
LY335979, RS-33295-198 (Zosuquidar)
Roche Palo Alto (Originator)
LY335979 (Zosuquidar) is a selective Pgp (P-glycoprotein) inhibitor with a Ki of 59 nM. LY335979 significantly enhanced the survival of mice implanted with Pgp-expressing murine leukemia (P388/ADR) when administered in combination with either daunorubicin, doxorubicin or etoposide.
LY335979 (Zosuquidar)
M.Wt: 636.99
Formula: C32H31F2N3O2.3HCl
Name: Zosuquidar trihydrochloride
Elemental Analysis: C, 60.34; H, 5.38; Cl, 16.70; F, 5.97; N, 6.60; O, 5.02
CAS : 167465-36-3
167354-41-8 (free base)
Roche Bioscience (Originator), Eli Lilly and Company (Licensee).
US5654304, WO1994024107A1, WO2000075121, US6570016
Drug Des Discov 1992, 9(1): 69, Bioorg Med Chem Lett 1995, 5(21): 2473, Drugs Fut 2003, 28(2): 125
Zosuquidar is currently under development. It is now in “Phase 3” of clinical tests in the United States. Its action mechanism consists of the inhibition of P-glycoproteins; other drugs with this mechanism include tariquidar and laniquidar. P-glycoproteins are proteins which convert the energy derived from the hydrolysis of ATP to structural changes in protein molecules, in order to perform coupling, thus discharging medicine from cells. If P-glycoprotein coded with the MDR1 gene manifests itself in cancer cells, it discharges much of the antineoplastic drugs from the cells, making cancer cells medicine tolerant, and rendering antineoplastic drugs ineffective. This protein also manifests itself in normal organs not affected by the cancer (such as the liver, small intestine, and skin cells in blood vessels of the brain), and participates in the transportation of medicine. The compound Zosuquidar inhibits this P-glycoprotein, causing the cancer cells to lose their medicine tolerance, and making antineoplastic drugs effective
Clinicial trials: Clinical report published in 2010 showed that zosuquidar did not improve outcome in older acute myeloid leukemia, in part, because of the presence P-gp independent mechanisms of resistance. (Blood. 2010 Nov 18;116(20):4077-85.)
Zosuquidar is a potent P-glycoprotein inhibitor, which binds with high affinity to P-glycoprotein and inhibits P-glycoprotein-mediated multidrug resistance (MDR). P-glycoprotein, encoded by the MDR-1 gene, is a member of the ATP-binding cassette superfamily of transmembrane transporters and prevents the intracellular accumulation of many natural product-derived cytotoxic agents
Zosuquidar
U.S. Patent No. 5,112,817 to Fukazawa et al. discloses certain quinoline derivatives useful as anticancer drug potentiators for the treatment of multidrug resistance. One of the initially promising active agents there-disclosed is MS-073, which has the following structure:
MS-073
U.S. Pat. Nos. 5,643,909 and 5,654,304 disclose a series of 10,11- methanobenzosuberane derivatives useful in enhancing the efficacy of existing cancer chemotherapeutics and for treating multidrug resistance. One such derivative having good activity, oral bioavailability, and stability, is zosuquidar, a compound of formula (2R)-anti-5-
3 – [4-( 10, 11 -difluoromethanodibenzosuber-5-yl)piperazin- 1 -yl]-2-hydroxypropoxy) quinoline.
Zosuquidar
Given the limitations of previous generations of MDR modulators, three preclinical critical success factors were identified and met for zosuquidar: 1) it is a potent inhibitor of P-glycoprotein; 2) it is selective for P-glycoprotein; and 3) no pharmacokinetic interaction with co-administered chemotherapy is observed.
Zosuquidar is extremely potent in vitro (Kj = 59 nM) and is among the most active modulators of P-gp-associated resistance described to date. Zosuquidar has also demonstrated good in vivo activity in preclinical animal studies. In addition, the compound does not appear to be a substrate for P-gp efflux, resulting in a relatively long duration of reversal activity in resistant cells even after the modulator has been withdrawn.
Another significant attribute of zosuquidar as an MDR modulator is the minimal pharmacokinetic (PK) interactions with several oncolytics tested in preclinical models. Such minimal PK interaction permits normal doses of oncolytics to be administered and also a more straightforward interpretation of the clinical results.
Zosuquidar is generally administered in the form of the trihydrochloride salt. Conventional zosuquidar trihydrochloride formulations include those containing zosuquidar (50 mg as free base), glycine (15 mg), and mannitol (200 mg) dissolved in enough water for injection, to yield a free base concentration of 5 mg/mL. The formulation is filled into vials and lyophilized to give a vial containing 50 mg of free base. For such formulations, a 30 mL vial size is necessary to contain 50 mg of thezosuquidar formulation. For a typical >200 mg dose of zosuquidar, multiple 50 mg vials are needed to contain the formulation, greatly increasing manufacturing costs and reducing convenience for the end user {e.g., a pharmacist). Modified Cyclodextrins
Cyclodextrins are cyclic oligomers of glucose; these compounds form inclusion complexes with any drug whose molecule can fit into the lipophile-seeking cavities of the cyclodextrin molecule. See U.S. Pat. No. 4,727,064 for a description of various cyclodextrin derivatives. Cyclodextrins of preferred embodiments can include α-, β-, and χ-cyclodextrins. The α-cyclodextrins include six glucopyranose units, the β- cyclodextrins include seven glucopyranose units, and the χ-cyclodextrins include eight glucopyranose units. The β -cyclodextrins are generally preferred as having a suitable cavity size for zosuquidar. Cyclodextrin can be in any suitable form, including amorphous and crystalline forms, with the amorphous form generally preferred. Cyclodextrins suitable for use in the formulations of preferred embodiments include the hydroxypropyl, hydroxyethyl, glucosyl, maltosyl, and maltotrosyl derivatives of β- cyclodextrin, carboxyamidomethyl-β-cyclodextrin, carboxymethyl-β-cyclodextrin, and diethylamino-β-cyclodextrin.
