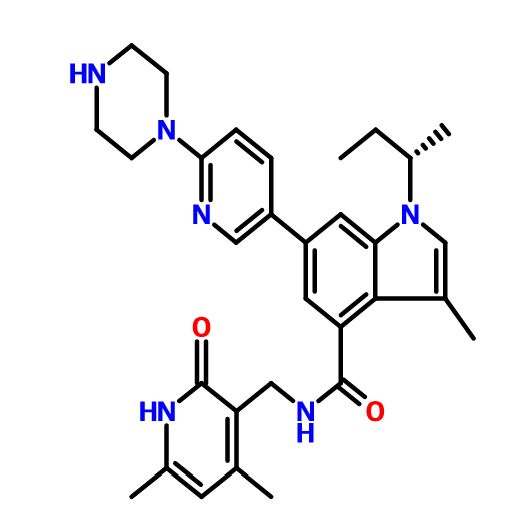
GSK-2816126
N-[(1,2-Dihydro-4,6-dimethyl-2-oxo-3-pyridinyl)methyl]-3-methyl-1-[(1S)-1-methylpropyl]-6-[6-(1-piperazinyl)-3-pyridinyl]-1H-indole-4-carboxamide, GSK 126, GSK 2816126, GSK 2816126A
N-[(4,6-Dimethyl-2-oxo-1,2-dihydro-3-pyridinyl)methyl]-3-methyl-1-((1S)-1-methylpropyl)-6-[6-(1-piperazinyl)-3-pyridinyl]-1H-indole-4-carboxamide
Phase I
| Formula |
C31H38N6O2
|
| Formula Wt. |
526.67
|
An histone-lysine n-methyltransferase EZH2 inhibitor potentially for the treatment of B-cell lymphoma.

Research Code GSK-2816126; GSK-126; 2816126
CAS No. 1346574-57-9
- Originator GlaxoSmithKline
- Class Antineoplastics
- Mechanism of Action EZH2 enzyme inhibitors; Histone modulators
- Phase I Diffuse large B cell lymphoma; Follicular lymphoma
- Preclinical Acute myeloid leukaemia
Most Recent Events
- 31 Mar 2014 Phase-I clinical trials in Follicular lymphoma (Second-line therapy or greater) in USA and United Kingdom (IV)
- 31 Mar 2014 Phase-I clinical trials in Diffuse large B cell lymphoma (Second-line therapy or greater) in USA and United Kingdom (IV)
- 16 Jan 2014 Preclinical trials in Diffuse large B cell lymphoma & Follicular lymphoma in United Kingdom (IV)
GSK-126 is an inhibitor of mutant EZH2, a histone methyltransferase (HMT) that exhibits point mutations at key residues Tyr641 and Ala677; this compound does not appreciably affect WT EZH2. EZH2 is responsible for modulating expression of a variety of genes. GSK-126 competes with cofactor S-adenylmethionine (SAM) for binding to EZH2. GSK-126 displays anticancer chemotherapeutic activity by inhibiting proliferation in in vitro and in vivo models of diffuse large B-cell lymphoma.

