
FLAGS AND HITS
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO FACEBOOK
...................................................................Join me on twitter
FACEBOOK
...................................................................Join me on twitter
 ..................................................................Join me on google plus
..................................................................Join me on google plus
 Googleplus
GoogleplusMYSELF

Ibuzatrelvir



Ibuzatrelvir
PF-07817883
CAS 2755812-39-4
| Molecular Weight | 489.49 |
|---|---|
| Formula | C21H30F3N5O5 |
- Ibuzatrelvir
- N-(Methoxycarbonyl)-3-methyl-L-valyl-(4R)-N-[(1S)-1-cyano-2-((3S)-2-oxopyrrolidin-3-yl)ethyl]-4-(trifluoromethyl)-L-prolinamide
- PF 07817883
- methyl N-[(2S)-1-[(2S,4R)-2-[[(1S)-1-cyano-2-[(3S)-2-oxopyrrolidin-3-yl]ethyl]carbamoyl]-4-(trifluoromethyl)pyrrolidin-1-yl]-3,3-dimethyl-1-oxobutan-2-yl]carbamate
- KZ2X7QH2VT
Ibuzatrelvir (development code PF-07817883) is an experimental antiviral drug being developed by Pfizer for the treatment of COVID-19.[1] It is a second-generation improvement over nirmatrelvir which has a similar chemical structure.[2] One of the disadvantages of nirmatrelvir is that it has low metabolic stability and must be given in combination with ritonavir (as Paxlovid) to limit its metabolic degradation in the body.[3] Ibuzatrelvir incorporates modifications to the chemical structure of nirmatrelvir that give it enhanced oral bioavailability, so it does not require coadministration with ritonavir.[3]
Ibuzatrelvir (PF-07817883), a second-generation, orally bioavailable, is SARS-CoV-2 main protease (Mpro and 3CLpro) inhibitor with improved metabolic stability. Ibuzatrelvir has demonstrated pan-human coronavirus antiviral activity and off-target selectivity profile in vitro and in preclinical animal studies. Ibuzatrelvir is well tolerated with a safety profile similar to placebo and prevents viral infection and transmission. Ibuzatrelvir can be used to inhibit COVID-19.
SCHEME
SIDECHAIN

MAIN

PATENT
WO2021250648 PFIZER
WO2023215910
PAPER
The Pfizer scientists described ibuzatrelvir’s medicinal chemistry campaign in a Journal of Medicinal Chemistry paper that was published in April 2024 (DOI: 10.1021/acs .jmedchem.3c02469).
https://pubs.acs.org/doi/10.1021/jacsau.4c00508
Ibuzatrelvir (1) was recently disclosed and patented by Pfizer for the treatment of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). It has received fast-track status from the USA Food and Drug Administration (FDA) and has entered phase III clinical trials as a possible replacement for Paxlovid. Like nirmatrelvir (2) in Paxlovid, this orally active drug candidate is designed to target viral main proteases (Mpro) through reversible covalent interaction of its nitrile warhead with the active site thiol of the chymotrypsin-like cysteine protease (3CL protease). Inhibition of Mpro hinders the processing of the proteins essential for viral replication in vivo. However, ibuzatrelvir apparently does not require ritonavir (3), which is coadministered in Paxlovid to block human oxidative metabolism of nirmatrelvir. Here, we report the crystal structure of the complex of ibuzatrelvir with the active site of SARS-CoV-2 Mpro at 2.0 Å resolution. In addition, we show that ibuzatrelvir also potently inhibits the Mpro of Middle East respiratory syndrome-related coronavirus (MERS-CoV), which is fortunately not widespread but can be dangerously lethal (∼36% mortality). Co-crystal structures show that the binding mode of the drug to both active sites is similar and that the trifluoromethyl group of the inhibitor fits precisely into a critical S2 substrate binding pocket of the main proteases. However, our results also provide a rationale for the differences in potency of ibuzatrelvir for these two proteases due to minor differences in the substrate preferences leading to a weaker H-bond network in MERS-CoV Mpro. In addition, we examined the reversibility of compound binding to both proteases, which is an important parameter in reducing off-target effects as well as the potential immunogenicity. The crystal structures of the ibuzatrelvir complexes with Mpro of SARS-CoV-2 and of MERS-CoV will further assist drug design for coronaviral infections in humans and animals.


General Boc-Deprotection and Coupling Procedure
This procedure was based on a literature procedure.1
The Boc-protected building block (1.0
equiv) was dissolved in 50/50 TFA/DCM and stirred for 1 h at room temperature. The reaction
mixture was then concentrated in vacuo and co-evaporated with DCM (5 × 5 mL). In a separate
RBF the carboxylic acid building block (1.0 equiv) and HATU (1.0 equiv) were dissolved in
DMF. HOAt (0.6 M in DMF) (0.1 equiv) and DIPEA (3.0 equiv) were added and the reaction
mixture was left to incubate at room temperature for 10 mins, as it turned yellow. The previously
concentrated Boc-deprotected building block was dissolved in DMF and added dropwise to the
incubating solution. The reaction mixture was capped under a blanket of argon and stirred at room
temperature for 2–3 h. The reaction mixture was diluted with 5 mL each of water and ethyl acetate
and the layers separated. The aqueous layer was extracted further with ethyl acetate (3 × 5 mL),
and all ethyl acetate layers combined and washed with sat. aq. NaHCO3 (10 mL), 1 M HCl (10
mL), water (2 × 10 mL) and brine (10 mL). It was then dried over Na2SO4, filtered, and
concentrated in vacuo to furnish the product.
Methyl ((S)-1-((2S,4R)-2-(((S)-1-cyano-2-((S)-2-oxopyrrolidin-3-yl)ethyl)carbamoyl)-4-
(trifluoromethyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)carbamate (1) Ibuzatrelvir
This known compound was synthesized according to the General Boc-Deprotection and
Coupling Procedure with building blocks 7 and 8. The characterization data matches the literature
report (IPN: WO2021250648A1). The crude material was obtained as a dark yellow sticky residue
that was then purified with flash column chromatography with an eluent of 92:8 EtOAc:MeOH.
The desired compound had an Rf
= 0.40 and was visible with KMnO4 stain. After concentration
of desired fractions, 1 was isolated as a clear, colorless oil that solidified to a white solid (0.051 g,
53%) This compound was isolated and used for all experiments as a mixture of diastereomers in a
ratio of about 2:1 and rotamers present, with only the major set of resonances reported, which are
for the desired isomer. It can be separated using high performance liquid chromatography (HPLC)
methods, as listed in the HPLC Separation of Ibuzatrelvir Diastereomers section.
IR (DCM cast film, vmax / cm–1) 3292, 3053, 2959, 2909, 2875, 1695, 1643, 1550, 1443, 1401,
1370, 1332, 1270, 1236, 1200, 1164, 1130
1H NMR (500 MHz, CDCl3) δH 8.32 (1H, d, J = 7.6 Hz), 6.22 (1H, br), 5.74 (1H, d, J = 9.3 Hz),
4.96 – 4.87 (1H, m), 4.54 (1H, dd, J = 8.6, 3.6 Hz), 4.30 (1H, d J = 9.9 Hz), 3.99 – 3.88 (2H, m),
3.65 (3H, s), 3.42 – 3.26 (2H, m), 2.66 – 2.57 (1H, m), 2.52 – 2.43 (1H, m), 2.40 – 2.28 (3H, m),
1.97 – 1.88 (1H, m), 1.84 – 1.75 (2H, m), 0.99 (9H, s)
13C {1H} NMR (125 MHz, CDCl3) δC 179.1, 171.4, 171.1, 156.9, 126.1 (q, J = 276.3 Hz), 118.3,
59.4, 58.9, 52.4, 47.3, 42.4 (q, J = 29.5 Hz), 40.4, 39.1 37.5, 35.6, 34.2, 28.2, 28.0, 26.3
SR: [α]D
26 = –35.71 (c = 0.21, DCM)
HRMS: (ESI) Calcd for C21H30F3N5NaO5 [M + Na]+
512.2091, found 512.2088



References
- ^ Allerton CM, Arcari JT, Aschenbrenner LM, Avery M, Bechle BM, Behzadi MA, et al. (August 2024). “A Second-Generation Oral SARS-CoV-2 Main Protease Inhibitor Clinical Candidate for the Treatment of COVID-19”. Journal of Medicinal Chemistry. 67 (16): 13550–13571. doi:10.1021/acs.jmedchem.3c02469. PMC 11345836. PMID 38687966.
- ^ Chen P, Van Oers TJ, Arutyunova E, Fischer C, Wang C, Lamer T, et al. (August 2024). “A Structural Comparison of Oral SARS-CoV-2 Drug Candidate Ibuzatrelvir Complexed with the Main Protease (Mpro) of SARS-CoV-2 and MERS-CoV”. JACS Au. 4 (8): 3217–3227. doi:10.1021/jacsau.4c00508. PMC 11350714. PMID 39211604.
- ^ Jump up to:a b Brewitz L, Schofield CJ (July 2024). “Fixing the Achilles Heel of Pfizer’s Paxlovid for COVID-19 Treatment”. Journal of Medicinal Chemistry. 67 (14): 11656–11661. doi:10.1021/acs.jmedchem.4c01342. PMC 11284777. PMID 38967233.
| Clinical data | |
|---|---|
| Other names | PF-07817883 |
| Routes of administration | Oral |
| Legal status | |
| Legal status | Investigational |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 2755812-39-4 |
| PubChem CID | 163362000 |
| DrugBank | 111 |
| ChemSpider | 128942571 |
| UNII | KZ2X7QH2VT |
| Chemical and physical data | |
| Formula | C21H30F3N5O5 |
| Molar mass | 489.496 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
- [1]. Owen, et al. Preparation of peptidomimetic nitriles as SARS-CoV-2 3CL protease inhibitors and methods for the treatment of COVID-19. World Intellectual Property Organization, WO2021250648 A1. 2021-12-16.[2]. Mahta Mortezavi, et al. Virologic Response and Safety After Oral Administration of Ibuzatrelvir, a Novel SARS-CoV-2 Mpro Inhibitor, in Non-Hospitalized Adults With Symptomatic COVID-19. European Congress of Clinical Microbiology and Infectious Disease (ECCMID) 2024; 2024 April 27-30.[3]. Westberg M, et al. An orally bioavailable SARS-CoV-2 main protease inhibitor exhibits improved affinity and reduced sensitivity to mutations[J]. Sci Transl Med. 2024 Mar 13;16(738):eadi0979.[4]. Allerton CMN, et al. A Second-Generation Oral SARS-CoV-2 Main Protease Inhibitor Clinical Candidate for the Treatment of COVID-19[J]. J Med Chem. 2024 Apr 30. [Content Brief]
////Ibuzatrelvir, PF 07817883, PF-07817883, PF07817883, KZ2X7QH2VT
Gamcemetinib



Gamcemetinib
CAS 1887069-10-4
CC-99677 , OS2IR8TV1O
| Molecular Weight | 469.94 |
|---|---|
| Formula | C22H20ClN5O3S |
- (10R)-3-[[2-Chloro-5-(ethoxymethyl)-4-pyrimidinyl]oxy]-9,10,11,12-tetrahydro-10-methyl-8H-[1,4]diazepino[5′,6′:4,5]thieno[3,2-f]quinolin-8-one (ACI)
- (10R)-3-{[2-chloro-5-(ethoxymethyl)pyrimidin-4-yl]oxy}-10-methyl-9,10,11,12-tetrahydro-8H-[1,4]diazepino[5′,6′:4,5]thieno[3,2-f]quinolin-8-one
- BMS 986371
- BMS-986371
- CC 99677
- CC-99677
(R)-3-((2-Chloro-5-(ethoxymethyl)pyrimidin-4-yl)oxy)-10-methyl-9,10,11,12-tetrahydro-8H-[1,4]diazepino[5′,6′:4,5]thieno[3,2-f]quinolin-8-one
- OriginatorCelgene Corporation
- ClassAnti-inflammatories
- Mechanism of ActionMAP-kinase-activated kinase 2 inhibitors
- Orphan Drug StatusNo
- 14 Nov 2024Efficacy and adverse events data from a phase II trial in Ankylosing Spondylitis presented at the ACR Convergence 2024 (ACR-2024)
- 27 Mar 2024Pharmacokinetics and adverse events data from a phase I trial (In volunteers) presented at the 125th Annual Meeting of the American Society for Clinical Pharmacology and Therapeutics 2024 (ASCPT-2024)
- 26 Oct 2023Discontinued – Phase-I for Inflammation (In volunteers) in USA, United Kingdom (PO) prior to October 2023 (Bristol-Myers Squibb pipeline, October 2023)
Gamcemetinib (CC-99677) is a potent, covalent, and irreversible inhibitor of the mitogen-activated protein (MAP) kinase-activated protein kinase-2 (MK2) pathway in both biochemical (IC50=156.3 nM) and cell based assays (EC50=89 nM). Gamcemetinib is extracted from patent WO2020236636, compound 1.
SCHEME
SIDECHAIN

SIDECHAIN

MAIN

REF
WO2018170203
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2018170203&_cid=P11-MBK7CL-38003-1



PATENT
WO2018170199 CELGENE
WO2018170203
US20160075720
WO2020236636, compound 1
////////////Gamcemetinib, BMS 986371, BMS-986371, CC 99677, CC-99677, OS2IR8TV1O
Alflutinib, Furmonertinib, Firmonertinib



FIRMOMERTINIB, Furmonertinib, Alflutinib
CAS 1869057-83-9
, AST 2818, UNII-A49A7A5YN4
N-[2-[[2-(Dimethylamino)ethyl]methylamino]-5-[[4-(1-methyl-1H-indol-3-yl)-2-pyrimidinyl]amino]-6-(2,2,2-trifluoroethoxy)-3-pyridinyl]-2-propenamide
N-[2-[2-(dimethylamino)ethyl-methylamino]-5-[[4-(1-methylindol-3-yl)pyrimidin-2-yl]amino]-6-(2,2,2-trifluoroethoxy)pyridin-3-yl]prop-2-enamide
C28H31F3N8O2 568.6 g/mol
2-Propenamide, N-[2-[[2-(dimethylamino)ethyl]methylamino]-5-[[4-(1-methyl-1H-indol-3-yl)-2-pyrimidinyl]amino]-6-(2,2,2-trifluoroethoxy)-3-pyridinyl]-
Alflutinib is under investigation in clinical trial NCT03452592 (Efficacy and Safety of Alflutinib in Locally Advanced or Metastatic Non-small Cell Lung Cancer Patients With T790M).
Firmonertinib is an orally available selective inhibitor of the epidermal growth factor receptor (EGFR) mutant form T790M, with potential antineoplastic activity. Upon administration, firmonertinib specifically binds to and inhibits the tyrosine kinase activity of EGFR T790M, a secondarily acquired resistance mutation. This prevents EGFR T790M-mediated signaling and leads to cell death in EGFR T790M-expressing tumor cells. EGFR, a receptor tyrosine kinase that is mutated in many tumor cell types, plays a key role in tumor cell proliferation and tumor vascularization. Compared to some other EGFR inhibitors, alflutinib may have therapeutic benefits in tumors with T790M-mediated drug resistance.
FIRMONERTINIB is a small molecule drug with a maximum clinical trial phase of III (across all indications) and has 4 investigational indications.
SCHEME

CONTD……..

