



| Patent | Submitted | Granted |
|---|---|---|
| BENZIMIDAZOLE MODULATORS OF VR1 [US2011190344] | 2011-08-04 | |
| BENZIMIDAZOLE MODULATORS OF VR1 [US2011190364] | 2011-08-04 | |
| Benzimidazole Modulators of VR1 [US7951829] | 2007-11-08 | 2011-05-31 |
Home » Articles posted by DR ANTHONY MELVIN CRASTO Ph.D (Page 435)
Author Archives: DR ANTHONY MELVIN CRASTO Ph.D
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on
ORGANIC SPECTROSCOPY INTERNATIONAL

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()




Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
UC Riverside’s Michael Pirrung announces development of TIR-199 for renal (kidney) cancer at conference in Dubai

The compound TIR-199 that holds much promise in the laboratory in fighting renal (kidney) cancer. feb19,2013
RIVERSIDE, Calif. — Chemists at the University of California, Riverside have developed a compound that holds much promise in the laboratory in fighting renal (kidney) cancer.
Named TIR-199, the compound targets the “proteasome,” a cellular complex in kidney cancer cells, similar to the way the drug bortezomib, approved by the Food and Drug Administration, targets and inhibits the proteasome in multiple myeloma cells, a cancer coming from bone marrow.
Michael Pirrung, a distinguished professor of chemistry at UC Riverside, announced the development of TIR-199 in a lecture he gave on Feb. 19 at the 5th International Conference on Drug Discovery and Therapy, held in Dubai, UAE.
Operating like the garbage dump of a cell, the proteasome breaks down proteins. Drugs that block the action of proteasomes are called proteasome inhibitors, and have been shown to have activity against a variety of cancer cell lines, albeit with mixed results. For example, bortezomib, though effective against multiple myeloma, has many side effects because cells other than bone marrow cells are affected.
“The novel feature of our new proteasome inhibitor, TIR-199, is that it is nearly as potent as bortezomib, but is selective in inhibiting the growth of only renal cancer cell lines,” Pirrung said. “It’s what makes TIR-199 attractive.”
The TIR-199 research project at UC Riverside began about four years ago after a multidisciplinary, international team reported on a class of compounds that act on the proteasome. These compounds are the “syringolin” natural products — such as a compound produced naturally by the wheat-infecting bacterium Pseudomonas syringae. TIR-199 is a synthetic relative of syringolin.

Michael Pirrung is a distinguished professor of chemistry at UC Riverside. Photo credit: I. Pittalwala, UC Riverside.

Structure of Syringolin A.
The ring structure of syringolin A is formed by the two nonproteinogenic amino acids 5-methyl-4-amino-2-hexenoic acid and 3,4-dehydrolysine. The α-amino group of the latter is joined by a peptide bond to a valine residue, which is linked to another valine residue via a urea moiety.
Researchers Begin Shigella Vaccine Trial , WRSs2 and WRSs3

FEB2013
PHASE 1 Safety and Immunogenicity of Two Live, Attenuated Oral Shigella Sonnei Vaccines WRSs2 and WRSs3
Phase 1, randomized, double-blind, placebo controlled, dose-escalation, inpatient study of single doses of S. sonnei. Enroll serial groups up to 90 subjects. Evaluate safety and tolerance of WRSs2 by monitoring presence and severity of clinical signs and symptoms, evaluate the immune response in blood and stool following ingestion of WRSs2
http://clinicaltrials.gov/show/NCT01336699
Shigellosis is one of those nasty bacterial diseases that follows the cringeworthy fecal-oral route to infect humans and other primates. Mild cases bring stomachaches; the severe end includes cramping, vomiting, fever, diarrhea, and it generally only gets more disgusting from there. While the disease can occur all over the world—estimates suggest ninety million cases of Shigellosis dysentery each year—the greatest mortality occurs in the third world. Hoping to stem transmission, or, at least, minimize the damage it causes, the World Health Organization has long called for a vaccine to stop Shigella infection.
And, today, scientists are one step closer. The National Institutes of Health announced that two Shigella vaccine have entered early-stage human clinical trials:
Researchers have launched an early-stage human clinical trial of two related candidate vaccines to prevent infection with Shigella, bacteria that are a significant cause of diarrheal illness, particularly among children. The Phase 1 clinical trial, funded by the National Institute of Allergy and Infectious Diseases (NIAID), part of the National Institutes of Health, will evaluate the vaccines for safety and their ability to induce immune responses among 90 healthy adults ages 18 to 45 years. The trial is being conducted at the Cincinnati Children’s Hospital Medical Center, one of the eight NIAID-funded Vaccine and Treatment Evaluation Units in the United States.

Researchers have launched an early-stage human clinical trial of two related candidate vaccines to prevent infection with Shigella, bacteria that are a significant cause of diarrheal illness, particularly among children. The Phase I clinical trial, funded by the National Institute of Allergy and Infectious Diseases (NIAID), part of the National Institutes of Health, will evaluate the vaccines for safety and their ability to induce immune responses among 90 healthy adults ages 18 to 45 years. The trial is being conducted at the Cincinnati Children’s Hospital Medical Center, one of the eight NIAID-funded Vaccine and Treatment Evaluation Units in the United States ….
…. Led by principal investigator Robert W. Frenck, Jr., M.D., director of clinical medicine at Cincinnati Children’s, the new clinical trial will evaluate two related candidate vaccines, known as WRSs2 and WRSs3, which have been found to be safe and effective when tested in guinea pigs and nonhuman primates. Both target Shigella sonnei, one of the bacteria’s four subtypes and the cause of most shigellosis outbreaks in developed and newly industrialized countries. Though neither candidate vaccine has been tested in humans, a precursor to both, known as WRSs1, was found to be safe and generated an immune response in small human trials in the United States and Israel. This early work was supported by NIAID, the U.S. Department of Defense and the Walter Reed Army Institute of Research. All three versions of the vaccine were developed by researchers at the Walter Reed institute.
Alexza gets EU thumbs-up for Adasuve, Loxapine

LOXAPINE
2-Chloro-11-(4-methylpiperazin-1-yl)dibenzo[b,f][1,4]oxazepine
FEBRUARY 22, 2013
Alexza Pharmaceuticals of the USA and partnerFerrer are celebrating after getting the green light for their inhaled antipsychotic Adasuve.
The European Commission has granted marketing authorisation to Adasuve (loxapine) for the rapid control of mild-to-moderate agitation in adults with schizophrenia or bipolar disorder. The approval is based on two Phase III studies involving over 650 patients which showed that the drug demonstrated statistically significant reductions in agitation from baseline compared to placebo.
The approval requires that patients receive regular treatment immediately after control of acute agitation symptoms, and that Adasuve is administered only in a hospital setting. Also, short-acting beta-agonist bronchodilator treatment should be available for treatment of possible severe respiratory side-effects.
Alexza chief executive Thomas King noted that Adasuve is the first authorised non-injectable therapy for these indications, noting that it will be launched on both sides of the Atlantic in the third quarter – US approval was granted in December.
Spain’s Ferrer has Adasuve rights in Europe, Latin America, Russia and the Commonwealth of Independent States countries. Chief executive Jordi Ramental said the focus for the initial EU launch will be Germany and Austria in 2013, adding that “at the same time, we are compiling the registration dossiers for the non-EU countries in our licensed territory”.
Links
Loxapine (Loxapac, Loxitane) is a typical antipsychotic medication, used primarily in the treatment of schizophrenia. It is a member of the dibenzoxazepine class and structurally related to clozapine (which belongs to the chemically akin class of dibenzodiazepines). Several researchers have argued that Loxapine may behave as an atypical antipsychotic.[1]
Loxapine may be metabolized by N-demethylation to amoxapine, a tetracyclic antidepressant.[2]
Dosage
The typical starting dosage is 10 mg twice daily; usual dose range 30–50 mg twice daily; maximum recommended dosage is 250 mg per day. The US Food and Drug Administration (FDA) has approved loxapine ( Adasuve , Alexza Pharmaceuticals) inhalation powder 10 mg for the acute treatment of agitation associated with schizophrenia or bipolar I disorder in adults. [4]

