
FLAGS AND HITS
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO FACEBOOK
...................................................................Join me on twitter
FACEBOOK
...................................................................Join me on twitter
 ..................................................................Join me on google plus
..................................................................Join me on google plus
 Googleplus
GoogleplusMYSELF

MORINIDAZOLE 吗啉硝唑


MORINIDAZOLE
吗啉硝唑
(迈灵达®
1- [3- (4-morpholinyl) -2-hydroxypropyl] -2-methyl-5- nitro -1H- imidazole
CAS 92478-27-8
Jiangsu Hansoh Pharmaceutical Co., Ltd.
Morinidazole was approved by China Food and Drug Administration (CFDA) on February 24, 2014. It was developed and marketed as a step Lingda ® by Hansoh Pharmaceutical.
A nitroimidazoles antibiotic used to treat bacterial infections including appendicitis and pelvic inflammatory disease.
![]()
Morinidazole is a nitroimidazoles antibiotic indicated for the treatment of bacterial infections including appendicitis and pelvic inflammatory disease (PID) caused by anaerobic bacteria.

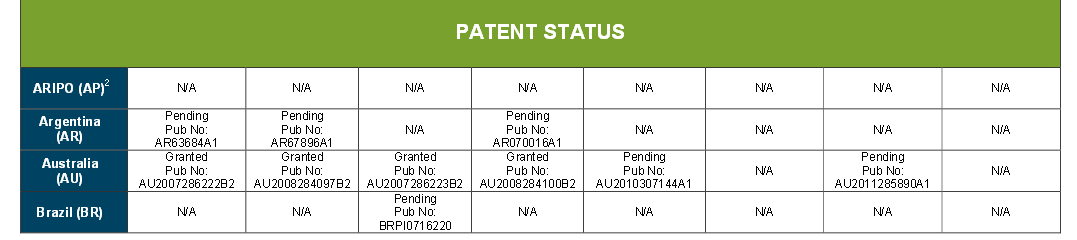
PATENT
WO2006058457A1.
http://www.google.com/patents/WO2006058457A1?cl=en
……………………….
PATENT
CN1981764A.
https://www.google.com/patents/CN1981764A?cl=en
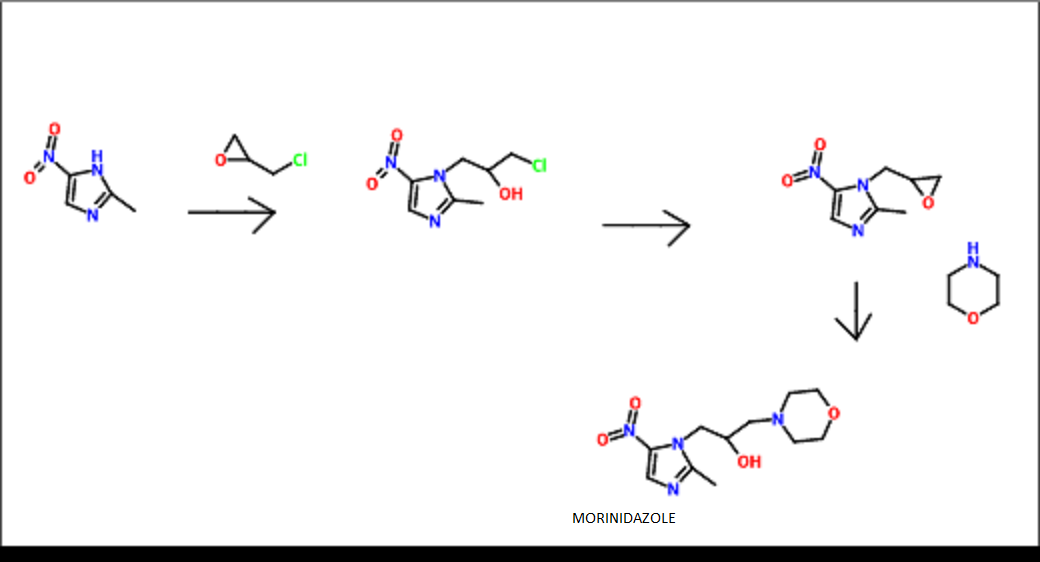
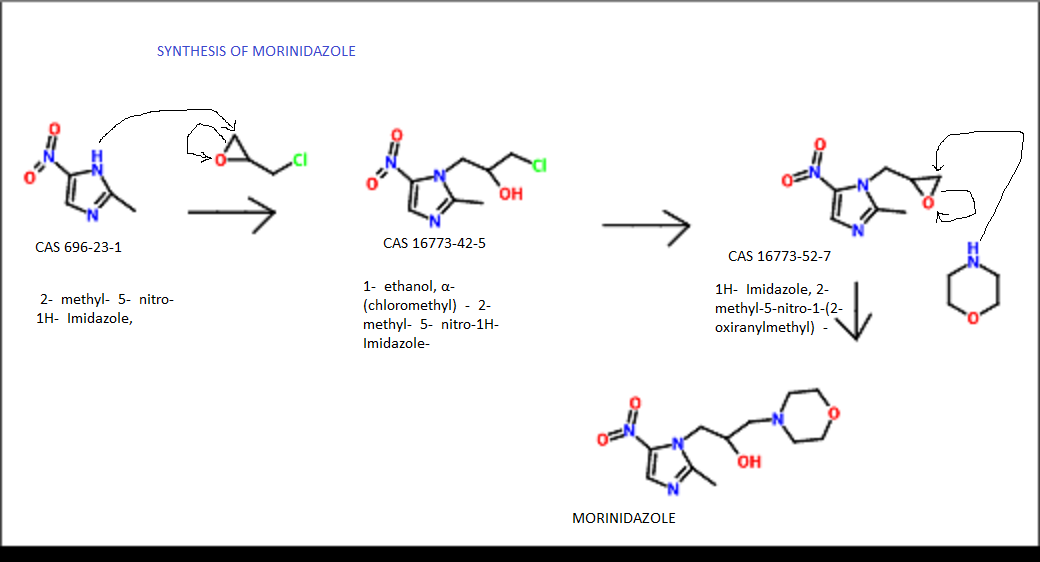
1- (2,3-epoxypropoxy yl) -2-methyl-5-nitro-imidazole (10g), morpholino (10g), 100ml of acetonitrile under reflux for 2 hours, vacuum recovery of acetonitrile, water was added 100ml, heating to the whole solution, filtered hot, let cool, filtering, washing and drying to obtain an off-white solid (11g).
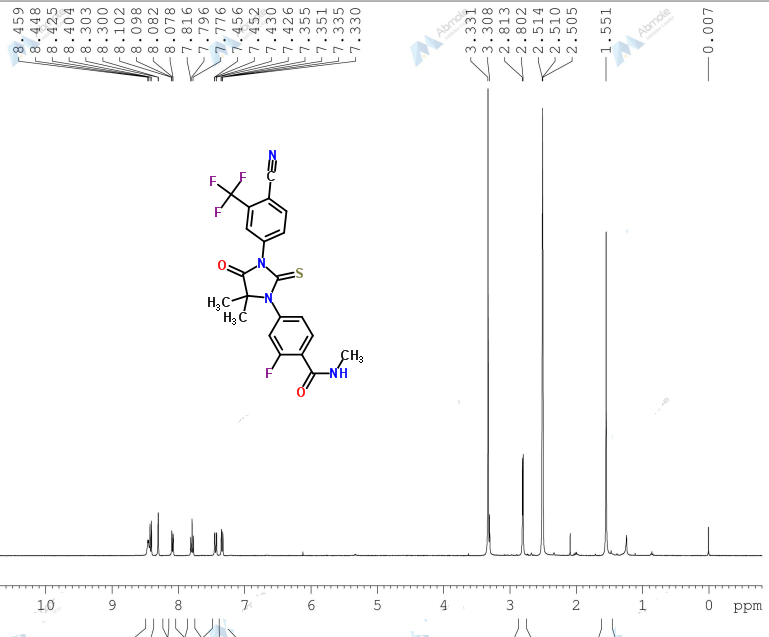
Proton nuclear magnetic resonance data: 1HNMR (CD3Cl) δ2.39 ~ 2.73 (6H, m) δ2.61 (3H, s) δ3.71 ~ 3.81 (4H, m) δ4.10 ~ 4.17 (2H, m) δ4 .63 ~ 4.66 (1H, m) δ8.00 (1H, s)
CN 102199147
http://www.google.com/patents/CN102199147A?cl=en
CN 1605586
https://www.google.com/patents/CN1605586A?cl=en
Example 7 Preparation of α- (morpholino-1-yl) methyl-2-methyl-5-nitroimidazole-1-ethanol according to Example 4 the same manner as in Preparation α- (morpholino-1-yl) methyl-2-methyl-5-nitroimidazole-1-ethanol, except for using morpholine instead of 4-hydroxypiperidine, prepared by the present invention Compound 7. Proton nuclear magnetic resonance data: 1HNMR (CD3Cl) δ2.39 ~ 2.73 (6H, m) δ2.61 (3H, s) δ3.71 ~ 3.81 (4H, m) δ4.10 ~ 4.17 (2H, m) δ4

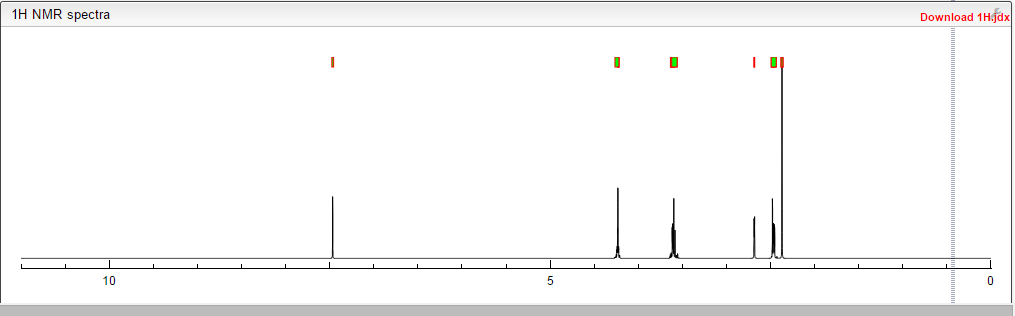
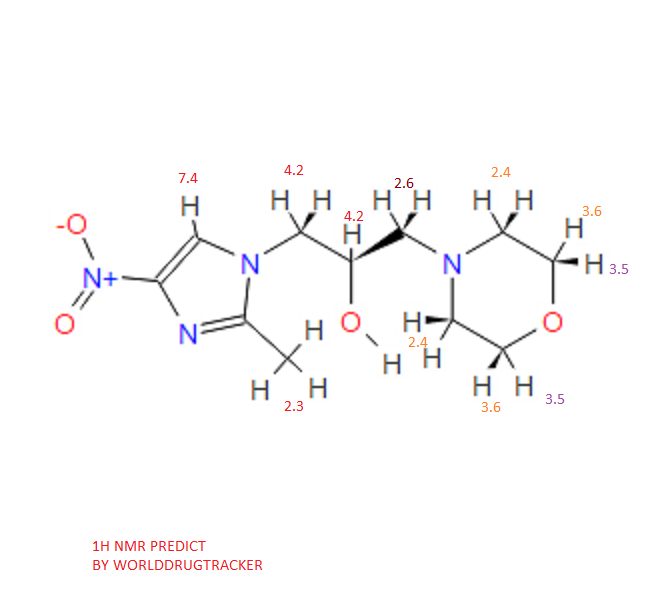
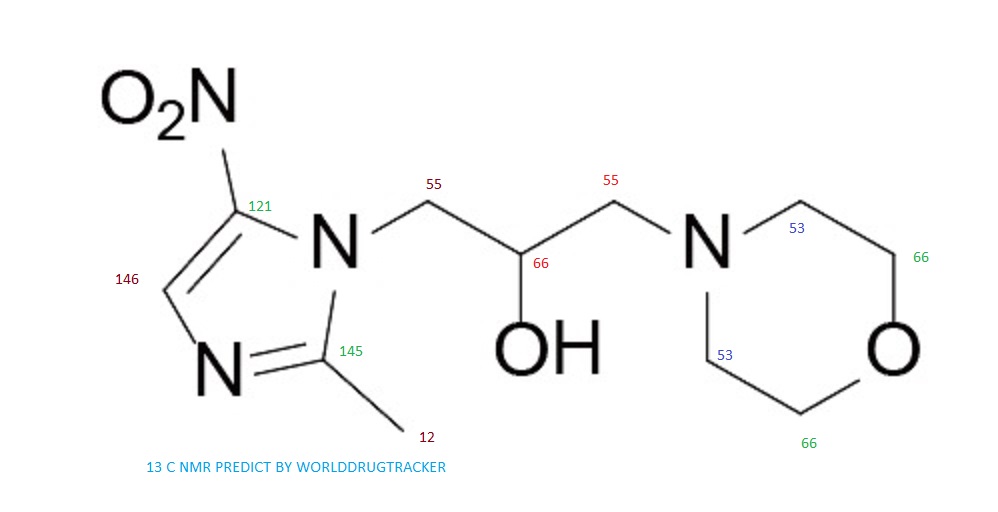
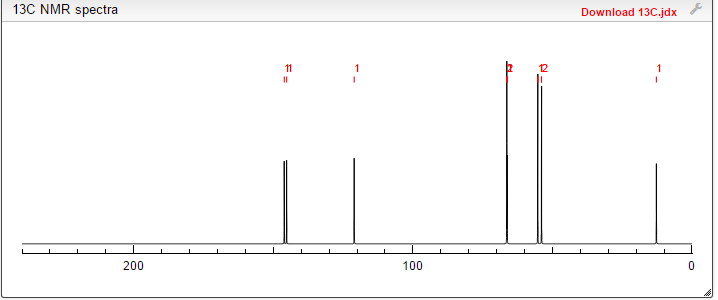
NMR PREDICT
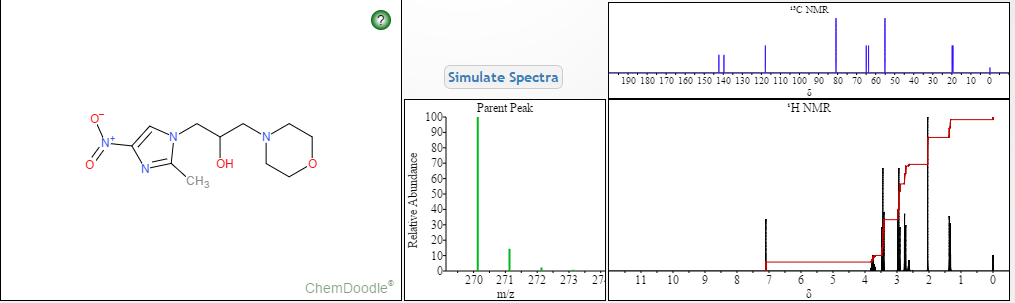
1H NMR PREDICT
13C NMR PREDICT
COSY

| CN1810815B | Mar 8, 2006 | Mar 16, 2011 | 陕西合成药业有限公司 | Nitroimidazole derivative for treatment |
| CN1903846B | Aug 15, 2006 | Jul 13, 2011 | 杨成 | Ornidazole derivative used for therapy, its preparation method and use |
| CN100387233C | Jun 9, 2006 | May 14, 2008 | 南京圣和药业有限公司 | Use of levo morpholine nidazole for preparing medicine for antiparasitic infection |
| CN100427094C | Dec 13, 2005 | Oct 22, 2008 | 江苏豪森药业股份有限公司 | Usage of alpha-(Morpholin-1-base) methyl-2-methyl-5-azathio-1-alcohol in preparation of anti-trichomoniasis and anti-ameba medicines |
| CN100540549C | Dec 15, 2005 | Sep 16, 2009 | 南京圣和药业有限公司 | Alpha-substituted-2-methyl-5-nitro-diazole-1-alcohol derivative with optical activity |
| WO2007079653A1 * | Dec 25, 2006 | Jul 19, 2007 | Junda Cen | OPTICALLY PURE α-SUBSTITUTED 2-METHYL-5-NITROIMIDAZOLE-1-ETHANOL DERIVATIVES |

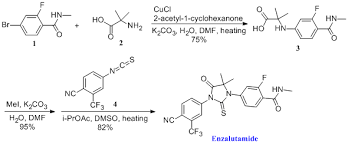
ENZALUTAMIDE
1H NMR PREDICT AND 13 C NMR PREDICT BELOW


Synthesis pics

……………………..
PATENT
http://www.google.com/patents/WO2015063720A1?cl=en

…………………………
PAPER
J Med Chem 2010, 53(7): 2779
http://pubs.acs.org/doi/full/10.1021/jm901488g
A structure−activity relationship study was carried out on a series of thiohydantoins and their analogues 14 which led to the discovery of 92 (MDV3100) as the clinical candidate for the treatment of hormone refractory prostate cancer.
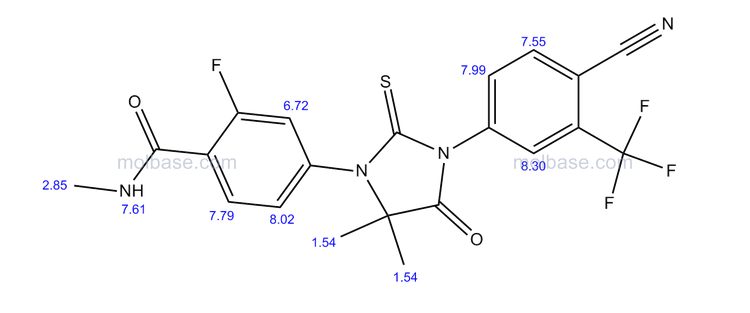
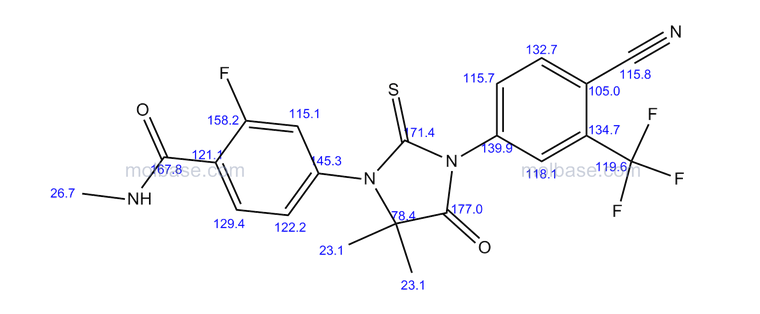
N-Methyl-4-[3-(4-cyano-3-trifluoromethylphenyl)-5,5-dimethyl-4-oxo-2-thioxoimidazolidin-1-yl]-2-fluorobenzamide, 92
REF
MEDIVATION PROSTATE THERAPEUTICS, INC.; JAIN, Rajendra, Parasmal; ANGELAUD, Remy; THOMPSON, Andrew; LAMBERSON, Carol; GREENFIELD, Scott Patent: WO2011/106570 A1, 2011 ; Location in patent: Page/Page column 46
Regents of the University of California Patent: US2007/254933 A1, 2007 ; Location in patent: Page/Page column 7 ;
WO2011/106570 A1,
J Med Chem 2010, 53(7): 2779
| WO2013067151A1 * | Nov 1, 2012 | May 10, 2013 | Medivation Prostate Therapeutics, Inc. | Treatment methods using diarylthiohydantoin derivatives |
| WO2014041487A2 * | Sep 11, 2013 | Mar 20, 2014 | Dr. Reddy’s Laboratories Limited | Enzalutamide polymorphic forms and its preparation |
| WO2014066799A2 * | Oct 25, 2013 | May 1, 2014 | Memorial Sloan-Kettering Cancer Center | Modulators of resistant androgen receptor |
| WO2014167428A3 * | Mar 5, 2014 | Feb 19, 2015 | Shilpa Medicare Limited | Amorphous 4-(3-(4-cyano-3-(trifluoromethyl)phenyl)-5,5-dimethyl-4-oxo-2-thioxoimidazolidin-1-yl)-2-fluoro-n-methylbenzamide |
| EP2536708A2 * | Feb 16, 2011 | Dec 26, 2012 | Aragon Pharmaceuticals, Inc. | Androgen receptor modulators and uses thereof |








From 2-fluoro-4-bromo – benzoic acid s-1, firstly the carboxylic acid is converted to acid chloride with SOCl2 s-2, and then methylamine to give the enamine compound s-3, and s-3 bromide aminoisobutyric acid Ullmann (Goldberg) in CuCl catalyzed reaction to give the compound s-4, followed by reaction of the carboxylic acid and methyl iodide to give the corresponding methyl ester compound s-5.
Aniline compound s-6 in the sulfur phosgene primary amine is converted to the isothiocyanate s-7.
Finally, the nitrogen atom of the compound s-5 attack isothiocyanate s-7, followed by transesterification ring closure to give the final Xtandi (Enzalutamide, uh miscellaneous Lu amine). Scheme: WO2011106570A1

//////////
Indian Generics 2016
The generic APIs market is expected to continue to rise faster than the branded/innovative APIs, by 7.7%/year to reach $30.3 billion in 2016. Asia-Pacific is expected to show the fastest growth rates (10.8%/year). The 24 fastest growing markets will include 11 in Asia-Pacific, seven in Eastern Europe and CIS, four in Africa-Middle East and two in Latin America (Figure ).

Figure – Top growth markets for generic APIs to 2016
By 2016, China will account for 27.7% of the global generic API merchant market, while the US will have fallen to 23.8%; the mature markets as a whole will see their share fall from 41.8% in 2012 to 36.9%. India will be the third largest, with a 7.2% share.
सुकून उतना ही देना प्रभू, जितने से जिंदगी चल जाये।औकात बस इतनी देना,कि औरों का भला हो जाये।………..P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
सुकून उतना ही देना प्रभू, जितने से जिंदगी चल जाये। औकात बस इतनी देना, कि औरों का भला हो जाये।
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE

Join me on Facebook
Googleplus amcrasto@gmail.com
amcrasto@gmail.com
 LIONEL MY SON
LIONEL MY SON
जिंदगी चल जाये।
औकात बस इतनी देना,
कि औरों का भला हो जाये।
Eisai’s lenvatinib 兰伐替尼 レンバチニブ gets FDA approval
 Lenvatinib Mesilate
Lenvatinib Mesilate
Eisai’s lenvatinib 兰伐替尼 レンバチニブ
See synthesis at https://newdrugapprovals.org/2014/08/04/eisais-lenvatinib-%E5%85%B0%E4%BC%90%E6%9B%BF%E5%B0%BC-%E3%83%AC%E3%83%B3%E3%83%90%E3%83%81%E3%83%8B%E3%83%96-to-get-speedy-review-in-europe/
Above post contains SYNTHESIS, spectrocopy predicts, etc
February 13, 2015
Release
The U.S. Food and Drug Administration today granted approval to Lenvima (lenvatinib) to treat patients with progressive, differentiated thyroid cancer (DTC) whose disease progressed despite receiving radioactive iodine therapy (radioactive iodine refractory disease).
The most common type of thyroid cancer, DTC is a cancerous growth of the thyroid gland which is located in the neck and helps regulate the body’s metabolism. The National Cancer Institute estimates that 62,980 Americans were diagnosed with thyroid cancer and 1,890 died from the disease in 2014. Lenvima is a kinase inhibitor, which works by blocking certain proteins from helping cancer cells grow and divide.
“The development of new therapies to assist patients with refractory disease is of high importance to the FDA,” said Richard Pazdur, M.D., director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “Today’s approval gives patients and healthcare professionals a new therapy to help slow the progression of DTC.”
Lenvima was reviewed under the FDA’s priority review program, which provides for an expedited review of drugs that, if approved, would provide significant improvement in safety or effectiveness in the treatment of a serious condition. The drug also received orphan product designation because it is intended to treat a rare disease. Lenvima is being approved approximately two months ahead of the prescription drug user fee goal date of April 14, 2015, the date when the agency was scheduled to complete its review of the application.
Lenvima’s efficacy was demonstrated in 392 participants with progressive, radioactive iodine-refractory DTC who were randomly assigned to receive either Lenvima or a placebo. Study results showed Lenvima-treated participants lived a median of 18.3 months without their disease progressing (progression-free survival), compared to a median of 3.6 months for participants who received a placebo. Additionally, 65 percent of participants treated with Lenvima saw a reduction in tumor size, compared to the two percent of participants who received a placebo. A majority of participants randomly assigned to receive the placebo were treated with Lenvima upon disease progression.
The most common side effects of Lenvima were high blood pressure (hypertension), fatigue, diarrhea, joint and muscle pain (arthralgia/myalgia), decreased appetite, decreased weight, nausea, inflammation of the lining of the mouth (stomatitis), headache, vomiting, excess protein in the urine (proteinuria), swelling and pain in the palms, hands and/or the soles of the feet (palmar-plantar erythrodysesthesia syndrome), abdominal pain and changes in voice volume or quality (dysphonia).
Lenvima may cause serious side effects, including cardiac failure, blood clot formation (arterial thromboembolic events), liver damage (hepatotoxicity), kidney damage (renal failure and impairment), an opening in the wall of the stomach or intestines (gastrointestinal perforation) or an abnormal connection between two parts of the stomach or intestines (fistula formation), changes in the heart’s electrical activity (QT Interval Prolongation), low levels of calcium in the blood (hypocalcemia), the simultaneous occurrence of headache, confusion, seizures and visual changes (Reversible Posterior Leukoencephalopathy Syndrome), serious bleeding (hemorrhage), risks to an unborn child if a patient becomes pregnant during treatment, and impairing suppression of the production of thyroid-stimulating hormone.
Lenvima is marketed by Woodcliff Lake, New Jersey-based Eisai Inc.
सुकून उतना ही देना प्रभू, जितने से जिंदगी चल जाये।औकात बस इतनी देना,कि औरों का भला हो जाये।………..P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
सुकून उतना ही देना प्रभू, जितने से जिंदगी चल जाये। औकात बस इतनी देना, कि औरों का भला हो जाये।
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
Googleplus amcrasto@gmail.com
LIONEL MY SONजिंदगी चल जाये।
औकात बस इतनी देना,
कि औरों का भला हो जाये।
ORGANIC SPECTROSCOPY INTERNATIONAL HAS 2 LAKH VIEWS

