PROUD Indian WORLD RECORD VIEWS holder on THIS BLOG, ………live, by DR ANTHONY MELVIN CRASTO, Worldpeaceambassador, Worlddrugtracker, Helping millions, 100 million hits on google, pushing boundaries,2.5 lakh plus connections worldwide, 45 lakh plus VIEWS on this blog in 227 countries, 7 CONTINENTS ……A 90 % paralysed man in action for you, I am suffering from transverse mylitis and bound to a wheel chair, [THIS BLOG HOLDS WORLD RECORD VIEWS ] A PROUD INDIAN
DR ANTHONY MELVIN CRASTO, Born in Mumbai in 1964 and graduated from Mumbai University, Completed his Ph.D from ICT, 1991,Matunga, Mumbai, India, in Organic Chemistry, The thesis topic was Synthesis of Novel Pyrethroid Analogues, Currently he is working with AFRICURE PHARMA, ROW2TECH, NIPER-G, Department of Pharmaceuticals, Ministry of Chemicals and Fertilizers, Govt. of India as ADVISOR, earlier assignment was
with GLENMARK LIFE SCIENCES LTD, as CONSUlTANT, Retired from GLENMARK in Jan2022 Research Centre as Principal Scientist, Process Research (bulk actives) at Mahape, Navi Mumbai, India. Total Industry exp 32 plus yrs, Prior to joining Glenmark, he has worked with major multinationals like Hoechst Marion Roussel, now Sanofi, Searle India Ltd, now RPG lifesciences, etc. He has worked with notable scientists like Dr K Nagarajan, Dr Ralph Stapel, Prof S Seshadri, etc, He did custom synthesis for major multinationals in his career like BASF, Novartis, Sanofi, etc., He has worked in Discovery, Natural products, Bulk drugs, Generics, Intermediates, Fine chemicals, Neutraceuticals, GMP, Scaleups, etc, he is now helping millions, has 9 million plus hits on Google on all Organic chemistry websites. His friends call him Open superstar worlddrugtracker. His New Drug Approvals, Green Chemistry International, All about drugs, Eurekamoments, Organic spectroscopy international,
etc in organic chemistry are some most read blogs He has hands on experience in initiation and developing novel routes for drug molecules
and implementation them on commercial scale over a 32 PLUS year tenure till date Feb 2023, Around 35 plus products in his career. He has good knowledge of IPM, GMP, Regulatory aspects, he has several International patents published worldwide . He has good proficiency in Technology transfer, Spectroscopy, Stereochemistry, Synthesis, Polymorphism etc., He suffered a paralytic stroke/ Acute Transverse mylitis in Dec 2007 and is 90 %Paralysed, He is bound to a wheelchair, this seems to have injected feul in him to help chemists all around the world, he is more active than before and is pushing boundaries, He has 100 million plus hits on Google, 2.5 lakh plus connections on all networking sites, 100 Lakh plus views on dozen plus blogs, 227 countries, 7 continents, He makes himself available to all, contact him on +91 9323115463, email amcrasto@gmail.com, Twitter, @amcrasto , He lives and will die for his family, 90% paralysis cannot kill his soul., Notably he has 38 lakh plus views on New Drug Approvals Blog in 227 countries......https://newdrugapprovals.wordpress.com/ , He appreciates the help he gets from one and all, Friends, Family, Glenmark, Readers, Wellwishers, Doctors, Drug authorities, His Contacts, Physiotherapist, etc
He has total of 32 International and Indian awards
The most common side effects include reactions at the injection site and nausea.[1]
Lenacapavir was approved for medical use in the European Union in August 2022.[1]
HIV/AIDS remains an area of concern despite the introduction of numerous successful therapies, mainly due to the emergence of multidrug resistance and patient difficulty in adhering to treatment regimens.1,2 Lenacapavir is a first-in-class capsid inhibitor that demonstrates picomolar HIV-1 inhibition as a monotherapy in vitro, little to no cross-resistance with existing antiretroviral agents, and extended pharmacokinetics with subcutaneous dosing.1,2,3,5
Lenacapavir was first globally approved by the European Commission to treat adults with multi-drug resistant HIV infection.7 It is currently being investigated in clinical trials in the US.
U.S. Patent Application No. 15/680,041 discloses novel compounds useful for treating a Retroviridae viral infection, including an infection caused by the HIV virus. One specific compound identified therein is a compound of formula I:
PATENTS
WO 2018/035359 A1
Different formulations and salts: WO 2019/035904 A1; WO 2019/035973 A1
I. Synthesis of Starting Materials and Intermediates
Example la: Preparation of (S)-1-(3,6-dibromopyridin-2-yl)-2-(3,5-difluorophenyl)ethan- 1-amine (VIII-02), or a co-crystal, solvate, salt, or combination thereof, and starting materials and/or intermediates therein
wherein R4 and R5 are each independently hydrogen, methyl, phenyl, benzyl, 4-nitrobenzyl, 4-chlorobenzyl, 4-brornobenzylamine, or 4-methoxybenzyl
Synthesis of 3,6-dibromopicolinaldehyde (1a)
[00553] A dry reaction flask with magnetic stir-bar was charged with 2,5-dibromopyridine (1.0 g). The flask was inerted under nitrogen, THF (4.2 mL) was added, and the thin slurry agitated. Separately, a dry glass reactor was charged with 2,2,6,6-tetramethylpiperidinylmagnesium chloride, lithium chloride complex (TMPMgCl●LiCl) (5.8 mL, 6.3 mmol). The TMPMgCl●LiCl solution was agitated and cooled to about -20 °C. The 2,5-dibromopyridine solution was added to the TMPMgCl●LiCl solution over about 30 min, maintaining a temperature below about -18 °C. Upon completing the addition, the flask was rinsed forward to the reactor with three additional portions of THF (1 mL x 2), and aged at about -20 for about 1 hour. A solution of N,N-dimethylformamide (1.6 mL, 20 mmol) in THF (1.6 mL) was added to the reactor over about 15 min. The reaction mixture was aged for a further 15 min. and quenched by the addition of a solution of acetic acid (1.9 mL, 34 mmol) in water (10 mL) over about 20 minutes, maintaining a temperature of no more than about 0 °C. To the reactor was added isopropyl acetate (10 mL) and the reaction mixture was warmed to about 20 °C. After aging for 30 min, the mixture was filtered through diatomaceous earth and the reactor rinsed with a mixture of isopropyl acetate (10 mL), saturated aqueous ammonium chloride (10 mL) and 0.2 M aqueous hydrochloric acid (10 mL). The reactor rinse was filtered and the pH of the combined reaction mixture was adjusted to about 8-9 by the addition of a 10% aqueous sodium hydroxide solution (about 6 mL). The mixture was filtered a second time to remove magnesium salts and transferred to a separatory funnel. The phases were separated and the aqueous phase was extracted with isopropyl acetate (3 x 10 mL). The combined organic extracts were washed with 50% saturated aqueous sodium chloride (20 mL), dried over anhydrous sodium sulfate, and filtered. The solution was concentrated to dryness by rotary evaporation and purified by chromatography (eluting with 0-100% ethyl acetate in heptane) to afford 3,6-dibromopicolinaldehyde (1a) as a solid. 1H NMR (400 MHz, DMSO-d6) δ 9.94 (q, J = 0.6 Hz, 1H), 8.19 (dq, J = 8.4, 0.6 Hz, 1H), 7.82 (dt, J = 8.4, 0.7 Hz, 1H). 13C NMR (101 MHz, DMSO-d6) δ 189.33, 148.59, 145.66, 140.17, 133.19, 120.27.
Synthesis of 3,6-dibromopicolinaldehyde (1a)
[00554] A solution of 2,5-dibromo-6-methylpyridine (8.03 g) in THF (81 mL) was cooled to about 0 °C. To this solution was charged tert-butyl nitrite (4.33 g), followed by a dropwise addition of potassium tert-butoxide (28 mL, 1.5 equiv, 20 wt% solution in THF). The reaction mixture was agitated at about 0 °C until the reaction was complete. The reaction mixture was diluted with THF (24 mL), and quenched with ammonium chloride (6.38 g, 119 mmol) in water (43 mL). The reaction mixture was distilled under vacuum to approximately 55 mL to afford a slurry, which was filtered and washed twice with water (2x 24 mL) to afford 1h. 1H NMR (400 MHz, DMSO-d6) δ 11.69 (s, 1H), 8.08 (d, J = 8.4 Hz, 1H), 7.67 (s, 1H), 7.61 (d, J = 8.5 Hz, 1H).
[00555] A solution of glyoxylic acid (407 L, 50 wt% in water) was heated to about 80 °C and in portions was charged with 1h (40.69 kg, 145.4 mol) . Reaction mixture was held at this temperature until the reaction was complete. The reaction mixture was cooled to about 20 °C, filtered, and the filter cake was washed with water until the filtrate had a pH ≥ 5, to afford 1a. 1H NMR (400 MHz, DMSO-d6) δ 9.95 (s, 1H), 8.22 (d, J = 8.4 Hz, 2H), 7.85 (d, J = 8.4 Hz, 1H).
Synthesis of (E)-N-benzhydryl-1-(3,6-dibromopyridin-2-yl)methanimine (1b-02)
[00556] Compound 1a (5.0 g, 18.0 mmol) in toluene (20 mL) was heated to about 50 °C and benzhydrylamine (3.47 g, 18.9 mmol) was charged in one portion and agitated at this temperature until the reaction was deemed complete. Methanol (61 mL) was charged and the reaction mixture was distilled to a volume of approximately 25 mL. Methanol (40 mL) was charged and the reaction mixture was distilled to a volume of approximately 30 mL. The resulting slurry was filtered and rinsed with two portions of methanol (15 mL each) and dried under vacuum to afford 1b-02.
[00557] Alternatively, compound 1a (10.0 g, 37.8 mmol) in 2-methyltetrahydrofuran (50 mL) was heated to about 50 °C and benzhydrylamine (7.28 g, 39.7 mmol) was charged dropwise. The reaction was agitated at this temperature until it was deemed complete. The reaction mixture was distilled to a volume of approximately 30 mL. To the reaction mixture was charged heptane (100 mL) and 1b-02 seed (59.3 mg, 0.138 mmol). The resulting slurry was filtered, rinsed with two portions of heptane (2x 20 mL), and dried under vacuum to afford 1b-02. 1H NMR (400 MHz, DMSO-d6) δ 8.73 (s, 1H), 8.12 (d, J = 8.4 Hz, 1H), 7.67 (d, J = 8.4 Hz, 1H), 7.44 – 7.40 (m,
Synthesis of (E)-N-benzhydryl-1-(3,6-dibromopyridin-2-yl)methanimine (1b-02)
[00558] 1a (2.00 g) was combined with isopropanol (7.6 mL) and agitated at ambient temperature. To this mixture was added potassium metabisulfite (0.96 g) in water (3.8 mL), dropwise. This mixture was agitated for at least 90 minutes and the resulting slurry was filtered. The filter cake was rinsed twice with isopropanol (6 mL then 12 mL) to afford 1i-1. 1H NMR (400 MHz, DMSO-d6) δ 7.92 (d, J = 8.3 Hz, 1H), 7.47 (d, J = 8.3 Hz, 1H), 5.48 – 5.38 (m, 2H).
[00559] li-1 (1.00 g) was combined with 2-methyltetrahydrofuran (3.5 mL) and agitated at ambient temperature. To this slurry was charged potassium hydroxide (443.8 mg, 7.91 mmol) in water (4 mL) and the biphasic mixture was agitated for 2 hours. The layers were separated and the aqueous layer was extracted with an additional portion of 2-methyltetrahydrofuran (3.5 mL). To the combined organics was charged benzhydrylamine (0.47 mL, 2.7 mmol). The reaction mixture was concentrated in vacuo (-300 mbar, 45 °C bath) to a volume of approximately 3 mL. Heptane (7 mL) was charged and the mixture was agitated. The resulting slurry was filtered to afford 1b-02 1H NMR (400 MHz, DMSO-d6) δ 8.73 (s, 1H), 8.12 (d, J = 8.4 Hz, 1H), 7.67 (d, J = 8.4 Hz, 1H), 7.44 – 7.40 (m, 4H), 7.38 – 7.32 (m, 4H), 7.28 – 7.22 (m, 2H), 5.88 (s, 1H).
Synthesis of (E)-N-benzhydryl-1-(3,6-dibromopyridin-2-yl)methanimine (1b-02)
[00560] Compound 1a (1.0 g) was added to a reactor, and toluene (6.0 mL) was added to the reactor. The mixture was agitated. Aminodiphenylmethane (0.73 g, 1.05 equiv.) was added to the reaction mixture. The jacket was heated to about 60 °C, and the mixture was allowed to age for about 1 hour. After about one hour, the mixture was carried forward to the next step. 1H NMR (400 MHz, DMSO-d6) δ 8.68 (s, 1H), 8.05 (d, J = 8.4 Hz, 1H), 7.60 (d, J = 8.4 Hz, 4H), 7.40 – 7.34 (m, 7H), 7.29 (td, J = 6.9, 6.5, 1.7 Hz, 5H), 7.22 – 7.16 (m, 3H), 5.81 (s, 1H).
Synthesis of N-(1-(3,6-dibromopyridin-2-yl)-2-(3,5-difluorophenyl)ethyl)-1,1-diphenylmethanimine (1d-02)
[00561] A solution of1b-02 in toluene (1.0 g in 3.8 mL) was stirred in a reactor at about 60 °C. Tetrabutylammonium bromide (0. 08 g, 0.10 equiv.) was added, 3,5-difluorobenzylbromide (0.60 g, 1.20 equiv.) was added, and potassium hydroxide (50% in water, 1.3 g, 5 equiv.) was added. The mixture was aged for about 4 hours and sampled for conversion. When the reaction was complete, the aqueous phase was removed, and water (3.1 mL) was added to the reactor. Contents were agitated and phases were allowed to settle. The aqueous phase was removed, and the toluene solution of1d-02 was carried forward to the next step. 1H NMR (400 MHz, Chloroform-d) δ 7.78 (dd, J = 8.6, 1.0 Hz, 1H), 7.64 – 7.60 (m, 2H), 7.59 – 7.53 (m, 1H), 7.49 (d, J = 8.3 Hz, 1H), 7.47 (s, 0H), 7.45 (s, 0H), 7.43 (d, J = 0.7 Hz, 0H), 7.41 – 7.34 (m, 3H), 7.33 (t, J = 1.4 Hz, 1H), 7.28 (t, J = 7.3 Hz, 2H), 7.22 (s, 0H), 7.18 (d, J = 8.3 Hz, 1H), 6.87 (dd, J = 7.7, 1.7 Hz, 2H), 6.55 (dt, J = 9.0, 2.3 Hz, 1H), 6.50 (dd, J = 7.0, 4.9 Hz, 3H), 5.26 (s, 0H), 5.16 (t, J = 6.9 Hz, 1H), 3.32 (dd, J = 13.2, 6.6 Hz, 1H), 3.16 (dd, J = 13.1, 7.2 Hz, 1H).
Synthesis of 1-(3,6-dibromopyridin-2-yl)-2-(3,5-difluorophenyl)ethan-1-amine (X) from N-(1-(3,6-dibromopyridin-2-yl)-2-(3,5-difluorophenyl)ethyl)-1,1-diphenylmethanimine (1d-02)
[00562] A solution of 1d-02 in toluene (1.0 g in 3.0 mL) was stirred in a reactor at about 60 °C. Sulfuric acid (0.93 g, 5 equiv.) was diluted into water (3.5 mL), and added to the reactor. The mixture was aged for about 4 hours. When the reaction was complete, the aqueous phase was removed. The aqueous phase was recharged to the reactor, and heptane (2.5 mL) was added. The mixture was agitated and agitation stopped and layers allowed to settle. The aqueous phase was removed, and heptane was discharged to waste. Toluene (5.0 mL) and potassium hydroxide (50% in water, 2.1 g, 10 equiv.) was added to the reactor. The aqueous acidic solution was added to the reactor. The mixture was agitated for about 10 minutes, and agitation stopped and phases allowed to settle. The aqueous phase was discharged to waste. Water (2.5 mL) was added to the reactor, and the mixture was agitated for about 5 minutes, and agitation was stopped and the phases were allowed to settle. The aqueous phase was discharged to waste. The toluene solution of 1-(3,6-dibromopyridin-2-yl)-2-(3,5-difluorophenyl)ethan-1-amine (X) was carried forward to the next step. 1H NMR (400 MHz, Chloroform-d) δ 7.60 (d, J = 8.3 Hz, 1H), 7.21 (d, J = 8.3 Hz, 1H), 6.74 – 6.67 (m, 2H), 6.66 – 6.58 (m, 1H), 4.57 – 4.45 (m, 1H), 3.02 (dd, J = 13.5, 5.2 Hz, 1H), 2.72 (dd, J = 13.5, 8.6 Hz, 1H), 1.77 (s, 3H).
Synthesis of (S)-1-(3.6-dibromopyridin-2-yl)-2-(3,5-difluorophenyl)ethan-1-amine (R)-2-hydroxy-2-phenyl acetate (VIII-03)
[00563] A solution of X in toluene (1.0 g in 7.1 mL) was stirred in a reactor at about 60 °C. The mixture was distilled to minimum volumes (2.9 mL), and methyl tert-butyl ether was added (7.1 mL). (R)-(-)-Mandelic acid (0.41 g, 1 equiv.) was added, and the mixture was cooled to about 0 °C. The newly formed slurry was filtered, providing (S)-1-(3,6-dibromopyridin-2-yl)-2-(3,5-difluorophenyl)ethan-1-amine (R)-2-hydroxy-2-phenylacetate (VIII-03). 1H NMR (400 MHz, DMSO-d6) δ 7.93 (d, J = 8.4 Hz, 1H), 7.49 (d, J = 8.4 Hz, 1H), 7.34 (d, J = 7.3 Hz, 2H), 7.28 – 7.14 (m, 4H), 7.01 (tt, J = 9.4, 2.3 Hz, 1H), 6.79 (d, J = 7.4 Hz, 3H), 4.77 (s, 1H), 4.55 (d, J = 6.6 Hz, 1H), 3.02 (s, 1H), 2.92 (d, J = 6.7 Hz, 2H), 1.05 (s, 2H).
Synthesis of (S)-1-(3,6-dibromopyridin-2-yl)-2-(3,5-difluorophenyl)ethan-1-amine N-acetyl-D- Leucine (VIII-04)
[00564] A reactor was charged with X (15.0 g), N-acetyl-D-leucine (8.28 g) and zinc oxide (0.311 g). Toluene (375 mL) was charged to the reactor followed by 2-pyridinecarboxaldehyde (183 μL). The mixture was aged at about 55 °C for about 6 hrs. and then held at about 35 °C for about 4 days. The mixture was cooled to about 0 °C and held for about 17 hrs. The product was isolated by filtration and the filter cake was washed with cold toluene (2 x 75 mL). The filter cake was re-charged to the reactor. Ethanol (150 mL) was added and the mixture distilled to remove residual toluene. Once the toluene was removed, the reactor volume was adjusted with ethanol to about 90 mL and the mixture was cooled to about 25 °C. Water (210 mL) was added over approximately 10 min. and the mixture aged for approximately 12 hrs. The slurry was filtered and the solids were dried to afford VIII-04. 1H NMR (400 MHz, DMSO-d6) δ 8.03 (d, J = 8.0 Hz, 1H). 7.95 (d, J = 8.3 Hz, 1H), 7.49 (d, 7 8.3 Hz, 1H), 7.03 (tt, J = 9.5, 2.4 Hz, 1H),
Example 1b: Preparation of alternative starting materials and intermediates for use in the formation of (S)-1-(3,6-dibromopyridin-2-yl)-2-(3,5-difliiorophenyl)ethan-1-amine (VIII), or a co-crystal, solvate, salt, or combination thereof
Synthesis of (R)-1-(3,6-dibromopyridin-2-yl)-2-(3,5-difluorophenyl)ethan-1-ol (XII)
[00565] A stainless steel autoclave equipped with a glass inner tube was charged with compound XI (1.00 g) and (A)-RuCY-XylBINAP (16 mg, 0.05 equiv.). EtOH (1.0 mL) and IPA (1.0 mL) followed by tert-BuOK (1.0 M solution in THE, 0.51 mL, 0.2 equiv.) were added to the autoclave. After being purged by H2, the autoclave was charged with 3 MPa of H2. The mixture was stirred at about 20 °C for about 10 h. To the mixture, cone. HCl aqueous solution was added and pH was adjusted to 2. 1H NMR (400 MHz, CDCl3): δ 7.72 ( d, J = 8.2 Hz, 1H), 7.33 (d, J = 8.2 Hz, 1H), 6.80 -6.72 (m, 2H), 6.68 (tt, J = 9.2, 2.4 Hz, 1H), 5.16 (dd, J = 8.2, 3.4 Hz, 1H), 3.60 (br, 1H), 3.12 (dd, J = 13.8, 3.4 Hz, 1H), 2.81 (dd, J = 13.8, 8.2 Hz,
Synthesis of N-(1-(3,6-dibromopyridin-2-yl)-2-(3,5-difluorophenyl)ethyl)-15-chloranimine (X-02)
[00566] Compound XIII (.0 g) was dissolved in THF (4.2 mL) and was cooled over an ice bath. Diphenylphosphoryl azide (0.66 mL, 1.2 equiv.) was added followed by DBU (0.46 mL, 1.2 equiv.) over about 25 min at below about 4 °C. The dark mixture was aged about 1 hour, and the cooling bath was removed. After about 2.5 hours age at RT, some starting material was still present so more diphenylphosphoryl azide (0.15 equiv.) and DBU (0.15 equiv.) were added after cooling over an ice bath. After about 2 hours, more diphenylphosphoryl azide (0.08 equiv.) and DBU (0.08 equiv.) were added. The reaction mixture was allowed to age overnight for about 16 h to allow the conversion to azide intermediate complete. The reaction mixture was cooled over an ice bath and triphenylphosphine (1.0 g, 1.5 equiv.) was added over about 15 min at about 6 °C). The cooling bath was removed after about 10 min and the reaction mixture was agitated for additional about 2.5 hours. To this reaction mixture was added water (0.18 mL, 4 equivalents) and the resulting mixture was aged for about 15 hours at room temperature. The mixture was diluted with EtOAc (5.0 mL) and was washed with water (4.2 mL + 2.0 mL). The aqueous layer was back extracted with EtOAc (4.0 mL) and the EtOAc layer was washed with water (1.0 mL). The organic layers were combined, concentrated via rotary evaporation and evaporated with EtOAc (4 x 4.0 mL) to dry. The residue was dissolved to a 50 ml solution in EtOAc, and cooled over an ice bath to become slurry. To the cold slurry 4N HCl/dioxane (0.76 mL, 1.2 equiv.) was added and the slurry was aged about 2 hours at room temperature. The solid product was filtered and the filter cake was rinsed with EtOAc and dried at about 35 to 50 °C under vacuum to give X-02.
[00567] Recrystallization: A portion of the above obtained X-02 (1.0 g) was mixed with EtOAc (10 mL) and was heated to 65 °C to afford thick slurry. The slurry was aged at about 65 °C for about 2 hours, and overnight at room temperature. The solids were filtered with recycling the mother liquor to help transfer the solids. The filter cake was rinsed with EtOAc, and dried overnight at about 50 °C vacuum to afford X-02. 1H NMR (300 MHz, DMSO-d) δ 8.78 (br s, 3 H), 8.06-8.02 (m, 1 H), 7.64-7.61 (m, 1 H), 7.15-7.08 (m, 1 H), 6.83-6.78 (m, 2 H), 4.87-4.82 (m, 1 H), 3.35-3.25 (m, 1 H), 3.17-3.05 (m, 1 H). 19F NMR (282.2 MHz, Chloroform-d) δ – 109.9-110.1 (m).
Synthesis of 1-(3,6-dibromopyridin-2-yl)-2-(3,5-difluorophenyl)ethyl methanesulfonate (XIII-A)
[00568] Compound XIII (1.0 g) and DMAP (0.1 equiv.) were dissolved in THF (4.5 mL) and cooled over an ice bath. Triethylamine (Et3N) (0.39 mL, 1.1 equiv.) was added followed by methanesulfonyl chloride (218 μL, 1.1 equiv.). The cooling bath was removed, and the mixture was aged about 1.5 hours at room temperature. The reaction mixture was cooled over an ice bath and quenched with water (10 mL). The mixture was diluted with EtOAc and the phases were separated. The aqueous phase was extracted with EtOAc, and the combined organic phase was dried (Na2SO4) and was passed through silica gel with EtOAc. The filtrate was concentrated to afford the mesylate (XIII-A). 1H NMR (300 MHz, Chloroform-d) δ 7.72-7.66 (m, 1 H), 7.38-7.32 (m, 1 H), 6.78-6.63 (m, 3 H), 6.17-6.13 (m, 1 H), 3.40-3.25 (m, 2 H), 2.87 (s, 3 H). 19F NMR (282.2 MHz, Chloroform-d) δ -109.3—109.5 (m).
Synthesis of 1-(3,6-dibromopyridin-2-yl)-2-(3,5-difluorophenyl)ethan-1-amine (X) from 1-(3,6- dibromopyridin-2-yl)-2-(3,5-difluorophenyl)ethyl methanesulfonate (XIII-A)
[00569] A glass pressure bottle was charged with the mesylate (XIII-A) (1.0 g), 28-30% ammonium hydroxide (19 mL) and MeOH (4.7 mL). The mixture was sealed and heated at about 70 °C for about 16 hours, and extracted with 2-MeTHF/ EtOAc. The organic layer was dried (Na2SO4) and purified by silica gel chromatography (10-60% EtOAc/hexanes) to afford racemic amine X. 1H NMR (300 MHz, Chloroform-d) δ 7.70-7.60 (m, 1 H), 7.30-7.20 (m, 1 H), 6.78-6.60 (m, 3 H), 4.46-4.58 (m, 1 H), 3.00-3.16 (m, 1 H), 2.70-2.80 (m, 1 H). 19F NMR (282.2 MHz, Chloroform-d) δ -110.3 – 110.4 (m).
Synthesis of (Z)-N-(1-(3,6-dibrornopyridin-2-yl)-2-(3,5-difluorophenyl)vinyl)acetamide (1f)
[00570] A glass reactor was charged with XI (1.0 g). Ethanol (5.0 mL) was added, and the slurry was agitated while hydroxylamine hydrochloride (0.88 g) was charged. Pyridine (1.0 mL) was added and the mixture heated at about 55-65 °C for about two hours. The mixture was cooled to about 20 °C, transferred to a flask, and concentrated to approximately 75 mL by rotary evaporation. The concentrate was returned to the reactor, rinsing through with isopropyl acetate (5.0 mL). Residue remaining in the flask was carefully (gas evolution) rinsed into the reactor with saturated aqueous sodium bicarbonate (5.0 mL). The bi-phasic mixture was agitated, the phases separated, and the organic extract washed with water (3.2 mL) and saturated sodium chloride (3.2 mL). The organic extract was dried over anhydrous sodium sulfate, filtered, and concentrated to dryness by rotary evaporation to yield 1e which was used without further purification.
[00571] A glass reactor was charged with iron powder (<10 micron, 0.30 g mmol) followed by acetic acid (1.6 mL) and acetic anhydride (0.72 mL). The slurry was de-gassed by holding the reactor contents under vacuum until bubbling was observed, and back-filled with nitrogen (3 cycles). The mixture was heated at 115-120 °C for 2 hours and cooled to 40 °C. Compound le from the previous step in isopropyl acetate (2.0 mL) was added over 30 min. Upon completing the addition, the temperature was raised to 45-65 °C and the mixture aged for about 2 hours. A slurry of diatomaceous earth (1.0 g) in isopropyl acetate (2.0 mL) was added, followed by toluene (2.0 mL). The slurry was filtered, hot, through a Buchner funnel and the reactor and filter cake were washed with warm isopropyl acetate (3 x 1.8 mL). The filtrate was transferred to a reactor and the solution washed with 0.5% aqueous sodium chloride (4.2 mL). Water (3.1 mL) was added to the reactor and the mixture was cooled to about 5 °C. The pH was adjusted to 7-9 with the addition of 50 wt% aqueous sodium hydroxide; following separation, the organic extract was warmed to room temperature and washed with aqueous 1% (w/w) sodium chloride NaCl (3.6 mL). The organic extract was discharged to a flask and dried over anhydrous sodium sulfate (ca. 0.8 g), filtered through diatomaceous earth, and concentrated to approximately 4 mL at 100 mmHg and 45 °C water bath. The warm solution was returned to the reactor, rinsing forward with isopropyl acetate to a produce a total volume of approximately 5.2 mL. This solution was heated further to 50 °C with agitation, cooled to about 35 °C, and seeded with pure 1f (0.006 g). Heptane (9.6 mL) was added over a period of about 4 hours, the solution was cooled to about 10 °C, and the product was isolated by filtration. The filter cake was washed with 33.3% iPAc in heptane (4.0 mL) and dried in a vacuum oven at 40 °C with nitrogen sweep for approximately 24 hours. Compound 1f, a mixture of geometric isomers (approximately 94:6 ratio) was isolated. Major isomer: 1H NMR (400 MHz, DMSO-d6) δ 9.96 (s, 1H), 8.04 (d, J = 8.4 Hz, 1H), 7.66 (d, J= 8.4 Hz, 1H), 7.05 (s, 1H), 6.97 (tt, J = 9.2, 2.2 Hz, 1H), 6.40 – 6.31 (m,
Synthesis of (S)-N-(1-(3,6-dibromopyridin-2-yl)-2-(3,5-difluorophenyl)ethyl)acetamide (1g)
[00572] Preparation of catalyst solution: A flask was charged with [IrCl(cod)((S)-segphos)] (110 mg) and the internal atmosphere was replaced with N2. EtOAc (200 mL) was added to the flask and the mixture was stirred until the catalyst solid was dissolved.
[00573] A stainless steel autoclave was charged with compound 1f (1.0 mg). EtOAc (16 mL) and followed by the catalyst solution prepared above (4.0 mL, 0.001 equiv.) were added to the autoclave. After being purged by H2, the autoclave was charged with 3 MPa of H2.The mixture was stirred at about 130 °C for about 6 hours and cooled to room temperature and H2 was vented out. The reaction mixture was purified by silica gel column chromatography (EtOAc/Hexane = 1/4 to 1/1) to afford 1g. 1H NMR (400 MHz, CD2Cl2): d 7.70 ( d, J = 8.0 Hz, 1H), 7.30 (d, J = 8.0 Hz, 1H), 6.68 (tt, J = 9.2, 2.4 Hz, 1H), 6.64 -6.58 (m, 2H), 6.49 (brd, j = 8.0 Hz, 1H), 5.74 (ddt, J = 8.0, 7.2, 6.4 Hz, 1H), 3.10 (dd, J = 13.6, 6.4 Hz, 1H), 2.99 (dd, J = 13.6, 7.2 Hz), 1.95 (s, 3H). 13C NMR (100 MHz, CD2Cl2): δ 169.5, 163.3 (dd, J = 246.0, 12.9 Hz), 159.1, 143.6, 141.4 (t, J = 9.1 Hz), 140.7, 129.1, 119.9, 112.9 (m), 102.6 (t, J= 25.1 Hz), 53.0, 41.3, 23.6. 19F NMR (376 MHz, CD2Cl2): δ -111.3 (m).
Synthesis of (S)-1-(3,6-dibromopyridin-2-yl)-2-(3,5-difluorophenyl)ethan-1-amine (VIII) from 1-(3,6-dibromopyridin-2-yl)-2-(3,5-difluorophenyl)ethan-1-one (XI), Method 1
[00574] A glass-lined reactor was charged with isopropylamine (about 18 g) and triethanolamine (3.8 g). Water (231 mL) was added and the pH was adjusted to about 7.5 by the addition of concentrated hydrochloric acid. A portion of the buffer solution (23 mL) was removed. The transaminase enzyme (2.5 g) was added to the reactor as a suspension in buffer solution (12 mL), followed by addition of pyridoxal phosphate monohydrate (50 mg) as a solution in buffer solution (12 mL). A solution of XI (1.0 g) in dim ethyl sulfoxide (23 mL) was added to the reactor and the mixture was heated at about 35 °C for about 48 hours with constant nitrogen sparging of the solution. The reaction mixture was cooled to about 20 °C the unpurified amine was removed by filtration. The filter cake was washed with water (3 x 7.7 mL) and the product was dried at about 60 °C under vacuum with nitrogen sweep to afford VIII.
Synthesis of (S)-1-(3.6-dibromopyridin-2-yl)-2-(3.5-difluorophenyl)ethan-1-amine (VIII) from 1-(3,6-dibromopyridin-2-yl)-2-(3,5-difluorophenyl)ethan-1-one (XI), Method 2
[00575] A stainless steel reactor was charged with XI (1.0 g) and p-toluenesulfonic acid (0.49 g). Ammonia (7 M in methanol, 3.7 mL) was added and the vessel was sealed and heated at about 60 °C for about 18 hours. The mixture was cooled to about 20 °C and sparged for about 30 min to remove excess ammonia. A solution of diacetato[(R)-5,5′-bis(diphenylphosphino)-4,4′-bi-1,3-benzodioxole]ruthenium(II) (0.10 g) in methanol (0.5 mL) was added to the reactor, which was sealed and heated at about 60 °C under a hydrogen atmosphere (400 psi) for a further about 6-10 hours. Upon cooling to about 20 °C the mixture was filtered through a plug of silica, rinsing with additional methanol (5.0 mL). Concentration of the filtrate by rotary evaporation affords VIII.
Example 1c: Preparation of 1-(3,6-dibromopyridin-2-yl)-2-(3,5-difluorophenyI)ethan-1-amine (X) by racemization of (S)-1-(3,6-dibromopyridin-2-yl)-2-(3,5-difluorophenyl)ethan-1-amine (VIII)
[00576] A vial was charged with zinc acetate (25 mol %), enantioenriched VIII (1.0 g, 92:8 enantiomer ratio), toluene (10 mL), and 2-formylpyridine (5 mol %). The vial was wanned to about 60 °C and stirred for about 4 h.
Example 2: Preparation of (S)-1-(3-bromo-6-(3-methyl-3-(methylsulfonyl)but-1-yn-1-yl)pyridin-2-yl)-2-(3,5-difluorophenyl)ethan-1-amine (VI)
[00577] A glass-lined reactor was charged with (S)-1-(3,6-dibromopyridin-2-yl)-2-(3,5-difluorophenyl)ethan-1-amine (R)-mandelic acid salt (VIII-03) (1.0 g), 3-methyl-3-(methylsulfonyl)but-1-yne (IX) (about 0.3 g), and dichlorobis(triphenylphosphine)palladium(II) (about 0.39 g). The reactor was evacuated and purged with nitrogen to inert. To this reactor was added 2-methyltetrahydrofuran (6.4 kg) and triethylamine (0.92 kg 5.0 equiv.). The reaction mixture was agitated at about 65-75 °C until the reaction was deemed complete by HPLC analysis. Upon cooling to about 30-40 °C the reaction mixture was discharged to another reactor and the parent reactor was rinsed with 2-methyltetrahydrofuran (4.6 g) and the resulting solution transferred to the receiving reactor. To the reactor was added water (5.0 g) and the biphasic mixture agitated at about 30-40 °C for about 30 min. Agitation was ceased and the mixture was allowed to layer for 30 min. The lower aqueous layer was discharged and the remaining organic solution held for about 15 hours. A solution of A-acetyl-L-cysteine (196 g) and sodium hydroxide (0.80 g) in water (11.8 g) was prepared. To the reactor was added approximately half of the N-acetyl-L-cysteine solution (6.7 g). The mixture was agitated at about 55-65 °C for about 30 min. The temperature was adjusted to about 30-40 °C and agitation was ceased. After about 30 min had elapsed, the lower aqueous phase was discharged. The remaining alkaline N-acetyl-L-cysteine solution (5.4 kg) was added and the mixture was heated, with agitation, to about 55-65 °C and held for about 30 min. The temperature was adjusted to about 30-40 °C and agitation was ceased. After about 30 min had elapsed, the lower aqueous phase was discharged. To the reactor was added a solution of sodium chloride (0.26 g) in water (4.9 g) and the mixture agitated at about 30-40 °C for about 30 min. Agitation was ceased and the biphasic mixture allowed to layer for about 30 min. The lower aqueous layer was discharged and the contents cooled to about 15-25 °C and held for about 16 hours. The mixture was concentrated at about 55-65 °C. The concentrated solution was cooled to about 30-40 °C and heptane (3.4 kg) was added over about 2 hours. The resulting slurry was cooled to about 20 °C and aged for about 20 h, and filtered. The filter cake was washed with 2-methyltetrahydrofuran/heptane (1:1 v/v,2 mL) and the solids dried in a vacuum oven at about 40 °C to yield (S)-1-(3-bromo-6-(3-methyl-3-(methylsulfonyl)but-1-yn-1-yl)pyridin-2-yl)-2-(3,5-difluorophenyl)ethan-1-amine (VI)). 1H NMR (400 MHz, DMSO-d6) δ 8.05 (d, J = 8.2 Hz, 1H), 7.42 (d, J = 8.2 Hz, 1H), 7.01 (tt, J = 9.5, 2.4 Hz, 1H), 6.97 – 6.84 (m, 2H), 4.41 (dd, J = 8.5, 5.2 Hz, 1H), 3.20 (s, 3H), 2.93 (dd, J = 13.3, 5.2 Hz, 1H), 2.79 (dd, J = 13.3, 8.5 Hz, 1H), 1.99 (s, 2H), 1.68 (s, 6H). 13C NMR (101 MHz, DMSO-d6) δ 162.25, 162.00 (dd, J = 245.2, 13.4 Hz), 143.88 (t, J= 9.4 Hz), 141.09, 139.72, 127.51, 120.08, 112.58 – 112.12 (m), 101.45 (t, J= 25.7 Hz), 87.94, 84.25, 57.24, 55.90, 42.57, 34.99, 22.19.
Example 2a: Preparation of 3-methyl-3-(methylsulfonyl)but-1-yne (IX)
[00578] Sodium methansulfmate (418.1 g), copper (II) acetate (26.6 g), N,N,N’,N’- Tetramethylethylenediamine (TMEDA, 34.0 g), and isopropyl acetate (2100 mL) were added to a reactor and the suspension was agitated at 20 – 25 °C. 3-Chloro-3-methylbut-1-yne (3-CMB,
300 g) was added slowly to maintain a constant temperature of about 20 – 25 °C. The reaction mixture was then heated to about 30 °C until the reaction was complete. The mixture was cooled to about 20 °C and washed twice with 5% aqueous sulfuric acid (600 mL). The combined
aqueous layers were then extracted with isopropyl acetate (600 mL). The combined organic layers were then washed with water (600 mL). The product was then isolated by crystallization from isopropyl acetate (900 mL) and n-heptane (1.8 kg) at about 0 °C. The wet cake was then washed with cold n-heptane to afford IX. 1H NMR (400 MHz, DMSO-d6) δ 3.61 (s, 1H), 3.07 (s, 3H), 1.55 (s, 6H); 13C NMR (10Q MHz, DMSO) d 82.59, 77.76, 56.95, 34.95, 22.77.
Example 3a: Preparation of (3bS,4aR)-3-(trifluoromethyI)-1,3b,4,4a-tetrahydro-5H-cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-5-one (XV) from lithium (Z)-2,2,2-trifluoro-1-(3-oxobicyclo[3.1.0]hexan-2-ylidene)ethan-1-olate (3a)
Synthesis of 3-(trifluoromethyl)-3b,4,4a,5-tetrahydro-1H-cyclopropa[3,4]cyclopenta[1,2-c]pyrazole (3b)
[00579] A reactor was charged with 3a (1.0 g) and AcOH (4.2 ml) and the resulting solution was adjusted to about 20 °C. Hydrazine hydrate (0.29 g, 1.4 equiv.) was added over about 60 min at about 17-25 °C and the reaction mixture was stirred for about 2 hours at about 20-25 °C, warmed up to about 45 to 50 °C over about 30 min, and aged at about 50 °C overnight. Water was slowly (5 mL) added at about 50 °C and product started to crystallize after addition of 5 mL of water. Another 5 mL of water was added at about 50 °C, and the slurry was cooled down to about 20 °C in about one hour and held overnight at about 20 °C. The solids were filtered, washed with water (4X 3 mL), and dried under vacuum at about 30 °C to yield 3b. 1H NMR (400 MHz, Chloroform-d) δ 2.99 (dd, J = 17.0, 6.1 Hz, 1H), 2.89 – 2.78 (m, 1H), 2.14 (dddd, J = 9.1, 7.9, 3.6, 2.5 Hz, 2H), 1.13 (td, J = 7.8, 5.1 Hz, 1H), 0.36 – 0.26 (m, 1H).
Isolation of (3bS,4aS)-3-(trifluoromethyl)-3b,4,4a,5-tetrahydro-1H-cyclopropa[3,4]cyclopenta[1,2-c]pyrazole (3c)
[00580] Chiral purification of 3b (1.0 g) was achieved using a 8×50 mm simulated moving bed (SMB) chromatography system and Chiralpak IG (20 μ particle size) stationary phase using acetonitrile as a mobile phase to afford 3c. 1H NMR (400 MHz, Chloroform-d) δ 3.00 (dd, J = 17.0, 5.7 Hz, 1H), 2.90 – 2.77 (m, 1H), 2.21 – 2.05 (m, 2H), 1.13 (td, J = 7.8, 5.1 Hz, 1H), 0.35 – 0.27 (m, 1H).
Synthesis of (3bS,4aR)-3-(trifluoromethyl)-1,3b,4,4a-tetrahydro-5H-cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-5-one (XV)
[00581] A reactor was charged with water (7 mL) and CuCl2 ● 2H2O (0.09 g, 0.1 equiv). To the reactor was added pyridine (0.42 g, 1 equiv.) and 3c. tert-Butylhydroperoxide (70% in water, 5.5 g, 8 equiv.) was added over about 0.5 hour. The reaction mixture was stirred at about 20 °C for about 2.5 days and quenched with aqueous sodium metabisulfite solution (0.73 g in 2.5 mL water). The quenched reaction mixture was extracted with isopropyl acetate (20 mL), and the aqueous layer was back extracted with isopropyl acetate (2.0 ml). The organic layers were combined and washed with aqueous ethylenediaminetetraacetic acid (EDTA) solution 0.16 g EDTA 10 ml in water), the aqueous layer was dropped, and the organic layer was further washed with aqueous EDTA solution (0.015 g EDTA in 20 ml water). The washed organic layer was concentrated to dryness. To the residue was added isopropyl acetate (2.0 ml) and heptane (2.0 mL). The solution was seeded and stirred overnight at about 20 °C, further diluted with heptane (2.0 mL), and the mixture was concentrated to dryness. The residue was suspended in heptane (4.0 mL) at about 40 °C. The solid was filtered and the filter cake was washed with heptane (1.0 mL) and dried at about 40 °C to yield XV. 1H NMR (400 MHz, Chloroform-d) δ 2.84 (dt, J = 6.8, 4.2 Hz, 1H), 2.71 – 2.64 (m, 1H), 1.79 – 1.67 (m, 2H).
Example 3b: Preparation of (3bS,4aR)-3-(trifluoromethyl)-1,3b,4,4a-tetrahydro-5H- cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-5-one (XV) from lithium (Z)-1-((1S,5R)-4,4- dimethoxy-3-oxobicyclo[3.1.0]hexan-2-ylidene)-2,2,2-trifluoroethan-1-olate (3d-02)
[00582] Hydrazine sulfate (0.45 g, 0.95 equiv.) and ketal lithium salt 3d-02 (1.0 g) were dissolved in ethylene glycol (9.5 mL), and the solution was heated to about 40 °C for about 16 hours. Reaction was cooled to room temperature and water (9.0 mL) was added. Reaction was polish filtered andThe filtrate was collected and to this receiving flask was added water (10 mL, 2x). Slurry was cooled in ice water bath for about five hours, and filtered. Solids were washed with ice water (10 mL, 2x), deliquored, and dried to afford XV. 1H NMR (400 MHz, CDCl3) δ 11.83 (bs, 1H), 2.93 – 2.77 (m, 1H), 2.77 – 2.58 (m, 1H), 1.86 – 1.57 (m, 2H). 19F NMR (376 MHz, CDCl3) δ -61.69. 13C NMR (101 MHz, CDCl3) δ 188.56, 144.08, 142.92, 121.82, 119.15, 36.28, 31.87, 14.15.
Example 3c: Preparation of (3bS,4aR)-3-(trifiuoromethyl)-1,3b,4,4a-tetrahydro-5H- cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-5-one (XV) from (1S,2S)-2-iodo-N-methoxy-N- methylcyclopropane-1-carboxamide (3f) and 1-(4-methoxybenzyl)-4-(4,4,5,5-tetramethyl- 1,3,2-dioxaborolan-2-yl)-3-(trifluoromethyl)-1H-pyrazole (3i) and preparation of starting materials and/or intermediates therein
Synthesis of (1S,2S)-2-iodo-N-methoxy-N-methylcyclopropane-1-carboxamide (3f)
[00583] Starting material iodoacid 3e is a mixture of 3e and cyclopropane carboxylic acid (des-iodo 3e) with mole ratio of 3e to des-iodo 3e of 2:1 by NMR. A mixture of 3e (1.0 g),
N,O-dimethyl hydroxyl amine-HCl (0.46 g) and carbonyl diimidazole (1.72 g) in THF was stirred overnight at room temperature. The reaction mixture was diluted with water, extracted with CH2Cl2, and concentrated to afford unpurified 3f (1.8 g). The unpurified 3f was purified by column chromatography to afford 3f which was a mixture of Wei nr eb amide 3f and des-iodo-3f (about 80:20 by HPLC).
Synthesis of 1-(4-methoxybenzyl)-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-3- (trifluoromethyl)-1H-pyrazole (3i)
[00584] To a suspension of NaH (60%, 0.31 g, 1.1 equiv.) in DMF (7.5 mL), a solution of 3g (1.0 g) in DMF (7.5 mL) was added dropwise over about 15 min at about 3 to 7 °C. The reaction mixture was stirred at room temperature for about 1 h and a solution of PMBCl (1.2 g, 1.05 equiv.) in DMF (4.2 mL) was added dropwise in about 25 min at room temperature. The reaction mixture was stirred at room temperature overnight, poured into water (17 mL), and extracted with diethyl ether (3×17 mL). The ether layers were combined and washed with water (2 x 17 mL) and brine (17 mL), dried over Na2SO4, and concentrated in vacuo to give unpurified 3h. Unpurified 3h was absorbed in silica gel (4.3 g) and purified by silica gel chromatography (eluting with 5-25% EtOAc in hexanes) to give 3h (1.5 g).
[00585] To solution of iodopyrazole 3h (1.0 g) in THF (8 mL) i-PrMgCl (2M in ether, 1.8 mL, 1.1 equiv.) was added dropwise over about 10 min at below about 5 °C. The resulting solution was stirred at about 0 °C for about 70 min and 2-methoxy-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (970 mg, 1.81 equiv.) was added at below about 6 °C. The reaction mixture was warmed up to room temperature, quenched by addition of saturated NH4Cl (20 mL), and
extracted with EtOAc (2 x 20 mL). The combined organic layer was washed with saturated NH4Cl (10 mL) and concentrated to unpurified oil, which was combined with the unpurified oil from a previous batch (prepared using 1.1 g of 3h), absorbed on silica gel (6 g), and purified via silica gel chromatography (eluting with 5-40% EtOAc/Hexanes,). Boronate 3i was obtained. 1H NMR (300 MHz, Chloroform-d) δ 7.60 (s, 1 H), 7.23-7.19 (m, 2 H), 6.90-6.85 (m, 2 H), 5.25
(s, 2 H), 3.81 (m, 3 H), 1.29 (s, 12 H).
Synthesis of (1R,2S)-N-methoxy-2-(1-(4-methoxybenzyl)-3-(trifluoromethyl)-1H-pyrazol-4-yl)-N-methylcyclopropane-1-carboxamide (3j)
[00586] A mixture of unpurified iodide 3f (1.0 g), boronate 3i (about 2.2 g), CsF (4.5 equiv.), Pd(OAc)2 (0.1 equiv.), and PPh3 (0.5 equiv.) in DMF (58 mL) was degassed by bubbling N2 and heated at about 87 °C for about 15 hours. The reaction mixture was diluted with water,
extracted with MTBE, concentrated and the unpurified product was purified by column chromatography to give 3j. 1H NMR (300 MHz, Chloroform-d) δ 7.18-7. 14 (m, 3 H), 6.86-6.82 (m, 2 H), 5.24-5.08 (m, 2 H), 3.77 (s, 3 H), 3.63 (s, 3 H), 3.05 (s, 3 H), 2.37-2.32 (m, 1 H), 1.50-1.42 (m, 1 H), 1.32-1.21 (m, 2 H).
Synthesis of (3bS,4aR)-1-(4-methoxybenzyl)-3-ftrifluoromethyl)-1,3b,4,4a-tetrahydro-5H-cyclopropa[3,4]cyclopenta91,2-c]pyrazol-5-one (3k)
[00587] Compound 3j (1.0 g) was treated with freshly prepared LDA (3.3 eq then 0.7 equiv.) at about -67 °C for about 2.5 hours. The reaction mixture was quenched with saturated NH4Cl (12.5 mL) and diluted with MTBE (63 mL). The organic layer was washed with brine, concentrated, and purified by column chromatography to give 3k. 1H NMR (300 MHz, Chloroform-d) δ 7.36-7.33 (m, 2 H), 6.86-6.83 (m, 2 H), 5.28 (s, 2 H), 3.78 (s, 3 H), 2.73-2.65
(m, 1 H), 2.60-2.53 (1 H), 1.70-1.61 (m, 2 H).
Synthesis of (3bS,4aR)-3-(trifluoromethyl)-1,3b,4,4a-tetrahydro-5H-cyclopropa[3,4]cyclopenta[1.2-c]pyrazol-5-one (XV)
[00588] A mixture of 3k (1.0 g) and TFA (5 mL) was heated at about 75 °C for about 3 hours and concentrated. The residue was dissolved in DCM (50 mL), washed with saturated NaHCO3 and brine, concentrated, and purified by column chromatography to give XV. 1H NMR (300 MHz, Chloroform-d) δ 2.86-2.80 (m, 1 H), 2.68-2.63 (m, 1 H), 1.77-1.65 (m, 2 H).
Example 3d: Resolution of 2-(2,2,2-trifluoroacetyl)bicyclo[3.1.0]hexan-3-one (3I) with quinine
[00589] A flask was charged with 3I (1.0 g), acetone (2.5 ml), and quinine (1.7 g, 0.65 equiv). The mixture was stirred at about 15 to 25 °C for about 18 hours and the solids were isolated by filtration and washed with acetone to provide the quinine salt 3n.
Example 4a: Preparation of ethyl 2-((3bS,4aR)-5-oxo-3-(trifluoromethyl)-3b,4,4a,5-tetrahydro-1H-cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-1-yl)acetate (XIV) from (3bS,4aR)-3-(trifluoromethyl)-1,3b,4,4a-tetrahydro-5H-cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-5-one (XV)
[00590] Acetonitrile (5 vol.) was added to a reactor containing XV (1.0 g). N,N-Diisopropylethylamine (0.80 g, 1.25equiv.) was added at about 0 °C. Ethyl bromoacetate (0.91 g, 1.1 equiv.) was added over about 1 hour at about 0 °C. The reaction was stirred at about 5 °C for about 30 minutes and warmed to about 10 °C. The reaction was stirred until complete as determined by HPLC, warmed to about 20 °C, and extracted with MTBE (2 vol.) and saturated NaCl (6 vol.). The aqueous layer was removed and the organic phase was concentrated and diluted with EtOH (3 vol.). The reaction was crystallized by the addition of H2O (7.8 vol.) at about 20 °C. The mixture was cooled to about 5 °C over about 2 hours and maintained at about 5 °C for about 0.5 hour. The mixture was filtered at about 5 °C and washed with cold water (4 vol). The product was dried at about 40 °C under vacuum to give XIV. 1H NMR (400 MHz, Chloroform-d) δ 4.97 (s, 2H), 4.31 – 4.17 (m, 2H), 2.77 (dddd, J= 6.4, 5.2, 2.9, 2.3Hz, 1H), 2.65 – 2.55 (m, 1H), 1.74 – 1.64 (m, 2H), 1.34 – 1.19 (m, 5H), 0.94 – 0.84 (m, 1H).