Schmutz, J.; Kunzle, F.; Hunziker, F.; Gauch, R.; Helv. Chim. Acta 1967, 50, 245.
- Glazer WM (1999). “Does loxapine have “atypical” properties? Clinical evidence”. The Journal of Clinical Psychiatry 60 (Suppl 10): 42–6. PMID 10340686.
- Cheung SW, Tang SW, Remington G (March 1991). “Simultaneous quantitation of loxapine, amoxapine and their 7- and 8-hydroxy metabolites in plasma by high-performance liquid chromatography”. Journal of Chromatography 564 (1): 213–21. doi:10.1016/0378-4347(91)80083-O. PMID 1860915.
- Sperry L, Hudson B, Chan CH (March 1984). “Loxapine abuse”. The New England Journal of Medicine 310 (9): 598. doi:10.1056/NEJM198403013100920. PMID 6694719.
- Harrison, Pam: Inhalant Approved for Agitation in Bipolar I, Schizophrenia. Medscape. Dec 24, 2012. “Clozapine and loxapine for schizophrenia”. Drug and Therapeutics Bulletin 29 (11): 41–2. May 1991. PMID 1747161.
- Chakrabarti A, Bagnall A, Chue P, et al. (2007). Chakrabarti, Abhijit. ed. “Loxapine for schizophrenia”. Cochrane Database of Systematic Reviews (Online) (4): CD001943. doi:10.1002/14651858.CD001943.pub2. PMID 17943763.
- Brennand, Kristen; Anthony Simone, Jessica Jou, Chelsea Gelboin-Burkhart, Ngoc Tran, Sarah Sangar, Yan Li, Yangling Mu, Gong Chen, Diana Yu, Shane McCarthy, Jonathan Sebat & Fred H. Gage (13 April 2011). “Modelling schizophrenia using human induced pluripotent stem cells”. Nature 473 (7346): 221–5. doi:10.1038/nature09915. PMID 21490598.
Janssen Research Development, LLC , JNJ 39439335, Mavatrep

Mavatrep; UNII-F197218T99; Mavatrep (USAN); JNJ-39439335; 956274-94-5;
2-(2-(2-(2-(4-trifluoromethylphenyl)vinyl)-1H-benzimidazol-5-yl)phenyl)propan-2-ol
(E)-2-(2-{2-[2-(4-trifluoromethyl-phenyl)-vinyl]-1H-benzimidazol-5-yl}-phenyl)-propan-2-ol
Phase I Musculoskeletal pain; Pain
- 01 Mar 2013 Janssen Research and Development completes a phase I trial in Japanese and Caucasian adult male volunteers in the US (NCT01631487)
- 01 Mar 2013 Janssen completes enrolment in its phase I trial for Pain (in volunteers) in the USA (NCT01631487)
- 05 Feb 2013 Janssen Research and Development initiates enrolment in a phase I trial for Pain (Japanese and Caucasian volunteers) in USA (NCT01631487)
- Originator Johnson & Johnson Pharmaceutical Research & Development
- Developer Janssen Research & Development
- Class Analgesics; Benzimidazoles; Small molecules
- Mechanism of Action TRPV1 receptor antagonists
| PHASE 1 Johnson & Johnson Pharmaceutical Research & Development, L.L.C. |
|
| Public title: | A Clinical Study to Investigate the Effect on Pain Relief of a Single Dose of JNJ-39439335 in Patients With Chronic Osteoarthritis Pain of the Knee |
http://clinicaltrials.gov/ct2/show/NCT01006304
http://apps.who.int/trialsearch/trial.aspx?trialid=NCT00933582
http://www.ama-assn.org/resources/doc/usan/mavatrep.pdf SEE STRUCTURE IN THIS FILE
MAVATREP IS JNJ-39439335
—
(E)-2-(2-(2-(4-(trifluoromethyl)styryl)-1H-benzo[d]imidazol-6-yl)phenyl)propan-2-ol hydrochloride
956282-89-6 CAS NO OF HCl SALT
Org. Process Res. Dev., Article ASAP
DOI: 10.1021/acs.oprd.5b00271, http://pubs.acs.org/doi/abs/10.1021/acs.oprd.5b00271

The process development of Mavatrep (1), a potent transient receptor potential vanilloid-1 (TRPV1) antagonist, is described. The two key synthetic transformations are the synthesis of (E)-6-bromo-2-(4-(trifluoromethyl)styryl)1H-benzo[d]imidazole (4) and the Suzuki coupling of 4 with 3,3-dimethyl-3H-benzo[c][1,2]oxaborol-1-ol (5). Compound 1a was prepared in four chemical steps in 63% overall yield.