ORGANIC SPECTROSCOPY INTERNATIONAL HAS 2 LAKH VIEWS
Read by one and all in academics and industry
link is ……http://orgspectroscopyint.blogspot.in/
I get minimum 1000 hits in a day and all across the world
सुकून उतना ही देना प्रभू, जितने से जिंदगी चल जाये।औकात बस इतनी देना,कि औरों का भला हो जाये।………..P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
सुकून उतना ही देना प्रभू, जितने से जिंदगी चल जाये। औकात बस इतनी देना, कि औरों का भला हो जाये।
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
Join me on Facebook
Googleplus amcrasto@gmail.com
LIONEL MY SONजिंदगी चल जाये।
औकात बस इतनी देना,
कि औरों का भला हो जाये।
DACLATASVIR, 达拉他韦 , Даклатасвир , داكلاتاسفير ,

Daclatasvir

Status: Launched 2014 (EU, Japan)
Originator: Bristol-Myers Squibb


Daclatasvir (USAN[1]) (formerly BMS-790052, trade name Daklinza) is a drug for the treatment of hepatitis C (HCV). It is was developed by Bristol-Myers Squibb and was approved in Europe on 22 August 2014.
Daclatasvir inhibits the HCV nonstructural protein NS5A.[2][3] Recent research suggests that it targets two steps of the viral replication process, enabling rapid decline of HCV RNA.[4]
Daclatasvir has been tested in combination regimens with pegylated interferon and ribavirin,[5] as well as with other direct-acting antiviral agents including asunaprevir[6][7][8][9] and sofosbuvir.[10][11]
It is on the World Health Organization’s List of Essential Medicines, a list of the most important medications needed in a basic health system.[12]

EUROPEAN MEDICINES AGENCY ADVISES ON COMPASSIONATE USE OF DACLATASVIR
- The first compassionate-use opinion for a hepatitis C treatment was adopted by the CHMP in October 2013.
- Sofosbuvir, which is part of this compassionate-use opinion, received a positive opinion from the CHMP recommending granting of a marketing authorisation at its November 2013 meeting.
- Daclatasvir is developed by Bristol-Myers Squibb and sofosbuvir is developed by Gilead.
|
1-6-2012
|
Anti-Viral Compounds
|
|
|
2-13-2009
|
CRYSTALLINE FORM OF METHYL ((1S)-1-(((2S)
-2-(5-(4′-(2-((2S)-1((2S)-2-((METHOXYCARBONYL)AMINO)-3-METHYLBUTANOYL)-2-PYRROLIDINYL) -1H-IMIDAZOL-5-YL)-4-BIPHENYLYL)-1H-IMIDAZOL-2-YL)-1-PYRROLIDINYL)CARBONYL) -2-METHYLPROPYL)CARBAMATE DIHYDROCHLORIDE SALT |
Synthesis
Daclatasvir dihydrochloride (Daklinza)
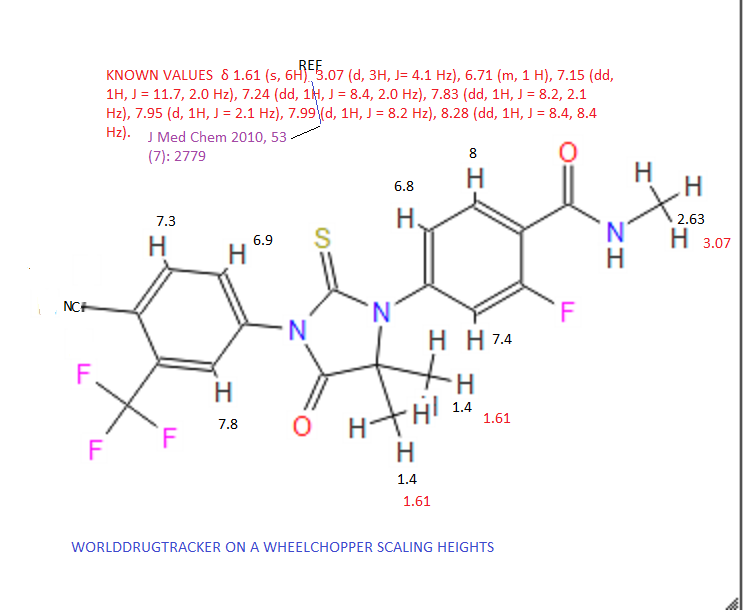
Daclatasvir dihydrochloride is a hepatitis C virus nonstructural 5A (NS5A) replication complex inhibitor which was first approved in Japan for the treatment of genotype 1 HCV patients who fail to respond to interferon plus ribavirin. The drug has also been approved for patients with untreated, chronic HCV who are eligible for interferon. Additionally, in Europe, daclatasvir was approved for use in combination with other products across genotype 1–4 HCV. Daclatasvir was discovered and developed by Bristol–Myers Squibb and a fascinating account describing the initiation of the program from a phenotypic screen and the medicinal chemistry strategy leading to the discovery of the compound has been recently reported.80 Daclatasvir has been prepared via two different routes81,82 and the process route is outlined in Scheme 11.83 Bromination of commercial 4,40-diacetylbiphenyl (58) gave 4,40-bis(bromoacetyl)biphenyl 59 in 82% yield. Alkylation of NBoc- L-proline (60) with 59 gave diester 61 which was treated with ammonium acetate to effect cyclization of the bis-ketoester to provide bis-imidazole 62 in 63% yield for the two steps. Acidic removal of the Boc protecting groups followed by recrystallization provided bis-pyrrolidine 63 in high yield. Acylation of 63 with N-(methoxycarbonyl)- L-valine (64) using N-(3-dimethylaminopropyl)-N0-ethylcarbodiimide(EDC) and 1-hydroxybenxotriazole hydrate (HOBT) provided declatasvir. The dihydrochloride salt was prepared and treated with Cuno Zet Carbon followed by crystallization from acetone
to give daclatasvir dihydrochloride (IX) in 74% yield.
80 Belema, M.; Meanwell, N. A. J. Med. Chem. 2014, 57, 5057.
81. Bachand, C.; Belema, M.; Deon, D. H.; Good, A. C.; Goodrich, J.; James, C. A.;
Lavoie, R.; Lopez, O. D.; Martel, A.; Meanwell, N. A.; Nguyen, V. N.; Romine, J.
L.; Ruediger, E. H.; Snyder, L. B.; St. Laurent, D. R.; Yang, F.; Langley, D. R.;
Wang, G.; Hamann, L. G. WO Patent 2008021927A2, 2008.
82. Belema, M.; Nguyen, V. N.; Bachand, C.; Deon, D. H.; Goodrich, J. T.; James, C.
A.; Lavoie, R.; Lopez, O. D.; Martel, A.; Romine, J. L.; Ruediger, E. H.; Snyder, L.
B.; St Laurent, D. R.; Yang, F.; Zhu, J.; Wong, H. S.; Langley, D. R.; Adams, S. P.;
Cantor, G. H.; Chimalakonda, A.; Fura, A.; Johnson, B. M.; Knipe, J. O.; Parker, D.
D.; Santone, K. S.; Fridell, R. A.; Lemm, J. A.; O’Boyle, D. R., 2nd; Colonno, R. J.;
Gao, M.; Meanwell, N. A.; Hamann, L. G. J. Med. Chem. 2014, 57, 2013.
83. Pack, S. K.; Geng, P.; Smith, M. J.; Hamm, J. WO Patent 2009020825A1, 2009.
PATENT
EXAMPLES

A 1 L, 3-neck round bottom flask, fitted with a nitrogen line, overhead stirrer and thermocouple, was charged with 20 g (83.9 mmol, 1 equiv) 1,1′-(biphenyl-4,4′-diyl)diethanone, 200 mL CH2Cl2 and 8.7 mL (27.1 g, 169.3 mmol, 2.02 quiv) bromine. The mixture was allowed to stir under nitrogen for about 20 hours under ambient conditions. The resulting slurry was charged with 200 mL CH2Cl2 and concentrated down to about 150 mL via vacuum distillation. The slurry was then solvent exchanged into THF to a target volume of 200 mL via vacuum distillation. The slurry was cooled to 20-25° C. over 1 hour and allowed to stir at 20-25° C. for an additional hour. The off-white crystalline solids were filtered and washed with 150 mL CH2Cl2. The product was dried under vacuum at 60° C. to yield 27.4 g (69.2 mmol, 82%) of the desired product : 1H NMR (400 MHz, CDCl3) δ 7.95-7.85 (m, 4H), 7.60-7.50 (m, 4H), 4.26 (s, 4H); 13C NMR (100 MHz, CDCl3) 6 191.0, 145.1, 133.8, 129.9, 127.9, 30.8; IR (KBr, cm−1) 3007, 2950, 1691, 1599, 1199; Anal calcd for C16H12Br2O2: C, 48.52; H, 3.05; Br, 40.34. Found: C, 48.53; H, 3.03; Br, 40.53 HRMS calcd for C16H13Br2O2 (M+H; DCI+): 394.9282. Found: 394.9292. mp 224-226° C.

A 500 mL jacketed flask, fitted with a nitrogen line, thermocouple and overhead stirrer, was charged with 20 g (50.5 mmol, 1 equiv) of Compound 2, 22.8 g (105.9 moles, 2.10 equiv) 1-(tert-butoxycarbonyl)-L-proline and 200 mL acetonitrile. The slurry was cooled to 20° C. followed by the addition of 18.2 mL (13.5 g, 104.4 mmol, 2.07 equiv) DIPEA. The slurry was warmed to 25° C. and allowed to stir for 3 hours. The resulting clear, organic solution was washed with 3×100 mL 13 wt % aqueous NaCl. The rich acetonitrile solution was solvent exchanged into toluene (target volume=215 mL) by vacuum distillation until there was less than 0.5 vol % acetonitrile.

The toluene solution of Compound 3 was charged with 78 g (1.011 moles, 20 equiv) ammonium acetate and heated to 95-100° C. The mixture was allowed to stir at 95-100° C. for 15 hours. After reaction completion, the mixture was cooled to 70-80° C. and charged with 7 mL acetic acid, 40 mL n-butanol, and 80 mL of 5 vol % aqueous acetic acid. The resulting biphasic solution was split while maintaining a temperature >50° C. The rich organic phase was charged with 80 mL of 5 vol % aqueous acetic acid, 30 mL acetic acid and 20 mL n-butanol while maintaining a temperature >50° C. The resulting biphasic solution was split while maintaining a temperature >50° C. and the rich organic phase was washed with an additional 80 mL of 5 vol % aqueous acetic acid. The rich organic phase was then solvent exchanged into toluene to a target volume of 215 mL by vacuum distillation. While maintaining a temperature >60° C., 64 mL methanol was charged. The resulting slurry was heated to 70-75° C. and aged for 1 hour. The slurry was cooled to 20-25° C. over 1 hour and aged at that temperature for an additional hour. The slurry was filtered and the cake was washed with 200 mL 10:3 toluene:methanol. The product was dried under vacuum at 70° C., resulting in 19.8 g (31.7 mmol, 63%) of the desired product: 1H NMR (400 MHz, DMSO-d6) δ 13.00-11.00 (s, 2H), 7.90-7.75 (m, 4H), 7.75-7.60 (m, 4H), 7.60-7.30 (s, 2H), 4.92-4.72 (m, 2H), 3.65-3.49 (m, 2H), 3.49-3.28 (m, 2H), 2.39-2.1 (m, 2H), 2.10-1.87 (m, 6H), 1.60-1.33 (s, 8H), 1.33-1.07 (s, 10H); 13C NMR (100 MHz, DMSO-d6) δ 154.1, 153.8, 137.5, 126.6, 125.0, 78.9, 78.5, 55.6, 55.0, 47.0, 46.7, 33.7, 32.2, 28.5, 28.2, 24.2, 23.5; IR (KBr, cm−1) 2975, 2876, 1663, 1407, 1156, 1125; HRMS calcd for C36H45N6O4 (M+H; ESI+): 625.3502. Found: 625.3502. mp 190-195° C. (decomposed).

To a 250 mL reactor equipped with a nitrogen line and overhead stirrer, 25.0 g of Compound 4 (40.01 mmol, 1 equiv) was charged followed by 250 mL methanol and 32.85 mL (400.1 mmol, 10 equiv) 6M aqueous HCl. The temperature was increased to 50° C. and agitated at 50° C. for 5 hours. The resulting slurry was cooled to 20-25° C. and held with agitation for about 18 hours. Filtration of the slurry afforded a solid which was washed successively with 100 mL 90% methanol/water (V/V) and 2×100 mL of methanol. The wet cake was dried in a vacuum oven at 50° C. overnight to give 18.12 g (31.8 mmol, 79.4%) of the desired product.
Recrystallization of Compound 5
To a 250 mL reactor equipped with a nitrogen line and an overhead stirrer, 17.8 g of Compound 5 from above was charged followed by 72 mL methanol. The resulting slurry was agitated at 50° C. for 4 hours, cooled to 20-25° C. and held with agitation at 20-25° C. for 1 hour. Filtration of the slurry afforded a crystalline solid which was washed with 60 mL methanol. The resulting wet cake was dried in a vacuum oven at 50° C. for 4 days to yield 14.7 g (25.7 mmol, 82.6%) of the purified product: 1H NMR (400 MHz, DMSO-d6) δ 10.5-10.25 (br, 2H), 10.1-9.75 (br, 2H), 8.19 (s, 2H), 7.05 (d, J=8.4, 4H), 7.92 (d, J=8.5, 4H), 5.06 (m, 2H), 3.5-3.35 (m, 4H), 2.6-2.3 (m, 4H), 2.25-2.15 (m, 2H), 2.18-1.96 (m, 2H); 13C NMR (100 MHz, DMSO-d6) δ 156.6, 142.5, 139.3, 128.1, 127.5, 126.1, 116.9, 53.2, 45.8, 29.8, 24.3; IR (KBr, cm−1) 3429, 2627, 1636, 1567, 1493, 1428, 1028. Anal calcd for C26H32N6Cl4: C, 54.75; H, 5.65; Cl, 24.86; Adjusted for 1.9% water: C, 53.71; H, 5.76; N, 14.46; Cl, 24.39. Found: C, 53.74; H, 5.72; N, 14.50; Cl, 24.49; KF=1.9. mp 240° C. (decomposed).

A 1 L jacketed flask equipped with a nitrogen line and an overhead stirrer was sequentially charged with 100 mL acetonitrile, 13.69 g (89.4 mmol, 2.5 equiv) hydroxybenzotriazole hydrate, 15.07 g (86 mmol, 2.4 equiv) N-(methoxycarbonyl)-L-valine, 16.46 g (85.9 mmol, 2.4 equiv) 1-(3-dimethyaminopropyl)-3-ethylcarbodiimide hydrochloride and an additional 100 mL acetonitrile. The resulting solution was agitated at 20° C. for 1 hour and charged with 20.4 g (35.8 mmol, 1 equiv) of purified Compound 5. The slurry was cooled to about 0° C. and 18.47 g (142.9 mmol, 4 equiv) diisopropylethylamine was added over 30 minutes while maintaining a temperature below 10° C. The solution was slowly heated to 15° C. over 3 hours and held at 15° C. for 12 hours. The resulting solution was charged with 120 mL 13 wt % aqueous NaCl and heated to 50° C. for 1 hour. After cooling to 20° C., 100 mL of isopropyl acetate was added. The biphasic solution was filtered through a 0.45 μm filter and the mixture split. The rich organic phase was washed with 2×240 mL of a 0.5 N NaOH solution containing 13 wt % NaCl followed by 120 mL 13 wt % aqueous NaCl. The mixture was then solvent exchanged into isopropyl acetate by vacuum distillation with a target volume of 400 mL. The resulting hazy solution was cooled to 20° C. and filtered through a 0.45 μm filter. The clear solution was then solvent exchanged into ethanol by vacuum distillation with a target volume of 140 mL. While maintaining a temperature of 50° C., 66.4 mL (82.3 mmol, 2.3 equiv) of 1.24M HCl in ethanol was added. The mixture was then charged with 33 mg (0.04 mmol, 0.001 equiv) of seed crystals of Compound (I) (see preparation below) and the resulting slurry was stirred at 50° C. for 3 hours. The mixture was cooled to 20° C. over 1 hour and aged at that temperature for an additional 22 hours. The slurry was filtered and the wet cake was washed with 100 mL of 2:1 acetone:ethanol. The solids were dried in a vacuum oven at 70° C. to give 22.15 g (27.3 mmol, 76.3%) of the desired product.

A solution of Compound (I) was prepared by dissolving 3.17 g of Compound (I) from above in 22 mL methanol. The solution was passed through a 47 mm Cuno Zeta Carbon® 53SP filter at ˜5 psig at a flow rate of ˜58 mL/min. The carbon filter was rinsed with 32 mL of methanol. The solution was concentrated down to 16 mL by vacuum distillation. While maintaining a temperature of 40-50° C., 15.9 mL acetone and 5 mg of seed crystals of Compound (I) (see procedure below) were added. The resulting slurry was then charged with 32 mL acetone over 30 minutes. The slurry was held at 50° C. for 2 hours, cooled to 20° C. over about 1 hour and held at 20° C. for about 20 hours. The solids were filtered, washed with 16 mL 2:1 acetone:methanol and dried in a vacuum oven at 60° C. to give 2.14 g (67.5%) of purified Compound (I):
1H NMR (400 MHz, DMSO-d6, 80° C.): 8.02 (d, J=8.34 Hz, 4 H), 7.97 (s, 2 H), 7.86 (d, J=8.34 Hz, 4 H), 6.75 (s, 2 H), 5.27 (t, J=6.44 Hz, 2 H), 4.17 (t, J=6.95 Hz, 2 H), 3.97-4.11 (m, 2 H), 3.74-3.90 (m, 2 H), 3.57 (s, 6 H), 2.32-2.46 (m, 2 H), 2.09-2.31 (m, 6 H), 1.91-2.07 (m, 2 H), 0.88 (d, J=6.57 Hz, 6 H), 0.79 (d, J=6.32 Hz, 6 H);
13C NMR (75 MHz, DMSO-d6): δ 170.9, 156.9, 149.3, 139.1, 131.7, 127.1, 126.5, 125.9, 115.0, 57.9, 52.8, 51.5, 47.2, 31.1, 28.9, 24.9, 19.6, 17.7;
IR (neat, cm−1): 3385, 2971, 2873, 2669, 1731, 1650.
Anal. Calcd for C40H52N8O6Cl2: C, 59.18; H, 6.45; N, 13.80; Cl, 8.73. Found C, 59.98; H, 6.80; N, 13.68; Cl, 8.77. mp 267° C. (decomposed).
Preparation of Seed Crystals of Compound (I)
A 250 mL round-bottom flask was charged with 6.0 g (10.5 mmol, 1 equiv) Compound 5, 3.87 g (22.1 mmol, 2.1 equiv) N-(methoxycarbonyl)-L-valine, 4.45 g (23.2 mmol, 2.2 equiv) 1-(3-dimethyaminopropyl)-3-ethylcarbodiimide hydrochloride, 0.289 g (2.14 mmol, 0.2 equiv) 1-hydroxybenzotriazole, and 30 mL acetonitrile. The resulting slurry was then charged with 7.33 mL (42.03 mmol, 4 equiv) diisopropylethylamine and allowed to stir at 24-30° C. for about 18 hours. The mixture was charged with 6 mL of water and heated to 50° C. for about 5 hours. The mixture was cooled and charged with 32 mL ethyl acetate and 30 mL water. The layers were separated and the rich organic layer was washed with 30 mL of 10 wt % aqueous NaHCO3, 30 mL water, and 20 mL of 10 wt % aqueous NaCl. The rich organic layer was then dried over MgSO4, filtered, and concentrated down to a residue. The crude material was then purified via flash chromatography (silica gel, 0-10% methanol in dichloromethane) to provide the free base of Compound (I).
The free-base of Compound (I) (0.03 g) was dissolved in 1 mL isopropanol at 20° C. Anhydrous HCl (70 μL, dissolved in ethanol, approximately 1.25M concentration) was added and the reaction mixture was stirred. To the solution was added methyl tert-butyl ether (1 mL) and the resulting slurry was stirred vigorously at 40° C. to 50° C. for 12 hours. The crystal slurry was cooled to 20° C. and filtered. The wet cake was air-dried at 20° C. A white crystalline solid (Form N-2 of Compound (I)) was obtained.
Clip
Daclatasvir synthesis: WO2009020828A1

Procedure:
Step a: A 1 L, 3 -neck round bottom flask, fitted with a nitrogen line, overhead stirrer and thermocouple, was charged with 20 g (83.9 mmol, 1 equiv) 1,1′-(biphenyl-4,4′-diyl)diethanone, 200 mL Dichloromethane and 8.7 mL (27.1g, 169.3 mmol, 2.02 equiv) bromine. The mixture was allowed to stir under nitrogen for about 20 hours under ambient conditions. The resulting slurry was charged with 200 mL Dichloromethane and concentrated down to about 150 mL via vacuum distillation. The slurry was then solvent exchanged into THF to a target volume of 200 mL via vacuum distillation. The slurry was cooled to 20-25 0C over 1 hour and allowed to stir at 20-25 0C for an additional hour. The off-white crystalline solids were filtered and washed with 150 mL Dichloromethane. The product was dried under vacuum at 60 0C to yield 27.4 g (69.2 mmol, 82%) of the desired product: 1H NMR (400 MHz, CDCl3) d 7.95-7.85 (m, 4H), 7.60-7.50 (m, 4H), 4.26 (s, 4H); 13C NMR 100 MHz, CDCl3) d 191.0, 145.1, 133.8, 129.9, 127.9, 30.8; IR (KBr, cm-1) 3007, 2950, 1691, 1599, 1199; Anal calcd for C16H12Br2O2: C, 48.52; H, 3.05; Br, 40.34. Found: C, 48.53; H, 3.03; Br, 40.53. HRMS calcd for C16H12Br2O2 (M + H; DCI+): 394.9282. Found: 394.9292. mp 224-226 0C.
Step b: A 500 mL jacketed flask, fitted with a nitrogen line, thermocouple and overhead stirrer, was charged with 20 g (50.5 mmol, 1 equiv) of Compound 2, 22.8 g (105.9 moles, 2.10 equiv) 1-(tert-butoxycarbonyl)-L-proline and 200 mL acetonitrile. The slurry was cooled to 20 0C followed by the addition of 18.2 mL (13.5 g, 104.4 mmol, 2.07 equiv) DIPEA. The slurry was warmed to 25 0C and allowed to stir for 3 hours. The resulting clear, organic solution was washed with 3 x 100 mL 13 wt% aqueous NaCl. The rich acetonitrile solution was solvent exchanged into toluene (target volume = 215 mL) by vacuum distillation until there was less than 0.5 vol% acetonitrile.
Step c: The toluene solution of Compound 3 was charged with 78 g (1.011 moles, 20 equiv) ammonium acetate and heated to 95-100 0C. The mixture was allowed to stir at 95-100 0C for 15 hours. After reaction completion, the mixture was cooled to 70- 80 0C and charged with 7 mL acetic acid, 40 mL n-butanol, and 80 mL of 5 vol% aqueous acetic acid. The resulting biphasic solution was split while maintaining a temperature > 50 0C. The rich organic phase was charged with 80 mL of 5 vol% aqueous acetic acid, 30 mL acetic acid and 20 mL n-butanol while maintaining a temperature > 50 0C. The resulting biphasic solution was split while maintaining a temperature > 50 0C and the rich organic phase was washed with an additional 80 mL of 5 vol% aqueous acetic acid. The rich organic phase was then solvent exchanged into toluene to a target volume of 215 mL by vacuum distillation. While maintaining a temperature > 60 0C, 64 mL methanol was charged. The resulting slurry was heated to 70-75 0C and aged for 1 hour. The slurry was cooled to 20-25 0C over 1 hour and aged at that temperature for an additional hour. The slurry was filtered and the cake was washed with 200 mL 10:3 toluene:methanol. The product was dried under vacuum at 70 0C, resulting in 19.8 g (31.7 mmol, 63%) of the desired product: 1H NMR (400 MHz, DMSO-^) d 13.00-11.00 (s, 2H), 7.90-7.75 (m, 4H), 7.75-7.60 (m, 4H), 7.60-7.30 (s, 2H), 4.92-4.72 (m, 2H), 3.65-3.49 (m, 2H), 3.49-3.28 (m, 2H), 2.39-2.1 (m, 2H), 2.10-1.87 (m, 6H), 1.60-1.33 (s, 8H), 1.33-1.07 (s, 10H); 13C NMR (100 MHz, DMSO-?fe) d 154.1, 153.8, 137.5, 126.6, 125.0, 78.9, 78.5, 55.6, 55.0, 47.0, 46.7, 33.7, 32.2, 28.5, 28.2, 24.2, 23.5; IR (KBr, cm-1) 2975, 2876, 1663, 1407, 1156, 1125; HRMS calcd for C36H45N6O4 (M + H; ESI+): 625.3502. Found: 625.3502. mp 190-195 0C (decomposed).
Step d: To a 250 mL reactor equipped with a nitrogen line and overhead stirrer, 25.0 g of Compound 4 (40.01 mmol, 1 equiv) was charged followed by 250 mL methanol and 32.85 mL (400.1 mmol, 10 equiv) 6M aqueous HCl. The temperature was increased to 50 0C and agitated at 50 0C for 5 hours. The resulting slurry was cooled to 20-25 0C and held with agitation for about 18 hours. Filtration of the slurry afforded a solid which was washed successively with 100 mL 90% methanoI/water (WV) and 2 x 100 mL of methanol. The wet cake was dried in a vacuum oven at 50 0C overnight to give 18.12 g (31.8 mmol, 79.4%) of the desired product.
CUT PASTE…….WO2009020825

Preparation of Compound (I)
A 1 L jacketed flask equipped with a nitrogen line and an overhead stirrer was sequentially charged with 100 mL acetonitrile, 13.69 g (89.4 mmol, 2.5 equiv) hydroxybenzotriazole hydrate, 15.07 g (86 mmol, 2.4 equiv) N-(methoxycarbonyl)- L-valine, 16.46 g (85.9 mmol, 2.4 equiv) l-(3-dimethyaminopropyl)-3- ethylcarbodiimide hydrochloride and an additional 100 mL acetonitrile. The resulting solution was agitated at 20 0C for 1 hour and charged with 20.4 g (35.8 mmol, 1 equiv) of purified Compound 7. The slurry was cooled to about 0 0C and 18.47 g (142.9 mmol, 4 equiv) diisopropylethylamine was added over 30 minutes while maintaining a temperature below 10 0C. The solution was slowly heated to 15 0C over 3 hours and held at 15 0C for 12 hours. The resulting solution was charged with 120 mL 13 wt% aqueous NaCl and heated to 50 0C for 1 hour. After cooling to 20 0C, 100 mL of isopropyl acetate was added. The biphasic solution was filtered through a 0.45 μm filter and the mixture split. The rich organic phase was washed with 2 x 240 mL of a 0.5 Ν NaOH solution containing 13 wt% NaCl followed by 120 mL 13 wt% aqueous NaCl. The mixture was then solvent exchanged into isopropyl acetate by vacuum distillation with a target volume of 400 mL. The resulting hazy solution was cooled to 20 0C and filtered through a 0.45 μm filter. The clear solution was then solvent exchanged into ethanol by vacuum distillation with a target volume of 140 mL. While maintaining a temperature of 50 0C, 66.4 mL (82.3 mmol, 2.3 equiv) of 1.24M HCl in ethanol was added. The mixture was then charged with 33 mg (0.04 mmol, 0.001 equiv) of seed crystals of Compound (I) (see preparation below) and the resulting slurry was stirred at 50 0C for 3 hours. The mixture was cooled to 20 0C over 1 hour and aged at that temperature for an additional 22 hours. The slurry was filtered and the wet cake was washed with 100 mL of 2: 1 acetone:ethanol. The solids were dried in a vacuum oven at 70 0C to give 22.15 g (27.3 mmol, 76.3%) of the desired product.

