
Home » Posts tagged 'FDA 2015' (Page 2)
Tag Archives: FDA 2015
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Uridine triacetate, ウリジントリアセタート FDA approves first emergency treatment for overdose of certain types of chemotherapy

December 11, 2015
Release
The U.S. Food and Drug Administration today approved Vistogard (uridine triacetate) for the emergency treatment of adults and children who receive an overdose of the cancer treatment fluorouracil or capecitabine, or who develop certain severe or life-threatening toxicities within four days of receiving these cancer treatments.
“Treating cancer requires not only selecting which drug may be most effective and well tolerated, but ensuring the correct dose is given at proper intervals. While rare, unintentional overdose can occur,” said Richard Pazdur, M.D., director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “Today’s approval is a first-of-its-kind therapy that can potentially save lives following overdose or life-threatening toxicity from these chemotherapy agents.”
Fluorouracil (taken by infusion) and capecitabine (taken orally) are similar types of chemotherapy that have been used for decades to treat several types of cancer, including breast and gastrointestinal cancers. An overdose of fluorouracil or capecitabine is rare, but when it occurs, the effects are serious and can be fatal.
Vistogard, taken orally, blocks cell damage and cell death caused by fluorouracil chemotherapy. Patients should take Vistogard as soon as possible after the overdose (whether or not they have symptoms) or early-onset (within four days) of severe or life-threatening toxicity. The patient’s health care provider will determine when he or she should return to the prescribed chemotherapy after treatment with Vistogard.
The efficacy and safety of Vistogard were studied in 135 adult and pediatric cancer patients who were treated in two separate trials and had either received an overdose of flourouracil or capecitabine, or had early-onset, unusually severe or life-threatening toxicities within 96 hours after receiving flourouracil (not due to an overdose). The studies’ primary measure was survival at 30 days or until chemotherapy could resume if prior to 30 days. Of those who were treated with Vistogard for overdose, 97 percent were still alive at 30 days. Of those treated with Vistogard for early-onset severe or life-threatening toxicity, 89 percent were alive at 30 days. In both studies, 33 percent of patients resumed chemotherapy in less than 30 days.
Vistogard is not recommended for treating non-emergency adverse reactions associated with flourouracil or capecitabine because Vistogard may lessen the efficacy of these drugs. The safety and efficacy of Vistogard initiated more than 96 hours following the end of treatment with flourouracil or capecitabine have not been established.
The most common side effects of treatment with Vistogard were diarrhea, vomiting and nausea.
The FDA granted Vistogard orphan drug designation, which provides financial incentives, like clinical trial tax credits, user fee waivers, and eligibility for market exclusivity to promote rare disease drug development. Vistogard was also granted priority review and fast track designations, which are distinct programs intended to facilitate and expedite the development and review of certain new drugs in light of their potential to benefit patients with serious or life-threatening conditions.
Vistogard is marketed by Wellstat Therapeutics Corporation based in Gaithersburg, Maryland.
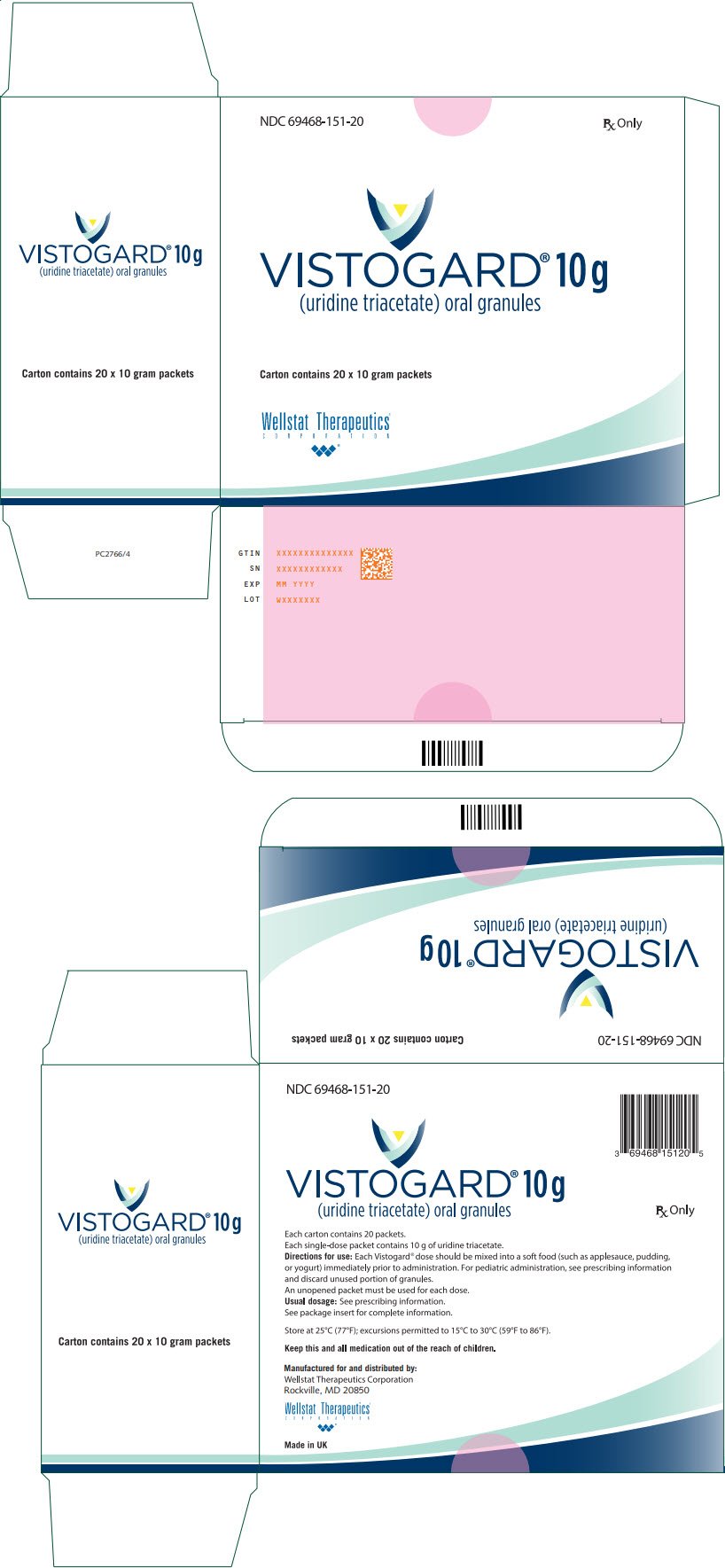
|
[(2R,3R,4R,5R)-3,4-bis(acetyloxy)-5-(2,4-dioxo-1,2,3,4-tetrahydropyrimidin-1-yl)oxolan-2-yl]methyl acetate
|
| Drug Name(s) | XURIDEN |
| FDA Application No. | (NDA) 208169 |
| Active Ingredient(s) | URIDINE TRIACETATE |
| Company | WELLSTAT THERAP |
| Original Approval or Tentative Approval Date | September 4, 2015 |
FDA APPROVAL SUMMARY
Chemotherapy induced poisoning, VISTOGARD, FDA 2015-12-11
Hereditary orotic aciduria, Xuriden, FIRST APPROVAL, 2015-09-04
|
|||||||
| External Identifiers |
|
|---|
Uridine triacetate is a drug used in the treatment of hereditary orotic aciduria[1] and to treat patients following an overdose ofchemotherapy drugs 5-fluorouracil or capecitabine, or in patients exhibiting early-onset, severe or life-threatening toxicity affecting the cardiac or central nervous system, and/or early-onset, unusually severe adverse reactions (e.g., gastrointestinal toxicity and/or neutropenia) within 96 hours following the end of 5-fluorouracil or capecitabine administration.[2][3]
Uridine triacetate was developed, manufactured and distributed by Wellstat Therapeutics and it is marketed in USA by BTG. Also, It was granted breakthrough therapy designation by FDA in 2015.
Uridine triacetate is a prodrug of uridine.[4]
Uridine triacetate, formerly known as vistonuridine, is an orally active prodrug of the naturally occurring nucleoside uridine. It is used for the treatment of hereditary orotic aciduria (Xuriden), or for the emergency treatment of fluorouracil or capecitabine overdose or toxicity (Vistogard). It is provided in the prodrug form as uridine triacetate as this form delivers 4- to 6-fold more uridine into the systemic circulation compared to equimolar doses of uridine itself. When used for the treatment or prevention of toxicity associated with fluorouracil and other antimetabolites, uridine triacetate is utilized for its ability to compete with 5-fluorouracil (5-FU) metabolites for incorporation into the genetic material of non-cancerous cells. It reduces toxicity and cell-death associated with two cytotoxic intermediates: 5-fluoro-2′-deoxyuridine-5′-monophosphate (FdUMP) and 5-fluorouridine triphosphate (FUTP). Normally, FdUMP inhibits thymidylate synthase required for thymidine synthesis and DNA replication and repair while FUTP incorporates into RNA resulting in defective strands. As a result, these metabolites are associated with various unpleasant side effects such as neutropenia, mucositis, diarrhea, and hand–foot syndrome. Like many other neoplastic agents, these side effects limit the doses of 5-FU that can be administered, which also affects the efficacy for treatment. By pre-administering with uridine (as the prodrug uridine triacetate), higher doses of 5-FU can be given allowing for improved efficacy and a reduction in toxic side effects [3]. It can also be used as a rescue therapy if severe side effects present within 96 hours after initiation of therapy. Uridine triacetate is also used for the treatment of hereditary orotic aciduria, also known as uridine monophosphate synthase deficiency. This rare congenital autosomal recessive disorder of pyrimidine metabolism is caused by a defect in uridine monophosphate synthase (UMPS), a bifunctional enzyme that catalyzes the final two steps of the de novo pyrimidine biosynthetic pathway. As a result of UMPS deficiency, patients experience a systemic deficiency of pyrimidine nucleotides, accounting for most symptoms of the disease. Additionally, orotic acid from the de novo pyrimidine pathway that cannot be converted to UMP is excreted in the urine, accounting for the common name of the disorder, orotic aciduria. Furthermore, orotic acid crystals in the urine can cause episodes of obstructive uropathy. When administered as the prodrug uridine triacetate, uridine can be used by essentially all cells to make uridine nucleotides, which compensates for the genetic deficiency in synthesis in patients with hereditary orotic aciduria. When intracellular uridine nucleotides are restored into the normal range, overproduction of orotic acid is reduced by feedback inhibition, so that urinary excretion of orotic acid is also reduced.
Marketed as the product Xuriden (FDA), uridine triacetate is indicated for the treatment of hereditary orotic aciduria. Marketed as the product Vistogard (FDA), uridine triacetate is indicated for the emergency treatment of adult and pediatric patients in the following situations: following a fluorouracil or capecitabine overdose regardless of the presence of symptoms; or who exhibit early-onset, severe or life-threatening toxicity affecting the cardiac or central nervous system, and/or early-onset, unusually severe adverse reactions (e.g., gastrointestinal toxicity and/or neutropenia) within 96 hours following the end of fluorouracil or capecitabine administration.

Uridine Triacetate was approved by the U.S. Food and Drug Administration (FDA) on Sep 4, 2015. It was developed by Wellstat Therapeutics, then marketed as Xuriden® by Wellstat Therapeutics in US. Then it was also approved by FDA for overdose of certain types of chemotherapy on Dec 11, 2015 and marketed as Vistogard®.
Uridine Triacetate is a prodrug of the nucleoside uridine used to treat hereditary orotic aciduria. Hereditary orotic aciduria is inherited from a recessive gene. The disease is due to a defective or deficient enzyme, which results in the body being unable to normally synthesize uridine, a necessary component of ribonucleic acid (RNA). Signs and symptoms of the disease include blood abnormalities (anemia, decreased white blood cell count, decreased neutrophil count), urinary tract obstruction due to the formation of orotic acid crystals in the urinary tract, failure to thrive, and developmental delays.
Xuriden® is approved as oral granules that can be mixed with food or in milk or infant formula, and is administered once daily. The starting dosage is 60 mg/kg once daily; the dose may be increased to 120 mg/kg (not to exceed 8 grams) once daily for insufficient efficacy.
Mechanism Of Action
Uridine triacetate is an acetylated form of uridine. Following oral administration, uridine triacetate is deacetylated by nonspecific esterases present throughout the body, yielding uridine in the circulation (Figure 1).
Figure 1: Uridine Triacetate Conversion to Uridine

URIDEN provides uridine in the systemic circulation of patients with hereditary orotic aciduria who cannot synthesize adequate quantities of uridine due to a genetic defect in uridine nucleotide synthesis.
Uridine triacetate is a synthetic uridine pro-drug that is converted to uridine in vivo. When used for the treatment or prevention of toxicity associated with fluorouracil and other antimetabolites, uridine triacetate is utilized for its ability to compete with 5-fluorouracil (5-FU) metabolites for incorporation into the genetic material of non-cancerous cells. It reduces toxicity and cell-death associated with two cytotoxic intermediates: 5-fluoro-2′-deoxyuridine-5′-monophosphate (FdUMP) and 5-fluorouridine triphosphate (FUTP). By pre-administering with uridine (as the prodrug uridine triacetate), higher doses of 5-FU can be given allowing for improved efficacy and a reduction in toxic side effects [A18578] such as neutropenia, mucositis, diarrhea, and hand–foot syndrome. Uridine triacetate is also used for replacement therapy in the treatment of hereditary orotic aciduria, also known as uridine monophosphate synthase (UMPS) deficiency. As a result of UMPS deficiency, patients experience a systemic deficiency of pyrimidine nucleotides, accounting for most symptoms of the disease. Additionally, orotic acid from the de novo pyrimidine pathway that cannot be converted to UMP is excreted in the urine, accounting for the common name of the disorder, orotic aciduria. Furthermore, orotic acid crystals in the urine can cause episodes of obstructive uropathy. When administered as the prodrug uridine triacetate, uridine can be used by essentially all cells to make uridine nucleotides, which compensates for the genetic deficiency in synthesis in patients with hereditary orotic aciduria.

