
FLAGS AND HITS
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO FACEBOOK
...................................................................Join me on twitter
FACEBOOK
...................................................................Join me on twitter
 ..................................................................Join me on google plus
..................................................................Join me on google plus
 Googleplus
GoogleplusMYSELF

PNQ 201 from Advinus for for potential treatment of IBD.
formula I

PNQ 201
STRUCTURE COMING……
Adenosine A2b receptor antagonist
Advinus Therapeutics Ltd
KEEP WATCHING THIS POST……………
PNQ-201 is a proprietary orally active A2B Adenosine receptor (A2BAdoR) antagonist, currently in pre-clinical development for potential treatment of IBD. Advinus is looking for partnering/co-development opportunities.
A2BAdenosine Receptor (A2BAdoR) Antagonist PNQ-201 for IBD
Inflammatory Bowel Disease (IBD), which includes ulcerative colitis (UC) and Crohn’s disease (CD), is a multifactorial disease of an etiology not fully understood. It includes chronic inflammation of the gut, characterized by dysfunction of mucosal immunity. Current oral therapies are ineffective, non-specific, and have significant adverse effects. As such, there is a large unmet medical need for the development of new and specific therapies for IBD.
Adenosine is a stimulator of pro-inflammatory effects in the gastro-intestinal tract. Adenosine regulates tissue function by activating its receptors: A1AdoR and A2AAdoR are high affinity receptors and A2BAdoR and A3AdoR are low affinity receptors. A2BAdoR is highly expressed in cecum and colon, with expression increased even further in epithelial cells in human and murine colitis. A2BAdoR, agonized by adenosine induces cytokine secretion at the mucosal surface, inflammatory cell infiltration into intestinal wall, focal crypt damage and ulceration. Therefore, A2BAdoR antagonists are expected to be beneficial in IBD patients.
PNQ-201 is a proprietary orally active A2BAdoR antagonist, currently in pre-clinical development for the potential treatment of IBD. PNQ-201 is a potent and selective A2B antagonist. It is selected for development on the basis of poor systemic bioavailability and high exposure in colon/cecum. Negligible systemic bioavailability and maximum exposure at the sites of action in the lower gastrointestinal tract is expected to offer maximum therapeutic benefits while minimizing potential side effects. PNQ-201 has shown a robust efficacy profile in standard models of IBD, namely, the mouse DSS-induced colitis model and the rat TNBS-induced colitis model. PNQ-201 was found to be safe in exploratory safety studies including a Drug Matrix Screen, mini-AMES test, and a 14- day repeat dose toxicology study in rats.
PATENT
Example 1 : 8-(l-Benzyl~lH-pyrazol-4-yl)-l-propyl-l,4,5,7-tetrahydro-purin-6-one

Step 1: l-Benzyl-lH-pyrazole-4-carboxylic acid (6-amino-2,4-dioxo-3-propyl-l,2,3,4-tetrahydro-pyrimidin-5-yl)-amide
A mixture of 5,6-diamino-3-propyl-lH-pyrimidine-2,4-dione (1.6g, 8.55mmol), 1-benzyl-lH-pyrazole-4-carboxylic acid (1.75g, 8.65mmol) in methanol (10ml) were cooled to 0 0C and added EDCLHCl (2.32g, 12.11mmol). The reaction mixture was stirred at 25 0C for 20 hours and the solvents were removed under reduced pressure. To this residue water (10ml) was added and the precipitate was filtered off, and was washed sequentially with cold water (20ml) and DCM (25ml) to obtain l-Benzyl-lH-pyrazole-4-carboxylic acid (6-amino-2,4-dioxo-3 -propyl- 1 ,2,3,4-tetrahydro-pyrimidin-5-yl)-amide (1.5 g, 47 %) as a pale yellow solid.
1HNMR^OOMHZ5 DMSO d6): δ 0.82 (t, J=7.6Hzs 3H); 1.46-1.51 (m, 2H); 3.64 (t, J=7.2Hz, 2H);^5.36 (s, 2H); 6.01 (s, 2H); 7.26-7.38 (m, 5H); 7.96 (s, IH); 8.31 (s, IH); 8.54 (s, IH); 10.43 (s, IH).
Step 2 : 8-(l-Benzyl-lH-pyrazol-4-yl)-2-chloro-l-propyH,7-dihydro-purin-6-one A mixture of l-benzyl-lH-pyrazole-4-carboxylicacid(6-amino-2,4-dioxo-3-propyl-l,2,3,4-tetrahydro-ρyrimidin-5-yl)-amide (0.5g, 13.5mmol)s POCl3 (10ml) and DMF (0.1ml) were heated at 125-130 0C for 20 hours. Reaction mixture was cooled to 20-25 0C. It was then concentrated under vacuum. The residue was triturated with diethyl ether, dried. The crude product was purified by column chromatography using silica gel (100-200 mesh) and 2 to 4 % methanol in DCM as an eluent to obtain 8-( 1 -Benzyl- 1 H-pyrazol-4-yl)-2-chloro-l -propyl- l,7-dihydro-purin-6-one (0.04g, 8%) as a pale brown solid.
1HNMR^OOMHZ5 DMSO d6): δ 0.93 (t, J=7.6Hz, 3H); 1.67-1.73 (m, 2H); 4.15 (t, J=7.6Hz, 2H); 5.42 (s, 2H); 7.29-7.39 (m, 5H); 8.14 (s, IH); 8.49 (s, IH); 13.68 (bs, IH). Step 3: 8-(l-Benzyl-lH-pyrazol-4-yl)-l-propyl-l,7-dihydro-purin-6-one
A mixture of 8-(l -benzyl- lH-pyrazol-4-yl)-2-chloro-l -propyl- l,7-dihydro-purin-6-one (0.035 g, 0.094 mmol), Pd\C (10%) (0.025g), in ethanol (20ml) were stirred under hydrogen atmosphere for 20 hours. Reaction mixture was filtered through celite bed washed with methanol (20ml), and the solvents were removed under vacuum. The crude product was purified by column chromatography using silica gel (100-200 mesh) and 2 to 4 % methanol in DCM as an eluent to obtain 8-(l-Benzyl-lH-pyrazol-4-yl)-l-propyl-l,7-dihydro-purin-6-one (0.012g, 39%) as off white solid.
1HNMR^OOMHZ, DMSO d6): δ 0.89 (t, J=7.2Hz, 3H); 1.66-1.72 (m, 2H); 3.94 (t, J=7.6Hz, 2H); 5.41 (s, 2H); 7.302-7.38 (m, 5H); 8.03 (s, IH); 8.16 (s, IH); 8.34 (s, IH).
PATENT
WO 2011055391
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2011055391
Preparation 1: 2-chloro-8-cyclopentyl-l-propyI-l, 7-dihydro-purin-6-one:

Step 1: Cyclopentane carboxylic acid (6-amino-2,4-dioxo-3-propyl-l,2,3,4-tetra hydro-pyriniidin-S-y -amide
To a solution of 5, 6-diamino-3-propyI-lH-pyrimidine-2, 4-dione (0.6 g, 2.72 mmol) in methanol (50 ml) was added cyclopentane carboxylic acid (0.310 g, 2.72 mmol). The reaction mixture was cooled to 0°C and then l-ethyl-3(3′-dimethylaminopropyl) carbodiimide hydrochloride (EDCI.HC1) (0.78 g, 4.1 mmol) was added. The resulting reaction mixture was stirred at room temperature overnight. The reaction mixture was concentrated under reduced pressure. The residue was dissolved in water. The solid was filtered and washed thoroughly with water followed by diethyl ether. The product obtained was dried under high vacuum. The crude product (0.40 g) was used for the next step without further purification.
Step 2: Preparation of 8-cyclopentyI-2-chloro-l-propyl-l, 7-dihydro-purin-6-one
To a suspension of cyclopentanecarboxylic acid (6-amino-2,4-dioxo-3-propyl- 1,2,3,4-tetrahydro-pyrimidin-5-yl)-amide (0.40 g, crude) obtained from step 1 in phosphorus oxychloride (25 ml) was added phosphorus pentachloride (0.10 g) and the resulting reaction mixture was refluxed overnight. Phosphorus oxychloride was evaporated under reduced pressure. The residue was slowly quenched with water. Ethyl acetate was added and the organic layer was separated and washed thoroughly with water followed by brine. The ethyl acetate layer was dried over anhydrous sodium sulphate and concentrated under vacuum. The crude product was purified by preparative TLC using dichloromethane, methanol (9:1) as the solvent system to give 0.075 g (19% over two steps) of the product as a white solid.
•H MR (400 MHz, DMSO d6): δ 0.9 (t, J = 8 Hz, 3H), 1.59-1.82 (m, 8H), 1.99 (m, 2H), 3.15 (t, J = 8 Hz, 1H), 4.12 (t, J = 8 Hz, 2H).
Preparations 2 to 7 were prepared following the experimental procedure as given for Preparation 1.
Preparation 2: 2-Chloro-8-cyclohexyl- 1 -propyl- 1 ,7-dihydro-purin-6-one,
Preparation 3: 2-Chloro-8-cyclopropyl-l -propyl- 1 ,7-dihydro-purin-6-one,
Preparation^ 2-Chloro-8-(hexahydro-2,5-methano-pentalen-3a-yl)-l -propyl- 1,7- dihydro-purin-6-one,
Preparation 5: 8-Bicyclo-[2.2.1]-hept-2-yl-2-chloro-l -propyl- 1, 7-dihydro-purin-6- one,
Preparation 6: 8-Adamantan-2-yl-2-chloro-l -propyl- 1, 7-dihydro-purin-6-one, Preparation7:3-[4-(2-Chloro-6-ox0-l-propyl-6,7-dihydro-lH-purin-8-yl)- bicyclo[2.2.2]oct-l-yl]-propionic acid.
Example 1: 8-Cyclopentyl-2-(3, 4-difluoro-phenoxy)-l-propyl-l, 7-dihydro-purin- 6-one:

To a solution of 8-cyclopentyl-2-chloro- 1 -propyl- l,7-dihydro-purin-6-one (0.06 g, 0.21 mmol) in N-methyl-2-pyrrolidone (0.2 ml) was added K2CO3 (0.044g, 0.32 mmol) followed by 3, 4-difluoro phenol and the reaction mixture was heated at 130 °C overnight. The reaction mixture was diluted with ethyl acetate and water. The layers were separated and ethyl acetate layer was washed with water. The ethyl acetate layer was dried over anhydrous sodium sulphate and concentrated under vacuum. The crude product was purified by preparative TLC using 3% methanol in DCM to give the product (0.015 g, 19 %) as a white solid.
‘HNMR (400 MHz, DMSO d6): δ 0.94 (t, J = 8 Hz, 3H), 1.59-1.74 (m, 6H), 1.94 (br.s, 2H), 3.12 (m, 2H), 4.09 (br. s, 2H), 7.21 (d, J = 8 Hz, 1H), 7.53-7.65 (m, 2H), 12.74 (br.s, 1H).
PNQ 370 useful in treating Parkinson’s disease from ADVINUS
2016

PNQ 370
Advinus Therapeutics Ltd
Adenosine A2a receptor antagonist
for treating disease or disorder susceptible to improvement by antagonism of A2A receptor.
Advinus Therapeutics is investigating PNQ-370, presumed to be lead from a series of small molecule therapeutics including PD-2 and PD-3, as adenosine A2a receptor antagonist, for the potential treatment of Parkinson’s disease . In November 2012, this drug was in preclinical development .
![]()
KEEP WATCHING THIS POST AS I ARRIVE AT THE STRUCTURE…………..








ONE OF THE ABOVE OR SIMILAR
INTRODUCTION
The effects of adenosine are mediated through at least four specific cell membrane receptors so far identified and classified as Ai, A2A, A2B and A3 belonging to G protein-coupled receptor family. The Ai and A3 receptors down-regulate cellular cAMP levels through their coupling to G protein, which inhibit adenylate cyclase. In contrast, A2A and A2B receptors couple to G protein that activate adenylate cyclase and increase intracellular levels of cAMP. Through these receptors, adenosine regulates the wide range of physiological functions.
Advances in understanding the role of adenosine and its receptors in physiology and pathophysiology, as well as new developments in medicinal chemistry of these receptors have identified potential therapeutic areas for drug development. With the combination of pharmacological data, using selective ligands and genetically modified mice, important progress has been made toward an understanding of the role of ARs in a variety of diseases, such as inflammatory conditions, sepsis, heart attack, ischemia-reperfusion injury, vascular injury, spinal cord injury, chronic obstructive pulmonary disease (COPD), asthma, diabetes, obesity, inflammatory bowel disease, retinopathy, and Parkinson’s Disease (PD).
Happy new year wishes 2016


Movement disorder constitutes a serious health problem, especially among the elderly. These movement disorders can often be the result of brain lesions. Disorders involving the basal ganglia which result in movement disorders include Parkinson’s disease, Huntington’s chorea and Wilson’s disease. Tremor, rigidity, akinesia and postural changes are four classic symptoms of Parkinson’s disease, it is also associated with depression, dementia and overall cognitive decline. Parkinson’s disease has a prevalence of 1 per 1000 of the total population and increases to 1 per 100 for those aged over 60 years. Degeneration of dopaminergic neurons in the substantia nigra and the subsequent reductions in the interstitial concentrations of dopamine in the striatum are critical to the development of Parkinson’s disease. About 80% of cells from the substantia nigra can be destroyed before the clinical symptoms of Parkinson’s disease become apparent
PD is a progressive, incurable disorder with no definite preventive treatment, although drugs are available to alleviate the symptoms and/or slow down the progress of the disease. Current therapy is based on dopamine replacement therapy, the most common drug treatments being dopaminomimetic agents, including L-DOPA, a dopamine precursor, as well as direct or indirect dopamine receptor agonists. L-DOPA is the mainstay in the treatment of PD, but because of tolerance problems and a wide range of adverse reactions, including involuntary movements and vomiting, a strong demand for new therapies exists. Among the various strategies, A2A AR blockers are considered a potential approach to treatment of the disease. Within the brain A2A ARs are richly expressed in the striatum, nucleus accumbens, and olfactory tubercle. A coexpression of A2A with D2 dopamine receptors has been reported in the GABAergic striatopallidal neurons where adenosine and dopamine agonists exert antagonistic effects in the regulation of locomotor activity. Activation of A2A ARs in striatopallidal neurons decreases the affinity of D2 receptors for dopamine, antagonizing the effects of D2 receptors.
The negative interaction between A2A and D2 receptors is at the basis of the use of A2A antagonists as a novel therapeutic approach in the treatment of PD. (Pharmacol. Ther. 2005, 105, 267). The recent discovery that the A2A can form functional heteromeric receptor complexes with other Gprote in-coupled receptors such as D2 and the mGlu5 receptors has also suggested new opportunities for the potential of A2A antagonists in PD. (J. Mol. Neurosci. 2005, 26, 209).
A2A knockout (KO) mice transient focal ischemia caused less neuronal damage in comparison to their wild-type (WT) littermates (J. Neurosci. 1999, 19, 9192.). Therefore, it seems that tonic activation of A2A ARs may be responsible for dangerous signal during injury, in contrast to the neuroprotective effects induced by endogenous Al activation. Recently, selective inactivation or reconstitution of A2A ARs in bone-marrow cells revealed their contribution to the development of ischemic brain injury (J.F. Nat. Med. 2004, 10, 1081) Blockade of A2A ARs has recently been implicated in the treatment of movement disorders such as Parkinson’s disease (Trends Pharmacol. Sci. 1997, 18, 338-344) and in the treatment of cerebral ischaemia (Life Sci. 1994, 55, 61-65).
The potential utility of A2A AR antagonists in the treatment of Parkinson’s disease has been reviewed (CNS drugs, 1998, 10, 31 1-320). One advantage of A2A AR antagonist therapy is that the underlying neurodegenerative disorder may also be treated ((Ann. N. Y. Acad. Sci. 1997, 825 (Neuroprotective Agents), 3048). In particular, blockade of A2A AR function confers neuroprotection against MPTP-induced neurotoxicity in mice (Neurosci. 2001, 21, RC143).
Alzheimer’s disease (AD) is a neurodegenerative disorder of the central nervous system manifested by cognitive and memory deterioration, a variety of neuropsychiatric symptoms, behavioral disturbances, and progressive impairment of daily life activities. Recent research suggests that adenosine receptors play important roles in the modulation of cognitive function. Epidemiological studies have found an association between coffee (a nonselective adenosine receptor antagonist) consumption and improved cognitive function in AD patients and in the elderly. Long-term administration of caffeine in transgenic animal models showed a reduced amyloid burden in brain with better cognitive performance.

Antagonists of adenosine A2A receptors mimic these beneficial effects of caffeine on cognitive function. Neuronal cell cultures with amyloid beta in the presence of an A2A receptor antagonist completely prevented amyloid beta-induced neurotoxicity. These findings suggest that the adenosinergic system constitutes a new therapeutic target for AD, and caffeine and A2A receptor antagonists may have promise to manage cognitive dysfunction in AD (Curr Neuropharmacol. 2009 September; 7(3): 207-216).
High expression of A2A ARs has been found in platelets, leukocytes, vascular smooth muscle, and endothelial cells with important implications in the regulation of inflammatory responses. It is now well established that stimulation of the A2A AR in immune cells induces anti-inflammatory effects, mostly due to its ability to increase cAMP levels, which has strong immunosuppressive effects (Trends Immunol. 2005, 26, 299). Stimulation of A2A ARs inhibits neutrophil adherence to the endothelium, degranulation of activated neutrophils and monocytes, plus superoxide anion generation. A2A ARs have been recently defined as sensors and terminators of proinflammatory activities. The strongest evidence for the key role of A2A in inflammation is derived by the elegant study using mice deficient in A2A ARs (Nature 2001, 414, 916).

In this model the lack of A2A subtype leads to increased tissue inflammation and damage, thus suggesting a negative and nonredundant regulatory role for the A2A AR. This model permits one to appreciate that adenosinergic regulation of immune cells is fundamental in normal physiological control of inflammation in vivo in spite of the fact that other Gs-protein-coupled receptors and cAMP elevating ligands are present, such as cathecolamines, prostaglandins, dopamine, and histamine (Trends Immunol. 2005, 26, 299). Interestingly, the A2A AR has been demonstrated to be involved in promotion of wound healing and angiogenesis in healing wounds (Am. J. Physiol. Regul. Integr. Comp. Physiol. 2005, 289, R283).
Moreover, it plays an active role in the pathogenesis of dermal fibrosis, suggesting a role for antagonists as novel therapeutic approach in the treatment and prevention of dermal fibrosis in diseases such as scleroderma (Arthritis Rheum. 2006, 54, 2632) as well as hepatic fibrosis (Br. J. Pharmacol. 2006 Aug; 148(8): 1 144-55). Studies also suggest that A2A receptor antagonists may be beneficial for social memory impairment and hypertension (Behav Brain Res. 2005 Apr 30;159(2):197-205), sepsis (J Immunol. 2006 May 1 ; 176(9): 5616-26), spinal cord injury and neuroprotection (J Neuroinflammation. 201 1 Apr 12;8:31), retinopathy (IVOS, Jan. 2000, vol. 41 (1), 230-243, depression (Neurology. 2003 Dec 9;61(1 1 Suppl 6):S82-7), narcolepsy and other sleep related disorders (Prog Neurobiol. 2007 Dec;83(5):332-47), attention-deficit hyperactivity disorder (ADHD) (Behav Pharmacol. 2009 Mar;20(2): 134-45; Clinical Genetics (2000), 58(1), 31-40 and references therein),

