






| Patent ID | Date | Patent Title |
|---|---|---|
| US2015290207 | 2015-10-15 | HETEROCYCLIC COMPOUNDS AND USES THEREOF |
| US2015225410 | 2015-08-13 | HETEROCYCLIC COMPOUNDS AND USES THEREOF |
| US2015111874 | 2015-04-23 | HETEROCYCLIC COMPOUNDS AND USES THEREOF |
FLAGS AND HITS
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
Join me on Linkedin
Join me on Researchgate
Join me on Facebook
 FACEBOOK
...................................................................Join me on twitter
FACEBOOK
...................................................................Join me on twitter
 ..................................................................Join me on google plus
..................................................................Join me on google plus
 Googleplus
Googleplus
FACEBOOK
...................................................................Join me on twitter
..................................................................Join me on google plus
GoogleplusMYSELF
DR ANTHONY MELVIN CRASTO Ph.D ( ICT, Mumbai)
, INDIA
36Yrs Exp. in the feld of Organic Chemistry,Working for AFRICURE PHARMA as ADVISOR earlier with GLENMARK PHARMA at Navi Mumbai, INDIA. Serving chemists around the world. Helping them with websites on Chemistry.Million hits on google,
NO ADVERTISEMENTS , ACADEMIC , NON COMMERCIAL SITE, world acclamation from industry, academia, drug authorities for websites, blogs and educational contribution, ........amcrasto@gmail.com..........+91 9323115463, Skype amcrasto64


Imidafenacin, イミダフェナシン

Imidafenacin
イミダフェナシン
Cas 170105-16-5
C20H21N3O, 319.408
APPROVED JAPAN 2015-07-29
|
4-(2-methyl-1H-imidazol-1-yl)-2,2-diphenylbutanamide
|
4-(2-methylimidazol-1-yl)-2,2-di(phenyl)butyramide
D06273
KRP-197
KRP-197;ONO-8025
ONO-8025
UNII:XJR8Y07LJO
Company:Kyorin (Originator), Ono (Originator)
4-(2-methyl-1-imidazolyl)- 2,2-diphenylbutyramide as a colorless needle: mp 189.0±190.0 C (from ethyl acetate:ethanol);
High MS (EI+) m/z calcd for C20H21N3O 319.1685, found 319.1671;
1 H NMR (400 MHz, CDCl3) d 2.23 (3H, s), 2.69±2.74 (2H, m), 3.77±3.82 (2H, m), 5.33 (1H, s), 5.49 (1H, s), 6.73 (1H, s), 6.85 (1H, s), 7.31±7.42 (10H, m).
イミダフェナシン
Imidafenacin

C20H21N3O : 319.4
[170105-16-5]
Imidafenacin
C20H21N3O : 319.4
[170105-16-5]
Imidafenacin (INN) is a urinary antispasmodic of the anticholinergic class. It’s molecular weight is 319.40 g/mol
| Imidafenacin (INN) is a urinary antispasmodic of the anticholinergic class. |
Kyorin and Ono have developed and launched imidafenacin, an oral M1 and M3 muscarinic receptor antagonist. Family members of the product case, WO9515951, expire in the US in 2019
Imidafenacin was approved by Pharmaceuticals Medical Devices Agency of Japan (PMDA) on Apr 18, 2007. It was marketed as Uritos® by Kyorin, and marketed as Staybla® by Ono.
Imidafenacin is a potent M1 and M3-subtype antagonist indicated for the treatment of urinary urgency, frequent urination and urgency urinary incontinence due to overactive bladder.
Uritos® is available as tablet for oral use, containing 0.1 mg of free Imidafenacin. The recommended dose is 0.1 mg twice daily, and it can be increased to 0.2 mg twice daily, if the efficacy was not enough.
Uritos® / Staybla®


MOA:Muscarinic acetylcholine receptor antagonist
Indication:Urinary incontinence; Urinary urgency and frequency

PAPER
Imidafenacin, the compound of formula (I), is an antimuscarinic agent marketed in Japan under the brand name Uritos® used to treat overactive bladder, a disease defined by the presence of urinary urgency, usually accompanied by frequency and nocturia, with or without urge incontinence. Overactive bladder dysfunction has a considerable impact on patient quality of life, although it does not affect survival.

(I)
Synthesis of 4-(2-methyl-1 -imidazolyl)-2,2-diphenylbutanamide is first disclosed in Japanese patent JP3294961 B2 as shown in Scheme 1 . 4-bromo-2,2-diphenylbutanenitrile (II) is reacted with three equivalents of 2-methylimidazol, in dimethylformamide and in the presence of triethylamine as a base, to afford 4-(2-methyl-1 H-imidazol-1 -yl)-2,2-diphenylbutanenitrile, compound of formula (III), which is purified by column chromatography and, further, converted into its hydrochloride salt and recrystallized. Then, compound (III) is hydrolyzed with an excess of 70% sulfuric acid at 140-150 °C, followed by basification and recrystallization to provide imidafenacin (I), in an overall yield of only 25% (as calculated by data provided in docume

(III) (I)
Scheme 1
This route of document JP3294961 B2 implies several drawbacks. Firstly, purification of intermediate (III) is carried out by means of chromatographic methods, which are generally expensive, environmentally unfriendly and time consuming. Secondly, the hydrolysis of the nitrile group is carried out under strong acidic conditions and high temperature not convenient for industrial application.
Japanese document JP2003-201281 discloses a process for preparing imidafenacin as shown in Scheme 2. 4-bromo-2,2-diphenylbutanenitrile (II) is reacted, with five equivalents of 2-methylimidazol, which acts also as a base, in dimethylsufoxide to provide intermediate (III), which after an isolation step is further reacted with phosphoric acid in ethanol to provide the phosphate salt of 4-(2-methyl-1 H-imidazol-1-yl)-2,2-diphenylbutanenitrile. Hydrolysis with potassium hydroxide, followed by purification with a synthetic adsorbent provides imidafenacin (I) in a moderate overall yield

(II) (I)
Scheme 2
The use of a synthetic adsorbent is associated with problems with operativities and purification efficiencies from the viewpoint of industrial production, therefore, the process disclosed in document JP2003-201281 is not suitable for industrial application.
EP1845091 A1 discloses a process for preparing imidafenacin, according to previous document JP2003-201281 , however the purification step is carried out by either preparing the hydrochloride or the phosphate salt of imidafenacin followed by neutralization as shown in Scheme 3. Purified imidafenacin is provided in low yield, overall yield of about 31 % (as calculated by data provided in document EP1845091 A1 ). This process has several disadvantages. Firstly, EP1845091 A1 states that the penultimate intermediate, the 4-(2-methyl-1 H-imidazol-1 -yl)-2,2-diphenylbutanenitrile phosphate is hygroscopic, which implies handling problems. Secondly, the additional steps carried out for purification increases the cost of the final imidafenacin process and the pharmaceutical compositions containing it, which already resulted in expensive medications.

(II) (I)
HCI or
H3PO4

purified (I) HCI or Ή3ΡΟ4
Scheme 3
The intermediate phosphate salt of 4-(2-methyl-1 H-imidazol-1 -yl)-2,2-diphenylbutanenitrile obtained and used in prior art processes is a solid form having needle-shaped crystals, which are difficult to filtrate. Moreover, said needle-shaped crystals are very hygroscopic and unstable and transform over time to other solid forms. In addition, the water absorbed by this solid form described in the prior art may react with the intermediate to generate further impurities.
Therefore, there is still a need to develop an improved industrially feasible process for the manufacture of imidafenacin in good purity and good yield, involving the use of stable intermediates having also improved handling characteristics.
Example 1 :
Preparation of 4-(2-methyl-1 H-imidazol-1 -yl)-2,2-diphenylbutanenitrile phosphate in solid Form I
4-bromo-2,2-diphenylbutanenitrile (II, 1.000 Kg, 3.33 mol) and 2-methylimidazol (1 .368 Kg, 16.66 mol) were heated in DMSO (0.8 L) at 100-105 °C for 7 hours. The solution was then cooled to 20-25 °C and toluene (2 L) and water (4 L) were added and stirred for 30 minutes. After phase separation, the aqueous layer was extracted with toluene (1 L). Organic layers were combined and washed twice with water (2 x 1 L). Distillation of toluene provided 4-(2-methyl-1 H-imidazol-1-yl)-2,2-diphenylbutanenitrile as a brown oil (0.915 Kg), which was, then, dissolved in dry acetone (3 L) and water (0.1 L), heated to 40-45°C and seeded with 4-(2-methyl-1 H-imidazol-1 -yl)-2,2-diphenylbutanenitrile phosphate. A solution of orthophosphoric acid (0.391 Kg, 3.39 mol) in acetone (2 L) was then added dropwise, maintaining temperature at 40-45 °C. Once the addition was finished, the reaction mixture was maintained 1 hour at 40-45 °C, cooled to 20-25 °C and stirred for 1 hour. The solid was filtered, washed with acetone (1 L), suspended in 2-propanol (10 L), heated at 80 °C and 2 L of solvent were distilled. The obtained suspension was then seeded with 4-(2-methyl-1 H-imidazol-1 -yl)-2,2-diphenylbutanenitrile phosphate solid Form I and maintained at 80 °C for 5 hours. The suspension was cooled down to 20-25°C, filtered off, washed with 2-propanol (1 L) and, finally, dried (45 °C, 0.5 torr, 12 hours).
Yield: 0.967 Kg (73%)
HPLC: 99.5 %
KF: 0.2 %
Optical microscopy: plate-shaped crystal habit as substantially in accordance to Figure 2.
PSD: D90 of 105 m
PXRD: Crystalline solid form as substantially in accordance to Figure 3.
DSC (10 °C/min): Endothermic peak with onset at 177 °C (-1 18 J/g), as substantially in accordance to Figure 4.
TGA (10 °C/min): Decomposition starting at 180 °C.
DVS: No significant weight gain up to 90% of relative humidity. At this humidity, a total increase of only 0.45% in weight was observed.
SCXRD: Crystal structure substantially in accordance to Figure 5. There are not water or solvent molecules in the crystal structure.
PATENT
https://www.google.com/patents/CN103351344A?cl=en
Overactive Bladder (symptomatic overactive bladder, 0AB) is a common chronic lower urinary tract dysfunction. Its incidence, United States and Europe over 75 year-old male incidence up to 42%, slightly lower incidence of women 31%; the incidence of domestic in Beijing 50 years of age for men was 16.4% for women over the age of 18 mixed The overall incidence of urinary incontinence and urge incontinence was 40.4 percent, seriously affecting the physical and mental health of the patient, reduced quality of life. Common antimuscarinic drugs in vivo and in vivo M receptor in some or all of binding with different affinities to improve the symptoms of OAB, but will also cause many side effects, such as dry mouth, constipation, cognitive impairment , tachycardia, blurred vision and so on. Imidafenacin have diphenylbutanoic amide structure, is a new high anticholinergic drugs, which selectively acts on the M3 and Ml receptors, blocking the contraction of the detrusor choline, so detrusor relaxation, reduce side effects of drugs. Meanwhile imidafenacin inhibit smooth muscle of the bladder and inhibiting acetylcholine free dual role, and selectivity for the bladder stronger than the salivary glands.
imidafenacin is a new diphenylbutanoic amides from Japan Ono Pharmaceutical Co., Ltd. jointly developed with Kyorin Pharmaceutical anticholinergics, structure (I) as follows:

The goods listed in June 2007 in Japan under the trade name: STAYBLA, chemical name: 4- (2-methyl-1-imidazolyl) _2,2- diphenylbutyric amide.
At present the preparation imidafenacin few reports, can be summed up as the following ways:
China Patent CN10699098 reported to bromoethyl diphenyl acetonitrile and 2-methylimidazole as a raw material, at 150 ° C condition, after the reaction DMF / triethylamine system, sulfuric acid hydrolysis reuse imidafenacin. The reaction equation is as follows:

BACKGROUND OF THE INVENTION This two-step method was 24% overall yield is too low, and the second step of the reaction is difficult to control. And the reaction product was purified by column chromatography required to obtain a purified product, is not conducive to industrial production.
Chinese patent CN101362721A referred to as the hydrolysis conditions for the preparation of sulfuric acid and organic acid mixed use imidafenacin yield have mentioned the smell.

Although this method increases the yield, but still more by-product of the reaction, the product is not easy purification.
Japanese Patent No. JP2005 / 023216 proposes hydrolysis under alkaline environment, and the use of products and solutions of salts hydrochloride salt and then purified product.

This method improves the yield of the second step of the hydrolysis reaction and simplified purification methods. But the need to use this method to purify salt activated carbon, and filtration devices require more stringent; and a need to be re-crystallized salt solution salt after the operation, a total of four steps of unit operations. Process more cumbersome and more stringent requirements for equipment, it is not conducive to industrial scale production. In addition, the product is dried for a long time, still remaining after solvent treatment product obtained, the purity of the product is still low.

DETAILED DESCRIPTION
The following typical examples are intended to illustrate the present invention, simple replacement of skill in the art of the present invention or improvement made in all part of the present invention within the protection of technical solutions.
Example 1
4- (2-methyl-1-imidazolyl) -2,2-diphenyl butanamide hydrobromide. The 16.5 g (52 mmol) 4- (2- methyl-1-imidazolyl) -2,2-diphenyl butyramide crude into 100 mL of isopropanol, stirring was added 8.0 mL hydrobromic acid and isopropyl alcohol mixed solution (volume ratio of 1: 1), the solid gradually dissolved, was nearly colorless and transparent liquid. After maintaining the reaction mixture was stirred for half an hour, the reaction mixture was added to 100 mL of ethyl acetate, stirred for I hour at room temperature, solid precipitated. Filtration, and the cake was rinsed with an appropriate amount of ethyl acetate. The solid was collected, 40 ° C drying oven and dried to constant weight to give 19.5 g white 4- (2-methyl-1-imidazolyl) -2,2-diphenyl butyramide hydrobromide, yield 98.9%. ?] \ 1 .228.4-229.00C0MS (m / z): 320 [M + 1] +. 1H-NMR (DMS0-1 / 6, 400 MHz) δ: 2.25 (3H, s), 2.73-2.74 (2H, m), 3.68-3.91 (2H, m), 6.81 (1H, s), 7.28-7.35 (I OH, m), 7.39 (1H, s), 7.49 (1H, d, /=2.4 Hz), 7.55 (1H, d, J = 2.2 Hz), 14.39 (1¾ br s).
Example 2
4- (2-methyl-1-imidazolyl) -2,2-diphenyl butyramide. -2,2-Diphenyl butyric acid amide acetate was dissolved in 900 mL of water to 19.5 g (0.051mmol) obtained in Example 1 4- (2-methyl-1-imidazolyl) embodiment. Extracted with 900mL diethyl ether solution, collecting the inorganic layer. Was added to an aqueous solution of 200 mL of ethanol, was added to the system with stirring in an aqueous solution of KOH 2mol / L, there is a solid precipitated. The reaction was stirred I h after filtration. Cake was washed with 40% ethanol solution rinse, rinsed with water several times. Collect the cake, put 40 ° C drying oven dried to constant weight to give 14.8 g white 4- (2-methyl-1-imidazolyl) -2,2-diphenyl methylbutanamide, yield 91.0% (total yield 90% two steps). Μ.p.192.3-193.00C (CN101076521A 191-193O). MS (m / z): 320 [M + l] +. 1H-NMR (DMSO-J6, 400MHz) δ: 2.11 (3Η, s), 2.69-2.73 (2H, m), 3.61-3.65 (2H, m), 6.75 (1H, d, J = L OMHz), 7.01 (1H, br s), 7.04 (1H, d, J = L 0 MHz), 7.34-7.49 (11H, m).
Example 3
4- (2-methyl-1-imidazolyl) -2,2-diphenyl butyramide. The 14.5 g (0.045mmol) obtained in Example 4- (2-methyl-1-imidazolyl) -2,2-diphenyl butanamide 2 was added 116 mL of ethyl acetate was slowly heated to reflux reflux for 30 min, cooled to room temperature for crystallization 5 h. Suction filtered, the filter cake was rinsed with a small amount of ethanol, collected cake was put 40 ° C drying oven and dried to constant weight to give 13.4 g white 4- (2-methyl-1-imidazolyl) -2,2- diphenyl methylbutanamide refined products, yield 92.4% (three-step total yield 83.1%). Mp192.5-193 (TC (CN101076521A 191_193 ° C) .MS (m / z):.. 320 [M + 1] + 1H-NMR (DMSO-J6, 400 MHz) δ
2.11 (3H, 7.01 (1H,
s), 2.69-2.73 (2H, br s), 7.04 (1H, d,
m), 3.61-3.65 (2H, m), 6.75 (1H, J = L 0 MHz), 7.34-7.49 (11H, m).

PATENT
CN103772286A.
imidafenacin (Imidafenacin) is a new diphenylbutanoic amides from Japan Ono Pharmaceutical Co., Ltd. jointly developed with Kyorin Pharmaceutical anticholinergic drugs, bladder is highly selective for the treatment of overactive bladder, in 2007 in June in Japan. Its chemical name is 4- (2-methyl -1H- imidazol-1-yl) -2,2-diphenyl butyramide chemical structure shown by the following formula I:

Reported in U.S. Patent No. US5932607 imidafenacin preparation method, the method is based on 4-bromo-2 ‘2 ~ phenyl butyronitrile, 2-methylimidazole, triethylamine as raw materials, with DMF as a solvent at 150 ° C reaction 30h, to give the intermediate 4- (2-methyl-imidazol-1-yl) -2,2-diphenyl-butyronitrile, 77% yield, then body 140 ~ 150 ° C with 70% sulfuric acid The resulting intermediate hydrolyzed to the amide, after completion of the reaction required excess soda and sulfuric acid, the reaction is as follows:

Which preclude the use of the dilute sulfuric acid hydrolysis, although succeeded in getting the product, but the yield is very low, only 32%, greatly increasing the production cost, mainly due to 70% sulfuric acid, the reaction is difficult to control amide phase, the product will continue to acid hydrolysis byproducts, resulting in decreased yield.
European Patent No. EP1845091 reports imidafenacin Another preparation method, the method using potassium hydroxide and isopropyl alcohol 4- (2-methyl-imidazol-1-yl) diphenyl _2,2- Hydrolysis of nitrile to amide phosphates, and the crude product was converted to the hydrochloride or phosphate, and recrystallized to remove impurities and then basified imidafenacin obtained, which reaction is as follows:

This method uses a lot of bases, product purification is too much trouble, and the total yield of 45%.
Chinese Patent Publication No. CN102746235 also disclosed imidafenacin preparation method of 4- (2-methyl-1-yl) -2,2-diphenyl phosphate or nitrile salt in methanol / ethanol, dimethyl sulfoxide, and the presence of a base, with hydrogen peroxide in 40 ~ 60 ° C under through improved Radziszewski the target compound, the reaction is as follows:

The method used in the hydrogen peroxide solution, but a solution of hydrogen peroxide has strong oxidizing, and has a certain corrosive, inhalation of the vapor or mist respiratory irritation strong, direct eye contact with the liquid may cause irreversible damage and even blindness, security It is not high on the human body and environmentally unfriendly. Alkaline environment, easily decomposed hydrogen peroxide, as the temperature increases, the decomposition reaction increased, and therefore reaction requires a large excess of hydrogen peroxide solution.

The method comprises the steps of: (1) 4-Bromo-2,2-diphenyl-butyronitrile is hydrolyzed to the amide under basic conditions; (2) The obtained 4-bromo-2,2-diphenylbutyric amide is reacted with 2-methylimidazole to give the desired product.
Example 1
2L reaction flask was added 400mL of dry tetrahydrofuran, under a nitrogen atmosphere was added 60% sodium hydride (82.8g, 2.06mol), stirred to obtain a gray turbid solution A. With 400mL dry tetrahydrofuran was sufficiently dissolved diphenyl acetonitrile (200g, 1.04mol), I, 2- dibromoethane (204.2g, 1.08mol), to give a colorless clear liquid B; 5 ~ 15 ° C, a solution of turbid solution B dropwise to solution A, 10 ~ 15 ° C the reaction was incubated 6h, TLC until the reaction was complete, to the reaction system a small amount of water was added dropwise until no bubbles. After addition of 800mL water, 400mL ethyl acetate and stirred, liquid separation, the organic layer was washed with water, saturated sodium chloride solution, respectively, and the organic layer was dried over anhydrous sodium sulfate, suction filtered, concentrated under reduced pressure to give a yellow liquid 310g.
[0018] The resulting yellow liquid with 800mL 90% ethanol and stirred to dissolve at 40 ° C, then cooling and crystallization, filtration, 45 ° C and concentrated under reduced pressure to give a white solid 232.8g, 75% yield.
Preparation of bromo-2,2-diphenyl 4_ butanamide: [0019] Example 2
3L reaction flask was added 4-bromo-2,2-diphenyl-butyronitrile (15 (^, 0.511101), 7501 ^ 6mol / L KOH solution, 750mL dimethylsulfoxide and heated to 100 ~ 120 ° C under stirring The reaction, the reaction lh, until the reaction was complete by TLC after cooling to 40 V, add 2000mL water, 2000mL of methylene chloride was stirred, liquid separation, the organic layer was washed with water, washed with saturated sodium bicarbonate and sodium chloride solution, separated, dried over anhydrous The organic layer was dried over sodium sulphate, filtration, concentrated under reduced pressure to give brown oily liquid 161.92g, 96% yield.
Preparation of bromo-2,2-diphenyl 4_ butanamide: [0020] Example 3
3L reaction flask was added 4-bromo-2,2-diphenyl-butyronitrile (150g, 0.5mol), 666mL 6mol / L NaOH solution, 750mL dimethylsulfoxide, the reaction mixture was stirred and heated to 100 ~ 120 ° C under The reaction lh, until the reaction was complete by TLC after cooling to 40 ° C, add water 2000mL, 2000mL of methylene chloride was stirred, liquid separation, the organic layer was washed with water, washed with saturated sodium bicarbonate and sodium chloride solution, separated, dried over anhydrous sulfate sodium organic layer was dried, filtration, concentrated under reduced pressure to give brown oily liquid 146.73g, 87% yield.
Preparation of bromo-2,2-diphenyl 4_ butanamide: [0021] Example 4
The reaction was stirred 3L reaction flask was added 4-bromo-2,2-diphenyl-butyronitrile (15 (^, 0.511101), 8331 ^ 36% Na2CO3 solution, 750mL dimethylsulfoxide and heated to 100 ~ 120 ° C under The reaction lh, until the reaction was complete by TLC after cooling to 40 ° C, add water 2000mL, 2000mL of methylene chloride was stirred, liquid separation, the organic layer was washed with water, washed with saturated sodium bicarbonate and sodium chloride solution, separated, dried over anhydrous The organic layer was dried over sodium sulphate, filtration, concentrated under reduced pressure to give brown oily liquid 153.48g, yield 91%.
`[0022] Example 5: 4- (2-methyl-imidazol _1_ -1H- yl) butyramide _2,2_ diphenyl (imidafenacin) Preparation 5L reaction flask was added 4-bromo-2 2-diphenyl butyric amide (160g, L 5mol), 2- methyl imidazole (123g,
1.5mol), triethylamine (50.6g, 0.5mol), potassium iodide (5g, 0.03mol), fully dissolved with 1000mL DMF solution was heated to 120 ° C at a reaction 5h, until completion of the reaction by TLC, heating was stopped, to be After cooling, water was added 3000mL system stirred 0.5h, filtration, washed with water until the filtrate is neutral, concentrated under reduced pressure and dried to give a brown solid 146.14g, a yield of 91%.
[0023] Example 6: 4- (2-methyl-imidazol _1_ -1H- yl) butyramide _2,2_ diphenyl (imidafenacin) Preparation 5L reaction flask was added 4-bromo-2, 2- diphenyl butyramide (160g, 0.5mol), 2- methyl imidazole (82.1g,
1.011101), triethylamine (50.68,0.5mol), potassium iodide (5g, 0.03mol), fully dissolved with 1000mL DMF solution was heated to 120 ° C at a reaction 5h, until completion of the reaction by TLC, heating was stopped, the system was cooled until After adding 3000mL water, stirring 0.5h, filtration, washed with water until the filtrate is neutral, concentrated under reduced pressure and dried to give a brown solid 120.45g, 80% yield.
[0024] Example 7: 4- (2-methyl-imidazol _1_ -1H- yl) butyramide _2,2_ diphenyl (imidafenacin) Preparation 5L reaction flask was added 4-bromo-2, 2- diphenyl butyramide (160g, 0.5mol), 2_ methylimidazole (164.2g,
2.011101), triethylamine (50.68,0.5mol), potassium iodide (5g, 0.03mol), fully dissolved with 1000mL DMF solution was heated to 120 ° C at a reaction 5h, until completion of the reaction by TLC, heating was stopped, the system was cooled until After adding water, stirring 3000mL
0.5h, suction filtered, washed with water until the filtrate was neutral, and concentrated under reduced pressure, and dried to give a brown solid 141.33g, yield 88%.
[0025] Example 8: 4- (2-methyl imidazole -1H- _1_ group) _2,2_ diphenylbutanoic amide (imidafenacin) refining up to 80g microphone said that new crude added 300mL of absolute ethanol, the system was warmed to reflux, refluxed
0.5h, after cooling the ethanol was distilled off to IOOmL about 500mL of ethyl acetate was added to precipitate a white solid, a small amount of ethyl acetate and wash the filter cake, 45 ° C and dried in vacuo to give 74.6g of white crystals, yield 93%.

