

Yet another atropisomeric kinase inhibitor, of Bruton’s tyrosine kinase (BTK), currently being evaluated in Phase II clinical trials for rheumatoid arthritis, comes from Bristol Myers-Squibb. BMS-986142 contains one point-chiral center and two atropisomeric chiral axes, making it a diastereomeric compound with eight possible isomers. The less stable atropisomeric axis has a half-life on the order of hours to days, which means it can’t be heated above about 45 °C without the compound morphing. To keep the molecule from racemizing, the team had to design its synthetic routes and analysis with a close eye on temperature.
Home » Posts tagged 'Bristol-Myers Squibb'
Tag Archives: Bristol-Myers Squibb
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on
ORGANIC SPECTROSCOPY INTERNATIONAL

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()




Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Varegacestat

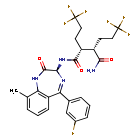
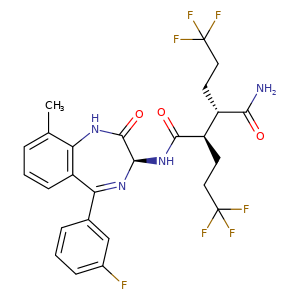
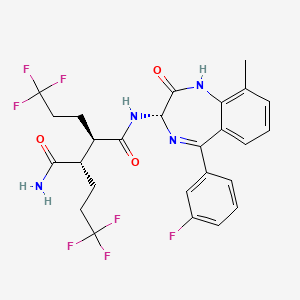
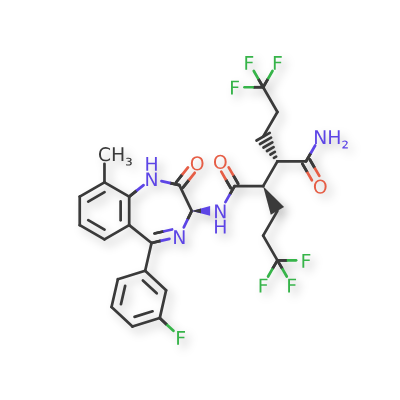
Varegacestat
CAS 1584647-27-7
MF C26H25F7N4O3 MW574.5
(2R,3S)-N1-[(3S)-5-(3-fluorophenyl)-9-methyl-2-oxo-2,3-dihydro-1H1,4-benzodiazepin-3-yl]-2,3-bis(3,3,3-trifluoropropyl)butanediamide
(2R,3S)-N1-((3S)-5-(3-FLUOROPHENYL)-2,3-DIHYDRO-9-METHYL-2-OXO-1H-1,4-BENZODIAZEPIN-3-YL)-2,3-BIS(3,3,3-TRIFLUOROPHENYL)BUTANEDIAMIDE
(2R,3S)-N1-((3S)-5-(3-FLUOROPHENYL)-9-METHYL-2-OXO-2,3-DIHYDRO-1H-1,4-BENZODIAZEPIN-3-YL)-2,3-BIS(3,3,3-TRIFLUOROPHENYL)BUTANEDIAMIDE
gamma-secretase inhibitor, antineoplastic, AL102, BMS 986115, LSK1L593UU, AL 102
BMS-986115 has been used in trials studying the treatment of Various Advanced Cancer.
Varegacestat is an orally bioavailable, gamma secretase (GS) and pan-Notch inhibitor, with potential antineoplastic activity. Upon administration, varegacestat binds to GS and blocks the proteolytic cleavage and release of the Notch intracellular domain (NICD), which would normally follow ligand binding to the extracellular domain of the Notch receptor. This prevents both the subsequent translocation of NICD to the nucleus to form a transcription factor complex and the expression of Notch-regulated genes. This results in the induction of apoptosis and the inhibition of growth of tumor cells that overexpress Notch. Overexpression of the Notch signaling pathway plays an important role in tumor cell proliferation and survival. The integral membrane protein GS is a multi-subunit protease complex that cleaves single-pass transmembrane proteins, such as Notch receptors, at residues within their transmembrane domains and leads to their activation
AL 102 (previously known as BMS 986115), was developed as an orally active a gamma-secretase and pan-Notch inhibitor. The drug participated in phase I clinical trials in solid tumor patients. The drug was safe and well-tolerated and stabilized disease for more than six months in 14% of patients, however, Bristol-Myers Squibb terminated the study because of the changes in the business objectives. Ayala, an Israeli biotech company, licensed rights for the development of AL 102 from Bristol-Myers Squibb. In December 2018, Ayala in collaborating with Novartis decided to investigate AL102 for treatment of multiple myeloma. Ayala studied AL102, an inhibitor of the Notch pathway, in blood cancers. It is known that the pathway regulates cell-fate determination during development and maintains adult tissue balance. Cumulative evidence indicates that Notch is overactive in multiple myeloma and participates in its onset and progression.
SYN
PATENTS
PATENT
https://www.google.com/patents/US20140087992
Example 1(2R,3S)—N-((3S)-5-(3-Fluorophenyl)-9-methyl-2-oxo-2,3-dihydro-1H-1,4-benzodiazepin-3-yl)-2,3-bis(3,3,3-trifluoropropyl)succinamide
Intermediate 1A: (2S,3R)-tert-Butyl 6,6,6-trifluoro-3-(((S)-5-(3-fluorophenyl)-9-methyl-2-oxo-2,3-dihydro-1H-benzo[e][1,4]diazepin-3-yl)carbamoyl)-2-(3,3,3-trifluoropropyl)hexanoate

In a 100 mL round-bottomed flask, a solution of Intermediate B-1 (1683 mg, 5.94 mmol), Et3N (1.656 mL, 11.88 mmol), and Intermediate S-1 in DMF (20 mL) was treated with o-benzotriazol-1-yl-N,N,N′,N′-tetramethyluronium tetrafluoroborate (3815 mg, 11.88 mmol) and stirred at room temperature for 1 hour. The reaction mixture was diluted with water and saturated aqueous NaHCO3. An off white precipitate formed and was filtered and washed with water. The resulting solid was dried on the filter under a stream of nitrogen to give Intermediate 1A (3.7 g, 99% yield). MS (ES): m/z=632.4[M+H+]; HPLC: RT=3.635 min Purity=98%. (H2O/MeOH with TFA, CHROMOLITH® ODS S5 4.6×50 mm, gradient=4 min, wavelength=220 nm). 1H NMR (400 MHz, methanol-d4) δ 7.53 (t, J=4.5 Hz, 1H), 7.46-7.30 (m, 3H), 7.28-7.23 (m, 1H), 7.23-7.18 (m, 2H), 5.37 (s, 1H), 2.88 (td, J=10.4, 3.4Hz, 1H), 2.60 (td, J=10.2, 4.1 Hz, 1H), 2.54-2.40 (m, 1H), 2.47 (s, 3H), 2.33-2.12 (m, 3H), 1.98-1.69 (m, 4H), 1.51 (s, 9H).
Intermediate 1B: (2S,3R)-6,6,6-Trifluoro-3-(((S)-5-(3-fluorophenyl)-9-methyl-2-oxo-2,3-dihydro-1H-benzo[e][1,4]diazepin-3-yl)carbamoyl)-2-(3,3,3-trifluoropropyl)hexanoic acid

In a 250 mL round-bottomed flask, a solution of Intermediate 1A (3.7 g, 5.86 mmol) in DCM (25 mL) was treated with TFA (25 mL) and the resulting pale orange solution was stirred at room temperature for 1.5 hours. The reaction mixture was then concentrated to give Intermediate 1B. HPLC: RT=3.12 min (H2O/MeOH with TFA, CHROMOLITH® ODS S5 4.6×50 mm, gradient=4 min, wavelength=220 nm). MS (ES): m/z=576.3 (M+H)+. 1H NMR (400 MHz, methanol-d4) δ 7.54 (t, J=4.5 Hz, 1H), 7.49-7.29 (m, 3H), 7.28-7.15 (m, 3H), 5.38 (br. s., 1H), 2.89 (td, J=10.3, 3.7 Hz, 1H), 2.67 (td, J=9.9, 4.2Hz, 1H), 2.56-2.38 (m, 1H), 2.48 (s, 3H), 2.34-2.13 (m, 3H), 2.00-1.71 (m, 4H).
Example 1
In a 250 mL round-bottomed flask, a solution of Intermediate 1B (4.04 g, 5.86 mmol) in THF (50 mL) was treated with ammonia (2M in iPrOH) (26.4 mL, 52.7 mmol), followed by HOBT (1.795 g, 11.72 mmol) and EDC (2.246 g, 11.72 mmol). The resulting white suspension was stirred at room temperature overnight. The reaction mixture was diluted with water and saturated aqueous NaHCO3. The resulting solid was filtered, rinsed with water and then dried on the filter under a stream of nitrogen. The crude product was suspended in 20 mL of iPrOH and stirred at room temperature for 20 min and then filtered and washed with iPrOH and dried under vacuum to give 2.83 g of solid. The solid was dissolved in refluxing EtOH (100 mL) and slowly treated with 200 mg activated charcoal added in small portions. The hot mixture was filtered through CELITE® and rinsed with hot EtOH. The filtrate was reduced to half volume, allowed to cool and the white precipitate formed was filtered and rinsed with EtOH to give 2.57 g of white solid. A second recrystallization from EtOH (70 mL) afforded Example 1 (2.39 g, 70% yield) as a white solid. HPLC: RT=10.859 min (H2O/CH3CN with TFA, Sunfire C18 3.5 μm, 3.0×150 mm, gradient=15 min, wavelength=220 and 254 nm); MS (ES): m/z=575.3 [M+H+]; 1H NMR (400 MHz, methanol-d4) δ 7.57-7.50 (m, 1H), 7.47-7.30 (m, 3H), 7.29-7.15 (m, 3H), 5.38 (s, 1H), 2.85-2.75 (m, 1H), 2.59 (td, J=10.5, 4.0 Hz, 1H), 2.53-2.41 (m, 4H), 2.31-2.10 (m, 3H), 1.96-1.70 (m, 4H).
PATENT
https://www.google.com/patents/WO2014047372A1?cl=en


Scheme 3


XII XI
Scheme 4

Intermediate S-l : (2R,3S)-3-(fert-Butoxycarbonyl)-6,6,6-trifluoro-2-(3,3,3- trifluoropropyl)hexanoic acid

Intermediate S-IA: 3,3,3-Trifluoro ropyl trifluoromethanesulfonate

[00180] To a cold (-25 °C) stirred solution of 2,6-lutidine (18.38 mL, 158 mmol) in DCM (120 mL) was added Tf20 (24.88 mL, 147 mmol) over 3 min, and the mixture was stirred for 5 min. To the reaction mixture was added 3,3,3-trifluoropropan-l-ol (12 g, 105 mmol) over an interval of 3 min. After 2 hr, the reaction mixture was warmed to room temperature and stirred for 1 hr. The reaction mixture was concentrated to half its volume, then purified by loading directly on a silica gel column (330g ISCO) and the product was eluted with DCM to afford Intermediate S-IA (13.74 g, 53%) as a colorless oil. 1H NMR (400 MHz, CDC13) δ ppm 4.71 (2 H, t, J= 6.15 Hz), 2.49-2.86 (2 H, m).
Intermediate S-1B: (4S)-4-Benzyl-3-(5,5,5-trifluoropentanoyl)-l,3-oxazolidin-2-one

[00181] To a stirring solution of 5,5,5-trifluoropentanoic acid (14.76 g, 95 mmol) and DMF (0.146 rriL) in DCM (50 mL) was slowly added oxalyl chloride (8.27 mL, 95 mmol). After 2h, the mixture was concentrated to dryness. A separate flask was changed with (S)-4-benzyloxazolidin-2-one (16.75 g, 95 mmol) in THF (100 mL) and then cooled to -78 °C. To the solution was slowly added n-BuLi (2.5M, 37.8 mL, 95 mmol) over 10 min, stirred for 10 min, and then a solution of the above acid chloride in THF (50 mL) was slowly added over 5 min. The mixture was stirred for 30 min, and then warmed to room temperature. The reaction was quenched with sat aq NH4C1. Next, 10% aq LiCl was then added to the mixture, and the mixture was extracted with Et20. The organic layer was washed with sat aq NaHC03 then with brine, dried (MgSC^), filtered and concentrated to dryness. The residue was purified by Si02 chromatography (ISCO, 330 g column, eluting with a gradient from 100% hexane to 100% EtOAc) to afford the product Intermediate S-IB; (25.25 g, 85%): 1H NMR (400 MHz, CDC13) δ ppm 7.32-7.39 (2 H, m), 7.30 (1 H, d, J= 7.05 Hz), 7.18-7.25 (2 H, m), 4.64-4.74 (1 H, m), 4.17-4.27 (2 H, m), 3.31 (1 H, dd, J= 13.35, 3.27 Hz), 3.00-3.11 (2 H, m), 2.79 (1 H, dd, J= 13.35, 9.57 Hz), 2.16-2.28 (2 H, m), 1.93-2.04 (2 H, m).
Intermediate S-IC: tert- utyl (3R)-3-(((4S)-4-benzyl-2-oxo-l,3-oxazolidin-3- yl)carbonyl)-6,6,6-trifluoroh xanoate

[00182] To a cold (-78 °C), stirred solution of Intermediate S-IB (3.03 g, 9.61 mmol) in THF (20 mL) was added NaHMDS (1.0M in THF) (10.6 mL, 10.60 mmol) under a nitrogen atmosphere. After 2 hours, tert-butyl 2-bromoacetate (5.62 g, 28.8 mmol) was added neat via syringe at -78 °C and stirring was maintained at the same temperature. After 6 hours, the reaction mixture was warmed to room temperature. The reaction mixture was partitioned between saturated NH4C1 and EtOAc. The organic phase was separated, and the aqueous phase was extracted with EtOAc (3x). The combined organics were washed with brine, dried (Na2s04), filtered and concentrated under reduced pressure. The residue was purified by flash chromatography (Teledyne ISCO
CombiFlash Rf, 5% to 100% solvent A/B = hexanes/EtOAc, REDISEP® Si02 120g). Concentration of the appropriate fractions provided Intermediate S-1C (2.79 g, 67.6%) as a colorless viscous oil: 1H NMR (400 MHz, CDC13) δ ppm 7.34 (2 H, d, J= 7.30 Hz), 7.24-7.32 (3 H, m), 4.62-4.75 (1 H, m, J= 10.17, 6.89, 3.43, 3.43 Hz), 4.15-4.25 (3 H, m), 3.35 (1 H, dd, J= 13.60, 3.27 Hz), 2.84 (1 H, dd, J= 16.62, 9.57 Hz), 2.75 (1 H, dd, J = 13.35, 10.07 Hz), 2.47 (1 H, dd, J= 16.62, 4.78 Hz), 2.11-2.23 (2 H, m), 1.90-2.02 (1 H, m), 1.72-1.84 (1 H, m), 1.44 (9 H, s).
Intermediate S-ID: (2R)-2-( -tert-Butoxy-2-oxoethyl)-5,5,5-trifluoropentanoic acid

[00183] To a cool (0 °C), stirred solution of Intermediate S-1C (2.17 g, 5.05 mmol) in THF (50 mL) and water (15 mL) was added a solution of LiOH (0.242 g, 10.11 mmol) and H202 (2.065 mL, 20.21 mmol) in H20 (2 mL). After 10 min, the reaction mixture was removed from the ice bath, stirred for lh, and then cooled to 0 °C. Saturated aqueous NaHCC”3 (25 mL) and saturated aqueous Na2s03 (25 mL) were added to the reaction mixture, and the mixture was stirred for 10 min, and then partially concentrated. The resulting mixture was extracted with DCM (2x), cooled with ice and made acidic with cone. HC1 to pH 3. The mixture was saturated with solid NaCl, extracted with EtOAc (3x), and then dried over MgS04, filtered and concentrated to a colorless oil to afford Intermediate S-ID, 1.2514g, 92%): 1H NMR (400 MHz, CDCI3) δ ppm 2.83-2.95 (1 H, m), 2.62-2.74 (1 H, m), 2.45 (1 H, dd, J= 16.62, 5.79 Hz), 2.15-2.27 (2 H, m), 1.88-2.00 (1 H, m), 1.75-1.88 (1 H, m), 1.45 (9 H, s). Intermediate S-l : (2R,3S)-3-(fert-Butoxycarbonyl)-6,6,6-trifluoro-2-(3,3,3- trifluoropropyl)hexanoic acid, and Intermediate S-1E: (2R,3R)-3-(tert-butoxycarbonyl)- 6,6,6-trifluoro-2-(3,3,3-trifluoropropyl)hexanoic acid
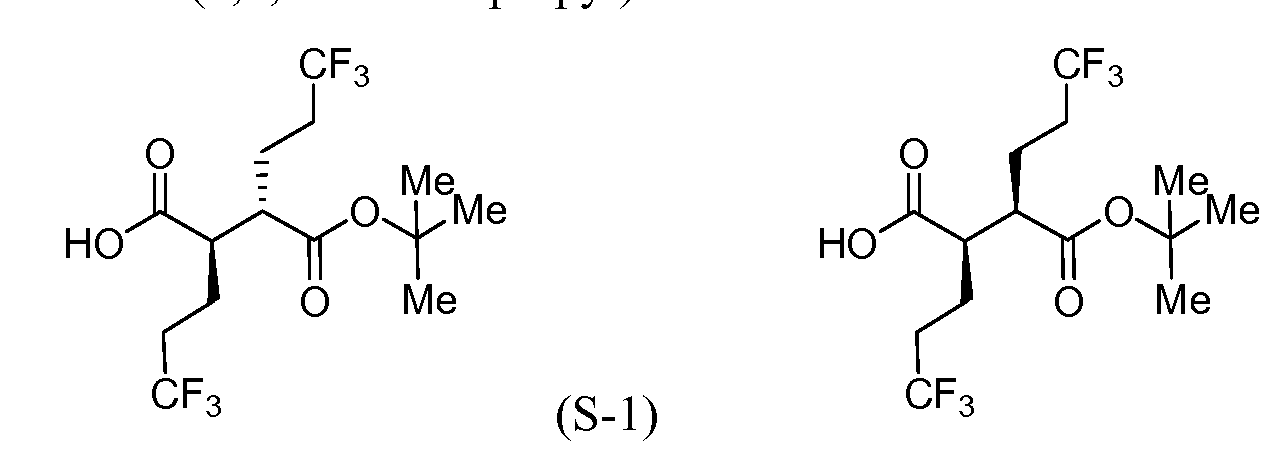
(S-1E)
[00184] To a cold (-78 °C) stirred solution of Intermediate S-1D (5 g, 18.50 mmol) in THF (60 mL) was slowly added LDA (22.2 mL, 44.4 mmol, 2.0M) over 7 min. After stirring for 2 hr, Intermediate S- 1 A (6.38 g, 25.9 mmol) was added to the reaction mixture over 3 min. After 60 min, the reaction mixture was warmed to -25 °C
(ice/MeOH/dry ice) and stirred for an additional 60 min at which time sat aq NH4C1 was added. The separated aqueous phase was acidified with IN HC1 to pH 3, and then extracted with Et20. The combined organic layers were washed with brine (2x), dried over MgS04, filtered and concentrated to provide a 1 :4 (II :I1E) mixture (as determined by 1H NMR) of Intermediate S-l and Intermediate S-1E (6.00 g, 89%) as a pale yellow solid. 1H NMR (500 MHz, CDC13) δ ppm 2.81 (1 H, ddd, J = 10.17, 6.32, 3.85 Hz), 2.63- 2.76 (1 H, m), 2.02-2.33 (4 H, m), 1.86-1.99 (2 H, m), 1.68-1.85 (2 H, m), 1.47 (9 H, s).
[00185] To a cold (-78 °C), stirred solution of a mixture of Intermediate S-l and Intermediate S-1E (5.97 g, 16.30 mmol) in THF (91 mL) was added LDA (19 mL, 38.0 mmol, 2.0M in THF/hexane/ethyl benzene) dropwise via syringe over 10 min (internal temperature never exceeded -65 °C, J-KEM® probe in reaction solution). The mixture was stirred for 15 min, and then warmed to room temperature (24 °C water bath), stirred for 15 min, and then cooled to -78 °C for 15 min. To the reaction mixture was added Et2AlCl (41 mL, 41.0 mmol, 1M in hexane) via syringe (internal temperature never exceeded -55 °C), and the mixture was stirred for 10 min, and then warmed to room temperature (24 °C bath) for 15 min and then back to -78 °C for 15 min. Meanwhile, a 1000 mL round bottom flask was charged with MeOH (145 mL) and precooled to -78 °C. With vigorous stirring the reaction mixture was transferred via cannula over 5 min to the MeOH. The flask was removed from the bath, ice was added followed by the slow addition of IN HC1 (147 mL, 147 mmol). Gas evolution was observed as the HC1 was added. The reaction mixture was allowed to warm to room temperature during which the gas evolution subsided. The reaction mixture was diluted with EtOAc (750 mL), saturated with NaCl, and the organic phase was separated, washed with a solution of potassium fluoride (8.52 g, 147 mmol) and IN HC1 (41 mL, 41.0 mmol) in water (291 mL), brine (100 mL), and then dried (Na2s04), filtered and concentrated under vacuum. 1H NMR showed the product was a 9: 1 mixture of Intermediate S-l and Intermediate S- 1E. The enriched mixture of Intermediate S-l and Intermediate S-1E (6.12 g, >99% yield) was obtained as a dark amber solid: 1H NMR (400 MHz, CDC13) δ ppm 2.64-2.76 (2 H, m), 2.04-2.35 (4 H, m), 1.88-2.00 (2 H, m), 1.71-1.83 (2 H, m), 1.48 (9 H, s).
Alternate procedure to make Intermediate S-l :
Intermediate S-IF: (2R,3 -1 -Benzyl 4-tert-butyl 2,3-bis(3,3,3-trifluoropropyl)succinate

[00186] To a stirred solution of a 9: 1 enriched mixture of Intermediate S-l and Intermediate S-1E (5.98 g, 16.33 mmol) in DMF (63 mL) were added potassium carbonate (4.06 g, 29.4 mmol) and benzyl bromide (2.9 mL, 24.38 mmol), the mixture was then stirred overnight at room temperature. The reaction mixture was diluted with EtOAc (1000 mL), washed with 10% LiCl (3×200 mL), brine (200 mL), dried (Na2S04), filtered, concentrated, and then dried under vacuum. The residue was purified by Si02 chromatography using a toluene:hexane gradient. Diastereomerically purified
Intermediate S-IF (4.81g, 65%) was obtained as a colorless solid: 1H NMR (400 MHz, chloroform-d) δ 7.32-7.43 (m, 5H), 5.19 (d, J= 12.10 Hz, 1H), 5.15 (d, J= 12.10 Hz, 1H), 2.71 (dt, J= 3.52, 9.20 Hz, 1H), 2.61 (dt, J= 3.63, 9.63 Hz, 1H), 1.96-2.21 (m, 4H), 1.69-1.96 (m, 3H), 1.56-1.67 (m, 1H), 1.45 (s, 9H).
Intermediate S-l : (2R,3S)-3-(fert-Butoxycarbonyl)-6,6,6-trifluoro-2-(3,3,3- trifluoropropyl)hexanoic acid

[00187] To a solution of Intermediate S-1F (4.81 g, 10.54 mmol) in MeOH (100 mL) was added 10% palladium on carbon (wet, Degussa type, 568.0 mg, 0.534 mmol) in a H2– pressure flask. The vessel was purged with N2 (4x), then purged with H2 (2x), and finally, pressurized to 50 psi and shaken overnight. The reaction vessel was
depressurized and purged with nitrogen. The mixture was filtered through CELITE®, washed with MeOH and then concentrated and dried under vacuum. Intermediate S-1 (3.81 g, 99% yield)) was obtained as a colorless solid: 1H NMR (400 MHz, chloroform-d) δ 2.62-2.79 (m, 2H), 2.02-2.40 (m, 4H), 1.87-2.00 (m, 2H), 1.67-1.84 (m, 2H), 1.48 (s, 9H).
Alternate procedure to make Intermediate S-1 :
Intermediate S-1 : (2R,3S)-3-(fert-Butoxycarbonyl)-6,6,6-trifluoro-2-(3,3,3- trifluoropropyl)hexanoic acid

[00188] Intermediate S-1 as a mixture with Intermediate S-IE was prepared in a similar procedure as above from Intermediate S-1D to afford a 1 :2.2 mixture of
Intermediate S-1 and Intermediate S-IE (8.60 g, 23.48 mmol), which was enriched using LDA (2.0 M solution in THF, ethyl benzene and heptane, 28.2 mL, 56.4 mmol) and diethyl aluminum chloride (1.0 M solution in hexane, 59 mL, 59.0 mmol) in THF (91 mL). After workup as described above, the resulting residue was found to be a 13.2: 1 (by 1H NMR) mixture of Intermediate S-1 and Intermediate S-IE, which was treated as follows: The crude material was dissolved in MTBE (43 mL). Hexanes (26 mL) were slowly charged to the reaction mixture while maintaining a temperature below 30 °C. The reaction mixture was stirred for 10 min. Next, tert-butylamine (2.7 mL, 1.1 eq) was charged slowly over a period of 20 minutes while maintaining a temperature below 30 °C. This addition was observed to be exothermic. The reaction mixture was stirred for 2 hrs below 30 °C and then filtered. The solid material was washed with 5:3 MTBE: hexane (80 mL), and the filtrate was concentrated and set aside. The filtered solid was dissolved in dichloromethane (300 mL), washed with IN HC1 (lOOmL), and the organic layer was washed with brine (100 mL x 2), and then concentrated under reduced pressure below 45 °C to afford Intermediate S-l (5.46 g, 64%).
A second alternate procedure for preparing Intermediate S-l :
Intermediate S-1G: tert- utyl 5,5,5-trifluoropentanoate

[00189] To a stirred solution of 5,5,5-trifluoropentanoic acid (5 g, 32.0 mmol) in THF (30 mL) and hexane (30 mL) at 0 °C, was added tert-butyl 2,2,2-trichloroacetimidate (11.46 mL, 64.1 mmol). The mixture was stirred for 15 min at 0 °C. Boron trifluoride etherate (0.406 mL, 3.20 mmol) was added and the reaction mixture was allowed to warm to room temperature overnight. To the clear reaction mixture was added solid NaHC03 (5 g) and stirred for 30 min. The mixture was filtered through MgSC^ and washed with hexanes (200 mL). The solution was allowed to rest for 45 min, and the resulting solid material was removed by filtering on the same MgSC^ filter again, washed with hexanes (100 mL) and concentrated under reduced pressure without heat. The volume was reduced to about 30 mL, filtered through a clean fritted funnel, washed with hexane (5 mL), and then concentrated under reduced pressure without heat. The resulting neat oil was filtered through a 0.45μιη nylon membrane filter disk to provide Intermediate S-1G (6.6 g, 31.4 mmol 98% yield) as a colorless oil: 1H NMR (400 MHz, CDC13) δ ppm 1.38 (s, 9 H) 1.74-1.83 (m, 2 H) 2.00-2.13 (m, 2 H) 2.24 (t, J= 7.28 Hz, 2 H). Intermediate S-1H: (4S)-4-(Propan-2-yl)-3-(5,5,5-trifluoropentanoyl)-l,3-oxazolidin-2- one

[00190] To a stirred solution of 5,5,5-trifluoropentanoic acid (5.04 g, 32.3 mmol) in DCM (50 mL) and DMF (3 drops) was added oxalyl chloride (3.4 mL, 38.8 mmol) dropwise over 5 min. The solution was stirred until all bubbling subsided. The reaction mixture was concentrated under reduced pressure to give pale yellow oil. To a separate flask charged with a solution of (4S)-4-(propan-2-yl)-l,3-oxazolidin-2-one (4.18 g, 32.4 mmol) in THF (100 mL) at -78 °C was added n-BuLi (2.5M in hexane) (13.0 mL, 32.5 mmol) dropwise via syringe over 5 min. After stirring for 10 min, the above acid chloride, dissolved in THF (20 mL), was added via cannula over 15 min. The reaction mixture was warmed to 0 °C, and was allowed to warm to room temperature as the bath warmed and stirred overnight. To the reaction mixture was added saturated NH4C1, and the mixture was extracted with EtOAc (2x). The combined organics were washed with brine, dried (Na2s04), filtered and concentrated under reduced pressure. The crude material was purified by flash chromatography (Teledyne ISCO CombiFlash Rf, 5% to 60% solvent A/B = hexanes/EtOAc, REDISEP® Si02 120g). Concentration of the appropriate fractions provided Intermediate S-1H (7.39 g, 86%) as a colorless oil: 1H NMR (400 MHz, CDC13) δ ppm 4.44 (1 H, dt, J= 8.31, 3.53 Hz), 4.30 (1 H, t, J= 8.69 Hz), 4.23 (1 H, dd, J= 9.06, 3.02 Hz), 2.98-3.08 (2 H, m), 2.32-2.44 (1 H, m, J= 13.91, 7.02, 7.02, 4.03 Hz), 2.13-2.25 (2 H, m), 1.88-2.00 (2 H, m), 0.93 (3 H, d, J= 7.05 Hz), 0.88 (3 H, d, J= 6.80 Hz).
Intermediate S-1I: (2S,3R)-tert-Butyl 6,6,6-trifluoro-3-((S)-4-isopropyl-2- oxooxazolidine-3-carbonyl)-2-(3,3,3-trifluoropropyl)hexanoate, and Intermediate S-U: (2R,3R)-tert-Butyl 6,6,6-trifluoro-3-((S)-4-isopropyl-2-oxooxazolidine-3-carbonyl)-2- (3 ,3 ,3 -trifluoropropyl)hexanoate

[00191] To a cold (-78 °C), stirred solution of diisopropylamine (5.3 mL, 37.2 mmol) in THF (59 mL) under a nitrogen atmosphere was added n-BuLi (2.5M in hexane) (14.7 mL, 36.8 mmol). The mixture was then warmed to 0 °C to give a 0.5M solution of LDA. A separate vessel was charged with Intermediate S-1H (2.45 g, 9.17 mmol). The material was azeotroped twice with benzene (the RotoVap air inlet was fitted with a nitrogen inlet to completely exclude humidity), and then toluene (15.3 mL) was added. This solution was added to a flask containing dry lithium chloride (1.96 g, 46.2 mmol). To the resultant mixture, cooled to -78 °C, was added the LDA solution (21.0 mL, 10.5 mmol) and the mixture was stirred at -78 °C for 10 min, then warmed to 0 °C for 10 min., and then cooled to -78 °C. To a separate reaction vessel containing Intermediate S-1G (3.41 g, 16.07 mmol), also azeotroped twice with benzene, was added toluene (15.3 mL), cooled to -78 °C and LDA (37.0 mL, 18.5 mmol) was added. The resulting solution was stirred at -78 °C for 25 min. At this time the enolate derived from the ester was transferred via cannula into the solution of the oxazolidinone enolate and stirred at -78 °C for an additional 5 min, at which time the septum was removed and solid powdered bis(2- ethylhexanoyloxy)copper (9.02 g, 25.8 mmol) was rapidly added to the reaction vessel and the septum was replaced. The vessel was immediately removed from the cold bath and immersed into a warm water bath (40 °C) with rapid swirling and with a concomitant color change from the initial turquoise to brown. The reaction mixture was stirred for 20 min, was then poured into 5% aqueous NH4OH (360 mL) and extracted with EtOAc (2x). The combined organics were washed with brine, dried (Na2s04), filtered and concentrated under reduced pressure. The residue was purified by flash chromatography (Teledyne ISCO CombiFlash Rf, 0% to 60% solvent A/B = hexanes/EtOAc, REDISEP® Si02 120g). Concentration of the appropriate fractions provided a mixture of Intermediate S- II and Intermediate S-1J (2.87 g, 66%) as a pale yellow viscous oil. 1H NMR showed the product was a 1.6: 1 mixture of diastereomers S-1LS-1J as determined by the integration of the multiplets at 2.74 and 2.84 ppm: 1H NMR (400 MHz, CDC13) δ ppm 4.43-4.54 (2 H, m), 4.23-4.35 (5 H, m), 4.01 (1 H, ddd, J= 9.54, 6.27, 3.51 Hz), 2.84 (1 H, ddd, J = 9.41, 7.28, 3.64 Hz), 2.74 (1 H, ddd, J= 10.29, 6.27, 4.02 Hz), 2.37-2.48 (2 H, m, J = 10.38, 6.98, 6.98, 3.51, 3.51 Hz), 2.20-2.37 (3 H, m), 1.92-2.20 (8 H, m), 1.64-1.91 (5 H, m), 1.47 (18 H, s), 0.88-0.98 (12 H, m). Intermediate S-1 : (2R,3S)-3-(fert-Butoxycarbonyl)-6,6,6-trifluoro-2-(3,3,3- trifluoropropyl)hexanoic acid, and Intermediate S-IE: (2R,3R)-3-(tert-Butoxycarbonyl)- 6,6,6-trifluoro-2-(3,3,3-trifluoropropyl)hexanoic acid

(S-IE)
[00192] To a cool (0 °C), stirred solution of Intermediate S-1I and Intermediate S-1 J (4.54 g, 9.51 mmol) in THF (140 mL) and water (42 mL) were sequentially added hydrogen peroxide (30% in water) (10.3 g, 91 mmol) and LiOH (685.3 mg, 28.6 mmol). The mixture was stirred for 1 hr. At this time the reaction vessel was removed from the cold bath and then stirred for 1.5 hr. To the reaction mixture were added saturated NaHC03 (45 mL) and saturated Na2s03 (15 mL), and then the mixture was partially concentrated under reduced pressure. The resulting crude solution was extracted with DCM (3x). The aqueous phase was acidified to pH~l-2 with IN HC1, extracted with DCM (3x) and then EtOAc (lx). The combined organics were washed with brine, dried (Na2s04), filtered and concentrated under reduced pressure to provide a mixture of Intermediates S-1 and S-IE (3.00 g, 86%) as a colorless oil: 1H NMR (400 MHz, CDC13) δ ppm 2.76-2.84 (1 H, m, diastereomer 2), 2.64-2.76 (3 H, m), 2.04-2.35 (8 H, m), 1.88- 2.00 (4 H, m), 1.71-1.83 (4 H, m), 1.48 (9 H, s, diastereomer 1), 1.46 (9 H, s,
diastereomer 2); 1H NMR showed a 1.7: 1 mixture of S-1E:S-1F by integration of the peaks for the t-butyl groups. Intermediate S-1 : (2R,3S)-3-(fert-Butoxycarbonyl)-6,6,6-trifluoro-2-(3,3,3- trifluoropropyl)hexanoic acid, and Intermediate S-IF: (2R,3R)-3-(fert-Butoxycarbonyl)- 6,6,6-trifluoro-2-(3,3,3-trifluoropropyl)hexanoic acid

[00193] To a cold (-78 °C) stirred solution of diisopropylamine (1.7 mL, 11.93 mmol) in THF (19 mL) under a nitrogen atmosphere was added n-BuLi (2.5M in hexanes) (4.8 mL, 12.00 mmol). The mixture was stirred for 5 min and then warmed to 0 °C. In a separate vessel, to a cold (-78 °C) stirred solution of the mixture of Intermediates S-1 and S-1E (1.99 g, 5.43 mmol) in THF (18 mL) was added the LDA solution prepared above via cannula slowly over 25 min. The mixture was stirred for 15 min, then warmed to room temperature (placed in a 24 °C water bath) for 15 min, and then again cooled to -78 °C for 15 min. To the reaction mixture was added Et2AlCl (1M in hexane) (11.4 mL, 11.40 mmol) via syringe. The mixture was stirred for 10 min, warmed to room
temperature for 15 min and then cooled back to -78 °C for 15 min. Methanol (25 mL) was rapidly added, swirled vigorously while warming to room temperature, and then concentrated to ~l/4 the original volume. The mixture was dissolved in EtOAc and washed with IN HC1 (50 mL) and ice (75 g). The aqueous phase was separated and extracted with EtOAc (2x). The combined organics were washed with a mixture of KF (2.85g in 75 mL water) and IN HC1 (13 mL) [resulting solution pH 3-4], then with brine, dried (Na2s04), filtered and concentrated under reduced pressure to give a 9: 1 (S-LS-1E) enriched diastereomeric mixture (as determined by 1H NMR) of Intermediate S-1 and Intermediate S-1E (2.13 g, >99%) as a pale yellow viscous oil: 1H NMR (400 MHz, CDC13) δ ppm 2.64-2.76 (2 H, m), 2.04-2.35 (4 H, m), 1.88-2.00 (2 H, m), 1.71-1.83 (2 H, m), 1.48 (9 H, s).
Intermediate S-2: (2R,3S)-3-(fert-Butoxycarbonyl)-6,6,6-trifluoro-2-(3- fluoropropyl)hexanoic acid

Intermediate S-2: (2R,3S)-3-(tert-Butoxycarbonyl)-7,7,7-trifluoro-2-(3,3,3- trifluoropropyl)heptanoic acid, and Intermediate S-2A: (2R,3R)-3-(tert-Butoxycarbonyl)- 7,7,7-trifluoro-2-(3,3,3-trifluoropropyl)heptanoic acid

(S-2A)
[00194] To a cold (-78 °C), stirred solution of Intermediate S-1D (1.72 g, 6.36 mmol) in THF (30 mL) was slowly added LDA (7.32 mL, 14.6 mmol) over 7 min. After stirring for 1 h, 4,4,4-trifluorobutyltrifluoromethanesulfonate (2.11 g, 8.11 mmol) was added to the reaction mixture over 2 min. After 15 min, the reaction mixture was warmed to -25 °C (ice/MeOH/dry ice) for lh, and then cooled to -78 °C. After 80 min, the reaction was quenched with a saturated aqueous NH4C1 solution (10 mL). The reaction mixture was further diluted with brine and the solution was adjusted to pH 3 with IN HC1. The aqueous layer was extracted with ether. The combined organics were washed with brine, dried over anhydrous magnesium sulfate, and concentrated under reduced pressure to provide a mixture of Intermediates S-2 and S-2A (2.29 g, 95%) as a colorless oil. 1H NMR (400MHz, chloroform-d) δ 2.83-2.75 (m, 1H), 2.64 (ddd, J = 9.9, 6.7, 3.6 Hz, 1H), 2.32-2.03 (m, 5H), 1.98-1.70 (m, 3H), 1.69-1.52 (m, 3H), 1.50-1.42 (m, 9H). 1H NMR showed a 1 :4.5 mixture (S-2:S-2A) of diastereomers by integration of the peaks for the t- Bu groups.
Intermediate S-2: (2R,3S)-3-(fert-Butoxycarbonyl)-7,7,7-trifluoro-2-(3,3,3- trifluoropropyl)heptanoic acid, and Intermediate S-2A: (2R,3R)-3-(tert-Butoxycarbonyl)- 7,7,7-trifluoro-2-(3,3,3-trifluoropropyl)heptanoic acid

[00195] A mixture of Intermediate S-2 and Intermediate S-2A (2.29 g, 6.02 mmol) was dissolved in THF (38 mL) to give a colorless solution which was cooled to -78 °C. Then, LDA (7.23 mL, 14.5 mmol) (2.0M in heptane/THF/ethylbenzene) was slowly added to the reaction mixture over 3 min. After stirring for 15 min, the reaction mixture was placed in a room temperature water bath. After 15 min the reaction mixture was placed back in a -78 °C bath and then diethylaluminum chloride (14.5 mL, 14.5 mmol) (1M in hexane) was added slowly over 5 min. The reaction mixture was stirred at -78 °C. After 15 min, the reaction mixture was placed in a room temperature water bath for 10 min, and then cooled back to -78 °C. After 15 min, the reaction was quenched with MeOH (30.0 mL, 741 mmol), removed from the -78 °C bath and concentrated. To the reaction mixture was added ice and HC1 (60.8 mL, 60.8 mmol) and the resulting mixture was extracted with EtOAc (2x 200 mL). The organic layer was washed with potassium fluoride (3.50g, 60.3 mmol) in 55 mL H20 and 17.0 mL of IN HC1. The organics were dried over anhydrous magnesium sulfate and concentrated under reduced pressure to provide an enriched mixture of Intermediate S-2 and Intermediate S-2A (2.25g, 98% yield) as a light yellow oil. 1H NMR (400MHz, chloroform-d) δ 2.83-2.75 (m, 1H), 2.64 (ddd, J= 9.9, 6.7, 3.6 Hz, 1H), 2.32-2.03 (m, 5H), 1.98-1.70 (m, 3H), 1.69-1.52 (m, 3H), 1.50-1.42 (m, 9H). 1H NMR showed a 9: 1 ratio in favor of the desired diastereomer Intermediate S-2.
Intermediate S-2B: (2R,3S)-1 -Benzyl 4-tert-butyl 2,3-bis(4,4,4-trifluorobutyl)succinate

[00196] To a stirred 9: 1 mixture of Intermediate S-2 and Intermediate S-2A (2.24 g, 5.89 mmoL) and potassium carbonate (1.60 g, 11.58 mmoL) in DMF (30 mL) was added benzyl bromide (1.20 mL, 10.1 mmoL)). The reaction mixture was stirred at room temperature for 19 h. The reaction mixture was diluted with ethyl acetate (400 mL) and washed with 10% LiCl solution (3 x 100 mL), brine (50 mL), and then dried over anhydrous magnesium sulfate, filtered and concentrated to dryness under vacuum. The residue was purified by flash chromatography (Teledyne ISCO CombiFlash 0%> to 100% solvent A/B = hexane/EtOAc, REDISEP® Si02 220 g, detecting at 254 nm, and monitoring at 220 nm). Concentration of the appropriate fractions provided Intermediate S-2B (1.59 g, 57.5%). HPLC: RT = 3.863 min (CHROMOLITH® SpeedROD column 4.6 x 50 mm, 10-90% aqueous methanol over 4 minutes containing 0.1% TFA, 4 mL/min, monitoring at 220 nm), 1H NMR (400MHz, chloroform-d) δ 7.40-7.34 (m, 5H), 5.17 (d, J= 1.8 Hz, 2H), 2.73-2.64 (m, 1H), 2.55 (td, J= 10.0, 3.9 Hz, 1H), 2.16-1.82 (m, 5H), 1.79-1.57 (m, 3H), 1.53-1.49 (m, 1H), 1.45 (s, 9H), 1.37-1.24 (m, 1H).
Intermediate S-2: (2R,3S)-3-(tert-Butoxycarbonyl)-6,6,6-trifluoro-2-(4,4,4- trifluorobutyl)hexanoic acid

[00197] To a stirred solution of Intermediate S-2B (1.59 g, 3.37 mmoL) in MeOH (10 mL) and EtOAc (10 mL) under nitrogen was added 10%> Pd/C (510 mg). The atmosphere was replaced with hydrogen and the reaction mixture was stirred at room temperature for 2.5 h. The palladium catalyst was filtered off through a 4 μΜ polycarbonate film and rinsed with MeOH. The filtrate was concentrated under reduced pressure to give intermediate S-2 (1.28 g, 99%). 1H NMR (400MHz, chloroform-d) δ 2.76-2.67 (m, 1H), 2.65-2.56 (m, 1H), 2.33-2.21 (m, 1H), 2.17-2.08 (m, 3H), 1.93 (dtd, J= 14.5, 9.9, 5.2 Hz, 1H), 1.84-1.74 (m, 2H), 1.70-1.52 (m, 3H), 1.48 (s, 9H).
Intermediate A- 1 : (2-Amino-3 -methylphenyl)(3 -fluorophenyl)methanone

Intermediate A-1 A: 2-Amino- -methoxy-N,3-dimethylbenzamide

[00198] In a 1 L round-bottomed flask was added 2-amino-3-methylbenzoic acid (11.2 g, 74.1 mmol) and Ν,Ο-dimethylhydroxylamine hydrochloride (14.45 g, 148 mmol) in DCM (500 mL) to give a pale brown suspension. The reaction mixture was treated with Et3N (35 mL), HOBT (11.35 g, 74.1 mmol) and EDC (14.20 g, 74.1 mmol) and then stirred at room temperature for 24 hours. The mixture was then washed with 10% LiCl, and then acidified with IN HCl. The organic layer was washed successively with 10%> LiCl and aq NaHC03. The organic layer was decolorized with charcoal, filtered, and the filtrate was dried over MgSC^. The mixture was filtered and concentrated to give 13.22 g (92% yield) of Intermediate A-1A. MS(ES): m/z = 195.1 [M+H+]; HPLC: RT = 1.118 min. (H20/MeOH with TFA, CHROMOLITH® ODS S5 4.6 x 50 mm, gradient = 4 min, wavelength = 220 nm); 1H NMR (500MHz, chloroform-d) δ 7.22 (dd, J= 7.8, 0.8 Hz, 1H), 7.12-7.06 (m, 1H), 6.63 (t, J= 7.5 Hz, 1H), 4.63 (br. s., 2H), 3.61 (s, 3H), 3.34 (s, 3H), 2.17 (s, 3H).
Intermediate A- 1 : (2-Amino-3 -methylphenyl)(3 -fluorophenyl)methanone

[00199] In a 500 mL round-bottomed flask, a solution of l-fluoro-3-iodobenzene (13.61 mL, 116 mmol) in THF (120 mL) was cooled in a -78 °C bath. A solution of n- BuLi, (2.5M in hexane, 46.3 mL, 116 mmol) was added dropwise over 10 minutes. The solution was stirred at -78 °C for 30 minutes and then treated with a solution of
Intermediate A-1 A (6.43 g, 33.1 mmol) in THF (30 mL). After 1.5 hours, the reaction mixture was added to a mixture of ice and IN HCl (149 mL, 149 mmol) and the reaction flask was rinsed with THF (5 ml) and combined with the aqueous mixture. The resulting mixture was diluted with 10% aq LiCl and the pH was adjusted to 4 with IN NaOH. The mixture was then extracted with Et20, washed with brine, dried over MgS04, filtered and concentrated. The resulting residue was purified by silica gel chromatography (220g ISCO) eluting with a gradient from 10% EtOAc/hexane to 30% EtOAc/hexane to afford Intermediate A-l (7.11 g, 94% yield) as an oil. MS(ES): m/z = 230.1 [M+H+]; HPLC: RT = 2.820 min Purity = 99%. (H20/MeOH with TFA, CHROMOLITH® ODS S5 4.6 x 50 mm, gradient = 4 min, wavelength = 220 nm).
Intermediate B-1 : (S)-3-Amino-5-(3-fluorophenyl)-9-methyl-lH-benzo[e][l,4]diazepin- 2(3H)-one

Intermediate B-1 A: (S)-Benzyl (5-(3-fluorophenyl)-9-methyl-2-oxo-2,3-dihydro benzo[e] [ 1 ,4]diazepin-3-yl)carbamate

(B-1A)
[00225] In a 1 L round-bottomed flask, a solution of 2-(lH-benzo[d][l,2,3]triazol-l- yl)-2-((phenoxycarbonyl)amino)acetic acid (J. Org. Chem., 55:2206-2214 (1990)) (19.37 g, 62.0 mmol) in THF (135 mL) was cooled in an ice/water bath and treated with oxalyl chloride (5.43 mL, 62.0 mmol) and 4 drops of DMF. The reaction mixture was stirred for 4 hours. Next, a solution of Intermediate A- 1 (7.11 g, 31.0 mmol) in THF (35 mL) was added and the resulting solution was removed from the ice/water bath and stirred at room temperature for 1.5 hours. The mixture was then treated with a solution of ammonia, (7M in MeOH) (19.94 mL, 140 mmol). After 15 mins, another portion of ammonia, (7M in MeOH) (19.94 mL, 140 mmol) was added and the resulting mixture was sealed under N2 and stirred overnight at room temperature. The reaction mixture was then concentrated to ~l/2 volume and then diluted with AcOH (63 mL) and stir at room temperature for 4 hours. The reaction mixture was then concentrated, and the residue was diluted with 500 mL water to give a precipitate. Hexane and Et20 were added and the mixture was stirred at room temperature for 1 hour to form an orange solid. Et20 was removed under a stream of nitrogen and the aqueous layer was decanted. The residue was triturated with 40 mL of iPrOH and stirred at room temperature to give a white precipitate. The solid was filtered and washed with iPrOH, then dried on a filter under a stream of nitrogen to give racemic Intermediate B-1A (5.4 g, 41.7%yield).
[00226] Racemic Intermediate B-1A (5.9 g, 14.3 mmol) was resolved using the Chiral SFC conditions described below. The desired stereoisomer was collected as the second peak in the elution order: Instrument: Berger SFC MGIII, Column: CHIRALPAK® IC 25 x 3 cm, 5 cm; column temp: 45 °C; Mobile Phase: C02/MeOH (45/55); Flow rate: 160 mL/min; Detection at 220 nm.
[00227] After evaporation of the solvent, Intermediate B-1A (2.73 g, 46% yield) was obtained as a white solid. HPLC: RT = 3.075 min. (H20/MeOH with TFA,
CHROMOLITH® ODS S5 4.6 x 50 mm, gradient = 4 min, wavelength = 220 nm).
Chiral HPLC RT: 8.661 min (AD, 60% (EtOH/MeOH)/heptane) > 99%ee. MS(ES): m/z = 418.3 [M+H+];1H NMR (500MHz, DMSO-d6) δ 10.21 (s, 1H), 8.38 (d, J= 8.3 Hz, 1H), 7.57-7.47 (m, 2H), 7.41-7.29 (m, 8H), 7.25-7.17 (m, 2H), 5.10-5.04 (m, 3H), 2.42 (s, 3H).
Intermediate B-l : (S)-3-Amino-5-(3-fluorophenyl)-9-methyl-lH-benzo[e][l,4]diazepin- 2(3H)-one.
[00228] In a 100 mL round-bottomed flask, a solution of Intermediate B-1A (2.73 g, 6.54 mmol) in acetic acid (12 mL) was treated with HBr, 33% in HOAc (10.76 mL, 65.4 mmol) and the mixture was stirred at room temperature for 1 hour. The solution was diluted with Et20 to give a yellow precipitate. The yellow solid was filtered and rinsed with Et20 under nitrogen. The solid was transferred to 100 mL round bottom flask and water was added (white precipitate formed). The slurry was slowly made basic with saturated NaHC03. The resulting tacky precipitate was extracted with EtOAc. The organic layer was washed with water, dried over MgS04, and then filtered and
concentrated to dryness to give Intermediate B-l (1.68 g, 91% yield) as a white foam solid. MS(ES): m/z = 284.2 [M+H+]; HPLC: RT = 1.72 min (H20/MeOH with TFA, CHROMOLITH® ODS S5 4.6 x 50 mm, gradient = 4 min, wavelength = 220 nm). 1H NMR (400MHz, DMSO-d6) δ 10.01 (br. s., 1H), 7.56-7.44 (m, 2H), 7.41-7.26 (m, 3H), 7.22-7.11 (m, 2H), 4.24 (s, 1H), 2.55 (br. s., 2H), 2.41 (s, 3H). [00229] The compounds listed below in Table 6 (Intermediates B-2 to B-3) were prepared according to the general synthetic procedure described for Intermediate B-l , using the starting materials Intermediate A- 10 and Intermediate A-4, respectively.
Example 1
(2R,3S)-N-((3S)-5-(3-Fluorophenyl)-9-methyl-2-oxo-2,3-dihydro-lH-l,4-benzodiazepin- 3-yl)-2, -bis(3,3,3-trifluoropropyl)succinamide

Intermediate 1A: (2S,3R)-tert-Butyl 6,6,6-trifluoro-3-(((S)-5-(3-fluorophenyl)-9-methyl- 2-0X0-2, 3-dihydro-lH-benzo[e][l,4]diazepin-3-yl)carbamoyl)-2-(3,3 ,3- trifluoropropyl)hexanoat

[00240] In a 100 mL round-bottomed flask, a solution of Intermediate B-l (1683 mg, 5.94 mmol), Et3N (1.656 mL, 11.88 mmol), and Intermediate S-l in DMF (20 mL) was treated with o-benzotriazol-l-yl-A .A .N’.N’-tetramethyluronium tetrafluoroborate (3815 mg, 11.88 mmol) and stirred at room temperature for 1 hour. The reaction mixture was diluted with water and saturated aqueous NaHC03. An off white precipitate formed and was filtered and washed with water. The resulting solid was dried on the filter under a stream of nitrogen to give Intermediate 1A (3.7 g, 99% yield). MS(ES): m/z =
632.4[M+H+]; HPLC: RT = 3.635 min Purity = 98%. (H20/MeOH with TFA,
CHROMOLITH® ODS S5 4.6 x 50 mm, gradient = 4 min, wavelength = 220 nm). 1H NMR (400MHz, methanol-d4) δ 7.53 (t, J = 4.5 Hz, 1H), 7.46-7.30 (m, 3H), 7.28-7.23 (m, 1H), 7.23-7.18 (m, 2H), 5.37 (s, 1H), 2.88 (td, J = 10.4, 3.4 Hz, 1H), 2.60 (td, J =
10.2, 4.1 Hz, 1H), 2.54-2.40 (m, 1H), 2.47 (s, 3 H), 2.33-2.12 (m, 3H), 1.98-1.69 (m, 4H), 1.51 (s, 9H). Intermediate IB: (2S,3R)-6,6,6-Trifluoro-3-(((S)-5-(3-fluorophenyl)-9-methyl-2-oxo-
2,3-dihydro-lH-benzo[e][l,4]diazepin-3-yl)carbamoyl)-2-(3,3,3-trifluoropropyl)hexanoic acid

[00241] In a 250 mL round-bottomed flask, a solution of Intermediate 1A (3.7 g, 5.86 mmol) in DCM (25 mL) was treated with TFA (25 mL) and the resulting pale orange solution was stirred at room temperature for 1.5 hours. The reaction mixture was then concentrated to give Intermediate IB. HPLC: RT = 3.12 min (H20/MeOH with TFA, CHROMOLITH® ODS S5 4.6 x 50 mm, gradient = 4 min, wavelength = 220 nm).
MS(ES): m/z = 576.3 (M+H)+. 1H NMR (400MHz, methanol-d4) δ 7.54 (t, J= 4.5 Hz, 1H), 7.49-7.29 (m, 3H), 7.28-7.15 (m, 3H), 5.38 (br. s., 1H), 2.89 (td, J= 10.3, 3.7 Hz, 1H), 2.67 (td, J= 9.9, 4.2 Hz, 1H), 2.56-2.38 (m, 1H), 2.48 (s, 3 H), 2.34-2.13 (m, 3H), 2.00-1.71 (m, 4H).
Example 1 :
[00242] In a 250 mL round-bottomed flask, a solution of Intermediate IB (4.04 g, 5.86 mmol) in THF (50 mL) was treated with ammonia (2M in iPrOH) (26.4 mL, 52.7 mmol), followed by HOBT (1.795 g, 11.72 mmol) and EDC (2.246 g, 11.72 mmol). The resulting white suspension was stirred at room temperature overnight. The reaction mixture was diluted with water and saturated aqueous NaHC03. The resulting solid was filtered, rinsed with water and then dried on the filter under a stream of nitrogen. The crude product was suspended in 20 mL of iPrOH and stirred at room temperature for 20 min and then filtered and washed with iPrOH and dried under vacuum to give 2.83 g of solid. The solid was dissolved in re fluxing EtOH(100 mL) and slowly treated with 200 mg activated charcoal added in small portions. The hot mixture was filtered through CELITE® and rinsed with hot EtOH. The filtrate was reduced to half volume, allowed to cool and the white precipitate formed was filtered and rinsed with EtOH to give 2.57 g of white solid. A second recrystallization from EtOH (70 mL) afforded Example 1 (2.39 g, 70% yield) as a white solid. HPLC: RT = 10.859 min (H20/CH3CN with TFA, Sunfire C18 3.5μπι, 3.0x150mm, gradient = 15 min, wavelength = 220 and 254 nm); MS(ES): m/z = 575.3 [M+H+]; 1H NMR (400MHz, methanol-d4) δ 7.57-7.50 (m, 1H), 7.47-7.30 (m, 3H), 7.29-7.15 (m, 3H), 5.38 (s, 1H), 2.85-2.75 (m, 1H), 2.59 (td, J= 10.5, 4.0 Hz, 1H), 2.53-2.41 (m, 4H), 2.31-2.10 (m, 3H), 1.96-1.70 (m, 4H).
SEE
WO2012129353A1 *Mar 22, 2012Sep 27, 2012Bristol-Myers Squibb CompanyBis(fluoroalkyl)-1,4-benzodiazepinone compounds
PAPER RELATED
Structure–activity relationships in a series of (2-oxo-1,4-benzodiazepin-3-yl)-succinamides identified highly potent inhibitors of γ-secretase mediated signaling of Notch1/2/3/4 receptors. On the basis of its robust in vivo efficacy at tolerated doses in Notch driven leukemia and solid tumor xenograft models, 12 (BMS-906024) was selected as a candidate for clinical evaluation.
Discovery of Clinical Candidate BMS-906024: A Potent Pan-Notch Inhibitor for the Treatment of Leukemia and Solid Tumors
Ashvinikumar V. Gavai*†, Claude Quesnelle†, Derek Norris†, Wen-Ching Han†, Patrice Gill†, Weifang Shan†, Aaron Balog†, Ke Chen§, Andrew Tebben†, Richard Rampulla†, Dauh-Rurng Wu†, Yingru Zhang†, Arvind Mathur†,Ronald White†, Anne Rose†, Haiqing Wang†, Zheng Yang†, Asoka Ranasinghe†, Celia D’Arienzo†, Victor Guarino†, Lan Xiao†, Ching Su†, Gerry Everlof†, Vinod Arora‡, Ding Ren Shen†, Mary Ellen Cvijic†, Krista Menard†, Mei-Li Wen†, Jere Meredith‡, George Trainor†, Louis J. Lombardo†, Richard Olson‡, Phil S. Baran§,John T. Hunt†, Gregory D. Vite†, Bruce S. Fischer†, Richard A. Westhouse†, and Francis Y. Lee†
†Bristol-Myers Squibb Research and Development, Princeton, New Jersey 08543, United States
‡Bristol-Myers Squibb Research and Development, 5 Research Parkway, Wallingford, Connecticut 06492, United States
§ Department of Chemistry, The Scripps Research Institute, 10550 North Torrey Pines Road, La Jolla, California 92037,United StatesACS Med. Chem. Lett.
, 2015, 6 (5), pp 523–527
DOI: 10.1021/acsmedchemlett.5b00001, http://pubs.acs.org/doi/abs/10.1021/acsmedchemlett.5b00001
*Phone: 609-252-5091. E-mail: ashvinikumar.gavai@bms.com.



AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

……
PAPER RELATED

An enantioselective synthesis of (S)-7-amino-5H,7H-dibenzo[b,d]azepin-6-one (S–1) is described. The key step in the sequence involved crystallization-induced dynamic resolution (CIDR) of compound 7 using Boc-d-phenylalanine as a chiral resolving agent and 3,5-dichlorosalicylaldehyde as a racemization catalyst to afford S–1 in 81% overall yield with 98.5% enantiomeric excess.
Crystallization-Induced Dynamic Resolution toward the Synthesis of (S)-7-Amino-5H,7H-dibenzo[b,d]-azepin-6-one: An Important Scaffold for γ-Secretase Inhibitors
Sukhen Karmakar†, Vijay Byri†, Ashvinikumar V. Gavai‡, Richard Rampulla‡, Arvind Mathur‡, and Anuradha Gupta*†
† Department of Discovery Synthesis, Biocon Bristol-Myers Squibb Research Centre, Biocon Park, Bommasandra IV Phase, Jigani Link Road, Bengaluru 560099, India
‡Bristol-Myers Squibb Company, P.O Box 4000, Princeton, New Jersey 08543-4000, United StatesOrg. Process Res. Dev.
, Article ASAP
DOI: 10.1021/acs.oprd.6b00207, http://pubs.acs.org/doi/suppl/10.1021/acs.oprd.6b00207
*E-mail: anuradha.gupta@syngeneintl.com.
| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2000007995A1 * | Aug 7, 1999 | Feb 17, 2000 | Du Pont Pharmaceuticals Company | SUCCINOYLAMINO LACTAMS AS INHIBITORS OF Aβ PROTEIN PRODUCTION |
| WO2000038618A2 * | Dec 23, 1999 | Jul 6, 2000 | Du Pont Pharmaceuticals Company | SUCCINOYLAMINO BENZODIAZEPINES AS INHIBITORS OF Aβ PROTEIN PRODUCTION |
| WO2001060826A2 * | Feb 16, 2001 | Aug 23, 2001 | Bristol-Myers Squibb Pharma Company | SUCCINOYLAMINO CARBOCYCLES AND HETEROCYCLES AS INHIBITORS OF Aβ PROTEIN PRODUCTION |
| US6737038 * | May 17, 2000 | May 18, 2004 | Bristol-Myers Squibb Company | Use of small molecule radioligands to discover inhibitors of amyloid-beta peptide production and for diagnostic imaging |
| US7053084 | Feb 17, 2000 | May 30, 2006 | Bristol-Myers Squibb Company | Succinoylamino benzodiazepines as inhibitors of Aβ protein production |
| US7456172 | Jan 13, 2006 | Nov 25, 2008 | Bristol-Myers Squibb Pharma Company | Succinoylamino benzodiazepines as inhibitors of Aβ protein production |
| US20030134841 * | Nov 1, 2002 | Jul 17, 2003 | Olson Richard E. | Succinoylamino lactams as inhibitors of A-beta protein production |
| US20120245151 * | Mar 22, 2012 | Sep 27, 2012 | Bristol-Myers Squibb Company | Bisfluoroalkyl-1,4-benzodiazepinone compounds |
//////////varegacestat, BMS-986115, BMS 986115, 3,5-dichlorosalicylaldehyde, Alzheimer’s disease, Boc-D-phenylalanine, CIDR;dibenzoazepenone, DKR; Notch inhibitors, Notch inhibitor, SAR, T-acute lymphoblastic leukemia, triple-negative breast cancer, γ-secretase inhibitor, PHASE 1, BMS, Bristol-Myers Squibb, Ashvinikumar Gavai, 1584647-27-7, LSK1L593UU, AL 102
Admilparant
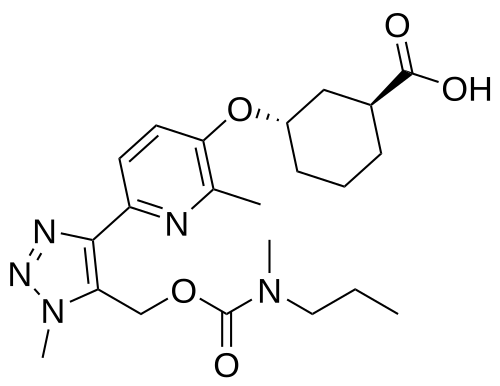
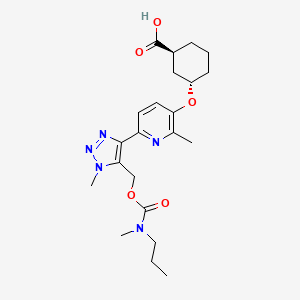
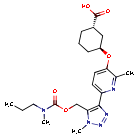
Admilparant, (BMS-986278)
CAS 2170126-74-4
MF C22H31N5O5 MW 445.5 g/mol
(1S,3S)-3-({2-methyl-6-[1-methyl-5-({[methyl(propyl)carbamoyl]oxy}methyl)-1H-1,2,3-triazol-4-l]pyridin-3-yl}oxy)cyclohexane-1-carboxylic acid
lysophosphatidic acid receptor 1 (LPA1) antagonist
- 4UN9AOU6G8
- BMS986278
- (1S,3S)-3-((2-Methyl-6-(1-methyl-5-(((methyl(propyl)carbamoyl)oxy)methyl)-1H-1,2,3-triazol-4-yl)pyridin-3-yl)oxy)cyclohexane-1-carboxylic acid
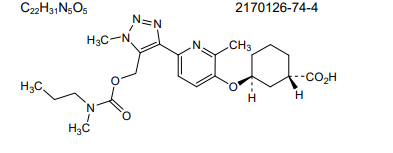
Admilparant is an investigational new drug being developed by Bristol-Myers Squibb for the treatment of idiopathic pulmonary fibrosis (IPF) and progressive pulmonary fibrosis (PPF). It is a first-in-class lysophosphatidic acid receptor 1 (LPA1) antagonist.[1][2]
As of 2024, admilparant is in Phase III clinical trials for both IPF and PPF.[2][3]
SYN
Publication Name: Journal of Medicinal Chemistry, Publication Date: 2021-10-28, PMID: 34709814
DOI: 10.1021/acs.jmedchem.1c01256
(1S,3S)-3-((2-Methyl-6-(1-methyl-5-(((methyl(propyl)carbamoyl)-oxy)methyl)-1H-1,2,3-triazol-4-yl)pyridin-3-yl)oxy)cyclohexane-1-carboxylic Acid (33). Compound 33 was prepared using the same
synthetic sequence as 25, except that intermediate 42 was reacted with
N-methylpropan-1-amine instead of 1-cyclobutyl-N-methylmethanamine. 1H NMR (500 MHz, DMSO-d6, 100 °C) δ 11.99−11.46 (m,1H), 7.82 (d, J = 8.3 Hz, 1H), 7.43 (d, J = 8.8 Hz, 1H), 5.65 (s, 2H),
4.89−4.62 (m, 1H), 4.10 (s, 3H), 3.12 (br t, J = 7.2 Hz, 2H), 2.79 (s,3H), 2.69 (tt, J = 9.4, 4.4 Hz, 1H), 2.44 (s, 3H), 2.03 (dt, J = 13.8, 4.5Hz, 1H), 1.92−1.86 (m, 1H), 1.86−1.79 (m, 2H), 1.74−1.68 (m, 1H),
1.68−1.58 (m, 2H), 1.58−1.51 (m, 1H), 1.43 (dq, J = 14.4, 7.1 Hz,2H), 0.76 (br t, J = 7.3 Hz, 3H). 13C NMR (126 MHz, DMSO-d6, 100°C) δ 175.4, 154.7, 150.1, 147.7, 143.9, 141.4, 129.6, 120.0, 118.6, 71.8,
54.5, 49.5, 37.4, 34.4, 33.4, 31.6, 28.7, 27.2, 19.8, 19.4, 18.6, 10.1. m/z446 [M + H]+
. HPLC/UV purity: 99.9% using the following reverse phase chromatographic conditions: Agilent HPLC; Phenomenex Kinetex-C-18; 100 (L) × 4.6 mm2 (i.d.) column; 2.6 μm particle size; wavelength, 220−380 nm; flow rate, 1.0 mL/min; temperature, 35°C; injection volume, 4 μL of 0.25 mg/mL in 1:1 MeCN:H2O; mobilephase A, H2O−0.05% TFA; mobile phase B, MeCN−0.05% TFA; gradient elution, starting at 10−80% B over 10 min and ending at 95% Bafter an additional 4 min; retention time = 8.28 min. Stereoisomeric purity was >99.5% using the following chiral chromatographic conditions: UPC2 Analytical SFC, ChromegaChiral CC4; 250 (L) ×4.6 mm2 (i.d.); 5 μm column; flow rate, 3 mL/min; temperature, 40 °C;injection volume, 10 μL of 0.25 mg/mL in MeCN:MeOH (1:1);mobile phase, 30% MeOH and 70% CO2 at 120 bar retention time =6.05 min. Accurate mass, [M + H]+ at m/z = 446.2398 (−2.03 ppmfrom theoretical for C22H32N5O5). [α]20D = +28.24° (MeOH, c = 0.51).
Elem. Anal. (theoretical): C, 59.31; H, 7.01; N, 15.72. Found: C, 59.35;H, 6.78; N, 15.69. UV (MeOH) at 254 nm (ε = 17,856), 290 nm (ε =7,519), and 296 nm (ε = 8,288). Concentration: adjusted for purity,
0.05154840 g/L or 0.0001157047 mol/L. Melting point = 152−154°C. Accurate mass, [M + H]+ at m/z 466.2398 (−2.03 ppm fromtheoretical for C22H32N5O5).
synthetic sequence as 25, except that intermediate 42 was reacted with N-methylpropan-1-amine instead of 1-cyclobutyl-N-methylmethanamine
a
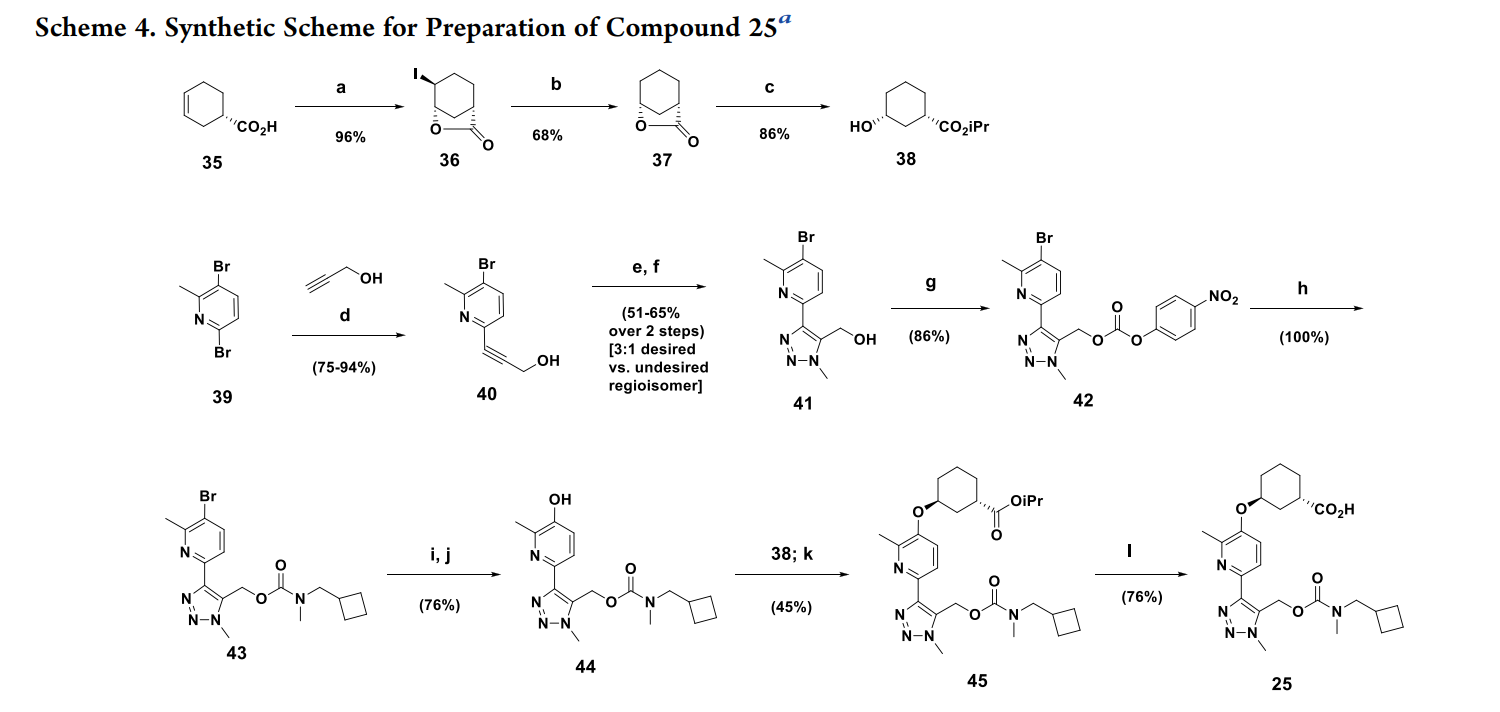
Reagents and conditions: (a) I2 (1.1 equiv)/KI (2.5 equiv)/NaHCO3 (3 equiv)/water (96%); (b) H2 (50 psi)/ Pd/C (cat)/Et3N (2 equiv)/EtOAc (68%); (c) CH3COCl (2.5 equiv)/iPrOH (87−95%); d) (Ph3P)2PdCl2 (5%)/ Et3N/CuI (5%)/RT (75−94%); (e) Ru(II)-(Ph3P)2(Me5Cyp)Cl (5%)/TMSCH2N3/dioxane 50 °C/15 h; (f) Bu4NF/0 °C to RT (51−65% over 2 steps; 3:1 desired:undesired regioisomer); (g) 4-nitrophenyl chloroformate/pyridine/CH2Cl2 (86%); (h) N-cyclobutyl N-methylamine/iPr2NEt/CH2Cl2 (100%); (i) B2(pin)2/KOAc/PdCl2(dppf)/THF/80 °C; (j) NaH2BO4/H2O/RT (76% over 2 steps); (k) 38; 1,1′-(azodicarbonyl)dipiperidine/Bu3P/toluene/50 °C (45%); (l)LiOH/H2O/MeOH (76%).
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=US208146892&_cid=P20-MFS2PF-83792-1
PATENT
- Carbamoyloxymethyl triazole cyclohexyl acids as lpa antagonistsPublication Number: US-2022249443-A1Priority Date: 2016-06-21
- Carbamoyloxymethyl triazole cyclohexyl acids as LPA antagonistsPublication Number: US-RE49352-EPriority Date: 2016-06-21Grant Date: 2023-01-03
- Carbamoyloxymethyl triazole cyclohexyl acids as LPA antagonistsPublication Number: AU-2021209334-B2Priority Date: 2016-06-21Grant Date: 2023-06-01
- Carbamoyloxymethyltriazole cyclohexylates as LPA antagonistsPublication Number: JP-7312295-B2Priority Date: 2016-06-21Grant Date: 2023-07-20
- Carbamoyloxymethyl triazole cyclohexyl acids as lpa antagonistsPublication Number: US-2023390249-A1Priority Date: 2016-06-21
- Carbamoyloxymethyltriazolylcyclohexanes as LPA antagonistsPublication Number: CN-109963843-BPriority Date: 2016-06-21Grant Date: 2022-03-11
- Carbamoyloxymethyltriazole cyclohexyl acid as LPA antagonistPublication Number: CN-114601830-APriority Date: 2016-06-21
- Carbamoyloxymethyl triazole cyclohexyl acid as an LPA antagonistPublication Number: KR-102377340-B1Priority Date: 2016-06-21Grant Date: 2022-03-21
- Carbamoyloxymethyl triazole cyclohexyl acids as lpa antagonistsPublication Number: KR-20220038537-APriority Date: 2016-06-21
- Carbamoyloxymethyl triazole cyclohexyl acids as lpa antagonistsPublication Number: KR-102463621-B1Priority Date: 2016-06-21Grant Date: 2022-11-03

AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
References
- “Admilparant (BMS-986278): Idiopathic Pulmonary Fibrosis Likelihood of Approval”. Pharmaceutical Technology. 25 December 2023. Retrieved 2024-11-23.
- Corte TJ, Behr J, Cottin V, Glassberg MK, Kreuter M, Martinez FJ, et al. (October 2024). “Efficacy and Safety of Admilparant, an LPA1 Antagonist in Pulmonary Fibrosis: A Phase 2 Randomized Clinical Trial”. American Journal of Respiratory and Critical Care Medicine. 211 (2): 230–238. doi:10.1164/rccm.202405-0977OC. PMID 39393084.
- Splete H (16 September 2024). “Admilparant Affects Biomarkers in Pulmonary Fibrosis”. Medscape. Retrieved 2024-11-23.
| Clinical data | |
|---|---|
| Other names | BMS-986278 |
| Identifiers | |
| IUPAC name | |
| CAS Number | 2170126-74-4 |
| PubChem CID | 132232205 |
| DrugBank | DB18011 |
| ChemSpider | 115009679 |
| UNII | 4UN9AOU6G8 |
| KEGG | D12657 |
| ChEMBL | ChEMBL5087506 |
| Chemical and physical data | |
| Formula | C22H31N5O5 |
| Molar mass | 445.520 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
References
- Zhou Y, Zhang Y, Zhao D, Yu X, Shen X, Zhou Y, Wang S, Qiu Y, Chen Y, Zhu F: TTD: Therapeutic Target Database describing target druggability information. Nucleic Acids Res. 2024 Jan 5;52(D1):D1465-D1477. doi: 10.1093/nar/gkad751. [Article]
/////////Admilparant, BMS 986278, PHASE 3, Bristol-Myers Squibb, idiopathic pulmonary fibrosis, 4UN9AOU6G8
BMS 262084
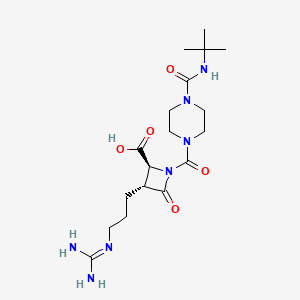
BMS-262084
CAS 253174-92-4
- Molecular FormulaC18H31N7O5
- Average mass425.483 Da
NII-I0IR71971G
I0IR71971G
(2S,3R)-1-[4-(tert-butylcarbamoyl)piperazine-1-carbonyl]-3-[3-(diaminomethylideneamino)propyl]-4-oxoazetidine-2-carboxylic acid(2S,3R)-1-{[4-(tert-butylcarbamoyl)piperazin-1-yl]carbonyl}-3-{3-[(diaminomethylidene)amino]propyl}-4-oxoazetidine-2-carboxylic acid
(2S,3R)-3-{3-[(Diaminomethylene)amino]propyl}-1-({4-[(2-methyl-2-propanyl)carbamoyl]-1-piperazinyl}carbonyl)-4-oxo-2-azetidinecarboxylic acid253174-92-4[RN]2-Azetidinecarboxylic acid, 3-[3-[(diaminomethylene)amino]propyl]-1-[[4-[[(1,1-dimethylethyl)amino]carbonyl]-1-piperazinyl]carbonyl]-4-oxo-, (2S,3R)-
Factor XIa inhibitors (thrombosis), BMS; Factor XIa inhibitors (thrombosis), Bristol-Myers Squibb; BMS-654457; Factor XIa inhibitors (cardiovascular diseases), BMS; BMS-724296
Novel crystalline forms of BMS-262084 as Factor XIa antagonist useful for treating cardiovascular diseases.
PHASE 2

PAPER
Bioorganic & Medicinal Chemistry Letters (2002), 12(21), 3229-3233.
https://www.sciencedirect.com/science/article/pii/S0960894X02006881
Abstract
A series of N1-activated C4-carboxy azetidinones was prepared and tested as inhibitors of human tryptase. The key stereochemical and functional features required for potency, serine protease specificity and aqueous stability were determined. From these studies compound 2, BMS-262084, was identified as a potent and selective tryptase inhibitor which, when dosed intratracheally in ovalbumin-sensitized guinea pigs, reduced allergen-induced bronchoconstriction and inflammatory cell infiltration into the lung.
BMS-262084 was identified as a potent and selective tryptase inhibitor that, when dosed intratracheally in ovalbumin-sensitized guinea pigs, reduced allergen-induced bronchoconstriction and inflammatory cell infiltration into the lung.

PAPER
https://pubs.acs.org/doi/10.1021/jo010757o
Journal of Organic Chemistry (2002), 67(11), 3595-3600.

A highly stereoselective synthesis of the novel tryptase inhibitor BMS-262084 was developed. Key to this synthesis was the discovery and development of a highly diastereoselective demethoxycarbonylation of diester 12 to form the trans-azetidinone 13. BMS-262084 was prepared in 10 steps from d-ornithine in 30% overall yield.
1 as a white powder (3.18 g, 99% yield). Mp: 213-215 °C dec. [α]25D = −65.9 (c 0.99, MeOH). 1H NMR (CD3OD): δ 4.17 (d, J = 3.29 Hz, 1H), 3.61−3.11 (m, 11H), 1.94−1.75 (m, 4H), 1.32 (s, 9H). 13C NMR (CD3OD): δ 176.6, 168.7, 159.4, 158.7, 152.3, 58.7, 53.2, 51.8, 46.5, 45.0, 41.8, 29.6, 27.4, 26.3. HRMS: calcd for C18H32N7O5(M+ + H) 426.2465, found 426.2470. IR (KBr): 3385, 3184, 1775, 1657, 1535, 1395, 1259, 1207, 996, 763 cm–1. Anal. Calcd for C18H31N7O5: C, 50.81, H, 7.34, N, 23.04. Found: C, 50.65, H, 7.42, N, 22.72. Chiral HPLC: ee 99.6%; Chiralpak OD column, 250 × 4.6 mm, 10 μm; mobile phase hexane/EtOH (85:15, v/v); isocratic at ambient temperature, 1.0 mL/min, 220 nm; concentration 0.25 mg/mL, 10 μL injection; RT = 18.6 min (enantiomer, RT = 15.7 min).


PATENT
WO2018133793
claiming macrocyclic compounds.
PATENT
WO-2020259366
Novel crystalline and solid forms of BMS-262084 (designates as monohydrate or 1.5 hydrate), processes for their preparation and compositions comprising them are claimed. BMS-262084 is disclosed to be Factor XIa antagonist, useful for treating cardiovascular diseases.MS-262084 (CAS number: 253174-92-4), the chemical name is (2S,3R)-1-[4-(tert-butylcarbamoyl)piperazine-1-carbonoyl]-3-[3- (Diaminomethylamino)propyl]-4-cyclopropanamide-2-carboxylic acid, also called compound (1) in the present invention, is developed by BMS (Bristol-Myers-Squibb) to treat cardiovascular diseases The drug, as an oral coagulation factor XIa inhibitor for thrombus, has the advantage of significantly reducing the risk of bleeding, and its structure is shown in formula (1):
Patent application WO 9967215A1 discloses the BMS-262084 compound, but the specific molecular formula of the solid substance obtained by the disclosed preparation process is C 18 H 31 N 7 O 5 ·1.56H 2 O, which is similar to the crystal of BMS-262084 described in this application. Type and amorphous water have different molecular weights.
“A stereoselective synthesis of BMS-262084 an azetidinone-based tryptase inhibitor” (Source: Journal of Organic Chemistry, 2002,67(11):3595-3600; Journal of Organic Chemistry,2002,67(11):3595-3600) It is mentioned that the preparation method of BMS-262084 is that hydrogenolysis under neutral conditions eliminates the benzene and Cbz protection groups, and obtains BMS-262084 (melting point 213-215℃). The inventors conducted experiments based on part of the contents disclosed in the document, and the test results obtained crystal form A and crystal form B. The X-ray powder diffraction patterns are shown in Figure 1 and Figure 2 respectively.Example 1
“A stereoselective synthesis of BMS-262084 an azetidinone-based tryptase inhibitor” (Source: Journal of Organic Chemistry, 2002,67(11):3595-3600; Journal of Organic Chemistry,2002,67(11):3595-3600) Only ethanol solvents are mentioned in the literature. Since no specific crystal refining process was provided, only part of the experiment was performed using ethanol solvent.
1) Ethanol solvent volatilization at room temperature: 50mg of BMS-262084 (amorphous) was added to 1.0 mL of ethanol solvent and completely dissolved at room temperature (about 25°C). After volatilizing at room temperature for two days, the solid product was obtained and its crystal form was tested. It is crystal form A, as shown in Figure 1. It is considered that it contains a small amount of amorphous form; but it is unstable and will undergo crystal transformation at room temperature. After standing for one day, the XRPD was tested, and it was found that it was converted to a mixture containing crystal form A, other crystal forms and amorphous forms.
2) Ethanol solvent high-temperature volatilization: 50mg BMS-262084 is added to 1.0mL ethanol solvent, completely dissolved at high temperature (about 60℃), and high-temperature volatilization is carried out in the open to obtain a solid product. The crystal form of the solid product is detected, and the crystal form is B (contains a lot of amorphous), see Figure 2.
SYN1
WO 9967215
The condensation of N-(tert-butyldimethylsilyl)-4-oxoazetidine-2(S)-carboxylic acid (I) with 1-chloro-3-iodopropane (II) by means of BuLi and triisopropylamine (TIA) in THF, followed by treatment with HCl, gives the 3(R)-(3-chloropropyl) derivative (III), which is treated with tetrabutylammonium azide and tetrabutylammonium iodide in DMF to yield the 3-azidopropyl derivative (IV). The reduction of (IV) with H2 over Pd/C in DMF affords the 3-aminopropyl compound (V), which is treated with 1-[N,N’-bis(benzyloxycarbonyl)-1H-pyrazole] (VI) in the same solvent to provide the protected 3-guanidinopropyl compound (VII). The esterification of (VII) with NaHCO3, tetrabutylammonium iodide and Bn-Br in DMF gives the benzyl ester (VIII), which is condensed with N-tert-butylpiperazine-1-carboxamide (IX) and phosgene by means of TEA in toluene to yield the protected precursor (X). Finally, this compound is debenzylated by hydrogenation with H2 over Pd/C in dioxane to give the target azetidine-carboxylic acid.
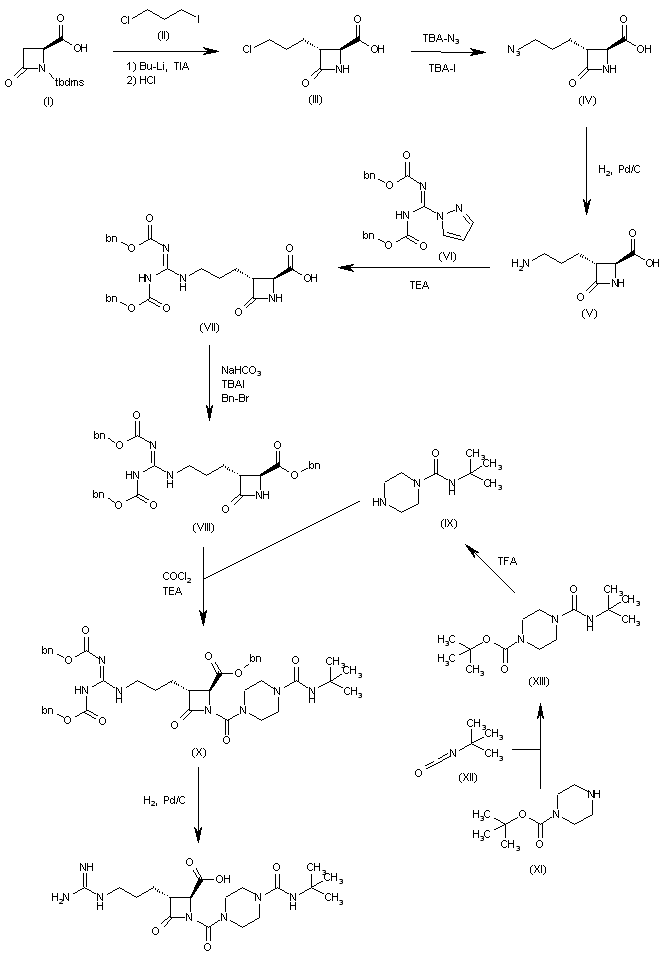
SYN 2
Ethyl nipecotate (I) was protected as the N-Boc derivative (II) and subsequently reduced to alcohol (III) by means of LiAlH4. Conversion of alcohol (III) into iodide (IV) was achieved by treatment with iodine and triphenylphosphine. The dianion of the chiral azetidinecarboxylic acid (V) was alkylated with iodide (IV) to furnish adduct (VI) as a diastereomeric mixture that was desilylated to (VII) using tetrabutylammonium fluoride. Benzyl ester (VIII) was then obtained by reaction of carboxylic acid (VII) with benzyl bromide and NaHCO3.
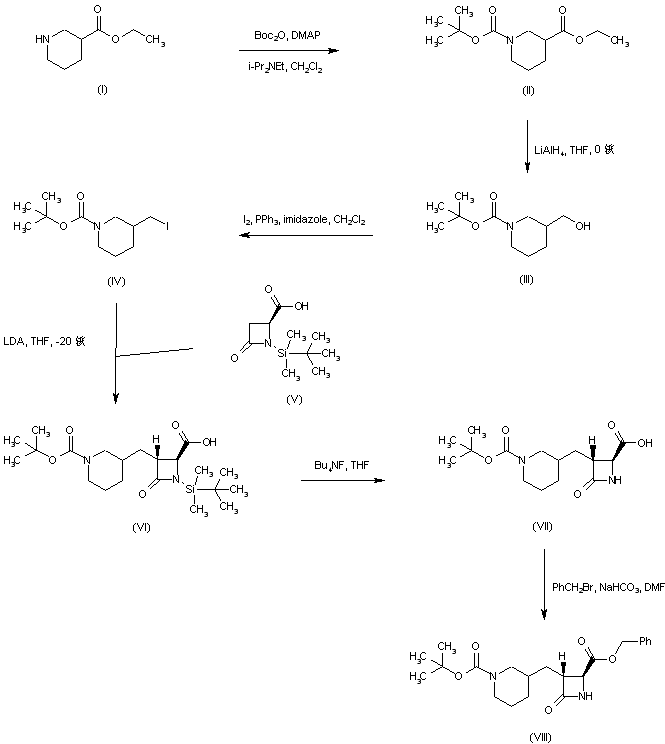
SYN 3
Coupling of 6-phenylhexanoic acid (X) with N-Boc-piperazine (IX) to give (XI), followed by acid deprotection of the Boc group of (XI), provided (6-phenylhexanoyl)piperazine (XII). This was converted to the carbamoyl chloride (XIII) upon treatment with phosgene. The condensation of carbamoyl chloride (XIII) with azetidinone (VIII) gave rise to the urea derivative (XIV). After acid cleavage of the Boc protecting group of (XIV), the resulting piperidine (XV) was condensed with N,N’-dicarbobenzoxy-S-methylisothiourea (XVI) in the presence of HgCl2, yielding the protected guanidine (XVII). This was finally deprotected by catalytic hydrogenolysis over Pd/C.
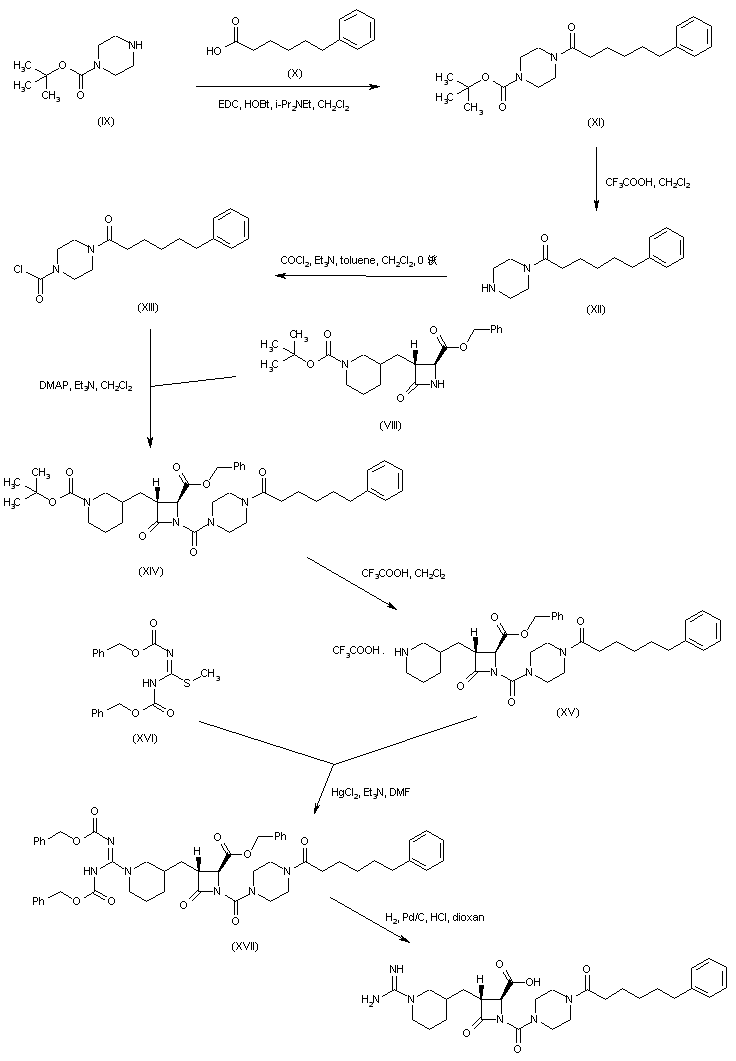
////////////////////////BMS-262084, BMS 262084, BMS 724296, Factor XIa inhibitors, thrombosis, Bristol-Myers Squibb, BMS 654457, PHASE 2
CC(C)(C)NC(=O)N1CCN(CC1)C(=O)N2C(C(C2=O)CCCN=C(N)N)C(=O)O
BMS 986142

BMS-986142
(2S,5R,3S)-6-fluoro-5-(3-(8-fluoro-1-methyl-2,4-dioxo-1,4-dihydroquinazolin-3(2H)-yl)-2-methylphenyl)-2-(2-hydroxypropan-2-yl)-2,3,4,9-tetrahydro-1H-carbazole-8-carboxamide
6-Fluoro-5-(R)-(3-(S)-(8-fluoro-l-methyl-2,4-dioxo-l,2-dihydroquinazolin-3(4H)-yl)-2- methylphenyl)-2-(S)-(2-hydroxypropan-2-yl)-2,3,4,9-tetrahydro-lH-carbazole-8- carboxamide
Molecular Formula, C32-H30-F2-N4-O4, Molecular Weight, 572.609, RN: 1643368-58-4
UNII: PJX9GH268R
- Originator Bristol-Myers Squibb
- Class Anti-inflammatories; Antirheumatics; Small molecules
- Mechanism of Action Agammaglobulinaemia tyrosine kinase inhibitors
- Phase II Rheumatoid arthritis; Sjogren’s syndrome
- 24 Jun 2018 Biomarkers information updated
- 07 Jun 2018 Bristol-Myers Squibb completes a phase II trial in Rheumatoid arthritis (Treatment-experienced) in Argentina, Austria, Belgium, Brazil, Canada, Chile, Colombia, Czech Republic, France, Germany, Israel, Italy, Japan, Mexico, Netherlands, Poland, Russia, South Africa, South Korea, Spain, Taiwan, USA (PO) (NCT02638948) (EudraCT2015-002887-17)
- 01 Oct 2016 Phase-II clinical trials in Sjogren’s syndrome in Puerto Rico (PO) (NCT02843659) after October 2016
- phase II clinical development at Bristol-Myers Squibb for the treatment of patients with moderate to severe rheumatoid arthritis and for the treatment of moderate to severe primary Sjogren’s syndrome.
BMS-986142 is a potent, selective, reversible BTK inhibitor. BMS-986142 shows BTK IC50 = 0.5nM; human WB IC50 = 90 nM. In molecule of BMS-986142, two atropisomeric centers were rotationally locked to provide a single, stable atropisomer, resulting in enhanced potency and selectivity as well as a reduction in safety liabilities. With significantly enhanced potency and selectivity, excellent in vivo properties and efficacy, and a very desirable tolerability and safety profile, BMS-986142 was advanced into clinical studies substituted tetrahydrocarbazole and 10 carbazole carboxamide compounds useful as kinase inhibitors, including the modulation of Bruton’s tyrosine kinase (Btk) and other Tec family kinases such as Itk. Provided herein are substituted tetrahydrocarbazole and carbazole carboxamide compounds, compositions comprising such compounds, and methods of their use. The invention further pertains to pharmaceutical compositions containing at least one compound 15 according to the invention that are useful for the treatment of conditions related to kinase modulation and methods of inhibiting the activity of kinases, including Btk and other Tec family kinases such as Itk, in a mammal. Protein kinases, the largest family of human enzymes, encompass well over 500 proteins. Btk is a member of the Tec family of tyrosine kinases, and is a regulator of 20 early B-cell development, as well as mature B-cell activation, signaling, and survival. B-cell signaling through the B-cell receptor (BCR) leads to a wide range of biological outputs, which in turn depend on the developmental stage of the B-cell. The magnitude and duration of BCR signals must be precisely regulated. Aberrant BCR- mediated signaling can cause disregulated B-cell activation and/or the formation of 25 pathogenic auto-antibodies leading to multiple autoimmune and/or inflammatory diseases. Mutation of Btk in humans results in X-linked agammaglobulinaemia (XLA). This disease is associated with the impaired maturation of B-cells, diminished immunoglobulin production, compromised T-cell-independent immune responses and marked attenuation of the sustained calcium signal upon BCR stimulation. 30 Evidence for the role of Btk in allergic disorders and/or autoimmune disease and/or inflammatory disease has been established in Btk-deficient mouse models. For example, in standard murine preclinical models of systemic lupus erythematosus (SLE), Btk deficiency has been shown to result in a marked amelioration of disease progression. Moreover, Btk deficient mice are also resistant to developing collagen-induced arthritis and are less susceptible to Staphylococcus-induced arthritis.
A large body of evidence supports the role of B-cells and the humoral immune system in the pathogenesis of autoimmune and/or inflammatory diseases. Protein-based therapeutics (such as RITUXAN®) developed to deplete B-cells, represent an important approach to the treatment of a number of autoimmune and/or inflammatory diseases. Because of Btk’s role in B-cell activation, inhibitors of Btk can be useful as inhibitors of B-cell mediated pathogenic activity (such as autoantibody production).
Btk is also expressed in mast cells and monocytes and has been shown to be important for the function of these cells. For example, Btk deficiency in mice is associated with impaired IgE-mediated mast cell activation (marked diminution of TNF-alpha and other inflammatory cytokine release), and Btk deficiency in humans is associated with greatly reduced TNF-alpha production by activated monocytes.
Thus, inhibition of Btk activity can be useful for the treatment of allergic disorders and/or autoimmune and/or inflammatory diseases including, but not limited to: SLE, rheumatoid arthritis, multiple vasculitides, idiopathic thrombocytopenic purpura (ITP), myasthenia gravis, allergic rhinitis, multiple sclerosis (MS), transplant rejection, type I diabetes, membranous nephritis, inflammatory bowel disease, autoimmune hemolytic anemia, autoimmune thyroiditis, cold and warm agglutinin diseases, Evans syndrome, hemolytic uremic syndrome/thrombotic thrombocytopenic purpura (HUS/TTP), sarcoidosis, Sj5gren’s syndrome, peripheral neuropathies (e.g., Guillain-Barre syndrome), pemphigus vulgaris, and asthma. In addition, Btk has been reported to play a role in controlling B-cell survival in certain B-cell cancers. For example, Btk has been shown to be important for the survival of BCR-Abl-positive B-cell acute lymphoblastic leukemia cells. Thus inhibition of Btk activity can be useful for the treatment of B-cell lymphoma and leukemia. In view of the numerous conditions that are contemplated to benefit by treatment involving modulation of protein kinases, it is immediately apparent that new compounds capable of modulating protein kinases such as Btk and methods of using these compounds should provide substantial therapeutic benefits to a wide variety of patients.
U.S. Patent No. 8,084,620 and WO 2011/159857 disclose tricyclic carboxamide compounds useful as kinase inhibitors, including the modulation of Btk and other Tec family kinases. There still remains a need for compounds useful as Btk inhibitors and yet having selectivity over Jak2 tyrosine kinase. Further, there still remains a need for compounds useful as Btk inhibitors that have selectivity over Jak2 tyrosine kinase and also have improved potency in the whole blood BCR-stimulated CD69 expression assay. Applicants have found potent compounds that have activity as Btk inhibitors. Further, applicants have found compounds that have activity as Btk inhibitors and are selective over Jak2 tyrosine kinase. Further still, applicants have found compounds that have activity as Btk inhibitors, are selective over Jak2 tyrosine kinase, and have improved potency in the whole blood BCR-stimulated CD69 expression assay. These compounds are provided to be useful as pharmaceuticals with desirable stability, bioavailability, therapeutic index, and toxicity values that are important to their drugability.
SYN

CLIP
Adventures in Atropisomerism: A Case Study from BMS – Not a Real Doctor
Scheme 2. Highlights from optimization of the first intermediate with axial chirality.


CLIP
https://cen.acs.org/pharmaceuticals/drug-development/Giving-atropisomers-another-chance/96/i33

During the discovery stage, BMS analytical chemist Jun Dai and the team developed methods to analyze the compounds’ isomers. She estimates that the researchers screened at least twice as many separation methods for atropisomers as they would have for normal chiral compounds because of the atropisomers’ potential for temperature-dependent conversion. “It was challenging but rewarding,” she says.
To determine the proportion of early atropisomers with half-lives of minutes to hours, the team ran high-performance liquid chromatography analysis at low temperature, chilling the column with ice or cooling equipment. Isolating some atropisomeric compounds required researchers to use ice-bath cooling during fraction collection and even solvent evaporation. The medicinal chemistry route to BMS-986142 required three chiral column purifications to obtain a single diastereomer with the best binding properties (J. Chromatogr. A 2017, DOI: 10.1016/j.chroma.2017.01.016).
Process synthesis, however, generally isn’t amenable to column chromatography steps, which can take weeks to months on a large scale. “To be honest, when I first saw it, I really wasn’t sure how we were going to make it,” says BMS chemist Thomas Razler, who led the process chemistry efforts to scale-up BMS-986142.
The researchers say extensive knowledge sharing between medicinal, analytical, and process teams about the atropisomeric compound was key to the program’s success. The process team took advantage of the fact that the diastereomeric forms of BMS-986142 had very different solubility profiles, enabling the chemists to replace all chiral chromatography with simpler crystallization steps and produce more than 200 kg of a single enantiomer and diastereomer (Org. Lett. 2018, DOI: 10.1021/acs.orglett.8b01218).
Although the final molecule is stable as a solid, the team says that in solution, the risk of racemization is higher. Citing ongoing work in that area of development, Razler declined to elaborate on how the molecule behaves in its formulation but notes the team hopes to publish that information next year. The atropisomerism is still an issue, he says, but a fascinating one.
Paper
Organic Letters, 20(13), 3736-3740; 2018
Adventures in Atropisomerism: Total Synthesis of a Complex Active Pharmaceutical Ingredient with Two Chirality Axes
Gregory Beutner  , Ronald Carrasquillo, Peng Geng, Yi Hsiao, Eric C. Huang, Jacob Janey, Kishta Katipally, Sergei Kolotuchin, Thomas La Porte, Andrew Lee, Paul Lobben, Federico Lora-Gonzalez, Brendan Mack, Boguslaw Mudryk, Yuping Qiu, Xinhua Qian, Antonio Ramirez, Thomas M. Razler* , Thorsten Rosner, Zhongping Shi, Eric Simmons , Jason Stevens, Jianji Wang, Carolyn Wei, Steven R. Wisniewski , and Ye Zhu
, Ronald Carrasquillo, Peng Geng, Yi Hsiao, Eric C. Huang, Jacob Janey, Kishta Katipally, Sergei Kolotuchin, Thomas La Porte, Andrew Lee, Paul Lobben, Federico Lora-Gonzalez, Brendan Mack, Boguslaw Mudryk, Yuping Qiu, Xinhua Qian, Antonio Ramirez, Thomas M. Razler* , Thorsten Rosner, Zhongping Shi, Eric Simmons , Jason Stevens, Jianji Wang, Carolyn Wei, Steven R. Wisniewski , and Ye Zhu
, Ronald Carrasquillo, Peng Geng, Yi Hsiao, Eric C. Huang, Jacob Janey, Kishta Katipally, Sergei Kolotuchin, Thomas La Porte, Andrew Lee, Paul Lobben, Federico Lora-Gonzalez, Brendan Mack, Boguslaw Mudryk, Yuping Qiu, Xinhua Qian, Antonio Ramirez, Thomas M. Razler* , Thorsten Rosner, Zhongping Shi, Eric Simmons , Jason Stevens, Jianji Wang, Carolyn Wei, Steven R. Wisniewski , and Ye ZhuChemical & Synthetic Development, Bristol-Myers Squibb Company, 1 Squibb Drive, New Brunswick, New Jersey 08901, United States
Org. Lett., 2018, 20 (13), pp 3736–3740
DOI: 10.1021/acs.orglett.8b01218
*E-mail: thomas.razler@bms.com

A strategy to prepare compounds with multiple chirality axes, which has led to a concise total synthesis of compound 1A with complete stereocontrol, is reported.


https://pubs.acs.org/doi/suppl/10.1021/acs.orglett.8b01218/suppl_file/ol8b01218_si_001.pdf
(2S,5R)-6-fluoro-5-(3-(8-fluoro-1-methyl-2,4-dioxo-1,4- dihydroquinazolin-3(2H)-yl)-2-methylphenyl)-2-(2-hydroxypropan-2-yl)-2,3,4,9- tetrahydro-1H-carbazole-8-carboxamide (1A).
1H NMR (500 MHz, DMSO-d6) 10.78 (s, 1H), 8.07 (br. s., 1H), 7.95 (d, J=7.8 Hz, 1H), 7.72 (dd, J=14.2, 8.0 Hz, 1H), 7.56 (d, J=10.8 Hz, 1H), 7.45 (br. s., 1H), 7.42 – 7.36 (m, 1H), 7.34 (d, J=6.9 Hz, 1H), 7.34 – 7.31 (m, 1H), 7.29 (dd, J=7.5, 1.3 Hz, 1H), 4.17 (s, 1H), 3.73 (d, J=8.0 Hz, 3H), 2.91 (dd, J=16.8, 4.4 Hz, 1H), 2.48 – 2.37 (m, 1H), 1.98 – 1.89 (m, 2H), 1.87 (d, J=11.0 Hz, 1H), 1.76 (s, 3H), 1.59 (td, J=11.5, 4.1 Hz, 1H), 1.20 – 1.12 (m, 1H), 1.11 (s, 6H). 13C NMR (125.8 MHz, DMSO-d6) 168.2 (d, J=1.8 Hz, 1C), 160.1 (d, J=3.6 Hz, 1C), 151.9 (d, J=228.9 Hz, 1C), 150.5 (d, J=41.8 Hz, 1C), 148.7 (d, J=205.3 Hz, 1C), 139.2, 135.1, 135.0, 134.8, 131.4, 130.6, 130.0 (d, J=7.3 Hz, 1C), 128.5, 127.1 (d, J=4.5 Hz, 1C), 125.7, 124.3 (d, J=2.7 Hz, 1C), 123.6 (d, J=8.2 Hz, 1C), 123.0 (d, J=23.6 Hz, 1C), 120.8 (d, J=20.0 Hz, 1C), 118.4, 115.3 (d, J=7.3 Hz, 1C), 108.8 (d, J=5.4 Hz, 1C), 106.7 (d, J=28.2 Hz, 1C), 70.4, 45.4, 34.3 (d, J=14.5 Hz, 1C), 27.1, 26.8, 24.8, 24.7, 22.1, 14.5. mp 222-225 °C. IR (neat) 3487, 3418, 3375, 2967, 1651, 1394, 756 cm-1; HRMS (ESI) m/z: calcd for C32H30F2N4O4 [M+H]+ 573.2308, found 573.2312.
Chiral HPLC Analysis: Gradient: Complex Start % B: 0 7 Min. 55% 11 Min. 55% 14 Min. 100% Stop Time: 17 min Flow Rate: 1.5 ml/min Wavelength1: 225 Wavelength2: 256 Solvent Pair: S194/S195 (TFA) Solvent A: A1=0.05%TFA Water:ACN (95:5) S194 Solvent B: B1=0.05%TFA Water:ACN (5:95) S195 Column 1 : 1: Chiralcel OX-3R 3um 4.6 x 150 mm SN = OX3RCD-TE001 Oven Temperature: 50


Clip
Adventures in Atropisomerism: Development of a Robust, Diastereoselective, Lithium-Catalyzed Atropisomer-Forming Active Pharmaceutical Ingredient Step
Steven R. Wisniewski* , Ronald Carrasquillo-Flores, Federico Lora Gonzalez, Antonio Ramirez, Matthew Casey, Maxime Soumeillant, Thomas M. Razler , and Brendan Mack
, Ronald Carrasquillo-Flores, Federico Lora Gonzalez, Antonio Ramirez, Matthew Casey, Maxime Soumeillant, Thomas M. Razler , and Brendan MackChemical and Synthetic Development, Bristol-Myers Squibb Company, One Squibb Drive, New Brunswick, New Jersey08903, United States
Org. Process Res. Dev., Article ASAP
DOI: 10.1021/acs.oprd.8b00246
*E-mail: steven.wisniewski@bms.com.

The final step in the route to BMS-986142, a reversible inhibitor of the BTK enzyme, involves the diastereoselective construction of a chiral axis during the base-mediated cyclization of the quinazolinedione fragment. Optimization of the reaction to minimize formation of the undesired atropisomer led to the discovery that the amount of base and nature of the counterion play a vital role in the diastereoselectivity of the reaction. The highest diastereoselectivities were observed with a catalytic amount of LiOt-Bu. Development of a crystallization to selectively purge the undesired atropisomer is reported. Interestingly, ripening of the crystalline API was observed and further investigated, leading to a significant increase in the purity of the active pharmaceutical ingredient.
(2S,5R)-6-fluoro-5-(3-(8-fluoro-1-methyl-2,4-dioxo-1,4- dihydroquinazolin-3(2H)-yl)-2-methylphenyl)-2-(2-hydroxypropan-2-yl)-2,3,4,9- tetrahydro-1H-carbazole-8-carboxamide 1A
white crystalline solid (80.52g, 6 wt % MeOH, 89.4% corrected yield).
1H NMR (500 MHz, DMSO-d6) 10.78 (s, 1H), 8.07 (br. s., 1H), 7.95 (d, J=7.8 Hz, 1H), 7.72 (dd, J=14.2, 8.0 Hz, 1H), 7.56 (d, J=10.8 Hz, 1H), 7.45 (br. s., 1H), 7.42 – 7.36 (m, 1H), 7.34 (d, J=6.9 Hz, 1H), 7.34 – 7.31 (m, 1H), 7.29 (dd, J=7.5, 1.3 Hz, 1H), 4.17 (s, 1H), 3.73 (d, J=8.0 Hz, 3H), 2.91 (dd, J=16.8, 4.4 Hz, 1H), 2.48 – 2.37 (m, 1H), 1.98 – 1.89 (m, 2H), 1.87 (d, J=11.0 Hz, 1H), 1.76 (s, 3H), 1.59 (td, J=11.5, 4.1 Hz, 1H), 1.20 – 1.12 (m, 1H), 1.11 (s, 6H).
13C NMR (125.8 MHz, DMSO-d6) 168.2 (d, J=1.8 Hz, 1C), 160.1 (d, J=3.6 Hz, 1C), 151.9 (d, J=228.9 Hz, 1C), 150.5 (d, J=41.8 Hz, 1C), 148.7 (d, J=205.3 Hz, 1C), 139.2, 135.1, 135.0, 134.8, 131.4, 130.6, 130.0 (d, J=7.3 Hz, 1C), 128.5, 127.1 (d, J=4.5 Hz, 1C), 125.7, 124.3 (d, J=2.7 Hz, 1C), 123.6 (d, J=8.2 Hz, 1C), 123.0 (d, J=23.6 Hz, 1C), 120.8 (d, J=20.0 Hz, 1C), 118.4, 115.3 (d, J=7.3 Hz, 1C), 108.8 (d, J=5.4 Hz, 1C), 106.7 (d, J=28.2 Hz, 1C), 70.4, 45.4, 34.3 (d, J=14.5 Hz, 1C), 27.1, 26.8, 24.8, 24.7, 22.1, 14.5.
mp 222-225 °C.
IR (neat) 3487, 3418, 3375, 2967, 1651, 1394, 756 cm-1;
HRMS (ESI) m/z: calcd for C32H30F2N4O4 [M+H]+ 573.2308, found 573.2312.
Chiral HPLC Analysis: Gradient: Complex Start % B: 0 7 Min. 55% 11 Min. 55% 14 Min. 100% Stop Time: 17 min Flow Rate: 1.5 ml/min Wavelength1: 225 Wavelength2: 256 Solvent Pair: S194/S195 (TFA) Solvent A: A1=0.05%TFA Water:ACN (95:5) S194 Solvent B: B1=0.05%TFA Water:ACN (5:95) S195 Column 1 : 1: Chiralcel OX-3R 3um 4.6 x 150 mm SN = OX3RCD-TE001 Oven Temperature: 50…..https://pubs.acs.org/doi/suppl/10.1021/acs.oprd.8b00246/suppl_file/op8b00246_si_001.pdf


PAPER
Discovery of 6-Fluoro-5-(R)-(3-(S)-(8-fluoro-1-methyl-2,4-dioxo-1,2-dihydroquinazolin-3(4H)-yl)-2-methylphenyl)-2-(S)-(2-hydroxypropan-2-yl)-2,3,4,9-tetrahydro-1H-carbazole-8-carboxamide (BMS-986142): A Reversible Inhibitor of Bruton’s Tyrosine Kinase (BTK) Conformationally Constrained by Two Locked Atropisomers
Bristol-Myers Squibb Research and Development, P.O. Box 4000, Princeton, New Jersey 08543, United States
J. Med. Chem., 2016, 59 (19), pp 9173–9200
DOI: 10.1021/acs.jmedchem.6b01088
Publication Date (Web): September 1, 2016
Copyright © 2016 American Chemical Society
*Phone: 609-252-6778. E-mail: scott.watterson@bms.com.