Alternative Preparation of Compound (I)
A jacketed reactor equipped with a mechanical agitator, a thermocouple and a nitrogen inlet was sequentially charged with 10 L acetonitrile, 0.671 kg (4.38 moles, 2.50 equiv) 1-hydroxybenzotriazole, 0.737 kg (4.21 moles, 2.40 equiv) N- (methoxycarbonyl)-L-valine and 0.790 kg (4.12 moles, 2.35 equiv) l-(3- dimethyaminopropyl)-3-ethylcarbodiimide hydrochloride. The mixture was agitated at 200C for 1 hour, cooled to 5 0C and charged with 1 kg (1.75 moles, 1.00 equiv) Compound 7. While maintaining a temperature < 10 0C, 0.906 kg (7.01 moles, 4 equiv) diisopropylethylamine was added. The mixture was heated to 15-20 0C over 2 hours and agitated for an additional 15 hours. After the reaction was complete, the mixture was washed once with 6.0 L 13 wt% aqueous NaCl, twice with 6.1 L (6.12 moles, 3.5 equiv) 1.0 M aqueous NaOH containing 13 wt% NaCl and once with 6.0 L 13 wt% aqueous NaCl. Water was then removed from the rich organic solution via azeotropic distillation. The mixture was cooled to 20 0C, agitated for 1 hour and filtered. The rich organic solution was then solvent exchanged into EtOH via vacuum distillation to a target volume of 5 L. While maintaining a temperature of 50 0C, 3.2 L (4.0 moles, 2.3 equiv) 1.25M HCl in EtOH was charged. The mixture was seeded with 1.6 g Compound (I) (see preparation below) and agitated at 50 0C for 3 hours. The resulting slurry was cooled to 20 0C and agitated for at least 3 hours. The product was collected by filtration and washed with 5 L 2: 1 acetone:
EtOH to give 1.29 kg (ca. 90 wt% product) of wet crude product. A reactor equipped with an overhead agitator, nitrogen inlet and thermocouple was charged with 1.11 kg of the above crude product and 7 L methanol. The resulting solution was treated with Cuno Zeta Carbon (TM) 55SP. The carbon was washed with 15 L MeOH and the combined filtrate and wash was concentrated down to 4 L via vacuum distillation. The concentrated solution was charged with 5 L acetone and seeded with 1.6 g Compound (I) (see preparation below) while maintaining a temperature of 50 0C. An additional 10 L acetone was charged and the resulting slurry was stirred at 50 0C for 3 hours. The slurry was cooled to 20 0C and allowed to agitate at 200C for 3 hours. The product was collected by filtration, washed with 5 L 2: 1 acetone: EtOH and dried under vacuum at 50-60 0C to give 0.900 kg (1.11 moles, 74% adjusted) of Compound (I)-

Carbon Treatment and Recrystallization of Compound (I) A solution of Compound (I) was prepared by dissolving 3.17 g of Compound (I) from above in 22 mL methanol. The solution was passed through a 47mm Cuno Zeta Carbon 53SP filter at ~5 psig at a flow rate of~58mL/min. The carbon filter was rinsed with 32 mL of methanol. The solution was concentrated down to 16 mL by vacuum distillation. While maintaining a temperature of 40-50 0C, 15.9 mL acetone and 5 mg of seed crystals of Compound (I) (see procedure below) were added. The resulting slurry was then charged with 32 mL acetone over 30 minutes. The slurry was held at 50 0C for 2 hours, cooled to 20 0C over about 1 hour and held at 20 0C for about 20 hours. The solids were filtered, washed with 16 mL 2: 1 acetone:methanol and dried in a vacuum oven at 60 0C to give 2.14 g (67.5%) of purified Compound (I):
1H NMR (400 MHz, DMSO-έfc, 80 0C): 8.02 (d, J=8.34 Hz, 4 H), 7.97 (s, 2 H), 7.86 (d, J=8.34 Hz, 4 H), 6.75 (s, 2 H), 5.27 (t, J=6.44 Hz, 2 H), 4.17 (t, J=6.95 Hz, 2 H), 3.97 – 4.11 (m, 2 H), 3.74 – 3.90 (m, 2 H), 3.57 (s, 6 H), 2.32 – 2.46 (m, 2 H), 2.09 – 2.31 (m, 6 H), 1.91 – 2.07 (m, 2 H), 0.88 (d, J=6.57 Hz, 6 H), 0.79 (d, J=6.32 Hz, 6 H);
13C NMR (75 MHz, DMSO-έfc): δ 170.9, 156.9, 149.3, 139.1, 131.7, 127.1, 126.5, 125.9, 115.0, 57.9, 52.8, 51.5, 47.2, 31.1, 28.9, 24.9, 19.6, 17.7;
IR (neat, cm“1): 3385, 2971, 2873, 2669, 1731, 1650.
Anal. Calcd for C40H52N8O6Cl2: C, 59.18; H, 6.45; N, 13.80; Cl, 8.73. Found C, 59.98; H, 6.80; N, 13.68; Cl, 8.77. mp 267 0C (decomposed).
Characteristic diffraction peak positions (degrees 2Θ + 0.1) @ RT, based on a high quality pattern collected with a diffractometer (CuKa) with a spinning capillary with 2Θ calibrated with a NIST other suitable standard are as follows: 10.3, 12.4, 12.8, 13.3, 13.6, 15.5, 20.3, 21.2, 22.4, 22.7, 23.7
Daclatasvir faces problems in USA
The US-FDA in 2014 issued a complete response letter for NS5A inhibitor daclatasvir saying it was unable to approve the drug because the marketing application was for its use in tandem with asunaprevir, an NS3/NS4A protease inhibitor discontinued in the US by BMS for commercial reasons. Daclatasvir is already on the market in Europe-where it is sold as Daklinza-and also in Japan where it was approved alongside asunaprevir in July as the country’s first all-oral HCV therapy. However, a delay in the large US market is clearly a major setback for BMS’ ambitions in hepatitis therapy.
To make the matter worse, US FDA has rescinded breakthrough therapy designation status from Bristol-Myers Squibb for Daclatasvir for the treatment of hepatitis C virus infection in Feb 2015.
PAPER
Makonen, B.; et. al. Hepatitis C Virus NS5A Replication Complex Inhibitors: The Discovery of Daclatasvir. J Med Chem 2014, 57(5), 2013–2032.
http://pubs.acs.org/doi/abs/10.1021/jm401836p
PATENT
http://www.google.com/patents/WO2008021927A2?cl=en
Example 24-23

methyl ((lS)-l-(((2S)-2-(5-(4′-(2-((2S)-l-((2S)-2-((methoxycarbonyl)amino)-3- methylbutanoyl)-2-pyrrolidinyl)-lH-imidazol-5-yl)-4-biphenylyl)-lH-imidazol-2-yl)-
1 -pyrrolidinyl) carbonyl) -2-methylpropyl) carbamate
A 50 mL flask equipped with a stir bar was sequentially charged with 2.5 mL acetonitrile, 0.344 g (2.25 mmol, 2.5 equiv) hydroxy benzotriazole hydrate, 0.374 g (2.13 mmol, 2.4 equiv) N-(methoxycarbonyl)-L-valine, 0.400 g (2.09 mmol, 2.4 equiv) 1 -(3 -dimethyaminopropyl)-3-ethylcarbodiimide hydrochloride and an additional 2.5 mL acetonitrile. The resulting solution was agitated at 20 0C for 1 hour and charged with 0.501 g (0.88 mmol, 1 equiv) Example A-le-4. The slurry was cooled to about 0 0C and 0.45 g (3.48 mmol, 4 equiv) diisopropylethylamine was added over 30 minutes while maintaining a temperature below 10 0C. The solution was slowly heated to 15 0C over 3 hours and held at 15 0C for 16 hours. The temperature was increased to 20 0C and stirred for 3.25 hours. The resulting solution was charged with 3.3 g of 13 wt% aqueous NaCl and heated to 50 0C for 1 hour. After cooling to 20 0C, 2.5 mL of isopropyl acetate was added. The rich organic phase was washed with 2 x 6.9 g of a 0.5 N NaOH solution containing 13 wt% NaCl followed by 3.3 g of 13 wt% aqueous NaCl. The mixture was then solvent exchanged into isopropyl acetate by vacuum distillation to a target volume of 10 mL. The resulting hazy solution was cooled to 20 0C and filtered through a 0.45 μm filter. The clear solution was then solvent exchanged into ethanol by vacuum distillation with a target volume of 3 mL. 1.67 mL (2.02 mmol, 2.3 equiv) of 1.21 M HCl in ethanol was added. The mixture was then stirred at 25 0C for 15 hours. The resulting slurry was filtered and the wet cake was washed with 2.5 mL of 2: 1 acetone:ethanol. The solids were dried in a vacuum oven at 50 0C to give 0.550 g (0.68 mmol, 77 %) of the desired product.
RecrystalHzation of Example 24-23
A solution of Example 24-23 prepared above was prepared by dissolving 0.520 g of the above product in 3.65 mL methanol. The solution was then charged with 0.078 g of type 3 Cuno Zeta loose carbon and allowed to stir for 0.25 hours. The mixture was then filtered and washed with 6 ml of methanol. The product rich solution was concentrated down to 2.6 mL by vacuum distillation. 7.8 mL acetone was added and allowed to stir at 25 0C for 15 h. The solids were filtered, washed with 2.5 mL 2: 1 acetone:ethanol and dried in a vacuum oven at 70 0C to give 0.406 g (57.0%) of the desired product as white crystals: 1H NMR (400 MHz, OMSO-d6, 80 0C): 8.02 (d, J=8.34 Hz, 4 H), 7.97 (s, 2 H), 7.86 (d, J=8.34 Hz, 4 H), 6.75 (s, 2 H), 5.27 (t, J=6.44 Hz, 2 H), 4.17 (t, J=6.95 Hz, 2 H), 3.97 – 4.11 (m, 2 H), 3.74 – 3.90 (m, 2 H), 3.57 (s, 6 H), 2.32 – 2.46 (m, 2 H), 2.09 – 2.31 (m, 6 H), 1.91 – 2.07 (m, 2 H), 0.88 (d, J=6.57 Hz, 6 H), 0.79 (d, J=6.32 Hz, 6 H); 13C NMR (75 MHz, DMSO- d6): δ 170.9, 156.9, 149.3, 139.1, 131.7, 127.1, 126.5, 125.9, 115.0, 57.9, 52.8, 51.5, 47.2, 31.1, 28.9, 24.9, 19.6, 17.7; IR (neat, cm“1): 3385, 2971, 2873, 2669, 1731, 1650. Anal. Calcd for C40H52N8O6Cl2: C, 59.18; H, 6.45; N, 13.80; Cl, 8.73. Found C, 59.98; H, 6.80; N, 13.68; Cl, 8.77. mp 267 0C (decomposed). Characteristic diffraction peak positions (degrees 2Θ ± 0.1) @ RT, based on a high quality pattern collected with a diffractometer (CuKa) with a spinning capillary with 2Θ calibrated with a NIST other suitable standard are as follows: 10.3, 12.4, 12.8, 13.3, 13.6, 15.5, 20.3, 21.2, 22.4, 22.7, 23.7
PAPER
Bioorganic & Medicinal Chemistry Letters (2015), 25(16), 3147-3150
http://www.sciencedirect.com/science/article/pii/S0960894X15005995

Scheme 1.
Synthetic route for the preparation of the target compounds 8a–8y. Reagents and conditions: (a) Br2, CH2Cl2, rt, overnight, 86%; (b) N-Boc-l-proline, MeCN, Et3N, rt, 2 h, 98%; (c) NH4OAc, toulene, 130 °C, 15 h, 85%; (d) 6 N HCl, MeOH, 50 °C, 4 h, 87%; (e) HATU, N-(methoxycarbonyl)-l-valine, DIPEA, rt, 14 h, 83%; (f) RCOCl, TEA, CH2Cl2, rt, 3 h, 64–87%.
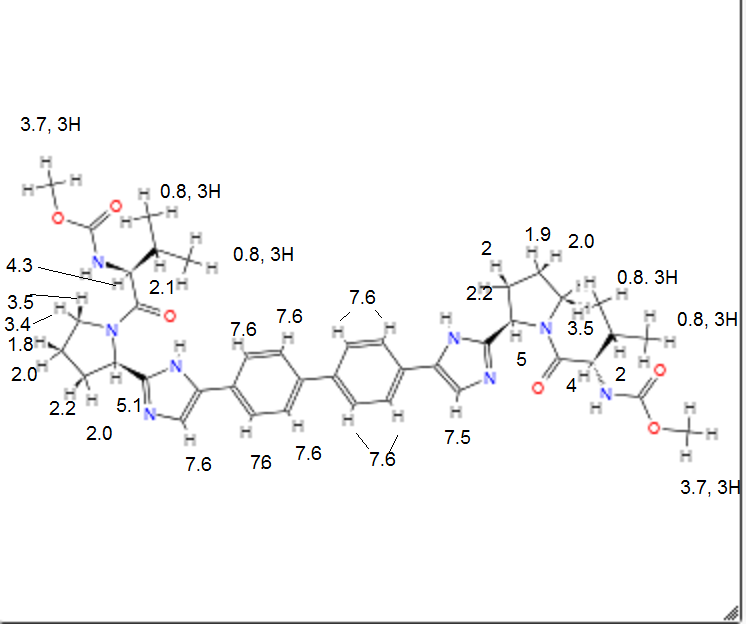
Dimethyl((2S,2’S)-((2S,2’S)-2,2′-(5,5′-([1,1′-biphenyl]-4,4′-diyl)bis(1H-imidazole-
5,2-diyl))bis(pyrrolidine-2,1-diyl))bis(3-methyl-1-oxobutane-2,1-
diyl))dicarbamate 7……………FREE BASE
To a solution of 5 (90 mg, 0.181 mmol), N-me-thoxycarbonyl-l-valine 6 (92 mg,0.525 mmol) and DIPEA (0.18 mL, 1.03 mmol) in DMF (5 mL) was added HATU(165.5 mg, 0.434 mmol). The resulting reaction was allowed to stir at room temperature for 15 h, the reaction mixture was filtered and the residue was partitioned between EtOAc and H2O, The aqueous phase was extracted with EtOAc, and the combined organic phase was dried (MgSO4), filtered, and concentrated in vacuo. The residue was purified by flash chromatography (silica gel; 5% Methanol /CH2Cl2) to
afford 7 (0.11 g, 83 %)as white solid.
1H NMR (DMSO-d6, 500 MHz) δ: 11.56 (s, 2H), 7.69-7.48 (m, 8H), 7.26-7.03 (m, 4H), 5.24-5.05 (m, 2H), 4.09-4.04 (m, 2H), 3.85-3.75 (m, 4H), 3.58 (s, 6H), 2.24-1.98 (m, 10H), 0.87 (d, J = 3.6 Hz, 12H).
Anal. calcd. (%) for C40H50N8O6: C 65.02, H 6.82, N 15.17; found: C 65.20, H 6.79, N 15.31.
ESI-MS m/z: 739.5 (M+H)+.
NMR PREDICT
1H NMR PREDICT


13C NMR PREDICT



COSY PREDICT

Patents
http://www.who.int/phi/implementation/ip_trade/daclatasvir_report_2014_09-02.pdf
Click on images to view
 Click on images to view
Click on images to view










Click on images to view
http://www.who.int/phi/implementation/ip_trade/daclatasvir_report_2014_09-02.pdf

Click on images to view
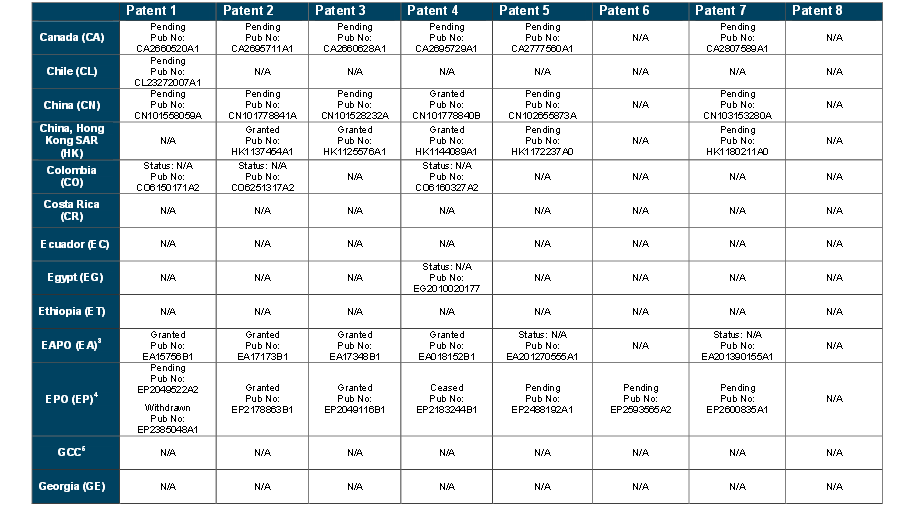
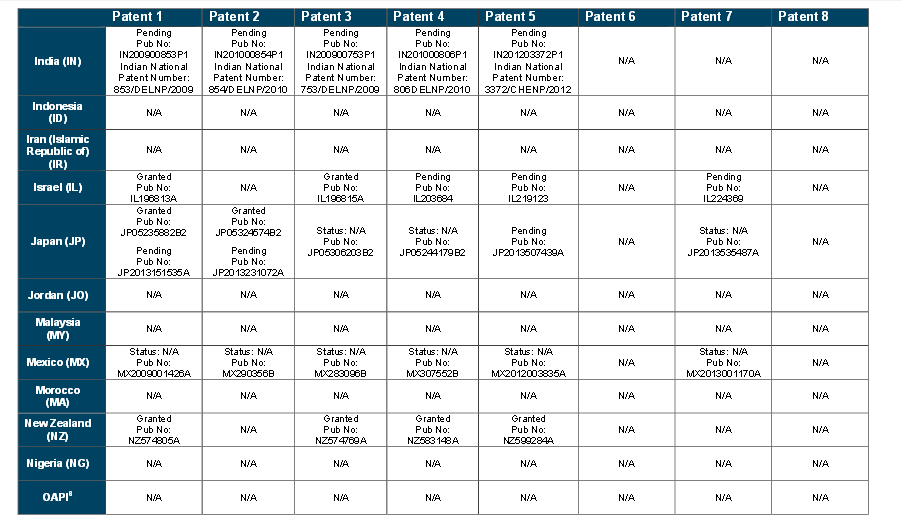
Click on images to view


Click on images to view
 |
|
| Names | |
|---|---|
| IUPAC name
Methyl [(2S)-1-{(2S)-2-[4-(4’-{2-[(2S)-1-{(2S)-2-[(methoxycarbonyl)amino]-3-methylbutanoyl}-2-pyrrolidinyl]-1H-imidazol-4-yl}-4-biphenylyl)-1H-imidazol-2-yl]-1-pyrrolidinyl}-3-methyl-1-oxo-2-butanyl]carbamate
|
|
| Other names
BMS-790052
|
|
| Identifiers | |
| 1009119-64-5 |
|
| ATC code | J05AX14 |
| ChEBI | CHEBI:82977 |
| ChEMBL | ChEMBL2023898 ChEMBL2303621 |
| ChemSpider | 24609522 |
| Jmol-3D images | Image |
| Properties | |
| C40H50N8O6 | |
| Molar mass | 738.89 g·mol−1 |
CLIP 1
Australian Government, National Measurement Institute
REFERENCE MATERIAL ANALYSIS REPORT
HPLC: Instrument: Shimadzu Binary pump LC-20AB, SIL-20 A HT autosampler
Column: X-Bridge C-18, 5.0 m (4.6 mm x 150 mm)
Column oven: 40 °C
Mobile Phase: A = Milli-Q water buffered at pH 10 with NH4
+ -OAc; B = MeCN
Gradient 0 min 35% B; 0-15 min 35% B; 15-18 min 35-75% B; 18-23 min 75% B.
Flow rate: 1.0 mL/min
Detector: Shimadzu SPD-M20A PDA operating at 310 nm
Relative peak area response of main component:
Initial analysis: Mean = 99.2%, s = 0.01%
Thermogravimetric analysis: Non volatile residue < 0.2% mass fraction . The volatile
content (e.g. organic solvents and/or water) could not be determined by
thermogravimetric analysis.
Karl Fischer analysis: Moisture content 0.6% mass fraction
QNMR: Instrument: Bruker Avance-III-500
Field strength: 500 MHz Solvent: DMSO-d6 (2.50 ppm)
Internal standard: Potassium hydrogen maleate (98.8% mass fraction)
Initial analysis: Mean (0.86 ppm) = 98.2%, s = 0.2%
LC-MS: Instrument: Thermo Scientific Dionex UltiMate 3000 Degasser,
Column: ZORBAX RRHD SB-C8, 2.1 x 50 mm, 1.8 μm (Agilent, 857700-906)
Column temp: 30.0 °C
Solvent system: Mobile phase A: 10 mM ammonium formate, 0.01% (v/v) formic acid in Milli-Q® water.
Mobile phase B: 0.01% (v/v) formic acid in acetonitrile.
Gradient from 90% A to 100% B
Flow rate: 0.25 mL/min
Sample prep: 2 mg/mL in MeOH with trace of formic acid
Injection volume: 10 L
Ionisation mode: Electrospray positive ion
Capillary voltage: 4.5 kV
Capillary temp: 360ºC Desolvation gas temperature: 300 ºC
Cone gas flow rate: 10 (arbitrary unit) Desolvation gas flow rate: 70 (arbitrary unit)
The retention time of daclatasvir is reported along with the major peak in the mass spectrum. The latter is reported as a mass/charge ratio.
9.98 min: 739.39545 (M+H+) m/z
HS-GC-MS: Instrument: Agilent 6890/5973/G1888
Column: DB-624, 30 m x 0.25 mm I.D. x 1.4 μm
Program: 50 C (5 min), 7 C/min to 120 C, 15 °C/min to 220 °C (8.3 min)
Injector: 150 C Transfer line temp: 280 C
Carrier: Helium, 1.2 mL/min Split ratio: 50/1
Solvents detected: Ethyl acetate
TLC: Conditions: Kieselgel 60F254. Ethyl acetate : methanol (95/5)
Single spot observed, Rf = 0.18. Visualisation with UV at 254 nm
The TLC was performed on the liberated free base.
IR: Instrument: Bruker Alpha FT-IR
Range: 4000-400 cm-1, neat
Peaks: 1723, 1697, 1643, 1523, 1439, 1235, 1099, 1024 cm-1
1H NMR: Instrument: Bruker Avance III 500
Field strength: 500 MHz Solvent: DMSO-d6 (2.50 ppm)
Spectral data: 0.77 (6H, d, J = 6.7 Hz), 0.83 (6H, d, J = 6.7 Hz), 2.01 (2H, m), 2.07 (2H, m), 2.12-2.27 (4H, m), 2.38 (2H, m), 3.54 (6H, s), 3.84 (2H, m), 3.97 (2H, m), 4.12 (2H, t, J = 7.7 Hz), 5.18 (2H, t, J = 7.0 Hz), 7.31 (2 N-H, d, J = 8.5 Hz), 7.94 (4H, d, J = 8.4 Hz), 7.99 (4H, d, J = 8.4 Hz), 8.16 (2H, s) ppm
Ethyl acetate estimated at 0.6% mass fraction was observed in the 1H NMR
13C NMR: Instrument: Bruker Avance III 500
Field strength: 126 MHz Solvent: DMSO-d6 (39.5 ppm)
Spectral data: 17.8, 19.6, 25.0, 29.0, 31.2, 47.3, 51.6, 52.9, 58.0, 115.1, 125.9, 126.6, 127.3, 131.8, 139.2, 149.4, 157.0, 171.1 ppm
Melting point: > 250 oC
Microanalysis: Found: C = 59.0%; H = 6.5%; N = 13.7% (August 2015)
Calc: C = 59.2%; H = 6.5%; N = 13.8% (Calculated for C40H50N8O6.2HCl)
REFERENCE
Australian NMI NATA Certification Daclatasvir – FixHepC
https://fixhepc.com/images/coa/NMI-NATA-Daclatasvir-Certification.pdf
Oct 7, 2015 – Compound Name: Daclatasvir dihydrochloride … Note: The assigned stereochemistry of this sample of daclatasvir has not …. Melting point:.
CLIP 2


