
FLAGS AND HITS
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO FACEBOOK
...................................................................Join me on twitter
FACEBOOK
...................................................................Join me on twitter
 ..................................................................Join me on google plus
..................................................................Join me on google plus
 Googleplus
GoogleplusMYSELF

Phase 2 , Sarepta, shows eteplirsen for Duchenne muscular dystrophy
ETEPLIRSEN
27 MAR 2013
The clock is ticking for Sarepta Therapeutics , a multitude of biotech investors and the boys who suffer from Duchenne muscular dystrophy.
Armed with promising Phase IIb data from a small study of eteplirsen involving only 12 patients with the lethal disease, Sarepta CEO Chris Garabedian is completing one of the most closely-watched high wire acts in the industry. At a time most companies would be focused solely on organizing a pivotal late-stage study, there’s intense speculation that the biotech will shoot for an accelerated approval with the data in hand. And it all comes down to their sit-down with the FDA to review mid-stage data.
Eteplirsen, also called AVI-4658, is an experimental drug, currently in clinical trials. It is designed for treatment of some mutations which cause Duchenne muscular dystrophy (DMD), a genetic degenerative muscle disease. Eteplirsen is a product of Sarepta Therapeutics Inc.
The drug is a Morpholino antisense oligomer which triggers excision of exon 51 during pre-mRNA splicing of the dystrophin RNA transcript. Skipping exon 51 changes the downstream reading frame of dystrophin;[1] giving eteplirsen to a healthy person would result in production of dystrophin mRNA which would not code for functional dystrophin protein but, for DMD patients with particular frameshifting mutations, giving eteplirsen can restore the reading frame of the dystrophin mRNA and result in production of functional (though internally-truncated) dystrophin.[2] Eteplirsen is given by intravenous infusion for systemic treatment of DMD.
Several clinical trials have been conducted to test eteplirsen, one in the UK involving local injection to the foot,[3][4] one in the UK involving systemic injection at low doses[5][6] and one in the USA at higher systemic doses[7] that progressed to a rollover extension study.[8]
- “Exon Skipping Quantification by qRT-PCR in Duchenne Muscular Dystrophy Patients Treated with the Antisense Oligomer Eteplirsen”. Hum Gene Ther Methods. 17 Oct 2012.
- “Morpholinos and Their Peptide Conjugates: Therapeutic Promise and Challenge for Duchenne Muscular Dystrophy.”. Biochim Biophys Acta. 1798 (12): 2296–303. 17 Feb 2010.
- Gary Roper/Manager Clinical Research Governance Organisation, Imperial College London. “Safety and Efficacy Study of Antisense Oligonucleotides in Duchenne Muscular Dystrophy”. ClinicalTrials.gov. US Government, NIH. Retrieved 30 October 2012.
- Lancet Neurol. 8 (10): 918–28. 25 Aug 2009.
- Professor Francesco Muntoni, University College of London Institute of Child Health. “Dose-Ranging Study of AVI-4658 to Induce Dystrophin Expression in Selected Duchenne Muscular Dystrophy (DMD) Patients”. ClinicalTrials.gov. US Government, NIH. Retrieved 30 October 2012.
- “Exon skipping and dystrophin restoration in patients with Duchenne muscular dystrophy after systemic phosphorodiamidate morpholino oligomer treatment: an open-label, phase 2, dose-escalation study.”. Lancet. 378 (9791): 595–605. 23 Jul 2011.
- Sarepta Therapeutics. “Efficacy Study of AVI-4658 to Induce Dystrophin Expression in Selected Duchenne Muscular Dystrophy Patients”. ClinicalTrials.gov. US Government, NIH. Retrieved 30 October 2012.
- Sarepta Therapeutics. “Efficacy, Safety, and Tolerability Rollover Study of Eteplirsen in Subjects With Duchenne Muscular Dystrophy”. ClinicalTrials.gov. US Government, NIH. Retrieved 30 October 2012.

