


 Click on images to view
Click on images to view















Ono Pharmaceutical Co has become the first company in the world to get an approval for a PD-1 checkpoint inhibitor, as regulators in Japan gave the green light to nivolumab, developed with Bristol-Myers Squibb, as a treatment for melanoma.
Home » Posts tagged 'Bristol-Myers Squibb' (Page 2)
Tag Archives: Bristol-Myers Squibb
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on
ORGANIC SPECTROSCOPY INTERNATIONAL

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()




Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
BRISTOL-MYERS SQUIBB’S TRICYCLOHEXADECAHEXAENE DERIVATIVES FOR USE IN THE TREATMENT OF HEPATITIS C VIRUS
 .
.
TRICYCLOHEXADECAHEXAENE DERIVATIVES FOR USE IN THE TREATMENT OF HEPATITIS C VIRUS
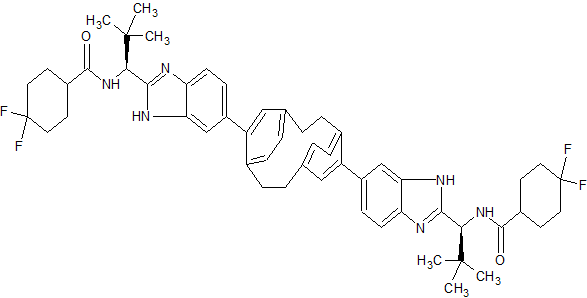
CAS 1663477-91-5
C54 H62 F4 N6 O2, 903.10
Cyclohexanecarboxamide, N,N‘-[tricyclo[8.2.2.24,7]hexadeca-4,6,10,12,13,15-hexaene-5,11-diylbis[1H-benzimidazole-6,2-diyl[(1S)-2,2-dimethylpropylidene]]]bis[4,4-difluoro-
WO2015026454, COMBINATIONS COMPRISING TRICYCLOHEXADECAHEXAENE DERIVATIVES FOR USE IN THE TREATMENT OF HEPATITIS C VIRUS
BRISTOL-MYERS SQUIBB COMPANY [US/US]; Route 206 and Province Line Road Princeton, New Jersey 08543 (US)
PATENT WO2015026454 [LINK]
WANG, Alan Xiangdong; (US).
LOPEZ, Omar D.; (US).
TU, Yong; (US).
BELEMA, Makonen; (US)
Example B-l
Example B-l Step a

To a solution of 4-bromobenzene-l,2-diamine (2.5 g, 13.37 mmol) in DCM (30 mL) was added (S)-2-((tert-butoxycarbonyl)amino)-3,3-dimethylbutanoic acid (3.09 g, 13.37 mmol), DIPEA (2.334 mL, 13.37 mmol) and HATU (5.08 g, 13.37 mmol). The reaction mixture was stirred at room temperature for 18 h. The reaction mixture was diluted with water and extracted with DCM. The organic phase was washed with brine, dried over Na2S04, filtered and concentrated. The crude material was purified by ISCO using 40 g Redisep silica column, CHCl3/MeOH as eluant to obtain (S)-tert-butyl ( 1 -((2-amino-4-bromophenyl)amino)-3 ,3 -dimethyl- 1 -oxobutan-2-yl) carbamate (1.82 g) as yellow solid. LC (Condition 1): Rt = 2.13 min. LC/MS: Anal. Calcd. for [M+H20]+ Ci7H27BrN204 : 402.12; found 402.2. 1H NMR (DMSO-d6, δ = 2.50 ppm, 400 MHz): δ 9.35 – 9.21 (m, 1 H), 7.07 (d, J= 8.5 Hz, 1 H), 6.91 (d, J= 2.0 Hz, 1 H), 6.80 – 6.60 (m, 1 H), 5.25 – 5.01 (m, 2 H), 4.07 – 3.89 (m, 1 H), 1.52 – 1.34 (m, 9 H), 1.02 – 0.86 (m, 9 H).
Example B-l, Step b

Acetic acid (15 mL) was added to (S)-tert-butyl (l-((2-amino-4-bromo phenyl)amino)-3,3-dimethyl-l-oxobutan-2-yl)carbamate (1.8 g, 4.50 mmol) and the reaction mixture was heated to 65 °C for overnight. The volatile component was removed in vacuo, and the residue was co-evaporated with dry CH2C12 (2 x 15 mL). The organic phase was washed with saturated NaHC03 solution, brine, dried over Na2S04 and concentrated to obtain (S)-tert-butyl (l-(6-bromo-lH-benzo[d] imidazol-2-yl)-2,2-dimethyl propyl)carbamate (1.68 g) as yellow solid. LC (Condition 1): Rt = 2.19 min. LC/MS: Anal. Calcd. for [M+H]+ Ci7H25BrN302 : 381.11; found 382.2. 1H NMR (DMSO-dg, δ = 2.50 ppm, 300 MHz): δ 12.46 – 12.27 (m, 1 H), 7.82 – 7.65 (m, 1 H), 7.59 – 7.41 (m, 1 H), 7.29 (dt, J= 1.9, 8.5 Hz, 1 H), 7.12 – 6.90 (m, 1 H), 4.64 (d, J= 9.8 Hz, 1 H), 1.44 – 1.27 (m, 9 H), 0.88 (br. s., 9 H).
-1 Step c

To a solution of (S)-tert-butyl (l-(6-bromo-lH-benzo[d]imidazol-2-yl)-2,2-dimethyl propyl) carbamate (1.57 g, 4.11 mmol) in dioxane (25 mL) was added bis (pinacolato)diboron (1.564 g, 6.16 mmol) and potassium acetate (1.209 g, 12.32 mmol). The reaction mixture was purged with argon for 10 min then PdCl2(dppf) (0.150 g, 0.205 mmol) was added to the above reaction mixture and again purged with argon for 5 min. The reaction mixture was heated to 90 °C for overnight. The reaction mixture was diluted with water (15 ml) and extracted with EtOAc (2 x 25 ml). The combined organic phase was washed with brine, dried over Na2S04 and concentrated in vacuo. The crude material was purified by ISCO using 40 g Redisep column, hexane/ethyl acetate as eluant to afford (S)-tert-butyl (2,2-dimethyl-l-(6-(4,4,5 ,5-tetramethyl- 1 ,3 ,2-dioxaborolan-2-yl)- 1 H-benzo[d]imidazol-2-yl)propyl) carbamate (1.35 g) as yellow solid. LC (Condition 1): Rt = 2.21 min. LC/MS: Anal. Calcd. for [M+H]+ C23H37BN304 : 430.29; found 430.4. 1H NMR (CD3OD, δ = 3.34 ppm, 400 MHz): δ 7.98 (s, 1 H), 7.65 (dd, J= 1.0, 8.5 Hz, 1 H), 7.53(d, J= 8.5 Hz, 1 H), 4.73 (br. s., 1 H), 1.37 (s, 12 H), 1.24 (m, 9 H), 1.01 (s, 9 H).
-1 Step d

To a solution of (S)-tert-butyl (2,2-dimethyl-l-(6-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-lH-benzo[d]imidazol-2-yl)propyl)carbamate (1.114 g, 2.59 mmol) and 4,16-dibromo[2,2]paracyclophane (0.38g, 1.038 mmol) in dioxane (10 mL) was added Cs2C03 (0.845 g, 2.59 mmol) in water (2 mL) and degassed for 10 min.
PdCl2(dppf) (0.038 g, 0.052 mmol) was added to the above reaction mixture and again degassed for 5 min. The reaction mixture was heated to 90 °C for 12 h. Then the reaction mixture was filtered to get Example B-1 Step d which was taken for next step without further purification. LC (Condition 1): Rt = 2.54 min. LC/MS: Anal. Calcd. for [M+H]+ ^0Η63Ν6Ο4 : 811.49; found 811.6. 1H NMR (DMSO-d6, δ = 2.50 ppm, 300 MHz): δ 12.36 (br. s., 2 H), 7.85 – 7.52 (m, 4 H), 7.32 (d, J= 7.9 Hz, 2 H), 7.05 (br. s., 2 H), 6.89 – 6.67 (m, 4 H), 6.54 (br. s., 2 H), 4.72 (d, J= 8.7 Hz, 2 H), 3.57 – 3.44 (m, 2 H), 3.07 (br. s., 2 H), 2.83 (br. s., 2 H), 2.65 (br. s., 2 H), 1.36 (s, 18 H), 1.08 – 0.91 (m, 18 H).
-1 Step e

HC1 in dioxane (4 mL, 24.00 mmol) was added to Example B-1 Step d (0.1 g,
0.102 mmol), and the reaction mixture was allowed to stir at RT for 2 h. Completion of the reaction was monitored by LCMS. The volatile component was removed in vacuo and the residue was washed with diethyl ether and dried to afford Example B-1 Step e (0.07 g) as yellow solid. LC (Condition 1): R, = 2.54 min. LC/MS: Anal.
Calcd. for [M+H]+ C40H47N6 : 611.39; found 611.4. 1H NMR (CD3OD, δ = 3.34 ppm, 400 MHz): δ 7.90 (d, J= 13.1 Hz, 2 H), 7.83 (d, J= 8.5 Hz, 2 H), 7.61 (d, J= 8.5 Hz, 2 H), 6.84 (d, J= 6.5 Hz, 2 H), 6.78 (s, 2 H), 6.70 – 6.65 (m, 2 H), 4.54 (d, J= 1.0 Hz, 2 H), 3.54 – 3.46 (m, 2 H), 3.18 – 3.10 (m, 2 H), 2.98 – 2.86 (m, 2 H), 2.71 (br. s., 2 H), 1.25 – 1.22 (m, 18 H).
To a solution of Example B-1 Step e (0.04 g, 0.053 mmol) in DMF (5 mL) was added 4,4-difluorocyclohexanecarboxylic acid (0.017 g, 0.106 mmol), DIPEA (0.055 mL, 0.317 mmol) and HATU (0.030 g, 0.079 mmol). After being stirred for 2 h at room temperature, the volatile component was removed in vacuo and the residue was dissolved in DCM (10 mL), washed with saturated solution of NH4C1, 10% NaHC03 solution, brine, dried over Na2S04 and concentrated in vacuo. The crude was purified by reverse phase HPLC purification to give Example B-1 as a white solid. LC (Condition 1): R, = 2.37 min. LC/MS: Anal. Calcd. for [M+H]+
C54H63F4N602: 903.49; found 903.4. 1H NMR (DMSO-d6, δ = 2.50 ppm, 400 MHz): δ 12.53 – 12.32 (m, 2 H), 8.41 – 8.21 (m, 2 H), 7.84 – 7.50 (m, 4 H), 7.43 – 7.24 (m, 2 H), 6.90 – 6.67 (m, 4 H), 6.60 – 6.44 (m, 2 H), 5.14 – 4.97 (m, 2 H), 3.44 (br. s., 2 H), 3.08 (br. s., 2 H), 2.93 – 2.77 (m, 2 H), 2.73 – 2.56 (m, 4 H), 2.20 – 1.98 (m, 3 H), 1.96 – 1.49 (m, 13 H), 1.02 (s, 18 H).
Starting materials can be obtained from commercial sources or prepared by well-established literature methods known to those of ordinary skill in the art. Acid precursors for the final step can be prepared according to the methods described in U.S. Patent Application Serial No. 13/933495, filed July 2, 2013.
LC/MS Condition 1
Column = Ascentis Express C18, 2.1 X 50 mm, 2.7 um
Solvent A = CH3CN (2%) + 10 mM NH4COOH in H20 (98%)
Solvent B = CH3CN (98%) + 10 mM NH4COOH in H20 (2%)
Start %B = 0; Final %B = 100
Gradient time = 1.4 min; Stop time = 4 min
Stop time = 4 min
Flow Rate = 1 mL/min; Wavelength = 220 nm
LC/MS Condition 2
Column = Waters BEH CI 8, 2.0 x 50 mm, 1.7 μιη
Slovent A = ACN (5%) + H20 (95%) containing 10 mM NH4OAc
Solvent B = ACN (95%) + H20 (5%) containing 10 mM NH4OAc
Start %B = 0; Final %B = 100
Gradient time = 3 min
Flow Rate = 1 mL/min
Wavelength = 220 nm
Temperature = 50 °C
LC/MS Condition 3
Column: Waters Phenomenex CI 8, 2.0 x 30 mm, 3 μιη particle
Mobile Phase A: 10% MeOH:90% Water :0.1%TFA
Mobile Phase B: 90% MeOH: 10% Water :0.1%TFA
Gradient: 0%B, 0-100% B over 3 minutes, then a 1 -minute hold at 100% B Flow: 0.8mL/min
Detection: 220 nm
Temperature: 40 °C
LC/MS Condition 4
Column: Waters BEH CI 8, 2.0 x 50 mm, 1.7 μιη particle
Mobile Phase A: 5:95 acetonitrile: water with 10 mM ammonium acetate Mobile Phase B: 95:5 acetonitrile: water with 10 mM ammonium acetate Gradient: 0%B, 0-100% B over 3 minutes, then a 0.5-minute hold at 100% B Flow: 1 mL/min
Detection: UV at 220 nm
Temperature: 50 °C
/////////1663477-91-5, BRISTOL-MYERS SQUIBB, TRICYCLOHEXADECAHEXAENE DERIVATIVES, TREATMENT OF HEPATITIS C VIRUS
FC1(F)CCC(CC1)C(=O)N[C@H](c2nc3ccc(cc3n2)c9cc4ccc9CCc5ccc(CC4)c(c5)c6ccc7nc(nc7c6)[C@@H](NC(=O)C8CCC(F)(F)CC8)C(C)(C)C)C(C)(C)C
BMS 986120
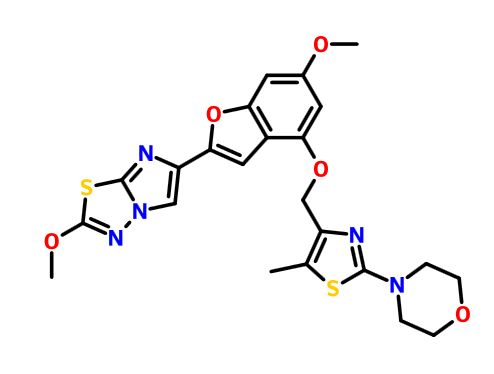
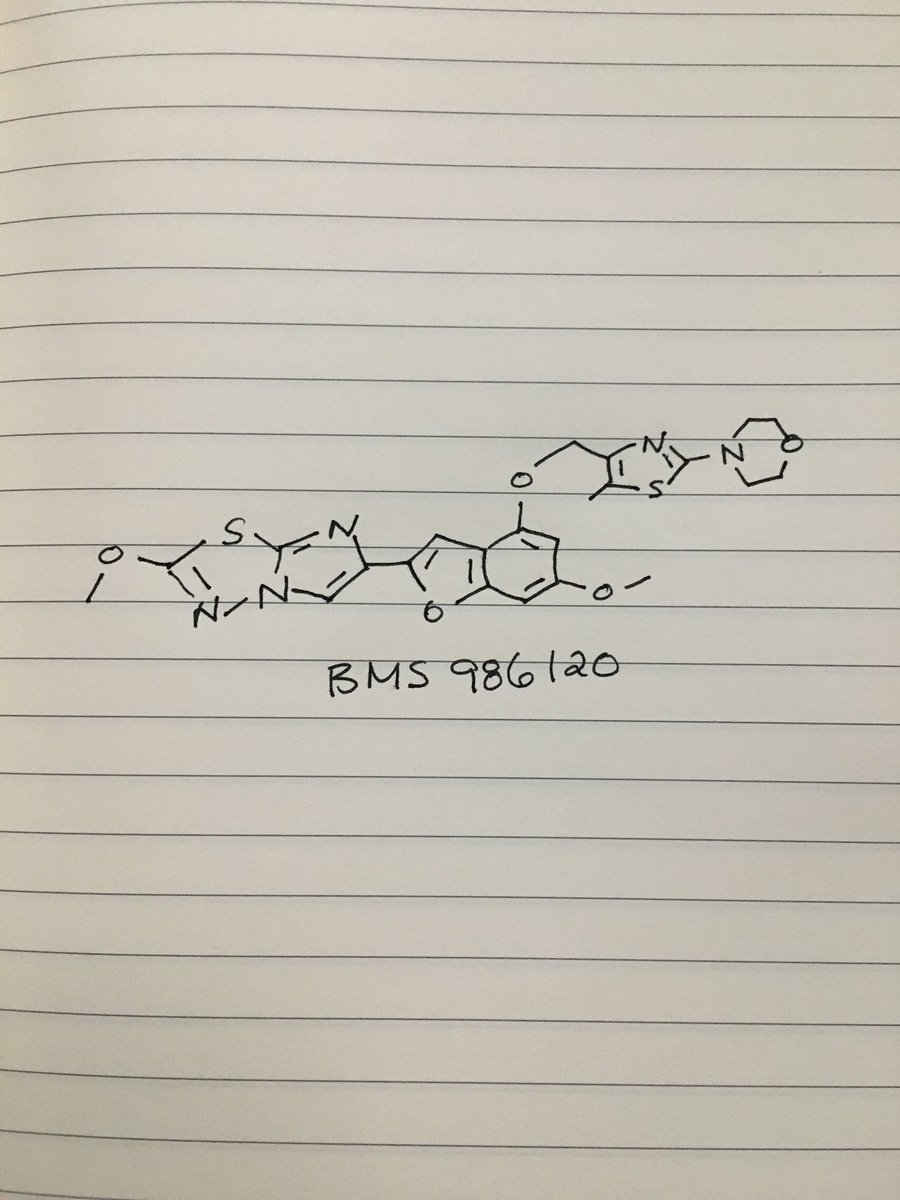 .
.
Picture credit….Bethany Halford
BMS 986120
Originator Bristol-Myers Squibb
Bristol-Myers Squibb Company, Université de Montréal
| Molecular Formula: | C23H23N5O5S2 |
|---|---|
| Molecular Weight: | 513.58922 g/mol |
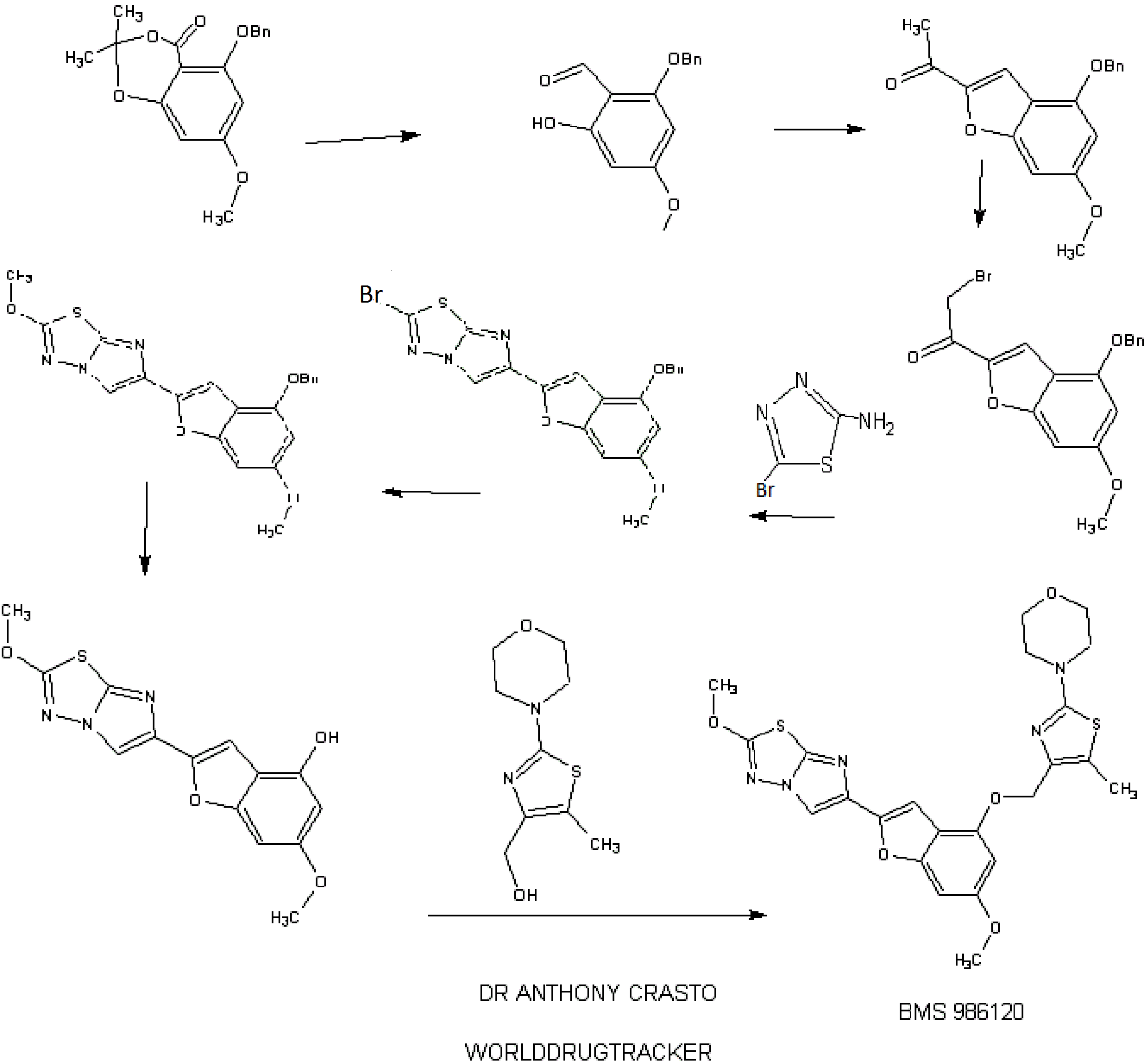
4-[4-[[6-methoxy-2-(2-methoxyimidazo[2,1-b][1,3,4]thiadiazol-6-yl)-1-benzofuran-4-yl]oxymethyl]-5-methyl-1,3-thiazol-2-yl]morpholine
4-(4-(((6-Methoxy-2-(2-methoxyimidazo[2,l-b][l,3,4]thiadiazol-6-yl)benzofuran-4-yl) oxy)methyl)-5-methylthiazol-2-yl)morpholine
Imidazo[2,1-b] -1,3,4-thiadiazole, 2-methoxy-6-[6-methoxy-4-[[5-methyl-2-(4-morpholinyl)-4- thiazolyl]methoxy]-2-benzofuranyl]-
CAS 1478712-37-6
Phase I Thrombosis
- 02 Apr 2015 Bristol-Myers Squibb plans a phase I trial in Thrombosis (In volunteers) in United Kingdom (NCT02439190)
- 01 Aug 2014 Preclinical trials in Thrombosis in USA (PO)
https://clinicaltrials.gov/ct2/show/NCT02208882
https://clinicaltrials.gov/ct2/show/NCT02439190
Class Imidazoles; Small molecules; Thiadiazoles
$BMY antithrombic compound
PATENT
http://www.google.com/patents/WO2013163279A1?cl=en
Thromboembolic diseases remain the leading cause of death in developed countries despite the availability of anticoagulants such as warfarin (COUMADIN®), heparin, low molecular weight heparins (LMWH), synthetic pentasaccharides, and antiplatelet agents such as aspirin and clopidogrel (PLAVIX®).
Current anti-platelet therapies have limitations including increased risk of bleeding as well as partial efficacy (relative cardiovascular risk reduction in the 20 to
30% range). Thus, discovering and developing safe and efficacious oral or parenteral antithrombotics for the prevention and treatment of a wide range of thromboembolic disorders remains an important goal.
Alpha-thrombin is the most potent known activator of platelet aggregation and degranulation. Activation of platelets is causally involved in atherothrombotic vascular occlusions. Thrombin activates platelets by cleaving G-protein coupled receptors termed protease activated receptors (PARs). PARs provide their own cryptic ligand present in the N-terminal extracellular domain that is unmasked by proteolytic cleavage, with subsequent intramolecular binding to the receptor to induce signaling (tethered ligand mechanism; Coughlin, S.R., Nature, 407:258-264 (2000)). Synthetic peptides that mimic the sequence of the newly formed N-terminus upon proteolytic activation can induce signaling independent of receptor cleavage. Platelets are a key player in atherothrombotic events. Human platelets express at least two thrombin receptors, commonly referred to as PARI and PAR4. Inhibitors of PARI have been investigated extensively, and several compounds, including vorapaxar and atopaxar have advanced into late stage clinical trials. Recently, in the TRACER phase III trial in ACS patients, vorapaxar did not significantly reduce cardiovascular events, but significantly increased the risk of major bleeding (Tricoci, P. et al, N. Eng. J. Med., 366(l):20-33 (2012). Thus, there remains a need to discover new antiplatelet agents with increased efficacy and reduced bleeding side effects.
There are several early reports of preclinical studies of PAR4 inhibitors. Lee, F-Y. et al., “Synthesis of l-Benzyl-3-(5′-hydroxymethyl-2′-furyl)indazole Analogues as Novel Antiplatelet Agents”, J. Med. Chem., 44(22):3746-3749 (2001) discloses in the abstract that the compound

58
“was found to be a selective and potent inhibitor or protease-activated receptor type 4 (PAR4)-dependent platelet activation. ”
Compound 58 is also referred to as YD-3 in Wu, C-C. et al, “Selective Inhibition of Protease-activated Receptor 4-dependent Platelet Activation by YD-3”, Thromb. Haemost., 87: 1026-1033 (2002). Also, see Chen, H.S. et al, “Synthesis and platelet activity”, J. Bioorg. Med. Chem., 16: 1262-1278 (2008).
EP1166785 Al and EP0667345 disclose various pyrazole derivatives which are useful as inhibitors of platelet aggregation.\
IB. 5-(Benzyloxy)-7-methoxy-2,2-dimethyl-4H-benzo[d][l,3]dioxin-4-one

A solution of 5-hydroxy-7-methoxy-2,2-dimethyl-4H-benzo[d][l,3]dioxin-4- one (30.00 g, 0.134 mol, see Kamisuki, S. et al, Tetrahedron, 60:5695-5700 (2004) for preparation) in N,N-dimethylformamide (400 mL) was treated with powdered anhydrous potassium carbonate (19.41 g, 0.14 mol) added all at once. The resulting mixture was stirred in vacuo for 10 min. and then flushed with nitrogen. The reaction flask was placed in a water bath (22 °C) and treated with benzyl bromide (24.03 g, 0.14 mol) added dropwise over 15 min. The resulting mixture was then stirred at 22 °C for 18 h (no starting material left by tic). The solid was filtered and washed with N,N- dimethylformamide. The filtrate was evaporated in vacuo and the residual oil was diluted with ethyl acetate (500 mL), washed with cold 0.1 N hydrochloric acid, saturated sodium bicarbonate and brine. After drying over anhydrous magnesium sulfate, evaporation of the solvent gave a thick syrup. Crystallization form ethyl acetate (50 mL) and hexane (150 mL) gave 35.17 g of 5-(benzyloxy)-7-methoxy-2,2-dimethyl-4H- benzo[d][l ,3]dioxin-4-one as large colorless prisms. Chromatography of the mother liquors on silica gel (4 x 13 cm, elution toluene – ethyl acetate 0-5%) gave 6.64 g of additional material to afford a total yield of 41.81 g (99%). HRMS(ESI) calcd for
Ci8Hi905 [M+H]+ m/z 315.1227, found 315.1386. 1H NMR (CDC13, 600 MHz) δ 1.68 (s, 6H), 3.77 (s, 3H), 5.19 (s, 2H), 5.19 (s, 2H), 6.04 (d, J = 2.03 Hz, 1H), 6.15 (d, J = 2.03 Hz, 1H), 7.27 (broad t, 1H), 7.36 (broad t, 2H), 7.52 (broad d, 2H).
1 C. 2-(Benzyloxy)-6-hydroxy-4-methoxybenzaldehyde

