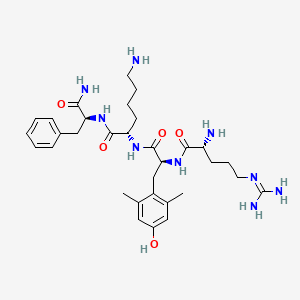

Elamipretide

H-D-Arg-Tyr(2,6-diMe)-Lys-Phe-NH2
D-arginyl-2,6-dimethyl-L-tyrosyl-L-lysyl-L-phenylalaninamide
(2S)-6-amino-2-[[(2S)-2-[[(2R)-2-amino-5-(diaminomethylideneamino)pentanoyl]amino]-3-(4-hydroxy-2,6-dimethylphenyl)propanoyl]amino]-N-[(2S)-1-amino-1-oxo-3-phenylpropan-2-yl]hexanamide
CAS 736992-21-5
Chemical Formula: C32H49N9O5
Molecular Weight: 639.8
- Elamipretide
- bendavia
- UNII-87GWG91S09
- 736992-21-5
- MTP 131
- RX 31
- SS 31
- 87GWG91S09
- L-Phenylalaninamide, D-arginyl-2,6-dimethyl-L-tyrosyl-L-lysyl-
- SS-31 peptide
- Arg-Dmt-Lys-Phe-NH2
- D-Arg-Dmt-Lys-Phe-NH2
- SS31 peptide
- Elamipretide [USAN:INN]
- MTP-131
- Elamipretide (USAN/INN)
- arginyl-2,’6′-dimethyltyrosyl-lysyl-phenylalaninamide
- CHEMBL3833370
- SCHEMBL15028020
- CTK2H1007
Elamipretide is a cardiolipin peroxidase inhibitor and mitochondria-targeting peptide, Improves Left Ventricular and Mitochondrial Function. In vitro: Elamipretide significantly increases enzymatic activities of both complexes to near normal levels.
Background Information
Elamipretide (also known as SS-31 and Bendavia)[1][2] is a small mitochondrially-targeted tetrapeptide (D-Arg-dimethylTyr-Lys-Phe-NH2) that appears to reduce the production of toxic reactive oxygen species and stabilize cardiolipin.[3]
Stealth Peptides, a privately held company, was founded in 2006 to develop intellectual property licensed from several universities including elamipretide; it subsequently changed its name to Stealth BioTherapeutics.[4][5]
Acute coronary syndrome; Age related macular degeneration; Cardiac failure; Corneal dystrophy; Diabetic macular edema; Lebers hereditary optic atrophy
- Originator Stealth Peptides
- Developer Stealth BioTherapeutics
- Class Eye disorder therapies; Ischaemic heart disorder therapies; Oligopeptides; Peptides; Small molecules
- Mechanism of Action Free radical scavengers; Mitochondrial permeability transition pore inhibitors
- Phase II/III Barth syndrome
-
- Phase II Acute kidney injury; Corneal disorders; Heart failure; Leber’s hereditary optic atrophy; Mitochondrial disorders; Reperfusion injury
- Phase I/II Diabetic macular oedema; Dry age-related macular degeneration; Mitochondrial myopathies
- Phase I Age-related macular degeneration
- No development reported Chronic heart failure; Diabetes mellitus; Eye disorders; Neurodegenerative disorders
Most Recent Events
- 29 Jun 2017 Initial efficacy and adverse events data from phase II MMPOWER-2 trial in Mitochondrial-myopathies released by Stealth
- 02 Jun 2017 Stealth BioTherapeutics completes a phase II trial in Heart failure in Germany and Serbia (SC) (NCT02814097)
- 01 May 2017 Phase-II/III clinical trials in Barth syndrome (In children, In adolescents, In adults, In the elderly) in USA (SC) (NCT03098797)
Novel crystalline salt (eg hydrochloride, mesylate and tosylate salts) forms of D-Arg-Dmt-Lys-Phe-NH2 (referred to as MTP-131 or elamipretide ) and composition comprising them are claimed. See WO2016190852 , claiming therapeutic compositions including chromanyl compounds, variants and analogues and uses thereof. Stealth BioTherapeutics (formerly known as Stealth Peptides) is developing elamipretide, which targets mitochondria, for the potential iv/sc treatment of cardiac reperfusion injury, acute coronary syndrome, acute kidney injury, mitochondrial myopathy, skeletal muscle disorders and congestive heart failure.