Lundbeck, Otsuka reach agreement to develop and commercialize Lu AE58054

Lu AE58054
467459-31-0
C20H19F5N2O, 398.37
2-(6-fluoro-1H-indol-3-yl)-N-(3-(2,2,3,3,3-pentafluoropropoxy)benzyl)ethanamine
1H-Indole-3-ethanamine, 6-fluoro-N-[[3-(2,2,3,3-tetrafluoropropoxy)phenyl]methyl]-
N-[2-(6-Fluoro-1H-indol-3-yl)ethyl]-3-(2,2,3,3-tetrafluoropropoxy)benzylamine
CAS No: 467458-02-2 hydrochloride, M.Wt: 434.83
Lu AE58054 Hydrochloride Formula: C20H20ClF5N2O

MAR26.2013
H. Lundbeck A/S (Lundbeck) and Otsuka Pharmaceutical Co., Ltd. (Otsuka) today announced a license and development agreement for Lu AE58054, a selective 5HT6receptor antagonist currently in development for the treatment of Alzheimer’s disease. Under the terms of the agreement, Lundbeck will grant Otsuka co-development and co-commercialization rights to Lu AE58054 in the U.S., Canada, East Asia including Japan, major European countries and Nordic countries.
Lu AE58054 is a potent and selective 5-HT6 receptor antagonist under development byLundbeck as an augmentation therapy for the treatment of cognitive deficits associated with Alzheimer’s disease and schizophrenia.[1][2]
Phase III trials recently began for Lu-AE58054, a novel 5-HT6 antagonist for Alzheimer’s disease (AD) from H Lundbeck and Otsuka. Lu-AE58054 already demonstrated significant improvement of cognitive function in an earlier phase II trial when administered with Aricept.
This orally available drug is an antagonist of the serotonin 6 (5-HT6) receptor. This receptor subtype is expressed primarily in the brain, particularly in the cerebral cortex and hippocampus, where it has been proposed to play a role in cognitive impairments associated with schizophrenia and Alzheimer’s disease. The 5-HT6 receptor antagonists are thought to enhance cholinergic, glutamatergic, noradrenergic, and dopaminergic neurotransmission. Apart from some affinity for adrenergic receptors, Lu AE58054 has been reported to be highly selective over other G-protein coupled receptors. The compound enters the brain and dose-dependently reversed deficits in a rat model of cognitive impairment (Upton et al., 2008; Arnt et al., 2010).
Lu AE58054 is being developed as a symptomatic adjunct to cholinesterase inhibitor treatment in Alzheimer’s disease. Lu AE58054 was originally discovered by Lilly, which licensed it to the biotechnology company Saegis for the development of cognitive impairment in thinking disorders such as schizophrenia. In 2006, Saegis was acquired by Lundbeck, which in October 2013 launched a global Phase 3 program in AD. This program consists of four trials planned to enroll a total of about 3,000 patients (see company press release).
No Phase 1 trials on this drug are listed in publicly available databases. In 2005, Saegis conducted a Phase 2a trial in 20 schizophrenia patients in the United States, evaluating the safety, tolerability, pharmacokinetics, and pharmacodynamics of giving this drug as an add-on to Risperidone (see company press release). In 2009 and 2010, Lundbeck conducted a Phase 2 trial in Europe and Asia to evaluate the compound as an adjuct to Risperidone for its effect on cognitive deficits in 124 patients with schizophrenia. Results were not published in the peer-reviewed literature, but development of Lu AE58054 for cognitive deficits in schizophrenia appears to have ended.
In 2010 and 2011, Lundbeck evaluated Lu AE58054 in a Phase 2 study in 278 patients with probable Alzheimer’s disease. Conducted in Australia, Canada, and Europe, the trial compared the effects of a six-month course of an undisclosed dose of Lu AE58054 with 10 mg/day of Donepezil to the same dose of placebo. In June 2012, the company announced that the trial had met its primary cognition endpoint as measured by the ADAS-cog. On secondary endpoints, such as measures of global status and activities of daily living, Lu AE58054 treatment showed trends for a benefit but fell short of achieving statistical significance (see Jun 2012 news story).
In July 2013, a Phase 1 study evaluating pharmacokinetics of single and multiple ascending doses in 42 healthy volunteers was added, and in October 2013, the first of four planned Phase 3 studies began. This study is set to enroll 930 patients with mild to moderate Alzheimer’s disease who are already taking a stable dose of 10 mg/day of Donepezil. The trial compares a six-month course of once-daily 30 or 60 mg capsules of drug to placebo for added benefit on cognition as measured by the ADAS-cog. Secondary outcomes will assess various aspects of global clinical function and behavior. No biomarkers are embedded in this trial, which is expected to last until September 2015. For all listed trials on Lu AE58054, seeclinicaltrials.gov.