CLIP
| EP 0733621; US 5932607; US 6103747; WO 9515951 |


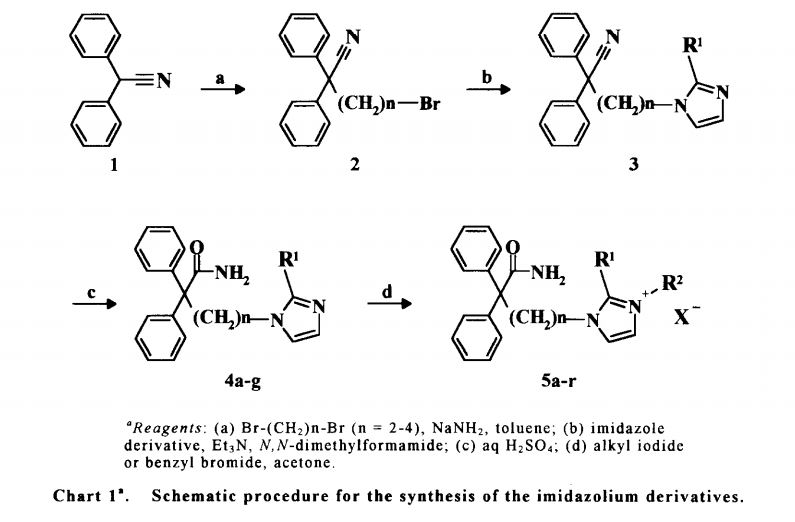
Alkylation of diphenylacetonitrile (I) with dibromoethane provided bromide (II). This was condensed with 2-methylimidazole (III) in the presence of Et3N in DMF to afford the substituted imidazole (IV). Finally, hydrolysis of the cyano group of (IV) with 70% sulfuric acid produced the target amide.

Treatment of acetonitrile derivative (I) with dibromoethane (II) in toluene in the presence of NaNH2 affords bromo compound (III), which is then condensed with imidazole derivative (IV) by means of Et3N in DMF to provide compound (V). Hydrolysis of the cyano group of (V) with aqueous H2SO4 yields amide derivative (VI), which is finally subjected to alkyl quaternization by reaction with bromobenzyl bromide (VI) in acetone to furnish the desired product.
Paper
Bioorganic & Medicinal Chemistry Letters 9 (1999) 3003-3008

PAPER
Bioorganic & Medicinal Chemistry 7 (1999) 1151±1161
4-(2-methyl-1-imidazolyl)- 2,2-diphenylbutyramide (2.02 g, 24%) as a colorless needle:
mp 189.0±190.0 C (from ethyl acetate:ethanol);
High MS (EI+) m/z calcd for C20H21N3O 319.1685, found 319.1671;
1 H NMR (400 MHz, CDCl3) d 2.23 (3H, s), 2.69±2.74 (2H, m), 3.77±3.82 (2H, m), 5.33 (1H, s), 5.49 (1H, s), 6.73 (1H, s), 6.85 (1H, s), 7.31±7.42 (10H, m).
PATENT
CN103880751A.
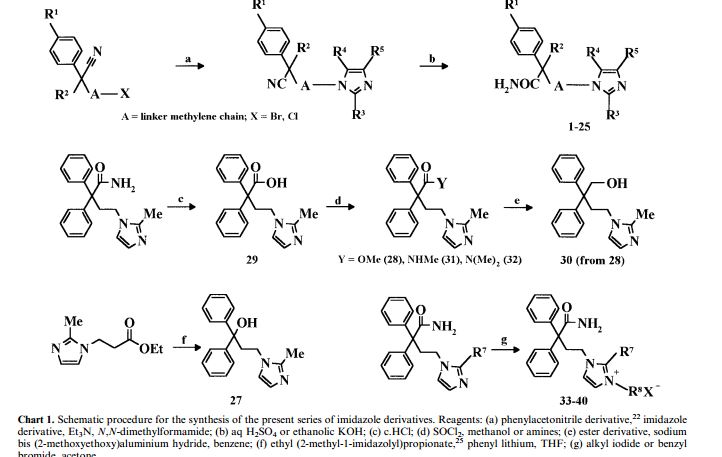
imidafenacin chemical name 4- (2-methyl–1H–1-yl) -2,2-diphenyl methylbutanamide (I).

In Patent JP93-341467, JP94-319355 and literature Bioorganic & Medicinal ChemistryLetters, 1999, vol.9,3003 – 3008 reported in the chemical synthesis routes to diphenyl acetonitrile (4) as the starting material,
Condensation and hydrolysis reaction step to give imidafenacin (1).

The new method is simple, mild reaction conditions, easy to control, good high yield and purity of the product, do not pollute the environment, suitable for industrial production.
[0012] The first method from 2-methylimidazole and I, 2- dibromoethane under phase transfer catalyst is tetrabutylammonium bromide (TBAB) and inorganic base catalyzed generate 1- (2-bromoethyl) – methyl -1H- imidazole (5), and diphenyl acetonitrile (4) a phase transfer catalyst and an inorganic base catalyzed condensation of 4- (2-methyl–1H- imidazol-1-yl) -2,2 – diphenylbutyronitrile hydrochloride (2), and then hydrolyzed to imidafenacin (I)


FIG. 1 imidafenacin IH-NMR spectrum
FIG. 2 imidafenacin 13C-NMR spectra

Examples I
1- (2-bromoethyl) -1H- -2_ methyl-imidazole (5) Preparation of
The 1,2_ dibromoethane (50ml), 2- methylimidazole (2.5g, 30.5mmol), tetrabutylammonium bromide (TBAB) (0.5g) and K2C03 (3.6g), K0H ( 4.6g) were added sequentially 100mL three-necked flask and stirred and heated to 50 ° C reaction 7h. Cooling to room temperature, the reaction solution was filtered, and the filtrate was washed with saturated aqueous sodium bicarbonate, dried over anhydrous sodium sulfate. Concentrated, added to a mixed solvent of isopropyl ether and ethyl acetate (3: 1) was stirred resolved crystal dissolved, to give the product 5.lg, yield 88.5%, mp.79_80 ° C.
Preparation of 4- (2-methyl-1-imidazolyl) -2,2-diphenyl-butyronitrile hydrochloride (2)
The diphenyl acetonitrile (5.8g, 30mmol) and 50% aqueous KOH (15ml), dimethyl sulfoxide (DMSO) (100ml), tetrabutylammonium bromide (TBAB) (0.9g) in toluene 50ml was added to the reaction flask and stirred for 0.5h in the 40 ° C. 1- (2-bromoethyl) -2-methyl -1H- imidazole (4) (5.lg, 27mmol), was heated to 20 ° C, the reaction was stirred, TLC tracking and monitoring the reaction was complete, the mixture was poured into 100mL water, extracted three times with ethyl acetate 240ml water phase. Washed three times with 300ml of water The organic phase was dried over anhydrous sodium sulfate, the organic phase was concentrated. Analytical crystal solution with hydrogen chloride ether solution, filtered crystals with a mixed solvent of isopropyl ether and recrystallized from ethyl acetate to give the condensation product of 4- (2-methyl-1-imidazolyl) -2,2-diphenylbutyric carbonitrile hydrochloride (2) of a white solid 7.lg, yield 77.8%, mp: 156.5-158 ° C. 1H-NMR (400MHz, CDCl3), δ (ppm): 7.35-7.42 (IOH, m), 6.90 (1H, s), 6.77 (1H, s), 3.90-3.94 (2H, m), 2.75-2.79 ( 2H, m), 2.25 (3H, s).
The preparation imidafenacin (I),
4- (2-methyl-1-imidazolyl) -2,2_ diphenyl butyronitrile hydrochloride (2) (8.78g, 26mmol) in 70% concentrated sulfuric acid (25ml) was added to the reaction bottle, the reaction was stirred at 90 ° C, the end of the reaction was monitored by TLC tracking. The reaction solution was poured into 120ml of water, solid sodium carbonate was added to adjust PH to weakly alkaline, sufficiently stirred. With 180ml of dichloromethane and 35ml of ethanol mixed solvent was extracted three times, the organic phase was washed with water, dried over anhydrous sodium sulfate, the organic phase was concentrated. The residue was mixed with a solvent of ethyl acetate and recrystallized from ethanol to give 4- (2-methyl-1-imidazolyl) 2,2-diphenyl butyramide 7.0g, yield 84.5%, mp: 188.0-190 (. TC.1H-NMR and 13C-NMR data are as follows (see Figure 1-2 spectra):
1H-NMR (CDC13,400ΜΗζ) δ: 2.209 (s, 3H, -CH3), 2.666-2.707 (t, 2H, -CH2-CH2-),
3.747-3.788 (t, 2H, -CH2-CH2 -), 5.341 (s, 1H, -NH -), 5.757 (s, 1H, -NH -), 6.699 (s, 1H, Ar-H)
, 6.828 (s, 1H, Ar-H), 7.287-7.390 (m, I OH, Ar-H).
[0030] 13C-NMR (CDC13,400MHz) δ: 12.17 (-CH3), 41.00 (-CH2 -), 43.74 (-CH2-), 59.44 (quaternary carbon, coupled with strong electron-withdrawing group), 119.08 (-C = C -), 126.95 (aromatic carbon), 127.88 (aromatic carbon), 128.52 (aromatic carbon), 129.10 (aromatic carbon), 142.61 (= CN), 144.54 (-C = N), 176.21 (carbonyl carbon).
Example 2
[0032] 1- (2-bromoethyl) -1H- -2_ methyl-imidazole (5) Preparation of
[0033] The 1,2_ dibromoethane (50ml), 2- methylimidazole (2.5g, 30.5mmol), tetrabutylammonium chloride (0.43g) and Na2CO3 (2.8g), NaOH (3.3g) followed by adding 100mL three-necked flask, stirred and heated to 40 ° C reaction 5h.Cooling to room temperature, the reaction solution was filtered, and the filtrate was washed with saturated aqueous sodium bicarbonate, dried over anhydrous sodium sulfate. Concentrated, added to a mixed solvent of isopropyl ether and ethyl acetate (3: 1) was dissolved with stirring parsing crystal give the product 4.9g, yield 85.1%, mp.79-80 ° C.
Preparation of [0034] 4- (2-methyl-1-imidazolyl) -2,2-diphenyl-butyronitrile hydrochloride (2)
[0035] A two phenylethyl chest (5.8g, 30mmol) and 50% aqueous NaOH (15ml), dimethylethylene Bitterness (DMSO) (100ml), tetrabutylammonium chloride (0.8g) was added to a toluene 50ml The reaction flask, stirred 0.5h in the 40 ° C. Join
1- (2-bromoethyl) -2-methyl -1H- imidazole (4) (5.lg, 27mmol), was heated to 60 ° C, the reaction was stirred, TLC tracking and monitoring the reaction was complete, the mixture was poured into 100mL of water and extracted three times with ethyl acetate 240ml water phase. Washed three times with 300ml of water The organic phase was dried over anhydrous sodium sulfate, the organic phase was concentrated. Solution of hydrogen chloride in ether solution with analytical crystal, crystals were filtered with a mixed solvent of isopropyl ether and recrystallized from ethyl acetate to give the condensation product of 4- (2-methyl-1-imidazolyl) -2,
2-phenyl-butyronitrile hydrochloride (2) as a white solid 7.0g, yield 76.8%, mp: 156.5-158 ° C. 1H-NMR (400MHz, CDCl3), δ (ppm): 7.35-7.42 (IOH, m), 6.90 (1H, s), 6.77 (1H, s), 3.90-3.94 (2H, m), 2.75-2.79 ( 2H, m), 2.25 (3H, s).
Preparation imidafenacin (I),
[0037] 4- (2-methyl-1-imidazolyl) -2,2_ diphenyl butyronitrile hydrochloride (2) (8.78g, 26mmol) in 70% concentrated sulfuric acid (25ml) was added to the reaction bottle, the reaction was stirred at 110 ° C, the end of the reaction was monitored by TLC tracking. The reaction solution was poured into 120ml of water, solid sodium carbonate was added to adjust PH to weakly alkaline, sufficiently stirred. With 180ml of dichloromethane and 35ml of ethanol mixed solvent was extracted three times, the organic phase was washed with water, dried over anhydrous sodium sulfate, the organic phase was concentrated. The residue was mixed with a solvent of ethyl acetate and recrystallized from ethanol to give 4- (2-methyl-1-imidazolyl) 2,2-diphenyl butyramide 7.2g, yield 86.8%, mp: 188.0-190 (. TC.1H-NMR and 13C-NMR data are as follows (see Figure 1-2 spectra):
1H-NMR (CDC13,400ΜΗζ) δ: 2.209 (s, 3H, -CH3), 2.666-2.707 (t, 2H, -CH2-CH2-),
3.747-3.788 (t, 2H, -CH2-CH2 -), 5.341 (s, 1H, -NH -), 5.757 (s, 1H, -NH -), 6.699 (s, 1H, Ar-H), 6.828 ( s, 1H, Ar-H), 7.287-7.390 (m, I OH, Ar-H).
13C-NMR (CDC13,400MHz) δ: 12.17 (-CH3), 41.00 (-CH2 -), 43.74 (-CH2-), 59.44 (quaternary carbon, coupled with strong electron-withdrawing group), 119.08 (-C = C -), 126.95 (aromatic carbon), 127.88 (aromatic carbon), 128.52 (aromatic carbon), 129.10 (aromatic carbon), 142.61 (= CN), 144.54 (-C = N), 176.21 (carbonyl carbon).
Example 3
[0041] 1- (2-bromoethyl) -1H- -2_ methyl-imidazole (5) Preparation of
[0042] The 1,2_ dibromoethane (50ml), 2- methylimidazole (2.5g, 30.5mmol), benzyltriethylammonium chloride (TEBA) (0.35g) and Na2CO3 (2.8g), Na0H (3.3g) were added sequentially 100mL three-necked flask, stirred and heated to 45 ° C reaction 4h. Cooling to room temperature, the reaction solution was filtered, washed with a saturated aqueous sodium bicarbonate paint filtrate was dried over anhydrous sodium sulfate. Concentrated, added to a mixed solvent of isopropyl ether and ethyl acetate (3: 1) was dissolved with stirring parsing crystal give the product 5.0g, yield 86.8%, mp.79-80. . .
Preparation of [0043] 4- (2-methyl-1-imidazolyl) -2,2-diphenyl-butyronitrile hydrochloride (2)
The diphenyl acetonitrile (5.8g, 30mmol) and 50% aqueous KOH (15ml), dimethyl sulfoxide (DMSO) (100ml), benzyltriethylammonium chloride (TEBA) (0.66g) 50ml Toluene was added to the reaction flask and stirred at 40 ° C under
0.5h0 was added 1- (2-bromoethyl) -2-methyl -1H- imidazole (4) (5.lg, 27mmol), was heated to 60 ° C, the reaction was stirred, TLC tracking and monitoring the reaction was complete, the mixture was poured into 100mL of water and extracted three times with ethyl acetate 240ml water phase. Washed three times with 300ml of water The organic phase was dried over anhydrous sodium sulfate, the organic phase was concentrated. Analytical crystal solution with hydrogen chloride ether solution, filtered crystals with a mixed solvent of isopropyl ether and recrystallized from ethyl acetate to give the condensation product of 4- (2-methyl-1-imidazolyl) -2,2-diphenylbutyric carbonitrile hydrochloride (2) as a white solid 7.0g, yield 76.8%, mp: 156.5-158. . . 1H-NmrgoomHzADCI3), δ (ppm): 7.35-7.42 (10H, m), 6.90 (1H, s), 6.77 (1H, s), 3.90-3.94 (2H, m), 2.75-2.79 (2H, m) , 2.25 (3H, s).
Preparation imidafenacin (I),
[0046] 4- (2-methyl-1-imidazolyl) -2,2_ diphenyl butyronitrile hydrochloride (2) (8.78g, 26mmol) in 70% concentrated sulfuric acid (25ml) was added to the reaction bottle, the reaction was stirred at 100 ° C, the end of the reaction was monitored by TLC tracking. The reaction solution was poured into 120ml of water, solid sodium carbonate was added to adjust PH to weakly alkaline, sufficiently stirred. With 180ml of dichloromethane and 35ml of ethanol mixed solvent was extracted three times, the organic phase was washed with water, dried over anhydrous sodium sulfate, the organic phase was concentrated. The residue was mixed with a solvent of ethyl acetate and recrystallized from ethanol to give 4- (2-methyl-1-imidazolyl) 2,2-diphenyl butyramide 7.lg, yield 85.5%, mp: 188.0-190. (TC.1H-NMR and 13C-NMR data are as follows (see Figure 1-2 spectra):
[0047] 1H-NMR (CDC13,400ΜΗζ) δ: 2.209 (s, 3H, -CH3), 2.666-2.707 (t, 2H, -CH2-CH2-),
3.747-3.788 (t, 2H, -CH2-CH2 -), 5.341 (s, 1H, -NH -), 5.757 (s, 1H, -NH -), 6.699 (s, 1H, Ar-H)
, 6.828 (s, 1H, Ar-H), 7.287-7.390 (m, I OH, Ar-H).
13C-NMR (CDC13,400MHz) δ: 12.17 (-CH3), 41.00 (-CH2 -), 43.74 (-CH2-), 59.44 (quaternary carbon, coupled with strong electron-withdrawing group), 119.08 (-C = C -), 126.95 (aromatic carbon), 127.88 (aromatic carbon), 128.52 (aromatic carbon), 129.10 (aromatic carbon), 142.61 (= CN), 144.54 (-C = N), 176.21 (carbonyl carbon).
Example 41- (2-bromoethyl) -1H- -2_ methyl-imidazole (5) Preparation of
[0051] The 1,2_ dibromoethane (50ml), 2- methylimidazole (2.5g, 30.5mmol), tetrabutylammonium bromide (TBAB) (0.5g) and K2C03 (3.6g), K0H ( 4.6g) were added sequentially 100mL three-necked flask, stirred and heated to 60 ° C reaction 4h.Cooling to room temperature, the reaction solution was filtered, and the filtrate was washed with saturated aqueous sodium bicarbonate, dried over anhydrous sodium sulfate. Concentrated, added to a mixed solvent of isopropyl ether and ethyl acetate (3: 1) was dissolved with stirring parsing crystal give the product 4.5g, yield 78.1%, mp.79_80 ° C.
Preparation of [0052] 4- (2-methyl-1-imidazolyl) -2,2-diphenyl-butyronitrile hydrochloride (2)
[0053] The diphenyl acetonitrile (5.8g, 30mmol) and 50% aqueous KOH (15ml), dimethyl sulfoxide (DMSO) (100ml), tetrabutylammonium bromide (TBAB) (0.9g) in toluene 50ml was added to the reaction flask and stirred for 0.5h in the 40 ° C. Plus Λ 1- (2- bromoethyl) -2-methyl -1H- imidazole (4) (5.lg, 27mmol), was heated to 100 ° C, the reaction was stirred, TLC tracking and monitoring the reaction was complete, the mixture was poured into 100mL of water and extracted three times with ethyl acetate 240ml water phase. Washed three times with 300ml of water The organic phase was dried over anhydrous sodium sulfate, the organic phase was concentrated. Analytical crystal solution with hydrogen chloride ether solution, filtered crystals with a mixed solvent of isopropyl ether and recrystallized from ethyl acetate to give the condensation product of 4- (2-methyl-1-imidazolyl) -2,2-diphenylbutyric carbonitrile hydrochloride (2) as a white solid 6.7g, yield 73.4%, mp: 156.5-158 ° C. 1H-NMR (400MHz, CDCl3), δ (ppm): 7.35-7.42 (IOH, m), 6.90 (1H, s), 6.77 (1H, s), 3.90-3.94 (2H, m), 2.75-2.79 ( 2H, m), 2.25 (3H, s).
The preparation imidafenacin (I),
4- (2-methyl-1-imidazolyl) -2,2_ diphenyl butyronitrile hydrochloride (2) (8.78g, 26mmol) in 70% concentrated sulfuric acid (25ml) was added to the reaction bottle, the reaction was stirred at 150 ° C at the end of the reaction was monitored TLC tracking. The reaction solution was poured into 120ml of water, solid sodium carbonate was added to adjust PH to weakly alkaline, sufficiently stirred. With 180ml of dichloromethane and 35ml of ethanol mixed solvent was extracted three times, the organic phase was washed with water, dried over anhydrous sodium sulfate, the organic phase was concentrated. The residue was mixed with a solvent of ethyl acetate and recrystallized from ethanol to give 4- (2-methyl-1-imidazolyl) 2,2-diphenyl butyramide 6.2g, yield 74.8%, mp: 188.0-190 (. TC.1H-NMR and 13C-NMR data are as follows (see Figure 1-2 spectra):
[0056] 1H-NMR (CDC13,400ΜΗζ) δ: 2.209 (s, 3H, -CH3), 2.666-2.707 (t, 2H, -CH2-CH2 -), 3.747-3.788 (t, 2H, -CH2-CH2 -), 5.341 (s, 1H, -NH -), 5.757 (s, 1H, -NH -), 6.699 (s, 1H, Ar-H)
, 6.828 (s, 1H, Ar-H), 7.287-7.390 (m, I OH, Ar-H).
13C-NMR (CDC13,400MHz) δ: 12.17 (-CH3), 41.00 (-CH2 -), 43.74 (-CH2-), 59.44 (quaternary carbon, coupled with strong electron-withdrawing group), 119.08 (-C = C -), 126.95 (aromatic carbon), 127.88 (aromatic carbon), 128.52 (aromatic carbon), 129.10 (aromatic carbon), 142.61 (= CN), 144.54 (-C = N), 176.21 (carbonyl carbon).
Example 5
1- (2-bromoethyl) -1H- -2_ methyl-imidazole (5) Preparation of
The 1,2_ dibromoethane (50ml), 2- methylimidazole (2.5g, 30.5mmol), tetrabutylammonium bromide (TBAB) (0.5g) and K2CO3 (3.6g), K0H ( 4.6g) were added sequentially 100mL three-necked flask, stirred and heated to 20 ° C reaction 10h. Cooling to room temperature, the reaction solution was filtered, washed with a saturated aqueous sodium bicarbonate paint filtrate was dried over anhydrous sodium sulfate. Concentrated, added to a mixed solvent of isopropyl ether and ethyl acetate (3: 1) was dissolved with stirring parsing crystal give the product 4.1g, yield 71.2%, mp.79-80. . .
Preparation of [0061] 4- (2-methyl-1-imidazolyl) -2,2-diphenyl-butyronitrile hydrochloride (2)
[0062] The diphenyl acetonitrile (5.8g, 30mmol) and 50% aqueous KOH (15ml), dimethyl sulfoxide (DMSO) (100mL), tetrabutylammonium bromide (TBAB) (0.9g) in toluene 50ml was added to the reaction flask and stirred at 20 ° C in Ih. Join
1- (2-bromoethyl) -2-methyl -1H- imidazole (4) (5.lg, 27mmol), was heated to 60 ° C, the reaction was stirred, TLC tracking and monitoring the reaction was complete, the mixture was poured into 100mL of water and extracted three times with ethyl acetate 240ml water phase. Washed three times with 300ml of water The organic phase was dried over anhydrous sodium sulfate, the organic phase was concentrated. Solution of hydrogen chloride in ether solution with analytical crystal, crystals were filtered with a mixed solvent of isopropyl ether and recrystallized from ethyl acetate to give the condensation product of 4- (2-methyl-1-imidazolyl) -2,
2-phenyl-butyronitrile hydrochloride (2) as a white solid 6.5g, yield 71.2%, mp: 156.5-158 ° C. 1H-NMR (400MHz, CDCl3), δ (ppm): 7.35-7.42 (IOH, m), 6.90 (1H, s), 6.77 (1H, s), 3.90-3.94 (2H, m), 2.75-2.79 ( 2H, m), 2.25 (3H, s).
[0063] Preparation of imidafenacin (I), [0064] 4- (2-methyl-1-imidazolyl) -2,2-diphenyl-butyronitrile hydrochloride (2) (8.78g, 26mmol ) and 70% of concentrated sulfuric acid (25ml) was added to the reaction flask, and stirred in at 50 ° C, the end of the reaction was monitored by TLC tracking. The reaction solution was poured into 120ml of water, solid sodium carbonate was added to adjust PH to weakly alkaline, sufficiently stirred. With 180ml of dichloromethane and 35ml of ethanol mixed solvent was extracted three times, the organic phase was washed with water, dried over anhydrous sodium sulfate, the organic phase was concentrated. The residue was mixed with a solvent of ethyl acetate and recrystallized from ethanol to give 4- (2-methyl-1-imidazolyl) 2,2-diphenyl butyramide 6.6g, yield 79.7%, mp: 188.0-190 (. TC.1H-NMR and 13C-NMR data are as follows (see Figure 1-2 spectra):
[0065] 1H-NMR (CDC13,400ΜΗζ) δ: 2.209 (s, 3H, -CH3), 2.666-2.707 (t, 2H, -CH2-CH2-),
3.747-3.788 (t, 2H, -CH2-CH2 -), 5.341 (s, 1H, -NH -), 5.757 (s, 1H, -NH -), 6.699 (s, 1H, Ar-H)
, 6.828 (s, 1H, Ar-H), 7.287-7.390 (m, I OH, Ar-H).
[0066] 13C-NMR (CDC13,400MHz) δ: 12.17 (-CH3), 41.00 (-CH2 -), 43.74 (-CH2-), 59.44 (quaternary carbon, coupled with strong electron-withdrawing group), 119.08 (-C = C -), 126.95 (aromatic carbon), 127.88 (aromatic carbon), 128.52 (aromatic carbon), 129.10 (aromatic carbon), 142.61 (= CN), 144.54 (-C = N), 176.21 (carbonyl carbon).
[0067] Example 6
[0068] 1- (2-bromoethyl) -1H- -2_ methyl-imidazole (5) Preparation of
[0069] The 1,2_ dibromoethane (50ml), 2- methylimidazole (2.5g, 30.5mmol), benzyltriethylammonium chloride (TEBA) (0.35g) and Na2CO3 (2.8g), Na0H (3.3g) were added sequentially 100mL three-necked flask and stirred and heated to 40 ° C reaction 8h. Cooling to room temperature, the reaction solution was filtered, washed with a saturated aqueous sodium bicarbonate paint filtrate was dried over anhydrous sodium sulfate. Concentrated, added to a mixed solvent of isopropyl ether and ethyl acetate (3: 1) was dissolved with stirring parsing crystal give the product 4.4g, yield 76.4%, mp.79-80. . .
Preparation of [0070] 4- (2-methyl-1-imidazolyl) -2,2-diphenyl-butyronitrile hydrochloride (2)
[0071] The diphenyl acetonitrile (5.8g, 30mmol) and 50% aqueous KOH (15ml), dimethyl sulfoxide (DMSO) (100ml), benzyltriethylammonium chloride (TEBA) (0.66g) 50ml Toluene was added to the reaction flask and stirred 0.5h0 1- (2-bromoethyl) -2-methyl -1H- imidazole (4) at 40 ° C (5.lg, 27mmol), was heated to 50 ° C, the reaction mixture was stirred, TLC tracking and monitoring the reaction was complete, the mixture was poured into 100mL of water and extracted three times with ethyl acetate. The aqueous phase was 240ml. Washed three times with 300ml of water The organic phase was dried over anhydrous sodium sulfate, the organic phase was concentrated. Analytical crystal solution with hydrogen chloride ether solution, filtered crystals with a mixed solvent of isopropyl ether and recrystallized from ethyl acetate to give the condensation product of 4- (2-methyl-1-imidazolyl) -2,2-diphenylbutyric carbonitrile hydrochloride (2) as a white solid 6.8g, yield 74.6%, mp: 156.5-158. . . 1H-NmrgoomHzADCI3), δ (ppm): 7.35-7.42 (10H, m), 6.90 (1H, s), 6.77 (1H, s), 3.90-3.94 (2H, m), 2.75-2.79 (2H, m) , 2.25 (3H, s).
[0072] Preparation imidafenacin (I),
[0073] 4- (2-methyl-1-imidazolyl) -2,2_ diphenyl butyronitrile hydrochloride (2) (8.78g, 26mmol) in 70% concentrated sulfuric acid (25ml) was added to the reaction bottle, the reaction was stirred at 80 ° C, the end of the reaction was monitored by TLC tracking. The reaction solution was poured into 120ml of water, solid sodium carbonate was added to adjust PH to weakly alkaline, sufficiently stirred. With 180ml of dichloromethane and 35ml of ethanol mixed solvent was extracted three times, the organic phase was washed with water, dried over anhydrous sodium sulfate, the organic phase was concentrated. The residue was mixed with a solvent of ethyl acetate and recrystallized from ethanol to give 4- (2-methyl-1-imidazolyl) 2,2-diphenyl butyramide 6.Sg, yield 81.8%, mp: 188.0-190. (TC.1H-NMR and 13C-NMR data are as follows (see Figure 1-2 spectra):
[0074] 1H-NMR (CDC13,400ΜΗζ) δ: 2.209 (s, 3H, -CH3), 2.666-2.707 (t, 2H, -CH2-CH2-),
3.747-3.788 (t, 2H, -CH2-CH2 -), 5.341 (s, 1H, -NH -), 5.757 (s, 1H, -NH -), 6.699 (s, 1H, Ar-H)
, 6.828 (s, 1H, Ar-H), 7.287-7.390 (m, I OH, Ar-H). [0075] 13C-NMR (CDC13,400MHz) δ: 12.17 (_CH3), 41.00 (-CH2 -), 43.74 (-CH2-), 59.44 (quaternary carbon, coupled with strong electron-withdrawing group), 119.08 (-C = C -), 126.95 (aromatic carbon), 127.88 (aromatic carbon), 128.52 (aromatic carbon), 129.10 (aromatic carbon), 142.61 (= CN), 144.54 (-C = N), 176.21 (carbonyl carbon).

