
Home » Posts tagged 'GMP' (Page 14)
Tag Archives: GMP
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
LUMACAFTOR an Orphan drug in clinical trial for oral the treatment of cystic fibrosis

Lumacaftor
3-[6-[1-(2,2-Difluoro-1,3-benzodioxol-5-yl)cyclopropylcarboxamido]-3-methylpyridin-2-yl]benzoic acid
3-{6-{[1-(2,2-difluoro-1,3-benzodioxol-5-yl)cyclopropanecarbonyl]amino}-3-methylpyridin-2-yl}benzoic acid
VRT-826809
VX-809
US patents: US8124781, US8461342
Indication:Cystic fibrosis
Developmental status:Phase III (US, UK, EU)
Developer:Vertex
| Vertex Pharmaceuticals |
| Company | Vertex Pharmaceuticals Inc. |
| Description | Small molecule cystic fibrosis transmembrane conductance regulator (CFTR) corrector |
| Molecular Target | Cystic fibrosis transmembrane conductance regulator (CFTR) |
| Mechanism of Action | CFTR stabilizer |
| Latest Stage of Development | Phase III |
| Indication | Cystic fibrosis (CF) |
| cas | 936727-05-8 |
http://www.ama-assn.org/resources/doc/usan/lumacaftor.pdf for all data
see……http://orgspectroscopyint.blogspot.in/2015/03/lumacaftor.html
Lumacaftor (USAN, codenamed VX-809) is an experimental drug for the treatment of cystic fibrosis being developed by Vertex Pharmaceuticals. The drug is designed to be effective in patients that have the F508del mutation in the cystic fibrosis transmembrane conductance regulator (CFTR), the defective protein that causes the disease. F508del, meaning that the amino acid phenylalanine in position 508 is missing, is found in about 60% of cystic fibrosis patients in Europe,[1] and in about 90% of persons with some mutation in the CFTR gene.
A corrector molecule, one of two new classes of ion channel modulators. The corrector modulators enhance the number of channels of the CFTR protein at the cell surface. in combination with ivacaftor in homozygous F508del pts
Results from a Phase II clinical trial indicate that patients with the most common form of genetic mutation causing cystic fibrosis—homozygous F508del—had a mean increase of 7.4% in lung function (FEV1) on a combination of lumacaftor and ivacaftor.[2]
VX-809 is an investigational corrector compound in a phase II clinical trial for oral the treatment of cystic fibrosis. The trial will evaluate single and multiple doses of VX-809 in healthy volunteers. This compound has resulted from a collaboration with the Cystic Fibrosis Foundation Therapeutics, Inc. (CFFT) . In 2010, orphan drug designation was assigned in the E.U. and the U.S. for the treatment of CF.
VX-809 may act to restore the function of the cystic fibrosis transmembrane conductance regulator (CFTR) protein, the defective cell membrane protein responsible for the progression of CF. VX-809 and other corrector compounds were designed to increase the amount of DF508-CFTR on the surface of cells lining the airway, which may result in an increase in chloride transport across the cell surface in patients with the DF508-CFTR mutation.
On January 11, 2013, the combination regimen of Lumacaftor (VX-809) and Kalydeco (Ivacaftor) was awarded by U.S. FDA with Breakthrough Therapy Designation as part of the agency’s efforts to accelerate the development and approval of drugs for serious and life-threatening disease.Breakthrough Therapy Designation for the combination regimen of VX-809 with ivacaftor was based on the Phase II combination data announced in 2012. Vertex Pharmaceuticals will report results from two Phase III trials (NCT01807949 (TRANSPORT) and NCT01807923 (TRAFFIC)) of the combination of Kalydeco + VX-809 in the middle of 2014. Positive data from TRAFFIC and TRANSPORT could open up a market with peak sales of approximately $6 billion, estimate analysts.

- 1 Merk; Schubert-Zsilavecz. Pharmazeutische Zeitung (in German) 156 (37): 24–27.
- 2 Wilschanski, M. (2013). “Novel therapeutic approaches for cystic fibrosis”. Discovery medicine 15 (81): 127–133. PMID 23449115

see……http://orgspectroscopyint.blogspot.in/2015/03/lumacaftor.htm
…………………………
PATENT
http://www.google.com/patents/EP2639222A1?cl=en
-
CFTR correctors useful in the treatment of cystic fibrosis. Such compounds include 3-(6-(1-(2,2-difluorobenzo[d][1,3]dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyridin-2-yl)benzoic acid (hereinafter “Compound 1”) which has the structure below:

-
Compound 1 and pharmaceutically acceptable compositions thereof are useful for treating or lessening the severity of a variety of CFTR mediated diseases.

-
Scheme 1. Synthesis of the acid chloride moiety.


Scheme 2. Synthesis of the amine moiety.

Scheme 3. Formation of an acid salt of 3-(6-(1-(2,2-difluorobcnzo[d][1,3]dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyridin-2-yl)benzoic acid.

-
Synthesis of 3-(6-(1-(2,2-difluorobenzord[d][1,3]dioxol-5-yl) cyclopropancearboxamido)-3-methylpyridin-2-yl)benzoic acid • HCl.
-
[0238]Acid Chloride Moiety
-
Synthesis of (2,2-difluoro-1,3-benzodioxol-5-yl)-methanol (Compound 18).

-
Commercially available 2,2-difluoro-1,3-benzodioxole-5-carboxylic acid (1.0 eq) is slurried in toluene (10 vol). Vitride® (2 eq) is added via addition funnel at a rate to maintain the temperature at 15-25 °C. At the end of addition the temperature is increased to 40 °C for 2 h then 10% (w/w) aq. NaOH (4.0 eq) is carefully added via addition funnel maintaining the temperature at 40-50 °C. After stirring for an additional 30 minutes, the layers are allowed to separate at 40 °C. The organic phase is cooled to 20 °C then washed with water (2 x 1.5 vol), dried (Na2SO4), filtered, and concentrated to afford crude Compound 18 that is used directly in the next step.
-
Synthesis of 5-chloromethyl-2,2-difluoro-1,3-benzodioxole (Compound 19).

-
Compound 18 (1.0 eq) is dissolved in MTBE (5 vol). A catalytic amount of DMAP (1 mol %) is added and SOCl2 (1.2 eq) is added via addition funnel. The SOCl2 is added at a rate to maintain the temperature in the reactor at 15-25 °C. The temperature is increased to 30 °C for 1 hour then cooled to 20 °C then water (4 vol) is added via addition funnel maintaining the temperature at less than 30 °C. After stirring for an additional 30 minutes, the layers are allowed to separate. The organic layer is stirred and 10% (w/v) aq. NaOH (4.4 vol) is added. After stirring for 15 to 20 minutes, the layers are allowed to separate. The organic phase is then dried (Na2SO4), filtered, and concentrated to afford crude Compound 19 that is used directly in the next step.
-
Synthesis of (2,2-difluoro-1,3-benzodioxol-5-yl)-acetonitrile (compound 20).

-
A solution of Compound 19 (1 eq) in DMSO (1.25 vol) is added to a slurry of NaCN (1.4 eq) in DMSO (3 vol) maintaining the temperature between 30-40 °C. The mixture is stirred for 1 hour then water (6 vol) is added followed by MTBE (4 vol). After stirring for 30 min, the layers are separated. The aqueous layer is extracted with MTBE (1.8 vol). The combined organic layers are washed with water (1.8 vol), dried (Na2SO4), filtered, and concentrated to afford crude compound 20 (95%) that is used directly in the next step.
-
Synthesis of (2,2-difluoro-1,3-benzodioxol-5-yl)-cyclopropanecarbonitrile (compound 21).