Full Text Article – European Journal of Pharmaceutical and Medical …
Nov 28, 2016 – Daclatasvir dihydrochloride (DCLD) is a new drug …. DSC thermogram of daclatasvirdihydrochloriderealed drug melting point at 273.600C as …
CLIP 3
DCV dihydrochloride (anhydrous) is a white to yellow, non hygroscopic powder which is highly soluble in water (>700mg/mL). Solubility is higher at low pH. In aqueous buffers over the physiological pH range (pH 1.2-6.8) solubility is very low (4mg/mL to 0.004 mg/mL) due to the slow formation of the less soluble hydrated form. Water content in the drug substance is adequately controlled by in process tests. The desired anhydrous crystalline form of DCV dihydrochloride (N-2) is consistently produced and has been shown to not change on storage.
[DOC]AusPAR Daclatasvir dihydrochloride – Therapeutic Goods Administration
https://www.tga.gov.au/sites/default/…/auspar-daclatasvir–dihydrochloride-151214.do…
Dec 14, 2015 – Australian Public Assessment Report for daclatasvir dihydrochloride …. Figure 1:Chemical structure of daclatasvir dihydrochloride. …… 24 weeks is based on a selected literaturereview mostly of studies in patients with GT-1.
CLIP 4
The structure of the active substance has been confirmed by UV, IR, Raman and 1 H and 13C NMR spectroscopy, MS spectrometry, and crystal X-Ray diffraction.
Daclatasvir is a white to yellow crystalline non-hygroscopic powder. It is freely soluble in water, dimethyl sulfoxide, methanol; soluble in ethanol (95%); practically insoluble in dichloromethane, tetrahydrofuran, acetonitrile, acetone and ethyl acetate.
Daclatasvir is a chiral molecule with four stereocenters (1,1’, 2, 2;) in the S configuration. The synthetic strategy and process design such as starting material and reagent selection, process parameters, and in-process controls ensure the desired configuration at each of the four chiral centers. In addition, the established control strategy minimizes epimerization and eliminates other diastereomeric impurity formation in each step.
Polymorphism has been observed for daclatasvir hydrochloride. Although two neat crystalline dihydrochloride salts, N1 and N-2 have been identified in screening studies, it has been confirmed that the form N-2 is the thermodynamically most stable polymorph and only this form produced by the proposed synthetic process.
Manufacture, characterisation and process controls
Daclatasvir dihydrochloride is synthesised in three main steps using three commercially available well defined starting materials with acceptable specifications. The synthesis involves an alkylation and formation of the imidazole ring, a coupling reaction and the formation of the hydrochloride salt.
As mentioned above, the synthetic process has been designed to ensure the correct configuration at each of the four chiral centres is achieved. In addition, it has been demonstrated that the stereogenic centres do not epimerize during normal or stressed processing conditions.
The manufacturing process has been developed using a combination of conventional univariate studies and elements of QbD such as risk assessment.
The characterisation of the active substance and its impurities are in accordance with the EU guideline on chemistry of new active substances. Potential and actual impurities were well discussed with regards to their origin and characterised. Adequate in-process controls are applied during the synthesis. The specifications and control methods for intermediate products, starting materials and reagents have been presented.
The active substance specification includes tests for: appearance, colour, identity (IR/Raman, HPLC), assay (HPLC), impurities (HPLC), residual solvents (GC), HCl content (titration), total inorganic impurities (ICP-MS), and particle size (laser light scattering). The absence of a test for chiral purity in the active substance specification has been adequately justified based on the stereochemical control during the synthetic process and demonstration that there is no epimerization during normal or stressed processing conditions. Similarly, since the N-2 form of daclatasvir hydrochloride is the thermodynamically most stable polymorph and, is consistently produced by the synthetic process and remained unchanged during storage under long-term or accelerated conditions, this parameter is not included in the specification
CLIP5
SEE
http://www.accessdata.fda.gov/drugsatfda_docs/nda/2015/206843Orig1s000ChemR.pdf
CLIP6
Daclatasvir dihydrochloride

References
- 1 Statement on a Nonproprietary Name Adopted by the USAN Council
- 2 Gao, Min; Nettles, Richard E.; Belema, Makonen; Snyder, Lawrence B.; Nguyen, Van N.; Fridell, Robert A.; Serrano-Wu, Michael H.; Langley, David R.; Sun, Jin-Hua; O’Boyle, Donald R., II; Lemm, Julie A.; Wang, Chunfu; Knipe, Jay O.; Chien, Caly; Colonno, Richard J.; Grasela, Dennis M.; Meanwell, Nicholas A.; Hamann, Lawrence G. (2010). “Chemical genetics strategy identifies an HCV NS5A inhibitor with a potent clinical effect”. Nature 465 (7294): 96–100. doi:10.1038/nature08960. PMID 20410884.
- 3 Bell, Thomas W. (2010). “Drugs for hepatitis C: unlocking a new mechanism of action”. ChemMedChem 5 (10): 1663–1665. doi:10.1002/cmdc.201000334. PMID 20821796.
- 4 Modeling shows that the NS5A inhibitor daclatasvir has two modes of action and yields a shorter estimate of the hepatitis C virus half-life. Guedj, J et al. Proceedings of the National Academy of Sciences. February 19, 2013.
- 5 AASLD: Daclatasvir with Pegylated Interferon/Ribavirin Produces High Rates of HCV Suppression. Highleyman, L. HIVandHepatitis.com. 6 December 2011.
- 6Preliminary Study of Two Antiviral Agents for Hepatitis C Genotype 1. Lok, A et al. New England Journal of Medicine. 366(3):216-224. January 19, 2012.
- 7“Bristol-Myers’ Daclatasvir, Asunaprevir Cured 77%: Study”. Bloomberg. Apr 19, 2012.
- 8AASLD: Daclatasvir plus Asunaprevir Rapidly Suppresses HCV in Prior Null Responders. Highleyman, L. HIVandHepatitis.com. 8 November 2011.
- 9High rate of response to BMS HCV drugs in harder-to-treat patients – but interferon-free prospects differ by sub-genotype. Alcorn, K. Aidsmap.com. 12 November 2012.
- 10AASLD 2012: Sofosbuvir + Daclatasvir Dual Regimen Cures Most Patients with HCV Genotypes 1, 2, or 3. Highleyman, L. HIVandHepatitis.com. 15 November 2012.
- 11Mark Sulkowski et al. (January 16, 2014). “Daclatasvir plus Sofosbuvir for Previously Treated or Untreated Chronic HCV Infection”. New England Journal of Medicine. doi:10.1056/NEJMoa1306218.
- 12“www.who.int” (PDF).
| WO2004005264A2 * | 7 Jul 2003 | 15 Jan 2004 | Axxima Pharmaceuticals Ag | Imidazole compounds for the treatment of hepatitis c virus infections |
| WO2008021927A2 * | 9 Aug 2007 | 21 Feb 2008 | Squibb Bristol Myers Co | Hepatitis c virus inhibitors |
| WO2008021928A2 * | 9 Aug 2007 | 21 Feb 2008 | Squibb Bristol Myers Co | Hepatitis c virus inhibitors |
| WO2008021936A2 * | 9 Aug 2007 | 21 Feb 2008 | Squibb Bristol Myers Co | Hepatitis c virus inhibitors |
सुकून उतना ही देना प्रभू, जितने से जिंदगी चल जाये।औकात बस इतनी देना,कि औरों का भला हो जाये।………..P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
सुकून उतना ही देना प्रभू, जितने से जिंदगी चल जाये। औकात बस इतनी देना, कि औरों का भला हो जाये।
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
Googleplus amcrasto@gmail.com
LIONEL MY SONजिंदगी चल जाये।
औकात बस इतनी देना,
कि औरों का भला हो जाये।
//////////
Ozanimod, RPC1063


cas 1306760-87-1
Ozanimod, RPC1063
Receptos, Inc. INNOVATOR
IUPAC/Chemical name: (S)-5-(3-(1-((2-hydroxyethyl)amino)-2,3-dihydro-1H-inden-4-yl)-1,2,4-oxadiazol-5-yl)-2-isopropoxybenzonitrile
Benzonitrile, 5-(3-((1S)-2,3-dihydro-1-((2-hydroxyethyl)amino)-1H-inden-4-yl)-1,2,4-oxadiazol-5-yl)-2-(1-methylethoxy)-
![]()
SMILES: N#CC1=CC(C2=NC(C3=CC=CC4=C3CC[C@@H]4NCCO)=NO2)=CC=C1OC(C)C
C23H24N4O3
Molecular Weight: 404.46
Elemental Analysis: C, 68.30; H, 5.98; N, 13.85; O, 11.87
Ozanimod is a selective sphingosine 1 phosphate receptor modulators and methods which may be useful in the treatment of S1P1-associated diseases. ozanimod, a sphingosine-1-phosphate receptor 1 (S1P1) agonist in Phase III studies as a treatment for ulcerative colitis and multiple sclerosis (MS). Although Novartis’s S1P1 modulator Gilenya has been available to treat MS since 2010,
Relapsing multiple sclerosis (RMS) is a chronic autoimmune disorder of the central nervous system (CNS), characterized by recurrent acute exacerbations (relapses) of neurological dysfunction followed by variable degrees of recovery with clinical stability between relapses (remission). The CNS destruction caused by autoreactive lymphocytes can lead to the clinical symptoms, such as numbness, difficulty walking, visual loss, lack of coordination and muscle weakness, experienced by patients. The disease invariably results in progressive and permanent accumulation of disability and impairment, affecting adults during their most productive years. RMS disproportionately affects women, with its peak onset around age 30. In the past, the treatments for RMS were generally injectable agents with significant side effects. There is a substantial market opportunity for effective oral RMS therapies with improved safety and tolerability profiles.
RPC1063 is a novel, orally administered, once daily, specific and potent modulator of the sphingosine 1-phosphate 1 receptor (S1P1R) pathway. The S1P1R is expressed on white blood cells (lymphocytes), including those responsible for the development of disease. S1P1R modulation causes selective and reversible retention, or sequestration, of circulating lymphocytes in peripheral lymphoid tissue. This sequestration is achieved by modulating cell migration patterns (known as “lymphocyte trafficking”), specifically preventing migration of autoreactive lymphocytes to areas of disease inflammation, which is a major contributor to autoimmune disease. S1P1R modulation may also involve the reduction of lymphocyte migration into the central nervous system (CNS), where certain disease processes take place. This therapeutic approach diminishes the activity of autoreactive lymphocytes that are the underlying cause of many types of autoimmune disease.
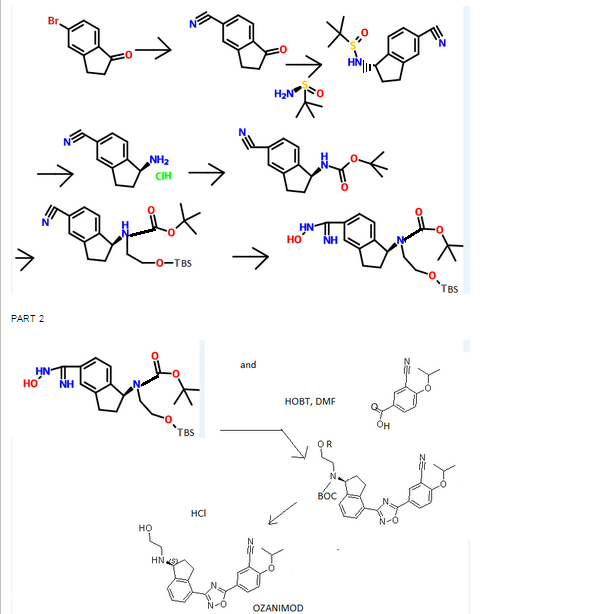
WO 2015066515
Scheme 3:

Reagents: (i) (a) MsCl, pyridine; (b) TsCl, pyridine; (c) NsCl, pyridine; (d) SOCl2, DCM; (e) SOCl2, pyridine, DCM; (f) NaN3, PPh3, CBr4; (ii) (a) DIEA, DMA, HNR’R”; (b) DIEA, NaBr or Nal, DMA, HNR’R”.
Enantiomerically enriched material can be prepared in the same manner outlined in Scheme 3 using the (R)- or (5)-indanols.
Scheme 4:

Reagents: (i) Zn(CN)2, Pd(PPh3)4, NMP; (ii) (i?)-2-methylpropane-2-sulfmamide, Ti(OEt)4, toluene; (iii) NaBH4, THF; (iv) 4M HCl in dioxane, MeOH; (v) Boc20, TEA, DCM; (vi) NH2OH HCl, TEA, EtOH; (vii) HOBt, EDC, substituted benzoic acid, DMF (viii) 4M HCl in dioxane; (ix) (a) R’-LG or R”-LG, where LG represents a leaving group, K2C03, CH3CN; (b) R -C02H or R2-C02H, HOBt, EDC, DMF or R -COCl or R2-COCl, TEA, DCM; (c) R -S02C1 or R3-S02C1, TEA, DCM (d) R2-CHO, HO Ac, NaBH4 or NaCNBH3 or Na(OAc)3BH, MeOH; (e) R -OCOCl or R2-OCOCl, DIEA, DMF; (f) HN(R5R5), CDI, TEA, DCM; (g) H2NS02NH2, Δ, dioxane; (h)
(R)-tert-butyl 2-(tert-butyldimethylsilyloxy)ethyl(4-cyano-2 ,3-dihydro- lH-inden- 1-yl)carbamate INT-16)

Prepared using General Procedure 9. To a flame-dried flask under N2 was added {R)-tert- vXy\ 4-cyano-2,3-dihydro-iH-inden-l-ylcarbamate INT-8 (8.3 g, 32.1 mmol) in anhydrous DMF (240 mL). The reaction mixture was cooled to 0°C and sodium hydride (3.8 g, 60% in oil, 160.6 mmol) was added portionwise. After stirring at 0°C for 2.75 h, (2-bromoethoxy)(tert-butyl)dimethylsilane (16.9 mL, 70.7 mmol) was added. The ice bath was removed after 5 mins and the reaction mixture was allowed to warm to room temperature. After 1.5 h, the reaction mixture was quenched by the slow addition of sat. NaHC03 at 0°C. Once gas evolution was complete the reaction was extracted with EA. The organic layers were washed with water and brine, dried over MgS04 and concentrated. The product was purified by chromatography (EA / hexanes) to provide 10.76 g (80%) of {R)-tert-bvXy\ 2-(tert-butyldimethylsilyloxy)ethyl(4-cyano-2,3-dihydro-iH-inden-l-yl)carbamate INT-16 as a colorless oil. LCMS-ESI (m/z) calculated for C23H36N203Si: 416.6; found 317.2 [M-Boc]+ and 439.0 [M+Na]+, tR = 4.04 min (Method 1). 1H NMR (400 MHz, CDC13) δ 7.46 (d, J = 7.6, 1H), 7.38- 7.32 (m, 1H), 7.33 – 7.18 (m, 1H), 5.69 (s, 0.5 H), 5.19 (s, 0.5 H), 3.70 (ddd, J = 48.8, 26.6, 22.9, 1.5 H), 3.50 – 3.37 (m, 1H), 3.17 (ddd, J = 16.7, 9.4, 2.2, 2H), 2.93 (m, 1.5 H), 2.45 (s, 1H), 2.21 (dd, J = 24.5, 14.5, 1H), 1.56 – 1.37 (bs, 4.5H), 1.22 (bs, 4.5H), 0.87 – 0.74 (m, 9H), -0.04 (dd, J = 26.6, 8.2, 6H). 13C NMR (101 MHz, CDC13) δ 155.03, 146.55, 145.54, 131.16, 130.76, [128.11, 127.03], 117.58, 109.20, 79.88, [63.93, 61.88], [61.44, 60.34], [49.73, 46.76], 30.30, 29.70, 28.44, 28.12, [25.87, 25.62], -5.43. (5)-tert-butyl 2-(tert-butyldimethylsilyloxy)ethyl(4-cyano-2,3-dihydro- 1 H-inden- 1 -yl)carbamate INT- 17 is prepared in an analogous fashion using INT-9.
(R)-tert-butyl 2-(tert-butyldimethylsilyloxy)ethyl (4-(N-hydroxycarbamimidoyl)-2,3-dihydro-lH-inden-l-yl)carbamate (INT-18)

Prepared using General Procedure 3. To a solution of (R)-tert-butyl 2-(tert-butyldimethylsilyloxy)ethyl(4-cyano-2,3-dihydro-iH-inden-l-yl)carbamate INT-16 (12.0 g, 28.9 mmol) in EtOH (120 mL), under an atmosphere of N2 was added hydroxylamine-HCl (6.0 g, 86.5 mmol) and triethylamine (13.4 mL, 9.7 g, 86.5 mmol). The reaction mixture was refluxed at 80°C for 4 h. The reaction mixture was cooled to room temperature and concentrated to dryness and then diluted with DCM (500 mL). The organic layer was washed with NaHC03, water, and brine. The combined organic layers were dried over MgSC^ and concentrated to produce 11.8 g of {R)-tert- vXy\ 2-(tert-butyldimethylsilyloxy) ethyl (4-(N-hydroxycarbamimidoyl)-2,3-dihydro-iH-inden-l-yl)carbamate INT-18 as a white foamy solid, which was used without purification in the next experiment. LCMS-ESI (m/z) calculated for C23H39N304Si: 449.7; found 350.2 [M-Boc]+ and 472.2 [M+Na]+, tR = 1.79 min (Method 1). 1H NMR (400 MHz, CDC13) δ 7.32 (t, J= 7.3 Hz, 1H), 7.21 – 7.07 (m, 2H), 5.69 (s, 0.5 H), 5.19 (s, 0.5 H), 4.89 (s, 2H), 3.85 – 3.50 (m, 2H), 3.31 (ddd, J = 12.2, 9.2, 2.5 Hz, 2H), 3.28 – 3.03 (m, 2H), 3.03 – 2.70 (m, 1H), 2.29 (t, J= 23.6 Hz, 1H), 1.43 (bs, 4.5H), 1.28 (bs, 4.5H), 1.16 – 1.04 (m, 1H), 0.90 – 0.71 (m, 9H), 0.08 – -0.14 (m, 6H). 13C NMR (101 MHz, CDC13) δ 170.99, [156.20, 155.62], 152.38, [144.53, 143.57], [141.82, 141.21], 129.61, 126.78, [126.59, 126.25], [125.02, 124.77], [79.91, 79.68], 64.04, 61.88, [61.57, 61.23], [46.03, 45.76], 30.76, 30.21, [28.53, 28.28], 25.95, [25.66, 25.29], 25.13, [18.28, 17.94], 3.72, -5.34. (S)-tert-butyl 2-(tert-butyldimethylsilyloxy)ethyl (4-(N-hydroxycarbamimidoyl)-2,3-dihydro-lH-inden-l-yl)carbamate INT-19 is prepared in an analogous fashion using INT- 17.
(R)-tert-butyl 2-(tert-butyldimethylsilyloxy)ethyl(4-(5-(3-cyano-4-isopropoxyphenyl)-l,2,4-oxadiazol-3-yl)-2,3-dihydro-lH-inden-l-yl)carbamate and (R)-tert-butyl 4-(5-(3-cyano-4-isopropoxyphenyl)-l,2,4-oxadiazol-3-yl)-2,3-dihydro-lH-inden-l-yl) (2-hydroxethyl) carbamate