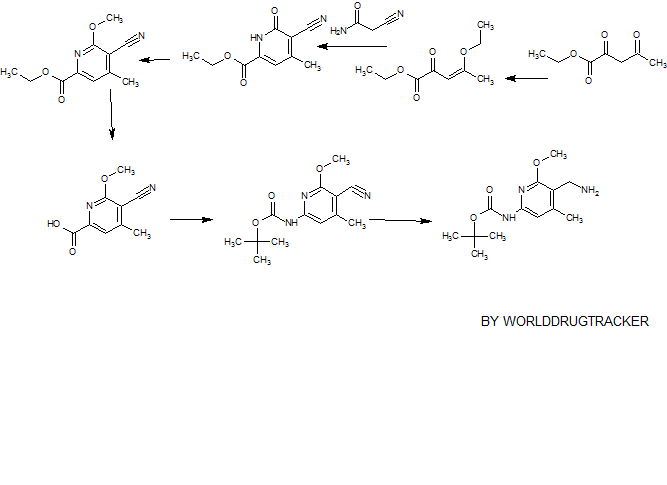
SYNTHESIS


PATENT
CN 105541801
https://www.google.com/patents/CN105541801A?cl=zh
Example 79: Add (S) in a three-necked flask 100 Qiu – bromo – Shu – (isobutyl) – N – ((4,6-dimethyl-2-oxo -l, 2- dihydropyridin-3-yl) methyl) -3-methyl-1 hydrogen – indole carboxamide (365mg, 0.82mmol), 2- (piperazin-1-yl) pyridine-5-boronic acid pinacol ester (309mg, 1.07mmol, 1 · 3eq), potassium phosphate (522mg, 2.46mmol, 3eq), water, and I, 4- diepoxy-hexadecane as the solvent. Then, under nitrogen was added [I, Γ- bis (diphenylphosphino) ferrocene] dichloropalladium (II) dichloromethane complex (53.9mg, 0.066mmo 1), and at 90 ° C reaction, to give the desired product after purification 400mg (92% yield). Goo NMR (500MHz, DMSO- (I6) JO.70-0 · 78 (ιή, 3H), 1.37-1.44 (m, 4H), 1.75-1.87 (m, 2H), 2.11 (s, 3H), 2.16 ( s, 3H), 2.22-2.27 (m, 3H), 2.77-2.85 (m, 4H), 3.41-3.49 (m, 4H), 4.35 (d, J = 5.31Hz, 2H), 4.56-4.68 (m, lH), 5.87 (s, 1H), 6.88 (d, J = 8.84Hz, 1H), 7.17 (d J = 1.52Hz, 1H), 7.26 (s, lH), 7.73 (d J = 1.26Hz, 1H) , 7.91 (dd, J = 8.84Hz, lH), 8.16 (t, J = 5.05Hz, lH), 8.50 (d, J = 2.53Hz, lH); 13C NMR (125MHz, DMSO- (I6) Sll .6 , 12.6,19.1, 19.9,21.7,30.4,35.9,46.3,46.9,52.4,107.6,108.2,108.5,110.6,116.9,122.6,123.8, 130.6,131.5,136.7,138.6,143.5,146.4,150.2,159.2,164.0 , 169.6.
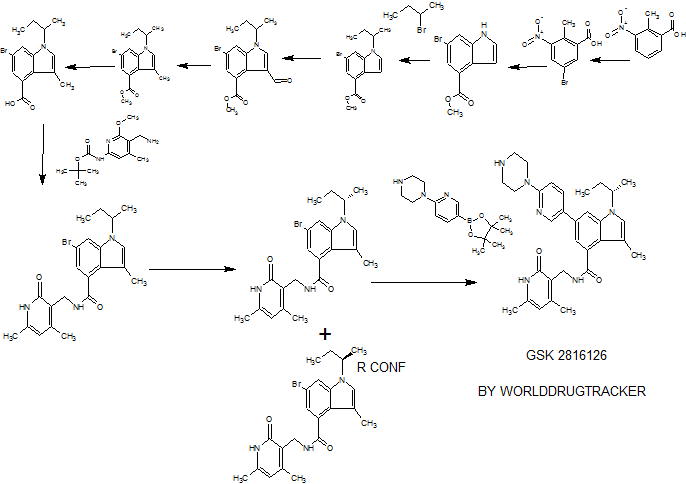
PATENT
WO 2013067296
Examples 267 and 268
(S)-6-bromo-1 -(sec-butyl)-N-((4,6-dimethyl-2-oxo-1 ,2-dihydropyridin-3-yl)methyl)-3- methyl-1 H-indole-4-carboxamide (Ex 267) and (R)-6-Bromo-1 -(sec-butyl)-N-((4,6- dimethyl-2-oxo-1 ,2-dihydropyridin-3-yl)methyl)-3-methyl-1 H-indole-4-carboxamide (Ex 268)
6-Bromo-1-(sec-butyl)-N-((4,6-dimethyl-2-oxo-1 ,2-dihydropyridin-3-yl)methy methyl-1 H-indole-4-carboxamide (racemic mixture, 1.9 g) was resolved by chiral HPLC (column : Chiralpak AD-H, 5 microns, 50 mm x 250 mm, UV detection :240 nM, flow rate: 100 mL/min, T = 20 deg C, eluent: 60:40:0.1 n-heptane:ethanol:isopropylamine
(isocratic)). For each run, 100 mg of the racemic compound was dissolved in 30 volumes (3.0 ml.) of warm ethanol with a few drops of isopropylamine added. A total of 19 runs were performed. Baseline resolution was observed for each run. The isomer that eluted at 8.3-10.1 min was collected (following concentration) as a white solid, which was dried at 50 °C (< 5 mm Hg) to afford 901 mg, and was determined to be the S isomer* (Ex. 267; chiral HPLC: >99.5% ee (no R isomer detected). The isomer that eluted at 10.8-13.0 min was collected as a white solid, which was dried at 50 °C (< 5 mm Hg) to afford 865 mg, and was determined to be the R isomer* (Ex. 268; chiral HPLC: 99.2% ee; 0.4% S isomer detected). 1H NMR and LCMS were consistent with the parent racemate.
