
FLAGS AND HITS
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO FACEBOOK
...................................................................Join me on twitter
FACEBOOK
...................................................................Join me on twitter
 ..................................................................Join me on google plus
..................................................................Join me on google plus
 Googleplus
GoogleplusMYSELF

Casdatifan


Casdatifan
CAS 2709069-30-5
MF C21H17F4NO3S, 439.4 g/mol
(5R,6S,8R)-3,5,6-trifluoro-8-[(1S,2R)-2-fluoro-1-hydroxy-7-(methanesulfonyl)-2,3-dihydro-1H-inden-4-yl]-5,6,7,8-tetrahydronaphthalene-1-carbonitrile
(5R,6S,8R)-3,5,6-trifluoro-8-[(1S,2R)-2-fluoro-1-hydroxy-7-methylsulfonyl-2,3-dihydro-1H-inden-4-yl]-5,6,7,8-tetrahydronaphthalene-1-carbonitrile
(5R,6S,8R)-3,5,6-trifluoro-8-[(1S,2R)-2-fluoro-1-hydroxy-7-methylsulfonyl-2,3-dihydro-1H-inden-4-yl]-5,6,7,8-tetrahydronaphthalene-1-carbonitrile
(5R,6S,8R)-3,5,6-trifluoro-8-[(1S,2R)-2-fluoro-1-hydroxy-7-methanesulfonyl-2, 3-dihydro-1H-inden-4-yl]-5,6,7,8-tetrahydronaphthalene-1-carbonitrile
hypoxia-inducible factor (HIF) inhibitor, antineoplastic, AB 521, DP73UWL6LE
Casdatifan is an orally bioavailable allosteric inhibitor of hypoxia inducible factor (HIF)-2alpha, with potential antineoplastic activity. Upon oral administration, casdatifan targets and allosterically binds to a hydrophobic pocket on HIF-2alpha leading to a confirmational change that prevents HIF-2alpha heterodimerization with HIF-1beta and binding to the hypoxia response element (HRE) binding site on DNA. This results in decreased transcription and expression of HIF-2alpha downstream target genes, many of which regulate tumor cell growth and survival. Blocking HIF-2alpha reduces the proliferation of HIF-2alpha-expressing tumor cells. HIF-2alpha, a heterodimeric transcription factor overexpressed under hypoxic conditions in many cancer cell types, promotes proliferation, progression and metastasis of tumors.
- A Phase 1 Study of AB521 Monotherapy and Combination Therapies in Renal Cell Carcinoma and Other Solid TumorsCTID: NCT05536141Phase: Phase 1Status: RecruitingDate: 2026-01-02
- A Relative Bioavailability Study and Food Effect Study of AB521 in Healthy Adult VolunteersCTID: NCT05999513Phase: Phase 1Status: CompletedDate: 2024-10-17
- A Study to Investigate the Efficacy and Safety of Volrustomig ± Casdatifan vs Nivolumab + Ipilimumab as 1L Treatment for Advanced ccRCCCTID: NCT07000149Phase: Phase 3Status: Active, not recruitingDate: 2025-11-14
- Study of Zanzalintinib (XL092) + AB521 and Zanzalintinib + AB521 + Nivolumab in Participants With Advanced Clear Cell Renal Cell Carcinoma (ccRCC) or Other Advanced Solid Tumors (STELLAR-009)CTID: NCT06191796Phase: Phase 1Status: TerminatedDate: 2025-06-12
- Drug-Drug Interaction Study of Casdatifan in Healthy Adult Participants (ARC-29)CTID: NCT06919991Phase: Phase 1Status: CompletedDate: 2025-11-13
SYN
https://pubs.acs.org/doi/10.1021/acs.oprd.4c00497




PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2021188769&_cid=P12-MKDEE0-87371-1
Example 215: (5R,6S,8R)-3,5,6-trifluoro-8-[(1S,2R)-2-fluoro-1-hydroxy-7-methanesulfonyl-2, 3-dihydro-1H-inden-4-yl]-5,6,7,8-tetrahydronaphthalene-1-carbonitrile



ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect

Phone : +91 897704 2010 / +91 9177075735
Email : info@anaxlab.com
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=US442743749&_cid=P12-MKDEE0-87371-1
Example 2: Synthesis of (5R,6S,8R)-3,5,6-trifluoro-8-[(1S,2R)-2-fluoro-1-hydroxy-7-methanesulfonyl-2,3-dihydro-1H-inden-4-yl]-5,6,7,8-tetrahydronaphthalene-1-carbonitrile

Step i: Synthesis of Compound 11

Product 10 of step h (37.85 g, 78.28 mmol, 1.0 equiv.) was dissolved in THF (400 mL) at 23° C. A solution of hydrochloric acid (320 mL, 6M) was added dropwise over 20 min, and the mixture was stirred at 30° C. for 4 h. After this time, the reaction reached completion, as shown by LC/MS (MeCN/H 2O—20%→100%, 6 min). The reaction mixture was diluted with water (1 L) and EtOAc (0.6 L), back-extracted twice with EtOAc, and washed with water, sat. sol. NaHCO 3, and brine. The organic layer was dried over Na 2SO 4, filtered, and concentrated. The material (32.25 g, 94%) was triturated with CH 2Cl 2 (45 mL) at 45° C., filtered, and washed with a minimum of cold CH 2Cl 2 and cold hexanes to afford 11 as a white crystalline solid (26.15 g, 76%, 12:1 dr). 1H NMR (400 MHZ, DMSO-d 6) δ 7.96 (ddd, J=8.3, 2.7, 1.3 Hz, 1H), 7.89 (dd, J=8.9, 2.7 Hz, 1H), 7.57 (d, J=8.1 Hz, 1H), 6.66 (d, J=8.1 Hz, 1H), 5.95 (ddd, J=51.2, 13.5, 2.2 Hz, 1H), 5.89 (d, J=5.6 Hz, 1H), 5.47 (ddd, J=10.0, 6.2, 4.9 Hz, 1H), 5.26 (qd, J=52.5, 5.4 Hz, 1H), 5.12 (tddd, J=47.4, 18.7, 10.3, 2.7 Hz, 1H), 4.83 (t, J=5.4 Hz, 1H), 3.30 (s, 3H), 3.28-3.13 (m, 2H), 2.71-2.60 (m, 1H), 2.02-1.85 (m, 1H). 19F NMR (376 MHZ, DMSO-d 6) δ −112.3, −179.6, −196.7, −199.4. ESI MS [M+Na] + for C 21H 17F 4NO 3SNa, calcd 462.0, found 461.9.
PAT
- Tetralin and tetrahydroquinoline compounds as inhibitors of hif-2alphaPublication Number: US-2023021476-A1Priority Date: 2020-03-19
- Tetralin and tetrahydroquinoline compounds as inhibitors of HIF-2αPublication Number: US-11407712-B2Priority Date: 2020-03-19Grant Date: 2022-08-09
- Tetralin and tetrahydroquinoline compounds as inhibitors of HIF-2alphaPublication Number: US-12103907-B2Priority Date: 2020-03-19Grant Date: 2024-10-01
- Tetralin and tetrahydroquinoline compounds as HIF-2α inhibitorsPublication Number: CN-115298165-APriority Date: 2020-03-19
- Tetralin and tetrahydroquinoline compounds as inhibitors of hif-2alphaPublication Number: US-2021317079-A1Priority Date: 2020-03-19
- Tetralin and tetrahydroquinoline compounds as inhibitors of hif-2alphaPublication Number: WO-2021188769-A1Priority Date: 2020-03-19
- Tetralin and tetrahydroquinoline compounds as inhibitors of hif-2alphaPublication Number: US-2024254079-A1Priority Date: 2020-03-19
- Process for preparing tetralin compoundsPublication Number: US-12145901-B1Priority Date: 2021-09-17Grant Date: 2024-11-19
- Tetralin and tetrahydroquinoline compounds as inhibitors of HIF-2alphaPublication Number: US-11787762-B2Priority Date: 2020-03-19Grant Date: 2023-10-17
- Tetrahydronaphthalene and tetrahydroquinoline compounds as HIF-2 alpha inhibitorsPublication Number: CN-119118872-APriority Date: 2020-03-19
- Tetralin and tetrahydroquinoline compounds as HIF-2α inhibitorsPublication Number: CN-115298165-BPriority Date: 2020-03-19Grant Date: 2024-09-17
- Tetralin and tetrahydroquinoline compounds as inhibitors of hif-2alphaPublication Number: US-2025214930-A1Priority Date: 2020-03-19
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect
Phone : +91 897704 2010 / +91 9177075735
Email : info@anaxlab.com



AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

……
////////Casdatifan, hypoxia-inducible factor (HIF) inhibitor, antineoplastic, AB 521, DP73UWL6LE
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions,, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect

Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
Cambritaxestat


Cambritaxestat
CAS 1979939-16-6
MFC25H22ClF3N4O2 MW502.9 g/mol
N-[(1S)-1-(4-chlorophenyl)ethyl]-3-[3-[[4-(trifluoromethoxy)phenyl]methyl]imidazo[4,5-b]pyridin-2-yl]propanamide
N-[(1S)-1-(4-chlorophenyl)ethyl]-3-(3-{[4-(trifluoromethoxy)phenyl]methyl}-3H-imidazo[4,5-b]pyridin-2-yl)propanamide
autotaxin inhibitor, antineoplastic, Orphan Drug, IOA 289, IOA-289, IOA289, LYY3P2KA27, CRT 0273750
- OriginatorCancer Research Technology; Merck & Co
- DeveloperiOnctura
- ClassAntifibrotics; Antineoplastics; Small molecules
- Mechanism of ActionAngiogenesis inhibitors; Cell proliferation inhibitors; ENPP2 protein inhibitors
- Orphan Drug StatusYes – Pancreatic cancer
- Phase I/IIPancreatic cancer
- Phase ISolid tumours
- PreclinicalNon-alcoholic steatohepatitis
- 14 Oct 2025Efficacy and adverse event data from a phase I/II trial in Pancreatic cancer released by iOnctura
- 04 Oct 2024Cambritaxestat is still in phase-I development in Solid-tumours (In volunteers) in Italy (PO, Capsule) (NCT05027568)
- 31 May 2024Efficacy and adverse event data from a phase I/II trial in Pancreatic cancer presented at the 60th Annual Meeting of the American Society of Clinical Oncology (ASCO-2024)
Cambritaxestat is an autotaxin inhibitor.
Cambritaxestat is an orally bioavailable small molecule inhibitor of autotaxin (ATX; ectonucleotide pyrophosphatase/phosphodiesterase family member 2; ENPP2), with potential antifibrotic and antineoplastic activities. Upon oral administration, cambritaxestat targets and binds to both the substrate pocket and the lysophosphatidic acid (LPA) carrier channel of ATX, thereby inhibiting the activity of ATX. This both directly inhibits the proliferation of tumor cells and reduces fibrosis in the tumor microenvironment (TME), allowing lymphocytes to infiltrate into the tumor and enhancing immune responses against tumor cells. ATX, a secreted glycoprotein with lysophospholipase D activity, hydrolyzes lysophosphatidylcholine (LPC) to LPA. LPA-mediated signaling plays an important role in cellular migration, proliferation and survival in fibrotic response. ATX and LPA are overexpressed in many tumors.
- A Study to Assess an ATX Inhibitor (IOA-289) in Healthy VolunteersCTID: NCT05027568Phase: Phase 1Status: CompletedDate: 2025-03-20
- A Study to Assess an ATX Inhibitor (IOA-289) in Patients with Metastatic Pancreatic CancerCTID: NCT05586516Phase: Phase 1/Phase 2Status: Active, not recruitingDate: 2025-03-20
SYN
WO2016/124939
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2016124939&_cid=P22-MKBYYZ-98558-1


SYN

WO2016/124939 describes various ATX inhibitor compounds and their use in the treatment of proliferative disorders in which ATX activity is implicated, including Compound 1.
Compound 1 is example 40 in WO2016/124939, which document is incorporated herein by reference in its entirety. WO2016/124939 describes over 200 examples. Compound 1’s structure is according to Formula I.