-
A mixture of compound 20 (1.0 eq), 50 wt % aqueous KOH (5.0 eq) 1-bromo-2-chloroethane (1.5 eq), and Oct4NBr (0.02 eq) is heated at 70 °C for 1 h. The reaction mixture is cooled then worked up with MTBE and water. The organic phase is washed with water and brine then the solvent is removed to afford compound 21.
-
Synthesis of 1-(2,2-difluoro-1,3-benzodioxol-5-yl)-cyclopropanecarboxylic acid (compound 22).

-
Compound 21 is hydrolyzed using 6 M NaOH (8 equiv) in ethanol (5 vol) at 80 °C overnight. The mixture is cooled to room temperature and ethanol is evaporated under vacuum. The residue is taken into water and MTBE, 1 M HCl was added and the layers are separated. The MTBE layer was then treated with dicyclohexylamine (0.97 equiv). The slurry is cooled to 0 °C, filtered and washed with heptane to give the corresponding DCHA salt. The salt is taken into MTBE and 10% citric acid and stirred until all solids dissolve. The layers are separated and the MTBE layer was washed with water and brine. Solvent swap to heptane followed by filtration gives compound 22 after drying in a vacuum oven at 50 °C overnight.
-
Synthesis of 1-(2,2-difluoro-1,3-benzodioxol-5-yl)-cyclopropanecarbonyl chloride (compound 7).

-
Compound 22 (1.2 cq) is slurried in toluene (2.5 vol) and the mixture heated to 60 °C. SOCl2 (1.4 eq) is added via addition funnel. The toluene and SOCl2 are distilled from the reaction mixture after 30 minutes. Additional toluene (2.5 vol) is added and distilled again.
-
Synthesis of 14C-(2,2-difluoro-1,3-benzodioxol-5-yl)-acetonitrile (compound 23).

-
A solution of Compound 19 (1 eq) in DMSO (1.25 vol) is added to a slurry of Na14 CN (1.4 eq) in DMSO (3 vol) maintaining the temperature between 30-40 °C. The mixture is stirred for 1 hour then water (6 vol) is added followed by MTBE (4 vol). After stirring for 30 min, the layers are separated. The aqueous layer is extracted with MTBE (1.8 vol). The combined organic layers are washed with water (1.8 vol), dried (Na2SO4), filtered, and concentrated to afford crude compound 23 that is purified by chromatography.
-
Synthesis of 14C-(2,2-difluoro-1,3-benzodioxol-5-yl)-cyclopropanecarbonitrile (compound 24).

-
A mixture of compound 23 (1.0 eq) and 1,2-dibromoethane (1.8 eq) in THF (3 vol) is cooled to -10 °C via external chiller. 1 M LHMDS in THF (2.5 eq) is added via an addition funnel and at a rate to maintain the temperature in the reactor below 10 °C. One hour after addition is complete, 20% w/v aq. citric acid (13 vol) is added via addition funnel maintaining the temperature in the reactor below 20 C. The external chiller is turned off and after stirring for 30 min the layers are separated. The organic layer is filtered and concentrated to afford crude compound 24 that is purified by chromatography.
-
Synthesis of 14C-1-(2,2-difluoro-1,3-benzodioxol-5-yl)-cyclopropanecarboxylic acid (compound 25).

-
Compound 24 is hydrolyzed using 6 M NaOH (8 equiv) in ethanol (5 vol) at 80 °C overnight. The mixture is cooled to room temperature and ethanol is evaporated under vacuum. The residue is taken into water and MTBE. 1 M HCl is added to the mixture and the organic layer is filtered and concentrated to afford compound 25.
-
Synthesis of 14C-1-(2,2-difluoro-1,3-benzodioxol-5-yl)-cyclopropanecarbonyl chloride (compound 26).

-
A mixture of Compound 25, 4-dimethylaminopyridine, and thionyl chloride (SOCl2) in CH2Cl2 is stirred to produce compound 26, which may be further reacted with compound 6 without isolation.
-
Amine Moiety
-
Synthesis of tert-butyl-3-(3-methylpyridin-2-yl)benzoate (compound 4).

-
2-Bromo-3-methylpyridine (1.0 eq) is dissolved in toluene (12 vol). K2CO3 (4.8 eq) is added followed by water (3.5 vol) and the mixture heated to 65 °C under a stream of N2 for 1 hour. 3-(t-Butoxycarbonyl)phenylboronic acid (1.05 eq) and Pd(dppf)Cl2-CH2Cl2 (0.015 eq) are then added and the mixture is heated to 80 °C. After 2 hours, the heat is turned off, water is added (3.5 vol) and the layers are allowed to separate. The organic phase is then washed with water (3.5 vol) and extracted with 10% aqueous methanesulfonic acid (2 eq MsOH, 7.7 vol). The aqueous phase is made basic with 50% aqueous NaOH (2 eq) and extracted with EtOAc (8 vol). The organic layer is concentrated to afford crude compound 4 (82%) that is used directly in the next step.
-
Synthesis of 2-(3-(tert-butoxycarbonyl)phenyl)-3-methylpyridine-1-oxide (compound 5).

-
Compound 4 (1.0 eq) is dissolved in EtOAc (6 vol). Water (0. 3 vol) is added followed by urea-hydrogen peroxide (3 cq). The phthalic anhydride (3 cq) is added portion-wise as a solid to maintain the temperature in the reactor below 45 °C. After completion of phthalic anhydride addition, the mixture is heated to 45 °C. After stirring for an additional 4 hours, the heat is turned off. 10% w/w aqueous Na2SO3 (1.5 eq) is added via addition funnel. After completion of Na2SO3 addition, the mixture is stirred for an additional 30 minutes and the layers separated. The organic layer is stirred and 10% w/w aq. Na2CO3 (2 eq) is added. After stirring for 30 minutes, the layers are allowed to separate. The organic phase is washed 13% w/v aq NaCl. The organic phase is then filtered and concentrated to afford crude compound 5 (95%) that is used directly in the next step.
-
Synthesis of tert-butyl-3-(6-amino-3-methylpyridin-2-yl)benzoate (compound 6).

-
A solution of compound 5 (1 eq) and pyridine (4 eq) in MeCN (8 vol) is heated to 70 °C. A solution of methanesulfonic anhydride (1.5 eq) in MeCN (2 vol) is added over 50 min via addition funnel maintaining the temperature at less than 75 °C. The mixture is stirred for an additional 0.5 hours after complete addition. The mixture is then allowed to cool to ambient. Ethanolamine (10 eq) is added via addition funnel. After stirring for 2 hours, water (6 vol) is added and the mixture is cooled to 10 °C. After stirring for NLT 3 hours, the solid is collected by filtration and washed with water (3 vol), 2:1 MeCN/water (3 vol), and MeCN (2×1.5 vol). The solid is dried to constant weight (<1% difference) in a vacuum oven at 50 °C with a slight N2 bleed to afford compound 6 as a red-yellow solid (53% yield).
-
Synthesis of 3-(6-(1-(2,2-difluorobenzo[d][1,3]dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyridin-2-yl)-t-butylbenzoate (compound 8).

