
Home » Uncategorized (Page 17)
Category Archives: Uncategorized
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Firibastat
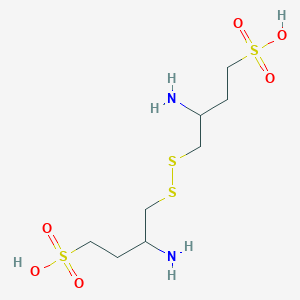
_648927-86-0.png)
Firibastat
- Molecular FormulaC8H20N2O6S4
- Average mass368.514 Da
368.5
RB 150
Qgc-001(racemate)
UNII-PD5EII1F9A
Firibastat, (+/-)-
PD5EII1F9A
3-amino-4-[(2-amino-4-sulfobutyl)disulfanyl]butane-1-sulfonic acid
1-Butanesulfonic acid, 4,4′-dithiobis(3-amino-
3-Amino-4-((2-amino-4-sulfo-butyl)disulfanyl)butane-1-sulfonic acid
cas 721392-96-7, RACEMIC
CAS 648927-86-0, (S)-3-amino-4-(((S)-2-amino-4-sulfobutyl)disulfaneyl)butane-1-sulfonic acid
фирибастат[Russian][INN]
فيريباستات[Arabic][INN]
(3S,3’S)-4,4′-Disulfanediylbis(3-aminobutane-1-sulfonic acid)
firibastatum
фирибастат
فيريباستات
非立巴司他[Chinese]
SCHEME
SEE AT END OF PAGE
PAPER
Journal of Labelled Compounds & Radiopharmaceuticals (2004), 47(13), 997-1005
PATENT
https://patents.google.com/patent/WO2020084131A1/en
PATENT
WO2012045849


EXAMPLES
Example 1: Synthesis of compound I from (S) ethyl 2-(benzyloxycarbonylamino) 4-(neopentyloxysulfonyl)butanoate
Step (a): (S) neopentyl 3-(benzylox carbonylamino) 4-hydroxybutane 1-sulfonate B
B
(S) ethyl 2-(benzyloxycarbonylamino) 4-(neopentyloxysulfonyl)butanoate A (41.55g, 100.0 mmol, 1.0 eq.) is added dropwise onto a 2M solution of LiBH4 in THF (50 mL, 44.8 g, 100.0 mmol, 1.0 eq.). The addition is performed at room temperature over a 3 hrs period. At the end of the addition, the mixture is stirred at room temperature until conversion is complete (A<1%). Addition of toluene, followed by hydrolysis with HC1, washings of the organic layer with NaHC03 and water, and concentration under vacuum lead to the desired product as a pale yellow oil in quantitative yield (ee = 98%), which slowly crystallises at room temperature in 4 or 5 days.
As B was found to have a very low melting point by DSC analysis, it was not possible to isolate it as a solid by simple crystallisation. It was decided to let it in solution and use it without further purification in the following step.
Step (b): (S) neopentyl 3-(benzyloxycarbonylamino) 4-(methylsulfonyloxy)butane 1-sulfonate
C
C
A solution of B (57.64 g, 154.34 mmol, 1.0 eq.) in toluene (115 mL, 2.0 vol.) is diluted with MTBE (173 mL, 3.0 vol.) at room temperature. Mesyl chloride (17.9 mL, 26.5 g, 231.50 mmol, 1.5 eq.) is then added at room temperature and the homogeneous mixture is cooled to 10°C. The addition of triethylamine (43.0 mL, 31.2 g, 308.67 mmol, 2.0 eq.) is performed at T<20°C. At the end of the addition, the mixture is stirred at 10°C until conversion is complete (B<1%). After hydrolysis with diluted HCl, the organic layer is washed with NaHC03, water and brine, followed by a partial concentration under reduced pressure. The corresponding mesylate is then crystallised by addition of heptanes (5.0 vol.) at 40°C. After cooling, filtration and drying, the expected product is isolated as a whitish solid in 92.5% yield and with a very high chemical purity (98%).
Step (c): (S) 2-(benzyloxycarbonylamino) 4-(neopentyloxysulfonyl)butyl thioacetate D
D
A solution of mesylate C (81.3 g, 180.05 mmol, 1.0 eq.) in acetone (203 mL, 2.5 vol.) is added dropwise to a suspension of potassium thioacetate (41.1 g, 360.1 mmol, 2.0 eq.) in acetone (203 mL, 2.5 vol.) at room temperature and over a period of 2 hrs. The reaction mixture is stirred at room temperature until conversion is complete (C<1%). After filtration of the salts and addition of toluene (4.0 vol.), acetone is removed by distillation under reduced pressure at 25°C. The solution is then treated with active charcoal and concentrated to 2.0 volumes. Slow addition of heptane (5.0 vol.) at room temperature, followed by cooling at 0°C, filtration and drying at 45°C, provides the expected product as a whitish solid in 78.2% yield and with a very high chemical purity (98%).
Step (d): (3S,3S’) neopentyl 4,4′-disulfanediylbis(3-(benzyloxycarbonylamino)butane 1-sulfonate) E
E
A solution of D (59.16 g, 137.1 mmol, 1.0 eq.) suspended in ethanol (203 mL, 2.5 vol.) is cooled to 0°C. 20% sodium hydroxide (25.1 mL, 150.8 mmol, 1.1 eq.) diluted with water
(16.9 mL, 0.285 vol.) is then added dropwise to the suspension by keeping the temperature below 10°C. The reaction mixture is warmed to room temperature and stirred until conversion is complete (D<1%). The intermediate thiol reacts at room temperature with a solution of iodine (20.9 g, 82.3 mmol, 0.6 eq.) in ethanol (118 mL, 2.0 vol.). The reaction is complete at the end of the addition of the oxidizing agent. After addition of a Na2S205 (13.0 g, 68.5 mmol, 0.5 eq.) aqueous solution (118 mL, 2.0 vol.) to reduce the excess of residual iodine, ethanol is removed by distillation under reduced pressure at 40°C. Addition of water (3.0 vol.) at room temperature, followed by cooling at 0°C, filtration and drying at 45-50°C, provides the expected dimer as a white solid in 98.3% yield and with a very high chemical purity (97.0%). The amount of iodide ions, coming from the reduction of iodine, is checked in the sample by potentiometric assay.
E°(Ag+/Ag(s))=0.80V
KsAgi=1.5.10“16
[AgNO3]=0.1N
Electrode: E=E°(Ag+/Ag(s))+0.061og[Ag+]
E=E°(Ag+/Ag(s))+0.061og (Ksi/[L])
Assay: [T] decreases and E increases
LOD=l mg
Four further washings with water are performed until no more iodide ions are detected. The results are presented in table 2.
Table 2.
Step (e): (3S,3S’) 4,4′-disulfanediylbis(3-aminobutane 1-sulfonic acid) compound I
4
Compound I
A solution of E (44.0 g, 56.6 mmol, 1.0 eq.) in TFA (220 mL, 5.0 vol.) and anisole (44 mL, 1.0 vol.) is heated to reflux (75°C) and the reaction mixture is stirred in these conditions until conversion is complete (E<1%). TFA is removed by distillation under reduced pressure at 50°C. Slow addition of MTBE (5.0 vol.) at room temperature makes the expected product precipitate. After trituration, filtration and washing with MTBE (1.0 vol.), the crude solid is suspended in methanol (220 mL, 5.0 vol.). New trituration, filtration and washing with MTBE (1.0 vol.), followed by drying under reduced pressure, provides compound I as a white solid in 92.5% yield.
NMR: 1H (solvent D20, 400 MHz, ppm): 4.70 (s, 6H, ¾); 3.77 (m, 2H, H2); 3.14 (dd, 2H, Hi); 2.98 (dd, 4H, H4); 2.86 (dd, 2H, Hi); 2.13 (m, 4H, H3). 13C (solvent D20, 100 MHz, ppm): 49.4 (2C, C2); 46.6 (2C, C4); 38.3 (2C, C ; 26.9 (2C, C3).
////////

AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
- OriginatorCNRS; INSERM; University Paris Descartes
- DeveloperQuantum Genomics
- ClassAmines; Aminopeptidases; Antihypertensives; Cardiovascular therapies; Disulfides; Heart failure therapies; Metalloexopeptidases; Small molecules; Sulfonic acids
- Mechanism of ActionGlutamyl aminopeptidase inhibitors
- Orphan Drug StatusNo
- New Molecular EntityYes
- Phase IIIHypertension
- Phase IIChronic heart failure; Left ventricular dysfunction
- 28 Mar 2022No recent reports of development identified for phase-I development in Hypertension(In volunteers) in United Kingdom (PO, Tablet)
- 25 Nov 2021Firibastat licensed to Teva in Israel
- 11 Oct 2021Quantum Genomics plans a phase III trial for Heart failure
////////Firibastat, фирибастат , فيريباستات , firibastatum, фирибастат ,فيريباستات ,非立巴司他 , rb 150, (+/-)-QGC-001, qgc 001,
C(CS(=O)(=O)O)C(CSSCC(CCS(=O)(=O)O)N)N

Enobosarm

Enobosarm
- Molecular FormulaC19H14F3N3O3
- Average mass389.328 Da
(2S)-3-(4-Cyanophenoxy)-N-[4-cyano-3-(trifluoromethyl)phenyl]-2-hydroxy-2-methylpropanamide
(2S)-3-(4-Cyanophénoxy)-N-[4-cyano-3-(trifluorométhyl)phényl]-2-hydroxy-2-méthylpropanamide
841205-47-8[RN]
GTx-024, MK 2866, Ostarine[Trade name]
Enobosarm, also known as ostarine or MK-2866, is an investigational selective androgen receptor modulator (SARM) developed by GTx, Inc. for the treatment of conditions such as muscle wasting and osteoporosis, formerly under development by Merck & Company.
Chemistry
According to a 2009 paper authored by GTx, “Readers are cautioned to note that the name ostarine is often mistakenly linked to the chemical structure of [S-4], which is also known as andarine. The chemical structure of ostarine has not been publicly disclosed.”[2] A 2009 review stated “Recently, GTx disclosed that compound 5 had advanced into clinical trials. The patent application described detailed data in an initial proof-of-concept Phase IIa clinical trial. It is not explicitly stated that compound 5 is Ostarine (MK-2866).[3]
As of 2012, the mechanism of action of Enobosarm is still being debated and requires further investigation.[4]
Enobosarm is in phase II clinical studies for the treatment of metastatic breast cancer. It has been in phase III clinical trials for the treatment of muscle wasting in patients with non-small cell lung cancer. However, this research has been discontinued.
Enobosarm was discovered by University of Tennessee, then licensed to GTx later. It was granted fast track designation by FDA in 2013 for treatment of muscle wasting in patients with non-small cell lung cancer. Route 1

2. US20100249228A1.
3. US2014080905A1.
4. US20070173546A1. Route 2

1. US20070123563A1.2. US20100249228A1.
////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
| Clinical data | |
|---|---|
| Other names | GTx-024; MK-2866; Ostarine; S-22[1] |
| Routes of administration | By mouth |
| ATC code | none |
| Legal status | |
| Legal status | US: Investigational New Drug |
| Pharmacokinetic data | |
| Elimination half-life | 24 hours[citation needed] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 841205-47-8 |
| PubChem CID | 11326715 |
| ChemSpider | 9501667 |
| UNII | O3571H3R8N |
| KEGG | D10221 |
| CompTox Dashboard (EPA) | DTXSID30233006 |
| Chemical and physical data | |
| Formula | C19H14F3N3O3 |
| Molar mass | 389.334 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| Melting point | 132 to 136 °C (270 to 277 °F) |
| showSMILES | |
| showInChI | |
History
GTx Incorporated was founded in Memphis in 1997 and licensed rights to enobosarm from the University of Tennessee Research Foundation; the SARM compounds were invented by James T. Dalton, Duane D. Miller, Karen A. Veverka and their research teams at Ohio State University, the University of Tennessee and GTx, respectively.[5]
By 2007, enobosarm was in a Phase II trial, and that year GTx signed an exclusive license agreement for its SARM program with Merck & Co.[6] The companies ended the deal in 2010.[7]
In August 2011, there was a double-blind, placebo controlled phase II trial that focused on elderly men and postmenopausal women which concluded that Enobosarm showed statistically significant improvements in total lean body mass and physical function without the negative side effects that are normally present with steroids.[8]
In August 2013, GTx announced that enobosarm had failed in two Phase III clinical trials to treat wasting in people with lung cancer.[9] The company had invested around $35 million in the development of the drug.[10] The company said at that time that it planned to pursue approval of enobosarm in Europe; the company was also still developing GTx-758 for castration-resistant prostate cancer.[11]
In 2016, GTx began Phase II trials, to see if enosobarm might be effective to treat stress urinary incontinence in women.[12]
In 2018, GTx announced the Phase II trials on Enobosarm’s efficacy on stress urinary incontinence[13] in women failed to achieve its primary endpoint in the ASTRID Trial.
Health effects
The FDA has warned that SARMs can have serious side effects ranging from risk of heart attack to stroke and liver damage.[14]
Society and culture
Doping
SARMs including Enobosarm may be and have been used by athletes to assist in training and increase physical stamina and fitness, potentially producing effects similar to anabolic steroids. For this reason, SARMs were banned by the World Anti-Doping Agency in January 2008, despite no drugs from this class yet being in clinical use, and blood tests for all known SARMs have been developed.[15][16] There are a variety of known cases of doping in sports with enobosarm by professional athletes.
Further information: List of doping in sport cases § Enobosarm
In May 2017, Dynamic Technical Formulations voluntarily recalled all lots of Tri-Ton, a dietary supplement that the USFDA tested and found to contain Enobosarm and andarine.[17]
In October 2018, UFC fighter Sean O’Malley tested positive for Enobosarm and was suspended by the Nevada State Athletic Commission and USADA for six months. O’Malley tested positive again on May 25, 2019 and was suspended for nine months by the same agencies. USADA determined that none of O’Malley’s positive tests were consistent with intentional use and he was allowed to compete at UFC 248 as long as he kept his levels below the threshold of 100 ng/ml.[18]
On January 7, 2019, the College National Football Championship was played between University of Alabama and Clemson University. Prior to the College Football National Championship game, three Clemson players who were suspended — Dexter Lawrence, Braden Galloway and Zach Giellaall — tested positive for a substance known as Enobosarm (ostarine). On June 23, 2019 Clemson did not release ostarine investigation findings, citing privacy law.[19]
In July 2019, National Football League player Taylor Lewan failed a drug test for Enobosarm, which Lewan claimed he ingested accidentally as an unlabeled ingredient in a supplement.[20]
On October 23, 2020, the Union Cycliste Internationale (UCI) announced that the Italian rider Matteo Spreafico has been notified of two adverse analytical findings (AAFs) for Enobosarm in two samples collected during the Giro d’Italia on 15–16 October 2020.[21]
On July 6, 2021, during the 2020 Summer Olympics, Brazil women’s national volleyball team player Tandara was temporarily suspended for testing positive for Ostarine. The test was carried out and identified by the Brazilian Doping Control Authority (ABDC).[22]
On August 12, 2021, after the 2020 Summer Olympics, Chijindu “CJ” Ujah, A member of the silver medal-winning British 4×100 relay team was temporarily suspended for testing positive for both Ostarine and S-23. The sample was collected post event by the International Testing Agency and confirmed two days later as positive. The case was referred to the anti-doping division of the Court of Arbitration for Sport.[23] Finally in February 2022, Great Britain were stripped of their silver medal.[24]
In October 2021, two Thoroughbred horses named Arafat and Komunist tested positive for ostarine after races at Woodbine Racetrack. In a decision of the Alcohol and Gaming Commission of Ontario issued May 30, 2022, the horses were declared unplaced in the races in question, and their trainer Robert Gerl was fined $100,000 (as well as forfeiting prize money) and suspended from racing for 20 years.[25]
In May 2022, National Football League Wide Receiver DeAndre Hopkins was suspended six games without pay by the NFL for violating the league’s performance-enhancing drug policy. According to Hopkins, he tested positive for ostarine.[26]
Wider use
In recent years, ostarine and related substances have increasingly become used by the general public as “gym supplements” such as pre-workout or lifestyle drugs, rather than as an aid to performance in athletic or bodybuilding competitions. In 2018, analysis of a fatberg from a sewer in central London showed ostarine to be the most abundant pharmaceutical drug detected, and was present at higher concentration than recreational drugs such as MDMA and cocaine. While this isolated result may not be representative of overall levels of use, for ostarine to be detectable in sewer deposits reflects significant levels of ostarine use in the area close to where the sample was collected.[27]
See also
References
- ^ “Enobosarm – GTx”. Adis Insight. Springer Nature Switzerland AG. Retrieved 25 April 2018.
- ^ Mohler ML, Bohl CE, Jones A, Coss CC, Narayanan R, He Y, et al. (June 2009). “Nonsteroidal selective androgen receptor modulators (SARMs): dissociating the anabolic and androgenic activities of the androgen receptor for therapeutic benefit”. Journal of Medicinal Chemistry. 52 (12): 3597–617. doi:10.1021/jm900280m. PMID 19432422.
- ^ Zhang X, Lanter JC, Sui Z (September 2009). “Recent advances in the development of selective androgen receptor modulators”. Expert Opinion on Therapeutic Patents. 19 (9): 1239–58. doi:10.1517/13543770902994397. PMID 19505196. S2CID 46186955. The first quoted sentence is cited to Published PCT application WO2008127717
- ^ Dubois V, Laurent M, Boonen S, Vanderschueren D, Claessens F (May 2012). “Androgens and skeletal muscle: cellular and molecular action mechanisms underlying the anabolic actions”. Cellular and Molecular Life Sciences. 69 (10): 1651–67. doi:10.1007/s00018-011-0883-3. PMID 22101547. S2CID 17276140.
- ^ WO 2005120483, Dalton JT, Mille DD, Veverka KA, “Selective androgen receptor modulators and methods of use thereof”, published 22 December 2005, assigned to University of Tennessee Research Foundation
- ^ Nagle M (7 November 2007). “Merck flexes muscle with GTx deal”. Outsourcing Pharma.
- ^ Swanekamp K (15 March 2010). “Merck And GTx Go Their Separate Ways”. Forbes.
- ^ Dalton JT, Barnette KG, Bohl CE, Hancock ML, Rodriguez D, Dodson ST, et al. (September 2011). “The selective androgen receptor modulator GTx-024 (enobosarm) improves lean body mass and physical function in healthy elderly men and postmenopausal women: results of a double-blind, placebo-controlled phase II trial”. Journal of Cachexia, Sarcopenia and Muscle. 2 (3): 153–161. doi:10.1007/s13539-011-0034-6. PMC 3177038. PMID 22031847.
- ^ “Enobosarm fails endpoints in Ph III study”. The Pharma Letter. 20 August 2013.
- ^ Sheffield M (April 4, 2014). “Steiner resigns from GTx”. Memphis Business Journal.
- ^ Garde D (4 April 2014). “GTx’s CEO finds the door as the company moves on from a PhIII failure”. FierceBiotech.
- ^ “GTx begins Phase II trial of enobosarm to treat women with stress urinary incontinence”. Drug Development Technology. 14 January 2016. Archived from the original on 22 June 2016.
- ^ “GTx’s Enobosarm Fails Phase II Trial in Stress Urinary Incontinence; Stock Plunges 90%+”. Genetic Engineering & Biotechnology News. Retrieved 1 August 2019.
- ^ “FDA In Brief: FDA warns against using SARMs in body-building products”. Retrieved 1 August 2019.
- ^ Thevis M, Kohler M, Schlörer N, Kamber M, Kühn A, Linscheid MW, Schänzer W (May 2008). “Mass spectrometry of hydantoin-derived selective androgen receptor modulators”. Journal of Mass Spectrometry. 43 (5): 639–50. Bibcode:2008JMSp…43..639T. doi:10.1002/jms.1364. PMID 18095383.
- ^ Thevis M, Kohler M, Thomas A, Maurer J, Schlörer N, Kamber M, Schänzer W (May 2008). “Determination of benzimidazole- and bicyclic hydantoin-derived selective androgen receptor antagonists and agonists in human urine using LC-MS/MS”. Analytical and Bioanalytical Chemistry. 391 (1): 251–61. doi:10.1007/s00216-008-1882-6. PMID 18270691. S2CID 206899531.
- ^ “Dynamic Technical Formulations, LLC. Issues a Voluntary Nationwide Recall of Tri-Ton Due to the Presence of Andarine and Ostarine”. U.S. Food & Drug Administration. May 19, 2017.
- ^ Raimondi M (January 22, 2020). “NSAC: Sean O’Malley can fight at UFC 248 in March after serving suspension”. ESPN. Retrieved June 9, 2020.
- ^ Needelman J (14 September 2020). “Clemson lineman suspended by ncaa for positive ostarine test opens up for first time”. Retrieved November 13, 2020.
- ^ Bieler D (25 July 2019). “Failed PED test has a highly paid offensive lineman sharing polygraph results”. Washington Post. Retrieved 25 July 2019.
One of the NFL’s highest-paid offensive linemen claimed Wednesday that he did not knowingly take a banned substance he says got him a four-game suspension — and he took a polygraph test in an attempt to prove it.
- ^ “UCI statement on Matteo Spreafico”. Union Cycliste Internationale (UCI). 22 October 2020. Retrieved 2020-10-23.
- ^ “Tandara é suspensa por “potencial violação” do antidoping e está fora das Olimpíadas”.
- ^ “Tokyo Olympics: Team GB 4x100m relay silver medallist CJ Ujah suspended for suspected doping violation”.
- ^ “CJ Ujah: Great Britain lose Tokyo Olympics relay medal after doping violation”. BBC. 18 February 2022.
- ^ “IN THE MATTER OF THE HORSE RACING LICENCE ACT, 2015, S.0.2015,c.38,Sched.9; AND IN THE MATTER OF Robert Gerl” (PDF). Retrieved 2 June 2022.
- ^ “Cardinals WR DeAndre Hopkins still hopes to reduce six-game suspension”. NFL.com. 23 June 2022.
- ^ Saner E (24 April 2018). “Why there are more gym supplements in a London fatberg than cocaine and MDMA”. The Guardian.
//////////GTx-024, MK 2866, Ostarine, enobosarm
O=C(NC1=CC=C(C#N)C(C(F)(F)F)=C1)[C@](C)(O)COC2=CC=C(C#N)C=C2
Betibeglogene autotemcel

