
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....

Cabotegravir, GSK 744,
PMDA APPROVED 2022/5/31, JAPAN
(3S,11aR)-N-(2,4-Difluorobenzyl)-6-hydroxy-3-methyl-5,7-dioxo-2,3,5,7,11,11a-hexahydro[1,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazine-8-carboxamide
3S, 1 1 aR)- N-[(2,4-difluorophenyl)methyl]-2,3,5,7, 1 1 , 1 1 a-hexahydro-6-hydroxy-3- methyl-5,7- dioxo-oxazolo[3,2-a]pyrido[1 ,2-d]pyrazine-8-carboxamide
OTHER ISOMER
(3R,11 aS)-N-[(2,4-Diflυorophenyl)methyl]-6-hydroxy-3-methyl-5,7-dioxo-2,3,5,7, 11, 11a-hexahydro[1 ,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazine-8-carboxamide
VIIV HEALTHCARE …INNOVATOR
-
GSK1265744, CAS 1051375-10-0, S-265744 LAP
-
C19-H17-F2-N3-O5
- 405.3553
- 744 LA
- GSK 1265744
- GSK 744
- GSK-1265744A
- GSK1265744
- GSK1265744A
- GSK744
- GSK744 LA
- GSK744 LAP
- S-265744
- S/GSK1265744
Product Ingredients
| INGREDIENT | UNII | CAS | INCHI KEY |
|---|---|---|---|
| Cabotegravir sodium | 3L12PT535M | 1051375-13-3 | AEZBWGMXBKPGFP-KIUAEZIZSA-M |
Cabotegravir, sold under the brand name Vocabria among others, is a antiretroviral medication used for the treatment of HIV/AIDS. It is available in the form of tablets and as an intramuscular injection, as well as in an injectable combination with rilpivirine under the brand name Cabenuva.[6][9]
It is an integrase inhibitor with a carbamoyl pyridone structure similar to that of dolutegravir.[10]
In December 2021, the U.S. Food and Drug Administration approved cabotegravir for pre-exposure prophylaxis (PrEP) in at-risk people under the brand name Apretude.[11]
GSK744 (also known as S/GSK1265744) is an investigational new drug under development for the treatment of HIV infection. It is anintegrase inhibitor, with a carbamoyl pyridone structure similar to dolutegravir. In investigational studies, the agent has been packaged into nanoparticles (GSK744LAP) conferring an exceptionally long half-life of 21–50 days following a single dose. In theory, this would make possible suppression of HIV with dosing as infrequently as once every three months.[1]
S-265744 LAP is in phase II clinical development at Shionogi-GlaxoSmithKline for the treatment of HIV infection. Phase III clinical trials had been ongoing for this indication; however, no recent development has been reported for this study.
Cabotegravir, or GSK1265744, is an HIV-1 integrase inhibitor that is prescribed with the non-nucleoside reverse transcriptase inhibitor, rilpivirine.4,6,7 Early research into cabotegravir showed it had lower oral bioavailability than dolutegravir,4 which resulted in the development of long acting monthly intramuscular injection formulation for cabotegravir.4,7
Cabotegravir was granted FDA approval on 21 January 2021 in combination with rilpivirine to treat HIV-1 infection in virologically suppressed individuals.8 While previously administered once monthly only, this combination product was granted FDA approval for dosing every two months on February 01, 2022 11 and without the need for an oral lead-in period prior.7
The human immunodeficiency virus (“HIV”) is the causative agent for acquired immunodeficiency syndrome (“AIDS”), a disease characterized by the destruction of the immune system, particularly of CD4+ T-cells, with attendant susceptibility to opportunistic infections, and its precursor Al DS-related complex (“ARC”), a syndrome characterized by symptoms such as persistent generalized lymphadenopathy, fever and weight loss. HIV is a retrovirus; the conversion of its RNA to DNA is accomplished through the action of the enzyme reverse transcriptase. Compounds that inhibit the function of reverse transcriptase inhibit replication of HIV in infected cells. Such compounds are useful in the prevention or treatment of HIV infection in humans.
A required step in HIV replication in human T-cells is the insertion by virally-encoded integrase of proviral DNA into the host cell genome. Integration is believed to be mediated by integrase in a process involving assembly of a stable nucleoprotein complex with viral DNA sequences, cleavage of two nucleotides from the 3′ termini of the linear proviral DNA and covalent joining of the recessed 3′ OH termini of the proviral DNA at a staggered cut made at the host target site. The repair synthesis of the resultant gap may be accomplished by cellular enzymes. There is continued need to find new therapeutic agents to treat human diseases. HIV integrase is an attractive target for the discovery of new therapeutics due to its important role in viral infections, particularly HIV infections. Integrase inhibitors are disclosed in WO2006/116724.
(3S, 1 1 aR)- N-[(2,4-difluorophenyl)methyl]-2,3,5,7, 1 1 , 1 1 a-hexahydro-6-hydroxy-3- methyl-5,7- dioxo-oxazolo[3,2-a]pyrido[1 ,2-d]pyrazine-8-carboxamide, a compound of formula (I), also referred to as compound (I), has proven antiviral activity against human immunodeficiency virus (HIV).

The present invention features pharmaceutical compositions comprising the active ingredient (3S, 1 1 aR)- N-[(2,4-difluorophenyl)methyl]-2,3,5,7, 1 1 , 1 1 a-hexahydro-6-hydroxy-3- methyl-5,7- dioxo-oxazolo[3,2-a]pyrido[1 ,2-d]pyrazine-8-carboxamide, or a pharmaceutically acceptable salt thereof, suitable for administration once monthly or longer.
Medical uses
Cabotegravir in combination with rilpivirine is indicated for the treatment of human immunodeficiency virus type-1 (HIV-1) in adults.[1][5] The combination injection is intended for maintenance treatment of adults who have undetectable HIV levels in the blood (viral load less than 50 copies/mL) with their current antiretroviral treatment, and when the virus has not developed resistance to non-nucleoside reverse transcriptase inhibitors (NNRTIs) and integrase strand transfer inhibitors.[5] The tablets are used to check whether a person tolerates the treatment before the injection therapy is started.[12][5]
The two medicines are the first antiretroviral drugs that come in a long-acting injectable formulation.[12]
Cabotegravir (Apretude) is indicated for use in at-risk people weighing at least 35 kilograms (77 lb) for pre-exposure prophylaxis (PrEP) to reduce the risk of sexually acquired HIV.[11]
Contraindications and interactions
Cabotegravir must not be combined with the drugs rifampicin, rifapentine, carbamazepine, oxcarbazepine, phenytoin or phenobarbital, which induce the enzyme UGT1A1.[5] These drugs significantly decrease cabotegravir concentrations in the body and thus may reduce its effectiveness.[9][5] Additionally, they induce the enzyme CYP3A4, which leads to reduced rilpivirine concentrations in the body.[5][13][14][15] Additionally, patients who are breastfeeding or plan to breastfeed should not take Cabotegravir because it is not known if it will pass within the breast milk.[16]
Adverse effects
The most common side effects of the injectable combination therapy with rilpivirine are reactions at the injection site (in up to 84% of patients) such as pain and swelling, as well as headache (up to 12%) and fever or feeling hot (in 10%). For the tablets, headache and a hot feeling were slightly less frequent. Less common side effects (under 10%) for both formulations are depressive disorders, insomnia, and rashes.[9]
Pharmacology
Mechanism of action
Cabotegravir is an integrase strand transfer inhibitor. This means it blocks the HIV’s enzyme integrase, thereby preventing its genome from being integrated into the human cells’ DNA.[9] As this is a necessary step for the virus to replicate, its further spread is hampered.[9]
Pharmacokinetics