PAT
- Autotaxin inhibitory compoundsPublication Number: US-10654846-B2Priority Date: 2015-02-06Grant Date: 2020-05-19
- Autotaxin inhibitory compoundsPublication Number: EP-3253737-B3Priority Date: 2015-02-06Grant Date: 2024-05-29
- Autotaxin inhibitor compoundsPublication Number: ES-2778898-T7Priority Date: 2015-02-06Grant Date: 2024-11-15
- Autotaxin inhibitory compoundsPublication Number: EP-3253737-A1Priority Date: 2015-02-06
- Home chemokine inhibiting compoundsPublication Number: CN-107428752-BPriority Date: 2015-02-06Grant Date: 2021-06-29
- Autotaxin inhibitory compoundsPublication Number: US-11453666-B2Priority Date: 2015-02-06Grant Date: 2022-09-27
- Autotaxin Inhibitory CompundsPublication Number: US-2020283435-A1Priority Date: 2015-02-06
- Autotaxin inhibitory compoundsPublication Number: WO-2016124939-A1Priority Date: 2015-02-06
- A pi3k-delta inhibitor for the treatment of pancreatic cancerPublication Number: WO-2022207648-A1Priority Date: 2021-03-29
- A pi3k-delta inhibitor for the treatment of pancreatic cancerPublication Number: EP-4313059-A1Priority Date: 2021-03-29
- A pi3k-delta inhibitor for the treatment of pancreatic cancerPublication Number: US-2024216385-A1Priority Date: 2021-03-29
- Autotaxin inhibitory compoundsPublication Number: EP-3253737-B1Priority Date: 2015-02-06Grant Date: 2020-01-08
- Autotaxin inhibitory compoundsPublication Number: US-2018016274-A1Priority Date: 2015-02-06
- Autotaxin (atx) inhibitor for the treatment of pancreatic cancerPublication Number: WO-2022258693-A1Priority Date: 2021-06-09
- Autotaxin (atx) inhibitor for the treatment of pancreatic cancerPublication Number: US-2025057820-A1Priority Date: 2021-06-09
- Autotaxin (atx) inhibitor for the treatment of pancreatic cancerPublication Number: EP-4351563-A1Priority Date: 2021-06-09
- Autotaxin (ATX) inhibitors for the treatment of pancreatic cancerPublication Number: CN-117295496-APriority Date: 2021-06-09
- PI3K-δ inhibitors for the treatment of pancreatic cancerPublication Number: CN-116997340-APriority Date: 2021-03-29
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
- Characterization and translational development of IOA-289, a novel autotaxin inhibitor for the treatment of solid tumorsPublication Name: Immuno-Oncology and TechnologyPublication Date: 2023-06PMCID: PMC10205783PMID: 37234285DOI: 10.1016/j.iotech.2023.100384
- The IUPHAR Guide to Immunopharmacology: connecting immunology and pharmacologyPublication Name: ImmunologyPublication Date: 2020-03-02PMCID: PMC7160657PMID: 32020584DOI: 10.1111/imm.13175
- Discovery of potent inhibitors of the lysophospholipase autotaxinPublication Name: Bioorganic & Medicinal Chemistry LettersPublication Date: 2016-11-15PMID: 27780639DOI: 10.1016/j.bmcl.2016.10.036
///////Cambritaxestat, autotaxin inhibitor, antineoplastic, Orphan Drug, IOA 289, IOA-289, IOA289, LYY3P2KA27, CRT 0273750
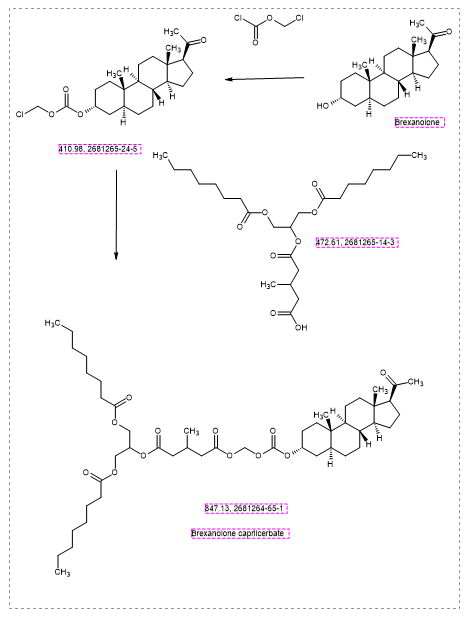
Brexanolone caprilcerbate



Brexanolone caprilcerbate
CAS 2681264-65-1
MFC48H78O12 MW 847.1 g/mol
1-O-[[(3R,5S,8R,9S,10S,13S,14S,17S)-17-acetyl-10,13-dimethyl-2,3,4,5,6,7,8,9,11,12,14,15,16,17-tetradecahydro-1H-cyclopenta[a]phenanthren-3-yl]oxycarbonyloxymethyl] 5-O-[1,3-di(octanoyloxy)propan-2-yl] 3-methylpentanedioate
1-[1,3-bis(octanoyloxy)propan-2-yl] 5-[({[(20-oxo-5α-pregnan3α-yl)oxy]carbonyl}oxy)methyl] 3-methylpentanedioate
GABAA receptor positive allosteric modulator, K3KLQ9T6WM, PHASE 2,
Brexanolone caprilcerbate (INNTooltip International Nonproprietary Name; developmental code names LYT-300, SPT-300) is an orally active prodrug of brexanolone (allopregnanolone) which is under development for the treatment of anxiety disorders.[1][2][3][4] It is a absorbed via the lymphatic system with oral administration.[5] The drug is being developed by Seaport Therapeutics and PureTech Health.[1][2] As of January 2025, it is in phase 2 clinical trials.[1]

SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=US335021515&_cid=P22-MKAJEO-33027-1
PAT
- Lipid prodrugs of neurosteroidsPublication Number: WO-2021159021-A1Priority Date: 2020-02-05
- Lipid prodrugs of neurosteroidsPublication Number: US-2023338552-A1Priority Date: 2020-02-05
- Lipid prodrugs of neurosteroidsPublication Number: US-2022395513-A1Priority Date: 2020-02-05
- Lipid prodrugs of neurosteroidsPublication Number: US-2021268115-A1Priority Date: 2020-02-05
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
References
- “Allopregnanolone prodrug”. AdisInsight. 28 January 2025. Retrieved 26 February 2025.
- “Delving into the Latest Updates on Brexanolone caprilcerbate with Synapse”. Synapse. 15 February 2025. Retrieved 26 February 2025.
- “Proposed INN: List 131 International Nonproprietary Names for Pharmaceutical Substances (INN)” (PDF). WHO Drug Information. 38 (2): 270. 2024.
brexanolonum caprilcerbas brexanolone caprilcerbate 1-[1,3-bis(octanoyloxy)propan-2-yl] 5-[({[(20-oxo-5α-pregnan3α-yl)oxy]carbonyl}oxy)methyl] 3-methylpentanedioate GABAA receptor positive allosteric modulator C48H78O12 2681264-65-1
- Carlini SV, Osborne LM, Deligiannidis KM (December 2023). “Current pharmacotherapy approaches and novel GABAergic antidepressant development in postpartum depression”. Dialogues in Clinical Neuroscience. 25 (1): 92–100. doi:10.1080/19585969.2023.2262464. PMC 10557560. PMID 37796239.
- Alashal N, Hussain N (2025). “Approach to the use of rescue medications in children for prolonged epileptic seizures in the community”. Paediatrics and Child Health. 35 (4): 113–117. doi:10.1016/j.paed.2025.01.004.
| Clinical data | |
|---|---|
| Other names | LYT-300; LYT300; SPT-300; SPT300; Allopregnanolone 3-O-caprilcerbate |
| Routes of administration | Oral[1] |
| Drug class | GABAA receptor positive allosteric modulator; Neurosteroid |
| Identifiers | |
| IUPAC name | |
| CAS Number | 2681264-65-1 |
| PubChem CID | 158098654 |
| UNII | K3KLQ9T6WM |
| Chemical and physical data | |
| Formula | C48H76O12 |
| Molar mass | 845.124 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
/////////////Brexanolone caprilcerbate, GABAA receptor positive allosteric modulator, K3KLQ9T6WM, PHASE 2,
Branosotine
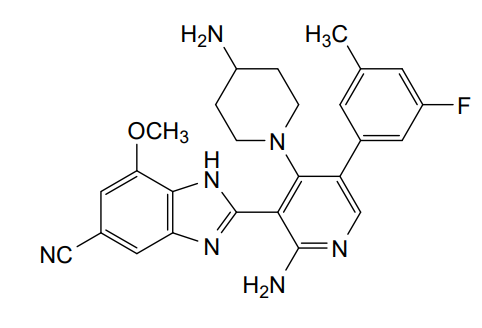
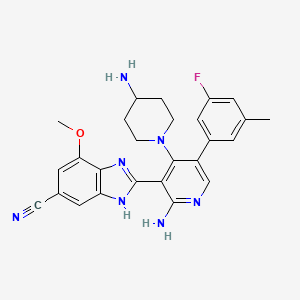
Branosotine
CAS 2412849-26-2
MF C26H26FN7O MW471.5 g/mol
2-[2-amino-4-(4-aminopiperidin-1-yl)-5-(3-fluoro-5-methylphenyl)-3-pyridinyl]-7-methoxy-3H-benzimidazole-5-carbonitrile
2-[2-amino-4-(4-aminopiperidin-1-yl)-5-(3-fluoro-5-
methylphenyl)pyridin-3-yl]-7-methoxy-1H-1,3-benzimidazole-5-
carbonitrile
somatostatin receptor agonist (veterinary use), 4L2VN6D3D8
Branosotine is a small molecule drug. The usage of the INN stem ‘-sotine’ in the name indicates that Branosotine is a non-peptidic somatostatin receptor agonist. Branosotine has a monoisotopic molecular weight of 471.22 Da.
SYN
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2020061046&_cid=P21-MK9408-98104-1
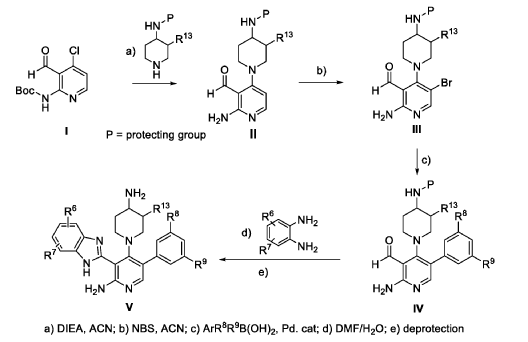
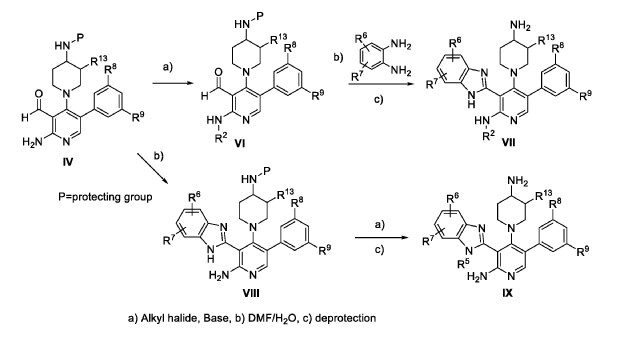
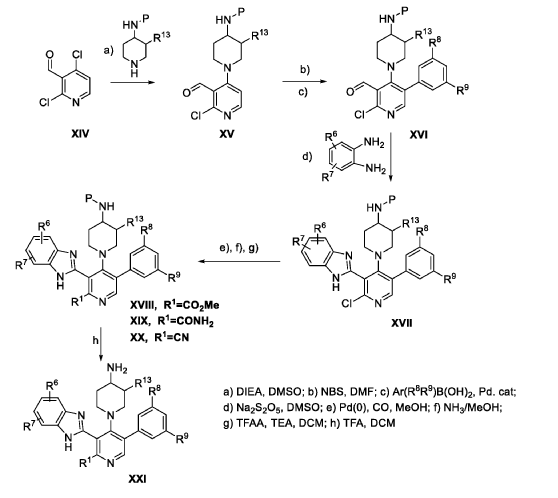
The following examples are provided for illustrative purposes only and not to limit the scope of the claims provided herein.
Example 1 : 2-[2-amino-4-(4-aminopiperidin-1-yl)-5-(3-fluoro-5-methylphenyl)pyridin- 3-yl]-4-methoxy-1H-1,3-benzodiazole-6-carbonitrile (1-1)
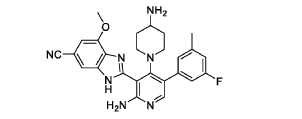