Also, the company is developing an oral formulation of elamipretide , which targets mitochondria and reduces the production of excess reactive oxygen species, for treating chronic heart failure. In January 2015, a phase II trial was ongoing . In July 2016, a phase II trial was initiated in Latvia, Spain and Hungary .
Further, the company is developing an ophthalmic formulation of elamipretide , a mitochondria targeting peptide, for treating ocular diseases including diabetic macular edema, age-related macular degeneration and fuchs’ corneal endothelial dystrophy and Leber’s hereditary optic neuropathy.
In April 2016, a phase II trial was initiated for LHON . Family members of the product case of elamipretide ( WO2007035640 ) hold protection in the EU until 2026 and expires in the US in 2027 with US154 extension.
Acute coronary syndrome; Age related macular degeneration; Cardiac failure; Corneal dystrophy; Diabetic macular edema; Lebers hereditary optic atrophy
SYNTHESIS

NEXT………………………



PATENT 2
ELAMIPRETIDE BY STEALTH
WO-2017156403
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2017156403&recNum=1&maxRec=&office=&prevFilter=&sortOption=&queryString=&tab=PCTDescription

; MTP-131; D-Arg-Dmt-Lys-Phe-Nth). Compound
1 has been shown to affect the mitochondrial disease process by helping to protect organs from oxidative damage caused by excess ROS production and to restore normal ATP production.
PATENT
US 20110082084
WO 2011091357
WO 2012129427
WO 2013059071
WO 2013126775
US 20140378396
US 20140093897
WO 2015134096
WO 2015100376
WO 2015060462
US 20150010588
PATENNT
WO 2015197723
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015197723
PROCESS FOR PREPARING
D-ARGINYL-2,6-DIMETHYL-L-TYROSYL-L-LYSYL-L-PHENYLALANINAMIDE
TECHNICAL FIELD
The invention relates to a process for solution-phase synthesis of D- Arginyl-2,6-dimethyl-L-tyrosyl-L-lysyl-L-phenylalaninamide (abbreviated H-D-Arg-(2,6-Dimethyl)Tyr-L-Lys-L-Phe-NH2, development code SS-31 , MTP-131 , X-31) of Formula (I), an active ingredient developed by Stealth BioTherapeutics under the investigational drug brand names Bendavia® and Ocuvia®, for both common and rare diseases including a mitochondrial targeted therapy for ischemia reperfusion injury.

Formula (I)
BACKGROUND
The product belongs to the class of so-called “Szeto-Schiller peptides”. Szeto-Sciller peptides or “SS peptides” are small, aromatic-cationic, water soluble, highly polar peptides, such as disclosed in US 6703483 and US 7576061 , which can readily penetrate cell membranes. The aromatic-cationic peptides include a minimum of two amino acids, and preferably include a minimum of four amino acids, covalently joined by peptide bonds. The maximum number of amino acids is about twenty amino acids covalently joined by peptide bonds. As described by EP 2012/2436390, optimally, the number of amino acids present in the SS peptides is four.
Bendavia® is being tested for the treatment of ischemia reperfusion injury in patients with acute myocardial infarction (AMI), for the treatment of acute kidney injury (AKI) and renal microvascular dysfunction in hypertension, for the treatment of skeletal muscle dysfunction, for the treatment of mitochondrial myopathy and for the treatment of chronic heart failure. Trials are ongoing to assess the Ocuvia’s potential to treat Leber’s Hereditary Optic Neuropathy (LHON) a devastating inherited disease that causes sudden blindness, often in young adults.
