolidin-4-one (2):
.
The reaction of (R)-3-chloro-4-(2,3-dihydroxypropoxy)-benzaldehyde (1 ) with 2-[(Z)-propylimino]-3-o-tolyl-thiazolidin-4-one can be performed under conditions which are typical for a Knoevenagel condensation. Such conditions are described in the literature for example in Jones, G., Knoevenagel Condensation in Organic Reaction, Wiley: New York, 1967, Vol. 15, p 204; or Prout, F. S., Abdel-Latif, A. A., Kamal, M. R., J. Chem. Eng. Data, 2012, 57, 1881-1886.
2-[(Z)-Propylimino]-3-o-tolyl-thiazolidin-4-one can be prepared as described in WO 2008/062376, preferably without the isolation and/or purification of intermediates such as the thiourea intermediate that occurs after reacting o-tolyl-iso-thiocyanate with n-propylamine. Preferably 2-[(Z)-propylimino]-3-o-tolyl-thiazolidin-4-one obtained according to WO 2008/062376 is also not isolated and/or purified before performing the Knoevenagel condensation, i.e. before reacting 2-[(Z)-propylimino]-3-o-tolyl-thiazolidin-4-one with (R)-3-chloro-4-(2,3-dihydroxypropoxy)-benzaldehyde (1 ), i.e. in a preferred embodiment compound (2) is prepared in a one-pot procedure analogous to that described in WO 2008/062376.
Example 1 : (2Z,5Z)-5-(3-Chloro-4-((R)-2,3-dihydroxypropoxy)benzylidene)-2-(propylimino)-3-(o-tolyl)thiazolidin-4-one
a) Preparation of (2Z,5Z)-5-(3-chloro-4-hydroxy-benzylidene)-2-propylimino-3-o-tolyl-thiazolidin-4-one:
Acetic acid solution: To acetic acid (149.2 mL) are added sodium acetate (1 1 .1 1 g, 2.00 eq.) and 3-chloro-4-hydroxybenzaldehyde (10.60 g, 1.00 eq.) at 20 °C. The mixture is stirred at 20 °C until complete dissolution (2 to 3 h).
n-Propylamine (4.04 g, 1.00 eq.) is added to a solution of o-tolyl-iso-thiocyanate (10 g, 1.00 eq.) in dichloromethane (100 mL) at 20 °C. The resulting pale yellow solution is agitated for 40 min at 20 °C before IPC (conversion specification≥ 99.0 %). The reaction is cooled to -2 °C. Bromoacetyl bromide (13.53 g, 1.00 eq.) is added and the resulting solution is stirred for 15 min at -2 °C. Pyridine (10.92 g, 2.05 eq.) is then added slowly at -2 °C. The intensive yellow reaction mixture is stirred for 15 min at -2 °C before IPC (conversion specification≥ 93.0 %). 70 mL of dichloromethane are distilled off under atmospheric pressure and jacket temperature of 60 °C. The temperature is adjusted to 42 °C and the acetic acid solution is added to the reaction mixture. The resulting solution is heated to 58 °C and stirred at this temperature for 15 h before IPC (conversion specification≥ 95 %). 25 mL of solvents are distilled off under vacuum 900 – 500 mbars and jacket temperature of 80 °C. The temperature is adjusted to 60 °C and water (80.1 mL) is added to the reaction mixture over 1 h. The resulting yellow suspension is stirred at 60 °C for 30 min. The suspension is cooled to 20 °C over 1 h and stirred at this temperature for 30 min.
The product is filtered and washed with a mixture of acetic acid (30 mL) and water (16 mL) and with water (50 mL) at 20 °C. The product is dried under vacuum at 50 °C for 40 h to afford a pale yellow solid; yield 25.93 g (78 %).
b) Preparation of crude (2Z,5Z)-5-(3-chloro-4-((R)-2,3-dihydroxypropoxy)benzylidene)-2-(propylimino)-3-(o-tolyl)thiazolidin-4-one:
To a suspension of (2Z,5Z)-5-(3-chloro-4-hydroxy-benzylidene)-2-propylimino-3-o-tolyl-thiazolidin-4-one (10.00 g, 1.00 eq.) in ethanol (47.2 mL) is added (R)-3-chloro-1 ,2-
propanediol (3.37 g, 1.18 eq.) at 20 °C. Potassium tert-butoxide (3.39 g, 1.13 eq.) is added in portions at 20 °C. The resulting fine suspension is stirred at 20 °C for 25 min before being heated to reflux (88 °C). The reaction mixture is stirred at this temperature for 24 h before IPC (conversion specification≥ 96.0 %). After cooling down to 60 °C, acetonitrile (28.6 mL) and water (74.9 mL) are added. The resulting clear solution is cooled from 60 °C to 0 °C over 2 h. During the cooling ramp, (2Z,5Z)-5-(3-chloro-4-((R)-2,3-dihydroxypropoxy)benzylidene)-2-(propylimino)-3-(o-tolyl)thiazolidin-4-one seeds of crystalline form C (0.010 g, 0.001 eq.; crystalline form C can be prepared as described in WO 2010/046835) are added at 50 °C. The suspension is heated from 0 °C to 50 °C, cooled to 0 °C over 6 h and stirred at this temperature for 12 h.
The product is filtered and washed with a mixture of acetonitrile (23.4 mL) and water (23.4 mL) at 0 °C. The product is dried under vacuum at 45 °C for 24 h to afford a pale yellow solid; yield 1 1.91 g (84 %).
c) Purification of (2Z,5Z)-5-(3-chloro-4-((R)-2,3-dihydroxypropoxy)benzylidene)-2-(propylimino)-3-(o-tolyl)thiazolidin-4-one:
Recrystallisation I: The crude (2Z,5Z)-5-(3-chloro-4-((R)-2,3-dihydroxypropoxy)benzylidene)-2-(propylimino)-3-(o-tolyl)thiazolidin-4-one (10 g) is dissolved in acetonitrile (30 mL) at 70 °C. The reaction mixture is cooled from 70 °C to 0 °C over 2 h. During the cooling ramp, (2Z,5Z)-5-(3-chloro-4-((R)-2,3-dihydroxypropoxy)benzylidene)-2-(propylimino)-3-(o-tolyl)thiazolidin-4-one seeds of crystalline form C (0.0075 g, 0.00075 eq.) are added at 50 °C. The suspension is heated up to 52 °C, cooled to 0 °C over 6 h and agitated at this temperature for 2 h. The product is filtered and washed with acetonitrile at -10 °C (2 x 12.8 mL).
Recrystallisation II: The wet product is dissolved in acetonitrile (27.0 mL) at 70 °C. The reaction mixture is cooled from 70 °C to 0 °C over 2 h. During the cooling ramp, (2Z,5Z)-5-(3-chloro-4-((R)-2,3-dihydroxypropoxy)benzylidene)-2-(propylimino)-3-(o-tolyl)thiazolidin-4-one seeds of crystalline form C (0.0075 g, 0.00075 eq.) are added at 50 °C. The suspension is heated up to 52 °C, cooled to 0 °C over 6 h and agitated at this temperature for 2 h. The product is filtered and washed with acetonitrile at -10 °C (2 x 1 1.3 mL).
Recrystallisation III: The wet product is dissolved in acetonitrile (24.3 mL) at 70 °C. The reaction mixture is cooled from 70 °C to 0 °C over 2 h. During the cooling ramp, (2Z,5Z)-5-(3-chloro-4-((R)-2,3-dihydroxypropoxy)benzylidene)-2-(propylimino)-3-(o-tolyl)thiazolidin-4- one seeds of crystalline form C (0.0075 g, 0.00075 eq.) are added at 50 °C. The suspension is heated up to 52 °C, cooled to 0 °C over 6 h and agitated at this temperature for 2 h. The product is filtered and washed with acetonitrile at -10 °C (2 x 10.1 mL).
Recrystallisation IV: The wet product is dissolved in acetonitrile (21.9 mL) at 70 °C. The reaction mixture is cooled from 70 °C to 0 °C over 2 h. During the cooling ramp, (2Z,5Z)-5-(3-chloro-4-((R)-2,3-dihydroxypropoxy)benzylidene)-2-(propylimino)-3-(o-tolyl)thiazolidin-4-one seeds of crystalline form C (0.0075 g, 0.00075 eq.) are added at 50 °C. The suspension is heated up to 52 °C, cooled to 0 °C over 6 h and agitated at this temperature for 2 h. The product is filtered and washed with acetonitrile at -10 °C (2 x 9.1 mL).
Recrystallisation V: The wet product is dissolved in acetonitrile (19.7 mL) at 70 °C. The reaction mixture is cooled from 70 °C to 0 °C over 2 h. During the cooling ramp, (2Z,5Z)-5-(3-chloro-4-((R)-2,3-dihydroxypropoxy)benzylidene)-2-(propylimino)-3-(o-tolyl)thiazolidin-4-one seeds of crystalline form C (0.0075 g, 0.00075 eq.) are added at 50 °C. The suspension is heated up to 52 °C, cooled to 0 °C over 6 h and agitated at this temperature for 2 h. The product is filtered and washed with acetonitrile at -10 °C (2 x 8.2 mL).
Recrystallisation VI: The wet product is dissolved in acetonitrile (23.9 mL) at 70 °C. Water (20 mL) is added at 70 °C. The reaction mixture is cooled from 70 °C to 0 °C over 2 h.
During the cooling ramp, (2Z,5Z)-5-(3-chloro-4-((R)-2,3-dihydroxypropoxy)benzylidene)-2- (propylimino)-3-(o-tolyl)thiazolidin-4-one seeds of crystalline form C (0.0075 g, 0.00075 eq.) are added at 50 °C. The suspension is heated up to 52 °C, cooled to 0 °C over 6 h and agitated at this temperature for 2 h. The product is filtered and washed twice with a mixture of acetonitrile (4.5 mL) and water (4.5 mL) at -10 °C.
The product is dried under vacuum at 45 °C for 24 h to afford a pale yellow solid; yield: 7.0 g (70 %).
Example 2: (R)-3-Chloro-4-(2,3-dihydroxypropoxy)-benzaldehyde
Potassium tert-butoxide (1 18 g, 1.20 eq.) is added to n-propanol (963 mL) followed by 3-chloro-4-hydroxybenzaldehyde (137 g, 1.00 eq.). To the mixture is added (R)-3-chloro-1 ,2-propanediol (126 g, 1.30 eq.). The suspension is heated to 90 °C and stirred at this temperature for 17 h. Solvent (500 mL) is distilled off at 120 °C external temperature and reduced pressure. Water is added (1.1 L) and solvent (500 mL) is removed by distillation. The turbid solution is cooled to 20 °C. After stirring for one hour a white suspension is obtained. Water (500 mL) is added and the suspension is cooled to 10 °C. The suspension is filtered and the resulting filter cake is washed with water (500 mL). The product is dried at 50 °C and reduced pressure to yield 149 g of a white solid (73%), which is (R)-3-chloro-4-(2,3-dihydroxypropoxy)-benzaldehyde in crystalline form A.
Example 3: (R)-3-Chloro-4-(2,3-dihydroxypropoxy)-benzaldehyde
Potassium tert-butoxide (8.60 g, 1.20 eq.) is added to n-propanol (70 mL) below 15 °C, the temperature is allowed to rise. After the addition the temperature is corrected again to below 15 °C before addition of 3-chloro-4-hydroxybenzaldehyde (10 g, 1 .00 eq.). The suspension is heated to 40 °C and stirred for 30 min. (R)-3-Chloro-1 ,2-propanediol (9.18 g, 1.30 eq.) is added at 40 °C. The resulting suspension is heated to 60 °C and stirred at this temperature for 15 h then heated to 94 °C till meeting the IPC-specification (specification conversion≥ 90.0 %). The mixture is cooled to 30 °C and n-propanol is partially distilled off (-50 mL are distilled off) under reduced pressure and a maximum temperature of 50 °C, the jacket temperature is not allowed to raise above 60 °C.
Water (81 mL) is added and a second distillation is performed under the same conditions (24 mL are distilled off). The mixture is heated till homogeneous (maximum 54 °C) and then cooled to 24 °C. At 24 °C the mixture is seeded with crystalline (R)-3-chloro-4-(2,3-dihydroxypropoxy)-benzaldehyde of form A (0.013 g, 0.00085 eq.). How to obtain the crystalline seeds is described in Examples 2 and 5. The reaction mixture is cooled to 0 °C over 7.5 h.
