
Home » Posts tagged 'APPROVALS 2022' (Page 5)
Tag Archives: APPROVALS 2022
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on
ORGANIC SPECTROSCOPY INTERNATIONAL

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Mavacamten
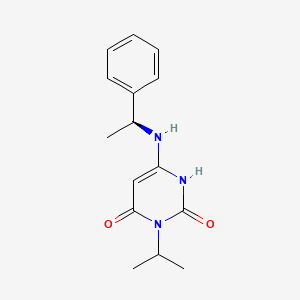 Mavacamten
SAR-439152; SAR 439152; SAR439152; MYK-461; MYK 461; MYK461; Mavacamten
(S)-3-isopropyl-6-((1-phenylethyl)amino)pyrimidine-2,4(1H,3H)-dione
cas 1642288-47-8
Chemical Formula: C15H19N3O2
Molecular Weight: 273.336
マバカムテン;
Mavacamten
SAR-439152; SAR 439152; SAR439152; MYK-461; MYK 461; MYK461; Mavacamten
(S)-3-isopropyl-6-((1-phenylethyl)amino)pyrimidine-2,4(1H,3H)-dione
cas 1642288-47-8
Chemical Formula: C15H19N3O2
Molecular Weight: 273.336
マバカムテン;
- UNII-QX45B99R3J
- QX45B99R3J
- HCM 1; MYK-461; SAR-439152
- Originator MyoKardia
- Class Cardiovascular therapies; Small molecules
- Mechanism of Action Myosin inhibitors
- Orphan Drug Status Yes – Hypertrophic cardiomyopathy
Highest Development Phases
- Phase III Hypertrophic cardiomyopathy
Most Recent Events
- 30 May 2018 Phase-III clinical trials in Hypertrophic cardiomyopathy in USA (PO) (NCT03470545)
- 08 May 2018 MyoKardia plans a long-term extension (LTE) trial of patients who complete the phase III EXPLORER-HCM or the phase II MAVERICK-HCM trial for Hypertrophic cardiomyopathy by the end of 2018
- 26 Apr 2018 MyoKardia initiates the PIONEER-OLE trial (an extension trial of phase II PIONEER trial) for Hypertrophic cardiomyopathy in USA (PO) (NCT03496168)
SYN CONSTRUCTION




SYN
J. Med. Chem. 2024, 67, 4376−4418
Mavacamten (Camzyos). Mavacamten (8) is a small molecule modulator of beta-cardiac myosin and reduces hypercontractility, a symptom of hypertrophic cardiomyopathy (HCM). Hypertrophic cardiomyopathy is a monogenetic cardiovascular disorder caused by mutations to sarcomere
proteins, specifically myosin, 61 1 in 200 people. 62 and may be found in as many as Mavacamten is thought to impact HCM in two ways: reduction of actin-myosin cross-bridge formation and inhibition of cardiac myosin ATPase. Both result in reduced contractility by modulating myosin function. 63
Mavacamten was developed by MyoKardia, Inc., a subsidiary of Bristol Myers Squibb. It was approved by the USFDA for treatment of adults with symptomatic New York Heart Association (NYHA) classes II−III obstructive hypertrophic cardiomyopathy.64
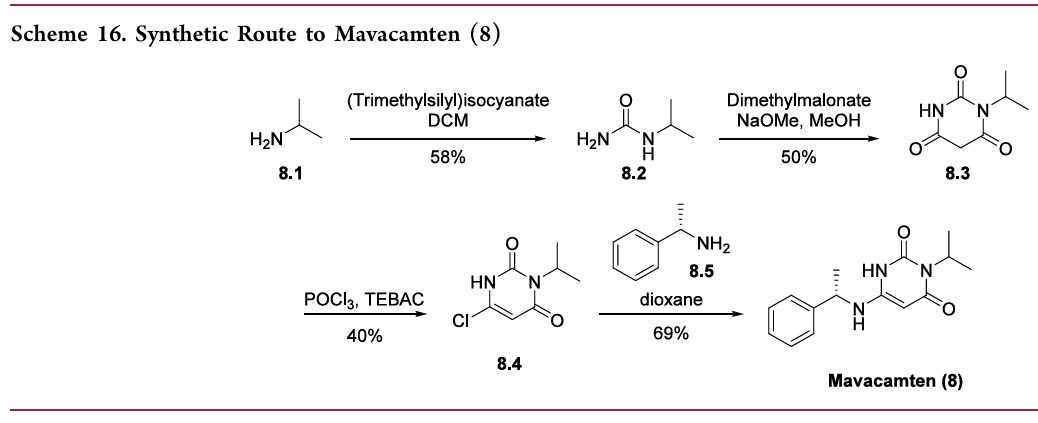
A synthetic route to mavacamten has been65 disclosed in a patent from MyoKardia and demonstrated on
the gram scale.
While synthesis of the molecule is straightforward, the conditions would likely require modification prior to manufacture on the kilogram scale. Starting with isopropyl amine (8.1), treatment with trimethylsilyl isocyanate provided urea 8.2 in 58% yield (Scheme 16). Cyclization with dimethyl malonate provided barbituric acid derivative 8.3 in 50% yield. Installation of the chiral amine side chain was achieved in a two-step sequence, first by chlorination usingneat POCl3 and triethylbenzyl ammonium chloride (TEBAC)
to access chloride 8.4 in 40% yield and then treatment with (S)-α-methylbenzylamine (8.5) to provide mavacamten (8) in 69% yield as a white solid.
(61) Green, E. M.; Wakimoto, H.; Anderson, R. L.; Evanchik, M. J.;
Gorham, J. M.; Harrison, B. C.; Henze, M.; Kawas, R.; Oslob, J. D.;
Rodriguez, H. M.; et al. A small-molecule inhibitor of sarcomere
contractility suppresses hypertrophic cardiomyopathy in mice. Science
2016, 351, 617−621.
(62) Tower-Rader, A.; Ramchand, J.; Nissen, S. E.; Desai, M. Y.
Mavacamten: a novel small molecule modulator of β-cardiac myosin
for treatment of hypertrophic cardiomyopathy. Expert Opin. Investig.
Drugs 2020, 29, 1171−1178.
(63) Kawas, R. F.; Anderson, R. L.; Ingle, S. R. B.; Song, Y.; Sran, A.
S.; Rodriguez, H. M. A small-molecule modulator of cardiac myosin
acts on multiple stages of the myosin chemomechanical cycle. J. Biol.
Chem. 2017, 292, 16571−16577.
(64) Keam, S. J. Mavacamten: First approval. Drugs 2022, 82, 1127−
1135.
(65) Semigran, M. J.; Lee, J. H.; Lambing, J.; Green, E.; Evanchik,
M. Mavacamten for use in the treatment of hypertrophic
cardiomyopathy. WO 2019028360, 2019.
PATENT
InventorJohan OslobRobert AndersonDanielle AubeleMarc EvanchikJonathan Charles FoxBrian KanePuping LuRobert McDowellHector RodriguezYonghong SongArvinder Sran
Current Assignee MyoKardia Inc Original AssigneeMyoKardia Inc
Priority date 2013-06-21
Example 1 Preparation of (S)-3-Isopropyl-6-((1-phenylethyl)amino)pyrimidine-2,4(1H,3H)-dione
Compound 1.1. Isopropylurea. To a stirred solution of isopropylamine (15.3 g, 0.258 mol, 1.0 equiv) in CH2Cl2 (200 mL) under argon at 0° C. was added dropwise trimethylsilyl isocyanate (30 g, 0.26 mol, 1.0 equiv). The resulting mixture was allowed to reach ambient temperature and stirred overnight. After cooling to 0° C., CH3OH (100 mL) was added dropwise. The resulting solution was stirred for 2 hours (h) at room temperature and then concentrated under reduced pressure. The crude residue was recrystallized from CH3OH:Et2O (1:20) to yield 15.4 g (58%) the title compound as a white solid. LC/MS: m/z (ES+) 103 (M+H)+.
Compound 1.2. 1-Isopropyl barbituric acid. To a stirred solution of 1.1 (14.4 g, 0.14 mol, 1.00 equiv) in CH3OH (500 mL) were added dimethyl malonate (19.55 g, 0.148 mol, 1.05 equiv) and sodium methoxide (18.9 g, 0.35 mol, 2.50 equiv). The resulting mixture was stirred overnight at 65° C. After cooling to ambient temperature and then to 0° C., the pH was carefully adjusted to 3 using aqueous concentrated HCl. The resulting mixture was concentrated under reduced pressure. The residue was taken up in EtOH (200 mL) and filtered. The filtrate was concentrated under reduced pressure and the residue was purified by silica gel column chromatography using CH2Cl2/CH3OH (20:1) as eluent to yield 16.8 g (50%) of the title compound as a white solid. LC/MS: m/z (ES+) 171 (M+H)+. 1 1H-NMR (300 MHz, d6-DMSO): δ 11.19 (s, 1H), 4.83 (m, 1H), 3.58 (s, 2H), 1.32 (d, J=6.0 Hz, 6H).
Compound 1.3. 6-chloro-3-isopropylpyrimidine-2,4(1H,3H)-dione. To a 100-mL round-bottom flask containing compound 1.2 (11.4 g, 66.99 mmol, 1.00 equiv) under argon were added triethylbenzylammonium chloride (21.3 g, 93.51 mmol, 1.40 equiv) and POCl3 (30 mL). The resulting mixture was stirred overnight at 50° C. After cooling to room temperature, the mixture was concentrated under reduced pressure. The residue was dissolved in CH2Cl2 (150 mL) followed by slow addition of H2O (100 mL). The phases were separated and the organic layer was washed with H2O (100 mL), dried with anhydrous Na2SO4, and concentrated under reduced pressure. The crude residue was purified by silica gel column chromatography using EtOAc/petroleum ether (1:1) as eluent to yield 5.12 g (40%) of the title compound as a light yellow solid. 1H-NMR (300 MHz, d6-DMSO): δ 12.22 (s, 1H), 5.88 (s, 1H), 4.95 (m, 1H), 1.34 (d, J=6.0 Hz, 6H).
Compound 1. (S)-3-Isopropyl-6-((1-phenylethyl)amino)pyrimidine-2, 4(1H,3H)-dione. To a solution of 6-chloro-3-isopropylpyrimidine-2,4(1H,3H)-dione (1.3, 1.0 g, 5.31 mmol) in 1,4-dioxane (20 mL) was added (S)-α-methylbenzylamine (Sigma-Aldrich, 1.43 g, 11.7 mmol, 2.2 equiv). The reaction mixture was stirred at 80° C. for 24 h. After cooling to ambient temperature, the mixture was concentrated under reduced pressure. The residual was taken up in EtOAc (70 mL) and washed with aqueous 1N HCl (2×50 mL) and brine (40 mL). The organic layer was dried with anhydrous Na2SO4 and then concentrated under reduced pressure to half the original volume to yield a precipitate. Hexane (20 mL) was added and the mixture was stirred at room temperature. The resulting solid was collected by filtration, washed with hexane (20 mL), and dried to yield 1.0 g (69%) of the title compound as a white solid. LC/MS: m/z (ES+) 274 (M+H)+. 1H-NMR (400 MHz, d6-DMSO): δ 9.77 (s, 1H), 7.32 (m, 4H), 7.24 (m, 1H), 6.50 (d, J=6.8 Hz, 1H), 4.87 (m, 1H), 4.52 (m, 1H), 4.31 (d, J=6.8 Hz, 1H), 1.37 (m, 3H), 1.24 (m, 6H). 1H NMR (400 MHz, CD3OD) δ ppm 7.39-7.20 (m, 5H), 5.01 (m, 1H), 4.48 (m, 1H), 1.49 (d, J=6.7 Hz, 3H), 1.36 (m, 6H).
PATENT
https://patents.google.com/patent/US9181200/zh-CN Genetic (heritable) hypertrophic cardiomyopathy (HCM) comprises a group of highly penetrant, monogenic, autosomal dominant myocardial diseases. HCM is caused by one or more of over 1,000 known point mutations in any one of the structural protein genes contributing to the functional unit of myocardium, the sarcomere. About 1 in 500 individuals in the general population are found to have left ventricular hypertrophy unexplained by other known causes (e.g., hypertension or valvular disease), and many of these can be shown to have HCM, once other heritable (e.g., lysosomal storage diseases), metabolic, or infiltrative causes have been excluded. [0004] Sarcomere gene mutations that cause HCM are highly penetrant, but there is wide variability in clinical severity and clinical course. Some genotypes are associated with a more malignant course, but there is considerable variability between and even within families carrying the same mutation. Sex differences have also been noted, with male patients generally more severely affected than female patients. While many patients with HCM report minimal or no symptoms for extended periods of time, HCM is a progressive disease with a significant cumulative burden of morbidity. Symptoms of effort intolerance predominate, and can be exacerbated by exercise and other maneuvers that increase heart rate and/or decrease preload. As with many other disorders, symptoms tend to worsen with age. By far the most prevalent clinical burden for patients with HCM is exertional dyspnea, which limits their activities of daily living and can be debilitating. [0005] Patients with HCM are often symptomatic in the absence of documented hemodynamic abnormalities like left ventricular outflow tract obstruction (with or without mitral regurgitation). Patients’ symptoms of exertional dyspnea can rapidly worsen with the onset of atrial fibrillation, a common complication of HCM that can precipitate acute pulmonary edema that increases the risk of systemic arterial thromboembolic disease, including stroke. Other adverse events associated with HCM include intolerance of hypovolemia or hypervolemia, and syncope. Concomitant coronary artery disease may confer a higher risk of acute coronary syndromes than in patients without HCM. Sudden cardiac death (SCD) in patients with HCM is both uncommon and difficult to predict but is a leading cause of non-traumatic death in young adults. For survivors of SCD, ICD placement is standard practice, and in other HCM patients risk profiling, while imprecise, is used to identify those for whom ICD placement for primary prevention is deemed prudent. [0006] Medical therapy for HCM is limited to the treatment of symptoms and does not address the fundamental, underlying cause of disease – disruptions in normal sarcomere function. Currently available therapies are variably effective in alleviating symptoms but typically show decreased efficacy with increasing disease duration. Patients are thus empirically managed with beta-blockers, non-dihydropyridine calcium channel blockers, and/or disopyramide. None of these agents carry labeled indications for treating HCM, and essentially no rigorous clinical trial evidence is available to guide their use. Compounding this unfortunate situation is the fact that no new medical therapies for HCM have been identified for many years. For patients with hemodynamically significant outflow tract obstruction (resting gradient >30mmHg), in appropriately selected patients surgical myectomy or alcohol septal ablation is usually required to alleviate the hemodynamic obstruction. Provided are new therapeutic agents and methods that remedy the long-felt need for improved treatment of HCM and related cardiac disorders.
Example 1. Preparation of (61-3-Isopropyl-6-((1-phenylethyl) amino) pyrimidine-2, 4(1H,3H)-dione.
 [0072] Compound 1.1. Isopropylurea. To a stirred solution of isopropylamine (15.3 g, 0.258 mol, 1.0 equiv) in CH2Cl2 (200 mL) under argon at 0 °C was added dropwise trimethylsilyl isocyanate (30 g, 0.26 mol, 1.0 equiv). The resulting mixture was allowed to reach ambient temperature and stirred overnight. After cooling to 0 °C, CH3OH (100 mL) was added dropwise. The resulting solution was stirred for 2 hours (h) at room temperature and then concentrated under reduced pressure. The crude residue was recrystallized from CH3OH:Et2O (1 :20) to yield 15.4 g (58%) the title compound as a white solid. LC/MS: m/z (ES+) 103 (M+H)+.
[0072] Compound 1.1. Isopropylurea. To a stirred solution of isopropylamine (15.3 g, 0.258 mol, 1.0 equiv) in CH2Cl2 (200 mL) under argon at 0 °C was added dropwise trimethylsilyl isocyanate (30 g, 0.26 mol, 1.0 equiv). The resulting mixture was allowed to reach ambient temperature and stirred overnight. After cooling to 0 °C, CH3OH (100 mL) was added dropwise. The resulting solution was stirred for 2 hours (h) at room temperature and then concentrated under reduced pressure. The crude residue was recrystallized from CH3OH:Et2O (1 :20) to yield 15.4 g (58%) the title compound as a white solid. LC/MS: m/z (ES+) 103 (M+H)+.
 [0073] Compound 1.2. 1-Isopropyl barbituric acid. To a stirred solution of 1.1 (14.4 g, 0.14 mol, 1.00 equiv) in CH3OH (500 mL) were added dimethyl malonate (19.55 g, 0.148 mol, 1.05 equiv) and sodium methoxide (18.9 g, 0.35 mol, 2.50 equiv). The resulting mixture was stirred overnight at 65 °C. After cooling to ambient temperature and then to 0 °C, the pH was carefully adjusted to 3 using aqueous concentrated HCl . The resulting mixture was concentrated under reduced pressure. The residue was taken up in EtOH (200 mL) and filtered. The filtrate was concentrated under reduced pressure and the residue was purified by silica gel column chromatography using CH2Cl2/CH3OH (20: 1) as eluent to yield 16.8 g (50%) of the title compound as a white solid. . LC/MS: m/z (ES+) 171 (M+H)+.1 1H-NMR (300 MHz, de-DMSO): 5 11.19 (s, 1H), 4.83 (m, 1H), 3.58 (s, 2H), 1.32 (d, J = 6.0 Hz, 6H).
[0073] Compound 1.2. 1-Isopropyl barbituric acid. To a stirred solution of 1.1 (14.4 g, 0.14 mol, 1.00 equiv) in CH3OH (500 mL) were added dimethyl malonate (19.55 g, 0.148 mol, 1.05 equiv) and sodium methoxide (18.9 g, 0.35 mol, 2.50 equiv). The resulting mixture was stirred overnight at 65 °C. After cooling to ambient temperature and then to 0 °C, the pH was carefully adjusted to 3 using aqueous concentrated HCl . The resulting mixture was concentrated under reduced pressure. The residue was taken up in EtOH (200 mL) and filtered. The filtrate was concentrated under reduced pressure and the residue was purified by silica gel column chromatography using CH2Cl2/CH3OH (20: 1) as eluent to yield 16.8 g (50%) of the title compound as a white solid. . LC/MS: m/z (ES+) 171 (M+H)+.1 1H-NMR (300 MHz, de-DMSO): 5 11.19 (s, 1H), 4.83 (m, 1H), 3.58 (s, 2H), 1.32 (d, J = 6.0 Hz, 6H).
 [0074] Compound 1.3. 6-chloro-3-isopropylpyrimidine-2,4(1H,3H)-dione. To a 100-mL round-bottom flask containing compound 1.2 (11.4 g, 66.99 mmol, 1.00 equiv) under argon were added triethylbenzylammonium chloride (21.3 g, 93.51 mmol, 1.40 equiv) and POCl3 (30 mL). The resulting mixture was stirred overnight at 50 °C. After cooling to room temperature, the mixture was concentrated under reduced pressure. The residue was dissolved in CH2Cl2 (150 mL) followed by slow addition of H2O (100 mL). The phases were separated and the organic layer was washed with H2O (100 mL), dried with anhydrous Na2SO4 , and concentrated under reduced pressure. The crude residue was purified by silica gel column chromatography using EtO Ac/petroleum ether (1 : 1) as eluent to yield 5.12 g (40%) of the title compound as a light yellow solid. 1H-NMR (300 MHz, d6-DMSO): δ 12.22 (s, 1H), 5.88 (s, 1H), 4.95 (m, 1H), 1.34 (d, J = 6.0 Hz, 6H).
[0074] Compound 1.3. 6-chloro-3-isopropylpyrimidine-2,4(1H,3H)-dione. To a 100-mL round-bottom flask containing compound 1.2 (11.4 g, 66.99 mmol, 1.00 equiv) under argon were added triethylbenzylammonium chloride (21.3 g, 93.51 mmol, 1.40 equiv) and POCl3 (30 mL). The resulting mixture was stirred overnight at 50 °C. After cooling to room temperature, the mixture was concentrated under reduced pressure. The residue was dissolved in CH2Cl2 (150 mL) followed by slow addition of H2O (100 mL). The phases were separated and the organic layer was washed with H2O (100 mL), dried with anhydrous Na2SO4 , and concentrated under reduced pressure. The crude residue was purified by silica gel column chromatography using EtO Ac/petroleum ether (1 : 1) as eluent to yield 5.12 g (40%) of the title compound as a light yellow solid. 1H-NMR (300 MHz, d6-DMSO): δ 12.22 (s, 1H), 5.88 (s, 1H), 4.95 (m, 1H), 1.34 (d, J = 6.0 Hz, 6H).
 [0075] Compound 1. (S)-3-Isopropyl-6-((1-phenylethyl) amino) pyrimidine-2,
4(1H,3H)-dione. To a solution of 6-chloro-3-isopropylpyrimidine-2,4(1H,3H)-dione (1.3,
1.0 g, 5.31 mmol) in 1,4-dioxane (20 mL) was added (S)-a-methylbenzylamine (Sigma- Aldrich, 1.43 g, 11.7 mmol, 2.2 equiv). The reaction mixture was stirred at 80 °C for 24 h. After cooling to ambient temperature, the mixture was concentrated under reduced pressure. The residual was taken up in EtOAc (70 mL) and washed with aqueous IN C1 (2 x 50 mL) and brine (40 mL). The organic layer was dried with anhydrous Na2SC”4 and then
concentrated under reduced pressure to half the original volume to yield a precipitate.
Hexane (20 mL) was added and the mixture was stirred at room temperature. The resulting solid was collected by filtration, washed with hexane (20 mL), and dried to yield 1.0 g (69%) of the title compound as a white solid. LC/MS: m/z (ES+) 274 (M+H)+. 1H-NMR (400 MHz, de-DMSO): δ 9.77 (s, 1H), 7.32 (m, 4H), 7.24 (m, 1H), 6.50 (d, J= 6.8 Hz, 1H), 4.87 (m,
1H), 4.52 (m, 1H), 4.31 (d, J=6.8 Hz, 1H), 1.37 (m, 3H ), 1.24 (m, 6H). 1H NMR (400 MHz, CD3OD) δ ppm 7.39-7.20 (m, 5H), 5.01 (m, 1H), 4.48 (m, 1H), 1.49 (d, J = 6.7 Hz, 3H), 1.36 (m, 6H).
[0075] Compound 1. (S)-3-Isopropyl-6-((1-phenylethyl) amino) pyrimidine-2,
4(1H,3H)-dione. To a solution of 6-chloro-3-isopropylpyrimidine-2,4(1H,3H)-dione (1.3,
1.0 g, 5.31 mmol) in 1,4-dioxane (20 mL) was added (S)-a-methylbenzylamine (Sigma- Aldrich, 1.43 g, 11.7 mmol, 2.2 equiv). The reaction mixture was stirred at 80 °C for 24 h. After cooling to ambient temperature, the mixture was concentrated under reduced pressure. The residual was taken up in EtOAc (70 mL) and washed with aqueous IN C1 (2 x 50 mL) and brine (40 mL). The organic layer was dried with anhydrous Na2SC”4 and then
concentrated under reduced pressure to half the original volume to yield a precipitate.
Hexane (20 mL) was added and the mixture was stirred at room temperature. The resulting solid was collected by filtration, washed with hexane (20 mL), and dried to yield 1.0 g (69%) of the title compound as a white solid. LC/MS: m/z (ES+) 274 (M+H)+. 1H-NMR (400 MHz, de-DMSO): δ 9.77 (s, 1H), 7.32 (m, 4H), 7.24 (m, 1H), 6.50 (d, J= 6.8 Hz, 1H), 4.87 (m,
1H), 4.52 (m, 1H), 4.31 (d, J=6.8 Hz, 1H), 1.37 (m, 3H ), 1.24 (m, 6H). 1H NMR (400 MHz, CD3OD) δ ppm 7.39-7.20 (m, 5H), 5.01 (m, 1H), 4.48 (m, 1H), 1.49 (d, J = 6.7 Hz, 3H), 1.36 (m, 6H).
[0072] Compound 1.1. Isopropylurea. To a stirred solution of isopropylamine (15.3 g, 0.258 mol, 1.0 equiv) in CH2Cl2 (200 mL) under argon at 0 °C was added dropwise trimethylsilyl isocyanate (30 g, 0.26 mol, 1.0 equiv). The resulting mixture was allowed to reach ambient temperature and stirred overnight. After cooling to 0 °C, CH3OH (100 mL) was added dropwise. The resulting solution was stirred for 2 hours (h) at room temperature and then concentrated under reduced pressure. The crude residue was recrystallized from CH3OH:Et2O (1 :20) to yield 15.4 g (58%) the title compound as a white solid. LC/MS: m/z (ES+) 103 (M+H)+.
[0073] Compound 1.2. 1-Isopropyl barbituric acid. To a stirred solution of 1.1 (14.4 g, 0.14 mol, 1.00 equiv) in CH3OH (500 mL) were added dimethyl malonate (19.55 g, 0.148 mol, 1.05 equiv) and sodium methoxide (18.9 g, 0.35 mol, 2.50 equiv). The resulting mixture was stirred overnight at 65 °C. After cooling to ambient temperature and then to 0 °C, the pH was carefully adjusted to 3 using aqueous concentrated HCl . The resulting mixture was concentrated under reduced pressure. The residue was taken up in EtOH (200 mL) and filtered. The filtrate was concentrated under reduced pressure and the residue was purified by silica gel column chromatography using CH2Cl2/CH3OH (20: 1) as eluent to yield 16.8 g (50%) of the title compound as a white solid. . LC/MS: m/z (ES+) 171 (M+H)+.1 1H-NMR (300 MHz, de-DMSO): 5 11.19 (s, 1H), 4.83 (m, 1H), 3.58 (s, 2H), 1.32 (d, J = 6.0 Hz, 6H).
[0074] Compound 1.3. 6-chloro-3-isopropylpyrimidine-2,4(1H,3H)-dione. To a 100-mL round-bottom flask containing compound 1.2 (11.4 g, 66.99 mmol, 1.00 equiv) under argon were added triethylbenzylammonium chloride (21.3 g, 93.51 mmol, 1.40 equiv) and POCl3 (30 mL). The resulting mixture was stirred overnight at 50 °C. After cooling to room temperature, the mixture was concentrated under reduced pressure. The residue was dissolved in CH2Cl2 (150 mL) followed by slow addition of H2O (100 mL). The phases were separated and the organic layer was washed with H2O (100 mL), dried with anhydrous Na2SO4 , and concentrated under reduced pressure. The crude residue was purified by silica gel column chromatography using EtO Ac/petroleum ether (1 : 1) as eluent to yield 5.12 g (40%) of the title compound as a light yellow solid. 1H-NMR (300 MHz, d6-DMSO): δ 12.22 (s, 1H), 5.88 (s, 1H), 4.95 (m, 1H), 1.34 (d, J = 6.0 Hz, 6H).
[0075] Compound 1. (S)-3-Isopropyl-6-((1-phenylethyl) amino) pyrimidine-2,
4(1H,3H)-dione. To a solution of 6-chloro-3-isopropylpyrimidine-2,4(1H,3H)-dione (1.3,
1.0 g, 5.31 mmol) in 1,4-dioxane (20 mL) was added (S)-a-methylbenzylamine (Sigma- Aldrich, 1.43 g, 11.7 mmol, 2.2 equiv). The reaction mixture was stirred at 80 °C for 24 h. After cooling to ambient temperature, the mixture was concentrated under reduced pressure. The residual was taken up in EtOAc (70 mL) and washed with aqueous IN C1 (2 x 50 mL) and brine (40 mL). The organic layer was dried with anhydrous Na2SC”4 and then
concentrated under reduced pressure to half the original volume to yield a precipitate.
Hexane (20 mL) was added and the mixture was stirred at room temperature. The resulting solid was collected by filtration, washed with hexane (20 mL), and dried to yield 1.0 g (69%) of the title compound as a white solid. LC/MS: m/z (ES+) 274 (M+H)+. 1H-NMR (400 MHz, de-DMSO): δ 9.77 (s, 1H), 7.32 (m, 4H), 7.24 (m, 1H), 6.50 (d, J= 6.8 Hz, 1H), 4.87 (m,
1H), 4.52 (m, 1H), 4.31 (d, J=6.8 Hz, 1H), 1.37 (m, 3H ), 1.24 (m, 6H). 1H NMR (400 MHz, CD3OD) δ ppm 7.39-7.20 (m, 5H), 5.01 (m, 1H), 4.48 (m, 1H), 1.49 (d, J = 6.7 Hz, 3H), 1.36 (m, 6H).