Prepared using General Procedure 4. To a solution of 3-cyano-4-isopropoxybenzoic acid (4.5 g, 21.9 mmol) in anhydrous DMF (100 mL) was added HOBt (5.4 g, 40.0 mmol) and EDC (5.6 g, 29.6 mmol). After 1 h, (R)-tert-butyl 2-(tert-butyldimethylsilyloxy)ethyl (4-(N-hydroxycarbamimidoyl)-2,3-dihydro-iH-inden-l-yl)carbamate INT- 18 (11.8 g, 26.3 mmol) was added and the reaction mixture was stirred at room temperature for 2 h. LCMS analysis showed complete conversion to the intermediate, (R)-tert-butyl 2-(tert-butyldimethylsilyloxy) ethyl (4-(N-(3-cyano-4-isopropoxybenzoyloxy) carbamimidoyl)-2,3-dihydro-7H-inden-l-yl)carbamate INT-20. The reaction mixture was then heated to 80°C for 12 h. The reaction mixture was cooled to room temperature and diluted with EA (250 mL). NaHC03 (250 mL) and water (350 mL) were added until all the solids dissolved. The mixture was extracted with EA and the organic layers washed successively with water and brine. The organic layers were dried over MgS04 and concentrated to produce 15.3 g of a mixture of (R)-tert-butyl 2-(tert-butyldimethylsilyloxy)ethyl(4-(5 -(3 -cyano-4-isopropoxyphenyl)-l,2,4-oxadiazol-3-yl)- 2,3-dihydro-iH-inden-l-yl) carbamate INT-21, and the corresponding material without the TBS protecting group, {R)-tert-bvXy\ 4-(5-(3-cyano-4-isopropoxyphenyl)-l,2,4-oxadiazol-3-yl)-2,3-dihydro-iH-inden-l-yl) (2-hydroxy ethyl) carbamate INT-22. The mixture was a brown oil, which could used directly without further purification or purified by chromatography (EA/hexane). INT-21: LCMS-ESI (m/z) calculated for C34H46N405Si: 618.8; found 519.2 [M-Boc]+ and 641.3 [M+Na]+, tR = 7.30 min (Method 1). 1H NMR (400 MHz, CDC13) δ 8.43 (d, J =
2.1, 1H), 8.34 (dd, J = 8.9, 2.2, 1H), 8.07 (d, J= 8.1, 1H), 7.46 – 7.26 (m, 2H), 7.12 (d, J = 9.0, 1H), 5.85 (s, 0.5H), 5.37 (s, 0.5H), 4.80 (dt, J = 12.2, 6.1, 1H), 3.92 – 3.32 (m, 3.5 H), 3.17 (s, 2H), 2.95 (s, 0.5 H), 2.62 – 2.39 (m, 1H), 2.38 – 2.05 (m, 1H), 1.53 (s, 4.5H), 1.48 (d, J = 6.1, 6H), 1.33 – 1.27 (m, 4.5H), 0.94 – 0.77 (m, 9H), 0.01 (d, J = 20.9, 6H). 13C NMR (101 MHz, DMSO) δ 173.02, 169.00, 162.75, [156.22, 155.52], [145.18, 144.12], [143.39, 142.76], 134.16, 133.89, 128.20, [128.01, 127.85], [127.04, 126.90], 126.43, 123.31, 116.93, 115.30, 113.55, 103.96, [79.95, 79.68], 72.73, 67.61, 63.42, [61.91, 61.77], 60.99, 46.11, 31.78, [30.47, 29.87], [28.55, 28.26], 25.93, 21.75, 18.30, 0.00, -5.37. INT-22: LCMS-ESI calculated for C28H32N405: 504.6; found 527.2 [M+Na]+, tR = 2.65 min (Method 1). 1H NMR (400 MHz, CDC13) δ 8.36 (d, J = 2.1, 1H), 8.27 (dd, J = 8.9, 2.2, 1H), 8.03 (d, J = 7.2, 1H), 7.35 – 7.26 (m, 2H), 7.06 (d, J = 9.0, 1H), 5.44 (s, 1H), 4.73 (dt, J= 12.2, 6.1, 1H), 3.64 (s, 2H), 3.44 (ddd, J= 17.5, 9.5,
3.2, 2H), 3.11 (dt, J = 17.4, 8.6, 3H), 2.54 – 2.38 (m, 1H), 2.04 (td, J = 17.6, 8.8, 1H), 1.50 – 1.24 (m, 15H).
(S)-tert-butyl 2-(tert-butyldimethylsilyloxy)ethyl(4-(5-(3-cyano-4-isopropoxyphenyl)-l,2,4-oxadiazol-3-yl)-2,3-dihydro-iH-inden-l-yl)carbamate INT-23 and {S)-tert- vXy\ 4-(5-(3-cyano-4-isopropoxyphenyl)-l,2,4-oxadiazol-3-yl)-2,3-dihydro-iH-inden-l-yl) (2-hydroxyethyl) carbamate INT-24 were made in an analogous fashion.
(S) IS DESIRED CONFIGURATION
……………………………………
(S)-tert-Butanesulfinamide
 CAS 343338-28-3
CAS 343338-28-3
 3-CYANO-4-ISOPROPOXYBENZOIC ACID;3-cyano-4-(propan-2-yloxy)benzoic acid;5-(1-hydroxyvinyl)-2-isopropoxybenzonitrile
3-CYANO-4-ISOPROPOXYBENZOIC ACID;3-cyano-4-(propan-2-yloxy)benzoic acid;5-(1-hydroxyvinyl)-2-isopropoxybenzonitrile
cas 258273-31-3
 (S)-1-Amino-2,3-dihydro-1H-indene-4-carbonitrile hydrochloride
(S)-1-Amino-2,3-dihydro-1H-indene-4-carbonitrile hydrochloride
cas 1306763-57-4 HCl, 1213099-69-4 FREE BASE
4-bromo-2,3-dihydro-1H-inden-1-one
cas 15115-60-3
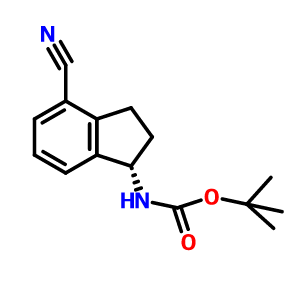 S CONFIGURATION
S CONFIGURATION
Carbamic acid, N-[(1S)-4-cyano-2,3-dihydro-1H-inden-1-yl]-, 1,1-dimethylethyl ester, cas 1306763-31-4
(S) IS DESIRED CONFIGURATION
……………….
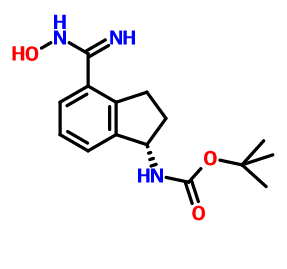
CAS 1306763-70-1, Carbamic acid, N-[(1S)-2,3-dihydro-4-[(hydroxyamino)iminomethyl]-1H-inden-1-yl]-, 1,1-dimethylethyl ester
…………………
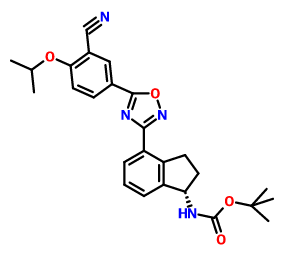
CAS 1306763-71-2, Carbamic acid, N-[(1S)-4-[5-[3-cyano-4-(1-methylethoxy)phenyl]-1,2,4-oxadiazol-3-yl]-2,3-dihydro-1H-inden-1-yl]-, 1,1-dimethylethyl ester
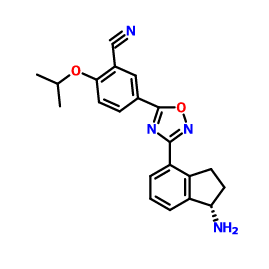
1306760-73-5, Benzonitrile, 5-[3-[(1S)-1-amino-2,3-dihydro-1H-inden-4-yl]-1,2,4-oxadiazol-5-yl]-2-(1-methylethoxy)-
………………………..
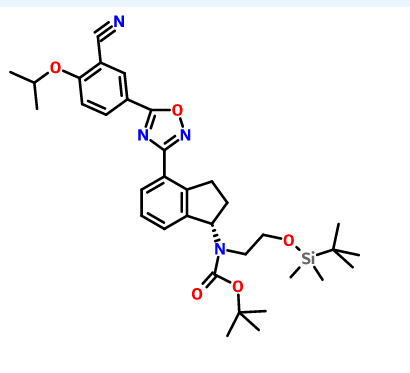
1306763-63-2,
………………….

86864-60-0, (2-Bromoethoxy)dimethyl-tert-butylsilane
Synthesis
……………………………………
WO 2011060392
http://www.google.com/patents/WO2011060392A1?cl=en
(R)-N-(4-cyano-2,3-dihydro-lH-indene-l-ylidene)-2-methylpropane-^
(INT-4

[0304] To l-oxo-2,3-dihydro-/H-indene-4-carbonitrile INT-1 (42.5 g, 0.27 mol) and (R)-2- methylpropane-2-sulfmamide (36.0 g, 0.30 mol) in toluene (530 mL) was added titanium tetraethoxide (84.1 mL, 92.5 g, 0.40 mol) and the reaction mixture was heated at 60°C for 12 h under N2. The crude (R)-N-(4-cyano-2,3-dihydro-lH-indene-l-ylidene)-2-methylpropane- 2-sulfinamide INT-4 was used directly in the next experiment. LCMS-ESI (m/z) calculated for C14Hi6N2OS: 260.3; found 261.1 [M+H]+, tR= 3.19 min.
[0305] (R)-N'((R)-4-cyano-2,3-dihydro-lH nden-l-yl)-2-n thylprop ne-2-sulfirmmide
(INT-5)

[0306] To a flask containing the crude suspension of (R)-N-(4-cyano-2,3-dihydro-iH-indene- l-ylidene)-2-methylpropane-2-sulfrnaniide INT -4 under N2 was added THF (1.0 L) and the reaction mixture cooled to -78°C. Sodium borohydride (40.9 g, 1.08 mol) was added portion- wise over 30 mins. (The internal temperature did not rise during the addition). The reaction mixture was stirred at -78°C for 30 mins, half out of the bath for 30 mins, then warmed to 0°C over 1 h. The 0°C reaction mixture was placed in an ice bath and quenched with brine (100 mL) followed by saturated sodium potassium tartrate (420 mL) and the Ti salts precipitated. The reaction mixture was diluted with EA (1.5 L) and stirred at room temperature overnight. The organic layers were decanted and washed successively with saturated NH4CI, water, and brine. The organic layers were dried over MgS04 and filtered through a pad of MgS04. The filtrate was concentrated to produce 52.9 g of crude (R)-N-((/?)-4-cyano-2,3-dihydro-lH- inden-l-yl)-2-methylpropane-2-sulfmamide INT-5 as a brown oil, which was used directly in the next step. LCMS-ESI (m/z) calculated for C14H18 2OS: 262.3; found 263.1 [M+H]+, tR = 2.99 min. 1H NMR (400 MHz, CDC13) δ 7.89 (d, J = 7.7, 1H), 7.56 (t, J = 6.8, 1H), 7.36 (t, J = 7.7, 1H), 4.97 (q, J = 7.5, 1H), 3.50 (d, J = 7.6, 1H), 3.22 (ddd, J = 16.9, 8.8, 3.9, 1H), 3.01 (dt, J = 22.4, 6.9, 1H), 2.70 – 2.53 (m, 1H), 2.15 – 1.95 (m, 1H), 1.33 – 1.20 (m, 9H).
[0307] (R)-l-amino-2,3-dihydro-lH-indene-l-yl)-4-carbonitrile (T^T-6)

[0308] To crude (R)-N-((R)-4-cyano-2,3-dihydro-iH-inden-l-yl)-2-methylpropane-2- sulfinamide INT-5 (52.9 g, 0.20 mol) in MeOH (200 mL) was added 4N HC1 in dioxane (152.0 mL, 0.60 mol) and the resulting yellow suspension was stirred at room temperature for 1.5 h. The crude reaction mixture was diluted with MeOH (500 mL) and filtered to remove some Ti by-products. The filtrate was concentrated and the resulting solid refluxed in acetonitrile (500 mL). The resulting white solid was collected to produce 13.0 g (31% over 3 steps) of the HC1 salt of (R)-l-amino-2,3-dihydro-7H-indene-l-yl)-4-carbonitrile INT-6. LCMS-ESI (m/z) calculated for Ci0H10N2: 158.2; found 142.0 [M-NH2]+, fR = 0.84 min. Ή NMR (400 MHz, DMSO) δ 8.61 (s, 3H), 7.96 (d, J = 7.7, 1H), 7.83 (d, J = 7.5, 1H), 7.52 (t, J = 7.7, 1H), 4.80 (s, 1H), 3.23 (ddd, J = 16.6, 8.7, 5.2, 1H), 3.05 (ddd, J = 16.6, 8.6, 6.3, 1H), 2.62 – 2.51 (m, 1H), 2.15 – 2.01 (m, 1H). 13C NMR (101 MHz, DMSO) δ 148.09, 141.15, 132.48, 130.32, 127.89, 117.27, 108.05, 54.36, 39.08, 29.64. The free base can be prepared by extraction with IN NaHC03and DCM. LCMS-ESI (m/z) calculated for Ci0H10N2: 158.2; found 142.0 [M-NH2]+, tR = 0.83 min. 1H NMR (400 MHz, CDC13) δ 7.52 – 7.38 (m, 2H), 7.23 (dd, 7 = 17.4, 9.8, 1H), 4.35 (t, J = 7.6, 1H), 3.11 (ddd, 7 = 16.8, 8.7, 3.2, 1H), 2.89 (dt, J = 16.9, 8.5, 1H), 2.53 (dddd, J = 12.8, 8.1, 7.3, 3.2, 1H), 1.70 (dtd, J = 12.8, 8.8, 8.0, 1H). 13C NMR (101 MHz, DMSO) δ 150.16, 146.67, 130.19, 128.74, 127.38, 117.77, 107.42, 56.86, 38.86, 29.14. Chiral HPLC: (R)-l-amino-2,3-dihydro-7H-indene-l-yl)-4-carbonitrile was eluted using 5% EtOH in hexanes, plus 0.05% TEA: 95% ee, ¾ = 23.02 min. The (S)- enantiomer INT-7 was prepared in an analogous fashion using (5)-2-methylpropane-2- sulfinamide. tR for (S)-enantiomer = 20.17 min.
[0309] (R)-tert-butyl 4-cyano-2,3-dihydro-lH-inden-l-ylcarbamate (INT-8)

[0310] To ( ?)-l-amino-2,3-dihydro-/H-indene-l-yl)-4-carbonitrile HC1 INT-6 (11.6 g, 59.6 mmol) in DCM (100 mL) at 0°C was added TEA (12.0 mL, 131.0 mmol). To the resulting solution was added a solution of Boc anhydride (14.3 g, 65.6 mmol) in DCM (30 mL) and the reaction mixture stirred at room temperature for 1.5 h. The reaction mixture was washed with brine, and the organic layers were dried over MgS04 and filtered. Additional DCM was added to a total volume of 250 mL and Norit (4.5 g) was added. The product was refluxed for 15 mins and the hot mixture filtered through a pad of celite / silica. The filtrate was concentrated and recrystallized from EA (50 mL) and hexane (150 mL) to produce 12.93 g (84%) of (/?)-tert-butyl 4-cyano-2,3-dihydro-iH-inden-l-ylcarbamate INT-8 as an off-white solid. LCMS-ESI (m/z) calculated for C15H18N202: 258.3; found 281.1 [M+Na]+, tR = 3.45 min. Elemental Analysis determined for C^H^^O^ C calculated = 69.74%; found = 69.98%. H calculated = 7.02%; found = 7.14%. N calculated = 10.84%; found = 10.89%. 1H NMR (400 MHz, CDC13) δ 7.64 – 7.49 (m, 2H), 7.34 (dt, / = 7.7, 3.8, 1H), 5.36 – 5.20 (m, 1H), 4.78 (d, J = 6.8, 1H), 3.20 (ddd, J = 16.9, 8.9, 3.3, 1H), 3.02 (dt, J = 25.4, 8.4, 1H), 2.82 – 2.53 (m, 1H), 1.88 (dq, J = 13.2, 8.6, 1H), 1.55 – 1.44 (m, 9H). 13C NMR (101 MHz, DMSO) δ 155.52, 146.68, 146.32, 130.89, 128.70, 127.63, 117.51, 107.76, 77.98, 55.09, 31.88, 29.11, 28.19. Chiral HPLC: (R)-tert-butyl 4-cyano-2,3-dihydro-lH-inden-l- ylcarbamate was eluted using 2.5% EtOH in hexanes: >99.9% ee, tR = 19.36 min. The (5)- enantiomer INT-9 was prepared in an analogous fashion using (S)-l-amino-2,3-dihydro-7H- indene-l-yl)-4-carbonitrile HC1. tR for (5)-enantiomer = 28.98 min.
General Procedure 3. Preparation oflndane Amide Oximes
[0311] To (R)- or (5)-tert-butyl 4-cyano-2,3-dihydro-7H-inden-l-ylcarbamate (1 eq) in EtOH
(0.56 M) was added hydroxylamine hydrochloride (3 eq) and TEA (3 eq) and the reaction mixture heated at 85°C for 1-2 h. The organic soluble amide oximes were isolated by removal of the solvent and partitioning between water and DCM. The water soluble amide oximes were chromatographed or used directly in the cyclization. Pure amide oximes can be obtained by recrystallization from alcoholic solvents.
[0312] (R)-tert-butyl 4-(N -hydroxy carbamimidoyl )-2, 3-dihydro-lH-inden-l -ylcarbamate
(INT-10)

[0313] Prepared using General Procedure 3. To (R)-tert-butyl 4-cyano-2,3-dihydro-iH- inden-1 -ylcarbamate INT-8 (15.0 g, 58.2 mmol) in EtOH (100 niL) was added hydroxylamine hydrochloride (12.1 g, 174.2 mmol) and TEA (17.6 mL, 174.2 mmol) and the reaction mixture heated at 85°C for 2 h. The solvents were removed and the resulting white solid was partitioned between water and DCM. The organic layers were dried over Na2S04, concentrated, and recrystallized from isopropanol (50 mL) to afford 14.4 g (85%) of (R)-tert- butyl 4-(N-hydroxycarbaniimidoyl)-2,3-dihydro-iH-inden-l-ylcarbamate INT-10 as white crystalline solid. LCMS-ESI (m/z) calculated for C15H21N303: 291.4; found 292.1 [M+H]+, ¾ = 2.04 min. 1H NMR (400 MHz, DMSO) δ 9.53 (s, 1H), 7.38 – 7.32 (m, 1H), 7.32 – 7.12 (m, 3H), 5.68 (s, 2H), 4.97 (q, J = 8.5, 1H), 3.07 (ddd, J = 16.6, 8.7, 2.6, 1H), 2.86 (dt, J = 16.8, 8.4, 1H), 2.30 (ddd, J = 12.6, 7.6, 3.6, 1H), 1.75 (dq, J = 12.3, 9.0, 1H), 1.44 (s, 9H). General Procedure 4. Cyclization to Indane Oxadiazole Amines
[0314] A solution of the appropriate acid (1 eq), HOBt (1.3 eq), and EDC (1.3 eq) in DMF
(0.08 M in acid) was stirred at room temperature under an atmosphere of N2. After the complete formation of the HOBt- acid complex (1-3 h), the (R)- or (5)-amide oxime (1.1 eq) was added to the mixture. After complete formation of the coupled intermediate (ca. 0.5- 2 h), the mixture was heated to 75-95°C until the cyclization was complete (8-12 h). The reaction mixture was diluted with saturated NaHC03 and extracted with EA. The combined organic extracts were dried, concentrated, and either purified by chromatography (EA/hexanes) or taken on directly. The oxadiazole was treated with HC1 (5N in dioxane, 5 eq) at 50-60°C for 0.5-6 h. The reaction mixture could be extracted (DCM /NaHC03), or the resulting HC1 salt concentrated, suspended in Et20, and collected. Pure indane amines can be obtained by recrystallization from alcoholic solvents or by chromatography.
( R)-tert-butyl 4-(5-( 3-cyano-4-isopropoxyphenyl)-l,2, 4-oxadiazol-3-yl )-2,3-dihydro-lH- inden-l-ylcarbamate (INT- 12)

[0315] Prepared using General Procedure 4. To a solution of 3-cyano-4-isopropoxybenzoic acid (7.74 g, 37.7 mmol) in DMF (50 mL) was added HOBt (6.02 g, 44.6 mmol) and EDC (8.53 g, 44.6 mmol) at room temperature. The reaction was stirred for 2 h until complete formation of the HOBt-acid complex. (R)-tert-butyl 4-(N-hydroxycarbamimidoyl)-2,3- dihydro-iH-inden-l-ylcarbamate INT-10 (10.0 g, 34.3 mmol) was added and the reaction mixture stirred at room temperature for 2 h until the formation of INT-11, (R)-tert-butyl 4- (N-(3-cyano-4-isopropoxybenzolyloxy) carbamimidoyl)-2,3-dihydro-iH-inden-l- ylcarbamate. The mixture was partitioned between EA and NaHC03 and the organic layer was collected and dried over MgS04. INT-11 (16.3 g, 34.0 mmol) was re-dissolved in DMF (50 mL) and the mixture was heated to 95°C for 12 hrs. The reaction was diluted with NaHC03 (200 mL) and extracted with EA (3 X 50 mL). The organic layer was dried over Na2S04and concentrated under reduced pressure to produce 12.8 g (81%) of (R)-tert-butyl 4- (5-(3-cyano-4-isopropoxyphenyl)- 1 ,2,4-oxadiazol-3-yl)-2,3-dihydro-iH-inden- 1-ylcarbamate INT-12 as a light brown solid and used without further purification in the next step. LCMS- ESI (m/z) calculated for C26H28N404: 460.5; found 483.2 [M+Na]+, tR = 4.25 min. Ή NMR (400 MHz, CDCI3) δ 8.43 (d, J = 2.1, 1H), 8.34 (dd, J = 8.9, 2.2, 1H), 8.09 (d, J = 7.6, 1H), 7.51 (d, / = 7.5, 1H), 7.39 (t, J = 7.6, 1H), 7.12 (d, J = 9.0, 1H), 5.28 (d, J = 8.2, 1H), 4.80 (hept, J = 6.0, 1H), 3.47 (ddd, J = 17.4, 8.9, 3.5, 1H), 3.27 – 3.03 (m, 1H), 2.68 (d, J = 8.7, 1H), 1.87 (td, J = 16.7, 8.5, 1H), 1.53 – 1.43 (m, 15H). 13C NMR (101 MHz, CDC13) δ 173.00, 168.82, 162.70, 155.68, 145.31, 142.96, 134.05, 133.83, 128.25, 127.21, 126.79, 123.09, 116.78, 115.24, 113.52, 103.87, 79.52, 72.70, 55.72, 33.86, 31.47, 28.39, 21.70. Chiral HPLC: (R)-tert-butyl 4-(5-(3-cyano-4-isopropoxyphenyl)-l,2,4-oxadiazol-3-yl)-2,3- dihydro-lH-inden-l-ylcarbamate was eluted using 20% /-PrOH in hexanes: >99.9% ee, ?R = 13.33 min. The (5)-enantiomer INT-13 was prepared in an analogous fashion using (S)-tert- butyl 4-cyano-2,3-dihydro-iH-inden-l-ylcarbamate using General Procedures 3 and 4 (tR for (Syenantiomer = 16.31 min).
( R )-5-( 3-(l -amino-2,3-dihydro-lH-inden-4-yl)-l,2, 4-oxadiazol-5-yl)-2-isopropoxy- benzonitrile h drochloride (Compound 49)

[0317] To (R)-tert-butyl 4-(5-(3-cyano-4-isopropoxyphenyl)-l,2,4-oxadiazol-3-yl)-2,3- dihydro-iH-inden-l-ylcarbamate(12.8 g, 27.8 mmol) in dioxane (200 mL) was added 4N HCl in dioxane (69 mL). The solution was heated to 55°C for 1 h, and product precipitated. Dioxane was removed and the resulting solid suspended in ether and collected. The material was recrystallized from MeOH (200 mL) to produce 8.11 g (81%) of (R)-5-(3-(l-amino-2,3- dihydro-iH-inden-4-yl)-l,2,4-oxadiazol-5-yl)-2-isopropoxybenzonitrile 49 as the HCl salt. LCMS-ESI (m/z): calcd for: C21H20N4O2: 360.4; found 383.2 [M+Na]+, tR = 2.49 min. Elemental Analysis and NMR spectra determined for C21H21N402C1 * 0.5 H20; C calculated = 62.14%; found = 62.25%. H calculated = 5.46%; found = 5.30%. N calculated = 13.80%; found = 13.84%. CI calculated = 8.73%; found = 8.34%. 1H NMR (400 MHz, DMSO) δ 8.71 (s, 3H), 8.49 (d, J = 2.3, 1H), 8.39 (dd, J = 9.0, 2.3, 1H), 8.11 (d, J = 7.6, 1H), 7.91 (d, J = 7.6, 1H), 7.55 (t, J = 8.5, 2H), 4.97 (hept, J = 6.1, 1H), 4.80 (s, 1H), 3.47 (ddd, J = 17.4, 8.7, 5.3, 1H), 3.23 (ddd, 7 = 17.4, 8.6, 6.4, 1H), 2.55 (ddd, 7 = 13.7, 8.3, 3.2, 1H), 2.22 – 1.97 (m, 1H), 1.38 (d, J = 6.0, 6H). 13C NMR (101 MHz, CDC13) δ 173.28, 167.98, 162.53, 143.69, 141.29, 134.59, 133.80, 128.93, 128.11, 127.55, 122.72, 115.87, 115.24, 114.91, 102.46, 72.54, 54.38, 31.51, 29.91, 21.47. Chiral HPLC of the free base: (R)-5-(3-(l-amino-2,3- dihydro-lH-inden-4-yl)-l,2,4-oxadiazol-5-yl)-2-isopropoxy benzonitrile was eluted using 15% i‘-PrOH in hexanes plus 0.3% DEA: > 99.9% ee, tR = 30.80 min.
(S)- 5-(3-(l-amino-2,3- dihydro-lH-inden-4-yl)-l,2,4-oxadiazol-5-yl)-2-isopropoxy-benzonitrile 50 was prepared in an analogous fashion from (S)-tert-b tyl 4-cyano-2,3-dihydro-lH-inden-l-ylcarbamate: >99.9% ee, tR for (5)-enantiomer = 28.58 min.
(R)-tert-butyl 2-(tert-butyldimethylsilyloxy)ethyl(4-cyano-2,3-dihydro-lH-inden-l- yl)carbamate ( -16)

[0366] Prepared using General Procedure 9. To a flame-dried flask under N2 was added (R)- tert-butyl 4-cyano-2,3-dihydro-iH-inden-l-ylcarbamate INT-8 (8.3 g, 32.1 mmol) in anhydrous DMF (240 mL). The reaction mixture was cooled to 0°C and sodium hydride (3.8 g, 60% in oil, 160.6 mmol) was added portionwise. After stirring at 0°C for 2.75 h, (2- bromoethoxy)(½rt-butyl)dimethylsilane (16.9 mL, 70.7 mmol) was added. The ice bath was removed after 5 mins and the reaction mixture was allowed to warm to room temperature. After 1.5 h, the reaction mixture was quenched by the slow addition of sat. NaHC03at 0°C. Once gas evolution was complete the reaction was extracted with EA. The organic layers were washed with water and brine, dried over MgS04 and concentrated. The product was purified by chromatography (EA / hexanes) to provide 10.76 g (80%) of (R)-teri-butyl 2-(tert- butyldimemylsilyloxy)emyl(4-cyano-2,3-dihydro-iH-inden-l-yl)carbamate INT-16 as a colorless oil. LCMS-ESI (m/z) calculated for C23H36N203Si: 416.6; found 317.2 [M-Boc]+ and 439.0 [M+Na]+, tR = 4.04 min (Method 1). 1H NMR (400 MHz, CDC13) δ 7.46 (d, J = 7.6, 1H), 7.38- 7.32 (m, 1H), 7.33 – 7.18 (m, 1H), 5.69 (s, 0.5 H), 5.19 (s, 0.5 H), 3.70 (ddd, J = 48.8, 26.6, 22.9, 1.5 H), 3.50 – 3.37 (m, 1H), 3.17 (ddd, J = 16.7, 9.4, 2.2, 2H), 2.93 (m, 1.5 H), 2.45 (s, 1H), 2.21 (dd, J = 24.5, 14.5, 1H), 1.56 – 1.37 (bs, 4.5H), 1.22 (bs, 4.5H), 0.87 – 0.74 (m, 9H), -0.04 (dd, J = 26.6, 8.2, 6H).13C NMR (101 MHz, CDC13) δ 155.03, 146.55, 145.54, 131.16, 130.76, [128.11, 127.03], 117.58, 109.20, 79.88, [63.93, 61.88], [61.44, 60.34], [49.73, 46.76], 30.30, 29.70, 28.44, 28.12, [25.87, 25.62], -5.43. (5)-tert-butyl 2-(tert- butyldimemylsilyloxy)emyl(4-cyano-2,3-dihydro-lH-inden-l-yl)carbamate INT-17 is prepared in an analogous fashion using INT -9. [0367] (R)-tert-butyl 2-(tert-butyldimethylsilyloxy)ethyl (4-(N-hydroxycarbamimidoyl)-2,3- dihydro-1 H-inden-1 -yl)carbamate (INT-18)