The product is filtered and washed with water (2 x 35 mL) and once with methyl tert-butyl ether (20 mL) at 5 °C. The product is dried under vacuum at 40 °C for 20 h to afford an off-white solid; yield: 10.6 g (72 %), which is (R)-3-chloro-4-(2,3-dihydroxypropoxy)-benzaldehyde in crystalline form A.
Example 4: (2Z,5Z)-5-(3-Chloro-4-((R)-2,3-dihydroxypropoxy)benzylidene)-2-(propylimino)- 3-(o-tolyl)thiazolidin-4-one
a) Preparation of crude (2Z,5Z)-5-(3-chloro-4-((R)-2,3-dihydroxypropoxy)benzylidene)-2-(propylimino)-3-(o-tolyl)thiazolidin-4-one:
n-Propylamine (5.23 g, 1.32 eq.) is added to a solution of o-tolyl-iso-thiocyanate (10 g, 1.00 eq.) in dichloromethane (100 mL) at 20 °C. The resulting pale yellow solution is agitated for 15 min at 20 °C before IPC (conversion specification≥ 99.0 %). The reaction is cooled to -2 °C. Bromoacetyl bromide (14.88 g, 1.10 eq.) is added and the resulting solution is stirred for 15 min at -2 °C. Pyridine (10.92 g, 2.05 eq.) is then added slowly at -2 °C. The intensive yellow reaction mixture is stirred for 15 min at -2 °C before IPC (conversion specification≥ 93.0 %). Dichloromethane is partially distilled off (66 mL are distilled off) under atmospheric pressure and jacket temperature of 60 °C. Ethanol (1 1 1.4 mL), sodium acetate (12.75 g, 2.30 eq.) and (R)-3-chloro-4-(2,3-dihydroxypropoxy)-benzaldehyde from Example 3 (14.38 g, 0.93 eq.) are added. The remaining dichloromethane and a part of ethanol are distilled off (49.50 mL are distilled off) under atmospheric pressure and jacket temperature up to 85 °C. The reaction mixture (orange suspension) is stirred for 3 – 5 h under reflux (78 °C) before IPC (conversion specification≥ 97.0 %).
Water (88.83 mL) is added and the temperature adjusted to 40 °C before seeding with micronized (2Z,5Z)-5-(3-chloro-4-((R)-2,3-dihydroxypropoxy)benzylidene)-2-(propylimino)-3-(o-tolyl)thiazolidin-4-one in crystalline form C (0.075 g, 0.0024 eq.). The reaction mixture is cooled to 0 °C over 5 h, heated up to 40 °C, cooled to 0 °C over 6 h and stirred at this temperature for 2 h.
The product is filtered and washed with a 1 :1 ethanohwater mixture (2 x 48 mL) at 0 °C. The product is dried under vacuum at 45 °C for 10 h to afford a pale yellow solid; yield: 24.71 g (86 %).
b) Purification of (2Z,5Z)-5-(3-chloro-4-((R)-2,3-dihydroxypropoxy)benzylidene)-2-(propylimino)-3-(o-tolyl)thiazolidin-4-one:
The crude (2Z,5Z)-5-(3-chloro-4-((R)-2,3-dihydroxypropoxy)benzylidene)-2-(propylimino)-3-(o-tolyl)thiazolidin-4-one (10 g) is dissolved in ethanol (40 mL) at 70 °C. The temperature is adjusted at 50 °C for seeding with micronised (2Z,5Z)-5-(3-chloro-4-((R)-2,3- dihydroxypropoxy)benzylidene)-2-(propylimino)-3-(o-tolyl)thiazolidin-4-one in crystalline form C (0.016 g, 0.0016 eq.). The reaction mixture is cooled from 50 °C to 0 °C over 4 h, heated up to 50 °C, cooled to 0 °C over 6 h and agitated at this temperature for 2 h.
The product is filtered and washed with ethanol at 0 °C (2 x 12.8 mL). The product is dried under vacuum at 45 °C for 10 h to afford a pale yellow solid; yield: 9.2 g (92 %).
Example 5: Preparation of crystalline seeds of (R)-3-chloro-4-(2,3-dihydroxypropoxy)- benzaldehyde
10 mg of (R)-3-chloro-4-(2,3-dihydroxypropoxy)-benzaldehyde of at least 99.5% purity by 1 H-NMR assay is dissolved in a 4 mL vial by adding 1 mL of pure ethanol (puriss p. a.). The solvent is allowed to evaporate through a small hole in the cap (approx. 2 mm of diameter) of the vial until complete dryness. The white solid residue is crystalline (R)-3-chloro-4-(2,3- dihydroxypropoxy)-benzaldehyde in crystalline form A. Alternatively, methanol or methylisobutylketone (both in puriss p. a. quality) is used. This procedure is repeated until sufficient seeds are made available.
“Multiple-dose tolerability, pharmacokinetics, and pharmacodynamics of ponesimod, an S1P1 receptor modulator: Favorable impact of dose up-titration”. The Journal of Clinical Pharmacology54: 179–88. Feb 2014. doi:10.1002/jcph.244. PMID24408162.
“Mass balance, pharmacokinetics and metabolism of the selective S1P1 receptor modulator ponesimod in humans”. Xenobiotica45: 139–49. Feb 2015. doi:10.3109/00498254.2014.955832. PMID25188442.
H. Spreitzer (29 September 2014). “Neue Wirkstoffe – Ponesimod”. Österreichische Apothekerzeitung (in German) (20/2014): 42.
“Oral ponesimod in patients with chronic plaque psoriasis: a randomised, double-blind, placebo-controlled phase 2 trial”. The Lancet384: 2036–45. Dec 2014. doi:10.1016/S0140-6736(14)60803-5. PMID25127208.
“Effect of Ponesimod, a selective S1P1 Receptor Modulator, on the QT Interval in Healthy Subjects”. Basic116: 429–37. May 2015.doi:10.1111/bcpt.12336. PMID25287214.
Ponesimod is a potent orally active, selective sphingosine-1-phosphate receptor 1 (S1P1) immunomodulator.
Ponesimod prevents lymphocytes from leaving lymph nodes, thereby reducing circulating blood lymphocyte counts and preventing infiltration of lymphocytes into target tissues. The lymphocyte count reduction is rapid, dose-dependent, sustained upon continued dosing, and quickly reversible upon discontinuation. Initial data suggest that ponesimod does not cause lymphotoxicity by destroying/depleting lymphocytes or interfering with their cellular function. Other blood cells e.g. cells of the innate immune system are largely unaffected. Ponesimod is therefore considered a promising new oral agent for the treatment of a variety of autoimmune disorders.
CURRENT STATUS
OPTIMUM (Oral Ponesimod versus Teriflunomide In relapsing MUltiple sclerosis) is a Phase III multi-center, randomized, double-blind, parallel-group, active-controlled superiority study to compare the efficacy and safety of ponesimod to teriflunomide in patients with relapsing multiple sclerosis (RMS). The study aims to determine whether ponesimod is more efficacious than teriflunomide in reducing relapses. The study is expected to enroll approximately 1’100 patients, randomized in 2 groups in a 1:1 ratio to receive ponesimod 20 mg/day or teriflunomide 14 mg/day, and is expected to last a little over 3 years. An additional study to further characterize the utility and differentiation of ponesimod in multiple sclerosis is being discussed with Health Authorities.
Ponesimod is also evaluated in a Phase II open-label, single-arm, intra-subject dose-escalation study to investigate the biological activity, safety, tolerability, and pharmacokinetics of ponesimod in patients suffering from moderate or severe chronic graft versus host disease (GvHD)inadequately responding to first- or second-line therapy. The study will also investigate the clinical response to ponesimod treatment in these patients. Approximately 30 patients will be enrolled to receive ponesimod in escalating doses of 5, 10, and 20 mg/day over the course of 24 weeks. The study is being conducted at approximately 10 sites in the US and is expected to last approximately 18 months.
AVAILABLE CLINICAL DATA
The decision to move into Phase III development was based on the Phase IIb dose-finding study with ponesimod in patients with relapsing-remitting multiple sclerosis. A total of 464 patients were randomized into this study and the efficacy, safety and tolerability of three ponesimod doses (10, 20, and 40 mg/day) versus placebo, administered once daily for 24 weeks.
The primary endpoint of this study was defined as the cumulative number of new gadolinium-enhancing lesions on T1-weighted magnetic resonance imaging (MRI) scans at weeks 12, 16, 20, and 24 after study drug initiation. A key secondary endpoint of this study was the annualized relapse rate over 24 weeks of treatment. Patients who completed 24 weeks of treatment were offered the opportunity to enter into an extension study. This ongoing trial is investigating the long-term safety, tolerability, and efficacy of 10 and 20 mg/day of ponesimod in patients with relapsing-remitting multiple sclerosis, in a double-blind fashion. The study continues to provide extensive safety and efficacy information for ponesimod in this indication, with some patients treated for more than 6 years.
The safety database from all studies with ponesimod now comprises more than 1,300 patients and healthy volunteers.
MILESTONES
2015 – Phase III program in multiple sclerosis initiated
2011 – Phase IIb dose-finding study in multiple sclerosis successfully completed
2006 – Entry-into-man
2004 – Preclinical development initiated
KEY SCIENTIFIC LITERATURE
Olsson T et al. J Neurol Neurosurg Psychiatr. 2014 Nov;85(11):1198-208. doi: 10.1136/jnnp-2013-307282. Epub 2014 Mar 21
The U.S. Food and Drug Administration today approved Axumin, a radioactive diagnostic agent for injection. Axumin is indicated for positron emission tomography (PET) imaging in men with suspected prostate cancer recurrence based on elevated prostate specific antigen (PSA) levels following prior treatment.
May 27, 2016
Release
The U.S. Food and Drug Administration today approved Axumin, a radioactive diagnostic agent for injection. Axumin is indicated for positron emission tomography (PET) imaging in men with suspected prostate cancer recurrence based on elevated prostate specific antigen (PSA) levels following prior treatment.
Prostate cancer is the second leading cause of death from cancer in U.S. men. In patients with suspected cancer recurrence after primary treatment, accurate staging is an important objective in improving management and outcomes.
“Imaging tests are not able to determine the location of the recurrent prostate cancer when the PSA is at very low levels,” said Libero Marzella, M.D., Ph.D., director of the Division of Medical Imaging Products in the FDA’s Center for Drug Evaluation and Research. “Axumin is shown to provide another accurate imaging approach for these patients.”
Two studies evaluated the safety and efficacy of Axumin for imaging prostate cancer in patients with recurrent disease. The first compared 105 Axumin scans in men with suspected recurrence of prostate cancer to the histopathology (the study of tissue changes caused by disease) obtained by prostate biopsy and by biopsies of suspicious imaged lesions. Radiologists onsite read the scans initially; subsequently, three independent radiologists read the same scans in a blinded study.
The second study evaluated the agreement between 96 Axumin and C11 choline (an approved PET scan imaging test) scans in patients with median PSA values of 1.44 ng/mL. Radiologists on-site read the scans, and the same three independent radiologists who read the scans in the first study read the Axumin scans in this second blinded study. The results of the independent scan readings were generally consistent with one another, and confirmed the results of the onsite scan readings. Both studies supported the safety and efficacy of Axumin for imaging prostate cancer in men with elevated PSA levels following prior treatment.
Axumin is a radioactive drug and should be handled with appropriate safety measures to minimize radiation exposure to patients and healthcare providers during administration. Image interpretation errors can occur with Axumin PET imaging. A negative image does not rule out the presence of recurrent prostate cancer and a positive image does not confirm the presence of recurrent prostate cancer. Clinical correlation, which may include histopathological evaluation of the suspected recurrence site, is recommended.
The most commonly reported adverse reactions in patients are injection site pain, redness, and a metallic taste in the mouth.
Axumin is marketed by Blue Earth Diagnostics, Ltd., Oxford, United Kingdom
The non-natural amino acid [ F]-l-amino-3-fluorocyclobutane-l-carboxylic acid
([18F]-FACBC, also known as [18F]-Fluciclovine) is taken up specifically by amino acid transporters and has shown promise for tumour imaging with positron emission tomography (PET).