REFERENCES
1: Green EM, Wakimoto H, Anderson RL, Evanchik MJ, Gorham JM, Harrison BC, Henze M, Kawas R, Oslob JD, Rodriguez HM, Song Y, Wan W, Leinwand LA, Spudich JA, McDowell RS, Seidman JG, Seidman CE. A small-molecule inhibitor of sarcomere contractility suppresses hypertrophic cardiomyopathy in mice. Science. 2016 Feb 5;351(6273):617-21. doi: 10.1126/science.aad3456. PubMed PMID: 26912705; PubMed Central PMCID: PMC4784435.Henagliflozin

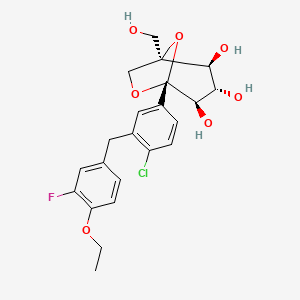

Henagliflozin, SHR-3824 ,
CAS 1623804-44-3
C22-H24-Cl-F-O7, 454.8756
PHASE 2 for the treatment of type 2 diabetes
China 20222, approvals 2022
HengRui (Originator)
| Jiangsu Hengrui Medicine Co Ltd |
UNII-21P2M98388; 21P2M98388; Henagliflozin; SHR3824; SHR-3824;

- HENAGLIFLOZIN PROLINE
- 4IO819SW6M
- 570.0 g/mol
- C27H33ClFNO9
- (1R,2S,3S,4R,5R)-5-[4-chloro-3-[(4-ethoxy-3-fluorophenyl)methyl]phenyl]-1-(hydroxymethyl)-6,8-dioxabicyclo[3.2.1]octane-2,3,4-triol;(2R)-pyrrolidine-2-carboxylic acid
In April 2016, Jiangsu Hengrui Medicine is developing henagliflozin (phase 2 clinical trial), a sodium-glucose cotransporter-2 (SGLT-2) inhibitor, for treating type 2 diabetes.
SGLT1 and SGLT2 inhibitors, useful for treating eg diabetes.
Henagliflozin proline is in phase II clinical trials by Jiangsu Hengrui (江苏恒瑞) for the treatment of type 2 diabetes.
1,6-dehydrated-1-C{4-chloro-3-[(3-fluoro-4-ethoxyphenyl)methyl]phenyl}-5-C-(hydroxymethyl)-β-L-idopyranose L-proline
(1 ^ 2345-5- [4-chloro-3 – [(4-ethoxy-3-fluoro-phenyl) – methyl] phenyl] -1- (hydroxymethyl) 6,8 – alcohol dioxide
(1R,2S,3S,4R,5R)-5-[4-chloro-3-[(4-ethoxy-3-fluorophenyl)methyl]phenyl]-1-(hydroxymethyl)-6,8-dioxabicyclo[3.2.1]octane-2,3,4-triol
Henagliflozin is a pharmaceutical drug for the treatment of type 2 diabetes.[1] In China, it is approved for adult patients with type 2 diabetes to improve the glycemic control.[2][3]
Henagliflozin, like other drugs of the gliflozin class, inhibits the transporter protein sodium/glucose cotransporter 2 (SGLT2) which leads to a reduction in blood glucose levels.[4]
Shanghai Hengrui Pharmaceutical Co., Ltd., 上海恒瑞医药有限公司, Jiangsu Hengrui Medicine Co., Ltd., 江苏恒瑞医药股份有限公司, Less «
- 01 May 2015 Jiangsu HengRui Medicine Co. initiates enrolment in a phase I drug interaction trial in volunteers in China (NCT02500485)
- 12 Feb 2015 Jiangsu HengRui Medicine plans a phase I trial for Type-2 diabetes mellitus in China (NCT02366377)
- 01 Feb 2015 Jiangsu HengRui Medicine initiates enrolment in a phase I trial for Type-2 diabetes mellitus in China (NCT02366351)
Henagliflozin is a novel sodium-glucose transporter 2 inhibitor and presents a complementary therapy to metformin for patients with T2DM due to its insulin-independent mechanism of action. This study evaluated the potential pharmacokinetic drug-drug interaction between henagliflozin and metformin in healthy Chinese male subjects. 2. In open-label, single-center, single-arm, two-period, three-treatment self-control study, 12 subjects received 25 mg henagliflozin, 1000 mg metformin or the combination. Lack of PK interaction was defined as the ratio of geometric means and 90% confidence interval (CI) for combination: monotherapy being within the range of 0.80-1.25. 3. Co-administration of henagliflozin with metformin had no effect on henagliflozin area under the plasma concentration-time curve (AUC0-24) (GRM: 1.08; CI: 1.05, 1.10) and peak plasma concentration (Cmax) (GRM: 0.99; CI: 0.92, 1.07). Reciprocally, co-administration of metformin with henagliflozin had no clinically significant on metformin AUC0-24 (GRM: 1.09, CI: 1.02, 1.16) although there was an 11% increase in metformin Cmax (GRM 1.12; CI 1.02, 1.23). All monotherapies and combination therapy were well tolerated. 4. Henagliflozin can be co-administered with metformin without dose adjustment of either drug.
PATENT
With the improvement of socio-economic development and living standards, worldwide rapid growth of diabetes, diabetes is usually divided into two kinds of diabetes type Ⅰ and type Ⅱ diabetes, more than 90% of type Ⅱ diabetes. Species has been listed diabetes drugs a lot, but so far, no drugs which can single-handedly blood glucose levels in patients with type Ⅱ diabetes in the long-term target range. In recent years, in-depth study of the pathogenesis of diabetes, for the treatment of type Ⅱ diabetes provide more and more ways, and sodium – glucose cotransporter 2 (sodium-glucose transporter 2, SGLT-2) inhibitors found for treatment of diabetes provides another new idea. SGLT-2 inhibitors in the treatment mechanism of inhibition of SGLT-2 activity by selective to lower blood sugar. Select the SGLT-2 as a target, partly because of its absolute weight of glucose absorption, and partly because it is only expressed in the kidney. The current study also found that the mechanism of SGLT-2 does not depend on the degree of abnormal function of β cells or insulin resistance, its effect is not as severe failure or insulin resistance and β-cell function decline.Therefore, it is reasonable that the SGLT-2 inhibitors for the treatment of type Ⅱ diabetes currently has good prospects.
WO2012019496 discloses SGLT-2 inhibitor of the following formula, and its chemical name is 1,6-anhydro -1-C- {4- chloro-3 – [(3-fluoro-4-ethoxyphenyl) methyl] phenyl} -5-C- (hydroxymethyl) -β-L- idose pyranose.
However, direct 1,6-anhydro -1-C- {4- chloro-3 – [(3-fluoro-4-ethoxyphenyl) methyl] phenyl} -5-C- (hydroxymethyl) – β-L- idose pyranose as a pharmaceutically active ingredient is not realistic, because a lower melting point (83 ℃), having a hygroscopicity, poor development of the form, therefore, to develop it into a stable form of the compound having the transformation very important.
Example 1
Take (1.0g, 2.2mmol) 1,6- dehydration -1-C- {4- chloro-3 – [(3-fluoro-4-ethoxyphenyl) methyl] phenyl} -5-C- ( hydroxymethyl) -β-L- Aidoo pyranose (prepared by the method disclosed in WO2012019496), in 7.20g ethanol addition was completed, stirring to dissolve. Was added at room temperature L- proline (0.2786g, 2.42mmol, 1.1eq), the addition was completed, the reaction was warmed at reflux for 10min, the reaction solution was clear, hot filtered and the filtrate was stirred to room temperature, there is a lot of white solid precipitated , allowed to stand overnight, filtered, and dried, to give the formula (I), compound as a white solid 1.14 g, yield 88%. X- ray diffraction spectrum of the crystalline sample is shown in Figure 1. The crystallization at about 5.41 (16.33) 7.69 (11.49), 10.22 (8.65) 12.04 (7.35), 12.46 (7.10), 14.42 (6.14), 17.30 (5.12), 18.79 (4.72), 19.38 (4.58), 20.24 (4.38), 22.73 (3.91), 24.58 (3.62), 27.55 (3.24), 28.82 (3.10) and 31.03 (2.88) at the characteristic peaks. DSC spectrum shown in Figure 2, has a melting endothermic peak 111.20 ℃, this is defined as a Form A polymorph.
PATENT
WO2012019496
https://www.google.com/patents/WO2012019496A1?cl=en
Example 4
(1 ^ 2345-5- [4-chloro-3 – [(4-ethoxy-3-fluoro-phenyl) – methyl] phenyl] -1- (hydroxymethyl) 6,8 – alcohol dioxide


