
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
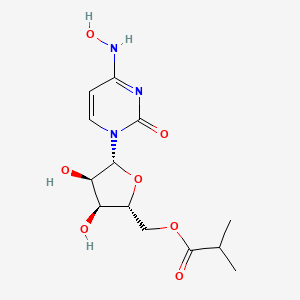
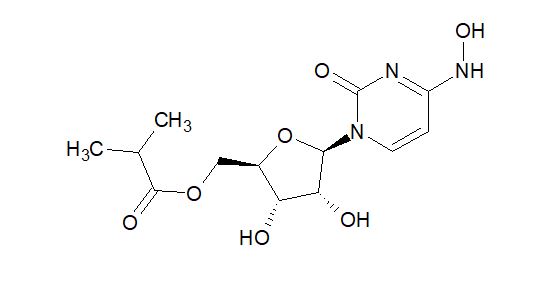
EIDD 2801
| Molecular Formula: | C13H19N3O7 |
|---|---|
| Molecular Weight: | 329.31 g/mol |
[(2R,3S,4R,5R)-3,4-dihydroxy-5-[4-(hydroxyamino)-2-oxopyrimidin-1-yl]oxolan-2-yl]methyl 2-methylpropanoate
UNII YA84KI1VEW
CAS 2349386-89-4
Molnupiravir (development codes MK-4482 and EIDD-2801) is an experimental antiviral drug which is orally active (can be taken orally) and was developed for the treatment of influenza. It is a prodrug of the synthetic nucleoside derivative N4-hydroxycytidine, and exerts its antiviral action through introduction of copying errors during viral RNA replication.[1][2] Activity has also been demonstrated against coronaviruses including SARS, MERS and SARS-CoV-2.[3]
The drug was developed at Emory University by the university’s drug innovation company, Drug Innovation Ventures at Emory (DRIVE). It was then acquired by Miami-based company Ridgeback Biotherapeutics, who later partnered with Merck & Co. to develop the drug further.
Safety Controversy
In April 2020, a whistleblower complaint by former Head of US Biomedical Advanced Research and Development Authority (BARDA) Rick Bright revealed concerns over providing funding for the further development of molnupiravir due to similar drugs having mutagenic properties (producing birth defects).[4] A previous company, Pharmasset, that had investigated the drug’s active ingredient had abandoned it. These claims were denied by George Painter, CEO of DRIVE, noting that toxicity studies on molnupiravir had been carried out and data provided to regulators in the US and UK, who permitted safety studies in humans to move forward in the spring of 2020. Also at this time, DRIVE and Ridgeback Biotherapeutics stated they planned future safety studies in animals.[5]
COVID-19
After being found to be active against SARS-CoV-2 in March 2020, molnupiravir was tested in a preliminary human study for “Safety, Tolerability, and Pharmacokinetics” in healthy volunteers in the UK and US.[6] In June 2020, Ridgeback Biotherapeutics announced it was moving to Phase II trials to test the efficacy of the drug as a treatment for COVID-19.[7] Two trials of small numbers of hospitalized and non-hospitalized patients in the US and the UK were underway in July.[8][9] In late July 2020, and without yet releasing any medical data, Merck, which had been partnering with Ridgeback Biotherapeutics on developing the drug, announced its intention to move molnupiravir to late stage trials beginning in September 2020.[10] On October 19 2020, Merck began a one year Stage 2/3 trial focused on hospitalized patients.[11]

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
PATENT
WO 2019113462
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019113462
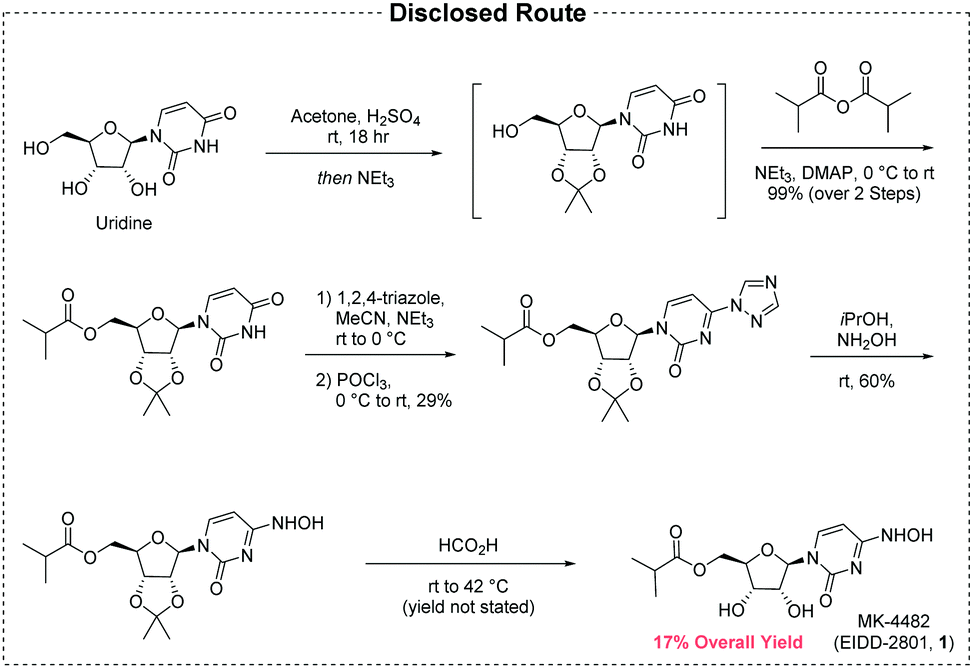
Example 10: Synthesis of EIDD-2801