-
Compound 7 is dissolved in toluene (2.5 vol based on acid chloride) and added via addition funnel to a mixture of compound 6 (1 eq), dimethylaminopyridine (DMAP, 0.02 eq), and triethylamine (3.0 cq) in toluene (4 vol based on compound 6). After 2 hours, water (4 vol based on compound 6) is added to the reaction mixture. After stirring for 30 minutes, the layers are separated. The organic phase is then filtered and concentrated to afford a thick oil of compound 8 (quantitative crude yield). MeCN (3 vol based on crude product) is added and distilled until crystallization occurs. Water (2 vol based on crude product) is added and the mixture stirred for 2 h. The solid is collected by filtration, washed with 1:1 (by volume) MeCN/water (2 x 1 vol based on crude product), and partially dried on the filter under vacuum. The solid is dried to constant weight (<1% difference) in a vacuum oven at 60 °C with a slight N2 bleed to afford 3-(6-(1-(2,2-difluorobenzo[d] [1,3]dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyridin-2-yl)-t-butylbenzoate as a brown solid.
-
Syntheisis of Syntheisis of 3-(6-(1-(2,2-difluorobenzo[d] [1,3]dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyridin-2-yl)benzoic acid • HCL salt (compound 9).

-
To a slurry of compound 8 (1.0 eq) in MeCN (3.0 vol) is added water (0.83 vol) followed by concentrated aqueous HCl (0.83 vol). The mixture is heated to 45 ± 5 °C. After stirring for 24 to 48 hours the reaction is complete and the mixture is allowed to cool to ambient. Water (1.33 vol) is added and the mixture stirred. The solid is collected by filtration, washed with water (2 x 0.3 vol), and partially dried on the filter under vacuum. The solid is dried to constant weight (<1% difference) in a vacuum oven at 60 °C with a slight N2 bleed to afford compound 9 as an off-white solid.















-
Synthesis of 3-(6-(1-(2,2-difluorobenzo[d][1,3]dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyridin-2-yl)benzoic acid (Compound 1).

-
A slurry of 3-(6-(1-(2,2-difluorobenzo[d][1,3]dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyridin-2-yl)benzoic acid • HCl (1 eq) in water (10 vol) is stirred at ambient temperature. A sample is taken after stirring for 24 hours. The sample is filtered and the solid washed with water (2 x). The solid sample is submitted for DSC analysis. When DSC analysis indicates complete conversion to Compound 1, the solid is collected by filtration, washed with water (2 x 1.0 vol), and partially dried on the filter under vacuum. The solid is dried to constant weight (<1% difference) in a vacuum oven at 60 °C with a slight N2 bleed to afford Compound 1 as an off-white solid (98% yield).
-
Synthesis of 3-(6-(1-(2,2-difluorobenzo[d][1,3]dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyridin-2-yl)benzoic acid (Compound 1) using water and base.


-
To a slurry of 3-(6-(1-(2,2-difluorobenzo[d][1,3]dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyridin-2-yl)benzoic acid • HCl (1 eq) in water (10 vol) stirred at ambient temperature is added 50% w/w aq. NaOH (2.5 eq). The mixture is stirred for NLT 15 min or until a homogeneous solution. Concentrated HCl (4 eq) is added to crystallize Compound 1. The mixture is heated to 60 °C or 90 °C if needed to reduce the level of the t-butylbenzoate ester. The mixture is heated until HPLC analysis indicates NMT 0.8% (AUC) t-butylbenzoate ester. The mixture is then cooled to ambient and the solid is collected by filtration, washed with water (3 x 3.4 vol), and partially dried on the filter under vacuum. The solid is dried to constant weight (<1% difference) in a vacuum oven at 60 °C with a slight N2 bleed to afford Compound 1 as an off-white solid (97% yield).
-
Synthesis of 3-(6-(1-(2,2-difluorobenzo[d][1,3]dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyridin-2-yl)benzoic acid (Compound 1) directly from benzoate.

-
A solution of 3-(6-(1-(2,2-difluorobenzo[d][1,3]dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyridin-2-yl)-t-butylbenzoate (1.0 eq) in formic acid (3.0 vol) is heated to 70 ± 10 °C. The reaction is continued until the reaction is complete (NMT 1.0% AUC 3-(6-(1-(2,2-difluorobenzo[d][1,3]dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyridin-2-yl)-t-butylbenzoate) or heating for NMT 8 h. The mixture is allowed to cool to ambient. The solution is added to water (6 vol) heated at 50 °C and the mixture stirred. The mixture is then heated to 70 ± 10 °C until the level of 3-(6-(1-(2,2-difluorobenzo[d][1,3]dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyridin-2-yl)-t-butylbenzoate is NMT 0.8% (AUC). The solid is collected by filtration, washed with water (2 x 3 vol), and partially dried on the filter under vacuum. The solid is dried to constant weight (<1% difference) in a vacuum oven at 60 °C with a slight N2 bleed to afford Compound 1 as an off-white solid.



-
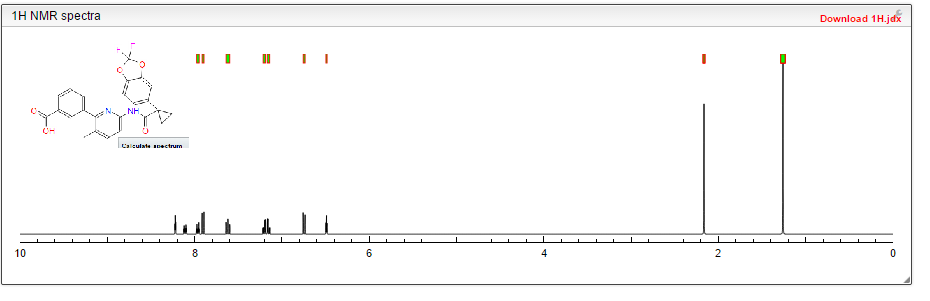
1HNMR spectra of Compound 1 are shown in Figures 9-11 (Figures 9 and 10 depict Compound 1 in Form I in a 50 mg/mL, 0.5 methyl cellulose-polysorbate 80 suspension, and Figure 11 depicts Compound 1 as an HCl salt).
-
Table 3 below recites additional analytical data for Compound 1.
-
Table 3.
Cmpd. No. LC/MS M+1 LC/RTmin NMR 1 453.3 1.93 H NMR (400 MHz, DMSO-d6) 9.14 (s, 1H), 7.99-7.93 (m, 3H), 7.80-7.78 (m, 1H), 7.74-7.72 (m, 1H), 7.60-7.55 (m, 2H), 7.41-7.33 (m, 2H), 2.24 (s, 3H), 1.53-1.51 (m, 2H), 1.19-1.17 (m, 2H)