first step
1-ethoxy-2-fluoro – benzene
A mixture of 2-fluoro-phenol 4a (6.7 g, 60 mmol) was dissolved in 66 mL of acetone, was added iodoethane (6.3 mL,
78 mmol) and potassium carbonate (12.4 g, 90 mmol), at reflux in an oil bath for 5 hours. The reaction solution was concentrated under reduced pressure, was added 100 mL of ethyl acetate and 60 mL of water, separated, the aqueous phase was extracted with ethyl acetate (30 mLx2), the organic phases combined, dried over anhydrous magnesium sulfate, filtered, and the filtrate was concentrated under reduced pressure, to give the title product 1-ethoxy-2-fluoro – benzene 4b (6.9 g, red oil). yield: 82.1%.
MS m / z (ESI): 280.2 [2M + 1]
The second step
(5-bromo-2-chloro – phenyl) – (4-ethoxy-3-fluoro-phenyl) – methanone A mixture of 5-bromo-2-chloro – benzoyl chloride 2a (12.4 g, 48.8 mmol) was dissolved a 100 mL of dichloromethane was added 1-ethoxy-2-fluoro – benzene 4b (6.84 g, 48.8 mmol), cooled to 0 ° C, was added portionwise aluminum (5.86 g, 44 mmol) chloride, 16 h. Was added dropwise under ice-cooling to the reaction mixture 20 mL of 2 M HCl solution, separated, the aqueous phase was extracted with 30 mL of dichloromethane, and the combined organic phase was dried over anhydrous magnesium sulfate, filtered, and the filtrate was concentrated under reduced pressure to give the title The product (5-bromo-2-chloro – phenyl) – (4-ethoxy-3-fluoro-phenyl) – methanone 4c (12.7 g, yellow solid), yield: 72.6%.
MS m / z (ESI): 358.9 [M + l] Step
(5 – bromo-2-chloro – phenyl) – (4-ethoxy-3-fluoro-phenyl) – methanol (5-Bromo-2-chloro – phenyl) – (4-ethoxy -3 – fluoro – phenyl) -methanone 4c (12.7 g, 35.5 mmol) was dissolved in methanol and a 100 mL of tetrahydrofuran (ν: ν = 1: 1) mixed solvent, under an ice bath was added portionwise sodium borohydride (2.68 g, 70 mmol), and reacted at room temperature for 30 minutes. Add 15 mL of acetone, the reaction solution was concentrated under reduced pressure, 150 mL of ethyl acetate was added to dissolve the residue, washed with saturated sodium chloride solution (50 mLx2). The combined organic phase was dried over anhydrous magnesium sulfate, filtered, and concentrated under reduced pressure The filtrate, to give the title product (5-bromo-2-chloro – phenyl) – (4-ethoxy-3-fluoro-phenyl) – methanol 4d (12.7 g, orange oil), was used directly without isolation next reaction.
the fourth step
4 – [(5-bromo-2-chloro-phenyl) – methyl] Small-ethoxy-2-fluoro – benzene (5-bromo-2-chloro – phenyl) – (4-ethoxy -3 – fluoro – phenyl) methanol 4d (12.7 g, 35.3 mmol) was dissolved in a 100 mL of dichloromethane was added triethylsilane (16.9 mL, 106 mmol), was added dropwise boron trifluoride etherate (8.95 mL, 70.6 mmol ), for 3 hours. Was added 50 mL of saturated sodium bicarbonate solution, separated, the aqueous phase was extracted with ethyl acetate (100 mLx2), the organic phases combined, dried over anhydrous magnesium sulfate, filtered, and the filtrate was concentrated under reduced pressure, purified by silica gel column chromatography to elute B surfactant system resulting residue was purified to give the title product 4 – [(5-bromo-2-chloro – phenyl) methyl] -1-ethoxy-2-fluoro – benzene 4e (10 g, as a pale yellow oil ) yield: 82.4%.
1H NMR (400 MHz, CDC1 3 ): δ 7.33-7.27 (m, 3H), 6.95-6.90 (m, 3H), 4.14 (q, 2H), 4.01 (s, 2H), 1.49 (t, 3H)
the fifth step
(2 3R, 4S, 5 ^ 6R) -2- [4- chloro-3 – [(4-ethoxy-3-fluoro-phenyl) – methyl] phenyl] -6- (hydroxymethyl) – 2-methoxy – tetrahydro-pyran-3,4,5-triol
4 – [(5-bromo-2-chloro – phenyl) methyl] -1-ethoxy-2-fluoro – benzene 4e (7.36 g, 21.4 mmol) was dissolved in 30 mL of tetrahydrofuran, cooled to -78 ° C, was added dropwise a solution of n-butyllithium in hexane (10.27 mL, 25.7 mmol), at -78 ° C to react 1 hour, a solution of 20 mL (3R, 4S, 5R, 6R) -3,4,5 – tris (trimethylsilyloxy) -6- (trimethylsilyloxy) tetrahydropyran-2-one 2f (llg, 23.6 mmol) in tetrahydrofuran at -78 ° C under reaction 2 h, 2.8 mL of methanesulfonic acid and 71 mL of methanol, the reaction at room temperature for 16 hours. Was added 100 mL of saturated sodium carbonate solution, the reaction solution was concentrated under reduced pressure, to the residue was added 50 mL of saturated sodium chloride solution, extracted with ethyl acetate (100 mLx3), organic phases were combined, dried over anhydrous magnesium sulfate, filtered, The filtrate was concentrated under reduced pressure, purified by silica gel column chromatography with eluent systems resulting A residue was purified to give the title product (2 3R, 4S, 5 6R) -2- [4- chloro-3 – [(4-ethoxyphenyl 3-fluoro-phenyl) – methyl] phenyl] -6- (hydroxymethyl) -2-methoxy – tetrahydro-pyran-3,4,5-triol 4f (5.7 g, white solid ) yield: 58.3%.
1H NMR (400 MHz, CD 3 OD): δ 7.56 (s, 1H), 7.48 (dd, 1H), 7.37 (dd, 1H), 6.95-6.87 (m, 3H), 4.08-4.07 (m, 4H) , 3.91 (m, 1H), 3.93-3.73 (m, 2H), 3.56-3.53 (m, 1H), 3.45-3.43 (m, 1H), 3.30 (s, 2H), 3.08 (s, 3H), 1.35 (t, 3H)
The sixth step
(2 3R, 4S, 5 6R) -6- [(tert-butyl (dimethyl) silyl) oxymethyl] -2- [4-chloro-3 – [(4-ethoxy-3-fluoro – phenyl) methyl] phenyl] -2-methoxy – tetrahydro-pyran-3,4,5-triol the (2 3R, 4S, 5 6R) -2- [4- chloro-3- [(4-ethoxy-3-fluoro-phenyl) – methyl] phenyl] -6- (hydroxymethyl) -2-methoxy – 4f tetrahydropyran-3,4,5-triol (5.7 g, 12.5 mmol) was dissolved in 50 mL of pyridine, followed by adding tert-butyldimethylsilyl chloride (2.26 g, 15 mmol) and 4-dimethylaminopyridine (305 mg, 2.5 mmol), for 16 hours. The reaction solution was concentrated under reduced pressure, was added 200 mL of ethyl acetate, washed with a saturated copper sulfate solution (50 mLx3). The combined organic phase was dried over anhydrous magnesium sulfate, filtered, and the filtrate was concentrated under reduced pressure to give the title product (2 3R, 4S, 5 6R) -6- [(tert-butyl (dimethyl) silyl) oxymethyl] -2- [4-chloro-3 – [(4-ethoxy-3-fluoro-phenyl) – methyl] phenyl] -2-methoxy – tetrahydro-pyran-3,4,5-triol 4g (7.14 g, colorless oil), without isolation directly used for the next reaction.
Seventh Step
[[(2R, 3R, 4S, 5R, 6 ^ -3,4,5- tris-benzyloxy-6- [4-chloro-3 – [(4-ethoxy-3-fluoro-phenyl) – methyl yl] phenyl] -6-methoxy – tetrahydropyran-2-yl] methoxy] – tert-butyl – dimethyl-silane (2 3R, 4S, 5 6R) -6- [(tert butyl (dimethyl) silyl) oxymethyl] -2- [4-chloro-3 – [(4-ethoxy-3-fluoro-phenyl) – methyl] phenyl] -2-methoxy yl – tetrahydro-pyran-3,4,5-triol 4g (7.14 g, 12.5 mmol) was dissolved in 100 mL N, N- dimethylformamide was added 60% sodium hydride under ice-cooling (2.5 g , 62.5 mmol), and reacted at room temperature for 40 minutes completed the opening force, was added benzyl bromide (7.5 mL, 62.5 mmol), reaction of 16 hours. 20 mL of methanol, the reaction solution was concentrated under reduced pressure, was added 200 mL of ethyl acetate and 50 mL of water to dissolve the residue, separated, the aqueous phase was extracted with ethyl acetate (50 mL), the organic phase was washed with water (50 mL), washed with saturated sodium chloride solution (50 mL), the combined organic phase was dried over anhydrous magnesium sulfate , filtered, and the filtrate was concentrated under reduced pressure to give the title product [[(2R, 3R, 4S, 5R, 6 ^ -3,4,5- tris-benzyloxy-6- [4-chloro-3 – [(4- ethoxy-3-fluoro-phenyl) – methyl] phenyl] -6-methoxy – tetrahydropyran-2-yl] methoxy] – tert-butyl – dimethylsilane 4h (10.5 g , yellow oil) yield: 99.8%.
Step Eight
[(2R, 3R, 4S, 5R, 6 -3,4,5- tris-benzyloxy-6- [4-chloro-3 – [(4-ethoxy-3-fluoro-phenyl) – methyl] phenyl] -6-methoxy – tetrahydropyran-2-yl] methanol
The [[(2R, 3R, 4S, 5R, 6 -3,4,5- tris-benzyloxy-6- [4-chloro-3 – [(4-ethoxy-3-fluoro-phenyl) – methyl yl] phenyl] -6-methoxy – tetrahydropyran-2-yl] methoxy] – tert-butyl – dimethylsilane 4h (10.52 g, 12.5 mmol) was dissolved in 50 mL of methanol dropwise add acetyl chloride CO.13 mL, 1.9 mmol), for 1 hour. The reaction solution was concentrated under reduced pressure, purified by silica gel column chromatography with eluent systems B resultant residue was purified to give the title product [(2R, 3R, 4S, 5R, 6 -3,4,5- tris-benzyloxy–6 – [4-chloro-3 – [(4-ethoxy-3-fluoro-phenyl) – methyl] phenyl] -6-methoxy – tetrahydropyran-2-yl] methanol 4i (7.6 g , yellow oil yield: 83.6%.
Step Nine
(2 ^ 3456 3,4,5-tris-benzyloxy-6- [4-chloro-3 – [(4-ethoxy-3-fluoro-phenyl) – methyl] phenyl] – 6-methoxy – tetrahydropyran-2-carbaldehyde
Oxalyl chloride (1.17 mL, 13.6 mmol) was dissolved in 20 mL of dichloromethane, cooled to -78 ° C, were added dropwise 20 mL of dimethyl sulfoxide (1.56 mL, 21.9 mmol) in methylene chloride and 50 mL [(2R, 3R, 4S, 5R, 6 -3,4,5- tris-benzyloxy-6- [4-chloro-3 – [(4-ethoxy-3-fluoro-phenyl) – methyl] phenyl] -6-methoxy – tetrahydropyran-2-yl] methanol 4i (7.6 g, 10.45 mmol) in methylene chloride, and reacted at -78 ° C for 30 min, triethylamine (7.25 mL, 52.3 mmol), 2 hours at room temperature was added 50 mL 1 M HCl solution, separated, the organic phase was washed with saturated sodium chloride solution (50 mL x 2), the aqueous phase was extracted with dichloromethane (50 mL), the combined organic phase was dried over anhydrous magnesium sulfate, filtered, and the filtrate was concentrated under reduced pressure to give the title product (2 ^ 3456 3,4,5-tris-benzyloxy-6- [4-chloro-3 – [(4 – ethoxy-3-fluoro-phenyl) – methyl] phenyl] -6-methoxy – tetrahydropyran-2-carbaldehyde 4j (7.58 g, colorless oil), was used directly without isolation next reaction.
The tenth step
(2S, 3 4S, 5R, 6 -3,4,5- tris-benzyloxy-6- [4-chloro-3 – [(4-ethoxy-3-fluoro-phenyl) – methyl] phenyl ] -2- (hydroxymethyl) -6-methoxy – tetrahydropyran-2-carbaldehyde
The (23456 3,4,5-tris-benzyloxy-6- [4-chloro-3 – [(4-ethoxy-3-fluoro-phenyl) – methyl] phenyl] – 6-methoxy – tetrahydropyran-2-carbaldehyde 4j (7.6 g, 10.45 mmol) was dissolved in 80 mL 1,4- dioxane, followed by adding 15.8 mL 37% aqueous formaldehyde and sodium hydroxide solution (31.35 mL, 31.35 mmol), reacted at 70 ° C for 16 h. Add 50 mL of saturated sodium chloride solution, extracted with ethyl acetate (50 mLx4), the organic phase was washed with saturated sodium bicarbonate solution (50 mL), washed with saturated sodium chloride solution (50 mL), the combined organic phase was dried over anhydrous magnesium sulfate, filtered, and the filtrate was concentrated under reduced pressure to give the title product (23,456 benzyloxy-3,4,5-tris – 6- [4-chloro-3 – [(4-ethoxy-3-fluoro-phenyl) – methyl] phenyl] -2- (hydroxymethyl) -6-methoxy – tetrahydropyran – 2- formaldehyde 4k (7.9g, as a colorless oil), without isolation directly used for the next reaction.
Step Eleven
[(3 4S, 5R, 6 -3,4,5- tris-benzyloxy-6- [4-chloro-3 – [(4-ethoxy-3-fluoro-phenyl) – methyl] phenyl] 2- (hydroxymethyl) -6-methoxy – tetrahydropyran-2-yl] methanol
The (23456 3,4,5-tris-benzyloxy-6- [4-chloro-3 – [(4-ethoxy-3-fluoro-phenyl) – methyl] phenyl] – 2- (hydroxymethyl) -6-methoxy – tetrahydropyran-2-carbaldehyde 4k (7.9 g, 10.45 mmol) was dissolved in 50 mL of tetrahydrofuran and methanol (v: v = 2: 3) mixed solvent , was added sodium borohydride (794 mg, 20.9 mmol), for 30 minutes. Add a small amount of acetone, the reaction solution was concentrated under reduced pressure, purified by silica gel column chromatography with eluent systems resulting A residue was purified to give the title product, 5R, 6 -3,4,5-tris-benzyloxy-6- [4-chloro-3 – [(4-ethoxy-3-fluoro-phenyl) – methyl] phenyl] -2- (hydroxymethyl ) -6-methoxy – tetrahydropyran-2-yl] methanol 4m (l.ll g, colorless oil). yield: 14.1%.
Step Twelve
[(12345 ^ -2,3,4-tris-benzyloxy-5- [4-chloro-3 – [(4-ethoxy-3-fluoro-phenyl) – methyl] phenyl] 6,8-dioxa-bicyclo [3.2.1] octane-1-yl] methanol
The [(3S, 4S, 5R, 6 -3,4,5- tris-benzyloxy-6- [4-chloro-3 – [(4-ethoxy-3-fluoro-phenyl) – methyl] benzene yl] -2- (hydroxymethyl) -6-methoxy – tetrahydropyran-2-yl] methanol 4m (l.ll g, 1.46 mmol) was dissolved in 20 mL of dichloromethane, cooled to -10 ° C, was added trifluoroacetic acid (0.23 mL, 3 mmol), and reacted at room temperature for 2 hours. 20 mL of saturated sodium bicarbonate solution, separated, the aqueous phase was extracted with dichloromethane (20 mL> <2), and the combined organic phase was dried over anhydrous magnesium sulfate, filtered, and the filtrate was concentrated under reduced pressure, purified by silica gel column chromatography with eluent systems B resultant residue was purified to give the title product [(1 2 3 4R, 5 -2,3,4- tris-benzyloxy-5- [4-chloro-3 – [(4-ethoxy-3-fluoro-phenyl) – methyl] phenyl] 6,8-dioxa-bicyclo [3.2.1] octane-1-yl] methanol 4nC830 mg, colorless oil). yield: 78.3%.
MS m / z (ESI): 742.3 [M + 18]
Thirteenth Step
(12345-5- [4-chloro-3 – [(4-ethoxy-3-fluoro-phenyl) – methyl] phenyl] -1- (hydroxymethyl) -6,8 dioxa-bicyclo [3.2.1] octane-2,3,4-triol
The [(1 2 3 4R, 5S) -2,3,4- tris-benzyloxy-5- [4-chloro-3 – [(4-ethoxy-3-fluoro-phenyl) – methyl] benzene yl] -6,8-dioxa-bicyclo [3.2.1] octane-1-yl] methanol 4n (830 mg, 1.14 mmol) was dissolved in 20 mL of tetrahydrofuran and methanol (v: v = l: l) the a mixed solvent of o-dichlorobenzene was added (1.3 mL, 1 1.4 mmol) and Pd / C (500 mg, 10%), purged with hydrogen three times, the reaction for 3 hours. The reaction solution was filtered, rinsed with a small amount of ethyl acetate, the filtrate was concentrated under reduced pressure, purified by silica gel column chromatography with eluent systems resulting A residue was purified to give the title product (1S, 2 3S, 4R, 5 -5- [ 4-chloro-3 – [(4-ethoxy-3-fluoro-phenyl) – methyl] phenyl] -1- (hydroxymethyl) -6,8-dioxa-bicyclo [3.2.1] octane-2,3,4-triol 4 (420 mg, white solid), yield: 81.0% MS m / z (ESI):. 472.2 [m + 18]
1H NMR (400 MHz, CD 3 OD): δ 7.47 (s, 1H), 7.42-7.35 (m, 2H), 6.95-6.87 (m, 3H), 4.16-4.14 (m, 1H), 4.06-4.02 ( m, 4H), 3.85-3.70 (m, 2H), 3.67-3.54 (m, 4H), 1.37 (t, 3H)
References
- Weng J, Zeng L, Zhang Y, Qu S, Wang X, Li P, et al. (August 2021). “Henagliflozin as add-on therapy to metformin in patients with type 2 diabetes inadequately controlled with metformin: A multicentre, randomized, double-blind, placebo-controlled, phase 3 trial”. Diabetes, Obesity & Metabolism. 23 (8): 1754–1764. doi:10.1111/dom.14389. PMID 33769656.
- Wang G (17 February 2022). “Monthly Report: New Drug Approvals in China, January 2022”. BaiPharm.
Henagliflozin Proline Tablets
- “Henagliflozin – Jiangsu HengRui Medicine”. AdisInsight. Springer Nature Switzerland AG.
- He X, Liu G, Chen X, Wang Y, Liu R, Wang C, et al. (July 2023). “Pharmacokinetic and Pharmacodynamic Interactions Between Henagliflozin, a Novel Selective SGLT-2 Inhibitor, and Warfarin in Healthy Chinese Subjects”. Clinical Therapeutics. 45 (7): 655–661. doi:10.1016/j.clinthera.2023.06.002. PMID 37451912.
 |
|
| Clinical data | |
|---|---|
| Trade names | Rui Qin; 瑞沁 |
| Other names | SHR3824; SHR-3824 |
| Legal status | |
| Legal status |
|
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| DrugBank | |
| UNII | |
| Chemical and physical data | |
| Formula | C22H24ClFO7 |
| Molar mass | |
////////Henagliflozin, SHR-3824 , PHASE 2, type 2 diabetes, UNII-21P2M98388, 21P2M98388, SHR 3824, SHR3824, approvals 2022, china 2022, Henagliflozin proline
CCOc1ccc(cc1F)Cc2cc(ccc2Cl)[C@]34[C@@H]([C@H]([C@@H]([C@](O3)(CO4)CO)O)O)O
SYN
Synthesis 2024, 56, 906–943
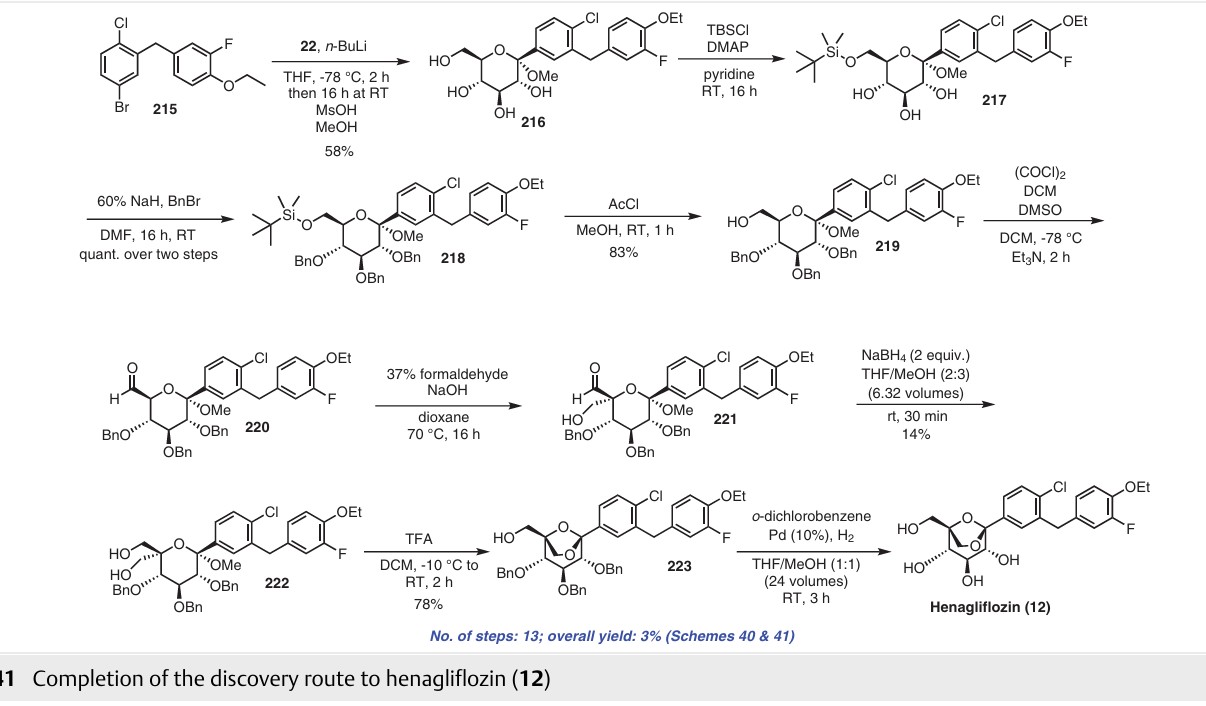
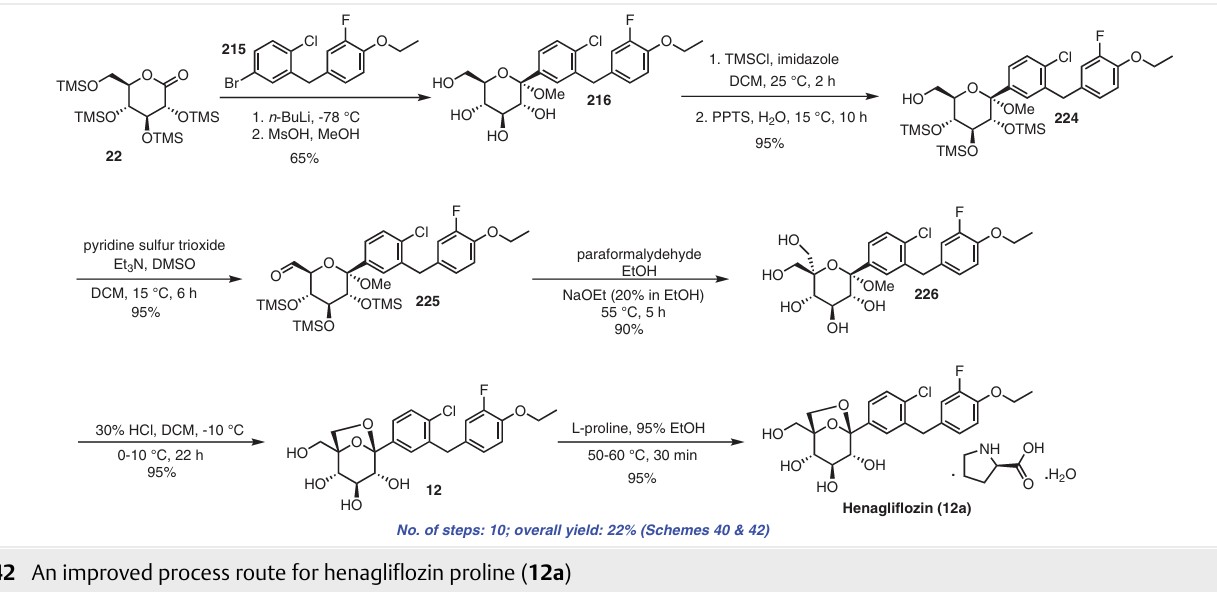
Henagliflozin (12) (also known as SHR3824), developed by Lexicon Pharmaceuticals (Princeton, NJ, USA), is a potent and selective SGLT inhibitor administered orally. In 2013, the first synthetic route for the preparation of henagliflozin (12) was described and claimed by two pharmaceutical companies: Shanghai Hengrui Pharmaceutical Co., Ltd., and Jiangsu Hengrui Medicine Co., Ltd. Several other C-aryl-glucoside-type derivatives were prepared and registered in the United States under patent application number US8609622B2.67 Among these derivatives, the synthesis of henagliflozin (12) was carried out using a thirteen-step process, resulting in an overall yield of 3% (Schemes 40 and 41). The process consisted of the formation of the key intermediate 215 starting from commercially available 2-fluorophenol (211). In the first step, phenolic compound 211 was converted into 212 in 82% yield using ethyl bromide and po
tassium carbonate in acetone. The Friedel–Crafts reaction of acid chloride 26c′ using AlCl3 in DCM afforded intermediate 213 in 72% yield, which was further reduced to 214 using NaBH4 in a mixture of THF/MeOH. Without further isolation, the reduction of 214 was carried out using Et3SiH and BF3·Et2O in DCM to give 215 (Scheme 40). The intermediate 215 was taken forward for lithium halogen exchange using n-BuLi followed by addition of the lithiated compound to O-silyl-protected compound 22 at
low temperature to afford a lactol intermediate. The obtained lactol intermediate was protected using
MsOH/MeOH to give the desired product 216 in 58% yield. Under the above conditions, deprotection of the O-silylgroups of the C-glucoside 22 was also observed. Further, under basic conditions, the secondary hydroxy group of 216 was silyl protected using tert-butyldimethylsilyl chloride (TBSCl) and DMAP to afford compound 217, which was treated with NaH and BnBr to give benzylated compound
218 in excellent yield. In methanol solution, deprotection of the silyl protecting group of compound 218 using acetylchloride afforded 219. Swern oxidation of the hydroxy compound 219 in the presence of oxalyl chloride and DMSO gave intermediate 220, which was used for the next step without isolation. The crude compound 220 was treated with NaOH and 37% formaldehyde solution to afford 221.
Dihydroxy intermediate 222 was then obtained in low yield via reduction of the aldehyde group of compound 221 with sodium borohydride in THF/MeOH mixture. Next, treatment of 222 with trifluoroacetic acid gave compound 223. Debenzylation of compound 223 was carried out by Pd/C
catalytic hydrogenation to afford the final product henaglifozin (12) (Scheme 41).
The highlight of the synthesis is the design of the route with minimal isolation stages and intermediates possessing unstable functional groups were subjected to subsequent transformations in situ. The drawbacks of the above synthetic process are the use of a protection and deprotection
strategy that led to low throughput and the final compound being obtained in low yield. Reduction of the aldehyde in 221 mediated by sodium borohydride resulted in a poor yield of product 222, and this procedure is not recommend ed for scale-up due to safety concerns. Additionally, the use
of palladium in the last step of the synthesis involves the risk of this toxic metal leaching into the final product. To address the issue with the discovery route, Yongjun and co-workers reported an alternative approach to obtain compound 12 (Scheme 42).68 The authors published the synthesis of henagliflozin proline (12a) starting from TMS protected D-glucolactone 22 and aglycone intermediate The diol 226 was obtained after carrying out a disproportionation reaction on the aldehyde using paraformaldehyde under strong alkaline conditions. Intramolecular etherification of diol 226 using 30% HCl gave henagliflozin
(12) in 95% yield, which was further treated with L-proline to give henagliflozin proline monohydrate 12a. The authors reported several advantages such as easy steps, cost-effective procedures, simple product purification and an overall method that was amenable for commercialization. This Addition of the aglycone intermediate 215 was carried out with 22 followed by mesylation of the OH group to provide 216 in 65% yield. Further, all the secondary hydroxy groups of intermediate 216 were selectively protected us ing TMSCl, imidazole and PPTS to give 224 in 95% yield. The free primary hydroxy group of 224 was oxidized using pyridine sulfur trioxide in triethylamine and DMSO to afford process involves 10 steps and gave an overall yield of 22% of henagliflozin proline (12a) (Schemes 40 and 42)
REF 67, 68
(67) Yang, F.; Tang, P. C.; Dong, Q.; Tu, W.; Fan, J.; Guan, D.; Shen, G.;Wang, Y.; Yuan, J.; Zhang, L. US8609622B2, 2013.
(68) Chun, K.; Peng, Z.; Qichao, L.; Bo, Z.; Zhen, W.; Guorong, Z.;Yongjun, T. CN 112375087A, 2020.
.
Pacritinib

Pacritinib
パクリチニブ;
| Formula |
C28H32N4O3
|
|---|---|
| CAS |
937272-79-2
|
| Mol weight |
472.5787
|
UPDATE FDA APPROVED 2/28/2022, Vonjo
To treat intermediate or high-risk primary or secondary myelofibrosis in adults with low platelets
A Jak2 inhibitor potentially for the treatment of acute myeloid Leukemia and myelofibrosis.
UNII-G22N65IL3O
пакритиниб
باكريتينيب
帕瑞替尼
ONX-0803; SB-1518
CAS No. 937272-79-2
472.57868 g/mol, C28H32N4O3
S*Bio Pte Ltd. and concert innovator
11-(2-pyrrolidin-1-ylethoxy)-14,19-dioxa-5,7,26-triazatetracyclo(19.3.1.1(2,6).1(8,12))heptacosa-1(25),2(26),3,5,8,10,12(27),16,21,23-decaene
| Pacritinib (SB1518) is a potent and selective inhibitor of Janus Kinase 2 (JAK2) and Fms-Like Tyrosine Kinase-3 (FLT3) with IC50s of 23 and 22 nM, respectively. | ||||||
UPDATED
Pacritinib, sold under the brand name Vonjo, is an anti-cancer medication used to treat myelofibrosis.[1][2] It is a macrocyclic Janus kinase inhibitor. It mainly inhibits Janus kinase 2 (JAK2) and Fms-like tyrosine kinase 3 (FLT3).
Common side effects include diarrhea, low platelet counts, nausea, anemia, and swelling in legs.[2]
Medical uses
Pacritinib in indicated to treat adults who have a rare form of a bone marrow disorder known as intermediate or high-risk primary or secondary myelofibrosis and who have platelet (blood clotting cells) levels below 50,000/µL.[1][2]
History
The effectiveness and safety of pacritinib were demonstrated in a study that included 63 participants with intermediate or high-risk primary or secondary myelofibrosis and low platelets who received pacritinib 200 mg twice daily or standard treatment.[2] Effectiveness was determined based upon the proportion of participants who had a 35% or greater spleen volume reduction from baseline to week 24.[2] Nine participants (29%) in the pacritinib treatment group had a 35% or greater spleen volume reduction, compared to one participant (3%) in the standard treatment group.[2]
The U.S. Food and Drug Administration (FDA) granted the application for pacritinib priority review, fast track, and orphan drug designations.[2]
Society and culture
Names
Pacritinib is the International nonproprietary name (INN).[3][4]
References
- ^ Jump up to:a b c “Enforcement Reports”. Accessdata.fda.gov. Retrieved 5 March 2022.
- ^ Jump up to:a b c d e f g h “FDA approves drug for adults with rare form of bone marrow disorder”. U.S. Food and Drug Administration. 1 March 2022. Retrieved 3 March 2022.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ World Health Organization (2010). “International nonproprietary names for pharmaceutical substances (INN). proposed INN: list 104” (PDF). WHO Drug Information. 24 (4): 386. hdl:10665/74579.
- ^ World Health Organization (2011). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 66”. WHO Drug Information. 25 (3). hdl:10665/74683.
External links
- “Pacritinib”. Drug Information Portal. U.S. National Library of Medicine.
OLD—
Pacritinib (INN[1]) is a macrocyclic Janus kinase inhibitor that is being developed for the treatment of myelofibrosis. It mainly inhibits Janus kinase 2 (JAK2). The drug is in Phase III clinical trials as of 2013.[2] The drug was discovered in Singapore at the labs of S*BIO Pte Ltd. It is a potent JAK2 inhibitor with activity of IC50 = 23 nM for the JAK2WT variant and 19 nM for JAK2V617F with very good selectivity against JAK1 and JAK3 (IC50 = 1280 and 520 nM, respectively).[3][4] The drug is acquired by Cell Therapeutics, Inc. (CTI) and Baxter international and could effectively address an unmet medical need for patients living with myelofibrosis who face treatment-emergent thrombocytopenia on marketed JAK inhibitors.[5]
Pacritinib is an orally bioavailable inhibitor of Janus kinase 2 (JAK2) and the JAK2 mutant JAK2V617F with potential antineoplastic activity. Oral JAK2 inhibitor SB1518 competes with JAK2 for ATP binding, which may result in inhibition of JAK2 activation, inhibition of the JAK-STAT signaling pathway, and so caspase-dependent apoptosis. JAK2 is the most common mutated gene in bcr-abl-negative myeloproliferative disorders; the JAK2V617F gain-of-function mutation involves a valine-to-phenylalanine modification at position 617. The JAK-STAT signaling pathway is a major mediator of cytokine activity.
Synthesis Reference
A245943 — William AD, Lee AC, Blanchard S, Poulsen A, Teo EL, Nagaraj H, Tan E, Chen D, Williams M, Sun ET, Goh KC, Ong WC, Goh SK, Hart S, Jayaraman R, Pasha MK, Ethirajulu K, Wood JM, Dymock BW: Discovery of the macrocycle 11-(2-pyrrolidin-1-yl-ethoxy)-14,19-dioxa-5,7,26-triaza-tetracyclo[19.3.1.1(2,6). 1(8,12)]heptacosa-1(25),2(26),3,5,8,10,12(27),16,21,23-decaene (SB1518), a potent Janus kinase 2/fms-like tyrosine kinase-3 (JAK2/FLT3) inhibitor for the treatment of myelofibrosis and lymphoma. J Med Chem. 2011 Jul 14;54(13):4638-58. doi: 10.1021/jm200326p. Epub 2011 Jun 15.
Pacritinib is an orally bioavailable inhibitor of Janus kinase 2 (JAK2) and the JAK2 mutant JAK2V617F with potential antineoplastic activity. Oral JAK2 inhibitor SB1518 competes with JAK2 for ATP binding, which may result in inhibition of JAK2 activation, inhibition of the JAK-STAT signaling pathway, and so caspase-dependent apoptosis. JAK2 is the most common mutated gene in bcr-abl-negative myeloproliferative disorders; the JAK2V617F gain-of-function mutation involves a valine-to-phenylalanine modification at position 617. The JAK-STAT signaling pathway is a major mediator of cytokine activity.


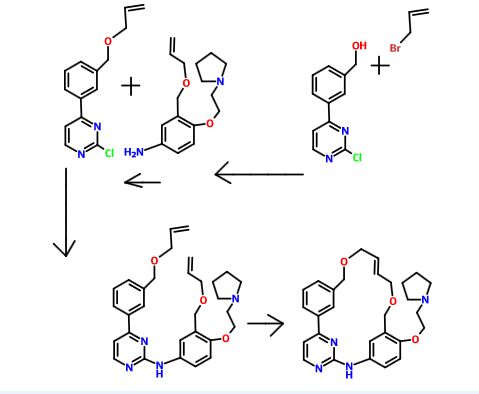
The compound 11-(2-pyrrolidin-1-yl-ethoxy)-14,19-dioxa-5,7,26-triaza-tetracyclo[19.3.1.1(2,6).1(8,12)]heptacosa-1(25),2(26),3,5,8,10,12(27),16,21,23-decaene (Compound I) was first described in PCT/SG2006/000352 and shows significant promise as a pharmaceutically active agent for the treatment of a number of medical conditions and clinical development of this compound is underway based on the activity profiles demonstrated by the compound.

