
FLAGS AND HITS
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO FACEBOOK
...................................................................Join me on twitter
FACEBOOK
...................................................................Join me on twitter
 ..................................................................Join me on google plus
..................................................................Join me on google plus
 Googleplus
GoogleplusMYSELF

AMIODARONE



In December 1985, amiodarone was approved by the FDA for the treatment of arrhythmias.[6] This makes amiodarone one of the few drugs approved by the FDA without rigorous randomized clinical trials.
Amiodarone is an antiarrhythmic agent used for various types of cardiac dysrhythmias, both ventricular and atrial. It was discovered in 1961. Despite relatively common side-effects, it is used in arrhythmias that are otherwise difficult to treat with medication.

A more recent synthesis of amiodarone reports the cyclisation of α-phenoxyhexanal 389 under acidic conditions to yield the substituted benzofuran 390 (Scheme 76). A Friedel–Crafts acylation next introduces the aryl ring at the 3-position. Demethylation, iodination and a final alkylation with a diethylaminoethane fragment yields amiodarone [115-117].
- 115 Witczak, M.; Kwiecień, H. Synth. Commun. 2005, 35, 2223–2230. doi:10.1080/00397910500182747
Return to citation in text: [1] - Wang, Z. J. Synthetic Process for 2-Butyl-3-(hydroxy-3,5-diiodobenzoyl)-benzofuran. Chin. Patent 1,858,042, Nov 8, 2006……….116
Return to citation in text: [1] - Ha, H. R.; Stieger, B.; Grassi, G.; Altorfer, H. R.; Follath, F. Eur. J. Clin. Pharmacol. 2000, 55, 807–814.doi:10.1007/s002280050701….117
![[1860-5397-7-57-i76]](http://www.beilstein-journals.org/bjoc/content/inline/1860-5397-7-57-i76.png?scale=2.0&max-width=1024&background=FFFFFF)
External links
- Siddoway LA (December 2003). “Amiodarone: guidelines for use and monitoring”. Am Fam Physician 68 (11): 2189–96. PMID 14677664.
- Amiodarone (MedicineNet.com)
- Amiodarone (FamilyPracticeNotebook.com)
- Amiodarone (The WorldWide Intensivist)
- U.S. National Library of Medicine: Drug Information Portal – Amiodarone
- Amiodarone (FDA MedWatch)
 |
|
| Systematic (IUPAC) name | |
|---|---|
| (2-{4-[(2-butyl-1-benzofuran-3-yl)carbonyl]-2,6-diiodophenoxy}ethyl)diethylamine | |
| Clinical data | |
| Trade names | Cordarone, Nexterone |
| AHFS/Drugs.com | monograph |
| MedlinePlus | a687009 |
| Pregnancy cat. |
|
| Legal status |
|
| Routes | oral or intravenous |
| Pharmacokinetic data | |
| Bioavailability | 20–55% |
| Metabolism | Liver |
| Half-life | 58 days (range 15-142 days) |
| Excretion | Primarily Hepatic and Biliary |
| Identifiers | |
| CAS number | 1951-25-3 |
| ATC code | C01BD01 |
| PubChem | CID 2157 |
| IUPHAR ligand | 2566 |
| DrugBank | DB01118 |
| ChemSpider | 2072 |
| UNII | N3RQ532IUT |
| KEGG | D02910 |
| ChEBI | CHEBI:2663 |
| ChEMBL | CHEMBL633 |
| Chemical data | |
| Formula | C25H29I2NO3 |
| Mol. mass | 645,31 g/mol |
AMIODARONE
|
|
Title: Amiodarone
CAS Registry Number: 1951-25-3
CAS Name: (2-Butyl-3-benzofuranyl)[4-[2-(diethylamino)ethoxy]-3,5-diiodophenyl]methanone
Additional Names: 2-butyl-3-benzofuranyl-4-[2-(diethylamino)ethoxy]-3,5-diiodophenyl ketone; 2-butyl-3-[3,5-diiodo-4-(b-diethylaminoethoxy)benzoyl]benzofuran
Molecular Formula: C25H29I2NO3
Molecular Weight: 645.31
Percent Composition: C 46.53%, H 4.53%, I 39.33%, N 2.17%, O 7.44%
Literature References: Benzofuran derivative with multiple electrophysiological effects. Prepn: FR 1339389; R. Tondeur, F. Binon, US 3248401 (1963, 1966 to Soc. Belge l’Azote Prod. Chim. Marly). Physicochemical properties: M. Bonati et al., J. Pharm. Sci. 73, 829 (1984). HPLC determn in plasma: M. De Smet, D. L. Massart, J. Pharm. Biomed. Anal. 6, 277 (1988). Comprehensive description: T. A. Plomp, Anal. Profiles Drug Subs. 20, 1-120 (1991). Review of pharmacology, clinical efficacy and safety: M. Chow, Ann. Pharmacother. 30, 637-643 (1996); B. N. Singh, Clin. Cardiol. 20, 608-618 (1997). Clinical trial in cardiac resuscitation: P. J. Kudenchuk et al., N. Engl. J. Med. 341, 871 (1999); to prevent atrial fibrillation: D. Roy et al., ibid. 342, 913 (2000).
Derivative Type: Hydrochloride
CAS Registry Number: 19774-82-4
Manufacturers’ Codes: L-3428
Trademarks: Amiodar (Sanofi Winthrop); Ancaron (Taisho); Cordarex (Sanofi Winthrop); Cordarone (Wyeth); Ortacrone (Sanofi Winthrop); Pacerone (Upsher-Smith); Tachydaron (AWD); Trangorex (Sanofi Winthrop)
Molecular Formula: C25H29I2NO3.HCl
Molecular Weight: 681.77
Percent Composition: C 44.04%, H 4.44%, I 37.23%, N 2.05%, O 7.04%, Cl 5.20%
Properties: Crystalline powder, mp 156°. Also reported as crystals from acetone, mp 159 ±2° (Bonati). Soly at 25° (g/100ml): chloroform 44.51; methylene chloride 19.20; methanol 9.98; ethanol 1.28; benzene 0.65; tetrahydrofuran 0.60; acetonitrile 0.32; 1-octanol 0.30; ether 0.17; 1-propanol 0.13; water 0.07; hexane 0.03 petroleum ether 0.001. Sparingly sol in isopropanol; slightly sol in acetone, dioxane, and carbon tetrachloride. pH (5% soln) 3.4-3.9. pKa (25°C) 6.56 ±0.06. uv max (methanol): 208, 242 nm (E1%1cm 662 ±8, 623 ±10).
Melting point: mp 156°; mp 159 ±2° (Bonati)
pKa: pKa (25°C) 6.56 ±0.06
Absorption maximum: uv max (methanol): 208, 242 nm (E1%1cm 662 ±8, 623 ±10)
Therap-Cat: Antiarrhythmic (class III).
Keywords: Antiarrhythmic.
|
Amiodarone
-
- ATC:C01BD01
- Use:antiarrhythmic
- Chemical name:(2-butyl-3-benzofuranyl)[4-[2-(diethylamino)ethoxy]-3,5-diiodophenyl]methanone
- Formula:C25H29I2NO3
- MW:645.32 g/mol
- CAS-RN:1951-25-3
- InChI Key:IYIKLHRQXLHMJQ-UHFFFAOYSA-N
- InChI:InChI=1S/C25H29I2NO3/c1-4-7-11-22-23(18-10-8-9-12-21(18)31-22)24(29)17-15-19(26)25(20(27)16-17)30-14-13-28(5-2)6-3/h8-10,12,15-16H,4-7,11,13-14H2,1-3H3
- EINECS:217-772-1
- LD50:178 mg/kg (M, i.v.); >4 g/kg (M, p.o.)
Derivatives
hydrochloride
- Formula:C25H29I2NO3 • HCl
- MW:681.78 g/mol
- CAS-RN:19774-82-4
Substance Classes
Synthesis Path
Trade Names
| Country | Trade Name | Vendor | Annotation |
|---|---|---|---|
| D | Cordarex | Sanofi-Aventis | |
| Cornaron | TAD Pharma | ||
| F | Corbionax Ge | Winthrop/Sanofi-Aventis | |
| Cordarone | Sanofi-Aventis | ||
| GB | Cordaron | Sanofi-Aventis | |
| I | Amiodar | Sigma-Tau | |
| Cordarone | Sanofi-Aventis | ||
| J | Ancaron | Sanofi-Aventis | |
| USA | Cordarone | Wyeth-Ayerst | as hydrochloride |
Formulations
- inj. sol. 150 mg/3ml; tabl. 100 mg, 200 mg
References
-
- FR 1 339 389 (Labaz; appl. 22.11.1962).
- US 3 248 401 (Labaz; 26.4.1966; D-prior. 24.11.1961).
-
2-butylbenzofuran:
- Buu-Hoï, N.P. et al.: J. Chem. Soc. (JCSOA9) 1964, 173.
PATENT
CN109053652-PREPARATION METHOD OF AMIODARONE HYDROCHLORIDE INTERMITTENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=CN235615504&_cid=P11-KL0AU0-06410-1
| Amiodarone hydrochloride is currently the most widely used antiarrhythmic drug. In addition, amiodarone hydrochloride has become the first choice for long-term medication for patients with arrhythmia due to its stable curative effect and minor side effects. In today’s society, people’s work rhythm is constantly increasing, the pressure they face is increasing, the number of patients with cardiovascular diseases is increasing, the demand for anti-arrhythmic drugs is greatly increasing, and the market share of amiodarone hydrochloride is bound to continue to increase. |
| 2-Butyl-3-(4-hydroxybenzoyl)benzofuran is an important intermediate of amiodarone hydrochloride. Patent CN104262304 uses compounds 1 and 2 as starting materials, and the resulting compound 3 is reacted with sodium methoxide as a base and toluene as a solvent to form compound 5; compound 5 is reacted with compound 7 under Lewis acid conditions to form compound 8; compound 8 Hydrolyzed under Lewis acid conditions, the reaction produces 2-butyl-3-(4-hydroxybenzoyl)benzofuran 9. |
| |
| This synthetic route has the following shortcomings: Compound 3 can be directly obtained without hydrolysis under the condition of sodium methoxide as base and toluene as solvent. However, the difficulty of demethylation is much greater than that of decarboxylation, resulting in the formation of compound 5. The conversion rate of the cyclization reaction is low, the by-products are many, and the separation is difficult; resulting in low total yield and increased cost. |
| The synthetic route reported in patent CN107382925, except that the route for preparing compound 5 from compound 3 is different, other reaction routes are the same, but the reaction conditions are different. In this reaction route, the aldehyde group of compound 3 is first protected with trimethyl orthoformate to produce compound 0, and then compound 0 is hydrolyzed to produce compound 4. Compound 4 is cyclized under the catalysis of p-toluenesulfonyl chloride to obtain compound 5. |
| |
| This route has the following disadvantages: when compound 3 is prepared to compound 5, trimethyl orthoformate is added to protect the aldehyde group, then the ester group is first hydrolyzed to form an acid, and then the protective group of the aldehyde group is removed; although side reactions of the aldehyde group can be reduced, but The reaction steps are added, and the trimethyl orthoformate is highly flammable and has potential safety hazards. The yield of compound 4 to compound 5 catalyzed by p-toluenesulfonyl chloride is low, and the cost is also increased. |
| Example 1: |
| Add 15.00kg (122.8mol, 1eq) compound 1 and 30.82kg (147.4mol, 1.2eq) compound 2 to 120.00kg (8w/w) ethyl acetate, add 24.00kg (73.7mol, 0.6eq) cesium carbonate, 1.00 kg (2.5 mol, 0.02 eq) methyl trioctyl ammonium chloride, stirred and heated to 75-85°C, reacted for 1 to 2 hours. After the reaction, the filtrate was filtered with suction, and the filtrate was washed with 60.00kg (4w/w) purified water, and the organic phase was concentrated to dryness under reduced pressure at 40°C to obtain 30.21kg (120.7mol) of compound 3 with a yield of about 98.3%. |
| 30.00kg (120.0mol, 1eq) of compound 3 was added to 14.40kg (360.0mol, 3eq) of 150.00kg (5w/w) aqueous solution of sodium hydroxide, and stirred at 20-30°C. After the reaction, add 1N dilute hydrochloric acid to adjust the pH to 4, add 30.00kg (1w/w) ethyl acetate for extraction, add 30.00kg (1w/w) for the organic phase and wash once with purified water, and add 30.00kg (1w/w) for the organic phase Wash with saturated sodium chloride aqueous solution, add 1.50kg (0.05w/w) anhydrous sodium sulfate to dry. The organic phase was concentrated to dryness under reduced pressure at 40° C. to obtain 27.30 kg (115.6 mol) of compound 4 with a yield of about 96.4%. |
| Add 27.00kg (114.3mol, 1eq) of compound 4 to 216.00kg (8.0w/w) of acetic anhydride, add 75.60kg (2.8w/w) of sodium acetate, raise the temperature to 90~100℃ for reaction, and react for 1~2 hours . After the reaction, the reaction solution was transferred to 270.00kg (10w/w) purified water, stirred for 2 hours, and then extracted with 81.00kg (3w/w) ethyl acetate. The organic phase was washed twice with 27.00kg (1.0w/w) purified water, and the organic phase was washed with 27.00kg (1.0w/w) saturated aqueous sodium chloride solution, and dried with 2.70kg (0.1w/w) anhydrous sodium sulfate. The organic phase was concentrated to dryness under reduced pressure at 40°C. 14.26kg (81.8mol) of compound 5 was obtained, and the yield was about 71.6%. 1 HNMR(400MHz,d DMSO )δ:0.92~0.96(t,3H,-CH 3 ),1.37~1.42(m,2H,-CH 2 CH 3 ),1.68~1.72(m,2H,-CH 2 CH 2 CH 3 ),2.77~2.81(t,2H,Ar-CH 2 -CH 2 ), 6.59 (s, 1H, -ArH), 7.20 ~ 7.24 (m, 2H, ArH), 7.49 ~ 7.56 (m, 2 H, ArH). 13 CNMR (400 Hz, DMSO) δ: 159.76, 154.46, 129.07, 123.61, 122.95, 120.72, 111.03, 102.41, 29.71, 27.79, 22.12, 14.05 See attached drawings 1-2. |
| Add 17.00kg (111.7mol, 1eq) of compound 6 to 34.00kg (2w/w) toluene, add 33.31kg (280.0mol, 2.5eq) of thionyl chloride, heat to 75~85℃, keep warm for 2~4 hour. After the reaction, the solvent was distilled off under reduced pressure at 65°C to obtain 18.99 kg (111.3 mol) of compound 7 as a colorless solution with a yield of about 97.4%. |
| Add 10.70kg (80.4mol, 1.0eq) of aluminum trichloride to 60.00kg of 1,2-dichloroethane at -20~-10℃, add 14.00kg (80.4mol, 1.0eq) of compound 5 under stirring, After stirring uniformly, 16.46kg (96.5mol, 1.2eq) of compound 7 is added at -20~-10°C, and the reaction is kept for 1~2 hours. After the reaction, the reaction solution was transferred to 90.00kg purified water, adjusted to pH 2 with dilute hydrochloric acid, separated into the organic phase and washed twice with 45.00kg purified water, the organic phase was washed with 45.00kg saturated sodium chloride aqueous solution, and 1.40kg was added. Dry with water sodium sulfate. The organic phase was concentrated under reduced pressure at 55°C to obtain 20.11 kg (65.0 mol) of compound 8. The yield is about 81.2%. |
| Add 20.00kg (64.9mol, 1.0eq) of compound 8 to 60.00kg (3w/w) of toluene, add 9.52kg (71.4mol, 1.1eq) of aluminum trichloride, and raise it to 80~90℃ to react for 2~4 hours . After the reaction, transfer the reaction solution to 90.00kg (4.5w/w) purified water, adjust the pH to 2 with dilute hydrochloric acid, separate the organic phase and wash 2 times with 50.00kg (2.5w/w) purified water, and add 50.00 for the organic phase kg (2.5w/w) saturated sodium chloride aqueous solution wash, add 2.00kg (0.1w/w) anhydrous sodium sulfate to dry. The organic phase was concentrated under reduced pressure at 65°C until a solid precipitated out, the temperature was lowered to 0°C, and the temperature was kept for 4 hours to crystallize. The wet product was filtered with suction and dried under vacuum at 80°C to obtain 16.60 kg (56.4 mol) of compound 9 as a white crystalline powder with a yield of about 86.9%. |
| 1 HNMR(400MHz,d DMSO )δ: 0.81~0.85(t,3H,-CH 3 ),1.24~1.29(m,2H,-CH 2 CH 3 ),1.68~1.70(m,2H,-CH 2 CH 2 CH 3 ),2.82~2.85(t,2H,Ar-CH 2 -CH 2 ), 6.91~7.72(m,8H,ArH), 10.46(m,1H,-OH). 13 CNMR(400 Hz,DMSO)δ:189.72,163.49,162.69,153.48,132.08,130.16,127.28,124.87,123.98 ,121.16,116.90,11 5.78,111.51,29.94,27.51,22.09,13.84. See attached drawings 3~4. |
| According to the operation of Example 1, the total yield was 46.6%. |
| Example 2: |
| Add 15.00kg (122.8mol, 1eq) of compound 1 and 30.82kg (147.4mol, 1.2eq) of compound 2 to 120.00kg (8w/w) ethyl acetate, add 10.19kg (73.7mol, 0.6eq) potassium carbonate, 1.00 kg (2.5 mol, 0.02 eq) methyl trioctyl ammonium chloride, stirred and heated to 75-85° C., reacted for 1 to 2 hours. After the reaction is completed, suction filtration, the filtrate is washed with 60.00kg (4w/w) purified water, and the organic phase is concentrated to dryness under reduced pressure at 40°C. Obtain 26.21kg (104.7mol) of compound 3, the yield is about 85.3%. |
| 26.00kg (103.9mol, 1eq) of compound 3 was added to 12.47kg (311.7mol, 3eq) of sodium hydroxide in 130.00kg (5w/w) methanol solution, and stirred at 20-30°C. After the reaction, it was concentrated to dryness under reduced pressure at 40°C. Add 130.00kg (5w/w) aqueous solution to dissolve, add 1N dilute hydrochloric acid to adjust the pH to 4, add 26.00kg (1w/w) ethyl acetate for extraction, add 26.00kg (1w/w) purified water for organic phase and wash once, organic phase Add 26.00kg (1w/w) saturated sodium chloride aqueous solution to wash, add 1.30kg (0.05w/w) anhydrous sodium sulfate to dry. The organic phase was concentrated to dryness under reduced pressure at 40°C. 23.50 kg (99.5 mol) of compound 4 was obtained, and the yield was about 95.8%. |
| Add 23.00kg (97.4mol, 1eq) of compound 4 to 184.00kg (8.0w/w) of acetic anhydride, add 64.40kg (2.8w/w) of sodium acetate, raise the temperature to 90~100℃ for reaction, and react for 1~2 hours . After the reaction, the reaction solution was transferred to 230.00kg (10w/w) purified water, stirred for 2 hours, and 69.00kg (3w/w) ethyl acetate was added for extraction. The organic phase was washed twice with 23.00kg (1.0w/w) purified water, the organic phase was washed with 23.00kg (1.0w/w) saturated sodium chloride aqueous solution, and 2.30kg (0.1w/w) was dried with anhydrous sodium sulfate. The organic phase was concentrated to dryness under reduced pressure at 40°C. 12.28kg (70.5mol) of compound 5 was obtained, and the yield was about 72.4%. |
| Add 15.00kg (98.6mol, 1eq) of compound 6 to 30.00kg (2w/w) of toluene, add 35.20kg (295.8mol, 3eq) of thionyl chloride, heat to 75~85℃, keep warm and react for 2~4 hours . After the reaction, the solvent was distilled off under reduced pressure at 65°C to obtain 16.63 kg (97.5 mol) of compound 7 as a colorless solution, with a yield of about 98.9%. |
| Add 27.56kg (206.7mol, 3.0eq) of aluminum trichloride to 60.00kg of 1,2-dichloroethane at -20~-10℃, add 12.00kg (68.9mol, 1.0eq) of compound 5 with stirring, After stirring uniformly, 14.11kg (82.7mol, 1.2eq) of compound 7 is added at -20~-10℃, and the reaction is kept for 1~2 hours. After the reaction, the reaction solution was transferred to 80.00kg purified water, adjusted to pH 2 with dilute hydrochloric acid, separated into the organic phase and washed twice with 40.00kg purified water, and the organic phase was washed with 40.00kg saturated sodium chloride aqueous solution, and 1.20kg was added. Dry with water sodium sulfate. The organic phase was concentrated under reduced pressure at 55°C to obtain 17.17 kg (55.7 mol) of compound 8, with a yield of about 80.8%. |
| Add 17.00kg (55.1mol, 1.0eq) of compound 8 to 51.00kg (3w/w) of toluene, add 22.04kg (165.3mol, 3.0eq) of aluminum trichloride, raise to 80~90℃ and react for 2~4 hours . After the reaction, the reaction solution was transferred to 76.50kg (4.5w/w) purified water, adjusted to pH 2 with dilute hydrochloric acid, separated, the organic phase was washed twice with 42.50kg (2.5w/w) purified water, and the organic phase was added 42.50kg (2.5w/w) saturated sodium chloride aqueous solution was washed, and 1.70kg (0.1w/w) anhydrous sodium sulfate was added for drying. The organic phase was concentrated under reduced pressure at 65°C until a solid precipitated out, the temperature was lowered to 0°C, and the temperature was kept for 4 hours to crystallize. After suction filtration, the wet product was dried in vacuum at 80° C. to obtain 13.81 kg (46.9 mol) of compound 9 as a white crystalline powder with a yield of about 85.1%. |
| According to the operation of Example 2, the total yield was 40.2%. |
| Example 3: |
| Add 15.00kg (122.8mol, 1eq) compound 1 and 30.82kg (147.4mol, 1.2eq) compound 2 to 120.00kg (8w/w) toluene, add 24.00kg (73.7mol, 0.6eq) cesium carbonate, 1.00kg (2.5mol, 0.02eq) methyl trioctyl ammonium chloride, stir and raise the temperature to 75~85℃, and react for 1~2 hours. After the reaction is completed, suction filtration, the filtrate is washed with 60.00kg (4w/w) purified water, and the organic phase is concentrated to dryness under reduced pressure at 40°C. 30.36 kg (121.3 mol) of compound 3 was obtained, and the yield was about 98.8%. |
| 30.00kg (120.0mol, 1eq) of compound 3 was added to 20.20kg (360.0mol, 3eq) of 150.00kg (5w/w) aqueous solution of potassium hydroxide, and stirred at 20-30°C. After the reaction, add 1N dilute hydrochloric acid to adjust the pH to 4, add 30.00kg (1w/w) ethyl acetate for extraction, add 30.00kg (1w/w) for the organic phase and wash once with purified water, and add 30.00kg (1w/w) for the organic phase Wash with saturated sodium chloride aqueous solution, add 1.50kg (0.05w/w) anhydrous sodium sulfate to dry. The organic phase was concentrated to dryness under reduced pressure at 40°C. 27.11 kg (114.7 mol) of compound 4 was obtained, and the yield was about 95.6%. |
| Add 27.00kg (114.3mol, 1eq) of compound 4 to 432.00kg (16.0w/w) of acetic anhydride, add 75.60kg (2.8w/w) of sodium acetate, raise the temperature to 90~100℃ for reaction, and react for 1~2 hours . After the reaction, the reaction solution was transferred to 540.00kg (20w/w) purified water, stirred for 5 hours, and 81.00kg (3w/w) ethyl acetate was added for extraction. The organic phase was washed twice with 27.00kg (1.0w/w) purified water, and the organic phase was washed with 27.00kg (1.0w/w) saturated aqueous sodium chloride solution, and dried with 2.70kg (0.1w/w) anhydrous sodium sulfate. The organic phase was concentrated to dryness under reduced pressure at 40°C. 14.57kg (83.6mol) of compound 5 was obtained, and the yield was about 73.1%. |
| Add 17.00kg (111.7mol, 1eq) of compound 6 to 34.00kg (2w/w) of toluene, add 35.20kg (295.8mol, 3eq) of thionyl chloride, heat to 75~85℃, keep the temperature and react for 2~4 hours . After the reaction, the solvent was distilled off under reduced pressure at 65°C to obtain 19.21 kg (112.6 mol) of compound 7 as a colorless solution, with a yield of about 100.0%. |
| Add 10.70kg (80.4mol, 1.0eq) of aluminum trichloride to 60.00kg of toluene at -20~-10℃, add 14.00kg (80.4mol, 1.0eq) of compound 5 with stirring, and after stirring, add at -20 16.46kg (96.5mol, 1.2eq) of compound 7 was added at ~-10°C, and the reaction was incubated for 1 to 2 hours. After the reaction, the reaction solution was transferred to 90.00kg purified water, adjusted to pH 2 with dilute hydrochloric acid, separated into the organic phase and washed twice with 45.00kg purified water, the organic phase was washed with 45.00kg saturated sodium chloride aqueous solution, and 1.40kg was added. Dry with water sodium sulfate. Add 10.72 kg (80.4 mol, 1.0 eq) of aluminum trichloride to the organic phase and raise it to 80-90° C. and react for 2 to 4 hours. After the reaction, the reaction solution was transferred to 90.00kg purified water, adjusted to pH 2 with dilute hydrochloric acid, separated into the organic phase and washed twice with 50.00kg purified water, the organic phase was washed with 50.00kg saturated sodium chloride aqueous solution, and 2.00kg Dry with water sodium sulfate. The organic phase was concentrated under reduced pressure at 65°C until a solid precipitated out, the temperature was lowered to 0°C, and the temperature was kept for 4 hours to crystallize. The wet product was filtered with suction and dried in vacuum at 80°C to obtain 14.47 kg (45.2 mol) of compound 9 as a white crystalline powder with a yield of 61.2%. |
| According to the operation of Example 3, the total yield was 42.3%. |
| Comparative Example 1 (CN104262304): |
| 700g (5.7mol, 1.0eq) of compound 1 was added to 2000g of ethyl acetate. After stirring, 1400g (6.7mol, 1.2eq) of compound 2 was slowly added. After the addition, 150g (0.46mol, 0.08eq) were added in sequence. Cesium carbonate and 50g (0.12mol, 0.02eq) methyl trioctyl ammonium chloride, and then slowly heated to 80 ℃, kept the reaction for 8 hours, at the end of the reaction, the ethyl acetate was evaporated under reduced pressure. After the evaporation, 2500g of toluene was added and controlled Warm to 20℃, continue to add 250g (4.6mol, 0.8eq) sodium methoxide, heat to reflux, keep the reaction for 5 hours; when the reaction is over, add dilute hydrochloric acid, adjust the pH to 7, stand still, separate the water phase; continue to add 600g Extract twice with water, discard the water phase, distill the organic phase under reduced pressure to remove the toluene, and collect the 135°C fraction. 383 g (2.2 mol) of compound 5 was obtained as a light yellow transparent liquid, and the yield was about 38.6%. |
| Add 360g (2.1mol, 1.0eq) of compound 5 to 2400g of toluene, heat to reflux, keep dehydration for 4 hours; after dehydration, cool to 15℃, add 420g (2.5mol, 1.2eq) of compound 7, 240g (1.8mol) , 0.86eq) zinc chloride, 60g (0.81mol, 0.4eq) N-nitrosodimethylamine, slowly increase the temperature to 80°C, keep the reaction for 10 hours; after the reaction is over, add dilute hydrochloric acid and adjust the pH to 1~2 , Continue to heat and stir for 2 hours, let stand, and discard the water phase; continue to add 450g of water to extract twice, discard the water phase, heat the organic phase to reflux, and keep dehydration for 4 hours; after dehydration, cool to 10°C, add 270g (2.1 mol, 1eq) Aluminum trichloride, slowly increase the temperature to 75°C, keep the reaction for 8 hours; after the reaction is over, add saturated sodium bicarbonate aqueous solution, adjust to pH 7, let stand, discard the water phase; add 360g saturated brine for extraction Three times, the water phase was discarded, the organic phase was distilled under reduced pressure, and it was steamed until a solid precipitated, and the temperature was reduced to 0°C, and the temperature was kept and stirred for 4 hours. After suction filtration, the wet product was vacuum dried at 80° C. for 8 hours to obtain 323 g (1.1 mol, 1 eq) of compound 9 as an off-white crystalline powder with a yield of about 52.4%. |
| According to the operation of Comparative Example 1, the total yield was 20.2%. |
| Comparative Example 2 (CN107382925): |
| 610g (5.0mol, 1.0eq) of compound 1 and 1728g (12.5mol, 2.5eq) of potassium carbonate were added to the mixture of 610kg (1w/w) DMF and 1830kg (3w/w) toluene, and heated to 60~ with stirring. Incubate at 70°C for half an hour, heat up to 80-100°C and add dropwise, 1098g (5.3mol, 1.05eq) of compound 2, react for 2 hours. After the reaction, 1830 g of water was added to wash twice, and the organic phase was concentrated to dryness under reduced pressure at 80°C. 1126 g (121.3 mol) of compound 3 was obtained, and the yield was about 90.1%. |
| 1100g (4.4mol, 1.0eq) of Compound 3 was added to 514g (4.8mol, 1.1eq) of trimethyl orthoformate and 7.6g (47mmol, 0.01eq) of p-toluenesulfonic acid in 11kg (10w/w) methanol solution, Stir at 20-30°C for 3 hours, add 2.4 g (44 mmol, 0.01 eq), and stir for 1 hour. Then it was concentrated to dryness under reduced pressure at 40°C. 1170 g (3.9 mol) of compound 0 was obtained, and the yield was about 89.7%. |
