
FLAGS AND HITS
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO FACEBOOK
...................................................................Join me on twitter
FACEBOOK
...................................................................Join me on twitter
 ..................................................................Join me on google plus
..................................................................Join me on google plus
 Googleplus
GoogleplusMYSELF

ND 2110
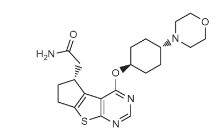
ND -2110
| Molecular Formula: | C21H28N4O3S |
|---|---|
| Molecular Weight: | 416.53702 g/mol |

2-[(3R)-12-{[(1r,4r)-4-(morpholin-4-yl)cyclohexyl]oxy}-7-thia-9,11-diazatricyclo[6.4.0.0²,⁶]dodeca-1(12),2(6),8,10-tetraen-3-yl]acetamide
1388894-17-4
- Molecular Weight416.54
ND-2110 is a potent and selective experimental inhibitor of IRAK4 described in patent WO2013106535 [2] and in a poster presented at the American College of Rheumatology meeting in 2012 (Abstract #1062 in Supplement: Abstracts of the American College of Rheumatology & Association of Rheumatology Health Professionals, Annual Scientific Meeting, November 9-4, 2012 Washington DC, Volume 64, Issue S10, Page S1-S1216).


PATENT
http://www.google.com/patents/WO2013106535A1?cl=en
Example 29: Synthesis of 2-((R)-4-(((lr,4R)-4-morpholinocyclohexyl)oxy)-6,7-

29.3 1-67
Synthesis of compound 29.1. 4-(Morpholin-4-yl)cyclohexan-l-ol (commercially available; 218 mg, 1.2 mmol, 1.50 equiv) was treated with NaH (60% dispersion in mineral oil, 128 mg, 3.2 mmol, 4 equiv) in freshly distilled tetrahydrofuran (15 mL) for 30 min at 0 °C in a water/ice bath under nitrogen. Then a solution of intermediate 25.1 (289 mg, 0.8 mmol, 1.00 equiv) in 5 mL of THF was added via syringe and the resulting solution was allowed to stir for an additional 3 h at 60 °C in an oil bath. The reaction was then quenched with saturated aqueous NH4CI and extracted with 3 x 50 mL of ethyl acetate. The combined organic layers were washed with brine, dried (Na2S04) and concentrated under vacuum. The residue was applied onto a silica gel column with ethyl acetate/petroleum ether (1:5-1:2) and purified to afford compound 29.1 (260 mg, 63%) as a colorless oil.
Synthesis of compound 29.2. To a solution of 29.1 (260 mg, 0.5 mmol, 1.0 equiv) in 10 mL of DCM was added 0.5 mL of concentrated hydrochloric acid in an ice/water bath. The resulting solution was stirred for 2 h and concentrated in vacuo. The residue was neutralized with saturated aqueous Na2C03 and extracted with 3 x 50 mL of ethyl acetate. The organic layers were combined, washed with brine, dried (Na2S04) and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel with DCM MeOH (15:1) to afford the desired alcohol 29.2 (185 mg, 91%) as a colorless oil. [00416] Synthesis of compound 29.3. Alcohol 29.2 (185 mg, 0.46 mmol, 1.00 equiv) was oxidized with dipyridinium dichromate (752 mg, 2.00 mmol, 4.36 equiv) in 50 mL of DMF for 24 h at room temperature. The resulting solution was diluted with water and extracted with 3 x 50 mL of mixed solutions of CHC¾/iso-PrOH. The organic layers were combined, dried (Na2S04) and concentrated under vacuum. The residue was applied onto a silica gel column with dichloromethane/methanol (5:1 to 1:1) and purified to afford 105 mg (55%) of acid 29.3 as a yellow oil.
[00417] Synthesis of Compound 1-67. A 50 mL round-bottom flask containing a solution of acid 29.3 (105 mg, 0.25 mmol, 1.00 equiv), NH4C1 (80 mg, 1.50 mmol, 6.00 equiv), EDCI (57 mg, 0.3 mmol, 1.2 equiv), 4-dimethylaminopyridine (37 mg, 0.3 mmol, 1.2 equiv) and HOBt (40 mg, 0.3 mmol, 1.2 equiv) in 5 mL of anhydrous DMF was stirred for 24 h at room temperature. The resulting solution was diluted with water and extracted with 4 x 50 mL of mixed solution of CHCl3:iso-PrOH. The combined organic layers were concentrated under vacuum. The crude product was purified by preparative HPLC (SHIMADZU) under the following conditions: column: SunFire Prep C18, 19*150mm 5um; mobile phase: water (0.05% NH4CO3) and CH3CN (6.0% CH3CN up to 50.0% in 25 min); UV detection at 254/220 nm. The product-containing fractions were collected and concentrated to give Compound 1-67 (22.5 mg) as a white solid. ¾ NMR (300 MHz, CD3OD) δ 8.43 (s, 1H), 5.27-5.20 (m, 1H), 3.80-3.70 (m, 5H), 3.29-3.27 (m, 1H), 3.12-2.90 (m, 2H), 2.73-2.67 (m, 5H), 2.49-2.42 (m, 1H), 2.32-2.19 (m, 4H), 2.10-2.06 (d, 2H), 1.67-1.46 (m, 4H). MS: m/z 417 (M+H)+.
PATENT
http://www.google.com/patents/WO2012097013A1
Example 29: Synthesis of 2-((R)-4-(((lr,4R)-4-morpholinocyclohexyl)oxy)-6,7- dihydro-5H-cyclopenta[4,5]thieno[2,3-d]pyrimidin-5-yl)acetamide.
Page 280 of 407
2009184-0008


33b
Synthesis of compound 31b. 4-(Morpholin-4-yl)cyclohexan-l-ol (commercially available; 218 mg, 1.2 mmol, 1.50 equiv) was treated with NaH NMR (60% dispersion in mineral oil, 128 mg, 3.2 mmol, 4 equiv) in freshly distilled tetrahydrofuran (15 mL) for 30 min at 0 °C in a water/ice bath under nitrogen. Then a solution of intermediate Hb (289 mg, 0.8 mmol, 1.00 equiv) in 5 mL of THF was added via syringe and the resulting solution was allowed to stir for an additional 3 h at 60 °C in an oil bath. The reaction was then quenched with saturated aqueous NH4CI and extracted with 3 x 50 mL of ethyl acetate. The combined organic layers were washed with brine, dried (Na2S04) and concentrated under vacuum. The residue was applied onto a silica gel column with ethyl acetate/petroleum ether (1 :5-1 :2) and purified to afford compound 31b (260 mg, 63%) as a colorless oil.
Synthesis of compound 32b. To a solution of 31b (260 mg, 0.5 mmol, 1.0 equiv) in 10 mL of DCM was added 0.5 mL of concentrated hydrochloric acid in an ice/water bath. The resulting solution was stirred for 2 h and concentrated in vacuo. The residue was neutralized with saturated aqueous Na2C( j and extracted with 3 x 50 mL of ethyl acetate. The organic layers were combined, washed with brine, dried (Na2S04) and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel with DCM/MeOH NMR ( 15: 1 ) to afford the desired alcohol 32b ( 185 mg, 91 %) as a colorless oil.
Synthesis of compound 33b. Alcohol 32b (185 mg, 0.46 mmol, 1.00 equiv) was oxidized with dipyridinium dichromate (752 mg, 2.00 mmol, 4.36 equiv) in 50 mL of DMF for
Page 281 of 407
2009184-0008 24 h at room temperature. The resulting solution was diluted with water and extracted with 3 x 50 mL of mixed solutions of CHCU/iso-PrOH. The organic layers were combined, dried (Na2S04) and concentrated under vacuum. The residue was applied onto a silica gel column with dichloromethane/methanol (5: 1 to 1 : 1 ) and purified to afford 105 mg (55%) of acid 33b as a yellow oil.
Synthesis of Compound. A 50 mL round-bottom flask containing a solution of acid 33b (105 mg, 0.25 mmol, 1.00 equiv), NH4C1 (80 mg, 1.50 mmol, 6.00 equiv), EDCI (57 mg, 0.3 mmol, 1.2 equiv), 4-dimethylaminopyridine (37 mg, 0.3 mmol, 1.2 equiv) and HOBt (40 mg, 0.3 mmol, 1.2 equiv) in 5 mL of anhydrous DMF was stirred for 24 h at room temperature. The resulting solution was diluted with water and extracted with 4 x 50 mL of mixed solution of CHCI3: iso-PrOH. The combined organic layers were concentrated under vacuum. The crude product was purified by preparative HPLC (SHIMADZU) under the following conditions: column: SunFire Prep C I 8, 19* 150mm 5um; mobile phase: water (0.05% Ν¾∞3) and CH3CN (6.0% CH3CN up to 50.0% in 25 min); UV detection at 254/220 nm. The product containing fractions were collected and concentrated to give the product (22.5 mg) as a white solid. Ή MR (300 MHz, CD3OD) δ 8.43 (s, 1H), 5.27-5.20 (m, 1H), 3.80-3.70 (m, 5H), 3.29-3.27 (m, 1 H), 3.12-2.90 (m, 2H), 2.73-2.67 (m, 5H), 2.49-2.42 (m, 1H), 2.32-2.19 (m, 4H), 2.10-2.06 (d, 2H), 1.67- 1.46 (m, 4H). MS: m/z 417 (M+H)+.
Paper
http://pubs.acs.org/doi/abs/10.1021/jm5016044
Recent Advances in the Discovery of Small Molecule Inhibitors of Interleukin-1 Receptor-Associated Kinase 4 (IRAK4) as a Therapeutic Target for Inflammation and Oncology Disorders
Miniperspective

IRAK4, a serine/threonine kinase, plays a key role in both inflammation and oncology diseases. Herein, we summarize the compelling biology surrounding the IRAK4 signaling node in disease, review key structural features of IRAK4 including selectivity challenges, and describe efforts to discover clinically viable IRAK4 inhibitors. Finally, a view of knowledge gained and remaining challenges is provided.
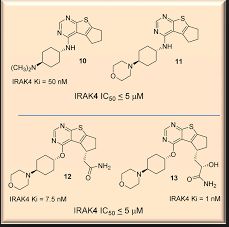
-
78 Romero, D. L.; Robinson, S.; Wessel, M. D.; Greenwood, J. R. IRAK Inhibitors and Uses Thereof. WO201401902, January 16, 2014.
-
Harriman, G. C.; Romero, D. L.; Masse, C. E.; Robinson, S.; Wessel, M. D.; Greenwood, J. R. IRAK Inhibitors and Uses Thereof. WO2014011911A2, January 16, 2014.
-
Harriman, G. C.; Wester, R. T.; Romero, D. L.; Masse, C. E.; Robinson, R.; Greenwood, J. R. IRAK Inhibitors and Uses Thereof. WO2014011906A2, January 16, 2014