A 1L round bottom flask was charged with uridine (25 g, 102.38 mmol) and acetone (700 mL). The reaction mixture was allowed to stir at rt. The slurry was then treated with sulfuric acid (0.27 mL, 5.12 mmol). Stirring was allowed to continue at rt for 18 hours. The reaction was quenched with 100 mL of trimethylamine and was used in the next step without further pruficication.
A 1L round bottom flask was charged with the reaction mixture from the previous reaction. Triethylamine (71.09 mL, 510.08 mmol) and 4-dimethylaminopyridine (0.62 g, 5.1 mmol) were then added. The flask was cooled using an ice bath and then 2-methylpropanoyl 2-methylpropanoate (17.75 g, 112.22 mmol) was slowly added. The reaction mixture was allowed to stir at rt until the reaction was complete. The reaction mixture was concentrated under reduced pressure, and the residue was dissolved in 600 mL ethyl acetate and washed with saturated aqueous bicarbonate solution x 2, water x 2 and brine x 2. The organics were dried over sodium sulfate and concentrated under reduced pressure to yield a clear colorless oil. The crude product was used in the next step without further purification.
A 1L round bottom flask was charged with the crude product from above (36 g, 101.59 mmol) and MeCN (406.37 mL). The reaction mixture was allowed to stir until all the starting material was dissolved. Next, 1,2, 4-triazole (50.52 g, 731.46 mmol) was added followed by the addition of N,N-diethylethanamine (113.28 mL, 812.73 mmol). The reaction mixture was allowed to stir at rt until all solids dissolved. The reaction was then cooled to 0°C using an ice bath. Phosphorous oxychloride (24.44 mL, 152.39 mmol) was added slowly. The slurry that formed was allowed to stir under argon while slowly warming to rt. The reaction was then allowed to stir until complete by TLC (EtOAc). The reaction was then quenched by the addition of lOOmL of water. The slurry then became a dark colored solution, which was
then concentrated under reduced pressure. The residue was dissolved in DCM and washed with water and brine. The organics were then dried over sodium sulfate, filtered, and concentrated under reduced pressure. The product was purified by silica gel chromatography (2 x 330 g columns). All fractions containing product were collected and concentrated under reduced pressure.
A 500 mL round bottom flask was charged with the product from the previous step (11.8 g, 29.11 mmol) and isopropyl alcohol (150 mL). The reaction mixture was allowed to stir at rt until all solids dissolved. Next, hydroxylamine (1.34 mL, 43.66 mmol) was added and stirring continued at ambient temperature. When the reaction was complete (HPLC) some solvent was removed under high vacuum at ambient temperature. The remaining solvent was removed under reduced pressure at 45°C. The resulting residue was dissolved in EtOAc and was washed with water and brine. The organics were dried over sodium sulfate, filtered, and concentrated under reduced pressure to yield oil. Crystals formed upon standing at rt. The crystals were collected by filtration, washed with ether x 3, and dried in vacuo to provide the product as a white solid.