Cabotegravir glucuronide, the main metabolite in human bile and urine[17]
When taken by mouth, cabotegravir reaches highest blood plasma levels after three hours. Taking the drug together with food slightly increases its concentrations in the blood, but this is not clinically relevant. After injection into the muscle, cabotegravir is slowly absorbed into the bloodstream, reaching its highest blood plasma levels after about seven days.[9]
Over 99% of the substance are bound to plasma proteins. The drug is inactivated in the body by glucuronidation, mainly by the enzyme UGT1A1, and to a much lesser extent by UGT1A9. More than 90% of the circulating substance are the unchanged cabotegravir, however. The biological half-life is 41 hours for the tablets and 5.6 to 11.5 weeks for the injection.[9]
Elimination has only been studied for oral administration: Most of the drug is eliminated via the faeces in unchanged form (47%). It is not known how much of this amount comes from the bile, and how much was not absorbed in the first place. (The bile actually contains the glucuronide, but this could be broken up again in the gut lumen to give the parent substance that is observed in the faeces.) To a lesser extent it is excreted via the urine (27%), almost exclusively as the glucuronide.[9]
Pharmacogenomics
UGT1A1 poor metabolizers have 1.3- to 1.5-fold increased cabotegravir concentrations in the body. This is not considered clinically significant.[9]
Chemistry
Cabotegravir is a white to off-white, crystalline powder that is practically insoluble in aqueous solutions under pH 9, and slightly soluble above pH 10. It is slightly acidic with a pKa of 7.7 for the enolic acid and 1.1 (calculated) for the carboxamide. The molecule has two asymmetric carbon atoms; only one of the four possible configurations is present in the medication.[18]
Formulation
In studies, the agent was packaged into nanoparticles (GSK744LAP) conferring a biological half-life of 21 to 50 days[citation needed] following a single dose. The marketed injection achieves its long half-life not via nanoparticles but with a suspension of the free cabotegravir acid. The tablets contain cabotegravir sodium salt.[18]
History
Cabotegravir was examined in the clinical trials HPTN 083 and HPTN 084.[19][20] In 2020, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) adopted a positive opinion, recommending the granting of a marketing authorization for the medicinal product Vocabria intended for the treatment of human immunodeficiency virus type 1 (HIV-1) infection in combination with rilpivirine injection.[21] The EMA also recommended marketing authorization be given for rilpivirine and cabotegravir injections to be used together for the treatment of people with HIV-1 infection.[12] Cabotegravir was approved for medical use in the European Union in December 2020.[8]
Society and culture
Names
Cabotegravir is the United States Adopted Name (USAN)[22] and the international nonproprietary name (INN).[23]
Research
Pre-exposure prophylaxis
In 2020, results for some studies were released showing success in using injectable cabotegravir for long-acting pre-exposure prophylaxis (PrEP) with greater efficacy than the emtricitabine/tenofovir combination being widely used for PrEP at the time.[24][25]
The safety and efficacy of cabotegravir to reduce the risk of acquiring HIV were evaluated in two randomized, double-blind trials that compared cabotegravir to emtricitabine/tenofovir, a once daily oral medication for HIV PrEP.[11] Trial 1 included HIV-uninfected men and transgender women who have sex with men and have high-risk behavior for HIV infection.[11] Trial 2 included uninfected cisgender women at risk of acquiring HIV.[11]
In Trial 1, 4,566 cisgender men and transgender women who have sex with men received either cabotegravir or emtricitabine/tenofovir.[11] The trial measured the rate of HIV infections among trial participants taking daily cabotegravir followed by cabotegravir injections every two months compared to daily oral emtricitabine/tenofovir.[11] The trial showed participants who took cabotegravir had 69% less risk of getting infected with HIV when compared to participants who took emtricitabine/tenofovir.[11]
In Trial 2, 3,224 cisgender women received either cabotegravir or emtricitabine/tenofovir.[11] The trial measured the rate of HIV infections in participants who took oral cabotegravir and injections of cabotegravir compared to those who took emtricitabine/tenofovir orally.[11] The trial showed participants who took cabotegravir had 90% less risk of getting infected with HIV when compared to participants who took emtricitabine/tenofovir.[11]
In December 2021, the U.S. Food and Drug Administration (FDA) approved cabotegravir for pre-exposure prophylaxis.[11] The FDA granted the approval of Apretude to Viiv.[11]
Methods for the preparation of a compound of formula (I) are described in WO 2006/1 16764, WO2010/01 1814, WO2010/068262, and WO2010/068253
WO 2006116764
http://www.google.com/patents/WO2006116764A1?cl=en

[Chemical formula 68] is UNDESIRED ISOMER………..amcrasto@gmail.com

Example Z-1:
(3R,11 aS)-N-[(2,4-Diflυorophenyl)methyl]-6-hydroxy-3-methyl-5,7-dioxo-2,3,5,7, 11, 11a
-hexahydro[1 ,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazine-8-carboxamide sodium salt.

(3R,11aS)-N-[(2,4-Diflυorophenyl)methyl]-3-methyl-5,7-dioxo-6-[(phenylmethyl)oxy]-2,
3,5,7,11,11a-hexahydro[1,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazine-8-carboxamide. To a solution of 16a (409 mg, 0.87 mmol) in dichloroethane (20 mL) was added (2R)-2-amino-1-propanol (0,14 mL, 1.74 mmol) and 10 drops of glacial acetic acid.
The resultant solution was heated at reflux for 2 h. Upon cooling, Celite was added
to the mixture and the solvents removed in vacuo and the material was purified via
silica gel chromatography (2% CH3OH/CH2CI2 gradient elution) to give
(3R),11aS)-N-[(2,4-difluorophenyl)methyl]-3-methyl-5,7-dioxo-6- [(phenylmethyl)oxy]-2,
3,5,7, 1 l , 11a-hexahydro[1,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazinc-8-carboxamide (396
mg, 92%) as a glass, JH NMR (CDCIo) δ 10.38 (m, 1 H), 8.42 (s, 1 H), 7,54-7,53 (m, 2
H), 7,37-7.24 (m, 4 H), 6.83-6,76 (m, 2 H), 5.40 (d, J = 10.0 Hz, 1 H), 5.22 (d, J = 10,0
Hz, 1 H), 5.16 (dd, J – 9,6, 6.0 Hz, 1 H), 4,62 (m, 2 H), 4.41 (m, 1 H), 4.33-4.30 (m, 2
H), 3.84 (dd, J= 12.0, 10.0 Hz, 1 H), 3.63 (dd, J= 8,4, 7.2 Hz, 1 H), 1.37 (d, J= 6.0 Hz,
3 H); ES+ MS: 496 (M+1).
b)
(3R, 11aS)-N-[(2,4-Difluorophenyl)methyl]-6-hydroxy-3-methyl-5,7-dioxo-2,3,5,7, 11, 1la
-hexahydro[1,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazine-8vcarboxamide sodium salt. To a
solution of
(37?, 11aS)-N-[(2,4-difluo]-ophenyl)methyl]-3-methyl-5,7-dioxo-6- [(phenylmethyl)oxy] -2,
3,5,7,11,11 a-hexahydro[1,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazine-8-carboxamide (396
mg, 0.80 mmol) in methanol (30 mL) was added 10% Pd/C (25 mg). Hydrogen was
bubbled through the reaction mixture via a balloon for 2 h. The resultant mixture
was filtered through Celite with methanol and dichloromethanc. The filtrate was
concentrated in vacuo to give
(3R, l] aS)-N-f(2,4-difliιorophenyl)methyl]-6-hydroxy-3-methyl-5,7-dioxo-2,3,5,7, υ , 11a- hexahydro[1,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazine-8-carboxamide as a pink tinted
white solid (278 mg, 86%), 1H NMR (ODCU) δ 11.47 (m, 1 H), 10.29 (m, 1 H), 8,32 (s,
1 H), 7.36 (m, 1 H), 6.82 (m, 2 H), 5.31 (dd, J – 9.6, 3.6 Hz, 1 H), 4.65 (m, 2 H),
4,47-4,38 (m, 3 H), 3.93 (dd, J= 12.0, 10.0 Hz, 1 H), 3,75 (m, 1 H), 1.49 (d, J= 5.6 Hz,
3 H); BS1 MS: 406 (M+ 1). The above material (278 mg, 0,66 mmol) was taken up
m cthanol (10 mL) and treated with 1 Nsodium hydroxide (aq) (0.66 mL, 0.66 mmol).
The resulting suspension was stirred at room temperature for 30 min, Ether was
added and the liquids were collected to provide the sodium salt of the title compound
as a white powder (291 mg, 99%).‘ 1H NMR (OMSO- do) δ 30.68 (m, 1 H), 7,90 (s, 1 H),
7.35 (m, 1 H), 7.20 (m, 1 H), 7,01 (m, 1 H), 5,20 (m, 1 H), 4,58 (m, I H), 4.49 (m, 2 H),
4.22 (m, 2 H), 3 74 (dd, J= 11.2, 10.4 Hz, 1 H), 3.58 (m, 1 H), 1.25 (d, J=- 4.4 Hz, 3 H)
Example Z-9-
(3£ 11aΛ^N-[(2.4-D-fluoroDhonyl)methyl] -6-hvdroxy-3-methyl-5.7-dioxo-2,3,5.7, n , 11 a
-hexahydro[1 ,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazino-8-carboxamide sodium salt.

