

The drug maker is seeing great signs in the development of treatment for multiple myeloma, a bone marrow cancer. The results from its Phase 3 of Kyprolis’ clinical trial shows that patients can live almost nine months longer without worsening symptoms. According to Amgen, about 70,000 people in the U.S. are living with the disease and 24,000 new cases are diagnosed every year. With the good clinical trial result, Amgen plans to begin regulatory submissions around the world next year. Dr. Pablo Cagnoni, president of Amgen’s subsidiary Onyx Pharmaceuticals said, “The results demonstrate that Kyprolis can significantly extend the time patients live without their disease progressing. The ability of novel therapies to produce deep and durable responses may, one day, transform this uniformly fatal disease to one that is chronic and manageable.” Male patients over the age of 65 have the highest risk of developing it.
FLAGS AND HITS
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
Join me on Linkedin
Join me on Researchgate
Join me on Facebook
 FACEBOOK
...................................................................Join me on twitter
FACEBOOK
...................................................................Join me on twitter
 ..................................................................Join me on google plus
..................................................................Join me on google plus
 Googleplus
Googleplus
FACEBOOK
...................................................................Join me on twitter
..................................................................Join me on google plus
GoogleplusMYSELF
DR ANTHONY MELVIN CRASTO Ph.D ( ICT, Mumbai)
, INDIA
36Yrs Exp. in the feld of Organic Chemistry,Working for AFRICURE PHARMA as ADVISOR earlier with GLENMARK PHARMA at Navi Mumbai, INDIA. Serving chemists around the world. Helping them with websites on Chemistry.Million hits on google,
NO ADVERTISEMENTS , ACADEMIC , NON COMMERCIAL SITE, world acclamation from industry, academia, drug authorities for websites, blogs and educational contribution, ........amcrasto@gmail.com..........+91 9323115463, Skype amcrasto64


Amgen’s Multiple Myeloma Drug Shows Promise in Phase 3 Trial
 Carfilzomib
Carfilzomib
Amgen’s Multiple Myeloma Drug Shows Promise in Phase 3 Trial
https://finance.yahoo.com/video/amgens-multiple-myeloma-drug-shows-195603222.html
Carfilzomib (marketed under the trade name Kyprolis, Onyx Pharmaceuticals, Inc.) is an anti-cancer drug acting as a selectiveproteasome inhibitor. Chemically, it is a tetrapeptide epoxyketone and an analog of epoxomicin.[1]
The U.S. Food and Drug Administration (FDA) approved it on 20 July 2012 for use in patients with multiple myeloma who have received at least two prior therapies, including treatment with bortezomib and an immunomodulatory therapy and have demonstrated disease progression on or within 60 days of completion of the last therapy. Approval is based on response rate. Clinical benefit, such as improvement in survival or symptoms, has not been verified.[2]
The abbreviation CFZ is common for referring to carfilzomib, but abbreviating drug names is not best practice in medicine.
Discovery, early development and regulatory approval
Carfilzomib is derived from epoxomicin, a natural product that was shown by the laboratory of Craig Crews at Yale University to inhibit the proteasome.[3] The Crews laboratory subsequently invented a more specific derivative of epoxomicin named YU101,[4] which was licensed to Proteolix, Inc. Craig Crews, Raymond Deshaies from Caltech, Phil Whitcome, the former CEO of Neurogen and Larry Lasky, a venture capitalist, founded Proteolix, and along with other researchers and scientists, advanced YU101. The scientists at Proteolix invented a new, distinct compound that had potential use as a drug in humans, known as carfilzomib. Proteolix advanced carfilzomib to multiple Phase 1 and 2 clinical trials, including a pivotal Phase 2 clinical trial designed to seek accelerated approval.[5]Clinical trials for carfilzomib continue under Onyx Pharmaceuticals, which acquired Proteolix in 2009.[5]
In January 2011, the FDA granted carfilzomib fast-track status, allowing Onyx to initiate a rolling submission of its new drug application for carfilzomib.[6] In December 2011, the FDA granted Onyx standard review designation,[7][8] for its new drug application submission based on the 003-A1 study, an open-label, single-arm Phase 2b trial. The trial evaluated 266 heavily-pretreated patients with relapsed and refractory multiple myeloma who had received at least two prior therapies, including bortezomib and either thalidomide or lenalidomide.[9] It costs approximately $10,000 per 28-day cycle, making it the most expensive FDA-approved drug for multiple myeloma.[10]
Mechanism
Carfilzomib irreversibly binds to and inhibits the chymotrypsin-like activity of the 20S proteasome, an enzyme that degrades unwanted cellular proteins. Inhibition of proteasome-mediated proteolysis results in a build-up of polyubiquinated proteins, which may cause cell cycle arrest, apoptosis, and inhibition of tumor growth.[1]
Clinical trials
Completed
A single-arm, Phase II trial (003-A1) of carfilzomib in patients with relapsed and refractory multiple myeloma showed that single-agent carfilzomib demonstrated a clinical benefit rate of 36 percent in the 266 patients evaluated and had an overall response rate of 22.9 percent and median duration of response of 7.8 months. The FDA approval of carfilzomib was based on results of the 003-A1 trial.[11]
In a Phase II trial (004), carfilzomib had a 53 percent overall response rate among patients with relapsed and/or refractory multiple myeloma who had not previously received bortezomib. This study also included a bortezomib-treated cohort. Results were reported separately.[12] This study also found prolonged carfilzomib treatment was tolerable, with approximately 22 percent of patients continuing treatment beyond one year. The 004 trial was a smaller study originally designed to investigate the impact of carfilzomib treatment in relationship to bortezomib treatment in less heavily pretreated (1-3 prior regimens) patients.[13]
A Phase II trial (005), which assessed the safety, pharmacokinetics, pharmacodynamics and efficacy of carfilzomib, in patients with multiple myeloma and varyi ng degrees of renal impairment, where nearly 50 percent of patients were refractory to both bortezomib and lenalidomide, demonstrated that pharmacokinetics and safety were not influenced by the degree of baseline renal impairment. Carfilzomib was tolerable and demonstrated efficacy.[14]
In another Phase II trial (006) of patients with relapsed and/or refractory multiple myeloma, carfilzomib in combination with lenalidomide and dexamethasone demonstrated an overall response rate of 69 percent.[15]
A Phase II trial (007) for multiple myeloma and solid tumors showed promising results.[16][17]
In Phase II trials of carfilzomib, the most common grade 3 or higher treatment-emergent adverse events were thrombocytopenia, anemia, lymphoenia, neutropenia, pneumonia, fatigue and hyponatremia.[18]
In a frontline Phase I/II study, the combination of carfilzomib, lenalidomide, and low-dose dexamethasone was highly active and well tolerated, permitting the use of full doses for an extended time in newly-diagnosed multiple myeloma patients, with limited need for dose modification. Responses were rapid and improved over time, reaching 100 percent very good partial response.[19]
Ongoing
A phase III confirmatory clinical trial, known as the ASPIRE trial, comparing carfilzomib, lenalidomide and dexamethasone versus lenalidomide and dexamethasone in patients with relapsed multiple myeloma is ongoing.[20] It is no longer recruiting and should report in 2014.


| Systematic (IUPAC) name | |
|---|---|
| (S)-4-Methyl-N-((S)-1-(((S)-4-methyl-1-((R)-2-methyloxiran-2-yl)-1-oxopentan-2-yl)amino)-1-oxo-3-phenylpropan-2-yl)-2-((S)-2-(2-morpholinoacetamido)-4-phenylbutanamido)pentanamide | |
| Clinical data | |
| Trade names | Kyprolis |
| Licence data | US FDA:link |
| Pregnancy cat. | D (US) |
| Legal status | ℞-only (US) |
| Routes | Intravenous |
| Identifiers | |
| CAS number | 868540-17-4 |
| ATC code | L01XX45 |
| PubChem | CID 11556711 |
| ChemSpider | 9731489 |
| KEGG | D08880 |
| ChEMBL | CHEMBL451887 |
| Synonyms | PX-171-007 |
| Chemical data | |
| Formula | C40H57N5O7 |
| Mol. mass | 719.91 g mol |


http://pubs.rsc.org/en/content/articlelanding/2013/np/c3np20126k/unauth#!divAbstract
The initial enthusiasm following the discovery of a pharmacologically active natural product is often fleeting due to the poor prospects for its ultimate clinical application. Despite this, the ever-changing landscape of modern biology has a constant need for molecular probes that can aid in our understanding of biological processes. After its initial discovery by Bristol-Myers Squibb as a microbial anti-tumor natural product, epoxomicin was deemed unfit for development due to its peptide structure and potentially labile epoxyketone pharmacophore. Despite its drawbacks, epoxomicin’s pharmacophore was found to provide unprecedented selectivity for the proteasome. Epoxomicin also served as a scaffold for the generation of a synthetic tetrapeptide epoxyketone with improved activity, YU-101, which became the parent lead compound of carfilzomib (Kyprolis™), the recently approved therapeutic agent for multiple myeloma. In this era of rational drug design and high-throughput screening, the prospects for turning an active natural product into an approved therapy are often slim. However, by understanding the journey that began with the discovery of epoxomicin and ended with the successful use of carfilzomib in the clinic, we may find new insights into the keys for success in natural product-based drug discovery.

References
- Carfilzomib, NCI Drug Dictionary
- “FDA Approves Kyprolis for Some Patients with Multiple Myeloma”. FDA. 2012-07-20. Retrieved 2013-07-23.
- Meng, L; Mohan, R.; Kwok, B.H.; Elofsson, M.; Sin, N.; Crews, C.M. (1999).“Epoxomicin, a potent and selective proteasome inhibitor, exhibits in vivo antiinflammatory activity”. Proc Natl Acad Sci USA 96 (18): 10403–8.doi:10.1073/pnas.96.18.10403. PMC 17900. PMID 10468620.
- Myung, J; Kim, K.B.; Lindsten, K.; Dantuma, N.P.; Crews, C.M. (2001). “Lack of proteasome active site allostery as revealed by subunit-specific inhibitors”. Mol Cell 7 (2): 411–20. doi:10.1016/S1097-2765(01)00188-5. PMID 11239469.
- ^ Jump up to:a b “Carfilzomib: From Discovery To Drug”. Chemical & Engineering News. 2012-08-27. Retrieved 2013-07-30.
- “Onyx multiple myeloma drug wins FDA fast-track status”. San Francisco Business Times. 2011-01-31. Retrieved 2011-09-01.
- “Beacon Breaking News – Carfilzomib to Get Standard, Not Priority, FDA Review”. The Myeloma Beacon. Retrieved 2012-02-27.
- “Fast Track, Accelerated Approval and Priority Review; Accelerating Availability of New Drugs for Patients with Serious Diseases”. FDA. Retrieved 2012-02-27.
- “PX-171-003-A1, an open-label, single-arm, phase (Ph) II study of carfilzomib (CFZ) in patients (pts) with relapsed and refractory multiple myeloma (R/R MM): Long-term follow-up and subgroup analysis”. ASCO 2011; Abstract 8027. 2011. Retrieved 2011-09-01.
- “FDA Approves Kyprolis (Carfilzomib) For Relapsed And Refractory Multiple Myeloma”. The Myeloma Beacon. Retrieved 2012-07-20.
- “Carfilzomib Prescribing Information”. NCI Drug Dictionary. Retrieved 2013-07-23.
- Vij, R (2012). “An open-label, single-arm, phase 2 study of single-agent carfilzomib in patients with relapsed and/or refractory multiple myeloma who have been previously treated with bortezomib”. Br J Haematol 158 (6): 739–748. doi:10.1111/j.1365-2141.2012.09232.x. PMID 22845873.
- Vij, R (2012). “An open-label, single-arm, phase ii (PX-171-004) study of single-agent carfilzomib in bortezomib-naive patients with relapsed and/or refractory multiple myeloma.”. Blood 119 (24): 5661–70. doi:10.1182/blood-2012-03-414359.PMID 22555973.
- Badros, AZ (2013). “Carfilzomib in multiple myeloma patients with renal impairment: pharmacokinetics and safety.”. Leukemia 27 (8): 1707–14. doi:10.1038/leu.2013.29.PMID 23364621.
- “European Hematology Association (EHA) 18th Congress. June 13-16, 2013.”. The Myeloma Beacon. 2013. Retrieved 2013-07-13.
- “Nikoletta Lendval, MD PhD et al. Phase II Study of Infusional Carfilzomib in Patients with Relapsed or Refractory Multiple Myeloma.”. Presented at: 54th ASH Annual Meeting and Exposition: December 2012. Retrieved 2013-07-23.
- “Phase II results of Study PX-171-007: A phase Ib/II study of carfilzomib (CFZ), a selective proteasome inhibitor, in patients with selected advanced metastatic solid tumors” – ASCO 2009; Abstract 3515.
- “Siegel DS, Martin T, Wang, M, et al. Results of PX-171- 003-A1, an open-label, single-arm, phase 2 study of carfilzomib in patients with relapsed and refractory multiple myeloma. Presented at: 52nd ASH Annual Meeting and Exposition; December 4-7, 2010; Orlando, Florida.”. OncLive.com. 2011-03-09. Retrieved 2011-09-01.
- “Final Results of a Frontline Phase 1/2 Study of Carfilzomib Lenalidomide, and Low-Dose Dexamethasone (CRd) in Multiple Myeloma (MM)”. ASH 20111; Abstract 631. Retrieved 2012-02-27.
- “Phase 3 Study Comparing Carfilzomib, Lenalidomide, and Dexamethasone (CRd) Versus Lenalidomide and Dexamethasone (Rd) in Subjects With Relapsed Multiple Myeloma”. ClinicalTrials.gov. 2011-08-04. Retrieved 2011-09-01.
External links
- “Carfilzomib Prescribing Information”. NCI Drug Dictionary.
Mangafodipir


Mangafodipir
CAS : 118248-94-5 (free acid); 155319-91-8 (hexahydrogen)
CAS Name: (OC-6-13)-[[N,N¢–1,2-Ethanediylbis[N-[[3-(hydroxy-kO)-2-methyl-5-[(phosphonooxy)methyl]-4-pyridinyl]methyl]glycinato-kN,kO]](8-)]manganate(6-)
Add Names: manganese(II)-N,N¢-dipyridoxylethylenediamine-N,N¢-diacetate-5,5-bis(phosphonate); manganese dipyridoxal diphosphate; MnDPDP
Manufacturers’ Codes: S-095
C22H24MnN4O14P2
685.33
Percent Composition: C 38.56%, H 3.53%, Mn 8.02%, N 8.18%, O 32.68%, P 9.04%

| Clinical data | |
|---|---|
| AHFS/Drugs.com | Micromedex Detailed Consumer Information |
| Pregnancy cat. | Not to be used |
| Routes | Intravenous infusion |
| Pharmacokinetic data | |
| Bioavailability | NA |
| Protein binding | 27% (manganese) Negligible (DPDP) |
| Half-life | 20 minutes (manganese) 50 minutes (DPDP) |
| Excretion | Renal and fecal (manganese) Renal (DPDP) |
| Identifiers | |
| ATC code | V08CA05 |
| PubChem | CID 3086672 |
| ChemSpider | 2343239 |
| UNII | N02W67RKJS |
| Chemical data | |
| Formula | C22H28MnN4O14P2 |
| Mol. mass | 689.362 g/mol |
Diagnostic Aid (MRI Contrast Agent)
Manganese dipyridoxal diphosphate trisodium salt, Mangafodipir trisodium, Win-59010-2, S-095, MnDPDP, Teslascan
Mangafodipir (sold under the brand name Teslascan as mangafodipir trisodium) is a contrast agent delivered intravenously to enhance contrast in magnetic resonance imaging (MRI) of the liver. It has two parts, paramagnetic manganese (II) ions and thechelating agent fodipir (dipyridoxyl diphosphate, DPDP). Normal liver tissue absorbs the manganese more than abnormal or cancerous tissue. The manganese shortens the longitudinal relaxation time (T1), making the normal tissue appear brighter in MRIs. This enhanced contrast allows lesions to be more easily identified.

The condensation of pyridoxal 5-phosphate (I) with ethylenediamine (II) in methanol by means of NaOH gives the corresponding diimine (III), which is reduced with hydrogen over Pt/C in methanol/water yielding the expected diamine (IV). The reaction of (IV) with bromoacetic acid (V) by means of NaOH in methanol/water affords the N,N’-diacetic acid derivative (VI), which is finally treated with MnCl2 in water containing NaOH.
References
Literature References:
Paramagnetic manganese (II) chelate designed as a tissue specific imaging agent taken up by normal liver parenchyma. Prepn: S. M. Rocklage, S. C. Quay, EP 290047; eidem, US 4933456 (1988, 1990 both to Salutar); idem et al.,Inorg. Chem. 28, 477 (1989).
Pharmacology, toxicity and image enhancement studies: G. Elizondo et al., Radiology 178, 73 (1991).
HPLC determn in plasma: K. G. Toft et al., J. Pharm. Biomed. Anal. 15, 973 (1997).
Series of articles on clinical studies, toxicology and physicochemical properties: Acta Radiol. 38, 626-789 (1997).
Review of use as contrast agent for liver lesion detection: N. M. Rofsky, J. P. Earls, MRI Clin. North Am. 4, 73-85 (1996).
Properties: LD50 i.v. in mice: 5.4 mmol/kg (Elizondo).
Toxicity data: LD50 i.v. in mice: 5.4 mmol/kg (Elizondo)
……………………………………
Mangafodipir Trisodium

C22H27MnN4Na3O14P2 ![]()
![]()
![]()
![]()
![]() 757.33
757.33
Trisodium trihydrogen (OC-6-13)-[[N,N¢-1,2-ethanediylbis[N-[[3-hydroxy-2-methyl-5-[(phosphonooxy)methyl]-4-pyridinyl]methyl]glycinato]](8-)] manganate(6-).
Trisodium trihydrogen (OC-6-13)-[[N,N¢-ethylenebis[N-[[3-hydroxy-5-(hydroxymethyl)-2-methyl-4-pyridyl]methyl]glycine] 5,5¢-bis(phosphato)](8-)]manganate(6-) ![]()
![]()
![]() [140678-14-4].
[140678-14-4].
Derivative Type: Trisodium salt
CAS Registry Number: 140678-14-4
Additional Names: Magnafodipir trisodium
Manufacturers’ Codes: Win-59010
Trademarks: Teslascan (Nycomed)
Molecular Formula: C22H27MnN4Na3O14P2
Molecular Weight: 757.32
Percent Composition: C 34.89%, H 3.59%, Mn 7.25%, N 7.40%, Na 9.11%, O 29.58%, P 8.18%
Properties: Pale yellow, triclinic hygroscopic crystals. d 1.537. uv max (water): 220, 257, 319 nm (e 37600, 10300, 13400). Soly (g/ml): 0.4596 water, 0.0230 methanol, 0.0008 ethanol, 0.0006 acetone, 0.0011 chloroform. Log P (1-octanol:water) -5.62; (1-butanol: water) -3.68. Prepd as 0.01 mmol/ml aqueous infusion: bright yellow, clear soln, pH 7.5. Viscosity (mPa.s): 1.0 at 20°, 0.7 at 37°. Osmolality (37°): 290 mosmol/kg. d20 1.01 g/ml.
Log P: Log P (1-octanol:water) -5.62; (1-butanol: water) -3.68
Absorption maximum: uv max (water): 220, 257, 319 nm (e 37600, 10300, 13400)
Density: d 1.537; d20 1.01 g/ml
Therap-Cat: Diagnostic aid (MRI contrast agent).
Keywords: Diagnostic Aid (MRI Contrast Agent).http://www.drugfuture.com/Pharmacopoeia/USP32/pub/data/v32270/usp32nf27s0_m47135.html
Eisai’s lenvatinib 兰伐替尼 レンバチニブ to get speedy review in Europe

Lenvatinib
For the treatment of patients with progressive radioiodine-refractory, differentiated thyroid cancer (RR-DTC).
CAS 417716-92-8,
CAS 857890-39-2 (lenvatinib mesylate)
E 7080, ER-203492-00, E7080, E 7080,
4-[3-Chloro-4-(cyclopropylaminocarbonyl)aminophenoxy]-7-methoxy-6-quinolinecarboxamide
Molecular Formula: C21H19ClN4O4
Molecular Weight: 426.85296
Eisai Co., Ltd INNOVATOR
European regulators have agreed to undertake an accelerated assessment of Eisai’s lenvatinib as a treatment for progressive radioiodine-refractory, differentiated thyroid cancer.
The drug, which carries Orphan Status in the EU, is to be filed “imminently” and could become the first in a new class of tyrosine kinase inhibitors, the drugmaker said.