Dr Rashmi Barbhaiya, CEO & Managing Director

… Dr Rashmi Barbhaiya, CEO & Managing Director and Dr Kasim Mookthiar, Chief Scientific Officer and SVP, Drug Discovery, Advinus Therapeutics …
Antagonists of the A2A receptor are potentially useful therapies for the treatment of addiction. Major drugs of abuse (opiates, cocaine, ethanol, and the like) either directly or indirectly modulate dopamine signaling in neurons particularly those found in the nucleus accumbens, which contain high levels OfA2A adenosine receptors. Dependence has been shown to be augmented by the adenosine signaling pathway, and it has been shown that administration of an A2A receptor antagonist redues the craving for addictive substances (“The Critical Role of Adenosine A2A Receptors and Gi βγ Subunits in Alcoholism and Addiction: From Cell Biology to Behavior”, by Ivan Diamond and Lina Yao, (The Cell Biology of Addiction, 2006, pp 291-316) and “Adaptations in Adenosine Signaling in Drug Dependence: Therapeutic Implications”, by Stephen P. Hack and Macdonald J. Christie, Critical Review in Neurobiology, Vol. 15, 235-274 (2003)). See also Alcoholism: Clinical and Experimental Research (2007), 31(8), 1302-1307.
A2A receptors may be beneficial for the treatment or prevention of disorders such as a movement disorder, for example, Parkinson’s disease or progressive supernuclear palsy, Restless leg syndrome, nocturnal myoclonus, cerebral ischaemia, Huntington’s disease, multiple system atrophy, corticobasal degeneration, Wilson’s disease or other disorders of basal ganglia which results in dyskinesias, post traumatic stress disorder. See for example WO200013682, WO200012409, WO2009156737, WO20091 1442, WO2008121748, WO2001092264, WO2007038284, WO2008002596, WO20091 1 1449, WO20091 1 1442, WO2008121748, WO2009156737, WO2003022283, WO2005044245, WO2008077557, WO20091 1 1449, WO2009705138, WO20091 1 1442, WO2007035542, WO20080870661, WO2008070529, WO20051 16026, WO2009055548, WO2007133983, WO2010045006, WO2010045015, WO2010045008 WO2009015236.
![]()

centre: Mr Ratan Tata, Chairman, Tata Sons, flanked by Dr Rashmi Barbhaiya (left), Managing Director and CEO, Advinus, and Mr R. Gopalakrishnan, …
ONE EXAMPLE………..
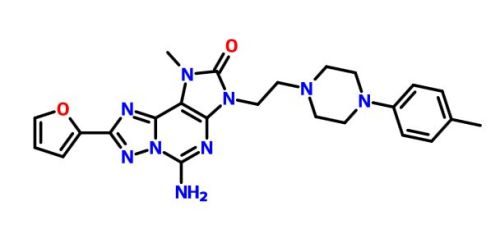
| Molecular Formula: | C26H31N9O4 |
|---|---|
| Molecular Weight: | 533.58224 g/mol |
 A1
A15-amino-8-(furan-2-yl)-3-[2-[4-[4-(2-methoxyethoxy)phenyl]piperazin-1-yl]ethyl]-1-methyl-[1,2,4]triazolo[5,1-f]purin-2-one
Example Al :
5-amino-8-(2-furyl)-3-[2-[4-[4-(2-methoxyethoxy)phenyl]piperazin- 1 -yl]ethyl]- 1 -methyl-[ 1 ,2,4]triazolo[5, 1 -f]purin-2-one
5-Amino-8-(2-furyl)-3-[2-[4-[4-(2-methoxyethoxy)phenyl]piperazin-l-
5-amino-8-(2-furyl)-3-[2-[4-[4-(2-methoxyethoxy)phenyl]piperazin- 1 -yl]ethyl]- 1 -methyl-[ 1 ,2,4]triazolo[5, 1 -f]purin-2-one
Step-1 : 2-[(2,5-Diamino-6-chloro-pyrimidin-4-yI)amino]ethanol
A mixture of 4,6-dichloropyrimidine-2,5-diamine (28g, 156mmol), ethanolamine (18ml, 312mmol) and ethanol (250ml) were heated at 100-1 10 °C for 16 hours. The mixture was cooled and solvent was removed. To the residue methanol (100ml) was added and stirred for 20 minutes. The solid was filtered off to obtain 2-[(2,5-diamino-6-chloro-pyrimidin-4-yl)amino]ethanol (22.0g, 70%).
‘H MR(400MHz, DMSO d6): δ 3.36-3.40 (m, 2H); 3.50-3.54 (m, 2H); 3.88 (bs, 2H); 4.74 (t, J=5.6Hz, 1H); 5.63 (bs, 2H); 6.51 (t, J=5.6Hz, 1H)
Step-2: 2-Amino-6-chloro-9-(2-hydroxyethyl)-7H-purin-8-one
A mixture of 2-[(2,5-diamino-6-chloro-pyrimidin-4-yl)amino]ethanol obtained in step 1 (l O.Og, 49.26mmol) in acetonitrile (400ml) were cooled to 0 °C. To this reaction mixture K2C03 (20.39gm, 147.7mmol) and 4-nitrophenyl chloroformate (19.8g, 98.52mmol)was added and stirred at 25-27 °C for 24 hours. This reaction mixture was filtered and washed with acetonitrile (300ml) and diethyl ether (300ml) respectively. Solid obtained was dried to obtain crude 2-amino-6-chloro-9-(2-hydroxyethyl)-7H-purin-8-one as a yellow solid. Small amount of crude material was purified by column chromatography to obtain pure product. ‘HNMR(400MHz, DMSO d6): δ 3.61-3.66 (m, 2H); 3.72-3.75 (m, 2H); 4.85 (t, J=6Hz, 1H); 6.60 (s, 2H); 1 1.21 (s, 1 H)
Step-3: 2-Amino-6-chloro-9-(2-hydroxyethyl)-7-methyl-purin-8-one
A mixture of 2-amino-6-chloro-9-(2-hydroxyethyl)-7H-purin-8-one obtained in step 2 (13g, 56.7mmol) , K2C03 (1 1.5g, 84mmol), methyl iodide (12g, 85.15mmol) and DMF (130ml) were stirred at 25-30 °C for 16 hours. The reaction mixture was concentrated and purified by column chromatography using 60-120 silica gel and 4% methanol in DCM as an eluent to obtain 2-amino-6-chloro-9-(2-hydroxyethyl)-7-methyl-purin-8-one (8g, 58%) as an off white solid.
‘HNMR(400MHz, DMSO d6): δ 3.42 (s, 3H); 3.65 (t, J=5.6Hz, 2H); 3.78 (t, J=5.6Hz, 2H); 4.85 (t, J=5.6Hz, 1H); 6.69 (bs, 2H).
Step-4: 2-Amino-6-hydrazino-9-(2-hydroxyethyl)-7-methyI-purin-8-one
A mixture of 2-amino-6-chloro-9-(2-hydroxyethyl)-7-methyl-purin-8-one obtained in step 3 (8g, 32.9mmol) , Hydrazine hydrate (16ml ,32.9mmol) and ethanol (300ml) were heated at 100-1 10 °C for 16 hours. The reaction mixture was concentrated and solid obtained was filtered off and dried to obtain 2-amino-6-hydrazino-9-(2-hydroxyethyl)-7-methyl-purin-8-one (7g, 89 %) as an off white solid.
‘HNMR(400MHz, DMSO d6): δ 3.37 (s, 3H); 3.58-3.61 (m, 2H); 3.71 (t, J=6Hz, 2H); 4.29 (bs, 2H); 4.87 (t, J=5.6Hz, 1H), 6.00 (bs, 2H); 7.63 (s, 1H).
Step-5: N’-[2-Amino-9-(2-hydroxyethyl)-7-methyl-8-oxo-purin-6-yl]furan-2-carbohydrazide
2-amino-6-hydrazino-9-(2-hydroxyethyl)-7-methyl-purin-8-one (4.5g, 18.18mmol) obtained in step 4, 2-furoic acid (2.53g, 22.5mmol), HOBT (2.53g, 18.8 mmol) and N-methylmorpholine were taken in dimethylformamide (40ml). l-Ethyl-3(3′-dimethylaminopropryl)carbodiimide hydrochloride (EDCI.HCl) (5.4g, 28.2mmol) was added to the reaction mixture and stirred at 25-27 °C for 14 hours. The reaction mixture was evaporated and residue was purified by column chromatography to obtain N’-[2-amino-9-(2-hydroxyethyl)-7-methyl-8-oxo-purin-6-yl]furan-2-carbohydrazide (5.3g, 84%) as an off white solid.
‘HNMR (400MHZ, DMSO d6): δ 3.43 (s, 3H); 3.59-3.63 (m, 2H); 3.74 (t, J=6Hz, 2H); 4.88 (t, J=5.6Hz, 1H); 5.98 (bs, 2H); 6.67 (bs, 1H); 7.25 (d, J=3.2Hz, 1H); 7.90 (s, 1H); 8.35 (s, 1H); 10.28 (s, lH).
Step-6: 5-Amino-8-(2-furyl)-3-(2-hydroxyethyl)-l-methyl-[l^,4]triazolo[5,l-flpurin-2-one
A mixture of N’-[2-amino-9-(2-hydroxyethyl)-7-methyl-8-oxo-purin-6-yl]furan-2-carbohydrazide obtained in step 5 (5.3g, 15.9mmol), Ν,Ο-bistrimethylsilylacetamide (27ml, 1 1 1.4mmol) and hexamethyldisilazane (83ml, 397mmol) were heated at 1 10-120 °C for 16 hours. The reaction mixture was quenched with methanol (100ml) and water (100ml) and organic volatiles were evaporated. The solid obtained was filtered off and washed with water (30ml) followed by diethyl ether (100ml) to obtain 5-amino-8-(2-furyl)-3-(2-hydroxyethyl)-l-methyl-[l,2,4]triazolo[5,l-f]purin-2-one (3.50g, 71%) as an off white solid.
‘HNMR (400MHZ, DMSO d6): δ 3.56 (s, 3H); 3.67-3.70 (m, 2H); 3.84-3.87 (m, 2H); 4.88 (t, J=5.6Hz, 1H); 6.73 (bs, 1H); 7.20 (bs, 1H); 7.79 (bs, 2H); 7.94 (bs, 1H).
Step-7: 2-[5-Amino-8-(2-furyl)-l-methyl-2-oxo-[l,2,4]triazolo[5,l-fJpurin-3-yl]ethyl 4-methylbenzenesulfonate
A mixture of 5-amino-8-(2-furyl)-3-(2-hydroxyethyl)-l -methyl-[l,2,4]triazolo[5, l-fJpurin-2-one obtained in step 6 (3.5g, l lmmol), p-toluene sulphonylchloride (5.2 g, 27mmol) were taken in pyridine (30ml)and stirred at 25-27 °C for 16 hours. To the reaction mixture hexane (100ml) was added and solid obtained was filtered off and washed with water (100ml) followed by hexane (100ml) to obtain 2-[5-amino-8-(2-furyl)-l-methyl-2-oxo-[l,2,4]triazolo[5, l-f]purin-3-yl]ethyl 4-methylbenzenesulfonate (4.1g, 78%) as a brown solid. ‘HNMR (400MHz, DMSO d6): δ 2.02 (s, 3H); 3.49 (s, 3H); 3.99 (t, J=4.8Hz, 2H); 4.71 (t, J=4.8Hz, 2H); 6.73-6.75 (m, 1H); 7.01 (d, J=8Hz, 2H); 7.23 (d, J=3.2Hz, 1H); 7.41 (d, J=8.4Hz, 2H); 7.78 (bs, 2H); 7.96 (d, J=1.2Hz, 1H).
Step-8: : 5-Amino-8-(2-furyl)-3-[2-[4-[4-(2-methoxyethoxy)phenyl]piperazin-l-yl]ethyl]-l-methyl-[l,2,4]triazolo[5,l-f)purin-2-one
A mixture of 2-[5-amino-8-(2-furyl)-l-methyl-2-oxo-[l ,2,4]triazolo[5, l-f]purin-3-yl]ethyl 4-methylbenzenesulfonate obtained in step 7 (0.25g, 0.533mmol), l-[4-(2-Methoxy-ethoxy)-phenyl]-piperazine (0.188g, 0.799mmol) and DIPEA (0.27ml, 1.599mmol) were taken in DMF (5ml) and stirred at 80 °C for 16 hours. To the reaction mixture water (100ml) was added and solid obtained was filtered off. The crude product was purified by column chromatography to obtain 5-amino-8-(2-furyl)-3-[2-[4-[4-(2-methoxyethoxy)phenyl]piperazin- 1 -yl]ethyl]- 1 -methyl-[ 1 ,2,4]triazolo[5, 1 -f]purin-2-one (0.135g, 47%) as an off white solid
‘HNMR (400MHz, DMSO d6): δ 2.60 (bs, 4H); 2.68 (t, J=6.4Hz, 2H); 2.96 (bs, 4H); 3.29 (s, 3H); 3.56 (s, 3H); 3.59-3.62 (m, 2H); 3.94-4.00 (m, 4H); 6.71 -6.73 (m, 1H); 6.79-6.86 (m, 4H); 7.19 (dd, J=3.2Hz, 1.2Hz, 1H); 7.80 (bs, 2H); 7.94 (bs, 1H).
ANOTHER……..
Example Gl: 5-Amino-l-ethyl-8-(2-furyl)-3-[2-[4-[4-(2-methoxyethoxy)phenyl]piperazin-l-yl]ethyl]-[l,2,4]triazolo[5,l-i]purin-2-one

Step-1 : 2-Amino-6-chloro-7-ethyl-9-(2-hydroxyethyl)purin-8-one
(Procedure is same as step-3 in example Al)
‘HNMR (400MHz, DMSO d6): δ 1.21 (t, J=7.2Hz, 3H); 3.64 (s, 2H); 3.78 (t, J=6Hz, 2H);
3.92 (q, J=7.2Hz, 2H); 4.92 (bs, I H); 6.7 (bs, 2H).
Step-2 : 2-Amino-7-ethyl-6-hydrazino-9-(2-hydroxyethyl)purin-8-one
(Procedure is same as step-4 in example Al)
‘ HNMR (400MHz, DMSO d6): δ 1.07 (t, J=6.8Hz, 3H); 3.59 (q, J=6Hz, 2H); 3.72 (t, J=6Hz,
2H); 3.91 (q, J=6.8Hz, 2H); 4.32 (bs, 2H); 4.86 (t, J=5.6Hz, IH); 5.99 (bs, 2H), 7.55 (bs, IH).
Step-3: N’-[2-Amino-7-ethyl-9-(2-hydroxyethyl)-8-oxo-purin-6-yl]furan- 2carbohydrazide (Procedure is same as step-5 in example Al)
Crude product was used in next step
Step-4: 5-Amino-l-ethyI-8-(2-furyl)-3-(2-hydroxyethyl)-[l,2,4]triazolo[5,l-flpurin-2-one
(Procedure is same as step-6 in example Al)
‘H MR (400MHZ, DMSO d6): δ 1.34 (t, J=7.2Hz, 3H); 3.67 (q, J=5.6Hz, 2H); 3.84 (t, J=5.6Hz, 2H); 4.01 (q, J=7.2Hz, 2H); 4.87 (t, J=6Hz, IH); 6.70 (bs, IH); 7.17 (d, J=2.8Hz, I H); 7.18 (bs, 2H); 7.92 (bs, IH).
Step-5: 2-[5-Amino-l-ethyl-8-(2-furyl)-2-oxo-[l,2,4]triazoIo[5,l-f|purin-3-yl]ethyl 4- methylbenzenesulfonate (procedure is same as step-7 in example Al)
lHNMR (400MHz, DMSO d6): δ 1.35 (t, J=7.2Hz, 3H); 2.00 (s, 3H); 3.95-4.00 (m, 4H); 4.47 (bs, 2H); 6.74 (s, IH); 7.00 (d, J=7.6Hz, 2H); 7.22 (s, IH); 7.42 (d, J=7.6Hz, 2H); 7.78 (bs, 2H); 7.97 (bs, IH).
Step-6: 5-Amino-l-ethyl-8-(2-furyl)-3-[2-[4-[4-(2-methoxyethoxy)phenyi]piperazin-l- yl]ethyl]-[l,2,4]triazolo[5,l-f]purin-2-one (procedure is same as step-8 in example Al)
HNMR(400MHz, DMSO d6): δ 1.35 (t, J=7.2Hz, 3H); 2.60 (bs, 4H); 2.68 (t, J=6.8Hz, 2H); 2.95 (bs, 4H); 3.28(s, 3H);3.61 (t, J=4.4Hz, 2H); 3.94-4.04 (m, 6H); 6.72 (dd, J=2Hz, 3.6Hz, I H); 6.78-6.85 (m, 4H); 7.19 (d, J=3.2Hz, IH); 7.81(bs, 2H); 7.94 (s, IH).
Representative compounds of the present disclosure were tested and had micromolar to nanomolar activity.

 A1 ABOVE
A1 ABOVE
A7 ABOVE

A9 ABOVE

A13 ABOVE
A31 ‘HNMR (400MHz, DMSO d6): δ 2.62 (bs,4H); 2.68 (t, J=6.8Hz, 2H); 2.85 (bs, 4H); 3.28 (s, 3H); 3.57 (s, 3H); 3.59-3.62 (m, 2H); o 3.95 (t, J=6.8Hz, 2H); 4.01-4.04 (m, 2H);
5-Amino-3-[2-[4-[2-fluoro-4-(2- 6.66-6.68 (m, 1H); 6.72 (dd, J=2 Hz,3.6Hz, methoxyethoxy)phenyl]piperazin-l-yl]ethyl]-8- 1H); 6.79 (dd, J=2.8Hz, 14Hz, 1H); 6.92 (t, (2-furyl)- 1 -methyl-[ 1 ,2,4]triazolo[5, 1 -f|purin-2- J=9.6Hz, 1H); 7.19 (d, J=3.2Hz, 1 H); 7.93 one (bs, 2H); 7.93-7.94 (m, 1H).
A31 ABOVE
A32 HNM (400MHz, DMSO d6): δ 2.59 (bs,
4H); 2.68(t, J=6.4Hz, 2H); 3.27(t, J=4.8Hz, 4H); 3.56 (s, 3H); 3.96 (t, J=6.4Hz, 2H);
0 6.72(dd, J=2Hz, 3.6Hz, 1H); 6.99 (d, J=8.8Hz,
4-[4-[2-[5-Amino-8-(2-furyl)-l-methyl-2-oxo- 2H); 7.19 (d, J=3.6Hz, 1H);7.56 (d, J=8.8Hz, [ 1 ,2,4]triazolo[5, 1 -f]purin-3-yl]ethyl]piperazin- 2H); 7.80 (bs, 2H); 7.93 (bs, lH).
l-yl]benzonitrile
A32 ABOVE
A36 ‘HNMR(400MHz, CDCI3): δ θ.09 (d,
J=4.4Hz, 2H); 0.50 (d, J=6.8Hz, 2H); 0.82- 0.89 (m, 1H); 2.24 (d, J=6.0Hz, 2H): 2.52- 2.72 (m, 8H); 2.80 (t, J=6.4Hz, 2H); 3.76 (s,
5-Amino-3-[2-[4-(cyclopropylmethyl)piperazin- 3H); 4.07 (t, J=6.8Hz, 2H); 5.89 (bs, 2H); l -yl]ethyl]-8-(2-furyl)-l-methyl- 6.61 (bs, 1H); 7.22 (d, J=2.4Hz, 1H); 7.64 (s, [ 1 ,2,4]triazolo[5, 1 -f]purin-2-one 1H).
A36 ABOVE
A38 ‘HNMR(400MHz, CDCI3): δ 2.62 . (t,
J=4.4Hz, 4H); 2.79 (t, J=6.4Hz, 2H); 2.81 (s, 6H); 3.22 (t, J=4.4Hz, 4H): 3.77 (s, 3H); 4.06 (t, J=6.8Hz, 2H); 5.74 (bs, 2H); 6.60 (dd,
4-[2-[5-Amino-8-(2-fiiryl)- 1 -methyl-2-oxo- J=2.0Hz, 3.2Hz, 1H); 7.24 (d, J=3.6Hz, 1H);
[ 1 ,2,4]triazolo[5, 1 -f]purin-3-yl]ethyl]-N,N- 7.65 (s, 1H).
dimethy l-piperazine- 1 -sulfonamide
A38 ABOVE
A39 ‘HNMR(400MHZ, DMSO d6): δ 1.89-1.94
im, 1H); 2.09-2.18 .(m, 1 H); 2.60 (bs, 4H); 2.67 (t, J=6.4Hz, 2H); 2.96 (bs, 4H); 3.56 (s, 3H); 3.69-3.85 (m, 4H); 3.95 (t, J=6.4Hz,
2H); 4.89 (bs, 1H); 6.72 (dd, J=2.0, 3.2Hz,
5-Amino-8-(2-furyl)-l -methyl-3-[2-[4-(4- 1H); 6.78 (d, J=9.2Hz, 2H); 6.85 (d, J=9.2Hz, tetrahydrofuran-3-yloxyphenyl)piperazin- 1 – 2H): 7.20 (d, J=3.2Hz, 1 H); 7.80 (bs, 2H); yl]ethyl]-[l ,2,4]triazolo[5,l-f]purin-2-one
7.93 (s, 1H).
A39 ABOVE
A42 ‘HNMR(400MHz, CDCI3): δ
2.26 (s,3H); 2.94-2.97 (m, 6H); 3.72 (s, 2H); 3.75 (s, 3H); 4.17 (t, J=6.4Hz, 2H); 5.74 (bs, 2H); 6.59 (dd, J=1.6Hz, 3.6Hz, 1H);7.13 (s, J=3.6Hz, IH); 7.21-7.24 (m, IH); 7.63 (s,
5-Amino-8-(2-furyl)-l-methyl-3-[2-(3-methyl- IH); 8.20 (bs, IH),
7,8-dihydro-5H- 1 ,6-naphthyridin-6-yl)ethyl]- [ 1 ,2,4]triazolo[5, 1 -f]purin-2-one
A42 ABOVE
A57 HNMR(400MHz, DMSO d6): δ 2.95 (t,
J=8Hz, 2H); 3.52 (s, 3H); 3.69 (s, 3H ), 3.97 (t, J=8Hz, 2H); 6.71 (dd, J=2Hz, 3.6Hz, I H );
5-Amino-8-(2-furyl)-3-[2-(4- 6.80 (dd, J=2Hz, 6.8Hz, 2H); 7.10 (d, methoxyphenyl)ethyl]- 1 -methyl- J=8.8Hz, 2H); 7.18 (dd, J=0.8Hz, 3.2Hz, I H );
[ 1 ,2,4]triazolo[5, 1 -f]purin-2-one 7.80 (bs, 2H), 7.94 (dd, J=lHz, 2Hz, I H ).
A57 ABOVE
A58 HNMR(400MHz, DMSO d6): δ 2.61 (bs,
4H); 2.68 (bs, 2H); 3.05(bs, 4H); 3.57 (s, 3H ), 3.96 (bs, 2H); 6.72 (bs, IH); 6.92 (d, J=8Hz, 2H); 7.01 (d, J=10Hz, 2H );7.03(d, J=148Hz, IH); 7.19 (bs , 1 H); 7.80 (bs, 2H); 7.94 (s,
5-amino-3-[2-[4-[4- IH).
(difluoromethoxy)phenyl]piperazin-l-yl]ethyl]- 8-(2-furyl)- 1 -methyl-[ 1 ,2,4]triazolo[5, 1 -fjpurin- 2-one
A58 ABOVE
A62 O ‘HNMR (400MHz, DMSO d6): δ 0.66-0.70
(m, 4H); 1.90-1.94 (m, lH); 2.41 (bs, 4H); 2.65 (t, J=6Hz, 2H); 3.38 (bs, 2H); 3.56 (bs, 5H); 3.93 (t, J=6.4 Hz, 2H); 6.71 (bs, 1H );
5-Amino-3-[2-[4- 7.19 (d, J=2.4Hz, 1H); 7.79 (bs, 2H); 7.93 (bs,
(cyclopropanecarbonyl)piperazin- 1 -yl]ethyl]-8- 1H).
(2-furyl)- 1 -methyl-[ 1 ,2,4]triazolo[5, 1 -fjpurin-2- one
A62 ABOVE
A63 ‘HNMR (400MHz, DMSO d6): δ 0.07-0.10
(m, 2H); 0.40-0.44 (m, 2H); 0.88-0.94 (m,lH); 2.21 (d, J=6.4Hz, 2H); 2.41-2.45 (m, 4H); 2.64 (t, J=6.4Hz, 2H); 3.38 (bs,4H); 3.56
5-Amino-3-[2-[4-(2- (s, 3H); 3.93 (t, J=6.4Hz, 2H); 6.72 (dd, cyclopropylacetyl)piperazin-l -yl]ethyl]-8-(2- J=2Hz,3.6 Hz, 1H); 7.19-7.20 (m, 1H); 7.80 fury 1)- 1 -methyl-[ 1 ,2,4]triazolo[5, 1 -fJpurin-2- (bs, 2H); 7.93 (d, J=0.8 Hz, 1H).
one
A63 ABOVE

C1 ABOVE

E1 ABOVE

D3 ABOVE
G1 ABOVE
G2
H2
M1
M2
M3
M6
ETC AS IN TABLE……………..