| WO2002096421A1 * | May 22, 2002 | Dec 5, 2002 | Neurogen Corp | 5-substituted-2-arylpyridines as crf1 modulators |
| WO2004072038A1 * | Feb 10, 2004 | Aug 26, 2004 | Vertex Pharma | Processes for the preparation of n-heteroaryl-n-aryl-amines by reacting an n-aryl carbamic acid ester with a halo-heteroaryl and analogous processes |
| WO2007056341A1 | Nov 8, 2006 | May 18, 2007 | Vertex Pharma | Heterocyclic modulators of atp-binding cassette transporters |
| see……http://orgspectroscopyint.blogspot.in/2015/03/lumacaftor.htm |
References
David Andrew Siesel;Processes for producing cycloalkylcarboxamido-pyridine benzoic acids,US patent number US8124781 B2 ;Also published as CA2707494A1, CN101910134A, EP2231606A2, EP2231606B1, EP2639222A1, EP2639223A1, EP2639224A1, US8592602, US20090176989, US20120190856, WO2009076142A2, WO2009076142A3;Filing date:Dec 4, 2008;Original Assignee:Vertex Pharmaceuticals Incorporated
David Andrew Siesel;Processes for producing cycloalkylcarboxamido-pyridine benzoic acids,US patent number US8461342 B2 ;Also published as US20100036130, US20120203006, US20130274477, WO2010138484A2, WO2010138484A3;Original Assignee:Vertex Pharmaceuticals Incorporated
Van Goor, Fredrick F. et al;Pharmaceutical compositions in the treatment of CFTR-mediated diseases such as cystic fibrosis;PCT Int. Appl., WO2011133956
Van Goor, Fredrick F. et al;Pharmaceutical compositions in the treatment of CFTR-mediated diseases such as cystic fibrosis.PCT Int. Appl., WO2011133951
Van Goor, Fredrick F. et al;Pharmaceutical compositions for treatment of CFTR-mediated diseases;PCT Int. Appl., WO2011133953
Verwijs, Marinus Jacobus et al;Preparation and pharmaceutical compositions of Lumacaftor for the treatment of cystic fibrosis and other diseases associated with CFTR mutations;PCT Int. Appl., WO2011127241
Keshavarz-Shokri, Ali et al;Preparation of Lumacaftor for therapeutical use;PCT Int. Appl., WO2011127290
Siesel, David;A process for the preparation of solid forms of (((difluorobenzodioxolyl)cyclopropanecarboxamido)methylpyridinyl)benzoic acid;U.S. Pat. Appl. Publ., US20100036130
Siesel, David;A process for the preparation of solid forms of (((difluorobenzodioxolyl)cyclopropanecarboxamido)methylpyridinyl)benzoic acid;PCT Int. Appl., WO2010138484
Young, Christopher;Dosage units of 3-(6-(1-(2,2-difluorobenzo[d][1,3]dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyridin-2-yl)benzoic acid;PCT Int. Appl., WO2010037066
Hadida-Ruah, Sara et al;paration of N-pyridinyl carboxamide derivatives as modulators of ATP-binding cassette transporters;U.S. Pat. Appl. Publ., 20080019915
Siesel, David;A process for the preparation of solid forms of (((difluorobenzodioxolyl)cyclopropanecarboxamido)methylpyridinyl)benzoic acid;PCT Int. Appl., WO2009076142
Hadida Ruah, Sara et al;Preparation of N-pyridinyl carboxamide derivatives as modulators of ATP-binding cassette transporters;PCT Int. Appl., WO2007056341
video on cystic fibrosis
second video
Update on 26 mar 2015
LUMACAFTOR
VX 809
| 3-[6-[[1-(2,2-difluoro-1,3-benzodioxol-5-yl)cyclopropanecarbonyl]amino]-3-methylpyridin-2-yl]benzoic Acid | |
| CAS No.: | 936727-05-8 |
|---|---|
| Synonyms: |
|
| Formula: | C24H18F2N2O5 |
| Exact Mass: | 452.11800 |
SMILLES…. Cc1ccc(nc1c2cccc(c2)C(=O)O)NC(=O)C3(CC3)c4ccc5c(c4)OC(O5)(F)F
NMR…………….http://file.selleckchem.com/downloads/nmr/S156503-VX-809-HNMR-Selleck.pdf
-
Synthesis of 3-(6-(1-(2,2-difluorobenzo[d][1,3]dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyridin-2-yl)benzoic acid (Compound 1) directly from benzoate.
-
A solution of 3-(6-(1-(2,2-difluorobenzo[d][1,3]dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyridin-2-yl)-t-butylbenzoate (1.0 eq) in formic acid (3.0 vol) is heated to 70 ± 10 °C. The reaction is continued until the reaction is complete (NMT 1.0% AUC 3-(6-(1-(2,2-difluorobenzo[d][1,3]dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyridin-2-yl)-t-butylbenzoate) or heating for NMT 8 h. The mixture is allowed to cool to ambient. The solution is added to water (6 vol) heated at 50 °C and the mixture stirred. The mixture is then heated to 70 ± 10 °C until the level of 3-(6-(1-(2,2-difluorobenzo[d][1,3]dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyridin-2-yl)-t-butylbenzoate is NMT 0.8% (AUC). The solid is collected by filtration, washed with water (2 x 3 vol), and partially dried on the filter under vacuum. The solid is dried to constant weight (<1% difference) in a vacuum oven at 60 °C with a slight N2 bleed to afford Compound 1 as an off-white solid.
-
1HNMR spectra of Compound 1 are shown in Figures 9-11 (Figures 9 and 10 depict Compound 1 in Form I in a 50 mg/mL, 0.5 methyl cellulose-polysorbate 80 suspension, and Figure 11 depicts Compound 1 as an HCl salt).
-
Table 3 below recites additional analytical data for Compound 1.
-
Table 3.
Cmpd. No. LC/MS M+1 LC/RTmin NMR 1 453.3 1.93 H NMR (400 MHz, DMSO-d6) 9.14 (s, 1H), 7.99-7.93 (m, 3H), 7.80-7.78 (m, 1H), 7.74-7.72 (m, 1H), 7.60-7.55 (m, 2H), 7.41-7.33 (m, 2H), 2.24 (s, 3H), 1.53-1.51 (m, 2H), 1.19-1.17 (m, 2H)

1H NMR PREDICT
![3-[6-[[1-(2,2-difluoro-1,3-benzodioxol-5-yl)cyclopropanecarbonyl]amino]-3-methylpyridin-2-yl]benzoic acid NMR spectra analysis, Chemical CAS NO. 936727-05-8 NMR spectral analysis, 3-[6-[[1-(2,2-difluoro-1,3-benzodioxol-5-yl)cyclopropanecarbonyl]amino]-3-methylpyridin-2-yl]benzoic acid H-NMR spectrum](https://i0.wp.com/pic11.molbase.net/nmr/nmr_image/2014-08-29/001/530/195/936727-05-8-1h.png)
13C NMR PREDICT
![3-[6-[[1-(2,2-difluoro-1,3-benzodioxol-5-yl)cyclopropanecarbonyl]amino]-3-methylpyridin-2-yl]benzoic acid NMR spectra analysis, Chemical CAS NO. 936727-05-8 NMR spectral analysis, 3-[6-[[1-(2,2-difluoro-1,3-benzodioxol-5-yl)cyclopropanecarbonyl]amino]-3-methylpyridin-2-yl]benzoic acid C-NMR spectrum](https://i0.wp.com/pic11.molbase.net/nmr/nmr_image/2014-08-29/001/530/195/936727-05-8-13c.png) CAS NO. 936727-05-8, 3-[6-[[1-(2,2-difluoro-1,3-benzodioxol-5-yl)cyclopropanecarbonyl]amino]-3-methylpyridin-2-yl]benzoic acid C-NMR spectral analysisCOSY PREDICT
CAS NO. 936727-05-8, 3-[6-[[1-(2,2-difluoro-1,3-benzodioxol-5-yl)cyclopropanecarbonyl]amino]-3-methylpyridin-2-yl]benzoic acid C-NMR spectral analysisCOSY PREDICT