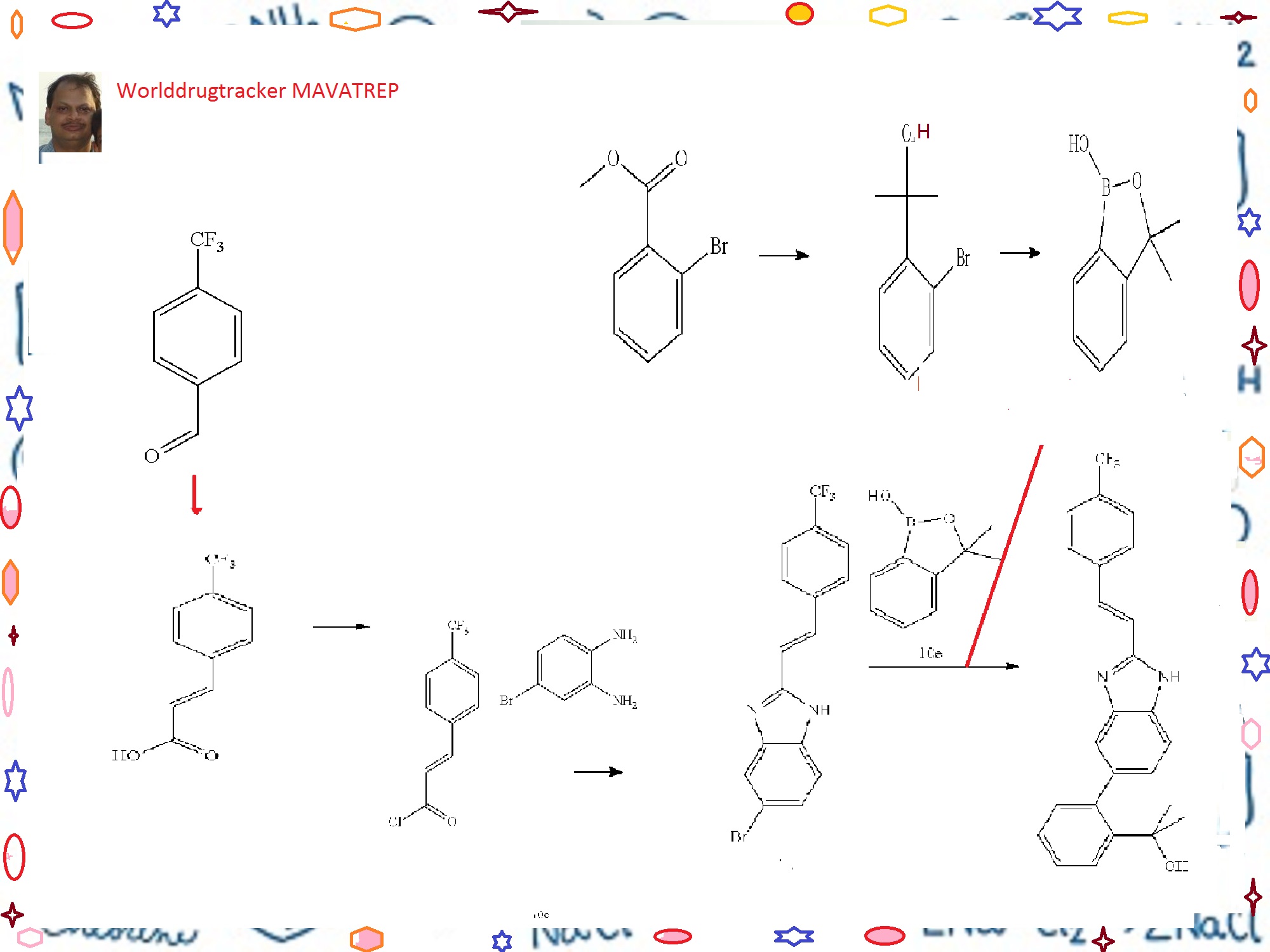
CLICK ON IMAGE FOR CLEAR VIEW
Example 10 (E)-2-(2-{2-[2-(4-trifluoromethyl-phenyl)-vinyl]-1H-benzimidazol-5-yl}-phenyl)-propan-2-ol(Cpd 18)
Step A. 3-(4-trifluoromethyl-phenyl)-acrylic acid
-
[0278]A solution of 4-trifluoromethylbenzaldehyde (7.7 mL, 57.7 mmol), malonic acid (12.0 g, 115.4 mmol), 0.567 μL piperidine (5.75 mmol) in 30 mL of pyridine was stirred at 70° C. for 18 h. The reaction solution was cooled to room temperature. Water (300 mL) was added and the resulting mixture was acidified to pH 4 (litmus) using concentrated hydrochloric acid to give a precipitate. The solid was filtered, and washed with water until the filtrate was neutral. The solid product was dried in vacuo to give the title Compound 10a as a white powder (11.2 g, 90%). 1HNMR (400 MHz, DMSO-d6) δ (ppm): 12.60 (bs, 1H), 7.92 (d, 2H, J=8.2 Hz), 7.77 (d, 2H, J=8.2 Hz), 7.66 (d, 1H, J=16.0 Hz), 6.70 (d, 1H, J=16.0 Hz).
-
[0000]

Step B. (E)-5-bromo-2-[2-(4-trifluoromethyl-phenyl)-vinyl]-1H-benzimidazole
-
[0279]A solution of Compound 10a (20.6 g, 95.4 mmol) in anhydrous methylene chloride (200 mL) was treated with oxalyl chloride (16.6 mL, 190 mmol) and “3 drops” of anhydrous dimethylformamide. The resulting solution was stirred at room temperature under an argon atmosphere for 18 h. The solvent was concentrated to give 3-(4-trifluoromethyl-phenyl)-acryloyl chloride Compound 10b as a solid, which was used without further purification in the next step.
-
[0280]To a solution of 4-bromo-benzene-1,2-diamine (16.1 g, 86.7 mmol) in acetic acid (100 mL) was added dropwise a solution of Compound 10b (assumed 95.4 mmol) in acetic acid (100 mL). The reaction mixture was stirred at 100° C. for 18 h. The reaction mixture was cooled to room temperature, and a mixture of ethyl acetate and hexanes 3:7 (500 mL) was added. The mixture was triturated at room temperature for 3 h to give a precipitate. The solid was filtered, and dried in vacuo to give the title Compound 10c (23.2 g, 73%). 1H NMR (400 MHz, DMSO-d6/CDCl3) δ (ppm): 8.45 (d, 1H, J=16.7 Hz), 7.84-7.90 (m, 1H), 7.74 (d, 2H, J=8.3
-
[0281]Hz), 7.56-7.62 (m, 3H), 7.50-7.52 (m, 1H), 7.34 (d, 1H, 16.7 Hz).
-
[0000]

Step C. 2-(2-bromo-phenyl)-propan-2-ol
-
[0282]To a solution of methyl 2-bromobenzoate (20.76 g, 96 mmol) in 120 mL of anhydrous ether under Argon at 0° C. was slowly added methylmagnesium bromide (77 mL, 3.26 M) at a rate that the internal temperature of the mixture was below 20° C. A white suspension resulted, and the mixture was stirred at room temperature for 2 h. The mixture was cooled in an ice-water bath. To the reaction mixture was very slowly added hydrochloric acid (400 mL, 0.5 M). The pH of the final mixture was adjusted to less than about 6 with few drops of 2M hydrochloric acid. The layers were separated, and the aqueous layer was extracted twice with ether. The organic layers were combined and dried over magnesium sulfate. The organic fraction was filtered, and the filtrate was concentrated to yield the title compound as a pale yellow liquid, which was distilled under vacuum to afford the title Compound 10d as a colorless liquid (16.9 g, 82%, b.p. about 65-70° C./0.3 mmHg). 1H NMR (400 MHz, CDCl3) δ (ppm): 7.67 (dd, 1H, J=1.7, 7.9 Hz), 7.58 (dd, 1H, J=1.3, 7.9 Hz), 7.30 (ddd, 1H, J=1.4, 7.4, 7.9 Hz), 7.10 (ddd, 1H, J=1.7, 7.4, 7.8 Hz), 2.77 (br s, 1H), 1.76 (s, 6H).
-
[0000]

Step D. 3,3-dimethyl-3H-benzo[c][1,2]oxaborol-1-ol
-
[0283]To a solution of n-BuLi (166 mL, 2.6 M, 432 mmol) in 200 mL of THF at −78° C. under argon was slowly added a solution of Compound 10d (42.2 g, 196 mmol) in 60 mL of THF at a rate that the internal temperature remained below −70° C. The mixture was stirred at −75° C. for 2 h. To the reaction mixture was then added triisopropylborate (59 mL, 255 mmol) in three portions. The mixture was allowed to warm slowly to room temperature overnight. The mixture was then cooled to 0° C., and was carefully quenched with dilute hydrochloric acid (250 mL, 2N). The mixture was then stirred at room temperature for 1 h. The pH of the mixture was checked and adjusted to acidic using additional 2N HCl if prophetic. The two layers were separated, and the aqueous layer was extracted twice with ether. The organic layers were combined, and dried with magnesium sulfate and filtered. The filtrate was concentrated under reduced pressure to yield a pale yellow oil. The residue was then diluted with ethyl acetate (400 mL) and, washed with 1N sodium hydroxide solution (150 mL×3). The basic aqueous layers were combined and acidified with 2N HCl. The clear solution turned cloudy when the acid was added. The mixture was extracted with ether (150 mL×3). The organic layers were combined and dried with magnesium sulfate. The solution was filtered, and the filtrate was concentrated under reduced pressure to yield the title Compound 10e as a colorless oil (26.2 g, 82%) which was used without further purification in the next step. 1H NMR (400 MHz, DMSO-d6) δ (ppm): 9.00 (s, 1H), 7.66 (dm, 1H, J=7.3 Hz), 7.45 (dt, 1H, J=1.1, 7.7 Hz), 7.40 (dm, 1H, J=7.6 Hz), 7.31 (dt, 1H, J=1.2, 7.1 Hz), 1.44 (s, 6H).
-
[0000]