A solution of 5-(benzyloxy)-7-methoxy-2,2-dimethyl-4H-benzo[d][l ,3]dioxin- 4-one (Example IB, 6.76 g, 21.5 mmol) in dichloromethane (120 mL) was cooled to -78 °C and treated with 43 mL (64.5 mmol) of a 1.5 M solution of diisobutylaluminum hydride in toluene added dropwise over 20 min. The resulting mixture was then stirred at -78 °C for 3 h. The reaction mixture was quenched by the careful addition of methanol (5 mL) added dropwise over 15 min, followed by IN hydrochloric acid (50 mL) added dropwise over 15 min. The cooling bath was then removed and an additional 150 mL of IN hydrochloric acid was added over 20 min. The mixture was then stirred at 22 °C for 2 h and diluted with dichloromethane (400 mL). The organic phase was collected and the aqueous phase (pH ~1) was extracted with dichloromethane (3 x 50 mL). The combined organic extracts were washed with brine, dried over anhydrous magnesium sulfate and concentrated in vacuo. The residual oil was diluted with tetrahydrofuran (70 mL), treated with 10 mL of 0.1N hydrochloric acid and stirred at 20 °C for 2 h. The reaction mixture was diluted with ethyl acetate (300 mL), washed with brine, dried over anhydrous magnesium sulfate, evaporated in vacuo to give a clear oil. Chromatography on silica gel (4 x 13 cm, elution toluene) gave 4.08 g (73% yield) of the title aldehyde as a clear oil which solidified on standing. LC (Method C): 2.237 min. HRMS(ESI) calcd for Ci5Hi504 [M+H]+ m/z 259.0965, found 259.1153. 1H NMR (CDC13, 600 MHz) δ 3.80 (s, 3H), 5.07 (s, 2H), 5.97 (d, J= 2.1 Hz, 1H), 6.01 (d, J= 2.1 Hz, 1H), 7.3 – 7.4 (m, 5 H), 10.15 (s, 1H), 12.49 (s, 1H).
ID. 1 -(4-(Benzyloxy)-6-methoxybenzofuran-2-yl)ethanone

A solution of 2-(benzyloxy)-6-hydroxy-4-methoxybenzaldehyde (Example 1C, 3.46 g, 13.4 mmol) in N,N-dimethylformamide (50 mL) was treated with powdered anhydrous cesium carbonate (4.58 g, 14.05 mmol) added all at once. The resulting mixture was stirred in vacuo for 10 min. and then flushed with nitrogen. The reaction flask was placed in a water bath (22 °C) and treated with chloroacetone (1.74 g, 18.7 mmol) added dropwise over 5 min. The resulting mixture was then stirred at 22 °C for 18 h (no starting aldehyde left by tic and formation of the intermediate alkylated aldehyde). The solid was filtered and washed with N,N-dimethylformamide. The filtrate was evaporated in vacuo and the residual oil was diluted with ethyl acetate (300 mL), washed with cold 0.1 N hydrochloric acid, saturated sodium bicarbonate and brine. After drying over anhydrous magnesium sulfate, evaporation of the solvent gave a thick syrup. This syrup was diluted with tetrahydrofuran (50 mL) and ethyl acetate (50 mL), treated p- toluenesulfonic acid monohydrate (0.2 g) and stirred at 20 °C for 1 h (tic indicated complete cyclization of the intermediate alkylated aldehyde to the benzofuran). The reaction mixture was diluted with ethyl acetate (300 mL), washed with saturated sodium bicarbonate and brine. After drying over anhydrous magnesium sulfate, evaporation of the solvent gave a thick syrup. Chromatography on silica gel (4 x 12 cm, elution toluene – ethyl acetate 2-4%) gave 3.51 g (88% yield) of the title benzofuran as a yellow solid. Recrystallization from ethyl acetate (10 mL) and hexane (20 mL) gave the title material as large yellow prisms (3.15 g). LC (Method D): 2.148 min. HRMS(ESI) calcd for Ci8Hiv04 [M+H]+ m/z 297.1121, found 297.1092. 1H NMR (CDC13, 600 MHz) δ 2.51 (s, 3H), 3.82 (s, 3H), 5.13 (s, 2H), 6.37 (d, J= 1.77 Hz, 1H), 6.63 (broad s, 1H), 7.34 (broad t, 1H), 7.39 (broad t, 2H), 7.44 (broad d, 2H), 7.55 (d, J = 0.7 Ηζ,ΙΗ). IE. l-(4-(Benzyloxy)-6-methoxybenzofuran-2-yl)-2-bromoethanone

A 250-mL, three-necked flask is equipped with a magnetic stirring bar and purged with a nitrogen atmosphere was charged with anhydrous tetrahydrofuran (25 mL) followed by 9.3 mL (9.3 mmol) of a 1M solution of lithium bis(trimethylsilyl)amide in tetrahydrofuran. The mixture was cooled to -78 °C and treated with a solution of l-(4- (benzyloxy)-6-methoxybenzofuran-2-yl)ethanone (Example ID, 2.40 g, 8.1 mmole) in tetrahydrofuran (20 mL) added dropwise over 10 min. The resulting mixture was then stirred at -78 °C for 45 min. Then chlorotrimethylsilane (1.18 mL, 9.31 mmol) was added dropwise over 5 min and the resulting solution was stirred at -78 °C for another 20 min. The cooling bath was then removed and the mixture is allowed to warm to room temperature over 30 min. The reaction mixture was then quenched by addition to a cold solution of ethyl acetate (200 mL), saturated sodium bicarbonate (30 mL) and ice. The organic phase was rapidly dried over anhydrous magnesium sulfate (magnetic stirring) and evaporated in vacuo to give the silyl enol ether as an oil which is co-evaporated with toluene (20 mL). The silyl enol ether was then dissolved in dry tetrahydrofuran (40 mL), cooled to -20 °C and treated with solid sodium bicarbonate (0.10 g) followed by N- bromosuccinimide (1.44 g, 8.1 mmol) added in small portions over 15 min. The reaction mixture was allowed to warm to 0 °C over 2h and then quenched by addition of ethyl acetate (300 mL) and saturated sodium bicarbonate. The organic phase was washed with brine, dried over anhydrous magnesium sulfate and evaporated to give an orange oil. Chromatography on silica gel (4 x 12 cm, elution toluene – ethyl acetate 0-5%) gave 2.62 g (86% yield) of the title bromomethylketone as a yellow solid. Recrystallization from ethyl acetate (10 mL) and hexane (20 mL) gave yellow prisms (2.30 g). LC (Method E): 1.977 min. HRMS(ESI) calcd for Ci8Hi6Br04 [M+H]+ m/z 375.0226, found 375.0277. 1H NMR (CDCls, 600 MHz) δ 3.84 (s, 3H), 4.33 (s, 2H), 5.14 (s, 2H), 6.38 (d, J = 1.76 Hz, 1H), 6.64 (broad s, 1H), 7.35 (broad t, 1H), 7.40 (broad t, 2H), 7.44 (broad d, 2H), 7.70 (s, 1H). 1 EE. 1 -(4-(Benzyloxy)-6-methoxybenzofuran-2-yl)-2-chloroethanone

Benzyltrimethylammonium dichloroiodate (117 g, 169 mmol) was added to a solution of l-(4-(benzyloxy)-6-methoxybenzofuran-2-yl)ethanone (Example ID, 50 g, 170 mmol) in THF (500 mL) in a 1 L multineck round bottom flask under nitrogen atmosphere. The reaction mixture was stirred at RT for 6 h, cooled to 0 °C and quenched with 10% NaHCC”3 solution. The organic layer was washed with 1 M sodium thiosulphate solution, water, and brine, dried over Na2S04, and concentrated in vacuo (bath temperature <45 °C). The residue was triturated with 5% EtOAc in pet. ether and dried to obtain the title chloromethylketone as a pale yellow solid (48 g, 130 mmol, 78%). 1H NMR (300 MHz, DMSO-d6) δ 3.84-3.82 (d, J =4.5Hz, 3H) 4.98 (s, 2H), 5.27(s, 2H), 6.62 -6.61 (d, J = 1.8Hz, 1H), 6.92-6.93 (m, 1H), 7.54-7.36 (m, 5H), 8.10-8.09 (d, J = 3Hz, 1H); MS m/z: [M+H]+ 331.0. IF. 6-(4-(Benzyloxy)-6-methoxybenzofuran-2-yl)-2-bromoimidazo[2, 1 – b] [ 1 ,3 ,4]thiadiazole

A mixture of l-(4-(benzyloxy)-6-methoxybenzofuran-2-yl)-2-bromoethanone (Example IE, 3.00 g, 8.0 mmol) and 5-bromo-l,3,4-thiadiazol-2-amine (1.65 g, 9.16 mmol) in isopropanol (100 mL) was heated in a pressure flask equipped with a magnetic stirring bar at 78-80 °C for 18 h (homogeneous after 20 min and then formation of a precipitate after 2 h). The cooled mixture is then transferred into five 20 mL microwave vials and then heated in a microwave apparatus to 150 °C for 30 min. Each vial was then diluted with dichloromethane (250 mL) washed with saturated sodium bicarbonate (25 mL) and brine (25 mL), dried over anhydrous magnesium sulfate. The fractions were combined and concentrated in vacuo. Chromatography of the orange-brown residual solid on silica gel (4 x 10 cm, slow elution with dichloromethane due to poor solubility) gave 2.96 g of the title imidazothiadiazole contaminated with some l-(4-(benzyloxy)-6- methoxybenzofuran-2-yl)ethanone. The solid material was triturated with ethyl acetate (20 mL), filtered, washed with ethyl acetate (10 ml) and dried in vacuo to give 2.34 g (64% yield) of pure title imidazothiadiazole as an off white solid which is used as such for the next step. LC (Method E): 2.188 min. HRMS(ESI) calcd for C2oHi5BrN303S [M+H]+ m/z 456.00175, found 456.00397. 1H NMR (CDC13, 600 MHz) δ 3.82 (s, 3H), 5.16 (s, 2H), 6.38 (d, J= 1.67 Hz, 1H), 6.66 (broad s, 1H), 7.15 (s, 1H), 7.31 (broad t, 1H), 7.38 (broad t, 2H), 7.45 (broad d, 2H), 8.02 (s, 1H).
Alternatively, Example IF, 6-(4-(benzyloxy)-6-methoxybenzofuran-2-yl)-2- bromoimidazo[2,l-b][l,3,4]thiadiazole, was prepared as follows:
A 1000-mL, three-necked flask equipped with a magnetic stirring bar and purged with a nitrogen atmosphere was charged with dry NMP (200 mL) followed by 1- (4-(benzyloxy)-6-methoxybenzofuran-2-yl)-2-chloroethanone (Example 1EE, 50 g, 150 mmol) and 5-bromo-l,3,4-thiadiazol-2-amine (27.2 g, 151 mmol). The resulting mixture was stirred at 80 °C for 8h. TLC (8:2 dichloromethane/pet. ether) and LC/MS showed intermediate uncyclized material (m/z 476) and the reaction mixture was stirred at 120 °C for 3h. The reaction mixture was cooled to RT, quenched with water and extracted with EtOAc (3X). The combined organic layers were washed with brine, dried over Na2S04, and concentrated in vacuo. The thick brown residue was purified by silica gel chromatography (0 to 100% dichloromethane in pet. ether) to give a brown solid. This material was triturated with EtOAc and dried to obtain the title imidazothiadiazole (24 g, 50 mmol, 33%>) as a light brown solid. (See the procedure set forth above for analytical data).
1 G. 6-(4-(Benzyloxy)-6-methoxybenzofuran-2-yl)-2-methoxyimidazo[2, 1 – b][l,3,4]thiadiazole

A solution of 6-(4-(benzyloxy)-6-methoxybenzofuran-2-yl)-2- bromoimidazo[2,l-b][l,3,4]thiadiazole (Example IF, 2.30 g, 5.04 mmol) in a mixture of dichloromethane (180 mL) and methanol (45 mL) was treated at 22 °C with 4.2 mL of a 25 wt.% solution of sodium methoxide in methanol (0.2 mmol) added in one portion. More methanol (45 mL) was added and the mixture was stirred for 1 h. The reaction mixture was quenched by the addition of 25 mL of IN hydrochloric acid followed by 20 ml of saturated sodium bicarbonate. The solvent was evaporated under reduced pressure and the residue was diluted with dichloromethane (400 mL), washed with brine, dried over anhydrous magnesium sulfate and evaporated in vacuo. Chromatography of the residue on silica gel (3 x 10 cm, elution with dichloromethane – ethyl acetate 0-4%) gave 1.70 g (83% yield) of the title compound as a white solid. This material was recrystallized from ethyl acetate (30 mL per gram, 80% recovery) to give white needles. LC (Method
D): 2.293 min. HRMS(ESI) calcd for C21H18N3O4S [M+H]+ m/z 408.1013, found 408.1024. 1H NMR (CDC13, 600 MHz) δ 3.81 (s, 3H), 4.18 (s, 3H), 5.16 (s, 2H), 6.37 (d, J = 1.75 Hz, 1H), 6.67 (broad s, 1H), 7.07 (s, 1H), 7.31 (broad t, 1H), 7.37 (broad t, 2H), 7.45 (broad d, 2H), 7.81 (s, 1H).
1H. 6-Methoxy-2-(2-methoxyimidazo[2,l-b][l,3,4]thiadiazol-6-yl)benzofuran-4-ol

A mixture of 6-(4-(benzyloxy)-6-methoxybenzofuran-2-yl)-2- methoxyimidazo[2,l-b][l,3,4]thiadiazole (Example 1G, 1.250 g, 3.06 mmol) and pentamethylbenzene (3.17 g, 21.4 mmol) in dichloromethane (200 mL) was cooled to -78 °C under a nitrogen atmosphere and then treated immediately (to avoid crystallization) with 8 mL (8 mmol) of a 1 M solution of boron trichloride in dichloromethane added dropwise over 3 min. The resulting mixture was stirred at -78 °C for 1 h. The reaction mixture was then quenched by the addition of a solution of sodium bicarbonate (6 g) in water (100 mL) added in one portion. The cooling bath was removed and the resulting mixture was stirred at room temperature for 1 h. The solid formed was filtered, washed successively with water (50 m) and dichloromethane (50 mL). The filter cake was allowed to soak with anhydrous ethanol (15 ml) and then sucked dry. The white solid obtained was then dried under vacuum for 24 h to give 0.788 g (80%> yield) of pure title material (> 95% by hplc). The combined filtrate and washings were diluted with dichloromethane (600 mL) and stirred in a warm water bath till the organic phase was clear with no apparent solid in suspension. The organic phase was collected, dried over anhydrous magnesium sulfate and rapidly filtered while still warm. The filtrate was evaporated and the residue (product and pentamethylbenzene) was triturated with toluene (20 mL), the solid collected and washed with toluene (20 mL) to give 0.186 g (19% yield, 99% combined yield) of title material as a tan solid (> 95% by hplc). LC (Method E): 1.444 min. HRMS(ESI) calcd for C14H12N3O4S [M+H]+ m/z 318.0543, found 318.0578. 1H NMR (DMSO-de, 600 MHz) 5 3.71 (s, 3H), 4.16 (s, 3H), 6.21 (d, J = 1.87 Hz, 1H), 6.61 (broad s, 1H), 6.95 (s, 1H), 8.29 (s, 1H), 9.96 (s, 1H).
Example 94
4-(4-(((6-Methoxy-2-(2-methoxyimidazo[2,l-b][l,3,4]thiadiazol-6-yl)benzofuran-4-yl) oxy)methyl)-5-methylthiazol-2-yl)morpholine

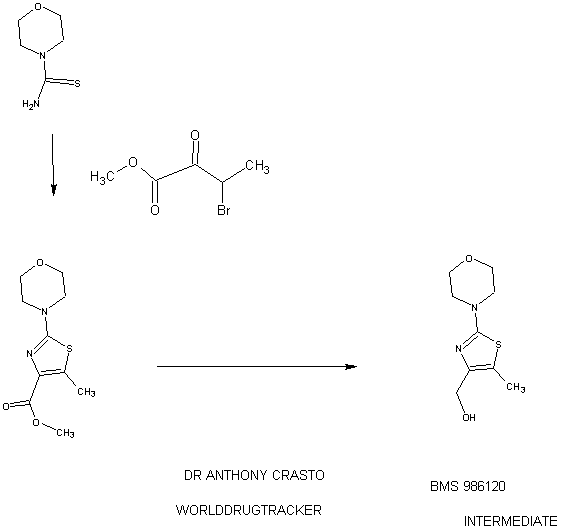
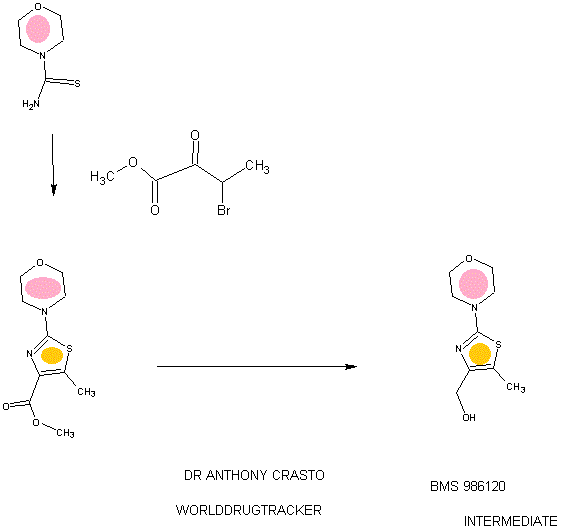
94 A. Methyl 5-methyl-2-morpholinothiazole-4-carboxylate [00258] A solution of methyl 2-bromo-5-methylthiazole-4-carboxylate (2.80 g, 11.86 mmol) and morpholine (4.5 mL, 51.7 mmol) in THF (10 mL) was heated at reflux under nitrogen for 18 h. The volatiles were then removed under reduced pressure and the crude product was purified on the ISCO using a REDISEP® 40 g column (0 to 40% EtOAc- DCM), to give the title compound (2.20 g, 77%) as a yellow solid. LCMS (APCI): calcd for CioHisNzOsS [M+H]+ m/z 243.07, found 243.1. 1H NMR (CDC13, 400 MHz) δ ppm: 3.89 (s, 3H), 3.77-3.83 (m, 4H), 3.41-3.47 (m, 4H), 2.64 (s, 3H). [00259] Alternatively, Example 94A, methyl 5-methyl-2-morpholinothiazole-4- carboxylate, was prepared as follows:
94AA. Methyl 3-bromo-2-oxobutanoate

A 5L 4-neck round bottom flask equipped with a mechanical stirrer, temperature thermocouple, condenser and a 1L addition funnel, was charged copper(II) bromide (962 g, 4310 mmol) and ethyl acetate (2 L). A solution of methyl 2-ketobutyrate (250 g, 2150 mmol) in CHC13 (828 mL) was added dropwise. A scrubber (400 mL 1 N NaOH) was connected and the reaction mixture was heated to reflux (75 °C). The reaction started as a dark green color and as heating progressed, it became a light green with a white precipitate forming. NMR after one hour at reflux indicated that the reaction was complete. The reaction was cooled to RT and filtered through a pad of CELITE®. The filtrate was concentrated to an oil, dissolved in methylene chloride (500 mL) and filtered again through CELITE®. The filtrate was then passed through a pad of silica gel and eluted with ethyl acetate. Concentration of the filtrate provided the title bromoketoester (399 g, 2040 mmol, 95%) as a yellow oil. 1H NMR (400MHz, CDC13) δ 5.18 (q, J = 6.7 Hz, 1H), 3.94 (s, 3H), 1.83 (d, J = 6.8 Hz, 3H). 94AAA. Morpholine-4-carbothioamide

To a solution of morpholine (199 g, 2280 mmol) in CHC13 (1 L) was added isothiocyanatotrimethylsilane (150 g, 1140 mmol) dropwise. A white precipitate formed almost immediately, and the reaction was stirred for 1 h at RT. The reaction was then filtered and the resulting solid was washed with additional CHC13 and dried in vacuo to give the title thiourea as a white solid. (137 g, 937 mmol, 82%). 1H NMR (400MHz, DMSO-de) δ 3.81 – 3.71 (m, 2H), 3.17 – 3.08 (m, 2H).
94 A. Methyl 5-methyl-2-morpholinothiazole-4-carboxylate

To a solution of morpholine-4-carbothioamide (Example 94 AAA, 175 g, 1200 mmol) in methanol (500 mL) was charged methyl 3-bromo-2-oxobutanoate (Example 94AA, 233 g, 1200 mmol). The reaction was then heated to reflux for 1 hour, cooled to RT, and filtered. The filtrate was concentrated and the crude product was purified on by silica gel chromatography. The title thiazole (206g, 850 mmol, 71%) was isolated as a yellow oil. (See the procedure set forth above for analytical data).
(5-Methyl-2-morpholinothiaz l-4-yl)methanol

The compound was prepared according to the protocol described for Example 92B. The crude product was purified on the ISCO using a REDISEP® Gold 24 g column (0 to 50% EtOAc-DCM) to give the title compound as a white solid (0.086 g, 51%). LCMS (APCI): calcd for C9Hi5N202S [M+H]+ m/z 215.08, found 215.1. 1H NMR (CDCI3, 400 MHz) δ ppm: 4.48 (d, J= 4.7 Hz, 2H), 3.77-3.83 (m, 4H), 3.37-3.43 (m, 4H), 2.30 (t, J= 4.7 Hz, 1H), 2.28 (s, 3H).
Example 94. 4-(4-(((6-Methoxy-2-(2-methoxyimidazo[2, 1 -b] [ 1 ,3,4]thiadiazol-6-yl) benzofuran-4-yl)oxy)methyl)-5 -methylthiazol-2-yl)morpholine

The title compound was prepared according to the protocol described for Example 86. The crude product was purified on the ISCO using a REDISEP® 4 g column (0 to 40% EtOAc-DCM) and the obtained solid was suspended in MeOH, sonicated, filtered and dried to give the title compound as an off-white solid (0.094 g, 53%). LC (Method C): 2.314 min. HRMS(ESI): calcd for C23H24N505S2 [M+H]+ m/z 514.122, found 514.126. 1H NMR (CDC13, 400 MHz) δ ppm: 7.83 (s, 1H), 7.06 (d, J = 0.8 Hz, 1H), 6.69 (d, J= 0.8 Hz, 1H), 6.50 (d, J= 2.0 Hz, 1H), 5.05 (s, 2H), 4.21 (s, 3H), 3.85 (s, 3H), 3.78- 3.84 (m, 4H), 3.39- 3.46 (m, 4H), 2.37 (s, 3H).
ABSTRACT
251st Am Chem Soc (ACS) Natl Meet (March 13-17, San Diego) 2016, Abst MEDI 263


| Patent ID | Date | Patent Title |
|---|---|---|
| US2015094297 | 2015-04-02 | IMIDAZOTHIADIAZOLE AND IMIDAZOPYRAZINE DERIVATIVES AS PROTEASE ACTIVATED RECEPTOR 4 (PAR4) INHIBITORS FOR TREATING PLATELET AGGREGATION |
////////BMS 986120, phase 1, Bristol-Myers Squibb , Imidazoles, Small molecules, Thiadiazoles, 1478712-37-6
c1(sc2nc(cn2n1)c3cc4c(cc(cc4o3)OC)OCc5nc(sc5C)N6CCOCC6)OC
CC1=C(N=C(S1)N2CCOCC2)COC3=C4C=C(OC4=CC(=C3)OC)C5=CN6C(=N5)SC(=N6)OC
BMS 955829

(4R,5R)-5-(2,5-difluorophenyl)-4-(5-(phenylethynyl)pyridin-3-yl)oxazolidin-2-one
(4R,5R)-5(2,5-Difluorophenyl)-4-(5-(phenylethynyl)-3-pyridinyl)-1,3-oxazolidin-2-one
(4R,5R)-5-(2,5-difluorophenyl)- 4-(5-(phenylethynyl)pyridin-3-yl)oxazolidin-2-one.
cas 1375751-08-8
Chemical Formula: C22H14F2N2O2
Exact Mass: 376.1023
Bristol-Myers Squibb Company INNOVATOR
BMS-955829 is a Positive allosteric modulators (PAMs). BMS-955829 shows high functional PAM potency, excellent mGluR5 binding affinity, low glutamate fold shift, and high selectivity for the mGluR5 subtype. BMS-955829 is a potent mGluR5 PAM (EC50 = 2.6 ± 1.0 nM; n = 6), devoid of inherent mGluR5 agonist activity (EC50 > 30μM). The measured binding Ki of BMS-955829 was found to be 1.6 nM, which was in good agreement with its functional potency.

SYNTHESIS AND INTERMEDIATES…….https://www.google.co.in/patents/WO2012064603A1?cl=en
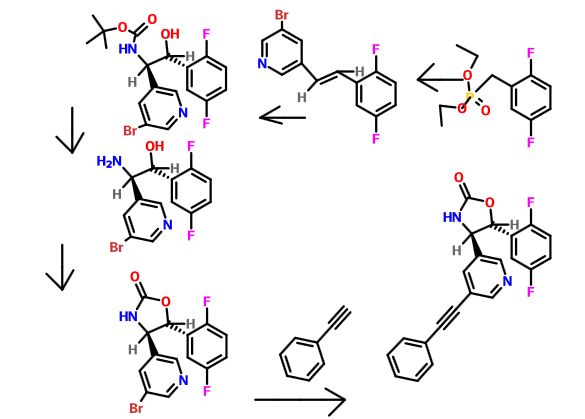
Intermediate 73

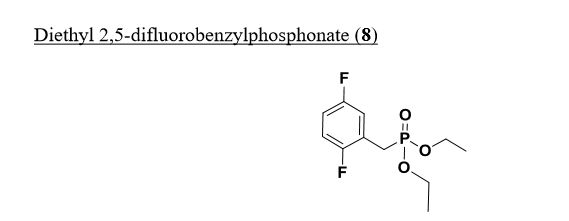
Diethyl 2,5-difluorobenzylphosphonate. A mixture of 2-(bromomethyl)-l,4- difluorobenzene (3 g, 14.49 mmol) and triethyl phosphite (7.72 ml, 43.5 mmol) was heated to 160 °C with stirring for 4 hours, cooled to ambient temperature and concentrated under high vacuum to remove most triethyl phosphite. The resulting residue was purified by column chromatography (20% to 30 % EtO Ac/Toluene) providing diethyl 2,5-difluorobenzylphosphonate (3.76 g, 13.52 mmol, 93 % yield) as colorless oil. ¾ NMR (500MHz, DMSO-d6) δ 7.30 – 7.10 (m, 3H), 4.05 – 3.91 (m, 4H), 3.31 – 3.20 (m, 2H), 1.18 (t, J=7.0 Hz, 6H). MS Anal. Calcd. for [M+H]+ CiiHieFzOsP: 265.2; found 265.3.