PAT
- Somatostatin modulators and uses thereofPublication Number: EP-4548974-A2Priority Date: 2018-09-18
- Somatostatin modulators and uses thereofPublication Number: EP-3853218-B1Priority Date: 2018-09-18Grant Date: 2025-02-19
- Somatostatin modulators and uses thereofPublication Number: TW-I852944-BPriority Date: 2018-09-18Grant Date: 2024-08-21
- Somatostatin modulator and its usePublication Number: JP-2022501342-APriority Date: 2018-09-18
- Somatostatin modulators and uses thereofPublication Number: US-11834462-B2Priority Date: 2018-09-18Grant Date: 2023-12-05
- Somatostatin modulators and uses thereofPublication Number: US-2022048924-A1Priority Date: 2018-09-18
- Somatostatin modulators and uses thereofPublication Number: US-2020283453-A1Priority Date: 2018-09-18
- Somatostatin modulators and their usesPublication Number: JP-7431813-B2Priority Date: 2018-09-18Grant Date: 2024-02-15
- Somatostatin modulators and uses thereofPublication Number: US-2023022513-A1Priority Date: 2018-09-18
- Somatostatin modulators for treating pituitary adenomasPublication Number: WO-2021076448-A1Priority Date: 2019-10-14
- Somatostatin modulators and uses thereofPublication Number: US-2020087318-A1Priority Date: 2018-09-18
- Somatostatin modulators and uses thereofPublication Number: US-10696689-B2Priority Date: 2018-09-18Grant Date: 2020-06-30
- Somatostatin modulators and uses thereofPublication Number: TW-202024095-APriority Date: 2018-09-18
- Somatostatin modulators and uses thereofPublication Number: US-11186590-B2Priority Date: 2018-09-18Grant Date: 2021-11-30
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
///////////Branosotine, somatostatin receptor agonist (veterinary use), 4L2VN6D3D8
Bosmolisib
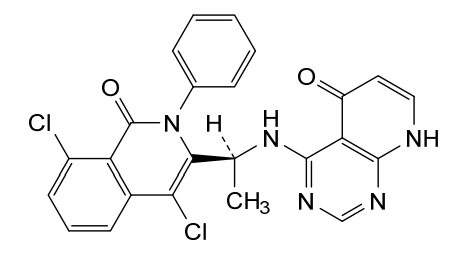
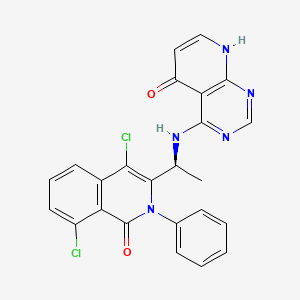
Bosmolisib
CAS 2055765-77-8
MF 2055765-77-8 MW478.3 g/mol
4-{[(1S)-1-(4,8-dichloro-1-oxo-2-phenyl-1,2-dihydroisoquinolin-3-yl)ethyl]amino}pyrido[2,3-d]pyrimidin-5(8H)-one
4-[[(1S)-1-(4,8-dichloro-1-oxo-2-phenylisoquinolin-3-yl)ethyl]amino]-8H-pyrido[2,3-d]pyrimidin-5-one
phosphatidylinositol 3-kinase (PI3K) inhibitor, antineoplastic, BR 101801, FJ5CTS1VNJ
- A Study of Bosmolisib (BR101801) in Participants With R/R PTCL.CTID: NCT07180771Phase: Phase 2Status: Not yet recruitingDate: 2025-09-18
- BR101801 in Adult Patients With Advanced Hematologic Malignancies(Phase I)CTID: NCT04018248Phase: Phase 1Status: CompletedDate: 2025-09-10
Bosmolisib is an orally bioavailable inhibitor of phosphoinositide 3-kinase delta (PI3-kinase subunit delta; PI3K-delta; PI3Kdelta) and DNA-dependent protein kinase (DNA-PK), with potential antineoplastic and immunomodulating activities. Upon oral administration, bosmolisib inhibits the activity of both PI3K-delta and DNA-PK. This prevents PI3K-mediated signaling pathways and may lead to the inhibition of cancer cell growth in PI3K-overexpressing tumor cells. Specifically, since PI3K regulates c-myc expression, inhibition of PI3K signaling may lead to a decrease in proliferation of c-myc-expressing tumor cells. Also, by inhibiting the activity of DNA-PK, bosmolisib interferes with the non-homologous end joining (NHEJ) process and prevents the repair of DNA double strand breaks (DSBs) caused by ionizing radiation or chemotherapeutic treatment. This increases chemo- and radiotherapy cytotoxicity by inhibiting the ability of tumor cells to repair damaged DNA. The PI3K pathway is upregulated in a variety of tumors and plays an important role in regulating cancer cell proliferation, growth, and survival. DNA-PK is activated upon DNA damage and plays a key role in repairing double-stranded DNA breaks. The enhanced ability of tumor cells to repair DSBs plays a major role in the resistance of tumor cells to chemo- and radiotherapy. In addition, bosmolisib is able to decrease Tregs and increase CD8 lymphocytes.
- OriginatorBoryung Pharmaceutical
- ClassAntineoplastics; Small molecules
- Mechanism of ActionDNA-activated protein kinase inhibitors; Phosphatidylinositol 3 kinase delta inhibitors; Phosphatidylinositol 3 kinase gamma inhibitors
- Phase IHaematological malignancies
- PreclinicalColorectal cancer
- 18 Sep 2025Boryung Pharmaceutical plans a phase II trial for Peripheral T Cell Lymphoma and Nodal T-follicular helper cell lymphoma (Second-line therapy or greater) in September 2025 (PO, Capsule) (NCT07180771)
- 06 Jan 2025Chemical structure information added.
- 09 Dec 2023Updated efficacy and adverse event data from a phase I trial in Hematological malignancies presented at the 65th American Society of Hematology Annual Meeting and Exposition (ASH-2023
SYN
WO 2016/204429.
SYN
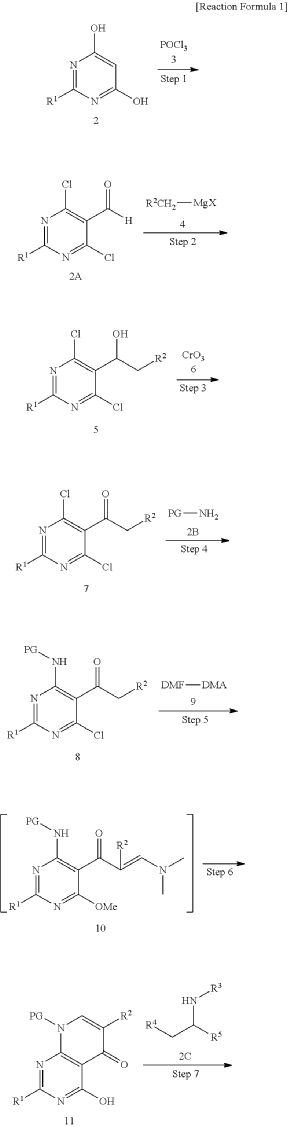
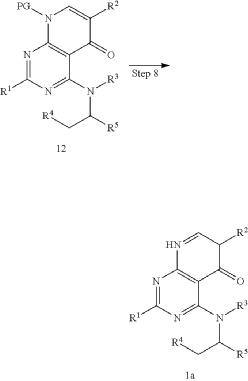
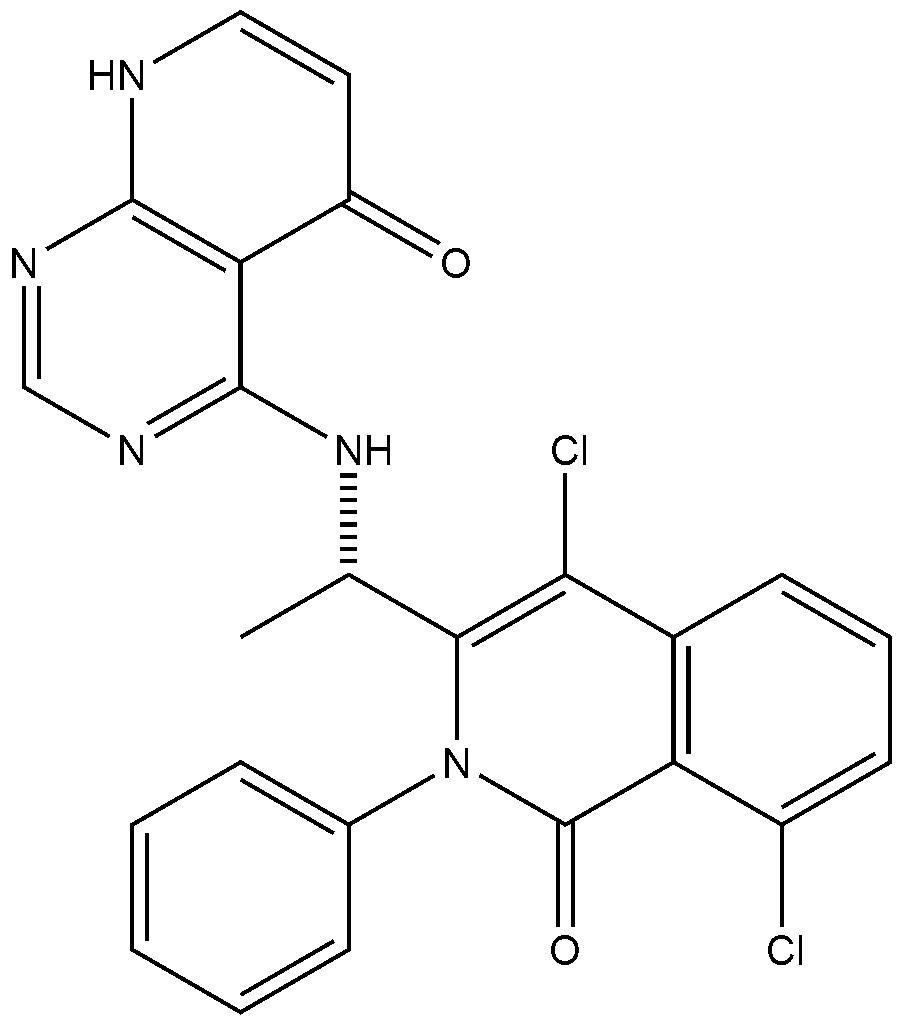
xample 1. Preparation of (S)-4-((1-(4,8-dichloro-1-oxo-2-phenyl-1,2-dihydroisoquinolin-3-yl)ethyl)amino)pyrido[2,3-d]pyrimidin-5(8H)-one
[116](S)-4-((1-(4,8-dichloro-1-oxo-2-phenyl-1,2-dihydroisoquinolin-3-yl)ethyl)amino)pyrido[2,3-d]pyrimidin-5(8H)-one (4-((1-(4,8-dichloro-1-oxo-2-phenyl-1,2-dihydroisoquinolin -3-yl)ethyl)amino)pyrido[2,3-d]pyrimidin-5(8H)-one) represented by the chemical formula 3 above was prepared by the same method as that described in Example 10 of International Patent Publication No.
WO 2016/204429.
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2016204429&_cid=P22-MK6A2W-95428-1
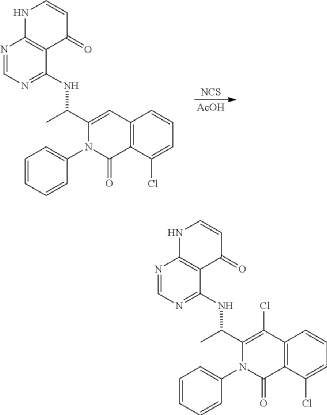
<Example 10> Preparation of (S)-4-((l-(4,8-dichloro-1-oxo-2-phenyl-1,2-dihydroisoquinolin-3-yl)ethyl)amino)pyrido [2,3-d]pyrimidin-5(8H)-one
In Example 5, 50 mg (0.113 vol) of (S)-4— ((1-(8-chloro-1—oxo-2-phenyl-1,2-dihydroisoquinolin-3-yl)ethyl)amino)pyrido [2, 3-d]pyrimidin-5(8H)-one prepared was dissolved in 2 mL of acetic acid, and then 17 mg (0.124 vol) of N—chlorosuccinimide (NCS) was added. The mixture was stirred at 50 ° C for 15 hours, filtered under reduced pressure, neutralized using an aqueous sodium bicarbonate solution, and then the organic layer extracted by adding dichloromethane and water was dried (Na 2 SO 4 ), filtered, concentrated under reduced pressure, and separated by column chromatography (SiO 2 , eluent: dichloromethane/methanol, 30/1 -> dichloromethane/methanol, 10/1) to afford 25 mg (0.052 mmol, 46% yield) of compound (S)— 4-((1— (4,8-dichloro-1-oxo-2-phenyl-1,2-dihydroisoquinolin-3-yl)ethyl)amino)pyrido[2, 3-d]pyramidin-5(8H)-one as a pale yellow solid.
LH NMR (300 MHz, CDC13) δ 10.99 (d, J = 4.8 Hz, 1Ή), 8.25 (s, 1H) , 7.95(dd, JJ = 1.9 Hz, J = 7.5 Hz, 1H), 7.75 (d, J = 7.8 Hz, 1H) , 7.46-7.62 (m, 6H), 7.20 (d, J = 6.7 Hz, 1H) , 6.3 (d, J = 7.5 Hz, 1H), 5.04 (t , J = 67.2 Hz, 1H) , 1.67 (d, J = 7.2 Hz, 3H) .
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=US214732247&_cid=P22-MK69S5-86256-1
Example 10: Preparation of (S)-4-((1-(4,8-dichloro-1-oxo-2-phenyl-1,2-dihydroisoquinoline-3-yl)ethyl)amino)pyrido[2,3-d]pyrimidine-5(8H)-one
| 50 mg (0.113 mmol) of (S)-4-((1-(8-chloro-1-oxo-2-phenyl-1,2-dihydroisoquinoline-3-yl)ethyl)amino)pyrido[2,3-d]pyrimidine-5(8H)-one prepared in Example 5 was dissolved in 2 mL of acetic acid, to which 17 mg (0.124 mmol) of N-chlorosuccinimide (NCS) was added, followed by stirring at 50° C. for 15 hours. The reaction mixture was filtered under reduced pressure. Saturated sodiumbicarbonate aqueous solution was added thereto, followed by neutralization. Dichloromethane and water were added thereto, followed by extraction. The extracted organic layer was dried (Na 2SO 4), filtered, and concentrated under reduced pressure. The residue was separated by column chromatography (SiO 2, eluent: dichloromethane/methanol, 30/1→dichloromethane/methanol, 10/1) to give 25 mg of the target compound (S)-4-((1-(4,8-dichloro-1-oxo-2-phenyl-1,2-dihydroisoquinoline-3-yl)ethyl)amino)pyrido[2,3-d]pyrimidine-5(8H)-one as a pale yellow solid (0.052 mmol, yield: 46%). |
PAT
- A pharmaceutical composition for preventing or treating a heteroaryl derivative or a pharmaceutically acceptable salt thereof, a method for producing the same, and a PI3 kinase-related disease containing the heteroaryl derivative as an active ingredient.Publication Number: JP-6808905-B2Priority Date: 2015-06-18Grant Date: 2021-01-06
- Heteroaryl derivative or pharmaceutically acceptable salt thereof, method of preparation thereof and pharmaceutical composition to prevent or treat diseases associated with PI3 kinases, which contains the same as active principlePublication Number: ES-2816050-T3Priority Date: 2015-06-18Grant Date: 2021-03-31
- Heteroaryl derivative or pharmaceutically acceptable salt thereof, preparation method therefor, and pharmaceutical compostion for preventing or treating diseases associated with pi3 kinases, containing same as active ingredientPublication Number: US-2018105527-A1Priority Date: 2015-06-18
- Heteroaryl derivative or pharmaceutically acceptable salt thereof, preparation method therefor, and pharmaceutical composition for preventing or treating diseases associated with pi3 kinases, containing same as active ingredientPublication Number: EP-3312175-B1Priority Date: 2015-06-18Grant Date: 2020-07-22
- Heteroaryl derivatives or pharmaceutically acceptable salts thereof, preparation method thereof and pharmaceutical composition for use in preventing or treating pi3 kinase related diseasesPublication Number: TW-I616446-BPriority Date: 2015-06-18Grant Date: 2018-03-01
- HETEROARYL DERIVATIVES OR PHARMACEUTICALLY ACCEPTABLE SALTS THEREOF, PROCESS FOR PRODUCING THE SAME, AND PHARMACEUTICAL COMPOSITIONS FOR PREVENTING OR TREATING PI3-KINASE RELATED DISEASES COMPRISING THE SAME AS THE ACTIVE INGREDIENTPublication Number: JP-2018522852-APriority Date: 2015-06-18
- Heteroaryl derivative or pharmaceutically acceptable salt thereof, preparation method therefor, and pharmaceutical composition for preventing or treating diseases associated with PI3 kinases, containing same as active ingredientPublication Number: US-10526337-B2Priority Date: 2015-06-18Grant Date: 2020-01-07
- Heteroaryl derivative or a pharmaceutically acceptable salt thereof, a method for production thereof and a pharmaceutical composition for preventing or treating diseases associated with pi3 kinases, containing said active substancePublication Number: RU-2719367-C2Priority Date: 2015-06-18Grant Date: 2020-04-17
- Heteroaryl derivative or pharmaceutically acceptable salt thereof, preparation method thereof, and pharmaceutical composition comprising same as active ingredient for preventing or treating PI3 kinase-associated diseasesPublication Number: CN-107690433-APriority Date: 2015-06-18
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
- PI3Kδ/γ inhibitor BR101801 extrinsically potentiates effector CD8+ T cell-dependent antitumor immunity and abscopal effect after local irradiationPublication Name: Journal for ImmunoTherapy of CancerPublication Date: 2022-03PMCID: PMC8921929PMID: 35288465DOI: 10.1136/jitc-2021-003762
- Synergistic radiosensitizing effect of BR101801, a specific DNA-dependent protein kinase inhibitor, in various human solid cancer cells and xenograftsPublication Name: American journal of cancer researchPublication Date: 2021PMCID: PMC8640799PMID: 34873471
/////////bosmolisib, phosphatidylinositol 3-kinase (PI3K) inhibitor, antineoplastic, BR 101801, FJ5CTS1VNJ
Beroterkib