WO 2014194245
WO 2014194201
WO 2014194242
WO 2013106535
WO 2012097013
| 1. Chaudhary D, Robinson S, Romero DL. (2015) Recent Advances in the Discovery of Small Molecule Inhibitors of Interleukin-1 Receptor-Associated Kinase 4 (IRAK4) as a Therapeutic Target for Inflammation and Oncology Disorders. J. Med. Chem., 58 (1): 96-110. |
| 2. Harriman GC, Wester RT, Romero DL, Robinson S, Shelley M, Wessel MD, Greenwood JR, Masse CE, Kapeller-Libermann R. (2013) Irak inhibitors and uses thereof. Patent number: WO2013106535C1CC(CCC1N2CCOCC2)OC3=C4C5=C(CCC5CC(=O)N)SC4=NC=N3. Assignee: Nimbus Iris, Inc.. Priority date: 18/07/2013. Publication date: 10/01/2012. |
http://nimbustx.com/sites/default/files/uploads/posters/irak4_nimbus_acr_poster_2012_small.pdf
///////ND-2110, ND 2110. IRAK4, NIMBUS, GTPL8802
NC(=O)CC1CCc2c1c1c(ncnc1s2)OC1CCC(CC1)N1CCOCC1
C1CC(CCC1N2CCOCC2)OC3=C4C5=C(CCC5CC(=O)N)SC4=NC=N3
CN-128 for the treatment of thelassemia and iron overload

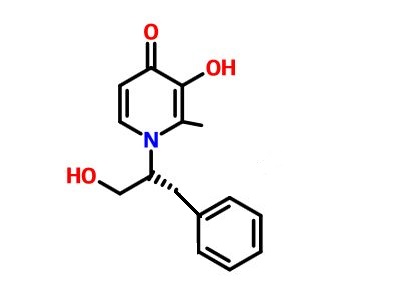
CN-128
(R)-3-Hydroxy-1-(1-hydroxy-3-benzyl propyl-2-)2-methyl pyridine-4(1H)-one
IND Filing
CN-128 is potentially for the treatment of thelassemia and iron overload.
| Zhejiang University, 浙江大学 |
![]()
CAS No. 1335282-04-6
- Molecular Weight, 259.30
Many diseases in humans and animals are caused by excessive accumulated metals, such as iron. Among such diseases, excess iron is accumulated in various tissues, which is called iron overload disorders, formerly known as siderosis Haemorrhagic. Excess iron has the following sources: 1) long-term blood transfusion; 2) the gastrointestinal system absorbing excess iron, because stimulated by diseases such as anemia. It is necessary to repeat transfusion for some patients with severe anemia, for example, β-thalassemia, as well as other anemia requiring transfusion therapy. Excessive iron absorption from the gastrointestinal tract usually occurs in hemochromatosis patients and in anemia patients who do not require blood transfusion, such as thalassemia intermedia. If iron overload disease is not treated, it will result in severe tissue damage, especially the liver, heart and endocrine organs, and ultimately lead to death. Iron chelators can remove and clear excess iron from such organs, relieve symptoms and reduce the corresponding mortality.
Desferrioxamine (DFO) is an effective iron chelator for a long time. However, in the treatment of the diseases mentioned above, the biggest disadvantage regarding DFO and its salts is its poor oral absorption capability. So, administration is achieved with a slow injection method (8∼12h/day), patients need to wear a portable drug delivery device during treatment, such as mounting the syringe on a mechanical pressing device. This method is inconvenient, and also expensive, which largely limits the utilization of DFO, especially for thalassemia-prone areas, such as Mediterranean, Middle East, and India &South East Asia, it plays no role in treatment of malaria in world-wide and sickle cell anemia in some African countries, which is a very serious problem to the populations there.
UK Patent No. 2, 13, 807, US Patent No. 4, 585, 780 and other scientific research have reported the treatment of iron overload symptoms by using 3-hydroxypyridin-4-one derivatives, especially in some pathological symptoms, such as thalassemia, sickle cell anemia, aplastic anemia in children, and idiopathic hemochromatosis, usually, treatment of the first three diseases includes frequent regular blood transfusion. 3-Hydroxypyridin-4-one derivatives, especially CP20 (commercial named Ferriprox) is employed to treat systemic iron overload disorders, and also to treat certain diseases associated with local iron overload distribution, although such patients do not show symptoms of systemic iron overload, i.e. inhibition free radical mediated reactions caused by excess iron ions in certain neurodegenerative diseases and cancer diseases. A serious limitation of CP20 is that the hydroxyl group at 3′ position is vulnerable to glycosylation, which reduces the half-life of this compound (approximately 2∼3 h). So it requires a high dosage, which is associated with obvious side effects.
EP0120669 discloses compounds with a 3-hydroxypyrid-4-one in which the H attached to the N atom is substituted by an aliphatic acyl group, or an aliphatic hydrocarbon group, these groups can be further substituted, but not by aromatic groups and their use against illnesses related to iron overload. Molenda et al. disclose in Journal of Medicinal Chemistry 1994, 37, pages 4363-4370 chiral 3-hydroxy-pyridin-4-one compound 6 as enhancing iron excretion.
US Patent No. 6, 465, 604 described a series of 3,5-diphenyl-1,2,4-triazole compounds, wherein including Exjade (commercial name), which has strong affinity to Fe(III), However, its active groups contain two negatively charged oxygen ions and a carboxyl group; it is a tridentate ligand while chelating Fe(III), which forms a Fe-L2 type complex, possessing three unit negative charges itself, that is bad for their discharge from cells/tissues. Moreover, one of the active groups is a nitrogen atom with a lone pair of electrons, Exjade may have a negative effect on the balance of Zn(II) in vivo, at the same time because it has two phenolic hydroxyl groups in different positions (forming intramolecular hydrogen bonds structure similar to cis/trans isomerization), it can be complexed to several zinc ions to form high molecular weight polymers complexes, which is not conducive to its discharge from the cells either.
Absorption, distribution, metabolism and excretion of chiral medicines are largely related to the 3D structures of their chiral centers. For drug absorption, chiral compounds entering cells via active transport mechanism are usually carried by special transport proteins, their recognition of enantiomers can be different, resulting in different absorption of enantiomers. For drug distribution, the binding effects of plasma protein and tissues are also somewhat stereoselective, leading to different in vivo distribution of enantiomers; for stereoselective of drug metabolism refers to when the substrate is biotransformated, the pathway and speed of enantiomer metabolism by biological systems can be different. One enantiomer may show ascendant metabolism, and therefore it is of great significance to the indicators including drug transformation and in vivo half-life. Glomerular filtration, tubular secretion and reabsorption of chiral drugs to clear the chiral drugs, having stereoselectivity, while the glomerular filtration rate is closely related to drug’s selectivity to binding plasma protein, so discharge style of enantiomers (urine / feces percentage) and the rate is also different.
Therefore, the qualitative difference of the interactions of a pair of enantiomers with various binding sites may exist or not, and the quantitive difference may exist (strong or weak), which results in the different activities between enantiomers. Thus the selection of optical enantiomers for medical use, requires a comprehensive study of metabolic activity, toxicology and pharmacokinetic properties etc. Thus the chiral nature of the 3-hydroxypyridin-4-one derivatives described in this patent has an important role on in vivo iron chelation.
The effectiveness of many oral 3-hydroxy-4-one derivatives drugs are subject to metabolic reaction of the 3-hydroxy moiety, which may be quickly glycosylated (see Reaction I). The hydroxypyridone after glycosylation loses the ability to chelate Fe(III). We can effectively inhibit glycosylation reaction by introducing hydroxyl groups to alkyl substituted residues on the pyridine ring. In addition, the partition coefficient of 3-hydroxy-pyridin-4-one derivatives has a great impact on the in vivo distribution and toxic effects. We have introduced various alkyl groups to the chiral point of the compound, in order to modify their lipophilicity, i.e. a phenyl group connected to the chiral point in compound IV-b while in IV-a it is a methyl group, and thus compound IV-b is relatively more lipophilic, and easier to penetrate through cell membranes of various tissues and critical barriers such as the blood-brain barrier and the placental barrier, thus affecting its in vivo distribution. Thus increase of hydroxyl groups can affect the intestinal absorption capacity, by introducing a large alkyl group, intestinal absorption of 3-hydroxy-pyridin-4-one derivatives can be enhanced.
Reaction I
Example7. (R)-3-Hydroxy-1-(1-hydroxy-3-benzyl propyl-2-)2-methyl pyridine-4(1H)-one, Number: CN128.
60 g 3-phenyloxy-2-methyl-4H-pyran-one(Example 1) was dissolved in 150 mL n-butanol, then 83.7 g D-phenylalaninol was added in. After thoroughly mixing, the solution was refluxed at 118°C for 36 h. After cooling and filtration, products were purified by silica gel column chromatography with Eluent ethanol: acetic ester=1:40. After Elution, light brown solid was obtained after rotary evaporation, which was then dissolved into 150 mL ethanol and 15 mL water, then it was hydrogenated and debenzylated with 5% Pd/C as catalyst, the solvent was removed under rotary evaporation, the remaining solid was recrystallized with methanol and ether, leading to 25.25 g light yellow solid. The yield was 35.1%. The free alkali’s 1HNMR (DMSO-d6): δ 2.00 (s, 3H), 2.98 (dd, J1=14, J2=5.5, 1H), 3.11 (dd, J1=14, J2=5, 1H), 3.73 (m, 2H), 4.54 (m, 1H), 6.21 (d, J=7, 1H), 7.17 (m, 5H), 7.87 (d, J=7.5, 1H).
PATENT
CN 102190644
http://www.google.com/patents/CN102190644B?cl=en
![]()