A known synthesis of [18F]-FACBC begins with the provision of the protected precursor compound 1 -(N-(t-butoxycarbonyl)amino)-3 –
[((trifluoromethyl)sulfonyl)oxy]-cyclobutane-l-carboxylic acid ethyl ester. This precursor compound is first labelled with [18F]-fluoride:
II before removal of the two protecting groups:
IT III
EP2017258 (Al) teaches removal of the ethyl protecting group by trapping the [18F]- labelled precursor compound (II) onto a solid phase extraction (SPE) cartridge and incubating with 0.8 mL of a 4 mol/L solution of sodium hydroxide (NaOH). After 3 minutes incubation the NaOH solution was collected in a vial and a further 0.8 mL 4 mol/L NaOH added to the SPE cartridge to repeat the procedure. Thereafter the SPE cartridge was washed with 3 mL water and the wash solution combined with the collected NaOH solution. Then 2.2 mL of 6 mol/L HCl was then added with heating to 60°C for 5 minutes to remove the Boc protecting group. The resulting solution was purified by passing through (i) an ion retardation column to remove Na+ from excess NaOH and Cl~ from extra HCl needed to neutralise excess of NaOH to get a highly acidic solution before the acidic hydrolysis step, (ii) an alumina column, and (iii) a reverse-phase column. There is scope for the deprotection step(s) and/or the
purification step in the production of [18F]-FACBC to be simplified.
Example 1: Synthesis of f FIFACBC
No-carrier- added [18F]fluoride was produced via the 180(p,n)18F nuclear reaction on a GE PETtrace 6 cyclotron (Norwegian Cyclotron Centre, Oslo). Irradiations were performed using a dual-beam, 30μΑ current on two equal Ag targets with HAVAR foils using 16.5 MeV protons. Each target contained 1.6 ml of > 96% [180]water (Marshall Isotopes). Subsequent to irradiation and delivery to a hotcell, each target was washed with 1.6 ml of [160]water (Merck, water for GR analysis), giving approximately 2-5 Gbq in 3.2 ml of [160]water. All radiochemistry was performed on a commercially available GE FASTlab™ with single-use cassettes. Each cassette is built around a one-piece-moulded manifold with 25 three-way stopcocks, all made of polypropylene. Briefly, the cassette includes a 5 ml reactor (cyclic olefin copolymer), one 1 ml syringe and two 5 ml syringes, spikes for connection with five prefilled vials, one water bag (100 ml) as well as various SPE cartridges and filters. Fluid paths are controlled with nitrogen purging, vacuum and the three syringes. The fully automated system is designed for single-step fluorinations with cyclotron-produced [18F]fluoride. The FASTlab was programmed by the software package in a step-by-step time-dependent sequence of events such as moving the syringes, nitrogen purging, vacuum, and temperature regulation. Synthesis of
[18F]FACBC followed the three general steps: (a) [18F]fluorination, (b) hydrolysis of protection groups and (c) SPE purification.
Vial A contained K222 (58.8 mg, 156 μπιοΐ), K2C03 (8.1 mg, 60.8 μπιοΐ) in 79.5% (v/v)
MeCN(aq) (1105 μΐ). Vial B contained 4M HC1 (2.0 ml). Vial C contained MeCN
(4.1ml). Vial D contained the precursor (48.4 mg, 123.5 μιηοΐ) in its dry form (stored at -20 °C until cassette assembly). Vial E contained 2 M NaOH (4.1 ml). The 30 ml product collection glass vial was filled with 200 mM trisodium citrate (10 ml). Aqueous
[18F]fluoride (1-1.5 ml, 100-200 Mbq) was passed through the QMA and into the 180-
H20 recovery vial. The QMA was then flushed with MeCN and sent to waste. The trapped [18F]fluoride was eluted into the reactor using eluent from vial A (730 μΐ) and then concentrated to dryness by azeotropic distillation with acetonitrile (80 μΐ, vial C). Approximately 1.7 ml of MeCN was mixed with precursor in vial D from which 1.0 ml of the dissolved precursor (corresponds to 28.5 mg, 72.7 mmol precursor) was added to the reactor and heated for 3 min at 85°C. The reaction mixture was diluted with water and sent through the tC18 cartridge. Reactor was washed with water and sent through the tC18 cartridge. The labelled intermediate, fixed on the tC18 cartridge was washed with water, and then incubated with 2M NaOH (2.0 ml) for 5 min after which the 2M NaOH was sent to waste. The labelled intermediate (without the ester group) was then eluted off the tC18 cartridge into the reactor using water. The BOC group was hydrolysed by adding 4M HC1 (1.4 ml) and heating the reactor for 5 min at 60 °C. The reactor content with the crude [18F]FACBC was sent through the HLB and Alumina cartridges and into the 30 ml product vial. The HLB and Alumina cartridges were washed with water (9.1 ml total) and collected in the product vial. Finally, 2M NaOH (0.9 ml) and water (2.1 ml) was added to the product vial, giving a purified formulation of [18F]FACBC with a total volume of 26 ml. Radiochemical purity was measured by radio-TLC using a mixture of MeCN:MeOH:H20:CH3COOH (20:5:5: 1) as the mobile phase. The radiochemical yield (RCY) was expressed as the amount of radioactivity in the [18F]FACBC fraction divided by the total used [18F]fluoride activity (decay corrected). Total synthesis time was 43 min.
The RCY of [18F]FACBC was 62.5% ± 1.93 (SD), n=4.
/////FDA, diagnostic imaging agent, recurrent prostate cancer, fda 2016, Axumin, marketed, Blue Earth Diagnostics, Ltd., Oxford, United Kingdom, fluciclovine F 18
Date of issue of marketing authorisation valid throughout the European Union
22/05/2017
Contact address:
Blue Earth Diagnostics Ltd
215 Euston Road
London NW1 2BE
United Kingdom
Manufacture, characterisation and process controls
The active substance fluciclovine (18F) is prepared from the precursor AH113487 by nucleophilic substitution
of a triflate group by 18F-fluoride, followed by two deprotection steps. Due to the short half-life of the 18Ffluorine
radioisotope, each batch is prepared on the day of clinical use.
The active substance is prepared in a proprietary automated synthesiser unit. The synthesiser module is
computer-controlled. A fluid path for synthesis is provided in the form of a single use cassette (FASTlab). The
cassette contains 3 reagent vials and 3 solid phase cartridges. Two other reagent vials are supplied
separately as they have a recommended storage temperature of 2-8°C. These 2 vials are inserted into the
cassette on the day of production.
Assessment report
EMA/237809/2017 Page 13/90
Fluciclovine (18F) is produced in a continuous operation from the precursor AH113487. Due to the radioactive
nature of the process, and the short half-life of [18F] fluorine, intermediates are not isolated and there is no
opportunity for operator intervention or in-process testing. Control of the synthesis of fluciclovine (18F) from
the precursor is achieved through the automated synthesis platform, which is pre-programmed with
synthesis parameters optimised for the process. On-board detectors record transfers of radioactivity through
the fluid path at critical points and monitor temperature and pressure as appropriate so that the operator
may track the progress of the synthesis.
The active substance fluciclovine (18F) progressses immediately to purification, formulation and dispensing as
the finished product within a single, continuous operation. Validation of the manufacturing process for
fluciclovine (18F) is therefore described as part of finished product validation.
The characterisation of the active substance is in accordance with the EU guideline on chemistry of new
active substances.
As mentioned, the manufacture of the active substance and finished product takes place in a single,
continuous process. The active substance is not isolated at any point. Therefore, relevant information about
impurities is given only for the finished product.
For the same reason, information for the container closure system is provided only for the finished product.http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/004197/WC500230836.pdf
Finerenone; UNII-DE2O63YV8R; BAY 94-8862; DE2O63YV8R; 1050477-31-0
update FDA approved, 7/9/2021, Kerendia, To reduce the risk of kidney and heart complications in chronic kidney disease associated with type 2 diabetes
This table compares inhibitory (blocking) concentrations (IC50, unit: nM) of three antimineralocorticoids. Mineralocorticoid receptor inhibition is responsible for the desired action of the drugs, whereas inhibition of the other receptors potentially leads to side effects. Lower values mean stronger inhibition.[1]
Aldosterone is a hormone that exerts manifold deleterious effects on the kidneys, blood vessels, and heart which can lead to pathophysiological consequences. Inhibition of the mineralocorticoid receptor (MR) is a proven therapeutic concept for the management of associated diseases. Use of the currently marketed MR antagonists spironolactone and eplerenone is restricted, however, due to a lack of selectivity in spironolactone and the lower potency and efficacy of eplerenone. Several pharmaceutical companies have implemented programs to identify drugs that overcome the known liabilities of steroidal MR antagonists. Herein we disclose an extended SAR exploration starting from cyano-1,4-dihydropyridines that were identified by high-throughput screening. Our efforts led to the identification of a dihydronaphthyridine, BAY 94-8862, which is a potent, selective, and orally available nonsteroidal MR antagonist currently under investigation in a clinical phase II trial.
100 mg (ca. 0:24 mmol) of the compound from Example 23A are initially charged in 3 ml DMF.Is 2.94 mg Then (0.024 mmol) of 4-N, N-dimethylaminopyridine and 340 ul of ammonia (28 wt .-% – solution in water, 2:41 mmol) and 3 h at 100 0 C temperature.After cooling, the crude product is purified directly by preparative HPLC (eluent: acetonitrile / water with 0.1% formic acid, gradient 20:80 → 95: 5).There are 32 mg (37% d. Th.) The title connection receive.
LC-MS (Method 3): R, = 1:57 min;MS (EIPOS): m / z = 365 [M + H] +
640 mg (1.69 mmol) of the compound from Example 27A are initially charged in 30 ml of ethyl acetate, 342 mg (2.11 mmol) l, r-carbonyldiimidazole and then stirred overnight at room temperature.A TLC check (silica gel; mobile phase: cyclohexane / ethyl acetate 1: 1 or dichloromethane / methanol 9: 1) shows complete conversion.The volatile components are removed on a rotary evaporator and the residue taken up in 20 ml DMF.Subsequently, 2.36 ml of ammonia (28 wt .-% – solution in water, 16.87 mmol) was added and the reaction mixture for 8 hours at 50 0 C temperature.The solvent is distilled off under reduced pressure and the residue purified by preparative HPLC (eluent: acetonitrile / water with 0.1% formic acid, gradient 20:80 -> 95: 5).This gives 368 mg (58% d. Th.) Of the title compound.
LC-MS (method 7): R t = 1.91 min;MS (EIPOS): m / z = 379 [M + H] +
e ‘f 4- (4-Cyano-2-methoxyphenyl) -5-ethoxy-2,7-dimethyl-l, 4-dihydro-l, 6-naphthyridine-3-carbox- amide [(-) – enantiomer and (+) – enantiomer \
The racemate of Example 2 can be separated on a preparative scale by chiral HPLC into its enantiomers [column: Chiralpak IA, 250 mm x 20 mm;Eluent: methyl tert-butyl ether / methanol 85: 15 (v / v);Flow: 15 ml / min;Temperature: 30 0 C;UV detection: 220 Dm].
(-) – Enantiomer:
HPLC: R, = 5.28 min, ee> 98% [column: Chiralpak IA, 250 mm x 4.6 mm;Eluent: methyl tert-butyl ether / methanol 80:20 (v / v);Flow: 1 ml / min;Temperature: 25 0 C;UV detection: 220 nm];
specific optical rotation (chloroform, nm 589, 19.8 ° C, c = 0.50500 g / 100 ml): -239.3 °.
A single crystal X-ray structural analysis revealed a ^ -configuration at C * for this enantiomer – atom.
(+) – Enantiomer:
HPLC: R = 4:50 min, ee> 99% [column: Chiralpak IA, 250 mm x 4.6 mm;Eluent: methyl tert-butyl ether / methanol 80:20 (v / v);Flow: 1 ml / min;Temperature: 25 ° C;UV detection: 220 nm];
specific optical rotation (chloroform, nm 589, 20 0 C, c = 0.51000 g / 100 ml): + 222.7 °.