Example 4b: Preparation of ethyl 2-((3bS,4aR)-5-oxo-3-(trifluoromethyl)-3b,4,4a,5-tetrahydro-1H-cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-1-yl)acetate (XIV) from (1R,5S)-bicyclo[3.1.0]hexan-2-one (4a)
Synthesis of (1R,5R)-2,2-dimethoxybicyclo[3.1.0]hexan-3-ol (4b-02)
[00591] Potassium hydroxide (KOH) (2.2 g, 3.50 equiv.) and anhydrous methanol (13 mL) were added to a reactor and the reaction mixture was warmed to about 55 °C and agitated until
KOH solids were dissolved completely. The mixture was adjusted to about 0 to 6 °C and compound 4a (1.0 g) was slowly added while maintaining the internal temperature at NMT 6 °C. The reaction mixture was agitated for about 45 min at about 0 to 6 °C. Diacetoxy iodobenzene (PhI(OAc)2, 5.0 g, 1.5 equiv.) was added over about 2 hours while maintaining the internal temperature at NMT 6 °C. The reaction mixture was agitated for NLT 1 hour at about 0 to 6 °C. Water (10 g) and heptane (10 mL) were added to the reaction mixture and the biphasic was agitated for NLT 30 min at about 19 to 25 °C The aqueous layer was separated and washed with heptane (10 mL). The combined organic layer was extracted twice with aqueous solution of methanol (MeOH, 10 mL) and water (5 g). The combined aqueous layer was concentrated under vacuum. The aqueous layer was extracted twice with DCM (15 mL and 5 mL). The combined organic layer was concentrated and dried under vacuum. The unpurified compound 4b-02 was obtained. 1H NMR (600 MHz, CDCl3): d 3.98 (d, 1H), 3.45 (s, 3H), 3.25 (s, 3H),
Synthesis of (1R,5R)-2,2-dimethoxybicyclo[3.1.0]hexan-3-one (4c-02)
[00592] Oxalyl chloride (0.96 g, 1.20 equiv.) and dichloromethane (10 mL) were added to a reactor and the mixture was cooled to about -78 °C. Dimethyl sulfoxide (DMSO, 1.2 g, 2.4 equiv.) was added over about 30 min while maintaining the internal temperature below about -60 °C. After agitation for about 5 min, the solution of compound 4b-02 (1.0 g) in dichloromethane (6 mL) was added over about 30 min while maintaining the internal temperature below about -60 °C and the reaction mixture was agitated for about 20 min at about -60 °C. Triethylamine (TEA, 3.1 g, 4.8 equiv.) was added over about 40 min at about -60 °C, and the reaction mixture was warmed to about 10 to 20 °C. Water (15 g) was added and the biphasic was agitated about 30 min at about 10 to 20 °C. After phase separation, the aqueous layer was back-extracted with dichloromethane (10 mL). Combined organic layer was concentrated until no distillate was observed. To the residue was added MTBE (1 mL), filtered and evaporated to afford unpurified compound 4c-02. 1H NMR (600 MHz, CDCl3): d 3.45 (s,
Synthesis of lithium (Z)-1-((1S,5R)-4,4-dimethoxy-3-oxobicyclo[3.1.0]hexan-2-ylidene)-2,2,2-trifluoroethan-1-olate (3d-02)
[00593] A reactor was charged with compound 4c-02 (1.0 g), ethyl trifluoroacetate (CF3COOEt, 0.91 g, 1.0 equiv.) and tetrahydrofuran (THF, 0.5 mL) and the reaction mixture was cooled to about -10 to 0 °C. The 1M solution of lithium bis(trimethylsilyl)amide (LiHMDS, 7.0 mL, 1.10 equiv.) was added over about 40 min while maintaining the internal temperature below about 0 °C. The reaction mixture was agitated for about 2 hours at about -10 to 0 °C until the reaction was complete. After then, the reaction mixture was wanned to about 20 °C followed by charging tert-butyl methyl ether (MTBE, 10 mL) and water (10 g). After agitating for about 30 min, the organic layer was separated and the aqueous layer was back-extracted twice with mixture of MTBE (6 mL) and THF (4 mL). The combi ned organic layer was concentrated until no distillate was observed. To the unpurified solids, THF (3 mL) and heptane (15 mL) were added at about 20 °C, and the reaction mixture was cooled to about 0 °C and agitated about 1 hour. The resulting slurry was filtered and wet cake was washed with heptane (7 g) and dried under vacuum at about 40 °C to afford compound 3d-02. 1H NMR (600
1.0 equiv.) and absolute ethanol (EtOH, 15 mL) were added to a reactor and the reaction mixture was cooled to about 0 – 5 °C. Sulfuric acid (H2SO4, 0.19 g, 0.50 equiv.) was added while maintaining the internal temperature below about 5 °C. Triethyl orthoformate (0.86 g, 1.50 equiv.) was added and the reaction mixture was agitated at about 0 to 5 °C for about 15 hours. The reaction mixture was warmed to about 20 to 25 °C and water (30 g) was added over about 15 minutes. The content was cooled to about 0 to 5 °C and agitated for about 1 hour. The slurry was filtered and wet cake was washed with water (5 g) and dried under vacuum at about 45 °C to afford XIV 1H NMR (600 MHz, CDCl3): d 4.97 (s, 1H), 4.23 (qd, 2H), 2.77 (quint. 1H), 2.60 (quint, 1H), 1.69 (m, 2H), 1.28 (t, 3H). 13C NMR (150 MHz, CDCl3): d 187.14, 165.98, 143.35, 143.12, 121.37, 119.59, 62.34, 51.83, 35.35, 31.72, 14.00, 13.73.
Example 4c: Kinetic resolution of ethyl 2-(5-oxo-3-(trifluoromethyl)-3b,4,4a,5-tetrahydro- 1H-cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-1-yl)acetate (XVII) to form ethyl 2- ((3bS,4aR)-5-oxo-3-(trifluoromethyl)-3b,4,4a,5-tetrahydro-1H- cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-1-yl)acetate (XIV)
[00595] Compound XVII (1.0 g), (R)-2-methyl-CBS-oxazaborolidine (0.0.05 g, 0.05 equiv.), and tetrahydrofuran (11.9 g) were combined and cooled to about 0 to 5 °C. A solution of borane dimethyl sulfide complex (0.14 g, 0.55 equiv.) in tetrahydrofuran (0.67 g) was added to the mixture, and the mixture was agitated at about 0 to 5 °C until the reaction was deemed complete. Methanol (1 mL) was added to the mixture at about 0 to 5 °C over about 1 h, and the mixture was adjusted to about 15 to 25 °C. The mixture was concentrated under vacuum and combined with tetrahydrofuran (2.7 g). The mixture was combined with 4-dimethylaminopyridine (0.18, 0.44 equiv.) and succinic anhydride (0.30 g, 0.87 equiv.) and agitated at about 15 to 25 °C until the reaction was deemed complete. The mixture was combined with tert-butyl methyl ether (5.2 g) and washed with 1 M aqueous HCl (6.7 g), twice with 5 wt % aqueous potassium carbonate (6.7 g each), and 5 wt % aq. sodium chloride (6.7 g). The organics were concentrated under reduced pressure to an oil which was dissolved in dichloromethane (0.1 g) and purified by flash column chromatography (2.0 g silica gel, 20:80 to 80:20 gradient of ethyl acetate:hexanes). The combined fractions were concentrated under vacuum to give XIV.
Example 4d: Preparation of (1R,5S)-bicyclo[3.1.0]hexan-2-one (4a)
[00596] 4-Tosyloxycyclohexanone (50 mg), (8α,9S)-6′-methoxycinchonan-9-amine trihydrochloride (16 mg), trifluoroacetic acid (28 μL), lithium acetate (49 mg), water (3.4 μL), and 2-methyltetrahydrofuran (0.75 mL) were combined in a vial. The mixture was agitated at about 20 °C until the reaction was complete. 4a was isolated by vacuum distillation. 1H NMR (400 MHz, CDCl3) δ2.05 (m, 5H), 1.74 (m, 1H), 1.18 (m, 1H), 0.91 (m, 1H).
Example 5: Synthesis of ethyl 2-((3bS,4aR)-3-(trifluoromethyl)-4,4a- dihydrospiro[cyclopropa[3,4]cyclopenta[1,2-c]pyrazole-5,2′-[1,3]dithiolane]-1(3bH)- yl)acetate (5h) from (1R,5R)-2,2-dimethoxybicyclo[3.1.0]hexan-3-ol (4b-02)
Synthesis of (1R,5R)-spiro[bicyclo[3.1.0]hexane-2,2′-[1,3]dithiolan1-3-ol (5d)
[00597] A mixture of ketal alcohol 4b-02 (1.0 g), ethanedi thiol (0.91 g), MeCN (7.5 ml) and BiCl3 (0.30 g) was agitated at r.t. overnight. The solids were removed by filtration and the filtrate was concentrated and the residue was further purified by flash column on silica gel to obtain the two isomers. Major product: 1H NMR (400 MHz, Chloroform-d) δ 3.82 (ddt, J = 6.1, 1.3, 0.6 Hz, 1H), 3.41 – 3.32 (m, 2H), 3.31 -3.23 (m, 1H), 3.14 – 3.06 (m, 1H), 2.71 (s, 1H),
Synthesis of (1R,5R)-spiro[bicyclo[3.1.0]hexane-2,2′-[1,3]dithiolan1-3-one (5e)
[00598] To a dried flask was sequentially added dithiolane alcohol 5d (1.0 g), CH2Cl2 (25 ml), anhydrous DMSO (8.5 ml), and tri ethylamine (3.5 ml) and the resulting mixture was aged at room temperature for about 21 hours. The reaction mixture was transferred to a separatory funnel, diluted with CH2Cl2 (30 ml), washed with 1 M HCl (25 ml), and water (25 ml). The CH2Cl2 layer was concentrated to a solid and further purify by flash column chromatography on silica gel eluted with gradient EtOAc/n-heptane (0-20%) to obtain 5e. 1H NMR (400 MHz, Chloroform-d) δ 3.57 (dddd, J = 10.5, 5.6, 4.3, 0.5 Hz, 1H), 3.49 – 3.41 (m, 1H), 3.39 – 3.28 (m, 2H), 3.10 (ddd, J = 18.3, 5.6, 2.2 Hz, 1H), 2.29 (d, J = 18.3 Hz, 1H), 1.89 (ddd, J = 8.0, 7.0, 3.9
Synthesis of lithium (Z)-2,2,2-trifluoro-1-((1R,5S)-3-oxospiro[bicyclo[3.1.0]hexane-2,2′-[1,3]dithiolan]-4-ylidene)ethan-1-olate (5f)
[00599] To a flask with dithiolane ketone 5e (1.0 g) under N2 was added anhydrous THF (8.8 ml), and the mixture was cooled to about -78 °C and followed by addition of LiHMDS (1 M in THF, 7.4 ml) over about 5 min. The resulting mixture was agitated at about -78 °C for about 0.5 hours, and ethyl trifluoroacetate (0.88 ml) was added. The resulting mixture was agitated at about -78 °C for about 10 minutes, at about 0 °C for about 1 hour, and at room temperature overnight. THF was removed under reduced pressure and the residue was crystallized in n-heptane (about 18 ml). The solid product was isolated by filtration, and the filter cake was rinsed with n-heptane (4.1 ml), and dried at about 50 °C under vacuum to provide 5f. 1H NMR (400 MHz, Acetonitrile-d3) δ 6.98 (s, 0H), 5.20 (s, 0H), 3.60 – 3.50 (m, 2H), 3.46 – 3.36 (m, 2H), 2.28 – 2.20 (m, 1H), 1.80 (ddd, J = 8.3, 7.2, 4.1 Hz, 1H), 1.39 (s, 1H), 1.03 (ddd, J = 8.3, 6.7, 4.8 Hz, 1H), 0.17 (ddd, J = 4.7, 4.2, 3.6 Hz, 1H).
Synthesis of (3bS,4aR)-3-(trifluoromethyl)-1,3b,4,4a-tetrahydrospiro[cvciopropa[3.4]cyclopenta[1,2-c]pyrazole-5,2′-[1,3]dithiolane] (5g)
[00600] To flask containing the dithiolane lithium salt 5f (1.0 g) was added water (10 ml), hydrazine hydrate (0.88 ml) and acetic acid (10 ml). The reaction mixture was heated at about 35 °C for about 2 hours, and at about 55 °C for about 2 hours. Water was removed under reduced pressure and the residue was diluted with acetic acid (20 ml) and heated at about 55 °C for about 0.5 hour and held at room temperature overnight. The reaction mixture was further heated at about 65 °C for about 20 hours, and cooled down and concentrated to remove volatile components by rotavap. The residue was triturated with water (50 ml) at about 0 °C and the solid residue was isolated and further washed with ice-cold water (2×10 ml). The solids were further dried to afford unpurified 5g. 1H NMR (400 MHz, Chloroform-d) δ 3.65 – 3.46 (m, 4H), 2.60 (dddd, J = 8.3, 5.6, 4.2, 0.7 Hz, 1H), 2.47 – 2.38 (m, 1H), 1.33 (dddd, J= 8.2, 7.4, 5.7, 0.7 Hz, 1H), 0.66 (dddd, J = 5.7, 4.3, 3.6, 0.7 Hz, 1H)
Synthesis of ethyl 2-((3bS,4aR)-3-(trifluoromethyl)-4,4a-dihydrospiro[cyclopropa[3,4]cyclopenta[1,2-c]pyrazole-5.2′-[1,3]dithiolane]-1(3bH)-yl)acetate
(5h) from (3bS,4aR)-3-(trifluoromethyl)-1,3b,4,4a-tetrahydrospiro[cyclopropa[3,4]cyclopenta[1,2-c]pyrazole-5,2′-[1,3]dithiolane] (5g)
[00601] A reactor was charged with dithiolane pyrazole 5g (1.0 g) and THF (15 ml). The contents were adjusted to about 0 to -5 °C and followed by addition of ethyl bromoacetate (0.44 ml, 1.1 equiv.). To the resulting mixture NaHMDS (2 M, 2.0 ml, 1.1 equiv.) was added over about 10 min via syringe pump at about -2.5 to 0 °C and the mixture was held for about 3 hours, a second portion of ethyl bromoacetate (0.050 ml, 0.12 equiv.) was added, and the mixture was aged for about 1 hour. The reaction mixture was quenched by excess water (2 ml) to form 5h.
Synthesis of ethyl 2-((3bS,4aR)-3-(trifluoromethyl)-4,4a-dihydrospiro[cyclopropa[3,4]cyclopenta[1,2-c]pyrazole-5,2′-[1,3]dithiolanel-1(3bH)-yl)acetate
(5h) from lithium (Z)-2,2,2-trifluoro-1-((1R,5S)-3-oxospiro[bicyclo[3.1.0]hexane-2.2′- [1,3]dithiolanl-4-ylidene)ethan-1-olate (5f)
[00602] A 100 ml flask was charged with ethanol (5 ml). The contents were cooled to about 0 °C and acetyl chloride (1.1 g, 4.0 equiv.) was added over about 10 min. The mixture was agitated at about 0 °C for about 20 minutes and at room temperature for about 20 minutes. To the freshly prepared HCl ethanol solution was added EHA.HCl (0.68 g, 1.2 equiv.) and dithiolane lithium salt 5f (1.0 g). The reaction mixture was heated at about 40 °C for about 22 hours. Ethanol was removed under reduced pressure, and the residue was partitioned between ethyl acetate (5 ml) and water (5 ml). The aqueous layer was discarded, and the organic layer was sequentially washed with aqueous NaHCO3 (5%, 5 ml) and brine (5%, 5 ml) and 5h was
Synthesis of ethyl 2-((3bS,4aR)-3-(trifluoromethyl)-4,4a-dihydrospiro[cyclopropa[3,4]cyclopenta[1,2-c]pyrazole-5,2′-[1,3]dithiolanel-1 (3bH)-yl)acetate (5h) from (1R,5R)-spiro[bicyclo[3.1.0]hexane-2.2′-[1,3]dithiolanl-3-one (5e)
[00603] 5e (756 mg) was charged to a vessel and dissolved in 2-methyltetrahydrofuran (7.6 mL). To this solution was charged ethyl trifluoroacetate (0.57 g) and the resulting solution was cooled to about 0 °C. Lithium hexamethyldisilazide (1.0 M solution in THF, 4.5 g) was charged over about 60 minutes and reaction was agitated until complete. A solution of sulfuric acid (2.0 g) in water (5.6 mL) was charged, then the reaction was warmed to about 20 °C and agitated for about 20 minutes. Layers were separated and aqueous layer was extracted twice with 2-methyltetrahydrofuran (5.3 mL). Combined organic layer was concentrated to about 0.4 mL and N,N-diisopropylamine (0.5 g) was charged. The product was crystallized by the addition of heptane (11 ml). The slurry was filtered and the filter cake was washed with heptane, then deliquored thoroughly, and dried to afford 5f-01. 1H NMR (400 MHz, Acetonitrile-d3) δ 7.84 (m, 2H), 3.58 (d, J = 8.7 Hz, 2H), 3.47 – 3.27 (m, 4H), 2.20 (s, 1H), 1.81 – 1.68 (m, 1H), 1.24 (dd, J = 6.5, 0.6 Hz, 12H), 0.99 (q, J = 6.5 Hz, 1H), 0.13 (s, 1H).
[00604] Acetyl chloride (1.02 g) was charged to a cooled reaction vessel containing ethanol (5.0 mL) at about 0 °C, then warmed to about 20 °C and agitated for about 30 minutes. In a separate vessel, 5f-01 (1.00 g), ethyl hydrazinoacetate hydrochloride (0.48 g), and lithium chloride (0.39 g) were combined, and the acetyl chloride/ethanol solution was charged to this mixture, followed by tri ethyl orthoformate (1.16 g). The mixture was heated to about 45 °C and agitated until reaction was complete. The reaction was then concentrated to 2 volumes and dichlorom ethane (5.0 mL) was added followed by water (5.0 mL). Layers were separated and organic layer was washed with 5 wt % aqueous sodium bicarbonate followed by 10 wt % aqueous sodium chloride to afford a solution of 5h in dichloromethane that was carried forward into the subsequent step. 1H NMR (400 MHz, DMSO-d6) δ 5.27 – 4.79 (m, 2H), 4.14 (qd, J =
Synthesis of (1R,5R)-spiro[bicyclo[3.1.0]hexane-2,2′-[1,3]dithiolan]-3-one (5e) from (1R,5R)-spiro[bicyclo[3.1.0]hexane-2,2′-[1,3]dithiolan]-3-one (5e) from (1R,5S)-bicyclo[3.1.0]hexan-2-one (4a)
[00605] Tert-butyl nitrite (1.31 g) was charged to a vessel containing 4a (1.00 g, 1.0 equiv) and tetrahydrofuran (5.0 mL) at about 20 °C. Potassium tert-butoxide (6.1 g, 1.7M in tetrahydrofuran) was charged over not less than 30 minutes. The mixture was then agitated until the reaction was complete. The reaction was quenched with aqueous citric acid (2.00 g in 10.00 g water) and extracted with dichloromethane (10.0 mL, 3x). This solution was partially concentrated and the product was isolated by the addition of heptane (6.0 mL). The slurry was filtered and the filter cake was washed with heptane (2.0 mL), then deliquored thoroughly to afford 4d 1H NMR (400 MHz, DMSO-d6) δ 12.26 (s, 1H), 2.73 (d, J = 18.5 Hz, 1H), 2.63 (ddd, J = 18.6, 5.3, 2.0 Hz, 1H), 2.17 – 2.01 (m, 2H), 1.34 (dddd, J= 9.2, 7.1, 4.9, 2.0 Hz, 1H), 0.77 (td, J= 4.6, 3.4 Hz, 1H).
[00606] 1,2-Ethanedithiol (0.41 g) was charged to a vessel containing a solution of 4d (0.50 g, 4.0 mmol) in glacial acetic acid (2.5 mL) at about 20 °C. para-toluenesulfonic acid monohydrate (0.15 g) was added and the mixture was agitated until the reaction was complete. The product was isolated by the addition of water (2 mL). The slurry was filtered and the filter cake was washed with water, then deliquored thoroughly to afford 5i. 1H NMR (400 MHz,
[00607] Para-toluenesulfonic acid (0.90 g) was charged to a vessel containing a suspension of 5i (0.50 g, 2.5 mmol) in methyl ethyl ketone (2.5 mL) and water (2.5 mL). The mixture was agitated at about 85 °C until the reaction was complete. The product was isolated from the reaction mixture by cooling to about 20 °C, adding water (2.50 mL), and cooling to about 0 °C. The slurry was filtered and the filter cake was washed with water, then deliquored thoroughly to afford 5e. 1H NMR (400 MHz, DMSO-d6) δ 3.55 – 3.37 (m, 3H), 3.28 – 3.13 (m, 1H), 3.03 (ddd, J = 18.5, 5.6, 2.2 Hz, 1H), 2.20 (d, J = 18.5 Hz, 1H), 1.84 (ddd, J = 8.0, 7.0, 3.8 Hz, 1H), 1.66 (tdd, J = 7.2, 5.6, 4.1 Hz, 1H), 1.03 (tdd, J = 7.9, 5.9, 2.1 Hz, 1H), 0.06 (dt, J = 6.0, 4.0 Hz, 1H).
Example 6: Preparation of 2-((3bS,4aR)-5,5-difluoro-3-(trifluoromethyl)-3b,4,4a,5-tetrahydro-1H-cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-1-yl)acetic acid (VII) from ethyl 2-((3bS,4aR)-5-oxo-3-(trifluoromethyl)-3b,4,4a,5-tetrahydro-1H-cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-1-yl)acetate (XIV)
Synthesis of ethyl 2-((3bS,4aR)-3-(trifluoromethyl)-4,4a-dihydrospiro[cyclopropa[3,4]cyclopenta[1,2-c]pyrazole-5,2′-[1,3]dithiolane]-1(3bH)-yl)acetate (5h) from ethyl 2-((3bS,4aR)-5-oxo-3-(trifluoromethyl)-3b,4,4a,5-tetrahydro-1H-cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-1-yl)acetate (XIV)
[00608] Dichloromethane (27 g) was added to a reactor containing XIV (1.0 g) and cooled to about 10 °C. To this was added 1,2-ethanedithiol (0.18 g, 1.2 equiv.). To this was added boron trifluoride acetic acid complex (3.3 g, 2.5 equivalents) over about 25 minutes, and the reaction mixture was agitated at about 20 °C until complete. A solution of calcium chloride dihydrate (0.80g, 0.78 equiv) in 0.10 N hydrochloric acid (16 g) was added over about 1 hour at about 10 °C, and the mixture was agitated for about 90 minutes at about 20 °C. The organic layer was washed successively with water (8 g) and sodium bicarbonate solution (5 wt/wt%). The organic layer was concentrated to afford 5h. 1H NMR (400 MHz, DMSO-d6) δ 5.27 – 4.79 (m, 2H),
Synthesis of ethyl 2-((3bS,4aR)-5,5-difluoro-3-(trifluoromethyl)-3b,4,4a,5-tetrahydro-1H-cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-1-yl)acetate (VII-A)
[00609] Dichloromethane (26 g) was added to a reactor containing 1,3-dibromo-5,5-dimethylhydantoin (DBDMH, 2.4 g, 3.1 equiv.) and cooled to about -10 °C. To this was added 70% hydrofluoric acid/pyridine complex (1.3 g, 17 equiv.), followed by a solution of 5h (1.0 g) in dichloromethane (3 g). The reaction was agitated at about 0 °C until complete. A solution of potassium hydroxide (3.7 g, 25 equivalents) and potassium sulfite (1 .9 g, 4 equiv.) in water (24 g) was added, maintaining an internal temperature of about 5 °C, and agitated for about 30 minutes at about 20 °C. Layers were separated and organic layer was washed with hydrochloric acid (1.1 g, 4 equiv.) in water (9.6 g). The organic layer was concentrated to afford VII-A. 1H NMR (400 MHz, DMSC-d6) δ 5.31 – 5.04 (m, 2H), 4.17 (q, J = 7.1 Hz, 2H), 2.78 – 2.57 (m,
Synthesis of 2-((3bS,4aR)-5,5-difluoro-3-(trifluoromethyl)-3b,4,4a,5-tetrahydro-1H- cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-1-yl)acetic acid (VII)
[00610] A reactor was charged with a solution of VII-A (1.0 g) in dichloromethane (18 g) and cooled to about 5 °C. To this was added ethanol (1.5 g), followed by potassium hydroxide (45 wt/wt%, 0.74 g, 2.0 equiv.). The reaction mixture was agitated at about 20 °C until complete. Water (3.7 g) was added and the reaction mixture was agitated for about 30 minutes. Organic layer was removed and reaction was cooled to about 10 °C. Dichloromethane (18 g) was added, followed by 2N hydrochloric acid (3.3 g, 2,2 equiv.). Reaction was warmed to about 20 °C and agitated for 10 minutes. Layers were separated and aqueous phase was washed with dichloromethane (18 g). Organic layers were combined and concentrated on rotary evaporator to afford VII. 1H NMR (400 MHz, DMSO-d6) δ 13.50 (s, 1H), 5.14 – 4.81 (m, 2H), 2.82 – 2.56 (m, 2H), 1.46 (dddd, J = 8.5, 7.1, 5.5, 1.4 Hz, 1H), 1.08 – 1.00 (m, 1H). 13C NMR (101 MHz, DMSO-d6) δ 168.16, 143.05 (t, J = 29.4 Hz), 134.40 (q, J = 38.9 Hz), 132.80, 121.11 (q, J = 268.4 Hz), 120.55 (t, J = 243.3 Hz), 52.54, 27.97 (dd, J = 34.7, 29.0 Hz), 23.81 (d, J = 2.5 Hz), 12.13 (t, J = 3.1 Hz). 19F NMR (376 MHz, DMSO-d6) δ -60.39 (d, J = 1.4 Hz), -79.83 (dd, J = 253.2, 13.1 Hz), -102.97 (dd, J= 253.2, 9.8 Hz).
Example 7: Preparation of 4-chloro-7-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1- (2,2,2-trifluoroethyl)-1H-indazol-3-amine (V-02) and its mesylated derivatives
Synthesis of 4-chloro-7-bromo-1-(2,2,2-trifluoroethyl)-1H-indazol-3-amine (V-A)
[00611] To a reactor was added tetrahydrofuran (THF, 275 kg) and diisopropyl amine (DIPA, 30 kg) and the mixture was cooled to about -35 °C. nButyl lithium (2.5 mol/L in hexanes, 74 kg) was charged slowly keeping the reaction temperature less than -30 °C. The mixture was agitated at-35 °C until the reaction was complete. 1-bromo-4-chloro-2-fluorobenzene (52 kg) was charged keeping reaction temperature less than 30 °C and the mixture was agitated at -35°C until reaction was complete. N,N-dimethylformamide (DMF, 36 kg) was charged keeping reaction temperature less than -30 °C and the mixture was agitated at about -35 °C until reaction was complete. Hydrochloric acid (HCl, 18 mass% in water, 147 kg) was charged keeping reaction temperature less than -5 °C. The reaction was warmed to about 0 °C, water (312 kg) was added, and the reaction was extracted with methyl tert-butyl ether (MTBE, 770 kg). The organic was warmed to about 20 °C and washed with brine (NaCl, 23.5 mass% in water, 1404 kg). The mixture was distilled to about 3-4 volumes and heptane was charged (354 kg). The product was isolated by distillation to 3-4 volumes. The slurry was filtered and washed with heptane (141 kg) and dried to afford 6a. 1H NMR (400 MHz, DMSO-d6) δ 10.23 (d, J = 1.2 Hz, 1H), 8.00 (dd, J = 8.7, 1.4 Hz, 1H), 7.44 (dd, J = 8.7, 1.4 Hz, 1H).
[00612] 6a (98.5 kg) was charged to a reactor containing acetic anhydride (105 kg) and acetic acid (621 kg) at 20 °C. The mixture was heated to about 45 °C and hydroxyl amine hydrochloride (31.5 kg) was charged. The reaction was heated to about 75 °C and agitated until the reaction was complete. The product was isolated from the reaction mixture by adding water (788 kg) at about 45 °C. The mixture was cooled to about 25 °C and then the slurry was filtered. The filtered cake was washed with water (197 kg,). The cake was dried to afford 6b. 1H NMR (400 MHz, DMSO-d6) δ 8.11 (dd, J= 8.8, 1.4 Hz, 1H), 7.58 (dd, J = 8.8, 1.4 Hz, 1H).
[00613] To a reactor was charged 6b (84 kg), isopropanol (318 kg), and water (285 kg).
Hydrazine hydrate (20 wt% in water, 178 kg) was charged and the mixture was heated to about 80 °C until the reaction was complete. The product was isolated by cooling the reaction to about 25 °C. The slurry was filtered and the filtered cake was washed with a mixture of isopropanol (127 kg) and water (168 kg). The wet solids were recharged to the reactor and water (838 g) was added. The mixture was agitated at about 25 °C and then filtered and washed with water
[00614] 6c (75 kg) was charged to a reactor containing N,N-dimethylformamide (75 kg). Potassium phosphate (99.8 kg) was charged to the reactor at about 25 °C and the mixture was agitated. 2,2,2-trifluoroethyl trifluoromethanesulfonate (74.3 kg) was charged at about 25 °C and the mixture was agitated until the reaction was complete. Water (375 kg) was charged and the mixture was agitated at about 20 °C. The slurry was filtered and washed with water (150 kg). N,N-dimethylformamide (424 kg) and the wet solid were charged to a reactor at about 20 °C.
The mixture was agitated at about 45 °C. 5 % hydrochloric acid (450 kg) was charged drop-wise to the mixture at about 45 °C. The mixture was cooled to about 25 °C. The slurry was filtered and washed with water (375 g). Water (375 kg) and the filtered solid were charged to a reactor at about 20 °C. The mixture was agitated for about 1 hour at about 20 °C. The slurry was filtered and washed with water (375 kg). The cake was dried to afford V-A. 1H NMR (400 MHz, DMSO-d6) δ 7.57 (d, J= 8.1 Hz, 1H), 6.98 (d, J = 8.1 Hz, 1H), 5.70 (s, 2H), 5.32 (q, J = 8.6 Hz,
2H).
Synthesis of 4-chloro-7-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1-(2,2,2-trifluoroethyl)- 1 H-indazol-3-amine (V-02)
[00615] A reactor containing tetrahydrofuran (27 g) and V-A (1.0 g) was cooled to about 0 °C. Chlorotrimethylsilane (7.6 g, 2.3 equiv) was added, followed by the slow addition of lithium bis(trimethylsilyl)amide (5.7 g, 1 M in THF, 2.1 equiv.). The mixture was stirred at about 0 °C until bistrimethylsilane protection was complete. The solution was washed with ammonium chloride in water (10 wt%, 52 g), toluene (44 g) was added, and the biphasic mixture was filtered through celite. The organic and aqueous phases were separated and the aqueous phase was washed with toluene (44 g). The organics were combined, washed with brine (58 g), and azeotropically distilled . The solution was cooled to about 0 °C, isopropylmagnesium chloride lithium chloride complex (2.7 g, 1.3 M in THF, 1.2 equiv.) was added and the reaction was stirred at about 0 °C until lithium halogen exchange was complete. Isopropoxyboronic acid pinacol ester (6.8 g, 1.2 equiv.) was added and the reaction was stirred at about 0°C until botylation was complete. At about 0 °C, The reaction was quenched with aqueous hydrochloric acid (52 g, 1 M), acetonitrile (16 g) was added, and the mixture was stirred until trimethylsilane deprotection was complete. The solution was extracted with ethyl acetate (45 g) and the organic was washed twice with brine (2 x 58 g). The solution was concentrated to low volumes (26 g), dim ethylformami de (47 g) was added, and the solution was concentrated again (51 g). The product was crystallized by the addition of water (50 g). The slurry was filtered and filter cake was washed with heptane (14 g). The solids were dried to afford V-02. 1H NMR (400 MHz, DMSO-d6) δ 7.70 (dd, J = 7.6, 1.0 Hz, 1H), 7.07 (dd, J = 7.6, 1.0 Hz, 1H), 5.58 (s, 2H), 5.46 (q, J = 9.1Hz, 2H), 1.32 (s, 12H).
Synthesis of 4-chloro-7-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1-(2,2,2-trifiuoroethyl)- 1 H-indazol-3-amine (V-02)
[00616] To a reactor was charged V-A (30 kg), bis(pinacolato)diboron (27.9 kg), bis(triphenylphosphine)palladium (II) dichloride (0.9 kg, 1.5 mol%), N,N-dimethylformamide (56 kg, 2 rel. vol.) and toluene (157 kg, 6 rel vol.). The mixture was heated to about 105 °C until the reaction was complete. The mixture was cooled to about 25 °C, filtered through celite (15 kg, 0.5 rel. wt.) and rinsed forward with ethyl acetate (270 kg, 10 rel vol.). PSA-17 palladium scavenger (3 kg, 10 wt%) was added and the mixture was stirred at about 45 °C. The mixture was filtered and the cake was washed with ethyl acetate (54 kg, 2 rel. vol.). The mixture was washed twice with lithium chloride (180 kg, 6 rel. vol.) and once with brine (NaCl, 23.5 mass% in water, 180 kg, 6 rel. vol.). The mixture was then concentrated to about 5-6 rel. vol. under vacuum, heated to about 45 °C then cooled to about 25 °C. Heptane (102 kg, 5 rel. vol.) was charged and the mixture was concentrated to about 4-5 rel. vol. The product was isolated by charging heptane (41 kg, 2 rel. vol.) and cooling the mixture to about 0 °C. The slurry was filtered and washed with heptane (41 kg, 2 rel. vol.). The wet solids were recharged to the reactor with ethyl acetate (27 kg, 1 rel. vol.) and heptane (82 kg, 4 rel. vol.), heated to about 65 °C, and then cooled to about 5 °C. The slurry was filtered and washed with heptane (41 kg, 2 rel. vol.). The cake was dried to afford V-02. 1H NMR (400 MHz, DMSO-d6) δ 7.70 (dd, J =
Synthesis of N-(4-chloro-7-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1-(2,2,2- trifluoroethyl)-1H-indazol-3-yl)-N-(methylsulfonyl)methanesulfonamide (V-04)
[00617] To a 100 mL reactor was added V-02 (5.00 g), 2-methyltetrahydrofuran (50 mL), and triethylamine (11.1 mL). The mixture was cooled to about 10 °C and methanesulfonyl chloride (2.58 mL, 33.3 mmol) was added to the mixture. The mixture was agitated at about 10 °C until reaction was complete. The mixture was concentrated to dryness and the residue was purified by column chromatography to afford V-04. 1H NMR (400 MHz, DMSO-d6) δ 7.96 (d, J = 7.7 Hz, 1H), 7.50 (d, J = 7.6 Hz, 1H), 5.95 (q, J = 8.8 Hz, 2H), 3.66 (s, 6H), 1.37 (s, 12H).
Synthesis of N-(4-chloro-7-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2)-1-(2,2,2,- trifluoroethyl)-1H-indazol-3-yl)methanesulfonamide (V-03)
[00618] To a 100 mL reactor was added V-02 (5.00 g), 2-methyltetrahydrofuran (50 mL), and triethylamine (11.1 mL, 79.6 mmol). The mixture was cooled to about 10 °C and methanesulfonyl chloride (2.58 mL) was added to the mixture. The mixture was agitated at about 10 °C until reaction was complete. To the mixture was added 2-methyltetrahydrofuran (21.5 g) and sodium hydroxide (0.43 g) and the mixture was agitated at about 25 °C until the reaction was complete. To the resulting solution was added 2-methyltetrahydrofuran (21.5 g), water (25 g) and acetic acid to achieve a pH of less than 7. The lower aqueous layer was then removed and the organic layer was washed with brine (5 wt%, 7.8g). The organic layer was then concentrated to dryness and the residue was purified by column chromatography to afford V-03. 1H NMR (400 MHz, DMSO-d6) δ 9.96 (s, 1H), 7.86 (d, J = 7.6 Hz, 1H), 7.34 (d, J = 7.6 Hz, 1H), 5.80 (q, J = 8.9 Hz, 2H), 3.22 (s, 3H), 1.36 (s, 12H).
Synthesis of N-(7-bromo-4-chloro-1-(2,2,2-trifluoroethyl)-1H-indazol-3-yl)-N- (methylsulfonyl)methanesulfonamide (V-06)
[00619] To a reactor was added V-A (3 g), 2-methyltetrahydrofuran (25.8 g), and triethylamine (7.6 mL). The mixture was cooled to about 10 °C, methanesulfonyl chloride (1.8 mL) was added, and the mixture was stirred until reaction was complete. The reaction mixture was washed with aqueous sodium chloride (30 mL) and the organic layer was evaporated to dryness. The residue was purified by column chromatography to afford V-06. 1H NMR (400 MHz, DMSO-d6) δ 7.83 (d, J = 8.0 Hz, 1H), 7.35 (d, J = 8.1 Hz, 1H), 5.79 (q, J = 8.5 Hz, 2H), 3.62 (s, 6H).
Synthesis of N-(7-bromo-4-chloro-1-(2,2,2-trifluoroethyl)-1H-indazol-3-yl)methanesulfonamide (V-05)
[00620] To a reactor was added V-02 (3 g), 2-methyltetrahydrofuran (30 mL), and triethylamine (7.6 mL). The mixture was cooled to about 10 °C, methanesulfonyl chloride (1.8 mL) was added, and the mixture was stirred until reaction was complete. The reaction mixture was washed with aqueous sodium chloride (30 mL) and the organic portion was concentrated to dryness.
[00621] To the resulting mixture (2.7g) was added 2-methyltetrahydrofuran (15 mL) and sodium hydroxide (1M in water, 15 mL). The mixture was stirred at about 20 °C until the reaction was complete. The aqueous layer was removed and the organic was washed with acetic acid (0.7M in water, 10 mL) and sodium chloride (5 wt% in water, 10 mL).The organic layer was then concentrated to dryness and the residue was purified by column chromatography to afford V-05. 1H NMR (400 MHz, DMSO-D6) δ 10.03 (s, 1H), 7.71 (dd, J = 8.0, 1.6 Hz, 1H), 7.20 (dd, J = 8.1, 1.6 Hz, 1H), 5.64 (q, J = 8.7 Hz, 3H), 3.19 (2, 3H).
Synthesis of N-(4-chloro-7-(4,4,5,5-tetramethyl-1,3,2,-dioxaborolan-2-yl)-1-(2,2,2- trifluoroethyl)-1H-indazol-3-yl)-N-(methylsulfonyl)methanesulfonamide (V-04)
[00622] To a reactor was charged V-06 (148 mg), bis(pinacolato)diboron (93 mg), potassium acetate (90 mg) and bis(triphenylphosphine)palladium (II) chloride (4.3 mg, 1.5 mol%). N,N- dimethylformamide (0.2 mL) and toluene (0.6 mL) were added and the reaction was heated to about 105 °C until completion. V-04 was formed. 1H NMR (400 MHz, DMSO-D6) δ 7.96 (d, J = 7.7 Hz, 1H), 7.50 (d, J= 7.6 Hz, 1H), 5.95 (q, J= 8.8 Hz, 2H), 3.66 (s, 6H), 1.37 (s, 12H).
Synthesis of N-(4-chloro-7-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1-(2,2,2- trifluoroethyl)-1H-indazol-3-yl)methanesulfonamide (V-03)
[00623] To a reactor was charged V-05 (124 mg), bis(pinacolato)diboron (93 mg), potassium acetate (90 mg) and bis(triphenylphosphine)palladium (II) chloride (4.3 mg, 1.5 mol%). N,N- dimethylform amide (0.2 mL.) and toluene (0.6 mL, 6 rel. vol.) were added and the reaction was heated to about 105 °C until completion. V-03 was formed. 1H NMR (400 MHz, DMSO-d6) δ
Example 8: Preparation of N-((S)-1-(3-bromo-6-(3-methyl-3-(methylsulfonyl)but-1-yn-1- yl)pyridin-2-yl)-2-(3,5-difluorophenyl)ethyl)-2-((3bS,4aR)-5,5-difluoro-3-(trifluoromethyl)- 3b,4,4a,5-tetrahydro-1H-cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-1-yl)acetamide (IV)
Synthesis of N-((S)-1-(3-bromo-6-(3-methyl-3-(methylsulfonyl)but-1-yn-1-yl)pyridin-2-yl)-2- (3,5-difluorophenyl)ethyl)-2-((3bS.4aR)-5,5-difluoro-3-(trifluoromethyl)-3b,4,4a,5-tetrahydro- 1H-cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-1-yl)acetamide (IV) from (S)-1-(3-bromo-6-(3- methyl-3-(methylsulfbnyl)but-1-yn-1-yl)pyridin-2-yl)-2-(3.5-difluorophenyl)ethan-1-amine (VI) Method 1
[00624] n-Propyl phosphonic anhydride (T3P, 3.1 g, 1.5 equiv.) was slowly added to a reactor containing amine VI (1.5 g), acid VII (1.0 g, 1.1 equiv.), triethylamine (Et3N, 0.5 g, 1.5 equiv.), and acetonitrile (MeCN, 8.0 g). The mixture was agitated at about 20 °C until the reaction was complete. The product was crystallized from the reaction mixture with DMF (0.63 g), and water (15 g). The slurry was filtered and the filter cake was washed with a mixture of acetonitrile and water (2 x 2.5 g). The cake was dried to afford IV. 1H NMR (400 MHz, DMSO-d6) δ9.19 (d, J = 8.3 Hz, 1H), 8.12 (d, J = 8.3 Hz, 1H), 7.50 (d, J = 8.3 Hz, 1H), 7.07 (tt, J = 9.4, 2.4 Hz, 1H),
Synthesis of N-((S)-1-(3-bromo-6-(3-methyl-3-(methylsulfonyl)but-1-yn-1-yl)pyridin-2-yl)-2-(3,5-difluorophenyl)ethyl)-2-((3bS,4aR)-5,5-difluoro-3-(trifluoromethyl)-3b,4,4a,5-tetrahydro-1H-cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-1-yl)acetamide (IV) from (S)-1-(3-bromo-6-(3-methyl-3-(methylsulfonyl)but-1-yn-1-yl)pyridin-2-yl)-2-(3,5-difluorophenyl)ethan-1-amine (VI) Method 2
[00625] N-methylmorpholine (NMM, 0.51 g, 2.3 equiv.) was added to a vessel containing amine VI (1.0 g), acid VII (1.0 g), 1-hydroxybenzotriazole hydrate (HOBt ● H2O, 0.17 g, 0.5 equiv.), N-(3-dimethylaminopropyi)-N’-ethylcarbodiimide (EDCI ● HCl, 0.52 g, 1.25 equiv.), and acetonitrile (MeCN, 7.8 g). The mixture was agitated at about 20 °C until the reaction was complete. The product was crystallized from the reaction mixture with DMF (2.8 g), and water (10 g). The slurry was filtered and the filter cake was washed with a mixture of acetonitrile and water. The cake was dried to afford IV. 1H NMR (400 MHz, DMSO-d6) δ9.19 (d, J = 8.3 Hz, 1H), 8.12 (d, J = 8.3 Hz, 1H), 7.50 (d, J = 8.3 Hz, 1H), 7.07 (tt, J = 9.4, 2.4 Hz, 1H), 6.96 – 6.87 (m, 2H), 5.52 (td), J = 8.8, 5.3 Hz, 1 H), 4.93 – 4.73 (m, 2H), 3.22 (s, 3H), 3.11 – 2.90 (m, 2H), 2.66 – 2.52 (m, 2H), 1.69 (s, 6H), 1.45 – 1.36 (m, 1H), 1.02 – 0.93 (m, 1H). 13C NMR (100 MHz, DMSO-d6): δ 164.42, 163.62, 163.49, 161.17, 161.04, 158.19, 142.92, 142.20, 142.10, 142.01, 141.63, 140.23, 134.11, 133.73, 132.14, 128.66, 122.23, 120.49, 119.56, 112.49, 112.25, 104.75, 102.25, 88.62, 84.20, 57.44, 53.85, 53.03, 35.21, 23.41, 22.46, 22.40, 11.79.
Example 9: Preparation of N-((S)-1-(3-(3-amino-4-chloro-1-(2,2,2-trifluoroethyl)-1H- indazol-7-yl)-6-(3-methyl-3-(methylsulfonyl)but-1-yn-1-yl)pyridin-2-yl)-2-(3,5- difluorophenyl)ethyl)-2-((3bS,4aR)-5,5-difluoro-3-(trifluoromethyl)-3b,4,4a,5-tetrahydro- 1H-cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-1-yl)acetamide (III)
Synthesis of compound III-03
[00626] To a reactor was added IV (1 .0 g), potassium bicarbonate (0.43 g, 1.3 equiv), dichlorobis(tricyclohexylphosphine)palladium(II) (28 mg, 2.5mol%), V-02 (0.67 g), butyl acetate (7.3 g) and water (2.1 g). The reactor was inerted and the mixture was agitated at about 85 °C (75-90 °C) until the reaction was complete. The mixture was cooled to about 40 °C and passed through celite (0.52 g). The celite cake was rinsed with butyl acetate (1.8 g). The filtrate and rinse were combined and this solution was washed twice with a mixture of N-acetyl-L-
cysteine (0.31 g) dissolved in water (5.2 g) and sodium hydroxide in water (5 wt%, 5.4 g). The organics were washed twice with sodium chloride in water (5 wt%, 11 g). The solution was azeotropically distilled into 1-propanol (3.3 g). To the propanol solution at about 50 °C was added methanesulfonic acid (0.31 g, 2.25 equiv.) and the product was crystallized using dibutyl ether (5.1 g). The slurry was cooled to about 10 °C, filtered, and the filter cake was washed with a 5:1 mixture of propanol in dibutyl ether (1.6 g). The solids were dried to afford III-03 1H NMR (400 MHz, DMSO-d6) δ 9.19 (d, J = 8.3 Hz, 2H), 7.84 – 7.69 (m, 4H), 7.11 (d, J = 7.7 Hz, 2H), 7.07 – 6.95 (m, 3H), 6.82 (d, J = 7.7 Hz, 2H), 6.54 – 6.40 (m, 4H), 4.90 (d, J = 16.4 Hz, 2H), 4.76 – 4.60 (m, 4H), 4.15 (dq, J = 16.6, 8.4 Hz, 2H), 3.75 (dt, J = 16.3, 8.7 Hz, 2H), 3.25 (s, 7H), 2.99 – 2.86 (m, 4H), 2.63 – 2.50 (m, 3H), 2.41 (s, 14H), 1.73 (d, J = 2.1 Hz, 13H), 0.93 (dd, J = 6.1, 3.9 Hz, 2H).
Synthesis of N-((S)-1-(3-(3-amino-4-chloro-1-(2,2,2-trifluoroethyl)-1H-indazol-7-yl)-6-(3-methyl-3-(methylsulfonyl)but-1-yn-1-yl)pyridin-2-yl)-2-(3,5-difluorophenyl)ethyl)-2- ((3bS,4aR)-5,5-difluoro-3-(trifluoromethyl)-3b,4,4a,5-tetrahydro-1H-cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-1-yl)acetamide (III)
[00627] Aqueous sodium hydroxide (0.2 M; 2.2 equivalents; 9.2 g) was added to a reactor containing III-03 (1.0 g) in MeTHF (8.3 g) at about 20 °C. The biphasic mixture was agitated for about 15 min, and the aqueous layer was removed. The organic layer was washed four times with 2.0 wt% aqueous sodium chloride (9.8 g) and was distilled. The solution containing III was used directly in the II process below. A sample was concentrated to dryness for analysis. 1H NMR (400 MHz, CDCl3): δ 7.44 ( m, 1H), 7.39 (br, 1H), 7.18 (m, 1H), 6.90 (m, 1H), 6.65 (m 1H), 4.10 (m, 2H), 3.72 (m, 4H), 2.78 (m 2H), 2.56 (br, 4H), 1.31 (s, 9H). 13C NMR (100 MHz, DMSO-d6): δ 176.88, 158.95, 141,06, 129.55, 112.79, 109.56, 106.83, 66.66, 65.73, 57.45,
54.12, 39.53, 27.63.
Example 10: Preparation of N-((S)-1-(3-(4-chloro-3-(N- (methylsulfonyl)methylsulfonamido)-1-(2,2,2-trifluoroethyl)-1H-indazol-7-yl)-6-(3-methyl-3-(methylsulfonyl)but-1-yn-1-yl)pyridin-2-yl)-2-(3,5-difluorophenyl)ethyl)-2-((3bS,4aR)-5,5-difluoro-3-(trifluoromethyl)-3b,4,4a,5-tetrahydro-1H-cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-1-yl)acetamide (II)
[00628] Methanesulfonyl chloride (0.32 g, 2.5 equivalents) was added to a reactor containing III (1.0 g), triethylamine (0.69 g, 6.0 equivalents), and MeTHF (11 g) at about 10 °C. The mixture was agitated at about 10 °C until the reaction was complete. The reaction mixture was washed with water (6.4 g) for about 15 minutes, and warmed to about 20 °C. The layers were separated and the organic layer was washed for about 15 minutes with 10 wt% aqueous sodium chloride (6.9 g). The layers were separated and the organic layer was used directly in the next step. An aliquot was concentrated to dryness for analysis. 1H NMR (400 MHz, δ6-DMSO; 9: 1 mixture of atropi somers): δ 9.20 (d, J = 7.9 Hz 1 H), 8.99* (d, J = 8.6 Hz, 1 H), 7.96* (d, J = 7.9 Hz, 1 H), 7.83 (d, J = 8.0 Hz, 1 H), 7.80* (d, J = 7,9 Hz, 1 H), 7.76 (d, J – 8.0 Hz, 1 H), 7.45 (d, J = 7.7 Hz, 1 H), 7.41* (d, J = 7.8 Hz, 1 H), 7.31* (d, J = 7.8 Hz, 1 H), 7.02 (tt, J = 9.4, 2.1 Hz,
Example 11: Preparation of N-((S)-1-(3-(4-chIoro-3-(methylsuIfonamido)-1-(2,2,2- trifluoroethyl)-1H-indazol-7-yl)-6-(3-methyl-3-(methylsulfonyl)but-1-yn-1-yl)pyridin-2-yl)- 2-(3,5-difluorophenyl)ethyl)-2-((3bS,4aR)-5,5-difluoro-3-(trifluoromethyl)-3b,4,4a,5- tetrahydro-1H-cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-1-yl)acetamide (I)
Synthesis of sodium (4-chloro-7-(2-((S)-1-(2-((3bS.4aR)-5,5-difluoro-3-(trifluoromethyl)- 3b,4,4a,5-tetrahydro-1H-cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-1-yl)acetamido)-2-(3,5- difluorophenyl)ethyl)-6-(3-methyl-3-(methylsulfonyl)but-1-yn-1-yl)pyridin-3-yl)-1-(2,2,2- trifluoroethyl)-1H-indazol-3-yl)(methylsulfonyl)amide (1-02)
[00629] Sodium hydroxide (1 M, 2.9 g, 3.0 equiv.) was added to a reactor containing II (1.0 g) and 2-methyltetrahydrofuran (8.4 g) at about 35 °C. The mixture was agitated until the reaction was deemed complete. The reaction mixture was adjusted to between about 20 and 40 °C and the bottom layer was removed. The organic layer was washed with water (2.9 g) for about 15 minutes, and the bottom layer was removed. The organic solvent was swapped for ethanol and the solution was concentrated to about 5 volumes and the temperature was adjusted to about 35 °C. n-Heptane (3.4 g) was slowly added, and the mixture was aged for about 12 hours. The solids were collected by filtration, and the filter cake was washed with ethanol/n- heptane (1:1). The resultant wet cake was dried under vacuum to afford 1-02. 1H NMR (400 MHz, DMSO-d6) δ 9.09 (d, J = 8.0 Hz, 1H), 8.93* (d, J = 8.5 Hz), 7.80 – 7.72* (m), 7.71 (s, 2H), 6.99 (tt, J = 9.5, 2.4 Hz, 1H), 6.94 (d, J = 7.6 Hz, 1H), 6.90* (d, J = 6.3 Hz), 6.69 (d, J = 7.6 Hz, 1H), 6.57 – 6.51* (m), 6.48 – 6.40 (m, 2H), 4.90 (d, J = 16.5 Hz, 1H), 4.77 (d, J = 16.4
88.14, 88.00, 84.69, 84.65, 57.33, 53.22, 52.96, 52.76, 52.44, 40.15, 39.94, 39.73, 39.52, 39.31, 39.10, 38.97, 38.89, 38.65, 35.10, 35.08, 27.86, 27.56, 27.52, 27.23, 23.19, 22.42, 22.41, 22.30, 22.28, 11.63. * Signals arising from minor atropisomer. 13C NMR data is reported for the mixture of atropisomers.