| Add 1100g (3.7mol, 1eq) of compound 0 to 2200g (2w/w) toluene, add 163g (4.1mol, 1.1eq) sodium hydroxide and 2200g (2w/w) water under stirring, 35~40 Stir and keep at ℃ for 4 hours. At the end of the reaction, 110g (0.1w/w) sodium chloride is added, and hydrochloric acid is added dropwise to adjust the pH to 1~2. The liquid was separated to obtain a toluene organic phase containing compound 4. Add 415g (4.1mol, 1.1eq) of triethylamine to the organic phase, raise it to 70~80℃ for reaction, add 782g (4.1mol, 1.1eq) of p-toluenesulfonyl chloride dropwise, and finish the reaction at 70~80℃ for 2 hours. A solution of 164g (4.1mol, 1.1eq) sodium hydroxide and 2200g (2w/w) water was added dropwise, and the reaction was kept at 70-80°C for 2 hours. Separate the liquids, wash the organic phase with 1100 g of 0.1M hydrochloric acid until neutral, and concentrate the organic phase to dryness at 80°C under reduced pressure. 453 g (2.6 mol) of compound 5 was obtained, and the yield was about 70.0%. |
| Add 409g (2.4mol, 1.0eq) compound 7, 336g (2.5mol, 1.05eq) aluminum trichloride to 60.00kg 1,2-dichloroethane at -20~-10℃, add 418g( 2.4 mol, 1.0 eq) Compound 5, incubated and reacted for 4 hours. Add 320g (2.4mol, 1.0eq) of aluminum trichloride to the organic phase, raise to reflux, and react under reflux for 6 hours. After the reaction, the reaction solution was transferred to 1000g purified water, the organic phase was separated and washed twice with 500g purified water, and the organic phase was purified by column chromatography to obtain 283g (1.0mol) of compound 9 as white crystalline powder with a yield of about 40.1%. . |
| According to the operation of Comparative Example 2, the total yield was 22.7%. |
| Description of the drawings |
| Figure 1 shows the proton nuclear magnetic resonance spectrum of compound 5; |
| Figure 2 shows the carbon nuclear magnetic resonance spectrum of compound 5; |
| Figure 3 shows the proton nuclear magnetic resonance spectrum of compound 9; |
| Figure 4 shows the carbon nuclear magnetic resonance spectrum of compound 9. |
PATENT
CN106946822
| Example 1: Preparation of 2-butyl-3-(4-hydroxybenzoyl)benzofuran |
| Step (1): Take 117.1g (0.5mol) of 1-(4-methoxyphenyl)-1,3-heptanedione and 56.1g (0.5mol) of acrolein dimer in organic solvent tetrahydrofuran (2L). ), 126.9 g (0.5 mol) of elemental iodine and 3.4 g (25 mmol) of zinc chloride were added, and the above reaction solution was stirred and reacted in a reactor equipped with magnetic stirring at 40°C for 2 hours. After the reaction, the acidic reaction system was neutralized to neutrality with saturated sodium thiosulfate and saturated sodium bicarbonate respectively; the aqueous phase obtained by liquid-liquid extraction with ethyl acetate; the organic phase was combined and concentrated under reduced pressure; ethyl acetate and Diethyl ether was recrystallized and separated to obtain 98.6 g (0.32 mol) of 2-butyl-3-(4-methoxybenzoyl)benzofuran. 1 H NMR(400MHz, CDCl 3 , TMS, 25℃) δ = 7.84 (d, J = 8.8 Hz, 2H), 7.47 (d, J = 8.2 Hz, 1H), 7.36 (d, J = 7.3 Hz, 1H), 7.26 (dd, J = 9.4,5.9Hz,1H), 7.18(t,J=7.5Hz,1H), 6.96(d,J=8.8Hz,2H), 3.89(s,3H), 2.91(t,J=7.6Hz,2H) ,1.80–1.71(m,2H),1.36(dd,J=15.0,7.4Hz,2H), 0.89ppm(t,J=7.4Hz,3H). 13 C NMR(100MHz,CDCl 3 ,25℃)δ=190.7,164.8,163.6,153.8,132.1,131.8,127.4,124.3,123.4,121.4,116.9,113.8,111.1,55.6,30.3,28.0,22.5,13.8ppm |
| |
| Step (2): Put 98.6g (0.32mol) of 2-butyl-3-(4-methoxybenzoyl)benzofuran obtained in step (1) into a reactor equipped with magnetic stirring. 2.1g (16mmol) of aluminum oxide was dissolved in 2L of acetonitrile; the mixture was stirred and reacted at 80°C for 5 hours. After the reaction, the acidic reaction system was neutralized with saturated sodium bicarbonate solution to neutrality; The aqueous phase obtained by extraction; the organic phases were combined and concentrated under reduced pressure; ethyl acetate and ether were recrystallized and separated to obtain 52.9 g (0.18 mol) of the 2-butyl-3-(4-hydroxybenzoyl)benzofuran. 1 H NMR(400MHz, CDCl 3 ,TMS,25℃)δ=10.46(s,1H),7.68(d,J=8.6Hz,2H), 7.62(d,J=8.1Hz,1H), 7.33(dd,J=15.7,7.9Hz, 2H), 7.24 (t, J = 7.4 Hz, 1H), 6.89 (d, J = 8.6 Hz, 2H), 2.80 (t, J = 7.5 Hz, 2H), 1.65 (dt, J = 15.0, 7.5 Hz, 2H),1.23(dd,J=14.7,7.3Hz,2H),0.80(t,J=7.4Hz,3H). 13 C NMR(100MHz,CDCl 3 ,25℃)δ=189.3,163.1,162.2,153.0,131.6,129.7,126.8,124.5,123.6,120.7,116.4,115.3,111.1,29.5,27.1,21.6,13.4ppm. |
| |
| The yield of Example 1 was 36%, wherein the yield of step (1) was 64%, and the yield of step (2) was 56%. |
| Example 2: Preparation of 2-butyl-3-(4-hydroxybenzoyl)benzofuran |
| Step (1): Take 58.5g (0.25mol) of 1-(4-methoxyphenyl)-1,3-heptanedione and 56.1g (0.5mol) of acrolein dimer in the organic solvent methylene chloride (2L), then add 165.8g (0.5mol) of carbon tetrabromide, BBr 3 6.26g (25mmol), the above reaction solution was stirred and reacted for 4 hours at 25°C in a reactor equipped with magnetic stirring. After the reaction, the acidic reaction system was neutralized with saturated sodium bicarbonate; the aqueous phase obtained by liquid-liquid extraction with dichloromethane; the organic phases were combined and concentrated under reduced pressure; ethyl acetate and ether were recrystallized to obtain 2-butane Benzyl-3-(4-methoxybenzoyl)benzofuran 49.3 g (0.16 mol). 1 H NMR(400MHz, CDCl 3 , TMS, 25℃) δ = 7.84 (d, J = 8.8 Hz, 2H), 7.47 (d, J = 8.2 Hz, 1H), 7.36 (d, J = 7.3 Hz, 1H), 7.26 (dd, J = 9.4,5.9Hz,1H), 7.18(t,J=7.5Hz,1H), 6.96(d,J=8.8Hz,2H), 3.89(s,3H), 2.91(t,J=7.6Hz,2H) ,1.80–1.71(m,2H),1.36(dd,J=15.0,7.4Hz,2H), 0.89ppm(t,J=7.4Hz,3H). 13 C NMR(100MHz,CDCl 3 ,25℃)δ=190.7,164.8,163.6,153.8,132.1,131.8,127.4,124.3,123.4,121.4,116.9,113.8,111.1,55.6,30.3,28.0,22.5,13.8ppm |
| |
| Step (2): Put 49.3g (0.16mol) of 2-butyl-3-(4-methoxybenzoyl)benzofuran obtained in step (1) into a reactor equipped with magnetic stirring. 1.1g (8mmol) of boron diethyl ether was dissolved in 2L of 1,2-dichloroethane; the mixture was stirred and reacted at 60℃ for 5 hours. After the reaction, the acidic reaction system was neutralized with saturated sodium bicarbonate solution The aqueous phase obtained by liquid-liquid extraction with ethyl acetate; the organic phases were combined and concentrated under reduced pressure; ethyl acetate and ether were recrystallized and separated to obtain the 2-butyl-3-(4-hydroxybenzoyl) benzo Furan 32.37g (0.11mol). 1 H NMR(400MHz, CDCl 3 ,TMS,25℃)δ=10.46(s,1H),7.68(d,J=8.6Hz,2H), 7.62(d,J=8.1Hz,1H), 7.33(dd,J=15.7,7.9Hz, 2H), 7.24 (t, J = 7.4 Hz, 1H), 6.89 (d, J = 8.6 Hz, 2H), 2.80 (t, J = 7.5 Hz, 2H), 1.65 (dt, J = 15.0, 7.5 Hz, 2H),1.23(dd,J=14.7,7.3Hz,2H),0.80(t,J=7.4Hz,3H). 13 C NMR(100MHz,CDCl 3 ,25℃)δ=189.3,163.1,162.2,153.0,131.6,129.7,126.8,124.5,123.6,120.7,116.4,115.3,111.1,29.5,27.1,21.6,13.4ppm. |
| |
| The yield of Example 2 was 44%, wherein the yield of step (1) was 64%, and the yield of step (2) was 68%. |
| Example 3: Preparation of 2-butyl-3-(4-methylbenzoyl)benzofuran |
| Step (1): Take 117.1g (0.5mol) of 1-(4-methoxyphenyl)-1,3-heptanedione, and dissolve 56.1g (0.5mol) of acrolein dimer in organic solvent ethanol (2L) ), then add 66.8g (0.5mol) of N-chlorosuccinimide, ZrCl 4 5.8 g (25 mmol), the above reaction solution was stirred and reacted in a reactor equipped with magnetic stirring at 70°C for 8 hours. After the reaction is completed, the acidic reaction system is neutralized with saturated sodium bicarbonate; the aqueous phase obtained by liquid-liquid extraction with ethyl acetate; the organic phases are combined and concentrated under reduced pressure; ethyl acetate and ether are recrystallized and separated to obtain 2-butane 104.7 g (0.34 mol) of phenyl-3-(4-methoxybenzoyl)benzofuran. 1 H NMR(400MHz, CDCl 3 , TMS, 25℃) δ = 7.84 (d, J = 8.8 Hz, 2H), 7.47 (d, J = 8.2 Hz, 1H), 7.36 (d, J = 7.3 Hz, 1H), 7.26 (dd, J = 9.4,5.9Hz,1H), 7.18(t,J=7.5Hz,1H), 6.96(d,J=8.8Hz,2H), 3.89(s,3H), 2.91(t,J=7.6Hz,2H) ,1.80–1.71(m,2H),1.36(dd,J=15.0,7.4Hz,2H), 0.89ppm(t,J=7.4Hz,3H). 13 C NMR(100MHz,CDCl 3 ,25℃)δ=190.7,164.8,163.6,153.8,132.1,131.8,127.4,124.3,123.4,121.4,116.9,113.8,111.1,55.6,30.3,28.0,22.5,13.8ppm |
| |
| Step (2): Put 104.7g (0.34mol) of 2-butyl-3-(4-methoxybenzoyl)benzofuran obtained in step (1) into a reactor equipped with magnetic stirring. 6.5g (34mmol) of boron chloride was dissolved in 2L acetonitrile; the mixture was stirred and reacted at 0°C for 1 hour. After the reaction, the acidic reaction system was neutralized with saturated sodium bicarbonate solution to neutrality; The aqueous phase obtained by extraction; the organic phases are combined and concentrated under reduced pressure; ethyl acetate and ether are recrystallized and separated to obtain 79.4 g (0.27 mol) of the 2-butyl-3-(4-hydroxybenzoyl)benzofuran. 1 H NMR(400MHz, CDCl 3 ,TMS,25℃)δ=10.46(s,1H),7.68(d,J=8.6Hz,2H), 7.62(d,J=8.1Hz,1H), 7.33(dd,J=15.7,7.9Hz, 2H), 7.24 (t, J = 7.4 Hz, 1H), 6.89 (d, J = 8.6 Hz, 2H), 2.80 (t, J = 7.5 Hz, 2H), 1.65 (dt, J = 15.0, 7.5 Hz, 2H),1.23(dd,J=14.7,7.3Hz,2H),0.80(t,J=7.4Hz,3H). 13 C NMR(100MHz,CDCl 3 ,25℃)δ=189.3,163.1,162.2,153.0,131.6,129.7,126.8,124.5,123.6,120.7,116.4,115.3,111.1,29.5,27.1,21.6,13.4ppm. |
| |
| The yield of Example 3 was 54%, wherein the yield of step (1) was 68%, and the yield of step (2) was 79%. |
| Example 4: Preparation of 2-butyl-3-(4-hydroxybenzoyl)benzofuran |
| Step (1): Take 117.1g (0.5mol) of 1-(4-methoxyphenyl)-1,3-heptanedione, and dissolve 112.0g (1.0mol) of acrolein dimer in organic solvent toluene (2L) ), then add 142.9g (0.5mol) of dibromoglycine, AlCl 3 5.8 g (50 mmol), the above reaction liquid was stirred and reacted in a reactor equipped with magnetic stirring at 100°C for 1 hour. After the reaction is completed, the acidic reaction system is neutralized with saturated sodium bicarbonate; the aqueous phase obtained by liquid-liquid extraction with ethyl acetate; the organic phases are combined and concentrated under reduced pressure; ethyl acetate and ether are recrystallized and separated to obtain 2-butane Group-3-(4-methoxybenzoyl)benzofuran 110.9g (0.36mol). 1 H NMR(400MHz, CDCl 3 , TMS, 25℃) δ = 7.84 (d, J = 8.8 Hz, 2H), 7.47 (d, J = 8.2 Hz, 1H), 7.36 (d, J = 7.3 Hz, 1H), 7.26 (dd, J = 9.4,5.9Hz,1H), 7.18(t,J=7.5Hz,1H), 6.96(d,J=8.8Hz,2H), 3.89(s,3H), 2.91(t,J=7.6Hz,2H) ,1.80–1.71(m,2H),1.36(dd,J=15.0,7.4Hz,2H), 0.89ppm(t,J=7.4Hz,3H). 13 C NMR(100MHz,CDCl 3 ,25℃)δ=190.7,164.8,163.6,153.8,132.1,131.8,127.4,124.3,123.4,121.4,116.9,113.8,111.1,55.6,30.3,28.0,22.5,13.8ppm |
| |
| Step (2): Put 110.9g (0.36mol) of 2-butyl-3-(4-methoxybenzoyl)benzofuran obtained in step (1) into a reactor equipped with magnetic stirring. 10.2g (72mmol) of boron diethyl ether was dissolved in 2L acetonitrile; the mixture was stirred and reacted at 80°C for 2 hours. After the reaction, the acidic reaction system was neutralized with saturated sodium bicarbonate solution to neutrality; The aqueous phase obtained by liquid extraction; the organic phases were combined and concentrated under reduced pressure; ethyl acetate and ether were recrystallized and separated to obtain the 2-butyl-3-(4-hydroxybenzoyl)benzofuran 82.3g (0.28mol) . 1 H NMR(400MHz, CDCl 3 ,TMS,25℃)δ=10.46(s,1H),7.68(d,J=8.6Hz,2H), 7.62(d,J=8.1Hz,1H), 7.33(dd,J=15.7,7.9Hz, 2H), 7.24 (t, J = 7.4 Hz, 1H), 6.89 (d, J = 8.6 Hz, 2H), 2.80 (t, J = 7.5 Hz, 2H), 1.65 (dt, J = 15.0, 7.5 Hz, 2H),1.23(dd,J=14.7,7.3Hz,2H),0.80(t,J=7.4Hz,3H). 13 C NMR(100MHz,CDCl 3 ,25℃)δ=189.3,163.1,162.2,153.0,131.6,129.7,126.8,124.5,123.6,120.7,116.4,115.3,111.1,29.5,27.1,21.6,13.4ppm. |
| |
| The yield of Example 4 was 56%, wherein the yield of step (1) was 72%, and the yield of step (2) was 78%. |
| Example 5: Preparation of 2-butyl-3-(4-methylbenzoyl)benzofuran |
| Step (1): Take 117.1g (0.5mol) of 1-(4-methoxyphenyl)-1,3-heptanedione, and dissolve 28g (0.25mol) of acrolein dimer in the organic solvent dichloromethane ( 2L), then add 159.8g (0.5mol) of liquid bromine, ZnCl 2 6.8 g (50 mmol), the above reaction solution was stirred and reacted for 8 hours at 25°C in a reactor equipped with magnetic stirring. After the reaction is completed, the acidic reaction system is neutralized with saturated sodium bicarbonate; the aqueous phase obtained by liquid-liquid extraction with ethyl acetate; the organic phases are combined and concentrated under reduced pressure; ethyl acetate and ether are recrystallized and separated to obtain 2-butane Group-3-(4-methoxybenzoyl)benzofuran 58.5g (0.19mol). 1 H NMR(400MHz, CDCl 3 , TMS, 25℃) δ = 7.84 (d, J = 8.8 Hz, 2H), 7.47 (d, J = 8.2 Hz, 1H), 7.36 (d, J = 7.3 Hz, 1H), 7.26 (dd, J = 9.4,5.9Hz,1H), 7.18(t,J=7.5Hz,1H), 6.96(d,J=8.8Hz,2H), 3.89(s,3H), 2.91(t,J=7.6Hz,2H) ,1.80–1.71(m,2H),1.36(dd,J=15.0,7.4Hz,2H), 0.89ppm(t,J=7.4Hz,3H). 13 C NMR(100MHz,CDCl 3 ,25℃)δ=190.7,164.8,163.6,153.8,132.1,131.8,127.4,124.3,123.4,121.4,116.9,113.8,111.1,55.6,30.3,28.0,22.5,13.8ppm |
| |
| Step (2): Put 58.5g (0.19mol) of 2-butyl-3-(4-methoxybenzoyl)benzofuran obtained in step (1) into a reactor equipped with magnetic stirring. 7.2g (38mmol) of sulfonic acid was dissolved in 2L of toluene; the mixture was stirred and reacted at 100°C for 4 hours. After the reaction, the acidic reaction system was neutralized with saturated sodium bicarbonate solution to neutrality; The aqueous phase obtained by extraction; the organic phases were combined and concentrated under reduced pressure; ethyl acetate and ether were recrystallized and separated to obtain 41.2 g (0.14 mol) of the 2-butyl-3-(4-hydroxybenzoyl)benzofuran. 1 H NMR(400MHz, CDCl 3 ,TMS,25℃)δ=10.46(s,1H),7.68(d,J=8.6Hz,2H), 7.62(d,J=8.1Hz,1H), 7.33(dd,J=15.7,7.9Hz, 2H), 7.24 (t, J = 7.4 Hz, 1H), 6.89 (d, J = 8.6 Hz, 2H), 2.80 (t, J = 7.5 Hz, 2H), 1.65 (dt, J = 15.0, 7.5 Hz, 2H),1.23(dd,J=14.7,7.3Hz,2H),0.80(t,J=7.4Hz,3H). 13 C NMR(100MHz,CDCl 3 ,25℃)δ=189.3,163.1,162.2,153.0,131.6,129.7,126.8,124.5,123.6,120.7,116.4,115.3,111.1,29.5,27.1,21.6,13.4ppm. |
| |
| The yield of Example 5 was 28%, wherein the yield of step (1) was 38%, and the yield of step (2) was 74% |
| In addition to the specific types of halogenated reagents, acid catalysts, and organic solvent raw materials used in the above embodiments, other halogenated reagents, acid catalysts, and organic solvents can also be used; wherein, the halogenated reagent in step (1) is preferably Liquid bromine, elemental iodine, N-iodosuccinimide, N-bromosuccinimide, N-chlorosuccinimide, 1,3-dichloro-5,5-dimethyl Any of hydantoin, dibromohydantoin, bromochlorohydantoin, carbon tetrabromide, and the acid catalyst is boron tribromide, boron trifluoride ether, aluminum chloride, hydrogen fluoride, zinc chloride, zirconium chloride At least one of the organic solvents is dichloromethane, 1,2-dichloroethane, acetonitrile, tetrahydrofuran, ethanol; the acid catalyst in step (2) is boron tribromide, trifluoride One of boron diethyl ether, aluminum chloride, hydrogen fluoride, sulfuric acid, and p-toluenesulfonic acid, and the organic solvent is any of methylene chloride, 1,2-dichloroethane, acetonitrile, tetrahydrofuran, ethanol, and toluene . The specific types of acid catalysts and organic solvents used in steps (1) and (2) of the present invention may be the same or different; in addition, the boiling point of the organic solvent used must be lower than the corresponding treatment temperature. |
| Unless otherwise specified, the various reaction raw materials in the present invention (eg, acrolein dimer, 1-(4-methoxyphenyl)-1,3-heptanedione, etc.) are commercially available The purity is preferably analytically pure. |
PATENT
CN109988131
https://patentscope.wipo.int/search/en/detail.jsf?docId=CN248953969&_cid=P11-KL0BBI-08827-1
| Amiodarone (Amiodarone), also known as amiodarone, was first introduced as a coronary artery dilator, and Rosenbanm was the first to use it in the treatment of anti-arrhythmia in 1976. Amiodarone is extremely toxic. The lethal dose of intravenous injection is 10 times that of the therapeutic dose. The large oral lethal dose is negligible. Long-term larger doses are safe. Amiodarone hydrochloride (ADHC) is the hydrochloride of amiodarone, which was first marketed in Italy in 1984, and its structure is as follows: |
| |
| Amiodarone hydrochloride is a class III antiarrhythmic drug. It is mainly used clinically for supraventricular and ventricular tachyarrhythmias. It is also used for various organic heart diseases and acute coronary syndromes. It can be used as a symptomatic The first-line treatment of atrial fibrillation with left ventricular insufficiency or chronic heart failure. At present, it has become the drug of choice for the prevention of AMI with ventricular tachyarrhythmia, post-infarction ventricular arrhythmia, heart failure with arrhythmia, and sudden cardiac death. |
| In the existing synthetic methods, amiodarone hydrochloride is all obtained by etherification and salt formation with 2-butyl-(4-hydroxy-3,5-diiodobenzoyl)benzofuran as the key intermediate. The structure of this key intermediate is as follows: |
| |
| As a key intermediate of amiodarone hydrochloride, its synthesis is relatively mature. The traditional synthetic route has been applied to industrial production. The route is as follows: |
| |
| This process has the following problems: 1. Long route, complicated steps, and various operations; 2. Low yield and high cost; 3. Many wastes and high waste liquid treatment cost. Therefore, this route is no longer suitable for the current industrial and environmental protection requirements, and more efficient and green methods need to be developed. |
| Patent CN104262304 and CN1858042 improved the above synthesis method and developed a new synthesis method, the route is as follows: |
| |
| This method has been greatly improved compared with the earlier process, but the yield and purity of 2-butylbenzofuran prepared by the route method are low, which leads to a greatly reduced yield and purity of amiodarone hydrochloride. CN107382925 continues to improve the above process, and the purity and yield have increased, but column chromatography purification is required, which is not suitable for industrial large-scale production. |
| Example 1: |
| 1. Preparation of intermediate 2-butylbenzofuran: |
| |
| Put 5.5 g K 2 CO 3 , 076 g of CuI and 0.74 g of TBAI were added to a 100 ml reaction flask containing 30 ml of toluene, and 4.4 g of 2-iodophenol, 2.5 ml of 1-hexyne and 60 mg of nickel catalyst were added to it, and replaced with nitrogen 3 Secondly, the reaction was kept at 50°C and stirred for 18-22 hours under nitrogen protection. The reaction solution was filtered, and the filtrate was washed with 40 ml of 5% NaOH aqueous solution and twice with 40 ml of water. The filtrate was concentrated under reduced pressure to obtain 3.12 g of a dark yellow solid, namely 2-butylbenzofuran, with a yield of 89.7%. |
| 2. Synthesis of intermediate 2-butyl-(4-methoxybenzoyl)benzofuran: |
| |
| Add 5.50 g of aluminum trichloride and 24 ml of dichloromethane to a 100 ml reaction flask, stir and lower the temperature to 0°C, and add 7.0 g of p-methoxybenzoyl chloride dropwise to it within 5°C, and keep warm after dropping. Stir for 1 hour. Dissolve 6.00 g of 2-butylbenzofuran in 24 ml of dichloromethane and add dropwise to the above reaction solution within 5°C of temperature control. After dropping, slowly raise the temperature to 25°C, keep the temperature for 2 hours, and complete the reaction. After cooling to room temperature, it was poured into ice water for separation, the aqueous phase was extracted with dichloromethane, the organic layers were combined, washed twice with water, and the organic phase was concentrated under reduced pressure to obtain 10.60 g of oil. |
| 3. Synthesis of 2-butyl-(4-hydroxybenzoyl)benzofuran: |
| |
| Add 10.00 g of 2-butyl-(4-methoxybenzoyl) benzofuran, 4.50 g of aluminum trichloride and 40 ml of toluene into a 100 ml reaction flask. The temperature is raised to reflux, and the reaction is kept warm for 6 hours. The reaction is complete . Cool down to 0℃, pour into ice water, separate the liquids, extract the aqueous phase with toluene, combine the organic phases, add equal volumes of water, adjust the pH to above 12 with NaOH solution, separate the liquids, adjust the pH to less than 3 with hydrochloric acid for the aqueous phase , Filtered, and the filter cake was vacuum dried to obtain 8.16 g of light yellow solid. |
| 4. Synthesis of 2-butyl-(4-hydroxy-3,5-diiodobenzoyl)benzofuran: |
| |
| Add 8 g of 2-butyl-(4-hydroxybenzoyl) benzofuran, 15.18 g of iodine, 8.26 g of potassium carbonate and 48 ml of ethanol to a 100 ml reaction flask, and heat to reflux with stirring, and keep the reaction temperature for 2 hours. The reaction is over. The temperature was lowered to room temperature, filtered, the filtrate was added dropwise to the sodium metabisulfite aqueous solution, after dripping, the mixture was kept and stirred for 0.5 hours, filtered, the filter cake was washed twice with water, and the solid was vacuum dried to obtain 14.61 g of off-white solid. |
| 5. Synthesis of 2-butyl-[4-[2-(diethylamino)hydroxyethyl]-3,5-diiodobenzoyl]benzofuran: |
| |
| Add 12.00 g of 2-butyl-(4-hydroxy-3,5-diiodobenzoyl) benzofuran and 120 ml of toluene into a 250 ml reaction flask. The temperature is raised to 60°C. After the solid is dissolved, add to it 4.94 g of 2-diethylaminochloroethane hydrochloride, 5.65 g of potassium carbonate and 8.50 g of water were heated to reflux, stirred and reacted for 8 hours, and the reaction was completed. The reaction solution was washed 3 times with water, 0.60 g activated carbon was added to the organic phase, the temperature was raised to reflux and stirred for 1 hour, and then filtered with suction. The filtrate was concentrated under reduced pressure until solids began to precipitate, and the temperature was reduced to 0°C for crystallization. The cold toluene was washed twice, and the solid was vacuum-dried at 80°C to obtain 13.97 g of a finished white solid, which was the finished product of amiodarone hydrochloride. |
| Compared with the existing production process, the preparation method of amiodarone hydrochloride in this embodiment simplifies the operation and improves the convenience of operation and the stability of the product. Through the control of the catalyst and material ratio, the purity and yield of each intermediate are improved, and the product does not require column chromatography to purify, which saves costs and improves production efficiency, which provides convenience for industrial large-scale production. |
| Example 2: |
| 1. Preparation of intermediate 2-butylbenzofuran: |
| Put 5.5 g K 2 CO 3 , 0.76 g of CuI and 0.74 g of TBAI were added to a 100 ml reaction flask containing 30 ml of toluene, and 4.4 g of 2-iodophenol, 2.5 ml of 1-hexyne and 190 mg of ruthenium catalyst were added to it, and replaced with nitrogen 3 Secondly, the reaction was kept at 50°C and stirred for 22-28 hours under nitrogen protection. The reaction solution was filtered, and the filtrate was washed with 40 ml of 5% NaOH aqueous solution and 40 ml of water twice. The filtrate was concentrated under reduced pressure to obtain 3.25 g of a dark yellow solid, which was 2-butylbenzofuran, with a yield of 93.4%. |
| The remaining steps are the same as in Example 1. |
| Example 3: |
| 1. Preparation of intermediate 2-butylbenzofuran: |
| Put 5.5 g K 2 CO 3 , 0.76 g of CuI and 0.74 g of TBAI were added to a 100 ml reaction flask containing 30 ml of toluene, and 4.4 g of 2-iodophenol, 2.5 ml of 1-hexyne and 89 mg of palladium catalyst were added to it, and replaced with nitrogen 3 Secondly, the reaction was kept at 40°C under nitrogen and stirred for 26-30 hours. The reaction solution was filtered, and the filtrate was washed with 40 ml of 5% NaOH aqueous solution and 40 ml of water twice. The filtrate was concentrated under reduced pressure to obtain 3.36 g of a dark yellow solid, which was 2-butylbenzofuran, with a yield of 96.6%. |
| The remaining steps are the same as in Example 1. |
| Example 4: |
| 1. Preparation of intermediate 2-butylbenzofuran: |
| Put 5.5 g K 2 CO 3 , 0.76 g of CuI and 0.74 g of TBAI were added to a 100 ml reaction flask containing 30 ml of toluene, and 4.4 g of 2-iodophenol, 2.5 ml of 1-hexyne and 100 mg of rhodium catalyst were added to it, and replaced with nitrogen 3 Next, the reaction was kept at 40°C under nitrogen protection and stirred for 20-24 hours. The reaction solution was filtered, and the filtrate was washed with 40 ml of 5% NaOH aqueous solution and 40 ml of water twice. The filtrate was concentrated under reduced pressure to obtain 2.93 g of dark yellow solid, which is 2-butylbenzofuran, with a yield of 84.2%. |
| The remaining steps are the same as in Example 1. |
| Example 5: |
| 1. Preparation of intermediate 2-butylbenzofuran: |
| Put 5.5 g K 2 CO 3 , 0.76 g of CuI and 0.74 g of TBAI were added to a 100 ml reaction flask containing 30 ml of toluene, and 4.4 g of 2-iodophenol, 2.5 ml of 1-hexyne and 80 mg of gold catalyst were added to it, and replaced with nitrogen 3 Secondly, the reaction was kept at 40°C and stirred for 20-28 hours under nitrogen protection. The reaction solution was filtered, and the filtrate was washed with 40 ml of 5% NaOH aqueous solution and 40 ml of water twice. The filtrate was concentrated under reduced pressure to obtain 3.07 g of dark yellow solid, which is 2-butylbenzofuran, with a yield of 88.2%. |
| The remaining steps are the same as in Example 1. |
?///////////
Mitsubishi Tanabe And EnVivo in Phase III Trial Of Alzheimer’s Disease Treatment MT-4666