Mitochondria are the cell’s powerhouse, responsible for more than 90% of the energy our bodies need to sustain life and support growth. The energetics from mitochondria maintains healthy physiology and prevents disease. In many common and rare diseases, dysfunctional mitochondria are a key component of disease progression.
D-Arginyl-2,6-dimethyl-L-tyrosyl-L-lysyl-L-phenylalaninamide is a cell-permeable and mitochondria-targeted peptide that showed antioxidant activity and was concentrated in the inner mitochondrial membrane. Compound (< 1 nM) significantly reduced intracellular reactive oxygen species, increased mitochondrial potential and prevented tBHP-induced apoptosis in both N2A and SH-SY5Y neuronal cell lines. In rats, intraperitoneal treatment (1 and 3 mg/kg) 1 day prior to unilateral ureteral obstruction and every day thereafter for 14 days significantly decreased tubular damage, macrophage infiltration and interstitial fibrosis. Compound (3 mg/kg i.p. qd for 2 weeks) also prevented apoptosis and insulin reduction in mouse pancreatic islets caused by streptozotocin.
Further studies performed in a G93A mouse model of amyotrophic lateral sclerosis (ALS) demonstrated that the compound (5 mg/kg/day i.p. starting at 30 days of age) led to a significant delay in disease onset.
Potentially useful for the treatment of ALS and may be beneficial in the treatment of aging and diseases associated with oxidative stress.
In the last few years the peptide H-D-Arg-(2,6-Dimethyl)Tyr-L-Lys-L-Phe-NH2, shown in Fig 1 , and its therapeutic activity have been disclosed and
claimed by in several patent applications.
EP 2436390, US 201 10245182 and US 201 10245183 claim topical anesthetic compositions for application to the skin for pain management or anti-skin aging agents, respectively, comprising Szeto-Schiller peptides; SS-31 is specifically claimed as active ingredient. Sequence of solid-phase synthesis is indicated as the preferred preparation process.
US 7718620 claims a process of treating or preventing ischemia-reperfusion injury of the kidney in a mammal by administrating an effective amount of an aromatic-cationic peptide. SS-31 is specifically claimed as active ingredient.
WO2005/001023 discloses a generical process and carrier complexes for delivering molecules to cells comprising a molecule and an aromatic cationic peptide of type D-Arg-Dmt-Lys-Phe-NH2. The tetrapeptide SS-31 is
specifically claimed as product useful for the process at claim 18.
WO2012/1741 17 and WO2014/210056 claim therapeutic compositions based on SS peptides and the aromatic-cationic peptide D-Arg-Dmt-Lys-Phe-NH2 as active agent.
WO 2013/086020, WO 2004/070054 and WO 2005/072295 provide processes for preventing mithochondrial permeability transition and reducing oxidative damage in a mammal, a removed organ, or a cell in need thereof and specifically claims the process wherein the peptide does not have mu-opioid receptor agonist activity, i.e., D-Arg-Dmt-Lys-Phe-NH2.
WO 2009/108695 discloses a process for protecting a kidney from renal injury which may be associated with decreased or blocked blood flow in the subject’s kidney or exposure to a nephrotoxic agent, such as a radiocontrast dye. The processes include administering to the subject an effective amount of an aromatic-cationic peptide to a subject in need thereof and one of the selected peptide is D-Arg-Dmt-Lys-Phe-NH2.
US 6703483 discloses a detailed procedure for the preparation of novel analogs of DALDA [H-Tyr-D-Arg-Phe-Lys-NH2], namely H-Dmt-D-Arg-Phe-Lys-NH2 using the solid-phase techniques and /?-methylbenzhydrylamine
resin and protocols that have been extensively used by inventor’s laboratory.
Most prior art processes for preparing the compound typically comprise conventionally performed peptide solid-phase synthesis with further purification by chromatography in order to obtain the requested purity for therapeutic use.