Reference:1. J. Am. Chem. Soc. 1953, 75, 2017-2019.
2. Angew. Chem. internat. Edit. 1971, 10, 75.
3. US3116282.
PATENT
Production Example 1

5.6 g of uracil and 0.1 g of ammonium sulfate were dissolved in 22.4 ml of 1,1,1,3,3,3-hexamethyldisilazane and reacted at 120° C. for 2.5 hours. After the completion of the reaction, the reaction mixture was distilled to give 11.8 g of 2,4-bis(trimethylsilyloxy)-1,3-diazine. 1H-NMR (400 MHz, in C2D6CO): δ=0.29 (s, 9H), 0.31 (s, 9H), 6.35 (d, J=5.6 Hz, 1H), 8.19 (d, J=5.5Hz, 1H)
Referential Example 11.21 g of 2,4-bis(trimethylsilyloxy)-1,3-diazine obtained in PRODUCTION EXAMPLE 1 and 1.15 g of 1,2,3,5-tetra-O-acetyl-β-D-ribofuranose were dissolved in 4.8 ml of acetonitrile and cooled to 5° C. Next, 0.94 g of SnCl4 was added dropwise thereinto at the same temperature. After stirring for 10 minutes at the same temperature, the mixture was heated to 50° C. and reacted for 3 hours. The reaction mixture was analyzed by HPLC. Thus, β-uridine triacetate was obtained with a reaction yield of 83%.
Example 1

0.93 g of 2,4-bis(trimethylsilyloxy)-1,3-diazine obtained in PRODUCTION EXAMPLE 1 and 0.92 g of 1,2,3,5-tetra-O-acetyl-β-D-ribofuranose were dissolved in 4.7 ml of acetonitrile and cooled to 4° C. Then 0.49 g of FeCl3 was added thereto at the same temperature. After stirring for 10 minutes at the same temperature, the mixture was heated to 50° C. and reacted. The reaction was monitored by HPLC. After the completion of the reaction, the reaction mixture was added dropwise at 4° C. into a cold aqueous solution of sodium hydrogencarbonate which had been preliminarily prepared. After filtering off the catalyst residue, the filtrate was separated and the aqueous layer was extracted with 20 ml portions of ethyl acetate thrice. The organic layers were combined, washed with a saturated aqueous solution of sodium chloride and dried over sodium sulfate. After distilling off the solvent, 1.2 g (purity 80%) of the target compound was obtained as a viscous white solid.
Namely, the target compound could be obtained at a yield comparable to REFERNTIAL EXAMPLE 1 wherein SnCl4 was employed as the catalyst. 1H-NMR (400 MHz, in CDCl3): δ=2.11 (s, 3H), 2.14 (s, 3H), 2.15 (s, 3H), 4.35 (m, 3H), 5.33 (m, 2H), 5.79 (d, J=8.2 Hz, 1H), 6.04 (d, J=4.9 Hz, 1H), 7.39 (d, J=8.2 Hz, 1H)

CLIP
December 11, 2015
Release
The U.S. Food and Drug Administration today approved Vistogard (uridine triacetate) for the emergency treatment of adults and children who receive an overdose of the cancer treatment fluorouracil or capecitabine, or who develop certain severe or life-threatening toxicities within four days of receiving these cancer treatments.
“Treating cancer requires not only selecting which drug may be most effective and well tolerated, but ensuring the correct dose is given at proper intervals. While rare, unintentional overdose can occur,” said Richard Pazdur, M.D., director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “Today’s approval is a first-of-its-kind therapy that can potentially save lives following overdose or life-threatening toxicity from these chemotherapy agents.”
Fluorouracil (taken by infusion) and capecitabine (taken orally) are similar types of chemotherapy that have been used for decades to treat several types of cancer, including breast and gastrointestinal cancers. An overdose of fluorouracil or capecitabine is rare, but when it occurs, the effects are serious and can be fatal.
Vistogard, taken orally, blocks cell damage and cell death caused by fluorouracil chemotherapy. Patients should take Vistogard as soon as possible after the overdose (whether or not they have symptoms) or early-onset (within four days) of severe or life-threatening toxicity. The patient’s health care provider will determine when he or she should return to the prescribed chemotherapy after treatment with Vistogard.
The efficacy and safety of Vistogard were studied in 135 adult and pediatric cancer patients who were treated in two separate trials and had either received an overdose of flourouracil or capecitabine, or had early-onset, unusually severe or life-threatening toxicities within 96 hours after receiving flourouracil (not due to an overdose). The studies’ primary measure was survival at 30 days or until chemotherapy could resume if prior to 30 days. Of those who were treated with Vistogard for overdose, 97 percent were still alive at 30 days. Of those treated with Vistogard for early-onset severe or life-threatening toxicity, 89 percent were alive at 30 days. In both studies, 33 percent of patients resumed chemotherapy in less than 30 days.
Vistogard is not recommended for treating non-emergency adverse reactions associated with flourouracil or capecitabine because Vistogard may lessen the efficacy of these drugs. The safety and efficacy of Vistogard initiated more than 96 hours following the end of treatment with flourouracil or capecitabine have not been established.
The most common side effects of treatment with Vistogard were diarrhea, vomiting and nausea.
The FDA granted Vistogard orphan drug designation, which provides financial incentives, like clinical trial tax credits, user fee waivers, and eligibility for market exclusivity to promote rare disease drug development. Vistogard was also granted priority review and fast track designations, which are distinct programs intended to facilitate and expedite the development and review of certain new drugs in light of their potential to benefit patients with serious or life-threatening conditions.
Vistogard is marketed by Wellstat Therapeutics Corporation based in Gaithersburg, Maryland.
CLIP
With support from Almac, Wellstat delivers for a rare disease.
Proximity of API and finished drug development helps uridine triacetate to market for two indications
By Rick Mullin
“The initial contact was a cold call by Almac in 2010 or 2011,” recalls Mike Bamat, senior vice president of R&D at Wellstat Therapeutics, a small drug company in Gaithersburg, Md. “There were probably a couple of calls. It was one of those things where timing is everything.”
Almac, a Craigavon, Northern Ireland-based pharmaceutical services company, was looking to get in on Wellstat’s development of uridine triacetate, a synthetic pyrimidine analog, as an antidote for fluorouracil and capecitabine toxicity and overdose in cancer patients receiving those chemotherapies. And the calls, which Almac records indicate followed some communication between the companies, happened to come just when Wellstat was looking to change service partners as it moved toward commercial development of the drug.

Uridine triacetate
Discovery: Wellstat Therapeutic’s research on the therapeutic potential of exogenous uridine leads to a determination that uridine triacetate is a safe means of delivering the agent
Applications: Treatment of hereditary orotic aciduria (HOA), an extremely rare disease in which the body does not produce uridine, causing overproduction of orotic acid; emergency treatment of toxic reaction to or overdose of the cancer treatments fluorouracil and capecitabine
Methods of action: Treating HOA, uridine triacetate restores intracellular nucleotide concentrations, normalizing orotic acid production; as a chemotherapy antidote, it increases intracellular levels of uridine to dilute fluorouracil and capecitabine
Years in development: Since 2008 for chemotherapy antidote, and 2013 for HOA
Approved: Xuriden for HOA, Sept. 4, 2015; Vistogard for chemotherapy antidote, Dec. 11, 2015
The job went to Almac, as did work that sprang up as the result of another phone call to Wellstat—this one from the U.S. Food & Drug Administration.
As Bamat explains, uridine triacetate caught FDA’s attention regarding another potential indication—an extremely rare and life-threatening disease called hereditary orotic aciduria, or HOA. A consequence of the body’s inability to produce uridine, a necessary component of ribonucleic acid, HOA can manifest in a range of symptoms including blood abnormalities, developmental delays, and urinary tract obstruction caused by overproduction of orotic acid. There have been 20 reported cases of HOA since the 1950s. Only four cases are currently known in the U.S., Bamat says, and likely fewer than 20 in the world.
Wellstat landed approvals for Xuriden, the HOA treatment, in September of last year and Vistogard, the chemotherapy antidote, in December.
The story of Xuriden centers on a raft of FDA incentives for super-rare diseases that enabled Wellstat to move forward on an expedited application for a drug that will never be made in any great volume. But bringing Xuriden and Vistogard to market may also be viewed as the story of a drug discovery firm becoming a commercial enterprise thanks to its partnership with a service provider.
As Wellstat began late-stage development of the chemotherapy antidote, its research partner at the time, QS Pharma, was acquired by the service firm WIL Research. The look and feel of the partnership changed, according to Bamat.
“We kind of lost the small, easy-to-work-with relationship we had with them,” he says. Wellstat also needed support on development and manufacturing of a finished drug product composed of granules delivered in packets or sachets. The drug is administered orally, usually sprinkled on food such as applesauce or yogurt.
Almac was deemed a good fit because of its experience with developing drugs in granule form for “sachet presentation,” a packaging method more common in Europe than in the U.S. The Northern Ireland firm’s ability to develop and manufacture the active pharmaceutical ingredient (API) and the drug product in one location—at its headquarters—would also prove to be a significant advantage.
The distance between Gaithersburg and Craigavon, however, was a concern, according to Bamat. “We debated it. Especially those of us who knew we would be going there,” he says. “We couldn’t just jump in a car and go. But we looked at a variety of things, including cost and value, and it was all very positive at Almac.”
According to David Downey, vice president of commercial operations at Almac, bringing Wellstat’s work on uridine triacetate to commercial production posed several challenges, the first being to secure supply of uridine starting material, which is extracted from sugar beets by Euticals, an Italian firm. Next was developing a method to control particle size in both the API and the finished product. Almac also had to validate process equipment as it scaled up production.
“Uridine triacetate is Wellstat’s first commercial product,” Downey says. “So we were provided with a process more fit for development than for commercial production.”
The basic formulation of a granule drug product is simple, according to Downey: The API and excipient are mixed in a dry blender. The challenge is developing an analytical regimen to assure the granules are blended uniformly. Meeting the challenge required a high level of coordination between API and drug product process development.
“Wellstat needed a partner that could support them from the API to the drug product,” Downey says. The physical proximity between the Almac facilities in Craigavon conducting API and drug product work was a key advantage, he claims.

