
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
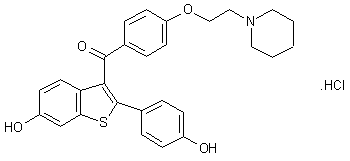
Keoxifene hydrochloride, Raloxifene hydrochloride, LY-139481(free base), LY-156758, Optruma, Loxifen, EvistaTitle: RaloxifeneCAS Registry Number: 84449-90-1CAS Name: [6-Hydroxy-2-(4-hydroxyphenyl)benzo[b]thien-3-yl][4-[2-(1-piperidinyl)ethoxy]phenyl]methanoneAdditional Names: keoxifeneManufacturers’ Codes: LY-139481Molecular Formula: C28H27NO4SMolecular Weight: 473.58Percent Composition: C 71.01%, H 5.75%, N 2.96%, O 13.51%, S 6.77%Literature References: Nonsteroidal, selective estrogen receptor modulator (SERM). Prepn: C. D. Jones, EP62503; idem,US4418068 (1982, 1983 both to Lilly); idemet al.,J. Med. Chem.27, 1057 (1984). Review of pharmacology and toxicology: J. Buelke-Sam et al.,Reprod. Toxicol.12, 217-221 (1998); of clinical pharmacology and pharmacokinetics: D. Hochner-Celnikier, Eur. J. Obstet. Gynecol. Reprod. Biol.85, 23-29 (1999); of clinical efficacy in osteoporosis: D. Agnusdei, ibid. 43-46. Clinical effect on risk of breast cancer: S. R. Cummings et al.,J. Am. Med. Assoc.281, 2189 (1999); on reduction of fracture risk: B. Ettinger et al.,ibid.282, 637 (1999).Properties: Crystals from acetone, mp 143-147°. uv max (ethanol): 290 nm (e 34000).Melting point: mp 143-147°Absorption maximum: uv max (ethanol): 290 nm (e 34000)
Derivative Type: HydrochlorideCAS Registry Number: 82640-04-8Manufacturers’ Codes: LY-156758Trademarks: Evista (Lilly)Molecular Formula: C28H27NO4S.HClMolecular Weight: 510.04Percent Composition: C 65.94%, H 5.53%, N 2.75%, O 12.55%, S 6.29%, Cl 6.95%Properties: Crystals from methanol/water, mp 258°. uv max (ethanol): 286 nm (e 32800).Melting point: mp 258°Absorption maximum: uv max (ethanol): 286 nm (e 32800)
Therap-Cat: Antiosteoporotic.Keywords: Antiosteoporotic; Selective Estrogen Receptor Modulator (SERM).
Raloxifene, sold under the brand name Evista among others, is a medication used to prevent and treat osteoporosis in postmenopausal women and those on glucocorticoids.[4] For osteoporosis it is less preferred than bisphosphonates.[4] It is also used to reduce the risk of breast cancer in those at high risk.[4] It is taken by mouth.[4]
Common side effects include hot flashes, leg cramps, swelling, and joint pain.[4] Severe side effects may include blood clots and stroke.[4] Use during pregnancy may harm the baby.[4] The medication may worsen menstrual symptoms.[5] Raloxifene is a selective estrogen receptor modulator (SERM) and therefore a mixed agonist–antagonist of the estrogen receptor (ER).[4] It has estrogenic effects in bone and antiestrogenic effects in the breasts and uterus.[4]
Raloxifene was approved for medical use in the United States in 1997.[4] It is available as a generic medication.[4][6] A month supply in the United Kingdom costs the NHS about 3.50 £ as of 2019.[6] In the United States the wholesale cost of this amount is about $16.[7] In 2017, it was the 330th most commonly prescribed medication in the United States, with more than 900 thousand prescriptions.[8
Medical uses
Raloxifene is used for the treatment and prevention of osteoporosis in postmenopausal women.[9] It is used at a dosage of 60 mg/day for both the prevention and treatment of osteoporosis.[10] In the case of either osteoporosis prevention or treatment, supplemental calcium and vitamin D should be added to the diet if daily intake is inadequate.[11]
Raloxifene is used to reduce the risk of breast cancer in postmenopausal women. It is used at a dosage of 60 mg/day for this indication.[10] In the Multiple Outcomes of Raloxifene (MORE) clinical trial, raloxifene decreased the risk of all types of breast cancer by 62%, of invasive breast cancer by 72%, and of invasive estrogen receptor-positive breast cancer by 84%.[12] Conversely, it does not reduce the risk of estrogen receptor-negative breast cancer.[12] There were no obvious differences in effectiveness of raloxifene in the MORE trial for prevention of breast cancer at a dosage of 60 mg/m2/day relative to 120 mg/m2/day.[12] In the Study of Tamoxifen and Raloxifene (STAR) trial, 60 mg/day raloxifene was 78% as effective as 20 mg/day tamoxifen in preventing non-invasive breast cancer.[13] Women with undetectable levels of estradiol (<2.7 pg/mL) have a naturally low risk of breast cancer and, in contrast to women with detectable levels of estradiol, do not experience significant benefit from raloxifene in terms of reduction of breast cancer risk.[12]
Contraindications
Raloxifene is contraindicated in lactating women or women who are or who may become pregnant.[14] It also may be of concern to women with active or past history of venous thromboembolic events, including deep vein thrombosis, pulmonary embolism, and retinal vein thrombosis.[15]
Side effects
Common side effects of raloxifene include hot flashes (25–28% vs. 18–21% for placebo),[12] vaginal dryness, and leg cramps (generally mild; 5.5% vs. 1.9% for placebo).[14][1][16] Raloxifene does not cause breast tenderness, endometrial hyperplasia, menstrual bleeding, or endometrial cancer.[17] It does not appear to affect cognition or memory.[15][12] Raloxifene is a teratogen; i.e., it can cause developmental abnormalities such as birth defects.
Raloxifene may infrequently cause serious blood clots to form in the legs, lungs, or eyes.[1] Other reactions experienced include leg swelling/pain, trouble breathing, chest pain, and vision changes. Black box warnings were added to the label of raloxifene in 2007 warning of increased risk of death due to stroke for postmenopausal women with documented coronary heart disease or at increased risk for major coronary events, as well as increased risk for deep vein thrombosis and pulmonary embolism.[14] The risk of venous thromboembolism with raloxifene is increased by several-fold in postmenopausal women (RR = 3.1).[18][12] Raloxifene has a lower risk of thromboembolism than tamoxifen.[13] In the MORE trial, raloxifene caused a 40% decrease in risk of cardiovascular events in women who were at increased risk for coronary artery disease, although there was no decrease in cardiovascular events for the group as a whole.[12]
A report in September 2009 from Health and Human Services’ Agency for Healthcare Research and Quality suggests that tamoxifen and raloxifene, used to treat breast cancer, significantly reduce invasive breast cancer in midlife and older women, but also increase the risk of adverse side effects.[19]
A recent human case report in July 2016 suggests that raloxifene may in fact, at some point, also stimulate breast cancer growth leading to a reduction of advanced breast cancer disease upon the withdrawal of the drug.[20]
Unlike other SERMs, such as tamoxifen, raloxifene has no risk of uterine hyperplasia or endometrial cancer (RR = 0.8).[1][18][13]
Raloxifene does not increase the incidence of breast pain or tenderness in postmenopausal women.[16][21]
Overdose
Raloxifene has been studied in clinical trials across a dosage range of 30 to 600 mg/day, and was well-tolerated at all dosages.[16]
Pharmacology
Pharmacodynamics
Mechanism of action
Raloxifene is a selective estrogen receptor modulator (SERM) and hence is a mixed agonist and antagonist of the estrogen receptor (ER) in different tissues.[4] It has estrogenic activity in some tissues, such as bone and the liver, and antiestrogenic activity in other tissues, such as the breasts and uterus.[4] Its affinity (Kd) for the ERα is approximately 50 pM, which is similar to that of estradiol.[16] Relative to estradiol, raloxifene has been reported to possess about 8 to 34% of the affinity for the ERα and 0.5 to 76% of the affinity for the ERβ.[22][23] Raloxifene acts as a partial agonist of the ERα and as a pure antagonist of the ERβ.[24][25] In contrast to the classical ERs, raloxifene is an agonist of the G protein-coupled estrogen receptor (GPER) (EC50 = 10–100 nM), a membrane estrogen receptor.[26][27]
Clinical effects
Raloxifene has antiestrogenic effects in the mammary glands in preclinical studies.[16] In accordance, raloxifene reduces breast density in postmenopausal women, a known risk factor for breast cancer.[28] It does not stimulate the uterus in postmenopausal women, and results in no increase in risk of endometrial thickening, vaginal bleeding, endometrial hyperplasia, or endometrial cancer.[29][16][21] At the same time, raloxifene has minimal antiestrogenic effect in the uterus in premenopausal women.[29] This may possibly be due to inadequate tissue exposure of the uterus to raloxifene in these estrogen-rich individuals.[29]
In premenopausal women, raloxifene increases levels of follicle-stimulating hormone (FSH) and estradiol.[12] Conversely, in postmenopausal women, raloxifene has been found to reduce levels of the gonadotropins, luteinizing hormone (LH) and FSH, while not affecting levels of estradiol.[12][29] Raloxifene also decreases prolactin levels in postmenopausal women.[29] In men, raloxifene has been found to disinhibit the hypothalamic–pituitary–gonadal axis (HPG axis) and thereby increase total testosterone levels.[30][31][32][33] Due to the simultaneous increase in sex hormone-binding globulin (SHBG) levels however, free testosterone levels often remain unchanged in men during therapy with raloxifene.[30]