Betibeglogene autotemcel
ベチベグロゲンアウトテムセル
2022/8/17, FDA APPROVED Zynteglo
Cellular therapy product
Treatment of betathalassemia
BB305 LVV
bb 1111
BB305 transduced SCD CD34+ HSCs bb1111
LentiGlobin BB305 LVV-transduced autologous SCD CD34+ HSCs bb1111
LentiGlobin drug product for SCD
LentiGlobin drug product for sickle cell disease
LentiGlobin for SCD bb1111
Betibeglogene autotemcel, sold under the brand name Zynteglo, is a medication for the treatment for beta thalassemia.[1][5][2] It was developed by Bluebird Bio and was given breakthrough therapy designation by the U.S. Food and Drug Administration in February 2015.[6][7]
The most common adverse reactions include reduced platelet and other blood cell levels, as well as mucositis, febrile neutropenia, vomiting, pyrexia (fever), alopecia (hair loss), epistaxis (nosebleed), abdominal pain, musculoskeletal pain, cough, headache, diarrhea, rash, constipation, nausea, decreased appetite, pigmentation disorder and pruritus (itch).[5]
It was approved for medical use in the European Union in May 2019,[2] and in the United States in August 2022.[5]
FDA Approves First Cell-Based Gene Therapy to Treat Adult and Pediatric Patients with Beta-thalassemia Who Require Regular Blood Transfusions
https://www.fda.gov/news-events/press-announcements/fda-approves-first-cell-based-gene-therapy-treat-adult-and-pediatric-patients-beta-thalassemia-whoFor Immediate Release:August 17, 2022
Today, the U.S. Food and Drug Administration approved Zynteglo (betibeglogene autotemcel), the first cell-based gene therapy for the treatment of adult and pediatric patients with beta-thalassemia who require regular red blood cell transfusions.
“Today’s approval is an important advance in the treatment of beta-thalassemia, particularly in individuals who require ongoing red blood cell transfusions,” said Peter Marks, M.D., Ph.D., director of the FDA’s Center for Biologics Evaluation and Research. “Given the potential health complications associated with this serious disease, this action highlights the FDA’s continued commitment to supporting development of innovative therapies for patients who have limited treatment options.”
Beta-thalassemia is a type of inherited blood disorder that causes a reduction of normal hemoglobin and red blood cells in the blood, through mutations in the beta-globin subunit, leading to insufficient delivery of oxygen in the body. The reduced levels of red blood cells can lead to a number of health issues including dizziness, weakness, fatigue, bone abnormalities and more serious complications. Transfusion-dependent beta-thalassemia, the most severe form of the condition, generally requires life-long red blood cell transfusions as the standard course of treatment. These regular transfusions can be associated with multiple health complications of their own, including problems in the heart, liver and other organs due to an excessive build-up of iron in the body.
Zynteglo is a one-time gene therapy product administered as a single dose. Each dose of Zynteglo is a customized treatment created using the patient’s own cells (bone marrow stem cells) that are genetically modified to produce functional beta-globin (a hemoglobin component).
The safety and effectiveness of Zynteglo were established in two multicenter clinical studies that included adult and pediatric patients with beta-thalassemia requiring regular transfusions. Effectiveness was established based on achievement of transfusion independence, which is attained when the patient maintains a pre-determined level of hemoglobin without needing any red blood cell transfusions for at least 12 months. Of 41 patients receiving Zynteglo, 89% achieved transfusion independence.
The most common adverse reactions associated with Zynteglo included reduced platelet and other blood cell levels, as well as mucositis, febrile neutropenia, vomiting, pyrexia (fever), alopecia (hair loss), epistaxis (nosebleed), abdominal pain, musculoskeletal pain, cough, headache, diarrhea, rash, constipation, nausea, decreased appetite, pigmentation disorder and pruritus (itch).
There is a potential risk of blood cancer associated with this treatment; however, no cases have been seen in studies of Zynteglo. Patients who receive Zynteglo should have their blood monitored for at least 15 years for any evidence of cancer. Patients should also be monitored for hypersensitivity reactions during Zynteglo administration and should be monitored for thrombocytopenia and bleeding.
This application was granted a rare pediatric disease voucher, in addition to receiving Priority Review, Fast Track, Breakthrough Therapy, and Orphan designations.
The FDA granted approval of Zynteglo to bluebird bio, Inc.
/////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
| Clinical data | |
|---|---|
| Trade names | Zynteglo |
| Other names | LentiGlobin BB305, autologous CD34+ cells encoding βA-T87Q-globin gene |
| License data | EU EMA: by INNUS DailyMed: Betibeglogene autotemcel |
| Pregnancy category | Contraindicated[1][2] |
| Routes of administration | Intravenous[3] |
| ATC code | B06AX02 (WHO) |
| Legal status | |
| Legal status | UK: POM (Prescription only) [1]US: ℞-only [3][4][5]EU: Rx-only [2]In general: ℞ (Prescription only) |
| Identifiers | |
| UNII | MEE8487RTP |
| KEGG | D11930 |
Medical uses
Betibeglogene autotemcel is indicated for the treatment of people twelve years and older with transfusion-dependent beta thalassemia (TDT) who do not have a β0/β0 genotype, for whom hematopoietic stem cell (HSC) transplantation is appropriate but a human leukocyte antigen (HLA)-matched related HSC donor is not available.[2]
Betibeglogene autotemcel is made individually for each recipient out of stem cells collected from their blood, and must only be given to the recipient for whom it is made.[2] It is given as an autologous intravenous infusion and the dose depends on the recipient’s body weight.[3][2]
Before betibeglogene autotemcel is given, the recipient receives conditioning chemotherapy to clear their bone marrow of cells (myeloablation).[2]
To make betibeglogene autotemcel, the stem cells taken from the recipient’s blood are modified by a virus that carries working copies of the beta globin gene into the cells.[2] When these modified cells are given back to the recipient, they are transported in the bloodstream to the bone marrow where they start to make healthy red blood cells that produce beta globin.[2] The effects of betibeglogene autotemcel are expected to last for the recipient’s lifetime.[2]
Mechanism of action
Beta thalassemia is caused by mutations to or deletions of the HBB gene leading to reduced or absent synthesis of the beta chains of hemoglobin that result in variable outcomes ranging from severe anemia to clinically asymptomatic individuals.[8] LentiGlobin BB305 is a lentiviral vector which inserts a functioning version of the HBB gene into a recipient’s blood-producing hematopoietic stem cells (HSC) ex vivo. The resulting engineered HSCs are then reintroduced to the recipient.[9][10]
History
In early clinical trials several participants with beta thalassemia, who usually require frequent blood transfusions to treat their disease, were able to forgo blood transfusions for extended periods of time.[11][12][13] In 2018, results from phase 1-2 trials suggested that of 22 participants receiving Lentiglobin gene therapy, 15 were able to stop or reduce regular blood transfusions.[14][15]
In February 2021, a clinical trial[16] of betibeglogene autotemcel in sickle cell anemia was suspended following an unexpected instance of acute myeloid leukemia.[17] The HGB-206 Phase 1/2 study is expected to conclude in March 2023.[16]
It was designated an orphan drug by the European Medicines Agency (EMA) and by the U.S. Food and Drug Administration (FDA) in 2013.[2][18] The Food and Drug Administration has also declared betibeglogene autotemcel a Regenerative Medicine Advanced Therapy.[19]
The safety and effectiveness of betibeglogene autotemcel were established in two multicenter clinical studies that included adult and pediatric particpiants with beta-thalassemia requiring regular transfusions.[5] Effectiveness was established based on achievement of transfusion independence, which is attained when the particpiant maintains a pre-determined level of hemoglobin without needing any red blood cell transfusions for at least 12 months. Of 41 particpiants receiving betibeglogene autotemcel, 89% achieved transfusion independence.[5]
Society and culture
Legal status
It was approved for medical use in the European Union in May 2019,[2] and in the United States in August 2022.[5]
Names
The international nonproprietary name (INN) is betibeglogene autotemcel.[20]
References
- ^ Jump up to:a b c “Zynteglo dispersion for infusion – Summary of Product Characteristics (SmPC)”. (emc). 12 May 2020. Retrieved 3 January 2021.[permanent dead link]
- ^ Jump up to:a b c d e f g h i j k l m “Zynteglo EPAR”. European Medicines Agency (EMA). 25 March 2019. Archived from the original on 16 August 2019. Retrieved 16 August 2019. Text was copied from this source which is © European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ Jump up to:a b c “Archived copy”. Archived from the original on 26 August 2022. Retrieved 26 August 2022.
- ^ “Zynteglo”. U.S. Food and Drug Administration. 17 August 2022. Archived from the original on 26 August 2022. Retrieved 26 August 2022.
- ^ Jump up to:a b c d e f g “FDA Approves First Cell-Based Gene Therapy to Treat Adult and Pediatric Patients with Beta-thalassemia Who Require Regular Blood Transfusions”. U.S. Food and Drug Administration (FDA) (Press release). 17 August 2022. Archived from the original on 21 August 2022. Retrieved 20 August 2022.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ “Ten things you might have missed Monday from the world of business”. The Boston Globe. 3 February 2015. Archived from the original on 1 August 2020. Retrieved 13 February 2015.
- ^ “Lentiviral vectors”. 27 June 2019. Archived from the original on 21 August 2022. Retrieved 8 July 2019.
- ^ Cao A, Galanello R (February 2010). “Beta-thalassemia”. Genetics in Medicine. 12 (2): 61–76. doi:10.1097/GIM.0b013e3181cd68ed. PMID 20098328.
- ^ Negre O, Bartholomae C, Beuzard Y, Cavazzana M, Christiansen L, Courne C, et al. (2015). “Preclinical evaluation of efficacy and safety of an improved lentiviral vector for the treatment of β-thalassemia and sickle cell disease” (PDF). Current Gene Therapy. 15 (1): 64–81. doi:10.2174/1566523214666141127095336. PMC 4440358. PMID 25429463. Archived (PDF) from the original on 19 July 2018. Retrieved 19 June 2018.
- ^ Thompson AA, Rasko JE, Hongeng S, Kwiatkowski JL, Schiller G, von Kalle C, et al. (2014). “Initial Results from the Northstar Study (HGB-204): A Phase 1/2 Study of Gene Therapy for β-Thalassemia Major Via Transplantation of Autologous Hematopoietic Stem Cells Transduced Ex Vivo with a Lentiviral βΑ-T87Q -Globin Vector (LentiGlobin BB305 Drug Product)”. Blood. 124 (21): 549. doi:10.1182/blood.V124.21.549.549. Archived from the original on 18 October 2019. Retrieved 13 February 2015.
- ^ Cavazzana-Calvo M, Payen E, Negre O, Wang G, Hehir K, Fusil F, et al. (September 2010). “Transfusion independence and HMGA2 activation after gene therapy of human β-thalassaemia”. Nature. 467 (7313): 318–322. Bibcode:2010Natur.467..318C. doi:10.1038/nature09328. PMC 3355472. PMID 20844535.
- ^ Winslow R (8 December 2015). “New Gene Therapy Shows Promise for Lethal Blood Disease”. The Wall Street Journal. Archived from the original on 2 March 2020. Retrieved 13 February 2015.
- ^ (8 December 2014) bluebird bio Announces Data Demonstrating First Four Patients with β-Thalassemia Major Treated with LentiGlobin are Transfusion-Free Archived 26 September 2015 at the Wayback Machine Yahoo News, Retrieved 17 May 2015
- ^ Thompson AA, Walters MC, Kwiatkowski J, Rasko JE, Ribeil JA, Hongeng S, et al. (April 2018). “Gene Therapy in Patients with Transfusion-Dependent β-Thalassemia”. The New England Journal of Medicine. 378 (16): 1479–1493. doi:10.1056/NEJMoa1705342. PMID 29669226.
- ^ Stein R (18 April 2018). “Gene Therapy For Inherited Blood Disorder Reduced Transfusions”. NPR. Archived from the original on 21 August 2022. Retrieved 4 March 2019.
- ^ Jump up to:a b Clinical trial number NCT02140554 for “A Phase 1/2 Study Evaluating Gene Therapy by Transplantation of Autologous CD34+ Stem Cells Transduced Ex Vivo With the LentiGlobin BB305 Lentiviral Vector in Subjects With Severe Sickle Cell Disease” at ClinicalTrials.gov
- ^ “Bluebird bio Halts Sickle Cell Trials After Leukemia Diagnosis”. BioSpace. Archived from the original on 27 June 2021. Retrieved 27 June 2021.
- ^ “Autologous CD34+ hematopoietic stem cells transduced with LentiGlobin BB305 lentiviral vector encoding the human BA-T87Q-globin gene Orphan Drug Designations and Approvals”. U.S. Food and Drug Administration (FDA). 18 March 2013. Archived from the original on 9 June 2020. Retrieved 8 June 2020.
- ^ “bluebird bio Announces Temporary Suspension on Phase 1/2 and Phase 3 Studies of LentiGlobin Gene Therapy for Sickle Cell Disease (bb1111)”. Bluebird Bio (Press release). 16 February 2021. Archived from the original on 27 June 2021. Retrieved 27 June 2021.
- ^ World Health Organization (2020). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 83”. WHO Drug Information. 34 (1): 34. Archived from the original on 15 July 2020.
////////////Betibeglogene autotemcel, FDA 2022, APPROVALS 2022, ベチベグロゲンアウトテムセル , Zynteglo, bluebird bio, bb 1111
BB305 transduced SCD CD34+ HSCs bb1111
LentiGlobin BB305 LVV-transduced autologous SCD CD34+ HSCs bb1111
LentiGlobin drug product for SCD
LentiGlobin drug product for sickle cell disease
LentiGlobin for SCD bb1111

NEW DRUG APPROVALS
one time
$10.00
ATISOBAN


ATOSIBAN
cas 90779-69-4
WeightAverage: 994.19
Monoisotopic: 993.441208989
Chemical FormulaC43H67N11O12S2
(2S)-5-amino-2-{[(2S)-1-[(4R,7S,10S,13S,16R)-13-[(2S)-butan-2-yl]-7-(carbamoylmethyl)-16-[(4-ethoxyphenyl)methyl]-10-[(1R)-1-hydroxyethyl]-6,9,12,15,18-pentaoxo-1,2-dithia-5,8,11,14,17-pentaazacycloicosane-4-carbonyl]pyrrolidin-2-yl]formamido}-N-(carbamoylmethyl)pentanamide
- Oxytocin, 1-(3-mercaptopropanoic acid)-2-(O-ethyl-D-tyrosine)-4-L-threonine-8-L-ornithine-
- 1,2-Dithia-5,8,11,14,17-pentaazacycloeicosane, cyclic peptide deriv.
- Antocile
- Antocin
- Antocin II
Product Ingredients
| INGREDIENT | UNII | CAS | INCHI KEY |
|---|---|---|---|
| Atosiban acetate | 0P5DNO7CEF | 914453-95-5 | SVDWBHHCPXTODI-QIWYXCRTSA-N |
- CAP-449
- CAP-476
- CAP-581
- F-314
- ORF 22164
- ORF-22164
- RW-22164
- RWJ 22164
- RWJ-22164
Atosiban, sold under the brand name Tractocile among others, is an inhibitor of the hormones oxytocin and vasopressin. It is used as an intravenous medication as a labour repressant (tocolytic) to halt premature labor. It was developed by Ferring Pharmaceuticals in Sweden and first reported in the literature in 1985.[5] Originally marketed by Ferring Pharmaceuticals, it is licensed in proprietary and generic forms for the delay of imminent preterm birth in pregnant adult women.
The most commonly reported side effect is nausea.[4]
Atosiban is an inhibitor of the hormones oxytocin and vasopressin. It is used intravenously to halt premature labor. Although initial studies suggested it could be used as a nasal spray and hence would not require hospital admission, it is not used in that form. Atobisan was developed by the Swedish company Ferring Pharmaceuticals. It was first reported in the literature in 1985. Atosiban is licensed in proprietary and generic forms for the delay of imminent pre-term birth in pregnant adult women.
Medical uses
Atosiban is used to delay birth in adult women who are 24 to 33 weeks pregnant, when they show signs that they may give birth pre-term (prematurely).[4] These signs include regular contractions lasting at least 30 seconds at a rate of at least four every 30 minutes,[4] and dilation of the cervix (the neck of the womb) of 1 to 3 cm and an effacement (a measure of the thinness of the cervix) of 50% or more.[4] In addition, the baby must have a normal heart rate.[4]
Pharmacology
Mechanism of action
Atosiban is a nonapeptide, desamino-oxytocin analogue, and a competitive vasopressin/oxytocin receptor antagonist (VOTra). Atosiban inhibits the oxytocin-mediated release of inositol trisphosphate from the myometrial cell membrane. As a result, reduced release of intracellular, stored calcium from the sarcoplasmic reticulum of myometrial cells and reduced influx of Ca2+ from the extracellular space through voltage-gated channels occur. In addition, atosiban suppresses oxytocin-mediated release of PGE and PGF from the decidua.[6]
In human preterm labour, atosiban, at the recommended dosage, antagonises uterine contractions and induces uterine quiescence. The onset of uterus relaxation following atosiban is rapid, uterine contractions being significantly reduced within 10 minutes to achieve stable uterine quiescence.
Other uses
Atosiban use after assisted reproduction
Atosiban is useful in improving the pregnancy outcome of in vitro fertilization-embryo transfer (IVF-ET) in patients with repeated implantation failure.[7] The pregnancy rate improved from zero to 43.7%.[8]
First- and second-trimester bleeding was more prevalent in ART than in spontaneous pregnancies. From 2004 to 2010, 33 first-trimester pregnancies with vaginal bleeding after ART with evident uterine contractions, when using atosiban and/or ritodrine, no preterm delivery occurred before 30 weeks.[9]
In a 2010 meta-analysis,[10] nifedipine is superior to β2 adrenergic receptor agonists and magnesium sulfate for tocolysis in women with preterm labor (20–36 weeks), but it has been assigned to pregnancy category C by the U.S. Food and Drug Administration, so is not recommended before 20 weeks, or in the first trimester.[9] A report from 2011 supports the use of atosiban, even at very early pregnancy, to decrease the frequency of uterine contractions to enhance success of pregnancy.[7]
Pharmacovigilance
Following the launch of atosiban in 2000, the calculated cumulative patient exposure to atosiban (January 2000 to December 2005) is estimated as 156,468 treatment cycles. To date, routine monitoring of drug safety has revealed no major safety issues.[11]
Regulatory affairs
Atosiban was approved in the European Union in January 2000 and launched in the European Union in April 2000.[12][4] As of June 2007, atosiban was approved in 67 countries, excluding the United States and Japan.[12] It was understood that Ferring did not expect to seek approval for atosiban in the US or Japan, focusing instead on development of new compounds for use in Spontaneous Preterm Labor (SPTL).[12] The fact that atosiban only had a short duration before it was out of patent that the parent drug company decided not to pursue licensing in the US.[13]
Systematic reviews
In a systematic review of atosiban for tocolysis in preterm labour, six clinical studies — two compared atosiban to placebo and four atosiban to a β agonist — showed a significant increase in the proportion of women undelivered by 48 hours in women receiving atosiban compared to placebo. When compared with β agonists, atosiban increased the proportion of women undelivered by 48 hours and was safer compared to β agonists. Therefore, oxytocin antagonists appear to be effective and safe for tocolysis in preterm labour.[14]
A 2014 systematic review by the Cochrane Collaboration showed that while atosiban had fewer side effects than alternative drugs (such as ritodrine), other beta blockers, and calcium channel antagonists, it was no better than placebo in the major outcomes i.e. pregnancy prolongation or neonatal outcomes. The finding of an increase in infant deaths in one placebo-controlled trial warrants caution. Further research is recommended.[15]
PATENT
WO 2021207870
Atosiban (Atosiban) is an oxytocin and vasopressin V1A combined receptor antagonist, which can be used as a competitive antagonist of cyclic peptide oxytocin receptors in the uterus, decidua and fetal membrane. Atosiban is a disulfide-bonded cyclic polypeptide composed of 9 amino acids. It is a modified oxytocin molecule at positions 1, 2, 4 and 8. The N-terminal of the peptide is 3-mercaptopropionic acid (thiol and [ Cys] 6 thiol forms a disulfide bond), the C-terminal is in the form of an amide, and the second amino acid at the N-terminal is ethylated [D-Tyr(Et)] 2 . Atosiban is generally present in medicines in the form of acetate salt, commonly known as atosiban acetate. Its chemical formula is C 45 H 71 N 11 O 14 S 2 , its molecular weight is 994.19, and its structural formula is as follows:
[0003]
[0004]
In the prior art, atosiban is usually synthesized by a solid-phase peptide synthesis (SPPS) method, an amino resin is used as a starting carrier resin, and protected amino acids are sequentially connected, and the obtained atosiban is oxidized and then cleaved to obtain atosiban. However, the above-mentioned existing process has high cost, generates a large amount of solvent waste, and is not easy to monitor during the cyclization process. In addition, the above-mentioned prior art has deficiencies in the overall yield of crude peptides. Moreover, due to the existence of D-Tyr(Et) in the structure of atosiban, Fmoc-D-Tyr(Et) easily undergoes a racemization reaction during the peptide attachment process, resulting in [Tyr(Et) 2 ]-A The impurity of tosiban, which is similar in polarity to atosiban itself, is difficult to completely remove through purification, thus affecting the quality of atosiban.
[table 0001]
| Amino acid name | alphabetic symbols |
| Glycine | Gly |
| Ornithine | Orn |
| Proline | Pro |
| cysteine | Cys |
| Asparagine | Asn |
| Threonine | Thr |
| Isoleucine | Ile |
| D-tyrosine (oxyethyl) | D-Tyr(ET) |
Table 3 List of intermediates and Fmoc protected amino acids
[0043]
[table 0002]
| Fmoc-Orn(Boc)-OH |
| Fmoc-Pro-OH |
| Fmoc-Cys(Trt)-OH |
| Fmoc-Asn-OH |
| Fmoc-Thr(tBu)-OH |
| Fmoc-Ile-OH |
| Fmoc-D-Tyr(ET)-OH |
| Fmoc-Gly Rink Resin |
| Fmoc-Orn(Boc)-Gly Rink Resin |
| Fmoc-Pro-Orn(Boc)-Gly Rink Resin |
| Fmoc-Cys(Trt)-Pro-Orn(Boc)-Gly Rink Resin |
| Fmoc-Asn-Cys(Trt)-Pro-Orn(Boc)-Gly Rink Resin |
| Fmoc-Thr(tBu)-Asn-Cys(Trt)-Pro-Orn(Boc)-Gly Rink Resin |
| Fmoc-Ile-Thr(tBu)-Asn-Cys(Trt)-Pro-Orn(Boc)-Gly Rink Resin |
| Fmoc-D-Tyr(RT)-Ile-Thr(tBu)-Asn-Cys(Trt)-Pro-Orn(Boc)-Gly Rink Resin |
| Mpa(Trt)-D-Tyr(ET)-Ile-Thr(tBu)-Asn-Cys(Trt)-Pro-Orn(Boc)-Gly Rink Resin |