It is well known that solid-phase synthesis followed by chromatographic purification is time consuming, very expensive and very difficult to be scaled up on industrial scale, so the need of developing a process for large scale production is obvious. The compound is isolated as organic acid salt, as acetate or trifluoro acetate.
eddy et al., Adv. Exp. Med. Biol, 2009, 61 1 , 473 generally describes the liquid-phase synthesis of antioxidant peptides of Figure 1 and similar others (SS-02, SS-20), involving routinely used side chain protecting groups for amino acid building blocks. The guanidine group was protected with NO2 and the ε-ΝΗ2 of Lys was protected by Cbz or 2-Cl-Cbz. These peptides were
synthesized using Boc/Cbz chemistry and BOP reagent coupling. Starting with the C-terminal Lys residue protected as H-Lys(2-Cl-Cbz)-NH2, (prepared
from the commercially available Boc-Lys(2-Cl-Cbz)-OH in two steps by amidation with NH4HCO3 in the presence of DCC/HOBt following a literature procedure [Ueyama et all, Biopolymers, 1992, 32, 1535, PubMed: 1457730], followed by exposure to TFA). Selective removal of the 2-Cl-Cbz in the
presence of the NO2 group was accomplished using catalytic transfer hydrogenolysis (CTH) [Gowda et al., Lett. Pept. Sci., 2002, 9, 153].
A stepwise procedure by standard solution peptide synthesis for preparation of potent μ agonist [DmtJDALDA and its conversion into a potent δ antagonist H-Dmt-Tic-Phe-Lys(Z)-OH by substitution of D-Arg with Tic to enhance the δ opioid agonist activity is described by Balboni et al., J. Med.
Chem., 2005, 48, 5608. A general synthetic procedure for a similar tetrapeptide ([Dmt-D-Arg-Phe-Lys-NH2 is described by Ballet et al., J. Med.
Chem. 2011, 54, 2467.
Similar DALDA analog tetrapeptides were prepared by the manual solid-phase technique using Boc protection for the a-amino group and DIC/HOBt or HBTU/DIEA as coupling agent [Berezowska et al., J. Med. Chem., 2009, 52, 6941 ; Olma et al., Acta Biochim. Polonica, 2001, 48, 4, 1 121 ; Schiller at al., Eur. J. Med. Chem., 2000, 35, 895].
Despite the high overall yield in the solid-phase approach, it has several drawbacks for the scale-up process such as:
a. the application of the highly toxic and corrosive hydrogen fluoride for cleavage of the peptide from the resin,
b. low loading (0.3-0.35 mmol/g of resin) proved necessary for successful end-step, and
c. use of excess amounts of reagents (3-fold of DIC, 2.4-fold of HOBt, etc.) on each step [ yakhovsky et al., Beilstein J. Org. Chem., 2008, 4(39), 1 , doi: 10.376/bjoc.4.39]
SUMMARY
The invention relates to a more efficient process avoiding either solid-phase synthesis or chromatographic purification, more suitable for large scale production. The process of the invention is described in Scheme A.
The following abbreviations are used:
Dmt = 2,6-dimethyl tyrosine; Z= benzyloxycarbonyl; MeSO3H = methane sulphonic acid; Boc = Tert-butyloxycarbonyl; NMM = N-methyl morpholine; TBTU= N,N,N’,N’-Tetramethyl-O-(benzotriazol- l-yl)uronium tetrafluoroborate; DMF = dimethyl formamide; TFA = trifluoroacetic acid
Scheme A shows the process for the solution phase synthesis of peptide
1 for assembly of the tetrapeptide backbone using O-Benzyl (Bzl) group and benzyloxycarbonyl (Z) group respectively, as the temporary protection for amino acids’ N-termini (Scheme Figure 2), followed by a final catalytic hydrogenolysis. The final product is isolated as organic acid salt, for example, acetic acid salt.