Pharmaceutical complexes including various cyclodextrins and cyclodextrin derivatives are disclosed in the following United States patents: U.S. Pat. No. 4,024,223; U.S. Pat. No. 4,228,160; U.S. Pat. No. 4,232,009; U.S. Pat. No. 4,351,846; U.S. Pat. No. 4,352,793; U.S. Pat. No. 4,383,992; U.S. Pat. No. 4,407,795; U.S. Pat. No. 4,424,209; U.S. Pat. No. 4,425,336; U.S. Pat. No. 4,438,106; U.S. Pat. No. 4,474,881; U.S. Pat. No. 4,478,995; U.S. Pat. No. 4,479,944; U.S. Pat. No. 4,479,966; U.S. Pat. No. 4,497,803; U.S. Pat. No. 4,499,085; U.S. Pat. No. 4,524,068; U.S. Pat. No. 4,555,504; U.S. Pat. No. 4,565,807; U.S. Pat. No. 4,575,548; U.S. Pat. No. 4,598,070; U.S. Pat. No. 4,603,123; U.S. Pat. No. 4,608,366; U.S. Pat. No. 4,659,696; U.S. Pat. No. 4,623,641; U.S. Pat No. 4,663,316; U.S. Pat. No. 4,675,395; U.S. Pat. No. 4,728,509; U.S. Pat. No. 4,728,510; and U.S. Pat. No. 4,751,095.
Chemically modified and substituted α-, β-, and χ-cyclodextrins are generally preferred over unmodified α-, β-, and χ-cyclodextrins due to improved toxicity and solubility properties. The degree of substitution of the hydroxy 1 groups of the glucopyranose units of the cyclodextrin ring can affect solubility. In general, a higher average degree of substitution of substituent groups in the cyclodextrin molecule yields a cyclodextrin of higher solubility.
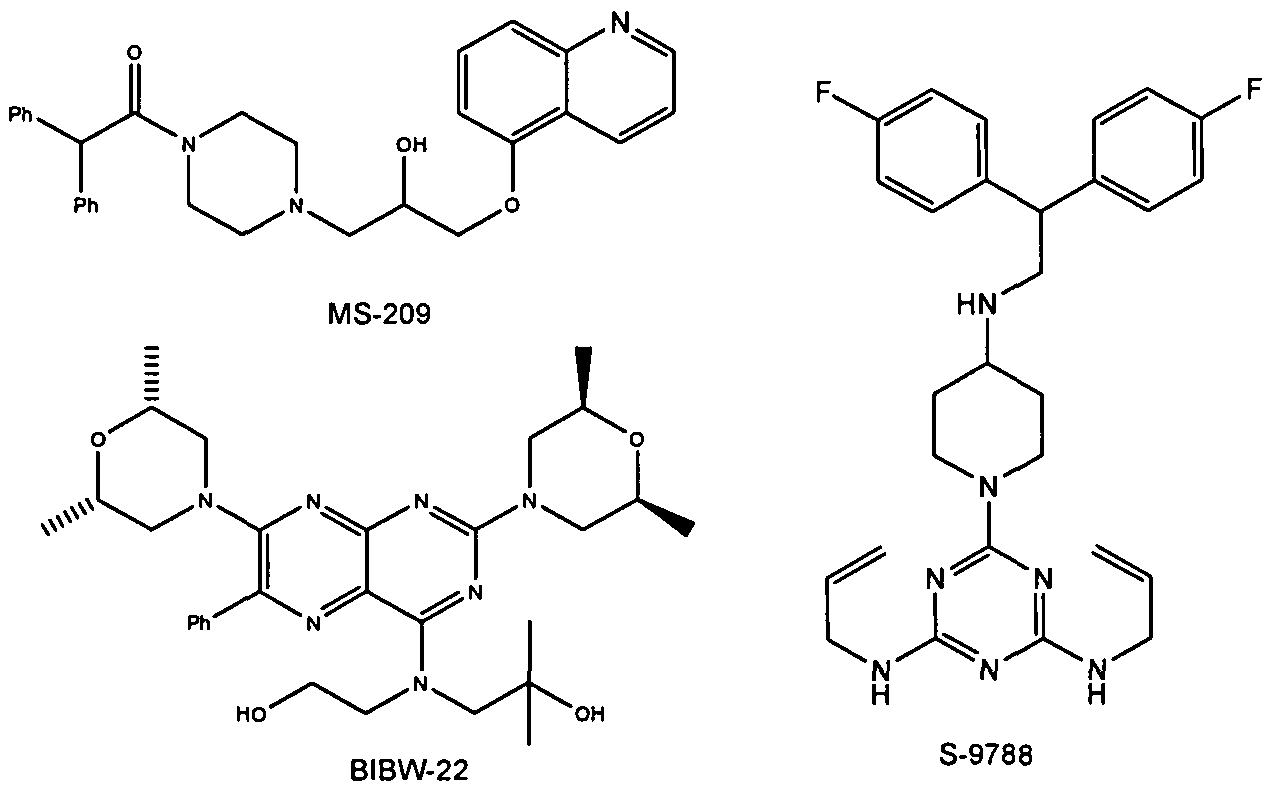
Examples for Pgp inhibitors are cyclosporine A, valpodar, elacridar, tariquidar, zosuquidar, laniquidar, biricodar, S-9788, MS-209, BIBW-22 (BIBW-22-BS) , toremifene, verapamil, dexverapamil , quinine, quinidine, trans- flupentixol, chinchonine and others (J. Roberts, C. Jarry (2003) : J. Med. Chem. 46, 4805 – 4817) . The list of inhibitors of P-glycoprotein is increasing (e.g. Wang et al . (2002) : Bioorg. Med. Chem. Lett. 12, 571 – 574) .