* The absolute configuration was determined by an independent synthesis of each enantiomer from the corresponding commercially available homochiral alcohols via Mitsunobu reaction. The sterochemical assignments were also consistent by vibrational circular dichroism (VCD) analysis.
Example 269
1-(sec-butyl)-N-((4,6-dimethyl-2-oxo-1 ,2-dihydropyridin-3-yl)methyl)-3-methyl-6-(6- (piperazin-1 -yl)pyridin-3-yl)-1 -indole-4-carboxamide

Added sequentially to a reaction vial were 6-bromo-1 -(sec-butyl)-N-((4,6-dimethyl- 2-OXO-1 , 2-dihydropyridin-3-yl)methyl)-3-methyl-1 H-indole-4-carboxamide (0.15 g, 0.338 mmol), 1-(5-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)pyridin-2-yl)piperazine (0.127 g, 0.439 mmol), and potassium phosphate (tribasic) (0.287 g, 1.350 mmol), followed by 1 ,4- Dioxane (3 mL) and water (0.75 mL). The suspension was stirred under N2 degassing for 10 min., and then added PdCI2(dppf)-CH2CI2adduct (0.028 g, 0.034 mmol). The reaction vial was sealed, placed into a heat block at 95 °C, and stirred for 1.5 h. The contents were removed from heating and allowed to cool to room temperature. The aq layer was removed from bottom of the reaction vial via pipette. The reaction mixture was diluted into EtOAc (20 mL) followed by addition of 0.2 g each of Thiol-3 silicycle resin and silica gel. The volatiles were removed in vacuo and the residue dried on hi-vac for 1 h. The contents were purified by silica gel chromatography (dry loaded, eluent : A:
Dichloromethane, B: 10% (2M Ammonia in Methanol) in Chloroform, Gradient B: 8- 95%). The obtained solid was concentrated from TBME and dried in vacuum oven at 45 °C for 18 h. The product was collected as 129 mg (70%). 1H NMR (400 MHz, DMSO-d6) δ ppm 0.73 (t, J=7.33 Hz, 3H), 1.40 (d, J=6.57 Hz, 3H), 1.80 (dq, J=10.07, 7.08 Hz, 2H), 2.1 1 (s, 3H), 2.14 – 2.19 (m, 3H), 2.24 (s, 3H), 2.76 – 2.85 (m, 4H), 3.41 – 3.49 (m, 4H), 4.35 (d, J=5.05 Hz, 2H), 4.54 – 4.67 (m, 1 H), 5.87 (s, 1 H), 6.88 (d, J=8.84 Hz, 1 H), 7.17 (d, J=1.26 Hz, 1 H), 7.26 (s, 1 H), 7.73 (d, J=1.26 Hz, 1 H), 7.91 (dd, J=8.84, 2.53 Hz, 1 H), 8.16 (t, J=5.05 Hz, 1 H), 8.50 (d, J=2.53 Hz, 1 H), 1 1.48 (br. s.,1 H) ; LCMS MH+ =527.3.