Raloxifene has estrogenic effects on liver protein synthesis.[12] It increases SHBG levels in both pre- and postmenopausal women as well as in men.[12][30] The medication decreases levels of total and low-density lipoprotein (LDL) cholesterol, C-reactive protein, apolipoprotein B, and homocysteine.[12][29] Conversely, it has little effect on levels of triglycerides and high-density lipoprotein (HDL).[12] Raloxifene has been shown to inhibit the oxidation of LDL cholesterol in vitro.[16] The medication has been found to decrease insulin-like growth factor 1 (IGF-1) levels in pre- and postmenopausal women as well as in men.[31] It has also been found to increase insulin-like growth factor binding protein 3 (IGFBP-3) levels in pre- and postmenopausal women.[12] Due to activation of estrogen receptors in the liver, raloxifene has procoagulatory effects, such as decreasing levels of fibrinogen and influencing levels of other coagulation factors.[12][29][16] For these reasons, raloxifene increases the risk of thrombosis.[12][29]
Raloxifene increases bone mineral density in postmenopausal women but decreases it in premenopausal women.[12] In the MORE trial, the risk of vertebral fractures was decreased by 30%, and bone mineral density was increased in the spine (by 2.1% at 60 mg, 2.4% at 120 mg) and femoral neck (2.6% at 60 mg, 2.7% at 120 mg).[18] It has been found to possess estrogenic effects in adipose tissue in postmenopausal women, promoting a shift from an android fat distribution to a gynoid fat distribution.[34][35] The medication has been found to increase levels of leptin, an adipokine.[12]
AbsorptionPharmacokinetics
The absorption of raloxifene is approximately 60%.[1][2] However, due to extensive first-pass metabolism, the absolute bioavailability of raloxifene is only 2.0%.[1][2] Raloxifene is rapidly absorbed from the intestines upon oral administration.[1] Peak plasma levels of raloxifene occur 0.5 to 6 hours after an oral dose.[1][2]
Distribution
Raloxifene is widely distributed throughout the body.[1] There is extensive distribution of raloxifene into the liver, serum, lungs, and kidneys.[1] The volume of distribution of raloxifene with a single 30 to 150 mg oral dose is approximately 2348 L.[1][36] Both raloxifene and its metabolites show high plasma protein binding (>95%), including to both albumin and α1 acid glycoprotein, but not to sex hormone-binding globulin.[1][2]
Metabolism
Raloxifene is metabolized in the liver and undergoes enterohepatic recycling.[2] It is metabolized exclusively by glucuronidation and is not metabolized by the cytochrome P450 system.[1][2] Less than 1% of radiolabeled material in plasma comprises unconjugated raloxifene.[2] The metabolites of raloxifene include several glucuronides.[1] The elimination half-life of raloxifene after a single dose is 27.7 hours (1.2 days), whereas its half-life at steady state at a dosage of 60 mg/day is 15.8 to 86.6 hours (0.7–3.6 days), with an average of 32.5 hours (1.4 days).[1][2] The extended half-life of raloxifene is attributed to enterohepatic recirculation and its high plasma protein binding.[1] Raloxifene and its glucuronide conjugates are interconverted by reversible metabolism and enterohepatic recycling, which prolongs the elimination half-life of raloxifene with oral administration.[2] The medication is deconjugated into its active form in a variety of tissues, including liver, lungs, spleen, bone, uterus, and kidneys.[1]
Elimination
Raloxifene is mainly excreted in bile and is eliminated in feces.[1][2] Less than 0.2% of a dose is excreted unchanged in urine and less than 6% of a dose is excreted in urine as glucuronide conjugates.[2]
Chemistry
See also: List of selective estrogen receptor modulators and Benzothiophene
Raloxifene hydrochloride has the empirical formula C28H27NO4S•HCl, which corresponds to a molecular weight of 510.05 g/mol. Raloxifene hydrochloride is an off-white to pale-yellow solid that is slightly soluble in water.[14]
Raloxifene is a benzothiophene derivative and is structurally distinct from the triphenylethylene SERMs like tamoxifen, clomifene, and toremifene.[37] It is the only benzothiophene SERM to have been marketed.[37] A benzothiophene SERM that was not marketed is arzoxifene (LY-353381).[38] Bazedoxifene (Duavee, Viviant) and pipendoxifene (ERA-923) are structurally related to raloxifene but are technically not benzothiophenes and instead are indoles.[38]
History
Raloxifene was approved in the United States for the prevention of postmenopausal osteoporosis in 1997, the treatment of postmenopausal osteoporosis in 1999, and to prevent or reduce the risk of breast cancer in certain postmenopausal women in 2007.[39][40][41][42] It received orphan designation in 2005.[39]
Society and culture

A bottle of raloxifene.
Names
Raloxifene is the generic name of the drug and its INN and BAN, while raloxifène is its DCF and raloxifene hydrochloride is its USAN, BANM, and JAN.[43][44][45][46] It has also been known by the name keoxifene.[43][44][46]
Raloxifene is sold mainly under the brand name Evista and to a lesser extent the brand name Optruma.[46][44] It is also sold under a variety of other brand names in various countries.[46]
Availability
Raloxifene is available widely throughout the world, including in the United States, Canada, the United Kingdom, Ireland, elsewhere throughout Europe, Australia, New Zealand, South Africa, Latin America, Southern, Eastern, and Southeastern Asia, and elsewhere in the world such as in Israel and Egypt.[46][44]
Raloxifene is provided in the form of 60 mg oral tablets.[10]
Controversy
An editorial in Lancet Oncology criticized the way that research about the medication for breast cancer prevention was released.[47]
Research
Clinical studies of raloxifene for metastatic breast cancer in women have been conducted but found little effectiveness at 60 mg/day in those previously treated with tamoxifen, though modest effectiveness has been observed at higher doses.[12][48] In contrast to tamoxifen, raloxifene is not approved for the treatment of breast cancer.[49]
Raloxifene has been studied in men for a variety of uses, such as for treatment of schizophrenia, prostate cancer, and osteoporosis.[50][51][52][53][54][33][32][55][56][57][58] It has been studied in combination with castration and bicalutamide, a nonsteroidal antiandrogen, for the treatment of prostate cancer.[58][55]
Raloxifene has been studied as an adjunct in the treatment of schizophrenia in postmenopausal women.[59] A 2017 meta-analysis concluded that it was safe and effective for this indication, although further studies with larger sample sizes are needed for confirmation.[59] It may be effective in women with less severe symptoms.[59]
A tissue-selective estrogen-receptor complex (TSEC) of estradiol and raloxifene has been studied in postmenopausal women.[60]
Raloxifene (60 mg/day) was reported to be effective in the treatment of pubertal gynecomastia in adolescent boys in a small retrospective chart review.[61][62][63] Other SERMs are also known to be effective in the treatment of gynecomastia.[64]
Raloxifene has been reported to augment the antidepressant effects of selective serotonin reuptake inhibitors (SSRIs).[65]
June 18th 2020, Exscalate4CoV, the private-public consortium supported by the EU’s Horizon 2020 programme for research and innovation, led by Dompé farmaceutici and currently representing 18 partners (including Fraunhofer Institute, CINECA, Chelonia Applied Science, Swiss Institute of Bioinformatics and others) has requested access to clinical trials for the use of Raloxifene in Covid 19 patients. Raloxifene, already proven effective against Mers and Sars in precliinical tests, has been indicated as effective against Sars-Cov2 by the “in-silico” research conducted by the consortium which has shown efficacy in countering the replication of the virus in cells. The IP for its use against Sars-Cov2 has already been protected on May 6 2020 in the name Dompé farmaceutici, Fraunhofer Institute and KU Leuven, to facilitate the largest possible access. Raloxifene would be used in mildly symptomatic Covid19 patients to halt the spread of infection. This result emerged from the first virtual (in silico) screening conducted on the Consortium’s supercomputers of more than 400.000 molecules (safe-in-man drugs and natural products) made available by Dompé farmaceutici and the partner Fraunhofer (IME) to the Consortium. The molecules were prioritized if in clinical stage or already on the market. 7.000 molecules with certain promising characteristics were tested.
