
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
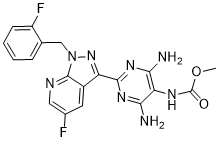
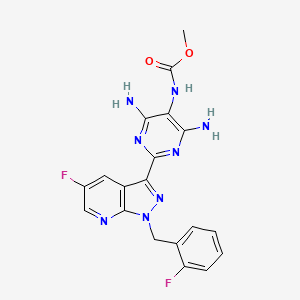
Vericiguat
BAY 102; BAY-1021189; MK-1242
1350653-20-1
Chemical Formula: C19H16F2N8O2
Molecular Weight: 426.3878
Vericiguat; 1350653-20-1; UNII-LV66ADM269; Methyl (4,6-diamino-2-(5-fluoro-1-(2-fluorobenzyl)-1H-pyrazolo[3,4-b]pyridin-3-yl)pyrimidin-5-yl)carbamate; BAY-1021189; LV66ADM269
Methyl (4,6-diamino-2-(5-fluoro-1-((2-fluorophenyl)methyl)-1H-pyrazolo(3,4-b)pyridin-3-yl(pyrimidin-5-yl)carbamate
methyl N-[4,6-diamino-2-[5-fluoro-1-[(2-fluorophenyl)methyl]pyrazolo[3,4-b]pyridin-3-yl]pyrimidin-5-yl]carbamate
Methyl{4,6-diamino-2-[5-fluoro-1-(2-fluorobenzyl)-1H-pyrazolo[3,4-b]pyridi- n-3-yl]pyrimidin-5-yl}carbamate
- Originator Bayer HealthCare Pharmaceuticals
- Developer Bayer HealthCare Pharmaceuticals; Merck & Co
- Mechanism of Action Guanylate cyclase stimulants
- Phase III Chronic heart failure
- Phase I Coronary artery disease
- 28 May 2018 Phase II VITALITY HFpEF trial for Chronic heart failure in Austria, USA, Belgium, Portugal, Canada, Spain, Hungary and Greece (PO) (EudraCT2018-000298-65) (NCT03547583)
- 17 May 2018 Phase-I clinical trials in Coronary artery disease (In adults, In the elderly) in Moldova and Germany (PO) (NCT03504982)
- 20 Apr 2018 Bayer in collaboration with Merck Sharp & Dohme Corp. plans a phase I trial for Coronary Artery Disease in the Netherlands, Moldova and Germany (NCT03504982)
Vericiguat, also known as BAY1021189 or BAY10-21189, is a potent and orally active sGC stimulator (Soluble Guanylate Cyclase Stimulator). Direct stimulation of soluble guanylate cyclase (sGC) is emerging as a potential new approach for the treatment of renal disorders. sGC catalyzes the formation of cyclic guanosine monophosphate (cGMP), deficiency of which is implicated in the pathogenesis of chronic kidney disease (CKD).