13C NMR PREDICT




| WO2002096421A1 * | May 22, 2002 | Dec 5, 2002 | Neurogen Corp | 5-substituted-2-arylpyridines as crf1 modulators |
| WO2004072038A1 * | Feb 10, 2004 | Aug 26, 2004 | Vertex Pharma | Processes for the preparation of n-heteroaryl-n-aryl-amines by reacting an n-aryl carbamic acid ester with a halo-heteroaryl and analogous processes |
| WO2007056341A1 | Nov 8, 2006 | May 18, 2007 | Vertex Pharma | Heterocyclic modulators of atp-binding cassette transporters |

http://www.google.co.in/patents/US8124781



Synthesis of 3-(6-(1-(2,2-difluorobenzo[d][1,3]dioxol-5-yl)cyclopropanecarboxamido)-3-methylpyridin-2-yl)benzoic Acid (Compound 1)

Synthesis of 3-(6-(1-(2,2-difluorobenzo[d][1,3]dioxol-5-yl)cyclopropanecarboxamido)-3-methylpyridin-2-yl)benzoic Acid (Compound 1) Using Water and Base

Synthesis of 3-(6-(1-(2,2-difluorobenzo[d][1,3]dioxol-5-yl)cyclopropanecarboxamido)-3-methylpyridin-2-yl)benzoic Acid (Compound 1) Directly from Benzoate

Compound 1
Compound 1 is used as the starting point for the other solid state forms and can be prepared by coupling an acid chloride moiety with an amine moiety according to Schemes 1-4.
Scheme 1. Synthesis of the acid chloride moiety.

1. NaCN
2. H20

socio

Scheme 1 depicts the preparation of l-(2,2-difluorobenzo[d][l,3]dioxol-5- yl)cyclopropanecarbonyl chloride, which is used in Scheme 3 to make the amide linkage of Compound 1.
The starting material, 2,2-difluorobenzo[d][l,3]dioxole-5-carboxylic acid, is commercially available from Saltigo (an affiliate of the Lanxess Corporation). Reduction of the carboxylc acid moiety in 2,2-difluorobenzo[d][l ,3]dioxole-5-carboxylic acid to the primary alcohol, followed by conversion to the corresponding chloride using thionyl chloride (SOCl2), provides 5-(chloromethyl)-2,2-difluorobenzo[d][l,3]dioxole, which is subsequently converted to 2-(2,2-difluorobenzo[d][l,3]dioxol-5-yl)acetonitrile using sodium cyanide. Treatment of 2-(2,2- difluorobenzo[d][l,3]dioxol-5-yl)acetonitrile with base and l-bromo-2-chloroethane provides 1- (2,2-difluorobenzo[d][l,3]dioxol-5-yl)cyclopropanecarbonitrile. The nitrile moiety in l-(2,2- difluorobenzo[d][l,3]dioxol-5-yl)cyclopropanecarbonitrile is converted to a carboxylic acid using base to give l-(2,2-difluorobenzo[d][l,3]dioxol-5-yl)cyclopropanecarboxylic acid, which is converted to the desired acid chloride using thionyl chloride.
Scheme 2. Alternative synthesis of the acid chloride moiety.

Touene, H20, 70 °C3 N HC1,
DMSO,
75 °C

Scheme 2 depicts an alternative synthesis of the requisite acid chloride. 5- bromomethyl-2,2-difluoro-l,3-benzodioxole is coupled with ethyl cyanoacetate in the presence of a palladium catalyst to form the corresponding alpha cyano ethyl ester. Saponification of the ester moiety to the carboxylic acid gives the cyanoethyl compound. Alkylation of the cyanoethyl compound with l-bromo-2-chloro ethane in the presence of base gives the cyanocyclopropyl compound. Treatment of the cyanocyclopropyl compound with base gives the carboxylate salt, which is converted to the carboxylic acid by treatment with acid. Conversion of the carboxylic acid to the acid chloride is then accomplished using a chlorinating agent such as thionyl chloride or the like.
Scheme 3. Synthesis of the amine moiety.

ptBu urea-hydrogen peroxide hthalic anhydride EtOAc, water

Scheme 3 depicts the preparation of the requisite tert-butyl 3-(6-amino-3- methylpyridin-2-yl)benzoate, which is coupled with l-(2,2-difluorobenzo[d][l,3]dioxol-5- yl)cyclopropanecarbonyl chloride in Scheme 3 to give Compound 1. Palladium-catalyzed coupling of 2-bromo-3-methylpyridine with 3-(tert-butoxycarbonyl)phenylboronic acid gives tert-butyl 3-(3-methylpyridin-2-yl)benzoate, which is subsequently converted to the desired compound. Scheme 4. Formation of an acid salt of 3-(6-(l-(2,2-difluorobenzo[d] [l,3]dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyridin-2-yl)benzoic acid.

Scheme 4 depicts the coupling of l-(2,2-difluorobenzo[d][l,3]dioxol-5- yl)cyclopropanecarbonyl chloride with tert-butyl 3-(6-amino-3-methylpyridin-2-yl)benzoate using triethyl amine and 4-dimethylaminopyridine to initially provide the tert-butyl ester of Compound 1.
……………………..
WO2010037066
http://www.google.im/patents/WO2010037066A2?cl=en
Syntheisis of 3-(6-(l-(2,2-difluorobenzo[d] [l,3]dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyridin-2-yl)benzoic acid • HCL salt.

HCl
To a slurry of 3-(6-(l-(2,2-difluorobenzo[d][l,3]dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyridin-2-yl)-t-butylbenzoate (1.0 eq) in MeCN (3.0 vol) is added water (0.83 vol) followed by concentrated aqueous HCl (0.83 vol). The mixture is heated to 45 ± 5 0C. After stirring for 24 to 48 hours the reaction is complete and the mixture is allowed to cool to ambient. Water (1.33 vol) is added and the mixture stirred. The solid is collected by filtration, washed with water (2 x 0.3 vol), and partially dried on the filter under vacuum. The solid is dried to constant weight (<1% difference) in a vacuum oven at 60 0C with a slight N2 bleed to afford 3-(6-(l-(2,2- difluorobenzo[d][l,3]dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyridin-2- yl)benzoic acid • HCl as an off-white solid.
Synthesis of 3-(6-(l-(2,2-difluorobenzo[d] [l,3]dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyridin-2-yl)benzoic acid (Compound 1 in Form I).

HCl

Compound 1 in Form I
A slurry of 3-(6-(l-(2,2-difluorobenzo[d][l,3]dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyridin-2-yl)benzoic acid • HCl (1 eq) in water (10 vol) is stirred at ambient temperature. A sample is taken after stirring for 24 hours. The sample is filtered and the solid washed with water (2 x). The solid sample is submitted for DSC analysis. When DSC analysis indicates complete conversion to Compound 1, the solid is collected by filtration, washed with water (2 x 1.0 vol), and partially dried on the filter under vacuum. The solid is dried to constant weight (<1% difference) in a vacuum oven at 60 0C with a slight N2 bleed to afford Compound 1 as an off-white solid (98% yield).
Synthesis of 3-(6-(l-(2,2-difluorobenzo[d] [l,3]dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyridin-2-yl)benzoic acid (Compound 1 in Form I) using water and base.


Compound 1 in Form I
To a slurry of 3-(6-(l-(2,2-difluorobenzo[d][l,3]dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyridin-2-yl)benzoic acid • HCl (1 eq) in water (10 vol) stirred at ambient temperature is added 50% w/w aq. NaOH (2.5 eq). The mixture is stirred for NLT 15 min or until a homogeneous solution. Concentrated HCl (4 eq) is added to crystallize Compound 1. The mixture is heated to 60 0C or 90 0C if needed to reduce the level of the t-butylbenzoate ester. The mixture is heated until HPLC analysis indicates NMT 0.8% (AUC) t-butylbenzoate ester. The mixture is then cooled to ambient and the solid is collected by filtration, washed with water (3 x 3.4 vol), and partially dried on the filter under vacuum. The solid is dried to constant weight (<1% difference) in a vacuum oven at 60 0C with a slight N2 bleed to afford Compound 1 as an off-white solid (97% yield).
Synthesis of 3-(6-(l-(2,2-difluorobenzo[d] [l,3]dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyridin-2-yl)benzoic acid (Compound 1 in Form I) directly from benzoate.