REF
https://patentscope.wipo.int/search/en/detail.jsf?docId=US201062358&_cid=P22-MBFXFH-62339-1
Example 3: N-{2-{[2-(dimethylamino)ethyl](methyl)amino}-6-(2,2,2-trifluoroethoxyl)-5-{[4-(1-methyl-H-indol-3-yl)pyrimidin-2-yl]amino}pyridin-3-yl}acrylamide
Step 1: Synthesis of N2-methyl-N2-[2-(dimethylamino)ethyl]-6-(2,2,2-trifluoroethoxyl)-N5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-yl]-3-nitropyridin-2,5-diamine
| The compound was synthesized in the same manner as those in Step 1 of Example 1 with a yield of 86%. MS m/z: 545 [M+1]. |
Step 2: Synthesis of N2-methyl-N2-[2-(dimethylamino)ethyl]-6-(2,2,2-trifluoroethoxyl)-N5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-yl]pyridin-2,3,5-triamine
| The compound was synthesized in the same manner as those in Step 2 of Example 2 with a yield of 56%. MS m/z: 515 [M+1]. |
Step 3: Synthesis of N-{2-{[2-(dimethylamino)ethyl](methyl)amino}-6-(2,2,2-trifluoroethoxyl)-5-{[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-yl]amino}pyridin-3-yl}acrylamide
| The compound was synthesized in the same manner as those in Step 3 of Example 1 with a yield of 23%. MS m/z: 569 [M+1]. |
PATENT
CN110606842
https://patentscope.wipo.int/search/en/detail.jsf?docId=CN280196686&_cid=P22-MBFXJY-67679-1
Patent application CN105315259A protects the compound of formula I and discloses its preparation method as follows:



| Example 1: Preparation of 6-chloro-3-nitro-2-(2,2,2-trifluoroethoxy)pyridine (XI-1) |
| Add toluene (24.0L) to the reactor, then add 2,6-dichloro-3-nitropyridine (3000g, 15.54mol), adjust the internal temperature between -20℃ and -10℃, and add sodium hydrogen (933g, 23.33mol) in batches. Add 2,2,2-trifluoroethanol (1586g, 16.00mol) toluene (6.0L) solution dropwise. React for 2h, and monitor the reaction end point by TLC and HPLC. After the reaction is completed, add 10% ammonium chloride solution (6.0L) dropwise. Let stand and separate. Wash the organic phase with water (6.0L) and concentrate under reduced pressure. Add ethyl acetate (0.3L), heat to 40-50℃, add n-heptane (2.7L) dropwise, cool to -15 to -5℃ after dripping, and continue crystallization for 3 hours, and filter with suction. Obtain 3017g of product solid, with a yield of 75.65%. |
| 1H NMR(500MHz,DMSO-d6)δ8.60(d,J=8.0Hz,1H),7.50(d,J=8.5Hz,lH),5.13(q,J=9.0Hz,2H); |
| 13C NMR(126MHz,DMSO-d6)δ153.20,151.09,139.34,132.67,123.38(q,J=277.2Hz),119.14,63.34(q,J=36Hz); |
| MS m/z:256.99[M+1]。 |
| Example 2: Preparation of 6-chloro-3-amino-2-(2,2,2-trifluoroethoxy)pyridine (X-1) |
| At room temperature, add acetonitrile (21.0L) and water (21.0L) to the reactor, start stirring, add 6-chloro-3-nitro-2-(2,2,2-trifluoroethoxy)pyridine (3017.0g, 11.76mol) obtained in Example 1, and add hydrosulfite (15.1Kg, 70.54mol). Control the temperature at 27-33°C to react for 2 hours. Add 36% concentrated hydrochloric acid (11.9Kg, 117.60mol) dropwise, and continue to react for 1.5 hours. Add solid sodium bicarbonate (12.8Kg, 12.96mol). Filter, separate the mother liquor, wash the organic phase with saturated brine (21.0L), and concentrate under reduced pressure to obtain an oily substance. Theoretically calculated for the next step reaction. |
| 1H NMR(500MHz,DMSO-d6)δ7.03(d,J=8.0Hz,1H),6.90(d,J=8.0Hz,1H),5.21(s,2H),4.93(q,J=9.0Hz,2H); |
| 13C NMR(126MHz,DMSO-d6)δ148.16,131.72,130.55,123.93(q,J=278.5Hz),121.02,118.42,61.72(q,J=34.0Hz); |
| MS m/z:227.01[M+1]。 |
| Example 3: Preparation of 6-chloro-3-(2,2,2-trifluoroacetamido)-2-(2,2,2-trifluoroethoxy)pyridine (IX-1) |
| At room temperature, dichloromethane (10.4 L) was added to the reaction kettle, stirring was started, 6-chloro-3-amino-2-(2,2,2-trifluoroethoxy)pyridine (2664 g, 11.76 mol) obtained in Example 2 was added, diisopropylethylamine (2279 g, 17.64 mol) was added, the temperature was controlled at -15 to -10°C, a dichloromethane (5.2 L) solution of trifluoroacetic anhydride (2963 g, 14.11 mol) was added dropwise, and stirring was continued for 20 minutes after the addition was completed. Water (13.0 L) was added dropwise, the layers were separated, the organic phase was concentrated under reduced pressure, and the next step reaction was theoretically calculated. |
| 1 H NMR(400MHz,DMSO-d6)δ11.23(s,7H),7.95(d,J8.0Hz,1H),7.34(d,J8.0Hz,1H),5.03(q,J8.9Hz,2H) |
| 13C NMR(101MHz,DMSO-d6)δ155.74(q,J=46.6Hz),155.60,145.37,140.24,124.01(q,J=278.8Hz),119.07,118.30,116.19(q,J=289.9Hz),62.99(q,J=35.4Hz); |
| MS m/z.322.99[M+1]。 |
| Example 4: Preparation of 6-chloro-5-nitro-3-(2,2,2-trifluoroacetamido)-2-(2,2,2-trifluoroethoxy)pyridine (VIII-1) |
| At room temperature, concentrated sulfuric acid (11.7 L) was added to the reaction kettle, stirring was started, 6-chloro-3-(2,2,2-trifluoroacetamido)-2-(2,2,2-trifluoroethoxy)pyridine (3.9 Kg, 11.76 mol) obtained in Example 3 was added, and potassium nitrate solid (1783.4 g, 17.64 mol) was added in batches. After the addition, stirring was continued for about 40 minutes. After monitoring the reaction, the temperature was lowered to control the internal temperature at 10-25°C, and dichloromethane (27.3 L) was added dropwise. Stirring was continued, stirring was continued for 45 minutes, and the layers were separated. The organic phase was taken and washed once with water (11.7 L). The organic phase was concentrated under reduced pressure and theoretically calculated for the next step reaction. |
| 1H NMR(500MHz,DMSO-d6)δ11.58(s,1H),8.78(s,1H),5.17(q,J=8.7Hz,2H); |
| 13C NMR(126MHz,DMSO-d6)δ155.89,155.43(q,J=37.8Hz),138.84,138.57,135.05,123.22(q,J=273.4Hz),118.47,115.51(q,J=278.5Hz),63.65(q,J=35.3Hz); |
| MS m/z:367.98[M+1]。 |
| Example 5: Preparation of 6-chloro-5-nitro-3-amino-2-(2,2,2-trifluoroethoxy)pyridine (VII-1) |
| At room temperature, methanol (13.0 L) was added to the reactor, 6-chloro-5-nitro-3-(2,2,2-trifluoroacetamido)-2-(2,2,2-trifluoroethoxy)pyridine (4322 g, 11.76 mol) obtained in Example 4 was added, p-toluenesulfonic acid monohydrate (3355 g, 17.64 mol) was added, the temperature was controlled at 60-65°C for 15 hours, and the methanol was removed under reduced pressure. Methyl tert-butyl ether (13.0 L) and water (6.5 L) were added, and the pH was adjusted to 7-8 with potassium carbonate. Layering was performed, the organic phase was washed once with water (8.6 L), separated, and concentrated under reduced pressure. n-heptane (21.5 L) was added, the temperature was controlled at 60-65°C and stirred for 1 hour, cooled to room temperature, filtered, and the filter cake was dried with air at 50°C for 18 hours to obtain 1475 g of the product. |
| The total yield of the five-step reaction from Example 1 to Example 5 is 34.9%. |
| 1H NMR(500 MHz,DMSO-d6)δ7.62(s,1H),5.92(s,2H),5.05(q,J=8.9Hz,2H). |
| 13C NMR(126MHz,DMSO-d6)δ149.30,139.53,132.84,123.46,123.44(q,J=278.5Hz),116.25,62.52(q,J=35.3Hz); |
| MS m/z:272.00[M+1]。 |
| Example 6: Preparation of 2-chloro-3-nitro-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (V-1) |
| Toluene (50 mL) was added to a 100 mL reaction bottle, and the compound of formula VII-1, 6-chloro-5-nitro-3-amino-2-(2,2,2-trifluoroethoxy)pyridine (5.0 g, 18.4 mmol), the compound of formula VI, 3-(2-chloropyrimidin-4-yl)-1-methyl-1H-indole (5.8 g, 23.8 mmol), p-toluenesulfonic acid monohydrate (1.8 g, 9.2 mmol) were added in sequence, and the reaction mixture was heated to 110-115°C and reacted for 24 hours. The temperature was lowered to 22°C, filtered by suction, and the filter cake was dried at 50°C for 20 hours to obtain the compound of formula V-1, 2-chloro-3-nitro-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (10.4 g, 74.7 HPLC area% purity). According to the HPLC purity conversion, the next step reaction was carried out. |
| 1H NMR(400MHz,DMSO-d6)δ9.43(s,1H),8.76(s,1H),8.46-8.45(d,J=5.4Hz,1H),8.39(s,1H),8.38-8.36(d,J=7.8Hz,1H),7.57-7.55(d,J=8.2Hz,1H),7.41-7.40(d,J=5.4Hz,1H),7.31-7.27(t,J=7.5Hz,1H),7.20-7.16(t,J=7.5Hz,1H),5.23-5.16(q,J=8.8Hz,2H),3.90(s,3H); |
| MS m/z:479.08[M+1]。 |
| Example 7: Preparation of 2-[2-(dimethylaminoethyl)methylamino]-3-nitro-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (IV) |
| Add N,N-dimethylformamide (30 mL) to a 250 mL reaction bottle, add the compound of formula V-1 obtained in Example 6, 2-chloro-3-nitro-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (10.4 g, 16.22 mmol), stir, add potassium carbonate (4.48 g, 32.44 mol), N,N,N’-trimethylethylenediamine (2.48 g, 24.33 mol) in sequence, heat the reaction mixture to 77-82°C, keep warm for 1-1.5 hours. Add water (60 mL), and cool to room temperature after addition. Filter by suction, transfer the filter cake to a 50 L reactor, add acetonitrile (40 mL), and heat to reflux for 2 hours. The mixture was cooled to room temperature and filtered with suction. The filter cake was dried at 50°C for 18 hours to give a compound of formula IV, 2-[2-(dimethylaminoethyl)methylamino]-3-nitro-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (6.7 g). The total yield of the two-step reaction with Example 6 was 66.8%. |
| 1H NMR(500MHz,DMSO-d6)δ8.62(s,1H),8.41(s,1H),8.26(s,2H),8.24(s,1H),7.48(d,J=8.2Hz,1H),7.21(t,J=7.6Hz,1H),7.16(d,J=5.3Hz,1H),7.05(t,J=7.3Hz,1H),5.04(q,J=8.9Hz,2H),3.84(s,3H),3.69(t,J=6.9Hz,2H),2.89(s,3H),2.55(t,J=6.9Hz,2H),2.17(s,6H); |
| 13C NMR(126MHz,DMSO-d6)δ162.15,160.55,156.99,154.98,148.42,137.53,132.83,132.68,125.50,123.58(q,J=279.7Hz),124.38,122.11,122.06,120.67,113.38,112.27,110.30,107.11,62.14(q,J=35.3Hz),56.10,49.51,45.34,45.33,39.35,32.98。 |
| MS m/z.:545.22[M+1]。 |
| Example 8: Preparation of 2-chloro-3-nitro-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine p-toluenesulfonate (V-1′) |
| Toluene (7.43 L) was added to a 20 L reactor, and compound VII-1 6-chloro-5-nitro-3-amino-2-(2,2,2-trifluoroethoxy)pyridine (743.0 g, 2.74 mol), compound VI 3-(2-chloropyrimidin-4-yl)-1-methyl-1H-indole (866.7 g, 3.56 mol), p-toluenesulfonic acid monohydrate (780.7 g, 4.10 mol) were added in sequence, stirred, and the reaction mixture was heated to 110-115°C and reacted for 36 hours. The temperature was controlled at 15-30°C, tetrahydrofuran (3.72 L) was added and stirred for 30 minutes. Filtered by suction, the filter cake was transferred to a 50 L reactor, tetrahydrofuran (4.46 L) was added, and heated to reflux for 3 hours. The temperature was lowered to 15-25°C, filtered, and the filter cake was dried at 50°C for 17 hours to obtain 2-chloro-3-nitro-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine p-toluenesulfonate (1719 g, 85.96 HPLC area% purity). The purity was calculated according to HPLC and used for the next step reaction. |
| Melting point: 216.0-218.3℃ |
| 1H NMR(500MHz,DMSO-d6)δ9.70(s,1H),9.21(s,1H),8.62(s,1H),8.40(d,J=6.2Hz,1H),8.24(d,J=7.8Hz,1H),7.59(d,J=8.3Hz,1H),7.50(d,J=6.5Hz,1H),7.49(d,J=8.3Hz,2H),7.32(t,J=7.6Hz,1H),7.18(t,J=7.5Hz,1H),7.12(d,J=7.9Hz,2H),5.17(q,J=8.8Hz,2H),3.91(s,3H),2.29(d,J=5.2Hz,3H); |
| 13C NMR(126MHz,DMSO-d6)δ166.66,157.35,155.72,147.40,140.87,139.90,139.72,138.59,135.83,130.09,129.99,129.98,129.97,127.39,127.38,127.37,127.15,125.22(q,J=278.5Hz),124.97,123.85,123.69,113.63,112.97,110.27,63.58(q,J=35.3Hz),35.57,22.81。 |
| Example 9: Preparation of 2-[2-(dimethylaminoethyl)methylamino]-3-nitro-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (IV) |
| Add N,N-dimethylformamide (5.14L) to a 50L reactor, add 2-chloro-3-nitro-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine p-toluenesulfonate (1714.0g, 2.261mol) obtained in Example 8, stir, add potassium carbonate (624.7g, 4.52mol), N,N,N’-trimethylethylenediamine (346.2g, 3.39mol) in sequence, heat the reaction mixture to 77-82°C, keep warm for 1-1.5 hours. Add water (10.28L), and cool to room temperature after adding. Filter by suction, transfer the filter cake to a 50L reactor, add acetonitrile (6.86L), and heat to reflux for 2 hours. The temperature was lowered to 15-25°C, filtered with suction, and the filter cake was dried at 50°C for 18 hours to obtain the compound of formula IV, 2-[2-(dimethylaminoethyl)methylamino]-3-nitro-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (1142 g). The total yield of the two-step reaction with Example 8 was 76.54%. |
| 1H NMR(500MHz,DMSO-d6)δ8.62(s,1H),8.41(s,1H),8.26(s,2H),8.24(s,1H),7.48(d,J=8.2Hz,1H),7.21(t,J=7.6Hz,1H),7.16(d,J=5.3Hz,1H),7.05(t,J=7.3Hz,1H),5.04(q,J=8.9Hz,2H),3.84(s,3H),3.69(t,J=6.9Hz,2H),2.89(s,3H),2.55(t,J=6.9Hz,2H),2.17(s,6H); |
| 13C NMR(126MHz,DMSO-d6)δ162.15,160.55,156.99,154.98,148.42,137.53,132.83,132.68,125.50,123.58(q,J=279.7Hz),124.38,122.11,122.06,120.67,113.38,112.27,110.30,107.11,62.14(q,J=35.3Hz),56.10,49.51,45.34,45.33,39.35,32.98。 |
| MS m/z:545.22[M+1]。 |
| Example 10: Preparation of 2-[2-(dimethylaminoethyl)methylamino]-3-nitro-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (IV) |
| Acetonitrile (10 mL) was added to a 50 L reactor, and 2-chloro-3-nitro-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine p-toluenesulfonate (1.0 g, 1.5 mmol) obtained in Example 8 was added, and stirred. Potassium carbonate (577 mg, 3 mmol) and N,N,N’-trimethylethylenediamine (320 mg, 2.25 mmol) were added in sequence. The reaction mixture was heated to 77-82°C and kept for 1-2 hours. Water (10 mL) was added and the temperature was cooled to room temperature after the addition. The product was filtered to give a compound of formula IV, 2-[2-(dimethylaminoethyl)methylamino]-3-nitro-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (629 mg) with a purity of 95.94%. The total yield of the two-step reaction with Example 8 was 77%. |
| Example 11: Preparation of 2-[2-(dimethylaminoethyl)methylamino]-3-amino-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (III’) |
| Add the compound of formula IV 2-[2-(dimethylaminoethyl)methylamino]-3-nitro-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (4.0 g, 7.34 mmol) to a 100 mL reaction bottle at room temperature, add tetrahydrofuran (27 mL) and water (13 mL), and stir for 10 to 20 minutes. Add hydrosulfite (9.6 g, 44.1 mmol) to the reactor in batches. After addition, continue stirring for 10 to 20 minutes. Control the temperature of the reactor to 30 to 35 ° C for reaction. The purity of the product compound of formula III’ was 64.68% after sampling the liquid phase after 2 hours of reaction. The reaction was continued until 17 hours after the reaction. 40 mL of water was added to the reaction solution, and the layers were separated by standing. The tetrahydrofuran phase was taken, and the aqueous phase was extracted twice with 100 mL of dichloromethane. The organic phases were combined, washed with saturated brine, separated by standing, and concentrated under reduced pressure to obtain 3.2 g of solid with a purity of 62.32%. |
| 1H NMR(500MHz,DMSO)δ10.67(s,1H),10.36(s,1H),8.82(s,1H),8.18(s,1H),8.01(s,1H),7.59(d,J=8.2Hz,1H),7.45(d,J=6.8Hz,1H),7.32(t,J=7.5Hz,1H),7.24(s,1H),4.97(q,J=8.7Hz,2H),3.93(s,3H),3.75(s,2H),3.41(s,2H),3.10(s,3H),2.78(s,6H); |
| MS m/z:515.24[M+1]。 |
| Example 12: Preparation of 2-[2-(dimethylaminoethyl)methylamino]-3-amino-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (III’) |
| In a 100mL single-mouth bottle, there is 2-[2-(dimethylaminoethyl)methylamino]-3-nitro-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (2.0g, 3.67mmol), palladium carbon (200mg), ethanol (20mL), hydrogen balloon replacement twice, hydrogen gas, magnetic stirring, room temperature overnight (17 hours). After the reaction is completed, suction filtration, the filtrate is taken, and it is concentrated to dryness under reduced pressure to obtain 2.1g of product with a purity of 56.93%. |
| Example 13: Preparation of 2-[2-(dimethylaminoethyl)methylamino]-3-amino-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (III’) |
| At room temperature, add the compound of formula IV 2-[2-(dimethylaminoethyl)methylamino]-3-nitro-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (1317.0 g, 2.42 mol) to a 50 L reactor, add tetrahydrofuran (8.8 L) and water (4.3 L), and stir for 10 to 20 minutes. Add hydrosulfite (2970.0 g, 14.52 mol) to the reactor in batches. After adding, continue stirring for 10 to 20 minutes. Control the temperature of the reactor to 40-45 ° C and react for 2 hours. Add concentrated hydrochloric acid (5882.2 g, 58.08 mol) to the reactor. After the addition is complete, heat to 42 to 47 ° C and react for 15 hours. Add 30% sodium hydroxide (2323.2g, 58.08mol) aqueous solution dropwise, and then add solid sodium bicarbonate (1219.7g, 14.52mol) in batches to adjust the pH value to 6-8. After stirring for 20 minutes, filter with suction, let the filtrate stand and separate. The organic phase is concentrated under reduced pressure to obtain 2-[2-(dimethylaminoethyl)methylamino]-3-amino-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine, with a purity of 97.1%. Calculated based on the theoretical yield of 100%, it is directly used in the next step reaction. |
| 1H NMR(500MHz,DMSO)δ10.67(s,1H),10.36(s,1H),8.82(s,1H),8.18(s,1H),8.01(s,1H),7.59(d,J=8.2Hz,1H),7.45(d,J=6.8Hz,1H),7.32(t,J=7.5Hz,1H),7.24(s,1H),4.97(q,J=8.7Hz,2H),3.93(s,3H),3.75(s,2H),3.41(s,2H),3.10(s,3H),2.78(s,6H); |
| MS m/z:515.24[M+1]。 |
| Example 14: Preparation of 2-[2-(dimethylaminoethyl)methylamino]-3-amino-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine dihydrochloride (III-1) |
| To the 2-[2-(dimethylaminoethyl)methylamino]-3-amino-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine obtained in Example 13, THF (5.3 L) and ethanol (4.0 L) were added, the temperature was raised to 50-70°C, and concentrated hydrochloric acid (617.8 g, 6.1 mol) was added dropwise. After the addition was completed, the mixture was cooled to room temperature and stirred for 12 hours. Filtered by suction, the filter cake was dried by air at 50°C to obtain 1507.4 g of a crude product. Methanol (6.0 L) and ethanol (4.5 L) were added to a 20 L reaction bottle, and the above crude product was added, the temperature was raised to 55-60 ° C, hot slurry was added for 1-2 hours, the temperature was lowered to room temperature, and suction was filtered to obtain 2-[2-(dimethylaminoethyl)methylamino]-3-amino-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine dihydrochloride (1335.6 g), the liquid phase purity was 99.80%, and the total yield of the two-step reaction with Example 13 was 94.0%. Melting point: 236.6-240.8 ° C. |
| 1H NMR(500MHz,DMSO-d6)δ10.67(s,1H),10.36(s,1H),8.82(s,1H),8.18(s,1H),8.01(s,1H),7.59(d,J=8.2Hz,1H),7.45(d,J=6.8Hz,1H),7.32(t,J=7.5Hz,1H),7.24(s,1H),4.97(q,J=8.7Hz,1H),3.93(s,3H),3.75(s,2H),3.41(s,2H),3.10(s,3H),2.78(s,6H); |
| 13C NMR(126MHz,DMSO-d6)δ166.81,153.27,152.17,150.76,138.61,138.16,138.15,125.46,124.94,123.83(q.J=278.5Hz),123.42,123.41,122.60,122.59,120.52,111.34,111.17,106.29,62.14(q,J=35.3Hz),53.53,46.28,42.27,42.26,40.92,33.67。 |
| Example 15: Preparation of 2-[2-(dimethylaminoethyl)methylamino]-3-amino-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine dihydrochloride (III-1) |
| At room temperature, add the compound of formula IV 2-[2-(dimethylaminoethyl)methylamino]-3-nitro-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (1136.0 g, 2.09 mol) to a 50 L reactor, add acetonitrile (7.95 L) and water (7.95 L), and stir for 10 to 20 minutes. Add hydrosulfite (2563.9 g, 12.50 mol) to the reactor in batches. After adding, continue stirring for 10 to 20 minutes. Control the temperature of the reactor to 35 to 40 ° C and react for 3 hours. Add concentrated hydrochloric acid (2505.3 g, 25.08 mol) to the reactor. After the addition is complete, heat to 35 to 45 ° C and react for 18 hours. 30% sodium hydroxide (1003.2 g, 25.08 mol) aqueous solution was added dropwise to adjust the pH value to 6-8. Solid sodium bicarbonate (1053.5 g, 12.54 mol) was added to adjust the pH value to 7-8. After stirring for 40 minutes, the mixture was filtered, the filtrate was allowed to stand, the layers were separated, and the organic phase was concentrated under reduced pressure. The purity of the liquid phase was detected to be 97.60%. |
| Add ethanol (5.68 L) to the product of the previous step, raise the temperature to 50-70°C, and drop concentrated hydrochloric acid (522 g, 5.23 mol). After the dropwise addition is completed, cool to room temperature and stir for 15 hours. Filter by suction, and air dry the filter cake at 50°C to obtain 2-[2-(dimethylaminoethyl)methylamino]-3-amino-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine dihydrochloride (780 g), with a liquid phase purity of 98.74%. |
| Example 16: Preparation of 2-[2-(dimethylaminoethyl)methylamino]-3-(3-chloro-propionamido)-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine hydrochloride (II-1) |
| 2-[2-(Dimethylaminoethyl)methylamino]-3-amino-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine dihydrochloride (1543.5 g, 2.63 mol) was added to a 50 L reactor, and dichloromethane (13.1 L) and triethylamine (532.2 g, 5.26 mol) were added. The mixture was stirred and cooled to -10 to -5 °C, and a solution of 3-chloropropionyl chloride (501.5 g, 3.95 mol) in dichloromethane (10.0 L) was added dropwise. After the addition is completed, keep warm and stir for 10 to 20 minutes, filter with suction, and the filter cake is formula II-12-[2-(dimethylaminoethyl)methylamino]-3-(3-chloro-propionamide)-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine hydrochloride wet product (2683.5g), which is calculated based on the theoretical yield of 100% and is directly used in the next reaction. |
| Melting point: 233.2-238.7℃ |
| 1H NMR(500MHz,DMSO-d6)δ10.18(s,1H),8.57(s,1H),8.42(s,1H),8.27(t,J=6.6Hz,2H),8.17(s,1H),7.51(d,J=8.1Hz,1H),7.26-7.22(m,1H),7.22-7.17(m,2H),4.99(q,J=9.1Hz,2H),3.91(d,J=6.3Hz,2H),3.89(s,3H),3.55(s,2H),3.13(s,2H),3.02(t,J=6.1Hz,2H),2.85(s,3H),2.64(s,6H); |
| 13C NMR(126MHz,DMSO-d6)δ168.41,161.88,160.22,157.34,148.05,146.73,137.62,133.25,130.86,125.43,124.09(q,J=279.2Hz),122.04,121.74,120.88,118.51,116.60,112.33,110.40,107.09,61.65(q,J=35.3Hz),54.90,40.96,40.95,40.60,38.71,32.96,32.95,32.94。 |
| Example 17: Preparation of 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamido-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (I, crude product) |