[0045]
According to the most preferred embodiment of the present invention, the method of the present invention comprises the following steps:
[0046]
The first step: Fmoc-Gly Rink resin can be directly purchased, which reduces the first step of synthesis and improves the synthesis efficiency;
[0047]
The second step: preparing a deprotection solution: the deprotection solution is a mixture of piperidine/N,N-dimethylformamide, preferably piperidine/N,N-dimethylformamide in a volume ratio of 1/4.
[0048]
The third step: preparation of Fmoc-Orn(Boc)-Gly Rink resin: deprotect the Fmoc-Gly Rink resin obtained in the first step, wash with DMF, add Fmoc-Orn(Boc)-OH in DMF solution, Condensation reaction is carried out under the condition of peptide coupling condensing agent to obtain Fmoc-Orn(Boc)-Gly Rink resin;
[0049]
The fourth step: preparation of Fmoc-Pro-Orn(Boc)-Gly Rink resin: the peptide resin obtained in the fourth step is deprotected and washed, and then reacted with Fmoc-Pro-OH under the condition of a peptide coupling agent to obtain Fmoc-Pro-Orn(Boc)-Gly Rink resin;
[0050]
The fifth step: preparation of Fmoc-Cys(Trt)-Pro-Orn(Boc)-Gly Rink resin. The peptide resin obtained in the fifth step is deprotected and washed, and then reacted with Fmoc-Cys(Trt)-OH under the condition of peptide coupling agent to obtain Fmoc-Cys(Trt)-Pro-Orn(Boc)-Gly Rink resin;
[0051]
The sixth step: preparation of Fmoc-Asn-Cys(Trt)-Pro-Orn(Boc)-Gly Rink resin. The peptide resin obtained in the sixth step is deprotected and washed, and then reacted with Fmoc-Asn-OH under the condition of peptide coupling agent to obtain Fmoc-Asn-Cys(Trt)-Pro-Orn(Boc)-Gly Rink resin ;
[0052]
The seventh step: preparation of Fmoc-Thr(tBu)-Asn-Cys(Trt)-Pro-Orn(Boc)-Gly Rink resin. The peptide resin obtained in the seventh step was deprotected and washed, and then reacted with Fmoc-Thr(tBu)-OH under the condition of a peptide coupling agent. Obtain Fmoc-Thr(tBu)-Asn-Cys(Trt)-Pro-Orn(Boc)-Gly Rink resin;
[0053]
The eighth step: preparation of Fmoc-Ile-Thr(tBu)-Asn-Cys(Trt)-Pro-Orn(Boc)-Gly Rink resin. The peptide resin obtained in the eighth step is deprotected and washed, and then reacted with Fmoc-Ile-OH under the condition of a peptide coupling agent to obtain Fmoc-Ile-Thr(tBu)-Asn-Cys(Trt)-Pro-Orn (Boc)-Gly Rink resin;
[0054]
The ninth step: preparation of Fmoc-D-Tyr(RT)-Ile-Thr(tBu)-Asn-Cys(Trt)-Pro-Orn(Boc)-Gly Rink resin. The peptide resin obtained in the ninth step is deprotected and washed, and then reacted with Fmoc-D-Tyr(ET)-OH under the condition of a peptide coupling agent to obtain Fmoc-D-Tyr(RT)-Ile-Thr(tBu )-Asn-Cys(Trt)-Pro-Orn(Boc)-Gly Rink resin;
[0055]
The tenth step: preparation of Mpa(Trt)-D-Tyr(ET)-Ile-Thr(tBu)-Asn-Cys(Trt)-Pro-Orn(Boc)-Gly Rink resin. The peptide resin obtained in the tenth step is deprotected and washed, and then reacted with Mpa(Trt) under the condition of a peptide coupling agent to obtain Mpa(Trt)-D-Tyr(ET)-Ile-Thr(tBu)-Asn -Cys(Trt)-Pro-Orn(Boc)-Gly Rink resin;
[0056]
The eleventh step: Mpa(Trt)-D-Tyr(ET)-Ile-Thr(tBu)-Asn-Cys(Trt)-Pro-Orn(Boc)-Gly Rink resin in TFA/TIS/EDT/H2O =90/54/10/5 TFA, cleaved for 3 hours, and filtered to obtain crude peptide solution;
[0057]
The twelfth step: sedimentation and washing of the crude peptide solution with methyl tert-butyl ether, centrifugation at 2000 rpm, and vacuum drying to obtain a pale yellow solid powder of atosiban linear crude peptide;
[0058]
The thirteenth step: prepare three solutions for atosiban cyclization: solution A-sodium acetate buffered aqueous solution, solution B-aqueous solution of linear peptide atosiban crude peptide acetic acid, solution C: 30%-60% hydrogen peroxide solution ;
[0059]
The fourteenth step: Mix the above three solutions of A, B, and C at 15-25 ° C, and stir for 1-3 hours after mixing, so that the Mpa at the 1st position and the Cys at the 6th position form a disulfide bond to obtain Cyclized atosiban crude peptide.
[0060]
Step fifteen: Purify crude atosiban by preparative high performance liquid chromatography with a water/acetonitrile gradient from 100% water to 100% acetonitrile in 20 minutes.
[0061]
The sixteenth step: freeze-dry the purified atosiban solution at -50 to -70° C. for 18-48 hours with a freeze dryer.
[0062]
The purity of atosiban obtained by the method of the invention is more than 99.5%, and the total product yield is 55%-65%.
[0063]
The advantage of the method for preparing atosiban of the present invention is:
[0064]
The traditional SPPS synthesis of atosiban usually produces a large amount of waste with high disposal costs. This process adopts high-temperature SPPS process and selects different condensing agent combinations, which is faster than the conventional SPPS process, the product purity can reach more than 99.9%, the purity is better than that of the conventional atosiban process, the impurity content is low, and the product quality is high. The total yield can reach 55%-65%.
Detailed ways
[0065]
The invention will now be described with reference to specific embodiments. It must be understood that these examples are merely illustrative of the invention and are not intended to limit the scope of the invention. Unless otherwise stated, percentages and parts are by weight. Unless otherwise specified, experimental materials and reagents used in the following examples were obtained from commercial sources.
[0066]
Example 1:
[0067]
Using Rink-Fmoc-Gly resin (40 g, substitution amount 0.61 mmol/g) as the starting material, the stepwise Fmoc-SPPS (solid phase peptide synthesis) method was used to synthesize the peptide. Fmoc deprotection was performed with piperidine in DMF (1:4 v/v). Subsequently, other amino acids in the sequence are connected in the following order, and the coupling reagents are N,N-diisopropylcarbodiimide, 2-(7-benzotriazole)-N,N,N’,N ‘-Tetramethylurea hexafluorophosphate mixed in a volume ratio of 1:1, Fmoc-Orn(Boc)-OH, Fmoc-Pro-OH, Fmoc-Cys(Trt)-OH, Fmoc-Asn-OH, Fmoc-Thr(tBu)-OH, Fmoc-Ile-OH, Fmoc-D-Tyr(ET)-OH, Mpa(Trt). Coupling and deprotection of amino acids were carried out at 90°C for 2-3 min and monitored with the Kaiser test. The peptide was cleaved with the lysing solution of TFA for 3 hours, precipitated and washed twice with methyl tert-butyl ether, and after centrifugal drying, the atosiban linear crude peptide was cyclized by the method of liquid phase synthesis, and the volume ratio was 1: 2:2 A solution-acetic acid-sodium acetate buffer aqueous solution (concentration is 30g/L), B solution-linear peptide atosiban crude peptide acetic acid aqueous solution and C solution: 60% hydrogen peroxide solution.
[0068]
The crude peptide yield was 85%. Crude atosiban was purified by preparative high performance liquid chromatography with a water/acetonitrile gradient from 100% water to 100% acetonitrile in 20 minutes. The purified atosiban solution is freeze-dried at -50 to -70° C. for 18 hours with a freeze dryer, the obtained atosiban has a purity of more than 99.5%, and the total product yield is 56%.
[0069]
Example 2:
[0070]
Using Rink-Fmoc-Gly resin (40 g, substitution amount 0.36 mmol/g) as the starting material, the stepwise Fmoc-SPPS (solid phase peptide synthesis) method was used to synthesize the peptide. Fmoc deprotection was performed with piperidine in DMF (1:4 v/v). Subsequently, the other amino acids in the sequence are connected in the following order, and the coupling reagents are N,N-tert-dicyclohexylcarbodiimide, 1-hydroxybenzotriazole and Oxyma, which are mixed in a volume ratio of 1:1:1 , Fmoc-Orn(Boc)-OH, Fmoc-Pro-OH, Fmoc-Cys(Trt)-OH, Fmoc-Asn-OH, Fmoc-Thr(tBu)-OH, Fmoc-Ile-OH, Fmoc-D- Tyr(ET)-OH, Mpa(Trt). Coupling and deprotection of amino acids were carried out at 90°C for 2-3 min and monitored with the Kaiser test. The peptide was cleaved with the lysing solution of TFA for 3 hours, precipitated with methyl tert-butyl ether and washed twice, and after centrifugal drying, the atosiban linear crude peptide was cyclized by the method of liquid phase synthesis, and the volume ratio was 1: 3:2 solution A-formic acid-sodium formate buffer aqueous solution (concentration 25g/L), solution B-linear peptide atosiban crude peptide formic acid aqueous solution and solution C: 30% hydrogen peroxide solution, and oxygen was introduced.
[0071]
The crude peptide yield was 83%. Crude atosiban was purified by preparative high performance liquid chromatography with a water/acetonitrile gradient from 100% water to 100% acetonitrile in 20 minutes. The purified atosiban solution is freeze-dried at -50 to -70° C. for 18 hours with a freeze dryer, the obtained atosiban has a purity greater than 99.5%, and the total product yield is 57%.
[0072]
Example 3:
[0073]
Using Rink-Fmoc-Gly resin (40 g, substitution amount 0.36 mmol/g) as the starting material, the stepwise Fmoc-SPPS (solid phase peptide synthesis) method was used to synthesize the peptide. Fmoc deprotection was performed with piperidine in DMF (1:4 v/v). Subsequently, other amino acids in the sequence were connected in the following order, and the coupling reagents were N,N-diisopropylethylamine, 2-(7-benzotriazole)-N,N,N’,N’- Two kinds of tetramethylurea hexafluorophosphate mixed in a 1:1 volume ratio, Fmoc-Orn(Boc)-OH, Fmoc-Pro-OH, Fmoc-Cys(Trt)-OH, Fmoc-Asn-OH, Fmoc- Thr(tBu)-OH, Fmoc-Ile-OH, Fmoc-D-Tyr(ET)-OH, Mpa(Trt). Coupling and deprotection of amino acids were carried out at 75°C for 2-3 min and monitored with the Kaiser test. The peptide was cleaved with the lysing solution of TFA for 3 hours, precipitated and washed twice with methyl tert-butyl ether, and after centrifugal drying, the atosiban linear crude peptide was cyclized by the method of liquid phase synthesis, and the volume ratio was 1: 2:3 solution A-sodium phosphate buffered aqueous solution (concentration 15g/L), solution B-linear peptide atosiban crude peptide phosphoric acid aqueous solution and solution C: DMSO aqueous solution (volume 1:1).
[0074]
The crude peptide yield was 80%. Crude atosiban was purified by preparative high performance liquid chromatography with a water/acetonitrile gradient from 100% water to 100% acetonitrile in 20 minutes. The purified atosiban solution is freeze-dried at -50 to -70 DEG C for 28 hours with a freeze dryer, the obtained atosiban has a purity of more than 99.5%, and the total product yield is 55%.
[0075]
Example 4:
[0076]
Using Rink-Fmoc-Gly resin (40 g, substitution amount 0.36 mmol/g) as the starting material, the stepwise Fmoc-SPPS (solid phase peptide synthesis) method was used to synthesize the peptide. Fmoc deprotection was performed with piperidine in DMF (1:3 by volume). Subsequently, the other amino acids in the sequence were connected in the following order, and the coupling reagents were selected from 2-oxime ethyl cyanoacetate, N,N-diisopropylcarbodiimide, and 1-hydroxybenzotriazole in a volume ratio of 1. :1:1 mix, Fmoc-Orn(Boc)-OH, Fmoc-Pro-OH, Fmoc-Cys(Trt)-OH, Fmoc-Asn-OH, Fmoc-Thr(tBu)-OH, Fmoc-Ile-OH , Fmoc-D-Tyr(ET)-OH, Mpa(Trt). Coupling and deprotection of amino acids were carried out at 80°C for 2-3 min and monitored with the Kaiser test. The peptide was cleaved with the lysing solution of TFA for 3 hours, precipitated with methyl tert-butyl ether and washed twice, and after centrifugal drying, the atosiban linear crude peptide was cyclized by the method of liquid phase synthesis, and the volume ratio was 1: 3:4 solution of A-trifluoroacetic acid-aqueous ammonia solution (concentration of 45 g/L), solution B-aqueous solution of linear peptide atosiban crude peptide trifluoroacetic acid and solution C: saturated aqueous iodine solution.
[0077]
The crude peptide yield was 78%. Crude atosiban was purified by preparative high performance liquid chromatography with a water/acetonitrile gradient from 100% water to 100% acetonitrile in 20 minutes. The purified atosiban solution is freeze-dried at -50 to -70° C. for 38 hours with a freeze dryer, the obtained atosiban has a purity of more than 99.5%, and the total product yield is 52%.
PATENT
WO/2022/141615
Atosiban Acetate Injection was first listed in Austria on March 23, 2000 under the trade name: Atosiban, a new type of anti-prematurity drug developed by Ferring GmbH, which is an oxytocin The analog is a competitive antagonist of oxytocin receptors in the uterus, decidua, and fetal membranes. It is a first-line drug recommended by the European Medical Association; it can inhibit the binding of oxytocin and oxytocin receptors, thereby directly inhibiting the effect of oxytocin. In the uterus, it can inhibit uterine contraction; it can also inhibit the hydrolysis of phosphatidylinositol.
Atosiban is a cyclic nonapeptide whose molecular formula is C 43 H 67 N 11 O 12 S 2 ; molecular weight is 994.19; CAS registration number is 90779-69-4; its peptide sequence is as follows:
Cyclo[Mpa-D-Tyr(Et)-Ile-Thr-Asn-Cys]-Pro-Orn-Gly-NH 2
In the Chinese patents with announcement numbers CN101314613B and CN101696236B, the solid-phase synthesis of atosiban uses Rink Amide AM Resin resin solid-phase coupling stepwise to obtain Mpa(Trt)-D-Tyr(Et)-Ile-Thr(tBu)- Asn(Trt)-Cys(Trt)-Pro-Orn(Boc)-Gly-Resin is directly oxidized in solid phase to generate disulfide bonds, and then cleaved to obtain atosiban. The Rink Amide AM Resin resin used in the prior art needs to be cracked under a strong acid environment, which is not conducive to product stability and has a greater operational risk; Mpr and Cys both have sulfhydryl groups, and the sulfhydryl groups have the ability to capture tBu to generate double tBu impurities, When the peptide resin after solid-phase oxidation is cleaved to remove the protective group and resin, due to the presence of tBu or tBu source Boc protective group, it requires high capture agent, which is not conducive to product quality control and reduces product yield.
The Chinese patent with publication number CN105408344B discloses a method for synthesizing atosiban starting from Fmoc-Orn-Gly-NH2, wherein Fmoc-Orn-Gly-NH2 is connected to trityl through the side chain of ornithine On the base resin, impurities can be effectively controlled. However, using dipeptide and trityl-type resin for coupling, the resin attached to the Orn side chain of the dipeptide increases the steric hindrance of the subsequent Pro coupling and prolongs the coupling time, which is easy to cause missing peptide impurities.
Example 1. Synthesis of Fmoc-Pro-Orn-Gly-NH 2 tripeptide
[0027]
Fmoc-Pro-OH (134.94 g, 400 mmol) and N-hydroxysuccinimide (46.00 g, 400 mmol) were weighed into 1600 ml of tetrahydrofuran, and stirred at room temperature. The temperature was controlled at about 5°C, and a solution of DCC (90.72g, 440mmol) in tetrahydrofuran (320ml) was slowly added and stirred at room temperature for 2.5h, filtered, concentrated and added to petroleum ether for recrystallization to precipitate a solid, washed and dried, and the obtained activated ester was The solid was dissolved in 400 ml of tetrahydrofuran, and H-Orn(Boc)-NH 2 (92.92 g, 400 mmol) was dissolved in 300 ml of tetrahydrofuran and slowly added dropwise to the above solution. After dropping, the reaction was continued at room temperature. Concentrate to dryness under reduced pressure, add N-hydroxysuccinimide (46.00 g, 400 mmol) and 1600 ml of tetrahydrofuran to dissolve, and stir at room temperature. The temperature was controlled at about 5°C, and a solution of DCC (90.72g, 440mmol) in tetrahydrofuran (320ml) was slowly added and stirred at room temperature for 2.5h, filtered, concentrated and added to petroleum ether for recrystallization to precipitate a solid, washed and dried, and the obtained activated ester was The solid was dissolved in 400 ml of tetrahydrofuran, and H-Gly-NH 2 (29.64 g, 400 mmol) was dissolved in 300 ml of tetrahydrofuran and slowly added dropwise to the above solution, and the reaction was continued at room temperature after dropping, and the monitoring of the raw materials was completed. The reaction was filtered, and the filtrate was concentrated under reduced pressure. Dry, add 1000 mL of 5% TFA/DCM solution to the reaction solution, continue to react for 1 h, and concentrate to dryness to obtain a yellow oil, which is recrystallized from isopropanol to obtain 171.56 g of white solid with a yield of 69%.
[0028]
Example 2. Synthesis of Fmoc-Pro-Orn (trityl resin)-Gly-NH 2 peptide resin with a degree of substitution of 0.42 mmol/g
[0029]
Trityl resin (37.5 g, 30 mmol, substitution degree: 0.80 mmol/g) was weighed into a solid-phase reaction synthesis column. 400 mL of dry DMF was added to swell for 30 min, and the DMF was removed. The resin was washed with 3*400 mL of dry DMF, and the DMF was removed. Fmoc-Pro-Orn-Gly-NH 2 (37.30 g, 60 mmol) prepared in Example 1 , DIEA (11.63 g, 90 mmol) were added, 100 mL of dry DMF was added to dissolve and clarified, added to the resin to react for 2 h, and methanol (9.61 mmol) was added. g, 300 mmol) reacted for 20 min, sucked dry, washed the resin with 3*400 mL of CH 2 Cl 2 , and removed CH 2 Cl 2 . The resin was taken out and dried under vacuum at 25-35° C. to obtain 52.14 g of Fmoc-Pro-Orn (trityl resin)-Gly-NH 2 resin with a measured substitution degree of 0.42 mmol/g.
[0030]
Example 3. Synthesis of Fmoc-Pro-Orn(2-CTC Resin)-Gly-NH 2 peptide resin with a degree of substitution of 0.50 mmol/g
[0031]
2-CTC Resin resin (30.0 g, 30 mmol, substitution degree: 1.00 mmol/g) was weighed into a solid-phase reaction synthesis column. 400 mL of dry DMF was added to swell for 30 min, and the DMF was removed. The resin was washed with 3*400 mL of dry DMF, and the DMF was removed. Fmoc-Pro-Orn-Gly-NH 2 (37.30 g, 60 mmol) prepared in Example 1 , DIEA (11.63 g, 90 mmol) were added, 100 mL of dry DMF was added to dissolve and clarified, added to the resin to react for 2 h, and methanol (9.61 mmol) was added. g, 300 mmol) reacted for 20 min, sucked dry, washed the resin with 3*400 mL of CH 2 Cl 2 , and removed CH 2 Cl 2 . The resin was taken out and dried under vacuum at 25-35° C. to obtain 43.80 g of Fmoc-Pro-Orn(2-CTC Resin)-Gly-NH 2 resin with a measured substitution degree of 0.50 mmol/g.
[0032]
Example 4. Synthesis of Fmoc-Pro-Orn (4-methyl-trityl resin)-Gly-NH 2 peptide resin with a degree of substitution of 0.50 mmol/g
[0033]
4-methyl-trityl resin (33.33 g, 30 mmol, substitution degree: 0.90 mmol/g) was weighed into a solid-phase reaction synthesis column. 400 mL of dry DMF was added to swell for 30 min, and the DMF was removed. The resin was washed with 3*400 mL of dry DMF, and the DMF was removed. Fmoc-Pro-Orn-Gly-NH 2 (37.30 g, 60 mmol) prepared in Example 1 , DIEA (11.63 g, 90 mmol) were added, 100 mL of dry DMF was added to dissolve and clarified, added to the resin to react for 2 h, and methanol (9.61 mmol) was added. g, 300 mmol) reacted for 20 min, sucked dry, washed the resin with 3*400 mL of CH 2 Cl 2 , and removed CH 2 Cl 2 . The resin was taken out and dried under vacuum at 25-35° C. to obtain 43.89 g of Fmoc-Pro-Orn (4-methyl-trityl resin)-Gly-NH 2 resin with a measured substitution degree of 0.50 mmol/g.
[0034]
Example 5. Synthesis of Fmoc-Pro-Orn (4-methoxy-trityl resin)-Gly-NH 2 peptide resin with a degree of substitution of 0.50 mmol/g
[0035]
4-Methoxy-trityl resin (30.0 g, 30 mmol, substitution degree: 1.00 mmol/g) was weighed into a solid-phase reaction synthesis column. 400 mL of dry DMF was added to swell for 30 min, and the DMF was removed. The resin was washed with 3*400 mL of dry DMF, and the DMF was removed. Fmoc-Pro-Orn-Gly-NH 2 (37.30 g, 60 mmol) prepared in Example 1 , DIEA (11.63 g, 90 mmol) were added, 100 mL of dry DMF was added to dissolve and clarified, added to the resin to react for 2 h, and methanol (9.61 mmol) was added. g, 300 mmol) reacted for 20 min, sucked dry, washed the resin with 3*400 mL of CH 2 Cl 2 , and removed CH 2 Cl 2 . The resin was taken out and dried under vacuum at 25-35° C. to obtain 43.69 g of Fmoc-Pro-Orn (4-methoxy-trityl resin)-Gly-NH 2 resin with a measured substitution degree of 0.50 mmol/g.
[0036]
Example 6. Synthesis of Atosiban Linear Peptide Resin 1
[0037]
Fmoc-Pro-Orn (trityl resin)-Gly-NH 2 (35.71 g) prepared in Example 2 was weighed into a solid-phase reaction synthesis column. 400 mL of DMF was added to swell for 30 min, and the DMF was removed. The resin was washed with 3*200 mL of dry DMF, and the DMF was removed. 200 mL of DBLK solution (20% piperidine/DMF solution, V/V) was added and deprotected twice, the first time was 5 min and the second time was 15 min. After deprotection, the resin was washed with 200 mL of DMF each time, and washed 6 times. After the fourth washing, a little resin was taken with a glass rod. The ninhydrin test was positive, indicating that Fmoc had been removed.
[0038]
Weigh 17.57g Fmoc-Cys(Trt)-OH and 4.86g HOBt, add 100mL DMF to dissolve, after complete dissolution, cool the solution to below 5°C, then add 5.68g DIC (pre-cooled to <0°C), Activated in the solution for about 3 to 5 minutes, the activated solution was added to the reaction column under control, and reacted at 20 to 35 °C for 2 to 3 hours. The ninhydrin test was negative. The reaction solution was removed, and 200 mL of DMF was added to wash the resin. 6 times. After washing, the washing liquid was removed to obtain Fmoc-Cys(Trt)-Pro-Orn (trityl resin)-Gly-NH 2 .
[0039]
Repeat the step of receiving the peptide and remove the Fmoc protective group. According to the amino acid sequence of atosiban, Fmoc-Cys(Trt)-Pro-Orn (trityl resin)-Gly-NH 2 was coupled to Fmoc- Asn-OH, Fmoc-Thr-OH, Fmoc-Ile-OH, Fmoc-D-Tyr(Et)-OH, Mpa(Trt)-OH give Mpa(Trt)-D-Tyr(Et)-Ile-Thr- Asn-Cys(Trt)-Pro-Orn (trityl resin)-Gly- NH2 . After washing with DMF, the washing solution was removed. The resin was washed with 200 ml of DCM each time, 4 times, 5 min/time, the DCM was removed, and the resin was vacuum-dried at room temperature (20-35° C.) until it was quicksand. The peptide resin was 48.72g after drying, and the resin weight gain was 89.0%.
[0040]
Example 7. Synthesis of atosiban linear peptide resin 2
[0041]
Fmoc-Pro-Orn(2-CTC Resin)-Gly-NH 2 (30.00 g) prepared in Example 3 was weighed into a solid-phase reaction synthesis column. 400 mL of DMF was added to swell for 30 min, and the DMF was removed. The resin was washed with 3*200 mL of dry DMF, and the DMF was removed. 200 mL of DBLK solution (20% piperidine/DMF solution, V/V) was added and deprotected twice, the first time was 5 min and the second time was 15 min. After deprotection, the resin was washed with 200 mL of DMF each time, and washed 6 times. After the fourth washing, a little resin was taken with a glass rod. The ninhydrin test was positive, indicating that Fmoc had been removed.
[0042]
Weigh 17.57g Fmoc-Cys(Trt)-OH and 13.65g HBTU, add 100mL DMF to dissolve, after complete dissolution, cool the solution to below 5°C, then add 5.82g DIEA (pre-cooled to <0°C), put Activated in the solution for about 3 to 5 minutes, the activated solution was added to the reaction column under control, and reacted at 20 to 35 °C for 2 to 3 hours. The ninhydrin test was negative. The reaction solution was removed, and 200 mL of DMF was added to wash the resin. 6 times. After washing, the washing solution was removed to obtain Fmoc-Cys(Trt)-Pro-Orn(2-CTC Resin)-Gly-NH 2 .
[0043]
Fmoc-D-Tyr(Et)-OH (86.30 g, 200 mmol) and N-hydroxysuccinimide (23.00 g, 200 mmol) were weighed into 800 ml of tetrahydrofuran, and stirred at room temperature. The temperature was controlled at about 5°C, and a solution of DCC (45.36g, 220mmol) in tetrahydrofuran (160ml) was slowly added and stirred at room temperature for 2.5h, filtered, concentrated and added to petroleum ether for recrystallization to precipitate a solid, washed and dried, and the obtained activated ester was The solid was dissolved in 200 ml of tetrahydrofuran, and H-Ile-OH (26.24 g, 200 mmol) was dissolved in 150 ml of tetrahydrofuran and slowly added dropwise to the above solution. After dropping, the reaction was continued at room temperature. The monitoring of the raw materials was completed. After filtration, the solution was concentrated under reduced pressure. , the concentrated solution was added to petroleum ether to separate out the solid, the solid was washed and then dried, recrystallized and dried with isopropanol to obtain 75.60 g of Fmoc-D-Tyr(Et)-Ile-OH with a yield of 75%.
[0044]
Repeat the step of receiving the peptide and removing the Fmoc protective group. According to the amino acid sequence of atosiban, sequentially couple Fmoc-Asn on Fmoc-Cys(Trt)-Pro-Orn(2-CTC Resin)-Gly-NH 2 -OH, Fmoc-Thr-OH, Fmoc-D-Tyr(Et)-Ile-OH, Mpa(Trt)-OH to give Mpa(Trt)-D-Tyr(Et)-Ile-Thr-Asn-Cys(Trt )-Pro-Orn( 2 -CTC Resin)-Gly-NH2 . After washing with DMF, the washing solution was removed. The resin was washed with 200 ml of DCM each time, 4 times, 5 min/time, the DCM was removed, and the resin was vacuum-dried at room temperature (20-35° C.) until it was quicksand. The peptide resin was 42.77g after drying, and the resin weight gain rate was 87.4%.
[0045]
Example 8. Synthesis of atosiban linear peptide resin 3
[0046]
Fmoc-Pro-Orn (4-methyl-trityl resin)-Gly-NH 2 (30.00 g) prepared in Example 4 was weighed into a solid-phase reaction synthesis column. 400 mL of DMF was added to swell for 30 min, and the DMF was removed. The resin was washed with 3*200 mL of dry DMF, and the DMF was removed. 200 mL of DBLK solution (20% piperidine/DMF solution, V/V) was added and deprotected twice, the first time was 5 min and the second time was 15 min. After deprotection, the resin was washed with 200 mL of DMF each time, and washed 6 times. After the fourth washing, a little resin was taken with a glass rod, and the ninhydrin test was positive, indicating that Fmoc had been removed.
[0047]
Weigh 17.57g Fmoc-Cys(Trt)-OH, 13.65g HBTU and 4.05g HOBt, add 100mL DMF to dissolve, after complete dissolution, cool the solution to below 5°C, then add 5.82g DIEA (pre-cooled to <0 ℃), activate in the solution for about 3-5min, add the activated solution to the reaction column, react at 20-35 ℃ for 2-3h, the ninhydrin test is negative, remove the reaction solution, add 200mL of DMF The resin was washed 6 times. After washing, the washing liquid was removed to obtain Fmoc-Cys(Trt)-Pro-Orn(4-methyl-trityl resin)-Gly-NH 2 .