Bruton’s tyrosine kinase (BTK), a nonreceptor tyrosine kinase, is a member of the Tec family of kinases. BTK plays an essential role in B cell receptor (BCR)-mediated signaling as well as Fcγ receptor signaling in monocytes and Fcε receptor signaling in mast cells and basophils, all of which have been implicated in the pathophysiology of autoimmune disease. As a result, inhibition of BTK is anticipated to provide an effective strategy for the clinical treatment of autoimmune diseases such as lupus and rheumatoid arthritis. This article details the structure–activity relationships (SAR) leading to a novel series of highly potent and selective carbazole and tetrahydrocarbazole based, reversible inhibitors of BTK. Of particular interest is that two atropisomeric centers were rotationally locked to provide a single, stable atropisomer, resulting in enhanced potency and selectivity as well as a reduction in safety liabilities. With significantly enhanced potency and selectivity, excellent in vivo properties and efficacy, and a very desirable tolerability and safety profile, 14f (BMS-986142) was advanced into clinical studies.
HPLC purity: 99.9%; tr = 11.05 min (Method A); 99.9%; tr = 10.72 min (Method B). Chiral purity: 99.8% ie;
Optical rotation: [α]D20 (c = 2.10, CHCl3) = +63.8°;
LCMS (ESI) m/z calcd for C32H30F2N4O4 [M + H]+ 573.2. Found: 573.5. Anal. calcd for C32H30F2N4O4, 0.72% H2O: C 65.56, H 5.42, N 9.55. Found: C 65.69, H 5.40, N 9.52.
1H NMR (500 MHz, DMSO-d6) δ 10.78 (s, 1H), 8.07 (br. s., 1H), 7.95 (d, J = 7.8 Hz, 1H), 7.72 (dd, J = 14.2, 8.0 Hz, 1H), 7.56 (d, J = 10.8 Hz, 1H), 7.45 (br. s., 1H), 7.42–7.36 (m, 1H), 7.34 (d, J = 6.9 Hz, 1H), 7.34–7.31 (m, 1H), 7.29 (dd, J = 7.5, 1.3 Hz, 1H), 4.17 (s, 1H), 3.73 (d, J = 8.0 Hz, 3H), 2.91 (dd, J = 16.8, 4.4 Hz, 1H), 2.48–2.37 (m, 1H), 1.98–1.89 (m, 2H), 1.87 (d, J = 11.0 Hz, 1H), 1.76 (s, 3H), 1.59 (td, J = 11.5, 4.1 Hz, 1H), 1.20–1.12 (m, 1H), and 1.11 (s, 6H). 1
3C NMR (126 MHz, DMSO-d6) δ 168.2 (d, J = 1.8 Hz, 1C), 160.1 (d, J = 3.6 Hz, 1C), 151.9 (d, J = 228.9 Hz, 1C), 150.5 (d, J = 41.8 Hz, 1C), 148.7 (d, J= 205.3 Hz, 1C), 139.2, 135.1, 135.0, 134.8, 131.4, 130.6, 130.0 (d, J = 7.3 Hz, 1C), 128.5, 127.1 (d, J = 4.5 Hz, 1C), 125.7, 124.3 (d, J = 2.7 Hz, 1C), 123.6 (d, J = 8.2 Hz, 1C), 123.0 (d, J = 23.6 Hz, 1C), 120.8 (d, J = 20.0 Hz, 1C), 118.4, 115.3 (d, J = 7.3 Hz, 1C), 108.8 (d, J = 5.4 Hz, 1C), 106.7 (d, J = 28.2 Hz, 1C), 70.4, 45.4, 34.3 (d, J = 14.5 Hz, 1C), 27.1, 26.8, 24.8, 24.7, 22.1, and 14.5.
19F-NMR (470 MHz, DMSO-d6) δ −121.49 (dt, J = 22.9, 11.4 Hz, 1F), and −129.56 (d, J = 11.4 Hz, 1F).
PATENT
WO 2014210085
Atropisomers are stereoisomers resulting from hindered rotation about a single bond axis where the rotational barrier is high enough to allow for the isolation of the individual rotational isomers. (LaPlante et al., J. Med. Chem., 54:7005-7022 (2011).)
Th compounds of Formula (A):
have two stereogenic axes: bond (a) between the tricyclic tetrahydrocarbazole/carbazole group and the phenyl group; and bond (b) between the asymmetric heterocyclic dione group Q and the phenyl group. Due to the non-symmetric nature of the substitutions on the rings connected by the single bonds labeled a and b, and due to limited rotation about these bonds caused by steric hindrance, the compounds of Formula (A) can form rotational isomers. If the rotational energy barriers are sufficiently high, hindered rotations about bond (a) and/or bond (b) occur at rates that are slow enough to allow isolation of the separated atropisomers as different compounds. Thus, the compounds of Formula (A) can form four rotational isomers, which under certain conditions, such as chromatography on a chiral stationary phase, can be separated into individual atropisomers. In solution, the compounds of Formula (A) can be provided as a mixture of four diastereomers, or mixtures of two pairs of diastereomers, or single atropisomers.
For the compounds of Formula (A), the pair of rotational isomers formed by hindered rotation about stereogenic axis (a) can be represented by the compounds of Formula (I) and Formula (B) having the structures:
The compounds of Formula (I) and the compounds of Formula (B) were found to be separable and stable in solution at ambient and physiological temperatures. Additionally, rotational isomers are formed by hindered rotation about stereogenic axis (b). These two atropisomers of the compounds of Formula (I) were also found to be separable and stable in solution at ambient and physiological temperatures.
Chiral compounds, such as the compounds of Formula (A), can be separated by various techniques including Supercritical Fluid Chromatography (SFC). SFC, which is form of normal phase HPLC, is a separation technique that uses super/subcritical fluid CO2 and polar organic modifiers such as alcohols as mobile phases. (White et al, J. Chromatography A, 1074: 175-185 (2005).
Example 28
6-Fluoro-5-(R)-(3-(S)-(8-fluoro-l-methyl-2,4-dioxo-l,2-dihydroquinazolin-3(4H)-yl)-2- methylphenyl)-2-(S)-(2-hydroxypropan-2-yl)-2,3,4,9-tetrahydro-lH-carbazole-8- carboxamide (single atropisomer)
(28)
Following the procedure used to prepare Example 27, (S)-5-bromo-6-fluoro-2-(2-hydroxypropan-2-yl)-2,3,4,9-tetrahydro- lH-carbazole-8-carboxamide (single enantiomer) [Intermediate 26] (0.045 g, 0.122 mmol) and 8-fluoro-l-methyl-3-(S)-(2-methyl-3-(4,4,5, 5-tetramethyl-l,3,2-dioxaborolan-2-yl)phenyl)quinazoline-2,4(lH,3H)-dione
[Intermediate 10] (0.065 g, 0.158 mmol) were converted into 6-fluoro-5-(3-(S)-(8-fluoro-1 -methyl-2,4-dioxo- 1 ,2-dihydroquinazolin-3(4H)-yl)-2-methylphenyl)-2-(S)-(2-
hydroxypropan-2-yl)-2,3,4,9-tetrahydro- lH-carbazole-8-carboxamide (mixture of two atropisomers) as a yellow solid (0.035 g, 49% yield). Separation of a sample of this material by chiral super-critical fluid chromatography, using the conditions used to separate Example 27, provided (as the first peak to elute from the column) 6-fluoro-5-(R)-(3-(S)-(8-fluoro-l-methyl-2,4-dioxo-l,2-dihydroquinazolin-3(4H)-yl)-2-methylphenyl)-2-(S)-(2-hydroxypropan-2-yl)-2,3,4,9-tetrahydro-lH-carbazole-8-carboxamide. The chiral purity was determined to be greater than 99.5%. The relative and absolute configurations were determined by x-ray crystallography. Mass spectrum m/z 573 (M+H)+. XH NMR (500 MHz, DMSO-d6) δ 10.77 (s, 1H), 8.05 (br. s., 1H), 7.94 (dd, J=7.9, 1.2 Hz, 1H), 7.56-7.52 (m, 1H), 7.43 (br. s., 1H), 7.40-7.36 (m, 1H), 7.35-7.30 (m, 2H), 7.28 (dd, J=7.5, 1.4 Hz, 1H), 4.15 (s, 1H), 3.75-3.70 (m, 3H), 2.90 (dd, J=16.8, 4.6 Hz, 1H), 2.47-2.39 (m, 1H), 1.93-1.82 (m, 3H), 1.74 (s, 3H), 1.57 (td, J=1 1.7, 4.2 Hz, 1H), 1.16-1.11 (m, 1H), and 1.10 (d, J=1.9 Hz, 6H). [a]D: +63.8° (c 2.1, CHC13). DSC melting point onset temperature = 202.9 °C (heating rate = 10 °C/min.).
The absolute configuration of Example 28 was confirmed by single crystal x-ray analysis of crystals prepared by dissolving the compound in excess methanol and slowly evaporating the solvent at room temperature to provide a di-methanol solvate (crystalline form M2-1). Unit cell dimensions: a = 9.24 A, b = 7.97 A, c = 22.12 A, a = 90.0°, β = 94.1°, γ = 90.0°; Space group: P2i; Molecules of Example 28/asymmetric unit: 1 ;
Volume/Number of molecules in the unit cell = 813 A3; Density (calculated) = 1.301 g/cm3. Fractional atomic coordinates at 173 K are given in Table 6, and a depiction of the structure is given in Figure 5.
Alternative Synthesis of Example 28:
A mixture of (S)-5-bromo-6-fluoro-2-(2-hydroxypropan-2-yl)-2,3,4,9-tetrahydro-lH-carbazole-8-carboxamide [Intermediate 1 1] (5.00 g, 13.54 mmol), 8-fluoro-l-methyl-3-(S)-(2-methyl-3-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)phenyl)quinazoline-2,4(lH,3H)-dione [Intermediate 10] (6.67 g, 16.25 mmol), tripotassium phosphate (2 M in water) (20.31 mL, 40.6 mmol), and tetrahydrofuran (25 mL) was subjected to 3 evacuate-fill cycles with nitrogen. The mixture was treated with l, l’-bis(di-/er/-butylphosphino)ferrocene palladium dichloride (0.441 g, 0.677 mmol) and the mixture was subjected to 2 more evacuate- fill cycles with nitrogen. The mixture was stirred at room temperature overnight, then was diluted with EtOAc, washed sequentially with water and brine, and dried and concentrated. The residue was purified by column chromatography on silica gel, eluting with EtOAc-hexanes (sequentially 50%, 62%, 75% and 85%), to provide 6-fluoro-5-(3-(8-fluoro-l-methyl-2,4-dioxo-l,2-dihydroquinazolin-3-(S)-3(4H)-yl)-2-methylphenyl)-2-(S)-(2-hydroxypropan-2-yl)-2,3,4,9-tetrahydro-lH-carbazole-8-carboxamide as a white solid (6.58 g, 85% yield).
Material prepared by this method (40.03 g, 69.9 mmol) was separated by chiral super-critical fluid chromatography to give (2S, 5R)-6-fluoro-5-(3-(8-fluoro-l-methyl-2,4-dioxo-l,2-dihydroquinazolin-3(4H)-yl)-2-methylphenyl)-2-(S)-(2-hydroxypropan-2-yl)-2,3,4,9-tetrahydro-lH-carbazole-8-carboxamide. Further purification was achieved by suspending this material in methanol, sonicating for 5 min, collection of the solid by filtration, rinsing the collected solid with methanol and drying at room temperature under reduced pressure to give a white solid (22.0 g, 90% yield).
2R ANALOGUE
Example 27
6-Fluoro-5-(R)-(3-(S)-(8-fluoro-l-methyl-2,4-dioxo-l,2-dihydroquinazolin-3(4H)-yl)-2- methylphenyl)-2-(R)-(2-hydroxypropan-2-yl)-2,3 ,4,9-tetrahydro- 1 H-carbazole-8- carboxamide (single atropisomer)
Preparation 27A: 6-Fluoro-5-(3-(S)-(8-fluoro-l-methyl-2,4-dioxo-l,2-dihydroquinazolin-3(4H)-yl)-2-methylphenyl)-2-(R)-(2-hydroxypropan-2-yl)-2,3,4,9-tetrahydro-lH-carbazole-8-carboxamide (mixture of 2 atropisomers)
A mixture of (R)-5-bromo-6-fluoro-2-(2-hydroxypropan-2-yl)-2,3,4,9-tetrahydro-lH-carbazole-8-carboxamide (single enantiomer) [Intermediate 25] (5.00 g, 13.5 mmol), 8-fluoro-l-methyl-3-(S)-(2-methyl-3-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)phenyl) quinazoline-2,4(lH,3H)-dione [Intermediate 10] (6.94 g, 16.9 mmol), 2 M aqueous K3PO4 (20.3 mL, 40.6 mmol) and THF (60 mL) was subjected to three evacuate-fill cycles with nitrogen. The mixture was treated with 1 , l’-bis(di-tert-butylphosphino) ferrocene palladium(II) chloride (441 mg, 677 μιηοΐ) and subjected to two more evacuate-fill cycles with nitrogen. The mixture was stirred at room temperature overnight. The mixture was diluted with EtOAc, washed sequentially with water and brine, and dried and concentrated. The residue was purified by column chromatography on silica gel, eluting with EtOAc-hexanes (sequentially 50%, 62%, 75% and 85%), to give 6-fluoro-5-(3-(S)-(8-fluoro-l-methyl-2,4-dioxo-l,2-dihydroquinazolin-3(4H)-yl)-2-methylphenyl)-2-(R)-(2-hydroxypropan-2-yl)-2,3,4,9-tetrahydro-lH-carbazole-8-carboxamide (mixture of two atropisomers) as an off-white solid (6.77 g, 87% yield). Mass spectrum m/z 573 (M+H)+. ¾ NMR (500 MHz, DMSO-d6) δ 10.79-10.74 (m, 1H), 8.05 (br. s., 1H), 7.98-7.93 (m, 1H), 7.76-7.69 (m, 1H), 7.57-7.51 (m, 1H), 7.43 (br. s., 1H), 7.40-7.26 (m, 4H), 4.19-4.13 (m, 1H), 3.74-3.68 (m, 3H), 2.94-2.84 (m, 1H), 2.49-2.35 (m, 2H), 1.92-1.80 (m, 3H), 1.76-1.68 (m, 3H), 1.62-1.52 (m, 1H), and 1.12-1.06 (m, 6H).
Example 27:
A sample of 6-fluoro-5-(3-(S)-(8-fluoro-l-methyl-2,4-dioxo-l,2-dihydroquinazolin-3(4H)-yl)-2-methylphenyl)-2-(R)-(2-hydroxypropan-2-yl)-2, 3,4,9-tetrahydro-lH-carbazole-8-carboxamide (mixture of two atropisomers) was separated by chiral super-critical fluid chromatography as follows: column: CHIRALPAK® AS-H (3 x 25 cm, 5 μιη); Mobile Phase: C02-MeOH (70:30) at 120 mL/min, 35 °C, 100 bar; sample preparation: 9 mg/mL in MeOH; injection: 1.7 mL. The first peak eluting from the column provided 6-fluoro-5-(R)-(3-(S)-(8-fluoro-l-methyl-2,4-dioxo-l,2-dihydroquinazolin-3(4H)-yl)-2-methylphenyl)-2-(R)-(2 -hydroxypropan-2-yl)-2, 3,4,9-tetrahydro-lH-carbazole-8-carboxamide. The chiral purity was determined to be greater than 99.5%. Mass spectrum m/z 573 (M+H)+. XH NMR (500 MHz, DMSO-d6) δ 10.76 (s, 1H), 8.05 (br. s., 1H), 7.96 (d, J=7.8 Hz, 1H), 7.72 (ddd, J=14.3, 8.0, 1.2 Hz, 1H), 7.55 (d, J=10.8 Hz, 1H), 7.44 (br. s., 1H), 7.40-7.36 (m, 1H), 7.35-7.28 (m, 3H), 4.18 (s, 1H), 3.72
PATENT
WO 2018118830
The present invention generally relates to processes for preparing a
tetrahydrocarbazole carboxamide compound.
Protein kinases, the largest family of human enzymes, encompass well over 500 proteins. Btk is a member of the Tec family of tyrosine kinases, and is a regulator of early B-cell development, as well as mature B-cell activation, signaling, and survival.
B-cell signaling through the B-cell receptor (BCR) leads to a wide range of biological outputs, which in turn depend on the developmental stage of the B-cell. The magnitude and duration of BCR signals must be precisely regulated. Aberrant BCR-mediated signaling can cause disregulated B-cell activation and/or the formation of pathogenic auto-antibodies leading to multiple autoimmune and/or inflammatory diseases. Mutation of Btk in humans results in X-linked agammaglobulinaemia (XLA). This disease is associated with the impaired maturation of B-cells, diminished immunoglobulin production, compromised T-cell-independent immune responses and marked attenuation of the sustained calcium signal upon BCR stimulation.
Evidence for the role of Btk in allergic disorders and/or autoimmune disease and/or inflammatory disease has been established in Btk-deficient mouse models. For example, in standard murine preclinical models of systemic lupus erythematosus (SLE), Btk deficiency has been shown to result in a marked amelioration of disease progression. Moreover, Btk deficient mice are also resistant to developing collagen-induced arthritis and are less susceptible to Staphylococcus-induced arthritis.
A large body of evidence supports the role of B-cells and the humoral immune system in the pathogenesis of autoimmune and/or inflammatory diseases. Protein-based therapeutics (such as Rituxan) developed to deplete B-cells, represent an important approach to the treatment of a number of autoimmune and/or inflammatory diseases. Because of Btk’s role in B-cell activation, inhibitors of Btk can be useful as inhibitors of B-cell mediated pathogenic activity (such as autoantibody production).
Btk is also expressed in mast cells and monocytes and has been shown to be important for the function of these cells. For example, Btk deficiency in mice is
associated with impaired IgE-mediated mast cell activation (marked diminution of TNF-alpha and other inflammatory cytokine release), and Btk deficiency in humans is associated with greatly reduced TNF-alpha production by activated monocytes.
Thus, inhibition of Btk activity can be useful for the treatment of allergic disorders and/or autoimmune and/or inflammatory diseases including, but not limited to: SLE, rheumatoid arthritis, multiple vasculitides, idiopathic thrombocytopenic purpura (ITP), myasthenia gravis, allergic rhinitis, multiple sclerosis (MS), transplant rejection, type I diabetes, membranous nephritis, inflammatory bowel disease, autoimmune hemolytic anemia, autoimmune thyroiditis, cold and warm agglutinin diseases, Evan’s syndrome, hemolytic uremic syndrome/thrombotic thrombocytopenic purpura (HUS/TTP), sarcoidosis, Sjogren’s syndrome, peripheral neuropathies (e.g., Guillain-Barre syndrome), pemphigus vulgaris, and asthma.
In addition, Btk has been reported to play a role in controlling B-cell survival in certain B-cell cancers. For example, Btk has been shown to be important for the survival of BCR-Abl-positive B-cell acute lymphoblastic leukemia cells. Thus inhibition of Btk activity can be useful for the treatment of B-cell lymphoma and leukemia.
Atropisomers are stereoisomers resulting from hindered rotation about a single bond axis where the rotational barrier is high enough to allow for the isolation of the individual rotational isomers. (LaPlante et al., J. Med. Chem. 2011, 54, 7005-7022).
US Patent 9,334,290 discloses substituted tetrahydrocarbazole and carbazole compounds useful as Btk inhibitors, including 6-fluoro-5-(R)-(3-(S)-(8-fluoro-l-methyl-2,4-dioxo-l,2-dihydroquinazolin-3(4H)-yl)-2-methylphenyl)-2-(S)-(2-hydroxypropan-2-yl)-2,3,4,9-tetrahydro-lH-carbazole-8-carboxamide as Example 28. 6-fluoro-5-(R)-(3-(S)-(8-fluoro-l-methyl-2,4-dioxo-l,2-dihydroquinazolin-3(4H)-yl)-2-methylphenyl)-2-(S)-(2-hydroxypropan-2-yl)-2,3,4,9-tetrahydro-lH-carbazole-8-carboxamide, referred to herein as Compound 8, has two stereogenic axes:
(i) bond “a” between the tricyclic tetrahydrocarbazole/carbazole group and the phenyl group; and (ii) bond “b” between the substituted tetrahydroquinazolinedione group and the phenyl group. Compound 8 has non-symmetric substitutions on the rings connected by the single bonds labeled “a” and “b”, and limited rotation about these bonds caused by steric hindrance. As the rotational energy barriers are sufficiently high, hindered rotations about bond (a) and bond (b) occur at rates that are slow enough to allow isolation of Compound 8 and the other atropisomers of Compound 8 as four individual diastereomeric atropisomer compounds. These four rotational isomers can be separated by
chromatography on a stationary phase to provide chiral mixtures of two atropisomers or individual atropisomers.
US Patent 9,334,290 discloses a multistep synthesis process for preparing the Compound 8. This process is shown schematically in Figures 2-4. The disclosed process includes three chiral separations from racemic mixtures including (i) a chiral separation of a racemic mixture of chiral enantiomers (FIG.2); (ii) chiral separation of a mixture of atropisomers along bond “b” between the substituted tetrahydroquinazolinedione group and the phenyl group (FIG.3); and chiral separation of a mixture of atropisomers along bond “a” between the tricyclic tetrahydrocarbazole/carbazole group and the phenyl group (FIG.4). In each one of these chiral separations, the maximum yield of the desired enantiomer or atropisomer from the racemic mixture is 50%.
There are difficulties associated with the adaptation of this multistep synthesis disclosed in US Patent 9,334,290 to a larger scale synthesis, such as production in a pilot plant or a manufacturing plant for commercial production. Additionally, it is desired to have a process that provides higher yields and/or reduces waste.
Applicants have discovered a synthesis process for the preparation of Compound 8 that provides higher yields, reduces waste, and/or is adaptable to large scale manufacturing.
he invention is illustrated by reference to the accompanying drawing described below.
FIG.1 shows the stereoselective synthesis scheme for the preparation of 6-fluoro-5-(R)-(3-(S)-(8-fluoro-l-methyl-2,4-dioxo-l,2-dihydroquinazolin-3(4H)-yl)-2-methylphenyl)-2-(S)-(2-hydroxypropan-2-yl)-2,3,4,9-tetrahydro-lH-carbazole-8-carboxamide, Compound 8, according to the processes of second aspect, the third aspect, and the first aspect of the invention.
FIG.2 shows the synthesis scheme disclosed in US 9,334,290 for the preparation of (S)-5-bromo-6-fluoro-2-(2-hydroxypropan-2-yl)-2,3,4,9-tetrahydro-lH-carbazole-8- carboxamide, Compound 5 (Intermediate 26 in US 9,334,290).
FIG.3 shows the synthesis scheme disclosed in US 9,334,290 for the preparation of 8-fluoro-l-methyl-3-(S)-(2-methyl-3-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl) phenyl)quinazoline-2,4(lH,3H)-dione, Intermediate 10 in US 9,334,290.
FIG.4 shows the synthesis scheme disclosed in US 9,334,290 for the preparation of Compound 8 from the coupling reaction of 8-fluoro-l -methyl-3-(S)-(2-methyl-3- (4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl) phenyl)quinazoline-2,4(lH,3H)-dione, Intermediate 10, and (S)-5-bromo-6-fluoro-2-(2-hydroxypropan-2-yl)-2,3,4,9-tetrahydro- lH-carbazole-8-carboxamide, Compound 5, to provide a racemic mixture of Example 27 in US 9,334,290; and the chiral separation of Example 27 to provide Compound 8.
wherein R is Ci-8 alkyl or benzyl;
in the presence of:
(i) one or more bases selected from lithium bases, sodium bases, potassium bases, cesium bases, l,8-diazabicycloundec-7-ene, and 1,1,3,3-tetramethylguanidine; and
(ii) a solvent selected from n-butyl acetate (nBuOAc), cyclopentyl methyl ether (CPME), dimethoxy ethane (DME), dimethylacetamide (DMAc), dimethylformamide (DMF), 1,4-dioxane, ethyl acetate (EtOAc), isobutyl acetate (iBuOAc), isopropyl acetate (IP Ac), isopropyl alcohol (IP A), methanol (MeOH), methyl acetate (MeOAc), methyl isobutyl ketone (MIBK), N-methyl-2-pyrrolidone (NMP), 2-methyltetrahydrofuran (MeTHF), tetrahydrofuran (THF), tetrahydropyran (THP), and mixtures thereof;
to provide said Compound 8.
Intermediate Al
2-amino-4 robenzoic acid
(Al)
5% Pt/C (50% water-wet) (60 g, 6 wt%) was charged to a nitrogen blanketed vessel containing isopropyl acetate (22 L) and 4-bromo-5-fluoro-2-nitrobenzoic acid (1.00 kg, 3.79 mol). The headspace was exchanged three times with nitrogen and followed three times with hydrogen. The reaction mixture was stirred at 25 °C under an atmosphere of hydrogen. After 40 hours, the reaction was complete and the headspace was exchanged three times with nitrogen. The reaction mixture was filtered. The reaction vessel and filter train were rinsed with isopropyl acetate (5 L). The combined organic layers were concentrated under reduced pressure to 5.0 L. The solvent was then exchanged to toluene under reduced pressure and the resulting solids were isolated by filtration, washed with toluene, and dried at 50 °C under reduced pressure to afford 0.59 kg (66% yield) of 2-amino-4-bromo-5-fluorobenzoic acid as a white to off-white crystalline solid.
Additional 2-amino-4-bromo-5-fluorobenzoic acid was obtained by washing the spent catalyst twelve times with 2.75: 1 w/w THF in water (9.0 L). Each portion of wash was allowed to soak the spent catalyst for 30 minutes. The filtrate was concentrated to 10 L. The resulting solids were isolated by filtration, washed with water (1.0 L), and dried at 40 °C under reduced pressure to afford 0.15 kg (17% yield) of 2-amino-4-bromo-5-fluorobenzoic acid as an off-white crystalline solid. ¾ NMR (400 MHz, DMSO-de) δ 8.74 (br s, 2H), 7.50 (d, J=9.6 Hz, 1H), 7.08 (d, J=6.1 Hz, 1H). 13C NMR (101 MHz, DMSO-de) 5 168.2, 149.5, 148.8, 147.2, 119.9, 117.0, 116.8, 114.8, 114.6, 109.1.
HPLC Conditions: Column: Waters X-bridge C-18 (150X4.6mm, 3.5μ); Column
Temeprature: 30 °C; Solvent A: 0.05% TFA in water: acetonitrile (95:05 v/v); Solvent B: 0.05%TFA in water: acetonitrile:methanol (05:75:20 v/v); Diluent: 0.25 mg/ml in acetonitrile; Gradient: %B: 0 min. 5%; 20 min. 95%; 25 min. 95%; 26 min. 5%; stop time 30 min; Flow Rate: 0.8 ml/min; Wavelength: 230 nm; The retention time of 2-amino-4-bromo-5-fiuorobenzoic acid was 13.2 min. The retention time of 4-bromo-5-fluoro-2-nitrobenzoic acid was 12.9 min.
Intermediate A2
4-bromo-5-fluoro- -hydrazinylbenzoic acid hydrochloride
A solution of sodium nitrite (100.0 g, 6.38 mol) and water (1.8 L) was slowly charged to a cold slurry (0 °C) of 2-amino-4-bromo-5-fluorobenzoic acid (1.00 kg, 4.27 mol) in water (2.2 L) containing 35% HCl (2.1 kg, 20.15 mol). The reaction mixture slurry was stirred at 0 °C for 5 hours. The resultant cold diazonium salt slurry was charged over 4 hours to a cold solution (0 °C) of sodium bisulfite (2.66 kg, 25.0 mol in water (7.5 L). The diazonium reaction vessel was rinsed with cold water (2.5 L). The rinse water was transferred slowly to the reaction mixture. After 40 minutes, the reaction mixture was warmed to 20 °C over one hour. The reaction mixture slurry was stirred at 20 °C for 3 hours. After 3 hours, the reaction mixture was slowly transferred to a 60 °C solution of 35% HCl (15.0 kg, 144.0 mol) and water (3.0 L). The vessel was rinsed with water (2.5 L); and transferred to 35% HCl and water reaction mixture. The reaction mixture was stirred at 60 °C for 2 hours. The product was isolated by filtration and washed with water (3.0 L). The wet cake was charged back to the reactor and was
slurried with isopropyl acetate (9.0 L) for 1 hour at 20 °C. The product was isolated by filtration, washed with isopropyl acetate (1.0 L), and dried at 45-50 °C under reduced pressure to afford 0.99 kg (81 % yield) of 4-bromo-5-fluoro-2-hydrazinylbenzoic acid hydrochloride as an off-white crystalline solid in 95% purity. ¾ NMR (400 MHz, DMSO-de) δ 10.04 (br s, 3H), 9.00 (br s, 1H), 7.74 (d, J=9.1 Hz, 1H), 7.61 (d, J=5.8 Hz, 1H). 13C NMR (101 MHz, DMSO-de) δ 167.3, 153.0, 150.6, 144.5, 119.2, 1 18.0, 114.6. HPLC analysis: Column: Zorbax Eclipse Plus C 18 3.5 um, 150 x 4.6 mm ID; Column Temeprature: 30 °C; Solvent A: 10 mM ammonium formate in water:MeOH (90: 10 v/v); Solvent B: MeOH : ACN (70:30 v/v); Diluent: 50% CH3CN(aq); Gradient: %B: 0 min. 0%; 15 min. 90%; 18 min. 100%; stop time 18 min; Flow Rate: 1.0 ml/min; Wavelength: 240 nm. The retention time of the diazonium salt intermediate was 3.7 min. The retention time of the mono-sulfamic acid intermediate was 5.2 min. The retention time of 4-bromo-5-fluoro-2-hydrazinylbenzoic acid hydrochloride was 8.0 min. The retention time of 2-amino-4-bromo-5-fluorobenzoic acid was 8.7 min.
INTERMEDIATE Bl
(3-amino-2-methylphenyl)boronic acid hydrochloride
A 500 mL ChemGlass reactor (Reactor A) was equipped with mechanical stirrer and a nitrogen inlet. To the reactor was added 150 ml of methyl tetrahydrofuran. Next, Pd(OAc)2 (241 mg, 0.02 eq) was added, followed by the addition of P(o-tolyl)3 ligand (654 mg, 0.04 eq). The containers holding the Pd(OAc)2 and P(o-tolyl)3 were rinsed with 15 ml of methyl tetrahydrofuran, and the rinse solvents were added to the reactor. The reactor was sealed, evacuated to less than 150 mbar, and filled with nitrogen gas. This was repeated an additional four times to reduce the oxygen level to below 400 ppm. The reaction mixture was stirred for 30 min. Next, 10 g (1.0 eq) of 3-bromo-2-methyl aniline was charged to the inerted reactor. The container that held the 3-bromo-2-methyl aniline was rinsed with 15 ml of Me-THF and added into the reactor. KOAc (15.6 g, 3 eq) was added to the reactor. A slurry formed. The reaction mixture was inerted by using three vacuum/nitrogen cycles to an oxygen endpoint of less than 400 ppm.
A second 500 ml ChemGlass reactor was charged with 150 mL of MeOH, followed by the addition of 7.2 g (1.5 eq) of B2(OH)4. The resultant slurry was agitated at 25 °C. After 30 min, the B2(OH)4 was fully dissolved. The homogeneous solution was inerted by using 5 vacuum/nitrogen purge cycles to reduce the oxygen level to less than 400 ppm. The B2(OH)4/MeOH solution was transferred to Reactor A under a nitrogen atmosphere.
The reactor was inerted using three vacuum/nitrogen cycles with agitation to reduce the oxygen level to less than 400 ppm. The batch was heated to 50 °C (internal batch temperature). A slurry was observed when the temperature reached 40 °C. After reacting for 3 hrs, HPLC analysis of the reaction mixture showed 0.2 AP starting material remained. N-acetyl cysteine (2.0 g, 0.2 g/g) was added to Reactor A. The reaction mixture was stirred at 50 °C (internal batch temperature) for 30 min. The reaction stream was concentrated through distillation to 5 ml/g (~ 50 ml). Methyl tetrahydrofuran (200 ml, 20 ml/g) was charged to the slurry. The slurry was then concentrated via distillation to 150 ml (15 ml/g). Methyl tetrahydrofuran (150 ml, 15 ml/g) was charged to the reaction mixture. The slurry was cooled to 20 °C (batch temperature). Brine (26 wt%, 25 ml, 2.5 ml/g) was charged followed by the addition of aqueous Na2C03 (20 wt%, 15 ml, 1.5 ml/g). The reaction mass was agitated at a moderate rate (50~75/min) for 30 min. Celite (1 g, 0.1 g/g) was charged to the bi-phasic solution. The resultant slurry was agitated for 30 min. The slurry was filtered and transferred to Reactor B. The Celite cake was washed with 10 ml of methyl tetrahydrofuran. The bottom, lean aqueous phase was split from the organic phase and discarded. Brine (26 wt%, 25 ml, 2.5 ml/g) was charged followed by the addition of aqueous Na2C03 (20 wt%, 15 ml, 1.5 ml/g) to the organic solution. The resultant bi-phasic solution was agitated at a moderate rate (75 rpm) for 30 min. The bottom, lean aqueous phase was split from the organic phase and discarded. B2(OH)4 analysis of the rich organic solution did not detect B2(OH)4.
In Reactor B, the rich organic phase was concentrated via distillation to 50 ml (5 ml/g). The concentrated solution was cooled to 0-5 °C (batch temp). Concentrated HC1 (1.06 kg, 2.0 eq) was charged to the solution over 30 min with the batch temperature maintained below 10 °C. Once the concentrated HC1 was added, a slurry formed. The
slurry was agitated for 2 h at 5 °C. The slurry was filtered. The wet cake was washed with methyl tetrahydrofuran (2 X 20 ml). The cake was collected and dried at 50 °C under 100 mbar vacuum for 6 h to afford 8.4 g of 3-amino-2-methylphenyl)boronic acid hydrochloride as a white solid (83.5 % yield). ¾ NMR (500 MHz, D20) δ 7.48-7.23 (m, 3H), 4.78 (br s, 5 H); 2.32 (s, 3H). 13C NMR (126 MHz, D2O) δ 135.2, 134.7, 130.1, 128.0, 124.3, 17.4.
HPLC analysis: Column: Zorbax Eclipse Plus CI 8 3.5 um, 150 x 4.6 mm ID; Solvent A: 10 mM ammonium formate in water: MeOH=90: 10); Solvent B: CH3CN: MeOH (30:70 v/v); Gradient: % B: 0 Min. 0%; 1 Min. 0%; 15 Min. 90%; 15.1 Min. 0%; Stop Time: 20 min; Flow Rate: 1 ml/min; wavelength: 240 nm. The retention time of (3-amino-2-methylphenyl)boronic acid hydrochloride was 4.4 min. The retention time of (3-amino-2-methylphenyl)boronic acid hydrochloride was 17.8 min.
Intermediate CI
7-fluoro-l-methylindoline-2,3-dione
N,N-dimethylformamide (540.0 mL, 6980 mmol, 100 mass%) was added to a 2-L ChemGlass reactor equipped with a mechanical agitator, a temperature probe, and a cooling/heating circulator. Next, 7-fluoroindoline-2,3-dione (135.0 g, 817.6 mmol, 100 mass%) was added at 25 °C and dissolved to form a dark red solution. The charging ports and the beaker that contained the 7-fluoroindoline-2,3-dione were washed with N,N-dimethylformamide (135.0 mL, 1750 mmol, 100 mass%) and the rinse solution was poured into the reactor. Next, cesium carbonate 60-80 mesh (203.66 g, 625.05 mmol, 100 mass%) was added portion-wise to the reaction mixture. The addition was exothermic and the temperature of the reaction mixture increased from 20 to 25.5 °C. The color of the reaction mixture changed from a dark red solution to a black solution. The reactor jacket temperature was set to 0 °C. Next, iodomethane (56.5 mL, 907 mmol, 100 mass%) was added slowly via an additional funnel at ambient temperature, (iodomethane
temperature) while maintaining the batch temperature at less than 30 °C. Upon stirring, the reaction was exothermic, reaching a temperature of 29.3 °C. The batch temperature decreased to 26.3 °C after 85% of iodomethane was added, and the reaction mixture turned from black to an orange. After the addition of the iodomethane was completed, the jacket temperature was raised to 25.5 °C. The reaction mixture was stirred at 25 °C for 2 hrs.
The reddish orange-colored reaction mixture was transferred to a 1 L Erlenmeyer flask. The reaction mixture was filtered through a ceramic Buchner funnel with a No.1 Whatman filter paper to remove solid CS2CO3 and other solid by-products. In addition to a light-colored powder, there were yellow to brown colored rod-shaped crystals on top of the cake, which were water soluble. The filtrate was collected in a 2-L Erlenmeyer flask. The solids cake was washed with N,N-dimethylformamide (100.0 mL, 1290 mmol, 100 mass%). The DMF filtrate was collected in a 2-L Erlenmeyer flask.
To a separate 5-L ChemGlass reactor was charged water (3000.0 mL, 166530 mmol, 100 mass%). Next, 1.66 g of 7-fluoro-l-methylindoline-2,3-dione was added as seed to the water to form an orange colored suspension. The DMF filtrate was charged to the 5-L reactor slowly while maintaining the batch temp, at less than 29 °C over a period of 60 min. Stirring was maintained at 290 rpm. The orange solids precipitated instantly. The 2-L Erlenmeyer flask was rinsed with N,N-dimethylformamide (55.0 mL, 711 mmol, 100 mass%) and charged to the 5-L reactor. The slurry was cooled to 25 °C and agitated at 200 rpm for 12 hrs. The mixture remained as a bright orange-colored suspension. The slurry was filtered over a No. l Whatman filter paper in a 9 cm diameter ceramic Buchner funnel to a 4L Erlenmeyer flask to provide a bright orange-colored cake. The cake was washed with 1200 mL of water via rinsing the 5000 mL reactor (400 mL x 2), followed by 300 mL of deionized water introduced directly on the orange cake. The wet cake was dried under suction for 40 min at ambient temperature until liquid was not observed to be dripping from the cake. The cake was introduced into a vacuum oven (800 mbar) with nitrogen sweeping at ambient temperature for 1 hr, at 40-45 °C for overnight, and at 25 °C for 1 day to provide 7-fluoro-l-methylindoline-2,3-dione (Q, 130.02 g, 725.76 mmol, 100 mass%, 88.77% yield) as a bright orange-colored solid. ¾ NMR (400 MHz, DMSO-de) δ 7.57 (ddd, J=12.0, 8.5, 1.0 Hz, 1H), 7.40 (dd, J=7.3, 1.0 Hz, 1H), 7.12 (ddd, J=8.5, 7.5, 4.0 Hz, 1H), 3.29 (d, J=3.0 Hz, 3H). 13C NMR (101 MHz, DMSO-de) δ 182.3, 158.2, 148.8, 146.4, 137.2, 125.9, 124.3, 120.6, 28.7.
Intermediate C2
3-fluoro-2-(methylamino)benzoic acid
To a 1-L three neck round bottom flask equipped with a mechanical overhead agitator, a thermocouple, and an ice-water bath was charged NaOH (5.0 N) in water (140.0 mL, 700 mmol, 5.0 mol/L) followed by deionized water (140.0 mL, 7771 mmol, 100 mass%) to form a colorless transparent solution (T = 20.2 °C). 7-fluoro-l-methylindoline-2,3-dione (R, 25 g, 139.55 mmol, 100 mass%) was charged portion-wise while controlling the batch temperature at less than 24 °C with an ice-water bath to provide cooling. 7-fluoro-l-methylindoline-2,3-dione was charged and 50 mL of water was used to rinse off the charging funnel, the spatula, and the charging port. The reaction mixture was a thick yellow-green hazy suspension. The yellow-greenish suspension was cooled to 5.0 °C with an ice-water bath. The mixture was stirred for 15 min. Next, hydrogen peroxide (50% wt.) in water (11.0 mL, 179 mmol, 50 mass%) was charged to a 60 mL additional funnel with deionized (4.0 mL, 220 mmol, 100 mass%). The concentration of H2O2 post dilution was ~ 36.7%. The dilute hydrogen peroxide solution was added over a period of 11 minutes to the 1 L round bottom flask cooled with an ice-water bath and stirred at 350 rpm. The reaction mixture color was observed to become lighter in color and less viscous after 5 mL of the peroxide solution was added. After adding 10 mL of peroxide solution, the reaction mixture became clear with visible solids. At the end of addition, the reaction mixture was a green-tea colored transparent solution. The ice-water bath was removed (batch temperature was 16.6 °C), and the transparent, greenish yellow reaction mixture was allowed to warm to ambient temperature (21.0 °C), stirred for 1 hr.
After the reaction was complete, (1.0 hr), the reaction mixture was cooled to 4.3 °C with an ice-water bath. The reaction mixture was neutralized by the addition 6.0 N HCl (aq.) over a period of 3 hours to minimize foaming and the exotherm, resulting in the formation of a yellow-green suspension. The ice-bath was removed and the quenched reaction mixture was stirred at ambient temperature for 20 min. The yellow-green colored reaction mixture was transferred to a 2 L separatory funnel. Dichloromethane (300.0 mL, 4680 mmol, 100 mass%) was charged to the separatory funnel via rinsing the 1 L 3-necked round bottom flask. The separatory funnel was shaken vigorously, then allowed to settle (phase split was fast). Gas evolution was minor. The top aqueous layer was dark amber in color. The bottom dichloromethane layer was tea-green in color. The bottom rich dichloromethane layer was transferred to a clean 1 L Erlenmeyer flask. Next, the 1 L three necked round bottom flask was rinsed again with dichloromethane (200.0 mL, 3120 mmol, 100 mass%). The dichloromethane rinse was added to the separatory funnel. The separatory funnel was shaken vigorously and allowed to settle (phase split was fast). The top aqueous layer was amber in color (lighter); the bottom
dichloromethane layer was lighter green. The bottom rich dichloromethane layer was transferred to the 1 L Erlenmeyer flask. Dichloromethane (200.0 mL, 3120 mmol, 100 mass%) was charged to the separatory funnel and the separatory funnel was shaken vigorously. The contents were allowed to settle (phase split was fast). The bottom rich dichloromethane layer was transferred to the same 1 L Erlenmeyer flask. Peroxide test strip showed > 10 mg/Liter peroxide concentration. The total volume of the aqueous layer was 540 mL.
In a separate 250-mL Erlenmeyer flask was added sodium thiosulfate
pentahydrate (20.0 g, 80.6 mmol, 100 mass%) followed by deionized water (180.0 mL, 9992 mmol, 100 mass%) to form a colorless solution (10% wt. solution). The sodium thiosulfate solution was added to the combined dichloromethane rich solution in the 1 L Erlenmeyer flask. The contents of the flask were stirred vigorously for 10 hrs at ambient temperature. Peroxide strip did not detect the presence of peroxides in the bottom DCM layer. The top Na2S203 layer was amber in color, the bottom dichloromethane layer was much lighter in color, but was still amber in color. After 10 hrs, the mixture was transferred to a 1 L separatory funnel. The top aqueous layer was discarded.
The dichloromethane solution was washed with 150.0 mL of saturated brine solution. After phase split, the bottom rich dichloromethane layer was transferred to a 1 L flask. The dichloromethane solution was distilled to approximately 150 mL to obtain an amber-colored solution. Next, dichloromethane (120 mL, 1872 mmol, 100 mass%) was added and the mixture was heated to 35-40 °C to fully dissolve the solids. The amber solution was filtered through a 0.45 micron PTFE membrane Zap Cap filtration unit into a 1 L flask. The filtrate was transferred into a 3-neck 1 L round bottom flask fitted with a thermocouple, a heating mantle, a mechanical agitator, and a condenser with a nitrogen inlet. To the flask was charged dichloromethane (120 mL, 1872 mmol, 100 mass%) via rinsing the 1 L flask. The contents of the flask were concentrated under reduced pressure to approximately 140 mL to afford a yellow-green-colored suspension. The mixture was heated to 40.5 °C (refluxing) with stirring at 155 rpm to form a green-colored suspension with white solid pieces. After refluxing for 5 min, heptane (100.0 mL, 683 mmol, 100 mass%) was charged to the above mixture. The batch temperature dropped from 41.3 °C to 33.8 °C and the reaction mixture was a suspension. The mixture was heated to 45 °C. The mixture remained as a suspension with supernatant being amber with white solids. The refluxing was mild. After 36 minutes, (batch temp. = 43.8 °C), heptane (120.0 mL, 819 mmol, 100 mass%) was added to the mixture. The batch temperature dropped to 38.0 °C. The reaction mixture was a suspension. The mixture was heated to 40-45 °C and seeded with 0.3 g of 3-fluoro-2-(methylamino)benzoic acid. The reaction mixture remained as a suspension with supernatant being amber and solid pieces of white color. At t = 1 h 25 min (T = 45.4 °C) heptane (100.0 mL, 683 mmol, 100 mass%) was charged to the mixture causing the temperature to drop to 41.0 °C. At t = 2 h l3 min, (T = 45.6 °C) additional heptane (100.0 mL, 683 mmol, 100 mass%) was added to the mixture causing temperature to drop to 41.7 °C. At t = 3 h 07 min, (T = 45.5 °C), the heating was stopped. The mixture was allowed to cool to 20-25 °C under a nitrogen blanket. The suspension was agitated at ambient temperature for 12 hrs. The mixture was filtered using No.1 Whatman filter paper fitted in a ceramic Buchner funnel to a 1 L Erlenmeyer flask. The solids were observed to settle quickly. The mother liquor was green in color. The bottom half of the round bottom flask was coated with a thin dark amber or brown film, which was water soluble. The 1 L round bottom flask was washed with 150 mL of heptane, and then the heptane was used to wash the collected off-white-colored solid.
The filter cake was allowed to dry at ambient temperature with suction for 10 min., then dried in a vacuum oven with nitrogen sweeping at 45-50 °C for 4 hrs, followed by drying at ambient temperature for 10 hrs, with nitrogen sweeping. 3-fluoro-2-(methylamino)benzoic acid (16.1 g) was isolated in 68.1 % yield. ¾ NMR (400 MHz, DMSO-de) δ 7.61 (d, J=7.7 Hz, IH), 7.23 (dq, J=7.9, 1.6 Hz, IH), 6.57 (td, J=8.0, 4.4 Hz, IH), 3.02 (d, J=6.8 Hz, 4H). 13C NMR (101 MHz, DMSO-de) δ 169.5, 153.1, 150.7, 141.8, 141.7, 127.4, 127.4, 120.9, 120.7, 114.8, 114.7, 114.4, 114.3, 32.8.
Intermediate C3
3-fluoro-2-(methyl(propoxycarbonyl)amino)benzoic
A 20 L jacketed glass reactor with an overhead mechanical agitator, a
thermocouple, a nitrogen inlet, a glass baffle, and a condenser rinsed with 4 liters of dichloromethane followed by nitrogen sweeping through bottom valve overnight. To the reactor was charged 3-fluoro-2-(methylamino)benzoic acid (1004.7 g, 5939.7 mmol, 100 mass%) followed by dichloromethane (6000 mL, 93400 mmol, 99.8 mass%) to form an off-white-colored suspension. Next, cesium carbonate (1035.2 g, 3170 mmol, 99.9 mass%) was added followed the addition of water (6000 g, 333056 mmol, 99 mass%) at ambient temperature. The batch temperature rose from 17.0 °C to 29.6 °C prior to addition of the water. Gas evolution was observed during the water charging. The colorless biphasic mixture was stirred for 15 min. The batch temperature was approximately 18.8 °C. Next, n-propyl chloroformate (806.0 g, 6445.4 mmol, 98 mass%) was charged to an addition funnel. The reaction mixture was cooled to 15.0 °C with a glycol circulator. The n-propyl chloroformate was added from the addition funnel to the mixture while maintaining the batch temperature between 15.0 and 20.0 °C over 1 hr with stirring at 156 rpm. At the end of the addition, the batch temperature was 18.1 °C. The jacket temperature was increased to 20 °C. The white milky reaction mixture was agitated for 90 minutes.
The agitation was stopped and the reaction mixture was allowed to settle for phase split for 50 min. The hazy, bottom rich dichloromethane layer split from the aqueous layer and was transferred to a carboy. Next, 500 g of anhydrous Na2S04 (s) and 100 g of 60-200 mesh silica gel was added to the dichloromethane solution of 3-fluoro-2-(methyl(propoxycarbonyl)amino)benzoic acid in the carboy. The dichloromethane solution was allowed to dry overnight.
The dichloromethane solution containing the 3-fluoro-2-(methyl
(propoxycarbonyl)amino)benzoic acid was transferred from the carboy to a clean 20 L reactor via a 10 micron Cuno® in-line filter under vacuum to remove solid Na2S04 and silica gel. The carboy was rinsed with 1 liter x 2 of dichloromethane to remove residual solids. The dichloromethane was distilled off in the 20 L reactor with the jacket temperature set at 32 °C, the batch temperature at 15 °C, and vacuum set to 200-253 torr. At the end of distillation, the crude product was a thick light-amber-colored syrup. The solution was concentrated to 3 L of dichloromethane, and refilled with 3 L of dichloromethane each time to a final fill volume of 6 L. Next, 1 liter of dichloromethane was charged via vacuum to the residue in the 20-L reactor. The solution of 3-fluoro-2-(methyl(propoxycarbonyl)amino)benzoic acid became hazier. The solution was filtered using a Buchner funnel with a No.1 filter paper into a new carboy. The reactor was rinsed with 500 mL x 2 of dichloromethane and the rinse was filtered through the same Buchner funnel. All the filtrates were combined in a carboy and stored at the ambient temperature under nitrogen. Yellow-colored solids were observed to settle at the bottom of the carboy. The solution of 3-fluoro-2-(methyl (propoxycarbonyl)amino)benzoic acid in dichloromethane was transferred back to the clean 20-L reactor via vacuum and a 1 micron Cuno® in-line filter. The filtrate was still slightly hazy. The carboy was rinsed with 300 mL x 3 of dichloromethane and the rinses were transferred to the reactor via the 1 micron Cuno® filter. The reactor walls were rinsed with 500-mL of dichloromethane. The dichloromethane solution was concentrated by distillation under reduced pressure until the volume was less than 2.0 liters.
The temperature of the reactor jacket was lowered to 30 °C. The vacuum was broken and the reactor was filed with nitrogen. To the reactor was added 2 liters of cyclohexane followed by 5.0 g of 3-fluoro-2-(methyl(propoxycarbonyl)amino)benzoic acid crystalline seed. The seeds did not dissolve. The mixture was allowed to stir at 30 °C for 5-10 min to form a thick slurry. Additional cyclohexane (2.0 L) was added over 2 minutes. The jacket temperature was lowered to 25 °C. The mixture was allowed to stir for 40 min. Additional cyclohexane (2.0 L) was added over 2 minutes. The j acket temperature was lowered to 23 °C. The suspension was maintained at 23 °C for 60 min. Additional cyclohexane (2.0 L) was added over 2 minutes. The suspension was stirred for 20 min. The jacket temperature was lowered to 19.0 °C. The suspension was maintained at 19-21 °C for 10 hrs. The slurry settled well after overnight aging. A sample of the supernatant was obtained and assessed for the loss based on 9.5 L total volume. The slurry was filtered to collect solids via a ceramic Buchner funnel with a No. l Whatman filter paper. The solids were crystalline and white when dry. The wet cake was washed with cyclohexane (~ 2000 mL x 3) followed by drying for 10 min. The cake volume was 4933 cm3. The wet cake was transferred to four Pyrex glass trays for heated drying. The drying was continued in a vacuum oven at ~ 35-40 °C with nitrogen sweeping for 12 hrs to afford 1302.9 g of 3-fluoro-2-(methyl(propoxycarbonyl)amino) benzoic acid in 85.9 % yield. ¾ NMR (400 MHz, DMSO-de) (3: 1 mixture of rotamers) δ 13.2 (br s, 1H), 7.72-7.67 (m, 1H), 7.58-7.52 (m, 1H), 7.49-7.43 (m, 1H), 4.06-3.95 (m, 0.50H), 3.90 – 3.80 (m, 1.50H) 3.12 (s 0.75H), 3.12 (s 2.25H), 1.67 – 1.58 (m, 0.50H), 1.42 – 1.34 (m5 1.50H), 0.93 (t, J=7.5 Hz, 0.75H), 0.67 (t, J=7.5 Hz, 2.25H). 13C NMR (101 MHz, DMSO-de) (mixture of rotamers) δ 165.8, 159.0, 156.6, 154.3, 131.6, 131.0, 128.7, 128.6, 126.3, 1 19.9, 119.7, 66.6, 66.4, 36.9, 36.4, 36.4, 21.8, 21.5, 10.0, 9.8.
HPLC Analysis: Column: Agilent ZORBAX Eclipse Plus C18 3.5um 4.6X150 mm; Column Temeprature: 40 °C; Solvent A: 0.01M NH4OOCH in water:MeOH (90: 10 v/v); Solvent B: O.OIM NH4OOCH in MeOH:CH3CN (70:30 v/v); Diluent: 0.25 mg/ml in acetonitrile; Gradient: %B: 0 min. 10%; 10 min. 30%; 20 min. 90%; 20.1 min. 10%; stop time 25 min; Flow Rate: 1.0 ml/min; Wavelength: 220 nm;
The retention time of 7-fluoro-l-methylindoline-2,3-dione was 10.7 minutes.
The retention time of 7-fluoroindoline-2,3-dione was 6.8 minutes. The retention time of 3-fluoro-2-(methylamino)benzoic acid was 5.9 minutes. The retention time of 3-fluoro-2-(methyl(propoxycarbonyl)amino)benzoic acid was 12.0 minutes.
Compound 1
(S)-3-(prop-l -en-2-yl)cyclohexan-l-one
Catalyst Preparation: Rhodium (I) (S)-(+)-5,5′-bis[di(3,5-di-tert-butyl-4-methoxyphenyl) phosphino] -4,4′-bi- 1 ,3-benzodioxole
Methanol (320 mL) was charged into a 0.5 L inerted reactor equipped with an overhead agitator, nitrogen sparging tube and an outlet connected to an oxygen meter. The reactor was inerted by sparging nitrogen subsurface through methanol until <300 ppm 02 was detected in the headspace. S-(+) DTBM-SEGPHOS (77.3 g, 65.6 mmol) and [Rh(cod)Cl]2 (15.4 g, 31 mmol) were charged and the nitrogen sparging continued until <300 ppm C was detected in the headspace. The mixture was agitated at room temperature under constant positive nitrogen pressure for 30 min by sweeping a low flow of nitrogen through the headspace. The initial yellow slurry gradually transformed into a deep-red solution containing a small amount of solids (excess ligand). The ligation completion was confirmed by 1P NMR by disappearance of the ligand peak at 13.1 ppm (s) and the appearance of the new singlets at 26.10 ppm and 27.01 ppm for the ligated species.
Synthesis of the Compound I
A 20 L jacketed Chemglass reactor, equipped with an overhead agitator, a thermocouple, nitrogen sparging tube, a sampling port, a condenser connected to the glycol supply and a nitrogen outlet connected sequentially to a bubbler, flow meter and an oxygen meter, was inerted using a vigorous nitrogen sweep. A Teledyne 3110 oxygen meter was used to monitor the progress of inertion. A vigorous nitrogen sweep was implemented prior to reagent charges until the oxygen reading was <300 ppm.
Heptane (4.0 L), 2-cyclohexen-l-one (1 kg, 10.4 M) in heptane (1.0 L), isopropenyl pinacol boronate (1.92 kg, 11.4 M, 1.1 eq) in heptane (1.0 L), DIPEA (0.91 L, 0.67 kg, 0.50 eq), a solution of 2,2-dimethy 1-1, 3 -propanediol (1.19 kg, 1.1 eq) in methanol (0.12L) in water (3 L), and additional heptane (2.55L) were sequentially charged to the reactor via vacuum. Nitrogen sparging subsurface through the agitated bi phasic mixture continued after the charges until an oxygen level of <300 ppm was
reached in the headspace prior to the catalyst charge. Then the nitrogen flow was reduced to maintain a slight positive pressure in the reactor.
The catalyst light slurry was transferred from the bottom value of the 0.5 L reactor’s bottom into the 20 L reactor through an inerted Teflon tubing by applying slight positive pressure of nitrogen. The contents of the small reactor was transferred including the excess of the undissolved solid.
The jacket was set to 60 °C on the 20 L reactor and the biphasic mixture was vigorously heated and agitated under nitrogen at 55-58 °C. After the transfer, the nitrogen flow was reduced to maintain a slight positive pressure and to minimize solvent loss. After completion of the reaction, the reaction mixture was cooled to 20-25 °C. The phases were separated and the organic phase was washed with IN HC1 aq (v=5.7 L, 0.55 eq) to remove DIPEA, and with water (2.5 L). Two back-extractions with heptane (2 x 2L) from the original aqueous phase were performed to bring back an additional 8 mol% of the product. All organic phases were combined and polished filtered back to the cleaned reactor. Heptane was removed under reduced pressure (30-40 °C at 45-55 torr) to give the crude product, which was transferred to a 2 L 4-necked round bottom flask, equipped with a mechanical stirrer, a thermocouple, a 30 cm Vigreaux column, a distillation adapter containing a thermocouple to measure the vapor temperature, a condenser (glycol) and a Teflon tubing attached to a receiver flask. Distillation was performed at a pressure of 10 torr with the main fraction containing the product boiling at 85-92 °C to afford 1.18 kg (85 mol % as is, 82.1 % corrected) of (S)-3-(prop-l-en-2-yl)cyclohexan-l-one. Chiral GC: Supelco AlphaDex 120 30 x 0.25 mm x 0.25 μπι, inlet 200 °C, split ratio 30: 1, carrier gas: helium, constant flow 1.9 mL/min, oven program: 80 °C to 110 °C at 2 °C /min, then 20 °C /min to 220 °C, detector: FID 250 °C; RT for the desired product: 14.4 min. Chemical purity: 97.1 GCAP. Chiral purity: ee = 99.6 %. ¾ NMR (CDCh): 1.57-1.70 (m, 12H), 1.75 (s, 3H), 1.91-1.96 (m, 1H), 2.05-2.12 (m, 1H), 2.26-2.46 (m, 5H), 4.73 (s, 1H), 4.78 (s, 1H).
Compound 2
(S,E)-4-bromo-5-fluoro-2-(2-(3-(prop-l-en-2-yl)cyclohexylidene)hydrazinyl)benzoic acid
(S)-3 -(prop- l -en-2-yl)cyclohexan-l -one (50.00 mL, 33.4 mmol, 0.667 mmol/mL) solution in heptane was added to a Chemglass reactor. Next, 75 mL of MeOH was added. The MeOH solution was distilled at 60 torr/50 °C jacket temperature and 75 mL of constant volume with the addition of 300 mL of MeOH. The contents of the reactor were cooled to 20 °C. 2-amino-4-bromo-5-fluorobenzoic acid (8.5415 g, 29.918 mmol) was added to the reactor. The reaction mixture was stirred at 20 °C. After, 30 minutes, the solid material was dissolved to form a clear brown solution. After 2.0 h, water (25.0 mL) was added over 25 min to the reaction mixture under slow agitation (RPM = 100). After an additional 1.0 h, the slurry was filtered (fast; < 3 seconds). The cake was washed with 2×25 mL of MeOH/H20 (3:2). The cake was dried at 55 °C under vacuum overnight to afford (S,E)-4-bromo-5-fluoro-2-(2-(3-(prop-l -en-2-yl)cyclohexylidene)
hydrazinyl)benzoic acid (10.5701 g; 95.7% yield). HPLC method: Column: Zorbax Eclipse plus 1.8 um C8 (4.6 X 50 mm); inj ection volume: 10 μί; Mobile Phase A: 0.05% TFA in acetonitrile: water (5 :95, v/v); Mobile Phase B: 0.05% TFA in water: acetonitrile (5:95, v/v); Gradient (%B) 0 min (30%), 14 min (100%), 15 min (30%); Flow Rate: 1.0 mL/min; Wavelength: 240 nm for IPC; Column temp: 25 °C; IPC Sample Prep:
Dissolved 10 of the reaction mixture and dilute with MeOH to 1.5 mL; HPLC results: Intermediate A2, 0.87 min; Compound 2, 9.97 min. ¾ NMR (400 MHz, DMSO-de) δ 13.54 (s, 1H), 10.76 (d, J = 26.5 Hz, 1H), 7.73 (appt triplet, J = 6.32 Hz, 1H), 7.64 (dd, J = 9.35, 1.26 Hz, 1H), 4.77-4.75 (m, 2H), 2.68-2.61 (m, 1H), 2.46-2.44 (m, 1H), 2.27-2.12 (m, 2H), 2.06-1.97 (m, 1H), 1.96-1.86 (m, 1H), 1.82-1.80 (m, 1H), 1.75-1.74 (m, 3H), 1.50-1.41 (m, 2H). 13C NMR (100 MHz, DMSO-de) δ 168.67, 152.76, 152.73, 150.71 , 148.41 , 148.38, 148.20, 145.10, 117.45, 117.21 , 116.45, 1 16.40, 1 15.76, 1 15.74, 1 15.54, 1 15.52, 109.64, 109.39, 108.88, 108.85, 108.83, 108.80, 44.80, 43.72, 34.22, 30.89, 30.08, 30.05, 25.42, 25.39, 24.15, 20.60, 20.44.
Compound 3
(S)-5-bromo-6-fluoro-2-(prop-l-en-2-yl)-2,3,4,9-tetrahydro-lH-carbazole-8-carboxylic acid
Zinc chloride (8.7858 g, 64.46 mmol) and (S,E)-4-bromo-5-fluoro-2-(2-(3-(prop- 1- en-2-yl)cyclohexylidene)hydrazinyl)benzoic acid (17.0011 g, 46.05 mmol) were added to a Chemglass reactor. Next, isopropyl acetate (170 mL) was added. The contents of the reactor were heated at 69.5 °C for 71 h and then cooled to room temperature. 2-MeTHF (205 mL) and HC1 (1 mol/L) in water (85 mL) were added. The reaction mixture was stirred at room temperature for 0.5 h. The layers were allowed to separate. The organic layer was washed with water (85 mL). The layers were separated and the organic layer was polish-filtered. The rich organic layer was distilled at 220 torr and 70 °C jacket temperature to 85 mL (5.0 mL/g (S,E)-4-bromo-5-fluoro-2-(2-(3-(prop-l-en-2-yl)cyclohexylidene)hydrazinyl) benzoic acid). Next, the solution was distilled at 120 mL (7.0 mL/g (S,E)-4-bromo-5-fluoro-2-(2-(3-(prop-l-en-2-yl)cyclohexylidene)hydrazinyl) benzoic acid) constant volume under 220 torr and 70 °C jacket temperature with continuous addition of acetonitrile (350 mL, 20 mL/g). Additional CFbCN was added to make the slurry volume = 153 mL (9.0 mL/g (S,E)-4-bromo-5-fluoro-2-(2-(3-(prop-l-en- 2- yl)cyclohexylidene) hydrazinyl)benzoic acid). The slurry was heated to 82 °C batch temperature. After 3.0 h, the slurry was cooled to 20 °C over 2.0 h. The slurry was stirred at 20 °C for an additional 14 h. The slurry was filtered and the cake was washed with acetonitrile (2 x 17 mL, 1.0 mL/g (S,E)-4-bromo-5-fluoro-2-(2-(3-(prop-l-en-2-yl)cyclohexylidene) hydrazinyl)benzoic acid). The wet cake was dried in a vacuum oven at a temperature range of 50-55 °C overnight to afford (S)-5-bromo-6-fluoro-2-(prop-l-en-2-yl)-2,3,4,9-tetrahydro-lH-carbazole-8-carboxylic acid (7.8991 g; 48.7% yield). HPLC method: Column: Agilent Zorbax Eclipse plus 1.8 μπι C8 (4.6 X 50 mm);
Injection Volume: 10 μί; Mobile Phase A: 0.05% TFA in acetonitrile: water (5:95, v/v); Mobile Phase B: 0.05% TFA in water: acetonitrile (5:95, v/v); Gradient (%B) 0 min
(30%), 14 min (100%), 15 min (100%); Flow Rate: 1.0 mL/min; Wavelength: 240 nm for IPC and Isolated product; Column temp: 25 °C; IPC Sample Prep: 1 mL/100 mL in tetrahydrofuran; Isolated Sample Prep: 0.25 mg/mL in tetrahydrofuran; HPLC results: Compound 3, 8.86 min; Compound 2, 10.0 min. ¾ NMR (400 MHz, DMSO-de) δ 13.41 (s, 1H), 11.03 (s, 1H), 7.45 (d, J = 9.85 Hz, 1H), 4.79 (appt d, J = 4.55Hz, 2H), 3.21-3.17 (m, 1H), 2.95 (dd, J = 17.18, 4.80 Hz, 1H), 2.91-2.83 (m, 1H), 2.61 (dd, J = 16.93, 10.61 Hz, 1H), 2.41-2.35 (m, 1H), 2.01-1.95 (m, 1H), 1.79 (s, 3H), 1.67-1.57 (m, 1H). 13C NMR (100 MHz, DMSO-de) δ 166.64, 166.61, 152.72, 150.42, 148.44, 139.96, 131.90, 127.44, 127.43, 112.40, 112.33, 109.67, 109.54, 109.39, 109.19, 109.14, 28.28, 27.79, 22.20, 20.69.
Compound 4
(S)-5-bromo-6-fluoro-2-(prop- -en-2-yl)-2,3,4,9-tetrahydro-lH-carbazole-8-carboxamide
Acetonitrile (70 mL) was added to a Chemglass reactor, followed by the addition of (S)-5-bromo-6-fluoro-2-(prop-l-en-2-yl)-2,3,4,9-tetrahydro-lH-carbazole-8-carboxylic acid (7.0150 g). Next, Ι,Γ-carbonyldiimidazole (4.2165 g, 26.004 mmol) was added. The reaction mixture was stirred (RPM = 100) for 5.0 hr at 20 °C. The slurry was cooled to 3 °C. Ammonia (30 mL, 200 mmol, 30 mass%) was added in less than 2 min. The slurry was stirred at 3 °C for 17.5 h. Water (70 mL) was added over 5 min. The slurry was stirred at 3 °C for 3 h. The slurry was filtered and the wet cake was washed with 2×50 mL of CH3CN/H2O (1 : 1). The wet cake was dried at 55 °C under vacuum overnight to afford (S)-5-bromo-6-fluoro-2-(prop-l-en-2-yl)-2,3,4,9-tetrahydro-lH-carbazole-8-carboxamide (5.2941 g; 75.8% yield). HPLC Method; Column: Agilent Zorbax Eclipse plus 1.8 μιη C8 (4.6 X 50 mm); Injection Volume: 10 μί; Mobile Phase A: 0.05% TFA in acetonitrile: water (5:95, v/v); Mobile Phase B: 0.05% TFA in water: acetonitrile (5:95, v/v); Gradient (%B) 0 min (0%), 8 min (100%), 10 min (100%); Flow Rate: 1.0 mL/min; Wavelength: 240 nm for IPC and Isolated product; Column temp: 25 °C; IPC Sample
Prep: Dissolved 10 of the reaction mixture into 1.0 mL 0.05 v% DBU/MeOH;
Product sample preparation: Dissolved product in MeOH at 1 mg/mL; HPLC results: Compound 4, 6.39 min; Compound 3, 6.80 min. ¾ NMR (400 MHz, DMSO-de) δ 11.05 (s, 1H), 8.11 (s, 1H), 7.59 (d, J = 10.36 Hz, 1H), 7.55 (br s, 1H), 4.78 (br s, 2H), 3.18 (br d, J = 14.65 Hz, 1H), 2.94 (dd, J = 16.93, 4.80 Hz, 1H), 2.88-2.82 (m, 1H), 2.62 (dd, J = 16.93, 10.61 Hz, 1H), 2.40-2.34 (m, 1H), 1.98 (d, J = 11.87 Hz, 1H), 1.78 (s, 3H), 1.66-1.56 (m, 1H). 13C NMR (100 MHz, DMSO-de) δ 167.64, 152.68, 150.38, 148.47, 139.47, 131.71, 127.02, 127.01, 115.36, 115.28, 109.53, 108.66, 108.61, 107.47, 107.19, 28.24, 27.87, 22.21, 20.67.
Compound 5
(S)-5-bromo-6-fluoro-2-(2-hydroxypropan-2-yl)-2,3,4,9-tetrahydro-lH-carbazole-8- carboxamide
Dichloromethane (100 mL) and (S)-5-bromo-6-fluoro-2-(prop-l-en-2-yl)-2,3,4,9-tetrahydro-lH-carbazole-8-carboxamide (PPP, 10.0016 g, 28.48 mmol) were added to a 250 mL Chemglass reactor. The slurry was cooled to 5 °C. Next, trifluoroacetic acid (14.68 g, 128.7 mmol) was added over 0.5 h with agitation (RPM = 250) while maintaining the internal temperature at less than 10 °C). The temperature was raised to 14 °C and the reaction mixture was stirred at 14 °C for 17.5 h. Next, 60 mL of MeOH was added to dissolve the thin slurry. The solution was cooled to -10 °C. The solution was distilled at 80 torr while the jacket temperature was gradually raised from -10 °C to 20 °C. The solution was distilled to about 60 mL volume. The internal temperature changed from -7 °C to -2 °C. The solution became a heavy slurry. The distillation was continued at 80 torr at 20 °C jacket temperature at 60 mL volume with the addition of 120 mL MeOH. The intemal temperature changed from -2 °C to 15 °C. The solution became a heavy slurry. The distillation became slow. The vacuum pressure was changed to 60 torr, and the distillation was continued with a 20 °C jacket temperature to 40 mL slurry volume. The batch temperature went from 12 °C to 13 °C.
MeOH (20 mL) was sprayed to wash solid crust off the reactor wall, but was not effective. Aqueous N¾ (30.0 mL, 400 mmol, 28 mass%) was sprayed to the slurry (pH = 10.59). Some solid crust on the upper reactor wall still remained. The slurry was stirred at 20 °C for 0.5 h (pH = 10.58), then heated to 70 °C in 15 min. All the solid crust on the upper reactor wall dissolved. Next, water (40 mL) was added over a period of 15 min. The solution remained as a clear solution at 70 °C.
The slurry was seeded with solid (S)-5-bromo-6-fluoro-2-(2 -hydroxy propan-2-yl)-2,3,4,9-tetrahydro-lH-carbazole-8-carboxamide (~ 5 mg). The seeds remained but there was little additional crystallization was observed at 70 °C. The slurry was heated at 70 °C (jacket temperature = 80 °C) for 0.5 h, and then cooled down to 20 °C in 0.5 h. At 65 °C the mixture became cloudy. The mixture was stirred at 20 °C for 65 h. The mixture was filtered. The cake was washed with 2×15 mL of MeOH/LhO (1 : 1). The wet cake was dried at 65 °C under vacuum for 24 h, giving (S)-5-bromo-6-fluoro-2-(2-hydroxypropan-2-yl)-2,3,4,9-tetrahydro-lH-carbazole-8-carboxamide (9.1741 g, 87.3% yield).
(S)-5-bromo-6-fluoro-2-(2-hydroxypropan-2-yl)-2,3,4,9-tetrahydro-lH-carbazole-8-carboxamide was recrystallization in MeOH/MTBE/n-Heptane (1 :4:8).
(S)-5-bromo-6-fluoro-2-(2-hydroxypropan-2-yl)-2,3,4,9-tetrahydro-lH-carbazole-8-carboxamide (8.0123 g) was added to a reactor. Next, MeOH (8.0 mL) and MTBE (32.0 mL) were added. The mixture was heated to 45 °C to dissolve the slurry. Heptane (64 mL) was added over a period of 15 min at 45 °C. The slurry was stirred at 45 °C for an additional 0.5 h and then cooled to 5 °C in 1.0 h. Stirring was continued at 5 °C for an additional 1.0 h. The slurry was filtered and the wet cake was washed with 2×20 mL of n-heptane. The wet cake was dried at 65 °C under vacuum for 16 h to afford (S)-5-bromo-6-fluoro-2-(2-hydroxypropan-2-yl)-2,3,4,9-tetrahydro-lH-carbazole-8-carboxamide (6.9541 g; 86.8%).
(S)-5-bromo-6-fluoro-2-(2-hydroxypropan-2-yl)-2,3,4,9-tetrahydro-lH-carbazole-8-carboxamide (8.0123 g) was added to a reactor. Next, MeOH (8.0 mL) and MTBE (32.0 mL) were added. The mixture was heated to 45 °C to dissolve the slurry. Heptane (64 mL) was added over a period of 15 min at 45 °C. The slurry was stirred at 45 °C for an additional 0.5 h and then cooled to 5 °C in 1.0 h. Stirring was continued at 5 °C for an additional 1.0 h. The slurry was filtered and the wet cake was washed with 2×20 mL of n-heptane. The wet cake was dried at 65 °C under vacuum for 16 h to afford (S)-5-bromo-6-fluoro-2-(2-hydroxypropan-2-yl)-2,3,4,9-tetrahydro-lH-carbazole-8-carboxamide (6.9541 g; 86.8%). HPLC method Column: Phenomenex Kinetex C18 2.6um 100A 4.6X150mm SN:538219-97; Injection Volume 5 μί; Mobile Phase A: 0.05% TFA in acetonitrile:water (5:95, v/v); Mobile Phase B: 0.05% TFA in
water: acetonitrile (5 :95, v/v); Gradient (%B) 0 min (32%), 5 min (38%), 1 1 min (38%), 18 min (68%), 22 min (68%), 30 min (90%), 31 min (100%); Flow Rate: 1.0 mL/min; Wavelength: 220 nm for IPC and Isolated product; Column temp: 25 °C; IPC Sample Prep: 1 μΙ71 mL in tetrahydrofuran; Isolated Sample Prep: 0.25 mg/mL in
tetrahydrofuran; HPLC results: Compound 5, 9.58 min; Compound 4, 19.98 min; ¾ NMR (400 MHz, DMSO-de) δ 10.99 (s, 1H), 8.10 (s, 1H), 7.57 (d, J = 10.36 Hz, 1H), 7.54 (br s, 1H), 4.27 (s, 1H), 3.26 (dd, J = 15.66, 4.29 Hz, 1H), 2.93 (dd, J = 17.18, 4.55 Hz, 1H), 2.76-2.68 (m, 1H), 2.44 (dd, J = 16.17, 1 1.87 Hz, 1H), 2.12 (br d, J = 1 1.12 Hz, 1H), 1.69-1.62 (m, 1H), 1.31 (ddd, J = 25.01, 12.38, 5.31 Hz, 1H), 1.14 (s, 6H). 13C
NMR (100 MHz, DMSO-de) δ 167.67, 152.64, 150.34, 140.46, 131.77, 127.03, 127.02, 1 15.28, 1 15.21, 109.09, 109.05, 107.30, 107.03, 101.43, 101.19, 70.37, 44.96, 27.17, 26.73, 24.88, 24.36, 22.85.
Compound 6
(2S)-5-(3-amino-2-methylphenyl)-6-fluoro-2-(2-hydroxypropan-2-yl)-2,3,4,9-tetrahydro- lH-carbazole-8-carboxamide
Catalyst activation
Into a 1 Liter Chemglass reactor (Reactor A) were added Me-THF (4 L/kg) followed by (R)-BINAP (0.0550 mol/mol, 7.45 mmol) and Pd(OAc)2 (0.0500 mol/mol, 6.77 mmol). Additional Me-THF (1 L/kg) was added. The mixture was stirred at 25 °C
for 1 h. Next, 4-bromo-3-fluoro-7-(l-hydroxy-l-methyl-ethyl)-6,7,8,9-tetrahydro-5H-carbazole-l-carboxamide (0.10 equiv, 13 mmol) was added into the mixture in Reactor A, followed by the addition of 2-methyltetrahydrofuran (0.50 L/kg) and water (0.5 L/kg).
The overhead space of Reactor A was sparged with nitrogen at 1 mL/second for 40 min at 25 °C. The resulting mixture was then stirred at 70 °C for 3 h under a positive pressure of nitrogen (1.05 atm). The resulting mixture containing the activated catalyst was cooled to
25 °C and kept at 25 °C under a positive pressure of nitrogen before use.
To a 500 mL Chemglass reactor (Reactor B) were added water (6 L/kg) followed by K3PO4 (6 equiv., 813 mmol). The addition was exothermic. The mixture was stirred till the base was fully dissolved. The overhead space of Reactor B was sparged with nitrogen at 1 mL/second for 60 min at 25 °C. The K3PO4 solution in Reactor B was then kept under a positive pressure of nitrogen before use.
To Reactor A, which contained the activated catalyst, was added 4-bromo-3-fluoro-7-(l-hydroxy-l-methyl-ethyl)-6,7,8,94etrahydro-5H-carbazole-l-carboxarnide (0.90 equiv., 122 mmol), followed by THF (2.5 L/kg). Then (3-amino-2-methyl-phenyl)boronic acid hydrochloride (1.15 equiv., 156 mmol) and MeOH (2 L/kg) were added to Reactor A. The overhead space of Reactor A was sparged with nitrogen at 1 mL/second for 40 min. Then the reaction mixture in Reactor A was cooled to -10 °C under a positive pressure of nitrogen.
The K3PO4 aqueous solution in Reactor B was then transferred into Reactor A via a cannula while both reactors were kept under a positive pressure of N2. The rate of transfer was controlled so that the inner temperature in Reactor A was below 0 °C throughout the operation.
The resulting biphasic reaction mixture was stirred at 5 °C under a positive pressure of nitrogen. After 2.5 h at 5 °C, HPLC analysis of the reaction mixture showed
0.3 AP starting material remained. The reaction mixture was then warmed to 25 °C and stirred at 25 °C for 30 min. HPLC analysis of the reaction mixture showed 0.0 AP starting material remained.
N-acetyl-L-cysteine (1 kg/kg, 306 mmol) and water (2.5 L/kg) were added into Reactor A. The resulting mixture was stirred at 40 °C for 2 h then cooled to 25 °C. The bottom layer (aqueous layer) was discharged and the top layer (organic layer) was retained in the reactor.
Afterwards, THF (1 L/kg) and NaCl solution (13 mass%) in water (7 L/kg) were added into Reactor A, and the resulting mixture was stirred at 25 °C for lh. The bottom layer (aqueous layer) was discharged and the top layer (organic layer) was retained in the reactor.
The organic layer was filtered through a polyethylene filter. Then the reactor was rinsed with Me-THF (0.50 L/kg). The rinse was filtered through the polyethylene filter and combined with the filtrate. The solution was transferred into a clean 1 L reactor (Reactor C).
The mixture in Reactor C was concentrated under reduced pressure to 8.8 L/kg. (2 L/kg solvent was removed by distillation). At 50 °C, n-BuOH (4 L/kg) was added slowly over 2 h. The mixture was then stirred at 50 °C for 2.5 h, and a slurry was obtained.
The solvent was swapped to n-BuOH through constant volume distillation. During this operation, n-BuOH (8 L/kg) was used and 8 L/kg solvent was removed from Reactor C. The resulting mixture was stirred at 55 °C for 1 h and cooled to 25 °C over 1 h.
The slurry in Reactor C was filtered. The reactor rinsed with n-BuOH (2 L/kg).
The cake was then washed with this reactor rinse, followed by heptane (8 L/kg). The product was dried under vacuum at 55 °C for 24 h to afford (2S,5R)-5-(3-amino-2-methylphenyl)-6-fluoro-2-(2-hydroxypropan-2-yl)-2,3,4,9-tetrahydro-lH-carbazole-8-carboxamide, which was isolated as an off-white solid powder (46.2 g, 86% yield).
HPLC analysis: (2S,5R)-5-(3-amino-2-methylphenyl)-6-fluoro-2-(2-hydroxypropan-2-yl)-2,3,4,9-tetrahydro-lH-carbazole-8-carboxamide: 98.1 AP (19.2 min); (2S,5S)-5-(3-amino-2-methylphenyl)-6-fluoro-2-(2-hydroxypropan-2-yl)-2,3,4,9-tetrahydro-lH-carbazole-8-carboxamide: 1.8 AP (19.9 min), (S)-6-fluoro-2-(2-hydroxypropan-2-yl)-2,3,4,9-tetrahydro-lH-carbazole-8-carboxamide: 0.1 AP (20.9 min). Column: Waters XBridge BEH C18 S-2.5um 150 X 4.6mm; Solvent A: 10 mM sodium phosphate buffer pH 7; Solvent B: CH3CN:MeOH (50:50 v/v); Gradient: % B: 0 Min. 5%; 4 Min. 30%; 41 Min. 95%; 47 Min. 95%; Stop Time: 48 min; Flow Rate: 0.7 ml/min wavelength: 240 nm. ¾ NMR (500 MHz, DMSO-de) δ 10.76 (s, 1H), 8.09 (br s, 1H), 7.54 (d, J=10.7 Hz, 1H), 7.47 (br s, 1H), 6.96 (t, J=7.7 Hz, 1H), 6.72 (d, J=7.9 Hz, 1H), 6.41 (d, J=7.3 Hz, 1H), 4.90 (s, 2H), 4.19 (s, 1H), 2.91 (br dd, J=16.6, 4.0 Hz, 1H), 2.50-2.39 (m, 1H), 2.05-1.93 (m, 1H), 1.88-1.75 (m, 5H), 1.64-1.53 (m, 1H), 1.21-1.11 (m, 1H), 1.09 (s, 6H). 13C NMR (126 MHz, DMSO-de) δ 169.0 (d, J=2.7 Hz), 152.5 (d, J=229.8 Hz), 146.7, 139.1,
134.4, 132.0, 127.7 (d, J=4.5 Hz), 125.6, 123.3 (d, J=20.0 Hz), 120.5, 119.2, 1 15.1 (d, J=7.3 Hz), 1 14.3, 109.5(d, J=4.5 Hz), 107.2 (d, J=27.3 Hz), 70.9, 45.9, 27.6, 27.2, 25.3, 25.0, 22.7, 14.7.
Compound 7
propyl (2-((3-((2S)-8-carbamoyl-6-fluoro-2-(2-hydroxypropan-2-yl)-2,3,4,9-tetrahydro- lH-carbazol-5-yl)-2-methylphenyl)carbamoyl)-6-fluorophenyl)(methyl)carbamate
N, N-Dimethylformamide (7.0 L, 7 L/kg) was charged into a reactor followed by the addition of (2S)-5-(3-amino-2-methylphenyl)-6-fluoro-2-(2-hydroxypropan-2-yl)-2,3,4,9-tetrahydro-lH-carbazole-8-carboxamide (1 kg, 2528 mmol, 1.0 eq.). 3-Fluoro-2-(methyl(propoxycarbonyl)amino)benzoic acid (0.774 kg, 3034 mmol, 1.2 eq.) was added to the reactor, followed by the addition of 1 -methylimidazole (0.267 kg, 3287 mmol, 1.3 eq) and methanesulfonic acid (0.122 kg, 1264 mmol, 0.5 eq.) at 20 °C. The reaction mixture was stirred for at 20 °C for 30 min to completely dissolve the reaction contents. The reaction mixture was cooled to 10 °C and EDAC (l-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride) (0.679 kg, 3540 mmol, 1.4 eq) was charged into the reactor. An exotherm of approximately 4 °C was observed. The reaction mixture was stirred at 10 °C for 4 h.
After 4 hrs, the reaction mixture was warmed to 20 °C. Isopropyl acetate (25 L, 25 L/kg) was added to the reaction mixture followed by 25 wt% aqueous sodium chloride solution (2.5 L, 2.5 L/kg) and 1.0 M aqueous hydrochloric acid (2.5 L, 2.5 L/kg). The reaction mixture was stirred for 30 min. The agitation was stopped and the bottom aqueous layer was separated. Water (5 L, 5 L/kg) was charged to the rich organic solution and stirred for 30 min. The agitation was stopped and the bottom aqueous layer was separated. Next, 2.5% aqueous sodium bicarbonate solution (10 L, 10 L/kg) was charged to the rich organic solution and stirred for 30 min. The agitation was stopped and the bottom aqueous layer was separated. Water (10 L, 10 L/kg) was charged to the rich organic solution and stirred for 30 min. The agitation was stopped and the bottom aqueous layer was separated. The rich organic solution was concentrated under reduced pressure (90 mbar and 40 °C jacket temperature) to 7 L/kg volume. Dichloromethane (5 L, 5 L/kg) was charged to the product rich isopropyl acetate solution at 20 °C. Seeds of propyl (2-((3-((2S)-8-carbamoyl-6-fluoro-2-(2-hydroxypropan-2-yl)-2,3,4,9-tetrahydro-lH-carbazol-5-yl)-2-methylphenyl)carbamoyl)-6-fluorophenyl)(methyl)carbamate (10 g, 1%) were charged and a thin slurry formed. Heptane (7 L, 7 L/kg) was charged to the above slurry slowly over 1 hr at 25 °C and stirred for another 1 h before cooling 20 °C over 30 min. The resultant slurry was stirred for 4-6 hrs at 20 °C. The slurry was filtered over a laboratory Buchner funnel. The wet cake was washed with a dichloromethane-heptane mixture (10:7 ratio, 12 vol). The wet cake was dried in a vacuum oven at 25 mm Hg vacuum and 50 °C until the residual heptane was <13 wt% in the solid to provide 1.5 kg of propyl (2-((3-((2S)-8-carbamoyl-6-fluoro-2-(2-hydroxypropan-2-yl)-2,3,4,9-tetrahydro-lH-carbazol-5-yl)-2-methylphenyl)carbamoyl)-6-fluorophenyl)(methyl) carbamate in 94% yield. The product was a mixture of four amide rotational isomers. ¾ NMR (400 MHz, DMSO-de) δ 10.79 (br s, 1H), 9.96 (m, 1H), 8.07 (br s, 1H), 7.50 (m, 6H), 7.29 (m, 1H), 7.09 (m, 1H), 4.15 (m, 1H), 3.89 (m, 2H), 3.19 (br s, 1H), 3.13 (br s, 2H), 2.90 (m, 1H), 2.44 (m, 1H), 1.97 (m, 3H), 1.82 (m, 3H), 1.50 (m, 3H), 1.26 (m, 5H), 1.09 (m, 7H), 0.85 (m, 4H), 0.70 (m, 2H). 13C NMR (101 MHz, DMSO-de) δ 168.33, 168.32, 164.85, 164.55, 159.38, 159.16, 156.93, 156.69, 154.90, 154.74, 153.14, 150.86, 139, 15, 139.11, 137.96, 137.89, 137.36, 137.23, 135.75, 135.68, 135.64, 134.77, 134.68, 132.57, 132.51, 132.46, 132.42, 131.50, 128.98 (m), 128.26 (m), 127.05, 127.01, 125.99, 125,76, 124.97, 124.83, 124.06, 121.48, 121.40, 121.28, 121.20, 117.90, 117.86, 117.70, 117.65, 115.19, 115.15, 115.12, 115.07, 108.69, 108.65, 106.87, 106.60, 70.39, 66.83, 66.80, 66.73, 45.32, 37.38, 37.15, 31.23, 28.35, 27.05, 26.68, 24.85, 24.61, 22.27, 22.07, 21.84, 21.75, 14.98, 14.93, 14.86, 14.84, 13.87, 10.11, 9.89.
HPLC Analysis: Column: Zorbax Eclipse Plus C18 3.5 um, 150 x 4.6 mm ID;
Solvent A: 10 mM ammonium formate in water-MeOH (90: 10); Solvent B: C¾CN :
MeOH (30:70 v/v); Gradient: % B: 0 Min. 50%; 25 Min. 81 %; 26 Min. 100%; 30 Min. 100%; Stop Time: 30 min; Flow Rate: 1 ml/min; Wavelength: 240 nm. The retention time of propyl (2-((3-((2S)-8-carbamoyl-6-fluoro-2-(2-hydroxypropan-2-yl)-2,3,4,9-tetrahydro-lH-carbazol-5-yl)-2-methylphenyl)carbamoyl)-6-fluorophenyl)(methyl) carbamate wasl4.6 min. The retention time of 3-fluoro-2-(methyl(propoxycarbonyl) amino)benzoic acid was 2.6 min. The retention time of (2S)-5-(3-amino-2-methylphenyl)-6-fluoro-2-(2-hydroxypropan-2-yl)-2,3,4,9-tetrahydro-lH-carbazole-8-carboxamide was 6.1 min.
Compound 8
6-fluoro-5-(R)-(3-(S)-(8-fluoro-l-methyl-2,4-dioxo-l ,2-dihydroquinazolin-3(4H)-yl)-2- methylphenyl)-2-(S)-(2-hydroxypropan-2-yl)-2,3,4,9-tetrahydro-lH-carbazole-8- carboxamide
(8)
To a 1 L round bottom flask with stir bar was added propyl (2-((3-((2S)-8-carbamoyl-6-fluoro-2-(2-hydroxypropan-2-yl)-2,3,4,9-tetrahydro-lH-carbazol-5-yl)-2-methylphenyl)carbamoyl)-6-fluorophenyl)(methyl)carbamate (100 g, 148 mmol, 93.5 mass%) followed by MeTHF (500 mL, 4990 mmol, 100 mass%). The mixture was stirred at room temperature for 10 minutes to ensure complete dissolution. Next, 150 mL of MeTHF was added, and an azeotropic distillation to remove water was performed at 50 °C and 70 torr. The KF was measured to be 424 ppm. This solution is termed the “Compound 8 solution.”
To a 2 L Chemglass reactor was charged MeTHF (2000 mL, 19900 mmol, 100 mass%) followed by lithium fert-butoxide (7.9 mL, 7.9 mmol, 1 mol/L). The KF of MeTHF was measured to be 622 ppm. The Compound 8 solution was added dropwise
over 2 hours at room temperature via a Simdos pump. After the addition was complete, the reaction mixture was maintained at temperature for 15 minute.
MeOH (200 mL, 4940 mmol, 100 mass%) was then added to the reactor followed by the addition of acetic acid (0.5 mL, 9 mmol, 100 mass%). The reaction mixture was distilled to 5 volumes of organics (60 mbar pressure, jacket temperature = 40 °C). After the distillation, acetone (150 mL, 2000 mmol, 100 mass%) was added to the thick slurry as the solution warmed to 35 °C. Once at 35 °C, MeOH (550 mL, 13600 mmol, 100 mass%) was charged to the reactor, re-dissolving the batch to provide a yellow solution. The reaction mixture was cooled over 1 hour to 20 °C resulting in crystallization of the product. Ten heat cycles were performed. Starting at 20 °C, the batch was heated to 35 °C over 45 minutes, held at 35 °C for 10 minutes, cooled 20 °C over 60 minutes, and held at 20 °C for 10 minutes. After the heat cycles, the slurry was maintained at room temperature for 1 hour at room temperature. Heptane (1100 mL, 7510 mmol, 100 mass%) was added over 4 hours at 20 °C with agitation via a Simdos pump. After the addition, the slurry aged to 20 °C overnight. The product was isolated by vacuum filtration and washed twice with MeOH (200 mL, 4940 mmol, 100 mass%). The product was dried on a filter with vacuum for 1.5 h to afford 6-fluoro-5-(R)-(3-(S)-(8-fluoro-l-methyl-2,4-dioxo-l,2-dihydroquinazolin-3(4H)-yl)-2-methylphenyl)-2-(S)-(2-hydroxypropan-2-yl)-2,3,4,9-tetrahydro-lH-carbazole-8-carboxamide at 89.4% corrected yield (80.52g, 6 wt % MeOH, Purity by HPLC: 99.32 AP; Retention time (11.65 min)).
¾ NMR (500MHz, DMSO-de) 10.78 (s, 1H), 8.07 (br. s., 1H), 7.95 (d, J=7.8 Hz, 1H), 7.72 (dd, J=14.2, 8.0 Hz, 1H), 7.56 (d, J=10.8 Hz, 1H), 7.45 (br. s., 1H), 7.42-7.36 (m, 1H), 7.34 (d, J=6.9 Hz, 1H), 7.34-7.31 (m, 1H), 7.29 (dd, J=7.5, 1.3 Hz, 1H), 4.17 (s, 1H), 3.73 (d, J=8.0 Hz, 3H), 2.91 (dd, J=16.8, 4.4 Hz, 1H), 2.48-2.37 (m, 1H), 1.98-1.89 (m, 2H), 1.87 (d, J=11.0 Hz, 1H), 1.76 (s, 3H), 1.59 (td, J=l 1.5, 4.1 Hz, 1H), 1.20-1.12 (m, 1H), 1.11 (s, 6H).
13C NMR (126MHz, DMSO-de) 168.2 (d, J=1.8 Hz, 1C), 160.1 (d, J=3.6 Hz, 1C), 151.9 (d, J=228.9 Hz, 1C), 150.5 (d, J=41.8 Hz, 1C), 148.7 (d, J=205.3 Hz, 1C), 139.2, 135.1, 135.0, 134.8, 131.4, 130.6, 130.0 (d, J=7.3 Hz, 1C), 128.5, 127.1 (d, J=4.5 Hz, 1C), 125.7, 124.3 (d, J=2.7 Hz, 1C), 123.6 (d, J=8.2 Hz, 1C), 123.0 (d, J=23.6 Hz, 1C), 120.8 (d, J=20.0 Hz, 1C), 118.4, 115.3 (d, J=7.3 Hz, 1C), 108.8 (d, J=5.4 Hz, 1C), 106.7 (d, J=28.2 Hz, 1C), 70.4, 45.4, 34.3 (d, J=14.5 Hz, 1C), 27.1, 26.8, 24.8, 24.7, 22.1, 14.5.
HPLC Analysis: Column: Chiralcel OX-3R 3um 4.6 x 150 mm; Oven
Temperature: 50 °C; Solvent A: 0.05%TFA Water/ ACN (95:5); Solvent B: 0.05%TFA Water/ ACN (5:95); Gradient % B: 0 Min. 0%; 7 Min. 55%; 11 Min. 55%; 14 Min. 100%; Stop Time: 17 Min.; Flow Rate: 1.5 ml/min; wavelength: 225 nm. (2-((3-((2S)-8-carbamoyl-6-fluoro-2-(2-hydroxypropan-2-yl)-2,3,4,9-tetrahydro-lH-carbazol-5-yl)-2-methylphenyl)carbamoyl)-6-fluorophenyl)(methyl)carbamate: 0.00 AP (9.85 min).
Alternative Preparation of Compound 8
To a 2.5 L Chemglass reactor with agitator were added 2-Me-THF (162.4 g, 1885 mmol, 100 mass%, 189 mL, 11.83) and DMF (179.5 g, 2456 mmol, 100 mass%, 190 mL, 15.41), followed by the addition of (2S)-5-(3-amino-2-methylphenyl)-6-fluoro-2-(2-hydroxypropan-2-yl)-2,3,4,9-tetrahydro-lH-carbazole-8-carboxamide (63.03 g, 63.03 mL, 159.4 mmol, 63.03 g), 3-fluoro-2-(methyl(propoxycarbonyl)amino)benzoic acid (44.77 g, 44.77 mL, 175.4 mmol, 44.77 g), and 1 -Me-Imidazole (16.99 g, 16.48 mL, 206.9 mmol, 16.99 g). With agitation, MSA (7.66 g, 5.23 mL, 79.7 mmol, 7.66 g) was added at -20 °C, and a slight exotherm to 26 °C was observed. The reaction mixture was cooled to 10 °C and ED AC (42.73 g, 42.73 mL, 222.9 mmol, 42.73 g) was added as a solid followed by a DMF rinse (60.4 g, 63.9 mL, 826 mmol, 60.4 g). The reaction mixture was aged overnight at 10 °C with agitation. An aliquot was taken and subjected to HPLC analysis to confirm reaction completion.
The batch temperature was increased to 15 °C, and 2-Me-THF (923.96 g, 10727 mmol, 100 mass%, 1080 mL, 67.31) was charged to the reactor, followed by a saturated aqueous brine solution (158 mL, 835.8 mmol, 26 mass%, 158 mL, 5.244) and an aqueous 2.0 M HCl solution (78 mL, 78 mmol, 1.0 mol/L, 78 mL, 0.49). The batch temperature was then increased to 20 °C. The biphasic mixture was agitated for 15 min and allowed to settle for 5 min. An saturated aqueous brine solution (157 mL, 830.5 mmol, 26 mass%, 157 mL, 5.211) and an aqueous 2.0 M HCl solution (78 mL, 78 mmol, 1.0 mol/L, 78 mL, 0.49) were then added to the reactor. The biphasic mixture was agitated for 15 min, allowed to settle for 5 min, and the aqueous layer was removed. Water (634.6 g, 35230 mmol, 100 mass%, 634.6 mL, 221.0) was then added to the reactor. The biphasic mixture was agitated for 15 min, allowed to settle for 5 min, and the aqueous layer was removed. Next, 10 w/w% aqueous NaHCC solution (164.2 g, 97.73 mmol, 5 mass%,
158.2 mL, 0.6132) and water (476.3 g, 26440 mmol, 100 mass%, 476.3 mL, 165.9) were added to the reactor. The biphasic mixture was agitated for 15 min, settled for 5 min, and the aqueous layer was removed. A saturated aqueous brine solution (752.9 g, 3349 mmol, 26 mass%, 633.2 mL, 21.02) was then added to the reactor. The biphasic mixture was agitated for 30 min, allowed to settle for 5 min, and the aqueous layer was removed.
The organic stream was distilled to 6 volumes (380 mL) at a pressure of 200 mbar, a jacket temperature of 60 °C, and a batch temperature of -35 °C. 2-Me-THF (765 g, 8881.6 mmol, 100 mass%, 891 mL, 55.73) was charged to the reactor. The organic solution was distilled to 6 volumes (380 mL) at a pressure of 200 mbar, a jacket temperature of 60 °C, and a batch temperature of -35 °C. 2-Me-THF (268.5 g, 3117 mmol, 100 mass%, 313 mL, 19.56) was charged to the reactor. The organic solution was distilled to 6 volumes (380 mL) at a pressure of 200 mbar, a jacket temperature of 60 °C, and a batch temperature of -35 °C. The concentrated stream was polish filtered through a 0.4 μιη PTFE filter. The reactor was rinsed with 2-Me-THF (134.6 g, 1563 mmol, 100 mass%, 157 mL, 9.806) and the rinse was passed through the PTFE filter. This solution was termed “organic solution.”
To a clean, dry, 2.5 L Chemglass reactor were added LiOtBu 1.0 M in THF (9.91 g, 11.2 mmol, 1 mol/L, 11.2 mL, 0.0700) and 2-Me-THF (1633.3 g, 18963 mmol, 100 mass%, 1900 mL, 119.0). The organic solution was charged to the reactor, with agitation, over 2 hours (at a rate of -100 mL/h) via a sim-dos pump. The reaction mixture was aged 10 minutes upon completion of the addition. An aliquot was taken and subjected to HPLC analysis to confirm reaction completion.
Acetic acid (1.03 g, 17.2 mmol, 100 mass%, 0.983 mL, 0.108) and methanol (150 g, 4681.41 mmol, 100 mass%, 189 mL, 29.37) were charged to the reactor. The organic stream was distilled to 16.5 vol Me-THF. Acetone (638.4 g, 10990 mmol, 100 mass%, 810 mL, 68.97) was added to the reactor and the organic stream was distilled to 9 vol at a pressure of 100 mbar and ajacket temperatures of less than 40 °C. The organic stream was heated to 35 °C, and methanol (400 g, 12483.8 mmol, 100 mass%, 505 mL, 78.33) was added. The stream was cooled to 20 °C to induce crystallization.
Heat cycles were performed for -15 h by heating the batch to 35 °C over 20 min, holding for 10 min, cooling to 20 °C over 20 min, and holding 10 min. After the heat cycles, heptane (686 g, 6846.10 mmol, 100 mass%, 1000 mL, 42.96) was added over 4 hours via a sim-dos pump. The slurry was aged for 2 h. The product was filtered, washed with methanol (152.2 g, 4750 mmol, 100 mass%, 192 mL, 29.81) to afford 6-fluoro-5-(R)-(3-(S)-(8-fluoro-l -methyl-2,4-dioxo-l,2-dihydroquinazolin-3(4H)-yl)-2-methylphenyl)-2-(S)-(2-hydroxypropan-2-yl)-2,3,4,9-tetrahydro-lH-carbazole-8-carboxamide (68.4 g, 1 19 mmol, 100 mass%, 75.0% Yield, 68.4 mL, 0.750).
Comparative Process Disclosed in US 9,334,290
Intermediates 25 and 26
(R)-5-Bromo-6-fluoro-2-(2-hydroxypropan-2-yl)-2,3,4,9-tetrahydro-lH-carbazole-8- carboxamide (1-25), and
(S)-5-Bromo-6-fluoro-2-(2-hydroxypropan-2-yl)-2,3,4,9-tetrahydro-lH-carbazole-8- -26)
A sample of racemic 5-bromo-6-fluoro-2-(2-hydroxypropan-2-yl)-2,3,4,9-tetrahydro-lH-carbazole-8-carboxamide [Intermediate 24] was separated by chiral supercritical fluid chromatography as follows: column: CHIRALPAK® OD-H (3 x 25 cm, 5μηι); Mobile Phase: CC -MeOH (70:30) at 150 mL/min, 40 °C. The first peak eluting from the column provided (R)-5-bromo-6-fluoro-2-(2-hydroxypropan-2-yl)-2,3,4,9-tetrahydro-lH-carbazole-8-carboxamide [Intermediate 25]. The second peak eluting from the column provided (S)-5-bromo-6-fluoro-2-(2-hydroxypropan-2-yl)-2,3,4,9-tetrahydro-lH-carbazole-8-carboxamide [Intermediate 26]. The mass spectra and ¾ NMR spectra of the two enantiomers were the same. Mass spectrum m/z 369, 371 (M+H)+. ¾ NMR (500 MHz, DMSO-de) δ 10.96 (s, 1H), 8.07 (br. s., 1H), 7.55 (d, J=10.3 Hz, 1H), 7.50 (br. s., 1H), 4.24 (s, 1H), 3.26 (dd, J=15.8, 4.4 Hz, 1H), 2.93 (dd, J=17.1, 4.6 Hz, 1H), 2.72 (t, J=11.7 Hz, 1H), 2.48-2.40 (m, 1H), 2.12 (d, J=9.2 Hz, 1H), 1.70-1.62 (m, 1H), and 1.32 (qd, J=12.4, 5.3 Hz, 1H).
Alternative SFC Separation to Give Intermediate 26:
CHIRALPAK® AD-H (3 x 25 cm, 5 μηι); Mobile Phase: C02-MeOH (55:45) at
150 mL/min, 40 °C. The first peak eluting from the column provided (S)-5-bromo-6- fluoro-2-(2-hydroxypropan-2-yl)-2,3,4,9-tetrahydro-lH-carbazole-8-carboxarnide
[Intermediate 26]. The second peak eluting from the column provided (R)-5-bromo-6- fluoro-2-(2-hydroxypropan-2-yl)-2,3,4,9-tetrahydro-lH-carbazole-8-carboxarnide
[Intermediate 25].
Example 28
6-Fluoro-5-(R)-(3-(S)-(8-fluoro-l-methyl-2,4-dioxo-l,2-dihydroquinazolin-3(4H)-yl)-2- methylphenyl)-2-(S)-(2-hydroxypropan-2-yl)-2,3,4,9-tetrahydro-lH-carbazole-8-
Following the procedure used to prepare Example 27, (S)-5-bromo-6-fluoro-2-(2- hydroxypropan-2-yl)-2,3,4,9-tetrahydro-lH-carbazole-8-carboxamide (single enantiomer) [Intermediate 26] (0.045 g, 0.122 mmol) and 8-fluoro-l-methyl-3-(S)-(2-methyl-3- (4,4,5, 5-tetramethyl-l,3,2-dioxaborolan-2-yl)phenyl)quinazoline-2,4(lH,3H)-dione
[Intermediate 10] (0.065 g, 0.158 mmol) were converted into 6-fluoro-5-(3-(S)-(8-fluoro- l-methyl-2,4-dioxo-l,2-dihydroquinazolin-3(4H)-yl)-2-methylphenyl)-2-(S)-(2- hydroxypropan-2-yl)-2,3,4,9-tetrahydro-lH-carbazole-8-carboxamide (mixture of two atropisomers) as a yellow solid (0.035 g, 49% yield). Separation of a sample of this material by chiral super-critical fluid chromatography, using the conditions used to separate Example 27, provided (as the first peak to elute from the column) 6-fluoro-5-(R)-(3-(S)-(8-fluoro-l-methyl-2,4-dioxo-l,2-dihydroquinazolin-3(4H)-yl)-2-methylphenyl)-2-(S)-(2-hydroxypropan-2-yl)-2,3,4,9-tetrahydro-lH-carbazole-8-carboxarnide. The chiral purity was determined to be greater than 99.5%. The relative and absolute configurations were determined by x-ray crystallography. Mass spectrum m/z 573 (M+H)+. ¾ NMR (500 MHz, DMSO-de) δ 10.77 (s, 1H), 8.05 (br. s., 1H), 7.94 (dd, J=7.9, 1.2 Hz, 1H), 7.56-7.52 (m, 1H), 7.43 (br. s., 1H), 7.40-7.36 (m, 1H), 7.35-7.30 (m, 2H), 7.28 (dd, J=7.5, 1.4 Hz, 1H), 4.15 (s, 1H), 3.75-3.70 (m, 3H), 2.90 (dd, J=16.8, 4.6 Hz, 1H), 2.47-2.39 (m, 1H), 1.93-1.82 (m, 3H), 1.74 (s, 3H), 1.57 (td, J=l 1.7, 4.2 Hz, 1H), 1.16-1.11 (m, 1H), and 1.10 (d, J=1.9 Hz, 6H). [a]D: +63.8° (c 2.1, CHCh). DSC melting point onset temperature = 202.9 °C (heating rate = 10 °C/min.).
Alternative Synthesis of Example 28:
A mixture of (S)-5-bromo-6-fluoro-2-(2-hydroxypropan-2-yl)-2,3,4,9-tetrahydro-lH-carbazole-8-carboxamide [Intermediate 26] (5.00 g, 13.54 mmol), 8-fluoro-l-methyl-3-(S)-(2-methyl-3-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)phenyl)quinazoline-2,4(lH,3H)-dione [Intermediate 10] (6.67 g, 16.25 mmol), tripotassium phosphate (2 M in water) (20.31 mL, 40.6 mmol), and tetrahydrofuran (25 mL) was subjected to 3 evacuate-fill cycles with nitrogen. The mixture was treated with l,l’-bis(di-fert-butylphosphino)ferrocene palladium dichloride (0.441 g, 0.677 mmol) and the mixture was subjected to 2 more evacuate-fill cycles with nitrogen. The mixture was stirred at room temperature overnight, then was diluted with EtOAc, washed sequentially with water and brine, and dried and concentrated. The residue was purified by column chromatography on silica gel, eluting with EtOAc-hexanes (sequentially 50%, 62%, 75% and 85%), to provide 6-fluoro-5-(3-(8-fluoro-l-methyl-2,4-dioxo-l,2-dihydroquinazolin-3-(S)-3(4H)-yl)-2-methylphenyl)-2-(S)-(2-hydroxypropan-2-yl)-2,3,4,9-tetrahydro-lH-carbazole-8-carboxamide as a white solid (6.58 g, 85% yield).
Material prepared by this method (40.03 g, 69.9 mmol) was separated by chiral super-critical fluid chromatography to give (2S, 5R)-6-fluoro-5-(3-(8-fluoro-l-methyl-2,4-dioxo-l,2-dihydroquinazolin-3(4H)-yl)-2-methylphenyl)-2-(S)-(2-hydroxypropan-2-yl)-2,3,4,9-tetrahydro-lH-carbazole-8-carboxamide. Further purification was achieved
by suspending this material in methanol, sonicating for 5 min, collection of the solid by filtration, rinsing the collected solid with methanol and drying at room temperature under reduced pressure to give a white solid (22.0 g, 90% yield).
REFERENCES
1: Watterson SH, De Lucca GV, Shi Q, Langevine CM, Liu Q, Batt DG, Beaudoin Bertrand M, Gong H, Dai J, Yip S, Li P, Sun D, Wu DR, Wang C, Zhang Y, Traeger SC, Pattoli MA, Skala S, Cheng L, Obermeier MT, Vickery R, Discenza LN, D’Arienzo CJ, Zhang Y, Heimrich E, Gillooly KM, Taylor TL, Pulicicchio C, McIntyre KW, Galella MA, Tebben AJ, Muckelbauer JK, Chang C, Rampulla R, Mathur A, Salter-Cid L, Barrish JC, Carter PH, Fura A, Burke JR, Tino JA. Discovery of 6-Fluoro-5-(R)-(3-(S)-(8-fluoro-1-methyl-2,4-dioxo-1,2-dihydroquinazolin-3(4H)-yl )-2-methylphenyl)-2-(S)-(2-hydroxypropan-2-yl)-2,3,4,9-tetrahydro-1H-carbazole-8-carboxamide (BMS-986142): A Reversible Inhibitor of Bruton’s Tyrosine Kinase (BTK) Conformationally Constrained by Two Locked Atropisomers. J Med Chem. 2016 Oct 13;59(19):9173-9200. PubMed PMID: 27583770.
(a) Watterson, S. H.; De Lucca, G. V.; Shi, Q.; Langevine, C. M.; Liu, Q.; Batt, D. G.; Bertrand, M. B.; Gong, H.; Dai, J.; Yip, S.; Li, P.; Sun, D.; Wu, D.-R.; Wang, C.; Zhang, Y.; Traeger, S. C.; Pattoli, M. A.; Skala, S.; Cheng, L.; Obermeier, M. T.; Vickery, R.; Discenza, L. N.; D’Arienzo, C. J.; Zhang, Y.; Heimrich, E.; Gillooly, K. M.; Taylor, T. L.; Pulicicchio, C.; McIntyre, K. W.; Galella, M. A.; Tebben, A. J.; Muckelbauer, J. K.; Chang, C.; Rampulla, R.; Mathur, A.; Salter-Cid, L.; Barrish, J. C.; Carter, P. H.; Fura, A.; Burke, J. R.; Tino, J. A. Discovery of 6-Fluoro-5-(R)-(3-(S)-(8-fluoro-1-methyl-2,4-dioxo-1,2-dihydroquinazolin-3(4H)-yl)-2-methylphenyl)-2-(S)-(2-hydroxypropan-2-yl)-2,3,4,9-tetrahydro-1H-carbazole-8-carboxamide (BMS-986142): A Reversible Inhibitor of Bruton’s Tyrosine Kinase (BTK) Conformationally Constrained by Two Locked Atropisomers. J. Med. Chem. 2016, 59, 9173, DOI: 10.1021/acs.jmedchem.6b01088
(b) De Lucca, G. V.; Shi, Q.; Liu, Q.; Batt, D. G.; Bertrand, M. B.; Rampulla, R.; Mathur, A.; Discenza, L.; D’Arienzo, C.; Dai, J.; Obermeier, M.; Vickery, R.; Zhang, Y.; Yang, Z.; Marathe, P.; Tebben, A. J.; Muckelbauer, J. K.; Chang, C. J.; Zhang, H.; Gillooly, K.; Taylor, T.; Pattoli, M. A.; Skala, S.; Kukral, D. W.; McIntyre, K. W.; Salter-Cid, L.; Fura, A.; Burke, J. R.; Barrish, J. C.; Carter, P. H.; Tino, J. A. Small Molecule Reversible Inhibitors of Bruton’s Tyrosine Kinase (BTK): Structure–Activity Relationships Leading to the Identification of 7-(2-Hydroxypropan-2-yl)-4-[2-methyl-3-(4-oxo-3,4-dihydroquinazolin-3-yl)phenyl]-9H-carbazole-1-carboxamide (BMS-935177). J. Med. Chem. 2016, 59, 7915, DOI: 10.1021/acs.jmedchem.6b00722
Watterson, S.H.; De Lucca, G.V.; Shi, Q.; et al.
Twisted road to the discovery of BMS-986142: Using conformationally locked atropisomers to drive potency in a reversible inhibitor of Brutonas tyrosine kinase (BTK)
255th Am Chem Soc (ACS) Natl Meet (March 18-22, New Orleans) 2018, Abst MEDI 6
Twisted road to the discovery of BMS-986142: Using conformationally locked atropisomers to drive potency in a reversible inhibitor of Brutonas tyrosine kinase (BTK)
255th Am Chem Soc (ACS) Natl Meet (March 18-22, New Orleans) 2018, Abst MEDI 6
////////////BMS-986142, BMS 986142, BMS986142, phase II, clinical development, Bristol-Myers Squibb, rheumatoid arthritis, primary Sjogren’s syndrome,
CN1C(=O)N(C(=O)c2cccc(F)c12)c3cccc(c3C)c4c(F)cc(C(=O)N)c5[nH]c6C[C@H](CCc6c45)C(C)(C)O
Linrodostat BMS 986205, ONO 7701
cas 2221034-29-1
- Linrodostat
- (2R)-N-(4-chlorophenyl)-2-(cis-4-(6-fluoroquinolin-4-yl)cyclohexyl)propanamide
- Linrodostat mesylate
- Linrodostat [USAN]
- UNII-OS7OBU191R
- OS7OBU191R
- Linrodostat mesylate [USAN]
- BMS-986205-04
- 2221034-29-1
- Cyclohexaneacetamide, N-(4-chlorophenyl)-4-(6-fluoro-4-quinolinyl)-alpha- methyl-, (alphaR,1alpha,4alpha)-, methanesulfonate (1:1)
Linrodostat; (2R)-N-(4-chlorophenyl)-2-(cis-4-(6-fluoroquinolin-4-yl)cyclohexyl)propanamide; Linrodostat mesylate; Linrodostat [USAN]; UNII-OS7OBU191R; OS7OBU191R