A 200 mL round bottom flask was charged with the product from the previous step (6.5 g, 17.6 mmol) and formic acid (100 mL, 2085.6 mmol). The reaction mixture was allowed to stir at rt overnight. The progress of the reaction was monitored by HPLC. The reaction mixture was concentrated under reduced pressure at 42°C to yield a clear, pale pink oil. Next, 30 mL of ethanol was added. Solvent was then removed under reduced pressure. MTBE (50 mL) was added to the solid and heated. Next, isopropyl alcohol was added and heating was continued until all solid material dissolved (5 mL). The solution was then allowed to cool and stand at rt.
A solid started to form after about lhr. The solids were collected by filtration, washed with MTBE, and dried in vacuo to yield the EIDD-2801 as a white solid. The filtrate was concentrated under reduced pressure to yield a sticky solid, which was dissolved in a small amount of isopropyl alcohol with heating. The solution was allowed to stand at rt overnight. A solid formed in the flask, which was collected by filtration, rinsed with isopropyl alcohol and MTBE, and dried in vacuo to an additional crop of desired product.
EIDD-2801 (25 g) was dissolved in 250 mL of isopropyl alcohol by heating to 70°C to give a clear solution. The warm solution was polish filtered and filtrate transferred to 2L three neck flask with overhead stirrer. It was warmed back to 70°C and MTBE (250 mL) was slowly added into the flask. The clear solution was seeded and allowed to cool slowly to rt with stirring for 18 hrs. The EIDD-2801 solid that formed was filtered and washed with MTBE and dried at 50°C under vacuum for l8hours. The filtrate was concentrated, redissolved in 50 mL isopropyl alcohol and 40 mL MTBE by warming to give clear solution and allowed to stand at rt to give a second crop of EIDD-2801.
Example 11: General synthesis for Deuteration
389 390
The lactone 389 (0.0325 mol) was added to a dry flask under an argon atmosphere and was then dissolved in dry THF (250 mL). The solution as then cooled to -78°C and a DIBAL-D solution in toluene (0.065 mol) was dropwise. The reaction was allowed to stir at -78°C for 3-4 hours. The reaction was then quenched with the slow addition of water (3 mL). The reaction was then allowed to stir while warming to rt. The mixture was then diluted with two volumes of diethyl ether and was then poured into an equal volume of saturated sodium potassium tartrate solution. The organic layer was separated, dried over MgSCri. filtered, and concentrated under reduced pressure. The residue was purified on silica eluting with hexanes/ethyl acetate. The resulting lactol 390 was then converted to an acetate or benzolyate and subjected to cytosine coupling conditions and then further elaborated to N-hydroxycytidine.
PATENT
WO 2019173602
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019173602
PAPER
ChemRxiv (2020), 1-3.
AND
ChemRxiv (2020), 1-2
PAPER
A Concise Route to MK-4482 (EIDD-2801) from Cytidine: Part 2
Synlett (2020), Ahead of Print.

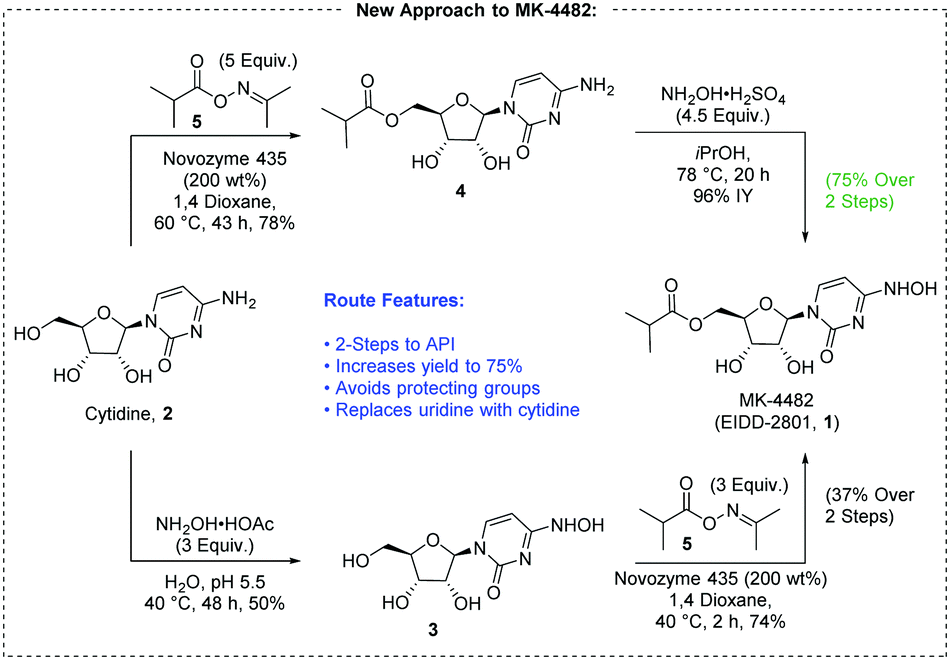
A new route to MK-4482 was developed. The route replaces uridine with the more available and less expensive cytidine. Low-cost, simple reagents are used for the chemical transformations, and the yield is improved from 17% to 44%. A step is removed from the longest linear sequence, and these advancements are expected to expand access to MK-4482 should it become a viable drug substance.