[0048]
Mpa(Trt)-OH (69.69 g, 200 mmol) and N-hydroxysuccinimide (23.00 g, 200 mmol) were weighed into 800 ml of tetrahydrofuran, and stirred at room temperature. The temperature was controlled at about 5°C, and a solution of DCC (45.36g, 220mmol) in tetrahydrofuran (160ml) was slowly added and stirred at room temperature for 2.5h, filtered, concentrated and added to petroleum ether for recrystallization to precipitate a solid, washed and dried, and the obtained activated ester was The solid was dissolved in 200 ml of tetrahydrofuran, and HD-Tyr(Et)-OH (41.85 g, 200 mmol) was dissolved in 150 ml of tetrahydrofuran and slowly added dropwise to the above solution. After dropping, the reaction was continued at room temperature. Concentrate under reduced pressure, add the concentrated solution to petroleum ether to precipitate a solid, wash the solid and then dry it, recrystallize and dry with isopropanol to obtain Mpa(Trt)-D-Tyr(Et)-OH 77.98g, yield 72%.
[0049]
Repeat the step of receiving the peptide and removing the Fmoc protective group, according to the amino acid sequence of atosiban, on Fmoc-Cys(Trt)-Pro-Orn (4-methyl-trityl resin)-Gly- NH 2 Fmoc-Asn-OH, Fmoc-Thr-OH, Fmoc-Ile-OH, Mpa(Trt)-D-Tyr(Et)-OH were sequentially coupled to obtain Mpa(Trt)-D-Tyr(Et)-Ile-Thr -Asn-Cys(Trt)-Pro-Orn(4-methyl-trityl resin)-Gly- NH2 . After washing with DMF, the washing solution was removed. The resin was washed with 200 ml of DCM each time, 4 times, 5 min/time, the DCM was removed, and the resin was vacuum-dried at room temperature (20-35° C.) until it was quicksand. The peptide resin was 42.91g after drying, and the resin weight gain rate was 88.3%.
[0050]
Example 9. Synthesis of atosiban linear peptide resin 4
[0051]
Fmoc-Pro-Orn (4-methoxy-trityl resin)-Gly-NH 2 (30.00 g) prepared in Example 5 was weighed into a solid-phase reaction synthesis column. 400 mL of DMF was added to swell for 30 min, and the DMF was removed. The resin was washed with 3*200 mL of dry DMF, and the DMF was removed. 200 mL of DBLK solution (20% piperidine/DMF solution, V/V) was added and deprotected twice, the first time was 5 min and the second time was 15 min. After deprotection, the resin was washed with 200 mL of DMF each time, and washed 6 times. After the fourth washing, a little resin was taken with a glass rod. The ninhydrin test was positive, indicating that Fmoc had been removed.
[0052]
Fmoc-Asn-OH (70.87 g, 200 mmol) and N-hydroxysuccinimide (23.00 g, 200 mmol) were weighed into 800 ml of tetrahydrofuran, and stirred at room temperature. The temperature was controlled at about 5°C, and a solution of DCC (45.36g, 220mmol) in tetrahydrofuran (160ml) was slowly added and stirred at room temperature for 2.5h, filtered, concentrated and added to petroleum ether for recrystallization to precipitate a solid, washed and dried, and the obtained activated ester was The solid was dissolved in 200 ml of tetrahydrofuran, and H-Cys(Trt)-OH (79.96 g, 200 mmol) was dissolved in 150 ml of tetrahydrofuran and slowly added dropwise to the above solution. After dropping, the reaction was continued at room temperature. Concentrate under reduced pressure, add the concentrated solution to petroleum ether to precipitate a solid, wash the solid and then dry, recrystallize and dry with isopropanol to obtain Fmoc-Asn-Cys(Trt)-OH 102.17g, yield 73%.
[0053]
Weigh 20.99g Fmoc-Asn-Cys(Trt)-OH and 13.65g HCTU, add 100mL DMF to dissolve, after complete dissolution, cool the solution to below 5°C, then add 5.82g DIEA (pre-cool to <0°C) , activate in the solution for about 3-5min, add the activated solution to the reaction column, react at 20-35°C for 2-3h, the ninhydrin test is negative, remove the reaction solution, add 200mL of DMF to wash the resin , wash 6 times. After washing, the washing liquid was removed to obtain Fmoc-Asn-Cys(Trt)-Pro-Orn(4-methoxy-trityl resin)-Gly-NH 2 .
[0054]
Repeat the step of receiving the peptide and removing the Fmoc protective group. According to the amino acid sequence of atosiban, in Fmoc-Asn-Cys(Trt)-Pro-Orn(4-methoxy-trityl resin)-Gly- Fmoc-Thr-OH, Fmoc-Ile-OH, Fmoc-D-Tyr(Et)-OH, Mpa(Trt)-OH were sequentially coupled on NH 2 to obtain Mpa(Trt)-D-Tyr(Et)-Ile- Thr-Asn-Cys(Trt)-Pro-Orn(4-methoxy-trityl resin)-Gly- NH2 . After washing with DMF, the washing solution was removed. The resin was washed with 200 ml of DCM each time, 4 times, 5 min/time, the DCM was removed, and the resin was vacuum-dried at room temperature (20-35° C.) until it was quicksand. The peptide resin was 42.28g after drying, and the resin weight gain was 84.0%.
[0055]
Example 10. Synthesis of atosiban crude peptide 1
[0056]
Configure 487.2ml of TFA/DCM=2/98 (V/V) lysis solution, cool to 5-10°C, add 48.72g of peptide resin prepared in Example 6 into the lysis solution, at room temperature (20-35°C) React for 5h, filter, wash the peptide resin twice with acetonitrile, 50ml/time, combine into the filtrate, spin the filtrate to dry, obtain a solid after drying, wash with isopropyl ether, filter, and dry under reduced pressure at 20-35°C to constant weight To obtain 14.77g of atosiban linear peptide, dissolve 14.30g of atosiban linear peptide in 0.75L of glacial acetic acid, add 6.75L of water to dilute, add 0.1M/L iodine ethanol solution dropwise until the solution changes color, react at room temperature for 1.0h, That is, the crude atosiban peptide is obtained, and its HPLC spectrum is shown in Figure 1.
[0057]
Example 11. Synthesis of atosiban crude peptide 2
[0058]
Configure TFA/DCM=5/95 (V/V) lysate 448.6ml, cool to 5~10℃, add 42.77g of peptide resin prepared in Example 7 into the lysate, at room temperature (20~35℃) React for 3h, filter, wash the peptide resin twice with acetonitrile, 50ml/time, combine into the filtrate, spin the filtrate, dry to obtain a solid, wash with isopropyl ether, filter, and dry under reduced pressure at 20-35°C to constant weight To obtain 14.21g of atosiban linear peptide, dissolve 14.21g of atosiban linear peptide in 1.5L of glacial acetic acid, add 6L of water to dilute, add 0.1M/L iodoethanol solution dropwise until the solution changes color, react at room temperature for 1.0h, that is The crude atosiban peptide was obtained, and its HPLC chromatogram was similar to that in Figure 1.
[0059]
Example 12. Synthesis of atosiban crude peptide 3
[0060]
Configure 450.5ml of TFA/DCM=20/80(V/V) lysis solution, cool to 5~10℃, add 45.05g of peptide resin prepared in Example 8 into the lysis solution, at room temperature (20~35℃) React for 2h, filter, wash the peptide resin twice with acetonitrile, 50ml/time, combine into the filtrate, spin the filtrate to dry, obtain a solid after drying, wash with isopropyl ether, filter, and dry under reduced pressure at 20-35°C to constant weight To obtain 14.63g of atosiban linear peptide, dissolve 14.63g of atosiban linear peptide in 1.5L of glacial acetic acid, add 6L of water to dilute, add 10% hydrogen peroxide solution, and react at room temperature for 1.0h to obtain atosiban Crude peptide, its HPLC chromatogram is similar to Figure 1.
[0061]
Example 13. Synthesis of atosiban crude peptide 4
[0062]
Configure TFA/DCM=1/99 (V/V) lysate 442.7ml, cool to 5~10℃, add 44.27g of peptide resin prepared in Example 9 into the lysate, at room temperature (20~35℃) React for 5h, filter, wash the peptide resin twice with acetonitrile, 50ml/time, combine into the filtrate, spin the filtrate to dry, obtain a solid after drying, wash with isopropyl ether, filter, and dry under reduced pressure at 20-35°C to constant weight To obtain 14.13 g of atosiban linear peptide, dissolve 14.13 g of atosiban linear peptide in 1.5 L of glacial acetic acid, add 6 L of water to dilute, add 30% hydrogen peroxide solution, and react at room temperature for 1.0 h to obtain atosiban Crude peptide, its HPLC chromatogram is similar to Figure 1.
[0063]
Example 14. Purification of atosiban crude peptide 1
[0064]
The atosiban crude peptide prepared in Example 10 was dissolved in 15% acetonitrile aqueous solution and filtered, purified by preparative reverse-phase HPLC (C18 column), transferred to salt, collected more than 99% of the fraction, concentrated and lyophilized to obtain 10.12g , the yield is 64%, the purity is 99%, and the HPLC spectrum of the obtained atosiban peptide is shown in Figure 2.
[0065]
Example 15. Purification of atosiban crude peptide 2
[0066]
The crude atosiban peptide obtained in Example 11 was dissolved in a 15% acetonitrile aqueous solution and filtered, purified by preparative reverse-phase HPLC (C18 column), transferred to salt, collected more than 99% of the fraction, concentrated and lyophilized to obtain 9.80 g , the yield is 62%, the purity is 99%, and the obtained atosiban peptide HPLC spectrum is similar to Figure 2.
[0067]
Example 16. Purification of atosiban crude peptide 3
[0068]
The crude atosiban peptide obtained in Example 12 was dissolved in a 15% acetonitrile aqueous solution and filtered, purified by preparative reverse-phase HPLC (C18 column), transferred to salt, collected more than 99% of the fraction, concentrated and lyophilized to obtain 10.28g , the yield is 65%, the purity is 99%, and the HPLC spectrum of the obtained atosiban peptide is similar to that in Figure 2.
[0069]
Example 17. Purification of atosiban crude peptide 4
[0070]
The crude atosiban peptide obtained in Example 13 was dissolved in 15% acetonitrile aqueous solution and filtered, purified by preparative reverse-phase HPLC (C18 column), transferred to salt, collected more than 99% of the fraction, concentrated and lyophilized to obtain 10.27g , the yield is 65%, the purity is 99%, and the HPLC spectrum of the obtained atosiban peptide is similar to that in Figure 2.
PATENT
https://patents.google.com/patent/US9434767B2/es
Atosiban is a nonapeptide which contains three non-natural amino acids: D-Tyr(Et), Mpa and Orn, and a pair of disulfide bonds looped between Mpa and Cys, the structural formula is:
c[Mpa-D-Tyr(Et)-Ile-Thr-Asn-Cys]-Pro-Orn-Gly-NH2.
By means of competing for oxytocin receptor with oxytocin, Atosiban can inhibit the combination between oxytocin and oxytocin receptor, and directly prevent the oxytocin from acting on uterus, and then inhibit the uterine contraction; as another hand, atosiban can also inhibit the hydrolysis of phosphatidylinositol and then block the generation of messenger and activity of Ca2+, with the decreasing of activity from oxytocin, the contraction of uterine is indirectly inhabited.
At present, there are many reports about synthesis process method in China and abroad A report in China shows that the inventor found a simple process by adopting solid phase oxidation, resulting in a low purity crude product, with low yield and low application value. The aforementioned reports about atosiban synthesis process reveal that most of them adopt the method using Boc solid phase synthetic and cleaving peptide with liquid ammonia, then oxidating with liquid phase oxidation, and purifying. Those respective processes result in “the three wastes” and are too complex for industrial production. See U.S. Pat. No. 4,504,469.
Example 1Preparing the Linear Atosiban Peptide Resin
(i) 6.25 g of Rink Amide resin (substitutability=0.8 mmol/g) is put into a reaction bottle, DMF is added into the bottle and washed twice, then swelled for 30 min with DMF. Fmoc protecting group of Rink Amide resin is removed with 30-40 ml of 20% DBLK, washed for 4 times with DMF, then washed twice with DCM after removal, the product is detected by ninhydrin detecting method, the resin is reddish-brown.
(ii) 4.46 g of Fmoc-Gly-OH and 2.43 g of HOBt dissolved in a suitable amount of DMF, which had been pre-activated with 3.05 ml DIC; the mixture is, added to the reaction bottle, and reacted for 2 h, the resin is negative by ninhydrin detecting method, after the reaction, the product is washed for 4 times with DMF, then washed twice with DCM, if the resin is positive, repeating the above condensation reaction until negative.
(iii) Fmoc-Orn(Boc)-OH, Fmoc-Pro-OH, Fmoc-Cys(Trt)-OH, Fmoc-Asn(Trt)-OH, Fmoc-Thr(tBu)-OH, Fmoc-Ile-OH, Fmoc-D-Tyr(ET)-OH and Mpa(Trt)-OH are coupled orderly.
Example 2Cleaving the Linear Atosiban Peptide Resin
5.15 g of linear atosiban is prepared by washing the linear atosiban peptide resin obtained from Example 1 for 3 times with 30 ml of methanol, adding the dry resin obtained to 150 ml of mixed solution with a volume ratio of TFA:H2O=95:5, reacting for 2 hours at 25° C. and filtering, washing the resin for 3 times with few trifluoroacetic acid, combining the filtrate and pouring into 1500 ml glacial ether, making rest for 2 hours, centrifugally separating the linear atosiban, washing for 3 times, and drying in a vacuum drier, MS: 995.3, HPLC: 91.5%, content: 65.5%, synthesis yield: 68%.
Example 3Oxidizing the Linear Atosiban
2.85 g of atosiban acetate is prepared by dissolving the linear atosiban obtained from Example 2 in 250 ml of 5% acetonitrile aqueous solution, adjusting the pH value to 8 to 9 with 30% ammonia water, adding 0.60 g of H2O2, reacting for 10 min at 25° C., monitoring with HPLC (HPLC: 75.6%), filtering after reaction, purifying filtrate by preparative RP-HPLC (column C18 or C8), transferring salt, and freeze-drying, MS: 994.5, HPLC: 99.4%.
Example 4Oxidizing the Linear Atosiban
3.01 g of atosiban acetate is prepared by dissolving the linear atosiban obtained from Example 2 in 250 ml of 10% acetonitrile aqueous solution, adjusting the pH value to 8 to 9 with 30% ammonia water, adding 0.85 g of H2O2, reacting for 30 min at 25° C., monitoring with HPLC (HPLC: 89.5%), filtering after reaction, purifying filtrate by preparative RP-HPLC (column C18 or C8), transferring salt, and freeze-drying, MS: 994.5, HPLC: 99.6%.
Example 5Oxidizing the Linear Atosiban
2.95 g of atosiban acetate is prepared by dissolving the linear atosiban obtained from Example 2 in 250 ml of 10% acetonitrile aqueous solution, adjusting the pH value to 8 to 9 with 30% ammonia water, adding 0.85 g of H2O2, reacting for 60 min at 25° C., monitoring with HPLC (HPLC: 83.5%), filtering after reaction, purifying filtrate by preparative RP-HPLC (column C18 or C8), transferring salt, and freeze-drying, MS: 994.5, HPLC: 99.4%.
The above is the further detailed description of the invention in conjunction with specific preferred examples, but it should not be considered that the specific examples of the invention are only limited to the these descriptions. For one of ordinary skill in the art, many deductions and replacements can be made without departing from the inventive concept. Such deductions and replacements should fall within the scope of protection of the invention.
Clips
https://www.mdpi.com/1420-3049/27/6/1920/htm
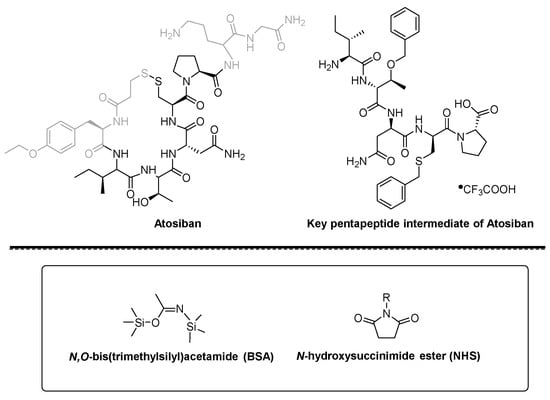
Figure 1. Structure of Atosiban, pentapeptide intermediate, BSA and NHS ester.
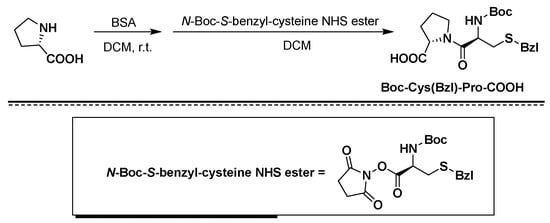
Figure 2. Synthesis of Boc-Cys(Bzl)-Pro-COOH using BSA/NHS as coupling agents.
//////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
Clinical trials
Atosiban vs. nifedipine
A 2013 retrospective study comparing the efficacy and safety of atosiban and nifedipine in the suppression of preterm labour concluded that atosiban and nifedipine are effective in delaying delivery for seven days or more in women presenting with preterm labour.[16] A total of 68.3% of women in the atosiban group remained undelivered at seven days or more, compared with 64.7% in the nifedipine group.[16] They have the same efficacy and associated minor side effects.[16] However, flushing, palpitation, and hypotension were significantly higher in the nifedipine group.[16]
A 2012 clinical trial compared tocolytic efficacy and tolerability of atosiban with that of nifedipine.[17] Forty-eight (68.6%) women allocated to atosiban and 39 (52%) to nifedipine did not deliver and did not require an alternate agent at 48 hours, respectively (p=.03).[17] Atosiban has fewer failures within 48 hours.[17] Nifedipine may be associated with a longer postponement of delivery.[17]
A 2009 randomised controlled study demonstrated for the first time the direct effects of atosiban on fetal movement, heart rate, and blood flow.[18] Tocolysis with either atosiban or nifedipine combined with betamethasone administration had no direct fetal adverse effects.[18]
Atosiban vs. ritodrine
Multicentre, controlled trial of atosiban vs. ritodrine in 128 women shows a significantly better tocolytic efficacy after 7 days in the atosiban group than in the ritodrine group (60.3 versus 34.9%), but not at 48 hours (68.3 versus 58.7%). Maternal adverse events were reported less frequently in the atosiban group (7.9 vs 70.8%), resulting in fewer early drug terminations due to adverse events (0 versus 20%). Therefore, atosiban is superior to ritodrine in the treatment of preterm labour.[19]
Brand names
In India it is marketed under the brand name Tosiban by Zuventus healthcare ltd.
References
- ^ “Atosiban International Drug Names”. Drugs.com. 10 April 2020. Retrieved 29 April 2020.
- ^ “Tractocile 7.5 mg/ml Solution for Injection – Summary of Product Characteristics (SmPC)”. (emc). Retrieved 29 April 2020.
- ^ “Tractocile 7.5 mg/ml Concentrate for Solution for Infusion – Summary of Product Characteristics (SmPC)”. (emc). 24 June 2013. Retrieved 29 April 2020.
- ^ Jump up to:a b c d e f g “Tractocile EPAR”. European Medicines Agency (EMA). Retrieved 29 April 2020. This article incorporates text from this source, which is in the public domain.
- ^ Akerlund M, Carlsson AM, Melin P, Trojnar J (1985). “The effect on the human uterus of two newly developed competitive inhibitors of oxytocin and vasopressin”. Acta Obstet Gynecol Scand. 64 (6): 499–504. doi:10.3109/00016348509156728. PMID 4061066. S2CID 25799128.
- ^ Sanu O, Lamont RF (2010). “Critical appraisal and clinical utility of atosiban in the management of preterm labor”. Ther Clin Risk Manag. 6: 191–199. doi:10.2147/tcrm.s9378. PMC 2861440. PMID 20463780.
- ^ Jump up to:a b Chou PY, Wu MH, Pan HA, Hung KH, Chang FM (June 2011). “Use of an oxytocin antagonist in in vitro fertilization-embryo transfer for women with repeated implantation failure: a retrospective study”. Taiwan J Obstet Gynecol. 50 (2): 136–40. doi:10.1016/j.tjog.2011.04.003. PMID 21791296.
- ^ Lan, VT; Khang, VN; Nhu, GH; Tuong, HM (September 2012). “Atosiban improves implantation and pregnancy rates in patients with repeated implantation failure”. Reprod Biomed Online. 25 (3): 254–60. doi:10.1016/j.rbmo.2012.05.014. PMID 22818095.
- ^ Jump up to:a b Wu, MY; Chen, SU; Yang, YS (December 2011). “Using atosiban in uterine contractions of early pregnancies after assisted reproduction”. J Formos Med Assoc. 110 (12): 800. doi:10.1016/j.jfma.2011.11.016. PMID 22248840.
- ^ Conde-Agudelo, A; Romero, R; Kusanovic, JP (2011). “Nifedipine in the management of preterm labor: a systematic review and metaanalysis”. Am J Obstet Gynecol. 204 (2): 134.e1–134.e20. doi:10.1016/j.ajog.2010.11.038. PMC 3437772. PMID 21284967.
- ^ Lamont, Ronald F; Kam, KY Ronald (March 2008). “Atosiban as a tocolytic for the treatment of spontaneous preterm labor”. Expert Review of Obstetrics & Gynecology. 3 (2): 163–174. doi:10.1586/17474108.3.2.163. ISSN 1747-4108.
- ^ Jump up to:a b c Lamont, Ronald F.; Kam, KY Ronald (2008). “Atosiban as a tocolytic for the treatment of spontaneous preterm labor”. Expert Review of Obstetrics & Gynecology. 3 (2): 163–174. doi:10.1586/17474108.3.2.163.
- ^ Lamont CD, Jørgensen JS, Lamont RF (September 2016). “The safety of tocolytics used for the inhibition of preterm labour”. Expert Opinion on Drug Safety. 15 (9): 1163–73. doi:10.1080/14740338.2016.1187128. PMID 27159501. S2CID 4937942.
It was for this reason and the fact that Tractocile (atosiban) only had a short duration before it was out of patent that the parent drug company decided not to pursue licensing in the USA.
- ^ Coomarasamy, A; Knox, EM; Gee, H; Khan, KS (November 2002). “Oxytocin antagonists for tocolysis in preterm labour — a systematic review”. Med Sci Monit. 8 (11): RA268–73. PMID 12444392.
- ^ Flenady, Vicki; Reinebrant, Hanna E.; Liley, Helen G.; Tambimuttu, Eashan G.; Papatsonis, Dimitri N. M. (6 June 2014). “Oxytocin receptor antagonists for inhibiting preterm labour” (PDF). The Cochrane Database of Systematic Reviews (6): CD004452. doi:10.1002/14651858.CD004452.pub3. ISSN 1469-493X. PMID 24903678.
- ^ Jump up to:a b c d Saleh SS, Al-Ramahi MQ, Al Kazaleh FA (January 2013). “Atosiban and nifedipine in the suppression of preterm labour: a comparative study”. J Obstet Gynaecol. 33 (1): 43–5. doi:10.3109/01443615.2012.721822. PMID 23259877. S2CID 20753923.
- ^ Jump up to:a b c d Salim R, Garmi G, Nachum Z, Zafran N, Baram S, Shalev E (December 2012). “Nifedipine compared with atosiban for treating preterm labor: a randomized controlled trial”. Obstet Gynecol. 120 (6): 1323–31. doi:10.1097/aog.0b013e3182755dff. PMID 23168756. S2CID 22487349.
- ^ Jump up to:a b de Heus R, Mulder EJ, Derks JB, Visser GH (June 2009). “The effects of the tocolytics atosiban and nifedipine on fetal movements, heart rate and blood flow”. J Matern Fetal Neonatal Med. 22 (6): 485–90. doi:10.1080/14767050802702349. PMID 19479644. S2CID 35810758.
- ^ Shim JY, Park YW, YoonBH, Cho YK, Yang JH, Lee Y, Kim A. “Multicentre, parallelgroup, randomised, single-blind study of the safety and efficacy of atosibanversus ritodrine in the treatment of acute preterm labour in Korean women. BJOG 2006Nov;113(11):1228-34.
External links
- “Atosiban”. Drug Information Portal. U.S. National Library of Medicine.
| Clinical data | |
|---|---|
| Trade names | Tractocile, Antocin, others[1] |
| AHFS/Drugs.com | UK Drug Information |
| License data | EU EMA: by INN |
| Routes of administration | Intravenous |
| ATC code | G02CX01 (WHO) |
| Legal status | |
| Legal status | AU: S4 (Prescription only)UK: POM (Prescription only) [2][3]EU: Rx-only [4]In general: ℞ (Prescription only) |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 90779-69-4 |
| PubChem CID | 5311010 |
| IUPHAR/BPS | 2213 |
| DrugBank | DB09059 |
| ChemSpider | 4470550 |
| UNII | 081D12SI0Z |
| KEGG | D03008 |
| ChEMBL | ChEMBL378642 |
| CompTox Dashboard (EPA) | DTXSID8048991 |
| ECHA InfoCard | 100.234.128 |
| Chemical and physical data | |
| Formula | C43H67N11O12S2 |
| Molar mass | 994.19 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI |
Publication numberPriority datePublication dateAssigneeTitle
US4504469A *1982-12-211985-03-12Ferring AbVasotocin derivatives
WO2006119388A2 *2005-05-032006-11-09Novetide, Ltd.Methods for the production of peptide having a c-terminal amide
CN102127146A *2010-12-242011-07-20深圳翰宇药业股份有限公司Method for preparing atosiban acetate
DK0710243T3 *1993-06-292000-10-16Ferring BvSynthesis of cyclic peptides
CN101357937B *2007-07-312012-11-07上海苏豪逸明制药有限公司Method for synthesizing atosiban acetate from solid phase polypeptide
CN101314613B *2008-05-082012-04-25吉尔生化(上海)有限公司Solid phase synthesis method for atosiban
CN102127146B *2010-12-242013-04-24深圳翰宇药业股份有限公司Method for preparing atosiban acetate
CN102584953B *2012-02-092014-01-01深圳翰宇药业股份有限公司Purification method for atosiban
CN104098650B *2013-04-152019-04-09中国医学科学院药物研究所The synthesis and application of the intermediate of Atosiban
GB201310921D0 *2013-06-192013-07-31Chemical & Biopharmaceutical Lab Of Patras S APeptide-resin conjugate and use thereof
CN105949283A *2016-06-072016-09-21海南合瑞制药股份有限公司Atosiban acetate impurities and preparation and detection methods
CN106279367B *2016-08-152019-06-04海南合瑞制药股份有限公司A kind of atosiban acetate crystal and preparation method thereof
CN107312072A *2017-06-202017-11-03浙江湃肽生物有限公司A kind of method of purifies and separates Atosiban
ApplicationPriority dateFiling dateTitle
CN201010604790.62010-12-24
CN2010106047906A2010-12-242010-12-24Method for preparing atosiban acetate
CN2010106047902010-12-24
PCT/CN2011/0844142010-12-242011-12-22Method for preparing atosiban acetate
Similar Documents
PublicationPublication DateTitle
US9434767B22016-09-06Method for preparing atosiban acetate
CN103497245B2015-05-13Method for synthesizing thymalfasin
DK2421887T32015-07-27A process for the preparation of degarelix
CN104974237B2019-02-12A kind of method of segment method synthesis in solid state ziconotide
CN101747426B2013-01-16Method for synthesizing pramlintide
CN102702320B2013-08-21Method for preparing eptifibatide
CN106589069B2018-07-17A kind of preparation method of oxytocin
US9394341B22016-07-19Eptifibatide preparation method
CN104231051A2014-12-24Preparation method for linaclotide
CN104177490B2017-02-08Method for preparing salmon calcitonin acetate by fragment condensation
CN101372505B2011-03-30Method for preparing desmopressin acetate
CN105418736A2016-03-23Preparation method of terlipressin through combination of solid and liquid
CN106084015B2020-01-31method for synthesizing carbetocin
CN103467573B2017-12-15A kind of preparation method of carbetocin
CN103214568B2014-09-24Solid phase method of secretin
CN103992389A2014-08-20Method for solid cyclizing synthesis of desmopressin
CN109021087A2018-12-18A kind of method that solid liquid phase combination prepares ziconotide
CN104761619A2015-07-08Desmopressin acetate solid phase preparation technology
CN106854235B2020-09-18Solid phase fragment method for synthesizing carbetocin
CN106554391B2021-11-05Method for synthesizing marine biological peptide Xen2174
CN107216374A2017-09-29A kind of synthetic method of ziconotide
CN103992401B2017-05-17Method for preparing exenatide
CN105037496B2018-12-25A kind of preparation method of eptifibatide
WO2017097194A12017-06-15Completely-solid-phase preparation method for carbetocin
CN103980350A2014-08-13Solid-phase cyclization synthesis method of Atosiban
////////////// ATOSIBAN, CAP-449, CAP-476, CAP-581, F-314, ORF 22164, ORF-22164, RW-22164, RWJ 22164, RWJ-22164
[H][C@]1(NC(=O)[C@@]([H])(NC(=O)[C@@H](CC2=CC=C(OCC)C=C2)NC(=O)CCSSC[C@H](NC(=O)[C@H](CC(N)=O)NC1=O)C(=O)N1CCC[C@H]1C(=O)N[C@@H](CCCN)C(=O)NCC(N)=O)[C@@H](C)CC)[C@@H](C)O