OR
Encenicline (EVP-6124, MT-4666)
EVP-6124 , MT-4666, α7-nAChR agonist, UNII-5FI5376A0X
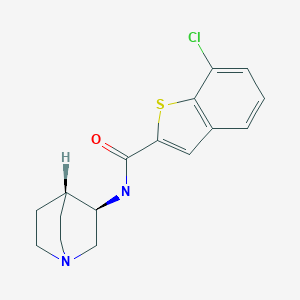
Chemical Name: (R)-7-chloro-N-quinuclidin-3-yl)benzo[b]thiophene-2-carboxamide
Therapy Type: Small Molecule
Target Type: Cholinergic System
CAS : 550999-75-2
- C16 H17 Cl N2 O S
- Benzo[b]thiophene-2-carboxamide, N-(3R)-1-azabicyclo[2.2.2]oct-3-yl-7-chloro-
- (R)-7-Chloro-N-(quinuclidin-3-yl)benzo[b]thiophene-2-carboxamide; EVP 6124
Condition(s): Alzheimer’s Disease, Schizophrenia
U.S. FDA Status: Alzheimer’s Disease (Phase 3), Schizophrenia (Phase 3)
Status in Select Countries: Investigational in Japan
Company: FORUM Pharmaceuticals Inc. (was EnVivo Pharmaceuticals), Mitsubishi Tanabe Pharma
Approved for: None AS ON SEPT 2014

CAS 550999-74-1
Benzo[b]thiophene-2-carboxamide, N-(3R)-1-azabicyclo[2.2.2]oct-3-yl-7-chloro-, monohydrochloride
(R)-7-Chloro-N-(quinuclidin-3-yl)benzo[b]thiophene-2-carboxamide hydrochloride
Mitsubishi Tanabe Pharma ..Encenicline-hydrochloride (EVP-6124) for Alzheimer’s disease by partner EnVivo Pharmaceuticals. Mitsubishi Tanabe has licensed EVP-6124 from EnVivo and is currently developing the drug under the code MT-4666.
The drug is a new alpha-7 potentiator intended to improve cognition in patients affected with Alzheimer’s disease. The drug is being tested in Phase III COGNITIV clinical trials in two categories: COGNITIV AD in patients with Alzheimer’s disease and COGNITIV CIAS in patients with cognitive impairment associated with schizophrenia.
Alzheimer’s disease affects five million people in the U.S. alone, or one in eight Americans over the age of 65. The disease is the sixth-leading cause of death in the country, with the number of affected patients expected to balloon to nearly triple by 2030. Alzheimer’s disease is a complex neurodegenerative disease that eventually leads to cellular loss and dysfunction in the brain resulting in decline of language skills and reasoning among others.
Phase III of COGNITIV AD clinical trial program involves about 1,600 patients with mild to moderate AD and who are presently receiving stable treatment with or have undergone previous acetylcholinesterase inhibitor treatment. The trials will be placebo-controlled, double-blind, and randomized. Patients in the trial will be randomized to receive either one of two doses of MT-4666 once daily against a placebo to assess safety and efficacy of the drug.
In the news release recently launched by EnVivo, CEO and president Deborah Dunsire said, “We are pleased to advance encenicline into Phase 3 clinical development in Alzheimer’s disease, a significant milestone for our company and promising step forward for patients who desperately need new therapies…Prior clinical studies of encenicline have demonstrated clinically significant improvements in cognitive function in patients with Alzheimer’s disease. For the millions of patients living with AD, we believe encenicline has the potential to make a meaningful difference.”
Encenicline hydrochloride is a partial, selective agonist of the α-7 nicotinic acetylcholine receptor (α7-nAChR). It is being developed for the treatment of cognitive deficits in schizophrenia and Alzheimer’s disease. Cholinergic function declines in Alzheimer’s, and currently approved acetylcholinesterase inhibitor therapies modestly improve cognitive deficits in patients with AD by way of boosting cholinergic transmission. The rationale of selective α7-nAChR agonists is that they will enhance cognition without causing side effects associated with overactivation of other nAChRs such as α4β2, or muscarinic AChRs. In rats, encenicline penetrates the blood-brain barrier and improves memory performance by potentiating the acetylcholine response. Encenicline has been reported to act as a co-agonist with acetylcholine. It sensitizes the α-7 nACh receptor to its natural ligand and renders sub-efficacious doses of AChEI drugs effective in restoring memory function in an object recognition task (Prickaerts et al., 2012).
This compound was originally developed at Bayer Healthcare and then licensed to Envivo Pharmaceuticals, which subsequently licensed development in Asia to Mitsubishi Tanabe Pharma Corporation. Envivo then changed its name to FORUM Pharmaceuticals Inc.
Encenicline is being tested in Alzheimer’s disease and schizophrenia. In Alzheimer’s, an ascending-dose Phase 1/2 study showed 0.1 to 1 mg/day of EVP-6124 to be safe and well-tolerated when given to 49 people with mild to moderate AD for 28 days. No serious side effects were reported. Secondary efficacy endpoints suggested that EVP-6124 given in addition to therapy with the acetyl cholinesterase inhibitors donepezil or rivastigmine appeared to improve attention, verbal fluency, and executive function as measured on tests in the CogState or NTB batteries (see conference news story). This study has posted results on clinicaltrials.gov.
A 24-week Phase 2 trial conducted in 409 people with mild to moderate Alzheimer’s disease in the United States and Eastern Europe compared 0.3, 1, and 2 mg of EVP-6124 per day to placebo, measuring cognition with ADAS-Cog as the primary outcome plus cognitive, functional, and psychiaric secondary outcomes. EVP 6124 was given as adjunct therapy to donepezil or rivastigmine. This trial was reported to have met its primary and most secondary endpoints, showing that people on the highest dose improved over baseline. EVP-6124 dose-dependently improved measures of attention, verbal and language fluency, and executive function. In this trial, all treatment groups initially improved, possibly due to a placebo effect, but by 12 weeks the groups separated and the placebo and low-dose groups declined (see conference news story). EVP-6124 was well-tolerated.
Mitsubishi Tanabe Pharma Corporation is conducting a Phase 2 trial for the treatment of Alzheimer’s disease in Japan.
In October 2013, two international Phase 3 trials began enrolling what are to be 790 patients in each trial with mild to moderate Alzheimer’s who are already taking an acetylcholinesterase inhibitor. The trials will compare two fixed, undisclosed add-on doses of EVP-6124 to placebo, all given as once-daily tablets for six months, for cognitive benefit as measured by the ADAS-Cog, clinical benefit as measured by the Clinical Dementia Rating Sum of Boxes (CDR-SB), as well as for safety and tolerability. Called COGNITIV AD, this Phase 3 program is is set to run through 2016.
For schizophrenia, a Phase 1 study comparing 0.3 and 1 mg/day of EVP-6124 to placebo in 28 people with the disease gave preliminary evidence for the compound’s safety, tolerability, and pharmacokinetics in this population. In addition, the compound yielded signals of bioactivity in the brain by way of EEG tests of evoked potentials, a measure of sensory gating affected in this disease. See study results on clinicaltrials.gov.
A subsequent 12-week Phase 2 trial compared 0.3 and 1 mg/day of EVP-6124 to placebo in 317 people with schizophrenia and measured safety and the compound’s efficacy on cognitive function. As presented at the American College of Neuropsychopharmacology meeting held in Hawaii December 2011, EVP 6124 met its primary endpoint of improvement on the CogState overall cognition index. The study also met secondary endpoints, showing improvement in clinical function as assessed by the Schizophrenia Cognition Rating Scale, and a decrease in negative symptoms (See company press release).
Two six-month, 700-patient Phase 3 studies, plus a six-month extention study, are ongoing. For all clinical trials of encenicline, see clincialtrials.gov.
http://www.google.com.ar/patents/WO2014051055A1?cl=pt-PT
Synthesis (hereinafter, the compound of Reference Example 25) carboxamide hydrochloride (Reference Example 25) (R) -7 – chloro-N-(quinuclidin-3 – – yl) benzo [b] thiophene-2:
[First Step]
Synthesis of carboxamide (R) -7 – chloro-N-(quinuclidin-3 – – yl) benzo [b] thiophene-2:
-N, N, N ‘, N’-tetra-7 – chloro-1 – benzothiophene -2 – – o-(yl benzotriazol-1) chloroform solution (210mg, 1.0mmol) of carboxylic acid in (10mL) was added (0.70mL, 4.0mmol) and (570mg, 1.5mmol), diisopropylethylamine methyl hexafluorophosphate, (R) – (200mg, 1.0mmol) amine hydrochloride – quinuclidine-3 was added, and the mixture was stirred at room temperature. 16 hours later, was added distilled water, 1.0N sodium hydroxide solution, and extracted with chloroform. Was washed with saturated brine and the organic layer was concentrated and then dried over anhydrous sodium sulfate. (Fuji Silysia Chemical amine silica gel DM1020, chloroform alone – chloroform / methanol = 90/10) on silica gel column chromatography of the crude product obtained was purified by the title compound; was obtained as a white solid (170mg 53%).
1 H-NMR (400MHz, DMSO-d 6)
δ :1.22-1 .38 (1H, m) ,1.53-1 .62 (2H, m) ,1.75-1 .82 (2H, m) ,2.63-2 .73 (4H , m) ,2.84-2 .94 (1H, m) ,3.07-3 .18 (1H, m) ,3.90-4 .00 (1H, m), 7.49 (1H, dd , J = 7.6,8.0 Hz), 7.59 (1H, d, J = 7.6Hz), 7.96 (1H, d, J = 8.0Hz), 8.31 (1H, s) ,8.62-8 .66 (1H, m).
MS (ESI): 321 [M + H] +
[Second Step]
Synthesis of the compound of Reference Example 25:

Ethyl acetate solution – solution of hydrogen chloride in ethyl acetate (170mg, 0.53mmol) of the (2.0mL) carboxamide – (R) -7 – chloro-N-(quinuclidin-3 – yl) benzo [b] thiophene-2 was added (4.0M, 0.20mL, 0.80mmol), and the mixture was stirred at room temperature. 10 minutes later, by which is filtered off and the resulting solid was washed with ethyl acetate and hexane, and dried, the compound of Reference Example 25; was obtained as a white solid (170mg 90%).
1 H-NMR (400MHz, DMSO-d 6)
δ :1.70-1 .78 (1H, m) ,1.86-1 .94 (2H, m) ,2.10-2 .19 (2H, m) ,3.18-3 .35 (5H , m) ,3.63-3 .72 (1H, m) ,4.27-4 .36 (1H, m), 7.50 (1H, d, J = 7.6,8.0 Hz), 7 .61 (1H, d, J = 7.6Hz), 7.98 (1H, d, J = 8.0Hz), 8.38 (1H, s) ,9.07-9 .10 (1H, m) ,9.80-9 .85 (1H, m).
MS (ESI): 321 [M + H] +
………………………………….
http://www.google.com/patents/EP1461335A1?cl=en
Example 69
N-[(3 R) – 1 – azabicyclo [2.2.2] oct-3-y 1]-7-chloro-1-benzothiophene-2-carboxamide hydrochloride DESIRED

x HCI
176.2 mg (0.83 mmol) of 7-chloro-l-benzothiophene-2-carboxylic acid, 150 mg (0.75 mmol)
R-3-Aminochinuklidin dihydrochloride, 343.7 mg (0.90 mmol) of HATU, 350.5 mg
(2.71 mmol) of N, N-diisopropylethylamine and 3.0 ml of DMF are reacted according to the general working procedure (variant B). The reaction mixture is purified by preparative HPLC. The product will be in a mixture of 4 M HCl solution in dioxane and methanol, and then concentrated. This gives 175.2 mg
(65.1% of theory) of the title compound.
1H NMR (200 MHz, DMSO-d 6): δ – 10.03 (s, IH, br), 9.17 (d, IH), 8.43 (s, IH), 7.98 (m, IH), 7.63 (m, IH ), 7.52 (dd, IH), 4.33 (m, IH), 3.77-3.10 (m, 6H), 2.28-
2.02 (m, 2H), 1.92 (m, 2H), 1.75 (m, IH) ppm.
HPLC: R t = 4.0 min (Method H)
MS (ESIpos): m / z = 321 (M + H) + (free base).
Example 70
N-[(3 S) – 1-azabicyclo [2.2.2] oct-3-yl]-7-chloro-1-benzothiophene-2-carboxamide hydrochloride UNDESIRED

x HCI
176.2 mg (0.83 mmol) of 7-chloro-l-benzothiophene-2-carboxylic acid, 150 mg (0.75 mmol) of S-3-Aminochinuklidin dihydrochloride, 343.7 mg (0.90 mmol) of HATU, 350.5 mg (2.71 mmol) of N, N- diisopropylethylamine and 3.0 ml of DMF are implemented according to the general procedure (Method B). The reaction mixture is purified by preparative HPLC. The product will be in a mixture of 4 M HCl solution in dioxane and methanol, and then concentrated. Obtained 231.9 mg (85.7% of theory) of the title compound. The analytical data are consistent with those of the enantiomeric compound from Example 69.
………………………………………
http://www.google.com.ar/patents/EP2727604A1?cl=en
(Reference Example 3)
Synthesis of (R)-7-Chloro-N-(quinuclidin-3-yl)benzo[b]thiophene-2-carboxamide hydrochloride (hereinafter referred to as the compound of Reference Example 3):
[First step]Synthesis of 7-Chloro-1-benzothiophene-2-carboxylic acid:

[Second step]Synthesis of (R)-7-Chloro-N-(quinuclidine-3-yl)benzo[b]thiophene-2-carboxamide:

1H-NMR (400 MHz, DMSO-d6)
δ: 1.22-1.38 (1H, m), 1.53-1.62 (2H, m), 1.75-1.82 (2H, m), 2.63-2.73 (4H, m), 2.84-2.94 (1H, m), 3.07-3.18 (1H, m), 3.90-4.00 (1H, m), 7.49 (1H, dd, J=7.6, 8.0 Hz), 7.59 (1H, d, J=7.6 Hz), 7.96 (1H, d, J=8.0 Hz), 8.31 (1H, s), 8.62-8.66 (1H, m).
MS (ESI) [M+H]+ 321
[Third step]Synthesis of Compound of Reference Example 3:
1H-NMR (400 MHz, DMSO-d6)
δ: 1.70-1.78 (1H, m), 1.86-1.94 (2H, m), 2.10-2.19 (2H, m), 3.18-3.35 (5H, m), 3.63-3.72 (1H, m), 4.27-4.36 (1H, m), 7.50 (1H, d, J=7.6, 8.0 Hz), 7.61 (1H, d, J=7.6 Hz), 7.98 (1H, d, J=8.0 Hz), 8.38 (1H, s), 9.07-9.10 (1H, m), 9.80-9.85 (1H, m).
MS (ESI) [M+H]+
321
| WO1991012254A1 * | 15 Feb 1991 | 17 Aug 1991 | Novo Nordisk As | Substituted urea compounds and their preparation and use |
| WO2004069141A2 * | 5 Feb 2004 | 19 Aug 2004 | Strakan Ltd | Transdermal granisetron |
| WO2004076449A2 * | 20 Feb 2004 | 10 Sep 2004 | Jozef Klucik | 3-substituted-2(arylalkyl)-1-azabicycloalkanes and methods of use thereof |
| WO2008019372A2 * | 7 Aug 2007 | 14 Feb 2008 | Amr Technology Inc | 2-aminobenzoxazole carboxamides as 5ht3 modulators |
| WO2008096870A1 * | 8 Feb 2008 | 14 Aug 2008 | Astellas Pharma Inc | Aza-bridged-ring compound |
| JPH0881374A * | Title not available |
|
MW: 357.3032 |
||
| 2 |
MW: 320.8423 |
|
| 3 |
MW: 375.318 |
Quarfloxin, Itarnafloxin , CX-3543….Inhibits rRNA biogenesis.