BMS 986205
(2R)-N-(4-Chlorophenyl)-2-[cis-4-(6-fluoro-4-quinolinyl)cyclohexyl]propanamide
Cyclohexaneacetamide, N-(4-chlorophenyl)-4-(6-fluoro-4-quinolinyl)-α-methyl-, cis-
Cyclohexaneacetamide, N-(4-chlorophenyl)-4-(6-fluoro-4-quinolinyl)-α-methyl-, cis-(αR)-
(i?)-N-(4-chlorophenyl)-2- c 5-4-(6-fluoroquinolin-4-yl)cyclohexyl)propanamide
CAS: 1923833-60-6
Phase III Head and neck cancer; Malignant melanoma
BMS-986205, ONO-7701, F- 001287
- Molecular Formula C24H24ClFN2O
- Average mass 410.912 Da
BMS986205, BMS 986205, ONO-7701
Cyclohexaneacetamide, N-(4-chlorophenyl)-4-(6-fluoro-4-quinolinyl)-α-methyl-, cis-(αR)-
A potent and selective IDO1 (indoleamine 2,3-dioxygenase 1) inhibitor.
| Alternate Name | (R)-N-(4-chlorophenyl)-2-((1s,4S)-4-(6-fluoroquinolin-4-yl)cyclohexyl)propenamide |
|---|---|
| Appearance | Crystalline solid |
| CAS # | 1923833-60-6 |
| Molecular Formula | C₂₄H₂₄ClFN₂O |
| Molecular Weight | 410.92
|
- Originator Bristol-Myers Squibb
- Developer Bristol-Myers Squibb; Ono Pharmaceutical
- Class Antineoplastics; Cyclohexanes; Quinolines; Small molecules
- Mechanism of Action Indoleamine-pyrrole 2,3-dioxygenase inhibitors
Highest Development Phases
- Phase II IHead and neck cancer; Malignant melanoma
- Phase I/II Cancer
- Phase I Solid tumours
Most Recent Events
- 01 Jun 2018Efficacy and adverse events data from a phase I/IIa trial in Bladder cancer (Combination therapy, Late-stage disease) presented at the 54th Annual Meeting of the American Society of Clinical Oncology (ASCO- 2018)
- 08 May 2018Bristol-Myers Squibb plans the CheckMate 9UT phase II trial for Bladder Cancer in USA, Canada, Italy, Mexico, Netherlands, Spain and United Kingdom , (NCT03519256)
- 30 Apr 2018Bristol-Myers Squibb withdraws a phase III trial for Non-small cell lung cancer (First-line therapy, Combination therapy, Late-stage disease) in USA, Austria, Australia, Brazil, Canada, Czech Republic, France, Germany, Greece, Italy, Japan, South Korea, Mexico, Spain, Switzerland, Taiwan and Turkey prior to enrolment (NCT03417037)
BMS , following its acquisition of Flexus Biosciences , and licensee Ono Pharmaceutical are developing linrodostat, a once-daily, indoleamine 2,3-dioxygenase 1 inhibitor for the potential oral treatment of cancer including renal cell carcinoma, muscle-invasive bladder cancer and melanoma. In October 2018, the trial was initiated in the US, Europe, Israel and Brazil.
WO2015031295 product pat
WO2016073770 first disclosed
WO2018209049
- WO 2016073770


Bristol-Myers Squibb, following its acquisition of Flexus Biosciences, is developing BMS-986205 (previously F- 001287), the lead from an immunotherapy program of indoleamine 2,3-dioxygenase 1 (IDO1) inhibitors for the potential treatment of cancer. In February 2016, a phase I/IIa trial was initiated .
BMS-986205 (ONO-7701) is being evaluated at Bristol-Myers Squibb in phase I/II clinical trials for the oral treatment of adult patients with advanced cancers in combination with nivolumab. Early clinical development is also ongoing at Ono in Japan for the treatment of hematologic cancer and for the treatment of solid tumors.
In April 2017, data from the trial were presented at the 108th AACR Annual Meeting in Washington DC. As of February 2017, the MTD had not been reached, but BMS-986205 plus nivolumab treatment was well tolerated, with only two patients discontinuing treatment due to DLTs. The most commonly reported treatment-related adverse events (TRAEs) were decreased appetite, fatigue, nausea, diarrhea, and vomiting. Grade 3 TRAEs were reported in three patients during the combination therapy; however, no grade 3 events were reported during BMS-986205 monotherapy lead-in. No grade 4 or 5 TRAEs were reported with BMS-986205 alone or in combination with nivolumab
Indoleamine 2,3-dioxygenase (IDO; also known as IDOl) is an IFN-γ target gene that plays a role in immunomodulation. IDO is an oxidoreductase and one of two enzymes that catalyze the first and rate-limiting step in the conversion of tryptophan to N-formyl-kynurenine. It exists as a 41kD monomer that is found in several cell populations, including immune cells, endothelial cells, and fibroblasts. IDO is relatively well-conserved between species, with mouse and human sharing 63% sequence identity at the amino acid level. Data derived from its crystal structure and site-directed mutagenesis show that both substrate binding and the relationship between the substrate and iron-bound dioxygenase are necessary for activity. A homolog to IDO (ID02) has been identified that shares 44% amino acid sequence homology with IDO, but its function is largely distinct from that of IDO. (See, e.g., Serafini P, et al, Semin. Cancer Biol, 16(l):53-65 (Feb. 2006) and Ball, H.J. et al, Gene, 396(1):203-213 (Jul. 2007)).
IDO plays a major role in immune regulation, and its immunosuppressive function manifests in several manners. Importantly, IDO regulates immunity at the T cell level, and a nexus exists between IDO and cytokine production. In addition, tumors frequently manipulate immune function by upregulation of IDO. Thus, modulation of IDO can have a therapeutic impact on a number of diseases, disorders and conditions.
A pathophysiological link exists between IDO and cancer. Disruption of immune homeostasis is intimately involved with tumor growth and progression, and the production of IDO in the tumor microenvironment appears to aid in tumor growth and metastasis. Moreover, increased levels of IDO activity are associated with a variety of different tumors (Brandacher, G. et al, Clin. Cancer Res., 12(4): 1144-1151 (Feb. 15, 2006)).
Treatment of cancer commonly entails surgical resection followed by chemotherapy and radiotherapy. The standard treatment regimens show highly variable degrees of long-term success because of the ability of tumor cells to essentially escape by regenerating primary tumor growth and, often more importantly, seeding distant metastasis. Recent advances in the treatment of cancer and cancer-related diseases, disorders and conditions comprise the use of combination therapy incorporating immunotherapy with more traditional chemotherapy and radiotherapy. Under most scenarios, immunotherapy is associated with less toxicity than traditional chemotherapy because it utilizes the patient’s own immune system to identify and eliminate tumor cells.
In addition to cancer, IDO has been implicated in, among other conditions, immunosuppression, chronic infections, and autoimmune diseases or disorders (e.g. , rheumatoid arthritis). Thus, suppression of tryptophan degradation by inhibition of IDO activity has tremendous therapeutic value. Moreover, inhibitors of IDO can be used to enhance T cell activation when the T cells are suppressed by pregnancy, malignancy, or a virus (e.g., HIV). Although their roles are not as well defined, IDO inhibitors may also find use in the treatment of patients with neurological or neuropsychiatric diseases or disorders (e.g., depression).
Small molecule inhibitors of IDO have been developed to treat or prevent IDO-related diseases. For example, the IDO inhibitors 1-methyl-DL-tryptophan; p-(3-benzofuranyl)-DL-alanine; p-[3-benzo(b)thienyl]-DL-alanine; and 6-nitro-L-tryptophan have been used to modulate T cell-mediated immunity by altering local extracellular concentrations of tryptophan and tryptophan metabolites (WO 99/29310). Compounds having IDO inhibitory activity are further reported in WO 2004/094409.
In view of the role played by indoleamine 2,3-dioxygenase in a diverse array of diseases, disorders and conditions, and the limitations (e.g., efficacy) of current IDO inhibitors, new IDO modulators, and compositions and methods associated therewith, are needed.
In April 2017, preclinical data were presented at the 108th AACR Annual Meeting in Washington DC. BMS-986205 inhibited kynurenine production with IC50 values of 1.7, 1.1 and > 2000 and 4.6, 6.3 and > 2000 nM in human (HeLa, HEK293 expressing human IDO-1 and tryptophan-2, 3-dioxygenase cell-based assays) and rat (M109, HEK293 expressing mouse ID0-1 and -2 cell-based assays) respectively. In human SKOV-3 xenografts (serum and tumor) AUC (0 to 24h; pharmacokinetic and pharmacodynamic [PK and PD])) was 0.8, 4.2 and 23 and 3.5, 11 and 40 microM h, respectively; area under the effect curve (PK and PD) was 39, 32 and 41 and 60, 63 and 76% kyn, at BMS-986205 (5, 25 and 125 mg/kg, qd×5), respectively
In April 2017, preclinical data were presented at the 253rd ACS National Meeting and Exhibition in San Francisco, CA. BMS-986205 showed potent and selective inhibition of IDO-1 enzyme (IC50 = 1.7nM) and potent growth inhibition in cellular assays (IC50 = 3.4 nM) in SKOV3 cells. A good pharmacokinetic profile was seen at oral and iv doses in rats, dogs and monkeys. The compound showed good oral exposure and efficacy in in vivo assays
Preclinical studies were performed to evaluate the activity of BMS-986205, a potent and selective optimized indoleamine 2, 3-dioxygenase (IDO)- 1inhibitor, for the treatment of cancer. BMS-986205 inhibited kynurenine production with IC50 values of 1.7, 1.1 and > 2000 and 4.6, 6.3 and > 2000 nM in human (HeLa, HEK293 expressing human IDO-1 and tryptophan-2, 3-dioxygenase cell-based assays) and rat (M109, HEK293 expressing mouse ID0-1 and -2 cell-based assays) respectively. BMS-986205 was also found to be potent when compared with IDO-1from other species (human < dog equivalent monkey equivalent mouse > rat). In cell-free systems, incubation of inhibitor lead to loss of heme absorbance of IDO-1 which was observed in the presence of BMS-986205 (10 microM), while did not observed with epacadostat (10 microM). The check inhibitory activity and check reversibility (24 h after compound removal) of BMS-986205 was found to be < 1 and 18% in M109 (mouse) and < 1 and 12% SKOV3 (human) cells, respectively. In human whole blood IDO-1, human DC mixed lymphocyte reaction and human T cells cocultured with SKOV3 cells- cell based assays, BMS-986205 showed potent cellular effects (inhibition of kynurenine and T-cell proliferation 3H-thymidine) with IC50 values of 2 to 42 (median 9.4 months), 1 to 7 and 15 nM, respectively. In human SKOV-3 xenografts (serum and tumor) AUC (0 to 24h; pharmacokinetic and pharmacodynamic [PK and PD])) was 0.8, 4.2 and 23 and 3.5, 11 and 40 microM h, respectively; area under the effect curve (PK and PD) was 39, 32 and 41 and 60, 63 and 76% kyn, at BMS-986205 (5, 25 and 125 mg/kg, qd×5), respectively. In vivo human-SKOV3 and hWB-xenografts, IC50 values of BMS-986205 were 3.4 and 9.4 NM, respectively. The ADME of BMS-986205 at parameters iv/po dose was 0.5/2, 0.5/1.5 and 0.5/1.2 mg/kg, respectively; iv/clearance was 27, 25 and 19 ml, min/kg, respectively; iv Vss was 3.8, 5.7 and 4.1 l/kg, respectively; t1/2 (iv) was 3.9, 4.7 and 6.6 h, respectively; fraction (po) was 64, 39 and 10%, respectively. At the time of presentation, BMS-986205 was being evaluated in combination with nivolumab.
The chemical structure and preclinical profile was presented for BMS-986205 ((2R)-N-(4-Chlorophenyl)-2-[cis-4-(6-fluoroquinolin-4-yl)cyclohexyl]propanamide), a potent IDO-1 inhibitor in phase I for the treatment of cancer. This compound showed potent and selective inhibition of IDO-1 enzyme (IC50 = 1.7nM) and potent growth inhibition in cellular assays (IC50 = 3.4 nM) in SKOV3 cells. The pharmacokinetic profile in rats dosed at 0.5 mg/kg iv and 2 mg/kg po, with clearance, Vss, half-life and bioavailability of 27 ml/min/kg, 3.8 l/kg, 3.9 h and 4%, respectively; in dogs at 0.5 iv and 1.5 po mg/kg dosing results were 25 ml/min/kg, 5.7 l/kg, 4.7 h and 39%; and, in cynomolgus monkeys with the same doses as dogs results were 19 ml/min/kg, 4.1 l/kg, 6.6 h and 10%, respectively. The compound showed good oral exposure and efficacy in in vivo assays.
BMS-986158: a BET inhibitor for cancerAshvinikumar Gavai of Bristol Myers Squibb (BMS) gave an overview of his company’s research into Bromodomian and extra-terminal domain (BET) as oncology target for transcriptional suppression of key oncogenes, such as MYC and BCL2. BET inhibition has been defined as strong rational strategy for the treatment of hematologic malignancies and solid tumors. From crystal-structure guided SAR studies, BMS-986158, 2-{3-(1,4-Dimethyl-1H-1,2,3-triazol-5-yl)-5-[(S)-(oxan-4-yl)(phenyl)methyl]-5H-pyrido[3,2-b]indol-7-yl}propan-2-ol, was chosen as a potent BET inhibitor, showing IC50 values for BRD2, BRD3 and BRD4 activity of 1 nM; it also inhibited Myc oncogene (IC50 = 0.5 nM) and induced chlorogenic cancer cell death. In vitro the compound also displayed significant cytotoxicity against cancer cells. When administered at 0.25, 0.5 and 1 mg/kg po, qd to mice bearing human lung H187 SCLC cancer xenograft, BMS-986158 was robust and showed efficacy as a anticancer agent at low doses. In metabolic studies, it showed t1/2 of 36, 40 and 24 min in human, rat and mice, respectively, and it gave an efflux ratio of 3 in Caco-2 permeability assay. In phase 1/II studies, BMS-986158 was well tolerated at efficacious doses and regimens, and drug tolerable toxicity at efficacy doses and regimens. Selective Itk inhibitors for inflammatory disordersThe development of highly selective Itk inhibitors for the treatment of diseases related to T-cell function, such as inflammatory disorders, was described by Shigeyuki Takai (Ono Pharmaceutical). Inhibitory properties of a hit compound, ONO-8810443, were modified via X-ray structure and Molecular Dynamics stimulation to get ONO-212049 with significant kinase selectivity (140-fold) against Lck, a tyrosine kinase operating upstream of Itk in the TCR cascade. Further modifications identified final lead compound ONO-7790500 (N-[6-[3-amino-6-[2-(3-methoxyazetidin-1-yl)pyridin-4-yl]pyrazin-2-yl]pyridin-3-yl]-1-(3-methoxyphenyl)-2,3-dimethyl-5-oxopyrazole-4-carboxamide), which selectively inhibited Itk (IC50 = < 0.004 microM) over Lck (IC50 = 9.1 microM; SI 2000-fold) and suppressed Jurkat T-cell proliferation (IC50 = 0.014 microM). This compound suppressed alphaCD3/CDP28 CD4+T-cell stimulation (IC50 = 0.074 microM) with selectivity over PMA/Ionomycin (IC50 = > 10 microM). ONO-7790500 also exhibited in vivo IL-2 inhibitory properties (62% inhibition at 30 mg/kg po) in mice. In pharmacokinetic studies in balb/c mice, the compound administered orally (10 mg/kg) showed a Cmax of 1420 ng/ml, AUClast of 11,700 ng*h/ml, t1/2 of 5.3 h and oral bioavailability of 68%. Administration iv at 0.3 mg/kg gave an AUC last of 610 ng*h/ml, t1/2 of 3.8 h, Vss of 1260 ml/kg and Cl of 5.1 ml/min/kg. ADMET data showed ONO-7790500 did not have relevant activity in cytochromes and hERG channels (IC50 > 10 microM) in toxicological studies, and gave a PAMPA value of 5.0 x 10(-6) cm/s. Fused imidazole and pyrazole derivatives as TGF-beta inhibitorsDual growth and differentiation factor-8 (GDF-8; also known as myostatin) and TGF-beta inhibitors were described. Both targets belong to TGF-beta superfamily consisting of a large group of structurally related cell regulatory proteins involved in fundamental biological and pathological processes, such as cell proliferation or immunomodulation. Myostatin (GDF8) is a negative regulator negative regulator of skeletal muscle growth and has also been related to bone metabolism. Investigators at Rigel Pharmaceuticals found that compounds designed to be GDF-8 inhibitors were able to inhibit TGF-beta as well, this could be an advantage for the treatment of diseases associated with muscle and adipose tissue disorders, as well as potentially immunosuppressive disorders. Jiaxin Yu from the company described new fused imidazole derivatives, of which the best compound was 6-[2-(2,4,5-Trifluorophenyl)-6,7-dihydro-5H-pyrrolo[1,2-a]imidazol-3-yl]quinoxaline. This compound was very potent at TGF-beta Receptor Type-1 (ALK5) inhibition with an IC50 value of 1nM. In an in vivo mouse assay this compound showed good activity at 59.7 mg/kg, po, and good plasma exposure; inhibition of GDF-8 and TGFbeta growth factors was 90 and 81.6 %, respectively.Rigel’s Ihab Darwish described a series of fused pyrazole derivatives, with the best compound being 6-[2-(2,4-Difluorophenyl)-5,6-dihydro-4H-pyrrolo[1,2-b]pyrazol-3-yl][1,2,4]triazolo[1,5-a]pyridine. This compound showed an IC50 of 0.06 and 0.23 microM for GDF-8 and TGFbeta, respectively, in the pSMAD (MPC-11) signaling inhibition test. The compound had a good pharmacokinetic profile, with 40% of bioavailability in mice after a 5-mg/kg po dose. An iv dose of 1 mg/kg showed t1/2 of 0.7 h and Vss of 1.0 l/h/kgDiscovery of selective inhibitor of IDO BMS-986205 for cancerIndoleamine-2,3-dioxygenase (IDO)-1 enzyme initiates and regulates the first step of the kynurenine pathway (KP) of tryptophan metabolism, and evidence has shown that overexpression of IDO-1 in cancer tumors is a crucial mechanism facilitating tumor immune evasion and persistence. The chemical structure and preclinical profile of BMS-986205 was presented by Aaron Balog from BMS. BMS-986205 ((2R)-N-(4-Chlorophenyl)-2-[cis-4-(6-fluoroquinolin-4-yl)cyclohexyl]propanamide), is a potent IDO-1 inhibitor in phase I for the treatment of cancer. This compound showed potent and selective inhibition of IDO-1 enzyme (IC50 = 1.7nM) and potent growth inhibition in cellular assays (IC50 = 3.4 nM) in SKOV3 cells. The pharmacokinetic profile in rats dosed at 0.5 mg/kg iv and 2 mg/kg po, with clearance, Vss, half-life and bioavailability of 27 ml/min/kg, 3.8 l/kg, 3.9 h and 4%, respectively; in dogs at 0.5 iv and 1.5 po mg/kg dosing results were 25 ml/min/kg, 5.7 l/kg, 4.7 h and 39%; and, in cynomolgus monkeys with the same doses as dogs results were 19 ml/min/kg, 4.1 l/kg, 6.6 h and 10%, respectively. The compound showed good oral exposure and efficacy in in vivo assays.Three further reports have been published from this meeting .The website for this meeting can be found at https://www.acs.org/content/acs/en/meetings/spring-2017.html.
SYNTHESIS
1 Wittig NaH
2 REDUCTION H2, Pd, AcOEt, 4 h, rt, 50 psi
3 Hydrolysis HCl, H2O, Me2CO, 2 h, reflux
4 4-Me-2,6-(t-Bu)2-Py, CH2Cl2, overnight, rt
5 SUZUKI AcOK, 72287-26-4, Dioxane, 16 h, 80°C
6 Heck Reaction, Suzuki Coupling, Hydrogenolysis of Carboxylic Esters, Reduction of Bonds, HYDROGEN
7 Et3N, THF, rt – -78°C , Pivaloyl chloride, 15 min, -78°C; 1 h, 0°C ,THF, 0°C – -78°C, BuLi, Me(CH2)4Me, 15 min, -78°C, R:(Me3Si)2NH •Na, THF, 10 min, -50°C , HYDROLYSIS, (PrP(=O)O)3, C5H5N, AcOEt, 5 min, rt

Product Patent
WO2016073770
Example 19
(i?)-N-(4-chlorophenyl)-2- c 5-4-(6-fluoroquinolin-4-yl)cyclohexyl)propanamide
Example 19 : (i?)-N-(4-chlorophenyl)-2-(cz5-4-(6-fluoroquinolin-4- yl)cyclohexyl)propanamide
[0277] Prepared using General Procedures K, B, E, L, M, N, and O. General Procedure L employed 2-(4-(6-fluoroquinolin-4-yl)-cyclohexyl)acetic acid (mixture of
diastereomers), and ( ?)-2-phenyl-oxazolidinone. General Procedure M employed the cis product and iodomethane. The auxiliary was removed following General Procedure N and the desired product formed employing General Procedure O with 4-chloroaniline.
Purified using silica gel chromatography (0% to 100% ethyl acetate in hexanes) to afford Example 19. 1H NMR of czs-isomer (400 MHz; CDC13): δ 9.14 (s, 1H), 8.70 (d, J= 4.6 Hz, 1H), 8.06 (dd, J= 9.2 Hz, J= 5.6 Hz, 1H), 7.58-7.64 (m, 3H), 7.45 (ddd, J= 9.3 Hz, J= 7.8 Hz, J= 2.7 Hz, 1H), 7.19-7.24 (m, 2H), 7.15 (d, J= 4.6Hz, 1H), 3.16-3.26 (m, 1H), 2.59-2.69 (m, 1H), 2.08-2.16 (m, 1H), 1.66-1.86 (m, 7H), 1.31-1.42 (m, 1H), 1.21 (d, J= 6.8Hz, 3H) ppm. m/z 411.2 (M+H)+.
PAPER
Bioorganic & Medicinal Chemistry Letters (2018), 28(3), 319-329.
https://www.sciencedirect.com/science/article/pii/S0960894X17312180

PATENT
WO 2018022992
PATENT
WO 2018071500
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2018071500&redirectedID=true
WO-2019006292
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019006292&tab=PCTDESCRIPTION&maxRec=1000
Improved methods for the preparation of substituted quinolinycyclohexylpropanamide compounds, such as linrodostat claiming substituted pyridine compounds as IDO1 inhibitors, useful for treating cancers.
Indoleamine 2,3 -di oxygenase (IDO; also known as IDOl) is an IFN-γ target gene that plays a role in immunomodulation. IDO plays a major role in immune regulation, and its immunosuppressive function manifests in several manners. A pathophysiological link exists between IDO and cancer. Disruption of immune homeostasis is intimately involved with tumor growth and progression, and the production of IDO in the tumor microenvironment appears to aid in tumor growth and metastasis. Moreover, increased levels of IDO activity are associated with a variety of different tumors (Brandacher, G. et al, Clin. Cancer Res. , 12(4): 1144-1151 (Feb. 15, 2006)). In addition to cancer, IDO has been implicated in, among other conditions, immunosuppression, chronic infections, and autoimmune diseases or disorders (e.g., rheumatoid arthritis).
Substituted quinolinylcyclohexylpropanamide pharmaceutical compounds that inhibit IDO and are useful for the treatment of cancer have been previously described. See, e.g., WO2016/073770. Improved methods of making such compounds, which reduce production costs and improve production safety, are, therefore, needed.
Scheme 4
[0076] The disclosure is also directed to methods of preparing intermediate compounds of formula IV. Methods to produce compounds of formula IV are depicted in Schemes 5 and 6.
Scheme 5
IX-A
Scheme 6
IX-B IV
Compounds of the disclosure that include one or more radioisotopes can be used in imaging. See, e.g., WO2018017529. For example, radiolabeled compounds of the disclosure can be used in Positron Emission Tomography (PET). Such methods are useful in the imaging of cancer in a subject. A preferred radiolabeled compound is
1
Pharmaceutically acceptable salts of [18F]-Compound 1 are also within the scope of the disclosure. An exemplary method for the preparation of [18F]-Compound 1 is depicted in Scheme below.
1 . reaction
[18F]-Compound 1
Example 9
(R)-N-(4-chlorophenyl)-2-((ls,4S)-4-(6-fluoroquinolin-4-yl)cyclohexyl)propanamide
[00258] To a 10 L glass-lined reactor under a blanket of nitrogen was charged 349 g Ν,Ν,Ν’,Ν’-tetramethylchloroformamidinium hexafluorophosphate (TCFH) and 2 L acetonitrile. 245 g N-methylimidazole was added followed by 0.3 L acetonitrile. 300 g (R)-2-((ls,4S)-4-(6-fluoroquinolin-4-yl)cyclohexyl)propanoic acid was added followed by 0.3 L acetonitrile. The mixture was held for 0.5 h then 139 g 4-chloroaniline charged followed by 0.4 L acetonitrile. The mixture was maintained at 20 °C until the reaction was deemed complete by HPLC analysis. The solution was then heated to 60°C, and 1.2 L water was charged. The solution was then cooled to 40 °C, seeds (3 g) were charged, and the resulting slurry was maintained for 1 h. The slurry was then cooled to 20 °C and 2.7 L water was charged. The slurry was filtered and the cake was washed three times with 3 L of 2: 1 water: acetonitrile. The cake was dissolved with 5.1 L ethyl acetate and the solution was distilled to a volume of 4.2 L at 41 °C under vacuum. The slurry was cooled to 20 °C, 4.14 g seeds were charged, and a solution of 95.7 g methanesulfonic acid in 2.9 L ethyl acetate was added. The slurry was then filtered and washed two times with 1.65 L ethyl acetate and dried under vacuum at 50°C to yield 445 g of (R)-N-(4-chlorophenyl)-2-((l s,4S)-4-(6-fluoroquinolin-4-yl)cyclohexyl)propanamide methanesulfonate as a white solid in 88% yield.
[00259] ¾ NMR (600 MHz, DMSO-de) δ 10.19 (s, IH), 9.24 (d, J=5.7 Hz, IH), 8.40 (dd, J=10.3, 2.6 Hz, IH), 8.33 (dd, J=9A, 5.3 Hz, IH), 8.09 (d, J=5.7 Hz, IH), 8.04 (t, J=8.6 Hz, IH), 7.71 – 7.64 (m, 2H), 7.37 – 7.30 (m, 2H), 3.64 (ddt, J=10.8, 7.3, 3.8 Hz, IH), 2.98 – 2.89 (m, IH), 2.43 (s, 3H), 2.05 – 1.60 (m, 9H), 1.14 (d, J=6.7 Hz, 3H); 13C NMR (126 MHz, DMSO-de) δ 175.0, 162.7, 161.1 , 145.4, 138.2, 136.8, 128.6, 128.1 , 126.7, 126.4, 123.3, 120.8, 119.8, 109.0, 39.8, 39.7, 38.6, 35.5, 28.3, 27.6, 27.2, 26.1 , 16.2 MS (ESI): calcd for C24H24CIFN2O
([M + H]+), 410.16; found, 410.15.
[00260] HPLC analysis: Column: Sigma-Aldrich Supelco Ascentis Express CI 8 2.7um, 150 x 4.6 mm ID; Solvent A: 0.05% TFA with MeCN:water (5/95 v/v); Solvent B: 0.05% TFA with MeCN: water (95/5 v/v); Gradient: %B: 0 Min. 15%; 1 Min. 15%; 13 Min. 55%; 19 Min. 65%; 24 Min. 100%; 24.1 15%; 28 Min. 15%; Stop Time: 24 Min; Flow Rate: 1.0 ml/min;
Column temperature: 30 °C; wavelength: 218 nm. The retention time (R)-N-(4-chlorophenyl)-2-((ls,4S)-4-(6-fluoroquinolin-4-yl)cyclohexyl)propanamide peak was 12.6 min.
Example 7
(R)-N-(4-chlorophenyl)-2-((ls, -4-(6-fluoroquinolin-4-yl)cyclohexyl)propanamide
[00252] To a 50 L glass-lined reactor under a blanket of nitrogen was charged 13.75 kg acetonitrile, then 2.68 Kg Ν,Ν,Ν’,Ν’-tetramethylchloroformamidinium hexafluorophosphate (TCFH) and rinsed with 2.0 Kg acetonitrile. 2.03 Kg N-methylimidazole was added followed by 1.95 Kg acetonitrile. 2.48 Kg (R)-2-((ls,4S)-4-(6-fluoroquinolin-4-yl)cyclohexyl)propanoic acid was added followed by 1.05 Kg acetonitrile. The mixture was held for 0.5 h then 1.21 Kg 4-chloroaniline charged followed by 1.0 Kg acetonitrile. The mixture was maintained at 20 °C until the reaction was deemed complete by HPLC analysis. The solution was then heated to 60°C, and 9.25 Kg water was charged. The solution was then cooled to 40 °C, the mixture was aged
for 1 h, seeds (32 g) were charged and rinsed with 1.15 Kg 2: 1 water: acetonitrile, and the resulting slurry was maintained for 1 h. The slurry was then cooled to 20 °C and 25.75 Kg water was charged. The slurry was filtered and the cake was washed three times with 6.9 Kg of 2: 1 water: acetonitrile. The cake was dried under vacuum at 50°C to yield 3.33 Kg of (R)-N-(4-chlorophenyl)-2-((ls,4S)-4-(6-fluoroquinolin-4-yl)cyclohexyl)propanamide hydrate as a white solid in 94.1% yield.
[00253] ¾ NMR (600 MHz, DMSO-de) δ 10.09 (s, 1H), 8.86 (d, J=4.5 Hz, 1H), 8.08 (dd, J=9.0, 5.6 Hz, 1H), 7.95 (dd, J=10.9, 2.6 Hz, 1H), 7.70 – 7.60 (m, 3H), 7.54 (d, J=4.5 Hz, 1H), 7.33 (d, J=9.0 Hz, 2H), 3.43 – 3.31 (m, 3H), 2.90 – 2.80 (m, 1H), 1.99 – 1.55 (m, 9H), 1.13 (d, J=6.8 Hz, 3H); 13C NMR (151 MHz, DMSO-de) δ 175.0, 159.9, 152.4, 149.7, 145.2, 138.1, 132.7, 128.5, 127.2, 126.7, 120.8, 119.0, 118.6, 107.2, 40.2, 37.4, 35.6, 28.5, 27.6, 27.4, 26.3, 16.1 ; HRMS (ESI); calcd for C24H24CIFN2O ([M + H]+), 411.1619; found 411.1649.
WO-2019006283
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019006283&redirectedID=true
Novel crystalline forms of linrodostat , its salts and hydrates, designated as Forms 1, 2 and 4 (first disclosed in WO2016073770 ), processes for their preparation and compositions comprising them are claimed. Also claims are their use for treating prostate cancer, liver cancer, brain cancer, bladder cancer, ovary cancer and breast cancer.
(R)-N-(4-chlorophenyl)-2-((l S,4S)-4-(6-fluoroquinolin-4-yl)cyclohexyl)propanami the below structure:
[0003] Compound 1 is a potent inhibitor of indoleamine 2,3-dioxygenase (IDO; also known as IDOl), which is an IFN-γ target gene that plays a role in immunomodulation.
Compound 1 is being investigated as a treatment for cancer and other diseases. Compound 1 has been previously described in WO2016/073770.
[0004] A compound, as a free base, hydrate, solvate, or salt, can exist in amorphous form and/or one or more crystalline forms, each having different physical properties, for example, different X-ray diffraction patterns (XRPD or PXRD) and different thermal behavior. The free base, hydrate, solvate, and salt forms of a compound can also differ with respect to their individual stabilities, processing, formulation, dissolution profile, bioavailability, and the like. [0005] New forms of (R)-N-(4-chlorophenyl)-2-((l S,4S)-4-(6-fluoroquinolin-4-yl)cyclohexyl)propanamide, having desirable and beneficial chemical and physical properties are needed. There is also a need for reliable and reproducible methods for the manufacture, purification, and formulation of Compound 1 (and its hydrates, solvates, salt,, and hydrated salt forms) to facilitate commercialization. The present disclosure is directed to these, as well as other important aspects.
REFERENCES
23-Feb-2015
Bristol-Myers Squibb To Expand Its Immuno-Oncology Pipeline with Agreement to Acquire Flexus Biosciences, Inc
Bristol-Myers Squibb Co; Flexus Biosciences Inc
17-Dec-2014
Flexus Biosciences, a Cancer Immunotherapy Company Focused on Agents for the Reversal of Tumor Immunosuppression (ARTIS), Announces $38M Financing
Flexus Biosciences Inc
2015106thApril 21Abs 4290
Potent and selective next generation inhibitors of indoleamine-2,3-dioxygenase (IDO1) for the treatment of cancer
American Association for Cancer Research Annual Meeting
Jay P. Powers, Matthew J. Walters, Rajkumar Noubade, Stephen W. Young, Lisa Marshall, Jan Melom, Adam Park, Nick Shah, Pia Bjork, Jordan S. Fridman, Hilary P. Beck, David Chian, Jenny V. McKinnell, Maksim Osipov, Maureen K. Reilly, Hunter P. Shunatona, James R. Walker, Mikhail Zibinsky, Juan C. Jaen
2017108thApril 04Abs 4964
Structure, in vitro biology and in vivo pharmacodynamic characterization of a novel clinical IDO1 inhibitor
American Association for Cancer Research Annual Meeting
John T Hunt, Aaron Balog, Christine Huang, Tai-An Lin, Tai-An Lin, Derrick Maley, Johnni Gullo-Brown, Jesse Swanson, Jennifer Brown
2017253rdApril 05Abs MEDI 368
Discovery of a selective inhibitor of indoleamine-2,3-dioxygenase for use in the therapy of cancer
American Chemical Society National Meeting and Exposition
Aaron Balog
April 2-62017
American Chemical Society – 253rd National Meeting and Exhibition (Part IV) – OVERNIGHT REPORT, San Francisco, CA, USA
Casellas J, Carceller V
////////////////PHASE 1, BMS 986205, 1923833-60-6, BMS-986205, ONO-7701,Bristol-Myers Squibb, Antineoplastics, F- 001287
C[C@H]([C@H]1CC[C@@H](C2=CC=NC3=CC=C(F)C=C23)CC1)C(NC4=CC=C(Cl)C=C4)=O
Wrapping up #MEDI‘s 1st time disclosures is Aaron Balog of @bmsnews talking about an IOD-1 inhibitor to treat cancer #ACSSanFran
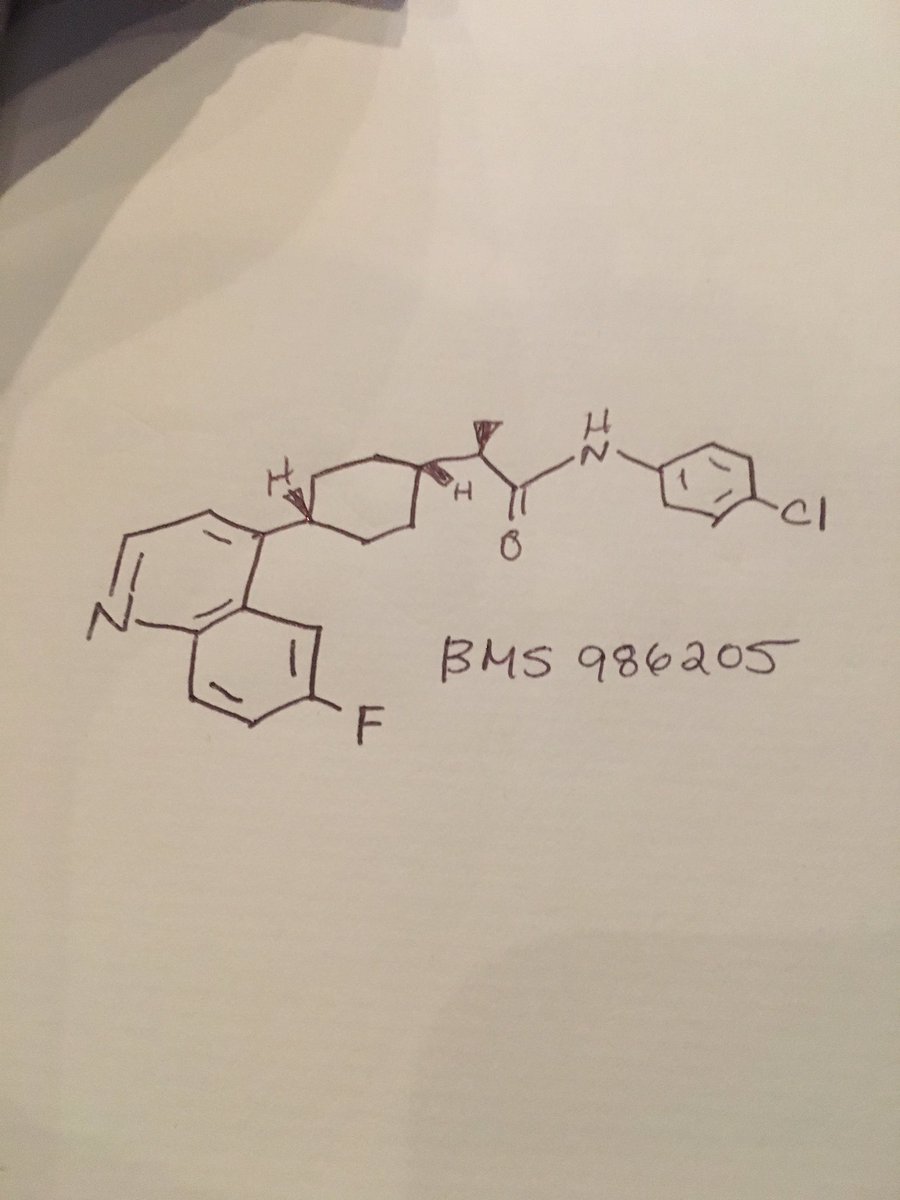
////////////////BMS986205, BMS 986205, BM-986205, ONO-7701, Phase III, Head and neck cancer, Malignant melanoma, 1923833-60-6, Linrodostat
CC(C1CCC(CC1)C2=C3C=C(C=CC3=NC=C2)F)C(=O)NC4=CC=C(C=C4)Cl
BMS-986020

BMS-986020
AM-152; BMS-986020; BMS-986202
cas 1257213-50-5
Chemical Formula: C29H26N2O5
Molecular Weight: 482.536
(R)-1-(4′-(3-methyl-4-(((1-phenylethoxy)carbonyl)amino)isoxazol-5-yl)-[1,1′-biphenyl]-4-yl)cyclopropane-1-carboxylic acid
Cyclopropanecarboxylic acid, 1-(4′-(3-methyl-4-((((1R)-1-phenylethoxy)carbonyl)amino)-5-isoxazolyl)(1,1′-biphenyl)-4-yl)-
1-(4′-(3-Methyl-4-(((((R)-1-phenylethyl)oxy)carbonyl)amino)isoxazol-5-yl)biphenyl-4-yl)cyclopropanecarboxylic acid
UNII: 38CTP01B4L
For treatment for pulmonary fibrosis, phase 2, The lysophosphatidic acid receptor, LPA1, has been implicated as a therapeutic target for fibrotic disorders
Lysophospholipids (LPs), including lysophosphatidic acid (LPA), sphingosine 1-phospate (S1P), lysophosphatidylinositol (LPI), and lysophosphatidylserine (LysoPS), are bioactive lipids that transduce signals through their specific cell-surface G protein-coupled receptors, LPA1-6, S1P1-5, LPI1, and LysoPS1-3, respectively. These LPs and their receptors have been implicated in both physiological and pathophysiological processes such as autoimmune diseases, neurodegenerative diseases, fibrosis, pain, cancer, inflammation, metabolic syndrome, bone formation, fertility, organismal development, and other effects on most organ systems.

- Originator Amira Pharmaceuticals
- DeveloperB ristol-Myers Squibb; Duke University
- Class Antifibrotics; Azabicyclo compounds; Carboxylic acids; Small molecules; Tetrazoles
- Mechanism of Action Lysophosphatidic acid receptor antagonists
- Orphan Drug Status Yes – Fibrosis
- Phase II Idiopathic pulmonary fibrosis
- Phase IPsoriasis
Most Recent Events
- 05 May 2016 Bristol-Myers Squibb plans a phase I trial for Psoriasis in Australia (PO, Capsule, Liquid) (NCT02763969)
- 01 May 2016 Preclinical trials in Psoriasis in USA (PO) before May 2016
- 14 Mar 2016 Bristol-Myers Squibb withdraws a phase II trial for Systemic scleroderma in USA, Canada, Poland and United Kingdom (PO) (NCT02588625)
BMS-986020, also known as AM152 and AP-3152 free acid, is a potent and selective LPA1 antagonist. BMS-986020 is in Phase 2 clinical development for treating idiopathic pulmonary fibrosis. BMS-986020 selectively inhibits the LPA receptor, which is involved in binding of the signaling molecule lysophosphatidic acid, which in turn is involved in a host of diverse biological functions like cell proliferation, platelet aggregation, smooth muscle contraction, chemotaxis, and tumor cell invasion, among others
![]()



PRODUCT PATENT
GB 2470833, US 20100311799, WO 2010141761
Hutchinson, John Howard; Seiders, Thomas Jon; Wang, Bowei; Arruda, Jeannie M.; Roppe, Jeffrey Roger; Parr, Timothy
Assignee: Amira Pharmaceuticals Inc, USA

John Hutchinson
PATENTS
WO 2011159632
WO 2011159635
PATENT
WO 2013025733
Synthesis of Compound 74
Synthetic Route (Scheme XLV)

Compound 74 Compound 74a
[0562] Compound XLV-1 was prepared by the same method as described in the synthesis of compound 1-4 (Scheme 1-A).
[0563] To a solution of compound XLV-1 (8 g, 28.08 mmol) in dry toluene (150 mL) was added compound XLV-2 (1.58 g, 10.1 mmol), triethylamine (8.0 mL) and DPPA (9.2 g, 33.6 mmol). The reaction mixture was heated to 80 °C for 3 hours. The mixture was diluted with EtOAc (50 mL), washed with brine, dried over Na2S04, filtered and concentrated. The residue was purified by column chromatography (PE/EA = 10 IX) to give compound XLV-3 (9.4 g, yield: 83 %). MS (ESI) m/z (M+H)+402.0.
[0564] Compound 74 was prepared analogously to the procedure described in the synthesis of Compound 28 and was carried through without further characterization.
[0565] Compound 74a was prepared analogously to the procedure described in the synthesis of Compound 44a. Compound 74a: 1HNMR (DMSO-d6 400MHz) δ 7.81 (d, J = 8.4 Hz, 2H), 7.41 (d, J = 8.4 Hz, 2H), 7.52 (d, J = 8.4 Hz, 2H), 7.29-7.32 (m, 7 H), 5.78 (q, 1 H), 2.15 (s, 3 H), 1.52 (d, J = 6.0 Hz, 3H), 1.28 (br, 2 H), 0.74 (br, 2 H). MS (ESI) m/z (M+H)+ 483.1.