The title compound was made in two steps using a similar process to that described
in example Z-I. 16a (510 mg, 1.08 mmol) and (2«5)-2-amino-1-propanol (0.17 mL, 2,17 mmol) were reacted in 1,2-dichloroethane (20 mL) with acetic acid to give
(3S, 11aR)-i\A[(2,4-diflιιorophenyl)methyl]-3-methyl-5,7-d.ioxo-6-[(phenylmethyl)oxy]-2,
3,5,7,11,1la-hexahydro[1,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazine-8-carboxamide (500
mg, 93%). This material was hydrogenated in a second step as described in example
Z- I to give
3S, 11a R)-7N-[(2,4-Diiαuorophenyl)methyl]-6-hydroxy-3-methyl-5,7-dioxo-2,3,5,7, 11, 11a-
hexahydro[1,3]oxazolo[3,2-a]pyrido[1,2-d]pyraziine-8-carboxamide (386 mg, 94%) as a
tinted white solid. Η NMR (CDCL3) δ 11.46 (m, 1 H), 10.28 (m, 1 H), 8.32 (s, 1 H),
7.35 (m, 1 H), 6.80 (m, 2 H), 5.30 (dd, J = 10.0, 4.0 Hz, 1 H), 4.63 (m, 2 H), 4.48-4.37
(m, 3 H), 3.91 (dd, J = 12.0, 10.0 Hz, 1 H), 3.73 (m, 1 H), 1.48 (d, J – 6.0 Hz, 3 H);
ES 1 MS: 406 (M+ 1). This material (385 mg, 0.95 mmol) was treated with sodium
hydroxide (0,95 mL, 1.0 M, 0.95 mmol) m ethanol (15 mL) as described in example Z-1
to provide its corresponding sodrum sail (381 mg, 94%) as a white solid. 1H NMR
(DMSO- Λ) δ 10.66 (m, 1 PI), 7.93 (s, 1 H), 7.33 (m, 1 H), 7.20 (m, 1 H), 7.01 (m, 1 H),
5.19 (m, 1 H), 4.59 (m, 1 H), 4 48 (m, 2 H), 4.22 (m, 2 H), 3,75 (m, 1 H), 3.57 (m, 1 H),
1.24 (d, J= 5 6 Hz, 3 H).
WO 2010068253
http://www.google.com/patents/WO2010068253A1?cl=en
Example A
The starting material of Example A is compound 8, which is identical to formula (Ia). Thus, Example A depicts a process in providing an intermediate for the compound of formula 17 below which is isomeric to the compound ZZ-2 at page 237 of WO 2006/116764 to Brian Johns et al.