| The wet product (2683.5 g) of Formula II 2-[2-(dimethylaminoethyl)methylamino]-3-(3-chloro-propionamide)-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine hydrochloride obtained in Example 16 was added to a 20L reactor, and acetonitrile (16.8L) and triethylamine (1329.3g, 13.15mol) were added, stirred, and heated to reflux for 4 hours. Cooled to room temperature, purified water (4.20L) was added, stirred at room temperature for 3-4 hours, and filtered. The filter cake was transferred to a 50L reactor, dichloromethane (17L) was added, and the pH value was adjusted to 7-8 with saturated sodium bicarbonate aqueous solution (17L). Liquid separation, the organic phase was transferred to a 20L reactor, activated carbon (84.3g) was added, refluxed for 1 hour, cooled to 20-30°C, and filtered. The filtrate was concentrated to dryness under reduced pressure to obtain the compound of formula I 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (1390g), with a total yield of 92.9% and a purity of 99.21% for the two-step reaction with Example 16. |
| 1H NMR(500MHz,DMSO-d6)δ9.96(s,1H),8.71(s,1H),8.44(s,1H),8.29(d,J=5.3Hz,1H),8.26(d,J=7.7Hz,1H),8.13(s,1H),7.51(d,J=8.2Hz,1H),7.24(t,J=7.2Hz,1H),7.20(d,J=5.3Hz,1H),7.15(t,J=7.2Hz,1H),6.51(dd,J=17.0,10.2Hz,1H),6.28(dd,J=17.0,1.8Hz,1H),5.78(dd,J=10.2,1.8Hz,1H),5.00(q,J=9.1Hz,2H),3.89(s,3H),3.18(t,J=6.5Hz,2H),2.87(s,3H),2.48(t,J=6.5Hz,2H),2.22(s,6H); |
| 13C NMR(126MHz,DMSO-d6)δ163.40,161.84,160.26,157.35,148.07,147.15,137.60,133.23,131.61,130.07,126.67,125.41,124.03(q,J=278.5Hz),122.00,121.68,120.80,118.39,116.13,112.36,110.37,107.02,61.29(q,J=35.3Hz),56.57,52.44,45.60,45.59,38.54,32.93; |
| MS m/z:569.25[M+1]。 |
| Example 18: Preparation of 2-[2-(dimethylaminoethyl)methylamino]-3-amino-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (III’) |
| At room temperature, add the compound of formula IV 2-[2-(dimethylaminoethyl)methylamino]-3-nitro-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (20.0 g, 36.73 mmol) to a 1L reaction bottle, add tetrahydrofuran (134 mL) and water (66 mL), and stir for 10 to 20 minutes. Add hydrosulfite (47.9 g, 220.38 mmol) to the reaction bottle in batches. After addition, continue stirring for 10 to 20 minutes. Control the internal temperature to 35-40°C and react for 3 hours. Add concentrated hydrochloric acid (89.3 g, 881.52 mmol) to the reaction bottle. After the addition is complete, heat to 42 to 47°C and react for 17 hours. 30% sodium hydroxide (35.26 g, 881.52 mmol) aqueous solution was added dropwise, and solid sodium bicarbonate (18.5 g, 220.38 mmol) was added in batches to adjust the pH value to 6-8. After stirring for 30 minutes, the mixture was filtered, and the filtrate was allowed to stand and separated. The organic phase was concentrated to dryness under reduced pressure to obtain 2-[2-(dimethylaminoethyl)methylamino]-3-amino-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (19.2 g) with a purity of 95.8% and a yield of 97.12%. |
| Example 19: Preparation of 2-[2-(dimethylaminoethyl)methylamino]-3-(3-chloro-propionamido)-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine hydrochloride (II-1) |
| Add 2-[2-(dimethylaminoethyl)methylamino]-3-amino-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (5 g, 9.72 mmol) to a 250 mL reaction bottle, add dichloromethane (42 mL), stir, protect with argon, cool to -5 to 0°C, and add 3-chloropropionyl chloride (1.851 g) and dichloromethane (33 mL) dropwise. After the addition is complete, the mixture is stirred for 10-20 minutes at a temperature maintained at room temperature. After the reaction is complete, the mixture is concentrated under reduced pressure to obtain 2-[2-(dimethylaminoethyl)methylamino]-3-(3-chloro-propionamido)-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine hydrochloride (7.0 g) with a purity of 85.67%. Melting point: 233.5-238.9°C. |
| 1H NMR(500MHz,DMSO-d6)δ10.18(s,1H),8.57(s,1H),8.42(s,1H),8.27(t,J=6.6Hz,2H),8.17(s,1H),7.51(d,J=8.1Hz,1H),7.26-7.22(m,1H),7.22-7.17(m,2H),4.99(q,J=9.1Hz,2H),3.91(d,J=6.3Hz,2H),3.89(s,3H),3.55(s,2H),3.13(s,2H),3.02(t,J=6.1Hz,2H),2.85(s,3H),2.64(s,6H); |
| 13C NMR(126MHz,DMSO-d6)δ168.41,161.88,160.22,157.34,148.05,146.73,137.62,133.25,130.86,125.43,124.09(q,J=279.2Hz),122.04,121.74,120.88,118.51,116.60,112.33,110.40,107.09,61.65(q,J=35.3Hz),54.90,40.96,40.95,40.60,38.71,32.96,32.95,32.94。 |
| Example 20: Preparation of 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamido-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (I, crude product) |
| The 2-[2-(dimethylaminoethyl)methylamino]-3-(3-chloro-propionamido)-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine hydrochloride obtained in Example 19 was added to a 250 mL reaction bottle, and acetonitrile (45 mL) and triethylamine (4.9 g) were added. The mixture was stirred magnetically and protected by argon. The temperature was raised to reflux in an oil bath. The reaction was allowed to react for 6 h. Water (23 mL) was added dropwise, and the mixture was naturally cooled to room temperature in an oil bath. The mixture was filtered with suction, and the filter cake was transferred to a 500 mL reaction bottle. Dichloromethane (100 mL) was added, and the pH value was adjusted to 7-8 with saturated aqueous sodium bicarbonate solution (100 mL). The liquids were separated and the organic phase was concentrated under reduced pressure. The solid was dried in an oven at 50°C to give 2-[2-(dimethylaminoethyl)methylamino]-3-(3-chloro-propionamide)-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (4.1 g) with a purity of 97.7%. The total yield of the two-step reaction with Example 19 was 74.17%. |
| Comparative Example 1: Preparation of 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (I, crude product) |
| 2-[2-(dimethylaminoethyl)methylamino]-3-amino-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (1 g, 1.94 mmol) was added to a 50 mL multi-necked flask, tetrahydrofuran (10 mL) was used as the solvent, argon was replaced three times, and stirring was maintained at 0-5°C under argon protection, and 3-chloropropionyl chloride (0.37 g, 2.92 mmol), the addition was completed in 15 minutes, and the mixture was stirred at 0-5°C for 1 hour. Sodium hydroxide (0.31 g, 7.77 mmol) and water (1 mL) were added to the reaction solution, and the temperature was raised to 65°C and stirred for 15 hours. Saturated ammonium chloride solution (10 mL) was added, and the liquids were separated. The organic phase was washed with saturated sodium bicarbonate solution (10 mL). The liquids were separated and the organic phase was concentrated to dryness to obtain 1.04 g of a yellow solid with a yield of 94.9% and a purity of 87.35%. |
| Comparative Example 2: Preparation of 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (I, crude product) |
| Add 2-[2-(dimethylaminoethyl)methylamino]-3-amino-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (5.0 g) to a 250 mL reaction bottle, add acetone (50 mL) and potassium carbonate (940 mg), stir, protect with argon, cool to -50°C, and add 3-chloropropionyl chloride (1.481 g) dropwise. After the addition is completed, the temperature is raised to -20°C and stirred for 30 minutes. A solution of sodium hydroxide (350 mg) and water (60 ml) is added dropwise over 10 minutes. The mixture is stirred at room temperature for 3 to 4 hours. The mixture is filtered and the filter cake is dried in an oven at 50°C to obtain a compound of formula II-1′, 2-[2-(dimethylaminoethyl)methylamino]-3-(3-chloro-propionamide)-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (4.28 g) with a purity of 64.18%. |
| 1H NMR(400MHz,DMSO-d 6 )δ10.32(s,1H),10.21(s,1H),8.54(s,1H),8.43(s,1H)8.29-8.28(d,J=5.1Hz,1H),8.28-8.26(d,J=6.2Hz,1H),8.19(s,1H),7.54-7.52(d,J=8.0Hz,1H),7.27-7.18(m,3H),5.77(s,2H),5.00(q,J=9.1Hz,1H),3.92(t,J=6.2Hz,1H),3.63(t,J=5.7Hz,2H),3.28(t,J=5.7Hz,2H),3.06-3.03(t,J=6.2Hz,2H),2.85(s,3H),2.74(s,6H). |
| MS m/z:605.23[M+1]。 |
| Add 2-[2-(dimethylaminoethyl)methylamino]-3-(3-chloro-propionamido)-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (4.28 g) to a 250 mL reaction bottle, add acetonitrile (45 ml) and triethylamine (3.606 g), stir magnetically, protect with argon, heat in an oil bath to reflux, and react for 6 h. Water (23 ml) was added dropwise, the temperature was naturally lowered in an oil bath and stirring was continued overnight (16 h), filtered with suction, and the solid was dried to obtain 2-[2-(dimethylaminoethyl)methylamino]-3-(3-chloro-propionamido)-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (3.3 g) with a purity of 95.13% and a two-step yield of 59.42%. |
| Example 21: Purification of 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamido-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (I) |
| The crude product (1390 g) of 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine was transferred to a 50L reactor, acetone (25.0L) was added, argon was replaced 3 times, the temperature was raised to 45-50°C, all the solids were dissolved, and purified water (6.95L) was added dropwise. After the addition was completed, the mixture was cooled to 20-25°C and stirred for 2 hours. The mixture was filtered and the filter cake was vacuum dried at 50°C for 24 hours to obtain purified 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (895g). The reaction yield is 66.7% and the purity is 99.89%. |
| Example 22: Purification of 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamido-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (I) |
| In a 250mL three-necked flask, add 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine crude product (5.0g), add ethyl acetate 100mL, heat to 70-75°C in an oil bath to dissolve all the solids, then cool naturally to 25°C in an oil bath, filter and dry to obtain purified 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (3.1g) with a purity of 99.73% and a yield of 62.0%. |
| Example 23: Purification of 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamido-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (I) |
| In a 250mL three-necked flask, add crude 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (5.0g), add ethyl acetate 100mL, heat to 70-75°C in an oil bath, dissolve all the solids, continue to stir for 30min, and drop 150mL of n-heptane. After the drop is complete, cool to 25°C in an oil bath, filter by suction, and dry to obtain purified 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (4.0g), with a purity of 99.32% and a yield of 80%. |
| Example 24: Purification of 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamido-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (I) |
| In a 250mL three-necked flask, add crude 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (5.0g), add acetonitrile 75mL, heat in an oil bath to 77-82°C, dissolve all the solids, and drop 25mL of water. After dripping, naturally cool to 25°C in the oil bath, filter with suction, and dry to obtain purified 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (4.3g), with a purity of 99.64% and a yield of 86%. |
| Example 25: Purification of 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamido-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (I) |
| In a 250mL three-necked flask, add crude 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (5.0g), add acetonitrile 75mL, heat in an oil bath to 77-82°C, dissolve all the solids, and continue to stir for 30min. Cool naturally to 25°C in the oil bath, filter with suction, and dry to obtain purified 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (4.0g), with a purity of 99.45% and a yield of 80%. |
| Example 26: Purification of 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamido-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (I) |
| In a 250mL three-necked flask, add crude 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamido-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (5.0 g), add 20mL of tetrahydrofuran, heat in an oil bath to 45-50°C to dissolve all the solids, continue to stir and maintain the temperature for 30 minutes, and add 40mL of n-heptane dropwise. After the addition was completed, the mixture was naturally cooled to 25°C in an oil bath, filtered and dried to obtain purified 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (4.23 g) with a purity of 99.51% and a yield of 84.6%. |
| Example 27: Purification of 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamido-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (I) |
| In a 250mL three-necked flask, add crude 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (5.0g), add 100mL of isopropanol, heat to 50°C in an oil bath, dissolve all the solids, and continue to stir for 30 minutes. Cool naturally to 22°C in the oil bath, filter with suction, and dry to obtain purified 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (4.25g), with a purity of 99.51% and a yield of 85%. |
| Example 28: Purification of 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamido-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (I) |
| In a 250mL three-necked flask, add crude 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (5.0g), add 75mL of methanol, and heat to 55-60°C in an oil bath to dissolve all the solids. Cool naturally to 17°C in the oil bath, stir overnight, filter with suction, and dry to obtain purified 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (3.55g), with a purity of 99.63% and a yield of 71%. |
| Example 29: Purification of 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamido-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (I) |
| In a 250mL three-necked flask, add crude 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (5.0 g), add 50mL of dichloromethane, heat in an oil bath to 40°C to dissolve all the solids, continue to stir and maintain the temperature for 30 minutes, and add 100mL of n-heptane dropwise. The mixture was naturally cooled to 15°C in an oil bath, stirred overnight, filtered and dried to obtain purified 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (3.78 g) with a purity of 99.56% and a yield of 75.6%. |
| Example 30: Purification of 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamido-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (I) |
| In a 250mL three-necked flask, add crude 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (5.0g), add 100mL of toluene, heat to 65°C in an oil bath, dissolve all the solids, and continue to stir for 30 minutes. Cool naturally to 20°C in the oil bath, filter with suction, and dry to obtain purified 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (3.27g), with a purity of 99.57% and a yield of 65.4%. |
| Example 31: Purification of 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamido-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (I) |
| In a 250mL three-necked flask, add 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine crude product (5.0g), add DMF50mL, heat to 80°C in an oil bath, dissolve all the solids, continue to stir for 30min, and drop 25mL of water. Naturally cool to 20°C in the oil bath, filter with suction, and dry to obtain purified 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (3.84g), with a purity of 99.77% and a yield of 76.8%. |
| Example 32: Purification of 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamido-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (I) |
| To a 25 mL single-necked bottle, add 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine crude product (1.0 g), add tetrahydrofuran (6 mL), protect with argon, heat in an oil bath to 40-45°C until all the solution is dissolved, continue to stir and keep warm for 30 min, cool naturally to 22°C in an oil bath, filter and obtain a solid. The solid was transferred to a crystallization dish and dried to obtain purified 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (622 mg) with a purity of 99.83% and a yield of 62.2%. |
| Example 33: Purification of 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamido-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (I) |
| To a 50 mL single-mouth bottle, add 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine crude product (1.0 g), add acetone (15 mL), protect with argon, heat in an oil bath to 45-50° C. until all the solution is dissolved, and then continue to stir and keep warm for 30 min, cool naturally to 22° C. in an oil bath, filter and dry to obtain purified 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (537 mg) with a purity of 99.83% and a yield of 53.7%. |
| Example 34: Purification of 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamido-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (I) |
| To a 50 mL single-mouth bottle, add 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine crude product (1.0 g), add tetrahydrofuran (8 mL), protect with argon, heat in an oil bath to 40-45° C. until all the solution is dissolved, and continue to stir and keep warm for 30 min. Add water (16 mL) dropwise, cool naturally to 21° C. in an oil bath, filter and dry to obtain purified 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (880 mg) with a purity of 99.68% and a yield of 88.0%. |
| Example 35: Purification of 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamido-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (I) |
| To a 100 mL single-mouth bottle, add crude 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (1.0 g), add ethanol (35 mL), protect with argon, heat in an oil bath to 75-80° C. until all the solution is dissolved, add water (10 mL) dropwise over 10 min, cool naturally to 20° C. in an oil bath, filter with suction, and dry to obtain purified 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (915 mg), with a yield of 91.5% and a purity of 99.49%. |
| Example 36: Purification of 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamido-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (I) |
| To a 50 mL single-mouth bottle, add 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine crude product (1.0 g), add xylene (20 mL), protect with argon, heat in an oil bath to 80° C. until all the solution is dissolved, cool naturally to 20° C. in an oil bath, filter with suction, and dry to obtain purified 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (798 mg), with a yield of 79.8% and a purity of 99.48%. |
| Example 37: Purification of 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamido-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (I) |
| In a 250mL three-necked flask, add 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine crude product (5.0g), add ethanol 125mL, heat in an oil bath to 75-80°C to dissolve all the solids, continue to stir and keep warm for 30min, then cool naturally to 25°C in an oil bath, filter and dry to obtain purified 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (3.7g) with a purity of 99.66% and a yield of 74%. |
| Example 38: Purification of 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamido-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (I) |
| To a 100 mL single-mouth bottle, add crude 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (1.0 g), add methanol (35 mL), protect with argon, heat in an oil bath to 80° C. until all the solution is dissolved, add water (10 mL) dropwise over 10 min, cool naturally to 20° C. in an oil bath, filter and dry to obtain purified 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (912 mg), with a yield of 91.2% and a purity of 99.53%. |
PATENT
CN110606842
WO2019238103
PAPER
https://www.nature.com/articles/s41401-020-0389-3
| NCT Number | Sponsor | Condition | Start Date | Phase |
|---|---|---|---|---|
| NCT02973763 | Allist Pharmaceuticals, Inc. | NSCLC | December 30, 2016 | Phase 1 |
| NCT03787992 | Allist Pharmaceuticals, Inc. | Locally Advanced or Metastatic EGFR Sensitising Mutation Positive Non-small Cell Lung Cancer | May 30, 2019 | Phase 3 |
| NCT03452592 | Allist Pharmaceuticals, Inc. | Advanced NSCLC Patients With T790M | April 30, 2018 | Phase 2 |
- [1]. Y. Shi, et al. P2.03-028 Third Generation EGFR Inhibitor AST2818 (Alflutinib) in NSCLC Patients with EGFR T790M Mutation: A phase1/2 Multi-Center Clinical Trial.[2]. Alexander I. Spira, et al. FURVENT: Phase 3 trial of furmonertinib vs chemotherapy as first-line treatment for advanced NSCLC with EGFR exon 20 insertion mutations (FURMO-004). Journal of Clinical Oncology. Volume 42, Number 16_suppl.
/////////FIRMOMERTINIB, Furmonertinib, Alflutinib, AST 2818, UNII-A49A7A5YN4, PHASE 2, CANCER
Lotilaner