Synthesis of N-((S)-(3-(4-chloro-3-(methylsulfonamido)-1-(2,2,2-trifluoroethyl)-1H-indazol-7-yl)-6-(3-methyl-3-(methylsulfonyl)but-1-yn-1-yl)pyridin-2-yl)-2-(3,5-difluorophenyl)ethyl)-2-((3bS,4aR)-5,5-difluoro-3-(trifluoromethyl)-3b,4,4a,5-tetrahydro-1H-cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-1-yl)acetamide (I) from sodium (4-chioro-7-(2-((S)-1-(2-((3bS.4aR)-5,5-difluoro-3-(trifluoromethyl)-3b,4,4a,5-tetrahydro-1H-cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-l-yl)acetamido)-2-(3.5-difluorophenyl)ethyl)-6-(3-methyl-3-(methylsulfonyl)but-1-yn-1-yl)pyridin-3-yl)-1-(2,2,2-trifluoroethyl)-1H-indazol-3-yl)(methylsulfonyl)amide (I-02)
[00630] Compound I-02 (1.0 g) and glacial acetic acid (2.1 g) were combined at about 20 °C and were agitated until dissolved. The resultant solution was transferred to a reactor containing water (15 g) over about 1 hour. The resultant slurry was further agitated for about one hour, and was filtered. The wet cake was washed with water (2 x 5 g), deliquored, and dried at about 60 °C under vacuum to provide I. 1H NMR (400 MHz, δ6-DMSO; 5:1 mixture of atropi somers) δ 10.11* (s), 10.00 (s, 1 H), 9.25 (d, J= 8.0 Hz, 1 H), 8.92* (d, J = 8.4 Hz), 7.90* (d, J = 7.6 Hz), 7.81 (d, J = 8.0 Hz, 1 H), 7.76 (d, J= 8.0 Hz, 1 H), 7.32 (d, J = 7.6 Hz, 1 H), 7.23* (d, J = 8.0 Hz), 7.19* (d, J = 8.0 Hz), 7.02 (tt, J = 9.4, 2,4 Hz, 1 H), 6.94* (m), 6.86 (d, J = 7.6 Hz, 1 H), 6.54* (m), 6.48 (m, 2 H), 4.92 (d, J = 16.4 Hz, 1 H), 4.77* (d, J = 16.4 Hz), 4.71 (d, J = 16.4 Hz, 1 H), 4.68* (m), 4.51 (dq, J = 16.4, 8.3 Hz, 1 H), 4.19* (dq, J = 16.4, 8.2 Hz), 3.96 (dq, J = 16.8,
41.4*, 41.2, 39.8, 38.7, 35.1, 27.5 (dd, J= 35.1, 29.0 Hz), 23.2, 22.4, 22.3, 22.2*, 11.6. * Signals arising from the minor atropisomer.
[00631] Alternatively, a premixed solution of acetic acid (1.5 g), ethanol (12 g), and water (0.3 g) were combined with Compound I-02 at 20 °C and were agitated until dissolved. The resultant solution was transferred to a reactor containing water (100 g) over about 30 minutes. The resultant slurry was further agitated for about one hour, and was filtered. The wet cake was washed with water (2 x 25 g), deliquored, and dried at about 60 °C under vacuum to provide I.
Synthesis of N-((S)-1-(3-(4-chloro-3-(methylsulfonamido)-1-(2,2,2-trifluoroethyl)-1H-indazol- 7-yl)-6-(3-methyl-3-(methylsulfonyl)but-1-yn-1-yl)pyridin-2-yl)-2-(3,5-difluorophenyl)ethyl)-2- ((3bS,4aR)-5,5-difluoro-3-(trifluoromethyl)-3b,44a,5-tetrahydro-1H-cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-1-yl)acetamide(I) from N-((S)-1-(3-(3-amino-4-chloro-1-(2,2,2-trifluoroethyl)-1H-indazol-7-yl)-6-(3-methyl-3-(methylsulfonyl)but-1-yn-1-yl)pyridin-2-yl)-2-(3,5-difluorophenyl)ethyl)-2-((3bS,4aR)-5,5-difluoro-3-(trifluoromethyl)- 3b,4,4a,5-tetrahydro-1H-cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-1-yl)acetamide (III)
[00632] A reactor was charged with III (1.0 g) followed by cyclopentyl methyl ether (2.0 mL). The contents were adjusted to about 80 °C. In a separate reactor, methanesulfonic acid anhydride (0.3g, 1.5 equiv.) was dissolved in cyclopentyl methyl ether (6 mL). The solution was added to the first reactor via a syringe pump over 5 h. Following addition, the reaction mixture was aged for 16 h. The reaction mixture was quenched with water (10 mL). UPLC analysis of the organic phase showed I with 94.8% purity.
Synthesis of N-((S)-1-(3-(4-chloro-3-(methylsulfonamido)-1-(2,2,2-trifluoroethyl)-1H-indazol- 7-yl)-6-(3-methyl-3-(methylsulfonyl)but-1-yn-1-yl)pyridin-2-yl)-2-(3,5-difluorophenyl)ethyl)-2- ((3bS,4aR)-5,5-difluoro-3-(trifluoromethyl)-3b,4,4a,5-tetrahydro-1H- cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-1-yl)acetamide (I) from N-((S)-1-(3-bromo-6-(3- methyl-3-(methylsulfonyl)but-1-yn-1-yl)pyridin-2-yl)-2-(3,5-difluorophenyl)ethyl)-2- ((3bS,4aR)-5,5-difluoro-3-(trifluoromethyl)-3b,4,4a,5-tetrahydro-1H- cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-1-yl)acetamide (IV)
[00633] To a 40 mL vial was added IV (1 .00 g), potassium bicarbonate (420 mg), palladium(II) chloride (4.9 mg, 2.0 mol%), cyclohexyl diphenylphosphine (13.4 mg, 3.6 mol%), V-03 (849 mg), 2-methyltetrahydrofuran (8.0 mL) and water (2.0 mL). The vial was inerted and the mixture was agitated at about 68 °C (65-73 °C) until the reaction was complete. The mixture was cooled to about 40 °C and the aqueous layer was removed. The organic layer was washed with aqueous acetic acid (5% w/v, 5.1 g). The organic was then concentrated to dryness and the residue was purified by column chromatography to afford I. 1H NMR (400 MHz, DMSO-d6) δ 10.12 (s, 0.2H), 10.00 (s, 1H), 9.25 (d, J = 8.2 Hz, 1H), 8.92 (d, J = 8.6 Hz, 0H),
Synthesis of N-((S)-1-(3-(4-chloro-3-(methylsulfonamido)-1-(2,2,2-trifluoroethyl)-1H-indazol- 7-yl)-6-(3-methyl-3-(methylsulfonyl)but-1-yn-1-yl)pyridin-2-yl)-2-(3.5-difluorophenyl)ethyl)-2- ((3bS,4aR)-5,5-difluoro-3-(trifluoromethyl)-3b,4,4a,5-tetrahydro-1H- cyclopropa[3,4]cvclopenta[1,2-c]pyrazol-1-yl)acetamide(I) from N-((S)-1-(3-bromo-6-(3- methyl-3-(methylsulfonyl)but-1-yn-1-yl)pyridin-2-yl)-2-(3,5-difluorophenyl)ethyl)-2- ((3bS,4aR)-5,5-difluoro-3-(trifluoromethyl)-3b,4,4a,5-tetrahydro-1H- cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-1-yl)acetamide (IV)
[00634] To a 40 mL vial was added IV (1.00 g), potassium bicarbonate (420 mg), palladium(II) chloride (4.9 mg, 2.0 mol%), cyclohexyl diphenylphosphine (13.4 mg, 3.6 mol%), V-04 (923 mg), 2-methyltetrahydrofuran (8.0 mL) and water (2.0 mL). The vial was inerted and the mixture was agitated at about 68 °C (65-73 °C) until the reaction was complete. The mixture was cooled to about 40 °C and the aqueous layer was removed. The organic was stirred with aqueous sodium hydroxide (5 % w/w, 6.3 g) at 40 °C until reaction was complete. The organic was washed with aqueous acetic acid (5% w/v, 5.1 g). The organic was then concentrated to dryness and the residue was purified by column chromatography to afford I. 1H NMR (400 MHz, DMSO-d6) δ 10.12 (s, 0.2H), 10.00 (s, 1H), 9.25 (d, J = 8.2 Hz, 1H), 8.92 (d, J = 8.6 Hz, 0H), 7.90 (d, J = 7.9 Hz, 0.1H), 7.85 – 7.71 (m, 2H), 7.52-7.50 (m, 0.1H), 7.32 (d, J = 7.7 Hz, 1H), 7.21 (q, J = 9.6 Hz, 0.4H), 7.11 – 6.97 (m, 1H), 6.94-6.89 (m, 0.2H), 6.86 (d, J =
As of 2021, it is in phase II/III clinical trials.[3] It is being investigated as a treatment for HIV patients infected with multidrug-resistant virus and as a twice-yearly injectable for pre-exposure prophylaxis (PrEP).[3][4]
Society and culture
Legal status
On 23 June 2022, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) adopted a positive opinion, recommending the granting of a marketing authorization for the medicinal product Sunlenca, intended for the treatment of adults with multidrug‑resistant human immunodeficiency virus type 1 (HIV‑1) infection.[5] The applicant for this medicinal product is Gilead Sciences Ireland UC.[5] Lenacapavir was approved for medical use in the European Union in August 2022.[1]
References
^ Jump up to:abcdef“Sunlenca EPAR”. European Medicines Agency (EMA). 22 June 2022. Archived from the original on 26 August 2022. Retrieved 25 August 2022. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
^ Jump up to:ab“Sunlenca: Pending EC decision”. European Medicines Agency. 23 June 2022. Archived from the original on 26 June 2022. Retrieved 26 June 2022. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
External links
“Lenacapavir”. Drug Information Portal. U.S. National Library of Medicine.
“Lenacapavir sodium”. Drug Information Portal. U.S. National Library of Medicine.
“Lenacapavir”. Clinical Info. National Institutes of Health.
Lenacapavir (Sunlenca). Lenacapavir was first approved in the EU in 2022 for use in combination with other antiretroviral(s) in adults with multidrug resistant HIV infection and for whom it is otherwise not possible to construct a suppressive antiviral regimen.1314Later in the year the drug also received approval in Canada and the US for similar indications. Lenacapavir received first-in-class designation as 15,16 the first approved drug known to inhibit the capsid of HIV-1, aprotein shell that encompasses the genetic material of the virus and is known to be involved in multiple stages of the HIV life cycle. Lenacapavir is available as both an oral and injectable therapy. As a slow-release, long lasting treatment for HIV-1, dosing may be required as infrequently as every 6months when taken in combination with other antiretrovirals, with the goal of improving patient adherence and medication compliance. Lenacapavir was developed by Gilead Sciences, Inc. and is currently being evaluated in a number of other clinical trials for both HIV-1 treatment and prevention. 13 The synthesis of lenacapavir is achieved by joining four advancedintermediates:17boronateester2.5,18 cyclopropane carboxylic acid 2.13a,17 chiral amino pyridine 2.19,17 and alkynyl sulfone2.20(Schemes2, 3, 4, and5, respectively).18 Thesynthesisof thesekeyintermediateshavebeendescribed inthepatent literatureemployingavarietyof routes17−22and is also exemplified on scales ranging fromsingle gram to multikilogramscale, albeit without reporting isolated yields. For thepurposeof this review, the largest reportedroute to each intermediate will be described. First, synthesis of boronateester2.5beganwitharylcyanofluorideintermediate 2.1 and reliedon a condensation/intramolecular cyclization processwithhydrazinehydrate in i-PrOH/water togenerate aminopyrazole2.2onmultikilogramscaleafter coolingand precipitation of the product from the reaction mixture (Scheme2).18Next, nucleophilicdisplacementof triflate2.3, facilitated by potassium phosphate in DMF, generated ultimately providing 2.13a as a single isomer to carry forward to lenacapavir. Chiral amine intermediate 2.19 was accessed from 3,6 dibromopicolinaldehyde (2.14), using (S)-2-ethylpropane-2 sulfinamide (2.15) as a chiral auxiliary for installation of the chiral amine (Scheme 4). 17 Condensation of aldehyde 2.14 and sulfinamide 2.15 in the presence of Cs2CO3 and NMP furnished chiral sulfinimine 2.16, which was precipitated from the reaction mixture by addition of water. The resulting solid was then treated with (3,5-difluorobenzyl)zinc bromide using a controlled addition protocol before HCl-mediated cleavage of the chiral auxiliary. Precipitation from the reaction mixture yielded the HCl salt of 2.18 as a single isomer, which was treated with Boc2O under aqueous basic conditions to yield the Boc-protected chiral amine 2.19. 17 It is important to note that while this chiral auxiliary-based method constitutes the largest scale route to 2.13a published to date, patent literature by Gilead has recently emerged, describing synthesis of similar analogs on a small scale using chiral salt-based resolution methods. The final steps in the synthesis of lenacapavir began with the Sonogashira cross coupling of advanced pyridyl bromide intermediate 2.19 and 3-methyl-3-(methylsulfonyl)but-1-yne (2.20) (Scheme 5). 17 After precipitation from the reaction mixture, the resulting product, 2.21, was subjected to Suzuki coupling with boronate ester 2.5, furnishing 2.22. Bis sulfonylation of amine 2.22 with MsCl in TEA and subsequent Boc group cleavage with TFA allowed for generation of 2.23 following a hexane wash and neutralization process. Finally, incorporation of the chiral cyclopropane fragment was made possible by HATU-mediated coupling of amine 2.23 and cyclopropane-carboxylic acid intermediate 2.13a in DMF and DIPEA to give a bis-substituted methylsulfonamide. Selective cleavage of one methanesulfonyl group was performed by addition of 2 N NaOH to the crude reaction mixture, completing the synthesis of lenacapavir (2).17
(13) Paik, J. Lenacapavir: First approval. Drugs 2022, 82, 1499− 1504. (14) Tuan, J.; Ogbuagu, O. Lenacapavir: a twice-yearly treatment for adults with multidrug-resistant HIV infection and limited treatment options. Expert Rev. Anti Infect. Ther. 2023, 21, 565−570. (15) Mullard, A. FDA approves first-in-class HIV capsid inhibitor. Nat. Rev. Drug Discovery 2023, 22, 90. (16) Dvory-Sobol, H.; Shaik, N.; Callebaut, C.; Rhee, M. S. Lenacapavir: A first-in-class HIV-1 capsid inhibitor. Curr. Opin. HIV AIDS 2022, 17, 15−21. (17) Graupe, M.; Henry, S. J.; Link, J. O.; Rowe, C. W.; Saito, R. D.; Schroeder, S. D.; Stefanidis, D.; Tse, W. C.; Zhang, J. R. Preparation of cyclopropacyclopentapyrazolylacetamide compounds useful for the prophylactic or therapeutic treatment of HIV virus infection. WO 2018035359, 2018. (18) Allan, K. M.; Batten, A. L.; Brizgys, G.; Dhar, S.; Doxsee, I. J.; Goldberg, A.; Heumann, L. V.; Huang, Z.; Kadunce, N. T.; Kazerani, S.; et al. Methods and intermediates for preparation of antiretroviral pyridine derivative useful for treatment of HIV-1 infections. WO 2019161280, 2019.
FDA APPROVED 2026, 4/20/2026, doravirine and islatravir, Idvynso
To treat HIV-1 infection (as a complete regimen) in adults to replace the current antiretroviral regimen in those who are virologically-suppressed on a stable antiretroviral regimen with no history of virologic treatment failure and no known substitutions associated with resistance to doravirine
Islatravir is known to be a nucleoside reverse transcriptase inhibitor, useful for treating HIV-1 and -2 infection and AIDS.
Islatravir (MK-8591, EFdA), useful for the treatment of eg HIV, AIDS and related diseases.
Merck & Co and Idenix , under license from Yamasa Shoyu , are developing islatravir, a nucleoside reverse transcriptase inhibitor, for the oral prevention and treatment of HIV-1 and HIV-2 infection; in July 2019, data from a phase IIb trial in patients with HIV-1 infection were presented.In August 2015, Merck licensed Codexis ‘ CodeEvolver® protein engineering platform technology to develop enzymes for use in the manufacture of the pharmaceutical products such as islatravir.
Islatravir (4′-ethynyl-2-fluoro-2′-deoxyadenosine, EFdA, or MK-8591) is an investigational drug for the treatment of HIV infection.[1]It is classified as a nucleoside reverse transcriptase translocation inhibitor (NRTTI).[2]Merck is developing a subdermal drug-eluting implant to administer islatravir.[3][4]
Biological activity
Islatravir has activity against HIV in animal models,[5] and is being studied clinically for HIV treatment and prophylaxis.[6] Islatravir is a nucleoside analog reverse transcriptase translocation inhibitor that unlike other such inhibitors, inhibits HIV through multiple mechanisms,[5] providing rapid suppression of the virus, when tested in macaques and mice.[7] Nevertheless, there are HIV strains resistant to islatravir and research is ongoing.[8]
PATENTS
WO2020014046 ,
PATENT
WO2020014047
PATENT
WO2020014050 (assigned to Codexis ), covering engineered phosphopentomutase (PPM) enzymes, useful in the synthesis of pharmaceutical compounds including islatravir.
4’-Ethynyl-2’-deoxy nucleoside analogs are known for activity against HIV, AIDS and related diseases.
One example of a 4’-ethynyl nucleoside analog is 4’-ethynyl-2-fluoro-2’-deoxyadenosine (EFdA, also known as MK-8591) which is a nucleoside reverse transcriptase translocation inhibitor that blocks HIV-l and SIV viral replication in vitro (Kawamoto, A., Kodama, E., Sarafianos S. F. et al, Int. J. Biochem. Cell Biol.; 40(l l):24lO-2O [2008]; Ohrui, H., Kohgo, S., Hayakawa, H. et al, Nucleosides, Nucleotides & Nucleic Acids, 26, 1543-1546
[2007]) and in vivo (Hattori, S., Ide, K., Nakata, H. et al. Antimicrobial. Agents and
Chemotherapy, 53, 3887-3893 [2009]). EFdA is claimed in US Patent No. 7,339,053 (referred to in the‘053 patent as 2,-deoxy-4’-C-ethynyl-2-fluoroadenosine). EFdA has the following chemical structure:
EFdA is metabolized in cells to its active triphosphate anabolite which inhibits HIV reverse transcriptase. In contrast to nucleoside reverse transcriptase inhibitors (NsRTIs) and nucleotide reverse transcriptase inhibitors (NtRTIs) currently available for the treatment of HIV infection which lack a 3′-OH group to block incorporation of incoming nucleotide, EFdA retains a 3′ OH group and acts as a chain terminator by preventing translocation of the primer template in the reverse transcriptase (RT) active site and preventing binding of incoming
deoxyribonucleotide triphosphates (dNTPs). In addition, the pucker of the modified ribose ring of EFdA is believed to contribute to inhibition of reverse transcriptase by placing the 3′-OH in a vector in which phosphotransfer from the incoming nucleotide is inefficient. (Michailidis E, et ak, Mechanism of inhibition of HIV-l reverse transcriptase by 4’-ethynyl-2-fluoro-2’-deoxyadenosine triphosphate, J Biol Chem 284:35681-35691 [2009]; Michailidis E, et ak, 4’-Ethynyl-2-fluoro-2’-deoxyadenosine (EFdA) inhibits HIV-l reverse transcriptase with multiple mechanisms, J Biol Chem 289:24533-24548 [2014] ).
In in-vitro HIV replication assays, EFdA is a potent antiretroviral and exhibits comparable antiviral activity against clinical isolates across all subtypes that have been evaluated. It is rapidly anabolized in both lymphoid derived cell lines and in peripheral blood mononuclear cells to the active triphosphate in vitro, and the intracellular half-life of EFdA Triphosphate (EFdA- TP) exceeds 72 hrs. (Stoddart, C. A., Galkina, et ak, Oral Administration of the Nucleoside EFdA (4’-Ethynyl-2-Fluoro-2’-Deoxyadenosine) Provides Rapid Suppression of HIV Viremia in Humanized Mice and Favorable Pharmacokinetic Properties in Mice and the Rhesus Macaque, Antimicrob Agents Chemother, 2015 Jul; 59(7): 4190-4198, Published online 2015 May 4).
EFdA has been shown to have efficacy in animal models of HIV infection including humanized mouse models and an SIV infected rhesus macaque model. Pharmacokinetic studies of orally administered EFdA in mouse and rhesus monkey have demonstrated rapid absorption and high plasma concentrations. A long intracellular half-life was demonstrated by the fact that isolated peripheral blood mononuclear cells from the rhesus macaque were refractory to SIV infection 24 hr after drug administration. (Ibid.)
Previous syntheses of 4’-ethynyl nucleoside analogs including EFdA suffer from modest stereoselectivity in the formation of the C-N bond between the ethynyl-deoxyribose sugar and the 2-fluoroadenine (also referred to as 2-fluoro-9H-purin-6-amine) nucleobase. The previous syntheses also require protecting groups to carry out the glycosylation reaction which reduces the efficiency of the syntheses.
The synthesis described in Kei Fukuyama, et ak, Synthesis of EFdA via a
Diastereoselective Aldol Reaction of a Protected 3-Keto Furanose, Organic Letters 2015, 17(4), pp. 828-831; DOI: 10.102 l/ol5036535) is a l4-step synthesis from D-glucose diacetonide that uses diastereoselective reactions to set the three stereocenters. The stereochemistry of the anomeric center is controlled by having a 2′-acetoxy directing group that is subsequently removed by hydrolysis and deoxygenation. This route requires 4 chromatographic purifications, and the stoichiometric use of a toxic organotin reagent for late-stage deoxygenation.
In another route (see Mark McLaughlin, et al., Enantioselective Synthesis of 4′-Ethynyl-2-fluoro-2′-deoxyadenosine (EFdA) via Enzymatic Desymmetrization, Organic Letters 2017, 19 (4), pp. 926-929), the fully-substituted 4′- carbinol is generated stereoselectively with an enzymatic desymmetrization. The 3 ‘-stereocenter is set with a catalytic asymmetric transfer hydrogenation, and the anomeric 1 ‘-linkage is established in modest stereoselectivity using substrate control, with an upgrade in stereochemical purity achieved by crystallization of an intermediate. This process requires 15 steps, requires the use of several protecting groups and generates the glycosyl linkage between the nucleobase and sugar fragments in low
stereoselectivity (1.8: 1).
A l2-step synthesis for making EFdA from R-glyceraldehyde acetonide is described in Kageyama, M., et al., Concise Synthesis of the Anti-HIV Nucleoside EFdA, Biosci. Biotechnol. Biochem, 2012 , 76, pp. 1219 -1225; and Enantioselective Total Synthesis of the Potent Anti-HIV Nucleoside EFdA, Masayuki Kageyama, et al., Organic Letters 2011 13 (19), pp. 5264-5266 [DOL 10.1021 / ol202116k] . The syntheses use the chiral starting material to set the 3′-stereocenter with moderate diastereoselectivity. After chromatographic separation of stereoisomers, the new stereocenter is used to guide a diastereoselective alkyne addition to set the fully-substituted 4′-stereocenter. The anomeric 1 ‘-position is established with little stereocontrol and requires chromatography to separate the anomers. This route requires chromatographic separation of diastereoisomers at two different stages and starts from an expensive chiral starting material.
Kohgo, S., et al., Design, Efficient Synthesis, and Anti-HIV Activity of 4′-C-Cyano- and 4′-C-Ethynyl-2′-deoxy Purine Nucleosides, Nucleosides, Nucleotides and Nucleic Acids, 2004, 23, pp. 671-690 [ DOL 10.1081/NCN-120037508] describes a synthetic route that starts from an existing nucleoside and modifies both the sugar and nucleobase portions. It is an 18-step synthesis starting from 2-amino-2′-deoxy adenosine with a low 2.5% overall yield.
It is known that enzymes such as purine nucleoside phosphorylase (PNP, EC 2.4.2.1) can form the glycosyl linkage in nucleosides and nucleoside analogs in high stereoselectivity and without the use of protecting groups. See for example the review: New Trends in Nucleoside Biotechnology, Mikhailopulo, I. A., Miroshnikov, A.I,. Acta Naturae 2010, 2, pp. 36-58.
However, the current scope of the sugar fragments capable of undergoing reaction catalyzed by PNP has been limited to the a- 1 -phosphates of natural ribose and deoxyribose along with a small number of analogs with small H, NH2, or F substituents at the C2’ and C3’ positions and replacements of the C5’ OH group. There have been no reports of successful glycosylation catalyzed by PNP using sugars with carbon substituents on the ring or any substitution at the C4’ position.
Access to the ribose and deoxyribose a- 1 -phosphate substrates for the PNP-catalyzed glycosylation has been demonstrated by translocation of the phosphate group from the 5’-hydroxyl to G -hydroxyl position with the enzyme phosphopentomutase (PPM, EC 5.4.2.7) (see Mikhailopulo, I. A., et al. supra). However, the scope of the sugars for which PPM is capable of catalyzing this reaction has been limited to ribose, arabinose, 2-deoxyribose, and 2,3-dideoxyribose. No examples have been reported of successful reaction with sugar phosphates containing any additional substituents.
Deoxyribose phosphate aldolase (DERA, EC 4.1.2.4) enzymes are known to catalyze the aldol addition of acetaldehyde to other short-chain aldehydes (see review: Stephen M. Dean, et al., Recent Advances in Aldolase-Catalyzed Asymmetric Synthesis, Adv. Synth. Catal. 2007, 349, pp. 1308 – 1320; DOI: 10. l002/adsc.200700115). However, no examples have been reported with aldehydes bearing a fully substituted carbon a to the aldehyde.
ETS Patent 7,229, 797 describes the formation of deoxyribonucleosides from the natural unsubstituted deoxyribose 1 -phosphate by use of purine nucleoside phosphorylase (PNP) and additionally using enzymes such as sucrose phosphorylase to remove the inorganic phosphate byproduct and drive the equilibrium. It does not disclose enzyme engineering for the creation of PNP enzymes that can generate nucleosides from the unnatural 4-ethynyl-D-2-deoxyribose 1-phosphate, nor that through engineering of PPM and DERA enzymes to act on unnatural substrates, 4-ethynyl-D-2-deoxyribose 1 -phosphate can be generated.
In view of the difficult and lengthy synthetic options developed to date for producing 4’-ethynyl nucleoside analogs, it would be desirable to develop an improved enzymatic synthesis for 4’-ethynyl nucleoside analogs such as EFdA that reduces the number of process steps, minimizes the use of protecting groups, improves the stereoselectivity of glycosylation and avoids the use of toxic materials.
Surprisingly, it has been found that PPM enzymes have some activity with the 3-atom ethynyl substituent at the 4’ position on ribose and that the PPM enzyme activity could be improved by introducing mutations into the enzymes to successfully develop a reaction for
isomerization of
4-ethynyl-D-2-deoxyribose 5-phosphate (6) to 4-ethynyl-D-2-deoxyribose 1 -phosphate (6.5) catalyzed by PPM to enable a more efficient method for production of 4’-ethynyl-2’-deoxy nucleosides.
Additionally, PNP enzymes have also been found to have some activity with the 3-atom ethynyl substituent at the 4 position on deoxyribose and that the PNP enzyme activity could be improved by introducing mutations into the enzymes to successfully develop a glycosylation reaction catalyzed by PNP to enable a more efficient method for production of 4’ -ethynyl -2’-deoxy nucleosides.
Even further improvement to the overall synthetic method came from the finding that
DERA enzymes, particularly the DERA from Shewanella halifaxensis, have activity for aldol reaction with 2-ethynyl-glyceraldehyde 3-phosphate which has a fully substituted a-carbon. This discovery allowed for the efficient synthesis of 4-ethynyl-D-2-deoxyribose 5-phosphate, a precursor to 4’-ethynyl-2’-deoxy nucleoside analogs, e.g., including EFdA.
SUMMARY OF THE INVENTION
The present invention involves the use of engineered enzymes in a novel enzymatic synthesis of 4’-ethynyl-2’-deoxy nucleoside analogs, including EFdA, that eliminates the use of protecting groups on intermediates, improves the stereoselectivity of glycosylation and greatly reduces the number of process steps needed to make said compounds compared to prior methods, among other process improvements. It further relates to novel intermediates which are an integral part of the enzymatic process.
The overall process is summarized in the following Scheme 1 and Scheme 2; the latter scheme provides an alternative method for making compound 5:
Scheme 1
kinase
p p y
Scheme 1A
kinase galactose oxidase
3 2X+ 9
2
p p y
It has been discovered that 4’-ethynyl-2’-deoxy nucleoside analogs such as EFdA can be synthesized employing a final step one-pot process by combining 4-ethynyl-D-2-deoxyribose 5-phosphate (6) with two enzymes, phosphopentomutase (PPM) [for example but not limited to SEQ ID NO.: 8] and purine nucleoside phosphorylase (PNP) [for example but not limited to SEQ ID NO.: 9, SEQ ID NO.: 15], as shown in Scheme 2.
Scheme 2
Scheme 2A
Several upstream intermediates used in the present process for the synthesis of the final product 4’-ethynyl-2’-deoxy nucleosides and analogs thereof are also made using enzymatic reaction methods as shown in Scheme 3; Scheme 3 A and Scheme 3B
Scheme 3
Scheme 3A
o2
pTsOH
deoxyribose
aldolase
Scheme 3B
Experimental Procedures
Preparation of 2-ethynyl-2-hvdroxypropane-l,3-diyl diacetate 12)
Method A:
To a -35 °C solution of diacetoxyacetone (1) (159 g, 914.0 mmol) in THF (1000 mL) was added 1600 mL of a 0.5 M solution of ethynyl magnesium chloride in THF maintaining the temperature below -20 °C. After the reaction reached completion, acetic acid (78 mL) in 400 mL methyl tert-butyl ether (MTBE) was added dropwise keeping the temperature below -20 °C. MTBE (800 mL) was then added and the mixture was warmed to room temp. Saturated NaCl in water (1000 mL) was added followed by saturated NH4CI solution in water (1050 mL). The organic layer was separated, dried over Na2SC>4 and evaporated to give compound (2) as an oil (160 g, 88%). 1H NMR (CDCI3, 500 MHz): d 4.26 (dd, 4 H), 2.55 (s, 1H), 2.14 (s, 6H).
Preparation of 2-ethynyl-propane-l,2,3-triol 13)
Method B:
To a solution of 2-ethynyl-2-hydroxypropane-l,3-diyl diacetate (2) (70 g, 350 mmol) in ethanol was added a 0.5M solution of sodium methoxylate in methanol (69.9 mL, 35.0 mmol) at room temperature (rt). The reaction was stirred at rt for 2 hours (h) to reach completion. The solvents were evaporated and the residue was re-dissolved in 100 mL water and extracted with 3 x 50 mL MTBE. The aqueous layer was sparged with nitrogen to remove residual solvents to give a 40.9% solution of 2-ethynyl-propane-l,2,3-triol (3) (108 g , 100% yield) as determined by nuclear magnetic resonance (NMR) (maleic acid as internal standard). lH NMR (D2O, 500 MHz): d 3.60 (dd, 4 H), 2.85 (s, 1H).
Alternate Preparations o ethynyl-glvcer aldehyde 14)
Method Cl:
In a stirred reactor, 2-ethynyl-propane-l,2,3-triol (3) (1.1 g, 9.47 mmol) in sodium phosphate buffer (30 mL, 100 mM, pH 7.0) containing antifoam 204 (Sigma A6426, 1 drop ~ 20 pL) was warmed to 30 °C with air sparging at 12.5 seem. Galactose oxidase (GOase, SEQ ID NO.: 1) (250 mg), Horseradish Peroxidase* (Type I, 5 mg) and bovine catalase** (5 mg) dissolved in sodium phosphate buffer (5 mL 100 mM, pH 7.0) were added to the reactor, followed by the addition of CuS04 aq. solution (100 mM, 150 pL). The reaction mixture was stirred at 600 rpm with air sparging for 47h to give (f?)-2-ethynyl-glyceraldehyde (4) in 47% conversion (by NMR) and 72% e.e. . (The product was not isolated). lH NMR (D2O, 500 MHz): d 4.29 (s, 1H), 3.65 (dd, 2H), 2.83 (s, 1H).
* Horse Radish Peroxidase: wild type peroxidase from horseradish Type I, commercially available from SIGMA (P8125), isolated from horseradish roots (Amoracia rusticana).
** Bovine catalase: heme-dependent catalase from bovine source, commercially available from Sigma (C1345)
Method C2:
In a stirred 100 L jacketed reactor charged with deionized water (56.2 kg), sodium dihydrogen phosphate (1.212 kg, 10 moles) was added. The pH was adjusted to 7.02 using 10 N sodium hydroxide solution (852.6 g) at 25 °C. The reactor was charged with Antifoam 204 (A6426, 10 mL), followed CuS04*5H20 (6.5 g). Galactose oxidase (451.2 g) (SEQ ID NO.: 10) was added and stirred for 15 min while sparged with air. Horseradish peroxidase* (200.2 g) and catalase** (502.6 g) were added and the reactor was rinsed with water (2.0 kg). Next 2-ethynyl-propane- 1,2, 3 -triol (3) solution in water (9.48%, 30.34 kg, 24.72 mol) was added followed by an additional portion of Antifoam 204 (A6426, 10 mL). The reaction was sparged with air and
stirred overnight to give 94.0 kg of (A)-2-ethynyl-glyceraldehyde (4) in 66% conversion (by NMR) and 84% e.e. Assay yield 60%: 1H NMR (D20, 500 MHz): d 4.29 (s, 1H), 3.65 (dd, 2H), 2.83 (s, 1H).
* Horse Radish Peroxidase: wild type peroxidase from horseradish purified, commercially available from Toyobo (PEO-301), isolated from horseradish roots (Amoracia rusticana).
** Bovine catalase: heme-dependent catalase from bovine source, commercially available from Sigma (C1345).
The above reaction was also performed using the galactose oxidase (SEQ ID NO.: 11) and the product (4) was obtained in 67% conversion (by NMR) and 88% e.e. and assay yield 59%: 1H NMR (D2O, 500 MHz): d 4.29 (s, 1H), 3.65 (dd, 2H), 2.83 (s, 1H).
Method C3:
In a 100 mL Easy Max vessel equipped with sparger and flow controller, water (82 mL) and PIPES potassium buffer (5mL, 0.5 M) were charged. The pH was adjusted to 7.5 using 5 M KOH solution at 25 °C. Antifoam 204 (200 pL) was added, followed by evolved galactose oxidase (SEQ ID NO.: 17, 450 mg enzyme powder) and copper(II) sulfate pentahydrate (100 pL, 100 mM). The reaction mixture was sparged with air at 125 standard cubic centimeters per minute (seem) for 15 min. Bovine catalase (Cl 345, Sigma-Aldrich, 150 mg, 2000-5000 U/mg, 0.75 MU) was charged, followed by horseradish peroxidase (HRP, Toyobo PEO-301, 100 mg,
130 U/mg, 1.3 kU) and the aqueous solution of 2-ethynyl-propane-l,2,3-triol (3) (25 wt%, 12 mL, 25.8 mmol). The reaction mixture was stirred at 30 °C with aeration at 125 seem and sampled using EasySampler over 20h to give 70% conversion and form compound (4) ((A)- 2-ethynyl-glyceraldehyde) in 58% assay yield and 99% e.e. lH NMR (D2O, 500 MHz): d 4.29 (s, 1H), 3.65 (dd, 2H), 2.83 (s, 1H). The crude reaction stream was carried directly into the subsequent phosphorylation step.
Method C4: Oxidation with immobilized galactose oxidase
Galactose
Oxidase
immobilized
3
Enzyme immobilization procedure:
Nuvia IMAC Ni-charged resin (16 mL based on settled volume) was added to a filter funnel and washed with binding buffer (10 column volumes, 160 mL; 500 mM sodium chloride, 50 mM sodium phosphate, 15 mM imidazole, pH 8.0) to remove the resin storage solution. In a vessel evolved galactose oxidase (SEQ ID NO.: 17, 2.00 g) lyophilized powders were resuspended in copper (II) sulphate solution (100 mM; 5.00 mL), followed by addition of binding buffer (50 mL) and the resin. The solution was mixed using rotating mixer at 20 °C for 5h. The resin was filtered and washed with binding buffer (10 column volumes, 160 mL) and potassium PIPES buffer (10 column volumes, 160 mL; 50 mM, pH 7.5) and it was used directly in a reaction. Reaction procedure:
In a 100 mL Easy Max vessel equipped with sparger and flow controller, water (82 mL) and PIPES potassium buffer (5mL, 1 M) were charged. The pH was adjusted to 7.5 using 5 M KOH solution at 25 °C. Antifoam 204 (200 pL) was added, followed by evolved galactose oxidase immobilized on the resin (SEQ ID NO.: 17, 750 mg enzyme powder per 6 mL resin) and copper(II) sulfate pentahydrate (100 pL, 100 mM). The reaction mixture was sparged with air at 125 standard cubic centimeters per minute (seem) for 15 min. Bovine catalase (C1345, Sigma-Aldrich, 210 mg, 2000-5000 U/mg, 1.05 MU) was charged, followed by horseradish peroxidase (HRP, Toyobo PEO-301, 100 mg, 130 U/mg, 1.3 kU) and the aqueous solution of 2-ethynyl-propane- 1,2, 3 -triol (3) (25 wt%, 13 mL, 29.4 mmol). The reaction mixture was stirred at 25 °C with aeration at 125 seem. After 22h the reaction reached 91% conversion to give 200 mM (//)-2-ethynyl-glyceraldehyde (4) solution (100 mL, 68% assay yield, 97% e.e. lH NMR (D2O, 500 MHz): d 4.29 (s, 1H), 3.65 (dd, 2H), 2.83 (s, 1H). The crude reaction stream was carried directly into the subsequent phosphorylation step.
Method C5: Optional Isolation of aldehyde via formation of aminal (8)
Step 1: Preparation of (S)-2-( \ .3-dibenzylimidazolidin-2-yl )but-3-yne- l 2-diol
A 100 L jacketed cylindrical vessel equipped with nitrogen bubbler, mechanical stirrer and thermocouple was charged with crude oxidase reaction stream containing (f?)-2-ethynyl-glyceraldehyde ((4), 26.0 kg, 1.85 wt% aldehyde, 3.64 mol) and inerted with N2 atmosphere. The aqueous solution was warmed to 20 °C and Af,A-di methyl dodecan- 1 -ami ne oxide (DDAO) (30 wt% in water, 798 g, 0.96 mol;) was added, followed by MTBE (55.3 kg, 76 L) and N,N -dibenzylethane-l, 2-diamine (1.55 kg, 6.43 mol). The brown, biphasic mixture was stirred overnight at 20 °C under nitrogen atmosphere. After 17 hours the stirring was stopped and the organic phase was removed and discarded. A light brown MTBE solution of fV)-2-( l ,3-dibenzylimidazolidin-2-yl)but-3-yne-l,2-diol (56.5 kg, 2.02 wt% aminal, 3.39 mmol, 93% assay yield) was obtained.
Six similar MTBE solutions were processed together in a single distillation and crystallization step (in total 374.4 kg of solution, containing 7.91 kg aminal).
A 50 L jacketed cylindrical vessel equipped with mechanical stirrer, distillation head (condenser at -20 °C) and thermocouple was charged with aminal solution (45 L). Vacuum was applied to the vessel (65-95 torr) and the jacket was set to 40 °C. Solvent was removed by distillation until a volume of 35 L had been reached. At this point, the internal temperature was 6.1 °C and an off-white solid had begun to crystallize. The remaining MTBE solution was slowly added, maintaining a constant volume of 35-40 L and an internal temperature of 0-10 °C. Once all the MTBE solution had been added the volume was decreased to 25 L. Distillation was halted, the vessel was inerted with nitrogen and the jacket temperature was decreased to 10 °C. The resulting pale yellow suspension was aged at this temperature for 2 hours and the solids were collected by filtration. The filter cake was washed with cold (-2 °C) MTBE (12.7 kg) and then dried under nitrogen flow for 7 hours. (5)-2-(l,3-dibenzylimidazolidin-2-yl)-but-3-yne-l,2-diol was obtained as an off-white crystalline solid (5.75 kg) lff NMR (500 MHz, DMSO-i¾) d 7.42 – 7.35 (m, 4H), 7.32 (td, J= 7.5, 1.6 Hz, 4H), 7.27 – 7.21 (m, 2H), 5.10 (t, J= 5.6 Hz, 1H), 5.03 (s, 1H), 4.28 (d, J= l3.3Hz, 1H), 4.16 (d, J= 13.3 Hz, 1H), 3.76 (s, 1H), 3.70 – 3.58 (m, 4H), 3.21 (d, J= 0.9 Hz, 1H), 2.90 – 2.80 (m, 2H), 2.60 – 2.51 (m, 2H).13C NMR (126 MHz, DMSO-i¾) d 140.0, 140.0, 128.5, 128.3, 128.2, 128.1, 126.8, 126.8, 88.6, 86.9, 75.0, 74.0, 66.4, 60.7, 60.5, 50.4, 50.3, 39.5. HR-MS (ESI) Aminal (M + H+) C21H25N202+ calculated 337.1911; found 337.1922.
Step 2: Prep l (8)
A 4 L jacketed cylindrical vessel equipped with nitrogen bubbler and mechanical stirrer was charged with of TsOH»H20 (12.0 g, 63.1 mmol), water (60 mL), (ri)-2-(l,3-dibenzylimidazolidin-2-yl)but-3-yne-l,2-diol (110 g, 327 mmol) and MTBE (1700 mL). The biphasic mixture was placed under nitrogen and the jacket temperature was set to 15 °C. A solution of TsOH»H20 (114 g, 599.3 mmol) in water (600 mL) was added dropwise over 1.5 hours with overhead stirring (200 rpm). After addition had completed, the jacket temperature was lowered to 5 °C and the resulting slurry was aged for 1 hour. The solids were removed by filtration and washed with cold water (270 mL). The biphasic solution was transferred to a separating funnel and the organic phase was removed and discarded. The aqueous phase was treated with DOWEX™ MARATHON™ A resin (hydroxide form, 11.0 g) and AMBERLYST® 15 resin (hydrogen form, 11.0 g) while sparging with N2 at a rate of 200 seem for 24 hours to remove residual MTBE. The resins were removed by filtration to give a colorless aqueous solution of (f?)-2-hydroxy-2-(hydroxymethyl)but-3-ynal (774 g, 4.6 wt% aldehyde, 82% yield). lH MR (500 MHz, D2O) d 5.01 (s, 1H), 3.77 (d, J= 11.7 Hz, 1H), 3.73 (d, J= 11.7 Hz, 1H), 2.92 (s, 1H). 13C NMR (126 MHz, D2O) d 129.4, 125.4, 90.3, 81.0, 76.0, 73.9, 65.3. HRMS
Alternate Preparations o ethvnyl-glvceraldehvde 3-phosphate (5):
Method Dl: Acetate kinase: ATP -regeneration system
Pantothenate kinase PanK
ATP
Acetate kinase
4 Acetate phosphate
5
In a stirred reactor, to a solution of adenosine diphosphate disodium salt (40 mg, 0.087 mmol) and magnesium chloride (38 mg, 0.400 mmol) in HEPES buffer (66 mM, pH 7.5, 30 mL) was added (i?)-2-ethynyl-glyceraldehyde (4) (1.9 mL, 210 g/L solution in water, 3.51 mmol), followed by acetate kinase (SEQ ID NO.: 3) (40 mg), and pantothenate kinase (SEQ ID NO.: 2) (120 mg). The reaction mixture was warmed to 25 °C and a solution of acetyl phosphate lithium potassium salt (1.3 g, 7.01 mmol) in HEPES buffer (50 mM, pH 7.5, 10 mL) was added dropwise over 4 hours, with pH maintained at 7.5 using 5M sodium hydroxide. The reaction was stirred for 18 hours to give (i?)-2-ethynyl-glyceraldehyde 3-phosphate (5) in 85% conversion (by HPLC) (The product was not isolated). iH NMR (D2O, 400 MHz): d 5.02 (s, 1H), 4.00 (dq, 2 H), 2.88 (s, 1H). LC-MS: (ES, m/z): calculated for C5H7O6P (M-H): 193.1; found 193.0.
Method D2: Pyruvate oxidase ATP -regeneration system
Pan
Pyruvate oxidase
Pyruvate
Phosphate
02
In a stirred reactor, a solution of sodium pyruvate (3.11 g, 28 mmol) and phosphoric acid (0.523 mL, 7.71 mmol) in 76 mL water pH 7.5 was charged with (i?)-2-ethynyl-glyceraldehyde (4) (3.8 mL, 210 g/L solution in water, 7.01 mmol), adenosine diphosphate disodium salt (80 mg, 0.174 mmol), thiamine pyrophosphate (40 mg, 0.086 mmol), flavin adenine dinucleotide disodium salt hydrate (64 mg, 0.077 mmol), and magnesium chloride (400 pL, 1 M solution in water, 0.4 mmol). The pH was re-adjusted to 7.5 with 5M aq sodium hydroxide and the reaction volume was re-adjusted to 80 mL with water. Acetate kinase (SEQ ID NO.: 3) (80 mg), pyruvate oxidase (SEQ ID NO.: 4) (80 mg, lyophilized cell free extract), pantothenate kinase (SEQ ID NO.: 2) (400 mg), and catalase (800 pL, ammonium sulfate suspension CAT-101, Biocatalytics) were added. The reaction was stirred at 500 rpm and 30 °C with air sparging for 72 hours to give (//)-2-ethynyl-glyceraldehyde 3 -phosphate 5 in 95% conversion (by HPLC) (The product was not isolated). lH NMR (D2O, 400 MHz): d 5.02 (s, 1H), 4.00 (dq, 2 H), 2.88 (s, 1H). LC-MS: (ES, m/z): calculated for C5H7O6P (M-H): 193.1; found 193.0.
The above reaction was also performed using the pantothenate kinase (SEQ ID NO.: 13) and the product 5 was obtained in 66% conversion. (The product was not isolated). iH NMR (D2O, 400 MHz): d 5.02 (s, 1H), 4.00 (dq, 2 H), 2.88 (s, 1H).
Method D3: Acetate kinase: ATP -regeneration system using immobilized enzymes
Panth
Acetate phosphate
Enzyme immobilization procedure:
NUVIA™ Immobilized Metal-ion Affinity Chromatography (IMAC) nickel-charged resin (168 mL based on settled volume) was added to a filter funnel and washed with binding buffer (1.6 L; 500 mM sodium chloride, 50 mM sodium phosphate, pH 8.0). In a vessel, pantothenate kinase
(8.4 g) (SEQ ID NO.: 12) and acetate kinase (2.8 g) (SEQ ID NO.: 3) were dissolved in binding buffer (500 mL). The washed resin was charged to the vessel and the solution was stirred for 4 hours at 20 °C. The resin was filtered and washed first with binding buffer (1.6 L) followed by piperazine-N,N’-bis(2-ethanesulfonic acid) (PIPES) buffer (840 mL; 50 mM, pH 6.5). The washed resin was used directly in the next step.
Reaction procedure:
To a 1 L reactor, a solution of (f?)-2-ethynyl-glyceraldehyde (4) in water (608.7 g, 4.6 wt%, 212 mmol) was charged and cooled to 5 °C. To the cooled solution piperazine-N,N’-bis(2-ethanesulfonic acid) (PIPES) buffer (32.7 mL, 1 M, pH 6.5, 32.7 mmol), magnesium chloride (9.33 mL, 1 M, 9.33 mmol), acetyl phosphate diammonium salt (51.8 g, 265 mmol), adenosine diphosphate disodium salt hydrate (1.17 g, 2.12 mmol), and water (192 mL) were added. The solution was allowed to stir and pH was adjusted to 6.4 using 5 N KOH. The reaction was warmed to 20 °C and 168 mL of resin with co-immobilized pantothenate kinase (SEQ ID NO.: 12) and acetate kinase (SEQ ID NO.: 3) was added. The reaction was stirred for 10 hours with 5 N KOH used to maintain a pH of 6.4 to give (f?)-2-ethynyl-glyceraldehyde 3-phosphate (5) in
92% conversion (by HPLC) and 91% yield (by 3 lp NMR with tetraphenylphosphonium chloride as internal standard) (the product was not isolated). lH NMR (D2O, 400 MHz): d 5.02 (s, 1H), 4.00 (dq, 2 H), 2.88 (s, 1H). LC-MS: (ES, m/z): calculated for C5H7O6P (M-H): 193.1; found 193.0.
Preparation of 4-ethynyl-D-2-deoxyribose 5-phosphate 16)
Method E:
To a solution of (f?)-2-ethynyl-glyceraldehyde 3-phosphate (5) (5, 20 mL, 5.3 mmol) in water, a solution of acetaldehyde in water (40 wt.%, 2.02 mL, 15.9 mmol) was added at room
temperature, followed by the addition of Deoxyribose-phosphate aldolase (DERA) (SEQ ID NO. : 6), 25 mg solution in triethanolamine hydrochloride buffer (1 mL, 1 M, pH 7.0). The reactor was sealed and the mixture was stirred overnight at 30 °C and 600 rpm to give 4-ethynyl-D-2-deoxyribose 5-phosphate (6) in 99% conv. and 99% e.e., 99% d.e. as a 1 : 1 anomer mixture (The product was not isolated) a-anomer: lH NMR (D2O, 600 MHz) 5 5.31 (t, 1H), 4.13 (t, 1H), 3.81-3.72 (m, 2H), 2.89 (s, 1H), 2.42-2.34 (m, 1H), 1.87-1.79 (m, 1H); 13c NMR (D2O, 151 MHz) 5 97.7 (s), 81.4 (d), 79.4 (s), 78.9 (s), 71.1 (s), 67.7 (d), 39.6 (s). b-anomer: 1H NMR
Alternate Preparations of (2 ?,3A,5 ?)-5-(6-amino-2-fluoro-9H-purin-9-yl)-2-ethynyl-2-(hydroxymethyl)tetrahydrofuran-3-ol monohydrate (7) [alternative name 4’-ethynyl-2-fluoro- 2’-deoxvadenosine or EFdAI
Method FI:
Ammonium ((2f?,3ri)-2-ethynyl-3,5-dihydroxytetrahydrofuran-2-yl)m ethyl hydrogen phosphate (1.00 g, 3.91 mmol) was dissolved in 10 mL of pH 7.5 buffer (100 mM triethanolamine ΉO containing 5 mM MnCl2). The solution pH was adjusted to 7.3 with 5 N NaOH. To the solution was added 2-fluoroadenine (0.599 g, 3.91 mmol) and sucrose (2.68 g, 7.82 mmol). The enzyme solution was prepared by dissolving phosphopentomutase (SEQ ID NO. : 8) (100 mg), purine nucleoside phosphorylase (SEQ ID NO.: 9) (50 mg), and sucrose phosphorylase (SEQ ID NO. :
7) (10 mg) in 10 mL of the pH 7.5 buffer. The enzyme solution was added to the reagent mixture and the resulting suspension was shaken at 40 °C. After 20 h, the suspension was cooled to 0 °C and filtered, rinsing with cold water. The solid was suction dried to give the title compound (1.12 g, 92%) as a single isomer.
The PPM and PNP enzymes used in this step were each derived from mutations starting from the enzymes from E. coli ( Escherichia coli). The sucrose phosphorylase (SP) used in this step was derived from Alloscardovia omnicolens ; SP derived from other organisms could also be used.
Method F2:
To an aqueous solution of (f?)-2-ethynyl-glyceraldehyde 3-phosphate (5) (950 mL, 157 mmol) containing piperazine-N,N’-bis(2-ethanesulfonic acid) (PIPES) buffer at a pH from about 5.5 to 6.0 was added triethanolamine (7.09 g, 47.5 mmol). The pH of the solution was adjusted from 7.1 to 7.6 using potassium hydroxide (8 mL, 8M). Manganese(II) chloride hydrate (0.592 g, 4.70 mmol) was added followed by sucrose (161 g, 470 mmol), giving a pH of 7.5 To the solution
was added the following enzymes: deoxyribose-phosphate aldolase (SEQ ID NO. : 14) (461 mg), sucrose phosphorylase (SEQ ID NO. : 7) (494 mg), phosphopentomutase (SEQ ID NO.: 8)(2.63 g), and purine nucleoside phosphorylase (SEQ ID NO. : 15) (659 mg). Once the enzymes were dissolved, 2-fluoroadenine (19.80 g, 125 mmol) was added. The reaction was heated to 35 °C and acetaldehyde was added (40 wt% in isopropyl alcohol, 29.8 mL, 235 mmol). After reacting for 2h, the mixture was seeded with EFdA crystalline product (0.96 g, 2 mol%). After reacting over 26 h at 35 °C, the slurry was cooled to 0 °C, and the solids were collected by filtration, washing with water two times (40 mL ea.). The solids were dried under a nitrogen sweep. Yield 43.2 g, 92 wt%, 96.2% corrected. ¾ NMR: (300 MHz, DMSO-d6, ppm): d 7.68 (br s, 2H), 7.32 (d, J = 2.0 Hz, 1H), 6.44 (t, J = 5.8 Hz, 1H), 5.52 (d, J = 5.6 Hz, 1H), 5.27 (t, J = 6.0 Hz, 1H), 4.44 (q, J = 6.4 Hz, 1H), 3.60 (q, J = 6.0 Hz, 1H), 3.53 (q, J = 6.4 Hz, 1H), 3.48 (s, 1H), 2.48-2.41 (m, 1H), 2.37-2.30 (m, 1H). 13C NMR (150.92 MHz, DMSO-d6, ppm) d 158.5 (d, JCF = 203.5), 157.6 (d, JCF = 21.2), 150.2 (d, JCF = 20.2), 139.7 (d, JCF = 2.4), 117.4 (d, JCF = 4.0), 85.1, 82.0, 81.4, 78.7, 70.1, 64.2, 38.1. LC-MS: (ES, m/z): calculated for C12H12FN5O3 (M+Na): 316.0822; found 316.0818.