“If you listen to our business development people, you’ll hear them use the term, ‘crossing car parks as opposed to crossing oceans,’ ” Downey says, explaining that many competitors who offer API and finished drug services run these operations thousands of kilometers apart from each other, sometimes on different continents.
Before it signed on with Almac, Wellstat had been working with uridine triacetate for about 10 years. Its focus on developing the antidote drug started in 2008. Branching into the HOA treatment, however, upped the stakes.
Clinical study development for an HOA therapy was expedited via a full house of regulatory incentives from FDA, according to Bamat. “We had orphan drug designation, rare pediatric designation, breakthrough therapy designation, and priority review,” he says. “So they really went all out in helping us develop this.”
Although Wellstat was interested in developing a life saving drug for children, it was concerned about paying for it, given the tiny market. “At that time, the rare pediatric disease priority review voucher program was just on the radar,” Bamat says. “FDA said, ‘Consider this new program. Maybe it’s a way that at some risk you could recoup some of your costs.’ We looked at it and were willing to take the risk.”
It paid off. Wellstat was able to sell its priority review voucher—which entitles a company that brings a rare pediatric drug to market to receive expedited review of a subsequent drug—to AstraZeneca last year for an undisclosed amount. Other vouchers sold in 2015 brought high sums, including $350 million for one that AbbVie bought from United Therapeutics in August.
Bamat says Wellstat is not likely to change focus after its success with uridine triacetate. It continues to investigate new indications for the compound and will likely work with Almac on anything going into commercial development.
He emphasizes the importance of maintaining an effective working relationship with an outsourcing partner. “My main consideration is that these are people we can really work with on a day-to-day, week-to-week basis,” Bamat says. “Will the communication be good? Will they be honest and transparent with us, and will we be the same for them? That was a key factor, and we felt it was a plus with Almac.”
 |
|
| Clinical data | |
|---|---|
| Trade names | Vistogard, Xuriden |
| Routes of administration |
Oral granules |
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Metabolism | Pyrimidine catabolic pathway |
| Onset of action | Tmax = 2-3 hours |
| Biological half-life | 2-2.5 hours |
| Excretion | Renal |
| Identifiers | |
| DrugBank | DB09144 |
| Chemical data | |
| Formula | C15H18Cl0N2O9S0 |
| Molar mass | 370.31 g·mol−1 |
References
- HIGHLIGHTS OF PRESCRIBING INFORMATION OF XURIDEN
- Jump up^ BTG Announces FDA Approval of VISTOGARD® (Uridine Triacetate) as Antidote to Overdose and Early Onset, Severe, or Life-Threatening Toxicities from Chemotherapy Drugs 5-Fluorouracil (5-FU) or Capecitabine
- Jump up^ “FDA Approved Drugs:Uridine Triacetate”. FDA. 2015-12-11. Retrieved 2016-04-29.
- “Uridine triacetate”. DrugBank.
| Patent ID | Date | Patent Title |
|---|---|---|
| US7807654 | 2010-10-05 | Compositions and methods for treatment of mitochondrial diseases |
| US2010222296 | 2010-09-02 | PYRIMIDINES, SUCH AS URIDINE, IN TREATMENTS FOR PATIENTS WITH BIPOLAR DISORDER |
| US7737128 | 2010-06-15 | Pyrimidines, such as uridine, in treatments for patients with bipolar disorder |
| US2010098678 | 2010-04-22 | Methods of Treatment of Mitochondrial Disorders |
| US2010041620 | 2010-02-18 | METHODS FOR IMPROVING FRONTAL BRAIN BIOENERGETIC METABOLISM |
| US2010041621 | 2010-02-18 | METHODS AND COMPOSITIONS FOR IMPROVING COGNITIVE PERFORMANCE |
| US7582619 | 2009-09-01 | Compositions and methods for treatment of mitochondrial diseases |
| US2008226684 | 2008-09-18 | METHOD AND PROCESS FOR THE PRODUCTION OF MULTI-COATED RECOGNITIVE AND RELEASING SYSTEMS |
| US7105498 | 2006-09-12 | Acylated uridine and cytidine and uses thereof |
| US6956028 | 2005-10-18 | Compositions and methods for treatment of mitochondrial diseases |
| Patent ID | Date | Patent Title |
|---|---|---|
| US2015307542 | 2015-10-29 | MODIFIED NUCLEIC ACID MOLECULES AND USES THEREOF |
| US2015167017 | 2015-06-18 | ALTERNATIVE NUCLEIC ACID MOLECULES AND USES THEREOF |
| US8821899 | 2014-09-02 | Method and process for the production of multi-coated recognitive and releasing systems |
| US8771713 | 2014-07-08 | Method and process for the production of multi-coated recognitive and releasing systems |
| US8741316 | 2014-06-03 | Highly porous, recognitive polymer systems |
| US2012294869 | 2012-11-22 | Methods for Treating Fatty Liver Disease |
| US2012078529 | 2012-03-29 | DETERMINING THE SEVERITY OF 5-FLUOROURACIL OVERDOSE |
| US8067392 | 2011-11-29 | Compositions and methods for treatment of mitochondrial diseases |
| US7915233 | 2011-03-29 | Compositions and methods for treatment of mitochondrial diseases |
| US7884202 | 2011-02-08 | Nucleobase Having Perfluoroalkyl Group and Process for Producing the Same |
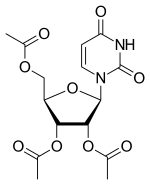
Uridine triacetate
- Molecular FormulaC15H18N2O9
- Average mass370.311 Da
ウリジントリアセタート
[(2R,3R,4R,5R)-3,4-diacetyloxy-5-(2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methyl acetate
FDA APPROVED2015/9/4 . AS Xuriden
Uridine triacetate (INN),[1] formerly known as vistonuridine, is an orally active tri-acetylated prodrug of uridine[2] used:
- in the treatment of hereditary orotic aciduria (brand name Xuriden /ˈzʊərədɛn/ ZOOR-ə-den);[3]
- to treat patients following an overdose of chemotherapy drugs 5-fluorouracil (5-FU) or capecitabine regardless of the presence of symptoms, or who exhibit early-onset, severe or life-threatening toxicity affecting the cardiac or central nervous system, and/or early-onset, unusually severe adverse reactions (e.g., gastrointestinal toxicity and/or neutropenia) within 96 hours following the end of fluorouracil or capecitabine administration (brand name Vistogard).[4][5][6]
Uridine triacetate was developed, manufactured and distributed by Wellstat Therapeutics. Also, It was granted breakthrough therapy designation by FDA in 2015.
Uridine Triacetate is a synthetic uridine pro-drug that is converted to uridine in vivo. Uridine, a pyrimidine nucleotide, has been used in a variety of diseases including depressive disorders and inherited myopathies. (NCI04)
Uridine triacetate, formerly known as vistonuridine, is an orally active prodrug of the naturally occurring nucleoside uridine. It is used for the treatment of hereditary orotic aciduria (Xuriden), or for the emergency treatment of fluorouracil or capecitabine overdose or toxicity (Vistogard). It is provided in the prodrug form as uridine triacetate as this form delivers 4- to 6-fold more uridine into the systemic circulation compared to equimolar doses of uridine itself. When used for the treatment or prevention of toxicity associated with fluorouracil and other antimetabolites, uridine triacetate is utilized for its ability to compete with 5-fluorouracil (5-FU) metabolites for incorporation into the genetic material of non-cancerous cells. It reduces toxicity and cell-death associated with two cytotoxic intermediates: 5-fluoro-2′-deoxyuridine-5′-monophosphate (FdUMP) and 5-fluorouridine triphosphate (FUTP). Normally, FdUMP inhibits thymidylate synthase required for thymidine synthesis and DNA replication and repair while FUTPincorporates into RNA resulting in defective strands. As a result, these metabolites are associated with various unpleasant side effects such as neutropenia, mucositis, diarrhea, and hand–foot syndrome. Like many other neoplastic agents, these side effects limit the doses of 5-FU that can be administered, which also affects the efficacy for treatment. By pre-administering with uridine (as the prodrug uridine triacetate), higher doses of 5-FU can be given allowing for improved efficacy and a reduction in toxic side effects [A18578]. It can also be used as a rescue therapy if severe side effects present within 96 hours after initiation of therapy. Uridine triacetate is also used for the treatment of hereditary orotic aciduria, also known as uridine monophosphate synthase deficiency. This rare congenital autosomal recessive disorder of pyrimidinemetabolism is caused by a defect in uridine monophosphate synthase (UMPS), a bifunctional enzyme that catalyzes the final two steps of the de novo pyrimidine biosynthetic pathway. As a result of UMPS deficiency, patients experience a systemic deficiency of pyrimidine nucleotides, accounting for most symptoms of the disease. Additionally, orotic acid from the de novo pyrimidine pathway that cannot be converted to UMP is excreted in the urine, accounting for the common name of the disorder, orotic aciduria. Furthermore, orotic acid crystals in the urine can cause episodes of obstructive uropathy. When administered as the prodrug uridine triacetate, uridine can be used by essentially all cells to make uridine nucleotides, which compensates for the genetic deficiency in synthesis in patients with hereditary orotic aciduria. When intracellular uridine nucleotides are restored into the normal range, overproduction of orotic acid is reduced by feedback inhibition, so that urinary excretion of orotic acid is also reduced.
References
- ^ “International Nonproprietary Names for Pharmaceutical Substances (INN). Recommended International Nonproprietary Names: List 65” (PDF). World Health Organization. p. 92. Retrieved 12 March 2017.
- ^ “Uridine triacetate — DrugBank Page”. 12 March 2017.
- ^ “Xuriden (uridine triacetate) Oral Granules. Full Prescribing Information” (PDF). Wellstat Therapeutics Corporation. Gaithersburg, MD 20878. Retrieved 12 March 2017.
- ^ “Vistogard (uridine triacetate) Oral Granules. Full Prescribing Information” (PDF). Wellstat Therapeutics Corporation. Gaithersburg, MD 20878. Retrieved 12 March 2017.
- ^ “BTG Announces FDA Approval of Vistogard® (Uridine Triacetate) as Antidote to Overdose and Early Onset, Severe, or Life-Threatening Toxicities from Chemotherapy Drugs 5-Fluorouracil (5-FU) or Capecitabine”. BTG International Ltd. 11 December 2015. Retrieved 12 March 2017.
- ^ “Approved Drugs — Uridine Triacetate”. U.S. Food and Drug Administration. Retrieved 12 March 2017.
External links
Patents
- US7776838
- US5968914
- US6258795
FDA Orange Book Patents
| FDA Orange Book Patents: 1 of 2 (FDA Orange Book Patent ID) | |
|---|---|
| Patent | 7776838 |
| Expiration | Aug 17, 2027 |
| Applicant | WELLSTAT THERAP |
| Drug Application | N208159 (Prescription Drug: VISTOGARD. Ingredients: URIDINE TRIACETATE) |
| FDA Orange Book Patents: 2 of 2 (FDA Orange Book Patent ID) | |
|---|---|
| Patent | 6258795 |
| Expiration | Jul 10, 2019 |
| Applicant | WELLSTAT THERAP |
| Drug Application | N208159 (Prescription Drug: VISTOGARD. Ingredients: URIDINE TRIACETATE) |
 |
|
| Clinical data | |
|---|---|
| Trade names | Vistogard, Xuriden |
| Routes of administration |
Oral granules |
| ATC code | |
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Metabolism | Pyrimidine catabolic pathway |
| Onset of action | Tmax = 2–3 hours |
| Elimination half-life | 2–2.5 hours |
| Excretion | Renal |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ECHA InfoCard | 100.021.710 |
| Chemical and physical data | |
| Formula | C15H18N2O9 |
| Molar mass | 370.31 g·mol−1 |
| 3D model (JSmol) | |
////////////Uridine triacetate, ウリジントリアセタート , FDA 2015, breakthrough therapy designation ,
|
CC(=O)OC[C@H]1O[C@H]([C@H](OC(C)=O)[C@@H]1OC(C)=O)N1C=CC(=O)NC1=O
|
FDA approves new oral therapy to treat ALK-positive lung cancer
December 11, 2015
Release
The U.S. Food and Drug Administration today approved Alecensa (alectinib) to treat people with advanced (metastatic) ALK-positive non-small cell lung cancer (NSCLC) whose disease has worsened after, or who could not tolerate treatment with, another therapy called Xalkori (crizotinib).
Lung cancer is the leading cause of cancer death in the United States, with an estimated 221,200 new diagnoses and 158,040 deaths in 2015, according to the National Cancer Institute. An ALK (anaplastic lymphoma kinase) gene mutation can occur in several different types of cancer cells, including lung cancer cells. ALK gene mutations are present in about 5 percent of patients with NSCLC. In metastatic cancer, the disease spreads to new parts of the body. In ALK-positive NSCLC metastatic patients, the brain is a common place for the disease to spread.
“Today’s approval provides a new therapy for a group of patients who would have few treatment options once their disease no longer responds to treatment with Xalkori,” said Richard Pazdur, M.D., director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “In addition to the primary effect on tumors in the lung, Alecensa clinical trials provide evidence of an effect on tumors that had spread to the brain, which is an important effect for clinicians to understand.”
Alecensa is an oral medication that blocks the activity of the ALK protein, which may prevent NSCLC cells from growing and spreading.
The safety and efficacy of Alecensa were studied in two single-arm clinical trials of patients with metastatic ALK-positive NSCLC whose disease was no longer controlled by treatment with Xalkori. Study participants received Alecensa twice daily to measure the drug’s effect on their lung cancer tumors. In the first study, 38 percent of participants experienced a partial shrinkage of their NSCLC tumors, an effect that lasted for an average of 7.5 months. In the second study, 44 percent of participants experienced a partial shrinkage of their NSCLC tumors, lasting for an average of 11.2 months. The trials also examined Alecensa’s effect on individuals’ brain metastases, a common occurrence in this population. Sixty-one percent of participants in the two trials who had measurable brain metastases experienced a complete or partial reduction in their brain tumors, lasting an average of 9.1 months.
The most common side effects of Alecensa are fatigue, constipation, swelling (edema) and muscle pain (myalgia). Alecensa may cause serious side effects, including liver problems, severe or life-threatening inflammation of the lungs, very slow heartbeats and severe muscle problems. Treatment with Alecensa may cause sunburn when patients are exposed to sunlight.
Alecensa was approved using the accelerated approval regulatory pathway, which allows the FDA to approve products for serious or life-threatening diseases based on evidence that the product has an effect on an outcome that is reasonably likely to predict clinical benefit. In the case of Alecensa, the tumor response to treatment, along with the duration of response, provided this evidence. Under the accelerated approval requirements, a confirmatory study is required to verify and describe the clinical benefit of Alecensa.
The FDA granted the Alecensa application breakthrough therapy designation and priority review status. These are distinct programs intended to facilitate and expedite the development and review of certain new drugs in light of their potential to benefit patients with serious or life-threatening conditions. Alecensa also received orphan drug designation, which provides incentives such as tax credits, user fee waivers and eligibility for exclusivity to assist and encourage the development of drugs for rare diseases.
Alecensa is marketed by Genentech, based in San Francisco, California. Xalkori is marketed by Pfizer, based in New York, New York.
Synthesis
Read also
/////////////////
FDA approves first recombinant von Willebrand factor to treat bleeding episodes

| Company | Baxalta Inc. |
| Description | Recombinant human von Willebrand factor (vWF) |
| Molecular Target | von Willebrand factor (vWF) |
| Mechanism of Action | |
| Therapeutic Modality | Biologic: Protein |
| Latest Stage of Development | Registration |
| Standard Indication | Bleeding |
| Indication Details | Treat and prevent bleeding episodes in von Willebrand disease (vWD) patients; Treat von Willebrand disease (vWD) |
| Regulatory Designation | U.S. – Orphan Drug (Treat and prevent bleeding episodes in von Willebrand disease (vWD) patients); EU – Orphan Drug (Treat and prevent bleeding episodes in von Willebrand disease (vWD) patients); Japan – Orphan Drug (Treat and prevent bleeding episodes in von Willebrand disease (vWD) patients) |
December 8, 2015
Release
The U.S. Food and Drug Administration today approved Vonvendi, von Willebrand factor (Recombinant), for use in adults 18 years of age and older who have von Willebrand disease (VWD). Vonvendi is the first FDA-approved recombinant von Willebrand factor, and is approved for the on-demand (as needed) treatment and control of bleeding episodes in adults diagnosed with VWD.
VWD is the most common inherited bleeding disorder, affecting approximately 1 percent of the U.S. population. Men and women are equally affected by VWD, which is caused by a deficiency or defect in von Willebrand factor, a protein that is critical for normal blood clotting. Patients with VWD can develop severe bleeding from the nose, gums, and intestines, as well as into muscles and joints. Women with VWD may have heavy menstrual periods lasting longer than average and may experience excessive bleeding after childbirth.
“Patients with heritable bleeding disorders should meet with their health care provider to discuss appropriate measures to reduce blood loss,” said Karen Midthun, M.D., director of the FDA’s Center for Biologics Evaluation and Research. “The approval of Vonvendi provides an additional therapeutic option for the treatment of bleeding episodes in patients with von Willebrand disease.”
The safety and efficacy of Vonvendi were evaluated in two clinical trials of 69 adult participants with VWD. These trials demonstrated that Vonvendi was safe and effective for the on-demand treatment and control of bleeding episodes from a variety of different sites in the body. No safety concerns were identified in the trials. The most common adverse reaction observed was generalized pruritus (itching).
The FDA granted Vonvendi orphan product designation for these uses. Orphan product designation is given to drugs intended to treat rare diseases in order to promote their development.
Vonvendi is manufactured by Baxalta U.S., Inc., based in Westlake Village, California.
FDA approves first drug to treat a rare enzyme disorder in pediatric and adult patients
![]()


December 8, 2015
Release
Today, the U.S. Food and Drug Administration approved Kanuma (sebelipase alfa) as the first treatment for patients with a rare disease known as lysosomal acid lipase (LAL) deficiency.
Patients with LAL deficiency (also known as Wolman disease and cholesteryl ester storage disease [CESD]) have no or little LAL enzyme activity. This results in a build-up of fats within the cells of various tissues that can lead to liver and cardiovascular disease and other complications. Wolman disease often presents during infancy (around 2 to 4 months of age) and is a rapidly progressive disease. Patients with Wolman disease rarely survive beyond the first year of life. CESD is a milder, later-onset form of LAL deficiency and presents in early childhood or later. Life expectancy of patients with CESD depends on the severity of the disease and associated complications. Wolman disease affects one to two infants per million births, and CESD affects 25 individuals per million births.
Today’s action involved approvals from two FDA centers. The Center for Veterinary Medicine (CVM) approved an application for a recombinant DNA (rDNA) construct in chickens that are genetically engineered (GE) to produce a recombinant form of human lysosomal acid lipase (rhLAL) protein in their egg whites. The FDA regulates GE animals under the new animal drug provisions of the Federal Food, Drug, and Cosmetic Act, because an rDNA construct introduced into an animal to change its structure or function meets the definition of a drug. The Center for Drug Evaluation and Research (CDER) approved the human therapeutic biologic (Kanuma), which is purified from those egg whites, based on its safety and efficacy in humans with LAL deficiency.
“LAL deficiency is a rare inherited genetic disorder that can lead to serious and life-threatening organ damage, especially when onset begins in infancy,” said CDER Director Janet Woodcock, M.D. “Using this technology, these patients for the first time ever have access to a treatment that may improve their lives and chances of survival.”
The new therapy, Kanuma, provides an rhLAL protein that functions in place of the missing, partially active or inactive LAL protein in the patient. Kanuma is produced by GE chickens containing an rDNA construct responsible for producing rhLAL protein in their egg whites. These egg whites are refined to extract the rhLAL protein that is eventually used to produce Kanuma and treat patients with LAL deficiency. The GE chickens are used only for producing the drug substance, and neither the chicken nor the eggs are allowed in the food supply.
Kanuma is approved for use in patients with LAL deficiency. Treatment is provided via intravenous infusion once weekly in patients with rapidly progressive LAL deficiency presenting in the first six months of life, and once every other week in all other patients.
CDER evaluated the safety and efficacy of Kanuma in an open-label, historically controlled trial in nine infants with rapidly progressive Wolman disease and in a double-blind, placebo-controlled trial in 66 pediatric and adult patients with CESD. In the trial in infants with Wolman disease, six of nine infants (67 percent) treated with Kanuma were alive at 12 months of age, whereas none of the 21 infants in the historical control group survived. In the trial in CESD patients, there was a statistically significant improvement in LDL-cholesterol levels and other disease-related parameters in those treated with Kanuma versus placebo after 20 weeks of treatment.
The most common side effects observed in patients treated with Kanuma are diarrhea, vomiting, fever, rhinitis, anemia, cough, headache, constipation, and nausea.
In its review of the GE chicken application, CVM assessed the safety of the rDNA construct, including the safety of the rDNA construct to the animals, as well as a full review of the construct and its stability in the genome of the chicken over several generations. No adverse outcomes were noted in the chickens. As required by the National Environmental Policy Act and its implementing regulations, CVM evaluated the potential environmental impacts of approval of the sponsor’s GE chickens and determined that the approval does not cause any significant impact on the environment, because the chickens are raised in highly secure indoor facilities.
“We reviewed all of the data to ensure that the hens do produce rhLAL in their egg whites, without suffering any adverse health effects from the introduced rDNA construct. The company has taken rigorous steps to ensure that neither the chickens nor the eggs will enter the food supply, and we have confirmed their containment systems by inspecting the manufacturing facilities,” said CVM Director Bernadette Dunham, D.V.M., Ph.D.
The FDA granted Kanuma orphan drug designation because it treats a rare disease affecting fewer than 200,000 patients in the United States. Orphan drug designation provides financial incentives for rare disease drug development such as clinical trial tax credits, user fee waivers, and eligibility for market exclusivity to promote rare disease drug development. Kanuma was also granted breakthrough therapy designation as it is the first and only treatment available for Wolman disease, the very severe infant form of the disease. The breakthrough therapy designation program encourages the FDA to work collaboratively with sponsors, by providing timely advice and interactive communications, to help expedite the development and review of important new drugs for serious or life-threatening conditions. The Kanuma application was also granted a priority review, which is granted to drug applications that show a significant improvement in safety or effectiveness in the treatment of a serious condition. The manufacturer of Kanuma was granted a rare pediatric disease priority review voucher –– a provision intended to encourage development of new drugs and biologics for the prevention and treatment of rare pediatric diseases.
Kanuma is produced by Alexion Pharmaceuticals Inc., based in Cheshire, Connecticut.