14


Example Aa After dissolution of mixture of 320 g of compound 8 (1.0 eq.) in 3.20 L of MeOH by heating, the solution was concentrated. To the residue, 1.66 L of MeCN, 5.72 mL of AcOH(0.1 eq.) and 82.6 g of (S)-2-Amino-propan-1-ol(1.1 eq.) were added and the mixture was heated to 70 °C, stirred at 70 0C for 4 h and concentrated. To the residue, 1.67 L of 2-propanol was added and the mixture was concentrated (twice). After cooling of the residue, filtration, washing with 500 mL of cold 2-propanol and drying provided 167 g of compound 14 (52% yield) as a crystal. 1H NMR(300 MHz1 CDCI3) δ 7.61-7.55 (m, 2H), 7.40-7.20 (m, 4H), 6.53 (d, J = 7.2, 1H), 5.46 (d, J = 10.5 Hz, 1H), 5.23 (d, J = 10.2 Hz, 1H), 5.20 (dd, J = 3.9, 9.6 Hz, 1H), 4.46- 4.34 (m, 1H)1 4.31 (dd, J = 6.6, 8.7 Hz, 1H)1 4.14 (dd, J = 3.9, 12.3 Hz1 1H)1 3.79 (dd, J = 9.9, 12.3 Hz1 1 H), 3.62 (dd, J = 6.9, 8.7 Hz1 1 H), 1.38 (d, J = 6.3 Hz1 3H).
Example Ab
To slurry of 156 g of compound 14 (1.0 eq.) in 780 ml_ of NMP was added 93.6 g of NBS(1.1 eq.) and the mixture was stirred at room temperature for 2.5 h. The reaction mixture was added to 3.12 L of H2O. Filtration, washing with 8.0 L of H2O and drying provided 163 g of compound 15 (84% yield) as a crystal.
1H NMR(300 MHz, DMSO-CT6) δ 8.37 (s, 1H), 7.55-7.50 (m, 2H), 7.42-7.25 (m, 3H), 5.34 (dd, J = 3.6, 9.9 Hz, 1H), 5.18 (d, J = 10.8 Hz, 1H), 5.03 (d, J = 10.5 Hz, 1H), 4.53 (dd, J = 3.6, 12.0 Hz, 1H)1 4.40-4.20 (m, 2H), 3.99 (dd, J = 9.9, 11.7 Hz1 1H), 3.64 (dd, J = 5.7, 8.1 Hz1 1 H)1 1.27 (d, J = 6.3 Hz1 3H).
Example Ac
Under carbon mono-oxide atmosphere, a mixture of 163 g of compound 15 (1.0 eq.), 163 mL of /-Pr2NEt(2.5 eq.), 68.4 ml_ of 2,4-difluorobenzylamine(1.5 eq.) and 22.5 g of Pd(PPh3)4(0.05 eq.) in 816 mL of DMSO was stirred at 90 0C for 7 h. After cooling, removal of precipitate, washing with 50 mL of DMSO and addition of 11.3 g of
Pd(PPh3)4(0.025 eq.), the reaction mixture was stirred at 90 0C for 2 h under carbon mono-oxide atmosphere again. After cooling, removal of precipitate and addition of 2.0 L of AcOEt and 2.0 L of H2O1 the organic layer was washed with 1.0 L of 1 N HCIaq. and 1.0 L of H2O (twice) and the aqueous layer was extracted with 1.0 L of AcOEt. The organic layers were combined and concentrated. Silica gel column chromatography of the residue provided 184 g of compound 16 (96% yield) as foam.
1H NMR(300 MHz, CDCI3) δ 10.38 (t, J = 6.3 Hz1 1H)1 8.39 (s, 1H)1 7.75-7.25 (m, 7H), 6.90-6.70 (m, 2H), 5.43 (d, J = 10.2 Hz, 1H), 5.24 (d, J = 10.2 Hz, 1H)1 5.19 (dd, J = 3.9, 9.9 Hz, 1H)1 4.63 (d, J = 6.0 Hz, 2H), 4.50-4.25 (m, 3H)1 3.86 (dd, J = 9.9, 12.3 Hz, 1H), , 3.66 (dd, J = 6.9, 8.4 Hz1 1 H), 1.39 (d, J = 6.0 Hz, 3H).
Example Ad
Under hydrogen atmosphere, a mixture of 184 g of compound 16 (1.0 eq.) and 36.8 g of 10%Pd-C in 3.31 L of THF and 0.37 L of MeOH was stirred for 3 h. After filtration of precipitate(Pd-C), washing with THF/MeOH(9/1 ) and addition of 36.8 g of 10% Pd-C, the mixture was stirred for 20 min under hydrogen atmosphere. After filtration of precipitate(Pd-C) and washing with THF/MeOH(9/1), the filtrate was concentrated. After 200 ml_ of AcOEt was added to the residue, filtration afforded crude solid of compound 17. The precipitates were combined and extracted with 4.0 L of CHCl3/MeOH(5/1). After concentration of the CHCI3ZMeOH solution and addition of 250 ml_ of AcOEt to the residue, filtration afforded crude solid of compound 17. The crude solids were combined and dissolved in 8.2 L of MeCN/H2O(9/1 ) by heating. After filtration, the filtrate was concentrated. To the residue, 1.5 L of EtOH was added and the mixture was concentrated (three times). After cooling of the residue, filtration and drying provided 132 g of compound 17 (88% yield) as a crystal. 1H NMR(300 MHz, DMSO-cfe) δ 11.47 (brs, 1H), 10.31 (t, J = 6.0 Hz, 1H), 8.46 (s, 1H), 7.40 (td, J = 8.6, 6.9 Hz, 1H), 7.24 (ddd, J = 2.6, 9.4, 10.6, 1H), 7.11-7.01 (m, 1H), 5.39 (dd, J = 4.1, 10.4 Hz, 1H), 4.89 (dd, J = 4.2, 12.3 Hz, 1H), 4.55 (d, J = 6.0 Hz, 2H), 4.40 (dd, J = 6.8, 8.6 Hz, 1H), 4.36-^.22 (m, 1H)1 4.00 (dd, J = 10.2, 12.3 Hz, 1H), 3.67 (dd, J = 6.7, 8.6 Hz, 1H), 1.34 (d, J = 6.3 Hz, 3H).
Example Ae
After dissolution of 16.0 g of compound 17 (1.0 eq.) in 2.56 L of EtOH and 0.64 L of H2O by heating, followed by filtration, 39 ml_ of 1N NaOHaq.(1.0 eq.) was added to the solution at 75 0C. The solution was gradually cooled to room temperature. Filtration, washing with 80 ml_ of EtOH and drying provided 13.5 g of compound 18 (80% yield) as a crystal.
1H NMR(300 MHz, DMSO-cfe) δ 10.73 (t, J = 6.0 Hz, 1H), 7.89 (s, 1H), 7.40-7.30 (m, 1H), 7.25-7.16 (m, 1H), 7.07-6.98 (m, 1H), 5.21 (dd, J = 3.8, 10.0 Hz, 1H), 4.58 (dd, J = 3.8, 12.1 Hz, 1H), 4.51 (d, J = 5.4 Hz, 2H), 4.3CM.20 (m, 2H), 3.75 (dd, J = 10.0, 12.1 Hz, 1H), 3.65-3.55 (m, 1H), 1.27 (d, J = 6.1 Hz, 3H).
………………
WO2010011814
http://www.google.st/patents/WO2010011814A1?cl=en&hl=pt-PT
Scheme 1

2a 2b
Scheme 2

Scheme 3

Scheme 4
phosphorylation


Scheme 5
Hydrogenolysis



The following examples are intended for illustratation only and are not intended to limit the scope of the invention in any way. Preparation 1 : (3S.11 af?VΛ/-r(2.4-DifluoroDhenvnmethyll-6-hvdroxy-3-methyl-5.7-dioxo- 2,3,5,7, 11 ,11 a-hexahydroM ,31oxazolor3,2-alpyridori ,2-c/1pyrazine-8-carboxamide sodium salt (compound 1 b, scheme 2).
I) MsCI, Et3N