To a 20 mL vial was added N-hydroxycytidine acetonide ester 5 (0.25 g, 96% purity) followed by formic acid (4 mL). The resultant solution was stirred at room temperature for 4 h 20 min. Solvent was removed under reduced pressure and fresh EtOH (5 mL) was added. The resultant solution was again concentrated under vacuum to afford an oil. Methyl tert-butyl ether and IPA (5 mL each) were successively added as described earlier for preparation of compound 4 and concentrated to give 0.205 g of crude material (77% assay yield, 79% purity). This material was purified by silica gel column chromatography in 8 % MeOH/ Chloroform to afford 130 mg of EIDD-2801 as a solid (60% isolated yield corrected for purity, 98% purity) 1H NMR (600 MHz, CD3OD): δ 6.91 (d, J = 8.2 Hz, 1H), 5.82 (d, J = 4.8 Hz, 1H), 5.61 (d, J = 8.2 Hz, 1H), 4.29 (d, J = 3.6 Hz, 2H), 4.14 (t, J = 4.9 Hz, 1H), 4.08 (p, J = 4.9 Hz, 2H), 2.62 (septet, J = 7.0 Hz, 1H), 1.19 (d, J = 7.0 Hz, 6H); 13C NMR (151 MHz, CD3OD): δ 178.6, 151.81, 146.44, 132.04, 99.84, 90.74, 82.88, 74.67, 71.80, 65.23, 35.45, 27.49, 19.65, 19.61.


One-Pot Transamination/Deprotection of 4 to EIDD-2801: To acetonide ester 4 (1.03 g, 77% Purity) in a 100 mL single neck round bottom flask was added hydroxylamine sulfate (1.09 g, 3.2 equiv.) followed by 40% IPA (20 mL prepared by mixing 12 mL of water and 8 mL of 99.5% IPA. The resultant solution was heated to 78˚C (internal temperature 72-73 ˚C) for 23 h upon which time HPLC showed the formation of EIDD-2801. Solvent was removed on a rotary evaporator and isopropanol (20 mL) was then added. The resulting slurry was sonicated for 5 minutes. The insoluble residue was then filtered and the filtrate concentrated under reduced pressure to afford crude material. (1.34 g, 38% purity, 69% assay yield). The resultant material was purified by silica gel chromatography (5-6% MeOH/DCM) to provide pure EIDD-2801 as two fractions (0.26 g, >99% purity, 36% corrected yield) as an yellow solid and 0.27 g (69.5% purity, 26% corrected yield) as a pinkish solid. The lower purity material was subjected to a second column purification again using 7% MeOH/ DCM to afford 0.137 g of material with 90% purity by NMR. The combined yield thus was estimated to be 53%. The 1H NMR spectrum of the product thus obtained matched the one obtained in the sequential approach as outlined above.
SYN
- A High‐Yielding Synthesis of EIDD‐2801 from Uridine,
Alexander Steiner, Desiree Znidar, Sándor B. Ötvös, David R. Snead, Doris Dallinger, C. Oliver Kappe,
Eur. J. Org. Chem. 2020.
https://doi.org/10.1002/ejoc.202001340





EIDD-2801 was isolated in 69% yield (307 mg) and ≥99% purity as a white
solid.
1H-NMR (300 MHz, MeOH-d4) δ 6.91 (d, J= 8.3 Hz, 1H), 5.82 (d, J= 4.8 Hz, 1H), 5.61 (d, J= 8.2 Hz, 1H), 4.29
(d, J= 3.6 Hz, 2H), 4.15-4.07 (m, 3H), 2.62 (sept, J= 7.0 Hz, 1H), 1.18 (d, J= 7.0 Hz 6H);
13C-NMR (75 MHz,
MeOH-d4Ϳ δ 178.2, 151.5, 146.1, 131.7, 99.5, 90.4, 82.5, 74.3, 71.5, 64.9, 35.1, 19.3, 19.3. The NMR data
is in agreement with previously published values.[2] HRMS (ESI, positive mode): m/z [M + H]+
Calcd for
[C13H20N3O7 +H]+
: 330.1296, found: 330.1297.