1:46 g (3.84 mmol) of the compound from Example 3oA are introduced into 50 ml of ethyl acetate, 777 mg (4.79 mmol) l, r-carbonyldiimidazole and then stirred overnight at room temperature.A TLC check (silica gel; eluent: ethyl acetate) shows complete conversion.The volatile components are removed on a rotary evaporator and the residue taken up in 20 ml DMF.Then 10.74 ml of ammonia (28 wt% solution in water, 76.8 mmol) was added and the reaction mixture heated for 30 minutes at 100 0 C.The solvent is distilled off under reduced pressure and the residue purified by preparative HPLC (eluent: acetonitrile / water with 0.1% formic acid, gradient 20:80 -> 95: 5).After concentrating the product fractions, the residue in 40 ml of dichloromethane / methanol (1: 1 v / v) and treated with 100 ml of ethyl acetate.The solvent is concentrated to a volume of about 20 ml, whereupon the product crystallized.The precipitate is filtered off and washed with a little diethyl ether.After drying at 40 0 C in a vacuum oven obtained 1:40 g (96%. Th.) The title connection.
LC-MS (Method 3): R, = 1.64 min;MS (EIPOS): m / z = 379 [M + H] +
e “M- (4-Cyano-2-methoxyphenyl) -5-ethoxy-2,8-dimethyl-l, 4-dihydro-l, 6-naphthyridine-3-carbox- amide [(-) – enantiomer and (+ ) enantiomer]
The racemate of Example 4 can be separated on a preparative scale by chiral HPLC into its enantiomers [column: 680 mm x 40 mm;Silica gel phase based on the chiral selector poly (N-methacryloyl-D-leucine dicyclopropylmethylamide; eluent: ethyl acetate; temperature: 24 ° C; flow: 80 ml / min; UV detection: 260 nm].
(-) – Enantiomer:
HPLC: R = 2:48 min, ee = 99.6% [column: 250 mm x 4.6 mm;Silica gel phase based on the chiral selector poly (N-methacryloyl-D-leucine dicyclopropylmethylamide; eluent: ethyl acetate; temperature: 24 ° C; flow: 2 ml / min; UV detection: 260 nm];
specific optical rotation (chloroform, nm 589, 19.7 ° C, c = 0.38600 g / 100 ml): -148.8 °.
A single crystal X-ray structure analysis showed this enantiomer S configuration at C * – atom.
(+) – Enantiomer:
HPLC: R = 4:04 min, ee = 99.3% [column: 250 mm x 4.6 mm;Silica gel phase based on the chiral selector poly (N-methacryloyl-D-leucine dicyclopropylmethylamide; eluent: ethyl acetate; temperature: 24 ° C; flow: 2 ml / min; UV detection: 260 nm];
specific optical rotation (chloroform, nm 589, 19.8 ° C, c = 0.36300 g / 100 ml): + 153.0 °.
The present invention relates to a novel and improved process for preparing 4- (4-Cyano-2-methoxyphenyl) -5-ethoxy-2,8-dimethyl-1, 4-dihydro- 1, 6-naphthyridine-3-carbox- amide of formula (I)
as well as the preparation and use of crystalline modification I of (4S) – 4- (4-Cyano-2-methoxyphenyl) -5-ethoxy-2,8-dimethyl-1, 4-dihydro- 1, 6-naphthyridine-3- carbox-amide of formula (I).
The compound of formula (I) acts as a non-steroidal mineralocorticoid receptor antagonist and can be used as agents for the prophylaxis and / or treatment of cardiovascular and renal diseases such as heart failure and diabetic nephropathy.
The compound of formula (I) and their preparation process are described in WO 2008/104306 and ChemMedChem 2012 7, described in 1385, in both publications a detailed discussion of research synthesis is disclosed. A disadvantage of the synthesis described there is the fact that this synthesis is not suitable for another large-scale process, since many steps in very high dilution, at very high reagent surpluses and thus run on a relatively low overall yield. Furthermore, many chromatographic cleanings are necessary, which are usually very expensive and require a high consumption of solvents, are costly and which should therefore be avoided if possible.Some stages can not be realized due to safety and procedural difficulties.
There is therefore a need for an industrially viable synthesis, reproducible in high overall yield, low production costs and high purity provides the compound of formula (I) and complies with all regulatory requirements in order to supply the clinical trials on drug and for subsequent regulatory submission to be used.
With the present invention a very efficient synthesis has been found, which allows to meet the above requirements.
In the publication ChemMedChem 2012 7, in which the research synthesis of the compound of formula (I) disclosed in 1385, the compound of formula (I), starting from vanillin prepared in 10 steps with an overall yield of 3.76% of theory , The compound of formula (I) was obtained by evaporation of the chromatography fractions as an amorphous solid, a defined process Kristalhsations- the stage for polymorphism-setting has not been described.
The following Scheme 1 shows the known process for preparing the compound of formula (I).
(II) (HI) (IV)
(V) (VI)
(XIII) (I)
Scheme 1: synthesis research of the compound of formula (I)
There are used 3 chromatographic purifications, and a chiral chromatography step to separate the enantiomers of the racemate of formula (XIII). The steps run partially in very high dilution and using very large amounts of reagent.
Thus, in particular the sequence of the preparation of the nitrile aldehyde intermediate (VI), which occupies a central role in the synthesis of atom not economically acceptable.
Furthermore, not to apply this process to an industrial scale, since [=> (IV) (III)] and excesses of acrylic acid tert-butyl ester are used for a very expensive reagents such as trifluoromethanesulfonic anhydride. When upscaling the Heck reaction (IV) => (V) formed in the boiler, a plastic similar residue resulting from the polymerization of acrylic acid tert.butyl ester used in excess. This is not acceptable in the technical implementation, there is a risk that there may be a Rührerbruch and it would lead to strong to remove residues in the agitators.
The subsequent cleavage of the double bond with sodium and the highly toxic osmium tetroxide is to be avoided since there is a delay of reaction and thereby caused to a strongly exothermic and connected with that comes a runaway under the test conditions described.
Scheme 2 illustrates the new process of the invention that the compound of formula (I) in 9 levels in 27.7% d. Th. Total yield without a chromatographic
Purification of intermediates supplies.
Scheme 2: According to the Invention for preparing the compound of formula (I).
Examples
example 1
Methyl 4-bromo-2-methoxybenzoate (XV)
3.06 kg (22.12 mol) potassium carbonate are placed in 1 acetone 3.6 and heated to reflux. To this suspension is metered in 1.2 kg of 4-bromo-2-hydroxybenzoic acid (5.53 mol) suspended in 7.8 1 of acetone and rinsed with 0.6 1 acetone. The mixture is heated for one hour under reflux (vigorous evolution of gas!). is boiled for 2.65 kg (21.01 mol) Dimethylsufat over 4 hours then metered. 2.5 hours then is stirred under reflux. The solvent is distilled off to a large extent (up to the stirrability) and returns to 12 1 toluene, then the remaining acetone is distilled off at 110 ° C. There are about 3 1 distillate distilled, these are supplemented by the addition of a further 3 1 toluene to approach. Allow to cool to 20 ° C and are 10.8 1 water were added and agitated vigorously. The organic phase is separated and the aqueous phase extracted again with 6.1 1 of toluene. The combined organic phases are washed with 3 1 of saturated sodium chloride solution, and the toluene phase is concentrated to about 4 first A quantitative analysis by evaporating a subset results converted a yield 1.306 kg (96.4% of theory). The solution is used directly in the next stage.
HPLC method A: RT about 11.9 min.
MS (EIPOS): m / z = 245 [M + H] +
H NMR (400 MHz, CD 2 C1 2 ): δ = 3.84 (s, 3H), 3.90 (s, 3H), 7:12 to 7:20 (m, 2H), 7.62 (d, 1H).
example 2
4-bromo-2-methoxybenzaldehyde (XVI)
It puts 1.936 kg (6.22 mol) 65% Red- Al solution in toluene with 1.25 1 of toluene at -5 ° C before. To this solution was dosed 0.66 kg (6.59 mol) of 1-methylpiperazine and rinsed with 150 ml of toluene, the temperature keeps you here from -7 to -5 ° C.. It is allowed for 30 minutes at 0 ° C. for. This solution is then dosed to a solution of 1.261 kg (5.147 mol) of methyl 4-bromo-2-methoxybenzoate (XV), dissolved in 4 1 of toluene, the temperature is maintained at – 8-0 ° C. Rinse twice with 0.7 1 of toluene and stirred for 1.5 hours at 0 ° C to. For working up, dosed to a 0 ° C cold aqueous sulfuric acid (12.5 1 water + 1.4 kg of conc. Sulfuric acid). The temperature should rise to a maximum of 10 ° C (slow dosage). The pH is, if necessary, by addition of further sulfuric acid to a pH of the first The organic phase is separated and extracted the aqueous phase with 7.6 1 of toluene. The combined organic phases are washed with 5.1 1 of water and then substantially concentrated and the residue taken up with 10 1 DMF. The mixture is concentrated again to about 5 1 volume. A quantitative analysis by evaporating a subset results converted a yield 1.041 kg (94.1% of theory). The solution is used directly in the next stage.
HPLC method A: RT approximately 12.1 min.
MS (EIPOS): m / z = 162 [M + H] +
X H-NMR (CDCl, 400MHz): δ = 3.93 (3H, s), 7.17 (2H, m), 7.68 (1H, d), 10:40 (1H, s)
example 3
4-formyl-3-methoxybenzonitrile (VI)
719 g (3.34 mol) of 4-bromo-2-methoxybenzaldehyde (XVI) as a solution in 4.5 1 of DMF with 313 g (0.74 mol) of potassium hexacyanoferrate (K4 [Fe (CN) 6]) and 354 g submitted (3.34 mol) of sodium carbonate and a further 1.2 1 of DMF and 3.8 g (0.017 mol) of palladium acetate. It is stirred for 3 hours at 120 ° C. Allow to cool to 20 ° C and are 5.7 1 water to approach. It is extracted with 17 1 ethyl acetate, and the aqueous phase is washed again with 17 1 of ethyl acetate to. The organic phases are combined and substantially concentrated with 5 1 of isopropanol was added and concentrated to about 2 1st The mixture is heated to boiling and dripping 2 1 of water.Allow to cool to 50 ° C and are again added 2 1 water. It is cooled to 3 ° C and stirred for one hour at this temperature. The product is filtered and washed with water (2 times 1.2 1) washed. It is dried at 40 ° C under vacuum.
1.035 kg (6.422 mol) of 4-formyl-3-methoxybenzonitrile (VI), 1.246 kg (8.028 mol) of 2-Cyanefhyl 3-oxobutanoate, 54.6 g (0.642 mol) of piperidine and 38.5 g (0.642 mol) of glacial acetic acid are heated under reflux on a water in 10 1 dichloromethane 6.5 hours. Allow to cool to room temperature and the organic phase was washed 2 times with 5 1 water. Subsequently, the dichloromethane phase is concentrated under atmospheric pressure and the still stirrable residue with 15.47 kg of 2-butanol and 0.717 kg (5.78 mol) of 4-amino-5-methylpyridone added. The residual dichloromethane is distilled off until an internal temperature of 98 ° C is reached. Then, 20 hours, heated under reflux. It is cooled to 0 ° C, can be 4 hours at this temperature is stirred and filtered off the product. It is dried at 40 ° C under vacuum to the carrier gas.
Yield: 2.049 kg (87.6% of theory based on 4-amino-5-methylpyridone, since this component is used in deficiency) of a slightly yellowish colored solid.
1.344 kg (8.34 mol) of 4-formyl-3-methoxy-benzonitrile (VI), 71 g (0.834 mol) piperidine and 50.1 g (0.834 mol) of glacial acetic acid are introduced into 6 1 of isopropanol at 30 ° C within 3 hours, a solution of 1.747 kg (11.26 mol) of 2-cyanoethyl 3-oxobutanoate metered in 670 ml of isopropanol. Stirring an hour after at 30 ° C. It is cooled to 0-3 ° C and stirred at 0.5 hours. the product is filtered off and washed 2 times with 450 ml of cold isopropanol to. For yield determination is under vacuum at 50 ° C. (2.413 kg, 97% of theory..); but it is usually due to the high yield continued to work directly with the isopropanol-moist product. For this, the product is taken up with 29 1 of isopropanol and 1.277 kg (7.92
mol) of 4-amino-5-methylpyridone added, followed by 24 internal temperature under about 1.4 bar overpressure in the closed vessel is heated at 100 ° C h. It is cooled by a ramp within 5 h at 0 ° C. stirred for 3 hours at 0 ° C. It is filtered off and washed with 2.1 1 of cold isopropanol. It is dried under vacuum at 60 ° C.