Description: IC50 Value: 0.83 nm [1] Lu AE58054 is an in-vitro potency and selectivity, in-vivo binding affinity and effect of the 5-HT (6) R antagonist in vitro:. Lu AE58054 displayed high affinity to the human 5-HT (6) receptor (5-HT (6) R) with a Ki of 0.83 nm. In a 5-HT (6) GTPgammaS efficacy assay Lu AE58054 showed no agonist activity, but demonstrated potent inhibition of 5-HT- . mediated activation Besides medium affinity to adrenergic alpha (1A) – and alpha (1B)-adrenoreceptors, Lu AE58054 demonstrated> 50-fold selectivity for more than 70 targets examined [1] in vivo: Orally administered Lu AE58054 potently inhibited striatal in. -vivo binding of the 5-HT (6) antagonist radioligand [(3) H] Lu AE60157, with an ED (50) of 2.7 mg / kg. Steady-state modelling of an acute pharmacokinetic/5-HT (6) R occupancy time-course experiment indicated a plasma EC (50) value of 20 ng / ml. Administration of Lu AE58054 in a dose range (5-20 mg / kg po) leading to above 65% striatal 5-HT (6) R binding occupancy in vivo, reversed cognitive impairment in a rat novel object recognition task induced after subchronic treatment for 7 d with phencyclidine (PCP 2 mg / kg bid, ip for 7 d, followed by 7 d drug free). The results indicate that Lu AE58054 is a selective antagonist of 5-HT (6) Rs with good oral bioavailability and robust efficacy in a rat model of cognitive impairment in schizophrenia [1] Clinical trial:. Lu-AE58054 Added to Donepezil for the Treatment for Moderate Alzheimer’s Disease Phage2.
Dementia is a clinical syndrome characterized by deficits in multiple areas of cognition that cannot be explained by normal aging, a noticeable decline in function, and an absence of delirium. In addition, neuropsychiatric symptoms and focal neurological findings are usually present, Dementia is further classified based on etiology. Alzheimer’s disease (AD) is the most common cause of dementia, followed by mixed AD and vascular dementia, vascular dementia, Lewy body dementia (DLB), and fronto- temporal dementia.
The incidence of Alzheimer’s disease is expected to increase through the year 2050 with an estimated prevalence of 1 1 to 16 million cases. Currently, two classes of medications are FDA approved for managing symptoms of AD – acetylcholinesterase inhibitors (AChEIs) and an N-methyl-D-aspartase (NMDA) receptor antagonist. AChEIs are commonly used as initial treatment on diagnosis. The AChEIs – donepezil, rivastigmine, galantamine, and tacrine – are indicated for mild-to-moderate AD; only donepezil is approved for the severe stage.
Despite the available therapies, there are no treatments to cure AD or to prevent or stop disease progression. Acetylcholinesterase inhibitors do not help everyone who has AhJieimers disease and in fact are not efficacious in many patients. Considering that AChEIs and memantine have only a modest symptomatic effect, and cannot prevent AD decline and slow disease progression, mere is a high unmet need for more effective symptomatic treatments and for a disease modifying/slowing therapies. The use of selective 5-HTe receptor antagonists to treat cognitive dysfunction has been suggested and is based on several lines of reasoning. For example, selective 5-HT« receptor antagonists have been shown to modulate cholinergic and glutamatergic neuronal function. The activity of selective 5-HT6 receptor antagonists has been demonstrated in animal models of cognitive function. Since the disclosure of the first selective 5-HT4 receptor antagonists, there have been several reports on the activity of these selective compounds in in-vivo models of cognitive function, N*(2-(6-fluoro-lH-indol-3-yI)ethyI)-3- (2,2,3,3-tetrafluoropropoxy)benzylamine (herein referred to as “Compound Γ) is a potent and selective1
RECTIFIED SHEET RULE 91 ISA/EP 5-HT6 receptor antagonist which has been in clinical development for treating cognition impairment associated with schizophrenia and as a treatment for AD.