Beroterkib
CAS 2095719-92-7
MF C29H31ClFN5O5 MW584.0 g/mol
(2R)-2-(6-{5-chloro-2-[(oxan-4-yl)amino]pyrimidin-4-yl}-1,3-dihydro-2H-1-oxoisoindol-2-yl) -N-[(1S)-1-(3-fluoro-5-methoxyphenyl)-2-hydroxyethyl]propanamide
(2R)-2-[5-[5-chloro-2-(oxan-4-ylamino)pyrimidin-4-yl]-3-oxo-1H-isoindol-2-yl]-N-[(1S)-1-(3-fluoro-5-methoxyphenyl)-2-hydroxyethyl]propanamide
(2R)-2-[5-[5-chloro-2-(oxan-4-ylamino)pyrimidin-4-yl]-3-oxo-1H-isoindol-2-yl]-N-[(1S)-1-(3-fluoro-5-methoxyphenyl)-2-hydroxyethyl]propanamide
extracellular signal-regulated kinases (ERK) inhibitor, antineoplastic, ASTX029, ASTX 029, 14FDK6ISC9, Beroterkib anhydrous, AT 35029
Beroterkib Anhydrous is the anhydrous form of beroterkib, an orally bioavailable inhibitor of the extracellular signal-regulated kinases (ERK) 1 and 2, with potential antineoplastic activity. Upon administration, beroterkib specifically binds to and inhibits both ERK 1 and 2, thereby preventing the activation of mitogen-activated protein kinase (MAPK)/ERK-mediated signal transduction pathways. This results in the inhibition of ERK-dependent tumor cell proliferation and survival. The MAPK/ERK pathway is often upregulated in a variety of tumor cell types and plays a key role in the proliferation, differentiation and survival of tumor cells.
- Study of ASTX029 in Subjects With Advanced Solid TumorsCTID: NCT03520075Phase: Phase 1/Phase 2Status: CompletedDate: 2025-07-03
- Phase I/II Study of a Combination of Decitabine and Cedazuridine (ASTX727) and ASTX029, an ERK Inhibitor, for Patients With RAS Pathway Mutant Myelodysplastic Syndromes and Myelodysplastic/Myeloproliferative NeoplasmsCTID: NCT06284460Phase: Phase 1/Phase 2Status: WithdrawnDate: 2024-10-24
- A Phase 1 Study to Evaluate the Effect of Food on Pharmacokinetics of ASTX029CTID: NCT04466514Phase: Phase 1Status: CompletedDate: 2024-08-02
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2017068412&_cid=P21-MK4TZX-17603-1
Example 685: (2R)-2-(6-{5-chloro-2-[(oxan-4-yl)amino]pyrimidin-4-yl}-1-oxo-2,3-dihydro- 1H-isoindol-2-yl)-N-[(1S)-1-(3-fluoro-5-methoxyphenyl)-2-hydroxyethyl]propanamide

