
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
AVOSENTAN

AVOSENTAN
N-[6-Methoxy-5-(2-methoxyphenoxy)-2-(4-pyridyl)pyrimidin-4-yl]-5-methylpyridine-2-sulfonamide
5-methyl-pyridine-2-sulfonic acid [6-methoxy-5-(2-methoxy-phenoxy)-2-(pyridin-4-yl)-pyrimidin-4-yl]-amide,
5-methyl-pyridine-2-sulfonic acid [6-methoxy-5-(2-methoxy-phenoxy)-2-pyridin-4-yl-pyrimidin-4-yl]-amide,
Endothelin ETA Receptor Antagonists
M.Wt: 479.51
Formula: C23H21N5O5S
Roche (Originator)
CAS No.: 290815-26-8
- RO 67-0565
- SPP 301
- UNII-L94KSX715K
PHASE 3
CLINICAL TRIALS
http://clinicaltrials.gov/search/intervention=spp301+OR+Avosentan
SPP-301 is an oral, once-daily, second-generation endothelin ETA receptor antagonist which had been in phase III clinical development at Speedel for the treatment of diabetic nephropathy. In December 2006, the company reported that the phase III trial had been stopped based on the recommendation from the trial’s Data Safety Monitoring Board (DSMB) to stop the trial following incidence of a significant imbalance in fluid retention in patients in the study arms. Speedel reported that the compound will be evaluated for potential new clinical development for the treatment of diabetic kidney disease and other indications.
Originally developed by Roche and specifically optimized for improved liver safety, SPP-301 was licensed to Speedel in October 2000. In 2003, Speedel exercised its option to license from Roche all rights to SPP-301, including exclusive worldwide rights for the full development and commercialization of the ETA antagonist. SPP-301 has fast track designation and has undergone a special protocol assessment (SPA) by the FDA. Speedel had been studying the drug for the treatment of hypertension.
AVOSENTAN
290815-26-8 CAS
PATENTS
2. WO 2004078104
3. WO 2005113543
4. WO 2007031501
5. WO 2008077916
Dutzler R, Ernstb B, Hediger MA, Keppler D, Mohr P, Neidhart W, Märki HP.Chimia (Aarau). 2010;64(9):662-6.
………………………
INTRODUCTION
-
5-methyl-pyridine-2-sulfonic acid [6-methoxy-5-(2-methoxy-phenoxy)-2-pyridin-4-yl-pyrimidin-4-yl]-amide corresponding to the formula

is an inhibitor of endothelin receptors. WO00/52007 describes the preparation of said compound which is crystallized from Me2Cl2.
-
Own investigations have shown that there exist two distinct crystalline forms, hereinafter referred to as form A and form B, as well as a number of further solvates, in particular the methanol, ethanol, isopropanol, dichloromethane, acetone, methyl ethyl ketone and tetrahydrofuran solvates.
-
It was further surprisingly found that the thermodynamically stable crystalline form – form B – can be prepared under controlled conditions and that said form B can be prepared with a reliable method in an industrial scale, which is easy to handle and to process in the manufacture and preparation of formulations.

………………..

4,6-Dichloro-5-(2-methoxy-phenoxy)-2-(pyridin-4-yl)-pyrimidine (described in EP 0 799 209) can be transformed to the intermediate of formula (III)—according to scheme 1—on reaction with an appropriate sulfonamide of formula (II), wherein R1 is as defined in claim 1, in a suited solvent such as DMSO or DMF at room temperature or at elevated temperature and in the presence of a suited base such as potassium carbonate.


EXAMPLE 1
[0064] a) To a solution of 6.9 g sodium in MeOH (300 ml) were added 14.52 g of 5-methyl-pyridine-2-sulfonic acid [6-chloro-5-(2-methoxy-phenoxy)-2-pyridin-4-yl-pyrimidin-4-yl]-amide at RT and the mixture was refluxed for 5 days until completion of the reaction according to TLC analysis. The reaction mixture was concentrated in vacuo to half its volume upon which the crude reaction product precipitated as a sodium salt. It was filtered off by suction and dried in a high vacuum. The solid was dissolved in water, which was then made acidic by addition of acetic acid. The precipitating free sulfonamide was extracted into Me2Cl2. The organic layer was dried over Mg2SO4, concentrated on a rotary evaporator, and the crystalline solid that had formed was filtered off. It was then dried in a high vacuum for 12 h at 120° C. to give the desired 5-methyl-pyridine-2-sulfonic acid [6-methoxy-5-(2-methoxy-phenoxy)-2-pyridin-4-yl-pyrimidin-4-yl]-amide as white crystals. Melting point 225-226° C. ISN mass spectrum, m/e 478.2 (M-1 calculated for C23H21N5O5S1: 478).
[0065] C23H21N5O5S1: Calc: C 57.61; H 4.41; N 14.61; S 6.69. Found: C 57.56; H 4.38; N 14.61; S 6.83
[0066] Preparation of the starting material:
[0067] b) 11.3 g of 4,6-dichloro-5-(2-methoxy-phenoxy)-2-pyridin-4-yl)-pyrimidine and 19.66 g of 5-methylpyridyl-2-sulfonamide potassium salt (preparations described in EP 0 799 209) were dissolved in DMF (255 ml) under argon. The solution was stirred for 2 h at 40° C. until completion of the reaction according to TLC analysis. The reaction mixture was cooled to RT and the solvent removed in a high vacuum. The residue was suspended in water (850 ml), acetic acid (85 ml) was added and the mixture was stirred for 30 minutes at RT. The solid that precipitated was collected by filtration and dried in a high vacuum at 60° C. for 16 h to give 5-methyl-pyridine-2-sulfonic acid [6-chloro-5-(2-methoxy-phenoxy)-2-pyridin-4-yl-pyrimidin-4-yl]-amide ( CHLORO STARTING MATERIAL) as yellow crystals. Melting point 177-179° C. ISN mass spectrum, m/e 482.2 (M-1 calculated for C22H18ClN5O5S1: 482).
……………………………….
http://www.google.com/patents/US6417360
EXAMPLE 1
a) To a solution of 6.9 g sodium in MeOH (300 ml) were added 14.52 g of 5-methyl-pyridine-2-sulfonic acid [6-chloro-5-(2-methoxy-phenoxy)-2-pyridin-4-yl-pyrimidin-4-yl]-amide at RT and the mixture was refluxed for 5 days until completion of the reaction according to TLC analysis. The reaction mixture was concentrated in vacuo to half its volume upon which the crude reaction product precipitated as a sodium salt. It was filtered off by suction and dried in a high vacuum. The solid was dissolved in water, which was then made acidic by addition of acetic acid. The precipitating free sulfonamide was extracted into Me2Cl2. The organic layer was dried over Mg2SO4, concentrated on a rotary evaporator, and the crystalline solid that had formed was filtered off. It was then dried in a high vacuum for 12 h at 120° C. to give the desired 5-methyl-pyridine-2-sulfonic acid [6-methoxy-5-(2-methoxy-phenoxy)-2-pyridin-4-yl-pyrimidin-4-yl]-amide as white crystals. Melting point 225-226° C. ISN mass spectrum, m/e 478.2 (M-1 calculated for C23H21N5O5S1: 478).
C23H21N5O5S1: Calc: C 57.61; H 4.41; N 14.61; S 6.69. Found: C 57.56; H 4.38; N 14.61; S 6.83
Preparation of the starting material:
b) 11.3 g of 4,6-dichloro-5-(2-methoxy-phenoxy)-2-pyridin-4-yl)-pyrimidine and 19.66 g of 5-methylpyridyl-2-sulfonamide potassium salt (preparations described in EP 0 799 209) were dissolved in DMF (255 ml) under argon. The solution was stirred for 2 h at 40° C. until completion of the reaction according to TLC analysis. The reaction mixture was cooled to RT and the solvent removed in a high vacuum. The residue was suspended in water (850 ml), acetic acid (85 ml) was added and the mixture was stirred for 30 minutes at RT. The solid that precipitated was collected by filtration and dried in a high vacuum at 60° C. for 16 h to give 5-methyl-pyridine-2-sulfonic acid [6-chloro-5-(2-methoxy-phenoxy)-2-pyridin-4-yl-pyrimidin-4-yl]-amide as yellow crystals. Melting point 177-179° C. ISN mass spectrum, m/e 482.2 (M-1 calculated for C22H18ClN5O5S1: 482).
…………………….
http://www.google.com/patents/EP0799209B1
SYNTHESIS OF
4,6-dichloro-5-(2-methoxy-phenoxy)-2-pyridin-4-yl)-pyrimidine
A BASIC STARTING MATERIAL FOR AVOSENTAN
- Preparation of the starting material
-
- b) 53.1 g of 4-cyano-pyridine (98%) are added all at once to a solution of 1.15 g of sodium in 200 ml of abs. MeOH. After 6 hours 29.5 g of NH4Cl are added while stirring vigorously. The mixture is stirred at room temperature overnight. 600 ml of ether are added thereto, whereupon the precipitate is filtered off under suction and thereafter dried at 50°C under reduced pressure. There is thus obtained 4-amidino-pyridine hydrochloride (decomposition point 245-247°C).
- c) 112.9 g of diethyl (2-methoxyphenoxy)malonate are added dropwise within 30 minutes to a solution of 27.60 g of sodium in 400 ml of MeOH. Thereafter, 74.86 g of the amidine hydrochloride obtained in b) are added all at once. The mixture is stirred at room temperature overnight and evaporated at 50°C under reduced pressure. The residue is treated with 500 ml of ether and filtered off under suction. The filter cake is dissolved in 1000 ml of H2O and treated little by little with 50 ml of CH3COOH. The precipitate is filtered off under suction, washed with 400 ml of H2O and dried at 80°C under reduced pressure. There is thus obtained 5-(2-methoxy-phenoxy)-2-(pyridin-4-yl)-pyrimidine-4,6-diol (or tautomer), melting point above 250°C.
- d) A suspension of 154.6 g of 5-(2-methoxy-phenoxy)-2-(pyridin-4-yl)-pyrimidine-4,6-diol (or tautomer) in 280 ml of POCl3 is heated at 120°C in an oil bath for 24 hours while stirring vigorously. The reaction mixture changes gradually into a dark brown liquid which is evaporated under reduced pressure and thereafter taken up three times with 500 ml of toluene and evaporated. The residue is dissolved in 1000 ml of CH2Cl2, treated with ice and H2O and thereafter adjusted with 3N NaOH until the aqueous phase has pH 8. The organic phase is separated and the aqueous phase is extracted twice with CH2Cl2. The combined CH2Cl2 extracts are dried with MgSO4, evaporated to half of the volume, treated with 1000 ml of acetone and the CH2Cl2remaining is distilled off at normal pressure. After standing in a refrigerator for 2 hours the crystals are filtered off under suction and dried at 50°C overnight. There is thus obtained 4,6-dichloro-5-(2-methoxy-phenoxy)-2-pyridin-4-yl)-pyrimidine, melting point 178-180°C.
…………………………
http://www.google.com/patents/WO2000052007A1
Preparation of the starting material:
5-methyl-pyridine-2-sulfonic acid [6-chloro-5-(2- methoxy-phenoxy)-2-pyridin-4-yl-pyrimidin-4-yl] -amide IE THE 6 CHLORO COMPD
b) 11.3 g of 4,6-dichloro-5-(2-methoxy-phenoxy)-2-pyridin-4-yl)-pyrimidine and 1 .66 g of 5-methylpyridyl-2-sulfonamide potassium salt (preparations described in EP 0 799 209) were dissolved in DMF (255 ml) under argon. The solution was stirred for 2 h at 40°C until completion of the reaction according to TLC analysis. The reaction mixture was cooled to RT and the solvent removed in a high vacuum. The residue was suspended in water (850 ml), acetic acid (85 ml) was added and the mixture was stirred for 30 minutes at RT. The solid that precipitated was collected by filtration and dried in a high vacuum at 60 °C for 16 h to give 5-methyl-pyridine-2-sulfonic acid [6-chloro-5-(2- methoxy-phenoxy)-2-pyridin-4-yl-pyrimidin-4-yl] -amide as yellow crystals. Melting point 177-179 °C. ISN mass spectrum, m/e 482.2 (M-l calculated for C22Hi8ClN5O5Sι: 482).

………………………………………………………………………………………….
NEXT

Example 1AVOSENTAN
a) To a solution of 6.9 g sodium in MeOH (300 ml) were added 14.52 g of
5-methyl-pyridine-2-sulfonic acid [6-chloro-5-(2-methoxy-phenoxy)-2-pyridin-4-yl- pyrimidin-4-yl] -amide at RT and the mixture was refluxed for 5 days until completion of the reaction according to TLC analysis. The reaction mixture was concentrated in vacuo to half its volume upon which the crude reaction product precipitated as a sodium salt. It was filtered off by suction and dried in a high vacuum. The solid was dissolved in water, which was then made acidic by addition of acetic acid. The precipitating free sulfonamide was extracted into Me2Cl2. The organic layer was dried over Mg SO , concentrated on a rotary evaporator, and the crystalline solid that had formed was filtered off. It was then dried in a high vacuum for 12 h at 120 °C to give the desired 5-methyl-pyridine-2-sulfonic acid [6- methoxy-5-(2-methoxy-phenoxy)-2-pyridin-4-yl-pyrimidin-4-yl] -amide as white crystals. Melting point 225-226 °C. ISN mass spectrum, m/e 478.2 (M-l calculated for
C23H21N5O5S1: Calc: C 57.61; H 4.41; N 14.61; S 6.69. Found: C 57.56; H 4.38; N 14.61; S 6.83
…………………………………………….