Intermediate 74

(E)-3-Bromo-5-(2,5-difluorostyryl)pyridine. To a stirred solution of diethyl 2,5-difluorobenzylphosphonate (63.5 g, 240 mmol) and 5-bromonicotinaldehyde (50.7 g, 264 mmol) in tetrahydrofuran (1923 ml) was added potassium tert-butoxide in tetrahydrofuran (312 ml, 312 mmol) at -10 °C. After three hours, the reaction mixture was allowed to warm to ambient temperature and stirring was continued for another 16 hours at which time the reaction mixture was diluted with ether (800 mL) and washed with H2O. The organic layer was dried over anhydrous magnesium sulfate, filered and concentrated to provide a yellow wax to which was added 300 mL of hexane and after sonication filtered to provide (is)-3-bromo-5-(2,5- difluorostyryl)pyridine (54 g, 173 mmol, 72.1%) as a white solid. XH NMR
(500MHz, DMSO-d6) δ 8.78 (d, J=1.8 Hz, IH), 8.63 (d, J=2.1 Hz, IH), 8.44 (t, J=2.0 Hz, IH), 7.67 (ddd, J=9.4, 6.0, 3.2 Hz, IH), 7.56 – 7.48 (m, IH), 7.46 – 7.40 (m, IH), 7.34 (td, J=9.6, 4.6 Hz, IH), 7.24 (tt, J=8.3, 3.6 Hz, IH). MS Anal. Calcd. for [M+H]+ Ci3H9BrF2N: 296.0; found 298.1


Intermediate 75

Tert-butyl (lR,2R)-l-(5-bromopyridin-3-yl)-2-(2,5-difluorophenyl)-2- hydroxyethylcarbamate. A solution of tert-butyl carbamate (4.18 g, 35.0 mmol) in propanol (39 ml) was sequentially treated with sodium hydroxide (1.376 g, 34.4 mmol) in water (72 ml) and tert-butyl hypochlorite (3.88 ml, 34.4 mmol). After 5 min of stirring, the reaction mixture was cooled to 0 °C. A solution of
(DHQD)2PHAL (0.555 g, 0.677 mmol) in propanol (39 ml), a solution of (E)-3- bromo-5-(2,5-difluorostyryl)pyridine (3.34 g, 11.28 mmol) in propanol (68 ml) , and potassium osmate dihydrate (0.166 g, 0.451 mmol) were sequentially added. The reaction mixture was stirred for three additional hours at 0 °C, warmed to ambient temperature and after an additional 16 hours the light yellow homogenous solution was quenched with saturated aqueous sodium sulfite (100 mL). The aqueous phase was extracted with ethyl acetate( 2 X 50 mL), the combined organic phases were washed with brine (100 mL), dried over anhydrous magnesium sulfate and concentrated to afford a residue which was purified via column chromatography (25% to 40 % EtO Ac/Hex) to provide tert-butyl (7R,2R)-l-(5-bromopyridin-3-yl)-2- (2,5-difluorophenyl)-2-hydroxyethylcarbamate (2.2991 g, 5.09 mmol, 45.1 % yield) as an optically enriched mixture of enantiomers. XH NMR (500MHz, DIVISOR) δ 8.56 (d, J=1.8 Hz, IH), 8.40 (s, IH), 8.03 (s, IH), 7.52 (d, J=9.5 Hz, IH), 7.25 (br. s., IH), 7.10 (t, J=5.6 Hz, 2H), 5.89 (d, J=4.9 Hz, IH), 5.03 (t, J=5.0 Hz, IH), 4.83 (dd, J=8.9, 5.2 Hz, IH), 1.40 – 1.34 (m, 9H), MS Anal. Calcd. for [M+H]+
Ci8H2oBrF2 203: 429.1; found 431.3.
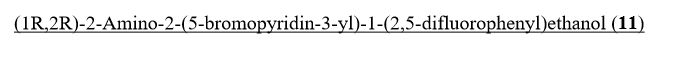
Intermediate 77

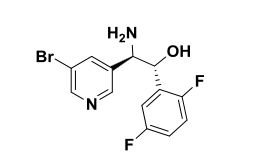
(lR,2R)-2-Amino-2-(5-bromopyridin-3-yl)-l-(2,5-difluorophenyl)ethanol To a stirred solution of tert-butyl tert-butyl (7R,2R,)-l-(5-bromopyridin-3-yl)-2-(2,5- difluorophenyl)-2-hydroxyethylcarbamate (2.30 g, 5.09 mmol) in methylene chloride (30 mL) was added HC1 in dioxane (30 ml, 120 mmol). The reaction mixture was placed in an oil bath set to 50 °C. After three hours, the reaction mixture was concentrated providing (7R,2R^-2-amino-2-(5-bromopyridin-3-yl)-l-(2,5- difluorophenyl)ethanol 2HC1 salt (2.10 g, 4.97 mmol, 98 % yield) as an optically enriched yellow wax. XH NMR (500MHz, DMSO-d6) δ 8.95 (d, J=3.7 Hz, 2H), 8.64 (d, J=2.4 Hz, 1H), 8.45 (d, J=1.5 Hz, 1H), 8.31 (t, J=2.0 Hz, 1H), 7.47 – 7.09 (m, 3H), 7.04 (td, J=9.2, 4.4 Hz, 1H), 5.29 (d, J=9.2 Hz, 1H), 4.57 (dd, J=9.0, 5.3 Hz, 1H). Anal. Calcd. for [M+H]+ Ci3H12BrF2N20: 329.0; found 331.2.

Intermediate 78

(4R,5R)-4-(5-Bromopyridin-3-yl)-5-(2,5-difluorophenyl)oxazotidin-2-one. To optically enriched (7R,2R)-2-amino-2-(5-bromopyridin-3-yl)-l-(2,5- difluorophenyl)ethanol, 2 HC1 (2.019 g, 4.82 mmol) in tetrahydrofuran (98 ml) was added diisopropylethylamine (2.95 ml, 16.87 mmol) and the resultant solution was stirred for ten mintues at ambient temperature, cooled to 0 °C and
carbonyldiimidazole (1.094 g, 6.75 mmol) was added. After an additional three hours at 0 °C the reaction mixture was warmed to ambient temperature and allowed to stir for another 16 hours. 2M ¾ in methanol (5ml) was added and after ten mintues the suspension was filtered and concentrated to a pink oil which was purified by column chromatography (25% to 40 % EtO Ac/Hex) providing (4R,5R)-4-(5- bromopyridin-3-yl)-5-(2,5-difluorophenyl)oxazolidin-2-one (1.353 g, 3.62 mmol, 75 % yield) as an optically enriched white solid. ¾ NMR (500MHz, DMSO-d6) δ 8.80 – 8.68 (m, 1H), 8.55 (d, J=2.1 Hz, 2H), 8.16 (t, J=2.1 Hz, 1H), 7.46 – 7.28 (m, 3H), 5.71 – 5.58 (m, 1H), 5.02 (d, J=6.7 Hz, 1H). MS Anal. Calcd. for [M+H]+ Ci4H10BrF2 2O2: 355.0; found 357.2.
Intermediate 79

(4R,5R)-4-(5-Bromopyridin-3-yl)-5-(2,5-difluorophenyl)oxazotidin-2-one. Method – 2 A mixture of tert-butyl ((lR,2R)-l-(54oromopyridin-3-yl)-2-(2,5- difluorophenyl)-2-hydroxyethyl)carbamate and tert-butyl ((lR,2R)-2-(5- bromopyridin-3-yl)-l-(2,5-difluorophenyl)-2-hydroxyethyl)carbamate (about 6: 1 ratio) (101 g, 236 mmol) in tetrahydrofuran (590 mL) was cooled to -7 °C with a methanol/ice bath. To this mixture was added a solution of 1 M potassium tert- butoxide in tetrahydrofuran (590 mL, 590 mmol) via an addition funnel while maintaining the internal temperature < 3 °C. The reaction mixture was stirred with a cooling bath for 30 min and then allowed to warm up to room temperature. After 20 h, the reaction was deemed complete by LC/MS. The reaction mixture was concentrated to dryness to give crude product. Another identical scale reaction was performed. The crude products of the two batches were combined to work up together. They were treated with ethyl acetate (1.75 L) and water (1.75 L). The layers were separated. The organic layer was washed with brine (1.75 L), dried (sodium sulfate), and evaporated to give 161.5 g of crude product as a brown solid. This was purified by ISCO to give 67.1 g (42% yield). LC/MS (ES+) 355/357 (M+H, 100; Br isotope pattern); XH NMR (400MHz, CDCl3) δ 8.75 (d, J=2.2 Hz, 1H), 8.53 (d, J=1.8 Hz, 1H), 7.97 (t, J=2.0 Hz, 1H), 7.29 – 7.23 (m, 1H), 7.18 – 7.09 (m, 2H), 6.40 (s, 1H), 5.56 (d, J=5.7 Hz, 1H), 4.84 (d, J=5.5 Hz, 1H); Calcd for
Ci4H9N2BrF202: C, 47.34; H, 2.55; N, 7.86; Br, 22.50; F, 10.69. Found: C, 47.29; H, 2.61; N, 7.87; Br, 22.40; F, 10.37. Note: Chiral HPLC of the above sample showed 4.7% of the enantiomer. The (4S, 55) enantiomer can be purged by recrystallization from methanol to give > 99.9 ee with 67% recovery.

WO2012064603
Scheme 1.

Pd(0)/Cu(l)/ TBAF Scheme 2.

cheme 4.

R’ = H, alkyl
Scheme 8.


cheme 11.

Scheme 12.


Scheme 14.

Scheme 15.

R” = H, alkyl R” = alkyl
cheme 16.

R’ = alky I
R” = alkyl

Scheme 17.

R’ = H, alkyl
R” = H, alkyl
Scheme 18.
R’ = H, alkyl R’ = H, alkyl

P T/US2011/059339

COMPD IS 185

Example 185

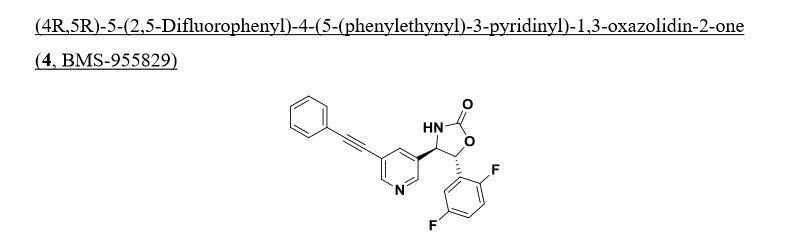
(4R, 5R)-5-(2, 5-difluorophenyl)-4-(5-(phenylethynyl)-3-pyridinyl)-l, 3-oxazolidin-2- one.
To a stirred solution of optically enriched (4R,5R)-4-(5-bromopyridin-3-yl)-5- (2,5-difluorophenyl)oxazolidin-2-one (1.25 g, 3.25 mmol) in triethylamine (70 mL) was added ethynylbenzene (0.592 mL, 5.28 mmol), copper(I) iodide (67 mg, 0.352 mmol), and triphenylphosphine (653 mg, 2.464 mmol). Nitrogen was bubbled through the mixture for 10 mintues before adding dichlorobis(triphenylphosphine)- palladium(II) (202 mg, 0.282 mmol) with continued nitrogen gas bubbling. After an additional 10 mintues the reaction mixtrue was heated to reflux for 16 hours, cooled to ambient temperature, diluted with EtOAc, washed with water (3X), brine, dried over magnesium sulfate, and concentrated in vacuo. Column chromatography (25% – -> 40% EtO Ac/Hex) provided optically enriched (4R,5R)-5-(2,5-difluorophenyl)-4- (5-(phenylethynyl)pyridin-3-yl)oxazolidin-2-one which was separated by chiral SFC chromatography (Chiralcel OJ-H preparative column, 30 x 250mm, 5μιη, Mobile Phase: 40% MeOH (0.1%DEA) in C02 @ 150Bar, Temp: 35°C, Flow rate: 70.0 mL/min. for 16 min, UV monitored @ 280 nM . tR = 9.23 min) to provide (1.38 g, 2.99 mmol, 85 % yield) of pure single enantiomer (4R,5R)-5-(2,5-difluorophenyl)- 4-(5-(phenylethynyl)pyridin-3-yl)oxazolidin-2-one.
‘H NMR (500 MHz, DMSO-i¾) δ ppm 8.77 (d, J=2.21 Hz, 1 H) 8.57 (s, 1 H) 8.56 (d, J=2.20 Hz, 1 H) 8.07 (t, J=2.05 Hz, 1 H) 7.58 – 7.66 (m, 2 H) 7.44 – 7.52 (m, 3 H) 7.39 – 7.45 (m, 1 H) 7.28 – 7.39 (m, 2 H) 5.67 (d, J=6.62 Hz, 1 H) 5.04 (d, J=6.62 Hz, 1 H). 13C NMR (126 MHz,
DMSO-i¾) δ ppm 157.28; 157.24 (d, J=240.70 Hz) 155.92 (d, J=245.20 Hz) 151.63; 147.70; 136.78; 135.02; 131.57; 129.43; 128.89; 126.63 (dd, J=14.99, 7.72 Hz) 121.51; 119.47; 117.83 (dd, J=23.60, 9.10 Hz) 117.50 (dd, J=24.50, 8.20 Hz); 114.60 (dd, J=26.34, 4.54 Hz); 92.86; 85.76; 78.12; 59.43;
LCMS (ESI) m/z calcd for C22H15F2N202: 377.11, found 377.20[M+H]+;
HRMS (ESI) m/z calcd for
C22H15F2N202: 377.1096, found 377.1096 [M+H]+.
SEE
WO2015054103, OXAZOLIDINONES AS MODULATORS OF MGLUR5
PAPER

Positive allosteric modulators (PAMs) of the metabotropic glutamate receptor subtype 5 (mGluR5) are of interest due to their potential therapeutic utility in schizophrenia and other cognitive disorders. Herein we describe the discovery and optimization of a novel oxazolidinone-based chemotype to identify BMS-955829 (4), a compound with high functional PAM potency, excellent mGluR5 binding affinity, low glutamate fold shift, and high selectivity for the mGluR5 subtype. The low fold shift and absence of agonist activity proved critical in the identification of a molecule with an acceptable preclinical safety profile. Despite its low fold shift, 4 retained efficacy in set shifting and novel object recognition models in rodents.
Discovery and Preclinical Evaluation of BMS-955829, a Potent Positive Allosteric Modulator of mGluR5
Bristol-Myers Squibb Research & Development, 5 Research Parkway, Wallingford, Connecticut 06492, United States
ACS Med. Chem. Lett., Article ASAP
DOI: 10.1021/acsmedchemlett.5b00450
Publication Date (Web): January 4, 2016
Copyright © 2016 American Chemical Society
http://pubs.acs.org/doi/abs/10.1021/acsmedchemlett.5b00450
http://pubs.acs.org/doi/suppl/10.1021/acsmedchemlett.5b00450/suppl_file/ml5b00450_si_001.pdf
SEE…………http://orgspectroscopyint.blogspot.in/2016/01/bms-955829.html
///////BMS 955829, mGluR5, positive allosteric modulator, schizophrenia, cognition, neurotoxicity, Bristol-Myers Squibb
FC1=CC=C(C=C1[C@H]([C@@H](C2=CC(C#CC3=CC=CC=C3)=CN=C2)N4)OC4=O)F
Sparsentan, PS433540, RE-021

Sparsentan (PS433540, RE-021)
- C32H40N4O5S
- Average mass592.749
FDA APPROVED 2023/2/17, Filspari
4′-((2-butyl-4-oxo-1,3-diazaspiro[4.4]non-1-en-3-yl)methyl)-N-(4,5-dimethylisoxazol-3-yl)-2′-(ethoxymethyl)-[1,1′-biphenyl]-2-sulfonamide
4′-[(2-Butyl-4-oxo-1.3-diazaspiro[4.41non-l-en-3-yl)methvn-N-(3,4- dimethyl-5-isoxazolyl)-2′-ethoxymethyl [ 1 , l’-biphenyll -2-sulfonamide
Sparsentan
PS433540; RE-021, formerly known as DARA
CAS :254740-64-2
4-[(2-butyl-4-oxo-1,3-diazaspiro[4.4]non-1-en-3-yl)methyl]-N-(4,5- dimethylisoxazol-3-yl)-2-(ethoxymethyl)biphenyl-2-sulfonamide
Mechanism of Action:acting as both an Endothelin Receptor Antagonist (ERA) and Angiotensin Receptor Blocker (ARB).
Indication: Focal Segmental Glomerulosclerosis (FSGS).Focal Segmental Glomerulosclerosis (FSGS) is a rare and severe nephropathy which affects approximately 50,000 patients in the United States. Most cases of FSGS are pediatric.
Development Stage: Phase II
Developer:Retrophin, Inc
- OriginatorBristol-Myers Squibb
- DeveloperRetrophin
- ClassAntihypertensives; Isoxazoles; Small molecules; Spiro compounds; Sulfonamides
- Mechanism of ActionAngiotensin type 1 receptor antagonists; Endothelin A receptor antagonists
- Orphan Drug Status Yes – Focal segmental glomerulosclerosis
-
- 09 Jan 2015 Sparsentan receives Orphan Drug status for Focal segmental glomerulosclerosis in USA
- 31 Dec 2013 Phase-II/III clinical trials in Focal segmental glomerulosclerosis in USA (PO)
- 07 May 2012I nvestigation in Focal segmental glomerulosclerosis in USA (PO)
Sparsentan is an investigational therapeutic agent which acts as both a selective endothelin receptor antagonist and an angiotensin receptor blocker. Retrophin is conducting the Phase 2 DUET trial of Sparsentan for the treatment of FSGS, a rare and severe nephropathy that is a leading cause of end-stage renal disease. There are currently no therapies approved for the treatment of FSGS in the United States. Ligand licensed worldwide rights of Sparsentan (RE-021) to Retrophin in 2012 .The Food and Drug Administration (FDA) has granted orphan drug designation for Retrophins sparsentan for the treatment of focal segmental glomerulosclerosis (FSGS) in January 2015.
In 2006, the drug candidate was licensed to Pharmacopeia by Bristol-Myers Squibb for worldwide development and commercialization. In 2012, a license was obtained by Retrophin from Ligand. In 2015, Orphan Drug Designation was assigned by the FDA for the treatment of focal segmental glomerulosclerosis.
Sparsentan, also known as RE-021, BMS346567, PS433540 and DARA-a, is a Dual angiotensin II and endothelin A receptor antagonist. Retrophin intends to develop RE-021 for orphan indications of severe kidney diseases including Focal Segmental Glomerulosclerosis (FSGS) as well as conduct proof-of-concept studies in resistant hypertension and diabetic nephropathy. RE-021, with its unique dual blockade of angiotensin and endothelin receptors, is expected to provide meaningful clinical benefits in mitigating proteinuria in indications where there are no approved therapies
Sparsentan, sold under the brand name Filspari, is a medication used for the treatment of primary immunoglobulin A nephropathy.[1] Sparsentan is an endothelin and angiotensin II receptor antagonist.[1][4] It is taken by mouth.[1]
The most common side effects include swelling of the extremities, low blood pressure, dizziness, high blood potassium, anemia, injury to the kidney, and increased liver enzymes in the blood.[5]
It was approved for medical use in the United States in February 2023.[5][6][7] The US Food and Drug Administration (FDA) considers it to be a first-in-class medication.[8]
PATENT
WO 2000001389
https://www.google.co.in/patents/WO2000001389A1?cl=en


Example 41
4′- [(2-Butyl-4-oxo- 1.3-diazaspiro [4.4! non- l-en-3-yl)methyll -N-(3.4- dimethyl-5-isoxazolyl)-2′-hydroxymethyl[l, l’-biphenyl! -2-sulfonamide

A. 4′-[(2-Butyl-4-oxo-1.3-diazaspiro[4.41non-l-en-3-yl)methyll-N-(3.4- dimethyl-5-isoxazolyl)-N-[(2-trimethylsilylethoxy)methyl]-2′- hydroxym ethyl [1, l’-biphenyl] -2-sulfonamide P14 (243 mg, 0.41 mmol) was used to alkylate 2-butyl-4-oxo-l,3- diazaspiro[4.4]non-l-ene hydrochloride according to General Method 4. 41A (100 mg, 35% yield) was isolated as a slightly yellow oil after silica gel chromatography using 1:1 hexanes/ethyl acetate as eluant. B. 4′- [(2-Butyl-4-oxo- 1 ,3-diazaspiro [4.41 non- l-en-3-yl)methvn -N-0.4- dimethyl-5-isoxazolyl)-2′-hydroxymethyl[l,l’-biphenyn-2- sulfonamide
Deprotection of 41A (100 mg, 0.14 mmol) according to General Method 8 (ethanol) gave the title compound as white solid in 46% yield following silica gel chromatography (96:4 methanol/chloroform eluant):
MS m/e 565 (ESI+ mode); HPLC retention time 3.21 min (Method A);
HPLC purity >98%.
Example 42
4′-[(2-Butyl-4-oxo-1.3-diazaspiro[4.41non-l-en-3-yl)methvn-N-(3,4- dimethyl-5-isoxazolyl)-2′-ethoxymethyl [ 1 , l’-biphenyll -2-sulfonamide

A. 4′- [(2-Butyl-4-oxo- 1 ,3-diazaspiro [4.41 non- l-en-3-yl)methyll -N-(3 ,4- dimethyl-5-isoxazolyl)-N-[(2-methoxyethoxy)methyll-2′- hvdroxym ethyl [1 , l’-biphenyl] -2-sulfonamide
Triethylsilane (6 ml) and TFA (6 ml) were added to a solution of 5F (960 mg, 1.5 mmol) in 15 ml dichloromethane at RT. The mixture was stirred at RT for 2 h and was then concentrated. The residue was taken up in ethyl acetate and was washed successively with aqueous sodium bicarbonate, water, and brine. The organic layer was dried over sodium sulfate and concentrated. The residue was chromatographed on silica gel using 100:2 dichloromethane/methanol to afford 42A (740 mg, 77%) as a colorless gum. Rf=0.13, silica gel, 100:5 dichloromethane/methanol. B. 4′- [(2-Butyl-4-oxo- 1.3-diazaspiro [4.41 non- l-en-3-yl)methyll -N-(3.4- dimethyl-5-isoxazolyl)-N-r(2-methoxyethoxy)methyll-2′- ethoxymethyl[l.l’-biphenyll-2-sulfonamide A mixture of 42A (100 mg, 0.15 mmol), iodoethane (960 mg, 6.1 mmol) and silver (I) oxide (180 mg, 0.77 mmol) in 0.7 ml DMF was heated at 40 ° C for 16 h.. Additional iodoethane (190 mg, 1.2 mmol) and silver (I) oxide (71 mg, 0.31 mmol) were added and the reaction mixture was heated at 40 ° C for an additional 4 h. The mixture was diluted with 1:4 hexanes/ethylacetate and was then washed with water and brine. The organic layer was dried over sodium sulfate and was then concentrated. The residue was chromatographed on silica gel using 200:3 dichloromethane/methanol as eluant to afford 42B (51mg, 49%) as a colorless gum. Rf=0.35, silica gel, 100:5 dichloromethane/methanol.
C. 4,-[(2-Butyl-4-oxo-1.3-diazaspirof4.41non-l-en-3-yl)methyll-N-(3.4- dimethyl-5-isoxazolyl )-2′-ethoxym ethyl [ 1. l’-biphenyll -2-sulfonamide
42B (51 mg) was deprotected according to General Method 7 to afford the title compound in 80% yield following preparative reverse-phase HPLC purification: white solid; m.p. 74-80 ° C (amorphous); IH NMR (CDCL, )δ0.87(tr, J=7Hz, 3H), 0.99(tr, J=7Hz, 3H), 1.32(m, 2H), 1.59(m, 2H), 1.75-2.02(m, 11H), 2.16(s, 3H), 2.35(m, 2H), 3.38 (m, 2H), 4.23(m, 2H), 4.73(s, 2H), 7.11-7.85 (m, 7H); MS m/e 593 (ESI+ mode); HPLC retention time 18.22 min. (Method E); HPLC purity >97%.
PATENT
WO 2001044239
http://www.google.co.in/patents/WO2001044239A2?cl=en
……………………
Dual angiotensin II and endothelin A receptor antagonists: Synthesis of 2′-substituted N-3-isoxazolyl biphenylsulfonamides with improved potency and pharmacokinetics
J Med Chem 2005, 48(1): 171
J. Med. Chem., 2002, 45 (18), pp 3829–3835
DOI: 10.1021/jm020138n
 BMS 248360 A DIFFERENT COMPD
BMS 248360 A DIFFERENT COMPDThe ETA receptor antagonist (2) (N-(3,4-dimethyl-5-isoxazolyl)-4‘-(2-oxazolyl)-[1,1‘-biphenyl]-2-sulfonamide, BMS-193884) shares the same biphenyl core as a large number of AT1 receptor antagonists, including irbesartan (3). Thus, it was hypothesized that merging the structural elements of 2 with those of the biphenyl AT1 antagonists (e.g., irbesartan) would yield a compound with dual activity for both receptors. This strategy led to the design, synthesis, and discovery of (15) (4‘-[(2-butyl-4-oxo-1,3-diazaspiro[4.4]non-1-en-3-yl)methyl]-N-(3,4-dimethyl-5-isoxazolyl)-2‘-[(3,3-dimethyl-2-oxo-1-pyrrolidinyl)methyl]-[1,1‘-biphenyl]-2-sulfonamide, BMS-248360) as a potent and orally active dual antagonist of both AT1 and ETAreceptors. Compound 15 represents a new approach to treating hypertension.