PATENT
CN 105399678
https://www.google.com/patents/CN105399678A?cl=en
CLIP
http://dmd.aspetjournals.org/content/35/9/1624/T3.expansion.html
TABLE 3
Chemical shifts of protons and carbons in 1H NMR and 13C NMR spectra of major (M-11b) and minor (M-11a) constituents of reference products obtained from imidafenacin
|
Position of Proton |
1H NMR Data (in D2O) |
||
|---|---|---|---|
| Major Constituent (M-11b)
|
Minor Constituent (M-11a)
|
||
| 1 | 2.18a (3Hb, sc) | 2.11a (3Hb, sc) | |
| 2 | 2.82 (2H, m) | 2.79 (2H, m) | |
| 3 | 3.45 (2H, m) | 3.41 (2H, m) | |
| 5 | 5.26 (1H, s) | 5.43-5.47d (1H, d, J = 8.1e) | |
| 6 | 5.33 (1H, s) | 5.43-5.47d (1H, d, J = 8.1e) | |
| 8, 9, and 10 | 7.39-7.49 (10H, m) | 7.40-7.48 (10H, m) | |
| 13
|
8.45 (1.3H, s)
|
8.45 (2H, s)
|
|
| Position of Carbon
|
13C-NMR Data (in D2O)xc
|
||
|---|---|---|---|
| Major Constituent (M-11b)
|
Minor Constituent (M-11a)
|
||
| 1 | 14.61a | 14.48a | |
| 2 | 39.04 | 38.49 | |
| 3 | 43.49 | 42.90 | |
| 4 | 61.95-61.99f | 61.95-61.99f | |
| 5 | 87.61 | 80.22 or 85.78f | |
| 6 | 93.10 | 80.22 or 85.78f | |
| 7 | 144.2-144.4f | 144.2-144.4f | |
| 8, 9, and 10 | 130.7-131.8f | 130.7-131.8f | |
| 11 | 170.8 | 169.5 | |
| 12 | 181.9-182.2f | 181.9-182.2f | |
| 13
|
173.8
|
173.8
|
|
-
↵ a Chemical shifts are reported in parts per million.
-
↵ b Intensities are represented as number of protons.
-
↵ c Multiplicity: s, singlet; d, doublet; m, multiplet.
-
↵ d These proton signals could not be distinguished.
-
↵ e Coupling constants (J) are given in Hertz.
-
↵ f These carbon signals could not be distinguished.

FIG. 1.
Chemical structures of [14C]imidafenacin and postulated metabolites, and their fragment ions. *, 14C labeled position; broken line, precursor and product ions obtained by collision-induced dissociation in LC/MS/MS.
| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| CN101076521A * | Dec 13, 2005 | Nov 21, 2007 | 杏林制药株式会社 | Process for producing muscarine receptor antagonist and intermediate therefor |
| CN102746235A * | Jul 20, 2012 | Oct 24, 2012 | 北京科莱博医药开发有限责任公司 | Improved method for preparing imidafenacin |
| CN103275007A * | May 27, 2013 | Sep 4, 2013 | 朱雪琦 | Pyrazole derivatives and preparation method thereof |
| US7351429 | 2008-04-01 | Oral solid preparation |
| US2008004247 | 2008-01-03 | Combinations of Statins with Bronchodilators |
| US2007270436 | 2007-11-22 | NOVEL AMINO- AND IMINO-ALKYLPIPERAZINES |
| US2007219237 | 2007-09-20 | Chromane Derivatives |
| US2007185129 | 2007-08-09 | ACID ADDITION SALTS OF THIENOPYRANCARBOXAMIDE DERIVATIVES |
| US2007092566 | 2007-04-26 | Oral sustained-release tablet |
| US2006188554 | 2006-08-24 | Transdermal absorption preparation |
| EP0733621 | 2002-05-15 | NOVEL IMIDAZOLE DERIVATIVE AND PROCESS FOR PRODUCING THE SAME |
| US6103747 | 2000-08-15 | Imidazole derivatives and process for preparing the same |
| US5932607 | 1999-08-03 | Imidazole derivatives and process for preparing the same |
| Patent ID | Date | Patent Title |
|---|---|---|
| US2015064232 | 2015-03-05 | TRANSDERMAL ABSORPTION PREPARATION |
| US8729056 | 2014-05-20 | Preventive and/or therapeutic agent of hand-foot syndrome |
| US8722133 | 2014-05-13 | Method for production of orally rapidly disintegrating tablet comprising imidafenacin as active ingredient |
| US2013211352 | 2013-08-15 | PERCUTANEOUSLY ABSORBED PREPARATION |
| US2013211353 | 2013-08-15 | PERCUTANEOUS ABSORPTION TYPE FORMULATION |
| US8343544 | 2013-01-01 | Oral sustained-release tablet |
| US2012289563 | 2012-11-15 | COMBINATIONS OF IMIDAFENACIN AND SALIVARY STIMULANTS FOR THE TREATMENT OF OVERACTIVE BLADDER |
| US8247415 | 2012-08-21 | Hydroxymethyl pyrrolidines as [beta]3 adrenergic receptor agonists |
| US8158152 | 2012-04-17 | Lyophilization process and products obtained thereby |
| US8124633 | 2012-02-28 | HYDROXYMETHYL ETHER HYDROISOINDOLINE TACHYKININ RECEPTOR ANTAGONISTS |
- Kobayashi F, Yageta Y, Segawa M, Matsuzawa S: Effects of imidafenacin (KRP-197/ONO-8025), a new anti-cholinergic agent, on muscarinic acetylcholine receptors. High affinities for M3 and M1 receptor subtypes and selectivity for urinary bladder over salivary gland. Arzneimittelforschung. 2007;57(2):92-100. [PubMed:17396619 ]
- Miyachi H, Kiyota H, Uchiki H, Segawa M: Synthesis and antimuscarinic activity of a series of 4-(1-Imidazolyl)-2,2-diphenylbutyramides: discovery of potent and subtype-selective antimuscarinic agents. Bioorg Med Chem. 1999 Jun;7(6):1151-61. [PubMed:10428387 ]
Reference
- Kobayashi F, Yageta Y, Segawa M, Matsuzawa S (2007). “Effects of imidafenacin (KRP-197/ONO-8025), a new anti-cholinergic agent, on muscarinic acetylcholine receptors. High affinities for M3 and M1 receptor subtypes and selectivity for urinary bladder over salivary gland”. Arzneimittelforschung. 57 (2): 92–100. doi:10.1055/s-0031-1296589. PMID 17396619.
- Miyachi H, Kiyota H, Uchiki H, Segawa M (June 1999). “Synthesis and antimuscarinic activity of a series of 4-(1-Imidazolyl)-2,2-diphenylbutyramides: discovery of potent and subtype-selective antimuscarinic agents”. Bioorg. Med. Chem. 7 (6): 1151–61.doi:10.1016/S0968-0896(99)00003-6. PMID 10428387.
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
4-(2-methyl-1H-imidazol-1-yl)-2,2-diphenylbutanamide
|
|
| Clinical data | |
| Routes of administration |
Oral |
| Legal status | |
| Legal status |
|
| Identifiers | |
| CAS Number | 170105-16-5 |
| ATC code | none |
| PubChem | CID 6433090 |
| ChemSpider | 4938278 |
| UNII | XJR8Y07LJO |
| ChEMBL | CHEMBL53366 |
| Chemical data | |
| Formula | C20H21N3O |
| Molar mass | 319.40 g/mol |
//////////イミダフェナシン , D06273, KRP-197, KRP 197, ONO-8025, ONO 8025, UNII:XJR8Y07LJO, Kyorin, Ono ,Imidafenacin, 170105-16-5, JAPAN 2015, Uritos® , Staybla®
JNJ 54166060

JNJ 54166060
JNJ-54166060; JNJ 54166060; JNJ54166060.
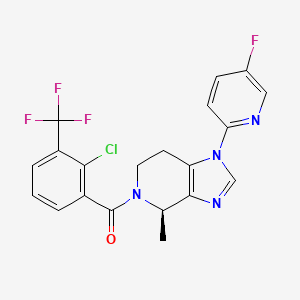
(R)-(2-chloro-3-(trifluoromethyl)phenyl)(1-(5-fluoropyridin-2-yl)-4-methyl-1,4,6,7-tetrahydro-5H-imidazo[4,5-c]pyridin-5-yl)methanone
[2-chloro-3-(trifluoromethyl)phenyl]-[(4R)-1-(5-fluoropyridin-2-yl)-4-methyl-6,7-dihydro-4H-imidazo[4,5-c]pyridin-5-yl]methanone
Methanone, [2-chloro-3-(trifluoromethyl)phenyl][(4R)-1-(5-fluoro-2-pyridinyl)-1,4,6,7-tetrahydro-4-methyl-5H-imidazo[4,5-c]pyridin-5-yl]-
CAS 1627900-41-7 OF R ISOMER DESIRED
CAS 1627900-42-8 OF S ISOMER
Chemical Formula: C20H15ClF4N4O
Exact Mass: 438.0871
JNJ-54166060 is a potent P2X7 antagonist. Bioactivity data of JNJ-54166060: rP2X7 IC50=4 nM; rP2X7 IC50=115nM; HLM/RLM = 0.35/0.64, ED50 = 2.3 mg/kg in rats. JNJ-54166060 shows high oral bioavailability and low-moderate clearance in preclinical species, acceptable safety margins in rats, and a predicted human dose of 120 mg of QD. Additionally, JNJ-54166060 possesses a unique CYP profile and was found to be a regioselective inhibitor of midazolam CYP3A metabolism.
The P2X7 receptor is a ligand-gated ion channel and is present on a variety of cell types, largely those known to be involved in the inflammatory and/ or immune process, specifically, macrophages and monocytes in the periphery and predominantly in glial cells (microglia and astrocytes) of the CNS. (Duan and Neary, Glia 2006, 54, 738-746; Skaper et al., FASEB J 2009, 24, 337-345;
Surprenant and North, Annu. Rev. Physiol. 2009, 71, 333-359). Activation of the P2X7 receptor by extracellular nucleotides, in particular adenosine triphosphate, leads to the release of proinflammatory cytokines IL-1 β and IL-18 (Muller, et. Al. Am. J. Respir. Cell Mol. Biol. 201 1 , 44, 456-464), giant cell formation
(macrophages/ microglial cells), degranulation (mast cells) and L-selectin shedding (lymphocytes) (Ferrari et al., J. Immunol. 2006, 176, 3877-3883; Surprenant and North, Annu. Rev. Physiol. 2009, 71, 333-359). P2X7 receptors are also located on antigen-presenting cells (keratinocytes, salivary acinar cells (parotid cells)), hepatocytes, erythrocytes, erythroleukaemic cells, monocytes, fibroblasts, bone marrow cells, neurones, and renal mesangial cells.
The importance of P2X7 in the nervous system arises primarily from experiments using P2X7 knock out mice. These mice demonstrate the role of P2X7 in the development and maintenance of pain as these mice were protected from the development of both adjuvant-induced inflammatory pain and partial nerve ligation induced neuropathic pain (Chessell et al., Pain 2005, 114, 386-396). In addition P2X7 knock out mice also exhibit an anti-depressant phenotype based on reduced immobility in forced swim and tail suspension tests (Basso et al., Behav. Brain Res. 2009, 798, 83-90.). Moreover, the P2X7 pathway is linked to the release of the pro-inflammatory cytokine, IL-1 β, which has been linked to precipitation of mood disorders in humans (Dantzer, Immunol. Allergy Clin. North Am. 2009, 29, 247-264; Capuron and Miller, Pharmacol. Ther. 201 1 , 730, 226-238). In addition, in murine models of Alzheimer’s disease, P2X7 was upregulated around amyloid plaques indicating a role of this target in such pathology as well (Parvathenani et al., J. Biol. Chem. 2003, 278, 13309-13317).
In view of the clinical importance of P2X7, the identification of compounds that modulate P2X7 receptor function represents an attractive avenue into the development of new therapeutic agents. Such compounds are provided herein.
PATENT
Example 11
(2-Chloro-3-(trifluoromethyl)phenyl)(1-(5-fluoropyridin-2-yl)-4-methyl-6,7-dihydro-1H-imidazo[4,5-c]pyridin-5(4H)-yl)methanone
Step A. (2-Chloro-3-(trifluoromethyl)phenyl)(1-(5-fluoropyridin-2-yl)-4-methyl-1H-imidazo[4,5-c]pyridin-5(4H)-yl)methanone
Step B. (2-Chloro-3-(trifluoromethyl)phenyl)(1-(5-fluoropyridin-2-yl)-4-methyl-6,7-dihydro-1H-imidazo[4,5-c]pyridin-5(4H)-yl)methanone
Example 40
(R*)-(2-chloro-3-(trifluoromethyl)phenyl)(1-(5-fluoropyridin-2-yl)-4-methyl-6,7-dihydro-1H-imidazo[4,5-c]pyridin-5(4H)-yl)methanone
Intermediate 1: 1-(5-Fluoropyridin-2-yl)-1H-imidazo[4,5-c]pyridine
Intermediate 12: 2-Chloro-3-(trifluoromethyl)benzoyl chloride
PAPER
Identification of (R)-(2-Chloro-3-(trifluoromethyl)phenyl)(1-(5-fluoropyridin-2-yl)-4-methyl-6,7-dihydro-1H-imidazo[4,5-c]pyridin-5(4H)-yl)methanone (JNJ 54166060), a Small Molecule Antagonist of the P2X7 receptor
†Janssen Pharmaceutical Research & Development, LLC, 3210 Merryfield Row, San Diego, California 92121 United States
‡Janssen Research & Development, Discovery Sciences, A Division of Janssen-Cilag, Jarama 75, 45007 Toledo, Spain
J. Med. Chem., Article ASAP
DOI: 10.1021/acs.jmedchem.6b00989, http://pubs.acs.org/doi/abs/10.1021/acs.jmedchem.6b00989
*Tel: 1-858-320-3306. E-mail: dswanso1@its.jnj.com.

The synthesis and SAR of a series of 4,5,6,7-tetrahydro-imidazo[4,5-c]pyridine P2X7 antagonists are described. Addressing P2X7 affinity and liver microsomal stability issues encountered with this template afforded methyl substituted 4,5,6,7-tetrahydro-imidazo[4,5-c]pyridines ultimately leading to the identification of 1 (JNJ 54166060). 1 is a potent P2X7 antagonist with an ED50 = 2.3 mg/kg in rats, high oral bioavailability and low-moderate clearance in preclinical species, acceptable safety margins in rats, and a predicted human dose of 120 mg of QD. Additionally, 1 possesses a unique CYP profile and was found to be a regioselective inhibitor of midazolam CYP3A metabolism.
(R)-(2-Chloro-3 (trifluoromethyl)phenyl)(1-(5-fluoropyridin-2-yl)-4-methyl-6,7-dihydro-1H-imidazo[4,5-c]pyridin-5(4H)-yl)methanone (1)The title compound was obtained as a single enantiomer by Chiral SFC purification performed using CHIRALCEL OD-H (5 μm, 250 × 20 mm) and a mobile phase of 72% CO2, 28% 1:1 EtOH/iPrOH. The enantiomeric purity was confirmed by analytical SFC using Whelk-al (S,S) (250 × 4.6 mm) and a mobile phase of 60% CO2, 40% MeOH over 7 min (100% single enantiomer, 4.03 min retention time) (0.16 g, 41%).
1H NMR (500 MHz, CDCl3) δ 8.43–8.32 (m, 1H), 7.94 (dd,J = 18.3, 11.2 Hz, 1H), 7.80–7.70 (m, 1H), 7.65–7.29 (m, 4H), 5.91–5.74 (m, 1H), 5.14–4.46 (m, 1H), 3.60–3.30 (m, 1H), 3.30–3.04 (m, 1H), 3.06–2.68 (m, 1H), 1.76–1.35 (m, 3H).
13C NMR (126 MHz, CDCl3) δ 165.95–165.79 (s), 139.33–139.10 (d, J = 8.8 Hz), 137.50–137.15 (m), 135.54–135.31 (m), 131.24–131.08 (s), 130.68–130.52 (s), 130.42–130.26 (s), 128.08–127.83 (t, J = 5.7 Hz), 127.50–127.13 (m), 126.23–125.85 (dd, J = 20.4, 7.6 Hz), 123.69–123.54 (s), 122.56–122.40 (s), 116.00–115.72 (m), 52.32–52.16 (s), 47.41–47.25 (s), 41.25–41.09 (s), 35.57–35.41 (s), 24.49–24.19 (m), 19.96–18.59 (m).
HRMS calc. for C20H13ClF4N4O [M + H]+ 439.0943, found 439.0957.
Specific rotation: [α]20D – 52.6 (c 0.5, CHCl3).
PATENT
https://www.google.com/patents/WO2014152604A1?cl=en
Example 40. (R* -(2-chloro-3-(trifluoromethyl phenyl (l-(5-fluoropyridin-2-yl -4-methyl- -dihydro-lH-imidazor4,5-c1pyridin-5(‘4H -yl methanone.

The title compound, absolute configuration unknown, was obtained as a single enantiomer by Chiral SFC purification of Example 11 performed using CHIRALCEL OD-H (5μιη, 250x20mm) and a mobile phase of 70% CO2, 30% EtOH. The enantiomeric purity was confirmed by analytical SFC using a CHIRALCEL OD-H (250×4.6mm) and a mobile phase of 70% CO2, 30% EtOH over 7 minutes. (100% single enantiomer, 2.29 min retention time). MS (ESI): mass calculated for C2oH15ClF4N40, 438.1; m/z found, 439.3 [M+H]+.
Example 11. (2-Chloro-3-(trifluoromethyl)phenyl)(l-(5-fluoropyridin-2-yl)-4-methyl-6,7- dihvdro-lH-imidazor4,5-c1pyridin-5(4H)-yl)methanone.

Step A. (2-Chloro-3 -(trifluoromethyl)phenylX 1 -(5 -fluoropyridin-2-yl)-4-methyl- 1 H- imidazor4.5-c1pyridin-5(‘4H)-yl)methanone.
To a solution of Intermediate 1 (0.70 g, 3.27 mmol) in THF (20 mL) was added
Intermediate 12 (0.87 g, 3.60 mmol) dropwise. The reaction was allowed to stir for 1 h then cooled to – 78 °C. To the cooled solution was added 3M MeMgBr in Et20 (1.31 mL, 3.92 mmoL) and the reaction was let come to room temperature. The mixture was then quenched with IN NaOH (50 mL) and extracted with EtOAc (3 x 30 mL). The organic layers were combined, dried (Na2S04), and concentrated. Chromatography of the resulting residue (Si02; MeOH (NH3):DCM) gave the title compound (770 mg, 54%). XH NMR (400 MHz, CDC13) δ 8.43 – 8.34 (m, 1H), 7.92 – 7.73 (m, 2H), 7.70 – 7.33 (m, 4H), 6.08 (dtd, J = 19.7, 11.7, 8.0 Hz, 3H), 1.54 (t, J = 7.0 Hz, 3H). MS (ESI): mass calculated for C2oH13ClF4 40, 436.07; m/z found 437.1 [M+H]+.
Step B. (2-Chloro-3-(trifluoromethyl)phenyl)(l-(5-fluoropyridin-2-yl)-4-methyl-6J- dihydro-lH-imidazo[4.5-clpyridin-5(4H)-yl)methanone.
To a solution of (2-chloro-3-(trifluoromethyl)phenyl)(l-(5-fluoropyridin-2-yl)-4-methyl- lH-imidazo[4,5-c]pyridin-5(4H)-yl)methanone (0.80 g, 1.83 mmol) in degassed EtOH (25 mL) was added 10% palladium on carbon (0.20 g, 0.19 mmol). The reaction was placed under an atmosphere of hydrogen and let stir for 48 h. The reaction was diluted with DCM and filtered through a pad of Celite ©. The solvent was concentrated and chromatography of the resulting residue (Si02; MeOH (NH3):DCM) gave the title compound (500 mg, 62%). ¾ NMR (500 MHz, CDC13) δ 8.45 – 8.30 (m, 1H), 7.94 (dd, J = 18.2, 10.7 Hz, 1H), 7.76 (d, J = 5.7 Hz, 1H), 7.67 – 7.43 (m, 3H), 7.43 – 7.30 (m, 1H), 5.81 (dd, J = 13.3, 6.7 Hz, 1H), 5.07 (d, J = 5.6 Hz, 1H), 4.52 (d, J = 6.7 Hz, 1H), 3.61 – 3.31 (m, 1H), 3.08 – 2.69 (m, 1H), 1.63 – 145 (m, 3H). MS (ESI): mass calculated for C20H15ClF4N4O, 438.08; m/z found 439.1 [M+H]+.
Intermediate 12: 2-Chloro-3-(trifluoromethyl)benzoyl chloride.

To a suspension of 2-chloro-3-(trifluoromethyl)benzoic acid (15 g, 67 mmol) and catalytic DMF (0.06 mL, 0.67 mmol) in DCM (150 mL) was added oxalyl chloride (6.8 mL, 80 mmol) dropwise. The reaction was let stir (vigorous bubbling) for 4 h and concentrated to an oily solid which became solid after overnight drying on high vacuum.
Intermediate 1 : l-(‘5-Fluoropvridin-2-vl)-lH-imidazor4,5-clpvridine.