-
In the development of a drug suitable for mass production and ultimately commercial use acceptable levels of drug activity against the target of interest is only one of the important variables that must be considered. For example, in the formulation of pharmaceutical compositions it is imperative that the pharmaceutically active substance be in a form that can be reliably reproduced in a commercial manufacturing process and which is robust enough to withstand the conditions to which the pharmaceutically active substance is exposed.
-
In a manufacturing sense it is important that during commercial manufacture the manufacturing process of the pharmaceutically active substance be such that the same material is reproduced when the same manufacturing conditions are used. In addition it is desirable that the pharmaceutically active substance exists in a solid form where minor changes to the manufacturing conditions do not lead to major changes in the solid form of the pharmaceutically active substance produced. For example it is important that the manufacturing process produce material having the same crystalline properties on a reliable basis and also produce material having the same level of hydration.
-
In addition it is important that the pharmaceutically active substance be stable both to degradation, hygroscopicity and subsequent changes to its solid form. This is important to facilitate the incorporation of the pharmaceutically active substance into pharmaceutical formulations. If the pharmaceutically active substance is hygroscopic (“sticky”) in the sense that it absorbs water (either slowly or over time) it is almost impossible to reliably formulate the pharmaceutically active substance into a drug as the amount of substance to be added to provide the same dosage will vary greatly depending upon the degree of hydration. Furthermore variations in hydration or solid form (“polymorphism”) can lead to changes in physico-chemical properties, such as solubility or dissolution rate, which can in turn lead to inconsistent oral absorption in a patient.
-
Accordingly, chemical stability, solid state stability, and “shelf life” of the pharmaceutically active substance are very important factors. In an ideal situation the pharmaceutically active substance and any compositions containing it, should be capable of being effectively stored over appreciable periods of time, without exhibiting a significant change in the physico-chemical characteristics of the active substance such as its activity, moisture content, solubility characteristics, solid form and the like.
-
In relation to 11-(2-pyrrolidin-1-yl-ethoxy)-14,19-dioxa-5,7,26-triaza-tetracyclo[19.3.1.1(2,6).1(8,12)]heptacosa-1(25),2(26),3,5,8,10,12(27),16,21,23-decaene initial studies were carried out on the hydrochloride salt and indicated that polymorphism was prevalent with the compound being found to adopt more than one crystalline form depending upon the manufacturing conditions. In addition it was observed that the moisture content and ratio of the polymorphs varied from batch to batch even when the manufacturing conditions remained constant. These batch-to-batch inconsistencies and the exhibited hygroscopicity made the hydrochloride salt less desirable from a commercial viewpoint.
-
Accordingly it would be desirable to develop one or more salts of 11-(2-pyrrolidin-1-yl-ethoxy)-14,19-dioxa-5,7,26-triaza-tetracyclo[19.3.1.1(2,6).1(8,12)]heptacosa-1(25),2(26),3,5,8,10,12(27),16,21,23-decaene which overcome or ameliorate one or more of the above identified problems.
PATENT
US 2011263616
http://www.google.com/patents/US20110263616
11-(2-pyrrolidin-1-yl-ethoxy)-14,19-dioxa-5,7,26triaza-tetra-cyclo[19.3.1.1(2,6).1(8,12)]heptacosa-1(25),2(26),3,5,8,10,12(27),16,21,23-decaene (Compound I) which have been found to have improved properties. In particular the present invention relates to the maleate salt of this compound. The invention also relates to pharmaceutical compositions containing this salt and methods of use of the salt in the treatment of certain medical conditions.

PATENT
http://www.google.com/patents/US8415338
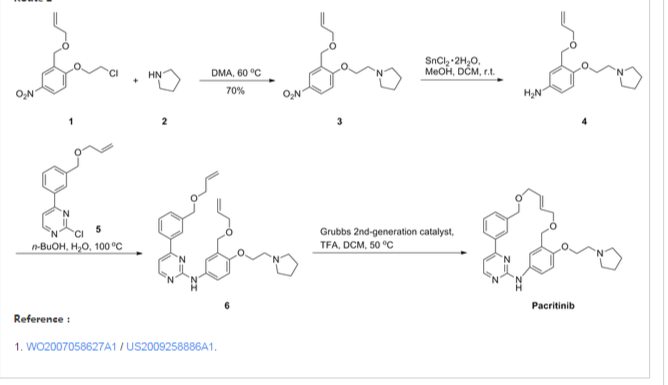
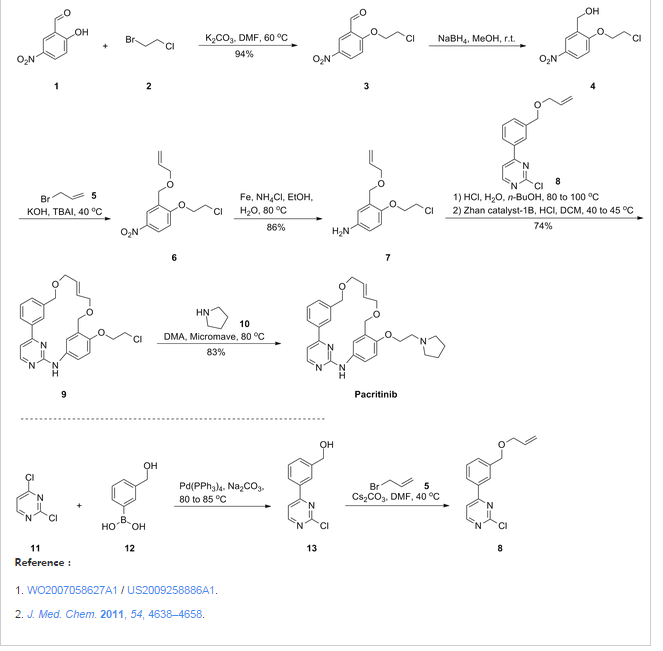
Representative Procedure for the Synthesis of Compounds Type (XVIIId) [3-(2-Chloro-pyrimidin-4-yl)-phenyl]-methanol (XIIIa2)

Compound (XIIIa2) was obtained using the same procedure described for compound (XIIIa1); LC-MS (ESI positive mode) m/z 221 ([M+H]+).
4-(3-Allyloxymethyl-phenyl)-2-chloro-pyrimidine (XVa2)

Compound (XVa2) was obtained using the same procedure described for compound (XVa1); LC-MS (ESI positive mode) m/z 271 ([M+H]+).
[4-(3-Allyloxymethyl-phenyl)-pyrimidin-2-yl]-[3-allyloxymethyl-4-(2-pyrrolidin-1-yl-ethoxy)-phenyl]-amine (XVIId1)

Compound (XVIId1) was obtained using the same procedure described for compound (XVIIb1); LC-MS (ESI positive mode) m/z 501.
Macrocycle Example 3 Compound 13

Compound (13) was obtained using the same procedure described for compound (1) HPLC purity at 254 nm: 99%; LC-MS (ESI positive mode) m/z 473 ([M+H]+); 1H NMR (MeOD-d4) δ 8.79 (d, 1H), 8.46 (d, 1H), 8.34-8.31 (m, 1H), 7.98-7.96 (m, 1H), 7.62-7.49 (m, 2H), 7.35 (d, 1H), 7.15-7.10 (m, 1H), 7.07-7.02 (m, 1H), 5.98-5.75 (m, 2H, 2×=CH), 4.67 (s, 2H), 4.67 (s, 2H), 4.39-4.36 (m, 2H), 4.17 (d, 2H), 4.08 (d, 2H), 3.88-3.82 (m, 2H), 3.70 (t, 2H), 2.23-2.21 (m, 2H), 2.10-2.07 (m, 2H).
PAPER
J MC 2011, 54 4638
http://pubs.acs.org/doi/abs/10.1021/jm200326p

Discovery of the activating mutation V617F in Janus Kinase 2 (JAK2V617F), a tyrosine kinase critically involved in receptor signaling, recently ignited interest in JAK2 inhibitor therapy as a treatment for myelofibrosis (MF). Herein, we describe the design and synthesis of a series of small molecule 4-aryl-2-aminopyrimidine macrocycles and their biological evaluation against the JAK family of kinase enzymes and FLT3. The most promising leads were assessed for their in vitro ADME properties culminating in the discovery of 21c, a potent JAK2 (IC50 = 23 and 19 nM for JAK2WT and JAK2V617F, respectively) and FLT3 (IC50 = 22 nM) inhibitor with selectivity against JAK1 and JAK3 (IC50 = 1280 and 520 nM, respectively). Further profiling of 21c in preclinical species and mouse xenograft and allograft models is described. Compound 21c(SB1518) was selected as a development candidate and progressed into clinical trials where it is currently in phase 2 for MF and lymphoma.
 Discovery of the Macrocycle 11-(2-Pyrrolidin-1-yl-ethoxy)-14,19-dioxa-5,7,26-triaza-tetracyclo[19.3.1.1(2,6).1(8,12)]heptacosa-1(25),2(26),3,5,8,10,12(27),16,21,23-decaene (SB1518), a Potent Janus Kinase 2/Fms-Like Tyrosine Kinase-3 (JAK2/FLT3) Inhibitor for the Treatment of Myelofibrosis and Lymphoma
Discovery of the Macrocycle 11-(2-Pyrrolidin-1-yl-ethoxy)-14,19-dioxa-5,7,26-triaza-tetracyclo[19.3.1.1(2,6).1(8,12)]heptacosa-1(25),2(26),3,5,8,10,12(27),16,21,23-decaene (SB1518), a Potent Janus Kinase 2/Fms-Like Tyrosine Kinase-3 (JAK2/FLT3) Inhibitor for the Treatment of Myelofibrosis and Lymphoma
S*BIO Pte. Ltd., 1 Science Park Road, #05-09, The Capricorn, Singapore Science Park II, Singapore 117528
J. Med. Chem., 2011, 54 (13), pp 4638–4658
DOI: 10.1021/jm200326p
Publication Date (Web): May 23, 2011
Copyright © 2011 American Chemical Society
(21c)
The title compound was synthesized from 21a and pyrrolidine (yield, 83%; mixture of trans/cis85:15 by NMR). LC-MS (ESI positive mode) m/z 473 ([M + H]+). HRMS: theoretical C28H32N4O3MW, 472.2474; found, 473.2547. 1H NMR (MeOD-d4): δ 8.79 (d, 1H), 8.46 (d, 1H), 8.34–8.31 (m, 1H, CH), 7.98–7.96 (m, 1H), 7.62–7.49 (m, 2H), 7.35 (d, 1H), 7.15–7.10 (m, 1H), 7.07–7.02 (m, 1H), 5.98–5.75 (m, 2H), 4.67 (s, 2H), 4.67 (s, 2H), 4.39–4.36 (m, 2H), 4.17 (d, 2H), 4.08 (d, 2H), 3.88–3.82 (m, 2H), 3.70 (t, 2H), 2.23–2.21 (m, 2H), 2.10–2.07 (m, 2H); chloride content (titration) 7.7% (1.18 equivs); water content (Karl Fischer) 6.1% (1.85 equivs); Anal. Calcd. for C28H32N4O3·1.18HCl·1.85H2O: C, 61.46; H, 6.46; N, 10.24; Cl, 7.65. Found: C, 61.99; H, 6.91; N, 10.25; Cl, 7.45.
References
1 “International Nonproprietary Names for Pharmaceutical Substances (INN) List 104” (PDF). WHO Drug Information 24 (4): 386. 2010.
2“JAK-Inhibitoren: Neue Wirkstoffe für viele Indikationen”. Pharmazeutische Zeitung (in German) (21). 2013.
3William, A. D.; Lee, A. C. -H.; Blanchard, S. P.; Poulsen, A.; Teo, E. L.; Nagaraj, H.; Tan, E.; Chen, D.; Williams, M.; Sun, E. T.; Goh, K. C.; Ong, W. C.; Goh, S. K.; Hart, S.; Jayaraman, R.; Pasha, M. K.; Ethirajulu, K.; Wood, J. M.; Dymock, B. W. (2011). “Discovery of the Macrocycle 11-(2-Pyrrolidin-1-yl-ethoxy)-14,19-dioxa-5,7,26-triaza-tetracyclo[19.3.1.1(2,6).1(8,12)]heptacosa-1(25),2(26),3,5,8,10,12(27),16,21,23-decaene (SB1518), a Potent Janus Kinase 2/Fms-Like Tyrosine Kinase-3 (JAK2/FLT3) Inhibitor for the Treatment of Myelofibrosis and Lymphoma”. Journal of Medicinal Chemistry 54 (13): 4638–58. doi:10.1021/jm200326p. PMID 21604762.
4Poulsen, A.; William, A.; Blanchard, S. P.; Lee, A.; Nagaraj, H.; Wang, H.; Teo, E.; Tan, E.; Goh, K. C.; Dymock, B. (2012). “Structure-based design of oxygen-linked macrocyclic kinase inhibitors: Discovery of SB1518 and SB1578, potent inhibitors of Janus kinase 2 (JAK2) and Fms-like tyrosine kinase-3 (FLT3)”. Journal of Computer-Aided Molecular Design 26 (4): 437–50. doi:10.1007/s10822-012-9572-z. PMID 22527961.
5http://www.pmlive.com/pharma_news/baxter_licenses_cancer_drug_from_cti_in_$172m_deal_519143
| US8153632 * | Nov 15, 2006 | Apr 10, 2012 | S*Bio Pte Ltd. | Oxygen linked pyrimidine derivatives |
| US8415338 * | Apr 4, 2012 | Apr 9, 2013 | Cell Therapeutics, Inc. | Oxygen linked pyrimidine derivatives |
| US20110294831 * | Dec 9, 2009 | Dec 1, 2011 | S*Bio Pte Ltd. | 11-(2-pyrrolidin-1-yl-ethoxy)-14,19-dioxa-5,7,26-triaza-tetracyclo[19.3.1.1(2,6).1(8,12)]heptacosa-1(25),2(26),3,5,8,10,12(27),16,21,23-decaene citrate salt |
| Patent | Submitted | Granted |
|---|---|---|
| OXYGEN LINKED PYRIMIDINE DERIVATIVES [US8153632] | 2009-03-19 | 2012-04-10 |
| ANTIVIRAL JAK INHIBITORS USEFUL IN TREATING OR PREVENTING RETROVIRAL AND OTHER VIRAL INFECTIONS [US2014328793] | 2012-11-30 | 2014-11-06 |
| OXYGEN LINKED PYRIMIDINE DERIVATIVES [US2013172338] | 2013-02-20 | 2013-07-04 |
| METHOD OF SELECTING THERAPEUTIC INDICATIONS [US2014170157] | 2012-06-15 | 2014-06-19 |
| CYCLODEXTRIN-BASED POLYMERS FOR THERAPEUTIC DELIVERY [US2014357557] | 2014-05-30 | 2014-12-04 |
| 11-(2-PYRROLIDIN-1-YL-ETHOXY)-14,19-DIOXA-5,7,26-TRIAZA-TETRACYCLO[19.3.1.1(2,6).1(8,12)]HEPTACOSA-1(25),2(26),3,5,8,10,12(27),16,21,23-DECAENE MALEATE SALT [US2011263616] | 2011-10-27 | |
| 11-(2-PYRROLIDIN-1-YL-ETHOXY)-14,19-DIOXA-5,7,26-TRIAZA-TETRACYCLO[19.3.1.1(2,6).1(8,12)]HEPTACOSA-1(25),2(26),3,5,8,10,12(27),16,21,23-DECAENE CITRATE SALT [US2011294831] | 2011-12-01 | |
| BIOMARKERS AND COMBINATION THERAPIES USING ONCOLYTIC VIRUS AND IMMUNOMODULATION [US2014377221] | 2013-01-25 | 2014-12-25 |
| Oxygen linked pyrimidine derivatives [US8415338] | 2012-04-04 | 2013-04-09 |
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
(16E)-11-[2-(1-Pyrrolidinyl)ethoxy]-14,19-dioxa-5,7,26-triazatetracyclo[19.3.1.12,6.18,12]heptacosa-1(25),2(26),3,5,8,10,12(27),16,21,23-decaene
|
|
| Clinical data | |
| Legal status |
|
| Routes of administration |
Oral |
| Identifiers | |
| ATC code | None |
| PubChem | CID: 46216796 |
| ChemSpider | 28518965 |
| ChEMBL | CHEMBL2035187 |
| Synonyms | SB1518 |
| Chemical data | |
| Formula | C28H32N4O3 |
| Molecular mass | 472.58 g/mol |


S*Bio Pte Ltd
Address: 1 Science Park Rd, Singapore 117528
Phone:+65 6827 5000

S*BIO Pte Ltd. provides research and clinical development services for small molecule drugs for the treatment of cancer in Singapore. The company’s products include JAK2 inhibitors, such as SB1518 for leukemia/myelofibrosis, lymphoma, and polycythemia; and SB1578 for RA/psoriasis. The company also offers SB939, a histone deacetylases for MDS/AML+combo, prostate cancer, sarcoma, pediatric tumor, and myelofibrosis; SB2602, a mTOR inhibitor; SB2343, a mTOR/PI3K inhibitor; and SB1317, a CDK/Flt3 inhibitor. The company was founded in 2000 and is based in Singapore. S*BIO Pte Ltd. operates as a subsidiary of Chiron Corporation Limited.
SEE……..http://apisynthesisint.blogspot.in/2016/01/pacritinib.html

AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
|
|
| Clinical data | |
|---|---|
| Trade names | Vonjo |
| Other names | SB1518 |
| License data |
|
| Routes of administration |
By mouth |
| ATC code | |
| Legal status | |
| Legal status | |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEMBL | |
| PDB ligand | |
| Chemical and physical data | |
| Formula | C28H32N4O3 |
| Molar mass | 472.589 g·mol−1 |
| 3D model (JSmol) | |
///////Vonjo, FDA APPTOVESD 2022, APPROVALS 2022, PACRITINIB, パクリチニブ, priority review, fast track, orphan drug, UNII-G22N65IL3O, пакритиниб , باكريتينيب , 帕瑞替尼 , SB 1518
c1cc2cc(c1)-c3ccnc(n3)Nc4ccc(c(c4)COC/C=C/COC2)OCCN5CCCC5
C1CCN(C1)CCOC2=C3COCC=CCOCC4=CC=CC(=C4)C5=NC(=NC=C5)NC(=C3)C=C2
Cabotegravir, GSK 744

Cabotegravir, GSK 744,
PMDA APPROVED 2022/5/31, JAPAN
(3S,11aR)-N-(2,4-Difluorobenzyl)-6-hydroxy-3-methyl-5,7-dioxo-2,3,5,7,11,11a-hexahydro[1,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazine-8-carboxamide
3S, 1 1 aR)- N-[(2,4-difluorophenyl)methyl]-2,3,5,7, 1 1 , 1 1 a-hexahydro-6-hydroxy-3- methyl-5,7- dioxo-oxazolo[3,2-a]pyrido[1 ,2-d]pyrazine-8-carboxamide
OTHER ISOMER
(3R,11 aS)-N-[(2,4-Diflυorophenyl)methyl]-6-hydroxy-3-methyl-5,7-dioxo-2,3,5,7, 11, 11a-hexahydro[1 ,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazine-8-carboxamide
VIIV HEALTHCARE …INNOVATOR
-
GSK1265744, CAS 1051375-10-0, S-265744 LAP
-
C19-H17-F2-N3-O5
- 405.3553
- 744 LA
- GSK 1265744
- GSK 744
- GSK-1265744A
- GSK1265744
- GSK1265744A
- GSK744
- GSK744 LA
- GSK744 LAP
- S-265744
- S/GSK1265744
Product Ingredients
| INGREDIENT | UNII | CAS | INCHI KEY |
|---|---|---|---|
| Cabotegravir sodium | 3L12PT535M | 1051375-13-3 | AEZBWGMXBKPGFP-KIUAEZIZSA-M |
Cabotegravir, sold under the brand name Vocabria among others, is a antiretroviral medication used for the treatment of HIV/AIDS. It is available in the form of tablets and as an intramuscular injection, as well as in an injectable combination with rilpivirine under the brand name Cabenuva.[6][9]
It is an integrase inhibitor with a carbamoyl pyridone structure similar to that of dolutegravir.[10]
In December 2021, the U.S. Food and Drug Administration approved cabotegravir for pre-exposure prophylaxis (PrEP) in at-risk people under the brand name Apretude.[11]
GSK744 (also known as S/GSK1265744) is an investigational new drug under development for the treatment of HIV infection. It is anintegrase inhibitor, with a carbamoyl pyridone structure similar to dolutegravir. In investigational studies, the agent has been packaged into nanoparticles (GSK744LAP) conferring an exceptionally long half-life of 21–50 days following a single dose. In theory, this would make possible suppression of HIV with dosing as infrequently as once every three months.[1]
S-265744 LAP is in phase II clinical development at Shionogi-GlaxoSmithKline for the treatment of HIV infection. Phase III clinical trials had been ongoing for this indication; however, no recent development has been reported for this study.
Cabotegravir, or GSK1265744, is an HIV-1 integrase inhibitor that is prescribed with the non-nucleoside reverse transcriptase inhibitor, rilpivirine.4,6,7 Early research into cabotegravir showed it had lower oral bioavailability than dolutegravir,4 which resulted in the development of long acting monthly intramuscular injection formulation for cabotegravir.4,7
Cabotegravir was granted FDA approval on 21 January 2021 in combination with rilpivirine to treat HIV-1 infection in virologically suppressed individuals.8 While previously administered once monthly only, this combination product was granted FDA approval for dosing every two months on February 01, 2022 11 and without the need for an oral lead-in period prior.7
The human immunodeficiency virus (“HIV”) is the causative agent for acquired immunodeficiency syndrome (“AIDS”), a disease characterized by the destruction of the immune system, particularly of CD4+ T-cells, with attendant susceptibility to opportunistic infections, and its precursor Al DS-related complex (“ARC”), a syndrome characterized by symptoms such as persistent generalized lymphadenopathy, fever and weight loss. HIV is a retrovirus; the conversion of its RNA to DNA is accomplished through the action of the enzyme reverse transcriptase. Compounds that inhibit the function of reverse transcriptase inhibit replication of HIV in infected cells. Such compounds are useful in the prevention or treatment of HIV infection in humans.
A required step in HIV replication in human T-cells is the insertion by virally-encoded integrase of proviral DNA into the host cell genome. Integration is believed to be mediated by integrase in a process involving assembly of a stable nucleoprotein complex with viral DNA sequences, cleavage of two nucleotides from the 3′ termini of the linear proviral DNA and covalent joining of the recessed 3′ OH termini of the proviral DNA at a staggered cut made at the host target site. The repair synthesis of the resultant gap may be accomplished by cellular enzymes. There is continued need to find new therapeutic agents to treat human diseases. HIV integrase is an attractive target for the discovery of new therapeutics due to its important role in viral infections, particularly HIV infections. Integrase inhibitors are disclosed in WO2006/116724.
(3S, 1 1 aR)- N-[(2,4-difluorophenyl)methyl]-2,3,5,7, 1 1 , 1 1 a-hexahydro-6-hydroxy-3- methyl-5,7- dioxo-oxazolo[3,2-a]pyrido[1 ,2-d]pyrazine-8-carboxamide, a compound of formula (I), also referred to as compound (I), has proven antiviral activity against human immunodeficiency virus (HIV).