Read more at: http://www.pharmatimes.com/Article/14-07-31/Eisai_s_lenvatinib_to_get_speedy_review_in_Europe.aspx#ixzz39OGhRHas
Lenvatinib was granted Orphan Drug Designation for thyroid cancer by the health authorities in Japan in 2012, and in Europe and the U.S in 2013. The first application for marketing authorization of lenvatinib in the world was submitted in Japan on June 2014. Eisai is planning to submit applications for marketing authorization in Europe and the U.S. in the second quarter of fiscal 2014.
Lenvatinib is an oral multiple receptor tyrosine kinase (RTK) inhibitor with a novel binding mode that selectively inhibits the kinase activities of vascular endothelial growth factor receptors (VEGFR), in addition to other proangiogenic and oncogenic pathway-related RTKs including fibroblast growth factor receptors (FGFR), the platelet-derived growth factor (PDGF) receptor PDGFRalpha, KIT and RET that are involved in tumor proliferation. This potentially makes lenvatinib a first-in-class treatment, especially given that it simultaneously inhibits the kinase activities of FGFR as well as VEGFR.
LENVATINIB BASE
COSY PREDICT
| Systematic (IUPAC) name | |
|---|---|
| 4-[3-chloro-4-(cyclopropylcarbamoylamino)phenoxy]-7-methoxy-quinoline-6-carboxamide | |
| Clinical data | |
| Legal status | ℞ Prescription only |
| Identifiers | |
| CAS number | |
| ATC code | None |
| PubChem | CID 9823820 |
| ChemSpider | 7999567 |
| UNII | EE083865G2 |
| Chemical data | |
| Formula | C21H19ClN4O4 |
| Mol. mass | 426.853 g/mol |
Lenvatinib (E7080) is a multi-kinase inhibitor that is being investigated for the treatment of various types of cancer by Eisai Co. It inhibits both VEGFR2 and VEGFR3 kinases.[1]
The substence was granted orphan drug status for the treatment of various types of thyroid cancer that do not respond toradioiodine; in the US and Japan in 2012 and in Europe in 2013[2] and is now approved for this use.

Clinical trials
Lenvatinib has had promising results from a phase I clinical trial in 2006[3] and is being tested in several phase II trials as of October 2011, for example against hepatocellular carcinoma.[4] After a phase II trial testing the treatment of thyroid cancer has been completed with modestly encouraging results,[5] the manufacturer launched a phase III trial in March 2011.[6]

Lenvatinib Mesilate
Molecular formula: C21H19ClN4O4,CH4O3S =523.0.
CAS: 857890-39-2.
UNII code: 3J78384F61.
About the Lenvatinib (E7080) Phase II Study
The open-label, global, single-arm Phase II study of multi-targeted kinase inhibitor lenvatinib (E7080) in advanced radioiodine (RAI)-refractory differentiated thyroid cancer involved 58 patients with advanced RAI refractory DTC (papillary, follicular or Hurthle Cell) whose disease had progressed during the prior 12 months. (Disease progression was measured using Response Evaluation Criteria in Solid Tumors (RECIST).) The starting dose of lenvatinib was 24 mg once daily in repeated 28 day cycles until disease progression or development of unmanageable toxicities.
2. About Thyroid Cancer
Thyroid cancer refers to cancer that forms in the tissues of the thyroid gland, located at the base of the throat or near the trachea. It affects more women than men and usually occurs between the ages of 25 and 65.
The most common types of thyroid cancer, papillary and follicular (including Hurthle Cell), are classified as differentiated thyroid cancer and account for 95 percent of all cases. While most of these are curable with surgery and radioactive iodine treatment, a small percentage of patients do not respond to therapy.
3. About Lenvatinib (E7080)
Lenvatinib is multi-targeted kinase inhibitor with a unique receptor tyrosine kinase inhibitory profile that was discovered and developed by the Discovery Research team of Eisai’s Oncology Unit using medicinal chemistry technology. As an anti-angiogenic agent, it inhibits tyrosine kinase of the VEGF (Vascular Endothelial Growth Factor) receptor, VEGFR2, and a number of other types of kinase involved in angiogenesis and tumor proliferation in balanced manner. It is a small molecular targeting drug that is currently being studied in a wide array of cancer types.
4-(3-chloro-4-(cyclopropylaminocarbonyl)aminophenoxy)-7-methoxy-6-quinolinecarboxamide (additional name: 4-[3-chloro-4-(N′-cyclopropylureido)phenoxy]-7-methoxyquinoline-6-carboxamide) is known to exhibit an excellent angiogenesis inhibition as a free-form product, as described in Example 368 of Patent Document 1. 4-(3-chloro-4-(cyclopropylaminocarbonyl)aminophenoxy)-7-methoxy-6-quinolinecarboxamide is also known to exhibit a strong inhibitory action for c-Kit kinase (Non-Patent Document 1, Patent Document 2).
However, there has been a long-felt need for the provision of a c-Kit kinase inhibitor or angiogenesis inhibitor that has high usability as a medicament and superior characteristics in terms of physical properties and pharmacokinetics in comparison with the free-form product of 4-(3-chloro-4-(cyclopropylaminocarbonyl)aminophenoxy)-7-methoxy-6-quinolinecarboxamide.
[Patent Document 1] WO 02/32872
[Patent Document 2] WO 2004/080462
[Non-Patent Document 1] 95th Annual Meeting Proceedings, AACR (American Association for Cancer Research), Volume 45, Page 1070-1071, 2004
………………………..
PATENT
http://www.google.co.in/patents/US8058474
EXAMPLES
Examples will now be described to facilitate understanding of the invention, but the invention is not limited to these examples.
Example 1Phenyl N-(2-chloro-4-hydroxyphenyl)carbamate

After suspending 4-amino-3-chlorophenol (23.7 g) in N,N-dimethylformamide (100 mL) and adding pyridine (23.4 mL) while cooling on ice, phenyl chloroformate (23.2 ml) was added dropwise below 20° C. Stirring was performed at room temperature for 30 minutes, and then water (400 mL), ethyl acetate (300 mL) and 6N HCl (48 mL) were added, the mixture was stirred and the organic layer was separated. The organic layer was washed twice with 10% brine (200 mL), and dried over magnesium sulfate. The solvent was removed to give 46 g of the title compound as a solid.
1H-NMR (CDCl3): 5.12 (1h, br s), 6.75 (1H, dd, J=9.2, 2.8 Hz), 6.92 (1H, d, J=2.8 Hz), 7.18-7.28 (4H, m), 7.37-7.43 (2H, m), 7.94 (1H, br s)
Example 21-(2-chloro-4-hydroxyphenyl)-3-cyclopropylurea

After dissolving phenyl N-(2-chloro-4-hydroxyphenyl)carbamate in N,N-dimethylformamide (100 mL), cyclopropylamine (22.7 mL) was added while cooling on ice and the mixture was stirred overnight at room temperature. Water (400 mL), ethyl acetate (300 mL) and 6N HCl (55 mL) were then added, the mixture was stirred and the organic layer was separated. The organic layer was washed twice with 10% brine (200 mL), and dried over magnesium sulfate. Prism crystals obtained by concentrating the solvent were filtered and washed with heptane to give 22.8 g of the title compound (77% yield from 4-amino-3-chlorophenol).
1H-NMR (CDCl3): 0.72-0.77 (2H, m), 0.87-0.95 (2H, m), 2.60-2.65 (1H, m), 4.89 (1H, br s), 5.60 (1H, br s), 6.71 (1H, dd, J=8.8, 2.8 Hz), 6.88 (1H, d, J=2.8 Hz), 7.24-7.30 (1H, br s), 7.90 (1H, d, J=8.8H)
Example 34-(3-chloro-4-(cyclopropylaminocarbonyl)aminophenoxy)-7-methoxy-6-quinolinecarboxamide

To dimethylsulfoxide (20 mL) were added 7-methoxy-4-chloro-quinoline-6-carboxamide (0.983 g), 1-(2-chloro-4-hydroxyphenyl)-3-cyclopropylurea (1.13 g) and cesium carbonate (2.71 g), followed by heating and stirring at 70° C. for 23 hours. After the reaction mixture was allowed to cool down to room temperature, water (50 mL) was added, and the produced crystals were collected by filtration to give 1.56 g of the title compound (88% yield).
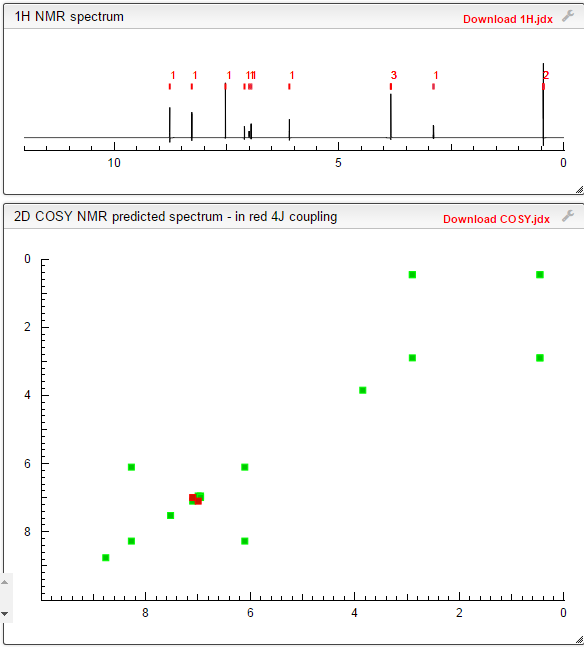
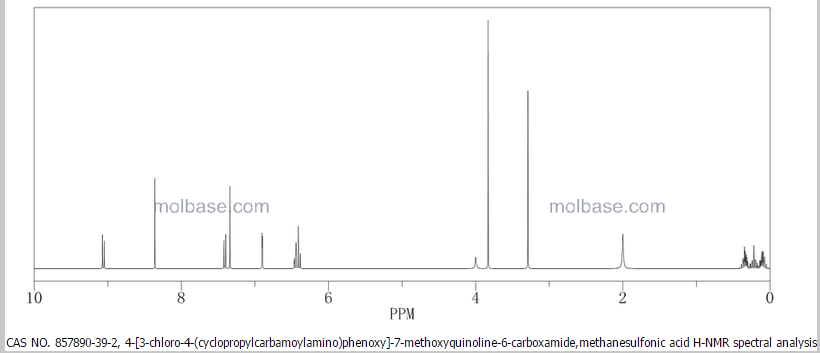
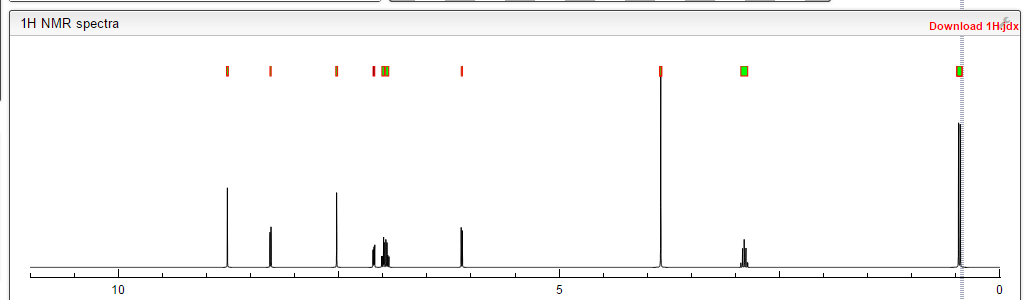
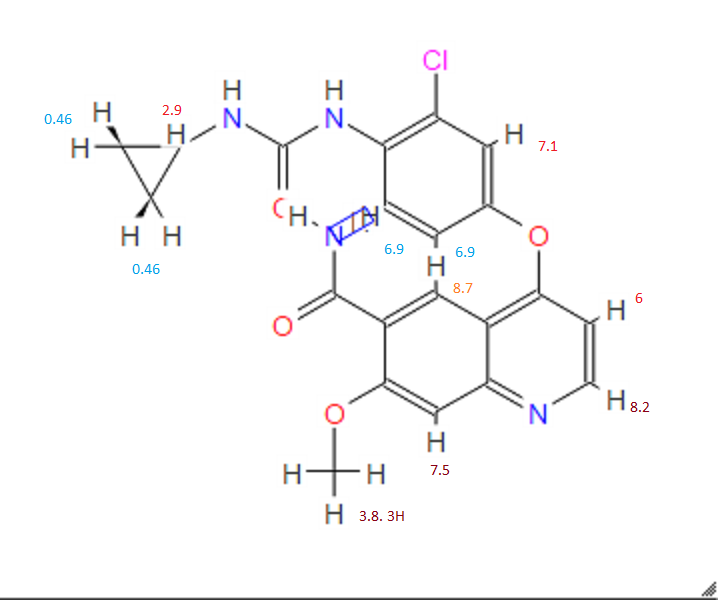
1H-NMR (d6-DMSO): 0.41 (2H, m), 0.66 (2H, m), 2.56 (1H, m), 4.01 (3H, s), 6.51 (1H, d, J=5.6 Hz), 7.18 (1H, d, J=2.8 Hz), 7.23 (1H, dd, J=2.8, 8.8 Hz), 7.48 (1H, d, J=2.8 Hz), 7.50 (1H, s), 7.72 (1H, s), 7.84 (1H, s), 7.97 (1H, s), 8.25 (1H, d, J=8.8 Hz), 8.64 (1H, s), 8.65 (1H, d, J=5.6 Hz)
Example 44-(3-chloro-4-(cyclopropylaminocarbonyl)aminophenoxy)-7-methoxy-6-quinolinecarboxamide
In a reaction vessel were placed 7-methoxy-4-chloro-quinoline-6-carboxamide (5.00 kg, 21.13 mol), dimethylsulfoxide (55.05 kg), 1-(2-chloro-4-hydroxyphenyl)-3-cyclopropylurea (5.75 kg, 25.35 mol) and potassium t-butoxide (2.85 kg, 25.35 mol) in that order, under a nitrogen atmosphere. After stirring at 20° C. for 30 minutes, the temperature was raised to 65° C. over a period of 2.5 hours. After stirring at the same temperature for 19 hours, 33% (v/v) acetone water (5.0 L) and water (10.0 L) were added dropwise over a period of 3.5 hours. Upon completion of the dropwise addition, the mixture was stirred at 60° C. for 2 hours, and 33% (v/v) acetone water (20.0 L) and water (40.0 L) were added dropwise at 55° C. or higher over a period of 1 hour. After then stirring at 40° C. for 16 hours, the precipitated crystals were collected by filtration using a nitrogen pressure filter, and the crystals were washed with 33% (v/v) acetone water (33.3 L), water (66.7 L) and acetone (50.0 L) in that order. The obtained crystals were dried at 60° C. for 22 hours using a conical vacuum drier to give 7.78 kg of the title compound (96.3% yield).
…………………………
SYNTHESIS
1H NMR PREDICT

13 C NMR PREDICT


…………………..
PATENT
http://www.google.co.in/patents/US7253286
EX 368

Example 368
4-(3-Chloro-4-(cyclopropylaminocarbonyl)aminophenoxy)-7-methoxy-6-quinolinecarboxamide
The title compound (22.4 mg, 0.052 mmol, 34.8%) was obtained as white crystals from phenyl N-(4-(6-carbamoyl-7-methoxy-4-quinolyl)oxy-2-chlorophenyl)carbamate (70 mg, 0.15 mmol) and cyclopropylamine, by the same procedure as in Example 11.
1H-NMR Spectrum (DMSO-d6) δ (ppm): 0.41 (2H, m), 0.66 (2H, m), 2.56 (1H, m), 4.01 (3H, s), 6.51 (1H, d, J=5.6 Hz), 7.18 (1H, d, J=2.8 Hz), 7.23 (1H, dd, J=2.8, 8.8 Hz), 7.48 (1H, d, J=2.8 Hz), 7.50 (1H, s), 7.72 (1H, s), 7.84 (1H, s), 7.97 (1H, s), 8.25 (1H, d, J=8.8 Hz), 8.64 (1H, s), 8.65 (1H, d, J=5.6 Hz).
The starting material was synthesized in the following manner.
Production Example 368-1Phenyl N-(4-(6-carbamoyl-7-methoxy-4-quinolyl)oxy-2-chlorophenyl)carbamate
The title compound (708 mg, 1.526 mmol, 87.4%) was obtained as light brown crystals from 4-(4-amino-3-chlorophenoxy)-7-methoxy-6-quinolinecarboxamide (600 mg, 1.745 mmol), by the same procedure as in Production Example 17.
1H-NMR Spectrum (CDCl3) δ (ppm): 4.14 (3H, s), 5.89 (1H, br), 6.50 (1H, d, J=5.6 Hz), 7.16 (2H, dd, J=2.4, 8.8 Hz), 7.22–7.30 (4H, m), 7.44 (2H, m), 7.55 (1H, s), 7.81 (1H, br), 8.31 (1H, d, J=8.8 Hz), 8.68 (1H, d, J=5.6 Hz), 9.27 (1H, s).
……………………
CRYSTALLINE FORM
http://www.google.co.in/patents/US7612208
Preparation Example 1
Preparation of 4-(3-chloro-4-(cyclopropylaminocarbonyl)aminophenoxy)-7-methoxy-6-quinolinecarboxamide (1)
Phenyl N-(4-(6-carbamoyl-7-methoxy-4-quinolyl)oxy-2-chlorophenyl)carbamate (17.5 g, 37.7 mmol) disclosed in WO 02/32872 was dissolved in N,N-dimethylformamide (350 mL), and then cyclopropylamine (6.53 mL, 94.25 mmol) was added to the reaction mixture under a nitrogen atmosphere, followed by stirring overnight at room temperature. To the mixture was added water (1.75 L), and the mixture was stirred. Precipitated crude crystals were filtered off, washed with water, and dried at 70° C. for 50 min. To the obtained crude crystals was added ethanol (300 mL), and then the mixture was heated under reflux for 30 min to dissolve, followed by stirring overnight to cool slowly down to room temperature. Precipitated crystals was filtered off and dried under vacuum, and then further dried at 70° C. for 8 hours to give the titled crystals (12.91 g; 80.2%).
Preparation Example 2Preparation of 4-(3-cloro-4-(cyclopropylaminocarbonyl)aminophenoxy)-7-methoxy-6-quinolinecarboxamide (2)
(1) Preparation of phenyl N-(2-chloro-4-hydroxyphenyl)carbamate

To a suspension of 4-amino-3-chlorophenol (23.7 g) in N,N-dimethylformamide (100 mL) was added pyridine (23.4 mL) while cooling in an ice bath, and phenyl chloroformate (23.2 mL) was added dropwise below 20° C. After stirring at room temperature for 30 min, water (400 mL), ethyl acetate (300 mL), and 6N-HCl (48 mL) were added and stirred. The organic layer was separated off, washed twice with a 10% aqueous sodium chloride solution (200 mL), and dried over magnesium sulfate. The solvent was evaporated to give 46 g of the titled compound as a solid.
- 1H-NMR Spectrum (CDCl3) δ(ppm): 5.12 (1H, br s), 6.75 (1H, dd, J=9.2, 2.8 Hz), 6.92 (1H, d, J=2.8 Hz), 7.18-7.28 (4H, m), 7.37-7.43 (2H, m), 7.94 (1H, br s).
(2) Preparation of 1-(2-chloro-4-hydroxyphenyl)-3-cyclopropylurea