/////////
n21c(nc4c(c1nc(n2)c3occc3)N(C(N4CCN5CCN(CC5)c6ccc(cc6)OCCOC)=O)C)N
CN1C2=C(N=C(N3C2=NC(=N3)C4=CC=CO4)N)N(C1=O)CCN5CCN(CC5)C6=CC=C(C=C6)OCCOC
CE-224535 for the treatment of rheumatoid arthritis and osteoarthritis


CE-224535
2-(4-Chloro-3-(3-(1-hydroxycycloheptyl)propanoyl)phenyl)-4-((2R)-2-hydroxy-3-methoxy-propyl)-1,2,4-triazine-3,5-dione
Benzamide, 2-chloro-5-(4,5-dihydro-4-((2R)-2-hydroxy-3-methoxypropyl)-3,5-dioxo-1,2,4-triazin-2(3H)-yl)-n-((1-hydroxycycloheptyl)methyl)-
2-chloro-N-[(1-hydroxycycloheptyl)methyl]-5-[4-[(2R)-2-hydroxy-3-methoxypropyl]-3,5-dioxo-1,2,4-triazin-2-yl]benzamide
Phase III
A P2X7 receptor antagonist potentially for the treatment of rheumatoid arthritis and osteoarthritis.
![]()
CE-224535
CAS No. 724424-43-5
mw 480.9, C22H29ClN4O6
DETAILS COMING…………….
US7407956
https://www.google.com.ar/patents/US7407956
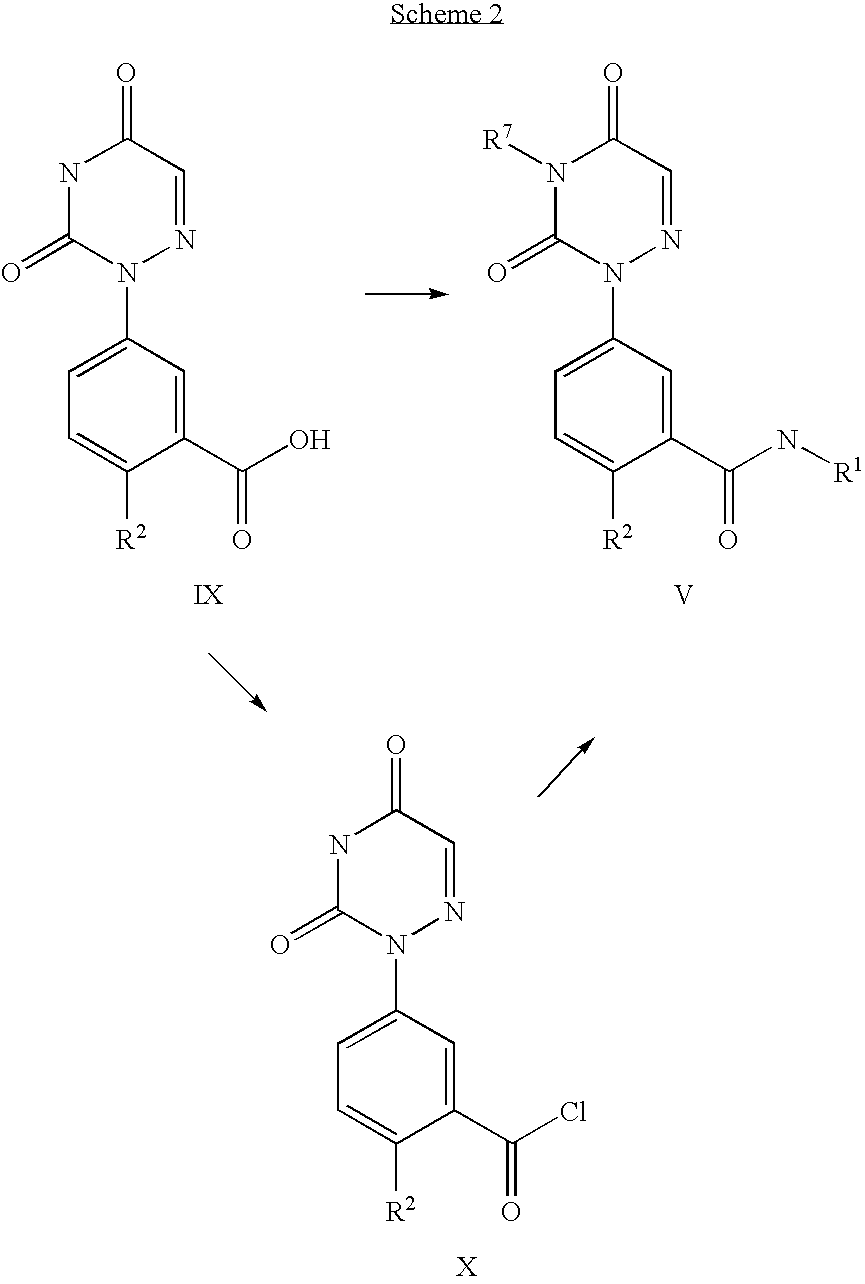
compounds of the formula I may be prepared according to the following reaction schemes and discussion. Unless otherwise indicated R1 through R7 in the reaction schemes and discussion that follows are as defined above.



| Patent | Submitted | Granted |
|---|---|---|
| Methods for preparing P2X7 inhibitors [US2005288288] | 2005-12-29 | |
| Combination therapies utilizing benzamide inhibitors of the P2X7 receptor [US2006018904] | 2006-01-26 | |
| Methods for preparing P2X7 inhibitors [US7235657] | 2005-12-29 | 2007-06-26 |
| Benzamide inhibitors of the P2X7 receptor [US7176202] | 2006-02-23 | 2007-02-13 |
| Benzamide Inhibitors of the P2X7 Receptor [US7671053] | 2009-02-12 | 2010-03-02 |
| Benzamide inhibitors of the P2X7 Ereceptor [US6974812] | 2004-09-16 | 2005-12-13 |
| Benzamide Inhibitors of The P2X7 Receptor [US7407956] | 2007-12-06 | 2008-08-05 |
/////////CE-224535, CE 224535
COC[C@@H](Cn1c(=O)cnn(c1=O)c2ccc(c(c2)C(=O)NCC3(CCCCCC3)O)Cl)O
GS 9883, Bictegravir an HIV-1 integrase inhibitor
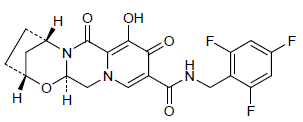

GS 9883, bictegravir
CAS 1611493-60-7
PHASE 3
HIV-1 integrase inhibitor
(2R,5S,13aR)-8-hydroxy-7,9-dioxo-N-[(2,4,6-trifluorophenyl)methyl]-2,3,4,5,7,9,13,13a-octahydro-2,5-methanopyrido[1′,2′:4,5]pyrazino[2,1-b][1,3]oxazepine-10-carboxamide
2,5-Methanopyrido(1′,2′:4,5)pyrazino(2,1-b)(1,3)oxazepine-10-carboxamide, 2,3,4,5,7,9,13,13a-octahydro-8-hydroxy-7,9-dioxo-N-((2,4,6-trifluorophenyl)methyl)-, (2R,5S,13aR)-
(2R,5S,13aR)-8-hydroxy-7,9-dioxo-N-(2,4,6-trifluorobenzyl)-2,3,4,5,7,9,13,13a-octahydro-2,5-methanopyrido[1′,2′:4,5]pyrazino[2,1-b][1,3]oxazepine-10-carboxamide
(2 ,5S,13aI )-8-hydroxy-7,9-dioxo-N-(2,4,6-trifluoroheoctahydro-2,5-methanopyrido[ 1 ‘,2’:4,5]pyrazino[2, 1 -b][ 1 ,3]oxazepine- 10-carboxamide
MF C21H18F3N3O5,
| MW | 449.37993 g/mol |
|---|
UNII-8GB79LOJ07; 8GB79LOJ07

BICTEGRAVIR
- 16 Nov 2015 Phase-III clinical trials in HIV-1 infections (Combination therapy, Treatment-naive) in USA (PO) (Gilead Pipeline, November 2015)
- 01 Jul 2015 Gilead Sciences completes a phase I trial in HIV-1 infections in USA and New Zealand (NCT02400307)
- 01 Apr 2015 Phase-I clinical trials in HIV-1 infections (In volunteers) in New Zealand (PO) (NCT02400307)
UPDATE Biktarvy (bictegravir/emtricitabine/tenofovir alafenamide); Gilead; For the treatment of HIV-1 infection in adults, Approved February 2018
Human immunodeficiency virus infection and related diseases are a major public health problem worldwide. Human immunodeficiency virus type 1 (HIV-1) encodes three enzymes which are required for viral replication: reverse transcriptase, protease, and integrase. Although drugs targeting reverse transcriptase and protease are in wide use and have shown effectiveness, particularly when employed in combination, toxicity and development of resistant strains have limited their usefulness (Palella, et al. N. Engl. J Med. (1998) 338:853-860; Richman, D. D. Nature (2001) 410:995-1001). Accordingly, there is a need for new agents that inhibit the replication of HIV and that minimize PXR activation when co-administered with other drugs.
Certain polycyclic carbamoylpyridone compounds have been found to have antiviral activity, as disclosed in PCT/US2013/076367. Accordingly, there is a need for synthetic routes for such compounds.
SYNTHESIS
WO 2014100323

PATENTS
xample 42
Preparation of Compound 42
(2 ,5S,13aI )-8-hydroxy-7,9-dioxo-N-(2,4,6-trifluorohe
octahydro-2,5-methanopyrido[ 1 ‘,2’:4,5]pyrazino[2, 1 -b][ 1 ,3]oxazepine- 10-carboxamide

42

Step 1
l-(2,2-dimethoxyethyl)-5-methoxy-6-(methoxycarbonyl)-4-oxo-l ,4-dihydropyridine-3-carboxylic acid (3.15 g, 10 mmol) in acetonitrile (36 mL) and acetic acid (4 mL) was treated with methanesuffhnic acid (0.195 mL, 3 mmol) and placed in a 75 deg C bath. The reaction mixture was stirred for 7 h, cooled and stored at -10 °C for 3 days and reheated to 75 °C for an additional 2 h. This material was cooled and carried on crude to the next step.
Step 2
Crude reaction mixture from step 1 (20 mL, 4.9 mmol) was transferred to a flask containing (lR,3S)-3-aminocyclopentanol (0.809 g, 8 mmol). The mixture was diluted with acetonitrile (16.8 mL), treated with potassium carbonate (0.553 g, 4 mmol) and heated to 85 °C. After 2 h, the reaction mixture was cooled to ambient temperature and stirred overnight. 0.2M HQ (50 mL) was added, and the clear yellow solution was extracted with dichloromethane (2×150 mL). The combined organic layers were dried over sodium sulfate, filtered and concentrated to 1.49 g of a light orange solid. Recrystallization from dichloimethane:hexanes afforded the desired intermediate 42 A: LC S-ESI (m/z): [M+H]+ calculated for Ci5Hi7N206: 321.1 1 ; found: 321.3.
Step 3
Intermediate 42-A (0.225 g, 0.702 mmol) and (2,4,6-trifluorophenyl)methanamine (0.125 g, 0.773 mmol) were suspended in acetonitrile (4 mL) and treated with N,N-diisopropylethylamine (DIPEA) (0.183 mmol, 1.05 mmol). To this suspension was added (dimethyiammo)- V,A/-dimethyi(3H-[l ,2,3]triazolo[4,5-&]pyridm~3-yiox.y)methammimum hexafluorophosphate (HATU, 0.294 g, 0.774 mmol). After 1.5 hours, the crude reaction mixture was taken on to the next step. LfJMS-ESlT (m/z): [M+H calculated for (\ ,l l.,, i \\:0< : 464.14; found: 464.2.
Step 4
To the crude reaction mixture of the previous step was added MgBr2
(0.258 g, 1.40 mmol). The reaction mixture was stirred at 50 °C for 10 minutes, acidified with 10% aqueous HC1, and extract twice with dichloromethane. The combined organic phases were dried over MgS04, filtered, concentrated, and purified by silica gel chromatography (EtOH/dichlormethane) followed by HPLC (ACN H2O with 0.1 % TFA modifier) to afford compound 42: 1H~ M (400 MHz, DMSO-</6) δ 12.43 (s, 1H), 10.34 (t, J = 5.7 Hz, IH), 8.42 (s, 1H), 7.19 (t, J = 8.7 Hz, 2H), 5.43 (dd, ./’ 9.5, 4.1 Hz, I H), 5.08 (s, i l l ). 4.66 (dd, ./ 12.9, 4.0 Hz, IH), 4.59 (s, 1 1 1 ). 4.56 4.45 (m, 2H), 4.01 (dd, J = 12.7, 9.7 Hz, IH), 1.93 (s, 4H), 1.83 (d, J —— 12.0 Hz, I H),
1.56 (dt, J = 12.0, 3.4 Hz, I H). LCMS-ESI+ (m/z): [M+H]+ calculated for { · Ί ί ] ΝΓ :Χ.¾ϋ : 450.13; found: 450.2.
PATENT
WO2015177537
PATENT
WO2015196116
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015196116&redirectedID=true
PATENT
WO2015196137
PATENT
http://www.google.com/patents/US20140221356
Example 42 Preparation of Compound 42 (2R,5S,13aR)-8-hydroxy-7,9-dioxo-N-(2,4,6-trifluorobenzyl)-2,3,4,5,7,9,13,13a-octahydro-2,5-methanopyrido[1′,2′:4,5]pyrazino[2,1-b][1,3]oxazepine-10-carboxamide

Step 1
-
1-(2,2-dimethoxyethyl)-5-methoxy-6-(methoxycarbonyl)-4-oxo-1,4-dihydropyridine-3-carboxylic acid (3.15 g, 10 mmol) in acetonitrile (36 mL) and acetic acid (4 mL) was treated with methanesulfonic acid (0.195 mL, 3 mmol) and placed in a 75 deg C. bath. The reaction mixture was stirred for 7 h, cooled and stored at −10° C. for 3 days and reheated to 75° C. for an additional 2 h. This material was cooled and carried on crude to the next step.
Step 2
-
Crude reaction mixture from step 1 (20 mL, 4.9 mmol) was transferred to a flask containing (1R,3S)-3-aminocyclopentanol (0.809 g, 8 mmol). The mixture was diluted with acetonitrile (16.8 mL), treated with potassium carbonate (0.553 g, 4 mmol) and heated to 85° C. After 2 h, the reaction mixture was cooled to ambient temperature and stirred overnight. 0.2M HCl (50 mL) was added, and the clear yellow solution was extracted with dichloromethane (2×150 mL). The combined organic layers were dried over sodium sulfate, filtered and concentrated to 1.49 g of a light orange solid. Recrystallization from dichlormethane:hexanes afforded the desired intermediate 42A: LCMS-ESI+ (m/z): [M+H]+ calculated for C15H17N2O6: 321.11; found: 321.3.
Step 3
-
Intermediate 42-A (0.225 g, 0.702 mmol) and (2,4,6-trifluorophenyl)methanamine (0.125 g, 0.773 mmol) were suspended in acetonitrile (4 mL) and treated with N,N-diisopropylethylamine (DIPEA) (0.183 mmol, 1.05 mmol). To this suspension was added (dimethylamino)-N,N-dimethyl(3H-[1,2,3]triazolo[4,5-b]pyridin-3-yloxy)methaniminium hexafluorophosphate (HATU, 0.294 g, 0.774 mmol). After 1.5 hours, the crude reaction mixture was taken on to the next step. LCMS-ESI+ (m/z): [M+H]+ calculated for C22H21F3N3O5: 464.14; found: 464.2.
Step 4
-
To the crude reaction mixture of the previous step was added MgBr2 (0.258 g, 1.40 mmol). The reaction mixture was stirred at 50° C. for 10 minutes, acidified with 10% aqueous HCl, and extract twice with dichloromethane. The combined organic phases were dried over MgSO4, filtered, concentrated, and purified by silica gel chromatography (EtOH/dichlormethane) followed by HPLC (ACN/H2O with 0.1% TFA modifier) to afford compound 42: 1H-NMR (400 MHz, DMSO-d6) δ 12.43 (s, 1H), 10.34 (t, J=5.7 Hz, 1H), 8.42 (s, 1H), 7.19 (t, J=8.7 Hz, 2H), 5.43 (dd, J=9.5, 4.1 Hz, 1H), 5.08 (s, 1H), 4.66 (dd, J=12.9, 4.0 Hz, 1H), 4.59 (s, 1H), 4.56-4.45 (m, 2H), 4.01 (dd, J=12.7, 9.7 Hz, 1H), 1.93 (s, 4H), 1.83 (d, J=12.0 Hz, 1H), 1.56 (dt, J=12.0, 3.4 Hz, 1H). LCMS-ESI+ (m/z): [M+H]+ calculated for C21H19F3N3O5: 450.13; found: 450.2.
PATENT
General Scheme I:

General Scheme II:



General Scheme II
General Scheme III:


General Scheme III
General Scheme IV:


G-1
General Scheme V:

II
EXAMPLES
In order for this invention to be more fully understood, the following examples are set forth. These examples are for the purpose of illustrating embodiments, and are not to be construed as limiting the scope of this disclosure in any way. The reactants used in the examples below may be obtained either as described herein, or if not described herein, are themselves either commercially available or may be prepared from commercially available materials by methods known in the art.
In one embodiment, a multi-step synthetic method for preparing a compound of Formula I is provided, as set forth below. In certain embodiments, each of the individual steps of the Schemes set forth below is provided. Examples and any combination of two or more successive steps of the below Examples are provided.
A. Acylation and amidation of Meldrum ‘s acid to form C-la:

[0520] In a reaction vessel, Meldrum’s acid (101 g, 1.0 equivalent) and 4-dimethylaminopyridine (1.8 g, 0.2 equivalents) were combined with acetonitrile (300 mL). The resulting solution was treated with methoxyacetic acid (6.2 mL, 1.2 equivalents). Triethylamine (19.4 mL, 2.0 equivalents) was added slowly to the resulting solution, followed by pivaloyl chloride (9.4 mL, 1.1 equivalents). The reaction was then heated to about 45 to about 50 °C and aged until consumption of Meldrum’s acid was deemed complete.
A separate reaction vessel was charged with acetonitrile (50 mL) and J-la (13.4 g, 1.2 equivalents). The resulting solution was treated with trifluoroacetic acid (8.0 mL, 1.5 equivalents), and then this acidic solution was added to the acylation reaction in progress at about 45 to about 50 °C.
The reaction was allowed to age for at least 18 hours at about 45 to about 50 °C, after which time the solvent was removed under reduced pressure. The crude residue was dissolved in ethyl acetate (150 mL), and the organic layer was washed with water. The combined aqueous layers were extracted with ethyl acetate. The combined organic layers were washed with saturated sodium bicarbonate solution, and the combined bicarbonate washes were back extracted with ethyl acetate. The combined organic layers were dried over magnesium sulfate, filtered, and concentrated under reduced pressure. The resulting crude material was purified twice via silica gel chromatography to yield C-la.
lH NMR (400 MHz, CDC13): δ 7.12 (br, 1H), 6.66 (app t, J= 8.1 Hz, 2H), 4.50 (app d, J= 5.7 Hz, 2H), 4.08 (s, 2H), 3.44 (s, 2H), 3.40 (s, 3H). 13C NMR (100 MHz, CDC13): δ 203.96, 164.90, 162.37 (ddd, J= 250.0, 15.7, 15.7 Hz), 161.71 (ddd, J = 250.3, 14.9, 10.9 Hz), 110.05 (ddd, J= 19.7, 19.7, 4.7 Hz), 100.42 (m), 77.58, 59.41, 45.71, 31.17 (t, J= 3.5 Hz). LCMS, Calculated: 275.23, Found: 275.97 (M).
I l l
B. Alkylation of C-la to form E-la:

A solution of C-la (248 mg, 1.0 equivalent) and 2-methyl tetrahydrofuran (1.3 niL) was treated with N,N-dimethylformamide dimethylacetal (0.1 mL, 1.1 equivalent) and stirred at room temperature overnight (~14 hours). The reaction was treated with aminoacetaldehyde dimethyl acetal (0.1 mL, 1.0 equivalents), and was allowed to age for about 2 hours, and then was quenched via the addition of 2 Ν HC1
(1.5 mL).
The reaction was diluted via the addition of ethyl acetate, and phases were separated. The aqueous layer was extracted with ethyl acetate. The combined organic layers were washed with brine, dried over magnesium sulfate, filtered, and concentrated under reduced pressure. The crude residue was purified via silica gel chromatography to yield E-la.
1H NMR (400 MHz, CDC13): δ 10.85 (s, 1H), 9.86 (s, 1H), 8.02 (d, J= 13.1 Hz, 1H), 6.65 (dd, J= 8.7, 7.7 Hz, 2H), 4.53 (d, J= 3.9 Hz, 2H), 4.40 (t, J= 5.1 Hz, 1H), 4.18 (s, 2H), 3.42 (s, 6H), 3.39 (m, 2H), 3.37 (s, 3H). 13C MR (100 MHz, CDC13): δ 193.30, 169.15, 162.10 (ddd, J= 248.9, 15.5, 15.5 Hz), 161.7 (ddd, J =
250.0, 14.9, 1 1.1 Hz), 161.66, 1 11.08 (ddd J= 19.9, 19.9, 4.7 Hz) 103.12, 100.29 (ddd, J= 28.1, 17.7, 2.3 Hz), 76.30, 58.83, 54.98, 53.53, 51.57, 29.89 (t, J= 3.3 Hz). LCMS, Calculated: 390.36, Found: 390.92 (M).
c. Cyclization of E-la to form F-la:

E-1a F-1a
] E-la (0.2 g, 1.0 equivalent), dimethyl oxalate (0.1 g, 2.5 equivalents) and methanol (1.5 mL) were combined and cooled to about 0 to about 5 °C. Sodium methoxide (0.2 mL, 30% solution in methanol, 1.75 equivalents) was introduced to the reaction slowly while keeping the internal temperature of the reaction below about 10 °C throughout the addition. After the addition was completed the reaction was heated to about 40 to about 50 °C for at least 18 hours.
After this time had elapsed, the reaction was diluted with 2 N HC1 (1.5 mL) and ethyl acetate (2 mL). The phases were separated, and the aqueous phase was extracted with ethyl acetate. The combined organic layers were washed with brine, dried over magnesium sulfate, filtered, and solvent was removed under reduced pressure. The resulting crude oil was purified via silica gel chromatography to afford F-la.
lR NMR (400 MHz, CDC13): δ 10.28 (t, J= 5.5 Hz, 1H), 8.38 (s, 1H), 6.66 – 6.53 (m, 2H), 4.58 (d, J= 5.6 Hz, 2H), 4.43 (t, J= 4.7 Hz, 1H), 4.00 (d, J= 4.7 Hz, 2H), 3.92 (s, 3H), 3.88 (s, 3H), 3.32 (s, 6H). 13C NMR (100 MHz, CDC13): δ 173.08, 163.81, 162.17, 162.14 (ddd, J= 249.2, 15.6, 15.6 Hz), 161.72 (ddd, J= 250.5, 15.0, 10.9 Hz), 149.37, 144.64, 134.98, 119.21, 1 10.53 (ddd, J= 19.8, 4.7, 4.7 Hz), 102.70, 100.22 (m), 60.68, 56.75, 55.61, 53.35, 30.64. LCMS, Calculated: 458.39, Found: 459.15 (M+H).
D. Alkylation and cyclization of C-la to form F-la:
1 . DMFDMA

C-1a NaOMe, MeOH, 40 °C F-1a
To a reaction vessel were added C-la (245 mg, 1.0 equivalent) and N,N-dimethylformamide dimethylacetal (0.5 mL, 4.3 equivalent). The reaction mixture was agitated for approximately 30 minutes. The reaction was then treated with 2-methyl tetrahydrofuran (2.0 mL) and aminoacetaldehyde dimethyl acetal (0.1 mL, 1.0 equivalent). The reaction was allowed to age for several hours and then solvent was removed under reduced pressure.
The resulting material was dissolved in methanol and dimethyl oxalate was added (0.3 g, 2.5 equivalents). The reaction mixture was cooled to about 0 to about 5 °C, and then sodium methoxide (0.4 mL, 30% solution in methanol, 1.75 equivalents) was introduced to the reaction slowly. After the addition was completed the reaction was heated to about 40 to about 50 °C.
After this time had elapsed, the reaction was cooled to room temperature and quenched via the addition of 2 Ν HC1 (1.5 mL). The reaction was then diluted with ethyl acetate, and the resulting phases were separated. The aqueous layer was extracted with ethyl acetate. The combined organic layers were dried over magnesium sulfate, filtered, and concentrated under reduced pressure. The crude residue was purified via silica gel chromatography to yield F-la.
lR NMR (400 MHz, CDC13): δ 10.28 (t, J= 5.5 Hz, 1H), 8.38 (s, 1H), 6.66 – 6.53 (m, 2H), 4.58 (d, J= 5.6 Hz, 2H), 4.43 (t, J= 4.7 Hz, 1H), 4.00 (d, J= 4.7 Hz, 2H), 3.92 (s, 3H), 3.88 (s, 3H), 3.32 (s, 6H). 13C NMR (100 MHz, CDC13): δ 173.08, 163.81, 162.17, 162.14 (ddd, J= 249.2, 15.6, 15.6 Hz), 161.72 (ddd, J= 250.5, 15.0, 10.9 Hz), 149.37, 144.64, 134.98, 119.21, 1 10.53 (ddd, J= 19.8, 4.7, 4.7 Hz), 102.70, 100.22 (m), 60.68, 56.75, 55.61, 53.35, 30.64. LCMS, Calculated: 458.39, Found: 459.15 (M+H).
E. Condensation of F-la with N-la to form G-la:

K2C03, MeCN, 75 °C
To a reaction vessel were added F-la (202 mg, 1.0 equivalent) and acetonitrile (1.4 mL). The resulting solution was treated with glacial acetic acid (0.2 mL, 6.0 equivalents) and methane sulfonic acid (0.01 mL, 0.3 equivalents). The reaction was then heated to about 70 to about 75 °C.
After 3 hours, a solid mixture of N-la (0.128g, 1.5 equivalents) and potassium carbonate (0.2 g, 2.7 equivalents) was introduced to the reaction at about 70 to about 75 °C. After the addition was completed, the reaction was allowed to progress for at least about 1 hour.
After this time had elapsed, water (1.4 mL) and dichloromethane (1.4 mL) were introduced to the reaction. The phases were separated, and the aqueous layer was extracted with dichloromethane. The combined organic layers were dried over magnesium sulfate, then were filtered and concentrated under reduced pressure. The resulting crude material was purified via silica gel chromatography to obtain G-la.
lR NMR (400 MHz, CDC13): δ 10.23 (t, J= 5.5 Hz, 1H), 8.39 (s, 1H), 6.60 (t, J= 8.1 Hz, 2H), 5.29 (dd, J= 9.5, 3.7 Hz, 2H), 4.57 (d, J= 5.4 Hz, 3H), 4.33 (dd, J = 12.8, 3.8 Hz, 1H), 4.02 – 3.87 (m, 1H), 3.94 (s, 3H), 2.06 – 1.88 (m, 4H), 1.78 (dd, J = 17.2, 7.5 Hz, 1H), 1.55 – 1.46 (m, 1H). 13C MR (100 MHz, CDC13): δ 174.53, 163.75, 162.33 (dd, J= 249.4, 15.7, 15.7 Hz), 161.86 (ddd, J= 250.4, 14.9, 10.9 Hz), 154.18, 154.15, 142.44, 129.75, 1 18.88, 1 10.58 (ddd, J= 19.8, 4.7, 4.7 Hz), 100.42 (m), 77.64, 74.40, 61.23, 54.79, 51.13, 38.31, 30.73, 29.55, 28.04. LCMS, Calculated: 463.14, Found: 464.15 (M+H).
Γ. Deprotection of G-la to form a compound of Formula la:
G-la (14 g) was suspended in acetonitrile (150 mL) and dichloromethane (150 mL). MgBr2 (12 g) was added. The reaction was heated to 40 to 50 °C for approximately 10 min before being cooled to room temperature. The reaction was poured into 0.5M HC1 (140 mL) and the layers separated. The organic layer was washed with water (70 mL), and the organic layer was then concentrated. The crude product was purified by silica gel chromatography (100% dichloromethane up to 6% ethanol/dichloromethane) to afford la.
REFERENCES
| Patent | Submitted | Granted |
|---|---|---|
| POLYCYCLIC-CARBAMOYLPYRIDONE COMPOUNDS AND THEIR PHARMACEUTICAL USE [US2014221356] | 2013-12-19 | 2014-08-07 |
| US9216996 | Dec 19, 2013 | Dec 22, 2015 | Gilead Sciences, Inc. | Substituted 2,3,4,5,7,9,13,13a-octahydropyrido[1′,2′:4,5]pyrazino[2,1-b][1,3]oxazepines and methods for treating viral infections |
see full gravir series at…………..http://medcheminternational.blogspot.in/p/ravir-series.html
//////////
C1CC2CC1N3C(O2)CN4C=C(C(=O)C(=C4C3=O)O)C(=O)NCC5=C(C=C(C=C5F)F)F
OR
c1c(cc(c(c1F)CNC(=O)c2cn3c(c(c2=O)O)C(=O)N4[C@H]5CC[C@H](C5)O[C@@H]4C3)F)F
![]()

BICTEGRAVIR, NEW PATENT, WO 2018005328, CONCERT PHARMA
WO2018005328) DEUTERATED BICTEGRAVIR
CONCERT PHARMACEUTICALS, INC.
TUNG, Roger, D.; (US)

Concert CEO Roger Tung
Novel deuterated forms of bictegravir is claimed. Gilead Sciences is developing the integrase inhibitor bictegravir as an oral tablet for the treatment of HIV-1 infection.
This invention relates to deuterated forms of bictegravir, and pharmaceutically acceptable salts thereof. In one aspect, the invention provides a compound of Formula (I) or a pharmaceutically acceptable salt thereof, wherein each of Y1, Y2, Y3, Y4a, Y4b, Y5a, Y5b, Y6, Y7a, Y7b, Y8, Y9, Y10a, Y10b, Y11a, and Y11b is independently hydrogen or deuterium; provided that if each Y1, Y2, Y3, Y4a, Y4b, Y5a, Y5b, Y6, Y7a, Y7b, Y8, Y9, Y10a, Y10b, and Y11 is hydrogen, then Y11b is deuterium.

Many current medicines suffer from poor absorption, distribution, metabolism and/or excretion (ADME) properties that prevent their wider use or limit their use in certain indications. Poor ADME properties are also a major reason for the failure of drug candidates in clinical trials. While formulation technologies and prodrug strategies can be employed in some cases to improve certain ADME properties, these approaches often fail to address the underlying ADME problems that exist for many drugs and drug candidates. One such problem is rapid metabolism that causes a number of drugs, which otherwise would be highly effective in treating a disease, to be cleared too rapidly from the body. A possible solution to rapid drug clearance is frequent or high dosing to attain a sufficiently high plasma level of drug. This, however, introduces a number of potential treatment problems such as poor patient compliance with the dosing regimen, side effects that become more acute with higher doses, and increased cost of treatment. A rapidly metabolized drug may also expose patients to undesirable toxic or reactive metabolites.
[3] Another ADME limitation that affects many medicines is the formation of toxic or biologically reactive metabolites. As a result, some patients receiving the drug may experience toxicities, or the safe dosing of such drugs may be limited such that patients receive a suboptimal amount of the active agent. In certain cases, modifying dosing intervals or formulation approaches can help to reduce clinical adverse effects, but often the formation of such undesirable metabolites is intrinsic to the metabolism of the compound.
[4] In some select cases, a metabolic inhibitor will be co-administered with a drug that is cleared too rapidly. Such is the case with the protease inhibitor class of drugs that are used to treat HIV infection. The FDA recommends that these drugs be co-dosed with ritonavir, an inhibitor of cytochrome P450 enzyme 3A4 (CYP3A4), the enzyme typically responsible for their metabolism (see Kempf, D.J. et al., Antimicrobial agents and chemotherapy, 1997, 41(3): 654-60). Ritonavir, however, causes adverse effects and adds to the pill burden for HIV patients who must already take a combination of different drugs. Similarly, the
CYP2D6 inhibitor quinidine has been added to dextromethorphan for the purpose of reducing rapid CYP2D6 metabolism of dextromethorphan in a treatment of pseudobulbar affect. Quinidine, however, has unwanted side effects that greatly limit its use in potential combination therapy (see Wang, L et al., Clinical Pharmacology and Therapeutics, 1994, 56(6 Pt 1): 659-67; and FDA label for quinidine at http://www.accessdata.fda.gov).
[5] In general, combining drugs with cytochrome P450 inhibitors is not a satisfactory strategy for decreasing drug clearance. The inhibition of a CYP enzyme’s activity can affect the metabolism and clearance of other drugs metabolized by that same enzyme. CYP inhibition can cause other drugs to accumulate in the body to toxic levels.
[6] A potentially attractive strategy for improving a drug’s metabolic properties is deuterium modification. In this approach, one attempts to slow the CYP-mediated metabolism of a drug or to reduce the formation of undesirable metabolites by replacing one or more hydrogen atoms with deuterium atoms. Deuterium is a safe, stable, non-radioactive isotope of hydrogen. Compared to hydrogen, deuterium forms stronger bonds with carbon. In select cases, the increased bond strength imparted by deuterium can positively impact the ADME properties of a drug, creating the potential for improved drug efficacy, safety, and/or tolerability. At the same time, because the size and shape of deuterium are essentially identical to those of hydrogen, replacement of hydrogen by deuterium would not be expected to affect the biochemical potency and selectivity of the drug as compared to the original chemical entity that contains only hydrogen.
[7] Over the past 35 years, the effects of deuterium substitution on the rate of metabolism have been reported for a very small percentage of approved drugs (see, e.g., Blake, MI et al, J Pharm Sci, 1975, 64:367-91; Foster, AB, Adv Drug Res 1985, 14:1-40 (“Foster”); Kushner, DJ et al, Can J Physiol Pharmacol 1999, 79-88; Fisher, MB et al, Curr Opin Drug Discov Devel, 2006, 9:101-09 (“Fisher”)). The results have been variable and unpredictable. For some compounds deuteration caused decreased metabolic clearance in vivo. For others, there was no change in metabolism. Still others demonstrated increased metabolic clearance. The variability in deuterium effects has also led experts to question or dismiss deuterium modification as a viable drug design strategy for inhibiting adverse metabolism (see Foster at p.35 and Fisher at p.101).
[8] The effects of deuterium modification on a drug’s metabolic properties are not predictable even when deuterium atoms are incorporated at known sites of metabolism. Only by actually preparing and testing a deuterated drug can one determine if and how the rate of metabolism will differ from that of its non-deuterated counterpart. See, for example, Fukuto et al. (J. Med. Chem.1991, 34, 2871-76). Many drugs have multiple sites where metabolism is possible. The site(s) where deuterium substitution is required and the extent of deuteration necessary to see an effect on metabolism, if any, will be different for each drug.
Exemplary Synthesis
[72] Deuterium-modified analogs of bictegravir can be synthesized by means known in the art of organic chemistry. For instance, using methods described in US Patent No.9,216,996 (Haolun J. et al., assigned to Gilead Sciences, Inc. and incorporated herein by reference), using deuterium-containing reagents provides the desired deuterated analogs.
[73] Such methods can be carried out utilizing corresponding deuterated and optionally, other isotope-containing reagents and/or intermediates to synthesize the compounds delineated herein, or invoking standard synthetic protocols known in the art for introducing isotopic atoms to a chemical structure.
[74] A convenient method for synthesizing compounds of Formula I is depicted in the Schemes below.
 [75] A generic scheme for the synthesis of compounds of Formula I is shown in Scheme 1 above. In a manner analogous to the procedure described in Wang, H. et al. Org. Lett.2015, 17, 564-567, aldol condensation of compound 1 with appropriately deuterated compound 2 affords enamine 3. Enamine 3 is then reacted with primary amine 4 to afford enamine 5, which then undergoes cyclization with dimethyl oxalate followed by ester hydrolysis to provide carboxylic acid 7.
[75] A generic scheme for the synthesis of compounds of Formula I is shown in Scheme 1 above. In a manner analogous to the procedure described in Wang, H. et al. Org. Lett.2015, 17, 564-567, aldol condensation of compound 1 with appropriately deuterated compound 2 affords enamine 3. Enamine 3 is then reacted with primary amine 4 to afford enamine 5, which then undergoes cyclization with dimethyl oxalate followed by ester hydrolysis to provide carboxylic acid 7.
[76] In a manner analogous to the procedure described in US 9,216,996, acetal deprotection of carboxylic acid 7 followed by cyclization with appropriately deuterated aminocyclopentanol 9 provides carboxylic acid intermediate 10. Amide coupling with appropriately deuterated benzylamine 11 followed by deprotection of the methyl ether ultimately affords a compound of Formula I in eight overall steps from compound 1.
[77] Use of appropriately deuterated reagents allows deuterium incorporation at the Y1, Y2, Y3, Y4a, Y4b, Y5a, Y5b, Y6, Y7a, Y7b, Y8, Y9, Y10a, Y10b, Y11a, and Y11bpositions of a compound of Formula I or any appropriate intermediate herein, e.g., about 90%, about 95%, about 97%, about 98%, or about 99% deuterium incorporation at any Y1, Y2, Y3, Y4a, Y4b, Y5a, Y5b, Y6, Y7a, Y7b, Y8, Y9, Y10a, Y10b, Y11a, and/or Y11b.
[78] Appropriately deuterated intermediates 2a and 2b, for use in the preparation of compounds of Formula I according to Scheme 1, may be prepared from corresponding deuterated reagents as exemplified in Scheme 2 below.
S h 2 S th i f C d 2 d 2b

[79] Synthesis of compound 2a (wherein Y3=H) by acetal formation of N,N-dimethylformamide (DMF) with dimethylsulfate has been described in Mesnard, D. et. al. J. Organomet. Chem.1989, 373, 1-10. Replacing DMF with N,N-dimethylformamide-d1 (98-99 atom % D; commercially available from Cambridge Isotope Laboratories) in this reaction would thereby provide compound 2b (wherein Y3=D).
[80] Use of appropriately deuterated reagents allows deuterium incorporation at the Y3 position of a compound of Formula I or any appropriate intermediate herein, e.g., about 90%, about 95%, about 97%, about 98%, or about 99% deuterium incorporation at Y3.
[81] Appropriately deuterated intermediates 4a-4d, for use in the preparation of compounds of Formula I according to Scheme 1, may be prepared from corresponding deuterated reagents as exemplified in Scheme 3 below.