2) DBU
P-1 P-2 P-3

a) Synthesis of 2-methyl-3-[(phenylmethvl)oxvl-4/-/-pvran-4-one (compound P-2). To a slurry of 2000 g of compound P-1(1.0 eq.) in 14.0 L of MeCN were added 2848 g of benzyl bromide(1.05 eq.) and 2630 g of K2CO3(1.2 eq.). The mixture was stirred at 80 0C for 5 h and cooled to 13°C. Precipitate was filtered and washed with 5.0 L of MeCN. The filtrate was concentrated and 3.0 L of THF was added to the residue. The THF solution was concentrated to give 3585 g of crude compound P-2 as oil. Without further purification, compound P-2 was used in the next step. 1H NMR(300 MHz, CDCI3) δ 7.60 (d, J = 5.7 Hz, 1 H), 7.4-7.3 (m, 5H), 6.37 (d, J = 5.7 Hz, 1 H), 5.17 (s, 2H), 2.09 (s, 3H).
b) Synthesis of 2-(2-hydroxy-2-phenylethyl)-3-[(phenylmethyl)oxy]-4H-pyran-4-one (compound P-3). To 904 g of the crude compound P-2 was added 5.88 L of THF and the solution was cooled to -60 0C. 5.00 L of 1.0 M of Lithium bis(trimethylsilylamide) in THF(1.25 eq.) was added dropwise for 2 h to the solution of compound 2 at -60 0C. Then, a solution of 509 g of benzaldehyde(1.2 eq.) in 800 ml. of THF was added at -60 0C and the reaction mixture was aged at -60 0C for 1 h. The THF solution was poured into a mixture of 1.21 L of conc.HCI, 8.14 L of ice water and 4.52 L of EtOAc at less than 2 0C.
The organic layer was washed with 2.71 L of brine (twice) and the aqueous layer was extracted with 3.98 L of EtOAc. The combined organic layers were concentrated. To the mixture, 1.63 L of toluene was added and concentrated (twice) to provide toluene slurry of compound P-3. Filtration, washing with 0.90 L of cold toluene and drying afforded 955 g of compound P-3 (74% yield from compound P-1 ) as a solid. 1H NMR(300 MHz, CDCI3) δ
7.62 (d, J = 5.7 Hz, 1 H), 7.5-7.2 (m, 10H), 6.38 (d, J = 5.7 Hz, 1 H), 5.16 (d, J = 11.4 Hz, 1 H), 5.09 (d, J = 11.4 Hz, 1 H), 4.95 (dd, J = 4.8, 9.0 Hz, 1 H), 3.01 (dd, J = 9.0, 14.1 Hz, 1 H), 2.84 (dd, J = 4.8, 14.1 Hz, 1 H).
c) Synthesis of 2-[(£)-2-phenylethenyl]-3-[(phenylmethyl)oxy]-4H-pyran-4-one (compound
P-4). To a solution of 882 g of compound P-3 (1.0 eq.) in 8.82 L of THF were added 416 g of Et3N(1.5 eq.) and 408 g of methanesulfonyl chloride(1.3 eq.) at less than 30 0C. After confirmation of disappearance of compound P-3, 440 ml. of NMP and 1167 g of DBU(2.8 eq.) were added to the reaction mixture at less than 30 0C and the reaction mixture was aged for 30 min. The mixture was neutralized with 1.76 L of 16% sulfuric acid and the organic layer was washed with 1.76 L of 2% Na2S03aq. After concentration of the organic layer, 4.41 L of toluene was added and the mixture was concentrated (tree times). After addition of 4.67 L of hexane, the mixture was cooled with ice bath. Filtration, washing with 1.77 L of hexane and drying provided 780 g of compound P-4 (94% yield) as a solid. 1H NMR(300 MHz, CDCI3) δ 7.69 (d, J = 5.7 Hz, 1 H), 7.50-7.25 (m, 10H), 7.22 (d, J = 16.2
Hz, 1 H), 7.03 (d, J = 16.2 Hz, 1 H), 6.41 (d, J = 5.7 Hz, 1 H), 5.27 (s, 2H). d) Synthesis of 4-oxo-3-[(phenylmethyl)oxy]-4H-pyran-2-carboxylic acid (compound P-5). To a mixture of 822 g of compound P-4 (1.0 eq.) and 1 1.2 g of RuCI3-nH2O(0.02 eq.) in 2.47 L of MeCN, 2.47 L of EtOAc and 2.47 L of H2O was added 2310 g of NalO4(4.0 eq.) at less than 25 0C. After aging for 1 h, 733 g of NaCIO2(S-O eq.) was added to the mixture at less than 25 0C. After aging for 1 h, precipitate was filtered and washed with 8.22 L of
EtOAc. To the filtrate, 1.64 L of 50% Na2S203aq, 822 ml. of H2O and 630 ml. of coc.HCI were added. The aqueous layer was extracted with 4.11 L of EtOAc and the organic layers were combined and concentrated. To the residue, 4 L of toluene was added and the mixture was concentrated and cooled with ice bath. Filtration, washing with 1 L of toluene and drying provided 372 g of compound P-5 (56% yield) as a solid. 1H NMR(300 MHz,
CDCI3) δ 7.78 (d, J = 5.7 Hz, 1 H), 7.54-7.46 (m, 2H), 7.40-7.26 (m, 3H), 6.48 (d, J = 5.7 Hz, 1 H), 5.6 (brs, 1 H), 5.31 (s, 2H).
e) Synthesis of 1-(2,3-dihydroxypropyl)-4-oxo-3-[(phenylmethyl)oxy]-1 ,4-dihydro-2- pyridinecarboxylic acid (compound P-6). A mixture of 509 g of compound P-5 (1.0 eq.) and
407 g of 3-amino-propane-1 ,2-diol(2.5 eq.) in 1.53 L of EtOH was stirred at 65 0C for 1 h and at 80 0C for 6 h. After addition of 18.8 g of 3-Amino-propane-1 ,2-diol(0.1 eq.) in 200 ml. of EtOH, the mixture was stirred at 80 0C for 1 h. After addition of 18.8 g of 3-amino- propane-1 ,2-diol (0.1 eq.) in 200 ml. of EtOH, the mixture was stirred at 80 0C for 30 min. After cooling and addition of 509 ml. of H2O, the mixture was concentrated. To the residue,
2.54 L of H2O and 2.54 L of AcOEt were added. After separation, the aqueous layer was washed with 1.02 L of EtOAc. To the aqueous layer, 2.03 L of 12% sulfuric acid was added at less than 12 0C to give crystal of compound P-6. Filtration, washing with 1.53 L of cold H2O and drying provided 576 g of compound P-6 (83% yield) as a solid. 1H NMR(300 MHz, DMSO-de) δ 7.67 (d, J = 7.5 Hz, 1 H), 7.5-7.2 (m, 5H), 6.40 (d, J = 7.5 Hz, 1 H), 5.07
(s, 2H), 4.2-4.0 (m, 1 H), 3.9-3.6 (m, 2H), 3.38 (dd, J = 4.2, 10.8 Hz, 1 H), 3.27 (dd, J = 6.0, 10.8 Hz, 1 H).
f) Synthesis of methyl 1-(2,3-dihydroxypropyl)-4-oxo-3-[(phenylmethyl)oxy]-1 ,4-dihydro-2- pyridinecarboxylate (compound P-7). To a slurry of 576 g of compound P-6 (1.0 eq.: 5.8% of H2O was contained) in 2.88 L of NMP were added 431 g of NaHCO3(3.0 eq.) and 160 ml. of methyl iodide(1.5 eq.) and the mixture was stirred at room temperature for 4 h. After cooling to 5 0C, 1.71 L of 2N HCI and 1.15 L of 20% NaClaq were added to the mixture at less than 10 0C to give crystal of compound 7. Filtration, washing with 1.73 L of H2O and drying provided 507 g of compound P-7 (89% yield) as a solid. 1H NMR(300 MHz, DMSO- cfe) δ 7.59 (d, J = 7.5 Hz, 1 H), 7.40-7.28 (m, 5H), 6.28 (d, J = 7.5 Hz, 1 H), 5.21 (d, J = 5.4 Hz, 1 H), 5.12 (d, J = 10.8 Hz, 1 H), 5.07 (d, J = 10.8 Hz, 1 H), 4.83 (t, J = 5.7 Hz, 1 H), 3.97 (dd, J = 2.4, 14.1 Hz, 1 H), 3.79 (s, 3H), 3.70 (dd, J = 9.0, 14.4 Hz, 1 H), 3.65-3.50 (m, 1 H), 3.40-3.28 (m, 1 H), 3.26-3.14 (m, 1 H).
g) Synthesis of methyl 1-(2,2-dihydroxyethyl)-4-oxo-3-[(phenylmethyl)oxy]-1 ,4-dihydro-2- pyridinecarboxylate (compound P-8). To a mixture of 507 g of compound P -7 (1.0 eq.) in