In November 2008, a multi-centre, randomised, double-blind, fixed-dose study (120 mg/day BID) was initiated to explore the efficacy and safety of Compound I as adjunctive treatment to risperidone in patients with schizophrenia. Overall improvement in schizophrenia symptoms was assessed by using the Positive and Negative Syndrome Scale (PANSS) total score. Compound I did not offer any treatment advantage over placebo as measured by the PANSS total score. In 2010, it was announced that there did not appear to be any treatment advantage over placebo in improving patients’ overall neurocognitive performance as assessed using the BACS composite Z-score and the PANSS cognitive subscale scores.
In 2012, it was reported that a randomized, double blind, placebo controlled trial conducted in Europe, Canada and Australia met its primary endpoint in the treatment of AD. Data demonstrated that Compound I plus 10 mg/day of donepezil significantly improved cognitive function in 278 patients with Alzheimer’s disease compared to placebo plus donepezil, when measured by Alzheimer’s Disease Assessment Scale-cognitive sub-scale (ADAS-cog). Compound I showed positive results in secondary endpoints including measures of global impression and daily living activities compared to donepezil treated patients.
The daily dose of 90 mg of Compound I in the AD study was administered three times daily (3 x 30 mg) to overcome the relative short half-life observed in subjects in previous clinical studies. An issue for that dose selection was to ensure that the maximum exposure level fell under the maximum exposure limit which had been established from non-clinical toxicology studies. Accordingly, a fixed dose of three times in the study was introduced.
As the 5-HT6 receptor is a novel target predominately localized in the brain, a key problem in the development is to determine the amount of receptor occupancy and the correlation with plasma exposure. With CNS targets, further challenges exist that revolve around whether a drug will pass the blood brain barrier and whether it will reach the target at a suitable concentration and for a sufficient length of receptor occupancy. Direct brain measures of 5 -HT6 receptor occupancy may be valuable to many decision-making processes during the development of centrally acting drugs targeted at 5-HT6 to ensure adequate proof-of-concept testing and to optimize dosing regimens. In humans, tools such as positron emission tomography (PET) with specific radiolabeled ligands have been used to quantitatively assess in-vivo occupancy of a number of neurotransmitter receptors, including those for dopamine, serotonin, and benzodiazepines (Talbot, et al., European Neuropsychopharmacology, 2002, 12, 503-511).
An effective PET ligand, [nC]-LuPET was developed and has since been successfully evaluated for human use. The ligand was subsequently used to determine the 5-HT6 receptor occupancy following multiple dose ranges of Compound I. In the assessment for receptor occupancy, human subjects were administered the compound for at least three days at several dosage regimens.
The inventors discovered that high receptor occupancies were observed after multiple dosages of Compound I and that receptor occupancy was maintained 24 hrs post dose. Data generated from a separate Phase I P study in the elderly and data generated from the above AD study have shown that the elimination half life of Compound I in the elderly population was longer (about 19 hours) compared to young healthy subjects (about 12 hours).
With these convergent discoveries, the inventors have identified improved methods of treating AD by introducing a new and improved dosage regime comprising once daily administration in a novel dosage range. Based on the findings described herein, the dose range contemplated is expected to be efficacious while providing exposure levels below the NOAEL, thus improving the safety ratio.
………………………
WO 2009073118
http://www.google.st/patents/WO2009073118A1?cl=en
General Synthetic Scheme

MeOH (-H2O); NaBH4 BoC2O



…………………………
WO 2002078693
http://www.google.com/patents/WO2002078693A2?cl=en
Example 402 N-(2-(6-Fluoro-lH-indol-3-yl)ethyl)-3-(2.2,3,3-tetrafluoropropoxy)benzylamine

Combine 6-fluorotryptamine hydrochloride (90 g, 0.419 mol) and water (900 ml). Add an aqueous solution of NaOH (2N, 230 ml) and dichloromethane (900 ml). After 1 hour, separate the organic layer, extract the aqueous layer with dichloromethane, combine the organic layers, wash water, dry over MgSO , and evaporate to a residue. Combine the residue and toluene (200 ml) and evaporate to give 78.45 g of a brown oil. Combine the above 78.45 g with another 41.4 g batch to provide 6-fluorotryptamine. Combine 6- fluorotryptamine (119.85) and ethanol (3.325 L), add 2,2,3,3- tetrafluoropropylbenzaldehyde (176 g, 0.745 moles, 1.2 equiv.) and 150 g of molecular sieve 3A. Heat to reflux. After 2 hours, cool to RT room temperature and add NaBH4 (35.2 g, 0.93 mol, 1.5 equiv.). After 1 hour, filter through celite and wash with 500 ml of ethanol. Evaporate the filtrate under reduced pressure to give an oily residue. Partition the residue between water and dichloromethane. Separate the layers, extract the aqueous later with dichloromethane, combine organic layers, wash with brine and dry over MgSO4. Filter and evaporate under reduced pressure to give the title compound. The HCI salt is formed as follows: Combine N-(2-(6-fluror-lH-indol-3-yl)ethyl)-
3-(2,2,3,3-tetrafluoropropyl)benzylamine (387 g, 0.97 moles) and diethyl ether (3.95 L) of at room temperature. Add dropwise a solution of HCl/Et2O (298 ml) over 15 minutes until the pH is about 3 to give a solid. Stir for 1 hour and collect the solid, wash with ether, and dry under reduced pressure for at 40°C to give the title compound as the hydrochloride.
……………………..
http://www.google.com/patents/US20130210829
Compound 6, also known as 2-(6-fluoro-1H-indol-3-yl)-N-(3-(2,2,3,3-tetrafluoropropoxy)benzyl)ethanamine,