Alternate Preparations of -2-ethvnyl-propane-l,2,3-triol 1 1-phosphate 19) :
Method Gl: Acetate kinase: ATP-regeneration system using enzymes SEQ. ID No.: 2 and SEQ. ID No.: 3
Panthotenate kinase PanK
ATP
Acetate kinase
Acetate phosphate
A 50 mL reactor was charged with a solution of 2-ethynyl-propane-l,2,3-triol (3) in water (9.29 g, 9.46 wt%, 7.57 mmol) potassium PIPES buffer (1.02 mL, 1 M, pH 6.5, 1.02 mmol), magnesium chloride (292 pL, 1 M, 0.292 mmol), acetyl phosphate diammonium salt (1.851 g, 89 wt%, 9.46 mmol), adenosine diphosphate disodium salt hydrate (ADP, 42 mg, 0.076 mmol, 0.01 eq), and water (28 mL). The pH was adjusted to 6.4 using 5 M KOH, the solution was warmed to 20 °C and evolved pantothenate kinase PanK SEQ. ID No.: 2 (264 mg) and acetate kinase AcK SEQ. ID No. : 3 (88 mg) were added. The reaction was stirred for 16 hours with pH maintained at 6.4 using 5 N KOH. The final reaction contents provided C.V)-2-ethynyl -propane- 1 ,2,3-triol 1-phosphate (9) in >95% e.e. and 99% conversion (by 31P NMR). The product was not isolated. ¾ NMR (D2O, 500 MHz) d 3.89 (m, 2H), 3.72 (d, 7= 11.6 Hz, 1 H), 3.65 (d, J= 11.6 Hz, 1H),
Method G2: Acetate kinase: ATP-regeneration system using enzyme SEQ. ID No.: 20 and enzyme SEQ. ID No.: 21
Panthotenate kinase PanK
– – ATP
Acetate kinase
Acetate phosphate
To a jacketed reactor aqueous solution 2-ethynyl-propane-l,2,3-triol (3) (11.47 kg, 8.7% wt, 8.61 mol) and water (7.5kg) was charged, followed by 1M BIS-TRIS methane buffer pH 6.5 (1L) and magnesium chloride (41.4 g). ATP (48g, 0.086 mol, 0.01 equivalent) and diammonium acetyl phosphate (2.021 kg, 89%, 10.33 mmol) were added, the solution was warmed up to 20 °C and the pH of the solution was re-adjusted to 6.8 using KOH (270.4 g). Evolved pantothenate kinase SEQ. ID No.: 20 (20.4 g) and evolved acetate kinase SEQ. ID No.: 21 (3 g) were then charged as solids. The reaction was stirred for at 20 °C for l6h during which pH dropped to 5.5.
Quantitative conversion of 2-ethynyl-propane-l,2,3-triol (3) was obtained as judged by ‘H and 31P NMR. Such prepared (ri)-2-ethynyl-propane-l,2,3-triol l-phosphate (9) solution (397 mM, 22.5 kg, 98% yield) was used in subsequent oxidation step without any further purification. ‘H NMR (D2O, 500 MHz) d 3.89 (m, 2H), 3.72 (d, 7= 11.6 Hz, 1 H), 3.65 (d, J= 11.6 Hz, 1H),
2.93 (s, 1H).
Method G3: Acetate kinase: ATP-regeneration system using enzyme SEQ. ID No.: 20 and enzyme SEQ. ID No.: 21 with deuterated compound (3) to assign absolute stereochemistry and demonstrate desymmetrizing phosphorylation.
Acetate phosphate
Z-d2, 95:5 er
Evolved pantothenate kinase SEQ. ID No. : 20 (100 pL of 10 g/L solution in water ) and evolved acetate kinase SEQ. ID No. : 21 (100 pL of 2g/L solution in water) were added to a solution containing diammonium acetyl phosphate (41 mg), 2-ethynyl-propane-l, l-72-l,2,3-triol ((A)- 3-d2, 20 mg, 170 pmol), magnesium chloride (10 pL of 1 M solution in water), ADP (10 pL of 100 g/L solution in water), and sodium phosphate buffer (10 pL of 1 M solution in water) in water (800 pL) at pH 6.5. The reaction was incubated for 24h at rt to give deuterated 2-ethynyl-propane-l,2,3-triol l-phosphate analogs (S)-9-(3,3-d2) and (S)-9-(l,l-d2) in 95:5 ratio and 99% overall yield. The ratio of phosphorylated compounds was determined by 31P NMR to be -95:5, confirming stereoselective phosphorylation of the 2-ethynyl-propane-l,2,3-triol (3) at the pro-(S) hydroxyl group (i.e. a desymmetrizing phosphorylation). 1H NMR (D2O, 500 MHz) d 3.89 (m, 2H), 3.72 (d, 7= 11.6 Hz, 1 H), 3.65 (d, J= 11.6 Hz, 1H), 2.93 (s, 1H). 13C NMR (D20, 126 MHz) d 82.9 (s), 75.1 (s), 71.0 (d, J= 6.9 Hz), 67.0 (d, J= 4.5 Hz), 64.7 (s).
Method G4: Acetate kinase: ATP-regeneration system using immobilized enzymes SEQ. ID No. : 20 and enzyme SEQ. ID No. : 21
Panthotenate kinase PanK
– – ATP
Acetate kinase
Acetate phosphate
Enzyme immobilization procedure:
Nuvia IMAC Ni-charged resin (75 mL based on settled volume) was added to a filter funnel and washed with water (9 column volumes, 3 x 225 mL) and binding buffer (1 column volume, 75mL; 500 mM sodium chloride, 50 mM sodium phosphate, 15 mM imidazole, pH 8.0). In a vessel pantothenate kinase (SEQ ID NO. : 20, 6.0 g) lyophilized powder was resuspended in binding buffer (200 mL) and the washed resin was added. The solution was mixed using rotating mixer at 25 °C for 6h. The resin was filtered and washed with binding buffer (6 column volumes, 6 x 225 mL) and BIS-TRIS buffer (8 column volumes, 600 mL; 50 mM, pH 6.2).
Reaction procedure:
An aqueous solution of 2-ethynyl-propane-l,2,3-triol (3) (574 g, 8.7% wt, 0.430 mol) and water (350 mL) was charged to a jacketed reactor, followed by 1M BIS-TRIS methane buffer pH 6.5 (50 mL) and magnesium chloride (2.033 g, 0.01 mol). ATP (2.37g, 0.0043 mol, 0.01 equivalent) and diammonium acetyl phosphate (101 g, 89%, 0.530 mmol, 1.2 eq) were added, the solution was warmed up to 20 °C and the pH of the solution was re-adjusted to 6.8 using 5 M KOH.
Resin with immobilized pantothenate kinase SEQ. ID No. : 20 (25 mL) and evolved acetate kinase SEQ. ID No. : 21 (0.15 g) were then charged as solids. The reaction was stirred for at 20 °C for l6h during which the pH dropped to 5.5. Quantitative conversion of 2-ethynyl-propane- I,2,3-triol (3) to (ri)-2-ethynyl-propane-l,2,3-triol l-phosphate (9) was obtained as judged by ¾ and 31P NMR. ¾ NMR (D20, 500 MHz) d 3.89 (m, 2H), 3.72 (d, J= 11.6 Hz, 1 H), 3.65 (d, J =
I I .6 Hz, 1H), 2.93 (s, 1H).
Alternate Preparations of (i?V2-ethvnyl-glvceraldehvde 3-phosphate 15):
Method HI: Immobilized galactose oxidases SEP ID No.: 16
Enzyme immobilization procedure:
Nuvia IMAC Ni-charged resin (10 mL based on settled volume) was added to a filter funnel and washed with binding buffer (10 column volumes, 100 mL; 500 mM sodium chloride, 50 mM sodium phosphate, 15 mM imidazole, pH 8.0) to remove the resin storage solution and give 16 g of washed resin. In a vessel evolved galactose oxidase (SEQ ID NO.: 16, 750 mg) lyophilized powders were resuspended in copper (II) sulphate solution (100 mM; 5.00 mL), followed by addition of binding buffer (20 mL) and the washed resin (3.0g). The solution was mixed using rotating mixer at 20 °C for 5h. The resin was filtered and washed with binding buffer (10 column volumes, 100 mL) and BIS-TRIS buffer (10 column volumes, 100 mL; 50 mM, pH 7.5) and it was used directly in the glycosylation reaction.
Reaction procedure:
The resin with immobilized galactose oxidase SEQ ID NO.: 16 (3.0 g) was added to a solution of S)-2-ethynyl-propane-l,2,3-triol l-phosphate (9, 5.4 mmol, 270 mM, 20 mL) in BIS-TRIS methane buffer (35 mM, pH adjusted to 7.2), followed by addition of copper (II) sulphate solution in water (30 pL, 100 mM) and horseradish peroxidase (PEO-301, 18 mg) and bovine catalase (C1345, 120 mg) resuspended in water (600 pL). The reaction was sealed with gas permeable membrane and shaken vigorously at 22 °C for 4 days to reach final conversion of 77% and give (f?)-2-ethynyl-glyceraldehyde 3 -phosphate (5) in 95% e.e. The enzyme resin was filtered off and the solution of the(f?)-2-ethynyl-glyceraldehyde 3-phosphate (5) was used
directly in the glycosylation reaction. iH NMR (D2O, 400 MHz): d 5.02 (s, 1H), 4.00 (dq, 2 H), 2.88 (s, 1H). LC-MS: (ES, m/z): calculated for C5H7O6P (M-H): 193.1; found 193.0.
Method H2: Immobilized galactose oxidases SEP ID No.: 17
Enzyme immobilization procedure:
Nuvia IMAC Ni-charged resin (10 mL based on settled volume) was added to a filter funnel and washed with binding buffer (10 column volumes, 100 mL; 500 mM sodium chloride, 50 mM sodium phosphate, 15 mM imidazole, pH 8.0) to remove the resin storage solution and give l6g of washed resin. In a vessel, evolved galactose oxidase (SEQ ID NO.: 16, 750 mg) lyophilized powders were resuspended in copper (II) sulphate solution (100 mM; 5.00 mL), followed by addition of binding buffer (20 mL) and the washed resin (3.0g). The solution was mixed using rotating mixer at 20 °C for 5h. The resin was filtered and washed with binding buffer (10 column volumes, 100 mL) and BIS-TRIS methane buffer (10 column volumes, 100 mL; 50 mM, pH 7.5) and it was used directly in the reaction.
Reaction procedure:
The resin with immobilized evolved galactose oxidase SEQ ID NO.: 17 (3.0 g) was added to a solution of (ri)-2-ethynyl-propane-l,2,3-triol l-phosphate (9, 5.4 mmol, 270 mM, 20 mL) in BIS-TRIS methane buffer (35 mM, pH adjusted to 7.2), followed by addition of copper (II) sulphate solution in water (30 pL, 100 mM) and horseradish peroxidase (PEO-301, 18 mg) and bovine catalase (C1345, 120 mg) resuspended in water (600 pL). The reaction was sealed with gas permeable membrane and shaken vigorously at 22 °C for 4 days to reach final conversion of 77% and give (i?)-2-ethynyl-glyceraldehyde 3-phosphate (5) in 95% e.e. The enzyme resin was filtered off and the solution of the (i?)-2-ethynyl-glyceraldehyde 3 -phosphate (5) was used directly in the glycosylation reaction. lH NMR (D2O, 400 MHz): d 5.02 (s, 1H), 4.00 (dq, 2 H), 2.88 (s, 1H). LC-MS: (ES, m/z): calculated for C5H7O6P (M-H): 193.1; found 193.0.
Method H3: Immobilized galactose oxidases SEQ ID No.: 18
Enzyme immobilization procedure:
Nuvia IMAC Ni-charged resin (3 mL based on settled volume) was added to a filter funnel and washed with binding buffer (10 column volumes, 30 mL; 500 mM sodium chloride, 50 mM sodium phosphate, 15 mM imidazole, pH 8.0) to remove the resin storage solution and give 2.4 g of washed resin. In a vial evolved galactose oxidase (SEQ ID NO.: 18, 75mg) lyophilized powders were resuspended in copper (II) sulphate solution (100 mM; 1.00 mL), followed by addition of binding buffer (5 mL) and the washed resin (400 mg). The solution was mixed using rotating mixer at 20 °C for 5h. The resin was filtered and washed with binding buffer (10 column volumes, 4 mL) and BIS-TRIS methane buffer (10 column volumes, 4 mL; 50 mM, pH 7.5) and it was used directly in a reaction.
Reaction procedure:
Immobilized evolved GOase SEQ ID NO.: 18 was added (400 mg) to a solution of (5)-2-ethynyl-propane-l,2,3-triol l-phosphate solution ((9), 5.4 mmol, 270 mM, 1 mL) in BIS-TRIS methane buffer (35 mM, pH adjusted to 7.2), , followed by addition of horseradish peroxidase (PEO-301, 1 mg) and catalase from Corynebacterium glutamicum (Roche, lyophilizate, #11650645103, 3 mg) resuspended in water (100 pL). The reaction was sealed with gas permeable membrane and shaken vigorously at 30 °C for 48h. Final conversion after 2 days reached 90% conversion and the (i?)-2-ethynyl-glyceraldehyde 3-phosphate (5) >99% e.e. The enzyme resin was filtered off and the solution of the (i?)-2-ethynyl-glyceraldehyde 3-phosphate (5) was used directly without further purification. lH NMR (D2O, 400 MHz): d 5.02 (s, 1H),
4.00 (dq, 2 H), 2.88 (s, 1H). LC-MS: (ES, m/z): calculated for C5H7O6P (MΉ): 193.1; found 193.0.
Method H4: Immobilized galactose oxidases SEP ID No.: 19
Enzyme immobilization procedure:
Nuvia IMAC Ni-charged resin (3 mL based on settled volume) was added to a filter funnel and washed with binding buffer (10 column volumes, 30 mL; 500 mM sodium chloride, 50 mM sodium phosphate, 15 mM imidazole, pH 8.0) to remove the resin storage solution and give 2.4 g of washed resin. In a vial evolved galactose oxidase (SEQ ID NO.: 19, 75mg) lyophilized powders were resuspended in copper (II) sulphate solution (100 mM; 1.00 mL), followed by addition of binding buffer (5 mL) and the washed resin (400 mg). The solution was mixed using rotating mixer at 20 °C for 5h. The resin was filtered and washed with binding buffer (10 column volumes, 4 mL) and BIS-TRIS methane buffer (10 column volumes, 4 mL; 50 mM, pH 7.5) and it was used directly in a reaction.
Reaction procedure:
Immobilized evolved GOase SEQ ID NO.: 18 was added (400 mg) to a solution of (5)-2-ethynyl-propane-l,2,3-triol l-phosphate solution (9, 5.4 mmol, 270 mM, 1 mL) in BIS-TRIS methane buffer (35 mM, pH adjusted to 7.2), , followed by addition of horseradish peroxidase (PEO-301, 1 mg) and catalase from Corynebacterium glutamicum (Roche, lyophilizate, #11650645103, 3 mg) resuspended in water (100 pL). The reaction was sealed with gas permeable membrane and shaken vigorously at 30 °C for 48h. Final conversion after 2 days reached 100% conversion and (i?)-2-ethynyl-glyceraldehyde 3 -phosphate (5) was obtained in >99% e.e. The enzyme resin was filtered off and the solution of the (i?)-2-ethynyl-glyceraldehyde 3-phosphate (5) was used directly without further purification. lH NMR (D2O, 400 MHz): d 5.02 (s, 1H), 4.00 (dq, 2 H), 2.88 (s, 1H). LC-MS: (ES, m/z): calculated for C5H7O6P (M-H): 193.1; found 193.0.
PATENT
CA 2502109
WO 2017053216
US 20200010834
US 20200010868
PAPER
Organic letters (2017), 19(4), 926-929.
Organic Letters (2017), 19(4), 926-929.
Journal of medicinal chemistry (2018), 61(20), 9218-9228.
Bioscience, Biotechnology, and Biochemistry (2020), 84(2), 217-227.
PAPER
Organic letters (2011), 13(19), 5264-6.
A concise enantioselective total synthesis of 4′-ethynyl-2-fluoro-2′-deoxyadenosine (EFdA), an extremely potent anti-HIV agent, has been accomplished from (R)-glyceraldehyde acetonide in 18% overall yield by a 12-step sequence involving a highly diastereoselective ethynylation of an α-alkoxy ketone intermediate.
Processes for preparing islatravir and its analogs comprising the reaction of a substituted tetrahydrofuran compound with purine nucleoside phosphorylase and a nucleobase, followed by stereochemical synthesis, glycosylation, reduction, oxidation and isolation are claimed. Also claimed are novel intermediates of islatravir and processes for their preparation and their use for the preparation of islatravir.
(2R,3S,5R)-5-(6-Amino-2-fluoropurin-9-yl)-2-ethynyl-2-(hydroxymethyl)- tetrahydrofuran-3-ol (1). To a stirred solution of 16 (66.2 mg, 0.115 mmol) in MeOH/CH2Cl2 (2:1, 1.5 mL) was added NH4F (85.1 mg, 2.30 mmol) at room temperature. After 16 h, MeOH (0.5 mL) was added, and the resulting mixture was stirred for an additional 27 h. To the mixture was added 10% NaOH in MeOH (1.5 mL) to adjust the pH of the mixture to ca. 10. After 10 min, Dowex 50W×8 (200– 400 mesh (H)) was added until the pH of the mixture reached ca. 4. To the resulting mixture was added CaCO3 (259 mg, 2.59 mmol), and the mixture was stirred for 30 min. The mixture was filtered through a pad of Celite, and the filtrate was concentrated in vacuo. The residue was purified by silica gel column chromatography (CHCl3/MeOH = 10:1) to give 29.3 mg (87%) of 1. Mp: 220.0–221.4 °C (dec.); [α] 25 D +12.4 (c 0.97, MeOH); IR: νmax 3315 (br m), 3179 (br m), 1690 (vs), 1356 (vs); 1 H NMR (600 MHz, DMSO-d6): δ 2.43 (1H, ddd, J = 13.2, 7.3, 7.3 Hz), 2.70 (1H, ddd, J = 13.2, 6.8, 5.1 Hz), 3.52 (1H, s), 3.54 (1H, dd, J = 11.7, 6.4 Hz), 3.65 (1H, dd, J = 11.7, 5.0 Hz), 4.57 (1H, m), 5.32 (1H, m), 5.60 (1H, m), 6.24 (1H, dd, J = 7.2, 5.1 Hz), 7.82 (1H, br s), 7.92 (1H, br s), 8.31 (1H, s); 13C NMR (150 MHz): δ 38.3, 64.4, 70.3, 79.2, 81.7, 82.2, 85.4, 117.6, 140.0, 150.4 (d, JCF = 20.7 Hz), 157.8 (d, JCF = 21.2 Hz), 158.8 (d, JCF = 203.4 Hz); HRMS (FAB): m/z calcd for C12H13FN5 O3, 294.1002; found, 294.1000 ([M+H]+ ).
EFdA (4′-ethynyl-2-fluoro-2′-deoxyadenosine), a nucleoside reverse transcriptase inhibitor with extremely potent anti-HIV activity, was concisely synthesized from (R)-glyceraldehyde acetonide in an 18% overall yield by a 12-step sequence involving highly diastereoselective ethynylation of an α-alkoxy ketone intermediate. The present synthesis is superior, both in overall yield and in the number of steps, to the previous one which required 18 steps from an expensive starting material and resulted in a modest overall yield of 2.5%.
PAPER
Bioscience, Biotechnology, and Biochemistry (2012), 76(6), 1219-1225.
Making small-molecule drugs usually goes something like this: set up a reaction, purify the intermediate, change a solvent, and repeat, repeat, repeat to get the final product. But there’s a lot of waste involved, which is why chemists stress the environmental benefits of an alternate approach: biocatalysis. Engineering enzymes to make reactions happen saves a lot of materials, minimizes chemical and hazardous waste, and even uses less plasticware and glassware. And not having to isolate intermediates saves time.
Some pharmaceutical companies are investigating biocatalysis at different points in their drug development pipelines, but mostly at one or two steps into the making of a small molecule. Scientists at Merck & Co. have taken this further—they are reporting an entire drug synthesis using a chain of nine enzymes, five of which had been engineered, to produce an experimental HIV drug at high yield in just a few steps (Science 2019, DOI: 10.1126/science.aay8484).
This biocatalytic cascade is turning heads. For the most part, scientists aren’t using biocatalysis to manufacture a compound so much as to develop it, says Princeton University chemist Todd Hyster. The Merck process stitches together nine enzymes to get good yields of the final product, which Hyster says is no small feat.
It literally took my breath away.
Alison Narayan, assistant professor, University of Michigan
“I was blown away,” Hyster says of the first time he saw Merck scientists talk about this work. “It’s something that was very complicated.”
Mark Huffman, a chemist who led the work at Merck with Anna Fryszkowska, says they turned to biocatalysis in order to overcome a couple of key hurdles in synthesizing some molecules. One is stereochemistry. Islatravir is a nucleoside that blocks the HIV enzyme reverse transciptase and traditionally, in medicinal chemistry, it’s been hard to get the stereochemistry of nucleosides right, Huffman says. But this is something enzymes are designed by nature to do. The other is preventing unwanted side reactions. A number of steps in the traditional chemical synthesis of islatravir put the compound’s functional groups at risk of being lopped off, so they must be protected. Huffman says enzymes are specific in the types of reactions they catalyze, so there’s little to no risk of an unwanted side reaction.
Scientists at Merck & Co. use nine enzymes, five of which are engineered, to biocatalytically make an HIV drug called islatravir without having to purify intermediates. Enzymes not engineered by Merck are not pictured.
On top of that, Huffman says, they are doing these reactions at neutral pH, in aqueous solvents, and at room temperature, which cuts down on electricity and the need for multiple bioreactors running under different conditions. Islatravir normally takes between 12 and 18 steps to make. With biocatalysis, the team has cut this down to three.
“You don’t have rigorous equipment requirements,” he says. “You’re usually running [these reactions] under much milder conditions.”
To run the cascade, the team started with 2-ethynylglycerol, and added a mixture of three enzymes to run one group of reactions. They then added more enzymes to drive a second set of reactions. Then, they remove the enzymes from the solution, which are immobilized and easy to filter out, and use four more enzymes to drive the final reactions that lead to islatravir. There are no intermediate purification steps. The overall yield is about 51% using biocatalysis, compared to yields of 7% and 15% using two more traditional syntheses.
To make their biocatalysts, the team surveyed natural enzymes, mostly from microbes, that interacted with the different intermediates in islatravir production. One of the reasons why Huffman says islatravir is an ideal small molecule to produce using biocatalysis is that most organisms have to make and break down nucleosides, so there are several natural enzymes found across multiple species. This gave the team a lot of starting material from which to alter amino acids and build the enzymes they needed to do their syntheses. By making adjustments to active sites and other areas of the enzymes, the team built five of the nine enzymes needed to make islatravir biochemically.
Huffman says that while islatravir is a good molecule to show that scientists can build large biocatalytic cascades, Merck is also looking at biocatalysis to make other small molecules and biologic drugs.
Alison Narayan, a biocatalysis chemist at the University of Michigan, calls Merck “bold” for putting the time, money, and people behind this change in production—it takes a lot of resources to try an entire synthesis via biocatalysis. And, she says, they’ve succeeded spectacularly. “It literally took my breath away,” Narayan says of her first exposure to this project in 2018. “I think it’s a huge accomplishment.”
She says that Merck’s islatravir work shows that industry is starting to appreciate what biocatalysis can do for their drug pipelines and their financial bottom lines. Alongside Merck, companies like GlaxoSmithKline and Pfizer are also exploring biocatalysis at different points in drug development and manufacturing.
“It’s an important proof of concept,” Narayan says. “This is a practical way to build molecules, and this will be the way that people will build molecules when you take into consideration efficiency, green-ness, and constructing an effective synthesis. Biocatalysis has a lot to offer.”
An investigational drug targeting the HIV virus is synthesized with nine enzymes
By Elaine O’Reilly and James Ryan
Natural biosynthesis assembles a vast array of complex natural products starting from a limited set of building blocks, under physiological conditions, and in the presence of numerous other biomolecules. Organisms rely on the extraordinary selectivity of enzymes and their ability to operate under similar reaction conditions, meaning that these catalysts are perfectly adapted to mediate cascade reactions. In these multistep processes, the product of one biocatalytic step becomes the substrate for the next transformation (Display footnote number:1-3). On page 1255 of this issue, Huffman et al. (Display footnote number:4) report the development of an impressive nine-enzyme biocatalytic cascade for the synthesis of the investigational drug islatravir for the treatment of human HIV.
This study represents a partnership between scientists from Merck and Codexis. These two companies have a history of successfully collaborating to develop biocatalysts for the synthesis of important pharmaceuticals. Almost a decade ago, they developed a chemoenzymatic route for the synthesis of the type 2 diabetes drug sitagliptin (Januvia), relying on a key enzyme-catalyzed transamination with a highly engineered (R)-selective transaminase (Display footnote number:5). The work was considered a landmark example of directed evolution and functioned to highlight the potential application of biocatalysis to revolutionize industrial chemical processes.
The cascade for synthesizing islatravir was inspired by the bacterial nucleoside salvage pathway, which recycles precious nucleosides by using three key enzymes: a purine nucleoside phosphorylase (PNP), a phosphopentomutase (PPM), and a deoxyribose-5-phosphate aldolase (DERA) (see the figure). However, to achieve the synthesis of the target molecule, Huffman et al. required the natural nucleoside degradative cascade to run in reverse. The reversible nature of enzymes is central to the design of this cascade and is one of the important features that sets biocatalysts apart from the majority of traditional chemical catalysts.
The success of the cascade developed by the team also relied on all three enzymes accepting non-natural substrates bearing a fully substituted carbon at the C-4 position of the 2-deoxyribose ring. The authors reconstructed the reverse nucleoside salvage pathway from a PNP and PPM found in Escherichia coli and a DERA from Shewanella halifaxensis. The native E. coli enzymes required engineering to improve their activity. The DERA displayed existing high activity and stereoselectivity for the formation of the desired sugar phosphate enantiomer, but it required engineering to improve its ability to operate at high substrate concentration.
One of the many advantages of performing biocatalytic cascade reactions is the effective displacement of unfavorable reaction equilibria that can be achieved through product removal. However, despite performing the PNP and PPM steps in tandem, the reaction proceeded with poor conversion, and the inorganic phosphate by-product inhibits the enzymes. An elegant solution to these issues was the inclusion of an auxiliary sucrose phosphorylase, along with its sugar substrate, which removed free phosphate and effectively displaced the reaction equilibrium toward product formation.
Having assembled enzymes for the three key steps in the cascade, Huffman et al. sought to develop a biocatalytic route for the synthesis of the DERA substrate 2-ethynylglyceraldehyde 3-phosphate. Extensive screening of a broad range of kinases resulted in the identification of pantothenate kinase (PanK) from E. coli, which displayed low levels of activity (∼1% conversion) toward the (R)-enantiomer of the target aldehyde. Despite the modest initial activity, directed evolution was successfully used to substantially improve the productivity and stability of this enzyme. Finally, after 12 rounds of evolution, the authors reversed the enantioselectivity and improved the activity, stability, and expression of a galactose oxidase variant for the desymmetrization of the starting substrate, 2-ethynylglycerol.
Opens in modal lightbox
GRAPHIC: A. KITTERMAN/SCIENCE
Viewable Image – engineering a biocatalytic cascade
Image Caption
GRAPHIC: A. KITTERMAN/SCIENCE
Advancements in protein engineering, rapid gene sequencing, and the availability of low-cost DNA synthesis have made it possible to alter the properties of enzymes and fine-tune them for biocatalytic applications (Display footnote number:6-8). The work by Huffman et al. is a milestone in cascade design, largely because of the number of biocatalysts operating in tandem and the engineering feat required to optimize five of the nine enzymes involved in the synthesis. It also highlights how biosynthetic or degradative pathways can be a source of inspiration for the design of efficient biocatalytic cascades and how sequences can be reconstituted using enzymes recruited from multiple sources—in this case, of bacterial, fungal, plant, and mammalian origin. The diverse role that biocatalysts can play is also exemplified in this work, where five engineered enzymes are directly involved in the synthesis of the target molecule, and four additional enzymes function to recycle coenzyme, remove inhibitory by-products, and maintain the correct oxidation state of the copper cofactor.
Previous approaches reported for the synthesis of islatravir relied on multistep syntheses and require protecting group manipulations and intermediate purification steps (Display footnote number:9, 10). The incorporation of a key biocatalytic step or steps has the potential to revolutionize synthetic design strategies by making possible transformations that are not accessible using solely chemical approaches (Display footnote number:11, 12). The application of enzymes in industry and the development of chemoenzymatic routes to complex molecules is now well established. However, multistep syntheses exclusively comprising biocatalytic transformations are rare (Display footnote number:13), and this contribution sets a new standard for the synthesis of complex molecules with enzymatic cascades.
School of Chemistry, University College Dublin, Belfield, Dublin 4, Ireland. Email: elaine.oreilly@ucd.ie.
REFERENCES AND NOTES
1. S. P. France, L. J. Hepworth, N. J. Turner, S. L. Flitsch, ACS Catal.7, 710 (2017).
2. S. Gandomkar, A. Żadło-Dobrowolska, W. Kroutil, ChemCatChem11, 225 (2019).
3. P. Both et al., Angew. Chem. Int. Ed.55, 1511 (2016).
4. M. A. Huffman et al., Science366, 1255 (2019).
5. C. K. Savile et al., Science329, 305 (2010).
6. F. H. Arnold, Angew. Chem. Int. Ed.57, 4143 (2018).
7. C. Zeymer, D. Hilvert, Annu. Rev. Biochem.87, 131 (2018).
8. C. A. Denard, H. Ren, H. Zhao, Curr. Opin. Chem. Biol.25, 55 (2015).
9. M. McLaughlin et al., Org. Lett.19, 926 (2017).
10. M. Kageyama, T. Nagasawa, M. Yoshida, H. Ohrui, S. Kuwahara, Org. Lett.13, 5264 (2011).
11. N. J. Turner, E. O’Reilly, Nat. Chem. Biol.9, 285 (2013).
12. M. Hönig, P. Sondermann, N. J. Turner, E. M. Carreira, Angew. Chem. Int. Ed. 56, 8942 (2017).
13. S. Wu et al., Nat. Commun.7, 11917 (2016).
ACKNOWLEDGMENTS
J.R. acknowledges the School of Chemistry, University College Dublin, for support.
References
^Kawamoto, A; Kodama, E; Sarafianos, SG; Sakagami, Y; Kohgo, S; Kitano, K; Ashida, N; Iwai, Y; Hayakawa, H; Nakata, H; Mitsuya, H; Arnold, E; Matsuoka, M (2008). “2′-deoxy-4′-C-ethynyl-2-halo-adenosines active against drug-resistant human immunodeficiency virus type 1 variants”. The International Journal of Biochemistry & Cell Biology. 40 (11): 2410–20. doi:10.1016/j.biocel.2008.04.007. PMID18487070.
Class Antiretrovirals; Nitriles; Pyridones; Small molecules; Triazoles
Mechanism of Action Non-nucleoside reverse transcriptase inhibitors
Phase III HIV-1 infections
Most Recent Events
16 Jul 2016 No recent reports of development identified for phase-I development in HIV-1-infections(Monotherapy, Treatment-naive) in Germany (PO, Tablet)
01 Jun 2016 Merck Sharp & Dohme completes a phase I pharmacokinetics trial in subjects requiring methadone maintenance therapy in USA (PO, Tablet) (NCT02715700)
01 May 2016 Merck completes a phase I trial in severe renal impairment in USA (NCT02641067)
Doravirine (MK-1439) is a non-nucleoside reverse transcriptase inhibitor under development by Merck & Co. for use in the treatment of HIV/AIDS. Doravirine demonstrated robust antiviral activity and good tolerability in a small clinical study of 7-day monotherapy reported at the 20th Conference on Retroviruses and Opportunistic Infections in March 2013. Doravirine appeared safe and generally well-tolerated with most adverse events being mild-to-moderate.[2][3]
Highly active antiretroviral therapy (HAART) is the standard of care for the treatment of HIV infection.Typically, this protocol recommends the combination of two nucleoside reverse-transcriptase inhibitors (NRTIs) with either a non-nucleoside reverse-transcriptase inhibitor (NNRTI), a ritonavir-boosted protease inhibitor or an integrase inhibitor.
NNRTI-based combinations have become first-line therapy mainly because of their demonstrated efficacies, convenient dosing regimen and relatively low toxicities. These inhibitors block the polymerase activity of the HIV reverse transcriptase by binding to an allosteric hydrophobic pocket adjacent to the active site. Efavirenz (1, ) is a first generation NNRTI that has been conveniently co-formulated with NRTIs tenofovir disoproxil fumarate (TDF) and emtricitabine (FTC) as a once-a-day fixed-dose combination (Atripla®). Although recommended for the therapy of treatment-naïve patients, efavirenz suffers from neurocognitive side effects,teratogenicity and exacerbation of hyperlipidemia. Moreover, the low barrier to genetic resistance of first generation NNRTIs led to the emergence of resistant viruses bearing mutations K103N and Y181C in patients failing therapy.
Figure .
Structures of marketed and lead NNRTIs.
Second generation NNRTIs etravirine (2) and rilpivirine (3) efficiently suppress the replication of the K103N resistant mutants as shown by an improved activity in cell culture assays . Etravirine (200 mg, bid) is approved for use in treatment-experienced adult patients with multi-drug resistance. With an improved pharmacokinetic profile, the close analog rilpivirine (25 mg, qd) was recently approved for use in treatment-naïve patients. Phase III data reveal that at the 96-week point, a rilpivirine/truvada® combination was better tolerated than efavirenz/truvada®.However, the virologic failure rate was twice as high for rilpivirine (14%) than it was for efavirenz (8%). For patients with viral load greater than 500,000 copies/mL, the response rate is 62% (rilpivirine) versus 81% (efavirenz). As a result, rilpivirine is not recommended for treating HIV patients with viral load >500,000 copies/mL. This difference in treatment durability could be explained by the much higher ratio of trough concentration over the antiviral activity for efavirenz versus rilpivirine.
Investigational next-generation, non-nucleoside reverse transcriptase inhibitor (NNRTI), at the 21st Conference on Retroviruses and Opportunistic Infections (CROI). Interim data demonstrating potent antiretroviral (ARV) activity for four doses (25, 50, 100 and 200 mg) of once-daily, oral doravirine in combination with tenofovir/emtricitabine in treatment-naïve, HIV-1 infected adults after 24 weeks of treatment were presented during a late-breaker oral session. Based on these findings as well as other data from the doravirine clinical program, Merck plans to initiate a Phase 3 clinical trial program for doravirine in combination with ARV therapy in the second half of 2014.
“Building on our long-standing commitment to the HIV community, Merck continues to evaluate new candidates we believe have the potential to make a meaningful difference in the lives of HIV patients,” said Daria Hazuda, Ph.D., vice president, Infectious Diseases, Merck Research Laboratories. “We look forward to advancing doravirine into Phase 3 clinical trials in the second half of 2014.”
Doravirine Clinical Data
This randomized, double-blind clinical trial examined the safety, tolerability and efficacy of once-daily doravirine (25, 50, 100 and 200 mg) in combination with once-daily tenofovir/emtricitabine versus efavirenz (600 mg), in treatment-naïve, HIV-1 infected patients. The primary efficacy analysis was percentage of patients achieving virologic response (< 40 copies/mL).
At 24 weeks, doravirine doses of 25, 50, 100, and 200 mg showed virologic response rates consistent with those observed for efavirenz at a dose of 600 mg. All treatment groups showed increased CD4 cell counts.
Proportion of Patients with Virologic
Response at 24 weeks (95% CI)
Mean CD4 Change
from Baseline (95% CI)
Treatment*
Dose (mg)
n/N
% <40
copies/mL
cells/μL
Doravirine
25
32/40
80.0 (64.6, 90.9)
158 (119, 197)
50
32/42
76.2 (60.5, 87.9)
116 (77, 155)
100
30/42
71.4 (55.4, 84.3)
134 (100, 167)
200
32/41
78.0 (62.4, 89.4)
141 (96, 186)
Efavirenz
600
27/42
64.3 (48.0, 78.4)
121 (73, 169)
Missing data approach:
Non-completer = Failure
Observed Failure
*In combination with tenofovir/emtricitabine
The incidence of drug-related adverse events was comparable among the doravirine-treated groups. The overall incidence of drug-related adverse events was lower in the doravirine-treated groups (n=166) than the efavirenz-treated group (n=42), 35 percent and 57 percent, respectively. The most common central nervous system (CNS) adverse events at week 8, the primary time point for evaluation of CNS adverse experiences, were dizziness [3.0% doravirine (overall) and 23.8% efavirenz], nightmare [1.2% doravirine (overall) and 9.5% efavirenz], abnormal dreams [9.0% doravirine (overall) and 7.1% efavirenz], and insomnia [5.4% doravirine (overall) and 7.1% efavirenz].
Based on the 24-week data from this dose-finding study, a single dose of 100 mg doravirine was chosen to be studied for the remainder of this study, up to 96 weeks.
About Doravirine
DORAVIRINE
Doravirine, also known as MK-1439, is an investigational next-generation, NNRTI being evaluated by Merck for the treatment of HIV-1 infection. In preclinical studies, doravirine demonstrated potent antiviral activity against HIV-1 with a characteristic profile of resistance mutations selected in vitro compared with currently available NNRTIs. In early clinical studies, doravirine demonstrated a pharmacokinetic profile supportive of once-daily dosing and did not show a significant food effect.
Merck’s Commitment to HIV
For more than 25 years, Merck has been at the forefront of the response to the HIV epidemic, and has helped to make a difference through our proud legacy of commitment to innovation, collaborating with the community, and expanding global access to medicines. Merck is dedicated to applying our scientific expertise, resources and global reach to deliver healthcare solutions that support people living with HIV worldwide.
About Merck
Today’s Merck is a global healthcare leader working to help the world be well. Merck is known as MSD outside the United States and Canada. Through our prescription medicines, vaccines, biologic therapies, and consumer care and animal health products, we work with customers and operate in more than 140 countries to deliver innovative health solutions. We also demonstrate our commitment to increasing access to healthcare through far-reaching policies, programs and partnerships. For more information, visit www.merck.com and connect with us on Twitter, Facebook and YouTube.
The compound 3 -chloro-5-( { 1 – [(4-methyl-5 -oxo-4,5 -dihydro- 1 H- 1 ,2,4-triazol-3 – yl)methyl]-2-oxo-4-(trifluoromethyl)-l,2-dihydropyridin-3-yl}oxy)benzonitrile has the following chemical structure.
Anhydrous 3 -chloro-5-( { 1 – [(4-methyl-5 -oxo-4,5 -dihydro- 1 H- 1 ,2,4-triazol-3 -yl)methyl] -2-oxo-4- (trifluoromethyl)-l,2-dihydropyridin-3-yl}oxy)benzonitrile is known to exist in three crystalline forms – Form I, Form II and Form III. The differential scanning calorimetry (DSC) curve for crystalline anhydrous Form II shows an endotherm with an onset at 230.8° C, a peak maximum at 245.2°C, and an enthalpy change of 3.7 J/g, which is due to polymorphic conversion of anhydrous Form II to anhydrous Form I, and a second melting endotherm with an onset at 283.1°C, a peak maximum at 284.8°C, and an enthalpy change of 135.9 J/g, due to melting of Anhydrous Form I. Alternative production and the ability of this compound to inhibit HIV reverse transcriptase is illustrated in WO 201 1/120133 Al, published on October 6, 201 1, and US 201 1/0245296 Al, published on October 6, 201 1, both of which are hereby incorporated by reference in their entirety.
The process of the present invention offers greater efficiency, reduced waste, and lower cost of goods relative to the methods for making the subject compounds existing at the time of the invention. Particularly, the late stage cyanation and methylation steps are not required.
The following examples illustrate the invention. Unless specifically indicated otherwise, all reactants were either commercially available or can be made following procedures known in the art. The following abbreviations are used:
EXAMPLE 1
Step 1
1 2
3-(Chloromethyl)-l-(2-methoxypropan-2-yl)-4-methyl-lH-l,2,4-triazol-5(4H)-one (2): A
100 ml round bottom flask equipped with stir bar and a nitrogen inlet was charged with 1 (5 g, 33.9 mmol) and (lS)-(+)-10-camphorsulfonic acid (0.39 g, 1.694 mmol) at ambient temperature. After 2,2-dimethoxy propane (36.0 g, 339 mmol) was charged at ambient temperature, the resulting mixture was heated to 45°C. The resulting mixture was stirred under nitrogen at 45°C for 18 hours and monitored by HPLC for conversion of the starting material (< 5% by HPLC). After the reaction was completed, the batch was taken on to the next step without further workup or isolation. ‘H NMR (CDCI3, 500 MHz): 4.45 (s, 2H), 3.35 (s, 3H), 3.21 (s, 3H), 1.83 (s, 6H).
Step 2
3-Fluoro-l-((l-(2-methoxypropan-2-yl)-4-methyl-5-oxo-4,5-dihydro-lH-l,2,4-triazol-3- yl)methyl)-4-(trifluoromethyl)pyridin-2(lH)-one (3): A mixture of 2 (100 mg, 93.1% purity, 0.49 mmol), pyridone (1 17 mg, 97.6% purity, 0.49 mmol) and K2CO3 (82 mg, 0.59 mmol) in DMF (0.5 ml) was aged with stirring at ambient temperature for 3h. After the reaction was completed, the batch was taken on to the next step without further work up or isolation.
Step 3
3-Chloro-5-((l-((l-(2-methoxypropan-2-yl)-4-methyl-5-oxo-4,5-dihydro-lH-l,2,4-triazol-3- yl)methyl)-2-oxo-4-(trifluoromethyl)-l,2-dihydropyridin-3-yl)oxy)benzonitrile (4): To a mixture of compound 3 in DMF (reaction mixture from the previous step) was added 3-chloro-5- hydroxybenzonitrile (1.77 g, 1 1.5 mmol) at ambient temperature. The resulting mixture was then heated to 95-100°C and held for 20 hours.
Upon completion (typically 18-20 hours), the reaction was cooled to room temperature, diluted with ethyl acetate and washed with water. The aqueous cut was back extracted with ethyl acetate. The organic layers were combined and then concentrated to an oil. MeOH (80 ml) was added and the resulting slurry was taken on to the next step. XH NMR (CDC13, 500 MHz): 7.60 (d, IH), 7.42 (s, IH), 7.23 (s, IH), 7.12 (s, IH), 6.56 (d, IH), 5.14 (s, 2H), 3.30 (s, 3H), 3.22 (s, 3H), 1.82 (s, 6H).
Step 4
4 5
3-Chloro-5-((l-((4-methyl-5-oxo-4,5-dihydro-lH-l,2,4-triazol-3-yl)methyl)-2-oxo-4- (trifluoromethyl)-l,2-dihydropyridin-3-yl)oxy)benzonitrile (5): To a solution of 4 (5.74 g., 1 1.53 mmol) in MeOH (from previous step) was added concentrated hydrochloric acid (lml, 12.18 mmol) at ambient temperature. The resulting mixture was agitated for 1 hour at room temperature.
The resulting solids were collected by filtration and dried under a nitrogen sweep, providing 5 as a white solid (2.63 g, 46% yield):XH NMR (DMSO, 400 MHz): 1 1.74 (S, IH), 7.92 (d, IH), 7.76 (s, IH), 7.61 (s, IH), 7.54 (s, IH), 6.69 (d, IH), 5.15 (s, 2H), 3.10 (s, 3H)
EXAMPLE 2
Step 1
Phenyl methylcarbamate: 40% Aqueous methylamine (500 g, 6.44 mol) was charged to a 2 L vessel equipped with heat/cool jacket, overhead stirrer, temperature probe and nitrogen inlet. The solution was cooled to -5 °C. Phenyl chloroformate (500.0 g, 3.16 mol) was added over 2.5 h maintaining the reaction temperature between -5 and 0 °C. On complete addition the white slurry was stirred for lh at ~0 °C.
The slurry was filtered, washed with water (500 mL) and dried under 2 sweep overnight to afford 465g (96%> yield) of the desired product as a white crystalline solid; 1H NMR (CDCI3, 500 MHz): δ 7.35 (t, J = 8.0 Hz, 2H), 7.19 (t, J = 8.0 Hz, 1H), 7.12 (d, J = 8.0 Hz, 2H), 4.95 (br s, 1H), 2.90 (d, J = 5 Hz, 3H).
Step 2
2-(2-Hydroxyacetyl)-N-methylhydrazinecarboxamide: Part A: Phenyl methylcarbamate (300 g, 1.95 mol) was charged to a 2 L vessel with cooling jacket, overhead stirrer, temperature probe, reflux condenser and nitrogen inlet. IPA (390 mL) was added at 23 °C. Hydrazine hydrate (119 g, 2.33 mol) was added and the slurry heated to 75 °C for 6 h.
Part B: On complete reaction (>99% conversion by HPLC), IPA (810 mL) and glycolic acid (222 g, 2.92 mol) were added and the mixture stirred at 83-85 °C for 10-12 h. The reaction mixture is initially a clear colorless solution. The mixture is seeded with product (0.5 g) after 4h at 83-85 °C. The slurry was slowly cooled to 20 °C over 2h and aged for lh.
The slurry was filtered and washed with IPA (600 mL). The cake was dried under 2 sweep to afford 241.8g (81% yield) of the desired product as a white crystalline solid: XH NMR (D20, 500 MHz): δ 4.11 (s, 2H), 2.60 (s, 3H).
Step 3
3-(Hydroxymethyl)-4-methyl-lH-l,2,4-triazol-5(4H)-one: 2-(2-Hydroxyacetyl)-N- methylhydrazinecarboxamide (130 g @ ~95wt%, 0.84 mol), w-propanol (130 mL) and water (130 mL) were charged to a 1 L vessel with jacket, overhead stirrer, temperature probe, reflux condenser and nitrogen inlet. Sodium hydroxide (pellets, 16.8 g, 0.42 mol) was added and the slurry warmed to reflux for 3h. The reaction mixture was cooled to 20 °C and the pH adjusted to 6.5 (+/- 0.5) using cone hydrochloric acid (28.3 mL, 0.34 mol). Water was azeotropically removed under vacuum at 40-50 °C by reducing the volume to -400 mL and maintaining that volume by the slow addition of n-propanol (780 mL). The final water content should be <3000 ug/mL. The resultant slurry (~ 400 mL) was cooled to 23 °C and heptane (390 ml) was added. The slurry was aged lh at 23 °C, cooled to 0 °C and aged 2h. The slurry was filtered, the cake washed with 1 :2 n-PrOH/heptane (100 mL) and dried to provide 125g (85% yield) of an off- white crystalline solid. The solid is ~73 wt% due to residual inorganics (NaCl): ‘H NMR (CD3OD, 500 MHz): δ 3.30 (s, 3H), 4.46 (s, 2H).
Step 4
3-(Chloromethyl)-4-methyl-lH-l,2,4-triazol-5(4H)-one (1): A mixture of 3- (Hydroxymethyl)-4-methyl-lH-l,2,4-triazol-5(4H)-one (54 g, at 73wt%, 307 mmol) in ethyl acetate (540 mL) was stirred at 45 °C. SOCI2 (26.9 mL, 369 mmol) was added over 30-45 min and aged at 50 °C for 2h. Monitor reaction progress by HPLC. On complete reaction (>99.5% by area at 210nm.), the warm suspension was filtered and the filter cake (mainly NaCl) was washed with ethyl acetate (108 mL). The combined filtrate and wash were concentrated at 50-60 °C under reduced pressure to approximately 150 mL. The resulting slurry was cooled to -10 °C and aged lh. The slurry was filtered and the filter cake washed with ethyl acetate (50 mL). The cake was dried under 2 sweep to afford 40. lg (86% yield) of the desired product as a bright yellow solid: ‘H NMR (CD3OD, 500 MHz): δ 3.30 (s, 3H), 4.58 (s, 2H).
EXAMPLE 3
3-fluoro-4-(trifluoromethyl)pyridin-2(lH)-one (2): To a 250 ml round bottom flask equipped with overhead stirring and a nitrogen inlet was added a mixture of sulfuric acid (24.31 ml, 437 mmol) and water (20.00 ml). To this was added 2,3-difluoro-4-(trifluoromethyl)pyridine (6.83 ml, 54.6 mmol) and the mixture was heated to 65 °C and stirred for 4 h. By this time the reaction was complete, and the mixture was cooled to room temperature. To the flask was slowly added 5M sodium hydroxide (43.7 ml, 218 mmol), maintaining room temperature with an ice bath. The title compound precipitates as a white solid during addition. Stirring was maintained for an additional lh after addition. At this time, the mixture was filtered, the filter cake washed with 20 mL water, and the resulting white solids dried under nitrogen. 3-fluoro-4- (trifluoromethyl)pyridin-2(lH)-one (2) was obtained as a white crystalline solid (9.4g, 51.9 mmol, 95 % yield): ¾ NMR (CDC13, 400 MHz): 12.97 (br s, 1H), 7.36 (d, 1H), 6.44 (m, 1H).
Ethyl 2-(3-chloro-5-cyanophenoxy)acetate (A): A 1L round bottom flask equipped with overhead stirring was charged with 3-chloro-5-hydroxybenzonitrile (50.0 g, 98 wt% purity, 319 mmol) and 15% aqueous DMF (200 mL DMF + 35.5 mL FLO). To the resulting solution was added diisopropylethylamine (61.3 mL, 99.0% purity, 1.1 equiv) and ethyl 2-bromoacetate (35.7 g, 98% purity, 1.15 equiv) at ambient temperature. The resulting solution was warmed to 50°C under nitrogen and aged for 12 h. Upon completion of the reaction the batch was cooled to 0- 5°C. To the clear to slightly cloudy solution was added 5% seed (3.8g, 16.0 mmol). H20 (64.5mL) was added to the thin suspension via syringe pump over 3h while maintaining the temp at 0-5 °C. Additional FLO (200mL) was added over lh while maintaining the temp at 0-5 °C. The final DMF/FLO ratio is 1 : 1.5 (10 vol). The resulting slurry was typically aged lh at 0-5 °C. The batch was filtered and the cake slurry washed with 2: 1 DMF/water (150 mL, 3 vol), followed by water (200 mL, 4 vol). The wet cake was dried on the frit with suction under a nitrogen stream at 20-25 °C; note: heat must not be applied during drying as product mp is 42 °C. The cake is considered dry when H20 is <0.2%. Obtained 73.4 g ethyl ester as a light tan solid, 96% yield (corrected), 99.5 LCAP: XH NMR (CDC13, 400 MHz) δ = 7.29 (s, 1H), 7.15 (s, 1H), 7.06 (s, 1H), 4.67 (s, 2H), 4.32 (q, 2H), 1.35 (t, 3H) ppm. Step 2 – Pyridone Synthesis
(2E/Z,4E)-Ethyl 2-(3-chloro-5-cyanophenoxy)-5-ethoxy-3-(trifluoromethyl)penta-2,4- dienoate (C): Ester A (25.01 g, 104.4 mmol, 1.00 equiv) was charged to toluene (113.43 g, 131 mL, 5.24 vol) and 4-ethoxy-l, l, l-trifluoro-3-buten-2-one (26.43 g, 157.2 mmol, 1.51 equiv) was added.
The flow reactor consisted of two feed solution inlets and an outlet to a receiving vessel. The flow reactor schematic is shown in Figure 1.
The ester solution was pumped to one flow reactor inlet. Potassium tert-pentoxide solution was pumped to the second reactor inlet. Trifluoroacetic anhydride was added continuously to the receiver vessel. Triethylamine was added continuously to the receiver vessel. The flow rates were: 13 mL/min ester solution, 7.8 mL/min potassium tert-pentoxide solution, 3.3 mL/min trifluoroacetic anhydride and 4.35 mL/min triethylamine.