Quarfloxin, Itarnafloxin
CAS: 865311-47-3.
Chemical Formula: C35H33FN6O3
Exact Mass: 604.25982
Molecular Weight: 604.67
Elemental Analysis: C, 69.52; H, 5.50; F, 3.14; N, 13.90; O, 7.94
Synonym: CX 3543; CX3543; CX-3543; QuarfloxacinTA1-1B
- CX 3543
- CX-3543
- Itarnafloxin
- Quarfloxacin
- Quarfloxin
- UNII-8M31J5031Q
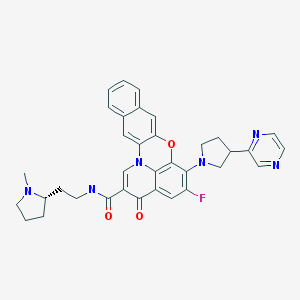
IUPAC/Chemical name:
5-fluoro-N-(2-((S)-1-methylpyrrolidin-2-yl)ethyl)-3-oxo-6-((R)-3-(pyrazin-2-yl)pyrrolidin-1-yl)-3H-benzo[b]pyrido[3,2,1-kl]phenoxazine-2-carboxamide.
-
5-Fluoro-N-(2-((2S)-1-methylpyrrolidin-2-yl)ethyl)-3-oxo-6-(3-(pyrazin-2- yl)pyrrolidin-1-yl)-3H-benzo(b)pyrido(3,2,1-kl)phenoxazine-2-carboxamide
-
3H-Benzo(b)pyrido(3,2,1-kl)phenoxazine-2-carboxamide, 5-fluoro-N-(2-((2S)- 1-methyl-2-pyrrolidinyl)ethyl)-3-oxo-6-(3-pyrazinyl-1-pyrrolidinyl)-
Quarfloxin, also known as Quarfloxacin and CX-3543, is a fluoroquinolone derivative with antineoplastic activity. Quarfloxin disrupts the interaction between the nucleolin protein and a G-quadruplex DNA structure in the ribosomal DNA (rDNA) template, a critical interaction for rRNA biogenesis that is overexpressed in cancer cells; disruption of this G-quadruplex DNA:protein interaction in aberrant rRNA biogenesis may result in the inhibition of ribosome synthesis and tumor cell apoptosis.
CX-3543, developed at Cylene Pharmaceuticals, is a multi-targeting oncogene inhibitor evaluated in phase II clinical studies for the treatment of low or intermediate grade neuroendocrine carcinoma, including carcinoid and islet cell cancer. In 2008, a trial for the treatment of chronic lymphocytic leukemia (CLL) was withdrawn prior to patient enrollment. In 2010, phase I clinical studies for the treatment of solid tumors and for the treatment of lymphoma were terminated upon observation that the modified dose schedule presented no advantage over previously studies schedule solid tumors.
CX-3543 was developed using the company’s Quadruplex Targeting technology which is based on quadruplex motifs in genomic DNA that regulate the expression of clusters of key oncogenes but not normal cellular genes. In 2013, the product was licensed to TetraGene by Cylene Pharmaceuticals on an exclusive, worldwide basis for development for the treatment of cancer. Cylene ceased operations in 2013.
Current developer: Cylene Pharmaceuticals Inc. phase 2
Clinical trial news: Quarfloxin is a ground-breaking small-molecule targeted cancer therapeutic derived from the validated fluoroquinolone class of drugs. Rationally designed to selectively inhibit ribosomal RNA (rRNA) biogenesis in cancer cells, quarfloxin disrupts the interaction between the Nucleolin protein and a G-quadruplex DNA structure in the ribosomal DNA (rDNA) template, a critical interaction for rRNA biogenesis and one that is amplified in cancer cells. As a result, quarfloxin selectively induces apoptotic cell death in cancers. Many commercialized cancer therapeutics act indirectly on rRNA Biogenesis through upstream modulators, but quarfloxin is the first agent to directly target this cancer-specific aberrant cell function. According to news released on June 19, 2011, Cylene Pharmaceuticals announced the initiation of a Phase II clinical trial of quarfloxin (CX-3543) in patients with carcinoid/neuroendocrine tumors (C/NET), which are malignant cancers arising from neural crest cells.
Cylene Pharmaceuticals today announced the initiation of a Phase II clinical trial of quarfloxin (CX-3543) in patients with carcinoid/neuroendocrine tumors (C/NET), which are malignant cancers arising from neural crest cells.
“Quarfloxin (CX-3543) is a small molecule that disrupts a protein:rDNA complex that forms in the abnormal nucleoli of cancer cells, thereby selectively inducing apoptotic cell death in cancers,” said Dr. William Rice, President and Chief Executive Officer of Cylene Pharmaceuticals. “Many commercialized cancer therapeutics act on or through the nucleolus, but quarfloxin is the first agent designed to directly target a key function within the nucleolus. Quarfloxin has been well tolerated in humans and has demonstrated signs of biological benefit for patients with C/NET in Phase I clinical trials. Moreover, biodistribution studies revealed that quarfloxin accumulates in the tissues in which C/NET arise.”
In this open-label Phase II trial, quarfloxin will be administered to patients with low or intermediate grade C/NET, including those receiving concomitant treatment with a stable dose of octreotide. This multi-centered study will include an assessment of improvements in patients’ symptoms and biochemical markers, in addition to RECIST tumor response measurements. The first patient was enrolled and treated at Front Range Cancer Specialists in Fort Collins, CO under the care of Robert Marschke Jr., M.D. This study is expected to enroll up to 25 patients at several leading cancer centers.
“The initiation of this Phase II trial with quarfloxin is a major milestone for Cylene, but more importantly, we hope that quarfloxin will be an effective treatment for cancer patients with limited therapeutic alternatives,” added Dr. Daniel Von Hoff, Cylene’s Co-Founder and Vice President, Medical Affairs. “Quarfloxin has demonstrated potent in vivo efficacy against a broad range of tumors and a considerable therapeutic window in preclinical antitumor models, and has a unique profile of concentrating in neural crest tissues. For these reasons, we are enthusiastic about offering a Phase II clinical trial for patients with carcinoid/neuroendocrine tumors.”
About Quarfloxin (CX-3543), a Nucleolus Targeting Agent (NTA)
Quarfloxin is a ground-breaking small-molecule targeted cancer therapeutic derived from the validated fluoroquinolone class of drugs. Rationally designed to selectively inhibit ribosomal RNA (rRNA) biogenesis in cancer cells, quarfloxin disrupts the interaction between the Nucleolin protein and a G-quadruplex DNA structure in the ribosomal DNA (rDNA) template, a critical interaction for rRNA biogenesis and one that is amplified in cancer cells. As a result, quarfloxin selectively induces apoptotic cell death in cancers. Many commercialized cancer therapeutics act indirectly on rRNA Biogenesis through upstream modulators, but quarfloxin is the first agent to directly target this cancer-specific aberrant cell function.
About Cylene Pharmaceuticals, Inc.
Cylene Pharmaceuticals is a biotech pharmaceutical company dedicated to the discovery, development and commercialization of targeted small-molecule drugs to treat life-threatening cancers. Cylene has created a diverse portfolio of product candidates, including novel inhibitors of cancer-linked serine/threonine kinases, as well as innovative Nucleolus Targeting Agents (NTAs) that target the abnormal nucleolus functions of cancer cells and selectively kill cancer cells. More information can be found athttp://www.cylenepharma.com.
………………………………………………………..
http://www.google.com/patents/US20060029950
To a series of solutions of the fluoroacid (0.5 mmol) in NMP (3.6 mL) was added the amines NHR1R2 (0.5-2.0 mmol) at room temperature. The vessels were sealed and heated on a 90° C. hotplate with constant stirring for 1-2 hours until the reactions were determined to be complete by HPLC/MS analysis. The reaction mixtures were allowed to cool to room temperature and water was added (20 mL). The resulting precipitates were collected by vacuum filtration and dried under vacuum. In cases where 1.0 equivalent of amine was used, the resulting reaction mixtures were used in the next step “as is.” The resulting solids or solutions were treated with HBTU (2.5 eq.) and DIEA in 3.6 mL NMP and allowed to stir for 30 minutes at room temperature under an inert atmosphere. These solutions were added to a series of amines NHR3R4 (2.5 equivalents) in a 96 well format (Whatman Uniplate, 2 mL) and allowed to react for 2 hours. Methanol was then added (50-100 μL) and the plate was filtered (Whatman Unifilter Polypropylene). The resulting liquids were directly chromatographed on reverse HPLC (Waters Xterra 19×50 mm) with mass directed collection (Micromass ZQ, Waters FCII). The fractions were analyzed for purity (MS TIC, UV) and dried by vacuum evaporation (Savant) with an average yield of 5-10 mg). Examples of substituted quinobenzoxazines analogs are described in Table 1.
Example 48Synthesis of CX-3092 and CX-3543
One method for synthesizing CX-3543 is shown below. As shown in Scheme 2, CX-3543 is synthesized in a convergent manner, assembling the substructures 1, 1A and 2A in the final two synthetic steps (Scheme 2), to form CX-3543 having a 50:50 ratio of RS and SS isomers. CX-3092 may be synthesized in a similar manner using a non-chiral form of 1A.


In more detail, pyrazinopyrrolidine 1A is synthesized via a [3+2] cycloaddition chemistry. Conversion of L-proline 7 to cyano-1-aminopyrrolidine 8 without loss of stereochemistry, followed by reduction provides the chiral 2-aminoethyl-1-methylpyrrolidine 2A in high yield. CX-3543 was found to have a formulated solubility of approximately 20 mg/mL.
Example 70This example describes a method for preparing a substituted benzoxazine analog from reaction of the corresponding ester with an amine, and aluminum chloride.

To a solution of 2,3,4,5-tetrafluorobenzoic acid (100 g, 510 mmol), in methylene chloride (0.5 L) was added oxalyl chloride (68 g, 540 mmol) and DMF (ca 3 drops) and the reaction mixture was allowed to stir at room temperature overnight allowing for the produced gasses to escape. The solvent was removed in vacuo and the vessel was placed on high vacuum (ca 0.5 mm Hg) for 2 hours to afford the acid chloride as a viscous oil (105 g) and was used in the subsequent reaction without further purification.

To a suspension of potassium ethyl malonate (97 g, 570 mmol) and magnesium chloride (55 g, 570 mmol) in acetonitrile and the suspension was chilled to 0° C. To this suspension was added the crude 2,3,4,5-benzoyl chloride (105 g, 520 mmol) over 5 minutes. Triethylamine was slowly added at a rate sufficient to keep the reaction temperature below 10° C. and the mixture was allowed to warm to room temperature and was stirred overnight. The solvent was removed in vacuo and replaced with toluene (300 mL) and 1N HCl (500 mL) was added and the mixture was allowed to stir for 1 hour. The organic layer was separated and washed with 1N HCl (100 mL) and brine (100 mL) and dried over sodium sulfate, filtering over a pad of silica gel (50×100 mm), eluting with ethyl acetate. The solvent was removed in vacuo and the resulting oil was dissolved in ethanol/water (9:1) and was allowed to crystallize overnight. The resulting crystals were Isolated by filtration, washing with ethanol/water (8:2) to afford the ketoester (43.75 g, 166 mmol) as a white crystalline solid.

To a 250 mL round bottom flask was added the tetrafluoroketoester (10.0 g, 37.9 mmol), triethylorthoformate (8.6 mL, 56.8 mmol) and acetic anhydride (7.15 mL, 75.8 mmol) and the reaction mixture was heated to 145° C. for 2 hours. The reaction was allowed to cool to room temperature and placed on high vacuum (ca 0.5 mm Hg) for 1 hour. The resulting oil was dissolved in ethanol (100 mL) and 2-amino-1-naphthol (6.02 g, 37.9 mmol) was added at room temperature and the solution became briefly clear and then product began to precipitate. The reaction was allowed to stir for 2 hours and was then filtered and washed with ethanol (100 mL) to afford the enamine as a yellow solid (12.5 g, 28.9 mmol).

To a solution of the enamine (12.13 g, 27.95 mmol) in dry DMF (50 mL) was added potassium carbonate (4.24 g, 1.1 eq.) and the mixture was heated to 90° C., with constant stirring, for 2 hours. The mixture was allowed to cool to room temperature without stirring and was allowed to remain at room temperature for an additional hour. The crystalline solid was collected by filtration, washing with water. Recrystallization from THF afforded the difluoroester as a white crystalline solid (9.3 g, 23.6 mmol).

To a solution of the difluoroester (1.0 g, 2.5 mmol) in NMP (10 mL) was added N-Boc-3-(2-pyrazino)pyrrolidine (870 mg, 3.5 mmol) and the mixture was heated to reflux for 3 hours. The reaction mixture was then allowed to cool to room temperature and the product was collected by filtration. Crystallization from THF afforded the pyrazine ester as a yellow solid (910 mg, 1.74 mmol).

To a solution of the pyrazine ester (250 mg, 0.48 mmol) and 2-(2-aminoethyl)-1-methylpyrrolidine (80 mg, 0.63 mmol) in methylene chloride at room temperature was added aluminum chloride (83 mg, 0.63 mmol) and the reaction mixture was allowed to stir for 2 hours. The solvent was removed in vacuo and saturated L-tartaric acid was added (5 mL) and the mixture was allowed to stir for 1 hour. Methylene chloride (10 mL) was then added and the mixture was basified with 1N NaOH. The organic layer was separated and washed with a saturated solution of Rochelle’s salt, brine and dried over sodium sulfate. The solvent was removed in vacuo and the resulting solid was dissolved in THF and filtered and the solvent was removed again. The crude solid was recrystallized in ethyl acetate to afford the amide as a yellow solid (225 mg, 0.37 mmol, 98.5% pure).
Example 71
This example describes a method for preparing a substituted benzoxazine analog from reaction of the corresponding carboxylic acid with an amine, and aluminum chloride.

The pyrazinoester (2.0 g, 3.8 mmol) was dissolved in ethanol (100 mL) and conc HCl was added (20 mL) and the mixture was refluxed overnight. The mixture was allowed to cool to room temperature and the solid was collected by vacuum filtration, washing with ethanol to afford the pyrazinoacid as a light tan powder (1.6 g, 3.2 mmol).

To a mixture of the fluoroaminoacid (1.6 g, 3.2 mmol) and HBTU (2.0 g, 5.3 mmol) in NMP (20 mL) was added N,N-diisopropyl-N-ethylamine (1.0 mL, 6 mmol) and the mixture was allowed to stir at room temperature, under argon, for 1 hour (the solution became clear). (S)-2-(2-aminoethyl)-1-methylpyrrolidine (Mizuno, A.; Hamada, Y.; Shioiri, T., Synthesis, 1980, 12 1007)(1.0 mL, 6.9 mmol) was added and the mixture was allowed to stir for 30 minutes. Water (200 mL) was added and the resulting solid was collected by vacuum filtration, washing with water, and dried to afford the pyrazine as a yellow solid. The yellow solid was purified on silica gel (10% MeOH/CH2Cl2 first eluting off impurities followed by eluting with 5% NH4OH/15% MeOH/CH2Cl2. The combined fractions were evaporated to afford the compound as a yellow solid. (1.2 g, 2.0 mmol, 85% pure).
……………………………..
http://www.google.com/patents/WO2007137000A2?cl=en
The present disclosure provides an improved method of treating cancer using a combination of a G-quadruplex-interactive compound that binds to G- quadruplexes in rDNA to release the nucleolin already bound to these G- quadruplexes together with a PARP inhibitor. This results in an increase in apoptosis in cancer cells. The PARP inhibitor can be administered to a patient (human or animal) in need of cancer treatment simultaneously or from 0.1 to 24 hours prior to or 0.1 to 24 after the administration of the G-quadruplex-interactive agent that releases the nucleolin bound to the G-quadruplex and triggers enhanced apoptosis of cancer cells, or the PARP inhibitor and the enhancer of nucleolin binding can be administered simultaneously, with each agent being administered in an amount sufficient to inhibit the growth and/or cell division of cancer (neoplastic) cells, and preferably to cause cancer cell death. In the methods provided herein, the PARP inhibitor can be benzamide (as specifically exemplified) or it can be 3- benzamide, 3-methoxybenzamide, carba-NAD+, nicotinamide, a dihydroisoquinolinone, an isoquinolinone such as 5-methyl-dihydroisoquinolinone, a benzimidazole-4-carboxamide, a 2-aryl-benzimidazole-4-carboxamide, a benzoxazole-4-carboxamide, an N,N-dimethylaminomethyl, pyrrolidinomethyl or bis- benzamide derivative, for example 1 ,5-di(3- carbamoylphenyl)aminocarbonyloxy)pentane, a phthalazinone, a quinazolinone, an isoindolinone, a phenanthhdinone, among others. The G-quadruplex-interactive agent that releases nucleolin from the rDNA bound to the G-quadruplexes and triggers apoptosis of cancer cells is desirably a substituted quinobenzoxazine analog; in an embodiment of the invention, it is CX-3543

(see also US Patent Publication 2006-0029950, which is incorporated by reference herein). This combination chemotherapy can be administered in a single dose, or it can be administered at intervals chosen by a medical or veterinary practitioner.
[0003] The present disclosure further provides compositions comprising a PARP inhibitor and a G-quadruplex-interactive compound that triggers the release of nucleolin from the G-quadruplexes in the rDNA and triggers apoptosis of cancer cells. The compositions desirably further comprises a pharmaceutically acceptable excipient, especially one which is compatible with intravenous administration in human patients. In an embodiment of the invention, the composition comprises benzamide and CX-3543.
……………………………
pubs.acs.org/doi/abs/10.1021/jm3013486

Nowadays, it has been demonstrated that DNA G-quadruplex arrangements are involved in cellular aging and cancer, thus boosting the discovery of selective binders for these DNA secondary structures. By taking advantage of available structural and biological information on these structures, we performed a high throughput in silico screening of commercially available molecules databases by merging ligand- and structure-based approaches by means of docking experiments. Compounds selected by the virtual screening procedure were then tested for their ability to interact with the human telomeric G-quadruplex folding by circular dichroism, fluorescence spectroscopy, and photodynamic techniques. Interestingly, our screening succeeded in retrieving a new promising scaffold for G-quadruplex binders characterized by a psoralen moiety.
……………………………..
US 20070293485
http://www.google.com/patents/US20070293485
…………………………..
see
US 20130005720
http://www.google.com/patents/US20130005720
…………………………….
see
WO 2004091504 or http://www.google.com/patents/EP1610759A2?cl=en
|
References: |
1. Bayes, M.; Rabasseda, X.; Prous, J. R., Gateways to clinical trials. Methods Find Exp Clin Pharmacol 2007, 29, (1), 53-71.
2. Brennan, A. B.; Long, C. J.; Bagan, J. W.; Schumacher, J. F.; Spiecker, M. M. Surface topographies for non-toxic bioadhesion control. US20100226943A1.
3. Drygin, D.; Siddiqui-Jain, A.; O’Brien, S.; Schwaebe, M.; Lin, A.; Bliesath, J.; Ho, C. B.; Proffitt, C.; Trent, K.; Whitten, J. P.; Lim, J. K. C.; Von, H. D.; Anderes, K.; Rice, W. G., Anticancer Activity of CX-3543: A Direct Inhibitor of rRNA Biogenesis. Cancer Res. 2009, 69, (19), 7653-7661.
4. Hurley, L. H.; Guzman, M. Combination cancer chemotherapy. WO2007137000A2, 2007.
5. Lim, J.; Whitten, J. P. Drug administration methods. WO2007143587A1, 2007.
6. Neidle, S., Human telomeric G-quadruplex: the current status of telomeric G-quadruplexes as therapeutic targets in human cancer. FEBS J. 277, (5), 1118-1125.
7. O’Brien, S.; Siddiqui-Jain, A. Targeting quadruplex sequences in human nucleic acids by identifying interacting quinoline and porphyrin derivatives. WO2007056113A2, 2007.
8. Ryckman, D. M.; Drygin, D.; Whitten, J. P.; Anderes, K.; Trent, K.; Darjania, L.; Haddach, M.; O’Brien, S.; Rice, W. G. Methods for treating aberrant cell proliferation disorders. US20080318938A1, 2008.
9. Tian, M.; Zhang, X.; Pan, R.; Zhao, C.; Tang, Y., Structure of G-quadruplex in the oncogene c-myc promoter and small ligands targeting the G-quadruplex. Huaxue Jinzhan 22, (5), 983-992.
10. Whitten, J. P.; O’Brien, S. Methods for treating ophthalmic disorders. US20080318939A1, 2008.
11. Whitten, J. P.; Pierre, F.; Regan, C.; Schwaebe, M.; Yiannikouros, G. P.; Jung, M. Preparation of fused quinolone analogs which inhibit cell proliferation and/or induce cell apoptosis. US20060074089A1, 2006.
12. Whitten, J. P.; Pierre, F.; Regan, C.; Schwaebe, M.; Yiannikouros, G. P.; Jung, M. Preparation of fused quinolone analogs which inhibit cell proliferation and/or induce cell apoptosis. WO2006034113A2, 2006.
13. Whitten, J. P.; Pierre, F.; Regan, C.; Schwaebe, M.; Yiannikouros, G. P.; Jung, M. Process for the preparation of benzothiazole and phenoxazine compounds. US20060063761A1, 2006.
14. Whitten, J. P.; Schwaebe, M.; Siddiqui-Jain, A.; Moran, T. Preparation of substituted quinobenzoxazine analogs as antitumor agents. US20060029950A1, 2006.
| “CX-3543, 386705” ANNUAL DRUG DATA REPORT, PROUS, BARCELONA, ES, vol. 27, no. 4, 2005, page 379, XP009092663 ISSN: 0379-4121 | ||
| 2 | * | CEPEDA, V. ET AL.: “Poly(ADP-Ribose) Polymerase-1 (PARP-1) Inhibitors in Cancer Chemotherapy” RECENT PATENTS ON ANTI-CANCER DRUG DISCOVERY, vol. 1, no. 1, January 2006 (2006-01), pages 39-53, XP007903584 ISSN: 1574-8928 |
| 3 | * | DATABASE INTEGRITY [Online] Prous science; DailyDrugNews.com (Daily Essentials) 22 July 2005 (2005-07-22), “CX-3543 begins phase I cancer trial” XP007903594 retrieved from INTEGRITY.PROUS.COM Database accession no. 386705 |
| 4 | * | JIN ET AL: “In vivo efficacy of CX-3543, a novel c-Myc oncogene inhibitor” PROCEEDINGS OF THE ANNUAL MEETING OF THE AMERICAN ASSOCIATION FOR CANCER RESEARCH, NEW YORK, NY, US, vol. 45, 2004, page ABS. LB-243, XP001537665 ISSN: 0197-016X |
| 5 | * | RICE WILLIAM G ET AL: “Design of CX-3543, a novel multi-targeting antitumor agent” PROCEEDINGS OF THE ANNUAL MEETING OF THE AMERICAN ASSOCIATION FOR CANCER RESEARCH, NEW YORK, NY, US, vol. 46, April 2005 (2005-04), pages 594-ABS. 2530, XP001536592 ISSN: 0197-016X |
| 6 | * | SHIOKAWA D ET AL: “Inhibitors of poly(ADP-ribose) polymerase suppress nuclear fragmentation and apoptotic-body formation during apoptosis in HL-60 cells” FEBS LETTERS, ELSEVIER, AMSTERDAM, NL, vol. 413, no. 1, 11 August 1997 (1997-08-11), pages 99-103, XP004261237 ISSN: 0014-5793 |
| 7 | * | VALERIOTE F ET AL: “SYNERGISTIC INTERACTION OF ANTICANCER AGENTS: A CELLULAR PERSPECTIVE” CANCER CHEMOTHERAPY REPORTS, vol. 59, no. 5, September 1975 (1975-09), pages 895-900, XP009019750 |
| S7910600 | Aug 29, 2008 | Mar 22, 2011 | Cylene Pharmaceuticals, Inc. | Therapeutic kinase modulators |
| US7956064 | Aug 31, 2007 | Jun 7, 2011 | Cylene Pharmaceuticals, Inc. | Fused tricyclic compounds as serine-threonine protein kinase and PARP modulators |
| US8481529 | May 9, 2007 | Jul 9, 2013 | The Arizona Board Of Regents On Behalf Of The University Of Arizona | Combination cancer chemotherapy |
| EP2023935A1 * | Jun 1, 2007 | Feb 18, 2009 | Cylene Pharmaceuticals, Inc. | Drug administration methods |
| WO2007137000A2 * | May 9, 2007 | Nov 29, 2007 | Univ Arizona | Combination cancer chemotherapy |
| WO2007143587A1 * | Jun 1, 2007 | Dec 13, 2007 | Cylene Pharmaceuticals Inc | Drug administration methods |
Piramal Drops Drug Discovery,…………. Pharmaceuticals: Risks and regulations convince the Indian company to reallocate resources

In a move that raises questions about the future of drug research in India, Piramal Enterprises will end its drug discovery activities. The decision—which involves possible job losses—will affect several hundred scientists, many of whom were recruited internationally to work in Mumbai in one of India’s most sophisticated pharmaceutical labs.
The company has been considered an Indian leader in drug research since opening its discovery labs in 2004. Within the firm, drug discovery was championed by the vice chairman, Swati A. Piramal, a medical doctor who also holds a master’s degree from the Harvard School of Public Health.
“After reevaluating the risk-benefits of new chemical entity research, the company decided to focus resources on our other areas of R&D with shorter development timelines and different risk profiles,” Piramal tells C&EN.
read all at
http://cen.acs.org/articles/92/i37/Piramal-Drops-Drug-Discovery.html

Piramal Enterprises, which sold off its domestic formulations business to Abbott in a multi-billion dollar deal a few years ago, is now shutting down its Mumbai-based R&D unit which would in effect bring to an end its early stage drug discovery business.
Separate media reports, citing Swati Piramal, part of the promoter group of the diversified firm and wife of group chief Ajay Piramal, said, the decision to move away from the drug discovery business was taken given the costs of basic research.
The company would now focus on molecules at an advanced stage of development; resources would be redeployed from basic research to the clinical unit.
Its other research facilities are located in Chennai, Hyderabad, Ahmedabad and Indore, which would continue to be functional.
Although Piramal Enterprises retains its exposure to healthcare as a sector, after selling the key pharma business, it is now more associated with financial services, including investments in infrastructure and real estate sectors.
In an unrelated development, the firm is forming a joint venture with Navin Fluorine International Limited, an Arvind Mafatlal Group company, to develop, manufacture and sell specialty fluorochemicals with a focus on applications in healthcare, according to a company release.
As per the agreement, Piramal Enterprises will hold 51 per cent of the equity share capital of the proposed joint venture company, whereas the remaining 49 per cent will be held by Navin.
In the first phase of development, the JV is expected to invest around Rs 120 crore in India for this project.
Mumbai-based Navin Fluorine has a turnover of around $100 million. It specialises in specialty fluorine. It had acquired UK-based Manchester Organics, a specialty fluorochemicals research company in 2011.

Read more at: http://www.livemint.com/Companies/7weGbimcrdp7lrKH0YaSnL/Piramal-to-exit-drug-discovery-business.html?utm_source=copy
LAPATINIB, GW572016, An EGFR-ErbB-2 inhibitor.