Scheme 2 a DIFFERENT COMPD
a (a) DIBAL, toluene; (b) NaBH4, MeOH; (c) (Ph)3P, CBr4, THF (51% from 9); (d) compound 7, NaH, DMF; (e) 1 N HCl; (f) compound 4, (Ph3P)4Pd, aqueous Na2CO3, EtOH/toluene; (g) 6 N aqueous HCl/EtOH (60% from 10); (h) 13, sodium triacetoxy borohydride, AcOH, (i) diisopropylcarbodiimide, CH2Cl2 (31% from 12).
PATENT
WO 2010135350
http://www.google.com/patents/WO2010135350A2?cl=en
Compound 1 :

Scheme IV

Scheme V

Formula IV 1
Scheme VII

Formula Vl

A solution of 2-(2,4-dimethylphenyl)benzenesulfonic acid (Compound 12) (0.5 g, 1.9 mmol) in 50 mL of anhydrous acetonitrile was prepared and transferred to a round-bottom flask. After flushing with nitrogen gas, N-bromosuccinimide (0.75 g, 4.2 mmol) was added followed by 50 mg (0.2 mmol) of benzoyl peroxide. The solution was heated at reflux for 3 hours. The solvent was removed in-vacuo and the resulting syrup purified by silica gel chromatography (1 :1 hexanes/EtOAc) to yield Compound 13 as a white solid. 1H NMR (500 MHz, CD3CN) 8.12 (d, J = 7.5 Hz, IH), 7.92 (t, J = 7.5 Hz, IH), 7.78 (d, J= 7.5 Hz, IH), 7.74-7.71 (m, 2H), 7.68-7.65 (m, 2H), 5.12 (s, 2H), 4.70 (s, 2H). Example 4 2-(4-Bromomethyl-2-ethoxymethylphenyl)benzenesulfonic acid (Compound 14)

A solution of 20 mg (0.058 mmol) of (l-bromomethylbenzo[3,4- d])benzo[l,2-f]-2-oxa-l,l-dioxo-l-thiocycloheptane (Compound 13) in ethanol was stirred at elevated temperature until the starting material was consumed to give crude product (compound 14) that was used directly in the next step without isolation or purification.
Example 5
2-(4-((2-Butyl-4-oxo-l,3-diazaspiro[4.4]non-l-en-3-yl)methyl>2- ethoxymethylphenyl)benzenesulfonic acid (Compound 15)

To the above ethanol solution of crude 2-(4-bromomethyl-2- ethoxymethylphenyl)benzenesulfonic acid (Compound 14) described in Example 4 was added approximately 25 mL of anhydrous DMF. The ethanol was removed from the system under reduced pressure. Approximately 15 mg (0.065 mmol) of 2-butyl-l,3- diazaspiro[4.4]non-l-en-4-one (compound 7 in Scheme IV) was added followed by 300 μL of a IM solution of lithium bis-trimethylsilylamide in THF. The solution was allowed to stir at room temperature for 3 hours. The solvents were removed under reduced pressure and the remaining residue purified by preparative RP-HPLC employing a Cl 8 column and gradient elution (H2O:MeCN) affording the title compound as a white solid; [M+H]+ calcd for C27H34N2O5S 499.21, found, 499.31 ; 1H NMR (500 MHz, CD3CN) 8.04 (t, J= 5.5 Hz, IH), 7.44-7.10 (m, 2H), 7.28 (s, IH), 7.22 (d, J= 8.0 Hz, 2H), 7.08- 7.04 (m, 2H), 4.74 (br s, 2H), 4.32 (d, J= 13.0 Hz IH), 4.13 (d, J= 13.0 Hz IH), 3.40- 3.31 (m, 2H), 2.66 (t, J= 8 Hz, 2H), 2.18-2.13 (m, 5H), 1.96-1.90 (m, 2H obscured by solvent), 1.48 (m, 2H), 1.27 (s, J= 7 Hz, 2H), 1.16 (t, J= 7 Hz, 3H), 0.78 (t, J= 7.5 Hz, 3H).
Example 6
2-(4-((2-Butyl-4-oxo-l,3-diazaspiro[4.4]non-l-en-3-yl)methyl>2- ethoxymethylphenyl)benzenesulfonyl chloride (Compound 16)

To a solution of DMF (155 μL, 2 mmol, 2 equiv.) in dichloromethane (5 mL) at 0 0C was added dropwise oxalyl chloride (175 μL, 2 mmol, 2 equiv.) followed by a dichloromethane (5 mL) solution of 2-(4-((2-butyl-4-oxo-l,3-diazaspiro[4.4]non-l- en-3-yl)methyl)-2-ethoxymethylphenyl)benzenesulfonic acid (Compound 15) (0.50 g, 1.0 mmol). The resulting mixture was stirred at 0 0C for ~2 hours, diluted with additional dichloromethane (25 mL), washed with saturated sodium bicarbonate solution (10 mL), water (10 mL), and brine (10 mL), dried over sodium sulfate, and then concentrated to give crude sulfonyl chloride (compound 16) that was used without purification.
Example 7
N-(3,4-Dimethyl-5-isoxazolyl)-2-(4-(2-butyl-4-oxo-l,3-diazospiro[4.4]non-l-en- 3yl)methyl-2-ethoxymethylphenyl)phenylsulfonamide (Compound 1)

[0062] To a solution of 5-amino-3,4-dimethylisoxazole (60 mg, 0.54 mmol) in THF at -60 °C was added dropwise potassium tert-butoxide (1 mL of 1 M solution) followed by a solution of crude 2-(4-((2-butyl-4-oxo-l,3-diazaspiro[4.4]non-l-en-3- yl)methyl)-2-ethoxymethylphenyl)benzenesulfonyl chloride (Compound 16) (0.28 g, 0.54 mmol) in THF (4 mL). The resulting mixture was stirred at about -60 °C for 1 hour, allowed to warm to room temperature overnight, and then quenched with IN HCl solution to about pH 4. Standard workup of extraction with ethyl acetate, washing with water, drying, and concentration provided the final compounds as a white solid. 1H NMR (400 MHz, CDCl3) 8.03 (dd, J = 8.0 and 1.2, IH), 7.60 (td, J = 7.5 and 1.5, IH), 7.50 (td, J = 7.7 and 1.5, IH), 7.36 (s, IH), 7.28 (d, J= 2.1, 1 H), 7.25 (dd, J = 7.5 and 1.2, IH), 7.09 (dd, J= 7.9 and 1.6, IH), 6.61 (bs, IH), 4.77 (AB quartet, J= 15.5 and 8.1, 2H), 4.18 (AB quartet, J= 12.0 and 35, 2H), 3.45-3.32 (m, 2H), 2.39 (t, J= 7.5, 2H), 2.26 (s, 3H), 2.02- 1.84 (m, 8H), 1.82 (s, 3H), 1.63 (quint, J = 7.5, 2H), 1.37 (sextet, J = 7.3, 2H), 1.07 (t, J = 7.0, 3H), and 0.90 (t J= 7.3, 3H).
Example 8 l-Bromo-2-ethoxymethyl-4-hydroxymethylbenzene (Compound 17)

To a solution of ethyl 4-bromo-3-ethoxymethylbenzoate (9.4 g, 33 mmol) in toluene (56 mL) at about -10 0C was added 51 g of a 20% diisobutylaluminum hydride solution in toluene (ca. 70 mmol). The reaction was stirred at the same temperature for about 30 minutes until the reduction was completed, and then quenched with icy 5% NaOH solution to keep the temperature below about 10 °C. Organic phase of the resulting mixture was separated and the aqueous phase was extracted with toluene. The combined organic phase was concentrated in vacuo to a final volume of ~60 mL toluene solution of l-bromo-2-ethoxymethyl-4-hydroxymethylbenzene (Compound 17) that was used in next step without purification.
Example 9 l-Bromo-2-ethoxymethyl-4-methanesulfonyloxymethylbenzene (Compound 18)

To a solution of 1 -bromo-2-ethoxymethyl-4-hydroxymethylbenzene (Compound 17) (8.4 g, 33 mmol) in toluene (60 mL) prepared in Example 8 at about -10 °C was added methanesulfonyl chloride (7.9 g, 68 mmol). The reaction was stirred at the same temperature for about 30 minutes until the reduction was completed, and then quenched with icy water to keep the temperature at about 0 °C. The organic layer was separated and washed again with icy water to provide a crude product solution of 1 – bromo-2-ethoxymethyl-4-methanesulfonyloxymethylbenzene (Compound 18) that was used without purification.
Example 10
1 -Bromo-4-((2-butyl-4-oxo- 1 ,3 -diazaspiro [4.4]non- 1 -en-3 -yl)methy l)-2- ethoxymethylbenzene bisoxalic acid salt (Compound 19)

To the crude solution of 1 -bromo-2-ethoxymethyl-4- methanesulfonyloxymethylbenzene (Compound 18) (1 1 g, 33 mmol) in toluene (80 mL) prepared in Example 9 was added a 75% solution of methyltributylammonium chloride in water (0.47 mL). The resulting mixture was added to a solution of 2-butyl-4-oxo-l,3- diazaspiro[4.4]non-l-ene (compound 7 in Scheme VI) (7.5 g, 32 mmol) in dichloromethane (33 mL) pretreated with a 10 M NaOH solution (23 mL). The reaction mixture was stirred at room temperature for 2 hours until compound 18 was not longer detectable by HPLC analysis and then was quenched with water (40 mL). After stirring about 10 minutes, the organic layer was separated and aqueous layer was extracted with toluene. The combined organic phase was washed with water and concentrated to a small volume. Filtration through a silica gel pad using ethyl acetate as solvent followed by concentration yielded 1 -bromo-4-((2-buty 1-4-oxo- 1 ,3 -diazaspiro [4.4]non- 1 -en-3 – yl)methyl)-2-ethoxymethylbenzene as a crude oil product.
The crude oil was dissolved in ethyl acetate (22 mL) and warmed to around 50 °C. Anhydrous oxalic acid (4.6 g) was added to the warm solution at once and the resulting mixture was stirred until a solution was obtained. The mixture was cooled gradually and the bisoxalic acid salt (compound 19) was crystallized. Filtration and drying provided pure product (compound 19) in 50-60% yield from ethyl 4-bromo-3- ethoxymethylbenzoate in 3 steps. 1H NMR (400 MHz, CDCl3) 12.32 (bs, 4H), 7.58 (d, J = 7.8, IH), 7.36 (s, IH), 7.12 (d, J= 7.8, IH), 4.90 (s, 2H), 4.56 (s, 2H), 3.68 (q, J= 7.5, 2H), 2.87-2.77 (m, 2H), 2.40-1.95 (m, 8H), 1.62-1.53 (m, 2H), 1.38-1.28 (m, 4H), and 1.82 (t, J= 7.5, 3H).
Example 11
N-(3,4-Dimethyl-5-isoxazolyl)-2-(4-(2-butyl-4-oxo-l,3-diazospiro[4.4]non-l-en- 3yl)methyl-2-ethoxymethylphenyl)phenylsulfonamide (Compound 1)

To a suspension of l-bromo-4-((2-butyl-4-oxo-l,3-diazaspiro[4.4]non- l-en-3-yl)methyl)-2-ethoxymethylbenzene bisoxalic acid salt (Compound 19) (5.0 g, 8.3 mmol) in toluene (20 niL) under nitrogen was added water (30 mL) and pH was adjusted to 8-9 by addition of a 2 M NaOH solution at room temperature. The organic phase was separated and mixed with 2-(N-(3,4-dimethyl-5-isoxazolyl)-N- methoxymethylamino)sulfonylphenylboronic acid pinacol ester (Scheme VII, Formula IX, where R8is methoxymethyl and M = boronic acid pinacol ester) (3.6 g, 8.5 mmol), bis(dibenzylideneacetone)palladium(0) (Pd(dba)2) (0.12 g), and a standard phosphine ligand. After a 2 M sodium carbonate solution was added, the reaction mixture was warmed to 70 0C and stirred until the reaction was complete by HPLC analysis. The reaction was cooled to room temperature and quenched with water, and then separated in phases. The organic phase was treated with activated carbon, filtered through a pad of silica gel, and was concentrated to afford a crude mixture.
The crude reaction mixture was dissolved in ethanol (40 mL) after palladium catalyst was removed and was treated with 6 M HCl solution (ca. 40 mL). The mixture was warmed to 75-80 °C and stirred for about 2 hours until the reaction was completed by HPLC analysis. After the mixture was cooled to room temperature, the pH of the mixture was adjusted to 8 by addition of 10 M NaOH solution. The mixture was stirred for 2 more hours and the pH was adjusted to 6 by adding 2 M HCl and the crystal seeds. Filtration of the crystalline solid followed by drying provided N-(3,4-dimethyl-5- isoxazolyl)-2-(4-(2-butyl-4-oxo-l,3-diazospiro[4.4]non-l-en-3yl)methyl-2- ethoxymethylphenyl)phenylsulfonamide (Compound 1) as a white solid.1H NMR (400 MHz, CDCIa) 8.03 (dd, J= 8.0 and 1.2, IH), 7.60 (td, J = 7.5 and 1.5, IH), 7.50 (td, J = 7.7 and 1.5, IH), 7.36 (s, IH), 7.28 (d, J= 2.1, 1 H), 7.25 (dd, J = 7.5 and 1.2, IH), 7.09 (dd, J= 7.9 and 1.6, IH), 6.61 (bs, IH), 4.77 (AB quartet, J= 15.5 and 8.1, 2H), 4.18 (AB quartet, J= 12.0 and 35, 2H), 3.45-3.32 (m, 2H), 2.39 (t, J= 7.5, 2H), 2.26 (s, 3H), 2.02- 1.84 (m, 8H), 1.82 (s, 3H), 1.63 (quint, J= 7.5, 2H), 1.37 (sextet, J= 7.3, 2H), 1.07 (t, J = 7.0, 3H), and 0.90 (t J= 7.3, 3H).
| US20040002493 * | Aug 20, 2001 | Jan 1, 2004 | Kousuke Tani | Benzoic acid derivatives and pharmaceutical agents comprising the same as active ingredient |
| US20070054806 * | Sep 6, 2006 | Mar 8, 2007 | Bayer Cropscience Gmbh | Novel sulfonamide-comprising solid formulations |
| US20070054807 * | Sep 8, 2006 | Mar 8, 2007 | Bayer Cropscience Gmbh | Storage-stable formulations of sulfonamides |
.
 |
|
| Clinical data | |
|---|---|
| Trade names | Filspari |
| Other names | RE-021, PS433540 |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a623018 |
| License data |
|
| Pregnancy category |
|
| Routes of administration |
By mouth |
| ATC code | |
| Legal status | |
| Legal status | |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| DrugBank | |
| UNII | |
| KEGG | |
| ChEBI | |
| ECHA InfoCard | 100.275.317 |
| Chemical and physical data | |
| 3D model (JSmol) | |
|
show
|
|
|
show
|
|
References
- ^ Jump up to:a b c d e f “Filspari- sparsentan tablet, film coated”. DailyMed. 17 February 2023. Retrieved 6 March 2023.
- ^ Jump up to:a b c d “Filspari EPAR”. European Medicines Agency (EMA). 22 February 2024. Retrieved 24 February 2024. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ Jump up to:a b “Filspari Product information”. Union Register of medicinal products. 23 April 2024. Retrieved 7 September 2024.
- ^ Chiu AW, Bredenkamp N (September 2023). “Sparsentan: A First-in-Class Dual Endothelin and Angiotensin II Receptor Antagonist”. The Annals of Pharmacotherapy. 58 (6): 645–656. doi:10.1177/10600280231198925. PMID 37706310. S2CID 261743204.
- ^ Jump up to:a b c d e f g h i j k l m n o p q “Drug Trials Snapshots: Filspari”. U.S. Food and Drug Administration (FDA). 17 February 2023. Retrieved 7 September 2024.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ “Travere Therapeutics Announces FDA Accelerated Approval of Filspari (sparsentan), the First and Only Non-immunosuppressive Therapy for the Reduction of Proteinuria in IgA Nephropathy” (Press release). Travere Therapeutics. 17 February 2023. Retrieved 17 February 2023 – via GlobeNewswire.
- ^ Syed YY (April 2023). “Sparsentan: First Approval”. Drugs. 83 (6): 563–568. doi:10.1007/s40265-023-01864-x. PMC 10232600. PMID 37022667.
- ^ New Drug Therapy Approvals 2023 (PDF). U.S. Food and Drug Administration (FDA) (Report). January 2024. Archived from the original on 10 January 2024. Retrieved 9 January 2024.
- ^ “PHARMACOPEIA LAUNCHES STUDY OF DARA COMPOUND | FDAnews”. http://www.fdanews.com.
- ^ “Ligand Licenses DARA Program to Retrophin”. investor.ligand.com. 21 February 2012.
- ^ https://www.fiercebiotech.com/biotech/retrophin-sheds-shkreli-connection-new-name-travere-therapeutics.
{{cite news}}: Missing or empty|title=(help) - ^ “Ongoing Non-malignant Hematological, Neurological, and Other Disorder Indications Accelerated Approvals”. U.S. Food and Drug Administration (FDA). 21 August 2024. Retrieved 7 September 2024.
- ^ “Travere Therapeutics Announces Full FDA Approval of Filspari (sparsentan), the Only Non-Immunosuppressive Treatment that Significantly Slows Kidney Function Decline in IgA Nephropathy” (Press release). Travere Therapeutics. 5 September 2024. Retrieved 7 September 2024 – via GlobeNewswire.
- ^ “Despite trial scare, Travere’s Filspari gains full FDA nod in kidney disease showdown with Novartis”. fiercepharma.com.
External links
- Clinical trial number NCT03762850 for “A Study of the Effect and Safety of Sparsentan in the Treatment of Patients With IgA Nephropathy (PROTECT)” at ClinicalTrials.gov
SYN
https://doi.org/10.1021/acs.jmedchem.4c02079
J. Med. Chem. 2025, 68, 2147−2182
Sparsentan (Filspari). Sparsentan (27), marketed by Travere Therapeutics, is an oral, dual endothelin angiotensin receptor antagonist that received accelerated USFDA approval in February 2023 for reducing proteinuria in adults with primary immunoglobulin A (IgA) nephropathy who are at risk of rapid
disease progression.205206,207 Also known as Berger’s disease, IgAnephropathy is an immune-complex mediated disease characterized by deposits of IgA in the kidneys, resulting in inflammation and damage which can eventually lead to kidney failure. Typical treatment of IgA nephropathy has focused
on supportive care to slow kidney decline, for example, lowering blood pressure, reducing proteinuria, and minimizing lifestyle risk factors; immunosuppressive therapy has also been utilized, though it is controversial and carries risks.208 Sparsentan is the first nonimmunosuppressive treatment for IgA nephropathy and has received first-in-class and orphan drug designations. Accelerated approval was based on reduction of proteinuria (which is a risk factor for disease progression) during interim
analysis in phase III clinical trials. 209 endothelin type A (ETASparsentan blocks ) and angiotensin II type 1 receptors(AT1), interrupting the signaling pathway that contributes to disease progression. 210
The structure of the drug combines 211,212 elements that target both of these receptor types.
213 Thesynthesis of sparsentan (27), as shown in Scheme 50 and Scheme 51, was disclosed by Retrophin Pharmaceuticals (now Travere Therapeutics). Its telescoped sequences and isolation of intermediates as salts suggest that this route may be suitable for large-scale manufacturing.
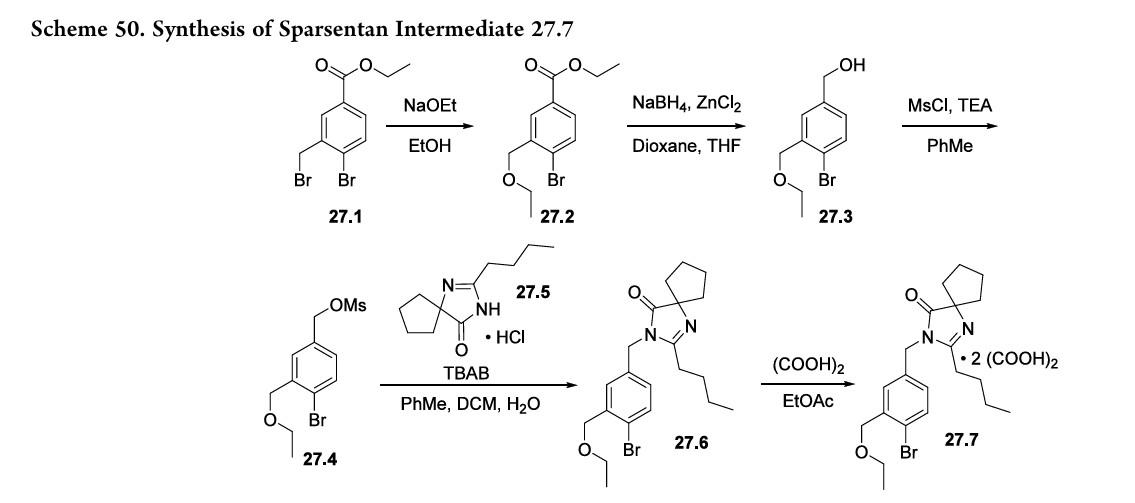
The synthesis of the spirocyclic imidazolinone intermediate 27.7 is shown in Scheme 50.
Displacement of the benzylic bromide in 27.1 with sodium ethoxide produced ether 27.2. Reduction of the ester with sodium borohydride and zinc chloride yielded alcohol 27.3 which was then converted to mesylate 27.4. Reaction with spirocyclic imidazolinone 27.5 under phase transfer conditions
yielded 27.6 whichwasisolatedasthebisoxalatesalt (27.7).The sequence from 27.1 to 27.7 is telescoped, and no yields were given in the patent.
The construction of the biphenyl framework is shown in Scheme 51. Treatment of aryl bromide 27.8 with n-BuLi and triisopropyl borate followed by reaction with pinacol yielded boronic ester 27.9. Intermediates 27.7 and 27.9 were coupled via a Suzuki reaction to form the biphenyl which was isolated as
the camphorsulfonate salt (27.10). The synthesis was finished with deprotection of the methoxymethyl group under acidic conditions followed by recrystallization from isopropanol and heptane to yield sparsentan (27).
(206) Donadio, J. V.; Grande, J. P. IgA nephropathy. N. Engl. J. Med.2002, 347, 738−748.
(207) Fabiano, R. C. G.; Pinheiro, S. V. B.; Simões e Silva, A. C.Immunoglobulin A nephropathy: a pathophysiology view. Inflammation Res. 2016, 65, 757−770.
(208) Floege, J.; Rauen, T.; Tang, S. C. W. Current treatment of IgAnephropathy. Springer Semin. Immunopathol. 2021, 43, 717−728.
(209) Rovin, B.H.; Barratt, J.; Heerspink, H. J. L.; Alpers, C. E.; Bieler,S.; Chae, D.-W.; Diva, U. A.; Floege, J.; Gesualdo, L.; Inrig, J. K.; et al.Efficacy and safety of sparsentan versus irbesartan in patients with IgA
nephropathy (PROTECT): 2-year results from a randomised, active controlled, phase 3 trial. Lancet 2023, 402, 2077−2090.
(210) Komers, R.; Plotkin, H. Dual inhibition of renin-angiotensin aldosterone system and endothelin-1 in treatment of chronic kidney disease. Am. J. Physiol.: Regul., Integr. Comp. Physiol. 2016, 310, R877−
R884.
(211) Murugesan, N.; Tellew, J. E.; Gu, Z.; Kunst, B. L.; Fadnis, L.;Cornelius, L. A.; Baska, R. A. F.; Yang, Y.; Beyer, S. M.; Monshizadegan, H.; et al. Discovery of N-isoxazolyl biphenylsulfonamides as potent dual
angiotensin II and endothelin A receptor antagonists. J. Med. Chem.2002, 45, 3829−3835.
(212) Murugesan, N.; Gu, Z.; Fadnis, L.; Tellew, J. E.; Baska, R. A. F.; Yang, Y.; Beyer, S. M.; Monshizadegan, H.; Dickinson, K. E.; Valentine,M.T.; et al. Dual angiotensin II and endothelin A receptor antagonists:
synthesis of 2′-substituted N-3-isoxazolyl biphenylsulfonamides withimproved potencyandpharmacokinetics. J. Med. Chem. 2005, 48, 171−179.
(213) Komers, R.; Shih, A. Biphenyl sulfonamide compounds for the treatment of kidney diseases or disorders. WO 2018071784, 2018.

//////////////Sparsentan, PS433540, RE-021, Bristol-Myers Squibb, ORPHAN DRUG, Retrophin, FDA 2023, APPROVALS 2023
O=S(C1=CC=CC=C1C2=CC=C(CN3C(CCCC)=NC4(CCCC4)C3=O)C=C2COCC)(NC5=NOC(C)=C5C)=O,
DACLATASVIR, 达拉他韦 , Даклатасвир , داكلاتاسفير ,

Daclatasvir
BMS-790052,
EBP 883; BMS 790052
THERAPEUTIC CLAIM Treatment of hepatitis C

CHEMICAL NAMES
1. Carbamic acid, N,N’-[[1,1′-biphenyl]-4,4′-diylbis[1H-imidazole-5,2-diyl-(2S)-2,1-
pyrrolidinediyl[(1S)-1-(1-methylethyl)-2-oxo-2,1-ethanediyl]]]bis-, C,C’-dimethyl ester
2. dimethyl N,N’-(biphenyl-4,4′-diylbis{1H-imidazole-5,2-diyl-[(2S)-pyrrolidine-2,1-
diyl][(1S)-1-(1-methylethyl)-2-oxoethane-2,1-diyl]})dicarbamate
MF C40H50N8O6
MW 738.9
SPONSOR Bristol-Myers Squibb
CODE BMS-790052
CAS 1009119-64-5
SMILES:CC(C)C(C(=O)N1CCCC1C2=NC=C(N2)C3=CC=C(C=C3)C4=CC=C(C=C4)C5=CN=C(N5)C6CCCN6C(=O)C(C(C)C)NC(=O)OC)NC(=O)OC
UNII-LI2427F9CI
Activity: Treatment of Hepatitis C; HCV Drug; Treatment of HCV; Inhibitor of NS5A
Status: Launched 2014 (EU, Japan)
Originator: Bristol-Myers Squibb
Status: Launched 2014 (EU, Japan)
Originator: Bristol-Myers Squibb
NMR
FDA APPROVAL……..July 24th, 2015
Daklinza (daclatasvir) is an NS5A inhibitor indicated for use in combination with sofosbuvir for the treatment of chronic hepatitis C virus (HCV) genotype 3 infection.