To a solution of phenyl N-(2-chloro-4-hydroxyphenyl)carbamate in N,N-dimethylformamide (100 mL) was added cyclopropylamine (22.7 mL) while cooling in an ice bath, and the stirring was continued at room temperature overnight. Water (400 mL), ethyl acetate (300 mL), and 6N-HCl (55 mL) were added thereto, and the mixture was stirred. The organic layer was then separated off, washed twice with a 10% aqueous sodium chloride solution (200 mL), and dried over magnesium sulfate. The solvent was evaporated to give prism crystals, which were filtered off and washed with heptane to give 22.8 g of the titled compound (yield from 4-amino-3-chlorophenol: 77%).
- 1H-NMR Spectrum (CDCl3) δ(ppm): 0.72-0.77 (2H, m), 0.87-0.95 (2H, m), 2.60-2.65 (1H, m), 4.89 (1H, br s), 5.60 (1H, br s), 6.71 (1H, dd, J=8.8, 2.8 Hz), 6.88 (1H, d, J=2.8 Hz), 7.24-7.30 (1H, br s), 7.90 (1H, d, J=8.8 Hz)
(3) Preparation of 4-(3-chloro-4-(cyclopropylaminocarbonyl)aminophenoxy)-7-methoxy-6-quinolinecarboxamide
To dimethyl sulfoxide (20 mL) were added 7-methoxy-4-chloroquinoline-6-carboxamide (0.983 g), 1-(2-chloro-4-hydroxyphenyl)-3-cyclopropylurea (1.13 g) and cesium carbonate (2.71 g), and the mixture was heated and stirred at 70° C. for 23 hours. The reaction mixture was cooled to room temperature, and water (50 mL) was added, and the resultant crystals were then filtered off to give 1.56 g of the titled compound (yield: 88%).
Preparation Example 3Preparation of 4-(3-chloro-4-(cyclopropylaminocarbonyl)aminophenoxy)-7-methoxy-6-quinolinecarboxamide (3)
7-Methoxy-4-chloroquinoline-6-carboxamide (5.00 kg, 21.13 mol), dimethyl sulfoxide (55.05 kg), 1-(2-chloro-4-hydroxyphenyl)-3-cyclopropylurea 5.75 kg, 25.35 mol) and potassium t-butoxide (2.85 kg, 25.35 mol) were introduced in this order into a reaction vessel under a nitrogen atmosphere. The mixture was stirred for 30 min at 20° C., and the temperature was raised to 65° C. over 2.5 hours. The mixture was stirred at the same temperature for 19 hours. 33% (v/v) acetone-water (5.0 L) and water (10.0 L) were added dropwise over 3.5 hours. After the addition was completed, the mixture was stirred at 60° C. for 2 hours. 33% (v/v) acetone-water (20.0 L) and water (40.0 L) were added dropwise at 55° C. or more over 1 hour. After stirring at 40° C. for 16 hours, precipitated crystals were filtered off using a nitrogen pressure filter, and was washed with 33% (v/v) acetone-water (33.3 L), water (66.7 L), and acetone (50.0 L) in that order. The obtained crystals were dried at 60° C. for 22 hours using a conical vacuum dryer to give 7.78 kg of the titled compound (yield: 96.3%).
1H-NMR chemical, shift values for 4-(3-chloro-4-(cyclopropylaminocarbonyl)aminophenoxy)-7-methoxy-6-quinolinecarboxamides obtained in Preparation Examples 1 to 3 corresponded to those for 4-(3-chloro-4-(cyclopropylaminocarbonyl)aminophenoxy)-7-methoxy-6-quinolinecarboxamide disclosed in WO 02/32872.
Example 5
A Crystalline Form of the Methanesulfonate of 4-(3-chloro-4-(cyclopropylaminocarbonyl)aminophenoxy)-7-methoxy-6-quinolinecarboxamide (Form A)
(Preparation Method 1)
In a mixed solution of methanol (14 mL) and methanesulfonic acid (143 μL, 1.97 mmol) was dissolved 4-(3-chloro-4-(cyclopropylaminocarbonyl)aminophenoxy)-7-methoxy-6-quinolinecarboxamide (700 mg, 1.64 mmol) at 70° C. After confirming the dissolution of 4-(3-chloro-4-(cyclopropylaminocarbonyl)aminophenoxy)-7-methoxy-6-quinolinecarboxamide, the reaction mixture was cooled to room temperature over 5.5 hours, further stirred at room temperature for 18.5 hours, and crystals were filtered off. The resultant crystals were dried at 60° C. to give the titled crystals (647 mg).
(Preparation Method 2)
In a mixed solution of acetic acid (6 mL) and methanesulfonic acid (200 μL, 3.08 mmol) was dissolved 4-(3-chloro-4-(cyclopropylaminocarbonyl)aminophenoxy)-7-methoxy-6-quinolinecarboxamide (600 mg, 1.41 mmol) at 50° C. After confirming the dissolution of 4-(3-chloro-4-(cyclopropylaminocarbonyl)aminophenoxy)-7-methoxy-6-quinolinecarboxamide, ethanol (7.2 mL) and seed crystals of a crystalline form of the methanesulfonate of 4-(3-chloro-4-(cyclopropylaminocarbonyl)aminophenoxy)-7-methoxy-6-quinolinecarboxamide (Form A) (12 mg) were added in this order to the reaction mixture, and ethanol (4.8 mL) was further added dropwise over 2 hours. After the addition was completed, the reaction mixture was stirred at 40° C. for 1 hour then at room temperature for 9 hours, and crystals were filtered off. The resultant crystals were dried at 60° C. to give the titled crystals (545 mg).
Example 6A Crystalline Form of the Methanesulfonate of 4-(3-chloro-4-(cyclopropylaminocarbonyl)aminophenoxy)-7-methoxy-6-quinolinecarboxamide (Form B)
A crystalline form of the acetic acid solvate of the methanesulfonate of 4-(3-chloro-4-(cyclopropylaminocarbonyl)aminophenoxy)-7-methoxy-6-quinolinecarboxamide (Form I) (250 mg) obtained in Example 10 was dried under aeration at 30° C. for 3 hours and at 40° C. for 16 hours to give the titled crystals (240 mg)…………MORE IN PATENT
……………………………..
PATENT
https://www.google.com/patents/WO2014098176A1?cl=en
According to the present invention 4- (3-chloro-4- (cyclopropylamino-carbonyl) aminophenoxy) -7-methoxy-6-quinolinecarboxamide amorphous is excellent in solubility in water.
Example 1 4- (3-chloro-4- (cyclopropylamino-carbonyl) aminophenoxy) -7-methoxy-6-quinolinecarboxamide manufacture of amorphous amide
4- (3-chloro-4- (cyclopropylamino-carbonyl) amino phenoxy) -7-methoxy-6-quinolinecarboxamide B-type crystals (Patent Document 2) were weighed to 300mg, is placed in a beaker of 200mL volume, it was added tert- butyl alcohol (tBA) 40mL. This was heated to boiling on a hot plate, an appropriate amount of tBA to Compound A is dissolved, water was added 10mL. Then, the weakened heated to the extent that the solution does not boil, to obtain a sample solution. It should be noted, finally the solvent amount I was 60mL. 200mL capacity eggplant type flask (egg-plant shaped flask), and rotated in a state of being immersed in ethanol which had been cooled with dry ice. It was added dropwise a sample solution into the interior of the flask and frozen. After freezing the sample solution total volume, to cover the opening of the flask in wiping cloth, and freeze-dried. We got an amorphous A of 290mg.
Patent Document 2: US Patent Application Publication No. 2007/0117842 Patent specification 
Amorphous A 13 C-solid state NMR spectrum in Figure 2, the chemical shifts and I are shown in Table 3.
[Table 3] *: peak of t- butyl alcohol

………………………..
Paper
ACS Medicinal Chemistry Letters (2015), 6(1), 89-94
http://pubs.acs.org/doi/full/10.1021/ml500394m
……………..
Paper
Journal of Pharmaceutical and Biomedical Analysis (2015), 114, 82-87
http://www.sciencedirect.com/science/article/pii/S0731708515002940
KEEP WATCHING WILL BE UPDATED………….most of my posts are updated regularly
References
- Matsui, J.; Funahashi, Y.; Uenaka, T.; Watanabe, T.; Tsuruoka, A.; Asada, M. (2008). “Multi-Kinase Inhibitor E7080 Suppresses Lymph Node and Lung Metastases of Human Mammary Breast Tumor MDA-MB-231 via Inhibition of Vascular Endothelial Growth Factor-Receptor (VEGF-R) 2 and VEGF-R3 Kinase”. Clinical Cancer Research 14 (17): 5459–65.doi:10.1158/1078-0432.CCR-07-5270. PMID 18765537.
- “Phae III trial shows lenvatinib meets primary endpoint of progression free surival benefit in treatment of radioiodine-refactory differentiated thyroid cancer”. Eisai. 3 February 2014.
- Glen, H; D. Boss; T. R. Evans; M. Roelvink; J. M. Saro; P. Bezodis; W. Copalu; A. Das; G. Crosswell; J. H. Schellens (2007). “A phase I dose finding study of E7080 in patients (pts) with advanced malignancies”. Journal of Clinical Oncology, ASCO Annual Meeting Proceedings Part I 25 (18S): 14073.
- ClinicalTrials.gov NCT00946153 Study of E7080 in Patients With Advanced Hepatocellular Carcinoma (HCC)
- Gild, M. L.; Bullock, M.; Robinson, B. G.; Clifton-Bligh, R. (2011). “Multikinase inhibitors: A new option for the treatment of thyroid cancer”. Nature Reviews Endocrinology 7 (10): 617–624.doi:10.1038/nrendo.2011.141. PMID 21862995.
- ClinicalTrials.gov NCT01321554 A Trial of E7080 in 131I-Refractory Differentiated Thyroid Cancer
UPDATES
EXTRAS……………
Martin Schlumberger et al. A phase 3, multicenter, double-blind, placebo-controlled trial of lenvatinib(E7080) in patients with 131I-refractory differentiated thyroid cancer (SELECT). 2014 ASCO Annual Meeting. Abstract Number:LBA6008. Presented June 2, 2014. Citation: J Clin Oncol 32:5s, 2014 (suppl; abstr LBA6008). Clinical trial information: NCT01321554.
Bando, Masashi. Quinoline derivative-containing pharmaceutical composition. PCT Int. Appl. (2011), WO 2011021597 A1
Tomohiro Matsushima, four Nakamura, Kazuhiro Murakami, Atsushi Hoteido, Yusuke Ayat, Naoko Suzuki, Itaru Arimoto, Pinche Hirose, Masaharu Gotoda.Has excellent characteristics in terms of physical properties (particularly, dissolution rate) and pharmacokinetics (particularly, bioavailability), and is extremely useful as an angiogenesis inhibitor or c-Kit kinase inhibitor. US patent number US7612208 Also published as: CA2426461A1, CA2426461C, CN1308310C, CN1478078A, CN101024627A, DE60126997D1, DE60126997T2, DE60134679D1, DE60137273D1, EP1415987A1, EP1415987A4, EP1415987B1, EP1506962A2, EP1506962A3, EP1506962B1, EP1777218A1, EP1777218B1 , US7612092, US7973160, US8372981, US20040053908, US20060160832, US20060247259, US20100197911, US20110118470, WO2002032872A1, WO2002032872A8.Publication date: Aug 7, 2007 Original Assignee: Eisai Co., Ltd
Funahashi, Yasuhiro et al.Preparation of urea derivatives containing nitrogenous aromatic ring compounds as inhibitors of angiogenesis. US patent number US7253286, Also published as:CA2426461A1, CA2426461C, CN1308310C, CN1478078A, CN101024627A, DE60126997D1, DE60126997T2, DE60134679D1, DE60137273D1, EP1415987A1, EP1415987A4, EP1415987B1, EP1506962A2, EP1506962A3, EP1506962B1, EP1777218A1, EP1777218B1, US7612092, US7973160, US8372981, US20040053908, US20060160832, US20060247259, US20100197911, US20110118470, WO2002032872A1, WO2002032872A8.Publication date:Aug 7, 2007. Original Assignee:Eisai Co., Ltd
Sakaguchi, Takahisa; Tsuruoka, Akihiko. Preparation of amorphous salts of 4-[3-chloro-4-[(cyclopropylaminocarbonyl)amino]phenoxy]-7-methoxy-6-quinolinecarboxamide as antitumor agents. PCT Int. Appl. (2006), WO2006137474 A1 20061228.
Naito, Toshihiko and Yoshizawa, Kazuhiro. Preparation of urea moiety-containing quinolinecarboxamide derivatives. PCT Int. Appl., WO2005044788, 19 May 2005
Itaru Arimoto et al. Crystal of salt of 4-[3-chloro-4-(cyclopropylaminocarbonyl)amino-phenoxy]-7-methoxy-6-quinolinecarboxamide or solvate thereof and processes for producing these. PCT Int. Appl. (2005), WO2005063713 A1 20050714.
|
10-23-2009
|
ANTITUMOR AGENT FOR UNDIFFERENTIATED GASTRIC CANCER
|
|
|
10-2-2009
|
ANTI-TUMOR AGENT FOR MULTIPLE MYELOMA
|
|
|
8-21-2009
|
ANTITUMOR AGENT FOR THYROID CANCER
|
|
|
8-14-2009
|
THERAPEUTIC AGENT FOR LIVER FIBROSIS
|
|
|
2-27-2009
|
USE OF COMBINATION OF ANTI-ANGIOGENIC SUBSTANCE AND c-kit KINASE INHIBITOR
|
|
|
9-5-2008
|
Medicinal Composition
|
|
|
8-8-2007
|
Nitrogen-containing aromatic derivatives
|
|
|
5-25-2007
|
Polymorph of 4-[3-chloro-4-(cyclopropylaminocarbonyl)aminophenoxy]-7-methoxy-6- quinolinecarboxamide and a process for the preparation of the same
|
|
|
7-21-2006
|
Nitrogen-containing aromatic derivatives
|
|
|
6-23-2006
|
Use of sulfonamide-including compounds in combination with angiogenesis inhibitors
|
|
11-16-2011
|
UREA DERIVATIVE AND PROCESS FOR PREPARING THE SAME
|
|
|
8-10-2011
|
c-Kit kinase inhibitor
|
|
|
7-6-2011
|
Nitrogen-Containing Aromatic Derivatives
|
|
|
12-24-2010
|
COMBINED USE OF ANGIOGENESIS INHIBITOR AND TAXANE
|
|
|
9-24-2010
|
COMBINATION OF ANTI-ANGIOGENIC SUBSTANCE AND ANTI-TUMOR PLATINUM COMPLEX
|
|
|
4-30-2010
|
METHOD FOR PREDICTION OF THE EFFICACY OF VASCULARIZATION INHIBITOR
|
|
|
4-16-2010
|
METHOD FOR ASSAY ON THE EFFECT OF VASCULARIZATION INHIBITOR
|
|
|
3-24-2010
|
Urea derivative and process for preparing the same
|
|
|
2-26-2010
|
COMPOSITION FOR TREATMENT OF PANCREATIC CANCER
|
|
|
2-26-2010
|
COMPOSITION FOR TREATMENT OF UNDIFFERENTIATED GASTRIC CANCER
|
| US7253286 * | 18 Apr 2003 | 7 Aug 2007 | Eisai Co., Ltd | Nitrogen-containing aromatic derivatives |
| US20040053908 | 18 Apr 2003 | 18 Mar 2004 | Yasuhiro Funahashi | Nitrogen-containing aromatic derivatives |
| US20040242506 | 9 Aug 2002 | 2 Dec 2004 | Barges Causeret Nathalie Claude Marianne | Formed from paroxetine hydrochloride and ammonium glycyrrhyzinate by precipitation, spray, vacuum or freeze drying, or evaporation to glass; solid or oil; masks the bitter taste of paroxetine and has a distinctive licorice flavor; antidepressants; Parkinson’s disease |
| US20040253205 | 10 Mar 2004 | 16 Dec 2004 | Yuji Yamamoto | c-Kit kinase inhibitor |
| US20070004773 * | 22 Jun 2006 | 4 Jan 2007 | Eisai R&D Management Co., Ltd. | Amorphous salt of 4-(3-chiloro-4-(cycloproplylaminocarbonyl)aminophenoxy)-7-method-6-quinolinecarboxamide and process for preparing the same |
| US20070078159 | 22 Dec 2004 | 5 Apr 2007 | Tomohiro Matsushima | Has excellent characteristics in terms of physical properties (particularly, dissolution rate) and pharmacokinetics (particularly, bioavailability), and is extremely useful as an angiogenesis inhibitor or c-Kit kinase inhibitor |
| US20070117842 * | 22 Apr 2004 | 24 May 2007 | Itaru Arimoto | Polymorph of 4-[3-chloro-4-(cyclopropylaminocarbonyl)aminophenoxy]-7-methoxy-6- quinolinecarboxamide and a process for the preparation of the same |
| EP0297580A1 | 30 Jun 1988 | 4 Jan 1989 | E.R. SQUIBB & SONS, INC. | Amorphous form of aztreonam |
| JP2001131071A | Title not available | |||
| JP2005501074A | Title not available | |||
| JPS6422874U | Title not available | |||
| WO2002032872A1 | 19 Oct 2001 | 25 Apr 2002 | Itaru Arimoto | Nitrogenous aromatic ring compounds |
| WO2003013529A1 | 9 Aug 2002 | 20 Feb 2003 | Barges Causeret Nathalie Claud | Paroxetine glycyrrhizinate |
| WO2004039782A1 | 29 Oct 2003 | 13 May 2004 | Hirai Naoko | QUINOLINE DERIVATIVES AND QUINAZOLINE DERIVATIVES INHIBITING AUTOPHOSPHORYLATION OF Flt3 AND MEDICINAL COMPOSITIONS CONTAINING THE SAME |
| WO2004080462A1 | 10 Mar 2004 | 23 Sep 2004 | Eisai Co Ltd | c-Kit KINASE INHIBITOR |
| WO2004101526A1 | 22 Apr 2004 | 25 Nov 2004 | Itaru Arimoto | Polymorphous crystal of 4-(3-chloro-4-(cyclopropylaminocarbonyl)aminophenoxy)-7-methoxy-6-qunolinecarboxamide and method for preparation thereof |
| WO2005044788A1 | 8 Nov 2004 | 19 May 2005 | Eisai Co Ltd | Urea derivative and process for producing the same |
| WO2005063713A1 | 22 Dec 2004 | 14 Jul 2005 | Itaru Arimoto | Crystal of salt of 4-(3-chloro-4-(cyclopropylaminocarbonyl)amino-phenoxy)-7-methoxy-6-quinolinecarboxamide or of solvate thereof and processes for producing these |
| WO2006030826A1 | 14 Sep 2005 | 23 Mar 2006 | Eisai Co Ltd | Medicinal composition |
UPDATE………….
1H NMR PREDICT OF LENVATINIB BASE




FDA expands approval of drug to treat Pompe disease to patients of all ages; removes risk mitigation strategy requirements