Lapatinib in 3d

LAPATINIB
-
N-(3-Chloro-4-((3-fluorobenzyl)oxy)phenyl)-6-(5-(((2-(methylsulfonyl)ethyl)amino)methyl)furan-2-yl)quinazolin-4-amine

 lapatinib
lapatinib
| Systematic (IUPAC) name | |
|---|---|
| N-[3-chloro-4-[(3-fluorophenyl)methoxy]phenyl]-6- [5-[(2-methylsulfonylethylamino)methyl]-2-furyl] quinazolin-4-amine |
|
| Clinical data | |
| Trade names | Tykerb, Tyverb |
| AHFS/Drugs.com | monograph |
| MedlinePlus | a607055 |
| Licence data | EMA:Link, US FDA:link |
| Pregnancy cat. | |
| Legal status | |
| Routes | Oral |
| Pharmacokinetic data | |
| Bioavailability | Variable, increased with food |
| Protein binding | >99% |
| Metabolism | Hepatic, mostly CYP3A-mediated (minor 2C19 and2C8 involvement) |
| Half-life | 24 hours |
| Excretion | Mostly fecal |
| Identifiers | |
| CAS number | 231277-92-2 388082-78-8 (ditosylate) |
| ATC code | L01XE07 |
| PubChem | CID 208908 |
| DrugBank | DB01259 |
| ChemSpider | 181006 |
| UNII | 0VUA21238F |
| Chemical data | |
| Formula | C29H26ClFN4O4S |
| Mol. mass | 581.058 g/mol |
Lapatinib (INN), used in the form of lapatinib ditosylate, (USAN) (Tykerb/Tyverb, GSK) is an orally active drug for breast cancerand other solid tumours.[1] It is a dual tyrosine kinase inhibitor which interrupts the HER2/neu and epidermal growth factor receptor(EGFR) pathways.[2] It is used in combination therapy for HER2-positive breast cancer. It is used for the treatment of patients with advanced or metastatic breast cancer whose tumors overexpress HER2 (ErbB2).[3]
Status
On March 13, 2007, the U.S. Food and Drug Administration (FDA) approved lapatinib in combination therapy for breast cancer patients already using capecitabine (Xeloda, Roche).[2][3] In January 2010, Tykerb received accelerated approval for the treatment of postmenopausal women with hormone receptor positive metastatic breast cancer that overexpresses the HER2 receptor and for whom hormonal therapy is indicated.[3]
Pharmaceutical company GlaxoSmithKline (GSK) markets the drug under the propriety names Tykerb (mostly US) and Tyverb (mostly Europe).[4] The drug currently has approval for sale and clinical use in the US,[2][4] Australia,[2] Bahrain,[2] Kuwait,[2] Venezuela,[2]Brazil,[5] New Zealand,[5][6] South Korea,[5] Switzerland,[4] Japan, Jordan, the European Union, Lebanon, India and Pakistan.[4]
On the 2nd of August 2013, India’s Intellectual Property Appellate Board revoked the patent for Glaxo’s Tykerb citing its derivative status, while upholding at the same time the original patent granted for Lapatinib.[7]
The drug lapatinib ditosylate is classified as S/NM (a synthetic compound showing competitive inhibition of the natural product) that is naturally derived or inspired substrate (Gordon M. Cragg, Paul G. Grothaus, and David J. Newman, Impact of Natural Products on Developing New Anti-Cancer Agents, Chem. Rev. 2009, 109, 3012–3043)
Lapatinib ditosylate, an ErB-1 and ErB-2 dual kinase inhibitor, was launched in the U.S. in 2007 for the treatment of advanced or metastatic HER2 (ErbB2) positive breast cancer in women who have received prior therapy, including Herceptin(R) (trastuzumab), in combination with Xeloda(R) (capecitabine). The compound was approved in 2007 in Switzerland and Australia and in 2009 in Canada, for this indication. Regulatory approval has also been obtained in Japan. In December 2007, a positive opinion was received in the E.U. In 2008, the CHMP issued a revised positive opinion confirming the positive benefit-risk profile for lapatinib following review by the CHMP of new data received in February 2008 from GlaxoSmithKline arising from a standard pharmacovigilance evaluation of clinical trial and post-marketing data. The CHMP confirmed that these data do not essentially change the positive benefit-risk profile for lapatinib in its proposed indication. In 2008, the MAA was approved in the E.U. and the product was subsequently commercialized in Germany. In 2009, regulatory applications were filed in the U.S. and the E.U. seeking approval for use of lapatinib as first-line treatment of patients with hormone-sensitive, metastatic (or advanced) breast cancer in combination with anti-hormonal therapy. In 2010, lapatinib was launched on the U.S. market as first-line treatment in combination with Femara(R) to treat hormone positive and HER2-positive advanced breast cancer in postmenopausal women for whom hormonal therapy is indicated. In 2010, the compound was approved and launched in the E.U. for the oral treatment of post-menopausal women with hormone receptor-positive, HER2 (ErbB2) over-expressing metastatic breast cancer and for whom chemotherapy is currently not intended, in combination with an aromatase inhibitor. In 2012, GlaxoSmithKline filed regulatory applications in the U.S. and the E.U. for the oral treatment of patients with HER2 (ErbB2)-positive metastatic breast cancer that has progressed on prior trastuzumab regimens, in combination with trastuzumab. In July 2012, GlaxoSmithKline withdrew this application in the U.S. In 2013, the product was approved for this indication in the E.U.
In terms of clinical development, the National Cancer Institute (US) is currently conducting phase II/III trials for the treatment of bladder cancer. Phase III trials are under way to evaluate the use of lapatinib as first-line treatment of breast cancer. The compound is also being evaluated for several oncologic indications in the treatment of brain, gallbladder, prostate, ovary, endometrium, bladder cancer, cervical and hepatobiliary cancers in collaboration with the National Cancer Institute (NCI). Lapatinib in combination with everolimus is also in early clinical studies for the treatment of lymphoma and non-Hodgkin’s lymphoma (NHL). A phase II combination trial is evaluating lapatinib for the treatment of advanced or metastatic colorectal cancer. The National Cancer Institute (NCI) is developing the compound in phase II trials for the treatment of peritoneal cancer, ovarian and ductal carcinoma in situ of the breast (DCIS), while Brown University is conducting combination trials with gemcitabine for the treatment of pancreas metastatic cancer, and Cedars-Sinai Medical Center is conducting phase II clinical trials for treatment for pituitary cancer. Phase III clinical study for the treatment of head and neck was terminated because the study didn´t meet primary endpoint.
Lapatinib was granted fast-track status by the FDA in 2005 for the treatment of refractory advanced or metastatic breast cancer patients who have documented ErbB-2 overexpression and who have failed previous therapy. In 2009, Orphan Drug Designation was received in the U.S. by GlaxoSmithKline for the treatment of ErbB2 positive gastric cancer and for the treatment of ErbB2 positive esophageal cancer.
Breast cancer
Lapatinib is used as a treatment for women’s breast cancer in treatment naive, ER+/EGFR+/HER2+ breast cancer patients(now often called “triple positive”) and in patients who have HER2-positive advanced breast cancer that has progressed after previous treatment with other chemotherapeutic agents, such as anthracycline, taxane-derived drugs, or trastuzumab (Herceptin, Genentech).
A 2006 GSK-supported randomized clinical trial on female breast cancer previously being treated with those agents (anthracycline, a taxane and trastuzumab) demonstrated that administrating lapatinib in combination with capecitabine delayed the time of further cancer growth compared to regimens that use capecitabine alone. The study also reported that risk of disease progression was reduced by 51%, and that the combination therapy was not associated with increases in toxic side effects.[11] The outcome of this study resulted in a somewhat complex and rather specific initial indication for lapatinib—use only in combination with capecitabine for HER2-positive breast cancer in women whose cancer have progressed following previous chemotherapy with anthracycline, taxanes and trastuzumab.

………………………………………………………..
Patent
Product patent
US6727256
or
http://www.google.co.in/patents/WO1999035146A1
………………………………………………..
W09935146 (GSK company, filed on 8 February 1999, I) propose a 2_ chlorine _4_ nitrophenol as the starting material, by addition, catalytic hydrogenation, replace, Suzuki coupling Union, such as reductive amination reaction was prepared by lapatinib

First, the method of the protected aldehyde group, deprotection after the completion of the coupling reaction for the reductive amination reaction, the reaction step so long; due to the use of expensive and highly toxic organic heteroaryl stannane reagent 5 – (_ 1,3-dioxolan-2 – yl) -2 – (tributylstannyl group) _ furan, intermediates for drugs and have greater safety and environmental risks; Furthermore, the process requires the synthesis of intermediate purified by column chromatography, post-processing is more complex.
………………………………………………..
CN102295638A (Qilu Pharmaceutical Co., Ltd., June 24, 2010 application) proposed a method of preparing lapatinib is mixture of 5 – formyl-furan-2 – boronic acid, N-[3 – chloro-4 – [(3 – fluorophenyl) methoxy] phenyl] -6 – iodo-4 – quinazolinamine 2 – methylsulfonyl – ethylamine and the catalyst to the solvent, Mr. into transitional intermediate, and then reducing agent such as sodium triacetoxy borohydride reduction to give the desired product, the synthesis route is as follows:

………………………………………………….
W02005120504A2 (Glaxo, in June 2005 I filed) proposed an alternative approach: a 4 – chloro-6 – iodine quinazoline as the starting material, with 5 – formyl-furan-2 – boric acid instead of highly toxic tin compounds alkylfuryl prepared lapatinib. The synthetic route is as follows:

………………………………………………………………….
Patent
http://www.google.com/patents/EP2550269A1?cl=en
Lapatinib has the structural formula (I) and chemical name N-[3- chloro-4-[(3-fluorophenyl)methoxy]phenyl]-6-[5-[(2-methylsulfonylethylamino)methyl]-2- furyl] quinazolin-4-amine.

BACKGROUND ART
Lapatinib is a tyrosine kinase inhibitor that is used as an orally administered drug as its ditosylate salt to treat certain types of advanced or metastatic breast cancer and other solid tumors. Lapatinib ditosylate was approved by the FDA in 2007 and the EMEA in 2008 and is marketed by GlaxoSmithKline (GSK) under the trade name of Tykerb® in the USA and Tyverb® in Europe.
Lapatinib substance is claimed in US 6,713,485 B2 and US 6,727,256 Bl and lapatinib ditosylate and its crystalline forms are claimed in US 7,157,466 B2. A synthesis of lapatinib that utilises a palladium mediated coupling of a substituted 4-anilino-6-iodo-quinazoline (II) with a 2- (tributylstannyl)furan (Ilia) is disclosed in US 6,727,256 Bl and is also presented in US 7,157,466 B2. In US 7,157,466 B2 a second generation approach was disclosed that utilises a palladium catalysed coupling of a substituted 4-anilino-6-iodo-quinazoline (II) with furan-2-yl-boronic acids (Illb). Following the palladium catalysed coupling reactions utilised in the two synthetic methods of US 6,727,256 Bl and US 7,157,466 B2, only one (US 7,157,466 B2) or two (US 6,727,256 Bl and US 7,157,466 B2) synthetic modification of the structure are utilised before the lapatinib substance is provided (Scheme 1). The EMEA’s COMMITTEE FOR MEDICINAL PRODUCTS FOR HUMAN USE (CHMP) has published guidelines titled GUIDELINE ON THE SPECIFICATION LIMITS FOR RESIDUES OF METAL CATALYSTS OR METAL REAGENTS and recommendations are presented for oral exposure to metals, including palladium. For a drug being consumed in quantities not exceeding a 10 g daily dose, a limit of 10 ppm (parts per million) concentration of palladium in the drug substance is recommended. Given this, there is still an unmet need for an alternative synthetic method that can be used for preparation of lapatinib in which the palladium mediated coupling step is performed early in the synthetic route, thereby being capable to provide .

Scheme 1
SUMMARY OF THE INVENTION
There are a number of ways that the levels of a metal, such as palladium, can be controlled in a drug substance through purging of the metal by treatment of the drug substance or its synthetic intermediates or both, including crystallisation, aqueous extraction, filtration through metal absorbent filter aids (Organic Process Research & Development 2005, 9, 198-205), precipitation of the metal from solution, chromatography, and treatment with metal scavenging reagents (Organic Process Research & Development 2003, 7, 733-742). By placing the palladium mediated coupling step downstream in the synthetic route, however, to take advantage of synthetic convergence, the opportunity to reduce the level of palladium in the drug substance is reduced. In contrast, however, by redesigning the synthetic route to move the palladium mediated coupling step upstream, further away from the drug substance, increases the opportunity to control the palladium level in the drug substance. Furthermore, by careful operational design (such as in a precipitation and crystallisation step), the palladium level in the intermediates can be consistently controlled. Given that there is a need, the present invention has addressed these two latter points and utilised them in a novel and efficient process for the manufacture of lapatinib and lapatinib ditosylate.

Scheme 2 – Synthesis of lapatinib and lapatinib ditosylate
In contrast to the prior art methods disclosure in US 6,727,256 Bl and US 7,157,466 B2, the present invention has performed a transition metal catalysed coupling reaction at the most upstream point in the synthetic route based on the utilization of commercially available starting materials SMla (6-iodoquinazolin-4(3H)-one) and SM2a (5-formylfuran-2-ylboronic acid), or their analogues SMI and SM2, to provide IM1. Thus, in one aspect of the present invention, lapatinib is made from a novel compound (IM1) (Scheme 2).
In another aspect of the present invention, a lapatinib ditosylate monohydrate is prepared by crystallizing lapatinib ditosylate in a mixture of water, DMSO and MeCN.
In another aspect of the present invention, novel compound IM1 is synthesized by the cross- coupling of commercially available SMla and SM2a, or their analogues SMI and SM2, in suitable solvents comprised of an organic solvent and water in the presence of a base and a catalyst formed from a transition metal and a ligand (scheme 3).

X = CI, Br, I, OTf Y = CHO, or CH(OR)2
BZ = B(OH)2, B(OR)2, [BF3]M or BR2
Scheme 3
Example
Example 1: Synthesis of 5-(4-oxo-3,4-dihydroquinazolin-6-yl)furan-2-carbaldehyde (IMl)

IM1
A 5:2 v/v mixture of DMSO and H20 (1400 mL) was degassed for 30 min at ambient temperature using nitrogen. 5-Formylfuran-2-ylboronic acid (SM2a; 26.8 g, 193 mmol) was added dissolved in this mixture. [HP(i-Bu)3] BF4 “ (840 mg, 2.94 mmol) and Pd(OAc)2 (680 mg, 2.94 mmol) was added and the mixture was stirred at ambient temperature under an atmosphere of nitrogen for 20 min. AcOK (18.8 g, 192 mmol) was added into the reactor and was stirred for 20 min at ambient temperature. 6-Iodoquinazolin-4(3 /)-one (SMla; 40 g, 147 mmol) was added and heated to 80±5°C (internal temperature) in an oil bath under nitrogen, Upon completion of the reaction (HPLC), the reaction mixture was hot-filtered, then hot water (400 mL, 80±5°C) was added into the filtrate. This was slowly cooled to 0-15°C (solid started to precipitate at 70°C (internal temperature) and was then filtered. The filter cake was washed with H20 (80 mL), then with MeCN (60 mL), and dried in vacuo at 60+5°C for 6 h to provide 5-(4-oxo-3,4-dihydroquinazolin-6-yl)-furan-2- carbaldehyde (IMl; 34.6 g, 144 mmol) with 99.7 % HPLC purity in 97.6% HPLC yield. XH NMR (300 MHz, de-DMSO): δ 7.47 (d, / = 3.8 Hz, 1H), 7.69 (d, / = 3.8 Hz, 1H), 7.77 (d, / = 8.6 Hz, 1H), 8.17 (s, 1H), 8.27 (dd, / = 8.6, 2.1 Hz, 1H), 8.52 (d, = 2.1 Hz, 1H), 9.66 (s, 1H); 13C NMR (75 MHz, CDC13): δ 110.5, 122, 6, 123.9, 126.0, 127.5, 129.0, 131.4, 147.1, 150.1, 152.7, 157.6, 161.2, 178,8; ESI-MS, Pos: [M+H]+ mJz 241; IR (cm 1): 1713, 1671, 1604,1462; m.p.: 267°C. See Figure 2 for the DSC/TGA of IMl; See Figure 3 for the X-ray powder diffraction pattern of IMl; Residual concentration of palladium: 230 ppm.
Example 2: Synthesis of 5-(4-chloroquinazolin-6-yl)furan-2-carbaldehyde hydrochloride
(IM2a.HCl)

I 1 reflux IM2a.HCI
Over a 1.5 hour period under an atmosphere of N2, SOCb (86.2 g) in MeCN (145 mL) was added dropwise into a mixture, that had been preheated at reflux for 0.5 h, of IM1 (29 g, 0.121 mol), MeCN (435 mL) and DMF (0.88 g) at reflux. The reaction was terminated when less than 2% (HPLC) of IM1 was remaining. If the reaction did not achieve complete reaction, extra SOCI2was added. The mixture was cooled to about 25±5°C (internal temperature), and was then filtered and washed with MeCN (58 mL) to give ca. 55 g of IM2a.HCl (moist with MeCN) with 82A purity by HPLC. IM2a.HCl: ¾ NMR (300 MHz, d6-DMSO): δ 9.68 (s, 1 H), 9.17 (s, 1H), 8.57 (d, / = 2.0 Hz, 1H), 8.46 (dd, J = 8.6, 2.1 Hz, 1H), 8.02 (d, / = 8.6 Hz, 1H), 7.74 (d, = 3.8 Hz, 1H), 7.60 (d, J = 3.8 Hz, 1H). See Figure 5 for the XH NMR spectrum of IM2a.HCl; 13C NMR (75 MHz, d6– DMSO) δ 179.0, 159. 6, 156.4, 152.9, 149.5, 141.0, 132.6, 129.2, 125.9, 123.2, 122.9, 122.7, 111.5;
IM2a.HCl was purified by column chromatography (eluent: ) to give pure IM2a. IM2a: lH NMR (300 MHz, d6-DMSO): δ 7.53 (d, / = 3.3 Hz, 1H), 7.68 (d, J = 3.3 Hz, 1H), 8.02 (d, / = 8.7 Hz, 1H), 8.42 (d, / = 8.4 Hz, 1H), 8.54 (d, / = 2.1 Hz, 1H), 8.90 (s, 1H), 9.64 (s, 1H); 13C NMR (75 MHz, CDCI3): δ 111.5, 122.8, 122.9, 123.7, 125.9, 129.1, 132.5, 142.1 , 149.3, 152.9, 156.6, 159.7, 179.1.
Example 3: Synthesis of 5-(4-(3-chloro-4-(3-fluorobenzyloxy)phenylamino)
– uinazolin-6-yl)furan-2-carbaldehyde hydrochloride (IM3.HC1)

A mixture of IM2a.HCl (moist with MeCN solvent, prepared from 29 g IM1, 0.120 mol) and 3-chloro-4-(3-fiuorobenzyloxy)aniline (SM3; 27.3 g, 0.108 mol) in MeCN (580 mL) was stirred under reflux, until HPLC analysis showed that the reaction was completed (about 2 h). The mixture was cooled to room temperature (25±5°C), filtered, and washed with MeCN (58 mL). A mixture of the moist crude solid IM3 and THF (870 mL) was treated with a 2.0 N aqueous NaOH (348 mL) and stirred for 3-4 h until most of the solid had dissolved. The mixture was filtered through diatomite and was washed with a saturated aqueous solution of NaCl (87 mL). The organic layer was treated with 10% aqueous HCI (174 mL) and stirred for 0.5 h. The resulting solid was filtered, washed with THF (87 mL), and dried in vacuo at 60+5°C for 16 h to give the crude IM3.HC1 (34 g, 0.067 mol, HPLC purity: 99%).
IM3.HC1: :H NMR (300 MHz, d6-DMSO): δ 9.69 (s, 1H), 9.52 (s, 1H), 8.94 (s, 1H), 8.50 (dd, / = 8.8, 1.7 Hz, 1H), 8.01 (d, / = 8.8 Hz, 1 H), 7.97 (d, J =2.5 Hz, 1H), 7.77 (d, / = 3.8 Hz, 1H), 7.73 (dd, = 9.0, 2.5 Hz, 1H), 7.69 (d, / = 3.8 Hz, 1H), 7.49 (td, 7 = 8.0, 6.1 Hz, 1 H), 7.41-7.28 (m, 3H), 7.20 (td, / = 8.4, 2.2 Hz, 1H), 5.31 (s, 2H).
Free base IM3 is obtained by column chromatography (eluting with EtOAc/DCM, 1:4, v/v). IM3 XH NMR (300 MHz, d6-DMSO): δ 5.28 (s, 2H), 7.19 (td, /= 8.7 Hz, 7 = 2.1 Hz 1H), 7.34 (m, 4H), 7.43 (d, 7 = 3.6 Hz , 1H), 7.49 (m, 1H), 7.73 (dd, 7 = 8.7 Hz 7 = 2.7 Hz, 1H), 7.76 (d, 7 = 3.6 Hz, 1H), 7.88 (d, 7 = 9 Hz, 1H), 8.07 (d, 7 = 2.1 Hz, 1H), 8.32 (dd, 7 = 4.43 Hz, 7 = 1.95 Hz, 1H), 8.95 (d, 7 = 1.5 Hz, 1H), 9.68 (s, 1H).
Example 4: Synthesis of N-(3-chloro-4-(3-fluorobenzyloxy)phenyl)-6-(5-((2- (methylsulfonyl)ethylamino)methyl)furan-2-yl)quinazolin-4-amine ditosylate (lapatinib ditosylate)

I
To a suspension of 2-(methylsulfonyl)ethanamine hydrochloride (SM4.HC1; 12.2 g, 76.7 mmol) in THF (600 mL) was added acetic acid (14.1 g, 235 mmol) followed by DIPEA (30.3 g, 235 mmol) were added. After stirred at ambient temperature for 0.5 h, ¾0 (4.2 g, 233 mmol) and IM3.HC1 (30.0 g, HPLC assay >99%, 58.7 mmol) were added. After being stirred at ambient temperature (20°C) for 4 h, sodium triacetoxyborohydride (37.4 g, 176 mmol) was added and the mixture was stirred at ambient temperature (20°C±5°C; external temperature) until HPLC showed the completion of the reaction. A 10% aqueous solution of sodium hydroxide (90 mL) was added and the mixture was stirred for 30 min. The organic phase was washed with 25% aqueous NH4C1 (60 mL), filtered, treated with -TsOH (40.4 g, 135 mmol) and heated to reflux for 2 h. The mixture was cooled to ambient temperature and stirred for 3 h at ambient temperature. The mixture was filtered, and the filter cake was washed twice with THF (120 mL each) and was then dried under vacuum at 70±5°C for 6 h to give 43 g (46.5 mmol) lapatinib ditosylate with 99.4% HPLC purity.
Lapatinib ditosylate [H NMR (300 MHz, d6-DMSO): δ 11.41(s, 2H), 9.33 (s, 3H), 9.04 (d, / = 1.3 Hz, 2H), 8.93 (s, 2H), 8.41 (dd, J =8.8, 1.6 Hz, 2H), 7.91 (d, J = 2.6 Hz, 2H), 7.54-7.41 (m, 9H), 7.37 – 7.27 (m, 6H), 7.25 (d, / = 3.4 Hz, 2H), 7.22 – 7.13 (m, 2H), 7.08 (dd, / = 8.4, 0.6 Hz, 8H), 6.87 ( d, / = 3.5 Hz, 2H), 5.29 (s, 4H), 4.46 (s, 4H), 3.65 – 3.51 (m, 4H), 3.51 – 3.38 (m, 4H), 2.26 (s, 12H).
A solution of lapatinib ditosylate was converted to its free base form, lapatinib, by washing a solution with aqueous NaOH followed by concentration. Lapatinib: XH NMR (300 MHz, d6-DMSO): δ 2.98 (t, / = 6.75 Hz, 1H), 3.04 (s, 1H), 3.29 (t, J = 6.6 Hz, 1H), 3.83 (s, 1H), 5.28 (s, 1H), 6.50 (d, / = 3.0 Hz, 1H), 7.08 (d, / = 3.3 Hz, 1H), 7.20 (m, 1H), 7.33 (m, 4H), 7.48 (m, 1H), 7.76 (m, 1H), 7.80 (d, 7 = 9 Hz, 1H), 8.04 (d, 7 = 2.75 Hz, 1H), 8.17 (dd, / = 8.7 Hz, / = 1.8 Hz, 1H), 8.56 (s, 1H), 8.75 (d, J = 1.8 Hz, 1H).
Example 5a: Purification of lapatinib ditosylate
Lapatinib ditosylate (5.0 g, 5.4 mmol, 96.5% HPLC purity with the maximum individual impurity at 0.8%) was dissolved in DMSO (10 mL) at 70°C (internal temperature). MeCN (10 mL) was added dropwise into the mixture at 70-80°C (internal temperature) and was stirred at this temperature for 1 h. Over a 4 h period the mixture was cooled to room temperature. MeCN (30 mL) was added dropwise, and the mixture was stirred for lh, then filtered and washed with MeCN (10 mL). The filter cake was dried under vacuum at 60°C for 16 h to give 4.0 g lapatinib ditosylate as crystalline Form 1 (as disclosed in US 7,157,466 B2) with 99.6% HPLC purity in 78% HPLC yield.
Example 5b. Purification of lapatinib ditosylate.
Lapatinib ditosylate (3 g, 3.25 mmol, 99.3% HPLC purity was dissolved in DMF (18 mL) at 80°C and stirred for 1 hour. The mixture was hot-filtered. MeCN (18 mL) was added into the filtrate at 80°C. The temperature was cooled to 70°C and crystal precipitated. The mixture was kept at 70°C for 1 h and then 60°C for 1 h. The mixture was further cooled to 0°C and stirred for 2 h. The crystals of lapatinib ditosylate were isolated by filtration and were dried at 40°C under vacuum overnight. Lapatinib ditosylate (2.5 g, 2.70 mmol, 83% yield) with 99.9% HPLC purity was obtained. XRPD analysis (figure 9) indicated that this was Form 2 as disclosed in WO 2009/079541 Al.
Example 6: Preparation of lapatinib ditosylate monohydrate Lapatinib ditosylate (2.0 g, 96.7% HPLC purity, 2.1 mmol) was dissolved in DMSO (5 mL) at 80°C (internal temperature) and the solution was filtered whilst the lapatinib ditosylate was still dissolved. A mixture of MeCN (5 mL, 2.5 P) and water (0.3 mL) was then added dropwise into the filtered solution at 70-80°C (internal temperature). The mixture was cooled at a rate of 10°C/h until 60°C, and was kept at 60°C for 2 h and was then slowly cooled down to 50°C. After being kept at 50°C for 1 h, MeCN (15 mL) was added, and then the mixture was cooled to 20-30°C and stirred at 20-30°C for 2 h. The slurry was filtered, washed with MeCN (6 mL) and the filter cake was dried in vacuo at 60°C for 4 h to give lapatinib ditosylate monohydrate (1.7 g, 99.4A% purity, 1.8 mmol). XRPD analysis (figure 10) indicated that this was the monohydrate crystalline form as disclosed in US 7,157,466 B2.