H-Phe-NH 2 + Boc-Lys(Z)-OH

Boc-Lys(Z)-Phe-NH 2
(IV)
(V) I MeS03H/CH2CI2
Boc-DMTyr(Bzl)-OH + MeS03H.H-Lys(Z)-Phe-NH 2
(

Boc-DMTyr(Bzl)-Lys(Z)-Phe-NH 2
(VIII)
I MeS03H/CH2CI2
Z-D-Arg-ONa + H-DMTyr(Bzl)-Lys(Z)-Phe-NH 2.MeS03H
(X) (IX)
TBTU/NMM/DMF
Z-D-Arg-DMTyr(Bzl)-Lys(Z)-Phe-NH
(XI)
I H2, Pd/C
X ACOH
H-D-Arg-DMTyr-Lys-Phe-NH
(I)
Scheme A
This process is a notable improvement with respect to the prior art and its advantages can be summarized as follows:
• The synthesis is performed in liquid phase allowing the scale up on industrial scale without need of special equipment; · The selection of the protecting group in the building blocks allows a straightforward synthesis with very simple deprotection at each step and minimize the formation of undesired by-product;
• Each intermediate can be crystallized allowing removal of impurities which are not transferred to the following step;
· The purity of each intermediate is very high and usually close to
99%.
EXAMPLES
Example 1: Preparation of Boc-Lys(Z)-Phe-NH2
Charge 200 mL of DMF, 44 g of Boc-Lys(Z)-OH and 15.6 g of H-Phe-NH2 in a flask. Stir the mixture at room temperature for 10 min. Add 19.2 g of
N-methylmorpholine and 32.1 g of TBTU successively at room temperature. Stir the mixture at room temperature for 1 h. Add 500 mL of water into the reaction mixture to precipitate the product at room temperature. Filter the mixture to isolate the solid product and wash the filter cake with water.
Transfer the filter cake into a flask containing 360 mL of ethyl acetate and heat the mixture at 50°C till all the solid is dissolved. Separate the organic phase of product and discard the small aqueous phase. Concentrate the organic phase at 40~45°C and under vacuum to remove the solvent till lots of solid is formed. Filter the residue to isolate the solid product. Transfer the filter cake into a flask containing 2000 mL of MTBE and heat the mixture at refluxing for 20 min. Then, cool down the mixture to room temperature. Filter the mixture to isolate the solid product. Dry the filter cake at 30 °C and under vacuum to give 35 g of solid product.
Example 2: Preparation of H-Lys(Z)-Phe-NH2.MeSC>3H
Charge 26.3 g of Boc-Lys(Z)-Phe-NH2, 200 mL of methylene chloride
and 9.6 g of methanesulfonic acid. Stir the mixture at 15-20 °C for 18 h. Add 100 mL of MTBE into the mixture and stir at 15-20 °C for 1 h. Filter the mixture to isolate the solid product. Dry the wet cake in air at room temperature to give 26.4 g of white solid product.
Example 3: Preparation of Boc-DMeTyr(Bzl)-Lys(Z)-Phe-NH2
Charge 8.4 g of Boc-DMeTyr(Bzl)-OH, 1 1 g of H-Lys(Z)-Phe-NH2.MeSO3H, 7.4 g of TBTU and 80 mL of THF in a flask. Stir the mixture
at room temperature for 15 min, and then cool down to 10°C. Add 6.36 g of N-methylmorpholine and stir the mixture at 20-25°C for 3 h. Add the reaction mixture into a flask containing 240 mL of water. Add 32 mL of methylene chloride into the mixture obtained in the previous operation of. Stir the resultant mixture at room temperature for 20 min. Filter the mixture to isolate the solid product and wash the filter cake with acetone (300 mL X 2). Dry the filter cake in air at room temperature to give 14.3 g of white solid product.
Example 4: Preparation of H-DMeTyr(Bzl)-Lys(Z)-Phe-NH2.MeS03H
Charge 14 g of Boc-BMeTyr(Bzl)-Lys(Z)-Phe-NH2, 280 mL of methylene chloride and 3.3 g of methanesulfonic acid in a flask. Stir the mixture at 18 ~ 22 °C for 10 h. Add 560 mL of heptanes into the mixture and stir the mixture at room temperature for 30 min. Filter the mixture to isolate the solid product. Dry the wet cake in air at room temperature to give 14 g of white solid product.