Paper
Development of a Concise Multikilogram Synthesis of LPA-1 Antagonist BMS-986020 via a Tandem Borylation–Suzuki Procedure
Michael J. Smith*  , Michael J. Lawler, Nathaniel Kopp, Douglas D. Mcleod, Akin H. Davulcu, Dong Lin, Kishta Katipally , and Chris Sfouggatakis
, Michael J. Lawler, Nathaniel Kopp, Douglas D. Mcleod, Akin H. Davulcu, Dong Lin, Kishta Katipally , and Chris Sfouggatakis
, Michael J. Lawler, Nathaniel Kopp, Douglas D. Mcleod, Akin H. Davulcu, Dong Lin, Kishta Katipally , and Chris SfouggatakisChemical and Synthetic Development, Bristol-Myers Squibb Company, One Squibb Drive, New Brunswick, New Jersey 08903, United States
Org. Process Res. Dev., Article ASAP
DOI: 10.1021/acs.oprd.7b00301
http://pubs.acs.org/doi/10.1021/acs.oprd.7b00301
*E-mail: michael.smith2@bms.com.

The process development for the synthesis of BMS-986020 (1) via a palladium catalyzed tandem borylation/Suzuki reaction is described. Evaluation of conditions culminated in an efficient borylation procedure using tetrahydroxydiboron followed by a tandem Suzuki reaction employing the same commercially available palladium catalyst for both steps. This methodology addressed shortcomings of early synthetic routes and was ultimately used for the multikilogram scale synthesis of the active pharmaceutical ingredient 1. Further evaluation of the borylation reaction showed useful reactivity with a range of substituted aryl bromides and iodides as coupling partners. These findings represent a practical, efficient, mild, and scalable method for borylation.
1H NMR (500 MHz, DMSO-d6) δ 1.19 (dd, J = 6.8, 3.8 Hz, 2H), 1.50 (dd, J = 6.8, 3.8 Hz, 2H), 1.56 (br s, 3H), 2.14 (br s, 3H), 5.78 (br s, 1H), 6.9–7.45 (br, 5H), 7.45 (br d, J = 8.3 Hz, 2H), 7.65 (d, J = 8.3 Hz, 2H), 7.79 (br d, 2H), 7.82 (br d, 2H), 8.87 (br s, 0.8H), 9.29 (s, 0.2H), 12.39 (br s, 1H). 13C NMR (126 MHz, DMSO-d6) δ 9.2, 15.8, 22.4, 28.3, 72.8, 113.8, 125.4, 125.6, 126.2, 126.3, 127.1, 127.7, 128.4, 130.9, 137.4, 140.0, 141.5, 142.2, 154.4, 159.6, 160.8, 175.2. HRMS (ESI+) Calculated M + H 483.19145, found 483.19095.
REFERENCES
1: Kihara Y, Mizuno H, Chun J. Lysophospholipid receptors in drug discovery. Exp
Cell Res. 2015 May 1;333(2):171-7. doi: 10.1016/j.yexcr.2014.11.020. Epub 2014
Dec 8. Review. PubMed PMID: 25499971; PubMed Central PMCID: PMC4408218.
//////////////BMS-986020, AM 152, BMS 986020, BMS 986202, Orphan Drug, BMS, Amira Pharmaceuticals, Bristol-Myers Squibb, Duke University, Antifibrotics, PHASE 2, pulmonary fibrosis
O=C(C1(C2=CC=C(C3=CC=C(C4=C(NC(O[C@H](C)C5=CC=CC=C5)=O)C(C)=NO4)C=C3)C=C2)CC1)O
BMS 986205


BMS 986205
(2R)-N-(4-Chlorophenyl)-2-[cis-4-(6-fluoro-4-quinolinyl)cyclohexyl]propanamide
Cyclohexaneacetamide, N-(4-chlorophenyl)-4-(6-fluoro-4-quinolinyl)-α-methyl-, cis-
Cyclohexaneacetamide, N-(4-chlorophenyl)-4-(6-fluoro-4-quinolinyl)-α-methyl-, cis-(αR)-
(i?)-N-(4-chlorophenyl)-2- c 5-4-(6-fluoroquinolin-4-yl)cyclohexyl)propanamide
CAS: 1923833-60-6
Phase 1 cancer
BMS-986205, ONO-7701, F- 001287
- Molecular Formula C24H24ClFN2O
- Average mass 410.912 Da
- Originator Bristol-Myers Squibb
- Class Antineoplastics
- 01 Feb 2016 Phase-I/II clinical trials in Cancer (Combination therapy, Late-stage disease, Second-line therapy or greater) in Canada (PO) (NCT02658890)
- 31 Jan 2016 Preclinical trials in Cancer in USA (PO) before January 2016
- 01 Jan 2016 Bristol-Myers Squibb plans a phase I/IIa trial for Cancer (Late-stage disease, Combination therapy, Second-line therapy or greater) in USA, Australia and Canada (PO) (NCT02658890)

Hilary Beck
 EX Principal Investigator, Company NameFLX Bio, Inc.,
EX Principal Investigator, Company NameFLX Bio, Inc.,
CURRENTLY Director, Medicinal Chemistry at IDEAYA Biosciences, IDEAYA Biosciences, The University of Texas at Austin
![]()

Brian Wong
Chief Executive Officer at FLX Bio, Inc.
Bristol-Myers Squibb, following its acquisition of Flexus Biosciences, is developing BMS-986205 (previously F- 001287), the lead from an immunotherapy program of indoleamine 2,3-dioxygenase 1 (IDO1) inhibitors for the potential treatment of cancer. In February 2016, a phase I/IIa trial was initiated .
BMS-986205 (ONO-7701) is being evaluated at Bristol-Myers Squibb in phase I/II clinical trials for the oral treatment of adult patients with advanced cancers in combination with nivolumab. Early clinical development is also ongoing at Ono in Japan for the treatment of hematologic cancer and for the treatment of solid tumors.
In April 2017, data from the trial were presented at the 108th AACR Annual Meeting in Washington DC. As of February 2017, the MTD had not been reached, but BMS-986205 plus nivolumab treatment was well tolerated, with only two patients discontinuing treatment due to DLTs. The most commonly reported treatment-related adverse events (TRAEs) were decreased appetite, fatigue, nausea, diarrhea, and vomiting. Grade 3 TRAEs were reported in three patients during the combination therapy; however, no grade 3 events were reported during BMS-986205 monotherapy lead-in. No grade 4 or 5 TRAEs were reported with BMS-986205 alone or in combination with nivolumab
Indoleamine 2,3-dioxygenase (IDO; also known as IDOl) is an IFN-γ target gene that plays a role in immunomodulation. IDO is an oxidoreductase and one of two enzymes that catalyze the first and rate-limiting step in the conversion of tryptophan to N-formyl-kynurenine. It exists as a 41kD monomer that is found in several cell populations, including immune cells, endothelial cells, and fibroblasts. IDO is relatively well-conserved between species, with mouse and human sharing 63% sequence identity at the amino acid level. Data derived from its crystal structure and site-directed mutagenesis show that both substrate binding and the relationship between the substrate and iron-bound dioxygenase are necessary for activity. A homolog to IDO (ID02) has been identified that shares 44% amino acid sequence homology with IDO, but its function is largely distinct from that of IDO. (See, e.g., Serafini P, et al, Semin. Cancer Biol, 16(l):53-65 (Feb. 2006) and Ball, H.J. et al, Gene, 396(1):203-213 (Jul. 2007)).
IDO plays a major role in immune regulation, and its immunosuppressive function manifests in several manners. Importantly, IDO regulates immunity at the T cell level, and a nexus exists between IDO and cytokine production. In addition, tumors frequently manipulate immune function by upregulation of IDO. Thus, modulation of IDO can have a therapeutic impact on a number of diseases, disorders and conditions.
A pathophysiological link exists between IDO and cancer. Disruption of immune homeostasis is intimately involved with tumor growth and progression, and the production of IDO in the tumor microenvironment appears to aid in tumor growth and metastasis. Moreover, increased levels of IDO activity are associated with a variety of different tumors (Brandacher, G. et al, Clin. Cancer Res., 12(4): 1144-1151 (Feb. 15, 2006)).
Treatment of cancer commonly entails surgical resection followed by chemotherapy and radiotherapy. The standard treatment regimens show highly variable degrees of long-term success because of the ability of tumor cells to essentially escape by regenerating primary tumor growth and, often more importantly, seeding distant metastasis. Recent advances in the treatment of cancer and cancer-related diseases, disorders and conditions comprise the use of combination therapy incorporating immunotherapy with more traditional chemotherapy and radiotherapy. Under most scenarios, immunotherapy is associated with less toxicity than traditional chemotherapy because it utilizes the patient’s own immune system to identify and eliminate tumor cells.
In addition to cancer, IDO has been implicated in, among other conditions, immunosuppression, chronic infections, and autoimmune diseases or disorders (e.g. , rheumatoid arthritis). Thus, suppression of tryptophan degradation by inhibition of IDO activity has tremendous therapeutic value. Moreover, inhibitors of IDO can be used to enhance T cell activation when the T cells are suppressed by pregnancy, malignancy, or a virus (e.g., HIV). Although their roles are not as well defined, IDO inhibitors may also find use in the treatment of patients with neurological or neuropsychiatric diseases or disorders (e.g., depression).
Small molecule inhibitors of IDO have been developed to treat or prevent IDO-related diseases. For example, the IDO inhibitors 1-methyl-DL-tryptophan; p-(3-benzofuranyl)-DL-alanine; p-[3-benzo(b)thienyl]-DL-alanine; and 6-nitro-L-tryptophan have been used to modulate T cell-mediated immunity by altering local extracellular concentrations of tryptophan and tryptophan metabolites (WO 99/29310). Compounds having IDO inhibitory activity are further reported in WO 2004/094409.
In view of the role played by indoleamine 2,3-dioxygenase in a diverse array of diseases, disorders and conditions, and the limitations (e.g., efficacy) of current IDO inhibitors, new IDO modulators, and compositions and methods associated therewith, are needed.
In April 2017, preclinical data were presented at the 108th AACR Annual Meeting in Washington DC. BMS-986205 inhibited kynurenine production with IC50 values of 1.7, 1.1 and > 2000 and 4.6, 6.3 and > 2000 nM in human (HeLa, HEK293 expressing human IDO-1 and tryptophan-2, 3-dioxygenase cell-based assays) and rat (M109, HEK293 expressing mouse ID0-1 and -2 cell-based assays) respectively. In human SKOV-3 xenografts (serum and tumor) AUC (0 to 24h; pharmacokinetic and pharmacodynamic [PK and PD])) was 0.8, 4.2 and 23 and 3.5, 11 and 40 microM h, respectively; area under the effect curve (PK and PD) was 39, 32 and 41 and 60, 63 and 76% kyn, at BMS-986205 (5, 25 and 125 mg/kg, qd×5), respectively
In April 2017, preclinical data were presented at the 253rd ACS National Meeting and Exhibition in San Francisco, CA. BMS-986205 showed potent and selective inhibition of IDO-1 enzyme (IC50 = 1.7nM) and potent growth inhibition in cellular assays (IC50 = 3.4 nM) in SKOV3 cells. A good pharmacokinetic profile was seen at oral and iv doses in rats, dogs and monkeys. The compound showed good oral exposure and efficacy in in vivo assays
Preclinical studies were performed to evaluate the activity of BMS-986205, a potent and selective optimized indoleamine 2, 3-dioxygenase (IDO)- 1inhibitor, for the treatment of cancer. BMS-986205 inhibited kynurenine production with IC50 values of 1.7, 1.1 and > 2000 and 4.6, 6.3 and > 2000 nM in human (HeLa, HEK293 expressing human IDO-1 and tryptophan-2, 3-dioxygenase cell-based assays) and rat (M109, HEK293 expressing mouse ID0-1 and -2 cell-based assays) respectively. BMS-986205 was also found to be potent when compared with IDO-1from other species (human < dog equivalent monkey equivalent mouse > rat). In cell-free systems, incubation of inhibitor lead to loss of heme absorbance of IDO-1 which was observed in the presence of BMS-986205 (10 microM), while did not observed with epacadostat (10 microM). The check inhibitory activity and check reversibility (24 h after compound removal) of BMS-986205 was found to be < 1 and 18% in M109 (mouse) and < 1 and 12% SKOV3 (human) cells, respectively. In human whole blood IDO-1, human DC mixed lymphocyte reaction and human T cells cocultured with SKOV3 cells- cell based assays, BMS-986205 showed potent cellular effects (inhibition of kynurenine and T-cell proliferation 3H-thymidine) with IC50 values of 2 to 42 (median 9.4 months), 1 to 7 and 15 nM, respectively. In human SKOV-3 xenografts (serum and tumor) AUC (0 to 24h; pharmacokinetic and pharmacodynamic [PK and PD])) was 0.8, 4.2 and 23 and 3.5, 11 and 40 microM h, respectively; area under the effect curve (PK and PD) was 39, 32 and 41 and 60, 63 and 76% kyn, at BMS-986205 (5, 25 and 125 mg/kg, qd×5), respectively. In vivo human-SKOV3 and hWB-xenografts, IC50 values of BMS-986205 were 3.4 and 9.4 NM, respectively. The ADME of BMS-986205 at parameters iv/po dose was 0.5/2, 0.5/1.5 and 0.5/1.2 mg/kg, respectively; iv/clearance was 27, 25 and 19 ml, min/kg, respectively; iv Vss was 3.8, 5.7 and 4.1 l/kg, respectively; t1/2 (iv) was 3.9, 4.7 and 6.6 h, respectively; fraction (po) was 64, 39 and 10%, respectively. At the time of presentation, BMS-986205 was being evaluated in combination with nivolumab.
The chemical structure and preclinical profile was presented for BMS-986205 ((2R)-N-(4-Chlorophenyl)-2-[cis-4-(6-fluoroquinolin-4-yl)cyclohexyl]propanamide), a potent IDO-1 inhibitor in phase I for the treatment of cancer. This compound showed potent and selective inhibition of IDO-1 enzyme (IC50 = 1.7nM) and potent growth inhibition in cellular assays (IC50 = 3.4 nM) in SKOV3 cells. The pharmacokinetic profile in rats dosed at 0.5 mg/kg iv and 2 mg/kg po, with clearance, Vss, half-life and bioavailability of 27 ml/min/kg, 3.8 l/kg, 3.9 h and 4%, respectively; in dogs at 0.5 iv and 1.5 po mg/kg dosing results were 25 ml/min/kg, 5.7 l/kg, 4.7 h and 39%; and, in cynomolgus monkeys with the same doses as dogs results were 19 ml/min/kg, 4.1 l/kg, 6.6 h and 10%, respectively. The compound showed good oral exposure and efficacy in in vivo assays.
BMS-986158: a BET inhibitor for cancerAshvinikumar Gavai of Bristol Myers Squibb (BMS) gave an overview of his company’s research into Bromodomian and extra-terminal domain (BET) as oncology target for transcriptional suppression of key oncogenes, such as MYC and BCL2. BET inhibition has been defined as strong rational strategy for the treatment of hematologic malignancies and solid tumors. From crystal-structure guided SAR studies, BMS-986158, 2-{3-(1,4-Dimethyl-1H-1,2,3-triazol-5-yl)-5-[(S)-(oxan-4-yl)(phenyl)methyl]-5H-pyrido[3,2-b]indol-7-yl}propan-2-ol, was chosen as a potent BET inhibitor, showing IC50 values for BRD2, BRD3 and BRD4 activity of 1 nM; it also inhibited Myc oncogene (IC50 = 0.5 nM) and induced chlorogenic cancer cell death. In vitro the compound also displayed significant cytotoxicity against cancer cells. When administered at 0.25, 0.5 and 1 mg/kg po, qd to mice bearing human lung H187 SCLC cancer xenograft, BMS-986158 was robust and showed efficacy as a anticancer agent at low doses. In metabolic studies, it showed t1/2 of 36, 40 and 24 min in human, rat and mice, respectively, and it gave an efflux ratio of 3 in Caco-2 permeability assay. In phase 1/II studies, BMS-986158 was well tolerated at efficacious doses and regimens, and drug tolerable toxicity at efficacy doses and regimens. Selective Itk inhibitors for inflammatory disordersThe development of highly selective Itk inhibitors for the treatment of diseases related to T-cell function, such as inflammatory disorders, was described by Shigeyuki Takai (Ono Pharmaceutical). Inhibitory properties of a hit compound, ONO-8810443, were modified via X-ray structure and Molecular Dynamics stimulation to get ONO-212049 with significant kinase selectivity (140-fold) against Lck, a tyrosine kinase operating upstream of Itk in the TCR cascade. Further modifications identified final lead compound ONO-7790500 (N-[6-[3-amino-6-[2-(3-methoxyazetidin-1-yl)pyridin-4-yl]pyrazin-2-yl]pyridin-3-yl]-1-(3-methoxyphenyl)-2,3-dimethyl-5-oxopyrazole-4-carboxamide), which selectively inhibited Itk (IC50 = < 0.004 microM) over Lck (IC50 = 9.1 microM; SI 2000-fold) and suppressed Jurkat T-cell proliferation (IC50 = 0.014 microM). This compound suppressed alphaCD3/CDP28 CD4+T-cell stimulation (IC50 = 0.074 microM) with selectivity over PMA/Ionomycin (IC50 = > 10 microM). ONO-7790500 also exhibited in vivo IL-2 inhibitory properties (62% inhibition at 30 mg/kg po) in mice. In pharmacokinetic studies in balb/c mice, the compound administered orally (10 mg/kg) showed a Cmax of 1420 ng/ml, AUClast of 11,700 ng*h/ml, t1/2 of 5.3 h and oral bioavailability of 68%. Administration iv at 0.3 mg/kg gave an AUC last of 610 ng*h/ml, t1/2 of 3.8 h, Vss of 1260 ml/kg and Cl of 5.1 ml/min/kg. ADMET data showed ONO-7790500 did not have relevant activity in cytochromes and hERG channels (IC50 > 10 microM) in toxicological studies, and gave a PAMPA value of 5.0 x 10(-6) cm/s. Fused imidazole and pyrazole derivatives as TGF-beta inhibitorsDual growth and differentiation factor-8 (GDF-8; also known as myostatin) and TGF-beta inhibitors were described. Both targets belong to TGF-beta superfamily consisting of a large group of structurally related cell regulatory proteins involved in fundamental biological and pathological processes, such as cell proliferation or immunomodulation. Myostatin (GDF8) is a negative regulator negative regulator of skeletal muscle growth and has also been related to bone metabolism. Investigators at Rigel Pharmaceuticals found that compounds designed to be GDF-8 inhibitors were able to inhibit TGF-beta as well, this could be an advantage for the treatment of diseases associated with muscle and adipose tissue disorders, as well as potentially immunosuppressive disorders. Jiaxin Yu from the company described new fused imidazole derivatives, of which the best compound was 6-[2-(2,4,5-Trifluorophenyl)-6,7-dihydro-5H-pyrrolo[1,2-a]imidazol-3-yl]quinoxaline. This compound was very potent at TGF-beta Receptor Type-1 (ALK5) inhibition with an IC50 value of 1nM. In an in vivo mouse assay this compound showed good activity at 59.7 mg/kg, po, and good plasma exposure; inhibition of GDF-8 and TGFbeta growth factors was 90 and 81.6 %, respectively.Rigel’s Ihab Darwish described a series of fused pyrazole derivatives, with the best compound being 6-[2-(2,4-Difluorophenyl)-5,6-dihydro-4H-pyrrolo[1,2-b]pyrazol-3-yl][1,2,4]triazolo[1,5-a]pyridine. This compound showed an IC50 of 0.06 and 0.23 microM for GDF-8 and TGFbeta, respectively, in the pSMAD (MPC-11) signaling inhibition test. The compound had a good pharmacokinetic profile, with 40% of bioavailability in mice after a 5-mg/kg po dose. An iv dose of 1 mg/kg showed t1/2 of 0.7 h and Vss of 1.0 l/h/kgDiscovery of selective inhibitor of IDO BMS-986205 for cancerIndoleamine-2,3-dioxygenase (IDO)-1 enzyme initiates and regulates the first step of the kynurenine pathway (KP) of tryptophan metabolism, and evidence has shown that overexpression of IDO-1 in cancer tumors is a crucial mechanism facilitating tumor immune evasion and persistence. The chemical structure and preclinical profile of BMS-986205 was presented by Aaron Balog from BMS. BMS-986205 ((2R)-N-(4-Chlorophenyl)-2-[cis-4-(6-fluoroquinolin-4-yl)cyclohexyl]propanamide), is a potent IDO-1 inhibitor in phase I for the treatment of cancer. This compound showed potent and selective inhibition of IDO-1 enzyme (IC50 = 1.7nM) and potent growth inhibition in cellular assays (IC50 = 3.4 nM) in SKOV3 cells. The pharmacokinetic profile in rats dosed at 0.5 mg/kg iv and 2 mg/kg po, with clearance, Vss, half-life and bioavailability of 27 ml/min/kg, 3.8 l/kg, 3.9 h and 4%, respectively; in dogs at 0.5 iv and 1.5 po mg/kg dosing results were 25 ml/min/kg, 5.7 l/kg, 4.7 h and 39%; and, in cynomolgus monkeys with the same doses as dogs results were 19 ml/min/kg, 4.1 l/kg, 6.6 h and 10%, respectively. The compound showed good oral exposure and efficacy in in vivo assays.Three further reports have been published from this meeting .The website for this meeting can be found at https://www.acs.org/content/acs/en/meetings/spring-2017.html.
SYNTHESIS
1 Wittig NaH
2 REDUCTION H2, Pd, AcOEt, 4 h, rt, 50 psi
3 Hydrolysis HCl, H2O, Me2CO, 2 h, reflux
4 4-Me-2,6-(t-Bu)2-Py, CH2Cl2, overnight, rt
5 SUZUKI AcOK, 72287-26-4, Dioxane, 16 h, 80°C
6 Heck Reaction, Suzuki Coupling, Hydrogenolysis of Carboxylic Esters, Reduction of Bonds, HYDROGEN
7 Et3N, THF, rt – -78°C , Pivaloyl chloride, 15 min, -78°C; 1 h, 0°C ,THF, 0°C – -78°C, BuLi, Me(CH2)4Me, 15 min, -78°C, R:(Me3Si)2NH •Na, THF, 10 min, -50°C , HYDROLYSIS, (PrP(=O)O)3, C5H5N, AcOEt, 5 min, rt
Patent
WO2016073770
Example 19
(i?)-N-(4-chlorophenyl)-2- c 5-4-(6-fluoroquinolin-4-yl)cyclohexyl)propanamide
Example 19 : (i?)-N-(4-chlorophenyl)-2-(cz5-4-(6-fluoroquinolin-4- yl)cyclohexyl)propanamide
[0277] Prepared using General Procedures K, B, E, L, M, N, and O. General Procedure L employed 2-(4-(6-fluoroquinolin-4-yl)-cyclohexyl)acetic acid (mixture of
diastereomers), and ( ?)-2-phenyl-oxazolidinone. General Procedure M employed the cis product and iodomethane. The auxiliary was removed following General Procedure N and the desired product formed employing General Procedure O with 4-chloroaniline.
Purified using silica gel chromatography (0% to 100% ethyl acetate in hexanes) to afford Example 19. 1H NMR of czs-isomer (400 MHz; CDC13): δ 9.14 (s, 1H), 8.70 (d, J= 4.6 Hz, 1H), 8.06 (dd, J= 9.2 Hz, J= 5.6 Hz, 1H), 7.58-7.64 (m, 3H), 7.45 (ddd, J= 9.3 Hz, J= 7.8 Hz, J= 2.7 Hz, 1H), 7.19-7.24 (m, 2H), 7.15 (d, J= 4.6Hz, 1H), 3.16-3.26 (m, 1H), 2.59-2.69 (m, 1H), 2.08-2.16 (m, 1H), 1.66-1.86 (m, 7H), 1.31-1.42 (m, 1H), 1.21 (d, J= 6.8Hz, 3H) ppm. m/z 411.2 (M+H)+.
REFERENCES
23-Feb-2015
Bristol-Myers Squibb To Expand Its Immuno-Oncology Pipeline with Agreement to Acquire Flexus Biosciences, Inc
Bristol-Myers Squibb Co; Flexus Biosciences Inc
17-Dec-2014
Flexus Biosciences, a Cancer Immunotherapy Company Focused on Agents for the Reversal of Tumor Immunosuppression (ARTIS), Announces $38M Financing
Flexus Biosciences Inc
2015106thApril 21Abs 4290
Potent and selective next generation inhibitors of indoleamine-2,3-dioxygenase (IDO1) for the treatment of cancer
American Association for Cancer Research Annual Meeting
Jay P. Powers, Matthew J. Walters, Rajkumar Noubade, Stephen W. Young, Lisa Marshall, Jan Melom, Adam Park, Nick Shah, Pia Bjork, Jordan S. Fridman, Hilary P. Beck, David Chian, Jenny V. McKinnell, Maksim Osipov, Maureen K. Reilly, Hunter P. Shunatona, James R. Walker, Mikhail Zibinsky, Juan C. Jaen
2017108thApril 04Abs 4964
Structure, in vitro biology and in vivo pharmacodynamic characterization of a novel clinical IDO1 inhibitor
American Association for Cancer Research Annual Meeting
John T Hunt, Aaron Balog, Christine Huang, Tai-An Lin, Tai-An Lin, Derrick Maley, Johnni Gullo-Brown, Jesse Swanson, Jennifer Brown
2017253rdApril 05Abs MEDI 368
Discovery of a selective inhibitor of indoleamine-2,3-dioxygenase for use in the therapy of cancer
American Chemical Society National Meeting and Exposition
Aaron Balog
April 2-62017
American Chemical Society – 253rd National Meeting and Exhibition (Part IV) – OVERNIGHT REPORT, San Francisco, CA, USA
Casellas J, Carceller V

Juan Jaen

Jordan Fridman
Chief Scientific Officer at FLX Bio, Inc.

Rekha Hemrajani
Chief Operating Officer at FLX Bio, Inc

Max Osipov
////////////////PHASE 1, BMS 986205, 1923833-60-6, BMS-986205, ONO-7701,Bristol-Myers Squibb, Antineoplastics, F- 001287
C[C@H]([C@H]1CC[C@@H](C2=CC=NC3=CC=C(F)C=C23)CC1)C(NC4=CC=C(Cl)C=C4)=O
Wrapping up #MEDI‘s 1st time disclosures is Aaron Balog of @bmsnews talking about an IOD-1 inhibitor to treat cancer #ACSSanFran
BMS-741672


BMS-741672
N-((1R,2S,5R)-5-(Isopropyl(methyl)amino)-2-((S)-2-oxo-3-(6-(trifluoromethyl)quinazolin-4-ylamino)pyrrolidin-1-yl)cyclohexyl)acetamide BMS-741672
N-((lR,2S,5R)-5-(isopropyl(methyl)amino)-2-((S)-2-oxo-3-(6- (trifluoromethyl)quinazolin-4-ylamino)pyrrolidin-l-yl)cyclohexyl)acetamide
N-((1R,2S,5R)-5-(isopropyl(methyl)amino)-2-((S)-2-oxo-3-(6-(trifluoromethyl)quinazolin-4-ylamino)pyrrolidin-1-yl)cyclohexyl)acetamide;
C25 H33 F3 N6 O2, 506.56
Acetamide, N-[(1R,2S,5R)-5-[methyl(1-methylethyl)amino]-2-[(3S)-2-oxo-3-[[6-(trifluoromethyl)-4-quinazolinyl]amino]-1-pyrrolidinyl]cyclohexyl]-
CAS 1004757-96-3
PHASE 2, , Treatment of Type 2 Diabetes, Agents for Neuropathic Pain
Chemokine CCR2 (MCP-1 Receptor) Antagonists

| Molecular Formula: | C25H33F3N6O2 |
|---|---|
| Molecular Weight: | 506.574 g/mol |

| Michael G. Yang, Robert J. Cherney | |
| Original Assignee | Bristol-Myers Squibb Company |
| Michael G. Yang, Robert J. Cherney, Martin G. Eastgate, Jale Muslehiddinoglu, Siva Josyula Prasad, Zili Xiao, | |
| Bristol-Myers Squibb Company |
- Originator Bristol-Myers Squibb
- Class Analgesics; Antihyperglycaemics
- Mechanism of Action CCR2 receptor antagonists
- Discontinued Diabetic neuropathies; Type 2 diabetes mellitus
Most Recent Events
- 10 Apr 2007 Preclinical trials in Inflammation in USA (unspecified route)
BMS-741672, 1 , is a highly selective CCR2 antagonist (IC50 = 1.4 nM) featuring a complex array of four stereocenters. The key synthetic challenge was efficient assembly of the densely functionalized 1,2,4-triaminocyclohexane (TACH) core in a minimum number of linear steps.

N-((1R,2S,5R)-5-(Isopropyl(methyl)amino)-2-((S)-2-oxo-3-(6-(trifluoromethyl)quinazolin-4-ylamino)pyrrolidin-1-yl)cyclohexyl)acetamide BMS-741672
Mp 161.3 °C.
1H NMR (400 MHz, CDCl3) δ 9.50–9.20 (1H), 9.04 (s, 1H), 8.68 (s, 1H), 8.41 (d, J = 7.1 Hz, 1H), 7.87 (s, 1H), 5.04 (dt, J = 1.3, 7.3 Hz, 1H), 4.9 (m, 1H), 4.07 (dt, J = 3.7, 12.9 Hz, 1H), 3.53 (dt, J = 1.4, 9.9 Hz, 1H), 3.44–3.30 (m, 2H), 2.39 (dq, J = 13.6, 8.4 Hz, 1H), 2.26 (m, 1H), 2.21 (s, 3H), 2.17 (q, J = 2.9 Hz, 1H), 2.03–1.91 (m, 5H), 1.71–1.54 (m, 5H), 1.04 (s, br., 6H).
13C NMR (100 MHz, d6-DMSO) δ 171.46, 169.49, 159.62, 156.92, 151.22, 129.28, 128.27 (q, 4JCF = 3 Hz), 125.78 (q, 2JCF = 32 Hz), 124.11 (q, 1JCF = 272 Hz), 121.57 (q, 3JCF = 4 Hz), 114.33, 54.83, 53.54, 52.36, 47.34, 46.94, 43.13, 30.76, 30.24, 26.94, 26.38, 23.28, 20.87, 17.65 (br.), 16.73 (br.).
13C NMR (100 MHz, CDCl3) δ 172.17. 170.73, 159.89, 156.91, 151.16, 128.68, 128.06 (q,4JCF = 3.0 Hz), 127.25 (q, 2JCF = 32 Hz), 123.98 (q, 1JCF = 272 Hz), 121.78 (q, 3JCF = 4 Hz), 115.11, 54.89, 53.21, 52.40, 47.40, 46.98, 43.72, 30.84, 30.70, 29.96, 27.80, 23.55, 19.96, 17.70 (2C).
LCMS (ESI, pos.): 508 (16.8), 507 (66.2), 254 (5.0). HR-ESI(pos)-MS: calcd for C25H34F3N6O2 507.2690 [M + H]+, found 507.2694.
IR (KBr): ν = 3428 (m, br.), 2966 (w), 1686 (s), 1635 (m), 1584 (s), 1540 (m), 1334 (m), 1307 (s), 1164 (m), 1121 (m), 870 (w), 845 (w).
[α]20D−187.9 (c 1.0, CHCl3).
Anal. Calcd for C25H33F3N6O2: C, 59.28; H, 6.57; F, 11.25; N, 16.59. Found: C, 59.21; H, 6.43; F, 11.07; N, 16.53.

PATENT
WO 2008014381
http://www.google.ch/patents/WO2008014381A2?cl=en&hl=de
EXAMPLE 1
N-((lR,2S,5R)-5-(isopropyl(methyl)amino)-2-((S)-2-oxo-3-(6- (trifluoromethyl)quinazolin-4-ylamino)pyrrolidin-l-yl)cyclohexyl)acetamide

[00212] Example 1, Step 1: (IR, 2S, 5R)-tert-Butyl 2-benzyloxycarbonylamino- 7-oxo-6-aza-bicyclo[3.2.1]octane-6-carboxylate (89.6 g, 0.24 mol, see: P. H. Carter, et al. PCT application WO 2005/021500) was dissolved in ethyl acetate (1.5 L) and the resulting solution was washed with sat. NaHCCh (2 x 0.45 L) and sat. NaCl (I x 0.45 L). The solution was dried (Na2SO4) and then filtered directly into a 3 -necked 3 L round-bottom flask. The solution was purged with direct nitrogen injection before being charged with 10% Pd/C (13.65 g) under nitrogen atmosphere. The flask was evacuated and back-filled with hydrogen; this was repeated twice more. Hydrogen was bubbled through the solution for 30 min and then the reaction was stirred under 1 atm H2 for 18 h. The flask was evacuated, back-filled with nitrogen, and charged with fresh catalyst (6 g of 10% Pd/C). Hydrogen was bubbled through the solution for 30 min and then the reaction was stirred under 1 atm H2 for 18 h. The flask was evacuated and back-filled with nitrogen. The mixture was filtered through Celite; the filter pad was then washed with ethyl acetate. The filtrate (-1.6 L EtOAc volume) was diluted with acetonitrile (0.3 L) and charged sequentially with Z-N-Cbz- methionine (68 g, 0.24 mol), TBTU (77 g, 0.24 mol), and Ν,Ν-diisopropylethylamine (42 mL, 0.24 mol). The reaction was stirred at room temperature for 4 h, during which time it changed from a suspension to a clear solution. The reaction was quenched with the addition of sat. NH4Cl (0.75 L) and water (0.15 L); the mixture was diluted further with EtOAc (0.75 L). The phases were mixed and separated and the organic phase was washed with sat. Na2Cθ3 (2 x 0.9 L) and sat. NaCl (1 x 0.75 L). The solution was dried (Na2SO4), filtered, and concentrated in vacuo to give (IR,2S,5R)- tert-butyl 2-((5)-2-(benzyloxycarbonylamino)-4-
(methylthio)butanamido)-7-oxo-6-aza-bicyclo[3.2.1]octane-6-carboxylate as an oil, which was taken into the next step without further purification. LC/MS for primary peak: [M-Boc+H]+ = 406.3; [M+Naf = 528.3. 1H-NMR (400 MHz, d4-Me0H): δ 7.36 (m, 5H), 5.11 (s, 2H), 4.32 (m, IH), 4.2 (m, IH), 4.0 (m, IH), 2.5 – 2.7 (m, 3H), 2.25 (m, IH), 2.11 (s, 3H), 2.05 (m, 4H), 1.9 (m, IH), 1.7 (m, 2H), 1.54 (s, 9H). Also present are EtOAc [1.26 (t), 2.03 (s), 4.12 (q)] and N,N,N,N-tetramethylurea [2.83
(S)].
[00213] Example 1, Step 2: A sample of (1^,25,5^)- tert-butyl 2-((5)-2- (benzyloxycarbonylamino)-4-(methylthio)butanamido)-7-oxo-6-aza- bicyclo[3.2. l]octane-6-carboxylate (0.24 mol assumed; see previous procedure) was dissolved in iodomethane (1,250 g) and stirred for 48 h at room temperature. The reaction was concentrated in vacuo. The residue was dissolved in dichloromethane and concentrated in vacuo. This was repeated twice more. The resultant sludge was dissolved in dichloromethane (0.4 L) and poured into a rapidly stirring solution of MTBE (4.0 L). The resultant yellow solids were collected via suction filtration and dried under high vacuum to afford the sulfonium salt (179 g). This material was taken into the next step without further purification. LC/MS for primary peak: [M- Me2S+H]+ = 458.4; [M]+ = 520.4. 1H-NMR (400 MHz, d4-Me0H): δ 7.35 (m, 5H), 5.09 (s, 2H), 4.33 (m, IH), 4.28 (m, IH), 3.98 (m, IH), 3.3 – 3.45 (m, 2H), 2.97 (s, 3H), 2.94 (s, 3H), 2.78 (m, IH), 2.0 – 2.3 (m, 4H), 1.7 (m, 2H), 1.52 (s, 9H). Also present are MTBE [1.18 (s), 3.2 (s)] and traces of N,N,N,N-tetramethylurea [2.81 (s)]. [00214] Example 1, Step 3: All of the sulfonium salt from the previous step (0.24 mol assumed) was dissolved in DMSO (2.0 L). The resultant solution was stirred under nitrogen at room temperature and charged with cesium carbonate (216 g) portionwise. The suspension was stirred at room temperature for 3 h and then filtered to remove the solids. The solution was divided into -0.22 L portions and worked up as follows: the reaction mixture (-0.22 L) was diluted with ethyl acetate (1.5 L) and washed successively with water (3 x 0.5 L) and brine (1 x 0.3 L). The organic phase was dried (Na2SO4), filtered, and concentrated in vacuo. The desired (\R,2S,5R)- tert-bvXyl 2-((S)-3-(benzyloxycarbonylamino)-2-oxopyrrolidin-l-yl)-7-oxo-6- azabicyclo[3.2.1]octane-6-carboxylate (90.8 g, 83%) was obtained as a microcrystalline foam, free from tetramethyl urea impurity. LC/MS for primary peak: [M-Boc+H]+ = 358.4; [M+Na]+ = 480.4. 1H-NMR (400 MHz, d4-MeOH): δ 7.35 (m, 5H), 5.12 (s, 2H), 4.35 (m, 2H), 4.2 (m, IH), 3.6 (m, IH), 3.3 (m, IH), 2.64 (m, IH), 2.28 – 2.42 (m, 2H), 2.15 (m, IH), 1.7 – 2.0 (m, 5H), 1.55 (s, 9H). If desired, this material can be isolated as a solid by dissolving in MTBE (1 volume), adding to heptane (3.3 volumes), and collecting the resultant precipitate.
[00215] Example 1, Step 4: A stirring solution of (\R,2S,5R)- tert-butyl 2-((S>3- (benzyloxycarbonylamino)-2-oxopyrrolidin-l-yl)-7-oxo-6-azabicyclo[3.2.1]octane-6- carboxylate (108 g, 0.236 mol) in THF (1 L) was charged with lithium hydroxide monohydrate (21.74 g, 0.519 mol). Water (0.3 L) was added slowly, such that the temperature did not exceed 20 0C. The reaction was stirred at room temperature overnight and the volatiles were removed in vacuo. The pH was adjusted to -4 through the addition of IN HCl (450 mL) and NaH2PO4. The resultant white precipitates were collected by filtration and washed with water (2 x 1 L). The solid was dissolved in dichloromethane (1.5 L) and water (~ 1 L). The organic layer was dried (Na2SO4), filtered, and concentrated in vacuo. The residue was dissolved in EtOAc (0.7 L) and the resultant solution was heated at reflux for 1 h. Solids separated after cooling to RT, and were collected via filtration. These solids were purified by recrystallization in isopropanol to afford the desired (\R,2S,5R)-2-((S)-3- (benzyloxycarbonylamino)-2-oxopyrrolidin-l-yl)-5-(tert- butoxycarbonylamino)cyclohexanecarboxylic acid as a white solid (104.5 g, 93% yield). LC/MS for primary peak: [M-tBu+H]+ = 420.2; [M-Boc+H]+ = 376.2; [M+H]+ = 476.2. 1H-NMR (400 MHz, d4-Me0H): δ 7.35 (m, 5H), 5.11 (s, 2H), 4.35 (m, 2H), 3.71 (m, IH), 3.45 – 3.6 (m, 2H), 2.99 (m, IH), 2.41 (m, IH), 2.15 (m, IH), 2.0 (m, 2H), 1.6 – 1.9 (m, 4H), 1.46 (s, 9H).
[00216] Example 1, Step 5: A 3 L round bottom flask was charged with (lR,25′,5R)-2-((5)-3-(benzyloxycarbonylamino)-2-oxopyrrolidin-l-yl)-5-(tert- butoxycarbonylamino)cyclohexanecarboxylic acid (75.5 g, 0.158 mol), EDOHCl (33.5 g, 0.175 mol), 1 -hydroxybenzotriazole (23.6 g, 0.175 mol), and dichloromethane (1 L). The reaction was stirred at room temperature for 2 h, during which time it changed from a white suspension to a clear solution. Ammonia (gas) was bubbled into the solution until the pH was strongly basic (paper) and the reaction was stirred for 10 min; this ammonia addition was repeated and the reaction was stirred for an additional 10 min. Water was added. The organic phase was washed with sat. NaHCθ3, NaH2PO4, and brine before being concentrated in vacuo. The residue was slurried with acetonitrile (0.5 L) and then concentrated in to give (lR,2S,5R)-2-((5)-3-(benzyloxycarbonylamino)-2-oxopyrrolidin-l-yl)-5-(tert- butoxycarbonylamino)cyclohexanecarboxamide as a white solid (75.9 g, -100%), which was used in the next step without further purification. LC/MS for primary peak: [M-Boc+H]+ = 375.3; [M+H]+ = 475.4; [M-tBu+H]+ = 419.3. 1H-NMR (400 MHz, Cl4-MeOH): δ 7.35 (m, 5H), 5.11 (s, 2H), 4.25 (m, 2H), 3.70 (m, IH), 3.6 (m, IH), 3.45 (m, IH), 2.91 (m, IH), 2.38 (m, IH), 2.12 (m, IH), 1.9 – 2.05 (m, 2H), 1.65 – 1.9 (m, 4H), 1.46 (s, 9H).
[00217] Example 1, Step 6: The reaction was run in three equal portions and combined for aqueous workup. A 5 L, 3-necked round bottom flask was charged with (lR,2S,5R)-2-((5)-3-(benzyloxycarbonylamino)-2-oxopyrrolidin-l-yl)-5-(tert- butoxycarbonylamino)cyclohexanecarboxamide (25.3 g, 53 mmol), acetonitrile (1.9 L), and 2.6 L of water/ice. The mixture was stirred and cooled to 0 0C. Iodobenzene diacetate (25.77 g, 80 mmol) was added and the reaction was stirred for 2 h; another 0.5 eq of iodobenzene diacetate was added. The reaction was stirred for 9 h (reaction temp < 10 0C). The mixture was charged with 8 eq N,N-diisopropylethylamine and 2 eq acetic anhydride. Over the next thirty minutes, 4 eq N,N-diisopropylethylamine and 2 eq acetic anhydride were added every ten minutes, until the reaction had proceeded to completion (HPLC). The acetonitrile was removed in vacuo; some solid separated from the residue, and this was collected by filtration. The remaining residue was extracted with dichloromethane (3 L, then 1 L). The organic phase was washed sequentially with water, sat. NaHCθ3, and brine. The collected solids were added to the organic phase, along with activated carbon (15 g). The mixture was stirred for 30 minutes at 40 0C before being filtered and concentrated in vacuo. The residue was dissolved in EtOAc (1 L), and the resultant solution was stirred at 75 0C for 1 h before being allowed to cool to room temperature. A solid separated and was collected by filtration. This solid was purified further by recrystallization: it was first dissolved in 0.5 L CH2CI2, then concentrated in vacuo, then re-crystallized from 1 L EtOAc; this was repeated three times. The solids obtained from the mother liquors of the above were recrystallized three times using the same method. The combined solids were recrystallized twice more from acetonitrile (0.7 L) to provide 66 g (84%) of tert-bυXyl (lR,3R,45)-3-acetamido-4-((5)-3-(benzyloxycarbonylamino)-2- oxopyrrolidin-l-yl)cyclohexylcarbamate (purity >99.5% by HPLC). LC/MS for primary peak: [M+H]+ = 489.4; [M-tBu+H]+ = 433.3. 1H-NMR (400 MHz, d4– MeOH): δ 7.3 – 7.4 (m, 5H), 5.11 (s, 2H), 4.35 (m, IH), 4.15 (m, IH), 4.04 (m, IH), 3.8 (m, IH), 3.6 (m, 2H), 2.44 (m, IH), 2.12 (m, IH), 1.87 – 2.05 (m, 4H), 1.87 (s, 3H), 1.55 – 1.7 (m, 2H), 1.46 (s, 9H). The stereochemical fidelity of the Hofmann rearrangement was confirmed through X-ray crystal structure analysis of this compound, as shown in Figure 1. [00218] Example 1, Step 7: A stirring solution of tert-butyl (\R,3R,4S)-3- acetamido-4-((5′)-3 -(benzyloxycarbonylamino)-2-oxopyrrolidin- 1 – yl)cyclohexylcarbamate (66 g, 0.135 mol) in dichloromethane (216 mL) was charged with trifluoroacetic acid (216 mL). The reaction was stirred for 2 h at room temperature and concentrated in vacuo. The residue was dissolved in methanol and the resultant solution was concentrated in vacuo; this was repeated once. Benzyl («S)-l-((l«S,2R,4R)-2-acetamido-4-aminocyclohexyl)-2-oxopyrrolidin-3-ylcarbamate was obtained as an oil and used directly in Step 8 below. LC/MS found [M + H]+ = 389.4. 1H-NMR (400 MHz, d4-MeOH): δ 7.3 – 7.4 (m, 5H), 5.12 (s, 2H), 4.41 (br. s, IH), 4.15 (m, IH), 4.00 (t, J= 9.3 Hz, IH), 3.81 (t, J= 9.1 Hz, IH), 3.65 (q, J= 8.4 Hz, IH), 3.3 – 3.4 (m, IH), 2.45 (m, IH), 1.95 – 2.24 (m, 5H), 2.00 (s, 3H), 1.6 – 1.8 (m, 2H). [00219] Example 1, Step 8: A stirring solution of benzyl (S)- 1-(( \S,2R,4R)-2- acetamido-4-aminocyclohexyl)-2-oxopyrrolidin-3-ylcarbamate (-0.135 mol) in methanol (675 mL) was charged sequentially with acetone (37.8 g, 4 eq), sodium acetate (33.2 g, 3 eq), and sodium cyanoborohydride (16.9 g, 2 eq). The mixture was stirred at room temperature for 6 h and filtered. The filtrate was dissolved in dichloromethane (1 L); this solution was washed with IN NaOH (1 L). The solids collected in the filtration were dissolved in IN NaOH (IL) at 0 0C and then extracted with dichloromethane (1 L). The organic extracts were combined and extracted with aqueous HCl (200 mL IN HCl + 800 mL water). The aqueous phase was basified with sat. NaHCO3 (500 mL) and then IN NaOH (100 mL) until pH 11. The aqueous phase was extracted with dichloromethane (2 L). The organic extracts were combined, dried (Na2SO4), filtered, and concentrated in vacuo to give benzyl (S)-I- ((lS,2R,4R)-2-acetamido-4-(isopropylamino)cyclohexyl)-2-oxopyrrolidin-3- ylcarbamate as an oil. LC/MS found [M + H]+ = 431.45. 1H-NMR (400 MHz, d4– MeOH): δ 7.3 – 7.4 (m, 5H), 5.12 (s, 2H), 4.31 (m, IH), 4.24 (t, J= 9.4 Hz, IH), 4.11 (m, IH), 3.61 (t, J= 9.1 Hz, IH), 3.52 (q, J= 8.6 Hz, IH), 3.04 (br. s, IH), 2.96 (sep, J= 6.3 Hz, IH), 2.40 (m, IH), 2.15 (m, IH), 1.92 (s, 3H), 1.7 – 1.9 (m, 5H), 1.65 (m, IH), 1.12 (app. dd, J= 6.3, 1.1 Hz, 6H).
[00220] Example 1, Step 9 (See Alternative Step 9, below): A stirring solution of benzyl (S)-I -((lS’,2R,4R)-2-acetamido-4-(isopropylamino)cyclohexyl)-2- oxopyrrolidin-3-ylcarbamate (-115 mmol) in dichloromethane (600 mL) was cooled to 0 0C and charged sequentially with formaldehyde (18.6 g, 37 wt% solution), triethylamine (23 mL), and sodium triacetoxyborohydride (28.7 g). The mixture was stirred at room temperature for 30 minutes and diluted with dichloromethane (up to 1.2 L). This solution was washed thrice with 500 mL sat. NaHCθ3 + NaOH (sat. NaHCO3, pH to 11 w/ IN NaOH). The organic layer was extracted with aq. HCl (200 mL IN HCl + 600 mL water). The aqueous phase was basified with sat. NaHCO3 (500 mL) and then IN NaOH (100 mL) until pH 11. The aqueous phase was extracted with dichloromethane (1.2 L). The organic extracts were combined, dried (Na2SO4), filtered, and concentrated in vacuo to give benzyl {S)-\-{{\S,2R,AR)-2- acetamido-4-(isopropyl(methyl)amino)cyclohexyl)-2-oxopyrrolidin-3-ylcarbamate as an oil, which was used directly in Step 10 below. LC/MS found [M + H]+ = 445.4. 1H-NMR (400 MHz, d4-MeOH): δ 7.3 – 7.4 (m, 5H), 5.12 (s, 2H), 4.33 (br s, IH), 4.25 (t, J= 9.2 Hz, IH), 4.11 (br s, IH), 3.5 – 3.6 (m, 2H), 2.77 (v br s, 2 H), 2.41 (m, IH), 2.26 (s, 3H), 2.0 – 2.1 (m, 2H), 1.92 (s, 3H), 1.7 – 1.9 (m, 5H), 1.10 (app. dd, J = 17, 6.4 Hz, 6H). [00221] Example 1, Step 10: To a solution of benzyl (S)- 1-(( 15″,2R,4R)-2- acetamido-4-(isopropyl(methyl)amino)-cyclohexyl)-2-oxopyrrolidin-3 -ylcarbamate (-0.115 mol) in methanol (600 mL) was added 10% Pd/C (6 g of 50% wet catalyst). The flask was evacuated and back-filled with hydrogen. The mixture was stirred under 1 atm H2 for 2 h and the catalyst was removed by filtration through Celite. The filtrate was concentrated in vacuo to provide N-((li?,25,5i?)-2-((S)-3-amino-2- oxopyrrolidin-l-yl)-5-(isopropyl(methyl)amino)cyclohexyl)acetamide as an oil, which was taken on to the next step without further purification. LC/MS found [M + H]+ = 311.47. 1H-NMR (400 MHz, (I4-MeOH): δ 4.39 (br s, IH), 4.00 (m, IH), 3.3 –
3.5 (m, 4H), 2.73 (m, IH), 2.38 (m, IH), 2.25 (s, 3H), 2.0 – 2.2 (m, 3H), 1.94 (s, 3H),
1.6 – 1.75 (m, 4H), 1.07 (app. dd, J= 21, 6.4 Hz, 6H). [00222] Example 1, Step 11: To a solution of N-((lR,25′,5R)-2-((S)-3-amino-2- oxopyrrolidin-l-yl)-5-(isopropyl(methyl)amino)cyclohexyl)acetamide (~35 g, 0.115 mol) in isopropanol (600 mL) was added 4-chloro-6-(trifluoromethyl)quinazoline (32 g, 0.138 mol, 1.2 eq, see: P.H. Carter et al, PCT application WO 2005/021500). The mixture was stirred at room temperature overnight before being charged with triethylamine (46 g, 0.46 mol, 4 eq). The mixture was stirred at 60 0C for 10 h. The solvent was removed under reduced pressure to give an oil. Azeotropic distillation with isopropanol was performed twice. The residue was dissolved in dichloromethane (600 mL) and extracted with water (250 mL, containing 4 eq acetic acid). Dichloromethane (600 mL) was added to the combined aqueous washes, and the mixture was cooled to 0 0C. Aqueous NaOH (50% by weight) was added with stirring until the pH reached 11. The water layer was extracted with dichloromethane twice (2 x 600 mL). The combined organic extracts were dried (Na2SO4), filtered, and concentrated in vacuo to give the amorphous free base of the title compound (99% purity by HPLC). LC/MS found [M+H]+ = 507.3. 1H-NMR (400 MHz, U4– MeOH): δ 8.82 (s, IH), 8.59 (s, IH), 8.05 (dd, J= 8.8, 1.8 Hz, IH), 7.9 (d, J= 8.7 Hz, IH), 5.28 (t, J= 8.6 Hz, IH), 4.58 (br s, IH), 4.06 (m, IH), 3.52 – 3.68 (m, 2H), 3.43 (m, IH), 2.76 (br s, IH), 2.55 (m, IH), 2.28 (s, 3H), 2.1 – 2.3 (m, 3H), 2.0 (s, 3H), 2.0 (m, IH), 1.65 – 1.8 (m, 3H), 1.09 (app. dd, J= 24, 6.4 Hz, 6 H).
Example 1, Alternative Step 9

[00223] Example 1, Alternative step 9a1: To a hydrogenator were charged ethyl (7R,SS)-S-((S)- l-phenyl-ethylamino)-l,4-dioxa-spiro[4.5]decane-7-carboxylate A- toluenesulfonate salt I A (1417 g, 2.8 moles, c.f : WO2004098516, prepared analogous to US Pat.6,835,841), ethanol (200 proof, 11.4 L), and 10% Pd/C catalyst (50% wet, 284 g). The mixture was inerted with nitrogen, then pressurized with hydrogen gas (45 psig) and agitated vigorously at approx. 40 0C until starting material was consumed (HPLC). The suspension was cooled, purged with nitrogen gas and the catalyst was removed by filtration while inerted. The spent catalyst was washed with ethanol (4.3 L). The filtrate and washings were combined and concentrated under vacuum to a volume of 2-3 L while maintaining the batch between 40°-60 0C. Isopropyl acetate (5 L) was charged and the mixture was concentrated to a volume of ~2 L until most ethanol was removed (<0.5%) and residual moisture content was <l,000 ppm. Batch volume was adjusted to -7.5 L by the addition of isopropyl acetate. The mixture was heated to 80 0C until clear, then cooled 65°-70 0C. Seed crystals of 1 (5 g) were added and the batch was cooled to 500C over 2 hours, then further cooled to 20 0C over 4 hours and held for ~10 hours. The resulting slurry was filtered and the cake was washed with isopropyl acetate (2 L). The product was dried under vaccum at -35 0C until volatiles were recduced below -1% (LOD). Ethyl (7R,85′)-8-amino-l,4-dioxa-spiro[4.5]decane-7-carboxylate 4-toluenesulfonate salt 1 was obtained as a white, crystalline solid (936 g, 83% yield; HPLC purity: 99.8%). 1H-NMR: (300MHz, CDCl3) 8.14-7.89 (brs, 3H), 7.75 (d, J 9.0Hz, 2H), 7.15 (d, J 8.0Hz, 2H), 4.22-4.04 (m, 2H), 4.01-3.77 (m, 4H), 3.55-3.43 (m, IH,), 3.20-3.13 (m, IH), 2.40-2.27 (m, 4H), 2.21-1.94 (m, 2H), 1.81-1.51 (m, 3H), 1.23 (t, J 7.0Hz, 3H); HPLC: Waters Xterra MS C18 4.6 mm x 150 mm Ld., 3.5μm particle size, 0.05% NH40H (5% ACN, 95% H2O, solvent A), to 0.05% NH4OH (95% ACN, 5% H2O, solvent B), 5% B to 20% B in 10 minutes, changed to 95% B in 25 minutes, and then changed to 5% B in 1 minute; 11.1 minutes (aminoester 1).

Example 1, Alternative Step 9a”: Aminoester 1 (63g, 0.16M, leq.; the product of reductive deprotection of a known compound – (See e.g. R. J. Cherney, WO 2004/098516 and G. V. Delucca & S. S. Ko, WO 2004/110993) was placed in a round bottom flask and MeCN (50OmL) was added. EDAC (33.1g, 0.17M, l. leq), HOBt-H2O (21.2g, 0.16M, l.Oeq) and N-Cbz-Z-methionine (46.7g, 0.17M, 1.05eq) were then added followed by TEA (48.OmL, 0.35M, 2.2eq). An exotherm to 38 0C was observed. The reaction mass was left to stir at RT. After 30mins, HPLC indicated complete conversion. The reaction mass was diluted with EtOAc (2.5L) and washed with KHCO3 (4x500mL, 20wt% aq. solution) and brine (50OmL). The organic phase was separated, dried over MgSO4 and concentrated. The residue was dissolved in TBME and reconcentrated to give ethyl (7R,85)-8- {(2S)-2-benzyloxycarbonylamino- 4-methylsulfanyl-butyr-yl-amino}-l,4-dioxa-spiro[4.5]decane-7-carboxylate 2 as a sticky semi-solid (76.2g, 98% yield, 93AP purity). 1H-NMR: (300MHz, CDCl3) δ 7.36-7.30 (m, 5H), 7.03 (d, J9.0Hz, IH), 5.66 (d, J 8.0Hz, IH), 5.10 (s, 2H), 4.35- 4.25 (m, 2H), 4.19-4.04 (m, 2H,), 3.98-3.86 (m, 4H), 2.87-2.80 (m, IH), 2.55-2.45 (m, 2H), 2.18 (dd, J 14.0Hz, 7.0Hz, IH), 2.08 (s, 3H), 2.05-1.67 (m, 6H), 1.26 (t, J 7.0Hz, 3H). HPLC: YMC-Pack Pro C18 5μm 4.6 x 150 mm, 0.05% TFA (20% MeOH, 80% H2O), to 0.05% TFA (20% MeOH, 80% MeCN), 0-100% lOmin gradient. lO.Olmin (Compound 2, 93.1 AP). HRMS: m/z 495.2166 [CaIc: C24H35N2O7S 495.2165].

2 3 [00224] Example 1, Alternative Step 9b: Methionine amide 2 (75.Og, 0.15M) was dissolved in MeI (225mL, 3mL/g) – some off gassing was noted but no exotherm. The reaction mass was left to stir in the dark for 16.5h. After this time a thick light yellow precipitate had formed. The flask was then evacuated to 200mmHg and some of the MeI removed. The remaining material was slurried in TBME (50OmL), after a 30min stir-out the slurry was filtered, the cake washed with TBME (50OmL). NMR analysis of this material indicated a small amount of MeI remaining. The cake was re-slurried in TBME (50OmL), filtered, washed with TBME (50OmL) and dried under vacuum to give [(35)-3-benzyloxycarbonylamino-3-{(7R,85′)-7- ethoxycarbonyl-l,4-di-oxa-spiro[4.5]dec-8-ylcarbamoyl}-propyl]-dimethylsulfonium iodide 3 as a free flowing off-white solid (93.5g, 97%, 99 area% purity). 1H-NMR: (300MHz, CDCl3) δ 7.75 (d, J 9.0Hz, IH), 7.38-7.27 (m, 5H), 6.40 (d, J 7.0Hz, IH), 5.10 (s, 2H), 4.76-4.65 (m, IH), 4.48-4.39 (m, IH), 4.14-3.85 (m, 6H), 3.84-7.73 (m, IH), 3.68-3.55 (m, IH), 3.21 (s, 3H), 3.12 (s, 3H), 2.90-2.83 (s, IH), 2.52-1.55 (m, 8H), 1.24 (t, J7.0Hz, 3H). HPLC: YMC-Pack Pro C18 5μm 4.6 x 150 mm, 0.05% TFA (20% MeOH, 80% H2O), to 0.05% TFA (20% MeOH, 80% MeCN), 0-100% lOmin gradient. 2.45min (I-), 8.14min (Compound 3, 43.6AP, I“ 54.6AP). HRMS: m/z 509.2341 [CaIc: C25H37N2O7S 509.2321].

[00225] Example 1, Alternative Step 9c: Cs2CO3 (61.5g, 0.19M, 1.5eq) was placed in an round bottom flask and anhydrous DMSO (2.4L) was added. Sulfonium salt 3 (80.Og, 0.13M, 1.Oeq) was then added portionwise. Once the addition was complete the reaction mass was left to stir in the dark for 2Oh. The reaction mass was then split in half and each half worked up separately: the reaction mass was diluted with EtOAc (2.0L) and washed with brine (2L), the organic phase was washed with brine (50OmL). The combined aq. layers were then washed EtOAc (50OmL). The combined organic phases were then washed with brine (3x750mL). The second half of the reaction mass was treated in an identical manner and the combined organics dried over MgSO4 and concentrated to give ethyl (7R,8S)-8-{(3S>3- Benzyloxycarbonylamino-2-oxo-pyrrolidin-l-yl}-l,4-dioxa-spiro[4.5]decane-7- carboxylate 4 as a light colored oil (56.5g, 0.13M, -100 area-% purity) pure by NMR analysis. 1H-NMR: (300MHz, CDCl3) δ 7.38-7.30 (m, 5H), 5.37 (br d, J4.0Hz, IH), 5.11 (s, 2H), 4.27-4.18 (m, IH), 4.17-3.82 (m, 8H), 3.32 (td, J 10.0Hz, 60.0Hz, IH), 3.23 (q, J5.0Hz, IH), 2.63-2.57 (m, IH), 2.42-2.25 (m, 2H), 1.94-1.68 (m, 5H), 1.25 (t, J 7.0Hz, 3H). HPLC: YMC-Pack Pro Cl 8 5μm 4.6 x 150 mm, 0.05% TFA (20% MeOH, 80% H2O), to 0.05% TFA (20% MeOH, 80% MeCN), 0-100% lOmin gradient. 8.99min (Compound 5, produced on column, 4.2AP), 9.48 (Compound 4, 74.3AP). HRMS: m/z 447.2127 [CaIc: C23H31N2O7 447.2131].

4 5
[00226] Example 1, Alternative Step 9d: Pyrrolidinone 4 (50.Og, 0.1 IM) was dissolved in acetone (50OmL) and IN HCl (50OmL) was added. The reaction mass was then heated to 65°C. After 20mins HPLC indicated complete reaction. The reaction mass was allowed to cool to RT and the acetone was removed on a rotary evaporator. During this distillation the product precipitated from solution as a white solid. This was isolated by filtration and the cake washed with water. The cake was then dried azeotropically with toluene (3x3OOmL) to give ethyl (\R,2S)-2-((3S)-3- Benzyloxycarbonylamino-2-oxo-pyrrolidin-l-yl)-5-oxo-cyclohexanecarboxylate 5 as a white solid (39.8g, 88%, 97 area-% purity). 1H-NMR: (300MHz, CDCl3) δ 7.37- 7.32 (m, 5H), 6.65 (br d, J4.0Hz, IH), 5.12 (s, 2H), 4.54-4.47 (m, IH), 4.34-4.26 (m, IH), 4.18 (dq, J 11.0Hz, 7.0Hz, IH), 4.09 (dq, J 11.0Hz, , 7.0Hz, IH), 3.36-3.20 (m, 3H), 2.70-2.35 (m, 6H), 2.05-1.96 (m, IH), 1.81 (quin., J l l.OHz, IH), 1.24 (t, J 7.0Hz, 3H). HPLC: YMC-Pack Pro C18 5μm 4.6 x 150 mm, 0.05% TFA (20% MeOH, 80% H2O), to 0.05% TFA (20% MeOH, 80% MeCN), 0-100% lOmin gradient. 8.95min (Compound 5). HRMS: m/z 403.1864 [CaIc: C2iH27N2O6403.1869].

[00227] Example 1, Alternative Step 9e: Cyclohexanone 5 (22.5g, 0.06M, leq), DMSO (3OmL) and Ti(O-ZPr)4 (33.7mL, 0.1 IM, 2.04eq) were placed in a round bottom flask. N-isopropyl-N-methylamine (11.6mL, 0.1 IM, 2.0eq) was then added in one portion. The mixture was left to stir for 30mins at room temperature before being cooled to <3°C in ice/water. MeOH (3OmL) was then added followed by the portionwise addition OfNaBH4 (4.33g, 0.1 IM, 2.04eq) – temperature kept <8°C. 30mins after the addition was completed the reaction mass was diluted with methylene chloride (30OmL) and then NaOH (IN, 4OmL). The resulting slurry was filtered through Celite, and the cake washed with methylene chloride (10OmL). The resulting liquor was concentrated under reduced pressure and the residue dissolved in EtOAc (50OmL). This solution was extracted with IN HCl (2x400mL), the combined aqueous layers were then basified with Na2CO3. Extraction with EtOAc (4x250mL) provided a clear and colorless organic phase which was dried over Na2SO4 and concentrated to give a white powder (24.6g, 96%, 7: 1 d.r.). This material was then slurried overnight in hexane (67OmL). The solid was isolated by filtration and dried under reduced pressure to give ethyl (lR,25′,5R)-2-((3S)-3-benzyloxycarbonylamino- 2-oxo-pyrrolidin-l-yl)-5-(isopropyl-methyl-amino)-cyclohexanecarboxylate 6 as a while solid (20.9g, 81%, 24: 1 d.r.). 1H-NMR: (300MHz, CDCl3) δ 7.37-7.28 (m, 5H), 5.55 (d, J4.5, IH), 5.10 (s, 2H), 4.42 (q, J4.5, IH), 4.23-4.12 (m, IH), 4.08 (dq, J 10.5, 7.0, IH), 4.02 (dq, J 10.5, 7.0, IH), 3.84 (t, J9.0, IH), 3.46-3.36 (m, IH), 3.04 (septet, J6.5, IH), 2.86-2.80 (m, IH), 2.63-2.48 (m, 2H), 2.17 (s, 3H, Me), 2.10-1.63 (m, 7H), 1.22 (t, J 7.0, 3H), 1.00 (d, J 6.5, 3H), 0.97 (d, J 6.5, 3H). HPLC: YMC- Pack Pro C18 5μm 4.6 x 150 mm, 0.01M NH4OAc (MeOH:water 20:80) to 0.01M NH4OAc (MeOH:water:MeCN 20:5:75) 10 to 100% 15min gradient. 8.23 (Compound 6), 8.88 (5-e/«-Compound 6). HRMS: 460.2798 [CaIc: C25H38N3O5 460.2811].