A solution of 5-azabenzimidazole (1.00 g, 8.40 mmol), 2-bromo-5-fluoropyridine (1.48 g, 8.40 mmol), copper (I) oxide (0.13 g, 0.84 mmol), 8-hydroxyquinoline (0.24 g, 1.68 mmol), and CS2CO3 (5.47 g, 16.8 mmol) in DMSO (4 mL) was irradiated in a microwave apparatus for 1 hour at 140 °C. The reaction was diluted with H2O (100 mL) and extracted with EtOAc (75 mL x 3). The organic layers were combined, dried (Na2S04), and concentrated. Chromatography of the resulting residue (S1O2; MeOH (NH3):DCM) gave the title compound (0.45 g, 25%). MS (ESI): mass calculated for C11H7FN4, 214.07; m/z found 215.1 [M+H]+.
| Patent ID | Date | Patent Title |
|---|---|---|
| US2016039809 | 2016-02-11 | P2X7 MODULATORS |
| US2015322062 | 2015-11-12 | P2X7 MODULATORS |
| US2014275015 | 2014-09-18 | P2X7 MODULATORS |
///////JNJ 54166060, JNJ-54166060, JNJ54166060, 1627900-42-8, P2X7 antagonists, 1627900-41-7
ClC1=C(C(N2CCC(N(C3=CC=C(F)C=N3)C=N4)=C4[C@H]2C)=O)C=CC=C1C(F)(F)F
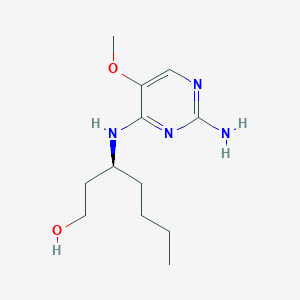
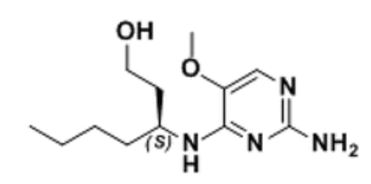
(3S)-3-[(2-amino-5-methoxypyrimidin-4-yl)amino]heptan-1-ol for the treatment of viral infections
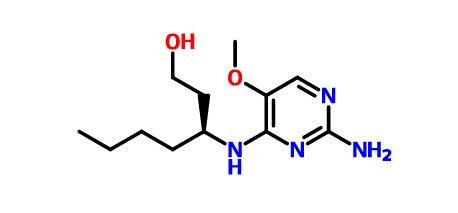
(3S)-3-[(2-amino-5-methoxypyrimidin-4-yl)amino]heptan-1-ol
JNJ ?
cas 1402802-45-2
WO 2012136834, WO 2014053595
for the treatment of viral infections
| Molecular Formula: | C12H22N4O2 |
|---|---|
| Molecular Weight: | 254.32868 g/mol |
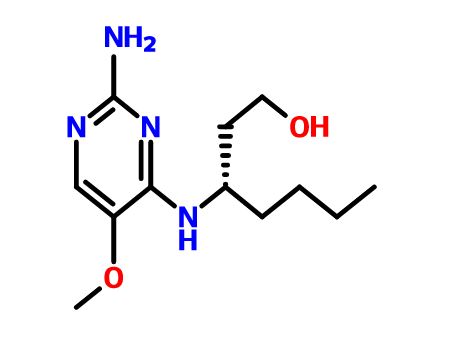
(3S)-3-[(2-amino-5-methoxypyrimidin-4-yl)amino]heptan-1-ol

Mark Hicken, Managing Director UK & Ireland, Janssen Ltd; Maggie Throup MP, Health Select Committee; Richard Mason, Head of Innovation, Johnson and Johnson; Dr Mark Lloyd Davies, Senior Director Government Affairs and Policy UK and Ireland, Johnson & Johnson
To the use of pyrimidine derivatives in the treatment of viral infections, immune or inflammatory disorders, whereby the modulation, or agonism, of toll-like-receptors (TLRs) is involved. Toll-Like Receptors are primary transmembrane proteins characterized by an extracellular leucine rich domain and a cytoplasmic extension that contains a conserved region. The innate immune system can recognize pathogen-associated molecular patterns via these TLRs expressed on the cell surface of certain types of immune cells. Recognition of foreign pathogens activates the production of cytokines and upregulation of co-stimulatory molecules on phagocytes. This leads to the modulation of T cell behaviour.
It has been estimated that most mammalian species have between ten and fifteen types of Toll-like receptors. Thirteen TLRs (named TLR1 to TLR13) have been identified in humans and mice together, and equivalent forms of many of these have been found in other mammalian species. However, equivalents of certain TLR found in humans are not present in all mammals. For example, a gene coding for a protein analogous to TLR10 in humans is present in mice, but appears to have been damaged at some point in the past by a retrovirus. On the other hand, mice express TLRs 11, 12, and 13, none of which are represented in humans. Other mammals may express TLRs which are not found in humans. Other non-mammalian species may have TLRs distinct from mammals, as demonstrated by TLR14, which is found in the Takifugu pufferfish. This may complicate the process of using experimental animals as models of human innate immunity.
For detailed reviews on toll-like receptors see the following journal articles. Hoffmann, J. A., Nature, 426, p 33-38, 2003; Akira, S., Takeda, K., and Kaisho, T., Annual Rev. Immunology, 21, p 335-376, 2003; Ulevitch, R. J., Nature Reviews: Immunology, 4, p 512-520, 2004.
Compounds indicating activity on Toll-Like receptors have been previously described such as purine derivatives in WO 2006/117670, adenine derivatives in WO 98/01448 and WO 99/28321, and pyrimidines in WO 2009/067081. However, there exists a strong need for novel Toll-Like receptor modulators having preferred selectivity, higher potency, higher metabolic stability, and an improved safety profile compared to the compounds of the prior art.
In the treatment of certain viral infections, regular injections of interferon (IFNα) can be administered, as is the case for hepatitis C virus (HCV), (Fried et. al. Peginterferon-alfa plus ribavirin for chronic hepatitis C virus infection, N Engl J Med 2002; 347: 975-82). Orally available small molecule IFN inducers offer the potential advantages of reduced immunogenicity and convenience of administration. Thus, novel IFN inducers are potentially effective new class of drugs for treating virus infections. For an example in the literature of a small molecule IFN inducer having antiviral effect see De Clercq, E.; Descamps, J.; De Somer, P. Science 1978, 200, 563-565.
IFNα is also given in combination with other drugs in the treatment of certain types of cancer (Eur. J. Cancer 46, 2849-57, and Cancer Res. 1992, 52, 1056). TLR 7/8 agonists are also of interest as vaccine adjuvants because of their ability to induce pronounced Th1 response (Hum. Vaccines 2010, 6, 1-14; Hum. Vaccines 2009, 5, 381-394).
SYNTHESIS
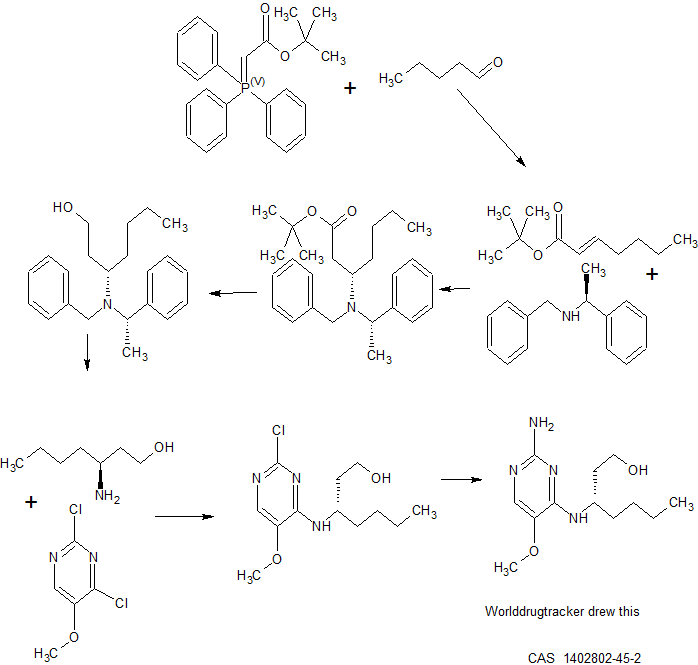
50d
patent
WO 2014053595
https://www.google.com/patents/WO2014053595A1?cl=en
Experimental Section.

B
Into a 50 ml_ vial equipped with a magnetic stir bar was placed 2,4-dichloro-5- methoxypyrimidine (2.0 g, 1 1.7 mmol), and acetonitrile (20 ml_), diisopropylethylamine (3.02 g, 23.4 mmol) and (S)-3-aminoheptanol (4.59 g, 35.1 mmol). The reaction mixture was allowed to stir 15 hours at room temperature. The solvents were removed under reduced pressure. The crude was purified via silica gel column chromatography using a dichloromethane to 10% methanol in dichloromethane gradient. The best fractions were pooled and the solvents were removed under reduced pressure to afford a white solid, B.

B C
To a thick wall glass vial equipped with a magnetic stir bar was added B (1 g, 3.66 mmol), NH3 (10 ml_, aq.), ammonium bicarbonate (3.34 g, 42.3 mmol) and copper(l) oxide (121 mg, 0.85 mmol). The vial was sealed and placed into an oil bath and heated to 150°C for 15 hours. The reaction mixture was extracted with dichloro- methane (3 x 25 ml_), the organic layers were pooled and dried over magnesium sulfate. The solids were removed by filtration and the solvents of the filtrate were removed under reduced pressure. Crude C was purified via HPLC.

c 1
C (463 mg, 1.82 mmol) was dissolved in THF (13 ml_) and cooled to -78°C. NaH (145 mg, 3.64 mmol, a 60% dispersion in mineral oil) was added in one portion and stirred at -78°C for 30 minutes. Isobutyryl chloride (389 μΙ_, 3.64 mmol) was added dropwise at -78°C and stirred for 10 minutes. The cooling bath was removed and the mixture was allowed to reach room temperature. The mixture was stirred at room temperature for 30 minutes. The mixture was quenched with water and concentrated in vacuo. The residue was purified by HPLC (RP Vydac Denali C18 10 μηι, 200 g, 5 cm, mobile phase 0.25% NH4HCO3 solution in water, acetonitrile), the desired fractions were collected, and the solvents were removed under reduced pressure to afford the pure product.
LC-MS m/z = 395 (M+H), Retention time 1.1 minutes, LC method A.
1H NMR (400 MHz, DMSO-d6) δ ppm 0.83 (t, J=6.90 Hz, 3 H) 0.98 – 1.07 (m, 12 H) 1.16 – 1.35 (m, 4 H) 1.44 – 1.62 (m, 2 H) 1.84 (q, J=6.78 Hz, 2 H) 2.45 (spt, J=7.00 Hz, 1 H) 2.96 (br. s., 1 H) 3.80 (s, 3 H) 3.92 – 4.07 (m, 2 H) 4.18 – 4.31 (m, 1 H) 6.69 (d, J=9.03 Hz, 1 H) 7.60 (s, 1 H) 9.49 (s, 1 H).
Synthetic scheme for the preparation of A


Preparati

valeraldehyde A2
To a solution of valeraldehyde (43 g, 500 mmol) in THF (1 L) was added A1 (200 g, 532 mmol) and the reaction mixture was stirred for 16 hours at room temperature. The solvents were evaporated and the residue was diluted in petroleum ether and filtered. The solvents of the filtrate were removed under reduced pressure and the residue was purified by silica chromatography using a petroleum ether to 3% ethyl acetate in petroleum ether gradient to give A2 (90 g) as a colorless oil.
1H NMR (400 MHz, CDCI3): δ ppm 6.81 -6.77 (m, 1 H), 5.68-5.64 (td, J=1.2Hz, 15.6 Hz, 1 H), 2.11-2.09 (m, 2H), 1.41 (s, 9H), 1.38-1.26 (m, 4H), 0.85-0.81 (t, J=7.2Hz, 3H).

/7-butyl lithium (290 mL, 725 mmol) was added to a stirred solution of A3 (165 g, 781 mmol) in THF (800 mL) at -78°C. The reaction mixture was stirred for 30 minutes then A2 (90 g, 488.4 mmol) in THF (400 mL) was added and the reaction was stirred for 2 hours at -78°C. The mixture was quenched with sat., aq. NH4CI solution and warmed to room temperature. The product was partitioned between ethyl acetate and water. The organic phase was washed with brine, dried and evaporated. The residue was purified by column chromatography eluting with 5% ethyl acetate in petroleum ether to afford a colorless oil, A4 (132 g).
1H NMR (400 MHz, CDCI3): δ ppm 7.36-7.16 (m, 10H), 3.75-3.70 (m, 2H), 3.43-3.39 (d, J=15.2Hz, 1 H), 3.33-3.15 (m, 1 H), 1.86-1.80 (m, 2H), 1.47-1.37 (m, 2H), 1.32 (s, 9H), 1.26-1.17 (m, 7H), 0.83-0.79 (t, J=7.2Hz, 3H).
Preparatio

A4 (130 g, 328 mmol) was dissolved in THF (1.5 L) and LAH (20 g, 526 mmol) was added at 0°C in small portions. The resulting mixture was stirred at the same temperature for 2 hours and then allowed to warm to room temperature. The mixture was quenched with a sat. aq. NH4CI solution. The product was partitioned between ethyl acetate and water. The organic phase was washed with brine, dried and evaporated. The combined organic layers were dried over sodium sulfate, the solids were removed via filtration and concentrated to afford crude A5 (100 g), which was used in the next step without further purification.
1H NMR (400 MHz, CDCI3): δ ppm 7.33-7.14 (m, 10H), 3.91 -3.86 (m, 1 H), 3.80-3.77 (d, J=13.6Hz, 1 H), 3.63-3.60 (d, J=13.6Hz, 1 H), 3.43-3.42 (m, 1 H), 3.15-3.10 (m, 1 H), 2.70-2.63 (m, 2H), 1.65-1.28 (m, 10H), 0.89-0.81 (m, 3H).
Preparation of A

A5 A
A solution of A5 (38 g, 116.75 mmol) and 10% Pd/C in methanol (200 ml_) was hydrogenated under 50 PSI hydrogen at 50°C for 24 hours. The reaction mixture was filtered and the solvent was evaporated to give A.
1H NMR (400 MHz, DMSO-d6): δ ppm 8.04 (s, 3H), 3.60-3.49 (m, 2H), 3.16-3.15 (m, 1 H), 1.71-1.67 (m, 2H), 1.60-1.55 (m, 2H), 1.33-1.26 (m, 4H), 0.90-0.87 (t, J=6.8Hz, 3H).
Analytical Method.
Compounds 1-8 in the table below were characterized by LC-MS according to the following LC-MS method.
Reverse phase UPLC (Ultra Performance Liquid Chromatography) was carried out on a bridged ethylsiloxane/silica hybrid (BEH) C18 column (1.7 μηι, 2.1 x 50 mm; Waters Acquity) with a flow rate of 0.8 ml/min. Two mobile phases (10 mM ammonium acetate in H20/acetonitrile 95/5; mobile phase B: acetonitrile) were used to run a gradient condition from 95 % A and 5 % B to 5 % A and 95 % B in 1.3 minutes and hold for 0.7 minutes. An injection volume of 0.75 μΙ_ was used. Cone voltage was 30 V for positive ionization mode and 30 V for negative ionization mode.
Paper

Toll-like receptor (TLR) 7 and 8 agonists can potentially be used in the treatment of viral infections and are particularly promising for chronic hepatitis B virus (HBV) infection. An internal screening effort identified a pyrimidine Toll-like receptor 7 and 8 dual agonist. This provided a novel alternative over the previously reported adenine and pteridone type of agonists. Structure–activity relationship, lead optimization, in silico docking, pharmacokinetics, and demonstration of ex vivo and in vivo cytokine production of the lead compound are presented.
Novel Pyrimidine Toll-like Receptor 7 and 8 Dual Agonists to Treat Hepatitis B Virus
†Janssen Infectious Diseases Diagnostics BVBA, Turnhoutseweg 30, 2340 Beerse, Belgium
‡Villapharma Research S.L., Parque Tecnológico de Fuente Álamo. Ctra. El Estrecho-Lobosillo, Km. 2.5, Av. Azul 30320 Fuente Álamo de Murcia, Murcia, Spain
J. Med. Chem., 2016, 59 (17), pp 7936–7949
DOI: 10.1021/acs.jmedchem.6b00747, http://pubs.acs.org/doi/suppl/10.1021/acs.jmedchem.6b00747
*For D.M.: phone, +32 6414 1019; E-mail, dmcgowan@its.jnj.com., *For F.H.: phone, +32 6414 1644; E-mail, fherschk@its.jnj.com.
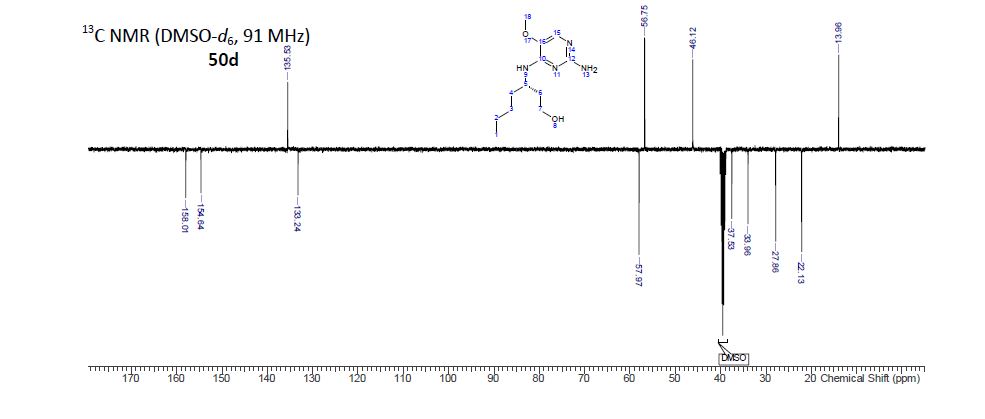
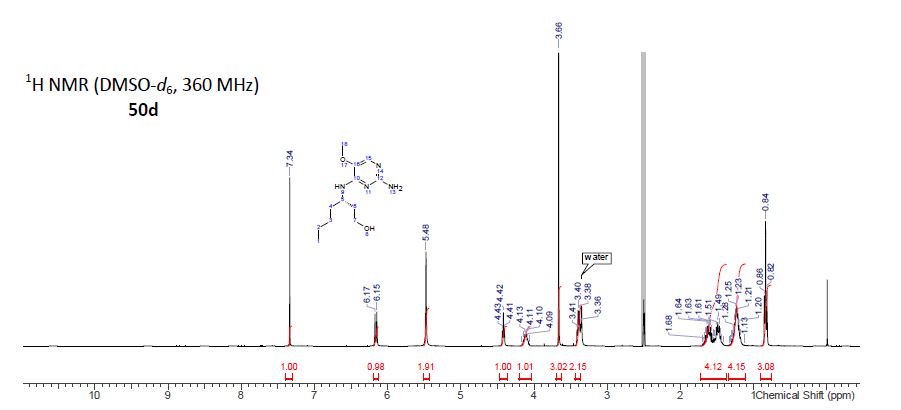
(S)-3-((2-Amino-5-methoxypyrimidin-4-yl)amino)heptan-1-ol (50d)
1H NMR (360 MHz, DMSO-d6) δ 7.34 (s, 1H), 6.16 (d, J = 9.15 Hz, 1H), 5.48 (s, 2H), 4.42 (t, J = 5.12 Hz, 1H), 4.04–4.20 (m, 1H), 3.66 (s, 3H), 3.36–3.44 (m, 2H), 1.37–1.73 (m, 4H), 1.11–1.35 (m, 4H), 0.84 (t, J = 6.95 Hz, 3H).
13C NMR (101 MHz, DMSO-d6) δ 158.0, 154.6, 135.5, 133.2, 58.0, 56.7, 46.1, 37.5, 34.0, 27.9, 22.1, 14.0.
ESI-HRMS (TOF) m/z: 255.1816 [M + H]+ (calcd for C12H22N4O2: 254.1743).
[α]D20 + 25.2 (c 0.81, DMF).
PATENT
David McGowan, Pierr Jeane-Marie Bernar Raboisson, Werner Embrechts, Tim Hugo Maria Jonckers, Stefaan Julien Last, Serge Maria Aloysius Pieters, Jaromir Vlach,
Preparation of Compounds.
Compounds of formula (I), where R1 is hydrogen atom are prepared according to scheme 1





Compounds of formula (I), when R1 is alkyl, cycloalkyl, trifluoromethyl, or alkoxy and where R2 is aromatic or aliphatic, can be prepared according scheme 4. This reaction scheme begins with a crossed-Claisen reaction where an acyl chloride reacts with ester intermediate A (shown in scheme 1) to form intermediates (G) as in scheme 3. From intermediate G, the reaction scheme follows the same pathway to the products as in scheme 3. This is a general scheme using methods known to a skilled person, see for instance The Journal of American Chemical Society volume 127, page 2854 (2005).
Synthetic Scheme for the Preparation of AA-9

A solution of AA-6 (38 g, 116.75 mmol) and 10% Pd/C in methanol (200 mL) was hydrogenated under 50 PSI hydrogen at 50° C. for 24 hours. The reaction mixture was filtered and the solvent was evaporated to give crude product AA-7 (17 g).
The crude product was dissolved in dichloromethane (200 mL), triethylamine (26.17 g, 259.1 mmol) and di-tert-butyl dicarbonate (84.7 g, 194.4 mmol) was added at 0° C. The resulting mixture was stirred at room temperature for 16 hours. The mixture was partitioned between dichloromethane and water. The organic phase was washed with brine, dried and evaporated. The residue was purified by silica gel chromatography eluting with 20% ethyl acetate in petroleum ether to give AA-8 (13 g) as colorless oil.
1H NMR (400 MHz, CDCl3): δ ppm 4.08-4.03 (br, 1H), 3.68 (m, 1H), 3.58-3.55 (m, 2H), 3.20-2.90 (br, 1H), 1.80-1.73 (m, 1H), 1.42-1.17 (m, 15H), 0.85-0.82 (t, J=6.8 Hz, 3H).
AA-8 (42 g, 0.182 mol) was dissolved in dioxane (200 mL) and dioxane/HCl (4M, 200 mL) was added at 0° C. The resulting mixture was stirred at room temperature for 2 h. The solvent was evaporated to afford the crude product. A dichloromethane/petroleum ether mixture (50 mL, 1:1, v/v) was added to the crude product, and the supernatant was decanted. This procedure was repeated two times to obtain an oil, AA-9 (26.6 g).
1H NMR (400 MHz, DMSO-d6): δ ppm 8.04 (s, 3H), 3.60-3.49 (m, 2H), 3.16-3.15 (m, 1H), 1.71-1.67 (m, 2H), 1.60-1.55 (m, 2H), 1.33-1.26 (m, 4H), 0.90-0.87 (t, J=6.8 Hz, 3H)
Procedure for Preparation of Intermediate C-1.Reaction Scheme:

A suspension of intermediate B-1 (160 g, 0.74 mol) in POCl3 (900 mL) was heated to 100° C. under N2 with stirring for 5 hours. The reaction mixture was cooled to room temperature. The excess POCl3 was removed under reduced pressure, the oil residue was poured into cold, sat. aq. NaHCO3 (2 L) that was stirred for 30 minutes. The mixture was extracted with ethyl acetate (3×1.5 L). The combined organic layers were separated and washed with brine (1 L), dried over sodium sulfate, the solids were removed via filtration, and the solvents of the filtrate were concentrated to afford intermediate C-1 (70 g) as a yellow solid. The product was used in the next step without further purification.
| Patent ID | Date | Patent Title |
|---|---|---|
| US2015274676 | 2015-10-01 | ACYLAMINOPYRIMIDINE DERIVATIVES FOR THE TREATMENT OF VIRAL INFECTIONS AND FURTHER DISEASES |
| US2014045849 | 2014-02-13 | PYRIMIDINE DERIVATIVES FOR THE TREATMENT OF VIRAL INFECTIONS |
| WO2006053109A1 * | Nov 10, 2005 | May 18, 2006 | Synta Pharmaceuticals Corp. | Heteroaryl compounds |
| WO2012136834A1 * | Apr 10, 2012 | Oct 11, 2012 | Janssen R&D Ireland | Pyrimidine derivatives for the treatment of viral infections |
| US5434157 * | Jan 21, 1993 | Jul 18, 1995 | The Upjohn Company | 6-aryl pyrimidine compounds and method for treating viral infections and inducing interferon production |
| Reference | ||
|---|---|---|
| 1 | * | ANGELICA M BELLO ET AL: “De Novo Design of Nonpeptidic Compounds Targeting the Interactions between Interferon-a and its Cognate Cell Surface Receptor“, JOURNAL OF MEDICINAL CHEMISTRY,, vol. 51, no. 9, 1 January 2008 (2008-01-01), pages 2734 – 2723, XP008157384, DOI: 10.1021/JM701182Y |
| 2 | DE CLERCQ, E.; DESCAMPS, J.; DE SOMER, P., SCIENCE, vol. 200, 1978, pages 563 – 565 | |
| 3 | FRIED: “Peginterferon-alfa plus ribavirin for chronic hepatitis C virus infection“, N ENGL J MED, vol. 347, 2002, pages 975 – 82 | |
/////////////// treatment of viral infections, Pyrimidine Toll-like Receptor 7, Toll-like Receptor 8 Dual Agonists, Treat Hepatitis B Virus, PRECLINICAL, 1402802-45-2, JNJ ?
n1c(nc(c(c1)OC)N[C@@H](CCCC)CCO)N
ORM 10921

ORM 10921
UNII-D26C95A960; D26C95A960; ORM-12741; ORM12741; ORM 12741; ORM-10921;
(1S,12bS)-1-(Methoxymethyl)-1-methyl-2,3,4,6,7,12b-hexahydro-1H-[1]benzofuro[2,3-a]quinolizine
(1S,12bS)-1-(methoxymethyl)-1-methyl-2,3,4,6,7,12b-hexahydro-[1]benzofuro[2,3-a]quinolizine
285.38, C18 H23 N O2
2H-Benzofuro[2,3-a]quinolizine, 1,3,4,6,7,12b-hexahydro-1-(methoxymethyl)-1-methyl-, (1S,12bS)-
cas 610782-82-6
| Belle David Din, Reija Jokela, Arto Tolvanen,Antti Haapalinna, Arto Karjalainen, Jukka Sallinen, Jari Ratilainen | |
| Applicant | Orion Corporation |


David Din Belle
Senior research scientist at Orion Corporation
https://fi.linkedin.com/in/david-din-belle-a2594115

Jari Ratilainen
https://fi.linkedin.com/in/jari-ratilainen-6a566218

https://fi.linkedin.com/in/reija-jokela-06499a1a
The basic drug substance candidate ORM10921 (MW = 285.38),
IUPAC name [1R*,12bR*)-(−)-1,3,4,6,7,12b-hexahydro-1-methoxymethyl-1-methyl-2H-benzofuro [2,3-a]quinolizine],
and its hydrochloric salt were synthesized by Orion Pharma, Finland.
The absolute configuration was assigned by optical rotation and later by single-crystal X-ray diffraction (see Supporting Information). The optical purity of the material was >97%.
- Originator Juvantia Pharma (CEASED); Orion
- Class Neuropsychotherapeutics
- Mechanism of Action Alpha 2c adrenergic receptor antagonists
Highest Development Phases
- Discontinued Major depressive disorder; Schizophrenia
Most Recent Events
- 10 May 2006 Discontinued – Phase-I for Schizophrenia in Finland (unspecified route)
- 10 May 2006 Discontinued – Preclinical for Depression in Finland (unspecified route)
- 15 Nov 2002 Preclinical trials in Schizophrenia in Finland (unspecified route)

Figure 1: Chemical structure of the study compound. Molecular Formula: C18H23NO2 · HCl · ½ H2O; Molecular Weights: 285.39 (free base), 321.85 (hydrochloride) 330.86 (hydrochloride hemihydrate). ORM-10921 · HCl is a single stereoisomer with the (1R*,12bR*) configuration.
The alpha adrenergic receptors can be divided on a pharmacological basis into alphal- and alpha2-adrenoceptors, which can both be further divided into subtypes. Three genetically encoded subtypes, namely alpha2A-, alpha2B- and alpha2C-adrenoceptors, have been discovered in human. Accordingly, alpha2- adrenoceptors in humans have been subdivided into three pharmacological subtypes known as alpha2A-, alpha2B- and alpha2C-adrenoceptors. A fourth, pharmacologically defined subtype, alpha2D, is known in rodents and in some other mammals, and it corresponds to the genetically defined alpha2A-adrenoceptors.
The alpha2-adrenoceptor subtypes have distinct tissue distributions and functional roles. For instance, while alpha2A-adrenoceptors are widely expressed in various tissues, alpha2C-adrenoceptors are concentrated in the CNS, and they appear to play a role in the modulation of specific CNS-mediated behavioural and physiological responses. Compounds that are non-specific to any of the above-mentioned alpha2 subtypes, and compounds that are specific to certain alpha2 subtypes, are already known. For example, atipamezole is a non-specific alpha2 antagonist. Atipamezole has been described in, for example, EP-A-183 492 (cf. p.13, compound XV) and Haapalinna, A. et al., Naunyn-Schmiedeberg’s Arch. Pharmacol. 356 (1997) 570-582. U.S. Patent No. 5,902,807 describes compounds that are selective antagonists for the alpha2C subtype and may be used in the treatment of mental illness, e.g. mental disturbance induced by stress. Such compounds include, for example, MK-912 and BAM- 1303. Furthermore, WO-A-99 28300 discloses substituted imidazole derivatives having agonist-like activity for alpha2B- or 2B/2C-adrenoceptors. hi addition, WO 01/64645 relates to derivatives of quinoline useful as alpha2 antagonists, as well as to selective alpha2C antagonist agents. The disclosures of all documents cited above in this paragraph are incorporated by reference herein.
Several arylquinolizine derivatives and related compounds have been described in the literature, some of which possess valuable pharmaceutical effects. For example, U.S. Patents No. 4,806,545 and 4,044,012 describe 1,1-disubstituted indolo[2,3-«]quinolizidines useful as vasodilators and antihypoxic agents. Further, substituted arylquinolizine derivatives, described for example in U.S. Patent No. 4,686,226 possessing alpha2-adrenoceptor antagonistic activity are useful for example as antidepressant, antihypertensive, or antidiabetic agents or platelet aggregation inhibitors. In addition, U.S. Patent No. 3,492,303 relates to indolo[2,3- α]quinolizidines useful as central nervous system depressants.
PATENT
WO 2003082866
https://www.google.com/patents/WO2003082866A1?cl=en
///////////
CC1(CCCN2C1C3=C(CC2)C4=CC=CC=C4O3)COC
How to document a Product Transfer? Example templates!
DRUG REGULATORY AFFAIRS INTERNATIONAL

All participants of the GMP training course “Product Transfer” will receive a special version of the Guideline Manager CD including documents and templates useable for site change projects.
Click
http://www.gmp-compliance.org/eca_mitt_05359_15221,Z-PEM_n.html
According to the European GMP-Rules, written procedures for tranfser activities and their documentation are required. For example, a Transfer SOP, a transfer plan and a report are now mandatory and will be checked during inspections.
As participant of the GMP education course “Product Transfer” in Berlin, from 25-27 October 2016 you will receive a special version of the Guideline Manager CD with a special section concerning product transfers. This section contains, amongst others, a Transfer SOP and a template for a Transfer Plan. Both documents are in Word format and can immediately be used after adoption to your own situation.