Vericiguat, discovered at Bayer, is the first soluble guanylate cyclase (sGC) stimulator. Vericiguat is currently being studied in a Phase III clinical program for the treatment of heart failure with reduced ejection fraction (HFrEF)
| ベルイシグアト Vericiguat  C19H16F2N8O2 : 426.38 [1350653-20-1] |
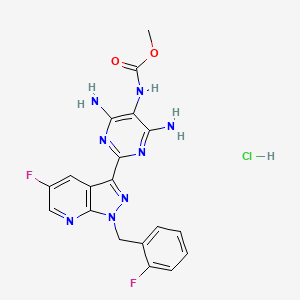
Vericiguat hydrochloride
cas 1350658-96-6
PHASE 3 MERCK/BAYER
| Chemical Names: | UNII-5G76IGF54K; 5G76IGF54K; ; 1350658-96-6; Carbamic acid, N-(4,6-diamino-2-(5-fluoro-1-((2-fluorophenyl)methyl)-1H-pyrazolo(3,4-b)pyridin-3-yl)-5-pyrimidinyl)-, methyl ester, hydrochloride (1:1); Methyl (4,6-diamino-2-(5-fluoro-1-(2-fluorobenzyl)-1H-pyrazolo(3,4-b)pyridin-3-yl)pyrimidin-5-yl)carbamate hydrochloride |
|---|---|
| Molecular Formula: | C19H17ClF2N8O2 |
| Molecular Weight: | 462.846 g/mol |

Clip
https://www.thieme-connect.com/products/ejournals/pdf/10.1055/s-0036-1590758.pdf
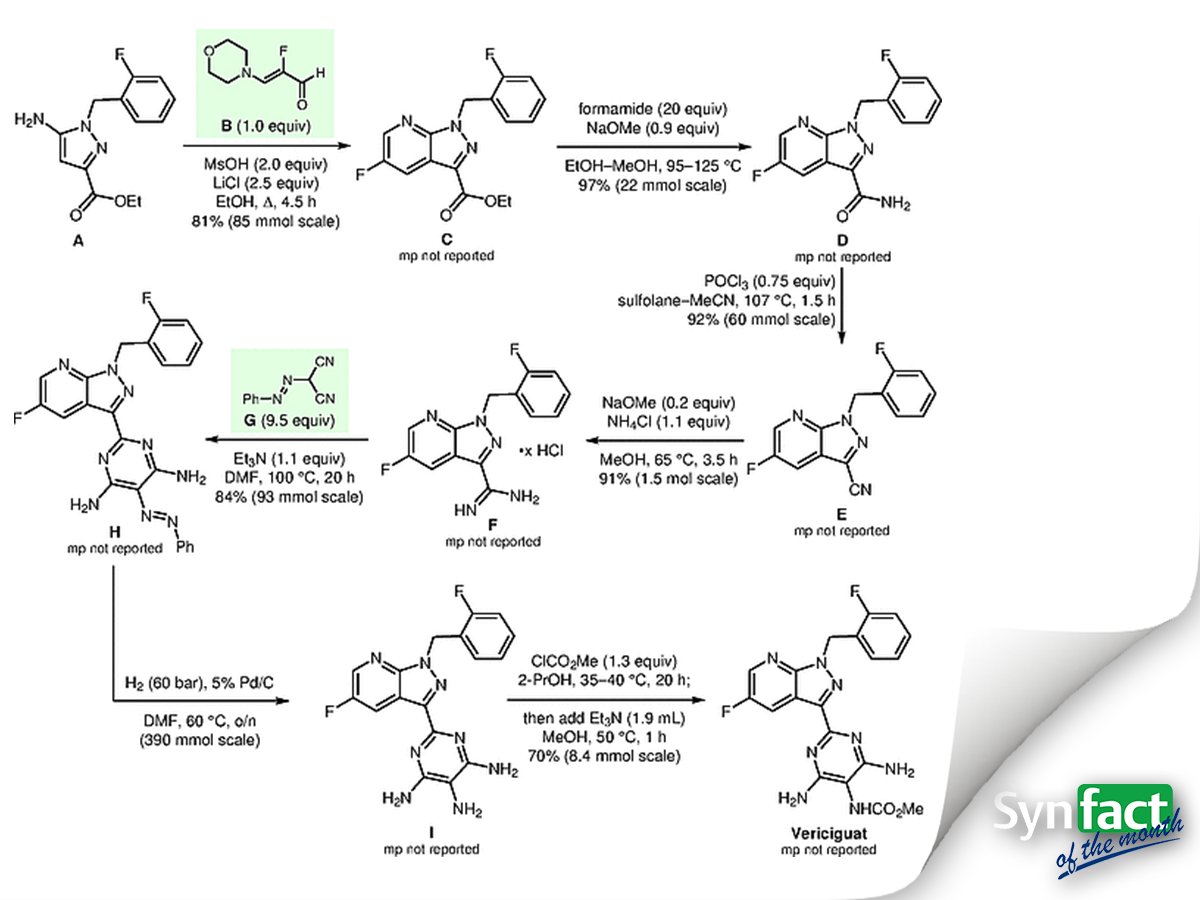
Significance: Vericiguat (BAY 1021189) is an orally available soluble guanylate cyclase (sGC) stimulator that has entered phase-three trials for the once-daily treatment of chronic heart failure. Key steps in the synthesis depicted are (1) construction of the 5-fluoro-(2-fluorobenzyl)-1H-pyrazolo[3,4-b]pyridine-3-carboxylate C by condensation of the 5-amino-1H-pyrazole-3-carboxylate A with the aldehyde B and (2) construction of the pyrimidine-4,5,6-triamine derivative H through reaction of [(E)-phenyldiazenyl]malononitrile (G) with amidine F.
Comment: Experimental details are provided for the noteworthy four-step synthesis (not shown) of the crystalline 2-fluoro-(3-morpholin-4-yl)acrylaldehyde B from commercially available 2,2,3,3- tetrafluoro-1-propanol. The synthesis of pyrazole A is described in a patent (A. Straub et al. WO 2000/006569 A1). The [(E)-phenyldiazenyl]malononitrile (G) was generated in situ by reaction of phenyldiazonium chloride with malononitrile.