A stirred solution of (R)-2-(6-(5-chloro-2-((oxan-4-yl)amino)pyrimidin-4-yl)-1-oxoisoindolin-2- yl)propanoic acid (70 mg, 0.168 mmol), (S)-2-amino-2-(3-fluoro-5-methoxyphenyl)ethanol, HCl (41 mg, 0.185 mmol) and triethylamine (0.094 ml, 0.672 mmol) in DMF (1 ml) was treated with TBTU (65 mg, 0.202 mmol) and stirred at room temperature overnight. The mixture was diluted with ethyl acetate (20 ml), was washed successively with 1M KHSO4 (10 ml), NaHCO3 (10 ml), brine (2x 10 ml) and then water (4x 10 ml), was dried (MgSO4) and evaporated. The residue was purified by chromatography (SiO2, 12 g column, 0- 5% EtOOH in EtOAc) to give a glass, which was triturated with ether (2 ml) to give a solid. The solid was collected by filtration, washed with ether (2x 1 ml) and dried under vacuum at 50°C overnight to give the titlecompound (64.3 mg, 64.3 %) as a cream solid. 1H NMR (DMSO, 400 MHz) δ 8.56 (1H, d), 8.44 (1H, s), 8.07 ‒ 8.00 (1H, m), 7.97 (1H, dd), 7.74 (1H, d), 7.61 (1H, s), 6.76 ‒ 6.64 (3H, m), 4.99 (1H, q), 4.91 (1H, t), 4.86 ‒ 4.70 (2H, m), 4.60 (1H, d), 4.00 ‒ 3.80 (3H, m), 3.76 (3H, s), 3.60 ‒ 3.47 (2H, m), 3.40 ‒ 3.33 (2H, m), 1.84 (2H, d), 1.59 ‒ 1.39 (5H, m). ). LCMS: [M+H]+ = 584.
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=US237389744&_cid=P21-MK4U5F-21416-1
Example 685: (2R)-2-(6-{5-chloro-2-[(oxan-4-yl)amino]pyrimidin-4-yl}-1-oxo-2,3-dihydro-1H-isoindol-2-yl)-N-[(1S)-1-(3-fluoro-5-methoxyphenyl)-2-hydroxyethyl]propanamide

SYN

PAT
- Conjugates comprising hydroxyalkyl starch and a cytotoxic agent and process for their preparationPublication Number: AU-2011276120-B2Priority Date: 2010-07-09Grant Date: 2013-12-19
- Conjugates comprising hydroxyalkyl starch and a cytotoxic agent and process for their preparationPublication Number: AU-2011276120-A1Priority Date: 2010-07-09
- Combustion modified flexible polyurethane foamPublication Number: GB-2124634-APriority Date: 1982-07-26
- Benzolactam compounds as protein kinase inhibitorsPublication Number: ES-2989326-T3Priority Date: 2015-10-21Grant Date: 2024-11-26
- Protein kinase inhibitor benzolactam compoundsPublication Number: CN-114948963-APriority Date: 2015-10-21
- Benzolactam compounds as protein kinase inhibitorsPublication Number: US-2024368136-A1Priority Date: 2015-10-21
- Protein Kinase Inhibitors Benzolactam CompoundsPublication Number: CN-108617166-BPriority Date: 2015-10-21Grant Date: 2022-05-17
- Benzolactam compounds as protein kinase inhibitorsPublication Number: CN-114948963-BPriority Date: 2015-10-21Grant Date: 2025-05-27
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
REF
- Discovery of ASTX029, a clinical candidate which modulates the phosphorylation and catalytic activity of ERK1/2Publication Name: Journal of Medicinal ChemistryPublication Date: 2021-10-06PMID: 34387469DOI: 10.1021/acs.jmedchem.1c00905
- ASTX029, a Novel Dual-mechanism ERK Inhibitor, Modulates Both the Phosphorylation and Catalytic Activity of ERKPublication Name: Molecular Cancer TherapeuticsPublication Date: 2021-07-30PMID: 34330842DOI: 10.1158/1535-7163.mct-20-0909
//////////////Beroterkib, extracellular signal-regulated kinases (ERK) inhibitor, antineoplastic, ASTX029, ASTX 029, 14FDK6ISC9, Beroterkib anhydrous, AT 35029
Balinatunfib



Balinatunfib
CAS 2248726-53-4
MF C27H24F2N6O2, 502.5 g/mol
(1R,11R)-5-[2-(1-aminocyclobutyl)pyrimidin-5-yl]-18-(difluoromethoxy)-12-methyl-2,9,12-triazapentacyclo[9.8.1.02,10.03,8.014,19]icosa-3(8),4,6,9,14(19),15,17-heptaen-13-one
(7R,14R)-11-[2-(1-Aminocyclobutyl)pyrimidin-5-yl]-1-(difluoromethoxy)-6-methyl-6,7-dihydro-7,14-methanobenzimidazo[1,2-b][2,5]benzodiazocin-5(14H)-one
(7R,14R)-11-[2-(1-aminocyclobutyl)pyrimidin-5-yl]-1-(difluoromethoxy)-6-methyl-6,7-dihydro-7,14-methano[1,3]benzimidazo[1,2-b][2,5]benzodiazocin-5(14H)-one
tumor necrosis factor (TNF) signaling inhibitor, SAR441566, SAR 441566, PLY98MAN4C
- OriginatorSanofi
- ClassAmines; Anti-inflammatories; Antipsoriatics; Antirheumatics; Azabicyclo compounds; Benzimidazoles; Cyclobutanes; Fluorinated hydrocarbons; Heterocyclic compounds with 4 or more rings; Ketones; Phenyl ethers; Pyrimidines; Small molecules
- Mechanism of ActionTumour necrosis factor alpha inhibitors
- Phase IICrohn’s disease; Psoriasis; Rheumatoid arthritis; Ulcerative colitis
- No development reportedInflammation
- 09 Dec 2025Sanofi plans a phase-I trial (In volunteers) in December 2025 (PO, Tablet), (NCT07272629)
- 29 Oct 2025Sanofi plans a phase II SPECIFI-IBD-LTS trial for Crohn’s Disease or Ulcerative Colitis ( Treatment-experienced) in unknown location (PO, Tablet) in December 2025 (NCT07222189)
- 16 Sep 2025Chemical structure information added.
- You need to be a logged in or subscribed to view this c
Balinatunfib (SAR441566) is an experimental drug which acts as a potent small molecule inhibitor of TNF. Rather than blocking TNF receptors, balinatunfib inactivates TNF directly by stabilising an inactive form of the TNF trimer which fails to bind to its target receptors. It is in early stage clinical trials for rheumatoid arthritis and other chronic autoimmune diseases.[1][2]
SYN



PAT
(WO 2016/050975,
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2016050975&_cid=P22-MK3F7M-67505-1
Intermediate 40

(1R,3R)-1-[2-bromo-6-(difluoromethoxy)phenyl]-7-chloro-2,3-dihydro-1H-pyrrolo[1,2-a]benzimidazol-3-amine
Intermediate 38 (5 g, 11.64 mmol) was suspended in toluene (22 mL) and cooled to 0°C before addition of diphenylphosphoryl azide (3.4 mL, 15 mmol) and 1,8-diazabicyclo[5.4.0]undec-7-ene (2.5 mL, 16 mmol). The mixture was allowed to warm up to r.t and stirred for 2 hours and subsequently at 45°C overnight. The reaction mixture was diluted with EtOAc (150 mL) and the organic phase washed with a saturated aqueous solution of ammonium chloride (50 mL) then a saturated solution of aqueous sodium bicarbonate (50 mL), and concentrated in vacuo. The crude residue thus obtained was solubilized in THF (100 mL) and water (10 mL), trimethylphosphine (17.46 mL, 17.46 mmol) was added and the reaction mixture stirred overnight. The mixture was concentrated in vacuo, partitioned between EtOAc (200 mL) and water (150 mL). The organic layer was extracted with 0.2M HCl aq (3 x 200 mL). The combined acid layer was stirred in an ice bath, whilst 10% NaOH solution was added with stirring until pH increased to 10. The stirred was continued for further 15 minutes to complete precipitation. The precipitate was filtered, rinsed with water (20 mL), then dried under suction for 10 minutes before drying under high vacuum overnight to afford 3.92 g (78%) of the title compound as an off white solid. LCMS basic: RT 1.96 min. (ES+) 428/430 (M+H)+
EXAMPLE 11

(7R, 14R)-11-chloro-1-(difluoromethoxy)-6,7-dihydro-7,14-methanobenzimidazo[1,2-b][2,5]benzodiazocin-5(14H)-one
Intermediate 40 (3.7 g, 8.6 mmol), activated molecular sieve 4A powder (1.2 g), potassium carbonate (1.5 equiv., 13 mmol) followed by dichloro[9,9-dimethyl-4,5-bis(diphenylphosphino)xanthene]palladium(II) (0.04 equiv., 0.35 mmol) were poured into the center of the 100 mL Glass Parr reaction vessel. 3 cycles of vacuum (~20 mmHg) followed by Argon were applied to the closed reactor.
Anhydrous dimethyl sulfoxide (35 mL) was added, followed by phenol 5M in DMSO (1.1 equiv., 9.5 mmol). The solution was degassed by 3 vacuum (~20 mmHg) / argon cycles followed by 3 cycles of vacuum / CO resulting in a final CO pressure of 1 bar.
The mixture was stirred and heated overnight at 100 °C under the CO atmosphere . The reaction was cooled to 30°C, the reactor vessel was opened and EtOAc (40 mL) was added. The resulting mixture was filtered on a pad of Celite, evaporated in vacuo to yield a green oil.
The residue thus obtained was taken up in EtOAc (100 mL) and the organic layer was washed with water, K2CO3 (saturated aqueous solution) and brine (saturated aqueous solution). The aqueous layer was then re-extracted with EtOAc (1 x 50 mL). The combined organic layers were dried over MgSO4, filtered and evaporated to dryness. The obtained green solid (3.65 g), was taken up in EtOAc, the insoluble material was filtered and rinsed with Et2O to afford 1.06 g (33.1%) of the title compound as a grey solid.
The filtrate can be purified by flash chromatography to provide additional product if required:
LCMS basic: MH+ m/z = 376, RT 1.90 minutes.
1H NMR (300 MHz, DMSO) δ 9.12 (d, 1 H, J = 6.7 Hz), 8.23 (dd, 1 H, J = 7.0, 2.4 Hz), 7.60 (m, 5 H), 7.20 (dd, 1 H, J = 8.7, 2.1 Hz), 6.29 (d, 1 H, J = 7.1 Hz), 4.87 (dd, 1 H, J = 6.7 Hz, 6.7 Hz), 3.46 (m, 1 H), 2.72 (d, 1 H, J = 13.4 Hz).
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=US283322316&_cid=P22-MK3EWF-57090-1
Intermediate 3