Figure 2: Structures of BIBW-22, MS-209 and S-9788
|
|
|
|
|
7-12-2000
|
10,11-methanodibenzosuberane derivatives
|
|
|
10-17-2007
|
Salt and crystalline forms of (2R)-anti-5-{3-[4-(10,11-difluoromethanodibenzosuber-5-yl)piperazin-1-yl]-2-hydroxypropoxy}quinoline
|
|
|
9-2-2009
|
Salt and crystalline forms of (2R)-anti-5-{3-[4-(10,11-difluoromethanodibenzosuber-5-YL)piperazin-1-YL]-2-hydroxypropoxy}quinoline
|
……………………

U.S. Pat. Nos. 5,643,909 and 5,654,304, incorporated herein by reference, disclose a series of 10,11-methanobenzosuberane derivatives useful in enhancing the efficacy of existing cancer chemotherapeutics and for treating multidrug resistance. (2R)-anti-5-{3-[4-(10,11-difluoromethanodibenzosuber-5-yl)piperazin-1-yl]-2-hydroxypropoxy}quinoline trihydrochloride disclosed therein, is currently under development as a pharmaceutical agent.
U.S. pat. No. 5,654,304 (‘304), incorporated by reference herein, discloses a series of 10,11-(optionally substituted)methanodibenzosuberane derivatives useful in enhancing, the efficacy of existing cancer chemotherapeutics and for treating multidrug resistance. (2R)-anti-5-{3-[4-(10,11-Difluoromethanodibenzosuber-5-yl)piperazin-1-yl]-2-hydroxypropoxy}quinolone trihydrochloride is disclosed in ‘304 and is currently under development as a pharmaceutical agent. WO00/75121 discloses Form I, a crystalline form of (2R)-anti-5-{3-[4-(10,11-difluoromethanodibenzosuber-5-yl)piperazin-1-yl]-2-hydroxypropoxy}quinolone trihydrochloride.
The art disclosed in U.S. Pat. No. 5,776,939, and U.S. Pat. No. 5,643,909 both incorporated herein by reference, and PCT Patent Applications (Publication numbers WO 94/24107 and 98/22112) teach the use of 1-formylpiperazine to introduce the piperazine group of the compound of formula II