FDA approves Praxbind, Idarucizumab the first reversal agent for the anticoagulant Pradaxa
October 16, 2015
Release
The U.S. Food and Drug Administration today granted accelerated approval to Praxbind (idarucizumab) for use in patients who are taking the anticoagulant Pradaxa (dabigatran) during emergency situations when there is a need to reverse Pradaxa’s blood-thinning effects.
“The anticoagulant effects of Pradaxa are important and life-saving for some patients, but there are situations where reversal of the drug’s effects is medically necessary,” said Richard Pazdur, M.D., director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “Today’s approval offers the medical community an important tool for managing patients taking Pradaxa in emergency or life-threatening situations when bleeding can’t be controlled.”
The FDA approved Pradaxa in 2010 to prevent stroke and systemic blood clots in patients with atrial fibrillation, as well as for the treatment and prevention of deep venous thrombosis and pulmonary embolism. Praxbind is the first reversal agent approved specifically for Pradaxa and works by binding to the drug compound to neutralize its effect. Praxbind solution is for intravenous injection.
The safety and effectiveness of Praxbind were studied in three trials involving a total of 283 healthy volunteers taking Pradaxa (i.e., people who did not require an anticoagulant). In the healthy volunteers who were given Praxbind, there was an immediate reduction in the amount of Pradaxa in participants’ blood (measured as unbound dabigatran plasma concentration) that lasted for a period of at least 24 hours. In this study, the most common side effect from use of Praxbind was headache.
Another trial included 123 patients taking Pradaxa who received Praxbind due to uncontrolled bleeding or because they required emergency surgery. In this ongoing trial, based on laboratory testing, the anticoagulant effect of Pradaxa was fully reversed in 89 percent of patients within four hours of receiving Praxbind. In this patient trial, the most common side effects were low potassium (hypokalemia), confusion, constipation, fever and pneumonia.
Reversing the effect of Pradaxa exposes patients to the risk of blood clots and stroke from their underlying disease (such as atrial fibrillation). The Praxbind labeling recommends patients resume their anticoagulant therapy as soon as medically appropriate, as determined by their health care provider.
Praxbind is approved under the FDA’s accelerated approval program, which allows the agency to approve drugs for serious conditions that fill an unmet medical need based on an effect on a surrogate or an intermediate clinical endpoint that is reasonably likely to predict a clinical benefit to patients. The program is designed to provide patients with earlier access to promising new drugs, but the company will be required to submit additional clinical information after approval to confirm the drug’s clinical benefit.
Praxbind and Pradaxa are both marketed by Boehringer Ingelheim of Ridgefield, Connecticut.
FDA approves new drug treatment for nausea and vomiting from chemotherapy

September 2, 2015
Release
The U.S. Food and Drug Administration approved Varubi (rolapitant) to prevent delayed phase chemotherapy-induced nausea and vomiting (emesis). Varubi is approved in adults in combination with other drugs (antiemetic agents) that prevent nausea and vomiting associated with initial and repeat courses of vomit-inducing (emetogenic and highly emetogenic) cancer chemotherapy.
Nausea and vomiting are common side effects experienced by cancer patients undergoing chemotherapy. Symptoms can persist for days after the chemotherapy drugs are administered. Nausea and vomiting that occurs from 24 hours to up to 120 hours after the start of chemotherapy is referred to as delayed phase nausea and vomiting, and it can result in serious health complications. Prolonged nausea and vomiting can lead to weight loss, dehydration and malnutrition in cancer patients leading to hospitalization.
“Chemotherapy-induced nausea and vomiting remains a major issue that can disrupt patients’ lives and sometimes their therapy,” said Amy Egan, M.D., M.P.H., deputy director of the Office of Drug Evaluation III in the FDA’s Center for Drug Evaluation and Research. “Today’s approval provides cancer patients with another treatment option for the prevention of the delayed phase of nausea and vomiting caused by chemotherapy.”
Varubi is a substance P/neurokinin-1 (NK-1) receptor antagonist. Activation of NK-1 receptors plays a central role in nausea and vomiting induced by certain cancer chemotherapies, particularly in the delayed phase. Varubi is provided to patients in tablet form.
The safety and efficacy of Varubi were established in three randomized, double-blind, controlled clinical trials where Varubi in combination with granisetron and dexamethasone was compared with a control therapy (placebo, granisetron and dexamethasone) in 2,800 patients receiving a chemotherapy regimen that included highly emetogenic (such as cisplatin and the combination of anthracycline and cyclophosphamide) and moderately emetogenic chemotherapy drugs. Those patients treated with Varubi had a greater reduction in vomiting and use of rescue medication for nausea and vomiting during the delayed phase compared to those receiving the control therapy.
Varubi inhibits the CYP2D6 enzyme, which is responsible for metabolizing certain drugs. Varubi is contraindicated with the use of thioridazine, a drug metabolized by the CYP2D6 enzyme, because use of the two drugs together may increase the amount of thioridazine in the blood and cause an abnormal heart rhythm that can be serious.
The most common side effects in patients treated with Varubi include a low white blood cell count (neutropenia), hiccups, decreased appetite and dizziness.
Varubi is marketed by Tesaro Inc., based in Waltham, Massachusetts.
FDA approves Praluent for the treatment of high LDL cholesterol
26 August 2015
Sanofi and Regeneron have announced that the US Food and Drug Administration (FDA) has approved Praluent® (alirocumab) Injection.

Praluent is indicated as an adjunct to diet and maximally tolerated statin therapy for the treatment of adults with heterozygous familial hypercholesterolemia or clinical atherosclerotic cardiovascular disease (ASCVD), who require additional lowering of low-density lipoprotein (LDL) cholesterol. The effect of Praluent on cardiovascular morbidity and mortality has not been determined.
////////Sanofi, Regeneron, US Food and Drug Administration, FDA, approved, Praluent® , alirocumab
FDA approves flibanserin first treatment for sexual desire disorder
FDA approves first treatment for sexual desire disorder
Addyi approved to treat premenopausal women
SEE FULL SYNTHESIS …CLICK HERE
The U.S. Food and Drug Administration today approved to treat acquired, generalized hypoactive sexual desire disorder (HSDD) in premenopausal women. Prior to Addyi’s approval, there were no FDA-approved treatments for sexual desire disorders in men or women.
August 18, 2015
Release
The U.S. Food and Drug Administration today approved Addyi (flibanserin) to treat acquired, generalized hypoactive sexual desire disorder (HSDD) in premenopausal women. Prior to Addyi’s approval, there were no FDA-approved treatments for sexual desire disorders in men or women.
“Today’s approval provides women distressed by their low sexual desire with an approved treatment option,” said Janet Woodcock, M.D., director of the FDA’s Center for Drug Evaluation and Research (CDER). “The FDA strives to protect and advance the health of women, and we are committed to supporting the development of safe and effective treatments for female sexual dysfunction.”
HSDD is characterized by low sexual desire that causes marked distress or interpersonal difficulty and is not due to a co-existing medical or psychiatric condition, problems within the relationship, or the effects of a medication or other drug substance. HSDD is acquired when it develops in a patient who previously had no problems with sexual desire. HSDD is generalized when it occurs regardless of the type of sexual activity, the situation or the sexual partner.
“Because of a potentially serious interaction with alcohol, treatment with Addyi will only be available through certified health care professionals and certified pharmacies,” continued Dr. Woodcock. “Patients and prescribers should fully understand the risks associated with the use of Addyi before considering treatment.”
Addyi can cause severely low blood pressure (hypotension) and loss of consciousness (syncope). These risks are increased and more severe when patients drink alcohol or take Addyi with certain medicines (known as moderate or strong CYP3A4 inhibitors) that interfere with the breakdown of Addyi in the body. Because of the alcohol interaction, the use of alcohol is contraindicated while taking Addyi. Health care professionals must assess the likelihood of the patient reliably abstaining from alcohol before prescribing Addyi.
Addyi is being approved with a risk evaluation and mitigation strategy (REMS), which includes elements to assure safe use (ETASU). The FDA is requiring this REMS because of the increased risk of severe hypotension and syncope due to the interaction between Addyi and alcohol. The REMS requires that prescribers be certified with the REMS program by enrolling and completing training. Certified prescribers must counsel patients using a Patient-Provider Agreement Form about the increased risk of severe hypotension and syncope and about the importance of not drinking alcohol during treatment with Addyi. Additionally, pharmacies must be certified with the REMS program by enrolling and completing training. Certified pharmacies must only dispense Addyi to patients with a prescription from a certified prescriber. Additionally, pharmacists must counsel patients prior to dispensing not to drink alcohol during treatment with Addyi.
Addyi is also being approved with a Boxed Warning to highlight the risks of severe hypotension and syncope in patients who drink alcohol during treatment with Addyi, in those who also use moderate or strong CYP3A4 inhibitors, and in those who have liver impairment. Addyi is contraindicated in these patients. In addition, the FDA is requiring the company that owns Addyi to conduct three well-designed studies in women to better understand the known serious risks of the interaction between Addyi and alcohol.
Addyi is a serotonin 1A receptor agonist and a serotonin 2A receptor antagonist, but the mechanism by which the drug improves sexual desire and related distress is not known. Addyi is taken once daily. It is dosed at bedtime to help decrease the risk of adverse events occurring due to possible hypotension, syncope and central nervous system depression (such as sleepiness and sedation). Patients should discontinue treatment after eight weeks if they do not report an improvement in sexual desire and associated distress.
The effectiveness of the 100 mg bedtime dose of Addyi was evaluated in three 24-week randomized, double-blind, placebo-controlled trials in about 2,400 premenopausal women with acquired, generalized HSDD. The average age of the trial participants was 36 years, with an average duration of HSDD of approximately five years. In these trials, women counted the number of satisfying sexual events, reported sexual desire over the preceding four weeks (scored on a range of 1.2 to 6.0) and reported distress related to low sexual desire (on a range of 0 to 4). On average, treatment with Addyi increased the number of satisfying sexual events by 0.5 to one additional event per month over placebo increased the sexual desire score by 0.3 to 0.4 over placebo, and decreased the distress score related to sexual desire by 0.3 to 0.4 over placebo. Additional analyses explored whether the improvements with Addyi were meaningful to patients, taking into account the effects of treatment seen among those patients who reported feeling much improved or very much improved overall. Across the three trials, about 10 percent more Addyi-treated patients than placebo-treated patients reported meaningful improvements in satisfying sexual events, sexual desire or distress. Addyi has not been shown to enhance sexual performance.
The 100 mg bedtime dose of Addyi has been administered to about 3,000 generally healthy premenopausal women with acquired, generalized HSDD in clinical trials, of whom about 1,700 received treatment for at least six months and 850 received treatment for at least one year.
The most common adverse reactions associated with the use of Addyi are dizziness, somnolence (sleepiness), nausea, fatigue, insomnia and dry mouth.
The FDA has recognized for some time the challenges involved in developing treatments for female sexual dysfunction. The FDA held a public Patient-Focused Drug Development meeting and scientific workshop on female sexual dysfunction on October 27 and October 28, 2014, to solicit perspectives directly from patients about their condition and its impact on daily life, and to discuss the scientific challenges related to developing drugs to treat these disorders. The FDA continues to encourage drug development in this area.
Consumers and health care professionals are encouraged to report adverse reactions from the use of Addyi to the FDA’s MedWatch Adverse Event Reporting program at www.fda.gov/MedWatch or by calling 1-800-FDA-1088.
Addyi is marketed by Sprout Pharmaceuticals, based in Raleigh, North Carolina.
////////
सुकून उतना ही देना प्रभू, जितने से जिंदगी चल जाये। औकात बस इतनी देना, कि औरों का भला हो जाये।
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE


Join me on twitter
 LIONEL MY SON
LIONEL MY SON
He was only in first standard in school when I was hit by a deadly one in a million spine stroke called acute transverse mylitis, it made me 90% paralysed and bound to a wheel chair, Now I keep him as my source of inspiration and helping millions, thanks to millions of my readers who keep me going and help me to keep my son happy