Step E. (E)-2-(2-{2-[2-(4-trifluoromethyl-phenyl)-vinyl]-1H-benzimidazol-5-yl}-phenyl)-propan-2-ol
-
[0284]To a mixture of Compound 10e (11.7 g, 71 mmol), Compound 10c (19.9 g, 54 mmol), sodium carbonate (46 g, 435 mmol) and PdCl2(dppf).CH2Cl2 (8.9 g, 11 mmol) in a 1 L round bottom flask equipped with water condenser was added 400 mL of anhydrous DME and 200 mL of water. The mixture was evacuated and filled with Argon three times. The mixture was heated to 100° C. for 20 h. The mixture was then cooled to room temperature. The biphasic system was transferred to a 1 L separatory funnel and the two layers were separated. The organic layer was washed with brine (2×300 mL). The aqueous layers were combined and extracted with ethyl acetate once (about 300 mL). The organic layers were combined, dried with sodium sulfate, and filtered. The volume of the filtrate was reduced to about 170 mL under reduced pressure. The mixture was then filtered through a pad of silica gel and the pad was washed with ethyl acetate until the filtrate did not contain any product. After concentration, a light pink/beige solid was obtained. The solid was triturated with 50 mL ethyl acetate, and the mixture was heated to 85° C. for 5 min. The mixture was slowly cooled to r.t., then cooled at 0° C. for 0.5 h. The mixture was filtered, and the solid was washed with cold ethyl acetate twice, and dried under vacuum at 40° C. to yield the title Compound 18 as a light beige solid (7.58 g, 33%). RP-HPLC 95% pure.
-
1H NMR (400 MHz, DMSO-d6) δ (ppm): 12.73 (m, 1H,), 7.90 (d, 2H, J=8.2 Hz), 7.85 (dd, 1H, J=8.0, 0.6 Hz), 7.78 (d, 2H, J=8.4 Hz), 7.74 (d, 1H, J=16.8 Hz), 7.59-7.47 (m, 1H), 7.41 (s, 1H), 7.37-7.32 (m, 2H), 7.21 (dt, 1H, J=1.2, 7.4 Hz), 7.06 (s, 1H), 7.02 (d, 1H, J=7.4 Hz), 4.85 (s, 1H), 1.21 (s, 6H).
-
Mass Spectrum (LCMS, APCI pos.) Calcd. For C25H21F3N2O: 423.2 (M+H). Found 423.3.
-
m.p. (uncorr.) 250-251° C.
Example 10.1 Scale Up Preparation of (E)-2-(2-{2-[2-(4-trifluoromethyl-phenyl)-vinyl]-1H-benzimidazol-5-yl}-phenyl)-propan-2-ol (Cpd 18) Step A. 3-(4-trifluoromethyl-phenyl)-acrylic acid
-
[0286]A 2-L 4-neck round bottom flask equipped with an air condenser/argon inlet, mechanical stirrer, thermocouple and a stopper was charged with 4-(trifluoromethyl)benzaldehyde (250 g, 196.2 mL, 1.44 mol), malonic acid (302.6 g, 2.87 mol), and pyridine (750 mL). An exotherm developed (about 38-40° C.), which was maintained for 30 min. Piperidine (14.202 mL, 143.58 mmol) was then added to the reaction and a second exotherm developed (Tmax about 42° C. after about 10 min.). The reaction was stirred for 30 min and then heated to 60° C. for 18 h (overnight). The reaction appeared to be complete by TLC, and was cooled to about 40° C., diluted into water (2 L; done to prevent reaction freezing), cooled to room temperature, and further diluted with water (4 L, 6 L total). The slurry was acidified to pH=2.0-3.0 with concentrated hydrochloric acid (about 675-700 mL). The material was stirred for 30 min., and a white solid was collected by filtration. The filter cake was washed with water until the filtrate was neutral (pH about 5.5-6, 2.5 L), air-dried in a Buchner funnel for 2 h, and then further dried in a vacuum oven at 60° C. overnight to provide 300.5 g (96%) of the title Compound 10a as a white solid.
Step B. (E)-5-bromo-2-[2-(4-trifluoromethyl-phenyl)-vinyl]-1H-benzimidazole
-
[0287]To a 5-L 4-neck round bottom flask equipped with a magnetic stirrer, argon inlet-argon outlet to a carbonate scrub, two stoppers, and a room temperature water bath was charged with 4-(trifluoromethyl)cinnamic acid (315 g, 1.46 mol) and dichloromethane (3.15 L) to give a slurry. To the slurry was added oxalyl chloride (151.71 mL, 1.75 mol) and DMF (1.13 mL, 14.57 mmol). Upon addition of DMF, gas evolution commenced, and the reaction was continued for about 3 h during which time a solution developed. When the reaction was complete (LC-MS), it was concentrated to dryness to give 342.4 g of 3-(4-trifluoromethyl-phenyl)-acryloyl chloride Compound 10b (>100%) as a yellow oily solid.
-
[0288]A 5-L 4-neck round bottom flask equipped with mechanical stirrer, thermocouple, air condenser with argon inlet, and a stopper was charged with 4-bromo-benzene-1,2-diamine (244 g, 1.27 mol) and acetic acid (2.13 L). To this solution was added a solution of Compound 10b (327 g, 1.39 mol) in toluene (237 mL). After this addition, the temperature spiked to 45° C. in about 30 seconds and then subsided. The reaction was then heated to 90° C. for 16 h (overnight). The reaction was cooled to 40° C., and poured into a mixed solution of EtOAc and heptane (about 1:3, 5.75 L) and a precipitate occurred. The resulting slurry was stirred for 3 h, and the solid was collected by filtration, washed with EtOAc:heptane (1:3, 3 L), and then dried in a vacuum oven (60° C.) to give 324.3 g (65%) of the title Compound 10c as a partial acetate salt.
Step C. 2-(2-bromo-phenyl)-propan-2-ol
-
[0289]A 12-Liter 4-neck flask equipped with a thermocouple, condenser, septum, addition funnel and overhead mechanical stirrer under argon was charged with methyl-2-bromobenzoate (226.5 g, 1.05 mol) and THF (1.6 L, 19.66 mol). The mixture was cooled to a temperature between 2 and 5° C. with stirring and held for 30 min. To the solution was slowly added methyl magnesium bromide in diethyl ether (3M, 1.05 L; 3.15 mol) via the addition funnel at a rate to maintain the reaction temperature below 15° C. An exotherm was observed during the addition, the reaction temperature warmed from 3 to 15° C. The addition of 1.05 L Grignard was complete in 4 h (approximate feed rate was 4.17 mL/min). The reaction mixture appeared to be off-white/yellow slurry. The reaction was allowed to warm to room temperature and stirred overnight (15 h). The reaction was sampled by HPLC/TLC and showed no starting material present. The ice bath was again applied to the reaction flask and a 0.5 M HCl solution (4.5 L; 2.25 mol) was slowly added over a period of 2 h. The temperature increased dramatically from 0 to 15° C. After the quench was complete, the reaction was stirred at room temperature for 30 min. Additional 2 N HCl (500 mL; 1.00 mol) was slowly added to maintain a pH less than 6. MTBE (1 L) was added to help with the phase split. The reaction was stirred at room temperature for 1 to 2 h to dissolve the solid material into the aqueous phase (most likely Mg(OH)2 which is very basic). The pH must be checked and adjusted with additional acid when necessary. The phases were separated and the aqueous layer was washed with an additional 1 L MTBE (2×500 mL). The organic phases were combined, washed with NaHCO3 solution (2×300 mL), dried over MgSO4, filtered and the filtrate was concentrated under vacuum to yield the title Compound 10d (220.83 g, 97.48% yield) as a clear yellow oil.
Step D. 3,3-dimethyl-3H-benzo[c][1,2]oxaborol-1-ol
-
[0290]A 12-Liter 4-neck round bottom flask equipped with a thermocouple, condenser, addition funnel and overhead mechanical stirrer under dry Argon was charged with anhydrous THF, (3 L) and chilled to −70 to −78° C. via a dry ice/acetone bath. n-Butyl lithium (2.5N in hexanes, 860 mL, 2.15 mol) was slowly added via addition funnel. An exotherm was observed as the temperature rose from −78 to −70° C. To the addition funnel was added a solution of Compound 10d (220 g, 979.97 mmol) in anhydrous THF (1 L). The 2-(2-bromophenyl)propan-2-ol solution was slowly added to the n-BuLi solution. The addition took 90 min in order to maintain a reaction temperature below −70° C. After the addition was complete, the reaction mixture was stirred at −70 to −75° C. for 30 min. The triethylborate (230 mL, 1.35 mol) was quickly added in 3 portions at −70° C. An exotherm was observed, the batch temperature rose from −70 to −64° C. The reaction was stirred at −70° C. and slowly warmed to room temperature over night. After the reaction was cooled to 0-5° C., the reaction was slowly quenched with 2 M HCl (1 L, 2.00 mol) added via the addition funnel while maintaining the batch temperature 0-5° C. The reaction mixture was stirred for 1 h. The aqueous phase pH was 9-10. The pH was then adjusted to acidic (4-5) with 2 M HCl (200 mL). The two phases were separated and the aqueous layer was extracted with MTBE (2×500 mL). The combined organic phases were dried with anhydrous magnesium sulfate. The solution was filtered and concentrated to yield a yellow oil. The yellow oil was diluted with MTBE (1.5 L) and washed with 1M NaOH (3×500 mL). The product containing basic aqueous phases were combined and acidified with 2 M HCl (800 mL) (the clear solution turns turbid with the addition of acid). After stirring the turbid solution for 15 min (pH=4-5) (Note 1), it was extracted with MTBE (2×500 mL). The organic phases were combined and dried over MgSO4. The solution was filtered and the filtrate was concentrated to yield the title Compound 10e as a clear yellow oil (121.78 grams, 77% yield).