NEW DRUG APPROVALS
TO MAINTAIN SUBSCRIPTION OF THIS BLOG
$10.00
Dr. D Srinivasa Reddy appointed Director CSIR-IICT Hyderabad India on 7th June 2022. A new assignment
Dr. D Srinivasa Reddy appointed Director CSIR-IICT Hyderabad India on 7th June 2022. A new assignment
This is on recommendation from search cum selection committee which met Prime minister who is president CSIR on 2nd may 2022
currently he is Director CSIR-IIIM jammu
we wish him all the best in New assignment
D. Srinivasa Reddy (DSReddy)


………….Srinivasa Reddy, Director, CSIR-IICT, Hyderabad, india
Tirzepatide
YXEGTFTSDY SIXLDKIAQK AFVQWLIAGG PSSGAPPPS

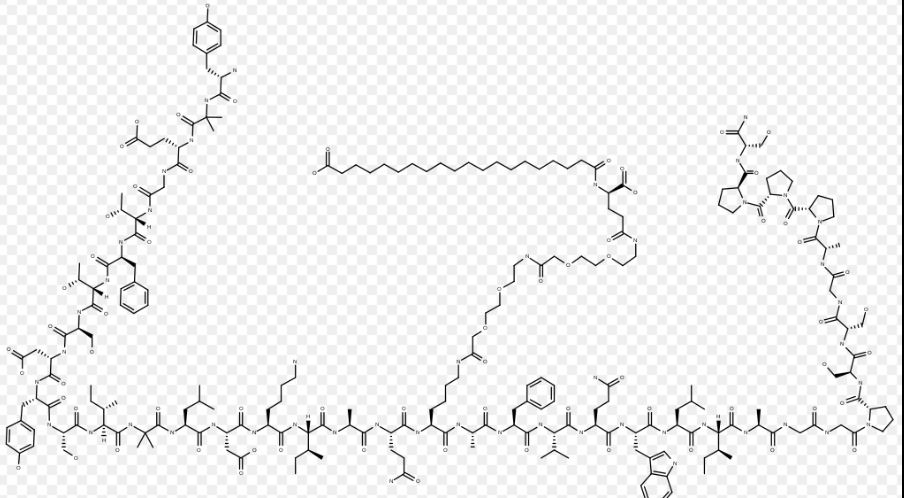


Tirzepatide
チルゼパチド
LY3298176,
| Formula | C225H348N48O68 |
|---|---|
| CAS | 2023788-19-2 |
| Mol weight | 4813.4514 |
FDA APPROVED 2022/5/13, Mounjaro
| Class | Antidiabetic agent GLP-1 receptor agonist |
|---|---|
| Efficacy | Antidiabetic, Gastric inhibitory polypeptide receptor agonist, Glucagon-like peptide 1 (GLP-1) receptor agonist |
| Disease | Type 2 diabetes mellitus |

Tirzepatide is an agonist of human glucose-dependent insulinotropic polypeptide (GIP) and human glucagon-like peptide-1 (GLP-1) receptors, whose amino acid residues at positions 2 and 13 are 2-methylAla, and the C-terminus is amidated Ser. A 1,20-icosanedioic acid is attached to Lys at position 20 via a linker which consists of a Glu and two 8-amino-3,6-dioxaoctanoic acids. Tirzepatide is a synthetic peptide consisting of 39 amino acid residues.
C225H348N48O68 : 4813.45
[2023788-19-2]
L-Serinamide, L-tyrosyl-2-methylalanyl-L-α-glutamylglycyl-L-threonyl-L-phenylalanyl-L-threonyl-L-seryl-L-α-aspartyl-L-tyrosyl-L-seryl-L-isoleucyl-2-methylalanyl-L-leucyl-L-α-aspartyl-L-lysyl-L-isoleucyl-L-alanyl-L-glutaminyl-N6-[(22S)-22,42-dicarboxy-1,10,19,24-tetraoxo-3,6,12,15-tetraoxa-9,18,23-triazadotetracont-1-yl]-L-lysyl-L-alanyl-L-phenylalanyl-L-valyl-L-glutaminyl-L-tryptophyl-L-leucyl-L-isoleucyl-L-alanylglycylglycyl-L-prolyl-L-seryl-L-serylglycyl-L-alanyl-L-prolyl-L-prolyl-L-prolyl-
Other Names
- L-Tyrosyl-2-methylalanyl-L-α-glutamylglycyl-L-threonyl-L-phenylalanyl-L-threonyl-L-seryl-L-α-aspartyl-L-tyrosyl-L-seryl-L-isoleucyl-2-methylalanyl-L-leucyl-L-α-aspartyl-L-lysyl-L-isoleucyl-L-alanyl-L-glutaminyl-N6-[(22S)-22,42-dicarboxy-1,10,19,24-tetraoxo-3,6,12,15-tetraoxa-9,18,23-triazadotetracont-1-yl]-L-lysyl-L-alanyl-L-phenylalanyl-L-valyl-L-glutaminyl-L-tryptophyl-L-leucyl-L-isoleucyl-L-alanylglycylglycyl-L-prolyl-L-seryl-L-serylglycyl-L-alanyl-L-prolyl-L-prolyl-L-prolyl-L-serinamide
Tirzepatide, sold under the brand name Mounjaro,[1] is a medication used for the treatment type 2 diabetes.[2][3][4] Tirzepatide is given by injection under the skin.[2] Common side effects may include nausea, vomiting, diarrhea, decreased appetite, constipation, upper abdominal discomfort and abdominal pain.[2]
Glucagon-like peptide-1 (GLP-1) and glucose-dependent insulinotropic polypeptide (GIP) are hormones involved in blood sugar control.[2] Tirzepatide is a first-in-class medication that activates both the GLP-1 and GIP receptors, which leads to improved blood sugar control.[2] Tirzepatide was approved for medical use in the United States in May 2022.[2]
SYN
https://pubs.acs.org/doi/10.1021/acs.oprd.1c00108

The large-scale manufacture of complex synthetic peptides is challenging due to many factors such as manufacturing risk (including failed product specifications) as well as processes that are often low in both yield and overall purity. To overcome these liabilities, a hybrid solid-phase peptide synthesis/liquid-phase peptide synthesis (SPPS/LPPS) approach was developed for the synthesis of tirzepatide. Continuous manufacturing and real-time analytical monitoring ensured the production of high-quality material, while nanofiltration provided intermediate purification without difficult precipitations. Implementation of the strategy worked very well, resulting in a robust process with high yields and purity.
PATENT
- WO2016111971
- US2020023040
- WO2019245893
- US2020155487
- US2020155650
- WO2020159949CN112592387
- WO2021066600CN112661815
- WO2021154593
- US2021338769