Example 13
Methyl{4,6-diamino-2-[5-fluoro-1-(2-fluorobenzyl)-1H-pyrazolo[3,4-b]pyridi- n-3-yl]pyrimidin-5-yl}carbamate
Method A:
4.0 g (77.0% by weight, 8.36 mmol) of the compound from Example 12 in 37.9 ml of isopropanol were heated to 35.degree. C. and then 0.84 ml (10.87 mmol) of methyl chloroformate was added dropwise. The mixture was stirred at 35.degree.-40.degree. C. for 20 h and heated to 50.degree. C., and 9.5 ml of methanol were added. Subsequently, 1.9 ml of triethylamine were added dropwise within 0.5 h and rinsed in with 1.3 ml of methanol, and the mixture was stirred at 50.degree. C. for 1 h. Thereafter, the reaction mixture was cooled to RT and stirred at RT for 1 h, and the solids were filtered off with suction, washed three times with 8 ml each time of ethanol, suction-dried and dried in a vacuum drying cabinet at 50.degree. C. under a gentle nitrogen stream. This gave 3.4 g of crude product. 3.0 g of the crude product were stirred in 8 ml of DMSO for 5 min, 13.0 ml of ethyl acetate and 50 mg of activated carbon were added, and the mixture was heated at reflux (84.degree. C.) for 15 min. The suspension was hot-filtered and the filter residue was washed with 1.9 ml of ethyl acetate.sup.1). 60 ml of ethyl acetate and 16 ml of ethanol were heated to 60.degree. C., and the combined filtrates were added dropwise and stirred at 60.degree. C. for 1.5 h. The suspension was cooled to RT within 25 min, stirred for a further 1.5 h, cooled further to 0.degree.-5.degree. C. and stirred for a further 1 h. The solids were filtered off with suction, washed twice with 6.4 ml each time of ethyl acetate, suction-dried and dried in a vacuum drying cabinet at 50.degree. C. under a gentle nitrogen stream. This gave 2.2 g (70.0% of theory) of the title compound. 1) According to the preparation process described, the di-dimethyl sulphoxide solvate is obtained at this point, and this is characterized in Tables 2 and 4 by the reflections in the x-ray diffractogram and bands in the IR spectrum.
MS (ESIpos): m/z=427 (M+H).sup.+
.sup.1H NMR (400 MHz, DMSO-d.sub.6): .delta.=3.62 (br s, 3H), 5.79 (s, 2H), 6.22 (br s, 4H), 7.10-7.19 (m, 2H), 7.19-7.26 (m, 1H), 7.32-7.40 (m, 1H), 7.67 and 7.99 (2 br s, 1H), 8.66 (m, 1H), 8.89 (dd, 1H) ppm.
The di-dimethyl sulphoxide solvate of the compound of the formula (I) has the advantage of much better filterability than the substance in the prior art. Furthermore, the preparation process via the di-dimethyl sulphoxide solvate of the compound of the formula (I) leads to a very high purity of the compound of the formula (I).
Method B:
4.0 g (10.8 mmol) of the compound from Example 12 Method B in 37.9 ml of isopropanol were heated to 35.degree. C. and then 1.1 ml (14.1 mmol) of methyl chloroformate were added dropwise. The mixture was stirred at 35.degree.-40.degree. C. for 16.5 h and cooled to RT, and 2.1 ml of aqueous ammonia (28%) were added. Subsequently, 4.2 ml of water were added and the mixture was stirred for 2.5 h. The solids were filtered off with suction, washed twice with 5 ml each time of water, suction-dried and dried in a vacuum drying cabinet at 50.degree. C. under a gentle nitrogen stream. This gave 4.4 g of crude product.
Method C:
4.0 g (10.8 mmol) of the compound from Example 12 Method B in 37.9 ml of isopropanol were heated to 35.degree. C. and then 1.1 ml (14.1 mmol) of methyl chloroformate were added dropwise. The mixture was stirred at 35.degree.-40.degree. C. for 16.5 h, and 9.5 ml of methanol were added at 50.degree. C. Subsequently, 2.42 ml of triethylamine were added dropwise within 20 min and rinsed in with 1.3 ml of methanol, and the mixture was stirred at 50.degree. C. for 1 h. Thereafter, the reaction mixture was cooled to RT and stirred at RT for 1 h, and the solids were filtered off with suction, washed three times with 8 ml each time of methanol, suction-dried and dried in a vacuum drying cabinet at 50.degree. C. under a gentle nitrogen stream. This gave 4.3 g of crude product.