Example 5: Preparation of Z-D-Arg-DMeTyr(Bzl)-Lys(Z)-Phe-NH2
Charge 6.34 g of Z-D-Arg-ONa, 100 mL of DMF and 2.0 g of methanesulfonic acid in a flask. Stir the mixture at room temperature till a clear solution was formed. Add 14 g of H-DMeTyr(Bzl)-Lys(Z)-Phe-NH2.MeSO3H and cool down the mixture to 10°C. Add 6.15 g of TBTU and
9.67 g of N-methylmorpholine successively. Stir the mixture at room temperature for 4 h. Add aqueous solution of LiOH prepared by dissolving 2.9 g of LiOH.L O in 8 mL of water. Stir the mixture for 30 min. Add the resultant mixture slowly into a flask containing 420 mL of water under stirring. Add 56 mL of methylene chloride into the mixture. Filter the mixture to isolate the solid product. Transfer the filter cake into a flask containing 150 mL of acetic acid, and heat the mixture at 35-40 °C till most of the solid was dissolved. Add 450 mL of MTBE into the mixture and cool down the mixture under stirring to room temperature. Filter the mixture to isolate the solid product. Dry the filter cake in air at room temperature to give 17.3 g of the white solid product.
Example 6 Preparation of H-D-Arg-DMeTyr-Lys-Phe-NH2.3AcOH
Charge 2.0 g of Z-D-Arg-DMeTyr(Bzl)-Lys(Z)-Phe-NH2, 20 mL of acetic acid and 5% Pd/C catalyst (which is obtained by washing 5.0 g of 5% Pd/C containing 60% of water with 30 mL of acetic acid) in a flask. Change the atmosphere of the flask with hydrogen. Stir the mixture at room temperature and pressure of 1 atm of hydrogen for 2 h. Filter the mixture to remove the Pd/C catalyst and wash the filter cake with 10 mL of acetic acid. Combine the filtrate and washing solution and concentrate the solution at 20°C and under vacuum to remove most the solvent. Add 100 mL of acetonitrile into the residue and stir the mixture at room temperature for 20 min. Filter the mixture to isolate the solid product. Dry the filter cake at room temperature and under vacuum to give 0.7 g of the white product.
PATENT
WO 2016001042
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2016001042&recNum=1&maxRec=&office=&prevFilter=&sortOption=&queryString=&tab=PCTDescription
References
| Patent ID
|
Patent Title
|
Submitted Date
|
Granted Date
|
| US2014294796 |
AROMATIC-CATIONIC PEPTIDES AND USES OF SAME |
2012-12-05 |
2014-10-02 |
| US2016264623 |
TETRAPEPTIDE COMPOUND AND METHOD FOR PRODUCING SAME |
2014-10-23 |
2016-09-15 |
| US2017081363 |
PHARMACEUTICALLY RELEVANT AROMATIC-CATIONIC PEPTIDES |
2014-12-23 |
|
| US2016340389 |
PHARMACEUTICALLY RELEVANT AROMATIC-CATIONIC PEPTIDES |
2014-12-23 |
|
| US2017129920 |
Process for Preparing D-Arginyl-2, 6-Dimethyl-L-Tyrosyl-L-Lysyl-L-Phenylalaninamide |
2015-06-24 |
REFERENCES
1: Alam NM, Mills WC 4th, Wong AA, Douglas RM, Szeto HH, Prusky GT. A mitochondrial therapeutic reverses visual decline in mouse models of diabetes. Dis Model Mech. 2015 Jul 1;8(7):701-10. doi: 10.1242/dmm.020248. Epub 2015 Apr 23. PubMed PMID: 26035391; PubMed Central PMCID: PMC4486862.
2: Szeto HH, Birk AV. Serendipity and the discovery of novel compounds that restore mitochondrial plasticity. Clin Pharmacol Ther. 2014 Dec;96(6):672-83. doi: 10.1038/clpt.2014.174. Epub 2014 Sep 4. Review. PubMed PMID: 25188726; PubMed Central PMCID: PMC4267688.