Daclatasvir dihydrochloride
1. Carbamic acid, N,N’-[[1,1′-biphenyl]-4,4′-diylbis[1H-imidazole-5,2-diyl-(2S)-2,1-
pyrrolidinediyl[(1S)-1-(1-methylethyl)-2-oxo-2,1-ethanediyl]]]bis-, C,C’-dimethyl ester,
hydrochloride (1:2)
2. dimethyl N,N’-(biphenyl-4,4′-diylbis{1H-imidazole-5,2-diyl-[(2S)-pyrrolidine-2,1-
diyl][(1S)-1-(1-methylethyl)-2-oxoethane-2,1-diyl]})dicarbamate dihydrochloride
MF C40H50N8O6 . 2 HCl, MW 811.8
SPONSOR Bristol-Myers Squibb
CODE BMS-790052-05
CAS 1009119-65-6
Daclatasvir (USAN[1]) (formerly BMS-790052, trade name Daklinza) is a drug for the treatment of hepatitis C (HCV). It is was developed by Bristol-Myers Squibb and was approved in Europe on 22 August 2014.
Daclatasvir inhibits the HCV nonstructural protein NS5A.[2][3] Recent research suggests that it targets two steps of the viral replication process, enabling rapid decline of HCV RNA.[4]
Daclatasvir has been tested in combination regimens with pegylated interferon and ribavirin,[5] as well as with other direct-acting antiviral agents including asunaprevir[6][7][8][9] and sofosbuvir.[10][11]
It is on the World Health Organization’s List of Essential Medicines, a list of the most important medications needed in a basic health system.[12]
Hepatitis C virus (HCV) is a major global health problem, with an estimated 150-200 million people infected worldwide, including at least 5 million in Europe (Pawlotsky, Trends Microbiol, 2004, 12: 96-102). According to the World Health Organization, 3 to 4 million new infections occur each year. The infection is often asymptomatic; however, the majority of HCV-infected individuals develop chronic infection (Hoof agle, Hepatology, 2002, 36: S21-S29; Lauer et al, N. Engl. J. Med., 2001, 345: 41-52; Seeff, Semin. Gastrointest., 1995, 6: 20-27). Chronic infection frequently results in serious liver disease, including fibrosis and steatosis (Chisari, Nature, 2005, 435: 930-932).
About 20% of patients with chronic HCV infection develop liver cirrhosis, which progresses to hepatocellular carcinoma in 5% of the cases (Hoofnagle, Hepatology, 2002, 36: S21-S29; Blonski et al, Clin. Liver Dis., 2008, 12: 661-674; Jacobson et al, Clin. Gastroenterol. Hepatol, 2010, 8: 924-933; Castello et al., Clin. Immunol, 2010, 134: 237-250; McGivern et al., Oncogene, 2011, 30: 1969-1983).
Chronic HCV infection is the leading indication for liver transplantations (Seeff et al., Hepatology, 2002, 36: 1-2). Unfortunately, liver transplantation is not a cure for hepatitis C; viral recurrence being an invariable problem and the leading cause of graft loss (Brown, Nature, 2005, 436: 973-978; Watt et al, Am. J. Transplant, 2009, 9: 1707-1713). No vaccine protecting against HCV is yet available. Current therapies include administration of ribavirin and/or interferon-alpha (IFN-Cc), two non-specific anti-viral agents.
Using a combination treatment of pegylated IFN-CC and ribavirin, persistent clearance is achieved in about 50% of patients with genotype 1 chronic hepatitis C. However, a large number of patients have contraindications to one of the components of the combination; cannot tolerate the treatment; do not respond to interferon therapy at all; or experience a relapse when administration is stopped. In addition to limited efficacy and substantial side effects such as neutropenia, haemo lytic anemia and severe depression, current antiviral therapies are also characterized by high cost.
To improve efficacy of standard of care (SOC), a large number of direct acting antivirals (DAAs) targeting viral polyprotein processing and replication have been developed (Hofmann et al, Nat. Rev; Gastroenterol. Hepatol., 2011, 8: 257-264). These include small molecule compounds targeting HCV nonstructural proteins including the HCV protease, polymerase and NS5A protein.
Although a marked improvement of antiviral response was observed when protease inhibitors were combined with SOC (Hofmann et al, Nat. Rev; Gastroenterol. Hepatol, 2011, 8: 257-264; Bacon et al, New Engl. J. Med., 2011, 364: 1207-1217; McHutchison et al, New Engl. J. Med., 2010, 362: 1292-1303; Poordad et al, New Engl. J. Med., 201 1, 364: 1195-1206; Hezode et al, New Engl. J. Med., 2009, 360: 1839-1850; Kwo et al, Lancet, 2010, 376: 705-716), toxicity of the individual compounds and rapid development of viral resistance in a substantial fraction of patients remain major challenges (Pawlotsky, Hepatology, 2011, 53: 1742-1751; Pereira et al, Nat. Rev. Gastroenterol. Hepatol., 2009, 6: 403-411; Sarrazin et al, Gastroenterol., 2010, 138: 447-462).
New therapeutic approaches against HCV are therefore still needed. HCV entry into target cells is a promising target for antiviral preventive and therapeutic strategies since it is essential for initiation, spread, and maintenance of infection (Timpe et al, Gut, 2008, 57: 1728-1737; Zeisel et al, Hepatology, 2008, 48: 299-307). Indeed, HCV initiates infection by attaching to molecules or receptors on the surface of hepatocytes.
Current evidence suggests that HCV entry is a multistep process involving several host factors including heparan sulfate (Barth et al, J. Biol. Chem., 2003, 278: 41003-41012), the tetraspanin CD81 (Pileri et al, Science, 1998, 282: 938-941), the scavenger receptor class B type I (SR-BI) (Zeisel et al, Hepatology, 2007, 46: 1722-1731; Bartosch et al, J. Exp. Med., 2003, 197: 633-642; Grove et al, J. Virol, 2007, 81 : 3162-3169; Kapadia et al, J. Virol, 2007, 81 : 374- 383; Scarselli et al, EMBO J., 2002, 21 : 5017-5025), Occludin (Ploss et al, Nature, 2009, 457: 882-886) and Claudin-1 (CLDN1), an integral membrane protein and a component of tight-junction strands (Evans et al, Nature, 2007, 446: 801-805).
Furthermore, Niemann-Pick CI -like cholesterol absorption receptor has been identified as a new hepatitis C virus entry factor (Sainz et al, Nature Medicine, 2012, 18: 281-285).
Daclatasvir (BMS-790052; EBP 883) is a first-in-class, highly-selective oral HCV NS5A inhibitor. NS5A is an essential component for hepatitis C virus (HCV) replication complex.Daclatasvir (BMS-790052; EBP 883)has broad genotype coverage and exhibits picomolar in vitro potency against genotypes 1a (EC50 50pm) and 1b (EC50 9pm).Daclatasvir (BMS-790052; EBP 883) produces a robust decline in HCV RNA (-3.6 logs after 48 hours from a single 100 mg) dosefollowing a single dose in patients chronically infected with HCV genotype 1.
It may be many years before the symptoms of hepatitis C infection appear. However, once they do, the consequences are significant: patients may have developed fibrosis, cirrhosis or even liver cancer, with the end result being liver failure. Even if diagnosed early, there’s no guarantee of a cure.
Only around half of patients respond to the standard therapy of an interferon plus the antiviral drug ribavirin, and while two add-on antiviral therapies were approved in 2011, the treatment period is long with no guarantee of a cure, and for non-responders treatment options remain limited.
A new drug with a different mechanism is being developed by Bristol-Myers Squibb, in conjunction with Pharmasset. Daclatasvir targets non-structural protein 5A, which is an important component of the viral replication process, although its precise role in this remains unclear. The drug is active in single oral doses, and may have potential as part of a treatment regimen that avoids the use of interferon, and in patients who do not respond to standard therapy.
In an open label Phase IIa study, 10 patients with chronic hepatitis C genotype 1b infection who did not respond to standard therapy were given daclatasvir in once daily 60mg doses, plus another experimental drug, BMS-790052, which is an NSP 3 protease inhibitor, in initial twice-daily 600mg doses, later reduced to 200mg twice a day.2 Nine patients completed 24 weeks of treatment, with the 10th discontinuing after 10 weeks. In those who completed the course, HCV RNA was undetectable at week 8, and remained so until the end of the trial, with all achieving a sustained virologic response. It was also undetectable post-treatment in the patient who discontinued.
Daclatasvir has also been investigated as monotherapy in a double blind, placebo-controlled, sequential panel, multiple ascending dose study.3 Thirty patients with chronic geno-type 1 hepatitis C infection were randomised to receive a 14 day course of the drug, in once daily doses of 1, 10, 30, 60 or 100mg, 30mg twice a day, or placebo. There was no evidence of antiviral activity in the placebo group, but the mean maximum decline of 2.8 to 4.1 log IU/ml. Most experienced viral rebound on or before day 7 of treatment, which was associated with viral variants that had previously been implicated in resistance development. It was well tolerated in all dose groups.
M. Gao et al. Nature 2010, 465, 96

22/11/2013
EUROPEAN MEDICINES AGENCY ADVISES ON COMPASSIONATE USE OF DACLATASVIR
Opinion concerns use in combination with sofosbuvir in patients with chronic hepatitis C in urgent need of therapy to prevent progression of liver disease
The European Medicines Agency’s Committee for Medicinal Products for Human Use(CHMP) has given an opinion on the use of daclatasvir in combination with sofosbuvir in the treatment of chronic (long-term) hepatitis C virus (HCV) infection, in a compassionate-use programme.
Compassionate-use programmes are set up at the level of individual Member States. They are intended to give patients with a life-threatening, long-lasting or seriously disabling disease with no available treatment options access to treatments that are still under development and that have not yet received amarketing authorisation. In this specific case, Sweden has requested an opinion from the CHMP on the conditions under which early access through compassionate use could be given to daclatasvir, for the use in combination with sofosbuvir, with or without ribavirin, for a specific patient population.
The recommended compassionate use is intended for adult patients at a high risk of their liver being no longer able to function normally (decompensation) or death within 12 months if left untreated, and who have a genotype 1 infection. Further, it is recognised that the potential benefit of such combination therapy may extend to patients infected with other HCV genotypes.
Daclatasvir and sofosbuvir are both first-in-class anti-viral medicines against HCV. These medicines have been studied in combination, with or without ribavirin, in aclinical trial which included treatment-naive (previously untreated) HCV genotype-1, -2 and -3 infected patients, as well as patients with genotype 1 infection who have previously failed telaprevir or boceprevir treatment. Results from the trial indicate high efficacy, also in those who have failed treatment with these protease inhibitors. Many such patients have very advanced liver disease and are in urgent need of effective therapy in order to cease the progression of liver injury.
This is the second opinion provided by the CHMP on compassionate use of medicines in development for the treatment of hepatitis C. Overall, it isthe fourth time compassionate use has been assessed by the CHMP.
The aim of the CHMP assessment and opinion on a compassionate-use programme for new medicinal products is to ensure a common approach, whenever possible, regarding the criteria and conditions of use under Member States’ legislation. The opinion provides recommendations to the EU Member States that are considering setting up such a programme, and its implementation is not mandatory. In addition to describing which patients may benefit from the medicine, it explains how to use it and gives information on safety.
The assessment report and conditions of use of daclatasvir in combination with sofosbuvir with or without ribavirin in this setting will be published shortly on the Agency’s website.
Notes
- The first compassionate-use opinion for a hepatitis C treatment was adopted by the CHMP in October 2013.
- Sofosbuvir, which is part of this compassionate-use opinion, received a positive opinion from the CHMP recommending granting of a marketing authorisation at its November 2013 meeting.
- Daclatasvir is developed by Bristol-Myers Squibb and sofosbuvir is developed by Gilead.
|
1-6-2012
|
Anti-Viral Compounds
|
|
|
2-13-2009
|
CRYSTALLINE FORM OF METHYL ((1S)-1-(((2S)
-2-(5-(4′-(2-((2S)-1((2S)-2-((METHOXYCARBONYL)AMINO)-3-METHYLBUTANOYL)-2-PYRROLIDINYL) -1H-IMIDAZOL-5-YL)-4-BIPHENYLYL)-1H-IMIDAZOL-2-YL)-1-PYRROLIDINYL)CARBONYL) -2-METHYLPROPYL)CARBAMATE DIHYDROCHLORIDE SALT |
Synthesis
Daclatasvir dihydrochloride (Daklinza)
Daclatasvir dihydrochloride is a hepatitis C virus nonstructural 5A (NS5A) replication complex inhibitor which was first approved in Japan for the treatment of genotype 1 HCV patients who fail to respond to interferon plus ribavirin. The drug has also been approved for patients with untreated, chronic HCV who are eligible for interferon. Additionally, in Europe, daclatasvir was approved for use in combination with other products across genotype 1–4 HCV. Daclatasvir was discovered and developed by Bristol–Myers Squibb and a fascinating account describing the initiation of the program from a phenotypic screen and the medicinal chemistry strategy leading to the discovery of the compound has been recently reported.80 Daclatasvir has been prepared via two different routes81,82 and the process route is outlined in Scheme 11.83 Bromination of commercial 4,40-diacetylbiphenyl (58) gave 4,40-bis(bromoacetyl)biphenyl 59 in 82% yield. Alkylation of NBoc- L-proline (60) with 59 gave diester 61 which was treated with ammonium acetate to effect cyclization of the bis-ketoester to provide bis-imidazole 62 in 63% yield for the two steps. Acidic removal of the Boc protecting groups followed by recrystallization provided bis-pyrrolidine 63 in high yield. Acylation of 63 with N-(methoxycarbonyl)- L-valine (64) using N-(3-dimethylaminopropyl)-N0-ethylcarbodiimide(EDC) and 1-hydroxybenxotriazole hydrate (HOBT) provided declatasvir. The dihydrochloride salt was prepared and treated with Cuno Zet Carbon followed by crystallization from acetone
to give daclatasvir dihydrochloride (IX) in 74% yield.
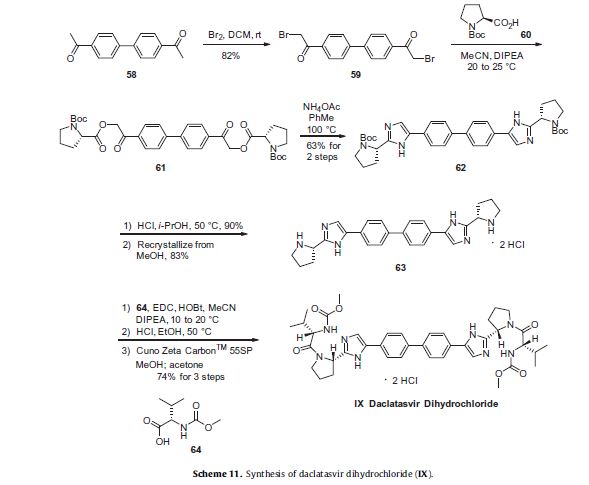
80 Belema, M.; Meanwell, N. A. J. Med. Chem. 2014, 57, 5057.
81. Bachand, C.; Belema, M.; Deon, D. H.; Good, A. C.; Goodrich, J.; James, C. A.;
Lavoie, R.; Lopez, O. D.; Martel, A.; Meanwell, N. A.; Nguyen, V. N.; Romine, J.
L.; Ruediger, E. H.; Snyder, L. B.; St. Laurent, D. R.; Yang, F.; Langley, D. R.;
Wang, G.; Hamann, L. G. WO Patent 2008021927A2, 2008.
82. Belema, M.; Nguyen, V. N.; Bachand, C.; Deon, D. H.; Goodrich, J. T.; James, C.
A.; Lavoie, R.; Lopez, O. D.; Martel, A.; Romine, J. L.; Ruediger, E. H.; Snyder, L.
B.; St Laurent, D. R.; Yang, F.; Zhu, J.; Wong, H. S.; Langley, D. R.; Adams, S. P.;
Cantor, G. H.; Chimalakonda, A.; Fura, A.; Johnson, B. M.; Knipe, J. O.; Parker, D.
D.; Santone, K. S.; Fridell, R. A.; Lemm, J. A.; O’Boyle, D. R., 2nd; Colonno, R. J.;
Gao, M.; Meanwell, N. A.; Hamann, L. G. J. Med. Chem. 2014, 57, 2013.
83. Pack, S. K.; Geng, P.; Smith, M. J.; Hamm, J. WO Patent 2009020825A1, 2009.
PATENT
EXAMPLES

A 1 L, 3-neck round bottom flask, fitted with a nitrogen line, overhead stirrer and thermocouple, was charged with 20 g (83.9 mmol, 1 equiv) 1,1′-(biphenyl-4,4′-diyl)diethanone, 200 mL CH2Cl2 and 8.7 mL (27.1 g, 169.3 mmol, 2.02 quiv) bromine. The mixture was allowed to stir under nitrogen for about 20 hours under ambient conditions. The resulting slurry was charged with 200 mL CH2Cl2 and concentrated down to about 150 mL via vacuum distillation. The slurry was then solvent exchanged into THF to a target volume of 200 mL via vacuum distillation. The slurry was cooled to 20-25° C. over 1 hour and allowed to stir at 20-25° C. for an additional hour. The off-white crystalline solids were filtered and washed with 150 mL CH2Cl2. The product was dried under vacuum at 60° C. to yield 27.4 g (69.2 mmol, 82%) of the desired product : 1H NMR (400 MHz, CDCl3) δ 7.95-7.85 (m, 4H), 7.60-7.50 (m, 4H), 4.26 (s, 4H); 13C NMR (100 MHz, CDCl3) 6 191.0, 145.1, 133.8, 129.9, 127.9, 30.8; IR (KBr, cm−1) 3007, 2950, 1691, 1599, 1199; Anal calcd for C16H12Br2O2: C, 48.52; H, 3.05; Br, 40.34. Found: C, 48.53; H, 3.03; Br, 40.53 HRMS calcd for C16H13Br2O2 (M+H; DCI+): 394.9282. Found: 394.9292. mp 224-226° C.

A 500 mL jacketed flask, fitted with a nitrogen line, thermocouple and overhead stirrer, was charged with 20 g (50.5 mmol, 1 equiv) of Compound 2, 22.8 g (105.9 moles, 2.10 equiv) 1-(tert-butoxycarbonyl)-L-proline and 200 mL acetonitrile. The slurry was cooled to 20° C. followed by the addition of 18.2 mL (13.5 g, 104.4 mmol, 2.07 equiv) DIPEA. The slurry was warmed to 25° C. and allowed to stir for 3 hours. The resulting clear, organic solution was washed with 3×100 mL 13 wt % aqueous NaCl. The rich acetonitrile solution was solvent exchanged into toluene (target volume=215 mL) by vacuum distillation until there was less than 0.5 vol % acetonitrile.

The toluene solution of Compound 3 was charged with 78 g (1.011 moles, 20 equiv) ammonium acetate and heated to 95-100° C. The mixture was allowed to stir at 95-100° C. for 15 hours. After reaction completion, the mixture was cooled to 70-80° C. and charged with 7 mL acetic acid, 40 mL n-butanol, and 80 mL of 5 vol % aqueous acetic acid. The resulting biphasic solution was split while maintaining a temperature >50° C. The rich organic phase was charged with 80 mL of 5 vol % aqueous acetic acid, 30 mL acetic acid and 20 mL n-butanol while maintaining a temperature >50° C. The resulting biphasic solution was split while maintaining a temperature >50° C. and the rich organic phase was washed with an additional 80 mL of 5 vol % aqueous acetic acid. The rich organic phase was then solvent exchanged into toluene to a target volume of 215 mL by vacuum distillation. While maintaining a temperature >60° C., 64 mL methanol was charged. The resulting slurry was heated to 70-75° C. and aged for 1 hour. The slurry was cooled to 20-25° C. over 1 hour and aged at that temperature for an additional hour. The slurry was filtered and the cake was washed with 200 mL 10:3 toluene:methanol. The product was dried under vacuum at 70° C., resulting in 19.8 g (31.7 mmol, 63%) of the desired product: 1H NMR (400 MHz, DMSO-d6) δ 13.00-11.00 (s, 2H), 7.90-7.75 (m, 4H), 7.75-7.60 (m, 4H), 7.60-7.30 (s, 2H), 4.92-4.72 (m, 2H), 3.65-3.49 (m, 2H), 3.49-3.28 (m, 2H), 2.39-2.1 (m, 2H), 2.10-1.87 (m, 6H), 1.60-1.33 (s, 8H), 1.33-1.07 (s, 10H); 13C NMR (100 MHz, DMSO-d6) δ 154.1, 153.8, 137.5, 126.6, 125.0, 78.9, 78.5, 55.6, 55.0, 47.0, 46.7, 33.7, 32.2, 28.5, 28.2, 24.2, 23.5; IR (KBr, cm−1) 2975, 2876, 1663, 1407, 1156, 1125; HRMS calcd for C36H45N6O4 (M+H; ESI+): 625.3502. Found: 625.3502. mp 190-195° C. (decomposed).

To a 250 mL reactor equipped with a nitrogen line and overhead stirrer, 25.0 g of Compound 4 (40.01 mmol, 1 equiv) was charged followed by 250 mL methanol and 32.85 mL (400.1 mmol, 10 equiv) 6M aqueous HCl. The temperature was increased to 50° C. and agitated at 50° C. for 5 hours. The resulting slurry was cooled to 20-25° C. and held with agitation for about 18 hours. Filtration of the slurry afforded a solid which was washed successively with 100 mL 90% methanol/water (V/V) and 2×100 mL of methanol. The wet cake was dried in a vacuum oven at 50° C. overnight to give 18.12 g (31.8 mmol, 79.4%) of the desired product.
Recrystallization of Compound 5
To a 250 mL reactor equipped with a nitrogen line and an overhead stirrer, 17.8 g of Compound 5 from above was charged followed by 72 mL methanol. The resulting slurry was agitated at 50° C. for 4 hours, cooled to 20-25° C. and held with agitation at 20-25° C. for 1 hour. Filtration of the slurry afforded a crystalline solid which was washed with 60 mL methanol. The resulting wet cake was dried in a vacuum oven at 50° C. for 4 days to yield 14.7 g (25.7 mmol, 82.6%) of the purified product: 1H NMR (400 MHz, DMSO-d6) δ 10.5-10.25 (br, 2H), 10.1-9.75 (br, 2H), 8.19 (s, 2H), 7.05 (d, J=8.4, 4H), 7.92 (d, J=8.5, 4H), 5.06 (m, 2H), 3.5-3.35 (m, 4H), 2.6-2.3 (m, 4H), 2.25-2.15 (m, 2H), 2.18-1.96 (m, 2H); 13C NMR (100 MHz, DMSO-d6) δ 156.6, 142.5, 139.3, 128.1, 127.5, 126.1, 116.9, 53.2, 45.8, 29.8, 24.3; IR (KBr, cm−1) 3429, 2627, 1636, 1567, 1493, 1428, 1028. Anal calcd for C26H32N6Cl4: C, 54.75; H, 5.65; Cl, 24.86; Adjusted for 1.9% water: C, 53.71; H, 5.76; N, 14.46; Cl, 24.39. Found: C, 53.74; H, 5.72; N, 14.50; Cl, 24.49; KF=1.9. mp 240° C. (decomposed).

A 1 L jacketed flask equipped with a nitrogen line and an overhead stirrer was sequentially charged with 100 mL acetonitrile, 13.69 g (89.4 mmol, 2.5 equiv) hydroxybenzotriazole hydrate, 15.07 g (86 mmol, 2.4 equiv) N-(methoxycarbonyl)-L-valine, 16.46 g (85.9 mmol, 2.4 equiv) 1-(3-dimethyaminopropyl)-3-ethylcarbodiimide hydrochloride and an additional 100 mL acetonitrile. The resulting solution was agitated at 20° C. for 1 hour and charged with 20.4 g (35.8 mmol, 1 equiv) of purified Compound 5. The slurry was cooled to about 0° C. and 18.47 g (142.9 mmol, 4 equiv) diisopropylethylamine was added over 30 minutes while maintaining a temperature below 10° C. The solution was slowly heated to 15° C. over 3 hours and held at 15° C. for 12 hours. The resulting solution was charged with 120 mL 13 wt % aqueous NaCl and heated to 50° C. for 1 hour. After cooling to 20° C., 100 mL of isopropyl acetate was added. The biphasic solution was filtered through a 0.45 μm filter and the mixture split. The rich organic phase was washed with 2×240 mL of a 0.5 N NaOH solution containing 13 wt % NaCl followed by 120 mL 13 wt % aqueous NaCl. The mixture was then solvent exchanged into isopropyl acetate by vacuum distillation with a target volume of 400 mL. The resulting hazy solution was cooled to 20° C. and filtered through a 0.45 μm filter. The clear solution was then solvent exchanged into ethanol by vacuum distillation with a target volume of 140 mL. While maintaining a temperature of 50° C., 66.4 mL (82.3 mmol, 2.3 equiv) of 1.24M HCl in ethanol was added. The mixture was then charged with 33 mg (0.04 mmol, 0.001 equiv) of seed crystals of Compound (I) (see preparation below) and the resulting slurry was stirred at 50° C. for 3 hours. The mixture was cooled to 20° C. over 1 hour and aged at that temperature for an additional 22 hours. The slurry was filtered and the wet cake was washed with 100 mL of 2:1 acetone:ethanol. The solids were dried in a vacuum oven at 70° C. to give 22.15 g (27.3 mmol, 76.3%) of the desired product.