Example 270
A/-[(4,6-dimethyl-2-oxo-1 ,2-dihydro-3-pyridinyl)methyl]-3-methyl-1 -[(1 S)-1 -methylpropyl]-6- [6-(1-piperazinyl)-3-pyridinyl]-1 H-indole-4-carboxamide
To a 30 mL microwave vial were added (S)-6-bromo-1 -(sec-butyl)-N-((4,6- dimethyl-2-oxo-1 ,2-dihydropyridin-3-yl)methyl)-3-methyl-1 H-indole-4-carboxamide (100 mg, 0.225 mmol), 1 -(5-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)pyridin-2-yl)piperazine (85 mg, 0.293 mmol), 1 ,2-Dimethoxyethane (DME) (3 mL), water (1.000 mL) and sodium carbonate (0.338 mL, 0.675 mmol), and the mixture was degassed for 5 min by bubbling nitrogen. PdCI2(dppf)-CH2CI2 adduct (14.70 mg, 0.018 mmol) was added and the tube was sealed. The mixture was irradiated (microwave) at 140 °C for 10 min. The mixture was concentrated and the residue was taken up into MeOH and filtered. The filtrate was purified using reverse-phase HPLC (eluent: 25%ACN/H20, 0.1 % NH4OH to 60%
ACN/H20, 0.1 % NH4OH ) to give 91 mg of product as off-white solid. 1 H NMR (400 MHz, DMSO-d6) δ ppm 0.70 – 0.78 (m, 3H), 1.37 – 1.44 (m, 3H), 1 .75 – 1.87 (m, 2H), 2.1 1 (s, 3H), 2.16 (s, 3H), 2.22 – 2.27 (m, 3H), 2.77 – 2.85 (m, 4H), 3.41 – 3.49 (m, 4H), 4.35 (d, J=5.31 Hz, 2H), 4.56 – 4.68 (m, 1 H), 5.87 (s, 1 H), 6.88 (d, J=8.84 Hz, 1 H), 7.17 (d, J=1.52 Hz, 1 H), 7.26 (s, 1 H), 7.73 (d, J=1.26 Hz, 1 H), 7.91 (dd, J=8.84, 2.53 Hz, 1 H), 8.16 (t, J=5.05 Hz, 1 H), 8.50 (d, J=2.53 Hz, 1 H); LCMS: 527.8 (MH+).
Example 271
A/-[(4,6-dimethyl-2-oxo-1 ,2-dihydro-3-pyridinyl)methyl]-3-methyl-1 -[(1 /?)-1-methylpropyl]- 6-[6-(1 -piperazinyl)-3-pyridinyl]-1 -indole-4-carboxamide
To a 30 mL microwave vial were added (R)-6-bromo-1-(sec-butyl)-N-((4,6- dimethyl-2-oxo-1 ,2-dihydropyridin-3-yl)methyl)-3-methyl-1 H-indole-4-carboxamide (100 mg, 0.225 mmol), 1 -(5-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)pyridin-2-yl)piperazine (85 mg, 0.293 mmol), 1 ,2-Dimethoxyethane (DME) (3 mL), water (1.000 mL) and sodium carbonate (0.338 mL, 0.675 mmol), and the mixture was degassed for 5 min by bubbling nitrogen. PdCI2(dppf)-CH2Cl2 adduct (14.70 mg, 0.018 mmol) was added and the tube was sealed. The mixture was irradiated (microwave) at 140 °C for 10 min. The mixture was concentrated and the residue was taken up into MeOH and filtered. The filtrate was purified using reverse-phase HPLC (eluent: 25%ACN/H20, 0.1 % NH4OH to 60%
ACN/H20, 0.1 % NH4OH ) to give 90 mg of product as off-white solid. 1 H NMR (400 MHz, DMSO-d6) δ ppm 0.73 (m, 3H), 1.41 (d, J=6.57 Hz, 3H), 1.81 (td, J=7.14, 2.91 Hz, 2H), 2.1 1 (s, 3H), 2.15 – 2.20 (m, 3H), 2.24 (s, 3H), 2.77 – 2.83 (m, 4H), 3.41 – 3.49 (m, 4H), 4.35 (d, J=5.05 Hz, 2H), 4.54 – 4.68 (m, 1 H), 5.87 (s, 1 H), 6.88 (d, J=8.84 Hz, 1 H), 7.17 (d, J=1.52 Hz, 1 H), 7.26 (s, 1 H), 7.73 (d, J=1.26 Hz, 1 H), 7.91 (dd, J=8.84, 2.53 Hz, 1 H), 8.16 (t, J=5.05 Hz, 1 H), 8.50 (d, J=2.27 Hz, 1 H); LCMS: 527.7 (MH+)