3: Dai W, Shi J, Gupta RC, Sabbah HN, Hale SL, Kloner RA. Bendavia, a mitochondria-targeting peptide, improves postinfarction cardiac function, prevents adverse left ventricular remodeling, and restores mitochondria-related gene expression in rats. J Cardiovasc Pharmacol. 2014 Dec;64(6):543-53. PubMed PMID: 25165999.
4: Eirin A, Ebrahimi B, Zhang X, Zhu XY, Woollard JR, He Q, Textor SC, Lerman A, Lerman LO. Mitochondrial protection restores renal function in swine atherosclerotic renovascular disease. Cardiovasc Res. 2014 Sep 1;103(4):461-72. doi: 10.1093/cvr/cvu157. Epub 2014 Jun 19. PubMed PMID: 24947415; PubMed Central PMCID: PMC4155472.
5: Liu S, Soong Y, Seshan SV, Szeto HH. Novel cardiolipin therapeutic protects endothelial mitochondria during renal ischemia and mitigates microvascular rarefaction, inflammation, and fibrosis. Am J Physiol Renal Physiol. 2014 May 1;306(9):F970-80. doi: 10.1152/ajprenal.00697.2013. Epub 2014 Feb 19. PubMed PMID: 24553434.
6: Brown DA, Hale SL, Baines CP, del Rio CL, Hamlin RL, Yueyama Y, Kijtawornrat A, Yeh ST, Frasier CR, Stewart LM, Moukdar F, Shaikh SR, Fisher-Wellman KH, Neufer PD, Kloner RA. Reduction of early reperfusion injury with the mitochondria-targeting peptide bendavia. J Cardiovasc Pharmacol Ther. 2014 Jan;19(1):121-32. doi: 10.1177/1074248413508003. Epub 2013 Nov 28. PubMed PMID: 24288396; PubMed Central PMCID: PMC4103197.
7: Birk AV, Chao WM, Bracken C, Warren JD, Szeto HH. Targeting mitochondrial cardiolipin and the cytochrome c/cardiolipin complex to promote electron transport and optimize mitochondrial ATP synthesis. Br J Pharmacol. 2014 Apr;171(8):2017-28. doi: 10.1111/bph.12468. PubMed PMID: 24134698; PubMed Central PMCID: PMC3976619.
8: Szeto HH. First-in-class cardiolipin-protective compound as a therapeutic agent to restore mitochondrial bioenergetics. Br J Pharmacol. 2014 Apr;171(8):2029-50. doi: 10.1111/bph.12461. Review. PubMed PMID: 24117165; PubMed Central PMCID: PMC3976620.
9: Zhao WY, Han S, Zhang L, Zhu YH, Wang LM, Zeng L. Mitochondria-targeted antioxidant peptide SS31 prevents hypoxia/reoxygenation-induced apoptosis by down-regulating p66Shc in renal tubular epithelial cells. Cell Physiol Biochem. 2013;32(3):591-600. doi: 10.1159/000354463. Epub 2013 Sep 6. PubMed PMID: 24021885.
10: Dai DF, Hsieh EJ, Chen T, Menendez LG, Basisty NB, Tsai L, Beyer RP, Crispin DA, Shulman NJ, Szeto HH, Tian R, MacCoss MJ, Rabinovitch PS. Global proteomics and pathway analysis of pressure-overload-induced heart failure and its attenuation by mitochondrial-targeted peptides. Circ Heart Fail. 2013 Sep 1;6(5):1067-76. doi: 10.1161/CIRCHEARTFAILURE.113.000406. Epub 2013 Aug 9. PubMed PMID: 23935006; PubMed Central PMCID: PMC3856238.
/////////////////////Elamipretide, SS-31, Bendavia, PEPTIDE
CC1=CC(=CC(=C1CC(C(=O)NC(CCCCN)C(=O)NC(CC2=CC=CC=C2)C(=O)N)NC(=O)C(CCCN=C(N)N)N)C)O

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....