[0368] Prepared using General Procedure 3. To a solution of (R)-iert-butyl 2-(tert- butyldimemylsilyloxy)ethyl(4-cyano-2,3-dmydro-/H-inden-l-yl)carbamate INT-16 (12.0 g, 28.9 mmol) in EtOH (120 mL), under an atmosphere of N2 was added hydroxylamine-HCl (6.0 g, 86.5 mmol) and triemylamine (13.4 mL, 9.7 g, 86.5 mmol). The reaction mixture was refluxed at 80°C for 4 h. The reaction mixture was cooled to room temperature and concentrated to dryness and then diluted with DCM (500 mL). The organic layer was washed with NaHC03, water, and brine. The combined organic layers were dried over MgS04 and concentrated to produce 11.8 g of (R)-tert-butyl 2-(tert-butyldimethylsilyloxy) ethyl (4-(N- hydroxycarbamimidoyl)-2,3-dihydro-iH-inden-l-yl)carbamate INT-18 as a white foamy solid, which was used without purification in the next experiment. LCMS-ESI (m/z) calculated for C23H39N304Si: 449.7; found 350.2 [M-Boc]+ and 472.2 [M+Na]+, ¾ = 1.79 min (Method 1). 1H NMR (400 MHz, CDC13) δ 7.32 (t, / = 7.3 Hz, 1H), 7.21 – 7.07 (m, 2H), 5.69 (s, 0.5 H), 5.19 (s, 0.5 H), 4.89 (s, 2H), 3.85 – 3.50 (m, 2H), 3.31 (ddd, / = 12.2, 9.2, 2.5 Hz, 2H), 3.28 – 3.03 (m, 2H), 3.03 – 2.70 (m, 1H), 2.29 (t, J = 23.6 Hz, 1H), 1.43 (bs, 4.5H), 1.28 (bs, 4.5H), 1.16 – 1.04 (m, 1H), 0.90 – 0.71 (m, 9H), 0.08 – -0.14 (m, 6H). 13C NMR (101 MHz, CDC13) 6 170.99, [156.20, 155.62], 152.38, [144.53, 143.57], [141.82, 141.21], 129.61, 126.78, [126.59, 126.25], [125.02, 124.77], [79.91, 79.68], 64.04, 61.88, [61.57, 61.23], [46.03, 45.76], 30.76, 30.21, [28.53, 28.28], 25.95, [25.66, 25.29], 25.13, [18.28, 17.94], 3.72, -5.34. ^-tert-butyl 2-(tert-butyldimethylsilyloxy)ethyl (4-(N- hydroxycarbamimidoyl)-2,3-dihydro-lH-inden-l-yl)carbamate INT-19 is prepared in an analogous fashion using INT-17. [0369] (R)-tert-butyl 2-( tert-butyldimethylsilyloxy)ethyl( 4-( 5-( 3-cyano-4-isopropoxyphenyl)- l,2,4-oxadiazol-3-yl)-2,3-dihydro-lH-inden-l-yl)carbamate and (R)-tert-butyl 4-(5-(3-cyano- 4-isopropoxyphenyl )-l,2, 4-oxadiazol-3-yl)-2,3-dihydro-lH-inden-l-yl) (2-hydroxethyl) carbamate

[0370] Prepared using General Procedure 4. To a solution of 3-cyano-4-isopropoxybenzoic acid (4.5 g, 21.9 mmol) in anhydrous DMF (100 mL) was added HOBt (5.4 g, 40.0 mmol) and EDC (5.6 g, 29.6 mmol). After 1 h, {R)-tert-buiy\ 2-(tert-butyldimethylsilyloxy)ethyl (4- (N-hydroxycarbamimidoyl)-2,3-dihydro-iH-inden-l-yl)carbamate INT-18 (11.8 g, 26.3 mmol) was added and the reaction mixture was stirred at room temperature for 2 h. LCMS analysis showed complete conversion to the intermediate, (R)-tert-b\xty\ 2-(tert- butyldimethylsilyloxy) ethyl (4-(N-(3-cyano-4-isopropoxybenzoyloxy) carbamimidoyl)-2,3- dihydro-7H-inden-l-yl)carbamate INT-20. The reaction mixture was then heated to 80°C for 12 h. The reaction mixture was cooled to room temperature and diluted with EA (250 mL). NaHC03 (250 mL) and water (350 mL) were added until all the solids dissolved. The mixture was extracted with EA and the organic layers washed successively with water and brine. The organic layers were dried over MgS04 and concentrated to produce 15.3 g of a mixture of (R)-tert-butyl 2-(tert-butyldimethylsilyloxy)ethyl(4-(5-(3-cyano-4-isopropoxyphenyl)- 1 ,2,4- oxadiazol-3-yl)- 2,3-dihydro-iH-inden-l-yl) carbamate INT-21, and the corresponding material without the TBS protecting group, (R)-tert-butyl 4-(5-(3-cyano-4- isopropoxyphenyl)-l,2,4-oxadiazol-3-yl)-2,3-dihydro-iH-inden-l-yl) (2-hydroxyethyl) carbamate INT -22. The mixture was a brown oil, which could used directly without further purification or purified by chromatography (EA hexane). INT-21: LCMS-ESI (m/z) calculated for C34H46N4O5S1: 618.8; found 519.2 [M-Boc]+ and 641.3 [M+Na]+, tR = 7.30 min (Method 1). Ή NMR (400 MHz, CDC13) δ 8.43 (d, J = 2.1, 1H), 8.34 (dd, J = 8.9, 2.2, 1H), 8.07 (d, J = 8.1, 1H), 7.46 – 7.26 (m, 2H), 7.12 (d, / = 9.0, 1H), 5.85 (s, 0.5H), 5.37 (s, 0.5H), 4.80 (dt, J = 12.2, 6.1, 1H), 3.92 – 3.32 (m, 3.5 H), 3.17 (s, 2H), 2.95 (s, 0.5 H), 2.62 – 2.39 (m, 1H), 2.38 – 2.05 (m, 1H), 1.53 (s, 4.5H), 1.48 (d, J = 6.1, 6H), 1.33 – 1.27 (m, 4.5H), 0.94 – 0.77 (m, 9H), 0.01 (d, J = 20.9, 6H). 1C NMR (101 MHz, DMSO) δ 173.02, 169.00, 162.75, [156.22, 155.52], [145.18, 144.12], [143.39, 142.76], 134.16, 133.89, 128.20, [128.01, 127.85], [127.04, 126.90], 126.43, 123.31, 116.93, 115.30, 113.55, 103.96, [79.95, 79.68], 72.73, 67.61, 63.42, [61.91, 61.77], 60.99, 46.11, 31.78, [30.47, 29.87], [28.55, 28.26], 25.93, 21.75, 18.30, 0.00, -5.37. INT-22: LCMS-ESI calculated for C28H32N4Os: 504.6; found 527.2 [M+Na]+, tR = 2.65 min (Method 1). Ή NMR (400 MHz, CDC13) δ 8.36 (d, J = 2.1, 1H), 8.27 (dd, / = 8.9, 2.2, 1H), 8.03 (d, / = 7.2, 1H), 7.35 – 7.26 (m, 2H), 7.06 (d, / = 9.0, 1H), 5.44 (s, 1H), 4.73 (dt, J = 12.2, 6.1, 1H), 3.64 (s, 2H), 3.44 (ddd, / = 17.5, 9.5, 3.2, 2H), 3.11 (dt, J = 17.4, 8.6, 3H), 2.54 – 2.38 (m, 1H), 2.04 (td, J = 17.6, 8.8, 1H), 1.50 – 1.24 (m, 15H). (5 -teri-butyl 2-(tert-butyldimethylsilyloxy)ethyl(4-(5-(3-cyano-4- isopropoxyphenyl)-l,2,4-oxadiazol-3-yl)-2,3-dihydro-iH-inden-l-yl)carbamate INT-23 and (S)-terf-butyl 4-(5-(3-cyano-4-isopropoxyphenyl)-l,2,4-oxadiazol-3-yl)-2,3-dihydro-iH- inden-l-yl) (2-hydroxyethyl) carbamate INT -24 were made in an analogous fashion.
[0371] (R)-5-(3-(l-(2-hydroxyethylamino)-2,3-dihydro-lH-inden-4-yl)-l,2,4-oxadi zol-^ 2-isopropoxybenzonitrile (Compound 85)

[0372] To a solution of (R)-tert-butyl 2-(tert-butyldimethylsilyloxy)ethyl(4-(5-(3-cyano-4- isopropoxyphenyl)-l,2,4-oxadiazol-3-yl)-2,3-dihydro-7H-inden-l-yl)carbamate INT-21 and (R)-tert-butyl 4-(5-(3-cyano-4-isopropoxyphenyl)- 1 ,2,4-oxadiazol-3-yl)-2,3-dihydro-iH- inden-l-yl) (2-hydroxethyl) carbamate INT-22 (13.9 g, 27.5 mmol) in dioxane (70 mL) at 0°C was added 4N HCl in dioxane (68.8 g, 275.4 mmol). The reaction mixture was warmed to room temperature and then heated to 50°C for 1 h. The resulting suspension was cooled to room temperature and Et20 (75 mL) was added. The precipitate was collected by filtration, washed with Et20 and dried to produce 10.5 g of an off-white solid. The HCl salt was recrystallized from MeOH (165 mL) to produce 5.98 g (56% overall yield from (R)-tert-butyl 2-(tert-butyldimethylsilyloxy)ethyl(4-cyano-2,3-dihydro-iH-inden-l-yl) carbamate) of (R)-5- (3-(l-(2-hydroxyethylamino)-2,3-dihydro-iH-inden-4-yl)-l,2,4-oxadiazol-5-yl)-2- isopropoxybenzonitrile 85 as a white solid. LCMS-ESI (m/z) calculated for C23H24N403: 404.5; found 405.4 [M+H]+, tR = 2.44 min. Ή NMR (400 MHz, DMSO) 5 9.25 (s, 2H), 8.53 (d, J = 2.3, 1H), 8.42 (dd, J = 9.0, 2.3, 1H), 8.17 (d, J = 7.7, 1H), 7.97 (d, J = 7.6, 1H), 7.63 – 7.50 (m, 2H), 5.28 (t, J = 5.0, 1H), 4.99 (hept, J = 6.1, 1H), 4.92 (s, 1H), 3.72 (q, J = 5.2, 2H), 3.57 – 3.43 (m, 1H), 3.27 (ddd, J = 17.6, 9.1, 5.0, 1H), 3.15-2.85 (m, J = 24.2, 2H), 2.53 (dtd, J = 9.0, 5.5, 5.3, 3.6, 1H), 2.30 (ddd, J = 13.4, 8.9, 4.6, 1H), 1.39 (d, J = 6.0, 6H). 13C NMR (101 MHz, DMSO) 6 173.25, 167.86, 162.47, 144.56, 139.13, 134.53, 133.77, 129.30, 128.93, 127.45, 122.83, 115.79, 115.15, 114.84, 102.40, 72.46, 61.04, 56.51, 46.38, 31.53, 27.74, 21.37. Elemental analysis for C23H25N403C1: C calc. = 62.65%; found = 62.73%; H calc. = 5.71%; found = 5.60%; N calc. = 12.71%; found = 12.64%; CI calc. = 8.04%; found = 8.16%. Chiral HRLC of the free base: (R)-5-(3-(l-(2-hydroxyemylamino)-2,3-dihydro-iH- inden-4-yl)-l,2,4-oxadiazol-5-yl)-2-isopropoxy – benzo-nitrile was eluted using 10% i‘-PrOH in hexanes plus 0.3% DEA: >99.9% ee, tR = 37.72 min.
(S)-5-(3-(l-(2-hydroxyethylamino)- 2,3-dihydro-iH-inden-4-yl)-l,2,4-oxadiazol-5-yl) -2-isopropoxy benzonitrile 86 was obtained in analogous fashion from (S)-tert-butyl 2-(tert-butyldimethylsilyloxy)ethyl(4-(5-(3- cyano-4-isopropoxyphenyl)- 1 ,2,4-oxadiazol-3-yl)-2, 3-dihydro-iH-inden- 1 -yl)carbamate INT-23 and (S)-tert-butyl 4-(5-(3-cyano-4-isopropoxyphenyl)-l,2,4-oxadiazol-3-yl)-2,3- dihydro-iH-inden-l-yl) (2-hydroxyethyl) carbamate INT-24: >99.9% ee, tR for (5)- enantiomer = 35.86 min.
(S) IS DESIRED CONFIGURATION
THE SYNTHESIS IS SUMMARISED BELOW
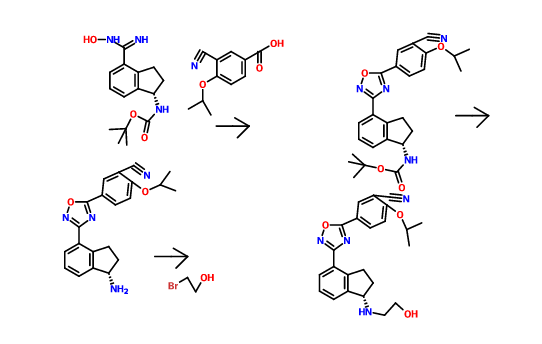
COSY PREDICT
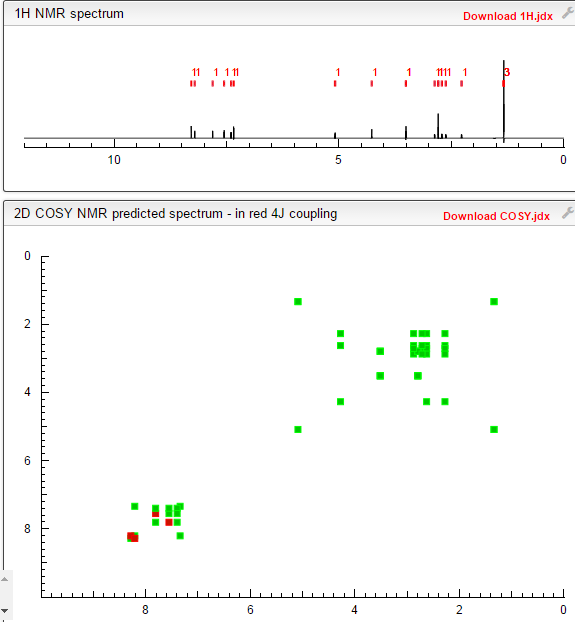
1H NMR PREDICT
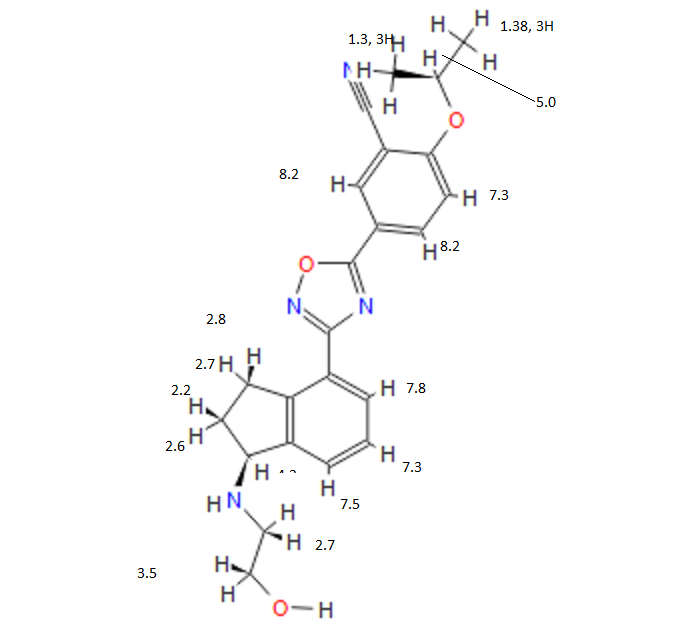
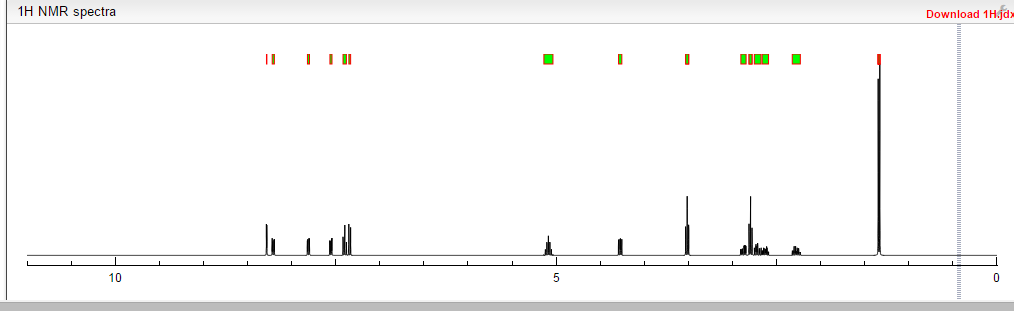
13C NMR PREDICT
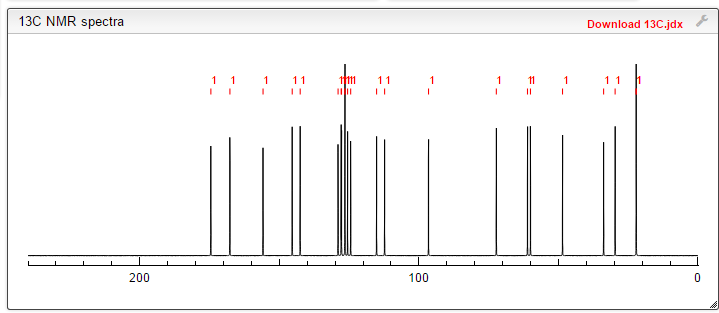
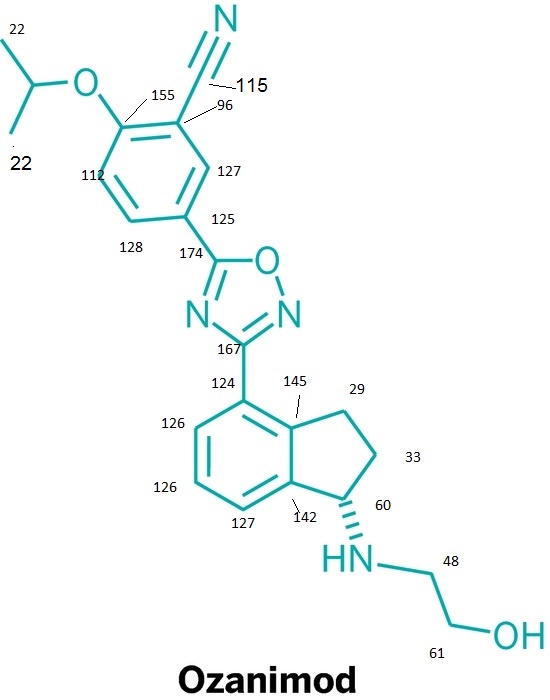
note——-(CH3 )2CH-O-AR appears at 72 ppm

////////
RIVAROXABAN 利伐沙班 ريفاروكسابان Ривароксабан SPECTRAL VISIT

 5-Chloro-N-{[(5S)-2-oxo-3-[4-(3-oxo-4-morpholinophenyl]oxazolidin-5-yl]methyl} thiophene-2-carboxamide
5-Chloro-N-{[(5S)-2-oxo-3-[4-(3-oxo-4-morpholinophenyl]oxazolidin-5-yl]methyl} thiophene-2-carboxamide
Originator and Manufacturer:Bayer
Marketer in the US: Johnson & Johnson
Sales: $1.3 billion (2013)
In September 2008, Health Canada granted marketing authorization for rivaroxaban for the prevention of venous thromboembolism(VTE) in people who have undergone elective total hip replacement or total knee replacement surgery.[8]
In September 2008, the European Commission granted marketing authorization of rivaroxaban for the prevention of venous thromboembolism in adults undergoing elective hip and knee replacement surgery.[9]
On July 1, 2011, the U.S. Food and Drug Administration (FDA) approved rivaroxaban for prophylaxis of deep vein thrombosis (DVT), which may lead to pulmonary embolism (PE), in adults undergoing hip and knee replacement surgery.[5]
On November 4, 2011, the U.S. FDA approved rivaroxaban for stroke prophylaxis in patients with non-valvular atrial fibrillation.

The drug compound having the adopted name “Rivaroxaban” has chemical name, 5-chloro-N-({(5S)-2-oxo-3-[4-(3-oxo-4-morpholinyl)phenyl]-l,3-oxazolidin-5- yljmethyl)-2-thiophenecarboxamide; and has the structural formula I,

Formula I
The commercial pharmaceutical product XARELTO® tablets, contains rivaroxaban as active ingredient. Rivaroxaban is a factor Xa inhibitor useful as oral anticoagulant. Rivaroxaban can be used for the prevention and treatment of various thromboembolic diseases, in particular of deep vein thrombosis (DVT), pulmonary embolism (PE), myocardial infract, angina pectoris and restenoses after angioplasty or aortocoronary bypass, cerebral stroke,
transitory ischemic attacks, and peripheral arterial occlusive diseases.
U.S. Patent No. 7, 157,456 describes Rivaroxaban and process for the preparation thereof. The process of US ‘456 for rivaroxaban involves reaction of 2-[(2S)-2-oxiranylmethyl]-lH-isoindole-l,3(2H)-dione with 4-(4-aminophenyl)-3-morpholinone to provide 2-((2R)-2-hydroxy-3- { [4-(3-oxo-4-morpholiny)phenyl]amino Jpropyl)- lH-isoindole- 1 ,3(2H)-dione, which on cyclization using Ν,Ν-carbonyl diimidazole to afford 2-({5S)-2-Oxo-3-[4-(3-oxo-4-morpholiny)phenyl]-l,3-oxazolidin-5-yl}methyl)-lH-isoindole-l,3(2H)-dione, which on reacted with methylamine followed by reaction with 5-chlorothiophene-2-carbonyl chloride to provide Rivaroxaban.
Various processes for the preparation of rivaroxaban, its intermediates, and related compounds are disclosed in U.S. Patent Nos. 7,585,860; 7,351,823, 7,816,355, and 8,101,609; patent application Nos. WO 2011/012321, WO 2012/156983, WO 2012/153155, WO 2013/053739, WO 2013/098833, WO 2013/156936, WO 2013/152168, WO 2013/120464, WO 2013/164833, US 2012/0283434 and US 2013/184457; and J. Med. Chem. 2005, 48, 5900-5908.