Compound II is a mixture of syn isomer (III)
and anti isomer (IV)
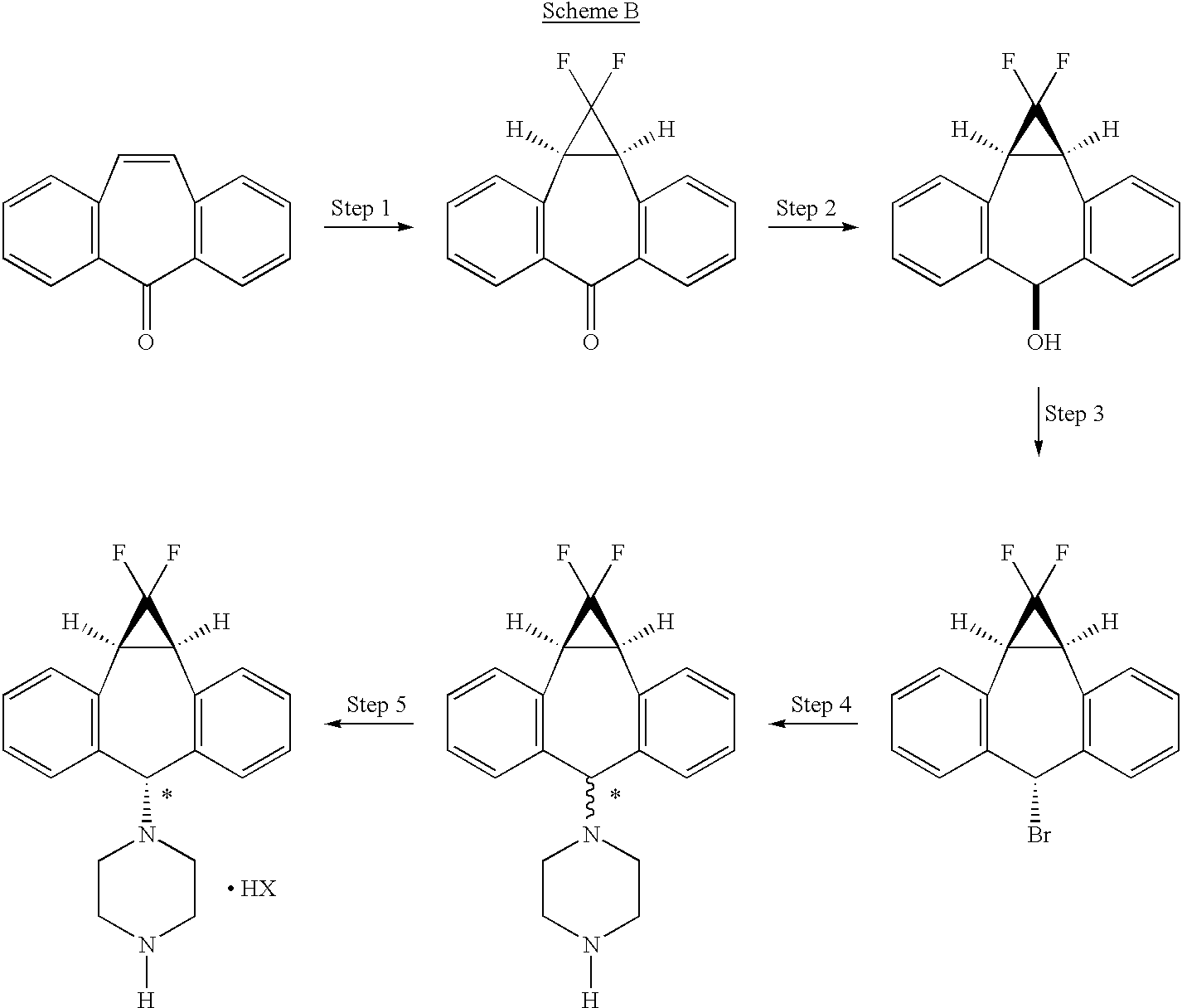
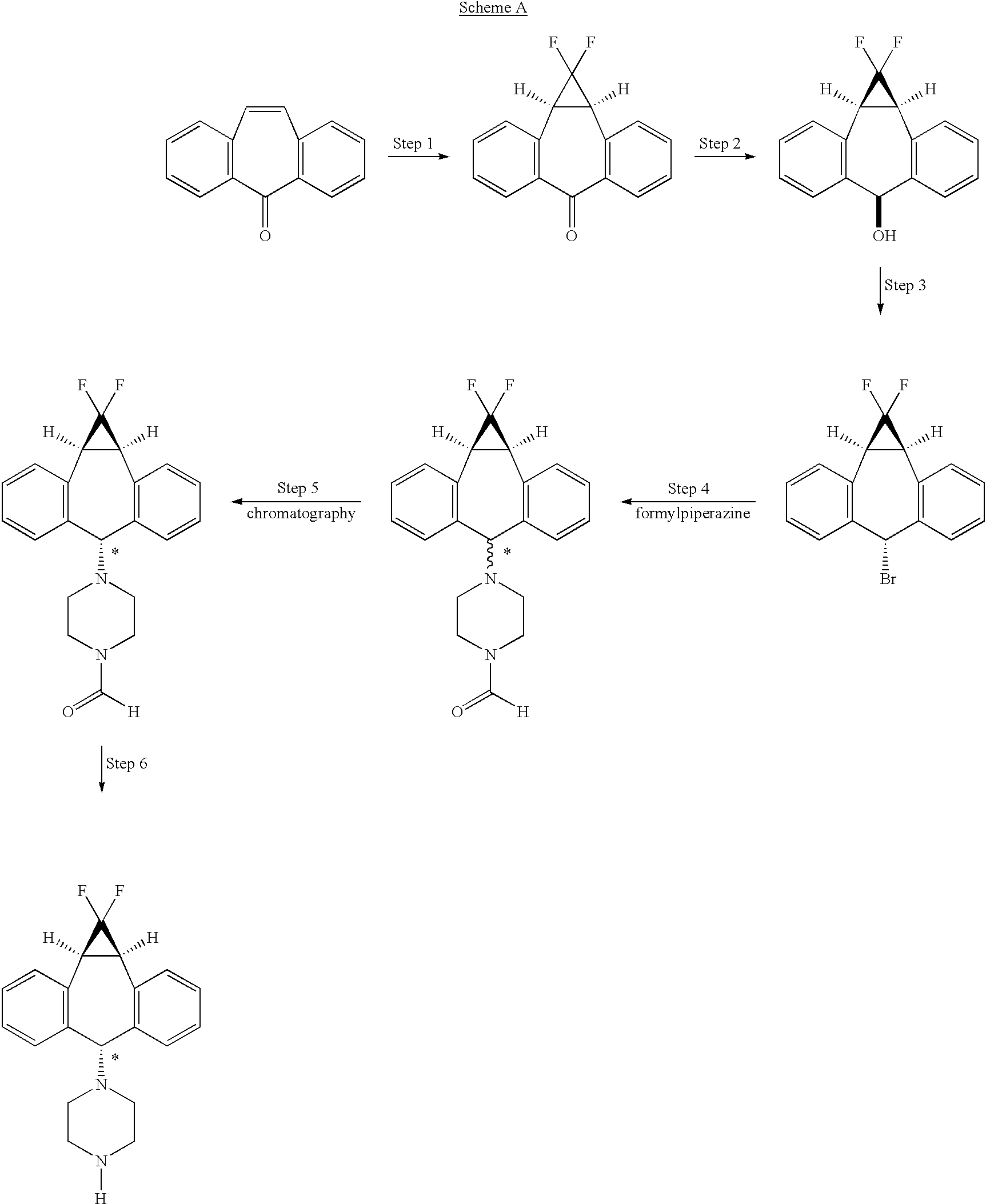
The process as disclosed in U.S. Pat. Nos. 5,643,909 and 5,654,304 (represented by scheme A, below) involves (a) chromatographic separation(s) of the formyl piperazine compound; and (b) deformylation of the formyl piperazine compound to provide compound IV.https://www.google.co.in/patents/US6570016?cl=en
The process of the present invention uses piperazine to react with the (1aα,6α,10bα)-6-halo-1,1-difluoro-1,1a,6,10b-tetrahydrodibenzo[a,e]cyclopropa[c]-cycloheptene compound or derivative, instead of formylpiperazine.
The process of the present invention is advantageous because piperazine is readily available in commercial quantities whereas 1-formylpiperazine, which was utilized in the process disclosed in U.S. Pat. No. 5,643,909 is often not readily available in commercial quantities. Additionally piperazine enjoys a significant cost advantage over 1-formylpiperazine.
The use of piperazine instead of 1-formylpiperazine is a significant advancement over the prior art because it obviates the need to deformylate or hydrolyze off the formyl group (step 6, scheme A), thereby providing fewer operational steps. U.S. Pat. No. 5,643,909 teaches the separation of the 1-formylpiperazine compounds by chromatography or repeated crystallization. The present invention obviates the need for chromatographic separations of the formylpiperazine diastereomeric addition compounds (see step 4, scheme A)
EXAMPLES
The following examples and preparations are illustrative only and are not intended to limit the scope of the invention in any way.