Method D:
6.9 g of the crude product were stirred in 18.4 ml of DMSO for 5 min, 30.0 ml of ethyl acetate and 115 mg of activated carbon were added, and the mixture was heated at reflux (84.degree. C.) for 15 min. The suspension was hot-filtered and the filter residue was washed with 4.4 ml of ethyl acetate. 138 ml of ethyl acetate were heated to 50.degree. C., and the combined filtrates were added dropwise and stirred at 45-50.degree. C. for 1 h. The suspension was cooled to 0.degree.-5.degree. C. within 1.5 h and stirred for a further 1 h. The solids were filtered off with suction, washed twice with 14.8 ml each time of ethyl acetate and suction-dried for 1 h. 6.4 g of the di-dimethyl sulphoxide solvate were obtained as a moist product.sup.1).
Method E:
2.0 g of the di-dimethyl sulphoxide solvate were stirred at reflux temperature in 40 ml of ethyl acetate and 11.1 ml of ethanol for 17 h, cooled to RT and stirred for a further 1 h. The solids were filtered off with suction, washed four times with 1.4 ml each time of ethyl acetate and dried in a vacuum drying cabinet at 50.degree. C. under a gentle nitrogen stream. This gave 1.4 g of the title compound present in polymorph I.
Method F:
0.5 g of the di-dimethyl sulphoxide solvate were stirred at reflux temperature in 12.5 ml of solvent for 17 h, cooled to RT and stirred for a further 1 h. The solids were filtered off with suction, washed with 2 ml of solvent and suction-dried for 30 min. This gave 0.3 g of the title compound present in polymorph I.
The following solvents were used:
1.) 9 ml of ethyl acetate/3.5 ml of ethanol/0.3 ml of water
2.) 12.5 ml of isopropanol
3.) 12.5 ml of isopropanol/0.3 ml of water
4.) 12.5 ml of methanol
5.) 12.5 ml of methanol/0.3 ml of water
6.) 12.5 ml of acetonitrile
7.) 12.5 ml of acetone
8.) 12.5 ml of tetrahydrofuran,
9.) 12.5 ml of methyl tert-butyl ether
Table 1 indicates the reflections of the x-ray diffractogram. Table 3 shows the bands of the IR spectrum.
The compound (I) in crystalline polymorph I is notable for higher stability and more particularly for the fact that it is stable in the micronization process and hence no conversion and recrystallization takes place.
The compound of the formula (I) can be prepared by processes described above. This affords the compound of the formula (I) in a crystal polymorph referred to hereinafter as polymorph I. Polymorph I has a melting point of 257.degree. C. and a characteristic x-ray diffractogram featuring the reflections (2 theta) 5.9, 6.9, 16.2, 16.5, 24.1 and 24.7, and a characteristic IR spectrum featuring the band maxima (in cm.sup.-1) 1707, 1633, 1566, 1475, 1255 and 1223 (Tables 1 and 3, FIGS. 1 and 5).
Surprisingly, four further polymorphs, a monohydrate, a dihydrate, a DMF/water solvate and a di-dimethyl sulphoxide solvate, and also a triacetic acid solvate of the compound of the formula (I) were found. The compound of the formula (I) in polymorph II melts at approx. 253.degree. C.; the compound of the formula (I) in polymorph III has a melting point of approx. 127.degree. C. Polymorph IV of the compound of the formula I melts at a temperature of 246.degree. C., while polymorph V has a melting point of 234.degree. C. The monohydrate contains approx. 4.1% water, the dihydrate contains 7.8% water, the DMF/water solvate contains 13.6% dimethylformamide and 0.9% water, the di-DMSO solvate contains 26.8% dimethyl sulphoxide and the triacetic acid solvate contains 29.7% acetate. Each of the crystalline forms mentioned has a characteristic x-ray diffractogram and IR spectrum (Tables 2 and 3, FIGS. 1-4, 6-14).