Step E. (E)-2-(2-{2-[2-(4-Trifluoromethyl-phenyl)-vinyl]-1H-benzimidazol-5-yl}-phenyl)-propan-2-ol
-
A 5-L 4-neck flask equipped with a thermocouple controller, condenser, overhead mechanical stirrer, Firestone Valve® and a nitrogen inlet/outlet was charged with dimethoxyethane (2 L), DI water (1 L) and sodium carbonate (230.9 g, 2.18 mol). The solution was degassed and purged with N2 three times. Compound 10e (71.7 g, 0.35 mol) and Compound 10c (100.0 g, 0.27 mol) were added to the degassed solution. The solution was degassed and purged with N2 three times. PdCl2(dppf) (44.48 g, 54.4 mmol) was added to the solution, and the solution was degassed and purged with N2 three times. The resulting two-phase suspension was heated to reflux for 18 h, and then cooled to room temperature. The reaction mixture was transferred to a 12-L separatory funnel, and the layers were separated. The organic layer was washed with brine (1 L). The two aqueous layers were combined and extracted with EtOAc (1 L). The combined organic layers were dried (Na2SO4), filtered, and the filtrate was concentrated to an oil. Two separate 100 g coupling reactions were combined and purified by chromatography in 10 successive chromatography runs on an ISCO preparative chromatography system (10×1.5 Kg SiO2, 5 column volumes of EtOAc, 250 mL/min flow rate). The combined fractions were transferred to two 22 L 4-neck round bottom flasks, and Silicycle Si-thiol functionalized silica gel (2 g) was added to each solution. The solutions were warmed to 40° C. and aged for 1 h. The solutions were filtered thru a medium glass funnel and washed with EtOAc (4 L) and combined. The filtrate was evaporated to a semi solid, which was transferred to a 2 L round bottom flask, to which EtOAc (0.4 L) was added. The resulting white precipitate slurry was cooled to −5° C. and stirred for 1 h. The slurry was filtered and washed twice with cold EtOAc (100 mL). The solids were dried in a vacuum oven at 40° C. for 40 h to afford 84.0 g (36.5% yield, 98.8 area % purity) of the title Compound 18 as a white solid. Anal. Calcd for C25H21N2OF3.0.04% H2O.0.15 mol MeOH: C, 70.48; H, 5.14: N, 6.42; F, 13.06 Found: C, 70.54; H, 4.83: N, 6.18; F, 13.33
Example 10.2 (E)-2-(2-{2-[2-(4-trifluoromethyl-phenyl)-vinyl]-1H-benzimidazol-5-yl}-phenyl)-propan-2-ol monosodium salt (Cpd 18)
-
A 5-L 4-neck flask equipped with a thermocouple controller, an overhead mechanical stirrer, and a nitrogen inlet/outlet was charged with (E)-2-(2-{2-[2-(4-trifluoromethyl-phenyl)-vinyl]-1H-benzimidazol-5-yl}-phenyl)-propan-2-ol. Compound 18 (125.0 g, 0.510 mol) and MeOH (1.25 L). A solution of sodium methoxide in methanol (0.5 M, 592 mL, 0.3 mol) was added. The reaction was heated to 65° C. for 30 min and all solids dissolved. The solution was cooled and evaporated to dryness. The foam was collected by scraping it out of the flask. The solids were placed in vacuum oven for 24 h at 40° C. to afford 139 g (about 100% isolated yield) of the title Compound 18 monosodium salt as a yellowish solid. 1H NMR (400 MHz, DMSO-d6) δ 7.80-7.84 (m, 3H), 7.74 (d, 2H, J=8.59 Hz), 7.65 (d, 1H, J=16.4 Hz), 7.40-7.44 (m, 2H), 7.25-7.37 (m, 2H), 7.16-7.20 (m, 1H), 7.01-7.05 (m, 1H), 6.84-6.87 (m, 1H), 1.23 (s, 6H). Mass Spectrum (LCMS, APCI pos.) Calcd. For C25H21F3N2O: 423.2 (M+H). Found 423.3. m.p. (uncorr.) 258-259° C.
Example 10.3 (E)-2-(2-{2-[2-(4-trifluoromethyl-phenyl)-vinyl]-1H-benzimidazol-5-yl}-phenyl)-propan-2-ol hydrochloride salt (Cpd 18)
-
A 250-mL separatory funnel was charged with (E)-2-(2-{2-[2-(4-trifluoromethyl-phenyl)-vinyl]-1H-benzimidazol-5-yl}-phenyl)-propan-2-ol. Compound 18 (1.0 g, 2.4 mmol) and EtOAc (20 mL). Aqueous HCl (1M, 20 mL) was added to the white slurry, and the separatory funnel was shaken. The solid product quickly dissolved, and a white precipitate started to form. The organic layer was transferred to a 100 mL round bottom flask equipped with a magnetic stir bar, and was stirred for 2 h. The thick slurry was filtered, rinsed with EtOAc (2×5 mL), and put into a vacuum oven at 40° C. for 36 h to afford 0.95 g (87.5%) of the title Compound 18 hydrochloride salt.
////////////Phase I, Musculoskeletal pain, Pain, Mavatrep, JNJ 39439335,
Sanofi Pasteur has received a positive opinion from the European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) recommending market approval for Sanofi Pasteur’s 6-in-1 pediatric vaccine Hexyon/Hexacima (DTaP-IPV-Hib-HepB vaccine).
FEB22,2013
French drug major Sanofi’s vaccines subsidiary Sanofi Pasteur has received a positive opinion from the European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) recommending market approval for Sanofi Pasteur’s 6-in-1 pediatric vaccine Hexyon/Hexacima (DTaP-IPV-Hib-HepB vaccine).
Hexyon/Hexacima is the only fully liquid, ready-to-use, 6-in-1 vaccine to protect infants against diphtheria, tetanus, pertussis (whooping cough), Hepatitis B, poliomyelitis and invasive infections caused by Haemophilus influenzae type b.
The new vaccine will be commercialized under the brand name Hexyon in Western European countries by Sanofi Pasteur MSD, the joint venture between US pharma giant Merck & Co and Sanofi Pasteur, and under the brand name Hexacima in Eastern European countries by Sanofi Pasteur.
“Availability of Hexyon/Hexacima ready-to-use, 6-in-1 pediatric vaccine will raise the standard of care of vaccination for millions of children. It reduces the number of vaccination visits for infants and it is more convenient for parents to complete the recommended vaccination schedule and thus better protect their children against six major childhood diseases,” said Olivier Charmeil, president and chief executive of Sanofi Pasteur, adding: “Upon licensure, we intend to introduce Hexyon/Hexacima vaccine in countries that are looking for improved and effective solutions for public immunization programs.”
Key benefits of Hexyon/Hexacima vaccine
According to Sanofi, the key benefits of Hexyon/Hexacima include the following:
• Hexyon/Hexacima is a fully liquid, ready-to-use vaccine; no reconstitution is needed prior to administration, which improves convenience for health care professionals. It is available in vial and pre-filled syringe presentations;
• by combining six vaccines into one, the vaccine reduces the number of injections, which improves comfort and vaccination compliance for infants, and
• the use of acP (acellular pertussis) antigens and IPV (inactivated poliovirus vaccine) improves safety and reduces reactogenicity as compared to wcP (whole cell pertussis)-containing vaccines and OPV (oral polio vaccine).
Assuming licensure, Hexyon/Hexacima would be indicated for primary and booster vaccination of infants from six weeks of age in accordance with official recommendations. The CHMP positive opinion is supported by results of multi-center clinical studies involving around 5,000 infants. Phase III clinical studies comparing Hexyon/Hexacima to licensed combination vaccines demonstrated that the vaccine is safe and induces a robust immune response against all six targeted diseases.
Medical Imaging Drugs Advisory Committee Recommends Approval of Guerbet NDA for Dotarem (gadoterate meglumine)