Preparation 1 R-1-(5-Quinolinyloxy)-2,3-epoxypropane
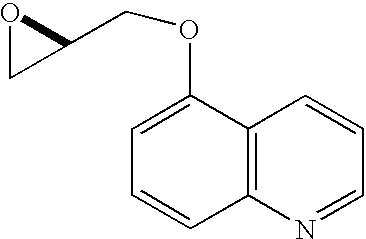
A mixture of 5-hydroxyquinoline (5.60 g, 38.6 mmol), R-glycidyl nosylate (10.0 g, 38.6 mmol), powdered potassium carbonate (11.7 g, 84.9 mmol), and N,N-dimethylformamide (100 mL) was stirred at ambient temperature until HPLC analysis (40% acetonitrile/60% of a 0.5% aqueous ammonium acetate solution, 1 mL/min, wavelength=230 nm, Zorbax RX-C8 25 cm×4.6 mm column) indicated complete disappearance of glycidyl nosylate (approximately 6 hours). The reaction mixture was filtered through paper and the filter cake was washed with 200 mL of a 3:1 mixture of MTBE and methylene chloride. The filtrate was washed with 200 mL of water and the aqueous layer was extracted four times with 100 mL of 3:1 MTBE/methylene chloride. The combined organic layers were dried over 30 grams of magnesium sulfate and the dried solution was then stirred with 50 grams of basic alumina for 30 minutes. The alumina was removed by filtration and the filter cake was washed with 200 mL of 3:1 MTBE/methylene chloride. The filtrate was concentrated to a volume of 100 mL, 300 mL of MTBE were added, and the solution was again concentrated to 80 mL. After heating to 50° C., the solution was treated with 160 mL of heptane dropwise over 15 minutes, allowed to cool to 40° C., and seeded, causing the formation of a crystalline precipitate. The mixture was stirred for two hours at ambient temperature and then at 0-5° C. for an additional 2 hours. The crystals were filtered, washed with cold heptane, and dried to provide 5.68 g (73.2%) of (2R)-1-(5-quinolinyloxy)-2,3-epoxypropane as white needles.
mp 79-81° C.;
[α]25 D−36.4° (c 2.1, EtOH);
1H NMR (500 MHz, CDCl3)δ 2.83 (dd, J=4.8, 2.7 Hz, 1H), 2.97 (m, 1H), 3.48 (m, 1H), 4.10 (dd, J=11.0, 6.0 Hz, 1H), 4.43 (dd, J=11.0, 2.7 Hz, 1H), 6.85 (d, J=7.8 Hz, 1H), 7.38 (dd, J=8.5 Hz, 4.1 Hz, 1H), 7.59 (m, 1H), 7.71 (d, J=8.5 Hz, 1H), 8.61 (m, 1H), 8.90 (m, 1H).
Example 1 (2R)-Anti-1-[4-(10,11-difluoromethano-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5-yl)-piperazin-1-yl]-3-qunolin-5-yloxy)-propan-2-ol Trihydrochloride
Preparation of the above compound is exemplified in the following preparative steps.
Step 1 1,1-Difluoro-1a,10b-dihydrodibenzo[a,e]cyclopropa[c]-cyclohepten-6 (1H)-one
A solution of sodium chlorodifluoroacetate (350 g) in diglyme (1400 mL) was added dropwise over 4 to 8 hours, preferably over 6 hours, to a solution of 5H-dibenzo[a,d]cyclo-hepten-5-one (25 g) in diglyme (500 mL), with stirring, and under nitrogen, maintaining the reaction temperature at 160°-165° C. The cooled reaction mixture was poured into water (1.8 L) and extracted with ether (1.8 L). The organic phase was washed with water, dried over sodium sulfate (Na2SO4), and evaporated. The residue was recrystallized from ethanol, then from acetone/hexane to give 14 g of 1,1-difluoro-1a,10b-dihydrodibenzo[a,e]cyclopropa[c]-cyclohepten-6(1H)-one.
mp 149.6° C.
Flash chromatography of the combined mother liquors on silica gel, eluting with 20% acetone/hexane, gave an additional 6.5 g of the target compound.
Step 2 (1aα,6β,10bα)-1,1-Difluoro-1,1a,6,10b-tetrahydrodibenzo[a,e]cyclopropa[c]cyclohepten-6-ol
A solution of 1,1-difluoro-1a,10b-dihydro-dibenzo[a,e]cyclopropa[c]cyclohepten-6(1H)-one (20.4 g) in tetrahydrofuran/methanol (1:2, 900 mL) was cooled in an ice bath. Sodium borohydride (12 g) was added in portions. The cooling bath was removed and the reaction mixture was stirred at ambient temperature for 2 hours, then poured into water. The product was filtered off, washed with water, and dried to give 20 g of (1aα,6β,10bα)-1,1-difluoro-1,1a,6,10b-tetrahydrodibenzo[a,e]cyclopropa[c]cyclohepten-6-ol (ii).
mp 230.1°-230.6° C.
Step 2A Combined Steps 1 and 2 Procedure (1aα,6β,10bα)-1,1-Difluoro-1,1a,6,10b-tetrahydrodibenzo[a,e]cyclopropa[c]cyclohepten-6-ol

To a solution of 103.1 g (0.500 mol) of 5H-dibenzo[a,d]cyclohepten-5-one (2) in 515 mL of triethylene glycol dimethyl ether heated to between 180° C. and 210° C. was added over 7 hours, 293.3 g (2.15 mol) of chlorodifluoroacetic acid lithium salt (as a 53% by weight solution in ethylene glycol dimethyl ether). The ethylene glycol dimethyl ether was allowed to distill from the reaction as the salt addition proceeded. The GC analysis of an aliquot indicated that all of the 5H-dibenzo[a,d]cyclohepten-5-one had been consumed. The reaction was cooled to ambient temperature and then combined with 400 mL of ethyl acetate and 75 g of diatomaceous earth. The solids were removed by filtration and washed with 300 mL of ethyl acetate. The washes and filtrate were combined and the ethyl acetate was removed by concentration under vacuum leaving 635 g of dark liquid. The dark liquid was cooled to 18° C. and to this was added, over 15 minutes, 6.62 g (0.175 mol) of sodium borohydride (as a 12% by wt solution in 14 M NaOH). After stirring for 2 h the reaction was quenched by careful addition of 900 mL of a 1:3.5:4.5 solution of conc. HCl-methanol-water. The suspension was stirred for 30 min and the crude product was collected by filtration, washed with 600 mL of 1:1 methanol-water and dried to 126.4 g of dark brown solid. The crude product was slurried in 600 mL of methylene chloride, filtered, washed twice with 150 mL portions of methylene chloride, and dried to 91.6 g (71%) of (1aα,6β,10bα)-1,1-difluoro-1,1a,6,10b-tetrahydrodibenzo[a,e]cyclopropa[c]-cyclohepten-6-ol. Gas Chromatography (GC) Conditions; Column: JW Scientific DB-1, Initial Temperature 150° C. for 5 min, 10° C./min ramp, Final temp 250° C. for 5 min. tR: intermediate, 11.5 min; reaction product (alcohol), 11.9 min; starting material, 12.3 minutes.