Charged toluene (50 mL, 2 vol) and potassium trifluoroacetate (0.64 g, 4.21 mmol, 0.04 equiv) to the receiver vessel. The flow reactor was submerged in a -10 °C bath and the pumps were turned on. The batch temperature in the receiver vessel was maintained at 5 to 10 °C throughout the run using a dry ice/acetone bath. After 13.5 min the ester solution was consumed, the reactor was flushed with toluene (10 mL) and the pumps were turned off.
The resulting yellow slurry was warmed to room temperature and aged for 4.5 h. Charged methanol (160 mL) to afford a homogeneous solution which contained 81.20 area percent diene C by HPLC analysis.
The solution of diene C (573 mL) was used without purification in the subsequent reaction. Cyclization, Diene C to E
3-Chloro-5-((2-oxo-4-(trifluoromethyl)-l,2-dihydropyridin-3-yl)oxy)benzonitrile (E): To a solution of diene C in PhMe/MeOH (573 mL; 40.69 g, 104.4 mmol theoretical C) was charged methanol (25 mL, 0.61 vol). Ammonia (32 g, 1.88 mol, 18 equiv based on theoretical C) was added and the solution was warmed to 60 °C. The reaction was aged at 60 °C for 18 h. The temperature was adjusted to 35-45 °C and the pressure was decreased maintain a productive distillation rate. The batch volume was reduced to -300 mL and methanol (325 mL, 8 vol) was charged in portions to maintain a batch volume between 250 and 350 mL. The heating was stopped and the system vented. The resulting slurry was cooled to room temperature and aged overnight.
The batch was filtered and the cake washed with methanol (3x, 45 mL). The wet cake was dried on the frit with suction under a nitrogen stream to afford 18.54 g of a white solid: XH NMR (DMSO-i/6, 500 MHz): δ 12.7 (br s, 1H), 7.73 (t, 1H, J= 1.5 Hz), 7.61-7.59 (m, 2H), 7.53 (t, 1H, J= 2.0 Hz), 6.48 (d, 1H, J= 7.0 Hz) ppm.
Step 3 – Chlorination, Alkylation and Isolation of 3-Chloro-5-({l-[(4-methyl-5-oxo-4,5-dihydro- lH-l,2,4-triazol-3-yl)methyl]-2-oxo-4-(trifluoromethyl)-l,2-dihydropyridin-3-yl}oxy)benzonitrile
3-(Chloromethyl)-4-methyl-lH-l,2,4-triazol-5(4H)-one: 3-(Hydroxymethyl)-4-methyl-lH- l,2,4-triazol-5(4H)-one (1.638 kg of 68wt%, 8.625 mol) and N-methylpyrrolidinone (8.9 L) was charged into a 30 L vessel. The suspension was aged for lOh at ambient temperature. The slurry was filtered through a 4L sintered glass funnel under 2 and the filter cake (mainly NaCl) was washed with NMP (2.23 L). The combined filtrate and wash had a water content of 5750 μg/mL. The solution was charged to a 75L flask equipped with a 2N NaOH scrubber to capture off-gasing vapors. Thionyl chloride (0.795 L, 10.89 mol) was added over lh and the temperature rose to 35 °C. HPLC analysis indicated that the reaction required an additional thionyl chloride charge (0.064 L, 0.878 mol) to bring to full conversion. The solution was warmed to 50 °C, placed under vacuum at 60 Torr (vented to a 2N NaOH scrubber), and gently sparged with subsurface N2 (4 L/min). The degassing continued for lOh until the sulfur dioxide content in the solution was <5 mg/mL as determined by quantitative GC/MS. The tan solution of 3-(chloromethyl)-4-methyl-lH-l,2,4-triazol-5(4H)-one in NMP weighed 13.0 kg and was assayed at 9.63 wt% providing 1.256 kg (97% yield).
3-chloro-5-((l-((4-methyl-5-oxo-4,5-dihydro-lH-l,2,4-triazol-3-yl)methyl)-2-oxo-4- (trifluoromethyl)-l,2-dihydropyridin-3-yl)oxy)benzonitrile: To a 75L flask was charged a 9.63wt% solution of 3-(chloromethyl)-4-methyl-lH-l,2,4-triazol-5(4H)-one in NMP (1 1.6 kg, 7.55 mol), 3 -chloro-5 -((2-oxo-4-(trifluoromethyl)- 1 ,2-dihydropyridin-3 -yl)oxy)benzonitrile (2.00 kg, 6.29 mol), NMP (3.8 L) and 2-methyl-2-butanol (6.0 L). To the resulting suspension was slowly added N,N-diisopropylethylamine (4.38 L, 25.2 mol) over 4h. The reaction was aged 18h at ambient temperature. The reaction is considered complete when HPLC indicates <1% 3 -chloro-5 -((2-oxo-4-(trifluoromethyl)-l,2-dihydropyridin-3-yl)oxy)benzonitrile remaining. The tan solution was quenched with acetic acid (1.26 L, 22.0 mol) and aged at ambient temperature overnight. The tan solution was warmed to 70 °C. Water (2.52 L) was added and the batch was seed with anhydrate Form II (134 g). The thin suspension was aged lh at 70 °C. Additional water (14.3 L) was added evenly over 7 h. The slurry was aged 2h at 70 °C and then slowly cooled to 20 °C over 5 h. The slurry was filtered and washed with 2 : 1 NMP/water (6 L), followed by water washes (6 L x 2). The filter cake was dried over a 2 sweep to give 2.53 kg (85% yield – corrected) of a white solid that was confirmed to be crystalline Form II by X-ray powder detraction analysis.
Phenyl methylcarbamate: 40% Aqueous methylamine (500 g, 6.44 mol) was charged to a 2 L vessel equipped with heat/cool jacket, overhead stirrer, temperature probe and nitrogen inlet. The solution was cooled to -5 °C. Phenyl chloroformate (500.0 g, 3.16 mol) was added over 2.5 h maintaining the reaction temperature between -5 and 0 °C. On complete addition the white slurry was stirred for lh at ~0 °C.
The slurry was filtered, washed with water (500 mL) and dried under a nitrogen sweep overnight to afford 465g (96% yield) of the desired product as a white crystalline solid; XH NMR (CDCI3, 500 MHz): δ 7.35 (t, J = 8.0 Hz, 2H), 7.19 (t, J = 8.0 Hz, 1H), 7.12 (d, J = 8.0 Hz, 2H), 4.95 (br s, 1H), 2.90 (d, J = 5 Hz, 3H).
Step 2
2-(2-Hydroxyacetyl)-N-methylhydrazinecarboxamide: Part A: Phenyl methylcarbamate (300 g, 1.95 mol) was charged to a 2 L vessel with cooling jacket, overhead stirrer, temperature probe, reflux condenser and nitrogen inlet. IPA (390 mL) was added at 23 °C. Hydrazine hydrate (119 g, 2.33 mol) was added and the slurry heated to 75 °C for 6 h.
Part B: On complete reaction (>99% conversion by HPLC), IPA (810 mL) and glycolic acid (222 g, 2.92 mol) were added and the mixture stirred at 83-85 °C for 10-12 h. The reaction mixture was initially a clear colorless solution. The mixture was seeded with product (0.5 g) after 4h at 83-85 °C. The slurry was slowly cooled to 20 °C over 2h and aged for lh. Seed was used to advance the crystallization, but the crystalline product can be precipitated and isolated without seed by allowing the solution to age at 83-85 °C for 4 hours.
The slurry was filtered and washed with IPA (600 mL). The cake was dried under a nitrogen sweep to afford 241.8g (81% yield) of the desired product as a white crystalline solid: XH NMR (D20, 500 MHz): δ 4.11 (s, 2H), 2.60 (s, 3H).
Step 3
3-(Hydroxymethyl)-4-methyl-lH-l,2,4-triazol-5(4H)-one: 2-(2-Hydroxyacetyl)-N-methylhydrazinecarboxamide (130 g @ ~95wt%, 0.84 mol), w-propanol (130 mL) and water (130 mL) were charged to a 1 L vessel with jacket, overhead stirrer, temperature probe, reflux condenser and nitrogen inlet. Sodium hydroxide (pellets, 16.8 g, 0.42 mol) was added and the slurry warmed to reflux for 3h. The reaction mixture was cooled to 20 °C and the pH adjusted to 6.5 (+/- 0.5) using concentrated hydrochloric acid (28.3 mL, 0.34 mol). Water was
azeotropically removed under vacuum at 40-50 °C by reducing the volume to -400 mL and maintaining that volume by the slow addition of n-propanol (780 mL). The final water content was <3000 ug/mL. The resultant slurry (~ 400 mL) was cooled to 23 °C and heptane (390 ml) was added. The slurry was aged lh at 23 °C, cooled to 0 °C and aged 2h. The slurry was filtered, the cake washed with 1 :2 n-PrOH/heptane (100 mL) and the filter cake was dried under a nitrogen sweep to provide 125g (85% yield) of an off-white crystalline solid. The solid was -73 wt% due to residual inorganics (NaCl): ¾ NMR (CD3OD, 500 MHz): δ 3.30 (s, 3H), 4.46 (s, 2H).
Step 4
3-(Chloromethyl)-4-methyl-lH-l,2,4-triazol-5(4H)-one (1): A mixture of 3-(Hydroxymethyl)-4-methyl-lH-l,2,4-triazol-5(4H)-one (54 g, at 73wt%, 307 mmol) in ethyl acetate (540 mL) was stirred at 45 °C. SOCl2 (26.9 mL, 369 mmol) was added over 30-45 min and aged at 50 °C for 2h. The reaction progress was monitored by HPLC. On complete reaction (>99.5% by area at 210nm), the warm suspension was filtered and the filter cake (mainly NaCl) was washed with ethyl acetate (108 mL). The combined filtrate and wash were concentrated at 50-60 °C under reduced pressure to approximately 150 mL. The resulting slurry was cooled to – 10 °C and aged lh. The slurry was filtered and the filter cake washed with ethyl acetate (50 mL). The cake was dried under a nitrogen sweep to afford 40. lg (86% yield) of the desired product as a bright yellow solid: XH NMR (CD3OD, 500 MHz): δ 3.30 (s, 3H), 4.58 (s, 2H).
EXAMPLE 2
Step 1 – Ethyl Ester Synthesis
Experimental Procedure;
A
Ethyl 2-(3-chloro-5-cyanophenoxy)acetate (A): A 1L round bottom flask equipped with overhead stirring was charged with 3-chloro-5-hydroxybenzonitrile (50.0 g, 98 wt% purity, 319 mmol) and 15% aqueous DMF (200 mL DMF + 35.5 mL Η20). To the resulting solution was added diisopropylethylamine (61.3 mL, 99.0% purity, 1.1 equiv) and ethyl 2-bromoacetate (35.7 g, 98% purity, 1.15 equiv) at ambient temperature. The resulting solution was warmed to 50°C under nitrogen and aged for 12 h. Upon completion of the reaction the batch was cooled to 0-5°C. To the clear to slightly cloudy solution was added 5% seed (3.8g, 16.0 mmol). H20 (64.5mL) was added to the thin suspension via syringe pump over 3h while maintaining the temperature at 0-5 °C. Additional H20 (200mL) was added over lh while maintaining the temp at 0-5 °C. The final DMF/H20 ratio is 1 : 1.5. The resulting slurry was aged lh at 0-5 °C. The batch was filtered and the cake slurry washed with 2: 1 DMF/water (150 mL), followed by water (200 mL). The wet cake was dried on the frit with suction under a nitrogen stream at 20-25 °C. The cake is considered dry when H20 is <0.2%. Obtained 73.4 g ethyl ester as a light tan solid, 96% yield: XH NMR (CDC13, 400 MHz) δ = 7.29 (s, 1H), 7.15 (s, 1H), 7.06 (s, 1H), 4.67 (s, 2H), 4.32 (q, 2H), 1.35 (t, 3H) ppm. Seed was used to advance the crystallization, but the crystalline product can be precipitated and isolated without seed by allowing the solution to age at 0-5 °C for at least about 2 hours.
Step 2 – Pyridone Synthesis
Synthetic Scheme;
Experimental Procedures;
Aldol Condensation
(2E/Z,4E)-Ethyl 2-(3-chloro-5-cyanophenoxy)-5-ethoxy-3-(trifluoromethyl)penta-2,4-dienoate (C): Ethyl 2-(3-chloro-5-cyanophenoxy)acetate (25.01 g, 104.4 mmol, 1.00 equiv) was charged to toluene (113.43 g, 131 mL) and 4-ethoxy-l, l,l-trifluoro-3-buten-2-one (26.43 g, 157.2 mmol, 1.51 equiv) was added.
The flow reactor consisted of two feed solution inlets and an outlet to a receiving vessel. The flow reactor schematic is shown in Figure 1.
The ester solution was pumped to one flow reactor inlet. Potassium tert-amylate solution was pumped to the second reactor inlet. Trifluoroacetic anhydride was added continuously to the receiver vessel. Triethylamine was added continuously to the receiver vessel.
Charged toluene (50 mL) and potassium trifluoroacetate (0.64 g, 4.21 mmol, 0.04 equiv) to the receiver vessel. The flow reactor was submerged in a -10 °C bath and the pumps were turned on. The batch temperature in the receiver vessel was maintained at 5 to 10 °C throughout the run using a dry ice/acetone bath. After 13.5 min the ester solution was consumed, the reactor was flushed with toluene (10 mL) and the pumps were turned off.
The resulting yellow slurry was warmed to room temperature and aged for 4.5 h. Charged methanol (160 mL) to afford a homogeneous solution which contained 81.20 LCAP diene .
The solution of diene (573 mL) was used without purification in the subsequent reaction.
Cyclization
3-Chloro-5-((2-oxo-4-(trifluoromethyl)-l,2-dihydropyridin-3-yl)oxy)benzonitrile (E): To a solution of diene in PhMe/MeOH (573 mL; 40.69 g, 104.4 mmol theoretical) was charged methanol (25 mL). Ammonia (32 g, 1.88 mol, 18 equiv based on theoretical) was added and the solution was warmed to 60 °C. The reaction was aged at 60 °C for 18 h. The temperature was adjusted to 35-45 °C and the pressure was decreased to maintain a productive distillation rate. The batch volume was reduced to -300 mL and methanol (325 mL) was charged in portions to maintain a batch volume between 250 and 350 mL. The heating was stopped and the system vented. The resulting slurry was cooled to room temperature and aged overnight.
The batch was filtered and the cake washed with methanol (3x, 45 mL). The wet cake was dried on the frit with suction under a nitrogen stream to afford 18.54 g of a white solid: XH NMR (DMSO-ifc, 500 MHz): δ 12.7 (br s, 1H), 7.73 (t, 1H, J= 1.5 Hz), 7.61-7.59 (m, 2H), 7.53 (t, 1H, J= 2.0 Hz), 6.48 (d, 1H, J= 7.0 Hz) ppm.
Step 3 – Chlorination, Alkylation and Isolation of 3-Chloro-5-({l-[(4-methyl-5-oxo-‘ dihydro-lH-l,2,4-triazol-3-yl)methyl]-2-oxo-4-(trifluoromethyl)-l,2-dihydropyridin-3-yl}oxy)benzonitrile
3-(Chloromethyl)-4-methyl-lH-l,2,4-triazol-5(4H)-one: 3-(Hydroxymethyl)-4-methyl-lH-l,2,4-triazol-5(4H)-one (1.638 kg of 68wt%, 8.625 mol) and N-methylpyrrolidinone (8.9 L) was charged into a 30 L vessel. The suspension was aged for lOh at ambient temperature. The slurry was filtered through a 4L sintered glass funnel under 2 and the filter cake (mainly NaCl) was washed with NMP (2.23 L). The combined filtrate and wash had a water content of 5750 μg/mL. The solution was charged to a 75L flask equipped with a 2N NaOH scrubber to capture off-gasing vapors. Thionyl chloride (0.795 L, 10.89 mol) was added over lh and the temperature rose to 35 °C. HPLC analysis indicated that the reaction required an additional thionyl chloride charge (0.064 L, 0.878 mol) to bring to full conversion. The solution was warmed to 50 °C, placed under vacuum at 60 Torr (vented to a 2N NaOH scrubber), and gently sparged with subsurface nitrogen (4 L/min). The degassing continued for lOh until the sulfur dioxide content in the solution was <5 mg/mL as determined by quantitative GC/MS. The tan solution of 3-(chloromethyl)-4-methyl-lH-l,2,4-triazol-5(4H)-one in NMP weighed 13.0 kg and was assayed at 9.63 wt% providing 1.256 kg (97% yield).
3-chloro-5-((l-((4-methyl-5-oxo-4,5-dihydro-lH-l,2,4-triazol-3-yl)methyl)-2-oxo-4-(trifluoromethyl)-l,2-dihydropyridin-3-yl)oxy)benzonitrile: To a 75L flask was charged a 9.63wt% solution of 3-(chloromethyl)-4-methyl-lH-l,2,4-triazol-5(4H)-one in NMP (1 1.6 kg, 7.55 mol), 3-chloro-5-((2-oxo-4-(trifluoromethyl)-l,2-dihydropyridin-3-yl)oxy)benzonitrile (2.00 kg, 6.29 mol), NMP (3.8 L) and 2-methyl-2-butanol (6.0 L). To the resulting suspension was slowly added N,N-diisopropylethylamine (4.38 L, 25.2 mol) over 4h. The reaction was aged 18h at ambient temperature. The reaction is considered complete when HPLC indicated <1% 3-chloro-5-((2-oxo-4-(trifluoromethyl)-l,2-dihydropyridin-3-yl)oxy)benzonitrile remaining. The tan solution was quenched with acetic acid (1.26 L, 22.0 mol) and aged at ambient temperature overnight. The tan solution was warmed to 70 °C. Water (2.52 L) was added and the batch was seeded with anhydrate Form II (134 g)(procedures for making anhydrate Form II are described in WO2014/052171). The thin suspension was aged lh at 70 °C. Additional water (14.3 L) was added evenly over 7 h. The slurry was aged 2h at 70 °C and then slowly cooled to 20 °C over 5 h. The slurry was filtered and washed with 2 : 1 NMP/water (6 L), followed by water washes (6 L x 2). The filter cake was dried under N2 to give 2.53 kg (85% yield) of a white solid that was confirmed to be crystalline Form II of the title compound by X-ray powder detraction analysis.
EXAMPLE 3
Ethyl 2-(3-chloro-5-cyanophenoxy)acetate (A):
70%
Step 3
Three step one pot sequence
Steps 1 and 2:
To an oven dried 250mL round bottom flask was added sodium 2-methylpropan-2-olate (12.85 g, 134 mmol) and BHT (0.641 g, 2.91 mmol) then added DMF (30mL). After lOmin, a light yellow solution resulted. 2-Phenylethanol (7.66 ml, 63.9 mmol) was added and the solution exothermed to 35 °C. The light yellow solution was warmed to 55 °C and then a solution of 3,5-dichlorobenzonitrile (10 g, 58.1 mmol) in DMF (15mL) was added over 2h via syringe pump. The resulting red-orange suspension was aged at 55-60 °C. After 2h, HPLC showed >98% conversion to the sodium phenolate.
Step 3:
The suspension was cooled to 10 °C, then ethyl 2-bromoacetate (8.70 ml, 78 mmol) was added over lh while maintaining the temperature <20 °C. The resulting mixture was aged at ambient temperature. After lh, HPLC showed >99% conversion to the title compound.
Work-up and isolation:
To the suspension was added MTBE (50mL) and H20 (50mL) and the layers were separated. The organic layer was washed with 20% aq brine (25mL). The organic layer was assayed at 12.5g (90% yield). The organic layer was concentrated to -38 mL, diluted with hexanes (12.5mL) and then cooled to 5 °C. The solution was seeded with 0.28g (2 wt%) of crystalline ethyl 2-(3-chloro-5-cyanophenoxy)acetate and aged 0.5h at 5 °C to give a free flowing slurry. Hexane (175mL) was added to the slurry over lh at 0-5 °C. The slurry was filtered at 0-5 °C, washed with hexane (50 mL) and dried under a nitrogen sweep to give 9.8g (70% yield) of the title compound as a white crystalline solid. Seed was used to advance the crystallization, but the crystalline product can be precipitated and isolated without seed by allowing the solution to age at 0-5 °C for at least about 2 hours.
Paper
Discovery of MK-1439, an orally bioavailable non-nucleoside reverse transcriptase inhibitor potent against a wide range of resistant mutant HIV viruses
Bioorg Med Chem Lett 2014, 24(3): 917
The optimization of a novel series of non-nucleoside reverse transcriptase inhibitors (NNRTI) led to the identification of pyridone 36. In cell cultures, this new NNRTI shows a superior potency profile against a range of wild type and clinically relevant, resistant mutant HIV viruses. The overall favorable preclinical pharmacokinetic profile of 36 led to the prediction of a once daily low dose regimen in human. NNRTI 36, now known as MK-1439, is currently in clinical development for the treatment of HIV infection.
Scheme 1.
Reagents and conditions: (a) K2CO3, NMP, 120 °C; (b) KOH, tert-BuOH, 75 °C; (c) Zn(CN)2, Pd(PPh3)4, DMF, 100 °C.
Scheme 3.
Reagents and conditions: (a) K2CO3, DMF, −10 °C; (b) MeI or EtI, K2CO3, DMF.
Scheme I depicts a method for preparing compounds of Formula I in which hydroxypyridine 1-1 is alkylated with chlorotriazolinone 1-2 to provide 1-3 which can be selectively alkylated with an alkyl halide (e.g., methyl iodide, ethyl iodide, etc.) to afford the desired 1-4. Scheme I
Scheme II depicts an alternative route to compounds of the present invention, wherein fluorohydroxypyridine II-l can be alkylated with chlorotriazolinone II-2 to provide the alkylated product II-3 which can be converted to the desired II-5 via nucleophilic aromatic substitution (S] fAr) using a suitable hydroxyarene II-4.
Scheme II
Hydroxypyridines of formula I-l (Scheme 1) can be prepared in accordance with Scheme III, wherein a SNAr reaction between pyridine III-l (such as commercially available 2- chloro-3-fluoro-4-(trifluoromethyl)pyridine) and hydroxyarene H-4 can provide chloropyridine III-2, which can be hydrolyzed under basic conditions to the hydroxypyridine I-l. Scheme III
Another method for preparing hydroxypyridines of formula I-l is exemplified in Scheme IV, wherein S Ar coupling of commercially available 2-chloro-3-fluoro-4- nitropyridone-N-oxide IV-1 with a suitable hydroxyarene II-4 provides N-oxide IV-2, which can first be converted to dihalides IV-3 and then hydro lyzed to hydroxypyridine IV-4. Further derivatization of hydroxypyridine IV-4 is possible through transition metal-catalyzed coupling processes, such as Stille or boronic acid couplings using a PdLn catalyst (wherein L is a ligand such as triphenylphosphine, tri-tert-butylphosphine or xantphos) to form hydroxypyridines IV-5, or amination chemistry to form hydroxypyridines IV-6 in which R2 is N(RA)RB.
Scheme IV
IV-1
– – Scheme V depicts the introduction of substitution at the five-position of the hydroxypyridines via bromination, and subsequent transition metal-catalyzed chemistries, such as Stille or boronic acid couplings using PdLn in which L is as defined in Scheme IV to form hydroxypyridines V-3, or amination chemistry to form hydroxypyridines V-4 in which R3 is N(RA)RB.
Scheme V
As shown in Scheme IV, fiuorohydroxypyridines II-l (Scheme II) are available from the commercially available 3-fluoroypridines VI- 1 through N-oxide formation and rearrangement as described in Konno et al., Heterocycles 1986, vol. 24, p. 2169.
Scheme VI
The following examples serve only to illustrate the invention and its practice. The examples are not to be construed as limitations on the scope or spirit of the invention.
The term “room temperature” in the examples refers to the ambient temperature which was typically in the range of about 20°C to about 26°C.
A mixture of the 3-bromo-5-chlorophenol (3.74 g; 18.0 mmol), 2-chloro-3-fluoro- 4-(trifluoromethyl)pyridine (3.00 g; 15.0 mmol) and 2CO3 (2.49 g; 18.0 mmol) in NMP (15 mL) was heated to 120°C for one hour, then cooled to room temperature. The mixture was then diluted with 250 mL EtOAc and washed with 3 x 250 mL 1 :1 H20:brine. The organic extracts were dried (Na2S04) and concentrated in vacuo. Purification by ISCO CombiFlash (120 g column; load with toluene; 100:0 to 0:100 hexanes:CH2Cl2 over 40 minutes) provided title compound (1-2) as a white solid. Repurification of the mixed fractions provided additional title compound. lH NMR (400 MHz, CDCI3): δ 8.55 (d, J = 5.0 Hz, 1 H); 7.64 (d, J = 5.0 Hz, 1 H);
To a suspension of 3-(3-bromo-5-chlorophenoxy)-2-chloro-4- (trifluoromethyl)pyridine (1-2; 3.48 g; 8.99 mmol) in lBuOH (36 mL) was added KOH (1.51 g; 27.0 mmol) and the mixture was heated to 75°C overnight, at which point a yellow oily solid had precipitated from solution, and LCMS analysis indicated complete conversion. The mixture was cooled to room temperature, and neutralized by the addition of -50 mL saturated aqueous NH4CI. The mixture was diluted with 50 mL H2O, then extracted with 2 x 100 mL EtOAc. The combined organic extracts were dried (Na2S04) and concentrated in vacuo. Purification by ISCO CombiFlash (120 g column; dry load; 100:0 to 90: 10 CH2Cl2:MeOH over 40 minutes) provided the title compound (1-3) as a fluffy white solid. lH NMR (400 MHz, DMSO): δ 12.69 (s, 1 H); 7.59 (d, J = 6.9 Hz, 1 H); 7.43 (t, J = 1.7 Hz, 1 H); 7.20 (t, J = 1.9 Hz, 1 H); 7.13 (t, J = 2.0 Hz, 1 H); 6.48 (d, J = 6.9 Hz, 1 H).
To a suspension of 3-(3-bromo-5-chlorophenoxy)-4-(trifluoromethyl)pyridin-2-ol (1-3; 3.25 g; 8.82 mmol) in NMP (29 mL) was added CuCN (7.90 g; 88 mmol) and the mixture was heated to 175°C for 5 hours, then cooled to room temperature slowly. With increased fumehood ventilation, 100 mL glacial AcOH was added, then 100 mL EtOAc and the mixture was filtered through Celite (EtOAc rinse). The filtrate was washed with 3 x 200 mL 1 : 1 H20:brine, then the organic extracts were dried (Na2S04) and concentrated in vacuo.
Purification by ISCO CombiFlash (120 g column; dry load; 100:0 to 90:10 CH2Cl2:MeOH over 40 minutes), then trituration of the derived solid with Et20 (to remove residual NMP which had co-eluted with the product) provided the title compound (1-4). lH NMR (400 MHz, DMSO): δ 12.71 (s, 1 H); 7.75 (s, 1 H); 7.63-7.57 (m, 2 H); 7.54 (s, 1 H); 6.49 (d, J = 6.9 Hz, 1 H).
The title compound was prepared as described in the literature: Cowden, C. J.; Wilson, R. D.; Bishop, B. C; Cottrell, I. F.; Davies, A. J.; Dolling, U.-H. Tetrahedron Lett. 2000, 47, 8661.
A suspension of the 3-chloro-5-{[2-hydroxy-4-(trifluoromethyl)pyridin-3- yl]oxy}benzonitrile (1-4; 2.00 g; 6.36 mmol), 5-(chloromethyl)-2,4-dihydro-3H-l,2,4-triazol-3- one (1-5; 0.849 g; 6.36 mmol) and K2CO3 (0.878 g; 6.36 mmol) in DMF (32 mL) was stirred for 2 hours at room temperature, at which point LCMS analysis indicated complete conversion. The mixture was diluted with 200 mL Me-THF and washed with 150 mL 1 : 1 : 1 H20:brine:saturated aqueous NH4CI, then further washed with 2 x 150 mL 1 : 1 H20:brine. The aqueous fractions were further extracted with 150 mL Me-THF, then the combined organic extracts were dried (Na2S04) and concentrated in vacuo. Purification by ISCO CombiFlash (80 g column; dry load; 100:0 to 90:10 EtOAc:EtOH over 25 minutes) provided the title compound (1-6) as a white solid. lH NMR (400 MHz, DMSO): δ 1 1.46 (s, 1 H); 1 1.39 (s, 1 H); 7.93 (d, J = 7.3 Hz, 1 H); 7.76 (s, 1 H); 7.58 (s, 1 H); 7.51 (s, 1 H); 6.67 (d, J = 7.3 Hz, 1 H); 5.02 (s, 2 H).
A solution of 3-chloro-5-({2-oxo-l -[(5-oxo-4,5-dihydro-lH-l,2,4-triazol-3- yl)methyl]- 4-(trifluoromethyl)-l ,2-dihydropyridin-3-yl}oxy)benzonitrile (1-6; 2.37 g; 5.76 mmol) and K2CO3 (0.796 g; 5.76 mmol) in DMF (58 mL) was cooled to 0°C, then methyl iodide (0.360 mL; 5.76 mmol) was added. The mixture was allowed to warm to room
temperature, and stirred for 90 minutes, at which point LCMS analysis indicated >95%
conversion, and the desired product of -75% LCAP purity, with the remainder being unreacted starting material and 6/s-methylation products. The mixture was diluted with 200 mL Me-THF, and washed with 3 x 200 mL 1 : 1 H20:brine. The aqueous fractions were further extracted with 200 mL Me-THF, then the combined organic extracts were dried (Na2S04) and concentrated in vacuo. The resulting white solid was first triturated with 100 mL EtOAc, then with 50 mL THF, which provided (after drying) the title compound (1-1) of >95% LCAP. Purification to >99% LCAP is possible using Prep LCMS (Max-RP, 100 x 30 mm column; 30-60% CH3CN in 0.6% aqueous HCOOH over 8.3 min; 25 mL/min). lH NMR (400 MHz, DMSO): δ 1 1.69 (s, 1 H); 7.88 (d, J = 7.3 Hz, 1 H); 7.75 (s, 1 H); 7.62 (s, 1 H); 7.54 (s, 1 H); 6.67 (d, J = 7.3 Hz, 1 H); 5.17 (s, 2 H); 3.1 1 (s, 3 H). EXAMPLE 1A
A mixture of the 3-chloro-l-iodophenol (208 g; 816.0 mmol), 2-chloro-3-fluoro-
4-(trifluoromethyl)pyridine (155 g; 777.0 mmol) and K2CO3 (161 g; 1 165.0 mmol) in NMP (1.5 L) was held at 60°C for 2.5 hours, and then left at room temperature for 2 days. The mixture was then re-heated to 60°C for 3 hours, then cooled to room temperature. The mixture was then diluted with 4 L EtOAc and washed with 2 L water + 1 L brine. The combined organics were then washed 2x with 500 mL half brine then 500 mL brine, dried over MgS04 and concentrated to afford crude 1A-2. lH NMR (500 MHz, DMSO) δ 8.67 (d, J = 5.0 Hz, 1 H), 7.98 (d, J = 5.0 Hz, 1 H), 7.63-7.62 (m, 1 H), 7.42-7.40 (m, 1 H), 7.22 (t, J = 2.1 Hz, 1 H).
To a suspension of 3-(3-chloro-5-iodophenoxy)-2-chloro-4- (trifluoromethyl)pyridine (1A-2; 421 g, 970 mmol) in t-BuOH (1 L) was added KOH (272 g, 4850 mmol) and the mixture was heated to 75°C for 1 hour, at which point HPLC analysis indicated >95% conversion. The t-BuOH was evaporated and the mixture diluted with water (7mL/g, 2.4L) and then cooled to 0°C, after which 12N HC1 (~240mL) was added until pH 5. This mixture was then extracted with EtOAc (20mL/g, 6.5L), back extracted with EtOAc 1 x 5mL/g (1.5L), washed 1 x water:brine 1 : 1 (l OmL/g, 3.2L), 1 x brine (lOmL/g, 3.2L), dried over MgS04, filtered and concentrated to afford a crude proudct. The crude product was suspended in MTBE (2.25 L, 7mL/g), after which hexanes (1 L, 3 mL/g) was added to the suspension over ten minutes, and the mixturen was aged 30minutes at room temperature. The product was filtered on a Buchner, rinsed with MTBE hexanes 1 :2 (2 mL/g = 640 mL), then hexanes
A solution of 3-(3-chloro-5-iodophenoxy)-4-(trifluoromethyl)pyridin-2-ol (1A-3; 190 g; 457 mmol) in DMF (914 mL) was degassed for 20 minutes by bubbling N2, after which CuCN (73.7 g; 823 mmol) was added, and then the mixture was degassed an additional 5 minutes. The mixture was then heated to 120°C for 17 hours, then cooled to room temperature and partitioned between 6 L MeTHF and 2 L ammonium buffer (4:3: 1 = NH4CI
sat/water/NH-iOH 30%). The organic layer washed with 2 L buffer, 1 L buffer and 1 L brine then, dried over MgS04 and concentrated. The crude solid was then stirred in 2.2 L of refluxing
MeCN for 45 minutes, then cooled in a bath to room temperature over 1 hour, aged 30 minutes, then filtered and rinsed with cold MeCN (2 x 400mL). The solid was dried on frit under N2 atm for 60 hours to afford title compound 1-4. lH NMR (400 MHz, DMSO): δ 12.71 (s, 1 H); 7.75 (s, 1 H); 7.63-7.57 (m, 2 H); 7.54 (s, 1 H); 6.49 (d, J = 6.9 Hz, 1 H).
Steps lA(d) and lA(e)
The title compound 1-1 was then prepared from compound 1-4 using procedures similar to those described in Steps 1(d) and 1(e) set forth above in Example 1.
Crystalline anhydrous Form II of doravirine, useful for the treatment of HIV-1 and HIV-2 infections. The compound was originally claimed in WO2008076223. Also see WO2011120133. Merck & Co is developing doravirine (MK-1439), for the oral tablet treatment of HIV-1 infection. As of April 2014, the drug is in Phase 2 trials.
CLIPS
The next-generation non-nucleoside reverse transcriptase inhibitor (NNRTI) doravirine (formerly MK-1439) showed potent antiretroviral activity and good tolerability in combination with tenofovir/FTC (the drugs in Truvada) in a dose-finding study presented at the 21stConference on Retroviruses and Opportunistic Infections (CROI) last week in Boston.
NNRTIs are generally well tolerated and well suited for first-line HIV treatment, but as a class they are susceptible to resistance. Pre-clinical studies showed that Merck’s doravirine has a distinct resistance profile and remains active against HIV with common NNRTI resistance mutations including K103N and Y181C.
As reported at last year’s CROI, doravirine reduced HIV viral load by about 1.3 log in a seven-day monotherapy study. Doravirine is processed by the CYP3A4 enzyme, but it is neither a CYP3A4 inducer nor inhibitor, so it is not expected to have major drug interaction concerns.
Javier Morales-Ramirez from Clinical Research Puerto Rico reported late-breaking findings from a phase 2b study evaluating the safety and efficacy of various doses of doravirine versus efavirenz (Sustiva) for initial antiretroviral therapy.
This study included 208 treatment-naive people living with HIV from North America, Europe and Asia. More than 90% were men, 74% were white, 20% were black and the median age was 35 years. At baseline, the median CD4 cell count was approximately 375 cells/mm3 and 13% had received an AIDS diagnosis. Study participants were stratified by whether their viral load was above (about 30%) or below 100,000 copies/ml; median HIV RNA was approximately 4.5 log10.
Morales-Ramirez reported 24-week results from part 1 of the study, which will continue for a total of 96 weeks. In this part, participants were randomly allocated into five equal-sized arms receiving doravirine at doses of 25, 50, 100 or 200mg once daily, or else efavirenz once daily, all in combination with tenofovir/FTC.
At 24 weeks, 76.4% of participants taking doravirine had viral load below 40 copies/ml compared with 64.3% of people taking efavirenz. Response rates were similar across doravirine doses (25mg: 80.0%; 50mg: 76.2%; 100mg: 71.4%; 200mg: 78.0%). More than 80% of participants in all treatment arms reached the less stringent virological response threshold of <200 copies/ml.
Both doravirine and efavirenz worked better for people with lower pre-treatment viral load in an ad hoc analysis. For people with <100,000 copies/ml at baseline, response rates (<40 copies/ml) ranged from 83 to 89% with doravirine compared with 74% with efavirenz. For those with >100,000 copies/ml, response rates ranged from 50 to 91% with doravirine vs 54% with efavirenz.
Median CD4 cell gains were 137 cells/mm3 for all doravirine arms combined and 121 cells/mm3 for the efavirenz arm.
Doravirine was generally safe and well tolerated. People taking doravirine were less than half as likely as people taking efavirenz to experience serious adverse events (3.0% across all doravirine arms vs 7.1% with efavirenz) or to stop treatment for this reason (2.4 vs 4.8%). Four people taking doravirine and two people taking efavirenz discontinued due to adverse events considered to be drug-related.
The most common side-effects were dizziness (3.6% with doravirine vs 23.8% with efavirenz), abnormal dreams (9.0 vs 7.1%), diarrhoea (4.8 vs 9.5%), nausea (7.8 vs 2.4%) and fatigue (6.6 vs 4.8%). Other central nervous system (CNS) adverse events of interest included insomnia (5.4 vs 7.1%), nightmares (1.2 vs 9.5%) and hallucinations (0.6 vs 2.4%). Overall, 20.5% of people taking doravirine reported at least one CNS side-effect, compared with 33.3% of people taking efavirenz.
People taking doravirine had more favourable lipid profiles and less frequent liver enzyme (ALT and AST) elevations compared with people taking efavirenz.
The researchers concluded that doravirine demonstrated potent antiretroviral activity in treatment-naive patients, a favourable safety and tolerability profile, and fewer drug-related adverse events compared with efavirenz.
Based on these findings, the 100mg once-daily dose was selected for future development and will be used in part 2 of this study, a dose-confirmation analysis that will enrol an additional 120 participants.
In the discussion following the presentation, Daniel Kuritzkes from Harvard Medical School noted that sometimes it takes longer for viral load to go down in people who start with a high level, so with further follow-up past 24 weeks doravirine may no longer look less effective in such individuals.
Reference
Morales-Ramirez J et al. Safety and antiviral effect of MK-1439, a novel NNRTI (+FTC/TDF) in ART-naive HIV-infected patients. 21st Conference on Retroviruses and Opportunistic Infections, Boston, abstract 92LB, 2014.
Merck Moves Doravirine Into Phase 3 Clinical Trials
Wednesday Mar 19 | Posted by: roboblogger | Full story: EDGE
Earlier this month, at the 21st Conference on Retroviruses and Opportunistic Infections , Merck indicated plans to initiate a Phase 3 clinical trial program for doravirine in combination with ARV therapy in the second half of 2014.
Doravirine is non-nucleoside reverse transcriptase inhibitor (NNRTI) currently in phase III clinical trials for the treatment of HIV infection. Herein we describe a robust kilo-scale synthesis for its manufacture. The structure and origin of major impurities were determined and their downstream fate-and-purge studied. This resulted in a redesign of the route to introduce the key nitrile functionality via a copper mediated cyanation which allowed all impurities to be controlled to an acceptable level. The improved synthesis was scaled to prepare ∼100 kg batches of doravirine to supply all preclinical and clinical studies up to phase III. The synthesis affords high-quality material in a longest linear sequence of six steps and 37% overall yield.
PAPER
Highly Efficient Synthesis of HIV NNRTI Doravirine
Gauthier, D. R., Jr.; Sherry, B. D.; Cao, Y.; Journet, M.; Humphrey, G.; Itoh, T.; Mangion, I.; Tschaen, D. M.Org. Lett.2015, 17, 1353, DOI: 10.1021/ol503625z………..http://pubs.acs.org/doi/full/10.1021/ol503625z
The development of an efficient and robust process for the production of HIV NNRTI doravirine is described. The synthesis features a continuous aldol reaction as part of a de novo synthesis of the key pyridone fragment. Conditions for the continuous flow aldol reaction were derived using microbatch snapshots of the flow process.
Doravirine (1). A 75 L flask was charged a 9.63 wt% solution of 16 in NMP (11.6kg, 7.55 mol). Charged 10(2.00 kg, 6.29 mol), NMP
(3.8 L) and 2-methyl-2-butanol (6.0 L). To theresulting suspension was slowly added N,N-diisopropylethylamine (4.38 L, 25.2 mol) over 4 h. The reaction was aged for 18 h at ambient temperature. The tan solution was quenched with acetic acid (1.26 L, 22.0 mol) and aged at ambient temperature overnight. The tan solution was warmed to 70 °C. Water (2.52 L) was added and the batch was seeded with crystalline 1 (134 g) to induce crystallization. The thin suspension was aged 1 h at 70 °C. Additional water (14.3 L)
was added over 7 h. The slurry was aged for 2 h at 70 °C and slowly cooled to 20 °C over 5 h. The slurry was filtered and washed with 2:1 NMP/water (6 L), followed by water (6 L x 2). The filter cake was dried under nitrogen to give 2.53 kg (85% yield) of 1 as a white solid.
MK-8718 is a potent, selective and orally bioavailable HIV protease inhibitor with a favorable pharmacokinetic profile with potential for further development.
A retrovirus designated human immunodeficiency virus (HIV), particularly the strains known as HIV type-1 (HIV-1) virus and type-2 (HIV-2) virus, is the etiological agent of acquired immunodeficiency syndrome (AIDS), a disease characterized by the destruction of the immune system, particularly of CD4 T-cells, with attendant susceptibility to opportunistic infections, and its precursor AIDS-related complex (“ARC”), a syndrome characterized by symptoms such as persistent generalized lymphadenopathy, fever and weight loss. This virus was previously known as LAV, HTLV-III, or ARV. A common feature of retrovirus replication is the extensive post-translational processing of precursor polyproteins by a virally encoded protease to generate mature viral proteins required for virus assembly and function. Inhibition of this processing prevents the production of normally infectious virus. For example, Kohl et al., Proc. Nat’l Acad. Sci. 1988, 85: 4686, demonstrated that genetic inactivation of the HIV encoded protease resulted in the production of immature, non-infectious virus particles. These results indicated that inhibition of the HIV protease represents a viable method for the treatment of AIDS and the prevention or treatment of infection by HIV.
Nucleotide sequencing of HIV shows the presence of a pol gene in one open reading frame [Ratner et al, Nature 1985, 313: 277]. Amino acid sequence homology provides evidence that the pol sequence encodes reverse transcriptase, an endonuclease, HIV protease and gag, which encodes the core proteins of the virion (Toh et al, EMBO J. 1985, 4: 1267; Power et al, Science 1986, 231 : 1567; Pearl et al, Nature 1987, 329: 351].
Several HIV protease inhibitors are presently approved for clinical use in the treatment of AIDS and HIV infection, including indinavir (see US 5413999), amprenavir (US5585397), saquinavir (US 5196438), ritonavir (US 5484801) and nelfmavir (US 5484926). Each of these protease inhibitors is a peptide-derived peptidomimetic, competitive inhibitor of the viral protease which prevents cleavage of the HIV gag-pol polyprotein precursor. Tipranavir (US 5852195) is a non-peptide peptidomimetic protease inhibitors also approved for use in treating HIV infection. The protease inhibitors are administered in combination with at least one and typically at least two other HIV antiviral agents, particularly nucleoside reverse transcriptase inhibitors such as zidovudine (AZT) and lamivudine (3TC) and/or non-nucleoside reverse transcriptase inhibitors such as efavirenz and nevirapine. Indinavir, for example, has been found to be highly effective in reducing HIV viral loads and increasing CD4 cell counts in HIV-infected patients, when used in combination with nucleoside reverse transcriptase inhibitors. See, for example, Hammer et al, New England J. Med. 1997, 337: 725-733 and Gulick et al, New England J. Med. 1997, 337: 734-739.
The established therapies employing a protease inhibitor are not suitable for use in all HIV-infected subjects. Some subjects, for example, cannot tolerate these therapies due to adverse effects. Many HIV-infected subjects often develop resistance to particular protease inhibitors. Furthermore, the currently available protease inhibitors are rapidly metabolized and cleared from the bloodstream, requiring frequent dosing and use of a boosting agent.
Accordingly, there is a continuing need for new compounds which are capable of inhibiting HIV protease and suitable for use in the treatment or prophylaxis of infection by HIV and/or for the treatment or prophylaxis or delay in the onset or progression of AIDS.
The title compound was prepared from 4-chlorocinnamic acid and 3,5- difluorophenylmagnesium bromide using the procedures given in steps 1-4 of Example 92.
The product from step 1 (105 mg, 0.31 mmol) and the product from step 4 of Example 89 (150 mg, 0.31 mmol) were dissolved in pyridine (1 mL) and the stirred solution was cooled to -10 °C in an ice/acetone bath. To the cold solution was added POCI3 dropwise (0.035 mL, 0.38 mmol). The mixture was stirred at -10 °C for 30 min. The reaction was quenched by the addition of saturated aqueous NaHC03 solution (1 mL) and the mixture was allowed to warm to ambient temperature. The mixture was diluted with water (10 mL) and extracted with dichloromethane (3 x 10 mL). The combined dichloromethane phases were dried (Na2S04), filtered, and the filtrate solvents were removed in vacuo. The residue was purified on a 12 g silica gel column using a gradient elution of 0-70% EtOAc:hexanes. Fractions containing product were combined and the solvents were removed in vacuo to give the title compound as a gum. (M+H)+ = 800.6.
The product from step 2 (150 mg, 0.19 mmol) and triphenylphosphine (74 mg, 0.28 mmol) were dissolved in THF (4 mL) and to the solution was added water (1 mL). The mixture was heated to reflux under a nitrogen atmosphere for 12 h. The mixture was cooled to ambient temperature and the solvents were removed in vacuo. The residue was purified on a 12 g silica gel column eluting with a gradient of 0-10% methanol: chloroform. Fractions containing product were combined and the solvents were removed in vacuo to give the title compound as a gum. (M+H)+ = 774.7. Step 4. ( S)- -(4-Chlorophenyl)-3,5-difluoro-N-(5-fluoro-4-{2-[(2R,5S)-5-({[(2,2,2- trifluoroethyl)carbamoyl]oxy}methyl)morpholin-2-yl]ethyl}pyridin-3-yl)-L-phenylala
The product from step 3 (60 mg, 0.078 mmol) was dissolved in a solution of 4M HCl in dioxane (1 mL, 4 mmol) and the solution was stirred at ambient temperature for 1 h. The solvent was removed under reduced pressure and the residue was dried in vacuo for 12 h to give an HCl salt of the title compound as a solid. LCMS: RT = 0.95 min (2 min gradient), MS (ES) m/z = 674.6 (M+H)+.
PAPER
A novel HIV protease inhibitor was designed using a morpholine core as the aspartate binding group. Analysis of the crystal structure of the initial lead bound to HIV protease enabled optimization of enzyme potency and antiviral activity. This afforded a series of potent orally bioavailable inhibitors of which MK-8718 was identified as a compound with a favorable overall profile.
Discovery of MK-8718, an HIV Protease Inhibitor Containing a Novel Morpholine Aspartate Binding Group
Discovery of MK-8718, an HIV Protease Inhibitor Containing a Novel Morpholine Aspartate Binding Group
Christopher J. Bungard*†, Peter D. Williams†, Jeanine E. Ballard†, David J. Bennett†, Christian Beaulieu‡, Carolyn Bahnck-Teets†, Steve S. Carroll†, Ronald K. Chang†, David C. Dubost†, John F. Fay†, Tracy L. Diamond†, Thomas J. Greshock†, Li Hao§, M. Katharine Holloway†, Peter J. Felock, Jennifer J. Gesell†, Hua-Poo Su†, Jesse J. Manikowski†, Daniel J. McKay‡, Mike Miller†, Xu Min†, Carmela Molinaro†, Oscar M. Moradei‡, Philippe G. Nantermet†, Christian Nadeau‡, Rosa I. Sanchez†, Tummanapalli Satyanarayana§, William D. Shipe†, Sanjay K. Singh§, Vouy Linh Truong‡, Sivalenka Vijayasaradhi§, Catherine M. Wiscount†, Joseph P. Vacca‡, Sheldon N. Crane‡, and John A. McCauley†
† Merck Research Laboratories, 770 Sumneytown Pike, PO Box 4, West Point, Pennsylvania 19486, United States
‡ Merck Frosst Centre for Therapeutic Research, 16711 TransCanada Highway, Kirkland, Quebec H9H 3L1, Canada
§ Albany Molecular Research Singapore Research Center, 61 Science Park Road #05-01, The Galen Singapore Science Park II, Singapore 117525
ACS Med. Chem. Lett., Article ASAP
DOI: 10.1021/acsmedchemlett.6b00135
Publication Date (Web): May 09, 2016
The U.S. Food and Drug Administration today approved Genvoya (a fixed-dose combination tablet containing elvitegravir, cobicistat, emtricitabine, and tenofovir alafenamide) as a complete regimen for the treatment of HIV-1 infection in adults and pediatric patients 12 years of age and older
The U.S. Food and Drug Administration today approved Genvoya (a fixed-dose combination tablet containing elvitegravir, cobicistat, emtricitabine, and tenofovir alafenamide) as a complete regimen for the treatment of HIV-1 infection in adults and pediatric patients 12 years of age and older.
The CDC estimates that 1.2 million persons ages 13 years and older are living with HIV infection, and that more than another 150,000 persons in this age range have HIV but are unaware of their infection. Over the past decade, the number of people living with HIV has increased, while the annual number of new HIV infections has remained relatively stable.
“Today’s approval of a fixed dose combination containing a new form of tenofovir provides another effective, once daily complete regimen for patients with HIV-1 infection,” said Edward Cox, M.D., director of the Office of Antimicrobial Products in the FDA’s Center for Drug Evaluation and Research.
Genvoya is approved for use in HIV-infected adults and children ages 12 years and older weighing at least 35 kilograms (77 pounds) who have never taken HIV therapy (treatment-naïve) and HIV-infected adults whose HIV-1 virus is currently suppressed. While Genvoya is not recommended for patients with severe renal impairment, those with moderate renal impairment can take Genvoya.
Genvoya’s safety and efficacy in adults were evaluated in 3,171 participants enrolled in four clinical trials. Depending on the trial, participants were randomly assigned to receive Genvoya or another FDA approved HIV treatment. Results showed Genvoya was effective in reducing viral loads and comparable to the other treatment regimens.
Genvoya contains a new form of tenofovir that has not been previously approved. This new form of tenofovir provides lower levels of drug in the bloodstream, but higher levels within the cells where HIV-1 replicates. It was developed to help reduce some drug side effects. Genvoya appears to be associated with less kidney toxicity and decreases in bone density than previously approved tenofovir containing regimens based on laboratory measures. Patients receiving Genvoya experienced greater increases in serum lipids (total cholesterol and low-density lipoprotein) than patients receiving other treatment regimens in the studies.
Genvoya carries a Boxed Warning alerting patients and health care providers that the drug can cause a buildup of lactic acid in the blood and severe liver problems, both of which can be fatal. The Boxed Warning also states that Genvoya is not approved to treat chronic hepatitis B virus infection. The most common side effect associated with Genvoya is nausea. Serious side effects include new or worsening kidney problems, decreased bone mineral density, fat redistribution and changes in the immune system (immune reconstitution syndrome). Health care providers are advised to monitor patients for kidney and bone side effects. Genvoya should not be given with other antiretroviral products and may have drug interactions with a number of other commonly used medications.
Genvoya is marketed by Gilead Sciences Inc. based in Foster City, California.
CAS NO. 161814-49-9, [(3S)-oxolan-3-yl] N-[(2S,3R)-4-[(4-aminophenyl)sulfonyl-(2-methylpropyl)amino]-3-hydroxy-1-phenylbutan-2-yl]carbamate
161814-49-9
Weight
505.224656557
Chemical Formula
C25H35N3O6S
Amprenavir is a protease inhibitor used to treat HIV infection.
Amprenavir (Agenerase, GlaxoSmithKline) is a protease inhibitor used to treat HIV infection. It was approved by the Food and Drug Administration on April 15, 1999, for twice-a-day dosing instead of needing to be taken every eight hours. The convenient dosing came at a price, as the dose required is 1,200 mg, delivered in eight very large gel capsules.
Production of amprenavir was discontinued by the manufacturer December 31, 2004; a prodrug version (fosamprenavir) is available.
Amprenavir is a protease inhibitor with activity against Human Immunodeficiency Virus Type 1 (HIV-1). Protease inhibitors block the part of HIV called protease. HIV-1 protease is an enzyme required for the proteolytic cleavage of the viral polyprotein precursors into the individual functional proteins found in infectious HIV-1. Amprenavir binds to the protease active site and inhibits the activity of the enzyme. This inhibition prevents cleavage of the viral polyproteins resulting in the formation of immature non-infectious viral particles. Protease inhibitors are almost always used in combination with at least two other anti-HIV drugs.