SYN
C. Oliver Kappe, Doris Dallinger, University of Graz, Austria, and colleagues have developed an improved synthesis of EIDD-2801 from uridine (pictured below) by strategically reordering the synthetic steps. The reaction sequence starts with the activation of uridine with 1,2,4-triazole and continues with a telescoped acetonide protection/esterification and a telescoped hydroxyamination/acetonide deprotection. Telescoped reaction sequences consist of two or more than one one-pot procedures that are performed back-to-back without a work-up step in-between. A continuous flow process was used for the final acetonide deprotection, which improved selectivity and reproducibility.
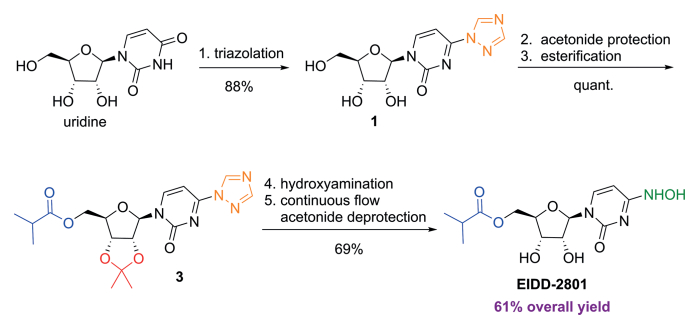
SYN
https://www.frontiersin.org/articles/10.3389/fphar.2020.01013/full
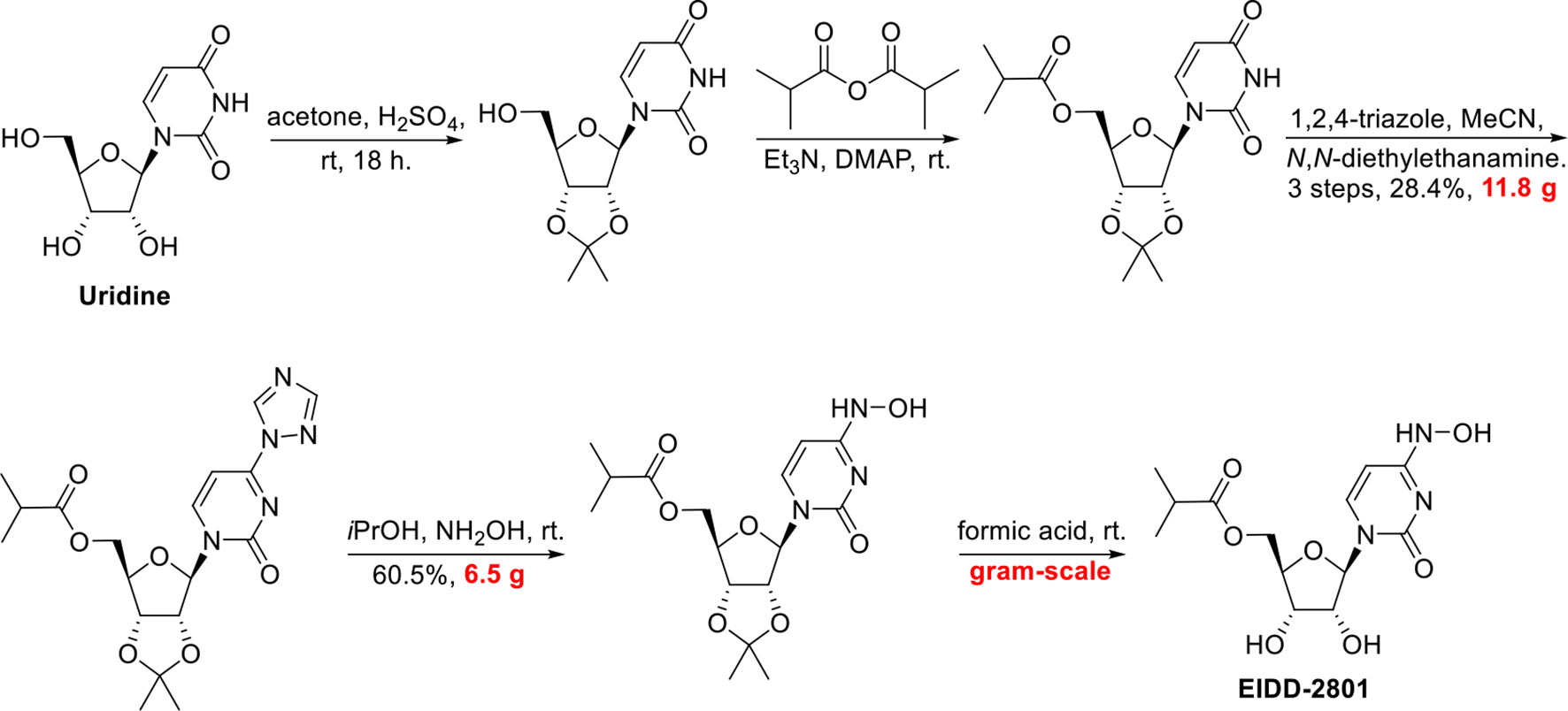
SYN
http://www.rsc.org/suppdata/d0/cc/d0cc05944g/d0cc05944g1.pdf
To a solution of 5’-O-isobutyrylcytidine 4 (1.0 g, 90% purity, 2.87 mmol, 1.0 eq) in 2-propanol (15 ml), hydroxylamine sulphate (2.12 g, 12.93 mmol, 4.5 eq.) was added and reaction was stirred for 20 h at 78 C. Upon completion, the reaction was cooled to room temperature. The organic layer (upper layer) was separated from biphasic reaction mixture. The aqueous layer was washed with 2-propanol (2 X 5 mL). The combined organic layer was concentrated using rotary evaporation and the crude was purified by column chromatography with a gradient of 2-15% methanol in dichloromethane to yield EIDD-2801 (1) as a white solid (963 mg, 94% purity, 96% yield). 1H NMR (600 MHz, D2O) δ 6.98 (d, J = 8.3 Hz, 1H), 5.87 (d, J = 5.0 Hz, 1H), 5.78 (d, J = 8.2 Hz, 1H), 4.39 – 4.33 (m, 3H), 4.28 (dd, J = 6.6, 3.4 Hz, 2H), 2.69 (hept, J = 7.0 Hz, 1H), 1.17 (d, J = 3.7 Hz, 3H), 1.16 (d, J = 3.7 Hz, 3H). 