Regulatory Guidance Documents like the WHO guideline on transfer of technology in pharmaceutical manufacturing and the EU/US…
View original post 36 more words
Analytical Lifecycle: USP “Statistical Tools”, Analytical Target Profile and Analytical Control Strategy
DRUG REGULATORY AFFAIRS INTERNATIONAL

The United States Pharmacopeia (USP) is currently undertaking further steps towards a comprehensive analytical lifecycle approach by publishing a draft of a new General Chapter <1210> Statistical Tools for Procedure Validation and two Stimuli Articles regarding Analytical Target Profile and AnalyticalControl Strategy in Pharmacopeial Forum. Read more about the life cycle concept for analytical procedures.
Following the recently announced elaboration of a new general chapter <1220> “The Analytical Procedure Lifecycle” the United States pharmacopeia (USP) is now proceeding in its approach for a comprehensive analytical lifecycle concept. A further step towards this approach is the draft of a new USP General Chapter <1210> Statistical Tools for Procedure Validation which has been published in Pharmacopeial Forum (PF) 42(5) in September 2016. Comment deadline is November 30, 2016.
Additionally, two Stimuli Articles regarding “Analytical Control Strategy” and “Analytical…
View original post 624 more words
Ibipinabant Revisited

Ibipinabant
| cas 464213-10-3; UNII-O5CSC6WH1T; BMS-646256; SLV-319; |
| Molecular Formula: | C23H20Cl2N4O2S |
|---|---|
| Molecular Weight: | 487.4015 g/mol |
(4S)-5-(4-chlorophenyl)-N-(4-chlorophenyl)sulfonyl-N’-methyl-4-phenyl-3,4-dihydropyrazole-2-carboximidamide
1H-Pyrazole-1-carboximidamide, 3-(4-chlorophenyl)-N’-[(4-chlorophenyl)sulfonyl]-4,5-dihydro-N-methyl-4-phenyl-, (4S)-
(4S)-3-(4-Chlorophenyl)-N-[(4-chlorophenyl)sulfonyl]-4,5-dihydro-N’-methyl-4-phenyl-1H-pyrazole-1-carboximidamide
1H-Pyrazole-1-carboximidamide, 3-(4-chlorophenyl)-N-[(4-chlorophenyl)sulfonyl]-4,5-dihydro-N‘-methyl-4-phenyl-, (4S)-
(-)-(4S)-N-Methyl-N’-((4-chlorophenyl)sulfonyl)-3-(4-chlorophenyl)-4,5-dihydro-4-phenyl-1 H-pyrazole-1 -carboxamidine
4S)-(−)-3-(4-Chlorophenyl)-N-methyl-N‘-[(4-chlorophenyl)sulfonyl]-4-phenyl-4,5-dihydro-1H-pyrazole-1-carboxamidine
It was originally developed by Solvay, which was acquired by Abbott in 2010.
SLV 319, UNII:O5CSC6WH1T, (S)-SLV 319, BMS 646256, JD 5001
- Originator Solvay
- Class Antipsychotics; Imides; Obesity therapies; Pyrazoles; Small molecules; Sulfonamides
- Mechanism of ActionCannabinoid receptor CB1 antagonists
Ibipinabant, also known as BMS-646256, JD-5001 and SLV-319, is a potent and highly selective CB1 antagonist. It has potent anorectic effects in animals, and was researched for the treatment of obesity, although CB1 antagonists as a class have now fallen out of favour as potential anorectics following the problems seen with rimonabant, and so ibipinabant is now only used for laboratory research, especially structure-activity relationship studies into novel CB1 antagonists
Ibipinabant (SLV319, BMS-646,256) is a drug used in scientific research which acts as a potent and highly selective CB1antagonist.[1] It has potent anorectic effects in animals,[2] and was researched for the treatment of obesity, although CB1 antagonists as a class have now fallen out of favour as potential anorectics following the problems seen with rimonabant, and so ibipinabant is now only used for laboratory research, especially structure-activity relationship studies into novel CB1 antagonists.[3][4][5]

| Inventors | Josephus H.M. Lange, Cornelis G Kruse,Jacobus Tipker, Jan Hoogendoorn |
| Applicant | Solvay Pharmaceuticals B.V. |
PATENT
WO 2002076949
https://www.google.com/patents/WO2002076949A1?cl=en
Example IV
(-)-(4S)-N-methyl-N’-((4-chlorophenyl)sulfonyl)-3-(4-chlorophenyl)-4,5- dihydro-4-phenyl-1 H-pyrazole-1 -carboxamidine
(-)-(4S)-N-Methyl-N’-((4-chlorophenyl)sulfonyl)-3-(4-chlorophenyl)-4,5-dihydro-4-phenyl-1 H-pyrazole-1 -carboxamidine (7.16 gram, 0.0147 mol)) ([α25 D] = -150°, c = 0.01 , MeOH) (melting point: 169-170 °C) was obtained via chiral chromatographic separation of racemic N-methyl-N’-((4-chlorophenyl)sulfonyl)-3- (4-chlorophenyl)-4,5-dihydro-4-phenyl-1 H-pyrazole-1 -carboxamidine (18 gram, 0.037 mol) using a Chiralpak AD, 20 μm chiral stationary phase. The mobile phase consisted of a mixture of hexane/ethanol (80/20 (v/v)) and 0.1 % ammonium hydroxide (25 % aqueous solution).
Example III N-Methyl-N’-((4-chlorophenyl)sulfonyl)-3-(4-chlorophenyl)-4,5-dihydro-4- phenyl-1 H-pyrazole-1 -carboxamidine
Part A: To a solution of N-((4-chlorophenyl)sulfonyl)carbamic acid methyl ester (CAS: 34543-04-9) (2.99 gram, 12.0 mmol) and pyridine (4 ml) in 1 ,4-dioxane (20 ml) is added 3-(4-chlorophenyl)-4,5-dihydro-4-phenyl-1 H-pyrazole (3.39 gram, 13.2 mmol) and the resulting mixture is stirred for 4 hours at 100 °C After concentration in vacuo the residue is dissolved in dichloromethane, successively washed with water, 1 N HCI and water, dried over anhydrous Na2SO4, filtered and concentrated in vacuo to a volume of 20 ml. Methyl-tert-butyl ether (60 ml) is added and the resulting solution is concentrated to a volume of 20 ml. The formed crystals are collected by filtration and recrystallised from methyl-te/τ-butyl ether to give 3-(4-chlorophenyl)-N-((4-chlorophenyl)sulfonyl)-4,5-dihydro-4-phenyl-1 H- pyrazole-1-carboxamide (4J5 gram, 76 % yield) Melting point: 211-214 °C
Part B: A mixture of 3-(4-chlorophenyl)-N-((4-chlorophenyl)sulfonyl)-4,5-dihydro- 4-phenyl-1 H-pyrazole-1 -carboxamide (3.67 gram, 7J5 mmol) and phosphorus pentachloride (1.69 gram, 8.14 mmol) in chlorobenzene (40 ml) is heated at reflux for 1 hour. After thorough concentration in vacuo, the formed N-((4- chlorophenyl)sulfonyl)-3-(4-chlorophenyl)-4,5-dihydro-4-phenyl-1 H-pyrazole-1- carboximidoyl chloride is suspended in dichloromethane and reacted with cold methylamine (1.5 ml). After stirring at room temperature for 1 hour, the mixture is concentrated in vacuo. The residue is crystallised from diethyl ether to give N-methyl-N’-((4-chlorophenyl)sulfonyl)-3-(4-chlorophenyl)-4,5-dihydro-4-phenyl- 1 H-pyrazole-1 -carboxamidine (2.29 gram, 61 % yield). Melting point: 96-98 °C(dec).
PATENT
WO 2008074816
https://google.com/patents/WO2008074816A1?cl=en
PAPER
An expedient atom-efficient synthesis of the cannabinoid CB1receptor inverse agonist ibipinabant
- Abbott Healthcare Products B.V., Chemical Design & Synthesis Unit, C.J. van Houtenlaan 36, 1381 CP Weesp, The Netherlands
http://www.sciencedirect.com/science/article/pii/S0040403911000955
http://dx.doi.org/10.1016/j.tetlet.2011.01.068

A novel synthetic route to the highly selective and orally active cannabinoid CB1 receptor inverse agonist ibipinabant is described which combines the use of inexpensive, commercially available reagents and mild reaction conditions with a high degree of atom-efficiency. The method is expected to enable the rapid synthesis of a variety of sulfonylguanidines.
PAPER
JD-5006 and JD-5037: Peripherally restricted (PR) cannabinoid-1 receptor blockers related to SLV-319 (Ibipinabant) as metabolic disorder therapeutics devoid of CNS liabilities
- Jenrin Discovery, 2515 Lori Lane North, Wilmington, DE 19810, USA
http://dx.doi.org/10.1016/j.bmcl.2012.08.004
http://www.sciencedirect.com/science/article/pii/S0960894X12009936

- Scheme 1.
Reagents and conditions: (a) ArylSO2NHCO2CH3, toluene, reflux, 3–6 h, 30–95%; (b) PCl5, chlorobenzene, reflux, 2–4 h, 50–90%; (c) NH2Me.HCl, Et3N, CH2Cl2, ice-bath to rt, 16–20 h, 30–70%; (d) X = CO2Et/X′ = CO2H: LiOH, H2O/THF(1:3), 50%; X′ = CO2H/X′ = CONH2, (1) IBCF, NMM, CH2Cl2, (2) NH3/THF, 60%; X′ = CO2H/X′ = CONHOH, (1) IBCF, NMM, CH2Cl2, (2) NH2OH·HCl, NMM, CH2Cl2, 40%; X = OCH2CO2Et/OCH2CO2H: LiOH, H2O/THF(1:3), rt, 50%; X′ = OCH2CO2H/X′ = OCH2CONH2: (1) IBCF, NMM, CH2Cl2, (2) NH3/THF, 30%; X = NO2:/X′ = NHCOCH3: (1) Fe/HCl, EtOH, H2O, reflux 1–2 h, 90%; (2) Ac2O, pyr, CH2Cl2, rt, 8 h, 90%; X = NO2:/X′ = NHCO2Et: (1) Fe/HCl, EtOH, H2O, reflux 1–2 h, 90%; (2) ClCO2Et, pyr, CH2Cl2, 0 °C to rt, 8 h, 90%
Clip
http://molpharm.aspetjournals.org/content/87/2/197.full.pdf
Paper
Lange et al (2005) Novel 3,4-diarylpyrazolines as potent cannabinoid CB1 receptor antagonists with lower lipophilicity. Bioorg.Med.Chem.Lett. 15 4794. PMID: 16140010.
http://www.sciencedirect.com/science/article/pii/S0960894X05010139
http://dx.doi.org/10.1016/j.bmcl.2005.07.054
Paper
Lange et al (2004) Synthesis, biological properties, and molecular modeling investigations of novel 3,4-diarylpyrazolines as potent and selective CB1 cannabinoid receptor antagonists. J.Med.Chem. 47 627. PMID:14736243.
A series of novel 3,4-diarylpyrazolines was synthesized and evaluated in cannabinoid (hCB1 and hCB2) receptor assays. The 3,4-diarylpyrazolines elicited potent in vitroCB1 antagonistic activities and in general exhibited high CB1 vs CB2 receptor subtype selectivities. Some key representatives showed potent pharmacological in vivo activities after oral dosing in both a CB agonist-induced blood pressure model and a CB agonist-induced hypothermia model. Chiral separation of racemic 67, followed by crystallization and an X-ray diffraction study, elucidated the absolute configuration of the eutomer 80 (SLV319) at its C4 position as 4S. Bioanalytical studies revealed a high CNS−plasma ratio for the development candidate 80. Molecular modeling studies showed a relatively close three-dimensional structural overlap between 80 and the known CB1 receptor antagonist rimonabant (SR141716A). Further analysis of the X-ray diffraction data of 80 revealed the presence of an intramolecular hydrogen bond that was confirmed by computational methods. Computational models and X-ray diffraction data indicated a different intramolecular hydrogen bonding pattern in the in vivo inactive compound 6. In addition, X-ray diffraction studies of 6 revealed a tighter intermolecular packing than 80, which also may contribute to its poorer absorption in vivo. Replacement of the amidine -NH2 moiety with a -NHCH3 group proved to be the key change for gaining oral biovailability in this series of compounds leading to the identification of 80

4S)-(−)-3-(4-Chlorophenyl)-N-methyl-N‘-[(4-chlorophenyl)sulfonyl]-4-phenyl-4,5-dihydro-1H-pyrazole-1-carboxamidine (80) and (4R)-(+)-3-(4-chlorophenyl)-N-methyl-N‘-[(4-chlorophenyl)sulfonyl]-4-phenyl-4,5-dihydro-1H-pyrazole-1-carboxamidine (81). Chiral preparative HPLC separation of racemic 67 (18 g, 0.037 mol) using a Chiralpak AD, 20 μm chiral stationary phase yielded 80 (7.16 g, 0.0147 mol) and 81 (7.46 g, 0.0153 mol), respectively. The mobile phase consisted of a mixture of n-hexane/ethanol (80/20 (v/v)) and 0.1% NH4OH (25% aqueous solution).
DESIRED
80: [  ] = −150°, c = 0.01, MeOH; mp 171−172 °C;
] = −150°, c = 0.01, MeOH; mp 171−172 °C;
] = −150°, c = 0.01, MeOH; mp 171−172 °C; 1H NMR (400 MHz, DMSO-d6) δ 2.94 (d, J = 4 Hz, 3H), 3.96 (dd, J = 11 and 4 Hz, 1H), 4.46 (t, J = 11 Hz, 1H), 5.05 (dd, J = 11 and 4 Hz, 1H), 7.20−7.35 (m, 5H), 7.45 (dt, J = 8 and 2 Hz, 2H), 7.53 (dt, J = 8 and 2 Hz, 2H), 7.77 (dt, J = 8 and 2 Hz, 2H), 7.82 (dt, J = 8 and 2 Hz, 2H), 8.19 (br d, J = 4 Hz, 1H);
HRMS (C23H21Cl2N4O2S) [M+H]+: found m/z 487.0768, calcd 487.0762. Anal. (C23H20Cl2N4O2S) C, H, N.
UNDESIRED
81: [  ] = + 150°, c = 0.01, MeOH; mp 171−172 °C; 1H NMR (400 MHz, DMSO-d6) δ 2.94 (d, J = 4 Hz, 3H), 3.96 (dd, J = 11 and 4 Hz, 1H), 4.46 (t, J = 11 Hz, 1H), 5.05 (dd, J = 11 and 4 Hz, 1H), 7.20−7.35 (m, 5H), 7.45 (dt, J = 8 and 2 Hz, 2H), 7.53 (dt, J = 8 and 2 Hz, 2H), 7.77 (dt,J = 8 and 2 Hz, 2H), 7.82 (dt, J = 8 and 2 Hz, 2H), 8.19 (br d, J = 4 Hz, 1H); HRMS (C23H21Cl2N4O2S) [M+H]+: found m/z 487.0749, calcd 487.0762. Anal. (C23H20Cl2N4O2S) C, H, N.
] = + 150°, c = 0.01, MeOH; mp 171−172 °C; 1H NMR (400 MHz, DMSO-d6) δ 2.94 (d, J = 4 Hz, 3H), 3.96 (dd, J = 11 and 4 Hz, 1H), 4.46 (t, J = 11 Hz, 1H), 5.05 (dd, J = 11 and 4 Hz, 1H), 7.20−7.35 (m, 5H), 7.45 (dt, J = 8 and 2 Hz, 2H), 7.53 (dt, J = 8 and 2 Hz, 2H), 7.77 (dt,J = 8 and 2 Hz, 2H), 7.82 (dt, J = 8 and 2 Hz, 2H), 8.19 (br d, J = 4 Hz, 1H); HRMS (C23H21Cl2N4O2S) [M+H]+: found m/z 487.0749, calcd 487.0762. Anal. (C23H20Cl2N4O2S) C, H, N.
] = + 150°, c = 0.01, MeOH; mp 171−172 °C; 1H NMR (400 MHz, DMSO-d6) δ 2.94 (d, J = 4 Hz, 3H), 3.96 (dd, J = 11 and 4 Hz, 1H), 4.46 (t, J = 11 Hz, 1H), 5.05 (dd, J = 11 and 4 Hz, 1H), 7.20−7.35 (m, 5H), 7.45 (dt, J = 8 and 2 Hz, 2H), 7.53 (dt, J = 8 and 2 Hz, 2H), 7.77 (dt,J = 8 and 2 Hz, 2H), 7.82 (dt, J = 8 and 2 Hz, 2H), 8.19 (br d, J = 4 Hz, 1H); HRMS (C23H21Cl2N4O2S) [M+H]+: found m/z 487.0749, calcd 487.0762. Anal. (C23H20Cl2N4O2S) C, H, N.Paper
Org. Process Res. Dev., 2012, 16 (4), pp 567–576
DOI: 10.1021/op2003024, http://pubs.acs.org/doi/full/10.1021/op2003024
Modeling-Based Approach Towards Quality by Design for the Ibipinabant API Step
This work presents a process modeling-based methodology towards quality by design that was applied throughout the development lifecycle of the ibipinabant API step. By combining mechanistic kinetic modeling with fundamental thermodynamics, the degradation of the API enantiomeric purity was described across a large multivariate process knowledge space. This knowledge space was then narrowed down to the process design space through risk assessment, target quality specifications, practical operating conditions for scale-up, and plant control capabilities. Subsequent analysis of process throughput and yield defined the target operating conditions and normal operating ranges for a specific pilot-plant implementation. Model predictions were verified via results obtained in the laboratory and at pilot-plant scale. Future efforts were focused on increasing fundamental process knowledge, improving model confidence, and using a risk-based approach to reevaluate the design space and selected operating conditions for the next scale-up campaign.
API process at the time of the first pilot-plant campaign

changed to

Process for the second pilot-plant implementation
Process parameter ranges and typical results from approximately 20 lab experiments conducted on the process shown in Scheme