HIV-1 Protease dimer with Amprenavir (sticks) bound in the active site. PDB entry 3nu3[1]
Country
Patent Number
Approved
Expires (estimated)
United States
5585397
1993-12-17
2013-12-17
United States
6730679
1997-11-11
2017-11-11
Background
Research aimed at development of renin inhibitors as potential antihypertensive agents had led to the discovery of compounds that blocked the action of this peptide cleaving enzyme. The amino acid sequence cleaved by renin was found to be fortuitously the same as that required to produce the HIV peptide coat. Structure–activity studies on renin inhibitors proved to be of great value for developing HIV protease inhibitors. Incorporation of an amino alcohol moiety proved crucial to inhibitory activity for many of these agents. This unit is closely related to the one found in the statine, an unusual amino acid that forms part of the pepstatin, a fermentation product that inhibits protease enzymes.
R.D. Tung, M.A. Murcko, G.R. Bhisetti, U.S. Patent 5,558,397 (1996). The scheme shown here is partly based on that used to prepare darunavir and fosamprenavir due to difficulty in deciphering the patent.
AGENERASE (amprenavir) is an inhibitor of the human immunodeficiency virus (HIV) protease. The chemical name of amprenavir is (3S)-tetrahydro-3-furyl N-[(1S,2R)-3-(4-amino-N-isobutylbenzenesulfonamido)-1-benzyl-2-hydroxypropyl]carbamate. Amprenavir is a single stereoisomer with the (3S)(1S,2R) configuration. It has a molecular formula of C25H35N3O6S and a molecular weight of 505.64. It has the following structural formula:
Amprenavir is a white to cream-colored solid with a solubility of approximately 0.04 mg/mL in water at 25°C.
AGENERASE Capsules (amprenavir capsules) are available for oral administration. Each 50- mg capsule contains the inactive ingredients d-alpha tocopheryl polyethylene glycol 1000 succinate (TPGS), polyethylene glycol 400 (PEG 400) 246.7 mg, and propylene glycol 19 mg. The capsule shell contains the inactive ingredients d-sorbitol and sorbitans solution, gelatin, glycerin, and titanium dioxide. The soft gelatin capsules are printed with edible red ink. Each 50- mg AGENERASE Capsule contains 36.3 IU vitamin E in the form of TPGS. The total amount of vitamin E in the recommended daily adult dose of AGENERASE is 1,744 IU.
Efficient and industrially applicable synthetic processes for precursors of HIV protease inhibitors (Amprenavir, Fosamprenavir) are described. These involve a novel and economical method for the preparation of a key intermediate, (3S)-hydroxytetrahydrofuran, from L-malic acid. Three new approaches to the assembly of Amprenavir are also discussed. Of these, a synthetic route in which an (S)-tetrahydrofuranyloxy carbonyl is attached to L-phenylalanine appears to be the most promising manufacturing process, in that it offers satisfactory stereoselectivity in fewer steps.
The reaction of N,N-dibenzyl-L-alaninal (I) with nitromethane, catalyzed by the chiral ammonium salt (II) and KF in THF gives the chiral nitroalcohol (III), which is reduced with NiCl2 and NaBH4 to yield the aminoalcohol (IV). The condensation of (IV) with isobutyraldehyde (V) affords the Schiff base (VI), which is reduced with NaBH4 to provide the secondary amine (VII). The reaction of (VII) with 4-nitrobenzenesulfonyl chloride (VIII) and TEA in dichloromethane furnishes the sulfonamide (IX), which is deprotected by hydrogenation with H2 over Pd/C in methanol, giving the diamino compound (X). Finally, this compound is condensed with 3(S)-tetrahydrofuryl (N-oxysuccinimidyl) carbonate (XI) by means of TEA in dichloromethane to afford the target carbamate.
Angew Chem. Int Ed Engl1999,38,(13-14):1931
……………………………………………………….
The reaction of the chiral epoxide (I) with isobutylamine (II) in refluxing ethanol gives the secondary amine (III), which is protected with benzyl chloroformate (IV) and TEA, yielding the dicarbamate (V). Selective deprotection of (V) with dry HCl in ethyl acetate affords the primary amine (VI), which is treated with 3(S)-tetrahydrofuryl N-succinimidinyl carbonate (VII) (prepared by condensation of tetrahydrofuran-3(S)-ol (VIII) with phosgene and N-hydroxysuccinimide (IX)) and DIEA in acetonitrile to provide the corresponding carbamate (X). The deprotection of (X) by hydrogenation with H2 over Pd/C in ethanol gives the secondary amine (XI), which is condensed with 4-nitrophenylsulfonyl chloride (XII) by means of NaHCO3 in dichloromethane/water to yield the sulfonamide (XIII). Finally, the nitro group of (XIII) is reduced with H2 over Pd/C in ethyl acetate to afford the target compound.
EP 0659181; EP 0885887; JP 1996501299; US 5585397; WO 9405639
……………………………………………………………
Patent
https://www.google.com/patents/WO1999048885A1?cl=ensynthesis of (3S)-tetrahydro-3-furyl N-[(1S,2R)-3(4-amino-N-isobutylbenzenesulphonamido)-1-benzyl-2-hydroxypropyl]carbamate, hereinafter referred to as the compound of formula (I), and to novel intermediates thereto.The compound of formula (I) has the following structure
and was first described in PCT patent publication number WO94/05639 at Example 168. Currently there is considerable interest in the compound of formula (I) as a new chemotherapeutic compound in the treatment of human immunodeficiency virus (HIV) infection and the associated conditions such as acquired immune deficiency syndrome (AIDS) and AIDS dementia.
There exists at the present time a need to produce large quantities of the compound of formula (I) for clinical investigation into the efficacy and safety of the compound as a chemotherapeutic agent in the treatment of HIV infections.
An ideal route for the synthesis of the compound should produce the compound of formula (I) in high yields at a reasonable speed and at low cost with minimum waste materials and in a manner that is of minimum impact to the environment in terms of disposing of waste-materials and energy consumption.
We have found a new process for the synthesis of the compound of formula (I) with many advantages over previously known routes of synthesis. Such advantages include lower cost, less waste and more efficient use of materials. The new process enables advantageous preparation of the compound of formula (I) on a manufacturing scale.
The route of synthesis of the compound of formula (I) described in the specification of WO94/05639 is specifically described therein in examples 39A, 51A, 51B, 51C, 51D, 167 and 168. The overall yield from these examples is 33.2% of theory.
Generally the route described in WO94/05639 involves protecting the amino alcohol of formula (A) (Ex.39)
wherein P is a protecting group to form a compound of formula (B);
wherein P and P′ are each independently a protecting group;
deprotecting the compound of formula (B) to form a compound of formula (C) (Ex 51A);
wherein P′ is a protecting group;
forming a hydrochloride salt of compound (C) (Ex 51B) then reacting with N-imidazolyl-(S)-tetrahydrofuryl carbamate to form the compound of formula (D) (Ex 51C);
wherein P′ is a protecting group;
deprotecting the compound of formula (D) (Ex 51D) wherein P′ is a protecting group to form the compound of formula (D) wherein P′ is H (Ex 51E); and coupling the resultant secondary amine on the compound of formula (D) to a p-nitrophenylsulphonyl group to form a compound of formula (E) (Ex 167);
the resultant compound of formula (E) is then reduced to form the compound of formula (I) (Ex 168).
In summary, the process disclosed in WO94/05639 for producing the compound of formula (I) from the compound of formula (A) comprises 6 distinct stages:
1) protecting,
2) deprotecting,
3) reacting the resultant compound with an activated tetrahydrofuranol group,
4) deprotecting,
5) coupling with a p-nitrophenylsulfonyl group, and
6) reducing the resultant compound to form a compound of formula (I).
Applicants have now found a process by which the compound of formula (I) may be prepared on a manufacturing scale from the same starting intermediate, the compound of formula (A), in only 4 distinct stages instead of 6. In addition to the associated benefits of fewer stages, such as savings in time and cost, the improved process reduces the number of waste products formed. Furthermore, product may be obtained in a higher yield, of approximately 50% of theory
.
EXAMPLESExample 1
(1S,2R)-tert-butyl N-[1-benzyl-2-hydroxy-3-(isobutylamino)propyl]carbamate (127.77 g, 379.7 mmol) was heated in toluene (888 ml) to 80° C. and triethylamine (42.6 g, 417.8 mmol) added. The mixture was heated to 90° C. and a solution of p-nitrobenzene sulphonyl chloride (94.3 g, 425.4 mmol) in toluene (250 ml) was added over 30 minutes then stirred for a further 2 hours. The resultant solution of the nosylated intermediate {(1S,2R)-tert-butyl N-[1-benzyl-2-hydroxy-3-(N-isobutyl- 4-nitrobenzenesulphonamido)propyl]carbamate } was then cooled to 80° C. The solution was maintained at approximately 80° C., and concentrated hydrochloric acid (31.4 ml, 376.8 mmol) was added over 20 minutes. The mixture was heated to reflux (approx 86° C.) and maintained at this temperature for an hour then a further quantity of concentrated hydrochloric acid (26.4 ml, 316.8 mmol) was added. Solvent (water and toluene mixture) was removed from the reaction mixture by azeotropic distillation (total volume of solvent removed approx 600 ml), and the resultant suspension was cooled to 70-75° C. Denatured ethanol (600 ml) was added, and the solution was cooled to 20° C. The mixture was further cooled to approximately −10° C. and the precipitate formed was isolated by filtration, washed with denatured ethanol (50 ml) and dried at approximately 50° C., under vacuum, for approximately 12 hours, to give (2R,3S)-N-(3-amino-2-hydroxy-4-phenylbutyl)-N-isobutyl-4-nitrobenzene sulphonamide hydrochloride (160 g; 73% of theory yield corrected for assay). NMR: 1H NMR (300Mhz, dmso-d6): 8.37(2H, d, J=9 Hz), 8.16(NH3+s), 8.06(2H, d, J=9 Hz), 7.31(5H, m), 5.65(1H, d, J=5 Hz), 3.95(1H, m), 3.39(2H, m), 2.95(5H, m), 1.90(1H, m), 0.77(6H, dd, J=21 Hz and 6 Hz).
1,1′-carbonyidiimidazole (27.66 kg, 170.58 mol) was added to ethyl acetate (314.3 kg) with stirring to give 3-(S)-tetrahydrofuryl imidazole-1-carboxylate. (S)-3-hydroxytetrahydrofuran (157 kg, 178.19 mol) was added over 30 minutes, washed in with ethyl acetate (9.95 kg), then the mixture was stirred for a further hour. (2R,3S)-N-(3-amino-2-hydroxy-4-phenylbutyl)-N-isobutyl-4-nitrobenzene sulphonamide hydrochloride (65.08 kg, 142.10 mol) was added and the mixture heated to reflux for approximately 22 hours. The solution was cooled slightly, and denatured ethanol (98 l) was added. The solution was stirred at 60° C. for 10 minutes then cooled and the product allowed to crystallise. The mixture was cooled to <10° C. and stirred for 2 hours. The product was isolated by filtration, washed with denatured ethanol (33 l) and dried at approximately 50° C., under vacuum to give (3S)-tetrahydro-3-furyl N-[(1S,2R)-1-benzyl-2-hydroxy-3-(N-isobutyl-4-nitrobenzene sulphonamido)propyl]carbamate in a yield of 82% of theory.
NMR: 1H NMR (500 Mhz, dmso-d6): 8.38(2H, d, J=9Hz), 8.06(2H, d, J=9 Hz), 7.20(6H, m), 5.02(1H, d, J=5 Hz), 4.94(1H, m), 4.35(EtOH, broad s), 3.71(EtOH, q), 3.65(1H, m), 3.60(1H, m), 3.51(2H, broad m), 3.40(2H, m), 3.15(1H, dd, J=8 Hz and 14 Hz), 3.07(1H, dd, J=8 Hz and 15 Hz), 2.94(2H, m), 2.48(1H, m), 2.06(1H, m), 1.97(1H, m), 1.78(1H, m), 1.05(EtOH, t), 0.83(6H, dd, J=7 Hz and 16 Hz).
Product from the above stage (80.0 g, 149.4 mmol) was hydrogenated in isopropanol (880 ml) with 5% palladium on carbon (16 g, of a wet paste) and hydrogen pressure (approx 0.5 to 1.5 bar) at 25-50° C. for approximately 5 hours. The mixture was cooled and the catalyst removed by filtration. The solution was distilled to a volume of approximately 320 ml and water (80 ml) was added. This solution was divided into two for the crystallisation step.
To half of the above solution, decolourising charcoal (2 g) was added, the mixture stirred at approximately 32° C. for 4 hours, then filtered. The filtercake was washed with isopropanol (20 ml) then further water (40 ml) was added to the filtrate. The solution was seeded to induce crystallisation and stirred for 5 hours. Water (130 ml) was added slowly over 1 hour then the mixture was stirred for 4 hours. The resultant slurry was cooled to approximately 20° C. and the product was isolated by filtration and washed with a 1:4 mixture of isopropano/water (120 ml). The product was dried at approximately 50° C., under vacuum, for approximately 12 hours to give (3S)-tetrahydro-3-furyl N-[(1S,2R)-3-(4-amino-N-isobutylbenzenesulphonamido)-1-benzyl-2-hydroxypropyl] carbamate (30.3 g; 80% of theory yield).
NMR: 1H NMR (300 Mhz, dmso-d6): 7.39(2H, d, J=9 Hz), 7.18(6H, m), 6.60(2H, d, J=9 Hz), 6.00(2H, s), 4.99(1H, d, J=6 Hz), 4.93(1H, ddt), 3.64(5H, m), 3.34(1H, m), 3.28(1H, dd, J=14 Hz and 3 Hz), 3.01(1H, m, J=14 Hz and 3 Hz), 2.91(1H, m), 2.66(2H, m), 2.50(1H, m), 2.05(1H, m), 1.94(1H, m), 1.78(1H, m), 0.81(6H, dd, J=16 Hz and 7 Hz). m/z: 506.2(M+H+)
Example 11Synthesis of Amprenavir (1)To a solution of carbamate nitro derivative 15 (0.05 g, 0.09 mmol) in 2 mL of EtOAc was added SnCl2.2H2O (0.1 g, 0.5 mmol) at 70° C. The reaction mixture was heated for 1 h until starting material disappeared and the solution cooled to room temperature. It was then poured into saturated aq. NaHCO3 solution and extracted with EtOAc. The organic extract was dried over anhyd. Na2SO4 and concentrated under reduced pressure. It was purified over chromatography using petroleum ether:EtOAc (3:2) to give amprenavir 1 (0.04 g, 90%).IR: (CHCl3, cm−1): υmax 757, 1090, 1149, 1316, 1504, 1597, 1633, 1705, 2960, 3371; 1H NMR (200 MHz, CDC3): δ 0.86 (d, J=5.7 Hz, 3H), 0.90 (d, J=6.6 Hz, 3H), 1.78-2.21 (m, 3H), 235-3.11 (m, 6H), 3.58-4.11 (m, 7H), 4.25 (s, 2H), 5.01 (br s, 1H), 5.07 (br s, 1H), 6.65 (d, J=8.4 Hz, 2H), 7.20-7.28 (m, 5H), 7.51 (d, J=8.4 Hz, 2H);13C NMR (50 MHz, CDC3): δ 19.9, 20.2, 27.3, 32.8, 35.4, 35.7, 53.8, 55.0, 58.6, 66.8, 72.6, 73.2, 75.3, 114.0, 125.9, 126.5, 1280.4, 129.5, 137.7, 150.9, 155.9;
Anal. Calcd for C25H35N3O6S: C, 59.39; H, 6.98; N, 8.31; S, 6.34. Found: C, 59.36; H, 6.81; N, 8.25; S, 6.29%.
FDA Approves Vitekta (elvitegravir) for HIV-1 Infection
September 24, 2014 — The U.S. Food and Drug Administration (FDA) has approved Vitekta (elvitegravir), an integrase strand transfer inhibitor for the combination treatment of human immunodeficiency virus type 1 (HIV-1) infection in treatment-experienced adults.
Elvitegravir (EVG, formerly GS-9137) is a drug used for the treatment of HIV infection. It acts as an integrase inhibitor. It was developed[1] by the pharmaceutical company Gilead Sciences, which licensed EVG from Japan Tobacco in March 2008.[2][3][4] The drug gained approval by U.S. Food and Drug Administration on August 27, 2012 for use in adult patients starting HIV treatment for the first time as part of the fixed dose combination known as Stribild.[5]
According to the results of the phase II clinical trial, patients taking once-daily elvitegravir boosted by ritonavir had greater reductions in viral load after 24 weeks compared to individuals randomized to receive a ritonavir-boosted protease inhibitor.[6]
。
Human immunodeficiency virus type 1 (HIV-1) is the causative agent of acquired immunodeficiency disease syndrome (AIDS). After over 26 years of efforts, there is still not a therapeutic cure or an effective vaccine against HIV/AIDS. The clinical management of HIV-1 infected people largely relies on antiretroviral therapy (ART). Although highly active antiretroviral therapy (HAART) has provided an effective way to treat AIDS patients, the huge burden of ART in developing countries, together with the increasing incidence of drug resistant viruses among treated people, calls for continuous efforts for the development of anti-HIV-1 drugs. Currently, four classes of over 30 licensed antiretrovirals (ARVs) and combination regimens of these ARVs are in use clinically including: reverse transcriptase inhibitors (RTIs) (e.g. nucleoside reverse transcriptase inhibitors, NRTIs; and non-nucleoside reverse transcriptase inhibitors, NNRTIs), protease inhibitors (PIs), integrase inhibitors and entry inhibitors (e.g. fusion inhibitors and CCR5 antagonists).
The life cycle of HIV-1. 1. HIV-1 gp120 binds to CD4 and co-receptor CCR5/CXCR4 on target cell; 2. HIV-1 gp41 mediates fusion with target cell; 3. Nucleocapsid containing viral genome and enzymes enters cells; 4. Viral genome and enzymes are released; 5. Viral reverse transcriptase catalyzes reverse transcription of ssRNA, forming RNA-DNA hybrids; 6. RNA template is degraded by ribonuclease H followed by the synthesis of HIV dsDNA; 7. Viral dsDNA is transported into the nucleus and integrated into the host chromosomal DNA by the viral integrase enzyme; 8. Transcription of proviral DNA into genomic ssRNA and mRNAs formation after processing; 9. Viral RNA is exported to cytoplasm; 10. Synthesis of viral precursor proteins under the catalysis of host-cell ribosomes; 11. Viral protease cleaves the precursors into viral proteins; 12. HIV ssRNA and proteins assemble under host cell membrane, into which gp120 and gp41 are inserted; 13. Membrane of host-cell buds out, forming the viral envelope; 14. Matured viral particle is released
Elvitegravir, also known as GS 9137 or JTK 303, is an investigational new drug and a novel oral integrase inhibitor that is being evaluated for the treatment of HIV-1 infection. After HIVs genetic material is deposited inside a cell, its RNA must be converted (reverse transcribed) into DNA. A viral enzyme called integrase then helps to hide HIVs DNA inside the cell’s DNA. Once this happens, the cell can begin producing genetic material for new viruses. Integrase inhibitors, such as elvitegravir, are designed to block the activity of the integrase enzyme and to prevent HIV DNA from entering healthy cell DNA. Elvitegravir has the chemical name: 6-(3-chloro-2-fluorobenzyl)-1-[(S)-1 -hydroxy -methyl-2- methylpropyl]-7-methoxy-4-oxo-1, 4-dihydroquinoline-3-carboxylic acid and has the following structural formula:
WO 2000040561 , WO 2000040563 and WO 2001098275 disclose 4-oxo-1 , 4-dihydro-3- quinoline which is useful as antiviral agents. WO2004046115 provides certain 4- oxoquinoline compounds that are useful as HIV Integrase inhibitors.
US 7176220 patent discloses elvitegravir, solvate, stereoisomer, tautomer, pharmaceutically acceptable salt thereof or pharmaceutical composition containing them and their method of treatment. The chemistry involved in the above said patent is depicted below in the Scheme A. Scheme-A
Toluene, DIPEA
SOCl2 ,COCl (S)-(+)-Valinol
Toluene
,4-Difluoro-5-iodo- benzoic acid
THF
dichlorobis(triphenylphosphine)
palladium argon stream,
Elvitegravir Form ] Elvitegravir (residue) US 7635704 patent discloses certain specific crystalline forms of elvitegravir. The specific crystalline forms are reported to have superior physical and chemical stability compared to other physical forms of the compound. Further, process for the preparation of elvitegravir also disclosed and is depicted below in the Scheme B. The given processes involve the isolation of the intermediates at almost all the stages.
Scheme B
2,
–
Zn THF,
CK Br THF CU “ZnBr dιchlorobis(trιphenylphos
phine)palladium
Elvitegravir WO 2007102499 discloses a compound which is useful as an intermediate for the synthesis of an anti-HIV agent having an integrase-inhibiting activity; a process for production of the compound; and a process for production of an anti-HIV agent using the intermediate.
WO 2009036161 also discloses synthetic processes and synthetic intermediates that can be used to prepare 4-oxoquinolone compounds having useful integrase inhibiting properties.
The said processes are tedious in making and the purity of the final compound is affected because of the number of steps, their isolation, purification etc., thus, there is a need for new synthetic methods for producing elvitegravir which process is cost effective, easy to practice, increase the yield and purity of the final compound, or that eliminate the use of toxic or costly reagents.
US Patent No 7176220 discloses Elvitegravir, solvate, stereoisomer, tautomer, pharmaceutically acceptable salt thereof or pharmaceutical composition containing them and ■ their method of treatment. US Patent No 7635704 discloses Elvitegravir Form II, Form III and processes for their preparation. The process for the preparation of Form Il disclosed in the said patent is mainly by three methods – a) dissolution of Elvitegravir followed by seeding with Form II, b) recrystallisation of Elvitegravir, and c) anti-solvent method.
The process for the preparation of Form III in the said patent is mainly by three methods – a) dissolution of Form Il in isobutyl acetate by heating followed by cooling the reaction mass, b) dissolution of Form Il in isobutyl acetate by heating followed by seeding with Form III, and c) dissolving Form Il in 2-propanol followed by seeding with Form III.
Amorphous materials are becoming more prevalent in the pharmaceutical industry. In order to overcome the solubility and potential bioavailability issues, amorphous solid forms are becoming front-runners. Of special importance is the distinction between amorphous and crystalline forms, as they have differing implications on drug substance stability, as well as drug product stability and efficacy.
An estimated 50% of all drug molecules used in medicinal therapy are administered as salts. A drug substance often has certain suboptimal physicochemical or biopharmaceutical properties that can be overcome by pairing a basic or acidic drug molecule with a counter- ion to create a salt version of the drug. The process is a simple way to modify the properties of a drug with ionizable functional groups to overcome undesirable features of the parent drug. Salt forms of drugs have a large effect on the drugs’ quality, safety, and performance. The properties of salt-forming species significantly affect the pharmaceutical properties of a drug and can greatly benefit chemists and formulators in various facets of drug discovery and development.
chemical synthesis from a carboxylic acid 1 starts after conversion to the acid chloride iodide NIS 2 , and with three condensation 4 . 4 and the amino alcohol 5 addition-elimination reaction occurs 6 , 6 off under alkaline conditions with TBS protected hydroxy get the ring 7 , 7 and zinc reagent 8 Negishi coupling occurs to get 9 , the last 9 hydrolysis and methoxylated
After isolation of the elvitegravir from the mixture of ethyl acetate-hexane, solvent from the filtrate was removed under reduced pressure. The resultant residue purified by column chromatography using a mixture of ethyl acetate-hexane (gradient, 20-80% EtOAc in hexane) as an eluent. Upon concentration of the required fractions, a thick solid was obtained which was further purified on slurry washing with ethyl acetate to get pure elvitegravir dimer impurity (13). The 1H-NMR, 13C-NMR and mass spectral data complies with proposed structure.
Abacavir (ABC) is a powerful nucleoside analog reverse transcriptase inhibitor (NRTI) used to treat HIV and AIDS. [Wikipedia] Chemically, it is a synthetic carbocyclic nucleoside and is the enantiomer with 1S, 4R absolute configuration on the cyclopentene ring. In vivo, abacavir sulfate dissociates to its free base, abacavir.
Abacavir (ABC) i/ʌ.bæk.ʌ.vɪər/ is a nucleoside analog reverse transcriptase inhibitor (NRTI) used to treat HIV and AIDS. It is available under the trade name Ziagen (ViiV Healthcare) and in the combination formulations Trizivir (abacavir, zidovudine andlamivudine) and Kivexa/Epzicom (abacavir and lamivudine). It has been well tolerated: the main side effect is hypersensitivity, which can be severe, and in rare cases, fatal. Genetic testing can indicate whether an individual will be hypersensitive; over 90% of patients can safely take abacavir. However, in a separate study, the risk of heart attack increased by nearly 90%.[1]
Viral strains that are resistant to zidovudine (AZT) orlamivudine (3TC) are generally sensitive to abacavir (ABC), whereas some strains that are resistant to AZT and 3TC are not as sensitive to abacavir.
Abacavir is a nucleoside reverse transcriptase inhibitor (NRTI) with activity against Human Immunodeficiency Virus Type 1 (HIV-1). Abacavir is phosphorylated to active metabolites that compete for incorporation into viral DNA. They inhibit the HIV reverse transcriptase enzyme competitively and act as a chain terminator of DNA synthesis. The concentration of drug necessary to effect viral replication by 50 percent (EC50) ranged from 3.7 to 5.8 μM (1 μM = 0.28 mcg/mL) and 0.07 to 1.0 μM against HIV-1IIIB and HIV-1BaL, respectively, and was 0.26 ± 0.18 μM against 8 clinical isolates. Abacavir had synergistic activity in cell culture in combination with the nucleoside reverse transcriptase inhibitor (NRTI) zidovudine, the non-nucleoside reverse transcriptase inhibitor (NNRTI) nevirapine, and the protease inhibitor (PI) amprenavir; and additive activity in combination with the NRTIs didanosine, emtricitabine, lamivudine, stavudine, tenofovir, and zalcitabine.
Abacavir is a carbocyclic synthetic nucleoside analogue and an antiviral agent. Intracellularly, abacavir is converted by cellular enzymes to the active metabolite carbovir triphosphate, an analogue of deoxyguanosine-5′-triphosphate (dGTP). Carbovir triphosphate inhibits the activity of HIV-1 reverse transcriptase (RT) both by competing with the natural substrate dGTP and by its incorporation into viral DNA. Viral DNA growth is terminated because the incorporated nucleotide lacks a 3′-OH group, which is needed to form the 5′ to 3′ phosphodiester linkage essential for DNA chain elongation.
Application
an antiviral agent, is used in the treatment of AIDS
Abacavir, (-) cis-[4-[2-amino-6-cyclopropylamino)-9H-purin-9-yl]-2-cyclopenten-yl]-1 – methanol, a carbocyclic nucleoside which possesses a 2,3-dehydrocyclopentene ring, is referred to in United States Patent 5,034,394 as a reverse transcriptase inhibitor. Recently, a general synthetic strategy for the preparation of this type of compound and intermediates was reported [Crimmins, et. al., J. Org. Chem., 61 , 4192-4193 (1996) and 65, 8499-8509-4193 (2000)].
Abacavir is the International Nonproprietary Name (INN) of {(1S,4R)-4-[2-amino-6-(cyclopropylamino)-9H-purin-9-yl]-cyclopent-2-enyl}methanol and CAS No. 136470-78-5. Abacavir and therapeutically acceptable salts thereof, in particular the hemisulfate salt, are well-known as potent selective inhibitors of HIV-1 and HIV-2, and can be used in the treatment of human immunodeficiency virus (HIV) infection.
The structure of abacavir corresponds to formula (I):
EP 434450-A discloses certain 9-substituted-2-aminopurines including abacavir and its salts, methods for their preparation, and pharmaceutical compositions using these compounds.
Different preparation processes of abacavir are known in the art. In some of them abacavir is obtained starting from an appropriate pyrimidine compound, coupling it with a sugar analogue residue, followed by a cyclisation to form the imidazole ring and a final introduction of the cyclopropylamino group at the 6 position of the purine ring.
According to the teachings of EP 434450-A , the abacavir base is finally isolated by trituration using acetonitrile (ACN) or by chromatography, and subsequently it can be transformed to a salt of abacavir by reaction with the corresponding acid. Such isolation methods (trituration and chromatography) usually are limited to laboratory scale because they are not appropriate for industrial use. Furthermore, the isolation of the abacavir base by trituration using acetonitrile gives a gummy solid (Example 7) and the isolation by chromatography (eluted from methanol/ethyl acetate) yields a solid foam (Example 19 or 28).
Other documents also describe the isolation of abacavir by trituration or chromatography, but always a gummy solid or solid foam is obtained (cf. WO9921861 and EP741710 ), which would be difficult to operate on industrial scale.
WO9852949 describes the preparation of abacavir which is isolated from acetone. According to this document the manufacture of the abacavir free base produces an amorphous solid which traps solvents and is, therefore, unsuitable for large scale purification, or for formulation, without additional purification procedures (cf. page 1 of WO 9852949 ). In this document, it is proposed the use of a salt of abacavir, in particular the hemisulfate salt which shows improved physical properties regarding the abacavir base known in the art. Said properties allow the manufacture of the salt on industrial scale, and in particular its use for the preparation of pharmaceutical formulations.
However, the preparation of a salt of abacavir involves an extra processing step of preparing the salt, increasing the cost and the time to manufacture the compound. Generally, the abacavir free base is the precursor compound for the preparation of the salt. Thus, depending on the preparation process used for the preparation of the salt, the isolation step of the abacavir free base must also be done.
EXAMPLE 21(-)-cis-4-[2-Amino-6-(cyclopropylmethylamino)-9H-purin-9-yl]-2-cyclopentene-1-methanol
The title compound of Example 7, (2.00 g, 6.50 mmol) was dissolved in 1,3-dimethyl-3,4,5,6-tetrahydro-2(1H)-pyrimidinone (Aldrich, 20 mL). Phosphoryl chloride (2.28 mL, 24.0 mmol) was added to the stirred, cooled (-10° C.) solution. After 3 minutes, cold water (80 mL) was added. The solution was extracted with chloroform (3×80 mL). The aqueous layer was diluted with ethanol (400 mL) and the pH adjusted to 6 with saturated aqueous NaOH. The precipitated inorganic salts were filtered off. The filtrate was further diluted with ethanol to a volume of 1 liter and the pH adjusted to 8 with additional NaOH. The resulting precipitate was filtered and dried to give the 5′-monophosphate of (±)-cis-4-[2-amino-6-(cyclopropylmethylamino)-9H-purin-9-yl]-2-cyclopentene-1-methanol as white powder (4.0 mmoles, 62% quantitated by UV absorbance); HPLC analysis as in Example 17 shows one peak. This racemic 5′ -monophosphate was dissolved in water (200 mL) and snake venom 5′-nucleotidase (EC 3.1.3.5) from Crotalus atrox (5,000 IU, Sigma) was added. After incubation at 37° C. for 10 days, HPLC analysis as in Example 17 showed that 50% of the starting nucleotide had been dephosphorylated to the nucleoside. These were separated on a 5×14 cm column of DEAE Sephadex A25 (Pharmacia) which had been preequilibrated with 50 mM ammonium bicarbonate. Title compound was eluted with 2 liters of 50 mM ammonium bicarbonate. Evaporation of water gave white powder which was dissolved in methanol, adsorbed on silica gel, and applied to a silica gel column. Title compound was eluted with methanol:chloroform/1:9 as a colorless glass. An acetonitrile solution was evaporated to give white solid foam, dried at 0.3 mm Hg over P2 O5 ; 649 mg (72% from racemate); 1 H-NMR in DMSO-d6 and mass spectrum identical with those of the racemate (title compound of Example 7); [α]20D -48.0°, [α]20436 -97.1°, [α]20365 -149° (c=0.14, methanol).
Anal. Calcd. for C15 H20 N6 O.0.10CH3 CN: C, 59.96; H, 6.72; N, 28.06. Found: C, 59.93; H, 6.76; N, 28.03.
Continued elution of the Sephadex column with 2 liters of 100 mM ammonium bicarbonate and then with 2 liters of 200 mM ammonium bicarbonate gave 5′-monophosphate (see Example 22) which was stable to 5′-nucleotidase.
…………………………………………
Синтез a)
Синтез b)
Preparation c)
Synthesis d)
An enantiopure β-lactam with a suitably disposed electron withdrawing group on nitrogen, participated in a π-allylpalladium mediated reaction with 2,6-dichloropurine tetrabutylammonium salt to afford an advanced cis-1,4-substituted cyclopentenoid with both high regio- and stereoselectivity. This advanced intermediate was successfully manipulated to the total synthesis of (−)-Abacavir.
Example 1: Preparation of crystalline Form I of abacavir base using methanol as solvent
[0026]
Abacavir (1.00 g, containing about 17% of dichloromethane) was dissolved in refluxing methanol (2.2 mL). The solution was slowly cooled to – 5 °C and, the resulting suspension, was kept at that temperature overnight under gentle stirring. The mixture was filtered off and dried under vacuum (7-10 mbar) at 40 °C for 4 hours to give a white solid (0.55 g, 66% yield, < 5000 ppm of methanol). The PXRD analysis gave the diffractogram shown in FIG. 1.
Abacavir, is the International Nonproprietary Name (INN) of {(1 S,4R)-4-[2- amino-6-(cyclopropylamino)-9H-purin-9-yl]-cyclopent-2-enyl}methanol, and CAS No. 136470-78-5. Abacavir sulfate is a potent selective inhibitor of HIV-1 and HIV-2, and can be used in the treatment of human immunodeficiency virus (HIV) infection.
The structure of abacavir hemisulfate salt corresponds to formula (I):
(I)
EP 434450-A discloses certain 9-substituted-2-aminopuhnes including abacavir and its salts, methods for their preparation, and pharmaceutical compositions using these compounds.
Different preparation processes of abacavir are known in the art. In some of them abacavir is obtained starting from an appropriate pyrimidine compound, coupling it with a sugar analogue residue, followed by a cyclisation to form the imidazole ring and a final introduction of the cyclopropylamino group at the 6 position of the purine ring. Pyrimidine compounds which have been identified as being useful as intermediates of said preparation processes include N-2-acylated abacavir intermediates such as N-{6- (cyclopropylamino)-9-[(1 R,4S)-4-(hydroxymethyl)cyclopent-2-enyl]-9H-purin- 2-yl}acetamide or N-{6-(cyclopropylamino)-9-[(1 R,4S)-4-
(hydroxymethyl)cyclopent-2-enyl]-9H-purin-2-yl}isobutyramide. The removal of the amino protective group of these compounds using acidic conditions is known in the art. According to Example 28 of EP 434450-A, the amino protective group of the N-{6-(cyclopropylamino)-9-[(1 R,4S)-4- (hydroxymethyl)cyclopent-2-enyl]-9H-purin-2-yl}isobutyramide is removed by stirring with 1 N hydrochloric acid for 2 days at room temperature. The abacavir base, after adjusting the pH to 7.0 and evaporation of the solvent, is finally isolated by trituration and chromatography. Then, it is transformed by reaction with an acid to the corresponding salt of abacavir. The main disadvantages of this method are: (i) the use of a strongly corrosive mineral acid to remove the amino protective group; (ii) the need of a high dilution rate; (iii) a long reaction time to complete the reaction; (iv) the need of isolating the free abacavir; and (v) a complicated chromatographic purification process.
Thus, despite the teaching of this prior art document, the research of new deprotection processes of a N-acylated {(1 S,4R)-4-[2-amino-6- (cyclopropylamino)-9H-purin-9-yl]-cyclopent-2-enyl}methanol is still an active field, since the industrial exploitation of the known process is difficult, as it has pointed out above. Thus, the provision of a new process for the removal of the amino protective group of a N-acylated {(1 S,4R)-4-[2-amino-6-
(cyclopropylamino)-9H-purin-9-yl]-cyclopent-2-enyl}methanol is desirable.
Example 1 : Preparation of abacavir hemisulfate
N-{6-(cyclopropylamino)-9-[(1 R,4S)-4-(hydroxymethyl)cyclopent-2-enyl]-9H- purin-2-yl}isobutyramide (6.56 g, 18.40 mmol) was slurried in a mixture of isopropanol (32.8 ml) and 10% solution of NaOH (36.1 ml, 92.0 mmol). The mixture was refluxed for 1 h. The resulting solution was cooled to 20-25 0C and tert-butyl methyl ether (32.8 ml) was added. The layers were separated and H2SO4 96% (0.61 ml, 11.03 mmol) was added dropwise to the organic layer. This mixture was cooled to 0-50C and the resulting slurry filtered off.
The solid was dried under vacuum at 40 0C. Abacavir hemisulfate (5.98 g, 97%) was obtained as a white powder.
Example 6: Preparation of abacavir
N-{6-(cyclopropylamino)-9-[(1 R,4S)-4-(hydroxymethyl)cyclopent-2-enyl]-9H- purin-2-yl}isobutyramide (1.0 g, 2.80 mmol) was slurried in a mixture of isopropanol (2 ml) and 10% solution of NaOH (1.1 ml, 2.80 mmol). The mixture was refluxed for 1 h. The resulting solution was cooled to 20-25 0C and tert-butyl methyl ether (2 ml) was added. The aqueous layer was discarded, the organic phase was cooled to 0-5 0C and the resulting slurry filtered off. The solid was dried under vacuum at 400C. Abacavir (0.62 g, 77%) was obtained as a white powder.
Example 7: Preparation of abacavir
N-{6-(cyclopropylamino)-9-[(1 R,4S)-4-(hydroxymethyl)cyclopent-2-enyl]-9H- purin-2-yl}isobutyramide (1.25 g, 3.51 mmol) was slurried in a mixture of isopropanol (2.5 ml) and 10% solution of NaOH (1.37 ml, 3.51 mmol). The mixture was refluxed for 1 h and concentrated to dryness. The residue was crystallized in acetone. Abacavir (0.47 g, 47%) was obtained as a white powder.
Example 8: Preparation of abacavir
N-{6-(cyclopropylamino)-9-[(1 R,4S)-4-(hydroxymethyl)cyclopent-2-enyl]-9H- purin-2-yl}isobutyramide (1.25 g, 3.51 mmol) was slurried in a mixture of isopropanol (2.5 ml) and 10% solution of NaOH (1.37 ml, 3.51 mmol). The mixture was refluxed for 1 h and concentrated to dryness. The residue was crystallized in acetonitrile. Abacavir (0.43 g, 43%) was obtained as a white powder.
Example 9: Preparation of abacavir
A mixture of N-{6-(cyclopropylamino)-9-[(1 R,4S)-4-(hydroxymethyl)cyclopent- 2-enyl]-9H-purin-2-yl}isobutyramide (10 g, 28 mmol), isopropanol (100 ml) and 10% solution of NaOH (16.8 ml, 42 mmol) was refluxed for 1 h. The resulting solution was cooled to 20-25 0C and washed several times with 25% solution of NaOH (10 ml). The wet organic layer was neutralized to pH 7.0-7.5 with 17% hydrochloric acid and it was concentrated to dryness under vacuum. The residue was crystallized in ethyl acetate (150 ml) to afford abacavir (7.2 g, 90%).
Example 10: Preparation of abacavir
A mixture of N-{6-(cyclopropylamino)-9-[(1 R,4S)-4-(hydroxymethyl)cyclopent- 2-enyl]-9H-purin-2-yl}isobutyramide (10 g, 28 mmol), isopropanol (100 ml) and 10% solution of NaOH (16.8 ml, 42 mmol) was refluxed for 1 h. The resulting solution was cooled to 20-25 0C and washed several times with 25% solution of NaOH (10 ml). The wet organic layer was neutralized to pH 7.0-7.5 with 17% hydrochloric acid and it was concentrated to dryness under vacuum. The residue was crystallized in acetone (300 ml) to afford abacavir (7.0 g, 88%).
or (1 S,4R)-4-[2-Amino-6-(cyclopropylamino)-9H-purin-9-yl]-2-cyclopentene-1 – methanol and its salts are nucleoside reverse transcriptase inhibitors. Abacavir sulfate is a nucleoside reverse transcriptase inhibitor and used in the treatment of human immunodeficiency virus infection. Abacavir sulfate and related compounds and their therapeutic uses are disclosed in US 5,034,394.
Crystalline forms of abacavir sulfate have not been reported in the literature. Moreover, the processes described in the literature do not produce abacavir sulfate in a stable, well-defined and reproducible crystalline form. It has now been discovered that abacavir sulfate can be prepared in three stable, well-defined and consistently reproducible crystalline forms.
Example 1
Abacavir free base (3.0 gm, obtained by the process described in example 21 of US 5,034,394) is dissolved in ethyl acetate (15 ml) and cone, sulfuric acid (0.3 ml) is added to the solution. Then the contents are stirred for 3 hours at 20°C and filtered to give 3.0 gm of form I abacavir sulfate. Example 2 Abacavir free base (3.0 gm) is dissolved in acetone (20 ml) and cone, sulfuric acid (0.3 ml) is added to the solution. Then the contents are stirred for 6 hours at 25°C and filtered to give 2.8 gm of form I abacavir sulfate.
Example 3 Abacavir free base (3.0 gm) is dissolved in acetonitrile (15 ml) and sulfuric acid (0.3 ml) is added to the solution. Then the contents are stirred for 2 hours at 25°C and the separated solid is filtered to give 3.0 gm of form II abacavir sulfate.
Example 4 Abacavir free base (3.0 gm) is dissolved in methyl tert-butyl ether (25 ml) and sulfuric acid (0.3 ml) is added to the solution. Then the contents are stirred for 1 hours at 25°C and the separated solid is filtered to give 3.0 gm of form II abacavir sulfate.
Example 5 Abacavir free base (3.0 gm) is dissolved in methanol (15 ml) and sulfuric acid (0.3 ml) is added to the solution. The contents then are cooled to 0°C and diisopropyl ether (15 ml) is added. The reaction mass is stirred for 2 hours at about 25°C and the separated solid is filtered to give 3.0 gm of form III abacavir sulfate
The present invention relates to a new process for the preparation of the chiral nucleoside analogue (1S, 4R)-4-[2-amino-6-(cyclopropylamino)-9H purin-9-yl]-2- cyclopentene-1 -methanol (compound of Formula (I)).
The compound of formula (I) is described as having potent activity against human immunodeficiency virus (HIV) and hepatitis B virus (HBV) in EPO34450.
Results presented at the 34th Interscience Conference on Antimicrobial Agents and Chemotherapy (October 4-7, 1994) demonstrate that the compound of formula I has significant activity against HIV comparable to, and if not better than, some current anti HIV drugs, such as zidovudine and didanosine.
Currently the compound of Formula (I) is undergoing clinical investigation to determine its safety and efficacy in humans. Therefore, there exists at the present time a need to supply large quantities of this compound for use in clinical trials.
Current routes of synthesising the compound of formula (I) involve multiple steps and are relatively expensive. It will be noted that the compound has two centres of asymmetry and it is essential that any route produces the compound of formula (I) substantially free of the corresponding enantiomer, preferably the compound of formula (I) is greater than 95% w/w free of the corresponding enantiomer.
Processes proposed for the preparation of the compound of formula (I) generally start from a pyrimidine compound, coupling with a 4-amino-2-cyclopentene-1- methanol analogue, cyciisation to form the imidazole ring and then introduction of the cyclopropylamine group into the 6 position of the purine, such routes include those suggested in EPO434450 and WO9521161. Essentially both routes disclosed in the two prior patent applications involve the following steps:-
(i) coupling (1S, 4R)-4-amino-2-cyclopentene-1 -methanol to N-(4,6-dichloro-5- formamido-2-pyrimidinyl) acetamide or a similar analogue thereof, for example N- (2-amino-4,6-dichloro-5-pyrimidinyl) formamide;
(ii) ring closure of the resultant compound to form the intermediate (1 S, 4R)-4- (2-amino-6-chloro-9H-purin-9-yl)-2-cyclopentene-1 -methanol;
(iii) substituting the halo group by a cyclopropylamino group on the 6 position of the purine ring.
The above routes are multi-step processes. By reducing the number of processing steps significant cost savings can be achieved due to the length of time to manufacture the compound being shortened and the waste streams minimised.
An alternative process suggested in the prior art involves the direct coupling of carbocyclic ribose analogues to the N atom on the 9 position of 2-amino-6-chloro purine. For example WO91/15490 discloses a single step process for the formation of the (1S, 4R)- 4-(2-amino-6-chloro-9H-purin-9-yl)-2-cyclopentene-1- methanol intermediate by reacting (1S, 4R)-4-hydroxy-2-cyclopentene-1 -methanol, in which the allylic hydroxyl group has been activated as an ester or carbonate and the other hydroxyl group has a blocking group attached (for example 1 ,4- bis- methylcarbonate) with 2-amino-6-chloropurine.
However we have found that when synthesising (1S, 4R)-4-(2-amino-6-chloro-9H- purin-9-yl)-2-cyclopentene-1- methanol by this route a significant amount of an N- 7 isomer is formed (i.e. coupling has occurred to the nitrogen at the 7- position of the purine ring) compared to the N-9 isomer desired. Further steps are therefore required to convert the N-7 product to the N-9 product, or alternatively removing the N-7 product, adding significantly to the cost. We have found that by using a transition metal catalysed process for the direct coupling of a compound of formula (II) or (III),
Example 1 (1 S. 4R)-4-[2-Amino-6-(cvclopropylamino)-9H purin-9-vπ-2-cvclopentene-1 – methanol
Triphenylphosphine (14mg) was added, under nitrogen, to a mixture of (1S.4R)- 4-hydroxy-2-cyclopentene -1 -methanol bis(methylcarbonate) (91 mg), 2-amino-6- (cyclopropylamino) purine (90mg), tris(dibenzylideneacetone)dipalladium (12mg) and dry DMF (2ml) and the resulting solution stirred at room temperature for 40 min.
The DMF was removed at 60° in vacuo and the residue partitioned between ethyl acetate (25ml.) and 20% sodium chloride solution (10ml.). The ethyl acetate solution was washed with 20% sodium chloride (2x12ml.) and with saturated sodium chloride solution, then dried (MgSO4) and the solvent removed in vacuo.
The residue was dissolved in methanol (10ml.), potassium carbonate (17mg) added and the mixture stirred under nitrogen for 15h.
The solvent was removed in vacuo and the residue chromatographed on silica gel
(Merck 9385), eluting with dichloromethane-methanol [(95:5) increasing to (90:10)] to give the title compound (53mg) as a cream foam.
Trizivir tablets 300 mg – abacavir in fixed combination with 150 mg of lamivudine and 300 mg zidovudine
ZIAGEN is the brand name for abacavir sulfate, a synthetic carbocyclic nucleoside analogue with inhibitory activity against HIV-1. The chemical name of abacavir sulfate is (1S,cis)-4-[2-amino-6-(cyclopropylamino)-9H-purin-9-yl]-2-cyclopentene-1-methanol sulfate (salt) (2:1). Abacavir sulfate is the enantiomer with 1S, 4R absolute configuration on the cyclopentene ring. It has a molecular formula of (C14H18N6O)2•H2SO4 and a molecular weight of 670.76 daltons. It has the following structural formula:
Abacavir sulfate is a white to off-white solid with a solubility of approximately 77 mg/mL in distilled water at 25°C. It has an octanol/water (pH 7.1 to 7.3) partition coefficient (log P) of approximately 1.20 at 25°C.
ZIAGEN Tablets are for oral administration. Each tablet contains abacavir sulfate equivalent to 300 mg of abacavir as active ingredient and the following inactive ingredients: colloidal silicon dioxide, magnesium stearate, microcrystalline cellulose, and sodium starch glycolate. The tablets are coated with a film that is made of hypromellose, polysorbate 80, synthetic yellow iron oxide, titanium dioxide, and triacetin.
ZIAGEN Oral Solution is for oral administration. Each milliliter (1 mL) of ZIAGEN Oral Solution contains abacavir sulfate equivalent to 20 mg of abacavir (i.e., 20 mg/mL) as active ingredient and the following inactive ingredients: artificial strawberry and banana flavors, citric acid (anhydrous), methylparaben and propylparaben (added as preservatives), propylene glycol, saccharin sodium, sodium citrate (dihydrate), sorbitol solution, and water.
In vivo, abacavir sulfate dissociates to its free base, abacavir. All dosages for ZIAGEN are expressed in terms of abacavir.
History
Abacavir was approved by the Food and Drug Administration (FDA) on December 18, 1998 and is thus the fifteenth approved antiretroviral drug in the United States. Its patent expired in the United States on 2009-12-26.
Links
US 5 089 500 (Burroughs Wellcome; 18.2.1992; GB-prior. 27.6.1988).
Synthesis a)
EP 434 450 (Wellcome Found .; 26.6.1991; appl. 21.12.1990; prior-USA. 22.12.1989).
Crimmins, MT et al .: J. Org. Chem. (JOCEAH) 61 4192 (1996).
EP 1 857 458 (Solmag; appl. 5.5.2006).
EP 424 064 (Enzymatix; appl. 24.4.1991; GB -prior. 16.10.1989).
Jump up^Mallal, S., Phillips, E., Carosi, G. et al. (2008). “HLA-B*5701 screening for hypersensitivity to abacavir”. New England Journal of Medicine358: 568–579.doi:10.1056/nejmoa0706135.
Jump up^Rauch, A., Nolan, D., Martin, A. et al. (2006). “Prospective genetic screening decreases the incidence of abacavir hypersensitivity reactions in the Western Australian HIV cohort study”. Clinical Infectious Diseases43: 99–102. doi:10.1086/504874.
Jump up^Heatherington et al. (2002). “Genetic variations in HLA-B region and hypersensitivity reactions to abacavir”. Lancet359: 1121–1122.
Jump up^Mallal et al. (2002). “Association between presence of HLA*B5701, HLA-DR7, and HLA-DQ3 and hypersensitivity to HIV-1 reverse-transcriptase inhibitor abacavir”. Lancet359: 727–732. doi:10.1016/s0140-6736(02)07873-x.
Jump up^Rotimi, C.N.; Jorde, L.B. (2010). “Ancestry and disease in the age of genomic medicine”. New England Journal of Medicine363: 1551–1558.
Jump up^Phillips, E., Mallal, S. (2009). “Successful translation of pharmacogenetics into the clinic”. Molecular Diagnosis & Therapy13: 1–9. doi:10.1007/bf03256308.
Jump up^Phillips, E., Mallal S. (2007). “Drug hypersensitivity in HIV”. Current Opinion in Allergy and Clinical Immunology7: 324–330. doi:10.1097/aci.0b013e32825ea68a.
Jump up^Swen JJ, Nijenhuis M, de Boer A et al. (May 2011). “Pharmacogenetics: from bench to byte–an update of guidelines”. Clin Pharmacol Ther.89 (5): 662–73.doi:10.1038/clpt.2011.34. PMID21412232.
Jump up^Shear, N.H., Milpied, B., Bruynzeel, D.P. et al. (2008). “A review of drug patch testing and implications for HIV clinicians”. AIDS22: 999–1007.doi:10.1097/qad.0b013e3282f7cb60.
Jump up^Ding X, Andraca-Carrera E, Cooper C et al. (December 2012). “No association of abacavir use with myocardial infarction: findings of an FDA meta-analysis”. J Acquir Immune Defic Syndr.61 (4): 441–7. doi:10.1097/QAI.0b013e31826f993c.PMID22932321.
Illing PT et al. 2012, Nature, doi:10.1038/nature11147
Carbocyclic nucleosides are synthetically the most challenging class of nucleosides, requiring multi-step and often elaborate synthetic pathways to introduce the necessary stereochemistry. There are two main strategies for the preparation of carbocyclic nucleosides. In the linear approach a cyclopentylamine is used as starting material and the heterocycle is built in a stepwise manner (see Scheme 1).
Scheme 1: Linear approach for the synthesis of abacavir.[5]
The more flexible strategy is a convergent approach: a functionalized carbocyclic moiety is condensed with a heterocycle rapidly leading to a variety of carbocyclic nucleosides. Initially, we started our syntheses from cyclopentadiene 1 that is deprotonated and alkylated with benzyloxymethyl chloride to give the diene 2. This material is converted by a hydroboration into cyclopentenol 3 or isomerized into two thermodynamically more stable cyclopentadienes 4a,b. With the protection and another hydroboration step to 5 we gain access to an enantiomerically pure precursor for the synthesis of a variety of carbocyclic 2’-deoxynucleosides e.g.:carba-dT, carba-dA or carba-BVDU.[6] The isomeric dienes 4a,b were hydroborated to the racemic carbocyclic moiety 6.