Lotilaner
- CAS 1369852-71-0
- Credelio
- XDEMVY
- lotilanerum
- 596.8 g/mol, C20H14Cl3F6N3O3S
- TP 03
- TP-03
- TP03
3-methyl-N-[2-oxo-2-(2,2,2-trifluoroethylamino)ethyl]-5-[(5S)-5-(3,4,5-trichlorophenyl)-5-(trifluoromethyl)-4H-1,2-oxazol-3-yl]thiophene-2-carboxamide
FDA Xdemvy, 7/25/2023, To treat Demodex blepharitis
Drug Trials Snapshot
Lotilaner, sold under the brand name Xdemvy, is an ectoparasiticide (anti-parasitic) medication used for the treatment of blepharitis (inflammation of the eyelid) caused by infestation by Demodex (tiny mites).[1][7] It is used as an eye drop.[1]
It was approved for medical use in the United States in July 2023.[1][7][8][9] The US Food and Drug Administration (FDA) considers it to be a first-in-class medication.[10]
SYN
https://patents.google.com/patent/WO2022016490A1/en
Scheme 1

Scheme 1 depicts coupling the compound of formula (5) with an appropriate amine to give lotilaner. An appropriate amine refers to either 2-amino-2′, 2′, 2′-trifluoroethyl-acetamide or the sequential reaction of glycine optionally carboxyl protected, followed by coupling with 2, 2, 2-trifluorethylamine. Such coupling reactions of carboxylic acids or activated carboxylic acid derivatives such as acid halides with amines to form amides are well known in the art. The use of carboxyl protected glycine, deprotection, and an amide coupling with 2, 2, 2-trifluorethylamine is likewise readily accomplished. See WO 2010/070068 and WO 2014/090918.
Example 1
(5S) -3- (5-Bromo-4-methyl-2-thienyl) -5- (3, 4, 5-trichlorophenyl) -5- (trifluoromethyl) -4H- isoxazole

Combined (Z/E) 1- (5-bromo-4-methyl-2-thienyl) -4, 4, 4-trifluoro-3- (3, 4, 5-trichlorophenyl) but-2-en-1-one (50.0 g, 104.5 mmol) and (R) – [ (2S) -1- [ [3, 5-bis (tert-butyl) phenyl] methyl] -5-vinyl-quinuclidin-1-ium-2-yl] – (6-methoxy-4-quinolyl) methanol bromide (0.11 eq. ) in dichloromethane (100 mL) and ethyl t-butyl ether (400 mL) . The reaction mixture was stirred at 30℃ for 30 minutes and then cooled to the range of-20℃ then slowly added a solution of hydroxylamine in water (50%, 40 mL, 313 mmol, 3.0 eq. ) and sodium hydroxide (34.5 mL, 345 mmol, 10 M, 3.3 eq. ) maintaining an internal temperature in the range of-15℃ to-20℃. After stirring 18 hours at-15℃ to-20℃ aqueous hydrochloric acid (1N, 500 mL) was added and the reaction mixture was stirred at 15℃ to 20℃ then the stirring was stopped and after 30 minutes the phases were separated. The organic layer was extracted with aqueous hydrochloric acid (1N, 75 mL) , the layers separated and the organic layer again extracted with aqueous hydrochloric acid (1N, 100 mL) . The organic layer was separated and extracted with saturated aqueous sodium bicarbonate (75 mL) and the layers were separated and again the organic layer was extracted with saturated aqueous sodium bicarbonate (100 mL) . The layers were separated and the organic layer was dried over sodium sulfate (10 g) . The organic layer was filtered, the cake washed with ethyl t-butyl ether (50 mL) and then montmorillonite clay (50 g) was added and the mixture was stirred at 10℃ to 20℃. After 2 hours the reaction mixture was filtered, the cake rinsed with ethyl t-butyl ether (50 mL) and the filtrate was concentrated to about 100 mL, twice added THF and concentrated again to about 100 mL, and then added THF (150 mL) to obtain the title compound as a solution in THF. The solution was evaluated by chiral HPLC which indicated 96.5%S-isomer and 3.5%R-isomer.
Example 2
3-Methyl-5- [ (5S) -5- (3, 4, 5-trichlorophenyl) -5- (trifluoromethyl) -4H-isoxazol-3- yl] thiophene-2-carboxylic acid

A 22%solution of (5S) -3- (5-bromo-4-methyl-2-thienyl) -5- (3, 4, 5-trichlorophenyl) -5- (trifluoromethyl) -4H-isoxazole (185.0 g, 374.8 mmol) in THF was cooled to 0℃ to 5℃. A solution of ethyl magnesium chloride in THF (2 M, 300 mL, 1.6 eq) was added dropwise maintaining an internal temperature below 10℃. The reaction mixture was stirred at 15℃ to 20℃ for 2 to 4 hours. Then carbon dioxide gas (58 g, 3.5 eq) was introduced subsurface at 0℃ to 5℃ after passing through concentrated sulfuric acid (50 mL) . The reaction mixture was stirred at 0℃ to 5℃ for 2 hours and an 8%aqueous sodium chloride solution (601 g) was added dropwise at below 10℃, followed by addition of 37%aqueous HCl solution (92.5 g) at below 0℃. The reaction mixture was stirred at 10℃ to 15℃ for 30 minutes then the stirring was stopped and after 30 minutes the phases were separated. The organic layer was concentrated to about 370 mL under vacuum, followed by three iterations of THF (1850 mL) addition and concentration under vacuum to about 370 mL to 555 mL. After confirming the reaction mixture was dry, three cycles of acetonitrile (925 mL) addition followed by vacuum concentration to about 555 mL to 740 mL were performed. The reaction mixture was heated to 75℃ and gradually cooled to 50℃ over one hour. Product seeds (1.85 g) were added at 50℃ and the reaction mixture was stirred at 50℃ for 30 minutes. The batch was gradually cooled to-10℃ over three hours and kept at-10℃ for two hours. The batch was filtered and the cake was washed with cold acetonitrile (93 to 185 mL) . 110 g of the title compound was obtained after drying the wet cake at 50℃ under vacuum for 12 hours. The product was evaluated by chiral HPLC which indicated>99.9%S-isomer.
Above-referenced product seeds were prepared as follows. A solution of (5S) -3- (5-bromo-4-methyl-2-thienyl) -5- (3, 4, 5-trichlorophenyl) -5- (trifluoromethyl) -4H-isoxazole (48.93 g, 99.1 mmol) in 300mL of THF was cooled to 0℃ to 5℃. A solution of ethyl magnesium chloride in THF (2 M, 80 mL) was added dropwise maintaining an internal temperature below 10℃. The reaction mixture was stirred at 15℃ to 20℃ for 2 to 4 hours. Then carbon dioxide gas (25 g, 3.5 eq) was introduced subsurface at 0℃ to 5℃ after passing through concentrated sulfuric acid (50 mL) . The reaction mixture was stirred at 0℃ to 5℃ for 6 hours and an 5%aqueous sodium chloride solution (157 g) was added dropwise at below 10℃, followed by addition of 37%aqueous HCl solution (25 g) at below 0℃. The reaction mixture was stirred at 10℃ to 15℃ for 30 minutes then the stirring was stopped and after 30 minutes the phases were separated. The organic layer was concentrated to remove the solvent. 50ml of heptane was added into the mixture then removed the solvent. The crude product was dissolved in 50mL of EA and 100mL of heptane at 40℃. Additional 1000mL of heptane was charged dropwise into the mixture slowly. Then the mixture was stirred at 40℃ for 15h. The mixture was filtered and the wet cake was obtained. The wet cake was slurried by acetone at 20℃. The mixture was filter and the wet cake was dried at 50℃ under vacuum for 3h to afford 9.7g of product. The product was evaluated by chiral HPLC which indicated>99.9%S-isomer.
Example 3
3-Methyl-N- [2-oxo-2- (2, 2, 2-trifluoroethylamino) ethyl] -5- [ (5S) -5- (3, 4, 5-trichlorophenyl) – 5- (trifluoromethyl) -4H-isoxazol-3-yl] thiophene-2-carboxamide

A solution of 3-methyl-5- [ (5S) -5- (3, 4, 5-trichlorophenyl) -5- (trifluoromethyl) -4H-isoxazol-3-yl] thiophene-2-carboxylic acid (101.5 g, 221.3 mmol) in DCM (1000 mL) was heated to 40℃. Thionyl chloride (50 g, 1.9 eq) was added dropwise and the reaction mixture was stirred at reflux for 2 to 4 hours. The reaction mixture was concentrated to 100 to 200 mL and DCM (500 mL) was added. Two more cycles of concentration followed by DCM addition were performed. In a separate vessel, a suspension of 2-amino-trifluoroethyl-acetamide HCl (50.26 g, 1.2 eq) in DCM (500 mL) was cooled to 0℃ to 5℃, triethylamine (70.15 g, 3.1 eq) was added, and the reaction mixture was stirred at 0℃ to 5℃ for 30 minutes. The acid chloride solution in DCM was then transferred to the reaction mixture containing 2-amino-trifluoroethyl-acetamide maintaining the internal temperature below 5℃. The reaction mixture was stirred at 0℃ to 5℃ for 2 to 4 hours. 1 N HCl (500 mL) was added dropwise and the reaction mixture was stirred at 15 to 25℃ for 30 minutes. The stirring was stopped and after 30 minutes the phases were separated. The organic layer was extracted with saturated sodium bicarbonate solution (1N, 1000 mL) , the layers separated and the organic layer extracted with water (1000 mL) . The layers were separated and the organic layer was concentrated under vacuum to 200 to 300 mL. Twice ethyl acetate (500 mL) was added and the batch was concentrated to 200 mL. The reaction mixture was heated to 55℃ and n-heptane (700 mL) was added dropwise at 55℃. Product seeds (1.0 g) was added and the reaction mixture was stirred at 55℃ for one hour. n-Heptane (1000 mL) was added dropwise and the mixture was stirred at 55℃ for three hours. The batch was gradually cooled to 35℃ over three hours, then to 20℃ over three hours. The batch was filtered and the cake was washed with n-heptane (200 mL) . 113 g of the title compound was obtained after drying at 50℃ under vacuum for 12 hours.
Above-referenced product seeds are prepared as follows. The crude product was dissolved in 7.9 wt-parts of cumene to obtain a solution at<150℃. Then 2.3 wt-parts of heptanes was added to the hot solution until slight haze was observed. The heating was turned off and the mixture was cooled to ambient temperature and stirred overnight. The desired polymorph G was obtained as powder after filtration and drying under vacuum, which was used as seeds to induce crystallization of polymorph G in future batches.
PATENT
https://patents.google.com/patent/WO2017176948A1/en
PATENT
CN112457267
https://patentscope.wipo.int/search/en/detail.jsf?docId=CN320823634&_cid=P10-MBBMZO-15915-1
REFSatish Kumar RangarajuSatish Kumar Rangaraju •
The first FDA-approved treatment for Demodex Blepharitis, a common eyelid disease caused by microscopic mites living in the eyelashes’ hair follicles. hashtag#Lotilaner
For more information blog:
https://lnkd.in/gFTeKEtv