Yield: 2.819 kg (88% of theory based on 4-amino-5-methylpyridone, since this component is used in deficiency) of a slightly yellowish colored solid.
2.142 kg (5.3 mol) of 2-cyanoethyl 4- (4-cyano-2-methoxyphenyl) -2,8-dimefhyl-5-oxo-l, 4,5,6-tetrahydro-l, 6-naphthyridin-3 carboxylate (X) and 4.70 kg (29 mol) of triethyl orthoacetate are dissolved in 12.15 1 of dimethylacetamide and 157.5 grams of concentrated sulfuric acid was added. The mixture is heated for 1.5 hours at 115 ° C and then cooled to 50 ° C. At 50 ° C are added dropwise to 30 minutes 12.15 1 water. After complete addition the Titelbelbindung (XI) is treated with 10 g seeded and further added dropwise to 12.15 1 of water over 30 minutes at 50 ° C. It is cooled to 0 ° C (ramp, 2 hours) and stirred for 2 hours at 0 ° C to. The product is filtered, washed 2 times each with 7.7 1 of water and dried in vacuo at 50 ° C.
Yield: 2114.2 g (92.2% of theory) of a slightly yellowish colored solid.
2.142 kg (5.3 mol) of 2-cyanoethyl 4- (4-cyano-2-methoxyphenyl) -2,8-dimethyl-5-oxo-l, 4,5,6-tetrahydro-l, 6-naphthyridin-3 carboxylate (X) and 2.35 kg (14.5 mol) of triethyl orthoacetate are in 3.21 kg NMP (l-methyl-2-pyrrolidone) and dissolved 157.5 g of concentrated sulfuric acid was added. The mixture is heated for 1.5 hours at 115 ° C and then cooled to 50 ° C. At 50 ° C are added dropwise to 30 minutes 2.2 1 water. After complete addition the Titelbelbindung (XI) is treated with 10 g seeded and dropped further 4.4 1 of water over 30 minutes at 50 ° C. It is cooled to 0 ° C (ramp, 2 hours) and stirred for 2 hours at 0 ° C to. The product is filtered off, washed 2 times each with 4 1 of water and dried under vacuum at 50 ° C.
Yield: 2180.7 g (95.1% of theory) of a slightly yellowish colored solid.
2.00 kg (4.624 mol) of 2-cyanoethyl 4- (4-cyano-2-methoxyphenyl) -5-ethoxy-2,8-dimethyl-l, 4-dihydro-l, 6-naphthyridine-3-carboxylate (XI ) are dissolved in a mixture of 12 1 THF and 6 1 of water and cooled to 0 ° C. To this solution, a sodium hydroxide solution is added in drops within 15 minutes at 0 ° C (prepared from 0.82 kg 45% aqueous. NaOH (9.248 mol) and 4.23 1 of water and stirred for 1.5 hours at 0 ° C to . The mixture is extracted 2 times with each 4.8 1 methyl tert-butyl and once with 4.8 1 of ethyl acetate. The aqueous solution is at 0 ° C with dilute hydrochloric acid (prepared from 0.371 kg 37% HCl and 1.51 1 water ) adjusted to pH 7. the mixture is allowed to warm to 20 ° C and adding an aqueous solution of 2.05 kg of ammonium chloride in 5.54 1 water. the mixture is stirred 1 hour at 20 ° C, the product filtered and 2 times with each each 1.5 1 water and washed once with 4 1 acetonitrile. It is dried at 40 ° C under vacuum to the carrier gas.
Yield: 1736.9 g (99% of theory..) Of an almost colorless powder (very slight yellow tinge).
2.00 kg (4.624 mol) of 2-cyanoethyl 4- (4-cyano-2-methoxyphenyl) -5-ethoxy-2,8-dimethyl-l, 4-dihydro-l, 6-naphthyridine-3-carboxylate (XI ) are dissolved in a mixture of 12 1 THF and 6 1 of water and cooled to 0 ° C. To this solution, a sodium hydroxide solution is added in drops within 15 minutes at 0 ° C (prepared from 0.82 kg 45% aqueous. NaOH (9.248 mol) and 4.23 1 of water and stirred for 1.5 hours at 0 ° C to . Add 5 L of toluene and 381.3 g Natiumacetat added and stirred vigorously. Allow to settle the phases and the organic phase is separated. the aqueous phase is adjusted with 10% hydrochloric acid to pH 6.9 (at about pH 9.5 is inoculated with 10 g of the title compound of). After completion of the precipitation of the product for one hour at 0 ° C is stirred and then filtered and washed twice with 4 1 of water and twice with 153 ml of toluene. the mixture is dried at 40 ° C under vacuum to carrier gas (nitrogen, 200 mbar. yield:.. 1719.5 g (98% of theory) of an almost colorless powder (very slight yellow tinge).
1.60 kg (4.22 mol) of 4- (4-Cyano-2-methoxyphenyl) -5-ethoxy-2,8-dimethyl-l, 4-dihydro-l, 6-naphthyridine-3-carboxylic-isäure ( XII) and 958 g (5.91 mol) of 1,1-carbodiimidazole be presented in 8 1 of THF and at 20 ° C 51 g (0.417 mol) of DMAP was added. Stirring for one hour at 20 ° C (gas evolution!) And then heated 2.5 hours 50 ° C. are added to this solution 2.973 kg (18.42 mol) of hexamethyldisilazane and boil for 22 hours under reflux. Man admits further 1.8 1 THF and cooled to 5 ° C. A mixture is prepared from 1.17 1 of THF and 835 g of water is metered in over 3 hours, so that the temperature is between 5 and 20 ° C remains. Then boiled for one hour under reflux, then cooled via a ramp (3 hours) at 0 ° C. and stirred for one hour at this temperature. The product is filtered off and washed 2 times with 2.4 1 THF and twice with 3.2 1 water. It is dried under vacuum at 70 ° C under a carrier gas.
Yield: 1.501 kg (. 94% of theory) of an almost colorless powder (very slight yellow tinge).
carbox-amide (I) as a solution in acetonitrile / Methariol 40:60
Enantiomeric separation on a SMB unit
As a feed solution a solution corresponding to a concentration is used consisting of 50 g racemic 4- (4-cyano-2-methoxyphenyl) -5-ethoxy-2,8-dimethyl-l, 4-dihydro-l, 6-naphthyridin-3 -carbox-amide (XIII) dissolved in 1 liter of a mixture of methanol / acetonitrile 60:40.
There is a SMB unit on a stationary phase: 20 chromatographed μιη Chiralpak AS-V. The pressure is 30 bar, as the eluent a mixture of methanol / acetonitrile 60:40 is used.
9.00 kg of 4- (4-Cyano-2-methoxyphenyl) -5-ethoxy-2,8-dimethyl-l, 4-dihydro-l, 6-naphthyridine-3-carbox-amide (XII) are dissolved in 180 1 a mixture dissolved consisting of methanol / acetonitrile 60:40 and chromatographed by SMB. After concentrating the product-containing fractions, 69.68 liters of a 6.2% solution (corresponding to 4.32 kg (4S) – 4- (4-Cyano-2-methoxyphenyl) -5-ethoxy-2,8-dimethyl- 1, 4-dihydro- 1, 6-naphthyridine-3-carbox-amide (I) as a solution in acetonitrile / methanol 40:60).
Yield: 4.32 kg (48% of theory.) Dissolved in 69.68 liters of acetonitrile / methanol 40:60 as a colorless fraction.
Enantiomeric purity:> 98.5% ee (HPLC, method D)
A sample is concentrated in vacuum to give: MS (EIPOS): m / z = 379 [M + H] +
64.52 liters of a 6.2% solution of Example 8 in a mixture Acetonitiril / methanol 40:60 (equal 4.00 kg of compound 1) (1.2 .mu.m) via a filter cartridge and then concentrated at 250 mbar applicable so that the solution is still stirrable. It added 48 1 of ethanol denatured with toluene and distilled again at 250 mbar to stirrability from (Umdestillation on ethanol). They gave an additional 48 1 of ethanol denatured with toluene and then distilled at atmospheric pressure to a total volume of about 14 1 from (jacket temperature 98 ° C). The mixture was cooled via a ramp (4 hours) to 0 ° C, stirred for 2 hours at 0 ° C and filtered by the product from. It was washed twice with 4 1 of cold ethanol and then dried in vacuo at 50 ° C.
Yield: 3.64 kg (91% of theory.) Of a colorless, crystalline powder
Enantiomeric purity: “99% ee (HPLC method D); Retention times / RRT: (4S) – 4- (4-Cyano-2-methoxyphenyl) -5-ethoxy-2,8-dimethyl-l, 4-dihydro-l, 6-naphthyridine-3-carbox-amide (1) ca. 11 min. RRT: 1.00; (4R) – 4- (4-Cyano-2-methoxyphenyl) -5-ethoxy-2,8-dimethyl-l, 4-dihydro-l, 6-naphthyridine-3-carbox-amide (I) is about 9 min ,RRT: 0.82
Purity:> 99.8% (HPLC method B) RT: about 6.7 min.
Content: 99.9% (against an external standard)
specific rotation (chloroform, 589 nm, 19.7 ° C, c = 0.38600 g / 100 ml): – 148.8 °.
Melting point: 252 ° C (compound of formula (I) in crystalline form of modification I)
Physico-chemical characterization of compound of formula (I) in crystalline form of modification I
Compound of formula (I) melts in crystalline form of modification I at 252 ° C, ΔΗ = 95 -113 Jg 1 (heating rate 20 K min 1 , Figure 1).
A depression of the melting point was observed as a function of the heating rate.
The melting point decreases at a lower heating rate (eg 2 K min “1 ) because decomposition occurs. There were no other phase transitions. A mass loss of about 0.1% was observed up to a temperature of 175 ° C.
References
Schubert-Zsilavecz, M, Wurglics, M, Neue Arzneimittel Herbst 2015(German)
Pitt, B; Anker, S. D.; Böhm, M; Gheorghiade, M; Køber, L; Krum, H; Maggioni, A. P.; Ponikowski, P; Voors, A. A.; Zannad, F; Nowack, C; Kim, S. Y.; Pieper, A; Kimmeskamp-Kirschbaum, N; Filippatos, G (2015). “Rationale and design of MinerAlocorticoid Receptor antagonist Tolerability Study-Heart Failure (ARTS-HF): A randomized study of finerenone vs. Eplerenone in patients who have worsening chronic heart failure with diabetes and/or chronic kidney disease”. European Journal of Heart Failure17 (2): 224–32.doi:10.1002/ejhf.218. PMID25678098.
Bakris, G. L.; Agarwal, R; Chan, J. C.; Cooper, M. E.; Gansevoort, R. T.; Haller, H; Remuzzi, G; Rossing, P; Schmieder, R. E.; Nowack, C; Kolkhof, P; Joseph, A; Pieper, A; Kimmeskamp-Kirschbaum, N; Ruilope, L. M.; Mineralocorticoid Receptor Antagonist Tolerability Study–Diabetic Nephropathy (ARTS-DN) Study Group (2015). “Effect of Finerenone on Albuminuria in Patients with Diabetic Nephropathy: A Randomized Clinical Trial”. JAMA314 (9): 884–94. doi:10.1001/jama.2015.10081. PMID26325557.
The US Food and Drug Administration (FDA) has approved Bayer HealthCare’s Gadavist (gadobutrol) injection as the first magnetic resonance contrast agent for evaluation of breast cancer in the US.
The agency has approved the new indication for Gadavist injection for intravenous use with magnetic resonance imaging of the breast to assess the presence and extent of malignant breast disease.
Approval is based on priority review of two Phase III studies with identical design (GEMMA-1 and GEMMA-2).