NEW DRUG APPROVALS
ONE TIME TO MAINTAIN THIS BLOG SUBSCRIPTION
$10.00
//////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
Medical uses
Tirzepatide in indicated to improve blood sugar control in adults with type 2 diabetes, as an addition to diet and exercise.[2]
Contraindications
Tirzepatide should not be used in people with a personal or family history of medullary thyroid cancer or in people with multiple endocrine neoplasia syndrome type 2.[2]
Adverse effects
Preclinical, phase I, and phase II trials have indicated that tirzepatide exhibits similar adverse effects to other established GLP-1 receptor agonists, such as GLP-1 receptor agonist dulaglutide. These effects occur largely within the gastrointestinal tract.[5] The most frequently observed adverse effects are nausea, diarrhoea and vomiting, which increased in incidence with the dosage amount (i.e. higher likelihood the higher the dose). The number of patients who discontinued taking tirzepatide also increased as dosage increased, with patients taking 15 mg having a 25% discontinuation rate vs 5.1% for 5 mg patients and 11.1% for dulaglutide.[6] To a slightly lesser extent, patients also reported reduced appetite.[5] Other side effects reported were dyspepsia, constipation, abdominal pain, dizziness and hypoglycaemia.[7][8]
Pharmacology
Tirzepatide is an analogue of gastric inhibitory polypeptide (GIP), a human hormone which stimulates the release of insulin from the pancreas. Tirzepatide is a linear polypeptide of 39 amino acids which has been chemically modified by lipidation to improve its uptake into cells and its stability to metabolism.[9] The compound is administered as a weekly subcutaneous injection.[10] It completed phase III trials globally in 2021.[11][12]
Mechanism of action
Tirzepatide has a greater affinity to GIP receptors than to GLP-1 receptors, and this dual agonist behaviour has been shown to produce greater reductions of hyperglycemia compared to a selective GLP-1 receptor agonist.[3] Signaling studies have shown that this is due to tirzepatide mimicking the actions of natural GIP at the GIP receptor.[13] However, at the GLP-1 receptor, tirzepatide shows bias towards cAMP (a messenger associated with regulation of glycogen, sugar and lipid metabolism) generation, rather than β-arrestin recruitment. This combination of preference towards GIP receptor and distinct signaling properties at GLP-1 suggest this biased agonism increases insulin secretion.[13] Tirzepatide has also been shown to increase levels of adiponectin, an adipokine involved in the regulation of both glucose and lipid metabolism, with a maximum increase of 26% from baseline after 26 weeks, at the 10 mg dosage.[3]
Chemistry
Structure
Tirzepatide is an analog of the human GIP hormone with a C20 fatty-diacid portion attached, used to optimise the uptake and metabolism of the compound.[9] The fatty-diacid section (eicosanedioic acid) is linked via a glutamic acid and two (2-(2-aminoethoxy)ethoxy)acetic acid units to the side chain of the lysine residue. This arrangement allows for a much longer half life, extending the time between doses, because of its high affinity to albumin.[14]
Synthesis
The synthesis of tirzepatide was first disclosed in patents filed by Eli Lilly and Company.[15] This uses standard solid phase peptide synthesis, with an allyloxycarbonyl protecting group on the lysine at position 20 of the linear chain of amino acids, allowing a final set of chemical transformations in which the sidechain amine of that lysine is derivatized with the lipid-containing fragment.
Large-scale manufacturing processes have been reported for this compound.[16]
History
Indiana-based pharmaceutical company Eli Lilly and Company first applied for a patent for a method of glycemic control using tirzepatide in early 2016.[15] The patent was published late that year. After passing phase 3 clinical trials, Lilly applied for FDA approval in October 2021 with a priority review voucher.[17]
Following the completion of the pivotal SURPASS-2 trial no. NCT03987919, the company announced on 28 April that tirzepatide had successfully met their endpoints in obese and overweight patients without diabetes.[18] Alongside results from the SURMOUNT-1 trial no. NCT04184622, they suggest that tirzepatide may potentially be a competitor for existing diabetic medication semaglutide, manufactured by Novo Nordisk.[19][20]
In industry-funded preliminary trials comparing tirzepatide to the existing diabetes medication semaglutide (an injected analogue of the hormone GLP-1), tirzepatide showed minor improvement of reductions (2.01%–2.30% depending on dosage) in glycated hemoglobin tests relative to semaglutide (1.86%).[21] A 10 mg dose has also been shown to be effective in reducing insulin resistance, with a reduction of around 8% from baseline, measured using HOMA2-IR (computed with fasting insulin).[3] Fasting levels of IGF binding proteins like IGFBP1 and IGFBP2 increased following tirzepatide treatment, increasing insulin sensitivity.[3] A meta-analysis published by Dutta et al. showed that over 1-year clinical use, tirzepatide was observed to be superior to dulaglutide, semaglutide, degludec, and insulin glargine with regards to glycemic efficacy and obesity reduction. Tirzepatide is perhaps the most potent agent developed to date to tackle the global problem of “diabesity“.[22]
Society and culture
Names
Tirzepatide is the international nonproprietary name (INN).[23]
References
- ^ Jump up to:a b “Highlights of prescribing information” (PDF). accessdata.fda.gov. FDA. May 2022. Retrieved 14 May 2022.
- ^ Jump up to:a b c d e f g h i “FDA Approves Novel, Dual-Targeted Treatment for Type 2 Diabetes”. U.S. Food and Drug Administration (FDA) (Press release). 13 May 2022. Retrieved 13 May 2022. This article incorporates text from this source, which is in the public domain.
- ^ Jump up to:a b c d e Thomas MK, Nikooienejad A, Bray R, Cui X, Wilson J, Duffin K, et al. (January 2021). “Dual GIP and GLP-1 Receptor Agonist Tirzepatide Improves Beta-cell Function and Insulin Sensitivity in Type 2 Diabetes”. The Journal of Clinical Endocrinology and Metabolism. 106 (2): 388–396. doi:10.1210/clinem/dgaa863. PMC 7823251. PMID 33236115.
- ^ Coskun T, Sloop KW, Loghin C, Alsina-Fernandez J, Urva S, Bokvist KB, et al. (December 2018). “LY3298176, a novel dual GIP and GLP-1 receptor agonist for the treatment of type 2 diabetes mellitus: From discovery to clinical proof of concept”. Molecular Metabolism. 18: 3–14. doi:10.1016/j.molmet.2018.09.009. PMC 6308032. PMID 30473097.
- ^ Jump up to:a b Min T, Bain SC (January 2021). “The Role of Tirzepatide, Dual GIP and GLP-1 Receptor Agonist, in the Management of Type 2 Diabetes: The SURPASS Clinical Trials”. Diabetes Therapy. 12 (1): 143–157. doi:10.1007/s13300-020-00981-0. PMC 7843845. PMID 33325008.
- ^ Frias JP, Nauck MA, Van J, Kutner ME, Cui X, Benson C, et al. (November 2018). “Efficacy and safety of LY3298176, a novel dual GIP and GLP-1 receptor agonist, in patients with type 2 diabetes: a randomised, placebo-controlled and active comparator-controlled phase 2 trial”. The Lancet. 392 (10160): 2180–2193. doi:10.1016/S0140-6736(18)32260-8. PMID 30293770.
- ^ Frias JP, Nauck MA, Van J, Benson C, Bray R, Cui X, et al. (June 2020). “Efficacy and tolerability of tirzepatide, a dual glucose-dependent insulinotropic peptide and glucagon-like peptide-1 receptor agonist in patients with type 2 diabetes: A 12-week, randomized, double-blind, placebo-controlled study to evaluate different dose-escalation regimens”. Diabetes, Obesity & Metabolism. 22 (6): 938–946. doi:10.1111/dom.13979. PMC 7318331. PMID 31984598.
- ^ Dahl D, Onishi Y, Norwood P, Huh R, Bray R, Patel H, Rodríguez Á (February 2022). “Effect of Subcutaneous Tirzepatide vs Placebo Added to Titrated Insulin Glargine on Glycemic Control in Patients With Type 2 Diabetes: The SURPASS-5 Randomized Clinical Trial”. JAMA. 327 (6): 534–545. doi:10.1001/jama.2022.0078. PMID 35133415.
- ^ Jump up to:a b Ahangarpour M, Kavianinia I, Harris PW, Brimble MA (January 2021). “Photo-induced radical thiol-ene chemistry: a versatile toolbox for peptide-based drug design”. Chemical Society Reviews. Royal Society of Chemistry. 50 (2): 898–944. doi:10.1039/d0cs00354a. PMID 33404559. S2CID 230783854.
- ^ Bastin M, Andreelli F (2019). “Dual GIP-GLP1-Receptor Agonists In The Treatment Of Type 2 Diabetes: A Short Review On Emerging Data And Therapeutic Potential”. Diabetes, Metabolic Syndrome and Obesity: Targets and Therapy. 12: 1973–1985. doi:10.2147/DMSO.S191438. PMC 6777434. PMID 31686879.
- ^ “Tirzepatide significantly reduced A1C and body weight in people with type 2 diabetes in two phase 3 trials from Lilly’s SURPASS program” (Press release). Eli Lilly and Company. 17 February 2021. Retrieved 28 October 2021 – via PR Newswire.
- ^ “Lilly : Phase 3 Tirzepatide Results Show Superior A1C And Body Weight Reductions In Type 2 Diabetes”. Business Insider. RTTNews. 19 October 2021. Retrieved 28 October 2021.
- ^ Jump up to:a b Willard FS, Douros JD, Gabe MB, Showalter AD, Wainscott DB, Suter TM, et al. (September 2020). “Tirzepatide is an imbalanced and biased dual GIP and GLP-1 receptor agonist”. JCI Insight. 5 (17). doi:10.1172/jci.insight.140532. PMC 7526454. PMID 32730231.
- ^ Østergaard S, Paulsson JF, Kofoed J, Zosel F, Olsen J, Jeppesen CB, et al. (October 2021). “The effect of fatty diacid acylation of human PYY3-36 on Y2 receptor potency and half-life in minipigs”. Scientific Reports. 11 (1): 21179. Bibcode:2021NatSR..1121179O. doi:10.1038/s41598-021-00654-3. PMC 8551270. PMID 34707178.
- ^ Jump up to:a b US patent 9474780, Bokvist BK, Coskun T, Cummins RC, Alsina-Fernandez J, “GIP and GLP-1 co-agonist compounds”, issued 2016-10-25, assigned to Eli Lilly and Co
- ^ Frederick MO, Boyse RA, Braden TM, Calvin JR, Campbell BM, Changi SM, et al. (2021). “Kilogram-Scale GMP Manufacture of Tirzepatide Using a Hybrid SPPS/LPPS Approach with Continuous Manufacturing”. Organic Process Research & Development. 25 (7): 1628–1636. doi:10.1021/acs.oprd.1c00108. S2CID 237690232.
- ^ Sagonowsky, Eric (26 October 2021). “As Lilly gears up for key 2022 launches, Trulicity, Taltz and more drive solid growth”. Fierce Pharma. Retrieved 9 April 2022.
- ^ Kellaher, Colin (28 April 2022). “Eli Lilly’s Tirzepatide Meets Main Endpoints in Phase 3 Obesity Study >LLY”. Dow Jones Newswires. Retrieved 29 April 2022 – via MarketWatch.
- ^ Kahan, Scott; Garvey, W. Timothy (28 April 2022). “SURMOUNT-1: Adults achieve weight loss of 16% or more at 72 weeks with tirzepatide”. healio.com. Retrieved 29 April 2022.
- ^ Taylor, Nick Paul (28 April 2022). “SURMOUNT-able: Lilly’s tirzepatide clears high bar set by Novo’s Wegovy in obesity”. FierceBiotech. Retrieved 29 April 2022.
- ^ Frías JP, Davies MJ, Rosenstock J, Pérez Manghi FC, Fernández Landó L, Bergman BK, et al. (August 2021). “Tirzepatide versus Semaglutide Once Weekly in Patients with Type 2 Diabetes”. The New England Journal of Medicine. 385 (6): 503–515. doi:10.1056/NEJMoa2107519. PMID 34170647. S2CID 235635529.
- ^ Dutta D, Surana V, Singla R, Aggarwal S, Sharma M (November–December 2021). “Efficacy and safety of novel twincretin tirzepatide a dual GIP and GLP-1 receptor agonist in the management of type-2 diabetes: A Cochrane meta-analysis”. Indian Journal of Endocrinology and Metabolism. 25 (6): 475–489. doi:10.4103/ijem.ijem_423_21.
- ^ World Health Organization (2019). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 81”. WHO Drug Information. 33 (1). hdl:10665/330896.
Further reading
- Bhagavathula AS, Vidyasagar K, Tesfaye W (September 2021). “Efficacy and Safety of Tirzepatide in Patients with Type 2 Diabetes Mellitus: A Systematic Review and Meta-Analysis of Randomized Phase II/III Trials”. Pharmaceuticals (Basel). 14 (10). doi:10.3390/ph14100991. PMC 8537322. PMID 34681215.
- Frías JP (November 2020). “Tirzepatide: a glucose-dependent insulinotropic polypeptide (GIP) and glucagon-like peptide-1 (GLP-1) dual agonist in development for the treatment of type 2 diabetes”. Expert Rev Endocrinol Metab. 15 (6): 379–394. doi:10.1080/17446651.2020.1830759. PMID 33030356.
- Ryan DH (September 2021). “Next Generation Antiobesity Medications: Setmelanotide, Semaglutide, Tirzepatide and Bimagrumab: What do They Mean for Clinical Practice?”. J Obes Metab Syndr. 30 (3): 196–208. doi:10.7570/jomes21033. PMC 8526285. PMID 34518444.
External links
- “Tirzepatide”. Drug Information Portal. U.S. National Library of Medicine.
- Clinical trial number NCT03954834 for “A Study of Tirzepatide (LY3298176) in Participants With Type 2 Diabetes Not Controlled With Diet and Exercise Alone (SURPASS-1)” at ClinicalTrials.gov
- Clinical trial number NCT03987919 for “A Study of Tirzepatide (LY3298176) Versus Semaglutide Once Weekly as Add-on Therapy to Metformin in Participants With Type 2 Diabetes (SURPASS-2)” at ClinicalTrials.gov
- Clinical trial number NCT03882970 for “A Study of Tirzepatide (LY3298176) Versus Insulin Degludec in Participants With Type 2 Diabetes (SURPASS-3)” at ClinicalTrials.gov
- Clinical trial number NCT03730662 for “A Study of Tirzepatide (LY3298176) Once a Week Versus Insulin Glargine Once a Day in Participants With Type 2 Diabetes and Increased Cardiovascular Risk (SURPASS-4)” at ClinicalTrials.gov
- Clinical trial number NCT04039503 for “A Study of Tirzepatide (LY3298176) Versus Placebo in Participants With Type 2 Diabetes Inadequately Controlled on Insulin Glargine With or Without Metformin (SURPASS-5)” at ClinicalTrials.gov
CLIP
FDA approves Lilly’s Mounjaro™ (tirzepatide) injection, the first and only GIP and GLP-1 receptor agonist for the treatment of adults with type 2 diabetes
May 13, 2022
Mounjaro delivered superior A1C reductions versus all comparators in phase 3 SURPASS clinical trials
While not indicated for weight loss, Mounjaro led to significantly greater weight reductions versus comparators in a key secondary endpoint
Mounjaro represents the first new class of diabetes medicines introduced in nearly a decade and is expected to be available in the U.S. in the coming weeks
INDIANAPOLIS, May 13, 2022 /PRNewswire/ — The U.S. Food and Drug Administration (FDA) approved Mounjaro™ (tirzepatide) injection, Eli Lilly and Company’s (NYSE: LLY) new once-weekly GIP (glucose-dependent insulinotropic polypeptide) and GLP-1 (glucagon-like peptide-1) receptor agonist indicated as an adjunct to diet and exercise to improve glycemic control in adults with type 2 diabetes. Mounjaro has not been studied in patients with a history of pancreatitis and is not indicated for use in patients with type 1 diabetes mellitus.
As the first and only FDA-approved GIP and GLP-1 receptor agonist, Mounjaro is a single molecule that activates the body’s receptors for GIP and GLP-1, which are natural incretin hormones.1
“Mounjaro delivered superior and consistent A1C reductions against all of the comparators throughout the SURPASS program, which was designed to assess Mounjaro’s efficacy and safety in a broad range of adults with type 2 diabetes who could be treated in clinical practice. The approval of Mounjaro is an exciting step forward for people living with type 2 diabetes given the results seen in these clinical trials,” said Juan Pablo Frías, M.D., Medical Director, National Research Institute and Investigator in the SURPASS program.
Mounjaro will be available in six doses (2.5 mg, 5 mg, 7.5 mg, 10 mg, 12.5 mg, 15 mg) and will come in Lilly’s well-established auto-injector pen with a pre-attached, hidden needle that patients do not need to handle or see.
The approval was based on results from the phase 3 SURPASS program, which included active comparators of injectable semaglutide 1 mg, insulin glargine and insulin degludec. Efficacy was evaluated for Mounjaro 5 mg, 10 mg and 15 mg used alone or in combination with commonly prescribed diabetes medications, including metformin, SGLT2 inhibitors, sulfonylureas and insulin glargine. Participants in the SURPASS program achieved average A1C reductions between 1.8% and 2.1% for Mounjaro 5 mg and between 1.7% and 2.4% for both Mounjaro 10 mg and Mounjaro 15 mg. While not indicated for weight loss, mean change in body weight was a key secondary endpoint in all SURPASS studies. Participants treated with Mounjaro lost between 12 lb. (5 mg) and 25 lb. (15 mg) on average.1
Side effects reported in at least 5% of patients treated with Mounjaro include nausea, diarrhea, decreased appetite, vomiting, constipation, indigestion (dyspepsia), and stomach (abdominal) pain. The labeling for Mounjaro contains a Boxed Warning regarding thyroid C-cell tumors. Mounjaro is contraindicated in patients with a personal or family history of medullary thyroid carcinoma or in patients with Multiple Endocrine Neoplasia syndrome type 2.1
“Lilly has a nearly 100-year heritage of advancing care for people living with diabetes – never settling for current outcomes. We’re not satisfied knowing that half of the more than 30 million Americans living with type 2 diabetes are not reaching their target blood glucose levels,” said Mike Mason, president, Lilly Diabetes. “We are thrilled to introduce Mounjaro, which represents the first new class of type 2 diabetes medication introduced in almost a decade and embodies our mission to bring innovative new therapies to the diabetes community.”
Mounjaro is expected to be available in the United States in the coming weeks. Lilly is committed to helping people access the medicines they are prescribed and will work with insurers, health systems and providers to help enable patient access to Mounjaro. Lilly plans to offer a Mounjaro savings card for people who qualify. Patients or healthcare professionals with questions about Mounjaro can visit www.Mounjaro.com or call The Lilly Answers Center at 1-800-LillyRx (1-800-545-5979).
Tirzepatide is also under regulatory review for the treatment of type 2 diabetes in Europe, Japan and several additional markets. A multimedia gallery is available on Lilly.com.
About the SURPASS clinical trial program
The SURPASS phase 3 global clinical development program for tirzepatide began in late 2018 and included five global registration trials and two regional trials in Japan. These studies ranged from 40 to 52 weeks and evaluated the efficacy and safety of Mounjaro 5 mg, 10 mg and 15 mg as a monotherapy and as an add-on to various standard-of-care medications for type 2 diabetes. The active comparators in the studies were injectable semaglutide 1 mg, insulin glargine and insulin degludec. Collectively, the five global registration trials consistently demonstrated A1C reductions for participants taking Mounjaro across multiple stages of their type 2 diabetes journeys, from an average around five to 13 years of having diabetes.2-8
- SURPASS-1 (NCT03954834) was a 40-week study comparing the efficacy and safety of Mounjaro 5 mg (N=121), 10 mg (N=121) and 15 mg (N=120) as monotherapy to placebo (N=113) in adults with type 2 diabetes inadequately controlled with diet and exercise alone. From a baseline A1C of 7.9%, Mounjaro reduced participants’ A1C by a mean of 1.8%* (5 mg) and 1.7%* (10 mg and 15 mg) compared to 0.1% for placebo. In a key secondary endpoint, from a baseline weight of 189 lb., Mounjaro reduced participants’ weight by a mean of 14 lb.* (5 mg), 15 lb.* (10 mg) and 17 lb.* (15 mg) compared to 2 lb. for placebo.2,3
- SURPASS-2 (NCT03987919) was a 40-week study comparing the efficacy and safety of Mounjaro 5 mg (N=470), 10 mg (N=469) and 15 mg (N=469) to injectable semaglutide 1 mg (N=468) in adults with type 2 diabetes inadequately controlled with ≥1500 mg/day metformin alone. From a baseline A1C of 8.3%, Mounjaro reduced participants’ A1C by a mean of 2.0%ꝉ (5 mg), 2.2%* (10 mg) and 2.3%* (15 mg) compared to 1.9% for semaglutide. In a key secondary endpoint, from a baseline weight of 207 lb., Mounjaro reduced participants’ weight by a mean of 17 lb.ꝉ (5 mg), 21 lb.* (10 mg) and 25 lb.* (15 mg) compared to 13 lb. for semaglutide.4,5
- SURPASS-3 (NCT03882970) was a 52-week study comparing the efficacy of Mounjaro 5 mg (N=358), 10 mg (N=360) and 15 mg (N=358) to titrated insulin degludec (N=359) in adults with type 2 diabetes treated with metformin with or without an SGLT-2 inhibitor. From a baseline A1C of 8.2%, Mounjaro reduced participants’ A1C by a mean of 1.9%* (5 mg), 2.0%* (10 mg) and 2.1%* (15 mg) compared to 1.3% for insulin degludec. From a baseline weight of 208 lb., Mounjaro reduced participants’ weight by a mean of 15 lb.* (5 mg), 21 lb.* (10 mg) and 25 lb.* (15 mg) compared to an increase of 4 lb. for insulin degludec.6
- SURPASS-4 (NCT03730662) was a 104-week study comparing the efficacy and safety of Mounjaro 5 mg (N=328), 10 mg (N=326) and 15 mg (N=337) to insulin glargine (N=998) in adults with type 2 diabetes inadequately controlled with at least one and up to three oral antihyperglycemic medications (metformin, sulfonylureas or SGLT-2 inhibitors), who have increased cardiovascular (CV) risk. The primary endpoint was measured at 52 weeks. From a baseline A1C of 8.5%, Mounjaro reduced participants’ A1C by a mean of 2.1%* (5 mg), 2.3%* (10 mg) and 2.4%* (15 mg) compared to 1.4% for insulin glargine. From a baseline weight of 199 lb., Mounjaro reduced weight by a mean of 14 lb.* (5 mg), 20 lb.* (10 mg) and 23 lb.* (15 mg) compared to an increase of 4 lb. for insulin glargine.7
- SURPASS-5 (NCT04039503) was a 40-week study comparing the efficacy and safety of Mounjaro 5 mg (N=116), 10 mg (N=118) and 15 mg (N=118) to placebo (N=119) in adults with inadequately controlled type 2 diabetes already being treated with insulin glargine, with or without metformin. From a baseline A1C of 8.3%, Mounjaro reduced A1C by a mean of 2.1%* (5 mg), 2.4%* (10 mg) and 2.3%* (15 mg) compared to 0.9% for placebo. From a baseline weight of 210 lb., Mounjaro reduced participants’ weight by a mean of 12 lb.* (5 mg), 17 lb.* (10 mg) and 19 lb.* (15 mg) compared to an increase of 4 lb. for placebo.8
*p<0.001 for superiority vs. placebo or active comparator, adjusted for multiplicity
ꝉp<0.05 for superiority vs. semaglutide 1 mg, adjusted for multiplicity
About Mounjaro™ (tirzepatide) injection1
Mounjaro™ (tirzepatide) injection is FDA-approved as an adjunct to diet and exercise to improve glycemic control in adults with type 2 diabetes mellitus. As the first and only FDA-approved GIP and GLP-1 receptor agonist, Mounjaro is a single molecule that activates the body’s receptors for GIP (glucose-dependent insulinotropic polypeptide) and GLP-1 (glucagon-like peptide-1). Mounjaro will be available in six doses (2.5 mg, 5 mg, 7.5 mg, 10 mg, 12.5 mg, 15 mg) and will come in Lilly’s well-established auto-injector pen with a pre-attached, hidden needle that patients do not need to handle or see.
PURPOSE AND SAFETY SUMMARY WITH WARNINGS
Important Facts About MounjaroTM (mown-JAHR-OH). It is also known as tirzepatide.
- Mounjaro is an injectable prescription medicine for adults with type 2 diabetes used along with diet and exercise to improve blood sugar (glucose).
- It is not known if Mounjaro can be used in people who have had inflammation of the pancreas (pancreatitis). Mounjaro is not for use in people with type 1 diabetes. It is not known if Mounjaro is safe and effective for use in children under 18 years of age.
Warnings
Mounjaro may cause tumors in the thyroid, including thyroid cancer. Watch for possible symptoms, such as a lump or swelling in the neck, hoarseness, trouble swallowing, or shortness of breath. If you have a symptom, tell your healthcare provider.
- Do not use Mounjaro if you or any of your family have ever had a type of thyroid cancer called medullary thyroid carcinoma (MTC).
- Do not use Mounjaro if you have Multiple Endocrine Neoplasia syndrome type 2 (MEN 2).
- Do not use Mounjaro if you are allergic to tirzepatide or any of the ingredients in Mounjaro.
Mounjaro may cause serious side effects, including:
Inflammation of the pancreas (pancreatitis). Stop using Mounjaro and call your healthcare provider right away if you have severe pain in your stomach area (abdomen) that will not go away, with or without vomiting. You may feel the pain from your abdomen to your back.
Low blood sugar (hypoglycemia). Your risk for getting low blood sugar may be higher if you use Mounjaro with another medicine that can cause low blood sugar, such as a sulfonylurea or insulin. Signs and symptoms of low blood sugar may include dizziness or light-headedness, sweating, confusion or drowsiness, headache, blurred vision, slurred speech, shakiness, fast heartbeat, anxiety, irritability, or mood changes, hunger, weakness and feeling jittery.
Serious allergic reactions. Stop using Mounjaro and get medical help right away if you have any symptoms of a serious allergic reaction, including swelling of your face, lips, tongue or throat, problems breathing or swallowing, severe rash or itching, fainting or feeling dizzy, and very rapid heartbeat.
Kidney problems (kidney failure). In people who have kidney problems, diarrhea, nausea, and vomiting may cause a loss of fluids (dehydration), which may cause kidney problems to get worse. It is important for you to drink fluids to help reduce your chance of dehydration.
Severe stomach problems. Stomach problems, sometimes severe, have been reported in people who use Mounjaro. Tell your healthcare provider if you have stomach problems that are severe or will not go away.
Changes in vision. Tell your healthcare provider if you have changes in vision during treatment with Mounjaro.
Gallbladder problems. Gallbladder problems have happened in some people who use Mounjaro. Tell your healthcare provider right away if you get symptoms of gallbladder problems, which may include pain in your upper stomach (abdomen), fever, yellowing of skin or eyes (jaundice), and clay-colored stools.
Common side effects
The most common side effects of Mounjaro include nausea, diarrhea, decreased appetite, vomiting, constipation, indigestion, and stomach (abdominal) pain. These are not all the possible side effects of Mounjaro. Talk to your healthcare provider about any side effect that bothers you or doesn’t go away.
Tell your healthcare provider if you have any side effects. You can report side effects at 1-800-FDA-1088 or www.fda.gov/medwatch.
Before using
- Your healthcare provider should show you how to use Mounjaro before you use it for the first time.
- Before you use Mounjaro, talk to your healthcare provider about low blood sugar and how to manage it.
Review these questions with your healthcare provider:
- Do you have other medical conditions, including problems with your pancreas or kidneys, or severe problems with your stomach, such as slowed emptying of your stomach (gastroparesis) or problems digesting food?
- Do you take other diabetes medicines, such as insulin or sulfonylureas?
- Do you have a history of diabetic retinopathy?
- Are you pregnant or plan to become pregnant or breastfeeding or plan to breastfeed? It is not known if Mounjaro will harm your unborn baby.
- Do you take birth control pills by mouth? These may not work as well while using Mounjaro. Your healthcare provider may recommend another type of birth control when you start Mounjaro or when you increase your dose.
- Do you take any other prescription medicines or over-the-counter drugs, vitamins, or herbal supplements?
How to take
- Read the Instructions for Use that come with Mounjaro.
- Use Mounjaro exactly as your healthcare provider says.
- Mounjaro is injected under the skin (subcutaneously) of your stomach (abdomen), thigh, or upper arm.
- Use Mounjaro 1 time each week, at any time of the day.
- Do not mix insulin and Mounjaro together in the same injection.
- If you take too much Mounjaro, call your healthcare provider or seek medical advice promptly.
Learn more
For more information, call 1-800-LillyRx (1-800-545-5979) or go to www.mounjaro.com.
This information does not take the place of talking with your healthcare provider. Be sure to talk to your healthcare provider about Mounjaro and how to take it. Your healthcare provider is the best person to help you decide if Mounjaro is right for you.
MounjaroTM and its delivery device base are trademarks owned or licensed by Eli Lilly and Company, its subsidiaries, or affiliates.
Please click to access full Prescribing Information and Medication Guide.
TR CON CBS MAY2022
About Lilly
Lilly unites caring with discovery to create medicines that make life better for people around the world. We’ve been pioneering life-changing discoveries for nearly 150 years, and today our medicines help more than 47 million people across the globe. Harnessing the power of biotechnology, chemistry and genetic medicine, our scientists are urgently advancing new discoveries to solve some of the world’s most significant health challenges, redefining diabetes care, treating obesity and curtailing its most devastating long-term effects, advancing the fight against Alzheimer’s disease, providing solutions to some of the most debilitating immune system disorders, and transforming the most difficult-to-treat cancers into manageable diseases. With each step toward a healthier world, we’re motivated by one thing: making life better for millions more people. That includes delivering innovative clinical trials that reflect the diversity of our world and working to ensure our medicines are accessible and affordable. To learn more, visit Lilly.com and Lilly.com/newsroom or follow us on Facebook, Instagram, Twitter and LinkedIn. P-LLY
Lilly Cautionary Statement Regarding Forward-Looking Statements
This press release contains forward-looking statements (as that term is defined in the Private Securities Litigation Reform Act of 1995) about Mounjaro™ (tirzepatide 2.5 mg, 5 mg, 7.5 mg, 10 mg, 12.5 mg and 15 mg) injection as a treatment to improve glycemic control in adults with type 2 diabetes, the timeline for supply of Mounjaro to become available, and certain other milestones and ongoing clinical trials of Mounjaro and reflects Lilly’s current beliefs and expectations. However, as with any pharmaceutical product or medical device, there are substantial risks and uncertainties in the process of research, development and commercialization. Among other things, there can be no guarantee that Mounjaro will be commercially successful, that future study results will be consistent with results to date, or that we will meet our anticipated timelines for the commercialization of Mounjaro. For further discussion of these and other risks and uncertainties, see Lilly’s most recent Form 10-K and Form 10-Q filings with the United States Securities and Exchange Commission. Except as required by law, Lilly undertakes no duty to update forward-looking statements to reflect events after the date of this release.
References
- Mounjaro. Prescribing Information. Lilly USA, LLC.
- Rosenstock, J, et. al. Efficacy and Safety of Once Weekly Tirzepatide, a Dual GIP/GLP-1 Receptor Agonist Versus Placebo as Monotherapy in People with Type 2 Diabetes (SURPASS-1). Abstract 100-OR. Presented virtually at the American Diabetes Association’s 81st Scientific Sessions; June 25-29.
- Rosenstock, J, et. al. (2021). Efficacy and safety of a novel dual GIP and GLP-1 receptor agonist tirzepatide in patients with type 2 diabetes (SURPASS-1): a double-blind, randomised, phase 3 trial. Lancet. 2021;398(10295):143-155. doi: 10.1016/S0140-6736(21)01324-6.
- Frías JP, Davies MJ, Rosenstock J, et al; for the SURPASS-2 Investigators. Tirzepatide versus semaglutide once weekly in patients with type 2 diabetes. N Engl J Med. 2021;385(6)(suppl):503-515. doi: 10.1056/NEJMoa2107519
- Frias, J.P. Efficacy and Safety of Tirzepatide vs. Semaglutide Once Weekly as Add-On Therapy to Metformin in Patients with Type 2 Diabetes. Abstract 84-LB. Presented virtually at the American Diabetes Association’s 81st Scientific Sessions; June 25-29.
- Ludvik B, Giorgino F, Jódar E, et al. Once-weekly tirzepatide versus once-daily insulin degludec as add-on to metformin with or without SGLT2 inhibitors in patients with type 2 diabetes (SURPASS-3): a randomised, open-label, parallel-group, phase 3 trial. Lancet. 2021;398(10300):583-598. doi: 10.1016/S0140-6736(21)01443-4
- Del Prato S, Kahn SE, Pavo I, et al; for the SURPASS-4 Investigators. Tirzepatide versus insulin glargine in type 2 diabetes and increased cardiovascular risk (SURPASS-4): a randomised, open-label, parallel-group, multicentre, phase 3 trial. Lancet. 2021;398(10313):1811-1824. doi: 10.1016/S0140-6736(21)02188-7
- Dahl D, Onishi Y, Norwood P, et al. Effect of subcutaneous tirzepatide vs placebo added to titrated insulin glargine on glycemic control in patients with type 2 diabetes: the SURPASS-5 randomized clinical trial. JAMA. 2022;327(6):534-545. doi:10.1001/jama.2022.0078
CLIP
Lilly’s tirzepatide delivered up to 22.5% weight loss in adults with obesity or overweight in SURMOUNT-1
April 28, 2022
Participants taking tirzepatide lost up to 52 lb. (24 kg) in this 72-week phase 3 study
63% of participants taking tirzepatide 15 mg achieved at least 20% body weight reductions as a key secondary endpoint
INDIANAPOLIS, April 28, 2022 /PRNewswire/ — Tirzepatide (5 mg, 10 mg, 15 mg) achieved superior weight loss compared to placebo at 72 weeks of treatment in topline results from Eli Lilly and Company’s (NYSE: LLY) SURMOUNT-1 clinical trial, with participants losing up to 22.5% (52 lb. or 24 kg) of their body weight for the efficacy estimandi. This study enrolled 2,539 participants and was the first phase 3 global registration trial evaluating the efficacy and safety of tirzepatide in adults with obesity, or overweight with at least one comorbidity, who do not have diabetes. Tirzepatide met both co-primary endpoints of superior mean percent change in body weight from baseline and greater percentage of participants achieving body weight reductions of at least 5% compared to placebo for both estimandsii. The study also achieved all key secondary endpoints at 72 weeks.
For the efficacy estimand, participants taking tirzepatide achieved average weight reductions of 16.0% (35 lb. or 16 kg on 5 mg), 21.4% (49 lb. or 22 kg on 10 mg) and 22.5% (52 lb. or 24 kg on 15 mg), compared to placebo (2.4%, 5 lb. or 2 kg). Additionally, 89% (5 mg) and 96% (10 mg and 15 mg) of people taking tirzepatide achieved at least 5% body weight reductions compared to 28% of those taking placebo.
In a key secondary endpoint, 55% (10 mg) and 63% (15 mg) of people taking tirzepatide achieved at least 20% body weight reductions compared to 1.3% of those taking placebo. In an additional secondary endpoint not controlled for type 1 error, 32% of participants taking tirzepatide 5 mg achieved at least 20% body weight reductions. The mean baseline body weight of participants was 231 lb. (105 kg).
“Obesity is a chronic disease that often does not receive the same standard of care as other conditions, despite its impact on physical, psychological and metabolic health, which can include increased risk of hypertension, heart disease, cancer and decreased survival,” said Louis J. Aronne, MD, FACP, DABOM, director of the Comprehensive Weight Control Center and the Sanford I. Weill Professor of Metabolic Research at Weill Cornell Medicine, obesity expert at NewYork-Presbyterian/Weill Cornell Medical Center and Investigator of SURMOUNT-1. “Tirzepatide delivered impressive body weight reductions in SURMOUNT-1, which could represent an important step forward for helping the patient and physician partnership treat this complex disease.”
For the treatment-regimen estimandiii, results showed:
- Average body weight reductions: 15.0% (5 mg), 19.5% (10 mg), 20.9% (15 mg), 3.1% (placebo)
- Percentage of participants achieving body weight reductions of ≥5%: 85% (5 mg), 89% (10 mg), 91% (15 mg), 35% (placebo)
- Percentage of participants achieving body weight reductions of ≥20%: 30% (5 mg, not controlled for type 1 error), 50% (10 mg), 57% (15 mg), 3.1% (placebo)
The overall safety and tolerability profile of tirzepatide was similar to other incretin-based therapies approved for the treatment of obesity. The most commonly reported adverse events were gastrointestinal-related and generally mild to moderate in severity, usually occurring during the dose escalation period. For those treated with tirzepatide (5 mg, 10 mg and 15 mg, respectively), nausea (24.6%, 33.3%, 31.0%), diarrhea (18.7%, 21.2%, 23.0%), vomiting (8.3%, 10.7%, 12.2%) and constipation (16.8%, 17.1%, 11.7%) were more frequently experienced compared to placebo (9.5% [nausea], 7.3% [diarrhea], 1.7% [vomiting], 5.8% [constipation]).
Treatment discontinuation rates due to adverse events were 4.3% (5 mg), 7.1% (10 mg), 6.2% (15 mg) and 2.6% (placebo). The overall treatment discontinuation rates were 14.3% (5 mg), 16.4% (10 mg), 15.1% (15 mg) and 26.4% (placebo).
Participants who had pre-diabetes at study commencement will remain enrolled in SURMOUNT-1 for an additional 104 weeks of treatment following the initial 72-week completion date to evaluate the impact on body weight and the potential differences in progression to type 2 diabetes at three years of treatment with tirzepatide compared to placebo.
“Tirzepatide is the first investigational medicine to deliver more than 20 percent weight loss on average in a phase 3 study, reinforcing our confidence in its potential to help people living with obesity,” said Jeff Emmick, MD, Ph.D., vice president, product development, Lilly. “Obesity is a chronic disease that requires effective treatment options, and Lilly is working relentlessly to support people with obesity and modernize how this disease is approached. We’re proud to research and develop potentially innovative treatments like tirzepatide, which helped nearly two thirds of participants on the highest dose reduce their body weight by at least 20 percent in SURMOUNT-1.”
Tirzepatide is a novel investigational once-weekly GIP (glucose-dependent insulinotropic polypeptide) receptor and GLP-1 (glucagon-like peptide-1) receptor agonist, representing a new class of medicines being studied for the treatment of obesity. Tirzepatide is a single peptide that activates the body’s receptors for GIP and GLP-1, two natural incretin hormones. Obesity is a chronic, progressive disease caused by disruptions in the mechanisms that control body weight, often leading to an increase in food intake and/or a decrease in energy expenditure. These disruptions are multifactorial and can be related to genetic, developmental, behavioral, environmental and social factors. To learn more, visit Lilly.com/obesity.
Lilly will continue to evaluate the SURMOUNT-1 results, which will be presented at an upcoming medical meeting and submitted to a peer-reviewed journal. Additional studies are ongoing for tirzepatide as a potential treatment for obesity or overweight.
About tirzepatide
Tirzepatide is a once-weekly GIP (glucose-dependent insulinotropic polypeptide) receptor and GLP-1 (glucagon-like peptide-1) receptor agonist that integrates the actions of both incretins into a single novel molecule. GIP is a hormone that may complement the effects of GLP-1 receptor agonists. In preclinical models, GIP has been shown to decrease food intake and increase energy expenditure therefore resulting in weight reductions, and when combined with GLP-1 receptor agonism, may result in greater effects on markers of metabolic dysregulation such as body weight, glucose and lipids. Tirzepatide is in phase 3 development for adults with obesity or overweight with weight-related comorbidity and is currently under regulatory review as a treatment for adults with type 2 diabetes. It is also being studied as a potential treatment for non-alcoholic steatohepatitis (NASH) and heart failure with preserved ejection fraction (HFpEF). Studies of tirzepatide in obstructive sleep apnea (OSA) and in morbidity/mortality in obesity are planned as well.
About SURMOUNT-1 and the SURMOUNT clinical trial program
SURMOUNT-1 (NCT04184622) is a multi-center, randomized, double-blind, parallel, placebo-controlled trial comparing the efficacy and safety of tirzepatide 5 mg, 10 mg and 15 mg to placebo as an adjunct to a reduced-calorie diet and increased physical activity in adults without type 2 diabetes who have obesity, or overweight with at least one of the following comorbidities: hypertension, dyslipidemia, obstructive sleep apnea or cardiovascular disease. The trial randomized 2,539 participants across the U.S., Argentina, Brazil, China, India, Japan, Mexico, Russia and Taiwan in a 1:1:1:1 ratio to receive either tirzepatide 5 mg, 10 mg or 15 mg or placebo. The co-primary objectives of the study were to demonstrate that tirzepatide 10 mg and/or 15 mg is superior in percentage of body weight reductions from baseline and percentage of participants achieving ≥5% body weight reduction at 72 weeks compared to placebo. Participants who had pre-diabetes at study commencement will remain enrolled in SURMOUNT-1 for an additional 104 weeks of treatment following the initial 72-week completion date to evaluate the impact on body weight and potential differences in progression to type 2 diabetes at three years of treatment with tirzepatide compared to placebo.
All participants in the tirzepatide treatment arms started the study at a dose of tirzepatide 2.5 mg once-weekly and then increased the dose in a step-wise approach at four-week intervals to their final randomized maintenance dose of 5 mg (via a 2.5 mg step), 10 mg (via steps at 2.5 mg, 5 mg and 7.5 mg) or 15 mg (via steps at 2.5 mg, 5 mg, 7.5 mg, 10 mg and 12.5 mg).
The SURMOUNT phase 3 global clinical development program for tirzepatide began in late 2019 and has enrolled more than 5,000 people with obesity or overweight across six clinical trials, four of which are global studies. Results from SURMOUNT-2, -3, and -4 are anticipated in 2023.
About Lilly
Lilly unites caring with discovery to create medicines that make life better for people around the world. We’ve been pioneering life-changing discoveries for nearly 150 years, and today our medicines help more than 47 million people across the globe. Harnessing the power of biotechnology, chemistry and genetic medicine, our scientists are urgently advancing new discoveries to solve some of the world’s most significant health challenges, redefining diabetes care, treating obesity and curtailing its most devastating long-term effects, advancing the fight against Alzheimer’s disease, providing solutions to some of the most debilitating immune system disorders, and transforming the most difficult-to-treat cancers into manageable diseases. With each step toward a healthier world, we’re motivated by one thing: making life better for millions more people. That includes delivering innovative clinical trials that reflect the diversity of our world and working to ensure our medicines are accessible and affordable. To learn more, visit Lilly.com and Lilly.com/newsroom or follow us on Facebook, Instagram, Twitter and LinkedIn. P-LLY
CLIP
Tirzepatide results superior A1C and body weight reductions compared to insulin glargine in adults with type 2 diabetes
Newly published data show that participants maintained A1C and weight control up to two years in SURPASS-4, the largest and longest SURPASS trial completed to dateNo increased cardiovascular risk identified with tirzepatide; hazard ratio of 0.74 observed for MACE-4 events
SURPASS-4 is the largest and longest clinical trial completed to date of the phase 3 program studying tirzepatide as a potential treatment for type 2 diabetes. The primary endpoint was measured at 52 weeks, with participants continuing treatment up to 104 weeks or until study completion. The completion of the study was triggered by the accrual of major adverse cardiovascular events (MACE) to assess CV risk. In newly published data from the treatment period after 52 weeks, participants taking tirzepatide maintained A1C and weight control for up to two years.
The overall safety profile of tirzepatide, assessed over the full study period, was consistent with the safety results measured at 52 weeks, with no new findings up to 104 weeks. Gastrointestinal side effects were the most commonly reported adverse events, usually occurring during the escalation period and then decreasing over time.
“We are encouraged by the continued A1C and weight control that participants experienced past the initial 52 week treatment period and up to two years as we continue to explore the potential impact of tirzepatide for the treatment of type 2 diabetes,” said John Doupis, M.D., Ph.D., Director, Diabetes Division and Clinical Research Center, Iatriko Paleou Falirou Medical Center, Athens, Greece and Senior Investigator for SURPASS-4.
Tirzepatide is a novel investigational once-weekly dual glucose-dependent insulinotropic polypeptide (GIP) and glucagon-like peptide-1 (GLP-1) receptor agonist that integrates the actions of both incretins into a single molecule, representing a new class of medicines being studied for the treatment of type 2 diabetes.
SURPASS-4 was an open-label global trial comparing the safety and efficacy of three tirzepatide doses (5 mg, 10 mg and 15 mg) to titrated insulin glargine in 2,002 adults with type 2 diabetes with increased CV risk who were treated with between one and three oral antihyperglycemic medicines (metformin, a sulfonylurea or an SGLT-2 inhibitor). Of the total participants randomized, 1,819 (91%) completed the primary 52-week visit and 1,706 (85%) completed the study on treatment. The median study duration was 85 weeks and 202 participants (10%) completed two years.
Study participants had a mean duration of diabetes of 11.8 years, a baseline A1C of 8.52 percent and a baseline weight of 90.3 kg. More than 85 percent of participants had a history of cardiovascular events. In the insulin glargine arm, the insulin dose was titrated following a treat-to-target algorithm with the goal of fasting blood glucose below 100 mg/dL. The starting dose of insulin glargine was 10 units per day, and the mean dose of insulin glargine at 52 weeks was 43.5 units per day.
About tirzepatide
Tirzepatide is a once-weekly dual glucose-dependent insulinotropic polypeptide (GIP) and glucagon-like peptide-1 (GLP-1) receptor agonist that integrates the actions of both incretins into a single novel molecule. GIP is a hormone that may complement the effects of GLP-1. In preclinical models, GIP has been shown to decrease food intake and increase energy expenditure therefore resulting in weight reductions, and when combined with a GLP-1 receptor agonist, may result in greater effects on glucose and body weight. Tirzepatide is in phase 3 development for blood glucose management in adults with type 2 diabetes, for chronic weight management and heart failure with preserved ejection fraction (HFpEF). It is also being studied as a potential treatment for non-alcoholic steatohepatitis (NASH).
About SURPASS-4 and the SURPASS clinical trial program
SURPASS-4 (NCT03730662) is a randomized, parallel, open-label trial comparing the efficacy and safety of tirzepatide 5 mg, 10 mg and 15 mg to insulin glargine in adults with type 2 diabetes inadequately controlled with at least one and up to three oral antihyperglycemic medications (metformin, sulfonylureas or SGLT-2 inhibitors), who have increased cardiovascular (CV) risk. The trial randomized 2,002 study participants in a 1:1:1:3 ratio to receive either tirzepatide 5 mg, 10 mg or 15 mg or insulin glargine. Participants were located in the European Union, North America (Canada and the United States), Australia, Israel, Taiwan and Latin America (Brazil, Argentina and Mexico). The primary objective of the study was to demonstrate that tirzepatide (10 mg and/or 15 mg) is non-inferior to insulin glargine for change from baseline A1C at 52 weeks in people with type 2 diabetes and increased CV risk. The primary and key secondary endpoints were measured at 52 weeks, with participants continuing treatment up to 104 weeks or until study completion. The completion of the study was triggered by the accrual of major adverse cardiovascular events (MACE). Study participants enrolled had to have a mean baseline A1C between 7.5 percent and 10.5 percent and a BMI greater than or equal to 25 kg/m2 at baseline. All participants in the tirzepatide treatment arms started the study at a dose of tirzepatide 2.5 mg once-weekly and then increased the dose in a step-wise approach at four-week intervals to their final randomized maintenance dose of 5 mg (via a 2.5 mg step), 10 mg (via steps at 2.5 mg, 5 mg and 7.5 mg) or 15 mg (via steps at 2.5 mg, 5 mg, 7.5 mg, 10 mg and 12.5 mg). All participants in the titrated insulin glargine treatment arm started with a baseline dose of 10 units per day and titrated following a treat-to-target algorithm to reach a fasting blood glucose below 100 mg/dL.
The SURPASS phase 3 global clinical development program for tirzepatide has enrolled more than 20,000 people with type 2 diabetes across 10 clinical trials, five of which are global registration studies. The program began in late 2018, and all five global registration trials have been completed.
About Diabetes
Approximately 34 million Americans2 (just over 1 in 10) and an estimated 463 million adults worldwide3 have diabetes. Type 2 diabetes is the most common type internationally, accounting for an estimated 90 to 95 percent of all diabetes cases in the United States alone2. Diabetes is a chronic disease that occurs when the body does not properly produce or use the hormone insulin.
| Clinical data | |
|---|---|
| Trade names | Mounjaro |
| Other names | LY3298176, GIP/GLP-1 RA |
| License data | US DailyMed: Tirzepatide |
| Routes of administration | subcutaneous |
| Drug class | Antidiabetic, GLP-1 receptor agonist |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only [1][2] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 2023788-19-2 |
| PubChem CID | 156588324 |
| IUPHAR/BPS | 11429 |
| DrugBank | DB15171 |
| ChemSpider | 76714503 |
| UNII | OYN3CCI6QE |
| KEGG | D11360 |
| ChEMBL | ChEMBL4297839 |
| Chemical and physical data | |
| Formula | C225H348N48O68 |
| Molar mass | 4813.527 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI |
////////////Tirzepatide, FDA 2022, APPROVALS 2022, Mounjaro, PEPTIDE, チルゼパチド , LY3298176,
UNIIOYN3CCI6QE
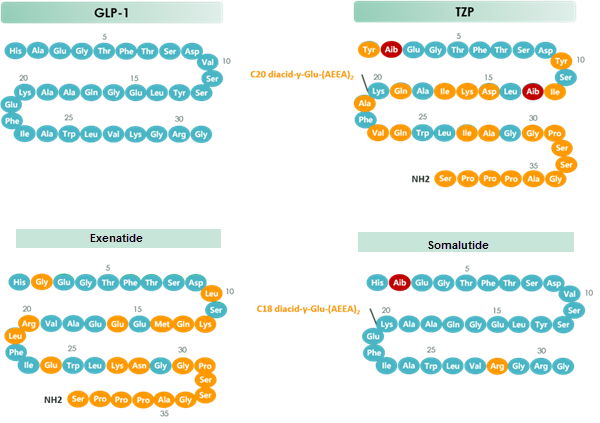
chart 1 Structure of GLP-1 & TZP & Exenatide & Somalutide
MONENSIN
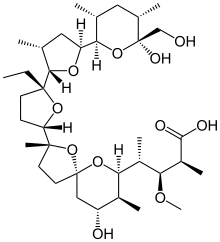
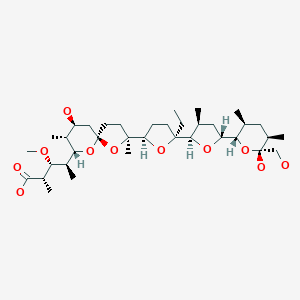
モネンシン;
MONENSIN
- Molecular FormulaC36H62O11
- Average mass670.871 Da
1,6-dioxaspiro[4.5]decane-7-butanoic acid, 2-[(2S,2’R,3’S,5R,5’R)-2-ethyloctahydro-3′-methyl-5′-[(2S,3S,5R,6R)-tetrahydro-6-hydroxy-6-(hydroxymethyl)-3,5-dimethyl-2H-pyran-2-yl][2,2′-bifuran]-5-yl]-9-hydroxy-β-methoxy-α,γ,2,8-tetramethyl-, (αS,βR,γS,2S,5R,7S,8R,9S)-
17090-79-8[RN]
241-154-0[EINECS]
(2S,3R,4S)-4-[(2S,5R,7S,8R,9S)-2-{(2S,2’R,3’S,5R,5’R)-2-Ethyl-5′-[(2S,3S,5R,6R)-6-hydroxy-6-(hydroxymethyl)-3,5-dimethyltetrahydro-2H-pyran-2-yl]-3′-methyloctahydro-2,2′-bifuran-5-yl}-9-hydroxy-2,8-di methyl-1,6-dioxaspiro[4.5]dec-7-yl]-3-methoxy-2-methylpentanoic acid
монензин[Russian]
مونانسين[Arabic]
莫能星[Chinese]
Antibiotic, Antifungal, Antiprotozoal