PAPER CONTAING SPECTRAL DATA
a College of Pharmaceutical Sciences, Zhejiang University of Technology, Hangzhou 310032, P. R. China
b Zhejiang Apeloa Medical Technology Co., Ltd, Dongyang 322118, P. R. China
available (R)-epichlorohydrin with 4-(morpholin-3-one)phenyl isocyanate catalysed by MgI2 etherate as the
key step, in 22% overall yield.
Keywords: (R)-epichlorohydrin, isocyanate, MgI2.etherate, rivaroxaban
STRUCTURE

SIMILARITY

Chemical structures of linezolid (top) and rivaroxaban (bottom). The shared structure is shown in blue.
IH NMR PREDICT
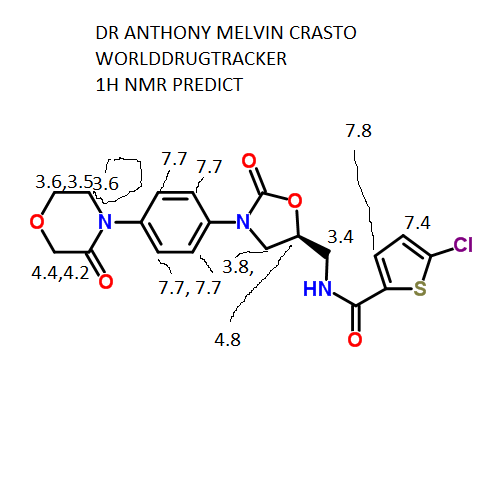
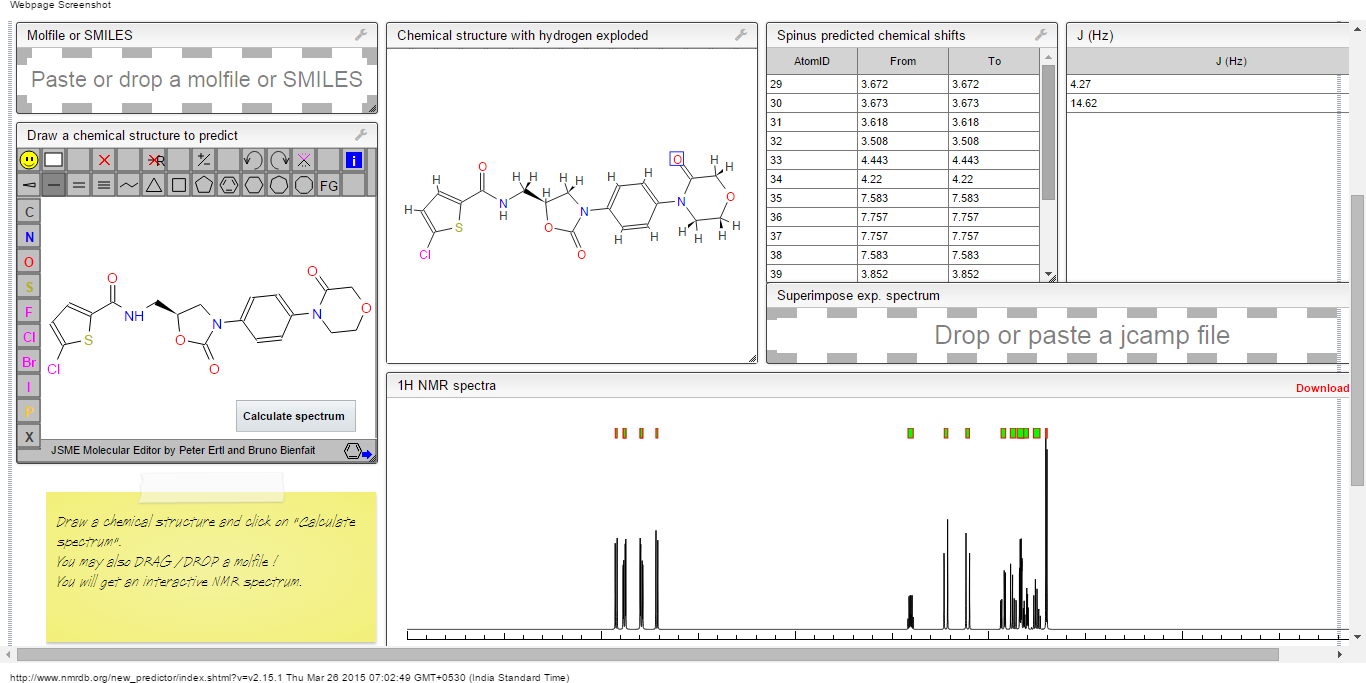

13 C NMR PREDICT


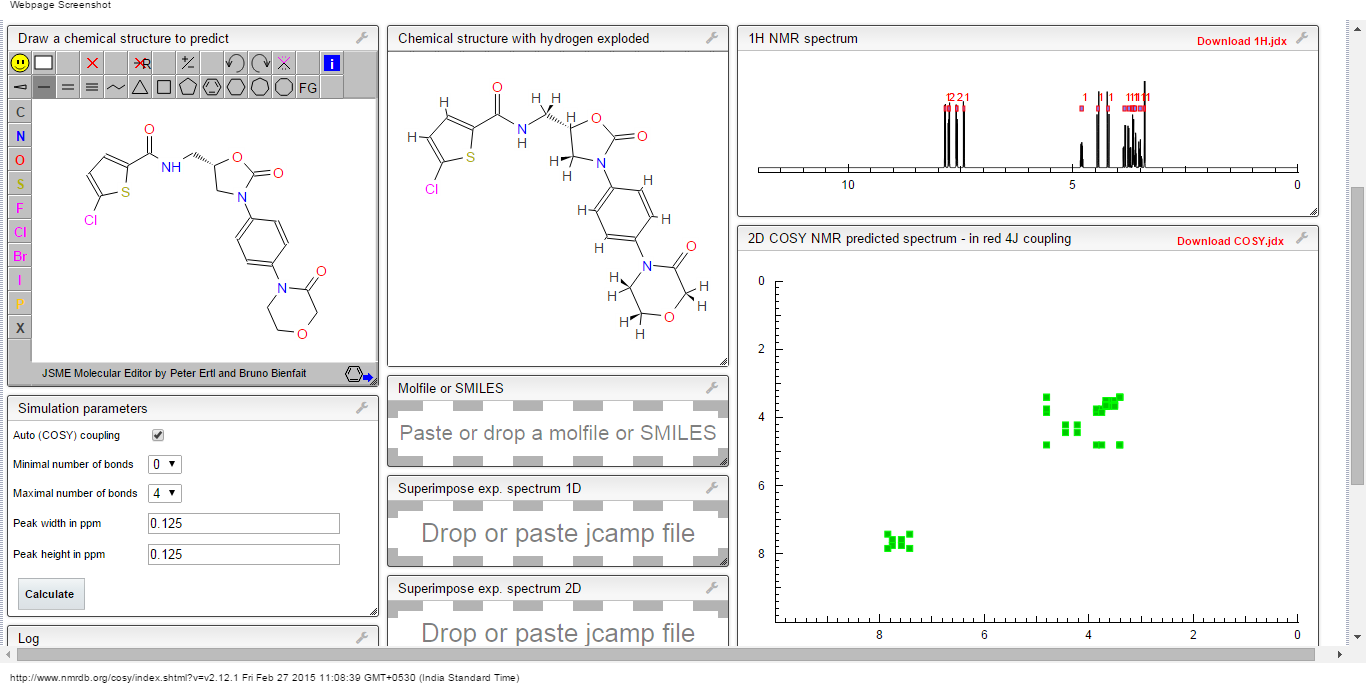
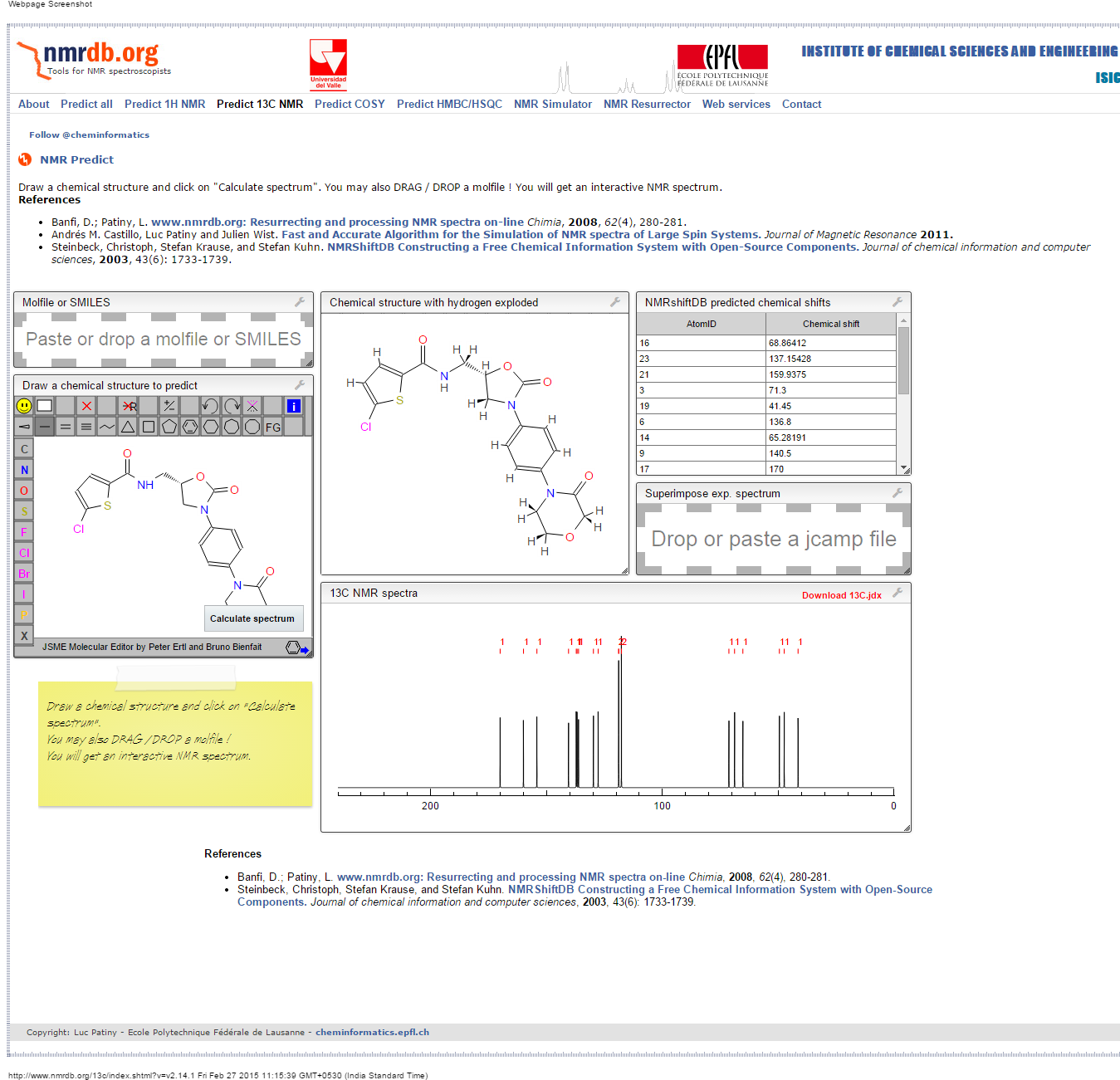

New patent WO-2015104605
Process for preparing rivaroxaban – comprising the reaction of a thioester compound and its salts with 4-{4-[(5S)-5-(aminomethyl)-2-oxo-1,3-oxazolidin-3-yl]phenyl}morpholine-3-one.
Wockhardt Ltd
The synthesis of (II) via intermediate (I) is described (example 7, page 15)
4-{4-[(5S)-5-(Aminomethyl)-2-oxo-1,3-oxazolidin-3-yl]phenyl}morpholine-3-one (formula III) is (I) and rivaroxaban is (II) (claim 1, page 16).
The present invention relates to a process for the preparation of Rivaroxaban and its novel intermediates, or pharmaceutically acceptable salts thereof. The present invention provides novel intermediates, which may be useful for the preparation of Rivaroxaban or its pharmaceutically acceptable salts thereof. The process of preparation by using novel intermediate is very simple cost effective and may be employed at commercial scale. The product obtained by using novel intermediate yield the Rivaroxaban of purity 99% or more, when measured by HPLC. The present invention especially relates to a process for the preparation of Rivaroxaban from thioester of formula II, or a pharmaceutically acceptable salt thereof, wherein R is leaving group.
process includes the step of , reacting thioester of formula IIA or pharmaceutically acceptable salt thereof
Formula IIA

with 4-{4-[(5S)-5-(aminomethyl)-2-oxo-l,3-oxazolidin-3-yl]phenyl}morpholine-3-one of formula III,
Formula III

Formula I
EXAMPLE 7: One pot process for Rivaroxaban
The triphenylphosphine (11.5g) and mercaptobenzothiazole disulphide (15.31g) were taken in methylene chloride and reaction mixture was stirred at 28°C -30°C for 1 hr. The 5-chlorothiophene-2-carboxylic acid (7.2g) and triethylamine (3.8 g) were added to the above reaction mixture. The reaction mixture is stirred at 0°C -25 °C for 1 hr. after 1 hr 4-{4-[(5S)-5-(aminomethyl)-2-oxo-l,3-oxazolidin-3-yl]phenyl}morpholine-3-one (lOg) and triethylamine (3.8g) were added. The resulting reaction mixture further stirred for 2 hrs. After completion of the reaction, water was added and stirred for 10 min. aqueous layer was separated and washed with methylene chloride. The organic layer was acidified to pH 6-7 with 2N hydrochloric acid and finally the organic layer was concentrated to get desired product. The product was purified and dried to yield Rivaroxaban.
Yield: 10.0 gm
Purity: 99.3 %
EXAMPLE 8: One pot process for Rivaroxaban
Exemplified procedure in example 7 with the replacement of solvent ethyl acetate and base potassium hydroxide were used to get the rivaroxaban.
EXAMPLE 9: One pot process for Rivaroxaban
Exemplified procedure in example 7 with the replacement of solvent acetonitile and base potassium carbonate were used, methylene chloride was added in the reaction mixture to extract the Rivaroxaban.
…………..
WO 01/47919 discloses ー species from 4_ (4_ aminophenyl) -3_ morpholinone (I) Preparation of rivaroxaban approach:

…………..
US 07/149522 discloses ー kind to 5_ chlorothiophenes _2_ carbonyl chloride (IV) is a method for preparing raw rivaroxaban in:

………….
http://www.google.com/patents/CN102786516A?cl=en

Preparation 6 rivaroxaban implementation

The 12.5 g (76.9 mmol) 5- chloro-thiophene-2-carboxylic acid was suspended in 35 g of toluene was heated to 80 で, at this temperature, a solution of 11.0 g (92.5 mmol) of thionyl chloride, reaction was continued for 30 min; then warmed to the boiling point of toluene was 120 ° C, and stirring was continued under reflux until cessation of gas; cooled to room temperature, the reaction mixture was concentrated under reduced pressure to remove excess thionyl chloride and toluene to give 5-chloro-thiophene-2-carbonyl chloride;
The 11.6 g (37.0 mmol) 4- {4 – [(5S) -5- (aminomethyl) -2-oxo-1,3-oxazolidin-3-yl] phenyl} morpholin-3 -one hydrochloride was added 40ml of water, was added 4. 64 g (43 8 mmol.) Na2CO3 stirred and dissolved; then added 50 ml of toluene, was added dropwise at 10 ° C under the mixture, the mixture is 8. 0 g ( 44. 4 mmol) 5- chloro-thiophene-2-carbonyl chloride was dissolved in 15 ml of toluene, 20 min the addition was complete, then stirring was continued at room temperature, TLC monitoring progress of the reaction, 2 h after completion of the reaction; and the filter cake washed with water and washed with acetone to give a pale yellow solid 19. 6 g, used directly ko acid recrystallization, as a white solid 15. 2 g,
mp 227. 2 – 228. 1 ° C, [a] D21 = -38 2 ° (. c = 0. 30, DMS0), rivaroxaban yield of 94%, the total yield of 87.5% 0
1H-NMR (DMSO) 8: 3. 61 (. 2 H, t, / = 5 4 Hz), 3. 71 (2 H, t, / = 5 4 Hz.), 3.85 (IH, m ), 3.97 (2 H, t, J = 4. 5 Hz), 4. 19 (3 H, t, / = 7. 5 Hz), 4.84 (IH, m), 7. 19 (IH, d, / = 4. 2Hz), 7.40 (2 H, d, /=9.0 Hz), 7. 57 (2 H, t, /=9.0 Hz), 7. 69 (IH, d, J = 4. 19 Hz), 8. 96 (IH, t, / = 5. 7 Hz).
…………………
WO2013120465
EXAMPLE 28 (preparation of rivaroxaban)

10 g of the salt prepared according to Example 18 were suspended in 75 ml of N- methylpyrolidone, the suspension was heated at 50°C, then 14 ml of triethylamine was added and the mixture was heated at 60°C. This was followed by addition of 15.7 ml of a solution of 5-chlorothiophene-2-carboxylic acid chloride in toluene (2.46 M) and the reaction mixture was stirred and heated at 55°C for 15 minutes, then slowly cooled below 30°C, 75 ml were added and the turbid solution was filtered. The clear filtrate was stirred at 50°C, which was followed by addition of 15 ml of water and 75 ml of ethanol and stirring for 1 hour under slow cooling. The separated product was filtered off, washed with water (15 ml, 60°C), ethanol (2 x 25 ml) and dried in vacuo. 9.1 g (yield 81%) of rivaroxaban in the form of an off-white powder with the melt, point of 229.5-231°C was obtained, HPLC 99.95%, content of the ( )-isomer below 0.03%.
1H NMR (250 MHz, DMSO-D6), δ (ppm): 3.61 (t, 2H, CH2); 3.71 (m, 2H, CH2); 3.85 and 4.19 (m, 2×1 H, CH2); 3.97 (m, 2H, CH2); 4.19 (s, 2H, CH2); 4.84 (pent, 1H, CH); 7.18 (d, 1H); 7.40 (m, 2H); 7.56 (m, 2H); 7.68 (d, 1H); 8.95 (bt, 1H, NH).
13C NMR (250 MHz, DMSO-D6), δ (ppm): 42.2; 47.4; 49.0; 63.4; 67.7; 71.3; 1 18.3; 125.9; 128.1 ; 128.4; 133.2; 136.4; 137.0; 138.4; 154.0; 160.8; 165.9.
MS (m/z): 436.0729 (M+H)+. ation)

The optical isomer of rivaroxaban with the (R)- configuration was obtained by a process analogous to Example 28 starting from the salt prepared according to Example 19. The yield was 76%, HPLC 99.90%, content of the (5)-isomer below 0.03%. The NMR and MS spectra were in accordance with Example 28.
……………………..
……………………
5- chloro-thiophene-2-chloride by condensation, bromide, with 4- (4-amino-phenyl) -3-morpholinone cyclization reaction rivaroxaban, the following reaction scheme 😦 References : W02005068456, US20070149522, DE10300111)

5- chloro-thiophene-2-chloride by condensation, oxidation, and 4- (4-amino-phenyl) -3-morpholinone cyclization reaction racemic rivaroxaban, since the epoxidation step is not give any stereoselectivity, the final chiral separation need to get rivaroxaban, the reaction scheme is as follows 😦 References: W0-0147919)

…………
4- (4- amino-phenyl) -3-morpholinone by condensation, cyclization, and potassium phthalimide after reaction with methyl chloroformate to give (S) -2 – hydroxy -3- (I, 3- dioxo – isoindoline-2-yl) propyl-4- (3-oxo –morpholino) phenyl carbamate, by condensation, methylamine and Ethanol action under profit rivaroxaban, the following reaction scheme (Ref: US20110034465):

……….
4- (4- amino-phenyl) -3-morpholinone (R) and – epichlorohydrin, in the DMF solvent phthalimide potassium salt was reacted with ammonia solution and then prepared to succeed amino compound, and 5-chloro-thiophene-2-chloride in pyridine catalyzed system benefit rivaroxaban, the following reaction scheme (Ref: W02009023233):

………….
4- (4- amino-phenyl) -3-morpholinone after condensation with (R) – epichlorohydrin, then the 5-chloro-thiophene-2-amide lithium chloride and tert-butyl the reaction of an alcohol potassium enrichment rivaroxaban, the following reaction scheme (Ref: US7816355):

……………….
3-chloro-1,2-propanediol by cyclization, the reaction with phthalimide, then with 4- (4-aminophenyl) -3-morpholinone reaction, CDI and hydrazine to give 4- {4- [(5S) -5- (aminomethyl) -2-oxo-1,3-oxazolidin-3-yl] phenyl} morpholin-3-one under the influence, in pyridine and under the action of tetrahydrofuran and 5-chloro-thiophene-2-chloride benefit rivaroxaban, the following reaction scheme (Reference: Gutcait, A. et al Tetrahedron:.. Asymmetry 1996, 7 (6), 1641-1648 Roehrig, .. S. et al J. Med Chem 2005,48 (19), 5900-5908)..:

…………..

http://www.google.com/patents/CN102702186A?cl=zh

Compound rivaroxaban Synthesis Example 7 formula (X), [0071] Example
[0072] Method One:

[0074] The compound of formula (VIII) of (180mg, 0. 618mmol), Ni chloride (5mL) and tris ko amine (187mg,
I. 85mmol) added to the reaction flask, stirred at room temperature for 10 minutes, cooled to 0 ° C, a solution of 5-chloro-2-thiophene chloride (224mg, 1.24mm0l), stirred at room temperature overnight; after the completion of the reaction, spin dry, rinse with anhydrous alcohol ko, filtered, washed ko anhydrous alcohol three times to obtain a white solid product rivaroxaban (215mg, embodiments of the total yield of 7,8 80%).
[0075] 1H-Mffi (DMSC) JOOMHz, δ d m):…. 3 61 (t, 2H, J = 5 6Hz), 3. 71 (t, 2H, J = 5 2Hz), 3 89 ( m, 1H), 3. 97 (t, 2H, J = 4. 4Hz), 4. 20 (m, 3H), 4. 85 (m, 1H), 7. 18 (d, 1H, J = 4. 0Hz), 7. 40 (d, 2H, J = 8. 8Hz), 7. 56 (d, 2H, J = 8. 8Hz), 7. 73 (d, 1H, J = 4. 0Hz).
The method of writing is:

[0078] The compound 5_ gas – oh -I- thiophene carboxylic acid (500mg, 3. 08mmol), MsCl (702mg, 6. 1 Bmmol) and sodium bicarbonate (. 517mg, 6 16mmol) was suspended in THF (20ml) in , heated to 60 ° C with stirring 45min, a large white suspension washed out; the reaction mixture was cooled to room temperature, the compound of formula VIII was added portionwise (800mg, 2 75mmol.), stirred for 5 hours, after completion of the reaction distilled THF, was added after the residue was cooled to room temperature, water (IOOml), at room temperature embrace Cheung 30min, filtered, and the filter cake washed with cold water, dried and added to a ko-ol (5ml) was heated at reflux for I hour. After cooling, stirred for 5 hours at room temperature After filtration to give the product of formula (X) of the compound rivaroxaban (719mg, 60%)
References
- “Xarelto: Summary of Product Characteristics”. Bayer Schering Pharma AG. 2008. Retrieved 2009-02-11.
- Abdulsattar, Y; Bhambri, R; Nogid, A (May 2009). “Rivaroxaban (xarelto) for the prevention of thromboembolic disease: an inside look at the oral direct factor xa inhibitor.”.P & T : a peer-reviewed journal for formulary management 34 (5): 238–44.PMID 19561868.
- Roehrig S, Straub A, Pohlmann J et al. (September 2005). “Discovery of the novel antithrombotic agent 5-chloro-N-({(5S)-2-oxo-3- [4-(3-oxomorpholin-4-yl)phenyl]-1,3-oxazolidin-5-yl}methyl)thiophene- 2-carboxamide (BAY 59-7939): an oral, direct factor Xa inhibitor”. Journal of Medicinal Chemistry 48 (19): 5900–8. doi:10.1021/jm050101d.PMID 16161994.
- Perzborn, Elisabeth; Roehrig, Susanne; Straub, Alexander; Kubitza, Dagmar; Misselwitz, Frank (17 December 2010). “The discovery and development of rivaroxaban, an oral, direct factor Xa inhibitor”. Nature Reviews Drug Discovery 10 (1): 61–75. doi:10.1038/nrd3185.
- “FDA Approves XARELTO® (rivaroxaban tablets) to Help Prevent Deep Vein Thrombosis in Patients Undergoing Knee or Hip Replacement Surgery” (Press release).Janssen Pharmaceutica. 2011-07-01. Retrieved 2011-07-01.
- Gómez-Outes, A; Terleira-Fernández, AI; Calvo-Rojas, G; Suárez-Gea, ML; Vargas-Castrillón, E (2013). “Dabigatran, Rivaroxaban, or Apixaban versus Warfarin in Patients with Nonvalvular Atrial Fibrillation: A Systematic Review and Meta-Analysis of Subgroups.”. Thrombosis 2013: 640723. doi:10.1155/2013/640723. PMC 3885278.PMID 24455237.
- Brown DG, Wilkerson EC, Love WE (March 2015). “A review of traditional and novel oral anticoagulant and antiplatelet therapy for dermatologists and dermatologic surgeons”.Journal of the American Academy of Dermatology 72 (3): 524–34.doi:10.1016/j.jaad.2014.10.027. PMID 25486915.
- “Bayer’s Xarelto Approved in Canada” (Press release). Bayer. 2008-09-16. Retrieved2010-01-31.
- “Bayer’s Novel Anticoagulant Xarelto now also Approved in the EU” (Press release).Bayer. 2008-02-10. Retrieved 2010-01-31.
- “Medication Guide–Xarelto” (PDF). http://www.fda.gov/. U.S. Food and Drug Administration. Retrieved 1 September 2014.
- “Xarelto Side Effects”. http://www.webmd.com/. WebMD. Retrieved 1 September2014.
- “Xarelto Side Effects Center”. http://www.rxlist.com/. RxList. Retrieved 1 September2014.
- Eriksson BI, Borris LC, Dahl OE et al. (November 2006). “A once-daily, oral, direct Factor Xa inhibitor, rivaroxaban (BAY 59-7939), for thromboprophylaxis after total hip replacement”. Circulation 114 (22): 2374–81.doi:10.1161/CIRCULATIONAHA.106.642074. PMID 17116766.
- Eriksson BI, Borris LC, Friedman RJ et al. (June 2008). “Rivaroxaban versus enoxaparin for thromboprophylaxis after hip arthroplasty”. The New England Journal of Medicine 358(26): 2765–75. doi:10.1056/NEJMoa0800374. PMID 18579811.
- Kakkar AK, Brenner B, Dahl OE et al. (July 2008). “Extended duration rivaroxaban versus short-term enoxaparin for the prevention of venous thromboembolism after total hip arthroplasty: a double-blind, randomised controlled trial”. Lancet 372 (9632): 31–9.doi:10.1016/S0140-6736(08)60880-6. PMID 18582928.
- Lassen MR, Ageno W, Borris LC et al. (June 2008). “Rivaroxaban versus enoxaparin for thromboprophylaxis after total knee arthroplasty”. The New England Journal of Medicine358 (26): 2776–86. doi:10.1056/NEJMoa076016. PMID 18579812.
- Turpie A, Bauer K, Davidson B et al. “Comparison of rivaroxaban – an oral, direct factor Xa inhibitor – and subcutaneous enoxaparin for thromboprophylaxis after total knee replacement (RECORD4: a phase 3 study) / European Federation of National Associations of Orthopaedics and Traumatology Annual Meeting; May 29 – June 1, 2008; Nice, France, Abstract F85”. Journal of Bone & Joint Surgery, British Volume 92–B (SUPP II): 329.
- Turpie AG, Lassen MR, Davidson BL et al. (May 2009). “Rivaroxaban versus enoxaparin for thromboprophylaxis after total knee arthroplasty (RECORD4): a randomised trial”.Lancet 373 (9676): 1673–80. doi:10.1016/S0140-6736(09)60734-0. PMID 19411100.
- ClinicalTrials.gov. “Randomized, Double-Blind Study Comparing Once Daily Oral Rivaroxaban With Adjusted-Dose Oral Warfarin for the Prevention of Stroke in Subjects With Non-Valvular Atrial Fibrillation”. Retrieved 2009-02-11.
- ClinicalTrials.gov. “MAGELLAN – Multicenter, Randomized, Parallel Group Efficacy Superiority Study in Hospitalized Medically Ill Patients Comparing Rivaroxaban with Enoxaparin”. Retrieved 2009-02-11.
- ClinicalTrials.gov. “Once-Daily Oral Direct Factor Xa Inhibitor Rivaroxaban in the Long-Term Prevention of Recurrent Symptomatic Venous Thromboembolism in Patients With Symptomatic Deep-Vein Thrombosis or Pulmonary Embolism. The Einstein-Extension Study”. Retrieved 2009-02-11.
- ClinicalTrials.gov. “Oral Direct Factor Xa Inhibitor Rivaroxaban In Patients With Acute Symptomatic Deep-Vein Thrombosis (DVT) Without Symptomatic Pulmonary Embolism: Einstein-DVT Evaluation”. Retrieved 2009-02-11.
- ClinicalTrials.gov. “Oral Direct Factor Xa Inhibitor Rivaroxaban In Patients With Acute Symptomatic Pulmonary Embolism (PE) With Or Without Symptomatic Deep-Vein Thrombosis: Einstein-PE Evaluation”. Retrieved 2009-02-11.
- ClinicalTrials.gov. “A Randomized, Double-Blind, Placebo-Controlled, Event-Driven Multicenter Study to Evaluate the Efficacy and Safety of Rivaroxaban in Subjects With a Recent Acute Coronary Syndrome”. Retrieved 2009-02-11.
- “Venous Thromboembolic Event (VTE) Prophylaxis in Medically Ill Patients (MAGELLAN)”. ClinicalTrials.gov. 11 March 2011. Retrieved 15 April 2011.
- Hughes, Sue (5 April 2011). “MAGELLAN: Rivaroxaban prevents VTE in medical patients, but bleeding an issue”. theheart.org. Retrieved 15 April 2011.
- “About the MAGELLAN Study”. Bayer HealthCare. Retrieved 15 April 2011.
- Bauersachs, M.D., Rupert; The EINSTEIN Investigators (December 23, 2010). “Oral Rivaroxaban for Symptomatic Venous Thromboembolism”. The New England Journal of Medecine 363 (26): 2499–2510. doi:10.1056/NEJMoa1007903. PMID 21128814. Retrieved 4 April 2011.
- “Oral Direct Factor Xa Inhibitor Rivaroxaban In Patients With Acute Symptomatic Deep-Vein Thrombosis Without Symptomatic Pulmonary Embolism: Einstein-DVT Evaluation”. clinicaltrials.gov. Retrieved 15 April 2011.
- European Medicines Agency (2008). “CHP Assessment Report for Xarelto (EMEA/543519/2008)” (PDF). Retrieved 2009-06-11.
- Turpie AG (January 2008). “New oral anticoagulants in atrial fibrillation”. European Heart Journal 29 (2): 155–65. doi:10.1093/eurheartj/ehm575. PMID 18096568.
| WO2013120465A1 * | Feb 18, 2013 | Aug 22, 2013 | Zentiva, K.S. | A process for the preparation of rivaroxaban based on the use of (s)-epichlorohydrin |
| WO2001047919A1 | Dec 11, 2000 | Jul 5, 2001 | Bayer Ag | Substituted oxazolidinones and their use in the field of blood coagulation |
| WO2004060887A1 | Dec 24, 2003 | Jul 22, 2004 | Bayer Healthcare Ag | Method for producing 5-chloro-n-({5s)-2-oxo-3-[4-(3-oxo-4-morpholinyl)-phenyl]-1,3-oxazolidin-5-yl}-methyl)-2-thiophene carboxamide |
| WO2007116284A1 | Mar 26, 2007 | Oct 18, 2007 | Pfizer Prod Inc | Process for preparing linezolid |
| WO2009023233A1 | Aug 14, 2008 | Feb 19, 2009 | Concert Pharmaceuticals Inc | Substituted oxazolidinone derivatives |
| WO2010043110A1 | Oct 9, 2009 | Apr 22, 2010 | Changzhou Multiple Dimension Institute Of Industry Technology Co., Ltd. | A preparation method of high-purity l-carnitine |
| WO2010082627A1 | Jan 15, 2010 | Jul 22, 2010 | Daiso Co., Ltd. | Process for producing 2-hydroxymethylmorpholine salt |
| WO2010124835A1 | Apr 27, 2010 | Nov 4, 2010 | Belte Ag | Aluminium-silicon diecasting alloy for thin-walled structural components |
| WO2011080341A1 | Jan 3, 2011 | Jul 7, 2011 | Enantia, S.L. | Process for the preparation of rivaroxaban and intermediates thereof |
| WO2011098501A1 | Feb 10, 2011 | Aug 18, 2011 | Sandoz Ag | Method for the preparation of rivaroxaban |
| WO2011102640A2 | Feb 16, 2011 | Aug 25, 2011 | Hanmi Holdings Co., Ltd. | Method for preparing sitagliptin and amine salt intermediates used therein |
| WO2012159992A1 * | May 18, 2012 | Nov 29, 2012 | Interquim, S.A. | Process for obtaining rivaroxaban and intermediate thereof |
| CN102786516A * | Aug 21, 2012 | Nov 21, 2012 | 湖南师范大学 | Method for synthesizing rivaroxaban |
| US7157456 | Dec 11, 2000 | Jan 2, 2007 | Bayer Healthcare Ag | Substituted oxazolidinones and their use in the field of blood coagulation |
| US7816355 * | Apr 28, 2009 | Oct 19, 2010 | Apotex Pharmachem Inc | Processes for the preparation of rivaroxaban and intermediates thereof |
| US20110034465 | Feb 10, 2011 | Apotex Pharmachem Inc. | Processes for the preparation of rivaroxaban and intermediates thereof |
| CN101560209A * | Apr 15, 2008 | Oct 21, 2009 | Shenyang, one hundred million Leo Pharmaceutical Co., Ltd. | Pyrimidine oxazolidinone compound and preparation method comprising |
| CN101619061A * | Aug 11, 2009 | Jan 6, 2010 | Shenyang Pharmaceutical University | Cyanopyridyl substituted oxazolidinone compounds |
| CN101821260A * | Aug 14, 2008 | Sep 1, 2010 | Consett Pharmaceuticals Ltd. | Substituted oxazolidinone derivatives |
| CN102250076A * | May 27, 2011 | Nov 23, 2011 | Hengdian Group homes Chemical Co., Ltd. | One kind of rivaroxaban Rivaroxaban intermediates and preparation methods |
| CN102250077A * | Jun 15, 2011 | Nov 23, 2011 | Zhejiang University | A method for intermediate and rivaroxaban Rivaroxaban for the synthesis of |
| CN102311400A * | Jun 29, 2010 | Jan 11, 2012 | Xiang really Biotechnology Co., Ltd. | Aminomethyl-3-aryl-2-oxazolidinone class method – Preparation of L-5- |
| CN102320988A * | Jun 3, 2011 | Jan 18, 2012 | Shanghai Institute of Organic Chemistry | 4- (4-aminophenyl) -3-morpholinone intermediate amide, the synthesis method and uses |
| EP2354128A1 * | Feb 10, 2010 | Aug 10, 2011 | Sandoz Ag | Method for the preparation of rivaroxaban |
| WO2010124385A1 * | Apr 28, 2010 | Nov 4, 2010 | Apotex Pharmachem Inc. | Processes for the preparation of rivaroxaban and intermediates thereof |
FROM THE NET
RIVAROXABAN 5-Chloro-N-{[(5S) 2-oxo-3 [4-(3-oxo-4 …
32 mins ago – RIVAROXABAN 5-Chloro-N-{[(5S) 2-oxo-3 [4-(3-oxo-4-morpholinophenyl]oxazolidin-5-yl]methyl} thiophene-2-carboxamide (Rivaroxaban) (1):1 rivaroxaban 1 …
WO 2015104605.new patent on Rivaroxaban, Wockhardt …
1 hour ago – WO 2015104605.new patent on Rivaroxaban, Wockhardt Ltd Process for preparing rivaroxaban – comprising the reaction of a thioester compound and its salts …

 |
|
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
(S)-5-chloro-N-{[2-oxo-3-[4-(3-oxomorpholin-4-yl)
phenyl]oxazolidin-5-yl]methyl} thiophene-2-carboxamide |
|
| Clinical data | |
| Trade names | Xarelto |
| AHFS/Drugs.com | Micromedex Detailed Consumer Information |
| Licence data | EMA:Link, US FDA:link |
| Pregnancy category |
|
| Legal status |
|
| Routes of administration |
oral |
| Pharmacokinetic data | |
| Bioavailability | 80% to 100%; Cmax = 2 – 4 hours (10 mg oral)[1] |
| Metabolism | CYP3A4 , CYP2J2 and CYP-independent mechanisms[1] |
| Biological half-life | 5 – 9 hours in healthy subjects aged 20 to 45[1][2] |
| Excretion | 2/3 metabolized in liver and 1/3 eliminated unchanged[1] |
| Identifiers | |
| CAS Registry Number | 366789-02-8 |
| ATC code | B01AX06 |
| PubChem | CID: 6433119 |
| IUPHAR/BPS | 6388 |
| DrugBank | DB06228 |
| ChemSpider | 8051086 |
| UNII | 9NDF7JZ4M3 |
| ChEMBL | CHEMBL198362 |
| Synonyms | Xarelto, BAY 59-7939 |
| Chemical data | |
| Formula | C19H18ClN3O5S |
| Molecular mass | 435.882 g/mol |

Rivaroxaban, a FXa inhibitor, is the active ingredient in XARELTO Tablets with the chemical name 5-Chloro-N-({(5S)-2-oxo-3-[4-(3-oxo-4-morpholinyl)phenyl]-1,3-oxazolidin-5yl}methyl)-2-thiophenecarboxamide. The molecular formula of rivaroxaban is C19H18ClN3O5S and the molecular weight is 435.89. The structural formula is:
 |
Rivaroxaban is a pure (S)-enantiomer. It is an odorless, non-hygroscopic, white to yellowish powder. Rivaroxaban is only slightly soluble in organic solvents (e.g., acetone, polyethylene glycol 400) and is practically insoluble in water and aqueous media.
Each XARELTO tablet contains 10 mg, 15 mg, or 20 mg of rivaroxaban. The inactive ingredients of XARELTO are: croscarmellose sodium, hypromellose, lactose monohydrate, magnesium stearate, microcrystalline cellulose, and sodium lauryl sulfate. Additionally, the proprietary film coating mixture used for XARELTO 10 mg tablets is Opadry® Pink and for XARELTO 15 mg tablets is Opadry® Red, both containing ferric oxide red, hypromellose, polyethylene glycol 3350, and titanium dioxide, and for XARELTO 20 mg tablets is Opadry® II Dark Red, containing ferric oxide red, polyethylene glycol 3350, polyvinyl alcohol (partially hydrolyzed), talc, and titanium dioxide.

DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
Googleplus amcrasto@gmail.com
////////SEE ABAN SERIES AT…………http://organicsynthesisinternational.blogspot.in/p/aban-series.html
VORICONAZOLE SPECTRAL VISIT

(ppm): 9.04 (1H), 8.84 (1H), 8.23 (1H), 7.61 (1H), 7.28 (1H), 7.17 (1H), 6.91 (1H),
5.97 (1H),
4.80 (1H),
4.34 (1H),
3.93 (1H),
1.1 (3H)………….US8263769
13 C NMR
DMSO-d6
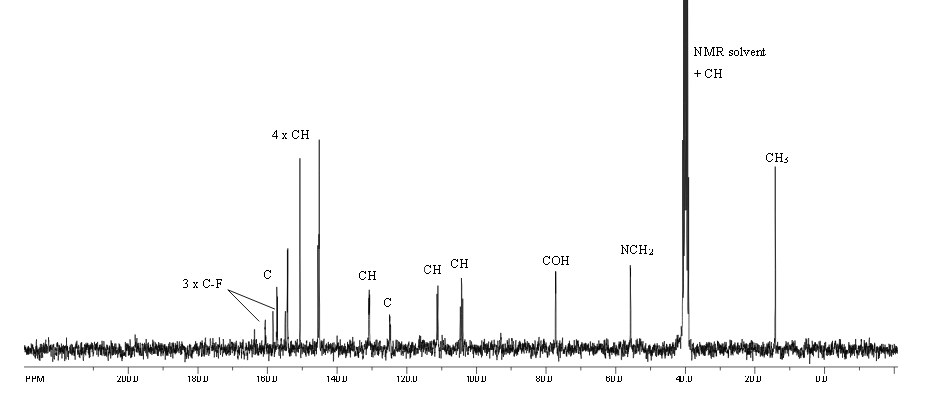

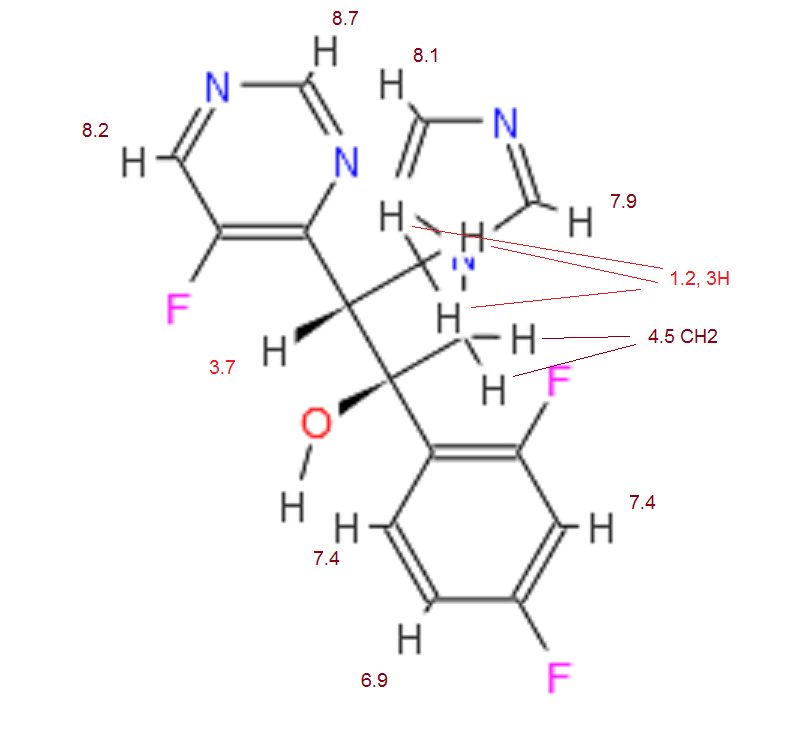

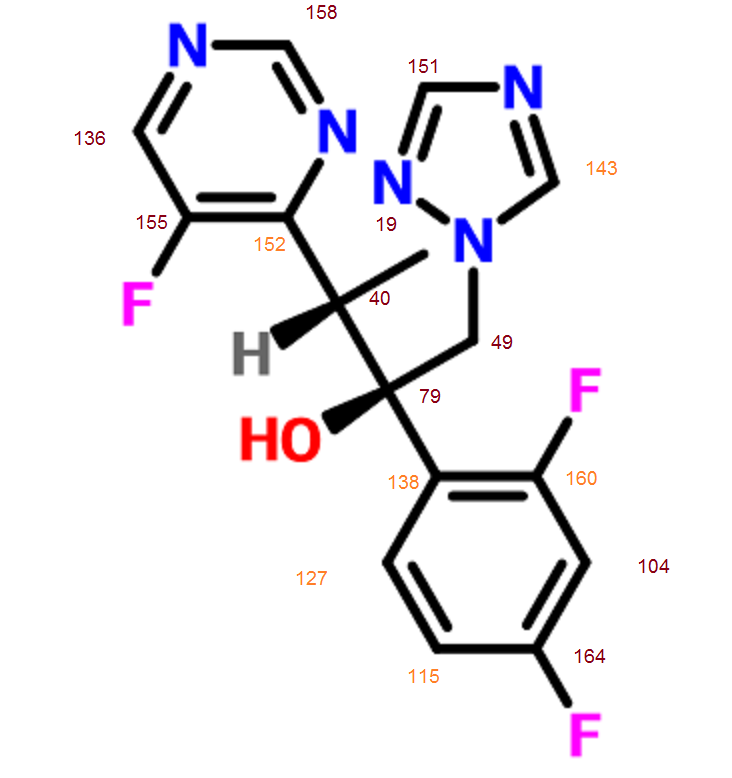

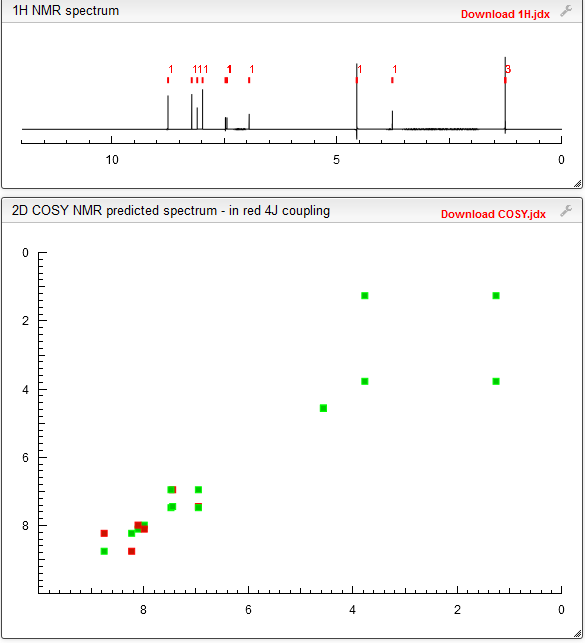

HPLC

………………….
PAPER
| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| US6586594 | 26 Jul 1996 | 1 Jul 2003 | Pfizer, Inc. | Preparation of triazoles by organometallic addition to ketones and intermediates therefor |
| CN1488630A | 8 Oct 2002 | 14 Apr 2004 | 张文更 | Method for preparing triazole antifungal agent |
| CN1814597A | 9 Dec 2005 | 9 Aug 2006 | 北京丰德医药科技有限公司 | New method for preparing voriconazole |
| EP0440372A1 | 24 Jan 1991 | 7 Aug 1991 | Pfizer Limited | Triazole antifungal agents |
| GB2452049A | Title not available | |||
| WO1993007139A1 | 1 Oct 1992 | 15 Apr 1993 | Pfizer Ltd | Triazole antifungal agents |
| WO1997006160A1 | 26 Jul 1996 | 20 Feb 1997 | Michael Butters | Preparation of triazoles by organometallic addition to ketones and intermediates therefor |
| WO2006065726A2 | 13 Dec 2005 | 22 Jun 2006 | Reddys Lab Ltd Dr | Process for preparing voriconazole |
| WO2007013096A1 | 26 Jun 2006 | 1 Feb 2007 | Msn Lab Ltd | Improved process for the preparation of 2r, 3s-2-(2,4-difluorophenyl)-3-(5-fluoropyrimidin-4-yl)-1-(1h-1,2,4-triazol-1-yl) butan-2-ol (voriconazole) |
| WO2007132354A2 | 29 Jan 2007 | 22 Nov 2007 | Medichem Sa | Process for preparing voriconazole, new polymorphic form of intermediate thereof, and uses thereof |
| WO2009024214A1 * | 10 Jul 2008 | 26 Feb 2009 | Axellia Pharmaceuticals Aps | Process for the production of voriconazole |
| WO2009084029A2 | 2 Dec 2008 | 9 Jul 2009 | Venkatesh Bhingolikar | Improved process for the preparation of (2r,3s)-2-(2,4- difluqrophenyl)-3-(5-fluoropyrimidin-4-yl)-1-(1h-1,2,4-triazol-1-yl) butan-2-ol |
| US8575344 * | 1 Feb 2011 | 5 Nov 2013 | Dongkook Pharmaceutical Co., Ltd. | Process for preparing voriconazole by using new intermediates |
| US20130005973 * | 1 Feb 2011 | 3 Jan 2013 | Dongkook Pharmaceutical Co., Ltd. | Process for preparing voriconazole by using new intermediates |
Reference |
|||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Butters et al., “Process Development of Voriconazole: A Novel Broad-Spectrum Triazole Antifungal Agent,” Organic Process Research & Development, 2001, vol. 5, pp. 28-36. | ||||||||||||||||||||||||||||||||||||
References:
Ergosterol biosynthesis inhibitor. Prepn: S. J. Ray, K. Richardson, EP 440372; eidem, US 5278175 (1991, 1994 both to Pfizer); R. P. Dickinson et al., Bioorg. Med. Chem. Lett. 6, 2031 (1996).
Mechanism of action: H. Sanati et al.,Antimicrob. Agents Chemother. 41, 2492 (1997). In vitro antifungal spectrum: F. Marco et al., ibid. 42, 161 (1998).
HPLC determn in plasma: R. Gage, D. A. Stopher, J. Pharm. Biomed. Anal. 17, 1449 (1998).
Review of pharmacology and clinical development: P. E. Verweij et al., Curr. Opin. Anti-Infect. Invest. Drugs 1, 361-372 (1999); J. A. Sabo, S. M. Abdel-Rahman, Ann. Pharmacother. 34, 1032-1043 (2000).
Clinical pharmacokinetics: L. Purkins et al., Antimicrob. Agents Chemother. 46, 2546 (2002).
Clinical comparison with amphotericin B: T. J. Walsh et al., N. Engl. J. Med. 346, 225 (2002).
MOLFILE
COPY ONLY BLUE SECTION
START
64684.mol
ChemDraw07261512442D
26 28 0 0 0 0 0 0 0 0999 V2000
0.5118 1.1267 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
-0.2030 0.7149 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
-0.9179 0.3031 0.0000 H 0 0 0 0 0 0 0 0 0 0 0 0
-0.6206 1.4298 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
-0.2030 2.1447 0.0000 N 0 0 0 0 0 0 0 0 0 0 0 0
-0.6206 2.8595 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
-1.4441 2.8595 0.0000 N 0 0 0 0 0 0 0 0 0 0 0 0
-1.8559 2.1447 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
-1.4441 1.4298 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
-1.8559 0.7149 0.0000 F 0 0 0 0 0 0 0 0 0 0 0 0
0.2088 0.0000 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
-0.5062 -0.4117 0.0000 O 0 0 0 0 0 0 0 0 0 0 0 0
0.9236 0.4118 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
0.9236 1.2411 0.0000 N 0 0 0 0 0 0 0 0 0 0 0 0
1.5928 1.7215 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
1.3354 2.5107 0.0000 N 0 0 0 0 0 0 0 0 0 0 0 0
0.5118 2.5107 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
0.2545 1.7215 0.0000 N 0 0 0 0 0 0 0 0 0 0 0 0
0.6205 -0.7149 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
0.2088 -1.4297 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
0.6205 -2.1446 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
1.4440 -2.1446 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
1.8559 -2.8595 0.0000 F 0 0 0 0 0 0 0 0 0 0 0 0
1.8559 -1.4297 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
1.4440 -0.7149 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
1.8559 0.0000 0.0000 F 0 0 0 0 0 0 0 0 0 0 0 0
1 2 1 0
2 3 1 1
2 4 1 0
2 11 1 0
4 5 1 0
4 9 2 0
5 6 2 0
6 7 1 0
7 8 2 0
8 9 1 0
9 10 1 0
11 12 1 1
11 13 1 0
11 19 1 0
13 14 1 0
14 15 1 0
14 18 1 0
15 16 2 0
16 17 1 0
17 18 2 0
19 20 1 0
19 25 2 0
20 21 2 0
21 22 1 0
22 23 1 0
22 24 2 0
24 25 1 0
25 26 1 0
M END
END
सुकून उतना ही देना प्रभू, जितने से जिंदगी चल जाये। औकात बस इतनी देना, कि औरों का भला हो जाये।
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
Join me on Facebook
Googleplus amcrasto@gmail.com
LIONEL MY SONजिंदगी चल जाये।
औकात बस इतनी देना,
कि औरों का भला हो जाये।
DR ANTHONY MELVIN CRASTO

ORGANIC SPECTROSCOPY