Compound 1 in Form I
A solution of 3-(6-(l-(2,2-difluorobenzo[d][l,3]dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyridin-2-yl)-t-butylbenzoate (1.0 eq) in formic acid (3.0 vol) is heated to 70 ± 10 0C. The reaction is continued until the reaction is complete (NMT 1.0% AUC 3-(6-(l-(2,2-difluorobenzo[d][l,3]dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyridin-2-yl)-t-butylbenzoate) or heating for NMT 8 h. The mixture is allowed to cool to ambient. The solution is added to water (6 vol) heated at 50 0C and the mixture stirred. The mixture is then heated to 70 ± 10 0C until the level of 3- (6-(l-(2,2-difluorobenzo[d][l,3]dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyridin- 2-yl)-t-butylbenzoate is NMT 0.8% (AUC). The solid is collected by filtration, washed with water (2 x 3 vol), and partially dried on the filter under vacuum. The solid is dried to constant weight (<1% difference) in a vacuum oven at 60 0C with a slight N2 bleed to afford Compound 1 in Form I as an off-white solid.
US OK for Forest/Pierre Fabre antidepressant fetzima, levomilnacipran

levomilnacipran
The US Food and Drug Administration has approved Forest Laboratories and Pierre Fabre’s Fetzima for major depressive disorder.
Fetzima (levomilnacipran extended-release), a once-daily serotonin and norepinephrine reuptake inhibitor, has been given the green light based on Phase III studies of adults with MDD and statistically significant and clinically meaningful improvement in depressive symptoms across three doses (40, 80, and 120 mg).
read all at
http://www.pharmatimes.com/Article/13-07-26/US_OK_for_Forest_Pierre_Fabre_antidepressant.aspx


Levomilnacipran (F2695) is an antidepressant currently under development by Forest Laboratories for the treatment of depression in the United States and Canada.[1][2][3] As of 2009 it is in phase III clinical trials.[4] Levomilnacipran is an active enantiomer of milnacipran and therefore has similar effects and pharmacology, acting as a serotonin-norepinephrine reuptake inhibitor.[2][5] On 20 January 2011, Forest and Pierre Fabre Medicament announced that levomilnacipran was no better than placebo in a late-stage clinical trial. Two other late-stage trials will be finished in mid-2011.
References
- “Future Treatments for Depression, Anxiety, Sleep Disorders, Psychosis, and ADHD — Neurotransmitter.net”.
- “Pierre Fabre Medicament and Forest Laboratories to Collaborate on Development and Commercialization of F2695 for Depression – FierceBiotech”.
- “News: Forest Buys CNS Disease-Related Drug for $75M Upfront.”.
- “Search of: F2695 – List Results – ClinicalTrials.gov”.
- Deprez D, Chassard D, Baille P, Mignot A, Ung HL, Puozzo C (1998). “Which bioequivalence study for a racemic drug? Application to milnacipran”. European Journal of Drug Metabolism and Pharmacokinetics 23 (2): 166–71. PMID 9725476.
Mavoglurant (AFQ-056) is an experimental drug candidate for the treatment of fragile X syndrome

Mavoglurant (AFQ-056) is an experimental drug candidate for the treatment of fragile X syndrome.[1] It exerts its effect as an antagonist of the metabotropic glutamate receptor 5 (mGLU5).[2]
Mavoglurant is under development by Novartis and is currently in Phase II and Phase III clinical trials.[1][3] If successful, it would be the first drug to treat the underlying disorder instead of the symptoms of fragile X syndrome.[4]

- P. Cole (2012). “Mavoglurant”. Drugs of the Future 37 (1): 7–12. doi:10.1358/dof.2012.37.1.1772147.
- Levenga, J; Hayashi, S; De Vrij, FM; Koekkoek, SK; Van Der Linde, HC; Nieuwenhuizen, I; Song, C; Buijsen, RA et al. (2011). “AFQ056, a new mGluR5 antagonist for treatment of fragile X syndrome”. Neurobiology of disease 42 (3): 311–7. doi:10.1016/j.nbd.2011.01.022. PMID 21316452.
- Jacquemont, S.; Curie, A.; Des Portes, V.; Torrioli, M. G.; Berry-Kravis, E.; Hagerman, R. J.; Ramos, F. J.; Cornish, K. et al. (2011). “Epigenetic Modification of the FMR1 Gene in Fragile X Syndrome is Associated with Differential Response to the mGluR5 Antagonist AFQ056”. Science Translational Medicine 3 (64): 64ra1. doi:10.1126/scitranslmed.3001708. PMID 21209411.
- “AFQ056 drug improves symptoms in Fragile X patients: Study”. news-medical.net. January 9, 2011.
Fragile X syndrome (FXS), Martin–Bell syndrome, or Escalante’s syndrome (more commonly used in South American countries), is a genetic syndrome that is the most widespread single-gene cause of autism and inherited cause of mental retardation among boys. It results in a spectrum of intellectual disabilities ranging from mild to severe as well as physical characteristics such as an elongated face, large or protruding ears, and large testes (macroorchidism), and behavioral characteristics such as stereotypic movements (e.g. hand-flapping), and social anxiety.
Fragile X syndrome is associated with the expansion of the CGG trinucleotide repeat affecting the Fragile X mental retardation 1 (FMR1) gene on the X chromosome, resulting in a failure to express the fragile X mental retardation protein (FMRP), which is required for normal neural development. Depending on the length of the CGG repeat, an allele may be classified as normal (unaffected by the syndrome), a premutation (at risk of fragile X associated disorders), or full mutation (usually affected by the syndrome).[1] A definitive diagnosis of fragile X syndrome is made through genetic testing to determine the number of CGG repeats. Testing for premutation carriers can also be carried out to allow for genetic counseling. The first complete DNA sequence of the repeat expansion in someone with the full mutation was generated by scientists in 2012 using SMRT sequencing.
There is currently no drug treatment that has shown benefit specifically for fragile X syndrome. However, medications are commonly used to treat symptoms of attention deficit and hyperactivity, anxiety, and aggression. Supportive management is important in optimizing functioning in individuals with fragile X syndrome, and may involve speech therapy, occupational therapy, and individualized educational and behavioral programs.
orphan drug designation EMA
http://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=EN&Expert=317238
Mavoglurant regulatory update 12/03/2012
ماووگلوران (به انگلیسی: Mavoglurant) یک ترکیب شیمیایی با شناسه پابکم ۹۹۲۶۸۳۲ است.
جستارهای وابسته[ویرایش]
Curis phase 1 Cancer Trial for CUDC-427 Begins