The present invention features pharmaceutical compositions comprising the active ingredient (3S, 1 1 aR)- N-[(2,4-difluorophenyl)methyl]-2,3,5,7, 1 1 , 1 1 a-hexahydro-6-hydroxy-3- methyl-5,7- dioxo-oxazolo[3,2-a]pyrido[1 ,2-d]pyrazine-8-carboxamide, or a pharmaceutically acceptable salt thereof, suitable for administration once monthly or longer.
Medical uses
Cabotegravir in combination with rilpivirine is indicated for the treatment of human immunodeficiency virus type-1 (HIV-1) in adults.[1][5] The combination injection is intended for maintenance treatment of adults who have undetectable HIV levels in the blood (viral load less than 50 copies/mL) with their current antiretroviral treatment, and when the virus has not developed resistance to non-nucleoside reverse transcriptase inhibitors (NNRTIs) and integrase strand transfer inhibitors.[5] The tablets are used to check whether a person tolerates the treatment before the injection therapy is started.[12][5]
The two medicines are the first antiretroviral drugs that come in a long-acting injectable formulation.[12]
Cabotegravir (Apretude) is indicated for use in at-risk people weighing at least 35 kilograms (77 lb) for pre-exposure prophylaxis (PrEP) to reduce the risk of sexually acquired HIV.[11]
Contraindications and interactions
Cabotegravir must not be combined with the drugs rifampicin, rifapentine, carbamazepine, oxcarbazepine, phenytoin or phenobarbital, which induce the enzyme UGT1A1.[5] These drugs significantly decrease cabotegravir concentrations in the body and thus may reduce its effectiveness.[9][5] Additionally, they induce the enzyme CYP3A4, which leads to reduced rilpivirine concentrations in the body.[5][13][14][15] Additionally, patients who are breastfeeding or plan to breastfeed should not take Cabotegravir because it is not known if it will pass within the breast milk.[16]
Adverse effects
The most common side effects of the injectable combination therapy with rilpivirine are reactions at the injection site (in up to 84% of patients) such as pain and swelling, as well as headache (up to 12%) and fever or feeling hot (in 10%). For the tablets, headache and a hot feeling were slightly less frequent. Less common side effects (under 10%) for both formulations are depressive disorders, insomnia, and rashes.[9]
Pharmacology
Mechanism of action
Cabotegravir is an integrase strand transfer inhibitor. This means it blocks the HIV’s enzyme integrase, thereby preventing its genome from being integrated into the human cells’ DNA.[9] As this is a necessary step for the virus to replicate, its further spread is hampered.[9]
Pharmacokinetics

Cabotegravir glucuronide, the main metabolite in human bile and urine[17]
When taken by mouth, cabotegravir reaches highest blood plasma levels after three hours. Taking the drug together with food slightly increases its concentrations in the blood, but this is not clinically relevant. After injection into the muscle, cabotegravir is slowly absorbed into the bloodstream, reaching its highest blood plasma levels after about seven days.[9]
Over 99% of the substance are bound to plasma proteins. The drug is inactivated in the body by glucuronidation, mainly by the enzyme UGT1A1, and to a much lesser extent by UGT1A9. More than 90% of the circulating substance are the unchanged cabotegravir, however. The biological half-life is 41 hours for the tablets and 5.6 to 11.5 weeks for the injection.[9]
Elimination has only been studied for oral administration: Most of the drug is eliminated via the faeces in unchanged form (47%). It is not known how much of this amount comes from the bile, and how much was not absorbed in the first place. (The bile actually contains the glucuronide, but this could be broken up again in the gut lumen to give the parent substance that is observed in the faeces.) To a lesser extent it is excreted via the urine (27%), almost exclusively as the glucuronide.[9]
Pharmacogenomics
UGT1A1 poor metabolizers have 1.3- to 1.5-fold increased cabotegravir concentrations in the body. This is not considered clinically significant.[9]
Chemistry
Cabotegravir is a white to off-white, crystalline powder that is practically insoluble in aqueous solutions under pH 9, and slightly soluble above pH 10. It is slightly acidic with a pKa of 7.7 for the enolic acid and 1.1 (calculated) for the carboxamide. The molecule has two asymmetric carbon atoms; only one of the four possible configurations is present in the medication.[18]
Formulation
In studies, the agent was packaged into nanoparticles (GSK744LAP) conferring a biological half-life of 21 to 50 days[citation needed] following a single dose. The marketed injection achieves its long half-life not via nanoparticles but with a suspension of the free cabotegravir acid. The tablets contain cabotegravir sodium salt.[18]
History
Cabotegravir was examined in the clinical trials HPTN 083 and HPTN 084.[19][20] In 2020, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) adopted a positive opinion, recommending the granting of a marketing authorization for the medicinal product Vocabria intended for the treatment of human immunodeficiency virus type 1 (HIV-1) infection in combination with rilpivirine injection.[21] The EMA also recommended marketing authorization be given for rilpivirine and cabotegravir injections to be used together for the treatment of people with HIV-1 infection.[12] Cabotegravir was approved for medical use in the European Union in December 2020.[8]
Society and culture
Names
Cabotegravir is the United States Adopted Name (USAN)[22] and the international nonproprietary name (INN).[23]
Research
Pre-exposure prophylaxis
In 2020, results for some studies were released showing success in using injectable cabotegravir for long-acting pre-exposure prophylaxis (PrEP) with greater efficacy than the emtricitabine/tenofovir combination being widely used for PrEP at the time.[24][25]
The safety and efficacy of cabotegravir to reduce the risk of acquiring HIV were evaluated in two randomized, double-blind trials that compared cabotegravir to emtricitabine/tenofovir, a once daily oral medication for HIV PrEP.[11] Trial 1 included HIV-uninfected men and transgender women who have sex with men and have high-risk behavior for HIV infection.[11] Trial 2 included uninfected cisgender women at risk of acquiring HIV.[11]
In Trial 1, 4,566 cisgender men and transgender women who have sex with men received either cabotegravir or emtricitabine/tenofovir.[11] The trial measured the rate of HIV infections among trial participants taking daily cabotegravir followed by cabotegravir injections every two months compared to daily oral emtricitabine/tenofovir.[11] The trial showed participants who took cabotegravir had 69% less risk of getting infected with HIV when compared to participants who took emtricitabine/tenofovir.[11]
In Trial 2, 3,224 cisgender women received either cabotegravir or emtricitabine/tenofovir.[11] The trial measured the rate of HIV infections in participants who took oral cabotegravir and injections of cabotegravir compared to those who took emtricitabine/tenofovir orally.[11] The trial showed participants who took cabotegravir had 90% less risk of getting infected with HIV when compared to participants who took emtricitabine/tenofovir.[11]
In December 2021, the U.S. Food and Drug Administration (FDA) approved cabotegravir for pre-exposure prophylaxis.[11] The FDA granted the approval of Apretude to Viiv.[11]
Methods for the preparation of a compound of formula (I) are described in WO 2006/1 16764, WO2010/01 1814, WO2010/068262, and WO2010/068253
WO 2006116764
http://www.google.com/patents/WO2006116764A1?cl=en

[Chemical formula 68] is UNDESIRED ISOMER………..amcrasto@gmail.com

Example Z-1:
(3R,11 aS)-N-[(2,4-Diflυorophenyl)methyl]-6-hydroxy-3-methyl-5,7-dioxo-2,3,5,7, 11, 11a
-hexahydro[1 ,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazine-8-carboxamide sodium salt.

(3R,11aS)-N-[(2,4-Diflυorophenyl)methyl]-3-methyl-5,7-dioxo-6-[(phenylmethyl)oxy]-2,
3,5,7,11,11a-hexahydro[1,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazine-8-carboxamide. To a solution of 16a (409 mg, 0.87 mmol) in dichloroethane (20 mL) was added (2R)-2-amino-1-propanol (0,14 mL, 1.74 mmol) and 10 drops of glacial acetic acid.
The resultant solution was heated at reflux for 2 h. Upon cooling, Celite was added
to the mixture and the solvents removed in vacuo and the material was purified via
silica gel chromatography (2% CH3OH/CH2CI2 gradient elution) to give
(3R),11aS)-N-[(2,4-difluorophenyl)methyl]-3-methyl-5,7-dioxo-6- [(phenylmethyl)oxy]-2,
3,5,7, 1 l , 11a-hexahydro[1,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazinc-8-carboxamide (396
mg, 92%) as a glass, JH NMR (CDCIo) δ 10.38 (m, 1 H), 8.42 (s, 1 H), 7,54-7,53 (m, 2
H), 7,37-7.24 (m, 4 H), 6.83-6,76 (m, 2 H), 5.40 (d, J = 10.0 Hz, 1 H), 5.22 (d, J = 10,0
Hz, 1 H), 5.16 (dd, J – 9,6, 6.0 Hz, 1 H), 4,62 (m, 2 H), 4.41 (m, 1 H), 4.33-4.30 (m, 2
H), 3.84 (dd, J= 12.0, 10.0 Hz, 1 H), 3.63 (dd, J= 8,4, 7.2 Hz, 1 H), 1.37 (d, J= 6.0 Hz,
3 H); ES+ MS: 496 (M+1).
b)
(3R, 11aS)-N-[(2,4-Difluorophenyl)methyl]-6-hydroxy-3-methyl-5,7-dioxo-2,3,5,7, 11, 1la
-hexahydro[1,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazine-8vcarboxamide sodium salt. To a
solution of
(37?, 11aS)-N-[(2,4-difluo]-ophenyl)methyl]-3-methyl-5,7-dioxo-6- [(phenylmethyl)oxy] -2,
3,5,7,11,11 a-hexahydro[1,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazine-8-carboxamide (396
mg, 0.80 mmol) in methanol (30 mL) was added 10% Pd/C (25 mg). Hydrogen was
bubbled through the reaction mixture via a balloon for 2 h. The resultant mixture
was filtered through Celite with methanol and dichloromethanc. The filtrate was
concentrated in vacuo to give
(3R, l] aS)-N-f(2,4-difliιorophenyl)methyl]-6-hydroxy-3-methyl-5,7-dioxo-2,3,5,7, υ , 11a- hexahydro[1,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazine-8-carboxamide as a pink tinted
white solid (278 mg, 86%), 1H NMR (ODCU) δ 11.47 (m, 1 H), 10.29 (m, 1 H), 8,32 (s,
1 H), 7.36 (m, 1 H), 6.82 (m, 2 H), 5.31 (dd, J – 9.6, 3.6 Hz, 1 H), 4.65 (m, 2 H),
4,47-4,38 (m, 3 H), 3.93 (dd, J= 12.0, 10.0 Hz, 1 H), 3,75 (m, 1 H), 1.49 (d, J= 5.6 Hz,
3 H); BS1 MS: 406 (M+ 1). The above material (278 mg, 0,66 mmol) was taken up
m cthanol (10 mL) and treated with 1 Nsodium hydroxide (aq) (0.66 mL, 0.66 mmol).
The resulting suspension was stirred at room temperature for 30 min, Ether was
added and the liquids were collected to provide the sodium salt of the title compound
as a white powder (291 mg, 99%).‘ 1H NMR (OMSO- do) δ 30.68 (m, 1 H), 7,90 (s, 1 H),
7.35 (m, 1 H), 7.20 (m, 1 H), 7,01 (m, 1 H), 5,20 (m, 1 H), 4,58 (m, I H), 4.49 (m, 2 H),
4.22 (m, 2 H), 3 74 (dd, J= 11.2, 10.4 Hz, 1 H), 3.58 (m, 1 H), 1.25 (d, J=- 4.4 Hz, 3 H)
DESIRED ISOMER………… ANY ERROR ………….amcrasto@gmail.com
Example Z-9-
(3£ 11aΛ^N-[(2.4-D-fluoroDhonyl)methyl] -6-hvdroxy-3-methyl-5.7-dioxo-2,3,5.7, n , 11 a
-hexahydro[1 ,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazino-8-carboxamide sodium salt.

The title compound was made in two steps using a similar process to that described
in example Z-I. 16a (510 mg, 1.08 mmol) and (2«5)-2-amino-1-propanol (0.17 mL, 2,17 mmol) were reacted in 1,2-dichloroethane (20 mL) with acetic acid to give
(3S, 11aR)-i\A[(2,4-diflιιorophenyl)methyl]-3-methyl-5,7-d.ioxo-6-[(phenylmethyl)oxy]-2,
3,5,7,11,1la-hexahydro[1,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazine-8-carboxamide (500
mg, 93%). This material was hydrogenated in a second step as described in example
Z- I to give
3S, 11a R)-7N-[(2,4-Diiαuorophenyl)methyl]-6-hydroxy-3-methyl-5,7-dioxo-2,3,5,7, 11, 11a-
hexahydro[1,3]oxazolo[3,2-a]pyrido[1,2-d]pyraziine-8-carboxamide (386 mg, 94%) as a
tinted white solid. Η NMR (CDCL3) δ 11.46 (m, 1 H), 10.28 (m, 1 H), 8.32 (s, 1 H),
7.35 (m, 1 H), 6.80 (m, 2 H), 5.30 (dd, J = 10.0, 4.0 Hz, 1 H), 4.63 (m, 2 H), 4.48-4.37
(m, 3 H), 3.91 (dd, J = 12.0, 10.0 Hz, 1 H), 3.73 (m, 1 H), 1.48 (d, J – 6.0 Hz, 3 H);
ES 1 MS: 406 (M+ 1). This material (385 mg, 0.95 mmol) was treated with sodium
hydroxide (0,95 mL, 1.0 M, 0.95 mmol) m ethanol (15 mL) as described in example Z-1
to provide its corresponding sodrum sail (381 mg, 94%) as a white solid. 1H NMR
(DMSO- Λ) δ 10.66 (m, 1 PI), 7.93 (s, 1 H), 7.33 (m, 1 H), 7.20 (m, 1 H), 7.01 (m, 1 H),
5.19 (m, 1 H), 4.59 (m, 1 H), 4 48 (m, 2 H), 4.22 (m, 2 H), 3,75 (m, 1 H), 3.57 (m, 1 H),
1.24 (d, J= 5 6 Hz, 3 H).
…………………..
WO 2010068253
http://www.google.com/patents/WO2010068253A1?cl=en
Example A
The starting material of Example A is compound 8, which is identical to formula (Ia). Thus, Example A depicts a process in providing an intermediate for the compound of formula 17 below which is isomeric to the compound ZZ-2 at page 237 of WO 2006/116764 to Brian Johns et al.

14


Example Aa After dissolution of mixture of 320 g of compound 8 (1.0 eq.) in 3.20 L of MeOH by heating, the solution was concentrated. To the residue, 1.66 L of MeCN, 5.72 mL of AcOH(0.1 eq.) and 82.6 g of (S)-2-Amino-propan-1-ol(1.1 eq.) were added and the mixture was heated to 70 °C, stirred at 70 0C for 4 h and concentrated. To the residue, 1.67 L of 2-propanol was added and the mixture was concentrated (twice). After cooling of the residue, filtration, washing with 500 mL of cold 2-propanol and drying provided 167 g of compound 14 (52% yield) as a crystal. 1H NMR(300 MHz1 CDCI3) δ 7.61-7.55 (m, 2H), 7.40-7.20 (m, 4H), 6.53 (d, J = 7.2, 1H), 5.46 (d, J = 10.5 Hz, 1H), 5.23 (d, J = 10.2 Hz, 1H), 5.20 (dd, J = 3.9, 9.6 Hz, 1H), 4.46- 4.34 (m, 1H)1 4.31 (dd, J = 6.6, 8.7 Hz, 1H)1 4.14 (dd, J = 3.9, 12.3 Hz1 1H)1 3.79 (dd, J = 9.9, 12.3 Hz1 1 H), 3.62 (dd, J = 6.9, 8.7 Hz1 1 H), 1.38 (d, J = 6.3 Hz1 3H).
Example Ab
To slurry of 156 g of compound 14 (1.0 eq.) in 780 ml_ of NMP was added 93.6 g of NBS(1.1 eq.) and the mixture was stirred at room temperature for 2.5 h. The reaction mixture was added to 3.12 L of H2O. Filtration, washing with 8.0 L of H2O and drying provided 163 g of compound 15 (84% yield) as a crystal.
1H NMR(300 MHz, DMSO-CT6) δ 8.37 (s, 1H), 7.55-7.50 (m, 2H), 7.42-7.25 (m, 3H), 5.34 (dd, J = 3.6, 9.9 Hz, 1H), 5.18 (d, J = 10.8 Hz, 1H), 5.03 (d, J = 10.5 Hz, 1H), 4.53 (dd, J = 3.6, 12.0 Hz, 1H)1 4.40-4.20 (m, 2H), 3.99 (dd, J = 9.9, 11.7 Hz1 1H), 3.64 (dd, J = 5.7, 8.1 Hz1 1 H)1 1.27 (d, J = 6.3 Hz1 3H).
Example Ac
Under carbon mono-oxide atmosphere, a mixture of 163 g of compound 15 (1.0 eq.), 163 mL of /-Pr2NEt(2.5 eq.), 68.4 ml_ of 2,4-difluorobenzylamine(1.5 eq.) and 22.5 g of Pd(PPh3)4(0.05 eq.) in 816 mL of DMSO was stirred at 90 0C for 7 h. After cooling, removal of precipitate, washing with 50 mL of DMSO and addition of 11.3 g of
Pd(PPh3)4(0.025 eq.), the reaction mixture was stirred at 90 0C for 2 h under carbon mono-oxide atmosphere again. After cooling, removal of precipitate and addition of 2.0 L of AcOEt and 2.0 L of H2O1 the organic layer was washed with 1.0 L of 1 N HCIaq. and 1.0 L of H2O (twice) and the aqueous layer was extracted with 1.0 L of AcOEt. The organic layers were combined and concentrated. Silica gel column chromatography of the residue provided 184 g of compound 16 (96% yield) as foam.
1H NMR(300 MHz, CDCI3) δ 10.38 (t, J = 6.3 Hz1 1H)1 8.39 (s, 1H)1 7.75-7.25 (m, 7H), 6.90-6.70 (m, 2H), 5.43 (d, J = 10.2 Hz, 1H), 5.24 (d, J = 10.2 Hz, 1H)1 5.19 (dd, J = 3.9, 9.9 Hz, 1H)1 4.63 (d, J = 6.0 Hz, 2H), 4.50-4.25 (m, 3H)1 3.86 (dd, J = 9.9, 12.3 Hz, 1H), , 3.66 (dd, J = 6.9, 8.4 Hz1 1 H), 1.39 (d, J = 6.0 Hz, 3H).
Example Ad
Under hydrogen atmosphere, a mixture of 184 g of compound 16 (1.0 eq.) and 36.8 g of 10%Pd-C in 3.31 L of THF and 0.37 L of MeOH was stirred for 3 h. After filtration of precipitate(Pd-C), washing with THF/MeOH(9/1 ) and addition of 36.8 g of 10% Pd-C, the mixture was stirred for 20 min under hydrogen atmosphere. After filtration of precipitate(Pd-C) and washing with THF/MeOH(9/1), the filtrate was concentrated. After 200 ml_ of AcOEt was added to the residue, filtration afforded crude solid of compound 17. The precipitates were combined and extracted with 4.0 L of CHCl3/MeOH(5/1). After concentration of the CHCI3ZMeOH solution and addition of 250 ml_ of AcOEt to the residue, filtration afforded crude solid of compound 17. The crude solids were combined and dissolved in 8.2 L of MeCN/H2O(9/1 ) by heating. After filtration, the filtrate was concentrated. To the residue, 1.5 L of EtOH was added and the mixture was concentrated (three times). After cooling of the residue, filtration and drying provided 132 g of compound 17 (88% yield) as a crystal. 1H NMR(300 MHz, DMSO-cfe) δ 11.47 (brs, 1H), 10.31 (t, J = 6.0 Hz, 1H), 8.46 (s, 1H), 7.40 (td, J = 8.6, 6.9 Hz, 1H), 7.24 (ddd, J = 2.6, 9.4, 10.6, 1H), 7.11-7.01 (m, 1H), 5.39 (dd, J = 4.1, 10.4 Hz, 1H), 4.89 (dd, J = 4.2, 12.3 Hz, 1H), 4.55 (d, J = 6.0 Hz, 2H), 4.40 (dd, J = 6.8, 8.6 Hz, 1H), 4.36-^.22 (m, 1H)1 4.00 (dd, J = 10.2, 12.3 Hz, 1H), 3.67 (dd, J = 6.7, 8.6 Hz, 1H), 1.34 (d, J = 6.3 Hz, 3H).
Example Ae
After dissolution of 16.0 g of compound 17 (1.0 eq.) in 2.56 L of EtOH and 0.64 L of H2O by heating, followed by filtration, 39 ml_ of 1N NaOHaq.(1.0 eq.) was added to the solution at 75 0C. The solution was gradually cooled to room temperature. Filtration, washing with 80 ml_ of EtOH and drying provided 13.5 g of compound 18 (80% yield) as a crystal.
1H NMR(300 MHz, DMSO-cfe) δ 10.73 (t, J = 6.0 Hz, 1H), 7.89 (s, 1H), 7.40-7.30 (m, 1H), 7.25-7.16 (m, 1H), 7.07-6.98 (m, 1H), 5.21 (dd, J = 3.8, 10.0 Hz, 1H), 4.58 (dd, J = 3.8, 12.1 Hz, 1H), 4.51 (d, J = 5.4 Hz, 2H), 4.3CM.20 (m, 2H), 3.75 (dd, J = 10.0, 12.1 Hz, 1H), 3.65-3.55 (m, 1H), 1.27 (d, J = 6.1 Hz, 3H).
………………
WO2010011814
http://www.google.st/patents/WO2010011814A1?cl=en&hl=pt-PT
Scheme 1

2a 2b
Scheme 2

Scheme 3

Scheme 4
phosphorylation


Scheme 5
Hydrogenolysis



The following examples are intended for illustratation only and are not intended to limit the scope of the invention in any way. Preparation 1 : (3S.11 af?VΛ/-r(2.4-DifluoroDhenvnmethyll-6-hvdroxy-3-methyl-5.7-dioxo- 2,3,5,7, 11 ,11 a-hexahydroM ,31oxazolor3,2-alpyridori ,2-c/1pyrazine-8-carboxamide sodium salt (compound 1 b, scheme 2).
I) MsCI, Et3N