Synonym(s):
Monensin A sodium salt
Empirical Formula (Hill Notation):C36H61NaO11
CAS Number:22373-78-0
Molecular Weight:692.85
Beilstein:4122200
Title: Monensin
CAS Registry Number: 17090-79-8
CAS Name: 2-[5-Ethyltetrahydro-5-[tetrahydro-3-methyl-5-[tetrahydro-6-hydroxy-6-(hydroxymethyl)-3,5-dimethyl-2H-pyran-2-yl]-2-furyl]-2-furyl]-9-hydroxy-b-methoxy-a,g,2,8-tetramethyl-1,6-dioxaspiro[4.5]decane-7-butyric acid
Additional Names: monensic acid (obsolete)
Manufacturers’ Codes: A-3823A
Molecular Formula: C36H62O11, Molecular Weight: 670.87
Percent Composition: C 64.45%, H 9.32%, O 26.23%
Literature References: Polyether antibiotic. Major factor in antibiotic complex isolated from Streptomyces cinnamonensis. Discovery and isolation: Haney, Hoehn, Antimicrob. Agents Chemother.1967, 349. Production: Haney, Hoehn, US3501568 (1970 to Lilly). Structure: Agtarap et al.,J. Am. Chem. Soc.89, 5737 (1967). Crystal structure studies: Lutz et al.,Helv. Chim. Acta53, 1732 (1970); ibid.54, 1103 (1971). Fermentation studies: Stark et al.,Antimicrob. Agents Chemother.1967, 353. Chemistry: Agtarap, Chamberlin, ibid. 359. Stereocontrolled total synthesis: T. Fukuyama et al.,J. Am. Chem. Soc.101, 262 (1979); D. B. Collum et al.,ibid.102, 2117, 2118, 2120 (1980). 13C-NMR study: J. A. Robinson, D. L. Turner, Chem. Commun.1982, 148. Biosynthesis: Day et al.,Antimicrob. Agents Chemother.4, 410 (1973). Review: Stark, “Monensin, A New Biologically Active Compound Produced by a Fermentation Process”, in Fermentation Advances, Pap. Int. Ferment. Symp., 3rd, 1968, D. Perlman, Ed. (Academic Press, New York, 1969) pp 517-540.
Properties: Crystals, mp 103-105° (monohydrate). [a]D +47.7°. pKa 6.6 (in 66% DMF). Very stable under alkaline conditions. Slightly sol in water; more sol in hydrocarbons; very sol in other organic solvents. LD50 of monensin complex in mice, chicks (mg/kg): 43.8 ± 5.2, 284 ± 47 orally (Haney, Hoehn).
Melting point: mp 103-105° (monohydrate)
pKa: pKa 6.6 (in 66% DMF)
Optical Rotation: [a]D +47.7°
Toxicity data: LD50 of monensin complex in mice, chicks (mg/kg): 43.8 ± 5.2, 284 ± 47 orally (Haney, Hoehn)
Derivative Type: Sodium salt
Trademarks: Coban (Elanco); Romensin (Elanco); Rumensin (Elanco)
Molecular Formula: C36H61NaO11, Molecular Weight: 692.85
Percent Composition: C 62.41%, H 8.87%, Na 3.32%, O 25.40%
Properties: mp 267-269°. [a]D +57.3° (methanol). Slightly sol in water; more sol in hydrocarbons; very sol in other organic solvents.
Melting point: mp 267-269°
Optical Rotation: [a]D +57.3° (methanol)
Therap-Cat-Vet: Coccidiostat. Feed additive to improve feed efficiency in ruminants.
Monensin is a polyether antibiotic isolated from Streptomyces cinnamonensis.[1] It is widely used in ruminant animal feeds.[1][2]
The structure of monensin was first described by Agtarap et al. in 1967, and was the first polyether antibiotic to have its structure elucidated in this way. The first total synthesis of monensin was reported in 1979 by Kishi et al.[3]
SYN

SYN
Production / synthesis Monensin is produced in vivo by Streptomyces cinnamonensis as a natural defense against competing bacteria. Monensin presents a formidable challenge to synthetic chemists as it possesses 17 asymmetric centers on a backbone of only 26 carbon atoms. Although its total synthesis has been described (e.g., Kishi et al., 1979), the high complexity of monensin makes an extraction from the bacterium the most economical procedure for its production. The total synthesis has 56 steps and a yield of only 0.26%. The chemical precursors are 2-allyl-1,3-propanediol and 2- (furan-2-yl)acetonitrile. The method used for synthesizing monensin is based on the principle of “absolute asymmetric synthesis”. Molecules are constructed out of prefabricated building blocks in the correct conformation, aiming for higher yields of the desired enantiomer. New stereocenters are also introduced. Using this method, monensin is assembled in two parts, a larger right side and a smaller left one. The penultimate step is connecting the left and the right halves of monensin, which are independently generated, in an Aldol-condensation. The two halves’ keto end groups (C7/ C8) are linked by eliminating a water molecule. The C7 atom is favored over the C1 atom, because it is more reactive. For catalyzing this step, Yoshito Kishi’s group used iPr2NMgBr (Hauser base) and THF to coordinate it at a temperature of − 78°C. Thus, they were able to isolate the molecule in the right conformation at a ratio of 8:1. Due to the low temperature required for a high yield of the correct enantiomer, the reaction is very solw. One of the most difficult steps is the last one: the connection of the spiro center. This is due to a characteristic feature of spiro compounds; they open and close very easily. Therefore, the conditions for forming the right conformation must be optimal in the last step of synthesis. The biosynthesis in a cell culture of Streptomyces cinnamonensis involves a complex medium containing, among other components, glucose, soybean oil, and grit. Cultivation is carried out for a week at a temperature of 30°C and under constant aeration. Product isolation requires filtration, acidification to pH3, extraction with chloroform and purification with activated carbon. In this way, a few grams per liter of monensin are produced and isolated. For crystallization, azeotropic distillation is necessary. In vivo, polyether backbones are assembled by modular polyketide synthases and are modified by two key enzymes, epoxidase and epoxide hydrolase, to generate the product. Precursors of the polyketide pathway are acetate, butyrate and propionate.
SYN
The final-stage aldol addition in Yoshito Kishi‘s 1979 total synthesis of monensin. (1979). “Synthetic studies on polyether antibiotics. 6. Total synthesis of monensin. 3. Stereocontrolled total synthesis of monensin”. J. Am. Chem. Soc. 101 (1): 262–263. DOI:10.1021/ja00495a066.

SYN
A polyether antibiotic, Monensin was the first member of this class of molecules to be structurally characterized.1 The structural features of these polyethers comprise of a terminal carboxylic acid, multiple cyclic ether rings (ex. Tetrahydrofuran and tetrahydropyran), a large amount of stereocenters and (for many of these molecules) one or more spiroketal moieties.2 Monensin was introduced into the market in 1971 and is used to fight coccidial infections in poultry and as an additive in cattle feed.3 Of the 26 carbon atom’s in Monensin’s backbone, 17 are stereogenic and six of those are contiguous. Coupled with a spiroketal moiety, three hydrofuran rings and two hydropyran rings, the molecule was an attractive synthetic target.
1. Agtarap, A.; Chamberlain, J.W.; Pinkerton, M.; Stein-rauf, L. J. Am. Chem. Soc. 1967, 89, 5737 2. Polyether Antibiotics : Naturally Occurring Acid Ionophores. Westley J.W.; Marcel Dekker: New York (1982) Vol. 1-2. 3. Stark, W.M. In Fermentation Advances, Perlman, D., Ed., Academic Press: New York, 1969, 517
Retrosynthetic Analysis of Monensin
//////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
Mechanism of action

The structure of the sodium (Na+) complex of monensin A.
Monensin A is an ionophore related to the crown ethers with a preference to form complexes with monovalent cations such as: Li+, Na+, K+, Rb+, Ag+, and Tl+.[4][5] Monensin A is able to transport these cations across lipid membranes of cells in an electroneutral (i.e. non-depolarizing) exchange, playing an important role as an Na+/H+ antiporter. Recent studies have shown that monensin may transport sodium ion through the membrane in both electrogenic and electroneutral manner.[6] This approach explains ionophoric ability and in consequence antibacterial properties of not only parental monensin, but also its derivatives that do not possess carboxylic groups. It blocks intracellular protein transport, and exhibits antibiotic, antimalarial, and other biological activities.[7] The antibacterial properties of monensin and its derivatives are a result of their ability to transport metal cations through cellular and subcellular membranes.[8]
Uses
Monensin is used extensively in the beef and dairy industries to prevent coccidiosis, increase the production of propionic acid and prevent bloat.[9] Furthermore, monensin, but also its derivatives monensin methyl ester (MME), and particularly monensin decyl ester (MDE) are widely used in ion-selective electrodes.[10][11][12]
In laboratory research, monensin is used extensively to block Golgi transport.[13][14][15]
Toxicity
Monensin has some degree of activity on mammalian cells and thus toxicity is common. This is especially pronounced in horses, where monensin has a median lethal dose 1/100th that of ruminants. Accidental poisoning of equines with monensin is a well-documented occurrence which has resulted in deaths.[16]
References
- ^ Jump up to:a b Daniel Łowicki and Adam Huczyński (2013). “Structure and Antimicrobial Properties of Monensin A and Its Derivatives: Summary of the Achievements”. BioMed Research International. 2013: 1–14. doi:10.1155/2013/742149. PMC 3586448. PMID 23509771.
- ^ Butaye, P.; Devriese, L. A.; Haesebrouck, F. (2003). “Antimicrobial Growth Promoters Used in Animal Feed: Effects of Less Well Known Antibiotics on Gram-Positive Bacteria”. Clinical Microbiology Reviews. 16 (2): 175–188. doi:10.1128/CMR.16.2.175-188.2003. PMC 153145. PMID 12692092.
- ^ Nicolaou, K. C.; E. J. Sorensen (1996). Classics in Total Synthesis. Weinheim, Germany: VCH. pp. 185–187. ISBN 3-527-29284-5.
- ^ Huczyński, A.; Ratajczak-Sitarz, M.; Katrusiak, A.; Brzezinski, B. (2007). “Molecular structure of the 1:1 inclusion complex of Monensin A lithium salt with acetonitrile”. J. Mol. Struct. 871 (1–3): 92–97. Bibcode:2007JMoSt.871…92H. doi:10.1016/j.molstruc.2006.07.046.
- ^ Pinkerton, M.; Steinrauf, L. K. (1970). “Molecular structure of monovalent metal cation complexes of monensin”. J. Mol. Biol. 49 (3): 533–546. doi:10.1016/0022-2836(70)90279-2. PMID 5453344.
- ^ Huczyński, Adam; Jan Janczak; Daniel Łowicki; Bogumil Brzezinski (2012). “Monensin A acid complexes as a model of electrogenic transport of sodium cation”. Biochim. Biophys. Acta. 1818 (9): 2108–2119. doi:10.1016/j.bbamem.2012.04.017. PMID 22564680.
- ^ Mollenhauer, H. H.; Morre, D. J.; Rowe, L. D. (1990). “Alteration of intracellular traffic by monensin; mechanism, specificity and relationship to toxicity”. Biochim. Biophys. Acta. 1031 (2): 225–246. doi:10.1016/0304-4157(90)90008-Z. PMC 7148783. PMID 2160275.
- ^ Huczyński, A.; Stefańska, J.; Przybylski, P.; Brzezinski, B.; Bartl, F. (2008). “Synthesis and antimicrobial properties of Monensin A esters”. Bioorg. Med. Chem. Lett. 18 (8): 2585–2589. doi:10.1016/j.bmcl.2008.03.038. PMID 18375122.
- ^ Matsuoka, T.; Novilla, M.N.; Thomson, T.D.; Donoho, A.L. (1996). “Review of monensin toxicosis in horses”. Journal of Equine Veterinary Science. 16: 8–15. doi:10.1016/S0737-0806(96)80059-1.
- ^ Tohda, Koji; Suzuki, Koji; Kosuge, Nobutaka; Nagashima, Hitoshi; Watanabe, Kazuhiko; Inoue, Hidenari; Shirai, Tsuneo (1990). “A sodium ion selective electrode based on a highly lipophilic monensin derivative and its application to the measurement of sodium ion concentrations in serum”. Analytical Sciences. 6 (2): 227–232. doi:10.2116/analsci.6.227.
- ^ Kim, N.; Park, K.; Park, I.; Cho, Y.; Bae, Y. (2005). “Application of a taste evaluation system to the monitoring of Kimchi fermentation”. Biosensors and Bioelectronics. 20 (11): 2283–2291. doi:10.1016/j.bios.2004.10.007. PMID 15797327.
- ^ Toko, K. (2000). “Taste Sensor”. Sensors and Actuators B: Chemical. 64 (1–3): 205–215. doi:10.1016/S0925-4005(99)00508-0.
- ^ Griffiths, G.; Quinn, P.; Warren, G. (March 1983). “Dissection of the Golgi complex. I. Monensin inhibits the transport of viral membrane proteins from medial to trans Golgi cisternae in baby hamster kidney cells infected with Semliki Forest virus”. The Journal of Cell Biology. 96 (3): 835–850. doi:10.1083/jcb.96.3.835. ISSN 0021-9525. PMC 2112386. PMID 6682112.
- ^ Kallen, K. J.; Quinn, P.; Allan, D. (1993-02-24). “Monensin inhibits synthesis of plasma membrane sphingomyelin by blocking transport of ceramide through the Golgi: evidence for two sites of sphingomyelin synthesis in BHK cells”. Biochimica et Biophysica Acta (BBA) – Lipids and Lipid Metabolism. 1166 (2–3): 305–308. doi:10.1016/0005-2760(93)90111-l. ISSN 0006-3002. PMID 8443249.
- ^ Zhang, G. F.; Driouich, A.; Staehelin, L. A. (December 1996). “Monensin-induced redistribution of enzymes and products from Golgi stacks to swollen vesicles in plant cells”. European Journal of Cell Biology. 71 (4): 332–340. ISSN 0171-9335. PMID 8980903.
- ^ “Tainted feed blamed for 4 horse deaths at Florida stable”. 2014-12-16.
| Names | |
|---|---|
| Preferred IUPAC name(2S,3R,4S)-4-[(2S,5R,7S,8R,9S)-2-{(2S,2′R,3′S,5R,5′R)-2-Ethyl-5′-[(2S,3S,5R,6R)-6-hydroxy-6-(hydroxymethyl)-3,5-dimethyloxan-2-yl]-3′-methyl[2,2′-bioxolan]-5-yl}-9-hydroxy-2,8-dimethyl-1,6-dioxaspiro[4.5]decan-7-yl]-3-methoxy-2-methylpentanoic acid | |
| Other namesMonensic acid | |
| Identifiers | |
| CAS Number | 17090-79-8 |
| 3D model (JSmol) | Interactive image |
| ChEBI | CHEBI:27617 |
| ChEMBL | ChEMBL256105 |
| ChemSpider | 389937 |
| ECHA InfoCard | 100.037.398 |
| E number | E714 (antibiotics) |
| KEGG | D08228 |
| PubChemCID | 441145 |
| UNII | 906O0YJ6ZP |
| CompTox Dashboard (EPA) | DTXSID4048561 |
| showInChI | |
| showSMILES | |
| Properties | |
| Chemical formula | C36H62O11 |
| Molar mass | 670.871 g/mol |
| Appearance | solid state, white crystals |
| Melting point | 104 °C (219 °F; 377 K) |
| Solubility in water | 3×10−6 g/dm3 (20 °C) |
| Solubility | ethanol, acetone, diethyl ether, benzene |
| Pharmacology | |
| ATCvet code | QA16QA06 (WHO) QP51AH03 (WHO) |
| Related compounds | |
| Related | antibiotics, ionophores |
| Related compounds | Monensin A methyl ester, |
| Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa). |
///////////MONENSIN, Elancoban, VETERINARY, Coccidiostat, A-3823A, A 3823A

NEW DRUG APPROVALS
ONE TIME TO SUSTAIN THIS BLOG SUBSCRIPTION AND KEEP GOING
$10.00
Ferric derisomaltose