Debiopharm and Aurigene dual c-src / jak inhibitors

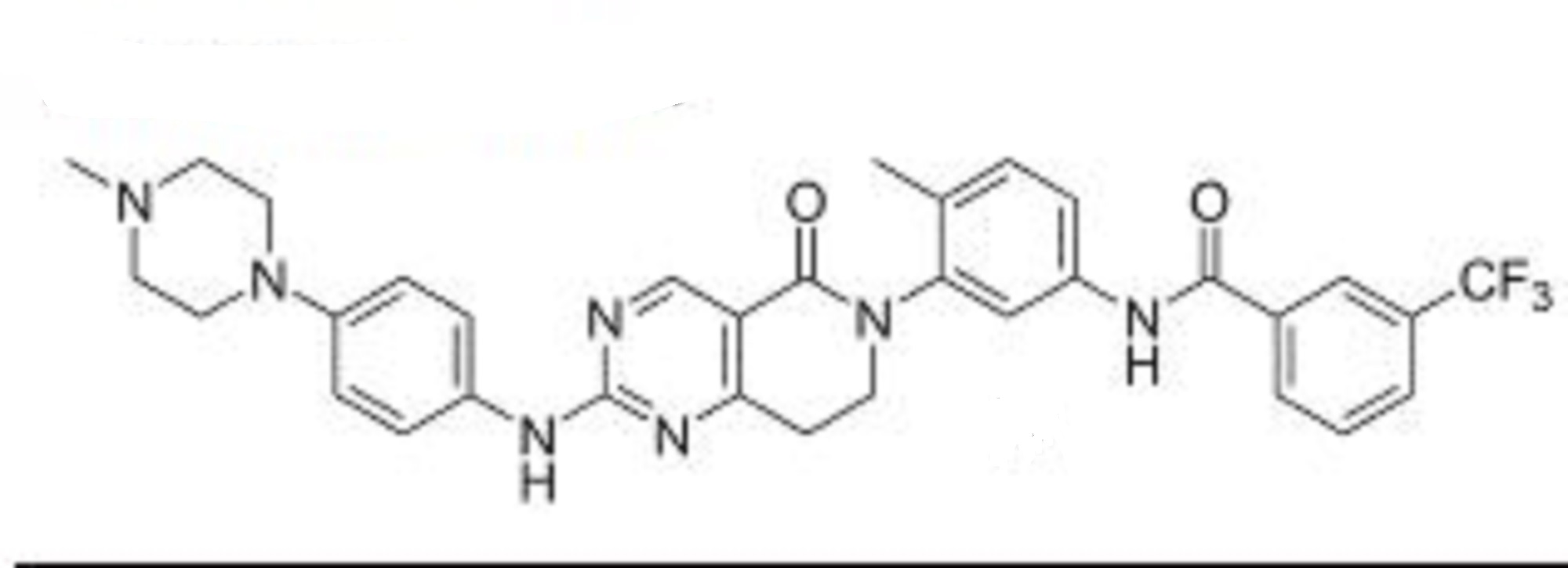
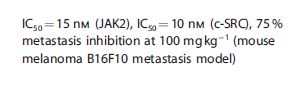
Debio 1142
Jak2 tyrosine kinase inhibitor; Src tyrosine kinase inhibitor
N-[4-methyl-3-[2-[4-(4-methylpiperazin-1-yl)anilino]-5-oxo-7,8-dihydropyrido[4,3-d]pyrimidin-6-yl]phenyl]-3-(trifluoromethyl)benzamide
| Molecular Formula: | C33H32F3N7O2 |
|---|---|
| Molecular Weight: | 615.64809 g/mol |
Debiopharm S.A., Aurigene Discovery Technologies Ltd.
ALLISTER Andrès MC, Maximilien Murone,Saumitra Sengupta, Shankar Jayaram Shetty
https://www.google.co.in/patents/WO2011101806A1?cl=en
Bicyclic compounds and their uses as dual c-src / jak inhibitors

Apr. 14 /PR Newswire/ –Debiopharm and Aurigene Sign Agreement for the Development and Commercialisation of Debio 1142, a Novel Inhibitor of an Undisclosed Oncology Pathway
LAUSANNE, Switzerland and BANGALORE, India, April 14, 2011 /PRNewswire/ — Debiopharm Group(TM) (Debiopharm), a global biopharmaceutical development specialist that focuses on serious medical conditions and particularly oncology, and Aurigene Discovery Technologies Ltd (Aurigene), a Bangalore-based drug discovery company, signed on March 23, 2011 an option and exclusive worldwide license agreement concerning the development and commercialisation of Debio 1142, a novel inhibitor of an undisclosed oncology pathway.
“We are very excited about this new collaboration with Aurigene. Their business model offers a one stop solution for structure guided drug design, lead optimisation and preclinical work. The Debio 1142 project aims at developing inhibitors targeting a key oncology pathway, which plays essential roles in various solid tumours, including resistance to chemotherapy” said Dr Rolland-Yves Mauvernay, president and founder of Debiopharm S.A.
“Coming as it does after a successful collaboration programme we already had with Debiopharm, and as a continuation of our close to 5 year association, the relationship between Debiopharm and Aurigene demonstrates the strategic fit between organisations with complimentary scientific skills. We are happy that we have the opportunity to continue to work with Debiopharm, in a unique business model that has been tailor-made to meet each partners’ needs” added CSN Murthy, CEO of Aurigene.
About Debiopharm Group
Debiopharm Group(TM) (Debiopharm) is a Swiss-based global biopharmaceutical group of companies with a focus on the development of prescription drugs that target unmet medical needs. The group in-licenses, develops and/or co-develops promising biological and small molecule drug candidates having reached clinical development phases I, II or III as well as earlier stage candidates. It develops its products for global registration and maximum commercial potential. The products are out-licensed to pharmaceutical partners for sales and marketing. Debiopharm Group is also active in the field of companion diagnostics with a view to progressing in the area of personalised medicine. Debiopharm independently funds the worldwide development of all of its products while providing expertise in pre-clinical and clinical trials, manufacturing, drug delivery and formulation, and regulatory affairs. For more information on Debiopharm Group(TM), please visit: http://www.debiopharm.com.
About Aurigene
Aurigene Discovery Technologies Limited is a Bangalore-based biotech focused on collaborative drug discovery with pharmaceutical and biotech companies on a risk-sharing basis. Aurigene has fully integrated drug discovery infrastructure, from Target to IND, along with strong in house structural biology and fragment based drug design capabilities. The company is engaged in over 20 discovery collaborations with US and European large and mid-pharma companies in Oncology, Inflammatory disorders and anti-infectives. For more information on Aurigene, please visit: http://www.aurigene.com.
PATENT
WO 2011101806
http://www.google.co.in/patents/WO2011101806A1?cl=en
PAPER
Journal of Chemical and Pharmaceutical Research (2014), 6(4), 1146-1152
http://jocpr.com/vol6-iss4-2014/JCPR-2014-6-4-1146-1152.pdf
REFERENCES
INDIAN PATENTS
7554/CHENP/2012
415/CHE/2010
https://www.debiopharm.com/our-business/pipeline.html
http://www.giiresearch.com/report/labd315710-debiopharm-international-sa-product-pipeline.html
| Patent ID | Date | Patent Title |
|---|---|---|
| US2013143895 | 2013-06-06 | BICYCLIC COMPOUNDS AND THEIR USES AS DUAL C-SRC / JAK INHIBITORS |
| US8440679 | 2013-05-14 | Bicyclic compounds and their uses as dual c-SRC / JAK inhibitors |
///////////Debio 1142, Jak2 tyrosine kinase inhibitor, Src tyrosine kinase inhibitor, Debio-1142, Debiopharm S.A., Aurigene Discovery Technologies Ltd, 1332328-01-4
c21cnc(nc1CCN(C2=O)c3c(ccc(c3)NC(=O)c4cc(ccc4)C(F)(F)F)C)Nc5ccc(cc5)N6CCN(CC6)C
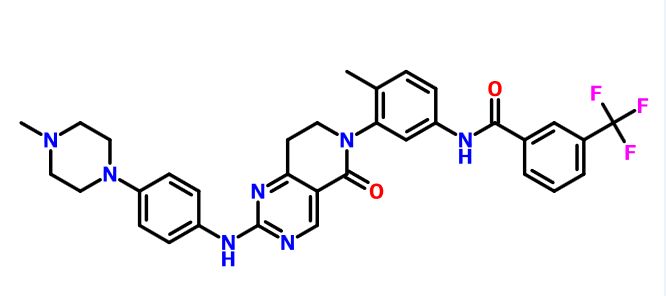
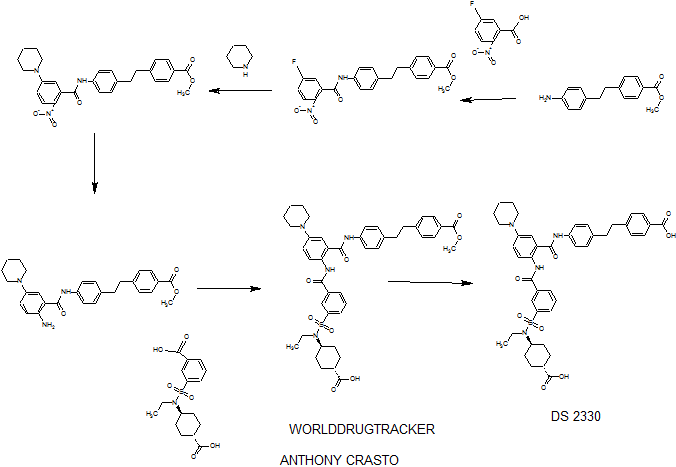
DS 2330 by Daiichi Sankyo
DS 2330
a trans compd
4-[2-(4-{[2-({3-[(trans-4-carboxy-cyclohexyl)(ethyl)sulfocarbamoyl]benzoyl}amino)-5-(piperidin-1-yl)benzoyl]amino}phenyl)ethyl]benzoic acid,
4- [2- (4 – {[2 – ({3 – [(trans-4-carboxy-cyclohexyl) (ethyl) sulfur carbamoyl] benzoyl} amino) -5- (piperidin-1-yl) benzoyl] amino} phenyl) ethyl] benzoate
- Originator Daiichi Sankyo Inc
- Class Hyperphosphataemia therapies
useful for treating hyperphosphatemia, DS-2330, a phosphorous lowering agent, being developed by Daiichi Sankyo, for treating hyperphosphatemia in chronic kidney disease. In April 2016, DS-2330 was reported to be in phase 1 clinical development.
- Phase IHyperphosphataemia
- 31 Oct 2015Phase-I clinical trials in Hyperphosphataemia in USA (unspecified route)
![]()
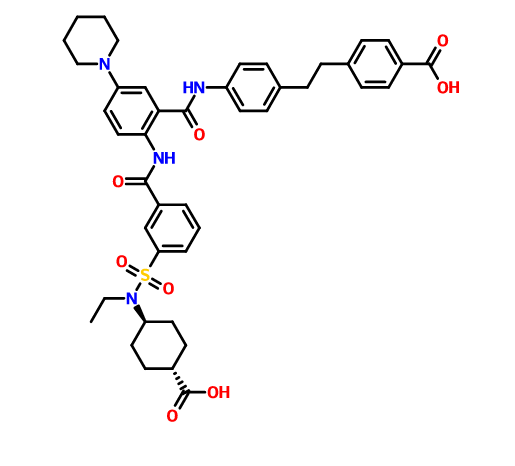
SEE WO2015108038,
PATENT
WO2014175317
http://www.google.com/patents/EP2990400A1?cl=en
PATENT
he problem is to provide a pharmaceutical for the prevention or treatment of hyperphosphatemia. The solution is a salt of a compound including formula (I), or a crystal of a hydrate thereof.
disodium 4- [2- (4 – {[2 – ({3 – [(trans-4-carboxy-cyclohexyl) (ethyl) sulfur carbamoyl] benzoyl} amino) -5- (piperidin-1-yl ) benzoyl] amino} phenyl) ethyl] benzoic acid trihydrate
Disodium 4- [2- (4 – { [2 – ({3 – [(trans-4-carboxylatocyclohexyl) (ethyl) sulfamoyl] benzoyl} amino) – 5- (piperidin-1-yl) benzoyl] amino} phenyl) ethyl] benzoate trihydrate
of α crystal
4- [2- (4 – {[2 – ({3 – [(trans-4-carboxy-cyclohexyl) (ethyl) sulfur carbamoyl] benzoyl} amino) -5- (piperidin-1-yl) benzoyl] amino} phenyl) ethyl] 1 mol / L NaOH aqueous solution to benzoic acid (1.2 g) (3.1 mL) was added and dissolved completely. After stirring at room temperature for 1 day was added acetonitrile (60 mL), at 40 ° C.
and stirred for further 1 day. The precipitated solid was collected by filtration, and 3 hours drying under reduced pressure at room temperature to give the title compound 1.1 g (85%).
in water (46.4 mL), 1-PrOH (72 mL), 4 mol / L NaOH aqueous solution (25.54 mL) was added, then filtered after stirring insolubles at room temperature, water / 1-PrOH: was washed with (3 7, 80 mL). The filtrate was heated up to 40 ℃, 1-PrOH the (160 mL) was added, and further seed crystal (α crystals, 0.2g) was added. Then the temperature was raised to 50 ℃, 1-PrOH (96 ml) was added, and the mixture was stirred overnight.Thereafter, 1-PrOH (480 ml) was added and after overnight stirring, was collected by filtration the precipitated solid was cooled to room temperature.Thereafter, and vacuum dried overnight at 40 ° C., to give the title compound 39.4 g (96%).
REFERENCES
////////////DS 2330, DS-2330, DAIICHI SANKYO, phase 1
O=C(O)[C@@H]1CC[C@H](CC1)N(CC)S(=O)(=O)c2cccc(c2)C(=O)Nc5ccc(cc5C(=O)Nc4ccc(CCc3ccc(cc3)C(=O)O)cc4)N6CCCCC6
OR
O=C(O)[C@@H]1CC[C@H](CC1)N(CC)S(=O)(=O)c2cccc(c2)C(=O)Nc5ccc(cc5C(=O)Nc4ccc(CCc3ccc(cc3)C(=O)O)cc4)N6CCCCC6
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
Join me on Facebook
Googleplus amcrasto@gmail.com
amcrasto@gmail.com
GDC-0084