Combine 6-fluorotryptamine hydrochloride (90 g, 0.419 mol) and water (900 ml). Add an aqueous solution of NaOH (2N, 230 ml) and dichloromethane (900 ml). After 1 hour, separate the organic layer, extract the aqueous layer with dichloromethane, combine the organic layers, wash water, dry over MgSO4, and evaporate to a residue. Combine the residue and toluene (200 ml) and evaporate to give 78.45 g of a brown oil. Combine the above 78.45 g with another 41.4 g hatch to provide 6-fluorotryptamine. Combine 6-fluorotryptamine (119.85) and ethanol (3.325 L), add 2,2,3,3-tetrafluoropropoxybenzaldehyde (176 g, 0.745 moles, 1.2 equiv.) and 150 g of molecular sieve 3A. Heat to reflux. After 2 hours, cool to RT room temperature and acid NaBH4 (35.2 g, 0.93 mol, 1.5 equiv.). After 1 hour, filter through celite and wash with 500 nil of ethanol. Evaporate the filtrate under reduced pressure to give an oily residue. Partition the residue between water and dichloromethane. Separate the layers, extract the aqueous later with dichloromethane, combine organic layers, wash with brine and dry over MgSO4. Filter and evaporate under reduced pressure to give the title compound.
The HCl salt is formed as follows: Combine N-(2-(6-fluoror-1H-indol-3-yl)ethyl)-3-(2,2,3,3-tetrafluoropropyl)benzylamine (387 g, 0.97 moles) and diethyl ether (3.95 L) of at room temperature. Add dropwise a solution of HCl/Et2O (298 ml) over 15 minutes until the pH is about 3 to give a solid. Stir for 1 hour and collect the solid, wash with ether, and dry under reduced pressure for at 40° C. to give the title compound as the hydrochloride.
- “U.S. Development Programs. – Lundbeck”.
- “Search of: Lu AE58054 – List Results – ClinicalTrials.gov”.
- WO 2002078693
- WO 2009073118
- Novel and Potent 5-Piperazinyl Methyl-N1-aryl Sulfonyl Indole Derivatives as 5-HT6 Receptor Ligands
ACS Medicinal Chemistry Letters (2010), 1(7), 340-344, http://pubs.acs.org/doi/full/10.1021/ml100101u - WO 2012002583
- WO 2013055386
- US 20130210829
- WO 2014037532
- Lundbeck expands its pipeline – initiating phase II clinical trials with a new compound for the treatment of schizophrenia
- Lundbeck initiates clinical phase II trials with Lu AE58054 as augmentation treatment in Alzheimer’s disease
References on Lu AE58054 Hydrochloride:
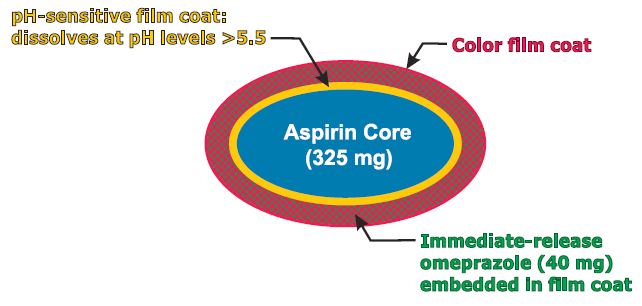
NDA PA32540, Pozen Submits NDA for Aspirin Alternatives
PA32540
PA-325/40 (EC-ASA 325 mg + IR omeprazole 40 mg)
enteric-coated aspirin (EC-ASA) 325 mg
MAR 27,2013
Pozen Inc. announced the submission of a new drug application to the U.S. Food and Drug Administration for the marketing approval of two potential cardiovascular drugs.
The move is a major step toward commercializing the drugs and means that the company thinks it has proven that the drugs are safe and effective.
Pozen’s PA32540 and PA8140 are both intended as alternatives to plain aspirin for the prevention of cardiovascular disease. Many people take aspirin to prevent heart problems, but long-term use of aspirin can cause ulcers.
Pozen’s drugs contain aspirin and the omeprazole, the active ingredient in heartburn drugs like Prilosec. The omeprazole is released as soon as the drug is taken and the aspirin is released over time.
Pozen is seeking approval for use in patients at risk of aspirin-induced ulcers.
Pozen CEO John Plachetka called the move “an important milestone for the drug” and said the company looks forward to completing a commercial deal with a partner “in the upcoming months.”
Pozen’s PA-325/40 is a combination pill containing enteric-coated aspirin 325 mg surrounded by a pH-sensitive film surrounded by an immediate-release omeprazole 40 mg.
SPOTLIGHT-Ganetespib Shows Potency Against Lung Cancer
Ganetespib
http://www.ama-assn.org/resources/doc/usan/ganetespib.pdf
A drug that indirectly impairs the function of several cancer-driving proteins, including anaplastic lymphoma kinase, may be an effective new treatment for patients with anaplastic lymphoma kinase-positive non-small cell lung cancer.
The drug, ganetespib, may also be effective for treating patients who have become resistant to the only FDA-approved targeted therapy for this disease, crizotinib, according to data published in Cancer Discovery, a journal of the American Association for Cancer Research.
“Lung cancer, a leading cause of death, is no longer thought of as a single disease, but rather as a group of diseases, each with a distinct genetic profile,” according to David Proia, Ph.D., associate director of cancer biology at Synta Pharmaceuticals Corporation, the company that funded the research. “This realization has paved the way for the design of new treatments tailored to the specific biological characteristics of a patient’s tumor.
“For example, patients with lung cancer caused by alterations in the anaplastic lymphoma kinase (ALK) protein typically respond well to crizotinib, which blocks that activity of the modified ALK and consequently kills off the cancer cells,” said Proia. “However, as is the case for many cancer drugs, most patients treated with crizotinib eventually become resistant to the drug.”
Proia and colleagues investigated ganetespib as an alternative treatment for ALK-positive non-small cell lung cancer (NSCLC). Ganetespib targets heat shock protein 90 (Hsp90), a chaperone for many different proteins, including ALK, to ensure proper functioning. When Hsp90 is blocked, ALK can no longer work properly and is destroyed by the cell. Once ALK is lost, the cancer cells die and the tumors shrink.
Ganetespib had 30 times greater potency than crizotinib against a cultured ALK-positive NSCLC cell line, resulting in the complete loss of ALK protein expression. In addition, the drug was active against ALK-positive lung cancer cell lines that had become resistant to the effects of crizotinib.
The researchers then compared ganetespib and crizotinib in mice xenografted with human ALK-positive NSCLC cancer cells. Ganetespib displayed greater antitumor activity and prolonged animal survival as compared to crizotinib. It was also shown that ganetespib had meaningful activity in a patient with ALK-driven NSCLC who had responded to, and then progressed, following crizotinib therapy.
“Ganetespib therapy represents a new option for treating ALK-dependent lung cancer in sequence with direct ALK inhibitors and/or for treating patients who relapse following direct ALK inhibitor therapy,” said Proia.
The U.S. Food and Drug Administration today approved Tecfidera (dimethyl fumarate) capsules to treat adults with relapsing forms of multiple sclerosis (MS).