IS DESCRIBED IN
http://www.google.com/patents/EP2331513A1?cl=en
ALSO

-
Diabetic nephropathy is the principle cause of end stage renal disease in the western world. It is a major cause of morbidity and mortality in Type-I Diabetes, but is an increasing problem in Type-II Diabetes and because the incidence of this is five times that of Type-I Diabetes, it contributes at least 50% of diabetics with end stage renal disease.
-
The initial stage of subtle morphologic changes in the renal glomeruli is followed by microalbuminuria. This is associated with a modestly rising blood pressure and an increased incidence of cardiovascular disease. There follows a continued increase in urinary protein excretion and declining glomerular filtration rate. Diabetic nephropathy has many possible underlying pathophysiological causes including metabolic, glycosylation of proteins, haemodynamics, altered flow/pressure in glomeruli, the development of hypertension and cytokine production; all of these are associated with the development of extracellular matrix and increased vascular permeability leading to glomerular damage and proteinuria.
| WO2005113543A1 * | May 12, 2005 | Dec 1, 2005 | Alexander Bilz | Crystalline forms of a pyridinyl-sulfonamide and their use as endothelin receptor antagonists |
| WO2007031501A2 * | Sep 11, 2006 | Mar 22, 2007 | Speedel Pharma Ag | Pyridylsulfonamidyl-pyrimidines for the prevention of blood vessel graft failure |
| WO2008077916A1 * | Dec 21, 2007 | Jul 3, 2008 | Ovidiu Baltatu | Pharmaceutical composition using aliskiren and avosentan |
| EP1454625A1 * | Mar 6, 2003 | Sep 8, 2004 | Speedel Development AG | Pyridylsulfonamidyl-pyrimidines for the treatment of diabetic nephropathies |
| EP1595880A1 * | May 13, 2004 | Nov 16, 2005 | Speedel Pharma AG | Crystalline forms of a pyridinyl-sulfonamide and their use as endothelin receptor antagonists |
| EP1938812A1 * | Dec 22, 2006 | Jul 2, 2008 | Speedel Pharma AG | Pharmaceutical composition using aliskiren and avosentan |
| US6951856 | Jul 10, 2001 | Oct 4, 2005 | Actelion Pharmaceuticals Ltd. | Arylethene-sulfonamides |
| US7402587 | May 12, 2005 | Jul 22, 2008 | Speedel Pharma Ag | Crystalline forms of a pyridinyl-sulfonamide and their use as endothelin receptor antagonists |
| WO1996019459A1 * | Dec 8, 1995 | Jun 27, 1996 | Volker Breu | Novel sulfonamides |
| EP0713875A1 * | Nov 13, 1995 | May 29, 1996 | F. Hoffmann-La Roche AG | Sulfonamides |
| EP0897914A1 * | Aug 10, 1998 | Feb 24, 1999 | F. Hoffmann-La Roche Ag | Process for the preparation of 2,5-disubstitued pyridines |
READ MORE ON SNTAN SERIES……http://medcheminternational.blogspot.in/p/sentan-series.html

TEGOBUVIR ..IN PHASE II FOR HEPATITIS C

TEGOBUVIR
A non-structural protein 5B polymerase inhibitor
for Treatment of chronic hepatitis C
5-[6-[2,4-Bis(trifluoromethyl)phenyl]pyridazin-3-ylmethyl]-2-(2-fluorophenyl)-5H-imidazo[4,5-c]pyridine
CHEMICAL NAMES
1. 5H-Imidazo[4,5-c]pyridine, 5-[[6-[2,4-bis(trifluoromethyl)phenyl]-3-pyridazinyl]methyl]-
2-(2-fluorophenyl)-
2. 5-({6-[2,4-bis(trifluoromethyl)phenyl]pyridazin-3-yl}methyl)-2-(2-fluorophenyl)-5H-
imidazo[4,5-c]pyridine
MOLECULAR FORMULA C25H14F7N5
MOLECULAR WEIGHT 517.4
MANUFACTURER Gilead Sciences, Inc.
CODE DESIGNATION
- GS 333126
- GS 9190
- GS-333126
- GS-9190
- Tegobuvir
- UNII-5NOK5X389M
CAS REGISTRY NUMBER 1000787-75-6
GS-9190, an RNA-directed RNA polymerase (NS5B) inhibitor, is in phase II clinical evaluation at Gilead for the treatment of hepatitis C virus (HCV) infection. A clinical trial with GS-9190 in combination with peginterferon alfa-2a and ribavirin and with GS-9451 or with GS-9256 in treatment-naive subjects with chronic genotype 1 HCV infection was discontinued due to serious adverse events.
Gilead (Originator)
Katholieke Universiteit Leuven (Originator)
……………………………………….
tegobuvir
PATENTS
WO 2005063744
WO 2008005519
WO 2009009001
WO 2010151488
WO 2010151487
WO 2010151472
WO 2011072370
WO 2011156757
WO 2012087596
WO 2013101550
Hebner CM, Han B, Brendza KM, Nash M, Sulfab M, Tian Y, Hung M, Fung W, Vivian RW, Trenkle J, Taylor J, Bjornson K, Bondy S, Liu X, Link J, Neyts J, Sakowicz R, Zhong W, Tang H, Schmitz U.
PLoS One. 2012;7(6):e39163. doi: 10.1371/journal.pone.0039163. Epub 2012 Jun 13.
Wong KA, Xu S, Martin R, Miller MD, Mo H.
Virology. 2012 Jul 20;429(1):57-62. doi: 10.1016/j.virol.2012.03.025. Epub 2012 Apr 28.
Zeuzem S, Buggisch P, Agarwal K, Marcellin P, Sereni D, Klinker H, Moreno C, Zarski JP, Horsmans Y, Mo H, Arterburn S, Knox S, Oldach D, McHutchison JG, Manns MP, Foster GR.
Hepatology. 2012 Mar;55(3):749-58. doi: 10.1002/hep.24744.
Shih IH, Vliegen I, Peng B, Yang H, Hebner C, Paeshuyse J, Pürstinger G, Fenaux M, Tian Y, Mabery E, Qi X, Bahador G, Paulson M, Lehman LS, Bondy S, Tse W, Reiser H, Lee WA, Schmitz U, Neyts J, Zhong W.
Antimicrob Agents Chemother. 2011 Sep;55(9):4196-203. doi: 10.1128/AAC.00307-11. Epub 2011 Jul 11.
- ……………………..
- http://www.google.com/patents/WO2013040492A2
- ompound 1 can be prepared using synthetic methods and intermediates like those described in US 7,754,720. Compound 1 can also be prepared as described in the following Example.
- Compound 1 is:

Compound 1 may also be referred to as 5-((6-(2,4-bis(trifluoromethyl)phenyl)pyridazin-3-yl)methyl)-2-(2-fluorophenyl)-5H-imidazo[4,5-c]pyridine, 5-[[6-[2,4-bis (trifluoromethyl)phenyl]pyridazin=3-yl]methyl]-2-(2-fluorophenyl).
- Example 1 : 5-({6-[2,4-bis(trifluoromethyl)phenyl]pyridazin-3-yl}methyl)-2-(2-fluorophenyl)-5H- imidazo[4,5-c]pyridi


Compound 103 was dissolved in dimethoxyethane (DME). To this solution was added 2,4-bis(trifluromethyl)phenylboronic acid 105 and a 2N aq. Na2C03 solution. To the resulting biphasic mixture was added Pd(PPh3)4 and the reaction was then heated at 80°C for 72 hrs. The reaction was cooled to room temperature and filtered through Celite and the Celite washed with EtOAc. The filtrate was concentrated in vacuo. The residue was purified on 6g Si02 using MeOH/CH2CI2 to elute compound. The compound thus obtained was contaminated with PPh3(0). The product was repurified on a 1 mm Chromatotron plate with 0 to 5%
MeOH/CH2CI2 in 1 % steps. The pure fractions were combined and concentrated in vacuo, then dried on high vacuum for 12 hrs. 11.8 mg of the free base of compound 1 was obtained with no PPh3 contamination. 1H NMR (300MHz,CD3OD) δ 6.20 (s, 2), 7.32 (m, 3), 7.52 (m, 1 ), 7.78 (d, 1), 7.89 (d, 1), 7.95 (s, 2), 8.15 (m, 3), 8.35 (d, 1), 9.12 (s, 1); LC/MS M+H = 518.
The intermediate compound 104 was prepared as follows, a. Preparation of Compound 10

101 102

To a solution of the commercially available starting material 101 in CHCI3, trichloroisocyanuric acid (TCCA) was added at 60°C. Then the solution was stirred for 1.5 hrs, cooled, and filtered with HiFlo-Celite. The filtrate was concentrated and dried with vacuum. The yield was 5.037 g of compound 102. b. Preparation of Compound 104.

102 104

To a solution of compound 103 in DMF (dimethylformamide), NaOH was added.
Compound 102 was dissolved in DMF (20 mL) and added to the solution slowly. The reaction was stirred for 3 hrs, was diluted with water and extracted with EtOAc. The organic layer was dried with Na2S0 . The solvent was removed and the product recrystallized with
dichloromethane. The yield was 5.7 g of compound 103.
- ……………………………
- US7754720
- Example 1a Synthesis of 5-({6-[2,4-bis(trifluoromethyl)phenyl]pyridazin-3-yl}methyl)-2-(2-fluorophenyl)-5H-imidazo[4,5-c]pyridineIn this method, dimethoxyethane or its related solvents, all having the general formula R1OR2O(R4O)aR3 wherein each of R1, R2, R3 and R4 are independently selected from C1-C6 alkyl and a is 0 or 1, have been found to be particularly advantageous over the conventional solvent DMF. Typically, each of R1, R2, R3 and R4 are independently C1-C2 alkyl and usually a is 0. C1-C6 alkyl includes fully saturated primary, secondary or tertiary hydrocarbon groups with 1 to 6 carbon atoms and thereby includes, but is not limited to methyl, ethyl, propyl, butyl, etc.Step 1



Compound MW Amount mmoles Equivalents SM 128.56 5 g 38.9 1 TCCA 232.41 3.62 g 15.6 0.4 CHCl3 130 ml To a solution of the commercially available starting material (SM) in CHCl3, trichloroisocyanuric acid (TCCA) was added at 60° C. Then the solution was stirred for 1.5 hrs., cooled down and filtered with HiFlo-Celite. The filtrate was concentrated and dried with vacuum. The yield was 5.037 g.
Step 2



Compound MW Amount mmoles Equivalents S.M. 163 5.073 g 31.12 1 Core 213.2 6.635 g 31.12 1 NaOH (10%) 40 1.245 g 31.12 1 DMF 320 ml To a solution of core (obtained as described in literature in DMF (dimethylformamide), NaOH was added. Then SM for this step (obtained from step 1) was dissolved in DMF (20 ml) and added to the solution slowly. The reaction was stirred for 3 hrs, was diluted with water and extracted with EtOAc. The organic layer was dried with Na2SO4. The solvent was removed and the product recrystallized with DCM (dichloromethane). The yield was 5.7 g.
Step 3



Compound MW Amount Moles Equivalents A 453.79 95 mg 0.209 1 DME 500 ul 2 N aq. Na2CO3 313ul 0.626 3 2,4-bisCF3– 257.93 80.9 mg 0.313 1.5 phenylboronic acid Pd(PPh3)4 1155 12 mg 0.0104 0.05 Compound A was dissolved in dimethoxyethane (DME). To this solution was added 2,4-bis(trifluromethyl)phenylboronic acid and a 2N aq. Na2CO3 solution. To the resulting biphasic mixture was added Pd(PPh3)4 and the reaction was then heated at 80° C. for 72 hrs. The reaction was cooled to room temperature and filtered through Celite and the Celite washed with EtOAc. The filtrate was concentrated in vacuo. The residue was purified on 6 g SiO2 using MeOH/CH2Cl2 to elute compound. The compound thus obtained was contaminated with PPh3(O). The product was repurified on a 1 mm Chromatotron plate with 0 to 5% MeOH/CH2Cl2 in 1% steps. The pure fractions were combined and concentrated in vacuo, then dried on high vacuum for 12 hrs. 11.8 mg of the free base of compound (1) was obtained with no PPh3 contamination.
1H NMR (300 MHz, CD3OD)
6.20 (s, 2)
7.32 (m, 3)
7.52 (m, 1)
7.78 (d, 1)
7.89 (d, 1)
7.95 (s, 2)
8.15 (m, 3)
8.35 (d, 1)
9.12 (s, 1)
LC/MS M+H=518
Example 1b Synthesis of 5-({6-[2,4-bis(trifluoromethyl)phenyl]pyridazin-3-yl}methyl)-2-(2-fluorophenyl)-5H-imidazo[4,5-c]pyridineThis example is directed to an additional method for making compound (1), employing the following schemes.

Methanesulfonic acid was added to 2-fluorobenzoic acid in a reactor with active cooling keeping T≦50° C. 3,4-Diaminopyridine was then added portionwise to this cooled slurry, keeping T≦35° C. The contents of the reactor were then heated to 50° C. Phosphorus pentoxide was added in a single charge. The reaction was then heated at 90-110° C. for at least 3 hours. The reaction was sampled for completion by HPLC analysis. The reaction was cooled to ambient temperature and water was added portionwise slowly to quench the reaction. The reaction was then diluted with water. In solubles were removed by filtration. The pH of the filtrate was adjusted to 5.5-5.8 with ammonium hydroxide. The reaction was allowed to self-seed and granulate for ˜4 hours at ambient temperature. The pH was then adjusted to 8.0-9.3 with ammonium hydroxide. The slurry was held at ambient temperature for at least 2 hours. The solids were isolated by filtration and washed with water, followed by IPE. The wet cake was dried in vacuo at not more than 60° C. until ≦1% water remains. The dry product is core (2).
Summary of Materials M.W. Wt. Ratio Mole ratio 3,4-Diaminopyridine 109.13 1.0 1.0 2-Fluorobenzoic acid 140.11 1.4 1.1 Methanesulfonic acid 96.1 7.0 8.0 Phosphorus pentoxide 141.94 1.3 1.0 Water 18.02 40 — Isopropyl ether 102.17 5.0 — Ammonium hydroxide 35.09 ~10 — 
A solution of compound (2a) in 1,2-dichloroethane was heated to 40-45° C. Trichloroisocyanuric acid was added and the mixture was heated at 60-70° C. for at least 2 hours. The reaction was sampled for completion by HPLC analysis. The reaction was cooled to ambient temperature. Celite was added to absorb insolubles, then solids were removed by filtration. The filtrate was washed with 0.5 N sodium hydroxide solution. The organic layer was concentrated to lowest stirrable volume and displaced with DMF. Core (2) and 10% aqueous sodium hydroxide solution were added. The reaction was stirred at ambient temperature for at least 8 hours. The reaction was sampled for completion by HPLC analysis. An additional 10% charge of 10% sodium hydroxide solution was added to the reaction. The reaction was then charged into water to isolate the crude product. After granulating for at least 1 hour, the solids were isolated and washed with water and isopropyl ether. Ethyl acetate was added and refluxed (internal T=70-77° C.) for 1-5 hours to dissolve product, then cooled to 18-23° C. slowly over 4-8 hours. The reactor contents were agitated at 18-23° C. for 8-20 hours and solids collected by filtration and rinsed with ethyl acetate. Low melt (i.e., DSC about 220 degrees C.) amorphous compound (1) was discharged. Amorphous compound (1) was dissolved in ethyl acetate by heating at reflux (internal T=70-77° C.) for 1-5 hours. Water content is controlled to about 0.2% by azeotropically removing water (with ethyl acetate the upper limit on water content is about 0.6% by weight; at about 0.9% by weight water the amorphous material will reprecipitate and crystals will not be obtained). The reactor contents are cooled slowly to 18-23° C. over 4-8 hours, then agitated at 18-23° C. for 8-20 hours and solids collected by filtration. The solids were rinsed with ethyl acetate and dried in vacuo at not more than 60° C. to obtain the dry crystalline compound (1).
Summary of Materials M.W. Wt. Ratio Mole ratio 3-chloro-6-methylpyridazine 128.56 1.0 1.0 2,4bis(trifluromethyl)phenylboronic 257.93 4.0 2.0 acid X-Phos 476.72 0.18 0.05 Palladium acetate 224.49 0.04 0.025 1,2-Dimethoxyethane 90.12 16.7 — Potassium carbonate 138.21 2.15 2.0 Water 18.02 7.8 — Copper iodide 190.45 0.037 0.025 Celite — 0.25 — Heptane 100.2 22.4 — Nuclear Magnetic Resonance (1H-, 13C-, and 19F-NMR) SpectraNuclear magnetic resonance (NMR) spectra of compound (1) is consistent with the proposed structure. The 13C, 19F, and 1H-NMR spectra of compound (1) in DMSO-d6 were measured using a Varian UnityInova-400 FT-NMR spectrometer. Spectra are shown in the table below. The NMR chemical shift assignments were established using 2D correlation experiments (COSY, HSQC, HMBC and HSQCTOCSY).
1H- and 13C-NMR Chemical Shift Assignments for Compound (1) Reference Standard
Atom δC/ppm (DMSO-d6) δF/ppm (DMSO-d6) δH/ppm (DMSO-d6) 1A 140.16 2A 128.32 (qa, JCF = 32 Hz) 3A 123.61, m 8.24 (m, 1 H) 4A 130.27 (q, JCF = 34 Hz) 5A 129.54 (q, JCF = 3 Hz) 8.22 (m, 1 H) 6A 133.36 7.88 (m, 1 H) 7A 123.20 (q, JCF = 273 Hz) −56.4b 8A 123.02 (q, JCF = 275 Hz) −62.0b 1B 158.76 2B 128.16 8.01 (d, 1 H, J = 8.4 Hz) 3B 126.20 7.95 (d, 1 H, J = 8.8 Hz) 4B 157.70 5B 60.49 6.17 (s, 2 H) 2C 131.86 8.31 (m, 1 H) 3C 112.63 7.86 (m, 1 H) 4C 155.44 6C 168.11 (d, JCF = 6 Hz) 8C 145.08 9C 133.06 9.25 (s, 1 H) 1D 123.11 (d, JCF = 10 Hz) 2D 160.46 (d, JCF = 254 Hz) −111.7 3D 116.59 (d, JCF = 22 Hz) 7.29 (m, 1 H) 4D 130.84 (d, JCF = 8 Hz) 7.46 (m, 1 H) 5D 124.13 (d, JCF = 4 Hz) 7.31 (m, 1 H) 6D 131.72 (d, JCF = 2 Hz) 8.35 (m, 1 H) amultiplicity, s: singlet, d: doublet, q: quartet, m: multiplet binterchangeable signals 
WANT TO KNOW ABOUT VIR SERIES CLICK
click
http://drugsynthesisint.blogspot.in/p/vir-series-hep-c-virus-22.html
AND
http://medcheminternational.blogspot.in/p/vir-series-hep-c-virus.html