Bayer HealthCare’s Gadavist (gadobutrol)
Bayer’s Gadavist injection cleared for breast cancer evaluation
UPDATE……. Gadoteridol 279.3 mg/ml for injection , CDSCO INDIA 29.07.2021
For intravenous use in magnetic reasonance imaging (MRI) in adults and pediatric patients over 2 years of age for whole body MRI including the head, neck, liver, breast, musculoskeletal system and soft tissue pathologies
The US Food and Drug Administration (FDA) has approved Bayer HealthCare’s Gadavist (gadobutrol) injection as the first magnetic resonance contrast agent for evaluation of breast cancer in the US.
It received marketing approval in Canada[1] and in the United States.[2][3][4]
As of 2007, it was the only GBCA approved at 1.0 molar concentrations.[5]
Gadobutrol is marketed by Bayer Schering Pharma as Gadovist, and by Bayer HealthCare Pharmaceuticals as Gadavist.[6]
Gadobutrol, SH-L-562, Gadovist
A different synthesis started from the previously reported tetraaza cyclopentaacenaphthylene (XV). Treatment of (XV) with a solution of piperazine at pH 6 gave rise to the bicyclic lactam (XVI). Alkylation of (XVI) with bromoacetic acid, followed by basic lactam hydrolysis furnished the tris(carboxymethyl) derivative (X), which was processed as in Scheme 3.
Argese, M.; Ripa, G. (Bracco SpA; Dibra SpA); 1,4,7,10-Tetraazabicyclo[8.2.2]tetradecan-2-one, a process for the preparation thereof and the use thereof for the preparation of tetraazamacrocycles. EP 0998476; JP 2002511884; WO 9905145
Gadobutrol, SH-L-562, Gadovist
In a related method for obtaining the precursor (V), epoxide (II) was condensed with the tosyl-protected tetraamine (XIII) in an autoclave at 170 C to give (XIV). The N-tosyl groups of (XIV) were then removed by treatment with lithium metal in liquid ammonia, yielding intermediate (III), which was then subjected to alkylation with bromoacetic acid, followed by acid hydrolysis
Platzek, J.; Gries, H.; Weinmann, H.-J.; Schuhmann-Giampieri, G.; Press, W.-R. (Schering AG); 1,4,7,10-Tetraazacyclododecane-butyl-triols, process for their preparation, and pharmaceutical agents containing these cpds.. DE 4009119; EP 0448191;
Gadobutrol, SH-L-562, Gadovist
The macrocyclic tetraamine (I) was protected as the triaminomethane derivative (VIII) by treatment with either triethyl orthoformate (4) or with dimethylformamide dimethylacetal (5). Alkylation of (VIII) with bromoacetic acid gave rise to the N-formyl N’,N”,N”’-tris(carboxymethyl) compound (IX). After basic hydrolysis of the formamide function of (IX), the resultant N-deprotected amine (X) was condensed with epoxide (II) to yield (XI). Further complexation with GdCl3 and ketal group hydrolysis led to the target compound
Murru, M.; Ripa, G.; Scala, A.; Viscardi, C.F.; Ausonio, M.; Scotti, C.; Cossuta, P. (Bracco SpA; Dibra SpA); A process for the preparation of macrocyclic chelants and the chelates thereof with paramagnetic metal ions. WO 9856775
This type of complexes with metal ions, in particular with paramagnetic metal ions; is used for the preparation of non-ionic contrast agents for the diagnostic technique known as magnetic resonance (MRI, Magnetic Resonance Imaging), among which are ProHance(R) (Gadoteridol, gadolinium complex of 10-(2-hydroxypropyl)-1,4,7,10-tetraazacyclododecane-1,4,7-triacetic acid), and Gadobutrol (gadolinium complex of [10-[2,3-dihydroxy-1-(hydroxymethyl)propyl]-1,4,7,10-tetraazacyclododecane-1,4,7-triacetic acid).
[0003]
Two different synthetic approaches are described in literature for the preparation of this kind of complexes, said approaches differing in the strategy taken to discriminate one of the four nitrogen atoms: the first one (Dischino et al., Inorg. Chem., 1991, 30, 1265 or EP 448191, EP 292689, EP 255471) is based on the selective protection of one of the nitrogen atoms by formation of the compound of formula (III), 5H,9bH-2a,4a,7-tetraazacycloocta[cd]pentalene, and on the subsequent hydrolysis to compound of formula (IV), 1-formyl-1,4,7,10-tetraazacyclododecane, followed by the carboxymethylation of the still free nitrogen atoms and by the deprotection and alkylation of the fourth nitrogen atom, according to scheme 1.
[0004]
The step from 1,4,7,10-tetraazacyclododecane disulfate (a commercially available product) to compound (III) is effected according to the conventional method disclosed in US 4,085,106, followed by formation of the compound of formula (IV) in water-alcohol medium.
[0005]
This intermediate is subsequently tricarboxymethylated with tert-butyl bromoacetate (TBBA) in dimethylformamide at 2.5°C and then treated with a toluene-sodium hydroxide diphasic mixture to give the compound of formula (V), 10-formyl-1,4,7,10-tetraazacyclododecane-1,4,7-triacetic, tris(1,1-dimethylethyl) ester, which is subsequently hydrolysed to compound of formula (II) in acidic solution.
[0006]
In the process described in WO 93/24469 for the synthesis of Gadobutrol, at first one of the nitrogen atoms is alkylated in conditions such as to minimize the formation of polyalkylated derivatives, then the monoalkylderivative is purified and carboxymethylated, according to scheme 2.
[0007]
The alkylation of 1,4,7,1,0-tetraazacyclododecane with the epoxide of formula (VI), 4,4-dimethyl-3,5,8-trioxabicyclo[5.1.0]octane, is carried out in anhydrous n-BuOH under reflux and the reaction mixture is extracted with water, evaporated to dryness and the residue is subsequently diluted with water and extracted with methylene chloride.
[0008]
The aqueous phase containing the mono-alkylated product (65% yield in Example 7 which reports the procedure for the preparation of 5 kg of Gadobutrol) is directly carboxymethylated at 70°C with chloroacetic acid, keeping pH 9.5 by addition of NaOH. The reaction mixture is adjusted to pH 1, concentrated to dryness and dissolved in methanol to remove the undissolved salts. The filtrate is then concentrated under vacuum, dissolved in water, and loaded onto a cation exchanger in the H+ form to fix the product. The subsequent elution with ammonia displaces the desired product, which is concentrated to small volume and subsequently complexed with gadolinium oxide according to conventional methods, and the resulting complex is purified by means of ion exchange resins. The overall yield is 42%.
[0009]
Although the first of these two processes could theoretically provide a higher yield, in that all the single steps (protection, carboxymethylation and deprotection) are highly selective, the complexity of the operations required to remove salts and solvents and to purify the reaction intermediates makes such theoretical advantage ineffective: the overall yield is in fact, in the case of Gadoteridol, slightly higher than 37%.
[0010]
The preparation of Gadobutrol according to the alternative process (WO 93/24469) provides a markedly better yield (72%) only on laboratory scale (example 2): example 7 (represented in the above Scheme 2) actually evidences that, when scaling-up, the yield of this process also remarkably decreases (42%).
[0011]
In addition to the drawback of an about 40% yield, both processes of the prior art are characterized by troublesome operations, which often involve the handling of solids, the use of remarkable amounts of a number of different solvents, some of them having undesirable toxicological or anyway hazardous characteristics.
[0012]
Moreover, the synthesis described by Dischino makes use of reagents which are extremely toxic, such as tert-butyl bromoacetate, or harmful and dangerous from the reactivity point of view, such as dimethylformamide dimethylacetal.
[0013]
An alternative to the use of dimethyl formamide dimethylacetal is suggested by J. Am. Chem. Soc. 102(20), 6365-6369 (1980), which discloses the preparation of orthoamides by means of triethyl orthoformate.
[0014]
EP 0596 586 discloses a process for the preparation of substituted tetraazacyclododecanes, among them compounds of formula (XII), comprising:
formation of the tricyclo[5.5.1.0] ring;
alkylation with an epoxide;
hydrolysis of the 10-formyl substituent;
reaction with an acetoxy derivative bearing a leaving group at the alpha-position.
[0015]
Nevertheless, this method requires quite a laborious procedure in order to isolate the product of step b).
[0016]
It is the object of the present invention a process for the preparation of the complexes of general formula (XII)
wherein
R1 and R2
are independently a hydrogen atom, a (C1-C20) alkyl containing 1 to 10 oxygen atoms, or a phenyl, phenyloxy group, which can be unsubstituted or substituted with a (C1-C5) alkyl or hydroxy, (C1-C5) alkoxy, carbamoyl or carboxylic groups,
Me3+
is the trivalent ion of a paramagnetic metal;
comprising the steps represented in the following Scheme 3:
The process of the present invention keeps the high selectivity typical of the protection/deprotection strategy described by Dischino in the above mentioned paper, while removing all its drawbacks, thus providing for the first time a reproducible industrial process for the preparation of the concerned compounds in high yields and without use of hazardous substances.
[0019]
The preparation of the gadolinium complex of 10-(2-hydroxypropyl)-1,4,7,10-tetraazacyclododecane-1,4,7-tri-acetic) acid (Gadoteridol), according to scheme 4, is particularly preferred:
in which the synthetic steps a), b), c), d), e), and f) have the meanings defined above and the epoxide of formula (XI) in step d) is propylene oxide.
[0020]
The preparation of the gadolinium complex of [10-[2,3-dihydroxy-1-(hydroxymethyl)propyl]-1,4,7,10-tetraazacyclododecane-1,4,7-triacetic) acid (Gadobutrol), according to the scheme 5, is also preferred.
in which the synthetic steps a), b), c), d), e), and f) have the meanings defined above and the epoxide of formula (XI) in step d) corresponds to the one of formula (VI), defined above.
[0021]
On the other hand, step a) of the process of the present invention involves the use of triethyl orthoformate in the presence of an acid catalyst, instead of dialkylformamide-dialkylacetal.
[0022]
Triethyl orthoformate can be added in amounts ranging from 105% to 200% on the stoichiometric value.
[0023]
The reaction temperature can range from 110 to 150°C and the reaction time from 5 to 24 h.
[0024]
The catalyst is a carboxylic acid having at least 3 carbon atoms, C3-C18, preferably selected from the group consisting of propionic, butyric and pivalic acids.
[0025]
Triethyl orthoformate is a less toxic and less expensive product than N,N-dimethylformamide-dimethylacetal and does not involve the formation of harmful, not-condensable gaseous by-products. Moreover, triethyl orthoformate is less reactive than N,N-dimethylformamide-dimethylacetal, which makes it possible to carry out the loading procedures of the reactives as well as the reaction itself in utterly safe conditions even on a large scale, allows to better monitor the progress of the reaction on the basis of such operative parameters as time and temperature, without checking the progress by gas chromatography, and makes dosing the reactive less critical, in that it can be added from the very beginning without causing the formation of undesired by-products: all that rendering the process suitable for the production of compound (III) on the industrial scale in easily reproducible conditions.
[0026]
The subsequent step b) involves the carboxymethylation of compound (III) in aqueous solution, using a haloacetic acid, to give compound (IX), i.e. the 10-formyl-1,4,7,10-tetraazacyclododecane-1,4,7-triacetic acid salt with an alkali or alkaline-earth metal, the salts of compound (IX) with sodium, potassium or calcium being most preferred.
Example 2
[0065]
[0066]
The procedure of Example 1 is followed until step C included, to obtain a solution of DO3A trisodium salt.
[0067]
pH is adjusted to 12.3 with conc. HCl and 57.7 kg (0.4 kmol) of 4,4-dimethyl-3,5,8-trioxabicyclo[5.1.0]-octane are added. After reaction for 4 h at 40°C and for 8 h at 80°C, the solution is cooled to 50°C, 120 kg of an aqueous solution containing 0.135 kmol of gadolinium trichloride are added. After 1 h the mixture is cooled at 17°C and acidified to pH 1.7 with conc. HCl, keeping this pH for 2 h. The solution is subsequently warmed to 50°C, pH is adjusted to 7 with sodium hydroxide, keeping these conditions for 1 h.
[0068]
After that, the resulting crude Gadobutrol is purified repeating exactly the same process as in steps E and F of Example 1.