Dimethyl fumarate
March 27, 2013
The U.S. Food and Drug Administration today approved Tecfidera (dimethyl fumarate) capsules to treat adults with relapsing forms of multiple sclerosis (MS).
MS is a chronic, inflammatory, autoimmune disease of the central nervous system that disrupts communication between the brain and other parts of the body. It is among the most common causes of neurological disability in young adults and occurs more frequently in women than men. For most people with MS, episodes of worsening function (relapses) are initially followed by recovery periods (remissions). Over time, recovery periods may be incomplete, leading to progressive decline in function and increased disability. MS patients often experience muscle weakness and difficulty with coordination and balance. Most people experience their first symptoms of MS between the ages of 20 and 40.
Dimethyl fumarate (DMF) is the methyl ester of fumaric acid. DMF was initially recognized as a very effective hypoxic cell radiosensitizer. Later, DMF combined with three other fumaric acid esters (FAE) was licensed in Germany as oral therapy for psoriasis (Fumaderm). Other diseases, such as necrobiosis lipoidica, granuloma annulare, and sarcoidosis were also found to respond to treatment with DMF in case reports or small patient series. Recently, phase III clinical trials found that DMF (BG-12) successfully reduced relapse rate and time to progression of disability in multiple sclerosis. DMF is thought to have immunomodulatory properties without significant immunosuppression.
In a non-medical use, DMF was applied as a biocide in furniture or shoes to prevent growths of mold during storage or transport in a humid climate. However, due to incidences of allergic reactions after skin contact the European Union banned DMF in consumer products since 1998, and since January 2009 the import of products containing DMF was also banned.
MHLW Japan approved Eisai’s Inovelon (rufinamide) as an adjunctive therapy to other antiepileptic drugs in the treatment of Lennox-Gastaut syndrome