(7R,14R)-11-Chloro-1-(difluoromethoxy)-6-methyl-6,7-dihydro-7,14-methanobenzimidazo[1,2-b][2,5]benzodiazocin-5(14H)-one
Intermediate 17

tert-Butyl (1-{5-[(7R,14R)-1-(difluoromethoxy)-6-methyl-5-oxo-5,6,7,14-tetrahydro-7,14-methanobenzimidazo[1,2-b][2,5]benzodiazocin-11-yl]pyrimidin-2-yl}cyclobutyl)-carbamate
EXAMPLE 6

(7R,14R)-11-[2-(1-Aminocyclobutyl)pyrimidin-5-yl]-1-(difluoromethoxy)-6-methyl-6,7-dihydro-7,14-methanobenzimidazo[1,2-b][2,5]benzodiazocin-5(14H)-one
To a solution of Intermediate 17 (18.0 g, 29.9 mmol) in 1,4-dioxane (25 mL) was added 4M hydrochloric acid in 1,4-dioxane (40 mL). The resulting mixture was stirred at room temperature for 1 h, then concentrated in vacuo. The residue was dissolved in water (500 mL) and washed with EtOAc (2×300 mL). The aqueous layer was basified to pH 9 with 2N aqueous sodium hydroxide solution, which resulted in precipitation of a solid. EtOAc (500 mL) was added and the mixture was stirred until all solids had dissolved. The residue was partitioned, then the aqueous layer was further extracted with EtOAc (500 mL). The combined organic layers were dried over Na 2SO 4 and filtered, then concentrated in vacuo and dried overnight under high vacuum. The foamy residue was suspended in a mixture of diethyl ether and hexane (150 mL), then stirred and shaken vigorously, before being concentrated in vacuo, to afford the title compound (12.4 g, 83%) as a white amorphous solid. δ H (400 MHz, DMSO-d 6) 9.05 (s, 2H), 8.32-8.22 (m, 1H), 7.91-7.66 (m, 3H), 7.62 (dd, J8.5, 1.8 Hz, 1H), 7.53-7.46 (m, 2H), 6.31 (d, J7.1 Hz, 1H), 5.26 (d, J 7.2 Hz, 1H), 3.52 (dt, J 14.2, 7.3 Hz, 1H), 3.36 (s, 3H), 2.84 (d, J 13.8 Hz, 1H), 2.63 (dtd, J11.5, 5.6, 2.5 Hz, 2H), 2.38 (s, 2H), 2.16-2.05 (m, 2H), 2.04-1.91 (m, 1H), 1.87-1.73 (m, 1H). LCMS (ES+APCI) [M-NH 2] − 486.0, RT 1.66 minutes (Method 2). LCMS (ES+) [M+H] + 503.0, RT 1.71 minutes (Method 1).
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2025008402&_cid=P22-MK3EWF-57090-1

(7R,14R)-1 l-[2-(l-aminocyclobutyl)pyrimidin-5-yl]-l-(difhroromethoxy)-6-methyl-6,7-dihydro-7, 14-methanobenzimidazo[l,2-b][2,5]benzodiazocin-5(14H)-one.
PAT
- Fused Pentacyclic Imidazole Derivatives as Modulators of TNF ActivityPublication Number: US-2021252012-A1Priority Date: 2017-04-25
- Fused pentacyclic imidazole derivatives as modulators of TNF activityPublication Number: KR-102565132-B1Priority Date: 2017-04-25Grant Date: 2023-08-08
- Fused Pentacyclic Imidazole Derivatives as Modulators of TNF ActivityPublication Number: US-2025127795-A1Priority Date: 2017-04-25
- Fused pentacyclic imidazole derivatives as modulators of TNF activityPublication Number: CN-110582495-BPriority Date: 2017-04-25Grant Date: 2022-04-01
- Fused Pentacyclic Imidazole DerivativesPublication Number: US-2017305932-A1Priority Date: 2014-10-03
- Fused pentacyclic imidazole derivatives as modulators of TNF activityPublication Number: CN-110582495-APriority Date: 2017-04-25
- Fused Pentacyclic Imidazole Derivatives as Modulators of TNF ActivityPublication Number: US-2023250105-A1Priority Date: 2017-04-25
- Fused pentacyclic imidazole derivatives as modulators of TNF activityPublication Number: US-10980814-B2Priority Date: 2017-04-25Grant Date: 2021-04-20
- Fused pentacyclic imidazole derivatives as modulators of tnf activityPublication Number: EP-3939980-A1Priority Date: 2017-04-25
- Process for preparing fused pentacyclic imidazole derivatives and uses thereof as modulators of tnf activityPublication Number: EP-3939980-B1Priority Date: 2017-04-25Grant Date: 2023-07-26
- Preparation of bridged pentacyclic imidazole derivatives as modulators of tnf activity, intermeditates and their preparationPublication Number: WO-2025068505-A1Priority Date: 2023-09-29
- DERIVATIVES OF COMBINED PENTACYCLIC IMIDAZOLES AS MODULATORS OF TNF ACTIVITYPublication Number: HR-P20211927-T1Priority Date: 2017-04-25
- Fused pentacyclic imidazole derivatives as modulators of tnf activityPublication Number: CA-3058980-A1Priority Date: 2017-04-25
- Fused pentacyclic imidazole derivatives as modulators of tnf activityPublication Number: EP-3615534-B1Priority Date: 2017-04-25Grant Date: 2021-09-15
- Fused Pentacyclic Imidazole Derivatives as Modulators of TNF ActivityPublication Number: US-2020046723-A1Priority Date: 2017-04-25
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
References
- Vugler A, O’Connell J, Nguyen MA, Weitz D, Leeuw T, Hickford E, et al. (2022). “An orally available small molecule that targets soluble TNF to deliver anti-TNF biologic-like efficacy in rheumatoid arthritis”. Frontiers in Pharmacology. 13 1037983. doi:10.3389/fphar.2022.1037983. PMC 9709720. PMID 36467083.
- Li Y, Ye R, Dai H, Lin J, Cheng Y, Zhou Y, et al. (January 2025). “Exploring TNFR1: from discovery to targeted therapy development”. Journal of Translational Medicine. 23 (1): 71. doi:10.1186/s12967-025-06122-0. PMC 11734553. PMID 39815286.
| Identifiers | |
|---|---|
| IUPAC name | |
| CAS Number | 2248726-53-4 |
| PubChem CID | 132042903 |
| IUPHAR/BPS | 13583 |
| ChemSpider | 129738176 |
| Chemical and physical data | |
| Formula | C27H24F2N6O2 |
| Molar mass | 502.526 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
//////////Balinatunfib, tumor necrosis factor (TNF) signaling inhibitor, SAR441566, SAR 441566, PLY98MAN4C
Atirmociclib



Atirmociclib
CAS 2380321-51-5
MF C22H27ClFN5O3,
463.9 g/mol
(3S,4R)-4-[[5-chloro-4-[7-fluoro-2-(2-hydroxypropan-2-yl)-3-propan-2-ylbenzimidazol-5-yl]pyrimidin-2-yl]amino]oxan-3-ol
(3S,4R)-4-({5-chloro-4-[4-fluoro-2-(2-hydroxypropan-2-yl)-1-(propan2-yl)-1H-1,3-benzimidazol-6-yl]pyrimidin-2-yl}amino)oxan-3-ol
1,5-anhydro-3-({5-chloro-4-[4-fluoro-2-(2-hydroxpropan-2-yl)-1-(propan-2-yl)-1H-benzimidazol-6-yl]pyrimidin-2-yl}amino)-2,3-dideoxy-D-threo-pentitol
D-threo-Pentitol, 1,5-anhydro-3-[[5-chloro-4-[4-fluoro-2-(1-hydroxy-1-methylethyl)-1-(1-methylethyl)-1H-benzimidazol-6-yl]-2-pyrimidinyl]amino]-2,3-dideoxy-
cyclin-dependent kinase (CDK) inhibitor, antineoplastic, PF 07220060, S743GOJ5LJ, CDK4/6-IN-6
Atirmociclib is an orally bioavailable inhibitor of cyclin-dependent kinase 4 (CDK4), with potential antineoplastic activity. Upon administration, atirmociclib selectively inhibits CDK4, which inhibits the phosphorylation of retinoblastoma protein (Rb) early in the G1 phase, prevents CDK-mediated G1-S-phase transition and leads to cell cycle arrest. This suppresses DNA replication and inhibits tumor cell proliferation. CDK4, a serine/threonine kinase, is upregulated in many tumor cell types and plays a key role in the regulation of both cell cycle progression from the G1-phase into the S-phase and tumor cell proliferation.
Atirmociclib (development code PF-07220060) is an investigational orally bioavailable and CDK4-specific inhibitor being developed by Pfizer for the treatment of various solid tumors, particularly hormone receptor-positive, HER2-negative breast cancer.[1][2] The safety and efficacy of atirmociclib have not been established, as it remains in clinical development as of September 2025.[3][4][5]
SYN
https://pubs.acs.org/doi/10.1021/acs.jmedchem.5c02137


PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=US275481329&_cid=P22-MK0K3I-13424-1

Example A94 (Scheme A-15): Preparation of 1,5-anhydro-3-({5-chloro-4-[4-fluoro-2-(2-hydroxypropan-2-yl)-1-(propan-2-yl)-1H-benzimidazol-6-yl]pyrimidin-2-yl}amino)-2,3-dideoxy-D-threo-pentitol

Step 8: Synthesis of 1,5-anhydro-3-({5-chloro-4-[4-fluoro-2-(2-hydroxypropan-2-yl)-1-(propan-2-yl)-1H-benzimidazol-6-yl]pyrimidin-2-yl}amino)-2,3-dideoxy-D-threo-pentitol (Example A94)
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=US275481329&_cid=P22-MK0KHW-23947-1