Research
Tarsus Pharmaceuticals has conducted phase II studies of lotilaner as a remedy to prevent tick bites in humans.[16][11]
References
- ^ Jump up to:a b c d e “Xdemvy- lotilaner ophthalmic solution solution/ drops”. DailyMed. 26 July 2023. Retrieved 23 August 2023.
- ^ Jump up to:a b c “Credelio- lotilaner tablet, chewable”. DailyMed. Archived from the original on 5 August 2021. Retrieved 4 August 2021.
- ^ Jump up to:a b c “Credelio- lotilaner tablet, chewable”. DailyMed. Archived from the original on 5 August 2021. Retrieved 4 August 2021.
- ^ Jump up to:a b “Credelio EPAR”. European Medicines Agency (EMA). 17 September 2018. Archived from the original on 27 January 2022. Retrieved 4 August 2021.
- ^ Jump up to:a b c “Lotimax EPAR”. European Medicines Agency. 13 March 2024. Retrieved 20 March 2024. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ “Lotimax Product information”. Union Register of veterinary medicinal products. 29 April 2024. Retrieved 29 August 2024.
- ^ Jump up to:a b “Novel Drug Approvals for 2023”. U.S. Food and Drug Administration (FDA). 25 July 2023. Archived from the original on 21 January 2023. Retrieved 5 August 2023.
- ^ “Xdemvy: FDA-Approved Drugs”. U.S. Food and Drug Administration (FDA). Archived from the original on 27 July 2023. Retrieved 5 August 2023.
- ^ “FDA Approves Xdemvy (lotilaner ophthalmic solution) 0.25% for the treatment of Demodex blepharitis” (Press release). Tarsus Pharmaceuticals. 25 July 2023. Retrieved 5 August 2023 – via GlobeNewswire.
- ^ New Drug Therapy Approvals 2023. U.S. Food and Drug Administration (FDA) (Report). January 2024. Archived from the original (PDF) on 10 January 2024. Retrieved 9 January 2024.
- ^ Jump up to:a b c “A Pill That Kills Ticks Is a Promising New Weapon Against Lyme Disease”. Wired. 15 March 2024. Retrieved 17 March 2024.
- ^ Jump up to:a b “Lotimax PI”. Union Register of medicinal products. 29 April 2024. Retrieved 17 June 2024.
- ^ Kuntz EA, Kammanadiminti S (November 2017). “Safety evaluation of lotilaner in dogs after oral administration as flavoured chewable tablets (Credelio)”. Parasites & Vectors. 10 (1): 538. doi:10.1186/s13071-017-2468-y. PMC 5664904. PMID 29089043.
- ^ “Freedom Of Information Summary, Supplemental New Animal Drug Application, NADA 141-494, Credelio, Lotilaner, Chewable Tablets, Dogs”. 3 September 2019. Archived from the original on 18 May 2022. Retrieved 1 December 2019.
- ^ Jump up to:a b “Credelio Plus EPAR”. European Medicines Agency. 19 February 2021. Archived from the original on 7 December 2022. Retrieved 4 August 2021. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ “Tarsus Announces Positive Topline Results from Carpo, a Phase 2a Proof-of-Concept “Tick-Kill” Trial Evaluating TP-05 (lotilaner) for the Prevention of Lyme Disease (press release)”. Tarsus Pharmaceuticals. 22 February 2024. Archived from the original on 17 March 2024. Retrieved 17 March 2024.
| Clinical data | |
|---|---|
| Trade names | Credelio, Xdemvy, others |
| Other names | TP-03 |
| AHFS/Drugs.com | Monograph |
| License data | US DailyMed: Lotilaner |
| Routes of administration | By mouth, eye drops |
| Drug class | Antiparasitic |
| ATC code | S01AX25 (WHO) QP53BE04 (WHO) |
| Legal status | |
| Legal status | US: ℞-only[1][2][3]EU: Rx-only[4][5][6] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 1369852-71-0 |
| PubChem CID | 76959255 |
| DrugBank | DB17992 |
| ChemSpider | 32699771 |
| UNII | HEH4938D7K |
| KEGG | D11212 |
| ChEBI | CHEBI:229657 |
| ChEMBL | ChEMBL3707310 |
| CompTox Dashboard (EPA) | DTXSID701027551 |
| Chemical and physical data | |
| Formula | C20H14Cl3F6N3O3S |
| Molar mass | 596.75 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
- Toutain CE, Seewald W, Jung M: Pharmacokinetics of lotilaner following a single oral or intravenous administration in cats. Parasit Vectors. 2018 Jul 13;11(1):412. doi: 10.1186/s13071-018-2966-6. [Article]
- Rufener L, Danelli V, Bertrand D, Sager H: The novel isoxazoline ectoparasiticide lotilaner (Credelio): a non-competitive antagonist specific to invertebrates gamma-aminobutyric acid-gated chloride channels (GABACls). Parasit Vectors. 2017 Nov 1;10(1):530. doi: 10.1186/s13071-017-2470-4. [Article]
- Lamassiaude N, Toubate B, Neveu C, Charnet P, Dupuy C, Debierre-Grockiego F, Dimier-Poisson I, Charvet CL: The molecular targets of ivermectin and lotilaner in the human louse Pediculus humanus humanus: New prospects for the treatment of pediculosis. PLoS Pathog. 2021 Feb 18;17(2):e1008863. doi: 10.1371/journal.ppat.1008863. eCollection 2021 Feb. [Article]
- FDA Approved Drug Products: XDEMVY (lotilaner) 0.25%, Ophthalmic Solution (July 2023) [Link]
- Business Wire: FDA Approves XDEMVY™ (lotilaner ophthalmic solution) 0.25% for the treatment of Demodex blepharitis [Link]
- EMA Medicines Overview: Credelio [Link]
///////////Lotilaner, Xdemvy, FDA 2023, APPROVALS 2023, Credelio, XDEMVY, lotilanerum
Fexagratinib
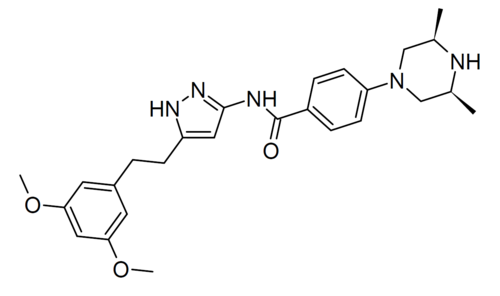
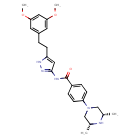
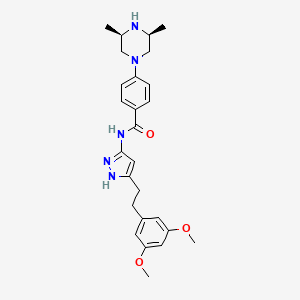
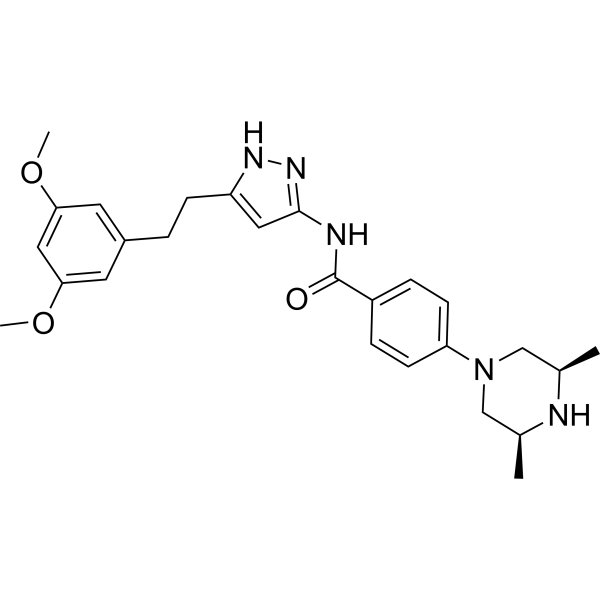
Fexagratinib
AZD 4547; ADSK091 cas 1035270-39-3
WeightAverage: 463.582
Monoisotopic: 463.258339943
Chemical FormulaC26H33N5O3
N-(5-(2-(3,5-DIMETHOXYPHENYL)ETHYL)-1H-PYRAZOL-3-YL)-4-((3R,5S)-3,5-DIMETHYLPIPERAZIN-1-YL)BENZAMIDE
N-{5-[2-(3,5-dimethoxyphenyl)ethyl]-1H-pyrazol-3-yl}-4-[(3R,5S)-3,5-dimethylpiperazin-1-yl]benzamide
- OriginatorAstraZeneca
- DeveloperAbbisko Therapeutics; AstraZeneca; Dust Diseases Authority; Institute of Respiratory Health; National Cancer Institute (USA); University of Glasgow; University of Leeds; University of Wisconsin-Madison
- ClassAntineoplastics; Benzamides; Phenyl ethers; Piperazines; Pyrazoles; Small molecules
- Mechanism of ActionType 1 fibroblast growth factor receptor antagonists; Type 3 fibroblast growth factor receptor antagonists; Type-2 fibroblast growth factor receptor antagonists
- Phase IIGastric cancer; Lymphoma; Multiple myeloma; Solid tumours; Urogenital cancer
- PreclinicalSkin cancer
- No development reportedLiver cancer
- DiscontinuedBladder cancer; Breast cancer; Glioblastoma; Head and neck cancer; Lung cancer; Mesothelioma; Non-small cell lung cancer; Oesophageal cancer
- 13 Sep 2024Pharmacodynamics data from the preclinical studies in Solid tumours presented at the 49th European Society for Medical Oncology Congress (ESMO-2024)
- 28 Feb 2024No recent reports of development identified for preclinical development in Liver-cancer in China (PO)
- 23 Jan 2024Preclinical trials in Solid tumours (Monotherapy) in China (PO) (Abbisko Therapeutics pipeline, January 2024)
Fexagratinib (AZD4547) is an experimental drug which acts as an inhibitor of the fibroblast growth factor receptors, having high affinity for FGFR1, FGFR2 and FGFR3 and weaker activity at FGFR4. It has reached clinical trials in humans against several forms of cancer, but has had only limited use as a medicine due to an unfavorable side effect profile, though it may have some applications in combination with other drugs. However it is still widely used in cancer research.[1][2][3][4][5]
SCHEME
SIDECHAIN
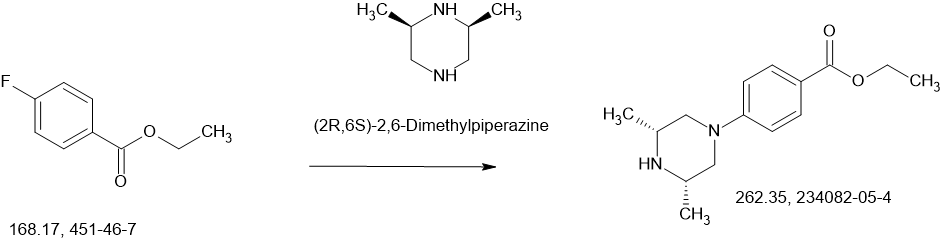
MAIN
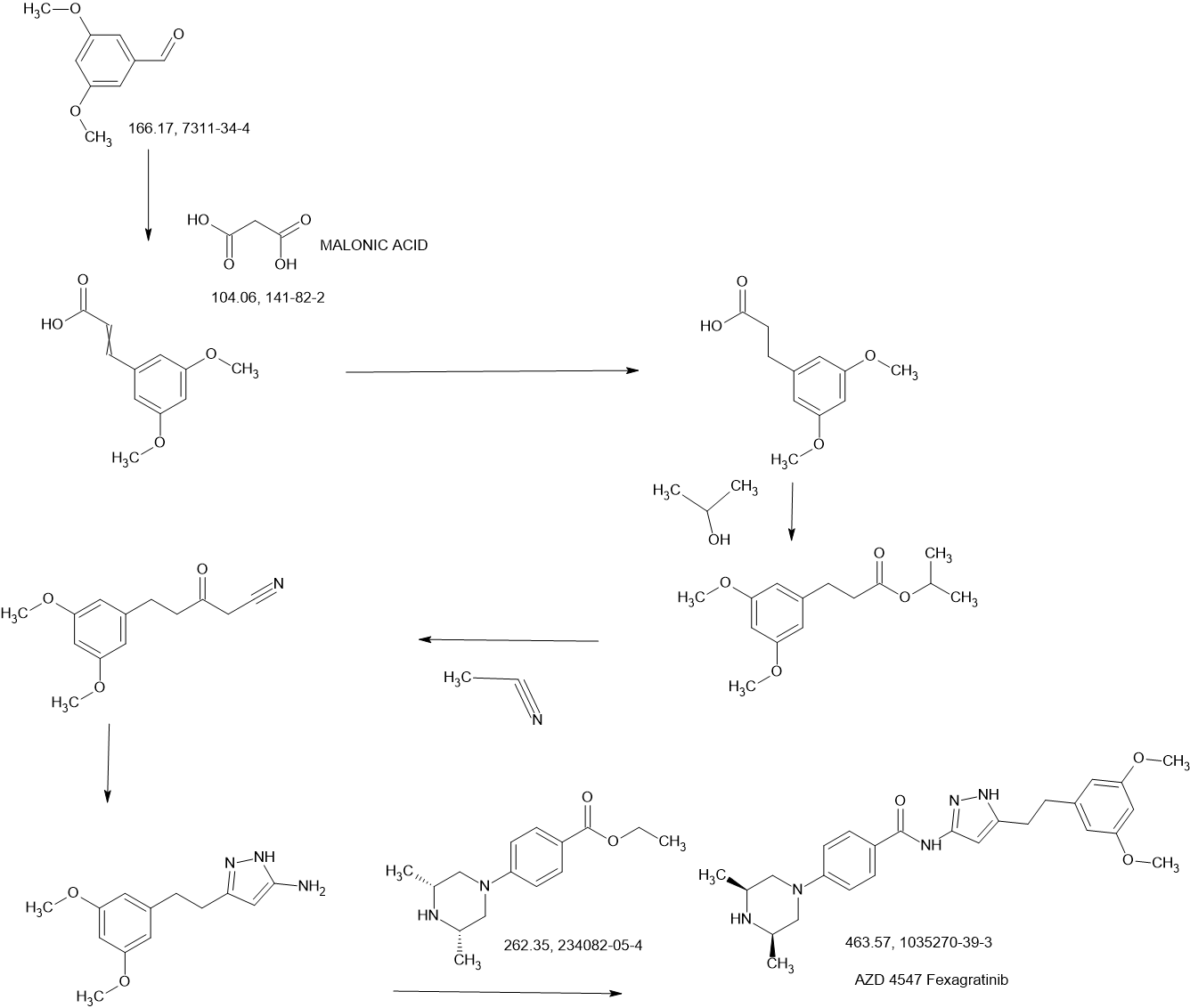
SYN
| At present, the preparation of AZD4547 mainly includes the following methods: |
| (1) Patent application WO2008075068A1 discloses a preparation method comprising the following steps: |
| |
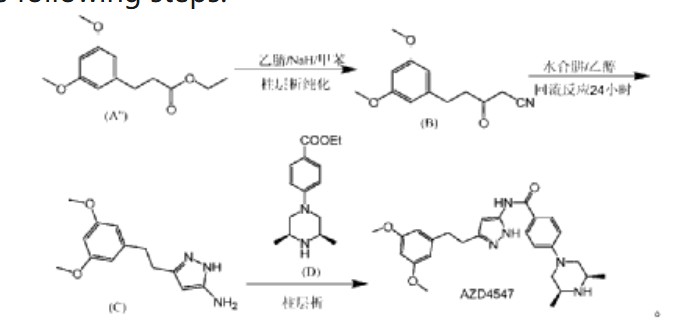
| In the preparation method, AZD4547 is prepared by three-step reactions using ethyl 3-(3,5-dimethoxyphenyl)propionate as a raw material, wherein the first step reaction needs to be purified by column chromatography, and the yield is only 42%; the second step reaction requires reflux reaction for 24 hours, hydrazine hydrate is prone to explosion in high-temperature reactions, and hydrazine hydrate is a highly toxic and genotoxic reagent, and direct high-temperature reaction is not friendly to humans and the environment; the third step reaction also requires column chromatography purification, and the total yield of the three-step reaction for preparing AZD4547 is only 21.08%; therefore, the multi-step reactions of the preparation method require column chromatography operations, have poor safety, low yield, are not suitable for industrialization, and cannot solve the problem of drug accessibility. |
| (2) Patent application CN111072638A discloses another preparation method, comprising the following steps: |
| |
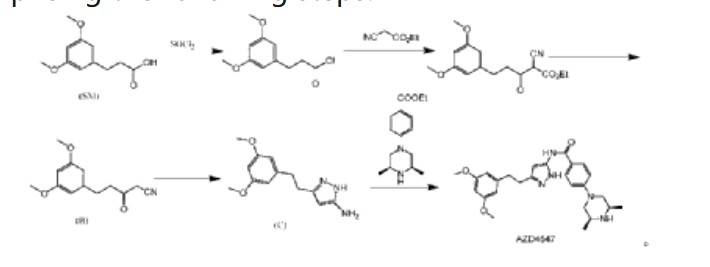
| In this preparation method, 3-(3,5-dimethoxyphenyl)propionic acid is used as the starting material, and AZD4547 is prepared through a five-step reaction with a total yield of 42.5%. In this preparation method, highly toxic reagent ethyl cyanoacetate and expensive reagents palladium carbon, stannous chloride, and Raney nickel are required, and it is not suitable for industrial production. |
| (3) In addition, patent application WO2016137506A1 discloses a method for preparing AZD4547 key intermediate 3-(3,5-dimethoxyphenethyl)-1H-pyrazole-5-amine, as follows: |
| |
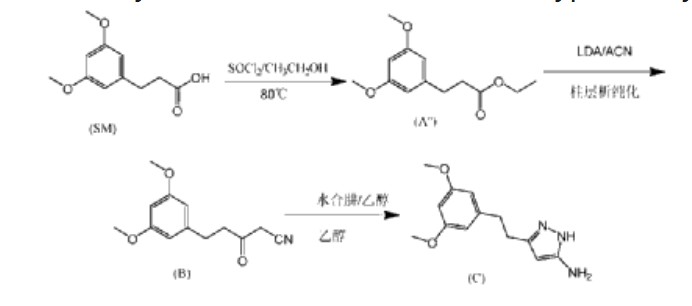
| In this preparation method, the first step of the reaction uses ethanol reflux reaction, the second step of the reaction uses a large amount of solvent, and needs to be reacted at an ultra-low temperature of -78°C. After the reaction is completed, column chromatography purification is required, which is not suitable for industrial application. |
CN115819239
https://patentscope.wipo.int/search/en/detail.jsf?docId=CN394502634&_cid=P11-MBA7JR-97597-1
| Example 1 |
| Add isopropanol (300 mL) and 3-(3,5-dimethoxyphenyl)propionic acid (60.0 g, 0.285 mol) to a 1L three-necked reaction bottle, raise the temperature to 40±5°C, and stir for 5 to 10 minutes to dissolve. Add SOCl dropwise at 40±5°C. 2 (37.3g, 0.314mol), the dropping time is ≥0.5 hours (the dropping process is obviously exothermic), after the dropping is completed, the temperature is raised to 60±5℃, the reaction is stirred for 1 hour, and the reaction of the raw materials is complete when HPLC is detected. The reaction solution is cooled to 35±5℃, the temperature is controlled below 50℃ and the solution is concentrated under reduced pressure until there is no obvious fraction, methyl tert-butyl ether (300mL) is added to dissolve, 5% potassium carbonate aqueous solution is added under ice bath to adjust the pH value of the reaction solution to 8-9, the temperature is controlled at 25±5℃ and stirred for 0.5 hours, the solution is allowed to stand and the organic phase is separated, washed with saturated brine, and the solution is concentrated under reduced pressure at 45℃ to dryness to obtain 72.1g of light yellow oily 3-(3,5-dimethoxyphenyl) propionic acid isopropyl ester, purity: 94%, yield: 94.3%. |
| 1HNMR(DMSO-d 6 ,400MHz)δ6.384-6.378(d,2H),6.318-6.306(t,1H),4.925-4.831(m,1H),3.706(s,6H),2.787-2.749(t,2H),2.571-2.533(t,2H),1.164-1.148(d,6H)。 |
| Example 2 |
| Under nitrogen protection, add isopropyl 3-(3,5-dimethoxyphenyl)propionate (20.0 g, 0.079 mol), anhydrous acetonitrile (80 ml), and anhydrous tetrahydrofuran (100 ml) to a 500 ml three-necked reaction bottle, cool the reaction solution to an internal temperature of about -20 ° C, slowly add lithium diisopropylamide (83 ml, 0.166 mol, 2M THF solution), and add the solution dropwise for about 25 minutes. Continue stirring for 5-10 minutes, and detect the reaction of the raw materials by HPLC. After the reaction mixture was completely dried, acetic acid solution (15 ml) was added to quench the reaction, the mixture was concentrated under reduced pressure, water (100 ml) was added, the pH was adjusted to neutral with 25% aqueous sodium carbonate solution, ethyl acetate (200 ml) was added for extraction (HPLC chart see Figure 2), the organic layer was concentrated under reduced pressure until there was no fraction, ethanol (200 ml) was added, stirred and slurried, filtered, and the filter cake was dried in vacuum at 45°C to obtain 14.8 g of 5-(3,5-dimethoxyphenyl)-3-oxopentanonitrile with a purity of 98% and a yield of 76%. |
| 1HNMR(DMSO-d 6 ,400MHz)δ6.370-6.364(s,2H),6.320-6.309(s,1H),4.038(s,2H),3.709(s,6H),2.851-2.815(t,2H),2.739-2.702(t,2H)。 |
| After preliminary separation, the HPLC, LCMS and 1 HNMR spectra of the impurity-containing mother liquor are shown in Figures 3-5. After analysis, the main impurity is generated by the self-polymerization of 5-(3,5-dimethoxyphenyl)-3-oxopentanonitrile, and the structure of the impurity compound [compound of formula (B’)] is as follows: |
| |
| Example 3 |
| Under nitrogen protection, 3-(3,5-dimethoxyphenyl)propionic acid isopropyl ester (11.29 g, 0.045 mol), anhydrous acetonitrile (40 ml) and anhydrous tetrahydrofuran (50 ml) were added to a 500 ml three-necked reaction bottle, the reaction solution was cooled to -20°C, diisopropylamide lithium tetrahydrofuran solution (47 ml, 0.094 mol) was slowly added dropwise, and the addition was completed in about 25 minutes. The reaction was continued with stirring for 5-10 minutes. HPLC detected that the raw material reaction was complete, anhydrous ethanol (20 ml) was added to quench the reaction, and 2-methyltetrahydrofuran (50 g) was added for extraction. The pH of the aqueous layer was adjusted to neutral with hydrochloric acid, filtered, and the filter cake was dried in vacuo at 45°C to obtain 9.28 g of 5-(3,5-dimethoxyphenyl)-3-oxopentanonitrile, purity: 99.5%, yield: 88.0%. |
| Example 4 |
| Under nitrogen protection, a tetrahydrofuran solution containing isopropyl 3-(3,5-dimethoxyphenyl)propionate (120.0 g, 0.4756 mol), anhydrous acetonitrile (380 g, 9.25 mol), and anhydrous tetrahydrofuran (270 g) were added to a 3L three-necked reaction flask. The mixture was stirred until the internal temperature dropped to about -20°C. At this temperature, a tetrahydrofuran solution of lithium diisopropylamide (500 ml, 1 mol) was slowly added dropwise. After the addition was completed, the mixture was stirred at about -20°C for 1 minute. -2 hours, HPLC detected that the raw material was completely converted, ethanol (474g) was added to the reaction to quench the reaction, and the reaction was concentrated under reduced pressure. Purified water was added, the internal temperature was controlled at 0-15°C, HCl was slowly added, the pH was adjusted to 7.0, and a large amount of solid was precipitated. The reaction was stirred for 30 minutes and filtered, and the mixture was rinsed with purified water and ethanol in turn. The mixture was dried in vacuo at 45°C to obtain 98.7g of 5-(3,5-dimethoxyphenyl)-3-oxopentanonitrile with a purity of 99.6% and a yield of 89.0%. |
| In addition, the inventors investigated the effects of the reaction raw materials, anhydrous acetonitrile, alkaline reagent, and reaction temperature on the reaction. The purity and reaction phenomena in the HPLC test were as follows: |
| |
| |
| From the above experimental investigation factors and experimental phenomena, it can be seen that the types of ester groups of different reaction raw materials, the amount of acetonitrile and the alkaline reagent have the following effects on the reaction: |
| (1) Effect of the type of ester group in the reaction raw materials on the reaction |
| When the reaction raw material is a compound of formula (A’) having a methyl ester group, a sticky mass will be formed during the reaction, affecting stirring, and the purity of the reaction is not high. Specifically, at the beginning of the reaction, a sticky mass appears in the reaction liquid, affecting stirring. As the reaction proceeds, the reaction liquid gradually becomes sticky, and even sticks to the wall, making it impossible to stir. |
| When the reaction raw material is a compound of formula (A”) having an ethyl ester group, the reaction purity is increased to 87%, but sticky lumps are still formed during the reaction, affecting stirring. The specific situation is similar to that when the reaction raw material is a compound of formula (A’) having a methyl ester group. |
| The appearance of viscous clumps during process scale-up can easily lead to incomplete reactions, and may even cause dangerous situations such as entanglement of stirring blades and burning of motors. Therefore, the above two preparation processes are not suitable for industrial scale-up production. |
| When the ester structure of the reaction raw material is changed to isopropyl ester, the reaction liquid is homogeneously clear without sticky micelles, and the reaction control purity is increased to more than 97%, which is suitable for industrial scale-up production. The inventors analyzed that the above experimental phenomenon may be due to the higher stability of the isopropyl ester structure, which reduces the formation of side reactions. |
| (2) Effect of acetonitrile dosage on the reaction |
| In Experiment 6 and Experiment 3 of the present invention, when the molar ratio of acetonitrile to the reaction raw material increased from 10eq to 20eq, the reaction control purity increased from 90.4% to 97.2%. |
| Comparing Experiment 2 and Experiment 3 in Experiment 1, when the molar ratio of acetonitrile to the reaction raw material increased from 1.2eq to 25eq, the reaction control purity increased from 60.8% to 92.8%. |
| (3) Effect of the selection and dosage of alkaline reagents on the reaction |
| From the above experimental results, it can be seen that the reaction control purity of NaHMDS, LDA and n-BuLi is relatively high. |
| The optimal molar ratio of alkaline reagent to reaction raw materials is 2.1eq. A molar ratio lower than 2eq may result in incomplete reaction. |
| Example 5 |
| Step 1: Synthesis of 5-(3,5-dimethoxyphenyl)-3-oxopentanonitrile (compound of formula (B)) |
| Under nitrogen protection, add isopropyl 3-(3,5-dimethoxyphenyl)propionate (20.0 g, 0.079 mol), anhydrous acetonitrile (80 ml), and anhydrous tetrahydrofuran (100 ml) into a 500 ml three-necked reaction bottle, cool the reaction solution to -20°C, slowly add lithium diisopropylamide (83 ml, 0.166 mol, 2M THF solution) dropwise, add for about 25 min, stir and react for 5-10 minutes, HPLC detection shows that the raw material reaction is complete, add anhydrous ethanol (40 ml), concentrate under reduced pressure to a viscous state, add anhydrous ethanol (60 ml) to prepare an ethanol solution, and directly put into the next step reaction. |
| Step 2: Preparation of 3-(3,5-dimethoxyphenethyl)-1H-pyrazole-5-amine (compound of formula (C)) |
| Add acetic acid (26.0 g, 0.436 mol), ethanol (100 ml), and 80% hydrazine hydrate (15.0 g, 0.238 mol) to a 500 ml three-necked reaction bottle, heat to an internal temperature of about 68°C, slowly add the product ethanol solution (18.5 g, 0.079 mol) obtained in step 1 to the mixed solution of acetic acid and hydrazine hydrate at this temperature, add for about 40 minutes, stir and react at an internal temperature of about 68°C for 1 hour, and HPLC detection shows that 5-(3,5-dimethoxyphenyl)-3-oxopentanonitrile is completely converted; the reaction solution is concentrated under reduced pressure, water (100 ml), ethyl acetate (200 ml), and about 25% Na 2 CO 3 (40ml) adjust the pH value of the water layer to 7-8; separate the water layer, wash the layers with saturated brine (20ml), concentrate the organic layer under reduced pressure until there is no fraction, add isopropyl acetate (100ml) and reduce the pressure to bring it to a viscous state, add isopropyl acetate (120ml) and heat to dissolve, cool and crystallize, filter at about 10°C, and dry under vacuum at 50°C to obtain 16.3g of 3-(3,5-dimethoxyphenethyl)-1H-pyrazole-5-amine with a purity of 99.6% and a total yield of 83% in two steps. |
| 1HNMR(DMSO-d 6 ,400MHz)δ6.370-6.364(s,2H),6.320-6.309(s,1H),4.038(s,2H),3.709(s,6H),2.851-2.815(t,2H),2.739-2.702(t,2H)。 |
| In addition, the inventors investigated the effect of the amount of acetic acid used in this step of the reaction on the reaction, and the purity was controlled by HPLC as follows: |
| |
| In addition, the inventors also investigated that the solid compound of formula (B) obtained after purification of the product in step 1 was reacted with hydrazine hydrate in the presence of acetic acid, and the compound of formula (B) was also completely converted to obtain a high-purity compound of formula (C). |
| Example 6 |
| 3-(3,5-dimethoxyphenethyl)-1H-pyrazole-5-amine (100.0 g, 0.4044 mol), ethyl 4-((3R,5S)-3,5-dimethylpiperazin-1-yl)benzoate (132.5 g, 0.5050 mol), and 2-methyltetrahydrofuran (1300 ml) were added to the reaction kettle, heated to 50-55°C and stirred for 1 hour, filtered through diatomaceous earth, and the filtrate was added to a clean reaction kettle, heated to atmospheric distillation with water, and the reaction temperature was controlled at 78°C-88°C, and 25% KO-tAm toluene solution (490.0 g) was slowly added dropwise for about 2 hours. After the addition was completed, the reaction temperature was adjusted to 83-88°C and stirred for 3-6 hours. Sampling was performed to detect whether the reaction of the raw materials was complete. The reaction system was cooled to 30-60°C, water (8 ml) was slowly added to quench the reaction, and the mixture was stirred at 30-60°C for 0.5 hour, then cooled to about 25°C, water (400 ml) was added, stirred and allowed to stand for stratification, the organic phase was separated, water (200 ml) was added, the mixture was heated to about 50°C and stirred for 0.5 hour, the water layer was separated, and this was repeated 2-3 times until the pH of the water layer was 7.0-9.5; the organic layer was concentrated under reduced pressure to remove part of the solvent, the residue was heated to 80-90°C and stirred for 1 hour, slowly cooled to 20-30°C, stirred for 2-5 hours, filtered, rinsed twice with ethyl acetate, and dried in vacuo at 45°C to obtain 155.6 g of a white amorphous solid product (AZD4547) with a purity of 98.5% and a yield of 83%. |
| 1HNMR(DMSO-d 6 ,400MHz)δ12.067(s,1H),10.275(s,1H),7.888-7.867(d,2H),6.943-6.922(d,2H),6.437-6.409(m,3H),6.317(s,1H),3.712-3.692(m,8H),2.861-2.803(m,6H),2.230-2.174(m,3H),1.036-1.020(d,6H)。 |
| Example 7 |
| 3-(3,5-dimethoxyphenethyl)-1H-pyrazole-5-amine (10.0 g, 0.040 mol), ethyl 4-((3R,5S)-3,5-dimethylpiperazin-1-yl)benzoate (12.1 g, 0.047 mol), anhydrous tetrahydrofuran (170 ml) were added to the reaction bottle, heated and distilled at atmospheric pressure until about 100 ml remained, cooled to -30°C to -20°C, and NaHMDS (0.125 mol, 63 ml, 2M THF solution) was slowly added dropwise. The temperature of the reaction system was controlled at about -25°C and stirred for 20 minutes. HPLC detected that the raw materials were basically reacted. Water (30 ml) was slowly added under temperature control to quench, and glacial acetic acid (about 10 ml) was added to neutralize. The temperature was raised to about 0°C and stirred, and 20% Na 2 CO 3 (10ml), concentrated under reduced pressure until there is no fraction, added ethyl acetate (120ml) to the residue, heated to about 45°C, stirred and separated, the organic phase was separated, added with saturated brine (30ml), washed once, concentrated under reduced pressure to leave about (30ml), then added ethyl acetate (30ml), concentrated under reduced pressure again, repeated twice, a large amount of solid precipitated, added ethyl acetate to a material volume of about 50ml, stirred at 0-10°C for 1 hour, filtered, and dried in vacuo at 45°C to obtain 16.87g of white amorphous solid product (AZD4547) with a purity of 99.8% and a yield of 91%. |
Heterocycles (2020), 100(2), 276-282 ,
CN111072638
PATENT
CN115819239
Nature Catalysis (2021), 4(5), 385-394
Shandong Huagong (2021), 50(7), 19-21
CN111072638
Heterocycles (2020), 100(2), 276-282
Physical Chemistry Chemical Physics (2020), 22(17), 9656-9663
Journal of Chemical Theory and Computation (2019), 15(2), 1265-1277
Journal of Medicinal Chemistry (2017), 60(14), 6018-6035
Bioorganic & Medicinal Chemistry Letters (2016), 26(20), 5082-5086
WO2016089208
WO2008075068
References
- ^ Gavine PR, Mooney L, Kilgour E, Thomas AP, Al-Kadhimi K, Beck S, et al. (April 2012). “AZD4547: an orally bioavailable, potent, and selective inhibitor of the fibroblast growth factor receptor tyrosine kinase family”. Cancer Research. 72 (8): 2045–2056. doi:10.1158/0008-5472.CAN-11-3034. PMID 22369928.
- ^ Katoh M, Nakagama H (March 2014). “FGF receptors: cancer biology and therapeutics”. Medicinal Research Reviews. 34 (2): 280–300. doi:10.1002/med.21288. PMID 23696246.
- ^ Katoh M (July 2016). “FGFR inhibitors: Effects on cancer cells, tumor microenvironment and whole-body homeostasis (Review)”. International Journal of Molecular Medicine. 38 (1): 3–15. doi:10.3892/ijmm.2016.2620. PMC 4899036. PMID 27245147.
- ^ Zengin ZB, Chehrazi-Raffle A, Salgia NJ, Muddasani R, Ali S, Meza L, et al. (February 2022). “Targeted therapies: Expanding the role of FGFR3 inhibition in urothelial carcinoma”. Urologic Oncology. 40 (2): 25–36. doi:10.1016/j.urolonc.2021.10.003. PMID 34840077.
- ^ Zarei P, Ghasemi F (2024). “The Application of Artificial Intelligence and Drug Repositioning for the Identification of Fibroblast Growth Factor Receptor Inhibitors: A Review”. Advanced Biomedical Research. 13: 9. doi:10.4103/abr.abr_170_23. PMC 10958741. PMID 38525398.
| Identifiers | |
|---|---|
| showIUPAC name | |
| CAS Number | 1035270-39-3 |
| PubChem CID | 51039095 |
| IUPHAR/BPS | 7707 |
| DrugBank | DB12247 |
| ChemSpider | 26333104 |
| UNII | 2167OG1EKJ |
| ChEBI | CHEBI:63453 |
| ChEMBL | ChEMBL3348846 |
| PDB ligand | 66T (PDBe, RCSB PDB) |
| CompTox Dashboard (EPA) | DTXSID80145887 |
| ECHA InfoCard | 100.206.232 |
| Chemical and physical data | |
| Formula | C26H33N5O3 |
| Molar mass | 463.582 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
////////////Fexagratinib, AZD 4547, ADSK091
ETRIPAMIL