[82] As described in Malik, M. S. et. al. Org. Prep. Proc. Int.1991, 26, 764-766, acetaldehyde is converted to alkylhalide 14a via reaction with chlorine gas and subsequent acetal protection with CaCl2 in methanol. As described in CN 103739506, reaction of 14a with aqueous ammonia and then sodium hydroxide provides primary amine 4a (wherein Y9=Y10a=Y10b=H). Replacing acetaldehyde with acetaldehyde-d1, acetaldehyde-2,2,2-d3, or acetaldehyde-d4 (all commercially available from CDN Isotopes with 98-99 atom % D) in the sequence then provides access to compounds 4b (Y9=D, Y10a=Y10b=H), 4c (Y9=H,
Y10a=Y10b=D) and 4d (Y9=Y10a=Y10b=D) respectively (Schemes 3b-d).
[83] Use of appropriately deuterated reagents allows deuterium incorporation at the Y9, Y10a, and Y10b positions of a compound of Formula I or any appropriate intermediate herein, e.g., about 90%, about 95%, about 97%, about 98%, or about 99% deuterium incorporation at any Y9, Y10a, and/or Y10b.
[84] Appropriately deuterated intermediates 9a-9d, for use in the preparation of compounds of Formula I according to Scheme 1, may be prepared from corresponding deuterated reagents as exemplified in Scheme 4 below.
 [85] Following the procedures described by Gurjar, M. et. al. Heterocycles, 2009, 77, 909-925, meso-diacetate 16a is prepared in 2 steps from cyclopentadiene. Desymmetrization of 16a is then achieved enzymatically by treatment with Lipase as described in Specklin, S. et. al. Tet. Lett.201455, 6987-6991, providing 17a which is subsequently converted to aminocyclopentanol 9a (wherein Y4a=Y4b=Y5a=Y5b=Y6=Y7a=Y7b=Y8=H) via a 3 step sequence as reported in WO 2015195656.
[85] Following the procedures described by Gurjar, M. et. al. Heterocycles, 2009, 77, 909-925, meso-diacetate 16a is prepared in 2 steps from cyclopentadiene. Desymmetrization of 16a is then achieved enzymatically by treatment with Lipase as described in Specklin, S. et. al. Tet. Lett.201455, 6987-6991, providing 17a which is subsequently converted to aminocyclopentanol 9a (wherein Y4a=Y4b=Y5a=Y5b=Y6=Y7a=Y7b=Y8=H) via a 3 step sequence as reported in WO 2015195656.
[86] As depicted in Scheme 4b, aminocyclopentanol 9b (Y4a=Y4b=Y5a=Y5b=Y6=Y7a=Y7b= Y8=D) is obtained through an analogous synthetic sequence using cyclopentadiene-d6 and performing the penultimate hydrogenation with D2 in place of H2. Cyclopentadiene-d6 is prepared according to the procedure described in Cangoenuel, A. et. al. Inorg. Chem.2013, 52, 11859-11866.
[87] Alternatively, as shown in Scheme 4c, the meso-diol obtained in Scheme 4a is oxidized to the diketone following the procedure reported by Rasmusson, G.H. et. al. Org. Syn.1962, 42, 36-38. Subsequent mono-reduction with sodium borodeuteride and CeCl3 then affords the D1-alcohol in analogy to the method described in WO 2001044254 for the all-protio analog using sodium borohydride. Reduction of the remaining ketone using similar conditions provides the meso-D2-diol in analogy to the method reported in Specklin, S. et. al. Tet. Lett.2014, 55, 6987-6991 for the all protio analog using sodium borohydride. The meso-D2-diol is then converted to 9c (Y4a=Y4b=Y5a=Y5b=Y7a=Y7b=H, Y6=Y8=D) following the same procedures outlined in Scheme 4a.
[88] Likewise, the meso-diol obtained in Scheme 4b may be converted to 9d
(Y4a=Y4b=Y5a=Y5b=Y7a=Y7b=D, Y6=Y8=H) in an analogous manner as depicted in Scheme 4d. The use of deuterated solvents such as D2O or MeOD may be considered to reduce the risk of D to H exchange for ketone containing intermediates.
[89] Use of appropriately deuterated reagents allows deuterium incorporation at the Y4a, Y4b, Y5a, Y5b, Y6, Y7a, Y7b, and Y8 positions of a compound of Formula I or any appropriate intermediate herein, e.g., about 90%, about 95%, about 97%, about 98%, or about 99% deuterium incorporation at any Y4a, Y4b, Y5a, Y5b, Y6, Y7a, Y7b, and/or Y8.
[90] Appropriately deuterated intermediates 11a-11d, for use in the preparation of compounds of Formula I according to Scheme 1, may be prepared from corresponding deuterated reagents exemplified in Scheme 5 below.
Scheme 5. Synthesis of Benzylamines 11a-11d

//////////////////
FDA approves new orphan drug Uptravi (selexipag) to treat pulmonary arterial hypertension

KEEPING WATCHING THIS POSTS FOR SYNTHESIS UPDATES
December 22, 2015
On December 21, the U.S. Food and Drug Administration approved Uptravi (selexipag) tablets to treat adults with pulmonary arterial hypertension (PAH), a chronic, progressive, and debilitating rare lung disease that can lead to death or the need for transplantation.
“Uptravi offers an additional treatment option for patients with pulmonary arterial hypertension,” said Ellis Unger, M.D., director of the Office of Drug Evaluation I in the FDA’s Center for Drug Evaluation and Research. “The FDA supports continued efforts to provide new treatment options for rare diseases.”
PAH is high blood pressure that occurs in the arteries that connect the heart to the lungs. It causes the right side of the heart to work harder than normal, which can lead to limitations on exercise ability and shortness of breath, among other more serious complications.
Uptravi belongs to a class of drugs called oral IP prostacyclin receptor agonists. The drug acts by relaxing muscles in the walls of blood vessels to dilate (open) blood vessels and decrease the elevated pressure in the vessels supplying blood to the lungs.
Uptravi’s safety and efficacy were established in a long-term clinical trial of 1,156 participants with PAH. Uptravi was shown to be effective in reducing hospitalization for PAH and reducing the risks of disease progression compared to placebo. Participants were exposed to Uptravi in this trial for a median duration of 1.4 years.
Common side effects observed in those treated with Uptravi in the trial include headache, diarrhea, jaw pain, nausea, muscle pain (myalgia), vomiting, pain in an extremity, and flushing.
Uptravi was granted orphan drug designation. Orphan drug designation provides incentives such as tax credits, user fee waivers, and eligibility for exclusivity to assist and encourage the development of drugs for rare diseases.
Uptravi is marketed by San Francisco-based Actelion Pharmaceuticals US, Inc.
Selexipag, Uptravi
475086-01-2 CAS
(C26H32N4O4S, Mr = 496.6 g/mol)
A prostacyclin receptor (PGI2) agonist used to treat pulmonary arterial hypertension (PAH).
NIPPON SHINYAKU….INNOVATOR
Selexipag (brand name Uptravi) is a drug developed by Actelion for the treatment of pulmonary arterial hypertension (PAH). Selexipag and its active metabolite, ACT-333679 (MRE-269) (the free carboxylic acid), are agonists of the prostacyclin receptor, which leads to vasodilation in the pulmonary circulation.[1]
The US FDA granted it Orphan Drug status[2] (for PAH). It was approved by the U.S. FDA on 22 December 2015.[2]

ACT-333679 or MRE-269, the active metabolite of selexipag




PATENT
US2012/101276
http://www.google.st/patents/US20120101276?hl=pt-PT&cl=en
The present invention relates to a crystal of 2-{4-[N-(5,6-diphenylpyrazin-2-yl)-N-isopropylamino]butyloxy}-N-(methylsulfonyl)acetamide (hereinafter referred to as “compound A”).

BACKGROUND OF THE INVENTION
Compound A has an excellent PGI2 agonistic effect and shows a platelet aggregation inhibitory effect, a vasodilative effect, a bronchodilative effect, a lipid deposition inhibitory effect, a leukocyte activation inhibitory effect, etc. (see, for example, in WO 2002/088084 (“WO ‘084”)).
Specifically, compound A is useful as preventive or therapeutic agents for transient ischemic attack (TIA), diabetic neuropathy, diabetic gangrene, peripheral circulatory disturbance (e.g., chronic arterial occlusion, intermittent claudication, peripheral embolism, vibration syndrome, Raynaud’s disease), connective tissue disease (e.g., systemic lupus erythematosus, scleroderma, mixed connective tissue disease, vasculitic syndrome), reocclusion/restenosis after percutaneous transluminal coronary angioplasty (PTCA), arteriosclerosis, thrombosis (e.g., acute-phase cerebral thrombosis, pulmonary embolism), hypertension, pulmonary hypertension, ischemic disorder (e.g., cerebral infarction, myocardial infarction), angina (e.g., stable angina, unstable angina), glomerulonephritis, diabetic nephropathy, chronic renal failure, allergy, bronchial asthma, ulcer, pressure ulcer (bedsore), restenosis after coronary intervention such as atherectomy and stent implantation, thrombocytopenia by dialysis, the diseases in which fibrosis of organs or tissues is involved [e.g., Renal diseases (e.g., tuburointerstitial nephritis), respiratory diseases (e.g., interstitial pneumonia (pulmonary fibrosis), chronic obstructive pulmonary disease), digestive diseases (e.g., hepatocirrhosis, viral hepatitis, chronic pancreatitis and scirrhous stomachic cancer), cardiovascular diseases (e.g, myocardial fibrosis), bone and articular diseases (e.g, bone marrow fibrosis and rheumatoid arthritis), skin diseases (e.g, cicatrix after operation, scalded cicatrix, keloid, and hypertrophic cicatrix), obstetric diseases (e.g., hysteromyoma), urinary diseases (e.g., prostatic hypertrophy), other diseases (e.g., Alzheimer’s disease, sclerosing peritonitis; type I diabetes and organ adhesion after operation)], erectile dysfunction (e.g., diabetic erectile dysfunction, psychogenic erectile dysfunction, psychotic erectile dysfunction, erectile dysfunction associated with chronic renal failure, erectile dysfunction after intrapelvic operation for removing prostata, and vascular erectile dysfunction associated with aging and arteriosclerosis), inflammatory bowel disease (e.g., ulcerative colitis, Crohn’s disease, intestinal tuberculosis, ischemic colitis and intestinal ulcer associated with Behcet disease), gastritis, gastric ulcer, ischemic ophthalmopathy (e.g., retinal artery occlusion, retinal vein occlusion, ischemic optic neuropathy), sudden hearing loss, avascular necrosis of bone, intestinal damage caused by administration of a non-steroidal anti-inflammatory agent (e.g., diclofenac, meloxicam, oxaprozin, nabumetone, indomethacin, ibuprofen, ketoprofen, naproxen, celecoxib) (there is no particular limitation for the intestinal damage so far as it is damage appearing in duodenum, small intestine and large intestine and examples thereof include mucosal damage such as erosion and ulcer generated in duodenum, small intestine and large intestine), and symptoms associated with lumbar spinal canal stenosis (e.g., paralysis, dullness in sensory perception, pain, numbness, lowering in walking ability, etc. associated with cervical spinal canal stenosis, thoracic spinal canal stenosis, lumbar spinal canal stenosis, diffuse spinal canal stenosis or sacral stenosis) etc. (see, for example, in WO ‘084, WO 2009/157396, WO 2009/107736, WO 2009/154246, WO 2009/157397, and WO 2009/157398).
In addition, compound A is useful as an accelerating agent for angiogenic therapy such as gene therapy or autologous bone marrow transplantation, an accelerating agent for angiogenesis in restoration of peripheral artery or angiogenic therapy, etc. (see, for example, in WO ‘084).
Production of Compound A
Compound A can be produced, for example, according to the method described in WO ‘084, and, it can also be produced according to the production method mentioned below.

Step 1:
6-Iodo-2,3-diphenylpyrazine can be produced from 6-chloro-2,3-diphenylpyrazine by reacting it with sodium iodide. The reaction is carried out in the presence of an acid in an organic solvent (e.g., ethyl acetate, acetonitrile, acetone, methyl ethyl ketone, or their mixed solvent). The acid to be used is, for example, acetic acid, sulfuric acid, or their mixed acid. The amount of sodium iodide to be used is generally within a range of from 1 to 10 molar ratio relative to 6-chloro-2,3-diphenylpyrazine, preferably within a range of from 2 to 3 molar ratio. The reaction temperature varies depending on the kinds of the solvent and the acid to be used, but may be generally within a range of from 60° C. to 90° C. The reaction time varies depending on the kinds of the solvent and the acid to be used and on the reaction temperature, but may be generally within a range of from 9 hours to 15 hours.
Step 2:
5,6-Diphenyl-2-[(4-hydroxybutyl(isopropyl)amino]pyrazine can be produced from 6-iodo-2,3-diphenylpyrazine by reacting it with 4-hydroxybutyl(isopropyl)amine. The reaction is carried out in the presence of a base in an organic solvent (e.g., sulfolane, N-methylpyrrolidone, N,N-dimethylimidazolidinone, dimethyl sulfoxide or their mixed solvent). The base to be used is, for example, sodium hydrogencarbonate, potassium hydrogencarbonate, potassium carbonate, sodium carbonate or their mixed base. The amount of 4-hydroxybutyl(isopropyl)amine to be used may be generally within a range of from 1.5 to 5.0 molar ratio relative to 6-iodo-2,3-diphenylpyrazine, preferably within a range of from 2 to 3 molar ratio. The reaction temperature varies depending on the kinds of the solvent and the base to be used, but may be generally within a range of from 170° C. to 200° C. The reaction time varies depending on the kinds of the solvent and the base to be used and on the reaction temperature, but may be generally within a range of from 5 hours to 9 hours.
Step 3:
Compound A can be produced from 5,6-diphenyl-2-[4-hydroxybutyl(isopropyl)amino]pyrazine by reacting it with N-(2-chloroacetyl)methanesulfonamide. The reaction is carried out in the presence of a base in a solvent (N-methylpyrrolidone, 2-methyl-2-propanol or their mixed solvent). The base to be used is, for example, potassium t-butoxide, sodium t-butoxide or their mixed base. The amount of N-(2-chloroacetyl)methanesulfonamide to be used may be generally within a range of from 2 to 4 molar ratio relative to 5,6-diphenyl-2-[4-hydroxybutyl(isopropyl)amino]pyrazine, preferably within a range of from 2 to 3 molar ratio. The reaction temperature varies depending on the kinds of the solvent and the base to be used, but may be generally within a range of from −20° C. to 20° C. The reaction time varies depending on the kinds of the solvent and the base to be used and on the reaction temperature, but may be generally within a range of from 0.5 hours to 2 hours.
The compounds to be used as the starting materials in the above-mentioned production method for compound A are known compounds, or can be produced by known methods.
PATENT
WO 2002088084
and
http://www.google.fm/patents/WO2009157398A1?cl=en
PAPER
Bioorganic and Medicinal Chemistry, 2007 , vol. 15, 21 p. 6692 – 6704
compd 31
PAPER
Bioorganic and Medicinal Chemistry, 2007 , vol. 15, 24 p. 7720 – 7725
 2a isthe drug
2a isthe drug
N-Acylsulfonamide and N-acylsulfonylurea derivatives of the carboxylic acid prostacyclin receptor agonist 1 were synthesized and their potential as prodrug forms of the carboxylic acid was evaluated in vitro and in vivo. These compounds were converted to the active compound 1 by hepatic microsomes from rats, dogs, monkeys, and humans, and some of the compounds were shown to yield sustained plasma concentrations of 1 when they were orally administered to monkeys. These types of analogues, including NS-304 (2a), are potentially useful prodrugs of 1.
http://www.sciencedirect.com/science/article/pii/S0968089607007614
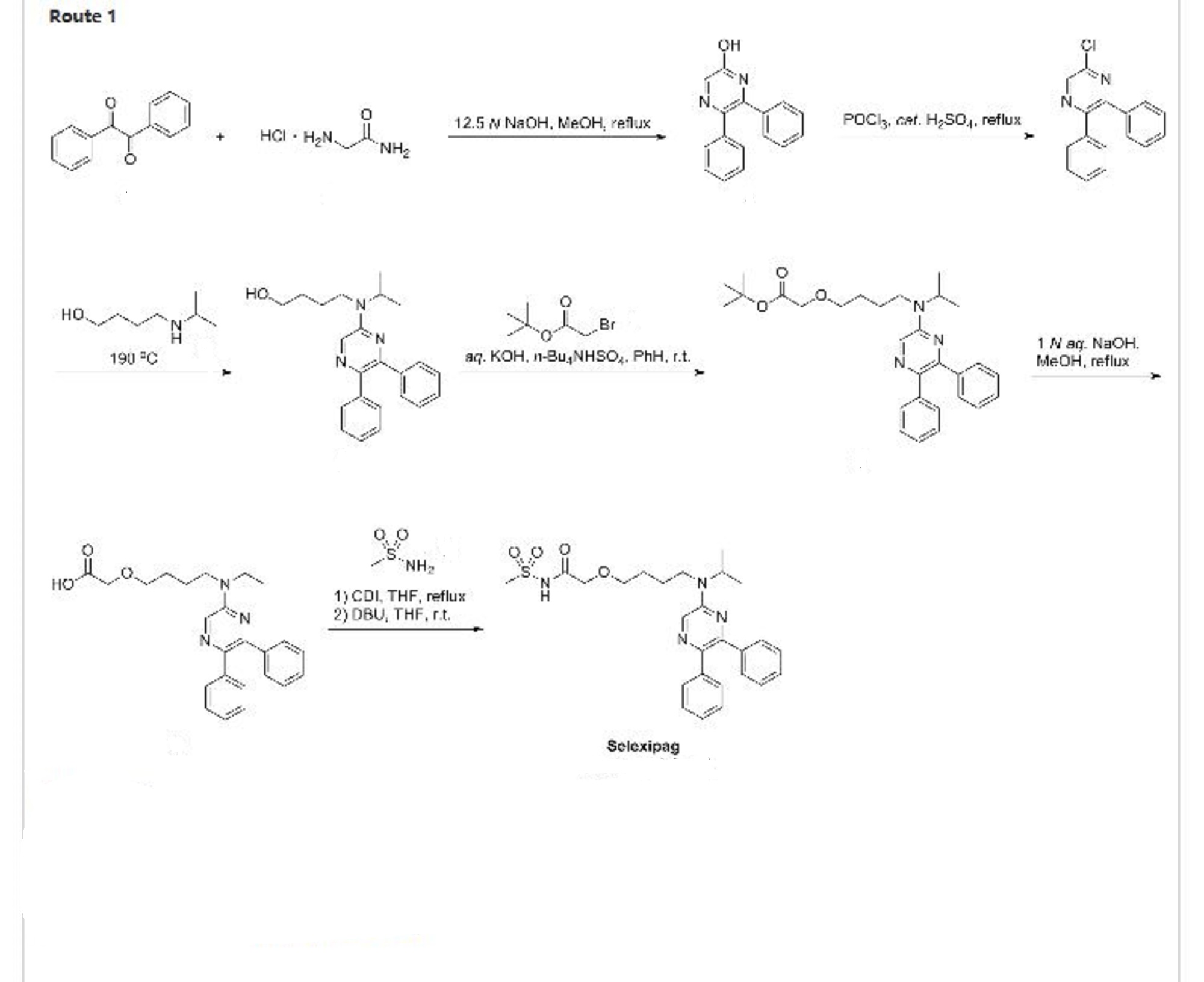
PATENT
Example 1 t- butylamine Form I crystal of the salt
Compound A (40 mg) with 0.5mL dimethoxyethane (hereinafter, referred to as. “DME”) was dissolved in, and t- butylamine (1.1 eq) were added, 25 1 ° C. at 8 it was stirred for hours. Thereafter, the reaction solution was added t- butyl methyl ether (1mL), at -20 ° C. 3 and held hours. It was collected by filtration the precipitated crystals produced, under reduced pressure, and dried, I-form crystals of t- butylamine salt ( 3 to afford 9.9mg). B Powder X-ray diffraction spectrum of type I crystal obtained t- butylamine salt using the apparatus shown in Figure 1.
Melting point: 152.5 ℃
elemental analysis (C 3 0 H 4 3 N 5 O 4 S + 0.0 3 H 2 as O)
calculated value (%) C: 6 3 .1 8 H: 7 . 6 1 N: 12 .2 8 measured value (%) C: 6 2. 8 5 H: 7 . 6 4 N: 12.52 1 H-NMR (DMSO-D 6 ): delta 8 .15 (s, 1H), 7 .55 – 7 . 8 0 (M, 2H), 7 .10- 7 . .45 (M, 10H), 4 7 . 0-4 8 5 (M, 1H), 3 . 6 6 (s, 2H), 3 .4 7 (t, 2H), 3 .45 (t, 2H), 2. 7 3 (s, 3 H), 1.50-1. 7 5 (M, 4H), 1.2 3 (s, 9H), 1.22 (D, 6 H)
Example 2 I-form crystal of the potassium salt
Compound A tetrahydrofuran with (40mg) 12mL (hereinafter, referred to as. “THF”) was dissolved in, 0.1M aqueous potassium hydroxide solution (1.1 eq) was added, 40 ℃ It was heated and stirred in for 15 minutes. After that, it was evaporated under reduced pressure, the solvent. The residue it was added ethyl acetate (200μL). While shaking the mixture heated to 50 ° C. 8 was allowed to cool to 25 ℃ over hours. After repeated two more times this step, at -20 ° C. 3 and held hours. The resulting precipitated crystals were collected by filtration under reduced pressure, and dried to obtain Form I crystal of the potassium salt. B Powder X-ray diffraction spectrum of type I crystal of the obtained potassium salt using the apparatus shown in Fig. 1 H-NMR (DMSO-D 6 ): delta 8 .14 (s, 1H), 7 .1 8 – 7 . 3 8 . (M, 10H), 4 7 . 2-4 8 4 (M, 1H) , 3 . 6 5 (s, 2H), 3 .4 7 (t, 2H), 3 .45 (t, 2H), 2. 7 2 (s, 3 H), 1.55-1. 7 0 ( M, 4H), 1.2 3 (D, 6 H)
Example 3 II-form crystals of the potassium salt
Compound A with (40mg) was dissolved in THF and 12mL, 0.1M aqueous potassium hydroxide solution (1.1 eq) was added and heated with stirring for 15 min at 40 ℃. After that, it was evaporated under reduced pressure, the solvent. The residue it was added ethyl acetate (200μL). While shaking the mixture heated to 50 ° C. 8 was allowed to cool to 25 ℃ over hours. This operation was repeated two more times, at -20 ° C. 3 and held hours. It was collected by filtration the precipitated crystals produced, under reduced pressure, after drying, 40 ℃, relative humidity 7 while 5% of thermo-hygrostat 7 left for days to give crystalline Form II of the potassium salt. B Powder X-ray diffraction spectrum of crystalline Form II of the resulting potassium salt using the apparatus Fig 3 is shown in.
Example 4 III type crystal of the potassium salt
Compound A , in addition to (100mg) acetonitrile (1mL), and stirred with heating, Compound A was dissolved, followed by cooling to 20 ℃. To a solution 3 .5M potassium hydroxide / ethanol solution (1.1 eq) was added and stirred for 200 minutes at 20 ℃. While stirring the mixture 7 after a heated stirring for 1 hour to 0 ° C., and then cooled to 10 ℃ over 10 hours. Further heated while the mixture 6 is heated to 0 ℃, t- butyl methyl ether (0. 3 after adding mL), cooled to 20 ℃ over 10 hours. It was collected by filtration the precipitated crystals produced, under reduced pressure, and dried, III type crystal of the potassium salt ( 7 to afford 5mg). The powder X-ray diffraction spectrum of the type III crystal of the obtained potassium salt using R unit is shown in FIG. Furthermore, in differential scanning calorimetry, of about 7 endothermic peak was observed at around 4 ° C..
Elemental analysis (C 2 6 H 3 1 N 4 O 4 . SK + 0 7 8 H 2 as O)
calculated value (%) C: 5 6 .91 H: 5.9 8 N: 10.21
measured value (%) C: 5 6 . 6 1 H: 5.55 N:. 10 3 6
EXAMPLE 5 IV-type crystal of the potassium salt
Compound A , in addition to (50mg) and ethyl acetate (1mL), and stirred with heating, Compound A was dissolved, followed by cooling to 20 ℃. To a solution 3 .5M potassium hydroxide / ethanol solution (2.2 eq) was added and 2 at 20 ° C. 3 and stirred for hours. It was collected by filtration the precipitated crystals produced, under reduced pressure, and dried to obtain Form IV crystal of the potassium salt (41mg). The powder X-ray diffraction spectrum of crystalline Form IV of the resulting potassium salt using R unit is shown in FIG. Furthermore, in differential scanning calorimetry, an endothermic peak was observed at around approximately 91 ℃.