[00228] Example 1, Alternative Step 9f: The aminoester 6 (9.76 g, 2.12 mmol) was dissolved in 2N HCl (80 mL), then heated to -55 0C under inert atmosphere. The reaction was stirred for 20 h, then cooled to room temperature. The reaction solution was washed twice with toluene (25 mL portions), neutralized to pH 6 – 7 by the addition of KOH pellets, then extracted eight times with methylene chloride (100 mL portions). The combined extracts were dried (Na2SO4), filtered, and concentrated under reduced pressure to 50 mL total volume. The concentrated solution was then slowly added to methyl tert-butyl ether (300 mL) over 15 min in an addition funnel with vigorous stirring. The resulting white slurry was stirred at ambient temperature for Ih, then cooled to 0 0C and stirred for Ih. The product was filtered, and washed twice with methyl tert-butyl ether (25 mL portions). Water from the wet cake was removed by azeotropic distillation with acetonitrile (300 mL). The product was dried under reduced pressure to provide (li?,25r,5R)-2-((35′)-3-Benzyloxycarbonylamino-2- oxo-pyrrolidin-l-yl)-5-(isopropyl-methyl-amino)-cyclohexanecarboxylic acid 7, (7.69 g, 84% yield) as a white foam. 1H-NMR: (400 MHz, 500C, CDCl3) δ 7.44-7.32 (m, 5H), 6.10 (broad s, IH), 5.19 (app s, 2H), 4.42 (dd, J= 15.6, 7.8 Hz, IH), 4.29-4.23 (m, IH), 3.68-3.60 (m, 2H), 3.33-3.27 (m, 2H), 3.20 (broad s, IH), 2.99 (broad s, IH), 2.51 (s, 3H), 2.49-2.45 (m, 3H), 2.33-2.31 (m, IH), 2.00 (ddd, J= 9.0, 8.6, 3.9 IH), 1.95-1.78 (m, 2H), 1.36-1.21 (m, 6H). LCMS: m/z 432.20 [CaIc: C23H34N3O5 432.25].
NHCbz



[00229] Example 1, Alternative Step 9g: Amino acid 7 (6.3g, 14.7mmol, l.Oeq) was dissolved in THF (8OmL) under N2 and NaH (584mg, 14.7mmol, l.Oeq, 60wt% dispersion in mineral oil) was added portionwise. When the addition was complete, and the evolution of gas had ceased, the reaction mass was concentrated under reduced pressure and the resulting solid azeotroped with toluene (50 mL) to give a white solid (KF 0.59wt%). This solid was slurried in toluene (100 mL) under N2and heated to 900C. DPPA (3.32 mL, 15.3 mmol, 1.05 eq) was added dropwise over ~2min. After ~5min all the solids had dissolved, after lOmins precipitation of a white solid was observed. After 30mins HPLC analysis indicated complete reaction. The reaction mass was allowed to cool to RT before being filtered, the cake was washed with toluene. The liquors where then slowly added into ACOH/AC2O (80/20, 168mL) solution at 900C. After 45mins HPLC still indicated some isocyanate. At 1.15h , the reaction mass was cooled to RT and diluted with toluene (10OmL) and water (10OmL). The organic layer was removed and the toluene washed with IN HCl
(10OmL). The combined aq. phases were then basified with K2Cθ3(s) and brought to pH 12 with NaOH (10N), keeping the temperature below 200C. The aq layer was then extracted with methylene chloride (4xl50mL), the combined organic layers dried over K2CO3 and concentrated to give benzyl (S)-l-((lS,2R,4R)-2-acetamido-4- (isopropyl(methyl)amino)cyclohexyl)-2-oxopyrrolidin-3-ylcarbamate 8 as a white foam (4.5g, 70%, 94AP purity). The 1H-NMR was identical to material obtained from the route described above (Example 1, Step 9). HPLC: YMC-Pack Pro Cl 8 5μm 4.6 x 150 mm, 0.05% TFA (20% MeOH, 80% H2O), to 0.05% TFA (20% MeOH, 80% MeCN), 0-100% lOmin gradient. 7.20min (Compound 8), 7.85min (urea dimer). HRMS: 445.2809 [CaIc: C24H37N4O4 445.2815].
Alternative Preparation of Example 1

2 3
[00230] Example 1, Alternative Preparation, Step 1: Ethyl (7R,85)-8-amino- l,4-dioxa-spiro[4.5]decane-7-carboxylate 4-toluenesulfonate salt 1 (450. Ig), was combined with l-ethyl-3-(3-dimethyl-amino-propyl)carbo-diimide hydrochloride (236.3g), 1-hydroxy benzotriazole hydrate (171.9g), N-carbobenzyloxy-Z -methionine (333.4g) and acetonitrile (3.1 L). To the stirred mixture was added triethylamine (249.5g) below 30 0C. Upon reaction completion (HPLC), the mixture was diluted with ethyl acetate (8.2 L) and washed with aqueous 25% potassium bicarbonate solution (2×4.5 L) followed by water (4.5 L). The organic phase was separated and concentrated under reduced pressure to obtain a solution of ethyl (7R,85)-8-((5)-2- benzyloxycarbonylamino-4-methylsulfanyl-butyrylamino)-l,4-dioxa- spiro[4.5]decane-7-carboxylate 2 (1.4 L). Methyl iodide (2.39 kg) was added, the vessel was shielded from light and the mixture was held under slow agitation for approx. 24 h. To the thick yellow precipitate was added methyl tert-butyl ether (2.7 L) and the mixture was held for approx. 1 h. The product was isolated by filtration and the cake was washed with methyl tert-butyl ether (2×1.4 L), then dried under vacuum, yielding [(5)-3-benzyloxy-carbonylamino-3-((7R,8«S’)-7-ethoxycarbonyl-l,4- dioxa-spiro[4.5]dec-8-ylcarbamoyl)-propyl]-dimethylsulfonium iodide 3 (671.4 g, -94% yield) as an off-white solid (HPLC purity 99.9%).

[00231] Example 1, Alternative Preparation, Step 2: Sulfonium salt 3 (619.4 g), and cesium carbonate (416.8 g) and anhydrous dimethyl sulfoxide (6.2 L) were combined in a reactor equipped with a scrubber to neutralize volatile sulfides.
Vigorous agitation was maintained until complete conversion was obtained (HPLC). Ethyl acetate (12.4 L) was added, followed by 20 % brine (3 L). The organic phase was separated, washed twice with brine (2×3 L) and evaporated to obtain a solution of ethyl (7R,8«S)-8-((«S)-3-benzyloxycarbonylamino-2-oxo-pyrrolidin-l-yl)-l,4-dioxa- spiro[4.5]decane-7-carboxylate 4 in ethyl acetate (~0.8 L). Acetone (2.55 L) was added, followed by aqueous 0.5 M hydrochloric acid solution (2.3 L). With good mixing, the solution was heated to 50 to 60 0C until conversion of 4 to ethyl (IR,2S)- 2-((5)-3-benzyloxycarbonylamino-2-oxo-pyrrolidin-l-yl)-5-oxo- cyclohexanecarboxylate 5 was complete (HPLC). The mixture was concentrated under reduced pressure while below 40 0C, cooled to -30 0C, and water (4.1 L) was added. The resulting slurry was cooled to 5 to 10 0C and agitated for ~1 hour. The product was filtered and the cake was washed with water (2×2.5 L). Upon deliquoring, the cake was dried to a constant weight below 40 0C in a vacuum oven. Cyclohexanone 5 (272g, 70% yield) was obtained (HPLC purity 98.7%).

[00232] Example 1, Alternative Preparation, Step 3: Cyclohexanone 5 (206 g) was dissolved in dichloromethane (1.1 L) and charged to a hydrogenator. Titanium tetraisopropoxide (218.2 g) and N-isopropyl N-methylamine (63.64 g) were added and the mixture was stirred at ambient temperature (23 to 25 0C) for at least 5 h. Platinum catalyst (5% Pt/S/C, 15 g, approx. 7.5 % relative to 5) was added and hydrogenation was performed at -30 psig for at least 6 h, yielding a mixture of ethyl (lR,25′,5R)-2-((5)-3-benzyloxycarbonylamino-2-oxo-pyrrolidin-l-yl)-5-(isopropyl- methyl-amino)-cyclohexanecarboxylate 6 and its 5-epz-isomer (-7%). The catalyst was removed by filtration and the filtrate was concentrated under reduced pressure to approx. -600 mL. Wet ethyl acetate (-3% water, 2.0 L) was added with vigorous agitation over a period of at least 1.5 h. Stirring was continued for at least an additional 6 h. The slurry was filtered. Filter cake was washed with ethyl acetate (1.0 L) and discarded. The combined filtrate and washings were concentrated to -400 mL. Toluene (2.0 L) was added and the solution was washed with 2M aqueous hydrochloric acid (2 x 400 mL). The aqueous layer was warmed to 50° to 60 0C for approx. 20 h or hydrolysis of 6 was deemed complete (HPLC). Aqueous sodium hydroxide solution was added to adjust to pH -10, and mixture was extracted with toluene (3×600 mL). The organic phase was discarded and pH was readjusted to ~6 by addition of aqueous hydrochloric acid. The aqueous phase was concentrated to -600 mL under reduced pressure and extracted with methylene chloride (at least 3×2.0 L). The combined methylene chloride layers were evaporated under reduced pressure and continuously replaced with THF to obtain a solution of (\R,2S,5R)-2- ((5*)-3-benzyloxycarbonylamino-2-oxo-pyrrolidin-l-yl)-5-(isopropyl-methyl-amino)- cyclohexane carboxylic acid 7 (-148 g) in THF (-4 L). Seed crystals of 8 were added, followed by 25 % solution of sodium methoxide in methanol (81.24 g) below 25 0C. The slurry was held for at least additional 16h with agitation. The product was isolated by filtration and the cake was washed with THF (4×200 mL) and dried to a constant weight in vacuo below 30 0C. Dry (lR,25′,5R)-2-((5)-3-benzyloxycarbonyl- amino-2-oxo-pyrrolidin-l-yl)-5-(isopropyl-methyl-amino)-cyclohexane-carboxylate sodium salt 8 was obtained (139g, -60% yield from 5).

[00233] Example 1, Alternative Preparation, Step 4: Aminoester sodium salt 8 (10Og), diphenyl phosphate (3.86g), tert-BuOH (1275 mL) and toluene (225 mL) were combined and heated to reflux under reduced pressure. Approx. 500 mL of distillate were collected and discarded while being continuously replaced with a solution of toluene in tert-BuOH. Vacuum was removed and distillate was switched to percolate through a column filled with molecular sieves and allowed to return to the vessel. After drying was complete, DPPA (52.4mL; dissolved in 60 mL toluene) was added slowly to the slurry at 80 0C. Upon complete conversion (HPLC), tert- BuOH was removed by vacuum distillation and continuously replaced with toluene. The mixture was cooled to room temperature and washed twice with 10% aqueous K2HPO4 (lx800mL, 1×400 mL) and water (40OmL). The organic phase was heated and concentrated in vacuo to approx. 27OmL. Vacuum was removed and heptane (1.1 L) was added slowly at approx. 80 0C, followed by seeds of 9 (~lg). The slurry was slowly cooled to room temperature and benzyl {(S)-l-[(lS,2R,4R)-2- tert- butoxycarbonylamino-4-(isopropyl-methyl-amino)-cyclo-hexyl]-2-oxo-pyrrolidin-3- yl} -carbamate 9 was isolated by filtration as a white solid (86.76g, 78% yield).

[00234] Example 1, Alternative Preparation, Step 5: The tert-Butyl carbamate 9 (5Og) was dissolved in Toluene (50OmL) and /-PrOH (15OmL). The resulting solution was then heated to 6O0C. Methanesulfonic acid (19.6mL) was added below 65°C. Upon reaction completion (HPLC), the mixture was cooled to RT and triethylamine (69.4mL) added slowly below 25°C. Acetic anhydride was then added below 25°C. After Ih acetic acid (25OmL) was added below 25°C. The toluene phase was discarded and 2-methyl-THF (50OmL) was added to the aqueous phase. The mixture was stirred vigorously and basified with NaOH (25% aqueous solution) to pH 12. The aqueous phase was discarded and the organic layer was washed with brine (25OmL). The organic layer was concentrated under reduced pressure and continuously replaced with /-PrOH. The solution was cooled and filtered to provide benzyl {(5′)-l-[(15r,2R,4R)-2-acetylamino-4-(isopropyl-methyl-amino)-cyclohexyl]-2- oxo-pyrrolidin-3-yl} -carbamate 10 in /-PrOH solution which was used directly in the hydrogenation.
[00235] Example 1, Alternative Preparation, Step 6: To a solution containing acetamide 10 (~61g) in /-PrOH (-625 mL) was added 10% Pd/C wet catalyst (2.5 g) and the suspension was hydrogenated at 30 psig and approx. 25 0C for at least 2 h. Upon completion (HPLC), the catalyst was removed by filtration and the filtrate was concentrated to approx. 550 mL. Water (8.8 mL) was added, followed by 5.6 N hydrochloric acid in /-PrOH solution (69.5 mL). The resulting slurry was held at room temperature overnight. The product was isolated by filtration and the cake was rinsed with /-PrOH (2×100 mL) and dried in vacuo to constant weight at -50 0C to give N-[(li?,25r,5R)-2-((5′)-3-amino-2-oxo-pyrrolidin-l-yl)-5-(isopropyl-methyl- amino)-cyclohexyl]-acetamide 11 (55.6 g, 97% yield) as its hydrochloric acid salt (73.6% free base assay, HPLC).
NH,
CL,
Example 1

[00236] Example 1, Alternative Preparation, Step 7: To 6-trifluoromethyl- quinazolin-4-ol 12 (20.1 g) in MeCN (400 mL) was added 5.5 M solution of sodium methoxide in methanol (17.0 mL). The resulting suspension was distilled under reduced pressure and continuously replaced by MeCN to remove methanol. To the slurry was added DMF (1.4 g), followed by oxalyl chloride (13.0 mL) below 50 0C. Upon reaction completion (HPLC), excess reagent was removed under reduced pressure to give -400 mL of slurry. The mixture was cooled to room temperature and washed with 10 % aqueous K2HPO4 (lxl.O L, 1×0.5 L) to afford 4-chloro-6- trifluoromethyl-quinazoline 13 (-21.2 g) in approx. 450 mL of wet MeCN solution, which was used directly in the subsequent coupling reaction (HPLC purity 99.8 %). [00237] Example 1, Alternative Preparation, Step 8: To a mixture of acetamide 11 (28.5 g, HCl salt, 73.6% free base assay), acetonitrile (100 mL), N,N,-di-isopropyl- N-ethylamine (61 mL) at room temperature was added a solution of 13 (-21.2 g) in MeCN (-450 mL). The homogeneous mixture was held overnight. Upon reaction completion (HPLC), the mixture was concentrated in vacuo to approx. 125 mL. A 9.5% aqueous solution of acetic acid (240 mL) was added and the aqueous phase was extracted with methylene chloride. The aqueous phase was separated and methyl tert- butyl ether (450 mL) was added, followed by 2N aqueous lithium hydroxide solution to adjust to pH >11.5. The organic layer was separated, washed with water and filtered. Approx. half of the ether phase was diluted with methyl tert-bvAyl ether (-250 mL) and concentrated in vacuo. Heptane (45 mL) was added slowly below 60 0C, followed by seed crystals of Example 1 (0.4 g). Additional heptane (125 mL) was added and the mixture was slowly cooled to room temperature and the resulting slurry was held overnight. The product was isolated by filtration, the cake was washed with heptane and dried in vacuo to constant weight to give N-((lR,25′,5R)-5- (isopropylamino)-2-((5′)-2-oxo-3-(6-(trifluoromethyl)-quin-azolin-4- ylamino)pyrrolidin-l-yl)cyclohexyl)acetamide 14 (15.0 g, 85% yield).
Crystallization Procedures for Example 1
[00238] Example 1, Production of bis-BSA salt and purification: The entirety of the amorphous free base from Example 1, Step 11 was dissolved in methanol (600 mL). The resultant solution was heated at 60 0C and charged with benzenesulfonic acid (2.5 eq). The mixture was cooled to room temperature and the resultant white solid was collected by filtration to yield the bis-benzene sulfonic acid salt of the title compound (95 g, 86%). This material was >99% pure by HPLC. This material was further purified by re-crystallization from 80/20 EtOH/H2θ, which provided the salt free from any residual methanol. HPLC purity = 99.8%. 1H ΝMR (500 MHz, D2O) δ ppm 8.75 (1 H, s), 8.66 (1 H, s), 8.25 (1 H, d, J=8.80 Hz), 7.90 (1 H, d, J=8.80 Hz), 7.75 (4 H, d, J=8.25 Hz), 7.43 – 7.57 (6 H, m), 5.42 (1 H, t), 4.33 – 4.44 (1 H, m), 4.09 – 4.19 (1 H, m), 3.83 – 3.91 (1 H, m), 3.74 – 3.83 (2 H, m), 3.61 (1 H, t, J=I 1.55 Hz), 2.75 (3 H, d, J=6.60 Hz), 2.61 – 2.70 (1 H, m), 2.31 – 2.44 (1 H, m), 2.20 – 2.27 (1 H, m), 2.17 (2 H, d, J=12.10 Hz), 1.94 – 2.04 (1 H, m, J=12.65 Hz), 1.90 – 1.95 (3 H, m), 1.72 – 1.91 (2 H, m), 1.37 (3 H, d, J=6.05 Hz), 1.29 (3 H, d, J=6.60 Hz). Differential scanning calorimetry utilized a heating rate of 10 °C/min and revealed a melting / decomposition endotherm with an onset temperature of 297.6 0C and a peak temperature at 299.1 0C. [00239] Example 1, Crystallization of the Free Base: A sample of the amorphous free base of N-((lR,25r,5R)-5-(isopropyl(methyl)amino)-2-((5′)-2-oxo-3- (6-(trifluoromethyl)quinazolin-4-ylamino)pyrrolidin- 1 -yl)cyclohexyl)acetamide ( 1 g) was dissolved in dichloromethane (5 mL). The solution was charged with heptane (30 mL) and then warmed to distill the dichloromethane. The solution was cooled to 40 0C; a white solid precipitated. The suspension was heated to 90 0C and stirred for 2 h. The suspension was cooled to room temperature and filtered to provide the pure free base of the title compound. No residual solvent was apparent by 1H-NMR.
![]()
PATENT
US 7671062
http://google.com/patents/US7671062
The present invention provides a novel antagonist or partial agonists/antagonists of MCP-1 receptor activity: N-((1R,2S,5R)-5-(isopropyl(methyl)amino)-2-((S)-2-oxo-3-(6-(trifluoromethyl)quinazolin-4-ylamino)pyrrolidin-1-yl)cyclohexyl)acetamide,

or a pharmaceutically acceptable salt, solvate or prodrug, thereof, having an unexpected combination of desirable pharmacological characteristics. Crystalline forms of the present invention are also provided. Pharmaceutical compositions containing the same and methods of using the same as agents for the treatment of inflammatory diseases, allergic, autoimmune, metabolic, cancer and/or cardiovascular diseases is also an objective of this invention. The present disclosure also provides a process for preparing compounds of Formula (I), including N-((1R,2S,5R)-5-(isopropyl(methyl)amino)-2-((S)-2-oxo-3-(6-(trifluoromethyl)quinazolin-4-ylamino)pyrrolidin-1-yl)cyclohexyl)acetamide:

wherein R1, R8, R9, R10, and
are as described herein. Compounds that are useful intermediates of the process are also provided herein.
1st embodiment, the disclosure provides a process for preparing a compound of formula IV, or a salt thereof:

Example 1 N-((1R,2S,5R)-5-(isopropyl(methyl)amino)-2-((S)-2-oxo-3-(6-(trifluoromethyl)quinazolin-4-ylamino)pyrrolidin-1-yl)cyclohexyl)acetamide

Example 1, Step 1: (1R,2S,5R)-tert-Butyl 2-benzyloxycarbonylamino-7-oxo-6-aza-bicyclo[3.2.1]octane-6-carboxylate (89.6 g, 0.24 mol, see: P. H. Carter, et al. PCT application WO 2005/021500) was dissolved in ethyl acetate (1.5 L) and the resulting solution was washed with sat. NaHCO3 (2×0.45 L) and sat. NaCl (1×0.45 L). The solution was dried (Na2SO4) and then filtered directly into a 3-necked 3 L round-bottom flask. The solution was purged with direct nitrogen injection before being charged with 10% Pd/C (13.65 g) under nitrogen atmosphere. The flask was evacuated and back-filled with hydrogen; this was repeated twice more. Hydrogen was bubbled through the solution for 30 min and then the reaction was stirred under 1 atm H2 for 18 h. The flask was evacuated, back-filled with nitrogen, and charged with fresh catalyst (6 g of 10% Pd/C). Hydrogen was bubbled through the solution for 30 min and then the reaction was stirred under 1 atm H2 for 18 h. The flask was evacuated and back-filled with nitrogen. The mixture was filtered through Celite; the filter pad was then washed with ethyl acetate. The filtrate (˜1.6 L EtOAc volume) was diluted with acetonitrile (0.3 L) and charged sequentially with L-N-Cbz-methionine (68 g, 0.24 mol), TBTU (77 g, 0.24 mol), and N,N-diisopropylethylamine (42 mL, 0.24 mol). The reaction was stirred at room temperature for 4 h, during which time it changed from a suspension to a clear solution. The reaction was quenched with the addition of sat. NH4Cl (0.75 L) and water (0.15 L); the mixture was diluted further with EtOAc (0.75 L). The phases were mixed and separated and the organic phase was washed with sat. Na2CO3 (2×0.9 L) and sat. NaCl (1×0.75 L). The solution was dried (Na2SO4), filtered, and concentrated in vacuo to give (1R,2S,5R)-tert-butyl 2-((S)-2-(benzyloxycarbonylamino)-4-(methylthio)butanamido)-7-oxo-6-aza-bicyclo[3.2.1]octane-6-carboxylate as an oil, which was taken into the next step without further purification. LC/MS for primary peak: [M-Boc+H]+=406.3; [M+Na]+=528.3. 1H-NMR (400 MHz, d4-MeOH): δ 7.36 (m, 5H), 5.11 (s, 2H), 4.32 (m, 1H), 4.2 (m, 1H), 4.0 (m, 1H), 2.5-2.7 (m, 3H), 2.25 (m, 1H), 2.11 (s, 3H), 2.05 (m, 4H), 1.9 (m, 1H), 1.7 (m, 2H), 1.54 (s, 9H). Also present are EtOAc [1.26 (t), 2.03 (s), 4.12 (q)] and N,N,N,N-tetramethylurea [2.83 (s)].
Example 1, Step 2: A sample of (1R,2S,5R)-tert-butyl 2-((S)-2-(benzyloxycarbonylamino)-4-(methylthio)butanamido)-7-oxo-6-aza-bicyclo[3.2.1]octane-6-carboxylate (0.24 mol assumed; see previous procedure) was dissolved in iodomethane (1,250 g) and stirred for 48 h at room temperature. The reaction was concentrated in vacuo. The residue was dissolved in dichloromethane and concentrated in vacuo. This was repeated twice more. The resultant sludge was dissolved in dichloromethane (0.4 L) and poured into a rapidly stirring solution of MTBE (4.0 L). The resultant yellow solids were collected via suction filtration and dried under high vacuum to afford the sulfonium salt (179 g). This material was taken into the next step without further purification. LC/MS for primary peak: [M-Me2S+H]+=458.4; [M]+=520.4. 1H-NMR (400 MHz, d4-MeOH): δ 7.35 (m, 5H), 5.09 (s, 2H), 4.33 (m, 1H), 4.28 (m, 1H), 3.98 (m, 1H), 3.3-3.45 (m, 2H), 2.97 (s, 3H), 2.94 (s, 3H), 2.78 (m, 1H), 2.0-2.3 (m, 4H), 1.7 (m, 2H), 1.52 (s, 9H). Also present are MTBE [1.18 (s), 3.2 (s)] and traces of N,N,N,N-tetramethylurea [2.81 (s)].
Example 1, Step 3: All of the sulfonium salt from the previous step (0.24 mol assumed) was dissolved in DMSO (2.0 L). The resultant solution was stirred under nitrogen at room temperature and charged with cesium carbonate (216 g) portionwise. The suspension was stirred at room temperature for 3 h and then filtered to remove the solids. The solution was divided into ˜0.22 L portions and worked up as follows: the reaction mixture (˜0.22 L) was diluted with ethyl acetate (1.5 L) and washed successively with water (3×0.5 L) and brine (1×0.3 L). The organic phase was dried (Na2SO4), filtered, and concentrated in vacuo. The desired (1R,2S,5R)-tert-butyl 2-((S)-3-(benzyloxycarbonylamino)-2-oxopyrrolidin-1-yl)-7-oxo-6-azabicyclo[3.2.1]octane-6-carboxylate (90.8 g, 83%) was obtained as a microcrystalline foam, free from tetramethyl urea impurity. LC/MS for primary peak: [M-Boc+H]+=358.4; [M+Na]+=480.4. 1H-NMR (400 MHz, d4-MeOH): δ 7.35 (m, 5H), 5.12 (s, 2H), 4.35 (m, 2H), 4.2 (m, 1H), 3.6 (m, 1H), 3.3 (m, 1H), 2.64 (m, 1H), 2.28-2.42 (m, 2H), 2.15 (m, 1H), 1.7-2.0 (m, 5H), 1.55 (s, 9H). If desired, this material can be isolated as a solid by dissolving in MTBE (1 volume), adding to heptane (3.3 volumes), and collecting the resultant precipitate.
Example 1, Step 4: A stirring solution of (1R,2S,5R)-tert-butyl 2-((S)-3-(benzyloxycarbonylamino)-2-oxopyrrolidin-1-yl)-7-oxo-6-azabicyclo[3.2.1]octane-6-carboxylate (108 g, 0.236 mol) in THF (1 L) was charged with lithium hydroxide monohydrate (21.74 g, 0.519 mol). Water (0.3 L) was added slowly, such that the temperature did not exceed 20° C. The reaction was stirred at room temperature overnight and the volatiles were removed in vacuo. The pH was adjusted to ˜4 through the addition of 1N HCl (450 mL) and NaH2PO4. The resultant white precipitates were collected by filtration and washed with water (2×1 L). The solid was dissolved in dichloromethane (1.5 L) and water (˜1 L). The organic layer was dried (Na2SO4), filtered, and concentrated in vacuo. The residue was dissolved in EtOAc (0.7 L) and the resultant solution was heated at reflux for 1 h. Solids separated after cooling to RT, and were collected via filtration. These solids were purified by recrystallization in isopropanol to afford the desired (1R,2S,5R)-2-((S)-3-(benzyloxycarbonylamino)-2-oxopyrrolidin-1-yl)-5-(tert-butoxycarbonylamino)cyclohexanecarboxylic acid as a white solid (104.5 g, 93% yield). LC/MS for primary peak: [M-tBu+H]+=420.2; [M-Boc+H]+=376.2; [M+H]+=476.2. 1H-NMR (400 MHz, d4-MeOH): δ 7.35 (m, 5H), 5.11 (s, 2H), 4.35 (m, 2H), 3.71 (m, 1H), 3.45-3.6 (m, 2H), 2.99 (m, 1H), 2.41 (m, 1H), 2.15 (m, 1H), 2.0 (m, 2H), 1.6-1.9 (m, 4H), 1.46 (s, 9H).
Example 1, Step 5: A 3 L round bottom flask was charged with (1R,2S,5R)-2-((S)-3-(benzyloxycarbonylamino)-2-oxopyrrolidin-1-yl)-5-(tert-butoxycarbonylamino)cyclohexanecarboxylic acid (75.5 g, 0.158 mol), EDC.HCl (33.5 g, 0.175 mol), 1-hydroxybenzotriazole (23.6 g, 0.175 mol), and dichloromethane (1 L). The reaction was stirred at room temperature for 2 h, during which time it changed from a white suspension to a clear solution. Ammonia (gas) was bubbled into the solution until the pH was strongly basic (paper) and the reaction was stirred for 10 min; this ammonia addition was repeated and the reaction was stirred for an additional 10 min. Water was added. The organic phase was washed with sat. NaHCO3, NaH2PO4, and brine before being concentrated in vacuo. The residue was slurried with acetonitrile (0.5 L) and then concentrated in to give (1R,2S,5R)-2-((S)-3-(benzyloxycarbonylamino)-2-oxopyrrolidin-1-yl)-5-(tert-butoxycarbonylamino)cyclohexanecarboxamide as a white solid (75.9 g, ˜100%), which was used in the next step without further purification. LC/MS for primary peak: [M-Boc+H]+=375.3; [M+H]+=475.4; [M-tBu+H]+=419.3. 1H-NMR (400 MHz, d4-MeOH): δ 7.35 (m, 5H), 5.11 (s, 2H), 4.25 (m, 2H), 3.70 (m, 1H), 3.6 (m, 1H), 3.45 (m, 1H), 2.91 (m, 1H), 2.38 (m, 1H), 2.12 (m, 1H), 1.9-2.05 (m, 2H), 1.65-1.9 (m, 4H), 1.46 (s, 9H).
Example 1, Step 6: The reaction was run in three equal portions and combined for aqueous workup. A 5 L, 3-necked round bottom flask was charged with (1R,2S,5R)-2-((S)-3-(benzyloxycarbonylamino)-2-oxopyrrolidin-1-yl)-5-(tert-butoxycarbonylamino)cyclohexanecarboxamide (25.3 g, 53 mmol), acetonitrile (1.9 L), and 2.6 L of water/ice. The mixture was stirred and cooled to 0° C. Iodobenzene diacetate (25.77 g, 80 mmol) was added and the reaction was stirred for 2 h; another 0.5 eq of iodobenzene diacetate was added. The reaction was stirred for 9 h (reaction temp<10° C.). The mixture was charged with 8 eq N,N-diisopropylethylamine and 2 eq acetic anhydride. Over the next thirty minutes, 4 eq N,N-diisopropylethylamine and 2 eq acetic anhydride were added every ten minutes, until the reaction had proceeded to completion (HPLC). The acetonitrile was removed in vacuo; some solid separated from the residue, and this was collected by filtration. The remaining residue was extracted with dichloromethane (3 L, then 1 L). The organic phase was washed sequentially with water, sat. NaHCO3, and brine. The collected solids were added to the organic phase, along with activated carbon (15 g). The mixture was stirred for 30 minutes at 40° C. before being filtered and concentrated in vacuo. The residue was dissolved in EtOAc (1 L), and the resultant solution was stirred at 75° C. for 1 h before being allowed to cool to room temperature. A solid separated and was collected by filtration. This solid was purified further by recrystallization: it was first dissolved in 0.5 L CH2Cl2, then concentrated in vacuo, then re-crystallized from 1 L EtOAc; this was repeated three times. The solids obtained from the mother liquors of the above were recrystallized three times using the same method. The combined solids were recrystallized twice more from acetonitrile (0.7 L) to provide 66 g (84%) of tert-butyl (1R,3R,4S)-3-acetamido-4-((S)-3-(benzyloxycarbonylamino)-2-oxopyrrolidin-1-yl)cyclohexylcarbamate (purity>99.5% by HPLC). LC/MS for primary peak: [M+H]+=489.4; [M-tBu+H]+=433.3. 1H-NMR (400 MHz, d4-MeOH): δ 7.3-7.4 (m, 5H), 5.11 (s, 2H), 4.35 (m, 1H), 4.15 (m, 1H), 4.04 (m, 1H), 3.8 (m, 1H), 3.6 (m, 2H), 2.44 (m, 1H), 2.12 (m, 1H), 1.87-2.05 (m, 4H), 1.87 (s, 3H), 1.55-1.7 (m, 2H), 1.46 (s, 9H). The stereochemical fidelity of the Hofmann rearrangement was confirmed through X-ray crystal structure analysis of this compound, as shown in FIG. 1.
Example 1, Step 7: A stirring solution of tert-butyl (1R,3R,4S)-3-acetamido-4-((S)-3-(benzyloxycarbonylamino)-2-oxopyrrolidin-1-yl)cyclohexylcarbamate (66 g, 0.135 mol) in dichloromethane (216 mL) was charged with trifluoroacetic acid (216 mL). The reaction was stirred for 2 h at room temperature and concentrated in vacuo. The residue was dissolved in methanol and the resultant solution was concentrated in vacuo; this was repeated once. Benzyl (S)-1-((1S,2R,4R)-2-acetamido-4-aminocyclohexyl)-2-oxopyrrolidin-3-ylcarbamate was obtained as an oil and used directly in Step 8 below. LC/MS found [M+H]+=389.4. 1H-NMR (400 MHz, d4-MeOH): δ 7.3-7.4 (m, 5H), 5.12 (s, 2H), 4.41 (br. s, 1H), 4.15 (m, 1H), 4.00 (t, J=9.3 Hz, 1H), 3.81 (t, J=9.1 Hz, 1H), 3.65 (q, J=8.4 Hz, 1H), 3.3-3.4 (m, 1H), 2.45 (m, 1H), 1.95-2.24 (m, 5H), 2.00 (s, 3H), 1.6-1.8 (m, 2H).
Example 1, Step 8: A stirring solution of benzyl (S)-1-((1S,2R,4R)-2-acetamido-4-aminocyclohexyl)-2-oxopyrrolidin-3-ylcarbamate (˜0.135 mol) in methanol (675 mL) was charged sequentially with acetone (37.8 g, 4 eq), sodium acetate (33.2 g, 3 eq), and sodium cyanoborohydride (16.9 g, 2 eq). The mixture was stirred at room temperature for 6 h and filtered. The filtrate was dissolved in dichloromethane (1 L); this solution was washed with 1N NaOH (1 L). The solids collected in the filtration were dissolved in 1N NaOH (1 L) at 0° C. and then extracted with dichloromethane (1 L). The organic extracts were combined and extracted with aqueous HCl (200 mL 1N HCl+800 mL water). The aqueous phase was basified with sat. NaHCO3 (500 mL) and then 1N NaOH (100 mL) until pH 11. The aqueous phase was extracted with dichloromethane (2 L). The organic extracts were combined, dried (Na2SO4), filtered, and concentrated in vacuo to give benzyl (S)-1-((1S,2R,4R)-2-acetamido-4-(isopropylamino)cyclohexyl)-2-oxopyrrolidin-3-ylcarbamate as an oil. LC/MS found [M+H]+=431.45. 1H-NMR (400 MHz, d4-MeOH): δ 7.3-7.4 (m, 5H), 5.12 (s, 2H), 4.31 (m, 1H), 4.24 (t, J=9.4 Hz, 1H), 4.11 (m, 1H), 3.61 (t, J=9.1 Hz, 1H), 3.52 (q, J=8.6 Hz, 1H), 3.04 (br. s, 1H), 2.96 (sep, J=6.3 Hz, 1H), 2.40 (m, 1H), 2.15 (m, 1H), 1.92 (s, 3H), 1.7-1.9 (m, 5H), 1.65 (m, 1H), 1.12 (app. dd, J=6.3, 1.1 Hz, 6H).
Example 1, Step 9 (See Alternative Step 9, below): A stirring solution of benzyl (S)-1-((1S,2R,4R)-2-acetamido-4-(isopropylamino)cyclohexyl)-2-oxopyrrolidin-3-ylcarbamate (˜115 mmol) in dichloromethane (600 mL) was cooled to 0° C. and charged sequentially with formaldehyde (18.6 g, 37 wt % solution), triethylamine (23 mL), and sodium triacetoxyborohydride (28.7 g). The mixture was stirred at room temperature for 30 minutes and diluted with dichloromethane (up to 1.2 L). This solution was washed thrice with 500 mL sat. NaHCO3+NaOH (sat. NaHCO3, pH to 11 w/1N NaOH). The organic layer was extracted with aq. HCl (200 mL 1N HCl+600 mL water). The aqueous phase was basified with sat. NaHCO3 (500 mL) and then 1N NaOH (100 mL) until pH 11. The aqueous phase was extracted with dichloromethane (1.2 L). The organic extracts were combined, dried (Na2SO4), filtered, and concentrated in vacuo to give benzyl (S)-1-((1S,2R,4R)-2-acetamido-4-(isopropyl(methyl)amino)cyclohexyl)-2-oxopyrrolidin-3-ylcarbamate as an oil, which was used directly in Step 10 below. LC/MS found [M+H]+=445.4. 1H-NMR (400 MHz, d4-MeOH): δ 7.3-7.4 (m, 5H), 5.12 (s, 2H), 4.33 (br s, 1H), 4.25 (t, J=9.2 Hz, 1H), 4.11 (br s, 1H), 3.5-3.6 (m, 2H), 2.77 (v br s, 2H), 2.41 (m, 1H), 2.26 (s, 3H), 2.0-2.1 (m, 2H), 1.92 (s, 3H), 1.7-1.9 (m, 5H), 1.10 (app. dd, J=17, 6.4 Hz, 6H).
Example 1, Step 10: To a solution of benzyl (S)-1-((1S,2R,4R)-2-acetamido-4-(isopropyl(methyl)amino)-cyclohexyl)-2-oxopyrrolidin-3-ylcarbamate (0.115 mol) in methanol (600 mL) was added 10% Pd/C (6 g of 50% wet catalyst). The flask was evacuated and back-filled with hydrogen. The mixture was stirred under 1 atm H2 for 2 h and the catalyst was removed by filtration through Celite. The filtrate was concentrated in vacuo to provide N-((1R,2S,5R)-2-((S)-3-amino-2-oxopyrrolidin-1-yl)-5-(isopropyl(methyl)amino)cyclohexyl)acetamide as an oil, which was taken on to the next step without further purification. LC/MS found [M+H]+=311.47. 1H-NMR (400 MHz, d4-MeOH): δ 4.39 (br s, 1H), 4.00 (m, 1H), 3.3-3.5 (m, 4H), 2.73 (m, 1H), 2.38 (m, 1H), 2.25 (s, 3H), 2.0-2.2 (m, 3H), 1.94 (s, 3H), 1.6-1.75 (m, 4H), 1.07 (app. dd, J=21, 6.4 Hz, 6H).
Example 1, Step 11: To a solution of N-((1R,2S,5R)-2-((S)-3-amino-2-oxopyrrolidin-1-yl)-5-(isopropyl(methyl)amino)cyclohexyl)acetamide (˜35 g, 0.115 mol) in isopropanol (600 mL) was added 4-chloro-6-(trifluoromethyl)quinazoline (32 g, 0.138 mol, 1.2 eq, see: P. H. Carter et al., PCT application WO 2005/021500). The mixture was stirred at room temperature overnight before being charged with triethylamine (46 g, 0.46 mol, 4 eq). The mixture was stirred at 60° C. for 10 h. The solvent was removed under reduced pressure to give an oil. Azeotropic distillation with isopropanol was performed twice. The residue was dissolved in dichloromethane (600 mL) and extracted with water (250 mL, containing 4 eq acetic acid). Dichloromethane (600 mL) was added to the combined aqueous washes, and the mixture was cooled to 0° C. Aqueous NaOH (50% by weight) was added with stirring until the pH reached 11. The water layer was extracted with dichloromethane twice (2×600 mL). The combined organic extracts were dried (Na2SO4), filtered, and concentrated in vacuo to give the amorphous free base of the title compound (99% purity by HPLC). LC/MS found [M+H]+=507.3. 1H-NMR (400 MHz, d4-MeOH): δ 8.82 (s, 1H), 8.59 (s, 1H), 8.05 (dd, J=8.8, 1.8 Hz, 1H), 7.9 (d, J=8.7 Hz, 1H), 5.28 (t, J=8.6 Hz, 1H), 4.58 (br s, 1H), 4.06 (m, 1H), 3.52-3.68 (m, 2H), 3.43 (m, 1H), 2.76 (br s, 1H), 2.55 (m, 1H), 2.28 (s, 3H), 2.1-2.3 (m, 3H), 2.0 (s, 3H), 2.0 (m, 1H), 1.65-1.8 (m, 3H), 1.09 (app. dd, J=24, 6.4 Hz, 6 H).
Example 1 Alternative Step 9

Example 1, Alternative step 9ai: To a hydrogenator were charged ethyl (7R,8S)-8-((S)-1-phenyl-ethylamino)-1,4-dioxa-spiro[4.5]decane-7-carboxylate 4-toluenesulfonate salt 1A (1417 g, 2.8 moles, c.f.: WO2004098516, prepared analogous to U.S. Pat. No. 6,835,841), ethanol (200 proof, 11.4 L), and 10% Pd/C catalyst (50% wet, 284 g). The mixture was inerted with nitrogen, then pressurized with hydrogen gas (45 psig) and agitated vigorously at approx. 40° C. until starting material was consumed (HPLC). The suspension was cooled, purged with nitrogen gas and the catalyst was removed by filtration while inerted. The spent catalyst was washed with ethanol (4.3 L). The filtrate and washings were combined and concentrated under vacuum to a volume of 2-3 L while maintaining the batch between 40°-60° C. Isopropyl acetate (5 L) was charged and the mixture was concentrated to a volume of ˜2 L until most ethanol was removed (<0.5%) and residual moisture content was <1,000 ppm. Batch volume was adjusted to ˜7.5 L by the addition of isopropyl acetate. The mixture was heated to 80° C. until clear, then cooled 65°-70° C. Seed crystals of 1 (5 g) were added and the batch was cooled to 50° C. over 2 hours, then further cooled to 20° C. over 4 hours and held for ˜10 hours. The resulting slurry was filtered and the cake was washed with isopropyl acetate (2 L). The product was dried under vaccum at ˜35° C. until volatiles were reduced below ˜1% (LOD). Ethyl (7R,8S)-8-amino-1,4-dioxa-spiro[4.5]decane-7-carboxylate 4-toluenesulfonate salt 1 was obtained as a white, crystalline solid (936 g, 83% yield; HPLC purity: 99.8%). 1H-NMR: (300 MHz, CDCl3) 8.14-7.89 (brs, 3H), 7.75 (d, J 9.0 Hz, 2H), 7.15 (d, J 8.0 Hz, 2H), 4.22-4.04 (m, 2H), 4.01-3.77 (m, 4H), 3.55-3.43 (m, 1H,), 3.20-3.13 (m, 1H), 2.40-2.27 (m, 4H), 2.21-1.94 (m, 2H), 1.81-1.51 (m, 3H), 1.23 (t, J 7.0 Hz, 3H); HPLC: Waters Xterra MS C18 4.6 mm×150 mm i.d., 3.5 μm particle size, 0.05% NH4OH (5% ACN, 95% H2O, solvent A), to 0.05% NH4OH (95% ACN, 5% H2O, solvent B), 5% B to 20% B in 10 minutes, changed to 95% B in 25 minutes, and then changed to 5% B in 1 minute; 11.1 minutes (aminoester 1).

Example 1, Alternative Step 9aii: Aminoester 1 (63 g, 0.16M, 1 eq.; the product of reductive deprotection of a known compound—(See e.g. R. J. Cherney, WO 2004/098516 and G. V. Delucca & S. S. Ko, WO 2004/110993) was placed in a round bottom flask and MeCN (500 mL) was added. EDAC (33.1 g, 0.17M, 1.1 eq), HOBt.H2O (21.2 g, 0.16M, 1.0 eq) and N-Cbz-L-methionine (46.7 g, 0.17M, 1.05 eq) were then added followed by TEA (48.0 mL, 0.35M, 2.2 eq). An exotherm to 38° C. was observed. The reaction mass was left to stir at RT. After 30 mins, HPLC indicated complete conversion. The reaction mass was diluted with EtOAc (2.5 L) and washed with KHCO3 (4×500 mL, 20 wt % aq. solution) and brine (500 mL). The organic phase was separated, dried over MgSO4 and concentrated. The residue was dissolved in TBME and reconcentrated to give ethyl (7R,8S)-8-{(2S)-2-benzyloxycarbonylamino-4-methylsulfanyl-butyr-yl-amino}-1,4-dioxa-spiro[4.5]decane-7-carboxylate 2 as a sticky semi-solid (76.2 g, 98% yield, 93 AP purity). 1H-NMR: (300 MHz, CDCl3) δ 7.36-7.30 (m, 5H), 7.03 (d, J 9.0 Hz, 1H), 5.66 (d, J 8.0 Hz, 1H), 5.10 (s, 2H), 4.35-4.25 (m, 2H), 4.19-4.04 (m, 2H,), 3.98-3.86 (m, 4H), 2.87-2.80 (m, 1H), 2.55-2.45 (m, 2H), 2.18 (dd, J 14.0 Hz, 7.0 Hz, 1H), 2.08 (s, 3H), 2.05-1.67 (m, 6H), 1.26 (t, J 7.0 Hz, 3H). HPLC: YMC-Pack Pro C18 5 μm 4.6×150 mm, 0.05% TFA (20% MeOH, 80% H2O), to 0.05% TFA (20% MeOH, 80% MeCN), 0-100% 10 min gradient. 10.01 min (Compound 2, 93.1 AP). HRMS: m/z 495.2166 [Calc: C24H35N2O7S 495.2165].

Example 1, Alternative Step 9b: Methionine amide 2 (75.0 g, 0.15M) was dissolved in MeI (225 mL, 3 mL/g)—some off gassing was noted but no exotherm. The reaction mass was left to stir in the dark for 16.5 h. After this time a thick light yellow precipitate had formed. The flask was then evacuated to 200 mmHg and some of the MeI removed. The remaining material was slurried in TBMF (500 mL), after a 30 min stir-out the slurry was filtered, the cake washed with TBMF (500 mL). NMR analysis of this material indicated a small amount of MeI remaining. The cake was re-slurried in TBMF (500 mL), filtered, washed with TBMF (500 mL) and dried under vacuum to give [(3S)-3-benzyloxycarbonylamino-3-{(7R,8S)-7-ethoxycarbonyl-1,4-di-oxa-spiro[4.5]dec-8-ylcarbamoyl}-propyl]-dimethylsulfonium iodide 3 as a free flowing off-white solid (93.5 g, 97%, 99 area % purity). 1H-NMR: (300 MHz, CDCl3) δ 7.75 (d, J 9.0 Hz, 1H), 7.38-7.27 (m, 5H), 6.40 (d, J 7.0 Hz, 1H), 5.10 (s, 2H), 4.76-4.65 (m, 1H), 4.48-4.39 (m, 1H), 4.14-3.85 (m, 6H), 3.84-7.73 (m, 1H), 3.68-3.55 (m, 1H), 3.21 (s, 3H), 3.12 (s, 3H), 2.90-2.83 (s, 1H), 2.52-1.55 (m, 8H), 1.24 (t, J 7.0 Hz, 3H). HPLC: YMC-Pack Pro C18 5 μm 4.6×150 mm, 0.05% TFA (20% MeOH, 80% H2O), to 0.05% TFA (20% MeOH, 80% MeCN), 0-100% 10 min gradient. 2.45 min (I−), 8.14 min (Compound 3, 43.6 AP, I−54.6 AP). HRMS: m/z 509.2341 [Calc: C25H37N2O7S 509.2321].

Example 1, Alternative Step 9c: Cs2CO3 (61.5 g, 0.19M, 1.5 eq) was placed in an round bottom flask and anhydrous DMSO (2.4 L) was added. Sulfonium salt 3 (80.0 g, 0.13M, 1.0 eq) was then added portionwise. Once the addition was complete the reaction mass was left to stir in the dark for 20 h. The reaction mass was then split in half and each half worked up separately: the reaction mass was diluted with EtOAc (2.0 L) and washed with brine (2 L), the organic phase was washed with brine (500 mL). The combined aq. layers were then washed EtOAc (500 mL). The combined organic phases were then washed with brine (3×750 mL). The second half of the reaction mass was treated in an identical manner and the combined organics dried over MgSO4 and concentrated to give ethyl (7R,8S)-8-{(3S)-3-Benzyloxycarbonylamino-2-oxo-pyrrolidin-1-yl}-1,4-dioxa-spiro[4.5]decane-7-carboxylate 4 as a light colored oil (56.5 g, 0.13M, ˜100 area-% purity) pure by NMR analysis. 1H-NMR: (300 MHz, CDCl3) δ 7.38-7.30 (m, 5H), 5.37 (br d, J 4.0 Hz, 1H), 5.11 (s, 2H), 4.27-4.18 (m, 1H), 4.17-3.82 (m, 8H), 3.32 (td, J 10.0Hz, 60.0 Hz, 1H), 3.23 (q, J 5.0 Hz, 1H), 2.63-2.57 (m, 1H), 2.42-2.25 (m, 2H), 1.94-1.68 (m, 5H), 1.25 (t, J 7.0 Hz, 3H). HPLC: YMC-Pack Pro C18 5 μm 4.6×150 mm, 0.05% TFA (20% MeOH, 80% H2O), to 0.05% TFA (20% MeOH, 80% MeCN), 0-100% 10 min gradient. 8.99 min (Compound 5, produced on column, 4.2 AP), 9.48 (Compound 4, 74.3 AP). HRMS: m/z 447.2127 [Calc: C23H31N2O7 447.2131].

Example 1, Alternative Step 9d: Pyrrolidinone 4 (50.0 g, 0.11M) was dissolved in acetone (500 mL) and 1N HCl (500 mL) was added. The reaction mass was then heated to 65° C. After 20 mins HPLC indicated complete reaction. The reaction mass was allowed to cool to RT and the acetone was removed on a rotary evaporator. During this distillation the product precipitated from solution as a white solid. This was isolated by filtration and the cake washed with water. The cake was then dried azeotropically with toluene (3×300 mL) to give ethyl (1R,2S)-2-((3S)-3-Benzyloxycarbonylamino-2-oxo-pyrrolidin-1-yl)-5-oxo-cyclohexanecarboxylate 5 as a white solid (39.8 g, 88%, 97 area-% purity). 1H-NMR: (300 MHz, CDCl3) δ 7.37-7.32 (m, 5H), 6.65 (br d, J 4.0 Hz, 1H), 5.12 (s, 2H), 4.54-4.47 (m, 1H), 4.34-4.26 (m, 1H), 4.18 (dq, J 11.0 Hz, 7.0 Hz, 1H), 4.09 (dq, J 11.0 Hz, 7.0 Hz, 1H), 3.36-3.20 (m, 3H), 2.70-2.35 (m, 6H), 2.05-1.96 (m, 1H), 1.81 (quin., J 11.0 Hz, 1H), 1.24 (t, J 7.0 Hz, 3H). HPLC: YMC-Pack Pro C18 5 μm 4.6×150 mm, 0.05% TFA (20% MeOH, 80% H2O), to 0.05% TFA (20% MeOH, 80% MeCN), 0-100% 10 min gradient. 8.95 min (Compound 5). HRMS: m/z 403.1864 [Calc: C21H27N2O6403.1869].

Example 1, Alternative Step 9e: Cyclohexanone 5 (22.5 g, 0.06M, 1 eq), DMSO (30 mL) and Ti(O-iPr)4 (33.7 mL, 0.11M, 2.04 eq) were placed in a round bottom flask. N-isopropyl-N-methylamine (11.6 mL, 0.11M, 2.0 eq) was then added in one portion. The mixture was left to stir for 30 mins at room temperature before being cooled to <3° C. in ice/water. MeOH (30 mL) was then added followed by the portionwise addition of NaBH4 (4.33 g, 0.11M, 2.04 eq)—temperature kept <8° C. 30 mins after the addition was completed the reaction mass was diluted with methylene chloride (300 mL) and then NaOH (1N, 40 mL). The resulting slurry was filtered through Celite, and the cake washed with methylene chloride (100 mL). The resulting liquor was concentrated under reduced pressure and the residue dissolved in EtOAc (500 mL). This solution was extracted with 1N HCl (2×400 mL), the combined aqueous layers were then basified with Na2CO3. Extraction with EtOAc (4×250 mL) provided a clear and colorless organic phase which was dried over Na2SO4 and concentrated to give a white powder (24.6 g, 96%, 7:1 d.r.). This material was then slurried overnight in hexane (670 mL). The solid was isolated by filtration and dried under reduced pressure to give ethyl (1R,2S,5R)-2-((3S)-3-benzyloxycarbonylamino-2-oxo-pyrrolidin-1-yl)-5-(isopropyl-methyl-amino)-cyclohexanecarboxylate 6 as a while solid (20.9 g, 81%, 24:1 d.r.). 1H-NMR: (300 MHz, CDCl3) δ 7.37-7.28 (m, 5H), 5.55 (d, J 4.5, 1H), 5.10 (s, 2H), 4.42 (q, J 4.5, 1H), 4.23-4.12 (m, 1H), 4.08 (dq, J 10.5, 7.0, 1H), 4.02 (dq, J 10.5, 7.0, 1H), 3.84 (t, J 9.0, 1H), 3.46-3.36 (m, 1H), 3.04 (septet, J 6.5, 1H), 2.86-2.80 (m, 1H), 2.63-2.48 (m, 2H), 2.17 (s, 3H, Me), 2.10-1.63 (m, 7H), 1.22 (t, J 7.0, 3H), 1.00 (d, J 6.5, 3H), 0.97 (d, J 6.5, 3H). HPLC: YMC-Pack Pro C18 5 μm 4.6×150 mm, 0.01M NH4OAc (MeOH:water 20:80) to 0.01M NH4OAc (MeOH:water:MeCN 20:5:75) 10 to 100% 15 min gradient. 8.23 (Compound 6), 8.88 (5-epi-Compound 6). HRMS: 460.2798 [Calc: C25H38N3O5 460.2811].

Example 1, Alternative Step 9f: The aminoester 6 (9.76 g, 2.12 mmol) was dissolved in 2N HCl (80 mL), then heated to ˜55° C. under inert atmosphere. The reaction was stirred for 20 h, then cooled to room temperature. The reaction solution was washed twice with toluene (25 mL portions), neutralized to pH 6-7 by the addition of KOH pellets, then extracted eight times with methylene chloride (100 mL portions). The combined extracts were dried (Na2SO4), filtered, and concentrated under reduced pressure to 50 mL total volume. The concentrated solution was then slowly added to methyl tert-butyl ether (300 mL) over 15 min in an addition funnel with vigorous stirring. The resulting white slurry was stirred at ambient temperature for Ih, then cooled to 0° C. and stirred for 1 h. The product was filtered, and washed twice with methyl tert-butyl ether (25 mL portions). Water from the wet cake was removed by azeotropic distillation with acetonitrile (300 mL). The product was dried under reduced pressure to provide (1R,2S,5R)-2-((3S)-3-Benzyloxycarbonylamino-2-oxo-pyrrolidin-1-yl)-5-(isopropyl-methyl-amino)-cyclohexanecarboxylic acid 7, (7.69 g, 84% yield) as a white foam. 1H-NMR: (400 MHz, 50° C., CDCl3) δ 7.44-7.32 (m, 5H), 6.10 (broad s, 1H), 5.19 (app s, 2H), 4.42 (dd, J=15.6, 7.8 Hz, 1H), 4.29-4.23 (m, 1H), 3.68-3.60 (m, 2H), 3.33-3.27 (m, 2H), 3.20 (broad s, 1H), 2.99 (broad s, 1H), 2.51 (s, 3H), 2.49-2.45 (m, 3H), 2.33-2.31 (m, 1H), 2.00 (ddd, J=9.0, 8.6, 3.9 1H), 1.95-1.78 (m, 2H), 1.36-1.21 (m, 6H). LCMS: m/z 432.20 [Calc: C23H34N3O5 432.25].

Example 1, Alternative Step 9g: Amino acid 7 (6.3 g, 14.7 mmol, 1.0 eq) was dissolved in THF (80 mL) under N2 and NaH (584 mg, 14.7 mmol, 1.0 eq, 60 wt % dispersion in mineral oil) was added portionwise. When the addition was complete, and the evolution of gas had ceased, the reaction mass was concentrated under reduced pressure and the resulting solid azeotroped with toluene (50 mL) to give a white solid (KF 0.59 wt %). This solid was slurried in toluene (100 mL) under N2and heated to 90° C. DPPA (3.32 mL, 15.3 mmol, 1.05 eq) was added dropwise over ˜2 min. After ˜5 min all the solids had dissolved, after 10 mins precipitation of a white solid was observed. After 30 mins HPLC analysis indicated complete reaction. The reaction mass was allowed to cool to RT before being filtered, the cake was washed with toluene. The liquors where then slowly added into AcOH/Ac2O (80/20, 168 mL) solution at 90° C. After 45 mins HPLC still indicated some isocyanate. At 1.15 h, the reaction mass was cooled to RT and diluted with toluene (100 mL) and water (100 mL). The organic layer was removed and the toluene washed with 1N HCl (100 mL). The combined aq. phases were then basified with K2CO3(s) and brought to pH 12 with NaOH (10N), keeping the temperature below 20° C. The aq layer was then extracted with methylene chloride (4×150 mL), the combined organic layers dried over K2CO3 and concentrated to give benzyl (S)-1-((1S,2R,4R)-2-acetamido-4-(isopropyl(methyl)amino)cyclohexyl)-2-oxopyrrolidin-3-ylcarbamate 8 as a white foam (4.5 g, 70%, 94AP purity). The 1H-NMR was identical to material obtained from the route described above (Example 1, Step 9). HPLC: YMC-Pack Pro C18 5 μm 4.6×150 mm, 0.05% TFA (20% MeOH, 80% H2O), to 0.05% TFA (20% MeOH, 80% MeCN), 0-100% 10 min gradient. 7.20 min (Compound 8), 7.85 min (urea dimer). HRMS: 445.2809 [Calc: C24H37N4O4445.2815].
Alternative Preparation of Example 1

Example 1, Alternative Preparation, Step 1: Ethyl (7R,8S)-8-amino-1,4-dioxa-spiro[4.5]decane-7-carboxylate 4-toluenesulfonate salt 1 (450.1 g), was combined with 1-ethyl-3-(3-dimethyl-amino-propyl)carbo-diimide hydrochloride (236.3 g), 1-hydroxy benzotriazole hydrate (171.9 g), N-carbobenzyloxy-L-methionine (333.4 g) and acetonitrile (3.1 L). To the stirred mixture was added triethylamine (249.5 g) below 30° C. Upon reaction completion (HPLC), the mixture was diluted with ethyl acetate (8.2 L) and washed with aqueous 25% potassium bicarbonate solution (2×4.5 L) followed by water (4.5 L). The organic phase was separated and concentrated under reduced pressure to obtain a solution of ethyl (7R,8S)-8-((S)-2-benzyloxycarbonylamino-4-methylsulfanyl-butyrylamino)-1,4-dioxa-spiro[4.5]decane-7-carboxylate 2 (1.4 L). Methyl iodide (2.39 kg) was added, the vessel was shielded from light and the mixture was held under slow agitation for approx. 24 h. To the thick yellow precipitate was added methyl tert-butyl ether (2.7 L) and the mixture was held for approx. 1 h. The product was isolated by filtration and the cake was washed with methyl tert-butyl ether (2×1.4 L), then dried under vacuum, yielding [(S)-3-benzyloxy-carbonylamino-3-((7R,8S)-7-ethoxycarbonyl-1,4-dioxa-spiro[4.5]dec-8-ylcarbamoyl)-propyl]-dimethylsulfonium iodide 3 (671.4 g, ˜94% yield) as an off-white solid (HPLC purity 99.9%).

Example 1, Alternative Preparation, Step 2: Sulfonium salt 3 (619.4 g), and cesium carbonate (416.8 g) and anhydrous dimethyl sulfoxide (6.2 L) were combined in a reactor equipped with a scrubber to neutralize volatile sulfides. Vigorous agitation was maintained until complete conversion was obtained (HPLC). Ethyl acetate (12.4 L) was added, followed by 20% brine (3 L). The organic phase was separated, washed twice with brine (2×3 L) and evaporated to obtain a solution of ethyl (7R,8S)-8-((S)-3-benzyloxycarbonylamino-2-oxo-pyrrolidin-1-yl)-1,4-dioxa-spiro[4.5]decane-7-carboxylate 4 in ethyl acetate (˜0.8 L). Acetone (2.55 L) was added, followed by aqueous 0.5 M hydrochloric acid solution (2.3 L). With good mixing, the solution was heated to 50 to 60° C. until conversion of 4 to ethyl (1R,2S)-2-((S)-3-benzyloxycarbonylamino-2-oxo-pyrrolidin-1-yl)-5-oxo-cyclohexanecarboxylate 5 was complete (HPLC). The mixture was concentrated under reduced pressure while below 40° C., cooled to ˜30° C., and water (4.1 L) was added. The resulting slurry was cooled to 5 to 10° C. and agitated for ˜1 hour. The product was filtered and the cake was washed with water (2×2.5 L). Upon deliquoring, the cake was dried to a constant weight below 40° C. in a vacuum oven. Cyclohexanone 5 (272 g, 70% yield) was obtained (HPLC purity 98.7%).

Example 1, Alternative Preparation, Step 3: Cyclohexanone 5 (206 g) was dissolved in dichloromethane (1.1 L) and charged to a hydrogenator. Titanium tetraisopropoxide (218.2 g) and N-isopropyl N-methylamine (63.64 g) were added and the mixture was stirred at ambient temperature (23 to 25° C.) for at least 5 h. Platinum catalyst (5% Pt/S/C, 15 g, approx. 7.5% relative to 5) was added and hydrogenation was performed at ˜30 psig for at least 6 h, yielding a mixture of ethyl (1R,2S,5R)-2-((S)-3-benzyloxycarbonylamino-2-oxo-pyrrolidin-1-yl)-5-(isopropyl-methyl-amino)-cyclohexanecarboxylate 6 and its 5-epi-isomer (˜7%). The catalyst was removed by filtration and the filtrate was concentrated under reduced pressure to approx. ˜600 mL. Wet ethyl acetate (˜3% water, 2.0 L) was added with vigorous agitation over a period of at least 1.5 h. Stirring was continued for at least an additional 6 h. The slurry was filtered. Filter cake was washed with ethyl acetate (1.0 L) and discarded. The combined filtrate and washings were concentrated to ˜400 mL. Toluene (2.0 L) was added and the solution was washed with 2M aqueous hydrochloric acid (2×400 mL). The aqueous layer was warmed to 50° to 60° C. for approx. 20 h or hydrolysis of 6 was deemed complete (HPLC). Aqueous sodium hydroxide solution was added to adjust to pH ˜10, and mixture was extracted with toluene (3×600 mL). The organic phase was discarded and pH was readjusted to ˜6 by addition of aqueous hydrochloric acid. The aqueous phase was concentrated to ˜600 mL under reduced pressure and extracted with methylene chloride (at least 3×2.0 L). The combined methylene chloride layers were evaporated under reduced pressure and continuously replaced with THF to obtain a solution of (1R,2S,5R)-2-((S)-3-benzyloxycarbonylamino-2-oxo-pyrrolidin-1-yl)-5-(isopropyl-methyl-amino)-cyclohexane carboxylic acid 7 (˜148 g) in THF (˜4 L). Seed crystals of 8 were added, followed by 25% solution of sodium methoxide in methanol (81.24 g) below 25° C. The slurry was held for at least additional 16 h with agitation. The product was isolated by filtration and the cake was washed with THF (4×200 mL) and dried to a constant weight in vacuo below 30° C. Dry (1R,2S,5R)-2-((S)-3-benzyloxycarbonyl-amino-2-oxo-pyrrolidin-1-yl)-5-(isopropyl-methyl-amino)-cyclohexane-carboxylate sodium salt 8 was obtained (139 g, ˜60% yield from 5).

Example 1, Alternative Preparation, Step 4: Aminoester sodium salt 8 (100 g), diphenyl phosphate (3.86 g), tert-BuOH (1275 mL) and toluene (225 mL) were combined and heated to reflux under reduced pressure. Approx. 500 mL of distillate were collected and discarded while being continuously replaced with a solution of toluene in tert-BuOH. Vacuum was removed and distillate was switched to percolate through a column filled with molecular sieves and allowed to return to the vessel. After drying was complete, DPPA (52.4 mL; dissolved in 60 mL toluene) was added slowly to the slurry at 80° C. Upon complete conversion (HPLC), tert-BuOH was removed by vacuum distillation and continuously replaced with toluene. The mixture was cooled to room temperature and washed twice with 10% aqueous K2HPO4 (1×800 mL, 1×400 mL) and water (400 mL). The organic phase was heated and concentrated in vacuo to approx. 270 mL. Vacuum was removed and heptane (1.1 L) was added slowly at approx. 80° C., followed by seeds of 9 (˜1 g). The slurry was slowly cooled to room temperature and benzyl {(S)-1-[(1S,2R,4R)-2-tert-butoxycarbonylamino-4-(isopropyl-methyl-amino)-cyclo-hexyl]-2-oxo-pyrrolidin-3-yl}-carbamate 9 was isolated by filtration as a white solid (86.76 g, 78% yield).

Example 1, Alternative Preparation, Step 5: The tert-Butyl carbamate 9 (50 g) was dissolved in Toluene (500 mL) and i-PrOH (150 mL). The resulting solution was then heated to 60° C. Methanesulfonic acid (19.6 mL) was added below 65° C. Upon reaction completion (HPLC), the mixture was cooled to RT and triethylamine (69.4 mL) added slowly below 25° C. Acetic anhydride was then added below 25° C. After 1 h acetic acid (250 mL) was added below 25° C. The toluene phase was discarded and 2-methyl-THF (500 mL) was added to the aqueous phase. The mixture was stirred vigorously and basified with NaOH (25% aqueous solution) to pH 12. The aqueous phase was discarded and the organic layer was washed with brine (250 mL). The organic layer was concentrated under reduced pressure and continuously replaced with i-PrOH. The solution was cooled and filtered to provide benzyl {(S)-1-[(1S,2R,4R)-2-acetylamino-4-(isopropyl-methyl-amino)-cyclohexyl]-2-oxo-pyrrolidin-3-yl}-carbamate 10 in i-PrOH solution which was used directly in the hydrogenation.
Example 1, Alternative Preparation, Step 6: To a solution containing acetamide 10 (˜61 g) in i-PrOH (˜625 mL) was added 10% Pd/C wet catalyst (2.5 g) and the suspension was hydrogenated at 30 psig and approx. 25° C. for at least 2 h. Upon completion (HPLC), the catalyst was removed by filtration and the filtrate was concentrated to approx. 550 mL. Water (8.8 mL) was added, followed by 5.6 N hydrochloric acid in i-PrOH solution (69.5 mL). The resulting slurry was held at room temperature overnight. The product was isolated by filtration and the cake was rinsed with i-PrOH (2×100 mL) and dried in vacuo to constant weight at ˜50° C. to give N-[(1R,2S,5R)-2-((S)-3-amino-2-oxo-pyrrolidin-1-yl)-5-(isopropyl-methyl-amino)-cyclohexyl]-acetamide 11 (55.6 g, 97% yield) as its hydrochloric acid salt (73.6% free base assay, HPLC).

Example 1, Alternative Preparation, Step 7: To 6-trifluoromethyl-quinazolin-4-ol 12 (20.1 g) in MeCN (400 mL) was added 5.5 M solution of sodium methoxide in methanol (17.0 mL). The resulting suspension was distilled under reduced pressure and continuously replaced by MeCN to remove methanol. To the slurry was added DMF (1.4 g), followed by oxalyl chloride (13.0 mL) below 50° C. Upon reaction completion (HPLC), excess reagent was removed under reduced pressure to give ˜400 mL of slurry. The mixture was cooled to room temperature and washed with 10% aqueous K2HPO4 (1×1.0 L, 1×0.5 L) to afford 4-chloro-6-trifluoromethyl-quinazoline 13 (˜21.2 g) in approx. 450 mL of wet MeCN solution, which was used directly in the subsequent coupling reaction (HPLC purity 99.8%).
Example 1, Alternative Preparation, Step 8: To a mixture of acetamide 11 (28.5 g, HCl salt, 73.6% free base assay), acetonitrile (100 mL), N,N,-di-isopropyl-N-ethylamine (61 mL) at room temperature was added a solution of 13 (˜21.2 g) in MeCN (˜450 mL). The homogeneous mixture was held overnight. Upon reaction completion (HPLC), the mixture was concentrated in vacuo to approx. 125 mL. A 9.5% aqueous solution of acetic acid (240 mL) was added and the aqueous phase was extracted with methylene chloride. The aqueous phase was separated and methyl tert-butyl ether (450 mL) was added, followed by 2N aqueous lithium hydroxide solution to adjust to pH>11.5. The organic layer was separated, washed with water and filtered. Approx. half of the ether phase was diluted with methyl tert-butyl ether (˜250 mL) and concentrated in vacuo. Heptane (45 mL) was added slowly below 60° C., followed by seed crystals of Example 1 (0.4 g). Additional heptane (125 mL) was added and the mixture was slowly cooled to room temperature and the resulting slurry was held overnight. The product was isolated by filtration, the cake was washed with heptane and dried in vacuo to constant weight to give N-((1R,2S,5R)-5-(isopropylamino)-2-((S)-2-oxo-3-(6-(trifluoromethyl)-quin-azolin-4-ylamino)pyrrolidin-1-yl)cyclohexyl)acetamide 14 (15.0 g, 85% yield).
Crystallization Procedures for Example 1Example 1, Production of bis-BSA salt and purification: The entirety of the amorphous free base from Example 1, Step 11 was dissolved in methanol (600 mL). The resultant solution was heated at 60° C. and charged with benzenesulfonic acid (2.5 eq). The mixture was cooled to room temperature and the resultant white solid was collected by filtration to yield the bis-benzene sulfonic acid salt of the title compound (95 g, 86%). This material was >99% pure by HPLC. This material was further purified by re-crystallization from 80/20 EtOH/H2O, which provided the salt free from any residual methanol. HPLC purity=99.8%. 1H NMR (500 MHz, D2O) δ ppm 8.75 (1H, s), 8.66 (1H, s), 8.25 (1H, d, J=8.80 Hz), 7.90 (1H, d, J=8.80 Hz), 7.75 (4H, d, J=8.25 Hz), 7.43-7.57 (6H, m), 5.42 (1H, t), 4.33-4.44 (1H, m), 4.09-4.19 (1H, m), 3.83-3.91 (1H, m), 3.74-3.83 (2H, m), 3.61 (1H, t, J=11.55 Hz), 2.75 (3H, d, J=6.60 Hz), 2.61-2.70 (1H, m), 2.31-2.44 (1H, m), 2.20-2.27 (1H, m), 2.17 (2H, d, J=12.10 Hz), 1.94-2.04 (1H, m, J=12.65 Hz), 1.90-1.95 (3H, m), 1.72-1.91 (2H, m), 1.37 (3H, d, J=6.05 Hz), 1.29 (3H, d, J=6.60 Hz). Differential scanning calorimetry utilized a heating rate of 10° C./min and revealed a melting/decomposition endotherm with an onset temperature of 297.6° C. and a peak temperature at 299.1° C.
Example 1, Crystallization of the Free Base: A sample of the amorphous free base of N-((1R,2S,5R)-5-(isopropyl(methyl)amino)-2-((S)-2-oxo-3-(6-(trifluoromethyl)quinazolin-4-ylamino)pyrrolidin-1-yl)cyclohexyl)acetamide (1 g) was dissolved in dichloromethane (5 mL). The solution was charged with heptane (30 mL) and then warmed to distill the dichloromethane. The solution was cooled to 40° C.; a white solid precipitated. The suspension was heated to 90° C. and stirred for 2 h. The suspension was cooled to room temperature and filtered to provide the pure free base of the title compound. No residual solvent was apparent by 1H-NMR.