ETRIPAMIL
CAS 1593673-23-4
AS ACETATE 512.64 CAS 2891832-59-8
HCL SALT 2560549-35-9
WeightAverage: 452.595
Monoisotopic: 452.267507647
Chemical FormulaC27H36N2O4
12/12/2025, FDA 2025, APPROVALS 2025
Benzoic acid, 3-[2-[[(4S)-4-cyano-4-(3,4-dimethoxyphenyl)-5-methylhexyl]methylamino]ethyl]-, methyl ester
methyl 3-[2-[[(4S)-4-cyano-4-(3,4-dimethoxyphenyl)-5-methylhexyl]-methylamino]ethyl]benzoate
- Methyl 3-[2-[[(4S)-4-cyano-4-(3,4-dimethoxyphenyl)-5-methylhexyl]methylamino]ethyl]benzoate
- (-)-MSP 2017
- MSP 2017
- OriginatorMilestone Pharmaceuticals
- DeveloperCorxel Pharmaceuticals; Milestone Pharmaceuticals
- ClassAmines; Antiarrhythmics; Benzoates; Esters; Ischaemic heart disorder therapies; Small molecules
- Mechanism of ActionCalcium channel antagonists
- PreregistrationParoxysmal supraventricular tachycardia
- Phase IIAtrial fibrillation
- Phase IUnspecified
- No development reportedAngina pectoris
- 14 May 2025Milestone Pharmaceuticals has patent protection for etripamil in the USA
- 28 Mar 2025Milestone pharmaceuticals plans to request a Type A meeting with USFDA to discuss the issues raised in the complete response letter
- 28 Mar 2025USFDA has issued a Complete Response Letter (CRL) regarding New Drug Application (NDA) for Etripamil for Paroxysmal supraventricular tachycardia
Etripamil has been used in trials studying the treatment of Paroxysmal Supraventricular Tachycardia (PSVT).
Etripamil (MSP-2017) is a short-acting, L-type calcium-channel antagonist. Etripamil inhibits calcium influx through slow calcium channels, thereby slowing AV node conduction and prolonging the AV node refractory period. Etripamil increases heart rate and decreases systolic blood pressure. Etripamil can be used in the study of paroxysmal supraventricular tachycardia (PSVT).
To treat episodes of paroxysmal supraventricular tachycardia
SCHEME
SIDE CHAIN