Ferric derisomaltose
WeightAverage: 562.297
Monoisotopic: 562.117975Chemical FormulaC18H34FeO16
Monover, JAPAN 2022, 2022/3/28
Monoferric (TN);
Monover (TN)
Anti-anemic, Hematinic, Supplement (iron)
CAS 1345510-43-1
デルイソマルトース第二鉄
- NS32
- WHO 9712
- UNII-AHU547PI9H
| Originator Company | Pharmacosmos |
| Active Companies | Nippon Shinyaku Co Ltd;Pharmacosmos A/S;Wasserburger Arzneimittelwerk Gmbh;Zealand University Hospital |
iron(3+) (2S,3R,4R,5R)-6-{[(2S,3R,4S,5S,6R)-3,4,5-trihydroxy-6-({[(2S,3R,4S,5S,6R)-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl]oxy}methyl)oxan-2-yl]oxy}hexane-1,2,3,4,5-pentol
- alpha-D-Glucan, (1-6)-, reduced, reaction products with iron hydroxide (Fe(OH)3)
Ferric derisomaltose is an iron injection used in the treatment of iron deficiency anemia.
Ferric derisomaltose, sold under the brand name Monoferric, is a medication for the treatment of iron deficiency anemia (IDA) in adults who have intolerance to oral iron or have had unsatisfactory response to oral iron or who have non-hemodialysis dependent chronic kidney disease (NDD-CKD).[1] It was approved for use in the United States in January 2020.[1][2][3] It is given intravenously.[1]
Iron deficiency is an extremely common condition and is the most frequent cause of anemia worldwide. Iron deficiency results when iron intake, iron stores, and loss of iron from the body do not adequately support production of erythrocytes, also known as red blood cells. Though it is generally considered non life-threatening, iron deficiency may considerably affect quality of life.3
Ferric derisomaltose is a form of iron used in the treatment of iron deficiency. This drug is a complex of iron (III) hydroxide and derisomaltose. The latter is an iron carbohydrate oligosaccharide that works to release iron. Ferric derisomaltose was developed by Pharmacosmos Therapeutics ad was granted FDA approval in January 2020.8,9 Clinical trials show that it is non-inferior to iron sucrose, another form of iron that is often administered in iron deficiency, and less likely to cause serious hypersensitivity that is associated with other forms of injectable iron.1,4
This drug is indicated for the treatment of iron deficiency anemia in adult patients who have experienced intolerance to oral iron preparations or insufficient clinical response to orally administered iron. Ferric derisomaltase is also indicated for patients with non-hemodialysis dependent chronic kidney disease.8 In Australia and United Kingdom, ferric derisomaltase is indicated for cases in which rapid delivery of iron is required.10,11
Iron deficiency is an extremely common condition and is the most frequent cause of anemia worldwide. Iron deficiency results when iron intake, iron stores, and loss of iron from the body do not adequately support production of erythrocytes, also known as red blood cells. Though it is generally considered non life-threatening, iron deficiency may considerably affect quality of life. Ferric derisomaltose is a form of iron used in the treatment of iron deficiency. This drug is a complex of iron (III) hydroxide and derisomaltose. The latter is an iron carbohydrate oligosaccharide that works to release iron. Ferric derisomaltose was developed by Pharmacosmos Therapeutics ad was granted FDA approval in January 2020. Clinical trials show that it is non-inferior to [iron sucrose], another form of iron that is often administered in iron deficiency, and less likely to cause serious hypersensitivity that is associated with other forms of injectable iron.
Monoferric is an iron replacement product containing ferric derisomaltose for intravenous infusion. Ferric derisomaltose is an iron carbohydrate complex with a matrix structure composed of interchanging layers of ferric hydroxide and the carbohydrate derisomaltose. Derisomaltose consists of linear, hydrogenated isomaltooligosaccharides with an average molecular weight of 1000 Da and a narrow molecular weight distribution that is almost devoid of mono-and disaccharides.
Ferric derisomaltose has an average molecular weight of 155,000 Da and has the following empirical formula:
{FeO(1-3X) (OH)(1+3X) (C6H5O73-)X}, (H20)T, –
(C6H10O6)R(-C6H10O5-)Z(C6H13O5)R, (NaCl)Y
X= 0.0311; T = 0.25; R = 0.14; Z = 0.49; Y = 0.14
Iron atoms placed in the electronegative cavities of the 3-D structure between and within the derisomaltose molecules. A schematic representation is presented below
 |
Monoferric is a sterile, dark brown, non-transparent aqueous solution with pH 5.0-7.0, containing ferric derisomaltose dissolved in water for injections and filled into Type I glass vials.
Each 1 mL of solution contains 100 mg of elemental iron as ferric derisomaltose in water for injection.
Each 1 mL of solution contains 100 mg of elemental iron as ferric derisomaltose in water for injection.
| Mkt. Status | Active Ingredient | Proprietary Name | Appl. No. | Dosage Form | Route | Strength | TE Code | RLD | RS | Applicant Holder |
|---|---|---|---|---|---|---|---|---|---|---|
| RX | FERRIC DERISOMALTOSE | MONOFERRIC | N208171 | SOLUTION | INTRAVENOUS | 1GM/10ML (100MG/ML) | RLD | RS | PHARMACOSMOS AS | |
| DISCN | FERRIC DERISOMALTOSE | MONOFERRIC | N208171 | SOLUTION | INTRAVENOUS | 100MG/ML (100MG/ML) | RLD | PHARMACOSMOS AS | ||
| DISCN | FERRIC DERISOMALTOSE | MONOFERRIC | N208171 | SOLUTION | INTRAVENOUS | 500MG/5ML (100MG/ML) | RLD | PHARMACOSMOS AS | ||
| Mkt. Status | Active Ingredient | Proprietary Name | Appl. No. | Dosage Form | Route | Strength | TE Code | >RLD | RS | Applicant Holder |
MONOFERRIC (FERRIC DERISOMALTOSE)
1GM/10ML (100MG/ML)
Marketing Status: Prescription
Active Ingredient: FERRIC DERISOMALTOSE
Proprietary Name: MONOFERRIC
Dosage Form; Route of Administration: SOLUTION; INTRAVENOUS
Strength: 1GM/10ML (100MG/ML)
Reference Listed Drug: Yes
Reference Standard: Yes
TE Code:
Application Number: N208171
Product Number: 003
Approval Date: Jan 16, 2020
Applicant Holder Full Name: PHARMACOSMOS AS
Marketing Status: Prescription
Patent and Exclusivity Information
Patent and Exclusivity for: N208171
Product 003
FERRIC DERISOMALTOSE (MONOFERRIC) SOLUTION 1GM/10ML (100MG/ML)
Patent Data
| Product No | Patent No | Patent Expiration | Drug Substance | Drug Product | Patent Use Code | Delist Requested | Submission Date |
|---|---|---|---|---|---|---|---|
| 003 | 8815301 | 08/14/2029 | DS | DP | U-2734 | 02/14/2020 | |
| 003 | 10414831 | 03/25/2029 | DS | DP | 02/14/2020 |
PATENT
AU2009342799B2
US10414831B2
US2012010166A1
US2014303364A1
///////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
References
- ^ Jump up to:a b c d “Monoferric- ferric derisomaltose solution”. DailyMed. 24 January 2020. Retrieved 16 February 2020.
- ^ “Monoferric approval letter” (PDF). U.S. Food and Drug Administration (FDA). 16 January 2020. Retrieved 16 February 2020. This article incorporates text from this source, which is in the public domain.
- ^ “Drug Approval Package: Monoferric Injection”. U.S. Food and Drug Administration (FDA). 7 May 2020. Retrieved 13 August 2020.
External links
- “Ferric derisomaltose”. Drug Information Portal. U.S. National Library of Medicine.
| Clinical data | |
|---|---|
| Trade names | Monoferric |
| AHFS/Drugs.com | Monograph |
| License data | US DailyMed: Ferric_derisomaltose |
| Routes of administration | Intravenous (IV) |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only [1] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 1345510-43-1 |
| PubChem CID | 86278348 |
| DrugBank | DB15617 |
| UNII | AHU547PI9H |
| KEGG | D11808 |
| Chemical and physical data | |
| Formula | C18H34FeO16+3 |
| Molar mass | 562.299 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI |
/////////////Ferric derisomaltose, デルイソマルトース第二鉄 , APPROVALS 2022, JAPAN 2022, NS32, WHO 9712
[Fe+3].OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO[C@H]1O[C@H](CO[C@H]2O[C@H](CO)[C@@H](O)[C@H](O)[C@H]2O)[C@@H](O)[C@H](O)[C@H]1O

NEW DRUG APPROVALS
one time
$10.00
Difamilast
Difamilast
FDA 2026, APPROVALS 2026, difamilast, Adquey, 2/12/2026, To treat mild to moderate atopic dermatitis
PMDA Moizerto, JAPAN APPROVED 2021/9/27
ジファミラスト
ディファミラスト;
地法米司特
N-({2-[4-(difluoromethoxy)-3-(propan-2-yloxy)phenyl]-1,3- oxazol-4-yl}methyl)-2-ethoxybenzamide
OPA-15406
| Formula |
C23H24F2N2O5
|
|---|---|
| CAS |
937782-05-3
|
| Mol weight |
446.4439
|
MM 36; MM-36-Medimetriks-Pharmaceuticals; Moizerto; OPA-15406
| Efficacy |
Anti-inflammatory, Phosphodiesterase IV inhibitor
|
|---|---|
| Comment |
Treatment of atopic dermatitis
|
OriginatorOtsuka Pharmaceutical Development & Commercialization- DeveloperMedimetriks Pharmaceuticals; Otsuka Pharmaceutical Development & Commercialization
- ClassBenzamides; Nonsteroidal anti-inflammatories; Oxazoles; Skin disorder therapies
- Mechanism of ActionType 4 cyclic nucleotide phosphodiesterase inhibitors
- RegisteredAtopic dermatitis
- 27 Sep 2021Registered for Atopic dermatitis (In adolescents, In children, In adults) in Japan (Topical)
- 11 Nov 2020Otsuka Pharmaceutical completes a phase III trial in Atopic dermatitis (In children, In adolescents, In adults) in Japan (Topical) (NCT03961529)
- 28 Sep 2020Preregistration for Atopic dermatitis in Japan (In children, In adolescents, In adults) (Topical)

Difamilast is under investigation in clinical trial NCT01702181 (A Safety Study to Evaluate the Use and Effectiveness of a Topical Ointment to Treat Adults With Atopic Dermatitis).
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019194211&_cid=P10-MLOK7B-25338-2
Synthesis of Oxazole Compound (Type A Crystal)
Compound (5) (white powder) was prepared in accordance with the method disclosed in Example 352 of PTL 1 (WO2007/058338).
SYN
https://www.chemicalbook.com/article/how-is-difamilast-synthesised.htm
Synthesis of difamilast commenced with the monobenzylated protocatechuic acid ethyl ester 15.1. Phenol 15.1 was first converted into the corresponding isopropyl ether, which was subsequently debenzylated under palladium-catalyzed hydrogenation conditions to generate the phenolic intermediate 15.3. Difluoromethylation of 15.3 was accomplished by introducing sodium chlorodifluoroacetate 15.4 in the presence of potassium carbonate at an elevated temperature. The decarboxylative C− O bond-forming reaction presumably proceeded via a difluorocarbene species. The difluoromethylated product was treated with acid followed by ester hydrolysis under a basic medium to furnish benzoic acid derivative 15.5. Benzoic acid 15.5 was subsequently transformed into benzamide 15.6 via a benzoyl imidazole intermediate. Condensation of benzamide 15.6 with 1-acetoxy-3-chloroacetone 15.7 produced an oxazole derivative, which was subsequently saponified and recrystallized from 50% aqueous MeOH to generate alcohol 15.8.
First, an activation−displacement process transformed alcohol 15.8 into bromide 15.9 via a mesylate intermediate. Alkyl bromide 15.9 was then treated with potassium phthalimide to incorporate the nitrogen center via an SN2-type displacement. Methylamine-mediated phthalimide deprotection and subsequent salt formation produced amine 15.11 as a hydrochloride salt in 69% yield over 3 steps. Finally, hydrochloride salt 15.11 was treated with aqueous sodium bicarbonate to generate a free amine, which was subjected to amide bond formation with 2-ethoxybenzoic acid 15.12 to deliver difamilast after recrystallization from aqueous EtOH.
PATENT
JP 2021059538
https://patentscope.wipo.int/search/en/detail.jsf?docId=JP322244172&_cid=P20-L1WXG6-04592-1
patcit 2 : International Publication No. 2014/034958 (Japanese Publication No. 2015-528433 )
patcit 3 : International Publication No. 2017/115780
Compound (5) (white powder) was prepared by the method described in Example 352 of Patent Document 1 (International Publication No. 2007/088383).
N−({2−[4−(difluoromethoxy)−3−isopropoxyphenyl]oxazol−4−yl}methyl)−2−ethoxybenzamide
: white powder.
1H NMR (400 MHz, CDCl3): δ = 8.56 (br s,
1H, NH), 8.23 (dd, J = 7.6 Hz, 1.6 Hz, 1H, ArH), 7.66 (s, 1H, ArH), 7.63 (d, J = 2.0 Hz, 1H, ArH), 7.58 (dd, J = 8.4 Hz, 2.0 Hz, 1H, ArH), 7.44−7.39 (m, 1H, ArH), 7.21 (d, J = 8.0 Hz, 1H, ArH), 7.08−7.04 (m, 1H, ArH), 6.94 (d, J = 8.0 Hz, 1H, ArH), 6.61 (t, J = 75.2 Hz, 1H, CHF 2), 4.68 (sept, J = 6.0 Hz, 1H, CH), 4.62
(d, J = 6.0 Hz, 2H, CH 2), 4.17 (q, J = 6.93, 2H, CH 2), 1.48 (t, J = 7.2 Hz, 3H,
CH 3), 1.39 (d, J = 5.6 Hz, 6H, 2CH 3).
Using the obtained B-type crystal as a seed crystal, it was examined to further prepare a B-type crystal. Specifically,
B-type crystals were prepared as follows according to the method described in Patent Document 3 (International Publication No. 2017/115780).
aqueous sodium hydroxide solution were added to the organic layer, the temperature was adjusted again to 40 to 50 ° C., the liquid was separated, and the organic layer was concentrated under reduced pressure. 50 mL of ethanol, 20 mL of water, 6 mL of a 25% aqueous sodium hydroxide solution, and 0.6 g of activated carbon were added to the concentrated residue, and the mixture was refluxed for 30 minutes. Activated carbon was removed by filtration, washed with 12 mL of ethanol, the filtrate was cooled, and 10 mg of B-type crystals (seed crystals) were added to precipitate crystals. Precipitated crystals were collected by filtration and dried at 60 ° C. to obtain 18.38 g (yield 88.18%) of crystals of compound (5).
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2007058338&_cid=P10-MLOK7P-25609-1
Example 352
Using the compound obtained in Example 347 and 2-bromopropane, white powdery N-[2-(4-difluoromethoxy-3-isopropoxyphenyl)oxazol-4-ylmethyl]-2-ethoxybenzamide was obtained following the procedure of Example 348.
1H-NMR (CDCl3) δ: 8.57 (1H, br s), 8.24 (1H, dd, J = 7.5, 1.8 Hz), 7.67 (1H, s), 7.65-7.57 (2H, m), 7.46-7.40 (1H, m), 7.26-7.21 (1H, m), 7.08 (1H, t, J = 7.5 Hz), 6.95 (1H, d, J = 8.4 Hz), 6.63 (1H, t, J = 75 Hz), 4.74-4.62 (3H, m), 4.19 (2H, q, J = 6.9 Hz), 1.49 (3H, t, J = 6.9 Hz), 1.40 (6H, d, J = 6.3 Hz)
PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2017115780
Compound (3) was produced in accordance with the following reaction scheme.
1H-NMR (CDCl 3) δ: 7.70 (2H,dd,J = 6.4 Hz,2.0 Hz),7.22 (1H,d,J = 9.2 Hz),6.66 (1H,t,J = 74.8 Hz),4.66(1H,sept,J = 6.0 Hz),1.39 (6H,d,J = 6.0 Hz).
Production Example 2: Production 2 of Compound (3)
Compound (3) was produced in accordance with the following reaction scheme.
Compound (7) was produced in accordance with the following reaction scheme.
Compound (11) was produced in accordance with the following reaction scheme.
20.00 g (66.8 mmol) of compound (7) and 17.28 g (134 mmol) of N,N-diisopropylethylamine were added to 300 ml of ethyl acetate, and the mixture was cooled. 11.48 g (100 mmol) of methanesulfonyl chloride was poured in and stirred at 10 to 30°C for 1 hour. 17.41 g (200 mmol) of lithium bromide was added thereto and reacted at 20 to 35°C for 1 hour. 100 ml of water was added to the reaction solution, and the mixture was partitioned, followed by concentration of the organic layer under reduced pressure. 300 ml of ethyl acetate was added to the concentrated residue to dissolve the residue, and the solution was again concentrated under reduced pressure. 200 ml of N,N-dimethylformamide and 17.33 g (93.6 mmol) of potassium phthalimide were added to the concentrated residue and reacted at 75 to 85°C for 1 hour. 200 ml of water was added to the reaction solution to precipitate crystals. The precipitated crystals were collected by filtration and dried at 80°C, thereby obtaining 25.90 g (yield: 90.5%) of compound (9) as a white powder.
15.00 g (35.0 mmol) of compound (9) was mixed with 30 ml of a 40% methylamine aqueous solution, 30 ml of methanol, and 75 ml of water, and reacted under reflux for 30 minutes. 150 ml of cyclopentyl methyl ether (CPME) and 15 ml of a 25% sodium hydroxide aqueous solution were added to the reaction solution, and the temperature was adjusted to 65 to 75°C, followed by partitioning. A mixture of 150 ml of water and 7.50 g of sodium chloride was added to the organic layer, and the temperature was adjusted to 65 to 75°C again, followed by partitioning. 3.75 ml of concentrated hydrochloric acid was added to the organic layer to precipitate crystals. The precipitated crystals were collected by filtration and dried at 60°C, thereby obtaining 11.95 g (yield: quant.) of compound (10) as a white powder.
13.30 g (39.7 mmol) of compound (10) was mixed with 3.83 g (37.8 mmol) of triethylamine and 108 ml of ethyl acetate, and stirred at 20 to 30°C for 1 hour. 9.78 g (58.9 mmol) of 2-ethoxybenzoic acid and 11.28 g (58.8 mmol) of 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (WSC) were added to the reaction solution, and reacted at 20 to 30°C for 1 hour. 54 ml of water and 5.4 ml of concentrated hydrochloric acid were added to the reaction solution, and the temperature was adjusted to 40 to 50°C, followed by partitioning. 54 ml of water and 5.4 ml of a 25% sodium hydroxide aqueous solution were added to the organic layer, and the temperature was adjusted to 40 to 50°C again. The mixture was partitioned, and the organic layer was concentrated under reduced pressure. 45 ml of ethanol, 18 ml of water, 5.4 ml of a 25% sodium hydroxide aqueous solution, and 0.54 g of activated carbon were added to the concentrated residue, and the mixture was refluxed for 30 minutes. The activated carbon was removed by filtration, and the filtrate was washed with 11 ml of ethanol. The filtrate was cooled, and a seed crystal was added thereto to precipitate crystals. The precipitated crystals were collected by filtration and dried at 35°C, thereby obtaining 12.88 g (72.6%) of compound (11) as a white powder.
PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019194211
*DIPEA: Diisopropylethylamine, CPME: Cyclopentyl methyl ether,
DMF: N,N-dimethylformamide, 2-EBA: 2-Ethoxybenzoic acid,
WSC: 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride
Analysis was conducted to further prepare the type B crystal using the obtained type B crystal as a seed crystal. More specifically, the type B crystal was prepared as follows, in accordance with the method disclosed in PTL 3 (WO2017/115780).
PATENT
WO2014034958A1
WO2007058338A2
WO2007058338A9
WO2007058338A3
US9181205B2
US2015239855A1
USRE46792E
US2020078340A1
US2017216260A1
US2019070151A1
US2009221586A1
US8637559B2
US2014100226A1
///////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
/////////////Difamilast, JAPAN 2021, APPROVALS 2021, ジファミラスト , MM 36, MM-36-Medimetriks-Pharmaceuticals, Moizerto, OPA-15406, OPA 15406, 地法米司特
O=C(NCC1=COC(C2=CC=C(OC(F)F)C(OC(C)C)=C2)=N1)C3=CC=CC=C3OCC
INTrmediate No.CAS No.DIFAM-001177429-27-5DIFAM-00293652-48-3DIFAM-0031574285-26-9DIFAM-00470-23-5DIFAM-0051574285-28-1DIFAM-0061574285-30-5DIFAM-0071574285-32-7DIFAM-0081574285-36-1DIFAM-0091574285-38-3DIFAM-010DIFAM-0111574285-40-7DIFAM-0121574285-43-0DIFAM-013134-11-2Difamilast937782-05-3
TOLDIMFOS SODIUM

TOLDIMFOS SODIUM
C9H12NNaO2P+ , 220.16
Toldimfos sodium
575-75-7
Sodium (4-(dimethylamino)-2-methylphenyl)phosphinate
UNII-6139240O1E
sodium;[4-(dimethylamino)-2-methylphenyl]-oxido-oxophosphanium
Sodium (4-(dimethylamino)-2-methylphenyl)phosphinate
sodium;[4-(dimethylamino)-2-methylphenyl]-oxido-oxophosphanium
Phosphinic acid, [4-(dimethylamino)-2-methylphenyl]-, sodium salt
Phosphinic acid, (4-(dimethylamino)-2-methylphenyl)-, sodium salt
Phosphinic acid, P-(4-(dimethylamino)-2-methylphenyl)-, sodium salt (1:1)
Toldimfos is an aromatic phosphorus compound which falls between phosphorous itself and phosphoric acid in the stages of oxidation. Toldimfos sodium is the sodium salt of 2- methyl-4-(dimethylamino)phenylphosphinic acid. It is used to treat and prevent diseases associated with parturition and peri-partum period, developmental and nutritional disorders in young animals, and bone growth disorders and tetany or paresis caused by calcium, magnesium, and phosphorus metabolism disorders. Toldimfos has been used as a human medicine since 1920. While it is no longer indicated for human use, it is used in horses, cattle, sheep, pigs, and goats, and administered by intravenous, intramuscular, or subcutaneous injection. No specific data on the pharmacodynamic action of toldimfos was submitted. The precise mode of action of toldimfos is unknown and it is questionable whether the effect of toldimfos is simply a matter of the substitution of deficient phosphorus. It appears more likely that the effect of toldimfos arises due to multiple stimulation of metabolism with the body.
Toldimfos sodium trihydrate
5787-63-3
Toldimfos [INN:BAN]
57808-64-7
Toldimfos Sodium
CAS Registry Number: 575-75-7
CAS Name: (4-Dimethylamino-o-tolyl)phosphonous acid sodium salt
Additional Names: sodium (4-dimethylamino-o-tolyl)phosphonate; p-dimethylamino-o-toluenephosphonous acid sodium salt
Trademarks: Foston (Hoechst); Tonofosfan (Hoechst)
Molecular Formula: C9H13NNaO2P, Molecular Weight: 221.17
Percent Composition: C 48.87%, H 5.92%, N 6.33%, Na 10.39%, O 14.47%, P 14.00%
Literature References: Prepd from N,N-dimethyl-m-toluidine and phosphorus trichloride: Benda, Schmidt, DE397813 (1924 to Cassella), Frdl.14, 1409.
Derivative Type: Trihydrate
CAS Registry Number: 5787-63-3
Properties: Scales, needles, or prisms from alc. Freely sol in cold water, hot alcohol.
Therap-Cat-Vet: Phosphorus source.
SYN

PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2011026571
PATENT
https://patents.google.com/patent/CN103830261A/en
China’s animal husbandry fast development is the important motivity that promotes China’s agricultural and rural economy development, improves farmers’ income.The disease relevant with Nutrition and Metabolism of serious harm animal health is in rising trend in recent years, the direct economic loss that raising poultry nutritive metabolic disease causes over ten billion to China’s animal husbandry every year, and indirect economic loss is difficult to estimate.
Due to the life-time service of chemicals, will cause some poultrys, poultry product drug residue is serious, this is harm humans healthy not only, also affecting the export of farm produce earns foreign exchange, therefore, tackle this problem, research and development are efficient, the new Nutrition and Metabolism medicine of low toxicity, wide spectrum will have huge market.
Toldimfos (Toldimfos Sodium) belongs to the nutritional supplementation medicine of phosphorus supplement, can be used as benzenephosphonic acid (Phenylphosphinicacid, BPA) succedaneum uses, can be used for treating the disease relevant with childbirth and perinatal stage of the food animals such as horse, cattle, pig, sheep, the diseases such as the bone lengthening obstacle being caused by calcium, magnesium, phosphorus metabolism obstacle.
Toldimfos has higher water solublity, mainly excretes through urine rapidly with the former medicine form of not metabolism in animal body, and the half-life is short, can in tissue, not accumulate.
Toldimfos is developed by German Hoechst company, the symptom such as since nineteen twenty, once physical weakness, chronic stress, depression, mental muscle power postoperative for human treatment, that catch was overtired.Now be not used in the mankind, be mainly used in animal.Its commodity are called onofosfan.
In sum, toldimfos, as a kind of nutritional supplementation medicine of new and effective noresidue, has wide market prospect in China.The development of this product will be made outstanding contributions to the sound development of China’s animal husbandry, remarkable economic and social benefits with application.
PATENT
https://patents.google.com/patent/WO2004003198A1/ja
///////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
//////////TOLDIMFOS SODIUM, HOECHST, Foston, Tonofosfan,
O.O.O.[Na+].CN(C)c1ccc(c(C)c1)P(=O)[O-]
NEW DRUG APPROVALS
ONE TIME
$10.00

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}