TELMISARTAN ..Actavis’ Generic Version of Micardis Receives FDA Approval
DUBLIN, Jan. 8, 2014 /PRNewswire/ — Actavis plc today announced that it has received approval from the U.S. Food and Drug Administration (FDA) on its Abbreviated New Drug Application (ANDA) for Telmisartan Immediate-Release Tablets, 20 mg, 40 mg and 80 mg, a generic equivalent to Boehringer Ingelheim’s Micardis. Actavis intends to launch the product immediately.
DANOPREVIR (ITMN-191) …..a peptidomimetic inhibitor of the NS3/4A protease of hepatitis C virus (HCV)

Danoprevir
Danoprevir(ITMN-191) is a peptidomimetic inhibitor of the NS3/4A protease of hepatitis C virus (HCV) with IC50 of 0.2-3.5 nM, inhibition effect for HCV genotypes 1A/1B/4/5/6 is ~10-fold higher than 2B/3A. Phase 2.
Array BioPharma (Originator)
| RG7227 |
| ITMN-191 |
| RO5190591 |
2H-Isoindole-2-carboxylic acid, 4-fluoro-1,3-dihydro-, (2R,6S,12Z,13aS,14aR,16aS)-
14a-[[(cyclopropylsulfonyl)amino]carbonyl]-6-[[(1,1-dimethylethoxy)carbonyl]amino]-
1,2,3,5,6,7,8,9,10,11,13a,14,14a,15,16,16a-hexadecahydro-5,16-
dioxocyclopropa[e]pyrrolo[1,2-a][1,4]diazacyclopentadecin-2-yl ester
2. (2R,6S,12Z,13aS,14aR,16aS)-14a-[(cyclopropylsulfonyl)carbamoyl]-6-{[(1,1-
dimethylethoxy)carbonyl]amino}-5,16-dioxo-
1,2,3,5,6,7,8,9,10,11,13a,14,14a,15,16,16a-hexadecahydrocyclopropa[e]pyrrolo[1,2-
a][1,4]diazacyclopentadecin-2-yl 4-fluoro-1,3-dihydro-2H-isoindole-2-carboxylate
Treatment of hepatitis C
MOLECULAR FORMULA C35H46FN5O9S
MOLECULAR WEIGHT 731.8
MANUFACTURER Genentech
CODE DESIGNATION R05190591
CAS REGISTRY NUMBER 850876-88-9, 916881-67-9
Danoprevir(ITMN-191) is a peptidomimetic
ITMN-191 (R-7227), a macrocyclic protease inhibitor, is in phase II clinical evaluation for the treatment of chronic hepatitis C virus (HCV) infection as monotherapy and in combination with Pegasys(R) (pegylated interferon alpha-2a) and Copegus(R) (ribavirin). The product candidate is also being evaluated in combination with R-7128 in treatment-naive patients infected with HCV genotype 1.
Danoprevir (ITMN-191; RG-7227), under development by InterMune Inc and Roche Holding AG, is a promising, potent NS3/4A protease inhibitor for the oral treatment of HCV infection. Preclinical data demonstrated that danoprevir binds with high affinity and dissociates slowly from the HCV NS3 protease, allowing high liver drug exposure with only modest plasma drug exposure.
In 2006, originator InterMune and licensee Roche entered into an exclusive worldwide collaboration agreement to develop and commercialize products from InterMune’s hepatitis C (HCV) protease inhibitor program, including ITMN-191. In 2010, the licensing agreement was terminated. Also in 2010, Roche acquired worldwide development and commercialization rights to R-7227 from InterMune. Preclinical pharmacokinetic results support the exploration of twice-daily oral dosing in HCV.
A phase Ib, ‘IFN-free’ clinical trial demonstrated that danoprevir, combined with the HCV polymerase inhibitor RG-7128 (Pharmasset Inc/Roche Holding AG), was effective in reducing HCV-RNA levels in a large proportion of treatment-naïve patients with HCV infection and in approximately half of previously non-responsive patients with HCV-1 infection, without resistance or safety concerns. In a phase IIb trial in treatment-naïve patients with HCV-1 infection, danoprevir plus pegylated IFNalpha2a and ribavirin resulted in undetectable levels of HCV-RNA in the majority of patients, without any evidence of viral resistance; however, the high-dose danoprevir arm was prematurely terminated because of grade 4 ALT elevations. Phase I trials have also demonstrated that ritonavir boosting improved the pharmacokinetic profile of danoprevir; therefore, at the time of publication, a phase IIb trial to evaluate ritonavir-boosted, low-dose danoprevir in combination with RG-7128 was planned. (source:
inhibitor of the NS3/4A protease of hepatitis C virus (HCV) with IC50 of 0.2-3.5 nM, inhibition effect for HCV genotypes 1A/1B/4/5/6 is ~10-fold higher than 2B/3A. Phase 2.
SODIUM SALT
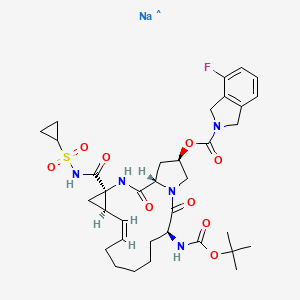
HERAPEUTIC CLAIM Treatment of hepatitis C
CHEMICAL NAMES
1. 2H-Isoindole-2-carboxylic acid, 4-fluoro-1,3-dihydro-, (2R,6S,12Z,13aS,14aR,16aS)-
14a-[[(cyclopropylsulfonyl)amino]carbonyl]-6-[[(1,1-dimethylethoxy)carbonyl]amino]-
1,2,3,5,6,7,8,9,10,11,13a,14,14a,15,16,16a-hexadecahydro-5,16-dioxocyclopropa
[e]pyrrolo[1,2-a][1,4]diazacyclopentadecin-2-yl ester, sodium salt (1:1)
2. sodium (cyclopropylsulfonyl){[(2R,6S,12Z,13aS,14aR,16aS)-6-{[(1,1-dimethylethoxy)
carbonyl]amino}-2-{[(4-fluoro-1,3-dihydro-2H-isoindol-2-yl)carbonyl]oxy}-5,16-dioxo-
1,2,3,6,7,8,9,10,11,13a,14,15,16,16a-tetradecahydrocyclopropa[e]pyrrolo[1,2-
a][1,4]diazacyclopentadecine-14a(5H)-yl]formyl}azanide
MOLECULAR FORMULA C35H45FN5NaO9S
MOLECULAR WEIGHT 753.8
SPONSOR Genentech
CODE DESIGNATION
- Danoprevir sodium
- ITMN-191
- R 7227 sodium
- R7227
- RO 5190591-001
- RO5190591-001
- UNII-217RJI972K
CAS REGISTRY NUMBER 916826-48-7
DANOPREVIR SODIUM
The HCV protease mediates the cleavage of the HCV polyprotein to release the functional proteins that are essential for viral propagation. The inhibition of the HCV protease activity is expected to block HCV replication in infected host cells. Numberous HCV protease inhibitors have been identified. Non- limiting examples of HCV protease inhibitors are described in U.S. Patent Application Pub. Nos. 20040106559, 20040180815, 20040266668, 2004038872, 20050090432, 20050267018, 20070054842, 20070281885, 2007299078, 20080032936, 20080125444, 20080279821, 20090111757, 20090148407, 20090202480, 20090269305, 20090285773, 20090285774, 20100081700, 20100144608, 2010018355, 20100183551, 20100221217, 20100260710, 20100286185 and 20110135604, and U.S. Patent Nos. 6608027, 6767991, 7091184, 7119072, 7544798, 7642235 and 7829665, as well as WO2007014919, WO2007014926, WO2008046860, WO2008095058,
………………………………
danoprevir
patents and journal ref
1. WO 2005037214..
2. WO 2005095403
3. WO 2007015824..
4. WO 2008128921
5. WO 2009080542
6. WO 2009142842
7. WO 2010015545
8. WO 2013079424
9. WO 2012062685
10.WO 2013106631
11. Concise asymmetric synthesis of a (1R,2S)-1-amino-2-vinylcyclopropanecarboxylic acid-derived sulfonamide and ethyl ester
Org Biomol Chem 2013, 11(39): 6796http://pubs.rsc.org/en/content/articlelanding/2013/ob/c3ob41394b/unauth#!divAbstract
12.J. Med. Chem., Article ASAP,DOI: 10.1021/jm400164c
Kazmierski WM, Hamatake R, Duan M, Wright LL, Smith GK, Jarvest RL, Ji JJ, Cooper JP, Tallant MD, Crosby RM, Creech K, Wang A, Li X, Zhang S, Zhang YK, Liu Y, Ding CZ, Zhou Y, Plattner JJ, Baker SJ, Bu W, Liu L.
J Med Chem. 2012 Apr 12;55(7):3021-6. doi: 10.1021/jm201278q. Epub 2012 Apr 3.
14 . Discovery of novel P3-oxo inhibitor of hepatitis C virus NS3/4A serine protease.
Duan M, Kazmierski W, Crosby R, Gartland M, Ji J, Tallant M, Wang A, Hamatake R, Wright L, Wu M, Zhang YK, Ding CZ, Li X, Liu Y, Zhang S, Zhou Y, Plattner JJ, Baker SJ.
Bioorg Med Chem Lett. 2012 Apr 15;22(8):2993-6. doi: 10.1016/j.bmcl.2012.02.039. Epub 2012 Feb 22.
……………….

(2R,6S,13aS,14aR,16aS,Z)-6-(tert-Butoxycarbonylamino)-14a-(cyclopropylsulfonylcarbamoyl)-5,16-dioxo-1,2,3,5,6,7,8,9,10,11,13a,14,14a,15,16,16a-hexadecahydrocyclopropa[e]pyrrolo[1,2-a][1,4]diazacyclopentadecin-2-yl 4-fluoroisoindoline-2-carboxylate (49)



For certain NS3 inhibitors shown in this section, additional chemical transformations are utilized to obtain the final products. The preparations of two such examples are described for compounds 153 and 154 below:

(2R,6S,13aS,14aR,16aS,Z)-6-(tert-botoxycarbonylamino)-2-(4-fluoroisoindoline-2-carbonyloxy)-5,16-dioxo-1,2,3,5,6,7,8,9,10,11,13a,14,14a,15,16,16a-hexadecahydrocyclopropa[e]pyrrolo[1,2-α][1,4]dizacyclopentadecine-14a-carboxylic acid (0.10 g, 0.16 mmol) and TEA (0.024 mL, 0.18 mmol) in THF (5 mL) was added ethyl carbonochlridate (0.016 mL, 0.17 mmol) at 0° C. The reaction was stirred at 0° C. for 2 hrs. Sodium boronhydride (0.012 g, 0.32 mmol) was added and the reaction was stirred at rt for 3 days. Water (5 mL) and ethyl acetate (10 mL) were added. The organic layer was separated, washed with brine and dried over sodium sulfate. After removal of solvent, the residue was purified by column chromatography (ethyl acetate) to give the product (0.060 g, 61.4%) as white solid. 1H NMR (400 MHz, d6-DMSO) δ 8.47 (b, 1H), 7.35 (m, 1H), 7.10-7.20 (m, 2H), 7.03 (m, 1H), 5.47 (m, 1H), 5.28 (b, 1H), 4.98 (m, 1H), 4.67 (b, 4H), 4.56 (m, 1H), 4.46 (m, 1H), 4.26 (m, 1H), 3.92 (m, 1H), 3.66 (m, 2H), 3.16 (m, 1H), 2.67 (m, 1H), 2.21 (m, 2H), 1.80 (m, 1H), 1.68 (m, 1H), 1.30 (m, 8H), 1.11-1.20 (m, 9H), 0.85 (m, 1H), 0.77 (m, 1H).

A solution of oxalyl chloride 90.045 mL, 0.089 mmol) in DCM (5 mL) at −78° C. was added a solution of DMSO (0.015 g, 0.020 mmol) in DCM (2 mL) dropwise over 2 ninytes. The reaction was stirred at −78° C. for 10 minutes and the a solution of (2R,6S,13aS,14aR,16aS,Z)-6-(tert-botoxycarbonylamino)-14a-(hydroxymethyl)-5,16-dioxo-1,2,3,5,6,7,8,9,10,11,13a,14,14a,15,16,16a-hexadecahydrocyclopropa[e]pyrrolo[1,2-α][1,4]dizacyclopentadecin-2-yl-4-fluoroisoindoline-2-carboxylate (0.050 g, 0.081 mmol) in DCM (2 mL) was added. After stirred at −78° C. for 40 min, TEA (0.051 mL, 0.37 mmol) was added. The reaction was warmed to rt, water (5 mL) was added. The organic layer was separated, washed with brine and dried over sodium sulfate. After removal of solvent, the residue was dissolved in MeOH (5 mL) and ammonium hydroxide (0.085 g, 2.45 mmol) and acetic acid (0.014 mL, 0.25 mmol) were added. The reaction stirred at rt for 3 minutes. NaCNBH3 90.015 g, 0.245 mmol) was added and stirred at rt for 30 minutes. The MeOH was removed. DCM (20 mL) and saturated sodium bicarbonate (5 mL) was added. The organic layer was separated, washed with brine and dried over sodium sulfate. After removal of solvent, the residue was dissolved in DCM (5 mL). TEA (0.017 mL, 0.122 mmol) was added and followed by the cyclopropanesulfonyl chloride (0.015 g, 0.098 mmol). The reaction was stirred at rt for 5 hrs. The solvent was removed. The residue was purified by column chromatography (ethyl acetate) to give the product (0.017 g, 28.2%) as white solid. 1H NMR (400 MHz, d6-DMSO) δ 8.52 (m, 1H), 7.35 (m, 1H), 7.02-7.20 (m, 4H), 5.56 (m, 1H), 4.99 (m, 1H), 4.97 (m, 1H), 4.67 (m, 2H), 4.66 (s, 2H), 4.46 (m, 1H), 4.24 (m, 1H), 3.92 (m, 1H), 3.67 (m, 1H), 3.46 (m, 1H), 2.74 (m, 1 h), 2.67 (m, 1H), 2.22 (m, 2H), 1.84 (m, 1H), 1.68 (m, 1H), 1.08-1.36 (m, 20H), 0.89 (m, 2H), 0.81 (m, 2H).