PATENT
WO 2011140324
Example 270
N-[(4,6-dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-3-methyl-l-[(15)-l-methylpropyl]-6-[6-(l-piperazinyl)-3-pyridinyl]-lH-indole-4-carboxamide

To a 30 niL microwave vial were added (S)-6-bromo-l-(sec-butyl)-N-((4,6-dimethyl-2-oxo-l,2-dihydropyridin-3-yl)methyl)-3 -methyl- lH-indole-4-carboxamide (100 mg, 0.225 mmol), l-(5-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)pyridin-2-yl)piperazine (85 mg, 0.293 mmol), 1 ,2-Dimethoxyethane (DME) (3 mL), water (1.000 mL) and sodium carbonate (0.338 mL, 0.675 mmol), and the mixture was degassed for 5 min by bubbling nitrogen. PdCi2(dppf)-CH2Ci2 adduct (14.70 mg, 0.018 mmol) was added and the tube was sealed. The mixture was irradiated (microwave) at 140 °C for 10 min. The mixture was concentrated and the residue was taken up into MeOH and filtered. The filtrate was purified using reverse-phase HPLC (eluent: 25%ACN/H20, 0.1% NH4OH to 60% ACN/H20, 0.1% NH4OH ) to give 91 mg of product as off-white solid. 1H NMR (400 MHz, DMSO-d6) δ ppm 0.70 – 0.78 (m, 3H), 1.37 – 1.44 (m, 3H), 1.75 – 1.87 (m, 2H), 2.11 (s, 3H), 2.16 (s, 3H), 2.22 – 2.27 (m, 3H), 2.77 – 2.85 (m, 4H), 3.41 – 3.49 (m, 4H), 4.35 (d, J=5.31 Hz, 2H), 4.56 – 4.68 (m, IH), 5.87 (s, IH), 6.88 (d, J=8.84 Hz, IH), 7.17 (d, J=1.52 Hz, IH), 7.26 (s, IH), 7.73 (d, J=1.26 Hz, IH), 7.91 (dd, J=8.84, 2.53 Hz, IH), 8.16 (t, J=5.05 Hz, IH), 8.50 (d, J=2.53 Hz, IH); LCMS: 527.8 (MH+).
Example 271
N-[(4,6-dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-3-methyl-l-[(li?)-l-methylpropyl]-6-[6-(l-piperazinyl)-3-pyridinyl]-l -indole-4-carboxamide

To a 30 mL microwave vial were added (R)-6-bromo-l-(sec-butyl)-N-((4,6-dimethyl-2-oxo-l,2-dihydropyridin-3-yl)methyl)-3 -methyl- lH-indole-4-carboxamide (100 mg, 0.225 mmol), l-(5-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)pyridin-2-yl)piperazine (85 mg, 0.293 mmol), 1 ,2-Dimethoxyethane (DME) (3 mL), water (1.000 mL) and sodium carbonate (0.338 mL, 0.675 mmol), and the mixture was degassed for 5 min by bubbling nitrogen. PdCl2(dppf)-CH2Cl2 adduct (14.70 mg, 0.018 mmol) was added and the tube was sealed. The mixture was irradiated (microwave) at 140 °C for 10 min. The mixture was concentrated and the residue was taken up into MeOH and filtered. The filtrate was purified using reverse-phase HPLC (eluent: 25%ACN/H20, 0.1% NH4OH to 60% ACN/H20, 0.1% NH4OH ) to give 90 mg of product as off-white solid. 1H NMR (400 MHz, DMSO-d6) δ ppm 0.73 (m, 3H), 1.41 (d, J=6.57 Hz, 3H), 1.81 (td, J=7.14, 2.91 Hz, 2H), 2.11 (s, 3H), 2.15 – 2.20 (m, 3H), 2.24 (s, 3H), 2.77 – 2.83 (m, 4H), 3.41 – 3.49 (m, 4H), 4.35 (d, J=5.05 Hz, 2H), 4.54 -4.68 (m, 1H), 5.87 (s, 1H), 6.88 (d, J=8.84 Hz, 1H), 7.17 (d, J=1.52 Hz, 1H), 7.26 (s, 1H), 7.73 (d, J=1.26 Hz, 1H), 7.91 (dd, J=8.84, 2.53 Hz, 1H), 8.16 (t, J=5.05 Hz, 1H), 8.50 (d, J=2.27 Hz, 1H); LCMS: 527.7 (MH+).