Step 3 Preparation of (1aα,6α,10b)-6-bromo-1,1-difluoro-1,1a,6,10b-tetrahydrodibenzo[a,e]cyclopropa-[c]cycloheptene
A slurry of (1aα,6β,10bα)-1,1-difluoro-1,1a,6,10b-tetrahydrodibenzo[a,e]cyclopropa[c]-cyclohepten-6-ol (3.0 g, 11.6 mmol, 1.0 equiv) in heptane (24 mL) was treated with 48% HBr (1.58 mL, 14.0 mmol, 1.2 equiv) and the reaction was heated at reflux with vigorous stirring for 2.5 hr. Solvent was then removed by atmospheric distillation (bp 95-98° C.) until approximately 9 mL of distillate was collected. The reaction was cooled and treated with EtOAc (15 mL), Na2SO4 and activated charcoal. The mixture was stirred at RT for 15 min and filtered through hyflo. The filter cake was washed with 50:50 EtOAc:heptane and the filtrate was concentrated in vacuo to provide the title product as a crystalline solid.
mp 119° C. (3.46 g corr., 93%);
1H NMR (500 MHz CDCl3) δ 7.20-7.41 (8H, m), 5.81 (1H, s), 3.41 (2H, d, J 12.5 Hz);
13CNMR (126 MHz CDCl3) δ 141.3, 141.2, 133.5, 130.1, 129.8, 128.3, 128.2, 112.9, 110.6, 110.5, 108.3, 53.6, 30.2, 30.1, 30.0.
Anal. Calcd. For C16H11BrF2: C, 59.84; H, 3.45. Found: C, 60.13; H, 3.50.
Step 3A Preparation of (1aα,6α,10bα)-6-Bromo-1-difluoro-1,1a,6,10b-tetrahydrodibenzo[a,e]cyclopropa[c]cycloheptene

To a stirred suspension of (1aα,6β,10bα)-1,1-difluoro-1,1a,6,10b-tetrahydrodibenzo[a,e]cyclopropa[c]-cyclohepten-6-ol, (18.4 g, 71.2 mmol) in 151 mL of methylene chloride which had been cooled to 10-17° C. was added phosphorous tribromide (9.6 g, 35.6 mmol) dropwise over 15 minutes. The cooling bath was removed and the reaction was stirred for 2 hours at ambient temperature. Analysis by gas chromatography indicated complete consumption of starting material. Cold water (92 mL) and activated carbon (1.84 g) were added and the resulting mixture was stirred for 30 minutes. The activated carbon was removed by filtration through Hyflo brand filter aid and the two phases were separated. The organic phase was washed with water (184 mL×2), brine (184 ml), dried over magnesium sulfate and concentrated to dryness under vacuum, affording 21.7 g (94.8%) of (1aα,6α,10bα)-6-bromo-1,1-difluoro-1,1a,6,10b-tetrahydrodibenzo[a,e]cyclopropa[c]cycloheptene.
1H NMR (CDCl3, 300 MHz) δ 3.36 (s, 1H), 3.40 (s, 1H), 5.77 (s, 1H), 7.16-7.38 (m, 8H).
Steps 4 and 5 (1aα,6α,10bα)-1-(1,1-Difluoro-1,1a,6,10b-tetrahydrodibenzo[a,e]cyclopropa[c]cyclohepten-6-yl)-piperazine, Hydrobromide Salt

To a solution of 237.5 g (0.739 mol) of (1aα,6α,10bα)-6-bromo-1,1-difluoro-1,1a,6,10b-tetrahydrodibenzo[a,e]-cyclopropa[c]cycloheptene in 3.56 L of acetonitrile was added 207.7 g (2.41 mol) of piperazine and the mixture was heated to reflux for 2 hours, at which time analysis by gas chromatography showed complete consumption of (1aα,6α,10bα)-6-bromo-1,1-difluoro-1,1a,6,10b-tetrahydrodibenzo[a,e]cyclopropa[c]cycloheptene (iii) and formation of a mixture of syn and anti piperazine compounds (III and IV) in an anti-syn ratio of 55:45. The reaction was cooled to about 7° C. and stirred for 30 minutes at that temperature. The reaction mixture was filtered to remove the precipitated syn-isomer (III) and the filter cake was washed with 250 mL of acetonitrile. The combined filtrate and wash were concentrated under vacuum to 262.4 grams of a foam which was dissolved