Hoffmann-La Roche and Genentech’s danoprevir/r (RG7227) is a twice-daily, ritonavir-boosted HCV protease inhibitor with activity against HCV genotypes 1, 4 and 6. DAUPHINE, an ongoing phase II trial in 421 treatment-naive people with HCV genotypes 1 and 4, is comparing doses (200, 100, and 50 mg danoprevir, boosted with 100 mg ritonavir, twice-daily) and response-guided therapy with danoprevir/r plus PEG-IFN/RBV. At 12 weeks after treatment completion, HCV RNA was undetectable in 86% of the highest-dosing arm, 77% of the 100 mg arm, and 65% of the 50 mg arm.
Response to treatment in the 200 mg dosing arm did not differ according to HCV subtype or IL28B genotype; at 12 weeks after treatment completion, 88% of people with HCV subtype 1a and an IL28B non-CC genotype had undetectable HCV RNA. Across all dosing arms, HCV RNA remained undetectable 12 weeks after treatment completion in 100% of people with HCV genotype 4.
In the response-guided therapy arm, 76% of early responders (who were treated for 12 weeks) and 67% of late responders (treated for 24 weeks) maintained undetectable HCV RNA 12 weeks after treatment completion, bringing the overall total to 72%.
One death occurred during the trial—from sudden heart attack, in a participant with preexisting diabetes and hypertension—it was considered unrelated to study drugs. Adverse events were reported in virtually all study participants. Side effects from ritonavir, which is used to boost danoprevir levels, increased the likelihood of more than one serious adverse event among people in the danoprevir/r arms (range 4–9% vs. 1% for placebo). The rate of danoprevir/r-related treatment discontinuations was similar to the rate of PEG-IFN/RBV-associated discontinuations (3–7%, and 3–8%, respectively).
Common side effects (experienced by more than 15% of study participants) included fatigue, fever, chills, weakness, nausea, diarrhea, itching, rash, hair loss, headache, aching muscles and joints, insomnia, cough, and appetite loss. Diarrhea was the only side effect associated with danoprevir/r. Adding danoprevir/r did not increase rates of rash or anemia (known side effects of other HCV protease inhibitors). Most grade 3 and grade 4 lab abnormalities were neutropenia, reported in 22% to 38% of study participants.
Interferon-free DAA Combinations
Danoprevir/r and Mericitabine, plus Ribavirin (HCV Genotypes 1 and 4)
Roche’s phase IIb study, INFORM-SVR, is combining response-guided therapy with danoprevir/r, a twice-daily ritonavir-boosted HCV protease inhibitor, and mericitabine, a twice-daily nucleoside polymerase inhibitor, with or without ribavirin for 12 to 24 weeks in non-cirrhotic people with HCV genotype 1. The original study design was modified after high relapse rates were observed in the 12-week treatment and ribavirin-free arms. Treatment was extended to 24 weeks, and ribavirin was given to all participants.
The majority of INFORM-SVR participants were male, had HCV genotype 1a, and non-CC genotypes. Of the 64 people treated for 24 weeks with all three drugs, 41% experienced SVR-12. People with HCV genotype 1b were more likely to achieve SVR-12 (71% versus 26% in HCV genotype 1a). In contrast, SVR-12 was more likely among people with non-CC genotypes (32% for CC versus 44% for non-CC), although only 4 people had HCV genotype 1b and CC genotype. Breakthrough rates were higher in people who did not receive ribavirin, and in HCV genotype 1a versus 1b. Resistance to danoprevir/r was observed in all patients who experienced viral breakthrough; mericitabine resistance was found in one person.
Almost all participants had more than one adverse event; a total of 567 mild-to-moderate events were reported among 83 people. The most common side effects, occurring in >10% of people were headache, fatigue, nausea, diarrhea, colds, insomnia, itching, weakness, dizziness, irritability, shortness of breath, cough, upset stomach, painful joints, and vomiting. As for laboratory abnormalities, one person experienced grade 3 anemia, four people had grade 3 lipid elevations, and one case each of grade 3 elevations in phosphate and lipase were observed.
A single serious adverse event, multiple myeloma, occurred 53 days after treatment completion and one person discontinued due to pain in the back of the throat (it was not specified whether or not this was a treatment-related adverse event).
- Everson G, Cooper C, Shiffman ML, et al. Rapid and sustained achievement of undetectable HCV RNA during treatment with ritonavir-boosted danoprevir/PEG-IFNa-2A/RBV in HCV genotype 1 or 4 patients: Dauphine week 36 interim analysis (Abstract 1177). Paper presented at: 47th Annual Meeting of the European Association for the Study of the Liver; 2012 April 18–22; Barcelona, Spain. Available from: http://mobile.ilcapp.eu/EASL_161/poster_24544/program.aspx. (Accessed 2012 June 25)
- Gane EJ, Pockros P, Zeuzem S, et al. Interferon-free treatment with combination of mericitabine and danoprevir/r with or without ribavirin in treatment-naïve HCV genotype-1 infected patients (Abstract 1412). 47th Annual Meeting of the European Association for the Study of the Liver; 2012 April 18–22; Barcelona, Spain. Available from:http://mobile.ilcapp.eu/EASL_161/poster_24848/program.aspx. (Accessed 2012 June 25)
Non- limiting examples of suitable HCV protease inhibitors include ACH-1095
(Achillion), ACH-1625 (Achillion), ACH-2684 (Achillion), AVL-181 (Avila), AVL-192 (Avila), BI-201335 (Boehringer Ingelheim), BMS-650032 (BMS), boceprevir, danoprevir, GS- 9132 (Gilead), GS-9256 (Gilead), GS-9451 (Gilead), IDX-136 (Idenix), IDX-316 (Idenix), IDX- 320 (Idenix), MK-5172 (Merck), narlaprevir, PHX-1766 (Phenomix), telaprevir, TMC-435 (Tibotec), vaniprevir, VBY708 (Virobay), VX-500 (Vertex), VX-813 (Vertex), VX-985 (Vertex), or a combination thereof. Non-limiting examples of suitable HCV polymerase inhibitors include ANA-598 (Anadys), BI-207127 (Boehringer Ingelheim), BILB-1941 (Boehringer Ingelheim), BMS-791325 (BMS), filibuvir, GL59728 (Glaxo), GL60667 (Glaxo), GS-9669 (Gilead), IDX-375 (Idenix), MK-3281 (Merck), tegobuvir, TMC-647055 (Tibotec), VCH-759 (Vertex & ViraChem), VCH-916 (ViraChem), VX-222 (VCH-222) (Vertex & ViraChem), VX-759 (Vertex), GS-6620 (Gilead), IDX-102 (Idenix), IDX-184 (Idenix), INX-189 (Inhibitex), MK-0608 (Merck), PSI-938 (Pharmasset), RG7128 (Roche), TMC64912 (Medivir), GSK625433 (Glaxo SmithKline), BCX-4678 (BioCryst), ALS-2200 (Alios BioPharma/Vertex), ALS-2158 (Alios BioPharma/Vertex), or a combination thereof. A polymerase inhibitor may be a nucleotide polymerase inhibitor, such as GS-6620 (Gilead), IDX-102 (Idenix), IDX-184 (Idenix), INX-189 (Inhibitex), MK-0608 (Merck), PSI-938 (Pharmasset), RG7128 (Roche), TMC64912 (Medivir), ALS-2200 (Alios BioPharma/Vertex), ALS-2158 (Alios BioPharma/Vertex), or a combination therefore. A polymerase inhibitor may also be a non- nucleoside polymerase inhibitor, such as ANA-598 (Anadys), BI-207127 (Boehringer Ingelheim), BILB-1941 (Boehringer Ingelheim), BMS-791325 (BMS), filibuvir, GL59728 (Glaxo), GL60667 (Glaxo), GS-9669 (Gilead), IDX-375 (Idenix), MK-3281 (Merck), tegobuvir, TMC-647055 (Tibotec), VCH-759 (Vertex & ViraChem), VCH-916 (ViraChem), VX-222 (VCH-222) (Vertex & ViraChem), VX-759 (Vertex), or a combination thereof. Non-limiting examples of suitable NS5A inhibitors include GSK62336805 (Glaxo SmithKline), ACH-2928 (Achillion), AZD2836 (Astra-Zeneca), AZD7295 (Astra-Zeneca), BMS-790052 (BMS), BMS- 824393 (BMS), GS-5885 (Gilead), PPI-1301 (Presidio), PPI-461 (Presidio), or a combination thereof. Non-limiting examples of suitable cyclophilin inhibitors include alisporovir (Novartis & Debiopharm), NM-811 (Novartis), SCY-635 (Scynexis), or a combination thereof. Non-limiting examples of suitable HCV entry inhibitors include ITX-4520 (iTherx), ITX-5061 (iTherx), or a combination thereof.
WO 2007015824WO 2003053349WO 2005095403WO 2005037214WO 2005095403WO 2005037214WO 2003053349WO 2007015824WO 2008128921
| US8048862 | 14 Apr 2009 | 1 Nov 2011 | Intermune, Inc. | Macrocyclic inhibitors of hepatitis C virus replication |
| US8119592 | 10 Oct 2006 | 21 Feb 2012 | Intermune, Inc. | Compounds and methods for inhibiting hepatitis C viral replication |
| US8232246 | 30 Jun 2009 | 31 Jul 2012 | Abbott Laboratories | Anti-viral compounds |
| US8299021 | 19 Apr 2012 | 30 Oct 2012 | Intermune, Inc. | Macrocyclic inhibitors of hepatitis C virus replication |
| US8420596 | 10 Sep 2009 | 16 Apr 2013 | Abbott Laboratories | Macrocyclic hepatitis C serine protease inhibitors |
| WO2013106631A1 | 11 Jan 2013 | 18 Jul 2013 | Abbvie Inc. | Processes for making hcv protease inhibitors |
Danoprevir Clinical Trial Information( data from http://clinicaltrials.gov)
| NCT Number | Recruitment | Conditions | Sponsor /Collaborators |
Start Date | Phases |
|---|---|---|---|---|---|
| NCT01331850 | Completed | Hepatitis C, Chronic | Hoffmann-La Roche | 2011-05 | Phase 2 |
| NCT01531647 | Completed | Healthy Volunteer | Hoffmann-La Roche | 2012-01 | Phase 1 |
| NCT01588002 | Completed | Healthy Volunteer | Hoffmann-La Roche | 2012-04 | Phase 1 |
| NCT01592318 | Active, not recruiting | Healthy Volunteer | Hoffmann-La Roche | 2012-05 | Phase 1 |
| NCT01749150 | Recruiting | Hepatitis C, Chronic | Hoffmann-La Roche | 2013-04 | Phase 2 |

WANT TO KNOW ABOUT VIR SERIES CLICK
click
http://drugsynthesisint.blogspot.in/p/vir-series-hep-c-virus-22.html
AND
http://medcheminternational.blogspot.in/p/vir-series-hep-c-virus.html
APREMILAST, … ORALLY ACTIVE PDE4 INHIBITOR

APREMILAST
PDE4 inhibitor
N-{2-[(1S)-1-(3-Ethoxy-4-methoxyphenyl)-2-(methylsulfonyl)ethyl]-1,3-dioxo-2,3-dihydro-1H-isoindol-4-yl}acetamide
(+)-2-[l-(3-ethoxy-4-methoxyphenyl)-2- methanesulfonylethyl]-4-acetylaminoisoindolin-l,3-dione,
(S)—N-{2-[1-(3-ethoxy-4-methoxy-phenyl)-2-methanesulfonylethyl]-1,3-dioxo-2,3-dihydro-1H-isoindol-4-yl}acetamide
(S)-N-{2-[1-(3-Ethoxy-4-methoxyphenyl)-2-methanesulfonylethyl]-1,3-dioxo-2,3-dihydro-1H-isoindol-4-yl}acetamide
Molecular Formula: C22H24N2O7S Molecular Weight: 460.50016
608141-41-9 CAS NO
Celgene (Originator)
CC-10004 (apremilast) is an oral compound that is being studied in multiple Phase III clinical trials for the treatment of psoriasis, psoriatic arthritis and other chronic inflammatory diseases. We successfully completed our early stage studies, demonstrating clinical activity and tolerability and meeting safety endpoints in a placebo controlled proof-of mechanism trial in moderate-to-severe psoriasis and psoriatic arthritis. With the initiation of six multi-center international clinical trials, we are advancing the clinical development of CC-10004.
CC-10004, , Apremilast (USAN), SureCN302992, Apremilast (CC-10004), QCR-202,
- Apremilast
- CC 10004
- CC-10004
- CC10004
- UNII-UP7QBP99PN
- CLINICAL TRIALS….http://clinicaltrials.gov/search/intervention=Apremilast+OR+CC-10004
Apremilast is an orally available small molecule inhibitor of PDE4 being developed byCelgene for ankylosing spondylitis, psoriasis, and psoriatic arthritis.[1][2] The drug is currently in phase III trials for the three indications. Apremilast, an anti-inflammatory drug, specifically inhibits phosphodiesterase 4. In general the drug works on an intra-cellular basis to moderate proinflammatory and anti-inflammatory mediator production.
 APREMILAST
APREMILAST
Apremilast is being tested for its efficacy in treating “psoriasis, psoriatic arthritis and other chronic inflammatory diseases such as ankylosing spondylitis, Behcet’s disease, and rheutmatoid arthritis.
“Apremilast is Celgene’s lead oral phosphodiesterase IV inhibitor and anti-TNF alpha agent in phase III clinical studies at Celgene for the oral treatment of moderate to severe plaque-type psoriasis and for the oral treatment of psoriatic arthritis.
Early clinical development is also ongoing for the treatment of acne, Behcet’s disease, cutaneous sarcoidosis, prurigo nodularis, ankylosing spondylitis, atopic or contact dermatitis and rheumatoid arthritis. No recent development has been reported for research for the treatment of skin inflammation associated with cutaneous lupus erythematosus.
In 2011, Celgene discontinued development of the compound for the management of vision-threatening uveitis refractory to other modes of systemic immunosuppression due to lack of efficacy.
Celgene had been evaluating the potential of the drug for the treatment of asthma; however, no recent development has been reported for this research. The drug candidate is also in phase II clinical development at the William Beaumont Hospital Research Institute for the treatment of chronic prostatitis or chronic pelvic pain syndrome and for the treatment of vulvodynia (vulvar pain).
In 2013, orphan drug designations were assigned to the product in the U.S. and the E.U. for the treatment of Behcet’s disease.
Celgene Corp has been boosted by more impressive late-stage data on apremilast, an oral drug for psoriatic arthritis, this time in previously-untreated patients.
The company is presenting data from the 52-week PALACE 4 Phase III study of apremilast tested in PsA patients who have not taken systemic or biologic disease modifying antirheumatic drugs (DMARDs) at the American College of Rheumatology meeting in San Diego. The results from the 527-patient trial show that at week 16, patients on 20mg of the first-in-class oral inhibitor of phosphodiesterase 4 (PDE4) achieved an ACR20 (ie a 20% improvement in the condition) response of 29.2% and 32.3% for 30mg aapremilast, compared with 16.9% for those on placebo.
After 52 weeks, 53.4% on the lower dose and 58.7% on 30mg achieved an ACR20 response. ACR50 and 70 was reached by 31.9% and 18.1% of patients, respectively, for apremilast 30mg. The compound was generally well-tolerated and discontinuation rates for diarrhoea and nausea were less than 2% over 52 weeks.
Commenting on the data, Alvin Wells, of the Rheumatology and Immunotherapy Center in Franklin, Wisconsin, noted that apremilast demonstrated long-term safety and tolerability and significant clinical benefit in treatment-naive patients. He added that “these encouraging results suggest that apremilast may have the potential to be used alone and as a first-line therapy”. Celgene is also presenting various pooled data from the first three trials in the PALACE programme which, among other things, shows that apremilast significantly improves swollen and tender joints.
Treatment for PSA, which affects about 30% of the 125 million people worldwide who have psoriasis, currently involves injectable tumour necrosis factor (TNF) inhibitors, notably AbbVie’s Humira (adalimumab) and Pfizer/Amgen’s Enbrel (etanercept), once patients have not responded to DMARDs (at least in the UK). While the biologics are effective, the side effect profile can be a concern, due to the risk of infection and tuberculosis and many observers believe that apremilast will prove popular with patients and doctors due to the fact that it is oral, not injectable.
Apremilast was filed for PsA with the US Food and Drug Administration in the first quarter and will be submitted on both sides of the Atlantic for psoriasis before year-end. The European filing will also be for PsA.
Apremilast impresses for Behcet’s disease
Celgene has also presented promising Phase II data on apremilast as a treatment for the rare inflammatory disorder Behcet’s disease. 71% of patients achieved complete response at week 12 in clearing oral ulcers
 APREMILAST
APREMILAST
- “Apremilast Palace Program Demonstrates Robust and Consistent Statistically Significant Clinical Benefit Across Three Pivotal Phase III Studies (PALACE-1, 2 & 3) in Psoriatic Arthritis” (Press release). Celgene Corporation. 6 September 2012. Retrieved 2012-09-10.
- “US HOT STOCKS: OCZ, VeriFone, Men’s Wearhouse, AK Steel, Celgene”. The Wall Street Journal. 6 September 2012. Retrieved 2012-09-06.
- Discovery of (S)-N-[2-[1-(3-ethoxy-4-methoxyphenyl)-2-methanesulfonylethyl]-1,3-dioxo-2,3-dihydro-1H-isoindol-4-yl] acetamide (apremilast), a potent and orally active phosphodiesterase 4 and tumor necrosis factor-alpha inhibitor.
Man HW, Schafer P, Wong LM, Patterson RT, Corral LG, Raymon H, Blease K, Leisten J, Shirley MA, Tang Y, Babusis DM, Chen R, Stirling D, Muller GW.
J Med Chem. 2009 Mar 26;52(6):1522-4. doi: 10.1021/jm900210d.
- Therapeutics: Silencing psoriasis.Crow JM.Nature. 2012 Dec 20;492(7429):S58-9. doi: 10.1038/492S58a. No abstract available.
- NMR…http://file.selleckchem.com/downloads/nmr/S803401-Apremilast-HNMR-Selleck.pdf
- WO 2003080049
- WO 2013126495
- WO 2013126360
- WO 2003080049
- WO 2006065814
- US2003/187052 A1 …..MP 144 DEG CENT
- US2007/155791
-
J. Med. Chem., 2008, 51 (18), pp 5471–5489DOI: 10.1021/jm800582j
-
J. Med. Chem., 2011, 54 (9), pp 3331–3347DOI: 10.1021/jm200070e