PAT
- 2-amino-pyridine or 2-amino-pyrimidine derivatives as cyclin dependent kinase inhibitorsPublication Number: KR-102661053-B1Priority Date: 2018-04-26Grant Date: 2024-04-26
- 2-amino-pyridine or 2-amino-pyrimidine derivatives as cyclin dependent kinase inhibitorsPublication Number: KR-20230152182-APriority Date: 2018-04-26
- Cyclin dependent kinase inhibitorsPublication Number: US-11220494-B2Priority Date: 2018-04-26Grant Date: 2022-01-11
- CYCLINE-DEPENDENT KINASE INHIBITORSPublication Number: PE-20201202-A1Priority Date: 2018-04-26
- Cyclin dependent kinase inhibitorsPublication Number: US-2022089580-A1Priority Date: 2018-04-26
- 2-amino-pyridine or 2-amino-pyrimidine derivatives as cyclin dependent kinase inhibitorsPublication Number: HR-P20250254-T1Priority Date: 2018-04-26
- Cyclin dependent kinase inhibitorsPublication Number: US-12378232-B2Priority Date: 2018-04-26Grant Date: 2025-08-05
- 2-amino-pyridine or 2-amino-pyrimidine derivatives as cyclin dependent kinase inhibitorsPublication Number: EP-3784664-B1Priority Date: 2018-04-26Grant Date: 2025-02-19
- 2-Amino-pyridine or 2-amino-pyrimidine derivatives as cyclin-dependent kinase inhibitorsPublication Number: CN-112313219-BPriority Date: 2018-04-26Grant Date: 2024-04-26
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
Mechanism of action
Atirmociclib is designed as a CDK4-specific inhibitor, distinguishing it from dual CDK4/6 inhibitors currently approved for cancer treatment.[6] The drug targets cyclin-dependent kinase 4, which plays a role in cell cycle regulation.[1][7][8]
Atirmociclib functions as a selective inhibitor of the CDK4/cyclin D complex, which plays a crucial role in cell cycle regulation.[4] The drug works by targeting the CDK4 kinase, rendering the retinoblastoma (Rb)/E2F transcription system inactive, which ultimately leads to cell cycle arrest in the G1 phase.[4] This mechanism is particularly effective in tumors that have lost Rb cell cycle-suppressive function, a common feature in various solid tumors.[5]
The selective nature of atirmociclib represents a significant advancement over existing dual CDK4/6 inhibitors.[6] By specifically targeting CDK4 while limiting CDK6 inhibition, atirmociclib is designed to maintain antitumor efficacy while potentially reducing dose-limiting hematologic toxicities, particularly neutropenia, which is believed to be primarily driven by CDK6 inhibition.[9]
Clinical development
Atirmociclib is currently being evaluated in clinical trials for the treatment of advanced solid tumors.[1] Clinical studies are ongoing with estimated completion dates extending to 2027–2028, reflecting the early stage of development for this investigational compound.[1]
Preclinical research published in Cancer Cell in March 2025 reported atirmociclib as a next-generation CDK4-selective inhibitor with enhanced anti-tumor activity and reduced predicted toxicity compared to FDA-approved dual CDK4/6 inhibitors, though these findings require validation in clinical studies.[6]
Preclinical studies
Preclinical research has demonstrated that atirmociclib exhibits enhanced anti-tumor activity compared to FDA-approved dual CDK4/6 inhibitors while showing reduced predicted toxicity.[6] Studies have shown that CDK4-selective inhibition can provide improved preclinical anti-tumor efficacy and safety profiles compared to dual CDK4/6 inhibition strategies.[10]
The preclinical development program has explored combination approaches with various therapeutic modalities, including endocrine therapy, CDK2 inhibition, HER2 antibodies, and immune checkpoint inhibitors.[6] These combination strategies are designed to counter resistance mechanisms to CDK4 inhibition and expand the potential therapeutic applications of cell cycle targeting therapy.[6]
Clinical trials
Atirmociclib has entered clinical development as part of Pfizer’s extensive oncology pipeline.[11] The clinical program is evaluating atirmociclib both as a single agent and in combination with other therapeutic approaches, particularly focusing on patients with hormone receptor-positive, HER2-negative breast cancer.[9][12][13][14][15][16][17]
Early clinical studies have included heavily pretreated patient populations, including those who have previously received CDK4/6 inhibitor therapy.[9] This approach allows for the evaluation of atirmociclib’s potential to overcome resistance to existing CDK4/6 inhibitors and provide therapeutic benefit in patients with limited treatment options.[9]
Safety profile and toxicity
One of the key differentiating features of atirmociclib is its potential for improved safety profile compared to existing dual CDK4/6 inhibitors.[6] The selective targeting of CDK4 while limiting CDK6 inhibition is specifically designed to reduce neutropenia, the most common dose-limiting toxicity associated with current CDK4/6 inhibitors.[18]
The rationale for this approach is based on preclinical evidence suggesting that neutropenia is primarily driven by CDK6 inhibition rather than CDK4 inhibition.[18] By selectively targeting CDK4, atirmociclib aims to maintain therapeutic efficacy while potentially allowing for higher or more sustained dosing without the dose-limiting hematologic toxicities that can compromise treatment outcomes with existing agents.[18]
Regulatory status
As of September 2025, atirmociclib remains an investigational drug that has not received approval from the FDA or other regulatory agencies.[5] The compound is part of Pfizer’s oncology development pipeline.[5]
References
- Pfizer (2 February 2025). A Phase 1/2A Study Evaluating the Safety, Tolerability, Pharmacokinetics, Pharmacodynamics, and Anti-Tumor Activity of Pf-07220060 as a Single Agent and as Part of Combination Therapy in Participants With Advanced Solid Tumors (Report). clinicaltrials.gov.
- Shapiro GI (March 2017). “The evolving role of cyclin-dependent kinase inhibitors in cancer management”. Clinical Advances in Hematology & Oncology. 15 (3): 174–177. PMID 28398270.
- “CDK4 inhibitor PF-07220060”. http://www.cancer.gov. 2 February 2011. Retrieved 3 September 2025.
- “Pfizer Pipeline”. Pfizer.
- “Atirmociclib PF-07220060”. Pfizer Oncology Development. Retrieved 3 September 2025.
- Chang J, Lu J, Liu Q, Xiang T, Zhang S, Yi Y, et al. (March 2025). “Single-cell multi-stage spatial evolutional map of esophageal carcinogenesis”. Cancer Cell. 43 (3): 380–397.e7. doi:10.1016/j.ccell.2025.02.009. PMID 40068596.
- Topacio BR, Zatulovskiy E, Cristea S, Xie S, Tambo CS, Rubin SM, et al. (May 2019). “Cyclin D-Cdk4,6 Drives Cell-Cycle Progression via the Retinoblastoma Protein’s C-Terminal Helix”. Molecular Cell. 74 (4): 758–770.e4. doi:10.1016/j.molcel.2019.03.020. PMC 6800134. PMID 30982746.
- Helsten T, Kato S, Schwaederle M, Tomson BN, Buys TP, Elkin SK, et al. (July 2016). “Cell-Cycle Gene Alterations in 4,864 Tumors Analyzed by Next-Generation Sequencing: Implications for Targeted Therapeutics”. Molecular Cancer Therapeutics. 15 (7): 1682–1690. doi:10.1158/1535-7163.MCT-16-0071. PMID 27196769.
- “ESMO 2024 – combos could be the way forward for CDK2”. ApexOnco. 15 September 2024.
- Palmer CL, Boras B, Pascual B, Li N, Li D, Garza S, et al. (March 2025). “CDK4 selective inhibition improves preclinical anti-tumor efficacy and safety”. Cancer Cell. 43 (3): 464–481.e14. doi:10.1016/j.ccell.2025.02.006. PMID 40068598.
- “Pfizer Highlights Diverse Oncology Portfolio and Combination Approaches at ESMO 2024”. Pfizer. 2024.
- Pfizer (12 August 2025). A Phase 1/2a Dose Escalation and Expansion Study to Evaluate Safety, Tolerability, Pharmacokinetic, Pharmacodynamic, and Anti-Tumor Activity of Pf-07248144 in Participants With Advanced or Metastatic Solid Tumors (Report). clinicaltrials.gov.
- Pfizer (2 July 2025). An Interventional Safety and Efficacy Phase 1/2, Open-Label Study to Investigate Tolerability, Pk, and Antitumor Activity of Vepdegestrant (Arv-47/Pf-07850327), an Oral Proteolysis Targeting Chimera, in Combination With Pf-07220060 in Participants Aged 18 Years and Older With Er+/her2- Advanced or Metastatic Breast Cancer (Report). clinicaltrials.gov.
- Pfizer (14 November 2024). A Phase 1/2, Open-Label, Multicenter, Dose Escalation and Dose Expansion Study to Evaluate the Safety, Tolerability, Pharmacokinetics, Pharmacodynamics, and Antitumor Activity of PF-07220060 in Combination With Pf-07104091 Plus Endocrine Therapy in Participants With Advanced Solid Tumors (Report). clinicaltrials.gov.
- Pfizer (17 June 2025). (FOURLIGHT-3) (Report). clinicaltrials.gov.
- Pfizer (13 March 2025). An Interventional, Open-Label, Randomized, Multicenter Phase 3 Study of PF-07220060 Plus Letrozole Compared to cdk4/6 Inhibitor Plus Letrozole in Participants Over 18 Years of Age With Hormone Receptor (Hr)-Positive, her2-Negative Advanced/Metastatic Breast Cancer Who Have Not Received Any Prior Systemic Anticancer Treatment for Advanced/Metastatic Disease (FOURLIGHT-1) (Report). clinicaltrials.gov.
- Pfizer (15 November 2024). An Interventional, Open-Label, Randomized, Multicenter, Phase 2 Study of Pf-07220060 Plus Letrozole Compared to Letrozole Alone in Postmenopausal Women 18 Years or Older With Hormone Receptor-Positive, her2-Negative Breast Cancer in the Neoadjuvant Setting (Report). clinicaltrials.gov.
- “Pfizer dials down its atirmociclib ambitions”. ApexOnco. 1 May 2025.
| Identifiers | |
|---|---|
| IUPAC name | |
| CAS Number | 2380321-51-5 |
| PubChem CID | 146219790 |
| ChemSpider | 115009592 |
| UNII | S743GOJ5LJ |
| KEGG | D12834 |
| ChEMBL | ChEMBL5187755 |
| Chemical and physical data | |
| Formula | C22H27ClFN5O3 |
| Molar mass | 463.94 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
///////////Atirmociclib, cyclin-dependent kinase (CDK) inhibitor, antineoplastic, PF 07220060, S743GOJ5LJ, CDK4/6-IN-6
Asaretoclax


Asaretoclax
CAS 2363074-01-3
MF C47H57F2N7O7S, MW 902.1 g/mol
4-[4-[[2-[3-(difluoromethyl)-1-bicyclo[1.1.1]pentanyl]-4,4-dimethylcyclohexen-1-yl]methyl]piperazin-1-yl]-N-[4-[(4-hydroxy-4-methylcyclohexyl)methylamino]-3-nitrophenyl]sulfonyl-2-(1H-pyrrolo[2,3-b]pyridin-5-yloxy)benzamide
2-((1H-pyrrolo[2,3-b]pyridin-5-yl)oxy)-4-(4-((2-(3-(difluoromethyl)bicyclo[1.1.1]pentan-1-yl)-4,4-dimethylcyclohex-1-en-1-yl)methyl)piperazin-1-yl)-N-((4-((((1r,4r)-4-hydroxy-4-methylcyclohexyl)methyl)amino)-3-nitrophenyl)sulfonyl)benzamide