SYN

Jones, Charles D.; Jevnikar, Mary G.; Pike, Andrew J.; Peters, Mary K.; Black, Larry J.; Thompson, Allen R.; Falcone, Julie F.; Clemens, James A. (1984). “Antiestrogens. 2. Structure-activity studies in a series of 3-aroyl-2-arylbenzo[b]thiophene derivatives leading to [6-hydroxy-2-(4-hydroxyphenyl)benzo[b]thien-3-yl]-[4-[2-(1-piperidinyl)ethoxy]phenyl]methanone hydrochloride (LY 156758), a remarkably effective estrogen antagonist with only minimal intrinsic estrogenicity”. Journal of Medicinal Chemistry 27 (8): 1057–66.doi:10.1021/jm00374a021. PMID 6431104.
syn 1
EP 0062053; GB 2097788
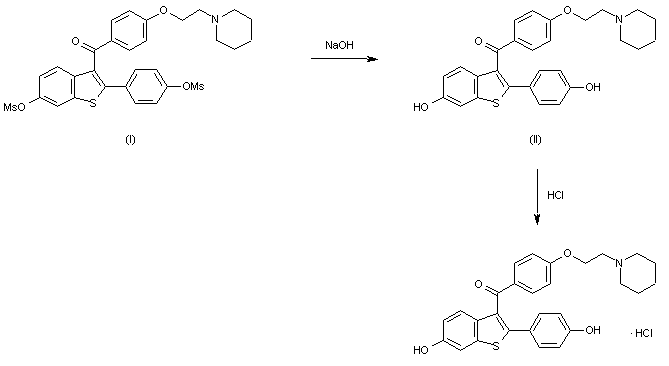
Keoxifene has been synthesized using the following process: A portion of 6-methanesulfonyloxy-2-(4-methanesulfonyloxyphenyl)-3-[4-(2-pipendinoethoxy)benzoyl]benzo[b]thiophene hydrochloride (I) was combined with denatured alcohol and 5N sodium hydroxide, and stirred under a nitrogen atmosphere. The reaction mixture was evaporated to dryness under vacuum, and the residue dissolved in water and washed with diethyl ether. The water layer was degassed under vacuum, and then nitrogen was bubbled through it to remove all traces of ether. The mixture was then acidified with 1N hydrochloric acid, and then made basic with excess sodium bicarbonate The precipitate was collected by filtration and washed with cold water to obtain crude product, which was purified on a column of silica gel. The column was eluted first with 700 ml of 5% methanol in chloroform, followed by 1l of 10% methanol in chloroform. The impurities came off first, and the product-containing fractions were combined and evaporated under vacuum to obtain a yellow oil. The oil was dissolved in acetone seeded and chilled in a freezer to obtain the purified product.
syn2
J Label Compd Radiopharm 1995,36(1),43

The synthesis of radiolabeled raloxifene has been reported: The esterification of 3,5-dibromo-4-hydroxybenzoic acid (I) with methanol/HCl gives the corresponding methyl ester (II), which is condensed with 1-(2-chloroethyl)piperidine (III) by means of K2CO3 in DMF yielding 3,5-dibromo-4-[2-(1-piperidyl)ethoxy]benzoic acid methyl ester (IV). The hydrolysis of (IV) with NaOH in methanol affords the corresponding free acid (V), which by treatment of SOCl2 in toluene is converted to the acyl chloride (VI). The Friedel-Crafts condensation of (VI) with 6-methoxy-2-(4-methoxyphenyl)benzo[b]thiophene (VII) by means of AlCl3 in dichloromethane gives [3,5-dibromo-4-[2-(1-piperidinyl)ethoxy]phenyl]-[6-methoxy-2-(4-methoxy phenyl)benzo[b]thien-3-yl]methanone (VIII), which is demethylated with AlCl3 and ethylmercaptane to dibromoraloxifene (IX). Finally, this compound is submitted to hydrogenolysis with tritium over Pd/C in methanol.
syn 3
Bioorg Med Chem Lett 1997,7(8),993
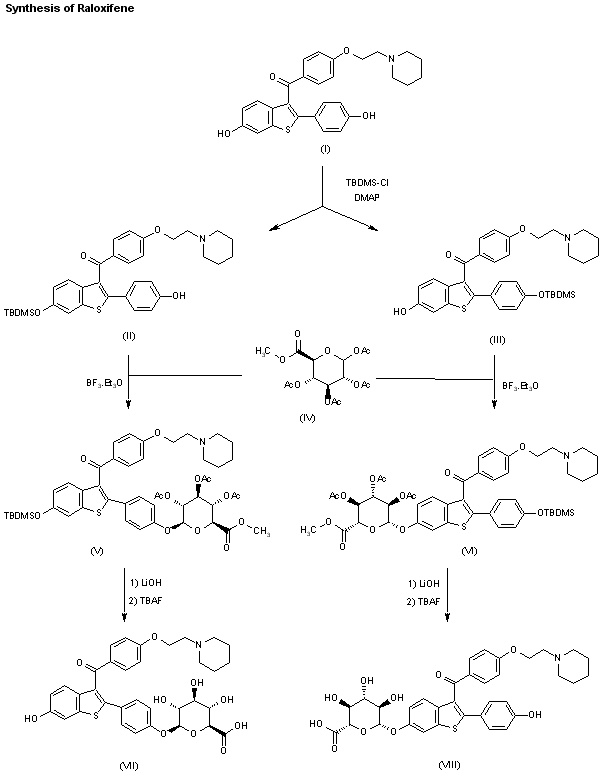
The two major metabolites of raloxifene, the glucuronide conjugates (VI) and (VIII) are synthesized as follows: The partial silylation of raloxifene (I) with tert-butyldimethylsilyl chloride (TBDMS-Cl) by means of dimethylaminopyridine (DMAP) in THF/DMF gives a mixture of the monosilylated compounds (II) and (III), which are separated by chromatography. Compounds (II) and (III) are independently condensed with methyl 1,2,3,4-tetra-O-acetyl-D-glucuronate (IV) by means of BF3.OEt2 in dichloromethane yielding protected glucuronides (V) and (VII), respectively. Finally, both compounds are deprotected by a treatment first with LiOH in dioxane to hydrolyzed the ester groups, and then with tetrabutylammonium fluoride in THF to eliminate the silyl groups, thus obtaining the desired metabolites (VI) and (VIII), respectively.
syn 4
Tetrahedron Lett 1999,40(28),5155
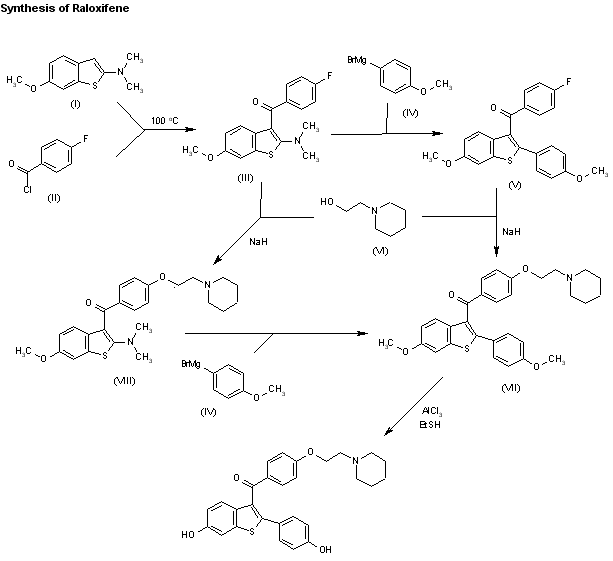
Two related new syntheses of raloxifene have been described: 1) The acylation of N-(6-methoxy-1-benzothiophen-2-yl)-N,N-dimethylamine (I) with 4-fluorobenzoyl chloride (II) by heating at 100 C in chlorobenzene gives the 3-acyl derivative (III), which is condensed with 4-methoxyphenylmagnesium bromide (IV) in THF yielding 3-(4-fluorobenzoyl)-6-methoxy-2-(4-methoxyphenyl)-1-benzothiophene (V). The condensation of (V) with 1-(2-hydroxyethyl)piperidine (VI) by means of NaH in DMF affords the ether (VII), which is finally demethylated with AlCl3 and ethanethiol. 2) The intermediate (III) can also be condensed first with 1-(2-hydroxyethyl)piperidine (VI) by means of NaH as before giving the piperidinoethyl ether (VIII), which is then condensed with the Grignard reagent (IV) affording the previously reported ether (VII).
syn
Org Chem Ind J, Volume: 14( 3)
A GREEN PROCESS FOR DEMETHYLATION REACTION IN SYNTHESIS OF RALOXIFENE HYDROCHLORIDEAuthors : Ramadas Chavakula *, Chakradhar Saladi J S, Narayana Rao Mutyalaa , Vijaya Raju Maddalaa and Raghu Babu Kb

A green process for demethylation reaction in synthesis of raloxifene hydrochloride by using aluminium chloride and odorless decanethiol as demethylation agent instead of aluminium chloride and ethanethiol (foul smell) under normal conditions is described.