Human glucosidase, prepro-α-[199-arginine,223-histidine] [1]
Alglucosidase alfa
C4435H6739N1175O1279S32
105270.8020
August 1, 2014
The U.S. Food and Drug Administration today announced the approval of Lumizyme (alglucosidase alfa) for treatment of patients with infantile-onset Pompe disease, including patients who are less than 8 years of age. In addition, the Risk Evaluation and Mitigation Strategy (REMS) known as the Lumizyme ACE (Alglucosidase Alfa Control and Education) Program is being eliminated.
Pompe disease is a rare genetic disorder and occurs in an estimated 1 in every 40,000 to 300,000 births. Its primary symptom is heart and skeletal muscle weakness, progressing to respiratory weakness and death from respiratory failure.
The disease causes gene mutations to prevent the body from making enough of the functional form of an enzyme called acid alpha-glucosidase (GAA). This enzyme is necessary for proper muscle functioning. GAA is used by the heart and muscle cells to convert a form of sugar called glycogen into energy. Without the enzyme action, glycogen builds up in the cells and, ultimately, weakens the heart and muscles. Lumizyme is believed to work by replacing the deficient GAA, thereby reducing the accumulated glycogen in heart and skeletal muscle cells.
Lumizyme, a lysosomal glycogen-specific enzyme, was approved by the FDA in 2010 with a REMS to restrict its use to treatment of patients with late (non-infantile) onset Pompe disease who are 8 years of age and older. The REMS was required to mitigate the potential risk of rapid disease progression in the infantile-onset Pompe disease patients and patients with late onset disease less than 8 years of age, and to communicate the risks of anaphylaxis, severe allergic reactions and severe skin and systemic immune mediated reactions to prescribers and patients.
At the time of Lumizyme’s approval, there were insufficient data to support the safety and efficacy of Lumizyme in the infantile-onset Pompe population, so Lumizyme was approved for use only in late onset Pompe disease patients who are at least 8 years of age. Pompe patients with infantile-onset disease and patients younger than 8 years of age continued treatment with Myozyme, which was already approved. Myozyme and Lumizyme, both manufactured by Genzyme Corporation, are produced from the same cell line at different production scales.
This approval provides access to Lumizyme for all Pompe disease patients, regardless of their age.
The FDA reviewed newly available information and determined that Lumizyme and Myozyme are chemically and biochemically comparable. Consequently, the safety and effectiveness of Lumizyme and Myozyme are expected to be comparable. In addition, a single-center clinical study of 18 infantile-onset Pompe disease patients, aged 0.2 to 5.8 months at the time of first infusion, provides further support that infantile-onset patients treated with Lumizyme will have a similar improvement in ventilator-free survival as those treated with Myozyme.
Because data were submitted supporting approval of Lumizyme for all Pompe patients, a REMS restricting its use to a specific age group is no longer necessary. While the risk of anaphylaxis, severe allergic reactions, and severe cutaneous and immune mediated reactions for Lumizyme still exist, these risks are comparable to Myozyme and are communicated in labeling through the Warnings and Precautions, and a Boxed Warning.
“REMS continue to be vital tools for the agency to employ as we work with companies to address the serious risks associated with drugs and monitor their appropriate and safe use in various health care settings,” said Janet Woodcock, M.D., director of the FDA’s Center for Drug Evaluation and Research. “The agency remains committed to exercising a flexible and responsible regulatory approach that ensures REMS programs are being effectively and efficiently used and not resulting in an unnecessary burden on health care professionals and patients.”
Health care professionals and patients should also be aware:
- The Warnings and Precautions section of the Lumizyme product label and the Clinical Studies section of the Lumizyme label have been updated to include the safety information of the drug in infantile-onset Pompe disease patients. This includes information from the currently approved Myozyme label and information from a new, uncontrolled study in which patients with infantile onset disease were treated with Lumizyme.
- Lumizyme is approved with a Boxed Warning because of the risk of anaphylaxis, severe allergic reactions, immune-mediated reactions and cardiorespiratory failure.
- Health care professionals should continue to refer to the drug prescribing information for the latest recommendations on prescribing Lumizyme and report adverse events to the FDA’s MedWatch program (http://www.fda.gov/Safety/MedWatch/default.htm).
- Distribution of Lumizyme will no longer be restricted. Health care professionals, healthcare facilities, and patients will no longer be required to enroll in the Lumizyme REMS program (Lumizyme ACE Program) to be able to prescribe, dispense, or receive Lumizyme.
The most commonly reported side effects for Lumizyme were infusion-related reactions and included severe allergic reactions, hives, diarrhea, vomiting, shortness of breath, itchy skin, skin rash, neck pain, partial hearing loss, flushing, pain in extremities, and chest discomfort.
Myozyme and Lumizyme are marketed by Cambridge, Massachusetts-based Genzyme.
The FDA, an agency within the U.S. Department of Health and Human Services, protects the public health by assuring the safety, effectiveness, and security of human and veterinary drugs, vaccines and other biological products for human use, and medical devices. The agency also is responsible for the safety and security of our nation’s food supply, cosmetics, dietary supplements, products that give off electronic radiation, and for regulating tobacco products.
| Country | Patent Number | Approved | Expires (estimated) |
|---|---|---|---|
| Canada | 2416492 | 2008-04-29 | 2021-07-10 |

>Alglucosidase alfa AHPGRPRAVPTQCDVPPNSRFDCAPDKAITQEQCEARGCCYIPAKQGLQGAQMGQPWCFF PPSYPSYKLENLSSSEMGYTATLTRTTPTFFPKDILTLRLDVMMETENRLHFTIKDPANR RYEVPLETPHVHSRAPSPLYSVEFSEEPFGVIVRRQLDGRVLLNTTVAPLFFADQFLQLS TSLPSQYITGLAEHLSPLMLSTSWTRITLWNRDLAPTPGANLYGSHPFYLALEDGGSAHG VFLLNSNAMDVVLQPSPALSWRSTGGILDVYIFLGPEPKSVVQQYLDVVGYPFMPPYWGL GFHLCRWGYSSTAITRQVVENMTRAHFPLDVQWNDLDYMDSRRDFTFNKDGFRDFPAMVQ ELHQGGRRYMMIVDPAISSSGPAGSYRPYDEGLRRGVFITNETGQPLIGKVWPGSTAFPD FTNPTALAWWEDMVAEFHDQVPFDGMWIDMNEPSNFIRGSEDGCPNNELENPPYVPGVVG GTLQAATICASSHQFLSTHYNLHNLYGLTEAIASHRALVKARGTRPFVISRSTFAGHGRY AGHWTGDVWSSWEQLASSVPEILQFNLLGVPLVGADVCGFLGNTSEELCVRWTQLGAFYP FMRNHNSLLSLPQEPYSFSEPAQQAMRKALTLRYALLPHLYTLFHQAHVAGETVARPLFL EFPKDSSTWTVDHQLLWGEALLITPVLQAGKAEVTGYFPLGTWYDLQTVPVEALGSLPPP PAAPREPAIHSEGQWVTLPAPLDTINVHLRAGYIIPLQGPGLTTTESRQQPMALAVALTK GGEARGELFWDDGESLEVLERGAYTQVIFLARNNTIVNELVRVTSEGAGLQLQKVTVLGV ATAPQQVLSNGVPVSNFTYSPDTKVLDICVSLLMGEQFLVSWC
| Systematic (IUPAC) name | |
|---|---|
| Human glucosidase, prepro-α-[199-arginine,223-histidine] [1] | |
| Clinical data | |
| AHFS/Drugs.com | monograph |
| Legal status | FDA approved for children[2] |
| Routes | Intravenous[2] |
| Identifiers | |
| CAS number | 420794-05-0 |
| ATC code | A16AB07 |
| DrugBank | DB01272 |
| UNII | DTI67O9503 |
| KEGG | D03207 |
| Chemical data | |
| Formula | C4758H7262N1274O1369S35[1] |
| Mol. mass | 105338 [1] |
Alglucosidase alfa (Lumizyme, Myozyme, Genzyme) is an enzyme replacement therapy (ERT) orphan drug for treatment of Pompe disease (Glycogen storage disease type II), a rare lysosomal storage disorder (LSD).[3] Chemically speaking, the drug is ananalog of the enzyme that is deficient in patients affected by Pompe disease, alpha-glucosidase. It is the first drug available to treat this disease.[2]
Status
Orphan drug pharmaceutical company, Genzyme, markets alglucosidase alfa as “Myozyme”. In 2006, the U.S. Food and Drug Administration (FDA) approved Myozyme as a suitable ERT treatment for children.[2] Some health plans have refused to subsidize Myozyme for adult patients because it lacks approval for treatment in adults, as well as its high cost (US$300,000/yr for life).[4]
On August 1, 2014 the U.S. Food and Drug Administration announced the approval of Lumizyme (alglucosidase alfa) for treatment of patients with infantile-onset Pompe disease, including patients who are less than 8 years of age. In addition, the Risk Evaluation and Mitigation Strategy (REMS) known as the Lumizyme ACE (Alglucosidase Alfa Control and Education) Program is being eliminated. [5]
Side effects
Common observed adverse reactions to alglucosidase alfa treatment are pneumonia, respiratory complications, infections and fever. More serious reactions reported includeheart and lung failure and allergic shock. Myozyme boxes carry warnings regarding the possibility of life-threatening allergic response.[2]
References
- ^ Jump up to:a b c American Medical Association (USAN). “Alglucosidase alfa” (Microsoft Word). STATEMENT ON A NONPROPRIETARY NAME ADOPTED BY THE USAN COUNCIL. Retrieved 18 December 2007.
- ^ Jump up to:a b c d e “FDA Approves First Treatment for Pompe Disease” (Press release). FDA. 2006-04-28. Retrieved 2008-07-07.
- Jump up^ Kishnani PS, Corzo D, Nicolino M et al. (2007). “Recombinant human acid [alpha]-glucosidase: major clinical benefits in infantile-onset Pompe disease”. Neurology 68 (2): 99–109.doi:10.1212/01.wnl.0000251268.41188.04. PMID 17151339.
- Jump up^ Geeta Anand (2007-09-18). “As Costs Rise, New Medicines Face Pushback”. Wall Street Journal (Dow Jones & Company). Retrieved 2008-07-07.
- Jump up^ cite press release |title=FDA expands approval of drug to treat Pompe disease to patients of all ages; removes risk mitigation strategy requirements |publisher=FDA |date=2014-08-14 |url=http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm407563.htm
External links
- Myozyme (alglucosidase alfa), Genzyme official website
- MTAP (Myozyme Temporary Access Program), Genzyme official website
MYOZYME (alglucosidase alfa), a lysosomal glycogen-specific enzyme, consists of the human enzyme acid α-glucosidase (GAA), encoded by the most predominant of nine observed haplotypes of this gene. MYOZYME is produced by recombinant DNA technology in a Chinese hamster ovary cell line. The MYOZYME manufacturing process differs from that for LUMIZYME®, resulting in differences in some product attributes. Alglucosidase alfa degrades glycogen by catalyzing the hydrolysis of α-1,4- and α-1,6- glycosidic linkages of lysosomal glycogen.
Alglucosidase alfa is a glycoprotein with a calculated mass of 99,377 daltons for the polypeptide chain, and a total mass of approximately 110 kilo Daltons, including carbohydrates. Alglucosidase alfa has a specific activity of 3 to 5 U/mg (one unit is defined as that amount of activity that results in the hydrolysis of 1 μmole of synthetic substrate per minute under the specified assay conditions). MYOZYME is intended for intravenous infusion. It is supplied as a sterile, nonpyrogenic, white to off-white, lyophilized cake or powder for reconstitution with 10.3 mL
Sterile Water for Injection, USP. Each 50 mg vial contains 52.5 mg alglucosidase alfa, 210 mg mannitol, 0.5 mg polysorbate 80, 9.9 mg sodium phosphate dibasic heptahydrate, 31.2 mg sodium phosphate monobasic monohydrate. Following reconstitution as directed, each vial contains 10.5 mL reconstituted solution and a total extractable volume of 10 mL at 5.0 mg/mL alglucosidase alfa. MYOZYME does not contain preservatives; each vial is for single use only.
FDA approves Jardiance to treat type 2 diabetes

Empagliflozin
For synthesis see https://newdrugapprovals.org/2013/12/19/empagliflozin/
August 1, 2014
The U.S. Food and Drug Administration today approved Jardiance (empagliflozin) tablets as an addition to diet and exercise to improve glycemic control in adults with type 2 diabetes.
Type 2 diabetes affects approximately 26 million people and accounts for more than 90 percent of diabetes cases diagnosed in the United States. Over time, high blood sugar levels can increase the risk for serious complications, including heart disease, blindness, and nerve and kidney damage.
“Jardiance provides an additional treatment option for the care of patients with type 2 diabetes,” said Curtis J. Rosebraugh, M.D., M.P.H., director of the Office of Drug Evaluation II in the FDA’s Center for Drug Evaluation and Research. “It can be used alone or added to existing treatment regimens to control blood sugar levels in the overall management of diabetes.”
Jardiance is a sodium glucose co-transporter 2 (SGLT2) inhibitor. It works by blocking the reabsorption of glucose (blood sugar) by the kidney, increasing glucose excretion, and lowering blood glucose levels in diabetics who have elevated blood glucose levels. The drug’s safety and effectiveness were evaluated in seven clinical trials with 4,480 patients with type 2 diabetes receiving Jardiance. The pivotal trials showed that Jardiance improved hemoglobin A1c levels (a measure of blood sugar control) compared to placebo.
Jardiance has been studied as a stand-alone therapy and in combination with other type 2 diabetes therapies including metformin, sulfonylureas, pioglitazone, and insulin. Jardiance should not be used: to treat people with type 1 diabetes; in those who have increased ketones in their blood or urine (diabetic ketoacidosis); and in those with severe renal impairment, end stage renal disease, or in patients on dialysis.
The FDA is requiring four postmarketing studies for Jardiance:
- Completion of an ongoing cardiovascular outcomes trial.
- A pediatric pharmacokinetic/pharmacodynamic study.
- A pediatric safety and efficacy study. As part of the safety and efficacy study, the effect on bone health and development will be evaluated.
- A nonclinical (animal) juvenile toxicity study with a particular focus on renal development, bone development, and growth.
Jardiance can cause dehydration, leading to a drop in blood pressure (hypotension) that can result in dizziness and/or fainting and a decline in renal function. The elderly, patients with impaired renal function, and patients on diuretics to treat other conditions appeared to be more susceptible to this risk.
The most common side effects of Jardiance are urinary tract infections and female genital infections.
Jardiance is distributed by Boehringer Ingelheim Pharmaceuticals, Inc., Ridgefield, Connecticut.
The FDA, an agency within the U.S. Department of Health and Human Services, protects the public health by assuring the safety, effectiveness, and security of human and veterinary drugs, vaccines and other biological products for human use, and medical devices. The agency also is responsible for the safety and security of our nation’s food supply, cosmetics, dietary supplements, products that give off electronic radiation, and for regulating tobacco products.
synthesis see https://newdrugapprovals.org/2013/12/19/empagliflozin/
NS 398 is a COX-2 inhibitor used in the study of the function of cyclooxygenases.

NS 398
N-[2-(Cyclohexyloxy)-4-nitrophenyl]methanesulfonamide
Taisho (Originator)
N-(2-cyclohexyloxy-4-nitrophenyl)methanesulfonamide.
123653-11-2, 123653-43-0 (Ca salt), 123653-44-1 (Na salt)
Cerebrovascular Diseases, Treatment of, NEUROLOGIC DRUGS, Stroke, Treatment of, Cyclooxygenase-2 Inhibitors
NS-398 is a COX-2 inhibitor used in the study of the function of cyclooxygenases.[2]
Selective cyclooxygenase-2 inhibitor (IC50 values are 3.8 and > 100 μM for COX-2 and COX-1 respectively). Orally active. Anti-inflammatory, anti-pyretic, analgesic and non-ulcerogenic in vivo. Induces apoptosis and cell cycle arrest
Cyclooxygenase (COX-2) has been recently suggested to play a role in hepatocarcinogenesis. However, the exact pathway by which COX-2 affects the growth of hepatocellular carcinoma (HCC) is not clear. This study investigated the effects of a specific COX-2 inhibitor, NS-398, on the cell proliferation and apoptosis of COX-2-expressing and non-expressing HCC cell lines.
In addition, the modulatory effect of NS-398 on apoptosis-regulating gene expression was examined. Semi-quantitative/quantitative reverse transcription-polymerase chain reaction and Western blot showed that Hep3B and HKCI-4 cells expressed COX-2 mRNA and protein, but HepG2 cells did not. NS-398 suppressed cell proliferation and induced apoptosis in the two COX-2-expressing cell lines in a dose-dependent manner, but not in HepG2 cells.
Fas ligand mRNA and protein expression were increased by the treatment with NS-398 (10 micro M) in COX-2-expressing cell lines. The expressions of Fas and Bcl-2 family genes (Bax, Bcl-2, Bcl-xL, Bcl-xS) were not affected by NS-398 treatment in all three cell lines. In conclusion, specific COX-2 inhibitor suppresses cell proliferation and induces apoptosis in HCC cell lines that express COX-2. Our finding suggests that COX-2 inhibition may offer a new approach for HCC chemoprevention.
| Identifiers | |
|---|---|
| CAS number | 123653-11-2 |
| PubChem | 4553 |
| Jmol-3D images | Image 1 |
| Properties | |
| Molecular formula | C13H18N2O5S |
| Molar mass | 314.36 g mol−1 |
| Appearance | Off-white solid |
| Solubility in water | Insoluble |
| Solubility in DMSO | 5 mg/mL |
| Hazards | |
| S-phrases | S22 S24/25 |

The condensation of 2-fluoronitrobenzene (I) with cyclohexanol (II) by means of NaH gives 2-(cyclohexyloxy)nitrobenzene (III), which is reduced with H2 over Pd/C in methanol yielding 2-(cyclohexyloxy)aniline (IV). The acylation of (IV) with methanesulfonyl chloride (V) in pyridine affords N-(2-cyclohexyloxy phenyl)methanesulfonamide (VI), which is finally nitrated with concentrated HNO3 in hot acetic acid.
EP 0317332
http://www.google.com/patents/EP0317332A2?cl=en
- Example 1
-
[0045](1) To 40 ml of a dioxane suspension containing 0.92 g of 60% sodium hydride was added 2.5 ml of cyclohexanol at room temperature over a 15-minute period, and the mixture was stirred at the same temperature for 1 hour and then at 50°C for 3.5 hours. The temperature of the reaction solution was returned to room temperature, 10 ml of a dioxane containing 3.2 g of 2-fluoronitrobenzene was added dropwise, and the mixture was stirred at room temperature overnight. The dioxane was evaporated, the residue was extracted with chloroform, and the chloroform layer was washed, in turn, with water and a saturated aqueous sodium chloride solution and then dried over anhydrous sodium sulfate. The solvent was evaporated to give an oil, which was then distilled under reduced pressure to give 3.8 g of 2-cyclohexyloxynitrobenzene.
b.p. 130 – 134°C/0.5 – 0.7 mmHg -
[0046](2) Fifty ml of a methanol solution containing 3.7 g of 2-cyclohexyloxynitrobenzene and 0.2 g of 5% palladium on carbon was stirred at room temperature under a hydrogen atmosphere for catalytic reduction. The catalyst was removed by filtration, and the filtrate was evaporated off to give 2.9 g of 2-cyclohexyloxyaniline as pale brown crystals.
m.p. 55 – 56°C -
[0047](3) To 20 ml of a pyridine solution containing 2.7 g of 2-cyclohexyloxyaniline was added dropwise 1.8 g of methanesulfonyl chloride under ice cooling with stirring. After completion of the addition, the mixture was stirred at room temperature for 2 hours. The reaction solution was poured into ice water and made acidic with dilute hydrochloric acid. The crystals which formed were collected by filtration, washed with water and dried to give 3.8 g of the crude crystals, which were then recrystallized from ethanol-n hexane to give 3.4 g of N-(2-cyclohexyloxyphenyl)methanesulfonamide.
m.p. 113 – 115°C -
[0048](4) To 20 ml of an acetic acid solution containing 3.4 g of N-(2-cyclohexyloxyphenyl)methanesulfonamide was added dropwise 1.5 g of 61% nitric acid on heating at 110°C over a 30-minute period, and then the mixture was stirred for 1 hour. The reaction solution was poured into ice water and neutralized with a dilute aqueous sodium hydroxide solution. The crystals which formed were collected by filtration, washed with water and dried to give 4.5 g of the crude crystals, which were then recrystallized from ethanol-n-hexane to give 3.3 g of N-(2-cyclohexyloxy-4-nitrophenyl)methanesulfonamide.
m.p. 136 – 137°C
| EP0093591A1 * | Apr 29, 1983 | Nov 9, 1983 | Eli Lilly And Company | Selective sulfonation process |
| FR2244473A1 * | Title not available | |||
| US3725451 * | Apr 13, 1970 | Apr 3, 1973 | Riker Laboratories Inc | Substituted benzoylhaloalkanesulfonanilides |
| US3840597 * | Jul 3, 1972 | Oct 8, 1974 | Riker Laboratories Inc | Substituted 2-phenoxy alkane-sulfonanilides |
| US3856859 * | Jun 8, 1973 | Dec 24, 1974 | Riker Laboratories Inc | Selective nitration process |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| EP1535614A2 * | Aug 22, 1997 | Jun 1, 2005 | University OofFlorida | Materials and methods for detection and treatment of immune system dysfunctions |
……………………………………………………..
The cortical collecting duct (CCD) is a major site of intrarenal prostaglandin E2 (PGE2) synthesis. This study examines the expression and regulation of the prostaglandin synthesizing enzymes cyclooxygenase-1 (COX-1) and -2 in the CCD. By indirect immunofluorescence using isoform-specific antibodies, COX-1 and -2 immunoreactivity was localized to all cell types of the murine M-1 CCD cell line. By immunohistochemistry, both COX-1 and COX-2 were localized to intercalated cells of the CCD on paraffin-embedded mouse kidney sections. When COX enzyme activity was measured in the M-1 cells, both indomethacin (COX-1 and -2 inhibitor) and the specific COX-2 inhibitor NS-398 effectively blocked PGE2 synthesis. These results demonstrate that COX-2 is the major contributor to the pool of PGE2synthesized by the CCD. By Western blot analysis, COX-2 expression was significantly upregulated by incubation with either indomethacin or NS-398. These drugs did not affect COX-1 protein expression. Evaluation of COX-2 mRNA expression by Northern blot analysis after NS-398 treatment demonstrated that the COX-2 protein upregulation occurred independently of any change in COX-2 mRNA expression. These studies have for the first time localized COX-2 to the CCD and provided evidence that the intercalated cells of the CCD express both COX-1 and COX-2. The results also demonstrate that constitutively expressed COX-2 is the major COX isoform contributing to PGE2synthesis by the M-1 CCD cell line. Inhibition of COX-2 activity in the M-1 cell line results in an upregulation of COX-2 protein expression.
http://jasn.asnjournals.org/content/10/11/2261.abstract
…………………………………………….
NS398 inhibits the growth of OSCC cells by mechanisms that are dependent and independent of suppression of PGE2 synthesis. Molecular targeting of COX-2, PGE2 synthase, or PGE2 receptors may be useful as a chemopreventive or therapeutic strategy for oral cancer.
http://clincancerres.aacrjournals.org/content/9/5/1885.full
…………………………………
References
- NS-398 at Sigma-Aldrich
- Wei Shen, Yong Li, Ying Tang, James Cummins and Johnny Huard (2005). “NS-398, a Cyclooxygenase-2-Specific Inhibitor, Delays Skeletal Muscle Healing by Decreasing Regeneration and Promoting Fibrosis”. American Journal of Pathology 167 (4): 1105–1117.doi:10.1016/S0002-9440(10)61199-6. PMC 1603662. PMID 16192645.
-
MORE References
Futaki et al (1993) NS-398, a novel non-steroidal anti-inflammatory drug with potent analgesic and antipyretic effects, which causes minimal stomach lesions. Gen.Pharmacol. 24 105. PMID: 8482483.
Futaki et al (1994) NS-398, a new anti-inflammatory agent, selectively inhibits prostaglandin G/H synthase/cyclooxygenase (COX-2) activity in vitro. Prostaglandins 47 55. PMID: 8140262.
Elder et al (2002) The MEK/ERK pathway mediates COX-2-selective NSAID-induced apoptosis and induced COX-2 protein expression in colorectal carcinoma cells. Int.J.Cancer 99 323. PMID: 11992399.
Gemoprost