GDC-0084
CAS#: 1382979-44-3
Chemical Formula: C18H22N8O2
Exact Mass: 382.1866
Synonym: RG7666; RG-7666; RG 7666; GDC-0084; GDC0084; GDC 0084.
IUPAC/Chemical Name: 5-(6,6-dimethyl-4-morpholino-8,9-dihydro-6H-[1,4]oxazino[4,3-e]purin-2-yl)pyrimidin-2-amine
| Latest Stage of Development | Phase I |
| Standard Indication | Brain cancer |
| Indication Details | Treat progressive or recurrent high-grade glioma |
| Regulatory Designation | |
| Partner | Genentech Inc. |
- Originator Genentech
- Class Antineoplastics; Small molecules
- Mechanism of Action 1 Phosphatidylinositol 3 kinase inhibitors
- 28 Jan 2015 Discontinued – Phase-I for Glioma in Spain (unspecified route)
- 28 Jan 2015 Discontinued – Phase-I for Glioma in USA (unspecified route)
- 01 Jan 2015 Genentech completes a phase I trial in Glioma in USA and Spain (NCT01547546)
GDC-0084, also known as RG7666, is a phosphatidylinositol 3-kinase (PI3K) inhibitor with potential antineoplastic activity. PI3K inhibitor GDC-0084 specifically inhibits PI3K in the PI3K/AKT kinase (or protein kinase B) signaling pathway, thereby inhibiting the activation of the PI3K signaling pathway. This may result in the inhibition of both cell growth and survival in susceptible tumor cell populations. Activation of the PI3K signaling pathway is frequently associated with tumorigenesis.
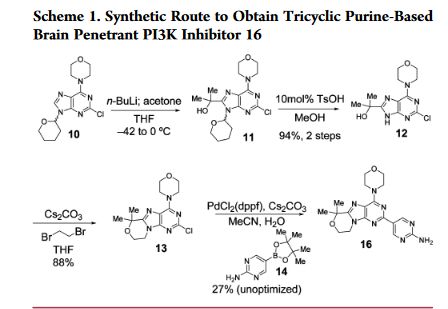
http://pubs.acs.org/doi/pdf/10.1021/acsmedchemlett.6b00005
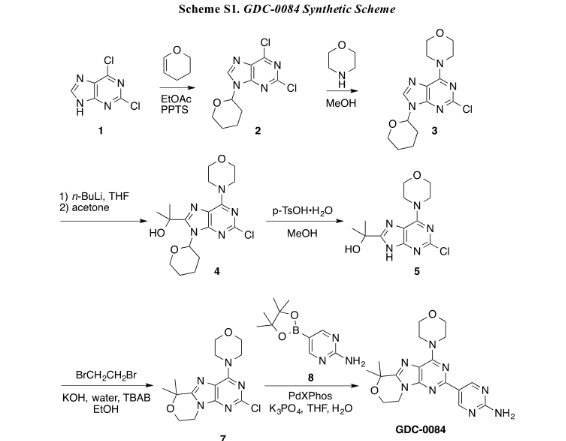