A solution of Compound (I) was prepared by dissolving 3.17 g of Compound (I) from above in 22 mL methanol. The solution was passed through a 47 mm Cuno Zeta Carbon® 53SP filter at ˜5 psig at a flow rate of ˜58 mL/min. The carbon filter was rinsed with 32 mL of methanol. The solution was concentrated down to 16 mL by vacuum distillation. While maintaining a temperature of 40-50° C., 15.9 mL acetone and 5 mg of seed crystals of Compound (I) (see procedure below) were added. The resulting slurry was then charged with 32 mL acetone over 30 minutes. The slurry was held at 50° C. for 2 hours, cooled to 20° C. over about 1 hour and held at 20° C. for about 20 hours. The solids were filtered, washed with 16 mL 2:1 acetone:methanol and dried in a vacuum oven at 60° C. to give 2.14 g (67.5%) of purified Compound (I):
1H NMR (400 MHz, DMSO-d6, 80° C.): 8.02 (d, J=8.34 Hz, 4 H), 7.97 (s, 2 H), 7.86 (d, J=8.34 Hz, 4 H), 6.75 (s, 2 H), 5.27 (t, J=6.44 Hz, 2 H), 4.17 (t, J=6.95 Hz, 2 H), 3.97-4.11 (m, 2 H), 3.74-3.90 (m, 2 H), 3.57 (s, 6 H), 2.32-2.46 (m, 2 H), 2.09-2.31 (m, 6 H), 1.91-2.07 (m, 2 H), 0.88 (d, J=6.57 Hz, 6 H), 0.79 (d, J=6.32 Hz, 6 H);
13C NMR (75 MHz, DMSO-d6): δ 170.9, 156.9, 149.3, 139.1, 131.7, 127.1, 126.5, 125.9, 115.0, 57.9, 52.8, 51.5, 47.2, 31.1, 28.9, 24.9, 19.6, 17.7;
IR (neat, cm−1): 3385, 2971, 2873, 2669, 1731, 1650.
Anal. Calcd for C40H52N8O6Cl2: C, 59.18; H, 6.45; N, 13.80; Cl, 8.73. Found C, 59.98; H, 6.80; N, 13.68; Cl, 8.77. mp 267° C. (decomposed).
Preparation of Seed Crystals of Compound (I)
A 250 mL round-bottom flask was charged with 6.0 g (10.5 mmol, 1 equiv) Compound 5, 3.87 g (22.1 mmol, 2.1 equiv) N-(methoxycarbonyl)-L-valine, 4.45 g (23.2 mmol, 2.2 equiv) 1-(3-dimethyaminopropyl)-3-ethylcarbodiimide hydrochloride, 0.289 g (2.14 mmol, 0.2 equiv) 1-hydroxybenzotriazole, and 30 mL acetonitrile. The resulting slurry was then charged with 7.33 mL (42.03 mmol, 4 equiv) diisopropylethylamine and allowed to stir at 24-30° C. for about 18 hours. The mixture was charged with 6 mL of water and heated to 50° C. for about 5 hours. The mixture was cooled and charged with 32 mL ethyl acetate and 30 mL water. The layers were separated and the rich organic layer was washed with 30 mL of 10 wt % aqueous NaHCO3, 30 mL water, and 20 mL of 10 wt % aqueous NaCl. The rich organic layer was then dried over MgSO4, filtered, and concentrated down to a residue. The crude material was then purified via flash chromatography (silica gel, 0-10% methanol in dichloromethane) to provide the free base of Compound (I).
The free-base of Compound (I) (0.03 g) was dissolved in 1 mL isopropanol at 20° C. Anhydrous HCl (70 μL, dissolved in ethanol, approximately 1.25M concentration) was added and the reaction mixture was stirred. To the solution was added methyl tert-butyl ether (1 mL) and the resulting slurry was stirred vigorously at 40° C. to 50° C. for 12 hours. The crystal slurry was cooled to 20° C. and filtered. The wet cake was air-dried at 20° C. A white crystalline solid (Form N-2 of Compound (I)) was obtained.
Clip
Daclatasvir synthesis: WO2009020828A1

Procedure:
Step a: A 1 L, 3 -neck round bottom flask, fitted with a nitrogen line, overhead stirrer and thermocouple, was charged with 20 g (83.9 mmol, 1 equiv) 1,1′-(biphenyl-4,4′-diyl)diethanone, 200 mL Dichloromethane and 8.7 mL (27.1g, 169.3 mmol, 2.02 equiv) bromine. The mixture was allowed to stir under nitrogen for about 20 hours under ambient conditions. The resulting slurry was charged with 200 mL Dichloromethane and concentrated down to about 150 mL via vacuum distillation. The slurry was then solvent exchanged into THF to a target volume of 200 mL via vacuum distillation. The slurry was cooled to 20-25 0C over 1 hour and allowed to stir at 20-25 0C for an additional hour. The off-white crystalline solids were filtered and washed with 150 mL Dichloromethane. The product was dried under vacuum at 60 0C to yield 27.4 g (69.2 mmol, 82%) of the desired product: 1H NMR (400 MHz, CDCl3) d 7.95-7.85 (m, 4H), 7.60-7.50 (m, 4H), 4.26 (s, 4H); 13C NMR 100 MHz, CDCl3) d 191.0, 145.1, 133.8, 129.9, 127.9, 30.8; IR (KBr, cm-1) 3007, 2950, 1691, 1599, 1199; Anal calcd for C16H12Br2O2: C, 48.52; H, 3.05; Br, 40.34. Found: C, 48.53; H, 3.03; Br, 40.53. HRMS calcd for C16H12Br2O2 (M + H; DCI+): 394.9282. Found: 394.9292. mp 224-226 0C.
Step b: A 500 mL jacketed flask, fitted with a nitrogen line, thermocouple and overhead stirrer, was charged with 20 g (50.5 mmol, 1 equiv) of Compound 2, 22.8 g (105.9 moles, 2.10 equiv) 1-(tert-butoxycarbonyl)-L-proline and 200 mL acetonitrile. The slurry was cooled to 20 0C followed by the addition of 18.2 mL (13.5 g, 104.4 mmol, 2.07 equiv) DIPEA. The slurry was warmed to 25 0C and allowed to stir for 3 hours. The resulting clear, organic solution was washed with 3 x 100 mL 13 wt% aqueous NaCl. The rich acetonitrile solution was solvent exchanged into toluene (target volume = 215 mL) by vacuum distillation until there was less than 0.5 vol% acetonitrile.
Step c: The toluene solution of Compound 3 was charged with 78 g (1.011 moles, 20 equiv) ammonium acetate and heated to 95-100 0C. The mixture was allowed to stir at 95-100 0C for 15 hours. After reaction completion, the mixture was cooled to 70- 80 0C and charged with 7 mL acetic acid, 40 mL n-butanol, and 80 mL of 5 vol% aqueous acetic acid. The resulting biphasic solution was split while maintaining a temperature > 50 0C. The rich organic phase was charged with 80 mL of 5 vol% aqueous acetic acid, 30 mL acetic acid and 20 mL n-butanol while maintaining a temperature > 50 0C. The resulting biphasic solution was split while maintaining a temperature > 50 0C and the rich organic phase was washed with an additional 80 mL of 5 vol% aqueous acetic acid. The rich organic phase was then solvent exchanged into toluene to a target volume of 215 mL by vacuum distillation. While maintaining a temperature > 60 0C, 64 mL methanol was charged. The resulting slurry was heated to 70-75 0C and aged for 1 hour. The slurry was cooled to 20-25 0C over 1 hour and aged at that temperature for an additional hour. The slurry was filtered and the cake was washed with 200 mL 10:3 toluene:methanol. The product was dried under vacuum at 70 0C, resulting in 19.8 g (31.7 mmol, 63%) of the desired product: 1H NMR (400 MHz, DMSO-^) d 13.00-11.00 (s, 2H), 7.90-7.75 (m, 4H), 7.75-7.60 (m, 4H), 7.60-7.30 (s, 2H), 4.92-4.72 (m, 2H), 3.65-3.49 (m, 2H), 3.49-3.28 (m, 2H), 2.39-2.1 (m, 2H), 2.10-1.87 (m, 6H), 1.60-1.33 (s, 8H), 1.33-1.07 (s, 10H); 13C NMR (100 MHz, DMSO-?fe) d 154.1, 153.8, 137.5, 126.6, 125.0, 78.9, 78.5, 55.6, 55.0, 47.0, 46.7, 33.7, 32.2, 28.5, 28.2, 24.2, 23.5; IR (KBr, cm-1) 2975, 2876, 1663, 1407, 1156, 1125; HRMS calcd for C36H45N6O4 (M + H; ESI+): 625.3502. Found: 625.3502. mp 190-195 0C (decomposed).
Step d: To a 250 mL reactor equipped with a nitrogen line and overhead stirrer, 25.0 g of Compound 4 (40.01 mmol, 1 equiv) was charged followed by 250 mL methanol and 32.85 mL (400.1 mmol, 10 equiv) 6M aqueous HCl. The temperature was increased to 50 0C and agitated at 50 0C for 5 hours. The resulting slurry was cooled to 20-25 0C and held with agitation for about 18 hours. Filtration of the slurry afforded a solid which was washed successively with 100 mL 90% methanoI/water (WV) and 2 x 100 mL of methanol. The wet cake was dried in a vacuum oven at 50 0C overnight to give 18.12 g (31.8 mmol, 79.4%) of the desired product.
CUT PASTE…….WO2009020825

Preparation of Compound (I)
A 1 L jacketed flask equipped with a nitrogen line and an overhead stirrer was sequentially charged with 100 mL acetonitrile, 13.69 g (89.4 mmol, 2.5 equiv) hydroxybenzotriazole hydrate, 15.07 g (86 mmol, 2.4 equiv) N-(methoxycarbonyl)- L-valine, 16.46 g (85.9 mmol, 2.4 equiv) l-(3-dimethyaminopropyl)-3- ethylcarbodiimide hydrochloride and an additional 100 mL acetonitrile. The resulting solution was agitated at 20 0C for 1 hour and charged with 20.4 g (35.8 mmol, 1 equiv) of purified Compound 7. The slurry was cooled to about 0 0C and 18.47 g (142.9 mmol, 4 equiv) diisopropylethylamine was added over 30 minutes while maintaining a temperature below 10 0C. The solution was slowly heated to 15 0C over 3 hours and held at 15 0C for 12 hours. The resulting solution was charged with 120 mL 13 wt% aqueous NaCl and heated to 50 0C for 1 hour. After cooling to 20 0C, 100 mL of isopropyl acetate was added. The biphasic solution was filtered through a 0.45 μm filter and the mixture split. The rich organic phase was washed with 2 x 240 mL of a 0.5 Ν NaOH solution containing 13 wt% NaCl followed by 120 mL 13 wt% aqueous NaCl. The mixture was then solvent exchanged into isopropyl acetate by vacuum distillation with a target volume of 400 mL. The resulting hazy solution was cooled to 20 0C and filtered through a 0.45 μm filter. The clear solution was then solvent exchanged into ethanol by vacuum distillation with a target volume of 140 mL. While maintaining a temperature of 50 0C, 66.4 mL (82.3 mmol, 2.3 equiv) of 1.24M HCl in ethanol was added. The mixture was then charged with 33 mg (0.04 mmol, 0.001 equiv) of seed crystals of Compound (I) (see preparation below) and the resulting slurry was stirred at 50 0C for 3 hours. The mixture was cooled to 20 0C over 1 hour and aged at that temperature for an additional 22 hours. The slurry was filtered and the wet cake was washed with 100 mL of 2: 1 acetone:ethanol. The solids were dried in a vacuum oven at 70 0C to give 22.15 g (27.3 mmol, 76.3%) of the desired product.

Alternative Preparation of Compound (I)
A jacketed reactor equipped with a mechanical agitator, a thermocouple and a nitrogen inlet was sequentially charged with 10 L acetonitrile, 0.671 kg (4.38 moles, 2.50 equiv) 1-hydroxybenzotriazole, 0.737 kg (4.21 moles, 2.40 equiv) N- (methoxycarbonyl)-L-valine and 0.790 kg (4.12 moles, 2.35 equiv) l-(3- dimethyaminopropyl)-3-ethylcarbodiimide hydrochloride. The mixture was agitated at 200C for 1 hour, cooled to 5 0C and charged with 1 kg (1.75 moles, 1.00 equiv) Compound 7. While maintaining a temperature < 10 0C, 0.906 kg (7.01 moles, 4 equiv) diisopropylethylamine was added. The mixture was heated to 15-20 0C over 2 hours and agitated for an additional 15 hours. After the reaction was complete, the mixture was washed once with 6.0 L 13 wt% aqueous NaCl, twice with 6.1 L (6.12 moles, 3.5 equiv) 1.0 M aqueous NaOH containing 13 wt% NaCl and once with 6.0 L 13 wt% aqueous NaCl. Water was then removed from the rich organic solution via azeotropic distillation. The mixture was cooled to 20 0C, agitated for 1 hour and filtered. The rich organic solution was then solvent exchanged into EtOH via vacuum distillation to a target volume of 5 L. While maintaining a temperature of 50 0C, 3.2 L (4.0 moles, 2.3 equiv) 1.25M HCl in EtOH was charged. The mixture was seeded with 1.6 g Compound (I) (see preparation below) and agitated at 50 0C for 3 hours. The resulting slurry was cooled to 20 0C and agitated for at least 3 hours. The product was collected by filtration and washed with 5 L 2: 1 acetone:
EtOH to give 1.29 kg (ca. 90 wt% product) of wet crude product. A reactor equipped with an overhead agitator, nitrogen inlet and thermocouple was charged with 1.11 kg of the above crude product and 7 L methanol. The resulting solution was treated with Cuno Zeta Carbon (TM) 55SP. The carbon was washed with 15 L MeOH and the combined filtrate and wash was concentrated down to 4 L via vacuum distillation. The concentrated solution was charged with 5 L acetone and seeded with 1.6 g Compound (I) (see preparation below) while maintaining a temperature of 50 0C. An additional 10 L acetone was charged and the resulting slurry was stirred at 50 0C for 3 hours. The slurry was cooled to 20 0C and allowed to agitate at 200C for 3 hours. The product was collected by filtration, washed with 5 L 2: 1 acetone: EtOH and dried under vacuum at 50-60 0C to give 0.900 kg (1.11 moles, 74% adjusted) of Compound (I)-

Carbon Treatment and Recrystallization of Compound (I) A solution of Compound (I) was prepared by dissolving 3.17 g of Compound (I) from above in 22 mL methanol. The solution was passed through a 47mm Cuno Zeta Carbon 53SP filter at ~5 psig at a flow rate of~58mL/min. The carbon filter was rinsed with 32 mL of methanol. The solution was concentrated down to 16 mL by vacuum distillation. While maintaining a temperature of 40-50 0C, 15.9 mL acetone and 5 mg of seed crystals of Compound (I) (see procedure below) were added. The resulting slurry was then charged with 32 mL acetone over 30 minutes. The slurry was held at 50 0C for 2 hours, cooled to 20 0C over about 1 hour and held at 20 0C for about 20 hours. The solids were filtered, washed with 16 mL 2: 1 acetone:methanol and dried in a vacuum oven at 60 0C to give 2.14 g (67.5%) of purified Compound (I):
1H NMR (400 MHz, DMSO-έfc, 80 0C): 8.02 (d, J=8.34 Hz, 4 H), 7.97 (s, 2 H), 7.86 (d, J=8.34 Hz, 4 H), 6.75 (s, 2 H), 5.27 (t, J=6.44 Hz, 2 H), 4.17 (t, J=6.95 Hz, 2 H), 3.97 – 4.11 (m, 2 H), 3.74 – 3.90 (m, 2 H), 3.57 (s, 6 H), 2.32 – 2.46 (m, 2 H), 2.09 – 2.31 (m, 6 H), 1.91 – 2.07 (m, 2 H), 0.88 (d, J=6.57 Hz, 6 H), 0.79 (d, J=6.32 Hz, 6 H);
13C NMR (75 MHz, DMSO-έfc): δ 170.9, 156.9, 149.3, 139.1, 131.7, 127.1, 126.5, 125.9, 115.0, 57.9, 52.8, 51.5, 47.2, 31.1, 28.9, 24.9, 19.6, 17.7;
IR (neat, cm“1): 3385, 2971, 2873, 2669, 1731, 1650.
Anal. Calcd for C40H52N8O6Cl2: C, 59.18; H, 6.45; N, 13.80; Cl, 8.73. Found C, 59.98; H, 6.80; N, 13.68; Cl, 8.77. mp 267 0C (decomposed).
Characteristic diffraction peak positions (degrees 2Θ + 0.1) @ RT, based on a high quality pattern collected with a diffractometer (CuKa) with a spinning capillary with 2Θ calibrated with a NIST other suitable standard are as follows: 10.3, 12.4, 12.8, 13.3, 13.6, 15.5, 20.3, 21.2, 22.4, 22.7, 23.7
Daclatasvir faces problems in USA
The US-FDA in 2014 issued a complete response letter for NS5A inhibitor daclatasvir saying it was unable to approve the drug because the marketing application was for its use in tandem with asunaprevir, an NS3/NS4A protease inhibitor discontinued in the US by BMS for commercial reasons. Daclatasvir is already on the market in Europe-where it is sold as Daklinza-and also in Japan where it was approved alongside asunaprevir in July as the country’s first all-oral HCV therapy. However, a delay in the large US market is clearly a major setback for BMS’ ambitions in hepatitis therapy.
To make the matter worse, US FDA has rescinded breakthrough therapy designation status from Bristol-Myers Squibb for Daclatasvir for the treatment of hepatitis C virus infection in Feb 2015.
PAPER
Makonen, B.; et. al. Hepatitis C Virus NS5A Replication Complex Inhibitors: The Discovery of Daclatasvir. J Med Chem 2014, 57(5), 2013–2032.
http://pubs.acs.org/doi/abs/10.1021/jm401836p
PATENT
http://www.google.com/patents/WO2008021927A2?cl=en
Example 24-23

methyl ((lS)-l-(((2S)-2-(5-(4′-(2-((2S)-l-((2S)-2-((methoxycarbonyl)amino)-3- methylbutanoyl)-2-pyrrolidinyl)-lH-imidazol-5-yl)-4-biphenylyl)-lH-imidazol-2-yl)-
1 -pyrrolidinyl) carbonyl) -2-methylpropyl) carbamate
A 50 mL flask equipped with a stir bar was sequentially charged with 2.5 mL acetonitrile, 0.344 g (2.25 mmol, 2.5 equiv) hydroxy benzotriazole hydrate, 0.374 g (2.13 mmol, 2.4 equiv) N-(methoxycarbonyl)-L-valine, 0.400 g (2.09 mmol, 2.4 equiv) 1 -(3 -dimethyaminopropyl)-3-ethylcarbodiimide hydrochloride and an additional 2.5 mL acetonitrile. The resulting solution was agitated at 20 0C for 1 hour and charged with 0.501 g (0.88 mmol, 1 equiv) Example A-le-4. The slurry was cooled to about 0 0C and 0.45 g (3.48 mmol, 4 equiv) diisopropylethylamine was added over 30 minutes while maintaining a temperature below 10 0C. The solution was slowly heated to 15 0C over 3 hours and held at 15 0C for 16 hours. The temperature was increased to 20 0C and stirred for 3.25 hours. The resulting solution was charged with 3.3 g of 13 wt% aqueous NaCl and heated to 50 0C for 1 hour. After cooling to 20 0C, 2.5 mL of isopropyl acetate was added. The rich organic phase was washed with 2 x 6.9 g of a 0.5 N NaOH solution containing 13 wt% NaCl followed by 3.3 g of 13 wt% aqueous NaCl. The mixture was then solvent exchanged into isopropyl acetate by vacuum distillation to a target volume of 10 mL. The resulting hazy solution was cooled to 20 0C and filtered through a 0.45 μm filter. The clear solution was then solvent exchanged into ethanol by vacuum distillation with a target volume of 3 mL. 1.67 mL (2.02 mmol, 2.3 equiv) of 1.21 M HCl in ethanol was added. The mixture was then stirred at 25 0C for 15 hours. The resulting slurry was filtered and the wet cake was washed with 2.5 mL of 2: 1 acetone:ethanol. The solids were dried in a vacuum oven at 50 0C to give 0.550 g (0.68 mmol, 77 %) of the desired product.
RecrystalHzation of Example 24-23
A solution of Example 24-23 prepared above was prepared by dissolving 0.520 g of the above product in 3.65 mL methanol. The solution was then charged with 0.078 g of type 3 Cuno Zeta loose carbon and allowed to stir for 0.25 hours. The mixture was then filtered and washed with 6 ml of methanol. The product rich solution was concentrated down to 2.6 mL by vacuum distillation. 7.8 mL acetone was added and allowed to stir at 25 0C for 15 h. The solids were filtered, washed with 2.5 mL 2: 1 acetone:ethanol and dried in a vacuum oven at 70 0C to give 0.406 g (57.0%) of the desired product as white crystals: 1H NMR (400 MHz, OMSO-d6, 80 0C): 8.02 (d, J=8.34 Hz, 4 H), 7.97 (s, 2 H), 7.86 (d, J=8.34 Hz, 4 H), 6.75 (s, 2 H), 5.27 (t, J=6.44 Hz, 2 H), 4.17 (t, J=6.95 Hz, 2 H), 3.97 – 4.11 (m, 2 H), 3.74 – 3.90 (m, 2 H), 3.57 (s, 6 H), 2.32 – 2.46 (m, 2 H), 2.09 – 2.31 (m, 6 H), 1.91 – 2.07 (m, 2 H), 0.88 (d, J=6.57 Hz, 6 H), 0.79 (d, J=6.32 Hz, 6 H); 13C NMR (75 MHz, DMSO- d6): δ 170.9, 156.9, 149.3, 139.1, 131.7, 127.1, 126.5, 125.9, 115.0, 57.9, 52.8, 51.5, 47.2, 31.1, 28.9, 24.9, 19.6, 17.7; IR (neat, cm“1): 3385, 2971, 2873, 2669, 1731, 1650. Anal. Calcd for C40H52N8O6Cl2: C, 59.18; H, 6.45; N, 13.80; Cl, 8.73. Found C, 59.98; H, 6.80; N, 13.68; Cl, 8.77. mp 267 0C (decomposed). Characteristic diffraction peak positions (degrees 2Θ ± 0.1) @ RT, based on a high quality pattern collected with a diffractometer (CuKa) with a spinning capillary with 2Θ calibrated with a NIST other suitable standard are as follows: 10.3, 12.4, 12.8, 13.3, 13.6, 15.5, 20.3, 21.2, 22.4, 22.7, 23.7
PAPER
Bioorganic & Medicinal Chemistry Letters (2015), 25(16), 3147-3150
http://www.sciencedirect.com/science/article/pii/S0960894X15005995

Scheme 1.
Synthetic route for the preparation of the target compounds 8a–8y. Reagents and conditions: (a) Br2, CH2Cl2, rt, overnight, 86%; (b) N-Boc-l-proline, MeCN, Et3N, rt, 2 h, 98%; (c) NH4OAc, toulene, 130 °C, 15 h, 85%; (d) 6 N HCl, MeOH, 50 °C, 4 h, 87%; (e) HATU, N-(methoxycarbonyl)-l-valine, DIPEA, rt, 14 h, 83%; (f) RCOCl, TEA, CH2Cl2, rt, 3 h, 64–87%.
Dimethyl((2S,2’S)-((2S,2’S)-2,2′-(5,5′-([1,1′-biphenyl]-4,4′-diyl)bis(1H-imidazole-
5,2-diyl))bis(pyrrolidine-2,1-diyl))bis(3-methyl-1-oxobutane-2,1-
diyl))dicarbamate 7……………FREE BASE
To a solution of 5 (90 mg, 0.181 mmol), N-me-thoxycarbonyl-l-valine 6 (92 mg,0.525 mmol) and DIPEA (0.18 mL, 1.03 mmol) in DMF (5 mL) was added HATU(165.5 mg, 0.434 mmol). The resulting reaction was allowed to stir at room temperature for 15 h, the reaction mixture was filtered and the residue was partitioned between EtOAc and H2O, The aqueous phase was extracted with EtOAc, and the combined organic phase was dried (MgSO4), filtered, and concentrated in vacuo. The residue was purified by flash chromatography (silica gel; 5% Methanol /CH2Cl2) to
afford 7 (0.11 g, 83 %)as white solid.
1H NMR (DMSO-d6, 500 MHz) δ: 11.56 (s, 2H), 7.69-7.48 (m, 8H), 7.26-7.03 (m, 4H), 5.24-5.05 (m, 2H), 4.09-4.04 (m, 2H), 3.85-3.75 (m, 4H), 3.58 (s, 6H), 2.24-1.98 (m, 10H), 0.87 (d, J = 3.6 Hz, 12H).
Anal. calcd. (%) for C40H50N8O6: C 65.02, H 6.82, N 15.17; found: C 65.20, H 6.79, N 15.31.
ESI-MS m/z: 739.5 (M+H)+.
NMR PREDICT
1H NMR PREDICT


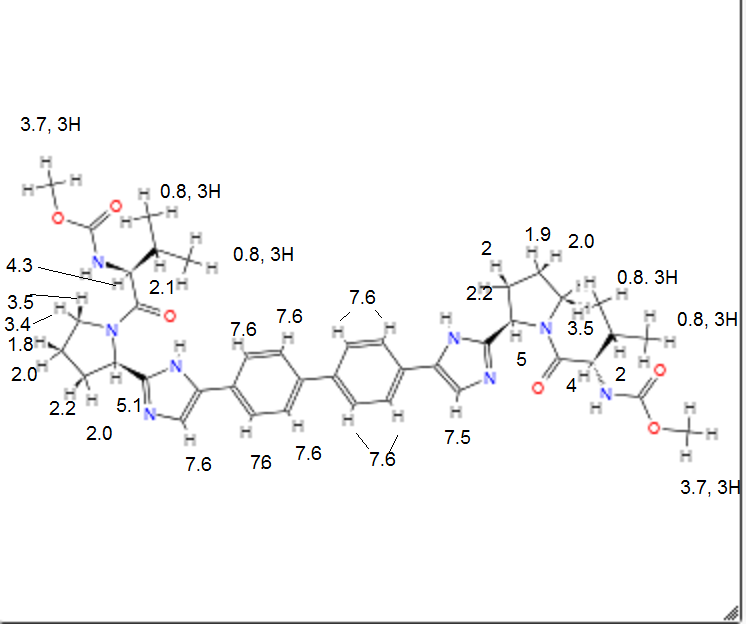
13C NMR PREDICT



COSY PREDICT

Patents
http://www.who.int/phi/implementation/ip_trade/daclatasvir_report_2014_09-02.pdf
Click on images to view
Click on images to view
http://www.who.int/phi/implementation/ip_trade/daclatasvir_report_2014_09-02.pdf

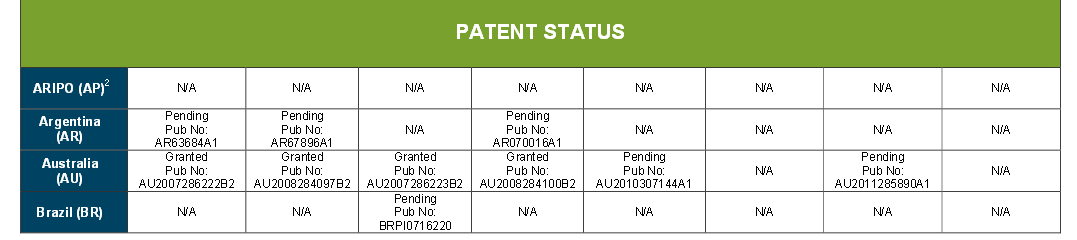
Click on images to view
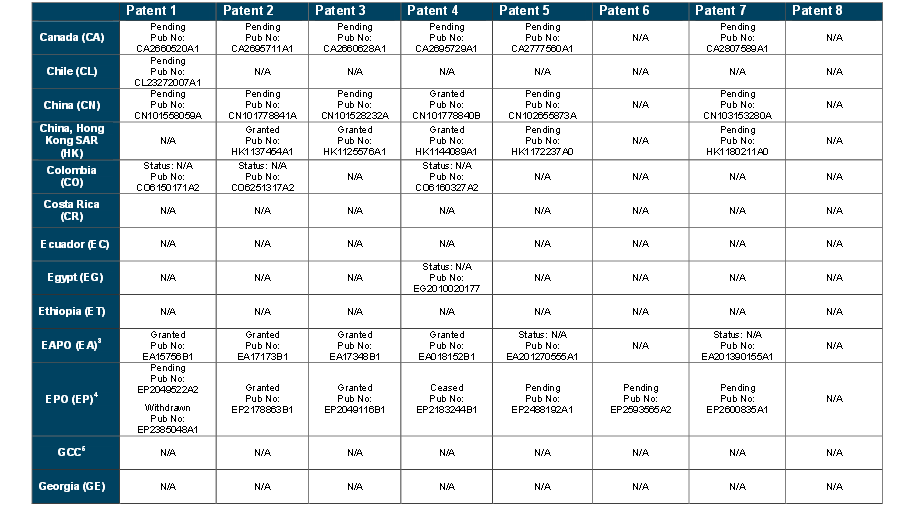
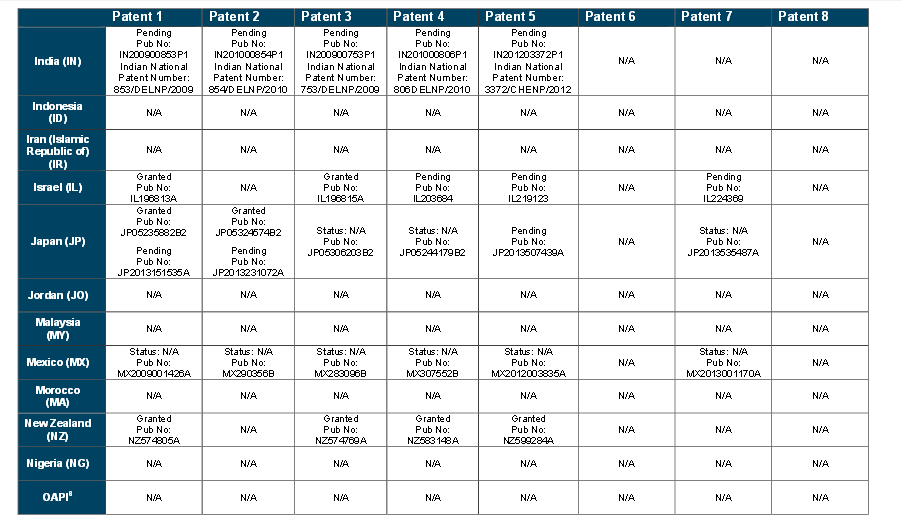
Click on images to view