Gemeprost, SC-37681, Ono-802, Cergem, Preglandin, Cervagem,
(E) -7 – [(1R, 2R, 3R-3-Hydroxy-2 – [(E) – (3R) -3-hydroxy-4,4-dimethyl-1-octenyl] -5-oxocyclopentyl] -2 -heptenoic acid methyl ester;
16,16-Dimethyl-DELTA2-trans-PGE1 methyl ester;
9-Oxo-11alpha, 15alpha-dihydroxy-16,16-dimethyl-2-trans, 13-trans-prostadiene-1-oic acid
Gemeprost (16, 16-dimethyl-trans-delta2 PGE1 methyl ester) is an analogue of prostaglandin E1.
Gemoprost, Preglandin (TN), SC-37681, AC1NQZPG, SureCN43075, Gemeprost (JAN/USAN/INN),
Molecular Formula: C23H38O5
Molecular Weight: 394.54482
Clinical use
It is used as a treatment for obstetric bleeding.
It is used with mifepristone to terminate pregnancy up to 24 weeks gestation. [1]
Side effects
Vaginal bleeding, cramps, nausea, vomiting, loose stools or diarrhea, headache, muscle weakness; dizziness; flushing; chills; backache; dyspnoea; chest pain; palpitations and mild pyrexia. Rare: Uterine rupture, severe hypotension, coronary spasms with subsequent myocardial infarctions
 |
|
| Systematic (IUPAC) name | |
|---|---|
| methyl (2E,11α,13E,15R)-11,15-dihydroxy-16,16-dimethyl-9-oxoprosta-2,13-dien-1-oate | |
| Clinical data | |
| AHFS/Drugs.com | International Drug Names |
| Legal status | ? |
| Routes | Pessary |
| Identifiers | |
| CAS number | 64318-79-2 |
| ATC code | G02AD03 |
| PubChem | CID 5282237 |
| ChemSpider | 4445416 |
| UNII | 45KZB1FOLS |
| KEGG | D02073 |
| Synonyms | methyl (E)-7-[(1R,2S,3R)-3-hydroxy-2-[(E,3R)-3-hydroxy-4,4-dimethyl-oct-1-enyl]-5-oxo-cyclopentyl]hept-2-enoate |
| Chemical data | |
| Formula | C23H38O5 |
| Mol. mass | 394.545 g/mol |

………………………………
http://www.chemdrug.com/databases/8_0_oqxuqtwlqgeukaaa.html

The reaction of 3-bromopropionic acid (I) with triphenylphosphine (II) in refluxing acetonitrile gives (2-carboxyethyl) -triphenylphosphonium bromide (III), which by a Wittig reaction with 2-oxa-3-hydroxy-6-syn- ( 3alpha-tetrahydropyranyloxy-4,4-dimethyl-1-trans-octen-1-yl) -7-anti-tetrahydropyranyloxybicyclo- [3.3.0] cis-octane (IV) (prepared according to reference 2) by means of sodium dimethylsulfinate in DMSO yields 9alpha-hydroxy-11alpha, 15alpha-bis (tetrahydropyranyloxy) -16,16-dimethyl-alpha-dinorprosta-5-cis-13-trans-dienoic acid (V). The reduction of (V) with H2 over Pd / C in methanol affords the 13-trans-prostenoic acid (VI), which is methylated with CH2N2 in ether yielding the methyl ester (VII). The reduction of (VII) with diisobutyl aluminum hydride in toluene affords the corresponding aldehyde (VIII) , which by a Wittig reaction with triethyl phosphonoacetate (IX) by means of NaH in THF is converted into 9alpha-hydroxy-11alpha, 15alpha-bis (tetrahydropyranyloxy) -16,16-dimethylprosta-2-trans-dienoic acid ethyl ester (X .) The hydrolysis of the ester (X) with KOH in ethanol-water gives the corresponding acid (XI), which is oxidized with CrO3, MnSO4 and H2SO4 in ether – water yielding the protected ketoacid (XII) The hydrolysis of (XII. ) with acetic acid-water at 80 C gives 9-oxo-11alpha, 15alpha-dihydroxy-16,16-dimethyl-prosta-2-trans-13-trans-dienoic acid (16,16-dimethyl-DELTA2-trans-PGE1 ) (XIII), which is finally methylated with CH2N2 in ether
References
- Bartley J, Brown A, Elton R, Baird DT (October 2001). “Double-blind randomized trial of mifepristone in combination with vaginal gemeprost or misoprostol for induction of abortion up to 63 days gestation”. Human reproduction (Oxford, England) 16 (10): 2098–102.doi:10.1093/humrep/16.10.2098. PMID 11574498. Retrieved 2008-10-29.
 |
|
: Gemeprost
CAS 64318-79-2
CAS Name: (2E,11a,13E,15R)-11,15-Dihydroxy-16,16-dimethyl-9-oxoprosta-2,13-dien-1-oic acid methyl ester
Additional Names: 16,16-dimethyl-trans-D2-PGE1 methyl ester
Manufacturers’ Codes: ONO-802
Trademarks: Cergem (Searle); Cervagem(e) (M & B); Preglandin (Ono)
Molecular Formula: C23H38O5
Molecular Weight: 394.54
Percent Composition: C 70.02%, H 9.71%, O 20.28%
Literature References:
Analog of prostaglandin E1, q.v. Prepn: M. Hayashi et al., DE 2700021; eidem, US 4052512 (both 1977 to Ono);
H. Suga et al., Prostaglandins 15, 907 (1978).
Effects on uterine contractility and steroid hormone plasma levels: K. Oshimaet al., J. Reprod. Fertil. 55, 353 (1979).
Effects on reproductive function: K. Matsumoto et al., Nippon Yakurigaku Zasshi 79, 15 (1982), C.A. 96, 98392 (1982).
Use in termination of first trimester pregnancy: O. Reiertsen et al., Prostaglandins Leukotrienes Med. 8, 31 (1982).
Therap-Cat: Abortifacient; oxytocic.
Keywords: Abortifacient/Interceptive; Oxytocic; Prostaglandin/Prostaglandin Analog
|
Latanoprost

Latanoprost
isopropyl-(Z)7[(1R,2R,3R,5S)3,5-dihydroxy-2-[(3R)-3-hydroxy-5-phenylpentyl]cyclopentyl]-5-heptenoate.
130209-82-4
XA41, PhXA34 [as 15 (R, S) -isomer], PhXA41, Xalatan
(Zanoni, G. et al., Tetrahedron 2010, 66, 7472)
Latanoprost (pronounced la-TA-noe-prost) ophthalmic solution is a medication administered into the eyes to control the progression of glaucoma or ocular hypertension by reducing intraocular pressure. It is a prostaglandin analogue (more specifically an analogue ofprostaglandin F2α[1]) that lowers the pressure by increasing the outflow of aqueous fluid from the eyes through the uvealsclearal tract.[2] Latanoprost is an isopropyl ester prodrug, meaning it is inactive until it is hydrolyzed by esterases in the cornea to the biologically active acid.[3]
It is also known by the brand name of Xalatan manufactured by Pfizer. Annual sales are approximately $1.6 billion. The patent for latanoprost expired in March 2011, and at least one generic version (manufactured by Mylan Inc.) is now widely available in the U.S. The Veterans Health Administration, part of the U.S. Department of Veterans Affairs, uses generic Latanoprost manufactured by Alcon Laboratories of Fort Worth, Texas distributed by Novartis generic brand Sandoz Pharmaceuticals.
Latanoprost was invented by Johan W. Stjernschantz and Bahram Resul, employees of the Pharmacia Corporation of Upsalla, Sweden.[4]
It is on the World Health Organization’s List of Essential Medicines, a list of the most important medication needed in a basic health system.[5]
 |
|
 |
|
| Systematic (IUPAC) name | |
|---|---|
| isopropyl (Z)-7-[(1R,2R,3R,5S)-3,5-dihydroxy-2- [(3R)3-hydroxy-5-phenylpentyl]-cyclopentyl] hept-5-enoate | |
| Clinical data | |
| Trade names | Xalatan |
| AHFS/Drugs.com | monograph |
| MedlinePlus | a697003 |
| Pregnancy cat. | C (US) |
| Legal status | ℞-only (US) |
| Routes | Topical (eye drops) |
| Pharmacokinetic data | |
| Half-life | 17 minutes |
| Identifiers | |
| CAS number | 130209-82-4 |
| ATC code | S01EE01 |
| PubChem | CID 5311221 |
| IUPHAR ligand | 1961 |
| DrugBank | DB00654 |
| ChemSpider | 4470740 |
| UNII | 6Z5B6HVF6O |
| KEGG | D00356 |
| ChEBI | CHEBI:6384 |
| ChEMBL | CHEMBL1051 |
| Chemical data | |
| Formula | C26H40O5 |
| Mol. mass | 432.593 g/mol |
Medical uses
Ocular hypertension
- In well-controlled clinical trials including patients with open-angle glaucoma or ocular hypertension (IOP ≥21 mm Hg), monotherapy with latanoprost reduced IOP levels by 22 to 39% over 1 to 12 months’ treatment. Latanoprost was significantly more effective than timolol 0.5% twice daily in 3 of 4 large (n = 163 to 267) randomised, double-blind trials. Latanoprost demonstrated a stable long-term IOP-lowering effect in 1- or 2-year continuations of these trials, with no sign of diminishing effect during prolonged treatment.[6]
- Meta analysis suggests that latanoprost is more effective than timolol in lowering IOP. However, it often causes iris pigmentation. While current evidence suggests that this pigmentation is benign, careful lifetime evaluation of patients is still justified.[7]
Closed-angle glaucoma
- Patients who had elevated IOP despite iridotomy and/or iridectomy (including patients of Asian descent), latanoprost was significantly more effective than timolol in two double-blind, monotherapy trials (8.2 and 8.8 mm Hg vs 5.2 and 5.7 mm Hg for latanoprost vs timolol at 12 and 2 weeks, respectively).[8]
Method of administration
One drop in the affected eye(s) once daily in the evening; do not exceed the once daily dosage because it has been shown that more frequent administration may decrease the intraocular-pressure (IOP) lowering effect[2]
Adverse effects[
Listed from most to least common:
- >5% to 15%: Blurred vision, burning and stinging, conjunctival hyperemia, foreign body sensation, itching, increased pigmentation of the iris causing (heterochromia), punctate epithelial keratopathy
- 4%: Cold or upper respiratory tract infections, flu-like syndrome
- 1-4%: Dry eyes, excessive tearing, eye pain, lid crusting, lid edema, lid erythema (hyperemia), lid pain, photophobia (light intolerance)
- 1 % – 2%: Chest pain, allergic skin reactions, arthralgia, back pain, myalgia, thickening of the eyelashes.(used,also bimatoprost,in cosmetic industry as eyelash growth enhancers)
- <1% (Limited to important or life-threatening): Asthma, herpes keratitis, iritis, keratitis, retinal artery embolus, retinal detachment, toxic epidermal necrolysis, uveitis, vitreous hemorrhage from diabetic retinopathy
- A single case report links latanoprost use to the progression of keratoconus.[9]
Concerns related to adverse effects:
- Bacterial keratitis: Inadvertent contamination of multiple-dose ophthalmic solutions, has caused bacterial keratitis.
- Ocular effects: May permanently change/increase brown pigmentation of the iris, the eyelid skin, and eyelashes. In addition, may increase the length and/or number of eyelashes (may vary between eyes); changes occur slowly and may not be noticeable for months or years. Long-term consequences and potential injury to eye are not known.
- Ocular disease: Use with caution in patients with intraocular inflammation, aphakic patients, pseudophakic patients with a torn posterior lens capsule, or patients with risk factors for macular edema. Safety and efficacy have not been determined for use in patients with angle-closure-, inflammatory-, or neovascular glaucoma.
Special populations
Contact lens wearers: Contains benzalkonium chloride which may be absorbed by contact lenses; remove contacts prior to administration and wait 15 minutes before reinserting
Contraindications
Hypersensitivity to latanoprost, benzalkonium chloride, or any component of the formulation
Drug Interactions
Bimatoprost: The concomitant use of Latanoprost and Bimatoprost may result in increased intraocular pressure. Risk D: Consider therapy modification
Nonsteroidal Anti-Inflammatory Agents: May diminish the therapeutic effect of Prostaglandins (Ophthalmic). Nonsteroidal Anti-Inflammatory Agents may also enhance the therapeutic effects of Prostaglandins (Ophthalmic). Risk C: Monitor therapy
Pregnancy
Prescription of Latanoprost is limited in human studies due to high incidence of abortion shown in animal experiments. Because of this, Latanoprost is classified as Risk factor C (Adverse events were observed in animal reproduction studies at maternally toxic doses)according to United States Food and Drug Administration’s use-in-pregnancy ratings.[10]Lactation Excretion in breast milk unknown/use caution. Breast-Feeding Considerations It is not known if latanoprost is excreted in breast milk. The manufacturer recommends that caution be exercised when administering latanoprost to nursing women.[2]
Storage
Latanoprost is a substance exhibiting thermal and solar instability. Concentration of latanoprost will decrease by 10% when stored at 50 and 70 degrees Celsius every 8.25 and 1.32 days respectively. Reaction with ultraviolet radiation will cause rapid degradation of Latanoprost. It is therefor important to store Latanoprost ideally in temperature below room temperature and free from sunlight in order to attain acceptable drug quality. [11]
Latanoprost is a prostaglandin F2α analogue. Its chemical name is isopropyl-(Z)7[(1R,2R,3R,5S)3,5-dihydroxy-2-[(3R)-3-hydroxy-5-phenylpentyl]cyclopentyl]-5-heptenoate. Its molecular formula is C26H40O5and its chemical structure is:

XALATAN Sterile Ophthalmic Solution (latanoprost ophthalmic solution) is supplied as a sterile, isotonic, buffered aqueous solution of latanoprost with a pH of approximately 6.7 and an osmolality of approximately 267 mOsmol/kg. Each mL of XALATAN contains 50 micrograms of latanoprost. Benzalkonium chloride, 0.02% is added as a preservative. The inactive ingredients are: sodium chloride, sodium dihydrogen phosphate monohydrate, disodium hydrogen phosphate anhydrous, and water for injection. One drop contains approximately 1.5 μg of latanoprostLatanoprost is a colorless to slightly yellow oil that is very soluble in acetonitrile and freely soluble in acetone, ethanol, ethyl acetate, isopropanol, methanol, and octanol. It is practically insoluble in water.
………………………….
http://www.google.com/patents/EP2495235A1?cl=en
When Latanoprost is the desired product the double bond on the side chain of compound 9a is hydrogenated to form compound 11, then by Wittig reaction with 4-carboxybutyltriphenylphosphonium bromide compound 11 is converted into Latanoprost acid 12. By conversion of the carboxylic acid into isopropyl ester, the final product Latanoprost is obtained:

EXAMPLE 16
(Z)-7-((1R,2R,3R,5S)-3,5-dihydroxy-2-((R)-3-hydroxy-5-phenylpentyl)cyclopentyl)hept-5-enoic acid (Latanoprost acid)
-

-
4-Carboxybutyltriphenylphosphonium bromide 15 (32.7 g, 0.074 mol) was suspended in tetrahydrofuran (75.0 mL) at 0°C under nitrogen atmosphere. A 1M solution of potassium tert-butoxide in tetrahydrofuran (296.0 mL, 0.296 mol) was added dropwise and the mixture turned into orange. After stirring for 45 minutes at 0°C the system was cooled to ―15°C. A solution of (3aR,4R,5R,6aS)-4-((R)-3-hydroxy-5-phenylpentyl)hexahydro-2H-cyclopenta[b]furan-2,5-diol (5.0 g, 0.016 mol) in tetrahydrofuran (23.0 mL) was added dropwise at a temperature lower than -10°C. After stirring overnight at -15°C no more starting was visible on TLC and water (100 mL) was added. The mixture was extracted with diisopropyl ether (70 mL) and after separation the aqueous phase was treated with 0.6 N HCl to pH 6.0. Three extractions with ethyl acetate (3x 125 mL) were then performed, each time adjusting the pH of the aqueous phase to 6.0. The combined organic layers were concentrated under vacuum at 35°C. An oil (13.87 g) was obtained which was used in the subsequent step without further purification.
-
1H-NMR {400 MHz, CDCl3, δ (ppm)}: 7.71 (m, 1H, Ph), 7.49 (m, 1H, Ph), 7.30-7.17 (m, 3H, Ph), 5.52-5.35 (m, 2H, -CH=CH-), 4.34 (bs, 4H, OH), 4.17 (m, 1H, -CH-OH(C-9)), 3.96 (m, 1H, -CH-OH (C-11)), 2.78 (m, 1H, -CH-OH (C-15)), 2.78 (m, 1H, -CH2Ph), 2.66 (m, 1H, -CH2Ph), 2.36-1.27 (m, 18H).
-
13C-NMR {400 MHz, CDCl3, δ (ppm)}: 176.5 (C), 142.2 (C), 130.8 (CH), 130.7 (CH), 129.4 (CH), 128.8 (CH), 128.7 (CH), 128.4 (CH), 125.7 (CH), 78.3 (CH), 74.2 (CH), 71.4 (CH), 52.2 (CH), 51.6 (CH), 42.4 (CH2), 38.8 (CH2), 35.2 (CH2), 33.4 (CH2), 32.0 (CH2), 29.1 (CH2), 26.6 (CH2), 26.4 (CH2), 24.7 (CH2).
-
HPLC-MS (ESI): [M+Na]+ = 413, [M+H]+ = 391.