Gadoterate meglumine STR- CREDIT PUBCHEM
Also known as: Magnescope, Magnescope (TN), AC1OCEY3, Meglumine gadoterate (JAN), EK-5504, D03355
Molecular Formula: C23H42GdN5O13
Molecular Weight: 753.85528
| Cas No. | 98059-18-8 |
| Name | 2-[4,7-bis(carboxylatomethyl)-10-(carboxymethyl)-1,4,7, 10-tetrazacyclododec-1-yl]acetate; gadolinium(3+); (2R,3R,4R,5S)-6-(methylamino)hexane-1,2,3,4,5-pentol |
MORE ABOUT STRUCTURE , CODE CAS NO, ETC- http://www.ama-assn.org/resources/doc/usan/gadoterate-meglumine.pdf
February 15, 2013 NDA FDA
Dotarem (gadoterate meglumine)
Company: Guerbet
Treatment for: Diagnostic
Dotarem (gadoterate meglumine) is a gadolinium-based contrast agent under review for use in magnetic resonance imaging (MRI).

VILLEPINTE, France, Feb. 14, 2013 Guerbet, the contrast agent specialist for medical imaging, today announced that the Medical Imaging Drugs Advisory Committee to US Food and Drug Administration (FDA) has voted unanimously by votes of 17 to 0 to recommend that FDA approve the New Drug Application (NDA) for Dotarem (gadoterate meglumine) for adults, and for pediatric use for children two years of age and older. The Committee voted 10 to 6 (with one member abstaining) not to recommend at this time approval of the indication for children under two years of age.
Dotarem is the only macrocyclic and ionic gadolinium-based contrast agent (GBCA) for the intravenous use with magnetic resonance imaging (MRI) in the brain (intracranial), spine and associated tissues in adults and pediatric patients to detect and visualize areas with disruption of the blood-brain barrier (BBB) and/or abnormal vascularity. The Guerbet NDA recommended dose is 0.1 mmol Gd/kg.

Gadoteric acid
Gadoteric acid (trade names Artirem, Dotarem) is a macrocycle-structured gadolinium-based MRI contrast agent. It consists of the organic acid DOTA as a chelating agent, and gadolinium (Gd3+), and is used in form of the meglumine salt.[1] The drug is approved and used in a number of countries worldwide.[2]
References
- Herborn, C. U.; Honold, E.; Wolf, M.; Kemper, J.; Kinner, S.; Adam, G.; Barkhausen, J. (2007). “Clinical Safety and Diagnostic Value of the Gadolinium Chelate Gadoterate Meglumine (Gd-DOTA)”. Investigative Radiology 42 (1): 58–62. doi:10.1097/01.rli.0000248893.01067.e5. PMID 17213750. edit
- Drugs.com: Gadoteric Acid