सुकून उतना ही देना प्रभू, जितने से
जिंदगी चल जाये।
औकात बस इतनी देना,
कि औरों का भला हो जाये।
Read all about Organic Spectroscopy on ORGANIC SPECTROSCOPY INTERNATIONAL
Eisai’s lenvatinib 兰伐替尼 レンバチニブ gets FDA approval
 Lenvatinib Mesilate
Lenvatinib Mesilate
Eisai’s lenvatinib 兰伐替尼 レンバチニブ
See synthesis at https://newdrugapprovals.org/2014/08/04/eisais-lenvatinib-%E5%85%B0%E4%BC%90%E6%9B%BF%E5%B0%BC-%E3%83%AC%E3%83%B3%E3%83%90%E3%83%81%E3%83%8B%E3%83%96-to-get-speedy-review-in-europe/
Above post contains SYNTHESIS, spectrocopy predicts, etc
February 13, 2015
Release
The U.S. Food and Drug Administration today granted approval to Lenvima (lenvatinib) to treat patients with progressive, differentiated thyroid cancer (DTC) whose disease progressed despite receiving radioactive iodine therapy (radioactive iodine refractory disease).
The most common type of thyroid cancer, DTC is a cancerous growth of the thyroid gland which is located in the neck and helps regulate the body’s metabolism. The National Cancer Institute estimates that 62,980 Americans were diagnosed with thyroid cancer and 1,890 died from the disease in 2014. Lenvima is a kinase inhibitor, which works by blocking certain proteins from helping cancer cells grow and divide.
“The development of new therapies to assist patients with refractory disease is of high importance to the FDA,” said Richard Pazdur, M.D., director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “Today’s approval gives patients and healthcare professionals a new therapy to help slow the progression of DTC.”
Lenvima was reviewed under the FDA’s priority review program, which provides for an expedited review of drugs that, if approved, would provide significant improvement in safety or effectiveness in the treatment of a serious condition. The drug also received orphan product designation because it is intended to treat a rare disease. Lenvima is being approved approximately two months ahead of the prescription drug user fee goal date of April 14, 2015, the date when the agency was scheduled to complete its review of the application.
Lenvima’s efficacy was demonstrated in 392 participants with progressive, radioactive iodine-refractory DTC who were randomly assigned to receive either Lenvima or a placebo. Study results showed Lenvima-treated participants lived a median of 18.3 months without their disease progressing (progression-free survival), compared to a median of 3.6 months for participants who received a placebo. Additionally, 65 percent of participants treated with Lenvima saw a reduction in tumor size, compared to the two percent of participants who received a placebo. A majority of participants randomly assigned to receive the placebo were treated with Lenvima upon disease progression.
The most common side effects of Lenvima were high blood pressure (hypertension), fatigue, diarrhea, joint and muscle pain (arthralgia/myalgia), decreased appetite, decreased weight, nausea, inflammation of the lining of the mouth (stomatitis), headache, vomiting, excess protein in the urine (proteinuria), swelling and pain in the palms, hands and/or the soles of the feet (palmar-plantar erythrodysesthesia syndrome), abdominal pain and changes in voice volume or quality (dysphonia).
Lenvima may cause serious side effects, including cardiac failure, blood clot formation (arterial thromboembolic events), liver damage (hepatotoxicity), kidney damage (renal failure and impairment), an opening in the wall of the stomach or intestines (gastrointestinal perforation) or an abnormal connection between two parts of the stomach or intestines (fistula formation), changes in the heart’s electrical activity (QT Interval Prolongation), low levels of calcium in the blood (hypocalcemia), the simultaneous occurrence of headache, confusion, seizures and visual changes (Reversible Posterior Leukoencephalopathy Syndrome), serious bleeding (hemorrhage), risks to an unborn child if a patient becomes pregnant during treatment, and impairing suppression of the production of thyroid-stimulating hormone.
Lenvima is marketed by Woodcliff Lake, New Jersey-based Eisai Inc.
सुकून उतना ही देना प्रभू, जितने से जिंदगी चल जाये।औकात बस इतनी देना,कि औरों का भला हो जाये।………..P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
सुकून उतना ही देना प्रभू, जितने से जिंदगी चल जाये। औकात बस इतनी देना, कि औरों का भला हो जाये।
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE


 Googleplus
Googleplus amcrasto@gmail.com
amcrasto@gmail.com
LIONEL MY SON
जिंदगी चल जाये।
औकात बस इतनी देना,
कि औरों का भला हो जाये।
DACLATASVIR, 达拉他韦 , Даклатасвир , داكلاتاسفير ,

Daclatasvir

Status: Launched 2014 (EU, Japan)
Originator: Bristol-Myers Squibb


Daclatasvir (USAN[1]) (formerly BMS-790052, trade name Daklinza) is a drug for the treatment of hepatitis C (HCV). It is was developed by Bristol-Myers Squibb and was approved in Europe on 22 August 2014.
Daclatasvir inhibits the HCV nonstructural protein NS5A.[2][3] Recent research suggests that it targets two steps of the viral replication process, enabling rapid decline of HCV RNA.[4]
Daclatasvir has been tested in combination regimens with pegylated interferon and ribavirin,[5] as well as with other direct-acting antiviral agents including asunaprevir[6][7][8][9] and sofosbuvir.[10][11]
It is on the World Health Organization’s List of Essential Medicines, a list of the most important medications needed in a basic health system.[12]

EUROPEAN MEDICINES AGENCY ADVISES ON COMPASSIONATE USE OF DACLATASVIR
- The first compassionate-use opinion for a hepatitis C treatment was adopted by the CHMP in October 2013.
- Sofosbuvir, which is part of this compassionate-use opinion, received a positive opinion from the CHMP recommending granting of a marketing authorisation at its November 2013 meeting.
- Daclatasvir is developed by Bristol-Myers Squibb and sofosbuvir is developed by Gilead.
|
1-6-2012
|
Anti-Viral Compounds
|
|
|
2-13-2009
|
CRYSTALLINE FORM OF METHYL ((1S)-1-(((2S)
-2-(5-(4′-(2-((2S)-1((2S)-2-((METHOXYCARBONYL)AMINO)-3-METHYLBUTANOYL)-2-PYRROLIDINYL) -1H-IMIDAZOL-5-YL)-4-BIPHENYLYL)-1H-IMIDAZOL-2-YL)-1-PYRROLIDINYL)CARBONYL) -2-METHYLPROPYL)CARBAMATE DIHYDROCHLORIDE SALT |
Synthesis
Daclatasvir dihydrochloride (Daklinza)
Daclatasvir dihydrochloride is a hepatitis C virus nonstructural 5A (NS5A) replication complex inhibitor which was first approved in Japan for the treatment of genotype 1 HCV patients who fail to respond to interferon plus ribavirin. The drug has also been approved for patients with untreated, chronic HCV who are eligible for interferon. Additionally, in Europe, daclatasvir was approved for use in combination with other products across genotype 1–4 HCV. Daclatasvir was discovered and developed by Bristol–Myers Squibb and a fascinating account describing the initiation of the program from a phenotypic screen and the medicinal chemistry strategy leading to the discovery of the compound has been recently reported.80 Daclatasvir has been prepared via two different routes81,82 and the process route is outlined in Scheme 11.83 Bromination of commercial 4,40-diacetylbiphenyl (58) gave 4,40-bis(bromoacetyl)biphenyl 59 in 82% yield. Alkylation of NBoc- L-proline (60) with 59 gave diester 61 which was treated with ammonium acetate to effect cyclization of the bis-ketoester to provide bis-imidazole 62 in 63% yield for the two steps. Acidic removal of the Boc protecting groups followed by recrystallization provided bis-pyrrolidine 63 in high yield. Acylation of 63 with N-(methoxycarbonyl)- L-valine (64) using N-(3-dimethylaminopropyl)-N0-ethylcarbodiimide(EDC) and 1-hydroxybenxotriazole hydrate (HOBT) provided declatasvir. The dihydrochloride salt was prepared and treated with Cuno Zet Carbon followed by crystallization from acetone
to give daclatasvir dihydrochloride (IX) in 74% yield.
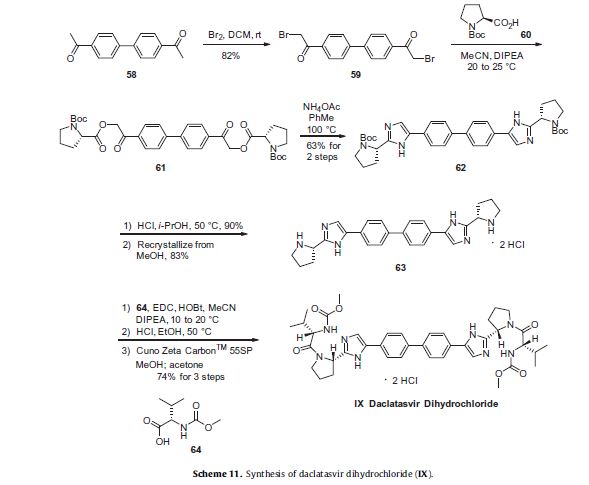
80 Belema, M.; Meanwell, N. A. J. Med. Chem. 2014, 57, 5057.
81. Bachand, C.; Belema, M.; Deon, D. H.; Good, A. C.; Goodrich, J.; James, C. A.;
Lavoie, R.; Lopez, O. D.; Martel, A.; Meanwell, N. A.; Nguyen, V. N.; Romine, J.
L.; Ruediger, E. H.; Snyder, L. B.; St. Laurent, D. R.; Yang, F.; Langley, D. R.;
Wang, G.; Hamann, L. G. WO Patent 2008021927A2, 2008.
82. Belema, M.; Nguyen, V. N.; Bachand, C.; Deon, D. H.; Goodrich, J. T.; James, C.
A.; Lavoie, R.; Lopez, O. D.; Martel, A.; Romine, J. L.; Ruediger, E. H.; Snyder, L.
B.; St Laurent, D. R.; Yang, F.; Zhu, J.; Wong, H. S.; Langley, D. R.; Adams, S. P.;
Cantor, G. H.; Chimalakonda, A.; Fura, A.; Johnson, B. M.; Knipe, J. O.; Parker, D.
D.; Santone, K. S.; Fridell, R. A.; Lemm, J. A.; O’Boyle, D. R., 2nd; Colonno, R. J.;
Gao, M.; Meanwell, N. A.; Hamann, L. G. J. Med. Chem. 2014, 57, 2013.
83. Pack, S. K.; Geng, P.; Smith, M. J.; Hamm, J. WO Patent 2009020825A1, 2009.
PATENT
EXAMPLES

A 1 L, 3-neck round bottom flask, fitted with a nitrogen line, overhead stirrer and thermocouple, was charged with 20 g (83.9 mmol, 1 equiv) 1,1′-(biphenyl-4,4′-diyl)diethanone, 200 mL CH2Cl2 and 8.7 mL (27.1 g, 169.3 mmol, 2.02 quiv) bromine. The mixture was allowed to stir under nitrogen for about 20 hours under ambient conditions. The resulting slurry was charged with 200 mL CH2Cl2 and concentrated down to about 150 mL via vacuum distillation. The slurry was then solvent exchanged into THF to a target volume of 200 mL via vacuum distillation. The slurry was cooled to 20-25° C. over 1 hour and allowed to stir at 20-25° C. for an additional hour. The off-white crystalline solids were filtered and washed with 150 mL CH2Cl2. The product was dried under vacuum at 60° C. to yield 27.4 g (69.2 mmol, 82%) of the desired product : 1H NMR (400 MHz, CDCl3) δ 7.95-7.85 (m, 4H), 7.60-7.50 (m, 4H), 4.26 (s, 4H); 13C NMR (100 MHz, CDCl3) 6 191.0, 145.1, 133.8, 129.9, 127.9, 30.8; IR (KBr, cm−1) 3007, 2950, 1691, 1599, 1199; Anal calcd for C16H12Br2O2: C, 48.52; H, 3.05; Br, 40.34. Found: C, 48.53; H, 3.03; Br, 40.53 HRMS calcd for C16H13Br2O2 (M+H; DCI+): 394.9282. Found: 394.9292. mp 224-226° C.

A 500 mL jacketed flask, fitted with a nitrogen line, thermocouple and overhead stirrer, was charged with 20 g (50.5 mmol, 1 equiv) of Compound 2, 22.8 g (105.9 moles, 2.10 equiv) 1-(tert-butoxycarbonyl)-L-proline and 200 mL acetonitrile. The slurry was cooled to 20° C. followed by the addition of 18.2 mL (13.5 g, 104.4 mmol, 2.07 equiv) DIPEA. The slurry was warmed to 25° C. and allowed to stir for 3 hours. The resulting clear, organic solution was washed with 3×100 mL 13 wt % aqueous NaCl. The rich acetonitrile solution was solvent exchanged into toluene (target volume=215 mL) by vacuum distillation until there was less than 0.5 vol % acetonitrile.

The toluene solution of Compound 3 was charged with 78 g (1.011 moles, 20 equiv) ammonium acetate and heated to 95-100° C. The mixture was allowed to stir at 95-100° C. for 15 hours. After reaction completion, the mixture was cooled to 70-80° C. and charged with 7 mL acetic acid, 40 mL n-butanol, and 80 mL of 5 vol % aqueous acetic acid. The resulting biphasic solution was split while maintaining a temperature >50° C. The rich organic phase was charged with 80 mL of 5 vol % aqueous acetic acid, 30 mL acetic acid and 20 mL n-butanol while maintaining a temperature >50° C. The resulting biphasic solution was split while maintaining a temperature >50° C. and the rich organic phase was washed with an additional 80 mL of 5 vol % aqueous acetic acid. The rich organic phase was then solvent exchanged into toluene to a target volume of 215 mL by vacuum distillation. While maintaining a temperature >60° C., 64 mL methanol was charged. The resulting slurry was heated to 70-75° C. and aged for 1 hour. The slurry was cooled to 20-25° C. over 1 hour and aged at that temperature for an additional hour. The slurry was filtered and the cake was washed with 200 mL 10:3 toluene:methanol. The product was dried under vacuum at 70° C., resulting in 19.8 g (31.7 mmol, 63%) of the desired product: 1H NMR (400 MHz, DMSO-d6) δ 13.00-11.00 (s, 2H), 7.90-7.75 (m, 4H), 7.75-7.60 (m, 4H), 7.60-7.30 (s, 2H), 4.92-4.72 (m, 2H), 3.65-3.49 (m, 2H), 3.49-3.28 (m, 2H), 2.39-2.1 (m, 2H), 2.10-1.87 (m, 6H), 1.60-1.33 (s, 8H), 1.33-1.07 (s, 10H); 13C NMR (100 MHz, DMSO-d6) δ 154.1, 153.8, 137.5, 126.6, 125.0, 78.9, 78.5, 55.6, 55.0, 47.0, 46.7, 33.7, 32.2, 28.5, 28.2, 24.2, 23.5; IR (KBr, cm−1) 2975, 2876, 1663, 1407, 1156, 1125; HRMS calcd for C36H45N6O4 (M+H; ESI+): 625.3502. Found: 625.3502. mp 190-195° C. (decomposed).