Rufinamide
mar26, 2013
The agency approved Eisai’s Inovelon (rufinamide) as an adjunctive therapy to other antiepileptic drugs in the treatment of Lennox-Gastaut syndrome
Rufinamide is an anticonvulsant medication. It is used in combination with other medication and therapy to treat Lennox–Gastaut syndrome and various other seizure disorders. Rufinamide, a triazole derivative, was developed in 2004 by Novartis Pharma, AG, and is manufactured by Eisai.
Rufinamide was approved by the US Food and Drug Administration on November 14, 2008 as adjunctive treatment of seizures associated with Lennox-Gastaut syndrome in children 4 years and older and adults. Its official FDA-approved labeling does not mention use in the treatment of partial seizures inasmuch as clinical trials submitted to the FDA were marginal. However, several recent clinical trials suggest that the drug has efficacy for partial seizures It is marketed under the brand name Banzel. It is also marketed in the European Union under the brand name Inovelon.
The mechanism of action of rufinamide is unknown. However, it is presumed to involve stabilization of the sodium channel inactive state, effectively keeping these ion channels closed. Although the direct mechanism of action may be different, several other antiepileptic agents also stabilize a sodium channel inactive state including phenytoin, carbamazepine, and lacosamide (stabilizes the slow inactive state).
MHLW Japan, GlaxoSmithKline and Genmab announced the approval of Arzerra (ofatumumab) by the MHLW for use in patients with relapsed/refractory CD20-positive chronic lymphocytic leukaemia.
mar 26, 2013
GlaxoSmithKline and Genmab announced the approval of Arzerra (ofatumumab) by the MHLW for use in patients with relapsed/refractory CD20-positive chronic lymphocytic leukaemia.
The thumbs-up triggers a milestone payment of 20 million Danish kroner to Genmab.
Ofatumumab(trade name Arzerra, also known as HuMax-CD20) is a human monoclonal antibody (for the CD20 protein) which appears to inhibit early-stage B lymphocyte activation. It is FDA approved for treating chronic lymphocytic leukemia that is refractory to fludarabine and alemtuzumab (Campath) and has also shown potential in treating Follicular non-Hodgkin’s lymphoma, Diffuse large B cell lymphoma, rheumatoid arthritis and relapsing remitting multiple sclerosis. Ofatumumab has also received conditional approval in Europe for the treatment of refractory chronic lymphocytic leukemia. This makes ofatumumab the first marketing application for an antibody produced by Genmab, as well as the first human monoclonal antibody which targets the CD20 molecule that will be available for patients with refractory CLL.
MHLW JAPAN has approved Nippon Shinyaku Co’s Regtect (acamprosate) for support maintenance of abstinence in patients with alcohol dependence

ACAMPROSATE
MAR 26 2013
MHLW JAPAN has approved Nippon Shinyaku Co’s Regtect (acamprosate) for support maintenance of abstinence in patients with alcohol dependence.
Acamprosate, also known as N-acetyl homotaurine and by the brand name Campral, is a drug used for treating alcohol dependence.
Acamprosate is thought to stabilize the chemical balance in the brain that would otherwise be disrupted by alcoholism, possibly by antagonizing glutamatergic N-methyl-D-aspartate receptors and agonizing gamma-aminobutyric acid (GABA) type A receptors. Reports indicate that acamprosate only works with a combination of attending support groups and abstinence from alcohol.Certain serious side effects include diarrhea, allergic reactions, irregular heartbeats, and low or high blood pressure, while less serious side effects include headaches, insomnia, and impotence. Acamprosate should not be taken by people with kidney problems or allergies to the drug.
Campral is manufactured and marketed in the United States by Forest Laboratories, while Merck KGaA markets it outside the US. It is sold as 333 mg white and odorless tablets of acamprosate calcium, which is the equivalent of 300 mg of acamprosate.
MAA, EU, Boehringer Ingelheim and Eli Lilly and Company announce acceptance of EMA application for investigational Type 2 Diabetes treatment empagliflozin