Raloxifene hydrochloride [1], is an estrogen agonist/antagonist, commonly referred to as a Selective Estrogen Receptor Modulator (SERM) [1,2] that belongs to the benzothiophene class of compounds. Raloxifene decreases the resorption of bone and reduces the biochemical markers of bone turnover to the premenopausal range [3–5]. Raloxifene hydrochloride may also lower the chance of developing a certain type of breast cancer (invasive breast cancer) in post-menopausal women [6,7]. It can be synthesized [3] directly from aroylation of 6-methoxy-2-(4-methoxyphenyl)benzo[b]thiophene [2] by the acid chloride(4) of 4-[2-(1-piperidinyl)ethoxy]benzoic acid hydrochloride [3] in the presence of AlCl3 followed by addition of ethanethiol (FIG. 1).
Experimental Section
4-[2-(1-Piperidinyl)ethoxy]benzoic acid hydrochloride [3] and 6-methoxy-2-(4-methoxyphenyl) benzo[b] thiophene [2] were prepared by procedures reported previously [3]. Decanethiol was from commercial source. All melting points are uncorrected and were determined in capillary tubes on an Electothermal melting point apparatus. 1H NMR spectra were recorded on a Brucker ADVANCE 400 MHz spectrometer, using DMSO-d6 as solvent and TMS as internal standard. Electrospray ionization mass spectroscopy was performed using an ion trap mass spectrometer (Model 6310 Agilent). All reactions were monitored and checked by Thin Layer Chromatography (TLC) using methanol and spots examined by a UV lamp.
Preparation of [6-hydroxy-2-(4-hydroxyphenyl)benzo[b]thiophen-3-yl][4-[2-(1-piperidyl)ethoxy]phenyl] methanone hydrochloride (Raloxifene hydrochloride) [1]
To a solution of 4-[2-(1-piperidinyl)ethoxy]benzoic acid hydrochloride (3) (14.3 g, 0.05 mol) in methylene dichloride (400 mL) and pyridine (0.5 mL) at 25ºC to 35ºC, thionyl chloride (23.8 g, 0.20 mol) was added dropwise under argon for 15-30 minute. The reaction mixture was stirred for 2 hr. at 40ºC to 45ºC. Excess thionyl chloride and solvent were removed in vacuum at 40◦C to afford 15.0 g of the crude acid chloride hydrochloride salt [4]. The crude solid acid chloride hydrochloride [4] was dissolved in methylene dichloride (150 mL), cooled to 0ºC to 10ºC, 6-methoxy-2-(4-methoxyphenyl)benzo[b] thiophene [2] (10.8 g, 0.04 mol) was added. Then, anhydrous aluminium chloride (37.0 g, 0.28 mol) was added portion wise over a period of 30 min and then the mixture was allowed to warm to 30ºC and stirred for 2 hr at 25-35ºC. Then decanethiol (28.0 g, 0.16 mol) was added and stirred for 2 hr. at 25-35ºC. The reaction mixture was quenched with mixture of methanol (100 mL), ice (200 g) and Conc. HCl (15 mL) and stirred for 1 hr. at 25-35ºC. The precipitated solid was collected, washed with water (100 mL X 2) and dried at 65ºC for 4 h to afford 20.0 g of crude compound 1, which was crystallized from methanol/water (23/1, vol/vol) to yield 13.6 g of compound 1 (53.3 %yield) as a white solid, MP 258-260°C, liter 3, 258°C ; 1H NMR: δ 1.34, 1.72 [2H, m, (CH2CH2)2CH2], 1.76 [4H, m, N(CH2CH2)2], 2.96 (2H, m, N-CH2), 3.43 [4H, m, N(CH2CH2)2], 4.44 (2H, m, O-CH2), 6.67 (2H, d, Ar), 6.85 (1H, d, Ar), 6.95 (2H, d, Ar), 7.18 (2H, d, Ar), 7.25 (1H, d, Ar), 7.35 (1H, s, Ar), 7.70 (2H, d, Ar), 9.77 (1H, s, OH), 9.82 (1H, s, OH), 10.16 (1H, brs, NH), MS (ESI): m/z 474.6 (M +H). “This procedure has been scaled up using 250g of compound 1.”
Results and Discussion
Commonly used thiols like ethanethiol and benzyl mercaptan in demethylation reactions have a foul smell making them difficult and unpleasant to use in the laboratory without fume hoods. The problem becomes even worse in industry on a large scale. Odorless substitutes are therefore always required. Few papers [8,9] discuss the use of long chain thiols to minimize odor, so we used this work as a basis for choosing a long chain thiol for our demethylation reaction. We now report a new, highly active demethylation reagent, an aluminum chloride and decanethiol, characterized by rapid action under mild conditions, easy workup of the reaction product, and high yield (FIG. 2.).

Figure 2: Synthesis of Raloxifene hydrochloride.
Conclusion
In conclusion, we have found that decanethiol is odorless thiol compared to ethanethiol. We believe that removing the foul-smelling thiols and use of these odorless thiols will greatly improve the greenchemistry.
References
- Grese TA, Dodge JA. Selective Estrogen Receptor Modulators (SERMs). Curr Pharm Des. 1998;4:71-92.
- Bryant HU, Dere WH. Selective estrogen receptor modulators: an alternative to hormone replacement therapy. Proc Soc Exp Biol Med. 1998;217:45-52.
- Jones CD, Jevnikar MG, Pike AJ, et al. Antiestrogens. 2. Structure-activity studies in a series of 3-aroyl-2-arylbenzo [b] thiophene derivatives leading to [6-hydroxy-2-(4-hydroxyphenyl) benzo [b] thien-3-yl]-[4-[2-(1-piperidinyl) ethoxy] phenyl] methanone hydrochloride (LY 156758), a remarkably effective estrogen antagonist with only minimal intrinsic estrogenicity. J Med Chem. 1984;27:1057-66.
- Sato M, Grese TA, Dodge JA, et al. Emerging therapies for the prevention or treatment of postmenopausal osteoporosis. J Med Chem. 1999;42:1-24.
- Draper MW, Flowers DE, Huster WJ, et al. A controlled trial of raloxifene (LY139481) HCl: impact on bone turnover and serum lipid profile in healthy postmenopausal women. J Bone Miner Res. 1996;11:835-42.
paper
https://www.sciencedirect.com/science/article/abs/pii/S0223523412001122





syn
https://www.tandfonline.com/doi/abs/10.1080/00397911.2014.943348?journalCode=lsyc20
Piperidine Nucleophilic Substitution Without Solvent: An Efficient Synthesis of RaloxifeneYewei Yang,Tao Zhang,Wenhai Huang &Zhenrong Shen Pages 3271-3276 |
Mild and high-yielding synthesis is described for raloxifene via piperdine nucleophilic substitution of a new raloxifene intermediate 3-aroyl-2-aryl-substituted benzo[b]thiophenes, which is obtained by acylation of para-substituted benzoyl chlorides and 2-arylbenzo[b]thiophenes. The key step is solvent free and offers valuable advantages, such as low cost, and is suitable for industrial production.