To a 250 mL reactor equipped with a nitrogen line and overhead stirrer, 25.0 g of Compound 4 (40.01 mmol, 1 equiv) was charged followed by 250 mL methanol and 32.85 mL (400.1 mmol, 10 equiv) 6M aqueous HCl. The temperature was increased to 50° C. and agitated at 50° C. for 5 hours. The resulting slurry was cooled to 20-25° C. and held with agitation for about 18 hours. Filtration of the slurry afforded a solid which was washed successively with 100 mL 90% methanol/water (V/V) and 2×100 mL of methanol. The wet cake was dried in a vacuum oven at 50° C. overnight to give 18.12 g (31.8 mmol, 79.4%) of the desired product.
Recrystallization of Compound 5
To a 250 mL reactor equipped with a nitrogen line and an overhead stirrer, 17.8 g of Compound 5 from above was charged followed by 72 mL methanol. The resulting slurry was agitated at 50° C. for 4 hours, cooled to 20-25° C. and held with agitation at 20-25° C. for 1 hour. Filtration of the slurry afforded a crystalline solid which was washed with 60 mL methanol. The resulting wet cake was dried in a vacuum oven at 50° C. for 4 days to yield 14.7 g (25.7 mmol, 82.6%) of the purified product: 1H NMR (400 MHz, DMSO-d6) δ 10.5-10.25 (br, 2H), 10.1-9.75 (br, 2H), 8.19 (s, 2H), 7.05 (d, J=8.4, 4H), 7.92 (d, J=8.5, 4H), 5.06 (m, 2H), 3.5-3.35 (m, 4H), 2.6-2.3 (m, 4H), 2.25-2.15 (m, 2H), 2.18-1.96 (m, 2H); 13C NMR (100 MHz, DMSO-d6) δ 156.6, 142.5, 139.3, 128.1, 127.5, 126.1, 116.9, 53.2, 45.8, 29.8, 24.3; IR (KBr, cm−1) 3429, 2627, 1636, 1567, 1493, 1428, 1028. Anal calcd for C26H32N6Cl4: C, 54.75; H, 5.65; Cl, 24.86; Adjusted for 1.9% water: C, 53.71; H, 5.76; N, 14.46; Cl, 24.39. Found: C, 53.74; H, 5.72; N, 14.50; Cl, 24.49; KF=1.9. mp 240° C. (decomposed).

A 1 L jacketed flask equipped with a nitrogen line and an overhead stirrer was sequentially charged with 100 mL acetonitrile, 13.69 g (89.4 mmol, 2.5 equiv) hydroxybenzotriazole hydrate, 15.07 g (86 mmol, 2.4 equiv) N-(methoxycarbonyl)-L-valine, 16.46 g (85.9 mmol, 2.4 equiv) 1-(3-dimethyaminopropyl)-3-ethylcarbodiimide hydrochloride and an additional 100 mL acetonitrile. The resulting solution was agitated at 20° C. for 1 hour and charged with 20.4 g (35.8 mmol, 1 equiv) of purified Compound 5. The slurry was cooled to about 0° C. and 18.47 g (142.9 mmol, 4 equiv) diisopropylethylamine was added over 30 minutes while maintaining a temperature below 10° C. The solution was slowly heated to 15° C. over 3 hours and held at 15° C. for 12 hours. The resulting solution was charged with 120 mL 13 wt % aqueous NaCl and heated to 50° C. for 1 hour. After cooling to 20° C., 100 mL of isopropyl acetate was added. The biphasic solution was filtered through a 0.45 μm filter and the mixture split. The rich organic phase was washed with 2×240 mL of a 0.5 N NaOH solution containing 13 wt % NaCl followed by 120 mL 13 wt % aqueous NaCl. The mixture was then solvent exchanged into isopropyl acetate by vacuum distillation with a target volume of 400 mL. The resulting hazy solution was cooled to 20° C. and filtered through a 0.45 μm filter. The clear solution was then solvent exchanged into ethanol by vacuum distillation with a target volume of 140 mL. While maintaining a temperature of 50° C., 66.4 mL (82.3 mmol, 2.3 equiv) of 1.24M HCl in ethanol was added. The mixture was then charged with 33 mg (0.04 mmol, 0.001 equiv) of seed crystals of Compound (I) (see preparation below) and the resulting slurry was stirred at 50° C. for 3 hours. The mixture was cooled to 20° C. over 1 hour and aged at that temperature for an additional 22 hours. The slurry was filtered and the wet cake was washed with 100 mL of 2:1 acetone:ethanol. The solids were dried in a vacuum oven at 70° C. to give 22.15 g (27.3 mmol, 76.3%) of the desired product.

A solution of Compound (I) was prepared by dissolving 3.17 g of Compound (I) from above in 22 mL methanol. The solution was passed through a 47 mm Cuno Zeta Carbon® 53SP filter at ˜5 psig at a flow rate of ˜58 mL/min. The carbon filter was rinsed with 32 mL of methanol. The solution was concentrated down to 16 mL by vacuum distillation. While maintaining a temperature of 40-50° C., 15.9 mL acetone and 5 mg of seed crystals of Compound (I) (see procedure below) were added. The resulting slurry was then charged with 32 mL acetone over 30 minutes. The slurry was held at 50° C. for 2 hours, cooled to 20° C. over about 1 hour and held at 20° C. for about 20 hours. The solids were filtered, washed with 16 mL 2:1 acetone:methanol and dried in a vacuum oven at 60° C. to give 2.14 g (67.5%) of purified Compound (I):
1H NMR (400 MHz, DMSO-d6, 80° C.): 8.02 (d, J=8.34 Hz, 4 H), 7.97 (s, 2 H), 7.86 (d, J=8.34 Hz, 4 H), 6.75 (s, 2 H), 5.27 (t, J=6.44 Hz, 2 H), 4.17 (t, J=6.95 Hz, 2 H), 3.97-4.11 (m, 2 H), 3.74-3.90 (m, 2 H), 3.57 (s, 6 H), 2.32-2.46 (m, 2 H), 2.09-2.31 (m, 6 H), 1.91-2.07 (m, 2 H), 0.88 (d, J=6.57 Hz, 6 H), 0.79 (d, J=6.32 Hz, 6 H);
13C NMR (75 MHz, DMSO-d6): δ 170.9, 156.9, 149.3, 139.1, 131.7, 127.1, 126.5, 125.9, 115.0, 57.9, 52.8, 51.5, 47.2, 31.1, 28.9, 24.9, 19.6, 17.7;
IR (neat, cm−1): 3385, 2971, 2873, 2669, 1731, 1650.
Anal. Calcd for C40H52N8O6Cl2: C, 59.18; H, 6.45; N, 13.80; Cl, 8.73. Found C, 59.98; H, 6.80; N, 13.68; Cl, 8.77. mp 267° C. (decomposed).
Preparation of Seed Crystals of Compound (I)
A 250 mL round-bottom flask was charged with 6.0 g (10.5 mmol, 1 equiv) Compound 5, 3.87 g (22.1 mmol, 2.1 equiv) N-(methoxycarbonyl)-L-valine, 4.45 g (23.2 mmol, 2.2 equiv) 1-(3-dimethyaminopropyl)-3-ethylcarbodiimide hydrochloride, 0.289 g (2.14 mmol, 0.2 equiv) 1-hydroxybenzotriazole, and 30 mL acetonitrile. The resulting slurry was then charged with 7.33 mL (42.03 mmol, 4 equiv) diisopropylethylamine and allowed to stir at 24-30° C. for about 18 hours. The mixture was charged with 6 mL of water and heated to 50° C. for about 5 hours. The mixture was cooled and charged with 32 mL ethyl acetate and 30 mL water. The layers were separated and the rich organic layer was washed with 30 mL of 10 wt % aqueous NaHCO3, 30 mL water, and 20 mL of 10 wt % aqueous NaCl. The rich organic layer was then dried over MgSO4, filtered, and concentrated down to a residue. The crude material was then purified via flash chromatography (silica gel, 0-10% methanol in dichloromethane) to provide the free base of Compound (I).
The free-base of Compound (I) (0.03 g) was dissolved in 1 mL isopropanol at 20° C. Anhydrous HCl (70 μL, dissolved in ethanol, approximately 1.25M concentration) was added and the reaction mixture was stirred. To the solution was added methyl tert-butyl ether (1 mL) and the resulting slurry was stirred vigorously at 40° C. to 50° C. for 12 hours. The crystal slurry was cooled to 20° C. and filtered. The wet cake was air-dried at 20° C. A white crystalline solid (Form N-2 of Compound (I)) was obtained.
Clip
Daclatasvir synthesis: WO2009020828A1

Procedure:
Step a: A 1 L, 3 -neck round bottom flask, fitted with a nitrogen line, overhead stirrer and thermocouple, was charged with 20 g (83.9 mmol, 1 equiv) 1,1′-(biphenyl-4,4′-diyl)diethanone, 200 mL Dichloromethane and 8.7 mL (27.1g, 169.3 mmol, 2.02 equiv) bromine. The mixture was allowed to stir under nitrogen for about 20 hours under ambient conditions. The resulting slurry was charged with 200 mL Dichloromethane and concentrated down to about 150 mL via vacuum distillation. The slurry was then solvent exchanged into THF to a target volume of 200 mL via vacuum distillation. The slurry was cooled to 20-25 0C over 1 hour and allowed to stir at 20-25 0C for an additional hour. The off-white crystalline solids were filtered and washed with 150 mL Dichloromethane. The product was dried under vacuum at 60 0C to yield 27.4 g (69.2 mmol, 82%) of the desired product: 1H NMR (400 MHz, CDCl3) d 7.95-7.85 (m, 4H), 7.60-7.50 (m, 4H), 4.26 (s, 4H); 13C NMR 100 MHz, CDCl3) d 191.0, 145.1, 133.8, 129.9, 127.9, 30.8; IR (KBr, cm-1) 3007, 2950, 1691, 1599, 1199; Anal calcd for C16H12Br2O2: C, 48.52; H, 3.05; Br, 40.34. Found: C, 48.53; H, 3.03; Br, 40.53. HRMS calcd for C16H12Br2O2 (M + H; DCI+): 394.9282. Found: 394.9292. mp 224-226 0C.
Step b: A 500 mL jacketed flask, fitted with a nitrogen line, thermocouple and overhead stirrer, was charged with 20 g (50.5 mmol, 1 equiv) of Compound 2, 22.8 g (105.9 moles, 2.10 equiv) 1-(tert-butoxycarbonyl)-L-proline and 200 mL acetonitrile. The slurry was cooled to 20 0C followed by the addition of 18.2 mL (13.5 g, 104.4 mmol, 2.07 equiv) DIPEA. The slurry was warmed to 25 0C and allowed to stir for 3 hours. The resulting clear, organic solution was washed with 3 x 100 mL 13 wt% aqueous NaCl. The rich acetonitrile solution was solvent exchanged into toluene (target volume = 215 mL) by vacuum distillation until there was less than 0.5 vol% acetonitrile.
Step c: The toluene solution of Compound 3 was charged with 78 g (1.011 moles, 20 equiv) ammonium acetate and heated to 95-100 0C. The mixture was allowed to stir at 95-100 0C for 15 hours. After reaction completion, the mixture was cooled to 70- 80 0C and charged with 7 mL acetic acid, 40 mL n-butanol, and 80 mL of 5 vol% aqueous acetic acid. The resulting biphasic solution was split while maintaining a temperature > 50 0C. The rich organic phase was charged with 80 mL of 5 vol% aqueous acetic acid, 30 mL acetic acid and 20 mL n-butanol while maintaining a temperature > 50 0C. The resulting biphasic solution was split while maintaining a temperature > 50 0C and the rich organic phase was washed with an additional 80 mL of 5 vol% aqueous acetic acid. The rich organic phase was then solvent exchanged into toluene to a target volume of 215 mL by vacuum distillation. While maintaining a temperature > 60 0C, 64 mL methanol was charged. The resulting slurry was heated to 70-75 0C and aged for 1 hour. The slurry was cooled to 20-25 0C over 1 hour and aged at that temperature for an additional hour. The slurry was filtered and the cake was washed with 200 mL 10:3 toluene:methanol. The product was dried under vacuum at 70 0C, resulting in 19.8 g (31.7 mmol, 63%) of the desired product: 1H NMR (400 MHz, DMSO-^) d 13.00-11.00 (s, 2H), 7.90-7.75 (m, 4H), 7.75-7.60 (m, 4H), 7.60-7.30 (s, 2H), 4.92-4.72 (m, 2H), 3.65-3.49 (m, 2H), 3.49-3.28 (m, 2H), 2.39-2.1 (m, 2H), 2.10-1.87 (m, 6H), 1.60-1.33 (s, 8H), 1.33-1.07 (s, 10H); 13C NMR (100 MHz, DMSO-?fe) d 154.1, 153.8, 137.5, 126.6, 125.0, 78.9, 78.5, 55.6, 55.0, 47.0, 46.7, 33.7, 32.2, 28.5, 28.2, 24.2, 23.5; IR (KBr, cm-1) 2975, 2876, 1663, 1407, 1156, 1125; HRMS calcd for C36H45N6O4 (M + H; ESI+): 625.3502. Found: 625.3502. mp 190-195 0C (decomposed).
Step d: To a 250 mL reactor equipped with a nitrogen line and overhead stirrer, 25.0 g of Compound 4 (40.01 mmol, 1 equiv) was charged followed by 250 mL methanol and 32.85 mL (400.1 mmol, 10 equiv) 6M aqueous HCl. The temperature was increased to 50 0C and agitated at 50 0C for 5 hours. The resulting slurry was cooled to 20-25 0C and held with agitation for about 18 hours. Filtration of the slurry afforded a solid which was washed successively with 100 mL 90% methanoI/water (WV) and 2 x 100 mL of methanol. The wet cake was dried in a vacuum oven at 50 0C overnight to give 18.12 g (31.8 mmol, 79.4%) of the desired product.
CUT PASTE…….WO2009020825

Preparation of Compound (I)
A 1 L jacketed flask equipped with a nitrogen line and an overhead stirrer was sequentially charged with 100 mL acetonitrile, 13.69 g (89.4 mmol, 2.5 equiv) hydroxybenzotriazole hydrate, 15.07 g (86 mmol, 2.4 equiv) N-(methoxycarbonyl)- L-valine, 16.46 g (85.9 mmol, 2.4 equiv) l-(3-dimethyaminopropyl)-3- ethylcarbodiimide hydrochloride and an additional 100 mL acetonitrile. The resulting solution was agitated at 20 0C for 1 hour and charged with 20.4 g (35.8 mmol, 1 equiv) of purified Compound 7. The slurry was cooled to about 0 0C and 18.47 g (142.9 mmol, 4 equiv) diisopropylethylamine was added over 30 minutes while maintaining a temperature below 10 0C. The solution was slowly heated to 15 0C over 3 hours and held at 15 0C for 12 hours. The resulting solution was charged with 120 mL 13 wt% aqueous NaCl and heated to 50 0C for 1 hour. After cooling to 20 0C, 100 mL of isopropyl acetate was added. The biphasic solution was filtered through a 0.45 μm filter and the mixture split. The rich organic phase was washed with 2 x 240 mL of a 0.5 Ν NaOH solution containing 13 wt% NaCl followed by 120 mL 13 wt% aqueous NaCl. The mixture was then solvent exchanged into isopropyl acetate by vacuum distillation with a target volume of 400 mL. The resulting hazy solution was cooled to 20 0C and filtered through a 0.45 μm filter. The clear solution was then solvent exchanged into ethanol by vacuum distillation with a target volume of 140 mL. While maintaining a temperature of 50 0C, 66.4 mL (82.3 mmol, 2.3 equiv) of 1.24M HCl in ethanol was added. The mixture was then charged with 33 mg (0.04 mmol, 0.001 equiv) of seed crystals of Compound (I) (see preparation below) and the resulting slurry was stirred at 50 0C for 3 hours. The mixture was cooled to 20 0C over 1 hour and aged at that temperature for an additional 22 hours. The slurry was filtered and the wet cake was washed with 100 mL of 2: 1 acetone:ethanol. The solids were dried in a vacuum oven at 70 0C to give 22.15 g (27.3 mmol, 76.3%) of the desired product.