Selexipag (C26H32N4O4S, Mr = 496.6 g/mol) ist ein Diphenylpyrazin-Derivat. Es wird in der Leber zum aktiven Metaboliten ACT-333679 (MRE-269) biotransformiert. Selexipag unterscheidet sich strukturell von Prostazyklin und anderen Prostazylin-Rezeptor-Agonisten.

References
- 1 Sitbon, O.; Morrell, N. (2012). “Pathways in pulmonary arterial hypertension: The future is here”. European Respiratory Review 21 (126): 321–327. doi:10.1183/09059180.00004812. PMID 23204120.
- 2 New Drug Approved for Rare Lung Disorder. PPN. 23 Dec 2015 Has link to GRIPHON study results
- Kuwano et al. NS-304, an orally available and long-acting prostacyclin receptor agonist prodrug. J Pharmacol Exp Ther 2007;322:1181-1188.
- Kuwano et al. A long-acting and highly selective prostacyclin receptor agonist prodrug, NS-304, ameliorates rat pulmonary hypertension with unique relaxant responses of its active form MRE-269 on rat pulmonary artery. J Pharmacol Exp Ther 2008;326:691-699.
- Simonneau G, Lang I, Torbicki A, Hoeper MM, Delcroix M, Karlocai K, Galie N. Selexipag, an oral, selective IP receptor agonist for the treatment of pulmonary arterial hypertension Eur Respir J 2012; 40: 874-880
- Mubarak KK. A review of prostaglandin analogs in the management of patients with pulmonary arterial hypertension. Respir Med 2010;104:9-21.
- Sitbon, O.; Morrell, N. (2012). “Pathways in pulmonary arterial hypertension: The future is here”. European Respiratory Review 21 (126): 321–327. doi:10.1183/09059180.00004812. PMID 23204120.
| Patent | Submitted | Granted |
|---|---|---|
| Methods of identifying critically ill patients at increased risk of development of organ failure and compounds for the treatment hereof [US8877710] | 2009-12-30 | 2014-11-04 |
| Form-I crystal of 2-{4-[N-(5,6-diphenylpyrazin-2-yl)-N-isopropylamino]butyloxy}-N-(methylsulfonyl)acetamide and method for producing the same [US8791122] | 2010-06-25 | 2014-07-29 |
| COMPOUNDS CAPABLE OF MODULATING/PRESERVING ENDOTHELIAL INTEGRITY FOR USE IN PREVENTION OR TREATMENT OF ACUTE TRAUMATIC COAGULOPATHY AND RESUSCITATED CARDIAC ARREST [US2015057325] | 2013-03-26 | 2015-02-26 |
| INHIBITION OF NEOVASCULARIZATION BY SIMULTANEOUS INHIBITION OF PROSTANOID IP AND EP4 RECEPTORS [US2014275200] | 2014-03-05 | 2014-09-18 |
| INHIBITION OF NEOVASCULARIZATION BY INHIBITION OF PROSTANOID IP RECEPTORS [US2014275238] | 2014-03-05 | 2014-09-18 |
| Fibrosis inhibitor [US8889693] | 2014-04-10 | 20 |
| Patent | Submitted | Granted |
|---|---|---|
| Heterocyclic compound derivatives and medicines [US7205302] | 2004-05-27 | 2007-04-17 |
| METHODS OF IDENTIFYING CRITICALLY ILL PATIENTS AT INCREASED RISK OF DEVELOPMENT OF ORGAN FAILURE AND COMPOUNDS FOR THE TREATMENT HEREOF [US2014322207] | 2014-07-11 | 2014-10-30 |
| THERAPEUTIC COMPOSITIONS CONTAINING MACITENTAN [US2014329824] | 2014-07-18 | 2014-11-06 |
| Sustained Release Composition of Prostacyclin [US2014303245] | 2012-08-10 | 2014-10-09 |
| COMPOUNDS CAPABLE OF MODULATING/PRESERVING ENDOTHELIAL INTEGRITY FOR USE IN PREVENTION OR TREATMENT OF ACUTE TRAUMATIC COAGULOPATHY AND RESUSCITATED CARDIAC ARREST [US2013261177] | 2011-09-30 | 2013-10-03 |
| METHODS OF TREATMENT OF PATIENTS AT INCREASED RISK OF DEVELOPMENT OF ISCHEMIC EVENTS AND COMPOUNDS HEREOF [US2013040898] | 2011-04-29 | 2013-02-14 |
| Substituted Diphenylpyrazine Derivatives [US2013005742] | 2010-08-06 | 2013-01-03 |
| USE OF FORM-I CRYSTAL OF 2–N-(METHYLSULFONYL)ACETAMIDE [US2014148469] | 2014-01-22 | 2014-05-29 |
| CRYSTALS OF 2- {4- [N- (5,6-DIPHENYLPYRAZIN-2-YL) -N-ISOPROPYLAMINO]BUTYLOXY}-N- (METHYLSULFONYL) ACETAMIDE [US2014155414] | 2014-01-22 | 2014-06-05 |
| PROSTACYCLIN AND ANALOGS THEREOF ADMINISTERED DURING SURGERY FOR PREVENTION AND TREATMENT OF CAPILLARY LEAKAGE [US2014044797] | 2012-03-30 | 2014-02-13 |
 |
|
| Names | |
|---|---|
| IUPAC name
2-{4-[(5,6-diphenylpyrazin-2-yl)(propan-2-yl)amino]butoxy}-N-(methanesulfonyl)acetamide
|
|
| Other names
ACT-293987, NS-304
|
|
| Identifiers | |
| 475086-01-2 |
|
| ChEMBL | ChEMBL238804 |
| ChemSpider | 8089417 |
| 7552 | |
| Jmol interactive 3D | Image |
| KEGG | D09994 |
| PubChem | 9913767 |
| UNII | P7T269PR6S |
| Properties | |
| C26H32N4O4S | |
| Molar mass | 496.6 g·mol−1 |
SEE……….http://apisynthesisint.blogspot.in/2015/12/fda-approves-new-orphan-drug-uptravi.html
//////////
CC(C)N(CCCCOCC(=O)NS(=O)(=O)C)C1=CN=C(C(=N1)C2=CC=CC=C2)C3=CC=CC=C3
New Drug Approvals blog by Dr Anthony Crasto hits ten lakh views in 211 countries
New Drug Approvals hits ten lakh views in 211 countries
|
 THANKS AND REGARD’S MOBILE-+91 9323115463
GLENMARK SCIENTIST , INDIA
web link 
|


//////////
AN 2898

AN2898
(5-(3,4-dicyanophenoxy)-1-hydroxy -1,3-dihydro-2,1-benzoxaborole)
1,2-Benzenedicarbonitrile, 4-((1,3-dihydro-1-hydroxy-2,1-benzoxaborol-5-yl)oxy)-,
AN-2898
cas: 906673-33-4
UNII: 6O60L94RMB,
MW 276.0581, MF C15 H9 B N2 O3
A PDE4 inhibitor potentially for the treatment of fungal infection.
AN-2898, a novel topical anti-inflammatory compound that inhibits phosphodiesterase 4 and 7 enzyme activit
PHASE 2 FUNGAL INFECTION, Anacor Pharmaceuticals for the treatment of atopic dermatitis
![]()
| Anacor Pharmaceuticals Inc. | |
| Description | Boron-containing small molecule phosphodiesterase-4 (PDE-4) inhibitor that reduces the production of tumor necrosis factor (TNF) alpha, IL-12 and IL-23 |
| Molecular Target | Phosphodiesterase-4 (PDE-4) |
| Mechanism of Action | Phosphodiesterase-4 (PDE-4) inhibitor |
| Therapeutic Modality | Small molecule |
AN2898 (5-(3,4-dicyanophenoxy)-1-hydroxy -1,3-dihydro-2,1-benzoxaborole) is a broad spectrum anti-inflammatory compound currently in development for the topical treatment of plaque and atopic psoriasis.
AN2898 inhibited phosphodiesterase 4 (PDE4) enzyme activity (IC50 0.060 μM) and the release of multiple cytokines including TNF-α (IC50 0.16 μM) from peripheral blood mononuclear cells (hPBMCs) stimulated by lipopolysaccharide (LPS) or phytohemag- glutinin.
Further, AN2898 was also found to inhibit IL-23 release (IC50 1.0 μM) from THP-1 cells stimulated by LPS and IFN-γ. Investigation of the structure-activity relation-ship around this compound was reported to identify a more potent dual TNF-α/IL-23 inhibitor
( ref………. Akama T, Antunes J, Freund Y, Kimura R, Dong C, Sanders V, et al. Structure-activity studies of novel oxaborole dual inhibitors of PDE4 and IL-23 release. 69th Annu Meet Soc Invest Dermatol (May 6-9, Montreal) 2009 Abst 282. ).
PATENT
WO 2007095638
https://www.google.co.in/patents/WO2007095638A2?cl=en
PATENT
WO 2006089067
http://www.google.co.in/patents/WO2006089067A2?cl=en
US 7582621
http://www.google.co.in/patents/US7582621
WO 2009111676
http://www.google.im/patents/WO2009111676A2?cl=en
WO 2007078340
http://www.google.im/patents/WO2007078340A2?cl=en
US 20070286822
http://www.google.com/patents/US20070286822
REFERENCES
1 Structure-activity studies led to the discovery of AN2898 in development for topical treatment of psoriasis and atopic dermatitis, J Am Acad Dermatol 2009, 60(3, Suppl. 1): Abst P1317
2 FEBS Letters (2012), 586(19), 3410-3414
See all Bboroles at………http://apisynthesisint.blogspot.in/p/borole-compds.html
/////////AN2898, AN 2898, ANACOR, BOROLE
B1(c2ccc(cc2CO1)Oc3ccc(c(c3)C#N)C#N)O
RO-28-1675 for Type 2 Diabetes

RO-28-1675
- (2R)-3-Cyclopentyl-2-[4-(methanesulfonyl)phenyl]-N-(thiazol-2-yl)propionamide
- Ro 028-1675
- Ro 0281675
- Ro 28-1675
3-Cyclopentyl-2(R)-[4-(methylsulfonyl)phenyl]-N-(2-thiazolyl)propionamide
| MW | 378.51 | .-70.4 °
Conc 0.027 g/100mL; chloroform, 589 nm; 23 °C
|
|
|---|---|---|---|
| Formula | C18H22N2O3S2 | ||
| CAS No | 300353-13-3 |
Glucokinase Activators
Ro 28-1675 (Ro 0281675) is a potent allosteric GK activator with a SC1.5 value of 0.24± 0.0019 uM.
Roche (Innovator)
PHASE 1 Type 2 DIABETES,
IC50 value: 0.24± 0.0019 uM (SC1.5) [1]
Target: Glucokinase activator
The R stereoisomer Ro 28-1675 activated GK with a SC1.5 of 0.24 uM, while the S isomer did not activated GK up to 10 uM. Oral administration of Ro 28-1675 (50 mg/Kg) to male C57B1/6J mice caused a statistically significant reduction in fasting glucose levels and improvement in glucose tolerance relative to the vehicle treated animals [1].
Comparison of rat PK parameters indicated that Ro 28-1675 displayed lower clearance and higher oral bioavailability compared to 9a.
Following a single oral dose, Ro 28-1675 reduced fasting and postprandial glucose levels following an OGTT, was well tolerated, and displayed no adverse effects related to drug administration other than hypoglycemia at the maximum dose (400 mg).
 .
.
RO-28-1675 as glucokinase activator.
Joseph Grimsby et al., of Roche have recently discovered activators of glucokinase that increase kcat and decrease the S0.5 for glucose, and these may offer a treatment for type II diabetes. Glucokinase (GK) plays a key role in whole-body glucose homeostasis by catalyzing the phosphorylation of glucose in cells that express this enzyme, such as pancreatic β cells and hepatocytes.
By screening of a library of 120,000 structurally diverse synthetic compounds, they found one small molecule that increased the enzymatic activity of GK. Chemical optimization of this initial molecule led to the synthesis of RO-28-0450 as a lead GK activator which is a class of antidiabetic agents that act as nonessential, mixed-type GK activators (GKAs) that increase the glucose affinity and maximum velocity (Vmax) of GK. RO-28-0450 is a racemic compound.
Activation of GK was exquisitely sensitive to the chirality of the molecule: The R enantiomer, RO-28-1675, was found to be a potent GKA, whereas the S enantiomer, RO-28-1674, was inactive. RO-28-1675 also reversed the inhibitory action of the human glucokinase regulatory protein (GKRP). The activators binding in a glucokinase regulatory site originally was discovered in patients with persistent hyperinsulinemic hypoglycemi.
The result of RO-28-1675 as a potent small molecule GKA may shed light to the chemical biologists to devise strategy for developing activators. Thus for a success to this end we must focus on highly regulated enzymes, or cooperative enzymes such as glucokinase, where nature has provided binding sites that are designed to modulate catalysis.
.SYNTHESIS


Paper

Glucokinase (GK) is a glucose sensor that couples glucose metabolism to insulin release. The important role of GK in maintaining glucose homeostasis is illustrated in patients with GK mutations. In this publication, identification of the hit molecule 1 and its SAR development, which led to the discovery of potent allosteric GK activators 9a and 21a, is described. Compound 21a (RO0281675) was used to validate the clinical relevance of targeting GK to treat type 2 diabetes.
Flash chromatography (Merck Silica gel 60, 70-230 mesh, 9/1, 3/1, and then 11/9 hexanes/ethyl acetate) afforded (2R)-3-cyclopentyl-2-(4-methanesulfonylphenyl)-N-thiazol-2-yl-propionamide (2.10 g, 74%) as a white foam.
[α] 23 589 = –70.4° (c=0.027, chloroform).
EI-HRMS m/e calcd for C18H22N2O3S2 (M+ ) 378.1072, found 378.1081.
1 H NMR (400 MHz, CHLOROFORM-d) δ ppm 10.48 (br. s., 1 H), 7.88 (d, J=8.6 Hz, 2 H), 7.53 (d, J=8.6 Hz, 2 H), 7.50 (d, J=3.5 Hz, 1 H), 7.06 (d, J=3.5 Hz, 1 H), 3.76 (t, J=7.7 Hz, 1 H), 3.03 (s, 3 H), 2.28 (dt, J=13.6, 7.7 Hz, 1 H), 1.88 – 1.98 (m, 1 H), 1.42 – 1.84 (m, 7 H), 1.07 – 1.19 (m, 2 H).
Anal. Calcd for C18H22N2O3S2: C, 56.94; H, 5.59; N, 7.28. Found: C, 57.12; H, 5.86; N, 7.40.
PATENT
WO 2000058293
http://www.google.com/patents/WO2000058293A2?cl=en
Example 3 (A) 3-CyclopentyI-2-(4-methanesulfonyl-phenyI)-N-thiazol-2-yI-propionamide

A solution of dπsopropylamine (3.3 mL, 23.5 mmol) in dry tetrahydrofuran (50 mL) and 1.3-dιmethyl-3,4,5,6-tetrahydro-2(lH)-pyπmιdιnone (10 mL) was cooled to -78°C under nitrogen and then treated with a 10M solution of n-butyllithium m hexanes (2.35 mL, 23 5 mmol) The yellow reaction mixture was stiπed at -78°C for 30 mm and then treated dropwise with a solution of 4-methylsulfonylphenylacetιc acid (2.40 g, 11.2 mmol) in a small amount of dry tetrahydrofuran. After approximately one-half of the 4- methylsulfonylphenylacetic acid m dry tetrahydrofuran was added, a precipitate formed Upon further addition of the remaining 4-methylsulfonylphenylacetιc acid in dry tetrahydrofuran, the reaction mixture became thick in nature After complete addition of the 4-methylsulfonylphenylacetιc acid in dry tetrahydrofuran, the reaction mixture was very thick and became difficult to stir An additional amount of dry tetrahydrofuran (20 mL) was added to the thick reaction mixture, and the reaction mixture was stirred at –
78 C for 45 mm, at which time, a solution of lodomethylcyclopentane (2.35 g, 11.2 mmol) in a small amount of dry tetrahydrofuran was added dropwise The reaction mixture was allowed to warm to 25°C where it was stiπed for 15 h. The reaction mixture was quenched with water (100 mL), and the resulting yellow reaction mixture was concentrated in vacuo to remove tetrahydrofuran. The aqueous residue was acidified to pH = 2 using concentrated hydrochloπc acid The aqueous layer was extracted with ethyl acetate The organic phase was dπed over magnesium sulfate, filtered, and concentrated in vacuo Flash chromatography (Merck Silica gel 60, 230-400 mesh, 1/3 hexanes/ethyl acetate) afforded 3-cyclopentyl-2-(4-methanesulfonyl-phenyl)propιonιc acid (1.80 g, 52%) as a white solid: mp 152-154°C; EI-HRMS m/e calcd for C15H20O4S (Nf) 296.1082, found 296.1080
A solution of 3-cyclopentyl-2-(4-methanesulfonyl-phenyl)propιonιc acid (4.91 g, 16.56 mmol) and tnphenylphosphine (6.52 g, 24.85 mmol) m methylene chloπde (41 mL) was cooled to 0°C and then treated with N-bromosuccinimide (5.01 g, 28.16 mmol) m small portions The reaction mixture color changed from light yellow to a darker yellow then to brown After the complete addition of N-bromosuccinimide, the reaction mixture was allowed to warm to 25°C over 30 min. The brown reaction mixture was then treated with 2-aminothiazole (4.98 g, 49.69 mmol). The resulting reaction mixture was stiπed at 25°C for 19 h. The reaction mixture was then concentrated in vacuo to remove methylene chloride. The remaining black residue was diluted with a 10% aqueous hydrochloric acid solution (400 mL) and then extracted with ethyl acetate (3 x 200 mL). The combined organic layers were washed with a saturated aqueous sodium chloride solution (1 x 200 mL), dried over sodium sulfate, filtered, and concentrated in vacuo. Flash chromatography (Merck Silica gel 60, 70-230 mesh, 3/1 hexanes/ethyl acetate then 1/1 hexanes/ethyl acetate) afforded 3-cyclopentyl-2-(4-methanesulfonyl-phenyl)-N-thiazol-2- yl-propionamide (4.49 g, 72%) as a white solid: mp 216-217°C; EI-HRMS m/e calcd for C18H22N2O3S2 (M+) 378.1072, found 378.1071.
Example 13
(2R)-3-Cyclopentyl-2-(4-methanesuIfonylphenyl)-N-thiazol-2-yl-propionamide