Scheme 2: Convergent approach for the synthesis of carba-dT.
The asymmetric synthesis route and the racemic route above are short and efficient ways to diverse carbocyclic D- or L-nucleosides (Scheme 2). Different heterocycles can be condensed to these precursors leading to carbocyclic purine- and pyrimidine-nucleosides. Beside α- and β-nucleosides, carbocyclic epi– andiso-nucleosides in the 2’-deoxyxylose form were accessable.[7]
What else is possible? The racemic cyclopentenol 6 can be coupled by a modified Mitsunobu-reaction.Moreover, this strategy offers the possibility of synthesizing new carbocyclic nucleosides by functionalizing the double bond before or after introduction of the nucleobase (scheme 3).[8]
Scheme 3: Functionalized carbocyclic nucleosides based on cyclopentenol 6.
Other interesting carbocyclic precursors like cyclopentenol 7 can be used to synthesize several classes of carbocyclic nucleoside analogues, e.g.: 2’,3’-dideoxy-2’,3’-didehydro nucleosides (d4-nucleosides), 2’,3’-dideoxynucleosides (ddNs), ribonucleosides, bicyclic nucleosides or even 2’-fluoro-nucleosides.
Scheme 4: Functionalized carbocyclic thymidine analogues based on cyclopentenol 7.
[1] V. E. Marquez, T. Ben-Kasus, J. J. Barchi, K. M. Green, M .C. Nicklaus, R. Agbaria, J. Am. Chem. Soc.2004,126, 543.
[2] A. D. Borthwick, K. Biggadike, Tetrahedron1992, 48, 571.
[3] H. Bricaud, P. Herdewijn, E. De Clercq, Biochem. Pharmacol.1983, 3583.
[4] P. L. Boyer, B. C. Vu, Z. Ambrose, J. G. Julias, S. Warnecke, C. Liao, C. Meier, V. E. Marquez, S. H. Hughes, J. Med. Chem.2009, 52, 5356.
[5] S. M. Daluge, M. T. Martin, B. R. Sickles, D. A. Livingston, Nucleosides, Nucleotides Nucleic Acids2000,19, 297.
[6] O. R. Ludek, C. Meier, Synthesis2003, 2101.
[7] O. R. Ludek, T. Kraemer, J. Balzarini, C. Meier, Synthesis2006, 1313.
[8] M. Mahler, B. Reichardt, P. Hartjen, J. van Lunzen, C. Meier, Chem. Eur. J.2012, 18, 11046-11062.
Novel hemi-succinate salt form of (2S)-2-tert-butoxy-2-(4-(2,3-dihydropyrano[4,3,2-de]quinolin-7-yl)-2-methylquinolin-3-yl)acetic acid (presumed to be BI-224436) and its crystalline forms is desc in WO-2014055618.
Gilead, under license from BI, was developing BI-224436 for the oral treatment of HIV infection. In September 2011, this drug had entered phase 1 trials. Picks up from WO2012138670, claiming a process for the preparation of the same drug. Also see the concurrently published WO2014055603. This compound is claimed specifically in WO2009062285 and generically in WO2007131350.
BI 224436 has antiviral EC50 values ranging between 4 and 15 nM against different HIV-1 laboratory strains and CC50 values >90 μM in different cells, including peripheral blood mononuclear cells. BI 224436 also has a low, 2.2-fold shift in antiviral potency in the presence of 50% human serum and by virtue of a steep dose-response curve slope, BI 224436 exhibits serum-shifted EC95 values ranging between 22 and 75 nM. Drug combination studies performed in cell-based antiviral assays have shown that BI 224436 displays, at the least, an additive effect in combination with any of the marketed antiviral classes including INSTIs. BI 224436 has drug-like ADME properties including a Caco-2 cell permeability of 14 .10-6 cm/sec, solubility > 24 mg/ml in the pH range 2.0-6.8 and low cytochrome P450 inhibition. Moreover BI 224436 shows excellent PK profiles in rat (CL=0.7% QH; F= 54%), monkey (CL= 23% QH; F= 82%) and dog (CL= 8%QH; F= 81%).
1a (600 g, 4.1 mol) was charged into a dry reactor under nitrogen followed by addition of Ac20 (1257.5 g, 12.3 mol, 3 eq.). The resulting mixture was heated at 40 °C at least for 2 hours. The batch was then cooled to 30 °C over 30 minutes. A suspension of 1b in toluene was added to seed the batch if no solid was observed. After toluene (600 ml_) was added over 30 minutes, the batch was cooled to -5— 10 °C and was held at this temperature for at least 30 minutes. The solid was collected by filtration under nitrogen and rinsed with heptanes (1200 ml_). After being dried under vacuum at room temperature, the solid was stored under nitrogen at least below 20 °C. The product 1 b was obtained with 77% yield. 1H NMR (500 MHz, CDCI3): δ = 6.36 (s, 1 H), 3.68 (s, 2H), 2.30 (s, 3H). Example 2
2a 2b
2a (100g, 531 mmol) and 1b (95 g, 558 mmol) were charged into a clean and dry reactor under nitrogen followed by addition of fluorobenzene (1000 mL). After being heated at 35-37 °C for 4 hours, the batch was cooled to 23 °C. Concentrated H2S04 (260.82 g, 2659.3 mmol, 5 eq.) was added while maintaining the batch temperature below 35 °C. The batch was first heated at 30-35 °C for 30 minutes and then at 40- 45 °C for 2 hours. 4-Methyl morpholine (215.19 g, 2127 mmol, 4 eq.) was added to the batch while maintaining the temperature below 50 °C. Then the batch was agitated for 30 minutes at 40-50 °C. eOH (100 mL) was then added while maintaining the temperature below 55 °C. After the batch was held at 50-55 °C for 2 hours, another portion of MeOH (100 mL) was added. The batch was agitated for another 2 hours at 50-55 °C. After fluorobenzene was distilled to a minimum amount, water (1000 mL) was added. Further distillation was performed to remove any remaining fluorobenzene. After the batch was cooled to 30 °C, the solid was collected by filtration with cloth and rinsed with water (400 mL) and heptane (200 mL). The solid was dried under vacuum below 50 °C to reach KF < 0.1%. Typically, the product 2b was obtained in 90% yield with 98 wt%. 1H NMR (500 MHz, DMSO- d6): δ = 10.83 (s, 1 H), 9.85 (s, bs, 1 H), 7.6 (d, 1 H, J
2b (20 g, 64 mmol) was charged into a clean and dry reactor followed by addition of THF (140 mL). After the resulting mixture was cooled to 0 °C, Vitride® (Red-AI, 47.84 g, 65 wt%, 154 mmol) in toluene was added while maintaining an internal temperature at 0-5 °C. After the batch was agitated at 5-10 °C for 4 hours, IPA (9.24 g, 153.8 mmol) was added while maintaining the temperature below 10 °C. Then the batch was agitated at least for 30 minutes below 25 °C. A solution of HCI in IPA (84.73 g, 5.5 M, 512 mmol) was added into the reactor while maintaining the temperature below 40 °C. After about 160 mL of the solvent was distilled under vacuum below 40 °C, the batch was cooled to 20-25 °C and then aqueous 6M HCI (60 mL) was added while maintaining the temperature below 40 °C. The batch was cooled to 25 °C and agitated for at least 30 minutes. The solid was collected by filtration, washed with 40 mL of IPA and water (1V/1V), 40 mL of water and 40 mL of heptanes. The solid was dried below 60 °C in a vacuum oven to reach KF < 0.5%. Typically, the product 3a was obtained in 90-95% yield with 95 wt%. 1H NMR (400 MHz, DMSO-d6): δ = 10.7 (s, 1 H), 9.68 (s, 1 H), 7.59 (d, 1 H, J = 8.7 Hz), 6.64 (, 1 H, J = 8.7 Hz), 6.27 (s, 1 H), 4.62 (bs, 1 H), 3.69 (t, 2H, J = 6.3 Hz), 3.21 (t, 2H, J = 6.3 Hz).
Example 4
3a (50 g, 174.756 mmol) and acetonitrile (200 mL) were charged into a dry and clean reactor. After the resulting mixture was heated to 65 °C, POCI3 (107.18 g, 699 mmol, 4 eq.) was added while maintaining the internal temperature below 75 °C. The batch was then heated at 70-75 °C for 5-6 hours. The batch was cooled to 20 °C. Water (400 mL) was added at least over 30 minutes while maintaining the internal temperature below 50 °C. After the batch was cooled to 20-25 °C over 30 minutes, the solid was collected by filtration and washed with water (100 mL). The wet cake was charged back into the reactor followed by addition of 1 M NaOH (150 mL). After the batch was agitated at least for 30 minutes at 25-35 °C, it was verified that the pH was greater than 12. Otherwise, more 6M NaOH was needed to adjust the pH >12. After the batch was agitated for 30 minutes at 25-35 °C, the solid was collected by filtration, washed with water (200 mL) and heptanes (200 mL). The solid was dried in a vacuum oven below 50 °C to reach KF < 2%. Typically, the product 4a was obtained at about 75-80% yield. H NMR (400 MHz, CDCI3): δ = 7.90 (d, 1 H, J = 8.4 Hz), 7.16 (s, 1 H), 6.89 (d, 1 H, J = 8.4 Hz), 4.44 (t, 2 H, J = 5.9 Hz), 3.23 (t, 2 H, J = 5.9 Hz). 13C NMR (100 MHz, CDCI3): δ = 152.9, 151.9, 144.9, 144.1 , 134.6, 1 19.1 , 1 17.0, 1 13.3, 1 1 1.9, 65.6, 28.3.
Example 5
4a 5a
Zn powder (54 g, 825 mmol, 2.5 eq.) and TFA (100 mL) were charged into a dry and clean reactor. The resulting mixture was heated to 60-65 °C. A suspension of 4a (100 g, 330 mmol) in 150 mL of TFA was added to the reactor while maintaining the temperature below 70 °C. The charge line was rinsed with TFA (50 mL) into the reactor. After 1 hour at 65±5 °C, the batch was cooled to 25-30 °C. Zn powder was filtered off by passing the batch through a Celite pad and washing with methanol (200 mL). About 400 mL of solvent was distilled off under vacuum. After the batch was cooled to 20-25 °C, 20% NaOAc (ca. 300 mL) was added at least over 30 minutes to reach pH 5-6. The solid was collected by filtration, washed with water (200 mL) and heptane (200 mL), and dried under vacuum below 45 °C to reach KF ≤ 2%. The solid was charged into a dry reactor followed by addition of loose carbon (10 wt%) and toluene (1000 mL). The batch was heated at least for 30 minutes at 45-50 °C. The carbon was filtered off above 35 °C and rinsed with toluene (200 mL). The filtrate was charged into a clean and dry reactor. After about 1000 mL of toluene was distilled off under vacuum below 50 °C, 1000 mL of heptane was added over 30 minutes at 40-50 °C. Then the batch was cooled to 0±5 °C over 30 minutes. After 30 minutes, the solid was collected and rinsed with 200 mL of heptane. The solid was dried under vacuum below 45 °C to reach KF≤ 500 ppm. Typically, the product 5a was obtained in about 90-95 % yield. 1H NMR (400 MHz, CDCI3): δ = 8.93 (m, 1 H), 7.91 (dd, 1 H, J = 1.5, 8 Hz), 7.17 (m 1 H), 6.90 (dd, 1 H, J = 1 .6, 8.0 Hz), 4.46-4.43 (m, 2 H), 3.28-3.23 (m, 2 H). 13C NMR (100 MHz, CDCI3): δ = 152.8, 151 .2, 145.1 , 141.0, 133.3, 1 18.5, 1 18.2, 1 14.5, 1 1 1.1 , 65.8, 28.4.
Example 6
5a 6a
5a (1.04 kg, 4.16 mol) and toluene (8 L) were charged into the reactor. The batch was agitated and cooled to -50 to -55 °C. BuLi solution (2.5 M in hexanes, 1.69 L, 4.23 mol) was charged slowly while maintaining the internal temperature between – 45 to -50 °C. The batch was agitated at -45 °C for 1 hour after addition. A solution of triisopropyl borate (0.85 kg, 4.5 mol) in MTBE (1 .48 kg) was charged. The batch was warmed to 10 °C over 30 minutes. A solution of 5 N HCI in I PA (1 .54 L) was charged slowly at 10 °C, and the batch was warmed to 20 °C and stirred for 30 minutes. It was seeded with 6a crystal (10 g). A solution of aqueous concentrated HCI (0.16 L) in IPA (0.16 L) was charged slowly at 20 °C in three portions at 20 minute intervals, and the batch was agitated for 1 hour at 20 °C. The solid was collected by filtration, rinsed with MTBE (1 kg), and dried to provide 6a (943 g, 88.7 % purity, 80% yield). 1H NMR (400 MHz, D20): δ 8.84 (d, 1 H, J = 4 Hz)
Iodine stock solution was prepared by mixing iodine (57.4 g, 0.23 mol) and sodium iodide (73.4 g, 0.49 mol) in water (270 mL). Sodium hydroxide (28.6 g, 0.715 mol) was charged into 220 mL of water. 4-Hydroxy-2 methylquinoline 7a (30 g, 0.19 mol) was charged, followed by acetonitrile (250 mL). The mixture was cooled to 10 °C with agitation. The above iodine stock solution was charged slowly over 30 minutes. The reaction was quenched by addition of sodium bisulfite (6.0 g) in water (60 mL). Acetic acid (23 mL) was charged over a period of 1 hour to adjust the pH of the reaction mixture between 6 and 7. The product was collected by filtration, washed with water and acetonitrile, and dried to give 7b (53 g, 98%). MS 286 [M + 1].
Example 8
7b 8a
4-Hydroxy-3-iodo-2-methylquinoline 7b (25 g, 0.09 mol) was charged to a 1-L reactor. Ethyl acetate (250 mL) was charged, followed by triethylamine (2.45 mL, 0.02 mol) and phosphorus oxychloride (12 mL, 0.13 mol). The reaction mixture was heated to reflux until complete conversion (~1 hour), then the mixture was cooled to 22 °C. A solution of sodium carbonate (3 .6 g, 0.3 mol) in water (500 mL) was charged. The mixture was stirred for 20 minutes. The aqueous layer was extracted with ethyl acetate (120 mL). The organic layers were combined and concentrated under vacuum to dryness. Acetone (50 mL) was charged. The solution was heated to 60 °C. Water (100 mL) was charged, and the mixture was cooled to 22 °C. The product was collected by filtration and dried to give 8a (25 g, 97.3 % pure, 91.4 % yield). MS 304 [M + 1].
(Note: 8a is a known compound with CAS # 1033931-93-9. See references: (a) J. Org Chem. 2008, 73, 4644-4649. (b) Molecules 2010, 15, 3171 -3178. (c) Indian J. Chem. Sec B: Org. Chem. Including Med Chem. 2009, 488(5), 692-696.)
Example 9
8a 9a
8a (100 g, 0.33 mol) was charged to the reactor, followed by copper (I) bromide dimethyl sulfide complex (3.4 g, 0.017 mol) and dry THF (450 mL). The batch was cooled to -15 to -12 °C. i-PrMgCI (2.0 M in THF, 173 mL, 0.346 mol) was charged into the reactor at the rate which maintained the batch temperature < -10 °C. In a 2nd reactor, methyl chlorooxoacetate (33 mL, 0.36 mol) and dry THF (150 mL) were charged. The solution was cooled to -15 to -10 °C. The content of the 1 st reactor (Grignard/cuprate) was charged into the 2nd reactor at the rate which maintained the batch temperature < -10 °C. The batch was agitated for 30 minutes at -10 °C. Aqueous ammonium chloride solution ( 0%, 300 mL) was charged. The batch was agitated at 20 – 25 °C for 20 minutes and allowed to settle for 20 minutes. The aqueous layer was separated. Aqueous ammonium chloride solution (10%, 90 mL) and sodium carbonate solution (10%, 135 mL) were charged to the reactor. The batch was agitated at 20 – 25 °C for 20 minutes and allowed to settle for 20 minutes. The aqueous layer was separated. Brine (10%, 240 mL) was charged to the reactor. The batch was agitated at 20 – 25 °C for 20 minutes. The aqueous layer was separated. The batch was concentrated under vacuum to -1/4 of the volume (about 80 mL left). 2-Propanol was charged (300 mL). The batch was concentrated under vacuum to -1/3 of the volume (about 140 mL left), and heated to 50 °C.
Water (70 mL) was charged. The batch was cooled to 20 – 25 °C, stirred for 2 hours, cooled to – 0 °C and stirred for another 2 hours. The solid was collected by filtration, washed with cold 2-propanol and water to provide 58.9 g of 9a obtained after drying (67.8 % yield). 1H NMR (400 MHz, CDCI3): δ 8.08 (d, 1 H, J = 12 Hz), 7.97 (d, 1 H, J = 12 Hz), 7.13 (t, 1 H, J = 8 Hz), 7.55 (t, 1 H, J= 8 Hz), 3.92 (s, 3H), 2.63 (s, 3H). 13C NMR (100 MHz, CDCI3): δ 186.6, 161.1 , 155.3, 148.2, 140.9, 132.0, 129.0, 128.8, 127.8, 123.8, 123.7, 53.7, 23.6.
Catalyst preparation: To a suitable sized, clean and dry reactor was charged dichloro(pentamethylcyclopentadienyl)rhodium (III) dimer (800 ppm relative to 9a, 188.5 mg) and the ligand (2000 ppm relative to 9a, 306.1 mg). The system was purged with nitrogen and then 3 ml. of acetonitrile and 0.3 ml_ of triethylamine was charged to the system. The resulting solution was agitated at room temperature for not less than 45 minutes and not more than 6 hours. Reaction: To a suitable sized, clean and dry reactor was charged 9a (1.00 equiv, 100.0 g (99.5 wt%), 377.4 mmol). The reaction was purged with nitrogen. To the reactor was charged acetonitrile (ACS grade, 4 L/Kg of 9a, 400 mL) and
triethylamine (2.50 equiv, 132.8 mL, 943 mmol). Agitation was initiated. The 9a solution was cooled to Tint= -5 to 0 °C and then formic acid (3.00 equiv, 45.2 mL, 1 132 mmol) was charged to the solution at a rate to maintain Tint not more than 20 °C. The batch temperature was then adjusted to Tint= -5 to -0 °C. Nitrogen was bubbled through the batch through a porous gas dispersion unit (Wiimad-LabGlass No. LG-8680-1 0, VWR catalog number 14202-962) until a fine stream of bubbles was obtained. To the stirring solution at Tint= -5 to 0 °C was charged the prepared catalyst solution from the catalyst preparation above. The solution was agitated at Tint= -5 to 0 °C with the bubbling of nitrogen through the batch until HPLC analysis of the batch indicated no less than 98 A% conversion (as recorded at 220 nm, 10-14 h). To the reactor was charged isopropylacetate (6.7 L/Kg of 9a, 670 ml_). The batch temperature was adjusted to Tint= 18 to 23 °C. To the solution was charged water (10 L/Kg of 9a, 1000 mL) and the batch was agitated at Tint= 18 to 23 °C for no less than 20 minutes. The agitation was decreased and or stopped and the layers were allowed to separate. The lighter colored aqueous layer was cut. To the solution was charged water (7.5 L/Kg of 9a, 750 mL) and the batch was agitated at Tint= 18 to 23 °C for no less than 20 minutes. The agitation was decreased and or stopped and the layers were allowed to separate. The lighter colored aqueous layer was cut. The batch was then reduced to 300 mL (3 L/Kg of 9a) via distillation while maintaining Text no more than 65 °C. The batch was cooled to Tint= 35 to 45 °C and the batch was seeded (10 mg). To the batch at Tint= 35 to 45 °C was charged heptane (16.7 L/Kg of 9a, 1670 mL) over no less than 1.5 hours. The batch temperature was adjusted to Tint= -2 to 3 °C over no less than 1 hour, and the batch was agitated at Tint= -2 to 3 °C for no less than 1 hour. The solids were collected by filtration. The filtrate was used to rinse the reactor (Filtrate is cooled to Tint= -2 to 3 °C before filtration) and the solids were suction dried for no less than 2 hours. The solids were dried until the LOD is no more than 4 % to obtain 82.7 g of 10a (99.6- 100 wt%, 98.5% ee, 82.5% yield). 1H-NMR (CDCI3, 400 MHz) δ: 8.20 (d, J= 8.4 Hz, 1 H), 8.01 (d, J= 8.4 Hz, 1 H), 7.73 (t, J= 7.4 Hz, 1 H), 7.59 (t, J= 7.7 Hz, H), 6.03 (s, 1 H), 3.93 (s, 1 H), 3.79 (s, 3H), 2.77 (s, 3H). 13C-NMR (CDCI3, 100 MHz) δ: 173.5, 158.3, 147.5, 142.9, 130.7, 128.8, 127.7, 127.1 , 125.1 , 124.6, 69.2, 53.4, 24.0.
Example 11
10a 6a
10a (2.45 kg, 96.8% purity, 8.9 mol), 6a (2.5 kg, 88.7% purity, 8.82 mol), tris(dibenzylideneacetone)dipalladium(0) (Pd2dba3, 40 g, 0.044 mol), (S)-3-ieri-butyl- 4-(2,6-dimethoxypheny1 )-2,3-dihydrobenzo[d][1 ,3]oxaphosphole (32 g, 0.01 1 mol), sodium carbonate (1.12 kg, 10.58 mol), 1-pentanol (16.69 L), and water (8.35 L) were charged to the reactor. The mixture was de-gassed by sparging with argon for 10-15 minutes, was heated to 60-63 °C, and was agitated until HPLC analysis of the reaction shows <1 A% (220 nm) of the 6a relative to the combined two atropisomer products (-15 hours). The batch was cooled to 18-23 °C. Water (5 L) and heptane (21 L) were charged. The slurry was agitated for 3 – 5 hours. The solids were collected by filtration, washed with water (4 L) and heptane/toluene mixed solvent (2.5 L toluene/5 L heptane), and dried. The solids were dissolved in methanol (25 L) and the resulting solution was heated to 50 °C and circulated through a CUNO carbon stack filter. The solution was distilled under vacuum to ~ 5 L. Toluene (12 L) was charged. The mixture was distilled under vacuum to ~ 5 L and cooled to 22 °C. Heptane (13 L) was charged to the contents over 1 hour and the resulting slurry was agitated at 20-25 °C for 3 – 4 hours. The solids were collected by filtration and washed with heptanes to provide 2.58 kg of 11a obtained after drying (73% yield). 1H NMR (400 MHz, CDCI3): δ 8.63 (d, 1 H, J = 8 Hz), 8.03 (d, 1 H, J = 12 Hz), 7.56 (t, 1 H, J = 8 Hz), 7.41 (d, 1 H, J = 8 Hz), 7.19 (t, 1 H, J = 8 Hz), 7.09 (m, 2H), 7.04 (d, 1 H, J = 8 Hz), 5.38 (d, 1 H, J = 8 Hz), 5.14 (d, 1 H, J = 8 Hz), 4.50 (t, 2H, J = 4 Hz), 3.40 (s, 3H), 3.25 (t, 2H, J = 4 Hz), 2.91 (s, 3H). 13C NMR (100 MHz, CDCI3): δ 173.6, 158.2, 154.0, 150.9, 147.3, 147.2, 145.7, 141.3, 132.9, 123.0, 129.4, 128.6, 127.8, 126.7, 126.4, 125.8, 1 18.1 , 1 17.3, 109.9, 70.3, 65.8, 52.3, 28.5, 24.0.
Example 12
11a 12a
To a suitable clean and dry reactor under a nitrogen atmosphere was charged 11a (5.47 Kg, 93.4 wt%, 1 .00 equiv, 12.8 mol) and fluorobenzene (10 vols, 51.1 kg) following by trifluoromethanesulfonimide (4 mol%, 143 g, 0.51 mol) as a 0.5 M solution in DCM (1.0 Kg). The batch temperature was adjusted to 35-41 °C and agitated to form a fine slurry. To the mixture was slowly charged i-butyl-2,2,2- trichloroacetimidate 12b as a 50 wt% solution (26.0 Kg of f-butyl-2,2,2- trichloroacetimidate (1 19.0 mol, 9.3 equiv), the reagent was -48-51 wt% with the remainder 52-49 wt% of the solution being – 1.8:1 wt:wt heptane: fluorobenzene) over no less than 4 hours at Tint= 35-41 °C. The batch was agitated at Tint= 35-41 °C until HPLC conversion (308 nm) was >96 A%, then cooled to Tint= 20-25 °C and then triethylamine (0.14 equiv, 181 g, 1 .79 mol) was charged followed by heptane (12.9 Kg) over no less than 30 minutes. The batch was agitated at Tint= 20-25 °C for no less than 1 hour. The solids were collected by filtration. The reactor was rinsed with the filtrate to collect all solids. The collected solids in the filter were rinsed with heptane (1 1 .7 Kg). The solids were charged into the reactor along with 54.1 Kg of DM Ac and the batch temperature adjusted to Tint= 70-75 °C. Water ( .2 Kg) was charged over no less than 30 minutes while the batch temperature was maintained at Tint= 65-75 °C. 12a seed crystals (34 g) in water (680 g) was charged to the batch at Tlnt= 65-75 °C. Additional water (46.0 Kg) was charged over no less than 2 hours while maintaining the batch temperature at Tint= 65-75 °C. The batch temperature was adjusted to Tint= 18-25 °C over no less than 2 hours and agitated for no less than 1 hour. The solids were collected by filtration and the filtrate used to rinse the reactor. The solids were washed with water (30 Kg) and dried under vacuum at no more than 45 °C until the LOD < 4% to obtain 12a (5.275 Kg, 99.9 A% at 220 nm, 99.9 wt% via HPLC wt% assay, 90.5% yield). 1H-NMR (CDCI3, 400
To a suitable clean and dry reactor under a nitrogen atmosphere was charged 12a (9.69 Kg, 21.2 mol) and ethanol (23.0 Kg). The mixture was agitated and the batch temperature was maintained at Τίηί= 20 to 25 °C. 2 M sodium hydroxide (17.2 Kg) was charged at Tint= 20 to 25 °C and the batch temperature was adjusted to Tint= 60- 65°C over no less than 30 minutes. The batch was agitated at Tint= 60-65°C for 2-3 hours until HPLC conversion was >99.5% area (12a is <0.5 area%). The batch temperature was adjuted to Tlnt= 50 to 55°C and 2M aqueous HCI (14.54 Kg) was charged. The pH of the batch was adjusted to pH 5.0 to 5.5 (target pH 5.2 to 5.3) via the slow charge of 2M aqueous HCI (0.46 Kg) at Tint= 50 to 55°C. Acetonitrile was charged to the batch (4.46 Kg) at Tint= 50 to 55°C. A slurry of seed crystals (1001 , 20 g in 155 g of acetonitrile) was charged to the batch at Tint= 50 to 55°C. The batch was agitated at Tint= 50 to 55°C for no less than 1 hour (1-2 hours). The contents were vacuum distilled to -3.4 vol (32 L) while maintaining the internal temperature at 45-55°C. A sample of the batch was removed and the ethanol content was determined by GC analysis; the criterion was no more than 10 wt% ethanol. If the ethanol wt% was over 10%, an additional 10% of the original volume was distilled and sampled for ethanol wt%. The batch temperature was adjusted to Tint= 18-22°C over no less than 1 hour. The pH of the batch was verified to be pH= 5 – 5.5 and the pH was adjusted, if necessary, with the slow addition of 2 M HCI or 2 M NaOH aqueous solutions. The batch was agitated at Tint= 18-22°C for no less than 6 hours and the solids were collected by filtration. The filtrate/mother liquid was used to remove all solids from reactor. The cake with was washed with water (19.4 Kg) (water temperature was no more than 20 °C). The cake was dried under vacuum at no more than 60 °C for 12 hours or until the LOD was no more than 4% to obtain 1001 (9.52 Kg, 99.6 A% 220 nm, 97.6 wt% as determined by HPLC wt% assay, 99.0% yield).
Compound (I), (2S)-2-tert-butoxy-2-(4-(2,3-dihydropyrano[4,3,2-de]quinolin-7-yl)-2- methylquinolin-3-yl)acetic acid, is an HIV non-catalytic site integrase inhibitor.
Compound (I) falls within the scope of the HIV inhibitors disclosed in WO
2007/131350. Compound (I) is disclosed specifically as compound no. 1144 in WO 2009/062285. Compound (I) can be prepared according to the general procedures found in WO 2007/13 350 and WO 2009/062285, which are hereby incorporated by reference.
Example 1
1 a 1b
1a (600 g, 4.1 mol) was charged into a dry reactor under nitrogen followed by addition of Ac20 (1257.5 g, 12.3 mol, 3 eq.). The resulting mixture was heated at 40 °C at least for 2 hours. The batch was then cooled to 30 °C over 30 minutes. A suspension of 1b in toluene was added to seed the batch if no solid was observed. After toluene (600 mL) was added over 30 minutes, the batch was cooled to -5 ~ -10 °C and was held at this temperature for at least 30 minutes. The solid was collected by filtration under nitrogen and rinsed with heptanes (1200 mL). After being dried under vacuum at room temperature, the solid was stored under nitrogen at least below 20 °C. The product 1b was obtained with 77% yield. 1H NMR (500 MHz, CDCI3): δ = 6.36 (s, 1 H), 3.68 (s, 2H), 2.30 (s, 3H).
Example 2
2a (100 g, 531 mmol) and 1 b (95 g, 558 mmol) were charged into a clean and dry reactor under nitrogen followed by addition of fluorobenzene ( 000 mL). After being heated at 35-37 °C for 4 hours, the batch was cooled to 23 °C. Concentrated H2S04 (260.82 g, 2659.3 mmol, 5 eq.) was added while maintaining the batch temperature below 35 °C. The batch was first heated at 30-35 °C for 30 minutes and then at 40- 45 °C for 2 hours. 4-Methyl morpholine (215.19 g, 2127 mmol, 4 eq.) was added to the batch while maintaining the temperature below 50 °C. Then the batch was agitated for 30 minutes at 40-50 °C. MeOH ( 00 mL) was then added while maintaining the temperature below 55 °C. After the batch was held at 50-55 °Cfor 2 hours, another portion of MeOH (100 mL) was added. The batch was agitated for another 2 hours at 50-55 °C. After fluorobenzene was distilled to a minimum amount, water (1000 mL) was added. Further distillation was performed to remove any remaining fluorobenzene. After the batch was cooled to 30 °C, the solid was collected by filtration with cloth and rinsed with water (400 mL) and heptane (200 mL). The solid was dried under vacuum below 50 °C to reach KF < 0.1 %. Typically, the product 2b was obtained in 90% yield with 98 wt%. 1H NMR (500 MHz, DMSO- cfe): δ = 10.83 (s, 1 H), 9.85 (s, bs, 1 H), 7.6 (d, 1 H, J = 8.7 Hz), 6.55 (d, 1 H, J = 8.7 Hz), 6.40 (s, 1 H), 4.00 (s, 2 H), 3.61 (s, 3 H).
Example 3
2b 3a
2b (20 g, 64 mmol) was charged into a clean and dry reactor followed by addition of THF (140 mL). After the resulting mixture was cooled to 0 °C, Vitride® (Red-AI, 47.84 g, 65 wt%, 154 mmol) in toluene was added while maintaining an internal temperature at 0-5 °C. After the batch was agitated at 5-10 °C for 4 hours, IPA (9.24 g, 153.8 mmol) was added while maintaining the temperature below 10 °C. Then the batch was agitated at least for 30 minutes below 25 °C. A solution of HCI in IPA (84.73 g, 5.5 M, 512 mmol) was added into the reactor while maintaining the temperature below 40 °C. After about 160 mL of the solvent was distilled under vacuum below 40 °C, the batch was cooled to 20-25 °C and then aqueous 6M HCI (60 mL) was added while maintaining the temperature below 40 °C. The batch was cooled to 25 °C and agitated for at least 30 minutes. The solid was collected by filtration, washed with 40 mL of IPA and water (1 V/1 V), 40 mL of water and 40 mL of heptanes. The solid was dried below 60 °C in a vacuum oven to reach KF < 0.5%. Typically, the product 3a was obtained in 90-95% yield with 95 wt%. 1H NMR (400 MHz, DMSO-c/e): 5 = 10.7 (s, 1 H), 9.68 (s, 1 H), 7.59 (d, 1 H, J = 8.7 Hz), 6.64 (, 1 H, J = 8.7 Hz), 6.27 (s, 1 H), 4.62 (bs, 1 H), 3.69 (t, 2H, J = 6.3 Hz), 3.21 (t, 2H, J = 6.3 Hz).
Example 4
3a 4a
3a (50 g, 174.756 mmol) and acetonitrile (200 mL) were charged into a dry and clean reactor. After the resulting mixture was heated to 65 °C, POC13 (107.18 g, 699 mmol, 4 eq.) was added while maintaining the internal temperature below 75 °C. The batch was then heated at 70-75 °C for 5-6 h. The batch was cooled to 20 °C. Water (400 mL) was added at least over 30 minutes while maintaining the internal temperature below 50 °C. After the batch was cooled to 20-25 °C over 30 minutes, the solid was collected by filtration and washed with water (100 mL). The wet cake was charged back into the reactor followed by addition of 1 M NaOH (150 mL). After the batch was agitated at least for 30 minutes at 25-35 °C, verify that the pH was greater than 12. Otherwise, more 6M NaOH was needed to adjust the pH >12. After the batch was agitated for 30 minutes at 25-35 °C, the solid was collected by filtration, washed with water (200 mL) and heptanes (200 mL). The solid was dried in a vacuum oven below 50 °C to reach KF < 2%. Typically, the product 4a was obtained at about 75-80% yield. 1H NMR (400 MHz, CDCI3): δ = 7.90 (d, 1 H, J = 8.4 Hz), 7.16 (s, 1 H), 6.89 (d, 1 H, J = 8.4 Hz), 4.44 (t, 2 H, J = 5.9 Hz), 3.23 (t, 2 H, J = 5.9 Hz). 13C NMR (100 MHz, CDCI3): δ = 152.9, 151.9, 144.9, 144.1 , 134.6, 119.1 , 1 17.0, 1 13.3, 1 1 1.9, 65.6, 28.3.
Example 5
4a 5a
Zn powder (54 g, 825 mmol, 2.5 eq.) and TFA (100 mL) were charged into a dry and clean reactor. The resulting mixture was heated to 60-65 °C. A suspension of 4a (100 g, 330 mmol) in 150 mL of TFA was added to the reactor while maintaining the temperature below 70 °C. The charge line was rinsed with TFA (50 mL) into the reactor. After 1 hour at 65±5 °C, the batch was cooled to 25-30 °C. Zn powder was filtered off by passing the batch through a Celite pad and washing with methanol (200 mL). About 400 mL of solvent was distilled off under vacuum. After the batch was cooled to 20-25 °C, 20% NaOAc (ca. 300 mL) was added at least over 30 minutes to reach pH 5-6. The solid was collected by filtration, washed with water (200 mL) and heptane (200 mL), and dried under vacuum below 45 °C to reach KF ≤ 2%. The solid was charged into a dry reactor followed by addition of loose carbon (10 wt%) and toluene (1000 mL). The batch was heated at least for 30 minutes at 45-50 °C. The carbon was filtered off above 35 °C and rinsed with toluene (200 mL). The filtrate was charged into a clean and dry reactor. After about 1000 mL of toluene was distilled off under vacuum below 50 °C, 1000 mL of heptane was added over 30 minutes at 40-50 °C. Then the batch was cooled to 0±5 °C over 30 minutes. After 30 minutes, the solid was collected and rinsed with 200 mL of heptane. The solid was dried under vacuum below 45 °C to reach KF≤ 500 ppm. Typically, the product 5a was obtained in about 90-95 % yield. 1H NMR (400 MHz, CDCI3): δ = 8.93 (m, 1 H), 7.91 (dd, 1 H, J = 1.5, 8 Hz), 7.17 (m 1 H), 6.90 (dd, 1 H, J = 1.6, 8.0 Hz), 4.46-4.43 (m, 2 H), 3.28-3.23 (m, 2 H). 13C NMR (100 MHz, CDCI3): δ = 152.8, 151 .2, 145.1 , 141.0, 133.3, 1 18.5, 1 18.2, 1 14.5, 1 1 1 .1 , 65.8, 28.4.
Example 6
5a (1.04 kg, 4.16 mol) and toluene (8 L) were charged into the reactor. The batch was agitated and cooled to -50 to -55 °C. BuLi solution (2.5 M in hexanes, 1.69 L, 4.23 mol) was charged slowly while maintaining the internal temperature between – 45 to -50 °C. The batch was agitated at -45 °C for 1 hour after addition. A solution of triisopropyl borate (0.85 kg, 4.5 mol) in MTBE (1.48 kg) was charged. The batch was warmed to 10 °C over 30 minutes. A solution of 5 N HCI in IPA (1.54 L) was charged slowly at 10 °C, and the batch was warmed to 20 °C and stirred for 30 minutes. It was seeded with 6a crystal (10 g). A solution of aqueous concentrated HCI (0.16 L) in IPA (0.16 L) was charged slowly at 20 °C in three portions at 20 minute intervals, and the batch was agitated for 1 hour at 20 °C. The solid was collected by filtration, rinsed with MTBE (1 kg), and dried to provide 6a (943 g, 88.7 % purity, 80% yield). 1H NMR (400 MHz, D20): δ 8.84 (d, 1 H, J = 4 Hz), 8.10 (m, 1 H), 7.68 (d, 1 H, J = 6 Hz), 7.09 (m, 1 H), 4.52 (m, 2H), 3.47 (m, 2H).
Example 7
7a 7b
Iodine stock solution was prepared by mixing iodine (57.4 g, 0.23 mol) and sodium iodide (73.4 g, 0.49 mol) in water (270 mL). Sodium hydroxide (28.6 g, 0.715 mol) was charged into 220 mL of water. 4-Hydroxy-2 methylquinoline 7a (30 g, 0.19 mol) was charged, followed by acetonitrile (250 mL). The mixture was cooled to 10 °C with agitation. The above iodine stock solution was charged slowly over 30 minutes. The reaction was quenched by addition of sodium bisulfite (6.0 g) in water (60 mL). Acetic acid (23 mL) was charged over a period of 1 hour to adjust the pH of the reaction mixture between 6 and 7. The product was collected by filtration, washed with water and acetonitrile, and dried to give 7b (53 g, 98%). MS 286 [M + 1].
7b 8a
4-Hydroxy-3-iodo-2-methylquinoline 7b (25 g, 0.09 mol) was charged to a 1 -L reactor. Ethyl acetate (250 mL) was charged, followed by triethylamine (2.45 mL, 0.02 mol) and phosphorus oxychloride (12 mL, 0.13 mol). The reaction mixture was heated to reflux until complete conversion (~1 hour), then the mixture was cooled to 22 °C. A solution of sodium carbonate (31.6 g, 0.3 mol) in water (500 mL) was charged. The mixture was stirred for 20 minutes. The aqueous layer was extracted with ethyl acetate (120 mL). The organic layers were combined and concentrated under vacuum to dryness. Acetone (50 mL) was charged. The solution was heated to 60 °C. Water (100 mL) was charged, and the mixture was cooled to 22 °C. The product was collected by filtration and dried to give 8a (25 g, 97.3 % pure, 91.4 % yield). MS 304 [M + 1].
(Note: 8a is a known compound with CAS # 1033931-93-9. See references: (a) J. Org Chem. 2008, 73, 4644-4649. (b) Molcules 2010, 15, 3171-3178. (c) Indian J. Chem. Sec B: Org. Chem. Including Med Chem. 2009, 48B(5), 692-696.)
8a (100 g, 0.33 mol) was charged to the reactor, followed by copper (I) bromide dimethyl sulfide complex (3.4 g, 0.017 mol) and dry THF (450 mL). The batch was cooled to – 5 to – 2 °C. i-PrMgCI (2.0 M in THF, 173 mL, 0.346 mol) was charged into the reactor at the rate which maintains the batch temperature < -10 °C.
In a 2nd reactor, methyl chlorooxoacetate (33 mL, 0.36 mol) and dry THF (150 mL) was charged. The solution was cooled to -15 to -10 °C. The content of the 1 st reactor (Grignard/cuprate) was charged into the 2nd reactor at the rate which maintained the batch temperature < -10 °C. The batch was agitated for 30 minutes at -10 °C. Aqueous ammonium chloride solution (10%, 300 mL) was charged. The batch was agitated at 20 – 25 °C for 20 minutes and allowed to settle for 20 minutes. The aqueous layer was separated. Aqueous ammonium chloride solution (10%, 90 mL) and sodium carbonate solution (10%, 135 mL) were charged to the reactor. The batch was agitated at 20 – 25 °C for 20 minutes and allowed to settle for 20 minutes. The aqueous layer was separated. Brine (10%, 240 mL) was charged to the reactor. The batch was agitated at 20 – 25 °C for 20 minutes. The aqueous layer was separated. The batch was concentrated under vacuum to -1/4 of the volume (about 80 mL left). 2-Propanol was charged (300 mL). The batch was concentrated under vacuum to -1/3 of the volume (about 140 mL left), and heated to 50 °C. Water (70 mL) was charged. The batch was cooled to 20 – 25 °C, stirred for 2 hours, cooled to -10 °C and stirred for another 2 hours. The solid was collected by filtration, washed with cold 2-propanol and water to provide 58.9 g of 9a obtained after drying (67.8 % yield). 1H NMR (400 MHz, CDCI3): δ 8.08 (d, 1 H, J = 12 Hz), 7.97 (d, 1 H, J = 12 Hz), 7.13 (t, 1 H, J = 8 Hz), 7.55 (t, 1 H, J = 8 Hz), 3.92 (s, 3H), 2.63 (s, 3H). 13C NMR (100 MHz, CDCI3): δ 186.6, 161.1 , 155.3, 148.2, 140.9, 132.0, 129.0, 128.8, 127.8, 123.8, 123.7, 53.7, 23.6.
Example 10
Catalyst preparation: To a suitable sized, clean and dry reactor was charged dichloro(pentamethylcyclopentadienyl)rhodium(lll) dimer (800 ppm relative to 9a, 188.5 mg) and the ligand (2000 ppm relative to 9a, 306.1 mg). The system was purged with nitrogen and then 3 ml_ of acetonitrile and 0.3 ml_ of triethylamine was charged to the system. The resulting solution was agitated at RT for not less than 45 minutes and not more than 6 hours.
Reaction: To a suitable sized, clean and dry reactor was charged 9a (1.00 equiv, 100.0 g (99.5 wt%), 377.4 mmol). The reaction was purged with nitrogen. To the reactor was charged acetonitrile (ACS grade, 4 L/Kg of 9a, 400 ml_) and
triethylamine (2.50 equiv, 132.8 ml_, 943 mmol). Agitation was initiated. The 9a solution was cooled to Tint= -5 to 0 °C and then formic acid (3.00 equiv, 45.2 ml_, 1 132 mmol) was charged to the solution at a rate to maintain Tint not more than 20 °C. The batch temperature was then adjusted to Tlnt= -5 to -0 °C. Nitrogen was bubbled through the batch through a porous gas dispersion unit (Wilmad-LabGlass No. LG-8680-1 10, VWR catalog number 14202-962) until a fine stream of bubbles was obtained. To the stirring solution at Jml= -5 to 0 °C was charged the prepared catalyst solution from the catalyst preparation above. The solution was agitated at Tint= -5 to 0 °C with the bubbling of nitrogen through the batch until HPLC analysis of the batch indicated no less than 98 A% conversion (as recorded at 220 nm, 10-14 h). To the reactor was charged isopropylacetate (6.7 L/Kg of 9a, 670 mL). The batch temperature was adjusted to Tint= 18 to 23 °C. To the solution was charged water (10 L/Kg of 9a, 1000 mL) and the batch was agitated at Tint= 18 to 23 °C for no less than 20 minutes. The agitation was decreased and or stopped and the layers were allowed to separate. The lighter colored aqueous layer was cut. To the solution was charged water (7.5 L/Kg of 9a, 750 mL) and the batch was agitated at Tint= 18 to 23 °C for no less than 20 minutes. The agitation was decreased and or stopped and the layers were allowed to separate. The lighter colored aqueous layer was cut. The batch was then reduced to 300 mL (3 L/Kg of 9a) via distillation while maintaining Text no more than 65 °C. The batch was cooled to Tint= 35 to 45 °C and the batch was seeded ( 0 mg). To the batch at Tint= 35 to 45 °C charged heptane (16.7 L/Kg of 9a, 1670 mL) over no less than 1.5 hours. Adjusted the batch temperature to Tint= -2 to 3 °C over no less than 1 hour, and agitated the batch at Tint= -2 to 3 °C for no less than 1 hour. Collected the solids by filtration. Used the filtrate to rinse the reactor (Filtrate is cooled to
-2 to 3 °C before filtration) and the solids were suction dried for no less than 2 hours. The solids were dried until the LOD was no more than 4 % to obtain 82.7 g of 10a (99.6-100 wt%, 98.5% ee, 82.5% yield). 1H- NMR (CDCI3, 400 MHz) δ: 8.20 (d, J= 8.4 Hz, 1 H), 8.01 (d, J= 8.4 Hz, 1 H), 7.73 (t, J= 7.4 Hz, 1 H), 7.59 (t, J= 7.7 Hz, 1 H), 6.03 (s, 1 H), 3.93 (s, 1 H), 3.79 (s, 3H), 2.77 (s, 3H). 13C-NMR (CDCI3, 100 MHz) δ: 173.5, 158.3, 147.5, 142.9, 130.7, 128.8, 127.7, 127.1 , 125.1 , 124.6, 69.2, 53.4, 24.0.
Example 11
10a 6a 11a
10a (2.45 kg, 96.8% purity, 8.9 mol), 6a (2.5 kg, 88.7% purity, 8.82 mol), tris(dibenzylideneacetone)dipalladium(0) (Pd2dba3, 40 g, 0.044 mol), (S)-3-iert-butyl-4-(2,6-dimethoxyphenyl)-2,3-dihydrobenzo[d][1 ,3]oxaphosphole (32 g, 0.01 1 mol), sodium carbonate (1.12 kg, 10.58 mol), 1 -pentanol (16.69 L), and water (8.35 L) were charged to the reactor. The mixture was de-gassed by sparging with argon for 10-15 minutes, was heated to 60-63 °C, and was agitated until HPLC analysis of the reaction shows <1 A% (220 nm) of the 6a relative to the combined two atropisomer products (-15 hours). The batch was cooled to 8-23 °C. Water (5 L) and heptane (21 L) were charged. The slurry was agitated for 3 – 5 hours. The solids were collected by filtration, washed with water (4 L) and heptane/toluene mixed solvent (2.5 L toluene/5 L heptane), and dried. The solids were dissolved in methanol (25 L) and the resulting solution was heated to 50 °C and circulated through a CUNO carbon stack filter. The solution was distilled under vacuum to ~ 5 L. Toluene (12 L) was charged. The mixture was distilled under vacuum to – 5 L and cooled to 22 °C. Heptane (13 L) was charged to the contents over 1 hour and the resulting slurry was agitated at 20-25 °C for 3 – 4 hours. The solids were collected by filtration and washed with heptanes to provide 2.58 kg of 11a obtained after drying (73% yield). 1H NMR (400 MHz, CDCI3): δ 8.63 (d, 1 H, J = 8 Hz), 8.03 (d, 1 H, J = 12 Hz), 7.56 (t, 1 H, J = 8 Hz), 7.41 (d, 1 H, J = 8 Hz), 7.19 (t, 1 H, J = 8 Hz), 7.09 (m, 2H), 7.04 (d, 1 H, J = 8 Hz), 5.38 (d, 1 H, J = 8 Hz), 5.14 (d, 1 H, J = 8 Hz), 4.50 (t, 2H, J = 4 Hz), 3.40 (s, 3H), 3.25 (t, 2H, J = 4 Hz), 2.91 (s, 3H). 13C NMR (100 MHz, CDCI3): δ 173.6, 158.2, 154.0, 150.9, 147.3, 147.2, 145.7, 141.3, 132.9, 123.0, 129.4, 128.6, 127.8, 126.7, 126.4, 125.8, 1 18.1 , 1 17.3, 109.9, 70.3, 65.8, 52.3, 28.5, 24.0.
To a suitable clean and dry reactor under a nitrogen atmosphere was charged 1a (5.47 Kg, 93.4 wt%, 1 .00 equiv, 12.8 mol) and fluorobenzene (10 vols, 51.1 kg) following by trifluoromethanesulfonimide (4 mol%, 143 g, 0.51 mol) as a 0.5 M solution in DCM (1.0 Kg). The batch temperature was adjusted to 35-41 °C and agitated to form a fine slurry. To the mixture was slowly charged i-butyt-2,2,2- trichloroacetimidate 12b as a 50 wt% solution (26.0 Kg of f-butyl-2,2,2- trichloroacetimidate (119.0 mol, 9.3 equiv), the reagent was -48-51 wt% with the remainder 52-49 wt% of the solution being ~ 1.8:1 wt:wt heptane: fluorobenzene) over no less than 4 hours at Tint= 35-41 °C. The batch was agitated at Tint= 35-41 °C until HPLC conversion (308 nm) was >96 A%, then cooled to Tlnt= 20-25 °C and then triethylamine (0.14 equiv, 181 g, 1.79 mol) was charged followed by heptane (12.9 Kg) over no less than 30 minutes. The batch was agitated at Tint= 20-25 °C for no less than 1 hour. The solids were collected by filtration. The reactor was rinsed with the filtrate to collect all solids. The collected solids in the filter were rinsed with heptane (1 1.7 Kg). The solids were charged into the reactor along with 54.1 Kg of DM Ac and the batch temperature adjusted to Tint= 70-75 °C. Water (1 1.2 Kg) was charged over no less than 30 minutes while the batch temperature was maintained at Tint= 65-75 °C. 12a seed crystals (34 g) in water (680 g) was charged to the batch at Tint= 65-75 °C. Additional water (46.0 Kg) was charged over no less than 2 hours while maintaining the batch temperature at Tint= 65-75 °C. The batch temperature was adjusted to Tint= 18-25 °C over no less than 2 hours and agitated for no less than 1 hour. The solids were collected by filtration and the filtrate used to rinse the reactor. The solids were washed with water (30 Kg) and dried under vacuum at no more than 45 °C until the LOD < 4% to obtain 12a (5.275 Kg, 99.9 A% at 220 nm, 99.9 wt% via HPLC wt% assay, 90.5% yield). H-NMR (CDCI3l 400 MHz) δ: 8.66-8.65 (m, 1 H), 8.05 (d, J= 8.3 Hz, 1 H), 7.59 (t, J= 7.3 Hz, 1 H), 7.45 (d, J= 7.8 Hz, 1 H), 7.21 (t, J= 7.6 Hz, 1 H), 7.13-7.08 (m, 3H), 5.05 (s, H), 4.63-4.52 (m, 2H), 3.49 (s, 3H), 3.41 -3.27 (m, 2H), 3.00 (s, 3H), 0.97 (s, 9H). 13C-NMR (CDCI3, 100 MHz) δ: 172.1 , 159.5, 153.5, 150.2, 147.4, 146.9, 145.4, 140.2, 131.1 , 130.1 , 128.9, 128.6, 128.0, 127.3, 126.7, 125.4, 1 17.7, 1 17.2, 109.4, 76.1 , 71.6, 65.8, 51.9, 28.6, 28.0, 25.4.
Example 13
To a suitable clean and dry reactor under a nitrogen atmosphere was charged 12a (9.69 Kg, 21.2 mol) and ethanol (23.0 Kg). The mixture was agitated and the batch temperature was maintained at Tjnt= 20 to 25 °C. 2 M sodium hydroxide (17.2 Kg) was charged at Tint= 20 to 25 °C and the batch temperature was adjusted to Tlnt= 60- 65°C over no less than 30 minutes. The batch was agitated at Tint= 60-65°C for 2-3 hours until HPLC conversion was >99.5% area (12a is <0.5 area%). The batch temperature was adjuted to Tint= 50 to 55°C and 2M aqueous HCI (14.54 Kg) was charged. The pH of the batch was adjusted to pH 5.0 to 5.5 (target pH 5.2 to 5.3) via the slow charge of 2M aqueous HCI (0.46 Kg) at Tint= 50 to 55°C. Acetonitrile was charged to the batch (4.46 Kg) at Τ,ηί= 50 to 55°C. A slurry of seed crystals (1001 , 20 g in 155 g of acetonitrile) was charged to the batch at Tint= 50 to 55°C. The batch was agitated at Tint= 50 to 55°C for no less than 1 hour (1-2 hours). The contents were vacuum distilled to -3.4 vol (32 L) while maintaining the internal temperature at 45-55°C. A sample of the batch was removed and the ethanol content was determined by GC analysis; the criterion was no more than 10 wt% ethanol. If the ethanol wt% was over 10%, an additional 10% of the original volume was distilled and sampled for ethanol wt%. The batch temperature was adjusted to Tint= 8-22°C over no less than 1 hour. The pH of the batch was verified to be pH= 5 – 5.5 and the pH was adjusted, if necessary, with the slow addition of 2 M HCI or 2 M NaOH aqueous solutions. The batch was agitated at Tint= 18-22°C for no less than 6 hours and the solids were collected by filtration. The filtrate/mother liquid was used to remove all solids from reactor. The cake with was washed with water (19.4 Kg) (water temperature was no more than 20 °C). The cake was dried under vacuum at no more than 60 °C for 12 hours or until the LOD was no more than 4% to obtain 1001 (9.52 Kg, 99.6 A% 220 nm, 97.6 wt% as determined by HPLC wt% assay, 99.0% yield). Example 14
Hydrochloride salt of Compound (I), Type A
Compound (I) (263 mg) was added to a vial of ethanol (1.5 ml_), and then 36.5% HCL aqueous solution (59 mg) was added. The mixture was heated to 70 °C; and stirred at this temperature until solid material was obtained. The mixture was cooled to 20 °C over a period of 10 hours. After cooling, isopropanol (400 μΙ_) was added over a period of 3 hours. The resulting solids were collected and characterized as the hydrochloride salt of Compound (I), Type A.