Alternative Preparation of Compound (I)
A jacketed reactor equipped with a mechanical agitator, a thermocouple and a nitrogen inlet was sequentially charged with 10 L acetonitrile, 0.671 kg (4.38 moles, 2.50 equiv) 1-hydroxybenzotriazole, 0.737 kg (4.21 moles, 2.40 equiv) N- (methoxycarbonyl)-L-valine and 0.790 kg (4.12 moles, 2.35 equiv) l-(3- dimethyaminopropyl)-3-ethylcarbodiimide hydrochloride. The mixture was agitated at 200C for 1 hour, cooled to 5 0C and charged with 1 kg (1.75 moles, 1.00 equiv) Compound 7. While maintaining a temperature < 10 0C, 0.906 kg (7.01 moles, 4 equiv) diisopropylethylamine was added. The mixture was heated to 15-20 0C over 2 hours and agitated for an additional 15 hours. After the reaction was complete, the mixture was washed once with 6.0 L 13 wt% aqueous NaCl, twice with 6.1 L (6.12 moles, 3.5 equiv) 1.0 M aqueous NaOH containing 13 wt% NaCl and once with 6.0 L 13 wt% aqueous NaCl. Water was then removed from the rich organic solution via azeotropic distillation. The mixture was cooled to 20 0C, agitated for 1 hour and filtered. The rich organic solution was then solvent exchanged into EtOH via vacuum distillation to a target volume of 5 L. While maintaining a temperature of 50 0C, 3.2 L (4.0 moles, 2.3 equiv) 1.25M HCl in EtOH was charged. The mixture was seeded with 1.6 g Compound (I) (see preparation below) and agitated at 50 0C for 3 hours. The resulting slurry was cooled to 20 0C and agitated for at least 3 hours. The product was collected by filtration and washed with 5 L 2: 1 acetone:
EtOH to give 1.29 kg (ca. 90 wt% product) of wet crude product. A reactor equipped with an overhead agitator, nitrogen inlet and thermocouple was charged with 1.11 kg of the above crude product and 7 L methanol. The resulting solution was treated with Cuno Zeta Carbon (TM) 55SP. The carbon was washed with 15 L MeOH and the combined filtrate and wash was concentrated down to 4 L via vacuum distillation. The concentrated solution was charged with 5 L acetone and seeded with 1.6 g Compound (I) (see preparation below) while maintaining a temperature of 50 0C. An additional 10 L acetone was charged and the resulting slurry was stirred at 50 0C for 3 hours. The slurry was cooled to 20 0C and allowed to agitate at 200C for 3 hours. The product was collected by filtration, washed with 5 L 2: 1 acetone: EtOH and dried under vacuum at 50-60 0C to give 0.900 kg (1.11 moles, 74% adjusted) of Compound (I)-

Carbon Treatment and Recrystallization of Compound (I) A solution of Compound (I) was prepared by dissolving 3.17 g of Compound (I) from above in 22 mL methanol. The solution was passed through a 47mm Cuno Zeta Carbon 53SP filter at ~5 psig at a flow rate of~58mL/min. The carbon filter was rinsed with 32 mL of methanol. The solution was concentrated down to 16 mL by vacuum distillation. While maintaining a temperature of 40-50 0C, 15.9 mL acetone and 5 mg of seed crystals of Compound (I) (see procedure below) were added. The resulting slurry was then charged with 32 mL acetone over 30 minutes. The slurry was held at 50 0C for 2 hours, cooled to 20 0C over about 1 hour and held at 20 0C for about 20 hours. The solids were filtered, washed with 16 mL 2: 1 acetone:methanol and dried in a vacuum oven at 60 0C to give 2.14 g (67.5%) of purified Compound (I):
1H NMR (400 MHz, DMSO-έfc, 80 0C): 8.02 (d, J=8.34 Hz, 4 H), 7.97 (s, 2 H), 7.86 (d, J=8.34 Hz, 4 H), 6.75 (s, 2 H), 5.27 (t, J=6.44 Hz, 2 H), 4.17 (t, J=6.95 Hz, 2 H), 3.97 – 4.11 (m, 2 H), 3.74 – 3.90 (m, 2 H), 3.57 (s, 6 H), 2.32 – 2.46 (m, 2 H), 2.09 – 2.31 (m, 6 H), 1.91 – 2.07 (m, 2 H), 0.88 (d, J=6.57 Hz, 6 H), 0.79 (d, J=6.32 Hz, 6 H);
13C NMR (75 MHz, DMSO-έfc): δ 170.9, 156.9, 149.3, 139.1, 131.7, 127.1, 126.5, 125.9, 115.0, 57.9, 52.8, 51.5, 47.2, 31.1, 28.9, 24.9, 19.6, 17.7;
IR (neat, cm“1): 3385, 2971, 2873, 2669, 1731, 1650.
Anal. Calcd for C40H52N8O6Cl2: C, 59.18; H, 6.45; N, 13.80; Cl, 8.73. Found C, 59.98; H, 6.80; N, 13.68; Cl, 8.77. mp 267 0C (decomposed).
Characteristic diffraction peak positions (degrees 2Θ + 0.1) @ RT, based on a high quality pattern collected with a diffractometer (CuKa) with a spinning capillary with 2Θ calibrated with a NIST other suitable standard are as follows: 10.3, 12.4, 12.8, 13.3, 13.6, 15.5, 20.3, 21.2, 22.4, 22.7, 23.7
Daclatasvir faces problems in USA
The US-FDA in 2014 issued a complete response letter for NS5A inhibitor daclatasvir saying it was unable to approve the drug because the marketing application was for its use in tandem with asunaprevir, an NS3/NS4A protease inhibitor discontinued in the US by BMS for commercial reasons. Daclatasvir is already on the market in Europe-where it is sold as Daklinza-and also in Japan where it was approved alongside asunaprevir in July as the country’s first all-oral HCV therapy. However, a delay in the large US market is clearly a major setback for BMS’ ambitions in hepatitis therapy.
To make the matter worse, US FDA has rescinded breakthrough therapy designation status from Bristol-Myers Squibb for Daclatasvir for the treatment of hepatitis C virus infection in Feb 2015.
PAPER
Makonen, B.; et. al. Hepatitis C Virus NS5A Replication Complex Inhibitors: The Discovery of Daclatasvir. J Med Chem 2014, 57(5), 2013–2032.
http://pubs.acs.org/doi/abs/10.1021/jm401836p
PATENT
http://www.google.com/patents/WO2008021927A2?cl=en
Example 24-23

methyl ((lS)-l-(((2S)-2-(5-(4′-(2-((2S)-l-((2S)-2-((methoxycarbonyl)amino)-3- methylbutanoyl)-2-pyrrolidinyl)-lH-imidazol-5-yl)-4-biphenylyl)-lH-imidazol-2-yl)-
1 -pyrrolidinyl) carbonyl) -2-methylpropyl) carbamate
A 50 mL flask equipped with a stir bar was sequentially charged with 2.5 mL acetonitrile, 0.344 g (2.25 mmol, 2.5 equiv) hydroxy benzotriazole hydrate, 0.374 g (2.13 mmol, 2.4 equiv) N-(methoxycarbonyl)-L-valine, 0.400 g (2.09 mmol, 2.4 equiv) 1 -(3 -dimethyaminopropyl)-3-ethylcarbodiimide hydrochloride and an additional 2.5 mL acetonitrile. The resulting solution was agitated at 20 0C for 1 hour and charged with 0.501 g (0.88 mmol, 1 equiv) Example A-le-4. The slurry was cooled to about 0 0C and 0.45 g (3.48 mmol, 4 equiv) diisopropylethylamine was added over 30 minutes while maintaining a temperature below 10 0C. The solution was slowly heated to 15 0C over 3 hours and held at 15 0C for 16 hours. The temperature was increased to 20 0C and stirred for 3.25 hours. The resulting solution was charged with 3.3 g of 13 wt% aqueous NaCl and heated to 50 0C for 1 hour. After cooling to 20 0C, 2.5 mL of isopropyl acetate was added. The rich organic phase was washed with 2 x 6.9 g of a 0.5 N NaOH solution containing 13 wt% NaCl followed by 3.3 g of 13 wt% aqueous NaCl. The mixture was then solvent exchanged into isopropyl acetate by vacuum distillation to a target volume of 10 mL. The resulting hazy solution was cooled to 20 0C and filtered through a 0.45 μm filter. The clear solution was then solvent exchanged into ethanol by vacuum distillation with a target volume of 3 mL. 1.67 mL (2.02 mmol, 2.3 equiv) of 1.21 M HCl in ethanol was added. The mixture was then stirred at 25 0C for 15 hours. The resulting slurry was filtered and the wet cake was washed with 2.5 mL of 2: 1 acetone:ethanol. The solids were dried in a vacuum oven at 50 0C to give 0.550 g (0.68 mmol, 77 %) of the desired product.
RecrystalHzation of Example 24-23
A solution of Example 24-23 prepared above was prepared by dissolving 0.520 g of the above product in 3.65 mL methanol. The solution was then charged with 0.078 g of type 3 Cuno Zeta loose carbon and allowed to stir for 0.25 hours. The mixture was then filtered and washed with 6 ml of methanol. The product rich solution was concentrated down to 2.6 mL by vacuum distillation. 7.8 mL acetone was added and allowed to stir at 25 0C for 15 h. The solids were filtered, washed with 2.5 mL 2: 1 acetone:ethanol and dried in a vacuum oven at 70 0C to give 0.406 g (57.0%) of the desired product as white crystals: 1H NMR (400 MHz, OMSO-d6, 80 0C): 8.02 (d, J=8.34 Hz, 4 H), 7.97 (s, 2 H), 7.86 (d, J=8.34 Hz, 4 H), 6.75 (s, 2 H), 5.27 (t, J=6.44 Hz, 2 H), 4.17 (t, J=6.95 Hz, 2 H), 3.97 – 4.11 (m, 2 H), 3.74 – 3.90 (m, 2 H), 3.57 (s, 6 H), 2.32 – 2.46 (m, 2 H), 2.09 – 2.31 (m, 6 H), 1.91 – 2.07 (m, 2 H), 0.88 (d, J=6.57 Hz, 6 H), 0.79 (d, J=6.32 Hz, 6 H); 13C NMR (75 MHz, DMSO- d6): δ 170.9, 156.9, 149.3, 139.1, 131.7, 127.1, 126.5, 125.9, 115.0, 57.9, 52.8, 51.5, 47.2, 31.1, 28.9, 24.9, 19.6, 17.7; IR (neat, cm“1): 3385, 2971, 2873, 2669, 1731, 1650. Anal. Calcd for C40H52N8O6Cl2: C, 59.18; H, 6.45; N, 13.80; Cl, 8.73. Found C, 59.98; H, 6.80; N, 13.68; Cl, 8.77. mp 267 0C (decomposed). Characteristic diffraction peak positions (degrees 2Θ ± 0.1) @ RT, based on a high quality pattern collected with a diffractometer (CuKa) with a spinning capillary with 2Θ calibrated with a NIST other suitable standard are as follows: 10.3, 12.4, 12.8, 13.3, 13.6, 15.5, 20.3, 21.2, 22.4, 22.7, 23.7
PAPER
Bioorganic & Medicinal Chemistry Letters (2015), 25(16), 3147-3150
http://www.sciencedirect.com/science/article/pii/S0960894X15005995

Scheme 1.
Synthetic route for the preparation of the target compounds 8a–8y. Reagents and conditions: (a) Br2, CH2Cl2, rt, overnight, 86%; (b) N-Boc-l-proline, MeCN, Et3N, rt, 2 h, 98%; (c) NH4OAc, toulene, 130 °C, 15 h, 85%; (d) 6 N HCl, MeOH, 50 °C, 4 h, 87%; (e) HATU, N-(methoxycarbonyl)-l-valine, DIPEA, rt, 14 h, 83%; (f) RCOCl, TEA, CH2Cl2, rt, 3 h, 64–87%.
Dimethyl((2S,2’S)-((2S,2’S)-2,2′-(5,5′-([1,1′-biphenyl]-4,4′-diyl)bis(1H-imidazole-
5,2-diyl))bis(pyrrolidine-2,1-diyl))bis(3-methyl-1-oxobutane-2,1-
diyl))dicarbamate 7……………FREE BASE
To a solution of 5 (90 mg, 0.181 mmol), N-me-thoxycarbonyl-l-valine 6 (92 mg,0.525 mmol) and DIPEA (0.18 mL, 1.03 mmol) in DMF (5 mL) was added HATU(165.5 mg, 0.434 mmol). The resulting reaction was allowed to stir at room temperature for 15 h, the reaction mixture was filtered and the residue was partitioned between EtOAc and H2O, The aqueous phase was extracted with EtOAc, and the combined organic phase was dried (MgSO4), filtered, and concentrated in vacuo. The residue was purified by flash chromatography (silica gel; 5% Methanol /CH2Cl2) to
afford 7 (0.11 g, 83 %)as white solid.
1H NMR (DMSO-d6, 500 MHz) δ: 11.56 (s, 2H), 7.69-7.48 (m, 8H), 7.26-7.03 (m, 4H), 5.24-5.05 (m, 2H), 4.09-4.04 (m, 2H), 3.85-3.75 (m, 4H), 3.58 (s, 6H), 2.24-1.98 (m, 10H), 0.87 (d, J = 3.6 Hz, 12H).
Anal. calcd. (%) for C40H50N8O6: C 65.02, H 6.82, N 15.17; found: C 65.20, H 6.79, N 15.31.
ESI-MS m/z: 739.5 (M+H)+.
NMR PREDICT
1H NMR PREDICT