Keywords: Friedel–Crafts acylation, green chemistry, nucleophiles, raloxifene, SERM

The improved synthesis of raloxifene 1 was accomplished as shown in Scheme 2. Methyl p-hydroxybenzoate 2, 1-bromo-2-chloroethane, and K2CO3 were refluxed in acetone, yielding compound 3 in 94% yield. Without prior purification, 3 was hydrolyzed to the corresponding p-substituted benzoyl acids 4 in 100% yield. The application of general reaction conditions of methanol as solvent and hydrochloric as acid would afford the substitution impurity 4-(2-methoxyethoxy)-benzoic acid. To control this impurity during reaction, various solvents such as alcohol, ethyl acetate, acetone, and tetrahydrofuran (THF) were screened, and THF gave the best result from the view of impurity formation and yield. Compound 4 is a solid and was easily isolated from THF by adding water. Then 4 was transferred to acid chlorides 5 and substantially reacted with benzothiophene 6 using AlCl3 in dichloromethane at 50 C to afford aroylated benzothiophene 7 in two steps, with yield of 95% (79% from method A[8] and 65.5% from method B[3]). With the requisite 7 in hand, we next examined piperidine nucleophilic substitution to produce the desired beno[b]thien-3-yl ketones 8. In general using reaction conditions A (acetone, NaI, K2CO3, reflux, 70%) and B (acetonitrile, NaI, K2CO3, reflux, 85%), impurity formation was observed from the beginning of the reaction. We screened various conditions and were delighted to found that using excess piperidine at reflux temperature gave negligible impurity formation. Piperidine was not only reagent but also solvent. The isolated product 8 was stable and was converted into the desired raloxifene 1 as reported. In conclusion, we have developed a viable alternative route for the synthesis of raloxifene. The new synthesis would have been better able to support the increase in bulk demand for this drug for the chemoprevention of breast cancer and novel formulations. Our synthetic route has several advantages: the use of difunctionalized coumpunds 5 as key intermediate makes Friedel–Crafts acylation and nucleophilic substitution highly efficient. The using of piperine as reagent and solvent avoids the large waste streams derived from neutralization reaction of sodium hydride. The cost of the new route is less than the current route of manufacture.
Preparation of [4-(2-Chloro-ethoxy)-phenyl]-[6-methoxy-2- (4-methoxy-phenyl)-benzo[b]thiophen-3-yl]-methanone (7) Under an N2 atmosphere, 5 was added to a mixture of 6 (20.25 g, 75 mmol) and AlCl3 (13.30 g, 100 mmol) in DCM (2 mL), and the mixture was stirred for 12 h. The reaction was monitored by TLC (n-hexane/EtOAc, 4:1). After the reaction was judged complete, the reaction mixture was allowed to cool. The crude mixture was poured into H2O and extracted with EtOAc. The organic layer was separated and concentrated. The residue was crystallized from EtOAc to give the product 7 (32.26 g, 95%): yellow solid crystals; mp 119–120 C; IR (KBr) nmax: 2960, 2835, 1647, 1599, 1472, 1251, 1169, 1032, 830 cm1 ; 1 H NMR (400 MHz, CDCl3) d 7.76 (d, J ¼ 8.8 Hz, 2H), 7.53 (d, J ¼ 8.8 Hz, 1H), 7.32 (d, J ¼ 8.4 Hz, 2H), 7.31 (s, 1H), 6.95 (dd, J ¼ 8.4, 2.4 Hz, 1H), 6.75 (dd, J ¼ 9.2, 7.2 Hz, 4H), 4.20 (t, J ¼ 4.0 Hz,2H), 3.87 (s, 3H), 3.78 (t, J ¼ 6.0 Hz, 2H), 3.74 (s, 3H); 13C NMR (100 MHz, CDCl3) d 193.1, 162.1, 159.7, 157.6, 142.7, 139.9, 133.8, 132.3, 130.9, 130.2, 130.1, 125.9, 123.9, 114.8, 114.1, 113.9, 104.4, 67.8, 55.6, 55.2, 41.5; MS (EI) m/z (%):452 (Mþ, 100.0), 437 (13.0), 297 (25.0), 183 (39.0), 121 (44.0). HRMS m/z (EI) calcd. for C25H22ClO4S: (MþH) þ: 453.0927; found: 453.0933.
Preparation of [6-Methoxy-2-(4-methoxy-phenyl)-benzo[b] thiophen-3-yl]-[4-(2-piperidin-1-yl-ethoxy)-phenyl]-methanone (8) Under an N2 atmosphere, a mixture of 7 (8.50 g, 19 mmol) and piperdine (30 ml) was stirred under reflux for 12 h. The reaction was monitored by TLC (n-hexane/EtOAc, 4:1). After the reaction was judged complete, the reaction mixture was allowed to cool. The mixture was concentrated for recovery of piperidine. EtOAc was added and the residue was washed with saturated NaHCO3 aqueous solution. The organic layer was separated and concentrated to give the product 8 (8.80 g, 94%): yellow viscous oil; IR (KBr) nmax: cm1 2933, 1645, 1597, 1535, 1501, 1470, 1249, 1164, 1030, 827; 1 H NMR (400 MHz, CDCl3) d7.76 (d, J ¼ 8.8 Hz, 2H), 7.52 (d, J ¼ 8.8 Hz, 1H), 7.33 (d, J ¼ 8.8 Hz, 2H), 7.30 (d, J ¼ 2.4 Hz, 1H), 6.94 (dd, J ¼ 8.8, 2.0 Hz, 1H), 6.75 (dd, J ¼ 7.2, 5.2 Hz, 4H), 4.08 (t, J ¼ 6.0 Hz, 2H), 3.86 (s, 3H), 3.73 (s, 3H), 2.71 (t, J ¼ 6.0 Hz, 2H), 2.46 (s, 4H), 1.60–1.54 (m, 4H), 1.43–1.41 (m, 2H).13C NMR (100 MHz, CDCl3) d 193.2, 163.0, 159.7, 157.6, 142.4, 140.1, 133.9, 132.3, 130.6, 130.4, 130.2, 126.0, 124.0, 114.8, 114.2, 114.1, 104.5, 66.3, 57.7, 55.6, 55.2, 55.1, 25.9, 24.1. MS (EI) m/z (%): 501 (Mþ, 100.0), 452 (12.0), 402 (21.0), 297 (24.0), 98 (100.0). HRMS m/z (EI) calcd. for C30H32NO4S: (MþH) þ: 502.2052; found: 502.2055.REFERENCES 1. Clemett, D.; Spencer, C. M. Drugs 2000, 60 (2), 379–411. 2. Land, S. R. JAMA 2007, 298 (9), 973–973. 3. Dadiboyena, S. Eur. J. Med. Chem. 2012, 51, 17–34. 4. Schmid, C. R.; Sluka, J. P.; Duke, K. M. Tetrahedron Lett. 1999, 40 (4), 675–678. 5. Bradley, D. A.; Godfrey, A. G.; Schmid, C. R. Tetrahedron Lett. 1999, 40 (28), 5155–5159. 6. Shinde, P. S.; Shinde, S. S.; Renge, A. S.; Patil, G. H.; Rode, A. B.; Pawar, R. R. Lett. Org. Chem. 2009, 6 (1), 8–10.7. Sach, N. W.; Richter, D. T.; Cripps, S.; Tran-Dube, M.; Zhu, H. C.; Huang, B. W.; Cui, J.; Sutton, S. C. Org. Lett. 2012, 14 (15), 3886–889. 8. Jones, C. D.; Jevnikar, M. G.; Pike, A. J.; Peters, M. K.; Black, L. J.; Thompson, A. R.; Falcone, J. F.; Clemens, J. A. J. Med. Chem. 1984, 27 (8), 1057–1066. 9. Grese, T. A.; Cho, S.; Finley, D. R.; Godfrey, A. G.; Jones, C. D.; Lugar, C. W.; Martin, M. J.; Matsumoto, K.; Pennington, L. D.; Winter, M. A.; Adrian, M. D.; Cole, H. W.; Magee, D. E.; Phillips, D. L.; Rowley, E. R.; Short, L. L.; Glasebrook, A. L.; Bryant, H. U. J. Med. Chem. 1997, 40 (2), 146–167.
synChapter 2 – 1-Substituted PiperidinesAuthor links open overlay panelRubenVardanyan
https://doi.org/10.1016/B978-0-12-805157-3.00002-8Piperidine-Based Drug DiscoveryHeterocyclic Drug Discovery2017, Pages 83-1011-Substituted Piperidines
Ruben Vardanyan, in Piperidine-Based Drug Discovery, 2017
Raloxifene (7685)
Raloxifene (Evista) (1.3.4) is a second-generation selective estrogen receptor modulator that functions as an estrogen antagonist on breast and uterine tissues, and an estrogen agonist on bone. Raloxifene is an antiresorptive agent, a new representative of a class of drugs that prevent the loss of bone mass, i.e., used to treat osteoporosis and similar diseases in postmenopausal women and those postmenopausal women at increased risk of invasive breast cancer [41–53].
It was shown that raloxifene can have some affect on cognition, mental health, sleep, and sexual function in menopausal women [54]. Raloxifene was used also as an adjuvant treatment in postmenopausal women with schizophrenia [55].
The first reported synthesis of the raloxifene scaffold consists in Friedel-Crafts aroylation in 1,2-dichloroethane and using AlCl3 as a catalyst by coupling of 4-(2-(piperidin-1-yl)ethoxy)benzoyl chloride (2.3.15) with benzothiophene derivative (2.3.16) followed by alkaline hydrolysis of mesyl groups, which give the desired raloxifene (2.3.4) [56–58] (Scheme 2.9).