An improved, efficient process with a significantly reduced process mass intensity (PMI) led to the multikilogram synthesis of a brain penetrant PI3K inhibitor GDC-0084. Highlights of the synthesis include a phase transfer catalyzed annulation in water, an efficient Suzuki-Miyaura cross-coupling of a chloropyrimidine with an arylboronic acid using a low palladium catalyst loading, and the development of a controlled crystallization to provide the API. The process delivered GDC-0084 with low levels of both impurities and residual metals.
Development of an Efficient, Safe, and Environmentally Friendly Process for the Manufacture of GDC-0084
//////GDC-0084
NC1=NC=C(C2=NC(N3CCOCC3)=C4N=C(C(C)(C)OCC5)N5C4=N2)C=N1
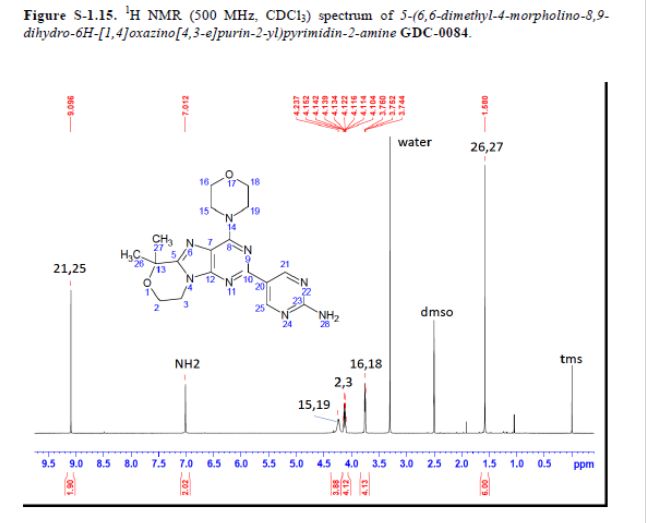
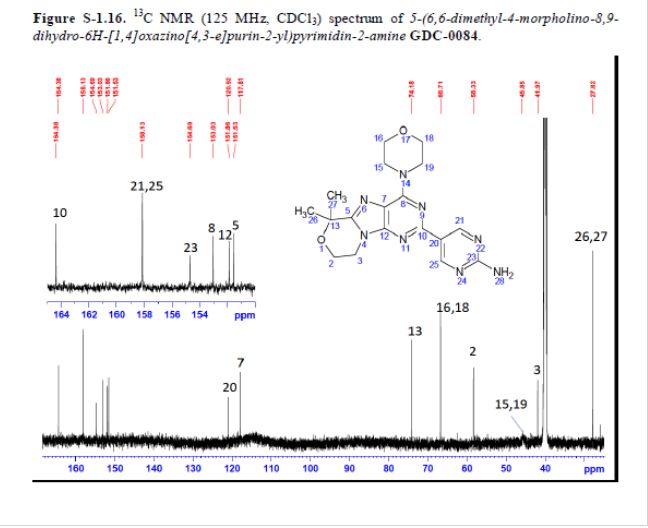
5-(6,6-Dimethyl-4-morpholino-8,9-dihydro-6H-[1,4]oxazino[4,3-e]purin-2-yl)pyrimidin-2-amine GDC-0084
mp 211 °C; 1H NMR (500 MHz, DMSO-d6) δ 9.09 (s, 2H), 7.03 (s, 2H), 4.32–4.17 (m, 4H), 4.17–4.04 (m, 4H), 3.84–3.65 (m, 4H), 1.58 (s, 6H); 13C NMR (125 MHz, DMSO-d6) δ 163.8, 157.6, 154.2, 152.5, 151.3, 151.0, 120.3, 117.3, 73.7, 66.2, 57.8, 45.2, 41.5, 27.3. HRMS [M + H]+calcd for C18H22N8O2 383.1938; found 383.1945.
-
The Discovery of Clinical Development Candidate GDC-0084, a Brain Penetrant Inhibitor of Class I Phosphoinositide 3-Kinases (PI3K) and mTOR.
Heffron, T.; Ndubaku, C.; Salphati, L.; Alicke, B.; Cheong, J.;Drobnick, J.; Edgar, K.; Gould, S.; Lee, L.; Lesnick, J.; Lewis, C.; Nonomiya, J.; Pang, j.; Plise, E.; Sideris,S.; Wallin, J.; Wang, L.; Zhang, X.; Olivero, A. ACS Med. Chem. Lett. 2016, , DOI: 10.1021/acsmedchemlett.6b00005
-
(a) Purine Derivatives Useful as PI3 Kinase Inhibitors. Goldsmith, P.; Hancox, T. C.; Hudson, A.; Pegg, N. A.; Kulagowski, J. J.; Nadin, A. J.; Price, S. PCT Int. Appl. WO 2009053716 A1 Apr 30, 2009.
(b) Preparation of Purine Derivatives with PI3K Inhibitory Activity and Methods of Use Thereof. Castanedo, G.; Chuckowree,I.; Folkes, A.; Sutherlin, D. P.; Wan, N. C. PCT Int. Appl. WO 2009146406 A1 Dec 3, 2009
The new Annex 16 “Certification by a Qualified Person and Batch Release” will become effective as of 15 April 2016.
DRUG REGULATORY AFFAIRS INTERNATIONAL
 The new Annex 16 is coming into Force
The new Annex 16 is coming into Force
The new Annex 16 “Certification by a Qualified Person and Batch Release” will become effective as of 15 April 2016. The contents will reflect the coming state of expectations regarding the batch release.
see
The new Annex 16 “Certification by a Qualified Person and Batch Release” will become effective as of 15 April 2016.
It is centrally pointed out that the main duty of a Qualified Person (QP) is the certification of batches. In this context, the QP must personally ensure that the responsibilities listed under Chapter 1.6 are fulfilled. Chapter 1.7 lists many other responsibilities to be guaranteed by the QP. However the related activities can be delegated and the QP can rely on the respective quality management systems. Yet, the “QP should have on-going assurance that this reliance is well founded” (1.7). The 21 responsibilites listed include amongst others:
- The…
View original post 406 more words
ICH Q3D implemented in the European Pharmacopoeia: Revision of Two General Monographs with Regard to Elemental Impurities
DRUG REGULATORY AFFAIRS INTERNATIONAL
 ICH Q3D implemented in the European Pharmacopoeia: Revision of Two General Monographs with Regard to Elemental Impurities
ICH Q3D implemented in the European Pharmacopoeia: Revision of Two General Monographs with Regard to Elemental Impurities
Two general monographs of the European Pharmacopoeia have been revised and published for comment in the newest “Pharmeuropa” edition. Read more about what you will have to consider in future with regard to the control of elemental impurities in pharmaceutical preparations, APIs and excipients.
see
In a press release dated 30 November 2015, the EDQM announced the revision of two general pharmacopoeial monographs: “Substances for pharmaceutical use” (2034) and “Pharmaceutical preparations” (2619). The decision was taken during the 153rd session of the European Pharmacopoeia Commission; the Commission follows its strategy for implementing the ICH Guideline Q3D “Guideline for Elemental Impurities” in the European Pharmacopoeia. A section “Elemental Impurities” has been added to both monographs which emphasizes that the provisions laid down in General Chapter 5.20 of the Pharmacopoeia (identical in wording with…
View original post 272 more words
I (Anthony Crasto) am Editorial Board member for our Journal of Analytical & Pharmaceutical Research


I am on editorial board ……… Editorial Board member for our Journal of Analytical & Pharmaceutical Research………http://medcraveonline.com/JAPLR/editorial-board
This is possible with your cooperation and support
read…….http://medcraveonline.com/JAPLR/JAPLR-02-00010.pdf
http://medcraveonline.com/JAPLR/JAPLR-02-00011.pdf
Tackling the Challenges with Poorly Soluble Drugs
http://medcraveonline.com/JAPLR/JAPLR-01-00001.pdf
BTI-320 (formerly PAZ320), Soluble mannan polysaccharides from Boston Therapeutics for the treatment of type 2 diabetes in combination with oral agents or insulin
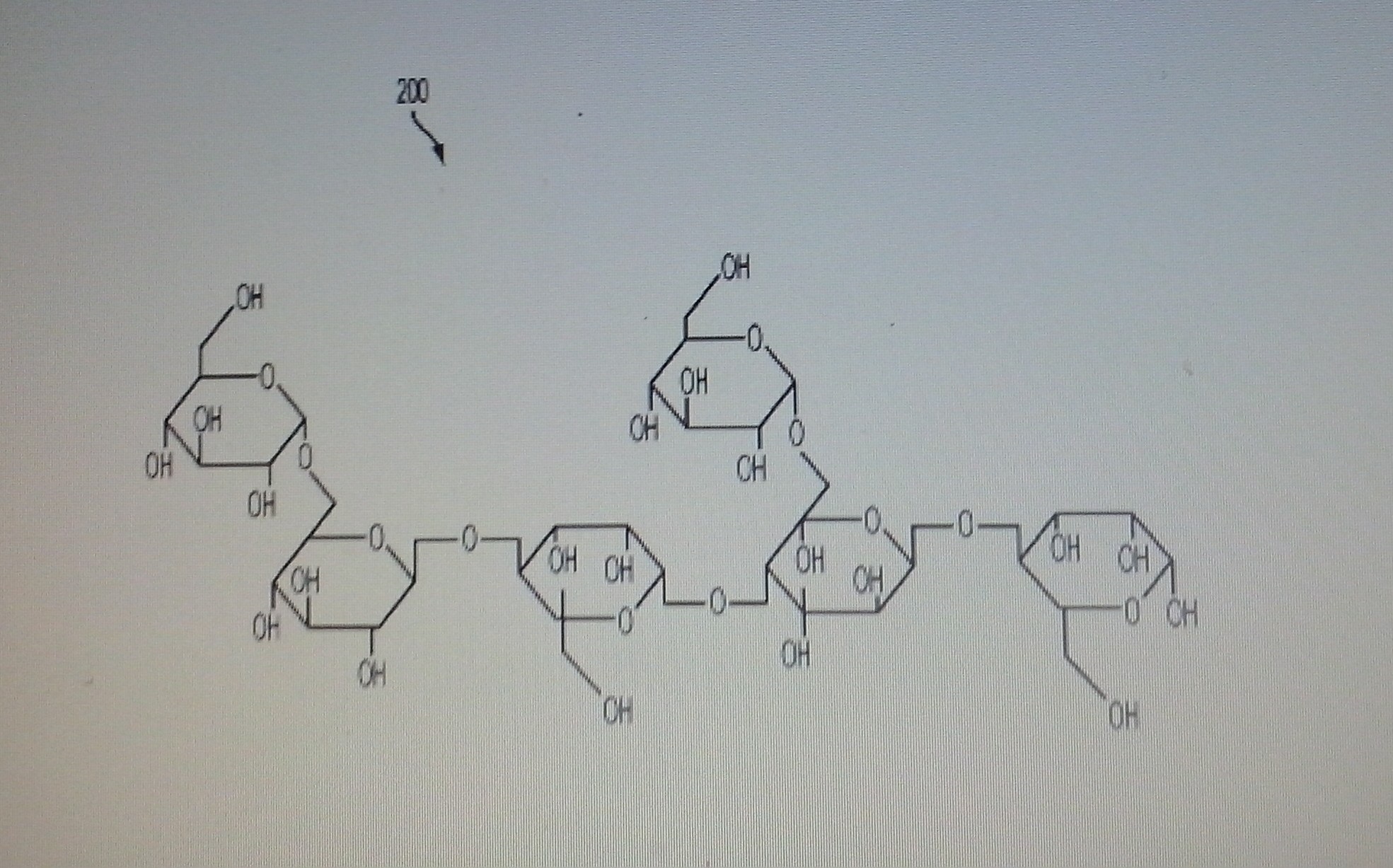
BTI-320 (formerly PAZ320)
PAZ 320
Non-insulin dependent diabetes
Alpha-glucosidase inhibitor; Hydrolase inhibitor; Sucrose alpha-glucosidase inhibitor
Composition of chemically purified (fractionation) soluble mannan polysaccharides from legume’s seeds
BTI-320 is in phase II clinical development at Boston Therapeutics for the treatment of type 2 diabetes in combination with oral agents or insulin, and also for the treatment of high-risk patients with pre-diabetes. A chewable tablet formulation is being developed. The product is already available as dietary supplement.
| Company | Boston Therapeutics Inc. |
| Description | Chewable polysaccharide that inhibits alpha glucosidase |
| Molecular Target | |
| Mechanism of Action | Alpha glucosidase inhibitor |
| Therapeutic Modality | Macromolecule: Polysaccharide |
| Latest Stage of Development | Phase II |
| Standard Indication | Diabetes |
| Indication Details | Treat Type II diabetes |