PAPER

A concise bulk synthesis of stereochemically complex CCR2 antagonist BMS-741672 is reported. A distinct structural feature is the chiral all-cis 1,2,4-triaminocyclohexane (TACH) core, which was assembled through consecutive stereocontrolled heterogeneous hydrogenations: efficient Pt-catalyzed reduction of a β-enaminoester, directed by (S)-α-methylbenzylamine as a low-cost chiral template, and reductive amination of a 3,4-cis-disubstituted cyclohexanone over sulfided Pt/C introduced a tert-amine, setting the third stereocenter in the all-cis cyclohexane core. The heterogeneous catalysts were recycled. Ester hydrolysis produced a γ-amino acid, isolated as its Na salt. A challenging Curtius reaction to introduce the remaining C–N bond at C-2 was strongly influenced by the presence of the basic tert-amine, providing a stereoelectronically highly activated isocyanate. Detailed mechanistic and process knowledge was required to enable clean trapping with an alcohol (t-BuOH) while avoiding formation of side products, particularly an unusual carbamoyl phosphate. Deprotection, N-acetylation, and uncatalyzed SNAr coupling with known 4-chloroquinazoline provided the final product. The resulting 12-step synthesis was used to prepare 50 kg of the target compound in an average yield of 82% per step.
Stereoselective Bulk Synthesis of CCR2 Antagonist BMS-741672: Assembly of an All-cis (S,R,R)-1,2,4-Triaminocyclohexane (TACH) Core via Sequential Heterogeneous Asymmetric Hydrogenations
Chemical and Synthetic Development, Bristol-Myers Squibb Company, One Squibb Drive, New Brunswick, New Jersey 08901, United States
Org. Process Res. Dev., Article ASAP
DOI: 10.1021/acs.oprd.6b00282


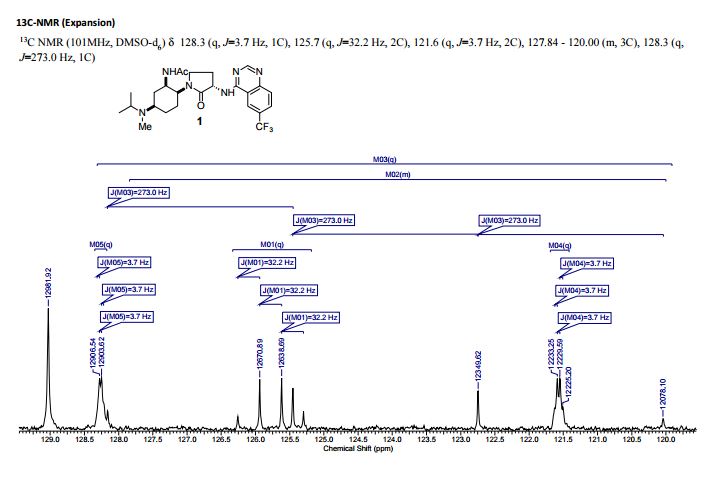

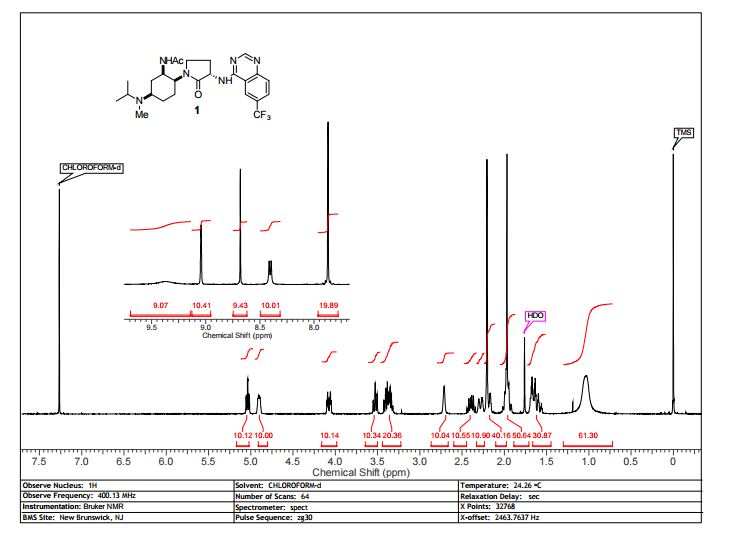
Patents
| Patent ID | Date | Patent Title |
|---|---|---|
| US7687508 | 2010-03-30 | CYCLIC DERIVATIVES AS MODULATORS OF CHEMOKINE RECEPTOR ACTIVITY |
| US7671062 | 2010-03-02 | N-((IR, 2S, 5R)-5-(ISOPROPYL(METHYL)AMINO)-2-((S)-2-0XO-3-(6-TRIFLUOROMETHYL)QUINAZOLIN-4-YLAMINO)PYRROLIDIN-1-YL)CYCLOHEXYL)ACETAMIDE AND OTHER MODULATORS OF CHEMOKINE RECEPTOR ACTIVITY, CRYSTALLINE FORMS AND PROCESS. |
Bristol-Myers Squibb, Paul Biondi Senior Vice President, Head of Business Development
About Bristol-Myers Squibb
Bristol-Myers Squibb is a global biopharmaceutical company whose mission is to discover, develop and deliver innovative medicines that help patients prevail over serious diseases. For more information, please visit www.bms.com or follow us on Twitter at http://twitter.com/bmsnews.
////////// ////////////////BMS-741672, BMS 741672, BRISTOL MEYER SQUIB, PHASE 2, type 2 Diabetes, Neuropathic Pain, Bristol-Myers Squibb
CC(C)N(C)[C@H]1C[C@@H](NC(C)=O)[C@H](CC1)N4CC[C@H](Nc3ncnc2ccc(cc23)C(F)(F)F)C4=O
BMS-986115

BMS-986115
CAS 1584647-27-7
(2R,3S)-N-((3S)-5-(3-Fluorophenyl)-9-methyl-2-oxo-2,3-dihydro-lH-l,4-benzodiazepin- 3-yl)-2, -bis(3,3,3-trifluoropropyl)succinamide
MW: 574.4945, C26-H25-F7-N4-O3, UNII: LSK1L593UU
10-Nitrooleate, CTK3B7458, CTK3C3167, 9-Octadecenoic acid, 10-nitro-, 875685-46-4, AG-L-63109, 9-Octadecenoic acid, 10-nitro-, (9E)-, 88127-53-1
FOR advanced solid tumors
- Originator Bristol-Myers Squibb
- Class Antineoplastics
- Mechanism of Action Amyloid precursor protein secretase inhibitors; Notch signalling pathway inhibitors
- Phase I Solid tumours
Most Recent Events
- 30 Aug 2016Bristol-Myers Squibb terminates a phase I trial for Solid tumours (late-stage disease, second-line therapy or greater) in USA, Australia and Canada (NCT01986218)
- 25 Jan 2016Bristol-Myers Squibb completes enrolment in its phase I trial for Solid tumours in USA, Australia and Canada (NCT01986218)
- 31 Dec 2013Phase-I clinical trials in Solid tumours (late-stage disease) in Canada & Australia (Oral)
DETAILS WILL BE UPDATED SOON………….
BMS-986115 is an orally bioavailable, gamma secretase (GS) and pan-Notch inhibitor, with potential antineoplastic activity. Upon administration, GS/pan-Notch inhibitor BMS 986115 binds to GS and blocks the proteolytic cleavage and release of the Notch intracellular domain (NICD), which would normally follow ligand binding to the extracellular domain of the Notch receptor. This prevents both the subsequent translocation of NICD to the nucleus to form a transcription factor complex and the expression of Notch-regulated genes. This results in the induction of apoptosis and the inhibition of growth of tumor cells that overexpress Notch. Overexpression of the Notch signaling pathway plays an important role in tumor cell proliferation and survival
| Ashvinikumar V. Gavai, George V. Delucca,Daniel O’MALLEY, Patrice Gill, Claude A. Quesnelle, Brian E. Fink, Yufen Zhao,Francis Y. Lee, | |
| Applicant | Bristol-Myers Squibb Company |

Ashvinikumar Gavai

Claude Quesnelle
Senior Research Investigator/Chemist at Bristol-Myers Squibb

RICHARD LEE


Patrice Gill
Research scientist at BMS

Dan O’Malley (Rice University)
Currently: Bristol-Myers Squibb
PICTURES WILL BE UPDATED………….
Useful for the treatment of conditions related to the Notch pathway, such as cancer and other proliferative diseases.
Notch signaling has been implicated in a variety of cellular processes, such as cell fate specification, differentiation, proliferation, apoptosis, and angiogenesis. (Bray, Nature Reviews Molecular Cell Biology, 7:678-689 (2006); Fortini, Developmental Cell 16:633-647 (2009)). The Notch proteins are single-pass heterodimeric transmembrane molecules. The Notch family includes 4 receptors, NOTCH 1-4, which become activated upon binding to ligands from the DSL family (Delta-like 1, 3, 4 and Jagged 1 and 2).
The activation and maturation of NOTCH requires a series of processing steps, including a proteolytic cleavage step mediated by gamma secretase, a multiprotein complex containing Presenilin 1 or Presenilin 2, nicastrin, APH1, and PEN2. Once NOTCH is cleaved, NOTCH intracellular domain (NICD) is released from the membrane. The released NICD translocates to the nucleus, where it functions as a transcriptional activator in concert with CSL family members (RBPSUH, “suppressor of hairless”, and LAG1). NOTCH target genes include HES family members, such as HES- 1. HES- 1 functions as transcriptional repressors of genes such as HERP 1 (also known as HEY2), HERP2 (also known as HEY1), and HATH1 (also known as ATOH1).
The aberrant activation of the Notch pathway contributes to tumorigenesis. Activation of Notch signaling has been implicated in the pathogenesis of various solid tumors including ovarian, pancreatic, as well as breast cancer and hematologic tumors such as leukemias, lymphomas, and multiple myeloma. The role of Notch inhibition and its utility in the treatment of various solid and hematological tumors are described in Miele, L. et al, Current Cancer Drug Targets, 6:313-323 (2006); Bolos, V. et al, Endocrine Reviews, 28:339-363 (2007); Shih, I.-M. et al, Cancer Research, 67: 1879- 1882 (2007); Yamaguchi, N. et al., Cancer Research, 68: 1881-1888 (2008); Miele, L., Expert Review Anti-cancer Therapy, 8: 1 197-1201 (2008); Purow, B., Current Pharmaceutical Biotechnology, 10: 154-160 (2009); Nefedova, Y. et al, Drug Resistance Updates, 1 1 :210-218 (2008); Dufraine, J. et al, Oncogene, 27:5132-5137 (2008); and Jun, H.T. et al, Drug Development Research, 69:319-328 (2008).
There remains a need for compounds that are useful as Notch inhibitors and that have sufficient metabolic stability to provide efficacious levels of drug exposure. Further, there remains a need for compounds useful as Notch inhibitors that can be orally or intravenously administered to a patient.
U.S. Patent No. 7,053,084 Bl discloses succinoylamino benzodiazepine compounds useful for treating neurological disorders such as Alzheimer’s Disease. The reference discloses that these succinoylamino benzodiazepine compounds inhibit gamma secretase activity and the processing of amyloid precursor protein linked to the formation of neurological deposits of amyloid protein. The reference does not disclose the use of these compounds in the treatment of proliferative diseases such as cancer.
Applicants have found potent compounds that have activity as Notch inhibitors and have sufficient metabolic stability to provide efficacious levels of drug exposure upon intravenous or oral administration. These compounds are provided to be useful as pharmaceuticals with desirable stability, bioavailability, therapeutic index, and toxicity values that are important to their drugability.
![]()

PATENTS
PATENT
https://www.google.com/patents/US20140087992
Example 1(2R,3S)—N-((3S)-5-(3-Fluorophenyl)-9-methyl-2-oxo-2,3-dihydro-1H-1,4-benzodiazepin-3-yl)-2,3-bis(3,3,3-trifluoropropyl)succinamide
Intermediate 1A: (2S,3R)-tert-Butyl 6,6,6-trifluoro-3-(((S)-5-(3-fluorophenyl)-9-methyl-2-oxo-2,3-dihydro-1H-benzo[e][1,4]diazepin-3-yl)carbamoyl)-2-(3,3,3-trifluoropropyl)hexanoate
In a 100 mL round-bottomed flask, a solution of Intermediate B-1 (1683 mg, 5.94 mmol), Et3N (1.656 mL, 11.88 mmol), and Intermediate S-1 in DMF (20 mL) was treated with o-benzotriazol-1-yl-N,N,N′,N′-tetramethyluronium tetrafluoroborate (3815 mg, 11.88 mmol) and stirred at room temperature for 1 hour. The reaction mixture was diluted with water and saturated aqueous NaHCO3. An off white precipitate formed and was filtered and washed with water. The resulting solid was dried on the filter under a stream of nitrogen to give Intermediate 1A (3.7 g, 99% yield). MS (ES): m/z=632.4[M+H+]; HPLC: RT=3.635 min Purity=98%. (H2O/MeOH with TFA, CHROMOLITH® ODS S5 4.6×50 mm, gradient=4 min, wavelength=220 nm). 1H NMR (400 MHz, methanol-d4) δ 7.53 (t, J=4.5 Hz, 1H), 7.46-7.30 (m, 3H), 7.28-7.23 (m, 1H), 7.23-7.18 (m, 2H), 5.37 (s, 1H), 2.88 (td, J=10.4, 3.4Hz, 1H), 2.60 (td, J=10.2, 4.1 Hz, 1H), 2.54-2.40 (m, 1H), 2.47 (s, 3H), 2.33-2.12 (m, 3H), 1.98-1.69 (m, 4H), 1.51 (s, 9H).
Intermediate 1B: (2S,3R)-6,6,6-Trifluoro-3-(((S)-5-(3-fluorophenyl)-9-methyl-2-oxo-2,3-dihydro-1H-benzo[e][1,4]diazepin-3-yl)carbamoyl)-2-(3,3,3-trifluoropropyl)hexanoic acid
In a 250 mL round-bottomed flask, a solution of Intermediate 1A (3.7 g, 5.86 mmol) in DCM (25 mL) was treated with TFA (25 mL) and the resulting pale orange solution was stirred at room temperature for 1.5 hours. The reaction mixture was then concentrated to give Intermediate 1B. HPLC: RT=3.12 min (H2O/MeOH with TFA, CHROMOLITH® ODS S5 4.6×50 mm, gradient=4 min, wavelength=220 nm). MS (ES): m/z=576.3 (M+H)+. 1H NMR (400 MHz, methanol-d4) δ 7.54 (t, J=4.5 Hz, 1H), 7.49-7.29 (m, 3H), 7.28-7.15 (m, 3H), 5.38 (br. s., 1H), 2.89 (td, J=10.3, 3.7 Hz, 1H), 2.67 (td, J=9.9, 4.2Hz, 1H), 2.56-2.38 (m, 1H), 2.48 (s, 3H), 2.34-2.13 (m, 3H), 2.00-1.71 (m, 4H).
Example 1
In a 250 mL round-bottomed flask, a solution of Intermediate 1B (4.04 g, 5.86 mmol) in THF (50 mL) was treated with ammonia (2M in iPrOH) (26.4 mL, 52.7 mmol), followed by HOBT (1.795 g, 11.72 mmol) and EDC (2.246 g, 11.72 mmol). The resulting white suspension was stirred at room temperature overnight. The reaction mixture was diluted with water and saturated aqueous NaHCO3. The resulting solid was filtered, rinsed with water and then dried on the filter under a stream of nitrogen. The crude product was suspended in 20 mL of iPrOH and stirred at room temperature for 20 min and then filtered and washed with iPrOH and dried under vacuum to give 2.83 g of solid. The solid was dissolved in refluxing EtOH (100 mL) and slowly treated with 200 mg activated charcoal added in small portions. The hot mixture was filtered through CELITE® and rinsed with hot EtOH. The filtrate was reduced to half volume, allowed to cool and the white precipitate formed was filtered and rinsed with EtOH to give 2.57 g of white solid. A second recrystallization from EtOH (70 mL) afforded Example 1 (2.39 g, 70% yield) as a white solid. HPLC: RT=10.859 min (H2O/CH3CN with TFA, Sunfire C18 3.5 μm, 3.0×150 mm, gradient=15 min, wavelength=220 and 254 nm); MS (ES): m/z=575.3 [M+H+]; 1H NMR (400 MHz, methanol-d4) δ 7.57-7.50 (m, 1H), 7.47-7.30 (m, 3H), 7.29-7.15 (m, 3H), 5.38 (s, 1H), 2.85-2.75 (m, 1H), 2.59 (td, J=10.5, 4.0 Hz, 1H), 2.53-2.41 (m, 4H), 2.31-2.10 (m, 3H), 1.96-1.70 (m, 4H).
PATENT
https://www.google.com/patents/WO2014047372A1?cl=en
Scheme 3
XII XI
Scheme 4
Intermediate S-l : (2R,3S)-3-(fert-Butoxycarbonyl)-6,6,6-trifluoro-2-(3,3,3- trifluoropropyl)hexanoic acid
Intermediate S-IA: 3,3,3-Trifluoro ropyl trifluoromethanesulfonate
[00180] To a cold (-25 °C) stirred solution of 2,6-lutidine (18.38 mL, 158 mmol) in DCM (120 mL) was added Tf20 (24.88 mL, 147 mmol) over 3 min, and the mixture was stirred for 5 min. To the reaction mixture was added 3,3,3-trifluoropropan-l-ol (12 g, 105 mmol) over an interval of 3 min. After 2 hr, the reaction mixture was warmed to room temperature and stirred for 1 hr. The reaction mixture was concentrated to half its volume, then purified by loading directly on a silica gel column (330g ISCO) and the product was eluted with DCM to afford Intermediate S-IA (13.74 g, 53%) as a colorless oil. 1H NMR (400 MHz, CDC13) δ ppm 4.71 (2 H, t, J= 6.15 Hz), 2.49-2.86 (2 H, m).
Intermediate S-1B: (4S)-4-Benzyl-3-(5,5,5-trifluoropentanoyl)-l,3-oxazolidin-2-one
[00181] To a stirring solution of 5,5,5-trifluoropentanoic acid (14.76 g, 95 mmol) and DMF (0.146 rriL) in DCM (50 mL) was slowly added oxalyl chloride (8.27 mL, 95 mmol). After 2h, the mixture was concentrated to dryness. A separate flask was changed with (S)-4-benzyloxazolidin-2-one (16.75 g, 95 mmol) in THF (100 mL) and then cooled to -78 °C. To the solution was slowly added n-BuLi (2.5M, 37.8 mL, 95 mmol) over 10 min, stirred for 10 min, and then a solution of the above acid chloride in THF (50 mL) was slowly added over 5 min. The mixture was stirred for 30 min, and then warmed to room temperature. The reaction was quenched with sat aq NH4C1. Next, 10% aq LiCl was then added to the mixture, and the mixture was extracted with Et20. The organic layer was washed with sat aq NaHC03 then with brine, dried (MgSC^), filtered and concentrated to dryness. The residue was purified by Si02 chromatography (ISCO, 330 g column, eluting with a gradient from 100% hexane to 100% EtOAc) to afford the product Intermediate S-IB; (25.25 g, 85%): 1H NMR (400 MHz, CDC13) δ ppm 7.32-7.39 (2 H, m), 7.30 (1 H, d, J= 7.05 Hz), 7.18-7.25 (2 H, m), 4.64-4.74 (1 H, m), 4.17-4.27 (2 H, m), 3.31 (1 H, dd, J= 13.35, 3.27 Hz), 3.00-3.11 (2 H, m), 2.79 (1 H, dd, J= 13.35, 9.57 Hz), 2.16-2.28 (2 H, m), 1.93-2.04 (2 H, m).
Intermediate S-IC: tert- utyl (3R)-3-(((4S)-4-benzyl-2-oxo-l,3-oxazolidin-3- yl)carbonyl)-6,6,6-trifluoroh xanoate
[00182] To a cold (-78 °C), stirred solution of Intermediate S-IB (3.03 g, 9.61 mmol) in THF (20 mL) was added NaHMDS (1.0M in THF) (10.6 mL, 10.60 mmol) under a nitrogen atmosphere. After 2 hours, tert-butyl 2-bromoacetate (5.62 g, 28.8 mmol) was added neat via syringe at -78 °C and stirring was maintained at the same temperature. After 6 hours, the reaction mixture was warmed to room temperature. The reaction mixture was partitioned between saturated NH4C1 and EtOAc. The organic phase was separated, and the aqueous phase was extracted with EtOAc (3x). The combined organics were washed with brine, dried (Na2s04), filtered and concentrated under reduced pressure. The residue was purified by flash chromatography (Teledyne ISCO
CombiFlash Rf, 5% to 100% solvent A/B = hexanes/EtOAc, REDISEP® Si02 120g). Concentration of the appropriate fractions provided Intermediate S-1C (2.79 g, 67.6%) as a colorless viscous oil: 1H NMR (400 MHz, CDC13) δ ppm 7.34 (2 H, d, J= 7.30 Hz), 7.24-7.32 (3 H, m), 4.62-4.75 (1 H, m, J= 10.17, 6.89, 3.43, 3.43 Hz), 4.15-4.25 (3 H, m), 3.35 (1 H, dd, J= 13.60, 3.27 Hz), 2.84 (1 H, dd, J= 16.62, 9.57 Hz), 2.75 (1 H, dd, J = 13.35, 10.07 Hz), 2.47 (1 H, dd, J= 16.62, 4.78 Hz), 2.11-2.23 (2 H, m), 1.90-2.02 (1 H, m), 1.72-1.84 (1 H, m), 1.44 (9 H, s).
Intermediate S-ID: (2R)-2-( -tert-Butoxy-2-oxoethyl)-5,5,5-trifluoropentanoic acid
[00183] To a cool (0 °C), stirred solution of Intermediate S-1C (2.17 g, 5.05 mmol) in THF (50 mL) and water (15 mL) was added a solution of LiOH (0.242 g, 10.11 mmol) and H202 (2.065 mL, 20.21 mmol) in H20 (2 mL). After 10 min, the reaction mixture was removed from the ice bath, stirred for lh, and then cooled to 0 °C. Saturated aqueous NaHCC”3 (25 mL) and saturated aqueous Na2s03 (25 mL) were added to the reaction mixture, and the mixture was stirred for 10 min, and then partially concentrated. The resulting mixture was extracted with DCM (2x), cooled with ice and made acidic with cone. HC1 to pH 3. The mixture was saturated with solid NaCl, extracted with EtOAc (3x), and then dried over MgS04, filtered and concentrated to a colorless oil to afford Intermediate S-ID, 1.2514g, 92%): 1H NMR (400 MHz, CDCI3) δ ppm 2.83-2.95 (1 H, m), 2.62-2.74 (1 H, m), 2.45 (1 H, dd, J= 16.62, 5.79 Hz), 2.15-2.27 (2 H, m), 1.88-2.00 (1 H, m), 1.75-1.88 (1 H, m), 1.45 (9 H, s). Intermediate S-l : (2R,3S)-3-(fert-Butoxycarbonyl)-6,6,6-trifluoro-2-(3,3,3- trifluoropropyl)hexanoic acid, and Intermediate S-1E: (2R,3R)-3-(tert-butoxycarbonyl)- 6,6,6-trifluoro-2-(3,3,3-trifluoropropyl)hexanoic acid
(S-1E)
[00184] To a cold (-78 °C) stirred solution of Intermediate S-1D (5 g, 18.50 mmol) in THF (60 mL) was slowly added LDA (22.2 mL, 44.4 mmol, 2.0M) over 7 min. After stirring for 2 hr, Intermediate S- 1 A (6.38 g, 25.9 mmol) was added to the reaction mixture over 3 min. After 60 min, the reaction mixture was warmed to -25 °C
(ice/MeOH/dry ice) and stirred for an additional 60 min at which time sat aq NH4C1 was added. The separated aqueous phase was acidified with IN HC1 to pH 3, and then extracted with Et20. The combined organic layers were washed with brine (2x), dried over MgS04, filtered and concentrated to provide a 1 :4 (II :I1E) mixture (as determined by 1H NMR) of Intermediate S-l and Intermediate S-1E (6.00 g, 89%) as a pale yellow solid. 1H NMR (500 MHz, CDC13) δ ppm 2.81 (1 H, ddd, J = 10.17, 6.32, 3.85 Hz), 2.63- 2.76 (1 H, m), 2.02-2.33 (4 H, m), 1.86-1.99 (2 H, m), 1.68-1.85 (2 H, m), 1.47 (9 H, s).
[00185] To a cold (-78 °C), stirred solution of a mixture of Intermediate S-l and Intermediate S-1E (5.97 g, 16.30 mmol) in THF (91 mL) was added LDA (19 mL, 38.0 mmol, 2.0M in THF/hexane/ethyl benzene) dropwise via syringe over 10 min (internal temperature never exceeded -65 °C, J-KEM® probe in reaction solution). The mixture was stirred for 15 min, and then warmed to room temperature (24 °C water bath), stirred for 15 min, and then cooled to -78 °C for 15 min. To the reaction mixture was added Et2AlCl (41 mL, 41.0 mmol, 1M in hexane) via syringe (internal temperature never exceeded -55 °C), and the mixture was stirred for 10 min, and then warmed to room temperature (24 °C bath) for 15 min and then back to -78 °C for 15 min. Meanwhile, a 1000 mL round bottom flask was charged with MeOH (145 mL) and precooled to -78 °C. With vigorous stirring the reaction mixture was transferred via cannula over 5 min to the MeOH. The flask was removed from the bath, ice was added followed by the slow addition of IN HC1 (147 mL, 147 mmol). Gas evolution was observed as the HC1 was added. The reaction mixture was allowed to warm to room temperature during which the gas evolution subsided. The reaction mixture was diluted with EtOAc (750 mL), saturated with NaCl, and the organic phase was separated, washed with a solution of potassium fluoride (8.52 g, 147 mmol) and IN HC1 (41 mL, 41.0 mmol) in water (291 mL), brine (100 mL), and then dried (Na2s04), filtered and concentrated under vacuum. 1H NMR showed the product was a 9: 1 mixture of Intermediate S-l and Intermediate S- 1E. The enriched mixture of Intermediate S-l and Intermediate S-1E (6.12 g, >99% yield) was obtained as a dark amber solid: 1H NMR (400 MHz, CDC13) δ ppm 2.64-2.76 (2 H, m), 2.04-2.35 (4 H, m), 1.88-2.00 (2 H, m), 1.71-1.83 (2 H, m), 1.48 (9 H, s).
Alternate procedure to make Intermediate S-l :
Intermediate S-IF: (2R,3 -1 -Benzyl 4-tert-butyl 2,3-bis(3,3,3-trifluoropropyl)succinate
[00186] To a stirred solution of a 9: 1 enriched mixture of Intermediate S-l and Intermediate S-1E (5.98 g, 16.33 mmol) in DMF (63 mL) were added potassium carbonate (4.06 g, 29.4 mmol) and benzyl bromide (2.9 mL, 24.38 mmol), the mixture was then stirred overnight at room temperature. The reaction mixture was diluted with EtOAc (1000 mL), washed with 10% LiCl (3×200 mL), brine (200 mL), dried (Na2S04), filtered, concentrated, and then dried under vacuum. The residue was purified by Si02 chromatography using a toluene:hexane gradient. Diastereomerically purified
Intermediate S-IF (4.81g, 65%) was obtained as a colorless solid: 1H NMR (400 MHz, chloroform-d) δ 7.32-7.43 (m, 5H), 5.19 (d, J= 12.10 Hz, 1H), 5.15 (d, J= 12.10 Hz, 1H), 2.71 (dt, J= 3.52, 9.20 Hz, 1H), 2.61 (dt, J= 3.63, 9.63 Hz, 1H), 1.96-2.21 (m, 4H), 1.69-1.96 (m, 3H), 1.56-1.67 (m, 1H), 1.45 (s, 9H).
Intermediate S-l : (2R,3S)-3-(fert-Butoxycarbonyl)-6,6,6-trifluoro-2-(3,3,3- trifluoropropyl)hexanoic acid
[00187] To a solution of Intermediate S-1F (4.81 g, 10.54 mmol) in MeOH (100 mL) was added 10% palladium on carbon (wet, Degussa type, 568.0 mg, 0.534 mmol) in a H2– pressure flask. The vessel was purged with N2 (4x), then purged with H2 (2x), and finally, pressurized to 50 psi and shaken overnight. The reaction vessel was
depressurized and purged with nitrogen. The mixture was filtered through CELITE®, washed with MeOH and then concentrated and dried under vacuum. Intermediate S-1 (3.81 g, 99% yield)) was obtained as a colorless solid: 1H NMR (400 MHz, chloroform-d) δ 2.62-2.79 (m, 2H), 2.02-2.40 (m, 4H), 1.87-2.00 (m, 2H), 1.67-1.84 (m, 2H), 1.48 (s, 9H).
Alternate procedure to make Intermediate S-1 :
Intermediate S-1 : (2R,3S)-3-(fert-Butoxycarbonyl)-6,6,6-trifluoro-2-(3,3,3- trifluoropropyl)hexanoic acid
[00188] Intermediate S-1 as a mixture with Intermediate S-IE was prepared in a similar procedure as above from Intermediate S-1D to afford a 1 :2.2 mixture of
Intermediate S-1 and Intermediate S-IE (8.60 g, 23.48 mmol), which was enriched using LDA (2.0 M solution in THF, ethyl benzene and heptane, 28.2 mL, 56.4 mmol) and diethyl aluminum chloride (1.0 M solution in hexane, 59 mL, 59.0 mmol) in THF (91 mL). After workup as described above, the resulting residue was found to be a 13.2: 1 (by 1H NMR) mixture of Intermediate S-1 and Intermediate S-IE, which was treated as follows: The crude material was dissolved in MTBE (43 mL). Hexanes (26 mL) were slowly charged to the reaction mixture while maintaining a temperature below 30 °C. The reaction mixture was stirred for 10 min. Next, tert-butylamine (2.7 mL, 1.1 eq) was charged slowly over a period of 20 minutes while maintaining a temperature below 30 °C. This addition was observed to be exothermic. The reaction mixture was stirred for 2 hrs below 30 °C and then filtered. The solid material was washed with 5:3 MTBE: hexane (80 mL), and the filtrate was concentrated and set aside. The filtered solid was dissolved in dichloromethane (300 mL), washed with IN HC1 (lOOmL), and the organic layer was washed with brine (100 mL x 2), and then concentrated under reduced pressure below 45 °C to afford Intermediate S-l (5.46 g, 64%).
A second alternate procedure for preparing Intermediate S-l :
Intermediate S-1G: tert- utyl 5,5,5-trifluoropentanoate
[00189] To a stirred solution of 5,5,5-trifluoropentanoic acid (5 g, 32.0 mmol) in THF (30 mL) and hexane (30 mL) at 0 °C, was added tert-butyl 2,2,2-trichloroacetimidate (11.46 mL, 64.1 mmol). The mixture was stirred for 15 min at 0 °C. Boron trifluoride etherate (0.406 mL, 3.20 mmol) was added and the reaction mixture was allowed to warm to room temperature overnight. To the clear reaction mixture was added solid NaHC03 (5 g) and stirred for 30 min. The mixture was filtered through MgSC^ and washed with hexanes (200 mL). The solution was allowed to rest for 45 min, and the resulting solid material was removed by filtering on the same MgSC^ filter again, washed with hexanes (100 mL) and concentrated under reduced pressure without heat. The volume was reduced to about 30 mL, filtered through a clean fritted funnel, washed with hexane (5 mL), and then concentrated under reduced pressure without heat. The resulting neat oil was filtered through a 0.45μιη nylon membrane filter disk to provide Intermediate S-1G (6.6 g, 31.4 mmol 98% yield) as a colorless oil: 1H NMR (400 MHz, CDC13) δ ppm 1.38 (s, 9 H) 1.74-1.83 (m, 2 H) 2.00-2.13 (m, 2 H) 2.24 (t, J= 7.28 Hz, 2 H). Intermediate S-1H: (4S)-4-(Propan-2-yl)-3-(5,5,5-trifluoropentanoyl)-l,3-oxazolidin-2- one
[00190] To a stirred solution of 5,5,5-trifluoropentanoic acid (5.04 g, 32.3 mmol) in DCM (50 mL) and DMF (3 drops) was added oxalyl chloride (3.4 mL, 38.8 mmol) dropwise over 5 min. The solution was stirred until all bubbling subsided. The reaction mixture was concentrated under reduced pressure to give pale yellow oil. To a separate flask charged with a solution of (4S)-4-(propan-2-yl)-l,3-oxazolidin-2-one (4.18 g, 32.4 mmol) in THF (100 mL) at -78 °C was added n-BuLi (2.5M in hexane) (13.0 mL, 32.5 mmol) dropwise via syringe over 5 min. After stirring for 10 min, the above acid chloride, dissolved in THF (20 mL), was added via cannula over 15 min. The reaction mixture was warmed to 0 °C, and was allowed to warm to room temperature as the bath warmed and stirred overnight. To the reaction mixture was added saturated NH4C1, and the mixture was extracted with EtOAc (2x). The combined organics were washed with brine, dried (Na2s04), filtered and concentrated under reduced pressure. The crude material was purified by flash chromatography (Teledyne ISCO CombiFlash Rf, 5% to 60% solvent A/B = hexanes/EtOAc, REDISEP® Si02 120g). Concentration of the appropriate fractions provided Intermediate S-1H (7.39 g, 86%) as a colorless oil: 1H NMR (400 MHz, CDC13) δ ppm 4.44 (1 H, dt, J= 8.31, 3.53 Hz), 4.30 (1 H, t, J= 8.69 Hz), 4.23 (1 H, dd, J= 9.06, 3.02 Hz), 2.98-3.08 (2 H, m), 2.32-2.44 (1 H, m, J= 13.91, 7.02, 7.02, 4.03 Hz), 2.13-2.25 (2 H, m), 1.88-2.00 (2 H, m), 0.93 (3 H, d, J= 7.05 Hz), 0.88 (3 H, d, J= 6.80 Hz).
Intermediate S-1I: (2S,3R)-tert-Butyl 6,6,6-trifluoro-3-((S)-4-isopropyl-2- oxooxazolidine-3-carbonyl)-2-(3,3,3-trifluoropropyl)hexanoate, and Intermediate S-U: (2R,3R)-tert-Butyl 6,6,6-trifluoro-3-((S)-4-isopropyl-2-oxooxazolidine-3-carbonyl)-2- (3 ,3 ,3 -trifluoropropyl)hexanoate
[00191] To a cold (-78 °C), stirred solution of diisopropylamine (5.3 mL, 37.2 mmol) in THF (59 mL) under a nitrogen atmosphere was added n-BuLi (2.5M in hexane) (14.7 mL, 36.8 mmol). The mixture was then warmed to 0 °C to give a 0.5M solution of LDA. A separate vessel was charged with Intermediate S-1H (2.45 g, 9.17 mmol). The material was azeotroped twice with benzene (the RotoVap air inlet was fitted with a nitrogen inlet to completely exclude humidity), and then toluene (15.3 mL) was added. This solution was added to a flask containing dry lithium chloride (1.96 g, 46.2 mmol). To the resultant mixture, cooled to -78 °C, was added the LDA solution (21.0 mL, 10.5 mmol) and the mixture was stirred at -78 °C for 10 min, then warmed to 0 °C for 10 min., and then cooled to -78 °C. To a separate reaction vessel containing Intermediate S-1G (3.41 g, 16.07 mmol), also azeotroped twice with benzene, was added toluene (15.3 mL), cooled to -78 °C and LDA (37.0 mL, 18.5 mmol) was added. The resulting solution was stirred at -78 °C for 25 min. At this time the enolate derived from the ester was transferred via cannula into the solution of the oxazolidinone enolate and stirred at -78 °C for an additional 5 min, at which time the septum was removed and solid powdered bis(2- ethylhexanoyloxy)copper (9.02 g, 25.8 mmol) was rapidly added to the reaction vessel and the septum was replaced. The vessel was immediately removed from the cold bath and immersed into a warm water bath (40 °C) with rapid swirling and with a concomitant color change from the initial turquoise to brown. The reaction mixture was stirred for 20 min, was then poured into 5% aqueous NH4OH (360 mL) and extracted with EtOAc (2x). The combined organics were washed with brine, dried (Na2s04), filtered and concentrated under reduced pressure. The residue was purified by flash chromatography (Teledyne ISCO CombiFlash Rf, 0% to 60% solvent A/B = hexanes/EtOAc, REDISEP® Si02 120g). Concentration of the appropriate fractions provided a mixture of Intermediate S- II and Intermediate S-1J (2.87 g, 66%) as a pale yellow viscous oil. 1H NMR showed the product was a 1.6: 1 mixture of diastereomers S-1LS-1J as determined by the integration of the multiplets at 2.74 and 2.84 ppm: 1H NMR (400 MHz, CDC13) δ ppm 4.43-4.54 (2 H, m), 4.23-4.35 (5 H, m), 4.01 (1 H, ddd, J= 9.54, 6.27, 3.51 Hz), 2.84 (1 H, ddd, J = 9.41, 7.28, 3.64 Hz), 2.74 (1 H, ddd, J= 10.29, 6.27, 4.02 Hz), 2.37-2.48 (2 H, m, J = 10.38, 6.98, 6.98, 3.51, 3.51 Hz), 2.20-2.37 (3 H, m), 1.92-2.20 (8 H, m), 1.64-1.91 (5 H, m), 1.47 (18 H, s), 0.88-0.98 (12 H, m). Intermediate S-1 : (2R,3S)-3-(fert-Butoxycarbonyl)-6,6,6-trifluoro-2-(3,3,3- trifluoropropyl)hexanoic acid, and Intermediate S-IE: (2R,3R)-3-(tert-Butoxycarbonyl)- 6,6,6-trifluoro-2-(3,3,3-trifluoropropyl)hexanoic acid
(S-IE)
[00192] To a cool (0 °C), stirred solution of Intermediate S-1I and Intermediate S-1 J (4.54 g, 9.51 mmol) in THF (140 mL) and water (42 mL) were sequentially added hydrogen peroxide (30% in water) (10.3 g, 91 mmol) and LiOH (685.3 mg, 28.6 mmol). The mixture was stirred for 1 hr. At this time the reaction vessel was removed from the cold bath and then stirred for 1.5 hr. To the reaction mixture were added saturated NaHC03 (45 mL) and saturated Na2s03 (15 mL), and then the mixture was partially concentrated under reduced pressure. The resulting crude solution was extracted with DCM (3x). The aqueous phase was acidified to pH~l-2 with IN HC1, extracted with DCM (3x) and then EtOAc (lx). The combined organics were washed with brine, dried (Na2s04), filtered and concentrated under reduced pressure to provide a mixture of Intermediates S-1 and S-IE (3.00 g, 86%) as a colorless oil: 1H NMR (400 MHz, CDC13) δ ppm 2.76-2.84 (1 H, m, diastereomer 2), 2.64-2.76 (3 H, m), 2.04-2.35 (8 H, m), 1.88- 2.00 (4 H, m), 1.71-1.83 (4 H, m), 1.48 (9 H, s, diastereomer 1), 1.46 (9 H, s,
diastereomer 2); 1H NMR showed a 1.7: 1 mixture of S-1E:S-1F by integration of the peaks for the t-butyl groups. Intermediate S-1 : (2R,3S)-3-(fert-Butoxycarbonyl)-6,6,6-trifluoro-2-(3,3,3- trifluoropropyl)hexanoic acid, and Intermediate S-IF: (2R,3R)-3-(fert-Butoxycarbonyl)- 6,6,6-trifluoro-2-(3,3,3-trifluoropropyl)hexanoic acid
[00193] To a cold (-78 °C) stirred solution of diisopropylamine (1.7 mL, 11.93 mmol) in THF (19 mL) under a nitrogen atmosphere was added n-BuLi (2.5M in hexanes) (4.8 mL, 12.00 mmol). The mixture was stirred for 5 min and then warmed to 0 °C. In a separate vessel, to a cold (-78 °C) stirred solution of the mixture of Intermediates S-1 and S-1E (1.99 g, 5.43 mmol) in THF (18 mL) was added the LDA solution prepared above via cannula slowly over 25 min. The mixture was stirred for 15 min, then warmed to room temperature (placed in a 24 °C water bath) for 15 min, and then again cooled to -78 °C for 15 min. To the reaction mixture was added Et2AlCl (1M in hexane) (11.4 mL, 11.40 mmol) via syringe. The mixture was stirred for 10 min, warmed to room
temperature for 15 min and then cooled back to -78 °C for 15 min. Methanol (25 mL) was rapidly added, swirled vigorously while warming to room temperature, and then concentrated to ~l/4 the original volume. The mixture was dissolved in EtOAc and washed with IN HC1 (50 mL) and ice (75 g). The aqueous phase was separated and extracted with EtOAc (2x). The combined organics were washed with a mixture of KF (2.85g in 75 mL water) and IN HC1 (13 mL) [resulting solution pH 3-4], then with brine, dried (Na2s04), filtered and concentrated under reduced pressure to give a 9: 1 (S-LS-1E) enriched diastereomeric mixture (as determined by 1H NMR) of Intermediate S-1 and Intermediate S-1E (2.13 g, >99%) as a pale yellow viscous oil: 1H NMR (400 MHz, CDC13) δ ppm 2.64-2.76 (2 H, m), 2.04-2.35 (4 H, m), 1.88-2.00 (2 H, m), 1.71-1.83 (2 H, m), 1.48 (9 H, s).
Intermediate S-2: (2R,3S)-3-(fert-Butoxycarbonyl)-6,6,6-trifluoro-2-(3- fluoropropyl)hexanoic acid
Intermediate S-2: (2R,3S)-3-(tert-Butoxycarbonyl)-7,7,7-trifluoro-2-(3,3,3- trifluoropropyl)heptanoic acid, and Intermediate S-2A: (2R,3R)-3-(tert-Butoxycarbonyl)- 7,7,7-trifluoro-2-(3,3,3-trifluoropropyl)heptanoic acid
(S-2A)
[00194] To a cold (-78 °C), stirred solution of Intermediate S-1D (1.72 g, 6.36 mmol) in THF (30 mL) was slowly added LDA (7.32 mL, 14.6 mmol) over 7 min. After stirring for 1 h, 4,4,4-trifluorobutyltrifluoromethanesulfonate (2.11 g, 8.11 mmol) was added to the reaction mixture over 2 min. After 15 min, the reaction mixture was warmed to -25 °C (ice/MeOH/dry ice) for lh, and then cooled to -78 °C. After 80 min, the reaction was quenched with a saturated aqueous NH4C1 solution (10 mL). The reaction mixture was further diluted with brine and the solution was adjusted to pH 3 with IN HC1. The aqueous layer was extracted with ether. The combined organics were washed with brine, dried over anhydrous magnesium sulfate, and concentrated under reduced pressure to provide a mixture of Intermediates S-2 and S-2A (2.29 g, 95%) as a colorless oil. 1H NMR (400MHz, chloroform-d) δ 2.83-2.75 (m, 1H), 2.64 (ddd, J = 9.9, 6.7, 3.6 Hz, 1H), 2.32-2.03 (m, 5H), 1.98-1.70 (m, 3H), 1.69-1.52 (m, 3H), 1.50-1.42 (m, 9H). 1H NMR showed a 1 :4.5 mixture (S-2:S-2A) of diastereomers by integration of the peaks for the t- Bu groups.
Intermediate S-2: (2R,3S)-3-(fert-Butoxycarbonyl)-7,7,7-trifluoro-2-(3,3,3- trifluoropropyl)heptanoic acid, and Intermediate S-2A: (2R,3R)-3-(tert-Butoxycarbonyl)- 7,7,7-trifluoro-2-(3,3,3-trifluoropropyl)heptanoic acid
[00195] A mixture of Intermediate S-2 and Intermediate S-2A (2.29 g, 6.02 mmol) was dissolved in THF (38 mL) to give a colorless solution which was cooled to -78 °C. Then, LDA (7.23 mL, 14.5 mmol) (2.0M in heptane/THF/ethylbenzene) was slowly added to the reaction mixture over 3 min. After stirring for 15 min, the reaction mixture was placed in a room temperature water bath. After 15 min the reaction mixture was placed back in a -78 °C bath and then diethylaluminum chloride (14.5 mL, 14.5 mmol) (1M in hexane) was added slowly over 5 min. The reaction mixture was stirred at -78 °C. After 15 min, the reaction mixture was placed in a room temperature water bath for 10 min, and then cooled back to -78 °C. After 15 min, the reaction was quenched with MeOH (30.0 mL, 741 mmol), removed from the -78 °C bath and concentrated. To the reaction mixture was added ice and HC1 (60.8 mL, 60.8 mmol) and the resulting mixture was extracted with EtOAc (2x 200 mL). The organic layer was washed with potassium fluoride (3.50g, 60.3 mmol) in 55 mL H20 and 17.0 mL of IN HC1. The organics were dried over anhydrous magnesium sulfate and concentrated under reduced pressure to provide an enriched mixture of Intermediate S-2 and Intermediate S-2A (2.25g, 98% yield) as a light yellow oil. 1H NMR (400MHz, chloroform-d) δ 2.83-2.75 (m, 1H), 2.64 (ddd, J= 9.9, 6.7, 3.6 Hz, 1H), 2.32-2.03 (m, 5H), 1.98-1.70 (m, 3H), 1.69-1.52 (m, 3H), 1.50-1.42 (m, 9H). 1H NMR showed a 9: 1 ratio in favor of the desired diastereomer Intermediate S-2.
Intermediate S-2B: (2R,3S)-1 -Benzyl 4-tert-butyl 2,3-bis(4,4,4-trifluorobutyl)succinate
[00196] To a stirred 9: 1 mixture of Intermediate S-2 and Intermediate S-2A (2.24 g, 5.89 mmoL) and potassium carbonate (1.60 g, 11.58 mmoL) in DMF (30 mL) was added benzyl bromide (1.20 mL, 10.1 mmoL)). The reaction mixture was stirred at room temperature for 19 h. The reaction mixture was diluted with ethyl acetate (400 mL) and washed with 10% LiCl solution (3 x 100 mL), brine (50 mL), and then dried over anhydrous magnesium sulfate, filtered and concentrated to dryness under vacuum. The residue was purified by flash chromatography (Teledyne ISCO CombiFlash 0%> to 100% solvent A/B = hexane/EtOAc, REDISEP® Si02 220 g, detecting at 254 nm, and monitoring at 220 nm). Concentration of the appropriate fractions provided Intermediate S-2B (1.59 g, 57.5%). HPLC: RT = 3.863 min (CHROMOLITH® SpeedROD column 4.6 x 50 mm, 10-90% aqueous methanol over 4 minutes containing 0.1% TFA, 4 mL/min, monitoring at 220 nm), 1H NMR (400MHz, chloroform-d) δ 7.40-7.34 (m, 5H), 5.17 (d, J= 1.8 Hz, 2H), 2.73-2.64 (m, 1H), 2.55 (td, J= 10.0, 3.9 Hz, 1H), 2.16-1.82 (m, 5H), 1.79-1.57 (m, 3H), 1.53-1.49 (m, 1H), 1.45 (s, 9H), 1.37-1.24 (m, 1H).
Intermediate S-2: (2R,3S)-3-(tert-Butoxycarbonyl)-6,6,6-trifluoro-2-(4,4,4- trifluorobutyl)hexanoic acid
[00197] To a stirred solution of Intermediate S-2B (1.59 g, 3.37 mmoL) in MeOH (10 mL) and EtOAc (10 mL) under nitrogen was added 10%> Pd/C (510 mg). The atmosphere was replaced with hydrogen and the reaction mixture was stirred at room temperature for 2.5 h. The palladium catalyst was filtered off through a 4 μΜ polycarbonate film and rinsed with MeOH. The filtrate was concentrated under reduced pressure to give intermediate S-2 (1.28 g, 99%). 1H NMR (400MHz, chloroform-d) δ 2.76-2.67 (m, 1H), 2.65-2.56 (m, 1H), 2.33-2.21 (m, 1H), 2.17-2.08 (m, 3H), 1.93 (dtd, J= 14.5, 9.9, 5.2 Hz, 1H), 1.84-1.74 (m, 2H), 1.70-1.52 (m, 3H), 1.48 (s, 9H).
Intermediate A- 1 : (2-Amino-3 -methylphenyl)(3 -fluorophenyl)methanone
Intermediate A-1 A: 2-Amino- -methoxy-N,3-dimethylbenzamide
[00198] In a 1 L round-bottomed flask was added 2-amino-3-methylbenzoic acid (11.2 g, 74.1 mmol) and Ν,Ο-dimethylhydroxylamine hydrochloride (14.45 g, 148 mmol) in DCM (500 mL) to give a pale brown suspension. The reaction mixture was treated with Et3N (35 mL), HOBT (11.35 g, 74.1 mmol) and EDC (14.20 g, 74.1 mmol) and then stirred at room temperature for 24 hours. The mixture was then washed with 10% LiCl, and then acidified with IN HCl. The organic layer was washed successively with 10%> LiCl and aq NaHC03. The organic layer was decolorized with charcoal, filtered, and the filtrate was dried over MgSC^. The mixture was filtered and concentrated to give 13.22 g (92% yield) of Intermediate A-1A. MS(ES): m/z = 195.1 [M+H+]; HPLC: RT = 1.118 min. (H20/MeOH with TFA, CHROMOLITH® ODS S5 4.6 x 50 mm, gradient = 4 min, wavelength = 220 nm); 1H NMR (500MHz, chloroform-d) δ 7.22 (dd, J= 7.8, 0.8 Hz, 1H), 7.12-7.06 (m, 1H), 6.63 (t, J= 7.5 Hz, 1H), 4.63 (br. s., 2H), 3.61 (s, 3H), 3.34 (s, 3H), 2.17 (s, 3H).
Intermediate A- 1 : (2-Amino-3 -methylphenyl)(3 -fluorophenyl)methanone
[00199] In a 500 mL round-bottomed flask, a solution of l-fluoro-3-iodobenzene (13.61 mL, 116 mmol) in THF (120 mL) was cooled in a -78 °C bath. A solution of n- BuLi, (2.5M in hexane, 46.3 mL, 116 mmol) was added dropwise over 10 minutes. The solution was stirred at -78 °C for 30 minutes and then treated with a solution of
Intermediate A-1 A (6.43 g, 33.1 mmol) in THF (30 mL). After 1.5 hours, the reaction mixture was added to a mixture of ice and IN HCl (149 mL, 149 mmol) and the reaction flask was rinsed with THF (5 ml) and combined with the aqueous mixture. The resulting mixture was diluted with 10% aq LiCl and the pH was adjusted to 4 with IN NaOH. The mixture was then extracted with Et20, washed with brine, dried over MgS04, filtered and concentrated. The resulting residue was purified by silica gel chromatography (220g ISCO) eluting with a gradient from 10% EtOAc/hexane to 30% EtOAc/hexane to afford Intermediate A-l (7.11 g, 94% yield) as an oil. MS(ES): m/z = 230.1 [M+H+]; HPLC: RT = 2.820 min Purity = 99%. (H20/MeOH with TFA, CHROMOLITH® ODS S5 4.6 x 50 mm, gradient = 4 min, wavelength = 220 nm).
Intermediate B-1 : (S)-3-Amino-5-(3-fluorophenyl)-9-methyl-lH-benzo[e][l,4]diazepin- 2(3H)-one
Intermediate B-1 A: (S)-Benzyl (5-(3-fluorophenyl)-9-methyl-2-oxo-2,3-dihydro benzo[e] [ 1 ,4]diazepin-3-yl)carbamate
(B-1A)
[00225] In a 1 L round-bottomed flask, a solution of 2-(lH-benzo[d][l,2,3]triazol-l- yl)-2-((phenoxycarbonyl)amino)acetic acid (J. Org. Chem., 55:2206-2214 (1990)) (19.37 g, 62.0 mmol) in THF (135 mL) was cooled in an ice/water bath and treated with oxalyl chloride (5.43 mL, 62.0 mmol) and 4 drops of DMF. The reaction mixture was stirred for 4 hours. Next, a solution of Intermediate A- 1 (7.11 g, 31.0 mmol) in THF (35 mL) was added and the resulting solution was removed from the ice/water bath and stirred at room temperature for 1.5 hours. The mixture was then treated with a solution of ammonia, (7M in MeOH) (19.94 mL, 140 mmol). After 15 mins, another portion of ammonia, (7M in MeOH) (19.94 mL, 140 mmol) was added and the resulting mixture was sealed under N2 and stirred overnight at room temperature. The reaction mixture was then concentrated to ~l/2 volume and then diluted with AcOH (63 mL) and stir at room temperature for 4 hours. The reaction mixture was then concentrated, and the residue was diluted with 500 mL water to give a precipitate. Hexane and Et20 were added and the mixture was stirred at room temperature for 1 hour to form an orange solid. Et20 was removed under a stream of nitrogen and the aqueous layer was decanted. The residue was triturated with 40 mL of iPrOH and stirred at room temperature to give a white precipitate. The solid was filtered and washed with iPrOH, then dried on a filter under a stream of nitrogen to give racemic Intermediate B-1A (5.4 g, 41.7%yield).
[00226] Racemic Intermediate B-1A (5.9 g, 14.3 mmol) was resolved using the Chiral SFC conditions described below. The desired stereoisomer was collected as the second peak in the elution order: Instrument: Berger SFC MGIII, Column: CHIRALPAK® IC 25 x 3 cm, 5 cm; column temp: 45 °C; Mobile Phase: C02/MeOH (45/55); Flow rate: 160 mL/min; Detection at 220 nm.
[00227] After evaporation of the solvent, Intermediate B-1A (2.73 g, 46% yield) was obtained as a white solid. HPLC: RT = 3.075 min. (H20/MeOH with TFA,
CHROMOLITH® ODS S5 4.6 x 50 mm, gradient = 4 min, wavelength = 220 nm).
Chiral HPLC RT: 8.661 min (AD, 60% (EtOH/MeOH)/heptane) > 99%ee. MS(ES): m/z = 418.3 [M+H+];1H NMR (500MHz, DMSO-d6) δ 10.21 (s, 1H), 8.38 (d, J= 8.3 Hz, 1H), 7.57-7.47 (m, 2H), 7.41-7.29 (m, 8H), 7.25-7.17 (m, 2H), 5.10-5.04 (m, 3H), 2.42 (s, 3H).
Intermediate B-l : (S)-3-Amino-5-(3-fluorophenyl)-9-methyl-lH-benzo[e][l,4]diazepin- 2(3H)-one.
[00228] In a 100 mL round-bottomed flask, a solution of Intermediate B-1A (2.73 g, 6.54 mmol) in acetic acid (12 mL) was treated with HBr, 33% in HOAc (10.76 mL, 65.4 mmol) and the mixture was stirred at room temperature for 1 hour. The solution was diluted with Et20 to give a yellow precipitate. The yellow solid was filtered and rinsed with Et20 under nitrogen. The solid was transferred to 100 mL round bottom flask and water was added (white precipitate formed). The slurry was slowly made basic with saturated NaHC03. The resulting tacky precipitate was extracted with EtOAc. The organic layer was washed with water, dried over MgS04, and then filtered and
concentrated to dryness to give Intermediate B-l (1.68 g, 91% yield) as a white foam solid. MS(ES): m/z = 284.2 [M+H+]; HPLC: RT = 1.72 min (H20/MeOH with TFA, CHROMOLITH® ODS S5 4.6 x 50 mm, gradient = 4 min, wavelength = 220 nm). 1H NMR (400MHz, DMSO-d6) δ 10.01 (br. s., 1H), 7.56-7.44 (m, 2H), 7.41-7.26 (m, 3H), 7.22-7.11 (m, 2H), 4.24 (s, 1H), 2.55 (br. s., 2H), 2.41 (s, 3H). [00229] The compounds listed below in Table 6 (Intermediates B-2 to B-3) were prepared according to the general synthetic procedure described for Intermediate B-l , using the starting materials Intermediate A- 10 and Intermediate A-4, respectively.
Example 1
(2R,3S)-N-((3S)-5-(3-Fluorophenyl)-9-methyl-2-oxo-2,3-dihydro-lH-l,4-benzodiazepin- 3-yl)-2, -bis(3,3,3-trifluoropropyl)succinamide
Intermediate 1A: (2S,3R)-tert-Butyl 6,6,6-trifluoro-3-(((S)-5-(3-fluorophenyl)-9-methyl- 2-0X0-2, 3-dihydro-lH-benzo[e][l,4]diazepin-3-yl)carbamoyl)-2-(3,3 ,3- trifluoropropyl)hexanoat
[00240] In a 100 mL round-bottomed flask, a solution of Intermediate B-l (1683 mg, 5.94 mmol), Et3N (1.656 mL, 11.88 mmol), and Intermediate S-l in DMF (20 mL) was treated with o-benzotriazol-l-yl-A .A .N’.N’-tetramethyluronium tetrafluoroborate (3815 mg, 11.88 mmol) and stirred at room temperature for 1 hour. The reaction mixture was diluted with water and saturated aqueous NaHC03. An off white precipitate formed and was filtered and washed with water. The resulting solid was dried on the filter under a stream of nitrogen to give Intermediate 1A (3.7 g, 99% yield). MS(ES): m/z =
632.4[M+H+]; HPLC: RT = 3.635 min Purity = 98%. (H20/MeOH with TFA,
CHROMOLITH® ODS S5 4.6 x 50 mm, gradient = 4 min, wavelength = 220 nm). 1H NMR (400MHz, methanol-d4) δ 7.53 (t, J = 4.5 Hz, 1H), 7.46-7.30 (m, 3H), 7.28-7.23 (m, 1H), 7.23-7.18 (m, 2H), 5.37 (s, 1H), 2.88 (td, J = 10.4, 3.4 Hz, 1H), 2.60 (td, J =
10.2, 4.1 Hz, 1H), 2.54-2.40 (m, 1H), 2.47 (s, 3 H), 2.33-2.12 (m, 3H), 1.98-1.69 (m, 4H), 1.51 (s, 9H). Intermediate IB: (2S,3R)-6,6,6-Trifluoro-3-(((S)-5-(3-fluorophenyl)-9-methyl-2-oxo-
2,3-dihydro-lH-benzo[e][l,4]diazepin-3-yl)carbamoyl)-2-(3,3,3-trifluoropropyl)hexanoic acid
[00241] In a 250 mL round-bottomed flask, a solution of Intermediate 1A (3.7 g, 5.86 mmol) in DCM (25 mL) was treated with TFA (25 mL) and the resulting pale orange solution was stirred at room temperature for 1.5 hours. The reaction mixture was then concentrated to give Intermediate IB. HPLC: RT = 3.12 min (H20/MeOH with TFA, CHROMOLITH® ODS S5 4.6 x 50 mm, gradient = 4 min, wavelength = 220 nm).
MS(ES): m/z = 576.3 (M+H)+. 1H NMR (400MHz, methanol-d4) δ 7.54 (t, J= 4.5 Hz, 1H), 7.49-7.29 (m, 3H), 7.28-7.15 (m, 3H), 5.38 (br. s., 1H), 2.89 (td, J= 10.3, 3.7 Hz, 1H), 2.67 (td, J= 9.9, 4.2 Hz, 1H), 2.56-2.38 (m, 1H), 2.48 (s, 3 H), 2.34-2.13 (m, 3H), 2.00-1.71 (m, 4H).
Example 1 :
[00242] In a 250 mL round-bottomed flask, a solution of Intermediate IB (4.04 g, 5.86 mmol) in THF (50 mL) was treated with ammonia (2M in iPrOH) (26.4 mL, 52.7 mmol), followed by HOBT (1.795 g, 11.72 mmol) and EDC (2.246 g, 11.72 mmol). The resulting white suspension was stirred at room temperature overnight. The reaction mixture was diluted with water and saturated aqueous NaHC03. The resulting solid was filtered, rinsed with water and then dried on the filter under a stream of nitrogen. The crude product was suspended in 20 mL of iPrOH and stirred at room temperature for 20 min and then filtered and washed with iPrOH and dried under vacuum to give 2.83 g of solid. The solid was dissolved in re fluxing EtOH(100 mL) and slowly treated with 200 mg activated charcoal added in small portions. The hot mixture was filtered through CELITE® and rinsed with hot EtOH. The filtrate was reduced to half volume, allowed to cool and the white precipitate formed was filtered and rinsed with EtOH to give 2.57 g of white solid. A second recrystallization from EtOH (70 mL) afforded Example 1 (2.39 g, 70% yield) as a white solid. HPLC: RT = 10.859 min (H20/CH3CN with TFA, Sunfire C18 3.5μπι, 3.0x150mm, gradient = 15 min, wavelength = 220 and 254 nm); MS(ES): m/z = 575.3 [M+H+]; 1H NMR (400MHz, methanol-d4) δ 7.57-7.50 (m, 1H), 7.47-7.30 (m, 3H), 7.29-7.15 (m, 3H), 5.38 (s, 1H), 2.85-2.75 (m, 1H), 2.59 (td, J= 10.5, 4.0 Hz, 1H), 2.53-2.41 (m, 4H), 2.31-2.10 (m, 3H), 1.96-1.70 (m, 4H).
SEE
WO2012129353A1 *Mar 22, 2012Sep 27, 2012Bristol-Myers Squibb CompanyBis(fluoroalkyl)-1,4-benzodiazepinone compounds
PAPER RELATED
Structure–activity relationships in a series of (2-oxo-1,4-benzodiazepin-3-yl)-succinamides identified highly potent inhibitors of γ-secretase mediated signaling of Notch1/2/3/4 receptors. On the basis of its robust in vivo efficacy at tolerated doses in Notch driven leukemia and solid tumor xenograft models, 12 (BMS-906024) was selected as a candidate for clinical evaluation.
Discovery of Clinical Candidate BMS-906024: A Potent Pan-Notch Inhibitor for the Treatment of Leukemia and Solid Tumors
†Bristol-Myers Squibb Research and Development, Princeton, New Jersey 08543, United States
‡Bristol-Myers Squibb Research and Development, 5 Research Parkway, Wallingford, Connecticut 06492, United States
§ Department of Chemistry, The Scripps Research Institute, 10550 North Torrey Pines Road, La Jolla, California 92037,United States
ACS Med. Chem. Lett., 2015, 6 (5), pp 523–527
DOI: 10.1021/acsmedchemlett.5b00001, http://pubs.acs.org/doi/abs/10.1021/acsmedchemlett.5b00001
*Phone: 609-252-5091. E-mail: ashvinikumar.gavai@bms.com.

Patent
http://www.google.co.in/patents/WO2012129353A1?cl=en
PATENT RELATED






PATENTS RELATED
Clip RELATED
For some disease targets, an indirect approach may be best. Or so Ashvinikumar V. Gavai and his colleagues atBristol-Myers Squibbfound in their quest toward a potential cancer drug. Gavai unveiled BMS-906024, which is an experimental—and slightly roundabout—treatment for a number of cancers, including breast, lung, and colon cancers, and leukemia.
Cancers have a tendency to relapse or to become resistant to treatments that once worked. Research at BMS and elsewhere had suggested that a family of proteins called Notch is implicated in that resistance and in cancer progression more generally. Gavai, director of oncology chemistry at BMS in Princeton, N.J., and his team set out to block Notch family signaling.
Notch family members lack enzymatic activity, so blocking them directly is difficult. Instead, BMS developed inhibitors of an enzyme that is essential for activating Notch signaling—γ-secretase.

Company: Bristol-Myers Squibb
Target: pan-Notch
Disease: breast, lung, colon cancer; leukemia
Interfering with Notch, even in this indirect way, can have detrimental effects on the gastrointestinal tract. Only two of the four Notch family members are linked to that side effect, Gavai says. But he and his team think their drug will be most effective if it acts on all four family members roughly equally—a so-called pan-Notch inhibitor. By selecting a molecule that’s well tolerated in animals and carefully scheduling doses of the drug in humans, it could be possible to minimize side effects, he says.
The BMS team relied on Notch signaling assays in leukemia and breast cancer cell lines to find leads. They soon learned that for their molecules to work, three chiral centers had to be in the S,R,Sconfiguration. After that, they strove to make the molecules last in the bloodstream. They removed an isobutyl group and tweaked some other parts of their candidate’s succinamide side chain. It was tough to retain both a long half-life and activity against Notch, Gavai told C&EN. “You’d optimize one and lose the other.”
His team threaded the needle with BMS-906024. Their studies with mice suggest that a dose of 4–6 mg once a week could be effective in people. That’s lower than doses being tested for other Notch-targeted agents, according to the website clinicaltrials.gov. The mouse studies also back the idea that Notch is involved in cancer drug resistance and suggest that Notch could be a target for taking on cancer stem cells, which are notoriously resistant to chemotherapy.
BMS-906024 is in Phase I clinical trials, both alone and in combination with other agents. Patients with colon, lung, breast, and other cancers are receiving intravenous doses of the compound to determine its safety and optimum dose ranges.