A gadolinium chelate paramagnetic contrast agent. When placed in a magnetic field, gadoterate meglumine produces a large magnetic moment and so a large local magnetic field, which can enhance the relaxation rate of nearby protons; as a result, the signal intensity of tissue images observed with magnetic resonance imaging (MRI) may be enhanced. Because this agent is preferentially taken up by normal functioning hepatocytes, normal hepatic tissue is enhanced with MRI while tumor tissue is unenhanced. In addition, because gadobenate dimeglumine is excreted in the bile, it may be used to visualize the biliary system using MRI.
FDA has approved a new use of Avastin® (bevacizumab) in combination with fluoropyrimidine-based irinotecan or oxaliplatin chemotherapy for people with metastatic colorectal cancer (mCRC).
Bevacizumab, CAS NO 216974-75-3
A MONOCLONAL ANTIBODY
January 23, 2013
Avastin (bevacizumab) is a recombinant humanized monoclonal IgG1 antibody that binds to and inhibits the biologic activity of human vascular endothelial growth factor (VEGF) in in vitro and in vivo assay systems. Bevacizumab contains human framework regions and the complementarity-determining regions of a murine antibody that binds to VEGF. Avastin has an approximate molecular weight of 149 kD. Bevacizumab is produced in a mammalian cell (Chinese Hamster Ovary) expression system in a nutrient medium containing the antibiotic gentamicin. Gentamicin is not detectable in the final product.
FDA has approved a new use of Avastin® (bevacizumab) in combination with fluoropyrimidine-based irinotecan or oxaliplatin chemotherapy for people with metastatic colorectal cancer (mCRC).
On January 23, 2013, the FDA has approved a new use of Avastin® (bevacizumab) in combination with fluoropyrimidine-based irinotecan or oxaliplatin chemotherapy for people with metastatic colorectal cancer (mCRC). The new indication will allow people who received Avastin plus an irinotecan or oxaliplatin containing chemotherapy as an initial treatment (first-line) for mCRC to continue to receive Avastin plus a different irinotecan or oxaliplatin containing chemotherapy after their cancer worsens (second-line treatment).
People who start on Avastin for mCRC can now stay on Avastin after their cancer worsens.
Bevacizumab (trade name Avastin, Genentech/Roche) is an angiogenesis inhibitor, a drug that slows the growth of new blood vessels. It is licensed to treat various cancers, including colorectal, lung, breast (outside the USA), glioblastoma (USA only), kidney and ovarian.
Bevacizumab is a humanized monoclonal antibody that inhibits vascular endothelial growth factor A (VEGF-A).[1] VEGF-A is a chemical signal that stimulates angiogenesis in a variety of diseases, especially in cancer. Bevacizumab was the first clinically available angiogenesis inhibitor in the United States.[citation needed]
Bevacizumab was approved by the U.S. Food and Drug Administration (FDA) for certain metastatic cancers. It received its first approval in 2004, for combination use with standard chemotherapy for metastatic colon cancer.[2] It has since been approved for use in certain lung cancers, renal cancers, and glioblastoma multiforme of the brain.
At one point bevacizumab was approved for breast cancer by the FDA, but the approval was revoked on 18 November 2011.[3][4]
- Los, M.; Roodhart, J. M. L.; Voest, E. E. (2007). “Target Practice: Lessons from Phase III Trials with Bevacizumab and Vatalanib in the Treatment of Advanced Colorectal Cancer”. The Oncologist 12 (4): 443–50. doi:10.1634/theoncologist.12-4-443. PMID 17470687.
- http://www.gene.com/gene/products/information/pdf/avastin-prescribing.pdf
- Pollack, Andrew (18 November 2011). “F.D.A. Revokes Approval of Avastin for Breast Cancer”. New York Times.
- “Cancer drug Avastin loses US approval”. BBC. November 18, 2011.
SEQUENCE
>1bj1_H|Fab-12, F(ab)-12, 12-IgG1, rhuMAb-VEGF|||VH-CH1 (VH(1-123)+CH1(124-215))|||||||231||||MW 24867.8|MW 24867.8| EVQLVESGGGLVQPGGSLRLSCAASGYTFTNYGMNWVRQAPGKGLEWVGWINTYTGEPTY AADFKRRFTFSLDTSKSTAYLQMNSLRAEDTAVYYCAKYPHYYGSSHWYFDVWGQGTLVT VSSASTKGPSVFPLAPSSKSTSGGTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVL QSSGLYSLSSVVTVPSSSLGTQTYICNVNHKPSNTKVDKKVEPKSCDKTHT >1bj1_L|Fab-12, F(ab)-12, 12-IgG1, rhuMAb-VEGF|||L-KAPPA (V-KAPPA(1-107)+C-KAPPA(108-213))|||||||214||||MW 23451.0|MW 23451.0| DIQMTQSPSSLSASVGDRVTITCSASQDISNYLNWYQQKPGKAPKVLIYFTSSLHSGVPS RFSGSGSGTDFTLTISSLQPEDFATYYCQQYSTVPWTFGQGTKVEIKRTVAAPSVFIFPP SDEQLKSGTASVVCLLNNFYPREAKVQWKVDNALQSGNSQESVTEQDSKDSTYSLSSTLT LSKADYEKHKVYACEVTHQGLSSPVTKSFNRGEC >1bj1_J|Fab-12, F(ab)-12, 12-IgG1, rhuMAb-VEGF|||L-KAPPA (V-KAPPA(1-107)+C-KAPPA(108-213))|||||||214||||MW 23451.0|MW 23451.0| DIQMTQSPSSLSASVGDRVTITCSASQDISNYLNWYQQKPGKAPKVLIYFTSSLHSGVPS RFSGSGSGTDFTLTISSLQPEDFATYYCQQYSTVPWTFGQGTKVEIKRTVAAPSVFIFPP SDEQLKSGTASVVCLLNNFYPREAKVQWKVDNALQSGNSQESVTEQDSKDSTYSLSSTLT LSKADYEKHKVYACEVTHQGLSSPVTKSFNRGEC >1bj1_K|Fab-12, F(ab)-12, 12-IgG1, rhuMAb-VEGF|||VH-CH1 (VH(1-123)+CH1(124-215))|||||||231||||MW 24867.8|MW 24867.8| EVQLVESGGGLVQPGGSLRLSCAASGYTFTNYGMNWVRQAPGKGLEWVGWINTYTGEPTY AADFKRRFTFSLDTSKSTAYLQMNSLRAEDTAVYYCAKYPHYYGSSHWYFDVWGQGTLVT VSSASTKGPSVFPLAPSSKSTSGGTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVL QSSGLYSLSSVVTVPSSSLGTQTYICNVNHKPSNTKVDKKVEPKSCDKTHT
FDA approves Kadcyla (ado-trastuzumab emtansine), a new therapy for patients with HER2-positive, late-stage (metastatic) breast cancer.


Structure of trastuzumab emtansine. An ADC is a three-block “engine” — antibody-linker-drug — and each part of the composite molecule has to be carefully selected and assembled. Considered as an armed-antibody, an ADC is a bi-dentate construction where both parts (antibody and drug) of the molecule combine their effect to ensure selectivity and potency. The role of the linker arm is of paramount importance demanding a fine tuning to execute the controlled release and delivery of the two active components in the tumor environment.
Feb. 22, 2013
FDA approves new treatment for late-stage breast cancer
The U.S. Food and Drug Administration today approved Kadcyla (ado-trastuzumab emtansine), a new therapy for patients with HER2-positive, late-stage (metastatic) breast cancer.
HER2 is a protein involved in normal cell growth. It is found in increased amounts on some types of cancer cells (HER2-positive), including some breast cancers. In these HER2-positive breast cancers, the increased amount of the HER2 protein contributes to cancer cell growth and survival.
Kadcyla is intended for patients who were previously treated with trastuzumab, another anti-HER2 therapy, and taxanes, a class of chemotherapy drugs commonly used for the treatment of breast cancer.
“Kadcyla is trastuzumab connected to a drug called DM1 that interferes with cancer cell growth,” said Richard Pazdur, M.D., director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “Kadcyla delivers the drug to the cancer site to shrink the tumor, slow disease progression and prolong survival. It is the fourth approved drug that targets the HER2 protein.”

Referred to as T-DM1 during clinical research, Kadcyla was reviewed under the FDA’s priority review program, which provides for an expedited six-month review of drugs that may provide safe and effective therapy when no satisfactory alternative therapy exists, or offer significant improvement compared to marketed products. Other FDA-approved drugs used to treat HER2-positive breast cancer include trastuzumab (1998), lapatinib (2007) and pertuzumab (2012).
Kadcyla, trastuzumab and pertuzumab are marketed by South San Francisco, Calif.-based Genentech, a member of the Roche Group. Lapatinib is marketed by GlaxoSmithKline, based in Research Triangle Park, N.C
ImmunoGen, Inc. a biotechnology company that develops anticancer therapeutics using its TAP technology, today announced that Roche has reported that the U.S. Food and Drug Administration (FDA) has granted marketing approval to Kadcyla for the treatment of people with HER2-positive metastatic breast cancer who have received prior treatment with Herceptin® (trastuzumab) and a taxane chemotherapy.
“This is a big day for the patients with this cancer and for ImmunoGen,” commented Daniel Junius, President and CEO. “In clinical testing, the findings with Kadcyla in this patient population have been impressive, and we’re delighted the product can now be used by practicing oncologists across the US. In addition to its importance from a medical perspective, commercialization of Kadcyla also marks the start of ImmunoGen earning royalty income.”