5.07 L of MeCN, 5.07 L of H2O and 9.13 g of AcOH(0.1 eq.) was added 390 g of NaIO4(1.2 eq.) and the mixture was stirred at room temperature for 2 h. After addition of 1.52 L of 10% Na2S2OsBq., the mixture was concentrated and cooled to 10 0C. Filtration, washing with H2O and drying provided 386 g of compound P-8 (80% yield) as a solid. 1H NMR(300 MHz, DMSO-d6) δ 7.62 (d, J = 7.5 Hz, 1 H), 7.42-7.30 (m, 5H), 6.33 (d, J = 6.0 Hz, 2H),
6.29 (d, J = 7.5 Hz, 1 H), 5.08 (s, 2H), 4.95-4.85 (m, 1 H), 3.80 (s, 3H), 3.74 (d, J = 5.1 Hz, 2H).
h) Synthesis of (3S, 11 aR)-3-methyl-6-[(phenylmethyl)oxy]-2,3, 1 1 ,1 1a- tetrahydro[1 ,3]oxazolo[3,2-a]pyrido[1 ,2-c/]pyrazine-5,7-dione (compound P-9). After dissolution of mixture of 320 g of compound P-8 (1.0 eq.) in 3.20 L of MeOH by heating, the solution was concentrated. To the residue, 1.66 L of MeCN, 5.72 ml. of AcOH(0.1 eq.) and 82.6 g of (S)-2-Amino-propan-1-ol(1.1 eq.) were added and the mixture was heated to 70 0C, stirred at 70 0C for 4 h and concentrated. To the residue, 1.67 L of 2-propanol was added and the mixture was concentrated (twice). After cooling of the residue, filtration, washing with 500 ml. of cold 2-propanol and drying provided 167 g of compound P-9 (52% yield) as a solid. 1H NMR(300 MHz, CDCI3) δ 7.61-7.55 (m, 2H), 7.40-7.20 (m, 4H), 6.53 (d, J = 7.2, 1 H), 5.46 (d, J = 10.5 Hz, 1 H), 5.23 (d, J = 10.2 Hz, 1 H), 5.20 (dd, J = 3.9, 9.6 Hz, 1 H), 4.46-4.34 (m, 1 H), 4.31 (dd, J = 6.6, 8.7 Hz, 1 H), 4.14 (dd, J = 3.9, 12.3 Hz, 1 H), 3.79 (dd, J = 9.9, 12.3 Hz, 1 H), 3.62 (dd, J = 6.9, 8.7 Hz, 1 H), 1.38 (d, J = 6.3 Hz, 3H).
i) Synthesis of (3 S, 1 1 aR)-8-bromo-3-methyl-6-[(phenylmethyl)oxy]-2,3, 11 ,11a- tetrahydro[1 ,3]oxazolo[3,2-a]pyrido[1 ,2-c/]pyrazine-5,7-dione (compound P-10). To slurry of 156 g of compound P-9 (1.0 eq.) in 780 ml. of NMP was added 93.6 g of NBS(1.1 eq.) and the mixture was stirred at room temperature for 2.5 h. The reaction mixture was added to 3.12 L of H2O. Filtration, washing with 8.0 L of H2O and drying provided 163 g of compound P-10 (84% yield) as a solid. 1H NMR(300 MHz, DMSO-d6) δ 8.37 (s, 1 H), 7.55- 7.50 (m, 2H), 7.42-7.25 (m, 3H), 5.34 (dd, J = 3.6, 9.9 Hz, 1 H), 5.18 (d, J = 10.8 Hz, 1 H), 5.03 (d, J = 10.5 Hz, 1 H), 4.53 (dd, J = 3.6, 12.0 Hz, 1 H), 4.40-4.20 (m, 2H), 3.99 (dd, J = 9.9, 1 1.7 Hz, 1 H), 3.64 (dd, J = 5.7, 8.1 Hz, 1 H), 1.27 (d, J = 6.3 Hz, 3H). j) Synthesis of (3S,1 1aS)-Λ/-[(2,4-difluorophenyl)methyl]-3-methyl-5,7-dioxo-6- [(phenylmethyl)oxy]-2,3,5,7, 11 ,1 1 a-hexahydro[1 ,3]oxazolo[3,2-a]pyrido[1 ,2-c/]pyrazine-8- carboxamide (compound P-11). Under carbon mono-oxide atmosphere, a mixture of 163 g of compound P-10 (1.0 eq.), 163 mL of /-Pr2NEt(2.5 eq.), 68.4 mL of 2,4- difluorobenzylamine(1.5 eq.) and 22.5 g of Pd(PPh3)4(0.05 eq.) in 816 mL of DMSO was stirred at 90 0C for 7 h. After cooling, removal of precipitate, washing with 50 mL of DMSO and addition of 1 1.3 g of Pd(PPh3)4(0.025 eq.), the reaction mixture was stirred at 90 0C for 2 h under carbon mono-oxide atmosphere again. After cooling, removal of precipitate and addition of 2.0 L of AcOEt and 2.0 L of H2O, the organic layer was washed with 1.0 L of 1 N HCIaq. and 1.0 L of H2O (twice) and the aqueous layer was extracted with 1.0 L of AcOEt.
The organic layers were combined and concentrated. Silica gel column chromatography of the residue provided 184 g of compound P-11 (96% yield) as foam. 1H NMR(300 MHz, CDCI3) δ 10.38 (t, J = 6.3 Hz, 1 H), 8.39 (s, 1 H), 7.75-7.25 (m, 7H), 6.90-6.70 (m, 2H), 5.43 (d, J = 10.2 Hz, 1 H), 5.24 (d, J = 10.2 Hz, 1 H), 5.19 (dd, J = 3.9, 9.9 Hz, 1 H), 4.63 (d, J = 6.0 Hz, 2H), 4.50-4.25 (m, 3H), 3.86 (dd, J = 9.9, 12.3 Hz, 1 H), 3.66 (dd, J = 6.9, 8.4 Hz,
1 H), 1.39 (d, J = 6.0 Hz, 3H).
k) Synthesis of (3S,1 1aR)-Λ/-[(2,4-difluorophenyl)methyl]-6-hydroxy-3-methyl-5,7-dioxo- 2,3,5,7, 11 ,11 a-hexahydro[1 ,3]oxazolo[3,2-a]pyrido[1 ,2-c/]pyrazine-8-carboxamide (compound 1a). Under hydrogen atmosphere, a mixture of 184 g of compound P-11 (1.0 eq.) and 36.8 g of 10%Pd-C in 3.31 L of THF and 0.37 L of MeOH was stirred for 3 h. After filtration of precipitate(Pd-C), washing with THF/MeOH(9/1 ) and addition of 36.8 g of 10% Pd-C, the mixture was stirred for 20 min under hydrogen atmosphere. After filtration of precipitate(Pd-C) and washing with THF/MeOH(9/1 ), the filtrate was concentrated. After 200 mL of AcOEt was added to the residue, filtration afforded crude solid of compound 1 a.
The precipitates were combined and extracted with 4.0 L of CHCI3/Me0H(5/1 ). After concentration of the CHCI3/MeOH solution and addition of 250 mL of AcOEt to the residue, filtration afforded crude solid of compound 1a. The crude solids were combined and dissolved in 8.2 L of MeCN/H2O(9/1 ) by heating. After filtration, the filtrate was concentrated. To the residue, 1.5 L of EtOH was added and the mixture was concentrated
(three times). After cooling of the residue, filtration and drying provided 132 g of compound 1a (88% yield) as a solid. 1H NMR(300 MHz, DMSO-d6) δ 11.47 (brs, 1 H), 10.31 (t, J = 6.0 Hz, 1 H), 8.46 (s, 1 H), 7.40 (td, J = 8.6, 6.9 Hz, 1 H), 7.24 (ddd, J = 2.6, 9.4, 10.6, 1 H), 7.11- 7.01 (m, 1 H), 5.39 (dd, J = 4.1 , 10.4 Hz, 1 H), 4.89 (dd, J = 4.2, 12.3 Hz, 1 H), 4.55 (d, J = 6.0 Hz, 2H), 4.40 (dd, J = 6.8, 8.6 Hz, 1 H), 4.36-4.22 (m, 1 H), 4.00 (dd, J = 10.2, 12.3 Hz, 1 H), 3.67 (dd, J = 6.7, 8.6 Hz, 1 H), 1.34 (d, J = 6.3 Hz, 3H).
I) Synthesis of (3S,1 1aR)-Λ/-[(2,4-difluorophenyl)methyl]-6-hydroxy-3-methyl-5,7-dioxo- 2,3,5,7, 11 ,11 a-hexahydro[1 ,3]oxazolo[3,2-a]pyrido[1 ,2-c/]pyrazine-8-carboxamide sodium salt (compound 1 b). After dissolution of 16.0 g of compound 1a (1.0 eq.) in 2.56 L of EtOH and 0.64 L of H2O by heating, followed by filtration, 39 ml. of 1 N NaOHaq.(1.0 eq.) was added to the solution at 75 0C. The solution was gradually cooled to room temperature. Filtration, washing with 80 ml. of EtOH and drying provided 13.5 g of compound 1b (80% yield) as a solid. 1H NMR(300 MHz, DMSO-d6) δ 10.73 (t, J = 6.0 Hz, 1 H), 7.89 (s, 1 H), 7.40-7.30 (m, 1 H), 7.25-7.16 (m, 1 H), 7.07-6.98 (m, 1 H), 5.21 (dd, J = 3.8, 10.0 Hz, 1 H), 4.58 (dd, J = 3.8, 12.1 Hz, 1 H), 4.51 (d, J = 5.4 Hz, 2H), 4.30-4.20 (m, 2H), 3.75 (dd, J = 10.0, 12.1 Hz, 1 H), 3.65-3.55 (m, 1 H), 1.27 (d, J = 6.1 Hz, 3H).
updates