A solution of ^-( ethanesulfonyl)phenyl acetic acid (43 63 g, 0.204 mol) in methanol (509 mL) was treated slowly with concentrated sulfunc acid (2 mL) The resulting reaction mixture was heated under reflux for 19 h The reaction mixture was allowed to cool to 25°C and then concentrated in vacuo to remove methanol The residue was diluted with ethyl acetate (800 mL) The organic phase was washed with a saturated aqueous sodium bicarbonate solution (1 x 200 mL), washed with a saturated aqueous sodium chlonde solution (1 x 200 mL), dned over sodium sulfate, filtered, and concentrated in vacuo Flash chromatography (Merck Silica gel 60, 70-230 mesh, 1/1 hexanes/ethyl acetate) afforded 4-(methanesulfonyl)phenyl acetic acid methyl ester (45.42 g, 98%) as a yellow oil which solidified to a cream colored solid upon sitting over time at 25°C mp 78-80°C, EI-HRMS m/e calcd for Cι0H12O4S (M+) 228 0456, found 228 0451.
A mechanical stiπer was used for this reaction A solution of dnsopropylamme (29.2 mL, 0.21 mol) in dry tetrahydrofuran (186 mL) and l,3-dιmethyl-3,4,5,6-tetrahydro- 2(lH)-pyπmιdιnone (62 mL) was cooled to -78°C and then treated with a 2.5M solution of n-butylhthium in hexanes (83 4 mL, 0.21 mol) The yellow-orange reaction mixture was stiπed at -78°C for 35 min and then slowly treated with a solution of 4- (methanesulfonyl)phenyl acetic acid methyl ester (45.35 g, 0.20 mol) in dry tetrahydrofuran (186 mL) and l,3-dιmethyl-3,4,5,6-tetrahydro-2(lH)-pyπmιdmone (62 mL) The reaction mixture turned dark in color. The reaction mixture was then stiπed at -78°C for 50 mm, at which time, a solution of lodomethylcyclopentane (50.08 g, 0.24 mol) in a small amount of dry tetrahydrofuran was added slowly. The reaction mixture was then stiπed at -78°C for 50 mm, and then allowed to warm to 25°C, where it was stirred for 36 h. The reaction mixture was quenched with water (100 mL), and the resulting reaction mixture was concentrated in vacuo to remove tetrahydrofuran The remaining residue was diluted with ethyl acetate (1.5 L). The organic phase was washed with a saturated aqueous sodium chloπde solution (1 x 500 mL), dned over sodium sulfate, filtered, and concentrated in vacuo Flash chromatography (Merck Silica gel 60, 70-230 mesh, 3/1 hexanes/ethyl acetate) afforded 3-cyclopentyl-2-(4- methanesulfonylphenyl)propιonιc acid methyl ester (41.79 g, 68%) as a yellow viscous oil EI-HRMS m/e calcd for Cι6H22O4S (M+) 310.1239. found 310.1230.
A solution of 3-cyclopentyl-2-(4-methanesulfonylphenyl)propιonιc acid methyl ester (50 96 g, 0.16 mol) in methanol (410 mL) was treated with a IN aqueous sodium hydroxide solution (345 mL, 0.35 mol). The reaction mixture was stirred at 25°C for 24 h. The reaction mixture was concentrated in vacuo to remove methanol. The resulting aqueous residue was acidified to pH = 2 with concentrated hydrochlonc acid and then extracted with ethyl acetate (5 x 200 mL) The combined organic layers were dned over sodium sulfate, filtered, and concentrated in vacuo to afford pure 3-cyclopentyl-2-(4- methanesulfonylphenyl)propιonιc acid (43 61 g, 90%) as a white solid which was used without further puπfication. mp 152-154°C, EI-HRMS m e calcd for C15H20O4S (M+) 296.1082, found 296.1080.
Two separate reactions were setup in parallel: (1) A solution of (R)-(+)-4-benzyl-2- oxazohdmone (3.67 g, 20.73 mmol) m dry tetrahydrofuran (35 mL) was cooled to -78°C and then treated with a 2.5M solution of n-butylhthium in hexanes (7.9 mL, 19.86 mmol). The resulting reaction mixture was stiπed at -78°C for 30 mm and then allowed to warm to 25°C, where it was stirred for 1.5 h (2) A solution of racemic 3-cyclopentyl-2-(4- methanesulfonylphenyl)propιonιc acid (5.12 g, 17.27 mmol) in dry tetrahydrofuran (35 mL) was cooled to 0°C and then treated with tnethylamme (2.8 mL, 19.86 mmol). The reaction mixture was stiπed at 0°C for 10 nun and then treated dropwise with tπmethylacetyl chlonde (2.6 mL, 20.73 mmol). The resulting reaction mixture was stiπed at 0°C for 2 h and then cooled to -78°C for the addition of the freshly prepared chiral oxazolidmone. The reaction mixture containing the oxazolidmone was then added to the cooled (-78°C) mixed anhydπde solution The resulting reaction mixture was stiπed as -78°C for 1 h and allowed to gradually warm to 25°C. The reaction mixture was then stiπed at 25°C for 3 d. The resulting reaction mixture was quenched with water (100 mL) and then concentrated in vacuo to remove tetrahydrofuran. The resulting aqueous residue was diluted with ethyl acetate (600 mL). The organic layer was washed with a saturated aqueous sodium chloπde solution (1 x 300 mL), dπed over sodium sulfate, filtered, and concentrated in vacuo Thin layer chromatography using 13/7 hexanes/ethyl acetate as the developing solvent indicated the presence of two products The higher moving product had a Rf =0.32 and the lower moving product had a Rf = 0.19. Flash chromatography (Merck Silica gel 60, 230-400 mesh, 9/1 then 13/7 hexanes/ethyl acetate) afforded two products: (1) The higher Rf product (4R, 2’S)-4-benzyl-3-[3- cyclopentyl-2-(4-methanesulfonylphenyl)propιonyl]-oxazohdm-2-one (2.12 g, 54%) as a white foam- mp 62-64°C; [c.]23 589 = +6.3° (c=0.24, chloroform); EI-HRMS m/e calcd for C25H29NO5S (M+) 455.1766, found 455.1757. (2) The lower Rf product (4R, 2R)-4- benzyl-3-[3-cyclopentyl-2-(4-methanesulfonylphenyl)propιonyl]-oxazolιdm-2-one (3.88 g, 99%) as a white foam: mp 59-61°C; [α]23 589 = -98.3° (c=0.35, chloroform); EI-HRMS m/e calcd for C25H29NO5S (M +) 455.1766, found 455.1753. The combined mass recovery from the two products was 6.00 g, providing a 76% conversion yield for the reaction
An aqueous solution of lithium hydroperoxide was freshly prepared from mixing a solution of anhydrous lithium hydroxide powder (707.3 mg, 16.86 mmol) m 5.27 mL of water with a 30% aqueous hydrogen peroxide solution (3.44 mL, 33.71 mmol). This freshly prepared aqueous lithium hydroperoxide solution was cooled to 0°C and then slowly added to a cooled (0°C) solution of (4R, 2’R)-4-benzyl-3-[3-cyclopentyl-2-(4- methanesulfonylphenyl)propιonyl]-oxazolιdm-2-one (3.84 g, 8.43 mmol) in tetrahydrofuran (33 mL) and water (11 mL). The reaction mixture was stiπed 0°C for 1.5 h The reaction mixture was then quenched with a 1.5N aqueous sodium sulfite solution (25 mL) The reaction mixture was further diluted with water (300 mL) The resulting aqueous layer was continuously extracted with diethyl ether until thm layer chromatography indicated the absence of the recovered chiral oxazolidmone in the aqueous layer The aqueous layer was then acidified to pH = 2 with a 10% aqueous hydrochlonc acid solution and extracted with ethyl acetate (300 mL) The organic extract was dned over sodium sulfate, filtered, and concentrated in vacuo to afford (2R)-3- cyclopentyl-2-(4-methanesulfonylphenyl)propιomc acid as a white solid (2.23 g, 89%) which was used without further puπfication Flash chromatography (Merck Silica gel 60, 70-230 mesh, 30/1 methylene chlonde/methanol then 10/1 methylene chlonde/methanol) was used to obtain a punfied sample for analytical data and afforded pure (2R)-3- cyclopentyl-2-(4-methanesulfonylphenyl)propιomc acid as a white foam- mp 62-64°C (foam to gel), [α]23 589 = -50.0° (c=0.02, chloroform), EI-HRMS m/e calcd for C15H20O4S (M+) 296 1082, found 296 1080
A solution of tnphenylphosphme (3.35 g, 12.79 mmol) m methylene chloπde (19 mL) was cooled to 0°C and then slowly treated with N-bromosuccmimide (2.28 g, 12.79 mmol) in small portions. The reaction mixture was stiπed at 0°C for 30 mm, and dunng this time penod, the color of the reaction mixture changed from light yellow to a darker yellow then to a purple color. The cooled purple reaction mixture was then treated with the (2R)-3-cyclopentyl-2-(4-methanesulfonylphenyl)propιonιc acid (2.23 g, 7.52 mmol) The resulting reaction mixture was then allowed to warm to 25°C over 45 mm, at which time, the reaction mixture was then treated with 2-amιnothιazole (1.88 g, 18.81 mmol) The resulting reaction mixture was stiπed at 25°C for 12 h. The reaction mixture was then concentrated in vacuo to remove methylene chloπde The remaining black residue was diluted with ethyl acetate (300 mL) and then washed well with a 10% aqueous hydrochlonc acid solution (2 x 100 mL), a 5% aqueous sodium bicarbonate solution (3 x 100 mL), and a saturated aqueous sodium chloride solution (1 x 200 mL). The organic layer was then dried over sodium sulfate, filtered, and concentrated in vacuo. Flash chromatography (Merck Silica gel 60, 70-230 mesh, 9/1, 3/1, and then 11/9 hexanes/ethyl acetate) afforded (2R)-3-cyclopentyl-2-(4-methanesulfonylphenyl)-N-thiazol-2-yl- propionamide (2.10 g, 74%) as a white foam: mp 78-80°C (foam to gel); [α]23 589 = -70.4° (c=0.027, chloroform); EI-HRMS m/e calcd for C18H22N2O3S2 (M+) 378.1072, found 378.1081.
REFERENCES
Glucokinase (GK) is a glucose sensor that couples glucose metabolism to insulin release. The important role of GK in maintaining glucose homeostasis is illustrated in patients with GK mutations. In this publication, identification of the hit molecule 1 and its SAR development, which led to the discovery of potent allosteric GK activators 9a and 21a, is described. Compound 21a (RO0281675) was used to validate the clinical relevance of targeting GK to treat type 2 diabetes.
http://www.nature.com/nrd/journal/v8/n5/fig_tab/nrd2850_T2.html
NMR…..http://www.medchemexpress.com/product_pdf/HY-10595/Ro%2028-1675-NMR-HY-10595-13569-2014.pdf
http://www.medchemexpress.com/product_pdf/HY-10595/Ro%2028-1675-Lcms_Ms-HY-10595-13569-2014.pdf
///////////RO-28-1675, Ro 0281675
O=C(Nc1nccs1)[C@H](CC2CCCC2)c3ccc(cc3)S(C)(=O)=O

Chemical structures of Roche’s glucokinase activators (GKAs) RO-28-1675 and piragliatin, as well as the related GKA 1.
PF-04191834 for Patients With Osteoarthritis Of The Knee


| Molecular Formula: | C22H23N3O2S |
|---|---|
| Molecular Weight: | 393.50192 g/mol |
5-Lipoxygenase (5-LO) inhibitor
AsthmaChronic osteoarthritis pain



- Example 1

4-(3-{[4-(1-methyl-1H-pyrazol-5-yl)phenyl]thio}phenyl)tetrahydro-2H-pyran-4 carboxamideStep 1: Preparation of 4-(3-bromophenyl)-tetrahydro-2H-pyran-4-carboxamide
-
4-(3-bromophenyl)tetrahydro-2H-pyran-4-carbonitrile made by the procedures described in EP 108114 (1.05 kg, 3.95 mole) was stirred in 98% H2SO4 (3.00 L) at room temperature for about 40 h. The mixture was then poured onto ice and the very fine suspension was filtered and washed with H2O thoroughly until pH of wash is neutral. The white solid was washed with hexanes and was then dried in vacuo at 35-40° C. to give 1119 g (99.8% yield) of product in 99.9% purity. LC/MS: 5%-100% CH3CN:H20-0.01% TFA gradient over 10 minutes: 4.68 min. (M+H)+. 1H NMR (400 MHz, DMSO-d6) δ ppm 7.50-7.49 (m, 1H), 7.43-7.40 (m, 1H), 7.36-7.30 (m, 1H), 7.27 (d, J=7.92 Hz, 1H) 7.06 (s, 1H), 5.00 (brs, 1H) 3.71 (dt, J=11.7, 3.7 Hz, 2H), 3.42 (t, J=10.7 Hz, 2H), 2.38 (d, J=13.6 Hz, 2H), 1.75 (td, J=12.2, 4.3 Hz, 2H).
Step 2: Preparation of 4-(3-(triisopropylsilylthio)phenyl)-tetrahydro-2H-pyran-4-carboxamide
-
Alternative 1
-
4-(3-Bromophenyl)-tetrahydro-2H-pyran-4-carboxamide prepared in step 1 (300 g (1.06 mole), sodium tert-butoxide (122 g, 1.27 mole), Pd(OAc)2 (4.74 g 0.0211 mole) and DIPPF (1,1-bis(diisopropylphosphino)ferrocene) (10.6 g 0.0253 mole) were placed in a flask which was evacuated and filled with N2 3 times. Anhydrous dioxane (2.3 L) was added and the mixture was stirred at room temperature for 1 h. To the mixture was added triisopropylsilane thiol (221 g 1.16 mole) and the resulting mixture was heated to reflux. Reflux was stopped after 1 h and the mixture was allowed to cool to room temperature. The mixture was then poured into ethyl acetate (7 L) which was then washed with H2O (2×4 L) and brine (2 L). The combined aqueous washes were back extracted with ethyl acetate (3 L) which was then washed with H2O (2×2 L) and brine (1 L). The combined organic layers were dried over MgSO4, filtered and concentrated to dryness. Ethyl acetate (0.5 L) was added to the solid and the mixture was stirred on the rotary evaporator to give a fine suspension. Hexanes (1.5 L) was then added and the suspension was allowed to stand for 1 hour. The solid was filtered, washed with 1:1 ethyl acetate-hexanes (1 L) and then hexanes. The resulting brown solid was dried in vacuo to give 334 g (80% yield) of the product in 99% purity. A second crop was obtained from the filtrate which was washed as before and dried to give an additional 15 g product for a total yield of 84%. LC/MS: 5%-100% CH3CN:H20-0.01% TFA gradient over 10 minutes: 9.35 min. 394.1 (M+H)+. 1H NMR (400 MHz, CDCl3) δ ppm 7.52-7.51 (m, 1H) 7.42-7.39 (m, 1H), 7.22-7.21 (m, 2H), 5.35 (brs, 1H), 5.13 (brs, 1H) 3.78-3.75 (m, 4H) 2.36-2.32 (m, 2H), 2.06-2.00 (m, 2H), 1.27-1.16 (m, 3H), 1.05 (d, J=7.25 Hz, 18H).
Step 2: Preparation of 4-(3-(triisopropylsilylthio)phenyl)-tetrahydro-2H-pyran-4-carboxamide
-
Alternative 2
-
Purge a 3-neck flask (overhead stirrer, nitrogen inlet, serum cap) with nitrogen. Add 4-(3-Bromophenyl)-tetrahydro-2H-pyran-4-carboxamide prepared in step 1 (10 g, 0.03519 mole). Add sodium t-butoxide (4.1 g, 0.04223 moles). Add anhydrous toluene. Toluene should be as dry as possible, <0.01% water by KF is sufficient. Initiate stirring. Purge the reaction mixture with 4 vacuum/nitrogen purge cycles, maintaining 60 torr vacuum for 30 seconds with each cycle. Add the thiol (9.1 g, 0.04223 moles) assuring that oxygen is not introduced into the vessel. Heat to 75° C. Add PdCl2(diphenyl-phosphino ferrocene) (0.258 g, 0.00035 moles). Continue heating to reflux (reaction temperature about 107° C.) for a minimum of 1 hour. The mixture should reach reflux within 30 minutes.
-
Cool the reaction mixture to 25° C. Add ethyl acetate (300 mL, 30 mL/g) and stir the resulting suspension for 30 min. Filter the suspension through celite (30 g). Rinse the celite with ethyl acetate for rinse (100 mL, of product to be rinsed), combining filtrates. Concentrate the filtrate via vacuum distillation at 70 torr at 30° C. until 80% of the filtrate volume has been removed. Add hexane (200 mL, 20 mL/g of product to be crystallized) for crystallization to the slurry over 5 minutes. Stir and cool the mixture to 5° C. Maintain the mixture at 5° C. for a minimum of 1 hour. Isolate product by filtration. Rinse the cake with hexane (100 mL, of product to be rinsed). Dry the cake on the filter to LOD of no more than 5%. Dry the solid at 45-50° C. under vacuum to an LOD of no more than 1.5%. Yield 12 grams (85% yield).
-
Any mL/g amount indicated above is referred to grams of bromo carboxamide.
Step 3: Preparation of 5-(4-bromophenyl)-1-methyl-1H-pyrazole
-
Alternative 1
-
A N,N′-dimethylformamide (15 mL) solution of 4-bromoacetophenone (10.60 g, 53.25 mmols) and N,N′-dimethylformamide dimethyl acetal (2.5 equivalents) was heated at 125 degrees Celcius for 3 hours. The dark red solution was cooled to room temperature. The volatiles were removed by rotary evaporation providing a red viscous oil. To this substance was added anhydrous N,N′-dimethylformamide (15 mL) and methyl hydrazine (7.6 g, 160 mmols, 3 equivalents). The mixture was stirred at room temperature for 1 hour and then heated at 75 degrees Celcius for 4 hours. The volatiles were removed by rotary evaporation and the crude residue was taken up in a small volume of methylene chloride. This red solution was applied to a cartridge of silica gel. The cartridge was eluted with a 20:80 mixture of ethyl acetate and hexanes, respectively. The appropriate fractions were combined and concentrated to produce 12.5 g of a white solid.
-
1H NMR (400 MHz, CHLOROFORM-d) δ ppm 3.87-3.95 (m, J=2.22 Hz, 3H) 6.29-6.36 (m, 1H) 7.31 (dd, J=8.36 Hz, 2H) 7.52-7.56 (m, 1H) 7.62 (dd, J=2.05 Hz, 2H).
Step 3: Preparation of 5-(4-bromophenyl)-1-methyl-1H-pyrazole
-
Alternative 2
-
4-bromoacetophenone (20.0 g; 0.10 mole) and N,N-dimethylformamide dimethylacetal (28.5 mL; 0.20 mole) were mixed together in DMF (12 mL) and heated to 110° C. for 4 hours. The methanol and water that were generated during the reaction were distilled (6.2 mL). The mixture was cooled to 25° C. Methyl t-butyl ether (100 mL) and methylhydrazine (21.2 mL; 0.40 moles) were added and the mixture was stirred over night. The reaction mixture was washed with 1 M aqueous ammonium chloride (3×40 mL) and water (40 mL). The organic phase was dried by azeotropic distillation using a Dean-Stark apparatus. As an alternative to distillation, the solution was dried through an anhydrous magnesium sulfate cartridge. The solution was filtered through a silica gel cartridge (60 g). The product was flushed from the cartridge with methyl t-butyl ether. The fraction(s) containing product were combined and concentrated to about 70 mL by distillation. Heptane (120 mL) was added and distillation was continued until the pot temperature reached 98.4° C. About 100 mL of distillate was collected. The mixture was cooled to 40° C. The mixture was seeded and the temperature was maintained at 40° C. for 30 minutes while crystallization was initiated. The mixture was slowly chilled to 0° C. over 90 minutes. The mixture was held at 0° C. for 30 minutes. The mixture was filtered and the solid was washed (3×) with chilled (0° C.) heptane. The solid was dried on the filter. A cream-colored, crystalline solid (16.3 g; 68% yield) was obtained. The NMR data of the title compound are as per alternative 1.
Step 4: Preparation of 4-(3-{[4-(1-methyl-1H-pyrazol-5-yl)phenyl]thio]phenyl) tetrahydro-2H-pyran-4-carboxamide
-
A mixture of 5-(4-bromophenyl)-1-methyl-1H-pyrazole (0.50 g, 2.10 mmols,), 4-{3-[(tri-isopropylsilyl)thio]phenyl}tetrahydro-2H-pyran-4-carboxamide (0.83 g, 2.10 mmols), Tetrakis(triphenylphosphine)palladium(0) (243 mg, 0.10 equivalents), bis[(2-diphenyl-phosphino)]phenyl ether (113 mg, 0.10 equivalents), and 1.0 M potassium tert-butoxide in THF (6.3 mmols, 3 equivalents) in iPrOH (15 mL) that contained 5% water was heated for 4 hours at 90 degrees Celcius in an atmosphere of nitrogen. The reaction mixture was cooled to room temperature and 7 mL of 1N HCl was added. The product was precipitated by the addition of water (30 mL). The precipitate was collected by suction filtration and washed with water (2×20 mL) and cold ethyl ether (4×20 mL). The tan brown solid was dissolved in a small volume of methylene chloride containing 1% methanol and applied to a 140 g cartridge of silica gel. The cartridge was eluted with an acetone:hexane gradient. The appropriate fractions were concentrated and triturated with methanol to produce a white solid (710 mg) as product. 1H NMR (400 MHz, DMSO-d6) δ ppm 1.75-1.84 (m, 3H) 2.40 (d, J=13.54 Hz, 3H) 3.43-3.51 (m, 1H) 3.72 (d, J=11.34 Hz, 3H) 3.84 (s, 3H) 6.40 (d, J=1.46 Hz, 1H) 7.02 (s, 1H) 7.22-7.30 (m, 2H) 7.34 (d, J=8.05 Hz, 1H) 7.38-7.43 (m, 2H) 7.45-7.52 (m, 3H). HRMS calc M+H, 394.1589, found 394.1630.
Step 4: Preparation of 4-(3-{[4-(1-methyl-1H-pyrazol-5-yl)phenyl]thio]phenyl) tetrahydro-2H-pyran-4-carboxamideScale-Up Alternative
-
4-{3-[(tri-isopropylsilyl)thio]phenyl}tetrahydro-2H-pyran-4-carboxamide (200 g, 0.51 moles), 5-(4-bromophenyl)-1-methyl-1H-pyrazole (126 g, 0.53 moles), and 2-methyltetrahydrofuran (2,000 mL, 10 mL/g of tips carboxamide) were put into the reactor and sparged with nitrogen while heating to 60° C. The sodium methoxide (244.0 mL, 1.07 moles, added as sodium methoxide in methanol solution 25% w/w) was added to the reactor and sparging was continued for another 30 minutes. PdCl2DPPF (3.7 g, 0.005 moles) was added to the reactor and the mixture was heated to 70° C. Once the amount of tips carboxamide was less than 1% of starting amount, the mixture was cooled to 0° C. The mixture was held at 0° C. for one hour. The mixture was filtered and the solid was washed with 2-methyltetrahydrofuran (3×2.5 mL/g). The solid was dried on the filter. The solid was returned to a clean reactor and triturated with water (2,000 mL, 10 mL/g) for two hours at 20° C. The mixture was filtered and the solid was washed with water (2,000 mL, 2×5 mL/g). The solid was dried on the filter. The solid was returned to a clean reactor with the Si-thiol (90.0 g, 0.5 g/g) and THF (about 12.8 L, 70 mL/g). The mixture was heated to 60-65° C. and held for two hours. The mixture was cooled to 25° C. and filtered. The Si-thiol was washed with THF (about 0.9 L, 5 mL/g). The solution was distilled to a concentration of 10 mL/g. The mixture was cooled to 25° C. and hexanes (422.5 mL, 5 mL/g) was added. The mixture was filtered and the solid was washed with hexanes (422.5 mL, 5 mL/g). The solid was dried in a vacuum oven at 70° C.
-
For 2-methyltetrahydrofuran and water, mL/g are referred to grams of tips carboxamide. For Si-thiol, tetrahydrofuran and hexanes, mL/g are referred to grams of title compound.
Step 5: Purification of 4-(3-{[4-(1-methyl-1H-pyrazol-5-yl)phenyl]thio]phenyl) tetrahydro-2H-pyran-4-carboxamide
Crude title compound (181.0 g, 1.0 eq.) obtained from step 4, scale-up version, was returned to a clean reactor with Si-thiol (0.5 g/g of title compound) and THF (75 mL/g of title compound). The mixture was heated to 60-65° C. and held overnight. The mixture was cooled to 25° C. and filtered. The Si-thiol was washed with THF (5 mL/g of title compound). The solution was distilled to a concentration of 10 mL/g. Product may cake on reactor wall during the distillation. The mixture was cooled to 25° C. Hexanes (5 mL/g of title compound) was added and the mixture was held for 30 minutes. The mixture was filtered and the solid was dried on the filter. The reactor was rinsed with methanol to remove residual THF. The solid was returned to the reactor with methanol (20 mL/g of title compound). The mixture was heated to reflux and held over night. The mixture was cooled to 20° C. and held for 2 hours. The mixture was filtered. The solid was dried in a vacuum oven at 70° C. 162 g of purified title compound was obtained (85% yield). The NMR data of the title compound are as per Step 4.
Any mL/g amount indicated above is referred to grams of crude title compound.
Book Title