…………………………………………………
http://www.google.com/patents/CN103159747A?cl=en


Example 3
[0029] Under a nitrogen atmosphere, 2 – furaldehyde diethyl acetal 950g, 9000mL of dry tetrahydrofuran and transferred to the flask, the system was cooled to _40 ° C, n-butyl lithium in tetrahydrofuran (3180mL, 2.2mol / L ) was added dropwise to the reaction system to maintain -4 (T-5 (TC stirred for 2.5 ~ 3h, then triisopropyl borate was added dropwise 1536mL, and stirred for Ih at _60 ° C, after the system was allowed to warm to room temperature, 384mL of glacial acetic acid was slowly added dropwise, followed by stirring for 30min, then dropping 156mL water was added to 3780mL of ethanol, 776mL of triethylamine were then added N_ [3_ chloro _4-[(3_ fluorophenyl) methoxy] phenyl] -6 – iodo-4 – quinazolinamine 1124g, 10% palladium on carbon 134g, and the reaction system was heated to reflux temperature, the reaction 14h. temperature was lowered to room temperature, the reaction mixture was filtered, the filter cake was washed with tetrahydrofuran, The filtrates were combined. To the filtrate was added 240g of triethylamine were then added 2 – (methylsulfonyl) ethylamine 390g and 450mL of methanol, and stirred at room temperature lh, then potassium borohydride was added 137.9g, room temperature for 1.5h, then ice under cooling, a 5N aqueous sodium hydroxide was added dropwise 3600mL, stirred at room temperature 15min, standing layered organic phase was separated, the organic phase p-toluenesulfonic acid was added dropwise 2400g / 3600mL of tetrahydrofuran was stirred for 40min, the solid was filtered and the filter cake was washed with tetrahydrofuran, and then recrystallized from methanol and dried in vacuo to obtain pure final two pairs of p-toluenesulfonic acid lapatinib 1185g. yield 70.8%, purity 98.1%. HNMR (DMSO) 2.27 Cs, 6H) , 3.11 (s, 3H), 3.50 (t, 2H), 3.60 (t, 2H), 4.47 (s, 2H), 5.32 (s, 2H), 6.90 (s, lH), 7.1 (d, J = 7.8 Hz, 4H), 7.19 (t, lH), 7.20 (t, lH), 7.22 (d, J = 3.2Hz, 1H) ,7.23-7 .25 (m, 3H),
7.56 (d, J = 8.0Hz, 4H), 7.62 (dd, Jl = 8.7Hz, J2 = 8.0Hz, 1H), 7.87 (s, 1H), 7.91 (d, J = 8.9Hz, 1H), 8.42 ( d, J = 8.7Hz, 1H), 8.93 (s, lH), 9.03 (s, lH), 9.32 (s, 1H), 11.34 (s, 1H).
…………………………….
PAPER
Beilstein J. Org. Chem. 2013, 9, 2265–2319.
http://www.beilstein-journals.org/bjoc/single/articleFullText.htm?publicId=1860-5397-9-265
GlaxoSmithKline’s lapatinib (3.38, Tykerb) is a novel dual kinase inhibitor used in the treatment of solid tumors such as those found in breast cancer and contains a quinazoline core structure. It consists of a 2,5-disubstituted furan ring, which is directly linked to the aminoquinazoline unit (Scheme 41). The quinazoline heterocycle was prepared starting from 5-iodoanthranilic acid (3.72) via initial condensation with formamidine acetate (3.73) followed by chlorination using oxalyl chloride or phosphorous oxychloride [101]. Performing a nucleophilic aromatic substitution on the chloride 3.74 with aniline 3.75renders the extended core of lapatinib. This intermediate (3.76) was then coupled with 5-formyl-2-furanoboronic acid (3.77) using standard Suzuki cross-coupling conditions. Finally, a reductive amination of the pendant aldehyde of3.78 with 2-(methylsulfonyl)ethylamine (3.79) furnishes the desired product lapatinib (Scheme 41).
![[1860-5397-9-265-i41]](http://www.beilstein-journals.org/bjoc/content/inline/1860-5397-9-265-i41.png?scale=2.0&max-width=1024&background=FFFFFF)
http://www.beilstein-journals.org/bjoc/single/articleFullText.htm?publicId=1860-5397-9-265
……………………………………………..
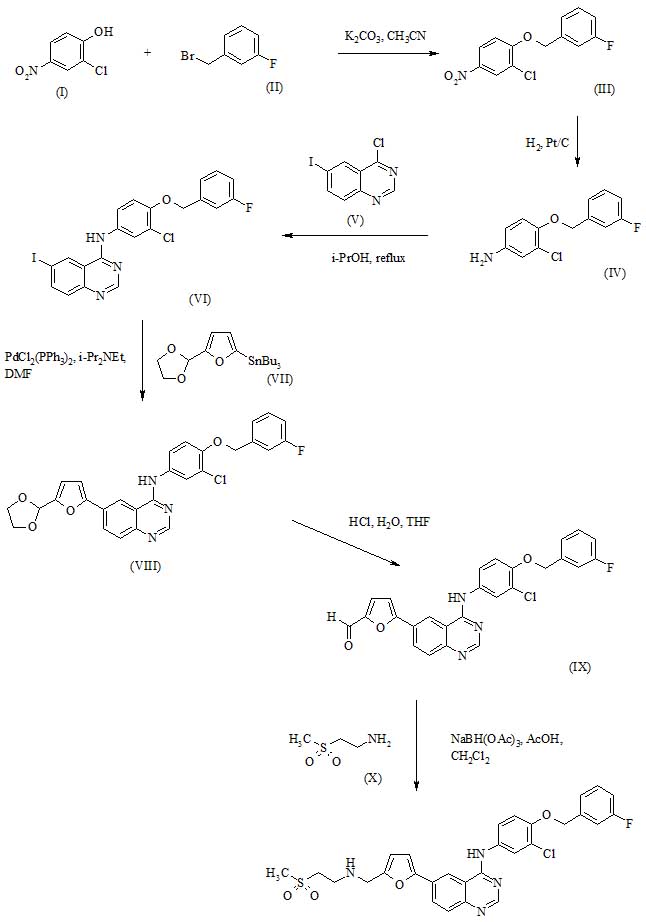
Guntrip SB, Lackey KE, Cockerill GS, Carter MC, Smith KJ Bicyclic heteroaromatic compounpds as protein tyrosine kinase inhibitors. EP 1047694; WO 9935146.

Quinazoline ditosylate salt compounds (US7157466)

A NOVEL PROCESS FOR THE PREPARATION OF Lapatinib AND ITS PHARMACEUTICALLY ACCEPTABLE SALTS ( WO 2010061400)
…………………………………………………….
Patent
Fresenius Kabi Oncology Ltd.WO 2013080218
Lahiri, Saswata; Gupta, Nitin; Singh, Hemant Kumar; Handa, Vishal; Sanghani, Sunil
6 JUNE 2013, http://www.google.com/patents/WO2013080218A1?cl=en
Lapatinib of Formula-(II), was first disclosed by SmithKline Beecham in US Patent No. 6,727,256.

The process for the preparation of Lapatinib of Formula-(II), disclosed in W099/35146, is given in the Scheme-I. Accordingly, 4-chloro-6-iodo-quinazoline of Formula-(IV), is reacted with 3-chloro-4-(3′-fluoro-benzyloxy)-aniline yielding N-[3- chloro-4-{(3′-fluorobenzyloxy) phenyl} ]-6-iodo-quinazoline of Formula-( l). The compound of the Formula-(l) reacts with 5-(l,3-dioxolan-2-yl)-2-(tributylstannyl)furan to get the compound of Formula-(2) which on deprotection with HC1, removes the 1,3- dioxolan-2-yl protecting group and liberates 5-(4-{3-chloro-4-(3-fluoro- benzyloxy)anilino}-6- quinazolinyl)-furan-2-carbaldehyde of Formula-(3). The compound of the Formula-(3) on reaction with 2-methanesulfonylethylamine, followed by reductive amination using sodium (triacetoxy)borohydride as the reducing agent gives the required compound Lapatinib of Formula-(II) as an organic residue, which is purified by column chromatography and subsequently converted into its hydrochloride salt (5).

Subsequently, US 7, 157,466 also discloses the preparation of Lapatinib and its ditosylate salt, which is given in Scheme-II.
Lapatinib ditosylate has been prepared by reacting the tosylate salt of 5-(4-[3- chloro-4-(3-fluorobenzyloxy)-anilino]-6-quinazolinyl)-furan-2-carbaldehyde of Formula (3) with 2-(methylsulfonyl)ethylamine in the presence of base (diisopropyl- ethylamine) followed by reduction with sodium triacetoxyborohydride to obtain Lapatinib base which is converted to Lapatinib ditosylate anhydrate by adding para- toulenesulfonic acid. Conversion to Lapatinib ditosylate monohydrate is carried out using THF/H20. Intercon vers ion to the anhydrate of the ditosylate salt and back to monohydrate is carried out with methanol and water respectively.

(lla)
WO201 1039759, filed by Natco Pharma also describes a process for the preparation of Lapatinib from 2-amino benzonitrile, as given in scheme-Ill. Firstly, 2- aminobenzonitrile (6) is reacted with iodine monochloride in acetic acid medium to form compound of Formula (7) which is recrystallized from mixture of hexane and toluene. The compound of Formula (1) is reacted with N,N-dimethylformamide dimethy|acetal in an organic solvent such as toluene or xylene to form novel compound of Formula (8). The compound of Formula (7) is then coupled with compound of Formula (8) in presence of acid catalyst such as trifluoroacetic acid, formic acid or acetic acid to form compound of Formula (3). The compound of Formula (3) is the subjected to Suzuki coupling with 5-formyl-2-furyl boronic acid in ethereal solvent in the presence of catalyst selected from palladium (II) acetate, palladium (II) chloride, and palladium on carbon to form aldehyde compound of Formula (4). The compound of Formula (4) is reacted with 2-methanesulphonyl ethylamine or its salt to produce imine compound of Formula (VI) which is reduced with sodium borohydride to form Lapatinib base (II). The crude Lapatinib base is purified by crystallization from organic solvents. The purified Lapatinib base is converted into Lapatinib ditosylate anhydrous by treating Lapatinib base in organic solvent with /7-toluenesulfonic acid monohydrate which is then recrystallized from aqueous alcohol to produce pharmaceutically acceptable Lapatinib ditosylate monohydrate. The process is depicted in Scheme-Ill.
-IH

Lapatinib (II) WO2010017387, filed by Teva relates to Lapatinib intermediates and process for the preparation of Lapatinib base and Lapatinib ditosylate. The application relates to highly pure intermediate of Formula (2), 3-chloro-4-(3-fluorobenzyloxy)aniline which is prepared by reducing a compound of Formula (1), 3-chloro-4-(3- fluorobenzyloxy)nitrobenzene, with iron and ammonium chloride system in the presence of a C1 -C4 alcohol and water at refluxing temperature. The application also relates to highly pure intermediate of Formula (3), N-[3-chloro-4-(3-fluorobenzyloxy)- phenyl]-6-iodoquinazolin-4-amine, which is prepared in one-pot process from compound of Formula (1 ) by reduction using iron and ammonium chloride system in presence of C1 -C4 alcohol and water. The compound of Formula (3) is reacted with 5- formyl-2-furanboronic acid in the presence of a palladium catalyst and a base in a polar organic solvent to obtain Lapatinib aldehyde base, compound of Formula (4). Optionally, Lapatinib aldehyde base is combined with /? oluenesulfonic acid to obtain Lapatinib aldehyde monotosylate, compound of Formula (5). The invention further provides a process for the preparation of Lapatinib base. Lapatinib aldehyde base or its salt is combined with methylsulfonylethylamine or its hydrochloride salt, acetic acid, an inorganic base in an organic solvent and a reducing agent (sodium triacetoxyborohydride) to form Lapatinib base. Lapatinib base is further purified by using organic solvents. Lapatinib base obtained is further converted to Lapatinib ditosylate. The process is depicted in scheme-IV.
Scheme-IV

Example-5
Preparation of Lapatinib Ditosylate
To a stirred mixture of Sodiumtriacetoxyborohydride (0.21 g) in Tetrahydrofuran (THF)(2.4 ml) was added N-(3-Chloro-4-(3-fluorobenzyloxy)phenyl)-6-(5-((2- (methylsulfonyl)ethylimino)- methyl)furan-2-yl)quinazolin-4-amine (0.2 g) in THF. The reaction mixture was stirred for 1 hour at 20-25 °C. Reaction was monitored by TLC and on completion of reaction, aqueous NaQH (0.16 g NaOH to 0.8 g demineralized water) was added. The organic layer was separated and added p- Toluenesulfonic acid (0.42) in THF (0.6 ml) and stirred for 3 hours. The solid was filtered and dried under vacuum at 60-65°C till constant weight.
Weight: 0.15 g
Yield: 46.9 %
Purity by HPLC: 96.16%
MS (ES+) m/z: 581 [M+H]+ & 583 [M+H+2]+
1H NMR (400 MHz; DMSO-d6): 2.28 (s, 6H), 3.14 (s, 3H), 3.44 (t, J=8.0 Hz, 2H), 3.55 (t, J=8.0 Hz, 2H), 4.46 (s, 2H), 5.31 (s, 2H), 6.89 (br s, 1H), 7.10 (d, J=7.2 Hz, 4H), 7.20 (m, 1H), 7.23 (br s, 1H), 7.31- 7.36 (m, 3H), 7.47 (d, J=7.2 Hz, 4H), 7.63 (d, J=8.8 Hz, IH), 7.89 (br s, IH), 7.92 (d, J=8.8 Hz, IH), 8.39 (d, J=8.8 Hz, IH), 8.89 (s, IH), 8.98 (s, IH), 9.28 (s, IH, NH), 11.18 (s, IH, NH).
………………………………………….
Patent
http://www.google.com/patents/WO2008024439A2?cl=en

…………………………………………..
Patent
http://www.google.co.in/patents/US7157466
The free base and HCl salts of the compounds of Formulae (I), (II), (III), and (IV), may be prepared according to the procedures of International Patent Application No. PCT/EP99100048, filed Jan. 8, 1999, and published as WO 99/35146 on Jul. 15, 1999, referred to above. A schematic of such procedures is presented in Scheme A following. The specific page references given are to WO 99/35146. The free base of the compound of formula II is used as an example of the general scheme.


The compound of formula (II), i.e., N-{3-Chloro-4-[(3-fluorobenzyl) oxy]phenyl}-6-[5-({[2-(methanesulphonyl) ethyl]amino}methyl)-2-furyl]-4-quinazolinamine ditosylate has been prepared in two distinct forms, an anhydrate form (Formula II′ in Scheme B) and a monohydrate form (Formula II″ in Scheme B). The relationship of these forms is illustrated in Scheme B below. The anhydrate form of N-{3-Chloro-4-[(3-fluorobenzyl) oxy]phenyl}-6-[5-({[2-(methanesulphonyl)ethyl]amino}methyl)-2-furyl]-4-quinazolinamine ditosylate may be prepared by (a) reacting the tosylate salt of 5-(4-[3-chloro-4-(3-fluorobenzyloxy)-anilino]-6-quinazolinyl)-furan-2-carbaldehyde (formula B in Scheme B) with 2-(methylsulfone)ethylamine in tetrahydrofuran in the presence of diisopropyl-ethylamine followed by (b) the introduction of this solution into to a slurry of sodium triacetoxyborohydride in tetrahydrofuran at room temperature, (c) adding 5N sodium hydroxide to adjust the pH to within a range of 10–11, (d) separating the organic tetrahydrofuran phase, and then (e) adding para-toulenesulfonic acid hydrate to the organic phase to provide the ditosylate anhydrate. Interconversion to the monohydrate and back to the anhydrate of the ditosylate salt compounds of the invention is as depicted in Scheme B. The tosylate salt of 5-(4-[3-chloro-4-(3-fluorobenzyloxy)-anilino]-6-quinazolinyl)-furan-2-carbaldehyde is prepared from the HCl salt of the carbaldehyde (Formula A of Scheme B). Preparation of N-{3-Chloro-4-[(3-fluorobenzyl) oxy]phenyl}-6-[5-({[2-(methanesulphonyl)ethyl]amino}methyl)-2-furyl]-4-quinazolinamine ditosylate and the anhydrate and monohydrate forms thereof are utilized as an example. As recognized by those skilled in the art, other compounds of formula I and anhydrate and hydrate forms thereof may be prepared by similar methods.

Compound A of Scheme B may be prepared by various synthetic strategies, other that the strategy recited in Scheme A above, utilizing the palladium(O) mediated coupling of quinazoline and substituted furan intermediates.
Example 8
Preparation of N-{3-Chloro-4-[(3-fluorobenzyl) oxy]phenyl}-6-[5-({[2-(methanesulphonyl)ethyl]amino}methyl)-2-furyl]-4-quinazolinamine ditosylate anhydrate (Anhydrate Form of Compound of Formula II)
To a 20 L reactor was added 13.3 vol of THF followed by 0.62 wt (2.93 mol) of NaBH(OAc)3. The 20 L reactor was set to maintain contents at 20° C. A second 20 L reactor was charged with 1000 grams, (1.55 mol) of 5-(4-[3-chloro-4-(3-fluorobenzyloxy)-anilino]-6-quinazolinyl)-furan-2-carbaldehyde 4-methyl benzenesulfonate prepared by the procedure of Example 7 and 6.7 vol of THF. To the THF solution of 5-(4-[3-chloro-4-(3-fluorobenzyloxy)-anilino]-6-quinazolinyl)-furan-2-carbaldehyde 4-methylbenzenesulfonate was added 0.325 vol (1.86 mol) diisopropylethylamine followed by 0.32 wt of 2-(methylsulfone)ethylamine, (321 g, 2.6 mol) and 0.15 vol of IPA. After 1 hour, the preformed imine/THF solution was transferred by vacuum to the stirred suspension of NaBH(OAC)3 in the first 20 L reactor over 10 minutes. After 90 minutes, 4 vol of 5N NaOH was added over 40 min via a pump. This solution was allowed to stir for 15 minutes after which the stirrer was switched off and the layers were allowed to separate. The aqueous layer was drained from the bottom of the reactor and the organic layer transferred to the empty 20 L reactor through a teflon-lined stainless steel jacketed transfer hose outfitted with an in-line 0.45 μm filter. To this solution was added a 2 vol THF solution of 4 wt (1180 g, 6.2 mole) of p-toluenesulfonic acid monohydrate over 5 min. A yellowish precipitate was observed to come out of solution and this was allowed to stir at room temperature for 12 hours. The reaction was drained from the bottom of the reactor and filtered through a ceramic filter lined with paper. The yellow filter cake was washed with 1 vol of a 95:5 THF water solution and allowed to air dry overnight. After suctioning dry for 12 hours, the yellow filter cake was transferred to two glass trays and placed in the drying oven (42° C.) under house vacuum (18 in Hg) with a nitrogen bleed. The two glass trays were removed from the oven and allowed to cool to room temperature and sampled accordingly. The isolated yield of N-{3-Chloro-4-[(3-fluorobenzyl) oxy]phenyl}-6-[5-({[2-(methane-sulphonyl)ethyl]amino}methyl)-2-furyl]-4-quinazolinamine ditosylate (anhydrate) was 1264 grams (1.3 wt, 88%; 1443 g Th) and was a yellow solid.
Approximately 50 mg of the product was transferred to a Karl Fisher Volumetric Moisture Apparatus (model DL35, Mettler, Hightstown, N.J.), which was operated according to the manufacturer’s instructions. The anhydrate water content was determined to be 0.31%.
Example 10Preparation of N-{3-Chloro-4-[(3-fluorobenzyl) oxy]phenyl}-6-[5-({[2-(methanesulphonyl)ethyl]amino}methyl)-2-furyl]-4-quinazolinamine ditosylate monohydrate (Monohydrate Form of Compound of Formula II)
A 20 L reactor was charged with 1 wt (930 g, 1.0 mol) of N-{3-Chloro-4-[(3-fluorobenzyl) oxy]phenyl}-6-[5-({[2-(methanesulphonyl)ethyl]amino}methyl)-2-furyl]-4-quinazolinamine ditosylate anhydrate prepared using the procedure of Example 8. To this was added 10 volumes of a pre-mixed 8:2 THF:deionized water solution and the reactor was heated to 65° C. Complete dissolution was observed at 50° C. The clear reaction mixture was transferred to another 20 L reactor through a stainless steel jacketed transfer hose that was equipped with an in-line 5.0 μm cartridge filter. The empty 20 L reactor and the filter line were washed with 0.2 vol of the pre-mixed 8:2 THF:deionized water solution. An additional 1 vol of pre-mixed 8:2 THF:deionized water solution was used to wash the material into the reaction mixture. The 20 L reactor was heated to ˜80° C. The reaction temperature was then ramped down to 55° C. over 2 hours and then to 45° C. over 10 hours. After 10 hours, the temperature was adjusted to 25° C. and the reaction mixture allowed to stir at room temperature for 45 minutes. The yellow precipitate was drained from the bottom of the 20 L reactor into a ceramic filter lined with paper. The flow was fast and smooth and the filter rate very good. The yellow filter cake was washed with 0.6 volumes of a pre-mixed 8:2 THF:deionized water solution and the yellow solid was air dried for 4 hours and placed into a glass tray. The glass tray was placed in a vacuum oven under house vacuum (˜18 in Hg) at 60° C. with a nitrogen bleed for 2 days. After removal from the oven, the material was sampled accordingly. The yield was 743 grams (0.8 wt, 80%; 930 g th) as a bright yellow, crystalline solid.
Approximately 50 mg of the product was transferred to a Karl Fisher Volumetric Moisture Apparatus (model DL35, Mettler, Hightstown, N.J.), which was operated according to the manufacturer’s instructions. The monohydrate water content was determined to be 1.99%, which is in agreement with the theoretical value of 1.92%.

Literature References:
Reversible dual inhibitor of ErbB1 and ErbB2 tyrosine kinases. Prepn: M. C. Carter et al., WO 9935146(1999 to Glaxo); eidem, US6727256 (2004 to SmithKline Beecham).
Mechanism of action study: W. Xia et al., Oncogene 21, 6255 (2002); and crystal structure in complex with epidermal growth factor receptor (EGFR, ErbB1): E. R. Wood et al., Cancer Res. 64, 6652 (2004).
In vitro antitumor activity in combination with anti-ErbB2 antibodies: W. Xia et al., Oncogene 24, 6213 (2005). Biologic effects on tumor growth: N. L. Spector et al., J. Clin. Oncol. 23, 2502 (2005).
Pharmacokinetics and clinical activity in metastatic carcinomas: H. A. Burris III et al., ibid. 5305.
Review of clinical development: T. E. Kim, J. R. Murren, IDrugs6, 886-893 (2003); H. A. Burris III, Oncologist 9, Suppl. 3, 10-15 (2004).
Lapatinib Ditosylate [USAN]
- Lapatinib ditosylate monohydrate
- Tykerb
- Tyverb
- UNII-G873GX646R
- KS-1300; 388082-78-8
-
N-(3-Chloro-4-((3-fluorobenzyl)oxy)phenyl)-6-(5-(((2-(methylsulfonyl)ethyl)amino)methyl)furan-2-yl)quinazolin-4-amine bis(4-methylbenzenesulfonate) monohydrate
Dosages/Routes/Forms
| Strength | Form/Route | Marketing Status | RLD | TE Code |
|---|---|---|---|---|
| EQ 250MG BASE | TABLET;ORAL | 1 | 1 |
Approval History
References
- Burris HA (2004). “Dual kinase inhibition in the treatment of breast cancer: initial experience with the EGFR/ErbB-2 inhibitor lapatinib”. Oncologist. 9 Suppl 3: 10–5.doi:10.1634/theoncologist.9-suppl_3-10. PMID 15163842.
- Higa GM & Abraham J (September 2007). “Lapatinib in the treatment of breast cancer”. Expert Review of Anticancer Therapy (log in required) (Future Drugs) 7(9): 1183–92. doi:10.1586/14737140.7.9.1183. PMID 17892419.
- Pazdur, Richard (14 January 2011). “FDA Approval for Lapatinib Ditosylate”.Womens Health (Lond Engl) (Cancer.gov) 6 (2): 173. doi:10.2217/whe.10.11.PMID 20187722.
- ^ Jump up to:a b c d “GlaxoSmithKline receives marketing authorisation in the EU for Tyverb (lapatinib), the first oral targeted therapy for ErbB2-positive breast cancer” (Press release). GlaxoSmithKline. 2008-06-12. Retrieved 2008-06-21.
- ^ Jump up to:a b c “GlaxoSmithKline Reports Positive New Data On Tykerb (lapatinib) At The 2007 American Society Of Clinical Oncology (ASCO) Annual Meeting” (Press release). Medical News Today. June 4, 2007. Retrieved December 2, 2008.
- “Data Sheet: TYKERB”. Medsafe. New Zealand Medicines and Medical Devices Safety Authority. March 12, 2008. Retrieved December 2, 2008.
- Jump up^ Kulkarni, Kaustubh (2 August 2013). “India revokes GSK cancer drug patent in latest Big Pharma blow”. Reuters (Mumbai, India: Reuters). Retrieved 2 August 2013.
- Wood, ER, Truesdale, AT, McDonald, OB, Yuan, D, Hassell, A, Dickerson, SH, Ellis, B, Pennisi, C et al. (2004). “A unique structure for epidermal growth factor receptor bound to GW572016 (Lapatinib): relationships among protein conformation, inhibitor off-rate, and receptor activity in tumor cells”. Cancer Research 64 (18): 6652–9. doi:10.1158/0008-5472.CAN-04-1168. PMID 15374980.
- Dr. Angel Rodriguez (April 2008). “New type of drug shrinks primary breast cancer tumors significantly in just six weeks; research provides leads to a new target in cancer treatment – the cancer stem cell”.
- Nelson MH, Dolder CR (February 2006). “Lapatinib: a novel dual tyrosine kinase inhibitor with activity in solid tumors”. Ann Pharmacother 40 (2): 261–9.doi:10.1345/aph.1G387. PMID 16418322.
- Jump up^ Geyer CE, Forster J, Lindquist D, et al. (December 2006). “Lapatinib plus capecitabine for HER2-positive advanced breast cancer”. N. Engl. J. Med. 355 (26): 2733–43.doi:10.1056/NEJMoa064320. PMID 17192538.
- J Burris HA, Hurwitz HI, Dees EC, et al. (August 2005). “Phase I safety, pharmacokinetics, and clinical activity study of lapatinib (GW572016), a reversible dual inhibitor of epidermal growth factor receptor tyrosine kinases, in heavily pretreated patients with metastatic carcinomas”. J. Clin. Oncol. 23 (23): 5305–13.doi:10.1200/JCO.2005.16.584. PMID 15955900.
- J NCI Cancer Drug Information. FDA Approval for Lapatinib Ditosylate (Tykerb®). Retrieved 27 January 2014.
- |url=http://www.bioportfolio.com/news/article/1492867/GSK-Tykerb-Tyverb-Phase-III-gastric-cancer-study-fails-to-meet-primary.html
External links
| WO1999035146A1 | Jan 8, 1999 | Jul 15, 1999 | Glaxo Group Ltd | Bicyclic heteroaromatic compounds as protein tyrosine kinase inhibitors |
| WO2010017387A2 | Aug 6, 2009 | Feb 11, 2010 | Teva Pharmaceutical Industries Ltd. | Lapatinib intermediates |
| WO2011039759A1 | Sep 29, 2009 | Apr 7, 2011 | Natco Pharma Limited | A new process for the preparation of lapatinib and its pharmaceutically acceptable salts |
| US6727256 | Jan 8, 1999 | Apr 27, 2004 | Smithkline Beecham Corporation | 4-aminoquinazoline derivatives as anticarcinogenic agents |
| US7157466 | Jun 28, 2001 | Jan 2, 2007 | Smithkline Beecham (Cork) Limited | Quinazoline ditosylate salt compounds |
| WO1998002434A1 * | Jul 11, 1997 | Jan 22, 1998 | Malcolm Clive Carter | Fused heterocyclic compounds as protein tyrosine kinase inhibitors |
| WO2007121279A2 * | Apr 12, 2007 | Oct 25, 2007 | Tona Morgan Gilmer | Cancer treatment method |
Hydrogenation in flow: homogenous and heterogeneous catalysts using Teflon AF-2400 to effect gas-liquid contact at elevated pressure

http://pubs.rsc.org/en/Content/ArticleLanding/2011/SC/c1sc00055a#!divAbstract
M. O’Brien, N. Taylor, A. Polyzos, I.R. Baxendale, S.V. Ley, Chem. Sci. 2011, 2, 1250-1257.
A Tube-in-Tube reactor/injector has been developed, based on a gas-permeable Teflon AF-2400 membrane, which allows both heterogeneous and homogeneous catalytic hydrogenation reactions to be efficiently carried out at elevated pressure in flow, thereby increasing the safety profile of these reactions. Measurements of the gas permeation through the tubing and uptake into solution, using both a burette method and a novel computer-assisted ‘bubble counting’ technique, indicate that permeation/dissolution follows Henry’s law and that saturation is achieved extremely rapidly. The same gas-permeable membrane has also been shown to efficiently effect removal of excess unreacted hydrogen, thus enabling further downstream reaction/processing.