Click on images to view
 |
|
| Names | |
|---|---|
| IUPAC name
Methyl [(2S)-1-{(2S)-2-[4-(4’-{2-[(2S)-1-{(2S)-2-[(methoxycarbonyl)amino]-3-methylbutanoyl}-2-pyrrolidinyl]-1H-imidazol-4-yl}-4-biphenylyl)-1H-imidazol-2-yl]-1-pyrrolidinyl}-3-methyl-1-oxo-2-butanyl]carbamate
|
|
| Other names
BMS-790052
|
|
| Identifiers | |
| 1009119-64-5 |
|
| ATC code | J05AX14 |
| ChEBI | CHEBI:82977 |
| ChEMBL | ChEMBL2023898 ChEMBL2303621 |
| ChemSpider | 24609522 |
| Jmol-3D images | Image |
| Properties | |
| C40H50N8O6 | |
| Molar mass | 738.89 g·mol−1 |
CLIP 1
Australian Government, National Measurement Institute
REFERENCE MATERIAL ANALYSIS REPORT
HPLC: Instrument: Shimadzu Binary pump LC-20AB, SIL-20 A HT autosampler
Column: X-Bridge C-18, 5.0 m (4.6 mm x 150 mm)
Column oven: 40 °C
Mobile Phase: A = Milli-Q water buffered at pH 10 with NH4
+ -OAc; B = MeCN
Gradient 0 min 35% B; 0-15 min 35% B; 15-18 min 35-75% B; 18-23 min 75% B.
Flow rate: 1.0 mL/min
Detector: Shimadzu SPD-M20A PDA operating at 310 nm
Relative peak area response of main component:
Initial analysis: Mean = 99.2%, s = 0.01%
Thermogravimetric analysis: Non volatile residue < 0.2% mass fraction . The volatile
content (e.g. organic solvents and/or water) could not be determined by
thermogravimetric analysis.
Karl Fischer analysis: Moisture content 0.6% mass fraction
QNMR: Instrument: Bruker Avance-III-500
Field strength: 500 MHz Solvent: DMSO-d6 (2.50 ppm)
Internal standard: Potassium hydrogen maleate (98.8% mass fraction)
Initial analysis: Mean (0.86 ppm) = 98.2%, s = 0.2%
LC-MS: Instrument: Thermo Scientific Dionex UltiMate 3000 Degasser,
Column: ZORBAX RRHD SB-C8, 2.1 x 50 mm, 1.8 μm (Agilent, 857700-906)
Column temp: 30.0 °C
Solvent system: Mobile phase A: 10 mM ammonium formate, 0.01% (v/v) formic acid in Milli-Q® water.
Mobile phase B: 0.01% (v/v) formic acid in acetonitrile.
Gradient from 90% A to 100% B
Flow rate: 0.25 mL/min
Sample prep: 2 mg/mL in MeOH with trace of formic acid
Injection volume: 10 L
Ionisation mode: Electrospray positive ion
Capillary voltage: 4.5 kV
Capillary temp: 360ºC Desolvation gas temperature: 300 ºC
Cone gas flow rate: 10 (arbitrary unit) Desolvation gas flow rate: 70 (arbitrary unit)
The retention time of daclatasvir is reported along with the major peak in the mass spectrum. The latter is reported as a mass/charge ratio.
9.98 min: 739.39545 (M+H+) m/z
HS-GC-MS: Instrument: Agilent 6890/5973/G1888
Column: DB-624, 30 m x 0.25 mm I.D. x 1.4 μm
Program: 50 C (5 min), 7 C/min to 120 C, 15 °C/min to 220 °C (8.3 min)
Injector: 150 C Transfer line temp: 280 C
Carrier: Helium, 1.2 mL/min Split ratio: 50/1
Solvents detected: Ethyl acetate
TLC: Conditions: Kieselgel 60F254. Ethyl acetate : methanol (95/5)
Single spot observed, Rf = 0.18. Visualisation with UV at 254 nm
The TLC was performed on the liberated free base.
IR: Instrument: Bruker Alpha FT-IR
Range: 4000-400 cm-1, neat
Peaks: 1723, 1697, 1643, 1523, 1439, 1235, 1099, 1024 cm-1
1H NMR: Instrument: Bruker Avance III 500
Field strength: 500 MHz Solvent: DMSO-d6 (2.50 ppm)
Spectral data: 0.77 (6H, d, J = 6.7 Hz), 0.83 (6H, d, J = 6.7 Hz), 2.01 (2H, m), 2.07 (2H, m), 2.12-2.27 (4H, m), 2.38 (2H, m), 3.54 (6H, s), 3.84 (2H, m), 3.97 (2H, m), 4.12 (2H, t, J = 7.7 Hz), 5.18 (2H, t, J = 7.0 Hz), 7.31 (2 N-H, d, J = 8.5 Hz), 7.94 (4H, d, J = 8.4 Hz), 7.99 (4H, d, J = 8.4 Hz), 8.16 (2H, s) ppm
Ethyl acetate estimated at 0.6% mass fraction was observed in the 1H NMR
13C NMR: Instrument: Bruker Avance III 500
Field strength: 126 MHz Solvent: DMSO-d6 (39.5 ppm)
Spectral data: 17.8, 19.6, 25.0, 29.0, 31.2, 47.3, 51.6, 52.9, 58.0, 115.1, 125.9, 126.6, 127.3, 131.8, 139.2, 149.4, 157.0, 171.1 ppm
Melting point: > 250 oC
Microanalysis: Found: C = 59.0%; H = 6.5%; N = 13.7% (August 2015)
Calc: C = 59.2%; H = 6.5%; N = 13.8% (Calculated for C40H50N8O6.2HCl)
REFERENCE
Australian NMI NATA Certification Daclatasvir – FixHepC
https://fixhepc.com/images/coa/NMI-NATA-Daclatasvir-Certification.pdf
Oct 7, 2015 – Compound Name: Daclatasvir dihydrochloride … Note: The assigned stereochemistry of this sample of daclatasvir has not …. Melting point:.
CLIP 2
Full Text Article – European Journal of Pharmaceutical and Medical …
Nov 28, 2016 – Daclatasvir dihydrochloride (DCLD) is a new drug …. DSC thermogram of daclatasvirdihydrochloriderealed drug melting point at 273.600C as …
CLIP 3
DCV dihydrochloride (anhydrous) is a white to yellow, non hygroscopic powder which is highly soluble in water (>700mg/mL). Solubility is higher at low pH. In aqueous buffers over the physiological pH range (pH 1.2-6.8) solubility is very low (4mg/mL to 0.004 mg/mL) due to the slow formation of the less soluble hydrated form. Water content in the drug substance is adequately controlled by in process tests. The desired anhydrous crystalline form of DCV dihydrochloride (N-2) is consistently produced and has been shown to not change on storage.
[DOC]AusPAR Daclatasvir dihydrochloride – Therapeutic Goods Administration
https://www.tga.gov.au/sites/default/…/auspar-daclatasvir–dihydrochloride-151214.do…
Dec 14, 2015 – Australian Public Assessment Report for daclatasvir dihydrochloride …. Figure 1:Chemical structure of daclatasvir dihydrochloride. …… 24 weeks is based on a selected literaturereview mostly of studies in patients with GT-1.
CLIP 4
The structure of the active substance has been confirmed by UV, IR, Raman and 1 H and 13C NMR spectroscopy, MS spectrometry, and crystal X-Ray diffraction.
Daclatasvir is a white to yellow crystalline non-hygroscopic powder. It is freely soluble in water, dimethyl sulfoxide, methanol; soluble in ethanol (95%); practically insoluble in dichloromethane, tetrahydrofuran, acetonitrile, acetone and ethyl acetate.
Daclatasvir is a chiral molecule with four stereocenters (1,1’, 2, 2;) in the S configuration. The synthetic strategy and process design such as starting material and reagent selection, process parameters, and in-process controls ensure the desired configuration at each of the four chiral centers. In addition, the established control strategy minimizes epimerization and eliminates other diastereomeric impurity formation in each step.
Polymorphism has been observed for daclatasvir hydrochloride. Although two neat crystalline dihydrochloride salts, N1 and N-2 have been identified in screening studies, it has been confirmed that the form N-2 is the thermodynamically most stable polymorph and only this form produced by the proposed synthetic process.
Manufacture, characterisation and process controls
Daclatasvir dihydrochloride is synthesised in three main steps using three commercially available well defined starting materials with acceptable specifications. The synthesis involves an alkylation and formation of the imidazole ring, a coupling reaction and the formation of the hydrochloride salt.
As mentioned above, the synthetic process has been designed to ensure the correct configuration at each of the four chiral centres is achieved. In addition, it has been demonstrated that the stereogenic centres do not epimerize during normal or stressed processing conditions.
The manufacturing process has been developed using a combination of conventional univariate studies and elements of QbD such as risk assessment.
The characterisation of the active substance and its impurities are in accordance with the EU guideline on chemistry of new active substances. Potential and actual impurities were well discussed with regards to their origin and characterised. Adequate in-process controls are applied during the synthesis. The specifications and control methods for intermediate products, starting materials and reagents have been presented.
The active substance specification includes tests for: appearance, colour, identity (IR/Raman, HPLC), assay (HPLC), impurities (HPLC), residual solvents (GC), HCl content (titration), total inorganic impurities (ICP-MS), and particle size (laser light scattering). The absence of a test for chiral purity in the active substance specification has been adequately justified based on the stereochemical control during the synthetic process and demonstration that there is no epimerization during normal or stressed processing conditions. Similarly, since the N-2 form of daclatasvir hydrochloride is the thermodynamically most stable polymorph and, is consistently produced by the synthetic process and remained unchanged during storage under long-term or accelerated conditions, this parameter is not included in the specification
CLIP5
SEE
http://www.accessdata.fda.gov/drugsatfda_docs/nda/2015/206843Orig1s000ChemR.pdf
CLIP6
Daclatasvir dihydrochloride
References
- 1 Statement on a Nonproprietary Name Adopted by the USAN Council
- 2 Gao, Min; Nettles, Richard E.; Belema, Makonen; Snyder, Lawrence B.; Nguyen, Van N.; Fridell, Robert A.; Serrano-Wu, Michael H.; Langley, David R.; Sun, Jin-Hua; O’Boyle, Donald R., II; Lemm, Julie A.; Wang, Chunfu; Knipe, Jay O.; Chien, Caly; Colonno, Richard J.; Grasela, Dennis M.; Meanwell, Nicholas A.; Hamann, Lawrence G. (2010). “Chemical genetics strategy identifies an HCV NS5A inhibitor with a potent clinical effect”. Nature 465 (7294): 96–100. doi:10.1038/nature08960. PMID 20410884.
- 3 Bell, Thomas W. (2010). “Drugs for hepatitis C: unlocking a new mechanism of action”. ChemMedChem 5 (10): 1663–1665. doi:10.1002/cmdc.201000334. PMID 20821796.
- 4 Modeling shows that the NS5A inhibitor daclatasvir has two modes of action and yields a shorter estimate of the hepatitis C virus half-life. Guedj, J et al. Proceedings of the National Academy of Sciences. February 19, 2013.
- 5 AASLD: Daclatasvir with Pegylated Interferon/Ribavirin Produces High Rates of HCV Suppression. Highleyman, L. HIVandHepatitis.com. 6 December 2011.
- 6Preliminary Study of Two Antiviral Agents for Hepatitis C Genotype 1. Lok, A et al. New England Journal of Medicine. 366(3):216-224. January 19, 2012.
- 7“Bristol-Myers’ Daclatasvir, Asunaprevir Cured 77%: Study”. Bloomberg. Apr 19, 2012.
- 8AASLD: Daclatasvir plus Asunaprevir Rapidly Suppresses HCV in Prior Null Responders. Highleyman, L. HIVandHepatitis.com. 8 November 2011.
- 9High rate of response to BMS HCV drugs in harder-to-treat patients – but interferon-free prospects differ by sub-genotype. Alcorn, K. Aidsmap.com. 12 November 2012.
- 10AASLD 2012: Sofosbuvir + Daclatasvir Dual Regimen Cures Most Patients with HCV Genotypes 1, 2, or 3. Highleyman, L. HIVandHepatitis.com. 15 November 2012.
- 11Mark Sulkowski et al. (January 16, 2014). “Daclatasvir plus Sofosbuvir for Previously Treated or Untreated Chronic HCV Infection”. New England Journal of Medicine. doi:10.1056/NEJMoa1306218.
- 12“www.who.int” (PDF).
| WO2004005264A2 * | 7 Jul 2003 | 15 Jan 2004 | Axxima Pharmaceuticals Ag | Imidazole compounds for the treatment of hepatitis c virus infections |
| WO2008021927A2 * | 9 Aug 2007 | 21 Feb 2008 | Squibb Bristol Myers Co | Hepatitis c virus inhibitors |
| WO2008021928A2 * | 9 Aug 2007 | 21 Feb 2008 | Squibb Bristol Myers Co | Hepatitis c virus inhibitors |
| WO2008021936A2 * | 9 Aug 2007 | 21 Feb 2008 | Squibb Bristol Myers Co | Hepatitis c virus inhibitors |
सुकून उतना ही देना प्रभू, जितने से जिंदगी चल जाये।औकात बस इतनी देना,कि औरों का भला हो जाये।………..P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
सुकून उतना ही देना प्रभू, जितने से जिंदगी चल जाये। औकात बस इतनी देना, कि औरों का भला हो जाये।
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
Join me on twitter
Join me on google plus  Googleplus
Googleplus
Googleplus amcrasto@gmail.com
amcrasto@gmail.com
 LIONEL MY SON
LIONEL MY SONHe was only in first standard in school when I was hit by a deadly one in a million spine stroke called acute transverse mylitis, it made me 90% paralysed and bound to a wheel chair, Now I keep him as my source of inspiration and helping millions, thanks to millions of my readers who keep me going and help me to keep my son happy

सुकून उतना ही देना प्रभू, जितने से
जिंदगी चल जाये।
औकात बस इतनी देना,
कि औरों का भला हो जाये।
जिंदगी चल जाये।
औकात बस इतनी देना,
कि औरों का भला हो जाये।
//////////
BMS-582949 in phase 2 for Treatment of Antipsoriatics , Rheumatoid arthritis
BMS 582949, PS-540446
UNII-CR743OME9E
CAS 623152-17-0
4-[5-(N-Cyclopropylcarbamoyl)-2-methylphenylamino]-5-methyl-N-propylpyrrolo[2,1-f][1,2,4]triazine-6-carboxamide
4-(5-(Cyclopropylcarbamoyl)-2-methylphenylamino)-5-methyl-N-propylpyrrolo[1,2-f][1,2,4]triazine-6-carboxamide
Bristol-Myers Squibb Company
M.Wt: 406.48
Cas : 623152-17-0 Formula: C22H26N6O2
BMS-582949 had been in phase II clinical trials at Bristol-Myers Squibb for the oral treatment of moderate to severe psoriasis and for the treatment of rheumatoid arthritis (RA) in combination with methotrexate and for the treatment of inflammation in atherosclerotic plaque. However, no recent development has been reported for this research.
…………………..
http://www.google.com/patents/WO2012031057A1?cl=en
The present invention generally relates to a method of treating resistant rheumatic disease, such as refractory rheumatoid arthritis, with a therapeutically effective amount of a dual action p38 inhibitor that is safe and well-tolerated. A dual action p38 kinase inhibitor is a compound that inhibits both activation of p38 kinase and p38 kinase activity in cells.
A large number of cytokines participate in the inflammatory response, including IL- 1 , IL-6, IL-8 and TNF-a. Overproduction of cytokines such as IL-1 and TNF-a are implicated in a wide variety of diseases, including inflammatory bowel disease, rheumatoid arthritis, psoriasis, multiple sclerosis, endotoxin shock, osteoporosis, Alzheimer’s disease, and congestive heart failure, among others. See e.g., Henry et al., Drugs Fut. , 24: 1345- 1354 ( 1999); Salituro et al., Curr. Med. Ckem., 6:807-823 (1999)]. Important mediators of proinflammatory cytokines such as TNFct and IL-1 β,. as well as cellular responses to such cytokines production, are the mitogen-activated protein (MAP) kinases, and in particular, p38 kinase. See e.g., Schieven, G.L., “The biology of p38 kinase: a central role in inflammation”, Current Topics in Medicinal Chemistry, 5 :921 – 928 (2005). Accordingly, modulation of p38 kinase may be useful in the treatment of inflammatory disease including rheumatic diseases such as rheumatoid arthritis (RA).
Compounds that reportedly inhibit p38 kinase and cytokines such as IL-1 and TNF-a for use in treating inflammatory diseases are disclosed in U.S. Patent Nos.
6,277,989 and 6, 130,235 to Scios, Inc; U.S. Patent. Nos. 6, 147,080 and 5,945,41 8 to Vertex Pharmaceuticals Inc; U.S. Patent Nos. 6,251 ,914, 5,977, 103 and 5,658,903 to Smith-Kline Beecham Corp.; U.S. Patent Nos. 5,932,576 and 6,087,496 to G.D. Searle & Co.; WO 00/56738 and WO 01 /27089 to Astra Zeneca; WO 01/34605 to Johnson & Johnson; WO 00/12497 (quinazoHne derivatives as p38 kinase inhibitors); WO 00/56738 (pyridine and pyrimidine derivatives for the same purpose); WO 00/12497 (discusses the relationship between p38 kinase inhibitors); and WO 00/12074 (piperazine and piperidine compounds useful as p38 inhibitors). Other compounds that inhibit p38 kinase are pyrrolotriazine aniline compounds, information on these compounds is disclosed in U.S. Patent Nos. 6,670,357; 6,867,300; 7,034, 151 ; 7, 160,883; 7,21 1,666; 7,253, 167; and U.S. Publication Nos. 2003/023283 1 (published Dec. 18, 2003); 2004/0229877 (published Nov. 1 8, 2004); 2005/0043306 (published Feb. 24, 2005; 2006/0003967 (published Jan. 5, 2006); 2006/0030708 (published Feb. 9, 2006); 2006/0041 124 (published Feb. 23, 2006); 2006/0229449 (published Oct. 12, 2006); 2006/0235020 (published Oct. 19, 2006); and 2007/0213300 (published Sept 13, 2007).
In particular, WO 2003/090912 (U.S. Patent Nos. 7, 160,883, 7,388,009, p38 inhibitor, BMS-582949 (Example 7,

including processes of making and uses thereof.
……………………
http://www.google.com/patents/WO2003090912A9?cl=en
Examples 4-22

Compounds having the formula (Id), above, wherein R4 has the values listed in the following Table, were prepared following the same procedure described for Example 3, using the appropriate amine in place of ra-butylamine.



…………………………
WO 2006020904
http://www.google.com.br/patents/WO2006020904A1?cl=en
EXAMPLE IA St


Part a.
A solution of Example 1 (0.86 g, 2.20 mmol, 1.0 eq.) in THF (4.0 mL) and 1 N aqueous NaOH (9.0 mL, 4.1 eq.) was stirred at 6O0C overnight. After cooling to RT, the reaction mixture was concentrated in vacuo but not to dryness. To the solution at O0C was added 1 N aqueous hydrochloric acid until it was acidic and the precipitate was collected and dried to afford crude Example IA acid (0.51 g, 64.0 % yield). HPLC Ret. t. = 2.400 min.; LC/MS (M+H) + = 366.06+. The filtrate was then extracted with EtOAc (3x) and the organic layers were combined, dried over sodium sulfate, and concentrated in vacuo to give Example IA acid (0.035 g, 4.4 % yield). Part b.

A solution of Part a. acid (0.026 g, 0.071 mmol, 1.0 eq.), EDC (0.021 g, 0.11 mmol, 1.5 eq.), HOBt (0.015 g, 0.11 mmol, 1.5 eq), ^-propylamine (0.015 mL, 0.15 mmol, 2.1 eq.) and DIPEA (0.040 mL, 0.23 mmol, 3.2 eq.) in DMF (0.20 mL) was shaken at RT overnight. Water (1 mL) was added and the precipitate collected by filtration, washed with water, and dried to give Example IA amide (0.021 g, 70% yield); HPLC Ret. t. = 2.883 min.; LC/MS (M+H)+ = 421.18 +.
EJiAMPLE 2 Direct Aminolysis Procedure

n-Buli/THF
Ester Compound I or Hexyllithium/THF
-^
,NH9

1. Aminolysis with hexyllithium
To a dried 100 ml flask was added THF (10 ml) under nitrogen, which was then cooled to -100C. Hexyllithium (2.3 M in hexane, 6.5 ml, 15.0 mmol) was added slowly (exothermic, temperature was up to 5°C), followed by dropwise addition of propylamine (1.01 g, 1.4 ml, 17.1 mmol) at such a rate to maintain the temperature below 5°C. The resulting mixture was stirred at O0C for 20 minutes. A suspension of ester compound I (1.0 g, 2.5 mmol) in THF (12 ml) was added over a 10 minute period (exothermic, T<5°C). After being stirred at 00C for 20 minutes, the mixture was allowed to warm to room temperature and stirred for 5 hours. Ester compound I was <0.1 AP at this point by HPLC analysis. The mixture was cooled to -50C. Acetic acid (2 ml) was added slowly to maintain the temperature <10°C. The resulting thick slurry was stirred at room temperature for 20 minutes, and then solvents were exchanged with DMF (15 ml) on a rotavapor. To the resulting yellow slurry, water (15 ml) was added slowly to keep T<25°C. During the addition of water, the slurry became a clear solution, and a new slurry was formed. The slurry was stirred at room temperature for overnight. In the morning the slurry was filtered and the solid was washed with DMF/water (1:1, 5 ml), water (5 ml) and acetone (5 ml). The cake was dried under vacuum at 55°C for 24 hours to afford 0.90 g of amide product II (yield: 87.2%) as a white solid. HPLC: 99.70 AP.
2. Aminolysis with n-butyllithium
To a dried 100 ml of flask was added THF (10 ml) under nitrogen and then cooled to -100C. n-Butyllithium (2.5 M in hexane, 6.0 ml, 15.0 mmol) was added slowly, followed by dropwise addition of propylamine (0.98 g, 16.5 mmol) at such a rate to keep the temperature below 00C. The resulting mixture was stirred at O0C for 20 minutes. A suspension of ester compound I (1.0 g, 2.5 mmol) in THF (12 ml) was added over a 10 minute period (T<5°C). After being stirred at O0C for 30 minutes, the mixture was allowed to warm to room temperature and stirred for overnight (~22h, Note 1). Compound I was not detected at this point by HPLC analysis. The mixture was cooled to -7°C. Acetic acid (2 ml) was added dropwise to maintain the temperature <10°C. The resulting thick slurry was stirred at 50C for 2 hours and at room temperature for 20 minutes, followed by evaporation on a rotavapor to give a wet yellow solid. To this solid was added acetone (10 ml) and water (20 ml). The slurry was stirred at room temperature for one and half hours. Filtration gave a white solid. This solid was washed with 35% acetone in water (10 ml), water (5 ml) and acetone (5 ml). The cake was dried under vacuum at 55°C for the weekend to afford 0.94g of amide product II (yield: 91.0%) as a white solid. HPLC: 99.76 AP. Note 1: Compound I was -0.056 AP at 2.5 hours.
……………………
WO 2003090912
http://www.google.com/patents/WO2003090912A1?cl=en
……………………..
Discovery of 4-(5-(Cyclopropylcarbamoyl)-2-methylphenylamino)-5-methyl-N-propylpyrrolo[1,2-f][1,2,4]triazine-6-carboxamide (BMS-582949), a clinical p38a MAP kinase inhibitor for the treatment of inflammatory diseases
J Med Chem 2010, 53(18): 6629
http://pubs.acs.org/doi/abs/10.1021/jm100540x
The discovery and characterization of 7k (BMS-582949), a highly selective p38α MAP kinase inhibitor that is currently in phase II clinical trials for the treatment of rheumatoid arthritis, is described. A key to the discovery was the rational substitution of N-cyclopropyl for N-methoxy in 1a, a previously reported clinical candidate p38α inhibitor. Unlike alkyl and other cycloalkyls, the sp2 character of the cyclopropyl group can confer improved H-bonding characteristics to the directly substituted amide NH. Inhibitor 7k is slightly less active than 1a in the p38α enzymatic assay but displays a superior pharmacokinetic profile and, as such, was more effective in both the acute murine model of inflammation and pseudoestablished rat AA model. The binding mode of 7k with p38α was confirmed by X-ray crystallographic analysis.