(From left, front row) Gavai, Weifeng Shan, (second row) Aaron Balog, Patrice Gill, Gregory Vite, (third row) Francis Lee, Claude Quesnelle, (rear row) Wen-Ching Han, Richard Westhouse.
Credit: Catherine Stroud Photography
http://cen.acs.org/articles/91/i16/BMS-906024-Notch-Signaling-Inhibitor.html

PAPER RELATED
An enantioselective synthesis of (S)-7-amino-5H,7H-dibenzo[b,d]azepin-6-one (S–1) is described. The key step in the sequence involved crystallization-induced dynamic resolution (CIDR) of compound 7 using Boc-d-phenylalanine as a chiral resolving agent and 3,5-dichlorosalicylaldehyde as a racemization catalyst to afford S–1 in 81% overall yield with 98.5% enantiomeric excess.
† Department of Discovery Synthesis, Biocon Bristol-Myers Squibb Research Centre, Biocon Park, Bommasandra IV Phase, Jigani Link Road, Bengaluru 560099, India
‡Bristol-Myers Squibb Company, P.O Box 4000, Princeton, New Jersey 08543-4000, United States
Org. Process Res. Dev., Article ASAP
DOI: 10.1021/acs.oprd.6b00207, http://pubs.acs.org/doi/suppl/10.1021/acs.oprd.6b00207
*E-mail: anuradha.gupta@syngeneintl.com.
| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2000007995A1 * | Aug 7, 1999 | Feb 17, 2000 | Du Pont Pharmaceuticals Company | SUCCINOYLAMINO LACTAMS AS INHIBITORS OF Aβ PROTEIN PRODUCTION |
| WO2000038618A2 * | Dec 23, 1999 | Jul 6, 2000 | Du Pont Pharmaceuticals Company | SUCCINOYLAMINO BENZODIAZEPINES AS INHIBITORS OF Aβ PROTEIN PRODUCTION |
| WO2001060826A2 * | Feb 16, 2001 | Aug 23, 2001 | Bristol-Myers Squibb Pharma Company | SUCCINOYLAMINO CARBOCYCLES AND HETEROCYCLES AS INHIBITORS OF Aβ PROTEIN PRODUCTION |
| US6737038 * | May 17, 2000 | May 18, 2004 | Bristol-Myers Squibb Company | Use of small molecule radioligands to discover inhibitors of amyloid-beta peptide production and for diagnostic imaging |
| US7053084 | Feb 17, 2000 | May 30, 2006 | Bristol-Myers Squibb Company | Succinoylamino benzodiazepines as inhibitors of Aβ protein production |
| US7456172 | Jan 13, 2006 | Nov 25, 2008 | Bristol-Myers Squibb Pharma Company | Succinoylamino benzodiazepines as inhibitors of Aβ protein production |
| US20030134841 * | Nov 1, 2002 | Jul 17, 2003 | Olson Richard E. | Succinoylamino lactams as inhibitors of A-beta protein production |
| US20120245151 * | Mar 22, 2012 | Sep 27, 2012 | Bristol-Myers Squibb Company | Bisfluoroalkyl-1,4-benzodiazepinone compounds |
//////////BMS-986115, BMS 986115, 3,5-dichlorosalicylaldehyde, Alzheimer’s disease, Boc-D-phenylalanine, CIDR;dibenzoazepenone, DKR; Notch inhibitors, Notch inhibitor, SAR, T-acute lymphoblastic leukemia, triple-negative breast cancer, γ-secretase inhibitor, PHASE 1, BMS, Bristol-Myers Squibb, Ashvinikumar Gavai, 1584647-27-7, UNII: LSK1L593UU
Cc1cccc2c1NC(=O)[C@H](N=C2c3cccc(c3)F)NC(=O)[C@H](CCC(F)(F)F)[C@H](CCC(F)(F)F)C(=O)N
BMS 906024


BMS 906024
cas 1401066-79-2
- MF C26H26F6N4O3
- MW 556.500
(2R,3S)-N-[(3S)-1-Methyl-2-oxo-5-phenyl-2,3-dihydro-1H-1,4-benzodiazepin-3-yl]-2,3-bis(3,3,3-trifluoropropyl)succinamide
Butanediamide, N1-((3S)-2,3-dihydro-1-methyl-2-oxo-5-phenyl-1H-1,4-benzodiazepin-3-yl)-2,3-bis(3,3,3-trifluorophenyl)-, (2R,3S)-
(2R,35)-N-((35)-l-Methyl-2-oxo-5-phenyl-2,3-dihydro-lH-l,4-benzodiazepin-3-yl)-3- (2,2,2-trifluoroethyl)-2-(3,3,3-trifluoropropyl)succinamide
| Claude Quesnelle, Soong-Hoon Kim, Francis Lee, Ashvinikumar Gavai | |
| Applicant | Bristol-Myers Squibb Company |

Claude Quesnelle
Senior Research Investigator/Chemist at Bristol-Myers Squibb
RICHARD LEE
BMS-906024 is a novel, potent Notch receptor inhibitor . Cancers have a tendency to relapse or to become resistant to treatments that once worked. A family of proteins called Notch is implicated in that resistance and in cancer progression more generally. BMS-906024 is in Phase I clinical trials, both alone and in combination with other agents. Patients with colon, lung, breast, and other cancers are receiving intravenous doses of the compound to determine its safety and optimum dose ranges.
New Phase I drug structure by Bristol-Myers Squibb disclosed at the spring 2013 American Chemical Society meeting in New Orleans to treat breast, lung, and colon cancers and leukemia.[1] The drug works as an pan-Notch inhibitor. The structure is one of a set patented in 2012,[2] and it currently being studied in clinical trials.[3][4]
useful for the treatment of conditions related to the Notch pathway, such as cancer and other proliferative diseases.
Notch signaling has been implicated in a variety of cellular processes, such as cell fate specification, differentiation, proliferation, apoptosis, and angiogenesis. (Bray, Nature Reviews Molecular Cell Biology, 7:678-689 (2006); Fortini, Developmental Cell 16:633-647 (2009)). The Notch proteins are single-pass heterodimeric transmembrane molecules. The Notch family includes 4 receptors, NOTCH 1-4, which become activated upon binding to ligands from the DSL family (Delta-like 1, 3, 4 and Jagged 1 and 2).
The activation and maturation of NOTCH requires a series of processing steps, including a proteolytic cleavage step mediated by gamma secretase, a multiprotein complex containing Presenilin 1 or Presenilin 2, nicastrin, APH1, and PEN2. Once NOTCH is cleaved, NOTCH intracellular domain (NICD) is released from the membrane. The released NICD translocates to the nucleus, where it functions as a transcriptional activator in concert with CSL family members (RBPSUH, “suppressor of hairless”, and LAG1). NOTCH target genes include HES family members, such as HES- 1. HES- 1 functions as transcriptional repressors of genes such as HERP 1 (also known as HEY2), HERP2 (also known as HEY1), and HATH1 (also known as ATOH1).
The aberrant activation of the Notch pathway contributes to tumorigenesis. Activation of Notch signaling has been implicated in the pathogenesis of various solid tumors including ovarian, pancreatic, as well as breast cancer and hematologic tumors such as leukemias, lymphomas, and multiple myeloma. The role of Notch inhibition and its utility in the treatment of various solid and hematological tumors are described in Miele, L. et al, Current Cancer Drug Targets, 6:313-323 (2006); Bolos, V. et al, Endocrine Reviews, 28:339-363 (2007); Shih, I.-M. et al, Cancer Research, 67: 1879- 1882 (2007); Yamaguchi, N. et al., Cancer Research, 68: 1881-1888 (2008); Miele, L., Expert Review Anti-cancer Therapy, 8: 1 197-1201 (2008); Purow, B., Current Pharmaceutical Biotechnology, 10: 154-160 (2009); Nefedova, Y. et al, Drug Resistance Updates, 1 1 :210-218 (2008); Dufraine, J. et al, Oncogene, 27:5132-5137 (2008); and Jun, H.T. et al, Drug Development Research, 69:319-328 (2008).
There remains a need for compounds that are useful as Notch inhibitors and that have sufficient metabolic stability to provide efficacious levels of drug exposure. Further, there remains a need for compounds useful as Notch inhibitors that can be orally or intravenously administered to a patient.
U.S. Patent No. 7,053,084 Bl discloses succinoylamino benzodiazepine compounds useful for treating neurological disorders such as Alzheimer’s Disease. The reference discloses that these succinoylamino benzodiazepine compounds inhibit gamma secretase activity and the processing of amyloid precursor protein linked to the formation of neurological deposits of amyloid protein. The reference does not disclose the use of these compounds in the treatment of proliferative diseases such as cancer.
Applicants have found potent compounds that have activity as Notch inhibitors and have sufficient metabolic stability to provide efficacious levels of drug exposure upon intravenous or oral administration. These compounds are provided to be useful as pharmaceuticals with desirable stability, bioavailability, therapeutic index, and toxicity values that are important to their drugability.
![]()
PAPER

Structure–activity relationships in a series of (2-oxo-1,4-benzodiazepin-3-yl)-succinamides identified highly potent inhibitors of γ-secretase mediated signaling of Notch1/2/3/4 receptors. On the basis of its robust in vivo efficacy at tolerated doses in Notch driven leukemia and solid tumor xenograft models, 12 (BMS-906024) was selected as a candidate for clinical evaluation.
Discovery of Clinical Candidate BMS-906024: A Potent Pan-Notch Inhibitor for the Treatment of Leukemia and Solid Tumors
†Bristol-Myers Squibb Research and Development, Princeton, New Jersey 08543, United States
‡Bristol-Myers Squibb Research and Development, 5 Research Parkway, Wallingford, Connecticut 06492, United States
§ Department of Chemistry, The Scripps Research Institute, 10550 North Torrey Pines Road, La Jolla, California 92037,United States
ACS Med. Chem. Lett., 2015, 6 (5), pp 523–527
DOI: 10.1021/acsmedchemlett.5b00001, http://pubs.acs.org/doi/abs/10.1021/acsmedchemlett.5b00001
*Phone: 609-252-5091. E-mail: ashvinikumar.gavai@bms.com.
(2R,3S)-N-((3S)-1-methyl-2-oxo-5-phenyl-2,3-dihydro-1H-1,4- benzodiazepin-3-yl)-2,3-bis(3,3,3-trifluoropropyl)succinamide
colorless solid: HPLC: RT = 9.60 min (HPLC Method D). Chiral LC/Analytical SFC conditions: Column: LuxCellulose-2 (0.46 x 25cm), Mobile phase: 10% methanol in CO2, Flow rate: 3 mL/min, wavelength: 220 nm; Temp.: 35C. RT = 9.21 min, Purity = 99.95%.
MS (ES): m/z = 557 [M+H]+ ;
1H NMR (400 MHz, DMSO-d6) 9.54 (1H, d, J = 7.28 Hz), 7.71 – 7.80 (1H, m), 7.68 (2H, d, J = 8.78 Hz), 7.50 – 7.62 (3H, m), 7.45 (2H, t, J = 7.28 Hz), 7.29 – 7.40 (2H, m), 7.15 (1H, s), 5.30 (1H, d, J = 7.28 Hz), 3.39 (3H, s), 2.74 – 2.86 (1H, m), 2.02 -2.32 (3H, m), 1.45 – 1.79 (4H, m);
[]D = -107.0° (5.73 mg/mL, DMSO).
Elemental analysis: Theoretical: C: 54.11%; H: 4.70%; N: 10.06%; Actual: C: 54.06%; H: 4.90%; N: 10.08%.
Karl Fisher Moisture: 0.48.
HPLC Method D: Sunfire C18 3.5um, 3.0x150mm column, solvent A: 5% acetonitrile – 95% water – 0.05% TFA, solvent B: 95% acetonitrile – 5% water – 0.05% TFA, flow=0.5 mL/min, gradient from 10%B to 100%B over 15min, 254 nm detector.
Patent
http://www.google.co.in/patents/WO2012129353A1?cl=en
Example 1
(2R,35)-N-((35′)-l-Methyl-2-oxo-5-phenyl-2,3-dihydro-lH-l,4-benzodiazepin-3-yl)-2,3- b -trifluoropropy l)succinamide
Preparation 1A: tert-Butyl 5, -trifluoropentanoate

[00219] To a stirred solution of 5,5,5-trifluoropentanoic acid (5 g, 32.0 mmol) in THF (30 mL) and hexane (30 mL) at 0 °C, was added tert-butyl 2,2,2-trichloroacetimidate (11.46 mL, 64.1 mmol). The mixture was stirred for 15 min at 0 °C. Boron trifluoride etherate (0.406 mL, 3.20 mmol) was added and the reaction mixture was allowed to warm to room temperature overnight. To the clear reaction mixture was added solid aHC03 (5 g) and stirred for 30 min. The mixture was filtered through MgS04 and washed with hexanes (200 mL). The solution was allowed to rest for 45 min, and the resulting solid material was removed by filtering on the same MgS04 filter again, washed with hexanes (100 mL) and concentrated under reduced pressure without heat. The volume was reduced to about 30 mL, filtered through a clean fritted funnel, washed with hexane (5 mL), and then concentrated under reduced pressure without heat. The resulting neat oil was filtered through a 0.45μηι nylon membrane filter disk to provide tert-butyl 5,5,5- trifluoropentanoate (6.6 g, 31.4 mmol 98% yield) as a colorless oil: XH NMR (400 MHz, CDC13) δ ppm 1.38 (s, 9 H) 1.74-1.83 (m, 2 H) 2.00-2.13 (m, 2 H) 2.24 (t, J=7.28 Hz, 2 H).
Preparation IB: (45)-4-(Propan-2- l)-3-(5,5,5-trifluoropentanoyl)-l,3-oxazolidin-2-one

[00220] To a stirred solution of 5,5,5-trifluoropentanoic acid (5.04 g, 32.3 mmol) in DCM (50 mL) and DMF (3 drops) was added oxalyl chloride (3.4 mL, 38.8 mmol) dropwise over 5 min and the solution was stirred until all bubbling subsided. The reaction mixture was concentrated under reduced pressure to give pale yellow oil. To a separate flask charged with a solution of (45)-4-(propan-2-yl)-l,3-oxazolidin-2-one (4.18 g, 32.4 mmol) in THF (100 mL) at -78 °C was added n-BuLi (2.5M in hexane) (13.0 mL, 32.5 mmol) dropwise via syringe over 5 min. After stirring for 10 min, the above acid chloride dissolved in THF (20 mL) was added via cannula over 15 min. The reaction mixture was warmed to 0 °C, and was allowed to warm to room temperature as the bath warmed and stirred overnight. To the reaction mixture was added saturated NH4CI, and then extracted with EtOAc (2x). The combined organics were washed with brine, dried (Na2S04), filtered and concentrated under reduced pressure. The crude material was purified by flash chromatography (Teledyne ISCO CombiFlash Rf, 5% to 60% solvent A/B=hexanes/EtOAc, REDISEP® S1O2 120g). Concentration of appropriate fractions provided Preparation IB (7.39 g, 86%) as a colorless oil: XH NMR (400 MHz, CDC13) δ ppm 4.44 (1 H, dt, J=8.31, 3.53 Hz), 4.30 (1 H, t, J=8.69 Hz), 4.23 (1 H, dd, J=9.06, 3.02 Hz), 2.98-3.08 (2 H, m), 2.32-2.44 (1 H, m, J=13.91, 7.02, 7.02, 4.03 Hz), 2.13-2.25 (2 H, m), 1.88-2.00 (2 H, m), 0.93 (3 H, d, J=7.05 Hz), 0.88 (3 H, d, J=6.80 Hz). Preparation 1C: (25′,3R)-tert-Butyl 6,6,6-trifluoro-3-((5)-4-isopropyl-2-oxooxazolidine- 3 -carbonyl)-2-(3 ,3,3 -trifluoropropyl)hexanoate, and
Preparation ID: (2R,3R)-tert-Butyl 6,6,6-trifluoro-3-((5)-4-isopropyl-2-oxooxazolidine- 3 -carbonyl)- -(3 ,3 ,3 -trifluoropropyl)hexanoate

(1 C) (1 D)
[00221] To a cold (-78 °C), stirred solution of diisopropylamine (5.3 mL, 37.2 mmol) in THF (59 mL) under nitrogen atmosphere was added n-BuLi (2.5M in hexane) (14.7 mL, 36.8 mmol), then warmed to 0 °C to give a 0.5M solution of LDA. A separate vessel was charged with Preparation IB (2.45 g, 9.17 mmol), the material was azeotroped twice with benzene (the RotoVap air inlet was fitted with nitrogen inlet to completely exclude humidity) then toluene (15.3 mL) was added. This solution was added to a flask containing dry lithium chloride (1.96 g, 46.2 mmol). To the resultant mixture, cooled to -78 °C, was added LDA solution (21.0 mL, 10.5 mmol) and stirred at -78 °C for 10 min, warmed to 0 °C for 10 min then recooled to -78 °C. To a separate reaction vessel containing Preparation 1A (3.41 g, 16.07 mmol), also azeotroped twice with benzene, was added toluene (15.3 mL), cooled to -78 °C and LDA (37.0 mL, 18.5 mmol) was added, the resulting solution was stirred at -78° for 25 min. At this time the enolate derived from the ester was transferred via cannula into the solution of the oxazolidinone enolate, stirred at -78 °C for an additional 5 min at which time the septum was removed and solid powdered bis(2-ethylhexanoyloxy)copper (9.02 g, 25.8 mmol) was rapidly added to the reaction vessel and the septum replaced. The vessel was immediately removed from the cold bath and immersed into a warm water bath (40 °C) with rapid swirling with a concomitant color change from the initial turquoise to brown. The reaction mixture was stirred for 20 min, was poured into 5% aqueous NH4OH (360 mL) and extracted with EtOAc (2x). The combined organics were washed with brine, dried (Na2S04), filtered and concentrated under reduced pressure. The residue was purified by flash chromatography (Teledyne ISCO CombiFlash Rf, 0% to 60% solvent A/B=hexanes/EtOAc, REDISEP® S1O2 120g). Concentration of appropriate fractions provided Preparation 1C (2.87 g, 66%) as pale yellow viscous oil. XH NMR showed the product was a 1.6: 1 mixture of diastereoisomers 1C: 1D as determined by the integration of the multiplets at 2.74 & 2.84 ppm: XH NMR (400 MHz, CDC13) δ ppm 4.43-4.54 (2 H, m), 4.23-4.35 (5 H, m), 4.01 (1 H, ddd, J=9.54, 6.27, 3.51 Hz), 2.84 (1 H, ddd, J=9.41, 7.28, 3.64 Hz), 2.74 (1 H, ddd, J=10.29, 6.27, 4.02 Hz), 2.37-2.48 (2 H, m, J=10.38, 6.98, 6.98, 3.51, 3.51 Hz), 2.20-2.37 (3 H, m), 1.92-2.20 (8 H, m), 1.64-1.91 (5 H, m), 1.47 (18 H, s), 0.88-0.98 (12 H, m). Preparation IE: (2R,35)-3-(tert-Butoxycarbonyl)-6,6,6-trifluoro-2-(3,3,3- trifluoropropyl)hexanoic acid, and
Preparation IF: (2R,3R)-3-(tert-Butoxycarbonyl)-6,6,6-trifluoro-2-(3,3,3- trifluoropropyl)hexanoic acid

(1 E) (1 F)
[00222] To a cool (0 °C), stirred solution of Preparation 1C and ID (4.54 g, 9.51 mmol) in THF (140 mL) and water (42 mL) was sequentially added hydrogen peroxide (30% in water) (10.3 g, 91 mmol) and LiOH (685.3 mg, 28.6 mmol) and the mixture was stirred for 1 hr. At this time the reaction vessel was removed from the cold bath and then stirred for 1.5 hr. The reaction was judged complete by HPLC. To the reaction mixture was added saturated NaHC03 (45 mL) and saturated a2S03(15 mL), and then partially concentrated under reduced pressure. The resulting crude solution was extracted with DCM (3x). The aqueous phase was acidified to pH~l-2 with IN HC1, extracted with DCM (3x) and EtOAc (lx). The combined organics were washed with brine, dried (Na2S04), filtered and concentrated under reduced pressure to provide a mixture of Preparation IE and IF (3.00 g, 86%) as colorless oil: XH NMR (400 MHz, CDC13) δ ppm 2.76-2.84 (1 H, m, diastereoisomer 2), 2.64-2.76 (3 H, m), 2.04-2.35 (8 H, m), 1.88-2.00 (4 H, m), 1.71-1.83 (4 H, m), 1.48 (9 H, s, diastereoisomer 1), 1.46 (9 H, s, diastereoisomer 2); XH NMR showed a 1.7: 1 mixture of 1E: 1F by integration of the peaks for the ?-butyl groups.
Preparation IE: (2R,35)-3-(tert-Butoxycarbonyl)-6,6,6-trifluoro-2-(3,3,3- trifluoropropyl)hexanoic acid, and
Preparation IF: (2R,3R)-3-(tert-Butoxycarbonyl)-6,6,6-trifluoro-2-(3,3,3- trifluoropropyl)hexanoic acid

(1 E) (1 F)
[00223] To a cold (-78 °C), stirred solution of diisopropylamine (1.7 mL, 11.93 mmol) in THF (19 mL) under nitrogen atmosphere was added n-BuLi (2.5M in hexanes) (4.8 mL, 12.00 mmol). The mixture was stirred for 5 min and then warmed to 0 °C. In a separate vessel, to a cold (-78 °C) stirred solution of the mixture of Preparation IE and IF (1.99 g, 5.43 mmol) in THF (18 mL) was added the LDA solution prepared above via cannula slowly over 25 min. The mixture was stirred for 15 min, then warmed to room temperature (placed in a 24 °C water bath) for 15 min, and then again cooled to -78 °C for 15 min. To the reaction mixture was added Et2AlCl (1M in hexane) (11.4 mL, 1 1.40 mmol) via syringe, stirred for 10 min, warmed to room temperature for 15 min and then cooled back to -78 °C for 15 min. Methanol (25 mL) was rapidly added, swirled vigorously while warming to room temperature, then concentrated to ~l/4 original volume. The mixture was dissolved in EtOAc and washed with IN HCl (50 mL) and ice (75 g). The aqueous phase was separated, extracted with EtOAc (2x). The combined organics were washed with a mixture of KF (2.85g in 75 mL water) and IN HCl (13 mL) [resulting solution pH 3-4], then with brine, dried (Na2S04), filtered and concentrated under reduced pressure to give a 9: 1 (IE: IF) enriched diastereoisomeric mixture (as determined by XH NMR) of Preparation IE and Preparation IF (2.13 g, >99%) as a pale yellow viscous oil: XH NMR (400 MHz, CDC13) δ ppm 2.64-2.76 (2 H, m), 2.04-2.35 (4 H, m), 1.88-2.00 (2 H, m), 1.71-1.83 (2 H, m), 1.48 (9 H, s). Preparation 1 G: (35)-3 -Amino- 1 -methyl-5-phenyl- 1 ,3 -dihydro-2H- 1 ,4-benzodiazepin-2- one, and
Preparation 1H: (3R)-3 -Amino- 1 -methyl-5-phenyl- 1 ,3-dihydro-2H- 1 ,4-benzodiazepin-2- one

(1G) (1 H)
[00224] Racemic 3-amino-l-methyl-5-phenyl-l,3-dihydro-2H-l,4-benzodiazepin-2- one (Rittle, K.E. et al, Tetrahedron Letters, 28(5):521-522 (1987)) was prepared according to the literature procedure. The enantiomers were separated under chiral-SFC conditions using the following method: CHIRALPAK® AS-H 5×25; Mobile phase: 30% MeOH+ 0.1% DEA in C02; Flow rate: 280 mL/min; Pressure: 100 bar; Temperature: 35 °C.
[00225] Obtained the S-enantiomer (Preparation 1G): HPLC: RT=1.75 min (30% MeOH + 0.1% DEA in C02 on CHIRALPAK® AS-H 4.6×250 mm, 3 mL/min, 35 °C, 100 bar, 230 nm, ΙΟμΙ injection); ¾ NMR (400 MHz, CDC13) δ ppm 7.58-7.63 (2 H, m), 7.55 (1 H, ddd, J=8.50, 7.1 1, 1.76 Hz), 7.40-7.47 (1 H, m), 7.34-7.40 (3 H, m), 7.31 (1 H, dd, J=7.81, 1.51 Hz), 7.14-7.22 (1 H, m), 4.46 (1 H, s), 3.44 (3 H, s), 3.42 (2 H, s); [a]D= -155° (c=1.9, MeOH) (Lit. Rittle, K.E. et al, Tetrahedron Letters, 28(5):521-522 (1987): [a]D=-236°).
[00226] Also obtained the R-enantiomer (Preparation 1H): HPLC: RT=1.71 min; [a]D=+165° (c=2.1, MeOH) (Lit [a]D= +227°).
Alternate procedure to make Preparation 1 G:
Preparation 1G»CSA salt: (35)-3-Amino-l-methyl-5-phenyl-l,3-dihydro-2H-l,4- benzodiazepin-2-one, (15)-(+)-10-camphorsulfonic acid salt

[00227] Preparation lG’CSA was prepared from racemic 3-amino-l-methyl-5-phenyl- l,3-dihydro-2H-l,4-benzodiazepin-2-one (9.98g, 37.6 mmol) (prepared according to the literature as shown above) according to the literature procedure (Reider, P.J. et al, J. Org. Chem., 52:955-957 (1987)). Preparation lG’CSA (16.91g, 99%) was obtained as a colorless solid: Optical Rotation: [a]D = -26.99° (c=l, H20) (Lit. [a]D = -27.8° (c=l,
H20))
Preparation II: tert-Butyl (25,,3R)-6,6,6-trifluoro-3-(((35)-l-methyl-2-oxo-5-phenyl-2,3- dihydro- 1 H- 1 ,4-benzodiazepin-3 -yl)carbamoyl)-2-(3 ,3 ,3 -trifluoropropyl)hexanoate, and Preparation 1J: tert-Butyl (2R,3R)-6,6,6-trifluoro-3-(((35)-l-methyl-2-oxo-5-phenyl-2,3- dihydro- 1 H- 1 ,4-benzodiazepin-3 -yl)carbamoyl)-2-(3 ,3 ,3-trifluoropropyl)hexanoate

(11) (U)
[00228] To a stirred solution of Preparation 1G (1.45 g, 5.47 mmol) and a 9: 1 mixture of Preparation IE and IF (1.989 g, 5.43 mmol) in DMF (19 mL) was added O- benzotriazol-l-yl-N,N,N’,N’-tetra-methyluronium tetrafluoroborate (1.79 g, 5.57 mmol) and triethylamine (3.0 mL, 21.52 mmol) and stirred overnight. The reaction was judged complete by LCMS. The reaction mixture was poured into water (125 mL) and the precipitated solid was collected by filtration, washed with water and air dried to provide an 8: 1 mixture of Preparation II and Preparation 1J (2.95 g, 89%) as a cream solid: MS (ES): m/z= 614 [M+H]+;XH NMR (400 MHz, CDC13) δ ppm 7.55-7.65 (3 H, m), 7.44- 7.52 (2 H, m), 7.35-7.45 (4 H, m), 5.52 (1 H, d, J=8.03 Hz), 3.48 (3 H, s), 2.63 (2 H, ddd, J=9.35, 3.95, 3.76 Hz), 2.14-2.25 (4 H, m), 1.90-2.03 (3 H, m), 1.69-1.82 (1 H, m), 1.51 (9 H, s).
Preparation IK: (25,,3R)-6,6,6-Trifluoro-3-(((35)-l-methyl-2-oxo-5-phenyl-2,3-dihydro- lH-l,4-benzodiazepin-3-yl)carbamoyl)-2-(3,3,3-trifluoropropyl)hexanoic acid, and
Preparation 1L: (2R,3R)-6,6,6-Trifluoro-3-(((35)-l-methyl-2-oxo-5-phenyl-2,3-dihydro- 1 H- 1 ,4-

(1 K) (1 L)
[00229] To a cool (0 °C), stirred solution of the above mixture of Preparation II and Preparation 1 J (2.95 g, 4.81 mmol) in DCM (20 mL) was added TFA (20 mL, 260 mmol). The reaction mixture was stirred for lhr, then allowed to warm to room temperature and stirred for 2.5 hr. The reaction was judged complete by LCMS. The reaction mixture was diluted with toluene (50 mL) and concentrated under reduced pressure. The residue mixture was redissolved in toluene (50 mL) and concentrated under reduced pressure then dried under high vacuum. The crude product was dissolved in DCM, S1O2 (15g) was added, concentrated, then was purified by flash chromatography (Teledyne ISCO CombiFlash Rf, 0% to 45% solvent A/B=DCM/EtOAc, REDISEP® S1O2 80g). Concentration of appropriate fractions provided a mixture of Preparation IK and Preparation 1L (2.00 g, 75%) as a cream solid: HPLC: RT=2.770 min
(CHROMOLITH® SpeedROD 4.6 x 50 mm (4 min grad) eluting with 10-90% aqueous MeOH over 4 minutes containing 0.1% TFA, 4 mL/min, monitoring at 254 nm); MS (ES): m/z= 558 [M+H]+; XH NMR (400 MHz, CDC13) δ ppm 8.32 (1 H, d, J=8.03 Hz), 7.65-7.71 (1 H, m), 7.50-7.60 (3 H, m), 7.41-7.49 (2 H, m), 7.39 (1 H, dd, J=7.91, 1.63 Hz), 7.23-7.35 (2 H, m), 5.59 (1 H, d, J=8.03 Hz), 3.51 (3 H, s), 2.81 (1 H, ddd, J=10.54, 6.90, 3.64 Hz), 2.67-2.76 (1 H, m), 2.22-2.33 (3 H, m), 1.99-2.12 (3 H, m), 1.85-1.94 (1 H, m), 1.79 (1 H, ddd, J=13.87, 7.84, 3.64 Hz). Example 1 :
[00230] To a stirred solution of an 8: 1 mixture of Preparation IK and Preparation 1L (3.46 g, 6.21 mmol) in DMF (25 mL) under nitrogen atmosphere was added ammonium chloride (3.32 g, 62.1 mmol), EDC (3.55 g, 18.52 mmol), HOBT (2.85 g, 18.61 mmol), and triethyl amine (16 mL, 1 15 mmol) and stirred overnight. The reaction was judged complete by LCMS. The reaction mixture was poured into water (200 mL) with vigorous swirling and then allowed to sit. The solid was collected by filtration, washed with water, allowed to dry to afford 3.6 g colorless solid. The solid was purified by preparative SFC chromatography (Lux-Cellulose-2 (3x25cm), 8% methanol in CO2, 140ml/min @220nm and 35 °C; Sample: 3.6g in 50cc methanol, conc.=70mg/ml, Stack injection:
0.5cc/9.2min). Fractions containing product were concentrated, dried overnight under vacuum. Obtained Example 1 (2.74 g, 79%) as a colorless solid (Crystal Form -1): HPLC: RT=9.601 min (H20/CH3CN with TFA, Sunfire CI 8 3.5um, 4.6x150mm, 4.6x150mm, gradient = 15 min, wavelength = 220 and 254 nm). MS (ES): m/z= 557 [M+H]+; XH NMR (400 MHz, DMSO-d6) δ ppm 9.54 (1 H, d, J=7.28 Hz), 7.71-7.80 (1 H, m), 7.68 (2 H, d, J=8.78 Hz), 7.50-7.62 (3 H, m), 7.45 (2 H, t, J=7.28 Hz), 7.29-7.40 (2 H, m), 7.15 (1 H, br. s.), 5.30 (1 H, d, J=7.28 Hz), 3.39 (3 H, s), 2.74-2.86 (1 H, m), 2.02-2.32 (3 H, m), 1.45-1.79 (4 H, m); [a]D = -107.0° (5.73 mg/mL, DMSO).
[00231] Crystal Form A-2 was prepared by adding approximately 1 mg of Example 1 to approximately 0.7 mL of acetone/acetonitrile/water solution (2:2: 1). A mixture of colorless needles and thin blades crystals were obtained after one day of slow evaporation of the solution at room temperature. The thin blade crystals were separated to provide crystal Form A-2.
[00232] Crystal Form EA-3 was prepared by adding approximately 1 mg of Example 1 to approximately 0.7 mL of ethyl acetate/heptane solution (1 : 1). Colorless blade crystals were obtained after three days of slow evaporation of the solution at room temperature.
[00233] Crystal Form THF-2 was obtained by adding approximately 5 mg of Example 1 to approximately 0.7 mL of THF/water solution (4: 1). Colorless blade-like crystals were obtained after one day of solvent evaporation at room temperature.
Alternate Procedure to Make Example 1 : Preparation 1M: 3,3,3-Trifluoropropyl trifluoromethanesulfonate

[00234] To a cold (-25 °C), stirred solution of 2,6-lutidine (18.38 mL, 158 mmol) in CH2CI2 (120 mL) was added Tf20 (24.88 mL, 147 mmol) over 3 min, and stirred for 5 min. To the reaction mixture was added 3,3,3-trifluoropropan-l-ol (12 g, 105 mmol) over an interval of 3 min. After 2 hr, the reaction mixture was warmed to room temperature and stirred for 1 hr. The reaction mixture was concentrated to half volume, then purified by loading directly on silica gel column (330g ISCO) and eluted with CH2C12. Obtained Preparation 1M (13.74 g, 53%) as a colorless oil. XH NMR (400 MHz, CDCI3) δ ppm 4.71 (2 H, t, J=6.15 Hz), 2.49-2.86 (2 H, m).
Preparation IN: (45)-4-Benzyl- -(5,5,5-trifluoropentanoyl)-l,3-oxazolidin-2-one

[00235] Preparation IN was prepared from 5,5,5-trifluoropentanoic acid (3.35 g, 21.46 mmol) and (45)-4-benzyl-l,3-oxazolidin-2-one (3.80 g, 21.46 mmol) by the general methods shown for Preparation IB. Preparation IN (5.67 g, 84%) was obtained as a colorless viscous oil: XH NMR (400 MHz, CDC13) δ ppm 7.32-7.39 (2 H, m), 7.30 (1 H, d, J=7.05 Hz), 7.18-7.25 (2 H, m), 4.64-4.74 (1 H, m), 4.17-4.27 (2 H, m), 3.31 (1 H, dd, J=13.35, 3.27 Hz), 3.00-3.1 1 (2 H, m), 2.79 (1 H, dd, J=13.35, 9.57 Hz), 2.16-2.28 (2 H, m), 1.93-2.04 (2 H, m).
Preparation 10: tert-Butyl (3R)-3-(((45)-4-benzyl-2-oxo-l,3-oxazolidin-3-yl)carbonyl)- 6,6,6-trifluorohexanoate

[00236] To a cold (-78 °C), stirred solution of Preparation IN (3.03 g, 9.61 mmol) in THF (20 mL) was added NaHMDS (1.0M in THF) (10.6 mL, 10.60 mmol) under nitrogen atmosphere. After 2 hours, tert-butyl 2-bromoacetate (5.62 g, 28.8 mmol) was added neat via syringe at -78 °C and stirring was maintained at the same temperature. After 6 hours, the reaction mixture was warmed to room temperature. The reaction mixture was partitioned between saturated NH4C1 and EtOAc. The organic phase was separated, and the aqueous was extracted with EtOAc (3x). The combined organics were washed with brine, dried (Na2S04), filtered and concentrated under reduced pressure. The residue was purified by flash chromatography (Teledyne ISCO CombiFlash Rf, 5% to 100% solvent A/B=hexanes/EtO Ac, REDISEP® Si02 120g). Concentration of appropriate fractions provided Preparation 10 (2.79 g, 67.6%) as a colorless viscous oil: XH NMR (400 MHz, CDC13) δ ppm 7.34 (2 H, d, J=7.30 Hz), 7.24-7.32 (3 H, m), 4.62- 4.75 (1 H, m, J=10.17, 6.89, 3.43, 3.43 Hz), 4.15-4.25 (3 H, m), 3.35 (1 H, dd, J=13.60, 3.27 Hz), 2.84 (1 H, dd, J=16.62, 9.57 Hz), 2.75 (1 H, dd, J=13.35, 10.07 Hz), 2.47 (1 H, dd, J=16.62, 4.78 Hz), 2.1 1-2.23 (2 H, m), 1.90-2.02 (1 H, m), 1.72-1.84 (1 H, m), 1.44 (9 H, s). -2-(2-tert-Butoxy-2-oxoethyl)-5,5,5-trifluoropentanoic acid

[00237] Preparation IP was prepared from Preparation 10 (2.79 g, 6.50 mmol) by the general methods shown for Preparation IE. Preparation IP (1.45 g, 83%) was obtained as a colorless oil: XH NMR (400 MHz, CDC13) δ ppm 2.83-2.95 (1 H, m), 2.62-2.74 (1 H, m), 2.45 (1 H, dd, J=16.62, 5.79 Hz), 2.15-2.27 (2 H, m), 1.88-2.00 (1 H, m), 1.75-1.88 (1 H, m), 1.45 (9 H, s). Preparation IE: (2R,35′)-3-(tert-Butoxycarbonyl)-6,6,6-trifluoro-2-(3,3,3- trifluoropropyl)hexanoic acid, and
Preparation IF: (2R,3R)-3-(tert-Butoxycarbonyl)-6,6,6-trifluoro-2-(3,3,3- trifluoropropyl)hexanoic acid

(1 E) (1 F)
[00238] To a cold (-78 °C), stirred solution of Preparation IP (5.44 g, 20.13 mmol) in THF (60 mL) was slowly added LDA (24.60 mL, 44.3 mmol) over 7 min. After stirring for 2 hr, Preparation 1M (6.44 g, 26.2 mmol) was added to the reaction mixture over 3 min. After 45 min, the reaction mixture was warmed to -25 °C bath (ice/MeOH/dry ice) for 1 hr, and then warmed to 0 °C. After 45 min, Preparation 1M (lg) was added and the reaction mixture was stirred for 20 min. The reaction was quenched with water and IN NaOH and was extracted with (¾(¾. The organic layer was again extracted with IN NaOH (2x) and the aqueous layers were combined. The aqueous layer was cooled in ice/water bath and then acidified with concentrated HCl to pH 2. Next, the aqueous layer was extracted with EtOAc. The combined organics were washed with brine, dried over anhydrous sodium sulphate, and concentrated under reduced pressure. The residue was dried under high vacuum to provide a 1 :5 (IE: IF) mixture (as determined by XH NMR) of Preparation IE and Preparation IF (5.925 g, 80%) as a pale yellow solid. XH NMR (500 MHz, CDC13) 8 ppm 2.81 (1 H, ddd, J=10.17, 6.32, 3.85 Hz), 2.63-2.76 (1 H, m), 2.02- 2.33 (4 H, m), 1.86-1.99 (2 H, m), 1.68-1.85 (2 H, m), 1.47 (9 H, s).
Preparation IE: (2R,35)-3-(tert-Butoxycarbonyl)-6,6,6-trifluoro-2-(3,3,3- trifluoropropyl)hexanoic acid, and
Preparation IF: (2R,3R)-3-(tert-Butoxycarbonyl)-6,6,6-trifluoro-2-(3,3,3- trifluoropropyl)hexanoic acid

(1 E) (1 F)
[00239] A mixture of Preparation IE and Preparation IF (64 mg, 1.758 mmol) was taken in THF (6 mL) to give a colorless solution which was cooled to -78 °C. Then, LDA (2.149 mL, 3.87 mmol) (1.8M in heptane/THF/ethylbenzene) was slowly added to the reaction mixture over 10 min. After stirring for 15 min the reaction mixture was placed in a room temperature water bath. After 15 min the reaction mixture was placed back in -78 °C bath and then diethylaluminum chloride (3.87 mL, 3.87 mmol) (1M in hexane) was added slowly over 5 min. The reaction mixture was stirred at -78 °C. After 15 min the reaction mixture was placed in a room temperature water bath for 10 min and then cooled back to -78 °C bath. After 15 min the reaction was quenched with MeOH (8 mL, 198 mmol), removed from the -78 °C bath and concentrated. To the reaction mixture was added ice and HC1 (16 mL, 16.00 mmol), followed by extraction with EtOAc (2x). The organic layer was washed with potassium fluoride (920 mg, 15.84 mmol) (in 25 mL FLO) and HC1 (4.5 mL, 4.50 mmol). The organics were dried over anhydrous magnesium sulphate and concentrated under reduced pressure to provide a 9: 1 (IE: IF) enriched mixture of Preparation IE and Preparation IF (540 mg, 1.583 mmol, 90% yield) as light yellow/orange solid. ¾ NMR (400 MHz, CDC13) δ ppm 2.64-2.76 (2 H, m), 2.04-2.35 (4 H, m), 1.88-2.00 (2 H, m), 1.71-1.83 (2 H, m), 1.48 (9 H, s). It was converted to Example 1 by the sequence of reactions as outlined above.
Alternate procedure to make Preparation IE:
Preparation 1Q: (2R,35)- -Benzyl 4-tert-butyl 2,3-bis(3,3,3-trifluoropropyl)succinate

(1Q) [00240] A clean and dry 5 L four neck round bottom flask equipped with mechanical stirring, thermometer socket and nitrogen bubbler at room temperature was charged with Ν,Ν-dimethyl formamide (2.07 L), a 1.2: 1 mixture of Preparation IE and Preparation IF (207 g, 0.5651 moles), potassium carbonate (1 17.1 g, 0.8476 moles) followed by benzyl bromide (116 g, 0.6781 moles) over 15-20 min. The reaction mixture was stirred for 2-3 hr. After completion of the reaction, the reaction mixture was concentrated to dryness at 50-55 °C under vacuum. Ethyl acetate (3.1 L, 30 Vol.) was charged into the concentrated reaction mass and then washed with water (2.07 L), brine (0.6 L) then dried over anhydrous sodium sulfate (207 g), filtered and concentrated to dryness at 40-45 °C under vacuum. The residue was dissolved in dichloromethane (1.035 L, 5 vol.) and then absorbed onto silica gel (60-120) (607 g, 3.0 w/w), then was purified with column chromatography using petroleum ether and ethyl acetate as solvents. After pooling several batches, Preparation 1Q (235 g) was obtained. HPLC purity: 99.77%, Preparation IE: (2R,35)-3-(tert-Butoxycarbonyl)-6,6,6-trifluoro-2-(3,3,3- trifluoropropyl)hexanoic acid

[00241] A clean and dry 2 L autoclave was charged with methanol (540 mL) and was purged with nitrogen for 5-10 minutes. To the autoclave was added 10% palladium on carbon (12 g, 20%), purged with nitrogen once again for 5-10 min then was charged with Preparation 1Q (60g, 0.1315 moles), the autoclave was flushed with methanol (60mL) and stirred for 4-6 hr at 20-25 °C under 5Kg hydrogen pressure. After completion of the reaction, the reaction mass was filtered through CELITE®, washed with methanol (180 mL), dried with anhydrous sodium sulfate (60 g), filtered and concentrated to dryness at 45-50 °C under vacuum. Obtained Preparation IE (45.8 g, 95%) as a colorless solid: HPLC purity: 98.9%.
Alternate procedure to make Preparation IE: Preparation IE: (2R,35)-3-(te^Butoxycarbonyl)-6,6,6-trifluoro-2-(3,3,3- trifluoropropyl)hexanoic acid

[00242] Preparation IE was prepared in a procedure identical as above from a mixture of Preparations IE and IF (200g, 0.5460 moles) using LDA (1.8 M solution in THF, ethyl benzene and heptane) (698mL, 2.3equiv.) and diethyl aluminum chloride (1.0 M solution in hexane) (1256mL, 2.3equiv) in THF (2.0L). After workup as explained above, the resulting residue was treated as follows: The crude material was added to a 2L four neck round bottom flask, followed by the addition of MTBE (1.0L) charged below 30 °C. The resulting mixture was stirred for 5-10 minutes to obtain a clear solution.
Hexanes (600mL) was charged to the reaction mixture at a temperature below 30 °C. The reaction mixture was stirred for 10 min. Next, tert-butylamine (43.8g, l. leq) was charged slowly over a period of 15 minutes below 30 °C. This addition was observed to be exothermic. The reaction mixture was stirred for 2 hrs below 30 °C and filtered. The solid material was washed with 5:3 MTBE: hexane (200mL), the filtrate was
concentrated and transferred to an amber color bottle. The filtered solid was dissolved in dichloromethane (2.0L), washed with IN HC1 (2.0), the organic layer was washed with brine (1.0L x 2), then was concentrated under reduced pressure below 45 °C. This material was found to be 91.12% pure. The material was repurified by the above t- butylamine crystallization purification procedure. Obtained Preparation IE (78 g, 39%): HPLC purity: 99.54%.
Alternate procedure to make Example 1 :
Preparation II: tert-Butyl (25,,3R)-6,6,6-trifluoro-3-(((35)-l-methyl-2-oxo-5-phenyl-2,3- dihydro- 1 H- 1 ,4-benzodiazepin-3 -yl)carbamoyl)-2-(3 ,3 ,3 -trifluoropropyl)hexanoate

[00243] A clean and dry 2 L four neck round bottom flask equipped with mechanical stirring, thermometer socket and nitrogen bubbler was charged with N,N- dimethylformamide (457 mL), Preparation IE (45.7g, 0.1248moles) and Preparation lG’CSA (62.08g, 0.1248moles) under nitrogen atmosphere at 20-25 °C. The reaction mixture was stirred for 15-20 minutes to make clear solution at 20-25 °C. To the reaction mixture was added TBTU (48.16g, 0.1498 moles) at 20-25 °C followed by triethylamine (50.51g, 0.4992 moles) over 15-20 minutes at 20-25 °C. The reaction mixture was stirred for 60-120 minutes at 20-25 °C under nitrogen atmosphere. After completion of the reaction, the reaction was quenched into water (1.37L, 30 Vol.) at 20-25 °C under stirring. The reaction mixture was stirred for 30 minutes at 20-25 °C. The reaction mixture was filtered and washed with water (228 mL). The resulting solid material was dissolved in ethyl acetate (457 mL), washed with water (2×137 mL), brine (137 mL), and then dried with anhydrous sodium sulfate (45.7g). Activated charcoal (9.14 g, 20%) was charged into the reaction mixture and stirred for 30 minutes. The mixture was filtered through CELITE® bed and 1 micron filter cloth, washed charcoal bed with ethyl acetate (137 mL), concentrated to 1.0 Vol. stage and then petroleum ether (457 mL, 10 Vol.) was charged and stirred for 30 minutes at 20-25 °C. The solid was collected by filtration, washed with petroleum ether (137 mL) and then dried under vacuum at 40-45 °C for 8 hr until loss on drying was less than 3.0%. Obtained Preparation II (65.2 g, 85%): HPLC purity: 98.26%.
Preparation IK: (25,,3R)-6,6,6-Trifluoro-3-(((35)-l-methyl-2-oxo-5-phenyl-2,3-dihydro- 1 H- 1 ,4-benzodiazepin-3 -yl)carbamoyl)-2-(3 ,3 ,3 -trifluoropropyl)hexanoic acid

[00244] A clean and dry 3 L four neck round bottom flask equipped with mechanical stirring, thermometer socket and nitrogen bubbler was charged with dichloromethane (980 mL) under nitrogen atmosphere followed by Preparation II (140 g, 0.2282 moles) at 20-25 °C. The reaction mixture was cooled to 0-5 °C and trifluoroacetic acid (980 mL) was charged slowly for 30-40 minutes. The resulting mixture was stirred for 2 hr at 0-5 °C under nitrogen atmosphere. The reaction temperature was raised to 20 to 25 °C, and the reaction mixture was stirred for 1-2 hr at 20 to 25 °C. After completion of the reaction, the reaction mixture was concentrated to dryness at 50 to 55 °C under vacuum. Toluene (3×700 mL,) was charged into the concentrated reaction mass, and then distilled off at 50 to 55 °C under vacuum. After complete concentration from toluene, ethyl acetate (280 mL) was charged into the reaction mass at 20 to 25 °C, stirred for 60 minutes, then the solid was collected by filtration, washed with ethyl acetate (140 mL), dried under vacuum at 50 to 55 °C for 12 hr until loss on drying was less than 2.0%. Obtained Preparation IK (106 g, 84%): HPLC purity: 98.43%.
Example 1 :
[00245] A reaction vessel was charged with Preparation IK (30 g, 53.81 mmol), HOBt (8.7g, 64.38 mmol), and THF (150 mL) at room temperature. To the homogeneous solution was added EDCI (12.4g, 64.68 mmol), stirred for 15 min, then cooled to 8 °C. To the reaction mixture was added ammonia (2M in IP A) (81 mL, 162 mmol) over 5 min so as to maintain a temperature below 10 °C. The resulting heavy slurry was stirred for 10 min, warmed to room temperature over 30 min, then stirred for 4 hr. At the completion of the reaction, water (230 mL) was slowly added over 15 min to maintain a temperature below 20 °C, and then stirred for 2 hr. The solid was collected by filtration, washed with water (3X60 mL), then dried under vacuum 48 hr at 55 °C. The above crude product was charged into a 1 L 3 -necked round flask. IP A (200 mL) was added, then heated to 80 °C resulting in a homogeneous solution. Water (170 mL) was slowly added (15 min) to maintain an internal temperature >75 °C. The resulting slurry was stirred and cooled to room temperature for 2 hr. The solid was collected by filtration, washed with water (2 X 50 mL), then dried under vacuum (55 °C for 24 h, and 30 °C for 48 h).
Obtained Example 1 (23.4 g, 78% yield): HPLC purity: 99.43%.
Example 2 NOT SAME
WITHOUT METHYL GROUP
(2R,35)-N-((35)-2-Oxo-5-phenyl-2,3-dihydro-lH-l,4-benzodiazepin-3-yl)-2,3-bis(3,3,3- trifluoropropyl)succinamide

Preparation 2A: (35)-3-Amino-5-phenyl-l,3-dihydro-2H-l,4-benzodiazepin-2-one, and Preparation 2B: -3-Amino-5-phenyl-l,3-dihydro-2H-l,4-benzodiazepin-2-one

(2A) (2B)
[00246] Racemic 3-amino-5-phenyl-l,3-dihydro-2H-l,4-benzodiazepin-2-one (J. Med. Chem., 49:231 1-2319 (2006), compound# 5) was prepared according to the literature procedure. The enantiomers were separated on Berger SFC MGIII Column: Lux 25X3 cm, 5cm; Mobile phase: 30% MeOH+ 0.1% DEA in C02; Flow rate: 150 mL/min;
Temperature: 40 °C; Detector wavelength: 250 nM. Obtained the S-enantiomer
Preparation 2A as a white solid: XH NMR (400 MHz, DMSO-d6) δ ppm 10.67 (1 H, br. s.), 7.58 (1 H, td, J=7.65, 1.76 Hz), 7.37-7.53 (5 H, m), 7.23-7.30 (2 H, m), 7.14-7.22 (1 H, m), 4.23 (1 H, s), 2.60 (2 H, br. s.); HPLC: RT=3.0625 min (30% MeOH + 0.1% DEA in C02 on OD-H Column, 3 mL/min, 35 °C, 96 bar, 230 nm, ΙΟμΙ inj); [a]D = -208.3° (5.05 mg/niL, MeOH). Also obtained the R-enantiomer Preparation 2B as an off white solid: HPLC: RT=3.970 min; [a]D = 182.1° (2.01 mg/mL, MeOH).
Preparation 2C: tert-Butyl (25,,3R)-6,6,6-trifluoro-3-(((35)-2-oxo-5-phenyl-2,3-dihydro- 1 H- 1 ,4-benzodiazepin-3 -yl)carbamoyl)-2-(3 ,3 ,3 -trifluoropropyl)hexanoate, and
Preparation 2D: tert-Butyl (2R,3R)-6,6,6-trifluoro-3-(((35)-2-oxo-5-phenyl-2,3-dihydro- 1 H- -benzodiazepin-3 -yl)carbamoyl)-2-(3 ,3 ,3 -trifluoropropyl)hexanoate

(2C) (2D)
[00247] Preparation 2C was prepared from Preparation 2A (564 mg, 2.244 mmol) and a mixture of Preparation IE and Preparation IF (822 mg, 2.244 mmol) according to the general procedure shown for Preparation II. Obtained Preparation 2C and Preparation 2D (1.31 g, 97%): HPLC: RT=3.443 min (CHROMOLITH® ODS 4.6 x 50 mm (4 min grad) eluting with 10-90% aqueous MeOH over 4 minutes containing 0.% TFA, 4 mL/min, monitoring at 220 nm); MS (ES): m/z= 600.3 [M+H]+.
Preparation 2E: (25′,3R)-6,6,6-Trifluoro-3-(((35)-2-oxo-5-phenyl-2,3-dihydro-lH-l,4- benzodiazepin-3-yl)carbamoyl)-2-(3,3,3-trifluoropropyl)hexanoic acid, and
Preparation 2F: (2R,3R)-6,6,6-Trifluoro-3-(((35)-2-oxo-5-phenyl-2,3-dihydro-lH-l,4- benzodiazepin-3-yl)carbamoyl)-2-(3,3,3-trifluoropropyl)hexanoic acid

(2E) (2F) [00248] A mixture of Preparation 2E and Preparation 2F was prepared from a mixture of Preparation 2C and Preparation 2D (1.3 lg, 2.185 mmol) by the general methods shown for Preparation IK. Obtained a mixture of Preparation 2E and Preparation 2F (1.18 g, 99%): HPLC: RT=2.885 min (CHROMOLITH® ODS 4.6 x 50 mm (4 min grad) eluting with 10-90% aqueous MeOH over 4 minutes containing 0.% TFA, 4 mL/min, monitoring at 220 nm). MS (ES): m/z= 544.2 [M+H]+.
Example 2:
[00249] Example 2 was prepared from a mixture of Preparation 2E and Preparation 2F (354 mg, 0.651 mmol) by the general methods shown for Example 1. After separation of the diastereoisomers, Example 2 was obtained (188 mg, 52%) as a white solid: HPLC: RT=9.063 min (H20/CH3CN with TFA, Sunfire C18 3.5um, 4.6x150mm, 4.6x150mm, gradient = 15 min, wavelength = 220 and 254 nm); MS (ES): m/z= 543 [M+H]+; XH NMR (400 MHz, DMSO-d6) δ ppm 10.87 (1 H, br. s.), 9.50-9.55 (1 H, m), 7.62-7.69 (2 H, m), 7.40-7.57 (5 H, m), 7.29-7.36 (2 H, m), 7.22-7.28 (1 H, m), 7.16 (1 H, br. s.), 5.25 (1 H, d), 3.30-3.32 (1 H, m), 2.75-2.86 (1 H, m), 2.44-2.48 (1 H, m), 2.06-2.34 (3 H, m), 1.51- 1.77 (4 H, m); [a]D = -114.4° (8.04 mg/mL, DMSO).
[00250] Crystal Form M2- 1 was prepared by adding approximately 1 mg of Example 2 to approximately 0.7 mL of MeOH/fluorobenzene solution (3 : 1). Colorless plate-like crystals were obtained after 2 days of solvent evaporation at room temperature.
PATENT

Example 1
(2R,3S)—N-((3S)-1-Methyl-2-oxo-5-phenyl-2,3-dihydro-1H-1,4-benzodiazepin-3-yl)-2,3-bis(3,3,3-trifluoropropyl)succinamide

Preparation 1A: tert-Butyl 5,5,5-trifluoropentanoate

Preparation 1B: (4S)-4-(Propan-2-yl)-3-(5,5,5-trifluoropentanoyl)-1,3-oxazolidin-2-one

Preparation 1C: (2S,3R)-tert-Butyl 6,6,6-trifluoro-3-((S)-4-isopropyl-2-oxooxazolidine-3-carbonyl)-2-(3,3,3-trifluoropropyl)hexanoate, and
Preparation 1D: (2R,3R)-tert-Butyl 6,6,6-trifluoro-3-((S)-4-isopropyl-2-oxooxazolidine-3-carbonyl)-2-(3,3,3-trifluoropropyl)hexanoate

Preparation 1E: (2R,3S)-3-(tert-Butoxycarbonyl)-6,6,6-trifluoro-2-(3,3,3-trifluoropropyl)hexanoic acid, and
Preparation 1F: (2R,3R)-3-(tert-Butoxycarbonyl)-6,6,6-trifluoro-2-(3,3,3-trifluoropropyl)hexanoic acid

Preparation 1E: (2R,3S)-3-(tert-Butoxycarbonyl)-6,6,6-trifluoro-2-(3,3,3-trifluoropropyl)hexanoic acid, and
Preparation 1F: (2R,3R)-3-(tert-Butoxycarbonyl)-6,6,6-trifluoro-2-(3,3,3-trifluoropropyl)hexanoic acid

Preparation 1G: (3S)-3-Amino-1-methyl-5-phenyl-1,3-dihydro-2H-1,4-benzodiazepin-2-one, and
Preparation 1H: (3R)-3-Amino-1-methyl-5-phenyl-1,3-dihydro-2H-1,4-benzodiazepin-2-one

Alternate Procedure to Make Preparation 1G
Preparation 1G•CSA salt: (3S)-3-Amino-1-methyl-5-phenyl-1,3-dihydro-2H-1,4-benzodiazepin-2-one, (1 S)-(+)-10-camphorsulfonic acid salt

Preparation 1I: tert-Butyl (2S,3R)-6,6,6-trifluoro-3-(((3S)-1-methyl-2-oxo-5-phenyl-2,3-dihydro-1H-1,4-benzodiazepin-3-yl)carbamoyl)-2-(3,3,3-trifluoropropyl)hexanoate, and
Preparation 1J: tert-Butyl (2R,3R)-6,6,6-trifluoro-3-(((3S)-1-methyl-2-oxo-5-phenyl-2,3-dihydro-1H-1,4-benzodiazepin-3-yl)carbamoyl)-2-(3,3,3-trifluoropropyl)hexanoate

Preparation 1K: (2S,3R)-6,6,6-Trifluoro-3-(((3S)-1-methyl-2-oxo-5-phenyl-2,3-dihydro-1H-1,4-benzodiazepin-3-yl)carbamoyl)-2-(3,3,3-trifluoropropyl)hexanoic acid, and
Preparation 1L: (2R,3R)-6,6,6-Trifluoro-3-(((3S)-1-methyl-2-oxo-5-phenyl-2,3-dihydro-1H-1,4-benzodiazepin-3-yl)carbamoyl)-2-(3,3,3-trifluoropropyl)hexanoic acid

Example 1
Alternate Procedure to Make Example 1
Preparation 1M: 3,3,3-Trifluoropropyl trifluoromethanesulfonate

Preparation 1N: (4S)-4-Benzyl-3-(5,5,5-trifluoropentanoyl)-1,3-oxazolidin-2-one

Preparation 1O: tert-Butyl (3R)-3-(((4S)-4-benzyl-2-oxo-1,3-oxazolidin-3-yl)carbonyl)-6,6,6-trifluorohexanoate

Preparation 1P: (2R)-2-(2-tert-Butoxy-2-oxoethyl)-5,5,5-trifluoropentanoic acid

Preparation 1E: (2R,3S)-3-(tert-Butoxycarbonyl)-6,6,6-trifluoro-2-(3,3,3-trifluoropropyl)hexanoic acid, and
Preparation 1F: (2R,3R)-3-(tert-Butoxycarbonyl)-6,6,6-trifluoro-2-(3,3,3-trifluoropropyl)hexanoic acid

Preparation 1E: (2R,3S)-3-(tert-Butoxycarbonyl)-6,6,6-trifluoro-2-(3,3,3-trifluoropropyl)hexanoic acid, and
Preparation 1F: (2R,3R)-3-(tert-Butoxycarbonyl)-6,6,6-trifluoro-2-(3,3,3-trifluoropropyl)hexanoic acid

Alternate Procedure to Make Preparation 1E
Preparation 1Q: (2R,3S)-1-Benzyl 4-tert-butyl 2,3-bis(3,3,3-trifluoropropyl)succinate

Preparation 1E: (2R,3S)-3-(tert-Butoxycarbonyl)-6,6,6-trifluoro-2-(3,3,3-trifluoropropyl)hexanoic acid

Alternate Procedure to Make Preparation 1E
Preparation 1E: (2R,3S)-3-(tert-Butoxycarbonyl)-6,6,6-trifluoro-2-(3,3,3-trifluoropropyl)hexanoic acid

Alternate Procedure to Make Example 1
Preparation 1I: tert-Butyl (2S,3R)-6,6,6-trifluoro-3-(((3S)-1-methyl-2-oxo-5-phenyl-2,3-dihydro-1H-1,4-benzodiazepin-3-yl)carbamoyl)-2-(3,3,3-trifluoropropyl)hexanoate

Preparation 1K: (2S,3R)-6,6,6-Trifluoro-3-(((3S)-1-methyl-2-oxo-5-phenyl-2,3-dihydro-1H-1,4-benzodiazepin-3-yl)carbamoyl)-2-(3,3,3-trifluoropropyl)hexanoic acid

Example 1
PATENTS
Clip
For some disease targets, an indirect approach may be best. Or so Ashvinikumar V. Gavai and his colleagues atBristol-Myers Squibbfound in their quest toward a potential cancer drug. Gavai unveiled BMS-906024, which is an experimental—and slightly roundabout—treatment for a number of cancers, including breast, lung, and colon cancers, and leukemia.
Cancers have a tendency to relapse or to become resistant to treatments that once worked. Research at BMS and elsewhere had suggested that a family of proteins called Notch is implicated in that resistance and in cancer progression more generally. Gavai, director of oncology chemistry at BMS in Princeton, N.J., and his team set out to block Notch family signaling.
Notch family members lack enzymatic activity, so blocking them directly is difficult. Instead, BMS developed inhibitors of an enzyme that is essential for activating Notch signaling—γ-secretase.
Company: Bristol-Myers Squibb
Target: pan-Notch
Disease: breast, lung, colon cancer; leukemia
Interfering with Notch, even in this indirect way, can have detrimental effects on the gastrointestinal tract. Only two of the four Notch family members are linked to that side effect, Gavai says. But he and his team think their drug will be most effective if it acts on all four family members roughly equally—a so-called pan-Notch inhibitor. By selecting a molecule that’s well tolerated in animals and carefully scheduling doses of the drug in humans, it could be possible to minimize side effects, he says.
The BMS team relied on Notch signaling assays in leukemia and breast cancer cell lines to find leads. They soon learned that for their molecules to work, three chiral centers had to be in the S,R,Sconfiguration. After that, they strove to make the molecules last in the bloodstream. They removed an isobutyl group and tweaked some other parts of their candidate’s succinamide side chain. It was tough to retain both a long half-life and activity against Notch, Gavai told C&EN. “You’d optimize one and lose the other.”
His team threaded the needle with BMS-906024. Their studies with mice suggest that a dose of 4–6 mg once a week could be effective in people. That’s lower than doses being tested for other Notch-targeted agents, according to the website clinicaltrials.gov. The mouse studies also back the idea that Notch is involved in cancer drug resistance and suggest that Notch could be a target for taking on cancer stem cells, which are notoriously resistant to chemotherapy.
BMS-906024 is in Phase I clinical trials, both alone and in combination with other agents. Patients with colon, lung, breast, and other cancers are receiving intravenous doses of the compound to determine its safety and optimum dose ranges.
(From left, front row) Gavai, Weifeng Shan, (second row) Aaron Balog, Patrice Gill, Gregory Vite, (third row) Francis Lee, Claude Quesnelle, (rear row) Wen-Ching Han, Richard Westhouse.
Credit: Catherine Stroud Photography
http://cen.acs.org/articles/91/i16/BMS-906024-Notch-Signaling-Inhibitor.html
clip

BMS-906024
Company: Bristol-Myers Squibb
Meant to treat: cancers including breast, lung, colon, and leukemia
Mode of action: pan-Notch inhibitor
Medicinal chemistry tidbit: The BMS team used an oxidative enolate heterocoupling en route to the candidate– a procedure from Phil Baran’s lab at Scripps Research Institute. JACS 130, 11546
Status in the pipeline: Phase I
Relevant documents: WO 2012/129353
PAPER
An enantioselective synthesis of (S)-7-amino-5H,7H-dibenzo[b,d]azepin-6-one (S–1) is described. The key step in the sequence involved crystallization-induced dynamic resolution (CIDR) of compound 7 using Boc-d-phenylalanine as a chiral resolving agent and 3,5-dichlorosalicylaldehyde as a racemization catalyst to afford S–1 in 81% overall yield with 98.5% enantiomeric excess.
† Department of Discovery Synthesis, Biocon Bristol-Myers Squibb Research Centre, Biocon Park, Bommasandra IV Phase, Jigani Link Road, Bengaluru 560099, India
‡Bristol-Myers Squibb Company, P.O Box 4000, Princeton, New Jersey 08543-4000, United States
Org. Process Res. Dev., Article ASAP
DOI: 10.1021/acs.oprd.6b00207, http://pubs.acs.org/doi/suppl/10.1021/acs.oprd.6b00207
*E-mail: anuradha.gupta@syngeneintl.com.
References
- Jump up^ C. Drahl, Liveblogging First-Time Disclosures of Drug Structures from #ACSNOLA, 2013, http://cenblog.org/the-haystack/2013/04/liveblogging-first-time-disclosures-of-drug-structures-from-acsnola/
- Jump up^ http://patentscope.wipo.int/search/en/detail.jsf?docId=WO2012129353
- Jump up^ http://clinicaltrials.gov/show/NCT01653470
- Jump up^ http://clinicaltrials.gov/show/NCT01292655
1. Quesnelle, Claude; Kim, Soong-Hoon; Lee, Francis; Gavai, Ashvinikumar. Bis(fluoroalkyl)-1,4-benzodiazepinone compounds as Notch receptor inhibitors and their preparation and use in the treatment of cancer. PCT Int. Appl. (2012), WO 2012129353 A1 20120927.
| Patent ID | Date | Patent Title |
|---|---|---|
| US2016060232 | 2016-03-03 | BIS(FLUOROALKYL)-1, 4-BENZODIAZEPINONE COMPOUNDS |
| US2016022723 | 2016-01-28 | COMBINATION THERAPY FOR THE TREATMENT OF PROLIFERATIVE DISEASES |
| US2016008316 | 2016-01-14 | USE OF DIANHYDROGALACTITOL AND ANALOGS OR DERIVATIVES THEREOF IN COMBINATION WITH PLATINUM-CONTAINING ANTINEOPLASTIC AGENTS TO TREAT NON-SMALL-CELL CARCINOMA OF THE LUNG AND BRAIN METASTASES |
| US2016009785 | 2016-01-14 | NOVEL FUSION MOLECULES AND USES THEREOF |
| US2015284342 | 2015-10-08 | BIS(FLUOROALKYL)-1, 4-BENZODIAZEPINONE COMPOUNDS |
| US2015232491 | 2015-08-20 | PRODRUGS OF 1, 4-BENZODIAZEPINONE COMPOUNDS |
| US8968741 | 2015-03-03 | Anti-CD22 antibodies and immunoconjugates and methods of use |
| US2014357605 | 2014-12-04 | BIS(FLUOROALKYL)-1, 4-BENZODIAZEPINONE COMPOUNDS |
| US8822454 | 2014-09-02 | Bisfluoroalkyl-1, 4-benzodiazepinone compounds |
| US8629136 | 2014-01-14 | Bisfluoroalkyl-1, 4-benzodiazepinone compounds |
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
(2R,3S)-N-[(3S)-1-Methyl-2-oxo-5-phenyl-2,3-dihydro-1H-1,4-benzodiazepin-3-yl]-2,3-bis(3,3,3-trifluoropropyl)succinamide
|
|
| Identifiers | |
| PubChem | CID 66550890 |
| ChemSpider | 28536138 |
| Chemical data | |
| Formula | C26H26F6N4O3 |
| Molar mass | 556.500 g/mol |
///////////////3,5-dichlorosalicylaldehyde, Alzheimer’s disease, Boc-D-phenylalanine, CIDR;dibenzoazepenone, DKR; Notch inhibitors, Notch inhibitor, SAR, T-acute lymphoblastic leukemia, triple-negative breast cancer, γ-secretase inhibitor, PHASE 1, BMS, Bristol-Myers Squibb, 1401066-79-2, Ashvinikumar Gavai
CN1c2ccccc2C(=N[C@@H](C1=O)NC(=O)[C@H](CCC(F)(F)F)[C@H](CCC(F)(F)F)C(=O)N)c3ccccc3

Patent US8377886 – Use of gamma secretase inhibitors and notch …
Figure US08377886-20130219-C00003. gamma secretase inhibitor

RO4929097 | γ-secretase inhibitor – Cellagen Technology
RO4929097 | γ-secretase inhibitor

YO-01027 (Dibenzazepine) | gamma-secretase inhibitor – Cellagen …
YO-01027 (Dibenzazepine) | gamma-secretase inhibitor

Semagacestat (LY450139) | Gamma-secretase inhibitor | Read Reviews …
Semagacestat (LY450139) Chemical Structure

{kind=link}