PATENT
http://www.google.co.in/patents/WO2012061675A1?cl=en
A composition of chemically purified soluble mannans from legumes’ seeds (e.g. Ceratonia siliqua, Cæsalpinia spinosa Trigonelle foenum-graecum, and Cyamopsis tetragonolobus) and their use in the assembly of palatable dietary supplements is disclosed herein. The fractionation process provides high-quality physiologically soluble, chemically modified and purified homogeneous size polysaccharide fibers, devoid of natural impurities, for example proteins, alkaloids, glycoalkaloids, and/or environmental impurities including heavy metals, agricultural residues and microbial toxins. This process provides hypoallergenic dietary fibers devoid of any potential allergens, cytotoxins, and gastrointestinal toxins. A sequential process for assembly of the soluble fibers with plurality of molecular weights to create a time controlled dissolution of the functional high and low molecular weight fibers for improving solubility and palatability with improved dietary performance in the oral and gastro-intestinal system is also disclosed herein.
Fig. 1 illustrates a block flow diagram of an embodiment of a method for recovering purified mannan polysaccharides;
Fig. 2 illustrates a chemical structure of a mannan polysaccharide;
Fig. 3 illustrates a block flow diagram of an embodiment of a method for recovering high molecular weight (HMW) purified mannan polysaccharides;
Fig. 4 illustrates a block flow diagram of an embodiment of a method for recovering low molecular weight (LMW) purified mannan polysaccharides;
REFERENCES
https://clinicaltrials.gov/show/NCT02060916
https://clinicaltrials.gov/show/NCT02358668
BTI-320, a nonsystemic novel drug to control glucose uptake into the bloodstream, functions as a competitive inhibitor of sugar hydrolyzing enzymes
75th Annu Meet Sci Sess Am Diabetes Assoc (ADA) (June 5-9, Boston) 2015, Abst 974-P
Boston Therapeutics’ Hong Kong Affiliate Advance Pharmaceutical’s BTI-320 Clinical Trial Reaches Mid-Point by Enrolling 30 Patients at the Chinese University of Hong Kong
Boston Therapeutics Press Release 2015, July 08
Insight into the molecular mechanism of action of BTI320, a non-systemic novel drug to control serum glucose levels in individuals with diabetes50th Annu Meet Eur Assoc Study Diabetes (EASD) (September 15-19, Vienna) 2014, Abst 545
////BTI-320, PAZ320, PHASE 2, BTI 320, PAZ 320, Macromolecule, Polysaccharide, Non-insulin dependent diabetes, Alpha-glucosidase inhibitor, Hydrolase inhibitor, Sucrose alpha-glucosidase inhibitor, phase II clinical development, Boston Therapeutics, Soluble mannan polysaccharides
Composition of chemically purified (fractionation) soluble mannan polysaccharides from legume’s seeds
POLYMER OF BELOW
CAS 9036-88-8, 51395-96-1
| refractive index : | 78.5 ° (C=1.4, H2O) |
Ailes;MANNAN;K-41K1;D-Mannan;NSC 174478;NSC 174479;NSC 174481;NSC 307194;NSC 174477;NSC 174473

| Chemical name: | 1,6-Anhydro-β-D-mannopyranose |
| Synonyms: | 1,6-Anhydro-D-mannose; 1,6-Anhydromannose; Mannosan; NSC 226600; |
| CAS Number: | 14168-65-1 |
| Possible CAS #: | NA |
| Molecular form.: | C₆H₁₀O₅ |
| Appearance: | White to Pale Beige Solid |
| Melting Point: | 182-184°C |
| Mol. Weight: | 162.14 |
Summary:
Mannans are major constitutents of hemicelluloses in plant tissue and are polymers composed of β(1→4)-linked mannose and glucose residues. Some contain galactopyranosyl side chains (see a galactomannan).
Slightly galactosylated mannans (4% galactose), considered as linear β(1→4)-D-mannans, have been isolated from the seed endosperm of vegetable ivory nut ( Phytelephas macrocarpa) and date ( Phoenix dactylifera) .
Glycan icon:

Child Classes: a 1,6-α-D-mannan backbone (0), a galactoglucomannan (0), a galactomannan (0), a glucomannan (0), a mannan oligosaccharide (1)
SMILES: C(O)C4(C(O[R1])C(O)C(O)C(OC3(C(O)C(O)C(OC2(C(O)C(O)C(OC1(C(O)C(O)C(O[R2])OC(CO)1))OC(CO)2))OC(CO)3))O4)
CAS:9036-88-8,
//////////
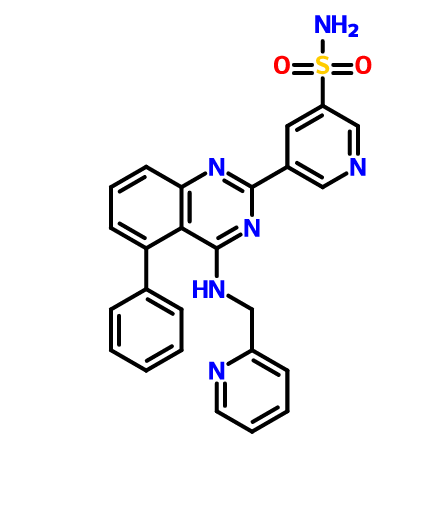
BMS 919373
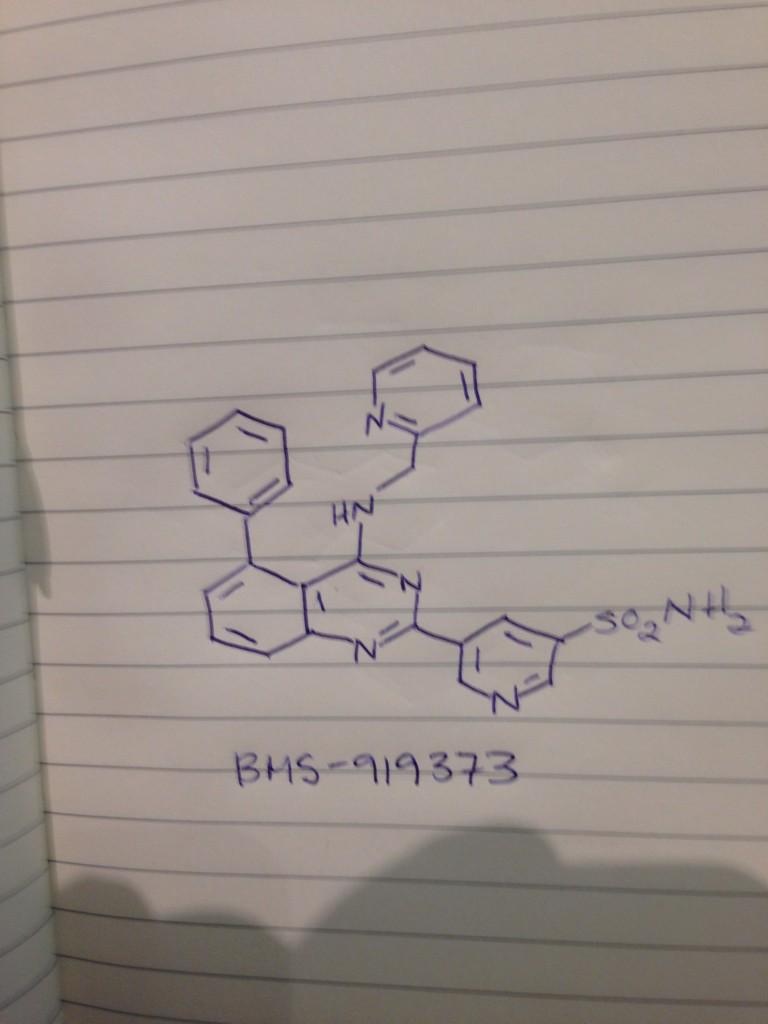 .
.
Bethany Halford on Twitter: “BMS-919373, from $BMS for …https://twitter.com/beth_halford/status/634105343719682048
Aug 19, 2015 – BMS–919373, from $BMS for atrial fibrillation #ACSBoston MEDI 1st disclosures @bmsnews pic.twitter.com/y3D4Yv2U7M.
BMS 919373
- Phase IIParoxysmal atrial fibrillation
- Phase IAcute coronary syndromes; Atrial fibrillation
-
- 01 Oct 2014Phase-I clinical trials in Atrial fibrillation in Canada (PO) (NCT02153437)
- 01 Jul 2014Phase-II clinical trials in Paroxysmal atrial fibrillation in Canada (PO) (NCT02156076)
- 01 Jul 2014Phase-II clinical trials in Paroxysmal atrial fibrillation in USA (PO)
- https://clinicaltrials.gov/ct2/show/NCT02153437
- https://clinicaltrials.gov/ct2/show/NCT02156076
- CAS HCL SALT 1272356-77-0
| Latest Stage of Development | Phase I |
| Standard Indication | Fibrillation |
| Indication Details | Treat atrial fibrillation |
Synthesis
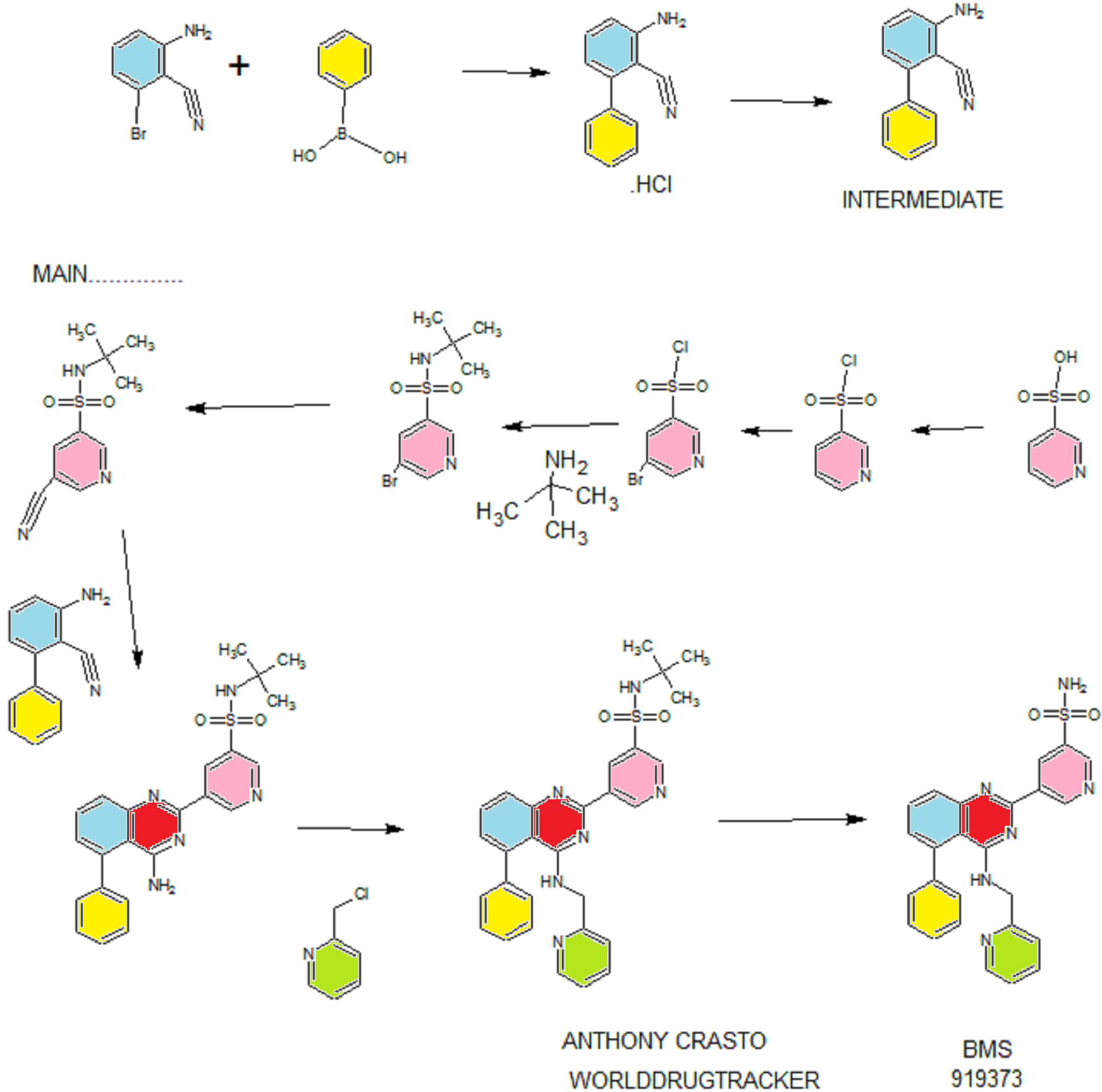
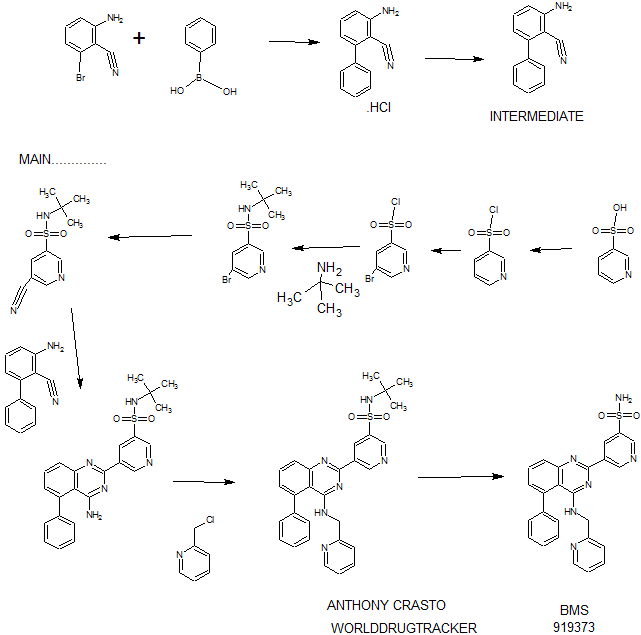
PATENT
WO 2011028741
http://www.google.co.in/patents/WO2011028741A1?cl=en
EXAMPLE 7
5-(5-Phenyl-4-(pyridin-2-ylmethylamino)quinazolin-2-yl)pyridine-3-sulfonamide