An Efficient and Highly Diastereoselective Synthesis of GSK1265744, a Potent HIV Integrase Inhibitor
Abstract

Bictegravir and dolutegravir are two recently approved integrase inhibitors for the treatment of HIV. A third inhibitor, cabotegravir, is in Phase 3 development. As a continuation of a series of articles on synthetic routes to newly approved drugs, the current article reviews the patent and journal literature regarding synthetic routes and final forms of these drug





References
- ^ Jump up to:a b c “Vocabria (cabotegravir) film-coated tablets Product Information”. TGA eBS. 12 June 2021. Retrieved 12 June 2021.
- ^ Jump up to:a b “Vocabria”. Therapeutic Goods Administration (TGA). 26 February 2021. Retrieved 8 September 2021.
- ^ “Summary for ARTG Entry: 323721 VOCABRIA cabotegravir (as sodium) 30 mg film-coated tablet, bottle”. Therapeutic Goods Administration. The G overnment of Australia. 13 August 2021.
- ^ “Vocabria Product information”. Health Canada. 25 April 2012. Retrieved 22 January 2021.
- ^ Jump up to:a b c d e f g “Vocabria- cabotegravir sodium tablet, film coated”. DailyMed. Retrieved 12 June 2021.
- ^ Jump up to:a b “FDA Approves First Extended-Release, Injectable Drug Regimen for Adults Living with HIV”. U.S. Food and Drug Administration (FDA) (Press release). 21 January 2021. Retrieved 21 January 2021.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ “Apretude- cabotegravir kit”. DailyMed. Retrieved 24 December 2021.
- ^ Jump up to:a b “Vocabria EPAR”. European Medicines Agency (EMA). Retrieved 22 January 2021. Text was copied from this source which is © European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ Jump up to:a b c d e f g h i “Vocabria: EPAR – Product information” (PDF). European Medicines Agency. 5 January 2021.
- ^ Borrell B (March 2014). “Long-acting shot prevents infection with HIV analogue”. Nature News. doi:10.1038/nature.2014.14819. S2CID 184399045.
- ^ Jump up to:a b c d e f g h i j k l m “FDA Approves First Injectable Treatment for HIV Pre-Exposure Prevention”. U.S. Food and Drug Administration (FDA) (Press release). 20 December 2021. Retrieved 21 December 2021. This article incorporates text from this source, which is in the public domain.
- ^ Jump up to:a b c “First long-acting injectable antiretroviral therapy for HIV recommended approval” (Press release). European Medicines Agency (EMA). 16 October 2020. Retrieved 16 October 2020. Text was copied from this source which is © European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ “Cabenuva- cabotegravir and rilpivirine kit”. DailyMed. Retrieved 12 June 2021.
- ^ “Edurant- rilpivirine hydrochloride tablet, film coated”. DailyMed. Retrieved 12 June 2021.
- ^ “Rilpivirine Monograph for Professionals”. Drugs.com. 24 September 2020. Retrieved 12 June 2021.
- ^ “A Treatment Option | CABENUVA (cabotegravir; rilpivirine)”. http://www.cabenuva.com. Retrieved 16 April 2022.
- ^ Patel M, Eberl HC, Wolf A, Pierre E, Polli JW, Zamek-Gliszczynski MJ (August 2019). “Mechanistic Basis of Cabotegravir-Glucuronide Disposition in Humans”. The Journal of Pharmacology and Experimental Therapeutics. 370 (2): 269–277. doi:10.1124/jpet.119.258384. PMID 31175220. S2CID 182950312.
- ^ Jump up to:a b “Vocabria: EPAR – Public assessment report” (PDF). European Medicines Agency. 5 January 2021.
- ^ “HPTN083 — Prevention Now”. HIV Prevention Trials Network. Retrieved 2 December 2017.
- ^ “HPTN084 — Prevention Now”. HIV Prevention Trials Network. Retrieved 2 December 2017.
- ^ “Vocabria: Pending EC decision”. European Medicines Agency (EMA). 16 October 2020. Retrieved 16 October 2020. Text was copied from this source which is © European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ “Adopted USANs” (PDF). American Medical Association. Retrieved 19 September 2014.
- ^ World Health Organization (2015). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 73”. WHO Drug Information. 29 (1): 70–1. hdl:10665/331088.
- ^ Ryan G (7 July 2020). “Injectable PrEP Is Even More Effective Than Daily Truvada”. Poz. Retrieved 9 November 2020.
- ^ Ryan G (9 November 2020). “For Women, Injectable Cabotegravir Is More Effective Than Truvada as PrEP”. Poz. Retrieved 9 November 2020.
External links
- “Cabotegravir”. Drug Information Portal. U.S. National Library of Medicine.
References
- PrEP GSK744 Integrase Administered Monthly Perhaps Quarterly Prevents HIV-Infection in Monkeys. 20th Conference on Retroviruses and Opportunistic Infections. Atlanta, GA March 3–6, 2013.
- http://www.natap.org/2013/CROI/croi_38.htm
SMILES [H][C@@]12CN3C=C(C(=O)NCC4=C(F)C=C(F)C=C4)C(=O)C(O)=C3C(=O)N1[C@@H](C)CO2
| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2006116764A1 * | Apr 28, 2006 | Nov 2, 2006 | Shionogi & Co | Polycyclic carbamoylpyridone derivative having hiv integrase inhibitory activity |
| US6919351 * | Oct 9, 2001 | Jul 19, 2005 | Merck & Co., Inc. | Aza-and polyaza-naphthalenyl-carboxamides useful as HIV integrase inhibitors |
| WO2012018065A1 * | Aug 4, 2011 | Feb 9, 2012 | Shionogi & Co., Ltd. | Process for preparing compound having hiv integrase inhibitory activity |
| WO2012151361A1 | May 3, 2012 | Nov 8, 2012 | Concert Pharmaceuticals Inc. | Carbamoylpyridone derivatives |
| WO2013038407A1 * | Sep 2, 2012 | Mar 21, 2013 | Mapi Pharma Ltd. | Amorphous form of dolutegravir |
| US8552187 | Dec 9, 2009 | Oct 8, 2013 | Shionogi & Co., Ltd. | Processes and intermediates for carbamoylpyridone HIV integrase inhibitors |
| US8580967 | Jul 23, 2009 | Nov 12, 2013 | Shionogi & Co., Ltd. | Methyl 3-(benzyloxy)-1-(2,2-dihydroxyethyl)-4-oxo-1,4-dihydropyridine-2-carboxylate and processes for the preparation thereof |
| US8624023 | Dec 8, 2009 | Jan 7, 2014 | Shionogi & Co., Ltd. | Synthesis of carbamoylpyridone HIV integrase inhibitors and intermediates |
SYN
Synthetic Reference
Dolutegravir HELP . ADDED TO AID HELP
Dolutegravir HELP . ADDED TO AID HELP
Dolutegravir HELP . ADDED TO AID HELP
Dolutegravir HELP . ADDED TO AID HELP
Aoyama, Yasunori; Hakogi, Toshikazu; Fukui, Yuki; Yamada, Daisuke; Ooyama, Takao; Nishino, Yutaka; Shinomoto, Shoji; Nagai, Masahiko; Miyake, Naoki; Taoda, Yoshiyuki; Yoshida, Hiroshi; Yasukata, Tatsuro. Practical and Scalable Synthetic Method for Preparation of Dolutegravir Sodium: Improvement of a Synthetic Route for Large-Scale Synthesis. Organic Process Research & Development. Volume 23. Issue 4. Pages 558-564. Journal; Online Computer File. (2019).
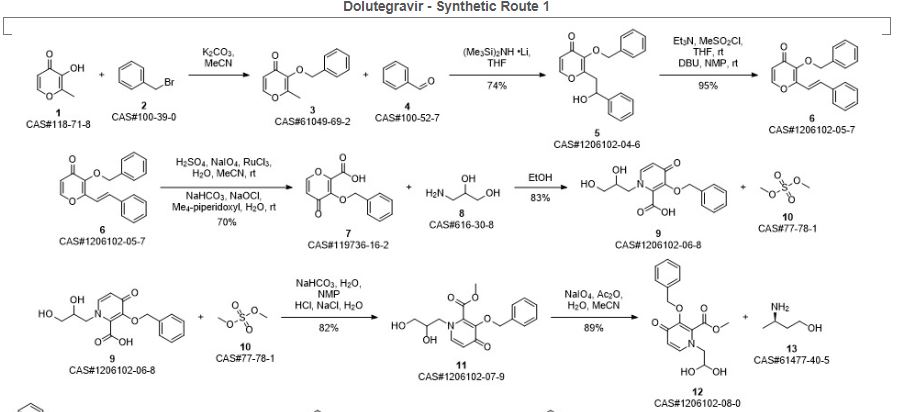
Synthetic Reference 2
Wang, Xianheng; Chen, Song; Zhao, Changkuo; Long, Liangye; Wang, Yuhe. Preparation of Dolutegravir Intermediate Diastereomer. Journal of Heterocyclic Chemistry. Volume 56. Issue 7. Pages 2063-2067. Journal; Online Computer File. (2019).