13C NMR (126 MHz, D2O) δ 18.1, 18.2, 33.9, 48.8, 63.6, 69.6, 72.5, 81.0, 88.5, 98.8, 131.1, 151.1, 179.8 ppm; LRMS: 330.1 [M+H]+ ; HRMS (ESI): calcd. for C13H19N3O7 [M+H]+ 330.1296, found 330.1302; Purity: 94% (assessed by qNMR).


https://pubs.rsc.org/en/content/articlehtml/2020/cc/d0cc05944g
 |
||
| Fig. 2 A new route to MK-4482 from cytidine. | ||
References
- ^ Toots M, Yoon JJ, Cox RM, Hart M, Sticher ZM, Makhsous N, et al. (October 2019). “Characterization of orally efficacious influenza drug with high resistance barrier in ferrets and human airway epithelia”. Science Translational Medicine. 11 (515): eaax5866. doi:10.1126/scitranslmed.aax5866. PMC 6848974. PMID 31645453.
- ^ Toots M, Yoon JJ, Hart M, Natchus MG, Painter GR, Plemper RK (April 2020). “Quantitative efficacy paradigms of the influenza clinical drug candidate EIDD-2801 in the ferret model”. Translational Research. 218: 16–28. doi:10.1016/j.trsl.2019.12.002. PMID 31945316.
- ^ Sheahan TP, Sims AC, Zhou S, Graham RL, Pruijssers AJ, Agostini ML, et al. (April 2020). “An orally bioavailable broad-spectrum antiviral inhibits SARS-CoV-2 in human airway epithelial cell cultures and multiple coronaviruses in mice”. Science Translational Medicine. 12 (541): eabb5883. doi:10.1126/scitranslmed.abb5883. PMC 7164393. PMID 32253226.
- ^ Halford, Bethany. “An emerging antiviral takes aim at COVID-19”. Retrieved 1 August 2020.
- ^ Cohen, Jon; Piller, Charles (13 May 2020). “Emails offer look into whistleblower charges of cronyism behind potential COVID-19 drug”. Science. Retrieved 1 August 2020.