The key intermediate – 6-methoxy-2-(4-methoxyphenyl)benzo[b]thiophene (2.3.16) – was prepared by the cyclization-rearrangement of 1-(4-methoxyphenyl)-2-((3-methoxyphenyl)thio)ethan-1-one (2.3.20) induced by polyphosphoric acid (PPA). This rearrangement (Kost rearrangement [59]) is general for 3-(R-substituted)indoles, -benzofurans, and -benzothiophenes, which are converted to the corresponding 2-isomers by heating with PPA.
The synthesis started from thiophenol (2.3.18) and bromoketone (2.3.19), which were coupled in presence of KOH in ethanol/water solution. Obtained (2.3.20) was heated with PPA to give a mixture that is easily separable by crystallization isomeric 2-phenylbenzo[b]thiophenes (2.3.21) and (2.3.22), where preferable, isomer (2.3.22) predominates. Cleavage of the methoxy groups in (2.3.22) was done conveniently with pyridine hydrochloride to give (2.3.23), which was easily converted to mesylate (2.3.16) with methanesulfonyl chloride in pyridine and 4-dimethylaminopyridine as a catalyst (Scheme 2.10).

The second reagent—4-(2-(piperidin-1-yl)ethoxy)benzoyl chloride (2.3.15)—was prepared starting with 4-hydroxybenzoate (2.3.24), which with 1-(2-chloroethyl)piperidine (2.3.25) in anhydrous DMF, and K2CO3 or sodium hydride, gave methyl 4-(2-(piperidin-1-yl)ethoxy)benzoate (2.3.26) hydrolyzed in MeOH/water NaOH solution. The acid (2.3.26) was converted to its chloride (2.3.15) with SOCl2 in 1,2-dichloroethane and a catalytic amount of DMF (Scheme 2.11).

Another novel convenient synthesis of raloxifene (2.3.4) have been proposed [60]. According to this method anisaldehyde (2.3.28) was transformed to corresponding cyanohydrin (2.3.29) using a mixture of sodium cyanide ethanol containing triethylamine through which HCl gas was passed over 30 minutes at 5–10°C.
Gaseous HCl was added to the solution of prepared cyanohydrin (2.3.29) in ethanol at room temperature over 30 minutes in order to give p-methoxybenzaldehyde cyanohydrin iminoether hydrochloride (2.3.30). Then, hydrogen sulfide was bubbled into a solution of the methyl imidate (2.3.30) and triethylamine in methanol at 0°C to give α-(4-methoxy phenyl)-α-hydroxy-N,N dimethylthioacetamide (2.3.31).
To the obtained α-hydroxythioamide (2.3.31) dissolved-in-methylene chloride methanesulfonic acid was slowly added, which transformed the starting material to 2-N,N-dimethylamino-6-methoxy benzo[β]thiophene (2.3.32).
The obtained 2-dimethylaminobenzothiophene (2.3.32) and known 4-(2-piperidinoethoxy)-benzoyl chloride (2.3.15) were partially dissolved in chlorobenzene and the mixture was warmed in a 100–105°C to give 2-(4-methoxyphenyl)-6-methoxy-3-[4-(piperidinoethoxy)benzoyl]-benzo[β]thiophene (2.3.33). 4-Methoxyphenylmagnesium bromide (2.3.34) in THF was added to chilled to 0°C prepared compound (2.3.33) in THF, which gave 2-(4-methoxyphenyl)-6-methoxy-3-[4-(piperidinoethoxy)benzoyl] benzo[β] thiophene (2.3.35). To the prepared benzothiophene (2.3.35) suspended in chlorobenzene was added AlCl3, followed by the addition of n-propanethiol, and the mixture was heated at 35°C. After the workup with aqueous HCl, the desired raloxifene (2.3.4) was separated [60] (Scheme 2.12).

There exist plenty of modifications for these two approaches, as reviewed in [61,62].
References
- ^ Jump up to:a b c d e f g h i j k l m n o p q r s t u v w Morello, Karla C.; Wurz, Gregory T.; DeGregorio, Michael W. (2003). “Pharmacokinetics of Selective Estrogen Receptor Modulators”. Clinical Pharmacokinetics. 42 (4): 361–372. doi:10.2165/00003088-200342040-00004. ISSN 0312-5963. PMID 12648026. S2CID 13003168.
- ^ Jump up to:a b c d e f g h i j k l m n o p q Hochner-Celnikier D (1999). “Pharmacokinetics of raloxifene and its clinical application”. Eur. J. Obstet. Gynecol. Reprod. Biol. 85 (1): 23–9. doi:10.1016/s0301-2115(98)00278-4. PMID 10428318.
- ^ Jeong, Eun Ju; Liu, Yong; Lin, Huimin; Hu, Ming (2005-03-15). “Species- and Disposition Model-Dependent Metabolism of Raloxifene in Gut and Liver: Role of UGT1A10”. Drug Metabolism and Disposition. ASPET. 33 (6): 785–794. doi:10.1124/dmd.104.001883. PMID 15769887. S2CID 24273998.
- ^ Jump up to:a b c d e f g h i j k l m “Raloxifene Hydrochloride Monograph for Professionals”. Drugs.com. American Society of Health-System Pharmacists. Retrieved 22 March 2019.
- ^ Yang, Z. D.; Yu, J.; Zhang, Q. (August 2013). “Effects of raloxifene on cognition, mental health, sleep and sexual function in menopausal women: a systematic review of randomized controlled trials”. Maturitas. 75 (4): 341–348. doi:10.1016/j.maturitas.2013.05.010. ISSN 1873-4111. PMID 23764354.
- ^ Jump up to:a b British national formulary : BNF 76 (76 ed.). Pharmaceutical Press. 2018. pp. 736–737. ISBN 9780857113382.
- ^ “NADAC as of 2019-02-27”. Centers for Medicare and Medicaid Services. Retrieved 3 March 2019.
- ^ “Raloxifene Hydrochloride – Drug Usage Statistics”. ClinCalc. Retrieved 11 April 2020.
- ^ “Raloxifene: MedlinePlus Drug Information”. medlineplus.gov. Retrieved 2018-11-07.
- ^ Jump up to:a b c Mosby (5 March 2013). Mosby’s Drug Reference for Health Professions – E-Book. Elsevier Health Sciences. pp. 1379–. ISBN 978-0-323-18760-2.
- ^ Ohta, Hiroaki; Hamaya, Etsuro; Taketsuna, Masanori; Sowa, Hideaki (January 2015). “Quality of life in Japanese women with postmenopausal osteoporosis treated with raloxifene and vitamin D: post hoc analysis of a postmarketing study”. Current Medical Research and Opinion. 31 (1): 85–94. doi:10.1185/03007995.2014.975339. ISSN 1473-4877. PMID 25299349. S2CID 24671531.
- ^ Jump up to:a b c d e f g h i j k l m n o p q r s t Fabian CJ, Kimler BF (March 2005). “Selective estrogen-receptor modulators for primary prevention of breast cancer”. J. Clin. Oncol. 23 (8): 1644–55. doi:10.1200/JCO.2005.11.005. PMID 15755972.
- ^ Jump up to:a b c Kirby I. Bland; Edward M. Copeland; V. Suzanne Klimberg; William J Gradishar (29 June 2017). The Breast E-Book: Comprehensive Management of Benign and Malignant Diseases. Elsevier Health Sciences. pp. 231–. ISBN 978-0-323-51187-2.
- ^ Jump up to:a b c d Raloxifene label Last updated 09/2007]
- ^ Jump up to:a b Gizzo S, Saccardi C, Patrelli TS, Berretta R, Capobianco G, Di Gangi S, Vacilotto A, Bertocco A, Noventa M, Ancona E, D’Antona D, Nardelli GB (2013). “Update on raloxifene: mechanism of action, clinical efficacy, adverse effects, and contraindications”. Obstet Gynecol Surv. 68 (6): 467–81. doi:10.1097/OGX.0b013e31828baef9. PMID 23942473. S2CID 9003157.
- ^ Jump up to:a b c d e f g h Goldstein, S. R. (2000). “A pharmacological review of selective oestrogen receptor modulators”. Human Reproduction Update. 6 (3): 212–224. doi:10.1093/humupd/6.3.212. ISSN 1355-4786. PMID 10874566.
- ^ Seeman E (2001). “Raloxifene”. J. Bone Miner. Metab. 19 (2): 65–75. doi:10.1007/s007740170043. PMID 11281162.
- ^ Jump up to:a b c Park, W (2002). “Selective estrogen receptor modulators (SERMS) and their roles in breast cancer prevention”. Trends in Molecular Medicine. 8 (2): 82–88. doi:10.1016/S1471-4914(02)02282-7. ISSN 1471-4914. PMID 11815274.