| TABLE 1 |
| X-ray diffractometry for polymorphs I to V |
FIGURES
FIG. 1: IR spectrum of the compound of the formula (I) in polymorphs I, II and III
FIG. 2: IR spectrum of the compound of the formula (I) in polymorphs IV, V and as the triacetic acid solvate
FIG. 3: IR spectrum of the compound of the formula (I) as the di-DMSO solvate, DMF/water solvate and monohydrate
FIG. 4: IR spectrum of the compound of the formula (I) as the dihydrate
FIG. 5: X-ray diffractogram of the compound of the formula (I) in polymorph I
FIG. 6: X-ray diffractogram of the compound of the formula (I) in polymorph II
FIG. 7: X-ray diffractogram of the compound of the formula (I) in polymorph III
FIG. 8: X-ray diffractogram of the compound of the formula (I) in polymorph IV
FIG. 9: X-ray diffractogram of the compound of the formula (I) in polymorph V
FIG. 10: X-ray diffractogram of the compound of the formula (I) as the triacetic acid solvate
FIG. 11: X-ray diffractogram of the compound of the formula (I) as the di-DMSO solvate
FIG. 12: X-ray diffractogram of the compound of the formula (I) as the DMF-water solvate
FIG. 13: X-ray diffractogram of the compound of the formula (I) as the monohydrate
FIG. 14: X-ray diffractogram of the compound of the formula (I) as the dihydrate
Example 11A
2-[5-Fluoro-1-(2-fluorobenzyl)-1H-pyrazolo[3,4-b]pyridin-3-yl]pyrimidine-4,5,6-triamine
Working Examples
Example 1
Methyl {4,6-diamino-2-[5-fluoro-1-(2-fluorobenzyl)-1H-pyrazolo[3,4-b]pyridin-3-yl]pyrimidin-5-yl}carbamate
| Patent ID | Title | Submitted Date | Granted Date |
|---|---|---|---|
| US2017273977 | SUBSTITUTED 5-FLUORO-1H-PYRAZOLOPYRIDINES AND THEIR USE |
2016-11-10
|
|
| US8921377 | Substituted 5-fluoro-1H-pyrazolopyridines and their use |
2013-03-27
|
2014-12-30
|
| US8420656 | Substituted 5-fluoro-1H-pyrazolopyridines and their use |
2012-01-26
|
|
| US9096592 | BICYCLIC AZA HETEROCYCLES, AND USE THEREOF |
2011-08-31
|
2014-05-29
|
| US2014038956 | Use of sGC stimulators, sGC activators, alone and combinations with PDE5 inhibitors for the treatment of systemic sclerosis (SSc). |
2011-05-24
|
2014-02-06
|
REFERENCES
1: Gheorghiade M, Greene SJ, Butler J, Filippatos G, Lam CS, Maggioni AP, Ponikowski P, Shah SJ, Solomon SD, Kraigher-Krainer E, Samano ET, Müller K, Roessig L, Pieske B; SOCRATES-REDUCED Investigators and Coordinators. Effect of Vericiguat, a Soluble Guanylate Cyclase Stimulator, on Natriuretic Peptide Levels in Patients With Worsening Chronic Heart Failure and Reduced Ejection Fraction: The SOCRATES-REDUCED Randomized Trial. JAMA. 2015 Dec 1;314(21):2251-62. doi: 10.1001/jama.2015.15734. PubMed PMID: 26547357.
2: Tschöpe C, Pieske B. [New therapy concepts for heart failure with preserved ejection fraction]. Herz. 2015 Apr;40(2):194-205. doi: 10.1007/s00059-015-4210-x. German. PubMed PMID: 25737289.
3: Stasch JP, Schlossmann J, Hocher B. Renal effects of soluble guanylate cyclase stimulators and activators: a review of the preclinical evidence. Curr Opin Pharmacol. 2015 Apr;21:95-104. doi: 10.1016/j.coph.2014.12.014. Epub 2015 Jan 31. Review. PubMed PMID: 25645316.
4: Pieske B, Butler J, Filippatos G, Lam C, Maggioni AP, Ponikowski P, Shah S, Solomon S, Kraigher-Krainer E, Samano ET, Scalise AV, Müller K, Roessig L, Gheorghiade M; SOCRATES Investigators and Coordinators. Rationale and design of the SOluble guanylate Cyclase stimulatoR in heArT failurE Studies (SOCRATES). Eur J Heart Fail. 2014 Sep;16(9):1026-38. doi: 10.1002/ejhf.135. Epub 2014 Jul 24. PubMed PMID: 25056511.
5: Stasch JP, Evgenov OV. Soluble guanylate cyclase stimulators in pulmonary hypertension. Handb Exp Pharmacol. 2013;218:279-313. doi: 10.1007/978-3-642-38664-0_12. Review. PubMed PMID: 24092345.
////////////////Vericiguat, BAY 102, BAY-1021189, MK-1242, ベルイシグアト , PHASE 3, MERCK, BAYER
COC(=O)NC1=C(N=C(N=C1N)C2=NN(C3=NC=C(C=C23)F)CC4=CC=CC=C4F)N