- ^ “COVID-19 First In Human Study to Evaluate Safety, Tolerability, and Pharmacokinetics of EIDD-2801 in Healthy Volunteers”. ClinicalTrials.gov. Retrieved 1 June 2020.
- ^ “Ridgeback Biotherapeutics Announces Launch of Phase 2 Trials Testing EIDD-2801 as Potential Treatment for COVID-19”. Business Wire. Retrieved 4 July 2020.
- ^ “A Safety, Tolerability and Efficacy of EIDD-2801 to Eliminate Infectious Virus Detection in Persons With COVID-19”. ClinicalTrials.gov. Retrieved 4 July 2020.
- ^ “The Effect of EIDD-2801 on Viral Shedding of SARS-CoV-2 (COVID-19)”. ClinicalTrials.gov. Retrieved 4 July 2020.
- ^ Court, Emma (31 July 2020). “Merck pushes ahead on COVID-19 treatment, vaccines”. Retrieved 31 July 2020.
- ^ ClinicaL trials register : Efficacy and Safety of Molnupiravir (MK-4482) in Hospitalized Adult Participants With COVID-19 (MK-4482-001)
Electron microscope image of SARS virus in a tissue culture isolate, courtesy of CDC Public Health Image Library.
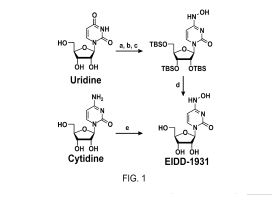
The drug EIDD-1931 was effective against SARS and MERS viruses in the laboratory, and a modified version (EIDD-2801) could potentially be valuable against 2019-nCoV.
https://news.emory.edu/stories/2020/02/coronavirus_eidd/index.html
Emory, collaborators testing antiviral drug as potential treatment for coronaviruses
An antiviral compound discovered at Emory University could potentially be used to treat the new coronavirus associated with the outbreak in China and spreading around the globe. Drug Innovation Ventures at Emory (DRIVE), a non-profit LLC wholly owned by Emory, is developing the compound, designated EIDD-2801.
In testing with collaborators at the University of North Carolina at Chapel Hill and Vanderbilt University Medical Center, the active form of EIDD-2801, which is called EIDD-1931, has shown efficacy against the related coronaviruses SARS (Severe Acute Respiratory Syndrome)- and MERS-CoV (Middle East Respiratory Syndrome Coronavirus). Some of the data was recently published in Journal of Virology.
EIDD-2801 is an oral ribonucleoside analog that inhibits the replication of multiple RNA viruses, including respiratory syncytial virus, influenza, chikungunya, Ebola, Venezuelan equine encephalitis virus, and Eastern equine encephalitis viruses.
“We have been planning to enter human clinical tests of EIDD-2801 for the treatment of influenza, and recognized that it has potential activity against the current novel coronavirus,” says George Painter, PhD, director of the Emory Institute for Drug Development (EIDD) and CEO of DRIVE. “Based on the drug’s broad-spectrum activity against viruses including influenza, Ebola and SARS-CoV/MERS-CoV, we believe it will be an excellent candidate.”
“Our studies in the Journal of Virology show potent activity of the EIDD-2801 parent compound against multiple coronaviruses including SARS and MERS,” says Mark Denison, MD, the Stahlman Professor of Pediatrics and director of pediatric infectious diseases at Vanderbilt University School of Medicine. “It also has a strong genetic barrier to development of viral resistance, and its oral bioavailability makes it a candidate for use during an outbreak.”
“Generally speaking, seasonal flu is still a much more common threat than this coronavirus, however, novel emerging coronaviruses represent a considerable threat to global health as evidenced by the new 2019-nCoV,” said Ralph Baric, PhD, an epidemiology professor at the University of North Carolina’s Gillings School of Global Public Health. “But the reason the new coronavirus is so concerning is that it’s much more likely to be deadly than the flu – fatal for about one in 25 people versus one in 1,000 for the flu.”
The development of EIDD-2801 has been funded in whole or in part with Federal funds from the National Institute of Allergy and Infectious Diseases (NIAID), under contract numbers HHSN272201500008C and 75N93019C00058, and from the Defense Threat Reduction Agency (DTRA), under contract numbers HDTRA1-13-C-0072 and HDTRA1-15-C-0075, for the treatment of Influenza, coronavirus, chikungunya, and Venezuelan equine encephalitis virus.