EXAMPLE 17(Z)-isopropyl 7-((1R,2R,3R,5S)-3,5-dihydroxy-2-((R)-3-hydroxy-5-phenylpentyl)cyclopentyl)hept-5-enoate (Latanoprost)
-

-
Latanoprost acid (6.78 g, corresponding to 0.008 mol) was dissolved in N,N-dimethylformamide (108 mL) and cesium carbonate (8.48 g, 0.026 mol) was added at room temperature. 2-Iodopropane (3.46 mL, 0.035 mol) was added and the suspension was stirred at 40°C for 3 hours, checking the conversion on TLC. The mixture was then allowed to reach 25°C and a mixture of ice (184 g), water (40 mL), sodium thiosulfate (1M, 18 mL), was added stirring at -5/0°C for 15 minutes. The mixture was extracted with tert-butylmethylether (285 mL) and the phases were separated. The aqueous phase was extracted twice with tert-butylmethyl ether (2x 200 mL) and the combined organic layers were washed with brine (176 mL, 130 mL). The organic phase was concentrated under reduced pressure at 25°C and the crude product was obtained as a yellow oil (6.60 g). Purification by column chromatography on silica gel was performed eluting with dichloromethane:methanol increasing the percentage of methanol from 0 to 5%. A second purification on silica gel afforded Latanoprost (2.36 g, 0.005 mol, 68% over two steps).
-
1H-NMR {400 MHz, CDCl3, δ (ppm)}: 7.32-7.19 (m, 5H, Ph), 5.45-5.51 (m, 2H, H-5 e H-6 vinyl), 5.0 (hept, J=6.3 Hz, 1H, CH3CHCH3), 4,18 (bs, 1H, CHOH), 3.95 (bs, 1H, CHOH), 3.67 (bs, 1H, CHOH), 2.76 (m, 2H, CH 2Ph), 1.23 (d, J=6.3Hz, 6H, C(CH3)2), 2.55-1.3, 21H).
-
HPLC-MS (ESI): [M+Na]+ = 455, [M+H]+ = 432.

……………………………….
http://www.google.com/patents/EP2454227A1?cl=en
Example 3
Synthesis of Latanoprost
ether MTBE



8c-iso

Latanoprost
Scheme 5. Synthesis of Latanoprost
Synthesis of Latanoprost from 12c:
As shown in Scheme 5 in Example 3, a 250 ml.3-necked round-bottom flask equipped with a magnetic bar, a temperature probe, rubber septa, and a nitrogen gas inlet was charged at room temperature with 7.3 g (19.6 mmol) of deprotected lactone 12c in 70 mL of 2-propanol and 1.6 g (39.2 mmol) of sodium hydride, 60%, in mineral oil. The reaction mixture was heated at 35 0C for 18 h and TLC analysis indicated complete reaction. The mixture was diluted with 60 mL of water and the pH was adjusted to 6 with 1 N HCI. The layers were separated and the aqueous layer was back extracted with 40 mL of 2-propanol four times. The combined organic layers were washed with 50 mL of brine, dried over sodium sulfate, filtered, and concentrated.
The material was dissolved in 60 mL of THF, 5.0 mL (33.3 mmol) of DBU and 3.3 mL (33.3 mmol) of iodopropane. The reaction mixture was stirred at room temperature for 18 h and TLC analysis indicated complete reaction. The mixture was diluted with 60 mL of ethyl acetate and 60 mL of water. The layers were separated and the aqueous layer was back extracted with 40 mL of ethyl acetate for two times. The combined organic layers were washed with 50 mL of brine, dried over sodium sulfate, filtered, and concentrated.
The material was purified by using reverse phase biotage, 70 : 30 ACN :
H2O to obtain 4.1 g (49% yield) of Latanoprost, confirmed by 1H NMR.
………………….

Wittig condensation of lactol (XIII) with (carboxybutyl) triphenylphosphonium bromide (XV) in the presence of potassium tert-butoxide produced the Z-olefin (XVI). Conversion of carboxylic acid (XVI) to the title isopropyl ester was then accomplished by alkylation with 2-iodopropane in the presence of DBU.
http://www.chemdrug.com/databases/8_0_xmmatmlqjiethrwn.html
………………………………
http://www.nature.com/nature/journal/v489/n7415/full/nature11411.html?WT.ec_id=NATURE-20120913

………………………

……………………………….
http://www.google.com/patents/WO2013186550A1?cl=en

(la) (Ic)

Example 6 – Experimental procedures for the synthesis of latanoprost
A synthesis of latanoprost is shown and described below.

latanoprost (77)
2-Phenethyloxirane, 61
m-CPBA,

A modified procedure of Woodward was used (Bernier, D. et al., The Journal of Organic Chemistry 2008, 73, 4229). A stirred solution of 4-phenyl-l-butene 62 (500 mg, 568 μΙ, 3.78 mmol) in CH2CI2 (20 ml) was cooled to 0 °C. m-CPBA (816 mg, 4.73 mmol) was added as a solid and the reaction mixture was stirred at 0 °C for 1.5 h, then r.t. for 24 h. The reaction mixture was poured into saturated K2C03 solution (50 ml) and extracted with CH2CI2 (2 x 50 ml). The combined organic phases were washed with saturated K2C03 solution (50 ml) before being dried (MgS04), filtered and concentrated to give a clear colourless liquid. This material was purified by column chromatography, eluting with petrol/EtOAc (9:1), to give the epoxide 61 (10.2 g, 91%) as a clear colourless liquid. The 13C, and IR data were consistent with the literature (Mitchell, J. M. et al., Journal of the American Chemical Society 2001, 123, 862; Elings, J. A. et al., European Journal of Organic Chemistry 1999, 1999, 837).
Rf = 0.42 (petrol :EtOAc, 9:1) vmax (neatycnrr1 3027, 2989, 2922, 2859, 1602, 1495, 1454, 1410, 835, 750, 699
*H NMR (400 MHz; CDCI3) δΗ = 1.83-1.99 (2 H, m, CH2), 2.53 (1 H, dd, J = 5.0, 2.7 Hz, CHH), 2.75-2.93 (2 H, m, CH2), 2.80 (1 H, dd, J = 5.0, 4.0 Hz, CHH), 3.01 (1 H, dddd, J = 6.5, 5.0, 4.0, 2.7 Hz, CH), 7.22-7.38 (5 H, m, Ar H’s)
13C NMR (100 MHz; CDCI3) 5C = 32.2 (CH2), 34.2 (CH2), 47.2 (CH2), 51.7 (CH), 126.0 (2 x ArCH), 128.3 (2 x ArCH), 128.4 (ArCH), 141.2 (ArC)
m/z (EI) 148.1 (M+, 10%), 130.1 (23%), 129.0 (18%), 118.1 (29%), 117.1 (83%), 115.0 (28%), 105.0 (22%), 104.0 (61%), 92.0 (22%), 91.0 (100%), 83.9 (37%), 77.0 (17%), 65.0 (31%)
(2S)-2-Phenethyloxirane, 63

A modified procedure of Jacobsen was used (Schaus, S. E. et al., Journal of the American Chemical Society 2002, 224, 1307). Racemic epoxide 61 (10.0 g, 67.5 mmol) was dissolved in THF (10 ml) and stirred at r.t.. (S^-i+J-^A/’-BisiS^-di-tert-butylsalicylidene)-!^- cyclohexanediaminocobalt(II) (204 mg, 0.34 mmol) was added and the resultant dark brown solution cooled to 0 °C. Acetic acid (77 μΙ, 1.35 mmol) and water (669 μΙ, 37.1 mmol) were added. The reaction was stirred at 0 °C for 1 h and then at r.t. for 23 h. The reaction mixture was concentrated under reduced pressure and purified by column chromatography (~200 g silica), eluting with petrol/EtOAc (9:1), to give the epoxide 3 as a dark red liquid. This was re- purified by column chromatography eluting with petrol/EtOAc (9.5:0.5 to 9:1), to give the epoxide 3 (4.62 g, 46%) as an orange liquid. The analytical data matched that of the racemic material described above. The enantioselectivity of the resolution was determined after subsequent conversion to the allylic alcohol 66. The optical rotation matched closely with that reported in the literature (Martynow, J. G. et al., European Journal of Organic Chemistry 2007, 2007, 689).
[a]D 21 -21.0 (c. 1.0, CHC ) (lit., [a]D 20 -22.5 (c. 1.0, CHCI3)) 5-Phenyl-l-penten-3-ol, 64

A modified procedure of Molander was used (Molander, G. A. et al., The Journal of Organic Chemistry 2009, 74, 1297). A stirred solution of hydrocinnamaldehyde 65 (2.50 g, 2.45 ml, 18.6 mmol) in THF (25 ml) was cooled to -78 °C. Vinyl magnesium bromide solution (1 M in THF) (22.4 ml, 22.4 mmol) was added dropwise over ~ 5 min. The reaction mixture was stirred at -78 °C for 1.5 h, then 0 °C for 3 h. The reaction mixture was poured into saturated NH4CI solution (50 ml) and extracted with Et20 (3 x 50 ml). The combined organic phases were washed with saturated NaCI solution (50 ml) before being dried (MgS04), filtered and concentrated to give a pale yellow liquid. This material was purified by column
chromatography, eluting with petrol/EtOAc (9: 1), to give the vinyl alcohol 64 (1.89 g, 63%) as a clear colourless liquid. The *H, 13C, and IR data were consistent with the literature (Molander, G. A. et al., The Journal of Organic Chemistry 2009, 74, 1297; Kim, J. W. et al., Chemistry – A European Journal 2008, 24, 4104). Rf = 0.40 (petrol :EtOAc, 4: 1)
max (CHC Vcnrr1 3335, 3026, 2923, 2859, 1496, 1454, 990, 922, 747, 698
*H NMR (400 MHz; CDCI3) δΗ = 1.55 (1 H, br.s, OH), 1.84-1.99 (2 H, m, CH2), 2.70-2.89 (2 H, m, CH2), 4.19 (1 H, app q, J = 6.0 Hz, CHO ), 5.20 (1 H, app dt, J = 10.5, 1.4 Hz, HC=C), 5.30 (1 H, app dt, J = 17.1, 1.4 Hz, HHC=C), 5.96 (1 H, ddd, J = 17.1, 10.5, 6.0 Hz, H2C=CH), 7.20-7.40 (5 H, m, ArCH’s)
13C NMR (100 MHz; CDCI3) 5C = 31.6 (CH2), 38.5 (CH2), 72.4 (HCOH), 114.9 (H2C=C), 125.8 (ArCH), 128.4 (2 x ArCH), 128.4 (2 x ArCH), 141.0 (H2C=Q, 141.8 (ArC)
m/z (EI) 162.1 (M+, 30%), 144.1 (52%), 129.1 (72%), 105.1 (68%), 92.1 (71%), 91.0 (100%), 57.0 (61%) 6D. (3S)-5-Phenyl-l-penten-3-ol, 66

A modified procedure of Falck was used (Alcaraz, L. et al., Tetrahedron Letters 1994, 35, 5449). A suspension of trimethylsulfonium iodide (18.2 g, 89.1 mmol) in anhydrous THF (220 ml) was stirred and cooled to -20 °C. 1.6 M n-BuLi (55.7 ml, 89.1 mmol) was added slowly and the reaction stirred at -20 °C for 1 h. A solution of epoxide 63 (4.40 g, 29.7 mmol) in anhydrous THF (50.0 ml) was added slowly. The reaction was stirred at -20 °C for 1 h and then allowed to warm to r.t. slowly. The reaction mixture was poured into water (200 ml) and extracted with Et20 (1 x 200 ml, 1 x 100 ml). The combined organic phases were washed with saturated NaCI solution (100 ml) before being dried (MgS04), filtered, and concentrated to give the crude material. This was purified by column chromatography (130 g silica), eluting with petrol/EtOAc (9:1), to give partially purified material. This was re-purified by column chromatography (50 g silica), eluting with petrol/EtOAc (9:1), to give allylic alcohol 66 (3.19 g, 66%) as a pale yellow liquid. The analytical data matched that described for the racemic material above.
[α]ο21 -11.0 (c. 1.0, CHCI3) (lit – Kanbayashi, N. et al., Angewandte Chemie International Edition 2011, 50, 5197, [a]D 25 -3.6 (for 85% ee (c. 0.4, CHCI3)))
Chiral-HPLC data: er = >99:1 (Chiralcel AD-H column, 210 nm, hexane/2-propanol: 98/2, flow rate: 0.5 mlVmin, room temperature; ¾: minor 41.0 min, major 43.7 min) 6E. tert-Butyl(dimethyl)[(lS)-l-phenethyl-2-propenyl]oxysilane, 67

67 A stirred solution of allylic alcohol 66 (3.00 g, 18.5 mmol) in CH2CI2 (53 ml) was cooled to 0 °C. Imidazole (2.27 g, 33.3 mmol) was added in one portion followed by t- butylchlorodimethylsilane (3.34 g, 22.2 mmol). The cooling bath was removed and the reaction mixture stirred at r.t. for 16 h before being poured into 10% aq. HCI (100 ml). The mixture was extracted with 40/60 petroleum ether (2 x 100 ml). The combined organics were washed with saturated NaCI solution (100 ml), dried (MgS04), filtered, and concentrated to give the crude material. This was purified by column chromatography, eluting with 40/60 petroleum ether, to give the protected alcohol 67 (4.68 g, 92%) as a colourless liquid. The *H NMR data and optical rotation matched that reported in the literature (Uenishi, J. i. et al., Organic Letters 2011, 13, 2350).
Rf = 0.25 (40/60 petroleum ether)
vmax (film)/cm-13064, 3027, 2952, 2929, 2886, 2856, 1497, 1472, 1462, 1455, 1361, 1251, 1122, 1083, 1030, 990, 921, 834, 774, 697
*H NMR (400 MHz; CDCI3) δΗ = 0.05 (3 H, s, SiCH3), 0.08 (3 H, s, SiCH3), 0.93 (9 H, s, C(CH3)3), 1.82 (2 H, m, CH2), 2.66 (2 H, m, CH2), 4.17 (1 H, m, OCH), 5.08 (1 H, ddd, J = 10.4, 1.5, 1.3 Hz, HA =CH), 5.19 (1 H, app dt, J = 17.2, 1.5 Hz, HHC=CH), 5.86 (1 H, ddd, J = 17.2, 10.4, 6.0 Hz, H2C=CH), 7.18 (3 H, m, ArH’s), 7.28 (2 H, m, ArH’s)
13C NMR (100 MHz; CDCI3) 5C = -4.8 (SiCH3), -4.3 (SiCH3), 18.3 (C(CH3)3), 25.9 (C(CH3)3), 31.5 (CH2), 39.8 (CH2), 73.3 (CHOSi), 114.0 (H2C=C), 125.7 (ArCH), 128.3 (2 x ArCH), 128.4 (2 x ArCH), 141.4 (H2C=Q, 142.5 (ArC).
[a]D” 12.0 (c. 1.0, CHCI3) (lit., [a]D 20 14.5 (c. 1.0, CHCI3)) 6F. (3S)-3-[l-(tert-Butyl)-l,l-dimethylsilyl]oxy-5-phenylpentan-l-ol, 68

67
A modified procedure of Denmark was used (Denmark, S. E. et al., Organic Letters 2005, 7, 5617). Compound 67 (2.00 g, 7.23 mmol) was added to a flame dried schlenk flask under N2. 9-BBN (0.5 M in THF) (15.9 ml, 7.96 mmol) was added via syringe and the resulting solution stirred at r.t. for 1 h. A further 1.1 eq. (15.9 ml, 7.96 mmol) of 9-BBN was added and the reaction stirred at r.t. for 2 h. Water (16.0 ml) and NaB03.4H20 (5.56 g, 36.2 mmol) were added and the reaction stirred at r.t. for 2 h. The reaction mixture was poured into saturated NH4CI solution (60 ml) and extracted with Et20 (3 x 100 ml). The combined organic phases were washed with sat. NaCI solution (100 ml), dried (MgS04), filtered, and concentrated to give the crude material. This was purified 3 times by column chromatography (twice eluting with petrol/EtOAc (6:1) and once with petrol/ EtOAc/Eti) (9:0.5:0.5)) to give the alcohol 68 (672 mg, 32%) as a clear colourless oil.
Rf = 0.18 (petrol :EtOAc, 9:1)
vmax (film)/cm-13351 (broad), 3063, 2950, 2928, 2885, 2856, 1496, 1471, 1462, 1454, 1360, 1253, 1092, 1057, 1028, 1005, 834, 773, 746, 698
*H NMR (400 MHz; CDCI3) δΗ = 0.09 (3 H, s, SiCH3), 0.10 (3 H, s, SiCH3), 0.92 (9 H, s, C(CH3)3), 1.75 (1 H, m, CHH), 1.83-1.94 (3 H, m, CH2, CHH), 2.32 (1 H, app t, J = 5.2 Hz, OH), 2.64 (2 H, m, CH2), 3.75 (1 H, app dq, J = 10.8, 5.5 Hz, OCHH), 3.87 (1 H, app ddt, J = 10.8, 8.1, 4.8 Hz, OCHtf), 3.99 (1 H, app qd, J = 6.1, 4.4 Hz, HCOTBDMS), 7.16-7.23 (3 H, m, ArCH’s), 7.27-7.33 (2 H, m, ArCH’s)
13C NMR (100 MHz; CDCI3) 5C = -4.7 (SiCH3), -4.4 (SiCH3), 18.0 (C(CH3)3), 25.8 (C(CH3)3), 31.7 (CH2), 37.8 (CH2), 38.7 (CH2), 60.1 (CH2), 71.2 (SiOCH), 125.8 (ArCH), 128.2 (2 x ArCH), 128.4 (2 x ArCH), 142.1 (ArC)
HRMS (ESI) calcd for Ci7H30O2SiNa [MNa+] 317.1907, found 317.1906
[a]D 23 23.0 (c. 1.0, CHCI3) 6G. tert-Butyl[(lS)-3-iodo-l-phenethylpropyl]oxydimethylsilane, 69

68 A modified procedure of Rychnovsky was used (Dalgard, J. E. et al., Organic Letters 2004, 6, 2713). Alcohol 68 (600 mg, 2.04 mmol) was added to a flame dried schlenk flask under N2. CH2CI2 (10 ml) was added via syringe and the resulting solution stirred at r.t..
Triphenylphosphine (695 mg, 2.65 mmol) and imidazole (222 mg, 3.26 mmol) were added as solids in one portion. Iodine (672 mg, 2.65 mmol) was added to the resulting solution. A slight exotherm was noted and the solution changed from a light yellow colour to a brown colour with the formation of a precipitate. The reaction was stirred at r.t. for 1 h. The reaction mixture was dry loaded onto silica (2 g) and purified by column chromatography (14 g silica), eluting with petrol to petrol/EtOAc (9: 1). This gave the iodide 69 (725 mg, 88%) as a clear, colourless oil.
Rf = 0.20 (40/60 petroleum ether)
ifiln /cnr^OeS, 3026, 2951, 2928, 2886, 2856, 1495, 1471, 1461, 1360, 1253, 1187, 1165, 1140, 1092, 1063, 1005, 975, 931, 833, 773, 697
*H NMR (400 MHz; CDCI3) δΗ = 0.09 (3 H, s, SiCH3), 0.10 (3 H, s, SiCH3), 0.92 (9 H, s, (C(CH3)3), 1.79 (2 H, m, CH2), 2.05 (2 H, m, CH2), 2.64 (2 H, m, CH2), 3.24 (2 H, m, CH2), 3.82 (1 H, quin., J = 5.7 Hz, OCH), 7.16-7.23 (3 H, m, ArCH’s), 7.27-7.33 (2 H, m, ArCH’s) 13C NMR (100 MHz; CDCI3) 5C = -4.3 (SiCH3), -4.3 (SiCH3), 3.0 (CH2), 18.1 (C(CH3)3), 25.9 (C(CH3)3), 31.3 (CH2), 38.7 (CH2), 40.8 (CH2), 71.7 (OCH), 125.8 (ArCH), 128.3 (2 x ArCH), 128.4 (2 x ArCH), 142.1 (ArC)
HRMS (ESI) calcd for Ci7H30OSiI [MH+] 405.1108, found 405.1105
[a]D 23 26.0 (c. 1.0, CHCI3)
6H. [(lR)-3-((3aR,4R,6aS)-2-Methoxy-5-(£)-l-[(l,l,l- trimethylsilyl)oxy]methylideneperhydrocyclopenta[d]furan-4-yl)-l- phenethylpropyl]oxy(tert-butyl)dimethylsilane, 70