in 450 mL of acetonitrile with heating. The solution was cooled to about 12° C. in an ice bath and stirred for 1 hour at that temperature. The precipitated syn-piperazine compound of formula (III) was filtered and washed with 125 ml of acetonitrile. The combined filtrate and wash were concentrated under vacuum to 194.1 g and dissolved in 1.19 L of ethyl acetate. The organic solution was washed sequentially with 500 mL portions of 1N sodium hydroxide, water, and saturated sodium chloride. The ethyl acetate solution was dried over sodium sulfate and concentrated to give 137.0 grams of residue which was dissolved in 1.37 L of methylene chloride and seeded with (1aα,6α,10bα)-1-(1,1-difluoro-1,1a,6,10b-tetrahydrodibenzo[a,e]cyclopropa[c]-cyclohepten-6-yl)-piperazine, hydrobromide salt, followed by the addition of 70.8 grams of 48% aqueous hydrobromic acid. The mixture was stirred for about 45 minutes, causing the anti-isomer to crystallize as its hydrobromide salt. The crystals were filtered, washed with methylene chloride, and dried to provide purified hydrobromide salt of compound (IVa), shown by HPLC to have an anti-syn ratio of 99.3:0.7. Treatment of the isolated hydrobromide salt of compound (IVa) with aqueous sodium hydroxide, extraction into methylene chloride, separation of the aqueous layer and concentration to dryness gave 80.1 grams (33.2% yield based on starting material) of (1aα,6α,10bα)-1-(1,1-difluoro-1,1a,6,10b-tetrahydrodibenzo[a,e]cyclopropa[c]-cyclohepten-6-yl)-piperazine as the free base. Acidification of a solution of the free base in 800 mL of methylene chloride by addition of 41.2 g of 48% hydrobromic acid as described above afforded 96.4 g of pure hydrobromide salt (title compound) with an anti-syn ratio of 99.8:0.2 (HPLC), mp 282-284° C. 1H NMR (DMSO-d6) δ 2.41 (m, 4H), 3.11 (m, 4H), 3.48 (d, J=12.4 Hz, 2H), 4.13 (s, 1H), 7.2 (m, 8H), 8.65 (bs, 2H). 13C NMR (DMSO-d6) δ 28.0, 42.9, 48.0, 75.1, 108.5, 112.9, 117.3, 127.5, 128.0, 128.6, 129.6, 132.4, 141.3. IR: (KBr) 3019, 2481, 1587, 1497, 1298 cm−1. Anal. Calcd for C20H21BrF2N2: C, 58.98; H, 5.20; N, 6.88. Found: C, 58.75; H, 5.29; N, 7.05.
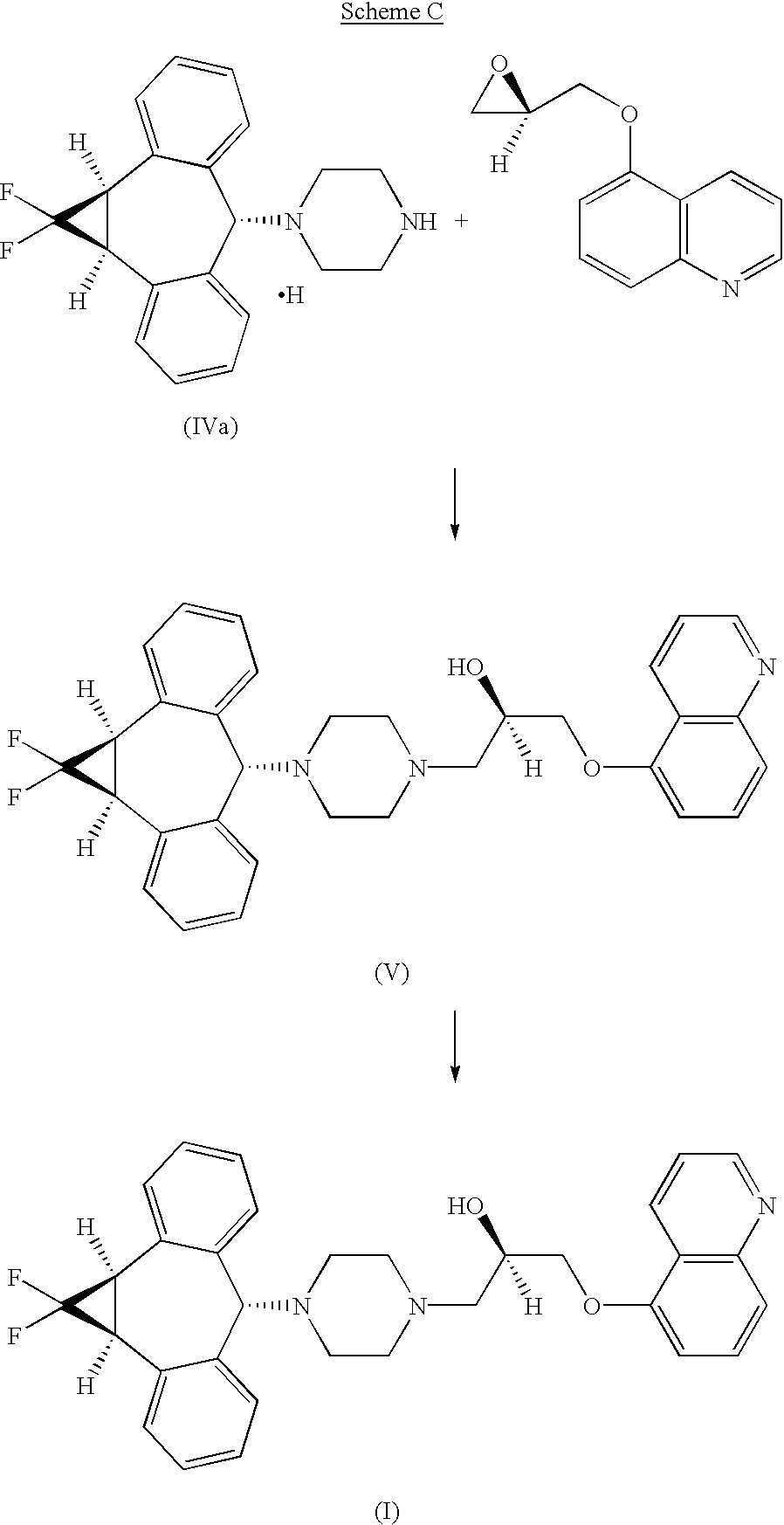
Step 6 Preparation of (2R)-Anti-1-[4-(10,11-difluoromethano-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5-yl)-piperazin-1-yl]-3-quinolin-5-yloxy)propan-2-ol Trihydrochloride
A suspension of (1aα,6α,10bα)-1-(1,1-difluoro-1,1a,6,10b-tetrahydrodibenzo[a,e]cyclopropa[c]-cyclohepten-6-yl)-piperazine, hydrochloride compound of formula IVa (5.41 g, 14.9 mmol) and powdered sodium carbonate (3.16 g, 29.8 mmol) in 54 mL of 3A ethanol was stirred at ambient temperature for 1 hour. R-1-(5-quinolinyloxy)-2,3-epoxypropane (3.00 g, 14.9 mmol) was added in one portion and the reaction mixture was heated to 65° C. for 19 hours. HPLC analysis (Gradient system with solvent A (acetonitrile) and solvent B (0.02M sodium monophosphate buffer containing 0.1% triethylamine adjusted to pH 3.5 with phosphoric acid) as follows: 0-12 min, 30% solvent A/70% solvent B; 12-30 min, linear gradient from 30% to 55% solvent A/70% to 45% solvent B; 30-35 min, 55% solvent A/45% solvent B, 1 mL/min, 1=240 nm, Synchropak SCD-100 25 cm×4.6 mm column) indicated the total consumption of the piperazinyl compound of formula (IV). The