CUDC-427, GDC-0917; RG-7459
Genentech Inc (Roche Holding AG)
Curis licenses GDC-0917 from Genentech
Curis Cancer Trial Begins
Curis Inc. has initiated patient dosing in a second Phase 1 dose-escalation study of CUDC-427 that is being conducted using a continuous, twice-daily oral dosing regimen in patients with advanced and refractory solid tumors or lymphoma.
FULL STORY
About CUDC-427 (GDC-0917)
CUDC-427 is an orally bioavailable small molecule that is designed to promote cancer cell death by antagonizing IAP proteins. IAP proteins are a family of functionally and structurally related proteins that promote cancer cell survival by inhibiting programmed cell death, also known as apoptosis, which is a normal process inherent in every cell. Using IAP proteins and other anti-apoptotic factors, cancer cells evade apoptosis in response to a variety of signals, including those provided by anti-cancer agents such as chemotherapy, or naturally occurring inflammatory and immune signals transmitted through members of the tumor necrosis factor, or TNF, family of factors. Evasion from apoptosis is a fundamental mechanism whereby human cancers develop resistance to standard anti-cancer treatments. IAP inhibitors such as CUDC-427 are designed to counteract the effects of IAP proteins, thus shifting the balance away from cancer cell survival and allowing apoptosis to proceed.
CUDC-427 was designed to mimic the endogenous IAP antagonist mitochondrial protein second mitochondria-derived activator of caspases/direct IAP-binding protein (Smac/DIABLO) that is released into the cytoplasm in response to pro-apoptotic stimuli. CUDC-427 has demonstrated single-agent and combination anti-tumor activity in mouse xenograft tumor models when administered orally on a daily schedule, and IND-enabling safety studies have shown it to be well tolerated when dosed daily by oral administration, potentially enabling sustained target inhibition.
In October 2010, an open-labeled, uncontrolled, dose-escalation, Phase I clinical trial of CUDC-427 (NCT01226277; IAM4914g) began in patients with refractory solid tumors or lymphoma. Genentech recently completed this Phase I clinical trial in which 42 people received daily oral doses of CUDC-427 for two weeks, followed by a one week rest period. This 21-day cycle is repeated until disease progression or study discontinuation for any other reason. The primary endpoints of the study include evaluating the safety and tolerability and the pharmacokinetics of CUDC-427 in people with solid tumors or lymphoma and determining the maximum-tolerated-dose and a potential recommended dose for further clinical studies. Secondary endpoints include a preliminary assessment of anti-tumor activity of CUDC-427 and evaluating pharmacodynamic markers. Genentech plans to present full study results at a medical conference in mid-2013. Please refer to http://www.clinicaltrials.gov for additional study details.
About Inhibitor of Apoptosis Proteins
Impairment of programmed cell death or apoptosis often contributes to the formation and progression of cancer, and evasion of apoptosis is one of the primary strategies by which cancer cells develop resistance to anticancer therapies. Inhibitor of apoptosis (IAP) proteins are a family of functionally and structurally related proteins which include X-linked IAP (XIAP), cellular IAPs (cIAP1 and cIAP2), and melanoma IAP (ML-IAP). They confer protection from death-inducing stimuli by exerting a range of biological activities that promote cancer cell survival and proliferation. Some even directly inhibit caspases, critical players in the execution of apoptosis.
Mutations, amplifications and chromosomal translocations of IAP genes are associated with various solid and hematologic cancer types, and increased IAP expression has been associated with an unfavorable prognosis and poor outcome for patients. As a consequence, IAP proteins are considered promising molecular targets for anticancer therapy.
Antibody Effective Against Norovirus

Antibody Effective Against Norovirus
Researchers have released data showing that a monoclonal antibody can neutralize human norovirus. Norovirus causes roughly 20 million cases of acute diarrhea and vomiting annually in the United States, alone. It is also responsible for roughly 800 deaths annually.
FULL STORY
What is Norovirus?

Norovirus is a stomach bug that sets in within 10 hours of transmission and usually lasts up to three days. It is completely different from the flu in that only your stomach is affected. While most people recover completely after three days, norovirus is more serious for young children, the elderly and people with other serious health conditions. Every year 70,000 people are hospitalized and 800 deaths are caused by the virus.
What are the symptoms?
The most common symptoms of norovirus include stomach pain, vomiting, diarrhea and nausea. Some people also experience a low-grade fever, headache and body ache. Because it is common to have continued vomiting and diarrhea during the three days of illness, dehydration is another concern for those affected.
How do you get it?
Norovirus is spread through direct contact with an infected person’s vomit or feces. Most commonly, unwashed hands can be attributed to spreading the virus through surfaces or food. The virus spreads quickly in enclosed spaces like cruise ships, nursing homes and schools.
What is the treatment?
Unfortunately, there are no medications to treat norovirus. Health care providers say the best thing to do is try to stay hydrated, rest and wait for the virus to run its course. People who are unable to keep fluids down may need to receive fluids intravenously.
How can you protect yourself?
Hand washing is the best defense against the norovirus, since no one is immune to the always-changing strains of the virus. However, new research has found hand sanitizers are not affective in killing the virus. Avoid direct contact with anyone who is infected and pay close attention to cleaning and preparing food. Also, anyone who is infected should not prepare food. Use disinfectants to wipe down all surfaces that have come in contact with someone who is infected. Also, launder infected clothes immediately on the longest wash cycle to help from spreading the virus.
Biosimilars applications under review by EMA – 2013 Q2
![]()
The European Medicines Agency (EMA) is the body responsible for approval of biosimilars within the EU. A legal framework for approving biosimilars was established in 2003. Approval of biosimilars is based on an abbreviated registration process, which allows biosimilars manufacturers to provide a reduced package of information compared to originator drugs, provided they can prove ‘similarity’ to the originator or ‘reference drug’.
read all at
http://www.gabionline.net/Biosimilars/General/Biosimilars-applications-under-review-by-EMA-2013-Q2
First biosimilar filgrastims launched in Japan


International nonproprietary name: Filgrastim
Chemical name: N-L- Methionyl colony-stimulating factor (human genetically engineered); non-glycated protein consisted of 175 amino acids.
Chemical name: N-L- Methionyl colony-stimulating factor (human genetically engineered); non-glycated protein consisted of 175 amino acids.
Filgrastim is a granulocyte colony-stimulating factor (G-CSF) analog used to stimulate the proliferation and differentiation of granulocytes.[1] It is produced by recombinant DNA technology. The gene for human granulocyte colony-stimulating factor is inserted into the genetic material of Escherichia coli. The G-CSF then produced by E. coli is different from G-CSF naturally made in humans.