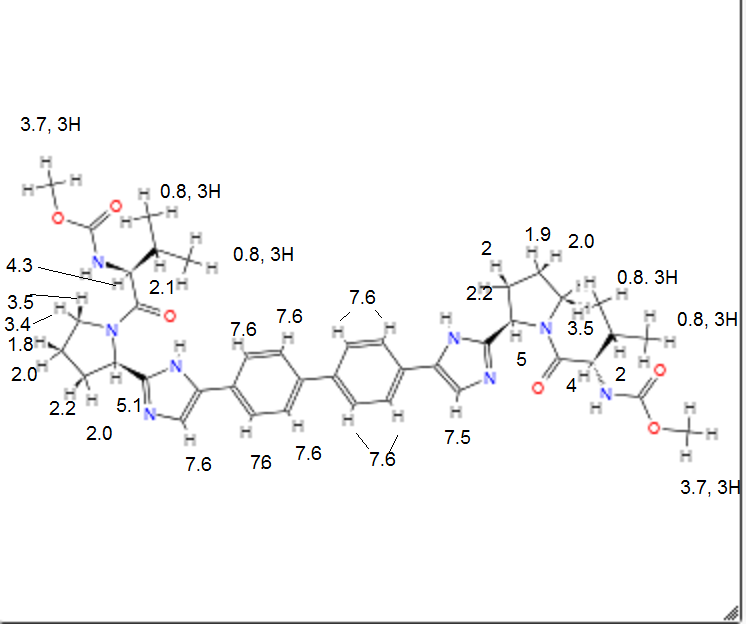
13C NMR PREDICT



COSY PREDICT

Patents
http://www.who.int/phi/implementation/ip_trade/daclatasvir_report_2014_09-02.pdf
Click on images to view
 Click on images to view
Click on images to view










Click on images to view
http://www.who.int/phi/implementation/ip_trade/daclatasvir_report_2014_09-02.pdf

Click on images to view
Click on images to view


Click on images to view
 |
|
| Names | |
|---|---|
| IUPAC name
Methyl [(2S)-1-{(2S)-2-[4-(4’-{2-[(2S)-1-{(2S)-2-[(methoxycarbonyl)amino]-3-methylbutanoyl}-2-pyrrolidinyl]-1H-imidazol-4-yl}-4-biphenylyl)-1H-imidazol-2-yl]-1-pyrrolidinyl}-3-methyl-1-oxo-2-butanyl]carbamate
|
|
| Other names
BMS-790052
|
|
| Identifiers | |
| 1009119-64-5 |
|
| ATC code | J05AX14 |
| ChEBI | CHEBI:82977 |
| ChEMBL | ChEMBL2023898 ChEMBL2303621 |
| ChemSpider | 24609522 |
| Jmol-3D images | Image |
| Properties | |
| C40H50N8O6 | |
| Molar mass | 738.89 g·mol−1 |
CLIP 1
Australian Government, National Measurement Institute
REFERENCE MATERIAL ANALYSIS REPORT
HPLC: Instrument: Shimadzu Binary pump LC-20AB, SIL-20 A HT autosampler
Column: X-Bridge C-18, 5.0 m (4.6 mm x 150 mm)
Column oven: 40 °C
Mobile Phase: A = Milli-Q water buffered at pH 10 with NH4
+ -OAc; B = MeCN
Gradient 0 min 35% B; 0-15 min 35% B; 15-18 min 35-75% B; 18-23 min 75% B.
Flow rate: 1.0 mL/min
Detector: Shimadzu SPD-M20A PDA operating at 310 nm
Relative peak area response of main component:
Initial analysis: Mean = 99.2%, s = 0.01%
Thermogravimetric analysis: Non volatile residue < 0.2% mass fraction . The volatile
content (e.g. organic solvents and/or water) could not be determined by
thermogravimetric analysis.
Karl Fischer analysis: Moisture content 0.6% mass fraction
QNMR: Instrument: Bruker Avance-III-500
Field strength: 500 MHz Solvent: DMSO-d6 (2.50 ppm)
Internal standard: Potassium hydrogen maleate (98.8% mass fraction)
Initial analysis: Mean (0.86 ppm) = 98.2%, s = 0.2%
LC-MS: Instrument: Thermo Scientific Dionex UltiMate 3000 Degasser,
Column: ZORBAX RRHD SB-C8, 2.1 x 50 mm, 1.8 μm (Agilent, 857700-906)
Column temp: 30.0 °C
Solvent system: Mobile phase A: 10 mM ammonium formate, 0.01% (v/v) formic acid in Milli-Q® water.
Mobile phase B: 0.01% (v/v) formic acid in acetonitrile.
Gradient from 90% A to 100% B
Flow rate: 0.25 mL/min
Sample prep: 2 mg/mL in MeOH with trace of formic acid
Injection volume: 10 L
Ionisation mode: Electrospray positive ion
Capillary voltage: 4.5 kV
Capillary temp: 360ºC Desolvation gas temperature: 300 ºC
Cone gas flow rate: 10 (arbitrary unit) Desolvation gas flow rate: 70 (arbitrary unit)
The retention time of daclatasvir is reported along with the major peak in the mass spectrum. The latter is reported as a mass/charge ratio.
9.98 min: 739.39545 (M+H+) m/z
HS-GC-MS: Instrument: Agilent 6890/5973/G1888
Column: DB-624, 30 m x 0.25 mm I.D. x 1.4 μm
Program: 50 C (5 min), 7 C/min to 120 C, 15 °C/min to 220 °C (8.3 min)
Injector: 150 C Transfer line temp: 280 C
Carrier: Helium, 1.2 mL/min Split ratio: 50/1
Solvents detected: Ethyl acetate
TLC: Conditions: Kieselgel 60F254. Ethyl acetate : methanol (95/5)
Single spot observed, Rf = 0.18. Visualisation with UV at 254 nm
The TLC was performed on the liberated free base.
IR: Instrument: Bruker Alpha FT-IR
Range: 4000-400 cm-1, neat
Peaks: 1723, 1697, 1643, 1523, 1439, 1235, 1099, 1024 cm-1
1H NMR: Instrument: Bruker Avance III 500
Field strength: 500 MHz Solvent: DMSO-d6 (2.50 ppm)
Spectral data: 0.77 (6H, d, J = 6.7 Hz), 0.83 (6H, d, J = 6.7 Hz), 2.01 (2H, m), 2.07 (2H, m), 2.12-2.27 (4H, m), 2.38 (2H, m), 3.54 (6H, s), 3.84 (2H, m), 3.97 (2H, m), 4.12 (2H, t, J = 7.7 Hz), 5.18 (2H, t, J = 7.0 Hz), 7.31 (2 N-H, d, J = 8.5 Hz), 7.94 (4H, d, J = 8.4 Hz), 7.99 (4H, d, J = 8.4 Hz), 8.16 (2H, s) ppm
Ethyl acetate estimated at 0.6% mass fraction was observed in the 1H NMR
13C NMR: Instrument: Bruker Avance III 500
Field strength: 126 MHz Solvent: DMSO-d6 (39.5 ppm)
Spectral data: 17.8, 19.6, 25.0, 29.0, 31.2, 47.3, 51.6, 52.9, 58.0, 115.1, 125.9, 126.6, 127.3, 131.8, 139.2, 149.4, 157.0, 171.1 ppm
Melting point: > 250 oC
Microanalysis: Found: C = 59.0%; H = 6.5%; N = 13.7% (August 2015)
Calc: C = 59.2%; H = 6.5%; N = 13.8% (Calculated for C40H50N8O6.2HCl)
REFERENCE
Australian NMI NATA Certification Daclatasvir – FixHepC
https://fixhepc.com/images/coa/NMI-NATA-Daclatasvir-Certification.pdf
Oct 7, 2015 – Compound Name: Daclatasvir dihydrochloride … Note: The assigned stereochemistry of this sample of daclatasvir has not …. Melting point:.
CLIP 2


Full Text Article – European Journal of Pharmaceutical and Medical …
Nov 28, 2016 – Daclatasvir dihydrochloride (DCLD) is a new drug …. DSC thermogram of daclatasvirdihydrochloriderealed drug melting point at 273.600C as …
CLIP 3
DCV dihydrochloride (anhydrous) is a white to yellow, non hygroscopic powder which is highly soluble in water (>700mg/mL). Solubility is higher at low pH. In aqueous buffers over the physiological pH range (pH 1.2-6.8) solubility is very low (4mg/mL to 0.004 mg/mL) due to the slow formation of the less soluble hydrated form. Water content in the drug substance is adequately controlled by in process tests. The desired anhydrous crystalline form of DCV dihydrochloride (N-2) is consistently produced and has been shown to not change on storage.
[DOC]AusPAR Daclatasvir dihydrochloride – Therapeutic Goods Administration
https://www.tga.gov.au/sites/default/…/auspar-daclatasvir–dihydrochloride-151214.do…
Dec 14, 2015 – Australian Public Assessment Report for daclatasvir dihydrochloride …. Figure 1:Chemical structure of daclatasvir dihydrochloride. …… 24 weeks is based on a selected literaturereview mostly of studies in patients with GT-1.
CLIP 4
The structure of the active substance has been confirmed by UV, IR, Raman and 1 H and 13C NMR spectroscopy, MS spectrometry, and crystal X-Ray diffraction.
Daclatasvir is a white to yellow crystalline non-hygroscopic powder. It is freely soluble in water, dimethyl sulfoxide, methanol; soluble in ethanol (95%); practically insoluble in dichloromethane, tetrahydrofuran, acetonitrile, acetone and ethyl acetate.
Daclatasvir is a chiral molecule with four stereocenters (1,1’, 2, 2;) in the S configuration. The synthetic strategy and process design such as starting material and reagent selection, process parameters, and in-process controls ensure the desired configuration at each of the four chiral centers. In addition, the established control strategy minimizes epimerization and eliminates other diastereomeric impurity formation in each step.
Polymorphism has been observed for daclatasvir hydrochloride. Although two neat crystalline dihydrochloride salts, N1 and N-2 have been identified in screening studies, it has been confirmed that the form N-2 is the thermodynamically most stable polymorph and only this form produced by the proposed synthetic process.
Manufacture, characterisation and process controls
Daclatasvir dihydrochloride is synthesised in three main steps using three commercially available well defined starting materials with acceptable specifications. The synthesis involves an alkylation and formation of the imidazole ring, a coupling reaction and the formation of the hydrochloride salt.
As mentioned above, the synthetic process has been designed to ensure the correct configuration at each of the four chiral centres is achieved. In addition, it has been demonstrated that the stereogenic centres do not epimerize during normal or stressed processing conditions.
The manufacturing process has been developed using a combination of conventional univariate studies and elements of QbD such as risk assessment.
The characterisation of the active substance and its impurities are in accordance with the EU guideline on chemistry of new active substances. Potential and actual impurities were well discussed with regards to their origin and characterised. Adequate in-process controls are applied during the synthesis. The specifications and control methods for intermediate products, starting materials and reagents have been presented.
The active substance specification includes tests for: appearance, colour, identity (IR/Raman, HPLC), assay (HPLC), impurities (HPLC), residual solvents (GC), HCl content (titration), total inorganic impurities (ICP-MS), and particle size (laser light scattering). The absence of a test for chiral purity in the active substance specification has been adequately justified based on the stereochemical control during the synthetic process and demonstration that there is no epimerization during normal or stressed processing conditions. Similarly, since the N-2 form of daclatasvir hydrochloride is the thermodynamically most stable polymorph and, is consistently produced by the synthetic process and remained unchanged during storage under long-term or accelerated conditions, this parameter is not included in the specification
CLIP5
SEE
http://www.accessdata.fda.gov/drugsatfda_docs/nda/2015/206843Orig1s000ChemR.pdf
CLIP6
Daclatasvir dihydrochloride

References
- 1 Statement on a Nonproprietary Name Adopted by the USAN Council
- 2 Gao, Min; Nettles, Richard E.; Belema, Makonen; Snyder, Lawrence B.; Nguyen, Van N.; Fridell, Robert A.; Serrano-Wu, Michael H.; Langley, David R.; Sun, Jin-Hua; O’Boyle, Donald R., II; Lemm, Julie A.; Wang, Chunfu; Knipe, Jay O.; Chien, Caly; Colonno, Richard J.; Grasela, Dennis M.; Meanwell, Nicholas A.; Hamann, Lawrence G. (2010). “Chemical genetics strategy identifies an HCV NS5A inhibitor with a potent clinical effect”. Nature 465 (7294): 96–100. doi:10.1038/nature08960. PMID 20410884.
- 3 Bell, Thomas W. (2010). “Drugs for hepatitis C: unlocking a new mechanism of action”. ChemMedChem 5 (10): 1663–1665. doi:10.1002/cmdc.201000334. PMID 20821796.
- 4 Modeling shows that the NS5A inhibitor daclatasvir has two modes of action and yields a shorter estimate of the hepatitis C virus half-life. Guedj, J et al. Proceedings of the National Academy of Sciences. February 19, 2013.
- 5 AASLD: Daclatasvir with Pegylated Interferon/Ribavirin Produces High Rates of HCV Suppression. Highleyman, L. HIVandHepatitis.com. 6 December 2011.
- 6Preliminary Study of Two Antiviral Agents for Hepatitis C Genotype 1. Lok, A et al. New England Journal of Medicine. 366(3):216-224. January 19, 2012.
- 7“Bristol-Myers’ Daclatasvir, Asunaprevir Cured 77%: Study”. Bloomberg. Apr 19, 2012.
- 8AASLD: Daclatasvir plus Asunaprevir Rapidly Suppresses HCV in Prior Null Responders. Highleyman, L. HIVandHepatitis.com. 8 November 2011.
- 9High rate of response to BMS HCV drugs in harder-to-treat patients – but interferon-free prospects differ by sub-genotype. Alcorn, K. Aidsmap.com. 12 November 2012.
- 10AASLD 2012: Sofosbuvir + Daclatasvir Dual Regimen Cures Most Patients with HCV Genotypes 1, 2, or 3. Highleyman, L. HIVandHepatitis.com. 15 November 2012.
- 11Mark Sulkowski et al. (January 16, 2014). “Daclatasvir plus Sofosbuvir for Previously Treated or Untreated Chronic HCV Infection”. New England Journal of Medicine. doi:10.1056/NEJMoa1306218.
- 12“www.who.int” (PDF).
| WO2004005264A2 * | 7 Jul 2003 | 15 Jan 2004 | Axxima Pharmaceuticals Ag | Imidazole compounds for the treatment of hepatitis c virus infections |
| WO2008021927A2 * | 9 Aug 2007 | 21 Feb 2008 | Squibb Bristol Myers Co | Hepatitis c virus inhibitors |
| WO2008021928A2 * | 9 Aug 2007 | 21 Feb 2008 | Squibb Bristol Myers Co | Hepatitis c virus inhibitors |
| WO2008021936A2 * | 9 Aug 2007 | 21 Feb 2008 | Squibb Bristol Myers Co | Hepatitis c virus inhibitors |
सुकून उतना ही देना प्रभू, जितने से जिंदगी चल जाये।औकात बस इतनी देना,कि औरों का भला हो जाये।………..P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
सुकून उतना ही देना प्रभू, जितने से जिंदगी चल जाये। औकात बस इतनी देना, कि औरों का भला हो जाये।
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
Googleplus amcrasto@gmail.com
LIONEL MY SONजिंदगी चल जाये।
औकात बस इतनी देना,
कि औरों का भला हो जाये।
//////////