70 Iodide 69 (1.32 g, 3.27 mmol, 1.1 eq.) was added via syringe to a flame dried schlenk flask (evacuated and purged with nitrogen several times and allowed to cool). Anhydrous Et20 (13.3 ml) was added via syringe and the resulting solution cooled to -78 °C. 1.63 M t-BuLi (4.01 ml, 6.54 mmol, 2.2 eq.) was added dropwise and the reaction mixture stirred at -78 °C for 2 h and -40 °C for 2 h before being cooled back to -78 °C. Meanwhile, thiophene (275 mg, 262 μΙ, 3.27 mmol, 1.1 eq.) was added via syringe to a flame dried schlenk flask (evacuated and purged with nitrogen several times and allowed to cool). Anhydrous THF (13.3 ml) was added via syringe and the resulting solution cooled to -30 °C. 1.63 M n-BuLi (2.01 ml, 3.27 mmol, 1.1 eq.) was added dropwise and the solution stirred at -30 °C for 30 min. CuCN (293 mg, 3.27 mmol, 1.1 eq.) was added as a solid, in one portion. The cooling bath was removed and the suspension allowed to warm to r.t. The resulting tan/brown solution of cuprate was added dropwise via syringe to the schlenk flask containing the alkyl lithium and anhydrous THF (13.3 ml) added. The mixture was stirred at -20 °C for 1 h to allow formation of mixed cuprate 71. This was cooled to -78 °C and a solution of enal 24 (500 mg, 2.97 mmol, 1.0 eq.) in anhydrous THF (13.3 ml) was added dropwise. The mixture was stirred at -78 °C for 1 h and then allowed to warm slowly to -20 °C. TMSCI (1.61 g, 1.89 ml, 14.9 mmol, 5.0 eq.) was added via syringe followed by NEt3 (1.80 g, 2.49 ml, 17.8 mmol, 6 eq.). The reaction was quenched by the addition of saturated NH4CI solution (50 ml) and extracted with Et20 (3 x 50 ml). The combined organic phases were washed with saturated NH4CI solution (50 ml) and saturated NaCI solution (50 ml) before being dried (MgS04), filtered, and concentrated to give the crude material as a yellow oil. This was used directly in the next step.
61. (3aR 4R,5R,6aS)-4-((3R)-3-[l-(tert-Butyl)-l,l-dimethylsilyl]oxy-5- phenylpentyl)-2-methoxyperhydrocyclopenta[d]furan-5-ol, 73

70 73 The crude material from the conjugate addition / trapping experiment, containing 70, was dissolved in CH2Cl2/MeOH (3: 1) (30 ml) and cooled to -78 °C. A stream of ozone was passed through the stirred solution. The reaction was monitored periodically by TLC in order to judge completion of the ozonolysis (judged by consumption of silyl enol ether). The reaction mixture was flushed with a stream of N2, for 15 min, to remove excess 03. NaBH4 (202 mg, 5.35 mmol) was added in one portion. The reaction mixture was stirred at -78 °C for 2 h before the cooling bath was removed and the reaction allowed to warm to r.t.. The reaction was stirred at r.t. for 1 h. NaBH4 (67.4 mg, 1.78 mmol) was added and the reaction stirred at r.t. for a further 15 min. The reaction mixture was poured into saturated NaCI solution (25 ml) and extracted with EtOAc (3 x 25 ml). The combined organic phases were dried (MgS04), filtered, and concentrated to give the crude product as a pale yellow oil. This was purified by column chromatography on silica, eluting with petrol/EtOAc (4: 1), giving the alcohol 73 (as an approximately 2:1 mixture of diastereoisomers) as a clear, colourless oil (800 mg, 62% (2 steps from enal 24)).
Rf = 0.23 (petrol :EtOAc, 4:1)
vmax (neatycnrr1 3434 (broad), 3026, 2928, 2856, 1496, 1471, 1454, 1360, 1343, 1254, 1098, 1053, 1004, 937, 833, 773, 698
1H NMR (400 MHz; CDCI3) 5H = (mixture of 2 diastereoisomers, signals of minor diastereoisomer indicated by *) 0.05 (3 H, s, CH3), 0.06* (3 H, s, CH3), 0.07 (3 H, s, CH3), 0.07* (3 H, s, CH3), 0.91 (9 H, s, C(CH3)3), 0.92* (9 H, s, C(CH3)3), 1.12-1.80 (7 H, m), 1.12- 1.80* (7 H, m), 1.90-2.38 (5 H, m), 1.90-2.38* (5 H, m), 2.53-2.75 (2 H, m, CH2), 2.53-2.75* (2 H, m, CH2), 3.32 (3 H, s, OCH3), 3.39* (3 H, s, OCH3), 3.72 (1 H, m, CHOTBDMS), 3.72* (1 H, m, CHOTBDMS), 3.79* (1 H, m, CHOH), 3.89 (1 H, m, CHOH), 4.55 (1 H, app td, J = 6.3, 2.5 Hz, CH), 4.64* (1 H, app td, J = 6.8, 2.7 Hz, CH), 5.06* (1 H, d, J = 5.5 Hz, OCHO), 5.11 (1 H, d, J = 4.9 Hz, OCHO), 7.19 (3 H, m, ArCH’s), 7.19* (3 H, m, ArCH’s), 7.29 (2 H, m, ArCH’s), 7.29* (2 H, m, ArCH’s)
13C NMR (100 MHz; CDCI3) 5C = (observed signals, mixture of 2 diastereoisomers) -4.42 (SiCH3), -4.41 (SiCH3), -4.33 (SiCH3), 18.1 (2 x C(CH3)3), 25.9 (2 x C(CH3)3), 29.3 (CH2), 30.3 (CH2), 31.7 (CH2), 35.1 (CH2), 38.8 (CH2), 39.9 (CH2), 40.0 (CH2), 41.1 (CH2), 42.7 (CH2),
46.5, 47.2, 54.4, 55.1, 55.3, 55.7, 71.8, 71.8, 79.3, 79.7, 82.5, 85.8, 106.5 (OCHOCH3), 108.0 (OCHOCH3), 125.7 (ArCH), 125.7 (ArCH), 128.3 (4 x ArCH), 128.3 (4 x ArCH), 142.6 (ArC), 142.6 (ArC). One SiCH3 and three CH2‘s could not be assigned due to overlapping signals. 6J. (3aR,4R,5R,6aS)-4-[(3R)-3-Hydroxy-5- phenylpentyl]perhydrocyclopenta[b]furan-2,5-diol, 74

Alcohol 73 (400 mg, 0.920 mmol) was stirred with 1.5% aqueous HQ / THF (3:2) (18 ml) at r.t. for 16 h. The mixture was neutralised with 1 M NaOH and extracted with CH2CI2 (5 x 30 ml). The combined organic phases were dried (MgS04), filtered, and concentrated to give the triol 74 and silanol by-product as a clear, colourless oil (~400 mg). This material was taken forward for the subsequent transformation without purification.

(4-Carboxybutyl)(triphenyl)phosphonium bromide 29 (2.45 g, 5.52 mmol) was added to a flame dried schlenk flask, under N2, and anhydrous THF (20.0 ml) added. The resulting suspension was cooled to 0 °C. KOt-Bu (1.24 g, 11.0 mmol) was added in one portion and the resulting orange mixture stirred at 0 °C for 40 min. A solution of crude triol 74 (282 mg, 0.920 mmol) in anhydrous THF (5.0 ml) was added dropwise via syringe. After complete addition the cooling bath was removed and the mixture was stirred at r.t. for 1.5 h. The reaction was quenched with H20 (30 ml) and washed with Et20 (2 x 30 ml) to remove triphenylphosphine oxide. The aqueous phase was made acidic with 1 M HQ (~10 ml) and extracted with CH2CI2 (5 x 25 ml). The combined organic phases were dried (MgS04), filtered, and concentrated to give the crude material as solids. These were placed on a sinter funnel and washed with petrol/EtOAc (1: 1) (4 x 20 ml) and then EtOAc (2 x 40 ml). The filtrate was concentrated under vacuum and purified by column chromatography on silica, eluting with CH2Cl2/MeOH (9.5:0.5 to 9:1) to give acid 75 (163 mg, 45% over 2 steps from alcohol 73) as a clear, colourless oil. The *Η data and optical rotation were consistent with the literature (Martynow, J. G. et al., European Journal of Organic Chemistry 2007, 2007, 689).
Rf = 0.27 (CH2CI2:MeOH, 9:1)
vmax (neatycnrr1 3338 (broad), 2930, 2857, 1704, 1452, 1407, 1254, 1028, 747, 699, 636 *H NMR (400 MHz; CDCI3) δΗ = 1.39 (2 H, m, CH2), 1.47-1.97 (10 H, m, 4 x CH2, 2 x CH), 2.07-2.48 (6 H, m, 3 x CH2), 2.67 (1 H, m, CH ), 2.80 (1 H, m, CH/-/), 3.60-4.85 (6 H, broad signal, 2 x OCH, 3 x OH, COOH), 3.72 (1 H, m, OCH), 5.40 (1 H, m, =CH), 5.49 (1 H, m, =CH), 7.15-7.24 (3 H, m, ArCH’s), 7.25-7.32 (2 H, m, ArCH’s)
[a]D 24 29.0 (c. 1.0, MeOH) (lit, [a]D 20 29.7 (c. 1.0, MeOH)) 6L. Isopropyl (Z)-7-(lR,2R,3R,5S)-3,5-dihydroxy-2-[(3R)-3-hydroxy-5- phenylpentyl]cyclopentyl-5-heptenoate, latanoprost, 77

A modified procedure of Zanoni and Vidari was used (Zanoni, G. et al., Tetrahedron 2010, 66, 7472). Carboxylic acid 75 (100 mg, 0.256 mmol) was dissolved in DMF (2.0 ml) and stirred at r.t.. Cs2C03 (125 mg, 0.384 mmol) was added in one portion followed by 2- iodopropane (51 μΙ, 0.512 mmol). The reaction was stirred at r.t. for 18 h. The reaction mixture was poured into 3% citric acid solution (10 ml) and extracted with TBME (4 x 10 ml). The combined organic phases were washed with 10% NaHC03 solution (10 ml) and saturated NaCI (2 x 10 ml) before being dried (MgS04), filtered, and concentrated to give the crude product as a clear, colourless oil (95 mg). This was purified by column chromatography (3 g silica), eluting with petrol/EtOAc (2: 1 to 1:2), to give latanoprost 77 (71 mg, 64 %) as a clear colourless oil. The IR, 13C, and optical rotation data were consistent with the literature (Zanoni, G. et al., Tetrahedron 2010, 66, 7472). Rf = 0.44 (EtOAc)
vmax (neatVcm“1 3360 (broad), 2980, 2931, 2857, 1712, 1495, 1454, 1374, 1311, 1247, 1180, 1106, 1030, 966, 910, 820, 731, 699
*H NMR (400 MHz; CDCI3) δΗ = 1.23 (6 H, d, J = 6.4 Hz, 2 x CH3), 1.30-1.90, (14 H, m, 5 x CH2, 2 x CH, 2 x OH), 2.07-2.39 (6 H, m, 3 x CH2), 2.45 (1 H, d, J = 5.5 Hz, OH), 2.63- 2.86 (2 H, m, CH2), 3.68 (1 H, br.s, CHO ), 3.95 (1 H, br.s, CHOH), 4.18 (1 H, br.s, CHO ), 5.01 (1 H, sept., J = 6.4 Hz, OCH(CH3)2), 5.35-5.52 (2 H, m, 2 x =CH), 7.16-7.24 (3 H, m, ArH’s), 7.25-7.32 (2 H, m, ArH’s)
13C NMR (125 MHz; CDCI3) 5C = 21.9 (2 x CH3), 24.9 (CH2), 26.6 (CH2), 26.8 (CH2), 29.6 (CH2), 32.1 (CH2), 34.0 (CH2), 35.7 (CH2), 39.0 (CH2), 42.5 (CH2), 51.8 (CH), 52.7 (CH), 67.6 (OCH), 71.2 (OCH), 74.5 (OCH), 78.6 (OCH), 125.7 (CH), 128.3 (2 x ArCH), 128.3 (2 x ArCH), 129.3 (CH), 129.5 (CH), 141.1 (ArC), 173.5 (C=0)
[a]D 23 33.0 (c. 1.0, MeCN) (lit, [a]D 20 32.7 (c. 1.0, MeCN))
References
- Ishikawa H, Yoshitomi T, Mashimo K, Nakanishi M, Shimizu K (February 2002). “Pharmacological effects of latanoprost, prostaglandin E2, and F2alpha on isolated rabbit ciliary artery”. Graefes Arch. Clin. Exp. Ophthalmol. 240 (2): 120–5. doi:10.1007/s00417-001-0412-4. PMID 11931077.
- Patel SS, Spencer CM (1996). “Latanoprost. A review of its pharmacological properties, clinical efficacy and tolerability in the management of primary open-angle glaucoma and ocular hypertension”. Drugs Aging 9 (5): 363–378. doi:10.2165/00002512-199609050-00007. PMID 8922563.
- Huttunen et al. (2011) Prodrugs—from Serendipity to Rational Design. Pharmacol Rev 63:750–771
- “Patent US5296504 – Prostaglandin derivatives for the treatment of glaucoma or ocular hypertension – Google Patents”.
- “WHO Model List of EssentialMedicines”. World Health Organization. October 2013. Retrieved 22 April 2014.
- Perry CM, McGavin JK, Culy CR, Ibbotson T (2003). “Latanoprost. An Update of its Use in Glaucoma and Ocular Hypertension”. Drugs Aging 20 (8): 1170–2229.PMID 12795627.
- Zhang WY, Wan Po AL, Dua HS, Azuara-Blanco A (2001). “Meta-analysis of randomised controlled trials comparing latanoprost with timolol in the treatment of patients with open angle glaucoma or ocular hypertension”. British Journal of Ophthalmology 85: 983–990. doi:10.1136/bjo.85.8.983. PMID 11466259.
- Aung T; Wong HT; Yip CC; et al. (2000). “Comparison of the intraocular pressure-lowering effect of latanoprost and timolol in patients with chronic angle closure glaucoma: a preliminary study.”. Ophthalmology 107 (6): 1178–83. doi:10.1016/s0161-6420(00)00073-7. PMID 10857840.
- Amano S, Nakai Y, Ko A, Inoue K, Wakakura M (2008). “A case of keratoconus progression associated with the use of topical latanoprost”. Japanese Journal of Ophthalmology 52 (4): 334–6. doi:10.1007/s10384-008-0554-6. PMID 18773275.
- De Santis, M., Lucchese, A., Carducci, B., Cavaliere, A., De Santis, L., & Merola, A. et al. (2004). Latanoprost exposure in pregnancy. American Journal Of Ophthalmology, 138(2), 305.pmid=15289149.1
- Morgan, P., Proniuk, S., Blanchard, J., & Noecker, R. (2001). Effect of temperature and light on the stability of latanoprost and its clinical relevance. Journal Of Glaucoma, 10(5), 401–405.
External links
- LATANOPROST solution [Greenstone LLC] (Nov 2011), Daily Med, U.S. National Library of Medicine, National Institutes of Health
Travoprost

Travoprost
cas 157283-68-6
[1R-[lα(Z),2β(lE,3R*),3α,5α]]-7-[3,5-Dihydroxy-2-[3-hydroxy-4-[3-(trifluoromethyl)phenoxy]-1 -butenyl]cyclopentyl]-5-heptenoic acid, 1 -methylethylester
(+)-16-m-trifluoromethylphenoxy tetranor Prostaglandin F2α isopropyl ester; (+)-Fluprostenol ispopropyl ester
(+)-(5Z,9α,1α,13E,15R)-trihydroxy-16-(3-(trifluoromethyl)phenoxy)-17,18,19,20-tetranor-prosta-5,13-dien-1-oic acid, isopropyl ester
(+) – Fluprostenol isopropyl ester,
CAS Name: (5Z)-7-[(1R,2R,3R,5S)-3,5-Dihydroxy-2-[(1E,3R)-3-hydroxy-4-[3-(trifluoromethyl)phenoxy]-1-butenyl]cyclopentyl]-5-heptenoic acid 1-methylethyl ester
Additional Names: (+)-16-[3-(trifluoromethyl)phenoxy]-17,18,19,20-tetranorprostaglandin F2a isopropyl ester; (+)-9a,11a,15-trihydroxy-16-(3-trifluoromethylphenoxy)-17,18,19,20-tetranor-5-cis-13-trans-prostadienoic acid isopropyl ester
Manufacturers’ Codes: AL-6221
Trademarks: Travatan (Alcon)
Percent Composition: C 62.39%, H 7.05%, F 11.39%, O 19.18%
Travatan, Travatan Z, AL-6221, Travatanz, Travatan Alcon, Travatan (TN), Travatan, Travoprost, Travoprost [USAN]
Molecular Formula: C26H35F3O6
Molecular Weight: 500.54771
Alcon (Originator)
Antiglaucoma Agents, OCULAR MEDICATIONS, Ophthalmic Drugs, Prostaglandins, Prostanoid FP Agonists
Properties: Colorless oil. [a]D20 +14.6° (c = 1.0 in methylene chloride). Very sol in acetonitrile, methanol, octanol, chloroform. Practically insol in water.
Optical Rotation: [a]D20 +14.6° (c = 1.0 in methylene chloride)
Therap-Cat: Antiglaucoma.