mixture was allowed to cool to room temperature, filtered through a plug of silica gel, and eluted with an additional 90 mL of ethanol. The eluent was concentrated to a volume of approximately 60 mL and heated to 65° C. with stirring. A solution of HCl in ethanol (16.1 g at 0.135 g/g of solution, 59.6 mmol) was added dropwise over 10 minutes and the resultant product solution was seeded, causing the trihydrochloride salt to precipitate. The mixture was allowed to cool to ambient temperature and stirred slowly (less than 100 RPM) for 2 hours. The precipitate was filtered, washed with ethanol, and dried in vacuo at 50° C. to give the crude trihydrochloride salt which was further purified by recrystallization from methanol/ethyl acetate to provide 7.45 g (78.4%) of (2R)-anti-1-[4-(10,11-difluoromethano-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5-yl)-piperazin-1-yl]-3-quinolin-5-yloxy)-propan-2-ol trihydrochloride.
Step 6a
The syn isomer compound of formula (III) isolated as described supra (combined steps 4 and 5), can be utilized to produce the corresponding syn-5-{3-[4-(10,11-difluoromethano-dibenzosuber-5-yl)piperazin-1-yl]-2-hydroxypropoxy}quinoline trihydrochloride (XII) essentially as shown below for the free base of the anti isomer (IVa)in step 6.
https://www.google.co.in/patents/US6570016?cl=en
………………………………………
http://www.google.it/patents/WO1994024107A1?cl=en
REACTION SCHEME 1
FormuIa 1
Formula 1
Formula 2 Formula 2
Formula 3
Formula 3
Formula 4
Formula I
……………………………………….
http://www.google.com/patents/WO2000075121A3

1HNMR (500 MHz DMSO-d6) δ9.41 (2H, br. s), 7.17-7.31 (8H, m), 4.17 (1H, s), 3.52 (2H, d, J=12.4 Hz), 3.11 (4H, br. s), 2.48-2.51 (4H, m)
13CNMR (126 MHz DMSO-d6) δ142.3, 133.4, 130.5, 129.6, 129.0, 128.4, 115.9, 113.6, 111.3, 76.2, 49.0, 43.6, 29.2, 29.1, 29.0; FD MS: m/e 326 (M+).
Anal. Calcd. For C20H21ClF2N2: C, 66.20; H, 5.83; N, 7.72.
Found: C, 66.08; H, 5.90; N, 7.72.
…………………………………………..
http://www.google.com/patents/US6570016?cl=fr


(2R)-Anti-1-[4-(10,11-difluoromethano-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5-yl)-piperazin-1-yl]-3-qunolin-5-yloxy)-propan-2-ol Trihydrochloride
……………….
Chemical Shift Data and Peak Assignments for the Crystal Forms.
https://www.google.co.in/patents/US7282585?pg=PA1&dq=US+7282585&hl=en&sa=X&ei=zN64UsC2FIaSrgfS8YGIBQ&ved=0CDcQ6AEwAA
Form II has a solid-state 13C NMR spectrum comprised of isotropic peaks at the following chemical shifts: 29.9, 50.1, 55.3, 62.0, 66.5, 72.0, 75.8, 104.8, 107.5, 108.2, 109.1, 110.2, 112.0, 118.4, 119.5, 120.1, 123.1, 128.7, 131.1, 133.0, 134.8, 136.4, 136.9, 139.9, 140.0, 142.3, 144.5, 146.6, 149.0, 144.2, 153.0 and 153.6 ppm.
Form III has a solid-state 13C NMR spectrum comprised of isotropic peaks at the following chemical shifts: 30.3, 50.4, 59.1, 63.2, 72.8, 77.2, 109.1, 110.2, 112.2, 112.8, 118.7, 119.5, 119.9, 121.0, 122.2, 123.0, 128.9, 130.6, 132.7, 134.0, 136.4, 140.0, 141.0, 141.8, 142.5, 143.3, 146.1, 153.1, 153.8 and 154.7 ppm.

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....

































 .
.













































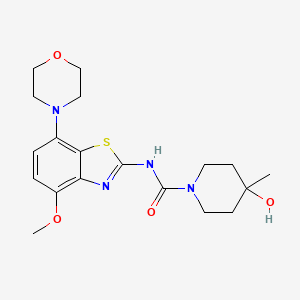
 THE ESTER OF EVACETRAPIB
THE ESTER OF EVACETRAPIB




























{kind=link}
{kind=link}
{kind=link}