Mr. Junius continued, “The efficacy and tolerability seen with Kadcyla underscores the transformative potential of our technology. Kadcyla is the most advanced of ten compounds with our TAP technology already in the clinic, with more in earlier stages of development. We are hopeful that in the future many different types of cancers will be routinely treated with TAP compounds.”
Genentech licensed from ImmunoGen exclusive rights to use the Company’s maytansinoid TAP technology to develop anticancer products targeting HER2.
 as a new therapy for patients with HER2-positive, late-stage (metastatic) breast cancer")
According to Genentech, Kadcyla will cost $9,800 per month, compared to $4,500 per month for regular Herceptin. The company estimates a full course of Kadcyla, about nine months of medicine, will cost $94,000. Thus, the cost of the drug is beyond the reach of many women unless they have an insurance plan.
”””””’
Afatinib
-
- Synonyms:BIBW 2992
- ATC:L01XE13
- Use:anticancer; tyrosine kinase inhibitor
- Chemical name:N-[4-[(3-chloro-4-fluorophenyl)amino]-7-[[(3S)-tetrahydro-3-furanyl]oxy]-6-quinazolinyl]-4-(dimethylamino)-2-butenamide; N-[(3-chloro-4-fluorophenyl)amino]-6-{[4-(N,N-dimethylamino)-1-oxo-2-buten-1-yl]amino}-7-((S)-tetrahydrofuran-3-yloxy)-quinazoline
- Formula:C24H25ClFN5O3
- MW:485.9 g/mol
- CAS-RN:439081-18-2; 850140-72-6
Derivatives
dimaleate
- Formula:C32H33ClFN5O11
- MW:718.1 g/mol
- CAS-RN:850140-73-7
Substance Classes
Synthesis Path
Substances Referenced in Synthesis Path
| CAS-RN | Formula | Chemical Name | CAS Index Name |
|---|---|---|---|
| 446-32-2 | C7H6FNO2 | 4-fluoro-anthranilic acid | |
| 162012-70-6 | C8H3ClFN3O2 | 4-chloro-7-fluoro-6-nitroquinazoline | |
| 367-21-5 | C6H5ClFN | 3-chloro-4-fluoroaniline | |
| 86087-23-2 | C4H8O2 | (S)-(+)-3-hydroxytetrahydrofuran | |
| 314771-76-1 | C18H16ClFN4O2 | N-(3-chloro-4-fluorophenyl)-7-((tetrahydrofuran-3-yl)oxy)quinazoline-4,6-diamine | |
| 13991-36-1 | C4H5BrO2 | bromocrotonic acid | |
| 3095-95-2 | C6H13O5P | diethylphophonoacetic acid | |
| 618061-76-0 | C24H27ClFN4O6P | Diethyl-{[4-((3-chloro-4-fluorophenyl)amino)-7-(((S)-tetrahydro- furan-3-yloxy)quinazolin-6-yl)carbamoyl]-methyl}phosphonate |
|
| 3616-56-6 | C8H19NO2 | (dimethylamino)-acetaldehyde diethylacetate |
Trade Names
| Country | Trade Name | Vendor | Annotation |
|---|---|---|---|
| USA | Gilotrif | Boehringer Ingelheim, 2013 | |
| EU | Giotrif | Boehringer Ingelheim, 2013 |
Formulations
- tabs.; 20, 30 and 40 mg
References
-
- a US 6 251 912 (American Cyanamid; 26.6.2001; appl. 29.7.1998; USA-prior. 1.8.1997).
- WO 0 250 043 (Boehringer Ingelheim; 27.6.2002; appl. 12.12.2001; DE-prior. 20.12.2000).
- US RE 43431 (Boehringer Ingelheim; 29.5.2012; appl. 18.8.2009; DE-prior. 20.12.2000).
- b US 8 426 586 (Boehringer Ingelheim; 1.2.2007; appl. 14.7.2006; DE-prior. 17.10.2003).
-
crystalline forms of Afatinib di-maleate:
- Solca, F. et al., J. Pharmacol. Exp. Ther., (2012) 343(2), 342-350.
- WO 2013 052157 (Ratiopharm/Teva; 11.4.2013; appl. 25.4.2012; USA-prior. 6.10.2011).
Natrol, Inc. Revitalizes Hair Technology with NuHair® FOAM, a Natural Solution for Male and Female Hair Rejuvenation
February 21, 2013, Natrol, Inc., a global leader in the nutrition industry and trusted manufacturer and marketer of superior quality supplements, has raised the bar for hair technology and created an alternate and innovative method to enhance hair rejuvenation among men and women with NuHair® FOAM. The product is hitting the marketplace following Natrol’s tablet hair-rejuvenation product being named the #1 selling supplement for hair growth.
NuHair® FOAM, now available at Walgreens nationwide, joins the NuHair line of dietary supplements that includes: Hair Regrowth Tablets designed for both men and women, Thinning Hair Serum, and DHT Blocker. The product was specifically created to protect against follicle damage, graying hair, and bring nourishment to the scalp and revitalize each strand to promote fuller, beautiful hair. It is also a 2-in-1 product, which has a light hold for styling.
“Natrol believes that hair rejuvenation begins at the root, and we’re thrilled to roll out NuHair® FOAM as the perfect product to stimulate that area,” said Stacy Dill, Natrol’s Senior Marketing Manager. “It works naturally, and with its robust styling ingredients as a bonus, provides a better alternative to what is already out there, without any extensive warning labels or side effects. NuHair® FOAM is simple…and can easily be a part of one’s daily regimen.”
NuHair® FOAM is formulated to work naturally with a natural blend of vitamins, herbs and extracts:
- Chamomile and Sage: Revitalizes the scalp, strengthens the texture of the hair, and promotes elasticity.
- Fo-Ti: Supports hair growth and may help prevent thinning and graying hair.
- Vitamin E: Stabilizes cell membranes in hair follicles to encourage proper growth.
- Vitamin B5: Penetrates the hair cuticle to retain moisture, leaving strands pliable, shinier and thicker.
- Shea Butter: Soothes dryness from root to tip, repairs breakage and mends split ends.
- Rosemary: Stimulates and improves hair group and may help darken gray hair.
- Nettle: Revitalizes and repairs brittle and damaged hair.
- Grape Seed Extract: Enhances hair growth and provides a rich, silky luster.
NuHair® belongs to Natrol, Inc.’s family of brands: Natrol, MRI, PROLAB, Promensil, Trinovin, Laci Le Beau, Shen Min, and Vedic Mantra.
NuHair® products are also available on Amazon.com, www.bodybuilders.com and other online retailers.
GSK/Isis rare disease drug moves into Phase II/III

feb20,2013
GlaxoSmithKline is paying out $7.5 million to partner Isis Pharmaceuticals as an antisense drug being developed for transthyretin amyloidosis, “a severe and rare genetic disease,” goes into a Phase II/III study.
TTR amyloidosis is characterised by progressive dysfunction of peripheral nerve and/or heart tissues and affects 50,000 patients worldwide and current treatments are limited. The 15-month study of the drug, known as ISIS-TTRRx, will involve some 200 patients with familial amyloid polyneuropathy who experience TTR build-up in their peripheral nerves and experience the loss of motor functions, such as walking.
Lynne Parshall, chief operating officer at Isis, said that “the rapid development of ISIS-TTRRx from a research-stage programme to a drug in late-stage clinical development in just over two years represents the strong commitment of both teams”. She added that the encouraging data from a Phase I study, “in which our drug was well tolerated and produced significant reductions in TTR protein, supported the advancement of ISIS-TTRRx directly into this registration-directed Phase II/III study”.