About DRIVE: DRIVE is a non-profit LLC wholly owned by Emory started as an innovative approach to drug development. Operating like an early stage biotechnology company, DRIVE applies focus and industry development expertise to efficiently translate discoveries to address viruses of global concern. Learn more at: http://driveinnovations.org/
Emory-discovered antiviral is poised for COVID-19 clinical trials
The nucleoside inhibitor has advantages over Gilead’s remdesivir but has yet to be tested in humans
Asmall-molecule antiviral discovered by Emory University chemists could soon start human testing against COVID-19, the respiratory disease caused by the novel coronavirus. That’s the plan of Ridgeback Biotherapeutics, which licensed the compound, EIDD-2801, from an Emory nonprofit.
EIDD-2801 works similarly to Gilead Sciences’ remdesivir, an unapproved drug that was developed for the Ebola virus and is being studied in five Phase III trials against COVID-19. Both molecules are nucleoside analogs that metabolize into an active form that blocks RNA polymerase, an essential component of viral replication.
But remdesivir can only be given intravenously, meaning it would be difficult to deploy widely. In contrast, EIDD-2801 can be taken in pill form, says Mark Denison, a coronavirus expert and director of the infectious diseases division at Vanderbilt Medical School. Denison partnered with Emory and researchers at the University of North Carolina to test the compound against coronaviruses.
EIDD-2801 has other promising features. Many antivirals work by introducing errors into the viral genome, but, unlike other viruses, coronaviruses can fix some mistakes. In lab experiments, EIDD-2801 “was able to overcome the coronavirus proofreading function,” Denison says.
He also notes that while remdesivir and EIDD-2801 both block RNA polymerase, they appear to do it in different ways, meaning they could be complementary.
Unlike remdesivir, EIDD-2801 lacks human safety data. Ridgeback founder and CEO Wendy Holman says she expects the US Food and Drug Administration to give the green light for a Phase I study in COVID-19 infections within “weeks, not months.”
“weeks, not months.”
 |
|
| Clinical data | |
|---|---|
| Other names | MK-4482, EIDD-2801 |
| Legal status | |
| Legal status |
|
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| UNII | |
| Chemical and physical data | |
| Formula | C13H19N3O7 |
| Molar mass | 329.31 g·mol−1 |
| 3D model (JSmol) | |
////////EIDD 2801, EMORY, CORONA VIRUS, COVID 19, mk 4482, molnupiravir, merck
CC(C)C(=O)OC[C@H]2O[C@@H](N1C=CC(=NC1=O)NO)[C@H](O)[C@@H]2O
NEW DRUG APPROVALS
ONE TIME
$10.00



| Application Id | Application Number | Application Date | Country | Title |
| US333828014 | 17170172 | 08.02.2021 | US | N4-HYDROXYCYTIDINE AND DERIVATIVES AND ANTI-VIRAL USES RELATED THERETO |
| US305251595 | 16755779 | 07.12.2018 | US | N4-HYDROXYCYTIDINE AND DERIVATIVES AND ANTI-VIRAL USES RELATED THERETO |
| WO2021159044 | PCT/US2021/016984 | 07.02.2021 | WO | N4-HYDROXYCYTIDINE AND DERIVATIVES AND ANTI-VIRAL USES RELATED THERETO |
| WO2021137913 | PCT/US2020/054857 | 08.10.2020 | WO | 4′-HALOGEN CONTAINING NUCLEOTIDE AND NUCLEOSIDE THERAPEUTIC COMPOSITIONS AND USES RELATED THERETO |




[…] EIDD 2801 — New Drug Approvals […]
LikeLike
I need to have product patent covering Molnupiravir
LikeLike
[…] concerns were raised about mutagenic mechanisms. For example, there was controversy when a whistleblower filed a complaint about the drug and the company (Ridgeback). Rick Bright, who was an executive at the U.S. […]
LikeLike
[…] not be deceived! We’ll look at ingredients soon. Highly likely it has the same ingredients as the […]
LikeLike
[…] https://newdrugapprovals.org/2020/03/28/eidd-2801/ […]
LikeLike
Reblogged this on DRUG REGULATORY AFFAIRS INTERNATIONAL.
LikeLike
[…] A previous company, Pharmasset, that had investigated the drug’s active ingredient had abandoned it. These claims were denied by George Painter, CEO of DRIVE, noting that toxicity studies on molnupiravir had been carried out and data provided to regulators in the US and UK, who permitted safety studies in humans to move forward in the spring of 2020. Also at this time, DRIVE and Ridgeback Biotherapeutics stated they planned future safety studies in animals. […]
LikeLike