B-cell lymphoma 2 (Bcl-2) inhibitor, antineoplastic, GY6FD5FXA3, HY 159817, ABT 263
Asaretoclax is an orally bioavailable inhibitor of the anti-apoptotic protein B-cell lymphoma 2 (Bcl-2), with potential pro-apoptotic and antineoplastic activities. Upon oral administration, asaretoclax targets, binds to and inhibits the activity of Bcl-2. This restores apoptotic processes in tumor cells. Bcl-2 is overexpressed in many cancers and plays an important role in the negative regulation of apoptosis; its expression is associated with increased drug resistance and tumor cell survival.
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=US309776623&_cid=P21-MJZ42N-73938-1
Example 34
2-((1H-pyrrolo[2,3-b]pyridin-5-yl)oxy)-4-(4-((2-(3-(difluoromethyl)bicyclo[1.1.1]pentan-1l-yl)-4,4-dimethylcyclohex-1-en-1-yl)methyl)piperazin-1-yl)-N-((4-((((1r,4r)-4-hydroxy-4-methylcyclohexyl)methyl)amino)-3-nitrophenyl)sulfonyl)benzamide
Intermediate 18
Intermediate 18
4-((((1r,4r)-4-hydroxy-4-methylcyclohexyl)methyl)amino)-3-nitrobenzenesulfonamide

Intermediate 18 was prepared following a procedure described in WO2014/165044A1. LC/MS (ESI) m/z 344.1 [M+H] +.
Intermediate 30
Intermediate 30
2-((1H-pyrrolo[2,3-b]pyridin-5-yl)oxy)-4-(4-((2-(3-(difluoromethyl)bicyclo[1.1.1]pentan-1-yl)-4,4-dimethylcyclohex-1-en-1-yl)methyl)piperazin-1-yl)benzoic Acid
| Step 1: Methyl 2-((1H-pyrrolo[2,3-b]pyridin-5-yl)oxy)-4-(4-((2-(3-(difluoromethyl)bicyclo[1.1.1]pentan-1-yl)-4,4-dimethylcyclohex-1-en-1-yl)methyl)piperazin-1-yl)benzoate (Intermediate 30-1) was prepared following the procedure described in Step 1, Route C for Intermediate 28 using Intermediate 24 in place of Intermediate 22. LCMS (ESI) m/z 591.2 [M+H] +. |


Example 34 was prepared following General Procedure A using Intermediate 30 and Intermediate 18. 1H NMR (400 MHz, DMSO-d 6) δ 11.70 (s, 1H), 11.40 (br s, 1H), 8.59-8.49 (m, 2H), 8.04 (d, J=2.0 Hz, 1H), 7.78 (d, J=8.8 Hz, 1H), 7.53-7.48 (m, 3H), 7.06 (d, J=9.2 Hz, 1H), 6.72 (d, J=7.2 Hz, 1H), 6.38 (s, 1H), 6.25 (s, 1H), 5.99 (t, J=56.8 Hz, 1H), 4.25 (s, 1H), 3.33-3.25 (m, 2H), 3.18-3.05 (m, 4H), 2.97 (s, 2H), 2.40-2.28 (m, 4H), 2.05-1.95 (m, 2H), 1.94 (s, 6H), 1.71-1.59 (m, 5H), 1.58-1.49 (m, 2H), 1.39-1.28 (m, 2H), 1.27-1.20 (m, 2H), 1.18-1.09 (m, 2H), 1.10 (s, 3H), 0.83 (s, 6H); LC/MS (ESI) m/z 902.6 [M+H] +.
SYN
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=US384526484&_cid=P21-MJZ3XL-69589-1
PAT
Publication Number: US-2021009543-A1
Priority Date: 2018-01-10
- Benzamide compoundsPublication Number: CN-118084904-APriority Date: 2018-01-10
- Benzamide compoundsPublication Number: EP-4556469-A1Priority Date: 2018-01-10
- Benzamide compounds as bci inhibitors for the treatment of hivPublication Number: EP-3740487-B1Priority Date: 2018-01-10Grant Date: 2025-01-08
- Benzamide compoundsPublication Number: US-11344546-B2Priority Date: 2018-01-10Grant Date: 2022-05-31
- Benzamide compoundsPublication Number: US-11318134-B2Priority Date: 2018-01-10Grant Date: 2022-05-03
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
/////////Asaretoclax, B-cell lymphoma 2 (Bcl-2) inhibitor, antineoplastic, GY6FD5FXA3, HY 159817, ABT 263
Doxecitine



Doxecitine
CAS951-77-9
MF C9H13N3O4
11/3/2025, FDA 2025, To treat thymidine kinase 2 deficiency in patients who start to show symptoms when they are 12 years old or younger
- CYTIDINE, 2′-DEOXY-
- dCYD
- DEOXYCYTIDINE
4-amino-1-[(2R,4S,5R)-4-hydroxy-5-(hydroxymethyl)oxolan-2-yl]-1,2-dihydropyrimidin-2-one
Doxecitine is a pyrimidine nucleoside used to treat thymidine kinase 2 deficiency.
Doxecitine is a synthetic form of the naturally occurring pyrimidine deoxyribonucleoside deoxycytidine. It is an essential component of the deoxyribonucleotide pool required for DNA synthesis and repair. Doxecitine is currently approved and marketed as a fixed-dose combination therapy with thymidine (KYGEVVI). This combination is the first and only approved treatment for Thymidine Kinase 2 deficiency.5,6
Deoxycytidine is a deoxyribonucleoside, a component of deoxyribonucleic acid. It is similar to the ribonucleoside cytidine, but with one hydroxyl group removed from the C2′ position. Deoxycytidine can be phosphorylated at C5′ of the deoxyribose by deoxycytidine kinase, converting it to deoxycytidine monophosphate (dCMP), a DNA precursor.[1] dCMP can be converted to dUMP and dTMP.
Doxecitine is the international nonproprietary name.[2]
SYN
Graham A. Mock, Douglas H. Lovern, “N.sup.4 -substituted 2′-deoxycytidine compounds, oligonucleotides including N.sup.4 -labeled 2′-deoxycytidines, and a process for making oligonucleotides with N-modified 2′-deoxycytidines.” U.S. Patent US5633364, issued April, 1995.US5633364
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=US37089691&_cid=P21-MJXONM-10154-1
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO1982003079&_cid=P21-MJXONM-10154-1
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
- Chow E, Miller L, Clearman A, Arnold P, Koenig MK, Russo SN: Doxecitine and doxribtimine treatment in an adult patient with thymidine kinase 2 deficiency. Mol Genet Metab. 2025 Aug;145(4):109159. doi: 10.1016/j.ymgme.2025.109159. Epub 2025 Jun 3. [Article]
- Mittur A, VanMeter SA, Orujov E, Glidden P: Pharmacokinetics and Safety of a 1:1 Mixture of Doxecitine and Doxribtimine: Open-label Phase 1 Single Ascending Dose and Food Effect Studies in Healthy Adults. Clin Ther. 2024 Jul;46(7):576-587. doi: 10.1016/j.clinthera.2024.06.006. Epub 2024 Jul 18. [Article]
- Lopez-Gomez C, Levy RJ, Sanchez-Quintero MJ, Juanola-Falgarona M, Barca E, Garcia-Diaz B, Tadesse S, Garone C, Hirano M: Deoxycytidine and Deoxythymidine Treatment for Thymidine Kinase 2 Deficiency. Ann Neurol. 2017 May;81(5):641-652. doi: 10.1002/ana.24922. Epub 2017 May 4. [Article]
- FDA Approved Drug Products: KYGEVVI (doxecitine and doxribtimine) powder, for oral solution (November 2025) [Link]
- UCB: New data on investigational therapy for thymidine kinase 2 deficiency presented at Muscular Dystrophy Association (MDA) 2025 Conference [Link]
- PR Newswire: U.S. FDA approves KYGEVVI™ (doxecitine and doxribtimine), the first and only treatment for adults and children living with thymidine kinase 2 deficiency (TK2d) [Link]
| Names | |
|---|---|
| IUPAC name2′-deoxycytidine | |
| Systematic IUPAC name4-Amino-1-[(2R,4S,5R)-4-hydroxy-5-(hydroxymethyl)oxolan-2-yl]pyrimidin-2(1H)-one | |
| Other namesdoxecitine | |
| Identifiers | |
| CAS Number | 951-77-9 |
| 3D model (JSmol) | Interactive image |
| ChEBI | CHEBI:15698 |
| ChEMBL | ChEMBL66115 |
| ChemSpider | 13117 |
| ECHA InfoCard | 100.012.231 |
| MeSH | Deoxycytidine |
| PubChem CID | 13711 |
| UNII | 0W860991D6 |
| CompTox Dashboard (EPA) | DTXSID70883620 |
| InChI | |
| SMILES | |
| Properties | |
| Chemical formula | C9H13N3O4 |
| Molar mass | 227.217 |
| Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa).Infobox references | |
References
- Staub M, Eriksson S (2006). “The Role of Deoxycytidine Kinase in DNA Synthesis and Nucleoside Analog Activation”. In Peters GJ (ed.). Deoxynucleoside Analogs In Cancer Therapy. Cancer Drug Discovery and Development. Humana Press. pp. 29–52. doi:10.1007/978-1-59745-148-2_2. ISBN 978-1-59745-148-2.
- World Health Organization (2022). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 87”. WHO Drug Information. 36 (1). hdl:10665/352794.
- Kim KW, Roh JK, Wee HJ, Kim C (2016). “Molecular Targeted Anticancer Drugs”. In Kim KW, Roh JK, Wee HJ, Kim C (eds.). Cancer Drug Discovery: Science and History. Springer Netherlands. pp. 175–238. doi:10.1007/978-94-024-0844-7_9. ISBN 978-94-024-0844-7.
- Guo M, Zhang L, Du Y, Du W, Liu D, Guo C, et al. (March 2018). “Enrichment and Quantitative Determination of 5-(Hydroxymethyl)-2′-deoxycytidine, 5-(Formyl)-2′-deoxycytidine, and 5-(Carboxyl)-2′-deoxycytidine in Human Urine of Breast Cancer Patients by Magnetic Hyper-Cross-Linked Microporous Polymers Based on Polyionic Liquid”. Analytical Chemistry. 90 (6): 3906–3913. doi:10.1021/acs.analchem.7b04755. PMID 29316399.
- “FDA approves 1st drug for thymidine kinase 2 deficiency”. U.S. Food and Drug Administration. 3 November 2025. Retrieved 4 November 2025.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain.
External links
- “Doxecitine ( Code – C420 )”. EVS Explore.
- MeSH 68003841
/////////doxecitine, deoxycytidine, CYTIDINE, 2′-DEOXY-, dCYD, FDA 2025, APPROVALS 2025
DR ANTHONY MELVIN CRASTO

ORGANIC SPECTROSCOPY


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}