2) DBU
P-1 P-2 P-3

a) Synthesis of 2-methyl-3-[(phenylmethvl)oxvl-4/-/-pvran-4-one (compound P-2). To a slurry of 2000 g of compound P-1(1.0 eq.) in 14.0 L of MeCN were added 2848 g of benzyl bromide(1.05 eq.) and 2630 g of K2CO3(1.2 eq.). The mixture was stirred at 80 0C for 5 h and cooled to 13°C. Precipitate was filtered and washed with 5.0 L of MeCN. The filtrate was concentrated and 3.0 L of THF was added to the residue. The THF solution was concentrated to give 3585 g of crude compound P-2 as oil. Without further purification, compound P-2 was used in the next step. 1H NMR(300 MHz, CDCI3) δ 7.60 (d, J = 5.7 Hz, 1 H), 7.4-7.3 (m, 5H), 6.37 (d, J = 5.7 Hz, 1 H), 5.17 (s, 2H), 2.09 (s, 3H).
b) Synthesis of 2-(2-hydroxy-2-phenylethyl)-3-[(phenylmethyl)oxy]-4H-pyran-4-one (compound P-3). To 904 g of the crude compound P-2 was added 5.88 L of THF and the solution was cooled to -60 0C. 5.00 L of 1.0 M of Lithium bis(trimethylsilylamide) in THF(1.25 eq.) was added dropwise for 2 h to the solution of compound 2 at -60 0C. Then, a solution of 509 g of benzaldehyde(1.2 eq.) in 800 ml. of THF was added at -60 0C and the reaction mixture was aged at -60 0C for 1 h. The THF solution was poured into a mixture of 1.21 L of conc.HCI, 8.14 L of ice water and 4.52 L of EtOAc at less than 2 0C.
The organic layer was washed with 2.71 L of brine (twice) and the aqueous layer was extracted with 3.98 L of EtOAc. The combined organic layers were concentrated. To the mixture, 1.63 L of toluene was added and concentrated (twice) to provide toluene slurry of compound P-3. Filtration, washing with 0.90 L of cold toluene and drying afforded 955 g of compound P-3 (74% yield from compound P-1 ) as a solid. 1H NMR(300 MHz, CDCI3) δ
7.62 (d, J = 5.7 Hz, 1 H), 7.5-7.2 (m, 10H), 6.38 (d, J = 5.7 Hz, 1 H), 5.16 (d, J = 11.4 Hz, 1 H), 5.09 (d, J = 11.4 Hz, 1 H), 4.95 (dd, J = 4.8, 9.0 Hz, 1 H), 3.01 (dd, J = 9.0, 14.1 Hz, 1 H), 2.84 (dd, J = 4.8, 14.1 Hz, 1 H).
c) Synthesis of 2-[(£)-2-phenylethenyl]-3-[(phenylmethyl)oxy]-4H-pyran-4-one (compound
P-4). To a solution of 882 g of compound P-3 (1.0 eq.) in 8.82 L of THF were added 416 g of Et3N(1.5 eq.) and 408 g of methanesulfonyl chloride(1.3 eq.) at less than 30 0C. After confirmation of disappearance of compound P-3, 440 ml. of NMP and 1167 g of DBU(2.8 eq.) were added to the reaction mixture at less than 30 0C and the reaction mixture was aged for 30 min. The mixture was neutralized with 1.76 L of 16% sulfuric acid and the organic layer was washed with 1.76 L of 2% Na2S03aq. After concentration of the organic layer, 4.41 L of toluene was added and the mixture was concentrated (tree times). After addition of 4.67 L of hexane, the mixture was cooled with ice bath. Filtration, washing with 1.77 L of hexane and drying provided 780 g of compound P-4 (94% yield) as a solid. 1H NMR(300 MHz, CDCI3) δ 7.69 (d, J = 5.7 Hz, 1 H), 7.50-7.25 (m, 10H), 7.22 (d, J = 16.2
Hz, 1 H), 7.03 (d, J = 16.2 Hz, 1 H), 6.41 (d, J = 5.7 Hz, 1 H), 5.27 (s, 2H). d) Synthesis of 4-oxo-3-[(phenylmethyl)oxy]-4H-pyran-2-carboxylic acid (compound P-5). To a mixture of 822 g of compound P-4 (1.0 eq.) and 1 1.2 g of RuCI3-nH2O(0.02 eq.) in 2.47 L of MeCN, 2.47 L of EtOAc and 2.47 L of H2O was added 2310 g of NalO4(4.0 eq.) at less than 25 0C. After aging for 1 h, 733 g of NaCIO2(S-O eq.) was added to the mixture at less than 25 0C. After aging for 1 h, precipitate was filtered and washed with 8.22 L of
EtOAc. To the filtrate, 1.64 L of 50% Na2S203aq, 822 ml. of H2O and 630 ml. of coc.HCI were added. The aqueous layer was extracted with 4.11 L of EtOAc and the organic layers were combined and concentrated. To the residue, 4 L of toluene was added and the mixture was concentrated and cooled with ice bath. Filtration, washing with 1 L of toluene and drying provided 372 g of compound P-5 (56% yield) as a solid. 1H NMR(300 MHz,
CDCI3) δ 7.78 (d, J = 5.7 Hz, 1 H), 7.54-7.46 (m, 2H), 7.40-7.26 (m, 3H), 6.48 (d, J = 5.7 Hz, 1 H), 5.6 (brs, 1 H), 5.31 (s, 2H).
e) Synthesis of 1-(2,3-dihydroxypropyl)-4-oxo-3-[(phenylmethyl)oxy]-1 ,4-dihydro-2- pyridinecarboxylic acid (compound P-6). A mixture of 509 g of compound P-5 (1.0 eq.) and
407 g of 3-amino-propane-1 ,2-diol(2.5 eq.) in 1.53 L of EtOH was stirred at 65 0C for 1 h and at 80 0C for 6 h. After addition of 18.8 g of 3-Amino-propane-1 ,2-diol(0.1 eq.) in 200 ml. of EtOH, the mixture was stirred at 80 0C for 1 h. After addition of 18.8 g of 3-amino- propane-1 ,2-diol (0.1 eq.) in 200 ml. of EtOH, the mixture was stirred at 80 0C for 30 min. After cooling and addition of 509 ml. of H2O, the mixture was concentrated. To the residue,
2.54 L of H2O and 2.54 L of AcOEt were added. After separation, the aqueous layer was washed with 1.02 L of EtOAc. To the aqueous layer, 2.03 L of 12% sulfuric acid was added at less than 12 0C to give crystal of compound P-6. Filtration, washing with 1.53 L of cold H2O and drying provided 576 g of compound P-6 (83% yield) as a solid. 1H NMR(300 MHz, DMSO-de) δ 7.67 (d, J = 7.5 Hz, 1 H), 7.5-7.2 (m, 5H), 6.40 (d, J = 7.5 Hz, 1 H), 5.07
(s, 2H), 4.2-4.0 (m, 1 H), 3.9-3.6 (m, 2H), 3.38 (dd, J = 4.2, 10.8 Hz, 1 H), 3.27 (dd, J = 6.0, 10.8 Hz, 1 H).
f) Synthesis of methyl 1-(2,3-dihydroxypropyl)-4-oxo-3-[(phenylmethyl)oxy]-1 ,4-dihydro-2- pyridinecarboxylate (compound P-7). To a slurry of 576 g of compound P-6 (1.0 eq.: 5.8% of H2O was contained) in 2.88 L of NMP were added 431 g of NaHCO3(3.0 eq.) and 160 ml. of methyl iodide(1.5 eq.) and the mixture was stirred at room temperature for 4 h. After cooling to 5 0C, 1.71 L of 2N HCI and 1.15 L of 20% NaClaq were added to the mixture at less than 10 0C to give crystal of compound 7. Filtration, washing with 1.73 L of H2O and drying provided 507 g of compound P-7 (89% yield) as a solid. 1H NMR(300 MHz, DMSO- cfe) δ 7.59 (d, J = 7.5 Hz, 1 H), 7.40-7.28 (m, 5H), 6.28 (d, J = 7.5 Hz, 1 H), 5.21 (d, J = 5.4 Hz, 1 H), 5.12 (d, J = 10.8 Hz, 1 H), 5.07 (d, J = 10.8 Hz, 1 H), 4.83 (t, J = 5.7 Hz, 1 H), 3.97 (dd, J = 2.4, 14.1 Hz, 1 H), 3.79 (s, 3H), 3.70 (dd, J = 9.0, 14.4 Hz, 1 H), 3.65-3.50 (m, 1 H), 3.40-3.28 (m, 1 H), 3.26-3.14 (m, 1 H).
g) Synthesis of methyl 1-(2,2-dihydroxyethyl)-4-oxo-3-[(phenylmethyl)oxy]-1 ,4-dihydro-2- pyridinecarboxylate (compound P-8). To a mixture of 507 g of compound P -7 (1.0 eq.) in
5.07 L of MeCN, 5.07 L of H2O and 9.13 g of AcOH(0.1 eq.) was added 390 g of NaIO4(1.2 eq.) and the mixture was stirred at room temperature for 2 h. After addition of 1.52 L of 10% Na2S2OsBq., the mixture was concentrated and cooled to 10 0C. Filtration, washing with H2O and drying provided 386 g of compound P-8 (80% yield) as a solid. 1H NMR(300 MHz, DMSO-d6) δ 7.62 (d, J = 7.5 Hz, 1 H), 7.42-7.30 (m, 5H), 6.33 (d, J = 6.0 Hz, 2H),
6.29 (d, J = 7.5 Hz, 1 H), 5.08 (s, 2H), 4.95-4.85 (m, 1 H), 3.80 (s, 3H), 3.74 (d, J = 5.1 Hz, 2H).
h) Synthesis of (3S, 11 aR)-3-methyl-6-[(phenylmethyl)oxy]-2,3, 1 1 ,1 1a- tetrahydro[1 ,3]oxazolo[3,2-a]pyrido[1 ,2-c/]pyrazine-5,7-dione (compound P-9). After dissolution of mixture of 320 g of compound P-8 (1.0 eq.) in 3.20 L of MeOH by heating, the solution was concentrated. To the residue, 1.66 L of MeCN, 5.72 ml. of AcOH(0.1 eq.) and 82.6 g of (S)-2-Amino-propan-1-ol(1.1 eq.) were added and the mixture was heated to 70 0C, stirred at 70 0C for 4 h and concentrated. To the residue, 1.67 L of 2-propanol was added and the mixture was concentrated (twice). After cooling of the residue, filtration, washing with 500 ml. of cold 2-propanol and drying provided 167 g of compound P-9 (52% yield) as a solid. 1H NMR(300 MHz, CDCI3) δ 7.61-7.55 (m, 2H), 7.40-7.20 (m, 4H), 6.53 (d, J = 7.2, 1 H), 5.46 (d, J = 10.5 Hz, 1 H), 5.23 (d, J = 10.2 Hz, 1 H), 5.20 (dd, J = 3.9, 9.6 Hz, 1 H), 4.46-4.34 (m, 1 H), 4.31 (dd, J = 6.6, 8.7 Hz, 1 H), 4.14 (dd, J = 3.9, 12.3 Hz, 1 H), 3.79 (dd, J = 9.9, 12.3 Hz, 1 H), 3.62 (dd, J = 6.9, 8.7 Hz, 1 H), 1.38 (d, J = 6.3 Hz, 3H).
i) Synthesis of (3 S, 1 1 aR)-8-bromo-3-methyl-6-[(phenylmethyl)oxy]-2,3, 11 ,11a- tetrahydro[1 ,3]oxazolo[3,2-a]pyrido[1 ,2-c/]pyrazine-5,7-dione (compound P-10). To slurry of 156 g of compound P-9 (1.0 eq.) in 780 ml. of NMP was added 93.6 g of NBS(1.1 eq.) and the mixture was stirred at room temperature for 2.5 h. The reaction mixture was added to 3.12 L of H2O. Filtration, washing with 8.0 L of H2O and drying provided 163 g of compound P-10 (84% yield) as a solid. 1H NMR(300 MHz, DMSO-d6) δ 8.37 (s, 1 H), 7.55- 7.50 (m, 2H), 7.42-7.25 (m, 3H), 5.34 (dd, J = 3.6, 9.9 Hz, 1 H), 5.18 (d, J = 10.8 Hz, 1 H), 5.03 (d, J = 10.5 Hz, 1 H), 4.53 (dd, J = 3.6, 12.0 Hz, 1 H), 4.40-4.20 (m, 2H), 3.99 (dd, J = 9.9, 1 1.7 Hz, 1 H), 3.64 (dd, J = 5.7, 8.1 Hz, 1 H), 1.27 (d, J = 6.3 Hz, 3H). j) Synthesis of (3S,1 1aS)-Λ/-[(2,4-difluorophenyl)methyl]-3-methyl-5,7-dioxo-6- [(phenylmethyl)oxy]-2,3,5,7, 11 ,1 1 a-hexahydro[1 ,3]oxazolo[3,2-a]pyrido[1 ,2-c/]pyrazine-8- carboxamide (compound P-11). Under carbon mono-oxide atmosphere, a mixture of 163 g of compound P-10 (1.0 eq.), 163 mL of /-Pr2NEt(2.5 eq.), 68.4 mL of 2,4- difluorobenzylamine(1.5 eq.) and 22.5 g of Pd(PPh3)4(0.05 eq.) in 816 mL of DMSO was stirred at 90 0C for 7 h. After cooling, removal of precipitate, washing with 50 mL of DMSO and addition of 1 1.3 g of Pd(PPh3)4(0.025 eq.), the reaction mixture was stirred at 90 0C for 2 h under carbon mono-oxide atmosphere again. After cooling, removal of precipitate and addition of 2.0 L of AcOEt and 2.0 L of H2O, the organic layer was washed with 1.0 L of 1 N HCIaq. and 1.0 L of H2O (twice) and the aqueous layer was extracted with 1.0 L of AcOEt.
The organic layers were combined and concentrated. Silica gel column chromatography of the residue provided 184 g of compound P-11 (96% yield) as foam. 1H NMR(300 MHz, CDCI3) δ 10.38 (t, J = 6.3 Hz, 1 H), 8.39 (s, 1 H), 7.75-7.25 (m, 7H), 6.90-6.70 (m, 2H), 5.43 (d, J = 10.2 Hz, 1 H), 5.24 (d, J = 10.2 Hz, 1 H), 5.19 (dd, J = 3.9, 9.9 Hz, 1 H), 4.63 (d, J = 6.0 Hz, 2H), 4.50-4.25 (m, 3H), 3.86 (dd, J = 9.9, 12.3 Hz, 1 H), 3.66 (dd, J = 6.9, 8.4 Hz,
1 H), 1.39 (d, J = 6.0 Hz, 3H).
k) Synthesis of (3S,1 1aR)-Λ/-[(2,4-difluorophenyl)methyl]-6-hydroxy-3-methyl-5,7-dioxo- 2,3,5,7, 11 ,11 a-hexahydro[1 ,3]oxazolo[3,2-a]pyrido[1 ,2-c/]pyrazine-8-carboxamide (compound 1a). Under hydrogen atmosphere, a mixture of 184 g of compound P-11 (1.0 eq.) and 36.8 g of 10%Pd-C in 3.31 L of THF and 0.37 L of MeOH was stirred for 3 h. After filtration of precipitate(Pd-C), washing with THF/MeOH(9/1 ) and addition of 36.8 g of 10% Pd-C, the mixture was stirred for 20 min under hydrogen atmosphere. After filtration of precipitate(Pd-C) and washing with THF/MeOH(9/1 ), the filtrate was concentrated. After 200 mL of AcOEt was added to the residue, filtration afforded crude solid of compound 1 a.
The precipitates were combined and extracted with 4.0 L of CHCI3/Me0H(5/1 ). After concentration of the CHCI3/MeOH solution and addition of 250 mL of AcOEt to the residue, filtration afforded crude solid of compound 1a. The crude solids were combined and dissolved in 8.2 L of MeCN/H2O(9/1 ) by heating. After filtration, the filtrate was concentrated. To the residue, 1.5 L of EtOH was added and the mixture was concentrated
(three times). After cooling of the residue, filtration and drying provided 132 g of compound 1a (88% yield) as a solid. 1H NMR(300 MHz, DMSO-d6) δ 11.47 (brs, 1 H), 10.31 (t, J = 6.0 Hz, 1 H), 8.46 (s, 1 H), 7.40 (td, J = 8.6, 6.9 Hz, 1 H), 7.24 (ddd, J = 2.6, 9.4, 10.6, 1 H), 7.11- 7.01 (m, 1 H), 5.39 (dd, J = 4.1 , 10.4 Hz, 1 H), 4.89 (dd, J = 4.2, 12.3 Hz, 1 H), 4.55 (d, J = 6.0 Hz, 2H), 4.40 (dd, J = 6.8, 8.6 Hz, 1 H), 4.36-4.22 (m, 1 H), 4.00 (dd, J = 10.2, 12.3 Hz, 1 H), 3.67 (dd, J = 6.7, 8.6 Hz, 1 H), 1.34 (d, J = 6.3 Hz, 3H).
I) Synthesis of (3S,1 1aR)-Λ/-[(2,4-difluorophenyl)methyl]-6-hydroxy-3-methyl-5,7-dioxo- 2,3,5,7, 11 ,11 a-hexahydro[1 ,3]oxazolo[3,2-a]pyrido[1 ,2-c/]pyrazine-8-carboxamide sodium salt (compound 1 b). After dissolution of 16.0 g of compound 1a (1.0 eq.) in 2.56 L of EtOH and 0.64 L of H2O by heating, followed by filtration, 39 ml. of 1 N NaOHaq.(1.0 eq.) was added to the solution at 75 0C. The solution was gradually cooled to room temperature. Filtration, washing with 80 ml. of EtOH and drying provided 13.5 g of compound 1b (80% yield) as a solid. 1H NMR(300 MHz, DMSO-d6) δ 10.73 (t, J = 6.0 Hz, 1 H), 7.89 (s, 1 H), 7.40-7.30 (m, 1 H), 7.25-7.16 (m, 1 H), 7.07-6.98 (m, 1 H), 5.21 (dd, J = 3.8, 10.0 Hz, 1 H), 4.58 (dd, J = 3.8, 12.1 Hz, 1 H), 4.51 (d, J = 5.4 Hz, 2H), 4.30-4.20 (m, 2H), 3.75 (dd, J = 10.0, 12.1 Hz, 1 H), 3.65-3.55 (m, 1 H), 1.27 (d, J = 6.1 Hz, 3H).
updates

An Efficient and Highly Diastereoselective Synthesis of GSK1265744, a Potent HIV Integrase Inhibitor
†Global API Chemistry, ‡MDR Chemical Science,§Analytical Sciences, GlaxoSmithKline, 709 Swedeland Road, King of Prussia, Pennsylvania 19406, United States
Org. Lett., Article ASAP
DOI: 10.1021/ol503580t
Publication Date (Web): January 23, 2015……..http://pubs.acs.org/doi/abs/10.1021/ol503580t
Copyright © 2015 American Chemical Society
*E-mail: huan.2.wang@gsk.com.
A novel synthesis of GSK1265744, a potent HIV integrase inhibitor, is described. The synthesis is highlighted by an efficient construction of the densely functionalized pyridinone core as well as a highly diastereoselective formation of the acyl oxazolidine moiety. The latter exploits the target molecule’s ability to chelate to Mg2+, a key feature in the integrase inhibitor’s mechanism of action.
PAPER
Abstract

Bictegravir and dolutegravir are two recently approved integrase inhibitors for the treatment of HIV. A third inhibitor, cabotegravir, is in Phase 3 development. As a continuation of a series of articles on synthetic routes to newly approved drugs, the current article reviews the patent and journal literature regarding synthetic routes and final forms of these drug





References
- ^ Jump up to:a b c “Vocabria (cabotegravir) film-coated tablets Product Information”. TGA eBS. 12 June 2021. Retrieved 12 June 2021.
- ^ Jump up to:a b “Vocabria”. Therapeutic Goods Administration (TGA). 26 February 2021. Retrieved 8 September 2021.
- ^ “Summary for ARTG Entry: 323721 VOCABRIA cabotegravir (as sodium) 30 mg film-coated tablet, bottle”. Therapeutic Goods Administration. The G overnment of Australia. 13 August 2021.
- ^ “Vocabria Product information”. Health Canada. 25 April 2012. Retrieved 22 January 2021.
- ^ Jump up to:a b c d e f g “Vocabria- cabotegravir sodium tablet, film coated”. DailyMed. Retrieved 12 June 2021.
- ^ Jump up to:a b “FDA Approves First Extended-Release, Injectable Drug Regimen for Adults Living with HIV”. U.S. Food and Drug Administration (FDA) (Press release). 21 January 2021. Retrieved 21 January 2021. This article incorporates text from this source, which is in the public domain.
- ^ “Apretude- cabotegravir kit”. DailyMed. Retrieved 24 December 2021.
- ^ Jump up to:a b “Vocabria EPAR”. European Medicines Agency (EMA). Retrieved 22 January 2021. Text was copied from this source which is © European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ Jump up to:a b c d e f g h i “Vocabria: EPAR – Product information” (PDF). European Medicines Agency. 5 January 2021.
- ^ Borrell B (March 2014). “Long-acting shot prevents infection with HIV analogue”. Nature News. doi:10.1038/nature.2014.14819. S2CID 184399045.
- ^ Jump up to:a b c d e f g h i j k l m “FDA Approves First Injectable Treatment for HIV Pre-Exposure Prevention”. U.S. Food and Drug Administration (FDA) (Press release). 20 December 2021. Retrieved 21 December 2021. This article incorporates text from this source, which is in the public domain.
- ^ Jump up to:a b c “First long-acting injectable antiretroviral therapy for HIV recommended approval” (Press release). European Medicines Agency (EMA). 16 October 2020. Retrieved 16 October 2020. Text was copied from this source which is © European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ “Cabenuva- cabotegravir and rilpivirine kit”. DailyMed. Retrieved 12 June 2021.
- ^ “Edurant- rilpivirine hydrochloride tablet, film coated”. DailyMed. Retrieved 12 June 2021.
- ^ “Rilpivirine Monograph for Professionals”. Drugs.com. 24 September 2020. Retrieved 12 June 2021.
- ^ “A Treatment Option | CABENUVA (cabotegravir; rilpivirine)”. http://www.cabenuva.com. Retrieved 16 April 2022.
- ^ Patel M, Eberl HC, Wolf A, Pierre E, Polli JW, Zamek-Gliszczynski MJ (August 2019). “Mechanistic Basis of Cabotegravir-Glucuronide Disposition in Humans”. The Journal of Pharmacology and Experimental Therapeutics. 370 (2): 269–277. doi:10.1124/jpet.119.258384. PMID 31175220. S2CID 182950312.
- ^ Jump up to:a b “Vocabria: EPAR – Public assessment report” (PDF). European Medicines Agency. 5 January 2021.
- ^ “HPTN083 — Prevention Now”. HIV Prevention Trials Network. Retrieved 2 December 2017.
- ^ “HPTN084 — Prevention Now”. HIV Prevention Trials Network. Retrieved 2 December 2017.
- ^ “Vocabria: Pending EC decision”. European Medicines Agency (EMA). 16 October 2020. Retrieved 16 October 2020. Text was copied from this source which is © European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ “Adopted USANs” (PDF). American Medical Association. Retrieved 19 September 2014.
- ^ World Health Organization (2015). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 73”. WHO Drug Information. 29 (1): 70–1. hdl:10665/331088.
- ^ Ryan G (7 July 2020). “Injectable PrEP Is Even More Effective Than Daily Truvada”. Poz. Retrieved 9 November 2020.
- ^ Ryan G (9 November 2020). “For Women, Injectable Cabotegravir Is More Effective Than Truvada as PrEP”. Poz. Retrieved 9 November 2020.
External links
- “Cabotegravir”. Drug Information Portal. U.S. National Library of Medicine.
References
- PrEP GSK744 Integrase Administered Monthly Perhaps Quarterly Prevents HIV-Infection in Monkeys. 20th Conference on Retroviruses and Opportunistic Infections. Atlanta, GA March 3–6, 2013.
- http://www.natap.org/2013/CROI/croi_38.htm
SMILES [H][C@@]12CN3C=C(C(=O)NCC4=C(F)C=C(F)C=C4)C(=O)C(O)=C3C(=O)N1[C@@H](C)CO2
| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2006116764A1 * | Apr 28, 2006 | Nov 2, 2006 | Shionogi & Co | Polycyclic carbamoylpyridone derivative having hiv integrase inhibitory activity |
| US6919351 * | Oct 9, 2001 | Jul 19, 2005 | Merck & Co., Inc. | Aza-and polyaza-naphthalenyl-carboxamides useful as HIV integrase inhibitors |
| WO2012018065A1 * | Aug 4, 2011 | Feb 9, 2012 | Shionogi & Co., Ltd. | Process for preparing compound having hiv integrase inhibitory activity |
| WO2012151361A1 | May 3, 2012 | Nov 8, 2012 | Concert Pharmaceuticals Inc. | Carbamoylpyridone derivatives |
| WO2013038407A1 * | Sep 2, 2012 | Mar 21, 2013 | Mapi Pharma Ltd. | Amorphous form of dolutegravir |
| US8552187 | Dec 9, 2009 | Oct 8, 2013 | Shionogi & Co., Ltd. | Processes and intermediates for carbamoylpyridone HIV integrase inhibitors |
| US8580967 | Jul 23, 2009 | Nov 12, 2013 | Shionogi & Co., Ltd. | Methyl 3-(benzyloxy)-1-(2,2-dihydroxyethyl)-4-oxo-1,4-dihydropyridine-2-carboxylate and processes for the preparation thereof |
| US8624023 | Dec 8, 2009 | Jan 7, 2014 | Shionogi & Co., Ltd. | Synthesis of carbamoylpyridone HIV integrase inhibitors and intermediates |
SYN
Synthetic Reference
Dolutegravir HELP . ADDED TO AID HELP
Dolutegravir HELP . ADDED TO AID HELP
Dolutegravir HELP . ADDED TO AID HELP
Dolutegravir HELP . ADDED TO AID HELP
Aoyama, Yasunori; Hakogi, Toshikazu; Fukui, Yuki; Yamada, Daisuke; Ooyama, Takao; Nishino, Yutaka; Shinomoto, Shoji; Nagai, Masahiko; Miyake, Naoki; Taoda, Yoshiyuki; Yoshida, Hiroshi; Yasukata, Tatsuro. Practical and Scalable Synthetic Method for Preparation of Dolutegravir Sodium: Improvement of a Synthetic Route for Large-Scale Synthesis. Organic Process Research & Development. Volume 23. Issue 4. Pages 558-564. Journal; Online Computer File. (2019).
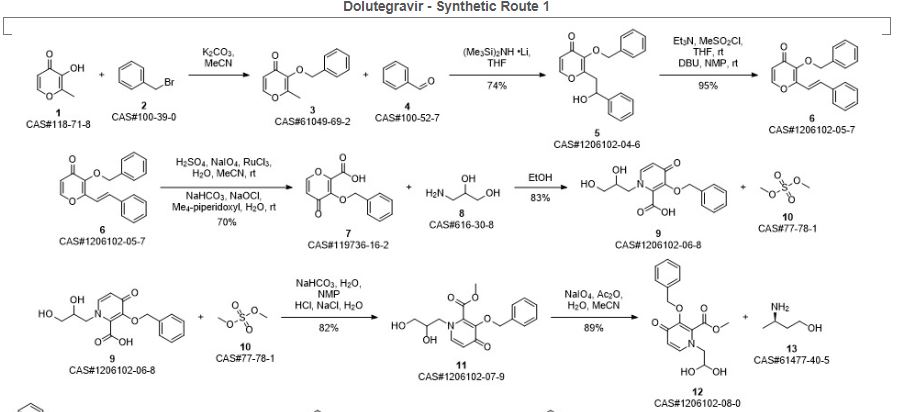
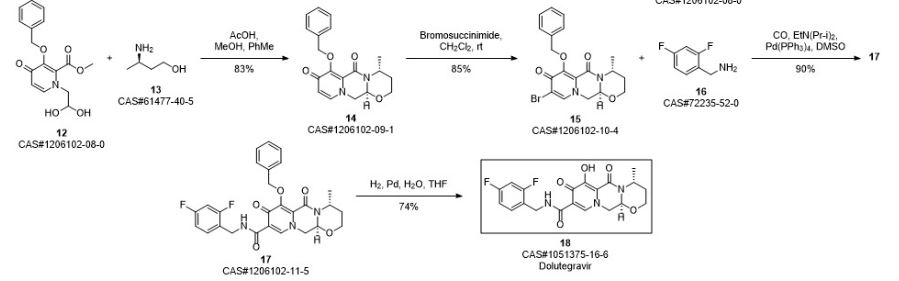
Synthetic Reference 2
Wang, Xianheng; Chen, Song; Zhao, Changkuo; Long, Liangye; Wang, Yuhe. Preparation of Dolutegravir Intermediate Diastereomer. Journal of Heterocyclic Chemistry. Volume 56. Issue 7. Pages 2063-2067. Journal; Online Computer File. (2019).

Synthetic Reference 3
Ziegler, Robert E.; Desai, Bimbisar K.; Jee, Jo-Ann; Gupton, B. Frank; Roper, Thomas D.; Jamison, Timothy F. 7-Step Flow Synthesis of the HIV Integrase Inhibitor Dolutegravir. Angewandte Chemie, International Edition. Volume 57. Issue 24. Pages 7181-7185. Journal; Online Computer File. (2018).

SYN 4
Synthetic Reference
Rajan, Srinivasan Thirumalai; Eswaraiah, Sajja; Reddy, Ghojala Venkat; Reddy, Sagyam Rajeshwar; Markandeya, Bekkam; Rajesham, Boge. Novel crystalline polymorph of sodium (4R,12aS)-9-{[(2,4-difluorophenyl)methyl]carbamoyl}-4-methyl-6,8-dioxo-3,4,6,8,12,12a-hexahydro-2H-pyrido[1′,2′:4,5]pyrazino[2,1-b][1,3]oxazin-7-olate and process for preparation thereof. Assignee MSN Research & Development Center, India. IN 201641037221. (2018).

Synthetic Reference 5
Sharma, Pramodkumar; Rao, Bhatraju Srinivasa; Deo, Keshav. A process for the preparation of Dolutegravir or its pharmaceutical acceptable salts thereof. Assignee Wockhardt Limited, India. IN 2015MU01007. (2016).