Figure 3. Ishikawa diagram for the API step, highlighting factors that potentially affect the enantiomeric purity of the product. Factors shown in blue were accounted for in the sulfonylation reaction and distillative crystallization models. Factors shown in red were not included in the models
table 3. Process parameter ranges and number of parameter levels utilized for model-based prediction of sulfonylation reaction conversion and degradation of API enantiopurity during the distillative crystallization
| process parameter | min. value | max. value | # of “levels” |
|---|---|---|---|
| sulfonylation reaction model | |||
| temp. (°C) | 5 | 35 | 7 |
| 4-chlorobenzenesulfonyl chloride (equiv) | 1.0 | 1.2 | 6 |
| conc. (mL/g) | 5 | 10 | 6 |
| reaction time (h) | 2 | 5 | 4 |
| distillative crystallization model | |||
| pressure (mbar) | 300 | 1013 | 6 |
| residual 2(AP) | 0.05 | 2.0 | 6 |
| distillation time (h) | 8 | 48 | 4 |
| distillation end point (wt % EtOH) | 90 | 98 | 3 |
REFERENCES
1: Schirris TJ, Ritschel T, Herma Renkema G, Willems PH, Smeitink JA, Russel FG. Mitochondrial ADP/ATP exchange inhibition: a novel off-target mechanism underlying ibipinabant-induced myotoxicity. Sci Rep. 2015 Sep 29;5:14533. doi: 10.1038/srep14533. PubMed PMID: 26416158; PubMed Central PMCID: PMC4586513.
2: Chorvat RJ, Berbaum J, Seriacki K, McElroy JF. JD-5006 and JD-5037: peripherally restricted (PR) cannabinoid-1 receptor blockers related to SLV-319 (Ibipinabant) as metabolic disorder therapeutics devoid of CNS liabilities. Bioorg Med Chem Lett. 2012 Oct 1;22(19):6173-80. doi: 10.1016/j.bmcl.2012.08.004. Epub 2012 Aug 20. PubMed PMID: 22959249.
3: Tomlinson L, Tirmenstein MA, Janovitz EB, Aranibar N, Ott KH, Kozlosky JC, Patrone LM, Achanzar WE, Augustine KA, Brannen KC, Carlson KE, Charlap JH, Dubrow KM, Kang L, Rosini LT, Panzica-Kelly JM, Flint OP, Moulin FJ, Megill JR, Zhang H, Bennett MJ, Horvath JJ. Cannabinoid receptor antagonist-induced striated muscle toxicity and ethylmalonic-adipic aciduria in beagle dogs. Toxicol Sci. 2012 Oct;129(2):268-79. doi: 10.1093/toxsci/kfs217. Epub 2012 Jul 21. PubMed PMID: 22821849.
4: Dawes J, Allenspach C, Gamble JF, Greenwood R, Robbins P, Tobyn M. Application of external lubrication during the roller compaction of adhesive pharmaceutical formulations. Pharm Dev Technol. 2013 Feb;18(1):246-56. doi: 10.3109/10837450.2012.705299. Epub 2012 Jul 20. PubMed PMID: 22813432.
5: Leane MM, Sinclair W, Qian F, Haddadin R, Brown A, Tobyn M, Dennis AB. Formulation and process design for a solid dosage form containing a spray-dried amorphous dispersion of ibipinabant. Pharm Dev Technol. 2013 Mar-Apr;18(2):359-66. doi: 10.3109/10837450.2011.619544. Epub 2012 Jan 23. PubMed PMID: 22268601.
6: Rohrbach K, Thomas MA, Glick S, Fung EN, Wang V, Watson L, Gregory P, Antel J, Pelleymounter MA. Ibipinabant attenuates β-cell loss in male Zucker diabetic fatty rats independently of its effects on body weight. Diabetes Obes Metab. 2012 Jun;14(6):555-64. doi: 10.1111/j.1463-1326.2012.01563.x. Epub 2012 Feb 24. PubMed PMID: 22268426.
7: Lynch CJ, Zhou Q, Shyng SL, Heal DJ, Cheetham SC, Dickinson K, Gregory P, Firnges M, Nordheim U, Goshorn S, Reiche D, Turski L, Antel J. Some cannabinoid receptor ligands and their distomers are direct-acting openers of SUR1 K(ATP) channels. Am J Physiol Endocrinol Metab. 2012 Mar 1;302(5):E540-51. doi: 10.1152/ajpendo.00258.2011. Epub 2011 Dec 13. PubMed PMID: 22167524; PubMed Central PMCID: PMC3311290.
8: Gamble JF, Leane M, Olusanmi D, Tobyn M, Supuk E, Khoo J, Naderi M. Surface energy analysis as a tool to probe the surface energy characteristics of micronized materials–a comparison with inverse gas chromatography. Int J Pharm. 2012 Jan 17;422(1-2):238-44. doi: 10.1016/j.ijpharm.2011.11.002. Epub 2011 Nov 10. PubMed PMID: 22100516.
9: Sinclair W, Leane M, Clarke G, Dennis A, Tobyn M, Timmins P. Physical stability and recrystallization kinetics of amorphous ibipinabant drug product by fourier transform raman spectroscopy. J Pharm Sci. 2011 Nov;100(11):4687-99. doi: 10.1002/jps.22658. Epub 2011 Jun 16. PubMed PMID: 21681752.
10: Gamble JF, Tobyn M, Dennis AB, Shah T. Roller compaction: application of an in-gap ribbon porosity calculation for the optimization of downstream granule flow and compactability characteristics. Pharm Dev Technol. 2010 Jun;15(3):223-9. doi: 10.3109/10837450903095342. PubMed PMID: 22716462.
11: Zhang H, Patrone L, Kozlosky J, Tomlinson L, Cosma G, Horvath J. Pooled sample strategy in conjunction with high-resolution liquid chromatography-mass spectrometry-based background subtraction to identify toxicological markers in dogs treated with ibipinabant. Anal Chem. 2010 May 1;82(9):3834-9. doi: 10.1021/ac100287a. PubMed PMID: 20387806.
12: Lange JH, van der Neut MA, den Hartog AP, Wals HC, Hoogendoorn J, van Stuivenberg HH, van Vliet BJ, Kruse CG. Synthesis, SAR and intramolecular hydrogen bonding pattern of 1,3,5-trisubstituted 4,5-dihydropyrazoles as potent cannabinoid CB(1) receptor antagonists. Bioorg Med Chem Lett. 2010 Mar 1;20(5):1752-7. doi: 10.1016/j.bmcl.2010.01.049. Epub 2010 Jan 20. PubMed PMID: 20137935.
References
- Lange, JH; Coolen, HK; Van Stuivenberg, HH; Dijksman, JA; Herremans, AH; Ronken, E; Keizer, HG; Tipker, K; et al. (2004). “Synthesis, biological properties, and molecular modeling investigations of novel 3,4-diarylpyrazolines as potent and selective CB(1) cannabinoid receptor antagonists”. Journal of Medicinal Chemistry. 47 (3): 627–43. doi:10.1021/jm031019q. PMID 14736243.
- Need, AB; Davis, RJ; Alexander-Chacko, JT; Eastwood, B; Chernet, E; Phebus, LA; Sindelar, DK; Nomikos, GG (2006). “The relationship of in vivo central CB1 receptor occupancy to changes in cortical monoamine release and feeding elicited by CB1 receptor antagonists in rats”.Psychopharmacology. 184 (1): 26–35. doi:10.1007/s00213-005-0234-x. PMID 16328376.
- Lange, JH; Van Stuivenberg, HH; Veerman, W; Wals, HC; Stork, B; Coolen, HK; McCreary, AC; Adolfs, TJ; Kruse, CG (2005). “Novel 3,4-diarylpyrazolines as potent cannabinoid CB1 receptor antagonists with lower lipophilicity”. Bioorganic & Medicinal Chemistry Letters. 15 (21): 4794–8. doi:10.1016/j.bmcl.2005.07.054. PMID 16140010.
- Srivastava, BK; Joharapurkar, A; Raval, S; Patel, JZ; Soni, R; Raval, P; Gite, A; Goswami, A; et al. (2007). “Diaryl dihydropyrazole-3-carboxamides with significant in vivo antiobesity activity related to CB1 receptor antagonism: synthesis, biological evaluation, and molecular modeling in the homology model”. Journal of Medicinal Chemistry. 50 (24): 5951–66. doi:10.1021/jm061490u. PMID 17979261.
- Srivastava, BK; Soni, R; Joharapurkar, A; Sairam, KV; Patel, JZ; Goswami, A; Shedage, SA; Kar, SS; et al. (2008). “Bioisosteric replacement of dihydropyrazole of 4S-(−)-3-(4-chlorophenyl)-N-methyl-N’-(4-chlorophenyl)-sulfonyl-4-phenyl-4,5-dihydro-1H-pyrazole-1-caboxamidine (SLV-319) a potent CB1 receptor antagonist by imidazole and oxazole”. Bioorganic & Medicinal Chemistry Letters. 18 (3): 963–8. doi:10.1016/j.bmcl.2007.12.036. PMID 18207393.
| Patent ID | Date | Patent Title |
|---|---|---|
| US9174965 | 2015-11-03 | Pyrimidinylpiperidinyloxypyridone analogues as GPR119 modulators |
| US2015133479 | 2015-05-14 | PYRIMIDINYLPIPERIDINYLOXYPYRIDONE ANALOGUES AS GPR119 MODULATORS |
| US8940716 | 2015-01-27 | Bicyclic heteroaryl compounds as GPR119 modulators |
| US8853205 | 2014-10-07 | Heteropyrrole analogs acting on cannabinoid receptors |
| US8729084 | 2014-05-20 | Benzofuranyl analogues as GPR119 modulators |
| US2014080788 | 2014-03-20 | NOVEL BICYCLIC NITROGEN CONTAINING HETEROARYL TGR5 RECEPTOR MODULATORS |
| US8513265 | 2013-08-20 | [6, 6] and [6, 7]-bicyclic GPR119 G protein-coupled receptor agonists |
| US8513424 | 2013-08-20 | Pyridone GPR119 G protein-coupled receptor agonists |
| US8476283 | 2013-07-02 | [6, 5]â??bicyclic GPR119 G protein-coupled receptor agonists |
| US8314095 | 2012-11-20 | [6, 6] and [6, 7]-bicyclic GPR119 G protein-coupled receptor agonists |
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
4S-(−)-3-(4-chlorophenyl)-N-methyl-N’-[(4-chlorophenyl)-sulfonyl]-4-phenyl-4,5-dihydro-1H-pyrazole-1-carboxamidine
|
|
| Identifiers | |
| CAS Number | 464213-10-3 |
| ATC code | none |
| PubChem | CID 9826744 |
| ChemSpider | 24765166 |
| UNII | O5CSC6WH1T |
| KEGG | D09349 |
| ChEMBL | CHEMBL158784 |
| Chemical data | |
| Formula | C24H22Cl2N4O2S |
| Molar mass | 501.427 |
///////// 464213-10-3, UNII-O5CSC6WH1T, BMS-646256, SLV-319, Ibipinabant, JD 5001, solvay, abbott
c2cc(Cl)ccc2C1=NN(C(NC)=NCS(=O)(=O)c3ccc(Cl)cc3)CC1c4ccccc4
IPI-549

IPI-549
CAS 1693758-51-8
MF : C30H24N8O2
Molecular Weight: 528.576
(S)-2-amino-N-(1-(8-((1-methyl-1H-pyrazol-4-yl)ethynyl)-1-oxo-2-phenyl-1,2-dihydroisoquinolin-3-yl)ethyl)pyrazolo[1,5-a]pyrimidine-3-carboxamide
2-amino-N-[(1S)-1-[8-[2-(1-methylpyrazol-4-yl)ethynyl]-1-oxo-2-phenylisoquinolin-3-yl]ethyl]pyrazolo[1,5-a]pyrimidine-3-carboxamide
| Latest Stage of Development | Phase I |
| Standard Indication | Solid tumors |
| Indication Details | Treat solid tumors |
- Originator Intellikine
- Developer Infinity Pharmaceuticals
- ClassAntineoplastics; Small molecules
- Mechanism of ActionPhosphatidylinositol 3 kinase delta inhibitors; Phosphatidylinositol 3 kinase gamma inhibitors
- Phase I Solid tumours

Most Recent Events
- 18 Apr 2016 Pharmacodynamics data from a preclinical study in Solid tumours presented at the 107th Annual Meeting of the American Association for Cancer Research (AACR-2016)
- 01 Dec 2015 Phase-I clinical trials in Solid tumours (Monotherapy, Combination therapy, Late-stage disease, Second-line therapy or greater) in USA (PO)
IPI-549 is a potent and selective phosphoinositide-3-kinase (PI3Kγ) Inhibitor as an Immuno-Oncology Clinical Candidate (Kd = 0.29 nM). Bioactivity data of IPI-549: biochemcial IC50 (nM) for PI3K isoform: 3200 (α); 3500 (β); 16 (γ); and >8400 (δ) respectively. Cellar IC50 (nM) of IPI549 for PI3K isoform: 250 (α); 240 (β); 1.6 (γ); and 180 (δ) respectively. IPI-549 shows >100-fold selectivity over other lipid and protein kinases. IPI-549 demonstrates favorable pharmacokinetic properties and robust inhibition of PI3K-γ mediated neutrophil migration in vivo and is currently in Phase 1 clinical evaluation in subjects with advanced solid tumors.
Patent
WO 2015051244
https://www.google.co.in/patents/WO2015051244A1?cl=en
Scheme 1

Scheme 2

Example 1

[00657] Compound 4 was prepared in 3 steps from compound A according to the following procedures:
Compound A was prepared according to Method A. It was coupled to 2-((tert-butoxycarbonyl)amino)pyrazolo[l,5-a]pyrimidine-3-carboxylic acid according to the following procedure: Compound A (27.4 mmol, 1.0 equiv), HOBt hydrate (1.2 equiv), 2-((tert-butoxycarbonyl)amino)pyrazolo[l,5-a]pyrimidine-3-carboxylic acid (1.05 equiv) and
EDC (1.25 equiv) were added to a 200 mL round bottomed flask with a stir bar. N,N-Dimethylformamide (50 mL) was added and the suspension was stirred at RT for 2 min. Hunig’s base (4.0 equiv) was added and after which the suspension became homogeneous and was stirred for 22h resulting in the formation of a solid cake in the reaction flask. The solid mixture was added to water (600 mL) and stirred for 3h. The resulting cream colored solid was filtered and washed with water (2 x 100 mL) and dried. The solid was then dissolved in methylene chloride (40 mL) after which trifluoroacetic acid (10 equiv, 20 mL) was added and the reaction was stirred for 30 min at RT after which there is no more starting material by LC/MS analysis. The solution was then concentrated and coevaporated with a mixture of methylene choride/ethanol (1 : 1 v/v) and then dried under high vacuum overnight. The resulting solid was triturated with 60 mL of ethanol for lh and then collected via vacuum filtration. The beige solid was then neutralized with sodium carbonate solution (100 mL) and then transferred to a separatory funnel with methylene chloride (350 mL). The water layer was extracted with an additional 100 mL of methylene chloride. The combined organic layers were dried over sodium sulfate, filtered and concentrated under vacuum to provide a pale yellow solid that was purified using flash silica gel chromatography (Combiflash, 24g column, gradient of 0-5% methanol/methylene chloride) to provide amide B. ESI-MS m/z: 459.4 [M+H]+.
[00658] Amide B was placed in a sealed tube (0.67 mmol, 1.0 equiv) followed by dichlorobis(acetonitrile)palladium (15 mol%), X-Phos (45 mol%), and cesium carbonate (3.0 equiv) Propionitrile (5 mL) was added and the mixture was bubbled with Ar for 1 min. 4-Ethynyl-l -methyl- lH-pyrazole (1.24 equiv) was added and the resulting orange mixture was sealed and stirred in an oil bath at 85 oC for 1.5h. The resulting brownish-black mixture was allowed to cool at which point there was no more SM by LC/MS analysis. The mixture was then filtered through a short plug of cotton using acetonitrile and methylene chloride. The combined filtrates were concentrated onto silica gel and purified using flash silica gel chromatography (Combiflash, 4g column, gradient of 0-5% methylene chloride/methanol). The resulting material was further purified by reverse phase HPLC (15-90%o acetonitrile with 0.1%o formic acid/water with 0.1%o formic water) to provide desired compound 4. ESI-MS m/z: 529.5 [M+H]+.
PAPER
WO 2015143012
https://www.google.com/patents/WO2015143012A1?cl=en
PAPER
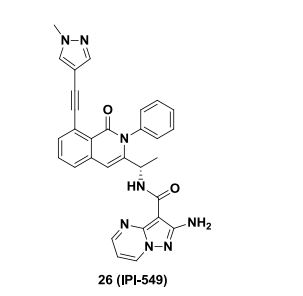
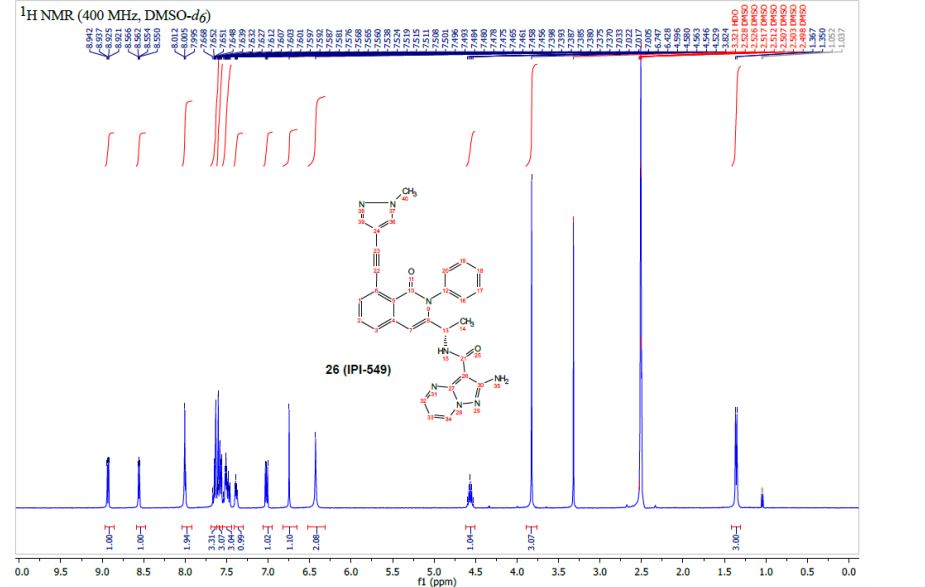
IPI-549 NMR 1H
IPI-549 13C NMR
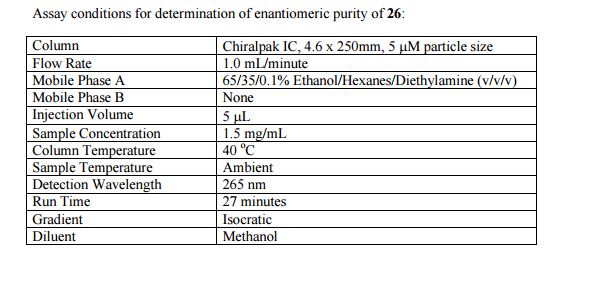
IPI-549 ASSAY


Compound 1 is coupled to 4-ethynyl-1-methyl-1H-pyrazole using the general procedure outlined above to provide compound 26 IPI-549, in 70% yield with >98% enantiomeric purity.
IPI-549
1H NMR (400 MHz, DMSO-d6) δ 8.92 (dd, J = 6.8, 1.7 Hz, 1H), 8.55 (dd, J = 4.5, 1.7 Hz, 1H), 8.00 (d, J=6.8 Hz, 1H), 8.00 (s, 1H), 7.69 – 7.54 (m, 5H), 7.53 – 7.43 (m, 3H), 7.41 – 7.35 (m, 1H), 7.01 (dd, J = 6.7, 4.5 Hz, 1H), 6.74 (s, 1H), 6.42 (s, 2H), 4.56 (quin, J = 6.8 Hz, 1H).), 3.82 (s, 3H), 1.35 (d, J = 6.8 Hz, 3H).
13C NMR (101 MHz, DMSO-d6) δ 162.73, 161.19, 160.93, 150.06, 147.51, 146.74, 141.05, 138.09, 137.81, 135.42, 133.66, 132.56, 131.90, 129.51, 129.24, 129.20, 129.17, 128.50, 126.16, 123.41, 123.31, 107.88, 102.44, 101.15, 90.40, 87.06, 85.94, 44.88, 38.62, 20.69.
ESI-HRMS: calcd for 529.2095 C30H25N8O2 (M+H)+ , found 529.2148.
[]D 22: +447.8o (c 1.007, DMSO)
COMPD1
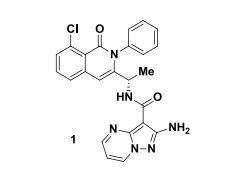
compound 1 in 95% yield.
1H NMR (400 MHz, CDCl3) 8.41 (dd, J = 6.8, 1.7 Hz, 1H), 8.37 (dd, J = 4.4, 1.7 Hz, 1H), 7.90 (d, J = 7.0 Hz, 1H), 7.50-7.34 (m, 5H), 7.34-7.27 (m, 2H), 6.76 (dd, J = 7.1, 4.9 Hz, 1H), 6.57 (s, 1H), 5.54 (broad s, 2H), 4.79 (quin, J = 6.9 Hz, 1H), 1.36 (d, J = 6.5 Hz, 3H);
ESI-HRMS: calcd for C24H20ClN6O2 459.1331 (M+H)+ , found 459.1386. HPLC Purity: 96% AUC.

Optimization of isoquinolinone PI3K inhibitors led to the discovery of a potent inhibitor of PI3K-γ (26 or IPI-549) with >100-fold selectivity over other lipid and protein kinases. IPI-549 demonstrates favorable pharmacokinetic properties and robust inhibition of PI3K-γ mediated neutrophil migration in vivo and is currently in Phase 1 clinical evaluation in subjects with advanced solid tumors.
Discovery of a Selective Phosphoinositide-3-Kinase (PI3K)-γ Inhibitor (IPI-549) as an Immuno-Oncology Clinical Candidate
Infinity Pharmaceuticals, Inc., 784 Memorial Drive, Cambridge, Massachusetts 02139, United States
ACS Med. Chem. Lett., 2016, 7 (9), pp 862–867
DOI: 10.1021/acsmedchemlett.6b00238
*E-mail: catherine.evans@infi.com., *E-mail: alfredo.castro@infi.com.
http://pubs.acs.org/doi/abs/10.1021/acsmedchemlett.6b00238
CLIP
Infinity Expands Pipeline with Addition of IPI-549, an Immuno-Oncology Development Candidate for the Treatment of Solid Tumors
– IPI-549, a Selective PI3K-Gamma Inhibitor, Targets the Immune-Suppressive Tumor Microenvironment –
– Preclinical Data for IPI-549 Presented at CRI-CIMT-EATI-AACR – The Inaugural International Cancer Immunotherapy Conference –
CAMBRIDGE, Mass.–(BUSINESS WIRE)–Infinity Pharmaceuticals, Inc. (NASDAQ: INFI) today announced the expansion of its pipeline with the addition of IPI-549, an orally administered immuno-oncology development candidate that selectively inhibits phosphoinositide-3-kinase gamma (PI3K-gamma), for the treatment of solid tumors. Preclinical data demonstrating the potential of IPI-549 to disrupt the immune-suppressive tumor microenvironment and enable a heightened anti-tumor immune response are being presented today at CRI-CIMT-EATI-AACR – The Inaugural International Cancer Immunotherapy Conference: Translating Science into Survival Meeting in New York City. IPI-549 was discovered at Infinity and is expected to enter Phase 1 clinical development in early 2016.
“Infinity is committed to developing first-in-class and best-in-class medicines, and the expansion of our pipeline with the addition of IPI-549 represents an important step toward fulfilling our vision of building a sustainable biopharmaceutical company that brings meaningful medicines to patients”
“Infinity is committed to developing first-in-class and best-in-class medicines, and the expansion of our pipeline with the addition of IPI-549 represents an important step toward fulfilling our vision of building a sustainable biopharmaceutical company that brings meaningful medicines to patients,” stated Vito Palombella, Ph.D., Infinity’s chief scientific officer. “Infinity’s ability to internally develop a selective PI3K-gamma inhibitor provides us with a unique opportunity to explore the impact that PI3K-gamma inhibition has on disrupting the tumor microenvironment. We look forward to initiating the first clinical study of IPI-549 in patients with solid tumors.”
“I have had the pleasure of collaborating with Infinity’s discovery team and am excited to have worked with IPI-549 in my laboratory,” Jedd Wolchok, M.D., Ph.D., chief of Melanoma and Immunotherapeutics Service, Lloyd J. Old/Ludwig Chair in Clinical Investigation Department of Medicine and Ludwig Center, at Memorial Sloan Kettering Cancer Center and the principal investigator for the planned Phase 1 clinical study of IPI-549. “IPI-549 is a novel, small molecule immuno-oncology agent, and I am looking forward to leading the Phase 1 study for this program.”
IPI-549 inhibits immune suppressive macrophages within the tumor microenvironment, whereas other immunotherapies such as checkpoint modulators more directly target immune effector cell function. As such, IPI-549 may have the potential to treat a broad range of solid tumors and represents a potentially complementary approach to restoring anti-tumor immunity in combination with other immunotherapies such as checkpoint inhibitors.
Preclinical Data for IPI-549 Presented at CRI-CIMT-EATI-AACR – The Inaugural International Cancer Immunotherapy Conference
Today at the AACR meeting in New York City Infinity researchers are presenting preclinical data for IPI-549 in a poster entitled, “The potent and selective phosphoinositide-3-kinase-gamma inhibitor, IPI-549, inhibits tumor growth in murine syngeneic solid tumor models through alterations in the immune suppressive microenvironment.”
In vitro data showed that IPI-549 blocks both the migration of murine myeloid cells and the differentiation of myeloid cells to the M2 phenotype, which is a type of myeloid cell known to promote cancer growth and suppress anti-tumor immune responses. In vivo data in murine solid tumor models demonstrated that IPI-549 treatment also decreased tumor-associated myeloid cells found in the immune suppressive microenvironment. Additionally, IPI-549 treatment increased the number of intratumoral CD8+T-cells, which are known to play a role in inhibiting tumor growth.
IPI-549 has demonstrated dose-dependent, single-agent, anti-tumor activity in multiple solid tumor models, including murine models of lung, colon and breast cancer. Additionally, mice treated with IPI-549 in combination with checkpoint inhibitors showed greater tumor growth inhibition than either treatment as a monotherapy. Preclinical in vivo data also demonstrated that T-cells are required for the anti-tumor activity of IPI-549, which is a hallmark of immunotherapy.
Further details about the IPI-549 development program will be provided at Infinity’s R&D Day on Tuesday, October 6, 2015. R&D Day will be held in New York City from 7:30 a.m. to 12:00 p.m. ET. The event will be webcast beginning at 8:00 a.m. ET and can be accessed in the Investors/Media section of Infinity’s website, www.infi.com. A replay of the event will also be available.
Infinity is also developing duvelisib, an investigational, oral, dual inhibitor of PI3K-delta and PI3K-gamma. The PI3K pathway is also known to play a critical role in regulating the growth and survival of certain types of blood cancers. The investigational agent is being evaluated in registration-focused studies, including DYNAMOTM, a Phase 2 study in patients with refractory indolent non-Hodgkin lymphoma, DYNAMO+R, a Phase 3 study in patients with previously treated follicular lymphoma, and DUOTM, a Phase 3 study in patients with relapsed/refractory chronic lymphocytic leukemia. Duvelisib is an investigational compound and its safety and efficacy have not been evaluated by the U.S. Food and Drug Administration or any other health authority.
About Infinity Pharmaceuticals, Inc.
Infinity is an innovative biopharmaceutical company dedicated to discovering, developing and delivering best-in-class medicines to people with difficult-to-treat diseases. Infinity combines proven scientific expertise with a passion for developing novel small molecule drugs that target emerging disease pathways. For more information on Infinity, please refer to the company’s website at www.infi.com.
Clip
IPI-549-01-A phase 1/1b first in human study of IPI-549, a PI3K-γ inhibitor, as monotherapy and in combination with pembrolizumab in subjects with advanced solid tumors.
Subcategory:
Category:
Developmental Therapeutics—Immunotherapy
Meeting:
Session Type and Session Title:
Poster Session, Developmental Therapeutics—Immunotherapy
Abstract Number: TPS3111
Poster Board Number:
Board #425a
Citation:
J Clin Oncol 34, 2016 (suppl; abstr TPS3111)
Abstract:
Background: IPI-549 is a potential first-in-class potent and selective PI3K-γ inhibitor being developed as a novel orally administered immuno-oncology therapeutic in multiple cancer indications. Preclinical studies demonstrate a role for PI3K-γ in polarization of immune suppressive myeloid cells in the tumor microenvironment. Inhibition of PI3K-γ by IPI-549 enhanced antitumor immune responses and inhibited tumor growth in syngeneic solid tumor preclinical models. In addition, IPI-549 in combination with immune checkpoint inhibitors showed increased tumor growth inhibition compared to each single agent in multiple pre-clinical models. These data served as the scientific foundation for initiating a clinical trial testing IPI-549 as a potential immuno-oncology therapy. This first-in-human clinical study will evaluate the safety and tolerability, and determine the recommended Phase 2 dose (RP2D) of IPI-549 when administered as a monotherapy and in combination with pembrolizumab (NCT02637531) in solid tumors. Methods: This multi-part Phase 1/1b open-label trial will initiate with monotherapy dose escalation cohorts consisting of an accelerated dose escalation phase followed by a standard phase with a 3+3 design. Evaluation of the PK, PD, and safety data in these cohorts will lead to the determination of the maximum tolerated dose (MTD) and RP2D of IPI-549 monotherapy. Subsequently, combination dose escalation cohorts will be initiated in which the combination of IPI-549 and pembrolizumab will be evaluated. Expansion cohorts evaluating the safety, PK, PD, and preliminary clinical activity of IPI-549 as a monotherapy and in combination with pembrolizumab will occur following the dose escalation phase. All subjects in the trial will have advanced and/or metastatic carcinoma or melanoma, and will have failed to respond to standard therapies. Combination expansion cohorts will recruit subjects with non-small cell lung cancer or melanoma who must have received an anti-PD-1 or anti-PD-L1 antibody as their most recent treatment. This trial is currently enrolling patients in the US. Clinical trial information: NCT02637531
REFERENCES
Discovery of a Selective Phosphoinositide-3-Kinase (PI3K)-γ Inhibitor (IPI-549) as an Immuno-Oncology Clinical Candidate
Catherine A. Evans, Tao Liu, André Lescarbeau, Somarajan J. Nair, Louis Grenier, Johan A. Pradeilles, Quentin Glenadel, Thomas Tibbitts, Ann M. Rowley, Jonathan P. DiNitto, Erin E. Brophy, Erin L. O’Hearn, Janid A. Ali, David G. Winkler, Stanley I. Goldstein, Patrick O’Hearn, Christian M. Martin, Jennifer G. Hoyt, John R. Soglia, Culver Cheung, Melissa M. Pink, Jennifer L. Proctor, Vito J. Palombella, Martin R. Tremblay, and Alfredo C. Castro
Publication Date (Web): July 22, 2016 (Letter)
DOI: 10.1021/acsmedchemlett.6b00238
///////immuno-oncology, IPI-549, isoform selectivity, neutrophil migration, phosphoinositide-3-kinase, PI3K-gamma inhibitor, IPI 549, IPI549. PRECLINICAL
O=C1N(C2=CC=CC=C2)C([C@@H](NC(C3=C(N=CC=C4)N4N=C3N)=O)C)=CC5=CC=CC(C#CC6=CN(C)N=C6)=C51
PF-04745637