…………………………………………
INTRODUCTION
2-[l-(3-ethoxy-4-methoxyphenyl)-2-methylsulfonylethyl]-4- acetylaminoisoindoline-l ,3-dione is a PDE4 inhibitor that is currently under investigation as an anti-inflammatory for the treatment of a variety of conditions, including asthma, chronic obstructive pulmonary disease, psoriasis and other allergic, autoimmune and rheumatologic conditions. S-enantiomer form of 2-[l-(3-ethoxy-4-methoxyphenyl)-2-methylsulfonylethyl]-4- acetylaminoisoindoline-l ,3-dione can be prepared by reacting (5)-aminosulfone 1 with intermediate 2.

Existing methods for synthesizing (S)-aminosulfone 1 involve resolution of the corresponding racemic aminosulfone by techniques known in the art. Examples include the formation and crystallization of chiral salts, and the use of chiral high performance liquid chromatography. See, e.g., Jacques, J., et al., Enantiomers, Racemates and Resolutions (Wiley Interscience, New York, 1981); Wilen, S. H., et al, Tetrahedron 33:2725 (1977); Eliel, E. L., Stereochemistry of Carbon Compounds (McGraw Hill, NY, 1962); and Wilen, S. H., Tables of Resolving Agents and Optical Resolutions p. 268 (E.L. Eliel, Ed., Univ. of Notre Dame Press, Notre Dame, IN, 1972). In one example, as depicted in Scheme 1 below, (5)-aminosulfone 1 is prepared by resolution of racemic aminosulfone 3 with N-Ac-L-Leu. Racemic aminosulfone 3 is prepared by converting 3-ethoxy-4-methoxybenzonitrile 4 to enamine intermediate 5 followed by enamine reduction and borate hydrolysis. This process has been reported in U.S. Patent
Application Publication No. 2010/0168475.

CH2CI2, NaOH

Scheme 1
The procedure for preparing an enantiomerically enriched or enantiomerically pure aminosulfone, such as compound 1, may be inefficient because it involves the resolution of racemic aminosulfone 3. Thus, a need exists as to asymmetric synthetic processes for the preparation of an enantiomerically enriched or enantiomerically pure aminosulfone, particularly for manufacturing scale production. Direct catalytic asymmetric hydrogenation of a suitable enamine or ketone intermediate is of particular interest because it eliminates the need for either classic resolution or the use of stoichiometric amount of chiral auxiliary, and thus, may be synthetically efficient and economical.
……………………………………….
SYNTHESIS OF KEY INTERMEDIATE
Example 1
Synthesis of 1 -(3-ethoxy-4-methoxyphenyl)-2-(methylsulfonyl)ethenamine

[00232] A slurry of dimethylsulfone (85 g, 903 mmol) in THF (480 ml) was treated with a
1.6M solution of n-butyllithium in hexane (505 ml, 808 mmol) at 0 – 5 °C. The resulting mixture was agitated for 1 hour then a solution of 3-ethoxy-4-methoxybenzonitrile (80 g, 451 mmol) in THF (240 ml) was added at 0 – 5 °C. The mixture was agitated at 0 – 5 °C for 0.5 hour, warmed to 25 – 30 °C over 0.5 hour and then agitated for 1 hour. Water (1.4 L) was added at 25 – 30 °C and the reaction mass was agitated overnight at room temperature (20 – 30 °C). The solid was filtered and subsequently washed with a 2: 1 mixture of water :THF (200 ml), water (200 ml) and heptane (2 x 200 ml). The solid was dried under reduced pressure at 40 – 45 °C to provide the product as a white solid (102 g, 83% yield); 1H NMR (DMSO-d6) δ 1.34 (t, J=7.0 Hz, 3H), 2.99 (s, 3H), 3.80 (s, 3H), 4.08 (q, J=7.0 Hz, 2H), 5.03 (s, 1H), 6.82 (s, 2H), 7.01 (d, J=8.5 Hz, 1H), 7.09 – 7.22 (m, 2H).
Example 2
Synthesis of (R)- 1 -(3-ethoxy-4-methoxyphenyl)-2-(methylsulfonyl)ethanamine

[00233] A solution of bis(l,5-cyclooctadiene)rhodium(I) trifluoromethanesulfonate (36 mg, 0.074 mmol) and (i?)-l-[(5)-2-(diphenylphosphino)ferrocenyl]ethyldi-tert-butylphosphine (40 mg, 0.074 mmol) in 25 mL of 2,2,2-trifluoroethanol was prepared under nitrogen. To this solution was then charged l-(3-ethoxy-4-methoxyphenyl)-2-(methylsulfonyl)ethenamine (2.0 g, 7.4 mmol). The resulting mixture was heated to 50 °C and hydrogenated under 90 psig hydrogen pressure. After 18 h, the mixture was cooled to ambient temperature and removed from the hydrogenator. The mixture was evaporated and the residue was purified by chromatography on a CI 8 reverse phase column using a water-acetonitrile gradient. The appropriate fractions were pooled and evaporated to -150 mL. To this solution was added brine (20 mL), and the resulting solution was extracted with EtOAc (3 x 50 mL). The combined organic layers were dried (MgS04) and evaporated to provide the product as a white crystalline solid (1.4 g, 70% yield); achiral HPLC (Hypersil BDS C8, 5.0 μπι, 250 x 4.6 mm, 1.5 mL/min, 278nm, 90/10 gradient to 80/20 0.1% aqueous TFA/MeOH over 10 min then gradient to 10/90 0.1% aqueous TFA/MeOH over the next 15 min): 9.11 (99.6%); chiral HPLC (Chiralpak AD-H 5.0 μιη Daicel, 250 x 4.6 mm, 1.0 mL/min, 280 nm, 70:30:0.1 heptane-z-PrOH-diethylamine): 7.32 (97.5%), 8.26 (2.47%); 1H NMR (DMSO-de) δ 1.32 (t, J= 7.0 Hz, 3H), 2.08 (s, 2H), 2.96 (s, 3H), 3.23 (dd, J= 3.6, 14.4 Hz, 1H), 3.41 (dd, J= 9.4, 14.4 Hz, 1H), 3.73 (s, 3H), 4.02 (q, J= 7.0 Hz, 2H), 4.26 (dd, J= 3.7, 9.3 Hz, 1H), 6.89 (s, 2H), 7.02 (s, 1H); 13C NMR (DMSO-d6) δ 14.77, 41.98, 50.89, 55.54, 62.03, 63.68, 111.48, 111.77, 118.36, 137.30, 147.93, 148.09. Example 3
Synthesis of (6 -l-(3-ethoxy-4-methoxyphenyl)-2-(methylsulfonyl)ethanamine N-Ac-L-Leu salt

[00234] A solution of bis(l,5-cyclooctadiene)rhodium(I) trifluoromethanesulfonate (17 mg, 0.037 mmol) and (5)-l-[(i?)-2-(diphenylphosphino)ferrocenyl]ethyldi-tert-butylphosphine (20 mg, 0.037 mmol) in 10 mL of 2,2,2-trifluoroethanol was prepared under nitrogen. To this solution was then charged l-(3-ethoxy-4-methoxyphenyl)-2-(methylsulfonyl)ethenamine (2.0 g, 7.4 mmol). The resulting mixture was heated to 50 °C and hydrogenated under 90 psig hydrogen pressure. After 18 h, the mixture was cooled to ambient temperature and removed from the hydrogenator. Ecosorb C-941 (200 mg) was added and the mixture was stirred at ambient temperature for 3 h. The mixture was filtered through Celite, and the filter was washed with additional trifluoroethanol (2 mL). Then, the mixture was heated to 55 °C, and a solution of N- acetyl-L-leucine (1.3 g, 7.5 mmol) was added dropwise over the course of 1 h. Stirring proceeded at the same temperature for 1 h following completion of the addition, and then the mixture was cooled to 22 °C over 2 h and stirred at this temperature for 16 h. The crystalline product was filtered, rinsed with methanol (2 x 5 mL), and dried under vacuum at 45 °C to provide the product as a white solid (2.6 g, 80% yield); achiral HPLC (Hypersil BDS Cg, 5.0 μιη, 250 x 4.6 mm, 1.5 mL/min, 278nm, 90/10 gradient to 80/20 0.1% aqueous TFA/MeOH over 10 min then gradient to 10/90 0.1% aqueous TFA/MeOH over the next 15 min): 8.57 (99.8%); chiral HPLC (Chiralpak AD-H 5.0 μιη Daicel, 250 x 4.6 mm, 1.0 mL/min, 280 nm, 70:30:0.1 heptane-z-PrOH-diethylamine): 8.35 (99.6%); 1H NMR (DMSO-<¾) δ 0.84 (d, 3H), 0.89 (d, J= 6.6 Hz, 3H), 1.33 (t, J= 7.0 Hz, 3H), 1.41 – 1.52 (m, 2H), 1.62 (dt, J= 6.7, 13.5 Hz, 1H), 1.83 (s, 3H), 2.94 (s, 3H), 3.28 (dd, J= 4.0, 14.4 Hz, 1H), 3.44 (dd, J= 9.1, 14.4 Hz, 1H), 3.73 (s, 3H), 4.02 (q, J= 6.9 Hz, 2H), 4.18 (q, J= 7.7 Hz, 1H), 4.29 (dd, J= 4.0, 9.1 Hz, 1H), 5.46 (br, 3H), 6.90 (s, 2H), 7.04 (s, 1H), 8.04 (d, J= 7.9 Hz, 1H); Anal. (C20H34N2O7S) C, H, N. Calcd C, 53.79; H, 7.67; N 6.27. Found C, 53.78; H, 7.57; N 6.18.
SUBSEQUENT CONVERSION
S-enantiomer form of 2-[l-(3-ethoxy-4-methoxyphenyl)-2-methylsulfonylethyl]-4- acetylaminoisoindoline-l ,3-dione can be prepared by reacting (5)-aminosulfone 1 with intermediate 2.
……………………………………
 APREMILAST
APREMILAST
GENERAL SYNTHESIS AND SYNTHESIS OF APREMILAST