REF
Tian X, Zhang S, Liu HM, et al. Histone lysine-specific methyltransferases and demethylases in carcinogenesis: new targets for cancer therapy and prevention. Curr Cancer Drug Targets. 2013 Jun 10;13(5):558-79. PMID: 23713993.
McCabe MT, Ott HM, Ganji G, et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature. 2012 Dec 6;492(7427):108-12. PMID: 23051747.
| WO2005034845A2 * |
Jul 13, 2004 |
Apr 21, 2005 |
Supergen, Inc. |
Compositions and methods for treatment of cancer |
| WO2007053114A1 * |
Oct 30, 2006 |
May 10, 2007 |
S*Bio Pte Ltd |
Method of predicting a response to hdac inhibitors |
| WO2010090723A2 * |
Feb 2, 2010 |
Aug 12, 2010 |
University Of Georgia Research Foundation, Inc. |
Methods of inhibiting fibrogenesis and treating fibrotic disease |
| US20110035336 |
May 1, 2008 |
Feb 10, 2011 |
Yigang Cai |
Rating change for a prepaid session based on movement of a mobile device |
| US20110035340 |
Aug 7, 2009 |
Feb 10, 2011 |
Fibre-Craft Materials Corp. |
Decorating system and method of marketing and enhancing a surface area using a decorating system |
| US20110035344 |
Aug 6, 2009 |
Feb 10, 2011 |
International Business Machines Corporation |
Computing mixed-integer program solutions using multiple starting vectors |
| US20110064664 * |
Oct 8, 2008 |
Mar 17, 2011 |
The Board Of Regents Of The University Of Texas System |
Methods and compositions involving chitosan nanoparticles |
| WO2015077194A1 * |
Nov 18, 2014 |
May 28, 2015 |
Bristol-Myers Squibb Company |
Inhibitors of lysine methyl transferase |
| WO2015132765A1 * |
Mar 6, 2015 |
Sep 11, 2015 |
Glaxosmithkline Intellectual Property (No.2) Limited |
Enhancer of zeste homolog 2 inhibitors |
| WO2015141616A1 * |
Mar 16, 2015 |
Sep 24, 2015 |
第一三共株式会社 |
1,3-benzodioxole derivative |
| WO2016066697A1 * |
Oct 28, 2015 |
May 6, 2016 |
Glaxosmithkline Intellectual Property (No.2) Limited |
Enhancer of zeste homolog 2 inhibitors |
| US9051269 |
Nov 19, 2012 |
Jun 9, 2015 |
Constellation Pharmaceuticals, Inc. |
Modulators of methyl modifying enzymes, compositions and uses thereof |
| US9085583 |
Feb 11, 2013 |
Jul 21, 2015 |
Constellation—Pharmaceuticals, Inc. |
Modulators of methyl modifying enzymes, compositions and uses thereof |
| US20150344459 * |
Dec 20, 2013 |
Dec 3, 2015 |
Epizyme, Inc. |
1,4-pyridone bicyclic heteroaryl compounds |
/////////GSK-2816126, GSK-126, 2816126, 1346574-57-9, GSK 126, GSK 126, GSK 2816126, GSK 2816126A
CC=5C=C(C)NC(=O)C=5CNC(=O)c1cc(cc2c1c(C)cn2[C@@H](C)CC)c3cnc(cc3)N4CCNCC4

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....