PF-04745637
cas 1917294-46-2
MW 509.00, MF C27 H32 Cl F3 N2 O2
Cyclopentanecarboxamide, 1-(4-chlorophenyl)-N-[2-[4-hydroxy-4-(trifluoromethyl)-1-piperidinyl]-3-phenylpropyl]-
rac-1-(4-Chlorophenyl)-N-f2-r4-hvdroxy-4-(trifluoromethyl)piperidin-1-vn-3-phenylpropyDcyclopentanecarboxamide
|
PRODUCT PATENT WO-2016067143-A1
|
| Applicants: | PFIZER INC. [US/US]; 235 East 42nd Street New York, New York 10017 (US) |
| Inventors: | SWAIN, Nigel Alan; (GB). PRYDE, David Cameron; (GB). RAWSON, David James; (GB). RYCKMANS, Thomas; (GB). SKERRATT, Sarah Elizabeth; (GB). AMATO, George Salvatore; (US). MARRON, Brian Edward; (US). REISTER, Steven Michael; (US). |

TrpA1 is a member of the Transient Receptor Potential (Trp) family of ion channels. It was first described as being activated in response to noxious cold. It is activated by a number of exogenous chemical compounds and some endogenous inflammatory mediators. It has also been reported to be activated in response to mechanical stress.
There is substantial evidence for the involvement of TrpA1 in the physiology of pain, including neuropathic and inflammatory pain, and in pruritus (itch). For example, see:
Bautista, D.M. et al., “TRPA 1: A Gatekeeper for Inflammation” , Annu. Rev. Physiol.2013, 75, 181-200;
Bishnoi, M. & Premkumar, L.S., “Changes in TRP Channels Expression in Painful
Conditions”, Open Pain Journal 2013, 6(Suppl. 1), 10-22;Brederson, J.-D. et al., “Targeting TRP channels for pain relief, Eur. J. Pharmacol.2013, 716, 61-76;
Radresa, O. et al., “Roles of TRPAI in Pain Pathophysiology and Implications for the Development of a New Class of Analgesic Drugs”, Open Pain Journal 2013, 6(Suppl. 1), 137-153; and Toth, B.I. & Biro, T., “TRP Channels and Pruritus” , Open Pain Journal 2013, 6(Suppl.1), 62-80.
There is a continuing interest in finding new compounds that interact with TrpA1.
 Nigel Swain
Nigel Swain
PATENT
E8 that is 1-(4-chlorophenyl)-/V-[2-(4-hydroxy-4-(trifluoromethyl)piperidin-1-yl)-3-phenylpropyl]-cyclopentanecarboxamide, or a pharmaceutically acceptable salt thereof. This compound is represented by formula (lE).

Example 1
rac-1-(4-Chlorophenyl)-N-f2-r4-hvdroxy-4-(trifluoromethyl)piperidin-1-vn-3-phenylpropyDcyclopentanecarboxamide

Method 1
To a solution of rac-1-(1-amino-3-phenylpropan-2-yl)-4-(trifluoromethyl)piperidin-4-ol (Preparation 2, 50 mg, 0.214 mmol) in DMF (1 mL) was added 1-(4-chlorophenyl)cyclopentanecarboxylic acid (37 mg, 0.165 mmol), DIPEA (0.035 mL, 0.198 mmol) and EDCI (38 mg, 0.198 mmol), followed by HOBt (30 mg, 0.198 mmol) and the reaction was stirred at room temperature for 18 hours. Water was added and the reaction stirred for a further 2 hours. DCM was added with further stirring for 1 hour followed by elution through a phase separation cartridge. The organic filtrate was concentrated in vacuo. The residue was dissolved in MeOH and treated with ethereal HCI with standing for 18 hours. The resulting suspension was filtered and triturated with EtOAc, heptanes and TBME to afford the title compound as the hydrochloride salt (69 mg, 82%).
1H NMR (400MHz, DMSO-d6): δ ppm 1.50-1.60 (m, 4H), 1.70-1.90 (m, 4H), 2.15-2.25 (m, 2H), 2.40-2.48 (m, 2H), 2.70-2.80 (m, 1 H), 3.05-3.25 (m, 6H), 3.47-3.62 (m, 2H), 6.38 (br s, 1 H), 7.20-7.40 (m, 9H), 7.80 (br m, 1 H).
MS m/z 509 [M+H]+
Example 1 may also be prepared according to the following method:
A mixture of 1-(4-chlorophenyl)cyclopentanecarboxylic acid (25.7 g, 114 mmol), 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium-3-oxid-hexafluoro phosphate (49.4 g, 130 mmol) and N,N-diisopropylethylamine (40 mL, 229 mmol) in DMF (475 mL) was stirred at room temperature for 15 minutes. To this mixture was added a solution of 1-(1-amino-3-phenylpropan-2-yl)-4-(trifluoromethyl)piperidin-4-ol (Preparation 2, 31.4 g, 104 mmol) in DMF (200 mL). The reaction was stirred at room temperature for 18 hours before partitioning between EtOAc (600 mL) and saturated aqueous sodium hydrogen carbonatesolution (600 mL). The aqueous layer was washed with EtOAc (2 x 600 mL). The combined organic layers were washed with water (600 mL), brine (600 mL), dried over sodium sulphate and concentrated in vacuo. The residue was purified using silica gel column chromatography eluting with 0: 1 to 1 : 1 EtOAc: heptanes to afford the title compound (44 g, 76%).
1H NMR (400MHz, CDCI3): δ ppm 1.35 (br s, 1 H), 1.49-1.85 (m, 6H), 1.90-1.99 (m, 2H), 2.25-2.55 (m, 7H), 2.56-2.70 (m, 1 H), 2.75-3.00 (m, 4H), 3.23-3.31 (m, 1 H), 5.87 (br s, 1 H), 7.07 (d, 2H), 7.16-7.30 (m, 7H).
MS m/z 509 [M+H]+
Examples 2 and 3
IS) and (R)-1-(4-Chlorophenyl)-N-f2-r4-hvdroxy-4-(trifluoromethyl)piperidin-1-vn-3-phenylpropyl)cyclopentanecarboxamide

Example 2
To a suspension of (S)-1-(1-amino-3-phenylpropan-2-yl)-4-(trifluoromethyl)piperidin-4-ol (Preparation 3, 70 mg, 0.232 mmol) and 1-(4-chlorophenyl)cyclopentanecarboxylic acid (57.3 mg, 0.255 mmol) in acetonitrile (0.8 mL) was added triethylamine (0.133 mL, 0.928 mmol) followed bypropylphosphonic anhydride (50% wt solution in EtOAc, 0.21 mL, 0.35 mmol). The reaction was stirred at room temperature for 1.5 hours after which the solution was purified directly by silica gel column chromatography eluting with 0-30% EtOAc in heptanes to afford the title compound (75 mg, 64%).
[a]D20 = +9.6 in DCM [20 mg/mL]
ee determination:
Column: ChiralTech AD-H, 250×4.6 mm, 5 micron.
Mobile phase A: CO2; Mobile phase B: MeOH with 0.2% ammonium hydroxide Gradient: 5% B at 0.00 mins, 60% B at 9.00 mins; hold to 9.5 mins and return to 5% B at 10 mins. Flow rate 3 mL/min.
Rt = 5.047 minutes, ee = 95%
Example 2 may also be prepared from rac-1-(4-chlorophenyl)-N-{2-[4-hydroxy-4- (trifluoromethyl)piperidin-1-yl]-3-phenylpropyl}cyclopentanecarboxamide(Example 1).
The racemate was separated into two enantiomers using preparative chiral chromatography as described below:
Chiralpak IA, 4.6x250mm, 5 micron.
Mobile phase: Hexane:DCM:EtOH:DEA 90:8:2:0.1
Flow rate: 1 mL/min
Rt = 8.351 minutes and Rt = 10.068 minutes
The first eluting isomer is Example 2: (S)-1-(4-chlorophenyl)-N-{2-[4-hydroxy-4-(trifluoromethyl)piperidin-1-yl]-3-phenylpropyl}cyclopentanecarboxamide. ee = 100% The second eluting isomer is Example 3: (R)-1-(4-chlorophenyl)-N-{2-[4-hydroxy-4-(trifluoromethyl)piperidin-1-yl]-3-phenylpropyl}cyclopentanecarboxamide. ee = 99.62% The compound of Example 2 prepared from the chiral separation method is identical by a-rotation and retention time to the compound of Example 2 prepared as the single enantiomer described above.
MS m/z 509 [M+H]+
1H NMR (400MHz, DMSO-d6): δ 1.30-1.80 (m, 10H), 2.20-2.30 (m, 1 H), 2.35-2.60 (m, 6H), 2.65-2.85 (m, 4H), 3.00-3.15 (m, 1 H), 5.50 (br s, 1 H), 6.95-7.00 (m, 1 H), 7.05-7.15 (m, 2H), 7.20-7.35 (m, 6H) ppm
PAPER
The discovery of a potent series of carboxamide TRPA1 antagonists
*Corresponding authors
aPfizer Worldwide Medicinal Chemistry, Neuroscience and Pain Research Unit, Portway Building, Granta Park, Great Abington, UK
bIcagen, Inc., 4222 Emperor Boulevard, Suite 350, Durham, USA
cNeuroscience and Pain Research Unit, Portway Building, Granta Park, Great Abington, UK
dPfizer Worldwide Medicinal Chemistry, Ramsgate Road, Sandwich, UK
ePfizer Worldwide Medicinal Chemistry, Neuroscience and Pain Research Unit, Groton, USA
Med. Chem. Commun., 2016, Advance Article
DOI: 10.1039/C6MD00387G, http://pubs.rsc.org/en/Content/ArticleLanding/2016/MD/C6MD00387G?utm_source=feedburner&utm_medium=feed&utm_campaign=Feed%3A+rss%2FMD+%28RSC+-+Med.+Chem.+Commun.+latest+articles%29#!divAbstract
. Please note PF-6667294 is Compound 4 and PF-4746537 is Compound 8.
A series of potent and selective carboxamide TRPA1 antagonists were identified by a high throughput screen. Structure–activity relationship studies around this series are described, resulting in a highly potent example of the series. Pharmacokinetic and skin flux data are presented for this compound. Efficacy was observed in a topical cinnamaldehyde flare study, providing a topical proof of pharmacology for this mechanism. These data suggest TRPA1 antagonism could be a viable mechanism to treat topical conditions such as atopic dermatitis.

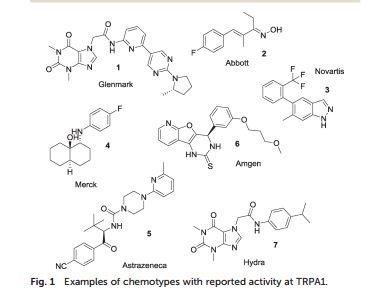
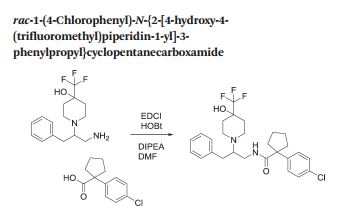
hydrochloride salt (69 mg, 82%). 1 H NMR (400 MHz, DMSO-d6): δ ppm 1.50–1.60 (m, 4H), 1.70– 1.90 (m, 4H), 2.15–2.25 (m, 2H), 2.40–2.48 (m, 2H), 2.70–2.80 (m, 1H), 3.05–3.25 (m, 6H), 3.47–3.62 (m, 2H), 6.38 (br s, 1H), 7.20–7.40 (m, 9H), 7.80 (br m, 1H). MS m/z 509 [M + H]+ .
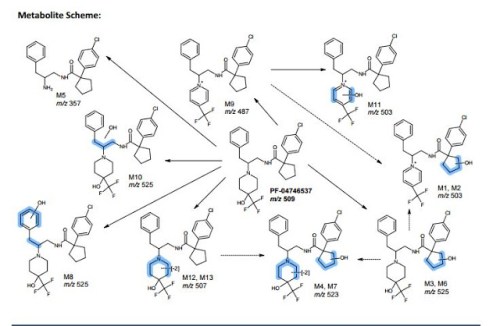
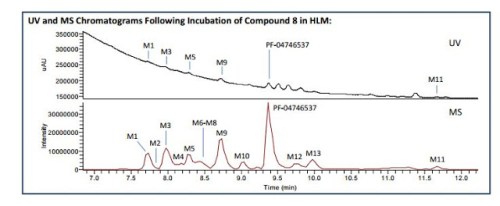

Discovery and development of TRPV1 antagonists
https://en.wikipedia.org/wiki/Discovery_and_development_of_TRPV1_antagonists
/////////////PF-04745637, PF 04745637, PF04745637, PFIZER, PRECLINICAL, TRPV1 antagonists, atopic dermatitis, 1917294-46-2
c1(ccccc1)CC(CNC(=O)C3(c2ccc(cc2)Cl)CCCC3)N4CCC(CC4)(O)C(F)(F)F
DDD 107498
DDD 107498, DDD 498
PATENT WO 2013153357, US2015045354
6-Fluoro-2-[4-(morpholinomethyl)phenyl]-N-(2-pyrrolidin-1-ylethyl)quinoline-4-carboxamide
6-Fluoro-2-[4-(4-morpholinylmethyl)phenyl]-N-[2-(1-pyrrolidinyl)ethyl]-4-quinolinecarboxamide
4-Quinolinecarboxamide, 6-fluoro-2-[4-(4-morpholinylmethyl)phenyl]-N-[2-(1-pyrrolidinyl)ethyl]-
CAS 1469439-69-7
CAS 1469439-71-1 SUCCINATE
| MF C27H31FN4O2 | |
| MW | 462.559043 g/mol |
|---|
- 6-fluoro-2-[4-(morpholin-4-ylmethyl)phenyl]-N-(2-pyrrolidin-1-ylethyl)quinoline-4-carboxamide
- Originator Medicines for Malaria Venture; University of Dundee
- Class Small molecules
- Mechanism of Action Protein synthesis inhibitors
Highest Development Phases
- No development reported Malaria
Most Recent Events
- 16 Jul 2016 No recent reports of development identified for preclinical development in Malaria in United Kingdom
- 01 Apr 2015 DDD 498 licensed to Merck Serono worldwide for the treatment of Malaria
| Inventors | Ian Hugh Gilbert, Neil Norcross, Beatriz Baragana Ruibal, Achim Porzelle |
| Original Assignee | University Of Dundee |

Prof Ian Gilbert:
Head of Biological Chemistry and Drug Discovery
BCDD, College of Life Sciences, University of Dundee, DD1 5EH, UK
Tel: +44 (0) 1382-386240
University of Dundee


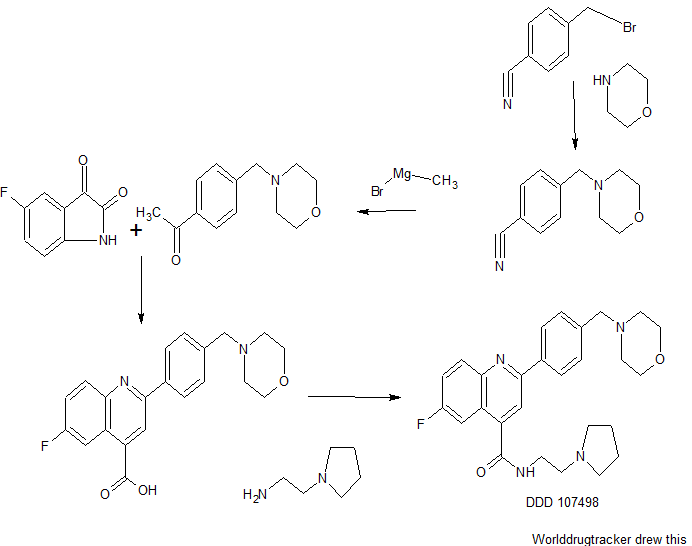
Merck Serono and MMV sign agreement to develop potential antimalarial therapy
Agreement further diversifies MMV’s partner base, strengthening our antimalarial research and development portfolio
01 April 2015
 Photo © Merck Serono
Photo © Merck Serono
Merck Serono, the biopharmaceutical business of Merck, and MMV announced today that an agreement has been signed for Merck Serono to obtain the rights to the investigational antimalarial compound DDD107498 from MMV. This agreement underscores the commitment of Merck Serono to provide antimalarials for the most vulnerable populations in need.
“This agreement strengthens our Global Health research program and our ongoing collaboration with Medicines for Malaria Venture,” said Luciano Rossetti, Executive Vice President, Global Head of Research & Development at Merck Serono. “MMV is known worldwide for its major contribution to delivering innovative antimalarial treatments to the most vulnerable populations suffering from this disease, and at Merck Serono we share this goal.”
DDD107498 originated from a collaboration between MMV and the University of Dundee Drug Discovery Unit, led by Prof. Ian Gilbert and Dr. Kevin Read. The objective of the clinical program is to demonstrate whether the investigational compound exerts activity on a number of malaria parasite lifecycle stages, and remains active in the body long enough to offer potential as a single-dose treatment against the most severe strains of malaria.
While development and commercialization of the compound is under Merck Serono’s responsibility, MMV will provide expertise in the field of malaria drug development, including its clinical and delivery expertise, and provide access to its public and private sector networks in malaria-endemic countries.
Merck Serono has a dedicated Global Health R&D group working to address key unmet medical needs related to neglected diseases, such as schistosomiasis and malaria, with a focus on pediatric populations in developing countries. Its approach is based on public-private partnerships and collaborations with leading global health institutions and organizations in both developed and developing countries.
“Working with partners like Merck Serono is critical to the progress of potential antimalarial compounds, like DDD107498, through the malaria drug pipeline,” said Dr. Timothy Wells, Chief Scientific Officer at MMV. “Their Global Health Program is gaining momentum and we need more compounds to tackle malaria, a disease that places a huge burden on the world’s most vulnerable populations. DDD107498 holds great promise and we look forward to working with the Merck Serono team through the development phase.”
According to the World Health Organization, there were an estimated 198 million cases of malaria worldwide in 2013, and an estimated 584,000 deaths, primarily in young children from the developing world. The launch of the not-for-profit research foundation, MMV, in 1999 and a number of collaborations and partnerships, including those with Merck Serono, has contributed to reducing the major gap in malaria R&D investment and subsequent dearth of new medicines.
“It’s hugely encouraging to see the German pharmaceutical industry increasing their engagement in the development of novel antimalarials,” said global malaria expert Prof. Dr. Peter Kremsner, Director of the Institute for Tropical Medicine at the University of Tübingen, Germany. “The Merck Serono and MMV collaboration to develop DDD107498 is a great step. It’s a compound that offers lots of promise so I’m excited to see how it progresses.


Scots scientists in ‘single dose’ malaria treatment breakthrough
An antimalarial drug that could treat patients was discovered by Dundee university scientists
Scientists have discovered an antimalarial compound that could treat malaria patients in a single dose and help prevent the spread of the disease from infected people.
The compound DDD107498 also has the potential to treat patients with malaria parasites resistant to current medications, researchers say.
Scientists hope it could lead to treatments and protection against the disease, which claimed almost 600,000 lives amid 200 million reported cases in 2013.
The compound was identified through a collaboration between the University of Dundee’s drug discovery unit (DDU) and the Medicines for Malaria Venture (MMV), a separate organisation.
The compound is now undergoing further safety testing with a view to entering human clinical trials within the next year.
Details of the discovery have been published in the journal Nature.
Professor Ian Gilbert, head of chemistry at the DDU, who led the team that discovered the compound, said: “The publication describes the discovery and profiling of this exciting new compound.
“It reveals that DDD107498 has the potential to treat malaria with a single dose, prevent the spread of malaria from infected people and protect a person from developing the disease in the first place.
“There is still some way to go before the compound can be given to patients. However, we are very excited by the progress that we have made.”
The World Health Organisation reports that there were 200 million clinical cases of malaria in 2013, with 584,000 people dying from the disease. Most of these deaths were children under the age of five and pregnant women.
MMV chief executive officer Dr David Reddy said: “Malaria continues to threaten almost half of the world’s population – the half that can least afford it.
“DDD107498 is an exciting compound since it holds the promise to not only treat but also protect these vulnerable populations.
“The collaboration to identify and progress the compound, led by the drug discovery unit at the University of Dundee, drew on MMV’s network of scientists from Melbourne to San Diego.”The publication of the research is an important step and a clear testament to the power of partnership.”
MMV selected DDD107498 to enter preclinical development in October 2013 following the recommendation of its expert scientific advisory committee.
Since then, with MMV’s leadership, large quantities of the compound have been produced and it is undergoing further safety testing with a view to entering human clinical trials within the next year.
Merck Serono has recently obtained the right to develop and, if successful, commercialise the compound, with the input of MMV’s expertise in the field of malaria drug development and access and delivery in malaria-endemic countries.
Dr Michael Chew from the Wellcome Trust, which provides funding for the DDU and MMV, said: “The need for new antimalarial drugs is more urgent than ever before, with emerging strains of the parasite now showing resistance against the best available drugs.
“These strains are already present at the Myanmar-Indian border and it’s a race against time to stop resistance spreading to the most vulnerable populations in Africa.
“The discovery of this new antimalarial agent, which has shown remarkable potency against multiple stages of the malaria lifecycle, is an exciting prospect in the hunt for viable new treatments.”
PAPER


Discovery of a Quinoline-4-carboxamide Derivative with a Novel Mechanism of Action, Multistage Antimalarial Activity, and Potent in Vivo Efficacy
† Drug Discovery Unit, Division of Biological Chemistry and Drug Discovery, School of Life Sciences, University of Dundee, Dundee, DD1 5EH, U.K.
‡ Cell and Molecular Biology, Department of Life Sciences, Imperial College, London, SW7 2AZ, U.K.
§ School of Medicine, University of California, San Diego, 9500 Gilman Drive, La Jolla, California 92093, United States
∥ Eskitis Institute, Griffith University, Brisbane Innovation Park, Nathan Campus, Brisbane, QLD 4111, Australia
⊥ Swiss Tropical and Public Health Institute, Swiss TPH, Socinstrasse 57, 4051 Basel, Switzerland
#University of Basel, CH-4003 Basel, Switzerland
∇Medicines for Malaria Venture, International Centre Cointrin, Entrance G, 3rd Floor, Route de Pré-Bois 20, P.O. Box 1826, CH-1215, Geneva 15, Switzerland
J. Med. Chem., Article ASAP
DOI: 10.1021/acs.jmedchem.6b00723
*K.D.R.: phone, +44 1382 388 688; e-mail, k.read@dundee.ac.uk., *I.H.G.: phone, +44 1382 386 240; e-mail,i.h.gilbert@dundee.ac.uk.