The hydrochloride salt of Compound (I), Type A was prepared analogously to the aforementioned procedure using methyl ethyl ketone, tetrahydrofuran, acetonitrile, ethyl acetate, dichloroethane and methyl-t-buyl ether instead of ethanol.
In December 2021, the U.S. Food and Drug Administration approved cabotegravir for pre-exposure prophylaxis (PrEP) in at-risk people under the brand name Apretude.[11]
GSK744 (also known as S/GSK1265744) is an investigational new drug under development for the treatment of HIV infection. It is anintegrase inhibitor, with a carbamoyl pyridone structure similar to dolutegravir. In investigational studies, the agent has been packaged into nanoparticles (GSK744LAP) conferring an exceptionally long half-life of 21–50 days following a single dose. In theory, this would make possible suppression of HIV with dosing as infrequently as once every three months.[1]
S-265744 LAP is in phase II clinical development at Shionogi-GlaxoSmithKline for the treatment of HIV infection. Phase III clinical trials had been ongoing for this indication; however, no recent development has been reported for this study.
Cabotegravir, or GSK1265744, is an HIV-1 integrase inhibitor that is prescribed with the non-nucleoside reverse transcriptase inhibitor, rilpivirine.4,6,7 Early research into cabotegravir showed it had lower oral bioavailability than dolutegravir,4 which resulted in the development of long acting monthly intramuscular injection formulation for cabotegravir.4,7
Cabotegravir was granted FDA approval on 21 January 2021 in combination with rilpivirine to treat HIV-1 infection in virologically suppressed individuals.8 While previously administered once monthly only, this combination product was granted FDA approval for dosing every two months on February 01, 2022 11 and without the need for an oral lead-in period prior.7
The human immunodeficiency virus (“HIV”) is the causative agent for acquired immunodeficiency syndrome (“AIDS”), a disease characterized by the destruction of the immune system, particularly of CD4+ T-cells, with attendant susceptibility to opportunistic infections, and its precursor Al DS-related complex (“ARC”), a syndrome characterized by symptoms such as persistent generalized lymphadenopathy, fever and weight loss. HIV is a retrovirus; the conversion of its RNA to DNA is accomplished through the action of the enzyme reverse transcriptase. Compounds that inhibit the function of reverse transcriptase inhibit replication of HIV in infected cells. Such compounds are useful in the prevention or treatment of HIV infection in humans.
A required step in HIV replication in human T-cells is the insertion by virally-encoded integrase of proviral DNA into the host cell genome. Integration is believed to be mediated by integrase in a process involving assembly of a stable nucleoprotein complex with viral DNA sequences, cleavage of two nucleotides from the 3′ termini of the linear proviral DNA and covalent joining of the recessed 3′ OH termini of the proviral DNA at a staggered cut made at the host target site. The repair synthesis of the resultant gap may be accomplished by cellular enzymes. There is continued need to find new therapeutic agents to treat human diseases. HIV integrase is an attractive target for the discovery of new therapeutics due to its important role in viral infections, particularly HIV infections. Integrase inhibitors are disclosed in WO2006/116724.
(3S, 1 1 aR)- N-[(2,4-difluorophenyl)methyl]-2,3,5,7, 1 1 , 1 1 a-hexahydro-6-hydroxy-3- methyl-5,7- dioxo-oxazolo[3,2-a]pyrido[1 ,2-d]pyrazine-8-carboxamide, a compound of formula (I), also referred to as compound (I), has proven antiviral activity against human immunodeficiency virus (HIV).
The present invention features pharmaceutical compositions comprising the active ingredient (3S, 1 1 aR)- N-[(2,4-difluorophenyl)methyl]-2,3,5,7, 1 1 , 1 1 a-hexahydro-6-hydroxy-3- methyl-5,7- dioxo-oxazolo[3,2-a]pyrido[1 ,2-d]pyrazine-8-carboxamide, or a pharmaceutically acceptable salt thereof, suitable for administration once monthly or longer.
Medical uses
Cabotegravir in combination with rilpivirine is indicated for the treatment of human immunodeficiency virus type-1 (HIV-1) in adults.[1][5] The combination injection is intended for maintenance treatment of adults who have undetectable HIV levels in the blood (viral load less than 50 copies/mL) with their current antiretroviral treatment, and when the virus has not developed resistance to non-nucleoside reverse transcriptase inhibitors (NNRTIs) and integrase strand transfer inhibitors.[5] The tablets are used to check whether a person tolerates the treatment before the injection therapy is started.[12][5]
The two medicines are the first antiretroviral drugs that come in a long-acting injectable formulation.[12]
Cabotegravir (Apretude) is indicated for use in at-risk people weighing at least 35 kilograms (77 lb) for pre-exposure prophylaxis (PrEP) to reduce the risk of sexually acquired HIV.[11]
Contraindications and interactions
Cabotegravir must not be combined with the drugs rifampicin, rifapentine, carbamazepine, oxcarbazepine, phenytoin or phenobarbital, which induce the enzyme UGT1A1.[5] These drugs significantly decrease cabotegravir concentrations in the body and thus may reduce its effectiveness.[9][5] Additionally, they induce the enzyme CYP3A4, which leads to reduced rilpivirine concentrations in the body.[5][13][14][15] Additionally, patients who are breastfeeding or plan to breastfeed should not take Cabotegravir because it is not known if it will pass within the breast milk.[16]
Adverse effects
The most common side effects of the injectable combination therapy with rilpivirine are reactions at the injection site (in up to 84% of patients) such as pain and swelling, as well as headache (up to 12%) and fever or feeling hot (in 10%). For the tablets, headache and a hot feeling were slightly less frequent. Less common side effects (under 10%) for both formulations are depressive disorders, insomnia, and rashes.[9]
Pharmacology
Mechanism of action
Cabotegravir is an integrase strand transfer inhibitor. This means it blocks the HIV’s enzyme integrase, thereby preventing its genome from being integrated into the human cells’ DNA.[9] As this is a necessary step for the virus to replicate, its further spread is hampered.[9]
When taken by mouth, cabotegravir reaches highest blood plasma levels after three hours. Taking the drug together with food slightly increases its concentrations in the blood, but this is not clinically relevant. After injection into the muscle, cabotegravir is slowly absorbed into the bloodstream, reaching its highest blood plasma levels after about seven days.[9]
Over 99% of the substance are bound to plasma proteins. The drug is inactivated in the body by glucuronidation, mainly by the enzyme UGT1A1, and to a much lesser extent by UGT1A9. More than 90% of the circulating substance are the unchanged cabotegravir, however. The biological half-life is 41 hours for the tablets and 5.6 to 11.5 weeks for the injection.[9]
Elimination has only been studied for oral administration: Most of the drug is eliminated via the faeces in unchanged form (47%). It is not known how much of this amount comes from the bile, and how much was not absorbed in the first place. (The bile actually contains the glucuronide, but this could be broken up again in the gut lumen to give the parent substance that is observed in the faeces.) To a lesser extent it is excreted via the urine (27%), almost exclusively as the glucuronide.[9]
Pharmacogenomics
UGT1A1 poor metabolizers have 1.3- to 1.5-fold increased cabotegravir concentrations in the body. This is not considered clinically significant.[9]
Chemistry
Cabotegravir is a white to off-white, crystalline powder that is practically insoluble in aqueous solutions under pH 9, and slightly soluble above pH 10. It is slightly acidic with a pKa of 7.7 for the enolic acid and 1.1 (calculated) for the carboxamide. The molecule has two asymmetric carbon atoms; only one of the four possible configurations is present in the medication.[18]
Formulation
In studies, the agent was packaged into nanoparticles (GSK744LAP) conferring a biological half-life of 21 to 50 days[citation needed] following a single dose. The marketed injection achieves its long half-life not via nanoparticles but with a suspension of the free cabotegravir acid. The tablets contain cabotegravir sodium salt.[18]
History
Cabotegravir was examined in the clinical trials HPTN 083 and HPTN 084.[19][20] In 2020, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) adopted a positive opinion, recommending the granting of a marketing authorization for the medicinal product Vocabria intended for the treatment of human immunodeficiency virus type 1 (HIV-1) infection in combination with rilpivirine injection.[21] The EMA also recommended marketing authorization be given for rilpivirine and cabotegravir injections to be used together for the treatment of people with HIV-1 infection.[12] Cabotegravir was approved for medical use in the European Union in December 2020.[8]
In 2020, results for some studies were released showing success in using injectable cabotegravir for long-acting pre-exposure prophylaxis (PrEP) with greater efficacy than the emtricitabine/tenofovir combination being widely used for PrEP at the time.[24][25]
The safety and efficacy of cabotegravir to reduce the risk of acquiring HIV were evaluated in two randomized, double-blind trials that compared cabotegravir to emtricitabine/tenofovir, a once daily oral medication for HIV PrEP.[11] Trial 1 included HIV-uninfected men and transgender women who have sex with men and have high-risk behavior for HIV infection.[11] Trial 2 included uninfected cisgender women at risk of acquiring HIV.[11]
In Trial 1, 4,566 cisgender men and transgender women who have sex with men received either cabotegravir or emtricitabine/tenofovir.[11] The trial measured the rate of HIV infections among trial participants taking daily cabotegravir followed by cabotegravir injections every two months compared to daily oral emtricitabine/tenofovir.[11] The trial showed participants who took cabotegravir had 69% less risk of getting infected with HIV when compared to participants who took emtricitabine/tenofovir.[11]
In Trial 2, 3,224 cisgender women received either cabotegravir or emtricitabine/tenofovir.[11] The trial measured the rate of HIV infections in participants who took oral cabotegravir and injections of cabotegravir compared to those who took emtricitabine/tenofovir orally.[11] The trial showed participants who took cabotegravir had 90% less risk of getting infected with HIV when compared to participants who took emtricitabine/tenofovir.[11]
In December 2021, the U.S. Food and Drug Administration (FDA) approved cabotegravir for pre-exposure prophylaxis.[11] The FDA granted the approval of Apretude to Viiv.[11]
Methods for the preparation of a compound of formula (I) are described in WO 2006/1 16764, WO2010/01 1814, WO2010/068262, and WO2010/068253
3,5,7,11,11a-hexahydro[1,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazine-8-carboxamide. To a solution of 16a (409 mg, 0.87 mmol) in dichloroethane (20 mL) was added (2R)-2-amino-1-propanol (0,14 mL, 1.74 mmol) and 10 drops of glacial acetic acid.
The resultant solution was heated at reflux for 2 h. Upon cooling, Celite was added
to the mixture and the solvents removed in vacuo and the material was purified via
silica gel chromatography (2% CH3OH/CH2CI2 gradient elution) to give
The title compound was made in two steps using a similar process to that described
in example Z-I. 16a (510 mg, 1.08 mmol) and (2«5)-2-amino-1-propanol (0.17 mL, 2,17 mmol) were reacted in 1,2-dichloroethane (20 mL) with acetic acid to give
The starting material of Example A is compound 8, which is identical to formula (Ia). Thus, Example A depicts a process in providing an intermediate for the compound of formula 17 below which is isomeric to the compound ZZ-2 at page 237 of WO 2006/116764 to Brian Johns et al.
14
Example Aa After dissolution of mixture of 320 g of compound 8 (1.0 eq.) in 3.20 L of MeOH by heating, the solution was concentrated. To the residue, 1.66 L of MeCN, 5.72 mL of AcOH(0.1 eq.) and 82.6 g of (S)-2-Amino-propan-1-ol(1.1 eq.) were added and the mixture was heated to 70 °C, stirred at 70 0C for 4 h and concentrated. To the residue, 1.67 L of 2-propanol was added and the mixture was concentrated (twice). After cooling of the residue, filtration, washing with 500 mL of cold 2-propanol and drying provided 167 g of compound 14 (52% yield) as a crystal. 1H NMR(300 MHz1 CDCI3) δ 7.61-7.55 (m, 2H), 7.40-7.20 (m, 4H), 6.53 (d, J = 7.2, 1H), 5.46 (d, J = 10.5 Hz, 1H), 5.23 (d, J = 10.2 Hz, 1H), 5.20 (dd, J = 3.9, 9.6 Hz, 1H), 4.46- 4.34 (m, 1H)1 4.31 (dd, J = 6.6, 8.7 Hz, 1H)1 4.14 (dd, J = 3.9, 12.3 Hz1 1H)1 3.79 (dd, J = 9.9, 12.3 Hz1 1 H), 3.62 (dd, J = 6.9, 8.7 Hz1 1 H), 1.38 (d, J = 6.3 Hz1 3H).
Example Ab
To slurry of 156 g of compound 14 (1.0 eq.) in 780 ml_ of NMP was added 93.6 g of NBS(1.1 eq.) and the mixture was stirred at room temperature for 2.5 h. The reaction mixture was added to 3.12 L of H2O. Filtration, washing with 8.0 L of H2O and drying provided 163 g of compound 15 (84% yield) as a crystal.
Under carbon mono-oxide atmosphere, a mixture of 163 g of compound 15 (1.0 eq.), 163 mL of /-Pr2NEt(2.5 eq.), 68.4 ml_ of 2,4-difluorobenzylamine(1.5 eq.) and 22.5 g of Pd(PPh3)4(0.05 eq.) in 816 mL of DMSO was stirred at 90 0C for 7 h. After cooling, removal of precipitate, washing with 50 mL of DMSO and addition of 11.3 g of
Pd(PPh3)4(0.025 eq.), the reaction mixture was stirred at 90 0C for 2 h under carbon mono-oxide atmosphere again. After cooling, removal of precipitate and addition of 2.0 L of AcOEt and 2.0 L of H2O1 the organic layer was washed with 1.0 L of 1 N HCIaq. and 1.0 L of H2O (twice) and the aqueous layer was extracted with 1.0 L of AcOEt. The organic layers were combined and concentrated. Silica gel column chromatography of the residue provided 184 g of compound 16 (96% yield) as foam.
Under hydrogen atmosphere, a mixture of 184 g of compound 16 (1.0 eq.) and 36.8 g of 10%Pd-C in 3.31 L of THF and 0.37 L of MeOH was stirred for 3 h. After filtration of precipitate(Pd-C), washing with THF/MeOH(9/1 ) and addition of 36.8 g of 10% Pd-C, the mixture was stirred for 20 min under hydrogen atmosphere. After filtration of precipitate(Pd-C) and washing with THF/MeOH(9/1), the filtrate was concentrated. After 200 ml_ of AcOEt was added to the residue, filtration afforded crude solid of compound 17. The precipitates were combined and extracted with 4.0 L of CHCl3/MeOH(5/1). After concentration of the CHCI3ZMeOH solution and addition of 250 ml_ of AcOEt to the residue, filtration afforded crude solid of compound 17. The crude solids were combined and dissolved in 8.2 L of MeCN/H2O(9/1 ) by heating. After filtration, the filtrate was concentrated. To the residue, 1.5 L of EtOH was added and the mixture was concentrated (three times). After cooling of the residue, filtration and drying provided 132 g of compound 17 (88% yield) as a crystal. 1H NMR(300 MHz, DMSO-cfe) δ 11.47 (brs, 1H), 10.31 (t, J = 6.0 Hz, 1H), 8.46 (s, 1H), 7.40 (td, J = 8.6, 6.9 Hz, 1H), 7.24 (ddd, J = 2.6, 9.4, 10.6, 1H), 7.11-7.01 (m, 1H), 5.39 (dd, J = 4.1, 10.4 Hz, 1H), 4.89 (dd, J = 4.2, 12.3 Hz, 1H), 4.55 (d, J = 6.0 Hz, 2H), 4.40 (dd, J = 6.8, 8.6 Hz, 1H), 4.36-^.22 (m, 1H)1 4.00 (dd, J = 10.2, 12.3 Hz, 1H), 3.67 (dd, J = 6.7, 8.6 Hz, 1H), 1.34 (d, J = 6.3 Hz, 3H).
Example Ae
After dissolution of 16.0 g of compound 17 (1.0 eq.) in 2.56 L of EtOH and 0.64 L of H2O by heating, followed by filtration, 39 ml_ of 1N NaOHaq.(1.0 eq.) was added to the solution at 75 0C. The solution was gradually cooled to room temperature. Filtration, washing with 80 ml_ of EtOH and drying provided 13.5 g of compound 18 (80% yield) as a crystal.
The following examples are intended for illustratation only and are not intended to limit the scope of the invention in any way. Preparation 1 : (3S.11 af?VΛ/-r(2.4-DifluoroDhenvnmethyll-6-hvdroxy-3-methyl-5.7-dioxo- 2,3,5,7, 11 ,11 a-hexahydroM ,31oxazolor3,2-alpyridori ,2-c/1pyrazine-8-carboxamide sodium salt (compound 1 b, scheme 2).
I) MsCI, Et3N
2) DBU
P-1 P-2 P-3
a) Synthesis of 2-methyl-3-[(phenylmethvl)oxvl-4/-/-pvran-4-one (compound P-2). To a slurry of 2000 g of compound P-1(1.0 eq.) in 14.0 L of MeCN were added 2848 g of benzyl bromide(1.05 eq.) and 2630 g of K2CO3(1.2 eq.). The mixture was stirred at 80 0C for 5 h and cooled to 13°C. Precipitate was filtered and washed with 5.0 L of MeCN. The filtrate was concentrated and 3.0 L of THF was added to the residue. The THF solution was concentrated to give 3585 g of crude compound P-2 as oil. Without further purification, compound P-2 was used in the next step. 1H NMR(300 MHz, CDCI3) δ 7.60 (d, J = 5.7 Hz, 1 H), 7.4-7.3 (m, 5H), 6.37 (d, J = 5.7 Hz, 1 H), 5.17 (s, 2H), 2.09 (s, 3H).
b) Synthesis of 2-(2-hydroxy-2-phenylethyl)-3-[(phenylmethyl)oxy]-4H-pyran-4-one (compound P-3). To 904 g of the crude compound P-2 was added 5.88 L of THF and the solution was cooled to -60 0C. 5.00 L of 1.0 M of Lithium bis(trimethylsilylamide) in THF(1.25 eq.) was added dropwise for 2 h to the solution of compound 2 at -60 0C. Then, a solution of 509 g of benzaldehyde(1.2 eq.) in 800 ml. of THF was added at -60 0C and the reaction mixture was aged at -60 0C for 1 h. The THF solution was poured into a mixture of 1.21 L of conc.HCI, 8.14 L of ice water and 4.52 L of EtOAc at less than 2 0C.
The organic layer was washed with 2.71 L of brine (twice) and the aqueous layer was extracted with 3.98 L of EtOAc. The combined organic layers were concentrated. To the mixture, 1.63 L of toluene was added and concentrated (twice) to provide toluene slurry of compound P-3. Filtration, washing with 0.90 L of cold toluene and drying afforded 955 g of compound P-3 (74% yield from compound P-1 ) as a solid. 1H NMR(300 MHz, CDCI3) δ
c) Synthesis of 2-[(£)-2-phenylethenyl]-3-[(phenylmethyl)oxy]-4H-pyran-4-one (compound
P-4). To a solution of 882 g of compound P-3 (1.0 eq.) in 8.82 L of THF were added 416 g of Et3N(1.5 eq.) and 408 g of methanesulfonyl chloride(1.3 eq.) at less than 30 0C. After confirmation of disappearance of compound P-3, 440 ml. of NMP and 1167 g of DBU(2.8 eq.) were added to the reaction mixture at less than 30 0C and the reaction mixture was aged for 30 min. The mixture was neutralized with 1.76 L of 16% sulfuric acid and the organic layer was washed with 1.76 L of 2% Na2S03aq. After concentration of the organic layer, 4.41 L of toluene was added and the mixture was concentrated (tree times). After addition of 4.67 L of hexane, the mixture was cooled with ice bath. Filtration, washing with 1.77 L of hexane and drying provided 780 g of compound P-4 (94% yield) as a solid. 1H NMR(300 MHz, CDCI3) δ 7.69 (d, J = 5.7 Hz, 1 H), 7.50-7.25 (m, 10H), 7.22 (d, J = 16.2
Hz, 1 H), 7.03 (d, J = 16.2 Hz, 1 H), 6.41 (d, J = 5.7 Hz, 1 H), 5.27 (s, 2H). d) Synthesis of 4-oxo-3-[(phenylmethyl)oxy]-4H-pyran-2-carboxylic acid (compound P-5). To a mixture of 822 g of compound P-4 (1.0 eq.) and 1 1.2 g of RuCI3-nH2O(0.02 eq.) in 2.47 L of MeCN, 2.47 L of EtOAc and 2.47 L of H2O was added 2310 g of NalO4(4.0 eq.) at less than 25 0C. After aging for 1 h, 733 g of NaCIO2(S-O eq.) was added to the mixture at less than 25 0C. After aging for 1 h, precipitate was filtered and washed with 8.22 L of
EtOAc. To the filtrate, 1.64 L of 50% Na2S203aq, 822 ml. of H2O and 630 ml. of coc.HCI were added. The aqueous layer was extracted with 4.11 L of EtOAc and the organic layers were combined and concentrated. To the residue, 4 L of toluene was added and the mixture was concentrated and cooled with ice bath. Filtration, washing with 1 L of toluene and drying provided 372 g of compound P-5 (56% yield) as a solid. 1H NMR(300 MHz,
e) Synthesis of 1-(2,3-dihydroxypropyl)-4-oxo-3-[(phenylmethyl)oxy]-1 ,4-dihydro-2- pyridinecarboxylic acid (compound P-6). A mixture of 509 g of compound P-5 (1.0 eq.) and
407 g of 3-amino-propane-1 ,2-diol(2.5 eq.) in 1.53 L of EtOH was stirred at 65 0C for 1 h and at 80 0C for 6 h. After addition of 18.8 g of 3-Amino-propane-1 ,2-diol(0.1 eq.) in 200 ml. of EtOH, the mixture was stirred at 80 0C for 1 h. After addition of 18.8 g of 3-amino- propane-1 ,2-diol (0.1 eq.) in 200 ml. of EtOH, the mixture was stirred at 80 0C for 30 min. After cooling and addition of 509 ml. of H2O, the mixture was concentrated. To the residue,
2.54 L of H2O and 2.54 L of AcOEt were added. After separation, the aqueous layer was washed with 1.02 L of EtOAc. To the aqueous layer, 2.03 L of 12% sulfuric acid was added at less than 12 0C to give crystal of compound P-6. Filtration, washing with 1.53 L of cold H2O and drying provided 576 g of compound P-6 (83% yield) as a solid. 1H NMR(300 MHz, DMSO-de) δ 7.67 (d, J = 7.5 Hz, 1 H), 7.5-7.2 (m, 5H), 6.40 (d, J = 7.5 Hz, 1 H), 5.07
f) Synthesis of methyl 1-(2,3-dihydroxypropyl)-4-oxo-3-[(phenylmethyl)oxy]-1 ,4-dihydro-2- pyridinecarboxylate (compound P-7). To a slurry of 576 g of compound P-6 (1.0 eq.: 5.8% of H2O was contained) in 2.88 L of NMP were added 431 g of NaHCO3(3.0 eq.) and 160 ml. of methyl iodide(1.5 eq.) and the mixture was stirred at room temperature for 4 h. After cooling to 5 0C, 1.71 L of 2N HCI and 1.15 L of 20% NaClaq were added to the mixture at less than 10 0C to give crystal of compound 7. Filtration, washing with 1.73 L of H2O and drying provided 507 g of compound P-7 (89% yield) as a solid. 1H NMR(300 MHz, DMSO- cfe) δ 7.59 (d, J = 7.5 Hz, 1 H), 7.40-7.28 (m, 5H), 6.28 (d, J = 7.5 Hz, 1 H), 5.21 (d, J = 5.4 Hz, 1 H), 5.12 (d, J = 10.8 Hz, 1 H), 5.07 (d, J = 10.8 Hz, 1 H), 4.83 (t, J = 5.7 Hz, 1 H), 3.97 (dd, J = 2.4, 14.1 Hz, 1 H), 3.79 (s, 3H), 3.70 (dd, J = 9.0, 14.4 Hz, 1 H), 3.65-3.50 (m, 1 H), 3.40-3.28 (m, 1 H), 3.26-3.14 (m, 1 H).
g) Synthesis of methyl 1-(2,2-dihydroxyethyl)-4-oxo-3-[(phenylmethyl)oxy]-1 ,4-dihydro-2- pyridinecarboxylate (compound P-8). To a mixture of 507 g of compound P -7 (1.0 eq.) in
5.07 L of MeCN, 5.07 L of H2O and 9.13 g of AcOH(0.1 eq.) was added 390 g of NaIO4(1.2 eq.) and the mixture was stirred at room temperature for 2 h. After addition of 1.52 L of 10% Na2S2OsBq., the mixture was concentrated and cooled to 10 0C. Filtration, washing with H2O and drying provided 386 g of compound P-8 (80% yield) as a solid. 1H NMR(300 MHz, DMSO-d6) δ 7.62 (d, J = 7.5 Hz, 1 H), 7.42-7.30 (m, 5H), 6.33 (d, J = 6.0 Hz, 2H),
h) Synthesis of (3S, 11 aR)-3-methyl-6-[(phenylmethyl)oxy]-2,3, 1 1 ,1 1a- tetrahydro[1 ,3]oxazolo[3,2-a]pyrido[1 ,2-c/]pyrazine-5,7-dione (compound P-9). After dissolution of mixture of 320 g of compound P-8 (1.0 eq.) in 3.20 L of MeOH by heating, the solution was concentrated. To the residue, 1.66 L of MeCN, 5.72 ml. of AcOH(0.1 eq.) and 82.6 g of (S)-2-Amino-propan-1-ol(1.1 eq.) were added and the mixture was heated to 70 0C, stirred at 70 0C for 4 h and concentrated. To the residue, 1.67 L of 2-propanol was added and the mixture was concentrated (twice). After cooling of the residue, filtration, washing with 500 ml. of cold 2-propanol and drying provided 167 g of compound P-9 (52% yield) as a solid. 1H NMR(300 MHz, CDCI3) δ 7.61-7.55 (m, 2H), 7.40-7.20 (m, 4H), 6.53 (d, J = 7.2, 1 H), 5.46 (d, J = 10.5 Hz, 1 H), 5.23 (d, J = 10.2 Hz, 1 H), 5.20 (dd, J = 3.9, 9.6 Hz, 1 H), 4.46-4.34 (m, 1 H), 4.31 (dd, J = 6.6, 8.7 Hz, 1 H), 4.14 (dd, J = 3.9, 12.3 Hz, 1 H), 3.79 (dd, J = 9.9, 12.3 Hz, 1 H), 3.62 (dd, J = 6.9, 8.7 Hz, 1 H), 1.38 (d, J = 6.3 Hz, 3H).
i) Synthesis of (3 S, 1 1 aR)-8-bromo-3-methyl-6-[(phenylmethyl)oxy]-2,3, 11 ,11a- tetrahydro[1 ,3]oxazolo[3,2-a]pyrido[1 ,2-c/]pyrazine-5,7-dione (compound P-10). To slurry of 156 g of compound P-9 (1.0 eq.) in 780 ml. of NMP was added 93.6 g of NBS(1.1 eq.) and the mixture was stirred at room temperature for 2.5 h. The reaction mixture was added to 3.12 L of H2O. Filtration, washing with 8.0 L of H2O and drying provided 163 g of compound P-10 (84% yield) as a solid. 1H NMR(300 MHz, DMSO-d6) δ 8.37 (s, 1 H), 7.55- 7.50 (m, 2H), 7.42-7.25 (m, 3H), 5.34 (dd, J = 3.6, 9.9 Hz, 1 H), 5.18 (d, J = 10.8 Hz, 1 H), 5.03 (d, J = 10.5 Hz, 1 H), 4.53 (dd, J = 3.6, 12.0 Hz, 1 H), 4.40-4.20 (m, 2H), 3.99 (dd, J = 9.9, 1 1.7 Hz, 1 H), 3.64 (dd, J = 5.7, 8.1 Hz, 1 H), 1.27 (d, J = 6.3 Hz, 3H). j) Synthesis of (3S,1 1aS)-Λ/-[(2,4-difluorophenyl)methyl]-3-methyl-5,7-dioxo-6- [(phenylmethyl)oxy]-2,3,5,7, 11 ,1 1 a-hexahydro[1 ,3]oxazolo[3,2-a]pyrido[1 ,2-c/]pyrazine-8- carboxamide (compound P-11). Under carbon mono-oxide atmosphere, a mixture of 163 g of compound P-10 (1.0 eq.), 163 mL of /-Pr2NEt(2.5 eq.), 68.4 mL of 2,4- difluorobenzylamine(1.5 eq.) and 22.5 g of Pd(PPh3)4(0.05 eq.) in 816 mL of DMSO was stirred at 90 0C for 7 h. After cooling, removal of precipitate, washing with 50 mL of DMSO and addition of 1 1.3 g of Pd(PPh3)4(0.025 eq.), the reaction mixture was stirred at 90 0C for 2 h under carbon mono-oxide atmosphere again. After cooling, removal of precipitate and addition of 2.0 L of AcOEt and 2.0 L of H2O, the organic layer was washed with 1.0 L of 1 N HCIaq. and 1.0 L of H2O (twice) and the aqueous layer was extracted with 1.0 L of AcOEt.
k) Synthesis of (3S,1 1aR)-Λ/-[(2,4-difluorophenyl)methyl]-6-hydroxy-3-methyl-5,7-dioxo- 2,3,5,7, 11 ,11 a-hexahydro[1 ,3]oxazolo[3,2-a]pyrido[1 ,2-c/]pyrazine-8-carboxamide (compound 1a). Under hydrogen atmosphere, a mixture of 184 g of compound P-11 (1.0 eq.) and 36.8 g of 10%Pd-C in 3.31 L of THF and 0.37 L of MeOH was stirred for 3 h. After filtration of precipitate(Pd-C), washing with THF/MeOH(9/1 ) and addition of 36.8 g of 10% Pd-C, the mixture was stirred for 20 min under hydrogen atmosphere. After filtration of precipitate(Pd-C) and washing with THF/MeOH(9/1 ), the filtrate was concentrated. After 200 mL of AcOEt was added to the residue, filtration afforded crude solid of compound 1 a.
The precipitates were combined and extracted with 4.0 L of CHCI3/Me0H(5/1 ). After concentration of the CHCI3/MeOH solution and addition of 250 mL of AcOEt to the residue, filtration afforded crude solid of compound 1a. The crude solids were combined and dissolved in 8.2 L of MeCN/H2O(9/1 ) by heating. After filtration, the filtrate was concentrated. To the residue, 1.5 L of EtOH was added and the mixture was concentrated
†Global API Chemistry, ‡MDR Chemical Science,§Analytical Sciences, GlaxoSmithKline, 709 Swedeland Road, King of Prussia, Pennsylvania 19406, United States
A novel synthesis of GSK1265744, a potent HIV integrase inhibitor, is described. The synthesis is highlighted by an efficient construction of the densely functionalized pyridinone core as well as a highly diastereoselective formation of the acyl oxazolidine moiety. The latter exploits the target molecule’s ability to chelate to Mg2+, a key feature in the integrase inhibitor’s mechanism of action.
Bictegravir and dolutegravir are two recently approved integrase inhibitors for the treatment of HIV. A third inhibitor, cabotegravir, is in Phase 3 development. As a continuation of a series of articles on synthetic routes to newly approved drugs, the current article reviews the patent and journal literature regarding synthetic routes and final forms of these drug
^“Adopted USANs”(PDF). American Medical Association. Retrieved 19 September 2014.
^World Health Organization (2015). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 73”. WHO Drug Information. 29 (1): 70–1. hdl:10665/331088.
Ziegler, Robert E.; Desai, Bimbisar K.; Jee, Jo-Ann; Gupton, B. Frank; Roper, Thomas D.; Jamison, Timothy F. 7-Step Flow Synthesis of the HIV Integrase Inhibitor Dolutegravir. Angewandte Chemie, International Edition. Volume 57. Issue 24. Pages 7181-7185. Journal; Online Computer File. (2018).
SYN 4
Synthetic Reference
Rajan, Srinivasan Thirumalai; Eswaraiah, Sajja; Reddy, Ghojala Venkat; Reddy, Sagyam Rajeshwar; Markandeya, Bekkam; Rajesham, Boge. Novel crystalline polymorph of sodium (4R,12aS)-9-{[(2,4-difluorophenyl)methyl]carbamoyl}-4-methyl-6,8-dioxo-3,4,6,8,12,12a-hexahydro-2H-pyrido[1′,2′:4,5]pyrazino[2,1-b][1,3]oxazin-7-olate and process for preparation thereof. Assignee MSN Research & Development Center, India. IN 201641037221. (2018).
Synthetic Reference 5
Sharma, Pramodkumar; Rao, Bhatraju Srinivasa; Deo, Keshav. A process for the preparation of Dolutegravir or its pharmaceutical acceptable salts thereof. Assignee Wockhardt Limited, India. IN 2015MU01007. (2016).
Synthetic Reference 6
Weaver, Jimmie Dean. Preparation of fluoroarenes via hydrogen bond directed photocatalytic hydrodefluorination of perfluoroarenes. Assignee The Board of Regents for Oklahoma State University, USA. WO 2018187336. (2018).
Vellanki, Sivaram Prasad; Nadella, Madumurthy; Bhalme, Mitali; Ramabhotla, Revathi Srinivas. Process for the preparation of dolutegravir, an integrase inhibitor for HIV-1 infection therapy. Assignee Mylan Laboratories Ltd., India. IN 2015CH00588. (2016).
SYN 9
Synthetic Reference
Sankareswaran, Srimurugan; Mannam, Madhavarao; Chakka, Veerababu; Mandapati, Srirami Reddy; Kumar, Pramod. Identification and Control of Critical Process Impurities: An Improved Process for the Preparation of Dolutegravir Sodium. Organic Process Research & Development. Volume 20. Issue 8. Pages 1461-1468. Journal; Online Computer File. (2016).

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....











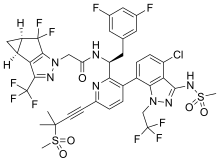
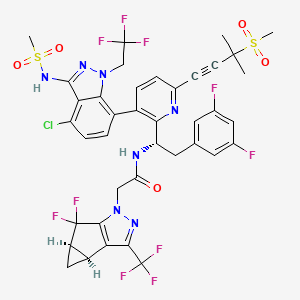






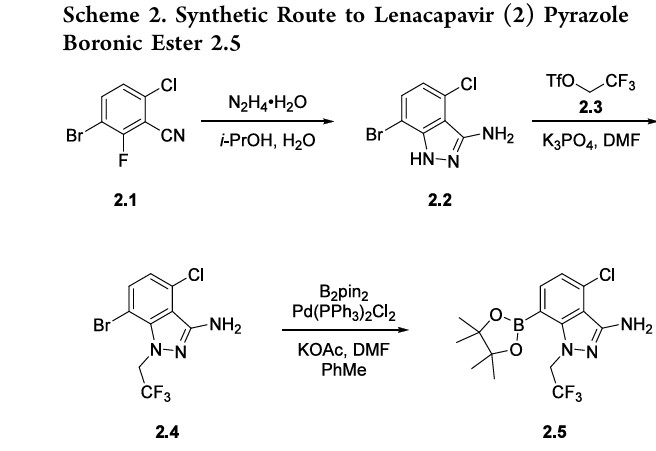







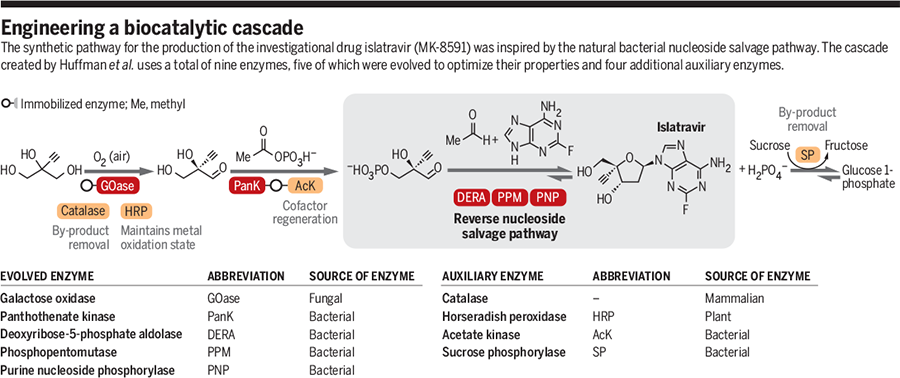




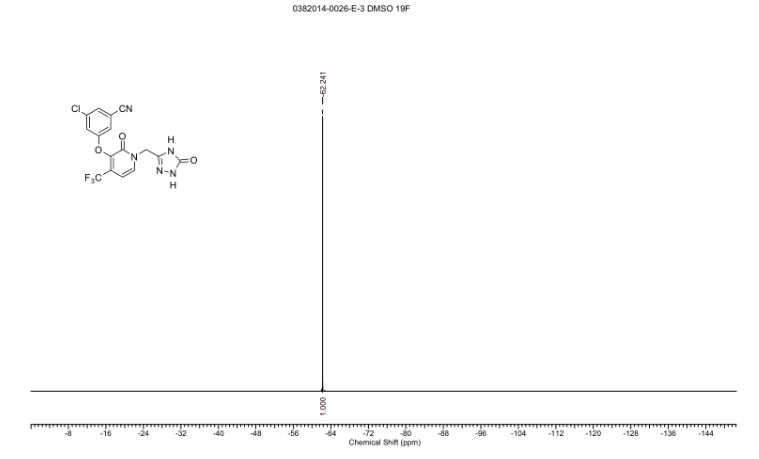
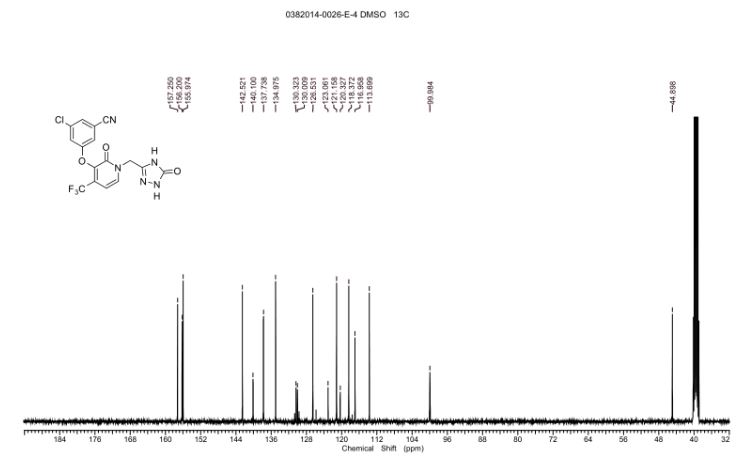
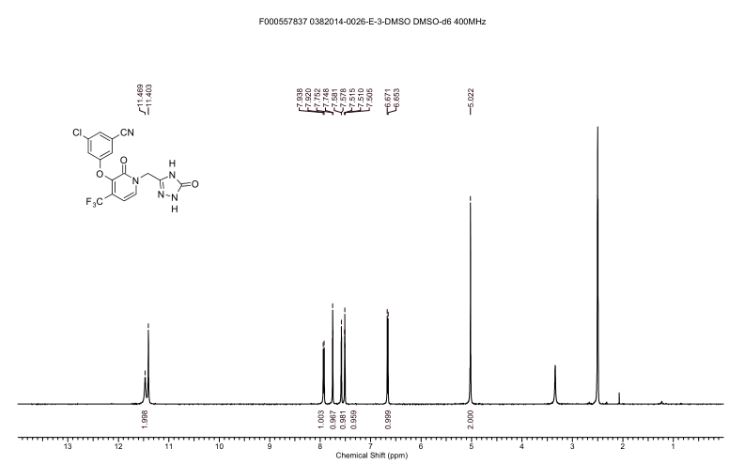
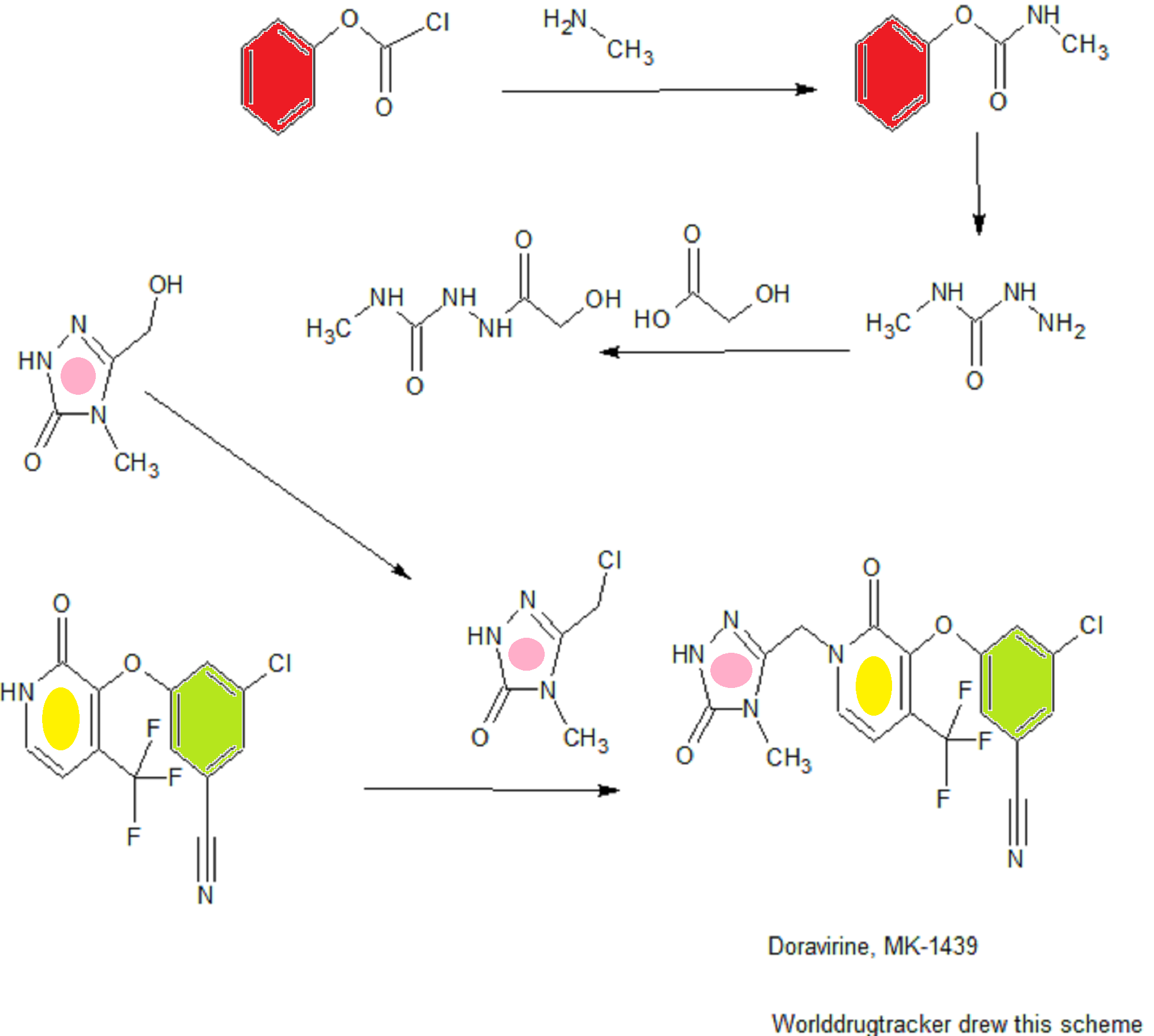
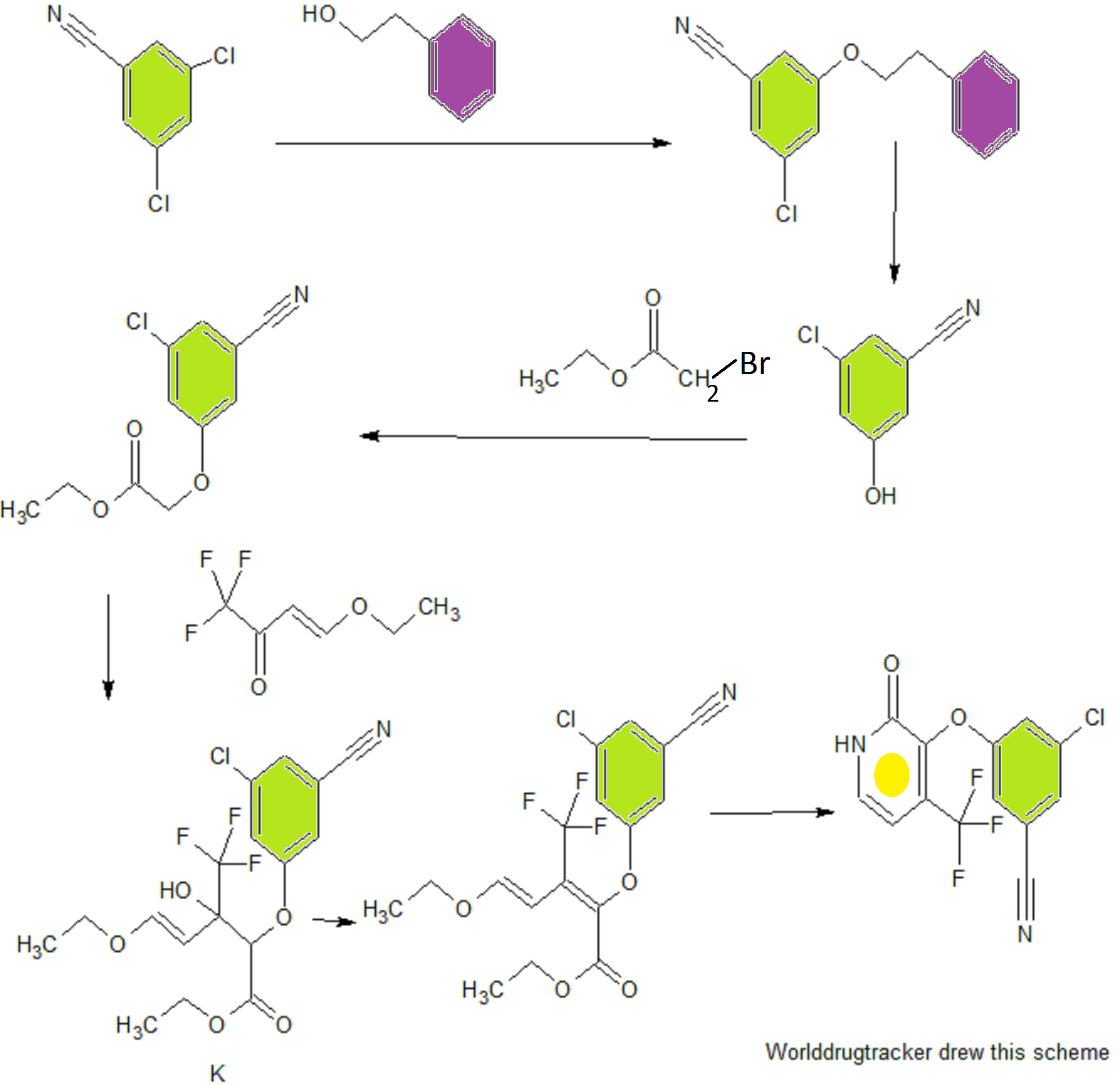

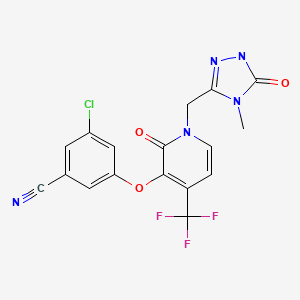





















































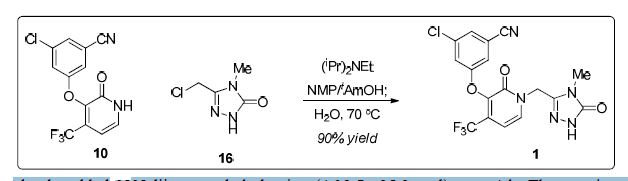
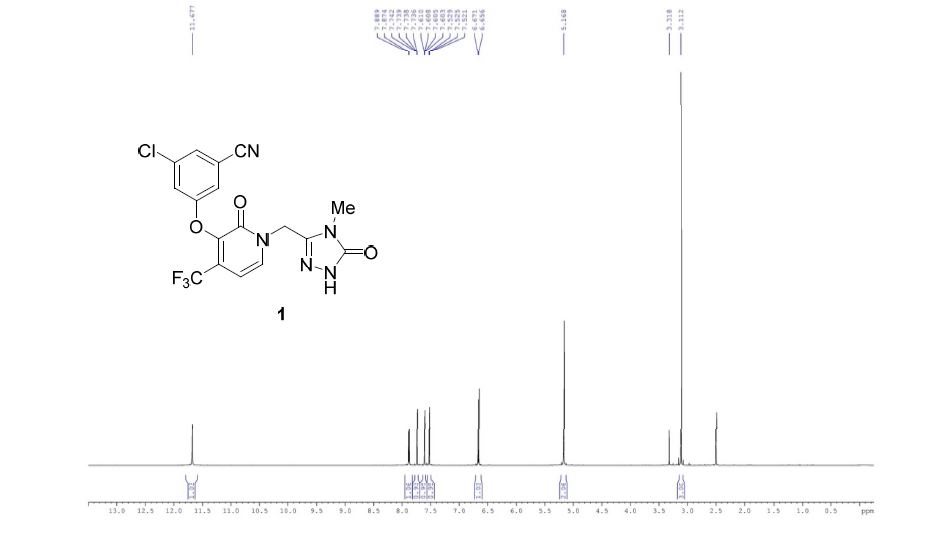




























![[(3S)-oxolan-3-yl] N-[(2S,3R)-4-[(4-aminophenyl)sulfonyl-(2-methylpropyl)amino]-3-hydroxy-1-phenylbutan-2-yl]carbamate NMR spectra analysis, Chemical CAS NO. 161814-49-9 NMR spectral analysis, [(3S)-oxolan-3-yl] N-[(2S,3R)-4-[(4-aminophenyl)sulfonyl-(2-methylpropyl)amino]-3-hydroxy-1-phenylbutan-2-yl]carbamate H-NMR spectrum](https://i0.wp.com/pic11.molbase.net/nmr/nmr_image/2014-07-23/000/115/669/161814-49-9-1h.png)
![[(3S)-oxolan-3-yl] N-[(2S,3R)-4-[(4-aminophenyl)sulfonyl-(2-methylpropyl)amino]-3-hydroxy-1-phenylbutan-2-yl]carbamate NMR spectra analysis, Chemical CAS NO. 161814-49-9 NMR spectral analysis, [(3S)-oxolan-3-yl] N-[(2S,3R)-4-[(4-aminophenyl)sulfonyl-(2-methylpropyl)amino]-3-hydroxy-1-phenylbutan-2-yl]carbamate C-NMR spectrum](https://i0.wp.com/pic11.molbase.net/nmr/nmr_image/2014-07-23/000/115/669/161814-49-9-13c.png)


























































































































.svg){kind=link}
{kind=link}