(apremilast)
[0145] Preparation of 3-Ethoxy-4-methoxybenzonitrile (Compound 2). 3-Ethoxy-
4-methoxybenzaldehyde (Compound 1, 10.0 gm, 54.9 mmol, Aldrich) and hydroxylamine hydrochloride (4.67 gm, 65.9 mmol, Aldrich) were charged to a 250 mL three-necked flask at room temperature, followed by the addition of anhydrous acetonitrile (50 mL). The reaction mixture was stirred at room temperature for thirty minutes and then heated to reflux (oil bath at 85 °C). After two hours of reflux, the reaction mixture was cooled to room temperature, and added 50 mL of deionized water. The mixture was concentrated under reduced pressure to remove acetonitrile and then transferred to a separatory funnel with an additional 80 mL of deionized water and 80 mL dichloromethane. The aqueous layer was extracted with dichloromethane (3 x 50 mL). The combined organic layers were washed successively with water (80 mL) and saturated sodium chloride (80 mL). The organic layer was dried over anhydrous sodium sulfate (approximately 20 gm). The organic layer was filtered and concentrated under reduced pressure to give a yellow oil. Purification by silica gel chromatography (0 to 1 % MeOH/DCM ) afforded 3-Ethoxy-4-methoxybenzonitrile
(Compound 2) as a white solid (7.69 gm, 79 % yield). MS (ESI positive ion) m/z 178.1 (M + 1). HPLC indicated >99% purity by peak area. 1H-NMR (500 MHz, DMSO-c¾: δ ppm 1.32 (t, 3H), 3.83 (s, 3H), 4.05 (q, 2H), 7.10 (d, J = 8.0 Hz, 1H), 7.35 (d, J = 2.0 Hz, 1H), 7.40 (dd, J = 2.0 Hz, 1H).
[0146] Preparation of l-(3-Ethoxy-4-methoxyphenyi)-2-
(niethylsulfonyl)ethanamine (Compound 3). Dimethyl sulfone (2.60 gm, 27.1 mmol, Aldrich) and tetrahydrofuran (10 mL, Aldrich) were charged to a 250 mL three-necked flask at room temperature. The mixture was cooled to 0 – 5 °C, and the solution gradually turned white. n-Butyllithium (10.8 mL, 27.1 mmol, 2.5 M solution in hexanes, Aldrich) was added to the flask at a rate such that the reaction mixture was maintained at 5 – 10 °C. The mixture was stirred at 0 – 5 °C for one hour, turning light-yellow. 3-Ethoxy-4-methoxybenzonitrile (Compound 2, 4.01 gm, 22.5 mmol) in tetrahydrofuran (8 mL) was then charged to the flask at a rate such that the reaction mixture was maintained at 0 – 5 °C. The mixture was stirred at 0 – 5 °C for another 15 minutes. After warming to room temperature, the reaction mixture was stirred for another 1.5 hours and then transferred to a second 250 mL three-necked flask containing a suspension of sodium borohydride (1.13 gm, 29.3 mmol, Aldrich) in
tetrahydrofuran (1 1 mL), maintained at – 5 – 0 °C for 30 minutes. Trifluoroacetic acid (“TFA,” 5.26 mL, 68.3 mmol, Aldrich) was charged to the flask at a rate such that the reaction mixture was maintained at 0 – 5 °C. The mixture was stirred at 0 – 5 °C for 40 minutes and an additional 17 hours at room temperature. The reaction mixture was then charged with 2.7 mL of deionized water over five minutes at room temperature. The mxiture was stirred at room temperature for 15 hours. Aqueous NaOH (10 N, 4.9 mL) was charged to the flask over 15 minutes at 45 °C. The mixture was stirred at 45 °C for two hours, at 60 °C for 1.5 hours, and at room temperature overnight. After approximately 17 hours at room temperature the mixture was cooled to 0 °C for thirty minutes and then concentrated under reduced pressure. The residual material was charged with deionized water (3 mL) and absolute ethanol (3 mL) and stirred at 0 – 5 °C for 2 hours. The mixture was filtered under vacuum, and the filtered solid was washed with cold absolute ethanol (3 x 5 mL), followed by deionized water until the pH of the wash was about 8. The solid was air dried overnight, and then in a vacuum oven at 60 °C for 17 hours to afford Compound 3 as a white solid (4.75 gm, 77 %). MS (ESI positive ion) m/z 274.1 (M + 1). Ή-NMR (500 MHz, DMSO-c¾): δ ppm 1.32 (t, J = 7.0 Hz, 3H), 2.08 (bs, 2H), 2.95 (s, 3H), 3.23 (dd, J = 4.0 Hz, 1H), 3.40 (dd, J = 9.5 Hz, 1H), 3.72 (s, 3H), 4.01 (q, J = 7.0 Hz, 2H), 4.25 (dd, J = 3.5 Hz, 1H), 6.88 (s, 2H), 7.02 (s, 1H).
[0147] Preparation of 4-Nitroisobenzofuran-l,3-dione (Compound 5). Into a 250 mL round bottom flask, fitted with a reflux condenser, was placed 3-nitrophthalic acid (21.0 gm, 99 mmol, Aldrich) and acetic anhydride (18.8 mL, 199 mmol, Aldrich). The solid mixture was heated to 85 °C, under nitrogen, with gradual melting of the solids. The yellow mixture was heated at 85 °C for 15 minutes, and there was noticeable thickening of the mixture. After 15 minutes at 85 °C, the hot mixture was poured into a weighing dish, and allowed to cool. The yellow solid was grinded to a powder and then placed on a cintered funnel, under vacuum. The solid was washed with diethyl ether (3 x 15 mL), under vacuum and allowed to air dry overnight, to afford 4-nitroisobenzofuran-l ,3-dione, Compound 5, as a light-yellow solid (15.8 gm, 82 %). MS (ESI positive ion) m/z 194.0 (M + 1). TLC: Rf = 0.37 (10% MeOH/DCM with 2 drops Acetic acid) Ή-NMR (500 MHz, DMSO-i¾: δ ppm 8.21 (dd, J = 7.5 Hz, 1H), 8.39 (dd, J = 7.5 Hz, 1H), 8.50 (dd, J = 7.5 Hz, 1 H).
[0148] Preparation of 2-(l-(3-Ethoxy-4-methoxyphenyI)-2-
(methylsulfonyl)ethyl)-4-nitroisoindoline-l,3-dione (Compound 6). Into a 2 – 5 mL microwave vial was added 4-nitroisobenzofuran-l ,3-dione (Compound 5, 0.35 gm, 1.82 mmol), the amino-sulfone intermediate (Compound 3, 0.50 gm, 1.82 mmol) and 4.0 mL of glacial acetic acid. The mixture was placed in a microwave at 125 °C for 30 minutes. After 30 minutes the acetic acid was removed under reduced pressure. The yellow oil was taken up in ethyl acetate and applied to a 10 gm snap Biotage samplet. Purification by silica gel chromatography (0 to 20 % Ethyl Acetate/Hexanes) afforded Compound 6 as a light-yellow solid (0.67 gm, 82 %). MS (ESI positive ion) m/z 449.0 (M + 1). TLC: Rf = 0.19
(EtOAc:Hexanes, 1 : 1). HPLC indicated 99% purity by peak area. Ή-NMR (500 MHz, DMSO-c¾: δ ppm 1.32 (t, 3H), 2.99 (s, 3H), 3.73 (s, 3H), 4.02 (m, 2H), 4.21 (dd, J = 5.0 Hz, 1H), 4.29 (dd, J = 10.0 Hz, 1H), 5.81 (dd, J = 5.0 Hz, 1H), 6.93 (d, J – 8.5 Hz, 1H), 7.00 (dd, J = 2.0 Hz, 1H), 7.10 (d, J = 2.5 Hz, 1H), 8.07 (t, J = 15.5 Hz, 1H), 8.19 (dd, J = 8.5 Hz, 1H), 8.30 (dd, J = 9.0 Hz, 1H).
[0149] Preparation of 4-Amino-2-(l-(3-ethoxy-4-methoxyphenyl)-2-
(methylsulfonyl)ethyl)isoindoline-l,3-dione (Compound 7). Compound 6 (0.54 gm, 1.20 mmol) was taken up in ethyl acetate / acetone (1 : 1 , 24 mL) and flowed through the H-cube™ hydrogen reactor using a 10 % Pd/C CatCart™ catalyst cartridge system (ThalesNano, Budapest Hungary). After eluting, the yellow solvent was concentrated under reduced pressure to give Compound 7 as a yellow foam solid (0.48 gm, 95 %). MS (ESI positive ion) m/z 419.1 (M + 1). 1H-NMR (500 MHz, DMSO-<¾): δ ppm 1.31 (t, J = 7.0 Hz, 3H), 2.99 (s, 3H), 3.72 (s, 3H), 4.04 (q, J = 7.0 Hz, 2H), 4.09 (m, 1H), 4.34 (m, 1H), 5.71 (dd, J = 5.5 Hz, 1H), 6.52 (bs, 2H), 6.92-6.98 (m, 3H), 7.06 (bs, 1 H), 7.42 (dd, J = 7.0 Hz, 1H).
[0150] Preparation of N-(2-(l-(3-ethoxy-4-methoxyphenyl)-2-
(methylsuIfonyl)ethyl)-l,3-dioxoisoindolin-4-yl)acetamide (Apremilast, Compound 8).
Into a 2-5 mL microwave vial was placed Compound 7 (0.18 gm, 0.43 mmol), acetic anhydride (0.052 mL, 0.53 mmol) and acetic acid (4 mL). The microwave vial was placed into a Biotage microwave and heated to 125 °C for 30 minutes. The solvents were removed under reduced pressure and the residue was purified by silica gel chromatography (0 to 5% MeOH/DCM) to afford apremilast (Compound 8) as a yellow oil (0.14 gm, 71%). HPLC indicated 94.6% purity by peak area.
1H-NMR (500 MHz, DMSO-c 6): δ ppm 1.31 (t, 3H), 2.18 (s, 3H), 3.01 (s, 3H), 3.73 (s, 3H), 4.01 (t, J = 7.0 Hz, 2H), 4,14 (dd, J = 4.0 Hz, 1H), 4.33 (m, 1H), 5.76 (dd, J = 3.0 Hz, 1H), 6.95 (m, 2H), 7.06 (d, J = 1.5 Hz, 1H), 7.56 (d, J = 7.0 Hz, 1H), 7.79 (t, J = 7.7 Hz, 1H), 8.43 (d, J = 8.5 Hz, 1H), 9.72 (bs, 1H).
……………………..
SYNTHESIS
5. EXAMPLES
Certain embodiments provided herein are illustrated by the following non-limiting examples.
5.1 PREPARATION OF (+)-2-[l-(3-ETHOXY-4-METHOXYPHENYL)-2- METHANESULFONYLETHYLJ-4- ACETYL AMINOISOINDOLIN-1,3- DIONE (APREMILAST)

5.1.1 Preparation of 3-aminopthalic acid
10% Pd/C (2.5 g), 3-nitrophthalic acid (75.0 g, 355 mmol) and ethanol (1.5 L) were charged to a 2.5 L Parr hydrogenator under a nitrogen atmosphere. Hydrogen was charged to the reaction vessel for up to 55 psi. The mixture was shaken for 13 hours, maintaining hydrogen pressure between 50 and 55 psi. Hydrogen was released and the mixture was purged with nitrogen 3 times. The suspension was filtered through a celite bed and rinsed with methanol. The filtrate was concentrated in vacuo. The resulting solid was reslurried in ether and isolated by vacuum filtration. The solid was dried in vacua to a constant weight, affording 54 g (84%> yield) of 3-aminopthalic acid as a yellow product. 1H-NMR (DMSO-d6) δ: 3.17 (s, 2H), 6.67 (d, 1H), 6.82 (d, 1H), 7.17 (t, 1H), 8-10 (brs, 2H). 13C-NMR(DMSO-d6) δ: 112.00, 115.32, 118.20, 131.28, 135.86, 148.82, 169.15, 170.09.
5.1.2 Preparation of 3-acetamidopthalic anhydride
A I L 3 -necked round bottom flask was equipped with a mechanical stirrer, thermometer, and condenser and charged with 3-aminophthalic acid (108 g, 596 mmol) and acetic anhydride (550 mL). The reaction mixture was heated to reflux for 3 hours and cooled to ambient temperature and further to 0-5. degree. C. for another 1 hour. The crystalline solid was collected by vacuum filtration and washed with ether. The solid product was dried in vacua at ambient temperature to a constant weight, giving 75 g (61% yield) of 3-acetamidopthalic anhydride as a white product. 1H-NMR (CDCI3) δ: 2.21 (s, 3H), 7.76 (d, 1H), 7.94 (t, 1H), 8.42 (d, 1H), 9.84 (s, 1H).
5.1.3 Resolution of 2-(3-ethoxy-4-methoxyphenyl)-l-(methylsulphonyl)- ethyl-2-amine
A 3 L 3 -necked round bottom flask was equipped with a mechanical stirrer, thermometer, and condenser and charged with 2-(3-ethoxy-4-methoxyphenyl)-l-(methylsulphonyl)-eth-2-ylamine (137.0 g, 500 mmol), N-acetyl-L-leucine (52 g, 300 mmol), and methanol (1.0 L). The stirred slurry was heated to reflux for 1 hour. The stirred mixture was allowed to cool to ambient temperature and stirring was continued for another 3 hours at ambient temperature. The slurry was filtered and washed with methanol (250 mL). The solid was air-dried and then dried in vacuo at ambient temperature to a constant weight, giving 109.5 g (98% yield) of the crude product (85.8% ee). The crude solid (55.0 g) and methanol (440 mL) were brought to reflux for 1 hour, cooled to room temperature and stirred for an additional 3 hours at ambient temperature. The slurry was filtered and the filter cake was washed with methanol (200 mL). The solid was air-dried and then dried in vacuo at 30°C. to a constant weight, yielding 49.6 g (90%> recovery) of (S)-2-(3-ethoxy-4- methoxyphenyl)-l-(methylsulphonyl)-eth-2-ylamine-N-acety 1-L-leucine salt (98.4% ee). Chiral HPLC (1/99 EtOH/20 mM KH2P04 @pH 7.0, Ultron Chiral ES-OVS from Agilent Technologies, 150 mm.times.4.6 mm, 0.5 mL/min., @240 nm): 18.4 min (S-isomer, 99.2%), 25.5 min (R-isomer, 0.8%)
5.1.4 Preparation of (+)-2-[l-(3-ethoxy-4-methoxyphenyl)-2- methanesulfonylethyl] -4-acetylaminoisoindolin- 1 ,3-dione
A 500 mL 3 -necked round bottom flask was equipped with a mechanical stirrer,
thermometer, and condenser. The reaction vessel was charged with (S)-2-(3-ethoxy-4- methoxyphenyl)-l-(methylsulphonyl)-eth-2-yl amine N-acetyl-L-leucine salt (25 g, 56 mmol, 98% ee), 3-acetamidophthalic anhydride (12.1 g, 58.8 mmol), and glacial acetic acid (250 mL). The mixture was refluxed over night and then cooled to <50°C. The solvent was removed in vacuo, and the residue was dissolved in ethyl acetate. The resulting solution was washed with water (250 mL x
2), saturated aqeous NaHC03 (250 mL.times.2), brine (250 mL.times.2), and dried over sodium sulphate. The solvent was evaporated in vacuo, and the residue recrystallized from a binary solvent containing ethanol (150 mL) and acetone (75 mL). The solid was isolated by vacuum filtration and washed with ethanol (100 mL.times.2). The product was dried in vacuo at 60°C. to a constant weight, affording 19.4 g (75% yield) of Compound 3 APREMILAST with 98% ee. Chiral HPLC (15/85 EtOH/20 mM KH2P04 @pH 3.5, Ultron Chiral ES-OVS from Agilent Technology, 150 mm x 4.6 mm, 0.4 mL/min., @240 nm): 25.4 min (S-isomer, 98.7%), 29.5 min (R-isomer, 1.2%).
1H-NMR (CDC13) δ: 1.47 (t, 3H), 2.26 (s, 3H), 2.87 (s, 3H), 3.68-3.75 (dd, 1H), 3.85 (s, 3H), 4.07-4.15 (q, 2H), 4.51-4.61 (dd, 1H), 5.84-5.90 (dd, 1H), 6.82-8.77 (m, 6H), 9.46 (s, 1H).
13C-NMR(DMSO-d6) δ: 14.66, 24.92, 41.61, 48.53, 54.46, 55.91, 64.51, 111.44, 112.40, 115.10, 118.20, 120.28, 124.94, 129.22, 131.02, 136.09, 137.60, 148.62, 149.74, 167.46, 169.14, 169.48.
…………………………………..
NMR
1H-NMR (CDCl3) δ: 1.47 (t, 3H), 2.26 (s, 3H), 2.87 (s, 3H), 3.68-3.75 (dd, 1H), 3.85 (s, 3H), 4.07-4.15 (q, 2H), 4.51-4.61 (dd, 1H), 5.84-5.90 (dd, 1H), 6.82-8.77 (m, 6H), 9.46 (s, 1H). 13C-NMR (DMSO-d6) δ: 14.66, 24.92, 41.61, 48.53, 54.46, 55.91, 64.51, 111.44, 112.40, 115.10, 118.20, 120.28, 124.94, 129.22, 131.02, 136.09, 137.60, 148.62, 149.74, 167.46, 169.14, 169.48.
…………….