Step 1. Preparatio -Bromopyridine-3 -sulfonamide

See also U.S. Publication Nos. 2006/217387 and 2006/375834, and J. Org. Chem., 54:389 (1989). A mixture of pyridine-3 -sulfonic acid (10.3 g, 64.8 mmol), phosphorous pentachloride (20.82 g, 100 mmol) and phosphorous oxychloride (10 mL, 109 mmol) was heated to reflux where it stirred for 4h. At the conclusion of this period, the reaction mixture was allowed to cool to room temperature. Once at the prescribed temperature, the reaction mixture was evaporated to dryness under reduced pressure to yield a residue. The residue was treated with bromine (6.00 mL, 1 16 mmol) and then heated to reflux where it stirred for 14h. After this time, the reaction mixture was cooled to 0 °C and then a saturated solution of NH4OH in ¾0 (40 mL) was slowly added. The resulting mixture was allowed to warm to room temperature where it stirred for 30 min. The reaction mixture was then filtered and the filter cake was washed with hexane to afford 5 -bromopyridine-3 -sulfonamide (6.0 g) as an off- white solid. The product was used without further purification. LCMS Method Q: retention time 0.75 min; [M+l] = 237.0.
Step 2. Preparation of pyridine-3-sulfonamide-5-ylboronic acid pinacol ester

See also WO2008/150827 Al and WO2008/144463. A mixture of 5- bromopyridine-3 -sulfonamide (1.5 g, 6.33 mmol), bis(pinacolato)diboron (2.41 g, 9.5 mmol) and potassium acetate (1.86 g, 19.0 mmol) in 1,4-dioxane (15 mL) was degassed with nitrogen for 15 min then (l, l’-bis(diphenylphosphino)- ferrocene)palladium (II) chloride dichloromethane complex (232 mg, 0.317 mmol) was added and the resulting mixture was degassed again with nitrogen for 10 min. At the conclusion of this period, the reaction mixture was heated in a microwave at 120 °C for 45 min. After this time, the reaction mixture was filtered through CELITE® and the filtrate was concentrated under reduced pressure to provide pyridine-3- sulfonamide-5-ylboronic acid pinacol ester (740 mg) as a brown solid. The product was used without further purification. XH NMR (400 MHz, DMSO-d6) δ (ppm): 8.83 (s, 1H), 8.80 (s, 1H), 8.26 (s, 1H), 7.56-7.74 (bs, 2H), 1.17 (s, 12H).
Step 3. Example 7

To a solution of 2-chloro-5-phenyl-N-(pyridin-2-ylmethyl)quinazolin-4- amine (150 mg, 0.43 mmol) in 1,4-dioxane (6 mL) and ¾0 (1 mL) under nitrogen was added pyridine-3-sulfonamide-5-ylboronic acid pinacol ester (185 mg, 0.65 mmol), and potassium carbonate (119 mg, 0.86 mmol). Upon completion of addition, the mixture was degassed with nitrogen for 15 minutes and then (1, 1′- bis(diphenylphosphino)ferrocene)palladium (II) chloride dichloromethane complex (31 mg, 0.043 mmol) was added. The resulting mixture was again degassed with nitrogen for 10 min. After this time, the mixture was heated to 90 °C where it stirred for 16h. At the conclusion of this period, the reaction mixture was allowed to cool to room temperature. Once at the prescribed temperature, the reaction mixture was quenched by the addition of water and then transferred to a separation funnel. The aqueous layer was extracted with ethyl acetate. The combined organic portions were washed with water and saturated NaCl, dried over Na2S04, filtered and concentrated under reduced pressure. The resulting concentrate was purified by preparative TLC using 5% methanol in dichloromethane to afford Example 7 (50 mg) as a brown solid. ‘H NMR (400 MHz, DMSO-d6) δ (ppm): 9.81 (s, 1H), 9.17 (s, 1H), 9.09 (s, 1H), 8.24 (d, J= 4.4 Hz, 1H), 7.94 (d, J=7.2 Hz, 1H), 7.86 (t, J= 7.6 Hz, 1Η),7.75-7.72 (t, J= 7.6 Hz, 3H), 7.59-7.51 (m, 5H), 7.34 (d, J=7.2 Hz, 2H), 7.24 (t, J=6.4 Hz, 1H), 6.98 (t, J= 3.2 Hz, 1H), 4.77 (d, J= 4.0 Hz, 2H). LCMS Method Q: retention time 1.39 min; [M+l] = 469.0. HPLC Method B: purity 98.1%, retention time = 8.74 min. [00120] Alternatively, Example 7 can be synthesized as follows:
Step 1. Preparation of 5-Bromo-pyridine-3-sulfonyl chloride

PC15 (2.95 Kg, 14.16 moles) and POCl3 (2.45 Kg, 15.98 moles) were added into pyridine-3 -sulfonic acid (1.5 Kg, 9.42 mol) in 10 L RB flask equipped with mechanical stirrer under inert atmosphere. The reaction mass was heated to 120- 125°C where it stirred for 18 h. After this time, the reaction progress was monitored by HPLC, which indicated the reaction was complete. Excess POCI3 was removed under vacuum to give a residue. The residue was cooled to ambient temperature and bromine (1.2 Kg, 7.5 moles) was added. Upon completion of addition, the resulting mixture was heated to 120-125°C where it stirred for 5 h. At the conclusion of this period, the reaction progress was monitored by HPLC, which indicated the reaction was complete. The reaction mixture was cooled to ambient temperature and then poured into ice-water (10 L), and the resulting mixture was extracted with DCM (10.5 Lx2). The DCM extracts were combined and the solvent was removed under vacuum to yield crude product (1.8 Kg, 74.4% yield).
Step 2. Preparation of 5-bromo-N-tert-butylpyridine-3 -sulfonamide

Crude 5 -bromopyridine-3-sulfonyl chloride from step 1 above was dissolved in THF (14 L, 8 vol) and then transferred to a 20 L RB flask equipped with mechanical stirrer under inert atmosphere. The solution was cooled to 0-5°C and tert- butyl amine (1.95 Kg, 26.66 moles) was added at 0-5°C. Upon completion of addition, the reaction mixture was warmed to ambient temperature where it stirred for 2 h. At the conclusion of this period, the reaction progress was monitored by HPLC, which indicated that the reaction was complete. The solvent was evaporated under vacuum to give a thick residue. The residue was dissolved in ethyl acetate (18 L, 12 vol). The organic layer was separated, washed with water (9 L, 5 vol) and then concentrated under vacuum to yield a residue. Hexanes (9 L, 5 vol) were added to the residue and the product precipitated out and was collected by filtration to yield a free flowing yellow solid (1.5 Kg, 54.28% overall yield). ¾ NMR (DMSO-D6, 400 MHz, δ ppm); 8.99 (d, J = 2Hz, 1H), 8.81 (d, J= 2 Hz, 1H), 8.29 (t, J= 2Hz, 1H). [M++l] = 293. Step 3. Preparation of 5-bromo-N-tert-butylpyridine-3 -sulfonamide