GMP Question & Answer Guide
DRUG REGULATORY AFFAIRS INTERNATIONAL

GMP Question & Answer Guide
The requirements defined in the GMP Guidelines often leave room for interpretation. However, regulators worldwide (EMA, FDA, TGA etc) sometimes publish frequently asked questions on GMP. In a new ECA document these Q&As are summarized in a single source. The Q&As are structured in 4 main GMP Areas (General GMPs, GMP for APIs, GMP for Medicinal Products, GMP for IMPs). The document contains 150 pages of Q&As and is available at no cost on the ECA Webpage. A first set of ECA Q&As have also been included and additional GMP Q&As are planned for the future. Here you can access the GMP Questions and Answers Guide
http://www.gmp-compliance.org/eca_gmp-guide.html
| GMP Question and Answer Guide „GMP Advisor“ |
| http://www.gmp-compliance.org/eca_gmp-guide.html |
| Searching for concrete answers to GMP questions is a time-consuming activity. The document we now offer is intended to provide a single source of information. We have summarized GMP questions… |
View original post 334 more words
Still a GMP problem? Or already a criminal act? Do we need more stringent measures and enforcement in certain situations?
DRUG REGULATORY AFFAIRS INTERNATIONAL
Still a GMP problem? Or already a criminal act? Do we need more stringent measures and enforcement in certain situations?
Sometimes EU and FDA Inspectors discover serious GMP deviations and fraud during an inspection. What are the consequences and do we need to think about additional measures? Please read more in our GMP News.
When GMP issues are discussed, different interpretations are possible. Sometimes, the implementation of GMP regulations and expectations can be a challenge. However, everyone involved should do his/her best to make sure that GMP has been put in place and that patient safety is ultimately guaranteed.
Now and again, companies may receive GMP Non-Compliance Statements from EU Inspectors or Warning Letters from US FDA Inspectors because of non-compliance issues identified during inspections. This is a serious situation for the companies involved. Organisational problems and frequently also gross mismanagement can be the reasons for these deviations. In…
View original post 436 more words
Commentary Regarding new USP Chapters and for Particulate Matter Guidance
DRUG REGULATORY AFFAIRS INTERNATIONAL
Commentary Regarding new USP Chapters and for Particulate Matter Guidance
There are new chapters in the USP regarding testing of subvisible particles. Chapter Subvisible Particulate Matter in Therapeutic Protein Injections <787> became official August 1, 2014. The informational chapter <1787> was developed to support chapter <787> and will be published in USP 38 in November and become official on May 1, 2015. Read more.
During the current (2010-2015) USP Expert Committee cycle, the Dosage Forms Expert Committee has developed both new and revised general chapters that provide guidance on particulate matter content of injectable drug products. For visible particles, methods are based upon human detection sensitivity as described in Visible Particulates in Injections <790>, which applies to all sterile injectable dosage forms. For subvisible particle content, which is based upon instrumental determination, new particulate matter guidance has been established specifically for sterile injectable biotherapeutic products.
The new…
View original post 490 more words
IMATINIB


 Imatinib
Imatinib
CAS No:- [152459-95-5]
IUPAC Name:- 4-[(4-Methyl-1-piperazinyl)methyl]-N-[4-methyl-3-[[4-(3-pyridinyl)-2-pyrimidinyl]amino]phenyl]benzamide
M. P.:- 211-213 °C
MW: 493.604
4-[(4-methylpiperazin-1-yl)methyl]-N-(4-methyl-3-{[4-(pyridin-3-yl)pyrimidin-2-yl]amino}phenyl)benzamide
-[(4-methylpiperazin-1-yl)methyl]-N-(4-methyl-3-{[4-(pyridin-3-yl)pyrimidin-2-yl]amino}phenyl)benzamide
N-(4-methyl-3-((4-(pyridin-3-yl)pyrimidin-2-yl)amino)phenyl)-4-((4-methylpiperazin-1-
yl)methyl)benzamide
Imatinib (INN), marketed by Novartis as Gleevec (Canada, South Africa and the USA) or Glivec (Australia, Europe and Latin America), and sometimes referred to by its investigational name STI-571, is a tyrosine-kinase inhibitor used in the treatment of multiple cancers, most notably Philadelphia chromosome-positive (Ph+) chronic myelogenous leukemia (CML).[1]
Like all tyrosine-kinase inhibitors, imatinib works by preventing a tyrosine kinase enzyme, in this case BCR-Abl, fromphosphorylating subsequent proteins and initiating the signalling cascade necessary for cancer growth and survival, thus preventing the growth of cancer cells and leading to their death by apoptosis.[2] Because the BCR-Abl tyrosine kinase enzyme exists only in cancer cells and not in healthy cells, imatinib works as a form of targeted therapy—only cancer cells are killed through the drug’s action.[3] In this regard, imatinib was one of the first cancer therapies to show the potential for such targeted action, and is often cited as a paradigm for research in cancer therapeutics.[4]
Imatinib has been cited as the first of the exceptionally expensive cancer drugs, costing $92,000 a year. Doctors and patients complain that this is excessive, given that its development costs have been recovered many times over, and that the costs of synthesizing the drug are orders of magnitude lower. In the USA, the patent protecting the active principle will expire on 4 January 2015 while the patent protecting the beta crystal form of the active principal ingredient will expire on 23 May 2019.[5]
The developers of imatinib were awarded the Lasker Award in 2009 and the Japan Prize in 2012.[6][7]

bcr-abl kinase (green), which causes CML, inhibited by imatinib (red; small molecule).
Medical uses
Imatinib is used to treat chronic myelogenous leukemia (CML), gastrointestinal stromal tumors (GISTs) and a number of othermalignancies.
Chronic myelogenous leukemia
The U.S. Food and Drug Administration (FDA) has approved imatinib as first-line treatment for Philadelphia chromosome-positive CML, both in adults and children. The drug is approved in multiple Philadelphia chromosome-positive cases of CML, including after stem cell transplant, in blast crisis, and newly diagnosed.[8]
Gastrointestinal stromal tumors
The FDA first granted approval for advanced GIST patients in 2002. On 1 February 2012, imatinib was approved for use after the surgical removal of KIT-positive tumors to help prevent recurrence.[9] The drug is also approved in unresectable KIT-positive GISTs.[8]
Other
The FDA has approved imatinib for use in adult patients with relapsed or refractory Philadelphia chromosome-positive acute lymphoblastic leukemia (Ph+ ALL), myelodysplastic/ myeloproliferative diseases associated with platelet-derived growth factor receptor gene rearrangements, aggressive systemic mastocytosis without or an unknown D816V c-KIT mutation, hypereosinophilic syndrome and/or chronic eosinophilic leukemia who have the FIP1L1-PDGFRα fusion kinase (CHIC2 allele deletion) or FIP1L1-PDGFRα fusion kinase negative or unknown, unresectable, recurrent and/or metastaticdermatofibrosarcoma protuberans.[8] On 25 January 2013, Gleevec was approved for use in children with Ph+ ALL.[10]
For treatment of progressive plexiform neurofibromas associated with neurofibromatosis type I, early research has shown potential for using the c-KIT tyrosine kinase blocking properties of imatinib.[11][12][13][14]
Legal challenge to generics
In 2007, imatinib became a test case through which Novartis challenged India’s patent laws. A win for Novartis would make it harder for Indian companies to produce generic versions of drugs still manufactured under patent elsewhere in the world. Doctors Without Borders argues a change in law would make it impossible for Indian companies to produce cheap generic antiretrovirals (anti-HIV medication), thus making it impossible for Third World countries to buy these essential medicines.[43] On 6 August 2007, the Madras High Court dismissed the writ petition filed by Novartis challenging the constitutionality of Section 3(d) of Indian Patent Act, and deferred to the World Trade Organization (WTO) forum to resolve the TRIPS compliance question. As of 2009 India has refused to grant patent exclusivity..
On April 01, 2013 Supreme Court of India dismissed the plea of Novartis for the grant of patent.

in germany
Mechanism of action

| Imatinib | |
|---|---|
| Drug mechanism | |

Crystallographic structure of tyrosine-protein kinase ABL (rainbow colored, N-terminus = blue, C-terminus = red) complexed with imatinib (spheres, carbon = white, oxygen = red, nitrogen = blue).[31]
|
|
| Therapeutic use | chronic myelogenous leukemia |
| Biological target | ABL, c-kit, PDGF-R |
| Mechanism of action | Tyrosine-kinase inhibitor |
| External links | |
| ATC code | L01XE01 |
| PDB ligand id | STI: PDBe, RCSB PDB |
| LIGPLOT | 1iep |
Imatinib is a 2-phenyl amino pyrimidine derivative that functions as a specific inhibitor of a number of tyrosine kinase enzymes. It occupies the TK active site, leading to a decrease in activity.
There are a large number of TK enzymes in the body, including the insulin receptor. Imatinib is specific for the TK domain inabl(the Abelson proto-oncogene), c-kit and PDGF-R (platelet-derived growth factorreceptor).
In chronic myelogenous leukemia, the Philadelphia chromosome leads to a fusion protein of abl with bcr(breakpoint cluster region), termed bcr-abl. As this is now aconstitutively active tyrosine kinase, imatinib is used to decrease bcr-abl activity.
The active sites of tyrosine kinases each have a binding site for ATP. The enzymatic activity catalyzed by a tyrosine kinase is the transfer of the terminal phosphate from ATP to tyrosine residues on its substrates, a process known as protein tyrosinephosphorylation. Imatinib works by binding close to the ATP binding site of bcr-abl, locking it in a closed or self-inhibited conformation, and therefore inhibiting the enzyme activity of the protein semi-competitively.[32] This fact explains why many BCR-ABL mutations can cause resistance to imatinib by shifting its equilibrium toward the open or active conformation.[33]
Imatinib is quite selective for bcr-abl – it does also inhibit other targets mentioned above (c-kit and PDGF-R), but no other knowntyrosine kinases. Imatinib also inhibits the abl protein of non-cancer cells but cells normally have additional redundant tyrosine kinases which allow them to continue to function even if abl tyrosine kinase is inhibited. Some tumor cells, however, have a dependence on bcr-abl.[34] Inhibition of the bcr-abl tyrosine kinase also stimulates its entry in to the nucleus, where it is unable to perform any of its normal anti-apoptopic functions.[35]
The Bcr-Abl pathway has many downstream pathways including the Ras/MapK pathway, which leads to increased proliferation due to increased growth factor-independent cell growth. It also affects the Src/Pax/Fak/Rac pathway. This affects the cytoskeleton, which leads to increased cell motility and decreased adhesion. The PI/PI3K/AKT/BCL-2 pathway is also affected. BCL-2 is responsible for keeping the mitochondria stable; this suppresses cell death by apoptosis and increases survival. The last pathway that Bcr-Abl affects is the JAK/STAT pathway, which is responsible for proliferation.[36]
synthesis
…………………………
Imatinib is known as an inhibitor of protein-tyrosine kinase and is indicated for the treatment of chronic myeloid leukemia (CML). Imatinib also has potential for the treatment of various other cancers that express these kinase including acute lymphocyte leukemia and certain solid tumors. It can also be used for the treatment of atherosclerosis, thrombosis, restenosis, or fibrosis. Thus, imatinib can also be used for the treatment of non-malignant diseases. Imatinib is usually administered orally in the form of a suitable salt, e.g., in the form of imatinib mesylate.
The chemical name of Imatinib is 4-(4-methyl piperazine -1- methyl) -N-4-methyl-3-[4- (3- pyridyl) pyrimidine-2-amino] – benzamide and is represented by the following structural formula:

(Imatinib)
Imatinib Mesylate is an inhibitor of signal transduction (STI571) invented by Novartis AG after 7 years of hard work; it is the first inhibitor of cancer signal transduction ratified in the whole world. It is sold by Novartis as Gleevec capsules containing imatinib mesylate in amounts equivalent to 100 mg or 400 mg of imatinib free base.
Imatinib Mesylate is the rare drug in America, European Union and Japan. In May 10, 2001, it was ratified by American Food and Drug Administration (FDA) to treat the chronic myelogenous leukemia patients. EP0564409 (US5521 184) describes the process for the preparation of imatinib and the use thereof, especially as an anti tumour agent.
There are generally two synthetic routes for synthesis of Imatinib, suitable for the industrial production. One synthetic process as described in scheme-I comprises using 2-methyl-5-nitroaniline as the raw material which is reacted with cyanamide to obtain guanidine; cyclization reaction with 3-dimethylamino-l-(3-pyridyl)-2-propylene-l- ketone; reduction step of nitro to amine and condensation reaction with 4- (Chloromethyl)benzoyl chloride and N-methylpiperazidine to obtain Imatinib (WO 2004/108669). -I

Scheme-2 describes the successful process for the synthesis of Imatinib using 4-methyl-3- nitroanilines as the raw material, comprising reacting 4-methyl-3-nitroanilines with 4- (Chloromethyl)benzoyl chloride and N-methyl piperazidine in turns; followed by reduction of nitro group to amino group; then reaction with cyanamide to obtain guanidine; finally cyclization reaction with 3- dimethyl amino- 1 -(3- pyridyl)-2- propylene-1 -ketone to obtain Imatinib (WO 03/066613). The said PCT application discloses the use of 4-4-(methyl piperazin-l-ylmethyl)-benzoic acid methyl ester as one of the raw material but rest of the reactants are different from that of N-(5-amino -2- methylphenyl)-4-(3-pyridyl)-2-pyrimidine amine in presence of trimethyl aluminium.
Scheme-2

Common feature of the processes for preparing imatinib according to (WO 2004/108669) and (WO03/066613) lies in use of cyanamide as a reagent. The main difference between the two routes is that the reaction sequence of cyclization of pyrimidine chain is different. Example 10 of PCT International Publication no. WO 2003/066613 is less applicable to industrial purposes. These include the reaction between N-(3-bromo-4-methyl-phenyl)-4- (4-methyl-piperazin-l -ylmethyl)-benzamide and 4-(3-pyridyl)-2-pyrimidineamine which uses a mixture of rac-BINAP (a phosphine oxide catalyst) and Pd2 (dba)3*CHCl3. These catalysts are very expensive, therefore, their use is unfit for commercial production.
CN1630648A describes a process comprising reaction of 3- bromine-4- methyl aniline with 4-(4-methyl-piperazin- methyl) methyl benzoate in presence of trimethyl-Aluminum to obtain N-(4-methyl-3-bromobenzene)-4-(4-methyl-piperazin- 1 -methyl)-benzamide, which further reacts with 2-amino-4-(3-pyridyl)- pyrimidine in presence of palladium as catalyst to obtain Imatinib.


The drawback of the above process is the use of trimethyl-Aluminum, which is flammable and reacts severely when comes in contact with water.
CN101016293A describes another process using N-(4-methyl-3-3- aminophenyl)-4-(4- methyl-piperazin-1 -methyl)- benzamide as the raw material. The said raw material is reacted with 2-halogen-4-(3-pyridyl)- pyrimidine to obtain Imatinib.

The process disclosed in CN 101016293 A comprises use of halogenated agent, such as phosphorus oxychloride, which is used to synthesize 2-halogeno-4- methyl- (3-pyridyl) – pyridine is lachrymator and corrosive and has great influence to the surroundings. EP0564409 describes a coupling reaction between N-(5-amino -2-methylphenyl)-4-(3- pyridyl)-2-pyrimidine amine and 4-(4-methyl piperazin-l-ylmethyl)-benzoyl chloride in the presence of high quantity of pyridine to starting reactant amine N-(5-amino -2- methylphenyl)-4-(3-pyridyl)-2-pyrimidine amine. The ratio of the pyridine to the said reactant is 138 which is equivalent to about 40 parts v/w. Use of such a large quantity of pyridine is unsafe as it is a toxic solvent according to ICH guidelines. The workup of the reaction comprises evaporation of the remaining pyridine and further processing, which finally involves chromatography for purification, which is highly undesirable on industrial scale because it is expensive and time consuming.

US2006/0149061 and US20060223817 also discloses a similar synthetic approach comprising the use of similar pyridine /starting amine ratio (140 equivalents which is equals about 41 parts v/w). The product obtained is purified by slurring in ethyl acetate.
WO2004/074502 describes a coupling reaction between N-(5-amino -2-methylphenyl)-4- (3-pyridyl)-2-pyrimidine amine and 4-(4-methyl piperazin-l-ylmethyl)-benzoyl chloride wherein solvent like dimethyl pharmamide , dimethyl acetamide, N-methyl pyrilidinone are used as solvents instead of pyridine. However the method described in this patent application lacks an advantage in that the coupling reaction produces the hydrohalide salt of imatinib, e.g. imatinib trihydrochloride monohydrate, which has to be treated with a base in order to afford the imatinib base, thus an extra step is required. Further, conventional methods for coupling N-(5-amino -2-methylphenyl)-4-(3-pyridyl)-2- pyrimidine amine require reaction with an acid halide, e.g. 4-(4-methyl piperazin-1- ylmethyl)-benzoyl chloride, which requires an additional production step that can involve harsh and/or toxic chlorinating agent.

WO2008/1 17298 describes a coupling reaction between N-(5-amino -2-methylphenyl)-4- (3-pyridyl)-2-pyrimidine amine and 4-(4-methyl piperazin-l-ylmethyl)-benzoyl chloride in presence of a base selected from potassium carbonate, sodium carbonate, potassium or sodium hydroxide. Use of potassium carbonate as base results into the formation of Imatinib dihydrochloride which ultimately requires an additional operation of neutralization by using excessive base to get imatinib.

WO2008/136010 describes a coupling reaction between N-(5-amino -2-methylphenyl)-4- (3-pyridyl)-2-pyrimidine amine and 4-(4-methyl piperazin-l-ylmethyl)-benzoyl chloride in presence of base potassium hydroxide resulting into 78.6% yield of crude imatinib base. Preparation of crude requires imatinib hydrochloride preparation during the workup which is then basified to get base in crude form. This also describes maleate salt preparation as mode of purification which is again basified to give pure Imatinib base.

US patent application 2004/0248918 discloses a different approach using N-(5-amino -2- methylphenyl)-4-(3-pyridyl)-2-pyrimidine amine and 4-(2-chloromethyl)-benzoyl chloride as raw material. The reaction for the preparation of Imatinib is carried out in tetrahydrofuran as a reaction solvent and in the presence of pyridine as a base. However the method described in this patent application lacks an advantage as purification of the product requires column chromatography using chloroform: methanol (3: 1 v/v), which is not suitable purification method when performing the reaction on large scale, followed by crystallizati

US patent application 2008/0103305 discloses a process comprising reacting N-(5-amino -2-methylphenyl)-4-(3-pyridyl)-2-pyrimidine amine or its alkyl derivative and an acid salt of 4-[(4-methyl-l-piperazinyl)-methyl] benzoyl derivative as given below in the scheme-3 using pyridine in an amount of about 2 to 10 volumes per gram of the said amine. However the drawback associated with this process is use of pyridine especially when reaction is performed on large scale. -3

………………………….
SYNTHESIS
Inverse synthetic analysis will be divided into four imatinib into fragment A has 1,3 – parents electrical, fragment B are 1,3 – parent nuclear, fragments A and B constitute a pyrimidine ring.
Compound 4 can be obtained in two ways, benzyl bromide 1 and secondary amines 2 by SN2 reaction, or the aldehyde 3 with a secondary amine 2 by reductive amination. Sodium cyanoborohydride electron withdrawing effect of the cyano group, thereby reducing the activity of the negative hydrogen, it may be present in acidic solution. Also in the acidic conditions of aldehydes and secondary amines imine positive ions, which is higher than the activity of aldehyde reduction.This is why the reductive amination reagent with inert negative and hydrogen under acidic conditions. 4 hydrolyzed ester with thionyl chloride into the acid chloride 5 . The reaction of aniline and cyanamide dinucleophile guanidine 7 . Compound 8 and DMF-DMA reaction electrophilic reagent parents 9 , 7 , and 9 ring closure under alkaline conditions to generate 10 . Finally, reduction, amidation, and a salt of imatinib mesylate generated.
………………………………..

An efficient, economic process has been developed for the production of imatinib with 99.99% purity and 50% overall yield from four steps. Formation and control of all possible impurities is described. The synthesis comprises the condensation of N-(5-amino-2-methylphenyl)-4-(3-pyridinyl)-2-pyrimidineamine with 4-(4-methylpiperazinomethyl)benzoyl chloride in isopropyl alcohol solvent in the presence of potassium carbonate to yield imatinib base.
…………………………
DOI: 10.1039/C2OB27003J
http://pubs.rsc.org/en/content/articlelanding/2013/ob/c2ob27003j#!divAbstract


Imatinib (1), nilotinib (2) and dasatinib (3) are Bcr-Abl tyrosine kinase inhibitors approved for the treatment of chronic myelogenous leukemia (CML). This review collates information from the journal and patent literature to provide a comprehensive reference source of the different synthetic methods used to prepare the aforementioned active pharmaceutical ingredients (API’s).

……………………..
|









![[1860-5397-9-265-i37]](http://www.beilstein-journals.org/bjoc/content/inline/1860-5397-9-265-i37.png?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-9-265-i38]](http://www.beilstein-journals.org/bjoc/content/inline/1860-5397-9-265-i38.png?scale=2.0&max-width=1024&background=FFFFFF)



Medicine for Blood Cancer
‘Imitinef Mercilet’ is a medicine which cures blood cancer.
Its available free of cost at “Adyar Cancer Institute in Chennai”.
Create Awareness. It might help someone.Cancer Institute in Adyar, Chennai
‘Imitinef Mercilet’ is apparently an alternative spelling of the drug Imatinib mesylate which is used in the treatment of some forms of leukemia along with other types of cancer. Imatinib, often referred to a “Gleevec”, has proved to be an effective treatment for some forms of cancers. However, “blood cancer” is a generalized term for cancers that affect the blood, lymphatic system or bone marrow. The three types of blood cancer are listed as leukemia, lymphoma, and multiple myeloma. These three malignancies require quite different kinds of treatments. While drugs (including Imatinib), along with other treatments such as radiation can help to slow or even stop the progress of these cancers, there is currently no single drug treatment that can be said to actually cure all such cancers.
Category: Cancer
Address: East Canal Bank Road , Gandhi Nagar
Adyar, Chennai -600020
Landmark: Near Michael School
Phone: 044-24910754 044-24910754 ,
044-24911526 044-24911526 , 044-22350241
Imatinib is a small molecule selectively inhibiting specific tyrosine kinases that has emerged recently as a valuable treatment for patients with advanced GIST. The use of imatinib as monotherapy for the treatment of GIST has been described in PCT publication WO 02/34727, which is here incorporated by reference. However, it has been reported that primary resistance to imatinib is present in a population of patients, for example 13.7% of patients in one study. In addition, a number of patients acquire resistance to treatment with imatinib. More generally this resistance is partial with progression in some lesions, but continuing disease control in other lesions. Hence, these patients remain on imatinib treatment but with a clear need for additional or alternative therapy.
Imatinib is 4-(4-methylpiperazin-1-ylmethyl)-N-[4-methyl-3-(4-pyridin-3-yl)pyrimidin-2-ylamino)phenyl]-benzamide having the formula I

The preparation of imatinib and the use thereof, especially as an anti-tumour agent, are described in Example 21 of European patent application EP-A-0 564 409, which was published on 6 Oct. 1993, and in equivalent applications and patents in numerous other countries, e.g. in U.S. Pat. No. 5,521,184 and in Japanese patent 2706682

The flow-based route required minimal manual intervention and was achieved despite poor solubility of many reaction components
UK chemists have used a combination of flow chemistry methods with solid-supported scavengers and reagents to synthesise the active pharmaceutical ingredient, imatinib, of the anticancer drug Gleevec. The method avoids the need for any manual handling of intermediates and allows the drug to be synthesised in high purity in less than a day.
Gleevec, developed by Novartis, is a tyrosine kinase inhibitor used for the treatment of chronic myeloid leukaemia and gastrointestinal stromal tumours.

READ ALL AT
http://www.rsc.org/chemistryworld/2013/01/flow-synthesis-anticancer-drug
 |
|
|---|---|
 |
|
| IMATINIB |

CREDIT
http://www.veomed.com/va041542042010

‘Wrapping’ Gleevec Fights Drug-Resistant Cancer, Study Shows
http://www.sciencedaily.com/releases/2007/05/070501115127.htm
The anti-cancer drug Gleevec® is far more effective against a drug-resistant strain of cancer when the drug wraps the target with a molecular bandage that seals out water from a critical area. This image shows the bandage (black box) on the modified version of the drug, WBZ-7. (Credit: Image courtesy of Rice University)
A new study in Cancer Research finds that the anti-cancer drug Gleevec® is far more effective against a drug-resistant strain of cancer when the drug wraps the target with a molecular bandage that seals out water from a critical area.


FIG 23.8 Optimization of imatinib as a chemotherapeutic agent. The discovery that 2-phenylaminopyrimidine inhibitors of PKC also inhibit the unrelated v-Abl oncogene turned attention to its potential use in the treatment of chronic myelogenous leukaemia. Starting with the 2-phenylaminopyrimidine backbone, addition of the benzamidine group increased activity against tyrosine kinases, the methyl group reduced its activity against PKC (so-called ‘ target hopping ’ ). Addition of a 3’-pyridyl group improved the activity in cellular assays. Subsequent addition of N -methylpiperazine increased water solubility and oral bioavailability, enabling the drug to survive the stomach and to enter the bloodstream.
……………………..
An automated flow-based synthesis of imatinib: the API of gleevec M.D. Hopkin, I.R. Baxendale, S.V. Ley, J.C.S. Chem. Commun.2010, 46, 2450-2452.