aReagents and conditions: (a) LiN(SiMe3)2, then Me2SO2/n-BuLi/BF3Et2O, −78 °C; (b) N-Ac-l-leucine, MeOH; (c) HOAc, reflux.
……………………
SARCOIDOSIS
Sarcoidosis is a disease of unknown cause. Sarcoidosis is characterized by the presence of granulomas in one or more organ systems. The most common sites of involvement are the lungs and the lymph nodes in the mediastinum and hilar regions. However, sarcoidosis is a systemic disease and a variety of organ systems or tissues may be the source of primary or concomitant clinical manifestations and morbidity. The clinical course of sarcoidosis is extremely variable, and ranges from a mild or even asymptomatic disease with spontaneous resolution to a chronic progressive disease leading to organ system failure and, in 1-5% of cases, death. See Cecil
Textbook of Medicine, 21st ed. (Goldman, L., Bennett, J. C. eds), W. B. Saunders Company, Philadelphia, 2000, p. 433-436.
While the cause of sarcoidosis is unknown, a substantial body of information suggests that immune mechanisms are important in disease pathogenesis. For example, sarcoidosis is
characterized by enhanced lymphocyte and macrophage activity. See Thomas, P.D. and
Hunninghake, G.W., Am. Rev. Respir. Dis., 1987, 135: 747-760. As sarcoidosis progresses, skin rashes, erythema nodosum and granulomas may form. Granulomas or fibrosis caused by sarcoidosis can occur throughout the body, and may affect the function of vital organs such as the lungs, heart, nervous system, liver or kidneys. In these cases, the sarcoidosis can be fatal. See
http://www.nlm.nih.gov/medlineplus/sarcoidosis.html (accessed November 12, 2009).
Moreover, a variety of exogenous agents, both infectious and non-infectious, have been hypothesized as a possible cause of sarcoidosis. See Vokurka et ah, Am. J. Respir. Crit. Care Med., 1997, 156: 1000-1003; Popper et al, Hum. Pathol, 1997, 28: 796-800; Almenoff et al, Thorax, 1996, 51 : 530-533; Baughman et al., Lancet, 2003, 361 : 1111-1118. These agents include mycobaceria, fungi, spirochetes, and the agent associated with Whipple’s disease. Id.
Sarcoidosis may be acute or chronic. Specific types of sarcoidosis include, but are not limited to, cardiac sarcoidosis, cutaneous sarcoidosis, hepatic sarcoidosis, oral sarcoidosis, pulmonary sarcoidosis, neurosarcoidosis, sinonasal sarcoidosis, Lofgren’s syndrome, lupus pernio, uveitis or chronic cutaneous sarcoidosis.
As the lung is constantly confronted with airborne substances, including pathogens, many researchers have directed their attention to identification of potential causative transmissible agents and their contribution to the mechanism of pulmonary granuloma formation associated with sarcoidosis. See Conron, M. and Du Bois, R.M., Clin. Exp. Allergy, 2001, 31 : 543-554; Agostini et al, Curr. Opin. Pulm. Med. , 2002, 8: 435-440.
Corticosteroid drugs are the primary treatment for the inflammation and granuloma formation associated with sarcoidosis. Rizatto et al. , Respiratory Medicine, 1997, 91 : 449-460. Prednisone is most often prescribed drug for the treatment of sarcoidosis. Additional drugs used to treat sarcoidosis include methotrexate, azathioprine, hydroxychloroquine, cyclophosphamide, minocycline, doxycycline and chloroquin. TNF-a blockers such as thalidomide and infliximab have been reported to be effective in treating patients with sarcoidosis. Baughman et al, Chest, 2002, 122: 227-232; Doty et al, Chest, 2005, 127: 1064-1071. Antibiotics have also been studied for the treatment of sarcoidosis, such as penicillin antibiotics, cephalosporin antibiotics, macrolide antibiotics, lincomycin antibiotics, and tetracycline antibiotics. Specific examples include minocycline hydrochloride, clindamycin, ampicillin, or clarithromycin. See, e.g., U.S. Patent Publication No. 2007/0111956.
There currently lacks a Food and Drug Administration-approved therapeutic agent for the treatment of sarcoidosis, and many patients are unable to tolerate the side effects of the standard corticosteroid therapy. See Doty et al, Chest, 2005, 127: 1064-1071. Furthermore, many cases of sarcoidosis are refractory to standard therapy. Id. Therefore, a demand exists for new methods and compositions that can be used to treat patients with sarcoidosis.
……………..
PATENTS
|
8-15-2012
|
PROCESSES FOR THE PREPARATION OF AMINOSULFONE COMPOUNDS
|
|
|
11-4-2011
|
HETEROCYCLIC COMPOUNDS AS PHOSPHODIESTERASE INHIBITORS
|
|
|
5-27-2011
|
Nanosuspension of a Poorly Soluble Drug via Microfluidization Process
|
|
|
5-28-2010
|
METHODS AND COMPOSITIONS USING PDE4 INHIBITORS FOR THE TREATMENT AND MANAGEMENT OF CANCERS
|

THANKS AND REGARD’S
DR ANTHONY MELVIN CRASTO Ph.D
GLENMARK SCIENTIST , NAVIMUMBAI, INDIA
did you feel happy, a head to toe paralysed man’s soul in action for you round the clock
need help, email or call me
I was paralysed in dec2007, Posts dedicated to my family, my organisation Glenmark, Your readership keeps me going and brings smiles to my family
TEDIGLUTIDE ..Glucagon-like peptide 2 (GLP-2) analog; protects small intestinal stem cells from radiation damage.


TEDUGLUTIDE
Glucagon-like peptide 2 (GLP-2) analog; protects small intestinal stem cells from radiation damage.
Gattex (teduglutide) is a recombinant analog of human glucagon-like peptide 2 for the treatment of adults with short bowel syndrome.
- (Gly2)GLP-2
- ALX 0600
- ALX-0600
- Gattex
- Gly(2)-GLP-2
- Teduglutide
- UNII-7M19191IKG
[Gly2]hGLP-2, [Gly2]-hGLP-2, ALX-0600,
Gattex, Revestive
| CAS number | 197922-42-2 |
|---|
L-histidylglycyl-L-α-aspartylglycyl-L-seryl-L-phenylalanyl-L-seryl-L-α-aspartyl-L-α-glutamyl-L-methionyl-L-asparaginyl-L-threonyl-L-isoleucyl-L-leucyl-L-α-aspartyl-L-asparaginyl-L-leucyl-L-alanyl-L-alanyl-L-arginyl-L-α-aspartyl-L-phenylalanyl-L-isoleucyl-L-asparaginyl-L-tryptophyl-L-leucyl-L-isoleucyl-L-glutaminyl-L-threonyl-L-lysyl-L-isoleucyl-L-threonyl-L-aspartic acid
| Formula | C164H252N44O55S |
|---|---|
| Mol. mass | 3752.082 g/mol |
Gattex, ALX-0600, (Gly2)GLP-2, Gly(2)-GLP-2, ALX 0600, [Gly2]GLP-2, Glucagon-like peptide II (2-glycine) (human), UNII-7M19191IKG
LAUNCHED 2013, NPS Pharmaceuticals
APPROVAL FDA
Company: NPS Pharmaceuticals, Inc.
Date of Approval: December 21, 2012 FDA
NDA 203441
POWDER; SUBCUTANEOUS GATTEX
U-1320=TREATMENT OF ADULT PATIENTS WITH SHORT BOWEL SYNDROME WHO ARE DEPENDENT ON PARENTERAL SUPPORT
| Patent No | Patent Expiry Date | Patent use code |
|---|---|---|
| 5789379 | Apr 14, 2015 | U-1320 |
| 7056886 | Sep 18, 2022 | U-1320 |
| 7847061 | Nov 1, 2025 | U-1320 |
| Exclusivity Code | Exclusivity_Date |
|---|---|
| ORPHAN DRUG EXCLUSIVITY | Dec 21, 2019 |
| NEW CHEMICAL ENTITY | Dec 21, 2017 |
SEE FDA
http://www.accessdata.fda.gov/drugsatfda_docs/label/2012/203441Orig1s000lbl.pdf
CLINICAL TRIALS
http://clinicaltrials.gov/search/intervention=Teduglutide+OR+ALX-0600
The active ingredient in GATTEX (teduglutide [rDNA origin]) for injection is teduglutide (rDNA origin), which is a 33 amino acid glucagon-like peptide-2 (GLP-2) analog manufactured using a strain of Escherichia coli modified byrecombinant DNA technology. The chemical name of teduglutide is L-histidyl-L-glycyl-L-aspartyl-L-glycyl-L-seryl-L-phenylalanyl-L-seryl-L-aspartyl-L-glutamyl-L-methionyl-L-asparaginyl-L-threonyl-L-isoleucyl-L-leucyl-L-aspartyl-L-asparaginyl-L-leucyl-L-alanyl-L-alanyl-L-arginyl-L-aspartyl-L-phenylalanyl-L-isoleucyl-L-asparaginyl-L-tryptophanyl-L-leucyl-L-isoleucyl-L-glutaminyl-L-threonyl-L-lysyl-L-isoleucyl-L-threonyl-L-aspartic acid. The structural formula is:
Figure 1: Structural formula of teduglutide

Teduglutide has a molecular weight of 3752 Daltons. Teduglutide drug substance is a clear, colorless to light-straw–colored liquid.
Each single-use vial of GATTEX contains 5 mg of teduglutide as a white lyophilized powder for solution for subcutaneous injection. In addition to the active pharmaceutical ingredient (teduglutide), each vial of GATTEX contains 3.88 mg L-histidine, 15 mg mannitol, 0.644 mg monobasic sodium phosphate monohydrate, 3.434 mg dibasic sodium phosphate heptahydrate as excipients. No preservatives are present.
At the time of administration the lyophilized powder is reconstituted with 0.5 mL of Sterile Water for Injection, which is provided in a prefilled syringe. A 10 mg/mL sterile solution is obtained after reconstitution. Up to 0.38 mL of the reconstituted solution which contains 3.8 mg of teduglutide can be withdrawn for subcutaneous injection upon reconstitution.
Teduglutide (brand names Gattex and Revestive) is a 36-membered polypeptide andglucagon-like peptide-2 analog that is used for the treatment of short bowel syndrome. It works by promoting mucosal growth and possibly restoring gastric emptying and secretion.[1] In Europe it is marketed under the brand Revestive by Nycomed. It was approved by the United States under the name Gattex on December 21, 2012.
Teduglutide is a proprietary analogue of glucagon-like peptide 2 (GLP-2) which was approved in the U.S. in December 2012 for the once-daily treatment of short-bowel syndrome in adults who are dependent on parenteral support. Commercial launch took place in 2013.The product was filed for approval in the E.U. in 2011 by Nycomed for this indication. In June 2012, a positive opinion was received in the E.U. and final approval was assigned in September 2012.
At NPS Pharmaceuticals, the compound is in phase III clinical development for this indication in pediatric patients and in phase II clinical studies for the treatment of Crohn’s disease. Preclinical studies are also ongoing at the company for the treatment of chemotherapy-induced enterocolitis and for the prevention and treatment of necrotizing enterocolitis (NEC) in preterm infants.
Teduglutide has been found to induce intestinal hyperplasia, reduce apoptosis and inflammation and improve cell barrier integrity in animal models. In 2001, orphan drug designation was assigned to teduglutide for the treatment of short-bowel syndrome.
In 2007, the compound was licensed to Nycomed for development and commercialization outside the U.S., Canada and Mexico for the treatment of gastrointestinal disorders. In 2012, the product was licensed to Neopharm by NPS Pharmaceuticals in Israel for development and commercialization for the treatment of gastrointestinal disorders.
The estimated prevalence of short bowel syndrome (SBS) patients with non-malignant disease requiring home parenteral nutrition (HPN) is at least 40 per million of the U.S. population. SBS usually results from surgical resection of some or most of the small intestine for conditions such as Crohn’s disease, mesenteric infarction, volvulus, trauma, congenital anomalies, and multiple strictures due to adhesions or radiation. Surgical resection may also include resection of all or part of the colon. SBS patients suffer from malabsorption that may lead to malnutrition, dehydration and weight loss. Some patients can maintain their protein and energy balance through hyperphagia; more rarely they can sustain fluid and electrolyte requirements to become independent from parenteral fluid.
Although long-term parenteral nutrition (PN) is life saving in patients with intestinal failure, it is expensive, impairs quality of life and is associated with serious complications such as catheter sepsis, venous occlusions and liver failure. Treatments that amplify absolute intestinal absorption, and eliminate or minimize the need for PN have great potential significance to SBS patients.
The endogenous meal-stimulated hormone, glucagon-like peptide-2 (GLP-2), raises considerable interest for SBS patients. GLP-2 functions to slow gastric emptying, reduce gastric secretions, increase intestinal blood-flow and stimulate growth of the small and large intestine. In animal studies, GLP-2 administration induces mucosal epithelial proliferation in the stomach and small and large intestine by stimulation of crypt cell proliferation and inhibition of enterocyte apoptosis.
SBS patients with end-jejunostomy and no colon have low basal GLP-2 levels and limited meal-stimulated GLP-2 secretion due to removal of GLP-2 secreting L-cells, which are located primarily in the terminal ileum and colon. This GLP-2 deficiency results in a minimal adaptive response following resection and could explain the gastric hypersecretion, rapid intestinal transit and lack of intestinal adaptation observed in these SBS patients.
Jeppesen et al. (Gastroenterology 2001; 120:806-815) have described positive benefit in an open-label study using pharmacologic doses of native GLP-2 in SBS jejunostomy patients. There was significant improvement in intestinal wet weight absorption and a more modest improvement in energy absorption that led to an increase in body weight, lean body mass and a rise in urinary creatinine excretion.
In contrast, SBS patients with colon-in-continuity have elevated basal endogenous GLP-2 levels resulting in an adaptive response to resection characterized by improved wet weight gain and energy absorption. The potential for added benefit of pharmacologic doses of GLP-2 receptor agonists in these patients is not obvious and has not been studied.

TEDUGLUTIDE
- Jeppesen PB (May 2012). “Teduglutide, a novel glucagon-like peptide 2 analog, in the treatment of patients with short bowel syndrome”. Therap Adv Gastroenterol 5 (3): 159–71. doi:10.1177/1756283X11436318. PMC 3342570. PMID 22570676.
- US 2013157954
- WO 2006050244
- WO 2005021022
- US 6586399
- WO 2002066062
- US 6297214
- US 2001021767
- WO 2001041779
- WO 1999058144
- WO 1998052600
Gattex Approved By FDA For Short Bowel Syndrome
 Gattex (teduglutide) has been approved by the U.S. Food and Drug Administration to be used in patients that have short bowel syndrome and require parenteral nutrition.
Gattex (teduglutide) has been approved by the U.S. Food and Drug Administration to be used in patients that have short bowel syndrome and require parenteral nutrition.
The drug, once it is in the market, will compete against two others that have been approved by the FDA for this type of patient population. Those two medications are Nutrestore (glutamine) and Zorbtive (Somatropin).
Short bowel syndrome comes on following the removal surgically of part of the large or small intestine or part of both. Patients who are affected must have parenteral nutrition due to the poor absorption they have of nutrients and fluids. Teduglutide is injected one time each day and improves the absorption making it less important to have nutrition assistance.
The advisory committee for the FDA voted unanimously in October to recommend the drug’s approval after seeing the results from a pair of clinical trials that showed the advantage teduglutide had over just a placebo in at least a reduction of 20% in the amount of parenteral nutrition at 6 months.
During the first clinical trial, 46% of the patients that took the drug saw a level of reduction, which was compared to only 6% who had taken only a placebo. In the other study, the figure increased to 63%, while the placebo rated was up to 30%
The side effects most common found in those who use teduglutide during the trials included nausea, reactions around the injection site, abdominal pain abdominal distension and headaches.
………..
| US5789379 | Jun 28, 1996 | Aug 4, 1998 | 1149336 Ontario Inc. | Glucagon-like peptide-2 analogs |
| US6077949 | Apr 24, 1997 | Jun 20, 2000 | Allelix Biopharmaceuticals, Inc. | Cloned glucagon-like peptide 2 receptors |
| US6184201 * | Apr 8, 1997 | Feb 6, 2001 | Nps Allelix Corp. | Intestinotrophic glucagon-like peptide-2 analogs |
| US7411039 | Oct 14, 2003 | Aug 12, 2008 | Novo Nordisk A/S | GLP-2 compounds, formulations, and uses thereof |
| EP1231219A1 | Apr 11, 1997 | Aug 14, 2002 | 1149336 Ontario Inc. | GLucagon-like peptide-2 analogs |
| WO1997039031A1 | Apr 11, 1997 | Oct 23, 1997 | Allelix Biopharma | Glucagon-like peptide-2 analogs |
| WO1997039091A1 | Apr 16, 1997 | Oct 23, 1997 | Burckett St Laurent James Char | Mid-chain branched surfactants |
| WO2002066511A2 | Feb 15, 2002 | Aug 29, 2002 | Conjuchem Inc | Long lasting glucagon-like peptide 2 (glp-2) for the treatment of gastrointestinal diseases and disorders |
THANKS AND REGARD’S
DR ANTHONY MELVIN CRASTO Ph.D
GLENMARK SCIENTIST , NAVIMUMBAI, INDIA
did you feel happy, a head to toe paralysed man’s soul in action for you round the clock
need help, email or call me
I was paralysed in dec2007, Posts dedicated to my family, my organisation Glenmark, Your readership keeps me going and brings smiles to my family
FDA grants fast-track status to Peregrine’s bavituximab for lung cancer treatment
FDA grants fast-track status to Peregrine’s bavituximab for lung cancer treatment
The US Food and Drug Administration (FDA) has granted fast-track designation for Peregrine Pharmaceuticals’ lead investigational immunotherapy ‘bavituximab’ to treat patients with second-line non-small cell lung cancer (NSCLC).