5 -Bromo-N-tert-butylpyridine-3 -sulfonamide (1.5 Kg, 5.11 moles) was dissolved in dimethylformamide (7.5 L, 5 vol) and the solution was added to a 20 L glass-lined reactor equipped with mechanical stirrer. The solution was degassed with nitrogen for 30 min. After this time, potassium ferrocyanide trihydrate (867 g, 2.05 moles), sodium carbonate (1.08 Kg, 10.189 moles), copper (I) iodide (73.2 g, 0.374 moles) and dichloro-bis (triphenylphosphine) palladium (II) (71.6 g, 0.102 moles) were added. Upon completion of addition, the reaction mixture was heated to 120- 125°C where it stirred for 4 h. At the conclusion of this period, the reaction progress was monitored by HPLC, which indicated the reaction was complete. The reaction mixture was cooled to ambient temperature and then filtered through a celite bed. Water (18 L, 12 vol) was added into the filtrate and the resulting mixture was extracted with ethyl acetate (7.5L*2). The organic layers were combined, washed with water and then concentrated to yield a thick residue. Hexanes (7.5 L, 5 vol) were added to the residue. The product precipitated out and was collected by filtration to yield a free flowing yellow solid (1.0 Kg, 82.8% yield, 89% purity by HPLC). ¾ NMR (DMSO-D6, 400 MHz, δ ppm); 9.21 – 9.24 (d,d J= 7.2Hz, 3.2Hz, 2H), 8.70-8.71(m,lH), 7.98 (s, lH). [M++l] = 239.2.
Step 4. Preparation of 3-aminobiphenyl-2-carbonitrile

2-Amino-6-bromo-benzonitrile (1.0 Kg, 5.07 moles) and toluene (10 L, 10 vol) were added to a 20 L glass-lined reactor equipped with mechanical stirrer under inert atmosphere. Potassium acetate (996 g, 10.16 moles) and phenylboronic acid (866, 7.10 moles) were added into the solution and the solution was degassed with nitrogen for 30 min. After this time, dichloro-bis (triphenylphosphine) palladium (II) (17.8 g, 0.025 moles) was added to the reaction mixture at ambient temperature. The mixture was heated to 110°C, where it stirred for 17 h. At the conclusion of this period, the reaction progress was monitored by HPLC, which indicated the reaction was completed. The reaction mixture was filtered through a celite bed. The filtrate was transferred back to the reactor and concentrated hydrochloric acid (-35%, 2 L, 2 vol) was charged to the reactor at ambient temperature. The HCl salt of the title compound precipitated out from the reaction and was collected by filtration. The HCl salt was transferred into the 20 L reactor and then made basic with 10% NaOH solution (pH 8-9). The resulting product was extracted with ethyl acetate (10 L, 10 vol). The ethyl acetate layer was washed with water (5 L, 5 vol) and then the solvent was evaporated under vacuum to give a residue. Hexanes (5 L, 5 vol) were added to the residue at 35-40°C, and the resulting slurry was cooled to ambient temperature. Once at the prescribed temperature, the product was collected by filtration to provide a pale yellow solid (802 g, 81.4%, 99% by HPLC). XH NMR (DMSO-D6, 400 MHz, δ ppm); 7.43-7.52 (m, 5H), 7.33-7.37 (m, 1H), 6.83 (d, J=8Hz, 1H), 6.62 (d, J=8Hz, 1H), 6.1 (s, 2H). ES-MS: [M++l] = 194.23.
Step 5. Preparation of 5-(4-amino-5-phenylquinazolin-2-yl)-N-tert-butylpyridine-3-

3-Aminobiphenyl-2-carbonitrile (1028 g, 5.30 moles), 5-bromo-N-tert- butylpyridine-3 -sulfonamide (1440 g, 5.55 moles) and 1,4-dioxane (10 L, 10 vol) were added to a 20 L glass-lined reactor equipped with mechanical stirrer. Sodium tert-butoxide (1.275 Kg 12.870 moles) was added to the solution portion-wise at 20- 30°C. Upon completion of addition, the reaction mixture was heated to reflux where it stirred for 2 h. At the conclusion of this period, the reaction progress was monitored by HPLC, which indicated the reaction was complete. The reaction mixture was cooled to 30-35°C and then poured into water (40 L, 40 vol). The resulting mixture was extracted with DCM (20 L*2). The DCM layers were combined, washed with water (10 L, 10 vol) and then dried over sodium sulfate. The solvent was evaporated under vacuum to give a residue. Isopropyl alcohol (1.2 L, 1.2 vol) was added to the residue at 40°C. The resulting precipitate slurry was cooled to 10-15°C and then stirred for 2 h. After this time, the precipitate was collected by filtration and dried at 50°C for 16 h to yield the product (1.9 Kg, 82.9% yield, 99% purity by HPLC). Ή NMR (DMSO-D6, 400 MHz, δ ppm); 9.72 (s, 1H), 9.11 (s, 2H), 7.83-7.94 (m, 4H), 7.49-7.60 (m, 5H), 7.31 (d,d /=6.8Hz,1.2Hz, 1H). ES-MS: [M++l] = 433.53.
Step 6. Preparation of N-tert-butyl-5-(5-phenyl-4-(pyridin-2-ylmethylamino) quinazolin-2-yl) pyridine-3 -sulfonamide

2-(Chloromethyl) pyridine hydrochloride (564 g, 3.44 moles) and dimethyl acetamide (7L, 7 vol) were added to a 20 L RB flask- 1 equipped with mechanical stirrer under inert atmosphere. The resulting solution was cooled to 0- 5°C and triethylamine (346.3, 3.44 moles) was added at 0-5°C. 5-(4-Amino-5- phenylquinazolin-2-yl)-N-tert-butylpyridine-3-sulfonamide (1.0 Kg. 2.306 moles) and dimethylacetamide (4 L, 4 vol) were added to a separate 20 L RB flask-2 equipped with mechanical stirrer under inert atmosphere. This solution was cooled to 0-5°C and sodium tert-butoxide (884 g, 9.24 moles) was added at 0-5°C. The resulting solution was stirred to affect dissolution and then transferred to the RB flask- 1 at 0- 5°C. Upon completion of addition, the reaction mixture was stirred at 0-5°C for 2 h. At the conclusion of this period, the reaction progress was monitored by HPLC, which indicated that the reaction was complete. The reaction mass was poured into water (60 L, 60 vol) with stirring. The crude product was collected by filtration and dried at 60°C for 12 h. After this time, the dried material was dissolved in THF (20 L, 20 vol). Upon dissolution, 6M HC1 in isopropyl alcohol (1 L, 1 vol) was added at 20-25°C. The crude HCL salt of the product was obtained a pale-yellow free flow solid (920 g, 71% yield, 93% purity by HPLC). The crude HC1 salt (1.345 Kg, 2.56moles), methanol (6.7 L, 5 vol) and dichloromethane (13.5 L, 10 vol) were added to a 20 L glass-lined reactor equipped with mechanical stirrer. The slurry was stirred for 20-30 min at 30°C. After this time, the solvent was distilled to 4 vol with respect to input under vacuum. The resulting slurry was cooled to 20-25°C, where stirred for 2 h. At the conclusion of this period, the slurry was filtered and dried at 50°C for 6 h to yield the product (1.1 Kg, 82% yield, 98% purity by HPLC). XH NMR (DMSO- D6, 400 MHz, δ ppm); 9.72 (s, 1H), 9.10-9.14 (m, 2H), 8.39 (s, 1H), 7.92-8.03 (m, 4H), 7.56-7.58 (m, 5H), 7.43-7.49 (m, 3H), 7.1 (bs, 1H), 4.88 (s, 2H), 1.17 (2, 9H).
Step 7. Example 7

N-tert-butyl-5-(5-phenyl-4-(pyridin-2-ylmethylamino) quinazolin-2-yl) pyridine-3 -sulfonamide (1.0 Kg, 1.9 moles) and concentrated hydrochloric acid (7 L, 7 vol) were added to a 20 L glass-lined reactor equipped with mechanical stirrer. The reaction mixture was heated to 90-100°C where it stirred for 1 h. At the conclusion of this period, the reaction progress was monitored by HPLC, which indicated the reaction was complete. The reaction mixture was cooled to 5-10°C and the pH was adjusted to 1.7 to 2.0 using 12% aqueous sodium hydroxide solution. Once at the prescribed pH, the crude HC1 salt of the product was collected by filtration. The HC1 salt filter cake and ethanol (5 L, 5 vol) were added to 10 L glass-lined reactor equipped with a mechanical stirrer. The resulting mixture was made basic to pH 7-8 at 20-25°C using triethyl amine (2.25 Kg, 22.23 moles). Once at the prescribed pH, the basic mixture was stirred for 2 h. After this time, the free base of product was filtered and washed with water (10 L, 10 vol) followed by ethanol (2L, 2 vol). The resulting product was dried at 50-55°C for 8 h to yield Example 7 (644 g, 72% yield, 99.9% purity by HPLC).
XH NMR (DMSO-D6, 400 MHz, δ ppm); 9.81 (d, J=2.0Hz, 1H), 9.18 (t, J=2Hz, 1H), 9.1 1 (d, J=2Hz, 1H), 8.23 (d, J=4.4Hz, 1H), 7.92-7.94 (m, 1H), 7.83-7.87 (m, 1H), 7.78 (s, 2H), 7.70-7.72 (m, 1H), 7.50-7.59 (m, 5H), 7.31-7.34 (m, 2H), 7.22-7.25 (m, 1H), 6.95 (t, J=4Hz, 1H), 4.76 (d, J=4Hz, 2H). ES-MS: [M++l] = 469.
/////////atrial fibrillation, Potassium channel Kv1.5 (KCNA5) inhibitor, IKur antagonist, Bristol-Myers Squibb Co., BMS 919373, BMS-919373, PHASE 2
NS(=O)(=O)c1cc(cnc1)c4nc2cccc(c2c(NCc3ccccn3)n4)c5ccccc5
DR ANTHONY MELVIN CRASTO

ORGANIC SPECTROSCOPY