Recovery of the product (Gadobutrol)
[0069]
The product-rich fraction is then thermally concentrated to a viscous residue and the residue is added with 350 kg of ethanol at 79°C.
[0070]
The resulting suspension is refluxed for 1 h, then cooled, centrifuged and dried under reduced pressure to obtain 66.0 kg of Gadobutrol (0.109 kmol), HPLC assay 99.5% (A%).
Overall yield: 79.1%
[0071]
The IR and MS spectra are consistent with the indicated structure.
600 MG TABLET ORAL, DRUGS FOR NEGLECTED DISEASES INITIATIVE
US FDA approves fexinidazole as the first all-oral treatment for sleeping sickness
POSTED ON JULY 19
The US Food and Drug Administration (FDA) has approved fexinidazole as the first all-oral treatment for both stages of the Trypanosoma brucei gambiense form of sleeping sickness (Human African trypanosomiasis) in patients 6 years of age and older and weighing at least 20 kg.
Fexinidazole was developed as part of an innovative partnership between the non-profit research and development organization Drugs for Neglected Diseases initiative (DNDi), which conducted the pivotal clinical trials for this treatment, in partnership with the National Sleeping Sickness Programs of the Democratic Republic of Congo (DRC) and Central African Republic (CAR), and Sanofi.
Sleeping sickness is a parasitic disease transmitted by the bite of an infected tse-tse fly. It affects mostly populations living in remote rural areas of sub-Saharan Africa, where about 65 million people are at risk of infection. Left untreated, sleeping sickness is almost always fatal. Through Sanofi’s collaboration the number of sleeping sickness cases reported to the WHO has been reduced by ~97% between 2001 and 2020. DNDi, Sanofi and partners are deeply committed to ensuring access to fexinidazole in all sleeping sickness-endemic countries.
Current treatment options for the disease are effective, but burdensome for patients and health workers due to the need for infusion or injection, requiring hospitalization, especially challenging for people living in remote areas.
“Having a simple, all-oral treatment for sleeping sickness is a dream come true for frontline clinicians,” said Dr Bernard Pécoul, DNDi Executive Director. “We are proud of this latest milestone in our long-term partnership with Sanofi, developed in close collaboration with researchers in countries hard-hit by sleeping sickness.”
Fexinidazole is indicated as a 10-day once-a-day treatment for Trypanosoma brucei gambiense sleeping sickness, the most common form of the disease found in West and Central Africa. Fexinidazole is the first all-oral treatment that works both for the first stage of the disease, as well as the second stage of the disease in which the parasites have crossed the blood-brain barrier, causing patients to suffer from neuropsychiatric symptoms.
“This FDA approval is a key milestone in Sanofi’s long-term commitment to fight sleeping sickness, started 20 years ago alongside the WHO through an ambitious partnership to combat Neglected Tropical Diseases” said Luc Kuykens, Senior Vice President, Sanofi Global Health unit. “Following the positive scientific opinion granted by the European Medicines Agency end 2018, the FDA approval is an important step to revitalize efforts to support the sustainable elimination of the disease”.
As a result of FDA approval, a Tropical Disease Priority Review Voucher (PRV) has been awarded to DNDi. The FDA Tropical Disease PRV Program was established in 2007 to incentivize development of new treatments for neglected tropical diseases, including sleeping sickness. Any benefits from the PRV will be shared between Sanofi and DNDi, which will enable continued investments in innovating for and ensuring access to new health tools for sleeping sickness and other neglected diseases. Sanofi commits to continue to provide the drug free-of-charge to the World Health Organization for distribution to affected countries, as part of a long-term collaboration with WHO.
About Sleeping sickness
Sleeping sickness, or human African trypanosomiasis (HAT), is usually fatal without treatment. Transmitted by the bite of an infected tse-tse fly, following a period with nonspecific symptoms, it evolves to cause neuropsychiatric symptoms, including abnormal behaviour, and a debilitating disruption of sleep patterns that have given this neglected disease its name. About 65 million people in sub-Saharan Africa are at moderate to very high risk of infection.
About DNDi
The Drugs for Neglected Diseases initiative (DNDi) is a collaborative, patient needs-driven, not-for-profit research and development (R&D) organization that develops safe, effective, and affordable treatments for sleeping sickness, leishmaniasis, Chagas disease, filarial infections, mycetoma, paediatric HIV, hepatitis C, and covid-19. Since its inception in 2003, DNDi has delivered eight new treatments, including nifurtimox-eflornithine combination therapy (NECT) for late-stage sleeping sickness, and fexinidazole, the first all-oral drug for sleeping sickness.
Fexinidazole was discovered by the German pharmaceutical company Hoechst AG, but its development as a pharmaceutical was halted in the 1980s.[5] Fexinidazole is now being studied through a collaboration between Sanofi and the Drugs for Neglected Diseases Initiative for the treatment of Chagas disease and human African trypanosomiasis (sleeping sickness).[6][7] Fexinidazole is the first drug candidate for the treatment of advanced-stage sleeping sickness in thirty years.[8]
Fexinidazole is currently in phase II/III clinical development at Drugs for Neglected Diseases Initiative for the oral treatment of African trypanosomiasis (sleeping sickness). In May 2009, Sanofi (formerly known as sanofi-aventis) licensed the drug candidate to Drugs for Neglected Diseases Initiative for the development, manufacturing and distribution as a treatment of human African trypanosomiasis. Once approved, the companies plan to make the drug available on a nonprofit basis.
Fexinidazole was originally developed by a German pharmaceutical company called Hoechst, now part of Sanofi; however, its development was abandoned in the 1980s when the company gave up its tropical disease programs. Fexinidazole is one of a class of drugs known as azoles, like fluconazole, that work against fungi and may work against cancer.
Onset of trypanosomiasis is caused by Trypanosoma protozoa and it is said that every year 200,000 to 300,000 of new patients of African sleeping sickness fall sick. At present the number of patients of African sleeping sickness cannot be confirmed due to the low reliability of the investigative data. According to the WHO, at least 150,000 people died of African sleeping sickness in 1996 and it is said that its aftereffect remains in not less than 100,000 people. Beyond that, enormous is the damage to domestic animals caused by a disease called as nagana, and several hundred thousands of cattle which are to be protein sources for people die every year. Further, in the area of about 10,000,000 km2of savanna equal to the United States of America, cattle-breeding is impossible due to Trypanosoma. Thus, African sleeping sickness remarkably damages the health and the economical development of African people, and this is the reason why the WHO adopts the trypanosomiasis as one of the infectious diseases that should be controlled.
African sleeping sickness is a protozoal infectious disease by Trypanosoma transmitted through tsetse flies and the protozoa appear in the blood stream in about 10 days after infection. In the initial period of infection the protozoa multiply in the blood stream and give fever, physical weakness, headache, a pain of muscles and joints and a feeling of itching to proceed. On entering the chromic period, the central nerve is affected to show symptoms such as mental confusion and systemic convulsion, and finally the patients lapse into lethargy and are led to death.
The trypanosomiasis of domestic animals has Trypanosoma brucei brucei, Trypanosoma evansi, Trypanosoma congolense and Trypanosoma vivax as pathogens and is a communicable disease which affects domestic animals such as horses, cattle, pigs and dogs and, in addition, mice, guinea pigs, rabbits and the like. Particularly, the loss of cattle and horses is greatest and almost fetal, and they are led to anemia, edema, weakening and the like and fall dead in one month after infection.
In treating trypanosomiasis, pentamidine, melarsoprol, eflornithine and the like are used and there was a feeling in the 1960s that its eradication might be possible. However, these drugs are old and are gradually losing their efficacy. Particularly, the resistance to melarsoprol of an arsenic agent causes a big problem and the situation is so dire that patients with no efficacy only await death and the development of novel antitrypanosoma agents are strongly desired.
Trypanosoma mainly lives in the blood stream of the human body. This bloodstream energy metabolism depends on the glycolytic pathway localized in the organelle characteristic of the protozoa which is called as glycosome and the so-called oxidative phosphorylation does not function. However, in order to efficiently drive this glycolytic pathway, the produced NADH has to be reoxidized, and the glycerol-3-phosphate oxidation system of mitochondria plays an important role in this reoxidation. The terminal oxidase of this oxidation system functions as a quinol oxidase having a reduced ubiquinone as an electron donor and has properties greatly different from those of cytochrome oxidase of an aerobic respiration system which the host has. Particularly, a remarkable point is that the terminal oxidase of the oxidation system is non-sensitive to the cyanide which quickly inhibits the cytochrome oxidase of the host. Then, many researchers centered around Western countries have tried to develop drugs targeting this cyanide resistant oxidase but effective drugs having a selective toxicity have not been obtained.
Under these circumstances the present inventors et al. found that isoprenoid based physiologically active substances of ascochlorin, ascofuranone and derivatives thereof, particularly ascofuranone specifically inhibits the glycerol-3-phosphate oxidation system of trypanosome at a very low concentration of the order of nM and filed a patent application (Japanese Patent Publication A No. : H09-165332). They also clarified that acofuranone exhibits a very strong multiplication inhibition effect in the copresence of glycerin (Molecular and Biochemical Parasitology, 81: 127-136, 1996).
In consideration of practical use of ascofuranone, it was found essential to discover agents which replace glycerin and exhibit an effect of the combined use in a small amount, and by using an alkaloid compound having an indole skeleton existing in a plant of the family Simaroubaceae together with ascofuranone, the prolongation of life and recovery effect in African seeping sickness was found and a patent application was filed (Japanese Patent Application No.: 2003-24643, Japanese Patent Publication A No.: 2004-23601).
Method for the preparation of fexinidazole, useful for the treatment of parasitic diseases, visceral leishmaniasis, chagas disease and human African trypanosomiasis. Family members of the product patent, WO2005037759, are expected to expire from October 2024. This to be the first application from Drugs for Neglected Diseases Initiative (DNDi) on this API. DNDi in collaboration with Sanofi, the Swiss Tropical & Public Health Institute and the University of Dundee, is developing fexinidazole, an antiparasitic agent, for treating human African trypanosomiasis (HAT) and visceral Leishmaniasis (VL). By June 2013, phase I clinical studies had been completed and at that time, DNDi was planning to initiate a phase II proof-of-concept study in VL patients in early 2013.
Chemotherapeutically active nitro compounds. 4,5-Nitroimidazoles. Part III
Synthesis
By condensation of 4 – (methylmercapto) phenol (II) with 1-mehtyl-2-chloromethyl-5-nitroimidazole (I) by means of K2CO3 in DMF (1,2) Description:. Crystals, mp 116 C. References: 1) Raether, W., Winkelman, E.; Chemotherapeutically active nitro compounds 4,5-Nitroimidazoles Part III Arzneim-Forsch 1978, 28 (5):. 739 2) Winkelmann,… E., Raether, W. (Hoechst AG); DE 2531303.
Winkelman, E.; Raether, W.;… Chemotherapeutically active nitro compounds 4,5-Nitroimidazoles Part III Arzneim-Forsch 1978, 28, 5, 739
Process for preparing fexinidazole – comprising the reaction of 1-methyl-2-hydroxymethyl-5-nitro-imidazole with methanesulfonyl chloride, followed by reaction with 4-methylmercapto-phenol, and further manipulative steps.
1-Methyl-2-hydroxymethyl-5-nitro-imidazole is (I) and 1-methyl-2-(4-methylmercapto-phenyloxymethyl)-5-nitro-imidazole (fexinidazole) is (II) (claim 1, page 12).
The synthesis of (II) via intermediate (I) is described (example 1, pages 6-8).
A process for preparing fexinidazole comprising the reaction of 1-methyl-2-hydroxymethyl-5-nitro-imidazole with methanesulfonyl chloride in the presence of a suspension of powdered alkaline carbonate (eg potassium carbonate) in an anhydrous organic solvent (eg acetone), followed by reaction with 4-methylmercapto-phenol, removal of hydrochloride salt, and isolation and purification is claimed. Also claimed is their use for treating parasitic diseases, visceral leishmaniasis, chagas disease, and human African trypanosomiasis. Fexinidazole is known to be an antiparasitic agent.
Topical chemotherapy for experimental murine African CNS-trypanosomiasis: the successful use of the arsenical, melarsoprol, combined with the 5-nitroimidazoles, fexinidazole or MK-436.