Hematopoietic growth factor. Interacting with receptors on the surface of hematopoietic cells it regulates production and release of neutrophils from the bone marrow to the peripheral blood. Dose dependant number growth of neutrophils with normal or increased functional activity is passing for 24 hours.
Filgrastim is marketed under several brand names, including Neupogen (Amgen), Imumax(Abbott Laboratories), Grafeel (Dr. Reddy’s Laboratories), Neukine (Intas Biopharmaceuticals), Emgrast (Emcure Pharmaceuticals), Religrast (Reliance Life Sciences), Zarzio (Sandoz), Nufil (Biocon) and others.
Apricus Biosciences is currently developing and testing a product under the brand nameNupen which can deliver filgrastim through the skin to improve post-chemotherapy recovery of neutrophil counts.
Filgrastim is also used to increase the number of hematopoietic stem cells in the blood before collection by leukapheresis for use in hematopoietic stem cell transplantation.Filgrastim is used to treat neutropenia,[2] stimulating the bone marrow to increase production of neutrophils. Causes of neutropenia include chemotherapy and bone marrow transplantation.
Filgrastim should not be used in patients with known hypersensitivity to E. coli-derived proteins.
The most commonly observed adverse effect is mild-to-moderate bone pain after repeated administration and local skin reactions at the site of injection.[3] Other observed adverse effects include serious allergic reactions (including a rash over the whole body, shortness of breath, wheezing, dizziness, swelling around the mouth or eyes, fast pulse, and sweating), ruptured spleen (sometimes resulting in death), alveolar hemorrhage, acute respiratory distress syndrome, and hemoptysis.[3] Severe sickle cell crises, in some cases resulting in death, have been associated with the use of filgrastim in patients with sickle cell disorders.[4]
Drug interactions between filgrastim and other drugs have not been fully evaluated. Drugs which may potentiate the release of neutrophils‚ such as lithium‚ should be used with caution.
Increased hematopoietic activity of the bone marrow in response to growth factor therapy has been associated with transient positive bone imaging changes; this should be considered when interpreting bone-imaging results.[5]
Filgrastim has not been studied in pregnant women and its effects on unborn babies is unknown. If taking filgrastim while pregnant, it is possible that traces of the drug could be found in the baby’s blood. It is not known if the drug can get into human breast milk.
- Beveridge, R. A.; Miller, J. A.; Kales, A. N.; Binder, R. A.; Robert, N. J.; Harvey, J. H.; Windsor, K.; Gore, I. et al. (1998). “A Comparison of Efficacy of Sargramostim (Yeast-Derived RhuGM-CSF) and Filgrastim (Bacteria-Derived RhuG-CSF) in the Therapeutic Setting of Chemotherapy-Induced Myelosuppression”. Cancer Investigation 16 (6): 366–373. doi:10.3109/07357909809115775.PMID 9679526. edit
- Crawford, J.; Glaspy, J. A.; Stoller, R. G.; Tomita, D. K.; Vincent, M. E.; McGuire, B. W.; Ozer, H. (2005). “Final Results of a Placebo-Controlled Study of Filgrastim in Small-Cell Lung Cancer: Exploration of Risk Factors for Febrile Neutropenia”. Supportive Cancer Therapy 3 (1): 36–46. doi:10.3816/SCT.2005.n.023. PMID 18632435. edit
- Neupogen “Neupogen: Patient Information Leaflet”. Amgen. Retrieved 24 June 2013.
- “NEUPOGEN® Patient Guide”. Amgen. Retrieved 24 June 2013.
- “Neupogen”. RxList. 4 June 2012. Retrieved 23 June 2013.
- Budiono Santoso; Chris J. van Boxtel; Boxtel, Christoffel Jos van (2001). Drug benefits and risks: international textbook of clinical pharmacology. New York: Wiley. ISBN 0-471-89927-5.
- “Neupogen information”. Retrieved 20 October 2005.
Genentech announced positive results from the Phase 3 CLL11 study, Leukemia Trial

Afutuzumab
Obinutuzumab (GA101)
Genentech announced positive results from the Phase 3 CLL11 study. At a pre-planned interim analysis, an independent data monitoring committee determined that the study met its primary endpoint showing that GA101 plus chlorambucil helped people live significantly longer without their disease worsening (progression-free survival; PFS) compared to Rituxan (rituximab) plus chlorambucil.
The CLL11 study is being conducted in cooperation with the German CLL Study Group (GCLLSG). These final data were reached well ahead of the target completion date in 2014 as a result of the magnitude of difference seen between the two study arms.
Afutuzumab is a monoclonal antibody being developed by Hoffmann-La Roche Inc. for the treatment of lymphoma.[1] It acts as an immunomodulator.[2][3] It was renamed obinutuzumab in 2009.[4]
Class/mechanism: Glyco-engineered anti-CD20 IgG1 type II monoclonal antibody. Engineered with a modified elbow hinge residue (valine instead of leucine at Kabat position 11) and a glyco-engineered Fc region, which is postulated to enhance its immunomodulatory effect.[1]
Route: IV
Extravasation: no information
For conciseness and simplicity, HemOnc.org currently will focus on treatment regimens and not list information such as: renal/hepatic dose adjustments, metabolism (including CYP450), excretion, monitoring parameters (although this will be considered for checklists), or manufacturer.
- Robak, T (2009). “GA-101, a third-generation, humanized and glyco-engineered anti-CD20 mAb for the treatment of B-cell lymphoid malignancies”. Current opinion in investigational drugs (London, England : 2000) 10 (6): 588–96. PMID 19513948.
- Statement On A Nonproprietary Name Adopted By The Usan Council – Afutuzumab, American Medical Association.
- International Nonproprietary Names for Pharmaceutical Substances (INN), World Health Organization.
- International Nonproprietary Names for Pharmaceutical Substances (INN), World Health Organization.

| FULL STORYread all |
FDA Approves New Drug to Treat Nephropathic Cystinosis
CHICAGO—The U.S. Food and Drug Administration (FDA) has recently approved PROCYSBI(cysteamine bitartrate), a delayed release capsule for treating nephropathic cystinosis in adults and children 6 years and older.
Ann and Robert H. Lurie Children’s Hospital of Chicago served as one of three United States sites for the landmark study and patients came from all over North America to be seen by lead investigator, Craig B Langman, M.D., The Isaac A Abt, M.D. professor of Kidney Diseases at Northwestern University Feinberg School of Medicine and head of Kidney Diseases at Lurie Children’s

Kyowa Hakko Kirin seeks MHLW Approval for Additional Indication for ATL, PTCL and CTCL of Mogamulizumab

Kyowa Hakko Kirin Co., Ltd. has been filed an application to Japan’s Ministry of Health, Labour and Welfare (“MHLW”) seeking approval for additional indication for untreated CCR4-positive adult T-cell leukemia-lymphoma (ATL), relapsed CCR4-positive peripheral T-cell lymphoma (PTCL) and cutaneous T-cell lymphoma (CTCL) of Mogamulizumab (brand name: POTELIGEO® Injection 20 mg).
read at…………
Mogamulizumab (USAN; trade name Poteligeo) is a humanized monoclonal antibodytargeting CC chemokine receptor 4 (CCR4). It has been approved in Japan for the treatment of relapsed or refractory adult T-cell leukemia/lymphoma.[1]
Mogamulizumab was developed by Kyowa Hakko Kirin Co., Ltd.[2] It has also been licensed to Amgen for development as a therapy for Asthma.[3]
- Subramaniam, J; Whiteside G, McKeage K, Croxtall J (18). “Mogamulizumab: First Global Approval”. Drugs 72 (9): 1293–1298. doi:10.2165/11631090-000000000-00000. Retrieved 10 September 2012.
- “Statement On A Nonproprietary Name Adopted By The USAN Council: Mogamulizumab”. American Medical Association.
- “Kyowa Hakko Kirin R&D Pipeline”. Kyowa Hakko Kirin. Retrieved 10 September 2012.

Poteligeo(mogamulizumab)-单克隆抗体
| 单克隆抗体Poteligeo(mogamulizumab)获得日本厚生劳动省批准治疗白血病-淋巴瘤 日本厚生劳动省批准Kyowa Hakko Kirin公司的Poteligeo治疗复发或难治性CC趋化因子受体4(CCR4,CD194)阳性的T细胞性白血病-淋巴瘤。厚生劳动省还批准了Kyowa公司这一抗体的两个诊断方法,用于测试IHC和FCM,从而确定最有可能对治疗有应答的患者亚群。Amgen公司拥有Poteligeo在除日本、韩国、中国大陆和台湾以外地区的所有非癌症适应症的开发和商业化独占权。Amgen公司正在进行本品用于治疗哮喘的Ⅰ期临床研究。 |