- ^ Agency for Healthcare Research and Quality, Rockville, MD. (September 2009). “Medications Effective in Reducing Risk of Breast Cancer But Increase Risk of Adverse Effects”. Retrieved 2009-09-14.
- ^ Lemmo, W (2016). “Anti-Estrogen Withdrawal Effect With Raloxifene? A Case Report”. Integrative Cancer Therapies. Published Online Before Print July 13 (3): 245–249. doi:10.1177/1534735416658954. PMC 5739193. PMID 27411856.
- ^ Jump up to:a b Haskell, Sally G. (2003). “Selective Estrogen Receptor Modulators”. Southern Medical Journal. 96 (5): 469–476. doi:10.1097/01.SMJ.0000051146.93190.4A. ISSN 0038-4348. PMID 12911186. S2CID 40607634.
- ^ Weatherman, Ross V; Clegg, Nicola J; Scanlan, Thomas S (2001). “Differential SERM activation of the estrogen receptors (ERα and ERβ) at AP-1 sites”. Chemistry & Biology. 8(5): 427–436. doi:10.1016/S1074-5521(01)00025-4. ISSN 1074-5521. PMID 11358690.
- ^ Escande A, Pillon A, Servant N, Cravedi JP, Larrea F, Muhn P, Nicolas JC, Cavaillès V, Balaguer P (May 2006). “Evaluation of ligand selectivity using reporter cell lines stably expressing estrogen receptor alpha or beta”. Biochem. Pharmacol. 71 (10): 1459–69. doi:10.1016/j.bcp.2006.02.002. PMID 16554039.
- ^ Greene, G. L.; Shiau, A. K.; Nettles, K. W. (2004). “A Structural Explanation for ERα/ERβ SERM Discrimination”. New Molecular Mechanisms of Estrogen Action and Their Impact on Future Perspectives in Estrogen Therapy (46): 33–45. doi:10.1007/978-3-662-05386-7_3. ISBN 978-3-662-05388-1. PMID 15248503.
- ^ Barkhem, Tomas; Carlsson, Bo; Nilsson, Yvonne; Enmark, Eva; Gustafsson, Jan-Åke; Nilsson, Stefan (1998). “Differential Response of Estrogen Receptor α and Estrogen Receptor β to Partial Estrogen Agonists/Antagonists”. Molecular Pharmacology. 54 (1): 105–112. doi:10.1124/mol.54.1.105. ISSN 0026-895X. PMID 9658195.
- ^ Prossnitz ER, Arterburn JB (July 2015). “International Union of Basic and Clinical Pharmacology. XCVII. G Protein-Coupled Estrogen Receptor and Its Pharmacologic Modulators”. Pharmacol. Rev. 67 (3): 505–40. doi:10.1124/pr.114.009712. PMC 4485017. PMID 26023144.
- ^ Petrie WK, Dennis MK, Hu C, Dai D, Arterburn JB, Smith HO, Hathaway HJ, Prossnitz ER (2013). “G protein-coupled estrogen receptor-selective ligands modulate endometrial tumor growth”. Obstet Gynecol Int. 2013: 472720. doi:10.1155/2013/472720. PMC 3863501. PMID 24379833.
- ^ Jeon-Hor, Chen; et al. (September 15, 2003). “Reduction of Breast Density Following Tamoxifen Treatment Evaluated by 3-D MRI: Preliminary Study”. Magn Reson Imaging. 29 (1): 91–8. doi:10.1016/j.mri.2010.07.009. PMC 3005955. PMID 20832226.
- ^ Jump up to:a b c d e f g h Draper, Michael W.; Chin, William W. (2003). “Molecular and Clinical Evidence for the Unique Nature of Individual Selective Estrogen Receptor Modulators”. Clinical Obstetrics and Gynecology. 46 (2): 265–297. doi:10.1097/00003081-200306000-00008. ISSN 0009-9201. PMID 12808380. S2CID 5132467.
- ^ Jump up to:a b c Corona G, Rastrelli G, Ratrelli G, Maggi M (February 2015). “The pharmacotherapy of male hypogonadism besides androgens”. Expert Opin Pharmacother. 16 (3): 369–87. doi:10.1517/14656566.2015.993607. PMID 25523084. S2CID 8891640.
- ^ Jump up to:a b Duarte FH, Jallad RS, Bronstein MD (November 2016). “Estrogens and selective estrogen receptor modulators in acromegaly”. Endocrine. 54 (2): 306–314. doi:10.1007/s12020-016-1118-z. PMID 27704479. S2CID 10136018.
- ^ Jump up to:a b Birzniece V, Sutanto S, Ho KK (April 2012). “Gender difference in the neuroendocrine regulation of growth hormone axis by selective estrogen receptor modulators”. J. Clin. Endocrinol. Metab. 97 (4): E521–7. doi:10.1210/jc.2011-3347. PMID 22319035.
- ^ Jump up to:a b Uebelhart B, Herrmann F, Pavo I, Draper MW, Rizzoli R (September 2004). “Raloxifene treatment is associated with increased serum estradiol and decreased bone remodeling in healthy middle-aged men with low sex hormone levels”. J. Bone Miner. Res. 19 (9): 1518–24. doi:10.1359/JBMR.040503. PMID 15312253. S2CID 36104038.
- ^ Xu, Beibei; Lovre, Dragana; Mauvais-Jarvis, Franck (2016). “Effect of selective estrogen receptor modulators on metabolic homeostasis”. Biochimie. 124: 92–97. doi:10.1016/j.biochi.2015.06.018. ISSN 0300-9084. PMID 26133657.
In healthy postmemopausal women, raloxifene treatment for one year prevented body weight gain and abdominal adiposity by promoting a shift from an android to gynoid fat distribution [46].
- ^ Francucci, C. M.; Pantaleo, D.; Iori, N.; Camilletti, A.; Massi, F.; Boscaro, M. (2014). “Effects of raloxifene on body fat distribution and lipid profile in healthy post-menopausal women”. Journal of Endocrinological Investigation. 28 (9): 623–631. doi:10.1007/BF03347261. ISSN 0391-4097. PMID 16218045. S2CID 28467435.
These results […] suggest, for the first time, that RLX promotes the shift from android to gynoid fat distribution, and prevents the uptrend of abdominal adiposity and body weight compared with untreated women.
- ^ Snyder KR, Sparano N, Malinowski JM (September 2000). “Raloxifene hydrochloride”. Am J Health Syst Pharm. 57 (18): 1669–75, quiz 1676–8. doi:10.1093/ajhp/57.18.1669. PMID 11006795.
- ^ Jump up to:a b Eric S. Orwoll; Michael Bliziotes (2 August 2002). Osteoporosis: Pathophysiology and Clinical Management. Springer Science & Business Media. pp. 320–. ISBN 978-1-59259-278-4.
- ^ Jump up to:a b Stuart Silverman; Bo Abrahamsen (29 December 2015). The Duration and Safety of Osteoporosis Treatment: Anabolic and Antiresorptive Therapy. Springer. pp. 24–. ISBN 978-3-319-23639-1.
- ^ Jump up to:a b Institute of Medicine; Board on Health Sciences Policy; Committee on Accelerating Rare Diseases Research and Orphan Product Development (3 April 2011). Rare Diseases and Orphan Products: Accelerating Research and Development. National Academies Press. pp. 113–. ISBN 978-0-309-15806-0.
- ^ Reducing Breast Cancer Risk with Drugs. Am Cncl on Science, Health. pp. 10–. GGKEY:CBEALLAHP8W.
- ^ Sydney Lou Bonnick (10 November 2007). Bone Densitometry for Technologists. Springer Science & Business Media. pp. 277–. ISBN 978-1-59259-992-9.
- ^ Jie Jack Li; Douglas S. Johnson (27 March 2013). Modern Drug Synthesis. John Wiley & Sons. pp. 2–. ISBN 978-1-118-70124-9.
- ^ Jump up to:a b J. Elks (14 November 2014). The Dictionary of Drugs: Chemical Data: Chemical Data, Structures and Bibliographies. Springer. pp. 1063–. ISBN 978-1-4757-2085-3.
- ^ Jump up to:a b c d Index Nominum 2000: International Drug Directory. Taylor & Francis. 2000. pp. 909–. ISBN 978-3-88763-075-1.
- ^ I.K. Morton; Judith M. Hall (31 October 1999). Concise Dictionary of Pharmacological Agents: Properties and Synonyms. Springer Science & Business Media. pp. 245–. ISBN 978-0-7514-0499-9.
- ^ Jump up to:a b c d e “Raloxifene”.
- ^ Thelancetoncology (2006). “A STARring role for raloxifene?”. Lancet Oncol. 7 (6): 443. doi:10.1016/S1470-2045(06)70701-X. PMID 16750489.