Empagliflozin
26 March 2013
Boehringer Ingelheim and Eli Lilly and Company today announced the European Medicines Agency (EMA) has accepted for review a marketing authorisation application (MAA) for the investigational sodium glucose cotransporter-2 (SGLT2) inhibitor, empagliflozin, for the treatment of Type 2 Diabetes (T2D) in adults. The acceptance of the MAA marks the beginning of the review process in the European Union for this potential oral diabetes treatment.
A New Drug Application (NDA) for empagliflozin was recently submitted to the Food and Drug Administration (FDA) in the United States for the treatment of Type 2 Diabetes mellitus (T2D) in adults.
Empagliflozin is part of a class of drugs being investigated for the reduction of blood glucose levels in adults with T2D. In clinical trials to date, SGLT2 inhibitors have been shown to reduce blood glucose by removing excess glucose independently of beta cell function and insulin resistance.
* Empagliflozin is an investigational compound. Its safety and efficacy have not yet been fully established
About Diabetes
An estimated 371 million people worldwide have Type 1 and Type 2 Diabetes. Type 2 Diabetes is the most common type, accounting for an estimated 90% of all diabetes cases.Diabetes is a chronic condition that occurs when the body either does not properly produce, or use, the hormone insulin.
Empagliflozin is drug which is being investigated in clinical trials for the oral treatment of type 2 diabetes by Boehringer Ingelheim and Eli Lilly and Company.[1][2] It is an inhibitor of the sodium glucose co-transporter-2 (SGLT-2), which is found almost exclusively in the proximal tubules of nephronic components in the kidneys. SGLT-2 accounts for about 90 percent of glucose reabsorption into the blood. Blocking SGLT-2 causes blood glucose to be eliminated through the urine via the urethra.[3][4]
SGLT-2 inhibitors such as empagliflozin reduce blood glucose by blocking glucose reabsorption in the kidney and thereby excreting glucose (i.e., blood sugar) via the urine.[5]
- Grempler R, Thomas L, Eckhardt M, Himmelsbach F, Sauer A, Sharp DE, Bakker RA, Mark M, Klein T, Eickelmann P (January 2012). “Empagliflozin, a novel selective sodium glucose cotransporter-2 (SGLT-2) inhibitor: characterisation and comparison with other SGLT-2 inhibitors”. Diabetes Obes Metab 14 (1): 83–90. doi:10.1111/j.1463-1326.2011.01517.x. PMID 21985634.
- “Empagliflozin”. clinicaltrials.gov. U.S. National Institutes of Health. Retrieved 22 September 2012.
- Nair S, Wilding JP (January 2010). “Sodium glucose cotransporter 2 inhibitors as a new treatment for diabetes mellitus”. J. Clin. Endocrinol. Metab. 95 (1): 34–42. doi:10.1210/jc.2009-0473. PMID 19892839.
- Bays H (March 2009). “From victim to ally: the kidney as an emerging target for the treatment of diabetes mellitus”. Curr Med Res Opin 25 (3): 671–81. doi:10.1185/03007990802710422. PMID 19232040.
- Abdul-Ghani MA, DeFronzo RA (September 2008). “Inhibition of renal glucose reabsorption: a novel strategy for achieving glucose control in type 2 diabetes mellitus”. Endocr Pract 14 (6): 782–90. PMID 18996802.
Kyowa Hakko Kirin, has received approval for NOURIAST tablets 20 mg (istradefylline), a novel antiparkinsonian agent, has been approved for manufacturing and marketing in Japan

Istradefylline (KW-6002) is a selective antagonist at the A2A receptor. It has been found to be useful in the treatment of Parkinson’s disease.[1] Istradefylline reduces dyskinesia resulting from long-term treatment with classical antiparkinson drugs such as levodopa. Istradefylline is an analog of caffeine.
References
Peter A. LeWitt, MD, M. Guttman, James W. Tetrud, MD, Paul J. Tuite, MD, Akihisa Mori, PhD, Philip Chaikin, PharmD, MD, Neil M. Sussman, MD (2008). “Adenosine A2A receptor antagonist istradefylline (KW-6002) reduces off time in Parkinson’s disease: A double-blind, randomized, multicenter clinical trial (6002-US-005)”. Annals of Neurology 63 (3): 295–302. doi:10.1002/ana.21315. PMID 18306243.
TUE 26 MAR 2013
DR ANTHONY MELVIN CRASTO

ORGANIC SPECTROSCOPY