AZASETRON..a selective 5-HT3 receptor antagonist
AZASETRON, NAZASETRON
N-(1-azabicyclo[2.2.2]octan-8-yl)-6-chloro-4-methyl-3-oxo-1,4-benzoxazine-8-carboxamide
6-chloro-3,4-dihydro-4-methyl-3-oxo-N-(3-quinuclidinyl)-2H-1,4-benzoxazine-8-carboxamide
123039-99-6 , 123040-69-7 (free base)
141922-90-9 HYD SALT
Y-25130 (hydrochloride)LAUNCHED 1994 AS HCL SALT FORM SEROTONE
a selective 5-HT3 receptor antagonist , ANTIEMETIC
- UNII-77HC7URR9Z
Mitsubishi Tanabe Pharma, JAPAN TOBACCO (Originator)
FOR..Nausea and Vomiting, Treatment of
Prokinetic Agents

Azasetron is an antiemetic which acts as a 5-HT3 receptor antagonist.
Chemical and Pharmaceutical Bulletin, 1992 , vol. 40, 3 p. 624 – 630, ENTRY 15 , MP 305 OF HCL SALT
Biological and Pharmaceutical Bulletin, 2006 , vol. 29, 9 p. 1931 – 1935, AS MERCK 903
US 4892872…
CN 101786963…
WO 1996001630..
WO 2006006595..
WO 2007134077..
CN 102526740, CN 102451166, US 4892872, JP 2008297277
US5773436 A1, WO2006/119295 A2, EP1336602 A1, US4892872 A1,
US2003/18008 A1, US2002/147197 A1, US4892872



The nitration of 5-chloro-2-hydroxybenzoic acid methyl ester (I) with nitric acid in sulfuric acid gives 5-chloro-2-hydroxy-3-nitrobenzoic acid methyl ester (II), which is reduced with Fe and NH4Cl in water yielding 3-amino-5-chloro-2-hydroxybenzoic acid methyl ester (III). The cyclization of (III) with chloroacetyl chloride (IV) by means of NaHCO3 in CHCl3 – water affords 6-chloro-3-oxo-3,4-dihydro-2H-1,4-benzoxazine-8-carboxylic acid methyl ester (V), which is methylated with methyl iodide and K2CO3 in DMF affording the corresponding 4-methyl derivative (VI). Hydrolysis of (VI) with ethanolic NaOH gives the corresponding acid (VII), which by treatment with refluxing SOCl2 is converted into its acyl chloride (VIII). Finally, this compound is condensed with 3-aminoquinuclidine (IX) by means of N-methylmorpholine (NMM) in CHCl3.
………………
SYNTHESIS
Azasetron hydrochloride (Azasetron hydrochloride) is a secondary cancer drugs, mainly synthetic route is as follows
[0003]

[0004] In EP 0313393; JP 1989207290; JP 1990005415; US 4892872; Eur.Pat.Appl., 313393,, Chem pharm Bull, 1992,40 (3) :624-630 reported the synthesis of intermediates III is with ammonium chloride, iron as the reducing agent, the nitro substrate was dissolved in toluene and added dropwise to a reduction of the media inside, the study found that this approach has a number of disadvantages, notably the reaction can not be completely, while post-processing is very troublesome, purity of the product obtained is poor, and difficult purification. Although the “Chinese Medicinal Chemistry” magazine, 10 (2) ,138-140; 2000, the author hydrochloride instead of ammonium chloride, but still need the nitro group was dissolved in toluene was added dropwise, in fact, the nitro compound in toluene the solubility is limited, and in the process will precipitate dropwise addition, there are also incomplete reaction, impurities, and complicated post-treatment problems.
[0005] In the “China Pharmaceutical Industry” magazine 34 (3) ,214-215; 2003 in order to restore hydrosulfite as reductant,
Azasetron preparation of intermediates, the steps are as follows:
[0015] (I) A mixture of 3 – nitro-5 – chloro-salicylate added glacial acetic acid was stirred for 30-40 minutes, add water, temperature 30 ~ 100 ° C, adding iron powder, 2.5-3 hour plus finished, the addition was completed, at a temperature of 30 ~ 100 ° C the reaction for 5 to 10 hours; give 3 – amino-5-chloro-salicylate mixture;
[0016] The mass ratio of the added material is 3 – nitro-5 – chloro-salicylate: acetic acid: water: iron = 1: 5 ~ 10: 5 ~ 10: 0.5 ~ I;
[0017] (2) obtained in the step ⑴ added to a mixture of glacial acetic acid, glacial acetic acid added in the amount of step ⑴ same amount of glacial acetic acid, 70 ° C was stirred for 30-35 minutes, filtered through celite to remove the iron sludge to obtain filter cake; this procedure to that step (I) the resulting product was dissolved in glacial acetic acid solvent sufficient, in order to facilitate post-processing; while removing impurities.
[0018] (3) The filter cake was washed with an organic solvent, the combined filtrate and stirred for 10-15 minutes, allowed to stand for 30-35 minutes, the organic layer was separated; aqueous phase was extracted once again with an organic solvent, the combined organic solution was washed three times , mention made three – amino -5 chloro methyl salicylate (intermediate III) sulphate solution.
[0019] (4) of step (3) of 3 – amino-5-chloro-salicylate was added a saturated NaHCO3 solution, saturated NaHCO3 solution and the amount of acetic acid in step ⑴ same volume, cooling to _1 ° C ~ 0 ° C, control the temperature -1 ° C_5 ° C was added dropwise acetyl chloride, drop Bi. Reaction 2-3 hours. To give 3 – (2 – chloro-acetylamino)-5-chloro-mixed solution of methyl salicylate;
[0020] The amount of chloroacetyl chloride is added in step ⑴ source material 3 – nitro-5 – chloro molar ratio of methyl salicylate
I: 0.9-0.95.
[0021] (5) obtained in the step ⑷ 3 – (2 – chloro-acetylamino) methyl salicylate _5 chloride mixture was heated to 30-40 ° C, stirred for 1-1.5 hours; organic layer was separated, water phase extracted once again with an organic solvent, the combined organic layer was washed with water three times, separated and the organic layer is directly subjected to atmospheric distillation, the organic solvent is distilled off, distillation was complete, methanol or ethanol as the crystallization solvent, 80 ° C under stirring for 2.5 hours under reflux , to give 3 – (2 – chloro-acetylamino) _5 chlorine in alcohol solution of methyl salicylate. This step is intended to 3 – (2 – chloro-acetylamino)-5-chloro methyl salicylate purification.
[0022] (6) in the step (5) cooling to room temperature, an alcoholic solution, crystallization, centrifugation, and washed with ethanol crystal; 80 ° C drying in needle-like crystals 3 – (2 – chloro-acetylamino) _5 Chlorine methyl salicylate (intermediate IV).
……………………………………………………
USEFUL PATENTS
|
6-31-1998
|
Use of serotonin antagonists for treating fibromyalgia
|
|
|
11-28-1997
|
COMPOSITIONS COMPRISING CONJUGATES OF CIS-DOCOSAHEXAENOIC ACID AND TAXOTERE
|
|
|
11-28-1997
|
DHA-PHARMACEUTICAL AGENT CONJUGATES DHA-PHARMACEUTICAL AGENT CONJUGATES
|
|
|
11-28-1997
|
CONJUGATES OF CIS-DOCOSAHEXAENOIC ACID AND PACLITAXEL
|
|
|
3-28-1997
|
5-HT3 RECEPTOR ANTAGONISTS FOR DYSKINESIA
|
|
|
2-19-1997
|
Nasally administrable compositions
|
|
|
1-26-1996
|
PREVENTIVE OR REMEDY FOR IRRITABLE BOWEL SYNDROME OR DIARRHEA
|
|
|
3-17-1994
|
Benzoxazine compounds and pharmaceutical use thereof.
|
|
|
1-10-1990
|
Benzoxazine compounds and pharmaceutical use thereof
|
|
12-17-1999
|
MULTIVALENT AGONISTS, PARTIAL AGONISTS, INVERSE AGONISTS AND ANTAGONISTS OF THE 5-HT3 RECEPTORS MULTIVALENT AGONISTS, PARTIAL AGONISTS, INVERSE AGONISTS AND ANTAGONISTS OF THE 5-HT>3< RECEPTORS MULTIVALENT AGONISTS, PARTIAL AGONISTS, INVERSE AGONISTS AND ANTAGONISTS OF THE 5-HT3 RECEPTORS
|
|
|
11-17-1999
|
Use of serotonin antagonists for treating fibromyalgia
|
|
|
11-5-1999
|
CNRE BINDING FACTORS AND USES THEREOF
|
|
|
7-23-1999
|
TRANSGLUTAMINASE LINKAGE OF AGENTS TO TISSUE TRANSGLUTAMINASE LINKAGE OF AGENTS TO TISSUE
|
|
|
7-7-1999
|
Taxane compounds and compositions
|
|
|
6-25-1999
|
ORAL DELIVERY FORMULATION
|
|
|
4-16-1999
|
MEDICAMENTS MEDICAMENTS
|
|
|
2-5-1999
|
CHEMOTHERAPY SYNERGISTIC AGENT
|
|
|
10-30-1998
|
USE OF 5HT3 ANTAGONISTS FOR PROMOTING INTESTINAL LAVAGE
|
|
|
8-19-1998
|
DHA-pharmaceutical agent conjugates of taxanes
|
|
8-7-2009
|
ALISKIREN MODULATION OF NEUROGENESIS
|
|
|
7-3-2003
|
USE OF 5HT3 ANTAGONISTS FOR PROMOTING INTESTINAL LAVAGE
|
|
|
9-18-2002
|
Inhibition of emetic effect of metformin with 5-HT3 receptor antagonists
|
|
|
11-8-2001
|
CONJUGATES OF CIS-DOCOSAHEXAENOIC ACID AND PACLITAXEL
|
|
|
6-14-2001
|
Nasally administrable compositions
|
|
|
12-22-2000
|
RECEPTOR AGONISTS AND ANTAGONISTS COMPOUND FOR USE AS A MEDICAMENT FOR TREATMENT OF DISORDERS INVOLVING BRONCHOCONTRACTION COMPOUND FOR USE AS A MEDICAMENT FOR TREATMENT OF DISORDERS INVOLVING BRONCHOCONTRACTION
|
|
|
12-15-2000
|
PHARMACEUTICAL COMPOSITION FOR INTRANASAL USE OF ACTIVE SUBSTANCES THAT ARE INSOLUBLE AND/OR HARDLY SOLUBLE IN WATER
|
|
|
11-31-2000
|
ANTI-TUMOR COMPRISING BOROPROLINE COMPOUNDS
|
|
|
11-30-2000
|
TRANSGLUTAMINASE LINKAGE OF AGENTS TO TISSUE
|
|
11-17-2000
|
FATTY ACID-N-SUBSTITUTED INDOL-3-GLYOXYL-AMIDE COMPOSITIONS AND USES THEREOF
|
|
|
10-12-2000
|
COMPOSITIONS COMPRISING CONJUGATES OF CIS-DOCOSAHEXAENOIC ACID AND TAXOTERE
|
|
|
9-15-2000
|
FATTY ACID-ANTICANCER CONJUGATES AND USES THEREOF FATTY ACID-ANTICANCER CONJUGATES AND USES THEREOF
|
|
|
7-20-2000
|
$g(b)2-ADRENERGIC RECEPTOR AGONISTS $g(b)2-ADRENERGIC RECEPTOR AGONISTS
|
|
|
7-7-2000
|
USE OF CD40 ENGAGEMENT TO ALTER T CELL RECEPTOR USAGE USE OF CD40 ENGAGEMENT TO ALTER T CELL RECEPTOR USAGE USE OF CD40 ENGAGEMENT TO ALTER T CELL RECEPTOR USAGE
|
|
|
6-28-2000
|
Taxanes
|
|
|
5-32-2000
|
$g(b)2-ADRENERGIC RECEPTOR AGONISTS
|
|
|
1-14-2000
|
NOVEL INDOLOCARBAZOLE DERIVATIVES USEFUL FOR THE TREATMENT OF NEURODEGENERATIVE DISEASES AND CANCER
|
|
|
1-12-2000
|
Indolocarbazole derivatives useful for the treatment of neurodegenerative diseases and cancer
|
|
|
12-30-1999
|
METHODS FOR IDENTIFYING NOVEL MULTIMERIC AGENTS THAT MODULATE RECEPTORS METHODS FOR IDENTIFYING NOVEL MULTIMERIC AGENTS THAT MODULATE RECEPTORS
|
………..AZASETRON
AZASETRON
NMR OF HCL SALT
http://file.selleckchem.com/downloads/nmr/S210601-Azasetron-hydrochloride-NMR-Selleck.pdf
…………….
SYNTHESIS
EXAMPLE 15
To a solution of 3.0 g of 3-aminoquinuclidine and 3.0 g of N-methylmorpholine in 60 ml of chloroform is added 6.2 g of 6-chloro-3,4-dihydro-4-methyl-3-oxo-2H-1,4-benzoxazine-8-carboxylic acid chloride under cooling and stirring followed by stirring for 2 hours. The resultant solution is washed with water, aqueous sodium hydrogen carbonate and then water, and dried over magnesium sulfate. After the solvent is distilled off under reduced pressure, the residue is recrystallized from ethanol-isopropyl ether and treated with ethanolic hydrochloric acid to give 6-chloro-3,4-dihydro-4-methyl-3-oxo-N-(3-quinuclidinyl)-2H-1,4-benzoxazine-8-carboxamide hydrochloride, melting at 281
………..
SYNTHESIS AND + AND – ISOMERS
EXAMPLE 36
A solution of 6 g of 6-chloro-3,4-dihydro-4-methyl-3-oxo-N-(3-quinuclidinyl)-2H-1,4-benzoxazine-8-carboxamide and 2.7 g of D-(-)-tartaric acid in 100 ml of methanol and 200 ml of ethanol is allowed to stand. The precipitated crystals are collected by filtration and recrystallized from methanol repeatedly to give the R-(+) isomer, [α].sub.D.sup.25 =+36.72 (c=1, chloroform), melting at 177 with ethanolic hydrochloric acid, and then the precipitated crystals are collected by filtration and dried to give the R-(+) isomer hydrochloride, [α].sub.D.sup.25 =+1.4 (c=1, water), melting at 309
By using L-(+)-tartaric acid in a similar manner, the R-(-) optical isomer, [α].sub.D.sup.25.5 =-36.76 (c=1, chloroform), melting at 176 [α].sub.D.sup.25.5 =-1.2 (c=1, water), melts at 310
THANKS AND REGARD’S
DR ANTHONY MELVIN CRASTO Ph.D
GLENMARK SCIENTIST , NAVIMUMBAI, INDIA
did you feel happy, a head to toe paralysed man’s soul in action for you round the clock
need help, email or call me
I was paralysed in dec2007, Posts dedicated to my family, my organisation Glenmark, Your readership keeps me going and brings smiles to my family