Synthetic Reference 3
Ziegler, Robert E.; Desai, Bimbisar K.; Jee, Jo-Ann; Gupton, B. Frank; Roper, Thomas D.; Jamison, Timothy F. 7-Step Flow Synthesis of the HIV Integrase Inhibitor Dolutegravir. Angewandte Chemie, International Edition. Volume 57. Issue 24. Pages 7181-7185. Journal; Online Computer File. (2018).

SYN 4
Synthetic Reference
Rajan, Srinivasan Thirumalai; Eswaraiah, Sajja; Reddy, Ghojala Venkat; Reddy, Sagyam Rajeshwar; Markandeya, Bekkam; Rajesham, Boge. Novel crystalline polymorph of sodium (4R,12aS)-9-{[(2,4-difluorophenyl)methyl]carbamoyl}-4-methyl-6,8-dioxo-3,4,6,8,12,12a-hexahydro-2H-pyrido[1′,2′:4,5]pyrazino[2,1-b][1,3]oxazin-7-olate and process for preparation thereof. Assignee MSN Research & Development Center, India. IN 201641037221. (2018).

Synthetic Reference 5
Sharma, Pramodkumar; Rao, Bhatraju Srinivasa; Deo, Keshav. A process for the preparation of Dolutegravir or its pharmaceutical acceptable salts thereof. Assignee Wockhardt Limited, India. IN 2015MU01007. (2016).

Synthetic Reference 6
Weaver, Jimmie Dean. Preparation of fluoroarenes via hydrogen bond directed photocatalytic hydrodefluorination of perfluoroarenes. Assignee The Board of Regents for Oklahoma State University, USA. WO 2018187336. (2018).

SYN 7
Synthetic Reference
Li, Xuguang; Chen, Shuiku; Zhu, Songlin; Zhang, Fangjie; Fang, Shuixia; Liu, Congjun; Zhu, Huifeng; Luo, Qi; Meng, Qingyue; Cui, Hao. Method for preparation of dolutegravir. Kaifeng Pharmaceutical (Group) Co., Ltd.; Henan Furen Pharmaceutical Technology Development Co., Ltd. CN 108299466. (2018).


SYN 8
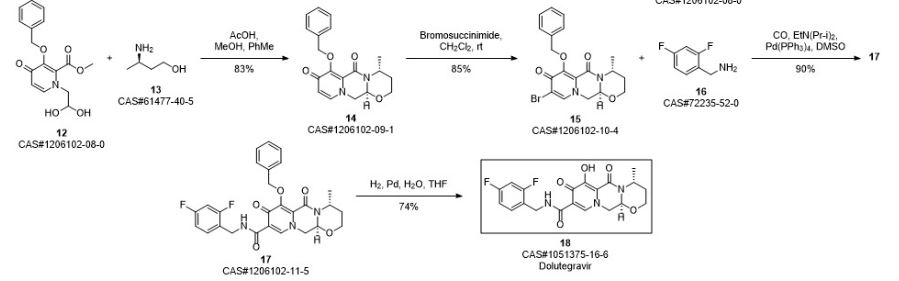
Synthetic Reference
Vellanki, Sivaram Prasad; Nadella, Madumurthy; Bhalme, Mitali; Ramabhotla, Revathi Srinivas. Process for the preparation of dolutegravir, an integrase inhibitor for HIV-1 infection therapy. Assignee Mylan Laboratories Ltd., India. IN 2015CH00588. (2016).
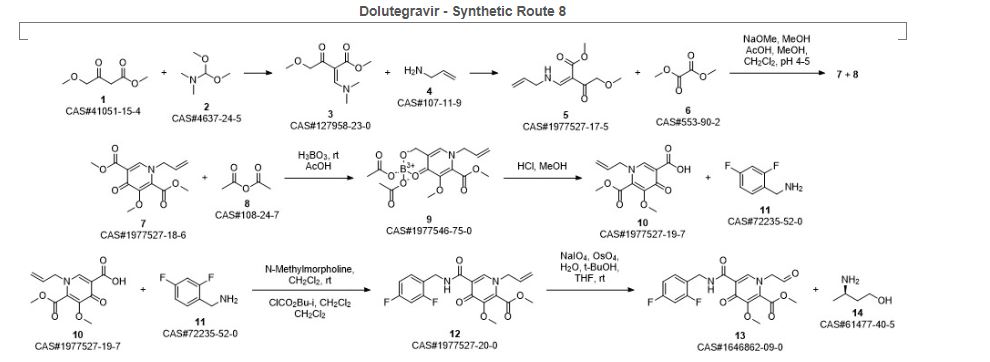

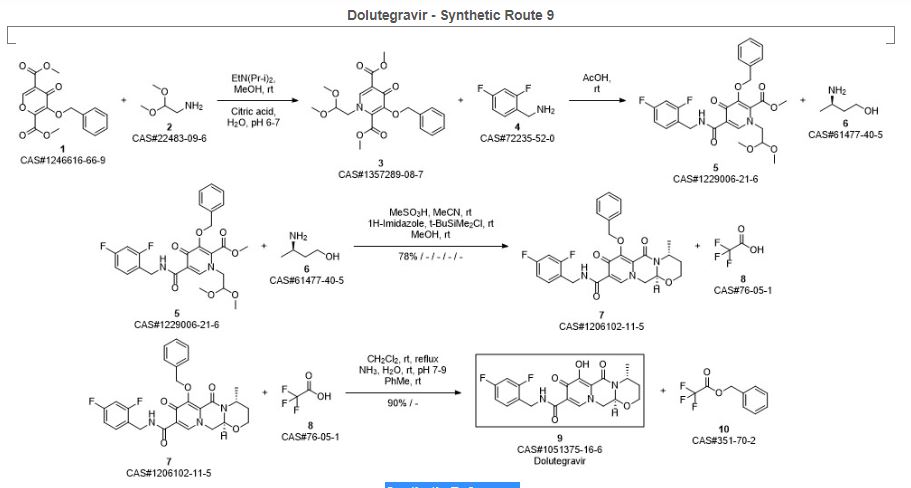
SYN 9
Synthetic Reference
Sankareswaran, Srimurugan; Mannam, Madhavarao; Chakka, Veerababu; Mandapati, Srirami Reddy; Kumar, Pramod. Identification and Control of Critical Process Impurities: An Improved Process for the Preparation of Dolutegravir Sodium. Organic Process Research & Development. Volume 20. Issue 8. Pages 1461-1468. Journal; Online Computer File. (2016).


.svg){kind=link}
{kind=link}
Ivan ont wait
LikeLike