Synthetic Reference 6
Weaver, Jimmie Dean. Preparation of fluoroarenes via hydrogen bond directed photocatalytic hydrodefluorination of perfluoroarenes. Assignee The Board of Regents for Oklahoma State University, USA. WO 2018187336. (2018).

SYN 7
Synthetic Reference
Li, Xuguang; Chen, Shuiku; Zhu, Songlin; Zhang, Fangjie; Fang, Shuixia; Liu, Congjun; Zhu, Huifeng; Luo, Qi; Meng, Qingyue; Cui, Hao. Method for preparation of dolutegravir. Kaifeng Pharmaceutical (Group) Co., Ltd.; Henan Furen Pharmaceutical Technology Development Co., Ltd. CN 108299466. (2018).


SYN 8
Synthetic Reference
Vellanki, Sivaram Prasad; Nadella, Madumurthy; Bhalme, Mitali; Ramabhotla, Revathi Srinivas. Process for the preparation of dolutegravir, an integrase inhibitor for HIV-1 infection therapy. Assignee Mylan Laboratories Ltd., India. IN 2015CH00588. (2016).
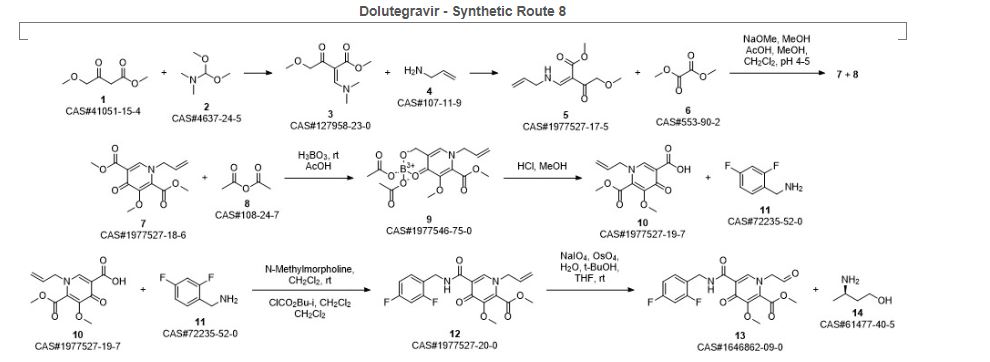

SYN 9
Synthetic Reference
Sankareswaran, Srimurugan; Mannam, Madhavarao; Chakka, Veerababu; Mandapati, Srirami Reddy; Kumar, Pramod. Identification and Control of Critical Process Impurities: An Improved Process for the Preparation of Dolutegravir Sodium. Organic Process Research & Development. Volume 20. Issue 8. Pages 1461-1468. Journal; Online Computer File. (2016).
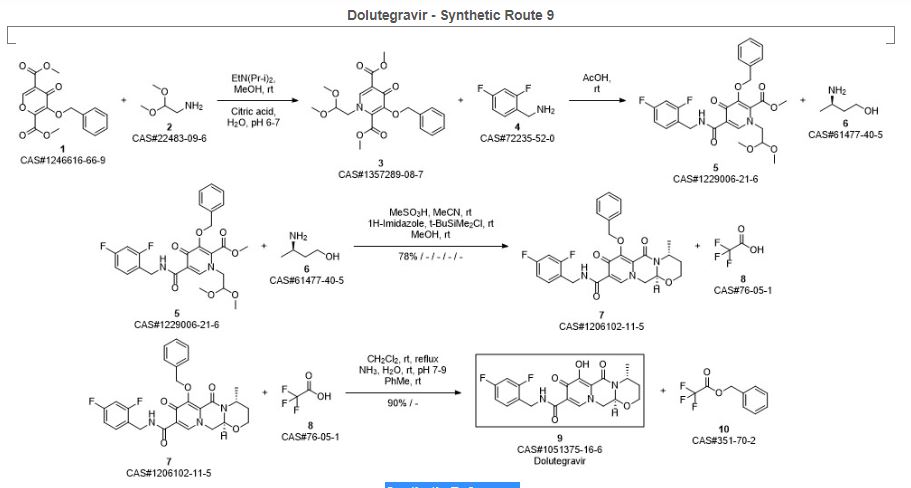
TOLVAPTAN

TOLVAPTAN
的合成
N-(4-{[(5R)-7-chloro-5-hydroxy-2,3,4,5-tetrahydro-1H-1-benzazepin-1-yl]carbonyl}-3-methylphenyl)-2-methylbenzamide
| Formula | C26H25ClN2O3 |
|---|---|
| Mol. mass | 448.941 g/mol |
150683-30-0 CAS NO
+ form 331947-66-1 Rform
OPC-41061
Otsuka…..innovator
UPDATE 2022
Tolvaptan sodium phosphate, Samtasu,
|
トルバプタンリン酸エステルナトリウム
|
| 2022/3/28 JAPAN PPROVED |
| Formula |
C26H24ClN2O6P. 2Na
|
|---|---|
| CAS |
|
| Mol weight |
572.8849
|
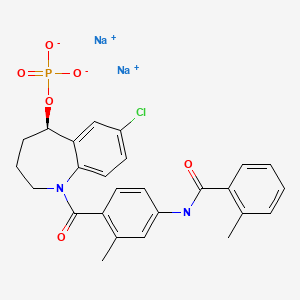
Tolvaptan sodium phosphate
disodium;[(5R)-7-chloro-1-[2-methyl-4-[(2-methylbenzoyl)amino]benzoyl]-2,3,4,5-tetrahydro-1-benzazepin-5-yl] phosphate
European Medicines Agency (EMA) Accepts Otsuka’s Marketing Authorisation Application (MAA) for Tolvaptan, an Investigational Compound for Autosomal Dominant Polycystic Kidney Disease (ADPKD)
•Tolvaptan was discovered by Otsuka in Japan and, if approved by the EMA, would become the first pharmaceutical therapy in Europe for patients with ADPKD
•ADPKD is an inherited genetic disease that causes cyst growth in the kidneys, which gradually impairs their functioning. There is no current pharmaceutical treatment option
•Otsuka’s development of tolvaptan as a treatment for ADPKD illustrates the company’s commitment to address significant patient needs for diseases that traditionally have not been a priority for the pharmaceutical industry
TOKYO–(BUSINESS WIRE)–Otsuka Pharmaceutical Co., Ltd. announced today that the European Medicines Agency (EMA) has accepted the submission of a marketing authorisation application (MAA) for the potential approval of tolvaptan for the treatment of autosomal dominant polycystic kidney disease (ADPKD). Phase III clinical trial results that form the basis of the regulatory filing were published in the New England Journal of Medicine.
http://www.pharmalive.com/ema-accepts-otsukas-maa-for-tolvaptan
Tolvaptan is a selective vasopressin V2-receptor antagonist with an affinity for the V2-receptor that is 1.8 times that of native arginine vasopressin (AVP).
Tolvaptan is (±)-4′-[(7-chloro-2,3,4,5-tetrahydro-5-hydroxy-1H-1-benzazepin-1-yl) carbonyl]-otolu-m-toluidide. The empirical formula is C26H25ClN2O3. Molecular weight is 448.94. The chemical structure is:
 |
SAMSCA tablets for oral use contain 15 mg or 30 mg of tolvaptan. Inactive ingredients include corn starch, hydroxypropyl cellulose, lactose monohydrate, low-substituted hydroxypropyl cellulose, magnesium stearate and microcrystalline cellulose and FD&C Blue No. 2 Aluminum Lake as colorant.
SEE NEW UPDATE AT END OF PAGE
Tolvaptan (INN), also known as OPC-41061, is a selective, competitive vasopressin receptor 2 antagonist used to treat hyponatremia (low blood sodium levels) associated withcongestive heart failure, cirrhosis, and the syndrome of inappropriate antidiuretic hormone(SIADH). Tolvaptan was approved by the U.S. Food and Drug Administration (FDA) on May 19, 2009, and is sold by Otsuka Pharmaceutical Co. under the trade name Samsca and in India is manufactured & sold by MSN laboratories Ltd. under the trade name Tolvat & Tolsama.
Tolvaptan is also in fast-track clinical trials[2] for polycystic kidney disease. In a 2004 trial, tolvaptan, when administered with traditional diuretics, was noted to increase excretion of excess fluids and improve blood sodium levels in patients with heart failure without producing side effects such as hypotension (low blood pressure) or hypokalemia(decreased blood levels of potassium) and without having an adverse effect on kidney function.[3] In a recently published trial (TEMPO 3:4 ClinicalTrials.gov number, NCT00428948) the study met its primary and secondary end points. Tolvaptan, when given at an average dose of 95 mg per day over a 3-year period, slowed the usual increase in kidney volume by 50% compared to placebo (2.80% per year versus 5.51% per year, respectively, p<0.001) and reduced the decline in kidney function when compared with that of placebo-treated patients by approximately 30% (reciprocal serum creatinine, -2.61 versus -3.81 (mg/mL)-1 per year, p <0.001)[4]
Tolvaptan was first approved by the U.S. Food and Drug Administration (FDA) on May 19, 2009, then approved by the European Medicines Agency (EMA) on August 3, 2009 and approved by Pharmaceuticals and Medical Devices Agency of Japan on Feb 4, 2013. It was developed and marketed as Samsca® by Otsuka in the US, DE and JP.
UPDATED
Tolvaptan is a selective vasopressin V2-receptor antagonist with an affinity for the V2-receptor that is 1.8 times that of native arginine vasopressin (AVP) and that is 29 times greater than for the V1a-receptor. When taken orally, 15 to 60 mg doses of tolvaptan antagonize the effect of vasopressin and cause an increase in urine water excretion that results in an increase in free water clearance (aquaresis), a decrease in urine osmolality, and a resulting increase in serum sodium concentrations. It is indicated for the treatment of clinically significant hypervolemic and euvolemic hyponatremia [serum sodium < 125 mEq/L or less marked hyponatremia that is symptomatic and has resisted correction with fluid restriction], including patients with heart failure, cirrhosis, and syndrome of inappropriate antidiuretic hormone (SIADH).
Samsca® is available as tablet for oral use, containing 7.5 mg/15 mg/30 mg of free Tolvaptan. The recommended starting dose is 15 mg once daily and it may be increased at intervals ≥ 24 hr to 30 mg once daily, and to a maximum of 60 mg once daily as needed to raise serum sodium.
| PATENT NUMBER | PEDIATRIC EXTENSION | APPROVED | EXPIRES (ESTIMATED) | |
|---|---|---|---|---|
| US5258510 | No | 1993-11-02 | 2010-11-02 |  |
| US5753677 | No | 1998-05-19 | 2020-05-19 | |
| US8501730 | No | 2013-08-06 | 2026-09-01 | |
| US5972882 | No | 1999-10-26 | 2018-12-14 | |
| US10905694 | No | 2021-02-02 | 2030-04-07 | |
Synthesis Reference
Bandi Parthasaradhi Reddy, “PROCESS FOR PREPARING TOLVAPTAN INTERMEDIATES.” U.S. Patent US20130190490, issued July 25, 2013.
US20130190490
Route 1


Reference:1. US5258510A.
Route 2


Reference:1. Bio. Med. Chemistry 2006, 14, 6165–6173.
Route 3


Reference:1. Bio. Med. Chem. Lett. 2007, 17, 6455–6458.
Route 4


Reference:1. CN102060769B.
Route 5


Reference:1. Org. Lett. 2014, 16, 6041−6043.
Route 6


Reference:1. Bio. Med. Chem. 1999, 7, 1743-1754.
2. WO2007026971A2.
SYN
SYN
Chemical synthesis:[5] 
Tolvaptan is chemically, N-[4-[(7-chloro-2,3,4,5-tetrahydro-5-hydroxy1H-1-benzazepin-1-yl)carbonyl]-3-methylphenyl]-2-methylbenzamide. Tolvaptan is represented by the following structure:
Tolvaptan, also known as OPC-41061, is a selective, competitive arginine vasopressin receptor 2 antagonist used to treat hyponatremia (low blood sodium levels) associated with congestive heart failure, cirrhosis, and the syndrome of inappropriate antidiuretic hormone (SIADH). Tolvaptan is sold by Otsuka Pharmaceutical Co. under the trade name Samsca.
Tolvaptan and its process for preparation were disclosed in U.S. Pat. No. 5,258,510.
Processes for the preparation of 7-chloro-2,3,4,5-tetrahydro-1H-1-benzazepin-5-one, 7-chloro-1-(2-methyl-4-nitrobenzoyl)-5-oxo-2,3,4,5-tetrahydro-1H-1-benzazepine and 7-chloro-1-[2-methyl-4-[(2-methylbenzoyl)amino]benzoyl]-5-oxo-2,3,4,5-tetrahydro-1H-1-benzazepine were reported in Bioorganic & medicinal chemistry 7 (1999), 1743-1754. According to the journal, 7-chloro-2,3,4,5-tetrahydro-1H-1-benzazepin-5-one can be prepared by reacting 7-chloro-4-ethoxycarbonyl-5-oxo-N-p-toluenesufonyl-2,3,4,5-tetrahydro-1H-1-benzazepine with acetic acid in the presence of hydrochloric acid and water to obtain 7-chloro-5-oxo-2,3,4,5-tetrahydro-1-p-toluenesulfonyl-1H-1-benzazepine, and then reacted with polyphospholic acid. According to the journal, 7-chloro-1-(2-methyl-4-nitrobenzoyl)-5-oxo-2,3,4,5-tetrahydro-1H-1-benzazepine can be prepared by reacting 7-chloro-5-oxo-2,3,4,5-tetrahydro-1H-1-benzazepine with 2-methyl-4-nitobenzoyl chloride in the presence of triethylamine.
According to the journal, 7-chloro-1-[2-methyl-4-[(2-methylbenzoyl)amino]benzoyl]-5-oxo-2,3,4,5-tetrahydro-1H-1-benzazepine can be prepared by reacting 1-(4-amino-2-methylbenzoyl)-7-chloro-5-oxo-2,3,4,5-tetrahydro-1H-1-benzazepine with 2-methylbenzoylchloride in the presence of triethylamine.
PCT publication no. WO 2007/026971 disclosed a process for the preparation oftolvaptan can be prepared by the reduction of 7-chloro-1-[2-methyl-4-(2-methylbenzoylamino)benzoyl]-2,3,4,5-tetrahydro-1H-1-benzazepin-5-one with sodium borohydride.
7-Chloro-2,3,4,5-tetrahydro-1H-1-benzazepin-5-one is a key intermediate for the preparation of tolvaptan.
Biooganic and Medicinal Chemistry I (2007) 6455-6458, Biooganic andMedicinal Chemistry 14 (2000) 2493-2495 reported in the literature of the intermediate 2 – carboxylic acid -5 – (2 – methyl-benzoylamino) toluene synthesis method,


5-Chloro-2-nitrobenzoic acid (I) was converted into methyl ester (II) using dimethyl sulfate and K2CO3 in acetone. The nitro group of (II) was then reduced with SnCl2 to afford aniline (III), which was protected as the p-toluenesulfonamide (IV) with tosyl chloride in pyridine. Alkylation of (IV) with ethyl 4-bromobutyrate (V) yielded diester (VI). Subsequent Dieckmann cyclization of (VI) in the presence of potassium tert-butoxide provided benzazepinone (VIIa-b) as a mixture of ethyl and methyl esters, which was decarboxylated to (VIII) by heating with HCl in AcOH. Deprotection of the tosyl group of (VIII) was carried out in hot polyphosphoric acid. The resulting benzazepinone (IX) was condensed with 2-methyl-4-nitrobenzoyl chloride (X) to give amide (XI). After reduction of the nitro group of (XI) to the corresponding aniline (XII), condensation with 2-methylbenzoyl chloride (XIII) provided diamide (XIV). Finally, ketone reduction in (XIV) by means of NaBH4 led to the target compound.
……………………………………….
PATENT

……………………………………………..
PATENT

Synthesis of Intermediate III: 1.
Example
2-methyl-4-nitrobenzoic acid (available from Alfa Aesar Tianjin Chemical Co., purity> 99%, 25g,
0.14mol) was added to a 250ml reaction flask, is reacted with thionyl chloride under reflux conditions for 3h, thionyl chloride was distilled off under reduced pressure to give 2-methyl-4-nitrobenzoyl chloride (26.Sg, light yellow oily liquid), without purification, was used directly in the next step.
Intermediate II (20g, 0.1moI) and 2_ methyl _4_ nitrobenzoylchloride (22.4g, 0.llmol) was added to a 250ml reaction flask. Dichloromethane (50ml), cooled to ice bath with stirring to dissolve O~5 ° C, was slowly added dropwise N- methylmorpholine (11.2g, 0.llmol), Bi dropwise with stirring while, at room temperature the reaction 4h. TLC [developing solvent: ethyl acetate – petroleum ether (I: I), hereinafter] is displayed after completion of the reaction, saturated aqueous sodium bicarbonate (20ml), stirred for lOmin, filtered, the filter cake with dichloromethane (15ml X 2 ) washing. The filtrate and washings were combined, washed with saturated sodium chloride solution (30ml X 3), dried over anhydrous sodium sulfate and filtered. The filtrate under reduced pressure to recover the solvent, the residue was recrystallized from anhydrous methanol to give a white powder 111 (27.5g, 75.2%), mp 154.8 ~155.6 ° C. Purity 97.9% (HPLC normalization method).
Synthesis of Intermediate IV:
Intermediate III (10g, 28mmol) was added to a 250ml reaction flask, concentrated hydrochloric acid (40ml) and ethanol (50ml), with stirring, was slowly added dropwise stannous chloride (20g, 88mmol) in ethanol (40ml) . Bi room temperature drops 5h. After TLC showed completion of the reaction, ethanol was distilled off under reduced pressure to about 70ml, the residue was -10 ° C -0 ° C allowed to stand overnight to cool. Filtered, and the filter cake was washed with water poured into water (40ml) in. Plus 20% sodium hydroxide solution (approximately 60ml) was adjusted to pH 9. Filtered, washed with ethanol and recrystallized to give a pale yellow powdered solid IV (6.3g, 68.7%), mp 190.4~191.1 ° C. Purity 97.2% (HPLC normalization method).
Synthesis of intermediate V:
Intermediate IV (5g, 15mmol) and triethylamine (2.3g, 23mmol) was added followed by IOOml reaction flask was added dichloromethane (30ml), stir until dissolved. Solution of o-methylbenzoyl chloride (2.8g, 18mmol), dropwise at room temperature completion of the reaction Ih0 TLC showed the reaction was complete was poured into ice-water (about 40ml) in, (20ml X 3) and extracted with dichloromethane, the combined organic phases, and saturated sodium chloride solution successively (25ml X 3), dried over anhydrous sodium sulfate and filtered with 5% hydrochloric acid (25ml X 3). The filtrate under reduced pressure to recover the solvent (about 50ml), dried over anhydrous methanol residue – petroleum ether (2: 1) and recrystallized to give white crystals of Intermediate V (6.2g, 90.9%), mp 121.1 ~123.6 ° C. Purity 98.6% (HPLC normalization method).
Synthesis of tolvaptan: Example 4
Intermediate V (5g, Ilmmol) IOOml added to the reaction flask, was added anhydrous methanol (25ml), stirred and then added portionwise sodium borohydride (0.65g, 17mmol) to the reaction mixture, addition was complete the reaction at room temperature lh. After TLC showed the reaction was complete, the methanol recovered under reduced pressure (approximately 20ml), the residue was added methylene chloride (25ml), (25mlX3) and washed with saturated sodium chloride solution. Anhydrous sodium sulfate and filtered, and the filtrate under reduced pressure to recover the solvent, the residue with absolute methanol – petroleum ether (2: 1) and recrystallized tolvaptan white crystals (4.85g, 96.6%), mp 220.1~221.5 ° C. Purity 99.2% (HPLC normalization method). ES1-HRMS (C26H25C1N203, m / z) found (calc): 447.1476 (447.1481) [MH] – “
…………..
PATENT
http://www.google.com/patents/WO2012046244A1?cl=en
Tolvaptan is chemically, N-[4-[(7-chloro-2,3,4,5-tetrahydro-5-hydroxylH-l- benzazepin- 1 -yl)carbonyl]-3-methylphenyl]-2-methylbenzamide. Tolvaptan is represented by the following structure:

Tolvaptan, also known as OPC-41061, is a selective, competitive arginine vasopressin receptor 2 antagonist used to treat hyponatremia (low blood sodium levels) associated with congestive heart failure, cirrhosis, and the syndrome of inappropriate antidiuretic hormone (SIADH). Tolvaptan is sold by Otsuka Pharmaceutical Co. under the trade name Samsca.
Tolvaptan and its process for preparation were disclosed in U.S. patent no. 5,258,510. Processes for the preparation of 7-chloro-2,3,4,5-tetrahydro-lH-l-benzazepin-5- one, 7-chloro-l-(2-methyl-4-nitrobenzoyl)-5-oxo-2,3,4,5-tetrahydro-lH-l-benzazepine and 7-chloro- 1 -[2-methyl-4-[(2-methylbenzoyl)amino]benzoyl]-5-oxo-2,3,4,5- tetrahydro-lH-l-benzazepine were reported in Bioorganic & medicinal chemistry 7 (1999), 1743-1754. According to the journal, 7-chloro-2,3,4,5-tetrahydro-lH-l- benzazepin-5-one can be prepared by reacting 7-chloro-4-ethoxycarbonyl-5-oxo-N-p- toluenesufonyl-2,3,4,5-tetrahydro-lH-l-benzazepine with acetic acid in the presence of hydrochloric acid and water to obtain 7-chloro-5-oxo-2,3,4,5-tetrahydro-l-p- toluenesulfonyl-lH-l-benzazepine, and then reacted with polyphospholic acid.
According to the journal, 7-chloro- 1 -(2 -methyl-4-nitrobenzoyl)-5-oxo-2,3,4,5- tetrahydro-lH-l-benzazepine can be prepared by reacting 7-chloro-5-oxo-2,3,4,5- tetrahydro-lH-l-benzazepine with 2-methyl-4-nitobenzoyl chloride in the presence of triethylamine.
According to the journal, 7-chloro- l-[2-methyl-4-[(2- methylbenzoyl)amino]benzoyl]-5-oxo-2,3,4,5-tetrahydro-lH-l-benzazepine can be prepared by reacting l-(4-amino-2-methylbenzoyl)-7-chloro-5-oxo-2,3,4,5-tetrahydro- lH-l-benzazepine with 2-methylbenzoylchloride in the presence of triethylamine.
PCT publication no. WO 2007/026971 disclosed a process for the preparation of tolvaptan can be prepared by the reduction of 7-chloro- l-[2-methyl-4-(2- methylbenzoylamino)benzoyl]-2,3,4,5-tetrahydro-lH-l-benzazepin-5-one with sodium borohydride.
7-Chloro-2,3,4,5-tetrahydro-lH-l-benzazepin-5-one is a key intermediate for the preparation of tolvaptan.
SYNTHESIS CONSTRUCTION