References
- Jump up^ Novartis Pharma AG. Gleevec® (imatinib mesylate) tablets prescribing information. East Hanover, NJ; 2006 Sep. Anon. Drugs of choice for cancer. Treat Guidel Med Lett. 2003; 1:41–52
- Jump up^ Goldman JM, Melo JV (October 2003). “Chronic myeloid leukemia–advances in biology and new approaches to treatment”. N. Engl. J. Med. 349 (15): 1451–64.doi:10.1056/NEJMra020777. PMID 14534339.
- Jump up^ Fausel, C. Targeted chronic myeloid leukemia therapy: Seeking a cure. Am J Health Syst Pharm 64, S9-15 (2007)
- Jump up^ Stegmeier F, Warmuth M, Sellers WR, Dorsch M (May 2010). “Targeted cancer therapies in the twenty-first century: lessons from imatinib”. Clin. Pharmacol. Ther. 87(5): 543–52. doi:10.1038/clpt.2009.297. PMID 20237469.
- Jump up^ “Novartis fails to patent Glivec (Gleevec) in India”.
- Jump up^ Rowley to receive Japan Prize for her role in the development of targeted cancer therapy Eurekalert, Press release, 24 January 2012
- Jump up^ Leukemia Drug and Magnet Material Net Japan Prizes by Dennis Normile, Science Insider, 25 January 2012
- ^ Jump up to:a b c “FDA Highlights and Prescribing Information for Gleevec(imatinib mesylate)”.
- Jump up^ “Prolonged Use of Imatinib in GIST Patients Leads to New FDA Approval”.
- Jump up^ “FDA approves Gleevec for children with acute lymphoblastic leukemia”. FDA News Release. US Food and Drug Administration. 25 January 2013. Retrieved 3 April 2013.
- Jump up^ Yang FC, Ingram DA, Chen S, Zhu Y, Yuan J, Li X, Yang X, Knowles S, Horn W, Li Y, Zhang S, Yang Y, Vakili ST, Yu M, Burns D, Robertson K, Hutchins G, Parada LF, Clapp DW (October 2008). “Nf1-dependent tumors require a microenvironment containing Nf1+/–and c-kit-dependent bone marrow”. Cell 135 (3): 437–48.doi:10.1016/j.cell.2008.08.041. PMC 2788814. PMID 18984156. Lay summary –Science Daily.
- Jump up^ “Gleevec NF1 Trial”. Nfcure.org. Retrieved 2013-04-03.
- Jump up^ “GIST in Neurofibromatosis 1”. Gistsupport.org. 2010-05-14. Retrieved 2013-04-03.
- Jump up^ “”Pilot Study of Gleevec/Imatinib Mesylate (STI-571, NSC 716051) in Neurofibromatosis (NF1) Patient With Plexiform Neurofibromas (0908-09)” (Suspended)”. Clinicaltrials.gov. Retrieved 2013-04-03.
- Jump up^ Droogendijk HJ, Kluin-Nelemans HJ, van Doormaal JJ, Oranje AP, van de Loosdrecht AA, van Daele PL (July 2006). “Imatinib mesylate in the treatment of systemic mastocytosis: a phase II trial”. Cancer 107 (2): 345–51. doi:10.1002/cncr.21996.PMID 16779792.
- Jump up^ Tapper EB, Knowles D, Heffron T, Lawrence EC, Csete M (June 2009). “Portopulmonary hypertension: imatinib as a novel treatment and the Emory experience with this condition”. Transplant. Proc. 41 (5): 1969–71.doi:10.1016/j.transproceed.2009.02.100. PMID 19545770.
- Jump up^ Boucher P, Gotthardt M, Li WP, Anderson RG, Herz J (April 2003). “LRP: role in vascular wall integrity and protection from atherosclerosis”. Science 300 (5617): 329–32.doi:10.1126/science.1082095. PMID 12690199.
- Jump up^ Lassila M, Allen TJ, Cao Z, Thallas V, Jandeleit-Dahm KA, Candido R, Cooper ME (May 2004). “Imatinib attenuates diabetes-associated atherosclerosis”. Arterioscler. Thromb. Vasc. Biol. 24 (5): 935–42. doi:10.1161/01.ATV.0000124105.39900.db.PMID 14988091.
- Jump up^ Reeves PM, Bommarius B, Lebeis S, McNulty S, Christensen J, Swimm A, Chahroudi A, Chavan R, Feinberg MB, Veach D, Bornmann W, Sherman M, Kalman D (July 2005). “Disabling poxvirus pathogenesis by inhibition of Abl-family tyrosine kinases”. Nat. Med.11 (7): 731–9. doi:10.1038/nm1265. PMID 15980865.
- Jump up^ He G, Luo W, Li P, Remmers C, Netzer WJ, Hendrick J, Bettayeb K, Flajolet M, Gorelick F, Wennogle LP, Greengard P (September 2010). “Gamma-secretase activating protein is a therapeutic target for Alzheimer’s disease”. Nature 467 (7311): 95–8.doi:10.1038/nature09325. PMC 2936959. PMID 20811458.
- Jump up^ “Alzheimer’s may start in liver – Health – Alzheimer’s Disease | NBC News”. MSNBC. Retrieved 2013-01-06.
- Jump up^ Holmes C, Boche D, Wilkinson D, Yadegarfar G, Hopkins V, Bayer A, Jones RW, Bullock R, Love S, Neal JW, Zotova E, Nicoll JA (July 2008). “Long-term effects of Abeta42 immunisation in Alzheimer’s disease: follow-up of a randomised, placebo-controlled phase I trial”. Lancet 372 (9634): 216–23. doi:10.1016/S0140-6736(08)61075-2. PMID 18640458.
- Jump up^ Eliminating Morphine Tolerance – Reformulated Imatinib 23 Feb 2012, 5:00 PST
- Jump up^ “GLIVEC Tablets – Summary of Product Characteristics (SPC)”. electronic Medicines Compendium. Novartis Pharmaceuticals UK Ltd.
- ^ Jump up to:a b c “Gleevec (imatinib) dosing, indications, interactions, adverse effects, and more”.Medscape Reference. WebMD. Retrieved 24 January 2014.
- Jump up^ “Imatinib”. Macmillan Cancer Support. Retrieved 26 December 2012.
- ^ Jump up to:a b Haberfeld, H, ed. (2009). Austria-Codex (in German) (2009/2010 ed.). Vienna: Österreichischer Apothekerverlag. ISBN 3-85200-196-X.
- Jump up^ Kerkelä R, Grazette L, Yacobi R, Iliescu C, Patten R, Beahm C, Walters B, Shevtsov S, Pesant S, Clubb FJ, Rosenzweig A, Salomon RN, Van Etten RA, Alroy J, Durand JB, Force T (August 2006). “Cardiotoxicity of the cancer therapeutic agent imatinib mesylate”. Nat. Med. 12 (8): 908–16. doi:10.1038/nm1446. PMID 16862153.
- Jump up^ Shima H, Tokuyama M, Tanizawa A, Tono C, Hamamoto K, Muramatsu H, Watanabe A, Hotta N, Ito M, Kurosawa H, Kato K, Tsurusawa M, Horibe K, Shimada H (October 2011). “Distinct impact of imatinib on growth at prepubertal and pubertal ages of children with chronic myeloid leukemia”. J. Pediatr. 159 (4): 676–81.doi:10.1016/j.jpeds.2011.03.046. PMID 21592517.
- ^ Jump up to:a b c d “GLIVEC (imatinib)” (PDF). TGA eBusiness Services. Novartis Pharmaceuticals Australia Pty Ltd. 21 August 2013. Retrieved 24 January 2014.
- Jump up^ PDB 1IEP; Nagar B, Bornmann WG, Pellicena P, Schindler T, Veach DR, Miller WT, Clarkson B, Kuriyan J (August 2002). “Crystal structures of the kinase domain of c-Abl in complex with the small molecule inhibitors PD173955 and imatinib (STI-571)”. Cancer Res. 62 (15): 4236–43. PMID 12154025.
- Jump up^ Takimoto CH, Calvo E. “Principles of Oncologic Pharmacotherapy” in Pazdur R, Wagman LD, Camphausen KA, Hoskins WJ (Eds)Cancer Management: A Multidisciplinary Approach. 11 ed. 2008.
- Jump up^ Gambacorti-Passerini CB, Gunby RH, Piazza R, Galietta A, Rostagno R, Scapozza L (February 2003). “Molecular mechanisms of resistance to imatinib in Philadelphia-chromosome-positive leukaemias”. Lancet Oncol. 4 (2): 75–85. doi:10.1016/S1470-2045(03)00979-3. PMID 12573349.
- Jump up^ Deininger MW, Druker BJ (September 2003). “Specific targeted therapy of chronic myelogenous leukemia with imatinib”. Pharmacol. Rev. 55 (3): 401–23.doi:10.1124/pr.55.3.4. PMID 12869662.
- Jump up^ Vigneri P, Wang JY (February 2001). “Induction of apoptosis in chronic myelogenous leukemia cells through nuclear entrapment of BCR-ABL tyrosine kinase”. Nat. Med. 7 (2): 228–34. doi:10.1038/84683. PMID 11175855.
- Jump up^ Weisberg E, Manley PW, Cowan-Jacob SW, Hochhaus A, Griffin JD (May 2007). “Second generation inhibitors of BCR-ABL for the treatment of imatinib-resistant chronic myeloid leukaemia”. Nature Reviews Cancer 7 (5): 345–56. doi:10.1038/nrc2126.PMID 17457302.
- Jump up^ Scheinfeld N, Schienfeld N (February 2006). “A comprehensive review of imatinib mesylate (Gleevec) for dermatological diseases”. J Drugs Dermatol 5 (2): 117–22.PMID 16485879.
- Jump up^ Klopp, T, ed. (2010). Arzneimittel-Interaktionen (in German) (2010/2011 ed.). Arbeitsgemeinschaft für Pharmazeutische Information. ISBN 978-3-85200-207-1.
- ^ Jump up to:a b Staff, Innovation.org (a project of the Pharmaceutical Research and Manufacturers of America)The Story of Gleevec
- Jump up^ Claudia Dreifus for the New York Times. November 2, 2009 Researcher Behind the Drug Gleevec
- ^ Jump up to:a b A Conversation With Brian J. Druker, M.D., Researcher Behind the Drug Gleevecby Claudia Dreifus, The New York Times, 2 November 2009
- Jump up^ Gambacorti-Passerini C (2008). “Part I: Milestones in personalised medicine—imatinib”. Lancet Oncology 9 (600): 600. doi:10.1016/S1470-2045(08)70152-9.PMID 18510992.
- Jump up^ Druker BJ, Lydon NB (January 2000). “Lessons learned from the development of an abl tyrosine kinase inhibitor for chronic myelogenous leukemia”. J. Clin. Invest. 105 (1): 3–7. doi:10.1172/JCI9083. PMC 382593. PMID 10619854.
- ^ Jump up to:a b c U.S. Patent 5,521,184
- Jump up^ “Imatinib Patent Family”. Espacenet. 1996. Retrieved 2014-07-23.
- ^ Jump up to:a b EP 0564409
- Jump up^ Staff, European Medicines Agency, 2004.EMEA Scientific Discussion of Glivec
- Jump up^ Note: The Indian patent application, which became the subject of litigation in India that gathered a lot of press, does not appear to be publicly available. However according todocuments produced in the course of that litigation (page 27), “The Appellant’s application under the PCT was substantially on the same invention as had been made in India.”
- ^ Jump up to:a b WO 9903854
- Jump up^ U.S. Patent 6,894,051
- Jump up^ FDA Orange Book; Patent and Exclusivity Search Results from query on Appl No 021588 Product 001 in the OB_Rx list.
- Jump up^ Novartis press release, May 10, 2001. [http://www.evaluategroup.com/Universal/View.aspx?type=Story&id=5838 FDA approves Novartis’ unique cancer medication Glivec®
- Jump up^ Cohen MH et al. Approval Summary for Imatinib Mesylate Capsules in the Treatment of Chronic Myelogenous Leukemia Clin Cancer Res May 2002 8; 935
- Jump up^ Margot J. Fromer for Oncology Times. December 2002. What’s in a Name? Quite a Lot When It Comes to Marketing & Selling New Cancer Drugs
- Jump up^ Novartis Press Release. April 30 2001Novartis Oncology Changes Trade Name of Investigational Agent Glivec(TM) to Gleevec(TM) in the United States
- Jump up^ Experts in Chronic Myeloid Leukemia. The price of drugs for chronic myeloid leukemia (CML) is a reflection of the unsustainable prices of cancer drugs: from the perspective of a large group of CML experts Blood. 2013 May 30;121(22):4439-42. PMID 23620577
- Jump up^ Andrew Pollack for the New York Times, April 25, 2013 Doctors Denounce Cancer Drug Prices of $100,000 a Year
- Jump up^ Schiffer CA (July 2007). “BCR-ABL tyrosine kinase inhibitors for chronic myelogenous leukemia”. N. Engl. J. Med. 357 (3): 258–65. doi:10.1056/NEJMct071828.PMID 17634461.
- Jump up^ As Pills Treat Cancer, Insurance Lags Behind, By ANDREW POLLACK, New York Times, 14 April 2009
- Jump up^ Living With a Formerly fatal Blood Cancer, By JANE E. BRODY, New York Times, 18 January 2010
- Jump up^ Patented Medicine Review Board (Canada). Report on New Patented Drugs – Gleevec.
- Jump up^ “pharmacychecker.com”. pharmacychecker.com. Retrieved 2013-04-03.
- Jump up^ Gardiner Harris and Katie Thomas for the New York Times. April 1 2013 Top court in India rejects Novartis drug patent
- Jump up^ Note: The Indian patent application No.1602/MAS/1998 does not appear to be publicly available. However according to the decision of the IPAB on 26 June 2009 (page 27) discussed below, “The Appellant’s application under the PCT was substantially on the same invention as had been made in India.”
- Jump up^ Staff, European Medicines Agency, 2004. EMEA Scientific Discussion of Glivec
- Jump up^ Indian Supreme Court Decision paragraphs 5-6
- Jump up^ Novartis v UoI, para 8-9
- ^ Jump up to:a b Shamnad Basheer for Spicy IP March 11, 2006First Mailbox Opposition (Gleevec) Decided in India
- Jump up^ Staff, LawyersCollective. September 6, 2011[http://www.lawyerscollective.org/news/archived-news-a-articles/126-novartis-case-background-and-update-supreme-court-of-india-to-recommence-hearing.html Novartis case: background and update – Supreme Court of India to recommence hearing
- Jump up^ R. Jai Krishna and Jeanne Whalen for the Wall Street Journal. April 1, 2013Novartis Loses Glivec Patent Battle in India
- Jump up^ Intellectual Property Appellate Board decision dated 26 June 2009, p 149
- Jump up^ W.P. No.24759 of 2006
- Jump up^ “Supreme Court rejects bid by Novartis to patent Glivec”.
- Jump up^ Novartis v UoI, Para 191
- Jump up^ Novartis v UoI, Para 24-25
- Jump up^ “How the Indian judgment will reverberate across the world”.
- Jump up^ “Patented drugs must be priced smartly”.
- Jump up^ Patent with a purpose, Prof. Shamnad Basheer, Indian Express, 3 April 2013
- Kevin Grogan for PharmaTimes. February 27, 2012 Novartis explains stance over India patent law challenge
- Berne Declaration. May 8, 2007 Short questions and answers about the court case initiated by Novartis in India
External links


2796.5(w), 1645.9(m), 1586.0(m), 1575.1(s), 1554.0(m), 1531.5(s), 1510.3(m), 1478.1(m),
1448.9(s), 1416.7(m), 1377.7(m), 1352.2(m), 1334.8(m), 1325.6(m), 1308.8(m), 1290.3(s),
1261.1(m), 1204.3(m), 1164.1(m), 1141.7(m), 1124.6(w), 1102.6(m), 1089.2(w), 1052.0(w),
1024.4(w), 1010.0(m), 992.5(w), 968.3(w), 924.5(w), 886.2(w), 857.9(w), 850.3(w),
807.8(m), 795.7(s), 748.1(m), 703.2(m), 690.1(m), 670.7(m);
10.14 (1 H, s, NH), 9.26 (1 H, d, J = 1.5 Hz, 2H-pyridin-3-yl), 8.95 (1 H, s, NH), 8.66 (1 H, dd,
J = 4.8 and 1.2 Hz, 6H-pyridin-3-yl), 8.49 (1 H, d, J = 5.1 Hz, 6H-pyridin-2-amine), 8.46 (1 H,
ddd, J = 7.9, 1.5 and 1.2 Hz, 4H-pyridin-3-yl), 8.06 (1 H, d, J = 1.5 Hz, 3H-2-aminotoluene),
7.89 (2 H, d, J = 8.1 Hz, 2H-benzamide), 7.50 (1 H, dd, J = 7.9 and 4.8 Hz, 5H-pyridin-3-yl),
7.46 (1 H, dd, J = 8.3 and 1.5 Hz, 5H-2-aminotoluene), 7.42 – 7.40 (3 H, m, 3H-benzamide
and 5H-pyridin-2-amine), 7.18 (1 H, d, J = 8.3 Hz, 6H-2-aminotoluene), 3.51 (2 H, s, CH2),
2.50 – 2.20 (8 H, m, piperazine CH2), 2.20 (3 H, s, CCH3), 2.13 (3 H, s, NCH3);
150 MHz) = 165.42(C), 162.72(C), 160.57(C), 158.99(CH), 151.44(CH), 148.48(CH),
142.52(C), 137.77(C), 136.60(C), 134.92(CH), 133.88(C), 132.66(C), 130.75(CH),
129.28(CH), 127.00(CH), 124.23(C), 123.71(CH), 115.35(CH), 113.19(CH), 108.32(CH),
62.49(CH2), 55.07(CH2), 53.10(CH2), 45.98(CH3), 17.65(CH3);
| WO2006024863A1 * | 2 Sep 2005 | 9 Mar 2006 | Cipla Ltd | Stable crystal form of imatinib mesylate and process for the preparation thereof |
| WO2006048890A1 * | 20 Oct 2005 | 11 May 2006 | Raja Jyotir Jani | Imatinib mesylate crystal form and process for preparation thereof |
| WO2006054314A1 * | 11 Aug 2005 | 26 May 2006 | Natco Pharma Ltd | Polymorphic forms of imatinib mesylate |
| WO2007023182A1 * | 24 Aug 2006 | 1 Mar 2007 | Novartis Ag | Delta and epsilon crystal forms of imatinib mesylate |
| WO2007059963A1 * | 23 Nov 2006 | 31 May 2007 | Novartis Ag | F,g,h,i and k crystal forms of imatinib mesylate |
| WO2007136510A2 * | 27 Apr 2007 | 29 Nov 2007 | Ivax Pharmaceuticals Sro | Polymorphic forms of imatinib mesylate and processes for their preparation as well as of amorphous imatinib mesylate and form alpha |
| WO2008150481A2 * | 29 May 2008 | 11 Dec 2008 | Sicor Inc | Processes for the preparation of crystalline form beta of imatinib mesylate |
| WO2010133976A2 | 24 May 2010 | 25 Nov 2010 | Actavis Group Ptc Ehf | Substantially pure imatinib or a pharmaceutically acceptable salt thereof |
| WO2011023146A1 | 19 Aug 2010 | 3 Mar 2011 | Zentiva, K.S. | Imatinib mesylate polymorphs generated by crystallization in aqueous inorganic salt solutions |
| WO2011049474A1 | 21 Oct 2010 | 28 Apr 2011 | Tomasz Kozluk | Salts of imatinib with tartaric acids |
| WO2011095835A1 | 22 Dec 2010 | 11 Aug 2011 | Actavis Group Ptc Ehf | Highly pure imatinib or a pharmaceutically acceptable salt thereof |
| WO2011099039A1 | 15 Feb 2011 | 18 Aug 2011 | Reliance Life Sciences Pvt. Ltd. | Process for the preparation of alpha form of imatinib mesylate |
| WO2011108953A1 | 4 Mar 2011 | 9 Sep 2011 | Tomasz Kozluk | PROCESS FOR PREPARATION OF POLYMORPHIC FORM α AND NEW POLYMORPHIC FORM OF IMATINIB MESYLATE ISOLATED IN THAT PROCESS |
| WO2011114337A1 | 15 Mar 2010 | 22 Sep 2011 | Natco Pharma Limited | Process for the preparation of highly pure crystalline imatinib base |
| WO2011157450A1 | 17 Jun 2011 | 22 Dec 2011 | Krka, D. D., Novo Mesto | New polymorphic form of imatinib base and preparation of salts thereof |
| WO2011158255A1 * | 14 Jun 2011 | 22 Dec 2011 | Aptuit Laurus Private Limited | Process for preparation of stable imatintb mesylate alpha form |
| WO2012014000A1 * | 30 Jul 2010 | 2 Feb 2012 | Narain Singh Awadesh | STABLE α-CRYSTAL FORM OF IMATINIB MESYLATE AND PREPARING PROCESS THEREOF |
| WO2013136141A1 * | 22 Aug 2012 | 19 Sep 2013 | Fresenius Kabi Oncology Ltd. | An improved process for the preparation of alpha form of imatinib mesylate |
| CN102321070A * | 27 Jul 2011 | 18 Jan 2012 | 江苏先声药物研究有限公司 | Method for preparing imatinib methylolsulfonate alpha crystal through inverse solvent recrystallization method |
| CN102321070B | 27 Jul 2011 | 22 May 2013 | 江苏先声药物研究有限公司 | Method for preparing imatinib methylolsulfonate alpha crystal through inverse solvent recrystallization method |
| CN102633775B | 6 Apr 2012 | 17 Jul 2013 | 江南大学 | Method for preparing alpha-crystal-form imatinib mesylate |
| DE102007021043B4 * | 4 May 2007 | 8 Apr 2010 | Chemagis Ltd. | Alpha-Form von Imatinib Mesylat und Verfahren zu seiner Herstellung |
| EP1988089A1 | 26 Oct 2007 | 5 Nov 2008 | Sicor, Inc. | Imatinib base, and imatinib mesylate and processes for preparation thereof |
| EP2009008A1 | 26 Oct 2007 | 31 Dec 2008 | Sicor, Inc. | Imatinib base, and imatinib mesylate and processes for preparation thereof |
| EP2311821A1 | 27 Apr 2007 | 20 Apr 2011 | Sicor, Inc. | Polymorphic form of Imatinib mesylate and processes for its preparation |
| EP2497464A2 | 20 Feb 2012 | 12 Sep 2012 | Adamed SP. Z O.O. | Pharmaceutical composition of imatinibe methanesulphonate and a process for its manufacture |
| EP2546248A1 * | 23 Nov 2006 | 16 Jan 2013 | Novartis AG | Crystal form H of imatinib mesylate |
| EP2578580A1 * | 23 Nov 2006 | 10 Apr 2013 | Novartis AG | G, I and K crystal forms of imatinib mesylate |
| EP2749557A1 * | 31 Dec 2012 | 2 Jul 2014 | Deva Holding Anonim Sirketi | Process for preparation of alpha polymorph of imatinib mesylate from IPA and THF solvate forms of imatinib mesylate |
| EP2829538A1 * | 27 Apr 2007 | 28 Jan 2015 | Sicor, Inc. | Polymorphic form of imatinib mesylate and process for its preparation |
| US7550591 | 2 May 2007 | 23 Jun 2009 | Chemagis Ltd. | 4-(4-methyl-piperazin-1-ylmethyl-benzoic acid with N-(5- amino-2-methylphenyl)-4-(3pyridyl)-2-pyrimidine-amine in the presence of a coupling reagent such as 2-(5-norborene-2,3- dicarboximido)- 1,1,3,3-tetramethyluronium tetrafluoroborate to produce imatinib, salt formation wtih methanesululfonic acid |
| US7879860 | 24 Aug 2006 | 1 Feb 2011 | Novartis Ag | Variations in melting points, hygroscopicities, solubilities, flow properties and/or thermodynamic stabilities |
| US7893076 | 23 Nov 2006 | 22 Feb 2011 | Novartis Ag | Crystalline form F of the methanesulfonic acid addition salt of Imatinib, 4-(4-methylpiperazin-1-ylmethyl)-N-[4-methyl-3-(4-(pyridin-3-yl)pyrimidin-2-ylamino)phenyl]-benzamide; suitable for topical, enteral, for example oral or rectal, or parenteral administration |
| US7947699 | 9 Jan 2009 | 24 May 2011 | Actavis Group Ptc Ehf | Anhydrous amorphous imatinib mesylate |
| US7977348 | 17 Apr 2008 | 12 Jul 2011 | Sicor Inc. | Polymorphic forms of imatinib mesylate and processes for preparation of novel crystalline forms as well as amorphous and form α |
| US8067421 | 27 Apr 2007 | 29 Nov 2011 | Sicor Inc. | Imatinib mesylate solvates with solvents selected from aliphatic alcohols, ethers, dioxolane, nitromethane, and acetic acid, and crystalline imatinib mesylate characterized by data selected from powder XRD patterns and solid state 13C NMR spectra; solubility in gastric juices |
| US8198289 | 14 Jan 2011 | 12 Jun 2012 | Novartis Ag | Crystal form H imatinib mesylate for pharmaceutical use |
| US8269003 | 2 Sep 2005 | 18 Sep 2012 | Cipla Limited | Stable crystal form of imatinib mesylate and process for the preparation thereof |
| US8414918 | 25 Sep 2008 | 9 Apr 2013 | Teva Pharmaceutical Industries Ltd. | Stable imatinib compositions |
| US8507515 | 15 Jul 2011 | 13 Aug 2013 | Novartis Ag | Crystalline form G of imatinib mesylate |
| US8592440 | 15 Jul 2011 | 26 Nov 2013 | Novartis Ag | Crystalline form I of imatinib mesylate |
| US8633213 | 15 May 2012 | 21 Jan 2014 | Novartis Ag | Crystalline form F of imatinib mesylate |
| US8846706 | 15 Jul 2011 | 30 Sep 2014 | Novartis Ag | Crystalline form K of imatinib mesylate |
| USRE43932 | 21 Sep 2011 | 15 Jan 2013 | Novartis Ag | Crystal modification of a N-phenyl-2-pyrimidineamine derivative, processes for its manufacture and its use |
—

THANKS AND REGARD’S
DR ANTHONY MELVIN CRASTO Ph.D
DR ANTHONY MELVIN CRASTO
ORGANIC SPECTROSCOPY