MAIN

SYN
US20180110752/ U.S. Patent No. 10,117,848,
EXAMPLES
Example 1: Synthesis methyl 3-(2-((4-cyano-4-(3,4-dimethoxyphenyl)-5-methylhexyl)(methyl)amino)ethyl)benzoate
Part I: Synthesis of 5-Bromo-2-(3,4-dimethoxyphenyl)-2-isopropylpentanenitrile
Part II: Synthesis of methyl 3-(2-(methylamino)ethyl)benzoate
Part III: Reaction of Compound II with Compound III Produced Compound I
| Analysis of the product by mass spectrometry revealed a peak with a mass-to-charge ratio (m/z) of 453, corresponding to the M+H molecular ion of compound I. |
Example 2: Concentrated Solution of Acetate Salt of Compound I
| A concentrated aqueous solution of the acetate salt of compound I is formed according to the following protocol: |
| This protocol readily can be adapted to provide a concentrated solution of the methanesulfonate salt of compound I. |
PRED BY CHIRAL SEPERATION
US20230065401
WO2016165014
EP4119137 chiral sepn done
[0034] In one embodiment the present invention is a kit for treating a cardiac arrhythmia (e.g., PSVT or atrial fibrillation), angina, or a migraine in a subject in need thereof wherein the kit comprises a nasal delivery system comprising two doses of a therapeutically effective amount of compound I having a structure according to the formula:
and instructions for nasally administering to the subject (i) a first dose, and, optionally, (ii) a second dose of an aqueous composition comprising a pharmaceutically acceptable acetate or methanesulfonate salt of compound I, or a racemate or enantiomer thereof, wherein the acetate or methanesulfonate salt of compound I, or the racemate or enantiomer thereof, is dissolved in the aqueous composition at a concentration of 350 mg/mL± 50 mg/mL, and wherein the second dose of the compound is to be administered between 5 minutes and 60 minutes after the first dose.
Cross ref U.S. Patent No. 10,117,848,
[0336]
- 1. A method of treating a cardiac arrhythmia in a subject in need thereof with a therapeutically effective amount of compound I having a structure according to the formula:
the method comprising nasally administering to the subject (i) a first dose, and (ii) a second dose of an aqueous composition comprising a pharmaceutically acceptable acetate or methanesulfonate salt of compound I, or a racemate or enantiomer thereof, wherein the acetate or methanesulfonate salt of compound I, or the racemate or enantiomer thereof, is dissolved in the aqueous composition at a concentration of 350 mg/mL ± 50 mg/mL, and wherein the second dose of the compound is administered between 5 minutes and 25 minutes after the first dose.
PATENT
Journal of the American College of Cardiology (2018), 72(5), 489-497
American Heart Journal (2022), 253, 20-29
Expert Opinion on Investigational Drugs (2020), 29(1), 1-4
EP4119137 WO2016165014
EP-2170050-B1
US-9737503-B2
US-4968717-A
EP-0231003-A2
- [1]. Stambler BS, et al. Etripamil Nasal Spray for Rapid Conversion of Supraventricular Tachycardia to Sinus Rhythm. J Am Coll Cardiol. 2018 Jul 31;72(5):489-497. [Content Brief][2]. Milestone Pharmaceuticals Announces USAN Approval of Generic Name “Etripamil” for its Phase 2 Clinical Development Product for the Treatment of Paroxysmal Supraventricular Tachycardia.[3]. Ascah A, et al. Cardiovascular and Pharmacokinetic Profiles of Intravenous Etripamil in Conscious Telemetered Cynomolgus Monkeys. Int J Toxicol. 2025 Apr 1:10915818251327963. [Content Brief][4]. Pion J, et al. Preclinical Safety Evaluation of Etripamil Nasal Spray in Cynomolgus Macaques (Macaca fascicularis) to Assess for Safety in Patients With Paroxysmal Supraventricular Tachycardia. Int J Toxicol. 2024 Sep-Oct;43(5):503-510. [Content Brief]
//////////ETRIPAMIL, (-)-MSP 2017, MSP 2017, FDA 2025, APPROVALS 2025
DR ANTHONY MELVIN CRASTO

ORGANIC SPECTROSCOPY


{kind=link}
{kind=link}
{kind=link}