4-(5-(Cyclopropylcarbamoyl)-2-methylphenylamino)-5-methyl-N-propylpyrrolo[1,2-f][1,2,4]triazine-6-carboxamide (7k)
A mixture of 4-(5-(cyclopropylcarbamoyl)-2-methylphenylamino)-5-methylpyrrolo[1,2-f][1,2,4]triazine-6-carboxylic acid (6b) (2.16 g, 5.91 mmol), n-propylamine (1.0 mL, 12.2 mmol), BOP (3.40 g, 7.69 mmol), and N-methylmorpholine (2.5 mL, 22.7 mmol) in DMF (10 mL) was stirred at 50 °C for 3 h. The mixture was poured into a mixture prepared from saturated NaHCO3 solution (60 mL) and water (60 mL). The precipitating product was collected by suction filtration was washed with water. This crude product was suspended into ethyl acetate (100 mL) and stirred at 70 °C for 1 h. Upon cooling to rt, the title compound (2.07 g, 86% yield) was collected as a white solid by suction filtration; 98% purity by HPLC. LCMS (EI)
m/z Calcd for C22H26N6O2 (M + H)+ = 407.21. Found: 407.22.
1H NMR (500 MHz, DMSO-d6) δ 8.49 (d, J = 3.6 Hz, 1H), 8.23 (s, 1H), 8.21 (s, 1H), 7.86 (s, 1H), 7.80 (s, 1H), 7.77 (d, J = 7.8 Hz, 1H), 7.42 (d, J = 7.8 Hz, 1H), 3.20 (m, 2H), 2.87 (m, 1H), 2.82 (s, 3H), 2.25 (s, 3H), 1.54 (m, 2H), 0.91 (t, J = 7.4 Hz, 3H), 0.68 (m, 2H), 0.59 (m, 2H).
13C NMR (125 MHz, DMSO-d6) δ 167.3, 164.45, 155.3, 148.7, 138.8, 137.1, 133.0, 130.6, 127.2, 125.8, 119.6, 118.8, 114.4, 113.3, 41.0, 23.6, 23.1, 18.5, 12.1, 12.0, 6.2.
EXAMPLE 3

Direct Aminolysis
Ester Compound I

Amide Product II
Method A:
A solution of n-propylamine (6.5 eq) in THF (20 ml/g of ester compound I) was cooled to — 5°C and was slowly treated with 2.5 M solution of n-butyllithium (6.1 eq). The mixture was stirred for 10 minutes. At the end of the period, a slurry of ester compound I (1 eq) in THF (14 ml/g of ester compound I) was cannulated into the performed Li-NHPr solution. The reaction mixture was warmed to 25°C and stirred till all of ester compound I was consumed (~ 3 hours). After the reaction was judged to be completed by HPLC, the reaction mixture was cooled to ~0°C and was slowly treated with acetic acid (5 ml/g of ester compound I). The slurry was then warmed to -2O0C and was stirred for 1 hour. At the end of the period, the solvent was distilled under vacuum to the minimum volume and the concentrated slurry was diluted with a solution of acetone (10 ml/g of ester compound I) and water (20 ml/g of ester compound I). The slurry was stirred for 1 hour and was cooled to ~5°C. The slurry was filtered and the cake was washed with acetone (5 ml/g of ester compound I). The cake was dried to give the amide product II (typically in 85% yield and 99 AP).
Method B:
A solution of n-propylamine (20 eq) in 2,2,2-trifmoroethanol (10 ml/g of ester compound I) was slowly treated with 2.5 M solution of n-butyllithium (1.5 eq). The mixture was stirred for 5 minutes. At the end of the period, the starting material, ester compound I, was added and the reaction mixture was warmed to 900C. The reaction mixture was held at 900C for 24 hours and was allowed to cool to ~20°C. The reaction mixture was then analyzed by HPLC. Typically, analysis indicated there was only 1.57 AP of starting material left.
Method C:
A solution of n-propylamine (2 eq) in methylene chloride (10 ml/g of ester compound I) at 200C was slowly treated with 2.0 M solution of trimethylaluminum (4 eq) in hexanes. The mixture was stirred for 15 minutes. At the end of the period, the starting material, ester compound 1 (1 eq), was added and the reaction mixture was warmed to 600C. The reaction mixture was held at 600C for 24 hours and was allowed to cool to ~20°C. The reaction mixture was then slowly quenched with aqueous HCl solution and analyzed by HPLC. Typically, analysis indicated there was 96.8AP of amide compound II product with 0.03 AP of the dipropylamide impurity.
…………………………………….
| WO2003090912A1 * | 15 abr. 2003 | 6 nov. 2003 | Squibb Bristol Myers Co | Pyrrolo-triazine aniline compounds useful as kinase inhibitors |
Liu C, Lin J, Everlof G, Gesenberg C, Zhang H, Marathe PH, Malley M, Galella MA, McKinnon M, Dodd JH, Barrish JC, Schieven GL, Leftheris K.
Bioorg Med Chem Lett. 2013 May 15;23(10):3028-33. doi: 10.1016/j.bmcl.2013.03.022. Epub 2013 Mar 15.
Freebern WJ, Bigwarfe TJ, Price KD, Haggerty HG.
J Immunotoxicol. 2013 Jan-Mar;10(1):106-17. doi: 10.3109/1547691X.2012.736427. Epub 2012 Nov 23.
Liu C, Lin J, Wrobleski ST, Lin S, Hynes J, Wu H, Dyckman AJ, Li T, Wityak J, Gillooly KM, Pitt S, Shen DR, Zhang RF, McIntyre KW, Salter-Cid L, Shuster DJ, Zhang H, Marathe PH, Doweyko AM, Sack JS, Kiefer SE, Kish KF, Newitt JA, McKinnon M, Dodd JH, Barrish JC, Schieven GL, Leftheris K.
J Med Chem. 2010 Sep 23;53(18):6629-39. doi: 10.1021/jm100540x.
BMS-582949: crystalline form of a p38alpha inhibitor? WO2008079857.
Norman P.
Expert Opin Ther Pat. 2009 Aug;19(8):1165-8. doi: 10.1517/13543770902816160.
| WO2000012074A2 | Aug 27, 1999 | Mar 9, 2000 | Sarvajit Chakravarty | Use of piperidines and/or piperazines as inhibitors of p38-alpha kinase | |
| WO2000012497A2 | Aug 27, 1999 | Mar 9, 2000 | Sarvajit Chakravarty | Quinazoline derivatives as medicaments | |
| WO2000056738A1 | Mar 17, 2000 | Sep 28, 2000 | Astrazeneca Ab | Pyridine and pyrimidine derivatives and their use as inhibitors of cytokine mediated disease | |
| WO2001027089A1 | Oct 10, 2000 | Apr 19, 2001 | Astrazeneca Ab | Pyrimidine derivatives | |
| WO2001034605A1 | Oct 27, 2000 | May 17, 2001 | Ortho Mcneil Pharm Inc | SUBSTITUTED 2-ARYL-3-(HETEROARYL)-IMIDAZO[1,2-a]PYRIMIDINES, AND RELATED PHARMACEUTICAL COMPOSITIONS AND METHODS | |
| WO2003090912A1 | Apr 15, 2003 | Nov 6, 2003 | Squibb Bristol Myers Co | Pyrrolo-triazine aniline compounds useful as kinase inhibitors | |
| US4200750 | Dec 8, 1977 | Apr 29, 1980 | Westwood Pharmaceuticals Inc. | 4-Substituted imidazo [1,2-a]quinoxalines | |
| US5658903 | Jun 3, 1996 | Aug 19, 1997 | Smithkline Beecham Corporation | Cytokine inhibitors | |
| US5932576 | May 22, 1998 | Aug 3, 1999 | G. D. Searle & Company | 3(5)-heteroaryl substituted pyrazoles as p38 kinase inhibitors | |
| US5945418 | Mar 20, 1997 | Aug 31, 1999 | Vertex Pharmaceuticals Incorporated | Administering to the mammal to inhibit a mammalian protein kinase p38 which causes cell proliferation, cell death and response to extracellular stimuli | |
| US5977103 | Jan 10, 1997 | Nov 2, 1999 | Smithkline Beecham Corporation | Substituted imidazole compounds | |
| US6087496 | Apr 1, 1999 | Jul 11, 2000 | G. D. Searle & Co. | Enzyme inhibitors | |
| US6130235 | Aug 3, 1998 | Oct 10, 2000 | Scios Inc. | Piperidine moieties coupled to indole, benzimidazole or benzotriazole. | |
| US6147080 | Jun 10, 1997 | Nov 14, 2000 | Vertex Pharmaceuticals Incorporated | Inhibitors of p38 | |
| US6251914 | Jul 1, 1998 | Jun 26, 2001 | Smithkline Beecham Corporation | Treating cytokine mediated diseases | |
| US6277989 | Mar 14, 2000 | Aug 21, 2001 | Scios, Inc. | Quinazoline derivatives as medicaments | |
| US6670357 | Nov 7, 2001 | Dec 30, 2003 | Bristol-Myers Squibb Company | Antiinflammatory agents | |
| US6867300 | Nov 6, 2002 | Mar 15, 2005 | Bristol-Myers Squibb Company | Methods for the preparation of pyrrolotriazine compounds useful as kinase inhibitors | |
| US7034151 | Feb 5, 2004 | Apr 25, 2006 | Bristol-Myers Squibb Company | 1,4-dihydro-4-oxo-pyrrolo[2,1-f][1,2,4]triazine-6-carboxylates; novel approach to the formation of the bicyclic heterocyclic ring system | |
| US7041501 | Oct 31, 2002 | May 9, 2006 | Bristol-Myers Squibb Company | Methods of screening for toxicity of test compounds | |
| US7160883 | Apr 22, 2003 | Jan 9, 2007 | Bristol-Myers-Squibb Company | Pyrrolo-triazine aniline compounds useful as kinase inhibitors | |
| US7211666 | Dec 22, 2004 | May 1, 2007 | Bristol-Myers Squibb Company | N-Cyclopropyl-4-[[5-[(methoxyamino)carbonyl]-2-methylphenyl]amino]-5-methylpyrrolo[2,1-f][1,2,4]triazine-6-carboxamide; aminating with chloramine to produce a pyrrole with a Nitrogen nitrogen bond; reacting with formamide, cyclizing to form the pyrrolotriazine core; kinase inhibitors | |
| US7253167 | Jun 29, 2005 | Aug 7, 2007 | Bristol-Myers Squibb Company | Tricyclic-heteroaryl compounds useful as kinase inhibitors | |
| US7388009 | Oct 3, 2003 | Jun 17, 2008 | Bristol-Myers Squibb Company | Heterocyclic drugs as enzyme inhibitors for Kinase enzymes or prodrugs | |
| US7462616 | Oct 24, 2006 | Dec 9, 2008 | Bristol-Myers Squibb Company | Pyrrolo-triazine aniline compounds useful as kinase inhibitors | |
| US7759343 | Oct 28, 2008 | Jul 20, 2010 | Bristol-Myers Squibb Company | Pyrrolo-triazine aniline compounds useful as kinase inhibitors | |
| US61379001 | Title not available | ||||
| US20030232831 | Apr 22, 2003 | Dec 18, 2003 | Alaric Dyckman | Aryl ketone pyrrolo-triazine compounds useful as kinase inhibitors | |
| US20040229877 | Oct 29, 2003 | Nov 18, 2004 | Katerina Leftheris | Administering pyrrolotriazine carboxamide and benzamide compounds for therapy of p38 kinase-associated conditions | |
| US20050043306 | Oct 3, 2003 | Feb 24, 2005 | Katerina Leftheris | Heterocyclic drugs as enzyme inhibitors for Kinase enzymes or prodrugs | |
| US20060003967 | Jun 28, 2005 | Jan 5, 2006 | Zhongping Shi | Method for preparing pyrrolotriazine compounds | |
| US20060030708 | Aug 5, 2005 | Feb 9, 2006 | Lobben Paul C | Methods for the preparation of pyrrolotriazine compounds | |
| US20060041124 | Oct 14, 2005 | Feb 23, 2006 | Bang-Chi Chen | Process for preparing pyrrolotriazine kinase inhibitors | |
| US20060229449 | Apr 3, 2006 | Oct 12, 2006 | Apurba Bhattacharya | Reacting with chloramine in presence of aqueous base, phase transfer catalyst; anti-cancer agents, kinase inhibitors | |
| US20060235020 | Apr 4, 2006 | Oct 19, 2006 | Soojin Kim | Process for preparing salts of 4-[[5-[(cyclopropylamino)carbonyl]-2-methylphenyl]amino]-5-methyl-N-propylpyrrolo[2,1-f][1,2,4]triazine-6-carboxamide and novel stable forms produced therein | |
| US20070213300 | Mar 6, 2007 | Sep 13, 2007 | Bristol-Myers Squibb Company | Pyrrolotriazine aniline prodrug compounds useful as kinase inhibitors |
Japan approves world’s first PD-1 drug, nivolumab
http://www.pharmatimes.com/Article/14-07-07/Japan_approves_world_s_first_PD-1_drug_nivolumab.aspx
old article cut paste

NIVOLUMAB
Anti-PD-1;BMS-936558; ONO-4538
PRONUNCIATION nye vol’ ue mab
THERAPEUTIC CLAIM Treatment of cancer
CHEMICAL DESCRIPTION
A fully human IgG4 antibody blocking the programmed cell death-1 receptor (Medarex/Ono Pharmaceuticals/Bristol-Myers Squibb)
MOLECULAR FORMULA C6362H9862N1712O1995S42
MOLECULAR WEIGHT 143.6 kDa
SPONSOR Bristol-Myers Squibb
CODE DESIGNATION MDX-1106, BMS-936558
CAS REGISTRY NUMBER 946414-94-4
Bristol-Myers Squibb announced promising results from an expanded phase 1 dose-ranging study of its lung cancer drug nivolumab
Nivolumab (nye vol’ ue mab) is a fully human IgG4 monoclonal antibody designed for the treatment of cancer. Nivolumab was developed by Bristol-Myers Squibb and is also known as BMS-936558 and MDX1106.[1] Nivolumab acts as an immunomodulator by blocking ligand activation of the Programmed cell death 1 receptor.
A Phase 1 clinical trial [2] tested nivolumab at doses ranging from 0.1 to 10.0 mg per kilogram of body weight, every 2 weeks. Response was assessed after each 8-week treatment cycle, and were evaluable for 236 of 296 patients. Study authors concluded that:”Anti-PD-1 antibody produced objective responses in approximately one in four to one in five patients with non–small-cell lung cancer, melanoma, or renal-cell cancer; the adverse-event profile does not appear to preclude its use.”[3]
Phase III clinical trials of nivolumab are recruiting in the US and EU.[4]
- Statement On A Nonproprietary Name Adopted By The USAN Council – Nivolumab, American Medical Association.
- A Phase 1b Study of MDX-1106 in Subjects With Advanced or Recurrent Malignancies (MDX1106-03), NIH.
- Topalian SL, et al. (June 2012). “Safety, Activity, and Immune Correlates of Anti–PD-1 Antibody in Cancer”. New England Journal of Medicine 366. doi:10.1056/NEJMoa1200690. Lay summary – New York Times.
- Nivolumab at ClinicalTrials.gov, A service of the U.S. National Institutes of Health.
The PD-1 blocking antibody nivolumab continues to demonstrate sustained clinical activity in previously treated patients with advanced non-small cell lung cancer (NSCLC), according to updated long-term survival data from a phase I trial.
Survival rates at one year with nivolumab were 42% and reached 24% at two years, according to the median 20.3-month follow up. Additionally, the objective response rate (ORR) with nivolumab, defined as complete or partial responses by standard RECIST criteria, was 17% for patients with NSCLC. Results from the updated analysis will be presented during the 2013 World Conference on Lung Cancer on October 29.
“Lung cancer is very difficult to treat and there continues to be a high unmet medical need for these patients, especially those who have received multiple treatments,” David R. Spigel, MD, the program director of Lung Cancer Research at the Sarah Cannon Research Institute and one of the authors of the updated analysis, said in a statement.
“With nivolumab, we are investigating an approach to treating lung cancer that is designed to work with the body’s own immune system, and these are encouraging phase I results that support further investigation in larger scale trials.”
In the phase I trial, 306 patients received intravenous nivolumab at 0.1–10 mg/kg every-other-week for ≤12 cycles (4 doses/8 week cycle). In all, the trial enrolled patients with NSCLC, melanoma, renal cell carcinoma, colorectal cancer, and prostate cancer.
The long-term follow up focused specifically on the 129 patients with NSCLC. In this subgroup, patients treated with nivolumab showed encouraging clinical activity. The participants had a median age of 65 years and good performance status scores, and more than half had received three or more prior therapies. Across all doses of nivolumab, the median overall survival was 9.9 months, based on Kaplan-Meier estimates.
In a previous update of the full trial results presented at the 2013 ASCO Annual Meeting, drug-related adverse events of all grades occurred in 72% of patients and grade 3/4 events occurred in 15%. Grade 3/4 pneumonitis related to treatment with nivolumab emerged early in the trial, resulting in 3 deaths. As a result, a treatment algorithm for early detection and management was developed to prevent this serious side effect.
Nivolumab is a fully human monoclonal antibody that blocks the PD-1 receptor from binding to both of its known ligands, PD-L1 and PD-L2. This mechanism, along with early data, suggested an associated between PD-L1 expression and response to treatment.
In separate analysis presented at the 2013 World Conference on Lung Cancer, the association of tumor PD-L1 expression and clinical activity in patients with NSCLC treated with nivolumab was further explored. Of the 129 patients with NSCLC treated with nivolumab in the phase I trial, 63 with NSCLC were tested for PD-L1 expression by immunohistochemistry (29 squamous; 34 non-squamous).
CHMP backs B-MS HCV drug and Lilly Lantus biosimilar
World News | June 29, 2014
The latest set of opinions from advisors to the European Medicines Agency include recommendations to approve six new medicines, including Bristol-Myers Squibb’s new hepatitis C drug and Eli Lilly’s biosimilar of the Sanofi diabetes blockbuster Lantus.
Bristol-Myers Squibb announced promising results from an expanded phase 1 dose-ranging study of its lung cancer drug nivolumab
NIVOLUMAB
Anti-PD-1;BMS-936558; ONO-4538
PRONUNCIATION nye vol’ ue mab
THERAPEUTIC CLAIM Treatment of cancer
CHEMICAL DESCRIPTION
A fully human IgG4 antibody blocking the programmed cell death-1 receptor (Medarex/Ono Pharmaceuticals/Bristol-Myers Squibb)
MOLECULAR FORMULA C6362H9862N1712O1995S42
MOLECULAR WEIGHT 143.6 kDa
SPONSOR Bristol-Myers Squibb
CODE DESIGNATION MDX-1106, BMS-936558
CAS REGISTRY NUMBER 946414-94-4
Bristol-Myers Squibb announced promising results from an expanded phase 1 dose-ranging study of its lung cancer drug nivolumab
Nivolumab (nye vol’ ue mab) is a fully human IgG4 monoclonal antibody designed for the treatment of cancer. Nivolumab was developed by Bristol-Myers Squibb and is also known as BMS-936558 and MDX1106.[1] Nivolumab acts as an immunomodulator by blocking ligand activation of the Programmed cell death 1 receptor.
A Phase 1 clinical trial [2] tested nivolumab at doses ranging from 0.1 to 10.0 mg per kilogram of body weight, every 2 weeks. Response was assessed after each 8-week treatment cycle, and were evaluable for 236 of 296 patients. Study authors concluded that:”Anti-PD-1 antibody produced objective responses in approximately one in four to one in five patients with non–small-cell lung cancer, melanoma, or renal-cell cancer; the adverse-event profile does not appear to preclude its use.”[3]
Phase III clinical trials of nivolumab are recruiting in the US and EU.[4]
- Statement On A Nonproprietary Name Adopted By The USAN Council – Nivolumab, American Medical Association.
- A Phase 1b Study of MDX-1106 in Subjects With Advanced or Recurrent Malignancies (MDX1106-03), NIH.
- Topalian SL, et al. (June 2012). “Safety, Activity, and Immune Correlates of Anti–PD-1 Antibody in Cancer”. New England Journal of Medicine 366. doi:10.1056/NEJMoa1200690. Lay summary – New York Times.
- Nivolumab at ClinicalTrials.gov, A service of the U.S. National Institutes of Health.
The PD-1 blocking antibody nivolumab continues to demonstrate sustained clinical activity in previously treated patients with advanced non-small cell lung cancer (NSCLC), according to updated long-term survival data from a phase I trial.
Survival rates at one year with nivolumab were 42% and reached 24% at two years, according to the median 20.3-month follow up. Additionally, the objective response rate (ORR) with nivolumab, defined as complete or partial responses by standard RECIST criteria, was 17% for patients with NSCLC. Results from the updated analysis will be presented during the 2013 World Conference on Lung Cancer on October 29.
“Lung cancer is very difficult to treat and there continues to be a high unmet medical need for these patients, especially those who have received multiple treatments,” David R. Spigel, MD, the program director of Lung Cancer Research at the Sarah Cannon Research Institute and one of the authors of the updated analysis, said in a statement.
“With nivolumab, we are investigating an approach to treating lung cancer that is designed to work with the body’s own immune system, and these are encouraging phase I results that support further investigation in larger scale trials.”
In the phase I trial, 306 patients received intravenous nivolumab at 0.1–10 mg/kg every-other-week for ≤12 cycles (4 doses/8 week cycle). In all, the trial enrolled patients with NSCLC, melanoma, renal cell carcinoma, colorectal cancer, and prostate cancer.
The long-term follow up focused specifically on the 129 patients with NSCLC. In this subgroup, patients treated with nivolumab showed encouraging clinical activity. The participants had a median age of 65 years and good performance status scores, and more than half had received three or more prior therapies. Across all doses of nivolumab, the median overall survival was 9.9 months, based on Kaplan-Meier estimates.
In a previous update of the full trial results presented at the 2013 ASCO Annual Meeting, drug-related adverse events of all grades occurred in 72% of patients and grade 3/4 events occurred in 15%. Grade 3/4 pneumonitis related to treatment with nivolumab emerged early in the trial, resulting in 3 deaths. As a result, a treatment algorithm for early detection and management was developed to prevent this serious side effect.
Nivolumab is a fully human monoclonal antibody that blocks the PD-1 receptor from binding to both of its known ligands, PD-L1 and PD-L2. This mechanism, along with early data, suggested an associated between PD-L1 expression and response to treatment.
In separate analysis presented at the 2013 World Conference on Lung Cancer, the association of tumor PD-L1 expression and clinical activity in patients with NSCLC treated with nivolumab was further explored. Of the 129 patients with NSCLC treated with nivolumab in the phase I trial, 63 with NSCLC were tested for PD-L1 expression by immunohistochemistry (29 squamous; 34 non-squamous).
Bristol-Myers Squibb announced promising results from phase 2b study of its rheumatoid arthritis drug clazakizumab
clazakizumab. BMS-945429, ALD518
Bristol-Myers Squibb, phase 2b study, rheumatoid arthritis,
NONPROPRIETARY NAME ADOPTED BY THE USAN COUNCIL
CLAZAKIZUMAB
PRONUNCIATION klaz” a kiz’ ue mab
THERAPEUTIC CLAIM Autoimmune diseases, rheumatoid arthritis
CHEMICAL NAMES
1. Immunoglobulin G1, anti-(human interleukin 6) (human-Oryctolagus cuniculus monoclonal BMS-945429/ALD518 heavy chain), disulfide with human-Oryctolagus cuniculus monoclonal BMS-945429/ALD518 κ-chain, dimer
2. Immunoglobulin G1, anti-(human interleukin-6 (B-cell stimulatory factor 2, CTL differentiation factor, hybridoma growth factor, interferon beta-2)); humanized rabbit monoclonal BMS-945429/ALD518 [300-alanine(CH2-N67>A67)]1 heavy chain (223-217′)-disulfide with humanized rabbit monoclonal BMS-945429/ALD518 light chain dimer (229-229”:232-232”)-bisdisulfide, O-glycosylated
MOLECULAR FORMULA C6426H9972N1724O2032S42
MOLECULAR WEIGHT 145.2 kDa
SPONSOR Bristol-Myers Squibb
CODE DESIGNATION BMS-945429, ALD518
CAS REGISTRY NUMBER 1236278-28-6
Monoclonal antibody
Type Whole antibody
Source Humanized
Target IL6
CAS number 1236278-28-6
Clazakizumab is a humanized monoclonal antibody designed for the treatment of rheumatoid arthritis.[1]
Clazakizumab was developed by Alder Biopharmaceuticals and Bristol-Myers Squibb.
Bristol-Myers Squibbalong with Alder Biopharmaceuticals, announced the presentation of efficacy and safety data from a Phase 2b dose-ranging study of subcutaneous (SC) clazakizumab in adults with moderate-to-severe rheumatoid arthritis (RA) and an inadequate response to methotrexate (MTX). Clazakizumab is a humanized anti-IL-6 monoclonal antibody that is directed against the IL-6 cytokine rather than its receptor.
In the Phase IIb study clazakizumab doses ranging from 25-200 mg monotherapy and in combination with MTX were studied vs. MTX alone. Adalimumab in combination with MTX was included as an active reference arm. All clazakizumab treatment arms, alone or in combination with MTX, demonstrated efficacy in controlling the signs and symptoms of RA, and met the predefined primary endpoint of a higher ACR20 response rate vs. MTX alone after 12 weeks of treatment. All clazakizumab treatment groups were also associated with improved ACR 20/50/70 response rates and HAQ-DI scores vs. MTX at week 24. Rates of low disease activity and remission with clazakizumab plus MTX, as measured by DAS28 CRP, CDAI and SDAI criteria were numerically greater for clazakizumab at 12 and 24 weeks than the active comparator.
The adverse event (AE) rates were similar across all clazakizumab arms. The most frequent AE for clazakizumab was dose-related injection site reactions. The most frequent reason for discontinuation due to AE in clazakizumab treated patients was laboratory abnormality, predominantly transaminase elevations, more frequent in MTX-containing arms. The most frequent serious adverse events (SAEs) were serious infections. Rates of serious infections were generally comparable for clazakizumab and adalimumab combination arms and were numerically greater than MTX alone.
“There is a great need for additional disease-modifying therapies that can provide more patients with deep and sustainable remission, helping preserve function and limit further joint damage,” said Paul Emery, MD, director of MSK Biomedical Unit at the Leeds Teaching Hospitals Trust in the United Kingdom. “Currently, less than 30% of RA patients experience sustained remission as defined by ACR criteria. Clazakizumab is an investigational therapy that neutralizes IL-6 signaling by blocking the IL-6 cytokine, and provides promising remission data that will need to be further investigated.”
Bristol-Myers Squibb has exclusive worldwide rights to develop and commercialize clazakizumab for all indications outside of cancer under a collaboration agreement with its discoverer, Alder Biopharmaceuticals.
clazakizumab
immunoglobulin G1-kappa, anti-[Homo sapiens IL6 (interleukin 6, IL-6)], humanized monoclonal antibody;
gamma1 heavy chain (1-450) [humanized VH (Homo sapiens IGHV3-66*01 (83.50%) -(IGHD)-IGHJ3*02 M123>L (115)) [8.8.14] (1-120) -Homo sapiens IGHG1*03 CH
gamma1 heavy chain (1-450) [humanized VH (Homo sapiens IGHV3-66*01 (83.50%) -(IGHD)-IGHJ3*02 M123>L (115)) [8.8.14] (1-120) -Homo sapiens IGHG1*03 CH