Conditions: (a) morpholine, Et3N, DCM, 16 h, 72% yield; (b) MeMgBr, toluene, reflux, 4 h and then a 10% aqueous HCl, reflux, 1 h, 70% yield; (c) NBS, benzoyl peroxide, dichlorobenzene, 140 °C, 16 h, 70% yield; (d) morpholine, K2CO3, acetonitrile, 40 °C, 16 h, 64% yield; (e) 5-fluoroisatin, KOH, EtOH, 120 °C, microwave, 20 min, 30–76% yield; (f) amine, CDMT, N-methylmorpholine, DCM, 20–61% yield.
// https://tpc.googlesyndication.com/pagead/js/r20160906/r20110914/abg.js//
A single-dose treatment against malaria worked in mice to cure them of the disease. The drug also worked to block infection in healthy mice and to stop transmission, according to a study published in Nature today. The fact that the drug can act against so many stages of malaria is pretty new, but what’s even more exciting is the compound’s mode of action: it kills malaria in a completely new way, researchers say. The feature would make it a welcome addition to our roster of antimalarials — a roster that’s threatened by drug resistance.
RESEARCHERS SIFTED THROUGH A LIBRARY OF ABOUT 4,700 COMPOUNDS TO FIND THIS ONE
Malaria is an infectious disease that’s transmitted through mosquito bites; it’s also a leading cause of death in a number of developing countries. Approximately 3.4 billion people live in areas where malaria poses a real threat. As a result, there were 207 million cases of malaria in 2012 — and 627,000 deaths. There are drugs that can be used to prevent malaria, and even treat it, but drug resistance is halting the use of certain treatments in some areas.
A long search
Searching for a new drug is all about trial and error. To find this particular compound, researchers sifted through a library of about 4,700 compounds, testing them to see if they were capable of killing the malaria parasite in a lab setting. When they found something that worked, they tweaked the drug candidate to see if it could perform more effectively. “We went through a lot of these cycles of testing and designing new compounds,” says Ian Gilbert, a medicinal chemist at the University of Dundee in the UK, and a co-author of the study. “Eventually we optimized to the compound which is the subject of the paper.” For now, that compound’s unwieldy name is DDD107498.
To make sure DDD107498 really had potential, the researchers tested it on mice that had already been infected with malaria. A single dose was enough to provoke a 90 percent reduction in the number of parasites in their blood. The scientists also gave the compound to healthy mice that were subsequently exposed to malaria. DDD107498 helped the mice evade infection with a single dose, but it’s unclear how long that effect would last in humans. Finally, the researchers looked at whether the compound could prevent the transmission from an infected mouse to a mosquito. A day after receiving the treatment, mice were put in contact with mosquitoes. The scientists noted a 91 percent reduction in infected mosquitoes.
“IT HAS THE ABILITY TO BE A ONE-DOSE [DRUG], IN COMBINATION WITH ANOTHER MOLECULE.”
“What’s exciting about this molecule is obviously the fact that it has the ability to be a one-dose [drug], in combination with another molecule to cure blood stage malaria,” says Kevin Read, a drug researcher also at the University of Dundee and a co-author of the study. The fact that the compound has the ability to block transmission and protect against infection is equally thrilling. But the way in which DDD107498 kills malaria might be its most interesting feature. It halts the production of proteins — which are necessary for the parasite’s survival. No other malaria drug does that right now, Read says. “So, in principle, there’s no resistance out there already to this mechanism.”
The drug hasn’t been tested in humans yet, so it may not be nearly as good in the field. But Read says DDD107498 looks promising. “From all the pre-clinical or non-clinical data we’ve generated, it is comparable or better than any of the current marketed anti-malarials in those studies.” And at $1 per treatment, the price of the drug should fall “within the range of what’s acceptable,” he says.
“It looks like an excellent study, and the results look very important,” says Philip Rosenthal, a malaria drug researcher at The University of California-San Francisco who didn’t participate in the study. This is a big shift for Rosenthal’s field. Five years ago, “we had very little going on in anti-malarial drug discovery,” he says. Now, there’s quite a bit going on for malaria researchers, and a number of promising compounds are moving along. DDD107498 “is another player, and it’s got a number of positive features,” he says.
OTHER TREATMENTS HAVE TO BE TAKEN FOR A FEW DAYS
One of the features is the drug’s potency. It’s very active against cultured malaria parasites, Rosenthal says. But what’s perhaps most intriguing about DDD107498 is that the drug works against the mechanism that enables protein synthesis the malaria parasite’s cells. No other malaria drug does that right now, Read says. “Considering challenges of treating malaria, which is often in rural areas and developing countries, a single dose would be a big plus,” he says. “In addition, because of it’s long half life, it may also work to prevent malaria with once a week dosing, which is also a benefit.”
Still, no drug is perfect. The data suggests that DDD107498 doesn’t kill malaria as quickly as some other drugs, Rosenthal says. And when the researchers tested it to see how long it might take for resistance to develop, the results weren’t as promising as he would like. The parasites figured out a way to become resistant to the compound “relatively easily,” he says. That shouldn’t be “deal-killer,” however. “Its slow onset of action probably means it should be combined with a faster-acting drug,” he says.
BUT IT’S SLOW-ACTING
The compound is going through safety testing now. If everything goes well, it should hit human trials within the next year, Read says. Chances are, it will have to be used in combination with other malaria drugs, Gilbert says. “All anti-malarials are given in combination because it slows down resistance.”
“When you’re treating infectious diseases, you know that drug resistance is always a potential problem, so having a number of choices to treat malaria is a good thing,” Rosenthal says. In this case, the drug’s new mode of action may hold lead to an entirely new weapon against malaria. “Obviously it’s got a long way to go,” Read says. But the compound is “very exciting,” nonetheless.
// https://tpc.googlesyndication.com/pagead/js/r20160906/r20110914/abg.js//
//
PATENT
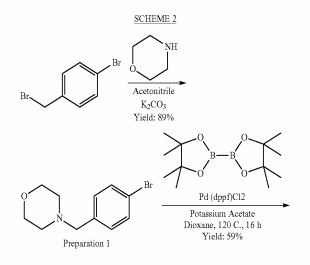
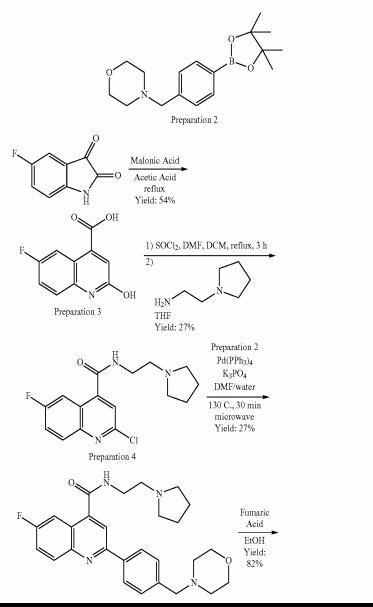
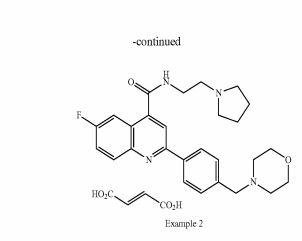
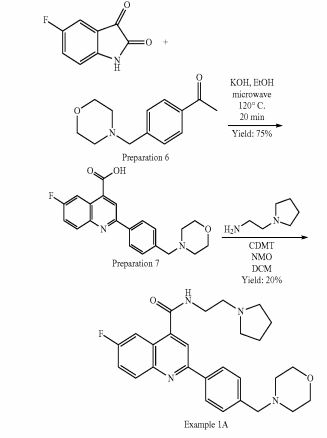
Example 16-Fluoro-2-[4-(morpholinomethyl)phenyl]-N-(2-pyrrolidin-1-ylethyl)quinoline-4-carboxamide, Example compound 1 in Scheme 2
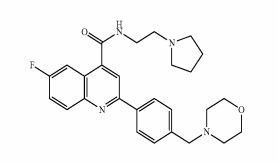
In a sealed microwave tube, a suspension of 2-chloro-6-fluoro-N-(2-pyrrolidin-1-ylethyl)quinoline-4-carboxamide (preparation 4) (2.00 g, 6 mmol), [4-(morpholinomethyl)phenyl]boronic acid, hydrochloride, available from UORSY, (3.20 g, 12 mmol), potassium phosphate (2.63 g, 12 mmol) and tetrakis(triphenylphosphine)palladium (0) (0.21 g, 0.19 mmol) in DMF/Water 3/1 (40 ml) was heated at 130° C. under microwave irradiation for 30 min. The reaction was filtered through Celite™ and solvents were removed under reduced pressure. The resulting residue was taken up in DCM (150 ml) and washed twice with NaHCO3 saturated aqueous solution (2×100 ml). The organic layer was separated, dried over MgSO4 and concentrate to dryness under reduced pressure. The reaction crude was purified by flash column chromatography using an 80 g silica gel cartridge and eluting with DCM (Solvent A) and MeOH (Solvent B) and the following gradient: 1 min hold 100% A, followed by a 30 min ramp to 10% B, and then 15 min hold at 10% B. The fractions containing product were pooled together and concentrated to dryness under vacuum to obtain the desired product as an off-white solid (1 g). The product was dissolved in methanol (100 ml) and 3-mercaptopropyl ethyl sulfide Silica (Phosphonics, SPM-32, 60-200 uM) was added. The suspension was stirred at room temperature over for 2 days and then at 50° C. for 1 h. After cooling to room temperature, the scavenger was filtered off and washed with methanol (30 ml). The solvent was removed under reduced pressure and the product was further purified by preparative HPLC. The fractions containing product were pooled together and freeze dried to obtain the desired product as a white solid (0.6 g, 1.3 mmol, Yield 20%).
1H NMR (500 MHz; CDCl3) δ 1.81-1.84 (m, 4H), 2.50-2.52 (m, 4H), 2.63 (brs, 4H), 2.82 (t, 2H, J=5.9 Hz), 3.61 (s, 2H), 3.71 (dd, 2H, J=5.4 Hz, J=11.4 Hz), 3.74-3.76 (m, 4H), 6.84 (brs, 1H), 7.52-7.57 (m, 3H), 7.97-8.00 (m, 2H), 8.13 (d, 2H, J=8.2 Hz), 8.21 (dd, 1H, J=5.5 Hz, J=9.2 Hz) ppm. 19F NMR (407.5 MHz; CDCl3) δ−111.47 ppm.
Purity by LCMS (UV Chromatogram, 190-450 nm) 99%, rt=5.7 min, m/z 463 (M+H)+ HRMS (ES+) found 463.2501 [M+H]+, C27H32F1N4O2 requires 463.2504.
Example 26-Fluoro-2-[4-(morpholinomethyl)phenyl]-N-(2-pyrrolidin-1-ylethyl)quinoline-4-carboxamide; fumaric acid salt, compound (IB) in Scheme 2
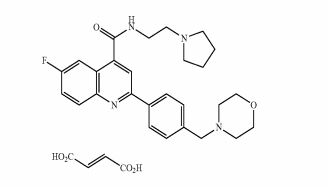
The starting free base (example 1) (0.58 g, 1 mmol) was dissolved in dry ethanol (10 ml) and added dropwise to a stirred solution of fumaric acid (0.15 g, 1 mmol) in dry ethanol (9 ml). The mixture was stirred at room temperature for 1 h. The white precipitate was filtered, washed with ethanol (20 ml) and then dissolved in 10 ml of water and freeze dried to obtain the desired salt as a white solid (0.601 g, 1 mmol, Yield 82%).
1H NMR (500 MHz; d6-DMSO) δ 1.83-1.86 (m, 4H), 2.41 (brs, 4H), 2.94 (brs, 4H), 3.03 (t, 2H, J=6.2 Hz), 3.57 (s, 2H), 3.60-3.65 (m, 6H), 6.47 (s, 2H), 7.51 (d, 2H, J=8.25), 7.74-7.78 (m, 1H), 8.06 (dd, 1H, J=2.9 Hz, J=10.4 Hz), 8.17 (dd, 1H, J=5.7 Hz, J=9.3 Hz), 8.24-8.26 (m, 3H), 9.24 (t, 1H, J=5.5 Hz) ppm. 19F NMR (407.5 MHz; d6-DMSO) δ-112.30 ppm.
Purity by LCMS (UV Chromatogram, 190-450 nm) 99%, rt=5.3 min, m/z 463 (M+H)+
Example 1AAlternative synthesis of 6-fluoro-2-[4-(morpholinomethyl)phenyl]-N-(2-pyrrolidin-1-ylethyl)quinoline-4-carboxamide, Example compound 1A in Scheme 4

To a stirred suspension of 6-fluoro-2-[4-(morpholinomethyl)phenyl]quinoline-4-carboxylic acid (preparation 7) (2.20 g, 6 mmol) in DCM (100 ml) at room temperature, 2-chloro-4,6-dimethoxy-1,3,5-triazine (CDMT) (1.26 g, 7 mmol) and 4-methylmorpholine (NMO) (1.33 ml, 12 mmol) were added. The reaction mixture was stirred at room temperature for 1 h and then 2-pyrrolidin-1-ylethanamine (0.77 ml, 6 mmol) was added and stirred at room temperature for further 3 h. The reaction mixture was washed with NaHCO3 saturated aqueous solution (2×100 ml) and the organic phase was separated, dried over MgSO4 and concentrated under reduced pressure. The resulting residue was absorbed on silica gel and purified by flash column chromatography using an 80 g silica gel cartridge and eluting with DCM (Solvent A) and MeOH (Solvent B) and the following gradient: 2 min hold 100% A followed by a 30 min ramp to 10% B and then 15 min hold at 10% B. The desired fractions were concentrated to dryness under vacuum to obtain the crude product as a yellow solid (95% purity by LCMS). The sample was further purified by a second column chromatography using a 40 g silica gel cartridge, eluting with DCM (Solvent A) and 10% NH3-MeOH in DCM (Solvent B) and the following gradient: 2 min hold 100% A, followed by a 10 min ramp to 23% B and then 15 min hold at 23% B. The desired fractions were concentrated to dryness under vacuum to obtain product as a white solid (1 g). Re-crystallisation form acetonitrile (18 ml) yielded the title compound as a white solid (625 mg, 1.24 mmol, 20%).
1H NMR (500 MHz; CDCl3) δ 1.81-1.84 (m, 4H), 2.50-2.52 (m, 4H), 2.63 (brs, 4H), 2.82 (t, 2H, J=5.9 Hz), 3.61 (s, 2H), 3.71 (dd, 2H, J=5.4 Hz, J=11.4 Hz), 3.74-3.76 (m, 4H), 6.84 (brs, 1H), 7.52-7.57 (m, 3H), 7.97-8.00 (m, 2H), 8.13 (d, 2H, J=8.2 Hz), 8.21 (dd, 1H, J=5.5 Hz, J=9.2 Hz) ppm.
1H NMR (500 MHz; d6-DMSO) δ 1.72-1.75 (m, 4H), 2.41 (brs, 4H), 2.56 (brs, 4H), 2.67 (t, 2H, J=6.6 Hz), 3.49-3.52 (m, 2H), 3.56 (s, 2H), 3.60-3.61 (m, 4H), 7.52 (d, 2H, J=8.3 Hz), 7.73-7.77 (m, 1H), 8.07 (dd, 1H, J=2.9 Hz, J=10.4 Hz), 8.18-8.21 (m, 2H), 8.26 (d, 2H, J=8.3 Hz), 8.85 (t, 1H, J=6.6 Hz) ppm.
13C NMR (125 MHz; d6-DMSO3) δ 23.2, 38.4, 53.2, 53.5, 54.5, 62.1, 66.2, 109.0, 109.1, 117.3, 120.1, 120.3, 124.1, 124.2, 127.1, 129.4, 132.2, 132.3, 136.8, 139.9, 142.8, 145.2, 155.3, 159.0, 161.0, 166.1 ppm.
19F NMR (500 MHz; d6-DMSO) δ-112.47 ppm.
Purity by LCMS (UV Chromatogram, 190-450 nm) 99%, rt=5.0 min, m/z 463 (M+H)+
PATENT
WO 2016033635
Patent
WO 2013153357
SCHEME 1

SCHEME 2

Preparation 4Yield: 54% Preparation 3
Yield: 27%

SCHEME 4 B

Yield: 72% Yield: 70% Preparation 6

Example 1 : 6-Fluoro-2-r4-(morpholinomethyl)phenyll-N-(2-pyrrolidin-1-ylethyl)quinoline- 4-carboxamide, Example compound 1 in Scheme 2

In a sealed microwave tube, a suspension of 2-chloro-6-fluoro-N-(2-pyrrolidin-1- ylethyl)quinoline-4-carboxamide (preparation 4) (2.00 g, 6 mmol), [4- (morpholinomethyl)phenyl]boronic acid, hydrochloride, available from UORSY, (3.20 g, 12 mmol), potassium phosphate (2.63 g, 12 mmol) and tetrakis(triphenylphosphine)palladium (0) (0.21 g, 0.19 mmol) in DMF/Water 3/1 (40 ml) was heated at 130°C under microwave irradiation for 30 min. The reaction was filtered through Celite™ and solvents were removed under reduced pressure. The resulting residue was taken up in DCM (150 ml) and washed twice with NaHC03 saturated aqueous solution (2 x 100 ml). The organic layer was separated, dried over MgS04and concentrate to dryness under reduced pressure. The reaction crude was purified by flash column chromatography using an 80 g silica gel cartridge and eluting with DCM (Solvent A) and MeOH (Solvent B) and the following gradient: 1 min hold 100% A, followed by a 30 min ramp to 10 % B, and then 15 min hold at 10% B. The fractions containing product were pooled together and concentrated to dryness under vacuum to obtain the desired product as an off-white solid (1 g). The product was dissolved in methanol (100 ml) and 3-mercaptopropyl ethyl sulfide Silica (Phosphonics, SPM-32, 60- 200 uM) was added. The suspension was stirred at room temperature over for 2 days and then at 50°C for 1 h. After cooling to room temperature, the scavenger was filtered off and washed with methanol (30 ml). The solvent was removed under reduced pressure and the product was further purified by preparative HPLC. The fractions containing product were pooled together and freeze dried to obtain the desired product as a white solid (0.6 g, 1.3 mmol, Yield 20%).
1 H NMR (500 MHz; CDCI3) δ 1.81-1.84 (m, 4H), 2.50-2.52 (m, 4H), 2.63 (brs, 4H), 2.82 (t, 2H, J = 5.9 Hz), 3.61 (s, 2H), 3.71 (dd, 2H, J = 5.4 Hz, J = 1 1.4 Hz), 3.74-3.76 (m, 4H), 6.84 (brs, 1 H), 7.52-7.57 (m, 3H), 7.97-8.00 (m, 2H), 8.13 (d, 2H, J = 8.2 Hz), 8.21 (dd, 1 H, J = 5.5 Hz, J = 9.2 Hz) ppm . 19 F NMR (407.5 MHz; CDCI3) δ -11 1.47 ppm. Purity by LCMS (UV Chromatogram, 190-450nm) 99 %, rt = 5.7 min, m/z 463 (M+H)+ HRMS (ES+) found 463.2501 [M+H]+, C27H32F1 N402 requires 463.2504.
Example 2: 6-Fluoro-2-[4-(morpholinomethyl)phenyl1-N-(2-pyrrolidin-1-ylethyl)quinoline- 4-carboxamide; fumaric acid salt, compound (IB) in Scheme 2

The starting free base (example 1) (0.58 g, 1 mmol) was dissolved in dry ethanol (10 ml) and added dropwise to a stirred solution of fumaric acid (0.15 g, 1 mmol) in dry ethanol (9 ml). The mixture was stirred at room temperature for 1 h. The white precipitate was filtered, washed with ethanol (20 ml) and then dissolved in 10 ml of water and freeze dried to obtain the desired salt as a white solid (0.601 g, 1 mmol, Yield 82%).
1 H NMR (500 MHz; d6-DMSO) δ 1.83-1.86 (m, 4H), 2.41 (brs, 4H), 2.94 (brs, 4H), 3.03 (t, 2H, J = 6.2 Hz), 3.57 (s, 2H), 3.60-3.65 (m, 6H), 6.47 (s, 2H), 7.51 (d, 2H, J = 8.25), 7.74-7.78 (m, 1 H), 8.06 (dd, 1 H, J = 2.9 Hz, J = 10.4 Hz), 8.17 (dd, 1 H, J = 5.7 Hz, J = 9.3 Hz), 8.24-8.26 (m, 3H), 9.24 (t, 1 H, J = 5.5 Hz) ppm. 19 F NMR (407.5 MHz; d6- DMSO) δ -112.30 ppm.
Purity by LCMS (UV Chromatogram, 190-450nm) 99 %, rt = 5.3 min, m/z 463 (M+H)+
Example 1A: Alternative synthesis of 6-fluoro-2-[4-(morpholinomethyl)phenyl1-N-(2- pyrrolidin-1-ylethyl)quinoline-4-carboxamide, Example compound 1A in Scheme 4

To a stirred suspension of 6-fluoro-2-[4-(morpholinomethyl)phenyl]quinoline-4-carboxylic acid (preparation 7) (2.20 g, 6 mmol) in DCM (100 ml) at room temperature, 2-chloro- 4,6-dimethoxy-1 ,3,5-triazine (CDMT) (1.26 g, 7 mmol) and 4-methylmorpholine (NMO) (1.33 ml, 12 mmol) were added. The reaction mixture was stirred at room temperature for 1 h and then 2-pyrrolidin-1-ylethanamine (0.77 ml, 6 mmol) was added and stirred at room temperature for further 3 h. The reaction mixture was washed with NaHC03 saturated aqueous solution (2x 100 ml) and the organic phase was separated, dried over MgS04 and concentrated under reduced pressure. The resulting residue was absorbed on silica gel and purified by flash column chromatography using an 80 g silica gel cartridge and eluting with DCM (Solvent A) and MeOH (Solvent B) and the following gradient: 2 min hold 100% A followed by a 30 min ramp to 10 %B and then 15 min hold at 10%B. The desired fractions were concentrated to dryness under vacuum to obtain the crude product as a yellow solid (95% purity by LCMS). The sample was further purified by a second column chromatography using a 40 g silica gel cartridge, eluting with DCM (Solvent A) and 10% NH3-MeOH in DCM (Solvent B) and the following gradient: 2 min hold 100% A, followed by a 10 min ramp to 23 % B and then 15 min hold at 23% B. The desired fractions were concentrated to dryness under vacuum to obtain product as a white solid (1 g). Re-crystallisation form acetonitrile (18 ml) yielded the title compound as a white solid (625 mg, 1.24 mmol, 20%).
1 H NMR (500 MHz; CDCI3) δ 1.81-1.84 (m, 4H), 2.50-2.52 (m, 4H), 2.63 (brs, 4H), 2.82 (t, 2H, J = 5.9 Hz), 3.61 (s, 2H), 3.71 (dd, 2H, J = 5.4 Hz, J = 1 1.4 Hz), 3.74-3.76 (m, 4H), 6.84 (brs, 1 H), 7.52-7.57 (m, 3H), 7.97-8.00 (m, 2H), 8.13 (d, 2H, J = 8.2 Hz), 8.21 (dd, 1 H, J = 5.5 Hz, J = 9.2 Hz) ppm .
1 H NMR (500 MHz; d6-DMSO) δ 1.72-1.75 (m, 4H), 2.41 (brs, 4H), 2.56 (brs, 4H), 2.67 (t, 2H, J = 6.6 Hz), 3.49-3.52 (m, 2H), 3.56 (s, 2H), 3.60-3.61 (m, 4H), 7.52 (d, 2H, J = 8.3 Hz), 7.73-7.77 (m, 1 H), 8.07 (dd, 1 H, J = 2.9 Hz, J = 10.4 Hz), 8.18-8.21 (m, 2H), 8.26 (d, 2H , J = 8.3 Hz), 8.85 (t, 1 H, J = 6.6 Hz) ppm.
13C NMR (125 MHz; d6-DMS03) 5 23.2, 38.4, 53.2, 53.5, 54.5, 62.1 , 66.2, 109.0, 109.1 , 1 17.3, 120.1 , 120.3, 124.1 , 124.2, 127.1 , 129.4, 132.2, 132.3, 136.8, 139.9, 142.8, 145.2, 155.3, 159.0, 161 .0, 166.1 ppm.
19 F NM R (500 MHz; d6-DMSO) δ -1 12.47 ppm.
Purity by LCMS (UV Chromatogram, 190-450nm) 99 %, rt = 5.0 min, m/z 463 (M+H)+
PAPER
A Quinoline Carboxamide Antimalarial Drug Candidate Uniquely Targets Plasmodia at Three Stages of the Parasite Life Cycle
Angewandte Chemie, International Edition (2015), 54, (46), 13504-13506
Angewandte Chemie, International Edition (2015), 54, (46), 13504-13506

Putting a stop to malaria: Phenotypic screening against malaria parasites, hit identification, and efficient lead optimization have delivered the preclinical candidate antimalarial DDD107498. This molecule is distinctive in that it has potential for use as a single-dose cure for malaria and shows a unique broad spectrum of activity against the liver, blood, and mosquito stages of the parasite life cycle.
Prof. P. M. O’Neill Department of Chemistry, University of Liverpool Liverpool, L69 7ZD (UK) E-mail: pmoneill@liverpool.ac.uk Prof. S. A. Ward Liverpool School of Tropical Medicine, Pembroke Place Liverpool, L3 5QA (UK)
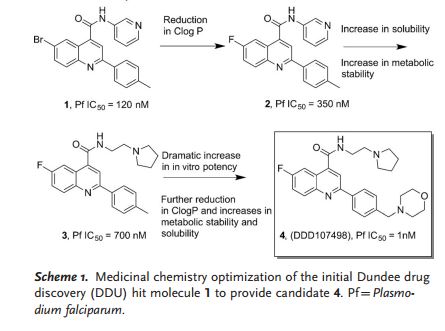
Professor Ian Gilbert FRSC
Design and synthesis of potential therapeutic agents

Position:
Professor of Medicinal Chemistry and Head of the Division of Biological Chemistry and Drug Discovery
Affiliation:
Address:
College of Life Sciences, University of Dundee, Dundee
Full Telephone:
+44 (0) 1382 386240, int ext 86240
Email:
Website:
Medicinal Chemistry
I am a medicinal chemist and my research interests are in the design and synthesis of potential drugs. The mainstay of my work is synthetic medicinal chemistry as part of the Drug Discovery Unit (DDU). Where possible we make extensive use of molecular modeling to guide our synthetic efforts. I have a particular interests in the following aspects of drug discovery:
Neglected diseases such as human African trypanosomiasis, leishmaniasis and malaria.
Chemical validation of drug targets, including novel targets for which there is little or no precedence for drug discovery.
Novel approaches to and paradigms for drug discovery.
Mode of action studies and target identification.
I am Head of Chemistry in the DDU. Our main focuses are on neglected diseases and novel drug targets. The neglected diseases we are tackling are malaria, tuberculosis and the kinetoplastid diseases. We use both target-based approaches and phenotypic approaches (whole parasite screening). We have had particular success in validating the enzyme N-myristoyltransferase as a drug target in human African trypanosomiasis, and in identifying and optimising phenotypic hits. In our novel targets area, we aim to validate novel areas of biology as potential drug targets.
Dr Neil Norcross
Position:
Medicinal Chemist
Affiliation:
Address:
College of Life Sciences, University of Dundee, Dundee
Full Telephone:
(0) , int ext
Email:

La investigadora asturiana Beatriz Baragaña, en La Pola. / PABLO NOSTI

Achim Porzelle
REFERENCES
///////////DDD107498, DDD 107498, PRECLINICAL, DUNDEE, MALARIA, DDD 498, Achim Porzelle, Ian Gilbert, MERCK SERENO, Beatriz Baragaña, Medicines for Malaria Venture, University of Dundee, Neil Norcross, 1469439-69-7, 1469439-71-1 , SUCCINATE
Fc1ccc2nc(cc(c2c1)C(=O)NCCN1CCCC1)-c1ccc(cc1)CN1CCOCC1
DR ANTHONY MELVIN CRASTO

ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on
ORGANIC SPECTROSCOPY INTERNATIONAL