- ^ Provinciali N, Suen C, Dunn BK, DeCensi A (October 2016). “Raloxifene hydrochloride for breast cancer risk reduction in postmenopausal women”. Expert Rev Clin Pharmacol. 9 (10): 1263–1272. doi:10.1080/17512433.2016.1231575. PMID 27583816. S2CID 26047863.
- ^ James F. Holland; Raphael E. Pollock (2010). Holland-Frei Cancer Medicine 8. PMPH-USA. pp. 743–. ISBN 978-1-60795-014-1.
- ^ Blum A, Hathaway L, Mincemoyer R, Schenke WH, Csako G, Waclawiw MA, Panza JA, Cannon RO (2000). “Hormonal, lipoprotein, and vascular effects of the selective estrogen receptor modulator raloxifene in hypercholesterolemic men”. Am. J. Cardiol. 85 (12): 1491–4, A7. doi:10.1016/s0002-9149(00)00802-x. PMID 10856400.
- ^ Doran PM, Riggs BL, Atkinson EJ, Khosla S (2001). “Effects of raloxifene, a selective estrogen receptor modulator, on bone turnover markers and serum sex steroid and lipid levels in elderly men”. J. Bone Miner. Res. 16 (11): 2118–25. doi:10.1359/jbmr.2001.16.11.2118. PMID 11697809. S2CID 28216610.
- ^ Dimaraki EV, Symons KV, Barkan AL (2004). “Raloxifene decreases serum IGF-I in male patients with active acromegaly”. Eur. J. Endocrinol. 150 (4): 481–7. doi:10.1530/eje.0.1500481. PMID 15080777.
- ^ Duschek EJ, Gooren LJ, Netelenbos C (2004). “Effects of raloxifene on gonadotrophins, sex hormones, bone turnover and lipids in healthy elderly men” (PDF). Eur. J. Endocrinol. 150 (4): 539–46. doi:10.1530/eje.0.1500539. PMID 15080785.
- ^ Smith MR, Fallon MA, Lee H, Finkelstein JS (2004). “Raloxifene to prevent gonadotropin-releasing hormone agonist-induced bone loss in men with prostate cancer: a randomized controlled trial”. J. Clin. Endocrinol. Metab. 89 (8): 3841–6. doi:10.1210/jc.2003-032058. PMID 15292315.
- ^ Jump up to:a b Ho TH, Nunez-Nateras R, Hou YX, Bryce AH, Northfelt DW, Dueck AC, Wong B, Stanton ML, Joseph RW, Castle EP (2017). “A Study of Combination Bicalutamide and Raloxifene for Patients With Castration-Resistant Prostate Cancer”. Clin Genitourin Cancer. 15 (2): 196–202.e1. doi:10.1016/j.clgc.2016.08.026. PMID 27771244. S2CID 19043552.
- ^ Khodaie-Ardakani MR, Khosravi M, Zarinfard R, Nejati S, Mohsenian A, Tabrizi M, Akhondzadeh S (2015). “A Placebo-Controlled Study of Raloxifene Added to Risperidone in Men with Chronic Schizophrenia”. Acta Med Iran. 53 (6): 337–45. PMID 26069170.
- ^ Weickert TW, Weinberg D, Lenroot R, Catts SV, Wells R, Vercammen A, O’Donnell M, Galletly C, Liu D, Balzan R, Short B, Pellen D, Curtis J, Carr VJ, Kulkarni J, Schofield PR, Weickert CS (2015). “Adjunctive raloxifene treatment improves attention and memory in men and women with schizophrenia”. Mol. Psychiatry. 20 (6): 685–94. doi:10.1038/mp.2015.11. PMC 4444978. PMID 25980345.
- ^ Jump up to:a b Fujimura, Tetsuya; Takayama, Kenichi; Takahashi, Satoru; Inoue, Satoshi (2018). “Estrogen and Androgen Blockade for Advanced Prostate Cancer in the Era of Precision Medicine”. Cancers. 10 (2): 29. doi:10.3390/cancers10020029. ISSN 2072-6694. PMC 5836061. PMID 29360794.
- ^ Jump up to:a b c Wang Q, Dong X, Wang Y, Li X (2017). “Raloxifene as an adjunctive treatment for postmenopausal women with schizophrenia: a meta-analysis of randomized controlled trials”. Arch Womens Ment Health. 21 (1): 31–41. doi:10.1007/s00737-017-0773-2. PMID 28849318. S2CID 4524617.
- ^ Carneiro AL, de Cassia de Maio Dardes R, Haidar MA (July 2012). “Estrogens plus raloxifene on endometrial safety and menopausal symptoms–semisystematic review”. Menopause. 19 (7): 830–4. doi:10.1097/gme.0b013e31824a74ce. PMID 22549172. S2CID 196380398.
- ^ Nordt CA, DiVasta AD (2008). “Gynecomastia in adolescents”. Curr. Opin. Pediatr. 20 (4): 375–82. doi:10.1097/MOP.0b013e328306a07c. PMID 18622190. S2CID 205834072.
- ^ Leung KC, Leung AC (2017). “Gynecomastia in Infants, Children, and Adolescents”. Recent Pat Endocr Metab Immune Drug Discov. 10 (2): 127–137. doi:10.2174/1872214811666170301124033. PMID 28260521.
- ^ Lawrence SE, Faught KA, Vethamuthu J, Lawson ML (July 2004). “Beneficial effects of raloxifene and tamoxifen in the treatment of pubertal gynecomastia”. J. Pediatr. 145 (1): 71–6. doi:10.1016/j.jpeds.2004.03.057. PMID 15238910.
- ^ Kanakis, G. A.; Nordkap, L.; Bang, A. K.; Calogero, A. E.; Bártfai, G.; Corona, G.; Forti, G.; Toppari, J.; Goulis, D. G.; Jørgensen, N. (2019). “EAA clinical practice guidelines—gynecomastia evaluation and management”. Andrology. 7 (6): 778–793. doi:10.1111/andr.12636. ISSN 2047-2919. PMID 31099174.
- ^ Sugiyama, Nobuhiro; Barros, Rodrigo P.A.; Warner, Margaret; Gustafsson, Jan-Åke (2010). “ERβ: recent understanding of estrogen signaling”. Trends in Endocrinology & Metabolism. 21 (9): 545–552. doi:10.1016/j.tem.2010.05.001. ISSN 1043-2760. PMID 20646931. S2CID 43001363.
Further reading
- Barrett-Connor E (2001). “Raloxifene: risks and benefits”. Ann N Y Acad Sci. 949 (1): 295–303. Bibcode:2001NYASA.949..295B. doi:10.1111/j.1749-6632.2001.tb04036.x. PMID 11795366. S2CID 41412601.
- Heringa M (2003). “Review on raloxifene: profile of a selective estrogen receptor modulator”. Int J Clin Pharmacol Ther. 41 (8): 331–45. doi:10.5414/cpp41331. PMID 12940590.
- Sporn MB, Dowsett SA, Mershon J, Bryant HU (2004). “Role of raloxifene in breast cancer prevention in postmenopausal women: clinical evidence and potential mechanisms of action”. Clin Ther. 26 (6): 830–40. doi:10.1016/s0149-2918(04)90127-0. PMID 15262454.
- Vogel VG (2009). “The NSABP Study of Tamoxifen and Raloxifene (STAR) trial”. Expert Rev Anticancer Ther. 9 (1): 51–60. doi:10.1586/14737140.9.1.51. PMC 2785111. PMID 19105706.
- Wickerham DL, Costantino JP, Vogel VG, Cronin WM, Cecchini RS, Ford LG, Wolmark N (2009). “The use of tamoxifen and raloxifene for the prevention of breast cancer”. Recent Results Cancer Res. Recent Results in Cancer Research. 181: 113–9. doi:10.1007/978-3-540-69297-3_12. ISBN 978-3-540-69296-6. PMC 5110043. PMID 19213563.
- Vogel VG (2011). “Update on raloxifene: role in reducing the risk of invasive breast cancer in postmenopausal women”. Breast Cancer: Targets and Therapy. 3: 127–37. doi:10.2147/BCTT.S11288. PMC 3846694. PMID 24367182.
- Yang ZD, Yu J, Zhang Q (2013). “Effects of raloxifene on cognition, mental health, sleep and sexual function in menopausal women: a systematic review of randomized controlled trials”. Maturitas. 75 (4): 341–8. doi:10.1016/j.maturitas.2013.05.010. PMID 23764354.
External links
- “Raloxifene”. Drug Information Portal. U.S. National Library of Medicine.
///////Keoxifene hydrochloride, Raloxifene hydrochloride, LY-139481, LY 156758, Optruma, Loxifen, Evista


{kind=link}
{kind=link}