Transition Metal-Catalyzed Couplings in Process Chemistry: Case Studies from the Pharmaceutical Industry
18. Development of Migita Couplings for the Manufacture of a 5-Lipoxygenase Inhibitor
Published Online: 19 JUL 2013
DOI: 10.1002/9783527658909.ch18
- 5-lipoxygenase inhibitor;
- isooctyl 3-mercaptopropionate;
- Migita couplings;
- one-pot process;
- triisopropylsilanethiol (TIPS-SH)
Summary
The biggest shortcoming of the medicinal chemistry route is the introduction of the sulfur source for the first of two Migita couplings. The authors felt that the initial Migita coupling was a better candidate for a kinetic study on the formation of impurity, as it was harder to maintain a constant concentration of active Pd for the second coupling with two sources of Pd/ligand in this step. As depicted in the mechanism of the Migita coupling, the catalytic cycle is composed of three steps: oxidative addition, transmetalation, and reductive elimination. This chapter develops a three-step, one-pot process for the synthesis of 5-lipoxygenase inhibitor via a sequence of two Migita couplings. This strategy employed cheap, odorless, and readily available isooctyl 3-mercaptopropionate as the sulfur source for the initial Migita coupling as a general alternative to the popular triisopropylsilanethiol (TIPS-SH) for the formation of diaryl thioethers.
PAPER

| Patent | Submitted | Granted |
|---|---|---|
| Pyrazole Analogs [US7772269] | 2008-05-29 | 2010-08-10 |
| Pyrazole Derivatives as 5-LO-Inhibitors [US8097733] | 2009-09-10 | 2012-01-17 |
| NOVEL TREATMENT FOR AGE RELATED MACULAR DEGENERATION AND OCULAR ISCHEMIC DISEASE ASSOCIATED WITH COMPLEMENT ACTIVATION BY TARGETING 5-LIPOXYGENASE [US2011269807] | 2011-11-03 | |
| TREATMENT AND PREVENTION OF DISEASES MEDIATED BY MICROORGANISMS VIA DRUG-MEDIATED MANIPULATION OF THE EICOSANOID BALANCE [US2014171445] | 2012-08-02 | 2014-06-19 |
TEVA’S CEP 1347, KT 7515 a MAP3K11 (MLK3) inhibitor potentially for the treatment of Parkinson’s disease.
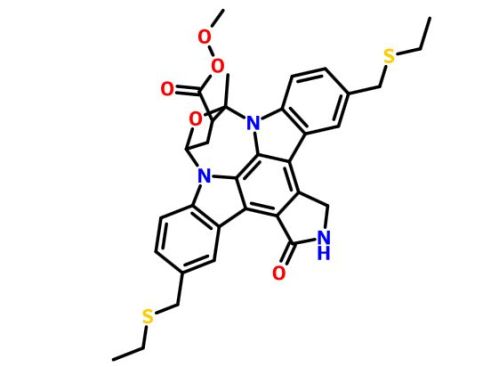

CEP-1347; KT-7515
(9S,10R,12R)-5-16-Bis[(ethylthio)methyl]-2,3,9,10,11,12-hexahydro-10-hydroxy-9-methyl-1-oxo-9,12-epoxy-1H-diindolo[1,2,3-fg:3′,2′,1′-kl]pyrrolo[3,4-i][1,6]benzodiazocine-10-carboxylic acid methyl ester
3,9-Bis(etsm)-K-252a; CEP1347; 3,9-Bis((ethylthio)methyl)-K-252a; AC1L31ZX
3,9-bis[(ethylthio)methyl]-K-252a
Phase III
A MAP3K11 (MLK3) inhibitor potentially for the treatment of Parkinson’s disease.
![]()

Scheme 1 a
a (a) Ac2O, DMAP, THF, room temperature, 93%; (b) Cl2CHOCH3, TiCl4, CH2Cl2, 66%; (c) NaBH4 CH3OH, CHCl3, 65%; (d) NaOCH3, CH3OH, ClCH2CH2Cl, room temperature, 90%; (e) ROH, CSA, CH2Cl2; (f) RSH, CSA, CH2Cl2.
Inhibitor of c-jun N-terminal kinase (JNK) signaling. Rescues motor neurons undergoing apoptosis (EC50 = 20 nM). Blocks Aβ-induced cortical neuron apoptosis (EC50 ~51 nM). Does not inhibit ERK1 activity. Neuroprotective.
Apoptosis has been proposed as a mechanism of cell death in Alzheimer’s, Huntington’s and Parkinson’s diseases and the occurrence of apoptosis in these disorders suggests a common mechanism.
Events such as oxidative stress, calcium toxicity, mitochondria defects, excitatory toxicity, and deficiency of survival factors are all postulated to play varying roles in the pathogenesis of the diseases.
However, the transcription factor c-jun may play a role in the pathology and cell death processes that occur in Alzheimer’s disease.
Parkinson’s disease (PD) is also a progressive disorder involving the specific degeneration and death of dopamine neurons in the nigrostriatal pathway. In Parkinson’s disease, dopaminergic neurons in the substantia nigra are hypothesized to undergo cell death by apoptotic processes.
The commonality of biochemical events and pathways leading to cell death in these diseases continues to be an area under intense investigation.
The current therapy for PD and AD remains targeting replacement of lost transmitter, but the ultimate objective in neurodegenerative therapy is the functional restoration and/or cessation of progression of neuronal loss.
a novel approach for the treatment of neurodegenerative diseases through the development of kinase inhibitors that block the active cell death process at an early transcriptional independent step in the stress activated kinase cascade.
In particular, preclinical data will be presented on the c-Jun Amino Kinase pathway inhibitor, CEP-1347/KT-7515, with respect to it’s properties that make it a desirable clinical candidate for treatment of various neurodegenerative diseases.
CEP-1347 is also known as KT-7515 and is being developed by Cephalon and Kyowa Hakko for treatment of Parkinson’s disease and cognitive disorders.
It is believed to be a JNK-MAP kinase inhibitor. CEP-1347 has the chemical name 9alpha,12alpha-Epoxy-5,16-bis(ethylsulfanylmethyl)-10beta-hydroxy-9-methyl-1-oxo-2,3,9,10,11,12alpha-hexahydro-1H-diindolo[1,2,3-fg:3′,2′,1′-kl]pyrrolo[3,4- i][1,6]benzodiazocine-10-carboxylic acid methyl ester and has the chemical structure as depicted in Formula 7.

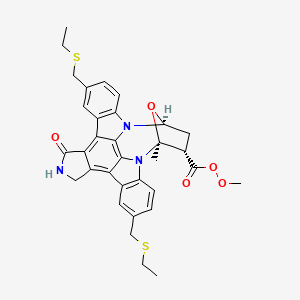


PATENT
https://google.com/patents/WO2005082920A1?cl=en
The compound with the structure outlined below is presently in clinical trials for Parkinson’s disease (Idrugs, 2003, 6(4), 377-383).

This compound is in the following referred to as Compound I. The chemical name of Compound I is [9S-(9α,10β,12α)]-5,16-Rw[(ethylthio)methyl]-2,3,9,10,l l,12-hexahydro- 10-hydroxy-9-methyl- 1 -oxo-9, 12-epoxy- 1 H-diindolo[l ,2,3 -fg:3 ‘,2’, 1 ‘-kl]ρyrrolo[3,4- i][l,6]benzodiazocine-10-carboxylic acid methyl ester.
The following references relate to Compound I, in particular to methods for its preparation [J.Med. Chem. 1997, 40(12), 1863-1869; Curr. Med. Chem. – Central Nervous System Agents, 2002, 2(2), 143-155] and its potential medical uses, mainly in diseases in the central nervous system (CNS), in particular for treatment of neurodegenerative diseases, e.g. Parkinson’s disease, Alzheimer’s disease, Huntington’s disease, peripheral neuropathy, AIDS dementia, and ear injuries such as noise-induced hearing loss [Progress in Medicinal Chemistry (2002), 40, 23-62; Bioorg. Med. Chem. Lett. 2002,12(2), 147-150; Neuroscience, Oxford, 1998, 86(2), 461-472; J. Neurochemistry (2001), 77(3), 849-863; J. Neuroscience (2000), 20(1), 43-50; J. Neurochemistry (2002), 82(6), 1424-1434; Hearing Research, 2002, 166(1-2), 33-43].
The following patent documents relate to Compound I, including its medical use and synthesis: WO 9402488, WO9749406, US 5621100, EP 0651754 and EP 112 932. By the known methods, Compound I is synthesized in a solid amorphous form. The inventors have now discovered 5 crystalline forms of Compound I (named alpha, beta, gamma, delta and epsilon) thereby providing an opportunity to improve the manufacturing process of Compound I and its pharmaceutical use. There exists a need for crystalline forms, which may exhibit desirable and beneficial chemical and physical properties. There also exists a need for reliable and reproducible methods for the manufacture, purification, and formulation of Compound I to permit its feasible commercialisation.
EXAMPLES
In the following the starting material ” Compound I” may, e.g., be prepared as described by Kaneko M. et al in J. Med. Chem. 1997, 40, 1863-1869.
Example 1. Preparation of crystalline alpha form of Compound I
Method I):
6.0 g amorphous Compound I was dissolved in 30 ml acetone. 0,6 g potassium carbonate was added and the suspension was stirred at room temperature for 1 hour before it was filtered to remove potential minor insoluble impurities and inorganic salts. The filter cake was washed with acetone. The filtrate was then evaporated on a rotary evaporator under reduced pressure at 60°C to a final volume of 10 ml to which 100 ml methanol was added slowly. The product separated as an oil, which almost dissolved on heating to reflux. Subsequently the residual insoluble impurities were removed by filtration. The filtrate was left with stirring at room temperature. A crystalline solid separated and was isolated by filtration. The filter cake was washed with methanol and dried in vacuo at 60°C overnight. Yield 2,83 g (47%), mp=182.4°C (DSC onset value), Weight loss by heating: 0.5%, Elemental analysis: 6.71%N, 63.93%C, 5.48%H, theoretical values corrected for 0.5% H2O: 6.79%N, 64.05%C, 5.43%H. XRPD analysis conforms with the alpha form. Method II):
5 g amorphous Compound I was dissolved in 25 ml acetone by gentle heating. 10 ml Methanol was added very slowly until the solution got turbid. The solution was allowed to cool to room temperature by natural cooling. The suspension was filtered and the filter-cake discarded. During filtration more material precipitated in the filtrate. The filtrate was heated until all material redissolves. Cold methanol was then added to the solution until precipitation was observed. The slightly turbid solution was then heated until all material was in solution. The solution was allowed to cool to room temperature, and the precipitate was removed by filtration. The second filter-cake was discarded. During the filtration some material separated in the filtrate. Heating redissolved the beginning crystallisation in the filtrate. Cold methanol was then added to the solution until precipitation was observed. The suspension was heated until a clear solution was obtained. The solution was allowed to reach room temperature by natural cooling. After a short period of time (15 min) precipitation begun. The precipitated pale yellow product was isolated by filtration and dried in vacuo at 50°C overnight. mp=188.9°C (DSC onset value), Weight loss by heating: 0.3%>, Elemental analysis: 6.53%N, 64.33%C, 5.43%H, theoretical values: 6.82%N, 64.37%C, 5.37%H. XRPD analysis conforms with the alpha form. Method III:
0.5g Compound I in a mixture of isopropyl acetate (10 mL) and water (0.6 mL) was heated to reflux with stirring. The compound was not completely dissolved so isopropyl acetate (10 mL) and water (0.6 mL) were added and heated to reflux. Stirring was stopped and the experiment was allowed to cool to room temperature. The crystalline product obtained were isolated by filtration and dried in vacuo at 40° C. Yield = 0.25g, mp = 183.7°C (DSC onset value). XRPD analysis conforms with the alpha form. Method IV: 0.5g Compound I in a mixture of ethyl acetate (10 mL) and water (0.4 mL) was heated to 70° C with stirring. The experiment was allowed to cool to room temperature. The crystalline product obtained were isolated by filtration and dried in vacuo at 40° C. XRPD analysis conforms with the alpha form.
PATENT
https://www.google.com/patents/US20050261762
PATENT
http://www.google.co.ug/patents/EP2004158A2?cl=en
CEP-1347 (KT7515) (Maroney et al. 1998; Roux et al. 2002).

PAPER
Neurotrophic 3,9-bis[(alkylthio)methyl]- and -bis(alkoxymethyl)-K-252a derivatives
J Med Chem 1997, 40(12): 1863
http://pubs.acs.org/doi/full/10.1021/jm970031d


The synthesis of the title compound used as the starting material was the indolocarbazole alkaloid K-252A (I). Compound (I) was protected as the diacetyl derivative (II) by treatment with Ac2O and DMAP. Formylation of (II) with dichloromethyl methyl ether in the presence of TiCl4 afforded dialdehyde (III), which was further reduced to diol (IV) using NaBH4 in MeOH-CHCl3. Condensation of diol (IV) with ethanethiol in the presence of camphorsulfonic acid furnished the bis-sulfanyl compound (V). The acetyl protecting groups of (V) were finally removed by treatment with sodium methoxide. Alternatively, diol (IV) was first deacetylated by treatment with NaOMe, and the deprotected bis(hydroxymethyl) compound (VI) was then condensed with ethanethiol to produce the title bis-sulfayl compound 8.
3,9-Bis[(ethylthio)methyl]-K-252a (8):
mp 163−165 °C;
IR (KBr) 1725, 1680 cm-1; FAB-MSm/z 615(M+);
1H-NMR (400 MHz, DMSO-d6) δ 1.23 (t, 6H, J = 7.3 Hz), 1.99 (dd, 1H, J = 4.8, 14.1 Hz), 2.132 (s, 3H), 2.489 (q, 2H, J = 7.3 Hz), 2.505 (q, 2H, J = 7.3 Hz), 3.37 (dd, 1H, J = 7.6, 14.1 Hz), 3.92 (s, 3H), 3.94 (s, 2H), 3.98 (s, 2H), 4.95 (d, 1H, J = 17.6 Hz), 5.02 (d, 1H, J = 17.6 Hz), 6.32 (s, 1H), 7.10 (dd, 1H, J = 4.8, 7.6 Hz), 7.450 (m, 2H), 7.84 (d, 1H, J = 8.5 Hz), 7.88 (d, 1H, J = 8.8 Hz), 7.95 (d, 1H, J = 1.0 Hz), 8.60 (s, 1H), 9.13 (d, 1H, J = 0.7 Hz);
HRFAB-MS calcd for C33H33N3O5S2 615.1862, found 615.1869. Anal. (C33H33N3O5S2·0.5H2O) C, H, N.
References
Maroney et al (1998) Motoneuron apoptosis is blocked by CEP-1347 (KT 7515), a novel inhibitor of the JNK signaling pathway. J.Neurosci. 18 104. PMID: 9412490.
Saporito et al (1998) Preservation of cholinergic activity and prevention of neuron death by CEP-1347/KT-7515 following excitotoxic injury of the nucleus basalis magnocellularis. Neuroscience 86 461. PMID: 9881861.
Bozyczko-Coyne et al (2001) CEP-1347/KT-7515, an inhibitor of SAPK/JNK pathway activation, promotes survival and blocks multiple events associated with Abeta-induced cortical neuron apoptosis. J.Neurochem. 77 849. PMID: 11331414.
| WO1994002488A1 * | Jul 26, 1993 | Feb 3, 1994 | Cephalon Inc | BIS-STAUROSPORINE AND K-252a DERIVATIVES |
| Reference | ||||
|---|---|---|---|---|
| 1 | * | KANEKO M ET AL: “Neurotrophic 3,9-Bis[(alkylthio)methyl]- and -Bis(alkoxymethyl)-K-252a Derivatives” JOURNAL OF MEDICINAL CHEMISTRY, AMERICAN CHEMICAL SOCIETY. WASHINGTON, US, vol. 40, no. 12, 1997, pages 1863-1869, XP002128804 ISSN: 0022-2623 cited in the application | ||
//////////CEP 1347, KT 7515 ,
CCSCC1=CC2=C(C=C1)N3C4CC(C(O4)(N5C6=C(C=C(C=C6)CSCC)C7=C8CNC(=O)C8=C2C3=C75)C)C(=O)OOC
DR ANTHONY MELVIN CRASTO
ORGANIC SPECTROSCOPY