Reference example 1 :
Preparation of methyl 5-chloro-2-nitrobenzoate
Potassium carbonate (515 gm) was added to a solution of 5-chloro-2-nitro benzoic acid (500 gm) in acetone (2750 ml) at room temperature. Dimethyl sulphate (306.5 gm) was added to the reaction mixture slowly and heated to reflux for 30 minutes. The reaction mass was filtered and then concentrated to obtain a residual mass. The residual mass was poured to the ice water and extracted with methylene chloride. The solvent was distilled off under reduced pressure to obtain a residual solid of methyl 5- chloro-2-nitrobenzoate (534 gm). Reference example 2:
Preparation of methyl 2-amino-5-chlorobenzoate
A mixture of methyl 5-chloro-2-nitrobenzoate (534 gm) as obtained in reference example 1 and concentrated hydrochloric acid (2250 ml) was added to ethyl acetate (1120 ml). To the reaction mixture was added a solution of tin chloride (1680 gm) in ethyl acetate (2250 ml). The reaction mass was stirred for 16 hours at room temperature and then poured to the ice water. The pH of the reaction mass was adjusted to 8.0 to 9.0 with aqueous sodium hydroxide solution (2650 ml). The separated aqueous layer was extracted with ethyl acetate and then concentrated to obtain a residual solid of methyl 2- amino-5-chlorobenzoate (345 gm). Reference example 3:
Preparation of methyl 5-chIoro-2-(N-p-toluenesulfonyl)aminobenzoate
To a solution of methyl-2-amino-5-chloro benzoate (345 gm) as obtained in reference example 2 in pyridine (1725 ml) was added p-toluenesulfonyl chloride (425 gm). The reaction mixture was stirred for 2 hours at room temperature and poured to the ice water. The separated solid was filtered and dried to obtain 585 gm of methyl 5- chloro-2-(N-p-toluenesulfonyl)aminobenzoate.
Reference example 4:
Preparation of methyl 5-chloro-2-[N-(3-ethoxycarbonyI)propyI-N-p- toluenesulfonyl] aminobenzoate
Methyl 5-chloro-2-(N-p-toluenesulfonyl)aminobenzoate (585 gm) as obtained in reference example 3, ethyl-4-bromo butyrate (369.6 gm) and potassium carbonate (664 gm) in dimethylformamide (4400 ml) were added at room temperature. The contents were heated to 120°C and maintained for 2 hours. The reaction mass was poured into water and filtered. The solid obtained was dried to give 726 gm of methyl 5-chloro-2-[N- (3 -ethoxycarbonyl)propyl-N-p-toluenesulfonyl] aminobenzoate.
Reference example 5:
Preparation of 7-chloro-4-ethoxycarbonyI-5-oxo-N-p-toluenesufonyl-2,3,4,5- tetrahydro-lH-l-benzazepine
To a heated mixture of potassium tetrabutoxide (363 gm) in toluene (1000 ml) at 70°C was added portion wise methyl 5-chloro-2-[N-(3-ethoxycarbonyl)propyl-N-p- toluenesulfonyl]aminobenzoate (726 gm) as obtained in reference example 4. The contents were heated to reflux and maintained for 30 minutes. The reaction mass was then cooled to room temperature and then poured to the ice water. The layers were separated and the aqueous layer was extracted with toluene. The solvent was distilled off under reduced pressure to obtain a residual solid of 7-chloro-4-ethoxycarbonyl-5-oxo-N- p-toluenesufonyl-2,3,4,5-tetrahydro-lH-l-benzazepine (455 gm).
Example 1:
Preparation of 7-chIoro-5-oxo-2,3,4,5-tetrahydro-lH-l-benzazepine
7-Chloro-4-ethoxycarbonyl-5-oxo-N-p-toluenesufonyl-2,3,4,5-tetrahydro- 1 H- 1 – benzazepine (455 gm) as obtained in reference example 5 was added to aqueous sulfuric acid (80%, 2275 ml). The contents heated to 75°C and maintained for 2 hours. The reaction mass was then cooled to room temperature and then poured to the ice water. The pH of the reaction mass was adjusted to 7.5 to 8.0 with sodium hydroxide solution (2575 ml). The solid obtained was collected by filtration and dried to give 160 gm of 7- chloro-5-oxo-2,3 ,4,5-tetrahydro- 1 H- 1 -benzazepine.
Example 2:
Preparation of 7-chIoro-l-(2-methyl-4-nitrobenzoyl)-5-oxo-2,3,4,5-tetrahydro-lH-l- benzazepine
7-Chloro-5-oxo-2,3,4,5-tetrahydro-lH-l -benzazepine (160 gm) as obtained in example 1 was dissolved in methylene dichloride (480 ml) and then added aqueous sodium bicarbonate solution (20%, 68.75 gm). The reaction mixture was then cooled to 0 to 5°C and then added 2-methyl-4-nitrobenzoylchloride (180 gm) slowly. The pH of the reaction mass was adjusted to 7.0 to 8.0 with aqueous sodium bicarbonate solution (170 ml). The layers were separated and the aqueous layer was extracted with methylene chloride. The solvent was distilled off under reduced pressure to obtain a residual mass. To the residual mass was dissolved in isopropyl alcohol (7300 ml) and maintained for 2 hours at reflux temperature. The separated solid was filtered and dried to obtain 250 gm of 7-chloro-l-(2-methyl-4-nitrobenzoyl)-5-oxo-2,3,4,5-tetrahydro-lH-l-benzazepine. Example 3:
Preparation of l-(4-amino-2-methylbenzoyl)-7-chIoro-5-oxo-2,3,4,5-tetrahydro-lH- 1-benzazepine
7-Chloro- 1 -(2-methyl-4-nitrobenzoyl)-5-oxo-2,3 ,4,5-tetrahydro- 1 H- 1 – benzazepine (250 gm) as obtained in example 2 was dissolved in methanol (575 ml) and then added a solution of tin chloride (630 gm) in methanol (1130 ml). The reaction mixture was stirred for 16 hours at room temperature and then poured to the ice water. The pH of the reaction mass was adjusted to 8.0 to 9.0 with sodium hydroxide solution (1250 ml). The layers were separated and the aqueous layer was extracted with ethyl acetate. The solvent was distilled off under vacuum to obtain a residual solid of l-(4- amino-2-methylbenzoyl)-7-chloro-5-oxo-2,3,4,5-tetrahydro- 1 H- 1 -benzazepine (185 gm).
Example 4:
Preparation of 7-chloro-l-[2-methyl-4-[(2-methylbenzoyl)amino]benzoyl]-5-dxo- 2,3,4,5-tetrahydro-lH-l-benzazepine
1 -(4-Amino-2-methylbenzoyl)-7-chloro-5-oxo-2,3 ,4,5-tetrahydro- 1 H- 1 – benzazepine (185 gm) as obtained in example 3 was dissolved in methylene chloride (4000 ml) and then added sodium bicarbonate solution (10%, 47.3 gm). The reaction mass was cooled to 0 to 5°C and then added 2-methyl benzoyl chloride (95.7 gm) slowly. -The pH of the reaction mass was adjusted to 7.0 to 8.0 with aqueous sodium bicarbonate solution (120 ml). The separated aqueous layer was extracted with methylene chloride and then concentrated to obtain a residual solid of 7-chloro-l-[2- methyl-4-[(2-methylbenzoyl)amino]benzoyl]-5-oxo-2,3,4,5-tetrahydro- 1 H- 1 – benzazepine (185 gm). Example 5:
Preparation of tolvaptan
7-Chloro- 1 -[2-methyl-4-[(2-methylbenzoyl)amino]benzoyl]-5-oxo-2,3,4,5- tetrahydro-lH-1 -benzazepine (63 gm) as obtained in example 4 was dissolved in methanol (570 ml) and then added sodium borohydride (2.07 gm) at room temperature. The reaction mass was stirred for 1 hour and pH of the reaction mass was adjusted to 6.0 to 7.0 with hydrochloric acid solution (1%, 630 ml). The separated solid was filtered and dried to obtain 57 gm of tolvaptan.
……………….

Process used to prepare Tolvaptan involves condensing 7-chloro-1, 2, 3, 4-tetrahydro-benzo[b]azepin-5-one with 2-methyl, 4-nitro benzoyl chloride, followed by reduction using SnCl2/HCl catalyst resulting in amine which is then condensed with o-toluoyl chloride followed by reduction with sodium borohydride to give Tolvaptan
///////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
SYN 1
Synthetic Reference
Cordero-Vargas, Alejandro; Quiclet-Sire, Beatrice; Zard, Samir Z. A flexible approach for the preparation of substituted benzazepines: Application to the synthesis of tolvaptan. Bioorganic & Medicinal Chemistry. Volume 14. Issue 18. Pages 6165-6173. 2006.

SYN 2
Synthetic Reference
Torisawa, Yasuhiro; Abe, Kaoru; Muguruma, Yasuaki; Fujita, Shigekazu; Ogawa, Hidenori; Utsumi, Naoto; Miyake, Masahiro. Process for preparation of benzoylaminobenzoylbenzazepinones by reaction of benzazepinones with benzoylaminophenyl halides in the presence of carbonylating agents. Assignee Otsuka Pharmaceutical Co.,

SYN 3
Synthetic Reference
Zard, Samir; Cordero Vargas, Alejandro; Sire, Beatrice. Improved process for the preparation of benzazepines and their derivatives. Assignee Centre National de la Recherche Scientifique CNRS, Fr.; Ecole Polytechnique. FR 2867187. (2005).

SYN 4
Synthetic Reference
Gao, Junlong; Li, Peng; Liu, Kai; Guo, Dapeng. Method for preparing high-purity Tolvaptan intermediate. Assignee Jiangsu Hengrui Medicine Co., Ltd., Peop. Rep. China. CN 108503586. (2018).

SYN 5
Synthetic Reference
Han, Shin; Jeon, Seong Hyeon; Lee, Shin Yoon. Improved method for preparing synthetic intermediates for tolvaptan. Assignee Hexa Pharmatec Co., Ltd., S. Korea. JP 2018012690. (2018).

SYN 6
Synthetic Reference
Guo, Xinfu; Wang, Qiang; Liu, Zhaoguo; Wang, Zhipeng. Preparation method of tolvaptan. Assignee Tianjin Taipu Pharmaceutical Co., Ltd., Peop. Rep. China. CN 106883175. (2017).

SYN 7
Synthetic Reference
Lixin, Juanzi; Li, Jianzhi; Ma, Xilai; Chi, Wangzhou; Liu, Hai; Hu, Xuhua; Zheng, Xiaoli; Zhai, Zhijun; Li, Jianxun. Process for the preparation of tolvaptan. Assignee Shanghai Tianci International Pharmaceutical Co., Ltd., Peop. Rep. China. CN 105753735. (2016).

STR8
Synthetic Reference
Patel, Dhaval J.; Shah, Tejas C.; Singh, Manoj Kumar. A process for the preparation of tolvaptan. Assignee Cadila Healthcare Limited, India. IN 2012MU01559. (2014).

STR9
Synthetic Reference
Sethi, Madhuresh Kumar; Rawat, Vijendrasingh; Thirunavukarasu, Jayaprakash; Yerramala, Raja Krishna; Kumar, Anish. Improved process for the preparation of tolvaptan. Assignee Matrix Laboratories Ltd., India. IN 2011CH01303. (2013).

/////////////////////

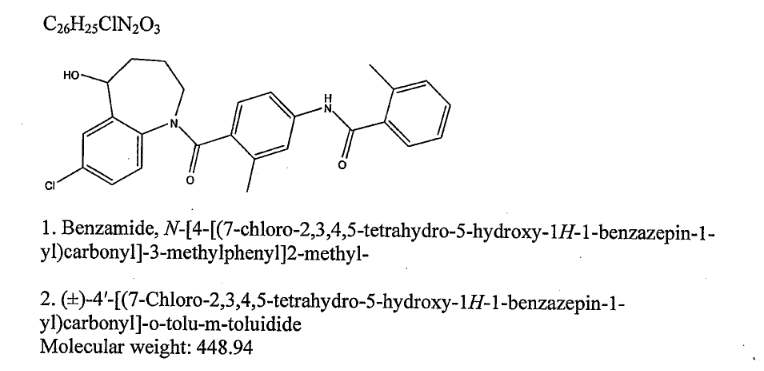

Title: Tolvaptan
CAS Registry Number: 150683-30-0
CAS Name: N-[4-[(7-Chloro-2,3,4,5-tetrahydro-5-hydroxy-1H-1-benzazepin-1-yl)carbonyl]-3-methylphenyl]-2-methylbenzamide
Additional Names: 7-chloro-5-hydroxy-1-[2-methyl-4-(2-methylbenzoylamino)benzoyl]-2,3,4,5-tetrahydro-1H-1-benzazepine
Manufacturers’ Codes: OPC-41061
Molecular Formula: C26H25ClN2O3
Molecular Weight: 448.94
Percent Composition: C 69.56%, H 5.61%, Cl 7.90%, N 6.24%, O 10.69%
Literature References: Nonpeptide arginine vasopressin V2 receptor antagonist. Prepn: H. Ogawa et al., WO 9105549; eidem, US 5258510 (1991, 1993 both to Otsuka); K. Kondo et al., Bioorg. Med. Chem. 7, 1743 (1999). Pharmacology: Y. Yamamura et al., J. Pharmacol. Exp. Ther. 287, 860 (1998). Clinical trial in heart failure: M. Gheorghiade et al., J. Am. Med. Assoc. 291, 1963 (2004).
Properties: Colorless prisms, mp 225.9°.
Melting point: mp 225.9°
Therap-Cat: In treatment of congestive heart failure.
Keywords: Vasopressin Receptor Antagonist.
- Shoaf S, Elizari M, Wang Z, et al. (2005). “Tolvaptan administration does not affect steady state amiodarone concentrations in patients with cardiac arrhythmias”. J Cardiovasc Pharmacol Ther 10 (3): 165–71. doi:10.1177/107424840501000304. PMID 16211205.
- Otsuka Maryland Research Institute, Inc.
- Gheorghiade M, Gattis W, O’Connor C, et al. (2004). “Effects of tolvaptan, a vasopressin antagonist, in patients hospitalized with worsening heart failure: a randomized controlled trial”. JAMA 291 (16): 1963–71. doi:10.1001/jama.291.16.1963. PMID 15113814.
- (2012) Tolvaptan in Patients with Autosomal Dominant Polycystic Kidney Disease
- Kondo, K.; Ogawa, H.; Yamashita, H.; Miyamoto, H.; Tanaka, M.; Nakaya, K.; Kitano, K.; Yamamura, Y.; Nakamura, S.; Onogawa, T.; et al.; Bioor. Med. Chem. 1999, 7, 1743.
- http://www.fda.gov/Safety/MedWatch/SafetyInformation/SafetyAlertsforHumanMedicalProducts/ucm350185.htm?source=govdelivery
- Gheorghiade M, Niazi I, Ouyang J et al. (2003). “Vasopressin V2-receptor blockade with tolvaptan in patients with chronic heart failure: results from a double-blind, randomized trial”. Circulation 107 (21): 2690–6. doi:10.1161/01.CIR.0000070422.41439.04.PMID 12742979.
G. R. Belum, V. R. Belum, S. K. Chaitanya Arudra, and B. S. N. Reddy, “The Jarisch-Herxheimer reaction: revisited,” Travel Medicine and Infectious Disease, vol. 11, no. 4, pp. 231–237, 2013.
H. D. Zmily, S. Daifallah, and J. K. Ghali, “Tolvaptan, hyponatremia, and heart failure,” International Journal of Nephrology and Renovascular Disease, vol. 4, pp. 57–71, 2011.
M. N. Ferguson, “Novel agents for the treatment of hyponatremia: a review of conivaptan and tolvaptan,” Cardiology in Review, vol. 18, no. 6, pp. 313–321, 2010.
H. Ogawa, H. Miyamoto, K. Kondo, et al., US5258510, 1993.
K. Kondo, H. Ogawa, H. Yamashita et al., “7-Chloro-5-hydroxy-1-[2-methyl-4-(2-methylbenzoylamino)benzoyl]-2,3,4,5- tetrahydro-1H-1-benzazepine (OPC-41061): a potent, orally active nonpeptide arginine vasopressin V2 receptor antagonist,” Bioorganic and Medicinal Chemistry, vol. 7, no. 8, pp. 1743–1754, 1999.
| WO2012046244A1 * | Aug 23, 2011 | Apr 12, 2012 | Hetero Research Foundation | Process for preparing tolvaptan intermediates |
| CN102060769A * | Dec 20, 2010 | May 18, 2011 | 天津药物研究院 | Preparation method of tolvaptan |
| CN102060769B | Dec 20, 2010 | Sep 18, 2013 | 天津药物研究院 | Preparation method of tolvaptan |
| US9024015 | Aug 23, 2011 | May 5, 2015 | Hetero Research Foundation | Process for preparing tolvaptan intermediates |
| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| CN101817783A | May 12, 2010 | Sep 1, 2010 | 天津泰普药品科技发展有限公司 | Method for preparing tolvaptan intermediate |
| WO2007026971A2 | Sep 1, 2006 | Mar 8, 2007 | Otsuka Pharma Co Ltd | Process for preparing benzazepine compounds or salts thereof |
| Reference | ||
|---|---|---|
| 1 | Cordero-Vargas, Alejandro | |
| 2 | Kondo, Kazumi et al.7-chloro-5-hydroxy-1-[2-methyl-4-(2-methylbenzoyl-amino)benzoyl]-2,3,4,5-tetrahydro-1H-1-benzazepine (OPC-41061): A potent, orally active nonpeptide arginine vasopressin V2 receptor antagonist.《Bioorganic & Medicinal Chemistry》.1999,1743-1757. | |
| 3 | Quiclet-Sire, Beatrice | |
| 4 | Torisawa, Yasuhiro et al.Aminocarbonylation route to tolvaptan.《Bioorganic & Medicinal Chemistry Letters》.2007,6455-6458. | |
| 5 | Zard, Samir Z.A flexible approach for the preparation of substituted benzazepines: Application to the synthesis of tolvaptan.《Bioorganic & Medicinal Chemistry》.2006,6165-6173. | |
///////////////
CC1=CC=CC=C1C(=O)NC2=CC(=C(C=C2)C(=O)N3CCCC(C4=C3C=CC(=C4)Cl)OP(=O)([O-])[O-])C.[Na+].[Na+]
////////////UPDATE 2022
-Tolvaptan_Structural_Formula_V1.svg) |
|
 |
|
| Clinical data | |
|---|---|
| Trade names | Samsca, Jinarc, Jynarque, others |
| Other names | OPC-41061 |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a609033 |
| License data | |
| Pregnancy category |
|
| Routes of administration |
By mouth |
| ATC code | |
| Legal status | |
| Legal status | |
| Pharmacokinetic data | |
| Bioavailability | Unknown (40% absorbed) |
| Protein binding | 99% |
| Metabolism | Liver (CYP3A4-mediated)[7] |
| Elimination half-life | 12 hours (terminal) |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| IUPHAR/BPS | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEMBL | |
| CompTox Dashboard (EPA) | |
| ECHA InfoCard | 100.219.212 |
| Chemical and physical data | |
| Formula | C26H25ClN2O3 |
| Molar mass | 448.95 g·mol−1 |
| 3D model (JSmol) | |
| |
|
Tolvaptan, sold under the brand name Samsca among others, is an aquaretic drug that functions as a selective, competitive vasopressin receptor 2 (V2) antagonist used to treat hyponatremia (low blood sodium levels) associated with congestive heart failure, cirrhosis, and the syndrome of inappropriate antidiuretic hormone (SIADH). Tolvaptan was approved by the U.S. Food and Drug Administration (FDA) on May 19, 2009, and is sold by Otsuka Pharmaceutical Co. under the trade name Samsca.[8] Tolvaptan, as Jynarque, was granted approval for medical use in the United States in April 2018.[9]
The U.S. Food and Drug Administration (FDA) granted tolvaptan a fast track designation for clinical trials investigating its use for the treatment of polycystic kidney disease.[10] The FDA granted Jynarque an orphan drug designation in April 2012, for the treatment of autosomal dominant polycystic kidney disease.[11]
Tolvaptan is available as a generic medication.[12]
Medical uses
Tolvaptan (Samsca) is indicated for the treatment of clinically significant hypervolemic and euvolemic hyponatremia.[13]
Tolvaptan (Jynarque) is indicated to slow kidney function decline in adults at risk of rapidly progressing autosomal dominant polycystic kidney disease (ADPKD).[14]
Side effects
The FDA has determined that tolvaptan should not be used for longer than 30 days and should not be used in patients with underlying liver disease because it can cause liver injury, potentially leading to liver failure.[15] When using to treat hyponatremia, it may cause too rapid correction of hyponatremia resulting in fatal osmotic demyelination syndrome.[16]
Pharmacology
Tolvaptan is a selective vasopressin V2 receptor antagonist.[13][14]
Chemistry
Tolvaptan is a racemate, a 1:1 mixture of the following two enantiomers:[17]
| Enantiomers of tolvaptan | |
|---|---|
-Tolvaptan_Structural_Formula_V1.svg) (R)-Tolvaptan CAS number: 331947-66-1 |
-Tolvaptan_Structural_Formula_V1.svg) (S)-Tolvaptan CAS number: 331947-44-5 |
References
- ^ “Samsca 15 mg tablets – Summary of Product Characteristics (SmPC)”. (emc). Retrieved 14 December 2020.
- ^ “Jinarc 15 mg tablets – Summary of Product Characteristics (SmPC)”. (emc). 21 April 2020. Retrieved 14 December 2020.
- ^ “Jynarque- tolvaptan kit Jynarque- tolvaptan tablet”. DailyMed. 31 March 2020. Retrieved 14 December 2020.
- ^ “Samsca- tolvaptan tablet”. DailyMed. 26 October 2020. Retrieved 14 December 2020.
- ^ “Samsca EPAR”. European Medicines Agency (EMA). Retrieved 14 December 2020.
- ^ “Jinarc EPAR”. European Medicines Agency (EMA). Retrieved 14 December 2020.
- ^ Shoaf S, Elizari M, Wang Z, et al. (2005). “Tolvaptan administration does not affect steady state amiodarone concentrations in patients with cardiac arrhythmias”. J Cardiovasc Pharmacol Ther. 10 (3): 165–71. doi:10.1177/107424840501000304. PMID 16211205. S2CID 39158242.
- ^ “Drug Approval Package: Samsca (Tolvaptan) Tablets NDA #022275”. U.S. Food and Drug Administration (FDA). 21 July 2009. Retrieved 15 August 2020. Lay summary (PDF).
{{cite web}}: Cite uses deprecated parameter|lay-url=(help) - ^ “Drug Approval Package: Jynarque (tolvaptan)”. U.S. Food and Drug Administration (FDA). 8 June 2018. Retrieved 15 August 2020.
- ^ “Otsuka Maryland Research Institute, Inc. Granted Fast Track Designation For Tolvaptan In PKD”. Medical News Today. Healthline Media UK Ltd. Retrieved 6 December 2018.
- ^ “Tolvaptan Orphan Drug Designations and Approvals”. U.S. Food and Drug Administration (FDA). 6 April 2012. Retrieved 15 August 2020.
- ^ “Drugs@FDA: FDA-Approved Drugs”. U.S. Food and Drug Administration (FDA). Retrieved 15 August 2020.
- ^ Jump up to:a b “Samsca- tolvaptan tablet”. DailyMed. 28 May 2019. Retrieved 15 August 2020.
- ^ Jump up to:a b “Jynarque- tolvaptan kit Jynarque- tolvaptan tablet”. DailyMed. 31 March 2020. Retrieved 15 August 2020.
- ^ “U.S. Food and Drug Administration.” Samsca (Tolvaptan): Drug Safety Communication. N.p., 30 Apr. 2013. Web. 1 June 2014. <http://www.fda.gov/Safety/MedWatch/SafetyInformation/SafetyAlertsforHumanMedicalProducts/ucm350185.htm>[dead link]
- ^ Goodman & Gilman’s the pharmacological basis of therapeutics. Brunton, Laurence L, Knollmann, Björn C, Hilal-Dandan, Randa (Thirteenth ed.). New York. 5 December 2017. ISBN 9781259584732. OCLC 994570810.
- ^ Rote Liste Service GmbH (Hrsg.): Rote Liste 2017 – Arzneimittelverzeichnis für Deutschland (einschließlich EU-Zulassungen und bestimmter Medizinprodukte). Rote Liste Service GmbH, Frankfurt/Main, 2017, Aufl. 57, ISBN 978-3-946057-10-9, S. 222.
Further reading
- Gheorghiade M, Niazi I, Ouyang J, et al. (2003). “Vasopressin V2-receptor blockade with tolvaptan in patients with chronic heart failure: results from a double-blind, randomized trial”. Circulation. 107 (21): 2690–6. doi:10.1161/01.CIR.0000070422.41439.04. PMID 12742979.
External links
- “Tolvaptan”. Drug Information Portal. U.S. National Library of Medicine.
Synthesis
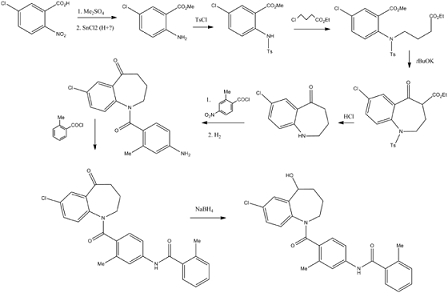
Fugure 1: Synthesis of Tolvaptan

NEW DRUG APPROVALS
HELP TO MAINTAIN THIS BLOG SUBSCRIPTION
$10.00
Title: Tolvaptan
CAS Registry Number: 150683-30-0
CAS Name: N-[4-[(7-Chloro-2,3,4,5-tetrahydro-5-hydroxy-1H-1-benzazepin-1-yl)carbonyl]-3-methylphenyl]-2-methylbenzamide
Additional Names: 7-chloro-5-hydroxy-1-[2-methyl-4-(2-methylbenzoylamino)benzoyl]-2,3,4,5-tetrahydro-1H-1-benzazepine
Manufacturers’ Codes: OPC-41061
Molecular Formula: C26H25ClN2O3
Molecular Weight: 448.94
Percent Composition: C 69.56%, H 5.61%, Cl 7.90%, N 6.24%, O 10.69%
Literature References: Nonpeptide arginine vasopressin V2 receptor antagonist. Prepn: H. Ogawa et al., WO 9105549; eidem, US 5258510 (1991, 1993 both to Otsuka); K. Kondo et al., Bioorg. Med. Chem. 7, 1743 (1999). Pharmacology: Y. Yamamura et al., J. Pharmacol. Exp. Ther. 287, 860 (1998). Clinical trial in heart failure: M. Gheorghiade et al., J. Am. Med. Assoc. 291, 1963 (2004).
Properties: Colorless prisms, mp 225.9°.
Melting point: mp 225.9°
Therap-Cat: In treatment of congestive heart failure.
Keywords: Vasopressin Receptor Antagonist.

.svg){kind=link}
{kind=link}