Ophthalmic solution used for the reduction of elevated intraocular pressure in patients with open-angle glaucoma or ocular hypertension who are intolerant of other intraocular pressure lowering medications or insufficiently responsive (failed to achieve target IOP determined after multiple measurements over time) to another intraocular pressure lowering medication.
Travoprost free acid is a selective FP prostanoid receptor agonist and is believed to reduce intraocular pressure by increasing the drainage of aqueous humor, which is done primarily through increased uveoscleral outflow and to a lesser extent, trabecular outflow facility.
Travoprost, an isopropyl ester prodrug, is a synthetic prostaglandin F2 alpha analogue that is rapidly hydrolyzed by esterases in the cornea to its biologically active free acid. The travoporst free acid is potent and highly selective for the FP prostanoid receptor.
Travoprost ophthalmic solution is a topical medication used for controlling the progression of glaucoma or ocular hypertension, by reducing intraocular pressure. It is a synthetic prostaglandin analog (or more specifically, an analog of prostaglandin F2α)[1][2] that works by increasing the outflow of aqueous fluid from the eyes.[3] It is also known by the brand names of Travatan and Travatan Z, manufactured by Alcon, and Travo-Z, manufactured by Micro Labs.
Travoprost is a synthetic prostaglandin F analogue. Its chemical name is [1R-[lα(Z),2β(lE,3R*),3α,5α]]-7-[3,5-Dihydroxy-2-[3-hydroxy-4-[3-(trifluoromethyl)phenoxy]-1 -butenyl]cyclopentyl]-5-heptenoic acid, 1 -methylethylester. It has a molecular formula of C26H35F3O6 and a molecular weight of 500.55. The chemical structure of travoprost is:
 |
Travoprost is a clear, colorless to slightly yellow oil that is very soluble in acetonitrile, methanol, octanol, and chloroform. It is practically insoluble in water.
TRAVATAN® (travoprost ophthalmic solution) 0.004% is supplied as sterile, buffered aqueous solution of travoprost with a pH of approximately 6.0 and an osmolality of approximately 290 mOsmol/kg.
TRAVATAN® contains Active: travoprost 0.04 mg/mL; Preservative: benzalkonium chloride 0.15 mg/mL; Inactives: polyoxyl 40 hydrogenated castor oil, tromethamine, boric acid, mannitol, edetate disodium, sodium hydroxide and/or hydrochloric acid (to adjust pH) and purified water.
| Country | Patent Number | Approved | Expires (estimated) |
|---|---|---|---|
| Canada | 2181172 | 2003-04-29 | 2015-12-19 |
| Canada | 2129287 | 2002-05-14 | 2014-08-02 |
| United States | 5631287 | 1994-12-22 | 2014-12-22 |
| United States | 6503497 | 1995-05-06 | 2012-05-06 |
|
7-25-2012
|
TOPICAL APPLICATION OF TRAVOPROST FOR COMBATING HAIR LOSS
|
|
|
12-28-2011
|
Stable prostaglandin-containing compositions
|
|
|
7-22-2011
|
IMPROVED PROCESS FOR THE PRODUCTION OF BIMATOPROST
|
|
|
6-3-2011
|
Process for the Preparation of Prostaglandin Analogues and Intermediates Thereof
|
|
|
9-17-2010
|
Compositions and Methods for Reducing Body Fat
|
|
|
5-28-2010
|
COMPLEXES OF PROSTAGLANDIN DERIVATIVES AND MONOSUBSTITUTED, CHARGED BETA-CYCLODEXTRINS
|
|
|
4-30-2010
|
AMINO ACID SALTS OF PROSTAGLANDINS
|
|
|
4-30-2010
|
AMINO ACID SALTS OF PROSTAGLANDINS
|
|
|
2-24-2010
|
Compositions and methods for reducing body fat
|
|
|
1-15-2010
|
Process for the Production of Prostaglandins and Prostaglandin Analogs
|
|
4-3-2009
|
METHOD FOR SCREENING OF PROSTAGLANDIN COMPOUNDS COMPRISING AN OPTIMAL FORMULATION FOR THE ENHANCEMENT OF HAIR GROWTH AND THE STIMULATION OF FOLLICULAR ANAGEN AND FORMULATIONS RESULTING THEREFROM
|
|
|
3-9-2005
|
9,11-cycloendoperoxide pro-drugs of prostaglandin analogues for treatment of ocular hypertension and glaucoma
|
|
|
10-8-2004
|
Use of cloprostenol and fluprostenol analogues to treat glaucoma and ocular hypertension
|
|
|
4-21-2004
|
Use of cloprostenol and fluprostenol analogues to treat glaucoma and ocular hypertension
|
Side effects
Possible side effects of this medication are:
- May cause blurred vision
- May cause eyelid redness
- May permanently darken eyelashes
- May cause eye discomfort
- May eventually cause permanent darkening of the iris to brown (heterochromia)
- May cause a temporary burning sensation during use
- May cause thickening of the eyelashes
- May cause inflammation of the prostate gland, restricting urine flow (BPH)
 |
|
| Systematic (IUPAC) name | |
|---|---|
| propan-2-yl 7-[3,5-dihydroxy-2-[3-hydroxy-4-[3-(trifluoromethyl) phenoxy]-but-1-enyl]-cyclopentyl]hept-5-enoate |
|
| Clinical data | |
| Trade names | Travatan |
| AHFS/Drugs.com | monograph |
| MedlinePlus | a602027 |
| Pregnancy cat. | C US |
| Legal status | Rx only (US) |
| Routes | Topical (eye drops) |
| Identifiers | |
| CAS number | 157283-68-6 |
| ATC code | S01EE04 |
| PubChem | CID 5282226 |
| DrugBank | DB00287 |
| ChemSpider | 4445407 |
| UNII | WJ68R08KX9 |
| Chemical data | |
| Formula | C26H35F3O6 |
| Mol. mass | 500.548 g/mol |


The condensation of 2- [3- (trifluoromethyl) phenoxy] acetyl chloride (I) with methylphosphonic acid dimethyl ester (II) by means of BuLi in THF gives 2-oxo-3- [3- (trifluoromethyl) phenoxy] propylphosphonic acid dimethyl ester (III), which is condensed with the known bicyclic aldehyde (IV) by means of BuLi in dimethoxyethane, yielding the unsaturated ketone (V). The reduction of (V) with zinc borohydride in dimethoxyethane affords the unsaturated alcohol (VI), which is treated with K2CO3 to give a diastereomeric mixture of unsaturated diols, resolved by chromatography to yield the chiral unsaturated diol (VII). The protection of (VII) with dihydropyran and TsOH in dichloromethane provides the bis (tetrahydropyranyl) ether (VIII), which by reduction of the lactone ring with diisobutylaluminum hydride in THF gives the lactol (IX). The condensation of (IX) with the phosphonium bromide (X) by means of NaH in DMSO yields the prostenoic acid (XI), which is esterified with isopropyl iodide and 1,8-diazabicyclo [5.4.0] undec-7-ene (DBU) in acetone to afford the corresponding isopropyl ester (XII). Finally, this compound is deprotected with acetic acid in hot THF / water.
http://www.chemdrug.com/databases/8_0_qkvreurfepijmjcf.html
…………………………………………..
Org Process Res Dev2002,6, (2): 138
http://pubs.acs.org/doi/abs/10.1021/op010097p

A commercial synthesis of the antiglaucoma agent, travoprost 2, is described. A total of 22 synthetic steps are required to provide the single enantiomer prostanoid, with the longest linear sequence being 16 steps from 3-hydroxybenzotrifluoride. The route is based upon a cuprate-mediated coupling of the single enantiomer vinyl iodide 13 and the tricyclic ketone 5, of high stereochemical purity, to yield the single isomer bicyclic ketone 15. A Baeyer−Villiger oxidation provides the lactone 16 as a crystalline solid, thus limiting the need for chromatographic purification. DIBAL-H reduction, Wittig reaction, esterification, and silyl group deprotection complete the synthesis of travoprost.
(5Z,13E)–(9S,11R,15R)-9,11,15-Trihydroxy-16-(m-trifluoromethylphenoxy-17,18,19,20-tetranor-5,13-prostadienoic Acid, Isopropyl Ester (2).
The silyl-protected compound (20a+b) (202 g, 277 mmol) ………..DELETED……………………………………… All relevant fractions were combined and concentrated to give the title compound 2 (97 g, 70%) as a colourless oil,  +14.6 (c 1.0, CH2Cl2); IR νmax (film) 3374 and 1727 cm–1; 1H NMR (400 MHz, CDCl3) δ 7.39 (1H, t, J = 8), 7.22 (1H, d, J = 8), 7.15 (1H, s), 7.08 (1H, d, J = 8), 5.70 (2H, m), 5.40 (2H, m), 4.98 (1H, heptet, J = 6.5), 4.52 (1H, m), 4.18 (1H, m), 3.97 (3H, m), 3.25 (2H, br s), 2.60 (1H, br s), 2.38 (1H, m), 2.30−1.96 (7H, m), 1.76 (1H, dd, J = 16, 4), 1.65 (2H, quintet, J = 7), 1.55 (1H, m), and 1.20 (6H, d, J = 6); 13C NMR (100 MHz, CDCl3) δ 173.57, 158.67, 135.45, 131.87 (q, J = 32), 130.02, 129.85, 129.75, 128.93, 123.89 (q, J = 270), 118.06, 117.82, 111.48, 77.77, 72.70, 71.99, 70.86, 67.72, 55.82, 50.24, 42.84, 34.00, 26.60, 25.48, 24.83, and 21.81; m/z (CI) 501 (MH+, 21), 321 (34), 303 (44), and 249 (100).
+14.6 (c 1.0, CH2Cl2); IR νmax (film) 3374 and 1727 cm–1; 1H NMR (400 MHz, CDCl3) δ 7.39 (1H, t, J = 8), 7.22 (1H, d, J = 8), 7.15 (1H, s), 7.08 (1H, d, J = 8), 5.70 (2H, m), 5.40 (2H, m), 4.98 (1H, heptet, J = 6.5), 4.52 (1H, m), 4.18 (1H, m), 3.97 (3H, m), 3.25 (2H, br s), 2.60 (1H, br s), 2.38 (1H, m), 2.30−1.96 (7H, m), 1.76 (1H, dd, J = 16, 4), 1.65 (2H, quintet, J = 7), 1.55 (1H, m), and 1.20 (6H, d, J = 6); 13C NMR (100 MHz, CDCl3) δ 173.57, 158.67, 135.45, 131.87 (q, J = 32), 130.02, 129.85, 129.75, 128.93, 123.89 (q, J = 270), 118.06, 117.82, 111.48, 77.77, 72.70, 71.99, 70.86, 67.72, 55.82, 50.24, 42.84, 34.00, 26.60, 25.48, 24.83, and 21.81; m/z (CI) 501 (MH+, 21), 321 (34), 303 (44), and 249 (100).
…………………………………………
http://www.google.com/patents/EP2495235A1?cl=en
-
In the case of Travoprost, compound 9 with A=3-(trifluoromethyl)phenoxy (in the following scheme, compound 9b) is converted into 10b, which in turn is converted into Travoprost by esterification of the carboxylic acid by reaction with 2-iodopropane, according to scheme 10:


……………………………
http://www.google.com/patents/EP2454227A1?cl=en

Example 2
Synthesis of Travoprost MTBE MTBE
C


7b
8b

9b-iso

Travoprost Scheme 4. Synthesis of Travoprost
References
- Alcon Laboratories, Inc. (September 2011). “TRAVATAN – travoprost solution”. DailyMed. Bethesda, MD: U.S. National Library of Medicine. Retrieved 2011-09-30.
- Alcon Laboratories, Inc. (September 2011). “TRAVATAN Z (travoprost) solution”. DailyMed. Bethesda, MD: U.S. National Library of Medicine. Retrieved 2011-09-30.
- AHFS Consumer Medication Information (2011-01-01). “Travoprost Ophthalmic”. MedlinePlus. Bethesda, MD: U.S. National Library of Medicine. Retrieved 2011-09-30.
More References:
Selective FP prostaglandin receptor agonist. Isopropyl ester of (+)-fluprostenol, q.v. General prepn (not claimed): J. W. Stjernschantz, EP 364417 (1989 to Pharmacia).
Large scale synthesis: L. T. Boulton et al., Org. Process Res. Dev. 6, 138 (2002).
Pharmacology: M. R. Hellberg et al., J. Ocul. Pharmacol. Ther. 17, 421 (2001).
LC/MS/MS determn in plasma: B. A. McCue et al., J. Pharm. Biomed. Anal. 28, 199 (2002). Ocular hypotensive effects in dogs: A. B. Carvalho et al., Vet. Ophthalmol. 9, 121 (2006).
Clinical trial in glaucoma or ocular hypertension: R. L. Fellman et al., Ophthalmology 109, 998 (2002); in combination with timolol: J. S. Schuman et al., Am. J. Ophthalmol. 140, 242-250 (2005).
- Ota T, Aihara M, Narumiya S, Araie M: The effects of prostaglandin analogues on IOP in prostanoid FP-receptor-deficient mice. Invest Ophthalmol Vis Sci. 2005 Nov;46(11):4159-63. PubMed: 16249494
- Thieme H, Schimmat C, Munzer G, Boxberger M, Fromm M, Pfeiffer N, Rosenthal R: Endothelin antagonism: effects of FP receptor agonists prostaglandin F2alpha and fluprostenol on trabecular meshwork contractility. Invest Ophthalmol Vis Sci. 2006 Mar;47(3):938-45. PubMed: 16505027
- Lim KS, Nau CB, O’Byrne MM, Hodge DO, Toris CB, McLaren JW, Johnson DH: Mechanism of action of bimatoprost, latanoprost, and travoprost in healthy subjects. A crossover study. Ophthalmology. 2008 May;115(5):790-795.e4. PubMed: 18452763
- Neacsu AM: [Receptors involved in the mechanism of action of topical prostaglandines] Oftalmologia. 2009;53(2):3-7. PubMed: 19697832
- Costagliola C, dell’Omo R, Romano MR, Rinaldi M, Zeppa L, Parmeggiani F: Pharmacotherapy of intraocular pressure – part II. Carbonic anhydrase inhibitors, prostaglandin analogues and prostamides. Expert Opin Pharmacother. 2009 Dec;10(17):2859-70. PubMed: 19929706
- Ferrari G, Scagliotti GV: Serum and urinary vascular endothelial growth factor levels in non-small cell lung cancer patients. Eur J Cancer. 1996 Dec;32A(13):2368-9. PubMed: 9038626
- Toris CB, Gabelt BT, Kaufman PL: Update on the mechanism of action of topical prostaglandins for intraocular pressure reduction. Surv Ophthalmol. 2008 Nov;53 Suppl1:S107-20. PubMed: 19038618
- Arranz-Marquez E, Teus MA: Prostanoids for the management of glaucoma. Expert Opin Drug Saf. 2008 Nov;7(6):801-8. PubMed: 18983226
- Chen X, Ji ZL, Chen YZ: TTD: Therapeutic Target Database. Nucleic Acids Res. 2002 Jan 1;30(1):412-5. PubMed: 11752352
Your Aunt can teach you Organic Opectroscopy

Organic spectroscopy should be brushed up and you get confidence
read my blog
ORGANIC SPECTROSCOPY INTERNATIONAL is the blog
Organic chemists from Industry and academics to interact on Spectroscopy techniques for Organic compounds ie NMR, MASS, IR, UV Etc. email me ……….. amcrasto@gmail.com
http://orgspectroscopyint.blogspot.in/ is the link
 amcrasto@gmail.com
amcrasto@gmail.com
Oleanolic acid spectral data and interpretation
 Oleanolic acid
Oleanolic acid(4aS,6aR,6aS,6bR,8aR,10S,12aR,14bS)-10-hydroxy-2,2,6a,6b,9,9,12a-heptamethyl-1,3,4,5,6,6a,7,8,8a,10,11,12,13,14b-tetradecahydropicene-4a-carboxylic acid
Oleanic acid, Caryophyllin, Astrantiagenin C, Giganteumgenin C, Virgaureagenin B, 3beta-Hydroxyolean-12-en-28-oic acid, OLEANOLIC_ACID
Molecular Formula: C30H48O3
Molecular Weight: 456.70032
http://orgspectroscopyint.blogspot.in/2014/08/oleanolic-acid-spectral-data-and.html

Ursolic acid [(3b)-3-Hydroxyurs-12-en-28-oic acid] rarely occurs without its isomer oleanolic acid [(3b)-3-Hydroxyolean-12-en-28-oic acid] They may occur in their free acid form, as shown in Figure 1, or as aglycones for triterpenoid saponins which are comprised of a triterpenoid aglycone linked to one or more sugar moieties. Ursolic and oleanolic acids are similar in pharmacological activity
A pentacyclic triterpene that occurs widely in many PLANTS as the free acid or the aglycone for many SAPONINS. It is biosynthesized from lupane. It can rearrange to the isomer, ursolic acid, or be oxidized to taraxasterol and amyrin.
MS
EIMS m/z (rel. int.) 456 [M]+ (5), 412 (3), 248 (100), 203 (50), 167 (25), 44 (51)
IR KBR
(KBr) 3500, 2950, 2850, 1715; 1H-NMR (250 MHz, pyridine-d5) δ: 5.49 (1H, s, H-12), 3.47 (1H, t, J = 8.0 Hz, H-3), 3.30 (1H, m, H-18), 1.12 (3H, s, CH3-27), 0.96 (3H, s, CH3-30), 0.91 (3H, s, CH3-25), 0.89 (3H, s, CH3-23), 0.87 (3H, s, CH3-24), 0.75 (3H, s, CH3-26)
http://orgspectroscopyint.blogspot.in/2014/08/oleanolic-acid-spectral-data-and.html
1H NMR
(250 MHz, pyridine-d5)δ: 5.49 (1H, s, H-12), 3.47 (1H, t, J = 8.0 Hz, H-3), 3.30 (1H, m, H-18), 1.12 (3H, s, CH3-27), 0.96 (3H, s, CH3-30), 0.91 (3H, s, CH3-25), 0.89 (3H, s, CH3-23), 0.87 (3H, s, CH3-24), 0.75 (3H, s, CH3-26) |

13 C NMR
(63 MHz, pyridine-d5) δ: 180.2 (C-28), 144.8 (C-13), 122.5 (C-12), 78.0 (C-3), 55.7 (C-5), 48.0 (C-9), 46.6 (C-8, 17), 42.1 (C-14), 39.7 (C-4), 39.4 (C-1), 37.3 (C-10), 33.2 (C-7), 32.9 (C-29), 32.4 (C-21), 30.9 (C-20), 28.7 (C-23), 27.2 (C-2), 26.9 (C-15), 26.1 (C-30), 23.7 (C-11), 23.6 (C-16), 18.7 (C-6), 17.4 (C-26), 16.5 (C-24), 15.5 (C-25) |
http://orgspectroscopyint.blogspot.in/2014/08/oleanolic-acid-spectral-data-and.html
http://www.google.com/patents/US20120237629
FIG. 4 shows the 1H NMR spectrum of oleanolic acid;
FIG. 5 shows the 13C NMR spectrum of oleanolic acid;
FIG. 6 shows the 13C DEPT NMR spectrum of oleanolic acid;
FIG. 7 shows the 1H 13C HSQC NMR spectrum of oleanolic acid;
see below

http://orgspectroscopyint.blogspot.in/2014/08/oleanolic-acid-spectral-data-and.html



EXAMPLE 2 Extraction and Isolation of Oleanolic Acid (9) and Maslinic Acid (10) from Cloves
Syzygium aromaticum dried buds or whole cloves were obtained commercially. The cloves (1.5 kg, whole) of Syzygium aromaticum were sequentially and exhaustively extracted with hexane and ethyl acetate to give, after solvent removal in vacuo, a hexane extract (68.8 g, 4.9%) and an ethyl acetate extract (34.1 g, 2.3%). A portion of the ethyl acetate extract (10.0 g), was subjected to chromatographic separation on silica gel (60-120 mesh) column (40×5.0 cm). Elution with hexane/ethyl acetate solvent mixtures (8:2→6:4) afforded pure oleanolic acid (9) (4.7 g, 1.06%), a mixture of oleanolic acid (9) and maslinic acid (10) (0.5 g), and pure maslinic acid (10) (0.25 g). The structures of oleanolic acid (9) and maslinic acid (10) (as 2,3-diacetoxyoleanolic acid) were confirmed by spectroscopic data analysis (1D and 2D 1H NMR and 13C NMR experiments) (FIGS. 4-7 and FIGS. 8-10, respectively).

THANKS AND REGARD’S
DR ANTHONY MELVIN CRASTO Ph.D
MOBILE-+91 9323115463
GLENMARK SCIENTIST , INDIA
web link
web link
ABOUTME, BRAND ANTHONYCRASTO, COLLECTION OF SITE LINKS, BRAVESITES,GRAVATAR, JIMDO, SKILLPAGES, MIXXT, APNACIRCLE, ZIC ZAC, ZING ME, SCOOP IT, scribd, Stumbleupon, Delicious, pinnterest, tumblr, Newsvine, SLIDESHARE, ACADEMIA.EDU, GOOGLE PLUS, FACEBOOK, TWITTER, ISSUU, DIIGO, YOLASITE, Authorstream, Symbaloo, ISSUU, BLOGLOVIN,FRIENDFEED, NEWSVINE
Congratulations! Your presentation titled “Anthony Cra sto Glenmark scientist, helping millions with websites” has just crossed MILLION views.
アンソニー 安东尼 Энтони 안토니 أنتوني
blogs are

New Drug Approvals, ALL ABOUT DRUGS, WORLD DRUG TRACKER
MEDICINAL CHEM INTERNATIONAL, DRUG SYN INTERNATIONAL
SCALEUP OF DRUGS, ALL FOR DRUGS ON WEB,
MY CHINA, VIETNAM AND JAPAN BLOGS
ICELAND, RUSSIA, ARAB
BOBRDOBR, BLAND ICELAND, 100zakladok, adfty
GROUPS
you can post articles and will be administered by me on the google group which is very popular across the world
OPD GROUPSPACES, SCOOP OCI, organic-process-development GOOGLE, TVINX, MENDELEY WDT,SCIPEOPLE OPD, EPERNICUS OPD, SYNTHETIC ORGANIC CHEMISTRYLinkedIn group, DIIGO OPD,LINKEDIN OPD, WDT LINKEDIN, WDTI ZING


DR ANTHONY MELVIN CRASTO
ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on
ORGANIC SPECTROSCOPY INTERNATIONAL