Tropical medicine & international health : TM & IH
Raether, W; Seidenath, H (1983). “The activity of fexinidazole (HOE 239) against experimental infections with Trypanosoma cruzi, trichomonads and Entamoeba histolytica”. Annals of Tropical Medicine and Parasitology77 (1): 13–26. PMID6411009.
Jennings, FW; Urquhart, GM (1983). “The use of the 2 substituted 5-nitroimidazole, Fexinidazole (Hoe 239) in the treatment of chronic T. brucei infections in mice”. Zeitschrift für Parasitenkunde69 (5): 577–581. doi:10.1007/bf00926669. PMID6636983.

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....











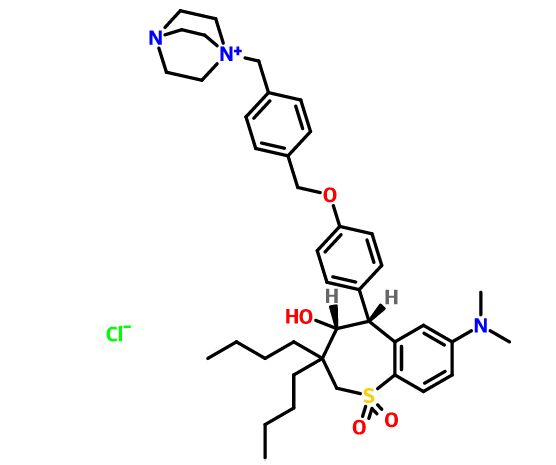




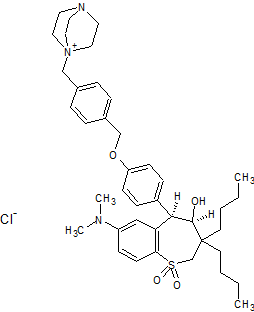
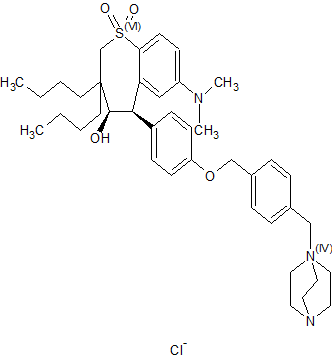















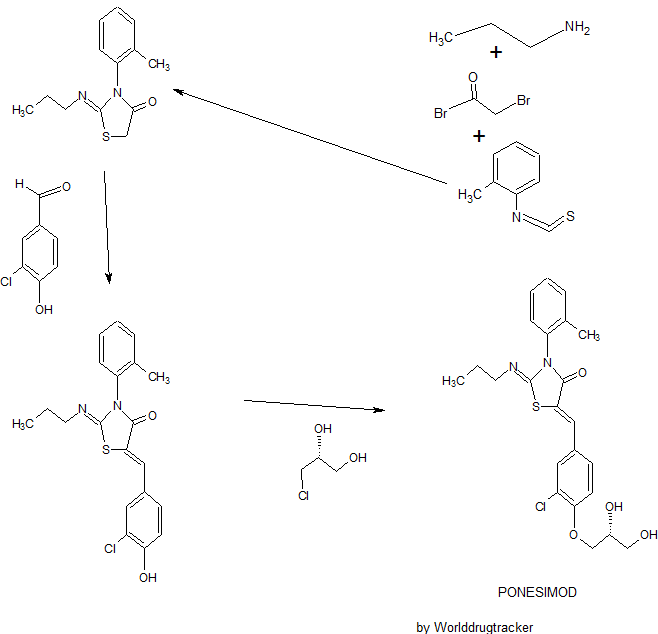
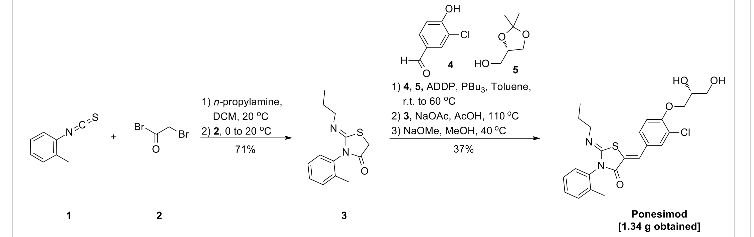

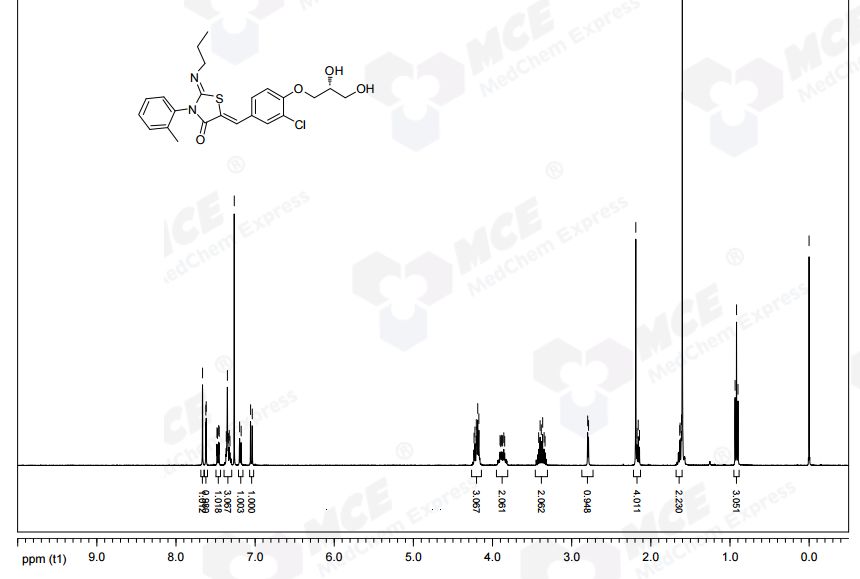
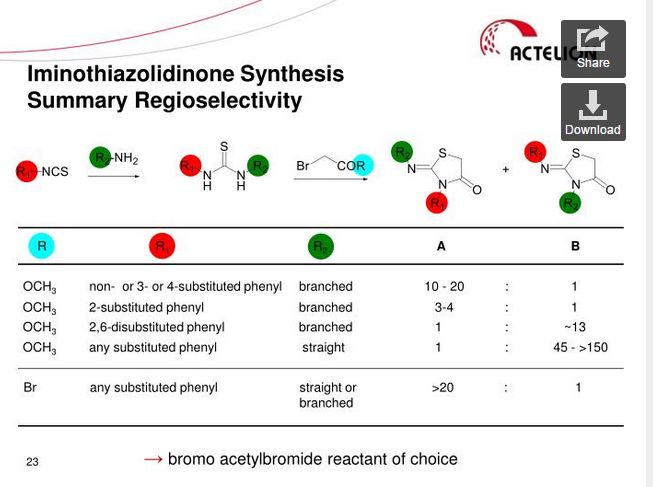
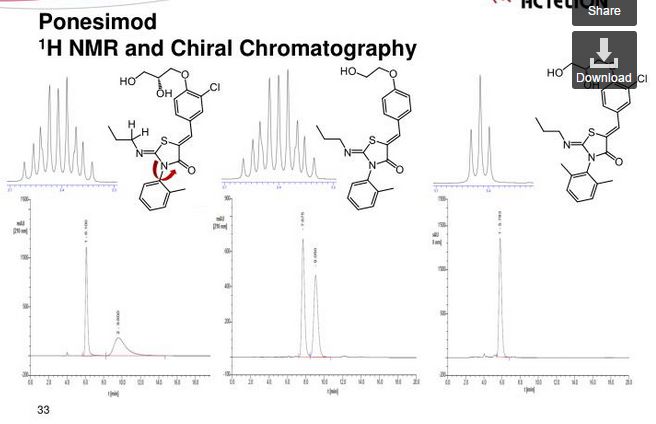














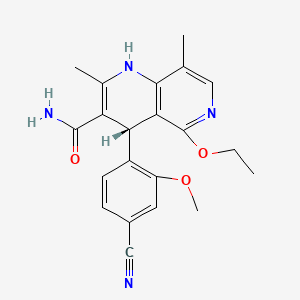
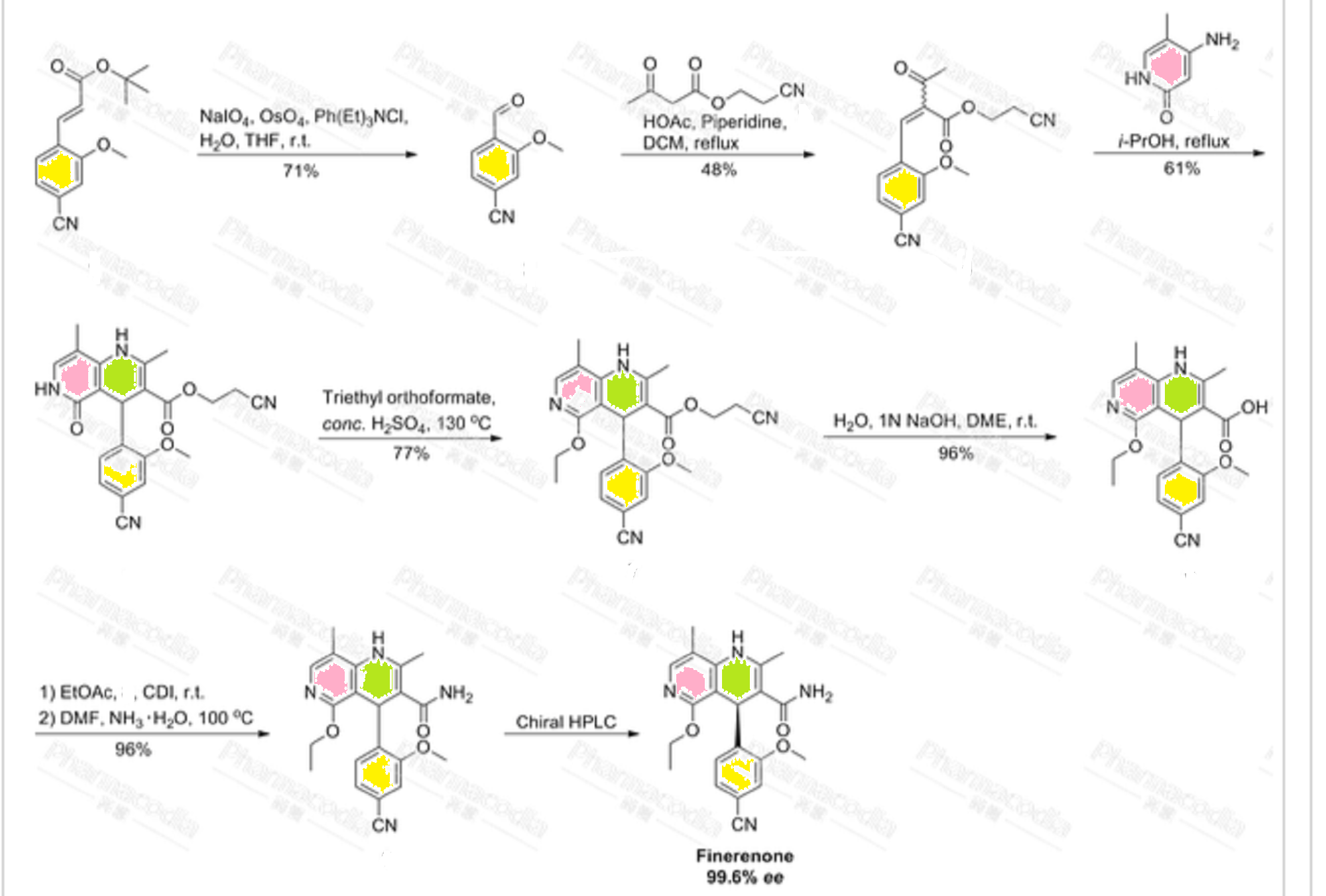





















 GADOBUTROL
GADOBUTROL




 WORLDCUP FOOTBALL WEEK 2014 BRAZIL
WORLDCUP FOOTBALL WEEK 2014 BRAZIL














